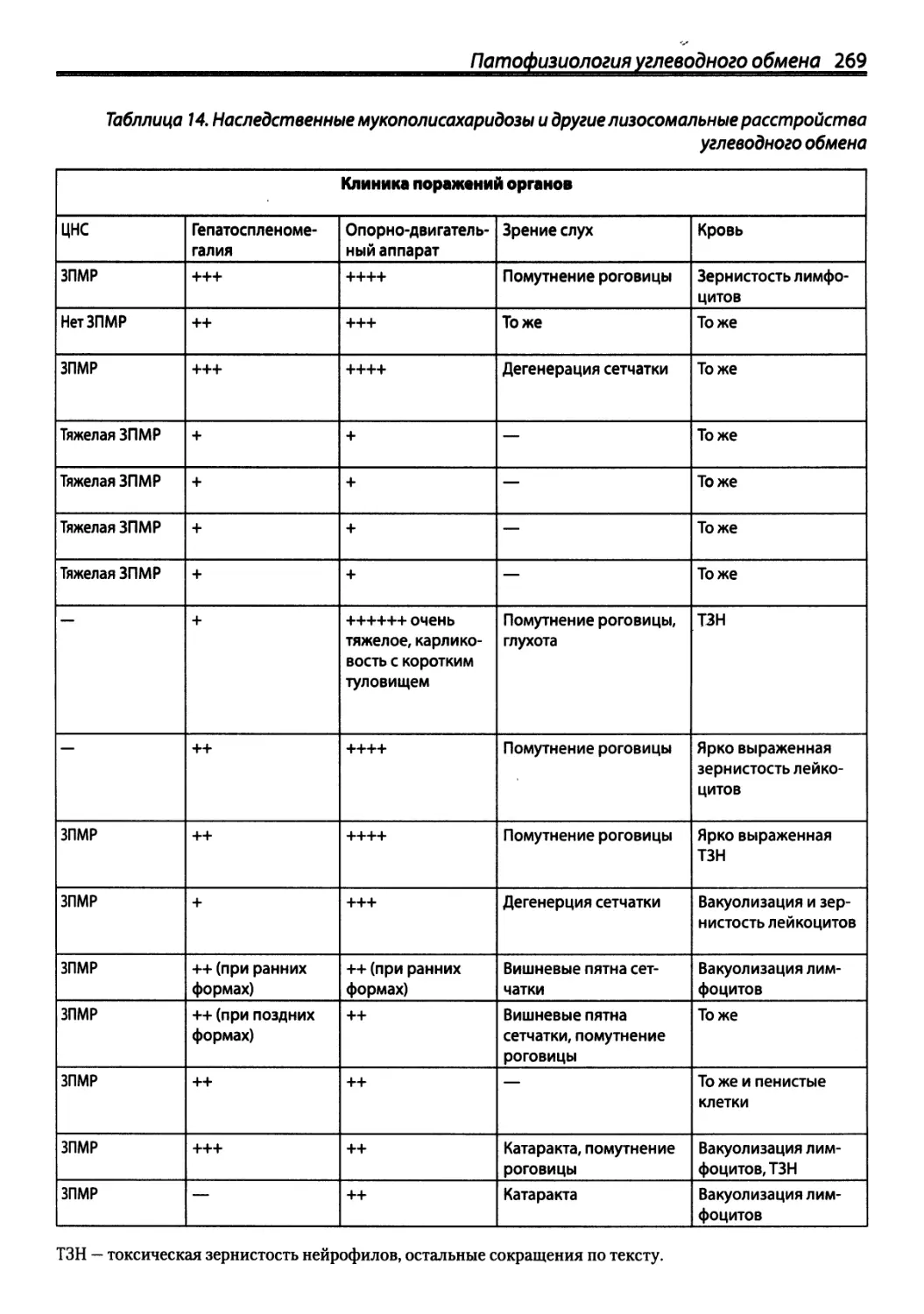
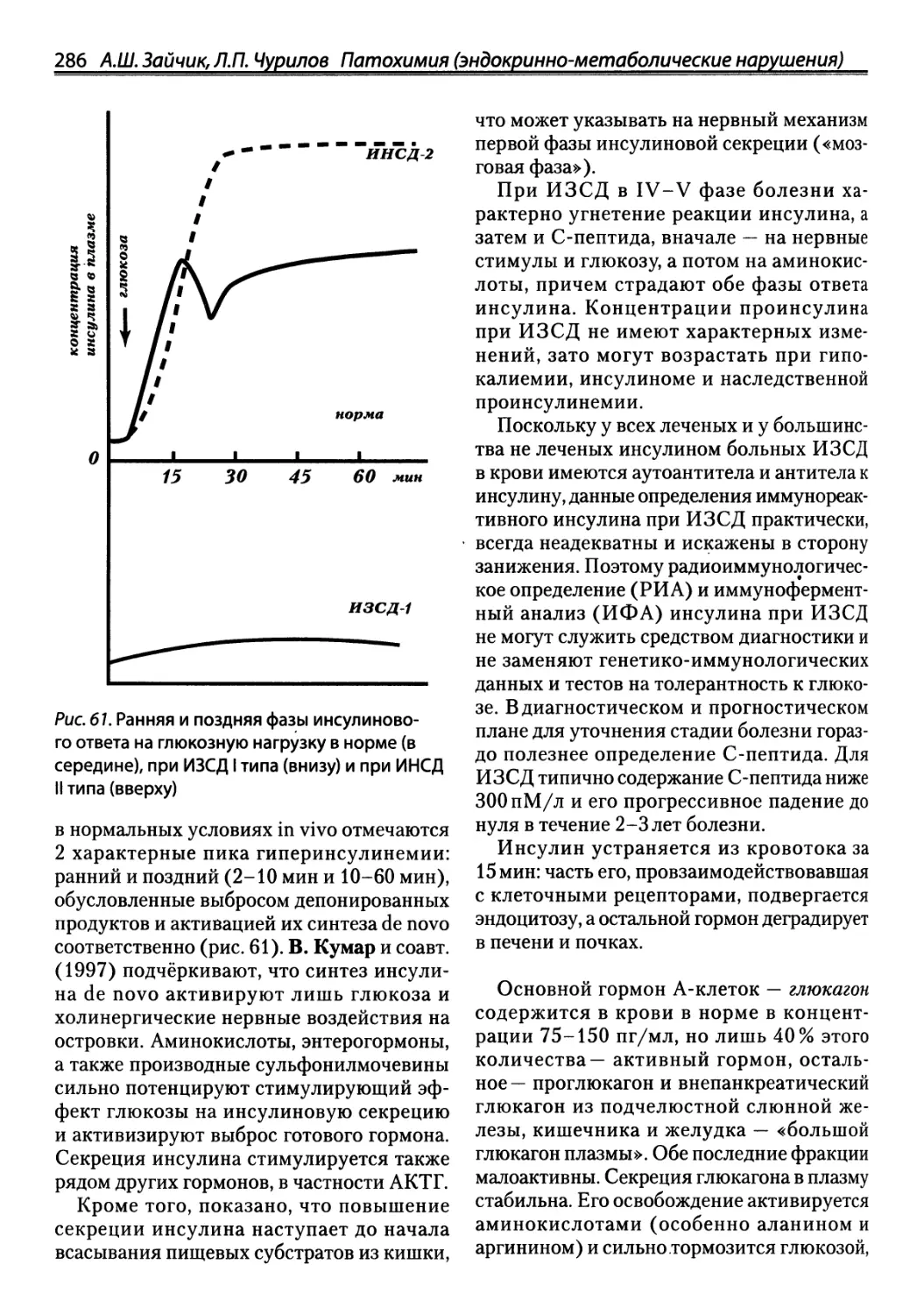
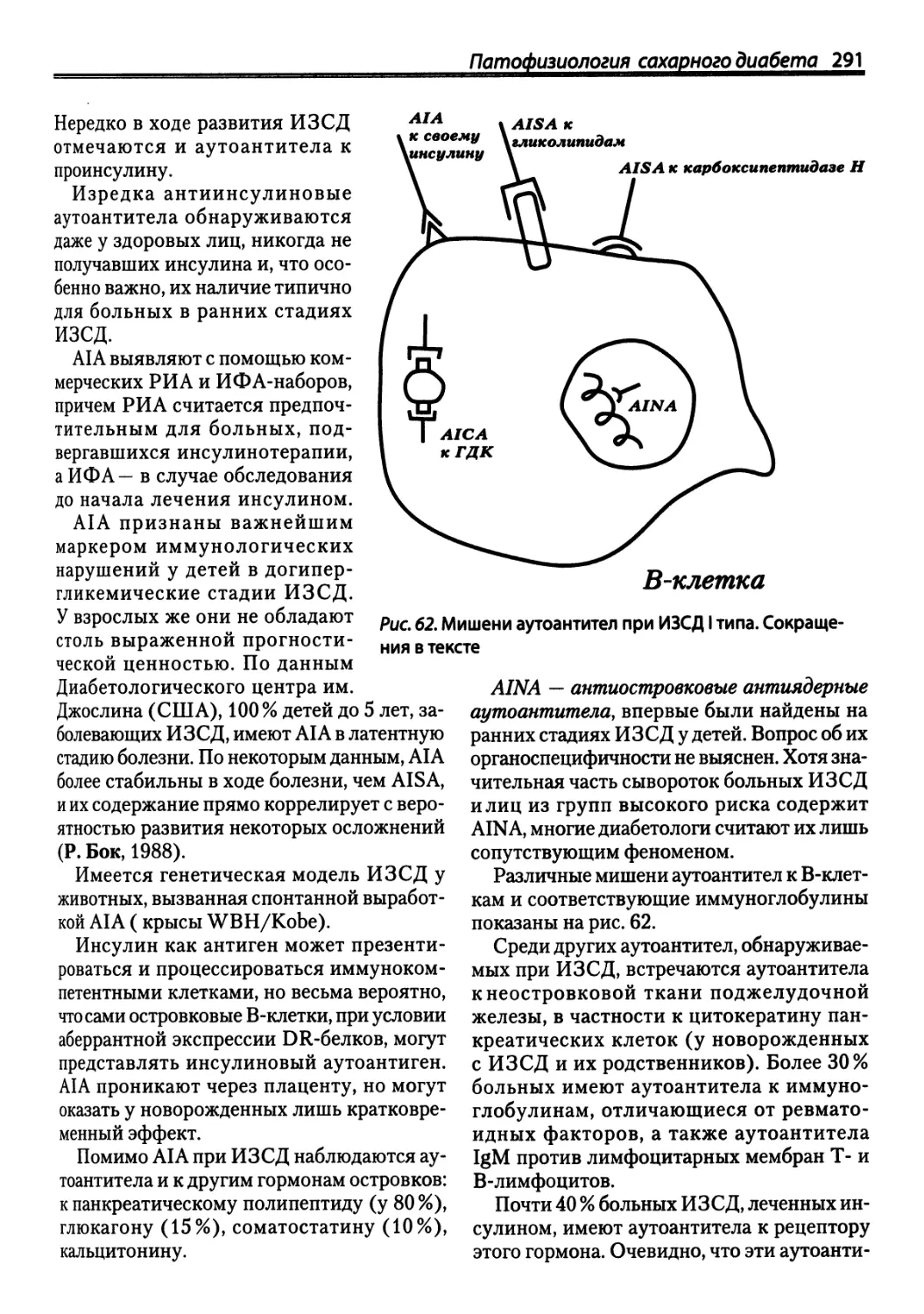
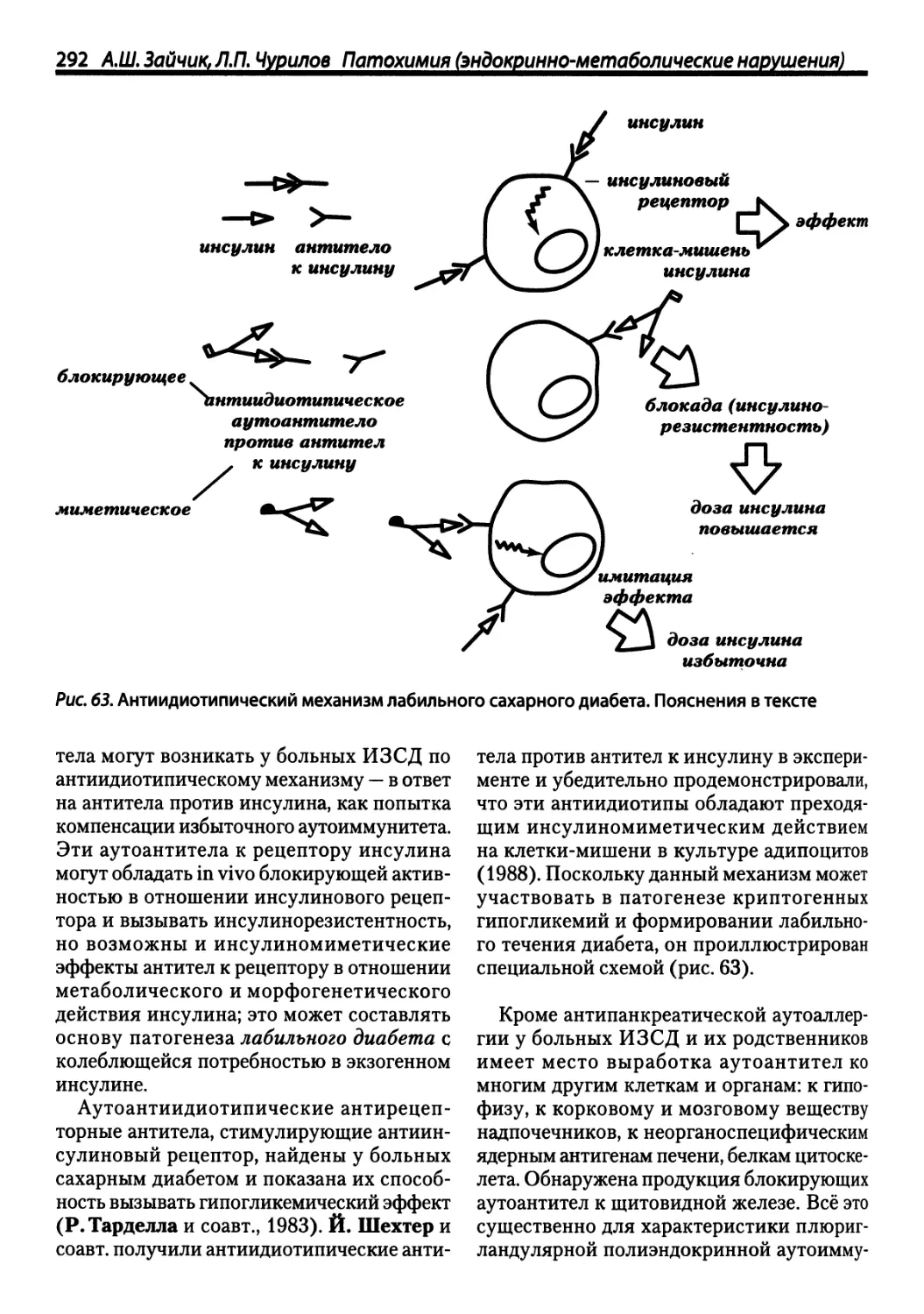
/













































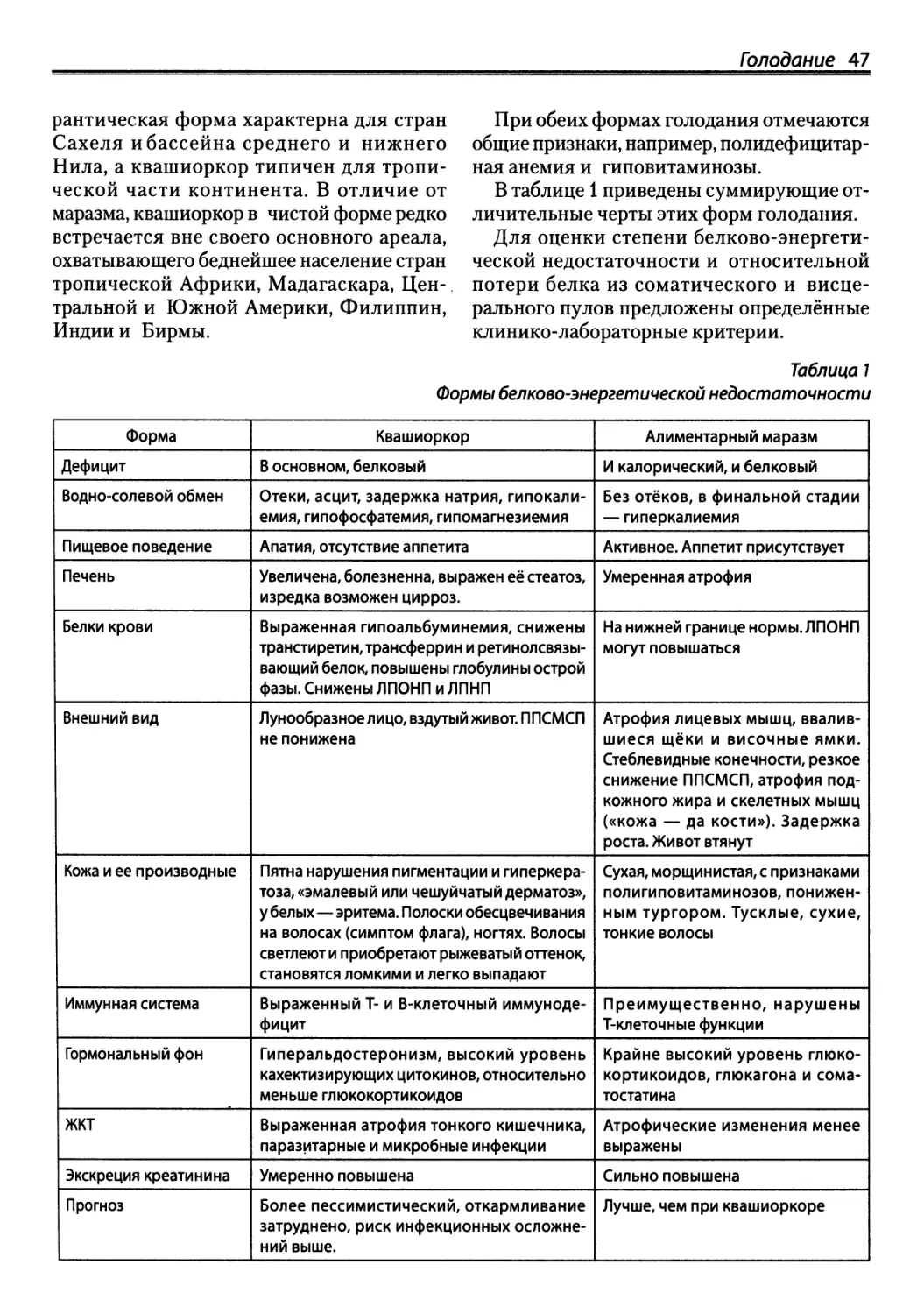
















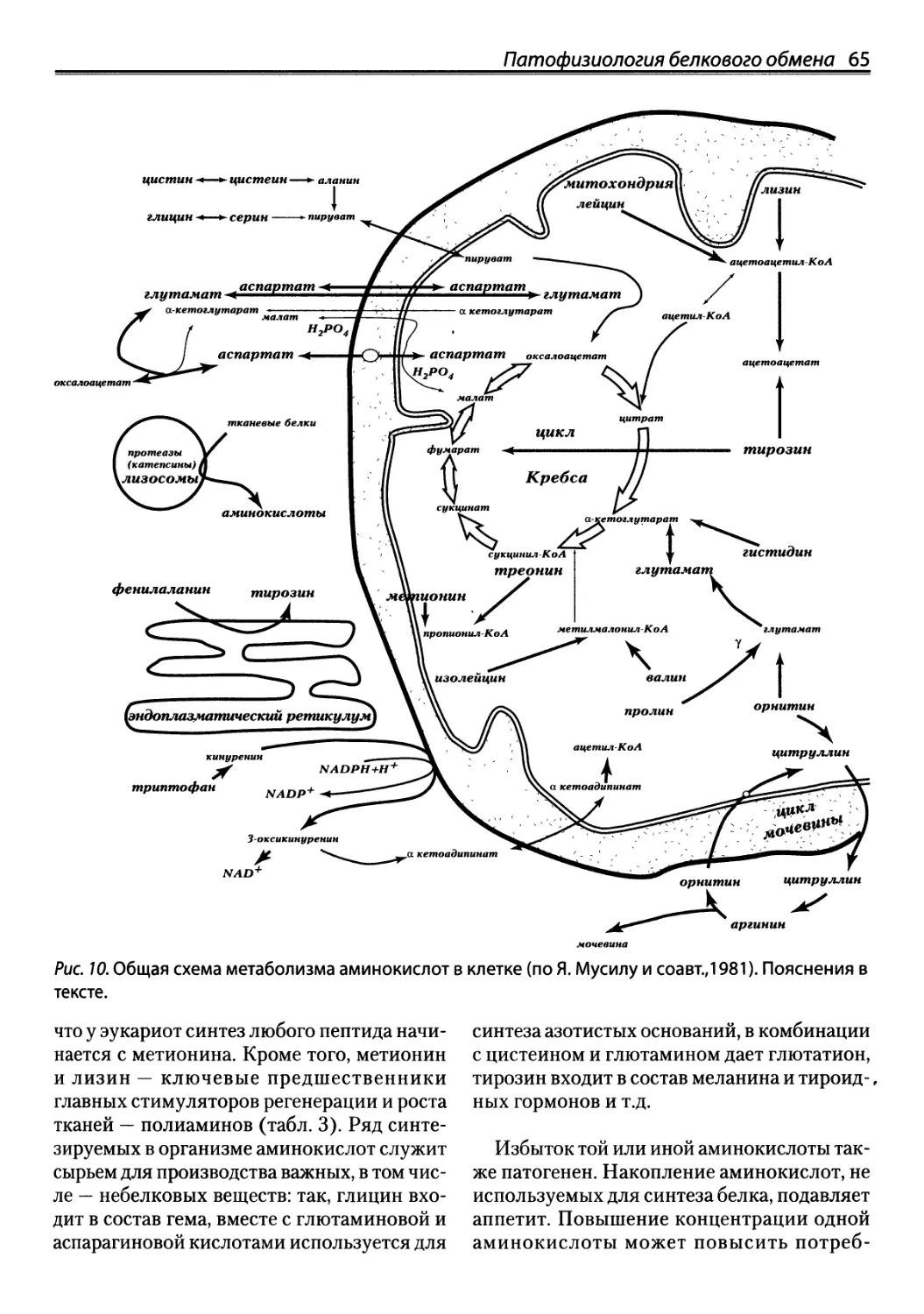












































































































































































































































































































































































































































































































































































































































































Автор: Зайчик А.Ш. Чурилов Л.П.
Теги: здравоохранение медицинские науки медицина эндокринология
ISBN: 978-5-93979-032-1
Год: 2007




Текст
Блестящему ученому
и прекрасному человеку—
нашему учителю Л. Р. Перельману
посвящаем эту книгу
А.Ш. Зайчик, Л.П. Чурилов
ПАТОЛОГИЧЕСКАЯ
ФИЗИОЛОГИЯ
Том 2
ПАТОХИМИЯ
(эндокринно-метаболические нарушения)
Рекомендовано Главным управлением учебных заведений
Министерства здравоохранения РФ
в качестве учебного пособия для студентов медицинских вузов
Учебник одобрен Межвузовским редакционно-издательским экспертным
советом по медицинской литературе Санкт-Петербурга и допущен для
преподавания патофизиологии и биохимии в медицинских высших учебных заведениях
Издание 3-е (дополненное и исправленное)
«ЭЛБИ-СПб»
Санкт-Петербург
2007
ББК55.4
319
Зайчик А. Ш., Чурилов Л. П. Патохимия (эндокринно-метаболические нарушения)
Учебник для студентов медицинских вузов. Изд. 3-е, дополненное и исправленное. — СПб.:
ЭЛБИ-СПб, 2007.-768 с, илл.
ISBN 978-5-93979-032-1
Учебник принадлежит к трехтомнику «Патофизиология» и представляет собой курс патофизиологии
эндокринной регуляции и метаболизма. Отдельные главы посвящены патофизиологии энергетического,
белкового, нуклеинового, углеводного, липидного, водно-минерального обменов, метаболизма витаминов и
микроэлементов, механизмам голодания и анабиоза. Детально описываются патофизиологические основы
эндокринологии включая как общую патофизиологию эндокринной системы, так и механизмы конкретных
эндокринопатий.
В ходе изложения анализируется широкий круг наследственных и приобретённых метаболических
заболеваний, как распространенных, так и редких. Представлены отдельные разделы, посвященные атеросклерозу,
ожирению, подагре, расстройствам питания, тезаурисмозам, метаболическому синдрому, остеопорозу и т.д.
Книга включает специальную главу о патофизиологии сахарного диабета.
Изложение носит междисциплинарный характер. Обильно представлен материал, позволяющий
читателю повторить аспекты биохимии, существенные для усвоения курса патофизиологии обмена веществ.
Широко затрагиваются смежные проблемы гигиены, диететики, витаминологии, иммунологии, экологии
человека и медицинской географии. Описание метаболических и эндокринных расстройств отвечает целям
и задачам клинической патофизиологии и перекликается с соответствующими разделами пропедевтики
внутренних болезней. Учебник содержит не только сводку классических фактов и теорий, но и новейшие
патофизиологические данные по всем затронутым аспектам. Впервые в учебном пособии подобного типа
дается детальная характеристика проблем, связанных с пищевыми антиоксидантами.
Учебник написан в соответствии с требованиями программы по патофизиологии в медвузах, но не
ограничен этими требованиями. Он рекомендован для преподавания как патофизиологии, так и биохимии
студентам-медикам и может представлять большой интерес для последипломного образования врачей и
системы усовершенствования преподавателей медвузов. Книга не только полностью освещает вопросы,
преподаваемые на лечебных факультетах, но и включает дополнительно обширный материал, касающийся
педиатрических и профилактических аспектов данного раздела патофизиологии.
Учебник иллюстрируют 49 таблиц и более 130 рисунков, приложение содержит справочные
материалы по нормальным биохимическим и эндокринологическим показателям, используемым в клинической
практике и по истории биохимии, имеются обширная библиография (более 800 источников) и краткий
предметный указатель.
Авторы выражают благодарность рецензенту данной книги зав. кафедрой физиологии и патологии
Санкт-Петербургской химико-фармацевтической академии проф. В.В. Давыдову.
ББК55.4
© ООО «ЭЛБИ-СПб», 2007 3 19
ISBN 978-5-93979-032-1 © А.Ш. Зайчик, Л.П. Чурилов, 2007
Оригинал-макет Н.Г. Философов
ООО «Медкнига "ЭЛБИ-СПб"».
194100, СПб., ул. Новолитовская, д. 5, литер А.
(812) 322-92-57; 322-92-58.
Подписано в печать 03.07.07. Формат 70X100 1/16.
Печать офсетная. Гарнитура Петербург. Объем 48 п. л. Тираж 2000 экз.
Заказ №1621.
Отпечатано по технологии CtP в ОАО «Печатный двор» им. А. М. Горького.
197110, Санкт-Петербург, Чкаловский пр., 15.
ПАТОХИМИЯ
(эндокринно-метаболические нарушения)
«Жизнь — это постоянная борьба против
тенденции к возрастанию энтропии. Синтез больших,
богатых информацией макромолекул, образование
клеток с их сложной структурой, развитие
организации — всё это мощные антиэнтропийные факторы.
Но поскольку, согласно второму закону
термодинамики, справедливому для всех явлений природы,
избежать возрастания энтропии нельзя, живые
организмы избрали наименьшее зло—они существуют в
стационарных состояниях, для которых характерна
минимальная скорость возрастания энтропии»
Арон Качальский
«Неравновесная термодинамика в биофизике»
«Мы пленники звёздных туманов —
В нас тот же горит водород...»
[Илья Зыков! «Мы»
Предисловие к 3-му изданию
Студент медицинского факультета
изучает такое великое множество разных
«химий», что студентам-химикам остаётся
лишь удивляться. Здесь и неорганическая
химия, и биоорганическая, физическая
и коллоидная, аналитическая,
биологическая и элементы судебной. В начале века
студентам-медикам преподавали даже
кристаллографию. Мы уже не говорим о
токсикологии и фармакологии, которые стоят
очень близко к химической проблематике,
об иммунохимии, без знакомства с которой
неполноценен курс иммунологии.
Спрашивается, зачем же нужна еще
какая-то «патохимия»?
Р^зве условия патологии отменяют в
организме действие химических принципов,
и, выражаясь словами Клода Бернара,
дом горит не по тем же самым химическим
и физическим законам, по которым он
строился?
Выбирая заглавие для второго тома
руководства по патофизиологии (см. первый
том серии — «Общая патофизиология»,
СПб.: ЭЛБИ-СПб. - 2005. - 656 с),
авторы руководствовались советом
известного американского биохимика Мюррея
Саффрана. В бытность деканом
факультета в Университете Огайо М. Саффран
написал проблемную статью о том, как
преподавать биохимию студентам-медикам
и высказал убеждение, что наиболее важна
для медиков не формально-статическая
сторона биохимии, а биохимическая логика
патологических процессов.
Данное издание не подменяет и не
дублирует курс биохимии и не является
пособием по клинической биохимии, оставляя
эту важнейшую задачу курсу лабораторной
диагностики.
Предмет книги — химические
механизмы патологических процессов. В
зарубежной литературе существует опыт
создания подобных руководств (см., например,
«Биохимия патологических процессов»
Я.Мусила, «Очерки по патологической
биохимии» Р.Хашена и Д.М. Шейха
и «Молекулярные основы патологии»
А. Хорста). В традиции российской высшей
медицинской школы до середины тридцатых
Предисловие 5
годов входило преподавание отдельного от
биохимии университетского курса
физиологической химии. За рубежом курс «Болезни
обмена веществ и питания» также
представляет собой самостоятельное по отношению
как к биохимии, так и к патофизиологии,
звено учебного плана.
Данный учебник освещает проблемы
патологической физиологии метаболизма,
включая типовые нарушения обмена белков,
нуклеиновых кислот, липидов, углеводов,
воды и минеральных веществ. Отдельные
разделы посвящаются патофизиологии
энергетического обмена, витаминологии,
патофизиологическим аспектам обмена
микроэлементов.
Поскольку основные регуляторные
механизмы обмена веществ связаны с
гормонами, обсуждение патофизиологии
метаболизма сопровождается детализированным
очерком патофизиологии эндокринной системы.
Во всех разделах руководства
анализируются как типовые синдромы, обусловленные
нарушениями обмена веществ (например,
гипергликемия, дегидратация, дислипо-
протеинемии, диспротеинозы и т.д.), так и
этиология и патогенез отдельных, наиболее
клинически и дидактически значимых
приобретённых и наследственных
метаболических заболеваний.
Авторы не сочли возможным
возвращаться в данном томе к патохимии некробиоза,
гипоксии, свободнорадикальных процессов,
апоптоза, воспаления, лихорадки, ответа
острой фазы и стресса, к общим вопросам
механизмов наследственных заболеваний.
Все эти аспекты составляют основное
содержание учебника «Общая
патофизиология». Там же читатель найдет и некоторые
общие сведения по иммунохимии, а также
по патофизиологии гипоталамо-гипофи-
зарно-надпочечниковой системы, которые
в настоящем издании лишь дополнены
частными аспектами. Вопросы патохимии
онкологических и гематологических
заболеваний рассмотрены в т. III. Клинико-пато-
физиологические аспекты метаболических
расстройств и основы экспериментальных
методов их моделирования и изучения
изложены в практикуме «Введение в
экспериментальную патологию» (в
дальнейшем — Практикум). Перекрестные ссылки
на тт. I и III данного учебника даются по их
последним изданиям 2005 г.
В последние годы широко обсуждается
вопрос об интеграции преподавания
патофизиологии и патологической анатомии.
Патология как наука и учебный предмет не
может быть сведена к патоморфологии или
даже представлена как двуединство
патофизиологии и патологической анатомии.
Полноправной составляющей современной
патологии является бурно
прогрессирующая патохимия. Ряд зарубежных авторов
современную патофизиологию
рассматривают еще шире — как патобиологию (3.
Ковач, 2005). Основоположником патохимии
в отечественной медицине был Виктор
Васильевич Пашутин (1845-1901),
изучивший химические изменения в клетках
при гипоксии и голодании, предсказавший
существование витаминов и создавший
первый в мире прибор для экспериментального
исследования энергетического метаболизма.
Его ученик Петр Михайлович Альбицкий
(1853-1922) развил патохимическое
направление. В замечательной работе «О
значении продуктов обмена веществ» (1919)
он предвосхитил современное понимание
сигнально-регуляторной роли
метаболитов, обосновал опередившую свое время
идею об отсутствии бесполезных
«шлаков» в метаболической системе организма.
Другой питомец Пашутина — Александр
Васильевич Репрев (1853-1930) — может
считаться основоположником изучения
патофизиологии эндокринной регуляции.
Классик отечественной медицины Ефим
Семенович Лондон (1868/69-1939)
основал как кафедру биохимии (в ЛГУ), так
и кафедру патофизиологии (в ЛПМИ) и
заложил традицию приоритетного
изучения вопросов эндокринно-метаболической
патологии, которой мы следовали, создавая
данный учебник.
Для врача организм — «черный ящик»
(И.Уилкинсон,1998). Научная медицина
6 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
шаг за шагом раскрывала, что и как
происходит внутри, откуда берутся энергия и
продукты выделения. История патохимии
восходит, вероятно, еще к Гиппократу с
его концепцией дискразий как основы
болезней. Но медицина не сразу осознала, что
« Человек — группа атомов, вполне зависимая
в своих движениях от всех сил, делений и
изменений целого...» (Ф. Ницше). Ведь живые
системы сложны и обладают своей
природной информатикой. В них огромную роль
играют не только элементы, но и связи
между ними. Интуитивно мыслители ощущали,
что кроме физики и химии в живых телах
есть что-то еще, но понятийного аппарата
кибернетики и информатики, позволяющего
материалистически описать
информационно-вещественный дуализм живых клеток
(см. т. I, гл. 2), не существовало, а мысль
Г. Гегеля «Живое не дает причине дойти до
её действия» долгое время истолковывалась
медиками виталистически. Со времён
трактата «Ней-цзин» (V—III век до нашей эры),
вплоть до Ганса Дриша (1867-1941),
считалось, что жизнь создается прибавлением к
физико-химическим процессам отдельного
от вещества упорядочивающего, целепо-
лагающего начала (энтелехии, жизненной
силы, энергии цы, бессмертной души — в
зависимости от авторской
культурно-этнической традиции). Знаковым событием
в истории патохимии послужил первый в
истории лабораторный синтез природного
метаболита из неорганических
компонентов. Ф. Вёлер перебросил этим мост между
организмом и лабораторией и взволнованно
сообщил: «Ямогу производить мочевину без
помощи почек и животных\» (1831). Именно
он создал теорию радикалов, углубившую
понимание животной химии, вместе с
Ю. Либихом, автором первого учебника
патохимии «Животная или органическая
химия в применении к физиологии и патологии»
(1841). Параллельно работами К. Бернара
(1853) были установлены закономерности
переваривания и всасывания липидов,
роль ЦНС и печени в поддержании уровня
глюкозы в крови, что привело к идее о
постоянстве внутренней химической среды как
основе здоровья (см. гл. 6-9). Пионер
клинической биохимии — Анри Бенс-Джонс
(1813-1873) — установил связь между
рядом нарушений состава биологических
жидкостей и болезнями. Развитие
медицинской химии в дальнейшем всё ускорялось
благодаря новым методам, что мы
наблюдаем и поныне. Современному врачу доступно
гораздо больше информации о химических
параметрах организма. Многие методы
автоматизировались, ультрамикромалые
количества тех или иных веществ
определяются поточно, без больших временных
затрат или уникального личного мастерства.
Но при интеграции медицинского знания,
создании целостного представления о
болезни и больном машина не может заменить
профессиональный интеллект врача. Все
чаще мы сталкиваемся с тем, что уровень
интеграции сведений о больном отстает
от достигнутой точности и
производительности анализаторов. Часто анализы
оценивают формально, вне связи с
индивидуализированными физикальными и
анамнестическими данными. Чем сильнее
методически становится клиническая
биохимия, тем больше значение патохимии
как части патофизиологии: только владея
ее принципами, врач может мыслить
целостно. Переиздание вызвано не только
востребованностью книги у читателей. За
последние годы накоплен опыт
преподавания студентам медицинского факультета
СПбГУ специального предмета «Введение
в эндокринно-обменную патологию», а
также последипломного преподавания
аспектов патохимии в Институте
эндокринологии СПбМАПО. Авторы благодарят
коллег и всех читателей книги за
высказанные пожелания и замечания, которые,
по возможности, были учтены при
переиздании. Посылайте свои отзывы,
пожелания и комментарии Л.П. Чурилову на
сайт Интернет-группы: http://tech.groups.
yahoo.com/group/pathophysio/.
Постараемся их учесть при подготовке
последующих изданий.
Глава 1
ОБЩИЕ АСПЕКТЫ
ПАТОФИЗИОЛОГИИ ОБМЕНА
ВЕЩЕСТВ
И РЕГУЛЯЦИИ МЕТАБОЛИЗМА
« Человек может жить, работать и сохранять здоровье,
как на большем, так и на меньшем уровне азота или энергии.
Важнейший вопрос заключается в том, какой уровень наиболее
благоприятен»
У. Этуотер, Ф. Бенедикт (1902)
Обмен веществ — сложная система
химических реакций, связанных между
собой через пластические компоненты,
энергетическое обеспечение и общие регуляторы.
Целями этих реакций являются извлечение
энергии, получение структурных блоков
и синтез полимеров, строение которых
соответствует индивидуальной генетической
программе организма, создание и
инактивация сигнальных молекул и разрушение
полимерных соединений, сконструированных
согласно чужим программам.
Биохимическая схема обмена веществ
включает цепи, каскады и циклы
химических превращений, связанные
метаболическими путями.
Как противоречивая равновесная
система разнонаправленных процессов,
обмен веществ не может быть весь изменен
в каком-то одном направлении, поэтому
выражения типа «обмен веществ усилился»,
«болезнь привела к снижению обмена
веществ» хотя и присутствуют в
профессиональном жаргоне медицинских работников,
являются неточными и неверными.
При любой форме патологии различные
химические реакции изменяются
разнонаправленно: так при инсулинзависимом
сахарном диабете усиливаются глюконеоге-
нез и распад гликогена, но тормозятся цикл
Кребса и пентозный путь. При фенилкето-
нурии тормозится образование меланина,
но усиливается продукция фенилэтиламина
и т.д.
Для регуляции обмена веществ эволю-
ционно сформировались различные
механизмы, влияющие на инструменты
метаболизма — то есть на каталитическую
активность энзимов и аффинитет
распознающих белков.
Эти формы контроля основаны на
химической сигнализации субстратами, ионами,
гормонами, нейротрансмиттерами, аутакои-
дами и антителами. Элементарные способы
химического контроля метаболизма
предусматривают внутреннюю и наружную ау-
токринную, юкстакринную и панокринную
регуляцию. У многоклеточных на этой
основе формируются паракринный и
специализированный паракринный тип хи-
8 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
мической сигнализации (см. т. I данного
руководства, с. 62). Их дальнейшее развитие
приводит к появлению эндокринных,
нейромедиаторных и нейроэндокринных
регуляторов, взаимодействующих между
собой. Регуляторные сигналы и антисигналы
образуют информационные сети, такие как
идиотип-антиидиотипическая сеть
иммунологических взаимодействий.
При осуществлении сигнальной
регуляции метаболизма активация и
инактивация ферментов может происходить
на посттрансляционном уровне, причем
аллостерическими регуляторами выступают
разные химические сигналы: ионы, гормоны,
аутакоиды и сами субстраты. Большое ре-
гуляторное значение может иметь адресное
закрепление ферментов в тех или иных
отсеках клетки и на соответствующих
биомембранах. Известную роль играет
возможность изменения с помощью гормонов
проницаемости клеточных мембран, что
позволяет варьировать условия протекания
метаболических процессов и доступность
метаболитов в различных частях клетки.
Играя первую скрипку в
метаболической регуляции одноклеточных, подобные
механизмы не исчезают, а сохраняют свою
актуальность и у высших организмов. Так,
например, именно аллостерическим
посттрансляционным путём кортикостероиды
активируют у человека триптофаноксиге-
назу.
Регуляторные сигнальные химические
воздействия могут непосредственно или
через посредников обращаться и к геному
клеток, изменяя репертуар и скорость
транскрипции генов. При этом происходит
комплементарное взаимодействие
химического регулятора с элементами
генетического аппарата, воспринимающими и
расшифровывающими сигнал. Сигнал способен
вызвать изменение транскрипции матриц,
обеспечивающих биосинтез ферментов или
рецепторов, которые используются в ходе
метаболического процесса. Кроме того,
можгут модулироваться скорость и
характер посттранскрипционной обработки
матриц и судьба долгоживущих РНК.
Наконец, химические биорегуляторы
способны вмешиваться и в процесс
трансляции, влияя на его инициацию и
эффективность.
Регуляторные механизмы обмена веществ
таковы, что при обеспечении поддержания
динамической оптимальности внутренней
среды (гомеореза) они могут минимальными
информационными воздействиями
обеспечивать осуществление целого ансамбля
запрограммированных
материально-энергетических сдвигов. Это позволяет метаболизму
быть гибким и достигать технологических
целей в разных условиях и при различном
сырье, при максимуме кпд и минимуме
побочных продуктов.
Интегральным результатом этого
является удивительное свойство организма,
сформулированное Гераклитом так:
«Человек подобен фонтану. Всё та же форма — но
всегда новая вода».
Регуляторные стереотипы метаболизма
сложились эволюционно. В связи с
быстрыми изменениями характера питания и образа
жизни современного человека
несоответствие между данными стереотипами и
требованиями, предъявляемыми к метаболизму,
способствует развитию эндокринно-мета-
болических заболеваний, превратившихся
в актуальную проблему медицины XXI
века. Их этиологии и патогенезу и посвящен
данный учебник.
В самом начале знакомства с
патофизиологией обмена веществ, уже в следующем
разделе книги, мы рассмотрим наиболее
общие показатели метаболизма, которые
характеризуют обмен веществ в целом
и претерпевают типовые изменения при
самых разных формах патологии.
Глава 2
энергетический метаболизм
и его нарушения
«Жизнь - слабый огонь, горящий без пламени»
Ж. Фернель
«О естественных составляющих медицины» (1542)
• Основной обмен: Исторические аспекты
• Термодинамические и биохимические основы биоэнергетики
• Условия измерения основного обмена
• Методы определения основного обмена и их принципы. Калориметрия
• Калорический эквивалент кислорода
• Дыхательный коэффициент и коэффициент Рубнера, их изменения при патологии
ОСНОВНОЙ ОБМЕН:
ИСТОРИЧЕСКИЕ АСПЕКТЫ
Поскольку все компоненты обмена веществ
сопровождаются тепловыми эффектами
той или иной направленности, наиболее
общие характеристики уровня обмена
будут энергетически значимыми.
Биотермодинамические показатели
функционирования, здорового и больного организма
издавна привлекали внимание патологов,
поскольку естественные науки
традиционно рассматривали теплоту как
производную движения молекул и трактовали
тепловые характеристики самых разных
процессов как наиболее фундаментальные
и интегративные. Еще Гиппократ писал
об избыточной теплоте, зарождающейся
в теле при болезни и сжигающей
болезнетворные начала.
Главная общая энергетическая
характеристика жизнедеятельности организма —
основной обмен (в зарубежной
литературе — BMR — basal metabolic rate или чаще
RMR — resting metabolic rate — уровень
метаболизма в покое).
Подчеркнем, что основной обмен — не
какой-то отдельный вид обмена веществ
наряду с липидным, белковым, нуклеиновым,
углеводным и водно-солевым. Это всего
лишь лабораторный показатель,
характеризующий сумму энерготрат организма
в определенных стандартных условиях,
приближенных к наиболее экономичному
режиму жизнедеятельности.
Данное понятие введено в медицину
в прошлом веке А. Магнус-Леви (1895)
в результате обстоятельных исследований
потребления кислорода и энергозатрат при
болезнях щитовидной железы. Этому более
двух столетий предшествовали этапы
эмпирического изучения «животной теплоты».
Почти сразу вслед за открытием У. Пристли
(1774) кислорода А. Лавуазье (1777)
постулировал, что источником энергии в
организме являются, исключительно, процессы
кислородного окисления пищевых веществ.
Он же отметил параллелизм изменений
потребления кислорода и теплопродукции
10 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
после приёма пищи. Особенно важным
наблюдением А.Лавуазье и А. Сегена
(1789-1790) было отсутствие зависимости
между интенсивностью доставки
кислорода к тканям (или составом газовой
смеси) и теплопродукцией. Создавалось
впечатление, что наибольшую роль для
определения скорости производства тепла
играют внутренние параметры организма:
«Вто время как горение мёртвого
органического вещества протекает ceteris paribus
тем живее, чем чище воздух, в котором оно
происходит, количество витального воздуха,
поглощаемого животным, помимо очень
маленьких колебаний, всегда одно и то же,
дышат ли они в чистом воздухе или в смеси
его с большим или меньшим количеством
азота». Авторы предполагали, что горение
субстратов и тепловыделение происходят,
в основном, прямо в лёгких. Но, вопреки
этому, ещё в конце XVI-ro века А. Борелли,
измерив температуру крови в различных
внутренних органах загнанного им на охоте
оленя, доказал, что тепло образуется
повсюду, хотя, не обладая знаниями об окислении,
он полагал его источником механическое
трение.
Ю.Либих (1803-1873) в 40-х годах XIX
века предложил классификацию
питательных веществ и предположил, что обмен
веществ регулируется процессом дыхания.
В том же столетии основоположники
научной диететики К. Фойт (1831-1908),
У.Этуотер (1844-1907) и Ф. Бенедикт
(1841-1902) ставят вопрос об оптимальном
для здоровья должном потреблении энергии
и основных пищевых веществ человеком
и определяют связь антропометрических
и энергетических показателей организма.
Становится ясно, что не дыхание управляет
метаболизмом, а наоборот — обмен веществ
определяет потребность в кислороде.
В1881-1886гг.В.В.Пашутини П.М.Аль-
бицкий конструируют первый в мире
прямой калориметр для непосредственного
определения изменений теплосодержания
в организме человека и животных.
Сопоставление данных прямых измерений
и непрямых расчётов теплообразования
по потреблению кислорода привело
развивавшего идеи Лавуазье М. Рубнера
(1854-1932) к принципу
пропорциональной зависимости потребления кислорода
и теплопродукции (1894). В дальнейшем
Э. Бюхнером (1907) и О. Мейергофом
(1923) было установлено существование
анаэробных путей энергетического
метаболизма (гликолиза и пентозного пути),
и стало ясно, что зависимость потребления
кислорода и теплопродукции носит более
сложный характер, а энергопроизводство,
в большей мере, определяется характером
используемых субстратов.
ТЕРМОДИНАМИЧЕСКИЕ
И БИОХИМИЧЕСКИЕ
ОСНОВЫ БИОЭНЕРГЕТИКИ
Для оценки и истолкования результатов
биотермодинамических измерений
необходимо использовать понятия изменения
энтальпии (АН), изменения энтропии (AS),
а также изменения свободной энергии
Гиббса (AG). Подробный теоретический
анализ этих термодинамических величин
содержится в ряде руководств (см.,
например, Э.Кальве, У. Прат, 1963), и тем
не менее мы считаем своим долгом для
удобства читателей перечислить
основные термодинамические характеристики,
использующиеся при анализе результатов
биоэнергетических исследований.
Изменение энтальпии (АН) определяется
как:
AE+PdV+VdP,
где АЕ — изменение полной внутренней
энергии системы, dV и dP — приращение
соответственно объема системы при
постоянном давлении и давления при
постоянном объеме.
Объем камеры для измерений основного
обмена и давление внутри нее постоянны,
поэтому АН=АЕ. При всяком
самопроизвольном процессе происходит перенос энергии
в системе таким образом, что ее способность
Энергетический метаболизм и его нарушения 11
к дальнейшему превращению (совершению
работы) стремится к минимуму, т.е.
система стремится к состоянию равновесия.
Мерой приближения системы к
равновесному состоянию служит энтропия — S. Это
совокупность всех тепловых потерь системы
в данном температурном интервале в
расчете на градус температуры (7):
Д5= AQ/T (кал-град-1).
Произведение TAS имеет размерность
энергии и сопоставимое АН и АЕ.
Все системы стремятся к минимальному
теплосодержанию, поэтому состояние
системы тем устойчивее, чем более экзотер-
мична реакция. С другой стороны, все
системы стремятся к максимуму энтропии, т. е.
наиболее вероятному состоянию,
соответствующему минимальной упорядоченности.
Общий критерий самопроизвольности
процесса требует соединения АН и TAS. Таким
критерием в термохимии служит функция
Tn66ca(G).
При постоянных давлении и объеме:
AG = AE-TAS,
AG — это та часть внутренней энергии
системы, которая не «энтропизована», т.е. годна
для совершения полезной работы. В
условиях калориметра приближение к равновесию
означает, что система стремится к
состоянию, исключающему обмен свободной
энергией (G стремится к нулю). Тогда AG<0.
При этом реакция течет самопроизвольно.
Самопроизвольно идут экзотермические
(АН< 0) и эндотермические (А#>0)
реакции, но последние лишь при условии:
TAS>AH>0.
Живые организмы существуют в
стационарных, удалённых от термодинамического
равновесия динамических состояниях, за
счёт постоянной утилизации химической
энергии пищевых субстратов для
совершения полезной работы. Они поддерживают
минимальную скорость нарастания
собственной энтропии (И.Р. Пригожий). Тем
не менее, организм не стоит за рамками
закона сохранения энергии, а его работа
имеет коэффициент полезного действия,
далёкий от 100%.
Следовательно, интегральную
характеристику энергетического метаболизма —
основной обмен — можно рассматривать
с точки зрения вышеизложенных
термодинамических закономерностей.
Для понимания того, как формируются
энергозатраты основного обмена,
необходимо вспомнить ряд положений
физиологической химии и биоэнергетики (рис. 1).
Организм получает энергию в результате
катаболизма пищевых веществ, в
нормальных условиях — в основном углеводов
и липидов. И те, и другие могут
запасаться, служить для энергетической
подпитки между приемами пищи и свободно
переходить друг в друга, причем липиды
запасаются долговременно. В обычных
условиях 99 % потребляемых углеводов
расходуется на производство свободной
энергии (А.Гайтон, 1989). Белки не могут
быть синтезированы только исходя из
липидов и углеводов, так как содержат
азот и незаменимые, то есть не
вырабатываемые заново аминокислоты. Кроме того,
нет специального резервного пула белков,
предназначенных для энергетических
целей. Поэтому, хотя отдельные глюкогенные
и кетогенные аминокислоты могут после
поступления в организм превращаться
в компоненты углеводов и липидов при
равновесном процессе дезаминирования, за
исключением этой части циркулирующего
пула аминокислот, белок и его компоненты
не используются нормальным организмом
в качестве топлива, а идут на обеспечение
синтеза азотсодержащих продуктов.
Массированное использование белка в
энергетических целях возможно лишь в крайне
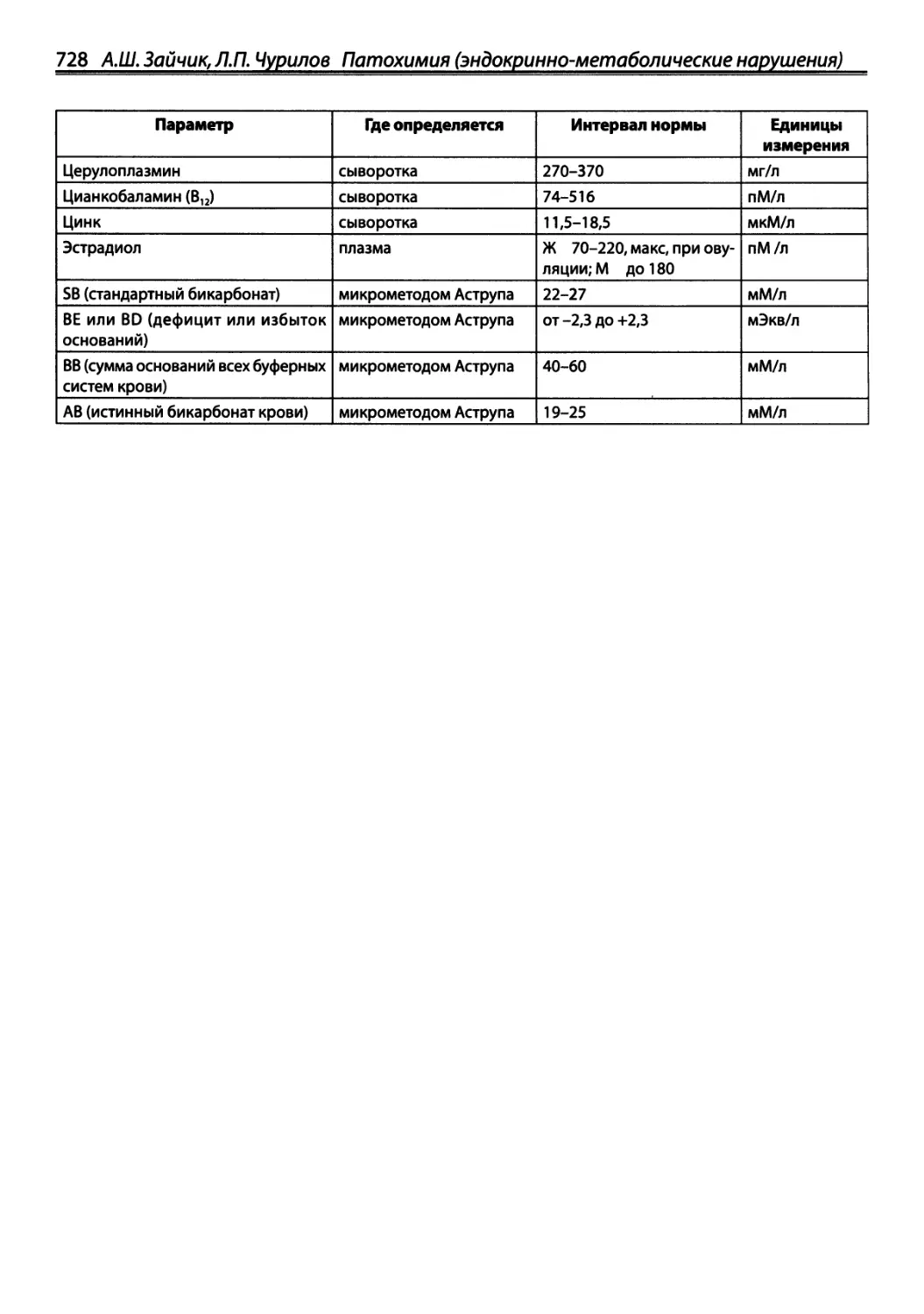
вынужденных ситуациях (например, при
стрессе (см. т. 1) и голодании — см. ниже).
Это заставляет вспомнить известную фразу
Д. И. Менделеева, сказанную касательно
утилизации нефти как топлива: «Кто же
топит печку ассигнациями!»
Катаболизм — трехэтапный процесс,
каждый из этапов приводит к освобождению
12 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
I этап
(в ЖКТ или
в лизосомах)
гидролитический
II этап
бескислородный
(в цитозоле)
III этап
аэробный
(митохондрии)
I углеводы I
т
| липиды |
| белки |
т
лактат
глицерин
хиерт
жирные
кислоты
аминокислоты __
аланин „
- лейцин
глутамат
тирозин
освобождаемая
энергия
<>
Рис. 7. Основные этапы катаболизма и их сравнительная энергетическая эффективность.
Сокрашения: Q^ — первично и вторично рассеянное тепло, А — полезная работа, КоА — коэнзим А
определенной энергии, которая частично
рассеивается в виде тепла сразу, не участвуя
в совершении работы. Данная компонента
носит название первично рассеянного тепла
и непосредственно идет на нагрев тела, а,
следовательно, сразу входит в тепловые
потери организма, в том числе и при
определении основного обмена.
Первый гидролитический этап
катаболизма проходит в ЖКТ и лизосомах без
участия кислорода и освобождает до 1 %
всей энергии субстратов. Эта энергия
полностью входит в первично рассеянное тепло
и не запасается. Второй этап —
бескислородного цитоплазматического
расщепления — представлен в клетках гликолизом и
аналогичными процессами распада липидов,
приводящими к получению универсального
катаболита — активного двухуглеродного
фрагмента ацетил-коэнзима А (Ф.А. Лип-
ман, 1953). Это приводит к освобождению
не менее 30 % всего теплосодержания
субстратов, при этом запасается около 43%, а
остальная часть тоже первично
рассеивается (56 000 калорий содержится в моле
глюкозы и 2 моля АТФ прибавляется при
ее распаде до пирувата, что соответствует
24000 калорий). В анаэробных условиях
практически единственным источником
энергии для организма служат углеводы,
тогда как в аэробных — спектр субстратов
расширяется.
Все клетки организма, за исключением
эмбриональных и злокачественных, а
также эритроцитов, в которых почти нет
митохондрий, в той или иной мере
проявляют способность подавлять анаэробный
распад глюкозы и активировать аэробное
митохондриальное окисление в условиях
доступности кислорода при исправных
митохондриях (эффект Л.Пастера).
Продукты второго этапа катаболизма, вовлекаясь
в митохондриальное окисление,
освобождают до 70 % всей своей химической энергии
Энергетический метаболизм и его нарушения 13
и распадаются до конечных
метаболитов — для углеводов и простых липидов это
будут С02 и Н20 (Х.А. Кребс, 1937).
Закономерности окислительного этапа
катаболизма были раскрыты благодаря изучению
О. Варбургом (1931) и А. Сент-Дьердьи
(1937) ансамбля дыхательных ферментов
и описанию А. Клодом и А. Ленинджером
(1948) строения и функции митохондрий.
Наконец, решающую роль сыграло
обнаружение АТФ, окислительного фосфорили-
рования (В.А. Белицер, В.А. Энгельгардт,
1930-1939) и хемиосмотического
механизма последнего процесса (П.Д. Митчелл,
1961-1966, В.П. Скулачёв, Е.С. Либерман,
1962-1975).
Аэробный, наиболее эффективный этап
катаболизма, может, в зависимости от
регулируемой степени сопряжения
окисления и фосфорилирования, приводить
к запасанию в виде макроэргических
соединений значительной части энергии — до
66 % (686 000 калорий энергии,
освобождаемых при полном окислении 1 моля
глюкозы дают возможность синтезировать до
38 молей АТФ, что соответствует 456 000
калорий), однако и здесь значительная
часть энергии сразу рассеивается, прямо
определяя уровень основного обмена.
Отметим, что срочная мобилизация теплоты,
сопровождаемая повышением
интенсивности свободного окисления, может сильно
увеличить эту долю, что используется при
адаптации к гипотермии, пробуждении от
зимней спячки и т.д. и контролируется
гормонами щитовидной железы и, отчасти,
надпочечников. Напротив, инсулин и
эндогенные опиаты, в частности динорфин
при зимней спячке и р-эндорфин — при
выходе из стресса, способствуют
уменьшению первичного рассеивания тепла и
максимально эффективному фосфорили-
рованию. Механизмы работы митохондрий
кратко рассмотрены в первом томе данного
руководства (т. I, с. 168-172), и нет
необходимости описывать их повторно.
Подчеркнём лишь, что часть энергии катаболизма
резервируется клетками в двух взаимо-
превращаемых формах — разности
потенциалов (например, протонового потенциала
внутренней митохондриальной мембраны)
и макроэргических фосфатных связей. Две
формы энергетической валюты организма
свободно конвертируются. Митохондрия
превращает протоновую разность
потенциалов в АТФ, а калий-натриевый насос,
например, осуществляет обратную
конвертацию АТФ в разность ионных потенциалов.
Энергия может передаваться, в
частности, при фосфорилирующей активации
тех или иных белков — участников
метаболизма, или же — при распространении
потенциалов вдоль проводящих структур.
Здесь большую роль играет промежуточный
характер АТФ, которая является «единым,
но не единственным» макроэргическим
носителем. Стандартная свободная энергия
гидролиза АТФ имеет среднее значение
(-7ккал/М1). В то же время, уфосфокре-
атина, например, она равна 10,5ккал/М,
а у гексозофосфатов от -5 до 3,3 ккал/М.
Именно это делает АТФ центральным
звеном передачи энергии (а значит —
способности к работе) от высокоэнергетических
соединений к фосфорилируемым «рабочим
молекулам» (А. Ленинджер, 1965). Фосфо-
рилирование белков на этом энергетическом
эскалаторе выступает как способ их
функциональной активации, ибо при этом растёт
активность каталитических и аффинность
распознающих молекул (Я. Квятковска,
1982). Поскольку нельзя расщепить менее
1 молекулы АТФ, значительная часть
энергии её гидролиза будет неизбежно теряться
в виде тепла при любой работе, так как
большинство сопряжённых реакций,
требующих затрат АТФ, используют на моль
продукта гораздо меньше того количества
энергии, которое даёт гидролиз 1 моля этого
топлива. Наиболее высокоэнергетические
начальные участники энергоконвейера,
такие, как креатин-фосфат, присутствуют
в организме в количествах на порядок
больших, чем АТФ, концентрация которой
1 В физиологических условиях — энергия
гидролиза АТФ несколько выше — 12 ккал/М.
14 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
строго постоянна, поскольку сильно влияет
на ход всех равновесных процессов.
В.А. Энгельгардт считал, что
координация функций многоклеточного организма
соответствует фундаментальному
принципу, согласно которому клетки производят
АТФ каждая, исключительно для себя.
Клетки не обмениваются готовыми макро-
эргическими фосфатами. Малые молекулы
утрачивают способность свободно выходить
из клетки, как только приобретают макро-
эргическую фосфатную связь, и остаются
внутри до их использования (Л. Певзнер,
1976). Внеклеточная АТФ служит хемоат-
трактантом лейкоцитов и воспринимается
как сигнал бедствия. Чтобы захватить одну
молекулу АТФ извне, клетка должна была
бы истратить 2 молекулы этого макроэрга.
Подобный разумный эгоизм предупреждает
паразитические взаимоотношения между
клетками организма и делает единственно
возможной формой энергетической
кооперации компонентов нашего тела обмен
полезной работой, а не экспорт и импорт
готовой АТФ.
Именно поэтому экзогенная АТФ не
оказывает прямого замещающего эффекта при
энергодефиците. Первооткрыватель АТФ
в одной из своих публичных лекций даже
Ql — первично рассеянная теплота
основной
обмен
Q2 — вторично-
рассеянная
теплота
Рис 2. Превращения энергии в организме.
Сокращения: 5—субстраты, Р—продукты,
Е—химическая энергия, 0—теплота, Д<р— разность
потенциалов, R-Ф—фосфатное макроэргическое соединение
употребил резкую фразу: «30 лет твержу
врачам — не колите АТФ, это бесполезно!
Колют». Впрочем, вполне реальная
эффективность курса инъекций АТФ в m. gluteus
maximus при гипоксии сердечной мышцы
присутствует, и определяется она, конечно
же, не прямым замещением, а
превращением макроэрга в пурины, попадающие
в системный и коронарный кровоток,
активирующие продукцию моноксида азота
и расширяющие венечные сосуды.
Затраты энергии необходимы организму
для поддержания жизнедеятельности, то
есть, по Э. Шредингеру (1972), для мини-
мализации темпов прироста собственной
энтропии и выполнения всех видов
полезной работы, а именно:
>• механической — как это происходит при
сокращении мышц или цитоскелета;
>- осмотической, для создания и
поддержания градиентов ионов, скажем: избытка
кальция в митохондриях;
>• электрической — как это достигается
при генерации потенциалов действия
(рис. 2).
Практически, все органы и системы
живого организма, совершающие работу,
можно представить как
преобразователи энергии, в которых
осуществляются взаимосвязанные
стационарные
процессы, в конечном итоге,
приводящие к
возрастанию энтропии. Так,
преобразование
химической энергии в
механическую происходит
в мышце, в
электрическую — в нервной
ткани, в
осмотическую — в почке или
на любой
плазматической мембране, в
световую — в
люминесцентном органе светляка.
Пластиды
преобразуют световую энергию
в химическую, сетчат-
П~Ф
м—■
механическая
[ осмотическая ^\
\(градиент-создающая) J
с
электрическая
f„работа роста" Л
Энергетический метаболизм и его нарушения 15
ка — световую в электрическую, в ухе
происходит трансформация механической
энергии в электрическую и т. д. (Т. Беннет,
Э. Фриден, 1967).
Несмотря на относительно высокую
эффективность биосистем (так, кпд здорового
сердца приближается к 43 % по сравнению
с 3-6 % у паровой машины), организм
подчиняется второму началу термодинамики
и не создает негэнтропию ни из чего. Во
всех преобразователях энергии,
функционирующих в организме, входные мощности не
меньше выходных. В необратимых
процессах входная мощность преобразователя
превышает выходную, в обратимых процессах
они равны. В итоге оказывается
справедливым уравнение Л. Онзагер (1932):
где: Ф — диссипативная функция; Т —
изменение абсолютной температуры; S —
энтропия; XJi и XJ2 — входная и выходная
мощность энергопреобразователя.
Поэтому при любом виде работы
существенная часть освобождаемой в организме
энергии рассеивается и превращается во
вторично рассеянную теплоту. Это дает
еще одну важную составляющую тепловых
потерь организма.
Поддерживать минимальную скорость
прироста собственной энтропии организм
может лишь за счёт увеличения энтропии
окружающей среды, расщепляя пищевые
биополимеры.
В состоянии здоровья эффективность
функционирования органов и систем
максимальна. Это означает, что практически
любое нарушение будет снижать кпд
вовлеченных в него органов (например, кпд
гипертрофированного миокарда меньше,
чем в норме). Следовательно, при всех
болезнях уровень тепловых потерь на
единицу достигнутого полезного эффекта будет,
вообще говоря, больше, чем при идеальном
здоровье. Поэтому в области патологии
питания существует понятие о калорических
затратах, связанных с заболеванием, и
априорно предполагается, что если болезнь не
ломает сами механизмы энергетического
обмена, то она вызывает определенную
прибавку к уровню энергозатрат. Так, при
ревматоидном артрите средней тяжести
такая прибавка на болезнь оценивается по
данным И.Х. Розенберга (1994) в 10% от
величины основного обмена индивида.
Высокая физическая активность
способна увеличить энергозатраты организма на
30-50 % (см. ниже). Это, однако, относится,
в основном, к физическому труду.
При крайнем физическом напряжении
уровень энергозатрат на короткое время
увеличивается в 8 раз. И даже столь
осмысленное и общественно полезное деяние, как
жевание резинки, по данным Л.Р. Перель-
мана (1937), способно дать 17 % прибавки
к базовым энергозатратам. Интересно, что
калорические затраты при физическом
труде не коррелируют со степенью утомления.
Многие виды работы вызывают сильное
утомление (например, прополка и другие
садово-огородные работы), но калорий
требуют мало. Высокие калорические затраты
характерны для любого физического труда,
связанного с поднятием тела и прямым
противодействием силе тяжести (бег, пог-
рузочно-разгрузочные работы).
В то же время в кажущемся
противоречии с известным принципом, рождённым
во время Египетского похода Наполеоном
(«Ослов и ученых — в середину!»), мозг,
даже в период напряженнейшей творческой
работы, стабильно окисляет 5-6 г глюкозы
в час, как и в покое или во время сна. Из
этого, однако, не следует, что ученых, в
отличие от шахтеров и футболистов, можно
не кормить. Просто, интенсивность и
эффект информационных процессов не может
меряться «дровяным» энергетическим
эквивалентом. Достаточно большое количество
энергии тратится просто, чтобы держать
мембраны нервных клеток под
электрическим напряжением потенциала покоя,
что эквивалентно простому поддержанию
сознания. При нехватке энергии в мозге
происходит деполяризация нейрональных
мембран и утрата их возбудимости. Этот
16 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
полиэтиологический синдром
энергодефицита и глубокой тканевой гипоксии ЦНС
проявляется утратой сознания и в клинике
носит название «кома». Очень многие
метаболические расстройства заканчиваются
комой. Они рассмотрены в соответствующих
разделах книги ниже.
Кроме того, эмоциональные эквиваленты
умственной деятельности способны, даже
без единого шевеления пальцем, вызвать
стресс, который приведет к значительному
увеличению энергозатрат и
теплопродукции вне мозга.
Помимо работы энергия требуется для
обеспечения анаболических процессов
и биосинтеза («энергия роста»). При
этом надо иметь в виду не только
энергию, непосредственно консервируемую
в форме химической связи между
мономерами, скажем, в пептидной связи двух
аминокислот. Если бы энергетический
эффект анаболизма исчерпывался этим,
то результирующая анаболизма была бы
эндотермической.
На деле для того, чтобы выработать
какое-либо вещество, нужное клетке,
метаболический путь должен быть, по существу,
необратимым1, т.е. помимо
формирования химических связей сопровождаться
значительным выделением свободной
энергии. Поэтому, даже включая различные
обратимые2 реакции, процессы анаболизма
нуждаются в необратимых и пусковых
стадиях, которые экзотермичны (рис.3).
Из-за этого, например, соединение пары
аминокислот в пептид требует расхода
минимум четырёх макроэргических связей. На
синтез одного моля пептидов, в
зависимости от их аминокислотного состава, тратится
от 0,5 до 4 ккал энергии. Поэтому усвоение
аминокислот и синтез из них белка требуют
1 Под необратимостью мы понимаем
невозможность вернуть систему в начальное состояние,
совершив то же количество работы, какое было
получено при прямом направлении процесса.
2 Те, которые при совершении точно такого же
количества работы, что было получено при прямом
направлении протекания реакции, возвращают
систему в исходное состояние.
расхода энергии углеводов и липидов
(калорические затраты на утилизацию белка).
Ведь клеточный аппарат анаболизма не
будет работать без энергозатрат, например,
инициация трансляции в рибосоме требует
распада ГТФ.
Вследствие этого, анаболизм не вычитает
из основного обмена, а прибавляет к нему.
Так, у детей в период интенсивного роста
требуется 5 ккал энергии из небелковых
источников на создание 1 г человеческой
ткани. У взрослых, в силу меньшей
интенсивности белкового анаболизма, этот
коэффициент меньше — около 4 ккал/г ткани.
Резкое ограничение потребления энергии
нарушает усвоение белка. Затраты АТФ
требуются и для синтеза липидов, и при глюко-
неогенезе, и даже для выработки мочевины,
хотя она и выводится из организма.
Итак, энергозатраты организма
определяются суммой первично рассеянной энергии
(более 50 % всей свободной энергии пищи),
вторично рассеянного тепла и
калорических затрат на анаболизм. При этом не более
25% свободной энергии пищи проходит
фазу превращения в полезную работу, но
и эта энергия, в конечном итоге, становится
теплом за исключением того, что
овеществляется в собственных биополимерах или
переходит в потенциальную и кинетичес-
Система
сопряжения
D
Ведомая
эндотермическая
реакция
анаболизма
Ведущая
экзотермическая
каталитическая IJ
реакция
+AGa
С
-AGk>+AGa
Рис. 3. Роль сопряженных реакций в
энергетическом обеспечении анаболизма и природа
«работы роста».
Сокращения: G — стандартная свободная энергия Гиббса
Энергетический метаболизм и его нарушения 17
кую энергии перемещенных индивидом
внешних объектов (перевезённой ослом
клади или стяга, водружённого альпинистом
на Эльбрусе).
Поэтому скорость тепловых потерь
является вполне адекватным показателем общего
уровня метаболизма.
УСЛОВИЯ ИЗМЕРЕНИЯ
ОСНОВНОГО ОБМЕНА
Если измерять тепловые потери организма
в период, когда не происходит
массированное усвоение новых пищевых субстратов,
и свести к минимуму рассеяние тепла,
связанное со всеми видами работы, то
в этих контролируемых условиях
теплопродукция становится основным путем потери
энергии телом, а главным источником этого
тепла может быть ранее запасенная энергия
(А. Уайт и соавт., 1981).
В этих условиях, измерение тепловых
потерь организма даёт относительно
точное представление об энергетических
потребностях тех процессов, которые
связаны с постоянными базовыми проявлениями
жизнедеятельности.
По определению основного обмена,
данному комитетом экспертов ФАО-ВОЗ,
основной обмен (00) — это лабораторный
показатель, отражающий «энергетические
затраты человека, который находится
в расслабленном и комфортном
состоянии по утрам вскоре после пробуждения
и спустя 14 часов после последнего
приёма пищи». При реальных клинических
определениях 00 условия могут
варьировать, но должны соответствовать
стандартным, которые предусматривают его
измерение:
> в состоянии полного мышечного и
психического покоя,
> натощак, т.е. через 12-18 ч после
последнего приема пищи,
> в горизонтальном положении,
> при температуре комфорта, что для
одетого человека составит 18-21 °С,
>- результаты должны экстраполироваться
на сутки по данным, полученным не
менее чем за 15 мин, а лучше — за 1 час.
Только, если эти условия обеспечены,
полученную величину можно трактовать
как 00. Поэтому вопросы типа: «Как
изменяется основной обмен во время бега?»
изначально некорректны. В подобных случаях
речь идёт об энергозатратах, а не об ОО.
Средняя величина 00 у взрослого
здорового 45-летнего мужчины, вес которого
70 килограмм (при идеальном весо-ростовом
соотношении), составит примерно 1785 ккал
тепла за сутки на все тело. Для женщины
того же возраста и веса должный средний
ОО — около 1679 ккал на всё тело за сутки
(Ф.Б.Тэлбот, 1938). Уместен вопрос:
велика ли эта цифра?
Скажем прямо — это немало! Если 1 кал
достаточна, чтобы нагреть при нормальных
условиях 1 г воды от 15 до 16 °С, значит,
даже ничего не делая, мы выделяем за сутки
энергию, способную вскипятить более 20
литров воды. Таким образом, даже в
состоянии откровенной обломовщины каждый из
нас остаётся мощным потребителем энергии.
С точки зрения биоэнергетики, жить — уже
значит работать] Вот почему
философичному И.И. Обломову кушать хотелось не
меньше, чем деятельному А.И. Штольцу.
Следует помнить об этом, чтобы
защищаться от психологической агрессии
некоторых рекламных утверждений. Например,
если сторонники спецдиет и сжигателей
жира утверждают, что, съев порцию
мороженого, Вы обязаны, дабы истратить
содержащуюся в ней энергию, полтора
часа стирать или 2 часа гладить (Д.Г. Кули,
1963), — в этом имеется передергивание:
человек — не калориметрическая бомба,
и окисление, например, грамма белков
в живом теле дает почти на 25 % меньше
свободной энергии, чем предсказывает
теория (4,1 вместо 5,3 ккал). Воспринимая
такие шокирующие подсчеты, надо помнить,
что кроме внешней работы (которая
действительно по калорическим затратам при
двухчасовом глажении белья эквивалентна
теплоте сгорания порции мороженого) во
18 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
время стирки, пилки, рубки, танцев в теле
человека продолжаются и все процессы,
составляющие основной метаболизм. Поэтому
реальные затраты энергии за 2 часа жизни
и работы гораздо выше.
00 — показатель, отражающий тот
уровень энергозатрат, который для данного
индивида в бодрствующем состоянии
минимально возможен. 00 — не абсолютный
минимум потерь тепла. При глубоком
естественном сне энерготраты падают еще
на 10-15%.
В то же время, уже при малой
физической активности, например, у постельного
больного, суточная прибавка на работу по
самообслуживанию составляет 400 ккал.
При обычном образе жизни и умеренной
физической нагрузке общая рабочая
прибавка может приблизиться к величине
основного обмена. Но даже в этих условиях
ОО составляет не менее 1/2 суточных
энергозатрат!
Некоторые эргономические расчеты
небезынтересны1. При экстремальной
физической нагрузке скорость энерготрат
может на секунды превысить уровень 00
в 200 раз, но лишь тренированные атлеты
могут удерживать уровень энерготрат в 50
раз больше 00 на протяжении нескольких
минут.
Так или иначе, изучение некоторых
данных эргономики говорит о том, что
процессы, ответственные за 00, составляют всегда
львиную долю от энерготрат организма.
1 Приводим характерные примеры
(по Дж. Дьюрнин, Р.Пэссмодру, 1967):
— набор текста на компьютере — 1,6 ккал/мин;
— игра на рояле —1,5 ккал/мин;
— починка и чистка обмундирование
военнослужащими —2,7 ккал/мин;
—«марш в джунглях — 6,5 ккал/мин;
— жатва серпом —4,5 ккал/мин;
— ковка молотом —6,9 ккал/мин;
— футбольный матч — > 7,5 ккал/мин;
Таким образом, некоторые виды активного
отдыха чрезвычайно энергоёмки, но рекордно
велики энергозатраты при глубокой ручной
вспашке — до 15,2 ккал/мин, в зависимости от
типа почвы.
Традиционно считается, что 00
обеспечивает уровень максимально экономичного
функционирования организма при
сохранении базовых анаболических процессов и
функциональной готовности. Вклад в 00
вносят сердечная деятельность,
перистальтика кишечника, работа системы дыхания,
поддержание всех электрических процессов
в клетках, терморегуляция,
энергорассеяние, сопровождающее анаболизм и т.п.
Но основной вклад в 00, как это
установлено в 80-е годы XX века, вносит
деятельность градиентсоздающих систем
клеток — электрохимическая работа.
Каждая клетка имеет плазматическую
мембрану и пока она жива, даже при
глубоком покое, на мембране поддерживается
калий-натриевый градиент, потенциал покоя,
а в ряде постоянно функционирующих
клеток — генерируются потенциалы действия.
Работа только одного фермента — натрий-
калиевой АТФ-азы, присутствующей во всех
клетках, отвечает за 30% энергозатрат
организма при полном покое.
Если с работой натрий-калиевого насоса
связана такая значительная составляющая
00, то, вполне понятно, что те регуляторы,
которые сильно влияют на активность
данного энзима, и будут наиболее мощно
изменять основной обмен. В этой связи
надо сразу же опровергнуть, как
архаичное имеющееся во многих руководствах
представление о том, что драматические
изменения величины О О при гипертирозе
и гипотирозе вызваны будто бы
разобщающим действием гормонов щитовидной
железы на окисление и фосфорилирование
(детальнее см. ниже в разделе
«Патофизиология эндокринной регуляции»).
Разобщающий эффект действительно
возможен, но лишь под влиянием
фармакологических доз этих гормонов, которые
на порядки превышают не только их
нормальный уровень, но и количество,
присутствующее в крови при самом тяжелом
тиротоксикозе.
Именно токсическая доза яда динит-
рофенола разобщает окисление и
фосфорилирование и вызывает усиление
Энергетический метаболизм и его нарушения 19
теплопродукции, вплоть до эндогенного
перегревания.
In vivo драматическое увеличение 00
при развитии гипертироза и его понижение
при компенсации гиперфункции
щитовидной железы, как и снижение 00 при
гипотирозе, связаны с тем, что тироидные
гормоны — самые сильные из всех
известных активаторов работы калий-натриевой
Ат-Фазы, практически во всех тканях,
особенно — в печени, скелетных мышцах,
почках, жировой ткани. Именно этот
механизм составляет основу несократительного
термогенеза у теплокровных, как впервые
доказали Ф. Исмаил-Бейги и А.С. Эй-
делмэн (1970). При гипертирозе основной
обмен остается повышенным и во сне,
и даже после приема морфия. При полном
атирозе степень падения 00 не превышает
35-40 процентов. Вероятно, этой величиной
и измеряется тироидзависимый вклад в
общую величину минимальных энергозатрат
организма.
Условия определения ОО требуют ряда
комментариев. Конечно же, температура
комфорта существенна, так как и
охлаждение, и перегревание организма требуют
расхода энергии на температурную
адаптацию и представляют стресс, а стрессор-
ные гормоны энерготраты увеличивают.
Любопытно, что у северян стандартные
величины 00 выше, чем у южан, вероятно,
из-за действия стереотипов хронической
холодовой адаптации.
Отдельных пояснений требует натощако-
вый принцип определения 00.
По-видимому, уже А. Лавуазье отметил,
что прием пищи вызывает увеличение
термогенеза организма. Оно начинается
примерно через 15-30 мин., достигает
максимума через 3-6 ч. и продолжается
около 12 ч.
Этот феномен ранее называли СДД —
специфическое динамическое действие пищи,
а теперь чаще именуют ТИД,
теплопродукция, индуцированная диетой.
По современным представлениям, его
механизм — комплексный. Оно включает
облигатный компонент, примерно
одинаковый для всех пищевых веществ и
обусловленный активацией деятельности ЖКТ
и энергозатратами на абсорбцию пищи.
Однако, у него есть также факультативный,
переменный компонент, связанный с
природой пищевых веществ и обусловленный
их действием как сигналов и их ролью как
субстратов.
М. Рубнер (1902) установил, что
наивысшее СДД присуще белкам. При их
употреблении энергозатраты возрастают
на 30-40 % против величины ОО, тогда как
для углеводов СДД составляет 5-6% 00
(для крахмала — 9 %), а для жиров — 2-2,5 %
00. A.M. Уголев в отдельных случаях
регистрировал нулевое СДД для жиров или
даже указывал на отрицательное изменение
00 после их приема (1991).
По мнению М.Рубнера и Г. Леска
(1931), это связано, прежде всего, с деза-
минированием глюкогенных аминокислот
(наиболее калоригенными оказались
глицин и аланин) и активацией
энергообразования, наступающей вследствие этого.
Белок не запасается, избыток аминокислот
переаминируется, а безазотистые остатки
идут на образование энергии, что может
вносить вклад в облигатное термогенное
действие белка. Кроме того, некоторые
этапы интермедиарного обмена самих
аминокислот требуют значительного распада
АТФ (А.Е.Браунштейн, 1957).
Показано, что ТИД, в основном,
генерирует энергию в печени и скелетных
мышцах.
Биофизиками было предложено
несколько объяснений механизмов ТИД включая
и довольно общие соображения, в
частности о его связи с нарастанием энтропии
системы при переваривании биополимеров.
Однако, еще О. Кестнер и Р. Плаут
(1924) указывали, что удаление
двенадцатиперстной кишки снижает ТИД и
приближает ТИД белков к таковому липидов
и углеводов. Н.В. Рязанцев в 1892 г.
обнаружил, что ТИД сохраняется при мнимом
кормлении, когда пища не попадает в ЖКТ
собаки, выходя через фистулу пищевода.
И.П.Павловым и Н.В.Рязанцевым был
20 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
сделан далеко опередивший время вывод о
том, что механизм ТИД — рефлекторный, а
не резорбционно-химический.
В связи с этим A.M. Уголев (1991)
предположил, что факультативный компонент
ТИД может рассматриваться как элемент
комплексной реакции организма на прием
пищи наряду с пищевым лейкоцитозом,
подавлением аппетита и другими
явлениями, управляемыми гормонами энтериновой
системы и гипоталамусом.
ТИД исчезал при удалении
двенадцатиперстной кишки. В то же время, при
сохранной кишке даже простое орошение
её слизистой кислым раствором (не
содержащим пищевых субстратов) вызывало
прирост основного обмена.
A.M. Уголеву удалось показать, что ТИД
во многом зависит от совместного действия
на терморегуляторные центры
гипоталамуса пептидов энтериновой системы (в
частности, секретина и холецистокинина),
которые он объединил под условным названием
«динэнтерин».
ТИД может рассматриваться как элемент
адаптации к пищевой нагрузке, он снижен
улице гипоталамическими дуоденальным
ожирением и стимулируется лептином (см.
гл. 7), уменьшен у детей,
предрасположенных к первичному ожирению (А.И. Клио-
рин, 1976) и составляет около 5 %
калорийности смешанной диеты, присутствуя,
частично, и при парэнтеральном кормлении.
Максимум увеличения теплопродукции при
приеме углеводов падает на момент высшей
точки алиментарной гипергликемии, а при
приеме белков — на периоды
максимального выведения мочевого азота.
Наличие высокого ТИД у белков
позволяет лицам с хорошей толерантностью
печени и почек к азотистой нагрузке
применять для контроля веса и похудения
высокобелковые диеты (например, такие
диеты разработаны для участников некоторых
программ НАСА).
Данное обстоятельство также убеждает,
что алиментарно-конституциональное
ожирение — не просто проблема баланса
калорий. Большое значение для его
развития имеет инадаптация гипоталамуса к
пищевой нагрузке, а в ряде случаев — и
нехватка полноценного белка по отношению
к избытку углеводов в диете (см. ниже
«Патология липидного обмена»).
Сбалансированная диета дает 6-10 %
прироста энергозатрат по отношению к 00
за счет ТИД.
Расчет 00 осуществляется на кг массы
тела или на м2 поверхности тела.
Последнее предпочтительнее, так как по правилу
М. Рубнера, чем больше отношение
поверхности тела к массе, или, чем для
млекопитающих меньше размер тела особи, тем
выше основной обмен, так как тем более
интенсивно тепловое рассеяние, требующее
гомойотермной компенсации. Подтверждая
это, М. Рубнер сопоставил ОО у двух собак,
вес которых отличался в 10 раз.
Теплообразование на кг веса разнилось в 2,5 раза
(и было больше у маленькой собаки). Но,
приведённые к 1 м2 величины были очень
близки (разброс порядка 14 %).
В связи с этим, видовые величины 00
в расчете на кг веса варьируют очень
значительно (так, у воробья они почти
в 14 раз выше, чем у индюка), а в расчёте
на м2 — намного более постоянны (от 900
до 1240 ккал в сутки у любых
теплокровных животных). Ряд авторов, в первую
очередь — Ф.Боненкамп, подчёркивали,
что поскольку потери тепла сильно зависят
от температуры кожи, а она находится под
влиянием выраженности
термоизолирующей жировой прослойки, величина ОО
в расчёте на кг будет очень значительно
колебаться именно в зависимости от того,
какую долю массы тела составляет жир.
Дополнительным соображением в пользу
ключевой роли этого показателя считалась
относительно малая окислительная
активность адипоцитов. Опыты отечественного
гигиениста М.Н. Шатерникова показали,
что после удаления курдюка потребление
кислорода у барана по абсолютной величине,
практически, не меняется. Сторонники
данного подхода объясняли разным
содержанием жира все различия О О включая половые,
Энергетический метаболизм и его нарушения 21
конституциональные и даже
патологические (например, его снижение при гипотиро-
зе и ожирении). Однако, в жировой ткани
идут активные анаэробные метаболические
процессы и совершается хемиосмотическая
работа. Кроме того, тепловые потери
сильно зависят и от сосудов кожи, а жировая
ткань, по современным данным, источник
лептина, кахексина, эстрогенов — то есть
важных регуляторов аппетита, метаболизма
и усвоения пищи, поэтому трактовка её
влияния на 00 не должна сводиться только
к термоизолирующему эффекту (см. ниже
«Устройство липостата»).
У взрослого человека 00 на м кв.
составляет в сутки порядка 900 ккал. (около
37ккал/м2в час).
Среди факторов, влияющих на основной
обмен, — возраст, пол, конституция,
состояние здоровья.
Влияние возраста, конечно, требует
учесть вышеописанный эффект «энергии
роста» и повышенные в раннем возрасте
затраты на терморегуляцию. По
классическим данным, ОО новорожденных
несколько ниже, чем взрослых — в первые дни
после рождения 27ккал/м2в час, к бмес.
он достигает уровня взрослых, а в период
1-3года— абсолютного онтогенетического
максимума (60-65 ккал/м2 в час, что выше
уровня взрослых почти в 2 раза), затем до
возраста 12-15 лет идет медленное
снижение, некоторый пубертатный прирост
и дальнейшее установление взрослого
уровня к 20 годам, что не случайно совпадает
с прекращением роста тела в длину.
До старости 00 у здорового индивида
относительно постоянен и у здоровых
престарелых людей, особенно женщин, может
несколько понижаться, конечно, если это не
маскируется патологическими прибавками
энергозатрат.
Влияние пола выражается в том, что 00
у женщин во всех возрастах, начиная с 1 дня
жизни, при прочих равных условиях, на 6-
10 % ниже, чем у мужчин. Беременность
прогрессивно увеличивает 00, кастрация
самцов и самок его понижает, в связи с этим,
нет основании думать, что половые отличия
00 — просто прямой результат действия
половых гормонов на метаболизм.
Как сказано выше, считалось, что
исключительное значение имеет разный
удельный вес мышечной и жировой ткани
у лиц разного пола, но у новорожденных он
одинаков, а 00 разный (150 и 136ккал/кг
у мальчиков и девочек, соответственно).
В.А.Геодакян указывает, что более низкий
00 у женщин свидетельствует о том, что
реактивность женского организма позволяет
ему достигать при одинаковых условиях
более экономичных режимов
функционирования и связывает это с наличием более
широкого выбора адаптационных программ
в женских клетках, по сравнению с геми-
зиготными по Х-хромосоме мужскими (см.
том1 настоящего руководства, стр. 8-70).
Таким образом, с биоэнергетической точки
зрения один из полов призван развивать
большую форсированную мощность, а
другой — реализовать более эффективные
способы утилизации энергии!
Авторам нравится это объяснение,
подчеркивающее большее энергетическое
совершенство женщин, тем более, что ряд
важных для энергетического обмена ферментов
кодируется именно в Х-хромосоме.
Очевидно, что 00 зависит от состояния
здоровья. Общая закономерность состоит
в том, что при подавляющем большинстве
болезней ОО возрастает. Особенно сильно
он растет при гипертирозе и существенно
увеличен при гиперкортицизме.
Вместе с тем, в отдельных ситуациях,
при нарушении самих механизмов
энергообразования и термогенеза возможно
и понижение ОО.
00 снижается при гипотирозе и гипокор-
тицизме из-за инактивации калий-натриевых
АТФаз.
00 повышен в 1-й и понижен во 2-й
период при голодании, снова возрастает
в заключительном периоде голодания,
снижен при неосложненном соматической
патологией первичном ожирении и у лиц,
искусственно сбросивших вес, при дости-
22 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
жении номинальной массы тела не
нормализуется.
Токсические дозы инсулина понижают
сократительный термогенез и увеличивают
долю запасаемой энергии, снижая ОО. При
зимней спячке ОО сильно снижается, что
вероятно связано с действием эндогенных
опиоидов, например, дерморфина,
выделяемых зимнеспящими животными (см. ниже
«Анабиоз и зимняя спячка»).
Нейропептид соматостатин сильно
снижает многие обменные процессы и тормозит
широкий спектр физиологических функций,
уменьшая ОО, что дает право некоторым
авторам называть его «пангибином» и
увязывать с механизмами анабиоза. Напротив,
при акромегалии ОО повышен (см.
«Патофизиология эндокринной регуляции» ниже).
К сильному увеличению энерготрат ведет
активация несократительного термогенеза,
достигаемая под влиянием избытка катехола-
минов симпатоадреналовой системы, в том
числе — при лихорадке (механизмы этих
изменений излагаются в т. 1 с. 371-372).
По правилу P.M. Рассела (1987), основной
обмен возрастает на 13% при повышении
температуры тела на 1 °С. Вообще, ОО
принципиально возрастает при любых серьёзных
расстройствах здоровья, если они не
повлекли за собой потом самих механизмов
регуляции энерготрат. Поэтому роль определения
ОО в клинике— не диагностическая.
Например, верификация диагноза гипертироза
осуществляется по концентрациям гормонов
щитовидной железы, а не с помощью
контроля уровня основного обмена. Однако, ОО
сохраняет практическую прогностическую
ценность (см. ниже).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
ОСНОВНОГО ОБМЕНА
И ИХ ПРИНЦИПЫ.
КАЛОРИМЕТРИЯ
Должный основной обмен может быть
оценен по таблицам Харриса и Бенедикта,
учитывающим пол, вес, рост и возраст
испытуемого. Для арифметического расчёта
должного ОО существуют формулы:
ООмужчин = 66+(13,7№)+
+5Я-6,8Д
ОО женщин = 655+9,5 W+
+ 1,8#-4,7Д
где W — вес в кг, А возраст в годах, Н —
рост в см.
По формуле Дрейера:
ОО = ОДЗЗЗл/W / К х А,
где W— вес в г, А — возраст, К — половой
коэффициент, равный у мужчин 0,1015, у
женщин — 0,1129.
Истинный ОО отличается от должного
и часто именно это отличие имеет
диагностическое или прогностическое значение.
Поэтому оценка должного О О не заменяет
определения фактического ОО.
Определение ОО называется
калориметрией (что не следует путать с
колориметрией — термин, относящийся к
определению концентрации вещества по цветности
его раствора).
Существуют 2 метода калориметрии —
прямщй и непрямщй. Их принципы ясны
из рис. 4.
При прямой калориметрии происходит
непосредственное измерение изменений
теплосодержания организма (энтальпии
-АН). Для этого используется калориметр.
Объект, находящийся в калориметре,
выделяет тепло. Создается тепловой поток,
направленный наружу. Если термостати-
ровать и изолировать камеру и направить
тепловой поток на нагревание
циркулирующего вокруг теплоносителя, то разность
температур теплоносителя будет отражать
теплопродукцию объекта. Наиболее
совершенные макрокалориметры,
сконструированные в XIX-M веке, использовали воду
как теплообменник и позволяли
определять ОО у человека, целиком помещенного
в калориметрическую камеру (калориметр
В.В.Пашутина).
Достоинством прямой калориметрии
является ее точность. Именно прямая ка-
Энергетический метаболизм и его нарушения 23
прямая калориметрия
AUB ► АНкДж
непрямая калориметрия
[02]хКЭ ►AKQ
прямой микрокалориметр непрямой калориметр (метаболятор)
Рис. 4. Принципы калориметрии и устройство прямого (слева) и непрямого (справа) калориметров.
Пояснения в тексте. U—напряжение, Н — энтальпия, Q — теплота, КЗ — калорический эквивалент,ЦК— дыхательный
коэффициент
лориметрия мышц позволила А.В. Хиллу
провести увенчанные Нобелевской премией
исследования энергетики мышечного
сокращения, а Дж. Гиббсу — определить КПД
изолированного сердца млекопитающих.
Однако, макрокалориметр — громоздкое
и неудобное сооружение.
Во второй половине XX столетия метод
прямой калориметрии был модифицирован
и послужил основой для создания такого
многообещающего медико-биологического
инструмента, каким является реакционная
микрокалориметрия (РМ). Она
предназначена для исследования изменений
энтальпии различных молекулярных и клеточных
систем при взаимодействии их компонентов.
Метод может быть применен в
адиабатических условиях, но в большинстве случаев
исследование ведется в изотермических
и близких к изотермическим условиях,
практически без изменения температуры
образцов. РМ оперирует количествами тепла
порядка микрокалорий. Но слово «микро»
подразумевает не просто
микрочувствительность. Высокочувствительные калориметры
были и прежде, но эта чувствительность
достигалась за счет большого объема
реакционного сосуда (~ 1 л) и большого расхода
реагентов. Современная РМ имеет дело
с микромолярными количествами
вещества и малыми объемами (до 10 мл). В этой
разновидности прямого
калориметрического анализа используется не целостный
организм, а его отдельные клетки — кровь,
эритроциты, лейкоциты, биоптаты жировой
и других тканей.
Микрокалориметрическая ячейка термостатирована и окружена
высокотеплопроводными термопарами —
спаями разнородных металлов (рис. 4).
Тепловой поток, создаваемый
жизнедеятельностью помещенных в нее клеток,
идет через термопары наружу и нагревает
эти спаи, что приводит к эффекту Ж.-Ш.
Пельтье — генерации разности
потенциалов, пропорциональной разности
температур. Эта ЭДС может быть зарегистрирована
чувствительным вольтметром и записана
в виде пика магнитным самописцем, причем
ее знак и величина будут соответствовать
изменению теплосодержания системы.
Основные достоинства РМ —
возможность исследования интактных биосистем,
прямой анализ биоматериала без его осо-
24 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
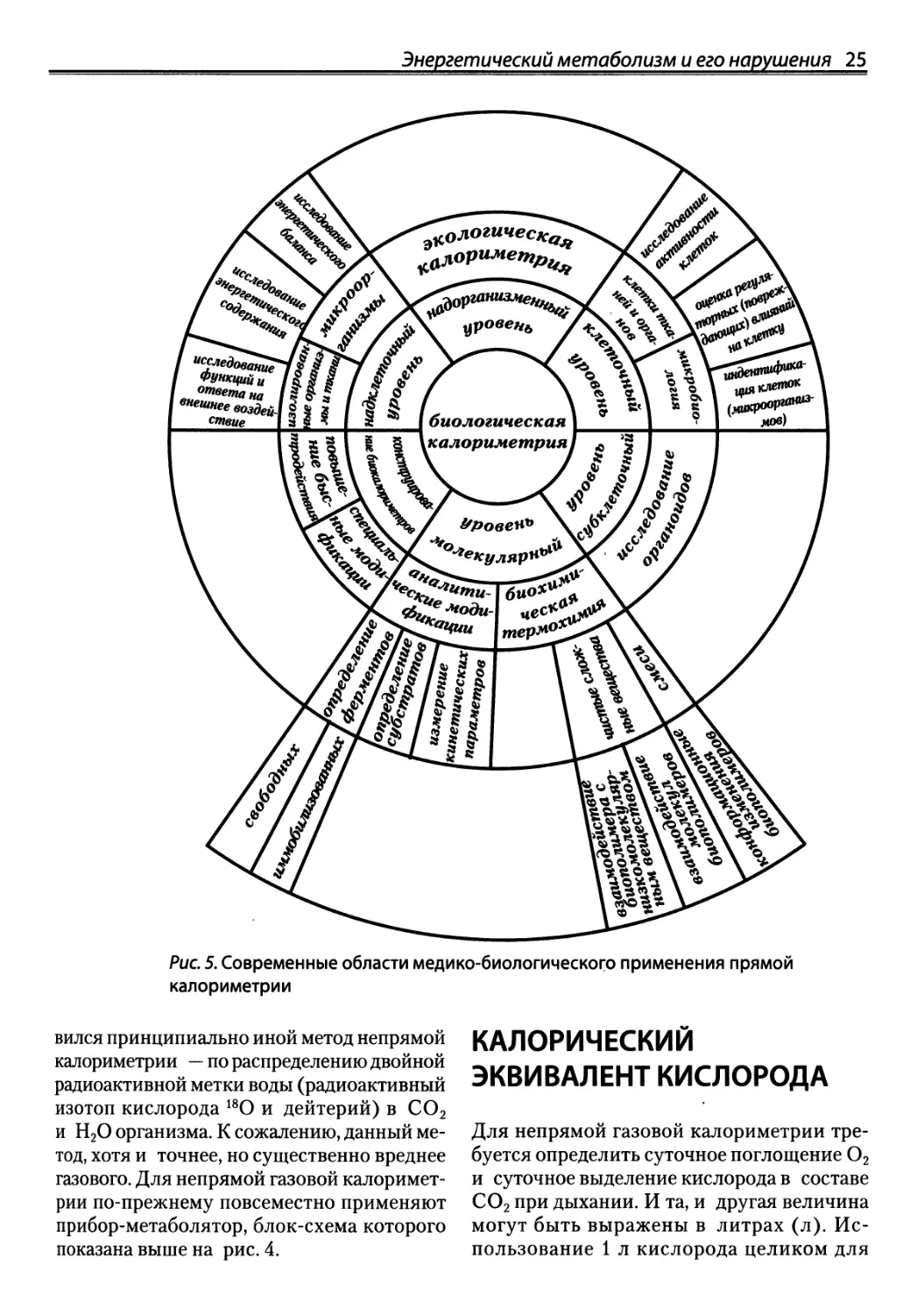
бой подготовки, получение интегральной
характеристики жизнедеятельности клеток
или фундаментального показателя всякой
биохимической реакции — динамики
изменений энтальпии.
Проблемы РМ — неспецифический
характер выходного параметра, что требует
многочисленных контролей; а также
большая длительность опытов: порой 12 ч уходит
на достижение температурного равновесия
микрокалориметрической ячейки. В
современных коммерческих приборах источники
артефактов сведены к минимуму.
Микрокалориметрия подтверждает
данные макроизмерений, в частности
применяется для отслеживания динамики 00
у больных с гипертирозом при лечении
по удельной теплопродукции эритроцитов
(К. Левин, 1974). В силу различий в
интенсивности гликолиза, а значит — и
теплопродукции у молодых и старых клеток,
она может использоваться клинически
в решении множества оригинальных
задач — например, в дифференциальной
диагностике анемий и даже для
регистрации бактериурии по тепловому
эффекту жизнедеятельности бактерий в моче.
Очень перспективным направлением
прямой реакционной микрокалориметрии
является изучение тепловых эффектов
высокоаффинных комплементарных
взаимодействий, например таких, как связывание
антигенов и антител, субстратов и
ферментов, биорегуляторов и их рецепторов
(А.Ш. Зайчик, Л.П. Чурилов; 1982, 1998).
В проточной модификации
микрокалориметрического метода взаимодействующие
агенты смешиваются в калориметрической
камере, а сопутствующие этому тепловые
эффекты сопоставляются с контрольными
теплотами разведения и записываются.
РМ может предоставлять информацию для
несерологической оценки активности
антисывороток и цитотоксических эффектов
(В.М. Пур, А.Э. Бизер, 1982). В наше время
прямая калориметрия разветвилась на
множество областей применения и
модификаций Расширяющееся поле применения
биокалориметрии отражено на рис. 5.
По традиции в клинике ОО измеряют
чаще методом непрямой калориметрии.
До недавнего времени существовал лишь
один метод непрямой калориметрии по
Крогу-Дугласу — на основе измерения
данных газообмена и их перерасчета в
теплопродукцию.
Этот метод основан на предположении
Лавуазье-Рубнера о пропорциональной
зависимости теплопродукции и поглощения
кислорода. Однако, многие экзотермические
процессы не являются кислородзависимы-
ми. Таков, например, гликолиз. При
фагоцитозе теплопродукция лейкоцита растет более
чем в 4 раза, а поглощение 02 увеличивается
ненамного — так как этот цитофизиологи-
ческий процесс обеспечивается, в
основном, анаэробно. Фагоцитоз не блокируется
цианидами. В связи с подобными фактами,
еще П.М.Альбицкийи П.П. Авроров
указывали на принципиальную погрешимость
непрямой калориметрии.
На деле график зависимости энерготрат
и поглощения кислорода представляет собой
не прямую, а гиперболу — следовательно, чем
выше истинный 00, тем в большей степени
непрямая газовая калориметрия занижает
00. Парадокс заключается в том, что в
клинике тест на ОО наиболее часто применялся
именно для исследования тех больных, у
которых ОО высок, а следовательно, как раз тогда,
когда непрямой метод дает наименее точные
результаты (болезнь фон Базедова).
Все это позволяет подтвердить вывод
о малом диагностическом значении ОО —
даже при патологии щитовидной железы
измерение уровня ее гормонов
характеризует функцию намного точнее и адекватнее.
В клинике тест на ОО применяется, в
основном, не с диагностической, а с
контрольно-прогностической целью — при всей
неточности его непрямого определения,
если доктор дважды в ходе лечения
определил в стандартных условиях одним и тем
же прибором ОО у одного пациента — это,
конечно же, позволяет путем сравнения
данных судить о направлении развития болезни
и эффекте лечения. В настоящее время,
с развитием изотопной диагностики, поя-
Энергетический метаболизм и его нарушения 25
Рис. 5. Современные области медико-биологического применения прямой
калориметрии
вился принципиально иной метод непрямой
калориметрии — по распределению двойной
радиоактивной метки воды (радиоактивный
изотоп кислорода 180 и дейтерий) в С02
и Н20 организма. К сожалению, данный
метод, хотя и точнее, но существенно вреднее
газового. Для непрямой газовой
калориметрии по-прежнему повсеместно применяют
прибор-метаболятор, блок-схема которого
показана выше на рис.4.
КАЛОРИЧЕСКИЙ
ЭКВИВАЛЕНТ КИСЛОРОДА
Для непрямой газовой калориметрии
требуется определить суточное поглощение 02
и суточное выделение кислорода в составе
С02 при дыхании. И та, и другая величина
могут быть выражены в литрах (л).
Использование 1 л кислорода целиком для
26 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
окисления пищевых субстратов дает разный
калорический эффект, в зависимости от
того, в какой пропорции утилизируются
клетками липиды, углеводы и
аминокислоты.
Поскольку в углеводах присутствует
сравнительно много кислорода и
соотношение О и Н соответствует продукту их
полного окисления — воде, кислород при
горении углеводов расходуется только на
окисление углерода. В типичной молекуле
липидов — соотношения иные и
собственного кислорода гораздо меньше. Экзогенный
кислород будет тратиться и на окисление
углерода, и на окисление водорода жиров
до воды.
При сжигании избытка любых
субстратов в литре 02 время горения и тепловой
эффект будут лимитироваться кислородом.
В этих условиях литр кислорода при горении
углеводов даст наибольший калорический
эффект — 5,047 ккал/л (5,01 для глюкозы и
5,06 — для крахмала), а при горении избытка
липидов —наименьший —4,68 ккал/л. При
горении избытка белков потребление 1 л 02
даст 4,82 ккал (4,6 ккал/л с учётом реальной
усвояемости белков в катаболических
процессах). При смешанной сбалансированной
диете у здоровых испытуемых данный
показатель приближается к 4,825 ккал/л.
Экспериментальные данные подтверждает
и теоретический расчет, исходя из
молекулярного веса глюкозы и количества энергии,
выделяемой при ее окислении на литр 02.
Тепловой эффект, полученный при
использовании 1 л 02 на окисление
избытка того или иного пищевого субстрата,
называется калорическим эквивалентом
02 для углеводов, липидов или белков
(КЭ). Калорический эквивалент 02 у
конкретного испытуемого, полученный
при непрямой газовой калориметрии,
помножается на суточное поглощение 02
в литрах и дает основной обмен в
тепловых единицах. Нельзя не отметить, что КЭ
варьирует у здорового человека при резкой
перемене состава пищи в довольно узких
пределах — порядка 4 % в обе стороны
от средней величины, типичной для
сбалансированной диеты (Л.Р. Перельман,
1937).
Калорический эквивалент не следует
путать с применяемым в диететике
калорийным (калорическим) коэффициентом белков,
жиров и углеводов. Последний
представляет собой теплотворную способность этих
субстратов и рассчитывается в ккал/г
субстрата при калориметрии сжигания в
полностью противоположной ситуации — при
избытке 02 и наличии ровно 1 г того или
иного субстрата. По сути дела это — теплота
сгорания пищевых веществ.
Поскольку в кислороде горят Си Н, то
имеющие наивысший молярный процент
этих атомов липиды дадут и самое большое
количество теплоты на грамм — 9,3 ккал
против 4,1 у углеводов и белков. Расчетное
значение калорического коэффициента
белков -5,3, но так как белок в организме
участвует в анаболических процессах,
фактическое значение меньше.
Теоретический подсчет должного
количества усвоенных за день белков, жиров
и углеводов по номинальному меню
бесполезен — это не дает информации о том,
в какой пропорции все горит! Не говоря
уже о несоответствии между теоретическим
и фактическим составом конкретных блюд
(а эти показатели, как известно не только
медикам, но и юристам, порой сильно
расходятся), мы не знаем, насколько полно
субстраты всасываются и как они
распределяются между катаболизмом и
анаболическими нуждами, окисляются ли они
до конца.
Достаточно посмотреть на такой
педиатрический объект, как младенец, слюнявчик
и щеки которого густо измазаны манной
кашей, чтобы понять, что принятые в
диететике расчёты, основанные на должном составе
продуктов питания, весьма условны, а
калорическую стоимость литра кислорода
сегодня и у данного индивида можно определить
только путем практического измерения.
Но что измерять? Параметр, измеряемый
непосредственно по данным газообмена
и используемый для выбора реального
калорического эквивалента, соответствую-
Энергетический метаболизм и его нарушения 27
щего данному индивидуальному паттерну
использования пищевых веществ, — это
дыхательный коэффициент (ДК). Каждому
ДК соответствует по номограмме
определенное значение КЭ, что и используют при
непрямой газовой калориметрии.
ДЫХАТЕЛЬНЫЙ
КОЭФФИЦИЕНТ
И КОЭФФИЦИЕНТ РУБНЕРА,
ИХ ИЗМЕНЕНИЯ
ПРИ ПАТОЛОГИИ
Согласно Л.Р. Перельману, Д К есть
отношение веса кислорода, выделенного организмом
в составе выдыхаемой углекислоты, к весу
поглощенного кислорода. По определению
это безразмерная величина, выраженная
в долях единицы.
ДК = С[02]/[02].
При чисто углеводном окислении
конечные продукты — только вода и двуокись
углерода. Для окисления углеводов на
каждый атом углерода требуется одна молекула
02 и получается одна же молекула С02,
причем 02 требуется только для окисления
С, следовательно, по правилу Авогадро,
объемы газов в числителе и знаменателе
формулы ДК будут равны и при полном
окислении чистых углеводов ДК=1.
Из расчета элементарной формулы
жиров средний ДК для них получается 0,707
(К.Цунц, Ф. Меринг, 1883).
Это меньше 1 так как известная часть
кислорода при окислении до конца жира
израсходована на образование воды
путем окисления Н, а кислород в
составе данного продукта распада не учтен
в выдохнутом С02. Поэтому на 100
молекул окисленного жира
углекислого газа образуется только 70 молекул.
Наконец, окисление белков идет с
образованием мочевины:
C=0(NH2)2.
Карбамид содержит О, полученный
при дыхании и учтенный в знаменателе
формулы ДК, но не выделяется лёгкими
в виде С02, а экскретируется почками
и в малой степени — потовыми железами.
Нуклеиновые кислоты метаболизируются
с выделением в составе мочи тригидрок-
сипуринового производного — мочевой
кислоты, представляющей у человека
конечный продукт их обмена. Наконец, при
окислении белков и нуклеиновых кислот
большая доля кислорода тратится на
окисление водорода, которого в этих субстратах
относительно больше, чем в углеводах. По
этим причинам ДК при белковой и нук-
леопротеидной диете не может быть равен
единице и составляет около 0,809.
Таким образом, практически определив
газообмен и рассчитав конкретный ДК для
данного индивида на реальной смешанной
диете, мы можем выбрать адекватный КЭ
и получить ОО по данным поглощения
кислорода. Поскольку, как уже сказано
выше, в норме значение белков как
энергосырья минимально (и может быть оценено
отдельно по показателям азотистого
баланса), колебания ДК между 1 и 0,7 будут
отражать соотношение углеводов и жиров в
окислительном катаболизме здорового
индивида. Так, сразу по насыщении любой пищей
он приближается к 1, так как немедленно
утилизуирются, исключительно, углеводы.
Через 10-12 ч голодания он понижается
вследствие перехода на утилизацию жира
из депо.
Значение ДК не исчерпывается его
служебной ролью при проведении непрямой
калориметрии. Он позволяет судить и о
некоторых типовых расстройствах интер-
медиарного обмена.
А. Биккель и О. Кауфман-Косла (1926)
ввели понятие дизоксидативной карбону-
рии — состояния, при котором часть
углерода не доокисляется до С02, а выходит из
организма с мочой в виде промежуточных
углеродокислородсодержащих продуктов.
Для полноты характеристики дизокси-
дативных состояний ДК сопоставляется
с другим показателем: коэффициентом
28 АЖ Зайчик, Я/7. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рубнера, или карбонурическим
показателем, равным соотношению общего углерода
и общего азота мочи:
>сРубнера ^мочи
/N
мочи*
Хотя основными формами экскреции
конечных продуктов белкового и
нуклеинового обмена у человека служат мочевина
(на 80%) и мочевая кислота, коэффициент
Рубнера в моче, в норме, выше, чем
соотношение углерода и азота в чистых
мочевине (0,43) и даже мочевой кислоте,
так как с мочой теряются более
углеродистые аминокислоты
(физиологический дизоксидабеильный белковый
углерод), составляющие 3-5% азота мочи
в норме, а также креатинин и порфирины.
При смешанной диете данный
коэффициент укладывается в пределы 0,6-0,7.
Усиленное белковое питание,
выраженный катаболизм белков, например при
состояниях с отрицательным азотистым
балансом (см. ниже — «Патофизиология
белкового обмена»), таких как тяжелый
стресс, последние дни голодания — могут
привести к снижению этого коэффициента,
а углеводистая диета, обусловливающая
усиление выведения гликргенных
аминокислот, а также состояния,
сопровождаемые гипераминоацидурией и выведением
избытка немочевинного азота (например,
печеночная недостаточность, голодание,
сахарный диабет, тяжелая мышечная
работа) — к его повышению. При голодании
(см. ниже) лишь в третий период из-за
усиления распада белков коэффициент
Рубнера снижается. В первый период он стабилен,
во второй идет его повышение.
Классическая форма дизоксидативной
карбонурии обуславливает при явном
сахарном диабете понижение, на фоне
неизмененной диеты ДК, при повышении
коэффициента Рубнера.
Интересно, что данное нарушение
общих обменных характеристик может быть
зарегистрировано даже до появления
выраженной глюкозурии и кетонурии,
безусловно, повышающих карбонурический
коэффициент. Большое значение имеет то,
что С. Джослин называл «голоданием среди
изобилия» — невозможность адекватной
утилизации углеводов при диабете ведет
к использованию жиров (см. ниже
«Патофизиология сахарного диабета»).
По-видимому, не только глюкозурия и
кетонурия, но и сильно выраженная амино-
ацидурия могут вызывать явление
дизоксидативной карбонурии.
Липогенная дизоксидативная карбонурия
сопровождает метаболическую ситуацию
2-го периода голодания с его кетоацидозом
на почве усиленного липолиза и
подавления использования ацетил-коэнзима А
в цикле Кребса из-за расхода кетокислот
в процессах глюконеогенеза,
стимулированных глюкагоном. При этом Д К достигает
1 в первые дни голодания и падает до 0,7
при переходе на липидные эндогенные
субстраты во 2 периоде голодания.
Л.Р. Перельман указывал, что
дизоксидативная карбонурия сопровождает и гипови-
таминозы по витаминам группы В.
Лихорадка характеризуется подъемом
ДК до 1 в фазу подъема температуры.
При длительных истощающих инфекциях
с продукцией большого количества ци-
токинов — ИЛ-1 и ФНОа, подавляющих
анаболические процессы в соматическом
белковом отсеке — скелетных мышцах
и других инсулинзависимых тканях. ДК
может упасть до 0,7, отражая усиление
катаболизма жиров и белков. В фазу падения
температуры ДК нормализуется. В разгар
лихорадки это сопровождается негативным
азотистым балансом и понижением
коэффициента Рубнера (см. т. I, гл. 13).
Особый интерес представляет позитивная
карбонурия — повышение коэффициента
Рубнера за счет ретенции азота и
уменьшения знаменателя при отсутствии дизокси-
дации углеводов и неизменном числителе.
Это бывает при выраженном
положительном азотистом балансе — беременности,
в период быстрого роста у детей, при
акромегалии и формирующемся гигантизме,
в фазу восстановления и усиленной
регенерации после тяжелых инфекций, стрессов
и травм.
Энергетический метаболизм и его нарушения 29
При развитии алиментарного ожирения
и паратрофии детей ДК может
парадоксально повышаться и составлять больше 1,
сигнализируя о массированном переходе
углеводов в жиры.
При этом из 100 г углеводов формируется
42 г жиров, имеющих меньшее
содержание О. Этот процесс освобождает 45 г С02,
поэтому часть С02при выдохе содержит
эндогенный кислород из состава субстратов,
который человек выдохнул, но не вдыхал.
Классический эксперимент М. Блейб-
трея (1901) по откорму рождественского
гуся позволил форсировать липогенез и
получить у птицы ДК=1,33. Известно, что
ДК= 1,01-1,08 — не редкость при
ожирении, паратрофии и откармливании после
голодания.
Рекордно низкие значения ДК отмечены
при зимней спячке — до 0,46, что
свидетельствует о задержке в организме или
выделении с калом кислородосодержащих веществ
и представляет уникальную особенность
анабиотического обмена веществ в
присутствии высоких уровней эндогенных опиатов
и соматостатина (см. ниже).
ЗНАЧЕНИЕ
ТЕРМОДИНАМИКИ
В МЕДИЦИНЕ
Таким образом, биомедицинская
термодинамика прошла солидный путь и обогатила
практическое здравоохранение ценными
методами исследования больного и
важными теоретическими концепциями. Как
получается, что «черный ящик» с неизвестным
устройством (см. «Предисловие» выше),
потребляя пищу, воду и воздух, производит
тепло, выполняет работу, поддерживает и
восстанавливает упорядоченность своей
индивидуальной структуры? Уже в
первой половине XVI столетия Рене Декарт
(1596-1650) сформулировал идею о том,
что в организме нет процессов вне рамок
физических и химических взаимодействий,
но всего 200 с небольшим лет назад даже
ведущие авторитеты
медико-биологических наук еще полагали, что воздух нужен,
исключительно, для охлаждения органов,
которые разогреваются виталистической
внутренней жизненной силой, а процессы,
происходящие в живом теле, принципиально
невоспроизводимы вне его (М.Ф.К. Биша,
1802). Однако, А. Лавуазье и П.С.Лаплас
сравнили теплопродукцию при горении
свечи и пребывании морской свинки в
ледяном калориметре и пришли к выводу
об аналогии между процессами
метаболизма и медленным горением (1780). Затем
А.Лавуазье и А. Сеген (1789) обнаружили
в опытах на себе, что чем выше физическая
активность, тем больше организм выделяет
С02, постулировав окислительную природу
животной теплоты. Но что горит и служит
ли энергетическим сырьем главный
компонент организма — белок? Спор об этом не
утихал более века, с тех пор как К. Бертолле
(1785) удалось установить, что при
гниении животных тканей выделяется аммиак,
а А.Ф. де Фуркруа (1799) — выделить из
мочи ее «эссенциальную соль» — мочевину.
Голландец Г. Мюлдер (1839) на основе идей
Ф. Вёлера и Ю. Либиха об органических
радикалах (см. выше «Предисловие») пришел
к выводу о существовании азотсодержащих
радикалов, из комбинации которых состоят
животные белки и предложил сам термин
«протеин». Но основатели концепции
азотистого баланса Ж.-Б. Буссиньоль и
Ж.-Б. Дюма (1839,1841), открыв усвоение
атмосферного азота у бобовых растений и
азотистое равновесие у животных (см. ниже
гл. 5), полагали, что животные и человек
лишь окисляют сложные вещества,
синтезированные растениями, добывая для себя
энергию. Им, однако, возразил Ю. Либих
(1841), который привлек прозаические
данные из сельскохозяйственной практики:
свиньи, вскармливаемые растительными
углеводами, жиреют, хотя для этого
углеводы надо не окислять, а восстанавливать.
Значит, метаболизм животных включает и
процессы редукции субстратов. В первом
руководстве по биохимии и патохимии,
справедливо подчеркивая роль белкового
30 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
метаболизма в норме и при патологии, барон
фон Либих вновь возвращается к старой
идее, что дыхание лишь охлаждает органы,
которые черпают энергию благодаря
распаду белка до мочевины, поскольку белки в
его понимании — и инструмент, и топливо
организма, а нехватка белка в питании и
изнашивание протеиновых машин — причина
многих болезней.
Замечательно, что не отрицая данных
А. Лавуазье о потреблении кислорода и
выделении углекислого газа при работе
тела, немецкий ученый объясняет их тем,
что при воздушном охлаждении белковой
машины в нее попадает опасный кислород,
способный вызвать «коррозию» белков,
поэтому жиры и углеводы требуются, чтобы
нейтрализовать этот эффект и сохранить
белки! Эта парадоксальная мысль через 100
с лишним лет возродится при
рассмотрении свободно-радикального повреждения
тканей и примет форму теории
«окислительного стресса».
Однако, шотландский врач Э. Смит (1862)
исследовал выделение мочевины в покое
и после тяжелого физического труда и не
нашел разницы, чем поколебал белковую
теорию энергетического обмена. На базе этих
данных немецкий глазной врач и создатель
закона сохранения энергии Г. Гельмгольц
(1861) рассчитал коэффициент полезного
действия организма человека как тепловой
машины и оценил эту величину в 25 %. Это
побудило искать аналогии между
организмом и паровой машиной, вследствие чего
швейцарцы А. Фик, С. Вислиценус и
Э.Франкланд (1866) опровергли теорию
Ю. Либиха с позиций биотермодинамики, в
опытах на себе показав, что энергия, которую
тратит человек с определённой массой тела
при подъеме на альпийскую вершину
известной высоты намного больше, чем могут дать
реально превращенные при этом в мочевину
белки, даже если бы кпд этого процесса
был бы равен 100%. Вывод тривиален для
современного читателя, но революционен
для медицины той эпохи: «Машина топится
углем, а не сама собою» — то есть, организм,
хотя и изнашивается при работе, но
функционирует не за счет самосожжения
структурных компонентов, а потребляя топливо,
отличное от его белков, именно: углеводы и
липиды. Огромную роль сыграло создание
У.О; Этуотером и Ф.Дж. Бенедиктом в
США непрямого дыхательного
калориметра, а В.В. Пашутиным в России — прямого
калориметра для человека. Наконец, в
1894 г. М.Рубнер доказал что выделяемая
организмом собаки теплота эквивалентна
теплоте сжигания всех потребленных ею
продуктов по данным газообмена и зыде-
ления мочевины. В 1902 г. в созданном ими
комбинированном калориметре У.О. Этуо-
тер и Ф.Дж. Бенедикт показали с высокой
точностью равенство теплоты, образуемой в
организме человека и при сжигании
потребленной им пищи. Кстати, попутно ими была
установлена калорическая значимость
алкоголя, а затем — и эквивалентность липидов
и углеводов в качестве топлива. А. Магнус-
Леви (1895), введя понятие основного
обмена, дал врачам мощный критерий для
интегральной оценки скорости метаболизма
в целом и, одновременно, начал объективно
оценивать функции щитовидной железы
еще до открытия ее гормонов. Во второй
половине XX века большим прорывом было
создание прямой микрокалориметрии. Врач
XXI столетия знает о методологических
ограничениях калориметрии, тем не менее,
она остается в арсенале клинической
патофизиологии. Термодинамические категории
используются общей нозологией в учении о
здоровье и болезни (см. т. I). Понятие кпд
является центральным для понимания
патогенеза таких синдромов, как сердечная и
дыхательная недостаточность. Знакомство
с патофизиологией энергетического обмена,
помимо теоретической и практической
ценности, важно для формирования
профессиональной психологии врача. Оно дает медику
иммунитет по отношению к псевдонаучным
спекуляциям на темы биоэнергетики,
которые имеют широкое хождение в современной
информационной среде, особенно в текстах
рекламно-коммерческого содержания, и
основываются на некорректном
использовании понятий биотермодинамики.
Глава 3
ГОЛОДАНИЕ
«Я определённо заметил, что стоит мне поголодать несколько дней
подряд, как мой мозг начинает словно бы вытекать и голова пустеет. Она
становится лёгкой и бесплотной, я больше не чувствую её у себя на плечах,
и мне кажется, что, когда я на кого-нибудь гляжу, глаза мои раскрываются
до невероятности широко».
К. Гамсун «Голод»
• История изучения голодания
• Этиология субстратно-энергетической недостаточности
• Классификация голодания
• Продолжительность жизни при голодании
• Периоды голодания и их эндокринно-метаболическая характеристика
• Квашиоркори алиментарный маразм
• Нейрогенные анорексия и булимия
• Откармливание после голодания
• Лечебное голодание
• Цикл голодание-насыщение
Голодание — патологический процесс,
обусловленный адаптацией к дефициту
калорий, пищевых субстратов и
незаменимых компонентов пищи. Голодание
(субстратно-энергетическая недостаточность)
может быть результатом неадекватного
питания, либо тех психических,
онкологических, гастроэнтерологических, эндокринных
заболеваний, которые делают приём и
усвоение пищи невозможным. При голодании
происходит, по выражению И.В.
Давыдовского (1969), атрофия тела.
ИСТОРИЯ ИЗУЧЕНИЯ
ГОЛОДАНИЯ
Первое научное описание голодания как
патологического процесса в медицинской
литературе дал английский автор Дж. Прингль
(1742) на материале массового голода во
время войны в Голландии. Прингль считал
патологические изменения,
зарегистрированные им у пациентов, особой «отёчной
болезнью». В XIX столетии классические
описания массового голодания в
колониальной Индии оставили английские врачи
Мак-Лид, Лоуэлл и Дэвидсон, считавшие
отёчную болезнь эпидемической.
В России основы патофизиологии
голодания в экспериментах на животных заложил
В.А. Манассеин (1869), которому
принадлежит классическая книга «Материалы
для вопроса о голодании». Закономерности,
касающиеся регуляции функций организма
при голодании, изучил П.М. Альбицкий
(1918). Этому автору принадлежит
важнейшая глобальная для всей патохимии идея,
что субстраты обмена веществ выполняют
не только роль
материально-энергетического сырья, но и роль
информационно-сигнальных регуляторов. Соответственно, по
Альбицкому, голодание — не просто
дефицит субстратов, но и определенный дефект
регуляции. Развивая это положение, авторы
школы П.М. Альбицкого—В.В. Пашутина
подчёркивали определение голодания, как
алиментарной дистрофии, указывая, что
это — «не чистая атрофия, но атрофия с из-
32 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
менением качества тканевого субстрата, что
и является дистрофическим изменением»
(Н.А. Краевский, 1943).
В годы блокады Ленинграда (1941 -1943 гг.)
массовая алиментарная дистрофия
подверглась изучению патологоанатомов и
патофизиологов. Значительный вклад в понимание
механизмов нарушения обмена веществ при
голодании внесли Л.Р.Перельман,
ставивший опыты по изучению патофизиологии
голодания на самом себе, и В.А. Свечников,
обобщивший огромное количество
клинических и патологоанатомических описаний
алиментарной дистрофии в монографии
«Болезнь голодания» (1947).
Эпикризы, написанные в те годы,
поражают трагическими, типичными для
блокадных ленинградских условий примерами:
«Больная Г-ва, 42 лет, бухгалтер,
находилась в железнодорожной больнице по
поводу заболевания алиментарной дистрофией
II степени и пеллагрой. Начало своего
заболевания больная связывает с тем, что она
недостаточно питалась и всю зиму, из-за
прекращения трамвайного движения, делала
ежедневно переходы в 12 километров.
Повторно ухудшение болезни наступило в
ноябре 1942 г., когда она работала на
лесозаготовках, и весной 1943 г., после того, как она
получила многочисленные ранения
осколками от попавшего в комнату
артиллерийского снаряда. Эта больная как бы
сконцентрировала в себе влияние многих
вредностей — недостаточного питания, физического
перенапряжения, переживаний военного
времени и травмы» (В.А. Свечников).
Во второй половине XX века
исследования патогенеза голодания вышли на новый
уровень в связи с раскрытием
гормональных, цитокиновых и нейропептидных
механизмов регуляции аппетита и насыщения.
Большой интерес в связи с развитием
космической медицины вызвала проблема
анабиоза (см. ниже), что привело к углублению
исследований гормонально-метаболических
особенностей эндогенного голодания при
зимней спячке животных.
Проблема голодания и доныне остаётся
комплексной, социально-медицинской.
Голод был постоянным спутником
различных бедствий и мощным фактором
естественной истории человека как вида, а
также социальной истории человечества.
Насколько эта проблема стара, настолько
же она актуальна для современного
человечества: обладая технологиями,
позволяющими накормить каждого жителя Земли,
цивилизация не внедряет социальных
механизмов решения этой задачи.
Характерно, что в 1998 г. Нобелевская
премия по экономике присуждена именно
исследователю проблемы голода —
индийскому учёному Амартья Кумар Сену за
работу «Нищета и голод: эссе о наделении
правами и их лишении», в которой автор
убедительно показывает, что причиной
голода в современном мире служит не
абсолютный дефицит продуктов питания,
а социальные факторы. Так, в период
засухи 1979-1984 гг. в Африке производство
продуктов питания в демократической
Зимбабве упало на 38%, но голода в этой
стране не было, а в то же время в
олигархических Эфиопии и Судане при сокращении
сельскохозяйственного производства всего
на 11% с небольшим, разразился страшный
голодомор. В Эфиопии, например, детская
смертность достигла чудовищного
показателя-247 на 1000.
В данном учебнике мы рассматриваем
голодание как медицинскую проблему, хотя
необходимо с самого начала подчеркнуть,
что пациент, не получающий адекватного
питания, очень часто представляет и
проблему социальную.
ПРИЧИНЫ ГОЛОДАНИЯ
Наиболее часто к
субстратно-энергетической недостаточности приводят (или ее
ускоренному развитию при недостаточном
питании способствуют) следующие
заболевания:
>- нарушения переваривания и/или
всасывания пищевых ингредиентов (синдромы
мальабсорбции и мальдигестии);
>* хронические и рецидивирующие
процессы, стимулирующие ответ острой
Голодание 33
фазы (преиммунный ответ — см. I том
данного руководства, гл. 13) включая
инфекции, лихорадку, онкологические
заболевания, некоторые аутоиммунные
болезни. При этих состояниях
освобождаются цитокины, придающие катабо-
лическую направленность метаболизму
(например, кахексии);
>- расстройства, связанные с потерей белка
и других нутриентов (нефротический
синдром, хронические обструктивные
болезни лёгких, кишечные свищи, плаз-
морея при ожоговой болезни, экссуда-
тивная энтеропатия, десквамативные
дерматиты и т.д.);
>* эндокринные болезни с нарушением
анаболизма и усиленным катаболизмом
(гипертироз, сахарный диабет);
>* психонейроэндокринные расстройства
с подавлением аппетита и извращением
пищевого поведения (anorexia nervosa —
см. ниже, психозы);
>* состояния повышенных нутритивных
потребностей (беременность, лактация,
детский и подростковый возраст, период
реконвалесценции от травм и острых
инфекционных болезней,
послеоперационный период);
>- алкоголизм, отравления антагонистами
витаминов и лекарствами;
>* продлённое парентеральное питание.
Голодание наблюдается при лечении
ограничением потребления калорий, а также
в режиме эндогенного голодания — при
спячке животных. Оно может быть
следствием ошибок в назначении и выполнении
парентерального питания. Однако в
громадном большинстве случаев как в прошлом,
так и в современной истории голодание
имеет экзогенный характер и социальную
подоплёку. Сюда относятся протестные
формы голодания, экстремальные условия, а
также все те случаи, когда индивиды
вынуждены голодать по социально-экономическим
причинам. Голоданию по самым скромным
оценкам ФАО-ВОЗ в конце XX столетия
подвержены на планете не менее 400 млн
детей и почти полмиллиарда взрослых. Их
количество за 15 лет возросло на четверть,
а доля детей с недостаточным питанием
в мире в целом (и в нашей стране, в
частности) в конце 90-х годов XX века стала
выше, чем в конце 60-х — вот факт, о
котором должны помнить все, кто считает,
что в истории существует категория
общественного прогресса! По данным В.В.
Коренной (1998), потребление рыбы в
России за период с 1987 по 1998 снизилось
на 2/3; мяса, птицы и сахара — на 50%;
колбас, маргарина и масла — на треть. При
этом, душевое потребление мяса в России
в 1997 г. не превысило и половины от
соответствующих показателей, характерных
для питания населения США, Германии и
Франции. Выборочные исследования
питания населения России показали, что около
25 % обследованных недоедают, а
распространённость гиповитаминозов и дефицита
микроэлементов приближается к 80%. Всё
это, к сожалению, не уменьшает
актуальности темы субстратно-энергетической
недостаточности для отечественных врачей.
От четверти до трети школьников Санкт-
Петербурга и Москвы имеют дефицит массы
тела (СВ. Мальцев, 1998, Ю.И.Строев и
соавт., 2005).
КЛАССИФИКАЦИЯ
ГОЛОДАНИЯ
Традиционно принято выделять виды и
формы голодания. По форме голодание
бывает с водой и абсолютное (т.е. без воды).
По виду выделяются полное, неполное
и частичное голодание.
При полном голодании поступление в
организм пищевых веществ полностью
прекращается. Вся поставка энергии и нутриентов
идёт из внутренних источников. Е.С.
Лондон называл полное голодание «эндогенным
питанием».
Подобные ситуации возникают в
эксперименте, при эндогенном голодании
зимнеспящих животных, при
категорическом отказе от еды, при ряде заболеваний,
34 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
делающих невозможным приём пищи, и по
социальным причинам.
При неполном голодании (недоедании)
основные пищевые вещества поступают
в организм. Их поступление
характеризуется калорически и пластически
недостаточным количеством. В эксперименте может
быть обеспечено «идеальное» неполное
голодание — с качественно правильным
соотношением макроингредиентов диеты
и с достаточной поставкой её
микроингредиентов. Но в реальности при неполном
голодании имеется, как правило, не только
калорическая, но и белково-витаминная
недостаточность. Основные причины
такого голодания — социальные. Существует
и медицинское неполное голодание — при
лечении и в результате заболеваний,
понижающих аппетит либо нарушающих
усвоение пищевых ингредиентов. Неполное
голодание — самая распространённая и
значимая в медико-социальном отношении
форма субстратно-энергетической
недостаточности.
Если распределить все случаи белково-
энергетической недостаточности в виде
спектра, то на одном полюсе окажутся
наблюдения, когда имеется выраженный
дефицит основного незаменимого компонента
пищи — белка, а калорический дефицит хотя
и присутствует, но не столь значителен. На
другом полюсе будут расстройства,
возникающие при полной калорической депри-
вации. В случае полной калорической де-
привации голодание чаще всего развивается
таким образом, что имеется длительная фаза
компенсации, когда
эндокринно-метаболические механизмы обеспечивают защиту
висцерального пула белка и мобилизуют
для энергетических нужд жиры и белки
соматического пула (кожи, жировой ткани
и скелетных мышц). Это проявляется в
виде безотёчной или марантической формы
голодания (алиментарный маразм). При
алиментарном маразме только в деком-
пенсированной фазе голодания страдает
висцеральный белковый пул.
Если же белковая недостаточность
развивается опережающими темпами, на фоне
обеспечения калорийности неполноценного
питания с помощью углеводов, то
компенсация может с самого начала оказаться
недостаточной в отношении щажения
висцерального белка. Тогда клиническая
картина голодания приближается к отёчной
(квашиоркорной) форме. Декомпенсация
наступает раньше, и общая тяжесть
процесса обусловливает при отёчном голодании
меньшую выживаемость. Реальные
вынужденные случаи неполного голодания могут
протекать согласно промежуточному
образцу, вначале больше напоминая марантичес-
кую форму, а при декомпенсации — кваши-
оркорную. Вариант протекания процесса
зависит не только от того, насколько и чем
покрываются калорические нужды, но и от
сочетанного действия нескольких
гормонов и аутокоидов в организме индивидов
с различной реактивностью (см. ниже «Ква-
шиоркор и алиментарный маразм»).
И полное, и неполное голодание Л.Р. Пе-
рельман объединял как голодание
количественное, в противоположность
качественному или частичному голоданию. Частичное
голодание, фактически, представляет собой
несбалансированное питание с дефицитом
или полным исключением из диеты того
или иного ингредиента (углеводов, белков,
жиров, конкретных микроэлементов или
витаминов, воды), в то время как
остальные нутриенты присутствуют в диете
в достаточном количестве, а калорические
нужды организма, в целом, покрываются.
По социальным и медицинским причинам
частичное голодание — это крайне
распространённое явление, особенно, если учесть,
что формально именно к данной категории
относятся разнообразные авитаминозы
и гиповитаминозы (см. ниже). Можно
говорить о голодании патологическом
(связанном с болезнями и вредными
экзогенными факторами), лечебном (применённом
в искусственных условиях, под контролем
медиков с терапевтическими целями), а
также о голодании физиологическом
(связанном с жизненными природными
циклами животных — у некоторых насекомых
Голодание 35
в стадии имаго, при зимней спячке у рыб,
земноводных, пресмыкающихся и
некоторых млекопитающих; у рыб на нересте;
при летней спячке у двоякодышащих рыб
в сухой сезон и т.д.). Наконец, если
потребление энергии повышено (например, при
стрессе и тяжёлой физической нагрузке), а
поступление калорий недостаточно то такое
голодание иногда называют ускоренным
(В.Ю. Шанин, 1996).
При этом гормоны стресса активируют
убиквитин-зависимый протеасомный нели-
зосомальный путь протеолиза в мышцах, что
способствует ускоренной потере белка
мышцами и глюконеогенезу в печени (П. Кумар,
М. Кларк, 2004).
ПРОДОЛЖИТЕЛЬНОСТЬ
ЖИЗНИ ПРИ ГОЛОДАНИИ
Несмотря на легенды о йогах, питающихся
флюидами Вселенной, все эксперименты
на животных и трагические наблюдения за
жертвами голодовок однозначно
показывают, что полное голодание раньше или позже
обязательно ведёт к необратимым
изменениям в органах и заканчивается
смертельно. Считается, что необратимые нарушения
наступают, и угроза гибели становится
неизбежной при потере примерно 50 % белка
тела, если нет сопутствующих голоданию
или спровоцированных им заболеваний
(А. Гайтон, 1989). При их наличии смерть
от инфекций и авитаминозов может
наступить и намного раньше. На протяжении
значительного срока голодание обратимо.
В истории зафиксировано весьма
продолжительное полное голодание добровольцев без
тяжёлых последствий. Профессиональный
добровольный голодалыцик Дж. Суччи
на 24-й день полного голодания фехтовал,
а на 50-й день совершал верховые
прогулки (1889). Предельная продолжительность
жизни у здорового человека среднего
возраста и упитанности при полном голодании
с водой, по-видимому, не превышает 80
дней (по Л.Р. Перельману, исходя из
среднесуточной потери в весе около 0,5%). На
79-й день протестного голодания умер один
из узников североирландской тюрьмы Лонг
Кеш в семидесятых годах. По данным
литературы, наиболее длительное полное
экспериментальное голодание человека средней
упитанности, завершившееся restitutio
ad integrum, продолжалось 50 дней (Дж.
Тэннер). В то же время, имеются
документальные свидетельства полного лечебного
голодания при крайне тяжёлом ожирении,
продолжавшегося более 200 дней.
Так как основной обмен связан с
поверхностью тела, а размеры тела — с
количеством жира, то мелкие животные при
голодании гибнут быстрее крупных (по
данным В.В. Пашутина: кролик — живёт
до 4 недель, собака — до 60 дней, куры — 34
дня, голуби — 13 дней, верблюд — много
месяцев). Холоднокровные животные, не тратя
энергии на поддержание температуры тела
и имея меньшую скорость метаболизма,
способны жить при эндогенном питании
много дольше гомойотермных (паук — 1 год,
улитка — 2 года, лягушки — 18 месяцев,
личинки паукообразных — до 5 лет — А. А.
Журавель, 1955).
Продолжительность голодания связана
с объёмом жировых запасов и
конституцией организма. Упитанные животные
переносят его дольше и с меньшими
последствиями, чем изначально истощенные
(П.П. Авроров). Гиперстеничные
индивиды более резистентны к голоданию, чем
астеники (М.В. Черноруцкий). По данным
большинства авторов, смерть наступает при
дефиците массы тела не менее 45-50% от
нормальной. Но имеются случаи выживания
после голодания с дефицитом массы от 50
до 60 % у животных и человека (В.А. Бал-
ковский, 1943). Так, В.В.Воронин (1948)
описывает собаку, успешно откормленную
после полного голодания с потерей 63 %
веса. Общепризнанным является факт
большей устойчивости к голоданию у женщин,
по сравнению с мужчинами того же
возраста и упитанности, а также у взрослых — по
сравнению с детьми и подростками.
Классические эксперименты Фалька
показали, что новорожденные щенята при
36 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
полном голодании имеют троекратно
большую суточную потерю веса и живут в 7 раз
меньше, чем годовалые собаки. В то же
время, старые собаки теряют ежесуточно в весе
вполовину меньше молодых и переживают
их в 2,5 раза.
В 1941 -1942 гг. в блокадном Ленинграде
О.Д. Иссерсон (1944) установил, что
наибольшая летальность наблюдалась в
группе молодёжи до 20 лет, а также у мужчин
трудоспособного возраста.
Крайне тяжело переносят голодание
маленькие дети. А.Б. Воловик в
Педиатрическом институте, ставшем в блокадные годы
центром реабилитации голодающих детей
(и занумерованном на фашистских картах как
особая «цель»!), отметил, что до 21,5% детей
с III степенью дистрофии умирали в первые
сутки после госпитализации, при этом у детей
младших возрастных групп алиментарная
дистрофия уже при средней степени тяжести
давала уровень смертности, близкий к тому,
который характерен для старших детей лишь
при тяжёлой дистрофии.
Голодание приводит к смерти
теплокровных животных тем быстрее, чем
дальше температура окружающей среды от
зоны комфорта. Дополнительный стресс
и физическая нагрузка отягощают течение
голодания.
Голодание без воды протекает тяжелее,
чем с водой, и приводит человека к гибели
от гиперосмолярной дегидратации за 4-7
дней. Л.Р. Перельман указывает, что при
этом суточная потеря воды парадоксально
велика и превышает уровень, характерный
для голодания с водой. По его мнению,
здесь большую роль играет более
энергичный катаболизм жиров, необходимый,
чтобы мобилизовать эндогенную воду. В то
же время, создаётся порочный круг, так как
продукты катаболизма требуют воды для
своего выведения. Таким образом, вода при
голодании оказывает «как бы сберегающее
действие на запасы организма», что
особенно характерно для голодания травоядных
и всеядных животных.
Точка необратимости при голодании
знаменуется резким увеличением
выведения продуктов азотистого распада, и даже
белка — с мочой. Согласно классической
концепции, это происходит в результате
полного истощения других энергетических
ресурсов (предсмертный или необратимый
распад белка — по Фрерихсу, 1848). С
современных позиций, кажется вероятным, что
усиление выведения немочевинного азота
в последние дни смертельного голодания
отражает распад нуклеиновых кислот
клеточных ядер при некрозе и апоптозе. Это
сопровождается продукцией мочевой
кислоты и свободных кислородных радикалов
(В.С.Новиков и соавт., 1995). Имеются
наблюдения о возможности откармливания
животных, начатого даже позже
терминального скачка кривой мочевого азота вверх
(П.П. Авроров, 1900). Следовательно,
граница между обратимой и необратимой
стадиями голодания относительна и
индивидуальна.
ПЕРЕРАСПРЕДЕЛЕНИЕ
РЕСУРСОВ
ПРИ ГОЛОДАНИИ
Голодание требует от организма общей
экономии энергетического и
пластического материала. Но, как и при инсулин-
зависимом сахарном диабете, при полном
голодании создаётся метаболическая
ситуация перераспределения ресурсов в пользу
инсулиннезависимых органов и тканей.
Инсулинзависимые структуры находятся
в положении наиболее обделённых. Хотя
продукция инсулина понижается, она не
прекращается. Однако,
гормонально-метаболическая картина голодания формируется
под знаком резкого преобладания действия
комплекса контринсулярных регуляторов.
Подобно ответу острой фазы при травмах
и воспалениях (см. т. I гл. 13), в
условиях голодания происходит мобилизация
энергоресурсов соматического компонента
тела — скелетных мышц и жировой ткани.
Аминокислоты и продукты липолиза
используются печенью для ресинтеза глюко-
Голодание 37
зы и образования кетоновых тел с целью
экономии белка висцеральных органов
и обеспечения энергетических
потребностей мозга.
Это ярко проявляется при голодании
в неравномерности весовой потери
отдельных органов, которая отмечена еще
классиками науки о питании. Особенно много
теряют в весе производные мезодермы
(С.М.Лукьянов, 1890), а также органы
и ткани, представляющие депо углеводов
и липидов. При среднем дефиците веса
на момент гибели 50-55 %, наибольшие
потери наблюдаются в жировой ткани,
редуцированной у погибших голодной смертью
почти на 99%. Наблюдается даже
исчезновение жира в липомах (А.Ф. Ефимова,
Е.З. Эльперин, 1944) и обратное развитие
липидных отложений в крупных артериях
(Д.М. Гротэль, 1944). Сальник и брыжейка
становятся тонкими
соединительно-тканными плёнками. Лишаются жира эпикард
и жёлтый костный мозг, что придаёт им
студенистый или слизистый вид. Скелетные
мышцы, которые также инсулинозависимы,
редуцируют свою массу на 70%. Очень
велики атрофические изменения в лимфо-
идных органах — вес селезёнки снижается
на 72 %. Во всех атрофированных органах
отмечается отложение липохрома, а в
селезёнке — гемосидероз (В.Г. Гаршин, 1944).
Печень теряет в весе 50-60%, слюнные
железы — 65 %, другие пищеварительные
органы, по данным разных авторов, — от 30
до 70 %, с наиболее выраженной атрофией
слизистой желудка и железистого аппарата
pancreas. В костях наблюдается
дистрофическая остеопатия, с остеопорозом и суб-
периостальными переломами.
Потеря в весе, приходящаяся на кровь
и кожу, примерно соответствует
относительной общей потере массы тела, при этом
происходит атрофия желёз — производных
кожи, истончение эпидермиса и утрата
сосочков кожи. В то же время, атрофия
жизненно важных инсулиннезависимых
органов бывает выражена в гораздо меньшей
степени. Мозг, надпочечники (особенно, их
мозговое вещество), глаз — по одним
данным (В.А. Свечников) — вообще не теряют
в весе (а в ряде случаев даже прибавляют),
по другим (А.А. Журавель , Ф.Г. Биддер
и А.А. Шмидт, 1852) — их весовые
потери минимальны (от 3 до 10%). При этом
спинной мозг теряет в весе больше, чем
головной, и проявляет больше признаков
дегенеративно-дистрофических изменений
(Л.Р. Перельман).
Весовые потери почек составляют 6-25 %,
что менее среднего в 2-9 раз. По данным
В.Д. Цинзерлинга, на почки умерших от
голода атрофические процессы вообще не
распространяются (1943).
Легкие теряют 18-20 % веса (Б.И.
Кадыков, 1966). Нет единого мнения по поводу
весовых потерь сердца и гонад. Одни
авторы, главным образом, на основании опытов
на животных, подчёркивают относительную
сохранность веса сердца (потери от 3,6 % по
А.А. Журавелю до 30 % — по В.А. Манас-
сеину). Другие отметили существенное
уменьшение веса этого органа и признаки
миокардиодистрофии у погибших от голода
людей (так, Н.А. Краевский (1943) находил
у жертв ленинградской блокады вес сердца
менее 130 г, что соответствует потере более
чем 50 % должной массы органа).
Среди желёз внутренней секреции
особенно сильно атрофируется щитовидная
(В.А. Свечников, 1947). Гонады в
некоторых опытах почти не теряют своего веса,
причём сексуальная способность
голодающих животных, особенно самцов,
сохраняется долго (Ф.Г. Биддер и А.А.Шмидт).
Однако, другие авторы (Манассеин,
Краевский, Адо) свидетельствуют о существенной
атрофии гонад (до 40%) и о нарушении,
особенно у голодающих людей половых
отправлений. Так, у голодающих женщин
отмечают аменорею, у мужчин — асперма-
тогенез. К счастью, имеются свидетельства,
что голод не оказывает необратимо
стерилизующего эффекта. Так, израильский врач
М. Дворецкий сообщил о крайне высокой
плодовитости в семьях, образованных
лицами, перенесшими алиментарную
дистрофию во время заключения в нацистских
концлагерях (1957).
38 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия(з
Фактом является привилегированное
положение плода при голодании по
отношению к материнскому организму. Хотя
недоедание приводит у беременных к рождению
детей с внутриутробной гипотрофией, но
весовые потери материнского организма,
относительно намного более значительны,
чем у плода (Рудольский, Дьяченко).
Недоедание ведет к сокращению и
прекращению лактации и снижению белковой
и жировой полноценности грудного
молока у кормящих женщин (Дж.Б. Мэйсон,
И.Х.Розенберг, 1994).
ПЕРИОДЫ ГОЛОДАНИЯ
И ИХЭНДОКРИННО-
МЕТАБОЛИЧЕСКАЯ
ХАРАКТЕРИСТИКА
Полное голодание делят по особенностям
обмена веществ и энергии на несколько
периодов. Это перекликается с клинической
классификацией на три степени тяжести
(соответствующие идущим друг за другом
периодам).
В классический период развития учения
о голодании патофизиологи основывали
свои заключения на наиболее общих
макропоказателях хода обменных процессов.
П.П. Авроров по удельной теплопродукции
выделял начальный период голодания,
характеризующийся ограничением окислений
и повышением дыхательного
коэффициента (ДК), наиболее длительный период
стабилизации удельной теплопродукции
на субнормальных уровнях при сниженном
ДК и непродолжительный терминальный
период активизации окислений с
повышением ДК. При этом, кривая выведения
мочевого азота была, до известной степени,
параллельна колебаниям удельной
теплопродукции. Полного параллелизма, однако,
не наблюдалось. Известно, что у здорового
человека 1-1,3 мг азота мочи приходится
на каждые 1000 калорий основного обмена.
Однако, при голодании происходит пере-
аминирование и окислительное дезами-
Эокринно-метаболические нарушения)
нирование аминокислот скелетных мышц,
позволяющее сберечь белок висцерального
пула. Резкое повышение выведения азота
запаздывает по отношению к
кратковременному начальному приросту Д К и относится
не к 1-му, а к 3-5 дням полного голодания.
Начиная со 2-й и примерно до 8-й недели
голодания, у человека выведение азота
значительно снижается и стабилизируется.
Затем следует терминальный скачок
выведения азота, о котором шла речь выше.
Клиницисты еще в XVIII веке выявили
сухую и отёчную формы голодания (см.
ниже «Квашиоркор и алиментарный
маразм»), но, несмотря на явные отличия
их симптомов, отметили их единое
стадийное течение. В настоящее время ясно,
что компенсация при голодании
обеспечивается использованием жира и
аминокислот соматического пула (жировой
ткани, скелетных мышц), а декомпенсации
соответствует начало интенсивного
использования белков висцеральных органов
на энергетические нужды. Сухая форма
голодания характеризуется относительно
длительным компенсированным периодом,
а отёчная — более коротким. С учётом этих
данных, может по-прежнему быть полезна
старая клиническая классификация
голодания по Л.И. Виленскому (1943), Г.Ф. Лангу
(1943), А.Л. Мясникову (1945), которая
подразумевает:
>- лёгкую или амбулаторную стадию,
соответствующую эндогенному питанию за
счёт углеводов и, главным образом,
жиров. При данной стадии имеется общая
слабость, угнетение психики, понижение
физических и умственных
способностей, сухость и бледность кожи, дефицит
массы до 20 %, могут быть эпизодические
преходящие отёки, но нет выраженных
симптомов гиповитаминозов.
Сохраняется чувство голода и нормальное
пищевое поведение, при откармливании
пища усваивается.
>■ сред нетяжёлую или стационарную
стадию, когда больной начинает
использовать белки крови и мышц. Дефицит веса
20-40%, при протекании белково-энер-
Голодание 39
гетическои недостаточности по квашиор-
корному типу (см. ниже) характерны
выраженные отёки. Могут быть адинамия,
эпизодически — поносы, присутствуют
симптомы полигиповитаминоза,
пищевое поведение может быть изменено до
апатии, чувство голода притуплено. При
откармливании усвояемость пищи
понижена и требуются специальные режимы
питания.
>- тяжёлую, необратимую стадию — с
дефицитом массы более 40%,
угрожающими жизни авитаминозами,
вторичными инфекциями, анасаркой, диареей,
далеко зашедшими
дегенеративно-дистрофическими изменениями внутренних
органов, полной неусвояемостью пищи
при попытках откармливания.
Данные современной патохимии
позволяют раскрыть механизмы эндокринно-ме-
таболической адаптации, лежащие в основе
периодизации голодания.
Процессы, которые происходят в
организме взрослого человека, если его внезапно
лишить пищи, характеризуются этапнос-
тью.
При голодании можно
выделить период экстренной
адаптации, период долговременной
стабильной адаптации и
период декомпенсации, каждый из
которых имеет свои особенные
черты.
Рассмотрим вначале период
экстренной адаптации (рис. 6).
Первое, что произойдет при
прекращении поступления
пищи,— снизится уровень
глюкозы в крови, а это приведёт
к снижению секреции
инсулина и повышению выработки
глюкагона клетками островков
Лангерганса. При выраженном
стрессе и при ускоренном
голодании секреция инсулина
может оставаться высокой,
однако превалирует действие
контринсулярных гормонов.
Повышение уровня глюкагона обусловит
стимуляцию киназы фосфорилазы в ге-
патоцитах, что, в свою очередь, приведет
к активации фосфорилазы и стимуляции
гликогенолиза. Не следует также забывать
и о таких эффектах глюкагона, как
торможение синтеза белков на рибосомах.
Дыхательный коэффициент в первые
часы голодания стремится к единице, кар-
бонурический, как правило, не изменяется.
Если пища не поступает, то запасы
гликогена в основном истощаются через 12-24 часа
и далее должны включиться иные
механизмы обеспечения потребностей в энергии.
Главный среди них — глюконеогенез.
Стимуляция этого процесса связана с голодным
стрессом.
Голодание приводит к возбуждению
центра аппетита в вентролатеральном
гипоталамусе из-за гипогликемии, а также, в
меньшей степени, в силу снижения притока
аминокислот в кровь. Играет роль и отсутствие
нервных импульсов от растянутых
пищеварительных органов, а также понижение
панкреатической секреции инсулина и
дуоденальной продукции холецистокинина,
стимулирующих в норме вентромедиальный
гипоталамический центр насыщения. При
| торможение1 синтеза белка \
глюконеогенез
ДК\ Нмочи\ c/N\)*+
транспорт
аминокислот
в висцеральный
отсек из
соматического
Рис. 6. Эндокринно-метаболические изменения в период
экстренной адаптации при голодании.
Сокращения:ЦК—дыхательный коэффициент, А/мочи — общий азот мочи,
GW—карбонурический коэффициент
40 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
голодании энтериновая система
активизирует выработку нейропептида Y, галанина
и мотилина, стимулирующих центр голода.
В дальнейшем высокая активность
центра голода и низкая — центра насыщения
поддерживается из-за снижения массы
жировой ткани и пониженной продукции
адипоцитами голодающего организма ано-
рексигенного гормона кахексина, а также
стимулятора чувства насыщения лептина
(см. также «Устройство липостата» ниже).
Таким образом, при голодании чувство
голода достигает мучительной интенсивности
и сохраняется в течение значительной
части компенсированного периода голодания
(пищевая доминанта — по А.А.
Богомольцу). Затем его интенсивность ослабевает,
вероятно, вследствие утомления центра
(Л.Р. Перельман) или же в силу продукции
при осложненном инфекциями голодании
анорексигенных цитокинов.
Длительное возбуждение центра голода,
как источник отрицательных эмоций, ведет
к активации амигдало-лимбических
образований, запускающих стресс (см. том I
настоящего руководства, гл. 18). Гормоны
стресса, вызванного голодом как
стрессором,— прежде всего, АКТГ, катехоламины,
глюкокортикоиды и вазопрессин —
довершают стимуляцию гликогенолиза и
активизируют глюконеогенез в печени.
Помимо того, что продолжающаяся
секреция глюкагона стимулирует гормон-
чувствительную липазу в гепатоцитах, что
per se способствует возрастанию уровня
окисляемых в гепатоцитах жирных кислот,
липаза, расщепляющая триацилглицериды
в липоцитах, активируется катехоламинами
и глюкокортикоидами, а ключевые
ферменты глюконеогенеза — глюкокортикоидами
и глюкагоном.
АКТГ при стрессе действует системно
и способствует липолизу в соматическом
жире, а также тормозит синтез мочевины
печенью, что влияет на усиление
использования аминокислот, как энергетического
материала. Под влиянием АКТГ в голодающем
организме растёт продукция СТГ,
оказывающего вместе с глюкокортикоидами,
катехоламинами, вазопрессином и глюкагоном
выраженный антиинсулиновый эффект.
СТГ опосредует свои анаболические
эффекты через систему соматомединов,
вырабатываемых печенью и другими тканями
и органами. Но при голодании
производство соматомединов (особенно, соматоме-
дина С) резко снижается. Липолитический
эффект СТГ, напротив, не связан с сомато-
мединовым посредником. Все это
обеспечивает катаболическое использование жиров
и аминокислот из соматического пула и
приоритетную поставку энергии в органы,
располагающие инсулиннезависимыми
транспортёрами глюкозы.
Глюкокортикоиды активизируют
превращение глюкогенных аминокислот в
глюкозу. Они тормозят синтез белка в скелетных
мышцах, соединительной ткани, лимфоид-
ных органах, коже и жировой ткани.
Активизируется под влиянием кортикостероидов
и транспорт аминокислот, прежде всего,
аланина, из мышц в гепатоциты,в
противоположность торможению их захвата в
тканях соматического отсека (см. т. I данного
руководства, стр. 554-556).
Вазопрессину, особенно важному
регулятору первого периода голодания,
принадлежит и еще одна способность —
активизировать липолиз и захват жирных кислот
печенью. Вместе с тем, считается, что этот
нонапептид тормозит синтез кетоновых тел
и противостоит непосредственному кето-
генному эффекту такого гормона голодного
организма, как глюкагон. Характерно, что
кетоз при голодании развивается не в
первые дни, а отсроченно, когда продукция
вазопрессина падает (о чём можно судить по
возрастающему в ходе голодания диурезу),
а уровень глюкагона остается высоким.
Таким образом, во 2-7 день голодания
доминирует стимуляция липолиза и
глюконеогенеза. Мозг в этот период по-прежнему,
как и до голодания, способен использовать
для энергетических нужд лишь глюкозу или
глюкогенные аминокислоты. На
церебральные энергетические нужды, в основном,
и направляются эквиваленты,
полученные от усиления глюконеогенеза. Чтобы
Голодание 41
обеспечить мозг энергией, органы и ткани,
способные использовать, непосредственно,
жирные кислоты и кетоновые тела в
качестве топлива (сердце, скелетные мышцы,
кора почек), переключаются на эти
продукты липолиза и конверсии кетогенных
аминокислот. Мозговое вещество почек, лим-
фоидные органы, периферические нервы
и другие органы, перешедшие на гликолиз,
поставляют пируват и лактатв гепатоциты,
которые, за счёт энергии утилизации жиров,
превращают эти метаболиты в реакциях
цикла Кори в глюкозу, питающую мозг.
Итак, начальный период экстренной
адаптации при голодании состоит в активации
гликогенолиза, полном использовании
запасов углеводов и стимуляции глюконеоге-
неза при переброске энергетических
эквивалентов из соматического отсека (жировая,
соединительная ткань и скелетные мышцы)
в висцеральный. Этот период нельзя
упрощенно характеризовать, как существование
за счет углеводов или углеводов и жиров,
так как в глюконеогенезе утилизируются
и аминокислоты соматического отсека
и крови. По данным Т. Аддиса и соавт.
(1940), мобилизация белка из печени, крови
и органов ЖКТ в первую неделю голодания
даже более активна, чем из скелетных мышц.
Процент калорий, получаемых из белка,
на 2-7 день голодания, согласно
классическим наблюдениям Рубнера, возрастает (до
11-8% против 5% —в 1-й день). Именно
вследствие вышеописанных гормонально-
метаболических сдвигов в первый период
голодания, основной обмен в начале этого
периода повышается, а затем прогрессивно
снижается. Биосинтез мочевины
уменьшается, доля немочевинного азота в моче
растёт. Начинается возрастание карбону-
рического показателя Рубнера. Продукция
мочевой кислоты в тканях понижается.
Существенного накопления кетоновых тел
в первые дни голодания еще не происходит.
Ежесуточные весовые потери в первый
период голодания максимальны.
Если нормальный взрослый мужчина
теряет в день с мочой до 3,1 г азота (с
фекалиями — до 2,5 и через кожу до 0,5 г),
то при голодании в первый период потери
азота достигают 12 г/сутки, а затем
понижаются примерно вдвое и стабилизируются
на этом уровне.
Снижение интенсивности процессов де-
заминирования и переаминирования
аминокислот, а также изменения в энергетике
мозга знаменуют переход ко второму
периоду — стабильной долговременной адаптации
при голодании (рис. 7).
Второй период голодания наиболее
длителен и определяет всю его возможную
продолжительность. Он начинается со 2-й недели
голодания. Главное, чем знаменуется его
начало — уменьшение использования аминокислот
с целью глюконеогенеза, нарастание
продукции и концентрации кетоновых тел и начало
прямого преимущественного использования
мозгом кетонов, в частности, р-оксибутирата
в качестве топлива. Кетоновые тела при этом
почти на 70 % покрывают энергетические
нужды мозга. Скорость глюконеогенеза падает
в 3-5 раз, суточные потери белка
стабилизируются, утилизация жира продолжается
высокими темпами. Гормональные изменения,
свойственные данному периоду голодания,
включают понижение продукции стрессорных
гормонов, при сохранении высокого глю-
кагон-инсулинового соотношения. Важной
особенностью эндокринно-метаболической
адаптации является изменение тироидной
секреции. При голодании продукция
тироксина и конверсия тироксина в
высокоактивный трийодтиронин снижаются. Возрастает
превращение тироксина в реверсированную
форму трийодтиронина. Как указано ниже
в разделе «Патофизиология эндокринной
системы», в тканях, чувствительных к Т4,
в основном, печени и почках, возможны
различные варианты дейодирования тироксина:
>- дейодирование наружного кольца,
которое приводит к образованию активного
трийодтиронина (Тз), то есть 3,5,3'-три
йод-Ь-тиронина. Т3 в 4 раза активнее Т4
в метаболическом отношении;
>- дейодирование внутреннего кольца
(ведущее к образованию реверсивного Тз или
рТ3, то есть 3,3',5'-трийод-Ь-тиронина).
42 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
глюконеогенез
АК0А
V^
дефицит
кетокислот
блокада
утилизации
АК0А
f глюкокортикоидов
4 конверсии Т4 в pTj
Рис 7. Эндокринно-метаболические изменения в
период долговременной адаптации при голодании.
Сокращения—согласно списку сокращений
Реверсивный рТз либо не обладает кало-
ригенным и метаболическим действием,
либо слабо противодействует влиянию Тз, а
потому уменьшает периферические
эффекты тироидных гормонов.
В норме примерно по 40 % дневной
продукции Т4 конверсирует в 2 эти формы
Т3. Но это возможно только при высокой
активности дейодиназы. Ее подавление
гормонами стресса при голодании ведет к
снижению образования активного Т3 и
смещению равновесия в сторону продукции
реверсивного рТ3. В результате происходит
существенное понижение интенсивности
работы калий-натриевых АТФаз всех
клеток тела, что выражается в значительном
(не менее 10%) уменьшении основного
обмена на единицу массы
тела —в течение большей
части данного периода
голодания. Мозг, гипофиз
и плацента оказываются
и здесь в
привилегированном положении, ибо
в них имеется изофер-
мент дейодиназы,
малочувствительный к такому
ингибированию.
Очевидна адаптивная роль этих
изменений при голодании.
Поскольку деиодирование
наружного кольца требует
участия восстановленной
формы НАДФ, именно
наличие этого
кофактора может определять
скорость реакции, а так
как НАДФН образуется
в ходе пентозного цикла
из глюкозы, активность
дейодирования Т4 в Т3
может понижаться при
голодании.
Конечно, снижение
активности
калий-натриевой АТФазы не проходит
бесследно для
организма. В сердце оно
обусловливает понижение
лабильности проводящей системы и
тенденцию к брадикардии. В почках снижается
натриевый градиент и падает способность
концентрировать мочу (Р.Б. Бэроун, 1987),
что приводит, даже при отсутствии в них
дегенеративных изменений, к полиурии
и поллакиурии, типичной для голодания
(В.А. Свечников, 1947). В ЖКТ это отчасти
определяет понижение перистальтики,
которая в начале голодания стимулировалась
мотилином и была очень активна. Данное
нарушение служит одним из механизмов
голодных запоров. Общее поведение
голодающего животного или человека меняется
в сторону меньшей спонтанной активности,
развиваются сонливость, апатия и
понижение умственных способностей, памяти
C/Nk
оо\
Nmohu ♦
ЛПОНП{
XHk
Голодание 43
и внимания. Понижается частота дыхания.
Экономия ресурсов во второй период
голодания выражается в уменьшении рассеяния
тепла, развивается относительная ишемия
кожи и падает ее температура, появляется
зябкость, снижается до нижней границы
нормы температура тела. По И.М. Флекелю,
у 41 % голодающих пациентов температура
тела была ниже 35 °С (1942). До известной
степени всё это напоминает
соответствующие изменения при гипотирозе, но на фоне
исхудания.
Значительный кетоацидоз ведет к ке-
тонурии и повышению карбонурического
коэффициента при понижении дыхательного
до 0,7 (а при окислении более насыщенных
жирных кислот — и ниже), что соответствует
метаболической ситуации превалирующего
окисления липидов. Считается, что, как
и при сахарном диабете, большое значение
для развития кетоза имеет невозможность
или ограниченность иных путей утилизации
ацетил-коэнзима А, полученного в результате
глюконеогенеза и распада жирных кислот.
Цикл трикарбоновых кислот не
справляется с его утилизацией, так как низкие
уровни оксалоацетата не позволяют вовлечь
в превращения достаточные количества
данного метаболита. Щавелево-уксусная
кислота использована в реакциях
глюконеогенеза, а её регенерация невозможна
в результате ослабления анаплеротического
превращения пирувата при угнетенном
гликолизе. Имеет значение и низкий уровень
активности ключевых ферментов цикла
Кребса в условиях гипоинсулинемии. В
конечном счете, и сердце, и мозг
приспосабливаются к удовлетворению значительной
доли своих энергопотребностей за счет
окисления кетоновых тел. Однако, ацидоз
не остаётся без последствий.
Одновременно с продолжающимся
уменьшением синтеза мочевины,
увеличивается выведение аммиака для нейтрализации
кислых продуктов кетоацидоза, истощаются
щелочные резервы организма. Атрофия
органов ведет к прогрессирующей потере
внутриклеточных калия, кальция и
фосфора с нарастанием их количества в моче.
Содержание хлоридов в моче, напротив,
падает. Продукция и выведение мочевой
кислоты, по сравнению с первым периодом,
нарастают.
Некоторое количество ацетилкоэнзима,
пока доступен НАДФН, может
переключаться на образование оксиметилглутарил-
коэнзима А и способствовать накоплению
холестерина. В результате, у многих
голодающих долго поддерживается гиперхолесте-
ринемия, которую А.Д. Адо (1994) считает
важным фактором в патогенезе голодной
гипертензии в период, предшествующий
глубокой алиментарной дистрофии.
Скорее всего, гиперхолестеринемия зависит
и от повышенной экскреции липопроте-
индов очень низкой плотности (ЛПОНП)
печенью, которая получает в этот период
голодания большую липидную нагрузку
и, по мере возможности, сохраняя баланс,
секретирует ЛПОНП, избегая стеатоза.
Не исключено, что при нефротическом
синдроме, когда белковая недостаточность
и гиперхолестеринемия также сочетаются,
могут действовать сходные
патогенетические механизмы.
Глубокая эндокринно-метаболическая
перестройка, связанная с режимом экономии,
затрагивает работу всех органов и систем,
обусловливая клиническую симптоматику
данного периода голодания.
Наблюдения за пациентами из
блокадного Ленинграда и 32 добровольцами,
находившимися в режиме неполного голодания
в течение 6 месяцев в так называемом
миннесотском эксперименте, показали, что
на протяжении второго периода голодания
происходит ряд закономерных изменений
в деятельности органов и систем:
>• Сократительная способность миокарда
понижается, имеется тенденция к ги-
потензии и резко возрастает риск
возникновения коллаптоидных состояний.
По данным Э.М. Гельштейна (1943),
в состоянии крайнего истощения
учащается внезапная сердечная смерть по
механизму острой аритмии.
>• Уменьшаются дыхательные объемы, и
формируется рестриктивный вариант
44 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
дыхательной недостаточности,
снижается вентиляционный ответ на гипоксию.
По данным Э.Н. Клёнова (1943), ЖЁЛ
в терминальной стадии алиментарной
дистрофии не превышала 1/3 нормы.
Угнетается работа мукоцилиарного
аппарата, что способствует респираторным
инфекциям.
>* В первом и самом начале второго
периода голодания отмечается
активизация секреции пищеварительных желёз
и слизистых желёз ЖКТ. Белки самих
пищеварительных соков при этом
перевариваются и аминокислоты
всасываются, участвуя в глюконеогенезе.
Возможно даже выделение в полость ЖКТ
липидов и углеводов, используемых при
этом своеобразном «самокормлении» для
перераспределения ресурсов в пользу
жизненно важных органов (И.П. Ра-
зенков, 1949; Б.И. Кадыков, 1966).
Этот процесс, однако, ускоряет атрофию
органов ЖКТ. В дальнейшем снижается
секреция желез желудочно-кишечного
тракта, кислотность желудка и
переваривающая способность
пищеварительных соков. Отмечается атрофия ворсин
тонкого кишечника с лимфоцитарной
инфильтрацией его слизистой
оболочки и понижением скорости всасывания
аминокислот и других субстратов.
Страдает мембранное пищеварение. Даже
в отсутствие характерных для далеко
зашедшей алиментарной дистрофии
инфекционных поражений кишечника
у больных могут развиваться мальаб-
сорбция и дистрофическая диарея, еще
больше усугубляя дефицит белка и
незаменимых компонентов пищи.
Присутствуют стеаторея, мальабсорбция
углеводов и дисбактериоз. Все это создает
большие сложности при осуществлении
откармливания голодающих.
>* Почки при голодании, если нет
сопутствующей патологии, например, гломеру-
лонефритов, относительно хорошо
сохраняют свою гистологическую структуру
и функции. Однако, описанные выше
особенности натриевого градиента
приводят к полиурии и поллакиурии. Весьма
характерна никтурия. Некоторые авторы
указывают на связь этих явлений с
поведением голодающих, которые зачастую
употребляют повышенные количества
воды. В конце второго периода голодания
диурез значительно уменьшается.
>* Мобилизация энергетических ресурсов,
происходящая за счёт соматического
отсека организма в пользу отсека
висцерального, сопровождается характерными
мышечными и костно-суставными
болями. Эти субъективные проявления ярко
описываются больными и накладывают
отпечаток на их поведение: «Вот уже
много недель я недоедал, и теперь
наступил ощутимый упадок сил. Когда мне,
так или иначе, удавалось раздобыть 5
крон, их хватало ненадолго, и поэтому я
не успевал оправиться до новой
голодовки. Хуже всего мне повиновались спина
и плечи; боль в груди была не слишком
сильна, и я мог унять её, хорошенько
прокашлявшись или подавшись всем
телом вперёд; а вот спина и плечи были
для меня сущим наказанием. Отчего
судьба так жестока?» (К. Гамсун).
>* Иммунная система поражается при
голодании особенно тяжело — как вследствие
пищевого дефицита, так и под влиянием
голодного стресса (см. том I данного
руководства, стр. 515). Следует учесть, что
вторичный иммунодефицит, связанный
с голоданием, приобретает особую
остроту по мере расходования белков
висцерального отсека организма. Поэтому
он в наибольшей мере характерен для
квашиоркорной формы белково-энерге-
тической недостаточности. Абсолютное
количество лимфоцитов в
периферической крови ниже 1200 на микролитр
считается характерным для голодания
и используется как индекс,
свидетельствующий о степени истощения
висцерального пула белка (P.M. Расселл,
1987). При марантической форме
голодания также имеются нарушения
иммунитета, особенно значительные
Голодание 45
в конце второго периода и в
терминальном периоде голодания. У голодающих
детей атрофируется вилочковая железа.
Имеется глубокий дефект Т-клеточных
функций, вплоть до анергии при кожных
пробах ГЗТ. Не удивительно, что опыт
медиков блокадного Ленинграда
продемонстрировал, что у голодающих очень
часто активизируется и диссеминирует
латентная туберкулёзная инфекция
(Г.С. Япольский, 1944). В-клеточные
функции, включая синтез антител, также
страдают, но в различной степени,
пропорционально потерям висцерального
белка. При марантических
(компенсированных) формах голодания синтез
иммуноглобулинов сохранён в большей мере,
чем при квашиоркорных (декомпенси-
рованных). Кратковременное голодание
подавляет аутоаллергию, как полагают, в
связи со снижением продукции лептина
(Дж. Матарезе и соавт., 2002).
Голодание сопровождается нейтропенией
и снижением подвижности фагоцитов,
но фагоцитоз осуществляется без
значительных нарушений. Имеется снижение
сывороточных концентраций многих
факторов комплемента и
неспецифических защитных белков, например, лизоци-
маи интерферонов. Атрофия и сухость
кожи, дегенерация слизистых оболочек,
иммунодепрессивные последствия
нехватки цинка и железа — дополняют
картину вторичного иммунодефицита.
При тяжелой алиментарной дистрофии
замедляется заживление ран. Инфекции
протекают без типичной температурной
реакции, гиперергические ответы
иммунной системы становятся невозможны.
Полиинфекционный синдром при
голодании включает бактериальные и
вирусные бронхиты, пневмонии,
энтероколиты (В.Д. Цинзерлинг, 1943). По данным
В.А. Свечникова, не менее 80 % умерших
от голода в годы блокады страдали
инфекционными поносами, в
частности, исключительное распространение
приобрела в блокадном Ленинграде
дизентерия. В то же время, практически не
наблюдались обострения бронхиальной
астмы, резко уменьшились проявления
атопических заболеваний (А.Д. Адо,
1994). Характерно, что в условиях
голодания сывороточная концентрация
глобулинов острой фазы повышена,
особенно, при квашиоркоре. Это
свидетельствует об участии цитокинов-индук-
торов преиммунного ответа в патогенезе
перераспределения энергоресурсов при
голодании, особенно, осложнённом
инфекциями.
>■ Со стороны красной крови при
голодании наблюдается полидефицитарная
* анемия, как правило, гипорегенератор-
ного характера, гипохромная или, реже,
нормохромная. Если резервы витамина
В12в организме голодающего были
незначительны, то возможно и развитие
гиперхромной анемии, но без типичного
значительного количества мегалобластов
(В.А. Свечников, 1947).
>* Функции печени при голодании
изменяются по-разному, в зависимости от
преобладания белковой либо
калорической недостаточности (см. ниже «Ква-
шиоркор и алиментарный маразм»).
По свидетельству А.А. Покровского
и Л.П. Крыстева (1977), при голодании
в печёночных клетках усиливаются
явления лизосомальной аутофагии,
редуцируются гладкий и шероховатый эндо-
плазматический ретикулюм, появляются,
в результате слияния митохондрий, их
гигантские формы. При квашиоркорной
форме голодания действие цитокинов
ведёт к раннему ограничению синтеза
альбумина, транстиретина, апопротеи-
нов и других печёночных белков. В то же
время, в печень приходит значительное
количество жирных кислот из жировой
ткани. Экскреция липидов с жёлчью
ограничена, так как ее продукция при
голодании падает. Выделение липидов
в кровь в форме ЛПОНП возможно
только при интенсивной продукции
апопротеинов липопротеидов и при
достаточном количестве метильных
донаторов (холина, бетаина, витамина
46 АЖ Зайчик, Л.П.Чурилов Патохимия (ai
В12). Так как эти условия не могут быть
выполнены, развивается дисбаланс
между поступлением и экскрецией ли-
пидов в печени, и может формироваться
стеатоз печени, рассматриваемый
многими авторами, как «мягкая» подострая
форма некробиоза гепатоцитов (см. том
I данного руководства, стр. 184). При
марантической форме голодания синтез
белка в печени страдает в меньшей
степени. Изменения в органе соответствуют
картине простой атрофии.
Терминальный период декомпенсации при
голодании наступает при потере 40-50 %
массы тела с утратой 100 % запасного жира
и почти 97 % жира внутренних органов. Он
характеризуется усилением распада белков
висцерального отсека организма.
Увеличивается выделение мочевого азота, как за
счёт немочевинных фракций, так и за счёт
мочевины. Отмечается возрастающая
потеря с мочой аминокислот, пептидов, калия,
фосфора. Гиперурикемия и уратурия
свидетельствуют об активном распаде нуклеоп-
ротеидов. Несколько повышается, в расчёте
на килограмм массы тела, основной обмен.
Дыхательный коэффициент приближается
к 0,8. Координирующая функция нервной
системы нарушается, следуют парезы и
параличи, в возникновении которых
определённую роль играют и гиповитаминозы.
КВАШИОРКОР
И АЛИМЕНТАРНЫЙ МАРАЗМ
Белково-калорическая недостаточность
известна в двух крайних формах, между
которыми существует значительное
количество промежуточных смешанных случаев. Эти
два вида — квашиоркор и алиментарный
маразм. Первый еще именуют
несбалансированной, а второй — сбалансированной
формой белково-энергетического дефицита.
Слово «квашиоркор» происходит из языка
западно-африканского народа га,
проживающего на территории современной Ганы. Оно
означает «болезнь первенца после рождения
ю-метаболические нарушения)
младшего». После отнятия от груди
первенец лишается источника полноценного
белка и его питание, субдостаточное в
калорическом отношении, становится,
преимущественно, углеводистым. Это приводит
к развитию характерной отёчной формы
белково-энергетической недостаточности,
сопровождаемой ранней потерей протеинов
из висцерального пула организма.
Алиментарный маразм
(мумифицированная или сухая форма алиментарной
дистрофии) характеризуется длительным
компенсированным течением, когда нутри-
енты мобилизуются из соматического отсека
тела, а висцеральный пул белка дольше
сохраняется. При этом значительной
степени достигает атрофия мышц и жировой
клетчатки, но долго не возникает заметных
отёков. У взрослых при полном голодании,
не осложнённом инфекциями, процесс
развивается, чаще всего, именно по этому
типу. Ещё раз подчеркнём, что квашиоркор
и маразм не являются «чистыми» формами
белкового и энергетического дефицита;
в реальности их симптоматика может
смешиваться у одного индивида на разных
стадиях голодания. Гормональные различия,
диктующие особенности этих двух форм,
по-видимому, включают значительный
избыток глюкокортикоидов при маразме
и отсутствие такого избытка с выраженным
вторичным гиперальдостеронизмом, а также
системными эффектами цитокинов, — при
квашиоркоре.
Квашиоркор, в силу относительной
сохранности углеводистого питания,
отличается более высоким содержанием инсулина,
при этой форме голодания отмечают также
прооксидантные сдвиги в редокс-состоянии
клеток, способствующие отекам.
Было бы ошибочно, полагать, что
квашиоркор и маразм — это чисто эндемические
формы голодания. Их, в принципе, можно
встретить в любых районах мира при
наличии, преимущественно, белковой или,
преимущественно, калорической
недостаточности. Однако, имеется и известная
природно-географическая приуроченность
этих форм голодания: так в Африке ма-
Голодание 47
рантическая форма характерна для стран
Сахеля и бассейна среднего и нижнего
Нила, а квашиоркор типичен для
тропической части континента. В отличие от
маразма, квашиоркор в чистой форме редко
встречается вне своего основного ареала,
охватывающего беднейшее население стран
тропической Африки, Мадагаскара,
Центральной и Южной Америки, Филиппин,
Индии и Бирмы.
При обеих формах голодания отмечаются
общие признаки, например, полидефицитар-
ная анемия и гиповитаминозы.
В таблице 1 приведены суммирующие
отличительные черты этих форм голодания.
Для оценки степени белково-энергети-
ческой недостаточности и относительной
потери белка из соматического и
висцерального пулов предложены определённые
клинико-лабораторные критерии.
Таблица 7
Формы белково-энергетической недостаточности
Форма
Дефицит
Водно-солевой обмен
Пищевое поведение
Печень
Белки крови
Внешний вид
Кожа и ее производные
Иммунная система
Гормональный фон
[жкт
1 Экскреция креатинина
Прогноз
Квашиоркор
В основном, белковый
Отеки, асцит, задержка натрия, гипокали-
емия, гипофосфатемия, гипомагнезиемия
Апатия, отсутствие аппетита
Увеличена, болезненна, выражен её стеатоз,
изредка возможен цирроз.
Выраженная гипоальбуминемия, снижены
транстиретин,трансферрин и ретинолсвязы-
вающий белок, повышены глобулины острой
фазы. Снижены ЛПОНП и ЛПНП
Лунообразное лицо, вздутый живот. ППСМСП
не понижена
Пятна нарушения пигментации и
гиперкератоза, «эмалевый или чешуйчатый дерматоз»,
у белых—эритема. Полоски обесцвечивания
на волосах (симптом флага), ногтях. Волосы
светлеют и приобретают рыжеватый оттенок,
становятся ломкими и легко выпадают
Выраженный Т- и В-клеточный
иммунодефицит
Гиперальдостеронизм, высокий уровень
кахектизирующих цитокинов, относительно
меньше глюкокортикоидов
Выраженная атрофия тонкого кишечника,
паразитарные и микробные инфекции
Умеренно повышена
Более пессимистический, откармливание
затруднено, риск инфекционных
осложнений выше.
Алиментарный маразм
И калорический, и белковый
Без отёков, в финальной стадии
— гиперкалиемия
Активное. Аппетит присутствует |
Умеренная атрофия
На нижней границе нормы. ЛПОНП
могут повышаться
Атрофия лицевых мышц,
ввалившиеся щёки и височные ямки.
Стеблевидные конечности, резкое
снижение ППСМСП, атрофия
подкожного жира и скелетных мышц
(«кожа — да кости»). Задержка
роста. Живот втянут
Сухая, морщинистая, с признаками
полигиповитаминозов,
пониженным тургором. Тусклые, сухие,
тонкие волосы
Преимущественно, нарушены
Т-клеточные функции
Крайне высокий уровень
глюкокортикоидов, глюкагона и сома-
тостатина
Атрофические изменения менее
выражены
Сильно повышена
Лучше, чем при квашиоркоре
48 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
На дефицит жира в организме указывает
толщина жировой складки над трёхглавой
мышцей плеча менее 5 мм у мужчин и менее
11 мм — у женщин.
Дефицит белка в соматическом отсеке
организма проявляется понижением
площади поперечного сечения мышц середины плеча
(ППСМСП в мм2). Данный индекс
вычисляют по формуле Мэйсона-Розенберга:
ППСМСП=(С-яТ)2/4я,
где С — окружность середины плеча, мм,
Т— кожная складка над трицепсом, мм.
Существуют специальные таблицы, по
которым ППСМСП ниже 10 перцентилей
от половозрастной нормы указывает на
мышечную атрофию.
Суточная экскреция креатинина с мочой
у мужчин среднего роста колеблется в
районе 1,4-1,6 г, у женщин — между 0,8 и 1,0
г. Её повышение свидетельствует о потере
белка соматического пула.
Динамометрия мышц кисти дает при этом
пониженные результаты, хорошо
коррелирующие со степенью атрофии мышц.
Состояние висцерального пула белка
оценивается по лабораторным показателям
крови: при его истощении уровень
альбумина падает ниже 35 г/л (латентный период
изменений — 20 дней); уровень трансфер-
рина — ниже 2,0 г/л (латентный период —
8-9 дней); уровень транстиретина — ниже
0,2 г/л (с латентным периодом — 2-3
дня); ретинолсвязывающего белка — ниже
0,03 г/л (латентный период — менее суток).
Косвенно, об истощении пула
висцерального белка свидетельствуют также лимфопения
и анергия при кожных аллергопробах.
НЕЙРОГЕННЫЕ АНОРЕКСИЯ
И БУЛИМИЯ
«Не делайте из еды культа».
И.А. Ильф, Е.П. Петров
«Золотой телёнок»
Одно из самых загадочных заболеваний,
провоцирующих эндогенное голодание, —
нейрогенная (психогенная) анорексия (НА).
Нейрогенная анорексия - гипоталамическое
расстройство, ведущее к нарушению
пищевого гомеостаза, патологическому пищевому
поведению, к отказу от пищи и к
уменьшению веса тела до состояния кахексии.
Случаи НА всегда привлекали не только
медицинское, но и повышенное
общественное внимание. Несомненно, описанные
в истории голодовки духовных подвижниц,
аскетов и столпников, отказывавшихся от
приема пищи, в ряде случаев представляли
собой патологическое пищевое поведение
больных НА. Научное описание НА у
18-летней пациентки дал впервые английский врач
XVII века Р. Мортон (1689). Много позже,
У. Галл (1874) предложил термин anorexia
nervosa. Фотография истощённой больной
с нейрогенной анорексией, впервые
опубликованная в 1913 г. М. Симмондсом, стала
одной из самых часто воспроизводимых
медицинских иллюстраций.
НА — болезнь юных девушек и
молодых женщин. 95 % больных женского пола
и у 80% болезнь наступает в первые 7
лет после прихода месячных. Болеют, как
правило, представительницы образованных
слоев населения — в колледжах США
частота недуга достигает 1 %, тогда как
средне-популяционная встречаемость не
превышает 3,27 на 100000. Подростковые
психологи отмечают тревожную тенденцию
учащения НА по мере торжества в mass
media нового идеала женской красоты и
социальности, близкого к «типу будущего»
по П. Матесу (см. т. I данного руководства,
стр. 95-96) — в 80-х годах XX века за 10 лет
частота НА выросла в 6,5 раз!
К НА существует полигенная
наследственная предрасположенность (среди
однояйцевых близнецов индекс конкордан-
тности в 5 раз выше). Определённый модус
воспитания, вырабатывающий повышенное
чувство долга, ориентирующий, скорее,
на соответствие определённому внешнему
стандарту или примеру, чем на
самоактуализацию, способствует проявлению НА.
Фактором риска является и
психоэмоциональный стресс, в частности, порождённый
Голодание 49
исходно повышенной массой тела и
подростковым кризисом самооценки.
Основными диагностическими
признаками НА служат:
> Дизморфофобия, при которой больная
воспринимает собственный вес как
повышенный, собственные формы — как
неизящные и испытывает навязчивое
стремление похудеть. Ощущение
избыточной массы тела не проходит, а
стремление похудеть не исчезает несмотря на
истощение.
> Аменорея (первичная или вторичная).
Эндокринной основой этого симптома
при НА является понижение
продукции гонадотропинов гипофиза и
инфантильная неотвечаемость аденогипофиза
на гипоталамический люлиберин.
> Несмотря на то, что больные хотят есть,
они отрицают или преуменьшают такие
интерорецептивные ощущения, как
голод, утомление, депрессию. Нередко
это сопровождается ритуализованными
упражнениями «for fitness». Характерной
психологической особенностью жертв
НА является большая зависимость от
мнений и оценок окружающих,
отсутствие уверенности в себе.
> Внешний вид больных нередко включает
проявления каротинемической ложной
желтухи. На теле имеются пушковые
волосы («лануго»).
> Из других нейроэндокринных
нарушений при НА типичны гиперпродукция
атриопептинов, гипофункция ренин-
ангиотензин-альдостероновой системы,
артериальная гипотензия и гипокаль-
циемия вплоть до тетании. Отмечаемая
эффективность блокаторов обратного
захвата серотонина в лечении НА наводит
на мысль о нарушении серотонинового
опосредования эффектов лептина на
продукцию стимулятора пищевого
поведения нейропептида Y у таких больных.
В основе патогенеза НА лежат нейроэн-
докринные нарушения. Столь
самоуничтожающая страсть к похуданию не может
возникнуть без участия гипоталамических
механизмов, ведь подбугорье — это отдел
мозга, порождающий страсти (см. т. I,
стр. 530-531). Известно участие
гипоталамуса, амигдалы и лимбических структур
в формировании пищевого поведения.
Так, при разрушении амигдалы возникает
исчезновение пищевых предпочтений, то
есть еда теряет положительные
эмоциональные корреляты (А. Гайтон, 1989).
В.Ю. Шанин (1996) считает, что НА связана
с застойным возбуждением,
охватывающим вентромедиальное ядро гипоталамуса.
Д.А. Дроуссмэн (1987) указывает на
изменение метаболизма андрогенов при НА
с избыточной продукцией этиохолонало-
на вместо тестостерона. Этиохолоналон
известен как активатор катаболических
процессов, ответа острой фазы и
метаболической мобилизации при лихорадке (см. т. I,
с. 550). При НА снижена продукция
эстрогенов, что вызывает остеопороз. Многие черты
эндокринно-метаболической картины НА
совпадают с типовыми изменениями,
наблюдаемыми и при других формах голодания (в
частности, нарушения тироидной и сомато-
тропиновой регуляции). Однако, продукция
вазопрессина понижена, а не увеличена как
при многих других формах голодания.
Наиболее интересной особенностью
гормональной регуляции при НА считается
обнаруженная у значительного числа
пациенток с этим диагнозом аномально высокая
концентрация кахексина в крови. Считается,
что кахексии может вырабатываться мононук-
леарами, адипоцитами и, возможно,
опухолями глиального происхождения. В
спинномозговой жидкости больных НА отмечается
повышенная концентрация
аппетит-подавляющего гормона холецистокинина (см. т. I,
стр. 537). Так или иначе, но история изучения
патогенеза НА показывает, что за
психическими симптомами стоят вполне материальные
патохимические механизмы,
преимущественно связанные с патологией гипоталамуса (см.
ниже)... Так, Ю.И. Строев и Е.Б. Карповская
(1998) сообщили о наличии микроаденом
гипофиза и пустого турецкого седла в типичных
случаях НА.
50 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Несмотря на лечение, болезнь
рецидивирует всю жизнь и может вызвать
гибель больных (больничная летальность
до 6%)— от истощения и сопутствующей
гипокалиемии, вызывающей аритмии. Как
пример эндогенного голодания приводим
классическое фото больной с тяжелой
формой кахексии на почве гипоталамических
нарушений, наблюдавшейся Г. Цондеком
(рис.8).
Более мягкой формой того же
расстройства, что и НА, считается нейрогенная
булимия. При булимии попытки сбросить вес не
характеризуются той степенью самоотре-
Рис 8. Эндогенное голодание. Кахексия у
больной вследствие гипоталамических нарушений,
(по Г. Цондеку, 1932).
чения и фанатизма, как при НА. Поэтому
через некоторое время соблюдения диеты
следует эпизод «запойной еды», когда
больная поглощает огромное количество пищи,
причём совсем не относящейся к
деликатесам. Рекордный уровень ежедневного
потребления энергии при этом превышает
50000ккал/сутки! После такого
пароксизма наступают раскаяние и попытки
«очиститься», в том числе — с помощью
индукции рвоты и применения
слабительных. Нейрогенная булимия была известна
еще древнеримским авторам, оставившим
нам описания симпосиев, на которых их
соплеменники после переедания
вызывали у себя рвоту, щекоча глотку птичьими
перьями. Среди современных завсегдатаев
фуршетов и презентаций описан
профессиональный синдром «социального едока»,
напоминающий мягкую форму булимии.
В отличие от НА, при нейрогенной
булимии вес держится в норме или
понижен слегка. В отличие от других форм
булимии, приступы повышенного
аппетита чередуются с периодами отказа от
еды и попытками «чисток». Нейрогенная
булимия встречается в несколько раз
чаще, чем НА. В развитых странах не
менее 10% населения испытывали хотя бы
лёгкие формы нейрогенной булимии. Ее
развитию способствуют наследственная
предрасположенность и особенности
семейного воспитания. В семьях, где
существовал культ здоровой еды, а праздники
содержали обильное застолье в качестве
центрального момента, формируется
отношение к еде, как к источнику большого
биологического и психического
удовлетворения, как к чему-то выходящему за
рамки обычного насыщения. Статистика
указывает на повышение частоты булимии
при таком семейном анамнезе.
Нейрогенная булимия связана с особенностями
продукции эндогенных опиатов после
акта еды. Указывалось также на дефицит
аппетит-подавляющих регуляторов се-
ротонина и холецистокинина в
спинномозговой жидкости пациенток на высоте
булимического приступа. При нейрогенной
Голодание 51
булимии, в отличие от НА, продукция ЛГ
в ответ на люлиберини секреция пролак-
тина повышены, половая функция более чем
у половины больных сохранна. Наиболее
интересной особенностью гормональной
регуляции при нейрогенной булимии может
оказаться гиперпродукция нейропептида Y,
регулятора пищевого поведения (Д.У. Фос-
тер, 1994). В целом, эта форма расстройства
пищевого поведения протекает более
благоприятно, чем НА.
Вместе с тем, эпизоды рвоты приводят
к эзофагиту, фарингиту, аспирационным
бронхолёгочным нарушениям и даже
синдрому Маллори-Вейсса и аритмиям,
которые могут вызвать смертельный исход. С
течением времени у больных прогрессирует
депрессия, при тяжелой булимии не менее
5-6% из них предпринимают суицидные
попытки. Интересно, что при НА процент
суицидальных попыток ниже (до 1%).
По-видимому, при НА пациентка следует
избранной программе поведения, находясь
в относительной гармонии с собой, а при
нейрогенной булимии налицо конфликт
поведенческих программ, порождающий
психопатогенные последствия. Оба
расстройства протекают у мужчин тяжелее,
чем у женщин, что установил еще в 1694 г.
Р. Мортон.
ОТКАРМЛИВАНИЕ
ПОСЛЕ ГОЛОДАНИЯ
Откармливание при голодании проходит
успешно при дефиците массы до 45-50%,
а в условиях применения парентерального
питания — и при более значительных
дефицитах (55-60%). Основной сложностью
является резкое снижение толерантности
атрофичных пищеварительных органов
к пище. При квашиоркорной форме
голодания, к тому же, часто отсутствует аппетит,
и имеется апатия.
Обмен веществ при откармливании имеет
свои особенности.
Прибавка в весе при откармливании идёт
быстрее, чем вес терялся в ходе
предшествующего голодания. Имеется положительный
азотистый баланс, а дыхательный
коэффициент возрастает выше единицы из-за
перехода углеводов в жиры (см. раздел
«Дыхательный коэффициент» выше).
Так как для осуществления анаболических
процессов в организме требуется
дополнительная энергия роста (см. выше, рис. 3),
уровень энергозатрат и скорость теплового
рассеяния при откармливании резко возрастают
и превышают норму. Может даже повыситься
температура тела. По-видимому, быстрый
рост объёма жировой ткани ведет к
гиперпродукции адипоцитами лептина, который
известен, как гипоталамический стимулятор
несократительного термогенеза, основного
обмена и теплового рассеяния. Увеличено при
откармливании и специфическое
динамическое действие пищи. Кроме азота, в организме
при откармливании задерживается фосфор,
что связано с восполнением запаса
макроэргов и нуклеопротеидов (Н.Н.Аничков,
1937). Некоторые авторы сообщают также о
задержке серы и хлоридов.
При первых нескольких повторных
голоданиях после откармливания вес
теряется медленнее, чем при начальном
голодании. Однако, ускорения откармливания
не наблюдается. Подобные проявления
тренировки не означают, что «искусство
голодать» может беспредельно повышать
резистентность организма к белково-энер-
гетической недостаточности. Установлено,
что при многократных циклах «голодание-
откармливание» у человека и ряда
животных (собаки, кролики, грызуны, птицы)
происходит накопление
дегенеративно-дистрофических изменений в висцеральных
органах, несмотря на то, что жировые
запасы откладываются эффективно. При
повторных откармливаниях процесс идет
не столь быстро. После определенного
количества циклов голодания-откармливания
способность выходить из состояния
истощения резко ограничивается, и животное
гибнет несмотря на попытки восстановить
его вес. Следовательно, глубокие
многократные повторные голодания переносятся
организмом плохо.
52 А.Ш. Зайчик, Л.П.Чурилов Патохимия
ЛЕЧЕБНОЕ ГОЛОДАНИЕ
Попытки лечебного ограничения питания
предпринимались еще врачами
древности—в античной Греции, Тибете и Индии.
Пифагор был энтузиастом этого метода, что
не помешало ему стать олимпийским
чемпионом по кулачному бою. У Авиценны мы
находим указания на возможность лечения
голодом при «сухих лихорадках». В XVIII веке
сторонниками гипокалорийных лечебных
диет, в частности, при лихорадочных
заболеваниях, были Дж. Броун и Ф. Бруссе,
которым приписывают авторство афоризма:
«Morbum nutris, non aegrotum» — «Болезнь
кормишь, но не больного». Им возражал
Р.Дж. Грейвс: «Вы уморите Ваших больных
голодом!» В XIX веке Л. Лучано создал для
объяснения лечебного эффекта голодания
«теорию регуляции», истолковывавшую
саногенное действие голода как результат
перехода на более экономичные механизмы
обмена. В.В. Пашутин считал, что лечебное
голодание способствует мобилизации
механизмов «активной и пассивной защиты».
Ветеринар Г. Зееланд показал, что при
повторных кратковременных голоданиях, когда
процесс ограничивается самой начальной
стадией, сельскохозяйственные животные
проявляют лучшую упитанность (высший
процент содержания плотных компонентов
в теле) по сравнению с получающими
постоянно-равномерное питание.
Во второй половине XX столетия, когда
в развитых странах возникла проблема
гиподинамии и переедания, и от 20% до 40%
населения во многих регионах оказались
носителями избыточного веса и связанных
с ним болезней, интерес к лечебному
голоданию обострился (П. Брэгг).
Авторы признают эффективность
лечебного голодания при многих заболеваниях.
В поддержку этого метода сошлёмся на
классическое положение Н.Н.Аничкова:
«В общем, следует считать, что при каждом
голодании подвергаются распаду, в первую
очередь, различные наименее деятельные
составные части клеток и тканей, вследствие
чего в некоторых случаях путём голодания
Эокринно-метаболические нарушения)
организму даётся возможность избавиться
от ненужных ему, частью уже отживших
элементов тканей. После голодания
наступает регенерация погибших клеток, и их
функциональные отправления происходят
особенно энергично. С этой точки зрения не
слишком продолжительные периоды
неполного голодания приносят известную пользу
организму» (1937). По Ю.С. Николаеву,
лечебное действие голодания, во многом,
связано с индукцией метаболического
ацидоза и адаптацией к нему (1978).
Голодание с целью лечения применяется,
прежде всего, при
алиментарно-конституциональном первичном ожирении. При этом
следует учесть, что при прекращении
голодания, достигнув формально нормативного
веса, пациент не становится здоровым. Так
как механизмы первичного ожирения
связаны с изменением установочной точки массы
тела гипоталамусом, при отклонении от неё
организм стремится вернуться к прежней
массе. Искусственное похудение
сопровождается гипотирозом, нарушением
температурной адаптации, депрессией, снижением
иммунитета (см. раздел «Ожирение»).
Лечебный эффект от более или менее
продолжительного медицинского голодания
может быть достигнут при аллергических
атопических заболеваниях, ряде
иммунопатологических кожных болезней, некоторых
нервно-психических расстройствах (в
частности, по данным Ю.С. Николаева — при
шизофрении и астено-невротических
синдромах), при гиперинсулинемическом
сахарном диабете, эссенциальной гипертен-
зии, подагре, ревматоидном полиартрите,
болезнях системы пищеварения (Ю.М. Гу-
бачёв, И.Г.Ефимова, 1994). Вместе с тем,
следует отметить нестойкость лечебных
эффектов голодания и необходимость
медицинского контроля этой процедуры.
Лечебное голодание необходимо осуществлять
таким образом, чтобы избегать гиповита-
минозов. Оно противопоказано при многих
заболеваниях и состояниях (беременность,
инсулинзависимый сахарный диабет, надпо-
чечниковая недостаточность, тиротоксикоз,
болезни печени, хронические инфекции
Голодание 53
и гельминтозы, злокачественные
новообразования, сердечная недостаточность,
обострение язвенной болезни, активный
ревматизм и аутоиммунные заболевания
соединительной ткани). Лечебное
голодание не должно превращаться в стрессорное
голодание (В.Ю. Шанин, 1996), имеющее, во
многом, иной гормональный фон. Поэтому
его рекомендуемая продолжительность не
превышает 15 дней включая начальный
период, период разгрузки и период
восстановления. Важной задачей при выходе из
лечебного голодания является постепенное
восстановление нормального питания,
учитывая неподготовленность ЖКТ после
голодания к использованию больших
количеств пищи.
В любом случае, голодание не может быть
рекомендовано как панацея (Г.П. Малахов,
1997) и не должно применяться в режиме
самолечения.
ЦИКЛ ГОЛОДАНИЕ-
НАСЫЩЕНИЕ
Сложные информационные взаимодействия
с участием ЖКТ, островков Лангерганса,
иммунонейроэндокринной системы,
жировых клеток и, в первую очередь,
гипоталамуса контролируют аппетит и чувство
насыщения, предупреждают переедание,
приспосабливают организм к эндогенному
питанию и сопрягают обмен веществ,
пищевое поведение и другие функции тела. Более
подробную характеристику этих
механизмов читатель найдет в главах 6,9 и 14 ниже.
Но именно сейчас, в заключение раздела,
посвященного голоданию, будет уместен их
краткий очерк.
При голодании выделяется гормон
дисперсных эндокриноцитов ЖКТ грелин,
который усиливает аппетит, действуя на ги-
поталамические структуры, производящие
нейропептид У и белок, связанный с агути.
Пища, особенно, углеводистая
(глюкоза— сильнее фруктозы) и белковая (и в
гораздо меньшей степени — жирная), а также
растяжение желудка — подавляют продукцию
грелина. В то же время, метаболические
последствия насыщения (поступление в кровь и
цереброспинальную жидкость глюкозы и рада
аминокислот, переход адипоцитов в «сытое»
состояние) вызывают продукцию инсулина в
В-клетках островкового аппарата
поджелудочной железы и лептина в жировой ткани.
Инсулин и лептин сдерживают выработку
грелина и, на уровне гипоталамуса, стимулируя
выделение нейронами насыщения некоторых
нейропептидов, а нейроглией — и цитокинов
{кокаин-амфетамин зависимый полипептид,
а-меланоцитостимулирутощий гормон, кислый
фактор роста фибробластов). При синергич-
ном действии на гипоталамические нейроны
непептидных регуляторов (2-бутен-4-олид
и 3,4-дигидроксибутаноил-у-лактон) - эти
эффекты подавляют пищевое поведение,
индуцируют чувство сытости, стимулируют
норадренергические механизмы,
обеспечивающие усиление траты калорий,
динамическое действие пищи, тироидную функцию,
ускорение катаболизма. В слаженном
оркестре массостата (липостата) свои партии
исполняют цитокины иммунной системы и
липоцитов - кахексии, ослабляющий эффекты
инсулина, но способствующий катаболизму и
понижению аппетита, а также адипонектин,
частичный антагонист кахексина (по
противовоспалительному, вазопротекторному и
инсулин-сенсибилизирующему эффектам),
который, впрочем, синергичен с кахексином
и лептином в снижении потребления пищи.
Гормон наполненного желудка оксинтомо-
дулин и пептид YY3_36, сокращенно — PYY,
производимый в тонком кишечнике, —
усиливают грелин-подавляющее действие инсулина
и лептина. Насыщению способствует также
гомолог PYY — панкреатический полипептид
островков Лангерганса, хоть он и не влияет
на продукцию грелина. К тому же, различные
гипоталамически контролируемые функции
(поддержание температуры тела, цикл сон-
бодрствование, эмоциональные реакции,
стресс, преиммунный и иммунный ответы)
тесно связаны с этими регуляторными
изменениями и накладывают на них свой
отпечаток. Так, недосыпание сдвигает грелин-леп-
тиновое равновесие в «грелиновую» сторону,
54 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
затрудняя насыщение, преиммунныи ответ
подавляет аппетит (см. т. I, гл. 13,18).
Стоит представить себе всю многокон-
турность и неоднозначность этих связей,
чтобы понять, почему метаболизм здорового
человека так устойчив при экстремальных
колебаниях объема и радикальных
изменениях характера поступающей пищи. Выше
шла речь о мощных механизмах адаптации
к голоданию. Основу дневного рациона
эскимоса может составлять тюлений жир,
у охотника банту — это кровь животных, а в
бассейне озера Танганьика традиционный
продукт — пирожки с прессованными
печеными москитами, коренные бутанцы на
высотах Гималаев поддерживают себя чаем
с ячьим маслом. И в любом случае, даже на
фоне экстремальных климатических
условий, такие разные виды питания позволяют
человеку сохранять здоровье, бодрость и
работоспособность.
Оборотной стороной медали является
большая устойчивость генетически
обусловленных нарушений в системе массостата к
лечению: если его общая установочная точка
сдвинута, как при нервной анорексии и
первичном ожирении, то многоконтурная
защита превращается в фактор, затрудняющий
коррекцию таких состояний. Так, например,
у многих лиц с нервной анорексией
концентрации грелина крайне высоки, но PYY
и панкреатического полипептида — также
повышены, парадоксально низкий уровень
грелина и PYY и высокий лептина —
сопровождает многие случаи первичного
ожирения. При лечении гиперлипопротеидемий
и ожирения методом экстракорпорального
липидафереза получен парадоксальный
эффект — булимия, связанная со
снижением уровня лептина (Г.К. Ляйтнер и соавт.,
2000).
Резистентность тех или иных
центральных и периферических структур к этим
сигналам может извратить работу всей
этой мощной системы. Поэтому терапия
заболеваний с недостаточной, либо
избыточной массой тела, включая голодание,
нервную анорексию-булимию и ожирение,
должна быть комплексной, поэтапной и
осторожной.
Глава 4
АНАБИОЗ И ЗИМНЯЯ СПЯЧКА
«Но не тем, холодным сном могилы...
Я б желал навеки так уснуть...
Чтоб в груди дремали жизни силы,
Чтоб дыша, вздымалась тихо грудь.»
М.Ю. Лермонтов, 1841
Анабиоз (В. Прейер, 1873) — состояние
организма, характеризующееся почти
полным, но обратимым прекращением
всех проявлений жизнедеятельности и,
в том числе, абсолютным эндогенным
голоданием.
Анабиоз у тихоходок и коловраток
открыл еще А. ван Левенгук в конце XVII
века. Первым анабиоз, как
общебиологическое явление, описал русский учёный
П.И.Бахметьев (1900).
Данное явление широко наблюдается у
семян растений. Имеются случаи успешного
проращивания семян, пребывавших в
анабиозе в китайских гробницах в течение
более чем 20 веков.
Среди животных истинный анабиоз
описан только у холоднокровных. Анабиоз
вызывается крайне неблагоприятными
условиями жизни (высыханием, гипоксией и,
особенно часто, гипотермией). При анабиозе
не только тормозится обмен веществ, но
происходит накопление эндогенных
антифризов — глицерина, других многоатомных
спиртов и особых гликопротеидов. Главная
цель подобной адаптации — избежать
внутриклеточной кристаллизации воды,
которая, безусловно, смертельна. Арктическая
жужелица Pterostihus brevicornis переносит
замерзание при -35 °С, повышая
концентрацию глицерина в своём теле до 22 %. Ното-
тениевые рыбы полярных вод располагают
специальными, гликопротеидами и липоп-
ротеидами криопротекторного действия,
сверхобогащёнными сульфгидрильными
группами. Эти вещества химически
содействуют таянию льда. Моллюски-мидии
могут удерживать внутриклеточную воду
с помощью неизвестных осмотически
сверхактивных молекул, препятствуя
внутриклеточной дегидратации при высушивании
и замерзании (П. Хочачка, Дж.
Семеро, 1977). Теплокровные не располагают
такими механизмами, и, вопреки идее
Дж. Хантера, высказанной в конце XVIII
века, и мечте фантастов, предсказывавших
межгалактические перелёты в состоянии
анабиоза, этот способ самоконсервации у
высших гомойотермных существ до сих пор
не получен.
56 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Своеобразным состоянием гипобиоза или
выраженного обратимого снижения
интенсивности жизненных процессов является
спячка холоднокровных и теплокровных
животных (гибернация). Это менее глубокое
торможение жизнедеятельности, чем
истинный анабиоз (Н.И. Калабухов, 1974). Она
не предусматривает столь резкого
изменения композиции цитоплазмы и накопления
криопротекторов, как при анабиозе.
Температура тела животного в спячке не такая
низкая, как при анабиозе. Однако, спячка
сопровождается, как и анабиоз,
абсолютным эндогенным голоданием. Животные,
создающие обильные запасы еды на зиму
(пчёлы, совы и многие грызуны), в спячку
не впадают.
Спячка бывает зимней (с осени до начала
весны) или летней (в жаркий сухой сезон).
В любом случае, это приспособление к
переживанию сезонной бескормицы и
экстремальных климатических факторов. Зимняя
спячка — яркое проявление особенностей
видовой реактивности и всегда вызывала
большой научный интерес у целых
поколений патофизиологов. В состоянии зимней
спячки, при сочетанном действии
гипотермии, гиперкапнии и гипоксемии,
резистентность животного по отношению ко многим
неблагоприятным воздействиям резко
повышается, хотя внешние проявления
реактивности заметно ослабевают (см. т. I, с. 50-51).
Этот парадокс, столь явно демонстрируемый
при гибернации, имеет фундаментальное
значение для всего учения о реактивности
(Н.Н. Сиротинин, 1981). Зимняя спячка
свойственна многим видам беспозвоночных,
принадлежащим к различным таксонам
(червям, моллюскам, членистоногим).
Среди позвоночных гибернация наблюдается у
многих пойкилотермных (рыб,
земноводных, пресмыкающихся). Для
млекопитающих и птиц спячка означает временный
переход к пойкилотермии. Гибернации
подвержены однопроходные (утконос, ехидна),
а также плацентарные млекопитающие из
отрядов грызунов и насекомоядных. Среди
высших млекопитающих зимнеспящими
являются: суслики, сурки, байбаки, ящуры,
бурундуки, тушканчики, сони, хомяки, ежи,
барсуки и летучие мыши. Среди птиц сюда
относятся стрижи и козодои. Классические
источники не причисляют к зимнеспящим
медведя и не признают его воспетый в
фольклоре берложный сон гибернацией,
поскольку температура тела у Топтыгина
во время физиологического зимнего сна,
практически, не падает (Л.Р. Перельман,
1937). Однако, в более поздней
литературе и медведь причислен к зимнеспящим
(Н.И. Калабухов, 1974).
Продолжительность зимней спячки
бывает различной — от 6 недель до 10
месяцев. Раз в несколько недель животное
пробуждается для выведения экскрементов,
а барсук встаёт для зимней охоты.
Гибернация начинается с падения
аппетита и уменьшения температуры тела.
При этом существенное снижение внешней
температуры вовсе необязательно: в
эксперименте не удаётся вызвать спячку «не
в сезон» простым понижением температуры
в помещении, и, напротив, «в сезон» спячка
начинается и в помещении с комнатной
температурой. Резко повысив температуру
в помещении, где содержится засыпающее
животное, можно нарушить ход спячки, но
не удаётся снять явления гипобиоза совсем.
Более того, многие зимнеспящие впадают
в гибернацию и летом: сурки, например,
в жаркий сухой сезон понижают
температуру тела до 15-16 градусов и впадают
в оцепенение. Гибернация, скорее, является
результатом генетически закреплённых
сезонных ритмов, возможно, связанных с
фотопериодизмом или зависящих от сезонной
доступности тех или иных видов корма,
нежели прямым следствием понижения
окружающей температуры.
У млекопитающих температура в норе
во время зимней спячки удерживается
около 8°С, причём, в холодном климате для
этого приходится рыть в 3-4 раза более
глубокие норы, чем в умеренном.
Примерно 8-9 градусов составляет и температура
тела животного в зимней спячке.
Зафиксированы и меньшие температуры, равные
окружающей (до +2°С — у сусликов). Од-
Анабиоз и зимняя спячка 57
нако, при падении температуры тела даже
на короткое время ниже 0°С наступает
пробуждение и активизация обмена веществ.
В этом отличие гибернации теплокровных
от гипобиоза и анабиоза холоднокровных
животных.
Гибернация отличается от экзогенного
полного голодания по многим параметрам.
Ежесуточные потери массы тела,
благодаря крайнему торможению
жизнедеятельности, столь малы, что продолжительность
обратимого голодания в спячке может быть в
несколько раз дольше, чем при бодрствовании.
Основной обмен в спячке падает
значительно ниже, чем при экзогенном
голодании.
Относительная потеря в весе различных
органов не вполне совпадает с
распределением потерь при экзогенном голодании.
Опыты Валантена показали, что, как и в
других случаях полного голодания,
больше всего теряет жировая ткань (свыше
99 % веса при средней потере общей массы
тела — 35%). Значительно теряют в весе
печень и органы ЖКТ (от 47 до 58%).
Весовые потери кожи находятся на уровне
средних. Однако, в отличие от экзогенного
голодания, значительна атрофия легких
диафрагмы (44-45%) и надпочечников
(45%). В то же время, скелетные мышцы
(30%), селезёнка (11%) и скелет (12%)
теряют относительно меньше, чем при
экзогенном голодании. Весовые потери сердца
и мозга, как и при экзогенном голодании,
ниже средних.
Эти данные говорят о том, что гибернация
также является примером эндогенного
питания за счёт жировых и гликогеновых
запасов. Однако, при ней белки — и внутренних
органов, и соматического отсека
—используются в гораздо меньшей степени, чем при
голодании экзогенном. По-видимому, имеет
значение тот факт, что при спячке
отсутствует стресс. Недаром надпочечники зимнеспя-
щих атрофируются. Глюкокортикоиды,
обеспечивающие при экзогенном голодании
использование аминокислот
соматического пула в глюконеогенезе, не обладают
столь большим метаболическим действием
в организме зимнеспящих. Понятно, что
и степень атрофии тимико-лимфатического
аппарата, и сопутствующие нарушения
иммунитета при гибернации, по сравнению
с экзогенным голоданием, не столь
значительны.
Отчасти, при оценке относительных
весовых потерь органов имеет значение и менее
интенсивное функционирование лёгких
и сердца при спячке (так, у спящего сурка
частота пульса 2-20 уд./мин, дыхания —
1-8 дых./мин), что обусловливает в этих
органах более значительные атрофические
изменения, чем при экзогенном голодании.
Так как зимняя спячка — голодание без
воды, при ней происходит относительная
дегидратация большинства органов за
исключением мозга. При гибернации,
несмотря на сгущение крови, её свёртываемость
и скорость тромбообразования понижены.
Важной гормональной особенностью
зимнеспящих, по Адлеру-Гудернатчу,
считается глубокое угнетение функций
и атрофия щитовидной железы. Степень
гипотироза, достигаемая при гибернации,
гораздо глубже, нежели при других формах
голодания. Это сопровождается гипопара-
тирозом и гипокальциемией.
При зимней спячке, по некоторым
данным, не снижено, а нормально или
повышено выделение инсулина. Вместе с тем,
поджелудочная железа зимнеспящих
выделяет, как и при других формах голодания,
избыток глюкагона. Весьма важной для
механизмов гибернации и анабиоза
является, по всей вероятности, способность
соматостатина тормозить потребление
глюкозы, основной обмен, функцию
щитовидной железы, ростовые и анаболические
процессы, электрофизиологическую
активность, функцию тромбоцитов и многие
другие проявления жизнедеятельности.
Поистине, этот регулятор, продукция
которого при спячке в островках Лангерганса
и в других клетках диффузной
эндокринной системы возрастает, выступает как
«пангибин», обеспечивающий глубокое
всестороннее ограничение интенсивности
жизнедеятельности (см. т. I, с. 534).
58 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
В последнее время установлено, что
зимняя спячка сопровождается
значительным увеличением продукции
эндогенных опиатов, в частности, эндорфинов
и дерморфина. Более того, инъекции
дерморфина способны вызвать спячку у
животных, которые в нее, обычно, не
впадают (т. I, с. 563). Опиаты при гибернации
обеспечивают торможение активности
систем дыхания и кровообращения,
увеличение эффективности фосфорили-
рования, частично — снижение аппетита
и температуры тела. Предполагается, что
механизмы, обеспечивающие повышение
резистентности организма в состоянии
зимней спячки и делающие ее
протекание относительно благоприятным, по
сравнению с иными формами
абсолютного голодания, во многом являются опиа-
тергическими. В.В. Пашутин усматривал
аналогию между зимней спячкой и
некоторыми состояниями у человека,
например, голоданием индийских факиров,
летаргией и кататоническим ступором.
Уместно напомнить, что, по крайней мере,
последнее состояние сопровождается
выраженной гиперпродукцией эндогенных
опиатов.
Эндокринные механизмы пробуждения
от зимней спячки включают активизацию
работы гипофиза, щитовидной железы и
мозгового вещества надпочечников. Под
влиянием их гормональных регуляторов
происходит активация калий-натриевых
АТФ-аз, а также экстренный распад бурого
жира в так называемой «сонной железе»,
скоплении бурой жировой клетчатки,
образуемом зимнеспящими в сезон
бодрствования. Резкое увеличение несократительного
термогенеза ведет к подъему температуры
тела и интенсификации метаболизма (см.
также т. I, стр. 384,572 — о буром жире и его
роли в температурной адаптации).
Зимняя спячка — фактор, влияющий на
сезонность некоторых зоонозов и антропозоо-
нозов. Её медицинское значение
определяется ещё и тем, что в хирургической практике
состояние, подобное зимней спячке, —
искусственная гибернация — воспроизводится
у человека для повышения резистентности
к хирургической травме методом нейролепт-
анальгезии с гипотермией.
Глава 5
ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО
ОБМЕНА
«Жизнь - есть способ существования белковых тел...»
Ф. Энгельс «Диалектика природы»
• Общие аспекты патологии белкового обмена
• Нарушения количественного поступления белка в организм
• Нарушения качественного состава белков
• Нарушения переваривания белков. Кишечная аутоинтоксикация
• Нарушения чрезмембранного транспорта аминокислот
• Аминоацидурия
• Общий обзор путей метаболизма аминокислот
• Гормональная регуляция белкового обмена и его нарушения при эндокринопатиях
• Нарушения межуточного обмена аминокислот и их производных
• Нарушения композиции белков плазмы
• Нарушения конечных этапов обмена белка
• Диспротеинозы
ОБЩИЕ АСПЕКТЫ
ПАТОЛОГИИ БЕЛКОВОГО
ОБМЕНА
Белок — важнейший пластический
компонент диеты, незаменимый источник
биогенного азота, необходимый для роста
и регенерации. В пересчёте на сухой вес
белки составляют 44 % массы тела. На долю
протеинов приходится более половины его
органических соединений (О.Эдхольм,
А.Бахаранч, 1965). В соматическом отсеке
тела (скелет, скелетные мышцы, кожа)
находится около 63 % белка тела, остальные
37% — в висцеральном отсеке. Важность
белка как пластического компонента
доказывается тем, что при хронической белковой
недостаточности в питании населения ряда
тропических регионов рост и развитие
целых народов могут замедляться. Известно,
что представители многих низкорослых
этносов тропической Африки, Азии,
Латинской Америки, переселяясь в раннем детстве
в развитые страны и следуя диете с
достаточной в качественном и количественном
отношении поставкой белка, приобретают
антропометрические показатели, сходные с
таковыми у коренного населения развитых
стран (Ж. де Кастро, 1950; Р. Котран и
соавт., 1997).
Белки — носители чужеродной
антигенной информации и должны расщепляться
при переваривании, утрачивая антиген-
ность, иначе их неполное расщепление
приведет к чрезкишечной сенсибилизации и
пищевой аллергии, либо аутоаллергическим
эксцессам (например, при спру, или при
провокации молочным альбумином и казеином
сахарного диабета у предрасположенных
индивидов— см. ниже «Патофизиология
сахарного диабета»).
Белки — рабочие инструменты,
исполняющие генетические программы организма.
Кажущееся разнообразие метаболических
функций белка иногда характеризуют в
учебной литературе целым букетом эпитетов
(ферментативная, гормональная, защитная,
антитоксическая, сократительная,
дыхательная, ростовая, транспортная, структурная
60 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия(:
и т.д.). На деле фундаментальной является
способность белков к комплементарному
взаимодействию — то есть способность
распознавать и распознаваться. По
классическому расчёту Э. Абдергальдена из 20
аминокислот возможно получение 2 432 902 008
176640 000 уникальных полипептидов!
На основе распознающей способности
сформирована другая важнейшая функция
белка — каталитическая. Строго говоря,
любому белку присуща рецепторно-сигнальная
или энзиматическая роль (а иногда — обе).
Все остальные функции являются
производными или представляют собой
дидактические переименования этих двух
(см. т. I данного руководства, стр. 139).
Именно белки, выполняя
каталитические и распознающие функции, реализуют
в виде обмена веществ индивидуальные
генетические программы, поэтому, строго
говоря, практически, любое наследственное
моногенное заболевание опосредуется
нарушением структуры и функции какого-то
ферментативного, либо распознающего
белка, что и лежит, по А.Э. Гарроду (1909), в
основе любого метаболического блока, даже
при наследственных нарушениях,
касающихся небелкового метаболизма —
например, обмена липидов или углеводов (см. т. I
данного руководства, стр. 137-141, а также
соответствующие разделы ниже).
Собственно нарушения обмена белков
включают:
>* нарушения количественного
поступления белка в организм (белковый
перекорм и белково-энергетическая
недостаточность как форма частичного
голодания);
>* нарушения качественного состава
белков (дефицит и избыток отдельных
аминокислот, причём последний может
приводить как к их антагонизму, так и
прямой токсичности);
>* нарушения переваривания белков в
ЖКТ;
>* нарушения чрезмембранного
транспорта аминокислот (расстройства
кишечного всасывания и аминокислот-
Эокринно-метаболические нарушения)
ные тубулопатии, синдром аминоациду-
рии);
>* нарушения промежуточного обмена
аминокислот (расстройства дезаминиро-
вания; взаимопревращения, то есть пере-
аминирования; декарбоксилирования);
>- нарушения композиции белков плазмы
(гипопротеинемии и диспротеинемии);
>* нарушения конечных этапов обмена
белка (аномалии и недостаточность цикла
мочевины).
НАРУШЕНИЯ
КОЛИЧЕСТВЕННОГО
ПОСТУПЛЕНИЯ БЕЛКА
В ОРГАНИЗМ
Как впервые показано русским учёным
Б.А. Словцовым (1898), белки не
депонируются в организме, то есть не имеют
лишённого специальных функций резервного
пула. При дефиците белка в диете организм
вынужден вовлекать в энергетический
метаболизм функциональные протеины, а при
избытке пищевого белка дополнительные
аминокислоты подвергаются
энергетической утилизации.
Азот теряется с мочой (60 %) и
фекалиями, а также через кожу. Взрослый мужчина
весом 70 кг в день лишается не менее 4,5 г
азота, что эквивалентно потере 0,4 г/кг
белка. Следует учесть, что не менее 1 г/кг
белка в сутки подвергается самообновлению
(ресинтезу).
Факториальный метод рекомендует
принимать за обязательную потерю белка
ее уровень, достигаемый после 6-10 дней
безбелковой диеты, адекватной в
энергетическом отношении, когда происходит
перегиб кривой суточных потерь азота и её
выход на плато (см. раздел «Голодание»).
В этот момент средние потери азота с мочой
у мужчин составляют 2 мг/ккал основного
обмена или примерно 46 мг азота на кг
массы тела (Н.С.Скримшоу и соавт., 1972).
В этих же условиях средние потери азота с
калом составляют 12 мг/кг, через кожу — 3
мг/кг, а потеря азота другими путями
(семяизвержение, отхаркивание, сплёвывание,
кровотечения и выдыхание аммиака) —
приравниваются к 2 мг/кг. По данным комитета
экспертов ФАО-ВОЗ (1974), для взрослых
минимальный балансовый уровень приема
азота с пищей, с учётом различной
усвояемости растительных и животных белков,
составляет не менее 77 мг/кг массы тела в
сутки — при смешанной и 93 мг/кг массы
тела в сутки — при вегетарианской диете.
Азот пересчитывается в белок при
среднем коэффициенте умножения 6,25
(для различных видов пищи коэффициент
варьирует от 5,18 — для миндаля, до 6,38 —
для твердых сыров). Основатель научной
диетологии К. Фойт щедро рекомендовал
суточную норму в 105 г усвояемого (118 г
реального) белка (1881). Характерно, что
этот автор ориентировался на исследования
диеты немецких семей средней
зажиточности. Его более «скаредные»
последователи обнаружили, что можно установить
азотистое равновесие в организме и на
брюквенно-картофельном питании, при
существенно меньшем потреблении белка
(67 г/сутки — по Р. Читтендену, 1909; и даже
26 г/сутки — согласно адептам
вегетарианства, М. Хиндхеде и соавторам, 1913). Но
методической ошибкой этих выкладок было
принятие азотистого равновесия за
эквивалент здоровья, что неверно. Следует учесть,
что простое восполнение ежесуточной
убыли белка — это лишь физиологический
минимум, но не гигиенический оптимум
потребности в нём. Так как существуют
незаменимые аминокислоты, то, оценивая
общую потребность в белке, необходимо
следовать принципу лимитирующего
минимума (Л.Б. Мендель, 1915) — только
количество белка, существенно
превосходящее физиологический минимум, может
гарантировать покрытие потребности в той
из незаменимых аминокислот, которой в
реальной диете меньше всего.
В связи с этим, рекомендуемый
минимальный прием белка взрослыми должен
быть между 1 и 1,5 г/кг в день, прием в те-
Патофизиология белкового обмена 61
чение нескольких дней менее 0,6 г/кг белка
уже вызывает белковую недостаточность.
Для лиц, занятых тяжёлым физическим
трудом, потребление рекомендуется
увеличить до 2 г/кг (X. Йошимура, 1961). У
грудных детей и в период полового
созревания потребность в белке наивысшая и
покрывается при приеме не менее 1,5, а
лучше — 2 г/кг веса белков. Потребность в
белке растет при лактации, беременности и
интенсивной регенерации и повышена при
наличии в организме быстро пролифериру-
ющих, например, лейкозных клонов клеток.
Установлено, что физический труд,
акклиматизация, стресс — повышают у здорового
человека потребность в белке. По мнению
Р. Пэссмора (1974), не менее 10 %
калорийного содержания диеты здорового человека
должно обеспечиваться белками. Нормы
питания, принятые в СССР, устанавливали
рекомендуемый уровень потребления белка
для мужчин 75-111 г/сутки, для женщин —
62-96 г/сутки при покрытии 12-14 %
суточной калорийности питания за счёт белков
(П.Е.Калмыков, М.Н. Логаткин, 1974).
Менее полноценный по аминокислотам
растительный и более трудно усвояемый
грибной белок могут удовлетворить
количественные потребности лишь при больших
дозах по сравнению с животным белком.
Еще М. Рубнеру принадлежит
категорическое мнение, что «Белки разного
происхождения имеют разную питательную ценность,
поэтому имеется столько минимумов белка,
сколько белков» (1902). Экспериментально
это впервые доказал К. Томас,
продемонстрировавший, что кукурузного белка для
установления азотистого равновесия потребно
в 3 раза больше, чем мясного (1909).
Считается, что в оптимальной диете от 50 до 60 %
суточного потребления белков должно быть
представлено животными протеинами.
Интегральным показателем общего
белкового метаболизма служит косвенно уже
упоминавшийся (см. раздел
«Патофизиология энергетического метаболизма» выше)
азотистый баланс (Ж.-Б.Буссиньоль, 1839).
Это разница между суточным количеством
поступающего с пищей азота и количеством
62 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
азота, выделенного за тот же период в
составе азотсодержащих компонентов мочи и кала
(мочевина, мочевая кислота, аминокислоты,
креатинин, соли аммония). У здорового
взрослого человека, вне состояний,
упомянутых выше, азотистый баланс нулевой. Для
его клинической оценки служит формула
М. Денке-Дж. Уилсона (1998):
Азотистый баланс (г) = Потребление
белка (г) / 6,25 - Азот мочевины мочи (г)
+ 2,5 г (эквивалент немочевинного азота и
потерь азота помимо мочи).
Положительный азотистый баланс может
быть не только в норме (при росте,
интенсивной регенерации, лактации и
беременности), но и при патологии — полицитемии,
крупных доброкачественных опухолях и
некоторых злокачественных клональных
процессах (если они не сопровождаются
значительным синтезом цитокинов-блокаторов
анаболизма), а также при гиперсекреции
гормона роста (см. ниже: «Гормон роста и
патофизиология ростовых и анаболических
процессов»).
Отрицательный азотистый баланс
сопровождает состояния с активированным глю-
конеогенезом (голодание, белково-энергети-
ческая недостаточность, инсулинзависимый
сахарный диабет, гиперкортицизм, стресс).
Особый типовой ответ представляет собой
переброска аминокислот из соматического
отсека (скелетные мышцы) в висцеральный
(печень и инсулиннезависимые ткани). Этот
адаптивный ответ сопровождает тяжелые
стрессы, например, экстремальные
состояния, и опосредуется гдюкокортикоидами,
адреналином и, частично, глюкагоном (см.
т. I, стр. 371-376).
Такие цитокины, как кахексии (ФНОа),
ИЛ-6 и ИЛ-1, при системных
воспалительных и септических процессах, инфекциях и
тяжелых злокачественных
новообразованиях, в ходе ответа острой фазы или преиммун-
ного ответа (см. т. I данного руководства, гл.
13) вызывают сходную переброску
аминокислот и отрицательный азотистый баланс
на фоне торможения синтеза ряда белков,
подавления аппетита и при наличии антиин-
сулинового действия. При этом, продукция
положительных глобулинов острой фазы
печенью и макрофагами нарастает. Интересно,
что кахексии — физиологический продукт
жировых клеток (см. ниже раздел
«Устройство липостата»). Все состояния,
сопровождаемые ответом острой фазы, требуют
увеличения суточного потребления белка
для сохранения азотистого баланса. При
этих состояниях альбумин-глобулиновый
коэффициент плазмы крови уменьшается
и становится, как правило, ниже нижней
границы нормы — 1,1. Нехватка любой
незаменимой аминокислоты также вызывает
отрицательный азотистый баланс по
причинам, рассмотренным ниже.
Избыточное поступление белков в
организм возможно при переедании,
несбалансированной диете, сахарном диабете,
некоторых поражениях гипоталамуса.
Общий перекорм белками в ветеринарной
и педиатрической практике ассоциируется с
ускорением темпов индивидуального
развития и психомоторного созревания (И.М.
Воронцов, 1994).
Имеются экспериментальные данные
на животных (Дж.Р. Слонакер, 1931),
свидетельствующие о том, что избыточный
приём белков приводит к ускоренному
росту и созреванию, но также коррелирует
с укорочением общей продолжительности
жизни в последующем. Некоторый
недостаток белка тормозил развитие подопытных
крыс, но удлинял продолжительность их
жизни (Р. Росс, 1961). Такое впечатление,
что преимущественное кормление белками
может ускорять ход биологических часов,
определяющих протекание онтогенеза.
Избыточное потребление белков
вызывает положительный азотистый баланс. Часть
принятого избыточного белка расходуется в
реакциях глюконеогенеза, увеличивая
теплопродукцию, часть задерживается в виде
циркулирующих аминокислот. Перекорм
белками не ведёт к развитию ожирения.
Субъективно при перекорме белками
возникает отвращение к белковой пище. При
значительном избытке пищевого белка
Патофизиология белкового обмена 63
создаётся повышенная функциональная
нагрузка на печень и почки, так как имеется
необходимость в нейтрализации
дополнительного аммиака и выведении мочевины.
Однако, старые данные Э. Ньюбурга и
соавт. (1925) об индукции гломерулонефри-
та у животных высокобелковой диетой
были опровергнуты (В.В. Воронин, 1948).
Считается, что длительное поступление
избытка белков в организм может
приводить к гипертрофии эпителия нефронов,
а если ЖКТ не справится с избытком
белка — возможно развитие гнилостной
диспепсии, дисбактериоза и аутоинтоксикации
ароматическими аминами, образуемыми
в результате бактериального кишечного
расщепления белков. Но, по-видимому,
некрозы гепатоцитов, ожирение печени и
снижение активности ряда печёночных и
почечных ферментов могут быть отмечены
в эксперименте лишь при очень высоком
потреблении белка (выше 70% от общей
калорийности — Ю.Н. Кремер, 1965).
Эскимосы и масаи, употребляя более 0,5 кг мяса
в день, не испытывают от этого каких-либо
специфических проблем со здоровьем, ведя
традиционный образ жизни.
Избыток отдельных аминокислот,
впрочем, патогенен (см. раздел «Нарушения
качественного состава белков»).
Белково-калорическая недостаточность,
как форма частичного голодания, известна
в двух крайних видах, между которыми
Рис. 9. Голодание у девочки
полуторагодовалого возраста (Верхняя Вольта)
существует значительное количество
промежуточных смешанных случаев (рис.9).
Эти два вида — квашиоркор и
алиментарный маразм. Первый еще именуют
несбалансированной, а второй — сбалансированной
формой белково-энергетического дефицита.
Квашиоркор, характеризующийся высоким
инсулино-кортизоловым соотношением и
отсутствием сберегания висцерального пула
белков, протекает острее, алиментарный
маразм имеет тенденцию к более
длительному течению. Подробная характеристика
этих нарушений, связанных с дефицитом
пищевого белка, дана выше в разделе
«Голодание». Дополнительно отметим здесь
только две детали, вместе представляющие
особенно разительный контраст.
Стоимость производства всех
аминокислот, эквивалентных суточной потребности
взрослого человека в белке, составляет
не более 60 американских центов. В то же
время, преквашиоркор или состояние
пограничного белкового дефицита, встречается
в 100 раз чаще, чем клинический
квашиоркор и охватывает более миллиарда людей
(П.Е.Калмыков, М.Н. Логаткин, 1974).
НАРУШЕНИЯ
КАЧЕСТВЕННОГО СОСТАВА
БЕЛКОВ
Из более чем 80 природных аминокислот
только 22 встречаются в пищевых белках. Из
них 12 (заменимые) — могут
синтезироваться в организме в достаточном количестве,
как у взрослых, так и у детей. Однако, при
их нехватке повышается энергетическая
стоимость белкового синтеза и снижается
эффективность метаболизма, а также растет
расход незаменимых компонентов белка.
8 аминокислот у человека: метионин,
лизин, триптофан, фенилаланин, лейцин,
изолейцин, треонин, валин — безусловно, не
могут быть синтезированы в достаточном
количестве и являются незаменимыми
(У. Роуз и соавт., 1948). В сущности, для
получения любой из них, кроме лизина и
64 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
треонина, необходимо поступление
соответствующего кетоскелета. Поэтому кето-
аналоги, при наличии достаточного
количества азота и нормального переаминирования,
могут превращаться в эти аминокислоты.
Таким образом, в 6 случаях незаменим (то
есть, не синтезируется в организме) именно
а-кетокислотный аналог, а не сама
аминокислота.
Тирозин понижает потребность в фени-
лаланине, а цистин — в другой
серосодержащей аминокислоте — метионине. С другой
стороны, тирозин в организме получается
только из фенилаланина, а цистин с цисте-
ином получают серу только из метионина.
Поэтому при учете поступления
незаменимых аминокислот цистин и метионин,
а также фенилаланин и тирозин попарно
суммируются.
У грудного ребенка не синтезируется в
достаточном количестве ещё и гистидин
(Л.Э. Хольт и соавт. 1960), а у многих
животных, например, крыс и кур — кроме
гистидина также и аргинин. Есть сведения о
недостаточности собственного синтеза
аргинина и у новорожденных детей (И.Б. Збар-
ский, 1974) Таким образом, в педиатрии
выделяют 10 эссенциальных аминокислот.
При парентеральном питании принято
нормировать еще и «полузаменимые» аланин и
пролин. В культурах in vitro человеческие
клетки нуждаются в 12 аминокислотах (10
«педиатрических» плюс цистин и тирозин).
Общая схема метаболизма аминокислот в
клетке представлена на рис. 10.
Потребность в незаменимых
ингредиентах белковой диеты, согласно
рекомендациям Комитета экспертов ФАО-ВОЗ,
представлена в нижеследующей таблице 2.
Обращает на себя внимание значительно
большая потребность растущего организма
в незаменимых аминокислотах.
Следовательно, дефицит любой из них
способен замедлить рост, не говоря уже о
специфических метаболических
нарушениях, провоцируемых их нехваткой.
Недостаток любой незаменимой аминокислоты,
помимо отрицательного азотистого баланса,
связанного с извлечением дефицитного
нутриента путём усиления катаболизма
тканевых белков, задержки роста и падения
веса, проявляется уменьшением утилизации
пищевого белка и потребления пищи. Белки,
дефицитные по незаменимым
аминокислотам, например кукурузный протеин — цеин,
не поддерживают азотистое равновесие.
В случае некоторых незаменимых
аминокислот действуют дополнительные
специальные факторы угнетения ростовых и
анаболических процессов. Так, известно,
Таблица 2
Потребность в незаменимых аминокислотах
Аминокислота
Гистидин
Изолейцин
Лейцин
Лизин
Метионин (с цистином)
Фенилаланин (с тирозином)
Треонин
Триптофан
Валин
Суточная
потребность взрослых
(мг/1 г
потребляемого белка)
0
18
25
22
24
25
13
6,5
18
Суточная потребность
грудных детей
(мг/1 г потребляемого
белка)
14
35
80
52
29
63
44
8,5
47
оксалоацетат
кинуренин
jf NADPH+H
триптофан NADP+
3-оксикинуренин
NAD+
мочевина
Рис. 10. Общая схема метаболизма аминокислот в клетке (по Я. Мусилу и соавт.,1981). Пояснения в
тексте.
что у эукариот синтез любого пептида
начинается с метионина. Кроме того, метионин
и лизин — ключевые предшественники
главных стимуляторов регенерации и роста
тканей — полиаминов (табл. 3). Ряд
синтезируемых в организме аминокислот служит
сырьем для производства важных, в том
числе — небелковых веществ: так, глицин
входит в состав гема, вместе с глютаминовой и
аспарагиновой кислотами используется для
Патофизиология белкового обмена 65
синтеза азотистых оснований, в комбинации
с цистеином и глютамином дает глютатион,
тирозин входит в состав меланина и тироид-,
ных гормонов и т.д.
Избыток той или иной аминокислоты
также патогенен. Накопление аминокислот, не
используемых для синтеза белка, подавляет
аппетит. Повышение концентрации одной
аминокислоты может повысить потреб-
66 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 3
Проявления дефицита незаменимых
аминокислот
Аминокислота
Гистидин
Изолейцин
Лейцин
Лизин
Метионин
(сцистином)
Фенилаланин
(с тирозином)
Аргинин
Триптофан
Валин
Треонин
Симптомы недостаточности
Дерматит, анемия, снижение
продукции гистамина, ухудшение
умственной деятельности
Поражение почек и щитовидной
железы, анемия, гипопротеине-
мия
Поражение почек и щитовидной
железы, гипопротеинемия
Анемия, миодистрофия, остеопо-
роз, поражение печени и лёгких,
головная боль, повышенная
чувствительность к шуму, анэструс у
крыс |
Ожирение и некрозы печени,
ускорение атеросклероза, надпо-
чечниковая недостаточность,
геморрагическое поражение почек,
дефицит холина и адреналина,
облысение
Нарушения тироидной функции и
недостаточность мозгового
вещества надпочечников
Нарушение сперматогенеза и
цикла мочевины
Пеллагра, катаракта, помутнение
роговицы, анемия, облысение,
гипопротеинемия, атрофия
семенников, рассасывание плода,
гиперплазия слизистой желудка
Расстройство координации
движений, гиперестезии
Отёки, падение веса
ность в других. Одна аминокислота, будучи
в избытке, может конкурентно подавлять
утилизацию других структурно сходных
(например, лейцин — изолейцина и валина).
Наконец, возможны и прямые токсические
эффекты избытка аминокислот. Например,
доказано стимулирующее действие гипер-
метионинемии и гипертирозинемии на
функции надпочечников.
Избыток метионина ведёт к нарастанию
концентрации гомоцистеина, что, в свою
очередь, может обусловить гемолитическую
анемию, гепатонекрозы, отставание в росте
и миокардиодистрофию.
Избыток триптофана в организме может
переходить в эндогенный канцероген — 3-ок-
сиантраниловую кислоту, повышающую
риск развития рака мочевого пузыря, а
также в серотонин, небезразличный для
ЦНС. Кроме того, возникает миокардио-
дистрофия.
Избыток гистидина может вызвать
задержку умственного и, особенно, речевого
развития.
При добавке избытка лейцина и лизина к
диете крыс-самок у них учащаются
выкидыши и внутриутробная гипотрофия плодов.
О пагубных последствиях избытка фе-
нилаланина при беременности говорится
ниже (см. раздел «Нарушения
межуточного обмена аминокислот»). Потребность
в отдельных аминокислотах меняется при
различных болезнях. Так, при карциноиде
резко повышено потребление триптофана,
превращаемого опухолью в серотонин; при
меланоме имеется усиленное превращение
тирозина в меланин и т.д (Н.Н.Лаптева,
1970).
НАРУШЕНИЯ
ПЕРЕВАРИВАНИЯ
БЕЛКОВ. КИШЕЧНАЯ
АУТОИНТОКСИКАЦИЯ
Начальные этапы переваривания белков
пептид-гидролазами происходят в рамках
полостного пищеварения и включают
гидролиз пептидных связей между
ароматическими и дикарбоновыми аминокислотами (при
участии пепсина и гастриксина желудочного
сока). В створаживании молока у маленьких
детей принимает участие ещё одна
желудочная протеаза — химозин (он же реннин).
При гипоацидных состояниях, если рН
желудочного содержимого не достигает хотя
бы 5, а лучше — 3 (и тем более — при полной
ахилии или тотальной резекции желудка,
Патофизиология белкового обмена 67
когда отсутствуют и соляная кислота, и
пепсин) желудочный этап переваривания
белка сильно замедляется. Без кислоты
нарушается набухание белков, активация
пепсиногена и снижается ферментативная
активность пепсина. Из-за
множественности протеолитических ферментов, даже при
полном отсутствии желудочного
пищеварения, не отмечается прекращения
переваривания белка, если нет сопутствующей
панкреатической недостаточности. Однако, как
указывал С.Я. Капланский (1966), при этом
сильно изменяется скорость протеолиза и
запаздывает появление в свободном виде
ряда аминокислот.
При выраженной недостаточности
желудочного пищеварения понижается скорость
освобождения и всасывания такой
незаменимой аминокислоты, как триптофан, а
также метаболически близкого к нему
тирозина, в норме освобождаемых, в основном,
уже в желудке. Аминокислотная смесь в
начальных отделах тонкого кишечника
обедняется триптофаном и тирозином. При этом
ухудшаются условия усвоения аминокислот
печенью, так как замедление поступления
триптофана лимитирует скорость синтеза
белка в гепатоцитах (С.Инуцука, 1956).
Доказано, что после резекции желудка
замедляется печёночное поглощение
аминокислот и повышается их концентрация в
крови (аминоацидемпя). Более того,
отмечается даже увеличенная потеря аминокислот
с мочой (аминоацидурия).
Пепсин желудочного сока — наиболее
сильная коллагеназа системы
пищеварения (А. Гайтон, 1989). Если его действие
нарушено, может происходить недоперева-
ривание коллагеновых составляющих мяса
и мясопродуктов, соединительно-тканные
прослойки экранируют мышечные волокна
мяса, которые также недостаточно
перевариваются, что проявляется изменениями
стула, описанными ниже.
В тонком кишечнике действует
панкреатический сок, содержащий проферменты:
трипсиноген, химотрипсиногены, прокар-
боксипептидазы А и В, проэластазу. Они
каскадно активируются, начиная с действия
кишечной энтерокиназы на трипсиноген,
причём получаемый трипсин активирует
остальные проэнзимы. Здесь происходит
расщепление пептидных связей с участием
аргинина и лизина, а также других
основных аминокислот (действует трипсин).
Химотрипсины разрушают пептидные
связи с участием тирозина, триптофана и
фенилаланина и, менее активно метионина
и лейцина. Эластаза расщепляет пептидные
связи нейтральных аминокислот, наиболее
активно — в эластине.
На последующем этапе экзопептидазы
щёточной каймы и гликокаликса энтеро-
цитов расщепляют до аминокислот
короткие пептиды, уже в рамках пристеночного
пищеварения. Это — адсорбированные
панкреатические карбоксипептидазы А
и В, действующие на С-концевые
аминокислотные остатки с алифатическими и
ароматическими боковыми цепями (А) или
на аргинин и лизин (В), а также кишечные
аминопептидазы (М и N), действующие
на N-концевые аминокислотные остатки.
Энтероциты содержат также дипептидазы,
довершающие дезинтеграцию пептидов,
уже в режиме внутриклеточного
пищеварения (A.M. Уголев, 1987). Значительное
торможение полостного кишечного этапа
переваривания белка не компенсируется
и даёт симптомы креатореи. В норме в
фекалиях имеются лишь переваренные
остатки мышечных волокон, имеющие вид
единичных желтоватых глыбок. При креа-
торее в кале присутствуют непереваренные
(цилиндрические, с прямыми углами) или
полупереваренные (цилиндрические, с
закруглёнными углами) мышечные
волокна. В большей или меньшей степени, они
сохраняют поперечную исчерченность.
Наиболее частые причины выраженной
креатореи — первичная или вторичная
панкреатическая недостаточность (при
панкреатитах, муковисцидозе и т.д.), закупорка вир-
сунгова протока при холелитиазе, а также
инактивация кишечного содержимого при
синдроме Цоллингера-Эллисона (из-за
быстрой эвакуации гиперацидного желудочного
секрета). К сожалению, одной из причин
68 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
угнетения продукции желудочных и
кишечных пептид-гидролаз является безбелковая
диета. В связи с этим, при квашиоркоре
(см. выше) переваривание белков быстро
угнетается, что препятствует эффективной
коррекции питания, замыкая своего рода
порочный круг. Креаторея может возникать
и при тотальной резекции желудка у лиц, не
придерживающихся специальной дробной
диеты, так как непереваренные белки
попадают в кишечник слишком обильной
порцией. Резкое ускорение моторики кишечника
также препятствует нормальному
перевариванию белка. Поэтому, некоторая креаторея
всегда наблюдается при ускоренной
кишечной перистальтике. При панкреатической
креаторее преобладают непереваренные,
желудочной — полупереваренные, а при
кишечной — переваренные мышечные волокна
(Л.В. Козловская, М.А. Мартынова, 1975).
Более экзотической причиной нарушения
гидролиза белка в кишечнике служат
ингибиторы пептид-гидролаз. Соя и другие
бобовые содержат их в большом количестве и
могут тормозить переваривание белка. При
очень выраженной недостаточности
желудочного и панкреатического пищеварения
креаторея сменяется лиентореей — когда в
фекалиях имеются крупные комки
непереваренной пищи.
Пристеночный этап переваривания
белков нарушается при дипептидазной
недостаточности. При этом затрагивается не только
мембранное пищеварение, но и всасывание
аминокислот.
Характерное нарушение переваривания
и всасывания определённых пептидов
свойственно нетропической форме спру
или целиакии (глютеновой энтеропатии).
Данное заболевание встречается с частотой
1/2000-1/3000, главным образом, у
европеоидов и негроидов. В некоторых странах
частота целиакии в несколько раз выше
(Италия).
В основе болезни лежит
иммунопатологический энтерит, обусловливающий лим-
фоидную инфильтрацию слизистой тонкого
кишечника, атрофию его ворсин и снижение
поверхности мембранного пищеварения и
всасывания. Этиологический агент
болезни — глиадин, нерастворимый в воде
компонент глютена и авенина, белков пшеницы и
других злаков (овёс, рожь, ячмень). Однако,
для развития целиакии необходимы
особенности реактивности: 90 % больных имеют
ген ГКГС DR3 или DR7,80 % - В8. Наиболее
тесно связано с целиакией носительство ал-
леля ГКГС DQw2. Предполагается, что при
наличии этих аллелей ГКГС презентация
пептидов глиадина кишечным лимфоцитам
может происходить особенным образом,
облегчая гиперергическую реакцию. Кроме
того, имеются данные о наличии у
пациентов изначальной недостаточности конечных
ферментов переваривания пептидов из
состава глиадина (Л.Н. Валенкевич, 1984).
Не разрушаясь до конца, эти пептиды
захватываются антигенпредставляющими
элементами и презентуются лимфоцитам, что
ведет к сенсибилизации. Надо учесть и лек-
тиноподобные потенции злаковых белков
как поликлональных иммуностимуляторов,
которые, подобно фитогемагглютининам,
могут стимулировать сразу многие клоны
лимфоидных клеток, в том числе аутореак-
тивные (см. т. I данного руководства, с. 501).
Определённую провоцирующую роль может
играть аденовирусная инфекция, поскольку
12-й серотип аденовируса располагает
пептидом Elb, перекрёстно реагирующим с
пептидом глиадина. Существуют тропические
формы спру, при которых, как раз,
постулируется провоцирующая роль инфекционных
агентов. При распространённом поражении
слизистой тонкого кишечника заболевание
характеризуется очень тяжёлым синдромом
мальабсорбции, в том числе нарушением
переваривания белков и всасывания
аминокислот. Это может повлечь за собой
истощение и гипопротеинемические отёки. При
целиакии повышен и риск возникновения
кишечных лимфом.
Данная форма патологии
иллюстрирует, насколько важным для эффективного
и безопасного усвоения белков является
взаимодействие конвейерного принципа
(полостное — пристеночное —
внутриклеточное пищеварение — всасывание) и
Патофизиология белкового обмена 69
эндоэкологического принципа (барьерная
роль ЖКТ и взаимоотношения его клеток с
микрофлорой). Более подробное изложение
роли этих принципов в деятельности ЖКТ
содержится в III томе данного руководства.
Здесь целиакия рассматривается с общих
позиций — лишь как пример нарушения
переваривания и всасывания белка.
При наследственном дефекте энтеро-
киназы каскадная активация трипсина и
других пептид-гидролаз полостного
пищеварения в тонком кишечнике нарушается.
Протеолитическая активность кишечного
содержимого резко падает. В результате
развивается эндогенное патологическое
белковое голодание организма, гипоста-
тура, гипотрофия, гипопротеинемия и
безбелковые отёки. Присутствуют анемия
и вторичный иммунодефицит (Л.Н. Ва-
ленкевич, 1984).
A.M. Уголев и соавт. (1991) установили,
что такой распространенный процесс, как
стресс — сопровождается выраженным
угнетением терминального, пристеночного
этапа переваривания дипептидов и
всасывания аминокислот. Угнетение мембранного
пищеварения, в том числе, в отношении
пептидов отмечено и при лихорадке. Цитокины,
играющие роль эндогенных пирогенов,
способны нарушать конечные этапы
переваривания и всасывания белков и других
нутриентов при иммунном ответе на
антигены и суперантигены, попадающие в
кишечник. Все эти состояния сопровождаются
синдромом мальдигестии-мальабсорбции.
Объективный метод регистрации степени
нарушения переваривания и всасывания
белков — определение общего азота кала
по Моделю. Здоровый взрослый
усваивает при физиологическом потреблении
белка более 90 % его количества. Поэтому
суточное выделение азота с фекалиями не
превышает 2,5 г. При нарушении усвоения
белка регистрируется азоторея с
выделением более 3 г фекального азота в сутки. При
панкреатической недостаточности, спру и,
особенно, язвенном колите, болезни Крона,
кишечном дивертикулёзе азоторея может
превышать 5 г/сутки.
Как и иные формы мальабсорбции,
нарушение переваривания и всасывания белков
осложняется осмотической диареей, дисбак-
териозом (по типу гнилостной диспепсии) и
аутоинтоксикацией продуктами
бактериального разрушения аминокислот. Это, прежде
всего, различные токсичные амины.
Понятие «каловая аутоинтоксикация»
существует в клинической медицине давно
и применяется для обозначения
нарушений самочувствия и функций внутренних
органов у пациентов с запорами и
гнилостной диспепсией. В классических трудах
В.В. Подвысоцкого содержится
представление о птомаинах, образуемых при гниении
белковой пищи (1905). Известны опыты
М. Ненцкого и И.П. Павлова (1892) с
«мясным отравлением» у собак с нарушенным
портальным кровообращением, которым
наложена фистула Н.Н. Экка (см. ниже
раздел «Нарушения конечных этапов белкового
обмена»). Именно наличие кишечных
токсинов имеется в виду в известном афоризме
И.И.Мечникова: «Чем длиннее толстые
кишки, тем жизнь короче!» (1908). В
настоящее время можно считать доказанной роль
кишечной микрофлоры в продукции целого
ряда токсических метаболитов белкового
происхождения. Здесь и
модифицированные нутриенты, и продукты собственной
жизнедеятельности микробов. Эти токсины
нейтрализуются печенью. При
нарушении переваривания белков и всасывания
аминокислот их продукция в кишечнике
может резко возрастать. Среди моноаминов
и полиаминов, вырабатываемых
кишечными бактериями и всасывающихся в кровь,
кадаверин, гистамин, октопамин, тирамин,
пирролидин, пиперидин, диметиламин,
серотонин, путресцин и многие другие. Из
триптофана, прямо под влиянием триптофа-
назы Е. Coli, а также через индолуксусную
кислоту, образуются циклическое иминосо-
единение индол, и его производные: скатол
(метилиндол), скатоксил (метоксииндол),
индоксил (гидроксииндол). Деградация
тирозина и тирамина кишечной
микрофлорой даёт фенол и крезол. Эти соединения
токсичны, обладают неприятным фекаль-
70 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ным запахом и, по некоторым данным, не
лишены канцерогенной активности. При
работе триптофаназы расщепляются также
и другие аминокислоты (серии, цистеин). В
ходе всех этих процессов выделяются
ядовитые сероводород, метилмеркаптан и аммиак.
Часть аминов превращается в альдегиды на
месте, с участием аминооксидаз кишечной
стенки. Однако, при этом также
освобождается аммиак. Поступая в систему v. porta,
производные фенола и индола
обезвреживаются здоровой печенью с образованием
парных соединений, образующиеся эфиры
сульфатов и глюкозидуронаты экскретиру-
ются с мочой. Показателем интенсивности
формирования некоторых вышеназванных
продуктов в кишечнике служат
концентрация пиперидина в крови (амины) и уровень
индоксил сульфата в моче (индолпроизвод-
ные), оцениваемый по содержанию его
калиевой соли — индикана. Что касается
аммиака, то его обезвреживание с образованием
карбамида — тоже важнейшая функция
печени, куда аммиак переносится, в основном,
в составе аминокислот и их амидов будучи
связан кетокислотами (см. ниже).
Из сказанного ясно, что бактериальное
расщепление непереваренных и невсосав-
шихся белковых продуктов в кишечнике
создает поток сигналов в виде биологически
активных веществ, устремляющихся по
портальному руслу.
В результате эволюционно сложившегося
симбиоза с кишечной микрофлорой,
организм толерантен к малым концентрациям
этих метаболитов и, более того, многие из
них включены в нормальные регуляторные
процессы в ЖКТ и за его пределами. Так,
пиперидин подавляет жизнедеятельность
некоторых гельминтов, фенол
бактерициден, путресцин служит
противовоспалительным агентом, гистамин вовлечён в
самые разные механизмы, от желудочной
секреции до регуляции микроциркуляции
и функций гипоталамо-гипофизарного
нейросекреторного комплекса, серотонин
участвует в регуляции стресса, мозгового
кровообращения, бодрствования и сна (см. т.
I данного руководства, с. 348,585) и т.д.
Увеличение поступления кишечных
аминов, аммиака и производных индола, по
мере исчерпания дезинтоксикационных
возможностей клеток самой кишечной стенки и
печени, может приводить к патологическим
последствиям.
Такие последствия наступают не только
при мальабсорбции и мальдигестии белков,
но также и при упорных запорах, и низкой
кишечной непроходимости, дисбактериозах.
Достоверно показано, что при нарушении
переваривания белков, например, при це-
лиакии, концентрация аминов в
портальной крови возрастает в несколько раз. При
кишечной аутоинтоксикации продуктами
гниения белков наступают колебания
артериального давления, иногда с
пульсирующей головной болью, понижение болевой
чувствительности, анемия, миокардиодист-
рофия, понижение аппетита, нарушение
желудочной секреции, а в тяжёлых случаях
возможны угнетение дыхания, сердечной
деятельности и кома.
Установлена корреляция между
накоплением отдельных токсических продуктов
бактериального расщепления аминокислот
и конкретными симптомами каловой
аутоинтоксикации.
Так, октопамин является ложным ней-
ротрансмиттером в ЦНС и, при
экспериментальном введении животным, вызывает
у них симптомы, весьма напоминающие
некоторые черты печёночных прекомы и
комы (извращение сна, хлопающий тремор,
характерные изменения ЭЭГ). Тирамин
вмешивается в регуляцию кровяного
давления и способен провоцировать гипертен-
зию. Серотонин известен своим влиянием
на мозговое кровообращение и связью с
патогенезом мигрени (см. т. I данного
руководства, стр. 585). Характерно, что головные
боли при упорных запорах сохраняются
даже при пересечённом спинном мозге.
Гистамин понижает порог резистентности к
анафилаксии, вызывает лейкотаксис и имеет
отношение к пищевому лейкоцитозу, кроме
того, по мнению А.М.Уголева, кишечный
гистамин способен оказывать влияние на
желудочную секрецию. Ряд аминов угнетает
Патофизиология белкового обмена 71
красное кроветворение. Пирролидин
вызывает депрессию.
Таким образом, протеин-мальдигестия
(и протеин-мальабсорбция) патогенны не
только в силу того, что они полностью или
частично лишают организм необходимых
нутриентов и калорий. Большое значение
имеет и информационная сторона
развивающихся нарушений: ложные сигналы,
поступающие из ЖКТ, отсутствие
физиологических сигналов (например, аминостати-
ческого действия продуктов переваривания
белков на аппетит) и, наконец энтеральная
антигенная и суперантигенная стимуляция
иммунной системы. Групповая ксантопро-
теиновая реакция Бехера с сывороткой
крови позволяет оценить степень
аутоинтоксикации продуктами кишечного гниения
белка. В нормальных условиях ее показатель
не превышает 20-30 ксантопротеиновых
единиц. При каловой аутоинтоксикации,
печёночной недостаточности и, особенно,
уремии он может значительно возрастать
(более 100 единиц).
НАРУШЕНИЯ
ЧРЕЗМЕМБРАННОГО
ТРАНСПОРТА
АМИНОКИСЛОТ
Трансмембранный перенос аминокислот
определяет эффективность их всасывания
в кишечнике, захвата печенью, реабсорб-
ции почками. Он имеет место в желчном
пузыре, мозге, скелете и мышцах, а также в
эритроцитах. Системы, участвующие в этом
транспорте, аналогичны в различных
органах. Они обеспечивают энергозависимый
транспорт аминокислот с участием HOHaNa+,
движущегося по градиенту концентраций,
без прямой затраты АТФ (A.M. Уголев,
1987). Аминокислотный транспортёр на
апикальной мембране энтероцитов работает
сопряженно с натриевым насосом базола-
теральной мембраны, создавая транзитный
перенос при сохранении натриевого
градиента. Независимый от Na+ транспорт
аминокислот также представлен в энтероцитах
и других клетках, но имеет второстепенное
значение. Механизмы транспорта
аминокислот в клетку множественны. По крайней
мере, для части из них, исключая
транспортёры пролина, большое значение имеет так
называемый у-глутамильный цикл (А. Уайт
исоавт., 1981).
В данном цикле аминокислоты
переносятся в клетку путём обратимого
формирования комплекса с трипептидом глу-
татионом за счёт гидролиза пептидной
связи последнего. После внутриклеточного
отщепления от комплекса аминокислоты
глутатион регенерирует с затратой АТФ
в трех последовательных реакциях. Всего
в данном цикле участвуют 6 ферментов, и
большую роль играет параллельный
транспорт натрия.
При нарушениях трансмембранного
переноса аминокислот механизмы и проявления
могут быть связаны с их кишечной
абсорбцией, обменом между кровью и печенью и
почечной реабсорбцией. Нарушения
транспорта аминокислот охватывают
селективные и групповые расстройства их кишечного
всасывания, равно как и тубулопатии с
нарушением реабсорбции ряда аминокислот
почечным канальцевым эпителием.
Эффективность всасывания
аминокислот при адекватном переваривании белков
превышает 98%. Меченые аминокислоты
появляются в крови уже через 10 минут
после приёма белка, а через 50 минут их
всасывание выходит на максимальную
скорость.
У взрослых всасываются только
аминокислоты. У новорожденных и грудных детей
первых 2-3 месяцев жизни, в особенности,
недоношенных — возможно и всасывание
коротких пептидов, в том числе —
антигенных. Некоторые белки, в частности,
иммуноглобулины в этом периоде онтогенеза
форсируют кишечный барьер. Именно этим
путём антитела материнского молозива
попадают в кровь новорожденных,
поддерживая пассивный иммунитет (Р. Воллер-
тюн, У.Мюллер, 1980). Характерно, что
молозиво содержит ингибитор трипсина,
72 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
предохраняющий иммуноглобулины от
быстрого гидролиза. При искусственном
вскармливании эти адаптивные особенности
могут оборачиваться против
новорожденного. Интактные пептиды коровьего молока
и яиц способны всосаться через кишечный
барьер и обеспечить энтеральную
сенсибилизацию. При раннем переходе на
смешанное и искусственное вскармливание
перекрёстная иммунная реактивность пептидов
из состава альбумина коровьего молока и
человеческого инсулина может
спровоцировать у носителей антигенов ГКГС D3/D4
аутоиммунный ответ против собственных
В-клеток, что приводит к инсулинзависи-
мому сахарному диабету (см. т. I данного
руководства, стр.502, а также ниже раздел
«Патофизиология сахарного диабета»).
Во всасывании аминокислот кишечником
(равно как и в их чрезмембранном переносе
другими клетками организма) участвуют
пермеазные системы, которые не
абсолютно индивидуализированы по отношению к
каждой аминокислоте. Насчитывают 5 таких
систем, работающих с группами химически
близких аминокислот.
>- Первая обеспечивает транспорт
относительно крупных нейтральных моноамино-
монокарбоновых аминокислот, к которым
относятся, по меньшей мере, 15 из числа
тех, что встречаются в пищевых белках.
>• Вторая специализируется на транспорте
двухосновных аминокислот (орнитин,
аргинин, лизин) и цистина.
>* Третья предназначена для перемещения
через биомембраны кислых дикарбо-
новых аминокислот (аспарагиновой,
глутаминовой).
>* Четвёртая занимается транспортом
наиболее малых по размеру молекул
глицина, оксипролина и пролина.
>* Существуют данные в пользу наличия
для циклической аминокислоты пролина
ещё одной, особой пермеазы.
Помимо группоспецифических пермеаз-
ных систем признают наличие
индивидуальных переносчиков для многих из
аминокислот. Кроме того, существует и система
внутриклеточного переноса коротких
пептидов, с их последующим внутриклеточным
гидролизом, дополняющая аминокислотный
транспорт.
Доказано, что аминокислоты могут
конкурировать за одну и ту же транспортную
систему.
Нарушения транспорта и интермедиарно-
го обмена аминокислот совокупно
называются аминоацидопатиями.
Нарушения обмена каждой отдельной
аминокислоты редки (частота фенилкето-
нурии — 1/10000 считается в этой группе
сравнительно большой), но поскольку их
много, суммарная частота аминоацидопатий
доходит до 0,5 % популяции.
Всего описано 10 различных
транспортных аминоацидопатий (см. также т. I
данного руководства, стр. 141-142). Из них 5
вызваны аномалиями группоспецифических
транспортных рецепторов (по
традиционной терминологии — пермеаз) и вовлекают
каждая несколько близких по строению
аминокислот.
Цистинурия, болезнь Хартнупа и
двухосновная аминоацидурия имеют
клинически значимые провления, а дикарбоксила-
миноацидурия и иминоглицинурия —
бессимптомны.
Другие 5 транспортных аминоацидопатий
(гиперцистинурия, гистидинурия, лизину-
рия, мальабсорбции триптофана и метиони-
на) являются субстрат-специфичными.
Все эти состояния аутосомно-рецессивные
моногенные наследственные болезни.
Наиболее часто встречается и наиболее важна
в клиническом отношении цистинурия
(частота 1/15000). Дело в том, что цистин
малорастворим (при концентрациях свыше
400 мг/л выпадает в осадок). При данном
заболевании он формирует камни почек,
мочевого пузыря и уретры. Болезнь имеет
3 генокопии. При I типе нет выраженной
гипераминоацидурии, при II типе, в
дополнение к мочевым симптомам, снижено
кишечное всасывание и увеличена почечная
экскреция орнитина, лизина и аргинина, а
не только цистина. III тип характеризуется
нарушенной почечной, но почти интактной
кишечной абсорбцией аминокислот.
Интересно, что пищевая нагрузка цистином при
цистинурии не ведёт к ухудшению мочевых
симптомов, так как эта аминокислота плохо
всасывается в кишечнике больных. Но при
поступлении цистеина он всасывается,
метаболизируется в цистин, который не ре-
абсорбируется почками, и это провоцирует
обострение мочевого синдрома.
Описана также особая наследственная ги-
перцистеинурия, при которой изолированно
нарушен транспорт цистеина.
Цистинурию дифференцируют от цисти-
ноза - другого нарушения транспорта
аминокислот, при котором понижена реабсорб-
ция сразу 16 аминокислот, принадлежащих
ко всем четырём транспортным группам. Не
страдает только транспорт пролина, окси-
пролина, фенилаланина, триптофана и
тирозина. При цистинозе мочевые цистиновые
камни являются, как раз, редкостью.
Считается, что присутствие других аминокислот
в моче солюбилизирует цистин. В тканях
же, особенно, в селезёнке, соединительной
ткани, печени, роговице — цистин может
откладываться. Описаны случаи, когда
цистиновые кристаллы формировали в
селезёнке островки весом более 1/12 общей массы
органа. В целом, при цистинозе сильнее
нарушен синтез белка и имеется более
глубокое отставание в физическом развитии,
чем при цистинурии. Считается, что при
цистинозе первичный дефект — системное
лизосомальное нарушение (дефект
транспортного белка лизосомальных мембран
цистинозина не дает эвакуировать цистин из
лизосом). Таким образом, это, своего рода,
первичный тезаурисмоз. Нарушение реаб-
сорбции аминокислот, неаминокислотных
компонентов первичной мочи и даже воды
развивается вторично, в силу расстройства
общих механизмов энергообеспечения
транспортных процессов в почках. Цистиноз
наблюдается изолированно и в структуре
синдрома Фанкони (см. т. III данного
руководства, раздел «Патофизиология почек», а
также данный раздел ниже). При детской и
подростковой форме течение болезни
благоприятнее, при поздней форме взрослых
Патофизиология белкового обмена 73
имеется прогрессирующая нефропатия с
исходом в нефросклероз.
Болезнь Хартнупа названа так по фамилии
семейства, где были больны четверо из
восьми детей. При данном заболевании (частота
1/24 000) нарушено всасывание и увеличена
экскреция крупномолекулярных
нейтральных аминокислот, вторично, вследствие
нехватки триптофана, развиваются нарушение
синтеза НАД и картина пеллагры (см.
«Патофизиология витаминного обмена» ниже).
В частности, пеллагроидные симптомы
представлены дерматитом, фотосенсибилизацией,
деменцией. Имеется мозжечковая атаксия.
В кишечнике увеличена продукция индол-
производных, что ведёт к индиканурии и
интоксикационным симптомам, описанным в
предыдущем разделе. Помимо полностью
манифестной генокопии (тип I) имеются
частично манифестная форма без кишечных
симптомов (тип И), а также латентное носи-
тельство (тип III).
При двухосновной аминоацидурии
(частота встречаемости 1/60000, повышена среди
финнов и франко-канадцев) не реабсор-
бируются аргинин, орнитин и лизин, но не
цистин. Камнеобразование нехарактерно.
Однако, развивается нарушение цикла
мочевины, гипераммониемия, задержка роста.
Имеются два подтипа заболевания, 1-й
может быть аутосомно-доминантным,
связан с дефектом транспорта на апикальной
мембране энтероцитов и сопровождается, в
дополнение к упомянутым симптомам, ещё
и умственным отставанием. При 2-м дефект
касается базолатеральной мембраны
энтероцитов и фибробластов. Больные не
переносят фенотиазиды и белковую нагрузку.
Мальабсорбция триптофана, как и
болезнь Хартнупа, ведёт к усиленному
образованию в кишечнике производных индола.
Наступает индиканурия и индолфекалия,
при которой испражнения и моча могут
приобретать голубоватый оттенок. Индо-
лопроизводные нарушают кроветворение
и вызывают поражение нервной системы.
Нередко отмечается кальциноз почек.
Метиониновая мальабсорбция
характеризуется умственным отставанием, задержкой
74 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
роста, гипопротеинемическими отеками —
вследствие дефицита этой незаменимой
аминокислоты и угнетения скорости
белкового синтеза. Больные имеют нарушения
образования меланина и, как правило, это
светлоглазые блондины. Они могут страдать
приступами одышки и судорогами.
Лизинурия сопровождается судорогами и
нарушением психомоторного развития.
При гистидинурии нарушаются синтез
гемоглобина и функции ЦНС.
Аутосомно-рецессивное изолированное
наследственное нарушение транспортной
системы дикарбоновых аминокислот (ас-
парагиновой и глутаминовой) протекает
доброкачественно и затрагивает как почки,
так и кишечник.
Наследственные нарушения могут
касаться вспомогательных ферментов,
регенерирующих глутатион в вышеупомянутом
универсальном звене транспорта многих
аминокислот — у-глутамильном цикле.
Такова, например, оксипролинурия, при
которой не расщепляется пирролидонкар-
боновая кислота, являющаяся побочным
продуктом при отщеплении аминокислот,
переносимых в клетки, от глутатионового
транспортера. Помимо самой этой кислоты,
в моче и фекалиях оказываются и многие
другие, чьё всасывание нарушено. Не
исключено, что и при цистинозе (см. выше)
действует сходный патогенетический
принцип, обусловливающий нарушение
транспорта многих аминокислот разных групп.
В транспорте ряда аминокислот участвует
фосфопиридоксалевая форма витамина В6,
в связи с чем их всасывание нарушается при
соответствующем гиповитаминозе.
Возможен конкурентный механизм
нарушения транспорта аминокислот, когда
из-за значительного избытка одной из них и
перегрузки транспортной системы страдает
реабсорбция (кишечная абсорбция) других,
переносимых той же пермеазой. Подобным
образом объясняют, например, нарушение
транспорта глицина и оксипролина при
пролинемии. При таких ситуациях
первопричиной, обычно, является повышение
концентрации одной или нескольких
аминокислот в крови в результате аномалии их
промежуточного метаболизма. Кишечный
транспорт, в отличие от почечного, чаще
всего не страдает.
Своеобразным нарушением транспорта
аминокислот, сопровождаемым вторичной
аминоацидурией, является синдром Лоу
(окуло-церебро-интестино-ренальный
синдром). Это рецессивное наследственное
заболевание, сцепленное с Х-хромосомой.
Нарушения состоят в генерализованном
дефекте транспорта аминокислот, по всей
вероятности, касающемся у-глутамильного
цикла. Имеется нарушение обмена
внутриклеточного посредника инозитолтрифосфа-
та. Кроме почечной реабсорбции нарушен
аммониогенез и, как следствие, имеется
ацидоз. Развивается выраженная задержка
физического и психомоторного развития,
двусторонняя врожденная катаракта и
глаукома, ведущие к слепоте. Отмечаются
гипотонус мышц и черепно-лицевая диз-
морфия.
При синдроме Фанкони (синонимы: ами-
нодиабет, синдром де Тони-Дебре-Фанко-
ни) имеется возрастание количества амино-
азота мочи в 30-40 раз (при отсутствии ги-
пераминоацидемии). Усиленное выведение,
практически, всех аминокислот с мочой и
полиурия сочетаются с почечным канальце-
вым ацидозом, гиперфосфатурией и
псевдорахитической остеомаляцией, а также с
почечной глюкозурией (при нормогликемии).
Предполагается, что в основе первичного
наследственного синдрома Фанкони могут
лежать дефекты тесных клеточных
контактов и апикальных мембран в канальцах неф-
рона и (или) расстройства энергетического
обеспечения реабсорбции. Синдром бывает
как аутосомно-рецессивным, так и ауто-
сомно-доминантным. Дифференцируются
взрослая и младенческая формы.
Вторичный полный и неполный синдром
Фанкони воспроизводится при галактозе-
мии, фруктозурии, цистинозе, тирозинемии
и болезни Коновалова-Вильсона.
При некоторых наследственных гипера-
миноацидуриях точная природа первичного
дефекта неизвестна. Так, аминоацидурия без
Патофизиология белкового обмена 75
поражения кишечной абсорбции
аминокислот характерна для аутосомно-доминантного
синдрома Людера-Шелдона и
сопровождается фосфатурией.
При тяжёлой аутосомно-рецессивной
аномалии — синдроме Роули-Розенберга,
сопровождаемой задержкой физического
развития, гипоплазией мышц, поражением
лёгких и правожелудочковой сердечной
недостаточностью, имеется дефект почечной
реабсорбции всех аминокислот.
Приобретённые нарушения транспорта
аминокислот характерны для отравления
тяжёлыми металлами (медью, ртутью,
свинцом, ураном, кадмием). Эндогенное
отравление медью при болезни
Коновалова-Вильсона (см. т. I, стр. 143, 161) также
сопровождается подобным нарушением и
выраженной аминоацидурией, как полагают,
вследствие вторичного угнетения почечной
цитохром-оксидазы (см. ниже
«Патофизиология обмена микроэлементов»).
Глюкоза и другие гексозы ингибируют
в высокой концентрации всасывание
аминокислот и способствуют как их задержке
в кишечнике, так и аминоацидурии (в том
числе, это происходит при сахарном
диабете, а также наследственных галактоземии и
фруктозурии).
Потеря кальция с мочой при выраженном
рахите и при наследственных нарушениях
активации и рецепции витамина D,
имитирующих рахит, тоже сопровождается
нарушением реабсорбции ряда аминокислот,
по-видимому, вследствие ингибирующего
действия парат-гормона на этот процесс при
вторичном гиперпаратирозе (см. ниже
«Патофизиология витаминного обмена»).
Почечный транспорт аминокислот
нарушен и при токсической нефропатии,
вызванной органическими ядами (щавелевой
кислотой, лизолом, фосфорными
соединениями).
До трёхмесячного возраста, система
трансмембранного переноса аминокислот
остается недостаточно мощной, что вызывает
физиологическую преходящую аминоаци-
дурию новорожденных. Практически у всех
грудных детей примерно до трёхмесячного
возраста существует преходящая имино-
урия, а до 6-месячного возраста — глици-
нурия. У некоторых новорожденных это
дополняется на первом месяце внеутробной
жизни недостаточностью реабсорбции
двухосновных аминокислот (лизин, аргинин,
орнитин) и цистина.
В заключение данного раздела отметим,
что аминоацидурия — синдром, который
может иметь различную этиологию —
заслуживает отдельного рассмотрения.
АМИНОАЦИДУРИЯ
Аминокислоты должны реабсорбировать-
ся тубулярным аппаратом почек на 99%,
главным образом, в начальном отделе
проксимальных извитых канальцев, причём
экскреция аминоазота в моче здоровых
индивидов старше года не превышает 2 мг/кг.
И. Тодоров (1961) указывает, что больше
всего в моче заменимых аминокислот и их
амидов — глицина, глютамина, аланина. В то
же время, реабсорбция многих незаменимых
аминокислот (аргинина, изолейцина,
лейцина) является стопроцентной.
Избыточное выведение с мочой
аминокислот и промежуточных продуктов их
обмена, в том числе и нефизиологических,
называется гипераминоацидурией
(аминоацидурией).
Не только первичные нарушения
системного, либо почечного транспорта
аминокислот, рассмотренные выше, могут приводить
к аминоацидурии.
Аминокислоты могут появляться в моче
в избытке (составляя более 3 % азота мочи)
вследствие приобретенных (например,
печеночная недостаточность) и наследственных
(например, тирозинемия) нарушений их
интермедиарного обмена.
Основные механизмы аминоацидурии
можно подразделить на 3 группы (Ч.Р. Скрай-
вер, 1987):
>* повышение концентрации
аминокислоты) в крови выше максимальных
возможностей почечной реасорбции
(предпочечная аминоацидурия). Сюда от-
76 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
носятся случаи перегрузочной аминоаци-
дурии при перекорме белками. Некоторые
перегрузочные приобретенные аминаци-
дурии могут быть следствием перекорма
белком при относительной
недостаточности витаминов (например, транзиторная
гиперфенилаланинтирозинемия
новорожденных, которая снимается уменьшением
дозы белка и применением фолиевой и
аскорбиновой кислоты). В этой же
группе аминоацидурия при наследственном
(фенилкетонурия) или приобретённом
(гиповитаминоз В6) нарушении процессов
дезаминирования и переаминирования
в печени (см. следующий раздел). Вообще,
основная предпочечная причина аминоа-
цидурии — печеночная недостаточность.
Нарушения дезаминирующего окисления
аминокислот и/или переаминирования
возможны также при белковых дефицитах
и голодании (из-за нехватки В6-зависимых
апоферментов), и при гаповитаминозах В1?
В2 и В5. Инсулинорезистентное ожирение
сопровождается избытком в крови и моче
ряда аминокислот (фенилаланин, лейцин,
изолейцин), внутриклеточное
проникновение которых в норме стимулируется
инсулином;
>■ конкурентное ингибирование одной
аминокислотой реабсорбции и метаболизма
других (смешанная аминоацидурия).
Примером может служить наступающая
при дефекте фермента аргининосукци-
нат-синтетазы цитру ллинемия, когда
выводится не только сам цитруллин,
концентрации которого в крови
чрезмерны, но и еще 5 аминокислот, имеющих
нормальные концентрации;
>• дефект транспортера или сопряженного
с ним энергетического процесса в самих
почечных канальцах (ренальная
аминоацидурия). Данное состояние может
развиваться при модификации
транспортных рецепторов или нарушении
энергообеспечения транспортных систем,
описанных в предыдущем разделе;
>* дефект апикальной мембраны клеток
почечного эпителия, приводящий к утечке
аминокислот из эпителиоцитов в просвет
канальца (СИ. Рябов, Ю.В. Наточин,
1997).
Аминоацидурия часто сопровождается
гипераминоацидемией и нарушением
всасывания аминокислот в кишечнике.
ПУТИ МЕТАБОЛИЗМА
АМИНОКИСЛОТ
При изучении данного и последующих
разделов книги, для облегчения восприятия
биохимических аспектов темы,
рекомендуется не только пользоваться
иллюстрациями в тексте, но и обращаться к известной
схеме «Метаболические пути» Д. Николсона
(http://iubmb.nicholson.org/).
В норме концентрация свободных
аминокислот в плазме крови невысока — 4-
8 мг/дл. Почти половина аминоазота крови
приходится на аланин. Приём аминокислот
в пищу и их внутривенное введение, при
здоровой печени, на удивление мало влияют
на этот показатель. В течение 5 минут почти
100% дозы оказывается в тканях. Главный
поглотитель аминокислот — печень,
менее активны почки. Классические данные
М. Фишлера показали, что для захвата
печенью всосанных в кишечнике аминокислот
не является принципиально важным, как
именно они попадут в печень — по v. porta
или по a. hepatica. После наложения экков-
ской фистулы аминоазот плазмы растёт
незначительно и ненадолго. Мозг поглощает
аминокислоты быстро, но избирательно,
предпочитая гистидин, глицин, аргинин,
метионин, глутамин, глутаминовую кислоту
и тирозин. В эритроцитах аминокислот в
2 раза больше, чем в плазме. Гиперамино-
ацидемия является показателем сниженной
утилизации аминокислот, прежде всего,
печёночной.
Печень, как главный орган белкового
метаболизма, выполняет ряд важных функций
(рис. 11). Среди них:
>* Переаминирование аминокислот, то есть
обратимый перенос их аминогруппы на
Патофизиология белкового обмена 11
а-кетокислоты без освобождения
аммиака. Этот процесс, открытый
отечественными биохимиками А.Е. Браунштейном
и М.Г. Крицман (1937), обеспечивает
образование структурно новых заменимых
аминокислот. В нём непосредственно
могут принимать участие все аминокис-
аминокислоты
периферическая
кровь
1. RCHCOOH-
- RCH2 + С02
I
NH2
RCHCOOH-
I
NH2
NH2
RCCOOH
II
NH
декарбоксилирование
HOH R-CCOOH+ NH3
-CCOOH +
О
окислительное
дезаминирование
3. RiCHCOOH + R2CCOOH
I
NH2
О
CH2OH
RiCCOOH + R2CHCOOH
NH2
переаминирование
CH
I
соон
NH2
I
CH
I
COOH
NH2
модификация
боковой цепи
н2о
RiCHCOOH + R2CHCOOH
I I
NH2 NH2
O
P
RiCHC-NH
NH2
-CHCOOH
I
R2
полимеризация
Puc.11. Основные реакции с участием аминокислот
(По Я. Мусилу и соавт., 1981). Пояснения в тексте.
лоты, кроме треонина, а также их амиды.
Центральную роль играет кофермент
трансаминаз — витамин В6 и, во многих
случаях, посредником служит глутамино-
вая кислота, которая, наряду со своим ке-
тоаналогом, а-кетоглутаровой кислотой,
используется для переноса аминогруппы
между различными кетоскеле-
тами. Благодаря переамини-
рованию печень обеспечивает
перераспределение аминного
азота и доводит пищевую смесь
до балансового оптимума, так
как этот процесс даёт
возможность понизить до требуемого
уровня концентрации любых
аминокислот, кроме треонина,
и повысить содержание любых
заменимых аминокислот, если
их не хватает. Оптимальная
смесь, благодаря этому,
переносится кровью во все органы.
Переаминирование — ключевое
звено взаимосвязи белкового
метаболизма с жировым и
углеводным. Кетокислоты могут
возникать из небелковых
предшественников. Аминокислоты
через переаминирование могут
терять аминный азот и
превращаться в кетокислоты, после
чего их углеродные фрагменты
могут входить в состав
глюкозы и гликогена, а перед этим
обнаруживаться в пирувате,
оксалате или а-кетоглутарате
(гликогенные
аминокислоты — например, аланин, валин
и еще 14 наименований).
Фрагменты других аминокислот
появляются в ацетоацетате и
ацетилкоэнзиме А (кетогенная
аминокислота лейцин) или
же как в гликогенных, так и в
кетогенных предшественниках
(изолейцин, фенилаланин,
тирозин, лизин). После переами-
нирования создаётся
возможность использовать продукты
78 А.Ш. Зайчик, Л.П.Чурилов Патохимия &
дезаминирования в глюконеогенезе и
для образования кетоновых тел и липи-
дов включая стероиды.
>• С переаминированием тесно связан
процесс окислительного дезаминирования,
осуществляемый аминооксидазами
печени. При этом аминокислоты
расщепляются до аммиака, воды и кетокислоты.
Как правило, сначала идёт В6-зависимое
переаминирование аминокислоты с
образованием глутаминовой кислоты,
которая и подвергается затем
окислительному дезаминированию. Равновесно
сопряженный с данным процессом путь
восстановительного аминирования ведет
к нейтрализации аммиака и
превращению кетокислоты в аминокислоту с
присоединением водорода, донором
которого выступают витамин В2-зависимые
флавиновые ферменты.
Для аминирования необходима
доступность кислот цикла Кребса. Глутамат,
образуемый при переаминировании, в
печёночных митохондриях может либо
окисляться через дезаминирование, либо
переаминируется с оксалоацетатом, давая
аспарагиновую кислоту — донора аминного
азота для производства мочевины (см. ниже
раздел «Нарушения конечных этапов
метаболизма белка»).
Направление равновесных процессов
переаминирования и аминирования-дезами-
нирования во многом зависит от наличия и
концентраций аминокислот и а-кетокислот.
При избытке аминного азота усиливается
превращение аминокислот в кетоаналоги, с
их последующей энергетической или
пластической утилизацией.
Во многих метаболических ситуациях
дикарбоновые кетокислоты становятся
малодоступны. Это бывает при нарушениях
цикла Кребса, гиповитаминозе Bt, тканевой
гипоксии, инсулиновой недостаточности,
а также, в особенности, при отравлении
аммиаком и печёночной недостаточности,
когда замедляется образование мочевины,
и аммиак в тканях связывает
дикарбоновые кетокислоты. Подобные изменения
ю-метаболические нарушения)
могут увеличить содержание свободных
аминокислот в крови, затормозить
переаминирование, направить ацетилкоэнзим А, не
утилизируемый циклом Кребса, на синтез
кетоновых тел, способствовать развитию
ацидоза (Н.Н.Лаптева, 1970). С другой
стороны, усиленное переаминирование в
клетках мозга при печёночной
недостаточности тормозит осуществление самого
цикла Кребса и вызывает тканевую
гипоксию нейронов, дефицит АТФ, нарушение
работы ионных насосов и, как следствие,
деполяризацию и невозбудимость нервных
клеток, что проявляется картиной
печёночной комы.
Ком а как синдром глубокого угнетения
ЦНС вследствие тканевой гипоксии
потому и универсальна по своей симптоматике,
несмотря на разные причины, что из-за
взаимосвязи цикла Кребса,
переаминирования и цикла мочевины, для клеток, в
частности нервных, любая глубокая
тканевая гипоксия оборачивается нарушением
переаминирования и первичной
нейтрализации аммиака, а нарушение нейтрализации
аммиака печенью, наоборот, истощает пул
дикарбоновых кислот и вторично
затрагивает цикл Кребса в ЦНС (подробнее см.
ниже раздел «Нарушения конечных этапов
метаболизма белка»). Эти взаимоотношения
подробно будут рассмотрены при
изложении основ патологии печени в т. III данного
руководства. Но уже здесь авторам кажется
уместным привести рис. 12, отражающий
взаимодействие двух циклов — мочевины и
Кребса — как двух колёс одного велосипеда,
из которых ни одно не способно
«вращаться» без исправного вращения второго!
>- Печень направляет аминокислоты на
синтез собственных белков, белков
плазмы крови и в другие органы (см. рис. 11,
вверху). Новые белки не встраивают
пептиды старых, а синтезируются всегда
заново. Очень быстрый ресинтез белка
из аминокислот постоянно происходит
в нервной и мышечной ткани, в железах
внутренней секреции, поставляющих
пептидные и аминокислотные
гормоны, а также во всех быстропролифе-
Патофизиология белкового обмена 79
карбамоилфосфат
АТФ
acnapmam
фумарагп
сукцинт
АКоА
цитрат
мочевина
Рис. 72. Взаимозависимость цикла Кребса и цикла мочевины
рирующих клетках. Экспортируемые
клеткой белки обмениваются быстрее
собственных. В то же время, скорость
обновления аминокислот в ряде
белков опорно-двигательного аппарата,
например, коллагене — минимальна.
Печень синтезирует с участием
аминокислот небелковые расходуемые
азотсодержащие соединения — пурины,
пиримидины, мочевую кислоту, креатин
и никотиновую кислоту (последнюю
у человека — в недостаточных
количествах). Продукты метаболизма этих
соединений или их избыток выводятся
почками, как и мочевина.
В печени, мозге, хромаффинной ткани
надпочечников, параганглиях,
диффузных нейроэндокриноцитах и многих
других тканях происходит декарбок-
силирование некоторых аминокислот
(гистидин, тирозин, триптофан, глу-
таминовая кислота и др.) с
образованием аминов (гистамина, тирамина,
окситирамина, триптамина, серотонина,
у-аминомасляной кислоты и т.д.). В свою
очередь, амины окисляются моноами-
ноксидазами тканей. Из-за нейротран-
смиттерной и гормональной роли этих
соединений, а также освобождения
активных кислородных радикалов при
их метаболизме данный путь обмена
аминокислот имеет большое значение
для регуляции гомеостаза (см. также
разделы «Нарушения переваривания
белков...», «Патофизиология
эндокринной регуляции»).
ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ
БЕЛКОВОГО ОБМЕНА И ЕГО
НАРУШЕНИЯ
ПРИ ЭНДОКРИНОПАТИЯХ
В данном разделе даётся краткая
характеристика эндокринной регуляции белкового
обмена, многие аспекты которой будут
существенно дополнены ниже, в разделе
«Патофизиология эндокринной регуляции».
80 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Основные гормоны, регулирующие
белковый обмен, — это СТГ, соматомедины, глюко-
кортикоиды, половые стероиды, тироидные
гормоны и пептидные гормоны островков
Лангерганса. При эндокринных нарушениях
с участием названных гормонов системный
белковый метаболизм закономерно и
значительно изменяется. Ниже приводится очерк,
обрисовывающий взаимодействие
основных гормональных регуляторов белкового
метаболизма. Попутно характеризуются
важнейшие нарушения обмена белков при
эндокринных заболеваниях (см. также
разделы, посвященные патологии отдельных
эндокринных желёз).
>* СТГ — способствует ускорению синтеза
белка, как в висцеральном отсеке тела
(печень, внутренние органы), так и в
соматическом (скелетные мышцы, кости,
хрящи). Усиливается включение
аминокислот и в белки лимфоидных органов
(у экспериментальных животных
инъекции СТГ даже способствуют развитию
лимфосарком!) Все это приводит при
акромегалии и гипофизарном гигантизме
к положительному азотистому балансу.
При гипофизэктомии и гипофизарном
нанизме азотистый баланс —
отрицательный. СТГ прямо увеличивает захват
аминокислот клетками, в частности,
мышечными волокнами. Продукция
мочевины в печени под влиянием СТГ
несколько понижается (Н.И. Лаптева,
1970). В то же время, гормон роста
повышает продукцию инсулиновых
рецепторов, а также освобождение инсулина
и глюкагона из островков Лангерганса.
По отношению к инсулину действие
СТГ, в целом, контринсулярное, однако
эти гормоны разделяют анаболический
эффект на белковый метаболизм. Гормон
роста обеспечивает свой
анаболический эффект на белковый обмен за счёт
активации катаболизма липидов, при
торможении использования глюкозы, а
инсулин — за счёт активации
использования экзогенной глюкозы. Во многих
случаях нехватка инсулина тормозит
эффективность СТГ в отношении
анаболизма белка. Тироидные гормоны,
инсулин и, особенно, андрогены пер-
миссивно способствуют эффектам СТГ
на метаболизм протеинов. В отсутствие
СТГ анаболический эффект андрогенов
минимален. СТГ опосредует своё
тканевое действие через семейство тканевых
соматомединов. Соматомедины и их
рецепторы имеют высокое сходство и
перекрёстную активность с инсулином,
(см. ниже раздел «Гормон роста и
патофизиология ростовых и анаболических
процессов»).
>■ Инсулин — анаболический гормон
белкового обмена, снижающий деградацию
и стимулирующий синтез белков как
во внутренних органах (печень, почки,
миокард), так и в соматическом отсеке
(скелетные мышцы, костный мозг). Его
секреция стимулируется белковой
пищей, особенно аргинином и кетогенной
аминокислотой лейцином. Механизмы
анаболизирующего действия инсулина
* на обмен белка включают:
>* Прямую стимуляцию сборки рибосом и
трансляции (по типу инициирующего
фактора).
>* Усиление активного транспорта
аминокислот в клетки (особенно, валина,
лейцина, изолейцина).
>• Торможение глюконеогенеза из
аминокислот в печени.
>- Ингибирование освобождения
аминокислот из клеток, особенно мышечных.
>* Трофические долговременные эффекты,
связанные с усилением синтеза ДНК,
РНК и митотической активности инсу-
линзависимых тканей.
Эффект инсулина на рост и белковый
синтез синергичен с соматотропным
гормоном. При сахарном диабете, особенно ин-
сулинопеническом, происходит угнетение
синтеза белка и усиление его катаболизма,
что ведет к таким симптомам, как гипер-
аминоацидемия, отрицательный азотистый
баланс, усиление экскреции мочевины.
Отчасти это обусловливает иммунодефицит
Патофизиология белкового обмена 81
при диабете, который, конечно же, связан и
с другими факторами (см. также т. I, стр. 515
и ниже в т. II раздел «Патофизиология
сахарного диабета»). Усиление распада
белков в инсулинчувствительных тканях при
диабете особенно характерно для скелетной
мускулатуры и сопровождается симптомами
мышечной слабости. Тироидные гормоны
стимулируют выработку инсулина, а
глюкокортикоиды -тормозят, но и те и другие
являются антагонистами многих его эффектов
(У.А. Содеман, Т.М. Содеман, 1985).
> Глюкагон, секреция которого в островках
Лангерганса стимулируется аргинином
и аланином, является антагонистом
инсулина по действию на белковый обмен
и вызывает понижение поглощения и
увеличение освобождения аминокислот
мышцами. В печени он стимулирует глю-
конеогенез и кетогенез, в том числе, из
аминокислот. Под влиянием глюкагона
в печени увеличивается образование
мочевины, синтез белка замедляется
на стадиях элонгации и терминации
полипептидных цепей, а скорость про-
теолиза растёт. Глюкагон тормозит
анаболические эффекты СТГ и, будучи
антагонистом инсулина, увеличивает его
освобождение из островков Лангерганса.
В условиях гиперглюкагонемии,
свойственной голоданию, многим формам ин-
сулинонезависимого сахарного диабета,
а также наблюдающейся при глюкаго-
номах — анаболизм белка тормозится, а
аминокислоты используются в
энергетических целях и для образования кетонов
(Л.К. Старосельцева, 1976). Бывают
гипоаминоацидемия и гипопротеине-
мия. Тироидные, глюкокортикоидные
гормоны и СТГ стимулируют продукцию
глюкагона у человека.
> Соматостатин — вырабатывается под
влиянием всех стимулов,
увеличивающих продукцию инсулина (в том
числе, аминокислот, особенно, лейцина и
аргинина). Он ингибирует эффекты и
продукцию инсулина, глюкагона и СТГ,
пролонгирует усвоение нутриентов,
в том числе аминокислот — тканями.
Соматостатин тормозит синтез белка,
но не стимулирует его распад. Он
считается одним из главных участников
метаболической перестройки при такой
белок-сберегающей форме эндогенного
голодания, как зимняя спячка (см. выше).
СТГ, тироидные гормоны и глюкагон
стимулируют его выделение, а
глюкокортикоиды, предположительно, тормозят.
>- Глюкокортикоиды стимулируют
превращение аминокислот в предшественники
глюконеогенеза, тормозят синтез белка и
ускоряют его деградацию в лимфоидной
ткани и соматическом отсеке организма,
но не в печени, где биосинтез многих
глобулинов и трансаминаз, наоборот,
усиливается. В этом качестве они принимают
участие в переброске аминокислот из
соматического отсека — в висцеральный
при стрессе, травмах, ответе острой фазы,
голодании (см. выше раздел «Голодание»
и в т. I — стр. 371-373). Так как
возрастание синтеза ряда печеночных белков не
уравновешивает усиления их распада в
соматическом отсеке, при гиперкорти-
цизме имеется гипераминоацидемия,
аминоацидурия, отрицательный
азотистый баланс, усиление биосинтеза
мочевины. Болезнь и синдром Ицен-
ко-Кушинга характеризуются всеми
этими проявлениями, что
сопровождается атрофией мышц, истончением кожи,
гипоплазией тимико-лимфатического
аппарата и остеопорозом. При раннем
дебюте заболевания даже рост больных
тормозится. При гиперкортицизме
замедлено заживление ран (см. ниже
«Патофизиология эндокринной
регуляции»). Действие глюкокортикоидов
антагонистическое по отношению к СТГ
и инсулину. В ситуации ответа острой
фазы они синергично взаимодействуют
при перераспределении энергетических
эквивалентов с глюкагоном, секрецию
которого увеличивают.
>■ Лндрогены проявляют СТГ-зависимый
анаболизирующий эффект на синтез
белка, особенно в мышцах, скелете, мужских
половых органах, коже и её производных,
А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
в меньшей степени — в почках и мозге.
Эстрогены — не менее активные
стимуляторы белкового синтеза в костях, но в
целом, их анаболическое действие более
прицельно и сконцентрировано на
женских половых органах и молочных железах.
При совместном действии с андрогенами
они взаимно антагонистичны по своему
действию на половые органы. По
некоторым данным, стимуляция синтеза белка в
соматическом отсеке организма и половых
органах при действии анаболических
стероидов не сопровождается адекватным
увеличением синтеза белка в печени. Ряд
авторов при больших дозах анаболических
стероидов сообщали даже о снижении
синтеза в печени альбумина (Н.Х. Аб-
дуллаев, 1966). К тому же, андрогенные
стероиды вызывают холестаз и могут в
фармакологических дозах способствовать
развитию гепатопатии, нивелирующей их
анаболическое действие на белковый
обмен в печени (В.В. Закусов, 1978). Все эти
особенности действия половых стероидов
на белковый обмен должны учитываться
при их применении в качестве анаболи-
зантов (см. также ниже «Патофизиология
эндокринной регуляции»).
>• Тироидные гормоны — обладают
уникальной способностью ускорять обновление
белков крови и внутренних органов, а
также соматического отсека тела. Это
достигается, по мнению Н.Н. Лаптевой
(1970), путём активации как
использования избытка свободных аминокислот, так
и катаболизма белков. Преобладающее
направление действия тироидных
гормонов на белковый метаболизм зависит
от «контекста», определяемого пермис-
сивным эффектом других эндокринных
регуляторов и доступностью различных
субстратов окисления, а также от
количества самих тироидных гормонов.
Анаболические функции преобладают
в условиях возбуждённого белкового
синтеза, в молодых растущих тканях и
при ограниченности поступления белка.
Катаболизм белка особенно
активируется при достаточном белковом питании,
при значительном повышении уровня
тироидных гормонов, а также при ответе
острой фазы. По современным данным,
тироидные гормоны располагают
ядерным, митохондриальным и цитозольным
рецепторами. Взаимодействие этих
регуляторов с ядерными рецепторами ведёт к
усилению транскрипции многих генов и,
в принципе, активирует синтез множества
функциональных белков, практически, во
всех органах и тканях, способствуя их
росту, а в ряде случаев — дифференцировке.
In vitro эти гормоны способны
активировать синтез белка в бесклеточной системе
на уровне трансляции. Считается, что этот
их эффект зависит от ускорения
связывания аминоацил-тРНК с полисомами и
in vivo играет большую роль в активации
синтеза быстро метаболизируемых белков
ряда органов, в первую очередь, мозга.
В печени тироидные гормоны значительно
активируют ферменты цикла мочевины и
увеличивают её продукцию. Митохонд-
риальный рецептор тироидных гормонов
опосредует стимуляцию ими синтеза ряда
функциональных белков этих органоидов.
Тироидные гормоны стимулируют захват
аминокислот многими клетками. В то же
время, цитозольное действие этих
гормонов, особенно, Т3, способствует активации
калий-натриевой АТФазы и расходу
энергии (см. выше раздел «Патофизиология
энергетического обмена»). В силу этого,
калорические потребности повышаются,
и активируется использование
энергетических субстратов, не исключая и
свободные аминокислоты. Однако стимуляции
глюконеогенеза из аминокислот под
влиянием тироидных гормонов не отмечается.
Тироидные гормоны стимулируют выброс
всех вышеназванных панкреатических
регуляторов белкового обмена, но
тормозят продукцию глюкокортикоидов. Они
синергичны в анаболическом действии с
СТГ и способствуют периферическому
метаболизму половых стероидов.
В связи с амбивалентностью действия
тироидных гормонов на белковый обмен
при гипотирозе развивается положительный
Патофизиология белкового обмена 83
азотистый баланс, увеличивается
концентрация белков (особенно, глобулинов) в
крови и их процентное содержание в ряде
органов. В интерстициальной жидкости
накапливается альбумин. Особенно
выражено накопление слизистых протеогликанов
в сердце, серозных оболочках и, прежде
всего, во всех слоях кожи. Это приводит к
изменению гидрофильных свойств кожи и
проявляется в виде её утолщения и
слизистого отека — микседемы (В.М. Орд, 1878),
характерной для выраженного гипотироза.
Интересно, что замедляется и
использование свободных аминокислот, вследствие
чего бывает гипераминоацидемия. При
врождённых формах гипотироза (синдром
Фаггё) развивается задержка роста,
психического и физического развития и
формируется одна из форм нанизма. Замедление
кругооборота быстро метаболизируемых
белков мозга при микседеме и гипотироид-
ном кретинизме может являться важным
механизмом, ответственным за ослабление
памяти, нарушение обучаемости и снижение
интеллекта у гипотироидных больных.
Гипертироз, особенно его крайний
вариант — тиротоксикоз (болезнь фон Базедова-
Грейвса) — напротив, сопровождается
ускорением самообновления белков организма.
При этом аминокислоты, по-видимому,
используются и в пластических, и в
энергетических целях. Возникает отрицательный
азотистый баланс, ускоряется катаболизм
мышечных протеинов, что проявляется
характерной креатинурией. Растет
активность тканевых и кровяных протеаз. В то
же время, есть данные о снижении
продукции трансаминаз печени при токсических
дозах тироидных гормонов (см. ниже «Ти-
ротропный гормон и нарушения тироидной
функции»).
На обмен некоторых аминокислот и
белков, в тех или иных тканях и органах,
оказывают специфическое влияние и другие
гормональные регуляторы.
Так, МСГ способствует, а мелатонин
препятствует активности тирозиназы и
синтезу из тирозина меланина. При
повышении образования проопиомеланокортина
(ПОМК), что наблюдается при первичной
недостаточности функций коркового
вещества надпочечников или болезни Аддисона,
возможно усиление пигментации кожи, т.к.
ПОМК содержит и освобождает при процес-
синге полную последовательность а-МСГ и
Р-МСГ (бронзовая болезнь).
Вместе с тем, при вторичной
недостаточности функций коры надпочечников,
связанной с первичной гипопродукцией ПОМК
в аденогипофизе, возможна депигментация
кожи («белый аддисонизм»). Детальнее эти
вопросы описаны в разделе «Основные
синдромы, связанные с дисфункцией
надпочечников» ниже).
Паратгормон в больших концентрациях
активирует катаболизм белка в костной
ткани, в малых дозах — стимулирует
продукцию белковой матрицы кости, а кальци-
тонин, наоборот, уменьшает рассасывание
костной ткани (см. раздел «Патофизиология
паращитовидных желёз...» ниже).
Гормоны тимуса увеличивают
анаболическое использование аминокислот лимфо-
идными клетками.
Катехоламины способствуют
мобилизации аминокислот соматического отсека
организма и использованию их печенью.
Гонадотропные гормоны активируют
образование трансаминаз печени и т.д.
Большое значение в патологии белкового
обмена придают наследственным и
приобретенным нарушениям обмена различных
аминокислот и их производных. Этиология
и патогенез наиболее важных подобных
расстройств рассматриваются в следующем
разделе.
НАРУШЕНИЯ МЕЖУТОЧНОГО
ОБМЕНА АМИНОКИСЛОТ
И ИХ ПРОИЗВОДНЫХ
Существуют наследственные и
приобретённые нарушения интермедиарного обмена
аминокислот. Вначале — об аномалиях,
вызванных мутациями.
84 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Рассматривая нарушения
взаимопревращения аминокислот, отметим, что
скорость их обмена в норме наиболее высока
в нервной ткани. Поэтому разнообразные
наследственные аминоацидопатии в
психоневрологической практике известны, как
одна из основных причин слабоумия.
В первую очередь, выделим наиболее
частую и хорошо изученную мозаичную
наследственную аминоацидопатию,
имеющую несколько генокопий и частоту
встречаемости 1/10000 (при частоте носи-
тельства — до 1/50 среди белых) — фенил-
пировиноградную олигофрению (см. также
т.1 данного руководства, стр. 138). Данное
расстройство аминокислотного обмена
открыто в 1934 г. А. Феллингом, аутосом-
но-рецессивно и чаще всего встречается в
скандинаво-балтийском регионе, а реже
всего поражает негров и евреев.
Большинство случаев фенилкетонурии
сопровождается дефицитом печеночного
фермента фенилаланин-4-гидроксилазы.
В полном соответствии с концепцией А. Гар-
рода это ведет к резкому увеличению
концентрации фенилаланина в крови. Недостаток
превращения фенилаланина в тирозин и
подавление избытком фенилаланина
активности тирозиназы поведет, в конечном итоге,
к дефициту тирозиновых и триптофановых
производных, включая пигмент меланин
(что делает кожу, глаза и волосы больных
светлыми), а также катехоламины (что
проявляется гипотензией) и серотонин (что
имеет отношение к развитию эпилептифор-
мных «салаамовых судорог» и тремора).
Избыток фенилаланина метаболизирует-
ся обходными путями (рис. 13, отражающий
основные пути метаболизма фенилаланина
и тирозина), повышается концентрация
его альтернативных продуктов
метаболизма, избыток которых выводится с мочой.
В крови и моче появляются в
существенных количествах фенилпировиноградная
и фенилмолочная кислоты, фенилацети-
лглутамин. Скрининг-тест на гиперфени-
лаланинемию — реакция Феллинга (проба с
полуторахлористым железом, дающая при
положительном результате зеленое
окрашивание) — выявляет в моче фенилпируват
и широко внедрена в практику неонатоло-
гии во многих странах. Для диагностики
используют и микробиологический тест
Гатри, при котором кровь больного с ги-
перфенилаланинемией поддерживает рост
ауксотрофного фенилаланинзависимого
штамма бактерий (Л.Г. Смит, 1987).
При фенилкетонурии активируются
минорные побочные пути метаболизма
фенилаланина, при этом образуются
метаболиты, практически отсутствующие в
норме (фенилэтиламин, ортофенилуксусная
кислота или фенилацетат — см. рис. 13). Эти
соединения рассматриваются как нейро-
токсины и способны нарушать метаболизм
липидов в мозге. В сочетании с дефицитом
некоторых нейромедиаторов (например,
серотонина) этот механизм считают
ответственным за прогрессирующее снижение
интеллекта у больных, выявляющееся через
несколько месяцев после рождения. Более
30 % из них имеют столь глубокую задержку
психомоторного развития, что не ходят, а
около 2/3 — не говорят. Слабоумие лишь
у 4-5 % этих пациентов остаётся на уровне
дебильности. У остальных коэффициент
интеллекта не достигает 60 и
регистрируется имбецильность или даже идиотизм
(Дж. Джервис, 1954). Фенилкетонурия —
одна из важнейших причин слабоумия у
человека. Для больных характерны также
наклонность к экземе и своеобразный
мышиный запах мочи и пота, обусловленный
фенилацетатом.
При классической форме уровень
фенилаланина в крови больше 16 мг/дл (1 мМ/л).
Основной метод лечения — ограничение
потребления фенилаланина с целью
снизить его концентрации в крови до уровня
3-12 мг/дл. Такое ограничение
практикуется до полового созревания. Взрослые могут
следовать менее строгой диете и потреблять
фенилаланин. Однако, в настоящее время
доказано, что избыток фенилаланина и
его минорные метаболиты тератогенны.
Поэтому, если избыток фенилаланина не
подвергается эффективному метаболизму,
то у женщин, страдающих фенилкетонури-
тгп
NADPH+H+
ДГП
NADP+
NH,
I
CHCOOH
o2 1
фенилаланингидро-
лаза, дигидроптерин-
редуктаза
CO,
фенилаланин
ОН
(! т-снхоон
\ / фенилпируват-
ЮН
гомогентизиновая
I кислота
o2-J
\оксидаза
HOOCC = СССНХСНХООН -
н ни 2||
° °
4-малеилацетоуксусная
кислота
J~\. '
NH
I
снсоон
переаминирование
с а-кетоглутаровой
О кислотой
тирозинамино-
траисфераза
ХА
ссоон
■II
о
снхснхоон
3|| 2
о
ацетоуксусная
кислота
п-оксифеиилпируват
г Н
-► ноосс =ссснхснхоо
Н II 2|| 2
о о
4-фумарилацетоуксусная
кислота
НООСС = ССООН
Н
фумарилацетоацетаза
фенилацетилглутамин
тирозинемия недоношенных,
хоукинсурия
фумаровая
кислота
хоризмовая
кислота
шикимовая
кислота
п-оксифениллактат
А
охронозный
пигмент
п-оксифенилпируват \
тирозиноз \
витамин С J
гомогентизат — """"^
ЯШ алкаптонурия
фумарилацетоацетат
й
тирозинемия I
ацетоацетат
4-диоксифениланин
тирозин) I ^альбинизм
■ I ^
антранилат ^^ w Ч 3,4
наследственный ± тирамин \
гипотироз Т л
моноиодтирозин фенол меланин I
дииодтирозин , пируват
тироксин * NH3 3,4-диоксифенилэтиламин
эритрозо-
4-фосфат
трииодтиронин
Р-енолпируват
^эпинефрин ^
норэпинефрин 1
- метаболиты, накапливаемые при ФКУ
- метаболиты, дефицитные при ФКУ
- места метаболического блока при различных энзимопатиях
Рис. 13. Метаболизм тирозина и фенилаланина и его нарушения. ФКУ-фенилкетонурия
86 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
ей, он может оказывать пагубное действие
на плод, вызывая множественные пороки
развития. При беременности больные фе-
нилкетонурией вынуждены возвращаться
к бесфенилаланиновой диете (В. Кумар и
соавт., 1997).
Болезнь вызвана генной мутацией в 12-й
хромосоме и имеет несколько генокопий.
Они связаны с различными мутантными
аллелями фенилаланингидроксилазы, а
также с дефектом другого фермента гид-
роксилирующей системы фенилаланина —
дигидроптеридинредуктазы, необходимой
для регенерации тетрагидроптерина, донора
электронов для фенилаланин-4-гидрокси-
лазной реакции.
Некоторые из мутантных аллелей фенил-
аланин-4-гидроксилазы обладают
пониженной, но выраженной активностью, и поэтому
в их присутствии болезнь протекает как
доброкачественная гиперфенилаланинемия — с
небольшим повышением уровня
фенилаланина в крови и без развития слабоумия.
Форма, обусловленная дефектом дигидро-
птеринредуктазы, охватывает от 5 до 10%
случаев болезни и, в отличие от
классической, не лечится ограничением употребления
фенилаланина. Она, таким образом, имеет
худший прогноз (злокачественная
гиперфенилаланинемия) и выявляется по снижению
уровня фенилаланина крови после пищевой
нагрузки тетрагидробиоптерином.
Алкаптонурия — аутосомно-рецессивная
болезнь, на примере которой сэр
Арчибальд Гаррод (1909) создал концепцию
метаболического блока. Причина
заболевания — дефект оксидазы промежуточного
продукта распада фенилаланина и
тирозина — гомогентизиновой кислоты. При
крайне незначительной частоте (не более
1/1000 000) болезнь наиболее часто
наблюдается в Северной Ирландии (1/200000).
Алкаптонурия — яркая иллюстрация того,
что генные мутации постоянно
воспроизводят в человеческих популяциях дефектные
аллели, несмотря на отбор, который, к тому
же, не слишком эффективен в отношении
болезней с таким поздним проявлением
(после 30 лет). Болезнь описана еще у
египетской мумии 3 500-летней давности (Дж.
Б. Вайнгарден, 1987). В норме фермент
пара-оксифенилпируватдезоксигеназа, в
присутствии витамина С, превращает в
гомогентизиновую кислоту полученный из
тирозина пара-оксифенилпируват. Затем,
гомогентизиновая кислота должна в почках
окислиться в присутствии Fe+2 и глутатиона
до 4-малеилацетоуксусной кислоты. Если
этот процесс тормозится, то накопление
гомогентизиновой кислоты ведет к ее
превращению полифенолоксидазой в хиноновые
полифенолы, составляющие так называемый
«охронозный пигмент», выводимый
почками (рис. 13). Это обусловливает потемнение
мочи больных на воздухе и при подщелачи-
вании. Хлорное железо при добавлении к
моче больных окрашивается в голубой цвет.
Пациенты, в буквальном смысле, выделяют
с мочой... фотопроявитель. При
попадании на фотобумагу подщелоченная моча
больного вызывает ее почернение!
Гомогентизиновая кислота ингибирует фермент
лизилгидроксилазу, участвующий в синтезе
коллагена, а охронозный пигмент {алкай-
тон) не полностью экскретируется с мочой,
откладывается в основном веществе хрящей
и других соединительно-тканных
образований и делает их хрупкими, что со временем
вызывает кальцификацию и дегенеративный
артрит позвоночника, а также крупных
суставов конечностей. Первыми проявлениями
болезни могут быть пигментация склер и
хрящей ушных раковин (С.Я. Капланский,
1966). На вскрытии хрящи скелета, гортани,
трахеи и другие соединительно-тканные
образования у больных бывают тёмными и даже
угольно-чёрными. Радикально болезнь не
лечится. Степень остеохондропатии можно
уменьшить, защищая активность лизилгид-
роксилазы большими дозами аскорбиновой
кислоты.
При лейцинозе (синоним: разветвлённо-
цепочечная кетонурия) нарушено
окисление разветвленных кетокислот,
производимых путем дезаминирования лейцина,
изолейцина и валина. В Северной Америке
Патофизиология белкового обмена 87
и Новой Зеландии, где традиционным
лакомством является кленовый сироп, его
запах знаком всем с детства. Поэтому там
эту болезнь аппетитно именуют: «maple
syrup urine disease». Поскольку в нашей
стране кленовый сироп непопулярен,
вероятно, можно рекомендовать пользоваться
рациональными названиями. Механизмы
данного и близких к нему заболеваний
и пути обмена лейцина представлены на
рис. 14. При данном аутосомно-рецессивном
заболевании, встречающемся с частотой
1/120 000-1/290 000, нарушено
окислительное декарбоксилирование вышеназванных
кетокислот. В результате накапливаются
и они, и их «материнские» аминокислоты.
Особенно патогенно накопление
лейцина. Дело в том, что данная
аминокислота — единственная чисто «кетогенная», то
есть в норме окисляемая до ацетоацетата
и ацетилкоэнзима А. Из-за большой роли
кетоновых тел в энергообеспечении мозга,
особенно при гипогликемии (см. выше
раздел «Голодание»), нарушения в окислении
лейцинопроизводных чреваты развитием
умственной отсталости, судорог, мышечной
ригидности. Отмечаются гипогликемия и
гипотония. Больные могут впадать в
летаргию. Имеются кетоацидоз, рвота, может
быть гемоцитопения (см. также раздел
«Нарушения промежуточного липидного
обмена» ниже).
Сходную с лейцинозом картину имеет
и связанное с дефектом изовалерил-ко-
фермент А-дегидрогеназы изолированное
нарушение обмена лейцина — изовалерата-
цидемия. При обеих болезнях нарушается
превращение кетокислот в липиды, что
вызывает нарушение продукции миелина,
а также страдает энергетика мозга.
Обнаружено понижение активности глутамат-дека-
рбоксилазы и образования у-аминомасляной
кислоты в мозге больных под влиянием
повышенных количеств разветвлённых
кетокислот.
У некоторых больных лейцинозом
дефектный фермент отвечает повышением
активности на сверхвысокие дозы витамина
В1# В большинстве случаев лечение сводится
к специальной диете с ограничением
потребления алифатических разветвлённых
аминокислот. К этому же семейству ами-
ноацидопатий относят гипервалинемию,
имеющую сходные проявления. Однако,
она вызвана дефектом валин-трансаминазы.
При всех указанных нарушениях обмена
разветвлённых аминокислот без лечения
смерть наступает за несколько месяцев.
Гомоцистинурия, прежде считавшаяся
отдельным наследственным заболеванием
(дефектом сериндегидратазы, то есть циста-
тионин-р-синтетазы), на деле оказалась
синдромом, имеющим у разных больных
неодинаковую этиологию (С.Х. Мадд,
1987). Его частота оценивается в 1/200000.
В большинстве случаев, в дефекте повинен,
действительно, вышеназванный фермент.
При этом гомоцистеин и серии (рис. 15) не
могут с должной скоростью образовывать
цистатионин. Гомоцистеин переходит в
гомоцистин, они накапливаются в крови,
и оба выделяются с мочой. Повышается и
концентрация в крови и моче метионина,
поскольку в норме часть его переходит в
гомоцистеин. Заболевание чувствительно к
терапии витамином В6, который активирует
метаболизм гомоцистеина.
К накоплению в крови и выделению с
мочой гомоцистина могут приводить, помимо
вышеописанного дефекта, также и любые
нарушения в протекании №-метилтетра-
гидрофолат-гомоцистеин-метилтрансфе-
разной реакции (А.Уайт и соавт., 1981).
Среди них:
>- дефект синтеза апофермента;
>• дефицит метилированной формы
витамина В12, являющегося коэнзимом
данной ферментативной системы. Это
состояние, в свою очередь, может иметь
двоякую причину и быть следствием
нарушения биосинтеза метилкобаламина
или его транспорта в лизосомах;
>• Недостаточная активность №10-метилен-
етрагидрофолат-редуктазы,
поставляющей в реакцию метильные группы.
Соответственно этому разнообразию,
часть больных с гомоцистинурией имеет бо-
СНз
I
СН3СНСН2СНСООН ■
траисаминаза
лейцин
I
NH2
СНз
I
-► СН3СНСН2ССООН
II
О
2-кетоизокапроновая
кислота
CoASH
NAD+
N
FADH2
СНз
I
CH3C=CHC-S-CoA
О
2-метилкротонил-СоА
изовалерил-СоА дегидрогеназа
СНз
I
CH3CHCH2C-S-CoA
О
изоеалерил-СоА
^- АТР, С02
«^ ADP + Р,
СНз
I
HOOCCH2C-CHC-S-CoA
II
О
3-метилглутарил-СоА
н2о
CH3C-S-CoA -<-
О
ацетил-СоА
СНз
-► HOOCCH2CCH2C-S-CoA
I II
НО О
3-окси-З-метилглутарил-СоА
J \
СН3ССН2СООН
о
ацетоуксусная
кислота
холестерин
каротины
кофермент Q
пируват
белки
изопреноидные
соединения
2-кетоизовалерат
ацетил-СоА
2-кетоизокапроновая \
кислота
3-окси-З-метилглутарил-СоА
\
блок при
лейцинозе
изовалерил-СоА \
ацетил-СоА\
блок при
изовале-
ратацидемии
ацетоацетат ■
-►- жирные
кислоты
Рис. 14. Метаболизм аминокислоте разветвлённой цепью и его нарушения (По Я. Мусилу и соавт., 1981)
Здесь и далее СоА — Коэнзим А
NH2
ch3-s-ch2<!:h2Chcooh
трансметилаза
►
метионин
NH2
HOCH2CH2CHCOOH
гомосерин
tcmeu,
транссульфураза
NH2
HSCH2CH2CHCOOH
NH2
SCH2CH2CHCOOH
трансаминаза
CoA-SH
NAD+
NADH+H+
co2
CH2CH2CCOOH —
fl
О
а-кетомасляная
кислота
^-Z
CH2CHCOOH
NH2
цистатионин
-► CH3CH2C-S-CoA
О
пропионил-СоА
C02, ATP
ADP, Pi
N
карбоксилаза
HOOCCH2CH2C-S-CoA -*
H
о
сукцинил-СоА
HOOCCH-S-CoA
метилмалонилмутаза \
CH2
метилмалонил-СоА
цистатионин-
синтетаза
„активный
метил**
S-аденозилметионин
I серии I цистатионин
а-кето-у-метилтио-
масляная кислота
| цистеин \
а-кето-у-оксимасляная
кислота
глюкоза
пируват
| -метаболический блок при гомоцистинурии
|| -метаболический блок при метилкобалами-
новой недостаточночти
HI -метаболический блок при цистатионинурии
Рис. 15. Метаболизм серосодержащих аминокислот и его нарушения (По Я. Мусилу и соавт., 1981)
90 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
лее тяжёлую клиническую картину,
резистентную к терапии витамином В6. В отличие
от классической формы у них не отмечается
и сопутствующей гиперметионинемии.
Напротив, содержание метионина в сыворотке
понижается. Характерной особенностью
больных, у которых гомоцистинурию
вызывает нарушение обмена метилкобаламина,
является метилмалонилацидурия, как и при
гиповитаминозах по фолиевой кислоте (см.
ниже раздел «Патофизиология витаминного
метаболизма»).
Данная наследственная аминоацидопатия
характеризуется развитием умственной
отсталости, эктопии хрусталиков,
остеопатии, остеопороза, особенно выраженного в
позвоночнике и длинных трубчатых костях.
Обычно наблюдается сколиоз, часто —
патологические переломы. Может развиваться
марфаноидность, так как нарушается
регуляция освобождения цитокина ТФР(3.
Очень своеобразной и, часто, решающей
для судьбы больных особенностью
клинической картины является тромбоэмболичес-
кий синдром. Отмечаются тромбозы
крупных и мелких артерий и вен. До 30-летнего
возраста не доживают около 5 % больных
с В6-зависимой формой и 22 % пациентов
с В6-резистентной гомоцистинурией. При
этом, типичные причины смерти —
тромбоэмболия лёгочной артерии и ишемический
инсульт.
Недавние исследования кровеносных
сосудов больных с гомоцистинурией выявили
у них резкое ускорение развития
атеросклероза. Гомоцистинурия внесена в список
«мягких» факторов риска атеросклероза.
Считается, что при гомоцистинурии
действуют несколько факторов,
ускоряющих атерогенез, в том числе, повышенная
продукция тромбоцитарных факторов
роста гладкомышечных клеток сосудов и
нарушение взаимодействия атерогенных
липопротеидов с апо-В-чувствительными
рецепторами клеток сосудистой стенки (см.
ниже «Атеросклероз»). Существует гипотеза
о значительной роли латентных нарушений
обмена гомоцистеина в патогенезе
атеросклероза. Возможо, они распространены
намного шире, чем клинически выявляемая
гомоцистинурия. Дефицит метилированной
формы витамина В12, нарушающий обмен
гомоцистеина и гомоцистина, способствует
и развитию атеросклероза.
Нарушение обмена серосодержащих
аминокислот характерно и для
цистатионинурии, когда поражается фермент гомосеринде-
гидратаза (она же — цистатионин-у-лиаза),
расщепляющая цистатионин до цистина,
а-кетобутирата и аммиака. При этом ткани,
кровь и моча насыщаются цистатиони-
ном. Заболевание протекает с развитием
некоторого умственного отставания, но не
ограничивает продолжительность жизни
больных так существенно, как
гомоцистинурия. Кроме того, витамин В6 в значительных
дозах оказывает при цистатионинурии
положительный эффект. Интересно, что
приобретённая форма цистатионинурии часто
сопутствует нейробластомам (С.Я. Каплан-
ский, 1966).
Многообразные наследственные
заболевания касаются обмена тирозина. Возможно,
самое известное из них — альбинизм,
описанный еще античными авторами. Причиной
этой болезни служат нарушения
образования пигмента меланина из тирозина
(см. рис. 13). Наиболее распространённая
аутосомно-рецессивная форма (средняя
частота заболевания 1/20 000, чаще всего
встречается в Центральной Америке — 1/150,
средня частота носительства 1/70)
основывается на дефекте медьсодержащего
фермента меланобластов тирозиназы, которая
должна превращать тирозин в 3,4-диоксифе-
нилаланин. Носитель полного альбинизма
(альбинос или лейкопат) имеет белую кожу
и волосы, розово-красные глаза. Вследствие
депигментации сетчатки альбиносы,
родопсин которых распадается ускоренно, плохо
видят днём (дневная слепота) и им присуща
фотофобия. Даже малые дозы солнечного
света могут вызывать у них фотодерматит.
Альбинизм поражает представителей всех
рас. Генокопиями альбинизма являются его
аутосомно-доминантная и сцепленая с
полом формы. Аутосомно-доминантная форма
Патофизиология белкового обмена 91
проявляется чаще всего в виде частичного
альбинизма.
Некоторые наследственные нарушения
тирозинового обмена вызывают гибель ге-
патоцитов, приводящую к врождённому и
раннему неонатальному циррозу.
Тирозиноз Медеса — заболевание, при
котором нарушена активность пара-окси-
фенилпируватдезоксигеназы. По другим
данным, вовлекается дефицит митохонд-
риальной печёночной тирозинаминотран-
сферазы. В отличие от алкаптонурии,
при данной патологии происходит прямо
противоположное — гомогентизиновая
кислота в печени вообще не образуется. Это
оборачивается значительно более тяжёлым
метаболическим расстройством, нежели её
избыток. Развиваются печёночная
недостаточность и нефропатия. В оригинальном
описании Дж. Медеса и соавторов (1932)
больной страдал также тяжёлой миастенией,
но нет достаточных оснований считать эту
аутоиммунную патологию закономерным
следствием тирозиноза.
Гипергпирозинемия I типа — заболевание,
вызванное дефектом фумарилацетоацетат-
гидролазы. В крови накапливаются тирозин
и метионин. Больные гибнут во
младенчестве от печёночной недостаточности и не-
фропатии. Аутопсия обнаруживает цирроз
печени и тубулонекроз. Гипергпирозинемия
II типа возникает вследствие дефицита
цитоплазматической тирозинаминотран-
сферазы печени. Она протекает не столь
фатально, но обусловливает выраженную
задержку психомоторного развития.
Хоукинсурия — нарушение активности
4-гидроксифенилпируватдезоксигеназы.
Она также сопровождается тирозинемией,
но, в отличие от всех вышеназванных амино-
ацидопатий, имеет аутосомно-доминантный
тип наследования. Болезнь
характеризуется задержкой психомоторного развития
(ЗПМР), степень которой варьирует в
зависимости от глубины нарушения обмена
глутатиона и формирования эпоксидных
метаболитов ароматических аминокислот.
Преходящая тирозинемия с тирозину-
рией развивается у недоношеных из-за
незрелости вышеназванного печеночного
медьсодержащего фермента. Так как
аскорбиновая кислота повышает активность
4-гидроксифенилпируватдезоксигеназы,
терапия витамином С способствует
быстрейшей коррекции обмена.
Из-за того, что тирозин — сырьё для
продукции тироидных гормонов, при
некоторых тирозинопатиях формируется
наследственный первичный гипотироз.
В этом отношении заслуживает упоминания
дефект йодтирозиндейодиназы. При
данном аутосомно-рецессивном заболевании
моно- и дийодтирозин не дейодируются и
йод секвестрируется в этих гормонально
неактивных метаболитах тирозина.
Развивается нехватка тироидных гормонов
(рис. 13) и компенсаторная гиперплазия
щитовидной железы до статуса зоба. В силу
врождённого характера гипотироза
нарушается развитие мозга и обучаемость, так
как тироидные гормоны — стимуляторы
аминоацил-т-РНК-синтетаз — существенны
для синтеза быстрообмениваемых пептидов
ЦНС. Отмечается кретинизм. Имеется
модель болезни на кроликах с аналогичным
дефектом. В изолятах, за счет эффекта
родоначальника и инбридинга, частота данного
дефекта возрастает, что не должно
неправильно интерпретироваться, как указание
на эндемичную геохимическую йодную
аномалию (см. ниже «Патофизиология
эндокринной регуляции»).
Большое клиническое значение имеет
наследственная оксалурия — причина раннего
оксалат-кальциевого нефроуролитиаза и
быстропрогрессирующей хронической
почечной недостаточности. При I типе этого
заболевания присутствует дефект перокси-
сомального фермента аланин-глиоксилат-
аминотрансферазы, при II типе — дефекты
редуктаз глиоксиловой кислоты. В обоих
вариантах аутосомно-рецессивные
дефекты приводят к накоплению глиоксиловой
кислоты и ее переходу в щавелевую
кислоту, кальциевые соли которой формируют
кристаллы в мочеполовой системе, а при I
типе — и в сосудах, сердце, костях. Витамин
В6 способствует облегчению оксалурии.
92 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
В таблице 4 можно найти краткую
характеристику еще нескольких наследственных
аномалий межуточного обмена
аминокислот.
Исключая эндокринопатии,
рассмотренные ниже, приобретённые нарушения обмена
аминокислот могут быть вызваны, прежде
всего, различными факторами,
расстраивающими процессы переаминирования.
При авитаминозе В6 (см. также ниже
«Патофизиология витаминного обмена»)
переаминирование и дезаминирование
аминокислот в печени полностью
нарушается. Такая ситуация возможна не только
при недостатке витамина В6 в диете, что
бывает довольно редко. Большое значение
имеют нарушения, связанные с действием
антагонистов трансаминаз, к которым
относятся фтивазид и его аналоги и циклосе-
рин. При терапии туберкулёза нарушение
трансаминирования, вследствие побочного
действия лекарств, усугубляет катаболи-
ческий эффект цитокинов, образуемых
при хроническом инфекционном процессе.
Недостаток синтеза апоферментов-тран-
саминаз сопровождает голодание и
печеночную недостаточность, как при развитии
цирроза, так и стеатоза печени и даже при
выраженном гепатите. При хроническом
алкоголизме потребность в витамине В6
растёт, а возможности трансаминирования
в печени сокращаются. Вследствие этого
у алкоголиков наблюдается зачастую ка-
таболический сдвиг белкового обмена и
отрицательный азотистый баланс. Как уже
упоминалось выше в разделе «Общий обзор
путей метаболизма аминокислот», дефицит
а-кетокислот также тормозит
переаминирование. Поэтому, нарушение данного
процесса (и аминирования-дезаминирования)
наблюдается при глубокой тканевой
гипоксии, гиповитаминозе^ и В2 и любом
торможении окислительно-восстановительных
ферментов цикла Кребса или ограничении
его пропускной способности.
Все вышеупомянутые состояния
характеризуются гипераминоацидемией,
аминоацидурией и увеличенной потерей
немочевинного азота с мочой.
Особое значение для диагностики имеют
ситуации, когда ферменты-аминотрансфе-
разы оказываются в увеличенном
количестве в плазме крови. Это свидетельствует об
усиленных процессах цитолиза в том или
ином органе (см. также т. I, стр. 153).
Отдельные трансаминазы содержатся в
различных органах в неодинаковых количествах.
Больше всего аспартатаминотрансферазы
(АСАТ) содержат кардиомиоциты, затем
по убыванию концентраций идут печень,
скелетные мышцы, почки и поджелудочная
железа. Аланинаминотрансфераза (АЛАТ)
присутствует в рекордном количестве в
печени, затем идут поджелудочная железа,
миокард и скелетная мускулатура.
Соответственно, повышение уровня АСАТ в
крови считается более характерным для
инфаркта миокарда, а нарастание активности
АЛАТ может свидетельствовать о цитолизе
гепатоцитов (А.Ф. Блюгер, 1976). Впрочем,
данные тесты неспецифичны.
Приобретенные нарушения могут
затрагивать и другие аспекты интермедиарного
метаболизма аминокислот.
Так, при алиментарной дистрофии,
подобно фенилкетонуриям, нарушается
окисление фенилаланина и могут возникать те
же токсические для ЦНС продукты, что и
при фенилпировиноградной олигофрении,
например, фенилпируват. Это заставляет
предполагать, что не только «Сытое брюхо к
ученью глухо», но и продолжительные
голодовки не благоприятствуют максимальному
проявлению умственных способностей.
Биосинтез многих заменимых
аминокислот имеет этапы, зависящие от присутствия
дериватов витамина никотиновой кислоты,
и страдает при пеллагре. Катаболизм
триптофана особенно чувствителен к витаминной
недостаточности и может тормозиться на
разных этапах ещё и при гиповитаминозах
В„ В2, В6, а также при гиперкортицизме.
Нарушение окисления тирозина
наблюдается при гипертирозе, цинге и дефиците
меди.
Биосинтез серина нарушается при гипо-
кортицизме и кастрации, а при превращении
серина в глицин центральную роль играет
Патофизиология белкового обмена 93
Таблица 4
Некоторые наследственные аминоацидопатии
Название
Гиперпролинемия
1типа
Гиперпролинемия
II типа
Гидроксипроли-
немия
Гистидинемия
Гипертреонинемия
Глицинурия
Гипераланинемия
Гиперлизинемия
Триптофанемия
Первичный
дефект
Пролинокси-
даза
Д'-пирролин-5-
де карбоксил ат-
дегидрогеназа
Гидроксипро-
лин-оксидаза
Гистидаза
Треонин-альдо-
лаза (?)
Глициназа
(некетогенная
форма) или аце-
тил-КоА-ацил-
трансфераза,
пропионил-
КоА-карбокси-
лаза
(кетогенная форма)
Липоатзависи-
мая пируватде-
гидрогеназа
Аминоадипин-
семиальдегид-
синтетаза (тип
1) или сахаро-
пин-редуктаза
(тип II).
Формамидаза
Тип
наследования
АР
АР
АР
АР 1/14000
АР
АР
АР
АР
АР
Патогенез
Не образуется из
пролина А'- пирролин-
5- декарбоксилат
Не образуется из А'-
пирролин-5-декарбок-
силата глутамин
Уровень гидроксипро-
лина в сыворотке, но не
в моче, увеличен
Не образуется урокаи-
новая кислота.
Накапливается имидазолпи-
руват
Нарушение
расщепления треонина
Нарушение
расщепления глицина,
глицинурия (некетогенная
форма). Нарушение
утилизации кетоновых
тел и глюкогенные
аминокислоты в моче
(кетогенная форма)
Подавлен аэробный
метаболизм пирувата
Лизинемия (I), лизин-
метинин-гомоцистеин-
емия (II)
Накопление
триптофана и индолкетонов
Проявления
Пролинемия. Иминоа-
цидурия
конкурентного типа. Нефропатия
Пролинемия. Иминоа-
цидурия
конкурентного типа.А'-пирролин-
5-декарбоксилатурия.
Нефропатия, судороги
Бессимптомна
Гистидинурия, ими-
дазолпируватурия,
ложноположительный
тест Феллинга.
Доброкачественное течение.
Редко — дефекты речи
Судороги
Глицин в
спинномозговой жидкости,
судороги и угнетение
ЦНС. При кетогенной
форме — кроме того,
кетоацидоз, цитопения,
ЗПМР
Лактатацидоз,
перемежающаяся атаксия,
слабоумие
Гипотонус мышц, ЗПМР
Благоприятное течение
94 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
витамин фолиевая кислота — и этот процесс
страдает при соответствующем
гиповитаминозе.
Большое значение в медицине имеют
приобретенные нарушения
переметилирования, при котором метильные группы
серина и метионина вовлекаются в
метаболизм, переносясь на другие акцепторы.
Метилтрансферазы, необходимые для этого,
могут использовать метилированную форму
тетрагидрофолиевой кислоты и витамин В12,
как метильные донаторы. Таким образом,
эти витамины необходимы для обмена
серосодержащих аминокислот — метионина и
цист(е)ина. Вместе с холином, бетаином, ко-
баламином метионин служит важным
источником метальных групп, необходимых при
синтезе ЛПОНП и ЛПВП печенью, и сам
входит в состав их апопротеинов.
Метильные группы из состава метионина
необходимы для формирования фосфатидилхолина,
используемого при построении оболочек
липопротеидов в печени. Без достаточного
количества фосфатидилхолина экскреция
липопротеидов печенью тормозится.
Кроме того, метильные донаторы участвуют в
построении триметилированного
производного аминокислоты лизина — карнитина.
Карнитин — челнок, транспортирующий в
митохондрию для окисления остатки длин-
ноцепочечных жирных кислот (см. рис. 44,
ниже). Вполне понятно, что при его нехватке
утилизация липидов печенью замедляется.
Таким образом, при дефиците метионина и
других вышеназванных метилдающих
соединений, получивших название липотроп-
ных веществ, происходит ожирение печени
(см. ниже «Нарушения промежуточного
обмена липидов»). Так как при этом нарушена
продукция антиатерогенных ЛПВП, можно
полагать, что этим объясняется взаимосвязь
между дефицитом метальных донаторов, го-
моцистинурией и ускорением атеросклероза
(см. выше).
Образование порфириновых соединений
из аминокислот и их производных (глицина
и сукцинил-КоА), необходимое для синтеза
гема, контролируется ферментом 5-амино-
левулинат-синтетазой, коферментом
которого служит витамин В6, а активный центр
содержит ион Mg2+. При гиповитаминозе
В6, в частности, вызванном алкоголизмом,
а также при отравлении свинцом,
конкурирующим с магнием активность данного
фермента нарушается, что ведет к
ослаблению синтеза порфиринов и сидеробласти-
ческим анемиям. Указанный фермент столь
чувствителен к РЬ2+, что его определение
рекомендовано для раннего скрининга
хронической свинцовой интоксикации
(сатурнизма) (см. т. III).
Процессы декарбоксилирования,
приводящие к образованию биогенных аминов
(гистамина, тирамина, дофамина, трипта-
мина и др.), особенно активно идут в
печени, мозге и хромаффинной ткани. Захват и
декарбоксилирование аминных
предшественников являются, по А. Дж.Э. Пирсу,
характерной чертой клеток диффузной
эндокринной системы (см. ниже
«Патофизиология эндокринной регуляции»). Всего
в организме человека образуется более 40
различных аминов.
Процессы синтеза аминов могут системно
усиливаться в организме при гипоксии и
голодании. Местное увеличение синтеза,
освобождения и инактивации катехолами-
нов, гистамина и серотонина свойственно
очагам воспаления, где они играют важную
роль в микроциркуляторных изменениях
и развитии отёка, а серотонин имеет также
отношение к гемостазу и стимуляции фиб-
роплазии. (см. т. I, стр. 347-349). При этом
организм не допускает системного действия
этих аминов, которое опасно. Системное
действие гистамина — важный компонент
патогенеза аллергического шока (т. I, стр.
114,285,432).
Злокачественные опухоли апудоцитар-
ного происхождения, находящиеся в
кишечнике, бронхах, поджелудочной железе и
других органах, могут синтезировать
большие количества серотонина (используя для
этой цели до 60 % суточного потребления
триптофана!).
Как правило, печень нейтрализует
серотонин с образованием неактивной
Патофизиология белкового обмена 95
5-гидроксииндолилуксусной кислоты,
экскретируемой в огромных количествах
с мочой. Но примерно в 1-2 % случаев
опухоль из энтерохромаффинных клеток,
локализованная, чаще всего, в аппендиксе,
метастазирует в печень. В силу этого, или
при наслоении печёночной
недостаточности, вызванной другими причинами,
серотонин перестает инактивироваться и
развивается карциноидный синдром (см.
также «Патофизиология эндокринной
регуляции» ниже).
Под влиянием системного действия
избытка данного производного триптофана
развиваются вазомоторные нарушения
(приливы крови к коже лица и эпизодический
цианоз, колебания артериального давления,
головные боли, астмоподобный бронхит,
усиленная перистальтика и
антиперистальтика с соответствующими
желудочно-кишечными симптомами и, самое характерное,
системный фиброз и отложение фибрина.
Нарушается фибринолиз и возникает
вторичная организация фибриновых депозитов
с образованием фибропластических очагов.
Последнее проявление включает фибриноз
клапанов сердца и фиброз эндокарда, ретро-
перитонеальный и тазовый фиброз жировой
клетчатки, фиброзные плевральные шварты
и отложение фибрина в аорте.
Серотонин, в присутствии церулоплазми-
на, способствует образованию хиноновых
производных фибрина, которые не
подвергаются фибринолизу. Кроме того, он
стимулирует коллагеногенез.
Под влиянием N-ацетилазы
шишковидной железы и оксииндол-ортометилтранс-
феразы, путём ацетилирования и
метилирования серотонина, возникает гормон
эпифиза мелатонин. Таким образом, для
адекватной гормональной функции
эпифиза, контролирующего суточные ритмы
и функцию гонад, существенным является
обмен и метионина, и триптофана (см. т. I,
стр. 79-80, а также ниже раздел
«Патофизиология шишковидной железы»). В этой
связи нелишне упомянуть, что при печеночной
недостаточности, когда обмен аминокислот
и серотонина нарушается, одним из
симптомов является извращение нормального
цикла сна и бодрствования.
Феохромоцитомы, симпатобластомы
и ганглионевромы могут синтезировать и
выделять в системный кровоток аномально
большие количества катехоламинов. В ткани
феохромоцитомы нарушен механизм
упаковки катехоламинов в аминосодержащих
гранулах. Это проявляется злокачественной
гипертензией, симптоматической
гипергликемией и может приводить к инсультам (см.
ниже раздел «Патофизиология мозгового
вещества надпочечников»).
Так как фосфопиридоксаль служит коэн-
зимом декарбоксилаз, продукция аминов,
нарушается при гиповитаминозе В6, что
ведёт к тяжелым неврологическим
расстройствам (см. раздел «Патофизиология
витаминного обмена»). Биосинтез ряда
аминов нарушается и при других гиповита-
минозах, так при нехватке витамина С
тормозится реакция превращения дофамина
в норадреналин.
При первичной идиопатической
болезни Паркинсона заторможено
образование дофамина из диоксифенилаланина
в мозге, так как его главные
поставщики — дофамин-секретирующие нейроны в
substantia nigra — подвергаются
дегенерации. Это вызывает глубокие нарушения в
деятельности стрио-паллидарной системы.
Систематическое применение 3,4-диокси-
фенилаланина (L-ДОФА), блокатора
печёночного декарбоксилирования
ароматических аминокислот, не затрагивает активность
декарбоксилаз мозга и позволяет (за счет
«экономии» субстратов и собственного
проникновения лекарства в ЦНС) поднять
концентрацию предшественника до уровня,
необходимого для увеличения продукции
дофамина мозгом.
Биогенные амины разрушаются
окислительными ферментами моноаминоксидаза-
ми (МАО). В условиях блокады МАО, при
терапии антидепрессантами способность
разрушать амины резко снижается.
Организм может стать избыточно
чувствительным к экзогенным аминам — например,
приём в пищу сыра и употребление некоторых
96 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
сортов красного вина, богатых тирамином,
на фоне терапии ингибиторами МАО ведет к
гипертензии. Снижение способности
разрушать амины наступает при хронической
почечной и печеночной недостаточности, что
ведет к вмешательству этих биологически
активных соединений в регуляторные
процессы, подобно их действию при кишечной
аутоинтоксикации (см. выше раздел
«Нарушения переваривания...»). Активность МАО
и других ферментов, инактивирующих
биогенные амины (например, катехоламин-
ортометилтрансферазы), понижается при
избытке тироидных гормонов, что вносит
свой вклад в развитие ряда симптомов
гипертироза. Так как один из продуктов
обмена тиамина является естественным
ингибитором МАО, при авитаминозе Bj
развиваются активация этого фермента и
нехватка биогенных аминов,
проявляющаяся депрессивными состояниями (см. ниже
«Патофизиология витаминного обмена»).
Окислительное дезаминирование гиста-
мина катализирует пиридоксаль-зависимый
фермент гистаминаза. Поскольку
физиологическое формирование плаценты при
беременности осуществляется с участием
анафилактических механизмов (см. т. I,
стр. 465-466), для компенсации усиленного
освобождения гистамина при беременности
развивается уникальный феномен: в 1000
раз возрастает гистаминазная активность
(В.З. Горкин, 1966). Уже на третий день
после родов она возвращается к норме. При
нехватке витамина В6, что не редкость в
организме беременной женщины, с её
повышенными витаминными потребностями,
формируется ослабление гистаминазной
активности, коррелирующее с клинической
картиной раннего токсикоза беременных.
Помимо моноаминов большое значение
в организме имеет адекватный обмен таких
аминокислотных дериватов, как
полиамины— путресцина, кадаверина, спермина,
спермидина. Эти соединения известны как
противовоспалительные медиаторы (см.
т.1, стр. 330, 332). Важнейший полиамин —
путресцин — возникает при декарбоксили-
ровании орнитина, а кадаверин — лизина.
Спермин представляет собой метионил-
путресцин, а спермидин — диметионилпут-
ресцин. Ключевым ферментом в продукции
полиаминов служит орнитиндекарбоксила-
за. Это самый активный в каталитическом
отношении фермент млекопитающих.
Установлено, что все основные ростостимулиру-
ющие воздействия, как гормональные, так и
опосредованные аутакоидами, замыкаются
на активацию образования полиаминов
в конкретных клетках-мишенях
посредством стимуляции орнитиндекарбоксилазы.
Продукция полиаминов возрастает в
опухолевых клетках и при регенерации ткани
(подробнее — см. ниже «Гормон роста и
патофизиология ростовых и анаболических
процессов»).
Итак, рассмотрение нарушений интерме-
диарного обмена аминокислот показывает,
что при этих формах патологии могут
вторично нарушаться липидный, углеводный,
водно-солевой, витаминный, нуклеиновый
метаболизмы. Это иллюстрирует
центральную роль адекватного метаболизма белка в
поддержании гомеостаза.
НАРУШЕНИЯ КОМПОЗИЦИИ
БЕЛКОВ ПЛАЗМЫ
Около 7 % массы плазмы крови составляют
различные белки. Специфика их обмена
заключается в том, что они возникают в
одних клетках и органах, а утилизируются,
как правило, в других. В связи с этим, плазма
крови — динамическая равновесная
система, и её протеинограмма, подобно зеркалу,
отражает состояние разных тканей.
Возможность исследования белковой композиции
плазмы в решающей степени была
достигнута благодаря разработке А. Тиселиусом
метода электрофореза на бумаге, с
подвижной границей, разделяющего плазменные
протеины. В 1948 г. это открытие, в связи с
его гигантским прикладным медицинским
значением, было удостоено Нобелевской
премии по химии.
Патофизиология белкового обмена 97
Данный метод (рис. 16) выявляет в
плазме крови здорового человека 5 основных
фракций (по убыванию электрофорети-
ческой подвижности) — альбуминовую
(54-58%, 35-45 г/л), а также 4 фракции
глобулинов — 0ц (6-7 %, 3-6 г/л), а2 (8-9 %,
4-9г/л),pt(13-14%, 6-11 г/л)иу(11-12%,
7-15 г/л).
Каждая из фракций представлена не
одним, а несколькими или даже многими,
совершенно разными по своей функции
конкретными белками, известными также под
своими особыми названиями. Они
объединяются в группы по признаку совместной
миграции в элекрическом поле.
Поэтому формулировки типа «антитела
и у-глобулины» тавтологичны, а вопросы
типа «Как влияет болезнь X на содержание
а-глобулинов?» — изначально не вполне
корректно поставлены и требуют уточнения,
какие конкретно белки имеются в виду.
Ниже следует таблица, напоминающая
наименования и основные характеристики
главных белков плазмы (в порядке
убывания электрофоретической подвижности), в
качестве введения к обсуждению нарушений
белковой композиции плазмы или диспро-
теинемий.
В таблице опущены белки с неясной
биологической функцией, минорные фракции
и фрагменты более крупных протеинов.
Факторы комплемента при электрофорезе
распределяются от а2-фракции (СIs, C9)
до у-глобулиновой фракции (Clq, C8).
Большинство их принадлежит к фракциям
р! (СЗ-С5) и р2 (С2, С6, С7). В таблицу
включен один пример. Информация о
свойствах и биологической роли этих
белков содержится в т. I данного руководства
(стр. 350-357).
Факторы свёртывания и противосвёрты-
вающей системы мигрируют, в большинстве,
в р^Рз-глобулиновой фракции. В таблице
дано по одному примеру. Подробная
характеристика этих плазменных белков имеется
в т.1, стр. 264-271.
Плазменные белки возникают, в
основном, в гепатоцитах и макрофагах, а у-глобу-
Y
t
1 Ц
: 1
Р
4
S
1
I,
аз а.
IIII
8
7
J
8 9
Рис 16. Нормальная протеинограмма плазмы
крови, полученная электрофоретическими
методами (по Ф.У. Патнему, 1975). а —
электрофорез с подвижней границей; б— на бумаге;
в — в крахмальном геле, г — иммуноэлект-
рофорез. Обозначения в тексте и на рисунке.
Стрелки помечают зону миграции антител
1 — (Злипопротеиды
2—трансферрин,
2' — трансферрин С
3 — а2-липопротеиды
4—а2-макроглобулин
5 — гаптоглобин
5'—гаптоглобин 1-2
6—церулоплазмин
7 — аггликопротеид
8 — альбумин
9—транстиретин
10 — IgG
11—(Зглипопротеиды
12 — аглипопротеиды
13 —IgA
14—IgM
лины — продукты плазматических клеток.
Синтез липопротеидов некоторых классов
связан с деятельностью клеток кишечника
(см. «Патофизиология липидного обмена»).
Отдельные протеины плазмы, например,
фактор фон Виллебранда, выделяются в
кровь и эндотелием. Использование этих
белков происходит в различных органах,
в зависимости от их метаболической роли:
это могут быть печень, почки, макрофаги и
другие клетки сосудистой стенки, энтеро-
циты. Главный плазменный белок альбумин
синтезируется гепатоцитами со скоростью
0,1-0,2 г/кг массы тела в сутки. Он
постоянно переходит в тканевую жидкость и воз-
98 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 5
Главные белки плазмы крови (курсивом набраны а2-глобулины, жирным шрифтом — fi-глобулины)
Белок
Транстиретин
Альбумин
сц-глико-проте-
ид кислый
сц-антитрипсин
аглипопроте-
иды
а,-фетоглобу-
лин
Ретинолсвязы-
вающий белок
Тироксинсвязы-
вающий белок
Транскортин
Церулоплазмин
Синонимы
Преальбумин
Орозомукоид
ЛПВП +
ЛПОВП
Альфа-фето-
проте ин
Трансретин
TBG, тирок-
син-свя-
зывающий
глобулин
Количество
в норме мг/дл
10-40
3500-4500
55-140
200-400
340-530
0,001
3-6
1-2
3-3,5
15-60
М*В«/
кД
55
66,2
40
54
150-
380
64
21
58
52
151
Функции, значение при патологии
Связывание и перенос Т4 и ретинолсвязы-
вающего белка. Понижается при белковом
голодании. Мутантная форма входит в состав
амилоида при семейных амилоидных поли-
нейропатиях и старческом амилоидозе
Основной вклад в онкотическое давление,
транспортёр билирубина, анионных лекарств,
жирных кислот, альдостерона, триптофана,
тема, кальция. Источник аминокислот,
утилизируется энтероцитами, почками. Отсутствует
при наследственной анальбуминемии.
Фракция раздвоена при бисальбуминемии I
Ингибитор протеаз и антиоксидант, уменьшен
при воспалении, повышается при
дезорганизации основного в-ва соединительной ткани,
стрессе, опухолях
Ингибитор протеаз, противовоспалительный
регулятор, понижен при генуинной эмфиземе
и первичном билиарном циррозе
Транспортёры холестерина, его эфиров и фос-
фолипидов. Дренажная функция в отношении
холестерина тканей. Понижаются при курении,
гиподинамии, холестазе, отсутствуют при
болезни о-ва Тэнжир, повышены у долгожителей,
при умеренном употреблении этанола
Эмбриональный белок, повышен при гепа-
томах, тератомах, беременности, гепатите
и циррозе. Стимулятор тромбоцитарного
фагоцитоза
Транспорт ретинола, комплексируется с
преальбумином, понижен при белковом
голодании
Связывание и перенос Т4
Транспортёр кортизола и кортикостерона
Транспортёр меди и цинка, регулятор обмена
меди в печени, антиоксидант, полиаминокси-
даза. Понижен при болезнях
Коновалова-Вильсона и Менкеса, положительный глобулин
острой фазы. Ферроксидаза, блокатор NO-
вазодилатации, окислитель ЛПНП,
регулятор обмена моноаминов в мозге, возможная
мишень ЛСД. Индуцируется у-ИНФ
Патофизиология белкового обмена 99
Продолжение таблицы 5
Белок
Гаптоглобины
1-1,2-12-2
а2-макроглобу-
лин
Интер-а-
трипсиновый
ингибитор
Трансферрин
Гемопексин
02-микрогло-
булин
С-реактивный
белок
Р,Е-глобулин
Фибриноген
Плазминоген
Р-липопроте-
иды
у-глобулин
Синонимы
Серомукоид
Сидерофилин
Цитохромо-
филин
С4 компонент
комплемента
Фактор 1
Профибрино-
лизин
ЛПОНПЛПНП
Иммуноглобулины
Количество
в норме мг/дл
100-220
160 300
120-260
150-420
20-70
200-320
50-100
0,2
<1
20-50
200-400
130-200
210-400
700-1500 см. т. I,
стр. 402
М.в.,
кД
100
200
400
725
160
76,5
57
11,8
118
206
340
143
5000-
13000,
2700-
4800
Разный по
классам.
Функции, значение при патологии
Связывают гемоглобин, способствуют его
реутилизации, препятствуют потере с
мочой, антиоксиданты, повышены при
ответе острой фазы, снижены при усиленном
внутрисосудистом гемолизе, наследственной
гипо(а)гаптоглобинемии
Антиоксидант, ингибитор эндопептидаз и
фибринолиза, повышен при ответе острой
фазы и у детей, при нефротическом синдроме.
Связывает инсулин
Антипротеаза, понижает фибринолиз
Транспортёр железа в макрофаги,
легкодоступное депо железа, прооксидант,
отрицательный глобулин острой фазы.
Понижен при атрансферринемии,
печёночной недостаточности. Повышен при
железодефиците и беременности
Связывет гем, способствует его
реутилизации. Антиоксидант. Положительный
глобулин острой фазы, понижен при усиленном
гемолизе
Компонент белков ГКГС 1 класса,
антиоксидант
Положительный глобулин острой фазы,
антиоксидант, прямая бактерицидная
активность в отношении S. pneumoniae.
Опсонин, хемоаттрактант
См. т. 1 стр. 341 -348. Медиатор воспаления,
эффектор иммунного ответа
1 фактор свёртывания (см. т. 1, с. 258-265).
Понижен при афибриногенемии, коагуло-
патии потребления (ДВС-синдром),
печеночной недостаточности
Предшественник плазмина. (т. 1, с. 264-
265)
Транспорт триглицеридов и холестерина.
Повышены при гиперлипопротеинемиях
II, III, IV, V типов. Снижены при
наследственной абеталипопротеинемии, печеночной
недостаточности
Продукты плазматических клеток.
Антитела. Повышены при инфекциях,
иммунопатологических заболеваниях, снижены при
В-клеточных и смешанных иммунодефици-
тах (см. т. 1, стр. 398,402)
100 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
вращается в кровь через лимфу. За 20 дней
весь альбумин крови успевает пройти этот
кругооборот. Утилизация альбумина идёт,
главным образом, в энтероцитах и, в
меньшей степени, в клетках почек, самой печени,
кожи и даже лёгких. Альбумин —
единственный из главных плазменных белков, не
является гликопротеидом и, может быть, именно
поэтому циркулирует в крови довольно
долго. Время жизни гликопротеидных белков
плазмы определяется отщеплением от них
остатков сиаловой кислоты ферментами
сиалидазами. После десиалирования белок
получает возможность связаться своими
обнажившимися галактозными остатками
с клеточными рецепторами — животными
лектинами, обеспечивающими его эндоци-
тоз. Определение уровня сиаловых кислот
плазмы, широко практиковавшееся в
клинической биохимии, по-видимому, зависит,
прежде всего, от скорости обновления
неальбуминовых плазменных белков.
Нормальное количество и фракционное
распределение белков* плазмы
обозначается, как эупротеинемия. Нарушения про-
теинограммы плазмы крови известны как
диспротеинемии.
К диспротеинемиям относятся
увеличение концентраций белков плазмы (гиперпро-
теинемии), уменьшение этих концентраций
(гипопротеинемии) и появление в плазме
необычных белков, в норме не
присутствующих там в сколько-нибудь значимых
количествах (парапротеинемии). Если
изменения относятся только к глобулиновым
фракциям, говорят о дисглобулинемиях.
Гиперпротеинемия может быть ложной
и отмечается при сгущении крови (гипер-
осмолярной дегидратации — см. ниже раздел
«Патофизиология водно-электролитного
обмена»). Истинная гиперпротеинемия
наблюдается только при парапротеинемиях
(см. ниже) и, практически всегда, касается
глобулиновых фракций, то есть является ги-
перглобулинемией. При усиленном синтезе
иммуноглобулинов и их отдельных цепей
патологическими клонами плазматических
клеток количество белка в плазме крови
может достигать 150-160 г/л, с резким
преобладанием глобулинов (миеломная болезнь,
макроглобулинемия Вальденстрёма).
Менее значительно количество
иммуноглобулинов может возрастать при действии
поликлональных иммуностимуляторов
(см.т.1,стр.404,432).
Псевдогипопротеинемия сопровождает
выраженную гемодилюцию. Истинные
гипопротеинемии могут иметь различную
этиологию.
Прежде всего, их делят на первичные
(наследственные) и вторичные
(приобретенные).
Первичная гипоальбуминемия
характерна для недоношеных детей, с их незрелой
печенью.
Наследственная аналъбуминемия - ауто-
сомно-рецессивное, крайне редкое
заболевание, проявляющееся гипопротеинемией за
счёт почти полного отсутствия альбумина
(менее 3 % белка плазмы). Компенсаторно
увеличено абсолютное содержание
глобулинов. Имеются вторичный гипераль-
достеронизм, нерезко выраженные отеки,
гипокальциемия, гиперхолестеринемия,
снижена концентрация Т4, ускорена СОЭ.
Характерны низкий рост и судорожные
припадки.
Врождённая агаммаглобулинемия
наблюдается при сцепленном с Х-хромосомой
рецессивном синдроме Брутона.
Содержание у-глобулинов падает ниже 1 г/л. Менее
значительная гипогаммаглобулинемия
характерна для простого вариабельного
иммунодефицита и других подобных расстройств
(см. т. I, стр. 506-514).
Первичную гипопротеинемию с
понижением содержания и альбумина, и
глобулинов, особенно их у-фракции, находят
при экссудативной энтеропатии у детей.
Пациенты страдают хронической
неинфекционной диареей и теряют через кишечник
большие количества лимфы и тканевой
жидкости с белком, причём обратное всасывание
аминокислот из кишечника значительно
нарушено. В некоторых случаях экссуда-
тивная энтеропатия является вторичной и
представляет собой паранеопластическое
явление, сопровождающее кишечные опухо-
ли, в частности, лимфомы, нарушающие
региональный лимфоотток. Ряд больных
имеет первичную целиакию или муковисцидоз.
Но у некоторых пациентов белок-теряющая
форма энтеропатии, безусловно, вызывается
неидентифицированным первичным
дефектом кишечной стенки, сопровождаемым
ускоренной деградацией плазменных белков
в энтероцитах.
Вторичные гипопротеинемии, в отличие
от первичных, встречаются гораздо чаще.
Как правило, гипоальбуминемия при них
не сопровождается гипоглобулинемией.
Напротив — в силу системного действия цито-
кинов, которое наблюдается при многих из
порождающих вторичные гипопротеинемии
заболеваний, формируются преиммунный
и иммунный ответ и происходит усиление
синтеза положительных глобулинов острой
фазы (см. т. I). Следовательно, вторичная
гипопротеинемия — это, чаще всего
гипоальбуминемия на фоне гиперглобулинемии.
Наиболее важные этиологические
факторы вторичных гипопротеинемии
следующие:
> Пищевая белковая недостаточность,
в частности — квашиоркор и
алиментарный маразм. Как уже указывалось в
предыдущем разделе, при квашиоркоре
гипопротеинемия существенно более
выражена, чем при марантической
форме голодания. Интересно, что снижение
концентрации альбумина в плазме
при голодании вызвано не усилением
прямого катаболизма этого белка, как
полагали классические авторы, а резким
ограничением его ресинтеза в печени из
аминокислот, которые направляются
на энергетические цели (Н.Н. Лаптева,
1970).
> Нарушения поступления аминокислот
из кишечника при адекватной диете
(см. выше «Нарушения переваривания
белков», «Нарушения чрезмембранного
транспорта аминокислот»), в том числе:
хронические и рецидивирующие
энтериты, в частности, целиакия,
инфекционные формы спру и наследственные
дефекты переваривания и всасывания
Патофизиология белкового обмена 101
белков, а также панкреатическая
недостаточность. Среди заболеваний этой
группы И. Вапцаров и соавт. (1978),
указывают на большую распространённость
и роль такой причины вторичной
гипопротеинемии, как латентные нетяжёлые
формы муковисцидоза.
>- Печёночная недостаточность
сопровождается нарушением функций этого
органа в белковом обмене. Тормозятся
переаминирование и восстановительное
аминирование, нарушается балансовая
функция в отношении смеси пищевых
аминокислот. Возникают гиперамино-
ацидемия, аминоацидурия и дефицит
белков печёночного происхождения в
плазме крови. Гипоальбуминемия
характерна для печеночной недостаточности
(и еще больше усиливается, если
поражение печени сопровождается нарушением
всасывания аминокислот в кишечнике,
как это бывает при некоторых формах
инфекционного гепатита А или при
асците). Помимо этого чувствительными
показателями белок-синтезирующей
функции печени служат концентрации
ряда глобулиновых плазменных белков.
Развиваются гипотрансферринемия,
гипопротромбинемия, гипофибриноге-
немия. Снижается продукция факторов
свёртывания V, VII, IX—XIII (см. т. I,
с. 267-269). Весьма быстро реагируют
на колебания белок-синтезирующей
функции печени такие фракции
плазмы, как ретинолсвязывающий белок и
транстиретин. Если болезнь, вызвавшая
угнетение функций печени, имеет
инфекционную или иммунопатологическую
природу, это закономерно
сопровождается гипербетагаммаглобулинемией, так
как действуют механизмы преиммунного
и иммунного ответа. Отдельные формы
патологии печени могут проявлять
специфические особенности
композиции белков плазмы. Так, концентрация
главного компонента сц-глобулиновой
фракции — антитрипсина — резко
понижена при наследственном первичном
билиарном циррозе.
102 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
>- Усиленные внекишечные потери белка
тоже чреваты гипопротеинемией.
Данная группа факторов включает, прежде
всего, интенсивные патологические
потери белка через почки. Почечная
потеря белка в норме пренебрежимо мала
и составляет не более 60-80 мг/сутки
(А.С. Чиж, 1983). Почечный фильтр
характеризуется наличием слоя
крупномолекулярных белков на
поверхности фенестр капиллярного эндотелия.
Эти белки затрудняют фильтрацию
более мелких протеиновых молекул.
К тому же, поры базальной мембраны
клубочка имеют диаметр меньший, чем
гидратированный диаметр молекулы
альбумина. Фильтрации препятствует
и одноимённый отрицательный заряд
нормальных плазменных белков, с одной
стороны, и структур фильтра (стенок
пор, сиалогликопротеинов ножек по-
доцитов), с другой, обусловливающий
электростатическое отталкивание между
ними. Поэтому, у здорового человека
первичная моча содержит небольшое
количество мелкодисперсных белков и
пептидов, которые проходят через поры
фильтрующей мембраны, однако,
практически все эти белки захватываются
путём пиноцитоза, гидролизуются до
аминокислот и реабсорбируются.
Выделение повышенных количеств белка с
мочой именуется протеинурией.
Протеинурия бывает реналъная, пре-
ренальная и постренальная. Подробное
обсуждение её механизмов относится к т.
III данного руководства (см. раздел
«Патофизиология почек» и «Практикум»). Для
целей данной части книги следует отметить,
что чаще всего встречаются ренальные
причины: так, при гломерулонефритах и
пиелонефритах та или иная степень протеинурии
характерна для более чем 99 % больных.
Наиболее важной характеристикой
протеинурии служит степень её селективности.
Почечные механизмы протеинурии
неоднозначны. Этот симптом может отражать:
1. Повышенную проницаемость
повреждённого клубочкового фильтра и
возрастание фильтрации протеиновых молекул
различного диаметра, превышающее
максимальную производительность
системы реабсорбции (например, при
остром диффузном гломерулонефрите).
2. Понижение реабсорбции белка в
проксимальных канальцах при неизменной
фильтрации (например, при
классическом первичном нефротическом
синдроме).
3. Увеличение выделения белка
эпителием канальцев (например, при, синдроме
Фанкони).
4. Комбинацию части или всех
вышеперечисленных механизмов (например, при
различных хронических
иммунопатологических поражениях почек).
Вполне понятно, что чем серьёзнее
поражение гломерулярного фильтра, тем меньше
селективность и больше в моче глобулинов,
особенно их крупномолекулярных фракций
(С.И.Рябов, Ю.В. Наточин, 1997). При
изолированном нарушении реабсорбции
степень селективности высока, и в
дефинитивной моче представлен, в основном, наиболее
мелкодисперсный белок — альбумин.
При этом в мочу проникают,
исключительно, белки с молекулярной массой до
100 кД и не менее 60% белка мочи
составляет альбуминовая фракция.
Соотношение альбумина и глобулинов в
моче — индекс селективности, позволяет
полуколичественно оценить степень
нарушения избирательности фильтрации, что имеет
диагностическое значение для дифференци-
ровки причин протеинурии (П. Маждраков
и соавт., 1975). Если протеинурия
неселективна, альбумин в моче составляет до 50 %,
представлены все основные фракции
плазменных белков и, как правило, не менее 10 %
белка мочи относится к у-глобулинам.
Среди преренальных причин потери
белка с мочой, имеющих тесную связь с
нарушениями протеинограммы плазмы,
можно указать на парапротеинемии (см.
ниже), когда в моче присутствуют
фрагменты антител — белки Бенс-Джонса. Среди
постренальных причин, в контексте целей
Патофизиология белкового обмена 103
данного раздела, заслуживают упоминания
гиперсекреция желез мочевыводящих
путей и протеинурия, смешанная с пиурией и
гематурией, которая может сопровождать
инфекции урогенитального тракта.
Массивная протеинурия, превышающая
3,5 г/сутки (и даже микропротеинурия при
длительном её существовании) может
вызвать значительное понижение уровня белка
в плазме. Наиболее выраженная массивная
высокоселективная протеинурия и гипоп-
ротеинемия (гипоальбуминемия) типичны
для нефротического синдрома. При тяжелом
нефротическом синдроме не редкость
падение концентрации общего плазменного
белка ниже 30 г/л. Это сочетается с гиперли-
попротеинемией (гиперхолестеринемией) и
нефротическими отёками, об особенностях
патогенеза которых сказано ниже в данном
разделе, а также в разделе «Патофизиология
водно-электролитного обмена». Более
детальная характеристика данного синдрома
содержится в т. III, раздел «Патофизиология
почек» и в «Практикуме».
Некишечные потери белка, приводящие
к снижению его плазменной концентрации,
могут происходить и через кожу —
выраженной плазмореей характеризуется,
прежде всего, ожоговая болезнь, при которой
теряются, в значительной мере, IgG. Сама
тяжесть прогноза при ожоговой болезни
связана со степенью развивающейся гипо-
гаммаглобулинемии и вторичного
иммунодефицита.
Эксфолиативный дерматит и синдромы
Лайелла и Стивенса-Джонсона также
приводят к гипопротеинемии из-за чрезкожной
потери белков в результате экссудации.
При образовании в организме
значительных гнойных экссудатов, например, в
результате эмпиемы, также возможна ги-
попротеинемия. Однако, здесь надо учесть,
что патогенез затяжных гнойных процессов
предусматривает системное действие ка-
хексина и других цитокинов, вызывающих
острофазный ответ (см. т. I), поэтому
ограничение синтеза альбумина при них
является более важным фактором, чем прямые
потери белка. По некоторым представлениям,
резко увеличенная проницаемость стенок
капилляров и венул при шоке, системном
действии медиаторов воспаления,
системных васкулитах — может приводить к потере
белков плазмы путём их перераспределения
между кровью и тканями. Но надо учесть,
что это возможно только при недостаточном
возврате белка через лимфатические пути.
При лимфодинамическом отёке, вызванном
филяриозами («элефантиаз»), поражённые
конечности не имеют возможности отдать
в системную циркуляцию избыток белка
из тканевой жидкости в силу окклюзии
лимфатических сосудов. При этом они
могут превратиться в депо белка, а в крови
возможна гипопротеинемия.
>* При синдроме Иценко-Кушинга
отмечается и глюкагономе гипопротеинемия,
которую связывают с действием еще
одного механизма — усиленного
катаболизма плазменного альбумина.
Патогенез явлений, вызванных
понижением количества плазменного альбумина,
зависит от его осмотической и лиганд-свя-
зывающей функций.
Альбумин — основной компонент плазмы,
обеспечивающий онкотическое давление, то
есть белковую фракцию её осмотического
давления. На втором месте стоят сц
-глобулины. Фибриноген онкотического давления,
практически, не оказывает. Онкотическая
фракция в 200 раз меньше по абсолютной
величине, чем суммарная осмоактивность
всех растворённых и взвешенных в плазме
частиц. Но именно она наиболее значима для
удержания жидкости в сосуде, ибо
онкотическое давление оказывают только частицы,
не проходящие свободно через капиллярную
стенку (см. т. I, стр. 216-220). Напомним,
что в норме плазменная концентрация белка
превышает тканевую интерстициальную
концентрацию, минимум, в 3 раза. Сами
белки создают давление около 19 мм. рт.ст.
и ещё около 9 мм рт. ст. добавляется в силу
удержания дополнительного натрия, из-за
эффекта Доннана. Онкотическое давление
ткани не превышает б мм рт. ст.
Гипопротеинемия характеризуется
безбелковыми отёками. Классическая концеп-
104 А.Ш. Зайчик, Л.П.Чурилов Патохимия
ция трактует их патогенез весьма просто — так
как уменьшается онкотическое давление
плазмы, равнодействующая старлинговских
векторов на границе плазмы и тканевой жидкости
меняет своё направление, околоравновесная
зона сдвигается в сторону посткапиллярных
венул, и происходит усиленная транссудация
натрия и воды во внесосудистый бассейн. По
этой концепции, снижение объема плазмы
ведет к вторичной волюмосберегающей
реакции организма и гиперальдостеронизму,
закрепляющему избыток натрия и воды.
Однако, накапливается всё больше
данных, свидетельствующих о том, что
патогенетические связи при этом не так
однозначны, как может показаться на первый взгляд.
И голодные, и печёночные, и нефротические
отёки в какой-то степени являются
безбелковыми. Но у каждой из этих
разновидностей отёка есть и свои особые механизмы,
взаимодействующие со старлинговской схемой
(см.также «Системные отеки» ниже).
Так, установлено, что при нефротических
отёках, в большинстве случаев, нет гипо-
волемии, у многих больных не повышена
функция ренин-ангиотензин-альдостероно-
вой системы и, наконец, практически мало
помогает инфузия альбумина!
Все это привело к созданию
неклассической концепции механизма нефротического
отёка (И.Г. Каюков, 1997). Согласно этой
точке зрения, первичной при данном типе
отёков является пониженная способность
почек экскретировать воду и натрий, их
малая отвечаемость по отношению к на-
трийуретическим стимулам и высокая
чувствительность к сигналам,
побуждающим к задержке воды и натрия. Переход
же альбумина, а вместе с ним — и какого-то
количества натрия и воды в интерстици-
альную жидкость рассматривается как
компенсаторный процесс, направленный на
то, чтобы избежать гиперволемии.
При печёночных отеках надо принять во
внимание снижение инактивации
альдостерона самой недостаточно эффективно
работающей печенью, что может создавать
вторичный гиперальдостеронизм до (и даже
без) уменьшения объема плазмы.
чдокринно-метаболические нарушения)
Наконец при любых безбелковых отёках
нельзя игнорировать тот важнейший факт,
что именно альбумин связывает более чем
50 % плазменного альдостерона и не даёт ему
проявлять свою биологическую активность.
Какова бы ни была первопричина гипопро-
теинемии, даже без первичного нарушения
старлинговского равновесия ограничение
альбуминового связывания альдостерона
может способствовать вторичному
гиперальдостеронизму и системным отёкам.
Гипоальбуминемия имеет и еще одно
важное патогенетическое следствие. Снижается
возможность плазмы связывать и инактиви-
ровать эндогенные и экзогенные токсины
(билирубин, лекарства и другие
ксенобиотики). Именно поэтому антитоксическая
резистентность уменьшена у лиц с низким
содержанием белка в плазме крови. Так,
недоношеные дети, в силу гипоальбумине-
мии, особенно чувствительны к патогенному
действию билирубина и имеют повышенный
риск билирубиновой энцефалопатии.
Многие белки, принадлежащие к а- и
Р-глобулинам плазмы, являются антиок-
сидантами, медиаторами воспаления и
проявляют бактерицидную активность (см.
выше табл. 5). При преиммунном ответе
фактор некроза опухолей, интерлейкин-1,
интерлейкин-6, интерлейкин-8 и некоторые
другие цитокины вызывают увеличение
синтеза гепатоцитами и макрофагами ряда
положительных глобулинов острой фазы.
При этом подавляется производство
альбумина и трансферрина (отрицательного
глобулина острой фазы). Биологический
смысл данной реакции заключается в
повышении антиокислительной резистентности
тканей, ограничении масштабов альтерации,
индукции гипоферремии и гипоцинкемии,
что снижает скорость размножения многих
бактерий. Побочным эффектом этих
изменений при воспалениях, инфекциях,
иммунопатологических процессах,
злокачественных новообразованиях служит ускорение
СОЭ, ухудшение реологических свойств
крови. Эти закономерности делают СОЭ
и альбумин-глобулиновый коэффициент
сыворотки (который в данных ситуациях
понижается) ценными неспецифическими
диагностическими показателями,
свидетельствующими об ответе острой фазы (см.
также более подробные сведения в т. I,
раздел «Преиммунный ответ и продромальный
синдром», гл. 13).
Помимо гипопротеинемий большое
клинико-диагностическое значение имеют
парапротеинемий.
Этиология парапротеинемий связана,
прежде всего, с неопластическими клональ-
ными пролиферациями мутантных линий
плазматических клеток. Эти клоны
синтезируют аномально большое количество
иммуноглобулинов или их отдельных цепей.
Наиболее важна среди парапротеинемий
поражающая до 15% от всех онкогемато-
логических больных миеломная болезнь
(болезнь Рустицкого-Калера) (см. т. III,
гл. 3). При данном заболевании имеются
множественные очаги — колонии мие-
ломных клеток в костях. Плазматические
клетки неопластического клона могут
вырабатывать (в порядке убывания частоты)
IgG, IgM, IgD, IgA, IgE; или же свободные
лёгкие, либо тяжёлые цепи
иммуноглобулинов, что вызывает в протеинограмме
плазмы необычный М-пик в у-фракции.
Патогенез болезни Рустицкого-Калера у
человека связан с экспрессией онкогена
с-тус и его транслокацией в 14-ю
хромосому, где он локализуется по соседству с
генами иммуноглобулинов. Особой формой
миеломной болезни, при которой
неопластические плазматические клетки секре-
тируют, исключительно, тяжёлые а-цепи
иммуноглобулинов, является
средиземноморская лимфома. Это заболевание
поражает лимфоидную ткань тонкого кишечника
и в настоящее время его принято именовать
иммунопролиферативная энтеропатия.
При плазмоцитоидной лимфоцитарной
лимфоме развивается гиперпродукция
неопластическими клетками IgM. Из-за
агглютинации тромбоцитов и эритроцитов
с участием IgM развиваются тромбоцитопе-
ния, кровоточивость, анемия, повышенная
вязкость крови. Иммуноглобулины особен-
Патофизиология белкового обмена 105
но легко агрегируют на холоде, что
обусловливает холодовой феномен Рейно.
Сама лимфома может быть небольшой
и клинически не распознаётся. В связи с
этим, данную патологию много лет считали
самостоятельной болезнью, именуя мак-
роглобулинемией Вальденстрёма.
При всех парапротеинемиях в моче
присутствуют легкие цепи
иммуноглобулинов — белки Бене-Джонса. При
парапротеинемиях, особенно, болезни
Рустицкого-Калера, часто развивается амилоидоз
(см. ниже «Диспротеинозы»).
К парапротеинемиям примыкает понятие
криоглобулинемии. Так именовали раньше
сборную группу нарушений композиции
глобулинов плазмы, при которых она имеет
тенденцию к формированию желе на холоде.
На самом деле, из парапротеинемий только
IgM-секретирующие миеломы и лимфомы
могут сопровождаться криоглобулинеми-
ей. Во многих случаях криоглобулинемия
определяется наличием низкоаффинных
Холодовых аутоантител класса IgM против
антигенов эритроцитов (холодовая
гемолитическая анемия) или антигенов плазмы
(ревматоидные факторы). Криоглобулином
неиммуноглобулиновой природы
является и фибронектин, присутствующий у
некоторых пациентов в плазме в аномально
больших количествах (см. также т. I, стр.
344-345, 68-469).
В заключение данного раздела уместно
сказать несколько слов о
патофизиологической роли ферментов плазмы крови. В норме
ллазменные ферменты, за исключением про-
теаз, участвующих в системах комплемента,
свёртывания, кининов и фибринолиза, не
играют физиологической роли, а служат
свидетелями определённой интенсивности
цитолиза в тех или иных органах.
При патологии, из-за деструкции клеток в
плазме могут оказываться повышенные
количества различных ферментов, что всегда
указывает на тот или иной цитолитический
синдром. Так, при панкреатите возрастает
специфично активность сывороточной
амилазы, при карциноме простаты очень
106 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
сильно увеличивается содержание кислой
фосфатазы, а при остеопатиях —
щелочной. Сорбитолдегидрогеназа и альдолаза
попадают в плазму, практически, только из
разрушаемых гепатоцитов, а уровень 5'-нук-
леотидазы повышается при поражении жел-
чевыводящих путей. Выше уже говорилось
о плазменных трансаминазах. Все эти
показатели используются в дифференциальной
диагностике.
Попадание в плазму большого количества
активных протеаз из-за острого
деструктивного панкреатита способно активировать
сторожевую полисистему крови и привести
к коллапсу и ДВС-синдрому (см. т. I, стр.
271-273).
НАРУШЕНИЯ КОНЕЧНЫХ
ЭТАПОВ ОБМЕНА БЕЛКА
Конечные этапы белкового обмена — это
вся совокупность превращений,
приводящих к формированию экскретируемых из
организма терминальных азотсодержащих
продуктов — аммиака, мочевины, мочевой
кислоты, креатинина, индикана, а также и
сам процесс их экскреции.
Равновесным показателем выведения и
образования всех этих продуктов служит
уровень остаточного (небелкового) азота
сыворотки крови. По И. Тодорову, эта
величина находится в пределах 15-40 мг/дл.
Остаточный азот по фракциям
распределяется в норме, как показано в табл. 6.
Таким образом, главная составная часть
остаточного азота — мочевина. Собственно
аммиак представлен в крови в связанном
состоянии. Его молярная концентрация
ничтожна и у здорового индивида составляет
менее 517мкмоль/л. В тканях равновесные
концентрации аммиака также крайне малы,
но скорости его образования и превращения
велики, что делает аммиак важнейшим
метаболитом белка.
Большие количества аммиака
формируются при окислительном и
неокислительном дезаминировании (рис. 17).
Аммиак — высокотоксичный продукт азотистого
метаболизма, к тому же он легко проникает
через липидные мембраны. Больше всего
аммиака формируют органы с наиболее
интенсивным обменом аминокислот и
биогенных аминов — лидером является мозг,
затем идут печень и ЖКТ (где этот процесс
сильно зависит от деятельности
микрофлоры и степени нарушения переваривания
белков, а также регулярности опорожнения
кишечника). Почки — также активные
продуценты аммиака, поскольку такой способ
регуляции кислотно-щелочного
равновесия, как аммониогенез, предусматривает
экскрецию иона аммония в мочу в обмен
на натрий, с целью компенсации ацидоза,
особенно хронического (см. ниже). До 6 % от
мочевого азота составляет именно продукт
аммониогенеза — ион аммония.
На месте своего образования, в тканях
и органах, аммиак подлежит немедленной
нейтрализации. Это достигается путём
аминирования а-кетокислот, прежде всего
а-кетоглутаровой, с образованием глута-
миновой кислоты (рис. 18, с. 108, вверху)
и, в особенности, путем превращения глу-
таминсинтетазой глутаминовой кислоты в
ее амид — глутамин. Он-то и служит, в ос-
Таблица 6
Остаточный азот сыворотки крови
и его составляющие
Компонент остаточного
азота
Азот мочевины
Азот аминокислот
Азот мочевой кислоты
Азот креатина
Азот креатинина
Индикан
Аммонийные соли
Азот пептидов и других
небелковых веществ
ИТОГО:
Содержание в норме
10-22 мг/дл
3,5-6,5 мг/дл
1,0-3,0 мг/дл
0,3-1,0 мг/дл
0,2-0,5 мг/дл
0,03-1 мг/дл
0,1-0,2 мг/дл
около 5,0 мг/дл,
15-40 мг/дл
Патофизиология белкового обмена 107
глутаминовая
кислота
2. а-аминокислота .
3. цистеин —^——
4. гистидин <
5. глицин*
6. глюкозамин-6-Р •
7. глутамин
2.
О
nh2-c-o-(D
карбамоилфосфат
NH2
новном, эквивалентом не-
протонированного NH3 при
синтетических процессах
и временным хранилищем
(транспортёром)
нетоксичной формы аммиака.
Глутамин образуется в
самой печени и испытывает
постоянный кругооборот
между нею и другими
органами. Избыток аммиака
из мозга достигает печени
в этой же транспортной
форме. Почки получают
глутамин для процесса
аммониогенеза также из
печени. Глутаминзависи-
мая аспарагинсинтетаза
может переводить аммиак
в состав аспарагина и
освобождать глутамат. Карба-
моилфосфат-синтетазы I и
II (рис.17), из которых
первая уникальна для печени,
а вторая имеется и в
других тканях с напряжённым
аминокислотным обменом, превращают,
соответственно, глутаминовую кислоту и
глутамин в карбамоилфосфат — активную
форму, переносящую аммиак в системы,
синтезирующие аргинин или пиримидины.
Через синтез и превращения аргинина
лежит путь аммиачного азота к мочевине
(карбамиду) — конечному выводимому продукту
его метаболизма у млекопитающих (рис. 18
внизу). Печень синтезирует подавляющую
часть мочевины, что было обнаружено в
результате опытов отечественных учёных
М.В. Ненцкого, С.С.Салазкина и И. За-
лесского в лаборатории И.П. Павлова.
Справедливости ради, надо отметить, что
незначительное количество мочевины
способен образовывать сам головной мозг.
Процесс образования карбамида
представляет цикл реакций (орнитиновый, он
же цитруллиновый цикл, в зарубежной
литературе — цикл Кребса-Гензелейта,
или цикл мочевины). Многие этапы этих
реакций требуют гидролиза макроэрги-
^ а-кетоглутаровая
кислота +
а-кетокислота +
-^- пируват +
урокаимовая
кислота +
глиоксалевая
кислота +
-► глюкоз а-6-Р +
глутаминовая
NH3
NH3
NH3
NH3
NH3
NH3
мочевина
пиримидины
глутамин
NH2-C(CH2)C COOH
О
пурины
глюкозамин
Рис. 17. Пути образования аммиака
ческих фосфатных связей. Считается, что
весь процесс объединения аммиака с С02 в
мочевину обходится печени в значительное
количество энергии — 4 макроэргических
связи на моль карбамида (рис. 17-18). Эти
расходы оправдываются тем, что в
результате ядовитейший аммиак становится частью
практически малотоксичной мочевины,
которая водорастворима и ведет себя, как
беспороговое вещество — то есть выводится
почкой пропорционально её образованию, а
не после преодоления какого-то
минимального уровня её плазменного содержания.
Если бы этот процесс не происходил, все
большее количество кетокислот (прежде
всего, а-кетоглутаровой и щавелевоуксус-
ной) связывалось бы с аммиаком и
расходовалось на его временную нейтрализацию,
без регенерации обратно в кетоформу. Рано
или поздно это привело бы к торможению
цикла Кребса и развитию тканевой
гипоксии, прежде всего, в органах, производящих
больше всего аммиака — в частности, в ЦНС.
а-амино-
кислота
I OKCl
а-кетокислота
FAD
оксидаза аминокислот
FADH2
V
слот I
Л
НгО
NH3
окислительное
дезаминирование
а-амино-
кислота
а-кетокислота
глутаминовая
кислота
NAD+
глутаматдегидрогеназа
а-кетоглутарат I NADH+H+
NH3
окислительное
дезаминирование с
переаминированием
У
ООН
карбомоил- /
фосфат /
Ф
NH2
(СН2)3
HCNH2
СООН
орнитин
J^
NH2
I
с=о
Л
NH
(СН2)з
HCNH2
СООН
цитруллин
СН2
HC-NH2
СООН
аспарагиновая
оксалоацетат
H2N-C = N4-I
1—^сн2
СООН
NH
I
' | 2'* аргининосукцинат
HCNH2
СООН
фумарат
аргинин
мочевина
цикл
мочевины
Рис. 18. Первичная нейтрализация аммиака (вверху) и цикл мочевины (внизу). Наследственные
дефекты образования мочевины обозначены цифрами 1-6, расшифровку см. в таблице 7
Патофизиология белкового обмена 109
Именно это и наблюдается при срыве моче-
винообразования в печени и проявляется
нарастающей тканевой гипоксией мозга,
дающей прекому и кому.
С другой стороны, большое значение
имеет постоянное выведение мочевины
почками. Опять-таки, ради точности отметим,
что около 1 % экскреции мочевины в норме
идёт через потовые железы, а при обильном
потении этот процент может стать
больше. Примерно 25% суточной продукции
мочевины диффундирует в кишечник, где
разлагается бактериями с образованием
аммиака и снова утилизируется. При почечной
недостаточности растет экстраренальное
выделение мочевины через кожу и
слизистые кишечника и желудка. У здорового
взрослого на обычной диете каждый день
почками выделяется 25-35 г этого
метаболита. Подчеркнем еще раз принципиальное
положение о малой токсичности карбамида.
Воспринимаемая обыденным сознанием
как яд (шлак), мочевина нужна организму,
который толерантен к достаточно большим
её концентрациям.
Уремия, как терминальная стадия
хронической почечной недостаточности, когда
конечные продукты азотистого обмена
не выводятся из орагнизма, также, по
современным данным, не является чисто
мочевинным отравлением, а представляет
результат совокупного действия на
организм более чем 200 других накапливаемых
продуктов, которые куда более вредоносны,
даже в малых, по сравнению с карбамидом,
концентрациях. Мочевина используется
почкой для осмотического диуреза, а
лейкоцитами как бактерицидный агент. Не
случайно, она реабсорбируется здоровыми
почками (при условии высокой скорости
мочетока и, особенно, при бедной белком
диете) примерно на 1/3 от всей ее
фильтрации и имеет клиренс порядка 75 мл/мин, в
отличие, например, от нереабсорбируемого
инулина (90-130 мл/мин).
Любопытно отметить, что мочевина
даже может служить источником пищевого
азота (в ветеринарии). П.Е. Калмыков и
М.Н. Логаткин отмечают, что мочевина
при пероральном введении и действии уре-
азы кишечных бактерий (а по некоторым
данным — и эндогенная мочевина) могут
использоваться организмом в особых
условиях, как источник азота для заменимых
аминокислот (1974). Хвалебную песнь
мочевине мы можем закончить упоминанием
о ней, как компоненте высокоэффективной
зубной пасты и жевательной резинки, о чём
с энтузиазмом сообщает реклама.
И всё же, избыточное накопление
карбамида не остаётся для организма без
последствий, как в силу его осмотического эффекта,
так и в связи с тем, что при уремии будет
снижаться активность аммиак-нейтрализу-
ющих реакций, приводящих к его синтезу:
ведь продукт из сферы действия реакции
не выводится.
В конечном итоге, и нарушение
выведения продуктов азотистого метаболизма, так
же как нарушение нейтрализации аммиака
в мочевину, приводит к дефициту
метаболитов цикла Кребса, тканевой гипоксии и
коме.
Следовательно, глубокие нарушения
энергообеспечения клеток возможны как
при срыве процессов мочевинообразования
(продукционная гиперазотемия), так и при
срыве ее выведения с мочой (ретенционная
гиперазотемия).
Главная причина продукционной
гиперазотемии — печёночно-клеточная
недостаточность. Ее механизмы подробно
обсуждаются в Практикуме и в т. III данного
руководства (раздел «Патология печени»).
Клинически, продукционной гиперазотемии
при этом соответствует симптомокомплекс
гепатогенной энцефалопатии — нарушение
ритмов сна и бодрствования, эмоциональная
лабильность, дисфория, хлопающий тремор,
изменения на ЭЭГ и даже бред, а при более
выраженной интоксикации немочевинными
азотистыми продуктами — клиника пре-
комы и комы, начиная с апатии, летаргии,
вплоть до глубокого сна, арефлексии и даже
исчезновения болевой реакции.
В данной главе следует отметить, что
из-за мозаичности функций гепатоцитов,
которые в неодинаковой степени вовлечены
110 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
в высокоаэробный энергозависимый
процесс мочевинообразования, далеко не все
поражения печени равно сопровождаются
продукционной гиперазотемией. Больной
острым гепатитом может, например, иметь
выраженную желтуху, но не проявлять
сколько-нибудь значительных признаков
печёночной энцефалопатии. Так как в мозге
более 30 % глутаминовой кислоты участвует
в переаминировании с янтарным
полуальдегидом, с образованием у-аминомасляной
кислоты (ГАМК), очевидно, что при
накоплении связанных форм аммиака этот
процесс будет усилен. Получены прямые
свидетельства кумуляции ГАМК — этого
тормозного нейромедиатора — в ЦНС
больных гепатогенной энцефалопатией.
Изменения остаточного азота при
продукционной гиперазотемии характеризуются
абсолютным и относительным возрастанием
его немочевинных фракций, прежде
всего — аммония, азота аминокислот и,
особенно, биогенных аминов (некоторые из них,
например, октопамин и тирамин — имеют
прямое отношение к симптомам
энцефалопатии, поскольку действуют на ЦНС как
лже-сигналы). Фракция мочевины в
остаточном азоте при этом убывает.
Другая причина продукционной
гиперазотемии — усиленный катаболизм белков
(при голодании или при значительном
перекорме белками). При этом содержание
азота аминокислот и аминов также
повышается, растёт и синтез мочевины, но при
нормальной функции почек не происходит
её накопления в крови и относительная доля
мочевинного азота может снизиться.
Ретенционная гиперазотемия
характеризует, прежде всего, почечную
недостаточность. Нормальные почки концентрируют
мочевину при ее выведении более, чем в
70 раз. При нарушениях фильтрации
клиренс мочевины снижается. Однако, из-за
повышения концентрации мочевины в плазме,
баланс ее длительное время может
оставаться нормальным при сниженном клиренсе,
так как суточное выведение и реабсорбция
будут соответствовать потребности.
При острой почечной недостаточности
(ОПН) в анурическую фазу, из-за резкого
снижения фильтрации, растут и уровень
остаточного азота, и азот мочевины. В
течение 5-7 дней, если не произойдёт
восстановления фильтрации, больной без лечения
погибает. Однако, этого срока и
достигаемого уровня гиперазотемии недостаточно,
чтобы обусловить фатальное отравление
азотистыми продуктами. Поэтому, в
анурическую стадию ОПН гибель наступает не
от уремии, а от нарушений водно-солевого
и кислотно-щелочного метаболизма (см.
Практикум и т. III, раздел «Патофизиология
почек»).
При хронической почечной
недостаточности (ХПН) прогрессирующий нефросклероз
приводит к гибели нефронов и с годами их
остается всё меньше. Компенсаторно,
принимая на себя функции погибших нефронов,
оставшиеся, рано или поздно, оказываются
в ситуации, когда скорость тока мочи в них
увеличивается до максимума и, даже
выделяя мочу, близкую по составу к первичной,
становится невозможно экскретировать все
вырабатываемые азотистые продукты, так
как низка их концентрация в моче. В исходе
ХПН уремия является неизбежной. При
высокой концентрации мочевина в кислой
среде образует с анионами хлора фосген,
участвующий в патогенезе уремической
интоксикации
(NH2-CO-NH2 + 2HC1 <-> ОСС12 + 2NH3).
Остаточный азот повышается до 200-
ЗООмг/дл. Возрастают как уровень
мочевины, так и немочевинных азотистых
компонентов, в частности, аммония, мочевой
кислоты и пептидов («средних молекул»).
Наиболее тяжелое и быстрое повышение
уровня остаточного азота свойственно
комбинированным нарушениям, когда страдают
и почечная, и печёночная функции. В этом
случае имеется смешанная, ретенционно-
продукционная гиперазотемия.
Подобные ситуации сопровождают шок
и шокоподобные состояния (глубокую
дегидратацию — особенно, гипоосмолярную
её форму, синдром длительного раздавли-
Патофизиология белкового обмена 111
вания). Особая форма смешанной печёноч-
но-почечной недостаточности известна как
гепато-реналъный синдром.
Это вторичная острая почечная
недостаточность при первичных болезнях печени.
Главным образом, она осложняет течение
острой и хронической печёночно-клеточной
недостаточности, острую
паренхиматозную желтуху, нередко бывает у больных с
выраженной портальной гипертензией. При
этом возникает резкое нарушение почечного
регионального кровотока и ограничение
фильтрации, ведущее кОПН.
Предполагается, что решающим фактором патогенеза
этого синдрома, крайне осложняющего
прогноз, является действие на почку
ложных нейромедиаторов — биогенных аминов,
не обезвреживаемых больной печенью.
Возможно, токсическое действие избытка
билирубина и секвестрация крови в
непарных органах брюшной полости также имеют
определённое значение.
Отдельного рассмотрения заслуживают
наследственные нарушения цикла
образования мочевины, которые представляют
собой одну из врождённых причин
задержки психомоторного развития (ПМР)
детей.
Выделяют не менее 6 наследственных
болезней с дефектами ферментов цитрул-
линового цикла (см. табл. 7) и блокадой их
активации. Для всех этих нарушений
характерны: снижение толерантности к белку,
эндогенное аммиачное отравление, задержка
ПМР и другие нервно-психические
симптомы. Гипераммониемия II типа сцеплена
с X хромосомой и доминантна, остальные
аутосомны и рецессивны. При некоторых
имеются аминоацидурия и гипераминоа-
цидемия. Практически все проявляются
от рождения и могут обусловить раннюю
смерть больных.
Таблица 7, характеризует
наследственные расстройства цикла мочевины. В левом
крайнем столбце, после названия
заболеваний в круглых скобках показано, какой
цифрой обозначен соответствующий дефект
на рис. 18 (стр. 108).
Таблица 7. Наследственные гипераммониемии
Заболевание
Дефицит ацетилглутами-
новой кислоты
Гипераммониемия
1типа(рис.18,1)
Гипераммониемия II типа
(рис. 18,2)
Цитруллинемия (рис.
18,3)
Аргининосукцинатурия
(рис. 18,4)
Гипераргининемия (рис.
18,5)
Гиперорнитинемии —
два типа (рис. 18, б).
Дефектный фермент
N-ацетилглутаматсинтета-
за
Митохондриальная карба-
моилфосфатсинтетаза
Митохондриальная ор-
нитин-карбамоилтранс-
фераза
Цитоплазматическая арги-
нин-сукцинатсинтетаза
Цитоплазматическая арги-
нинсукцинатлиаза
Аргиназа
Р-орнитин-2-оксо-ацил-
трансфераза или
транспортер орнитина в
митохондриях
Проявления
Задержка ПМР. Гипераммониемия.
Задержка ПМР. Гипераммониемия. Непереносимость
белка.
Задержка ПМР. Гипераммониемия. Непереносимость
белка, вторичная оротацидурия (карбамоилфосфат
диффундирует в цитозоль и идёт на пиримидиновый
ситнтез).
Задержка ПМР. Гипераммониемия. Вторичная
оротацидурия. Возможны поздние формы болезни. Частота
повышена у японцев.
Задержка ПМР. Гипераммониемия. Судороги,
атаксия, гепатомегалия, гипертермия фиброз печени,
цитруллинемия и аргининсук-цинатемия. Ломкость
и сухость волос, их пучковый рост.
Задержка ПМР. Гипераммониемия. Мышечные
спазмы, вторичная конкурентная цистинурия
Гипераммониемия, гомоцитруллинемия, гиперор-
нитинемия («синдром трёх Г»). Иногда — хориоре-
тинальная атрофия.
112 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Хотя подавляющая часть мочевого азота
входит в состав мочевины (в норме 6-18 г
азота в сутки) и аммония (0,4-1,0 г/сутки), у
здоровых индивидов экскретируются также
креатинин (0,3-0,8 г/сутки), мочевая
кислота (0,08-0,2 г/сутки), пептиды (0,3-0,7 г/
сутки), аминокислоты (0,08-0,15 г/сутки),
гиппуровая кислота (0,04-0,08 г/сутки),
индикан (0,005-0,025 г/сутки).
При патологии могут нарушаться и
процессы экскреции немочевинных
компонентов мочевого азота. О протеинурии,
индиканурии и гипераминоацидурии речь
шла выше. Нарушения выведения
остальных немочевинных азотистых продуктов
обсуждаются в данном разделе.
Креатинин представляет собой
ангидридную форму креатина — производного
аминокислот глицина и аргинина, которое
образуется в результате их переамидиро-
вания, через гуанидинуксусную кислоту.
Последняя метилируется, причём донором
метильной группы выступает еще одна
аминокислота — метионин.
Креатин активно образуется в печени,
миокарде и мышцах. В форме креатинфосфата
он несёт макроэргическую фосфатную связь
и принимает участие в транспорте энергии от
митохондрий к миофибриллам. В норме
креатин фильтруется, но почти полностью реаб-
сорбируется. Креатинин же экскретируется
путем фильтрации и секреции, практически,
без реабсорбции. Почка концентрирует
креатинин примерно в 70 раз.
Поскольку креатин — метаболит, в
основном, мышечного происхождения, степень
креатининурии пропорциональна
мышечной массе, и для оценки
метаболической значимости этого симптома принято
учитывать креатининовый коэффициент
(КрК)- количество креатинина в моче в мг
за 24 ч на 1 кг массы тела.
В норме у мужчин КрК равен 18-32, а
уженщин — 10-25. У атлетов КрК выше,
аутучных и астеников — снижен. При
ограничении фильтрации в почках падает
и выведение креатинина, в связи с чем
его клиренс оценивают для диагностики
почечной недостаточности. Уровень
креатинина в плазме связан обратной линейной
зависимостью с числом функционирующих
клубочков, поэтому его повторное
определение обладает при ХПН прогностической
значимостью, позволяя предсказать сроки
наступления ее терминальной стадии (см.
Практикум).
Креатинурия в норме характерна только
для новорожденных и беременных женщин.
У здоровых взрослых лиц креатина в моче
крайне мало. Увеличение выведения
креатина с мочой происходит при мышечной
атрофии, в том числе, при наследственных мио-
дистрофиях, генерализованной миастении,
миозитах. Такая же картина характерна для
голодания (см. выше), гипертироза и любых
пролонгированных состояний с
отрицательным азотистым балансом. Считается, что
повышение выведения креатина при данных
ситуациях сопровождается уменьшением
экскреции креатинина и отражает потерю
белка соматическим отсеком организма. При
сахарном диабете и акромегалии, чаще всего,
имеется и креатинурия, и креатининурия.
Гиппуровая кислота, производное
глицина и бензойной кислоты, образуется в
печени. При печеночной недостаточности
ее выделение снижается, на чем основана
одна из функциональных печеночных проб
с нагрузкой бензойной кислотой. Последняя
имеется не только в естественных фруктах
и ягодах, но и используется, как консервант
и компонент косметики. В связи с этим,
выделение гиппуровой кислоты может сильно
зависеть от особенностей диеты и быта.
Мочевая кислота, хотя и может включать
азот из аминокислот, представляет собой
конечный продукт пуринового обмена, поэтому
патология ее выделения обсуждается ниже в
соответствующем разделе (см.
«Патофизиология нуклеинового метаболизма»).
ДИСПРОТЕИНОЗЫ
При рассмотрении патологии белкового
обмена все классические руководства прежних лет
уделяли большое внимание так называемым
«белковым дистрофиям» или «диспротеино-
Патофизиология белкового обмена 113
зам» («cell degenerations of albuminous origin»).
Данный термин восходит к работам
родоначальника современной патологии Р. Вирхова
(1853). Не будет ошибкой расшифровать
основной смысл термина «диспротеино-
зы», как «процессы накопления в клетках и
межклеточном веществе количественно и
качественно измененных продуктов обмена
белков» (по Н.Е.Ярыгину, В.В. Серову,
1977). Однако, по сравнению с вирховскими
временами, когда основным инструментом
патолога был световой микроскоп, понимание
данного термина и спектр обозначаемых им
нарушений сильно изменились. Прежде
данное понятие подкреплялось лишь
морфологическим описанием наблюдаемых при тех или
иных диспротеинозах однотипных картин.
Так возникли представления о
существовании в этой группе клеточных диспротеинозов
(среди которых традиционно числили мутное
набухание, гиалиново-капельную дистрофию,
гидропическую или баллонирующую
дистрофию, роговую дистрофию) и внеклеточных
диспротеинозов (к которым относили муко-
идное и фибриноидное набухание, амилоидоз
и внеклкточный гиалиноз). Однако, развитие
патохимии показало некоторую
искусственность такого объединения.
Дело в том, что за одними из этих
терминов стоит отложение одного химически
конкретного белка (например, при
ороговении— экспрессия генов кератина). Другие
термины (например, амилоидоз) обозначают
отложение регулярного по структуре
комплекса нескольких белков, близкого состава
и происхождения, хотя и варьирующих по
композиции (причём выявляются типичные
для всех разновидностей
физико-химические и ультраструктурные особенности
депонируемого белкового комплекса).
И в первом, и во втором случаях,
поскольку речь идет о химически и физически
определённых субстанциях и своеобычных
процессах, выделение того или иного дис-
протеиноза выглядит оправданным как с
точки зрения патоморфологии, так и
патофизиологии этих феноменов.
Но ряд явлений, ранее включавшихся
в группу белковых дистрофий, оказался
результатом неприуроченных к какому-то
конкретному протеину или белковому
комплексу типовых изменений,
отражающих ход гипоксического некробиоза
(мутное набухание, гидропическая
дистрофия), или возникающих вследствие
альтерации межклеточного вещества при
воспалении и иммунопатологических
процессах (мукоидное набухание,
фибриноидное набухание).
В этих случаях рассматривать такие
«дистрофии» отдельно от патофизиологических
процессов, которые за ними стоят, под
другими именами, означало бы не следовать
принципу У. Оккама, который говорил, что
при познании: «Не должно создавать
лишних сущностей». Это было уместно, лишь
пока патохимия таких процессов оставалась
совершенно неизученной.
Наконец, некоторые феномены
(гиалиноз) оказались не химическими понятиями,
а лишь удобными условными
обозначениями, фиксирующими любые белковые
отложения определённого внешнего вида.
Из-за описанных объективных
гносеологических сложностей, в рамках многих
пособий по патофизиологии, стремящихся
к краткости, данные вопросы стали вообще
опускать, мотивируя это тем, что они не
патофизиологические, а патоанатомические.
Но, на деле, каждый патологический
процесс в равной мере служит объектом
рассмотрения обеих равноуважаемых ветвей
патологии! Наши многолетние
преподавательские наблюдения показывают, что
в сознании студентов, изучающих данный
вопрос, из-за разницы в терминологии
курсов патофизиологии и патологической
анатомии, могут возникать ложные
представления. Это либо приводит к
ошибочной уверенности, что диспротеинозы как
патофизиологический феномен вообще не
существуют; либо у обучаемых укореняется
архаичная терминология в этой области,
безо всякой связи с современными патохи-
мическими обозначениями.
Всё это заставило нас заключить главу
«Патофизиология белкового обмена»
разделом, освещающим диспротеинозы.
114 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Однако, мы следовали вышеизложенным
соображениям о содержании термина «дис-
протеинозы».
В связи с этим, механизмы мутного
набухания («зернистой дистрофии») трактуются
как корреляты ранней стадии обратимой
гипоксии клетки и рассмотрены в т. I
настоящего руководства, в разделе «Механизмы
гипоксического некробиоза» (с. 182-183).
Механизмы баллонирующей (гидропической)
дистрофии истолковываются как корреляты
более глубокой клеточной гипоксии (там же,
с. 184 и далее). Мукоидное набухание и фиб-
риноидное набухание интерпретируются как
начальные и глубокие, сопровождаемые
коагуляцией, стадии альтерации межклеточного
вещества при воспалении и рассматриваются
в соответствующем разделе I тома (с. 295).
Механизмы гиалиново-капельной дистрофии
и её связь с патологией цитоскелета и
агрегацией промежуточных филаментов, отражены
также в I томе (с. 162,185,187).
Накопление сложных белков, например,
липопротеидов, гликопротеидов —
рассматривается ниже в соответствующих разделах
данного тома (см. ниже «Патофизиология
липидного обмена», «Патофизиология
сахарного диабета»). Что касается накопления
хромопротеидов, которое традиционно
входит в содержание раздела «Диспротеинозы»
в патоанатомических учебниках, то в связи
с разнородностью химических и
патофизиологических процессов, приводящих к
накоплению окрашенных включений, мы
сочли логичным не объединять эту тему по
частному внешнему признаку окрашенности
включений, а диспергировать этот материал
по соответствующим разделам курса (см.
Практикум и т. III).
Так, нарушения обмена тирозин-трип-
тофановых пигментов затронуты выше, в
разделе «Нарушения межуточного обмена
аминокислот и их производных».
Нарушения метаболизма липидогенных пигментов
отнесены к разделу «Патология липидного
обмена». Накопление ферритина, гемоси-
дероз, порфирии и расстройства обмена
других железосодержащих пигментов будут
рассмотрены в разделе, посвященном
микроэлементам, и в т. III. Билирубинопатии
также будут рассматриваться в томе III и в
соответствующих разделах частной
патофизиологии (Практикум).
Таким образом, в данном разделе, на наш
взгляд, есть необходимость коснуться лишь
патофизиологии амилоидоза и роговой
дистрофии. Мы также дадим толкование пато-
анатомического понятия «гиалиноз».
Амилоидоз — патофизиологический
процесс, относительно которого есть явные
свидетельства, что в большинстве случаев он
связан с определёнными расстройствами в
иммунном аппарате.
При аутопсиях амилоидоз той или иной
локализации обнаруживается с частотой от
0,1 % — в Японии до почти 2 % — в странах
Пиренейского полуострова и в России. Как
системное нарушение, амилоидоз не может
быть отнесён только к тому отдельному
органу (органам), где обнаруживаются
отложения амилоида.
Амилоид — патологический белковый
комплекс, который на окрашенных
гематоксилином и эозином препаратах выглядит
как розовый прозрачный материал,
депонированный между клетками в различных
тканях и органах тела. Он обнаруживается
при большом числе разнообразных
синдромов и болезней.
Поскольку амилоидное перерождение
подолгу протекает без клинических
проявлений и иногда имеет загадочные причины,
его диагностическое распознавание, в
конечном счете, зависит от морфологической
идентификации этой особой субстанции в
соответствующих биоптатах.
Все типы амилоида обладают
следующими общими тинкториальными
характеристиками:
>- Добавление йода окрашивает амилоид
на свежеприготовленных срезах ткани в
коричневый или желто — красный цвет,
который преобразуется в синий или
фиолетовый после обработки разбавленной
серной кислотой.
>• При окраске гематоксилином и эозином в
световом микроскопе амилоид выглядит
Патофизиология белкового обмена 115
как аморфная, эозинофильная, розовая,
прозрачная, внеклеточная субстанция.
При прогрессирующем накоплении
происходит атрофия соседних с амилоидом
структур от давления.
> При окраске красителем конгорот
амилоид в препарате кажется красным с
яблочно-зеленым двойным лучепреломлением,
заметным, если ткань рассматривается в
поляризованном свете. Это используют
при дифференцировке амилоида от
других белковых депозитов, имеющих
вид гиалина (см. ниже — например, от
коллагена, фибрина, гликопротеидов).
> При окраске метиловым фиолетовым,
амилоид проявляет метахромазию, что
означает развитие цвета, отличного от
присущего самому красителю (розовый
вместо фиолетового).
> Амилоид метится различными
антисыворотками против его компонентов при
иммуноморфологическом анализе и даёт
люминесценцию с рибофлавинами S и Т.
История изучения амилоидоза началась
с описания врачом Т. Бонетусом
«деревянистой селезёнки» (1675). К. Рокитанским
амилоидоз был описан под именем «сальной
болезни» (1844) внутренних органов (ему
же принадлежат исторические термины
«восковая печень», «саговая селезёнка»).
Через 9 лет Р.Вирхов гистологически
охарактеризовал амилоид, открыл
некоторые его тинкториальные свойства и ввёл
само название этого диспротеиноза (1893).
Р. Вирхову посинение йодированного
амилоида при обработке препарата серной
кислотой напомнило известное свойство
крахмала (греч. ацоА,оо). Так укоренился
термин «амилоид», хотя впоследствии
доказали, что в данной субстанции нет крахмала.
В XIX веке после опытов Ф.-В. Бёрч-Хирш-
фельда (1888), воспроизводившего
амилоидоз у кроликов с помощью хронической
гнойной инфекции, сформировалась теория
бактериально-токсического
происхождения амилоида (В.В.Подвысоцкий, 1905).
Пониманию природы амилоидоза, как
диспротеиноза, способствовала разработка
М.Н. Кучинским и Н.П. Кравковым (1894)
неинфекционной модели этого процесса (с
помощью инъекций мышам и кроликам ка-
зеината натрия) и доказательство последним
белковой природы чистого амилоида (1897).
В развитие этой концепции, в XX столетии
появляется представление о роли иммуно-
комплексного процесса в формировании
амилоида (теория X. Лёшке-Э. Леттерера,
1927). Вместе с тем, так как амилоидоз
бывает и при агаммаглобулинемии, данная
теория не могла, как казалось,
удовлетворительно объяснить возникновения всех
его форм. Поэтому сформировались
представления о локальном клеточном генезе
амилоида (Дж.Тейлум, 1954), согласно
которой амилоид — комплекс разнородных
белков, которые возникают внутри клеток
ретикулярного происхождения («амилои-
добластов») в преамилоидную фазу, а
собираются в амилоид вне клеток, в амилоидную
фазу процесса. Сторонникам данной теории
удалось зарегистрировать образование
амилоида даже в культурах ткани. Роль клеток
макрофагального ряда (амилоидобластов)
в генезе фибриллярного компонента
амилоида при вторичном амилоидозе признают и
поныне (В.В. Серов и соавт., 1986).
Наконец, теория органопротеидоза
(В.Кальи, 1961) обогатила представления
об амилоидозе идеей, что решающее
значение имеет аномальный, мутантный характер
компонентов, составляющих амилоид.
Подлинное проникновение в механизмы
амилоидоза было обеспечено только с
развитием иммуногистохимии и электронной
микроскопии.
Как уже упоминалось, амилоид — это не
отдельное химическое вещество, но группа
белков, структурно взаимодействующих
между собой характерным образом.
Известны 2 главных химических типа амилоида и
несколько минорных. Из-за этого
локализация и характер отложений амилоида у разных
больных весьма изменчивы. В прошлом
термин «первичный амилоидоз» использовался,
чтобы описать амилоидное перерождение у
пациентов, не имевших какой-либо выяв-
11 б А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
ленной основной болезни,
способствовавшей его развитию.
Больные с различными хроническими
воспалительными болезнями
классифицировались как имеющие «вторичный амилоидоз».
В последнее время, внимание
исследователей было приковано к химическому
составу и происхождению амилоида, и, насколько
возможно, эти аспекты послужили основой
для новой его классификации. Амилоидоз
не должен рассматриваться как отдельная
болезнь; скорее, это — группа нарушений,
объединяемых по принципу отложения
сходно устроенных белковых комплексов.
Ультраструктура амилоида куда
сложнее и интереснее, чем можно ожидать по
его аморфному светомикроскопическому
виду.
Аморфные депозиты, фактически, в
значительной степени, составлены из нераз-
ветвленных фибрилл различной длины (до
800 нм), диаметром приблизительно от 7,5
до 10 нм. Эта ультраструктура идентична
при всех типах амилоидоза.
Фибриллы могут располагаться по
отдельности, в пучках с латеральными связями,
или в сцепленной сетчатой структуре.
Инфракрасная спектроскопия и рентгеновская
кристаллография показали, что эти
фибриллы всегда образуют типичную уникальную
структуру, известную как «р-складчатая
листоподобная структура». Ни один
другой фибриллярный белок млекопитающих
подобной микроархитектуры не имеет, так
что вышеназванная структура — абсолютная
диагностическая характеристика амилоида.
Плотно упакованная структура этих листков
чрезвычайно устойчива к протеолизу, таким
образом обеспечивая важное условие для
накопления амилоида в тканях. И.В.
Давыдовский указывает, что амилоид даже
устойчив к гниению (1968). Он не вымывается
из тканей солевыми растворами при
физиологических концентрациях электролитов.
Однако, амилоид можно экстрагировать
дистиллированной водой, что используется
при его химических анализах.
В дополнение к неразветвлённым
фибриллам, в амилоиде всегда присутствует другой
минорный белок, известный как Р-компонент.
Под электронным микроскопом он выглядит
как палочка, а его поперечный разрез — это
цМЦН
HtnitHnWw»
Рис 19. Ультраструктурные компоненты амилоида (по Дж. Гленнеру и соавт.,1980).
Слева—фибриллярный компонент амилоида (Ам), справа вверху - палочковидные образования Р-компонента,
внизу—нефибриллярный Р-компонент в поперечном разрезе '
пятиугольная, пышкообразная структура,
имеющая наружный диаметр приблизительно
9 нм и внутренний диаметр 4 нм. Каждый
пятиугольник составлен, в свою очередь, из пяти
шаровидных субъединиц (рис. 19).
Традиционно, амилоидоз разделяют на
системные и местные формы.
Системный, или общий вариант,
характеризуется множественной поражённостью
органов. Он подразделяется на первичный
амилоидоз, который связан с различными
иммунодискразиями (моноклональными
гаммапатиями — см. раздел «Нарушения
композиции белков плазмы»), и вторичный
амилоидоз, осложнение основных
хронических воспалительных или деструктивных
процессов. Наследственный или семейный
амилоидоз составляет отдельную, хотя и
гетерогенную группу, с несколькими
отличительными особенностями вовлечения
органов.
При первичном амилоидозе наиболее часто
вовлекаются почки, сердце, печень, селезенка,
лимфоузлы, суставы и лучезапястные связки,
кожа, нервы и язык. Вторичный подтип может
вовлекать печень, селезенку, надпочечные
железы и почки (по образцу амилоидоза
при хронических заразных болезнях,
например, проказе) или поражать сердце, ЖКТ
и язык — подобно первичному амилоидозу
(при иммунопатологических расстройствах,
например, при ревматоидном артрите).
В отличие от традиционной
клинической классификации изучение химической
природы амилоида методами иммуногис-
тохимии породило новый, основанный на
составе амилоида, подход к его систематике
(Дж.Н.Бюксбаум, 1987).
Имеются 2 главных элемента амилоидных
фибрилл. Оба, так или иначе, связаны с
иммунной системой, также как и
нефибриллярный компонент амилоида. Это факт
дидактически позволяет обсуждать амилоидоз
в разделе, посвященном иммунопатологии,
поэтому данный процесс вкратце
упоминается в I томе настоящего руководства (т. I,
с.377,399,440).
Приблизительно 90 % амилоидного
материала состоит из фибриллярного компонен-
Патофизиология белкового обмена 117
та, остальные 10 % приходятся на Р-
компонент, который является гликопротеидом.
Были идентифицированы два химически
отличных главных типа амилоидного
фибриллярного компонента (AL и АА):
— Первый, так называемый AL (amyloidlight
chain-related) формируется при участии
плазматических клеток (В-иммуноцитов)
и содержит, в большинстве случаев,
полные легкие цепи иммуноглобулинов, или
же, иногда — их N-концевые фрагменты.
Чаще представлены >--цепи, особенно
А,У1-подгруппы, и гораздо реже — х-цепи.
Таким образом, AL связан с В-клеточными
дискразиями. Типичный пример —
рассмотренная в разделе «Нарушения композиции
белков плазмы» и в т. III миеломная болезнь
(также известная как болезнь Рустицкого-Ка-
лера). Напомним, что это — моноклональная
злокачественная опухоль плазматических
клеток, которые и производят составные
части амилоида типа AL. Опухоль
первоначально развивается в костном мозге, позже
формируя множественные остеолитические
очаги в скелете. Параллельно амилоидозу
формируется моноклональная гаммапа-
тия с диспротеинемией и патологическим
М-пиком на электрофореграмме сыворотки.
Клон при миеломной болезни может
производить не только полные молекулы
иммуноглобулинов, но также и изолированные
легкие цепи, появляющиеся в сыворотке и
моче в более чем 70 % случаев и известные,
по фамилии первооткрывателя, как белки
Бене-Джонса (ББД). До 15 % пациентов при
миеломной болезни развивают амилоидоз,
все они Б БД-положительны (см. с. 100).
Существуют прямые доказательства
идентичности пептидов ББД и фибриллярного
компонента AL. Можно in vitro создать
фибриллярный осадок, который имеет типичную
ультраструктуру волокнистого компонента
амилоида, путём ограниченного протеолиза
естественных ББД (Дж.Дж. Гленнер, 1980).
Помимо миеломной болезни, В-клеточные
лимфомы, в том числе сопровождаемые мак-
роглобулинемией Вальденстрёма и
болезнью тяжёлых цепей, равно как и солитарные
плазмацитомы — также могут быть ослож-
118 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
нены иммунодискразией, моноклональнои
гаммапатией и амилоидозом типа AL.
Известно, что некоторые стимулы
(возможно, мутагены) вызывают экспрессию
онкогенов в В-клетках и их безудержную
клональную пролиферацию или утрату ими
способности к апоптозу. Одним из
последствий персистирования такого клона
является чрезмерный синтез растворимых
амилоидных белков-предшественников (лёгких
цепей иммуноглобулинов). Подмечено, что
чем злокачественнее сам клон, тем меньше
синтез амилоида и, наоборот — миеломы и
плазмацитомы, растущие не быстро и
имеющие, в силу малой злокачественности, сами
по себе, субклиническое течение —
производят много амилоида, и амилоидоз при них
выглядит как идиопатическое первичное
заболевание, а протекает тяжело.
Неизвестно, однако, почему не у всех,
а лишь у сравнительно небольшой части
индивидов с гиперпродукцией свободных
легких цепей, формируется амилоидоз?
Очевидно, дело здесь в особенностях
групповой и индивидуальной реактивности.
Существует концепция различного ами-
лоидогенного потенциала легких цепей,
произведенных и обработанных
различными индивидами.
Она гласит, что некоторые минимальные
различия легких цепей, полученных от
различных В-клонов (может быть, в результате
нелетальных соматических мутаций), или
же некоторые особенности последующего
катаболизма легких цепей (возможно, их
неодинаковый процессинг в макрофагах) —
способны обусловить самосборку типовых
«Р-складчатых листоподобных структур»
и производство амилоидных депозитов.
Большое значение может иметь
специфичность эпитопов производимых дискрасти-
ческих иммуноглобулинов. По-видимому,
их аутоиммунная направленность может
способствовать их отложению в тканях. Не
исключено, что аутоантитела, способные
связывать ингибиторы протеаз или сами
являющиеся такими ингибиторами, могли
бы внести вклад в формирование амилоида,
резистентного к протеолизу.
Так или иначе, амилоидоз, вызванный
амилоидом типа AL, произведенным В-клет-
ками, называется первичным. При этом мие-
лома или лимфома часто протекает латентно.
Однако, детальное исследование может
выявлять лимфоцитоз, умеренное
увеличение числа плазматических клеток в костном
мозге, моноклональные иммуноглобулины
и/или их легкие цепи в сыворотке крови и в
моче, хотя и без явных признаков опухоли.
Итак, первичный амилоидоз —
очевидно, иммунопатологическое расстройство,
потому что он всегда зависит от нарушения
функций и пролиферации иммуноглобули
н-синтезирующих клеток.
В странах Северной Америки именно
первичный AL-амилоидоз встречается
наиболее часто.
Другой тип амилоидоза по главному
фибриллярному компоненту, обозначается
АА (amyloid-associated). А А является
уникальным белком неиммуноглобулиновой
природы (8,5 кД молекулярной массы),
предшественник которого (18кД)
синтезируется в печени, по-видимому, в основном,
макрофагами (А.С. Кохен, 1994).
Белок АА не имеет структурной
гомологии с иммуноглобулинами или какими-либо
известными белками. У него 76
аминокислотных остатков. Амилоидный
предшественник в сыворотке обозначается SAA
(serum associated amyloid). Этот белок
циркулирует в составе а{-глобулинов, в комплексе
с ЛПВП третьего подкласса. SAA имеет 3
формы (SAAt_3) и принадлежит к
положительным глобулинам острофазного ответа
(т. I, гл. 13). В амилоидогенезе участвуют
продукты неполного протеолиза SAA,
создаваемые макрофагами, которые захватывают
этот дериваты этого белка в составе
иммунных комплексов. Таким образом, как и для
AL-амилоида, для АА- амилоида типична
связь с длительной активацией иммунной
системы и необходима обработка и сборка
их компонентов в активных макрофагах.
АА белок характерен для вторичного
амилоидоза. У большинства больных имеется
очевидная клиническая связь с первичным
Патофизиология белкового обмена 119
хроническим воспалением инфекционного
или иммунопатологического
происхождения. В современных классификациях это
называется также реактивным амилоидозом.
Было время, когда туберкулёз, бронхо-
эктатическая болезнь, хронические пие-
лонефирит и остеомиелит, лепра и другие
инфекции — служили главной основой
вторичного амилоидоза. Но, с внедрением
эффективной антибактериальной
химиотерапии, доля подобных причин сократилась.
В настоящее время, вторичный амилоидоз
осложняет системные иммунокомплексные
и другие системные иммунопатологические
болезни, например, ревматоидный артрит,
дерматомиозит, склеродермию, а также
воспалительные аутоиммунные болезни
ЖКТ, особенно, неспецифический
язвенный колит. Хронический тропический
инфекционный колит также может обусловить
продукцию амилоида типа АА. Хронические
инфекции кожи, связанные с внутрикож-
ным и подкожным введением лекарств,
особенно, наркотиков (например, героина),
по всей вероятности, ответственны за очень
высокую частоту вторичного амилоидоза у
наркоманов.
Реактивный системный амилоидоз может
также возникать в связи с неиммуноцитар-
ными неопластическими процессами. Два
наиболее типичных примера — рак почки и
лимфогранулематоз.
Среди всех этих причин вторичного
амилоидоза, наиболее эпидемиологически
значимым во многих странах (США, Канаде,
Польше, России) является ревматоидный
артрит.
При всех вышеназванных болезнях
сывороточный уровень SAA, а иногда — и
продукция АА хронически и очень сильно
увеличены.
Следует отметить, что SAA — белок
острой фазы. В пределах 24 часов после начала
острого воспаления происходит увеличение
его сывороточной концентрации в 1 000
раз. Это увеличение вызвано действием
интерлейкинов (ИЛ), в частности — ИЛ-1
(и также ИЛ-6), производимых
макрофагами. Данные цитокины стимулируют синтез
SAA в печени. При хронической деструкции
ткани и затяжном воспалении уровни SAA
длительно остаются высокими, возможно,
вследствие продлённой активации
макрофагов (В. Кумар и соавт., 1997).
Однако, увеличенное производство SAA,
само по себе, еще не достаточно для
амилоидного перерождения. Амилоидоз не
является обязательным последствием любого
хронического воспаления. Считается, что
SAA обычно разлагается до растворимых
продуктов под действием макрофагальных
и моноцитарных серинэстераз.
У лиц, наклонных к амилоидозу, этот
процесс тормозится, и из-за наследственных
особенностей метаболизма SAA,
появляются его нерастворимые формы. Точный
характер подобного дефекта (или дефектов)
доселе неизвестен.
В любом случае, справедливо будет
отметить, что вторичный амилоидоз осложняет
продлённый или избыточный ответ острой
фазы. Иными словами, и эта форма
амилоидоза тесно связана с функциями иммунной
системы, а именно, с преиммунным ответом
(см. т. I, гл. 13).
АА-компонент амилоида депонируется
также при важнейшем из наследственных
вариантов вторичного амилоидоза,
сопровождая семейную средиземноморскую
лихорадку. При этой аутосомно-рецессивной
болезни, распространенной среди лиц
армянского, турецкого и арабского
происхождения, а особенно — среди евреев-сефардов,
отмечается мутация белка пирина (маренос-
трина)} имеются периодические приступы
лихорадки, лейкоцитоз, ускорение СОЭ,
полисерозит и боли в суставах, эритема.
Симптомокомплекс вызван освобождением
цитокинов и сопровождается острофазным
ответом. При этом нарушении описаны
также дефицит ингибиторов анафилотоксинов,
нарушения обмена лейкотриенов и
снижение супрессорной активности лимфоцитов.
АА амилоид накапливается в интиме-медии
артериальных и венозных сосудов, а также
в селезенке, лимфоузлах, надпочечниках,
120 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
интерстиции легких и в почках таких
пациентов. Заболевание эпизодически
встречается и у жителей других регионов. Однако,
к вторичному амилоидозу, как результату
данного расстройства, особенно наклонны
именно жители Восточного
Средиземноморья. Интересно, что амилидоз типа АА
зафиксирован и у родственников этих
больных, которые никогда не страдали явными
приступами первичной болезни. Ингибитор
клеточного деления и некоторых функций
цитоскелета колхицин оказался эффективен
при ряде форм амилоидоза, в том числе, в
особенности — при семейной
средиземноморской лихорадке. Это позволяет ставить
вопрос о связи патогенеза вторичного
амилоидоза с функциями клеточного
цитоскелета (Дж.Н. Бюксбаум, 1987).
Кроме АА и AL компонентов, описанных
выше, несколько других биохимически
отличных белков были найдены в амилоидных
депозитах при различных, относительно
более редких клинических формах болезни:
>* Транстиретин (преальбумин) —
нормальный сывороточный белок, который
связывает и транспортирует тироксин и ретинол
(см. выше табл. 5). Мутантная форма
транстиретина (А^,.) депонируется при
некоторых наследственных (именуемых
AF — amyloid familiar) формах
амилоидоза— семейной амилоидной нейропатии
и семейной амилоидной миокардиодист-
рофии. В отличие от средиземноморской
семейной лихорадки это — аутосомно-до-
минантныерастройства. Нейропатические
формы описаны в различных популяциях
(чаще всего в таких странах, как
Португалия, Япония, США, Израиль, Швеция,
Финляндия) и проявляются полинейропа-
тией вегетативных и соматических нервов.
Форма с поражением сердечно-сосудистой
системы описана в Дании и ведёт к
сердечной недостаточности.
В каждом из этих случаев транстиретин,
депонированный в тканях, отличается от его
нормального аналога лишь единственной
аминокислотой. Вовлекается, наиболее
часто, замена метионина на валин в
положении 30.
Другая мутантная форма транстиретина
ответственна за накопление амилоида при
подтипе амилоидоза, связанного со
старением (старческом сердечном амилоидозе,
или AScl — amyloid senile cardiac 1st subtype).
Существуют и иные разновидности сениль-
ного амилоидоза (панкреатическая,
мозговая — см. ниже), но при них нет накопления
транстиретина. Транстиретиновый
фибриллярный компонент амилоида (ATTR),
в отличие от других, может образовываться
внеклеточно, без обязательного
конструирующего участия фагоцитов.
Наследственным является и
исландский сосудистый амилоидоз, описанный у
пациентов с этого приполярного острова.
Особенностью механизма данной формы
семейного амилоидоза служит накопление
уникального фибриллярного
компонента — AFu
, который представляет собой
ингибитор лизосомальных пептид-гидролаз
цистатин. Это — единственный вид
амилоидоза, при котором расшифрован механизм,
обеспечивающий уникальную
резистентность амилоида к перевариванию.
>• Белок р2-микроглобулин, составная часть
антигенов ГКГ класса I и нормальной
сыворотки, недавно был идентифицирован
как фибриллярный компонент амилоида,
депонированного в тканях пациентов,
подвергавшихся длительному или
многократному гемодиализу. В амилоиде
данный глобулин присутствует в виде
мономеров и димеров (Ар2т). При
хронической почечной недостаточности
сывороточные концентрации этого белка
всегда увеличены. Предполагается, что
даный протеин не отфильтровывается
должным образом при процедуре
гемодиализа. В некоторых выборках до 70 %
пациентов на продлённом диализе
развивали амилоидные депозиты в
синовиальной оболочке, суставах и сухожильных
влагалищах.
>- р2 — белок амилоида (ASp2), пептид
молекулярной массой 4 кД (также
называемый А4) составляет ядро бляшек,
найденных в мозге при болезни Альцхай-
мера, а также присутствует в амилоиде,
депонированном в стенках мозговых
кровеносных сосудов при этом крайне
распространенном гериатрическом
заболевании.
Вызываемый накоплением данного белка
амилоидоз иногда именуют «старческим
мозговым». Сенильный мозговой амилоидоз
некоторые авторы (Д.Я. Гольдгабер, 2003)
этиологически связывают с прионовой
инфекцией (см. т. I, стр. 61,439-440). Амилоид
обнаружен и при других болезнях, прионо-
вая этиология которых несомненна (скрепи
у овец, синдром Крейтцфельда — Якоба у
людей). Но амилоид при этих прионопатиях
не содержит компонента А4 (P.O. Мессинг,
1997).
А4 белок, как полагают, получается путём
вычленения 28 аминокислот из намного
большего по массе (40 кД)
предшественника— протеина АРР, который имеет
характеристики интегрального мембранного
гликопротеида, представлен в крови и
цереброспинальной жидкости и кодируется в
21-й хромосоме.
Амилоидоз может быть местным,
когда депозиты ограничены единственным
органом (очагом). С такими локальными
узловыми депозитами амилоида наиболее
часто приходится сталкиваться в легком,
гортани, коже, мочевом пузыре, языке и
периорбитальной области. Как необычный
случай, описан местный амилоидоз
мужского полового члена. Только местные формы
амилоидоза обратимы, т.е. дают амилоидо-
клазию.
Тип амилоидного перерождения, также
как химическая природа амилоида, при
местных формах могут варьировать. Иногда
это — узел, окруженный плазматическими
клетками, и амилоид содержит тип AL,
подобно первичному системному амилоидозу
(Р. Котран и соавт., 1994).
Разнообразие форм амилоидоза
дополняется местными эндокринными
разновидностями. Эти амилоидные депозиты
происходят в результате накопления му-
тантных форм прогормонов диффузных
эндокриноцитов: прокальцитонина — в
Патофизиология белкового обмена 121
медуллярных карциномах щитовидной
железы (AEt), проинсулина— в инсулиномах
железы поджелудочной (АЕр).
При местном кожном амилоидозе,
наблюдаемом в лихеноидных поражениях
кожи, имеется тип AD амилоида. Его
фибриллярный компонент имеет неизвестное
(возможно, кератиновое) происхождение.
Так как дерматологически кожный амилоид
выглядит по-разному, условно выделяют
его макулярную (ADm), папулёзную (ADp)
и нодулярную (ADn) форму.
>■ Нефибриллярный Р-компонент
амилоида отличается от фибриллярного,
является универсальным для всех
химических подтипов амилоида и тесно
связан с амилоидными фибриллами.
Обнаружена идентичность Р-компонента
и сывороточного сц-гликопротеида.
Данный оц-глобулин имеет молекулярную
массу 180-220 кД и высокогомологичен
С-реактивному белку (см. выше табл. 5).
По-видимому, как и его гомолог, он
является острофазным белком и его синтез
усиливается при преиммунном ответе.
Р-компонент, будучи гликопротеидом,
обеспечивает положительную реакцию
амилоида с перодной кислотой и реактивом Шиффа
(PAS-реакцию), что долго маскировало
неуглеводную природу амилоида.
Как уже указывалось, амилоидоз может
протекать субклинически.
Основные клинические проявления
зависят от локализации и размера депозитов,
но, как правило, включают увеличение того
или иного органа (гепатомегалия, спле-
номегалия, кардиомегалия, макроглоссия
и т.д.). Амилоидно измененные органы
больше подвержены механическим
травмам, например сосуды при амилоидной
вазопатии часто кровоточат (И.В.
Давыдовский, 1969).
Типичным проявлением амилоидоза
почек служит протеинурия, а исходом
является хроническая почечная недостаточность.
Амилоидоз сердца ведёт к суправентрику-
лярным аритмиям, блокадам, застойной
сердечной недостаточности. Амилоидное
перерождение органов ЖКТ может вызы-
122 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 8. Классификация амилоидоза
Амилоидный синдром
Первичный амилоидоз
Вторичный амилоидоз
Гемодиализный
амилоидоз
Старческий амилоидоз:
Сердечный
Церебральный
Панкреатический
Наследственный
амилоидоз:
Нейропатический
Миокардиодистрофичес-
кий
Нефропатический
Сосудистый
Местный эндокринный
В медуллярной
карциноме щитовидной железы
В опухолях клеток
островков Лангерганса
Местный кожный
Папулёзный
Макулярный
Нодулярный
Предшественник
фибриллярного
компонента амилоида
Иммуноглобулины и их
цепи
SAA
р2-микроглобулин
Тип и подтип
фибриллярного компонента
амилоида
AL
АА
Ар2,„
Примечание
При моноклинальных
иммунодискразиях
При усиленном преиммун-
ном и иммунном ответе
ПриХПН
Транстиретин
АРР
Проинсулин
Транстиретин
Транстиретин
SAA
Цистатин
ASC1
ASP(A4)
ASP
AF
AFDa
AA
AFHGHWa
Вовлекает и лёгкие
При б-ни Альцхаймера
Может обусловить
вторичный ИЗСД
АД. Типичное начало в
20-50 лет, чаще всего
регистрируется у
португальцев
АД. Начало — после 10
лет. Регистрируется чаще
у датчан
При семейной
средиземноморской
периодической лихорадке (АР)
«Исландский». Вызывает
кровоизлияния в мозг
Прокальцитонин
Проинсулин, амилин
AEt
AEP
Вызывает вторичные
эндокринные р-ва
Вызывает вторичные
эндокринные р-ва
(кератин?)
(кератин?)
(кератин?)
ADP
ADm
ADn
Сопровождается лихени-
фикацией кожи
Сопровождается лихени-
фикацией кожи
Сопровождается лихени-
фикацией кожи |
Патофизиология белкового обмена 123
вать гастроинтестинальные кровотечения,
мальдигестию и мальабсорбцию.
Амилоидная полинейропатия манифестирует
нарушениями проведения импульсов по
вегетативным и соматическим нервам. Как
диагностическая проба, ранее применялось
введение больным in vivo красителя конго-
рот, к которому амилоид обладает особым
сродством. В настоящее время лабораторная
диагностика амилоидного перерождения
основывается на иммуногистохимическом
исследовании биоптатов.
Таким образом, амилоидоз — диспротеи-
ноз, характеризующийся накоплением в
тканях кристаллоидного белкового комплекса,
устойчивого к протеолизу. Он развивается
в тесной связи с нарушением иммунного и
преиммунного ответа.
В заключение приводим таблицу 8,
характеризующую клинико-химическую
классификацию амилоидоза (по Дж.Н. Бюксбау-
му с изменениями).
Термин «гиалиноз, гиалиновый» — по
всей вероятности, один из самых часто
употребляемых в патологии.
Гиалинозом (от yvaXxvoq —
стекловидный) в патологии, по традиции, условно
именуется накопление в соединительной
ткани (включая строму органов и стенки
сосудов) любого неамилоидного белкового,
полупрозрачного гомогенного, плотного,
эозинофильного материала. Таким образом,
в отличие от амилоидоза, гиалиноз лишен
физико-химической специфичности.
Старые авторы считали гиалиноз предстадией
амилоидоза (А.А.Максимов, 1896; Е.Ще-
пилевский, 1899). Наделе речь просто идёт о
том, что одни и те же иммунопатологические
процессы могут способствовать
депонированию и амилоидного, и неамилоидного
белка.
Следовательно, к гиалинозу может
привести видоизменение многих разных белков.
Основные механизмы его образования —
плазматическое пропитывание (например,
при диабетической ангиопатии и
гипертоническом артериосклерозе), а также местная
денатурация и коагуляция белков при
воспалении и иммунопатологических процессах с
неорганоспецифическими аутоантителами (в
рубцах, фиброзных швартах, при
организации тромбов и сгустков свернувшейся крови,
после фибриноидного набухания).
В первом случае гиалин содержит белки
плазмы крови и иммунные комплексы. Во
втором также не исключено их наличие, но
основными составляющими служат
фибрин, видоизмененные волокнистые
соединительнотканные белки и гликопротеиды
аморфного вещества. Гиалиновые массы не
обладают тинкториальными и иммунохими-
ческими свойствами амилоида (см. выше)
и дифференцируются от него. Наибольшее
клиническое значение имеет гиалиноз
мелких артерий и артериол, участвующий в
развитии диабетических ангиопатии (см. ниже)
и поздних стадий гипертонической болезни.
Гиалинизация пропотевающих через
сосудистую стенку белков и откладываемых в
ней иммунных комплексов сопровождается
не только утолщением, но и
мультипликацией базальных мембран. Гиалиновый
артериосклероз сосудов клубочков почек является
характерной находкой при синдроме Ким-
мелыитиля-Уилсона (см. «Патофизиология
сахарного диабета»). Внеклеточный гиалин
легко может подвергаться кальцификации.
Классическая патологическая анатомия
подчёркивает, что внутриклеточный гиалин
при гиалиново-капельной дистрофии и
внеклеточный гиалин не имеют общности,
кроме внешнего вида и созвучия
традиционных названий (В.В. Серов).
Нам, однако, кажется что в связи с малой
определённостью самого понятия
«внеклеточный гиалин» рассмотрение в данном
разделе процессов, приводящих к появлению
в клетках обильных включений, имеющих
гиалиновый вид, не будет серьёзным
нарушением стиля и традиций.
В I томе уже рассматривались тельца
Меллори — гиалиновые включения в гепа-
тоцитах при алкогольной гепатопатии и
некоторых других токсических поражениях
печени. Механизм их образования связан с
агрегацией белка прекератина, входящего в
124 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рис. 20. Роговая дистрофия кожи у девушки 20
лет (по В.В. Подвысоцкому, 1905)
состав промежуточных филаментов
клеточного скелета (т. I, с. 147,161-162).
При интенсивном синтезе
иммуноглобулинов в плазматических клетках
обнаруживаются гиалиновые тельца Русселя
(агрегаты фрагментов антител).
При повышенной проницаемости
почечного фильтра, интенсивная реабсорбция
белка из первичной мочи ведет к
формированию гиалиновых капель в клетках
эпителия канальцев.
Вирусные белки при инфекциях (эн-
теровирусном везикулярном стоматите,
цитомегаловирусной инфекции и др.)
могут формировать характерные гиалиновые
капли в различных клетках.
Таким образом, и внутриклеточный
гиалин может иметь совершенно разное
происхождение.
Роговая дистрофия заключается в
избыточном образовании кератина при
ускорении клеточого цикла и апоптотической
гибели кератиноцитов эпидермиса. Это может
приводить к гиперкератозу (гиперплазии и
утолщению stratum corneum) и дискератозу
(образование кератина в любых клетках
кожи ниже stratum granulosum) и
наблюдается, в частности, при наследственном
ихтиозе. Роговой дистрофии способствует
гиповитаминоз А (см. ниже
«Патофизиология витаминного обмена»).
Гиперкератоз характерен для
доброкачественных опухолей из базальных клеток
эпидермиса (себорейного гиперкератоза, ке-
ратоакантомы и, особенно, для предракового
состояния — актинокератоза). В.В.Подвы-
соцкий (1905) приводит интереснейшее
уникальное наблюдение 20-летней пациентки с
редкой формой рогового перерождения кожи,
которое мы воспроизводим здесь (рис. 20).
Гены кератина имеются во всех клетках.
В связи с этим, возможна метаплазия неоро-
говевающего эпителия — в ороговевающий
и появление рогового вещества там, где оно
в норме не встречается. Примерами могут
служить лейкоплакия слизистых оболочек
и образование «раковых жемчужин» в
карциномах. При кератинизации слизистых
типичен паракератоз (гиперплазия
кератиноцитов с сохранением в них ядер).
Глава 6
ПАТОФИЗИОЛОГИЯ
НУКЛЕИНОВОГО ОБМЕНА
«Генетическая информация не записана на медных скрижалях и не
выбита на камне, а хранится в форме молекулы ДНК, столь хрупкой, что при
выделении её из живой клетки и перенесении в раствор она легко
распадается... если раствор просто взболтать или засосать в пипетку».
А. Ленинджер, 1978
• Информационные и патохимические аспекты нуклеинового метаболизма
• Пути обмена пуринов
• Патология обмена пуринов
• Этиология подагры
• Патогенез подагры
• Другие нарушения пуринового обмена
• Нарушения обмена пиримидиновых оснований
• Кристаллические артриты
В данном разделе рассматриваются
нарушения обмена нуклеиновых кислот как
метаболитов. Основное внимание уделено
патохимии метаболизма пуринов и пири-
мидинов, главных составных частей ДНК
и РНК.
ИНФОРМАЦИОННЫЕ
И ПАТОХИМИЧЕСКИЕ
АСПЕКТЫ НУКЛЕИНОВОГО
МЕТАБОЛИЗМА
Нуклеиновые кислоты —
информационные биополимеры, кодирующие
индивидуальный набор генетических программ.
Поэтому еще в разделах, посвященных роли
наследственности в определении
реактивности организма и значению повреждений
программного аппарата клетки в патологии,
мы косвенным образом касались нарушений
их обмена (см. т. I, стр. 123-152). В данном
томе нуклеиновые кислоты
рассматриваются как химические вещества,
участники метаболизма — то есть, прежде всего,
со структурно-энергетических позиций.
С формальной точки зрения, практически,
любые болезни, вызванные мутациями, надо
считать нарушениями обмена нуклеиновых
кислот. Но традиционно под нарушениями
нуклеинового метаболизма понимают
расстройства синтеза и распада пуриновых и
пиримидиновых нуклеотидов.
Среди пуриновых нарушений самые
распространенные и практически
значимые — гиперурикемия, поражающая от 2
до 18% населения в разных популяциях,
а также результат крайне выраженного
гиперурикемического синдрома — подагра,
имеющая частоту в разных регионах мира
от 0,13%, до почти 10%. Подагра поражает
каждого двадцатого пациента с болезнями
суставов и лежит в основе 5-8% всех
случаев мочекаменной болезни.
Некоторые нарушения обмена
пиримидиновых нуклеотидов не менее
распространены — например, р-аминоизобутиратурия
наблюдается у 10% европеоидов и,
практически, у каждого представителя монголоид-
126 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Гон1
N^?4icHa
цитозин
пуриновыи
остаток
N^
I
урацил
пиримидиновый
остаток
^. ^ N-гликозид
НОСН2
1\н н/
н\^/н
НО 4R
V" R
J
носн2
$
конфигурация
ЮН»
0У
\н н/
н\^/н
НО 1R
^-конфигурация
Рис.21.Структура нуклеотидов
ной расы. К счастью, эта мутация по своим
последствиям нейтральна.
В связи с большей клинической
значимостью расстройств пуринового обмена они
обсуждаются первыми.
ПУТИ ОБМЕНА ПУРИНОВ
Нуклеиновые кислоты организма
наполовину состоят из пуриновых нуклеотидов,
каждый из которых, помимо пентозы и
фосфорной кислоты, содержит пуриновое
азотистое основание. Пуриновые
основания — производные пурина. Пурин —
ароматическое гетероциклическое соединение,
образуемое из двух колец — имидазола и
пиримидина. Последний также
представляет собой ароматический гетероцикл в виде
шестичленного азот-углеродного кольца с
тремя двойными связями. Имидазол,
в свою очередь, пятичленное
азот-углеродное кольцо (рис.21).
В составе нуклеотидов
встречается два главных пуриновых
основания — аденин (А) и гуанин (Г).
Изредка в нуклеиновых кислотах
обнаруживаются минорные
метилированные пурины — 2-метил аденин
и 1-метилгуанин. По некоторым
данным, усиленное метилирование
генов препятствует их активной
экспрессии.
N-гликозиды пуриновых
оснований, лишенные фосфорной кислоты,
называются нуклеозидами (аденозин,
гуанозин). Таким образом, нуклео-
тиды — их фосфорнокислые эфиры.
Дифосфатные эфиры аденозина и
гуанозина известны как нуклеозид-
пирофосфаты (АДФ и ГДФ).
Наконец, трифосфат аденозина— это
знакомая каждому
студенту-медику со школьной скамьи адено-
зинтрифосфорная кислота (АТФ).
Трифосфатом гуанозина является
гуанозинтрифосфорная кислота
(ГТФ). Таким образом, роль пуринов
не исчерпывается их пластическими
функциями в составе РНК и ДНК.
Главные участники энергетических
процессов в клетке, связующее звено между
реакциями, протекающими с выделением и
с потреблением энергии — это именно три-
фосфорибонуклеозиды (см. выше).
Механизм биосинтеза пуриновых
нуклеотидов таков, что азотные атомы для их колец
происходят из амидных групп глутамина
(N-3, N-9) и аспарагина (N-1), а также из
глицина (N-9). Углеродные компоненты
колец берутся из глицина (С-4, С-5), из
формиата (С-2, С-8), а также из углекислого
газа (С-6). Вначале образуется на основе
фосфорибозил-пирофосфата
ациклическая форма нуклеотида с открытой цепью.
Только затем кольцо замыкается в
пуриновыи цикл. Скорость биосинтеза пуринов
регулируется по типу обратной связи
количеством конечных продуктов. Кроме того,
Патофизиология нуклеинового обмена 12 7
избыток АТФ способствует
синтезу гуаниловой, а избыток
ГТФ — адениловой кислоты.
Накопление ГМФ подавляет
продукцию АМФ (рис.22).
При распаде ДНК и РНК
образуются мононуклеотиды,
которые могут реутилизиро-
ваться для их нового синтеза.
Дальнейший ферментативный
гидролиз мононуклеотидов
даёт свободные азотистые
основания. Затем они
распадаются в соответствии с общей
последовательностью,
показанной на рис. 23. Разные
животные доводят распад
пуринов до различных конечных
стадий.
У беспозвоночных — это
аммиак, у пластинчатожаберных
моллюсков и рыб — мочевина
и, реже, аллантоиновая
кислота, у бронхоногих
моллюсков, черепах и
млекопитающих — аллантоин, наконец, у
человека, приматов, птиц,
ящериц и змей, а также у... героя
знаменитого мультфильма —
дога-далматинца — конечным
продуктом пуринового обмена
служит мочевая кислота.
Интересно, что такие разные живые
существа, как пауки и свиньи,
имея недостаточно гуаниндеза
рибоза-5*-фосфат
АТФ
~| ФРПФ |
глутамин
/V
£ /
/
4Г '
фосфорибозил
амин
I
1
Т
|
▼
| САИКАР |
S
ч s
\
1
» и
1
/
7
ингибирувание
\
2,8-дигидрокси-
аденин
Рис. 22. Пути пуринового обмена.
Сокращения: ФРПФ — фосфорибозилпирофосфат; САИКАР — сукцинил-амино-
миназы, экскретируют пурины имидазолкарбоксиамид-риботид; АИКАР — аминоимидазолкарбоксиамид-ри-
ботид; ФЕРМЕНТЫ: 1 —ФРПФ-синтетаза; 2 — амидофосфорибозилтрансфераза;
3 — аденилосукцинат-лиаза; 4 — АМФ-дезаминаза; 5 — 5'-нуклеотидаза;
6—аденозиндезаминаза, 7 — пуриннуклеозидфосфорилаза; 8 — гипоксан-
тин-фосфорибозилтрансфераза (ГФРТ); 9 — аденин-фосфорибозилтрансфераза;
10 — ксантиноксидаза.
Человек выводит около 1,5 г мочевой кис-
в виде гуанина.
С банально-энергетической,
утилитарной точки зрения
непонятно, почему эволюционно
более сложные животные все
более и более недоокисляют
пурины, теряя их во все более сложной форме?
Вероятно, за этим кроются какие-то
этапные эволюционные изменения в
метаболизме этих живых существ, и, по крайней мере,
мочевая кислота нужна им для каких-то
неэнергетических целей (см. также т. I,
стр. 108-109).
лоты в день, причем не менее 60 %
происходит из эндогенных пуринов, остальное — из
пуринов пищи. У 95 % мужского населения
концентрация мочевой кислоты в плазме
лежит между 2,2 и 7,5мг/дл, у женщин до
менопаузы эстрогены обеспечивают более
эффективную экскрецию уратов, поэтому
128 ALU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
статистическая норма содержания мочевой
кислоты у них ниже и находится между 2,1
и 6,6 мг/дл. У детей обоего пола уровень
мочевой кислоты существенно ниже (около
3-4 мг/дл), кроме первой недели жизни,
когда он подвержен действию транзиторной
физиологической гиперурикемии
новорожденных (см. т.1, с. 108). У подростков
среднее содержание уратов растет (до 5,1 мг/дл
у юношей и 4,1 — у девушек).
Нормальная судьба мочевой кислоты в
организме характеризуется следующими
данными. Хотя пурины синтезируются
и деградируют в каждой клетке, мочевая
кислота может быть образована только под
действием фермента ксантиноксидазы. У
человека этот энзим имеется лишь в печени
и энтероцитах, где и идет переработка ксан-
тина в мочевую кислоту.
Мочевая кислота — слабокислый
продукт, поэтому она, секретируясь в кровь,
образует соли-ураты — со щелочными
катионами (на 98 % — с натрием), которые
связываются оцаз-глобулинами плазмы
только на 5 % и выводятся на 2/3
почками, а на 1/3 — через тонкий кишечник. В
кишечнике бактерии разрушают кислоту,
последовательно, с помощью ферментов
уриказы,аллантоиназы,аллантоиказы и,
наконец, уреазы — до аммиака и глиок-
салевой кислоты. При гиперурикемии и
нарушениях почечной экскреции уратов
этот путь выведения конечных продуктов
пуринового и азотистого метаболизма
усиливается. Считается, что усиленное
кишечное выведение и бактериальное
превращение мочевой кислоты и мочевины
имеет отношение к возникновению
язвенных поражений ЖКТ при уремии.
Плазма крови при 37 °С насыщается
уратом натрия уже при концентрации
6,8 мг/дл, то есть верхняя граница
нормального содержания уратов в плазме у
мужчин находится, примерно, на уровне
насыщения. Однако, ураты в крови не
кристаллизуются даже при превышении
этого порога в 8-10 раз, оставаясь в
состоянии супернасыщенного раствора,
благодаря действию каких-то неидентифи-
цированных солюбилизаторов. Подобные
растворы, при действии каких-то
факторов, меняющих их свойства, могут легко
давать кристаллизацию. Вместе с тем, в
тканях и моче ураты кристаллизуются
и при меньших концентрациях. Г. Шаде
считал, что большое значение имеет
изомерная форма урата натрия, так как его
лактимы растворяются труднее лактамов.
По всеобщему признанию, наибольшее
сродство к уратам испытывают хрящевая
ткань и интерстиций почек.
В экскреции уратов почками участвуют
все три основные парциальные функции.
После фильтрации 98 % уратов реабсор-
бируется, половина этого количества
повторно секретируется в первичную
мочу и 40-44 % секретированных уратов
реабсорбируется вновь. Таким образом,
8-12% первоначально отфильтрованного
количества уратов выводится с
дефинитивной мочой. В моче кристаллизации
уратов способствует кислый рН. При рН=5
в моче можно растворить от 6 до 15 мг/дл
уратов без кристаллизации, подщелачива-
ние мочи с кристаллами уратов вызывает
их исчезновение, что используется как
экспресс-тест, но уже при рН=7 насыщение
мочи требует более чем 158 мг/дл уратов.
Именно поэтому все виды ацидоза, в том
числе — кетоацидоз и лактат-ацидоз
(например, наступающий при алкоголизации,
осложняющий сахарный диабет, а также
сопровождающий физическое утомление),
способствуют обострениям подагры.
Ураты легче выпадают в осадок при
температурах ниже 37 °С. При 32 °С
(температура коленного сустава) растворимость уратов
уменьшается на треть, при 29°С
(температура голеностопа) — наполовину. Наиболее
«холодным» суставом организма является
первый предплюснофаланговый сустав
стопы. Не случайно, именно он
поражается у 80 % больных подагрой, обострениям
которой способствует, в частности, и
переохлаждение. Описаны также подагрические
шишки в хрящах других подверженных
переохлаждению органов — ушных раковин
(рис. 25).
Патофизиология нуклеинового обмена 129
1
птицы
приматы
^ мочевая
кислота
\
млекопитающие
моллюски
рептилии
^ w аллантоиновая w
кислота
беспозвоночные
гуанин
пауки
\Ш\ н2о
рыбы
Ш\ н2о он [од он [од он
Н NH3 н н мочеваяН
аденин гипоксантин ксантин кислота
мочеваяН
кислота
(енольная форма)
О
H2N
н н
аллантоиновая
COOHI
ВТ г*
о^ V н
H2N ^c-i—N
— I I I
o^vN^4N/c^(
H H
аллантоин
Oo "
/ HlST^^l N
« / I JI
to X (AjAnA
T н н
кислота
глиокса-
левая
кислота
~А
мочевая
кислота
(кетоформа)
NH2
2С=0 -
I
NH2
мочевина
-► 2NH3+2C02
Рис. 23. Пути деградации пуринов и конечные продукты их обмена у животных
НАРУШЕНИЯ
ПУРИНОВОГО ОБМЕНА
Описание нарушений пуринового обмена
постоянно требует отсылок к рис. 22, с. 127
а все сокращения наименований
субстратов и ферментов пуринового метаболизма,
равно как и цифровые обозначения в тексте
данного раздела, соответствуют подписи к
этому рисунку.
Главный клинический синдром,
вызванный расстройством пуринового
обмена, — это подагра. Под подагрой понимают
гетерогенную группу нарушений
пуринового метаболизма, проявляющихся
выраженной гиперурикемией, приступами артрита
(как правило, моносуставного характера),
отложением кристаллов моногидрата мо-
чекислого натрия в тканях (подагрические
шишки — tophi unci), уратурической нефро-
патией и мочекаменной болезнью (рис. 25).
Подагра (среднемировая частота 1,3-
3,7%)— результат крайне выраженного
синдрома гиперурикемии (среднемировая
частота 2-18 %), то есть повышенного
накопления в крови мочевой кислоты. Ги-
перурикемическим считается уровень выше
420 мкМ/л или 7 мг/дл мочевой кислоты в
крови. Каждый миллиграмм мочевой кисло-
130 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
90 А
80 А
70 А
60
50 J
40 J
30 J
20 J
10 А
пораженность
мужчин
подагрой
средний возраст
58 лет
— — - средний возраст
49 лет
уровень
мочевой кислоты
в плазме, мг %
Рис 24. Пораженность подагрой у мужчин при
различном уровне мочевой кислоты в плазме и разном возрас
те (по Халлу-Залокару)
ты на децилитр плазмы выше этого уровня
существенно повышает пораженность
подагрой, особенно, у пожилых (рис. 24).
Подагра известна с античных времен.
Само ее название восходит к сочетанию
греческих слов «нога» и «жестокий». Ф. Рабле
оставил столь яркие описания проявлений
этого заболевания у героев «Гаргантюа и
Пантагрюэля», что создается впечатление
о более значительной распространенности
подагры в эпоху Ренессанса и барокко,
нежели сейчас. Первым ясно указал на
специфичность тканевого отложения урата
натрия при этом заболевании классик
британской медицины сэр Арчибальд Б. Гаррод
в своей статье 1859 г. «Истинная природа и
суть подагры». На основании данных
патологической анатомии он постулировал,
что ураты при подагре откладываются в
суставах и вокруг них в 100% случаев и
являются не свидетелями или
следствием, а патогенетической основой
процесса воспаления. Вскоре
была создана экспериментальная
модель подагры на добровольцах
и экспериментальных животных.
Артрит с характерной
симптоматикой воспроизводился микро-
иньекцией в сустав кристаллов
урата натрия (модель К. Фрей-
двейлера,1899). Развивая эти
исследования по моделированию
подагры, Д. Мак-Карти и Дж.
Холландер (1961) установили,
что подагрический синовиит
развивается у подопытных животных
при условии взаимодействия
лейкоцитов и любых кристаллов,
подобных уратам натрия по
физико-химическим свойствам
(обладающих полианионной
поверхностью). Например, урат натрия
(моногидрат) мог быть заменен
пирофосфатом кальция (дигидра-
том). Впоследствии было описано
заболевание человека, вызванное
отложением пирофосфата
кальция и артритом, — псевдоподагра
илихондрокальциноз (см. с. 142).
При хондрокальцинозе, как
правило, не поражается излюбленный подагрой
I предплюснофаланговый сустав, а артрит
носит полисуставной,
самоограничивающийся характер. Однако, само
взаимодействие лейкоцитов и медиаторов сторожевой
полисистемы с кристаллами служит
причиной воспаления в обоих случаях. У
домашних свиней найден спонтанный аналог
подагры — гуаниновая артропатия, механизмы
КОТ9Р0Й также включают взаимодействие
кристаллов пуринов и лейкоцитов.
Весьма интересны эпидемиологические
и медико-географические аспекты подагры
и гиперурикемии. Подагра — полигенное
заболевание с пороговым эффектом,
определяемым факторами, влияющими на
суточный кругооборот пуринов — диетой,
приемом алкоголя, физической
активностью, локальными геохимическими
особенностями. По крайней мере 2 из нескольких
Патофизиология нуклеинового обмена 131
аллелей, предрасполагающих
к гиперурикемии, находятся в
Х-хромосоме (гены гипоксан-
тинфосфорибозилтрансферазы,
ГФРТ — номер 8 на рис. 22; атак-
же фосфорибозилпирофосфат-
синтетазы, ФРПФ — номер 1 на
рис. 22). Кроме того, эстрогены
оказывают антиурикемическое
действие. Поэтому 95 % больных
подагрой — мужчины (рис.24).
Подагра — заболевание лиц
среднего и пожилого возраста, хотя
при наличии моногенных
дефектов пуринового обмена она
может формироваться ускоренно
и поражать молодых.
У коренного населения Новой
Зеландии, маори, наивысшая
частота подагры в мире — 10%,
а гиперурикемия присутствует
более чем у половины маори.
Поражённость подагрой сильно
повышена у аборигенов
Марианских островов (что на севере
Микронезии) и у филиппинцев,
проживающих в США, но не
среди филиппинцев самих
Филиппин, где, вероятно, сказывается
менее насыщенная пуринами
диета. В Европе частота подагры
исторически понижалась в
периоды войн и бедствий, возрастая в
благополучные эпохи
повышения жизненного уровня. В
Японии частота подагры неуклонно
нарастает в послевоенные годы,
по мере приближения диеты населения к
европейской.
Продукция мочевой кислоты в организме
повышается при активации молибденоза-
висимого фермента ФРПФ — синтетазы
(рис. 22,1). Интересно, что регионы с
молибденовыми аномалиями имеют сравнительно
высокую частоту гиперурикемии. В бывшем
СССР наивысшая частота подагры
отмечалась в Армении, как раз в тех регионах,
где повышено геохимическое содержание
молибдена. В США этим же отличаются
Рис 25. Этиология, патогенез и проявления подагры
(по Ф. Неттеру из CIBA Medical llustrations Collection).
Внизу слева — подагрический артрит I предплюснофалангового сустава,
справа — рентгенограмма той же области, в середине слева—уратная не-
фропатия, справа tophi urici суставов кисти; вверху слева—tophi unci в хряще
ушной раковины, справа—tophi urici в области локтевых суставов
некоторые районы Дальнего Запада (см.
ниже «Патофизиология обмена
микроэлементов»).
Заболеваемость подагрой выше у лиц с
нервно-артритическим диатезом,
проявления которого сами основаны на действии
гиперурикемии (т. I, с. 107-109). Подагра
и гиперурикемия чаще встречаются у лиц
с высоким социальным и образовательным
статусом и в течение нескольких столетий
считались «недугом аристократов». Разу-
132 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
меется, дело здесь не только в различиях
диеты, которая у обеспеченных слоев
населения содержит больше пуринов. Большое
значение имеет влияние самой мочевой
кислоты на уровень умственной активности
и интеллектуальную работоспособность
индивидов. Так как мочевая кислота (тригид-
роксиксантин) и кофеин (триметилксантин)
являются близкими структурными
аналогами (см. т. I, стр. 107), то гиперурикемия
оказывает определённый допинг-эффект*
на умственную деятельность. На обширном
статистическом эпидемиологическом
материале и в прямых исследованиях по
измерению коэффициента интеллекта и уровня
мочевой кислоты — показано, что между
гиперурикемией и повышенной умственной
активностью имеется явная связь.
Отечественный генетик В.П. Эфроимсон,
основываясь на архивных историях болезни и других
исторических документах, обнаружил среди
лиц, которых энциклопедии относят к гениям,
около 40 % носителей достоверных и высоко
вероятных признаков гиперурикемии (1984).
Подробнее вопрос о связи между
гиперурикемией и интеллектуальной активностью
рассмотрен в т. I (стр. 108-109).
В 80 % случаев подагра развивается как
первичное заболевание, связанное либо с
аддитивно-полигенными нарушениями, либо
реже — с моногенными ферментативными
дефектами. У остальных пациентов
подагрический синдром вторичен по отношению
к различным заболеваниям, повышающим
продукцию и понижающим экскрецию
уратов. С некоторыми расстройствами
первичная и вторичная подагра анамнестически
ассоциируется наиболее часто. Связи между
ней и сахарным диабетом, гипертензией,
атеросклерозом, ГЛП, ожирением, стеатозом
печени эпидемиологически столь сильны, а
патогенетически — столь многофакторны,
что некоторые авторы, начиная с Ж.-П.
Камю (1966), считают все эти нарушения
проявлениями единого конституционально
обусловленного синдрома — так
называемого метаболического Х-синдрома или, по
современным данным, системного дефекта
натрий-водородных трансмембранных
противопереносчиков (рис. 35, стр. 172), см.
также гл. 14.
этиология подагры
Этиология подагры, фактически, совпадает
с этиологией гиперурикемии. Последняя
полиэтиологична и, в основном, вызывается
тремя группами причин:
>* повышенным образованием мочевой
кислоты;
>* пониженным выведением мочевой
кислоты и уратов;
>• комбинацией первого и второго
механизмов.
Разграничение между разными
вариантами гиперурикемии не всегда возможно.
Однако, известной информативностью в этом
отношении обладает тест с введением
специальной беспуриновой диеты. Если пациент с
гиперурикемией отвечает на беспуриновую
диету выраженным (до 3,6мг/дл и ниже)
снижением уровня мочевой кислоты, то
гиперурикемия, скорее всего, была вызвана
гиперпродукцией этого метаболита, а если
нет — более вероятен вариант с понижением
экскреции уратов.
Повышенная продукция мочевой
кислоты бывает результатом действия различных
механизмов.
>- Диетическая перегрузка экзогенными
пуринами может вызывать гиперурикемию,
так как более 50% пуринов РНК и 20%
пуринов ДНК, употребленной в пищу,
оказывается в виде уратов в моче. Пу-
риновая нагрузка противопоказана при
гиперурикемии и, тем более — подагре.
Избыток пуринов содержат животные
продукты, особенно, икра, молоки (более
ЮОмг/100 г), печень (94 мг/100 г),
почки (80 мг/100 г) и другие внутренности
(50-80 мг/100 г), анчоусы, морские
моллюски, сваренные из этих продуктов
бульоны (16 мг/100 г). Немало пуринов
в рыбе, особенно консервированной
(от 46 мг/100 г в свежем судаке — до
83мг/100 г в шпротах и 118 мг/100 г — в
сардинах в масле). Меньше пуринов
содержит мясо (от 27 до 41 мг/100 г), еще
меньше их в яйцах. Темные сорта пива
и красное вино очень богаты пуринами,
присутствуют они и в других
алкогольных напитках за исключением водки.
Пурины имеются в кофе, какао,
шоколаде и чае, а также кофеиносодержащих
прохладительных напитках. Молоко и
сыры, крупы, фрукты и овощи бедны
пуринами. Из растительных продуктов на
первом месте бобовые (14-19 мг/100 г), а
особенно выделяется пуриновым
содержанием чечевица — 58 мг/100 г. В связи
со всеми этими особенностями, при
подагре и гиперурикемии назначается
полноценная молочно-растительная
ощелачивающая диета (в России — стол
№ 6 по М.И. Певзнеру).
> Важной причиной моногенной
наследственной формы первичной подагры
служит дефицит фермента гипоксан-
тин-фосфорибозилтрансферазы(ГФРТ,
см. №8 на рис. 22). Этот энзим в норме
реутилизирует избыток фосфорибозил-
пирофосфата (ФРПФ), вновь превращая
его в нуклеозиды гуанозинмонофосфат
(ГМФ) и инозинмонофосфат (ИМФ)
и не дает гипоксантину и гуанину стать
мочевой кислотой. ГМФ и ИМФ инги-
бируют продукцию эндогенных
пуринов из ФРПФ, кроме того, сам ФРПФ
утилизируется при их образовании — и
всё это сдерживает темпы образования
мочевой кислоты.
При дефиците ГФРТ происходит резкое
усиление продукции эндогенных пуринов.
Полная и частичная формы сцепленного с
Х-хромосомой дефицита ГФРТ известны,
соответственно, как синдромы Леша-Ни-
хена (вар. Найхена) и Келли-Зигмиллера.
Поражаются, практически исключительно,
мальчики.
При полном дефиците (синдром Легиа-
Найхена), кроме гиперурикемии, подагры,
и уролитиаза отмечаются ЗПМР, хореоате-
тоз, центральные спастические параличи,
Патофизиология нуклеинового обмена 13 3
мазохистическое поведение (больные,
например, способны самих себя искусать), а
также другие неврологические нарушения.
Продукция мочевой кислоты возрастает в
6-200 раз, а суточная экскреция уратов с
мочой повышается с 10 мг/кг до 47 мг/кг.
Синдром имеет несколько генокопии.
Поэтому, в одних семьях больше нарушается
обмен гуанина, в других — гипоксантина. При
такой генокопии как «неполный дефицит
ГФРТ» — синдром Келли-Зигмиллера —
подагра и уролитиаз не сопровождаются
тяжелыми неврологическими нарушениями.
Характерно, что при семейной подагре,
по некоторым представлениям,
воспроизводится именно эта генокопия, так как,
несмотря на наличие общих симптомов
(гиперурикемии и ее последствий),
клиника подагры отличается от классического
синдрома Леша-Нихена отсутствием как
ЗПМР (больные даже проявляют высокие
темпы умственного развития), так и другой
неврологической симптоматики (см. также
т. I, стр. 139).
>* Повышенная активность ФРПФ-син-
тетазы (№ 1 на рис. 22), которая вы-
зывана мутацией её аллостерического
участка, делающей затруднительным
регуляторное ингибирование энзима,
ведет к ускоренному синтезу ФРПФ.
Данный фермент также кодируется в
Х-хромосоме. Избыток ФРПФ приводит
к субстратной активации амидо-фосфо-
рибозилтрансферазы (АФРТ, N° 2 на
рис.22). АФРТ — ключевой
регулируемый фермент основного пути биосинтеза
пуринов. Ее активация сопровождается
резким усилением продукции
эндогенных пуринов. Уже в юношеском возрасте
у носителей данной аномалии
развиваются подагра и уролитиаз.
>* Ускоренный или высокий в абсолютном
отношении уровень распада пуриновых
нуклеотидов также способен привести
к гиперурикемии. Сюда относится как
ускоренный распад клеточных ядер и
нуклеиновых кислот, так и значительное
увеличение утилизации макроэргичес-
134 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
ких нуклеотид-фосфатов. Ускоренной
гибелью клеток и распадом
нуклеиновых кислот сопровождаются процессы
массированного апоптоза и некробиоза,
особенно в полиплоидных клетках.- Это
наблюдается при адаптации плода к
внеутробной жизни (быстрое
изменение количества клеток в крови ведет к
транзиторной гиперурикемии и урату-
рии). К подобным результатам может
привести терапия раковых и лейкозных
больных цитостатиками, которые по
данной причине нередко комбинируют
с блокаторами образования мочевой
кислоты. Ускоренная гибель клеток
вызывает гиперурикемию при истинных
полицитемиях, гемобластозах, псориазе,
рабдомиолизе, болезни Пэджета,
гемолитических процессах. B.C. Новиков и
соавт. (1996) указывают, что при
образовании мочевой кислоты ксантиноксидаза
формирует также супероксидные
анионы. Поэтому распад нуклеиновых кислот
во всех случаях приводит к нарастанию
потенциала активных кислородных
радикалов, обусловливающих дальнейшее
повреждение клеток. Ввиду этого
рекомендуется использование блокаторов
синтеза мочевой кислоты при лечении
пациентов с синдромом длительного
сдавления, что способствует не только
купированию гиперурикемии, но и
ограничению дальнейшей альтерации.
>- Ускоренный распад АТФ и других нук-
леотидных макроэргов происходит при
гликогенозах III, V и VII типа, судорогах,
гипоксических состояниях (в том числе,
вдыхании табачного дыма), интенсивных
физических упражнениях, гипертирозе.
Все эти состояния обостряют подагру
и усугубляют гиперурикемию.
Продукция пуринов увеличена при ожирении
(см. ниже, «Патофизиология липидного
обмена»). Уровни триглицеридов и
мочевой кислоты в плазме проявляют
сильную положительную корреляцию
(Г. Галлер, М.Ганефельд, 1979).
Метаболическая связь между ними, по
Х.Грейлингу (1969), объясняется тем,
что как синтез жиров, так и продукция
ФРПФ требуют НАДФН и зависят
от интенсивности функционирования
пентозного шунта. По новым данным,
при ожирении предполагается дефект в
работе водород-натриевых транспортных
систем, снижающий их эффективность и
приводящий к перерасходу макроэргов и
продукции дополнительных пуринов. В
самое последнее время выяснена
защитная роль апопротеина В и содержащих
его ЛПНП и ЛПОНП при подагре. Ад-
сорбируясь на поверхности кристаллов
уратов, апо-В препятствует активации
этими кристаллами лейкоцитов и меди-
аторных систем воспаления. Не
исключено, таким образом, что ГЛП при
гиперурикемии может иметь компенсаторное
значение (см. также гл. 14). Мочевая
кислота снижает продукцию N0 и
усиливает пролиферацию гладкомышечных
клеток в артериях, что способствует
гипертензии и атеросклерозу.
Еще Л.Н. Карлик (1936) отмечал, что
при введении пациентам, страдающим
подагрой, и здоровым людям, находящимся
на одинаковой беспуриновой диете, равной
дозы нуклеината натрия, больные выводят
мочевую кислоту в 2-2,5 раза дольше.
Следовательно, при подагре нарушена не только
продукция, но и экскреция уратов.
Сниженная экскреция уратов
присутствует более чем в 98 % случаев гиперурикемии
и бывает вызвана различными причинами:
>■ Как первичная, так и вторичная гиперу-
рикемия нередко связана с понижением
фильтрации мочевой кислоты (почечная
недостаточность, уремия). Гиперури-
кемия ответственна за ряд симптомов
уремии, например артрит. Довольно
характерно сочетание по дагры и поликис-
тоза почек. Нефропатия беременных
сочетается с гиперурикемией. В некоторых
случаях, по-видимому, гиперурикемия
первична по отношению к хронической
почечной недостаточности.
>- Сниженная секреция мочевой кислоты
отмечается при всех видах ацидоза
включая салицилатный, кетоновый,
лактатный и алкоголь-индуцирован-
ный, поскольку органические кислоты
конкурируют с мочевой за почечные
переносчики. Секреция уратов и
других веществ нарушена при отравлении
свинцом и бериллием, циклоспориновой
нефропатии, гипотирозе и др.
> Повышенная дистальная реабсорбция
тоже может обусловить задержку уратов.
Считается, что именно поэтому гиперу-
рикемия сопровождает внеклеточную
дегидратацию, несахарный диабет, терапию
салуретиками. Существуют синергичес-
кие взаимоотношения между реабсорб-
цией кальция и уратов. По крайней мере,
и тот, и другой процессы существенно
усилены при гиперпаратирозе и при
саркоидозе, когда имеется эндогенный
гипервитаминоз D. Реабсорбция магния
и уратов, возможно, антагонистична,
так как при синдроме Барттера имеется
ослабление первого и параллельное
усиление второго процесса.
> Во многих случаях нарушение
выведения уратов имеется, но механизмы его
комплексные или до конца не изучены.
Примером может служить гиперурике-
мия при синдроме Дауна.
При гиперурикемии могут параллельно
действовать и повышение образования
мочевой кислоты, и снижение её экскреции.
Наиболее важные причины такой синерги-
ческой ситуации приведены ниже.
> Дефицит глюкоза-6-фосфатазы или
болезнь фон Гирке (гликогеноз I типа)
приводит к нарушению освобождения
глюкозы в ЭПР печени из глюкоза-6-фосфата.
Возникает тенденция к гипогликемии,
требующая ускоренного распада
макроэргов, в том числе нуклеозидтрифосфа-
тов. Формируется избыток пуриновых
метаболитов. Низкая концентрация
нуклеозидмонофосфатов ведет к растор-
маживанию АФРТ. Продукция мочевой
кислоты увеличивается. Вдобавок,
формируется лактат-ацидоз, что снижает
Патофизиология нуклеинового обмена 135
экскрецию уратов. При этом
заболевании, как и во многих других случаях, ги-
перурикемия соседствует с выраженной
гипертриглицеридемией. Подагра
развивается у носителей данного гликогеноза
уже в юности (см. ниже
«Патофизиология углеводного обмена»).
>- Неолерантность к фруктозе из-за
наследственного дефекта фруктозо-1-фос-
фат-альдолазы приводит к накоплению
фруктозо-1-фосфата. Пурины из состава
усиленно используемой АТФ
превращаются в ураты. Выведение уратов
затормаживается из-за лактат-ацидоза и
почечного канальцевого ацидоза. Так как гете-
розиготность по данному аллелю очень
распространена (от 1/80 до 1/250), то
фруктозоальдолазная недостаточность,
возможно, должна рассматриваться как
одна из частых причин гиперурикемии.
>• Фактически, комплексным гиперури-
кемическим действием на продукцию и
выведение мочевой кислоты обладает и
алкоголизация. Алкоголь, принимаемый
с пищей, не только сам зачастую несет
дополнительные пурины (особенно,
темное пиво и красное вино), но и
ограничивает экскрецию уратов с мочой, а также
подкисляет мочу, способствуя ретенции
пищевых пуринов и кристаллизации
мочевых и тканевых уратов.
>• Характерным сочетанием пониженного
выведения уратов и их повышенной
продукции отличаются шок и шокопо-
добные состояния, особенно,
травматического происхождения. Одновременно
действуют цитолиз, нуклеолиз,
усиленный в условиях гипоксии распад АТФ и
острая почечная недостаточность.
ПАТОГЕНЕЗ ПОДАГРЫ
Патогенез подагры касается механизмов
накопления уратов в хрящевой ткани и
почках, а также механизмов провоцируемого
ими воспаления. Рис. 25, с. 131, отражает
основные проявления этого недуга.
136 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Подагра в типичных случаях дебютирует
после 40 лет и проходит 4 стадии.
- Латентная гиперурикемическая стадия
протекает без клинических симптомов и
проявляется лишь выявляемой
лабораторным анализом гиперурикемией.
Многие гиперурикемики не успевают развить
клинически выраженную подагру.
- Дебютная стадия представляет собой
острый моноартрит, который
самоограничивается и проходит, даже без
лечения, за 10-14 дней. Моноартрит
начинается внезапно, на фоне
предшествующей гиперурикемии и повышенного
содержания уратов в синовиальной
жидкости. Поражается почти всегда один
сустав (у женщин бывают формы с
полисуставным началом). Наиболее
характерно вовлечение предплюснофалангового
сустава большого пальца стопы, который
находится в относительно «холодной»
периферической зоне и испытывает
максимальную удельную нагрузку при
ходьбе. Подагрический моноартрит
характеризуется острейшими
признаками воспаления — отеком, краснотой,
ограничением функции, повышением
местной температуры, а часто — и общей
лихорадкой. Боль при подагре бывает
настолько мучительна, что этот недуг по
праву считается одним из самых
болезненных. Острый приступ длится 24-72
часа. Артрит сопровождается периартри-
том, вовлекающим связки, сухожилия и
суставные сумки. Стартовым сигналом
к началу острого артрита служит
резкое увеличение концентрации уратов в
синовиальной жидкости или снижение
солюбилизирующего эффекта.
Приступы могут провоцироваться перееданием,
алкоголем, лекарствами, стрессом,
переохлаждением, физической нагрузкой,
дегидратацией, травмами. Главным
механизмом острого артрита служит
реакция сторожевой полисистемы плазмы
и полиморфонуклеарных лейкоцитов, в
первую очередь — нейтрофильных, на
полианионные кристаллы уратов. Ураты
активируют классический и
альтернативный пути комплемента, фактор Хагемана,
а через них — и всю контактную систему
полипептидных медиаторов, включая
кинины, свёртывание, фибринолиз (см.
т. I, стр. 339-341). Хемоаттрактанты С5а
и С3а привлекают полиморфонуклеары.
Взаимодействие уратовых кристаллов,
особенно, покрытых
иммуноглобулинами G и М, с полиморфонуклеарами и
фагоцитами, вообще, приводит к
освобождению и активации ряда
воспалительных медиаторов, в первую очередь
нейтрофильного лейкотриена В4. Гибель
нейтрофилов при фагоцитозе
кристаллов ведет к освобождению активных
кислородных радикалов, а также особого
кристаллозависимого хемотактичес-
кого фактора (11,5кД), дефензинов и
огромного количества лизосомальных
гидролаз. Кристаллозависимый хемоат-
трактант лейкоцитов, возможно, один из
главнейших участников этого «пожара»,
поскольку доказано, что успешно
применяемый для лечения подагры блокатор
цитоскелета колхицин специфически
устраняет именно высвобождение этого
хемотактического фактора.
Макрофаги, фагоцитируя ураты и клеточный
дебрис, активируются и выделяют ци-
токины ИЛ-1, ИЛ-6, ИЛ-8, кахексии, а
также простагландины. Это усиливает
воспаление и приводит к выделению
синовиоцитами коллагеназ,
поддерживающих альтерацию. Самоограничение
симптомов артрита зависит от выработки
противовоспалительных медиаторов,
главным образом, макрофагального
и синовиоцитарного происхождения.
Большое значение может иметь
обволакивание поверхности кристаллов уратов
апопротеином В, который проникает
в воспаленную синовию из плазмы в
составе липопротеидов в результате
повышения сосудистой проницаемости.
Имеются наблюдения,
свидетельствующие, что мембраны, обогащенные
ХН и андрогенами, менее устойчивы
к повреждающему действию уратов, а
эстрогены, наоборот, сообщают мембра-
Патофизиология нуклеинового обмена 13 7
нам при контакте с этими кристаллами
дополнительную резистентность. Это
коррелирует с наблюдениями о связи
подагры и ГЛП и о преимущественном
поражении подагрой мужчин.
Межприсгпупный период подагры
характеризуется отсутствием симптомов
острого артрита. Несмотря на это
сохраняется гиперурикемия, происходит
отложение уратов в тканях, прогрессирует
нефропатия, и бывают повторные атаки
артрита, которые вовлекают тот же и
новые суставы. Как и первый приступ,
повторные возникают в ответ на тот же
разнообразный спектр провоцирующих
агентов.
В следующем периоде — хронического
продуктивного артрита — с течением
времени (как правило, по прошествии
8-10 лет от первого приступа) в
пораженных суставах и вокруг них образуются в
результате гранулёматозного воспаления
подагрические шишки — tophi urici. Это
деформирует суставы. Кожа над ними
может изъязвляться, имеется персис-
тирующий полиартрит с умеренным
болевым синдромом, хотя сами шишки
на ощупь вне приступа парадоксально
безболезненны. Главные механизмы
этого периода подагры связаны с
хронической активацией макрофагов, внутри
которых персистируют
фагоцитированные кристаллы. Это ведет к
накоплению цитокинов, формирующих своего
рода подагрическую гранулёму — очаг
хронического пролиферативного
воспаления (см. т. I, стр. 361-364, 483-485).
В центре тофуса находится скопление
кристаллических или аморфных
уратов, вокруг обнаруживаются гигантские
клетки, напоминающие классические
макрофаги гранулём, образованных
вокруг инородных тел. Здесь же
реактивные фибробласты и мононуклеарные
участники хронического воспаления.
Интересно, что гранулёматоз при
подагре, по-видимому, может вовлекать и
аутоаллергические механизмы ГЗТ. По
крайней мере, установлено (С. Трипати,
1982), что хроническая гиперурикемия
провоцирует аутоиммунный инсулит, а
мочевая кислота служит иммунозави-
симым диабетогенным фактором. Исход
воспаления связан с формированием
фиброза синовии, эрозией суставного
хряща, анкилозами. Изредка тофусы
могут возникать и вне суставов и пери-
суставных тканей. Описаны
подагрические шишки в костях, под кожей и даже в
сердце.
При подагре накопление мочевой кислоты
и уратов ведет, вследствие кристаллизации,
к образованию камней в почках. Уратурия
может отмечаться с первого периода
болезни, но выраженный уронефролитиаз и урат-
ная нефропатия свойственны лишь поздней
тяжелой подагре. Уролитиазу способствуют
кислая реакция мочи и инфекция. Более
20 % больных подагрой имеют камни в
почках, при гиперурикемии выше 12 мг/дл этот
показатель увеличен до 50%. Характерно,
что сами камни состоят, главным образом, из
мочевой кислоты, в чистом виде или вместе
с оксалатом кальция (84%). Урат натрия
не участвует в камнеобразовании. Мочевая
кислота вступает в перекристаллизацию
совместно с оксалатами, кроме того
последние формируются из глицина, который
участвует и в пуринообразовании. Пуриновая
нагрузка провоцирует оксалурию. По этим
причинам подагрикам не рекомендуются
продукты, вызывающие оксалурию (какао,
шоколад, свёкла, ревень, петрушка и иная
зелень). Клюквенный морс, содержащий бен-
зоат натрия, способен изменить метаболизм
глицина, уменьшить оксалурию и
кристаллизацию оксалатов и мочевой кислоты,
так как метаболизм глицина направляется
бензоатом в сторону гиппуровой кислоты
(В.А. Таболин и соавт., 1977).
Сами кристаллы мочевой кислоты и
оксалатов могут закупоривать канальцы.
Взаимодействие уратов с медиаторами
воспаления и лейкоцитами в почке, как и
в суставе, ведет к воспалению.
Формируется интерстициальныи и пирамидальный
нефрит. Как и в суставах, в почке могут
возникать гранулёматозные поражения и
138 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
даже подагрические шишки. Присоединение
инфекционного компонента способствует
развитию хронического пиелонефрита.
Клинически, течение нефропатии может
быть латентным, но могут отмечаться про-
теинурия и гипертензия. Исходом являются
нефросклероз и хроническая почечная
недостаточность, причем по мере ее
формирования уровень гиперурикемии растет и артрит
отягощается. Без лечения 25-30 % больных
подагрой умирают от уремии.
ДРУГИЕ НАРУШЕНИЯ
ПУРИНОВОГО ОБМЕНА
Характерную для подагры хроническую
уратную нефропатию с исходом во
вторично сморщеную почку и хроническую
почечную недостаточность не следует
смешивать с острой мочекислой обтурационной
нефропатией (мочекислый инфаркт). Это
осложнение развивается при массированной
гиперпродукции мочевой кислоты на фоне
сопутствующей дегидратации или
ограничения фильтрации. Происходит закупорка
собирательных трубок и почечных дисталь-
ных сосудов уратами и микротромбами.
В тяжелых случаях развивается острая
почечная недостаточность. Синдром типичен
для осложнений цитолитической терапии
гемобластозов и других неопластических
заболеваний, но может последовать и при
шокоподобных состояниях, в частности,
тяжелой дегидратации, сопряжённой с
физической нагрузкой. Для течения
собственно подагры это острое нарушение не
характерно.
Гипоурикемия — поражает не менее 0,2 %
людей. При этом уровень мочевой кислоты
в сыворотке аномально низок— до 2мг/
дл. Данное метаболическое отклонение,
само по себе, чаще течет бессимптомно.
Не отмечено и каких-либо существенных
особенностей интеллектуальной сферы у
гипоурикемических индивидов, что
находится в противоречии с представлениями
о «допинговом» эффекте мочевой кислоты
на умственную активность. Гипоурикемия
связана с понижением выработки мочевой
кислоты (например, из-за цирроза печени)
или, чаще всего, с повышением ее выведения
почками. Повышенная экскреция уратов с
мочой может быть следствием первичого
изолированного дефекта ее реабсорбции
или проявляться в структуре более широких
нарушений (синдром Фанкони, цистиноз,
отравление тяжелыми металлами).
Гипоурикемия также бывает следствием несба-
Таблица 9
Редкие наследственные нарушения пуринового обмена
(в скобках—номера ферментов на рис. 22, с. 127)
Дефектный фермент
Аденин-фосфорибозилтрансфераза(9)
Аденил-сукцинатлиаза (3)
Миоаденилат-дезаминаза (4)
Аденозин-дезаминаза (б)
Пурин ну клеотидфосфорил аза (7)
Основные проявления
Дигидроксиадениновые рентгенопрозрачные камни в почках.
Частота гетерозигот 0,4%, гомозигот менее 1/35000
Аутизм и ЗПМР, судороги
Миальгии, миопатия, лактатацидоз и отсутствие освобождения
аммония при физической нагрузке, повышение сывороточной
креатинфосфокиназы. Приобретенная форма сопровождает
миозиты и миопатии
Остеохондродисплазия грудной клетки, гипоплазия тимуса,
тяжелый комбинированный иммунодефицит, гемолитическая анемия,
лимфопения (см. т. I, с. 483)
Т-клеточный иммунодефицит, гипоплазия тимуса, гемоцитопения,
парезы, гипотонус, гипоурикемия (подробнее см. т. I, с. 482)
Патофизиология нуклеинового обмена 139
лансированного парентерального питания с
перегрузкой глицином.
Гипоурикемия наступает при дефекте
ксантиноксидазы и сочетанном дефиците
ксантиноксидазы и сульфитоксидазы
(совокупная примерная частота — 1/45 000).
Это приводит к накоплению гипоксантина
и ксантина, отложению рентгенопрозрачных
ксантиновых конкрементов в почках. Могут
быть миозит, судороги при физических
усилиях и рецидивирующий полиартрит.
Сочетание с сульфитоксидазной
недостаточностью менее благоприятно и добавляет
в клиническую картину синдрома
неврологические нарушения — нистагм, энофтальм,
судороги, подвывих хрусталика. Обе
мутации аутосомно-рецессивны и связаны с
дефицитом молибдензависимого кофактора
ксантин-и сульфитоксидазы. Гипоурикемия
сопровождает и некоторые наследственные
иммунодефициты (табл. 9).
Другие редкие аутосомно-рецессивные
нарушения обмена пуринов
охарактеризованы ниже в специальной таблице.
НАРУШЕНИЯ ОБМЕНА
ПИРИМИДИНОВЫХ
ОСНОВАНИЙ
Пиримидиновые основания включают
урацил, цитозин, тимин и минорные
основания 5-метилцитозин и 5-оксиметил-
цитозин (рис.21, выше на стр. 126). Путь
биосинтеза пиримидиновых нуклеотидов
несколько проще, чем у пуринов, но сходен
с пуриновым, а на начальной части имеет
общие звенья. Как и в пуриновом
биосинтезе, свободные основания не образуются
в качестве промежуточных продуктов. Из
предшественников с открытой цепью
возникает пиримидиновое кольцо, после чего (а
не до этого, как у пуринов) к нему
подсоединяется D-рибоза. Атомы пиримидинового
гексацикла берутся из карбамоил-фосфата
и аспарагина с глутамином. Важнейшим
промежуточным метаболитом при синтезе
пиримидинов служит оротовая кислота,
представляющая собой 6-карбоксиурацил. К
ней-то и присоединяется рибозный остаток.
Эти превращения схематично показаны на
рис. 26.
Пиримидиновые нуклеотиды входят в
состав ДНК и РНК (урацил — только в
состав РНК). Кроме того, они, как и
пурины, формируют нуклеотидтрифосфаты и
нуклеотидпирофосфаты, используемые как
макроэргические донаторы, в частности, в
транспортных системах, при образовании
парных соединений и синтезе сложных
липидов.
Катаболизм пиримидинов идет по пути,
отображенному на рис. 27, приводя к
образованию р-аминокислот.
Клинически важнейшим из нарушений
пиримидинового обмена считают
наследственную оротацидурию. При данном
заболевании нарушен синтез уридина de novo, что
превращает этот нормальный метаболит для
таких пациентов в незаменимый компонент
диеты. Это дефект оротатфосфорибозил-
трансферазы (ОФРТ 1 на рис. 26) и ороти-
дин-5'-фосфат-декарбоксилазы (0-5-ФД К 2
на рис. 26 ). Оба фермента — части белкового
комплекса, кодируемого в длинном плече
третьей хромосомы, и имеют общий пептид.
У детей с данным аутосомно-рецессивным
расстройством замедляется рост и
возникает тяжелая мегалобластическая анемия,
резистентная к витаминотерапии (см.
«Патофизиология витаминного обмена»).
Имеется лейкопения со сдвигом ядерной
формулы нейтрофилов вправо. Отмечается
иммунодефицит с поражением, в основном,
Т-клеточных функций. Избыток оротовой
кислоты выводится с мочой и, из-за малой
растворимости этого метаболита,
формирует кристаллурию, причем конкременты
могут закупорить даже мочеточники или
уретру. Менее значительная оротацидурия,
без тяжелого гематологического синдрома,
бывает и при дефектах ФРПФ-синтетазы, а
также при нарушении цикла мочевины —
дефиците орнитин-транскарбамоилазы, как
полагают, из-за избытка неметаболизиро-
ванного карбамоилфосфата (см. выше
«Нарушения конечных этапов обмена белка»).
глутамин
со2
Рис. 26. Пути биосинтеза пиримидинов.
Сокращения: ФРПФ — фосфорибозилпирофосфат; ЦТФ — цитидинтрифосфат; УТФ — уридинтрифосфат; УДФ — уридиндифос-
фат. Обозначения: 1 — оротатфосфорибозилтрансфераза (ОФРТ), 2 — оротидин-5'-фосфат-декарбоксилаза (0-5-ФДК)
I нуклеиновые кислоты
НАДФН *НАДФ+
+
HNt
О*
Хметилцитозин I
+н2о
-NH»
I тимин |
НАДФНf
+ НАДФ+
Н+ W
I метилмалонат
| дигидроротимин [__2—►! N-карб,
амоилизомасляная кислота
U
|Р- аминоизомасляная кислота
—_____—^^_ ____ +NH3 +С°2
+"2^ ^ \М-карбамоилпропионовая кислота \ > |р-длонмн[ .ш^жж +qq
н | дигидроурацил\
Рис.27. Катаболизм пиримидиновых нуклеотидов
Патофизиология нуклеинового обмена 141
Лечение подагры аллопуринолом приводит
к симптоматической оротацидурии, так как
его метаболиты ингибируют 0-5-ФДК.
Как уже указывалось выше, 10% белых
и, практически, все монголоиды выделяют
с мочой р-аминоизомасляную кислоту. Ами-
ноизобутиратурия — нейтральная мутация,
приводящая к нарушению трансамини-
рования этой кислоты в метилмалоновый
полуальдегид и нарушению катаболизма
тимина. Она не влечет никаких болезненных
последствий.
Дефект пиримидин-5'-нуклеотидазы
проявляется гемолитической анемией, причем, в
эритроцитах накапливается избыток немета-
болизированных рибонуклеотидов, в
основном, цитидиновых и уридиновых, который
морфологически проявляет себя в виде
базофильной пунктации. Наследственная
форма синдрома аутосомно-рецессивна,
однако отравление свинцом ингибирует этот
фермент и дает такую же гематологическую
симптоматику (см. т. III).
КРИСТАЛЛИЧЕСКИЕ
АРТРИТЫ
Подагра — заболевание, наложившее
значительный отпечаток на всю культуру
человечества. Имеется в виду как историческая
деятельность жертв подагры,
подстегиваемых эндогенным допинг-эффектом, так
и отражение мучений пациентов в
литературе и искусстве. Достаточно вспомнить
лишь некоторые примеры. Древнеримский
мыслитель Сенека в эпоху Нерона
сетовал: «Тяжёлые времена, исчезла старая
добродетель. Даже женщины стали болеть
подагрой». В средневековой Европе Томас
Мор писал другу Эразму Роттердамскому:
«У тебя камни в почках, а у меня — подагра:
мы женились на родных сестрах». Одно из
самых блестящих описаний клиники
болезни, принадлежащее перу «английского
Гиппократа» Томаса Сиденгема, начинается
со слов: «Подагра чаще поражает мудрых,
чем глупцов, что может служить
утешением для меня». А Уильям Шекспир призвал
главную «героиню» этого раздела учебника
в помощницы в борьбе с коррупцией:
«Подагра злая, скрючь седых вельмож, пусть, как
их честность, и они хромают!».
Однако, подагра - лишь представитель
большого класса заболеваний, связанных
с патогенным действием кристаллов
эндогенного происхождения. В тени подагры от
внимания медиков притаились во многом
сходные с нею недуги, объединяемые в
практическом здравоохранении как
кристаллические артриты или кристаллические арт-
ропатии. Их в ревматологии ранее строго
противопоставляли инфект-артритам, но к
настоящему времени выяснено, что
воспаление может быть вызвано комбинированным
действием кристаллов и микроорганизмов.
Более того, кристаллизация солей
кальция и фосфора в почках и суставах может
запускаться жизнедеятельностью
кальцинирующих нанобактерий, открытых финскими
патологами О. Кайяндером и Н. Чифчиоглу
в 1998 г. и присутствующих практически в
любых кальцинатах организма, что
предполагает участие инфекционного агента в
этиологии уронефролитиаза и
кристаллических артритов.
Большинство из кристаллоартритов
связано с нарушением нуклеинового и/или
аминокислотного обмена, и будет логичным
в заключение глав 5-6 этого учебника дать
их краткую характеристику.
Кристаллические артропатии —
заболевания, дифференциальная диагностика
которых требует от медика смежных
геологических знаний по кристаллографии. Для их
распознавания применяются
поляризационная микроскопия, рентгеноструктурный
анализ, а иногда и электронная
микроскопия. К тому же, при ХПН, гиперпаратирозе
и некоторых других нарушениях
комбинируется отложение в тканях кристаллов сразу
нескольких видов и возникают смешанные
кристаллоартриты, ранее трактовавшиеся
классической медициной как подагра.
Наибольшее после подагры значение
имеют псевдоподагра (хондрокальциноз — см.
выше с. 130), а также апатитная болезнь и
оксалоз (оксалурия - см. выше с. 91-92).
142 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
При псевдоподагре откладываются
кристаллы пирофосфата кальция дигидрата (в
виде палочек, ромбов и прямоугольников),
поражаются суставной хрящ, сухожилия,
связки и синовия. Процессу способствуют
цитокины ТФРР и ЭФР, а также
ускоренный распад АТФ, значение имеет дефицит
антикристаллизующих гликопротеидов
гиалинового хряща. Кроме подагроидного
синдрома типичны остеопороз и остеоар-
троз, связанные с нетипичным для подагры
кальцинозом гиалинового хряща, бывают
продуктивный синовит, как при
ревматоидном артрите и обызвествление хрящей и
связок позвоночника, как при болезни
Бехтерева, Болезнь чаще поражает пожилых,
но имеются наследственно обусловленные
юношеские формы (с дефектами генов в
5-й и 8-й хромосомах). Псевдоподагра часто
сочетается с гиперпаратирозом и гипофос-
фатазией (см. гл. 13), гемохроматозом и
гипомагнезиемией (гл. 12), бывает у
спортсменов после операций на мениске,
сопровождает наследственные хондродисплазии
(гл. 13) и, нередко, протекает параллельно
подагре. Некоторые этнические группы
(словацкие венгры, испано-чилийцы,
японцы, англичане, голландцы, шведы, евреи и
тунисцы) имеют повышенную поражен-
ность псевдоподагрой.
Апатитная болезнь - результат
накопления кристаллов гидроксиапатита,
нормального минерального компонента костей,
зубов и эпифизарного песка, в суставах и
околосуставных тканях. Она бывает чаще
у пожилых, на фоне деформирующего
остеоартроза, гиперпаратироза, ХПН (см.
гл. 13), а также при
иммунопатологических заболеваниях соединительной ткани с
неорганоспецифическими аутоантителами
(склеродермия, дерматомиозит и др. — см.
т. I, гл. 16). Некоторые формы апатитной
болезни считаются самостоятельными
заболеваниями («милуокское плечо») Кристаллы
при апатитной болезни крайне мелкие,
образуют округлые скопления и, в отличие
от остальных кристаллических артритов, не
обладают двойным лучепреломлением, зато
красятся в красный цвет ализарином S.
При оксалурии (оксалозе) поражаются, в
основом, почки (см. гл. 5), но при
длительном течении возможны артриты и периарт-
риты. Первичные оксалурии описаны выше
в гл. 5, как нарушения аминокислотного
обмена. Вторичная оксалурия с тяжелым
артритом может осложнять течение ХПН
при гемодиализе и перитонеальном
диализе, причем этому способствует применение
аскорбиновой кислоты, дающей в организме
оксалаты (см. гл. 11). Подагра и оксалурия
закономерно сочетаются (см. выше).
Кристаллы оксалата кальция чаще всего имеют
трехгранную бипирамидальную форму.
При изучении данного раздела для
ознакомления с проявлениями кристаллических
артропатий рекомендуется использовать
цветной плакат из комплекта «Общая
патология» под редакцией Л.П. Чурилова (2007)
«Повреждение клетки. 2. Смешанные
дистрофии. Нарушение обмена сложных белков и
минеральных веществ».
Глава 7
НАРУШЕНИЯ ЛИПИДНОГО
ОБМЕНА
«Без атерогенных липопротеидов не будет атеросклероза»
А.Н. Климов, 1984
• Введение
• Липиды организма и пищи. Алиментарная липидная недостаточность
• Нарушения переваривания и всасывания липидов
• Транспорт липидов в организме и его нарушения
• Гиперлипопротеидемии и другие дислипопротеидемии
• Атеросклероз
• Накопления и мобилизации липидов
• Липостат и патофизиология первичного ожирения
• Истощение и кахексия
• Лизосомальные болезни накопления липидов
• Кетоз и стеатоз печени
ВВЕДЕНИЕ
Начиная с 70-х годов XX века, эпицентр
активности в исследованиях патологии
обмена веществ переместился в область
нарушений липидного обмена и остается там
поныне. Об этом свидетельствуют как число
публикаций, так и объёмы финансирования.
Причина этого проста. Наследственные
и приобретенные нарушения липидного
обмена — самые частые метаболические
расстройства у населения развитых в
медицинском отношении стран мира. По данным
ВОЗ, не менее 10% населения Земли (а в
Европе и Северной Америке — более 20%)
страдают какой-либо дислипопротеидемией.
Семейная наследственная гиперхолесте-
ринемия в некоторых регионах поражает
1-2% населения и является самым частым
наследственным заболеванием европеоидов
вообще. Но дислипопротеинемии — только
одна из многочисленных групп расстройств
липидного метаболизма. По некоторым
оценкам, в Европе до 50 % женщин и 20 %
мужчин имеют ту или иную форму и степень
ожирения. Около половины от общего
уровня смертности, а в России — существенно
больше, обусловлено сердечно-сосудистыми
заболеваниями. Патогенетической основой
для ишемической болезни сердца,
инсультов, ряда форм артериальной гипертензии
служит атеросклероз, центральным звеном
механизма и важнейшим фактором риска
которого является именно нарушение
липидного обмена.
В 60-х годах XX века не было
современных знаний по патофизиологии липидного
обмена, и, как следствие, научно
обоснованной системы профилактики атеросклероза
не существовало ни в одной стране мира.
В результате к 1968 г. развитые страны
оказались перед лицом пандемии атеросклероза,
только в СССР и США вместе взятых
уносившей почти по 1100 000 жизней ежегодно.
144 АШ. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Но когда появились и были внедрены в
практику профилактической и лечебной
медицины новые сведения о механизмах
нарушений липидного метаболизма, это в
корне изменило ситуацию в тех странах,
где общество организационно и финансово
обеспечило использование данных
теоретических достижений. К концу 80-х годов
XX века в Северной Америке было
достигнуто (по сравнению с пиковым уровнем)
почти шестидесятипроцентное понижение
смертности от ИБС и уменьшение
частоты инфаркта миокарда на 1/3. Таким
образом, именно на основе использования
достижений патофизиологии липидного
обмена был получен самый впечатляющий
за всю историю успех профилактической
медицины в борьбе с неинфекционной
патологией. Сказанного достаточно, чтобы
обосновать большое значение данного
раздела патологии в медико-гигиеническом
образовании.
Патология липидного обмена находится
в фокусе постоянного внимания не только у
терапевтов и специалистов по превентивной
медицине, но и у педиатров. Дело в том, что
атеросклероз — длительный многолетний
процесс. С завершающими стадиями этого
заболевания и его осложнениями, в
большинстве случаев, имеют дело терапевты и
гериатры. Но дебют атеросклероза и
первичное взаимодействие сосудистой стенки
с патогенными липопротеидами относятся
к началу второго десятилетия жизни!
Соответственно, вопросы ранней диагностики
атерогенных расстройств липидного
обмена и превентивной терапии не могут быть
решены вне педиатрии и подростковой
медицины. Добавим к этому, что не менее
10% детей в экономически благополучных
странах имеют избыточный вес, страдают
от ожирения, и их число нарастает. Если
учесть, что существует не менее десятка
наследственных сфинголипидозов,
нарушающих раннее психомоторное развитие детей,
то станет совершенно ясно, что детальное
освещение патологии обмена липидов
требуется будущему педиатру, как и студенту
любого медицинского факультета.
ЛИПИДЫ ОРГАНИЗМА
И ПИЩИ. АЛИМЕНТАРНАЯ
ЛИПИДНАЯ
НЕДОСТАТОЧНОСТЬ
Липиды — большая группа органических
соединений, для которых характерны:
- нерастворимость в воде
- растворимость в эфире, бензоле, ацетоне,
хлоруглеродистых растворителях
- наличие в молекулах высших алкильных
радикалов
- построение молекул по типу сложных
эфиров с участием разных жирных
кислот и спиртов.
Фактически, группа липидов весьма
велика и гетерогенна, в нее входят многие
распространенные в живых организмах
соединения, в том числе рассмотренные в
специальном разделе жирорастворимые
витамины, каротиноиды, а также спирты
и высшие углеводороды. Вместе с тем,
любое определение липидов вряд ли можно
считать абсолютно корректным.
Например, по критерию нерастворимости в воде
мы вынуждены были бы исключить из
липидов лизолецитины, если бы
стремились к формальной строгости.
Разнообразие липидов представлено на рисунке 28.
Простые липиды содержат в своём составе
только углерод, водород и кислород. Если
они гидролизуются, то гидролиз разлагает
их на жирные кислоты и спирты.
Сложные липиды, в дополнение к
вышеозначенным элементам, содержат азот,
а часто— еще фосфор и серу. Их можно
разложить гидролитически на жирные
кислоты, спирты и соединения других классов
(азотистые основания, углеводы,
фосфорную кислоту).
Примерами простых липидов могут
служить: жирные кислоты, глицериды (в
частности, триацилглицериды, то есть
нейтральные жиры), липоспирты (холестерин,
ретинол, кальциферол), эфиры холестерина,
воска (ланолин, спермацет).
Нарушения липидного обмена 145
ЛИПИДЫ
ПРОСТЫЕ
-ZTT3I
Жирные
кислоты
Сквален
Ацилглицериды
Воска
| Сульфатиды |
Лип о с пир ты
Ретинол
I Кальциферол I
]
Эфиры
холестерина
Фосфолипиды
| Цере6розиды~\ \ Сфинголипиды^
Глицерофосфо-
липиды
Примеры сложных липидов —
фосфолипиды (включая глицеролипиды и
сфинголипиды), а также гликолипиды (в
том числе, ганглиозиды, сульфатиды и
цереброзиды).
Так как липиды содержат и
гидрофильные полярные (ионные) группировки, и
неполярные гидрофобные углеводородные
группы, их неотъемлемым свойством,
выраженным то в слабой (нейтральные
жиры), то в очень сильной (фосфолипиды)
степени, служит амфифильность. Слабо
амфифильные липиды депонируются в
безводной фазе. Сильно амфифильные
липиды могут служить клеточными
перегородками, так как образуют в водном
растворе бислой, в котором гидрофобные
остатки обращены внутрь, а
гидрофильные— в водную фазу. Хотя толщина
формируемых ими биомембран не
превышает 10 нм, но их площадь феноменально
огромна — при весе печени крысы около
6 г плазматические мембраны ее
клеток занимают сотни квадратных метров
(Л.Д.Бергельсон, 1975).
Рис. 28. Классификация липидов.
Исходя из этих особенностей липидов
можно указать на следующие их основные
функции в организме:
- резервно-энергетическая функция,
обеспечиваемая триглицеридами;
- мембранообразующая функция, в
основном, связанная с метаболизмом глицеро-
фосфолипидов;
- рецепторно-посредниковая функция,
обеспечиваемая, главным образом, гли-
косфинголипидами;
- регуляторно-сигнальная функция,
связанная, в первую очередь, с липидными
спиртами (стероидами).
Резервно-энергетическая функция,
выполняемая триглицеридами, делает
нейтральный жир долговременным резервом
калорий. Поскольку запасы относительно
быстро мобилизуемого гликогена не
превышают нескольких десятков граммов, а
чистого жира у здоровых взрослых — 16-23%
массы тела, а также из-за того, что
теплотворная способность (калорический
коэффициент) жира более чем вдвое превышает
соответствующий показатель углеводов (см.
выше «Патофизиология энергетического
обмена»), можно считать жир основным
энергоресурсом организма. Для того чтобы
запасти 10 ккал энергии, достаточно
отложить всего 1,3 г жира. К тому же, тригли-
146 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
цериды, в отличие от гликогена, запасают
энергию в негидратированной форме. Из
общего количества жира в 10-12 кг около
2 кг распределено в виде включений по
различным, не специализированным в
отношении депонирования липидов тканям
(конституциональный жир). Остальной жир
находится в адипоцитах, причем примерно
половина — в подкожно-жировой клетчатке,
а половина— в сальниках и жировой ткани
висцерального отсека тела. Указанные
количества жира могут дать от 75000 до
90 000 килокалорий и покрыть
минимальные энергетические нужды организма на
протяжении более чем 40 дней (см. выше
«Голодание»). Для энергетических нужд
некоторые органы, в основном, используют
жирные кислоты (миокард), другие
способны утилизировать и дериваты липидов
(кетоновые тела), и глюкозу (мозг), а
третьи питаются при интенсивной нагрузке, в
основном, глюкозой, а при обычной —
частично, жирными кислотами (скелетные
мышцы). Скопления нейтрального жира
играют и дополнительную защитную роль:
теплосберегающую, электроизоляционную,
механическую. Спермацет, депонируемый-
кашалотом на голове, позволяет ему
наносить механические таранящие удары и
избегать при этом сотрясения мозга.
Похудевшие испытуемые имеют пониженное
электросопротивление кожи. Ярким
примером недостаточности
резервно-энергетической и депонирующей функции липидов
служит состояние недоношенных детей.
Плод питается через плаценту непрерывно,
находится в термостатированной среде,
защищен от травм и поэтому обходится
без больших жировых прослоек. Некоторое
накопление жира происходит только в
последние 1,5 месяца внутриутробной жизни, и
недоношенные рождаются на свет без них.
Так как у недоношенных очень малы
накопления нейтрального жира, они нуждаются
в учащенном кормлении, характеризуются
ускоренной теплоотдачей и
затруднениями при обеспечении гомойотермности,
а также более подвержены механическим
травмам.
Большая часть резервных триглицеридов
человека — смешанные, то есть в них
присутствуют 2 или все 3 различных остатка
жирных кислот. Жиры человека содержат
жирные кислоты с чётным числом атомов
углерода, при этом в триацилглицеридах
широко представлены мононенасыщенные
ацилы, особенно, в среднем положении.
Так, олеиновая кислота, вообще, является
в триглицеридах преобладающей. Поэтому,
переходная температура жиров человека
невелика и близка к 37 °С, а его показатель
непредельности — йодное число — равен 64
(для сравнения, в конопляном масле — 150,
а в сливочном маргарине — 63). Избыточное
накопление жиров в жировой ткани под
кожей, в сальниках и брыжейке наблюдается
при ожирении (см. ниже). Жировая
трансформация — это накопление нейтральных
жиров вне адипоцитов, чаще всего — в
клетках печени (см. ниже «Нарушения
промежуточного обмена липидов: кетоз и стеатоз
печени»), реже в миокарде и почках.
Мембранообразующую роль в организме
играют, прежде всего, фосфолипиды -
сложные эфиры многоатомных спиртов
глицерина (глицерофосфолипиды), либо
сфингозина (сфингофосфолипиды) с
высшими жирными и фосфорной кислотами.
Фосфолипиды (кроме снабженных
остатками циклического шестиатомного спирта
инозита фосфатидилинозитов) содержат
в своей структуре и различные
азотсодержащие соединения — аминокислоту серии
и, особенно, аминоспирты этаноламин и
холин, который в глицерофосфолипидах
преобладает (до 30% веса лецитина), а в
сфингофосфолипидах служит
единственным азотистым компонентом. Спирты для
синтеза фосфолипидов нужны организму
в макроколичествах. Холин синтезируется
в организме лишь при соблюдении ряда
условий, в частности, в отсутствие
дефицита метионина, фолацина, кобаламина и
пиридоксина. Инозит в изобилии имеется
в растительных продуктах (фитины) и во
всех животных (инозит-фосфатиды), кроме
того, его синтез кишечной микрофлорой
превышает потребность человека. Поэтому
Нарушений липидного обмена 147
у людей не описан дефицит инозита, хотя у
подопытных крыс его исключение из диеты
приводит к дерматиту и задержке роста, а
также периорбитальной алопеции (симптом
«очков»). На этих основаниях, оба вещества
относят к витаминоподобным (см. далее
раздел «Патофизиология витаминного
обмена»).
Фосфолипиды высокоамфифильны и в
воде дают мицеллярные структуры. В
биомембранах клеток они образуют бислой,
а на поверхности липопротеидных частиц
представлены монослоем. Согласно ныне
принятой жидкостно-мозаичной модели
С. Сингера и Дж. Николсона (1972),
клеточные мембраны организованы наподобие
динамического фосфолипидного «озера» с
белковыми «островами»,
перемещающимися в нём. Фосфолипиды мембран могут
претерпевать флип-флоп переходы — из одного
слоя в другой, а также латеральную
диффузию, в том числе — при контакте соседних
клеток или же клеток и липопротеидных
частиц — из мембраны в мембрану.
Липидный состав различных клеточных
мембран неодинаков. Так, сфингозидов
крайне много в мембранах нервной ткани
и миелине. Митохондриальные мембраны
особенно богаты дифосфатидйлглицери-
дом кардиолипином, единственным из
фосфолипидов, обладающим антигенными
свойствами. Аутоантитела к кардиолипину
обнаруживаются при многих системных
иммунопатологических болезнях
соединительной ткани, в частности, при системной
красной волчанке. Кардиолипин -
липидный компонент хроматина, он
взаимодействует с ДНК и влияет на экспрессию генов,
представляя собой объект исследований
липономики - учения об информационной
роли липидов в клетке.
Во всех мембранных структурах фосфо-
липидам сопутствует холестерин,
являющийся стабилизатором клеточной
мембраны. Они связаны в слоях гидрофобными
и ионными взаимодействиями. Фосфоли-
пидные бислой несимметричны. Наружный
монослой обогащен фосфатидилхолином,
внутренний — другими фосфолипидами.
Дополнительное обогащение наружного
слоя фосфатидилхолином и потеря им
сфинголипидов наблюдаются при
активации тромбоцитов и других клеток крови и
способствуют контактному запуску
мембраной механизмов коагуляции и агрегации
(ранее эта функция усиления асимметрии
фосфолипидного бислоя при активации
клеток крови носила условное название
«кровяного тромбопластина» — см. также
в т. I стр. 259).
Мембранообразующая функция
липидов чрезвычайно важна для всех видов и
аспектов метаболизма. Именно благодаря
биомембранам осуществляется компарт-
ментализация клеток, то есть их деление
на отсеки. В разных отсеках клетки
одновременно проходят, не мешая друг другу,
процессы, требующие различных условий,
или даже противоположно направленные.
Пока клетка жива, градиенты веществ по обе
стороны мембран поддерживаются, порой,
ценой больших энергозатрат. Таким
образом, фосфолипиды, в кооперации с
холестерином и мембранными белками, выполняют
важную барьерную функцию и регулируют
процессы активного переноса веществ.
Так например, производное масляной
кислоты — окситриметиламмоний-бутират,
известный как карнитин — транспортирует
ацилы жирных кислот внутрь митохондрий,
а при его нехватке развивается нарушение
их р-окисления и дефицит энергии (см.
далее рис. 43, стр. 228).
В состав яда многих змей, ос и пауков
входят фосфолипазы, чьё токсическое (в
частности, гемолитическое) действие
основано как раз на разрушении мембранных
липидов и нарушении трансмембранных
градиентов. Фосфолипиды мембран должны
постоянно обновляться. При этом
используются компоненты, транспортируемые в
составе липопротеидов. При
наследственном отсутствии ЛПНП и ЛПОНП (абета-
липопротеинемия, см. ниже) стабильность
мембран понижается. Это проявляется, в
частности, в ускоренном гемолизе и
изменении формы эритроцитов (акантоцитоз).
При любой альтерации клеточных мембран
148 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
сами они служат источником биологически
активных соединений — эйкозаноидов,
координирующих местные типовые
патологические процессы, развивающиеся в ответ на
повреждение. О патологии биомембран см.
также т.1, стр.154-159, раздел
«Повреждение плазмолеммы».
Рецепторно-посредниковая роль
позволяет липидам участвовать в распознавании
химических сигналов и их доведении до
внутриклеточных эффекторов. В
осуществлении этой функции первостепенное место
отводится сложным липидам — церебрози-
дам и ганглиозидам. Цереброзиды содержат
спирт сфингозин, высшие жирные кислоты
(цереброновую, нервоновую, лигноцерино-
вую), а также остатки гексоз— галактозы
либо глюкозы. Ганглиозиды имеют еще
более разнообразный и сложный состав,
так как помимо этих компонентов содержат
нейраминовую кислоту и гетероолигоса-
хариды. Роль гликолипидов определяется
тем, что, практически, любой клеточный
рецептор содержит, наряду с белковой,
также и ганглиозидную часть и фиксируется в
мембране именно с ее участием. Гликоли-
пиды, например, цитолипин Н, обладают
антигенными свойствами. Они участвуют
в определении иммунологических свойств
клеточных мембран, в частности, в
формировании агглютиногенов системы АВО.
При патологии возможно формирование
аутоантител к ганглиозидной части
клеточных рецепторов, что приводит к нарушению
информационных процессов в клетке.
Примером служат аутоантитела против
ганглиозидной части рецепторов ТТГ, которые
стимулируют эти рецепторы на поверхности
фолликулярных клеток щитовидной железы
и ответственны за развитие гипертироза при
болезни фон Базедова (см. также т. I данного
руководства, с. 116-117). Провоцирующую
роль в развитии подобных аутоиммунных
рецепторных болезней играют некоторые
бактериальные инфекции, так как глико-
сфинголипиды бактериальных антигенов
зачастую вызывают перекрестный
иммунный ответ. Так, при болезни фон Базедова
подобную роль играет Yersinia enterocolitica
(см. также ниже «Нарушения эндокринной
регуляции»).
Существует целый класс наследственных
болезней накопления — гликосфинголипи-
дозов, когда из-за дефекта лизосомальных
ферментов нарушается обмен различных
сложных липидов. Подробнее они
рассматриваются ниже, в специальном разделе.
Однако, обращает на себя внимание, что
характерной общей чертой этих
разнообразных синдромов служит именно
нарушение различных видов рецепции, что может
проявляться слепотой, неврологическими
расстройствами, дефектами почечной реаб-
сорбции и т.д.
Роль передатчиков сигналов в клетке
по праву разделяют липидные
посредники — главным образом, производные
фосфатидилинозитола и простагландины.
Инозитфосфатиды осуществляют важные
функции в системе передачи
внутриклеточных сигналов вместе с G-белками (см.
т. I, стр. 121-122). Ряд этих белков способен
активировать семейство мембранных ино-
зитолфосфатаз, известных под условным
названием «фосфолипаза С». Под влиянием
этих ферментов мембранный фосфатиди-
линозитол-4,5-дифосфат расщепляется
с образованием инозитолтрифосфата и
диацилглицерина. Эти продукты
активируют, через повышение концентрации
ионизированного кальция в цитоплазме, а
также непосредственно, ряд протеинкиназ
С, которые, в свою очередь, фосфорилируют
многие клеточные белки. Этим путем могут
активироваться белки цитоскелета, а также
дерепрессироваться генетические
программы, связанные с ростом и размножением
клетки. Параллельно происходит усиленное
образование простагландинов. Липидные
посредники играют большую роль в
осуществлении ответа на гормональную
стимуляцию, например, в отсроченных аспектах
реакции адренокортикоцитов на АКТГ. Они
опосредуют и действие многих факторов
роста. Значение липономного посредника
при патологии клеточного роста
иллюстрируется тем фактом, что форболовые эфиры,
компонент кротонового масла, вмешиваются
Нарушения липидного обмена 149
в данные процессы и усиливают активацию
протеинкиназы С. Именно это позволяет им
действовать в качестве коканцерогенов не
будучи мутагенами (см. ниже «Нарушения
эндокринной регуляции» и т. III).
Ярким примером роли липидных
посредников при патологии служит и
опосредование пирогенного и острофазного эффектов
интерлейкинов через простагландины (т. I,
гл. 13).
Антиапоптогенные сигналы в клетках
передаются через сфингомиелиновый
посредник, включающий рецепторы ростовых
факторов и сопряженные с ними сфинго-
миелиназы, с образованием церамидов и
активацией церамид-зависимых протеинкиназ
(см. т. I, с. 201-202).
Сигнальная роль присуща таким липи-
дам, как стероиды. Это рассмотренный в
разделе «Патофизиология витаминного
обмена» витамин D, а также гормоны коры
надпочечников — минералокортикоиды,
глюкокортикоиды и производимые как в
надпочечниках, так и в гонадах половые
гормоны. По некоторым данным, и другие
клетки, в частности, мезенхимального и
лимфоидного происхождения, способны к
выработке стероидных гормонов. Так, они,
по-видимому, могут образовываться в
тимусе и макрофагах. Все стероидные гормоны
являются производными холестерина и
могут возникать из этого готового стероида,
переносимого липопротеидами из печени и
кишечника к месту гормонообразования.
Уже данный факт доказывает, что
холестерин (ХН) — не яд, и не враг рода
человеческого, как порой утверждают в упрощенной
популярной литературе, а нужнейший, в
меру безвредный метаболит. А ведь ХН еще
и является предшественником жёлчных
кислот. Подробное рассмотрение
закономерностей продукции стероидных гормонов
и нарушений этих процессов содержится в т.
I стр. 51 и в данной книге (раздел
«Патофизиология эндокринной регуляции»). Здесь
необходимо напомнить основные функци
онально-структурные характеристики их
главного предшественника — холестерина
(рис. 29). ХН — один из основных «героев»
2Г
з!>
1
А
11^
9
Тв
12
С
^Т*Г
Ji
&
и
17
D
.16
15
циклопентанпергидро-
фенантреновое ядро
стероидов
холестерин
но'
Рис. 29. Структура холестерина
в патофизиологии XX столетия, и уместно
будет посвятить его химии специально
несколько строк.
Холестерин (ХН) известен с XVIII
столетия как компонент жёлчных камней,
благодаря работам А.Ф. де Фуркруа (1789). От
греческих слов «жёлчь» и «твёрдый»
происходит и его название (М. Шеврёль, 1816).
Спиртовая природа холестерина и
возможность его этерификации были доказаны
М. Бертло (1859). Но только в начале
нашего столетия, благодаря А. Виндаусу, была
уточнена его химическая структура (1932).
ХН— производное циклопентана и гидра-
тированного фенантрена. Он имеет гидрок-
сильную группу в положении 3,
непредельную связь в положении 5-6 и боковую цепь
при 17-м углеродном атоме. ХН имеется и в
некоторых растительных тканях. Однако, в
растениях преобладают фитостерины (стиг-
мастерин и р-ситостерол). Они отличаются
от ХН только строением боковой цепи при
17-м атоме углерода. Благодаря сходству с
ХН ситостерин ингибирует его всасывание
в кишечнике и может использоваться как ги-
полипидемическое средство. Суточное
потребление ХН находится в диапазоне от 0,2
до 0,5 г. Одновременно, 0,5 г ХН окисляется
в жёлчные кислоты, столько же теряется с
150 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
фекалиями в виде нейтральных
насыщенных стеринов (особенно, копростерина), а
около 0,1 г выделяют сальные железы. В то
же время, в организме синтезируется в день
более 1 г этого стероида. Наиболее богаты
ХН яичные желтки (1,6-2 г%), сливочное
масло (0,2 г%), сметана и сливки.
ХН отличается жесткой
пространственной структурой. Каждое из циклогексано-
вых колец его молекулы может пребывать
в двух пространственных конформаци-
ях — «кресла» и «ванны». В природном
ХН все кольца находятся в наиболее
устойчивой конформации «кресла». Все клетки
организма содержат ХН в составе своих
мембран и принципиально способны
синтезировать его. Поэтому общее содержание
ХН в теле огромно — более 300 г.
Вследствие своих пространственных свойств,
ХН способен влиять на фазовые переходы
мембранных липидов и их
жидкокристаллические свойства. Мембранные фосфоли-
пиды находятся в жидкокристаллическом
подвижном виде при температурах выше
переходной, а при более низких —
проявляют свойства твердокристаллического
тела. ХН понижает температуру
плавления для фосфолипидов с насыщенными
жирными кислотами и повышает ее — для
фосфолипидных молекул, содержащих
ненасыщенные ацилы (а таких в мембранах
большинство). В результате, ХН понижает
жидкостность и проницаемость
биомембран и участвует в обеспечении их барьерной
функции, влияет на активность
мембранных ферментов. Он защищает мембраны
от электрического пробоя, а, по некоторым
данным, препятствует аутоокислению
мембранных липидов и дезинтеграции
мембран (см. выше, а также в т. I, стр. 154).
Полиеновые антибиотики удаляют ХН из
плазматических мембран. Это ведет к
цитолизу, в частности, гемолизу. Вместе с тем,
избыток ХН меняет свойства мембраны
таким образом, что затрудняется работа
кальциевых насосов. При этом
концентрация свободного цитоплазматического
кальция растет, что опасно для клетки.
При перегрузке плазматической
мембраны ХН, если нет возможности избавиться
от него путем латеральной диффузии в
мембрану липопротеидной частицы,
клетка вступает в деление, чтобы увеличить
площадь мембран и нормализовать
содержание мембранного ХН. Этот механизм
играет известную роль при стимуляции
пролиферативного ответа ряда клеток на
накопление холестеринсодержащего
материала при атеросклерозе и ксантоматозе
(см. ниже). В теле человека преобладает
свободный холестерин, которого почти в
3 раза больше, чем этерифицированного.
Интересно, что мозг, эритроциты и жёлчь
содержат только свободный холестерин, а
мышцы — лишь 7 % ХН в виде эфиров. Зато
в надпочечниках и гонадах (83 %), а также в
плазме (70 %) резко преобладают эфиры
холестерина (ЭХ), немало их и в макрофагах.
По мнению Ю.М. Лопухина (1986), можно
говорить не только о патогенности избытка
холестерина в организме (холестериноз,
предрасполагающий к развитию
атеросклероза), но и о патогенности общего
недостатка холестерина (холестеринодефицит,
оборачивающийся повышением риска
опухолевых и вирусных заболеваний). Эти две
формы нарушения обмена ХН касаются его
общего содержания в организме.
Возможны также нарушения содержания ХН в
крови (гиперхолестеринемии и гипохолестери-
немии). Наконец, большое диагностическое
значение имеют синдромы накопления ХН
в отдельных органах и тканях (первичные
и вторичные болезни депонирования ХН).
Эти нарушения подробно
рассматриваются в разделе «Гиперлипопротеинемии
и другие дислипопротеинемии», а также
в разделе «Патология депонирования и
мобилизации липидов». В данном разделе
приводится классификация холестерино-
патий (по Ю.М. Лопухину-А.С. Фокину,
с изменениями):
I. Общие холестеринопатии.
1.1. Холестериноз
1.2. Холестеринодефициты
И. Дисхолестеринемии
Нарушения липидного обмена 151
И.1. Гиперхолестеринемии
II. 1.1. Первичные
- семейная наследственная гиперхолесте-
ринемия
- гиперхолестеринемия при семейной
гипертриглицеридемии
- семейная дисбеталипопротеинемия
II. 1.2. Вторичные
- механическая желтуха
- первичный билиарный цирроз печени
- сахарный диабет
- гипотироз
- нефротический синдром
- гиперкортицизм
- моноклональные гаммапатии
- гепатома
- беременность
- применение пероральных
контрацептивов
- отравление ДДТ
II.2. Гипохолестеринемии
П.2.1. Первичные
- семейная абеталипопротеинемия
- семейная гипобеталипопротеинемия
- семейная анальфалипопротеинемия
П.2.2. Вторичные
- печёночно-клеточная недостаточность
- анемия
- гипертироз
III. Локальный холестериноз
III. 1. Первичные болезни накопления ХН
- болезнь Вольмана (первичный ксанто-
матоз)
- Танжерская болезнь
- р-ситостеринемия
- церебросухожильный ксантоматоз
- метахроматическая лейкодистрофия
- первичное ожирение
Ш.2. Вторичные болезни накопления ХН
- холелитиаз
- гистиоцитоз X, в том числе болезнь Хэн-
д а- Ш юл ера- Крисчена
- рассеянный склероз
- болезнь Ниманна-Пика
- наружный экссудативный ретинит
- вторичное ожирение.
Оценивая роль липидов в
жизнедеятельности организма, нельзя забывать о том,
что для оптимального роста и
нормального функционирования человеку нужны
жирорастворимые витамины (см. ниже
«Патофизиология витаминного обмена»), а
также не синтезируемые в организме
полиненасыщенные жирные кислоты. Уже одно
это делает липиды важнейшим компонентом
пищи, хотя подавляющее большинство
липидов может возникать в самом организме
из углеводов и аминокислот, поэтому
наличие липидов, по словам Г.П. Конради, «само
по себе, в пище не является необходимым».
Тем не менее, гигиенически рекомендуется
поступление липидов на уровне 30 %
суточного калоража, причем доля растительных
липидов от этого общего количества
должна быть не менее 30% (К.С. Петровский,
1975).
Процесс преобразования избытка
углеводов и аминокислот в липиды проходит в
печени и, в меньшем объеме, в жировой
ткани. Его эффективность, в расчёте по
энергетическому эквиваленту, приближается к
85 % (А. Гайтон, 1994). Преобразование идёт
через ацетил-КоА (рис. 43). Для успешного
хода процесса необходимы эффективное
осуществление гликолиза и пентозного пути
распада углеводов. Ведь эти катаболические
процессы обеспечивают синтез липидов
энергетически (НАДФН из реакций
пентозного шунта) и пластически (глицерин из
реакций гликолиза). Трансформация нели-
пидных веществ через ацетил-КоА в липиды
невозможна без адекватного действия
инсулина. При нехватке инсулина (например,
инсулинзависимом сахарном диабете) липо-
генез из ацетил-КоА тормозится, что ведет
к накоплению промежуточных продуктов
метаболизма жирных кислот — кетоновых
тел. В этом один из важнейших механизмов
152 АЖ Зайчик, Л.П. Чурилов Патохимия
кетоза и кахексии при инсулинзависимом
сахарном диабете (см. разделы «Кетоз и
стеатоз печени» и «Патофизиология
углеводного обмена» ниже). Трансформация
углеводов в липиды ограничена, так как
один из ключевых ферментов этого
процесса — фосфофруктокиназа — ингибируется
избытком жирных кислот. При
соматической мутации регуляторного участка данного
энзима возникают липомы (см. также т. I
данного руководства, стр. 141).
При успешном ходе описанных
преобразований у здорового человека даже
полугодовое пребывание на безжировой диете не
ведет к патологическим последствиям, так
как жирорастворимые витамины и
ненасыщенные жирные кислоты поставляются
из депо, а иные липиды синтезируются из
нелипидных предшественников (П.Е.
Калмыков, М.Н. Логаткин, 1974). Более
длительная или постоянная диета с сильным
ограничением липидов ведет к расстройству
здоровья, возникает алиментарная липид-
ная недостаточность.
Основные проявления алиментарной ли-
пидной недостаточности снимаются
введением полиненасыщенных жирных кислот.
В организме человека они не
синтезируются, и поэтому некоторые авторы
характеризовали их как «витамин F» — от
английского fat — жир. Впрочем, так как
потребность в них составляет от 4 до 8 г в
сутки, они не подпадают под критерии
истинных витаминов. Не менее 1 %, а у детей,
беременных и кормящих женщин — до 2 %
калорийности суточного рациона должны
поступать с эссенциальными полиеновыми
кислотами. Обязательность потребления
этих ингредиентов пищи выяснена в 1929 г.
(Дж. Бурр, М. Бурр) на крысах.
Проявления дефицита «витамина F» включают
задержку роста, алопецию, дерматит, некроз
хвоста, гиперемию и отек почек.
Характерной особенностью состояния эпидермиса
при нехватке полиеновых жирных кислот
является роговая дистрофия и
гиперкератоз, которые ранее считали типичными
лишь для авитаминоза А (Д.Б. Джелифф,
1967). Изменяется жирнокислотный состав
(эндокринно-метаболические нарушения)
фосфолипидов митохондриальных мембран
печени, облегчается разобщение фосфори-
лирования и окисления. Установлено, что
при дефиците эссенциальных полиеновых
кислот возникает гиперхолестеринемия и
ослабляется дренажная функция ЛПВП в
отношении холестерина тканей, липопро-
теиды без эссенциальных жирных кислот
менее устойчивы к окислению и другим
преобразованиям, облегчающим их накопление
в сосудистой стенке (см. ниже). Поэтому
относительная нехватка полиеновых кислот
в диете ускоряет развитие атеросклероза.
Так как они существенны для образования
печеночных фосфолипидов, их дефицит
ослабляет липотропное действие холина,
метионина и ряда витаминов, способствуя
стеатозу печени. Эссенциальные жирные
кислоты взаимодействуют с витамином В6,
который участвует как коэнзим в переходе
линолевой кислоты в арахидоновую. Ара-
хидоновая кислота излечивает такие
проявления дефицита эссенциальных жирных
кислот, как гиперхолестеринемия и
задержка роста, но для предотвращения дерматита
нужны и другие полиеновые кислоты.
Линолевая кислота содержит 18 атомов
углерода и имеет 2 двойных связи в
положениях 9 и 12. Она составляет 10 % жирных
кислот в адипоцитах и до 13% — в крови.
В фосфолипидах человека ее содержание
еще выше — до 30%, а в кардиолипинах
более 80 %. Огромное метаболическое
значение линолевой кислоты заключается в том,
что она этерифицирует более половины всех
эфиров холестерина. При наличии в диете
линолевой кислоты, остальные полиеновые
кислоты, рассматриваемые ниже, могут
возникать из нее в организме.
Линоленовая кислота, также 18-углерод-
ная, имеет 3 непредельных связи (в тех же
позициях, что и линолевая, а также в 15
положении). Ее количество в организме
минимально, но ее у-линоленовое производное
является важным промежуточным
продуктом превращения полиненасыщенных
жирных кислот. И линолевая, и линоленовая
кислота в пище, главным образом,
содержится в растительных продуктах. Особенно
Нарушения липидного обмена 15 3
богато ими конопляное и льняное масло.
В первом преобладает линолевая, во
втором — линоленовая кислота. Подсолнечное,
кукурузное и хлопковое масла — обильный
источник линолевой кислоты, но линоле-
новой не содержат. В кокосовом, наоборот,
нет линолевой.
Арахидоновая кислота - 20-ти
углеродное соединение с четырьмя непредельными
связями (в позициях 5, 8, 11 и 14). Входит
в состав фосфолипидов мембран, эфиров
холестерина и некоторых триглицеридов.
В печени составляет до 8 % жирнокислот-
ного состава. Поступает только из животных
продуктов, в частности, свиного жира, мяса,
печени и рыбы. Является предшественницей
важнейших аутокоидов — эйкозаноидов,
участвующих в воспалении, преиммунном
и иммунном ответе, модуляции и
опосредовании эффекта других биорегуляторов
(см. т. I, стр. 155-158,359-361). В организме
может возникать на основе линолевой
кислоты, при наличии пиридоксина.
Тимиодоновая кислота (C20-D-5,8,ll,17-
эйкозапентаеновая) содержится в животных
морепродуктах и, особенно, рыбьем (в
частности, скумбриевом) жире. Особенностью ее
метаболической роли считают возможность
ее преобразования непосредственно в про-
стациклин (Pgl2), обладающий антитром-
богенным и дезагрегационным действием, в
то же время, тромбоксаны из нее получены
быть не могут. Диета, богатая тимиодоновой
кислотой, снижает риск развития тромбоза и
тормозит прогрессирование атеросклероза.
НАРУШЕНИЯ
ПЕРЕВАРИВАНИЯ
И ВСАСЫВАНИЯ ЛИПИДОВ
Выдающемуся русскому врачу
С.П.Боткину (1860) принадлежит открытие, что
основным местом переваривания и
всасывания липидов является тонкий кишечник.
Читателям-педиатрам напомним, что у
новорожденных и грудных детей имеется
активная желудочная липаза, способная
переваривать эмульгированные жиры
грудного молока. Этому способствуют низкая
кислотность младенческого желудочного
сока (рН=5) и адаптивное усиление
синтеза желудочной липазы при естественном
вскармливании. Более того, доказано, что
у младенцев клетками слизистой оболочки
корня языка и глотки при сосании также
секретируется липаза, продолжающая свое
действие в желудке. В остальных случаях,
эффективное переваривание липидов
возможно только после превращения пищевых
липидов в эмульсию, при участии
компонентов жёлчи. Однако, и у взрослых
сохраняется небольшая липазная активность в
желудочном соке, поэтому можно считать
доказанным, что «затравочный гидролиз»
начинается в желудке, а порции жирных
кислот, поступая в кишечник, инициируют
там эмульгирование липидов.
Основные липолитические ферменты
кишечника имеют, как установил К. Бернар
(1856) панкреатическое происхождение.
Это, прежде всего, поджелудочная липаза,
которая при оптимуме рН=8-9 расщепляет
в триглицеридах крайние а-эфирные
связи, причём получаются жирные кислоты
ир-моноглицерид. Любопытно, что при
этом, практически, не освобождается
глицерин, хотя во многих учебниках традиционно
указывают, что «нейтральные жиры
расщепляются до глицерина и жирных кислот в
кишечнике». Липаза действует на поверхности
эмульсионных липидных капель. Поэтому
ее активность зависит от наличия в тонком
кишечнике жёлчи, содержащей
эмульгаторы. Ведущими эмульгаторами являются
парные жёлчные кислоты — гликохолевая,
таурохолевая, гликохенодезоксихолевая и
таурохенодезоксихолевая.
Разрыхлению пищевого комка
способствует и углекислый газ, образуемый при
нейтрализации желудочной кислоты
кишечным бикарбонатом. Инициирующую
роль при эмульгировании играют моногли-
церидные остатки и первые образовавшиеся
молекулы жирных кислот. В их присутствии
жёлчные кислоты образуют соли, которые
локализуются на поверхности капелек жира
154 АШ. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
и не дают тем сливаться. Поверхность,
доступная липазе, резко увеличивается.
Панкреатическая фосфолипаза А2
расщепляет сложноэфирную связь в р-положении
фосфолипида. Образуются жирная кислота
и лизофосфолипид, который не менее
активен, чем компоненты жёлчи, как эмульгатор.
Лизофосфолипаза поджелудочного сока
разрушает его до водорастворимого глицерил-
фосфохолина, всасываемого в кровь, и
жирных кислот. Эфиры холестерина расщепляет
перед всасыванием холестеринэстераза
панкреатического и кишечного сока.
Всасывание различных липидов в
кишечнике проходит неодинаково.
Эмульсионные капельки размером до полумикрона
проходят через стенку кишечника без
предварительного гидролиза. Более крупные
капли подвергаются гидролизу и
всасываются с участием жёлчных кислот. Из фос-
фолипидов, ХН и жёлчных кислот жёлчи,
с одной стороны, а также жирных кислот
и моноглицеридов пищи — с другой —
образуются смешанные мицеллы. Мицеллы
абсорбируют дополнительный ХН пищи
и жирорастворимые витамины. Размер
мицелл и гидрофильный поверхностный
слой, богатый холеинатами, позволяет им
всасываться. Механизмы всасывания
включают мицеллярную диффузию без затраты
энергии, а также пиноцитоз и молекулярную
латеральную диффузию липидов с
поверхности мицеллы в мембрану энтероцита.
Жёлчные кислоты при всасывании
покидают поверхность мицеллы и участвуют в
энтерогепатической рециркуляции. Всего
15-18 г жёлчных кислот за сутки могут
обеспечить эмульгирование и всасывание
более 100 г жиров, то есть они совершают
не менее 5-8 циклов в день, при потерях
до 0,5 г, компенсируемых ресинтезом из
холестерина печени. Кишечно-печёночный
кругооборот жёлчных кислот играет важную
роль не только для всасывания липидов, но
и для регуляции метаболизма ХН, а также
для оптимального функционирования
гепатоцитов. Жёлчные кислоты
улучшают кровоснабжение печени, осмотически
стимулирют образование жёлчи и
способствуют ее насыщению фосфолипидами и
ХН (холеретическое действие), усиливают
перистальтику кишечника. Холестаз
приводит не только к нарушению переваривания
и всасывания липидов, но и к изменениям
транспорта холестерина кровью (гиперхо-
лестеринемии).
Наиболее короткие жирные кислоты с
длиной углеродистого скелета до 10 атомов,
например, масляная — всасываются вне
мицелл, прямо в кровь воротной вены. Этот
процесс особенно важен у грудных детей,
так как молоко содержит много короткоце-
почечных жирных кислот. Ненасыщенные
жирные кислоты и те, что имеют чётное
число атомов углерода, всасываются
быстрее других.
В энтероцитах происходит ресинтез
липидов. При ресинтезе образуются на основе
абсорбированных моноглицеридов и жирных
кислот, образованных de novo, нейтральные
жиры и фосфолипиды характерного для
человека строения. Происходит обмен аци-
лами жирных кислот между экзогенными и
собственными (кишечными и печеночными
всосавшимися) липидами. Способность
трансформировать липиды в стенке
кишечника ограничена. Экспериментально
доказано, что при перекорме экзогенными жирами,
особенно, после голодания, часть жиров без
изменений проходит кишечный барьер и
откладывается в адипоцитах. Так, в жировой
ткани животного организма могут
появляться даже растительные жиры (A.M. Лебедев,
1885). Однако, состав жиров других клеток,
кроме адипоцитов, от пищевого жира не
зависит. ХН частично этерифицируется в
стенке кишечника. После этого в гладком
эндоплазматическом ретикулюме энтероци-
тов идет важнейший процесс формирования
стабильных липопротеидных комплексов
крупного размера — хиломикронов (ХМ).
ХМ содержат много триглицеридов, а также
фосфолипиды и ХН. Для образования
гидрофильной оболочки частиц ХМ энтероцит
синтезирует особый белок — апопротеин В.
Готовые ХМ диффундируют через
мембрану млечных сосудов в лимфу и по грудному
лимфатическому протоку входят в кровь,
Нарушения липидного обмена 155
придавая молочный цвет лимфе и плазме
крови, непосредственно после насыщения.
Вышеописанные сложные процессы
нарушаются при нескольких расстройствах:
- Из-за отсутствия жёлчи в кишечнике
(синдром ахолии).
- Из-за нарушения поступления в
кишечник панкреатического сока. Отметим,
что ухудшению эмульгирования и
переваривания жиров способствует и низкая
кислотность желудочного сока.
- Из-за первичной мальабсорбции (целиа-
кия, тропическая спру, болезнь Уиппла,
другие хронические энтериты,
гиповитаминоз по фолиевой кислоте).
- При приеме значительных количеств
тугоплавких липидов животного
происхождения (например, бараньего жира),
особенно у детей.
- При ускоренной перистальтике
кишечника.
- При ингибирующем действии
антибиотиков (неомицин, хлортетрациклин)
и блокаторов фосфорилирования (мо-
нойодацетат, флоридзин) на функции
энтероцитов.
- При гипокортицизме, в связи с потерей
натрия и осмотическими нарушениями.
- При избытке двухвалентных
щёлочноземельных катионов в пище и воде, что
способствует образованию
труднорастворимых кальций-магниевых солей
жирных кислот. Задержка липидов, в
частности, ХН в просвете кишечника
достигается и с помощью
ионообменных смол, связывающих жёлчные
кислоты (холестирамин, квестрол,
холестипол).
Во всех случаях нарушения
переваривания и всасывания липидов появляется
стеаторея. При стеаторее стул становится
частым и липким, из-за ахолии — часто
глинистым на вид, содержит липидные капли
и беловатые комочки мыл (кальциевых и
магниевых солей жирных кислот). При
хронической стеаторее вторично развивается
гиповитаминоз по жирорастворимым
витаминам (см. выше). Довольно закономерны
коагулопатия и остеопороз.
Если стеаторея вызвана ахолией, то она
сопровождается обесцвечиванием кала.
Стул содержит не всосавшиеся, но
полупереваренные (за счёт действия мыл) липиды.
При панкреатической стеаторее нет ахолии,
кроме того, нарушено переваривание и
всасывание и других, нелипидных компонентов
пищи (см. выше, в разделе
«Патофизиология белкового обмена» о креаторее). При
нарушении переваривания и всасывания
липидов возможно понижение содержания
ХМ и других липопротеидов, а значит —
триглицеридов и ХН — в лимфе и плазме
крови. Блокаду всасывания липидов и ХН с
помощью неомицина, холестирамина и
растительных стероидов используют в терапии
гиперлипопротеинемий (см. ниже).
Компенсаторным механизмом при
нарушении абсорбции липидов в верхних
отделах тонкой кишки является активизация их
всасывания в нижних отделах.
транспорт липидов
В ОРГАНИЗМЕ
И ЕГО НАРУШЕНИЯ
Суммарное содержание всех липидов в
плазме крови — от 4 до 8 г/л. Но большинство
липидов нерастворимы в воде. Поэтому в
плазме крови, в кругообороте между
органами и тканями они существуют лишь в солю-
билизированной форме. Около 5 % жирных
кислот плазмы в неэтерифицированной
форме (НЭЖК) образуют растворимые
комплексы с альбумином. Альбумин имеет
2 высокоаффинных и еще 3 менее аффинных
участка связывания НЭЖК на каждой
белковой молекуле. Эта транспортная система,
в основном, участвует в переносе НЭЖК
из депо к месту утилизации. Остальные
липиды транспортируются в виде особых
комплексов со специализированными
транспортными белками — апопротеинами.
Липопротеиды (ЛИ)— это
надмолекулярные образования, основанные на нековален-
156 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
тных связях, то есть частицы. Липопротеид-
ные частицы транспортируют экзогенные
и эндогенные липиды между местами их
всасывания и/или образования, утилизации
и депонирования. На системе ЛП лежит не
только снабжение тканей липидами, но и
обратный процесс, то есть дренирование
избытка тканевых липидов. Важной
дополнительной ролью ЛП является транспорт
жирорастворимых гормонов и витаминов.
Частицы ЛП имеют сферическую форму
и состоят из гидрофильной оболочки и
гидрофобного ядра. Гидрофильная
оболочка — это вязкий мозаичный монослой,
содержащий полярные фосфолипиды и ХН,
а также апопротеины. Она обеспечивает
растворимость ЛП и (благодаря сигнальной
роли апопротеинов) определяет пути
метаболизма и судьбу каждого класса липопро-
теидных частиц.
Гидрофобное ядро содержит основной
груз такой транспортной частицы: это могут
быть неполярные ЭХ и триглицериды (ТГ)
в разных соотношениях, в зависимости от
класса ЛП (рис. 30). Вязкость и
электронная плотность ядра меньше, чем оболочки.
В присутствии избытка ненасыщенных
жирных кислот в фосфолипидах и эфи-
рах холестерина,
микровязкость оболочки и ядра
частиц ЛП понижается,
что способствует более
динамичному обмену
составными частями между Л П и
клеточными мембранами.
Существуют не менее
пяти классов ЛП,
различных по свойствам и
метаболической роли. Их главное
информационное отличие
заключается в апопротеи-
новой композиции. В связи
с этим, одни из них богаты
ТГ, другие ХН и третьи —
фосфолипидами.
Основные ТГ
переносятся в составе хиломикронов
(ХМ) и липопротеидов
очень низкой плотности
(ЛПОНП). ХН обильнее всего присутствует
в составе липопротеидов промежуточной
плотности (ЛППП) и липопротеидов
низкой плотности (ЛПНП). Наконец, ли-
попротеиды высокой плотности (ЛПВП)
представляют группу, наиболее богатую
фосфолипидами. Содержание ЭХ
значительно и в ЛПНП, и в ЛПВП.
Разделение ЛП на классы достигается
при ультрацентрифугировании в
градиенте плотности, так как они имеют разную
скорость флотации (Sf). ХМ, даже при
обычном центрифугировании или стоянии
плазмы в пробирке, всплывают. Остальные
ЛП тонут с разной скоростью, наивысшей
у ЛПВП.
ЛП различаются также по электрофоре-
тической подвижности. При электрофорезе
на ацетилцеллюлозе и бумаге наиболее
крупные по размеру ХМ остаются на старте.
ЛПОНП формируют пре-р-ЛП фракцию,
ЛППП перемещаются в так называемой
«медленной р», а ЛПНП- представляют
Р-ЛП фракцию, ЛПВП мигрируют как
а-ЛП.
Основные физико-химические свойства
ЛП этих классов суммированы в
таблице 10.
апо В
ТГ
ЭХ
6 "ч
нэх
апопротеины А, С
ЭХ
25-70 им
2 им
ФЛ
9,5-11 им
ЛПНП
ЛПВП
Рис 30. Строение липопротеидных частиц. Слева — ЛПОНП.
Справа — ЛПВП. Сокращения: ЭХ — эфиры холестерина,
НЭХ — неэтерифицированный холестерин, ТГ — триглицериды,
апо — апопротеины, ФЛ — фосфолипиды, ХС — холестерин,
нм — нанометры (по А.Н. Климову и соавт., 1984)
Нарушения липидного обмена 157
Таблица 10. Физико-химические свойства ЛП разных классов
Показатель
Гидрати-
рованная
плотность
(г/мл)
Скорость
флотации Sf
(е 1063 г/мл).
Мол. масса кД
Диаметр
частиц (нм)
% белка
% фосфоли-
пидов
%ХН
%ЭХ
%тг
Содержание
натощак в
| плазме (г/л)
Что
переносят
Апопротеины
Электрофо-
ретическая
| подвижность
Направления
транспорта
Гиперлипо-
| протеидемии
Атерогенный
| потенциал
ХМ
<0,95
>400
106-107
100-1100
1-2
3-8
2
2
86-94
ТГпищи
В48, Е, СМИ,
AMI
На старте
Из
кишечника в ткани и
печень
IV
Минимальный
ЛПОНП
0,95-1,006
20-00
5x103-13x104
28-100
7-10
12-18
3-7
10-13
50-60
0,8-1,5
Эндогенные
ТГ
В48,С1-1И,Е
Пре-Р
Излечение
ткани
IV,V
Малый
ЛППП
1,006-1,019
12-20
4-5x103
25-30
14-18
11-17
6-12
29-33
24-34
0,2-0,75
ЭХ,ТГ
В100,Е,СШ
Медленные Р
Продукт
метаболизма
ЛПОНП
Ш,ПЬ
Высокий
ЛППП
1,019-1,063
0-12
2,7-4,8x103
21-25
22-35
22
8
38
7-10
3,2-4,5
ХН,ЭХ
В100
Р
Продукт
метаболизма
ЛППП
II a, lib
Очень
высокий
ЛПВП2
1,063-1,125
2,3-3,8x102
9,5-10
33
29
7
23
8
0,5 (м) 1,7 (ж)
ХН,ЭХ, фосфо-
липиды
AI-IVCI-III
а
Дренаж
тканей,
транспорт в
печень
Гипер-а
Антиатеро-
генный
лпвпз I
1,125-1,210
1,5-2,1x102
7-7,5
59
20
2
13
6
2,2 (м) 2,6 (ж)
ЭХ, фосфо-ли-
пиды
AI-IV, D,F
а
Дренаж
тканей,
транспорт в
печень
Гипер-а
Антиатеро-
генный
Из представленной таблицы видно, что
только ЛПОНП, ЛППП и ЛПНП
непосредственно превращаются друг в друга при
метаболизме. ХМ и ЛПВП переносят ли-
пиды на других участках их кругооборота,
и липидный материал переходит из них в
другие ЛП только в печени или путем
латеральной диффузии — из одной частицы
в другую. Более того, одни и те же липиды
в составе разных ЛП неэквивалентны по
своему значению, ибо находятся на разных
путях и стадиях метаболизма.
Кругооборот ЛП в организме
характеризуется в норме следующими особенностями
(рис.31).
Хиломикроны (ХМ) известны с 1774 г.,
когда английский врач У. Хьюсон обнаружил
белесоватый вид крови при кровопускании
и установил, что причиной этого является
абсорбционная липемия. В 1920 г. С. Кэйдж
локализовал ХМ под микроскопом после
приема жирной пищи, как «танцующие в
сыворотке частицы, диаметром в несколько раз
меньше эритроцитов», и дал им современное
158 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
эндогенный путь
——ооо
ЛПОНП X ЛПНП
П5г-
0
МЫШЦЫ
ЖИРОВАЯ ТКАНЬ
жировая ткань
мышцы
Рис. 3 7. Кругооборот липопротеидов в организме. ЛПЛ — липопротеиновая липаза, ФЛИП — фос-
фолипиды, апоапопротеины; остальные сокращения — как в списке сокращений. Пояснения — в
тексте.
название. ХМ образуются вэнтероцитах,
если ТГ пищи содержат длинноцепочечные
жирные кислоты, и когда превалирует
всасывание липидов в лимфу. Ключевую роль в
сборке частиц играет апопротеин В48. В них
также широко представлены апопротеины
С I—III и имеются апопротеины AI-II
кишечного и печеночного происхождения.
Однако, свежесекретированные частицы
ХМ, практически лишены апопротеинов С и
А и приобретают их в результате контакта с
челночными ЛПВП уже в плазме крови.
ХМ — это первый транспортер
экзогенных пищевых липидов, прежде всего, ТГ, на
пути через лимфу в кровь. Их
метаболические превращения известны как экзогенный
путь кругооборота Л П.
С кровью ХМ переносятся, в первую
очередь, в правое сердце и легкие, а затем в
большой круг кровообращения. При этом
они все время теряют значительные
количества ТГ за счёт липопротеид-липолиза
и их гидрофобное ядро заметно «худеет».
ХМ превращаются в остаточные частицы,
в которых фосфолипиды, ХН и
апопротеины находятся в относительном избытке.
Интересно, что атерогенный потенциал
остаточных частиц — высокий, хотя сами ХМ
неатерогенны. На поверхности похудевшей
частицы ХМ возникают складки
избыточной оболочки, которые могут отрываться
от частицы, замыкаться в особые малые
богатые фосфолипидами и апопротеинами
С, Е и А «насцентные диски» и служат затем
основой для наполнения гидрофобными
липидами и образования ЛПВП. Механизм
эффекта, обеспечивающего просветление
липемической плазмы, которое интенсивно
идет уже в первые 15 мин и за12-14ч после
приема жирной пищи полностью убирает
из плазмы ХМ, обеспечивается ферментом
липопротеиновой липазой (ЛПЛ). Этот
энзим капиллярной стенки освобождается
в плазму в ответ на жировую нагрузку и
гепарин. Наибольшую липолитическую
активность проявляют капилляры жиро-
Нарушения липидного обмена 159
вой ткани,мышц, легких и сердца, кроме
того ЛПЛ выделяется в печени, селезёнке,
почках, лактирующей молочной железе и
диафрагме. Очевидно, что это связано с
интенсивным отложением ТГ в адипоцитах,
с секрецией липидной эмульсии в молоко
и с энергетикой миокарда и диафрагмы,
использующих в топливных целях много
жирных кислот. Любопытно, что в
легких процесс частичного катаболизма ХМ
(активный липодиэрез) играет ключевую
роль для обеспечения высокой активности
альвеолярных макрофагов и существенно
необходим для синтеза фосфолипидов
сурфактанта. В связи с этим, при лёгочных
инфекциях благотворно действует жировая
диета. Еще народные знахари применяли
барсучий и медвежий жир и собачье сало
при чахотке. Такая процедура, как
искусственный лечебный пневмоторакс,
опосредует свой эффект не только через
возникающую в спавшемся лёгком венозную
гиперемию и усиление фибропластических
процессов. Известное значение имеет и
усиление недыхательных функций лёгких при
снижении вентиляции. Традиционное
питание северных народов, находящихся под
воздействием климатических факторов
повышенного риска бронхита и пневмонии, не
случайно богато жирами. Эта особенность
экологии повышает резистентность чукчей,
эскимосов и других представителей малых
реликтовых этносов к бронхолегочной
патологии. К сожалению, чтобы осознать это,
понадобился печальный опыт
«окультуривания» советского и американо-канадского
Севера, когда форсированный переход на
европеизированную диету и образ жизни,
несмотря на формально «улучшенные
условия существования», привел (в
рамках обеих противоположных социальных
систем) к значительному возрастанию
патологической пораженности болезнями
дыхательной системы у аборигенов. Конечно,
здесь сыграло роль и учащение контактов с
носителями новых для северных изолятов
штаммов инфекционных возбудителей.
Но, по крайней мере, во многих случаях
для индивидов оказывается биологически
выгодно оставаться в рамках привычной
экологии.
Активность ЛПЛ стимулируется
инсулином и СТГ. У человека более 80%
липогенеза в адипоцитах идет на основе
готовых жирных кислот, поставляемых
ЛПЛ-реакцией и только 20 % синтезируется
из углеводных предшественников в самих
жировых клетках.
Гепарин не является кофактором ЛПЛ,
но запускает ее секрецию. Коэнзимную
роль для ЛПЛ выполняет компонент ХМ,
апопротеин С-И. Вместе с тем, апопротеин
C-III, наоборот, ингибирует ЛПЛ. Таким
образом, от соотношения С-И и С-Ш может
зависеть скорость просветления
постгепариновой плазмы. Альбумин, подхватывая и
удаляя из сферы действия реакции НЭЖК,
также значительно ускоряет просветление
липемической плазмы. В связи с этими
фактами, липемия очень часто сопровождается
тромбофилитическим состоянием. Более
того, гипоальбуминемия, свойственная
голоданию и нефротическому синдрому,
протекает с задержкой катаболизма ХМ и других
ЛП в плазме и гиперлипопротеидемиями.
Кроме ХМ, ЛПЛ аналогично действует и
на Л П ОН П. Остаточные частицы ХМ
теряют апопротеины С и А, переходящие на
частицы ЛПВП. Через обмен апопротеинов
ЛПВП могут регулировать скорость
катаболизма ХМ и ЛПОНП, так как служат
челноком, снабжающим богатые триглицеридами
ЛП активаторами ЛПЛ апопротеинами С. В
конце концов, остатки ХМ приобретают из
состава ЛПВП апопротеин Е, который
способствует их захвату печенью через особый
апо-Е-чувствительный и комбинированный,
апо-В/Е-чувствительный рецепторы, и
подвергаются рецепторному эндоцитозу в
гепатоциты, где расщепляются. При этом
ХН и другие липиды поставляются в
печень, которая использует их для продукции
жёлчных кислот и прямой экскреции липи-
дов в жёлчь, для собственных пластических
и энергетических нужд и для продукции
ЛПОНП.
Имеются данные, что высокая активность
ЛПЛ в капиллярах адипоцитов способе-
160 АЖ Зайчик, Л.П. Чурилов Патохимия
твует ожирению. Установлено, что при
задержке катаболизма ХМ, из-за низкой
активности ЛИЛ, развивается продлённая
или стабильная хиломикронемия (см. ниже
о гиперлипопротеидемиях I и V типа). При
голодании активность ЛИЛ мышц растет, а
в жировой ткани — уменьшается.
Попадание молочной лимфы из грудного
лимфатического протока в плевральную
полость дает при травмах грудной клетки
хилоторакс, а фистула между кишечной
лимфатической системой и мочевыводящи-
ми путями проявляется хилурией (липиду-
рией), при которых экссудат и моча имеют
характерный вид молока.
Хиломикронемия повышает риск
тромбоза и эмболии и ведет к панкреатонекрозу
и панкреатитам (см. ниже).
Эндогенный путь кругооборота ЛП
связан с продукцией в печени ЛПОНП, их
дальнейшими превращениями и утилизацией.
ЛПОНП выделены в 1955 г. Р. Гавелом.
Это очень изменчивый класс ЛП,
включающий достаточно широкий спектр частиц,
нагруженных ТГ в неодинаковой степени и
секретируемых печенью в кровь.
Возможно, что минимальные количества ЛПОНП
возникают и в энтероцитах. При высоком
содержании ЛПОНП плазма не дает слив-
кообразного отстоя, как при
гиперхиломикронемии, но опалесцирует. Для продукции
ЛПОНП в печени необходим интенсивный
синтез фосфолипидов, создающих их
гидрофильную оболочку. Чем больше поступает
в печень липидного материала (НЭЖК из
жировой ткани и из точек действия ЛПЛ;
остатки ХМ; ЛПВП, дренирующие ткани),
тем больше необходимо синтезировать
фосфолипидов и создавать частиц ЛПОНП.
Печень также синтезирует липиды,
подлежащие включению в ЛПОНП, из углеводов и,
в меньшей степени, аминокислот. Глюкагон
способствует поступлению ацилов длинно-
цепочечных жирных кислот в митохондрии
и их сгоранию, активируя ацил-карнитин-
трансферазу. Инсулин, наоборот, тормозит
эту транспортную систему и направляет
ацилы жирных кислот на синтез липидов.
Пищевая нагрузка вызывает усиленный
чдокринно-метаболические нарушения)
катаболизм АКоА и накопление цитрата,
который непосредственно стимулирует
синтез НЭЖК в гепатоцитах и выход в кровь
частиц ЛПОНП. Метильные донаторы, они
же — липотропные факторы (холин, бетаин,
метионин, кобаламин) способствуют
образованию фосфолипидов и секретируемых
ЛПОНП. Метильные акцепторы
(никотиновая и гуанидинуксусная кислоты) могут
быть антилипотропными и способствуют
ретенции липидов в гепатоцитах.
Существенна также достаточная степень
обеспечения этих процессов энергией в форме
НАДФН, что нарушается при затрате
энергии на цели дезинтоксикации, при
различных отравлениях. При нарушении
баланса между поступлением липидов в
печень, построением частиц ЛПОНП и
их секрецией в кровь возникает ожирение
(стеатоз) печени (см. ниже раздел «Кетоз
и стеатоз печени», а также в т. I, стр. 107,
184). Стеатоз печени может протекать
как с повышением содержания ЛПОНП в
плазме (если печень максимально
интенсифицирует их сборку в ответ на усиление
поступления липидов и углеводов в гепато-
циты), так и с понижением уровня ЛПОНП
в плазме (если ретенция липидов связана
с угнетением сборки частиц в гепатоцитах
при интоксикациях). Решающее значение
для продукции частиц ЛПОНП в печени
имеет синтез их уникального апопротеи-
на — В100, который представляет собой
видоизмененный димер апо-В48.
Судьба ЛПОНП в крови связана с их
катаболизмом под влиянием ЛПЛ,
действующей и на ХМ. ЛПОНП теряют ТГ, которые
утилизуются адипоцитами и мышцами,
лишаются апопротеинов Е и С, но апо-ВЮО
полностью остается в них. Поэтому 96%
всего апо-ВЮО находится у людей в ЛПНП,
а на долю их предшественников приходится
лишь 4%. При кормлении избытком ХН в
ЛПОНП резко возрастает количество апо-
протеина Е и концентрация ХН.
В ходе катаболизма ЛПОНП
относительный процент ЭХ и ХН в липопротеидных
частицах возрастает и ЛПОНП переходят
вЛППИ
Нарушения липидного обмена 161
, Уровень ЛППП в плазме повышается при
нарушении катаболизма ЛПОНП. Этому
способствует замедленная потеря из состава
ЛП мутантного апопротеина Е (см. ниже — о
гиперлипопротеидемии III типа). У многих
плотоядных видов млекопитающих и у
грызунов ЛППП, в основном, захватываются
гепатоцитами при участии рецептора
апопротеина В100 и нацело метаболизируются.
Часть ЛППП, не захваченная печенью, при
дальнейшем катаболизме с участием как
ЛПЛ, так и секретируемой в кровь
аналогичной печеночной липазы, образует ЛПНП.
ЛПНП — это наиболее богатая ЭХ и ХН,
самая атерогенная фракция ЛП плазмы
человека. У человека, приматов, птиц, свиньи и
травоядных, в отличие от крыс и некоторых
других видов, довольно большая часть ЛППП
избегает печеночного эндоцитоза. Это
приводит к относительно высокому содержанию
холестерина ЛПНП у всех этих видов (до
75 % от общего уровня всех форм плазменного
ХН). Данная особенность учитывается при
моделировании атеросклероза (см. ниже).
Благодаря высокой плотности рецепторов
для апо-В 100 на многих клетках, в частности,
адренокортикоцитах, гепатоцитах, почечном
эпителии, клетках гонад, лимфоцитах,
клетках сосудистой стенки, ЛПНП поставляют
холестерин для метаболических нужд всем
этим потребителям. Адренокортикоциты,
гонады и гепатоциты предпочитают для
образования физиологически значимых
дериватов стероидов пользоваться готовым
ХН из ЛПНП, хотя способны синтезировать
его и заново из АКоА.
В клетки мозга липиды поставляют,
в основном, ЛППП, так как там шире
представлены рецепторы к имеющемуся
у этих ЛП апопротеину Е. В итоге катабо-
лических превращений апо-В-зависимых
ЛП, более 80% их утилизируют печень и
другие вышеназванные клетки, путем
регулируемого рецепторного эндоцитоза, через
апо-В 100/Е-чувствительные рецепторы.
Остальная часть ЛП захватывается
макрофагами путём фагоцитоза и деградирует в
этих «клетках-мусорщиках», причём в этом
участвуют иные рецепторы.
В 1984 г. М. С. Браун и Дж. Гольдстайн
были удостоены Нобелевской премии по
медицине за раскрытие механизмов
рецепторного эндоцитоза ЛП клетками и
последующих событий, адаптирующих клетки к
нагрузке ХН. Суть этих механизмов,
вкратце, сводится к следующему (рис.32).
После ассоциации апопротеина В или
апопротеина Е с соответствующим
рецептором клеток-потребителей ЛП (к которым
относятся, в частности, гладкомышечные
клетки и фибробласты сосудистой стенки,
а также гепатоциты) происходит интерна-
лизация комплекса рецептор-липопротеид.
Комплекс попадает в лизосомы, где
диссоциирует. Рецептор переносится обратно на
плазматическую мембрану и рециркулирует
с новыми молекулами ЛПНП, ЛППП или
ЛПОНП. ЭХ подвергаются действию холес-
терин-эстеразы (кислой липазы), а
свободный ХН включается в клеточные мембраны.
Фосфолипиды подвергаются действию
фосфолипаз, а апопротеин деградирует до
аминокислот с участием протеаз. В норме,
будучи «информирована» о захвате ЛП
частицы, клетка не допускает перегрузки ХН,
запуская гомеостатические механизмы:
>• Усиливается деградация компонентов
ЛП в лизосомах, как описано выше.
>• ХН подавляет продукцию новых
клеточных апо-В/Е-чувствительных
рецепторов, и клетка «глохнет» по отношению к
новым частицам ЛПНП.
>* Снижается продукция собственного
клеточного ХН, так как ингибируется
фермент р-окси-р-метил-глутарил-коэн-
зим-А-редуктаза.
>• Происходит реэтерификация избытка
ХН с помощью активации тканевой
ацил-холестерин-ацил-трансферазы
(АХАТ), которая и переводит ХН в ЭХ.
>* Главным механизмом, предохраняющим
ткань от избытка ХН, служит дренажная
система ЛПВП. В работе этого
механизма участвуют насцентные диски,
образованные из оболочки ХМ, частицы
ЛПВП, собранные в печени и кишечнике
на основе апопротеинов А, а также фер-
162 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
HMG-KoA
редуктаза
структурный
ХН
мембран
насцентные
диски
Рис 32. Взаимодействие клеточных рецепторов с апопротеин-В-зависимыми ЛП, рецепторный
эндоцитоз частиц ЛП и его регуляторные последствия. Лиз - лизосома, HMG-KoA-редуктаза —
р-окси-р-метилглутарилкоэнзим-А-редуктаза, апо-апопротеин, остальные обозначения — по
тексту. Пояснения в тексте
ментлецитин-холестерин-ацил-трансфе-
раза (ЛХАТ), открытый Дж. Гломсетом
(1962).
Насцентные диски приобретают в плазме
крови синтезируемые в печени и в энтеро-
цитах апопротеины АI—IV, а также
насыщаются апопротеинами СI—II и Е. Апопротеин
Е связывает избыток ХН тканей во время
пассажа дисков через сосудистую стенку,
который они свободно совершают благодаря
своему очень малому диаметру. Известную
роль при этом играет перфузия
имеющихся у крупных артерий vasa vasorum. Все
апопротеины С и апопротеины А обладают
значительным сродством к фосфолипидам.
Поэтому диски обогащаются фосфолипи-
дами и превращаются в частицы ЛПВП.
Апопротеины A-I, и в меньшей степени, C-I
служат коэнзимами фермента ЛХАТ. На
поверхности частиц ЛПВП сывороточная
ЛХАТ катализирует этерификацию ХН за
счёт ацилов жирных кислот из состава фос-
фолипидов оболочки ЛПВП (рис. 30). ХН
превращается в ЭХ, которые уходят внутрь
частицы ЛПВП, в состав ядра. В конечом
итоге, вместе с ЛПВП эти липиды попадают
в печень. Часть ЭХ переходит прямо в
плазме с ЛПВП на ЛПОНП и ЛППП, а затем
обнаруживается вновь в составе ЛПНП.
Фосфолипиды, использованные ЛХАТ,
превращаются в лизолецитин, который, до
определенной степени насыщения,
связывается альбумином и переносится в печень. В
печени ЛПВП катаболизируются (рис.31).
Постоянное протекание ЛХАТ-реакции
обеспечивает стабильность частиц ЛПВП
Нарушения липидного обмена 163
и дренирует ткани, а возможно, и апо-
В-зависимые липопротеиды плазмы, от
избытка ХН. Апопротеины С, переходя
через ЛПВП с ХМ на ЛПОНП, способствуют
нормальной скорости ЛПЛ-реакции.
У новорожденных активность ЛХАТ
понижена, аЛПВПиЛПНП перегружены
свободным ХН. Но это не приводит к
атеросклерозу, так как растущие ткани организма
младенцев ведут интенсивный мембрано-
генез и утилизируют огромные количества
ХН, не нарушая свойств биомембран.
При наследственной недостаточности
ЛХАТ — болезни острова Тэнжир
(«танжерской») — замедлено образование ЭХ,
а ЛПВП отсутствуют, либо имеют не
нормальную сферическую, а дискоидную
форму, близкую к насцентным частицам.
Из-за нарушения поставки апопротеинов
С в апо-В-зависимые ЛП замедляется
катаболизм ХМ и ЛПОНП с помощью
ЛПЛ-реакции. Подробную характеристику
тэнжирской болезни см. ниже в разделе
«Гиперлипопротеидемии и другие дисли-
попротеидемии». В связи с ключевой ролью,
которую ЛПВП играют при дренировании
тканей от избытка ХН, и с их челночным
участием в транспорте апопротеинов,
важных для катаболизма других ЛП, ЛПВП
считаются антиатерогенными факторами.
Понижение уровня ЛПВП при
атеросклерозе и ИБС было впервые отмечено Д.
Барром (1951) и подтверждено Дж. Гофманом
(1966). Повышение концентрации ЛПВП у
видов, резистентных к
экспериментальному холестериновому атеросклерозу, было
открыто Т.Н. Ловягиной и Э.Б. Банысов-
ской (1970). Впоследствии, А.Н. Климов
и соавт. (1984) сообщили о достоверно
более высоком уровне ЛПВП у кавказских
долгожителей, не страдающих
атеросклерозом. Антиатерогенный потенциал ЛПВП
подробно обсуждается ниже, в разделе
«Атеросклероз».
Нами рассмотрены, в основных чертах,
главные пути транспорта нормальных ЛП
в здоровом организме. Но нельзя
игнорировать и роль таких потенциальных ловушек
ЛП, какими могут быть мононуклеарные
фагоциты.
Принципиально возможен, особенно при
высокой концентрации и видоизменении
ЛП, а также в условиях цитокиновой
активации макрофагов (например, с помощью
моноцитарного колониестимулирующего
фактора — М-КСФ), нерегулируемый
прямой эндоцитоз липопротеидных частиц.
В этом процессе участвуют, по-видимому,
«мусорные» рецепторы, чувствительные к
производным Л П — перекисным дериватам,
а также к гликозилированным и ацетилиро-
ванным ЛП, в изобилии возникающим при
патологии. В макрофаги могут попадать и
иммунные комплексы с участием ЛП.
Вышеописанные тонкие регуляторные
механизмы, запускаемые при физиологической
ассоциации интактных ЛП частиц с апо-В/
Е-чувствительными рецепторами, при этом
не срабатывают или осуществляются в
недостаточной степени. Состояния,
сопровождаемые обильным накоплением и усиленной
модификацией ЛП (кормление избытком
холестерина, сахарный диабет, блокада
естественных путей регулируемого эндоци-
тоза Л П и дренирования тканей), приводят к
резкой активации нерегулируемого захвата
ЛП, посредством «мусорных» рецепторов
и механизмов фагоцитоза. Эти процессы
играют ключевую роль при формировании
скоплений макрофагальных клеток,
перегруженных Л П — ксантом. Ксантоматоз может
охватывать различные элементы системы
мононуклеарных фагоцитов. Например,
при болезни Хэнда-Шюлера-Кристиана,
включаемой современными
классификациями в число синдромов, принадлежащих к
гистиоцитозу X, происходит моноклональ-
ная пролиферация клеток макрофагального
ряда в твердой мозговой оболочке и костях
черепа. Мутантные макрофаги накапливают
эфиры ХН. Формируются желто-оранжевые
дефекты, заметные на рентгенограммах
черепа. Очаги вторичного ксантоматоза
могут причинять больным гипофизарно-
гипоталамические расстройства, вплоть до
диэнцефального ожирения и несахарного
диабета (см. «Нарушения эндокринной ре-
164 АЖ Зайчик, Л.П.Чурилов Патохимия
гуляции»), вызывают глухоту, нарушения
зрения, пучеглазие, расшатывание зубов
(И.В. Давыдовский, 1969).
Так как ксантомы — важные внешние
признаки многих гиперлипопротеинемий,
более подробно ксантоматоз
рассматривается в разделе «Гиперлипопротеидемии и
другие дислипопротеидемии», а здесь имеются
соответствующие иллюстрации (рис. 33).
Нерегулируемый захват видоизмененных
ЛИ интимацитами и гладкомышечными
клетками сосудистой стенки ведет к
формированию пенистых клеток и представляет
собой ключевое событие атерогенеза (см.
«Атеросклероз»).
В организме, как в результате
наследуемых мутаций, так и под влиянием
эпигенетических приобретенных механизмов, могут
формироваться минорные необычные
фракции Л П, метаболизм которых отличается от
описанных выше типовых закономерностей.
Нередко носительство таких минорных Л П
связано с возрастанием риска
атеросклероза и ксантоматоза. Они рассматриваются в
следующих двух разделах.
ГИПЕРЛИПОПРОТЕИДЕМИИ
И ДРУГИЕ
ДИСЛИПОПРОТЕИДЕМИИ
Гиперлипопротеидемиями (ГЛП) называются
нарушения образования, транспорта и
утилизации ЛП, сопровождаемые повышением
плазменного уровня ХН и/или ТГ. Термин
«гиперлипопротеидемия» заменил
историческое понятие «гиперлипидемия», так как
в плазме нет свободных липидов. Понятия
«гиперхолестеринемия» — как повышение
содержания в плазме крови общего
холестерина (суммирующее свободный и этери-
фицированный ХН) и «гипертриглицериде-
мия» — сохраняют свою употребительность,
так как при различных ГЛП возможно
накопление в плазменных ЛП, преимущественно,
ХН, ТГ, либо и того, и других.
ГЛП — самые распространенные
нарушения обмена веществ, особенно у европе-
(эндокринно-метаболические нарушения)
оидов и негроидов. Выше уже приводились
данные о пораженности населения разных
стран этими заболеваниями,
вовлекающими в отдельных популяциях от 10 до 25%
индивидов.
Дислипопротеидемии — более широкое
понятие, включающее, в частности, все ГЛП.
К этой группе относятся также гиполипо-
протеидемии, алипопротеидемии и
комбинированные расстройства (с избытком
одних и дефицитом других ЛП).
Роль ГЛП и дислипопротеидемии в
медицине определяется тем, что эти заболевания
длительно могут протекать бессимптомно,
но в конце концов сильно влияют на
вероятность, скорость развития и тяжесть таких
угрожающих жизни недугов, какими являются
атеросклероз, панкреатит и их осложнения.
Ранний диагноз и налаженная терапия ГЛП
могут реально удлинить жизнь пациентов
и улучшить ее качество. Различные
дислипопротеидемии коррелируют с разной
ожидаемой предстоящей продолжительностью
жизни, что имеет большое прогностическое
значение. Так как ранние внешние признаки
или стигмы ГЛП отмечаются со стороны
многих разных органов, врач любой узкой
специальности, например,
оториноларинголог, окулист или дерматолог, а не только
терапевт или педиатр, должен быть хорошо
информирован о дислипопротеидемиях
(см. рис. 33).
Обсуждая вопрос о
дислипопротеидемиях, необходимо подчеркнуть, что
уровень ЛП в крови зависит от совместного
действия многих разных генов (например,
генов ЛПЛ, ЛХАТ и других ферментов,
генов апопротеинов и т.д.). Ряд этих генов
характеризуется множественным аллелиз-
мом. Это делает генетическую вариацию
концентраций ЛП плазмы очень широкой.
Мало того, множество факторов экологии
человека и экзогенных воздействий
способны разнонаправленно менять концентрации
ЛП в рамках генетически обусловленных
варьирующих норм реакции.
Поэтому, до 1/3 ГЛП является
наследственными (первичными), причём среди
генетически обусловленных ГЛП ветре-
Рис.33. Внешние признаки ксантоматоза при гиперлипопротеинемиях. а — ксантелазмы век (ГЛП
На у пациентки 50 лет); 6 — туберозные ксантомы на локте (ГЛП III у женщины 55 лет); в — ли-
пидная дуга роговицы (ГЛП На у подростка); г — сухожильная ксантома ахиллова сухожилия
(ГЛП На у пациента 20 лет); д — эруптивные ксантомы сгибательной поверхности плеча (ГЛП Va у
пациентки 37 лет); — по Г. Галлеру и соавт., 1979.
166 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
чаются как моногенные менделирующие
формы, так и полигенные, которым
свойственно мультифакториальное
наследование с пороговым эффектом по
некоторым экзогенным факторам, например,
составу питания. Около 2/3 ГЛП считаются
приобретенными (вторичными) и
сопровождают развитие многих заболеваний.
Что же касается нормального содержания
в плазме ЛП, ХН и ТГ, то реальные цифры,
приведёные в таблице в приложении А, столь
широко варьируют, что разумным
представляется указать не на чисто статистическую
норму, которая соответствует обычно 90 %
или 95 % популяции, а на желаемый уровень,
который будет, как правило, уже и ниже.
Зато он отражает содержание липидов
плазмы крови у лиц с минимальной скоростью
прогрессирования атеросклероза.
С учетом этих соображений, наиболее
авторитетные источники (Дж. Гольдстайн,
1994; П. Чандрасома, К. Тэйлор; 1998) дают
следующие желаемые уровни
максимального содержания ХН, ТГ и ЛП, выше которых
можно говорить о ГЛП (см. также табл. А
в приложении):
>- Уровень общего ХН плазмы менее
5,2мМ/л (200 мг/дл) связан с низкой
степенью риска атеросклероза. Уровень
от 5,2 до 6,0 мМ/л является пограничным
и говорит о средней степени риска. Его
можно расценивать, как умеренную гипер-
холестеринемию. При уровне ХН выше 6,0
мМ/л (240 мг/дл) имеется выраженная
гиперхолестеринемия и высокий риск
атеросклероза. У детей желаемый уровень
общего ХН должен составлять до 180 мг/дл,
а ХН в составе ЛПНП до 110мг/дл.
>» Уровень общих ТГ плазмы ниже 1,6 мМ/л
(140 мг/дл) связан с низкой степенью
риска атеросклероза. Уровень ТГ от 1,6
до 2,2 мМ/л (140-200 мг/дл) допустим и
может расцениваться как начальная
степень гипертриглицеридемии. Уровень
свыше 2,2 до 2,8 мМ/л (200-250 мг/дл)
нежелателен и представляет
повышенный риск для здоровья. Уровень более
2,8 мМ/л (250 мг/дл) является
выраженной гипертриглицеридемией и означает
высокий риск той или иной патологии, в
зависимости от типа ГЛП.
>- Уровень ЛПНП составляет 130-
430 мг/дл (1,3-4,3 г/л) и прямо
коррелирует со степенью риска атеросклероза.
Цифры выше 4,3 г/л характерны для
ряда атерогенных ГЛП.
>• Желательно иметь достаточно высокий
уровень ЛПВП, в связи с их антиате-
рогенной ролью. Физиологический
уровень ЛПВП для мужчин находится
в пределах 125-425 мг/дл (1,3-4,3 г/л),
а у женщин — выше из-за высокого
содержания эстрогенов, способствующих
синтезу ЛПВП, и составляет в норме
250-660 мг/дл (2,5-6,6 г/л). Снижение
уровня ЛПВП ниже этих цифр
умеренно повышает риск атеросклероза,
при уменьшении уровня ЛПВП ниже
1,05 г/л у мужчин и 1,30 г/л — у женщин,
риск атеросклероза становится высоким.
Наоборот гиперальфалипопротеидемии
ассоциируются с понижением риска. Для
точной оценки степени риска, связанного
с метаболизмом ЛПВП, рекомендуется
определять ХН в составе ЛПВП
отдельно. Имеется способ осаждения ХМ,
ЛПНП, ЛППП и ЛПОНП из плазмы
по М.Бурштейну-Ж.Самай (1959),
инкубацией с гепарином или
синтетическими гепариноидами в присутствии
катионов кальция или сульфата магния.
При этом вышеназванные ЛП образуют
с гликозаминогликанами комплексы и
оседают, а определение ХН в супернатан-
те даёт представление о его содержании
в ЛПВП. Уровень ХН в ЛПВП должен
превышать 35 мг/дл (0,9 мМ/л). Более
низкие цифры коррелируют с
повышенным риском атеросклероза и ИБС в
восемь раз сильнее, чем общий ХН
плазмы и в 4 раза сильнее, чем ХН в составе
ЛПНП.
>■ В качестве интегрального критерия
вышеперечисленных липидологических
показателей А.Н. Климов (1977)
предложил холестериновый коэффициент
атерогенности (ХКА).
ХКА= ХН(общий)-ХН(ЛПВП)/
/ХН(ЛПВП).
> В норме ХКА растет с возрастом.
Желаемый ХКА составляет у
новорожденных — 1,0; у женщин детородного
возраста — 2,2; у мужчин до 40 лет — 2,5-3;
у мужчин после 40 до 60 лет — 3-3,5.
В группе долгожителей (старше 90 лет)
он оказался менее 3,0. При ИБС
величина ХКА, как правило, более 4 и нередко
достигает 6. В зарубежной литературе
аналогичную прогностическую роль
отводят величине ХН в составе ЛПНП,
определяемой по следующей формуле:
ХН (ЛПНП) = {ХН(общий)- •
-ХН (ЛПВП)}-ТГ(общие)/5.
> Величины ХН (ЛПНП) менее 3,4 мМ/л
(130 мг/дл) желательны, поскольку
связаны с пониженным риском
атеросклероза. Уровень от 3,4 до 4,1 мМ/л (до
159 мг/дл) считается пограничным, а
при превышении 4,2 мл (160 мг/дл) риск
атеросклероза повышен.
> Измеряя все эти интегральные
показатели, следует помнить, что нередко наличие
даже небольших количеств аномальных
минорных ЛП, серьёзно не затрагивая
общие липидологические индексы,
приводит к существенному ускорению
процессов атерогенеза.Таковы, например,
ЛП X и, особенно, ЛП (а), носительство
которого повышает риск атеросклероза в
десятки раз, даже без выраженной общей
гиперхолестеринемии. Эти ситуации
обсуждаются отдельно ниже.
ГЛП были основательно изучены и
классифицированы по паттерну липопротеидов
плазмы на 5 основных типов (6 подтипов) в
конце 60-х годов XX века Д.С. Фредерик-
соном. Дополненная Ж. Бомоном
классификация ГЛП была в начале 70-х годов
прошлого века принята ВОЗ.
В настоящее время ясно, что различные
наследственные дефекты и приобретенные
болезни могут приводить к сходной картине
изменений содержания ЛП разных классов.
Нарушения липидного обмена 167
В то же время, при одном и том же
генетическом заболевании могут затрагиваться
концентрации не одного, а нескольких
классов Л П. Поэтому, различные типы ГЛП
по классификации ВОЗ — не отдельные
болезни, а характерные симптомокомплексы,
отражающие те или иные паттерны
повышения уровня разных Л П.
Краткая характеристика ГЛП
представлена ниже в табл. 11.
Первичная гиперлипопротеидемия Imuna
или болезнь М. Бюргера-О. Грютца
описана в 1932 г. и связана с наследственным
аутосомно-рецессивным дефектом ЛПЛ.
В крови накапливается значительное
количество ХМ из-за блока их катаболизма.
ХМ провоцируют тромбоз и ишемические
микронекрозы, которые особенно типичны
для поджелудочной железы. Характерны
наблюдаемые с раннего детства
абдоминальные колики. Формируется хронический
рецидивирующий панкреатит. Бывают случаи
молниеносных смертельных обострений.
Большое значение имеет частичный
гидролиз ХМ панкреатической липазой, который
проходит в микроциркуляторном русле
органа. Лизолецитин и жирные кислоты
в избытке оказывают на панкреатические
клетки местное токсическое действие,
связанное с детергентным эффектом и
разрушением клеточных мембран.
На коже видны характерные стигмы
заболевания — желтовато-розовые папулы на
плечах, спине, ягодицах (рис. 33 д). Это
результат фагоцитоза ХМ гистиоцитами дермы
и образования эруптивных ксантом.
Макрофаги тоже перегружаются ХМ, что ведет к
гепатоспленомегалии и появлению пенистых
клеток в костном мозге. На бледном глазном
дне видны белые сосуды, что известно как
lipemia retinalis. В плазме, которая натощак
остается мутной и дает сливкообразный
слой при стоянии, повышен уровень ТГ, но
ХН в норме. ГЛП I не реагирует на гепарин.
Уровень апопротеина С II остается в норме.
Так как ХМ не проникают через эндотелий,
атеросклероз не ускоряется.
Приобретенная фенокопия ГЛП I
формируется у больных с аутоиммунными заболе-
168 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 11. Гиперлипопротеинемии.1
Тип
Наследование
Частота, ЛП
плазмы
Ключевое
звено
патогенеза
Стигмы
ХН
тг
ХН/ТГ
Плазма
Клиника
Лечение
Вторичные
формы
1
АР
1/1000000,
ХМ
Дефицит Л ПЛ
Эруптивные
ксантомы,
роговичная
дуга
N/ft
ftftft
<0,09
Сливкооб-
разный слой
при отстое,
прозрачна
после отстоя
Панкреатит.
Абдоминальные колики,
гепатоспле-
номегалия.
Неатеро-
генна
Диета
Системная
красная
волчанка
2а
АД
0,2%,ЛПНП
Дефект
рецептора
апоВ/Е
Сухожильные
ксантомы
ftftft
N
>3,5
Прозрачна
Ранняя
тяжелая ИБС.
Нет СД, Вес в
норме
Гемфибро-
зил, ниацин,
Ловастатин.
Холестира-
мин
Гипотироз,
гиперкор-
тицизм,
Пищевая
перегрузка
ХН и ТГ,
перемежающаяся
порфирия,
диабет
26
АД
2%ЛПНП,
ЛПОНП
Повышенная
продукция
апо-В
Ксантоматоз
нетипичен
ftft
ft
1-2
Опалесци-
рует, не дает
сливкообраз-
ного отстоя
Ранний
тяжелый
атеросклероз; стеатоз
печени,
ожирение, СД
Диета,
гемфибро-
зил, Ниацин,
Ловастатин.
Холестира-
мин. d-Тиро-
ксин
Гипотироз,
гиперкор-
тицизм,
Пищевая
перегрузка
ХН и ТГ,
перемежающаяся
порфирия,
диабет
3
АР (с
пороговым эффектом
диеты)
< 1/10000,
ЛППП,
Остаточные ХМ
Аномальный
апо-Е
Ладонные и
туберозные
ксантомы,
следы от колец
ftft
ftft
>0,45
Мутноватая,
незначительный сливкооб-
разный слой
Ускорение
атеросклероза,
Облитерирую-
щие
заболевания
конечностей, инсульты,
стеатоз печени,
диабет, гиперу-
рикемия
Гемифиброзил,
клофибрат,
РР, эстрогены
у пациенток.
Диета
Холестаз,
Гипотироз. Мо-
ноклональные
гаммапатии
4
АД
>2,5 %,
ЛПОНП
Повышенная
продукция
ТГ, ЛПОНП
печени
Лицо Луи-
Филиппа,
lipemia
retinalis
ft
ftftft
<1
Опалесци-
рует, сливок
при отстое не
даёт
Ожирение,
стеатоз
печени, диабет, ги-
перурикемия,
ретинопатия,
атеросклероз
несколько
ускорен
Гемфибро-
зил, Ниацин,
клофибрат,
гепарин-диета с
ограничением
углеводов и
насыщенных
жиров
Диабет,
Алкоголизм.
Стресс,
Оральная
контрацепция, гликоге-
нозI типа
5
АР
<0,02%,
ЛПОНПХМ
Дефект апо-
СИ
Эруптивные
ксантомы
ft
ftft
0,09-0,15
Опалесци-
рует, при
отстое, дает
сливкооб-
разныйслой
Комбинация
1nlV
Диета,
клофибрат
KaKlnV
1 Условные обозначения: Наел — тип наследования первичной формы. Частота — указана для первичной
формы, АР-аутосомно-рецессивный. АД — аутосомно-доминантный. ЛП — классы липопротеидов,
уровень которых в плазме повышается. СД — сахарный диабет. Остальные сокращения — как в тексте.
Нарушения липидного обмена 169
ваниями соединительной ткани, особенно
часто — при системной красной волчанке.
Антитела против гликозаминогликанов при
этих болезнях нарушают процесс
гепариновой активации ЛПЛ.
Первичная гиперлипопротеинемия 5 типа
развивается при аутосомно-рецессивном
отсутствии апопротеина С II, важного
кофактора ЛПЛ. В отличие от ГЛП I, в крови
накапливаются оба главных субстрата
ЛПЛ — ХМ и ЛПОНП. Основные симптомы
болезни сходны с ГЛП I. Однако, панкреатит
бывает менее тяжелым и клиника болезни
развивается гораздо позже, во взрослом
состоянии. Больные не имеют апо-С II.
Гепарин не эффективен. При введении свежей
донорской сыворотки здоровых лиц,
изобилующей этим апопротеином, наступает
быстрое, но временное облегчение. Эруптивные
ксантомы менее выражены, чем при ГЛП I
(рис. ЗЗд). Вторичная приобретенная ГЛП,
соответствующая паттерну ГЛП V,
бывает при гликогенозе Гирке, алкоголизме и
использовании пероральных
противозачаточных средств, если печень пациентов
вырабатывает очень много ЛПОНП.
Возможно, имеет место вторичное торможение
активности ЛПЛ избытком ХМ.
Первичная гиперлипопротеинемия На
типа развивается при распространенном и
смертельно опасном наследственном
заболевании — семейной гиперхолестеринемии (см.
также I том, стр. 119,127,141). Она поражает
не менее 0,2 % населения и является аутосом-
но-доминантной. Даже гетерозиготы имеют
повышение уровня ХН в 2-3 раза, а
гомозиготы — в 6-8 раз. У гетерозигот болезнь ведет
к развитию ИБС в 3-4-й декадах жизни.
Самый ранний достоверно зафиксированный в
истории случай инфаркта миокарда на почве
коронарного атеросклероза наблюдался в
Германии у ребенка 18 месяцев,
гомозиготного по гену данного заболевания. Каждый 20-й
инфаркт миокарда происходит у носителя
ГЛП На, и к 65 годам более 80% мужчин,
имеющих дефектный ген, уже переносят, по
крайней мере, один инфаркт.
Для заболевания характерен выраженный
ксантоматоз, связанный с отложением в
макрофагах ЭХ. Наиболее типичны (рис.
ЗЗг) ксантомы ахиллова сухожилия и
сухожилия четырехглавой мыщцы бедра.
Они не наблюдаются при других ГЛП, а
при данной — имеют место у 3/4 больных.
Ксантоматоз поражает веки («ксантелаз-
мы», рис. 33а), кожу локтей иколеней
(«бугорчатые ксантомы», рис.ЗЗб), роговицу
(«корнеальная дуга», появляющаяся до 45
лет, рис.ЗЗв).
У гомозигот присутствуют плоские
ксантомы в местах травматизации кожи и
межпальцевых складках.
Именно хорошее знание глазных и
кожных проявлений ксантоматоза,
сопутствующего грозным нарушениям липидного
обмена, позволяет гадалкам, хиромантам,
а также народным иридодиагностам делать
пророческие предсказания безвременной
болезни и ранней кончины. Современный
доктор достаточно вооружен, чтобы
изменить судьбу таких больных — тем выше его
ответственность, но значение кожно-глазной
и сухожильной симптоматики дислипопро-
теидемий от этого не уменьшается.
Кроме ИБС, больным угрожает
приобретенный аортальный стеноз из-за
ксантоматоза и атеросклероза иолулунных
клапанов.
Патогенез болезни связан с рецепторным
блоком. Рецептор ЛПНП либо
отсутствует, либо дефектен и не проявляет и 15%
нормальной аффинности. У некоторых
пациентов рецептор связывает ЛПНП, но не
осуществляет эндоцитоз. В последнее время,
методом полимеразной цепной реакции,
выявлены отдельные больные с клиникой ГЛП
На и нормальными рецепторами ЛПНП. У
них отсутствует всего одна аминокислота
в положении 3 500 в самом апопротеине В,
в связи с чем этот лиганд не связывается с
клеточным рецептором.
У всех больных ЛПОНП не
утилизируются печенью, к тому же печень снижает и
захват Л ППП, которые усиленно переходят
в ЛПНП. Избыток ЛПНП инфильтрирует
сосудистую стенку, подвергается окислению
и иным видам трансформации, стимулирует
тромбоциты к выделению ростовых факто-
170 АЖ Зайчик, Л.П. Чурилов Патохимия
ров и захватывается неспецифически
макрофагами и интимацитами. Это ведет к
нерегулируемому накоплению ХН в клетках,
их пенистой трансформации, ксантоматозу
и атеросклерозу (см. ниже).
У пациентов при ГЛП Па увеличен
уровень ХН, но ТГ в норме, а плазма
прозрачна. Характерно отсутствие ожирения и
диабета.
Лечение включает диету, обедненную
холестерином и обогащенную ненасыщенными
жирами. Назначают большие дозы витамина
РР, холестирамин, а в самых тяжелых
случаях — даже блокаторы синтеза эндогенного
холестерина (ловастатин, симвастатин, пар-
вастатин), несмотря на их миотоксичность.
Имеются попытки применять для лечения
данной аномалии метаболизма Л П
пересадку здоровой донорской печени.
Приобретенные состояния с ГЛП На
разнообразны. Синтез рецепторов ЛПНП
угнетается глюкокортикоидами и стимулируется
тиреоидными гормонами. Гиперкортицизм,
хронический стресс и гипотироз
способствуют накоплению ЛПНП. При нефротическом
синдроме отмечается иногда секреция
печенью ЛПНП. При гепатомах ЛПНП
накапливаются, так как отсутствует должное
ингибирование синтеза ХН в гепатоцитах
избытком этих ЛП. ГЛП На наблюдается
при острой перемежающейся порфирии,
по-видимому, из-за ингибирования порфи-
ринами рециркуляции рецептора ЛПНП.
При нейрогенной анорексии (см. выше
«Голодание») ЛПНП накапливаются на фоне
снижения экскреции ХН и его производных
с жёлчью, что связано с нарушением холе-
цистокининовой регуляции. Достоверно
установлено, что диета, перегруженная ХН
и ТГ ведет к ингибированию продукции
рецепторов ЛПНП и может повышать риск
развития приобретенной ГЛП данного типа.
При синдроме Вернера ГЛП данного типа
комбинируется с инсулинорезистентнос-
тью (см. ниже «Патофизиология сахарного
диабета»).
Первичная гиперлипопротеидемия II б
типа развивается примерно у 10%
больных с генетическими дефектами рецептора
чдокринно-метаболические нарушения)
ЛПНП, вероятно, у тех, кто несет мутант-
ные аллели, не связывающие ни ЛПНП, ни
ЛПОНП. Большинство больных с этим
паттерном ГЛП представляют другое наследс-
твеное заболевание — семейную смешанную
гиперлипидемию.
Это аутосомно-доминантное
расстройство. Предполагается, что у пациентов
повышены выработка апопротеина В и секреция
печенью ЛПОНП. Возможно, у них снижена
аффинность апопротеина В к рецептору,
или имеются особенности взаимодействия
ЛПЛиЛП.
В отличие от предыдущего расстройства,
повышены и уровень ХН, и концентрация
ТГ, однако, в обоих случаях — не так
катастрофически, как это происходит с ХН при
ГЛП II а.
Атеросклероз значительно ускорен, и не
менее 10% больных, перенесших инфаркт
миокарда, имеют это расстройство липид-
ного обмена. Ускоренному атеросклерозу
способствует понижение продукции
апопротеина АН и ЛПВП, наблюдаемое у части
таких больных. Однако, ксантоматоз менее
выражен, а сухожильных ксантом вообще не
отмечается. Бывают ксантелазмы. Имеется
избыточный вес и часто — гипертензия, ги-
перурикемия, стеатоз печени. Хотя данная
ГЛП моногенна, пенетрантность мутантного
гена сильно зависит от диеты и образа
жизни, и болезнь отягощается при переедании
холестерина, насыщенных жиров, диабете,
курении и алкоголизме. По неясным
причинам, некоторые больные с мутациями того
же гена развивают ГЛП IV или ГЛП II а.
Приобретенными фенокопиями
болезни являются ГЛП у некоторых больных с
гиперкортицизмом, нефротическим
синдромом и всех больных с гипофизарной
карликовостью, обусловленной изолированным
дефицитом СТГ. У последних увеличена
секреция печенью ЛПОНП и их конверсия
в ЛПНП.
Гиперлипопротеинемия III типа в ее
первичном варианте известна как болезнь
устранения остаточных частиц или
болезнь широкой р-полосы (см. характерную
электрофоретическую картину распреде-
Нарушения липидного обмена 171
ления JIU плазмы при этой и других ГЛП
на рис. 34). Современное название этого
наследственного аутосомно-рецессивного
заболевания — дисбеталипопротеинемия.
Известно, что оно поражает гомозигот
по одному из мутантных аллелей апопро-
теина Е. Наиболее типичен генотип Е2/Е2.
Экспрессия генов зависит от экзогенных
факторов, прежде всего, диеты и
присутствия генов других дислипопротеинемий,
ограничивающих компенсацию.
Патологический генотип имеется почти у 1 %
населения, но клинически больных примерно
в 100 раз меньше. Болезнь проявляется в
третьем десятилетии жизни. Характерен
выраженный прирост концентрации ЛППП.
Это остатки ЛПОНП, обогащенные ЭХ,
и потому имеющие при электрофорезе
р-подвижность, вместо характерной для
ЛПОНП пре-р-подвижности. В более
редких случаях нарушен и катаболизм
остаточных частиц ХМ. В плазме сильно увеличен
уровень ХН и, иногда, имеется умеренная
гипертриглицеридемия. Наблюдается
отчетливый ксантоматоз. Патогномоничными
считаются ладонные и подошвенные ксан-
томы, поражающие, в частности, складки
ладони и образующие оранжево-желтые
следы в области давления колец, перстней,
в межпальцевых складках. Обнаружить след
под обручальным кольцом всегда считалось
в народе недоброй приметой. Прогноз при
ГЛП III это подтверждает. Часто бывают
туберозные ксантомы на локтях и коленях
(рис. 336), менее специфичны ксантелаз-
мы век. Характерен ускоренный тяжелый
атеросклероз, не только коронарной, но
и церебральной, а также абдоминальной
локализаций. Имеются не только
проявления ИБС, но и перемежающаяся хромота, а
также ишемическая энцефалопатия, вплоть
до гангрены и инсультов. Болезнь нередко
сопровождается тучностью, диабетом, сте-
атозом печени. В последние годы описан
иной аллельный вариант наследственной
ГЛП III с аутосомно-доминантным
наследованием.
Приобретенные аналоги ГЛП III
зарегистрированы при гипотирозе и моноклональ-
N
На lib III IV
W
S3
SJSI
Ч N vj 4J
Рис 34. Результаты разделения ЛП нормальной
плазмы (N) и плазмы больных с ГЛП I-V типов
(обозначены соответствующими римскими
цифрами) методом электрофореза на бумаге.
Полосы ЛП: 1 — старт и ХМ;
2 — р-ЛП; 3 — пре-р-ЛП; 4 —■ а-ЛП
ных гаммапатиях. В последнем случае ау-
тоантитела связывают остаточные частицы
ХМ и ЛПОНП и мешают их нормальному
катаболизму. При холестазе и обтурацион-
ной желтухе ЭХ не выводятся из печени и
появляются перегруженные ими липопроте-
иды X, по свойствам близкие к остаточным
частицам ГЛП III. ЛП X также высокоате-
рогенны. Они отличаются от всех других
нормальных и патологических ЛП тем,
что окружены фосфолипидным бислоем а
не монослоем. Очевидно, по этой причине
в них больше ХН и фосфолипидов, чем в
ЛПОНП, и они имеют плотность ЛПНП,
хотя электрофоретическая подвижность
их меньше, чем у р-ЛП («тонущие пре-р»).
Дисковидные ЛП X легко слипаются по
типу монетных столбиков, ухудшая
реологические свойства крови в микроциркуля-
торном русле.
Близкая к ГЛП III типа картина липопро-
теидного спектра развивается при тэнжирс-
кой болезни (см. ниже).
Гиперлипопротеидемия IV типа — самая
распространенная дислипопротеидемия.
Наследственный первичный вариант этого
синдрома известен как семейная
гипертриглицеридемия. Это аутосомно-доминантное
расстройство, проявляющееся после
полового созревания. У пациентов повышен
уровень ТГ натощак, хотя и не так сильно,
как при ГЛП I и V типов. Уровень ХН в
норме. Ксантоматоза нет, но часто имеется
характерное отложение жира на лице и
172 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
шее — лицо Луи-Филиппа, знакомое по
историческим карикатурам О. Домье. Для
больных IV типом ГЛП часто характерен
метаболический синдром, уже упоминавшийся
выше в главе «Патофизиология
нуклеинового метаболизма» и предусматривающий
ожирение (как правило, андроидного типа),
инсулинорезистентность и скрытый
сахарный диабет II типа, гиперурикемию, часто
гипертензию и стеатоз печени. Развитие
атеросклероза при этом несколько
ускоряется, в особенности, если выражены
сахарный диабет, гипертензия и гиперурикемия.
Данный симптомокомплекс иногда именуют
метаболическим Х-синдромом (рис. 35).
Есть сведения, что он связан с дефектами
водород-натриевого противопереносчика
клеточных мембран (подробнее см. раздел
«Нарушения накроления и мобилизации
липидов...» ниже, а также гл. 14 и
Практикум).
В связи с большой частотой ГЛП IV, до
6 % больных с инфарктом миокарда имеют
данное нарушение спектра Л П.
Предполагают, что решающую атерогенную роль играют
сопутствующие проявления
метаболического Х-синдрома и снижение концентрации
ЛПВП, характерное для ГЛП III. При ГЛП
IV поражаются атеросклерозом и артерии
Рис. 35. Наиболее характерные проявления
метаболического Х-синдрома.
нижних конечностей. Из-за липидоза
сетчатки и диабета может страдать зрение,
нередко понижается потенция.
К сожалению, до сих пор мало
точных данных о патогенезе именно этой,
практически, очень часто наблюдаемой
ГЛП. Предполагается, что у больных либо
имеется моногенное повышение синтеза
апопротеина В, либо существует первичная
гиперпродукция ЛПОНП в печени, с
которой не справляются механизмы, связанные
с их катаболизмом. Возможно, что имеется
усиленное превращение углеводов
печенью в жиры, например, связанное с малой
чувствительностью печеночной фосфоф-
руктокиназы к обратному ингибированию
продуктами этого синтеза. Указывалось и
на сниженную скорость поглощения НЭЖК
адипоцитами таких больных.
Фенокопии ГЛП IV бывают очень часто.
Именно по этому типу нередко нарушается
спектр ЛП при алкоголизме, хроническом
стрессе, остром гепатите, уремии,
акромегалии, гликогенозе I типа фон Гирке,
сахарном диабете, врожденных и
приобретенных липодистрофиях, применении
эстрогеновых пероральных контрацептивов
и р-адреноблокаторов, гемодиализе, моно-
клональных гаммапатиях.
В заключение данного раздела
необходима характеристика редких аномалий спектра
ЛП, которые входят в понятие «дислипо-
протеидемии», но не включены в
классификацию ГЛП, приведенную выше.
Абеталипопротеинемия, или болезнь
Ф.Бассена-А.Корнцвейга (также
известная как наследственный акантоци-
тоз) — представляет собой аутосомно-до-
минантный генетический дефект продукции
апопротеина В. Данный апопротеин
отсутствует, не выявляется и апо-В-зависимых
ЛПОНП, ЛППП, ЛПНП и ХМ. ЛПВП
синтезируются и даже переносят часть ТГ
из кишечника, подменяя отсутствующие
ХМ. Болезнь совместима с жизнью, но
влечет ряд серьёзных расстройств: атаксию,
гипорефлексию, нарушения проприорецеп-
Нарушения липидного обмена 173
торной чувствительности. Обнаруживается
демиелинизирующая дегенерация бокового
и заднего канатиков спинного мозга и
пирамидного тракта. Типичны мальабсорбция,
стеаторея, пигментная ретинопатия и
шиловидные выбухания на поверхности
эритроцитов — акантоцитоз (греч. ar|avua — шип),
сопровождаемые гемолитической анемией.
Всё это возникает вследствие понижения
содержания ХН в клеточных мембранах и
полигиповитаминоза по жирорастворимым
витаминам (см. ниже и в т. III).
Гипобеталипопротеидемия, описываемая
у 3-6% населения, вероятно, представлена
гетерозиготами по какому-то другому ауто-
сомно-рецессивному аллелю, снижающему
продукцию апопротеина В. Возможно,
что к числу этих лиц принадлежат также
индивиды с фенокопиями. Синтез апо-В
подавляется при гипертирозе.
Концентрация соответствующих ЛП может снизиться
при наличии аутоантител к апопротеину В.
Носители данной дислипопротеидемии не
имеют аномалий всасывания и транспорта
ТГ, ХН и жирорастворимых витаминов.
По данным К. Глюка и соавт. (1976), у них
на 9-12 лет увеличена продолжительность
жизни за счет торможения развития
сердечно-сосудистых заболеваний, связанных
с атеросклерозом.
Аналъфалипопротеидемия — аутосомно-
рецессивная патология, связанная с отстус-
твием синтеза апопротеинов А и продукции
ЛПВП. Известна также как танжерская
болезнь (по названию островка у берегов
США, где она была описана Д.С. Фредерик-
соном в 1961 г., а не марокканского города
Танжер, как иногда неверно переводят). У
больных нарушен, прежде всего, транспорт
ЭХ. Уровень общего ХН и фосфолипидов в
сыворотке снижен, а ТГ натощак повышен.
ЭХ захватываются макрофагами,
формируется ксантоматоз, имеется накопление ЭХ
в селезенке, печени, лимфоидных органах.
Отмечают гепатоспленомегалию и лимфа-
денопатию. Патогномоничным признаком
считается оранжево-жёлтый цвет
увеличенных миндалин. Могут быть катаракта,
полинейропатия и ретинит, но поскольку
нет накопления свободного ХН,
атеросклероз, насколько известно из наблюдений за
носителями этой аномалии, существенно
не ускоряется.
Семейная наследственная
недостаточность ЛХАТ — редкое заболевание,
приводящее к нарушению перехода ХН в состав
ЛПВП и его этерификации. Связана с
ускорением развития атеросклероза. Уровень
ХН в плазме в разной степени повышен, а
ЭХ — понижен. Имеются гемолитическая
анемия и прогрессирующий нефросклероз.
Церебросухожилъный ксантоматоз —
наследственный дефект продукции жёлчных
кислот из ХН. Отсутствие жёлчной
экскреции надлежащих количеств ХН ведет
к накоплению ХН и его патологического
деривата холестанола, что способствует ксан-
томатозу необычной локализации, с
поражением головного мозга. Имеются мозжечковая
атаксия, слабоумие, парезы и катаракта.
При ситостеринемии патологически
повышено всасывание ХН и растительных
стероидов в кишечнике увеличен их уровень
в плазме. Имеется ксантоматоз сухожилий.
Болезнь Вольмана — аутосомно-рецессив-
ный дефицит холестерин-эстеразы (лизосо-
мальной кислой липазы). Имеет раннюю,
младенческую, и позднюю (взрослую)
форму. Характеризуется накоплением ЭХ
и, частично, ТГ в макрофагах и лимфоцитах.
Это ведет к гепатоспленомегалии, задержке
роста, фиброзу печени, кальцификации
надпочечников. Могут быть варикозные
расширения пищевода. Обнаруживаются
пенистые клетки, имеется повышение общего
ХН. При данном заболевании ксантоматоз
классифицируют как первичный. Однако,
поскольку нет накопления свободного ХН
и продолжается функционирование
дренажных механизмов, существенного ускорения
атеросклероза может и не наблюдаться.
Впрочем, есть сообщения о значительной
скорости развития атеросклероза у
некоторых пациентов с болезнью Вольмана, что
К. де Дюв (1974) и В. Кумар и соавт. (1997)
считают важным доказательством роли
недостаточности лизосомального гидролиза
ЭХ в генезе атеросклероза.
174 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Липопротеин (а) — патологический
минорный ЛП, носительство которого
чрезвычайно повышает риск атеросклероза. Это
разновидность Л ПНП, имеющая необычный
апопротеин (а), связанный с апопротеином
В100. Впервые ГЛП (а) была описана в
Норвегии (А.Берг, 1964, К. Энхольм и соавт.,
1972) у представителей рода, крайне тяжело
пораженного наследственной склонностью
к ИБС. ЛП (а) имеет плотность ЛПНП,
но электрофоретическую мобильность
ЛПОНП (ранее именовались медленные
Р-ЛП или тонущие пре-р-ЛП).
Роль апопротеина (а), составляющего 20 %
белка в ЛП (а), заключается в том, что один
из доменов этого мутантного белка — домен
4, повторенный в апо (а) 37 раз, совпадает с
аналогичным доменом в молекуле профиб-
ринолизина. Это позволяет рассмотреть апо
(а) как структурный аналог и ингибитор
плазминогена. ГЛП (а) увеличивает риск
атеросклероза и тромбоза даже при невысоком
общем ХН. Концентрация Л П (а),
практически, не зависит от диеты, а определяется лишь
генами. Существует 6 аллелей, комбинации
которых контролируют уровень апо (а), и
поэтому в популяции имеется 1000-кратная
вариация по уровню ЛП (а). Носительство
ЛП (а) повышает риск ИБС в 10 раз
сильнее, чем высокий уровень ЛПНП. 90 % лиц
с нетипичными формами ИБС и 25% всех
лиц с ИБС моложе 60 лет имеют повышение
уровня этого минорного Л П. Установлено,
что ЛП (а) — прямой индуктор
пролиферации ГМК, активатор тромбоцитов и
свертывания, а не только ингибитор фибринолиза.
Предполагается, что сохранение в ряду
поколений подобной мутации связано с ее
известной адаптивной ценностью при острых
повреждениях: у носителей ЛП (а) быстрее
заживают раны и активнее действует система
гемостаза. Р. Лоун установил близость ЛП
(а) и фактора роста гепатоцитов.
Апопротеин (а) — белок, накопление
которого наиболее тесно связано с высоким
риском атеросклероза (см. ниже).
Гипоальфалипопротеидемия наблюдается
у 3-8% индивидов с конституционально
низким или вторично пониженным уровнем
ЛПВП в плазме. Конституциональное
понижение может зависеть от низкого уровня
синтеза апопротеинов А. Нефротический
синдром ведет к потере ЛПВП с мочой и
гипоальфалипопротеидемии. Показано, что
при недостатке ЛПВП ускоряется развитие
атеросклероза и ИБС. В настоящее время
считается, что при гипоальфалипопротеи-
демиях большое значение имеют мутации
генов апопротеинов Al, A4 и СШ,
находящихся в 11 -й хромосоме.
Гиперальфалипопротеидемия
представляет собой повышение концентрации ЛПВП
и встречается не менее чем у 4-8 %
населения. Первичная наследственная форма
связана с усилением продукции апопротеина
A-I. Вероятно, имеются и формы, связанные
с повышенной активностью ЛПЛ и
ускоренной деградацией апо-В-зависимых ЛП.
Концентрации ЛПНПиЛПОНПу
носителей гиперальфалипопротеинемии чаще
всего понижены. Липиды ЛПВП у них богаче
полиеновыми кислотами. Большой интерес
представляет возможность развития
приобретённых гиперальфалипопротеинемии,
так как Н.Г. Никульчева и соавт. (1982)
показали пониженную заболеваемость ИБС
у носителей этой особенности ЛП спектра, а
упоминавшееся выше исследование К.
Глюка и соавт. установило у них увеличение
продолжительности жизни на 5-7 лет.
Различные авторы, занимавшиеся этой
проблемой, выявили немало факторов,
коррелирующих с повышенным содержанием
ЛПВП у людей. Далеко не все эти
закономерности укладываются в упрощенные
представления о здоровом образе жизни,
знакомые всем нам со времен «Пионерской
зорьки», хотя некоторые выглядят прямым
подтверждением хрестоматийных правил.
Эйкозапентаеновые кислоты,
изобилующие в морепродуктах, повышают уровень
ЛПВП. Гиперальфалипопротеидемия
отмечена у марафонцев, футболистов,
альпинистов и ряда других физкультурников,
а вот гиподинамия способствует
понижению уровня ЛПВП, что позволяет с новой
силой оценить «свежую» мысль о том, что
« Спорт — это здоровье!».
Нарушения липидного обмена 175
Ряд токсинов, стимулирующих
гиперплазию гладкого эндоплазматического
ретикулюма в печени, как это ни
парадоксально, тоже увеличивает содержание
ЛПВП. К ним относятся извечный враг
человечества — алкоголь1, а также его новые
враги, изобретенные наукой и
индустрией — хлорорганические пестициды, метил-
меркаптановые соединения и некоторые
эстрогенсодержащие лекарства. Авторы
далеки от того, чтобы перефразировать
цитированную выше бессмертную фразу и
выдать лозунг «Спирт — это здоровье». Так,
чего доброго, можно дойти до
безответственных советов ради профилактики
атеросклероза закусить пестицидами и нюхнуть из
трубы целлюлозно-бумажного комбината. А
дальше легко совсем погрузиться в
кощунство и усомниться в вечных истинах гигиены,
например в том, что «Лес — твой друг!», а
«Муха — твой враг!». Отрекаемся от ереси,
хотя и отмечаем, что в профессиональном
мышлении нет места категории
«абсолютного зла».
Как бы там ни было, повышение уровня
ЛПВП и понижение риска ИБС при
употреблении малых доз этилового алкоголя,
по сравнению с совершенно непьющими
испытуемыми, было зарегистрировано
бельгийскими эпидемиологами, которые
даже установили, что эффект утрачивается,
а затем и переходит в противоположный,
если суточная доза превышает 50 г этанола
и возрастает далее. Да, тонкая материя —
человеческий организм... Нелишне будет
напомнить, что при хроническом
алкоголизме развивается гиперлипопротеидемия
1 На страницах этой книги уже неоднократно
шла речь о метаболических эффектах спиртных
напитков. Авторы подчеркивают, что интерес к
этой проблеме вызван совсем не желанием
развлечь читателя, а естественен для российской
медицины и представляет собой наследование
классической традиции — ведь еще
основоположник отечественной экспериментальной
медицины И.М. Сеченов посвятил свою
докторскую диссертацию именно сравнению влияния
различных спиртных напитков на организм
человека.
IV типа, совсем по другому влияющая на
ход атерогенеза. Более того, если у пациента
атеросклероз сочетается с гипертензией, что
достаточно типично, то даже малые дозы
алкоголя, по отношению к гипертензии, будут
фактором риска.
Повышению уровня ЛПВП способствует
и сексуальная активность. Это
позволило автору одного из западноевропейских
гигиенических плакатов начертать на
знамени профилактики сердечно-сосудистой
патологии «три S (sports, spiritus, sex)», что
было, возможно, некоторым упрощением
проблемы. А вот курение сигарет, безо
всяких упрощений, связано со снижением
уровня ЛПВП (см. ниже). Итак,
поддержание высокого уровня ЛПВП облегчается
при некотором, если можно так выразиться,
раблезианстве.
Может быть поэтому в эпоху позднего
Ренессанса, при докторе Ф. Рабле,
атеросклероз не был столь распространен, несмотря
на широко практиковавшееся чревоугодие.
АТЕРОСКЛЕРОЗ
«Есть еще такая болезнь —
склероз!»
А.Н. Стругацкий, Б.Н. Стругацкий
«Понедельник начинается в субботу»
Одна из самых значимых проблем
здравоохранения — борьба с атеросклерозом и
его последствиями, сердечно-сосудистыми
заболеваниями. Так как атеросклероз
развивается под воздействием хронических
нарушений липидного обмена, патофизиологии
этой болезни посвящена в структуре данной
главы особая часть.
Определение атеросклероза
и его место среди поражений
артерий
Атеросклероз — хроническое
прогрессирующее заболевание крупных и средних
эластических и мышечно-эластических
артерий (но не артериол), характеризу-
176 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ющееся пролиферативно-синтетическим
ответом ряда клеток сосудистой стенки и
крови — гладкомышечных (ГМК),
макрофагов, тромбоцитов, фибробластов — на
патологические (качественно своеобразные
и/или количественно избыточные) липоп-
ротеиды, с формированием в интиме атером
(фиброзно-липидных бляшек). Прогрес-
сирование атером приводит к вовлечению
медии и к осложнениям (изъязвление,
кальциноз, тромбоз и эмболия, аневризмы,
кровотечения). Атеромы становятся
источником вазоконстрикторных эйкозаноидов.
Извращается реакция пораженного сосуда
на вазомоторные стимулы, и появляется
тенденция к спазмам, а вазодилатация
затрудняется. Следствием этих процессов
являются ишемические поражения органов.
Атеросклероз как специфическую
разновидность артериосклероза, при котором
происходит отложение липидов в
сосудистой стенке крупных артерий эластического
типа, впервые выделил Ф. Маршан (1904).
Само наименование процесса
происходит от греческих слов <х8ерг| — «кашица»
и окХерос, — «твёрдый».
Артериосклероз— более широкое понятие, известное
с 1833 г. (Й.Ф. Лобштейн) и включающее
все процессы, при которых происходят
фиброз и сужение артериальных сосудов
любого калибра. Помимо атеросклероза, в
эту категорию включают артериосклероз
Мёнкеберга и артериолосклероз
(гиалиновую и гиперпластическую разновидности).
В отличие от атеросклероза, эти процессы
первично охватывают медию, наблюдаются
в средних и малых артериях мышечного
типа (артериосклероз Мёнкеберга) или даже
в артериолах (артериолосклероз), а также
не связаны с накоплением липопротеидов.
Артериосклероз Мёнкеберга, как правило,
сопряжен с кальцификацией или даже осси-
фикацией медии мышечных артерий (чаще
всего, в конечностях и половых органах), но
эти изменения не затрагивают эндотелий и
значительно меньше сказываются на
кровотоке, чем атеромы. Гиалиновый
артериолосклероз типичен для диабетической мик-
роангиопатии и последствий хронической
гипертензии, особенно, в малых артериях
и артериолах почек (см. ниже
«Патофизиология сахарного диабета»).
Гиперпластический артериолосклероз наблюдается при
наиболее тяжелых хронических гипертензиях
и ведет к фибриноидному некрозу артериол.
Таким образом, отличия атеросклероза от
других видов артериосклероза
существенны, хотя эти процессы в средних артериях
могут комбинироваться у одного и того
же пациента. Так как атеросклероз имеет
наибольшее эпидемиологическое значение
и, единственный среди данных процессов,
этиологически прямо связан с нарушением
липидного обмена, то в этом разделе он
рассматривается детально, а остальные виды
артериосклероза подлежат лишь краткой
сравнительной характеристике.
Эпидемиология и медицинская
география атеросклероза
Атеросклероз — одна из главнейших
медицинских проблем XX столетия. В Европе
и Северной Америке данное заболевание,
его прямые последствия и осложнения
служат ведущей причиной смертности
населения. На протяжении более чем 70 лет в
развитых странах отмечалось
прогрессирующее учащение и отягощение проявлений
атеросклероза и их «омоложение». Так, в
США за 40 лет, с 1930 по 1970, смертность
от атеросклероза коронарных артерий
возросла в 40раз (Ж. Леким, 1974)! Доля
этой болезни в структуре смертности в
отдельных странах в 60-70-е годы превышала
60%. Лишь в 80-х годах эту тенденцию,
благодаря успехам в изучении
патофизиологии атеросклероза и рационально
организованной профилактике, удалось несколько
затормозить, а в ряде стран — в частности, в
США и Канаде — даже добиться ощутимой
обратной динамики смертности от
атеросклероза (минус 58 % за 20 лет!) Для России,
Великобритании, скандинавских и многих
других стран и поныне
эпидемиологическая картина остается, практически, столь
же тревожной, как и в 70-е годы XX века.
Более половины людей умирает именно в
результате этого недуга.
Нарушения липидного обмена 111
Чтобы оттенить всё эпидемиологическое
значение атеросклероза, перечислим те
основные нозологические единицы, которые
являются его прямым последствием.
> Ишемическая болезнь сердца (ИБС), к
которой, по определению ВОЗ,
принадлежат различные формы стенокардии,
инфаркт миокарда, атеросклеротичес-
кий кардиосклероз и синдром внезапной
сердечной смерти. Только один инфаркт
миокарда в США, например,
ответственен за 25 % смертей.
> Ишемическая болезнь мозга, грозным
проявлением которой служит острое
нарушение мозгового кровообращения —
инсульт. Улиц, страдающих
церебральным атеросклерозом, проявляется также
ишемическая энцефалопатия.
> Ишемическое заболевание конечностей,
включая перемежающуюся хромоту,
гангрену нижних конечностей и такое
тяжелое проявление, как синдром Леригиа
(тромбоз подвздошных артерий).
> Ишемическая болезнь кишечника,
обусловливающая атонические состояния и
другие расстройства функций ЖКТ у
пожилых пациентов.
> Атеросклероз почечных артерий,
приводящий к первично сморщенной почке и
развитию хронической почечной
недостаточности.
Медицинская география и глобальная
эпидемиология атеросклероза
характеризуются рядом важных особенностей.
В мировом масштабе, в разгар эпидемии, в
конце 70-х годов XX века, наивысшая по-
раженность атеросклерозом наблюдалась у
населения Шотландии, Финляндии,
Швеции, Дании, Англии, Уэльса и Ирландии. Во
всех этих странах смертность только от ИБС
была между 300 и 400 на 100 000 населения!
Очень высокая пораженность
характеризовала Австралию и Новую Зеландию, США,
Голландию, Исландию, Чехию, Венгрию,
Россию (особенно, северо-западную),
некоторые другие страны, прилегающие к
Балтийскому морю, Канаду (везде смертность
от ИБС между 200 и 300 на 100 000, в США,
в частности, 242 — в 1968 г.), Как уже
упоминалось выше, с 1968 г. в Северной
Америке наметилась стойкая, удерживающаяся
последние 40 лет тенденция к понижению
смертности от атеросклероза. Так, в 1990 г. в
США смертность от ИБС упала до 102 на 100
000 населения. К сожалению, эта тенденция
не захватила Россию и страны скандинаво-
балтийского региона, где ситуация остается
примерно той же, что и 30 лет назад. И в 70-х
годах, и сейчас значительно более низкие
показатели пораженности ИБС отмечены
в Средиземноморье и Латинской Америке.
Весьма низкая пораженность
атеросклерозом наблюдается в странах Африки, в Азии
(кроме Японии, где она на уровне Южной
Европы), в то же время, инсульт, особенно,
ишемический, в Японии, как раз,
встречается в 4-5 раз чаще, чем в Финляндии и США
(И.К. Шхвацабая, Г. Андерс и соавт., 1977).
Итак, атеросклероз чаще всего встречается
у белого городского населения Северной
и Средней Европы и Северной Америки.
Он поражает и негроидов, но сравнительно
редок среди монголоидов. В происхождении
межпопуляционных различий по частоте
атеросклероза и его осложнений роль
климата минимальна. Эскимосы живут
севернее, чем датчане и шведы, но имеют очень
низкую частоту ИБС. Значительно больше
пораженность этой болезнью зависит,
по-видимому, от особенностей питания и образа
жизни населения,
социально-экономических факторов. Показана прямая корреляция
между уровнем потребления холестерина,
насыщенных жиров и сахара и частотой
атеросклероза. Зарегистрировано обратное
соотношение между количеством в диете
непредельных жиров (особенно, содержащих
со-ненасыщенные жирные кислоты),
пищевых волокон, антиоксидантов и
смертностью от ИБС. Однако, и эти закономерности
не абсолютны — восточно-африканский
этнос масаев использует диету с рекордно
высоким содержанием холестерина (до
1,5 г/сутки на человека), а атеросклероз у
этих скотоводов и охотников саванны даже
в глубокой старости находится лишь на
начальных стадиях, причем ИБС у них прак-
178 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
тически не встречается. Роль образа жизни
доказывается тем, что у мигрантов-выходцев
из регионов с низкой распространенностью
атеросклероза, например, японцев и арабов,
при переселении в США пораженность
атеросклерозом увеличивается и приближается
к характерной для аборигенов (Т. Гордон,
1957, Э. Бирман, 1994). Роль социальных
факторов может быть проиллюстрирована
данными А.Н. Климова и соавторов,
которые обнаружили, что в СССР и в США, в
конце 70-х годов XX века влияние уровня
образования на частоту инфаркта миокарда
у мужчин-жителей мегаполисов было
противоположным. В цитадели империализма
интеллигенция оказалась несколько менее
подверженной инфарктам, чем
неквалифицированные рабочие. Но в стране
победившего пролетариата тенденция была
обратной. Очевидно, огромное значение имеет
разная распространенность патологических
аллелей генов апопротеинов и неодинаковая
частота наследственных гиперлипопротеи-
демий у населения разных регионов. Так,
ген ЛП (а) наиболее распространен именно
в высокопораженных атеросклерозом
скандинавских популяциях. Семейная
наследственная гиперхолестеринемия очень часто
встречается среди англо-саксов и кельтских
народов (до 2 % носителей дефектного гена),
но практически отсутствует среди монголов,
что коррелирует с распространенностью
ИБС, являющейся главной причиной
смертности представителей англо-саксонских
этносов, но казуистически редкой среди
потомков Чингисхана. Особую
доказательность имеют исследования частоты
атеросклероза у представителей разных этнических
общин, проживающих на одной территории.
Красноречивый пример — заболеваемость
инфарктом миокарда в
городе-государстве Сингапур. Несмотря на идентичность
климато-географических и экологических
факторов сингапурские индийцы
поражаются инфарктом в 10 раз чаще местных
китайцев и в 8 раз чаще местных малайцев
(Н.Саха и соавт., 1973). Именно
особенности эпидемиологии атеросклероза
послужили одной из главных основ для заключения
экспертов о том, что уровень общественного
здоровья определяется, в первую очередь,
особенностями образа жизни, экологии и
генофонда популяций, а только во вторую
очередь зависит от состояния системы
здравоохранения. Можно заключить, что
атеросклероз — мультифакториальное
заболевание, при котором играет роль действие
комплекса факторов, связанных с питанием,
образом жизни, экологическими условиями
и, в первую очередь, генетическими
особенностями организма.
Этиология атеросклероза.
Модели и факторы риска
После идентификации атеросклероза
Ф. Маршаном, как формы
артериосклероза, связанной с липидными отложениями в
сосудах, внимание исследователей привлёк
вопрос: «Какие именно липиды
депонируются в артериях при атеросклерозе?».
Дж. Адами и Л. Ашофф доказали, что это
двоякопреломляющие липиды, в которых
много холестерина (1906). Еще до детального
исследования химии атеросклеротических
поражений, русский ученый А.И. Игнатов-
ский (1908) выступил с идеей, что причиной
атеросклероза служит перегрузка какими-то
компонентами, присутствующими в
животной пище. Путем кормления морских свинок
избытком яичных желтков ему удалось
получить у животных сходные с атеросклерозом
поражения аорты. Однако, он приписал
этиологическую роль в этом процессе «токсинам»
белкового происхождения. Н.В. Стуккей и
Н.В. Веселкин (1910, 1912) отвергли роль
белков и лецитина и указали на решающее
значение каких-то липидов, отличных от
нейтрального жира. В 1910 г. А.Виндаусу
удалось точно ответить на вопрос о главном
компоненте атеросклеротических липидных
отложений, доказав, что атероматозные
бляшки содержат, главным образом,
холестерин и его эфиры.
21 сентября 1912 г. в Санкт-Петербурге
произошло следующее этапное событие
в становлении учения об атеросклерозе.
С.С. Халатов на заседании Общества врачей
Нарушения липидного обмена 179
доложил о создании первой в мире
экспериментальной модели атеросклероза. Болезнь
была воспроизведена у травоядных
животных — кроликов — путём продолжительного
скармливания им кристаллического
холестерина (С.С. Халатов, 1912, Н.Н. Аничков,
С.С. Халатов, 1912). Холестерин вводился
алиментарно по 0,5 г/кг массы тела в день. За
1-3 месяца возникала выраженная гиперхо-
лестеринемия, а к 4-6 месяцам — заметные
морфологические изменения (липидные и
фиброзные бляшки).
На этом основании авторы в своей ин-
фильтрационной теории атерогенеза
вначале охарактеризовали атеросклероз, как
проявление экзогенного холестериноза.
Согласно оригинальной трактовке Н.Н. Аничкова,
инфильтрационный механизм состоит в
том, что плазменные липиды, содержащие
холестерин, просачиваются по
направлению от эндотелия к адвентиции. В норме
они используются для энергетических
целей или удаляются через лимфатическую
систему. При избытке поступления липи-
дов и, особенно, холестерина, механизмы
удаления не поспевают за накоплением
и формируется липидоз — первая стадия
атеросклероза. В дальнейшем авторы
решающую роль стали придавать соотношению
«липоидов крови с лецитином и белками
плазмы», вплотную подойдя к идее о роли
свойств и количества липопротеидов в
атерогенезе (1935). Н.Н.Аничкову (1915)
принадлежит заострённая формулировка
«Без холестерина не может быть
атеросклероза», подчеркивающая роль
данного фактора риска как наиглавнейшего.
Забегая несколько вперед, надо отметить,
что модель атеросклероза по С.С. Халато-
ву-Н.Н. Аничкову, хоть и была успешно
воспроизведена на морских свинках,
голубях, курах (А.Л. Кац и соавт., 1942),
хомяках, свиньях и обезьянах (К. Тейлор и соавт.,
1959), не осталась в экспериментальной
патологии единственной. Большое значение
имела неудача попыток воспроизвести эту
модель на плотоядных животных. Более
того, даже среди кроликов в опытах
родоначальников холестериновой модели
обнаруживалось 10-13 % «нуллеров», не
развивавших гиперхолестеринемии и атеросклероза,
несмотря на кормление. На сегодняшний
день разных экспериментальных моделей
атеросклероза известно, по крайней мере,
восемь. Это, само по себе, свидетельствует о
полиэтиологической природе
атеросклероза. Однако, роль дислипопротеидемий в его
происхождении столь велика, что А.Н.
Климов, уже в современную эпоху (1984), счел
возможным сохранить канонический
принцип Н.Н. Аничкова, лишь уточнив, что:
«Без атерогенных липопротеидов не будет
атеросклероза».
Другие известные экспериментальные
модели атеросклероза кратко
охарактеризованы ниже.
- Модель А. Стайнера-Ф. Кендалла
(1946) — первая алиментарная
холестериновая модель на плотоядных
животных — собаках. В течение долгого
времени не удавалось получить алиментарный
холестериновый атеросклероз у
плотоядных животных. Причина неудач не была
случайной — плотоядные обладают
высоким уровнем дренажных антиатероген-
ных ЛПВП в крови. Стайнер и Кендалл
решили эту задачу, параллельно вызвав
гипотироз у собак метилтиоурацилом.
- Модель Г. Виганда-Х. Мальмроса
(1960) также алиментарная.
Однако в диету травоядных (кроликов)
вводился не холестерин, а
насыщенные триглицериды. При этом было
принципиально доказано, что
холестерин в атероматозных отложениях
может быть эндогенным, полученным
из экзогенных предшественников.
Противопоставление моделей Виган-
да-Мальмроса и Халатова-Аничкова
оказалось несостоятельным, так как
было доказано, что при избыточном
введении в организм любого
компонента ядра липопротеидов усиливается
синтез всех липидов, необходимых для
формирования их частиц (см. выше
«Транспорт липидов...»), а значит, при
обеих моделях развиваются и гипертри-
глицеридемия, и гиперхолестеринемия.
180 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Существует и стрессорная (нейрогенная)
модель атеросклероза. Дело в том, что
теоретические взгляды авторов прототи-
пической алиментарной модели
атеросклероза — Н.Н. Аничкова и С.С.Халато-
ва — разошлись. Если первый придавал
большое значение экзогенной
холестериновой нагрузке и инфильтратив-
ному механизму, то второй считал, что
атеросклероз возникает и в результате
нарушения нейроэндокринных
механизмов обмена эндогенного холестерина,
модифицировав инфильтративную теорию
в инфилыпративно-комбинационную.
Первые доказательства правоты С.С.
Халатова были получены Н.Т.
Шутовой и П.В. Горизонтовым (1940),
продемонстрировавшими, что различные
воздействия на ЦНС вызывают глубокие
нарушения холестеринового обмена,
даже без избытка этого стероида в диете.
П.В. Горизонтов и С.С. Халатов
выдвигали гипотезу о роли ЦНС как депо,
снабжающего избытком холестерина
кровь при нервном напряжении. В годы
Второй мировой войны чешскому
патологоанатому Ф. Благе удалось показать,
что у умерших узников нацистского
концлагеря Дахау, подвергавшихся
тяжелейшим хроническим стрессам,
выраженный атеросклероз присутствовал,
даже несмотря на резкое обеднение их
тюремной диеты холестерином и
насыщенными жирами (1958). Позже, в
результате развития исследований школы
С.С. Халатова, патофизиологи
Ленинградского Педиатрического института
Н.Т. Шутова, П.С. Хомуло и А.С.
Фокин (1958,1981) создали оригинальную
модель воспроизведения атеросклероза
у травоядных и плотоядных животных
с помощью хронического стресса (по
терминологии того периода «нервного
напряжения и перенапряжения»), или
путём комбинации нейрогенного
фактора и эндокринных нарушений (гипотиро-
за, либо кастрации), без алиментарной
липидной нагрузки. Стрессорной по
своему содержанию была и модель
атеросклероза, полученная у кроликов по
В.В. Тявокину — путём
продолжительной иммобилизации животных.
- Модель атеросклероза по Я. Ватанабэ
(1980) — генетическая. Этим японским
ветеринаром была выведена чистая
линия гомозиготных кроликов с дефектом
рецептора апо-В/Е-зависимых липоп-
ротеидов, аналогичным наблюдаемому
у пациентов с семейной
наследственной гиперхолестеринемией (ГЛП Па).
У животных, безо всякой липидной
нагрузки, уровень холестерина в крови
был повышен в 6-13 раз, по сравнению
с нормальным. Тяжелый атеросклероз,
очень похожий по морфологии на
человеческий, развивался уже на протяжении
2-3 мес. жизни, а через 5 месяцев
регистрировалась ИБС. Животные гибли
от инфаркта миокарда, не доживая до
трехлетнего возраста. Модель Вата-
набе явилась ярким подтверждением
справедливости изложенных в разделах
«Транспорт липидов...» и «Гиперлипоп-
ротеидемии...» представлений М. Брауна
и Дж. Гольдстайна о взаимодействии
клеток сосудистой стенки и атерогенных
липопротеидов.
- Атеросклероз получен у трансгенных
мышей, которым пересажен ген
аномального апопротеина Е (В. Кумар и соавт.,
1997). Это еще раз доказывает значение
наследственных дислипопротеидемий в
генезе данного заболевания.
Ряд авторов разработал модели
атеросклероза, основанные на парентеральном
введении атерогенных липопротеидов. Эти
модели важны как свидетельство
определенной роли иммунологических процессов
и повреждения липопротеидных частиц в
развитии атеросклероза.
- Венгерские авторы С. Гёро и М. Би-
хари-Варга, а также отечественные
патологи Ю.Н. Зубжицкий, А.В. На-
горнев и А.Н.Климов (1979)
показали возможность воспроизведения
атеросклеротических поражений
сосудов у кроликов путем иммунизации
Нарушения липидного обмена 181
их липопротеидами. Оказалось, что
атеросклероз возникает и при
ежедневном парэнтеральном введении
в течение 6 месяцев липопротеидов
(ЛПНП и ЛПОНП) больных кроликов,
получавших пищевую
холестериновую нагрузку, — здоровым животным
(А.Н.Климов и соавт., 1967).
- Существует модель атеросклероза X. Баум-
гартнера (1964), основанная на
ускорении атеросклеротического процесса в
сосуде, деэндотелизированном с
помощью катетера с надувным резиновым
баллоном. Автор считал основным
фактором увеличение инфильтрации сосуда
липопротеидами при снятии эндотели-
ального барьера. Впоследствии было
отмечено, что при этом происходит также
артериит и освобождение цитокинов, в
том числе — тромбоцитарных факторов
роста, способствующих пролифератив-
но-склеротическому ответу сосудистой
стенки, а максимальное накопление
липопротеидов идет как раз не пассивным
путем, при отсутствующем эндотелии,
а в фазу регенерации эндотелиоцитов,
при усилении активного трансцитоза
липопротеидных частиц (А.Н.
Климов, Н.Г. Никульчева, 1984). Важно
отметить, что развитие атеросклероза по
данной модели тормозится при введении
животному антитромбоцитарной
сыворотки (X. Мур, 1976).
- Некоторые другие модели
атеросклероза, оттеняющие роль тромбогенных
факторов и моноклональных процессов
в его развитии, охарактеризованы ниже,
в разделе «Патогенез атеросклероза».
Основным результатом моделирования
атеросклероза и множества
эпидемиологических исследований было выделение
факторов риска этой болезни.
В настоящее время установлена связь
атеросклероза с 246 различными факторами
риска. Некоторые из них, практически, не
подлежат профилактической коррекции
или отмене — например, принадлежность
к мужскому полу, наличие ряда
неизлечимых генетических заболеваний (некоторых
дислипопротеидемий, гомоцистинурии,
порфирии). Ничего не поделаешь и с такой
отчетливой тенденцией, как учащение и
отягощение атеросклероза с возрастом, хотя
следует согласиться с Д.Ф. Чеботаревым
и В.В. Фролькисом (1967), что нельзя
отождествлять возрастные и собственно
атеросклеротические сосудистые
изменения. Однако, многие важные факторы риска
поддаются превентивным воздействиям,
поэтому для врача важно быть
информированным об этиологии и ранних признаках
атеросклероза.
Главными факторами риска
атеросклероза, по данным многолетних
эпидемиологических популяционных исследований
(У.Б.Кеннел и соавт., 1976), считаются
пяты
>• дислипопротеидемий (как
наследственные, так и приобретенные),
>- гипертензия (особенно, у лиц старше
50 лет),
>• курение (притом, в первую очередь,
именно сигарет),
>• сахарный диабет (особенно, инсулинне-
зависимый тип),
>• принадлежность к мужскому полу
(кроме возрастных групп после 75 лет).
В списке основных «мягких» факторов
риска — ожирение (особенно,
абдоминального типа), гиподинамия, хронический
стресс и соревновательно-стрессорный тип
жизнедеятельности по А. Фридмену (см.
т. I, стр. 94,529), гиперурикемия, переедание
сладкого, гиперинсулинизм, гипергомо-
цистинемия и фолациновый
гиповитаминоз, гипервитаминоз D, использование
пероральных противозачаточных средств,
мягкая вода, геохимические особенности,
связанные с обеспеченностью организма
рядом микроэлементов, тромбофилитические
состояния и т.д.
Ниже ряд факторов риска атеросклероза
будет охарактеризован последовательно.
Дислипопротеидемий как фактор риска
атеросклероза подробно рассмотрены в
182 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
предыдущем разделе. Кратко напомним, что
наиболее значимы:
>- высокое содержание апопротеина (а);
>• генетические аномалии других апопроте-
инов, в частности — апопротеина Е — при
ГЛП III, апопротеинов, кодируемых 11
хромосомой, — при гипоальфалипопро-
теинемиях;
>* высокий уровень ЛПНП (при ГЛП На
и Нб типа), ЛППП (при ГЛП III типа),
ЛПОНП (при ГЛП II б типа, а также, в
меньшей степени, ГЛП IV и V типа) и,
наконец, остаточных частиц ХМ (при
ГЛП III типа);
>• понижение уровня ЛПВП.
Важно учесть, что атерогенны не только
наследственные ГЛП, но и их
приобретенные фенокопии. По этой причине скорость
развития атеросклероза повышена при
гипотирозе, нефротическом синдроме,
холестазе, гиперкортицизме, порфирии и
других состояниях, сопровождающихся
вторичными ГЛП.
ГЛП с повышением общего и свободного
ХН гораздо атерогеннее, чем те, при которых
повышается только уровень ТГ, хотя и
последние не лишены атерогенного потенциала.
ЭХ неатерогенны. Нельзя недооценить
значение хорошо доказанного положения,
что патологические качественные
модификации липопротеидных частиц — окисление,
гликозилирование, ацетилирование,
обогащение лизолецитином и свободным ХН,
присоединение к апопротеинам аутоанти-
тел — значительно усиливают атерогенность
ЛП, так как способ их взаимодействия с
клетками сосуда при этом меняется.
Атерогенность дислипопротеидемий усиливается
при недостатке полиеновых и со-3-ненасы-
щенных жирных кислот, а также антиокси-
дантов в ЛП, поскольку липиды таких ЛП
более подвержены окислительным и иным
трансформациям.
Многие факторы риска, перечисленные
отдельно, частично опосредуют свой ате-
рогенный потенциал через
дислипопротеидемий. Таковы, например, вызывающие
ГЛП: сахарный диабет, стресс, ожирение,
гиперурикемия, углеводистая диета,
противозачаточные средства, а также
гиподинамия и курение. Все они провоцируют
гипоальфалипопротеидемию. Д. Кричев-
ский установил связь между снижением
содержания волокон клетчатки в пище и
частотой атеросклероза (1988), что
объясняется способностью пищевых волокон
стимулировать развитие эубактериальной
флоры кишечника, снижающей всасывание
холестерина и облегчающей его выведение.
Все эти факторы обладают, по-видимому, и
собственным атерогенным действием,
независимым от дислипопротеидемий.
Гипертензия как фактор риска
атеросклероза впервые фундаментально рассмотрена
выдающимся отечественным терапевтом
А.Л. Мясниковым, в его классическом труде
«Гипертоническая болезнь и атеросклероз»
(1965). Он расценивал атеросклероз и
гипертоническую болезнь как «весьма близкие
по своему происхождению заболевания...
возможно даже представляющие собой
варианты течения процесса с общей этиологией».
В связи с этим, им подчеркивалась роль
общих патогенетических звеньев этих болезней
(хронический стресс), а также их взаимное
влияние. Гипертензия повышает риск
развития атеросклероза в связи с усилением
инфильтрации сосудистой стенки ЛП при
повышенном давлении. Большое значение
имеет и повреждение эндотелия гемодина-
мическими факторами, необходимое для
начальной активизации проникновения ЛП
в сосудистую стенку и для стимуляции тром-
боцитарных факторов атерогенеза.
Гипертензия способствует гипертрофии гладкомышеч-
ных клеток сосуда (ГМК) и выработке в них
соединительнотканных белков, а возможно,
и гиперплазии ГМК— одному из основных
событий атерогенеза. С другой стороны,
атеросклероз, особенно каротидных и почечных
артерий, нарушает работу стабилизирующих
механизмов регуляции артериального
давления и способствует гипертензии.
Вероятность ИБС растет
пропорционально повышению артериального кровяного
Нарушения липидного обмена 183
давления, причем какого-либо критического
уровня давления нет. При артериальном
давлении 160/95 мужчины имеют риск ИБС в 5
раз больше, чем при давлении 140/90.
Весьма существенное значение имеет повышение
диастолического давления. Гипертензия
в возрастных группах старше 45 лет более
важна как фактор риска ИБС и инсульта,
чем дислипопротеидемия. Доказано, что
успешное лечение гипертензии понижает
скорость развития атеросклероза и риск его
осложнений. В связи с взаимоотношениями
гипертензии и атеросклероза, большой
интерес представляют новые данные о
существовании однотипного нарушения — патологии
водород-натриевого противопереносчика
(hydrogen-sodium antiport) при ряде ате-
рогенных нарушений — эссенциальной
гипертензии, гиперурикемии, андроидном
ожирении, инсулинорезистентности — в
структуре так называемого Х-синдрома (см.
выше рис. 35 и подробнее гл. 14). Очевидно,
при нарушении транспорта этих катионов
меняется характер взаимодействия клеток
сосудистой стенки и ЛП частиц.
Курение как фактор риска атеросклероза
опосредует своё пагубное действие
несколькими путями. Во-первых, как уже
отмечалось выше, у курящих меньше продукция
антиатерогенных ЛПВП. Затем, курение
приводит к повреждению эндотелия
компонентами дыма, например угарным газом.
У курящих в повышенных титрах
обнаружены аутоантитела к эндотелию. Нельзя
сбрасывать со счетов и хорошо известное
наличие в табачном дыме мутагенов, ввиду
существования данных о роли соматических
мутаций ГМК в атерогенезе. Важным
фактором, усугубляющим атерогенез, служит
гипоксия, индуцированная курением и
связанная с выраженной карбоксигемог-
лобинемией. Показано, что гипоксия
вызывает ускорение развития алиментарного
холестеринового атеросклероза у кроликов
и обезьян, стимулирует пролиферацию
ГМК в культуре клеток и даже способна
(очевидно, будучи стрессором) при
хроническом действии спровоцировать у
животных атеромоподобные поражения артерий.
Гипоксия снижает скорость лизосомальной
деградации ЛПНП. Комплекс этих
механизмов влияет на ситуацию таким образом, что
лица, выкуривающие больше 10 сигарет в
день, имеют утроенный риск атеросклероза.
При выкуривании пачки сигарет в день
табачная зависимость увеличивает смертность
на 70-100%, по сравнению с некурящими
того же возраста, а смертность от ИБС — на
200%. Остается только повторить вслед за
В.В. Маяковским: «Курить бросим— яд
в папиросе!». Правда, сравнительные
исследования наиболее чётко
продемонстрировали повышение риска атеросклероза и
ИБС именно при курении сигарет. Курение
трубки и сигар оказалось в этих отношениях
менее опасным, может быть, из-за меньшей
ингаляции продуктов неполного сгорания
табака. Особенно опасным, в отношении
ускорения развития ИБС, является курение
сигарет для женщин в возрасте старше 35
лет, принимающих пероральные
противозачаточные средства. При курении особенно
возрастает скорость развития атеросклероза
артерий нижних конечностей, например,
подколенных. Не только атеросклероз, но и
артериосклероз в этих сосудах тоже
характерен для курящих. Частота облитерирующих
заболеваний артерий нижних конечностей
среди некурящих в десятки и даже в сотни
раз ниже.
Важным комплексным фактором риска
атеросклероза является сахарный диабет
(СД). При наличии сахарного диабета риск
ИБС в среднем возрастает не менее чем в 2
раза. Вероятность развития ишемических
артериопатий нижних конечностей у
пациентов, страдающих СД, почти в 150 раз
выше, чем у лиц с нормальным обменом
глюкозы! Если в норме принадлежность
к женскому полу в 3-5 раз снижает риск
развития основных сердечных осложнений
атеросклероза в репродуктивном возрасте,
то при наличии сахарного диабета частота
ИБС и инфаркта миокарда у мужчин и
женщин этого возраста выравнивается. Не более
5 % лиц без сахарного диабета и целых 75 %
184 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
больных СД страдают от ранних проявлений
атеросклероза, начинающегося в возрасте
до 40 лет. Полагают, что при длительности
явного сахарного диабета 10 лет и более,
независимо от его типа, все больные имеют
достаточно выраженный атеросклероз. Ате-
рогенным является любой сахарный диабет,
но особенно атеросклеротические
поражения характерны для инсулиннезависимого
сахарного диабета II типа (ИНСД II). Не
менее 1/2 больных этой формой болезни
умирает от последствий ускоренного
атеросклероза, который в диабетологии известен
как диабетическая макроангиопатия (см.
ниже «Патофизиология сахарного
диабета»). Инфаркт миокарда — самая частая
причина смерти пациентов с ИНСД (37 %).
Механизмы ускорения атеросклероза при
СД множественны.
Липидный метаболизм при сахарном
диабете, вкратце, характеризуется
следующим (подробно см. ниже «Патофизиология
сахарного диабета»).
— Инсулин способствует отложению жира
в жировой ткани и снижению его
утилизации. Механизмы не сводятся только
к сбереганию жира путем усиления
катаболизма экзогенной глюкозы.
Поэтому, при инсулинзависимом сахарном
диабете (ИЗСДI), из-за дефицита
инсулина будет развиваться исхудание, а при
ИНСД, вследствие избытка инсулина,
ожирение.
- При ИЗСД и ИНСД резко увеличена
скорость липолиза и содержание не-
этерифицированных жирных кислот в
крови. Жиры, освобождаемые адипо-
цитами, при этом ресинтезируются в
гепатоцитах, и поэтому обе формы СД
приводят к стеатозу печени. При ИЗСД
ожирение печени наблюдается,
несмотря на общее исхудание. Липиды могут
составлять до 1/3 веса печени больных
диабетом, в частности, при так
называемом синдроме Мориака. Вследствие
этого диабет, особенно, ИНСД, часто
сопровождается ГЛПIV типа (с
накоплением в крови Л ПОНП). Из-за снижения
стимулируемой инсулином активности
липопротеиновой липазы при ИЗСД
может быть и ГЛП I типа (персистирование
хиломикронов), равно как и сочетание I
и IV типов, классифицируемое как ГЛП
V типа.
Большое значение имеет обнаруженное
при ИЗСД повышение концентрации
особо атерогенного ЛП (а), стимулирующего
тромбогенез и коагуляцию.
Но, гиперлипопротеидемия лишь
частично объясняет ускорение атеросклероза при
сахарном диабете.
Данный недуг служит фактором риска
атеросклероза не только в силу ГЛП, но и
из-за присущих ему гипергликемии,
особенностей гемостаза, артериального кровяного
давления. Определенное значение, при ин-
сулиннезависимом типе диабета, имеет сам
гиперинсулинизм. Атерогенны и некоторые
применяемые при диабете лекарства.
- В условиях гипергликемии белки, в том
числе ЛП плазмы крови и сосудистой
стенки, легко подвергаются гликирова-
нию (см. раздел «Патофизиология
углеводного обмена»). ЛПНП, подвергнутые
неэнзиматическомугликозилированию,
по сравнению с нативными ЛПНП,
гораздо более активно захватываются
клетками сосудистой стенки, особенно,
макрофагами, через «мусорные»
рецепторы, без должного адаптивного ответа
на поступающий избыток ХН.
Отложение избытка холестеринсодержа-
щего материала в сосудистой стенке
ускоряется. В то же время, гликирование ЛПВП
приводит к укорочению времени их жизни
и снижению концентрации, соответственно,
тормозится дренаж ХН.
Гликированные молекулы волокнистых
белков соединительной ткани сосудистой
стенки захватывают и фиксируют Л ПОНП
и ЛПНП более активно, чем у индивидов без
сахарного диабета.
По мнению X. Стайнера (1985), сама
глюкоза может иметь и атерогенное действие,
независимое от гликирования компонентов
сосудистой стенки: в культуре эндотелиоци-
тов высокие концентрации глюкозы ингиби-
Нарушения липидного обмена 185
руют пролиферацию. Не исключено, что это
нарушает регенерацию микроповреждений
эндотелия у больных сахарным диабетом и
способствует инфильтрации артерий липоп-
ротеидами и тромбогенезу.
- ГиперинсулинеМия — важный
самостоятельный фактор риска атеросклероза,
аддитивный по отношению к
гипергликемии, гиперхолестеринемии, гипертен-
зии — и независимый от них. X. Стайнер
(1985) приводит данные, что избыток
инсулина ведет к ускорению развития
атеросклероза даже при отсутствии
гипергликемии и сахарного диабета,
например, у больных с инсулиномами.
Считается, что избыток инсулина
ускоряет пролиферацию клеток сосудистой
стенки. Инсулин способствует задержке
натрия и воды в организме, а значит и
гипертензии, ускоряющей атерогенез.
При гиперинсулинемии увеличивается
продукция ЛПОНПиЛППП, инсулин
ускоряет поглощение ЛПНП
человеческими фибробластами в культуре клеток.
Инсулин оказывает в культуре клеток
гонад прямой стероидогенный эффект.
Имеются данные, что атерогенное
действие присуще, в основном, избытку
инсулина при подкожном введении, так
как гормон при этом не проходит через
печень.
- Дополнительным важным фактором,
сопрягающим диабет и атеросклероз,
может быть своеобразие состояния
системы гемостаза при сахарном диабете.
Отмечается избыточная склонность к
тромбообразованию. Активация
тромбоцитов может приводить к появлению
в плазме и стенке сосудов медиаторов
пролиферации, усиливающих митоге-
нез гладкомышечных клеток и синтез
ими компонентов базальных мембран:
коллагена, гликозаминогликанов. Все
это ускоряет образование атером.
Показано, что тромбоциты больных сахарным
диабетом, в отличие от тромбоцитов
здоровых лиц, способны выделять
факторы, вызывающие усиленную
пролиферацию гладкомышечных клеток сосудов
(Д. Стаут; 1984). Сыворотка крови
больных сахарным диабетом содержит эти
факторы и усиливает коллагеногенез
в культивируемых гладкомышечных
клетках. Уровень ростовых факторов в
сыворотке больных ИЗСД
пропорционален степени гипергликемии.
Хроническая гипергликемия может увеличить
синтез тромбоксана А2 и подавить
продукцию простациклина тромбоцитами
и сосудистой стенкой. Это способствует
тромбофилитическому синдрому,
увеличенной адгезивности и агрегации
тромбоцитов при сахарном диабете. В аортах
и коронарных артериях животных и
людей, страдающих сахарным диабетом,
снижена продукция антитромбогенного
вазодилататора N0 (окиси азота) и
увеличено производство эндотелина-1,
оказывающего констрикторный эффект
на сосуды и стимулирующего митотичес-
кую активность гладкомышечных клеток
при атерогенезе. Поэтому, атеросклеро-
тические бляшки у больных не только
ускоренно формируются, но и чаще
приводят к более выраженным
сосудистым спазмам, в частности, коронарным
приступам. Снижение скорости
кровотока и частые резкие перепады уровня
сахара при СД приводят к мобилизации
адреналина, спазмированию сосудистых
стенок и гипоксии, вносящей свой вклад
в патогенез осложнений атеросклероза.
Д. Стаут (1980) указывает на
применение сорбита как на дополнительный
фактор риска атеросклероза при ИНСД,
поскольку сорбитол усиливает
пролиферацию гладкомышечных клеток и их
пенистую трансформацию.
Повсеместно атеросклероз быстрее
развивается у мужчин. Принадлежность к
сильному полу считается значительным фактором
риска во всех возрастных группах до 65 лет.
Между 35 и 55 годами мужчины в США,
например, в 5 раз чаще умирают от ИБС, чем
женщины. Риск несколько выравнивается в
группах, соответствующих
климактерическому периоду, а после 65 лет он одинаков,
причем частота неинфарктных форм ИБС
186 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
у женщин становится даже чуть выше,
чем у их сверстников-мужчин. Несмотря
на известный вклад, который вносится в
эту «половую асимметрию» атеросклероза
несколько большим распространением
курения, гипертензии, соревновательно-
стрессирующего образа жизни среди
мужчин, решающее значение имеет различие в
концентрациях половых стероидов.
Эстрогены способствуют продукции антиатеро-
генных ЛПВП, уровень которых у мужчин
существенно ниже. Андрогены увеличивают
уровень ЛПНП. Кастрация у мужчин и у
самцов подопытных животных приводит
к снижению коэффициента атерогенности
плазменных ЛП. Эстрогены
способствуют ускорению окисления ХН печенью, а
андрогены тормозят эти процессы (Д.
Поповичи, В. Сэхляну, 1969). Под влиянием
эстрогенов количество рецепторов ЛПНП в
печени растет (см. также ниже «Нарушения
эндокринной регуляции»).
Не совсем ясно, как увязать с данными
об антиатерогенности женских половых
гормонов свидетельства гинекологов о
стимулирующем действии эстрогенов на
развитие гладкомышечных опухолей и
гиперплазии гладких мышц матки — ведь
известно, что источником таких миом часто
бывают именно сосудистые гладкомышеч-
ные клетки (Г.А. Савицкий, А.Г. Савицкий,
1994), а при атеросклерозе большое
значение также имеет, как раз, гиперплазия и
пенистая трансформация мигрировавших в
интиму гладкомышечных клеток (см. ниже).
Возможно, эстрогены и патологические Л П
включают различные и, во многом,
альтернативные пути пролиферативного ответа
этих клеток-мишеней.
Тучность повышает риск развития
атеросклероза, особенно у молодых людей, в
возрастных группах до 50 лет. Ее действие
связывают с ГЛ П, гиподинамией, гипертен-
зией, гиперлептинемией и инсулинорезис-
тентностью, которые сопряжены с
ожирением (см. следующий раздел).
Низкая жесткость воды способствует
активизации кальций-задерживающих и
натрий-сберегающих механизмов клеток и
известна как фактор риска гипертензии, а
через нее — и атеросклероза. В Европе
наивысшей частотой ИБС отличаются районы
с самой мягкой водой. Среди них —
Финляндия, Карелия и Санкт-Петербург.
Диетические особенности как фактор
риска атеросклероза по ходу изложения уже
неоднократно затрагивались в
разрозненном виде. Нет необходимости повторяться,
поэтому здесь лишь подытожим, что
атеросклерозу способствуют диеты, богатые
холестерином, насыщенными жирами и
легкоусвояемыми углеводами, а также
бедные полиеновыми кислотами, пищевыми
волокнами, антиоксидантами и
некоторыми незаменимыми факторами (В. Кумар и
соавт., 1997).
О важности незаменимых полиеновых
кислот уже говорилось. В настоящее
время большое внимание привлекает вопрос
о гомоцистинемии как эндотелиотокси-
ческом атерогенном факторе. В связи с
этим, считается, что нормальная пищевая
обеспеченность метионином, фолацином,
кобаламином, витамином В6 — то есть
факторами, существенными для обмена
цистеина и цистина, способствует
предупреждению атеросклероза (см. также
разделы «Патофизиология белкового обмена»
выше и «Патофизиология витаминного
обмена» — ниже). В то же время, есть да-
ные об атерогенности гипервитаминоза D.
Гиполипопротеидемическая активность
витамина РР доказана фармакологически
и используется в терапии.
Многие специалисты указывают на
ускорение окислительной трансформации
ЛП и атерогенеза при нехватке пищевых
антиоксидантов, особенно,
жирорастворимых (токоферолы, ликопен и др. — см. ниже,
раздел «Редокс-витамины»).
Наконец, к темпам атерогенеза,
определенно, имеют отношение некоторые
микроэлементы. Л.Р. Ноздрюхина (1977)
приводит убедительные данные об атерогенности
дефицита ванадия. Известно, что районы с
биогеохимической нехваткой селена,
входящего в активный центр супероксиддисмута-
зы (ключевого элемента антиоксидантной
Нарушения липидного обмена 187
системы клеток) характеризуются высокой
пораженностью атеросклерозом и низкой
средней продолжительностью жизни. На
первом месте и в отношении низкого
содержания селена, и по пораженности
атеросклерозом стоит, в частности, Восточная
Финляндия. Есть сведения о роли избытка
свинца и недостатка хрома в повышении
риска развития атеросклероза. Но, в целом,
роль микроэлементов в атерогенезе изучена
всё еще недостаточно. Более подробная ее
характеристика имеется ниже в разделе
«Патофизиология обмена микроэлементов»).
Патогенез атеросклероза
Еще в XVIII веке А. Галлер обнаружил
«жировые отложения в стенке артерий»,
которые назвал атеромами (1755). Задолго
до выделения атеросклероза как
самостоятельной нозологической формы, К.Ро-
китанский, описав атеросклеротические
поражения интимы аорты, истолковал их
как результат «инкрустации» фибрина и
компонентов тромбов в сосудистую стенку
из крови — теория диспрозии (1850).
Замечательными догадками чешского патолога
были идея о плазменному а не сосудистом
происхождении «наслоений», которые он
обнаружил (и доселе важнейшим звеном
атерогенеза считается именно плазменная
инфильтрация сосудистой стенки липоп-
ротеидами!), а также мысль о связи между
свертыванием крови — и генезом этих
образований. До известной степени, это было
предвосхищение разработанной позже и
вошедшей в современную интегральную
концепцию тромбогенной теории
атерогенеза (Ж.Б. Дьюгид, 1946; см. ниже).
Р. Вирхов, напротив, придерживался
мнения, что атеросклеротические изменения
вызываются внутренними процессами в самой
сосудистой стенке и считал важными для
их возникновения механизмы воспаления
(1856,1892). Он даже дал процессу название
«деформирующий узловой артериит». По
его теории, происходит нутритивное
раздражение клеток интимы, а затем — дистрофия
и жировая дегенерация. Ю. Конхайм (1877),
развивая эти взгляды, трактовал
атеросклероз как медленно протекающее хроническое
воспаление сосудистой стенки, с исходом в
склероз, жировую дистрофию и кальциноз.
В своей последующей эволюции, учение об
атеросклерозе унаследовало вполне
обоснованную мысль основателя целлюлярной
патологии о решающем вкладе самих клеток
сосудистой стенки в происхождение атером,
а также о большой роли воспалительных
цитокинов, активированных макрофагов и
факторов роста при атерогенезе. И поныне
существует взгляд на атеросклероз, как
особую форму хронического продуктивного
воспаления, сопровождаемого отложением
ЯП (В.В. Мейер, 1949; X. Брент, 1969) или
же индуцированного ЛИ (В.Ю. Шанин,
1995; В. Кумар и соавт., 1997).
Таким образом, время показало, что в
чем-то правы были оба непримиримых
полемиста и современные концепции
трансформировали элементы и взглядов К. Роки-
танского, и теории Р. Вирхова.
Большое значение в становлении
теоретических представлений о механизмах
атеросклероза имели представления Р.
Тома (1886), придававшего решающую роль
в атерогенезе гладким мышцам сосудов и
А. Йореса (1904), верно определившего
интиму как место локализации начальных
поражений и ключевых событий при
атеросклерозе. После выделения Ф. Маршаном
атеросклероза как формы
артериосклероза, связанной с накоплением липидов
(1904), центр иследований переместился в
область установления природы липидных
отложений в атеромах. Как уже отмечалось
выше, при обсуждении
экспериментальных моделей атеросклероза, усилиями
Дж. Адами, Л. Ашоффа (1906) и, особенно,
А. Виндауса (1910) было доказано, что
основной липид-участник атерогенеза — ХН.
А. И. Игнатовский высказал предположение
о пищевом происхождении избытка
липидов при атерогенезе (1908). С.С. Халатов и
Н.Н. Аничков экспериментально доказали,
что пищевой ХН может играть центральную
патогенетическую роль в развитии
атеросклероза (1911). В результате этого появилась
188 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
инфилътрационная теория атерогенеза
Н.Н.Аничкова. Согласно этой концепции,
атеросклероз развивается в результате
превышения поступления пищевого ХН,
используемого для энергетических целей
бессосудистой зоной артерии (интимой и
внутренней третью медии), над его
утилизацией, при недостаточности дренажной
функции vasa vasorum и лимфатических
сосудов. Это ведет сначала к «липоидозу»,
а затем к реакции на него. Морфогенез
атеросклероза был детально изучен, с позиций
данной теории, В.Д. Цинзерлингом (1958) и
другими авторами, в результате чего
сложилось применявшееся много лет классическое
деление атеросклеротических поражений
на последовательно сменяющие друг друга
стадии: липидные пятна, атеросклероти-
ческие бляшки (липидные и фиброзные) и
осложненные поражения, включая атеро-
кальциноз и изъязвление бляшек.
Впоследствии появились представления, развитые
А. Л. Мясниковым, А. М. Вихертом и соавт.
(1962-1965) о существовании еще и ранней,
долипидной стадии, включающей изменения
эндотелия и предшествующей липоидозу.
Сложилось деление микроскопической
картины атеросклеротических поражений на
долипидные изменения, липидоз, липосклероз,
атероматоз и атерокальциноз.
Уже в ранний период развития инфиль-
трационной теории ее авторы писали о
важности для атерогенеза не столько самой
концентрации ХН, сколько его соотношений
с лецитином и белками плазмы,
предвосхищая открытие роли ЛП в судьбе ХН (1925).
Позже, Н. Н. Аничков и, в первую очередь,
С.С. Халатов (1946) трансформировали
инфильтративную теорию в инфильтратив-
но-комбинационную, придающую решающее
значение в атерогенезе не пищевому ХН,
а судьбе эндогенного ХН, которая
нарушается в зависимости от белок-липидных
взаимоотношений в крови и от расстройства
действия нейроэндокринных регуляторных
механизмов. В 50-60-х годах XX столетия,
после выделения Дж. Гофманом (1954,
1961) различных классов ЛП и открытий
Д.С. Фредериксона (1967), описавшего
(эндокринно-метаболические нарушения)
атерогенные и неатерогенные гиперлипоп-
ротеидемии (см. выше), стало ясно, что
инфильтрация сосудистой стенки ХН
происходит в составе ЛП, которые и определяют
метаболический результат действия ХН на
клетки сосудистой стенки. Инфильтратив-
но-комбинационную теорию атерогенеза,
применительно к представлениям о ЛП,
дополнил и развил Дж. Пэйдж (1954).
Благодаря исследованиям М.Д. Хост (1971),
которая определила происхождение
характерных для атеросклеротических цораже-
ний пенистых клеток, стало ясно, что ответ
гладкомышечных элементов артерий на ЛП,
их миграция, пролиферация, осуществление
ими эндоцитоза ЛП и их синтетическая
активность имеют центральное значение для
морфогенеза атеросклероза.
В конце 40-х годов Ж.Б. Дьюгид (1946,
1949) выступил с точкой зрения,
возрождающей тромбогенную теорию атерогенеза.
Он считал эволюцию атером процессом
инкрустации и организации микротромбов
и фибриновых масс. Автор опирался на
данные об изменениях эндотелия на ранних
стадиях развития атеросклероза,
предшествующих липоидозу. Впоследствии ряд
новых открытий подтвердил право тромбоген-
ной теории на актуальность и показал, что
тромботические факторы, по крайней мере,
должны расцениваться как неотъемлемое
патогенетическое звено атерогенеза, даже
при признании примата патологических
ЛП как пускового фактора всего процесса.
Здесь громадное значение имели
наблюдения за реципиентами внутрисердечных
аутотрансплантатов v. saphenae magnae, в
которых повреждение эндотелия высоким
коронарным давлением и последующий
атеросклероз развиваются безо всякой ГЛП.
Более того, сторонникам тромбогенной
теории удалось показать, что у
карликовых лабораторных свиней, вообще говоря,
подверженных атеросклерозу по модели
Н.Н. Аничкова-С.С.Халатова, процесс
тормозится, и успешного моделирования
не получается, если имеются болезнь фон
Виллебранда и нарушение функций
тромбоцитов. Развитие экспериментального ате-
Нарушения липидного обмена 189
росклероза замедляется также лекарствами,
ингибирующими тромбоцитарные функции
(дипиридомол) и антитромбоцитарной
сывороткой (Т. Шимамото, 1974). В 70-х годах
Р. Росс и А. Гломсет сообщили о том, что
тромбоциты при взаимодействии с ЛП
больных атеросклерозом могут выделять факторы
роста для гладкомышечых клеток и, таким
образом, нашелся мост, связывающий
воедино две, до этого непримиримые, казалось бы,
альтернативные теории атерогенеза.
Современный (синтетический) вариант
инфильтративно-комбинационной теории
атеросклероза появился из-за
необходимости учесть факты, добытые в русле развития
тромбогенной теории, а также сведения о
долипидных изменениях в интиме, не
отказываясь от краеугольного положения «без
атерогенных ЛП не будет атеросклероза»,
которое было с новой силой
подтверждено благодаря работам Дж. Гольдстайна и
М.С.Брауна (1974-1984), раскрывших
механизмы рецепторно-эндоцитотических
взаимодействий ЛП и клеток сосудистой
стенки (см. выше). Эту концепцию
впервые в развернутом виде сформулировали
Р.Росс, А. Гломсет, А. Готто и Р. Джексон
и охарактеризовали ее как «теорию ответа
на повреждение» (1976).
Предваряя её подробную характеристику,
приводим современную классификацию
атеросклеротических поражений у
человека, разработанную в 1995 г. Х.К. Стэри и
принятую кардиологическими обществами
ряда стран:
Тип I — начальные поражения,
характеризуются изменениями в эндотелии и
наличием отдельных пенистых клеток мак-
рофагального происхождения.
Тип II — липидные полоски,
характеризуются, преимущественно, внутриклеточным
депонированием липидов в скоплениях
пенистых клеток макрофагального и глад-
комышечного генеза.
Тип III— переходные поражения, сходные
с II, но имеющие некоторое количество
внеклеточных липидных депозитов.
Тип IV — атеромы, располагают
значительным ядром внеклеточных липидов.
Тип V— фиброатеромы, отличаются
наличием фиброзной «крышки» над липидным
ядром, могут кальцифицироваться или
бывают, преимущественно, фиброзными.
Фиброатеромы растут за счет
пролиферации гладкомышечных элементов и синтеза
ими коллагена, эластина и гликозаминог-
ликанов.
Тип VI — осложненные поражения,
имеют поверхностные дефекты, вторичное
тромбообразование, растут при участии
механизмов кровоизлияний и интрамураль-
ного тромбоза, часто проникают в медию.
Возникают не только из поражений типа V,
но и типа IV.
Клинические симптомы осложнений
атеросклероза характерны только для V-VI
типов, а иногда могут быть при богатых
липидами и активными макрофагами
поражениях типа IV. 1-Ш типы поражений, как
правило, протекают субклинически.
Патоморфологические основы теории
ответа на повреждение и стоящие за
морфогенезом поражений механизмы в
настоящее время представляются следующим
образом.
Атеросклероз охватывает в наибольшей
степени ряд артериальных сосудов, в
которых сильнее всего выражена механическая
нагрузка на стенку. В первую очередь, это
абдоминальная аорта, находящаяся, по
меткому выражению В. Кумара и соавт. (1997),
«между молотом пульса и наковальней
позвоночника». Затем, по убывающей степени,
идут артерии коронарные, подколенная,
бедренная и тибиальные, грудная аорта и ее
дуга, сонные артерии и артерии виллизиева
круга.
В пораженных артериях могут быть
выделены долипидная, ранняя липидная,
поздняя и осложненная стадии атерогенеза.
Ранние поражения наблюдаются с начала
первой декады жизни. Это долипидные
изменения интимы (которые обнаруживают
даже в артериях годовалых детей), липидные
пятна и липидные полоски.
Долипидные изменения неспецифичны, то
есть могут вызываться как самой гиперли-
190 А.Ш. Зайчик, Л.П.Чурилов Патохимия
попротеидемией (Р. Джеррити, 1987), так
и другими факторами: гипертензией,
колебаниями гемодинамических характеристик
потока крови в местах бифуркации изгибов,
сужений и ветвления сосудов, около
полулунных клапанов («shear stress»), курением,
иммунопатологическими реакциями,
гипоксией, вирусной инфекцией,
бактериальными эндотоксинами, гипервитаминозом
D, другими ядами, например, свинцом и
кадмием. Большое значение придают
прямой эндотелиотоксичности гомоцистеина,
поскольку при гипергомоцистеинемии и
гиповитаминозе по фолацину и другим
витаминам, участвующим в утилизации этого
метаболита, атеросклероз резко ускоряется
(Р. Кларк и соавт., 1991). Так или иначе,
существенно то, что для запуска атерогенеза не
нужно явной десквамации эндотелия.
Достаточно активации эндотелия и экспрессии на
его поверхности, под воздействием многих
вышеперечисленных факторов, в частности,
лизофосфатидилхолина патологических Л П
частиц, молекул клеточной адгезии (см. т. I,
стр. 303-310).
Это создает на поверхности эндотелия
зоны повышенной клейкости и
проницаемости. Внешне данный процесс может
проявляться в разрыхлении и истончении
(вплоть до исчезновения) защитного гли-
кокаликса на поверхности эндотелиоцитов.
Межэндотелиальные щели расширяются.
Везикуляция цитоплазмы эндотелиальных
клеток свидетельствует об усилении эндо-
цитоза и трансцитоза. Субъэндотелиальный
слой интимы отекает, происходит
разъединение его клеток и волокон. Некоторые
авторы, рассматривая механизмы долипидной
стадии, подчеркивали роль накопления в
интиме гликозаминогликанов, способных
связываться в комплексы с ЛП, и появление
плоских пристеночных наслоений
лейкоцитов и тромбоцитов над окнами активного
эндотелия.
Происходит не только адгезия, но и
усиленное проникновение моноцитов в интиму,
где они ведут себя как активированные
макрофаги. Макрофаги и эндотелий, а при
десквамации эндотелия на ранних или
(эндокринно-метаболические нарушения)
последующих стадиях атерогенеза — также
и активированные тромбоциты —
вырабатывают ряд медиаторов воспаления, среди
которых эндогенные окислители и факторы
роста, хемоаттрактанты и ингибиторы
миграции гладкомышечных клеток. Все это, в
совокупности, представляет по X. Стэри
и соавт. (1995) I тип изменений — стадию
начальных поражений.
Под влиянием эндогенных окислителей,
ЛПНП и ЛПОНП, проникновение
которых в интиму усилилось еще на стадии
дисфункции эндотелия, подвергаются
трансформации, в частности, окислению
и взаимодействию с продуктом активации
кровяных пластинок — малоновым ди-
альдегидом. Эти производные ЛП (а при
действии других факторов риска — также
гликированные и ацетилированные ЛП),
усиленно захватываются «мусорными»
рецепторами макрофагов и мигрирующих
из медии гладкомышечных клеток,
трансформирующихся под действием местных
цитокинов и ЛП в макрофагоподобные
миоинтимациты. Макрофаги при этом
приобретают пенистый вид.
ГМК тоже способны к
нерегулируемому захвату ЛП атерогенных классов, без
участия апо-В-чувствительных рецепторов
(Дж. Гольдстайн и соавт., 1976). Именно этот
вариант выступает на первый план при
дефектах специфической рецепторной системы
регуляции эндоцитоза ЛП, в частности:
- Отсутствии, дефиците, блокаде апо-В-
рецепторов, задержке интернализации
комплексов ЛП-рецептор.
- Резко повышенном притоке ЛП в
сосудистую стенку.
- Видоизменении ЛП частиц.
- Недостаточном выведении ЛП
материала из стенки сосуда.
Трансформацию гладких миоцитов
(ГМК) в перегруженные липидами клетки
удается получить in vitro, под действием
ЛП больных ИБС, но не ЛП здоровых
доноров (А. Фишер-Дзога и соавт., 1974).
Но in vivo, благодаря наличию цитокинов,
Нарушения липидного обмена 191
происходит классический массовый переход
ГМК в макрофагоподобные клетки, которые
насыщаются ЛП путем нерегулируемого эн-
доцитоза, исчерпывают ресурсы лизосомаль-
ной холестеринэстеразы и ацил-холестерин-
ацилтрансферазы ГЭР, нагружаются сначала
ЭХ (в которых сперва преобладает олеат), а
затем — и свободным ХН. Это приводит к
появлению пенистых клеток — характерных
элементов липидных пятен и полосок. Ли-
пидные пятна— это не возвышающиеся над
поверхностью эндотелия, желтоватые точки
диаметром до 1,5 мм, мягкой консистенции.
К началу второй декады жизни они могут
переходить в удлиненные образования
шириной 3 мм и длиной до 15 мм, иногда слегка
приподнятые над эндотелием. Это липид-
ные полоски. И те, и другие представляют
скопления пенистых клеток миоцитарного
и макрофагального происхождения, в
которых изобилуют внутриклеточные липиды,
богатые олеатом ХН. Рядом находятся
лимфоциты (как CD4+, так и CD8+). В данной
стадии не отмечается больших скоплений
внеклеточных липидов, хотя могут
появляться, вследствие гибели пенистых клеток,
их отдельные экстрацеллюлярные капли.
Миоинтимациты не синтезируют
значительных количеств компонентов межклеточного
вещества, но по периферии образований
пролиферируют.
Данная картина соответствует, по X. Стэ-
ри, II типу изменений, она сопровождается
ростом поражений за счет аккумуляции Л П,
и, даже при отсутствии дополнительных
факторов риска, достигает яркой
выраженности во второй-третьей декадах жизни.
Интегральные механизмы, приводящие к
формированию липидных полосок,
представлены на рис. 36., с. 178.
Эта стадия изменений встречается очень
часто в артериях молодых людей и даже
детей — причем и в тех регионах, где
осложнения атеросклероза являются редкостью.
Следовательно, лишь меньшинство этих
поражений переходит в более глубокие
стадии процесса и дает атеромы. Остальные
могут персистировать на этой стадии или
подвергаться обратному развитию.
В последнее время появились
наблюдения, реанимирующие цитированные выше
взгляды Р. Тома, согласно которым лишь
те липидные пятна и полоски переходят в
глубокие атеросклеротические поражения,
которые находятся над зонами
изначальной адаптивной мышечно-эластической
гиперплазии интимы. Подобные участки
Дж. Робертсон обнаружил еще в 1960 г.,
практически, у всех плодов старше 14-неде-
льного возраста, а А.С. Дауд (1964) показал,
что они возникают в участках наибольшей
биомеханической нагрузки и посчитал
их locus minoris resistentiae для будущего
прогрессирования атером. Очевидно, это
стигмы повышенной местной продукции
трансформирующих клетки интимы цито-
кинов.
Последующие стадии поражений
относятся уже к глубоким или поздним.
К ним принадлежат переходные поражения
(промежуточный тип III по X. Стэри или
липосклероз — по традиционной
терминологии Н.Н. Аничкова-В.Д. Цинзерлинга),
атеромы (атероматозный тип TVno X.
Стэри), фиброатеромы и фиброзные бляшки
(варианты фиброатероматозного типа V
по X. Стэри). Решающими отличиями
глубоких поражений являются:
>- Наличие значительных скоплений
внеклеточных липидов (начиная со стадии
III). Постепенно отложение экстрацел-
люлярных липидов увеличивается, и в
них повышается доля свободного ХН и
ЭХ близких к плазменным по составу
— линолеатов ХН. На стадии IV атерома
уже располагает липидным ядром.
>* Активная пролиферация и гибель
пенистых клеток, накопление межклеточного
вещества и формирование фиброзной
«крышечки» над ядром липидов
(начиная со стадии V). Ядра и крышки могут
быть в одной фиброатероме
множественными. В фиброатеромах, где
преобладают пенистые клетки
макрофагального генеза и макрофаги, внеклеточных
липидов много. Если пенистые клетки,
в основном, миоинтимацитарного
происхождения, фиброзные изменения могут
192 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
липидная полоска
моноцит
кровь
эндотелий
пенистые
клетки
цитокинъф тромбоциты v делящиеся
\$
лимфоцит г г -
^^^^^ факторы! г
^"\3 иМтокины роста jgu мигрирующие
медия
ГМК
Рис. 36. Механизмы формирования ранних атеросклеротических поражений в артерии
(по Д. Хайджару и А. Николсену, 1995). Обозначения и комментарии — в тексте
преобладать, и бляшка выглядит, как
фиброзная. Характерна также неоваску-
ляризация по периферии поражения и со
стороны медии, куда поздние поражения
могут «прорываться».
Все поздние стадии анатомически имеют
поверхность, выступающую в просвет
артерии. Они, в различной степени,
нарушают гемодинамику и снижают тром-
борезистентность сосуда, как вследствие
механических причин и экспрессии
молекул клеточной адгезии, так и в связи
с продукцией из липидов атеромы под
воздействием макрофагальной липоок-
сигеназы лейкотриенов, обладающих
вазоконстрикторной и тромбогенной
активностью. Это может
рекрутировать новые тромбоциты и лейкоциты,
добавляющие свои цитокины и иные
медиаторы, которые усиливают проли-
феративно-синтетический ответ клеток
интимы.
Трансформация ранних поражений в
поздние формы требует продолжения
инфильтрации JIU и может быть заторможена или
даже обращена вспять при нормализации
липидного обмена, но зависит в огромной
степени от спектра и количества
вырабатываемых в интиме цитокинов. Наибольший
процент трансформации ранних поражений
в глубокие характерен для коронарных
артерий и брюшной аорты. При наличии дисли-
попротеидемий подобный переход
ускоряется, и его масштабы расширяются. Самые
ранние сроки появления III типа — третья
декада жизни, IV тип формируется, в
быстро прогрессирующих случаях, начиная с
середины третьей декады; а к V типу первые
поражения подходят у лиц с выраженным
действием атерогенных факторов риска, уже
начиная с четвертого десятилетия жизни.
Цитокины усиливают миграцию мио-
цитов в интиму, их активацию, захват ЛП,
заставляют миоинтимациты пролифериро-
вать и синтезировать большие количества
гликозаминогликанов, гликопротеидов и
волокнистых белков (эластина и
коллагена). Особое значение в атерогенезе имеют
следующие регуляторы:
> ИЛ-1, ФНОа и моноцитарный хемотак-
тический белок I (МХБ-I) рекрутируют
в интиму из крови новые лейкоциты и
тромбоциты, усиливают адгезивность и
продукцию цитокинов эндотелием.
> Ряд цитокинов клеток интимы и крови
(среди них — лептин интерлейкин-1,
эндотелин-I, тромбин, ЛП (а), тромбо-
цитарные факторы роста гладких мышц,
основной фактор роста фибробластов,
трансформирующий фактор роста а
(ТФРа), фактор роста эпидермиса)
стимулируют пролиферацию и эндоцитоти-
ческую активность, а также биосинтез
белков и гликозаминогликанов
соединительной ткани в миоинтимацитах.
Тромбоцитарный фактор роста служит
мощным хемоаттрактантом миоцитов
медии, стимулируя их приток в интиму.
Данный цитокин выделяют не только
кровяные пластинки, но также
активированные эндотелиоциты и макрофаги.
Поэтому прогрессия липидных пятен
и полосок в атеромы и фиброатеромы
может проходить как с потерей эндоте-
лиального слоя и значительным тром-
бообразованием, так и под сохраненным
эндотелием, при минимальном участии
тромбоцитов (Р. Росс, 1986). Более того,
ген данного цитокина экспрессируется
Нарушения липидного обмена 193
в самих пенистых клетках миоинти-
мацитарного происхождения, поэтому
миоинтимациты, фактически, после
пенистой трансформации стимулируют
поддержание клеточного пула атером
аутокринным образом.
>• Экспрессия «мусорных» рецепторов на
поверхности клеток интимы
усиливается под действием отдельных цитокинов
(в частности, макрофагального колони-
естимулирующего фактора — М-КСФ),
а также самих видоизмененных ЛП.
>* Большое значение имеет снижение на
этой стадии процесса местной
продукции гепариноподобных ингибиторов
пролиферации, кейлонов и
трансформирующего фактора роста р (ТФРР).
Понижается продукция окиси азота, которая
в норме служит антипролиферативным
регулятором для ГМК артерий.
Детальная характеристика регуляторов
пролиферации, участвующих в
атерогенезе, дается в I томе данного руководства
(с. 332-337).
Реакция клеток интимы на перегрузку
патологическими ЛП может
расцениваться как адаптивная (А.Н. Климов, 1984;
А.В. Попов и соавт., 1978). Пенистые клетки
поглощают Л П в обход регулируемых путей,
связанных с физиологическим рецепторным
эндоцитозом через апо-В-чувствительные
рецепторы. Механизмы ауторегуляции,
описанные в разделе, посвященном
транспорту Л П, — понижение активности синтеза
собственного ХН, уменьшение экспрессии
апо-В-рецепторов, усиленная этерификация
и реэтерификация ХН, лизосомальный
гидролиз компонентов Л П частиц — не
включаются или исчерпывают свои возможности.
Дренажные механизмы сосудистой
стенки, связанные с ЛПВП, также используются
предельно, относительно их ограниченной у
лиц с предрасположенностью к
атеросклерозу мощности, или испытывают затруднения
в связи с тем, что часть ЛП иммобилизиру-
ется в составе нерастворимых комплексов в
сосудистой стенке.
194 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
В этих условиях клетки интимы
подвергаются опасности из-за избытка свободного
ХН в их мембранах. Часть из них некротизи-
руется, так как их мембраны понижают свою
жидкостность и проницаемость и меняют
фазовое состояние, а значит — страдают
метаболические возможности всей клетки
(Дж. Форрест и соавт., 1977). ХН из состава
некротизированых клеток входит в липид-
ное ядро атером в виде игольчатых
кристаллов, но может повторно захватываться
макрофагами.
Чтобы избежать критического повышения
холестеринизации мембран (при полном
исчерпании возможностей к образованию ЭХ),
миоинтимациты вступают в активную
пролиферацию, деля пул ХН между дочерними
элементами. Итак, в атеромах накопление ли-
пидов переходит в фазу изменения свойств
клеточных мембран. Поздние поражения
насыщены свободным ХН, олеатами ХН и
мембранным фосфолипидом — сфингоми-
елином (У.Портмен, В. Александер, 1972).
Сходным образом, как проявление
несовершенной адаптации, трактуется и
усиленный синтез пенистыми клетками
гликозаминогликанов и протеогликанов, а
также коллагена, эластина и гликопротеидов
межклеточного вещества.
Исследованиями С. Сринивазана (1977),
а также А.В. Попова, А.С. Кузнецова,
В.Т.Лозовского (1978) и других авторов
показано, что гликозаминогликаны (ГАГ)
связывают атерогенные ЛП в комплексы,
с участием кальция, магния и других
двухвалентных катионов. Наиболее активны
гепарин, гепарансульфат, хондроитинсуль-
фаты. Этот процесс происходит, главным
образом, у базальной мембраны и в подъ-
эндотелиальном слое, а ГМК населяют, в
основном, медию. Показано, что гепарин
может вытеснять ЛПНП из анионных
участков их фиксации на мембранах ГМК.
Все это, возможно, предохраняет ГМК от
притока новых порций ХН. Комплексы ЛП
и ГАГ могут ассоциироваться с эластином,
коллагеном и фибриногеном. Таким
образом, структуры фиброзной «крышки»
атером могут рассматриваться как аварийный
щит, с помощью которого интима пытается
предохранить клетки от нерегулируемого
захвата ХН-содержащего материала или
экранировать липидное ядро от действия
макрофагов. Адаптивная ценность фиброзных
изменений доказывается прижизненными
наблюдениями за состоянием пораженных
различными поздними атеросклеротичес-
кими изменениями коронарных артерий.
Эти данные получены методом коронарной
артериографии и допплеровской ультрасо-
нографии и обсуждаются У. Фъюстером
(1992). Доказано, что средние и малые по
размеру бляшки, богатые липидами,
вызывают спазм коронарных сосудов и приступы
ИБС намного чаще, чем большие фиброзные
бляшки, богатые ГАГ и эластином. Причина
этого кроется в том, что бляшка
провоцирует окклюзию сосуда не механически, в силу
своего размера и геометрии, а патохимичес-
ки - как источник лейкотриенов и тромбок-
санов. Эти липидные медиаторы
высвобождаются при контакте активных макрофагов
с липидами атером. Поэтому, нестабильная
стенокардия, инфаркт миокарда и внезапная
сердечная смерть чаще наблюдаются при
наличии бляшек, богатых липидами, или при
разрыве фиброзных капсул фиброатером.
Фиброзные бляшки чаще дают стабильные
формы стенокардии.
Еще Д. Холландер (1976) показал, что
после введения обезьяне меченых ЛПНП
большая часть недеградировавших частиц
в течение часа соединяется в стенке
артерий с коллагеном (20%), эластином (15%),
сульфатированными ГАГ (10 %), и высказал
предположение, что последствия такой
фиксации зависят от свойств комплексов и
могут быть неоднозначными. Большую роль
здесь могут играть свойства ЛП части
комплексов. Так, только при взаимодействии
липопротеидов больных ИБС людей, но
не липопротеидов плазмы крови здоровых
лиц, с компонентами сосудистой стенки
венесуэльским исследователям удалось
получить стойкие комплексы (А. Камехо
и соавт., 1976).
Как и всякая аварийная адаптация, про-
лиферативно-фиброзные механизмы не
Нарушения липидного обмена 195
свободны от побочных эффектов,
усугубляющих некоторые аспекты поражений.
ЛП иммобилизируются и задерживаются
в очаге поражения, вне доступности для
дренажных механизмов. Накопление
межклеточного вещества соединительной
ткани благоприятствует образованию
фиброзных бляшек. По данным А. Робера
и соавт. (1969), ЛП и свободные жирные
кислоты, связанные с эластином,
способствуют разволокнению эластических
волокон, оказывают мылоподобный эффект,
облегчают разрывы фиброзной крышки.
Детергентное действие липидов на эластин
и комплексообразование ГАГ и ЛП
приводят к атерокальцинозу.
Комплексообразование ГАГ и ЛП, в конечном итоге,
ослабляет антигемостатическое действие ГАГ
и снижает тромборезистентность сосуда.
Доказано, что при наличии атером любого
вида артерии менее способны к генерации
окиси азота и вазодилятаторным ответам,
но более склонны к вазоконстрикции. В
общем, считается, что
комплексообразование ЛП с хондроитинсульфатами,
особенно хондроитинсульфатом В, приводит
к формированию крупномолекулярных,
связанных со склеропротеинами
комплексов, трудно выводимых из сосуда. В
то же время, комплексообразование ЛП с
гепарином и гиалуроновой кислотой дает
комплексы с более благоприятными для
адаптации сосудистой стенки свойствами
(С. Леви и соавт., 1975).
Все вышеописанные фибропластические
реактивные процессы изменяют атеромы и
фиброатеромы и приводят к появлению
осложненных поражений (тип VI по X. Стэри^,
к которым относятся кальцификация (атеро-
кальциноз), расщепление или изъязвление
бляшки (приводящее к липидной эмболии,
наслаивающемуся тромбозу,
тромбоэмболии и окклюзии сосудов), разрыв сосудов,
новообразованных по периферии атеромы
(что ведет к интрамуральному тромбозу, а
также кровоизлияниям). Эволюция бляшки
в крупнейших артериях может закончиться
прорывом в медию и формированием ате-
росклеротической аневризмы (чаще всего,
дистальнои части аорты, ниже места отхож-
дения почечных артерий).
События атерогенеза по-своему
классифицировали и именовали многие авторы.
Дополняя классификацию стадий процесса
по Н.Н. Аничкову — В.Д. Цинзерлингу и
по X. Стэри, которые рассмотрены выше,
в заключение данного раздела, с целью
напоминания общей динамики развития
атеросклеротических поражений, приводим
и краткую характеристику трёх этапов
развития атеросклероза по У. Фыостеру (1992):
I — стадия эндотелиального повреждения.
Характеризуется утолщением и
альтерацией эндотелия, активацией и прилипанием
макрофагов, усиленной липопротеидной
инфильтрацией интимы.
II — стадия поражения интимы.
Характеризуется активацией тромбоцитов,
присутствием ряда цитокинов, миграцией из медии
ГМК, пенистой трансформацией интимаци-
тов, их пролиферацией и синтезом элементов
межклеточного вещества соединительной
ткани, формируется фиброзная капсула.
III — стадия интимо-медийного
поражения. Характеризуется появлением
разрывов капсулы, геморрагии, активным
неоангиогенезом. Формируются кальциноз,
интрамуральный и пристеночный тромбоз, в
крупнейших сосудах — аневризмы.
Итак, в современном понимании,
атеросклероз представляет собой процесс, который
начинается с изменений эндотелия крупных
артерий. Эти изменения вызываются как
патологическими Л П, так и иными факторами.
Они приводят к миграции в интиму из крови
и медии клеток, которые, под влиянием ЛП
и местных цитокинов, претерпевают
пенистую трансформацию, пролиферируют, ведут
себя как активные макрофаги и синтезируют
компоненты межклеточного вещества. Это
приводит к появлению и эволюции атером,
изменяющих тромборезистентность сосуда
и его ответ на регуляторы кровотока. В
конечном результате развиваются ишеми-
ческие осложнения.
Характеристика механизмов ИБС и
других осложнений атеросклероза лежит за
196 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
пределами тематики данной книги и входит
в т. III данной серии и Практикум.
Справедливости ради, надо отметить что
имеются альтернативные теории
атерогенеза, возникшие в стороне от материнской
инфильтративно-комбинационной
концепции и не интегрированные в теорию
ответа на повреждение. Мы не относим к
ним тромбогенную теорию К. Рокитанс-
кого-Ж.Б. Дьюгида, поскольку она вошла
в ныне существующие синтетические
представления и оказалась вполне согласуемой
с липидными теориями.
Это, прежде всего, концепции,
основанные на идее о первичности патологической
пролиферации гладкомышечных клеток при
атеросклерозе.
Моноклональная гипотеза принадлежит
американским авторам (Э.П. Бендитт
и Дж.М. Бендитт; 1973, 1988).
Согласно этим представлениям, атеросклероз
— клональный неопластический процесс,
который возникает в результате
соматической мутации (мутаций) в
артериальных гладкомышечных клетках, по типу
доброкачественных лейомиом. Расселение
мутантных клеток с током крови ведет к
распространению атеросклероза, по
аналогии с метастазированием. В поддержку
этой гипотезы говорят факты,
свидетельствующие, что во многих (но не во всех!)
атеромах пенистые клетки — потомство
одной клетки-предшественницы, то есть
они моноклональны или олигоклональны.
Мутагенами, по мнению основателей
теории, могут быть окисленные производные
ХН, экзогенные гетероциклические
соединения, особенно, растворимые в липидах, а
также вирусы.
Атеромы у экспериментальных
животных в аорте получены под воздействием
мутагена бензантрацена. Онкогенный вирус
куриной болезни Марека, ответственный
за возникновение у птиц лимфом и
поражений нервной системы, способствует в
аорте атеросклеротическим поражениям
(М. Майески, 1985). Тромбин индуцирует
в ГМК экспрессию онкогена c-fos. Впрочем,
чдокринно-метаболические нарушения)
все эти факты совместимы и с теорией
ответа на повреждение.
Из пенистых клеток человеческих атером
выделялась м-РНК онкогенного вируса
герпеса.
Здесь уместно охарактеризовать
инфекционные теории атерогенеза. К.Г. Фабрикант
интерпретировал некоторые атеросклероти-
ческие поражения у животных как результат
герпетической инфекции (1985). Помимо
подозрений о роли вирусной инфекции в
генезе атеросклероза, имеются
эпидемиологические и иммунологические данные,
позволяющие предполагать, что известную
роль в провоцировании атерогенеза могут
играть некоторые формы хламидиоза и их
поздние иммунопатологические
осложнения, поражающие эндотелий.
Некоторые инфекционно-иммунопато-
логические васкулиты, поражающие
эндотелий, способствуют атеросклерозу (см.
болезнь Кавасаки, т. I данного руководства,
стр. 252,307).
К моноклональной гипотезе близка
теория очагового клонального старения как
причины атерогенеза. Известно, что еще
И.В.Давыдовский (1969) отождествлял
некоторые проявления атеросклероза и
возрастные изменения сосудов. Теория
клонального старения постулирует, что из-за
старения и гибели клеток медии, в которых
накапливаются соматические мутации,
прекращается или ослабевает выработка этим
слоем артерий локальных антипролифера-
тивных сигналов, сдерживающих
размножение интимацитов. Это и служит толчком к
началу атерогенеза (Э. Бирман, 1994).
Г. Селье (1960) и Дж. Стэмлер (1961)
считали атеросклероз болезнью адаптации
(социальной эволюции) и придавали
решающую роль в его развитии хроническому
стрессу, в частности, вызванному факторами
цивилизации. Выше уже было показано, что
стрессорная концепция атерогенеза вполне
интегрируется в липидную теорию, а тем
более — в концепцию ответа на повреждение.
К. де Дюв (1974) был автором лизосо-
мальной теории атеросклероза, отводившей
ему место среди других лизосомальных бо-
Нарушения липидного обмена 197
лезней накопления. Наблюдения за
больными первичным ксантоматозом Вольмана не
вполне подтверждают эту гипотезу. К тому
же, накопления липидов в пенистых клетках
оказались, по большей части, внелизосо-
мальными, цитоплазматическими.
Атеросклероз — полиэтиологическое
заболевание. Однако, материалы данного
раздела показывают, сколь велика и в его
этиологии, и патогенезе роль нарушений
липопротеидного метаболизма.
НАРУШЕНИЯ ПРОЦЕССОВ
НАКОПЛЕНИЯ
И МОБИЛИЗАЦИИ ЛИПИДОВ
«Жировая клетка -
Страшная кокетка.
В мыслях о приданом
Красится Суданом.
В профиль перстневидна —
А внутри липидна.
Вот ведь что обидно!»
А.Г. Кнорре «Гистоазбука»
Мы уже отмечали громадное значение
липидов, как долговременного депо энергии.
Втриглицеридах тела здорового
человека весом 70 кг одновременно заключено
астрономическое количество энергии в
140000000 калорий (140000 ккал). Это в
100 раз больше, чем может дать весь
гликоген организма.
Для того, чтобы адекватно
распоряжаться жировыми запасами и поддерживать
постоянство массы тела, жировая ткань и
гипоталамус обмениваются сложными
гормональными сигналами, от которых зависят
аппетит, усвоение пищи, расход энергии и
вес. Жировая ткань, составляющая в норме
15-20 % от массы тела у мужчин и 20-25 %
у женщин, это не инертный складской
отсек организма, а метаболически активное
образование, где постоянно протекают
балансовые процессы липогенеза и липолиза,
контролируемые нейроэндокринной
системой. Правильное взаимодействие жировой
ткани и гипоталамуса обеспечивает эффект
массостата (липостата), то есть гомеостаз
массы тела (Ю.И. Строев, Л.П. Чурилов,
2007).
При недостаточном, по отношению к
энергетическим потребностям, поступлении
калорий с пищей происходит дополнительная
утилизация жировых запасов и истощение.
Истощение и некоторые его специфические
формы, в частности, нейрогенная анорексия,
рассматривались выше в разделе
«Патофизиология голодания». В данной части книги
нет необходимости повторяться.
Хроническое превышение калорического
содержания потребляемой еды над
энергозатратами организма ведет к накоплению
дополнительных триглицеридов в жировой
ткани и, в конце концов, имеет своим
результатом ожирение. Теоретически, достаточно
превышения на 100 ккал в день (эквивалент
трех грецких орехов), чтобы к концу года
прибавить 4,5 кг.
Определение, классификация
и эпидемиология ожирения
Ожирение — не особенность конституции
организма. Оно не должно восприниматься
профессионалом-медиком сквозь призму
предрассудков бытового мышления, как
предосудительный результат дурных привычек
или, наоборот, как общий признак здоровья
и плещущего через край благополучия.
Ожирение — это патологический
избыток триглицеридов в организме. Первичное
ожирение представляет собой
полиэтиологическую болезнь, зависящую от нарушения
адипоцитарно-гипоталамических
информационных взаимодействий, при котором
установочная точка массостата (липостата)
смещается вверх. Прежде первичное
ожирение характеризовали как алиментарно-кон-
ституционально-гиподинамическое (М. Га-
нефельд, 1979). В классической немецкой
литературе имеется для него даже термин
«ленивая полнота» — Faulfettsucht). Авторы
198 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
не считают такое определение
недопустимым. Однако, оно не должно создавать
ложно-упрощенного впечатления, что первичное
ожирение — это всего лишь, по шутливому
определению Р. Котрана, «синдром
двойного невставания» то есть отказа больного
вовремя встать из-за стола и покинуть кресло
с целью активного отдыха.
Дело в том, что превышение потребления
энергии над тратами — условие
первичного ожирения. Но его ключевой механизм
состоит в конкретных нарушениях
гормональной связи между жировой тканью и
гипоталамусом, раскрытых в середине 90-х
годов XX века. Именно из-за этих
нарушений меняются пищевое поведение
больного, его психология и тенденция к выбору
определенного образа жизни. Чтобы
правильно воспринять этиологию первичного
ожирения надо не забывать, что психика и
поведение не трансцендентны и не
вкладываются в человека в виде готовой программы
воспитателями, хотя именно такие взгляды
распространены вне профессиональной
медицины. Психика базируется на «соматике»,
а поведение детерминировано генами через
нейроэндокринный аппарат и метаболизм.
Следовательно, первичное ожирение
правильнее трактовать как самостоятельную
нейроэндокринную болезнь, причины
которой заключены в специфическом
нарушении оси «гипоталамус-адипоциты».
В настоящее время получены также данные
о том, что первичное ожирение гетерогенно
и имеет, по крайней мере, 2 типа.
Главная отличительная черта первичного
ожирения — относительная или абсолютная
лептиновая недостаточность (см. ниже).
Эволюция наших представлений о
нозологической специфике ожирения очень
напоминает динамику взглядов в отношении
сахарного диабета — и в дальнейшем мы
увидим, что это не случайно.
Вторичное ожирение — синдром,
возникающий при наличии в организме каких-либо
расстройств, усиливающих запасание и
ослабляющих темпы расходования тригли-
церидов, на фоне изначально нормальных
сигнальных взаимоотношений адипоцитов
и гипоталамуса. Большинство форм
вторичного ожирения носит симптоматический
характер и порождается различными эн-
докринопатиями. Понятия «диэнцефальное
ожирение» и «гипоталамическое ожирение»
нельзя смешивать с первичным ожирением.
Они традиционно применяются к формам
вторичного ожирения. После раскрытия
гипоталамических аспектов патогенеза
первичного ожирения можно сказать, что мы
имеем дело с характерной ситуацией, когда
традиционный язык медицины отстает при
описании новых понятий. De facto любые
формы ожирения протекают с нарушением
гипоталамических функций (С.А. Бугрова,
1985) — см. подробнее раздел «Нарушения
эндокринной регуляции».
Критерии ожирения трудно поддаются
унификации: иронизируя по этому поводу,
Р.Котран и соавт. (1994) замечают, что
«диагноз ожирения доктор ставит пациенту,
столь же толстому как он сам, если только
пациент ему не нравится». Если прежде при
антропометрии ориентировались на весо-
ростовое соотношение и формулу
идеального веса Р. Брока (рост минус 100), а также
на измерение подкожно-жировых складок и
различных окружностей (например, талии
и бёдер), то ныне пользуются таблицами
М.Мёра и Г.Кёлера (1969), вводящими
поправку на телосложение, а наиболее
ценным индексом считают индекс массы тела
(отношение его веса в килограммах к
квадрату роста в м2). По рекомендации
немецкого специалиста Й. Майера(1958),
принято считать, что патологическим является
избыток массы тела над идеальной в 20%
и более — для мужчин и в 25 % и более - у
женщин. Именно при таком избытке
реальной массы тела над массой,
соответствующей 85-му перцентилю популяции,
происходит существенное увеличение пораженности
рядом болезней и смертности. Если масса
тела больше должной на 50 % для мужчин
и 70 % для женщин, это считается тяжелым
ожирением. При нормальном индексе массы
тела около 25 кг/м2 верхняя граница зоны,
где нет выраженного изменения смертности,
соответствует примерно 27 кг/м2.
Нарушения липидного обмена 199
По таким критериям получается,' что
ожирение распространено эпидемически
широко. В среднем, в экономически
развитых странах, по статистике ВОЗ, около 30 %
взрослых людей имеют ту или иную форму
и степень ожирения (А.Ю. Барановский,
Н.В. Ворохобина, 2007). В СССР в начале
80-х годов таких пациентов было до 26 %. Во
многих европейских государствах, особенно,
в Германии и Чехии, ситуация с ожирением
еще сложнее — по данным С. Ошанцовой и
К. Гейды (1972), масса тела 30 % мужчин и
50 % женщин была патологически
избыточна. Для США в конце 90-х годов В. Кумар
и соавт. (1997) приводят уровень поражен-
ности ожирением до 30 % среди мужского,
и до 40 % — женского населения.
Количество случаев ожирения особенно велико в
возрастных группах после 50 лет. Среди
детей ожирение и его младенческая
предшественница -- паратрофия — тоже очень
распространены (до 10%). В некоторых
регионах Швеции до половины подростков
имеют избыточный вес (А.-М. Док, 2003).
Патофизиология и клиника ювенильного
ожирения подробно рассматриваются в
недавно вышедшей монографии «Ожирение у
подростков» (Ю.И. Строев и соавт., 2006).
Из-за широкой распространенности
и большой социально-психологической
значимости ожирение всегда было
актуально для врачей. Гиппократ посвятил
этой болезни специальный раздел «Как
похудеть» в своем «Трактате о здоровом
образе жизни». Ожирение как проблему
баланса, требующую комплексного подхода
в лечении, трактовали классики медицины
Запада (Клавдий Гален, Целий Аурелиан)
и Востока (Ибн Сина).
Тучность как болезнь не сводится к
причинению косметического, психологического
и физического дискомфорта. Она, как уже
отмечалось выше, сокращает
продолжительность жизни, увеличивая смертность,
которая среди лиц с повышенным индексом
массы тела, в среднем, возрастает на 50%.
Подсчитано, что минимальная смертность
наблюдается при значениях индекса
массы тела от 23 до 25 кг/м2. В то же время, в
диапазонах ниже 20 и выше 30 она быстро
растет. Так например, у мужчин первые
10% избыточного веса оборачиваются 14%
увеличением смертности, при избытке
веса 20 % смертность растет на 25 %, а при
30%-на42%.
Ожирение — признанный фактор
риска для атеросклероза и его осложнений,
гипертензии, варикозной болезни и
тромбофлебита, холелитиаза, артритов,
остеохондроза, плоскостопия, подагры, пиквик-
ского синдрома (приступы гиповентиляции
и сонливости, вплоть до апноэ во сне),
стеатоза печени и многих других болезней.
При ожирении имеется относительный
иммунодефицит, преимущественно,
связанный с нарушением Т-клеточных функций и
фагоцитоза, повышена частота грибковых
и стрептококковых кожных заболеваний, а
также гирсутизма. Показано, что при
ожирении у женщин учащаются рак эндометрия,
яичника, шейки матки, жёлчного пузыря и,
в небольшой степени, в популяциях с
низкой исходной пораженностью — также рак
грудной железы. У мужчин сходная
закономерность прослежена для рака простаты
и прямой кишки. Особенно тесно связано
ожирение с инсулиннезависимым сахарным
диабетом, для которого оно является одним
из непременных патогенетических звеньев и
участвует в развитии характерной для этого
недуга инсулинорезистентности (см. ниже
«Патофизиология углеводного обмена»).
На рис. 37 показаны патогенетические
взаимосвязи между ожирением и другими, часто
сопутствующими ему болезнями. Однако,
разные формы ожирения далеко не
одинаковы по степени и спектру своей пагубности
для здоровья.
Классификация ожирения, помимо
деления на первичную и вторичную форму,
предусматривает разграничение
гипертрофической и гиперпластической разновидностей,
а также андроидного (яблочного), гипоидного
(грушевидного) и смешанного типов.
При гипертрофическом типе количество
жировых клеток остается нормальным, а
накопление жира идет путем увеличения в
200 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
инфаркт]
миокарда\
Рис 37. Ожирение как фактор риска различных
заболеваний. Сокращения — как в тексте
каждой из них размера жировой капли.
Нормальный ее объём составляет около 0,3 мкл.
При гипертрофии он может возрастать
максимально до 1 мкл. Соответственно,
гипертрофические формы ожирения, которые
в целом имеют более позднее начало, при
максимальной тяжести сопровождаются
избытком веса в 3-3,3 раза и отличаются
большим средним диаметром адипоцитов
(Й.Д. Фаульхаберисоавт., 1969).
При гиперпластическом варианте
ожирения адипоциты не достигают предельной
величины, но их количество больше
нормы. В этих условиях избыток массы может
достичь гигантских величин (до 1000%).
В 1978 г. в одной из клиник США скончался
от дыхательной недостаточности больной
Дж.М. весом 635 кг при росте 185 см.
Вариант ожирения легко устанавливается по
изучении биоптата жировой ткани (В. Ле-
онгарт, 1973). Характерно, что
гиперпластическое ожирение начинается намного
раньше, чем гипертрофическое. Во взрослом
организме дифференцировка фибробла-
стических
клеток-предшественников в новые
адипоциты представляет
собой редкое исключение.
Но, до рождения и в
раннем грудном периоде она
вполне возможна (см.
следующий раздел). Поэтому
считается, что в развитии
гиперпластического
ожирения огромное значение
имеет наследственность,
определяющая пролиферативные
возможности этих клеток. Справедливости
ради, надо отметить, что и в
последующем некоторые периоды
(подростковый, преклимактерический)
характеризуются оживлением пролиферативной
активности преадипоцитов. Более того,
очень большой избыток калорий может
индуцировать их пролиферацию.
Поэтому гиперпластические проявления
бывают и при позднем ожирении у
. взрослых. Ряд факторов
перинатального и раннего грудного периода способен
обеспечить своего рода импринтинг в
отношении функционирования липоста-
та, описанного ниже, и морфологических
параметров жировой ткани. Увеличению
количества адипоцитов могут содействовать
сахарный диабет у беременной женщины
(и диабетическая фетопатия), переедание
у беременных, паратрофия грудных
детей и даже переедание в подростковом и
преклимактерическом периодах. Все это
эпидемиологически связано с
гиперпластическими и смешанными формами ожирения,
имеющими менее благоприятный прогноз.
Вот почему правильную диету беременных
и грудное вскармливание надо считать
мерами профилактики ожирения (Я. Татонь,
1981).
Андроидный, или яблочный, тип
ожирения характеризуется отложением жира
на животе и верхней части туловища, а
также в абдоминальных внутренностях.
При гиноидном или грушевидном типе
жир откладывается на бедрах, ягодицах и в
нижней части туловища. Первый тип более
Нарушения липидного обмена 201
характерен для мужчин, второй — для
женщин. Это зависит от различного влияния
мужских и женских половых гормонов на
распределение а2-катехоламиновых
рецепторов в разных отсеках жировой ткани. При
андроидном ожирении, которое у женщин
наблюдается при относительно высокой
продукции андрогенов, более характерны
атеросклероз, стеатоз печени, инсулиноре-
зистентность. Важно отметить, что жировая
ткань различных отсеков и сама отличается
по своей гормональной функции. Так как
при гиноидном ожирении продукция
эстрогенов в адипоцитах более значительна, то
сказывается антиатерогенное действие этих
гормонов, и поэтому гиноидное ожирение не
приводит к столь выраженному повышению
риска атеросклероза и его осложнений, как
это делает андроидное. Смешанное
ожирение комбинирует признаки андроидного и
ганоидного, но характеризуется высокой
патогенностью, как и андроидное.
Выделяют также тучность с преимущественным
накоплением висцерального жира (в
сальниках, брыжейке, ретроперитонеальном
пространстве) и подкожного жира (в
гиподерме). Висцеральное ожирение сочетается
с андроидным и сопряжено с большим
риском развития вторичных расстройств
функций внутренних органов. Дело в том,
что висцеральные адипоциты более
чувствительны к катехоламинам и выделяют
больше контринсулярного фактора кахекси-
на. Именно они вносят наибольший вклад в
развитие инсулинорезистентности у тучных
пациентов. Мало того, продукты липолиза
и собственной эндокринной деятельности
висцеральных адипоцитов
транспортируются по портальной вене прямо в печень,
сильно влияя на метаболизм гепатоцитов, а
периферические адипоциты посылают свои
продукты и сигналы в системный кровоток.
Впрочем, так как гиноидное ожирение
нередко носит частично гиперпластический
характер, оно более резистентно к
диетотерапии. Андроидное и гиноидное ожирение
дифференцируют на основе измерения
соотношения окружностей талии и бедер,
которое в норме составляет 0,7-0,8.
Итак, более патогенными считаются
гиперпластическое, андроидное, висцеральное
ожирение, а более благоприятными,
соответственно, гипертрофическое, гиноидное,
субкутанное.
Характерным примером малопатогенного
ожирения служит тучность борцов-сумои-
стов. Несмотря на избыток веса, мастера
сумо долго сохраняют относительно
хорошее здоровье. Самый «весомый» борец в
истории японского сумо — Конисики —
весит 248 кг.
По мнению В.Р. Лингаппа (1995), здесь
сказывается тот факт, что при занятиях
классическим сумо атлет не испытывает
гиподинамии, а прирост веса связан
исключительно со специальной традиционной
диетой, обогащенной, между прочим, по-
лиеновыми и ©-ненасыщенными жирными
кислотами. В отличие от гиперкалориче-
ски-гипердинамического состояния борцов
сумо, гиподинамические формы ожирения,
которые мы наблюдаем, например, у
заядлых телезрителей-любителей сладкого,
ведут к накоплению, преимущественно,
висцерального жира.
По предложению В. Риса (1970)
используют и внешнюю зональную
типологию (фенотииическая классификация).
Однако, поскольку нет полного
параллелизма между фенотипами и этиологией,
определения, звучащие как «тип рейтуз»,
«тип блузы», «стволовой тип» —
закрепились только в профессиональном жаргоне
эндокринологов и диетологов. Тем не
менее, сама по себе разная локализация
жировых отложений при различных формах
первичного и, особенно, вторичного
ожирения до сих пор остается далёкой от
исчерпывающего обьяснения. Считается, что
большое значение имеет пермиссивный
эффект разных гормонов на экспрессию
определяющих интенсивность отложения
жира адренорецепторов в тех или иных
отсеках жировой ткани. Принимая эту точку
зрения^ приходится признать, что
адипоциты разной локализации гетерогенны.
Возможно, их гетерогенность связана не
только с разным ответом на гормональные
202 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
сигналы, но и с клональными различиями
в их собственной эндокринной функции.
Тогда возникает вопрос о возможном
адаптивном смысле того или иного
паттерна отложения жира при различных
эндокринопатиях. Не вырабатывают ли,
например, адипоциты шеи, лица и верхней
части туловища аутакоиды, выполняющие
компенсаторную роль в отношении гипер-
кортицизма? Тогда становится
оправданным именно знаменитый «кушингоидный»
тип зонального отложения жира при этом
синдроме.
В любом случае, ясно, что зональные
особенности ожирения могут представлять
большой теоретический интерес. Очевидно,
что локализация жира не менее, а, скорее
всего, более важна, чем абсолютное
количество накопленных триглицеридов.
ЛИПОСТАТ
И ПАТОФИЗИОЛОГИЯ
ПЕРВИЧНОГО ОЖИРЕНИЯ
Липостат (массостат) — условное название
системы, контролирующей постоянство веса
тела. Как и для других балансовых констант
организма — температуры, осмолярности
тканевой жидкости, кровяного давления
центральным контролирующим звеном в
системе, регулирующей массу тела, служит
гипоталамус. Липостатический гомеостаз
обеспечивается путем прямых и обратных
сигнальных взаимодействий между
гипоталамусом и жировой тканью (с её
гормонами), а также гипоталамусом и ЖКТ (с его
энтериновой гормональной системой).
Гипоталамус как отдел промежуточного
мозга, занятый поддержанием балансовых
констант организма, уже был описан выше
(см. том I, стр. 531-536, а также краткое
описание механизмов аппетита в данной
книге, раздел «Нейрогенная анорексия и
булимия»). В данном разделе необходима
детализированная характеристика пищевых
центров подбугорья, порождающих такие
аффективные проявления деятельности
липостата, как чувство голода, аппетит1 и
чувство насыщения.
Идею о существовании «пищевого
центра», как автоматического регулятора
потребления пищи, впервые высказал И.П. Павлов
(1911). Он считал, что в коре больших
полушарий и нижележащих отделах ЦНС
имеется функциональное объединение
клеток, чувствительных к степени
наполнения желудка и химическим сигналам, и
обеспечивающих контроль поступления
пищи. Ранние представления об обратной
афферентации в системе контроля
пищевого поведения связывали сигнал сытости со
степенью растяжения желудка и обратной
афферентацией от механических процессов,
сопутствующих обработке пищи во рту и ее
пассажу через глотку и пищевод
{механическая теория регуляции аппетита).
Установлено, что эти факторы действительно
вносят вклад в краткосрочные механизмы
регуляции потребления пищи (А. Гайтон,
1989).
Впоследствии сложились различные
теории аппетита, выдвигавшие на первый план
роль того или иного тормозного
химического сигнала, связанного с потреблением пищи
и ингибирующего активность «пищевого
центра» (см. также выше раздел
«Нейрогенная анорексия и булимия»).
>- По аминоацидостатической теории,
торможение аппетита и пищевого поведения
зависит от аминокислотного сигнала
сытости. Действительно, многие
аминокислоты и их амиды (глутамин, глицин,
1 Аппетит — положительно эмоционально
окрашенное стремление к приему пищи в отличие
от чувства голода — отрицательно
эмоционально окрашенного влечения к приему пищи.
В формировании эмоционально-поведенческой
стороны аппетита, голода и насыщения, кроме
гипоталамуса, принимают участие
амигдалоидные структуры, перегородка, гиппокамп, поясная
извилина, орбитальная кора. Но гипоталамусу,
по выражению A.M. Уголева, «при всех
обстоятельствах, принадлежит роль основного звена,
участвующего в формировании пищевой
мотивации».
аланин) понижают аппетит и могут
служить медиаторами или модуляторами в
нервной передаче.
> По дегидратационной теории насыщение
зависит от возникновения гемоконцент-
рации. По мнению A.M. Уголева (1991),
хотя переход жидкости из крови и тканей
в ЖКТ при интенсивном пищеварении
имеет место, трудно объяснить с позиций
данной концепции, почему гемодилюция
при питье воды не вызывает чувства
голода.
> Термостатическая теория Дж.Л. Стро-
минджера-Дж.Р. Бробека (1953)
постулировала, что животные едят, дабы не
остывать, а прекращают еду, чтобы не
перегреться. По этой концепции, эффект
насыщения тем выше, чем выше
специфическое динамическое действие пищи
и температура тела.
> Согласно метаболической теории
аппетита А.М. Уголева-В.Н. Черниговско-
го-В.Г. Кассиля (1962), регулирующий
сигнал должен быть метаболически
универсален и генерироваться при
питании любыми видами пищи. На роль
таких сигналов могут претендовать
кетокислоты цикла Кребса, поскольку
последний является общим конечным
звеном катаболизма всех пищевых
веществ. В подтверждение известной
роли, принадлежащей этому механизму
в происхождении чувства сытости, было
показано, что цитрат натрия вызывает
у голодающих крыс наиболее раннее и
длительное торможение аппетита.
Полвека спустя после публикации
основополагающей статьи И.П. Павлова «О
пищевом центре», индийский патофизиолог
Б.К. Ананд (1961) установил локализацию
центра голода (аппетита) и центра
насыщения, соответственно, в вентро-латеральных
и вентро-медиальных ядрах подбугорья.
Вентро-медиальный центр сытости связан
с центром аппетита синапсами,
передающими тормозные импульсы. Звенья,
определяющие эмоционально-поведенческие
корреляты приема пищи, располагаются в
Нарушения липидного обмена 203
кортикальной части лимбической системы
(поясная извилина, гиппокамп, инфраор-
битальная область), а также в миндалине,
разрушение которой вызывает психическое
безразличие к виду и характеру
предлагаемой пищи («пищевая слепота»).
Благодаря этим открытиям, было
создано несколько экспериментальных моделей
ожирения и кахексии:
- при электролитическом разрушении у
грызунов вентро-латеральных ядер
гипоталамуса развивается стойкая анорексия,
животное впадает в кахексию и умирает
от голодания (экспериментальная
кахексия по Б. Ананду);
- электролитическое разрушение вентро-
медиальных ядер вызывает
пролонгированное пищевое возбуждение,
агрессивное поведение и булимию. Гиперфагия
приводит к развитию ожирения, вплоть
до 10-кратного избытка веса. После этого
животное становится менее активным
(модель гипоталамического ожирения, по
Б. Ананду, воспроизводится у крыс,
мышей, кошек и приматов). У подопытных
животных не только имеется полифагия,
но и усиливается преобразование
углеводов в жиры, снижается уровень
специфического динамического действия пищи
(СМ. Лейтес и соавт., 1966);
- модель ожирения у мышей,
индуцированного ауротиоглюкозой (CeHnAuSOs) —
по Р.Х. Драхмену — Дж. Теппермену
(1954). Однократная внутрибрюшинная
инъекция ауротиоглюкозы голодающим
мышам в дозе 1 мг/г массы тела ведет у
половиы подопытных животных к гипер-
фагии, усилению трансформации
углеводов в жиры, понижению термогенеза,
вызванного приемом пищи, и ожирению.
При этом в гипоталамусе наблюдают
дегенерацию нейронов вентро-медиальных
ядер. Повреждения возникают также в
гиппокампе и других отделах ЦНС. На
крысах модель не воспроизводится, так
как эти животные гибнут от
ауротиоглюкозы при достаточно невысоких дозах.
Зато имеются клинические наблюдения
за пациентами, у которых гиперфагия и
204 А.Ш. Зайчик, Л.П.Чурилов Патохимия
ожирение развились после лечения по
поводу ревматоидного артрита
препаратами коллоидного золота.
Наблюдения за результатами этих
попыток моделировать расстройства липостати-
ческих функций привели к предположению,
что вентро-медиальные ядра располагают
глюкозным рецептором, стимуляция
которого может индуцировать чувство насыщения,
а ауротиоглюкоза связывается с этим
рецептором и нарушает его работу. Сложилась
глюкостатическая теория краткосрочной
регуляции потребления пищи, аргументом
в пользу которой явилась невозможность
вызвать экспериментальное ожирение ауро-
тиосорбитолом, ауротиоглицерином и ауро-
тиокапроновой кислотой. Глюкостатическая
теория долго доминировала в этой области,
тем более, что гормональные сдвиги,
приводящие к вторичному ожирению (гипотироз,
гиперкортицизм, ИНСДII типа, острое
повреждение гипоталамуса), характеризуются
также и гипергликемией. При хронических
поражениях гипоталамуса отмечается,
напротив, гипогликемия. Было показано, что
не столько абсолютный уровень глюкозы,
сколько артерио-венозная разница по
глюкозе возбуждает глюкостатические гипота-
ламические рецепторы. При насыщении она
резко положительна, а при голодании падает
до отрицательных величин.
Критически относясь к вышеназванным
теориям контроля потребления пищи,
А.М. Уголев и В.Г. Кассиль (1961) писали,
что «...в процессе эволюции аппетит
формируется не как реакция на уже возникшее
истощение пищевых ресурсов, но как
механизм, задолго предупреждающий такое
истощение. Теории, связывающие голод и
аппетит с исчерпанием запасов, несмотря
на подкупающую простоту, должны быть
отвергнуты. Аппетит не следует за
исчерпанием депо пищевых веществ, но предваряет
и не допускает его... Многочисленные
раздражители, формирующие состояние голода
и аппетита имеют до известной степени
сигнальный характер. Это обусловливает
пластичность и нестандартность управления
процессами питания».
чдокринно-метаболические нарушения)
При такой постановке вопроса, выглядит
естественным предположение, что аппетит
должен появляться в момент перехода от
питания экзогенными нутриентами к эндо-
трофии, то есть утилизации жира из депо.
Это ставит в центр липостата активную
сигнальную роль гормонов. В дальнейшем
на этой основе возникла липостатическая
теория аппетита, основанная на
представлении, что адипоциты, переходя из
состояния накопления жира в фазу его траты
генерируют некий сигнал, воспринимаемый
центром насыщения. Долгое время его
природа оставалась неизвестна. Поиск
гормонального сигнала, воздействующего на
гипоталамус при индукции насыщения, был
продолжен. Выяснилось несколько важных
обстоятельств, связанных с эндокринными
функциями ЖКТ:
>■ Кишечные гормоны энтериновой
системы выделяются в ответ на прием пищи и
подавляют чувство аппетита (A.M.
Уголев, 1962). Особенно активны гормоны
двенадцатиперстной кишки. Одна из
гормонально активных фракций
экстрактов двенадцатиперстной кишки,
содержащая сигнальные пептиды,
подавляющие активность центра голода,
была названа арэнтерином. А.С. Хохлов
и А.М. Уголев показали, что аппетит-ре-
гулирующий энтериновый гормон имеет
белковую природу и молекулярную
массу около 100 кД (1978). Анорексигенное
действие арэнтерина — наиболее
продолжительное среди различных энтери-
новых гормонов. Механизм его —
центральный, так как оно не предупреждается
субдиафрагмальной ваготомией. Однако,
арэнтерин так и не был выделен в чистом
виде.
Сильным ингибитором чувства
голода, аппетита и пищевой активности
служит дуоденальный гормон холеци-
стокинин (Х.Купманс и соавт., 1972).
Потребление воды холецистокинин не
затрагивает. Данный гормон обладает
центральным тормозным действием
на вентролатеральный гипоталамус, а
также действует опосредованно, через
рецепторы и афферентные волокна
абдоминальной вагусной системы.
> Менее активными ингибиторами центра
голода являются бомбезин, соматоста-
тин, сатиетин, нейротензин, кортико-
либерин, тиролиберин, вазоактивный
интестинальный полипептид и инсулин,
имеющие рецепторы в центре
насыщения. Дериват тиролиберина — гистидил-
пролин дикетопиперазин — понижает
аппетит, вызывает истощение у крыс и
обнаружен (Л.Э. Морли и соавт., 1981) в
крови больных психогенной анорексией
(см. выше). Эндорфины и энкефалины,
а также соматолиберин, наоборот,
стимулируют аппетит. Все эти регуляторы
вырабатываются как в энтериновой
системе, так и в ЦНС.
> Энтериновые гормоны, понижающие
возбуждение центра голода,
выделяются в ответ на попадание пищи или даже
просто— кислого желудочного сока — в
двенадцатиперстную кишку.
Исследования энтериновой регуляции насыщения
позволили создать еще одну
экспериментальную модель кахексии и ожирения —
дуоденопривную модель. Еще в 1916 г.
основатель кафедры патофизиологии
Педиатрического института
Е.С.Лондон доказал, что двенадцатиперстная
кишка млекопитающих — жизненно
важный для них орган и выделяет в кровь
«какие-то витально необходимые
организму субстанции», поскольку
животные неизменно гибли при ее удалении, но
выживали, если оставался неудалённым
хотя бы небольшой участок
дуоденальной слизистой. Усовершенствовав
технику дуоденектомии по Е.С. Лондону,
А.М. Уголев (1952) добился
малотравматичного удаления этого органа. У
кошек при удалении (но не при изоляции
от пищеварительного канала)
двенадцатиперстной кишки, в первые полтора
месяца развивалась прогрессирующая
кахексия. Животные теряли до
половины массы тела и многие из них гибли. У
выживших кошек спонтанно наступала
вторая фаза, характеризующаяся
полифагией и сначала восстановлением
массы тела (через 8-12 недель после
операции), а затем — прогрессирующим
ожирением. Кошки жили 1,5-2 года и
набирали за это время более чем 30 %-й
избыток массы тела. Характерно, что в
отсутствие двенадцатиперстной кишки
наблюдались дегенеративные
изменения с задержкой нейросекрета в ядрах
гипоталамуса, гипотироз, нарушения
структуры коркового вещества
надпочечников, исчезновение
специфического динамического действия пищи
(см. выше раздел «Нарушения
энергетического обмена»). У собак, которые
имеют более диффузное расположение
эндокриноцитов вдоль тонкого
кишечника, роль данной кишки не столь
исключительна, и они развивают при
дуоденэктомии менее значительную
потерю в весе в раннюю фазу, а в
позднюю фазу не испытывают ожирения.
Все это позволило рассматривать
двенадцатиперстную кишку как «гипофиз
брюшной полости» (А.М. Уголев, 1991)
и укрепило положения о нейропептид-
ной регуляции липостата.
Ряд аппетит-регулирующих субстанций
был идентифицирован выдающимся
японским патофизиологом Ю. Омурой
(1966). Это неглюкозные регуляторы,
индуцирующие путём центрального
воздействия на соответствующие нейроны
подбугорья чувство сытости или чувство
голода. Субстанцией голода оказался
3,4,5-тригидроксипентаноил-у-лактон.
Напротив, 2-бутен-4-олид и 3,4-дигид-
роксибутаноил-у-лактон действуют на
гипоталамус, как факторы сытости.
Курьёзным фактом из истории данных
исследований является обнаружение
последнего вещества в орбитальном
спецпайке астронавтов НАСА. Это
незапланированное диетологами
обстоятельство вызывало у покорителей космоса
орбитальную анорексию, волновавшую
специалистов Центра управления
полётами. Ютака Омура установил механизм
орбитальной анорексии, которая исчезла
206 АЖ Зайчик, Я/7. Чурилов Патохимия (эндокринно-метаболические нарушения)
после корректировки состава
питательных смесей. По последним данным
Ю.Омуры и его школы (2005), важным
сигналом сытости и координатором
иммунонейроэндокринного ответа
организма на прием пищи служит кислый
фактор роста фибробластов (к-ФРФ).
Концентрация глюкозы в
цереброспинальной жидкости (ЦСЖ) удваивается
по сравнению с нормальным уровнем
(2,5 мМ/л) во время приема пищи.
Отвечая на это увеличение, эпендимоциты
III мозгового желудочка освобождают
к-ФРФ в ЦСЖ и головной мозг.
Освобожденный к-ФРФ сначала подавляет
потребление пищи, тормозя боковые
гипоталамические нейроны центра
аппетита. Во-вторых, этот цитокин
активирует СА1 нейроны морского конька,
таким образом облегчая возникновение
пространственных навыков и память,
вразрез с известной пословицей «Сытое
брюхо к ученью глухо!». Затем он
стимулирует гипоталамо-гипофизарно-над-
почечниковый аппарат и центробежные
симпатические импульсы. Фагоцитоз
макрофагов и другие
иммунобиологические функции также усиливаются под
действием к-ФРФ (см. т. I, с. 334-335) .
Таким образом, облегчается иммунный
ответ организма. Увеличение
концентрации глюкозы непосредственно действует
как сигнал насыщения, тормозя боковые
подбугорные нейроны и способствуя
разрядам вентромедиальных нейронов
насыщения. Затем глюкоза инициирует
СА1 нейроны гиппокампа, облегчая
возникновение пространственных навыков
и память. Очевидно, потребление пищи
необходимо не только для поддержания
метаболизма, но также для модуляции
высшей нервной деятельности и
иммунного ответа. Исследования Омуры
поставили цитокины в ряд важнейших
регуляторов аппетита и массы тела. Как
показано ниже, некоторые из них (лептин,
кахексии, адипонектин, орексин) играют во
взаимодействии метаболизма, иммунитета
и потребления пищи центральную роль.
>* Аппетит и насыщение подвержены
действию ряда нейротрансмиттеров.
Норадреналин, известный как липоли-
тический активатор, тормозит аппетит;
вызывают чувство насыщения и другие
Р-адреномиметики, что используется
при применении амфетаминов в качестве
анорексигенов. Менее активен в
качестве анорексигена серотонин. Впрочем, у
мышей с полифагией и наследственным
ожирением Л.Г. Тикотт и соавт. (1995)
обнаружили именно малую
эффективность серотонинового рецептора типа
2С при пострецепторной передаче се-
ротониновых сигналов. Это связано с
особенностями посттранкрипционного
редактирования структуры РНК-овой
матрицы этого рецептора. Прозек и
другие лекарства, блокирующие реабсорб-
цию серотонина в синаптической щели,
помогают при полифагии подобного
характера (Дж. Скотт, 1997). Дофаминер-
гические и ГАМК-эргические влияния
стимулируют аппетит. Повышают
пищевую активность и а2-адреномиметики,
известные также как активаторы липоге-
неза.
Подлинный прорыв в понимании нейро-
пептидных и липостатических механизмов
контроля массы тела наступил в 90-х годах
XX века в результате открытий,
характеризуемых В. Кумаром и соавторами (1997) как
«захватывающие».
Прежде чем подробно охарактеризовать их
сущность (отражённую также на рис. 38),
необходимо напомнить современные
представления о гистофизиологии жировой ткани.
Жировая ткань принадлежит к
соединительным тканям со специальными
свойствами, вместе с ретикулярной и
пигментной тканями, а также слизистой тканью
вартонова студня и стекловидного тела
(В.Л. Быков, 1998). Клетки жировой
ткани — адипоциты— формируются из
преадипоцитов, которые, в свою очередь,
являются потомками фибробластов. Диффе-
ренцировка преадипоцитов в новые
адипоциты интенсивно идет в последний триместр
внутриутробной жизни и продолжается до
Нарушения липидного обмена 207
POMC/CART-нейроны
NPY/AGRP-нейроны
адипоциты
липогенез
продукция лептина
Рис. 38. Устройство липостата и механизмы первичного ожирения. Сокращения: ГПП-1 — глюкаго-
ноподобный пептид I, СНС — симпатическая нервная система (CART-кокаин-амфетамин —
ассоциированный пептид), аМСГ — а-меланоцитостимулирующий гормон, РР — панкреатический
полипептид YYP-, остальные сокращения — по тексту.
3-го месяца постнатального периода. При
дифференцировке преадипоциты экспрес-
сируют маркерный фермент липопротеи-
новую липазу (ЛПЛ), утрачивают отростки,
синтезируют коллагены IV и XI типов,
некоторые белки цитоскелета и обретают
несколько жировых капель, которые затем
в белом жире сливаются в одну, а в буром —
остаются раздельными. При голодании,
теряя жир, адипоциты вновь приобретают
фибробластоподобный вид, а при
откармливании процесс их дифференцировки как бы
повторяется. Дифференцировке адипоцитов
способствуют соматотропный гормон и ин-
сулиноподобный фактор роста I. Тироидные
гормоны сдерживают этот процесс.
Адипоциты динамично реагируют на
изменения эндокринно-метаболической
ситуации, так как имеют обширный
набор поверхностных нейромедиаторных и
гормональных рецепторов. В этих клетках
непрерывно идут равновесные процессы
липогенеза и липолиза.
Липогенез характеризуется следующими
особенностями. Адипоцитарный фермент
ЛПЛ (см. также выше, раздел «Транспорт
липидов в организме и его нарушения»)
диффундирует на поверхность эндотели-
альных клеток капилляров жировой ткани
и осуществляет гидролиз ТГ из состава
различных ЛП кишечного и печеночного
происхождения. Активности этого энзима
208 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
способствует
инсулин. Трансцитоз
жирных кислот первичный
через эндотелио- рецептор
циты приводит к
их появлению в межклеточном
пространстве, откуда их захватывают
жировые клетки. В них идет реэтери-
фикация глицерина и формирование
собственных ТГ. При переедании и
первичном ожирении активность Л ПЛ растет, а при
голодании снижается. Существует модель
ожирения на животных с наследственно
повышенной активностью ЛПЛ.
Собственный синтез жиров из глюкозы в
адипоцитах также имеет место. Его мощным
стимулятором служит инсулин,
увеличивающий захват жировой тканью глюкозы,
активность фосфофруктокиназы,
пропускную способость фосфотриозного и пентоз-
ного пути метаболизма. Глюкагон влияет на
эти процессы противоположным образом.
Липогенез, как уже упоминалось выше,
стимулируется и а2-адреномиметиками.
Глюкокортикоиды увеличивают и
липогенез, и липолиз, причем суммарный эффект
их действия различен в адипоцитах разной
локализации, в силу пермиссивного
действия катехоламинов на жировые клетки,
имеющие неодинаковый набор адреноре-
цепторов. При гиперкортицизме подкожный
жир откладывается на лице, шее и в верхней
части туловища. На конечностях количество
жира, напротив, уменьшается.
Мобилизация жира из депо —
липолиз— это процесс, подверженный сложной
ступенчатой нейроэндокринной
регуляции (рис.39). Его ключевым ферментом
является внутриадипоцитарная гормо-
нозависимая липаза (ГЛ). Она подлежит
активирующему фосфорилированию, с
участием ц-АМФ-зависимых протеинки-
наз. Аденилатциклазы, необходимые для
активации, стимулируются
взаимодействием нескольких биорегуляторов с ади-
поцитарными рецепторами. Прежде всего
активирует липолиз норадреналин, через
Рз-адренорецепторы. У лиц, склонных к
первичному ожирению, уменьшена экспрес-
модифицированный
рецептор
v
неактивная
аденилатциклаза
активная
аденилатциклаза
Ч
АТФ
активная
протеинкиназа
ц-АМФ
\
неактивная
лцпаза
неактивная
протеинкиназа
активная
липаза
\
ТГ
Гл+ЖК
Рис. 39. Регуляция липолиза в адипоцитах.
Гл — глицерин, ЖК — жирные кислоты;
остальные сокращения — как в тексте
сия р3-адренорецепторов на адипоцитах.
При хроническом применении некоторых
Р-адреноблокаторов, например, в лечении
сердечных заболеваний, врачи могут
сталкиваться со вторичным ожирением как
побочным эффектом от понижения
интенсивности липолиза. Норадреналин освобождается
из симпатических нервных окончаний,
диффундируя к адипоцитам по
межклеточным промежуткам, и действует гормоно-
подобно, через кровь, после секреции его в
мозговом веществе надпочечников.
В буром жире норадренергические
нервные терминали особенно обильно
представлены и, по некоторым данным, образуют
синаптоподобные структуры на самих бурых
адипоцитах. Это, безусловно, обеспечивает,
наряду с особым устройством митохондрий,
большей поверхностью жировых капель и
наличием мощного разобщителя окисления
Нарушения липидного обмена 209
и фосфорилирования — термогенина — все
те уникальные функции бурого жира,
которые связаны с залповым липолизом (см. т. I,
стр. 106, 371, 541-542). Есть сведения, что
при развитии первичного ожирения в
организме компенсаторно возрастает процент
бурого жира, в котором калоригенез более
активен (Н. Ротвелл и соавт., 1982).
Повышение симпатоадреналовой
активности в промежутках между приемом
пищи способствует увеличению липолиза.
Липолиз в адипоцитах стимулируется
гормонами щитовидной железы и аденоги-
пофиза — липотропином (ЛПГ) и другими
дериватами проопиомеланокортина (АКТГ,
МСГ), соматотропином (СТГ), лютеотро-
пином (ЛТГ), а также тиротропином (ТТГ).
Отчасти поэтому при гипопитуитаризме,
не затрагивающем функции гипоталамуса,
возможно развитие вторичного ожирения.
Жирные кислоты и глицерин
освобождаются в плазму. Первые связываются
альбумином для транспорта в гепатоциты. Но
около 40 % глицерина реэтерифицируется
с жирными кислотами, поставляемыми
ЛПЛ. Интересно, что у лиц с
наклонностью к первичному ожирению значительно
большая доля глицерина подвергается
реэтерификации в ТГ, а меньшая —
освобождается. В связи с этим, существовала
даже гипотеза, что нормальная активность
липостата и его правильная с
антропометрической точки зрения установка,
относительно массы тела, достигаются лишь
при адекватном уровне глицерина в крови.
Делались, в русле этих представлений,
попытки глицеринотерапии первичного
ожирения. Роль глицерина как ведущего
сигнала обратной афферентации в липос-
тате, не была подтверждена.
Как бы там ни было, адипоциты больных
первичным ожирением с большей
«неохотой» расстаются с ТГ. Основным тормозным
регулятором ГЛ является инсулин. Поэтому
гиперинсулинизм как при сахарном диабете
II типа, так и при инсулиномах сопряжен
с вторичным ожирением. Есть данные об
антилиполитической роли простагландинов
и никотиновой кислоты.
Много лет адипоцитам отводилась в
липостате роль пассивных накопителей,
лишь повинующихся нейроэндокринным
сигналам. Но еще А. А. Богомолец указал на
значение конституции в регуляции
потребления пищи и, особенно, подчеркивал роль
пастозной (липоматозной) конституции
с ее обилием рыхлой, тонковолокнистой,
гидрофильной соединительной ткани — в
генезе ожирения. Это можно считать первой
концепцией, оценивающей первичную роль
особенностей жировой ткани в механизмах
алиментарно-конституционального
ожирения (Н.Б. Медведева, 1937). В дальнейшем
липостатическая теория развивалась
следующим образом.
В начале 90-х годов XX века стало ясно,
что адипоциты — важный источник ци-
токинов. В частности, было обнаружено,
что они вырабатывают факторы роста
гемопоэтических клеток, участвующие в
поддержании их адекватного размножения
и дифференцировки в костном мозге. Затем
стало ясно, что в «сытом» состоянии
адипоциты выделяют кахексии (ФНОа), который
понижает ответ тирозиновых протеинкиназ
жировой и мышечной ткани на инсулин и
таким образом сильно тормозит липогенез.
Тормозятся и другие эффекты инсулина, что
делает кахексии важным контринсулярным
фактором. Возможно, данный регуляторный
механизм должен предупредить чрезмерный
липогенез при «переполнении» адипоцитов
(см. рис. 38).
Кахексии участвует в патогенезе ин-
сулинорезистентности у тучных. Ранее
считалось, что, по отношению к тучности,
гиперинсулинизм всегда первичен. Затем
появилась точка зрения, что при первичном
ожирении, по достижении определенного
предельного размера адипоцитов, инсу-
линовые рецепторы на их поверхности
настолько разобщаются в пространстве,
что молекулы инсулина не могут
осуществить их перекрестное связывание, нужное
для передачи эффекта инсулина внутрь
клеток. Таким образом, признали
возможность первичного характера тучности, по
отношению к инсулинорезистентности (см.
210 АЖ Зайчик, Я/7. Чурилов Патохимия (эндокринно-метаболические нарушения)
ниже «Патофизиология углеводного
обмена»). Наконец, современная точка зрения
связывает инсулинорезистентность при
тучности с повышенной продукцией кахек-
сина в адипоцитах (Дж.С. Хотамизлиджил
и соавт., 1983).
Действуя на печень и, возможно,
гипоталамус, кахексии снижает аппетит и
усиливает катаболизм. Поэтому данный пептид,
особенно, вырабатывающийся в
висцеральных адипоцитах, служит существенным
элементом липостатической регуляции.
Антагонистом кахексина служит
уникальный противовоспалительный и инсулин-
сенсибилизирующий цитокин жировых
клеток — адипонектин, открытый Н.Э. Ше--
рером и соавторами (1995). Адипонектин
вырабатывается только адипоцитами^он
повышает чувствительность тканей к
инсулину, способствует подавлению глюконеоге-
неза в печени и захвату глюкозы мышцами.
Чем ниже продукция адипонектина в
висцеральном жире, тем выше индекс массы
тела. Адипонектин - гомолог Сlq фрагмента
комплемента, сильный
противовоспалительный медиатор, тормозит ответ острой
фазы и атерогенез, причем в сосудистой
стенке его специфическим лигандом служит
Т-кадхерин. Адипонектин препятствует
развитию ожирения, инсулинорезистентности
и сахарного диабета II типа. Он
способствует снижению потребления пищи и
увеличению калоригенеза на уровне гипоталамуса.
Мыши КО, лишенные гена адипонектина,
развивают спонтанно инсулинорезистен-
тный сахарный диабет. Ген адипонектина*
возможно, является дефектным у лиц,
предрасположенных к метаболическому
синдрому (см. выше с. 169-170, а также ниже
гл. 9,14). Высказано предположение, что это
комплексное метаболическое расстройство,
ассоциированное с инсулинорезистент-
ностью, СД, ожирением, гиперурикемией,
гипертензией и ускоренным атеросклерозом
зависит от недостаточной эффективности
адипонектиновой регуляции (Дж. П. Уайт-
хед и соавт., 2005).
Адипоциты, главным образом в
периферическом жире нижней половины тела,
обладают высокой активностью фермента
ароматазы и синтезируют значительные
количества эстрогенов, превращая в них
андрогенные предшественники.
Следовательно, жировая ткань — элемент половой
системы организма (см. ниже «Нарушения
эндокринной регуляции»). Более того, для
мужчин всех возрастов, а также девочек
перед пубертатом и пожилых женщин — это
важнейший источник женских половых
гормонов. Нельзя не учитывать этих фактов
при рассмотрении вопроса о связи ожирения
с расстройствами половой функции и
психофизиологической ориентации.
С этой точки зрения становятся
понятными некоторые поведенческие особенности
больных с полифагией и булимией (почти
исключительно, молодых женщин — см.
выше «Нейрогенная анорексия и булимия»),
которые буквально «сексуализуют» акт еды,
испытывая к ней неодолимое влечение, или,
наоборот, испытывают страсть к
истощению. То, что с точки зрения фрейдистского
подхода истолковывается как «фрустрация
оральной фазы, отказ от беременности,
стремление вернуться в детство», на деле
имеет конкретную гормональную
подоплёку. Установлено, что секреция эндогенных
опиатов в процессе еды, создающая
положительное эмоциональное подкрепление
процесса приема пищи, значительно
усилена у лиц обоего пола с наклонностью к
первичному ожирению. Аркуатное ядро
гипоталамуса — место рецепции лепти-
на — участвует также в продукции гонадо-
тропинов. Не случайно некоторые варианты
гипоталамического гипогонадизма, как мы
увидим в дальнейшем, в разделе
«Нарушения эндокринной регуляции»,
сопровождаются и гиперлептинемическим ожирением.
Тенденция к повышенному содержанию
эстрогенов может быть следствием активной
продукции женских половых гормонов в
избыточной жировой ткани мужчин. При этом
может изменяться их поведение, и
происходит отход от традиционных маскулинных
поведенческих стереотипов. Наблюдается
гинекомастия. У мужчин тяжелое ожирение
может негативно сказываться на потенции.
Избыток эстрогенов тормозит секрецию
лютропина и возникает гипогонадотропный
гипогонадизм с понижением уровня
тестостерона. Вместе с тем, у женщин продукция
эстрогенов в адипоцитах и само наличие
достаточно выраженной жировой ткани,
по-видимому, существенны для
поддержания нормальной половой функции. По
крайней мере, менструации у девочек, чей
вес не достиг определенной критической
точки (48 кг — по Р. Фриш и соавт., 1974),
не начинаются, даже если возраст пубертата
пройден. Часто при очень значительном
ожирении у женщин фертильность не
страдает. Известны многочисленные случаи
ожирения, наступившего во время и после
беременности и родов. Но потеря жировой
ткани у женщин при голодании сказывается
на половой функции сильнее, чем у мужчин,
вызывая дисменорею и аменорею. В то же
время, есть данные о стимулирующем
действии производимых местными адипоцитами
эстрогенов на развитие эстроген-зависимых
опухолей молочной железы и матки.
Половая функция у женщин при ожирении
характеризуется рядом особенностей.
У тучных девочек менструации начинаются
раньше, так как они набирают критическую
массу в более раннем возрасте, однако,
долго не устанавливаются и носят часто
нерегулярный характер. У тучных женщин
повышена частота развивающегося на почве
гиперинсулинемии поликистоза яичников
(см. ниже «Нарушения эндокринной
регуляции»), а менопауза наступает раньше.
Поликистоз яичников характеризуется тем,
что при усиленной конверсии андрогенов
в эстрогены, женские половые железы
начинают вырабатывать мужских половых
гормонов значительно больше нормы. Этот
синдром ответственен и за гирсутизм, часто
присутствующий у тучных пациенток. Но
истинной вирилизации не происходит, так
как образование эстрогенов очень
значительно.
Первичное ожирение характеризуется
множеством гормональных и
метаболических особенностей. Как уже отмечалось,
у больных имеются гиперинсулинемия,
Нарушениялипидного обмена 211
инсулинорезистентность, а кроме того —
понижены стимулированная секреция СТГ
и чувствительность тканей к тироидным
гормонам; увеличена конверсия тироксина
в более активный трийо^тиронин, снижено
количество реверсированного трийодти-
ронина, часто имеется гиперкортицизм,
несмотря на отсутствие синдрома Кушинга.
Довольно характерны гиперурикемия, ги-
пернатриемия, гипергидратация и
наклонность к ацидозам. В фазу активного набора
веса повышен основной обмен, снижено
специфическое динамическое действие
пищи, увеличены калорические затраты на
выполнение стандартной нагрузки. Но
главные гормональные особенности первичного
ожирения оставались неизвестны вплоть до
самого последнего времени. Лишь недавно
было выяснено, что в основе первичного
ожирения лежит нарушение
взаимоотношений между периферическим и центральным
компонентами липостата — жировой тканью
и гипоталамусом. Гипотезы о
существовании неких «адипсинов», выделяемых
жировой тканью и влияющих на ЦНС, имели
хождение и ранее. Но в 1994 г. был
обнаружен пептидный гормон адипоцитов лептпин,
который вырабатывается адипоцитами в
«сытом» состоянии. Лептпин вначале был
открыт (Й.Жэнь и соавт., 1994) как анорек-
сигенный метаболический гормон, который
секретируется жировой тканью и уменьшает
потребление пищи. Сейчас известно, что он
также влияет на иммунную, нервную,
эндокринную системы и участвует в гемопоэзе,
ангиогенезе и остеогенезе (Писарева СВ.
и соавт., 2004). Название лептин происходит
от греческого «А£ЯТО<;» — тонкий, изящный.
Ген лептина находится в длинном плече
7q31 хромосомы. Этот гидрофобный
протеин с молекулярным весом 16 кДа состоит из
167 аминокислот и принадлежит к
семейству цитокинов. Известно, что его фрагмент
22-56 ингибирует потребление пищи,
фрагмент 116-167 лишь незначительно влияет
на её прием, а 57-92 фрагмент влияния на
аппетит не оказывает (Терещенко И.В.,
2001). Множественные функции лептина
связаны с его прямым воздействием на
212 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
гипоталамус, эндокринную, иммунную и
сердечно-сосудистую системы.
Общее количество лептина в организме
пропорционально массе жировой ткани и
физиологически выше у женщин, нежели
у мужчин (рис. 38). Выработка лептина в
адипоцитах стимулируется инсулином и,
в меньшей степени,глюкокортикоидами
и зависит от размеров жировых клеток.
Лептин проникает в гипоталамус и
избирательно рецептируется вентромедиаль-
ными ядрами. Там имеются две первичные
популяции нейронов, вырабатывающие
либо нейропептид Y и белок, связанный
с агути (NPY/AGRP-нейроны), либо про-
опиомеланокортин и кокаин-амфетамин-
ассоциированный полипептид (РОМС/
CART-нейроны), которые экспрессируют
длинную (OB-Rb) форму лептинового
рецептора и оказывают противоположные
эффекты на энергетический метаболизм. NPY
(нейропептид У) и AGRP — белок,
связанный с агути — стимулируют прием пищи и
снижают теплопродукцию. Напротив, $-ме-
ланоцитостимулирующий гормон (фрагмент
проопиомеланокортина — РОМС), а также
CART {кокаин-амфетамин-зависимый
пептид) ингибируют прием пищи и
увеличивают потребление энергии (см. выше
специфическое динамическое действие
пищи с. 17-18), обращаясь к норадренерни-
ческим нисходящим симпатическим путям.
В конце концов, через р3-адренорецепторы
активируется липолиз и калоригенез в
жировой ткани. Лептин и инсулин,
количество которых в крови зависит от числа
и метаболического состояния адипоцитов,
ингибируют NPY/AGRP нейроны и
стимулируют POMC/CART-нейроны. Снижение
уровня лептина и инсулина способствует
активации NPY/AGRP и торможению
POMC/CART-нейронов. Таким образом
осуществляется действие лептина на
пищевое поведение и энергетический метаболизм
(Д. Каммингс, У. Шварц, 2003).
Очевидно, другие трансгипофизарные
и парагипофизарные влияния,
стимулирующие липолиз, например, продукция ЛТГ
и тироидная функция, также могут
изменяться при действии лептина. Лептин имеет
рецепторы на тироцитах и стимулирует их
рост и функцию. В то же время, трийодти-
ронин снижает его продукцию (К. Новак и
соавт., 2002; М. Корбониц, 1998). Под
влиянием лептина растут потребление кислорода
и основной обмен. Становится понятным,
почему при ожирении во многих случаях
основной обмен и термогенез, вызванные
приемом пищи, снижены. Вполне объясним,
с этих позиций, и хорошо известный факт
понижения основного обмена во втором
периоде голодания, по мере истощения
жировых запасов (см. выше «Голодание»).
Все это может быть связано с лептиновой
недостаточностью.
Недосыпание и алкоголь понижают
продукцию лептина, а вот гипоксия, курение и
никотин, наоборот - потенцируют действие
лептина на гипоталамус или увеличивают
выработку лептина. Это связано с хорошо
известной прибавкой в весе у
бросающих курить. Лептин - важный иммуно-
регулятор, медиатор ГЗТ и, в частности,
ГЗТ-опосредованных аутоаллергических
реакций. Он определенно участвует в
патогенезе таких ГЗТ-зависимых заболеваний,
как аутоиммунный тироидит, рассеянный
склероз, инсулинзависимый сахарный
диабет. Этот цитокин способствует диффе-
ренцировке CD4+ лимфоцитов в Т-хелперы
1 типа (Th-1), участники ГЗТ, и
соответствующих видов аутоаллергии (т. 1, с. 428).
Не случайно, ожирение сочетается нередко
с подобными аутоиммунными
заболеваниями, а краткосрочное голодание оказывает
при этом благоприятный эффект (подробнее
см. СВ. Писарева и соавт., 2003). Лептин
стимулирует атерогенез и способствует
развитию гипертрофии миокарда при
сердечной недостаточности, что связывает
ожирение и сердечно-сосудистые
заболевания (П.Х. Шульце, Ю. Кратч, 2005). Вуг. 1
уже шла речь о роли лептина в регуляции
аксиального роста производных мезодермы
(с. 97). Ниже в разделе «Остеопороз...»
представлена более детальная характеристика
роли лептина в регуляции костной массы.
Лептин стимулирует продукцию контраре-
Нарушения липидного обмена 213
гуляторного, по отношению к неиропептиду
Y, глюкагоноподобного пептида /,
подавляющего аппетит и пищевое поведение.
Именно нейропептид Y оказался главным
гуморальным триггером аппетита и чувства
голода. Нейропептид Y стимулирует
пищевое поведение, поиск пищи у животных,
трансгипофизарную и парагипофизарную
модуляцию работы ряда эндокринных
желез, потребление пищи, продукцию
инсулина, и, наконец, накопление жира в
адипоцитах. Характерно, что при ИЗСД,
который сопровождается инсулинопениеи
и исхуданием, секреция нейропептида Y
понижена. Все периферические и центральные
влияния, снижающие аппетит, например,
холецистокининовые, результируются через
снижение продукции нейропептида Y. Эти
достижения составили основу более
глубокого понимания механизмов первичного и
гипоталамического вторичного ожирения.
В частности, стала ясна природа еще двух
экспериментальных моделей ожирения
(в сочетании с сахарным диабетом — см.
ниже), полученных на линейных мышах.
Обе модели — генетические.
> Мыши ob/ob (линия Цукера)
спонтанно развивают ожирение, так как имеют
дефект гена лептина. Их адипоциты не
вырабатывают адекватного количества
лептина в ответ на инсулиновый стимул
(лептинопеническая форма ожирения).
Без лептина у мышей спонтанно
формируется ненасытный аппетит, возникают
гипотермия и гипометаболизм, а масса
тела в 2-3 раза превышает норму. При
добавлении лептина или при вживлении
гена лептина аппетит у мышей
снижается, они худеют, а потребление кислорода
и температура тела — нормализуются.
> Мыши db/db вырабатывают лептин
нормально, а его концентрации в крови у
таких мышей даже резко повышены. Но
гипоталамические нейроны аркуатного
ядра у этих грызунов лишены
адекватного количества лептиновых рецепторов.
Соответственно, их гипоталамус
вырабатывает избыток нейропептида Y и
резистентен к лептину (гиперлептинемическая
форма ожирения).
Существование генетических моделей
ожирения коррелирует с представлениями
о решающей роли наследственных факторов
в формировании наклонности к первичному
ожирению у человека.
Первичное ожирение (рис. 40) можно
рассматривать, как аддитивно-полигенную
болезнь с пороговым эффектом по диете.
О.Гюнтер и К. Готтшальк (1962)
указывали, что если болен один из родителей, шансы
заболеть ожирением у ребенка составляют
около 56 %. Дети из семей, где тучными были
и мать, и отец, впоследствии развивают
ожирение в 78 % случаев. В то же время у
детей родителей, не болевших ожирением,
оно развивается не более чем в 14 % случаев.
Известно, что первичным ожирением
значительно чаще заболевают носители гена ГКГС
В18(В.Ю.Шанин, 1995) (см. также с. 236).
Изучение лептиновой регуляции у лиц с
первичным ожирением показало, что около
20 % подобных тучных больных имеют
абсолютную лептиновую недостаточность.
Более 80 % пациентов, страдающих первичным
ожирением, характеризуются выраженной
гиперлептинемией. Таким образом, у них
можно предполагать наличие первичной
лептинорезистентности или
относительной недостаточности действия лептина на
гипоталамус. Во втором случае, несмотря
на наличие лептина, центр голода
продолжает активную выработку нейропептида Y.
По-видимому, эта нечувствительность может
отражать дефекты лептинового рецептора,
пострецепторные нарушения передачи
сигнала, генетические изъяны эффекторов лептина
в гипоталамусе и вне его — в частности —
аномалии CART или р3-адренорецептора, либо
другие расстройства в функции лептинза-
висимых звеньев липостата (В.М. Кейттал,
Арки Р.А. 2001).
Признавая полигенную основу
первичного ожирения и его гетерогенность, а также
ключевую роль лептиновой
недостаточности в механизмах этого расстройства
липидного обмена, мы, тем не менее, не можем не
подчеркнуть, что любое ожирение — вопрос
214 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рис 40. Первичное андроидное ожирение III
степени тяжести у больного Е.Д. (по А. Цаневу,
1968)
баланса. При первичном ожирении липог
стат, из-за особенностей функционирования
оси гипоталамус-адипоциты, переносит
установочную точку массы, тела на более
высокий уровень, воспринимая имеющееся
в организме количество жировой ткани, как
недостаточное. Однако, сам набор
избыточной массы невозможен без превышения
поступления энергии над её затратами, что
и реализуется через изменения поведения
больного, соответствующие состоянию его
нейропептидной регуляции.
Роль алиментарно-гиподинамического
фактора проявляется и в том, что
ограничивая потребление калорий и увеличивая
их трату, индивид может вернуться к
антропометрически должному весу. Не только
абсолютное количество потребляемой и
рассеиваемой энергии, но и качественный
состав, а также режим приема пищи имеют
значение при определении риска ожирения.
Показано, что ожирению способствует
недостаток белка в питании, ослабляющий
специфическое динамическое действие пищи
(о причинах — см. раздел «Энергетический
метаболизм...»). Как правило, ожирение,
при равном калорическом избытке, быстрее
развивается если перерывы между приемом
пищи велики, а порции обильны (Дж. Брей
и соавт., 1972). Очевидно, при таком режиме
питания дольше удерживается высокая
концентрация инсулина. Ожирению
способствует никтофагия — смещение максимума
пищевой активности на поздние вечерние
часы. Здесь проявляется характерная для
расстройств гипоталамической функции
тенденция к нарушению суточных ритмов.
, Рассматривая подходы к коррекции
тучности, необходимо подчеркнуть важнейшую
патофизиологическую установку,
касающуюся лечения первичного ожирения. Простая
нормализация веса у такого пациента не
делает его здоровым. По словам видного
немецкого диетолога X. Эделя (1963):
«Диета является неотъемлемым основным
условием лечения ожирения в каждом
случае». Однако, это не этиотропное лечение и
даже не лечение, адресованное ключевому
звену патогенеза! Дело обстоит так же, как
при лечении лихорадки, когда повышается
установочная точка температурного гомеос-
таза. Сбить температуру — еще не значит
вылечить. Если причина болезни продолжает
действовать, гипоталамус вновь вернет
температуру к патологической установочной
точке, при отмене жаропонижающих.
Разумеется, при первичном ожирении
многие из расстройств метаболизма после
Нарушения липидного обмена 215
нормализации веса корректируются
(например, уменьшается или совсем проходит инСу-
линорезистентность, купируется пиквшсскии
синдром, уменьшается или совсем исчезает
ГЛП). Тем не менее, больной с диагнозом
«первичное ожирение» и с нормализованным
весом не излечен. У него сохраняются многие
обменные нарушения, прежде всего, главное
из них — лептиновая недостаточность. Его
адипоциты, по-прежнему, неохотно
расстаются с жирными кислотами и глицерином
- показано, что в них, в отличие от жировых
клеток нормальных индивидов, гораздо выше
процент реэтерификации жирных кислот,
освобожденных при липолизе. У него,
по-прежнему, повышена активность Л ПЛ жировой
ткани, снижена реакция центров насыщения
на серотонин, а адипоцитов — на р-адрено-
миметики, нарушена рецепция инсулина в
гипоталамусе, а при гиперпластическом и
смешанном ожирении — увеличено число
адипоцитов и т.д.
Кроме того, ряд функциональных
показателей у больных ожирением может при
снижении массы тела ухудшаться.
Гипоталамус не считает анатомически нормальный
вес оптимальным для индивида и реагирует
на похудение.
В ответ на антропометрическую
«нормализацию» веса возникает понижение
продукции тиротропина, относительный
гипотироз, снижается холодовая адаптация.
Многие похудевшие до нормального веса
пациенты жалуются на чувство зябкости. При
падении веса еще больше снижается
основной обмен. У пациентов, особенно женщин,
возможно нарушение половых функций.
Причина расстройств овариально-менстру-
ального цикла у похудевших пациенток с
ожирением, очевидно, связана со снижением
эстрогенопродуцирующей функции
адипоцитов. Строя свой образ жизни и питание
вопреки программе, навязываемой
гипоталамусом, больные ведут более сознательную,
но отнюдь не более свободную жизнь. А ведь
здоровье, в том числе и
психоэмоциональное, основано на внутренней свободе!
Многие похудевшие пациенты испытывают
дисфорию, отмечаются обсессивные
неврозы. Вероятно, механизмы этого связаны с
понижением выработки опиатных пептидов
из-за тех ограничений, которые существуют
в их пищевом поведении и образе жизни.
Кроме того, для таких пациентов типична
усиленная выработка кортикостероидов.
У ряда похудевших больных отмечается
тенденция к лейкопении, брадикардии и
гипотонии. При резком похудении может
снизиться иммунитет.
Некоторые психосоматические
особенности похудевших лиц с первичным
ожирением напоминают картину, наблюдаемую
при психогенной анорексии (см. выше,
раздел «Голодание»).
Следовательно, адекватное лечение
больного с первичным ожирением возможно
лишь под наблюдением врача и не сводится
к диетотерапии и лечебной
гимнастике. В последнее время большие надежды
связывают с введением методов лечения,
основанных на применении выделенного
в чистом виде лептина. Лептин излечивает
мышей ob/ob, он успешно применялся для
лечения некоторых наследственных форм
низкорослости, сопровождаемой ожирением
у детей, однако еще нет достаточного опыта
его клинического применения.
Итак, в пределах функциональной
системы липостата активную роль играют
и гипоталамус, и энтериновая система, и
надпочечники, и островки Лангерганса, и
жировая ткань. Их сигнальное
взаимодействие обеспечивает постоянство массы тела и
адекватность приема пищи метаболическим
потребностям организма.
С точки зрения схемы липостатической
регуляции, представленной на рис. 38,
первичное ожирение трактуется как
проявление абсолютной, либо относительной
лептиновой недостаточности, на алиментар-
но-гиподинамическом фоне.
Этиология, патогенез
и разнообразие вторичного
ожирения
Традиционно вторичным считается
ожирение, наступающее как синдром при развитии
каких-то первичных нейроэндокринных
21 б АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
нарушений. В настоящее время можно
уточнить, что к вторичному ожирению не
относятся формы, связанные с изначальным
дефектом продукции лептина и экспрессии
лептинового рецептора.
Вторичное ожирение ранее делилось на
церебральные и гормональные формы (М. Га-
нефельд, 1979). Однако, церебральные
формы также нейроэндокринны по своей
природе, поэтому некорректно
противопоставлять их гормональным.
С учётом этого, кажется более
оправданным делить вторичное ожирение на гипота-
ламо-гипофизарные {центральные) формы
и связанные с дисфункциями других
эндокринных желез {периферические) формы.
Разумеется, и такое деление нельзя назвать
строгим — например, гиперкортицизм, как
причина вторичного ожирения, может иметь
как центральное, так и периферическое
происхождение.
К центральным формам относится
вторичное ожирение при:
- синдроме Лоуренса-Муна- Барде- Бид-
ла-Стюарта
- синдроме Прадера-Вилли
- адипозогенитальной дистрофии Бабин-
ского-Фрёлиха
- синдроме персистирующей лактореи-
аменореи
- синдроме Стюарта-Морганьи-Морре-
ля
- синдроме Ольстрема
- синдроме Карпентера
- базофильной аденоме (адренокорти-
котропиноме) гипофиза
- изолированном гипопитуитаризме
(например, синдроме Шихена (Шиена))
- синдроме Пархона
- гипофизарном нанизме
- юношеском диспитуитаризме
- отравлении коллоидным золотом и
другими гипоталамическими ядами
- травмах, опухолях, инсультах, гемо-
бластозах и энцефалитах с
повреждением вентромедиального гипоталамуса.
К периферическим формам принадлежит
вторичное ожирение при:
- гиперкортицизме, в том числе синдроме
Иценко-Кушинга
- ИНЗДПтипа
- гипогонадйзме
- гиперинсулинизме
- дуоденальной недостаточности в поздней
фазе
- тимико-лимфатическом статусе
- синдроме поликистозных яичников.
Центральные формы вторичного
ожирения наиболее трудно дифференцируются
с первичным. Одной из важных считается
стимуляционная проба с люлиберином.
У больных первичным ожирением он, как
и в норме, повышает продукцию пролакти-
на — в пределах 0,5 ч после в/в инъекции
не менее, чем на 200-300 мЕ/л. Пациенты с
гипоталамическим вторичным ожирением
имеют и общие нарушения работы
гипоталамуса, кроме характерной для многих
случаев первичного ожирения лептино-
резистентности. Поэтому они, чаще всего,
в ответ на люлиберин не дают должного
прироста секреции пролактина. У женщин
тест должен быть приурочен к 5-7 дню ова-
риально-менструального цикла, если нет
аменореи. Из-за повреждения аркуатного
ядра, практически все формы гипоталами-
ческого ожирения протекают с некоторым
гипогонадотропным гипогонадизмом.
Кроме того, для них характерна полифагия.
Некоторые формы гипоталамического
ожирения настолько типичны, что известны
как отдельные нозологические единицы.
Синдром Прадера-Вилли — анеуплоид-
ная хромосомная аномалия, связанная с
делецией участка ql2 в длинном плече 15-й
хромосомы. Нарушение относится к числу
так называемых «синдромов смежных
генов», когда мозаичная клиническая картина
объясняется моносомией по группе рядом
лежащих генов, ответственных за контроль
очень разнородных признаков. Его картина
включает тяжелое ожирение, задержку
психомоторного развития, гипогонадо-
Нарушения липидного обмена 217
тропный гипогонадизм, гипотонию, гипо-
пигментацию кожи, уменьшенные размеры
кистей и стоп — акромикрию, косоглазие
и искривление позвоночника. Имеются
полифагия и сахарный диабет. Отмечаются
термолабильность, гипорефлексия и
снижение болевой чувствительности. Слюна
вязкая, характерны остеопения, косоглазие
и миопия. Могут быть гинекомастия и
кожные стрии. Синдром носит врождённый
характер, в некоторых родословных
наследуется и вызывает большой теоретический
интерес как один из первых распознанных
примеров так называемого «геномного
импринтинга». Данное эпигенетическое
явление представляет собой маркировку
материнских и отцовских аллельных генов
в ходе гаметогенеза, возможно, с помощью
дифференцированного метилирования
ДНК. В результате у потомства отцовская
и материнская гомологичные хромосомы
и соответствующие аллельные гены будут
экспрессироваться по-разному, хотя
классическая генетика учит, что диплоидные
клетки выбирают для активной экспрессии
одну из пары гомологичных хромосом, без
различия ее происхождения, по случайному
принципу. У больных синдромом Праде-
ра-Вилли всегда делеция поражает только
отцовскую хромосому. Такая же делеция
строго гомологичного участка
материнской хромосомы 15 дает совершенно иной
симптомокомплекс — болезнь счастливой
марионетки (синдром Эйнджелмена). При
этом заболевании нет ожирения, а
клиническая картина характеризуется атаксией,
судорогами, насильственным смехом и
напоминает синдром Прадера-Вилли только
в отношении умственной отсталости. Это
противоречит классическому
представлению о том, что происхождение хромосомы
от того или иного из родителей не должно
влиять на ее экспрессию в соматических
диплоидных клетках.
Синдром Лоуренса-Муна-Барде-Бидла
(по старой терминологии добавляли ещё и
Стюарта) — знаком каждому, изучавшему
эндокринологию студенту как самый
длинный эпоним — пяти специалистов едва
хватило для описания одной нозологической
единицы! При данном синдроме сочетаются
пигментная ретинопатия, умственная
отсталость, черепной дизостоз, цирроз печени,
полидактилия и синдактилия,
инсулинорезистентность, гипергликемия и вторичное
гипоталамическое стволовое ожирение с ги-
погонадотропным гипогонадизмом. Могут
быть малые анамалии почек и проявления
хронического интерстициального нефрита.
Синдром является наследственным, ауто-
сомно-рецессивным. Некоторые авторы
(по выражению А. Мотульского (1987) —
«пуристы») разграничивают*синдромы
Лоуренса-Муна (протекает без ожирения,
но со спастической параплегией) и Бар-
де-Бидла (без параплегии, но с ожирением).
Синдром Стюарта-Морганъи-Мореля,
который ряд авторов дифференцируют от
предыдущего нарушения, характеризуется
гиперостозом костей черепа и заращением
их пазух, возможно, вследствие
аутоиммунного синусита. Очевидно, сам процесс
или вторичные нарушения кровоснабжения
затрагивают и гипоталамо-гипофизарный
нейросекреторный комплекс, поскольку
имеются ожирение, несахарный диабет,
сильные головные боли. Нередко
обнаруживаются микроаденомы гипофиза.
Поражаются девочки. Отмечают гиперэстрогению
и гиперандрогенизм, гипертрихоз,
«яблочный» тип отложения жира — на животе.
Характерны инсулинорезистентность и
сахарный диабет.
Синдром Ольстрема — наследственная
аутосомно-рецессивная аномалия, которая
сочетает ожирение, инсулинорезистентный
сахарный диабет, прогрессирующую не-
фропатию, а также более тяжелую, чем при
предыдущем расстройстве, раннюю форму
пигментной ретинопатии и раннюю глухоту.
Задержка психического развития и
полидактилия для этого синдрома не типичны. Так
как пациенты имеют питрессин-резистен-
тный несахарный диабет и гипогонадизм,
предполагается, что у них присутствует
поражение гипоталамуса и гипофиза, либо
множественная резистентность
тканей-мишеней к пептидным гормонам (о формах
218 АЖ Зайчик, ЛЛ. Чурилов Патохимия
Рис. 41. Случай адипозо-генитальной
дистрофии у девушки 16 лет. Рост 145 см. Сильно
снижено зрение (по О. Хиршу, 1911)
чдокринно-метаболические нарушения)
вторичного ожирения, сопровождаемых СД,
см. также ниже в разделе «Патофизиология
сахарного диабета»).
Синдром Карпентера — наследственная
аутосомно-рецессивная акроцефалопо-
лисиндактилия. Сочетается с задержкой
психического развития, гипогонадизмом,
ожирением.
Адипозо-гениталъная дистрофия Бабин-
ского-Фрёлиха — во многих отношениях
отличается от вышеописанных вариантов
церебрального ожирения (рис. 41).
Поражаются, в основном, мальчики. Однако
тип наследования не установлен, бывают
больные женского пола, и считается, что
чаще всего это приобретенное заболевание.
Его этиология наиболее часто связана с
негормонообразующими опухолями ги-
поталамической локализации. Описаны и
случаи, спровоцированные травмой головы.
Повреждение аркуатного ядра ведет к
полифагии и гипогонадизму. Ожирение, как
правило, ганоидного типа. Характерны
полные конечности. Заболевание протекает без
развития сахарного диабета, более того —
толерантность к углеводам повышена. Из-за
частого вовлечения в процесс гипофиза
страдает продукция вазопрессина, липо-
тропного и других тропных гормонов. На
почве гипопитуитаризма проявляется
типичная тенденция к гипогликемии натощак,
нередко развивается несахарный диабет.
У пациентов чаще нормальный интеллект.
Могут быть поражения зрения.
Синдром персистирующей лактореи-
аменореи (различные формы известны
также как синдромы Хиари-Фроммеля,
Форбс-Олбрайта, Аргонза-дель Кастильо,
Алхумада-дель Кастильо) представляет
собой результат гиперпролактинемии,
создаваемой гиперплазией пролактот-
рофов или пролактиномами гипофиза,
которые могут быть рентгенологически
обнаруживаемыми или незаметными и
развиваются после беременности и родов,
или же — спонтанно. Большую роль играет
дефицит ингибирующих дофаминергичес-
ких влияний гипоталамуса на гипофиз.
Избыток пролактина тормозит секрецию
гонадолиберина, в связи с чем страдает
половая функция. Поражаться могут оба
пола. У мужчин развивается ожирение по
гиноидному или смешанному типу,
импотенция и гинекомастия. У женщин
характерны лакторея, аменорея и инфертиль-
ность (30 % женского бесплодия связаны
сданным нарушением). Подробнее этот
синдром охарактеризован среди
заболеваний аденогипофиза. В данном разделе
важно отметить, что у 60 % пациенток и
значительного большинства пациентов
обнаруживается умеренное ожирение (у
женщин — с гирсутизмом). Однако
описаны и случаи, сопровождаемые кахексией,
что, по видимому, зависит от особенностей
топографии растущей опухоли (см. также
ниже «Патофизиология гонадотропной
функции и гонад»).
Синдром Пархона (SIADH) —
неадекватно повышенная секреция вазопрессина — в
своем идиопатическом варианте вызван
микровазопрессиномами гипоталамуса.
Ожирение при данном заболевании может
быть ложным, так как отмечается
внутриклеточная гипергидратация и значительная
задержка жидкости в организме, чаще без
выраженных отёков (см. раздел
«Патофизиология гипоталамо-гипофизарного
нейросекреторного комплекса»). Истинное
ожирение развивается, если вазопрессинома
нарушает работу аркуатного ядра и других
элементов, связанных с гипоталамической
частью липостата. При ложном ожирении,
в отличие от истинного, масса тела очень
лабильна и зависит от сдвигов
водно-солевого баланса.
Юношеский диспитуитаризм {синдром
ожирения с розовыми стриями, синдром
Симпсона-Пэйджа) представляет собой
одну из важнейших нозологических форм в
практике подростковой эндокринологии.
Его подробную характеристику можно
обнаружить в новейшем руководстве
«Эндокринология подростков» (Ю.И. Строев,
Л.П.Чурилов,2004).
Синдром упоминается ниже, в разделе
«Патофизиология
гипоталамо-гипофизарного нейросекреторного комплекса».
Нарушениялипидного обмена 219
Для целей данной части книги важно
подчеркнуть, что юношеский диспитуитаризм,
несмотря на внешнее сходство с картиной
синдрома Иценко-Кушинга и многими
формами диэнцефального ожирения, протекает
доброкачественно. При этом расстройстве
нет опухолей и повреждений,
обусловливающих необратимые нарушения в гипо-
таламо-гипофизарном нейросекреторном
аппарате. Поэтому ожирение, зависящее от
преходящей гиперпродукции соматотроп-
ного гормона и АКТГ, не сопровождается
гипогонадизмом, задержкой психического
или физического развития. Напротив, рост
и созревание таких подростков ускорены, а
раннее начало половой жизни благотворно
влияет на их жировой обмен и способствует
нормализации веса. Ожирение носит
смешанный или гиноидный характер, однако и
гиноидность контуров фигуры, и ложная
гинекомастия у юношей с диспитуитаризмом
не свидетельствуют о каких-либо аномалиях
полового развития, так как сохраняется
нормальная продукция гонадотропинов и
андрогенов. Так как в жировой клетчатке
отмечаются разрывы соединительнотканных
прослоек и формирование жировых кист,
наблюдаются розовые стрии. Наибольшую
опасность представляет сочетающаяся с
этой формой ожирения транзиторная ги-
пертензия, симптоматическое проявление
гиперинсулинизма и избытка надпочечни-
ковых гормонов. Она может персистировать
и переходить в пограничную гипертензию
и гипертоническую болезнь. Подростки
имеют несколько повышенную продукцию
АКТГ и кортикостероидов всех трёх групп.
Но ожирение не характеризуется типичной
для болезни и синдрома Иценко-Кушинга
строгой зональностью. Конечности
остаются полными. В плане дифференциальной
диагностики с иными формами ожирения,
важно учесть ложный характер
гинекомастии и розовый (а не фиолетовый, как при
болезни Иценко-Кушинга) цвет тонких стрий.
Механизм ожирения связан с собственным
липогенетическим эффектом АКТГ и глю-
кокортикоидов, а также с инсулинотропным
действием АКТГ на островки Лангерганса.
220 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Определенное значение имеет и повышение
аппетита под действием глюкокортикои-
дов. Из-за угнетения глюкокортикоидами
экспрессии рецептора ЛПНП, ожирение
при данной болезни сопровождается
выраженной атерогенной ГЛП (см. выше).
При гипофизарном нанизме ожирение
может развиваться из-за сочетанного
выпадения продукции СТГ и липотропного
гормона гипофиза, которые усиливают
липолиз, однако оно не обязательно для
картины этого синдрома.
При болезни Иценко-Кушинга
(подробно — см. раздел «Патофизиология гипота-
ламо-гипофизарного нейросекреторного
комплекса») имеется гиперпродукция
АКТГ, а в классических случаях — кор-
тиколиберина и АКТГ, вызывающая
вторичный гиперкортицизм. Ожирение
при данном заболевании поражает более
90 % пациентов и имеет очень типичную
клиническую картину. Запоминается
лунообразное багровое лицо пациента,
отложение жира в области шеи, верхней
половины туловища, на животе. Над
шейными позвонками формируется жировой
«буйволиный горбик». Конечности
истончаются. На коже живота, бёдер и плеч
появляются грубые
красновато-фиолетовые стрии, иногда с геморрагическими
экстравазатами. В отличие от иных форм
ожирения со стриями, развивается
выраженный остеопороз. У женщин могут
быть явления частичной вирилизации, а у
мужчин — напротив, тенденция к
феминизации (см. рис. 115). Интересно, что у 8%
больных ожирения не происходит, хотя
типичное перераспределение жира
наблюдается. Вероятно, большое значение имеет
способность глюкокрртикоидов
стимулировать выработку лептина в адипоцитах,
особенно периферического жира.
Интоксикации как причина
церебрального ожирения известны со времен
обследования так называемых «штирийских мышья-
коедов» — членов австрийской секты,
практиковавшей самоотравление малыми дозами
мышьяка. У многих из них формировалось
ожирение с гипогонадизмом. Наиболее
специфично развитие поражения вентро-
медиального гипоталамуса и центрального
ожирения для отравления ауротиоглюкозой
и другими препаратами коллоидного золота
(см. выше). Отравления ртутью также могут
сопровождаться ожирением. Ожирение
бывает при хронической передозировке ряда
р-адренергических средств, а у некоторых
пациентов — сульфаниламидов. Однако, в
этих случаях механизмы могут быть
периферическими (см. предыдущий раздел).
Опухоли (наиболее часто — кранио-
фарингиомы), а также кровоизлияния и
травмы подбугорья могут быть причиной
полифагического ожирения, если
разрушают вентромедиальные центры насыщения.
Вероятно, опухоли, продуцирующие ней-
ропептид Y, могут приводить к тому же,
за счет гиперпродукции этого регулятора.
Чтобы черепно-мозговая травма вызвала
«прицельное» повреждение подбугорья, ее
биомеханизм должен быть весьма
своеобразным. Авторы наблюдали случай диэнце-
фального ожирения у школьницы, 12 лет,
развившегося в течение года после
краниальной травмы, причиненной при зажатии
головы дверью. Двойной симметричный
удар, по-видимому, вызвал гидравлические
волны, суммировавшиеся как раз в области
подбугорья.
Периферическое вторичное ожирение
зависит от эндокринопатий. При синдроме
Иценко-Кушинга патогенез ожирения и его
клиника, практически, идентичны болезни
Иценкр-Кушинга, но этиология связана
с кортикостеромой коры надпочечников
или эктопической секрецией АКТГ. При
кушингоидном синдроме гиперкортицизм
и ожирение могут быть ятрогенными и
наступать в результате передозировки глю-
кокортикоидов.
Ожирение характерно для гипогонадизма.
Оно наступает при кастрации и ложном
гермафродитизме, может сопровождать
такие гоносомные аномалии, как синдром
Шерешевского-Тёрнера у женщин и
синдром Клайнфелтера у мужчин. Возможно,
известную роль в патогенезе гипогонадных
Нарушения липидного обмена 221
форм вторичного ожирения играет
продукция и метаболизм половых стероидов в
адипоцитах.
При гипотирозе снижается скорость,
расходования калорий, что лежит в основе
вторичного ожирения. Изменения жирового
обмена сочетаются с отёками,
усиливающими впечатление тучности (Ф. Ксавье
Пи-Суньер, 1987).
Тучность характерна для инсулиннеза-
висимого сахарного диабета. Ее механизм,
подробно охарактеризованный ниже, в
разделе «Патофизиология сахарного
диабета», включает липогенетическое действие
гиперинсулинизма. Считается, что
клетки-мишени инсулина у больных обладают
различными пострецепторными, а иногда
и рецепторными дефектами,
понижающими ответ на инсулин. Большое значение
имеет недостаток продукции адипонектина
и избыток кахексина, снижающие
чувствительность к инсулину. Тучность — важное
патогенетическое звено в развитии
порочного круга, делающего адипоциты еще более
инсулинорезистентными и закрепляющего
гипергликемию.
Инсулинома островкового аппарата
поджелудочной железы или эктопическая ин-
сулинома сопровождается гиперсекрецией
инсулина без диабета. Имеются приступы
гипогликемии натощак, легко купируемые
сахаром. Типичны головные боли, бывает
гипертензия. Характерны странности в
поведении (например, нередко
наблюдаются повышенная двигательная активность,
приплясывания и т.д.).
Опасаясь гипергликемического шока,
больные увеличивают потребление
легкоусвояемой углеводистой пищи. Полифагия
и липогенетический эффект инсулина ведут
к своеобразному ожирению, как правило,
«андроидного» типа. Другие гормонооб-
разующие опухоли островков Лангерганса
(глюкагонома, соматостатинома), наоборот,
сопровождаются исхуданием (см. ниже
«Истощение и кахексия»).
Как уже отмечалось выше, полифагичес-
кое ожирение может возникать в поздней
фазе после удаления двенадцатиперстной
кишки из-за расстройства аппетитподавля-
ющей функции энтериновых гормонов.
Тенденция к избыточному весу
характерна для гиперплазии тимуса. Интересно, что
как и тяжелое первичное ожирение, status
thymicolymphaticus характеризуется
наклонностью к апноэ во сне (Д.Я. Шурыгин,
1966). Возможно, известную роль в генезе
ожирения при гиперплазии вилочковой
железы играют сопутствующая тироидная
гипоплазия и иммунопатологические
нарушения.
Дополнительные сведения по
вторичному ожирению эндокринного генеза можно
почерпнуть ниже, в гл. 13 «Нарушения
эндокринной регуляции» и в монографиях
(Ю.И. Строев, Л.П.Чурилов, 2007).
Местные формы накопления
триглицеридов
Избыточное накопление триглицеридов
может носить локальный характер. Это, в
первую очередь, происходит при
липоматозе. Липома представляет собой монокло-
нальную доброкачественную опухоль из
жировых клеток. В мутантных адипоцитах
имеется дефект регуляторного участка
фермента фосфофруктокиназы, в результате
чего активность этого энзима,
способствующего переходу углеводов в жиры, не
подлежит ингибированию избытком жирных
кислот. Адипоциты гипертрофируются.
Рядом авторов, в том числе, А.С. Фокиным
(1959) описаны гигантские липомы, весом
7-14 кг (см. т. III, гл. 3). Одновременно у
пациента бывает более 140 липом. Известна
семейная аутосомно-доминантная форма
множественного липоматоза, при которой
неинкапсулированные липомы либо
располагаются на шее в виде воротника {болезнь
Маделунга — тип I), либо захватывают
туловище и нижние и верхние конечности, щадя
их дистальные отделы (тип II). При типе
I липоматоз средостения может вызывать
нарушения вентиляции. При системном
липоматозе отмечаются пострецепторный
дефект в активации гормонозависимой
липазы, повышенная активность ЛПЛ и
222 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
изменения нервов пораженных областей.
Нередко больные наклонны к алкоголизму.
Существуют также тазовая и эпидуральная
формы липоматоза, последняя часто связана
с различными формами гиперкортицизма.
При болезни Деркума (adipositas dolorosa)
скопления жира диаметром 5-10 см
возникают в подкожной клетчатке живота, бёдер
и верхних конечностей и воспаляются.
Отмечается интерстициальный неврит.
Боль вызвана парестезиями и вялотекущим
воспалением. Некроза в узлах не отмечают.
Поскольку в узлах Деркума имеются
гранулёмы и гигантские клетки, в настоящее
время предполагают, что это хронический
иммунопатологический процесс по типу
ГЗТ, вероятно, спровоцированный ауто-
аллергией к компонентам жировой ткани,
либо инфекцией адипоцитов. Болезнь
Деркума свойственна пациентам с общим
ожирением. Она сопровождается
эмоциональной лабильностью, утомляемостью,
иногда — снижением интеллекта и
множественными дисфункциями эндокринных
желез, в частности, гипотирозом.
Обсудив патофизиологию ожирения и
местных форм накопления жира, хотелось
бы в заключение отметить важную роль
профилактики в борьбе с эпидемией тучности.
До тех пор, пока в массовом общественном
сознании полнота, покой и калорийное
питание будут ассоциироваться с
благополучием, трудно ожидать улучшения ситуации
с ожирением в богатых странах мира.
ИСТОЩЕНИЕ И КАХЕКСИЯ
Истощение (ср. англ. «inanition»)
характеризуется понижением жировых запасов
организма. При этом существенное повышение
смертности начинается при 20-25 %
дефиците индекса массы тела (ниже 20кг/м2).
Крайняя степень истощения, с потерей
более чем 50 % от нормальной массы тела
называется кахексией.
Кахексия может быть экзогенной — при
голодании. Этот тип кахексии подробно
чдокринно-метаболические нарушения)
рассмотрен в соответствующей части
книги выше. Ниже речь пойдёт об эндогенных
формах кахексии, которые разнообразны по
своей этиологии и патогенезу. Церебральная
или психогенная форма наблюдается при
нейрогенной анорексии— булимии. Это
заболевание, в патогенезе которого особенно
важна гиперпродукция кахексина и других
пептидов, угнетающих активность центров
голода и стимулирующих катаболизм, уже
было детально рассмотрено выше в
отдельном разделе книги. В последнее время
появились данные, что кахексии
препятствует действию эндотелиальной липопроте-
иновой липазы и переходу жирных кислот
в адипоциты.
Кахексия и афагия развиваются при
нарушении продукции нейропептида Y в
гипоталамических вентро-латеральных
ядрах. Это может быть результатом некроза
соответствующих клеток под действием
травм, опухолей, нарушений
кровообращения. При болезни Симмондса развивается
гипопитуитаризм с сочетанным поражением
вентро-латерального гипоталамуса. Это
ведет к анорексии и крайнему истощению
(см. рис. 8, с. 46). Отметим, что сам по себе
гипопитуитаризм, без гипоталамических
нарушений, к кахексии не ведет и может даже
сопровождаться ожирением. Известное
значение при гипоталамо-гипофизарной форме
кахексии имеет и вторичная надпочечни-
ковая недостаточность, развивающаяся у
таких больных.
При нейрогенной анорексии
гиперпродукция кахексина может считаться
первичной. Однако, так как этот цитокин
продуцируется во время
иммунопатологических реакций, ГЗТ и хронических гра-
нулёматозных процессов, лихорадок, при
активации противоопухолевого иммунитета
— то не удивительно, что все эти состояния
сопровождаются вторичной гиперкахекси-
немией, снижением аппетита, усилением
катаболизма и истощением жировых
запасов. Примерами могут служить раковая
кахексия, раневое и ожоговое истощение,
кахексия при туберкулёзе, некоторых
аутоиммунных болезнях. При онкологических
заболеваниях причины кахексии
множественны. Кроме кахексина обнаружен протеог-
ликан, синтезируемый в организме раковых
больных, который ускоряет расщепление
белка в мышцах и других тканях больных.
Какое-то значение придают и эффекту
метаболического обкрадывания организма
хозяина опухолевыми клонами, которые
значительно менее эффективно используют
энергоресурсы (см. т. III, гл. 3).
Установлено, что тканевые макрофаги
увеличивают продукцию кахексина и при
хронической венозной гиперемии. Именно
это служит одним из ведущих механизмов
кахексии при хронической сердечной
недостаточности. Дополнительными
факторами, усугубляющими истощение у таких
больных, считаются застой и нарушение
всасывания в кишечнике, нарушения
усвоения пищевых веществ в застойной печени,
рвота и анорексия при интоксикации
сердечными гликозидами, а также увеличение
основного обмена. Кахексии стимулирует
гипертрофию миокарда.
Важной причиной кахексии служит
гипоинсулинизм. При инсулинопеническом
сахарном диабете выпадают анаболические
функции инсулина в отношении липидов и
белков. Поэтому, истощение является
неотъемлемой составной частью клинической
картины этого недуга (см. ниже,
«Патофизиология сахарного диабета»). К исхуданию
приводят гиперглюкагонемия при глюка-
гономах и гиперсоматостатинемия — при
соматостатиномах. В обоих случаях
понижается продукция и возрастает расщепление
жиров в адипоцитах.
Крайнее истощение развивается при
хронической недостаточности функций
коркового вещества надпочечников {болезни
Аддисона), в связи с гипокортицизмом. При
этом имеет значение отсутствие липогенети-
ческого действия глюкокортикоидов.
Однако, отчасти снижение веса здесь обусловлено
и обезвоживанием, характерным для таких
больных из-за усиленных потерь натрия.
Кахексия присутствует при тяжелых
длительных поражениях ЖКТ, связанных
с мальабсорбцией. Отчасти, это вызвано на-
Нарущения липидного обмена 223
рушением усвоения пищи, кроме того имеет
значение повышение продукции кахексина
при инфекционных, онкологических и
аутоиммунных поражениях ЖКТ,
Комплексную природу имеет, вероятно,
истощение, развивающееся в результате
гипоплазии тимуса или
экспериментальной неонатальной тимэктомии («wasting
syndrome»). В его развитии усматривают
роль неидентифицированных
анаболических факторов тимического происхождения.
Вместе с тем, возникающий дри;этам;имму-
нодефицит, с его инфекционными
осложнениями, сам способен вызвать истощение.
Таким образом, в патогенезе
большинства форм истощения, не связанного с
экзогенным голоданием, в настоящее время
решающее значение придают не простому
дефициту калорий, а нарушениям
сигнальных взаимоотношений с участием кахексина
и других регуляторов.
Местные формы утраты
жировых запасов
Существуют также локальные и
региональные разновидности утраты жировых
запасов—липоатрофии. Среди них большой
интерес представляют болезнь Барраке-
ра-Симмондса, генерализованная липоат^
рофия, семейная аутосомно-доминантная
липодистрофия и некоторые другие местные
формы.
Утрата жировой ткани очагового
характера наступает и при остром некротгмеском
панникулите.
Генерализованная липоатрофия (липоат-
рофический диабет) характеризуется
приобретенной или наследственной аутосом-
но-рецессивной атрофией периферической
и висцеральной жировой ткани (щадится
только околосуставной, эпидуральный, рет-
роорбитальный, ладонный и подошвенный
жир, а при приобретенных формах —
лицевой). Болезнь сопровождается гиперлипо-
протеидемией V типа, стеатозом печени,
иногда вторичным ксантоматозом и всегда—
ицсулинорезистентным сахарным диабетом.
Приобретенные формы провоцируются
224 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
инфекциями, особенно часто мононуклео-
зом, корью и коклюшем, могут проявиться
при болезни фон Базедова и беременности.
Подобный набор провоцирующих
факторов и частое воспалительное начало
с симптомами панникулита заставляют
предположить роль аутоиммунного
поражения адипоцитов в патогенезе болезни, тем
более, что аутоантитела к жировым клеткам
обнаружены при болезни фон Базедова и
некоторых других предрасполагающих к
липоатрофии расстройствах. У больных
липодистрофией иногда фиксируются
высокие титры аутоантител к инсулиновым
рецепторам. Адипоциты теряют жир и
приобретают фибробластоподобный вид.
При генерализованной форме
предполагают наличие в крови липид-мобилизующих
факторов (возможно, это кахексии или
аутоантитела). Не исключен дефицит
естественного ингибитора гормонозависимой
липазы или выпадение липогенетического
действия инсулина. Блокада действия
инсулина носит комбинированный рецептор-
но-пострецепторный характер. Поскольку
инсулинорезистентность в печени и мышцах
выражена меньше, чем в жировой ткани,
кетоацидоз при данной аномалии
встречается редко.
При парциальных формах пересадка
жировой ткани со здоровых участков в
атрофичные приводит к их вовлечению в
атрофию, обратная трансплантация ведет
к прекращению потери триглицеридов в
аутотрансплантате. Это говорит о
заинтересованности аутокринных и паракринных
механизмов в патогенезе расстройства:
возможно, в пораженных участках действует
местный аутокоид-ингибитор липогенеза
или стимулятор липолиза. При данной
форме липодистрофии имеется ускорение
роста в препубертатном периоде, зависящее
от имитации эффектов инсулиноподобного
фактора роста I избытком инсулина. У
женщин наблюдаются гирсутизм, поликистоз
яичников, у лиц обоего пола часты
поражения почек и бывает снижен интеллект. Сте-
атоз печени может закончиться циррозом.
Регулярно отмечается неспецифический
кожный симптом инсулинорезистентнос-
ти acanthosis nigricans в виде бархатистых
темных пятен на коже. Интересно, что
очаговой липоатрофии способствует
подкожное введение инсулина при ИЗСДI, а ее
очаги нередко образуются именно в области
инъекций.
Болезнь Барракера-Симмондса
(приобретенная парциальная липодистрофия)
поражает, почти исключительно, женщин.
Заболевание характеризуется чаще всего
прогрессирующей нисходящей атрофией
жировой ткани, начиная с головы («голова
мертвеца») вниз, вплоть до охвата всей
верхней половины туловища. Жир на бёдрах и
ягодицах не только сохраняется, но и может
дополнительно откладываться. Болезнь,
скорее всего, носит иммунопатологический
характер, ибо при ней регулярно
обнаруживают дефицит факторов комплемента (С3), а
также нефритогенные аутоантитела класса
IgG против конвертазы его альтернативного
пути (см.т.1, стр.357). Заболевание может
развиваться на фоне склеродермии и других
аутоиммунных поражений соединительной
ткани. Одна из форм сочетается с
нарушениями формирования нижнечелюстной кости
и ключиц, задержкой физического и
психомоторного развития и алопецией.
Известны и другие локальные липоатро-
фические синдромы: аутосомно-доминант-
ная липоатрофия конечностей, сочетанная с
синдромом Ригера (пороки развития зубов и
глаз); опоясывающая абдоминальная
липоатрофия у грудных детей, циркулярная и
полуциркулярная липоатрофия конечностей и т.д.
Панникулит представляет собой острое
воспаление жировой клетчатки,
охватывающее очаги в пределах ее долек. Панникулит
протекает остро, заканчивается некрозом
жировой ткани, сопровождается иногда
лихорадкой, ответом острой фазы и лейкемоид-
ной реакцией. В развитии этого нарушения
большое значение имеет дефицит
ингибиторов протеаз, в частности, сц-антитрипсина
и, возможно, адипонектина. Панникулит
может наблюдаться в структуре системных
иммунопатологических и гематологических
расстройств с аутоантителами — коллаге-
Нарушения липидного обмена 225
нозов (особенно системной красной
волчанки и склеродермии), парапротеинемий
и лимфом. Ему способствуют панкреатит
(при котором появляются циркулирующие
липаза и фосфолипаза А2), гистиоцитоз
(при котором жировая ткань может
инфильтрироваться активными макрофагами).
Описаны случаи панникулита и без этих
системных болезней — спровоцированные
холодом, травмами, флегмоной
новорожденных и даже противокоревой вакцинацией.
Наиболее тяжелая форма некротического
панникулита — диссеминированный некроз
жировой ткани — наблюдается при остром
панкреатите и распадающихся опухолях
поджелудочной железы. Ее характерным
осложнением подчас служит гипокальци-
емия из-за связывания кальция мылами,
образующимися в разрушаемой жировой
клетчатке (см. ниже «Патофизиология пара-
щитовидных желез и кальциево-фосфорно-
магниевого обмена»).
Понятие панникулита не следует
путать с лжетермином щеллюлигп». Этим
отсутствующим в Международной
классификации болезней и некорректным, с
точки зрения общей патологии, словом в
профессиональном жаргоне косметологов
и среди пациентов иногда обозначают гипо-
тпироидную дермопатпию (или «гиноидноую
липодистпрофию» — см. А.Ю. Барановский,
Н.В.Ворохобина, 2007).
ЛИЗОСОМАЛЬНЫЕ БОЛЕЗНИ
НАКОПЛЕНИЯ ЛИПИДОВ
В отличие от ТГ другие липиды не
выполняют резервно-энергетических функций,
поэтому их накопление всегда отражает
замедление катаболизма и происходит при
тезаурисмозах — лизосомальных энзима-
тических дефектах (см. также т. I, стр. 141,
167). Известно более дюжины
наследственных лизосомальных болезней
накопления сфинголипидов и гликолипидов, а
также болезнь накопления редкой жирной
кислоты — фитановой. На рис. 42 пред-
глобозид
Cer-Glc-Gal-GalNAc
гексозаминидаза А и В
(болезнь Сандхоффа)
церамидтригексозид Cer—Glc-J-Gal—Gal
а-галактозидаза
(болезнь Фабри)
лактозилцерамид Cer—Glc—Gal
fi-глюкозидаза
(лактозилцерамидоз)
глюкоцереброзид Cer—Glc
$ глюкозидаза
(болезнь Гоше)
церамид
Cer—Glc—Gal—GalNAc—Gal ганглиозид Gmi
NANA
- + - $-галактозидаза
I (G М1-ганглиозидоз)
Cer—Glc—Gal—GalNAc ганглиозид GM2
J NANA
— гексозаминидаза А
(болезнь Тея-Сакса)
Cer-Glc-Gal
I
ганглиозид GMj
NANA
галакто-
цереброзид
— Cer-Gal-«-
сфингомиелиназа
(болезнь Ниманна-Пика)
с фигомиелин
сульфатид
-Cer-Gal-OS03H
Сег мг
\ $-глюкозидаза сульфатаза
-1- (болезнь (мета хроматине екая
Краббе) лейкодистрофия)
Cer-P-Cholin
Рис 42. Наследственные болезни накопления сфинголипидов. Р — фосфат, Сег — церамид,
Glc — глюкоза, Gal — галактоза, GalNAc — ацетилгалактозамин, GM — ганглиозиды, Cholin -
лин, NANA — ацетиламин. Пояснения в таблице 12 (по А. Трюбсбаху и соавт., 1979)
-хо-
226 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Болезнь
Тея-Сакса (Gm2-raHnw-
озидоз)
Gml-ганглиозидоз
Гоше (гликозилцерами-
доз)
Краббе (галактозилцера-
мидоз
Метахроматическая
лейкодистрофия (суль-
фатидоз)
Фабри (тригексозилце-
рамидоз
Доусона-Стейна (Лакто-
зилцерамидоз
Ниманна-Пика (сфинго-
миелиноз)
Зандхоффа (Gm2-raHr-
лио-зидоз, вариант
Фарбера (церамидоз)
Муколипидозы 11—IV III—
псевдосиндром Хюрлер
Рефсума (фитаноз)
Пораженный энзим
Гексозаминидаза А
р-галактозидаза
Р-глюкоцереброзидаза
Галактозилцерамид-3-галакто-
зидаза
Арилсульфатаза А или ее
белок-активатор 1
а-галактозидаза А
Лактозил-церамид-р-галакто-
зидаза
Сфингомиелиназа (при А и
В-подтипах), или неизвестный
этап ХН обмена при С-подтипе
Гексозаминидазы А и В (подтип
Тея-Сакса)
Церамидаза
УДФ-М-ацетил-глюкозамингли-
копротеин-фосфотрансфераза
(II, Ш) или ганглиозиднейрами-
нидаза (IV)
а-оксидаза р-метилированных
жирных кислот
Накапливаемый
липид
Спл2-ганглиозид
Gml-ганглиозид
гликозилцерамид
Галактозилцерамид
Галактозилсульфа-
тиды
Тригексозилцерамид
Лактозилцерамид
Сфингомиелин, ХН
Ст2-гаН-глиозид,
глобозид
Церамид
Гликолипиды
Фитановая к-та
Орган накопления
Мозг, сетчатка
Внутренние органы,
мозг, скелет, клетки
крови
Печень, селезенка,
лимфоузлы
Мозг
Мозг, почки
Почки, кожа, глаз,
сосуды
Печень, мозг, почки,
клетки крови
Селезенка, печень,
мозг
Мозг, почки
Кожа, суставы, глаз,
мозг J
Скелет, глаз, клетки
крови, мозг (IV)
Сердце, печень,
селезенка, нервы, глаз
ставлены метаболические блоки в обмене
сфинголипидов, возникающие при таких
нарушениях.
Большинство болезней аутосомно-рецес-
сивны (единственное исключение
представляет сцепленная с Х-хромосомой, но часто
проявляющаяся у женщин болезнь Фабри),
практически всегда поражение затрагивает
органы с наиболее интенсивным обменом
сфинголипидов и гликолипидов — мозг и
богатую макрофагами печень (селезёнку).
Краткая характеристика этих
заболеваний приведена ниже в табл. 12.
Для диагностики многих из этих
болезней используются методы биопсии
кожи, почек, печени и даже мозга, а также
изучение лейкоцитов на предмет поиска
специальных включений, образующихся
из-за накопления липидов во вторичных
лизосомах. При световой микроскопии
идентифицируются пенистые клетки
Нарушения липидного обмена 227
Таблица 12. Наследственные лизосомальные болезни накопления липидов
Поражение ЦНС
Тяжелая ЗПМР,
слепота судороги
Тоже
ЗПМР, атаксия при
ювенильной форме
ЗПМР, лейкодистро-
фия
То же, при взрослой
форме деменция и
психоз
Полинейропатия
ЗПМР, атаксия,
тремор, слепота
ЗПМР, атаксия,
судороги
ЗПМР, слепота,
судороги
Незначительное
умственное отставание
ЗПМР, при III
-минимальная
Полинейропатия,
атаксия
Гепагоспле-
номегалия
—
++++
++++
гиперспле-
низм
_
++
++++
—
+/-
-/+00-
(III. IV)
+
Глазные, скелетные,
гематологические с-мы
Вишневые пятна
сетчатки
Вишневые пятна
сетчатки, помутнение
роговицы, остеодиспла-
зия, пенистые клетки
Остеодисплазия,
пенистые клетки
Атрофия зрительного
нерва
Атрофия зрительного
нерва
Катаракта, помутнение
роговицы
Атрофия зрительного
нерва, пенистые клетки
Вишневые пятна
сетчатки, макулярная
дегенерация, пенистые
клетки
Вишневые пятна
сетчатки
Умеренная макулярная
дегенерация
Помутнение роговицы,
вакуолизация нейтрофи-
лов (II), лимфоцитов (III),
остеодисплазия (II, III)
Гемералопия,
пигментный ретинит, остеопатия
Специфические проявления
Макроцефалия, гиперакузия, поражает
евреев-ашкенази (частота гена 1/30)
Грубое лицо, макроглоссия. Мукополи-
сахаридурия
Патологические переломы,
псевдоостеомиелит, клетки Гоше, чаще бывает у
евреев-ашкенази (1/1000)
Раздражительность, повышение белка в
ЦСЖ, гигантские глиальные глобоидные
клетки в белом в-ве
Повышение белка в ЦСЖ
Гипогидроз, тромбофилитический
диатез, кожные ангиокератомы, суставные
боли
Повышена кислая фосфатаза
сыворотки, анемия
Голубые гистиоциты, коричневая
кожа, инфильтраты в легких, клетки
Ниманна-Пика в крови, поражает чаще
евреев-ашкенази
Макроцефалия, гиперакузия, поражает
евреев-ашкенази
Артропатия, повышение белка в ЦСЖ
Грубое лицо, включения в
культивируемых фибробластах (l-клетки), клапанные
пороки (III), чаще среди
евреев-ашкенази (IV)
Ихтиоз, повышение белка в ЦСЖ,
сердечная недостаточность. Хлорофилл
усугубляет болезнь
Ниманна-Пика в крови и Тея-Сакса — в
пораженных органах, 1-клетки — при му-
колипидозе 2-го типа и т.д.
Клетки Гоше — это гистиоциты ретикуло-
эндотелиальных органов с характерной
цитоплазмой в виде мятой папиросной бумаги.
Электронная микроскопия биоптатов
позволяет различать в растянутых лизосомах
пластинчатые включения в виде зеброидных
телец (при болезни Ниманна-Пика),
штабелей (при болезни Гоше), завитков (при
болезни Тея-Сакса). Об I-клетках шла речь
вт.1, с. 167.
Рекомбинантные технологии вносят
новые перспективы в дело лечения липид-
ных тезаурисмозов. В настоящее время
примерно за 300000 американских
долларов в год пациенты с болезнью Гоше
могут получать очищенную плацентарную
форму отсутствующего у них энзима (так
называемую аглюцеразу), что ведет к
значительному улучшению их состояния. Есть
228 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
надежда, что рекомбинан-
тный фермент окажется
несколько дешевле. И
все же, даже в богатейших
странах, профилактика
данных болезней
остается более реалистической
задачей здравоохранения,
чем их лечение. Для этого
применяют амниоцентез
плодов в группах высокого
риска. Так как выявление
гетерозигот, особенно для
самой распространенной
из этой группы — болезни
Тея-Сакса, представляет
наиболее эффективный
способ исключить
рождение больных детей, в
США была реализована
программа сплошного
скрининга популяции
высокого риска — евреев-
ашкенази— на предмет
носительства дефектных
аллелей. Скрининг выявил
в группе исключительно
высокую частоту
носительства — 1/30.
КЕТОЗ И СТЕАТОЗ
ПЕЧЕНИ
углеводы
жирные кислоты
аминокислоты
ацетил-КоА
ацето-ацетил КоА
ацетил-КоА
i
оксиметил-
глутарил-КоА
окисление
через цикл
трикарбоновых
кислот
синтез
жирных
кислот
образование
кетоновых
тел
синтез
холестерина
НАД НАДН
Н
СО,
7 S^- ч_/ ?Нз
ОН-С-СНз ► ОН=С-СН3 ^ > О-С
I -< I ~* I
сн2 сн2 сн3
I _ I _
соо соо
$ оксимасляная
кислота
ацетоацетат
ацетон
Рис. 43. Интеграция липидного, углеводного и белкового обмена
и метаболическая судьба ацетил-коэнзима А. Сокращения —
как в тексте. Внизу — кетоновые тела
Под нарушениями промежуточного обмена
липидов понимают, в первую очередь,
расстройство интеграции липидного и
углеводного метаболизма, проявляющееся в виде
синдрома кетоза.
Липидный и углеводный обмен, а также
превращения ряда аминокислот, у
млекопитающих тесно интегрированы (рис.43).
Поступающие в печень жирные кислоты
в сытом состоянии (при высоком уровне
цитрата и инсулин-глюкагоновом
соотношении) направляются, главным образом,
на ресинтез ТГ, а в голодном — при низком
уровне цитрата и инсулин-глюкагоновом
соотношении — становятся объектом
окисления.
Центральную роль в интеграции
различных видов обмена играет метаболическая
судьба общего универсального катаболита
всех этих веществ — ацетил-КоА (АКоА).
Печень вырабатывает постоянно
значительные количества АКоА путем окисления
глюкозы и жирных кислот, а также
дериватов аминокислот. Часть АКоА тратится на
энергетические нужды в цикле Кребса, для
чего необходимо достаточное количество
оксалоацетата и других кетокислот этого
метаболического пути. Другая часть может
переходить в малонил-КоА и жирные кис-
Нарушения липидного обмена 229
лоты, а также стероиды, если синтез этих
липидов обеспечен пластически (глицерин
из реакций гликолиза) и энергетически
(НАДФН2 из пентозного пути), и если
имеются в достатке витамины-коферменты
этих процессов. Значительное количество
АКоА переходит в печени в ацетоацетат,
который деацилируется и поступает в кровь,
так как свободно проходит через мембрану
гепатоцитов, которые его не утилизируют.
Существенная часть ацетоацетата
восстанавливается в нелетучую р-оксимасляную
кислоту, а небольшое количество декарбок-
силируется в летучий ацетон (рис. 43 — низ).
Вне печени, в лёгких — кетоновые тела могут
формироваться лишь в незначительной
степени (СМ. Лейтес, 1945). Ацетоацетат
и его метаболиты могут использоваться для
энергетических нужд нервной тканью, а
также мышцами, легкими и почками, особенно,
в условиях голодания. При беременности
они утилизируются плацентой и плодом.
Человек способен окислять ацетоацетат в
количествах до 35 мг/кг массы тела в час.
Таким образом, несмотря на значительную
продукцию кетоновых тел, в норме они
быстро используются, а их концентрация
в крови находится на динамически низком
уровне (обычно до 3 мг/дл, верхняя граница
нормы — 10 мг/дл или 1 MEq/л).
Следовательно, кетоновые тела — это нормальные
метаболиты. Они даже играют важную роль
интегрального звена метаболизма. Но их
содержание в крови не должно быть
большим. Если кетоновые тела накапливаются в
избытке (до 10 MEq/л), то существенные их
количества появляются в моче. Кетонемия,
кетонурия и связанные с ними расстройства
функций ЦНС, ЖКТ, кислотно-основного
и водно-солевого равновесия составляют
картину кетоацидоза. Классические
источники включают в понятие «кетоз» еще
и запах ацетона, присутствующего в
выдыхаемом воздухе. Однако установлено,
что здоровая легочная ткань утилизирует
недоокисленный ацетон и не пропускает
его в альвеолы. Таким образом, запах
ацетона при кетозе либо свидетельствует о
сопутствующих поражениях респираторной
системы у больного, либо исходит от пота,
мочи, других выделений — но не
собственно выдыхаемого воздуха. Поэтому, запах
ацетона для синдрома кетоза не обязателен
(например, при алкогольном кетозе он часто
отсутствует).
При тяжелой гиперкетонемии кетоновые
тела возрастают в концентрации в 10-20 раз
(до 1772-3544 мкмоль/л или 100-200 мг/дл
при норме 177,2 мкмоль/л или 10 мг/дл)
(Т.Содеман, С. Содеман, 1986).
Накопление кетоновых тел может быть
связано как с ускорением их образования,
так и с нарушением их утилизации.
Ускорение образования кетоновых тел
является результатом усиленного липоли-
за и поступления в печень избытка СЖК.
наружная
поверхность
внутренней
митохондриальной
мембраны
тиокиназа
НЭЖК —рт—^ацилКоА
АТФ АМФ КоА
КоА ФФН
Ж
внутренняя
поверхность
внутренней
митохондриальной
мембраны
митохондриальный
матрикс
карнитин -^^ I ^рг- ацил-КоА
ацил-карнитин ^SI ^n*. КоА ^^
^-окисление
карнитин-
пальмитиновая
ацилтрансфераза I
карнитин-
пальмитиновая
ацилтрансфераза II
Рис 44. Утилизация ацил-КоА в митохондриях и роль карнитина
230 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
СЖК в печени формируют ацил-КоА,
который может проникать внутрь
митохондрий при участии фермента карнитин-
пальмитоил-трансферазы 1. (КПТ 1) и
транслоказы (рис.44). Карнитинпальми-
тоил-трансфераза2 внутри митохондрий
освобождает карнитиновый челнок, и
ацил-коэнзим А подвергается р-окислению
до ацетил-коэнзима А — универсального
катаболита.
В нормальном состоянии КПТ-1
малоактивна. При избытке глюкагона и дефиците
инсулина кетокислоты, включая малонил-
коэнзим А, используются в реакциях глюко-
неогенеза, а их продукция из глюкозы падает.
При снижении концентрации малонил-ко-
энзима А он перестает ингибировать КПТ-1,
и ацилы жирных кислот направляются в
избытке в митохондрии. К тому же глюкагон
прямо активирует карнитин-ацилтрансфера-
зу 1 путем увеличения количества
доступного карнитина. Вследствие этого образуется
избыток ацетилкоэнзима А. Таким образом,
повышение активности глюкагона будет
способствовать кетозу (см. также ниже
«Патофизиология сахарного диабета»).
Прямого пути вовлечения АКоА в глюконе-
огенез у животных нет. Но при нормальном
метаболизме существуют
множественные альтернативные пути использования
АКоА:
>• АКоА может вовлекаться в реакцию
изоцитратной конденсации с оксалоаце-
татом и сгорать в цикле Кребса;
>• АКоА может участвовать в ресинтезе
жирных кислот и жиров ;
>* АКоА может участвовать в стероидоге-
незе;
>* наконец, АКоА может образовывать
кетоновые тела.
Инсулин способствует реализации всех
некетогенных путей использования АКоА.
Поэтому, пока продукция инсулина
адекватна, кетоза не бывает, даже при высоком
уровне глюкагона, например,
обусловленном глюкагономой.
При отсутствии инсулина все пути
использования избытка АКоА оказываются
неэффективными или блокированными за
исключением тех, которые ведут к кетозу.
Так как кетоновые тела — кислоты
умеренной силы, они связывают катионы натрия и
вызывают ацидоз (см. ниже
«Патофизиология водно-электролитного обмена»).
Более детально эти все аспекты кетоза
рассматриваются на конкретном примере
диабетического кетоацидоза ниже (гл. 9).
Таким образом, главная метаболическая
ситуация, приводящая к кетозу —
ускоренный распад липидов при ограниченной
метаболической доступности углеводов
и кетокислот цикла Кребса, особенно, в
условиях высокого соотношения глюка-
гон/инсулин.»
Поскольку избыток ацетил-КоА может
быть получен при метаболизме как липидов,
так и других веществ (кетогенной
аминокислоты лейцина, углеводов, нуклеотидов),
кетоацидоз возникает не только в результате
нарушений липидного обмена.
Причины кетоацидоза, специальные
формы которого рассмотрены в разных разделах
данной книги, могут быть различными и
включают:
- декомпенсированный инсулинзависи-
мый сахарный диабет (см.
«Патофизиология углеводного обмена»);
- голодание, которое вызывает, как
правило, умеренный кетоацидоз во II период
своего развития (см. выше специальный
раздел «Голодание»);
- жировую диету, обедненную
углеводами. При этом, как и в двух предыдущих
случаях, углеводы недоступны и резко
усилено окислительное использование
жиров. Это ведет к гиперпродукции
ацетоацетата. Организм может
адаптироваться к данной экстремальной диете
и не развивать кетоза, несмотря на
усиленное потребление липидов, если такая
диета вводится постепенно и существует
долго. Примером служит диета народов
Крайнего Севера, которые не развивают
кетоза, так как их мышцы и нервная
ткань способны окислять огромные
количества кетоновых тел. В случае
Нарушения липидного обмена 231
внезапного перехода на жировое питание
срабатывает старое правило М. Рубнера:
«Жиры горят в пламени углеводов» — то
есть отмечается недоокисление ацето-
ацетата и его недоокисленные продукты
циркулируют и выводятся в виде
кетоновых тел;
метилмалонилацидурию, которая
свойственна гиповитаминозам по кобаламину
и затрудняет утилизацию АКоА в мало-
нил-КоА (см. раздел «Патофизиология
витаминного обмена»);
аминоацидопатии с нарушением обмена
разветвленных аминокислот (см. раздел
«Патофизиология белкового обмена»);
алкогольное отравление, при котором
механизмы кетоза комбинированные.
Кетоацидоз наблюдается у
хронических алкоголиков, пораженных
алкогольной гепатопатией и панкреатитом,
после значительной выпивки, не
сопровождавшейся должной закуской.
Имеет место массированный липолиз,
под влиянием выброса гормонов
гипофиза, для которых алкоголь является
своего рода триггером. Вместе с тем,
максимально возможная продукция
инсулина и метаболические потенции
печени и других органов у таких
пациентов недостаточны для утилизации
всего образованного ацетоацетата, а
уровень глюкагона высок. В результате
накапливается избыток р-оксимасля-
ной кислоты, который и формирует
кетоз. Ацидозу способствует гиперлак-
тацидемия, возникающая из-за
восстановления пирувата в лактат избытком
НАДН, а также кислые метаболиты
самого алкоголя;
отравление ацетоном, хотя как причина
экзогенного кетоза оно встречается редко;
подагру и гиперурикемию (см.
«Патофизиология нуклеинового метаболизма»);
стресс, который способствует активации
липолиза и использованию кетогенных
аминокислот, может приводить к кетозу
только при относительной инсулиновой
недостаточности;
гликогеноз, особенно I типа — болезнь
Гирке, при которой недостаточно
освобождение глюкозы из печени в кровь и
компенсаторно усилено использование
липидов (см. «Патофизиология
углеводного обмена»);
- аммиачное отравление, например, при
печеночной и почечной недостаточности,
когда аммоний может нейтрализовать
кетокислоты цикла Кребса и прервать
утилизацию в нём АКоА (см.
«Патофизиология белкового обмена»).
Еще один аспект интермедиарного
метаболизма липидов, который уже затрагивался
при рассмотрении их транспорта — это
ретенция липидов на «центральной станции»
их метаболизма — в печени. В данном разделе
уместна будет краткая интегративная
характеристика стеатоза (ожирения) печени (см.
также т. I, с. 107,184 и раздел
«Патофизиология витаминного обмена» в данном томе).
Печень — орган, получающий и секре-
тирующий липидный материал
несколькими путями (см. выше рис. 31). В печень
поступают комплексы альбумин-жирные
кислоты, несущие липиды из жировой
ткани (до 1/3 суточной продукции СЖК).
Здесь же оказываются захваченные гепа-
тоцитами ЛПВП, содержащие
дренированные липиды из тканей.. Печень подвергает
конечному катаболизму ЛПНП и остатки
ХМ, а также метаболизирует ЛПОНП и
ЛППП плазмы крови. Наконец, коротко-
цепочечные жирные кислоты и жёлчные
кислоты, реабсорбированные в кишечнике,
поступают в печень в составе портальной
крови. Кроме того гепатоциты способны
сами синтезировать жирные кислоты (в
основном — насыщенные), глицерин, триг-
лицериды, фосфолипиды — из углеводов, а
при необходимости — и аминокислот. Для
эндогенного ХН печень является местом
синтеза более чем на 80%.
Представление о масштабах переработки липидов в
нормальной печени даёт такая цифра: 200
г СЖК ежесуточно усваивается печенью
здорового взрослого человека. В силу этого
в нормальной печени до 10 % массы
составляют липиды (причем 5 % приходится на
232 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
ТГ). Печень отдает липиды, секретируя их
в кровь, в составе ЛПОНП, возникающих
только в ней, а также экскретируя ХН, его
дериваты — жёлчные кислоты и фосфо-
липиды — с жёлчью в кишечник. Липиды
в печени могут окисляться нацело или до
ацетил-Ко А, причем транспорт кетоновых
тел из печени в другие органы (см. выше)
тоже можно считать путём утилизации
продуктов липидного метаболизма.
Если содержание ТГ в печени превышает
10 %, а гистологически жировые капли (хотя
бы малого диаметра) выявляются более
чем в половине гепатоцитов -то говорят о
стеатозе печени (синонимы: ожирение или
жировая трансформация печени). Термин
«жировая инфильтрация» является
устаревшим, так как показано, что накопление ТГ в
печени — результат не только инфильтрации
при усиленной мобилизации жира из депо
(экзоцеллюлярный механизм, по А.Н.
Лебедеву, 1883), но и следствие ускоренного
собственного синтеза, недостаточной
собственной утилизации, а главное — нарушения
правильной секреции (эндоцеллюлярный
механизм, по В.К. Линдеману, 1900).
Термин «жировое перерождение» относится к
ситуации, когда в строме печени появляются
истинные адипоциты.
Любое ожирение печени — проблема
баланса, создаваемого на путях поступления
липидов в орган и их утилизации/секреции.
Лимитирующим фактором в достижении
такого баланса служит доступность
энергоресурсов и метильных донаторов,
необходимых для синтеза ЛПОНП в гепатоцитах.
Эпидемиологическая пораженность сте-
атозом печени очень велика. Немецкий
патологоанатом X. Фишер (1967), по данным
аутопсий клинически здоровых мужчин,
погибших от случайных причин, оценивает
ее почти в 10 %. В клинике, биопсии печени
свидетельствуют о ее жировой
трансформации у 10-38% больных, причем, наиболее
часто гепатостеатоз сопровождает
проявления «метаболического синдрома»,
включающего атеросклероз, ГЛП, общее (чаще,
андроидное) ожирение, сахарный диабет,
гиперурикемию, гипертензию (см. выше рис.
чдокринно-метаболические нарушения)
35 и гл. 14). Уже есть данные об общей
патогенетической основе всех этих нарушений,
при каждом из которых отмечается не
только изменение липидного метаболизма, но,
как уже отмечалось, и системное понижение
функциональных возможностей водород-
натриевого противопереносчика клеточных
мембран, а часто - и дефицит адипонектина.
Следует отметить и другой важный фактор
риска. Не только при алкоголизме, но даже
при умеренном употреблении алкоголя,
наблюдается обратимая
микровезикулярная жировая трансформация гепатоцитов.
Голодание, особенно, квашиоркор,
сопровождается стеатозом печени (см. выше
«Голодание»). Наконец, количество ядов и
лекарств, способных вызвать стеатоз
печени, исчисляется десятками, и даже первая
экспериментальная модель этого синдрома
была получена А.Н. Лебедевым при
отравлении собак желтым фосфором.
Таким образом, среди причин стеатоза
печени, по убывающей частоте, фигурируют
сахарный диабет, ожирение, ГЛП,
злоупотребление алкоголем, другие отравления,
голодание и гиповитаминозы, беременность,
а также изредка — наследственные дефекты
окисления жирных кислот (последнее
относится к стеатозу печени, отмеченному на
вскрытии при синдроме внезапой смерти
младенцев).
Выделяют острый и хронический
стеатоз печени. Ожирение печени может быть
микровезикулярным — когда липидные
капли придают гепатоциту пенистый вид,
но не смещают ядро (например, в I стадии
алкогольной гепатопатии); а также макро-
везикулярным — когда капли, по размеру
сопоставимые с ядром, смещают его.
Микровезикулярное ожирение считается менее
глубокой стадией, макровезикулярное
также обратимо, но при персистировании
вызвавших его причин, по существу, может
протекать как форма жирового некробиоза,
с гибелью гепатоцитов, их фагоцитозом,
активацией макрофагов и формированием
фиброза и даже цирроза органа (как это
происходит во II—III стадии алкогольной
гепатопатии).
Нарушения липидного обмена 233
Патогенез ожирения печени может быть
связан преимущественно с увеличением
поступления липидов в гепатоциты,
снижением темпов сборки и секреции ЛПОНП,
торможением гепатоцитарного окисления
липидов, а также (чаще всего)
комбинацией этих факторов. Характерно, что при
преобладании ретенционных механизмов
стеатоза печени содержание ЛПОНП в
плазме невысокое или пониженное, тогда
как при доминировании инфильтрационных
механизмов оно повышается.
> Увеличение поступления липидов в
печень имеет место при ГЛП, особенно, I,
IV и V типов. Этот механизм
присутствует и при быстрой утрате жировых
запасов, например, в результате общего или
углеводного голодания, а также при
ожирении. При инсулинзависимом диабете
липолиз в жировой ткани превышает
липогенез в силу отсутствия инсулина.
При инсулиннезависимом диабете поток
СЖК в печень возрастает из-за гиперг-
люкагонемии. Экстренная мобилизация
СЖК из депо свойственна действию
многих регуляторов— АДГ и оксито-
цина, гормонов аденогипофиза, коры
и мозгового вещества надпочечников,
гонад, щитовидной железы. Поэтому,
она происходит при стрессе, адаптации
к холоду, физической нагрузке, половом
акте, активном росте — например, в
организме беременных. Острое
микровезикулярное ожирение печени наблюдается
в последнем триместре беременности у
больных с преэклампсией.
Установлено, что даже однократный
прием алкоголя способствует залповому
освобождению многих из
вышеперечисленных липид-мобилизующих факторов.
Этот «эффект спускового крючка» ведет к
дополнительному притоку СЖК в печень
как фактору патогенеза алкогольного
ожирения печени.
> Снижение утилизации липидов в печени
происходит как результат торможения
окисления СЖК, нарушения сборки
частиц ЛПОНП и их секреции в кровь. Этот
механизм стеатоза является ведущим
для жировой трансформации печени
при разнообразных интоксикациях. Если
действует токсин в его нативном
состоянии — наиболее тяжело поражаются
гепатоциты на периферии долек, а при
влиянии продуктов биодеградации ядов
поражение может раньше отмечаться
во внутренней, центрипетальной зоне
дольки.
Гепатотропные яды и их метаболиты
могут нарушать работу карнитинового
челнока, обеспечивающего окисление
СЖК в митохондриях печени. Именно так
действует токсин гипоглицин А -
компонент незрелых плодов Карибского дерева
акии. Метаболиты гипоглицина связывают
необратимо карнитин и АКоА и
останавливают окисление жирных кислот, приводя к
кетозу, ацетонемической рвоте и острому
микровезикулярному стеатозу печени
(«ямайская рвотная болезнь»).
Гепатотропные яды также способны давать
активные кислородные радикалы, вызывающие
нероксидацию липидов. Это мешает
нормальной сборке частиц ЛПОНП. В связи со
своим прооксидаитным действием, стеатоз
печени вызывают хлороформ, дихлорэтан,
четырёххлористый углерод, желтый
фосфор, мышьяк — что следует учитывать в
профпатологии. По-видимому, сходным
образом влияет и йодсодержащий амиода-
рон. Многие лекарства, дающие в печени
эпоксидные и эпоксидные метаболиты, при
своей биотрансформации также способны
провоцировать стеатоз печени. В последние
годы большое внимание привлекает синдром
Рея — острая жировая трансформация
печени, почек и миокарда с печеночной
недостаточностью и энцефалопатией. Синдром
развивается у детей на почве применения
салицилатов и парацетамола, особенно при
вирусных инфекциях — гриппе и ветряной
оспе — и даёт почти 50 % летальность. У
больных отмечается замедленное
разрушение салицилатов и блокада митохондрий ге-
патоцитов. Описаны спорадические случаи
криптогенного синдрома Рея без
применения вышеназванных лекарств.
234 А.Ш. Зайчик, Л.П.Чурилов Патохимия
Угнетение синтеза белка в печени
токсинами может вести к дефициту апопротеинов
и отсутствию основы для сборки ЛПОНП.
Токсин бледной поганки, а-аманитин,
блокируя синтез м-РНК и белка, также приводит
к стеатозу и некрозу гепатоцитов. Острое
ожирение печени в прошлом было
характерным осложнением терапии высокими
дозами тетрациклина, нарушающего синтез
белка. Другие лекарства, опасные по
стеатозу печени: вальпроевая кислота, витамин
РР, метотрексат и иные цитостатики.
Способы их действия различны и могут включать
комбинации вышеназванных вариантов
нарушения использования ТГ, а для витамина
РР действует механизм акцепции метилов,
рассмотренный ниже (см. «Патофизиология
витаминного обмена»). При передозировке
глюкокортикоидов (равно как и при других
случаях гиперкортицизма) возможны
усиление синтеза ТГ в печени и ее макровези-
кулярный стеатоз.
Ранее алкоголь также относили к числу
ядов, формирующих токсический стеатоз
печени. Однако, этиловый спирт, как агент,
вызывающий стеатоз печени, обладает
меньшей прямой токсичностью, чем многие
вышеперечисленные яды. По современным
данным, его действие — комплексное и
включает, кроме непосредственного
токсического понижения утилизации АКоА в
цикле Кребса, еще и другие механизмы:
>- массированную мобилизацию жира из
депо (см. выше);
>• «экономию» жирных кислот из-за
калорической ценности алкоголя, который
даёт при окислении большое количество
энергии (7,1 ккал/г);
>- поражение промежуточных филаментов
цитоскелета (внутриклеточный
«алкогольный гиалин» или тельца Маллори) и
последующее нарушение
внутриклеточного транспорта и секреции ЛПОНП;
>* увеличение вязкости липидных мембран
и монослоёв, затрудняющее сборку
частиц ЛПОНП;
>■ насыщение гепатоцита
восстановительными эквивалентами НАДН в связи с
чдокринно-метаболические нарушения)
активной деятельностью алкогольдегид-
рогеназы и ацетальдегиддегидрогеназыи
сдвиг редокс-состояния клеток в сторону,
благоприятствующую синтезу липидов;
>- провокацию аутоиммунных процессов,
поражающих гепатоциты;
>- генерацию активных перекисных
метаболитов, повреждающих ЛПОНП и
гепатоцит;
>■ последствия алкогольного дефицита
липотропных витаминов В6и В12.
По вышеназванным причинам,
употребление более чем 80 г этанола в день опасно, с
точки зрения развития стеатоза печени. При
приеме добровольцами 160 г этанола
ежедневно, на 21 день биопсия печени выявила
жировую трансформацию данного органа у
всех этих мужественных подвижников науки.
Так как повседневный прием алкоголя
очень распространен, он является главным
экзогенным токсическим агентом,
вызывающим жировую трансформацию печени.
Существуют представления о стеатозе
печени в результате эндогенной
интоксикации. Старые авторы при этом ссылались на
стеатоз печени у септических и чахоточных
больных. Достоверно известно, что
производные гуаиидина при уремии, будучи
метальными акцепторами, нарушают
окисление жирных кислот и сборку ЛПОНП в
печени и, действительно, вызывают ее
стеатоз по механизму эндогенного отравления.
Для утилизации СЖК при участии
содержащего три метальные группы карнитина, а
также для сборки ЛПОНП, фосфолипиды
которых содержат холин, требуется
адекватное обеспечение метальными остатками.
Все вещества, являющиеся донаторами
метальных групп будут, следовательно,
способствовать секреции ЛПОНП и
окислению СЖК в печени. Наоборот, акцепторы
метилов могут понижать скорость этих
процессов. Метальные донаторы объединяются
под условным названием «липотропные
вещества», а акцепторы известны как «анти-
липотропные факторы». Главный липотроп-
ный фактор — холин, который сам входит
в состав фосфолипидов. Все агенты, спо-
собствующие прямо или косвенно синтезу
холина, в том числе — прямые поставщики
метилов, тоже липотропны (бетаин, мети-
онин, и, в меньшей степени — карнитин,
витамин В12, фолацин и витамин В6). Липот-
ропными считаются также инозит,
структурный компонент инозит-фосфатидов и
треонин, участвующий в метаболизме ме-
тионина, хотя в них и нет метильных групп.
Холин, рассматривавшийся ранее как
витамин группы В, ныне исключен из группы
витаминов, так как выполняет пластическую
функцию, требуется человеку в макродозах,
и в связи с тем, что его нехватка в диете не
дает патологических симптомов, если нет
сопутствующего дефицита белка. Однако,
это важное витаминоподобное вещество,
основной метальный донатор и компонент
фосфатидилхолина. У крыс сочетанный
дефицит холина и незаменимых
аминокислот ведет не только к ожирению печени, но
также к геморрагической нефропатии и к
анемии.
Холин изобилует в яичном желтке, мясе,
икре, рыбе и, особенно печей и, мозге и
других субпродуктах. Некоторые растительные
масла (соевое) тоже служат его богатым
источником. Метионин (известный в форме
метилметионина как «витамин £/») имеется
в достатке в мясо-молочных продуктах,
особенно, твороге, а также капустном соке.
Выводы об оптимальном составе закуски
при профилактике алкогольного стеатоза
печени очевидны.
Наиболее активными антилипотропными
факторами служат антиметаболит мети-
онина — этионин, никотиновая кислота и
гуанидинуксусная кислота. Последняя, как
уже говорилось выше, накапливается при
хронической почечной недостаточности.
Окислению жирных кислот в печени (но
не экскреции ЛПОНП) способствует также
пантотеновая кислота.
Дополнительные сведения о липотропных
и антилипотропных факторах представлены
также в разделе «Патофизиология
витаминного обмена». Таким образом, при общем и
белковом голодании, недостаточности
незаменимых аминокислот и гиповитаминозах,
Нарушения липидного обмена 235
а также при передозировке витамина РР
важным фактором, приводящим к стеатозу
печени, служит дефицит липотропных и
избыток антилипотропных влияний.
В 1930 г. Л. Драгштедт и в 1951 г.
Г.Л. Шрейберг показали, что клетки
выводных протоков поджелудочной железы
содержат вещество, которое оказывает ли-
потропное действие на печень. Это начало
именовали «липокаическим», а затем, по
инициативе СМ. Лейтеса, — «липокаином».
Было показано, что активный компонент
экстрактов имеет пептидную природу, но
липокаин, рассматривавшийся как гормон
экзогенной части панкреатической железы,
так никогда и не был выделен в чистом
виде, хотя его существование до сих пор
признаётся многими авторами.
Предполагается, что дефицит липокаина может
иметь важное значение в развитии стеатоза
печени при поражениях экзокринной части
поджелудочной железы, в связи с чем о нем
упоминают практически все отечественные
руководства.
Липотропные факторы применяются в
терапии и профилактике различных форм
ожирения печени, а антилипотропные — для
купирования гиперлипопротеидемий по
ЛПОНП.
Хроническое ожирение печени от любой
причины приводит к гепатомегалии, может
сопровождаться патологическими
печёночными пробами (бромсульфалеиновой, реже
тимоловой). Увеличивается активность
глютамико-пировиноградной трансами-
назы, щелочной фосфатазы. Вместе с тем,
даже далеко зашедшее ожирение печени
обратимо, может протекать бессимптомно и
представляет сравнительно благоприятный
вариант некробиоза гепатоцитов.
Однако, стеатоз может заканчиваться гибелью
печеночных клеток, а во многих случаях
протекает параллельно и во взаимодействии
с более агрессивными формами поражения
этого органа, чреватыми выраженным
некрозом и фиброзом (пример — алкогольная
гепатопатия).
Острое ожирение печени часто сочетается
с массированным некрозом ее паренхимы
236 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
и острой печеночно-клеточнои
недостаточностью (пример — упомянутый выше
синдром Рея).
Нарушения липидного обмена — главная
метаболическая причина смертности
населения. Поэтому предложено значительное
количество дорогостоящих способов лекарс-
твенной терапии и даже метаболической
хирургии для их коррекции. Расширяется
практика операций еюноилеошунтирова-
ния, мегалипосакции и
предпринимаются даже пересадки печени при тяжелых
гиперлипопротеидемиях, атеросклерозе
и морбидном ожирении (Л.В. Лебедев,
Ю.ИСедлецкий, 1987). Вместе с^тем,
поскольку имеется тесная субстратная связь
липидного и углеводного обмена, общие
гены, контролирующие как массу тела, так
и эффективность инсулиновой регуляции
(например, недавно открытый ген FTO,
дупликации которого предопределяют
наклонность к ожирению и инсулинорезистен-
тности), поскольку эндокринная функция
жировой ткани предусматривает контроль
метаболизма углеводов липоцитарными
гормонами и адипокинами — патология
липидного обмена не может рассматриваться
в отрыве от нарушений углеводного.
Интермедиарный обмен липидов и
углеводов тесно переплетён. Это делает
логичным, обсудив механизмы кетоза и жировой
трансформации, перейти к следующей главе
книги, посвященной патологии углеводного
метаболизма.
Глава 8
ПАТОФИЗИОЛОГИЯ
УГЛЕВОДНОГО ОБМЕНА
(Написано при участии В.И. Утехина)
«Едва ли можно без вреда для всяких тканей тела лишить человека
доставки хотя бы небольшого количества углеводов»
В.В. Пашутин
Курс общей и экспериментальной патологии. Т. II, (1902)
• Углеводы пищи и организма. Пищевой углеводный дисбаланс
• Переваривание и всасывание углеводов. Поступление глюкозы в клетку
• Нарушения всасывания и первичного преобразования углеводов
• Катаболизм углеводов и его нарушения
• Анаболизм углеводов и его нарушения
• Особенности обмена углеводов в различных тканях
• Гликогенозы иагликогенозы
• Гликозаминогликаны и мукополисахаридозы
УГЛЕВОДЫ ПИЩИ
МОРГАНИЗМА.
ПИЩЕВОЙ УГЛЕВОДНЫЙ
ДИСБАЛАНС
Углеводы — основные поставщики энергии
в питании человека. Как правило, на их
долю приходится более 50 % калорийности
и почти 3/4 веса суточного рациона.
Углеводы, потребляемые с пищей, представлены
главным образом крахмалом (резервный
полисахарид растений, подобный по структуре
гликогену животных). Гликоген живЪтных
продуктов имеет в питании сравнительно
малое значение, однако в печени на него
приходится до 20 % сырого веса. В рационе
«цивилизованного» общества широко
представлена сахароза — дисахарид, состоящий
из глюкозы и фруктозы. Важным
компонентом молочных продуктов является лактоза,
составляющая в них до 5 %. В меде и фруктах
содержатся небольшие количества глюкозы
и фруктозы. В винограде и хурме фруктозой
и глюкозой представлены все сахаристые
вещества. Мальтозу, мало встречающуюся в
природе, поставляют продукты, где крахмал
частично гидролизован, например солод
и пиво. Выше уже шла речь о роли
пищевых волокон (см. «Нарушения липидного
обмена») и пектиновых веществ, которые
необходимы для поддержания эубактериоза
кишечника, участвуют в связывании и
нейтрализации ксенобиотиков и тоже
представлены углеводами (полисахаридами).
В составе гликогена клетки организма
запасают некоторое количество энергии (не
менее 500 г этого полисахарида или более
2 000 ккал) в качестве быстромобилизуе-
мого резерва, необходимого в промежутках
между поступлением пищи. Не будь этой
формы резервирования глюкозы — ее
накопление в клетке приводило бы к гиперо-
смолярности цитоплазмы. Однако гликоген
238 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
быстро обновляется. В связи с оперативным
расходованием углеводов потребность в
них особенно возрастает при физических
нагрузках.
Нельзя недооценивать и структурную
роль углеводов. Основу межклеточного
вещества соединительной ткани, наиболее
широко представленной в организме
субстанции, составляют гликозаминогликаны.
Сотни белков человека, в том числе энзимы,
транспортёры, гормоны являются гликопро-
теидами. Триозы необходимы для
производства липидов, а пентозы — компоненты
нуклеиновых кислот. Углеводы — часть
антигенной среды организма, поскольку
ими представлены полисахаридные
антигены (например, эритроцитарные агглю-
тиногены), а также в связи с их участием
в построении антигенов гликолипидных
и гликопептидных. Комплекс Гольджи и
внутриклеточные транспортные системы,
согласно гипотезе Г. Блёбеля, используют
для маркировки и сортировки молекул
полисахаридный код (см. т. I, стр. 164).
Большое значение при местных типовых
патологических процессах имеют
полисахаридные медиаторы воспаления, носители
важнейших противовоспалительных и анти-
тромбогенных свойств (т. I, стр. 360). Глю-
курониды являются важной формой дето-
ксикации эндогенных ядов и ксенобиотиков.
Таким образом, данный класс органических
веществ, впервые определённых тартусским
химиком К.Э. Шмидтом как «соединения,
содержащие кроме углерода также кислород
с водородом в пропорциях молекул воды»
(1844), участвует во всех многообразных
аспектах метаболизма, не являясь просто
«клеточными дровами».
Формально углеводы не считаются
незаменимыми компонентами пищи. У
приматов единственное производное углеводов,
которое должно вводиться извне, витамин С
или аскорбиновая кислота (см. ниже раздел
«Патофизиология витаминного обмена»).
Остальные углеводы могут возникать из
липидов и аминокислот. Но этот процесс
сопровождается повышением генерации
кислых эквивалентов и азотистых
продуктов распада, которые, по выражению
В.В. Пашутина, «далеко не индифферентны,
если появляются в большом количестве»,
поэтому только при доступности пищевых
углеводов метаболизм протекает
оптимально. Еще Н.Б. Медведева (1941)
экспериментально показала, что голодный кетоацидоз
у человека исчезает при введении 50 г
сахара. Следовательно, можно говорить о
пищевой углеводной недостаточности. При
исключении углеводов из диеты возникает
тенденция к алиментарной гипогликемии,
компенсируемая усилением глюконеоге-
неза. Это может приводить к кетозу и
ускоренному расходованию белка. Ослабляется
образование печенью парных соединений
с участием глюкуроновой кислоты, что
снижает антитоксическую резистентность.
Наоборот, введение углеводов в организм
неспецифически улучшает детоксикацион-
ную функцию печени. Стресс, как процесс,
обеспечивающий повышение устойчивости
жизненно важных органов и тканей к острой
гипоксии, в значительной мере основан на
мобилизации и ускоренном транспорте в эти
ткани и органы глюкозы. Все эти аспекты
имеются в виду, когда говорят о пищевой
роли углеводов, как строго нормируемых
веществ (П.Е. Ко л маков, М.Н. Логаткин,
1974). Считается, что сбалансированная
диета взрослого должна содержать около
124 г углеводов на 1000 ккал рациона в
сутки (К.С. Петровский, 1975). При этом
желательно, чтобы легкоусвояемые дисаха-
риды составляли не более 25 % этого
количества. Переедание легкоусвояемых
углеводов — важный фактор риска ожирения и
атеросклероза (см. «Нарушения липидного
обмена»). Если рацион перегружен ими (что
нередко случается с диетой современного
человека из-за так называемой «пищевой
реакции на стресс»), то это ведет к
вторичной относительной недостаточности ряда
расходуемых при утилизации углеводов
витаминов (Bj, B2, PP, липоевой кислоты),
к повышению потребностей в белке и
некоторых микроэлементах (Mn, Mg, Mo, Fe).
Эти последствия перегрузки углеводами
рассмотрены подробнее в разделах, посвя-
щенных патологии витаминного и
минерального обмена.
Ниже вначале обсуждаются
метаболические пути углеводного обмена, а также его
нарушения, вызванные изолированными
ферментативными дефектами.
Затем представлен отдельный раздел,
посвященный главному интегральному
нарушению регуляции углеводного и всех других
видов метаболизма — сахарному диабету (СД).
ПЕРЕВАРИВАНИЕ
И ВСАСЫВАНИЕ УГЛЕВОДОВ.
ПОСТУПЛЕНИЕ ГЛЮКОЗЫ
В КЛЕТКУ
Полисахариды пищи перевариваются
амилазой слюны (которая, как это ни странно,
полностью отсутствует у некоторых близких
родичей человека, например бабуинов).
В желудочном соке нет амилолитической
активности, поэтому на данном этапе
пищеварения продолжает действовать внутри
пищевого комка лишь слюнная а-амилаза.
Возможен также незначительный
кислотный гидролиз крахмала. Основная угле-
водпереваривающая активность в ЖКТ
представлена панкреатической а-амилазой.
В результате в кишечнике оказывается смесь
мальтозы, изомальтозы и глюкозы. При
панкреатической недостаточности этот
процесс страдает и в фекалиях обнаруживаются
в значительных количествах гранулы
крахмала. Амилаза атакует только а-1,4-связи,
поэтому целлюлоза и растительные пенто-
заны остаются незатронутыми гидролизом
и попадают в толстый кишечник. Там под
действием бактериальных ферментов они
частично деградируют с образованием
органических кислот, спиртов и С02. Продукты
их бактериального расщепления важны как
стимуляторы перистальтики. Олигосахари-
ды и дисахариды завершают свой гидролиз в
тонком кишечнике (вплоть до
проксимальной части подвздошной кишки),
подвергаясь действию олигосахаридаз (сахараза,
Патофизиология углеводного обмена 239
мальтаза, изомальтаза, лактаза, гетерога-
лактозидаза, р-галактозидаза, трегалаза).
Олигосахаридазы — ферменты мембранного
и внутриклеточного пищеварения. Их
активность представлена в щеточной кайме
и внутри энтероцитов. Недостаточность
отдельных дисахаридаз, как и нередко
встречающаяся недостаточность всего изомаль-
тазно-сахаридазного комплекса, является
важной причиной синдрома мальабсорбции
(см. ниже). Интересно что при ответе острой
фазы, в частности, на фоне лихорадки,
происходит снижение интенсивности
мембранного переваривания олигосахаридов, так как
некоторые цитокины угнетают экспрессию
дисахаридаз. Нормальные моносахаридные
продукты работы олигосахаридаз —
галактоза, глюкоза, фруктоза и, менее активно, ман-
ноза, ксилоза и арабиноза — всасываются в
кровь. Для галактозы, фруктозы и глюкозы
действует система активного ко-транспорта.
Этот ко-транспорт — натрий-зависимый.
Ион натрия входит в энтероцит по
градиенту, обеспечивая всасывание моносахаридной
молекулы против градиента.
Калий-натриевый насос, тратя АТФ, восстанавливает
натриево-калиевый градиент. Аналогичный
ко-транспорт против градиента существует
и в почках. Благодаря такому механизму
клетки, выстилающие просвет кишечника и
почек, способны поглощать глюкозу и
другие моносахариды даже при очень низкой их
концентрации в химусе и моче
соответственно. Остальные моносахариды всасываются
под действием особой системы облегченного
транспорта, которая может подключаться
и к абсорбции вышеназванных трех
моносахаридов, если их количества в просвете
кишки велики. Многие яды нарушают
процесс натрий-зависимого активного вса->
сывания моносахаридов, что используется
при моделировании различных состояний
в экспериментальной патологии.
Флоридзин — ядовитый гликозид —
ингибитор всасывания Сахаров. Под его
действием глюкоза и другие сахара перестают
абсорбироваться в кишечнике и почке, но
транспорт натрия не прекращается.
Флоридзин применяют для воспроизведения
240 А.Ш. Зайчик, Л.П.Чурилов Патохимия
на животных глюкозурии. Так как при
этом нарушается поступление глюкозы в
клетки, модель иногда неточно называют
«флоридзиновый диабет». Однако глико-
зид не повреждает островки Лангерганса
и не вызывает инсулинорезистентности,
то есть воспроизводит симптомы, а не
болезнь. Правильнее считать, что фло-
ридзином моделируются эссенциальная
почечная глюкозурия и глюкозо-галактоз-
ная мальабсорбция. Оба эти редких ауто-
сомно-рецессивных заболевания описаны
у человека.
Эссенциальная почечная глюкозурия
имеет немалое клиническое значение, так
как имитирует ряд симптомов СД: наличие
восстанавливающих моносахаридов в моче,
полиурию, полидипсию. Причина этого
нарушения — мутация гена натрий-зависимого
флоридзин-чувствительного переносчика
моносахаридов в хромосоме 6 и резкое
понижение почечного порога реабсорбции для
глюкозы. По Н.Г. Левински (1994), в норме
глюкоза полностью реабсорбируется, если
ее концентрация в плазме до 10 мМ/л или
200мг/дл. По критериям Марбла, почечная
глюкозурия отличается от диабетической,
так как не сопровождается гипергликемией,
носит постоянный характер, не завися от
диеты, протекает на фоне нормальной или
уплощенной сахарной кривой и отсутствия
других метаболических нарушений. Одна из
форм данной аномалии (тип А) ограничена
почечным поражением, а при другой (тип
В) отмечается также кишечная
мальабсорбция глюкозы и галактозы (см. следующий
раздел).
Уабаин — гликозид наперстянки, ингиби-
рующий работу калий-натриевого насоса.
В связи с блокадой транспорта
одновалентных катионов и перенос глюкозы становится
невозможным.
После всасывания в кровь во всех
процессах, приводящих к поступлению глюкозы в
клетки, решающую роль играют белки —
переносчики глюкозы. Переносчики глюкозы
необходимы, поскольку молекула глюкозы
гидрофильна, а плазматическая мембрана
гидрофобна.
чдокринно-метаболические нарушения)
Первый переносчик был выделен из
эритроцитов человека. Он представляет собой
полипептидную цепь из 492
аминокислотных остатков, которые организованы в 25
сегментов:
>- 13 сегментов, по преимуществу,
гидрофильны (располагаются внутри и вне
клетки);
>• 12 сегментов, преимущественно, гидро-
фобны (обладают сродством к липидно-
му компоненту, располагаются внутри
плазматической мембраны).
Полипептидная цепь белка изгибается
зигзагом, пересекая мембрану 12 раз.
Белок-переносчик существует в двух
формах: одна связывает глюкозу на
внеклеточной стороне мембраны, другая — на
внутриклеточной. Выделяют четыре этапа
транспорта глкжозы через мембрану:
>• молекула глюкозы присоединяется к
связывающему участку, обращенному
наружу;
>■ конформация образовавшегося
комплекса переносчик-глюкоза изменяется
таким образом, что молекула глюкозы
оказывается в связывающем участке,
который обращен внутрь клетки;
>- глюкоза отделяется от переносчика,
переходя в содержимое клетки;
>• освободившийся переносчик принимает
конформацию, в которой участок,
связывающий глюкозу, обращен наружу
(возвращается в исходную форму).
В итоге: конформационные изменения
исключают контакт вне- и внутриклеточного
отсеков клетки, то есть глюкоза
переносится, а пора — «закрыта».
Считается, что подобный модус
организации белка-переносчика обусловлен
размерным сходством глюкозы и некоторых
ионов, в частности натрия, и направлен на
предотвращение внутриклеточного
проникновения ионов вместе с глюкозой при
ее транспортировке по градиенту
концентраций.
Переносчики глюкозы обозначаются
«GluT» и пронумерованы по порядку их
Патофизиология углеводного обмена 241
обнаружения. На сегодняшний день
известно пять переносчиков глюкозы. Эти
пять белков приблизительно на 50 % своей
структуры имеют:
> сходные аминокислотные
последовательности;
> складчатую организацию внутри
плазматической мембраны.
Все пять осуществляют трансмембранный
перенос глюкозы в тканях, по ее
концентрационному градиенту.
- GluT 1 содержится в больших
количествах в эндотелии, выстилающем
кровеносные сосуды гематоэнцефаличес-
кого барьера. Он служит
для обеспечения
стабильного потока глюкозы в
мозг, нуждающийся в ее
постоянной поставке. В
меньших количествах
GluT 1 присутствует в
других тканях, где он
поставляет глюкозу в клетки,
когда они находятся в
сравнительно
неактивном состоянии.
- GluT 2 обнаружен в
органах, выделяющих
глюкозу в кровь. Глюкоза
необходима всем клеткам, но
способностью гидроли-
зовать глюкозо-6-фосфат
до глюкозы (рис. 45,
этап 2) обладают только
гепатоциты, энтероци-
ты и почечный каналь-
цевый эпителий.
Соответственно кишечник,
печень, почки и выделяют
в кровь глюкозу.
Данный переносчик найден
также в В-клетках
панкреатических островков.
Интенсивность переноса
глюкозы GluT 2 зависит
от её содержания в крови.
Следовательно,
изменения уровня глюкозы в
процессе приема пищи и при физической
нагрузке компенсируются клетками
печени и В-клетками через GluT 2. Очень
важно, что именно при участии GluT 2
глюкоза переходит в кровяное русло
из энтероцитов, после ее всасывания в
кишечнике.
GluT 3 имеется в нейронах мозга.
Обладая большим, чем GluT 1, сродством
к глюкозе, он гарантирует постоянный
приток глюкозы к нервным клеткам.
GluT 4 — главный переносчик глюкозы в
мышцах и адипоцитах. Сокращающаяся
мышца нуждается в постоянном прито-
структурные полисахариды
\4
I 5
глюкозо-1-фосфат ^Г*^
t -со2
\глюкозо-6-фосфат *•
фруктозо-6-фосфат
фруктозо-1,6-дифосфат
пентозы
глицеро-3-
фосфат
(\
33\ 132
глицерин
- триозофосфат
фосфоенолпируеат
пиру ват
— — а-кетоглутарат
', Ацетил-КоА+оксалоацетат
жирные кислоты аспартат
цитрат
Рис. 45. Пути обмена углеводов. Цифровые обозначения и
пояснения — по тексту
242 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
ке глюкозы, а в жировой ткани глюкоза
может вовлекаться в синтез липидов.
80 % утилизации данного моносахарида
в условиях глюкозной нагрузки
приходится на миоциты, в которых глюкоза
превращается в гликоген. Особенность
данного транспортёра состоит в том, что
этот белок перемещается из внутренних
компартментов клетки к ее поверхности
и обратно.
— GluT 5 встречается главным образом в
тонкой кишке. Подробности его
функционирования пока неизвестны.
Семейство этих белков-переносчиков
глюкозы по градиенту концентраций
отличается от переносчиков, транспортирующих
моносахариды при их кишечном и почечном
всасывании, против концентрационного
градиента.
Работа белков-транспортеров глюкозы
контролируется гормонами, и в первую
очередь инсулином.
Наиболее выражен ответ на инсулин у
GluT 4 , в мышцах и жировой ткани.
Происходит это следующим образом:
>• Часть инсулинового рецептора,
выступающая наружу, связывает инсулин.
>* Часть инсулинового рецептора,
выступающая внутрь клетки, фосфорилирует за
счет АТФ один или более
белков-мишеней, с образованием фосфорилированно-
го белка(ов) и АДФ.
>• Как полагают, такой фосфорилирован-
ный белок служит сигналом для
перераспределения транспортёра глюкозы:
белок-переносчик глюкозы, в составе
содержащих его везикул, направляется
к плазматической мембране клетки и
включается в состав плазматической
мембраны (ПМ), обеспечивая транспорт
глюкозы.
>- Молекулы белка-переносчика глюкозы
возвращаются внутрь клетки, отшнуро-
вываясь в составе мембранных впячи-
ваний ПМ с последующим транспортом
образовавшихся везикул в околоядерное
пространство.
>• При снижении концентрации инсулина
цикл прерывается и
белок-переносчик глюкозы накапливается в составе
внутрицитоплазматических везикул.
Различные нарушения в работе
транспортёров глюкозы, в том числе
наследственные дефекты этих белков, могут
лежать в основе некоторых
генетических моделей инсулиннезависимого
сахарного диабета.
>* У некоторых линий спонтанно
заболевших диабетом мышей обнаружено
сниженное содержание GluT 2 в остров-
ковых В-клетках. При этом аномально
низкий уровень переносчика GluT 2 в
островковых В-клетках коррелирует с
ослабленной секрецией инсулина в ответ
на повышение содержания глюкозы в
крови.
>■ Некоторые экспериментальные
животные-модели инсулинорезистентного
СД, по-видимому, имеют дефект хотя бы
одного из белков-переносчиков глюкозы
в ПМ миоцитов. У больных инсулин-
независимым СД уровень отложения
гликогена в мышцах в 2 раза меньше, чем
у здоровых.
Можно предполагать и другие типы пос-
трецепторных дефектов в работе
транспортёров глюкозы при СД — на этапе
сигнализации о переброске переносчиков к мембране
или на стадиях циркуляции переносчика
в цитоплазме клетки, отшнуровывания от
мембраны, включения в состав мембраны
и т.д. Более подробно эти вопросы
обсуждаются ниже, в разделе «Патофизиология
сахарного диабета».
НАРУШЕНИЯ ВСАСЫВАНИЯ
И ПЕРВИЧНОГО
ПРЕОБРАЗОВАНИЯ
УГЛЕВОДОВ
Синдромы мальабсорбции углеводов
представляют собой «ферментный блок»,
который образуется либо на этапе их всасыва-
Патофизиология углеводного обмена 243
ния, либо на ранних стадиях их межуточного
обмена.
Нарушения кишечного всасывания
относятся к категории наследственных аутосом-
но-рецессивных ферментопатий. Выделяют
синдромы, связанные с недостатком натрий-
зависимого ко-транспортёра моносахаридов,
а также дисахаридаз — лактазы, инвертазы
или же мальтазы. Имеется и смешанный
синдром (олиго- или дисахаридазная
недостаточность).
Ведущими типовыми симптомами такого
рода патологии всегда являются:
> бродильный понос;
> эксикоз;
> вторичная гипотрофия.
Понос вызывают нерасщепленные диса-
хариды или невсосавшиеся моносахариды,
поступившие в более дистальные отделы
кишечника. Изменение осмотического
давления, а также кислые продукты
бактериального брожения «невостребованных»
макроорганизмом углеводов вызывают
усиленную перистальтику, увеличение
объема кишечного содержимого,
нарушение всасывания воды и оформления
каловых масс.
При глюкозо-галактозной мальабсорбции
дефицит натрий-зависимого
ко-транспортёра глюкозы и галактозы в кишечнике
приводит у детей к водянистому поносу после
приема пищи, с возрастом толерантность к
этим моносахаридам улучшается. Болезнь
может сопровождаться почечной глюкозу-
рией(см.выше).
При врожденной алактазии, как и при и
врожденном нарушении всасывания
глюкозы и галактозы, симптомы появляются
уже после первых прикладываний к груди.
При врожденном недостатке инвертазы или
мальтазы симптоматика проявляется при
переходе на искусственное вскармливание,
т. е. с введением в организм сахара и
крахмала. Нелеченные синдромы мальабсорбции
осложняются хроническим дисбактериозом
и задержкой физического развития детей.
С возрастом у всех экспрессия гена лак-
тазы снижается под действием
компонентов смешанного питания, причём у сотен
миллионов людей это происходит чересчур
быстро и1 радикально. Взрослые люди с
недостатком лактазы испытывают метеоризм
и диарею в результате употребления молока.
Носителей этой аномалии немало среди лиц
африканского и азиатского происхождения.
Средняя частота данного нарушения у
белого населения в Северной Америке и странах
Европы — не более 7-12%, на полуострове
Индостан — до 18 %. Но на Ближнем Востоке
она приближается к 70 %, а в Китае — к 80 %,
в Африке южнее Сахары достигает 97%.
В то же время у пигмеев 95 % популяции
сохраняют высокую лактазную активность.
A.M. Уголев указывал, что лактазная
недостаточность характерна для выходцев из
зон, традиционно исторически не связанных
с молочным скотоводством, а частота ее
определяется тем, как потреблялось молоко
на заре становления тех или иных этносов
(1991). При нарушении продукции сахаразы
(инвертазы) возникает непереносимость
сахара и сахарозосодержащих продуктов. При
сахарной нагрузке не происходит подъёма
гликемической кривой. При мальтазной и
изомальтазной недостаточности диспепсию
провоцируют крахмал, крупы, а также пиво
и другие напитки на основе солода. Описана
наследственная недостаточность трегалазы,
при которой диспепсию вызывают
содержащие трегалозу грибы и т.д.
Синдромы мальабсорбции иногда могут
предполагать и «ферментный блок» в
межуточном обмене углеводов.
Эти нарушения встречаются
относительно нечасто, но представляют серьезную
опасность в связи с токсическим действием
некоторых промежуточных продуктов
обмена углеводов, которые при этом
накапливаются.
Галактоземия — классический пример
тяжелейшего синдрома, развивающегося
на почве токсичности предшественника, не
претерпевающего должного
метаболического превращения. Этиология и патогенез
галактоземии были раскрыты благодаря
работам выдающегося американского
биохимика Г. Калькара (1956,1960).
244 ALU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
галактодегидрогеназа
НАДН
НАДФН + *
.--I
галактоза
/ альдо-
4 редуктаза
]__=2»-.
галактоновая кислота + ксилулоза + COi\
галактокиназный дефицит (катаракта)
галактокиназа
галактитол]
галактозо-1-фосфат
УДФ-глюкоза
/
классическая
галактоземия
(катаракта, ЗПМР,
полиорганное
поражение)
галактозо-1-фосфат-уридил-трансфера за
УДФ-галактоза + глюкозо-1-фосфат
впимеразный
дефицит (мягкое течение)
J
УДФ-глюкоза
УДФ-галактозо-4-зпимераза
Рис. 46. Пути обмена галактозы и его расстройства.
Штриховыми линиями показаны альтернативные
пути метаболизма. Пояснения — в тексте
Галактоза — продукт переваривания
лактозы. По всасывании она превращается в
глюкозу. Пути ее метаболического
преобразования в гепатоцитах показаны на рис.46.
Возможных причин синдрома галактоземии,
который поражает одного из 40 000
новорожденных, бывает три:
1) недостаток галактокиназы, которая в
норме превращает галактозу в
галактозо-1-фосфат (частота 1/500000);
2) недостаток галактозо-1-фосфат-ури-
дилтрансферазы, которая превращает
галактозо-1-фосфат в УДФ-галактозу
(частота 1/40 000);
3) недостаток УДФ-галакто-4-эпимеразы,
преобразующей УДФ-галактозу в УДФ-
глюкозу (частота менее 1/1000000).
В организме больного с дефицитом
галактокиназы накапливаются нерасщепленная
галактоза и ее побочные метаболиты. При
патологии избыток галактозы превращается
в спирт галактитол (дульцитол),
оказывающий токсическое влияние на хрусталик, где
дульцитол накапливается и осмотически
привлекает воду. Изменяется солевой
состав хрусталика и его белки
денатурируются, вследствие чего возникает катаракта
в молодом возрасте. Катаракта возможна
даже у плодов матерей с галактоземией,
употреблявших молоко во время
беременности. Все больные с врожденной и ранней
младенческой катарактой подозрительны на
наличие галактоземии.
Классическая тяжелая галактоземия
связана с дефектом галактозо-1-фосфат-
уридил-трансферазы. Кроме поражения
хрусталика при этой форме патологии
галактозо-1-фосфат оказывает
токсическое влияние на нейроны, гепатоциты и
нефроциты; угнетает активность ряда
ферментов печени, почек и других органов, в
том числе активность фосфоглюкомутазы
и глюкоза-6-фосфат-дегидрогеназы. Как
результат поражения нейронов возможно
возникновение задержки психомоторного
развития (ЗПМР).
Для понимания патогенеза поражений
ЦНС следует помнить, что галактоза —
важный компонент гликолипидов мозга.
У больного имеет место тенденция к
гипогликемии, истощаются запасы гликогена
в гепатоцитах, происходит компенсаторная
мобилизация жиров, развивается стеатоз, а
также некроз гепатоцитов и цирроз печени.
В почках и кишечнике избыток галактозы
и ее метаболитов ингибирует транспорт
аминокислот.
У женщин под влиянием метаболитов
галактозы формируется гипергонадо-
тропная первичная овариальная
недостаточность.
Патофизиология углеводного обмена 245
Лабораторно кроме гипогликемии при
галактоземии обнаруживаются:
> резкое увеличение содержания
галактозы в крови (до 1г/л)
> билирубинемия
> галактозурия
> протеинурия
> гипераминоацидурия
> повышение содержания гликозилиро-
ванного гемоглобина.
С возрастом, как правило, активность
отсутствующей у младенцев УДФ-галак-
топирофосфорилазы возрастает, что
позволяет вновь превращать галактозо-1-фосфат
вУДФ-галактозу и включать её в обмен
посредством эпимеризации. Поэтому чаще
всего течение болезни облегчается.
Классическая галактоземия представлена многими
аллельными генокопиями. При некоторых из
них у гомозигот активность галактозо-1 -фос-
фат-уридил-трансферазы лишь понижена до
50% нормы, что не дает клинических
симптомов (это нарушение известно как вариант
Дуарте). Гетерозиготное носительство генов
галактоземии очень распространено — им
поражены от 0,8 до 1,3 % населения. Вариант
Дуарте при скрининге обнаруживался с
частотой до 10 %! В то же время обнаружено,
что активность галактозо-1 -фосфат-уридил-
трансферазы при синдроме Дауна, наоборот,
повышена в 1,5 раза.
Эпимеразный дефект — самая редкая
разновидность галактоземии. По имеющимся
нескольким наблюдениям (исключительно
у швейцарцев и японцев), считается, что
картина полиорганного поражения при ней
менее тяжёлая.
Исключение из рациона до определенной
поры молока и других источников
галактозы позволяет предотвратитьфазвитие
патологических признаков. Кроме того, при
галактоземии помогает лечение
прогестероном и тестостероном, которые индуцируют
УДФ-галактопирофосфорилазу,
открывающую альтернативный путь вовлечения
галактозо-1-фосфата в метаболизм.
Проблема заключается в том, что сохранность
интеллекта можно гарантировать только,
если лечение начато в первые 2 месяца
жизни. Поэтому большое значение имеет
ранняя диагностика. Пренатальный диагноз
возможен путем изучения концентрации
галактитола в амниотической жидкости
и активности ферментов в фибробластах,
взятых путём амниоцентеза. Постнатально
активность галактозо-1 -фосфат-уридил-
трансферазы можно проконтролировать в
эритроцитах.
Имеются и такие наследственные
болезни, при которых извращается обмен
фруктового сахара. Нормальные пути
метаболизма фруктозы (ФРУ) представлены
на рис. 47.
гликоген
глюкоза-1-фосфат
блокада
X Y
1 фосфоглюкомутаза
глюкоза 6 фосфат
фруктоза
фруктокиназа
/*
фруктоза 1фосфат
доброкачественная
семейная
фруктозурия
/
непереносимость
фруктозы
фруктоза-1-фосфат-альдолаза
глицеральдегид фосфодиоксиацетон
Рис.47. Метаболизм фруктозы и его нарушения. Пояснения в тексте
246 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Непереносимость фруктозы —
относительно нередкая аутосомно-рецессивная
форма патологии. Причина — отсутствие
альдолазы фруктозо-1-фосфата. Следствием
отсутствия альдолазы фруктозо-1-фосфата
является накопление фруктозо-1-фосфата,
который тормозит активность фосфоглю-
комутазы. Последняя превращает глюкозо-
1-фосфат в глюкозо-6-фосфат и
обеспечивает конверсию продукта реакции,
катализируемой фосфорилазой, в глюкозо-б-фосфат.
Поэтому при фруктоземии происходит
торможение распада гликогена на этапе
глюкозо-1-фосфата и последующее
развитие гипогликемии, усиление мобилизации
липидов, формирование стеатоза печени и
гиперурикемии (см. выше
«Патофизиология нуклеинового обмена»).
Клинически при переводе детей на
смешанное или искусственное вскармливание
после приема фруктового сока или фруктов
возникают слабость, дрожь, судороги,
потливость, рвота, нарушение сознания. Могут
отмечаться транзиторная желтуха, протеи-
нурия. Возможны гепатоспленомегалия и
цирроз печени. Из-за ускоренного распада
АТФ и понижения экскреции уратов в
условиях метаболического ацидоза характерна
гиперурикемия, в молодом возрасте
формируется подагра. Понижение активности
фруктозо-1 -фосфатальдолазы свойственно
и болезни Тея-Сакса (см. «Нарушения ли-
пидного обмена»).
Непереносимость фруктозы протекает
тяжело и способна обусловить смертельные
исходы. От данного нарушения следует
отличать другой недуг, известный как
доброкачественная эссенциальная фруктозурия
(частота 1/130000).
Последняя характеризуется повышенным
содержанием фруктозы в крови и моче, что
может привести к постановке ошибочного
диагноза — сахарный диабет. Однако эта
форма патологии, в отличие от предыдущей,
важного клинического значения не имеет.
Дело в том, что она вызвана отсутствием в
почках, печени и энтероцитах фруктокина-
зы. В результате фруктоза не превращается
во фруктозо-1-фосфат и в свободном виде
выделяется с мочой. Так как токсичного
избытка фруктозо-1-фосфата не создаётся,
а ферменты печени, кишечника и почек не
блокируются, у больных не бывает
гипогликемии, проявлений печёночной и почечной
недостаточности. Рис. 47 представляет
механизмы патогенеза двух данных болезней.
Эссенциальная пентозурия, поражающая
исключительно евреев и ливанцев, в этих
этнических группах имеет частоту 1/2 000-
1/5 000. Это аутосомно-рецессивный дефект
фермента L-ксилулозо-редуктазы. Больные
выделяют до 4 г ксилулозы в сутки с мочой,
что может привести к ложному диагнозу
сахарного диабета. Однако ксилулоза — это
пелтоза, не дающая реакции с глюкозоокси-
дазой, что используется для дифферециров-
ки с глюкозурией. Сама по себе пентозурия
не вызывает существенных клинических
проявлений.
КАТАБОЛИЗМ УГЛЕВОДОВ И
ЕГО НАРУШЕНИЯ
Гепатоциты — главное место превращений
углеводов пищи. Сахара из клеток
кишечника попадают в кровеносные капилляры,
впадающие в воротную вену, по которой
моносахариды пищи приносятся в печень.
Глюкоза как моносахарид через
переносчик GluT-2 проникает в гепатоцит, где сразу
подвергается фосфорилированию (в ходе
реакции 1 рис. 45). Фосфорилирование
глюкозы — единственная реакция в организме,
где она участвует как таковая. У плода этим
занимается печеночная гексокиназа. Она
ингибируется продуктом реакции глюко-
зо-6-фосфатом. У грудных детей старше 1
месяца и у взрослого экспрессируется для
той же реакции иной фермент — глюкоки-
наза. Гексокиназа у взрослых сохраняется
в непаренхиматозных печеночных клетках.
Глюкокиназа не ингибируется продуктом
реакции и улавливает глюкозу при любом
уровне ее фосфатов в клетке. Эстрогены
репрессируют, а андростероиды
стимулируют экспрессию глюкокиназы. У негров
США и Карибских островов, креолов Мав-
Патофизиология углеводного обмена 247
рикия и японцев описан наследственный,
высокопенетрантныйаутосомно-доминант-
ный дефект глюкокиназы, кодируемой в
хромосоме 7. При наличии этого дефекта
воспроизводится картина инсулиннеза-
висимого сахарного диабета (MODY или
диабет взрослых в юности). Глюкоза не
удерживается в печени, возникает
гипергликемия и понижение отвечаемости В-клеток
на глюкозу, избыток глюкозы в адипоцитах
переводится в жиры, развивается тучность.
В последнее время появились данные о
значительно большей, чем предполагалось
ранее, частоте и разнообразии
встречаемости данной аномалии (см. ниже
«Патофизиология сахарного диабета»).
В норме фосфатные эфиры глюкозы и
фруктозы (образованные после реакций
1-4 и 8-10, рис. 45) уже не могут легко
преодолеть плазматическую мембрану,
фосфорилирование этих Сахаров создает
как бы ловушку для них, при этом реакции
гликолиза и пентозного цикла
эффективно изолируются в
пределах внутриклеточного
отсека. Чтобы выпустить глюкозу
назад, в кровь, гепатоциты, эн-
тероциты и нефроциты
располагают глюкозо-6-фосфатазой,
гидролизующей ее эфир.
Создание в печени большого
запаса недиффундирующей
глюкозы без
сопутствующего осмотического набухания
органа возможно благодаря
способности гепатоцита
превращать избыток углеводов
в большой ветвистый
нерастворимый полимер —
гликоген (рис.48). После приема
богатой углеводами пищи эта
резервная форма глюкозы
может составить до 1/5 массы
печени (в среднем составляя
5%), этот запас эквивалентен
70-90 г глюкозы. В мышцах
гликогена по абсолютному
количеству в 2 раза больше,
чем в печени. Однако там он
накапливается до уровня 1 % массы ткани и
метаболизируется более быстро. Гликоген
имеется также в макрофагах, жировой
ткани, нефроцитах, нейронах, нейроглиальных
и других клетках.
Контроль глюкокиназной и гексоки-
назной реакций превращения глюкозы в
глюкозо-6-фосфат осуществляется через
активирование этих ферментов инсулином,
который при этом «конкурирует за глюкоки-
назу с глюкокортикоидами».
Соответственно эта реакция хронически недостаточна
при сахарном диабете и гиперкортицизме и
временно ослаблена при стрессе, что вносит
вклад в развитие стрессорной
гипергликемии.
Распад гликогена, или гликогенолиз —
важный механизм мобилизации эндогенной
глюкозы, который подлежит сложной
регуляции и ступенчатой активации.
Гликогенолиз происходит в ответ на
повышение потребности в глюкозе. Разрыв
Рис. 48. Структура гликогена (по ЕЛ. Розенфельду, 1966).
Буквенные обозначения: а — наружные цепи, 6 — глюкоз-
ный остаток (1-4 связь), в — внутренние цепи (в фигурных
скобках), г — точки ветвления (1 -6 связь)
248 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
связей между мономерами глюкозы в цепи
гликогена путем присоединения по этим
связям неорганического фосфата
осуществляет активная фосфорилаза а. Фосфори-
лаза Ъ превращается в активный гликоге-
нолитический фермент фосфорилазу а под
действием фосфорилирования, которое
осуществляет киназа фосфорилазы Ь.
Последняя, в свою очередь, для активации
должна фосфорилироваться. Это происходит с
участием ее 8-единицы, представляющей
собою кальмодулин. На эту субъединицу при
активации процесса должна подействовать
ц-АМФ-кальций-зависимая протеинкиназа,
активируемая через гормональные
рецепторы адреналином, глюкагоном и/или тиро-
идными гормонами. Действие фосфорилазы
направлено на линейные 1,4-участки
молекулы полимера, вплоть до точек ветвления.
Гидролиз же 1,6-связей с освобождением
глюкозы катализирует фермент гликоген-
в-глюкано-гидролаза ( «сучкоруб» или
«debranching enzyme»); оставшаяся часть
«гликогенового дерева» становится
пригодной для дальнейшего распада при участии
фосфорилазы.
Продукт действия фосфорилазы — глю-
козо-1-фосфат (рис 45, реакция 6), который
под влиянием фосфоглюкомутазы (там же,
реакция 3) может вновь превратиться в
глюкозо-6-фосфат.
Гормональный контроль гликогенолиза
осуществляется несколькими
регуляторами:
>• глюкагоном (клетки-мишени: гепатоци-
ты и липоциты; интенсивность реакции
усиливается через активирование кина-
зы фосфорилазы);
>- катехоламинами (клетки-мишени: гепа-
тоциты, миоциты, липоциты; механизм
влияния — цАМФ — зависимая
активация киназы фосфорилазы);
>• тироидными гормонами
(клетки-мишени — все гликогенсодержащие
элементы, механизм — стимуляция
гликогенолиза);
>- инсулином — тормозит гликогенолиз,
переводя фосфорилазу в неактивную форму.
Гипоксия сопровождается
мобилизацией гликогеновых запасов, а исчезновение
гранул гликогена представляет признак
ранней стадии гипоксического некробиоза.
Важной группой наследственных
нарушений углеводного метаболизма считаются
гликогенозы — расстройства отложения и
мобилизации животного крахмала. Они
будут охарактеризованы ниже, в отдельном
разделе, после ознакомления с механизмами
синтеза гликогена.
Полученные из глюкозы и гликогена экзо-
и эндогенные фосфатные эфиры гексоз
вовлекаются в гликолиз (ри£. 45, реакции
11-13). В гепатоците 2/3 глюкозы
окисляются по этому пути, который становится
доминирующим источником энергии в
любых клетках, в условиях гипоксии, а в ма-
лодифференцированных и злокачественных
клетках преобладает над аэробным распадом
при любом содержании кислорода.
Гликолиз (он же фосфотриозный путь
или шунт Эмбдена-Мейерхофа) — путь
цитоплазматического превращения 1 моля
глюкозы в 2 моля лактата с образованием 2
молей АТФ в анаэробных условиях (если
кислорода в цитоплазме много, то
гликолиз идет до пирувата (рис. 45, реакция 13).
Последовательность этапов гликолиза
представлена в общем виде — выше, на рис. 45 и
подробнее на рис. 49).
Гликолиз — катаболический путь
огромной важности, поскольку:
>* обеспечивает энергией клеточные
реакции фосфорилирования, синтез белка и
другие процессы, требующие энергии;
>- промежуточные продукты
гликолиза — фосфотриозы — могут быть
использованы при синтезе жиров (через
НАДФН-зависимое восстановление для
образования а-глицерофосфата);
>• пируват может быть использован для
синтеза аланина, аспартата и других
соединений (через образование оксалоацетата);
>- пируват может служить источником
оксалоацетата в анаплеторическом пути
пополнения щавелевоуксусной кислоты
для цикла Кребса (см. далее);
Патофизиология углеводного обмена 249
> благодаря гликолизу доступность
кислорода и максимальная производительность
митохондрий не лимитируют
максимально возможную мощность, развиваемую
мышцами, что исключительно важно при
кратковременных предельных нагрузках.
Гликолиз делится на две фазы. На первой
стадии глюкоза фосфорилируется и делится
на две молекулы глицеральдегид-3-фосфа-
та, что требует затраты двух молей АТФ.
На этом этапе в процесс могут вовлекаться
другие гексозы, пентозы и глицерин — после
соответствующего фосфорилирования.
Второй этап сопряжён уже не с расходом,
а с запасанием энергии. Глицерофосфат
подвергается НАД-зависимому окислению,
а его дериваты отдают свои макроэргические
фосфатные группы АДФ с образованием
АТФ, причем формируются 4 молекулы
этого макроэрга. Энергетическая
эффективность гликолиза, по измерениям на модели
эритроцитов, превышает 50%.
Гормональный контроль реакций
гликолиза осуществляется через несколько
ключевых этапов, то есть практически
необратимых реакций, при которых сильно
уменьшается свободная энергия системы. Таких
этапов в гликолизе три, включая гексокиназ-
ную (реакция 1, рис. 49), фосфофруктокина-
зную (4) и пируваткиназную (12) реакции.
Инсулин стимулирует гликолиз через:
> активацию гексо(глюко)киназной
реакции (см. выше);
> стимуляцию фосфофруктокиназы
(ФФК), причём этот механизм (рис.
49, 4) считается наиболее важным.
ФФК соединяет фруктозо-6-фосфат и
АТФ, в результате чего формируются
фруктозо-1,6-дифосфат и АДФ. Жирные
кислоты и катионы водорода являются
ингибиторами этой реакции, а
интенсивность её протекания решающим образом
сказывается на всей пропускной
способности гликолиза. При гипоксическом
некробиозе зрелых клеток после перехода
на гликолиз накопление лактата и ацидоз
выключают ФФК. Это лимитирует гли-
колитическую адаптацию к гипоксии
(см. т. I, стр. 181-182). ФФ# эмбрио-
глюкоза
АТР'
ADP-
глюкозо-6-фосфат
глицер альдегид-^
3-фосфат *^
o-6-i
\\
ЗО-б
1,6-дщ
фруктозо 6 фосфат
АТР'
ADP-
фруктозо-1,6-дифосфат
2NAD+
2NADH
диоксиащтонфосфат
-дифосфоглицерин
2ADP
2 АТР
2,3-фосфоглицерин
лфосфог.
1\>
W
10
2,2-фосфоглицерин
It'
alt-
тоградная
2 фосфоенолпироеиноградная кислота
2ADP-
2 АТР*
2 пировиноградная кислота
2NADH'
2NAD+
2 молочная кислота
Рис. 49. Гликолиз. Пояснения в тексте.
Нумерация реакций — как на рис. 45.
нальных клеток, в отличие от зрелых,
некислотоингибируемый фермент.
Поэтому у плода и новорожденных, а
также детёнышей позвоночных животных
гликолиз в условиях гипоксии протекает
дольше и эффективнее, чем у взрослых.
Отсюда широко известный феномен
повышенной резистентности незрелого
организма к патогенному действию
острой гипоксии, описанный Н.Н. Сироти-
ниным (1981). Высокая интенсивность
ФФК-зависимой реакции способствует
250 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
поставке пластических и
энергетических эквивалентов для синтеза липидов.
В целях балансирования гликолиза и
глюконеогенеза ФФК ингибируется
избытком АКоА, жирных кислот, цитрата.
Мутации регуляторного участка ФФК,
делающие ее резистентной к ингибиро-
ванию цитратом и жирными кислотами,
найдены в липоцитах, формирующих
липомы, а также предполагаются при
некоторых случаях общего ожирения
и ГЛП (см. «Нарушения липидного
обмена»). Существует наследственная
аутосомно-рецессивная
недостаточность мышечного изофермента ФФК.
При этом гликолиз в скелетных мышцах
поддерживается за счет синтеза в них
небольшого количества печёночной
ФФК, однако имеется неустойчивость
к физическим нагрузкам. Обратная по
отношению к ФФК-зависимому этапу
гликолиза реакция 5 (рис. 49)
катализируется особым ферментом — фруктозо-
1,6-дифосфатазой. Это важный этап в
реакциях глюконеогенеза (см. ниже) и
объект воздействия при некоторых
моделях сахарного диабета;
>• стимуляцию пируваткиназы (ПК),
превращающей фосфоэнолпируват путём
его фосфорилирования в пиру ват.
Наследственный дефект пируваткиназы у
человека известен как одна из причин
гемолитической анемии, провоцируемой
окислителями (см. т. III).
На гликолиз влияют и другие
гормоны. Соматотропин (СТГ) — ингибитор
ферментов гликолиза. Тироидные
гормоны — стимуляторы активности ключевых
ферментов фосфотриозного пути.
Пентозный путь (прямое окисление
глюкозы, фосфоглюконатный или апото-
мический путь, гексозомонофосфатный
шунт) — катаболический цитоплазматичес-
кий путь обмена глюкозы, дополняющий
гликолиз. В гепатоците 1/3 глюкозы
окисляется по этому пути. Еще более активен он
в лактирующей молочной железе, а также
стероидпродуцирующих органах, жировой
ткани и лейкоцитах. В мышцах резко
преобладает гликолиз. В ходе пентозного цикла
происходит прямое превращение 1 моля
глюкозы в 6 молей С02 и образованием 12
молей НАДФН2 в анаэробных условиях.
Реакции апотомического пути представлены
на рис. 50.
Пентозный цикл обеспечивает:
>* энергетические эквиваленты для
биосинтетических реакций, таких как важные
этапы синтеза жирных кислот и
стероидов, требующие участия НАДФН;
>• потребности клетки в пентозах (рибозе
и дезоксирибозе) при синтезе нуклео-
тидов и нуклеиновых кислот, поскольку
рибулозо-5-фосфат является его
промежуточным продуктом.
В тканях млекопитающих пентозный
цикл практически необратим и протекает
лишь в направлении распада глюкозы.
Классик фармакологии А. Лабори (1971)
придавал большое значение балансу между
двумя дополняющими друг друга путями
анаэробного катаболизма глюкозы. Он
указывал, что пентозный шунт активируется
при подготовке к делению клетки, а
гликолиз — при интенсивной функции зрелых
клеток. Синтез липидов обеспечивается
пентозным шунтом энергетически, а
гликолизом — пластически. В отношении синтеза
нуклеиновых кислот можно говорить об
обратной комбинации (пластические
компоненты — из пентозного пути, а энергия — из
гликолиза), хотя оба пути равно
необходимы для обеспечения анаболизма липидов и
нуклеопротеидов.
Гормональный контроль пентозного
цикла осуществляет инсулин через стимуляцию
ряда его этапов, в том числе:
>* окислительного этапа цикла, повышая
активность глюкозо-6-фосфат- дегидро-
геназы, энзима, переводящего глюкозо-
6-фосфат в 6-фосфоглюконат;
>- неокислительного этапа цикла через
стимуляцию транскетолазы.
Тироидные гормоны стимулируют на
транскрипционном и на
посттранскрипционном уровне активность дегидрогеназ
Патофизиология углеводного обмена 251
2Н
с ,
н-6-он
(NADP)HO"V-H 9
■ н-с-он
н-с 1
Н2-С-ОРО:
ОН"
С ОО"
н-б-он
но-i-H
- н-с-он
н-с-он
6-Р-глюкозо-
лактон
н,
6-Р-глюконат
-ОР03
пентозо-Р- уу
3-эпимераза.^^
н '■''"'
рибулозо-5-Р
\пентозо-Р-
эпимераза
транскетолаза
но-
е^н27с-онм
№c = o^
гНО-С-НЛ
н-с-он
С-ОН;
b-i-o:
н-с-он
н2-с-оро3
ксилулозо-5-Р
н-с=о
н-с-он
н-с-он
н-с-он
н2-£-оро:
рибозо-5-Р
трансальдолаза
транскетолаза
zzH-C^O =
= Н-С-ОН=
-ОН
ОР032
фруктозо-6-Р
н2-с-
= н-с-он==
-ОРОз2
= н-с-(
1н2-с-(
эритрозо-4-Р
фруктозо 1,6-Р2
альдолаза
Рис. 50. Пентозный шунт. Р-фосфат, остальные
обозначения и пояснения — в тексте.
Штриховкой показана судьба фрагментов Сахаров
(по А. Уайту, 1981)
глюкозо-6-фосфата и 6-фосфоглюконата в
печени.
Один из центральных «перекрёстков»
углеводных и иных метаболических путей
связан с судьбой пировиноградной кислоты
(ПВК). Важная роль пирувата в обмене
веществ показана на рис. 51. Образовавшийся
в ходе гликолиза пируват путем диффузии
проникает в митохондрии, где пируватде-
гидрогеназная система ферментов в
практически необратимой реакции превращает его
в ацетил-Ко А (окислительное декарбокси-
лирование ПВК). Пируватдегидрогеназный
мультиферментный комплекс, описанный
н-
-с=о
н-с-он
Н2-С-ОРОз
iiH2-c-OH=
ffic=o
^но-у-н
zzzH-C-OH
=н-с-он
=н-с-он;
= н2-6-
ОР03
седогептулозо-7-Р
глицеральдегид-3-Р
н2-с-он
с=о
Н2-С-ОРОз
диоксиацетон-Р
Л. Дж. Ридом и соавт. (1969),
пространственно напоминает сложный многогранник
и представляет собой одну из красивейших
структур в биохимии. Он содержит 3
фермента (24 молекулы пируватдегидрогеназы,
12 молекул дегидролипоилдегидрогеназы и 1
молекулу дегидролипоилтрансацетилазы) и
5 витаминных кофакторов (тиамин, липое-
вую кислоту, никотинамид, рибофлавин, а
также пантотенат). Таким образом, работа
данного звена метаболизма чувствительна
к витаминному голоданию (см. ниже
«Патофизиология витаминного обмена»). При
многих отравлениях функционирование
этого «живого поливитамина» тормозится.
252 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Классическим антипируватдегидрогеназ-
ным ядом является мышьяк. Отравление
мышьяком и арсенитами, из-за
универсальности данного метаболического процесса,
отражается на множестве органов, клеток и
тканей — от ЦНС до эритроцитов.
Контроль активности пируватдегидро-
геназного комплекса осуществляется как
инсулином, так и другими регуляторами.
Инсулин увеличивает эффективность
превращения ПВК в ацетил Ко-А,
инактивация комплекса происходит как при
гипоксии, так и при избытке АТФ.
Ацетил Ко-А, полученный из ПВК,
может использоваться для синтеза липидов,
кетоновых тел, производства ацетилхолина
и ацетилирования биогенных аминов. Но
главная его часть, в норме, утилизуется в
цикле Кребса.
Цикл трикарбоновых кислот (цикл
Кребса, цикл лимонной кислоты) — аэробный,
локализованный в матриксе митохондрий
процесс, в ходе которого совершается
превращение двууглеродных соединений
(например, ацетата) до двух молей С02. При
этом соединения лишаются своего
водорода, который направляется в цепь тканевого
дыхания, где окисляется до воды. Другим
конечным продуктом аэробного распада
углеводов служит С02 (см. также т. I, стр.
168-169).
глюкозо-6-фосфат
О
молочная
кислота
пировиноградная
кислота
яблочная
кислота
^
Рис 51. Метаболическая судьба пирувата. Пояснения в
тексте
Итоговое уравнение одного оборота
цикла Кребса можно записать следующим
образом:
Ацетил-КоА -> +2С02 + КоА + 8е~
(где е — свободные электроны).
Иначе говоря, в ходе цикла Кребса
(см. схему на рис. 52) субстрат (ацетил-КоА)
подвергается декарбоксилированию и
дегидрированию. При этом водород
акцептируется переносчиками, такими как НАД,
ФАД. Атомы водорода становятся
субстратами (донорами электронов) в реакциях
переноса электронов по дыхательной цепи и
окислительного фосфорилирования. Кроме
того, в ходе цикла при деацилировании сук-
цината происходит образование АТф без
участия дыхательной цепи — субстратное
фосфорилирование. Множество ядов
нарушают работу цикла Кребса. Так, например,
аммиак, связывая его кетокислоты,
способен резко уменьшить продукцию АТФ в
клетке (см. «Нарушения конечных этапов
белкового обмена»). Вообще, цикл Кребса
и цикл мочевины иногда образно называют
«двухколесным велосипедом» (рис. 12, с.
71). Имеется в виду, что нарушение в
работе одного из них немедленно сказывается
на другом. Общим метаболитом для них
является фумарат, а С02, образуемый в
цикле Кребса, утилизуется в орнитиновом.
АТФ генерируется циклом
Кребса и служит источником
энергии для цикла мочевины.
Наконец, оба цикла связаны
и такими метаболитами, как
производные янтарной кис-
1ланин лоты. Таким образом,
аммиачная интоксикация ведет к
тканевой гипоксии, а блокада
клеточного дыхания нарушает
нейтрализацию
азотсодержащих продуктов распада.
Фторорганические
соединения (например, фторацетат),
превращаясь во фторцитрат,
блокируют ключевой фермент
трех реакций цикла Кребса,
щавелевоуксусная
кислота
ацетоуксусная
кислота
Мобилизация
ацетил-КоА
Аминокислоты
Углеводы
\
Пируват
Жирные
кислоты
Цикл трикарбоновых
кислот
Перенос электронов
и окислительное
фосфорилирование
Флавопротеид
АДф\фн-+ I—ЧАГФ1
Кофермент Q
Цитохром о
АДФ+ФН—^ I—ЧЛГФ1
Цитохром с
\
Цитохром а
адф+ф„—И—Члгф!
2Н++1/202
Рис. 52. Мобилизация ацетилкоэнзима А, цикл трикарбоновых кислот и дыхательная цепь
(по А. Ленинджеру, 1976). Пояснения в тексте
254 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
оксалоацетат 4-AKqA
цитратсинтетаза
■► цитрат+KoA-SH
сукцинит -
СДГ
ИДГ
Рис. 53. Стадии цикла Кребса, прямо или косвенно
стимулируемые инсулином. Сокращения: СДГ — сукцинатдегид-
рогеназа; ИДГ — изоцитратдегидрогеназа, KoA-SH — ко-
энзимА
аконитазу, ингибируя дыхание.
Южноафриканское растение Dichapetalum cymoscum,
которое на местном языке африкаанс
именуют гифблаар, за счет содержания фторуксус-
ной кислоты сильно ядовито.
Фундаментальное значение для
приспособления организма к изменению условий
внешней среды и состояния его собственных
физиологических систем имеет регуляция
работы цикла Кребса. Цель такой
регуляции— гарантировать стабильное
производство АТФ при переменном сырье. АДФ
и АМФ являются стимуляторами цикла
трикарбоновых кислот, а избыток АТФ —
ингибитором. Другие продукты цикла
Кребса, например НАДН, также понижают
его пропускную способность, а субстраты,
наоборот, активируют цикл. Ключевой
регулируемой стадией цикла Кребса является
синтез лимонной кислоты путем
конденсации ацетил-КоА с оксалоацетатом,
катализируемой цитрат-синтетазой,
чувствительной к соотношениям АТФ/АД Ф+АМФ
и НАД+/НАДН.
В конце оборота цикла оксалоацетат
регенерирует. Показано, что истощение
тканевых запасов оксалоацетата и в известной
степени других кетокислот этого пути ведет
к прекращению работы цикла лимонной
кислоты. Именно так обстоит дело, когда
эти кетокислоты отвлекаются в качестве
субстратов в реакции глюконеогенеза при
ИЗСД (см. ниже «Патофизиология
сахарного диабета») или связаны аммиаком (см.
«Патофизиология белкового обмена»).
Среди возмещающих, анап-
леротических, реакций,
направленных на пополнение
промежуточных продуктов цикла
Кребса, важнейшей является
ферментативное карбоксилиро-
вание пирувата до оксалоацетата,
протекающее только в матриксе
митохондрий под влиянием пи-
руваткарбоксилазы.
Это уникальная реакция,
единственная, при которой не-
фотосинтезирующие клетки
человека фиксируют в
органической форме митохондриальный
С02\ Стимуляция или угнетение этой
реакции определяется тремя моментами:
>• количеством оксалоацетата: если его
мало — имеет место стимуляция, если
много — происходит угнетение;
>• количеством ацетил-КоА: при малом его
содержании наступает угнетение, при
изобилии — стимуляция;
>* обеспеченностью витамином Н и
марганцем, поскольку биотин прямо
участвует в фиксации двуокиси углерода,
а Мп входит в активный центр энзима
(см. «Патофизиология витаминного
обмена», «Патофизиология обмена
микроэлементов»).
Гормональный контроль цикла Кребса
осуществляется прежде всего инсулином
(рис.53). Многие авторы отрицают
возможность прямого действия инсулина на
экспрессию и активность ферментов
рассматриваемого метаболического пути. Но,
по крайней мере, косвенно через субстраты
инсулин в состоянии повышать активность:
>* цитратсинтетазы;
>• изоцитратдегидрогеназы (ИДГ);
>* сукцинатдегидрогеназы (СДГ).
Ферменты цикла Кребса, кроме СДГ,
локализованной во внутренней митохондриаль-
ной мембране, находятся во внутримитохон-
дриальном матриксе. В цитоплазме
представлена активность отдельных энзимов цикла,
но там они участвуют в иных метаболических
реакциях. Кроме цикла Кребса, работа мито-
Патофизиология углеводного обмена 255
хондрий подразумевает окислительное фос-
форилирование (см. также т.1, стр. 168-174
и данный том выше, раздел
«Энергетический метаболизм и его нарушения»). Ниже
даётся лишь краткое рабочее описание этого
заключительного этапа метаболического
пути взятых из углеводов (или других
энергетических эквивалентов) электронов (см.
рис.52 —внизу).
Последовательность реакций переноса
водорода и электронов (дыхательная цепь)
локализована на внутренней стороне
внутренней мембраны митохондрий. Эта цепь
включает:
> НАДН — дегидрогеназы
> ФАДН — дегидрогеназы (флавопроте-
иды)
> негемовые железосодержащие белки
> коферментО,
> цитохромы в, с1? с, а, а3
> и, наконец, финальный акцептор
электронов — кислород.
Легко заметить, что дыхательная цепь
зависит от функционирования нескольких
витаминов и микроэлементов, а гиповитами-
нозы и дефицит железа и других металлов
в организме будут оборачиваться тканевой
гипоксией (см. ниже разделы
«Патофизиология витаминного обмена»,
«Патофизиология обмена микроэлементов»).
Окислительное фосфорилирование —
процесс, при котором выделение энергии
afa счет окисления субстратов сопряженно с
синтезом АТФ.
' Первым пунктом сопряжения является
участок, где два электрона и два водорода
от восстановленного НАД переносятся на
флавопротеиды.
Восстановленная форма цитохрома в
действует в качестве восстановителя ге-
мацитохромов с, что представляет собой
второй пункт сопряжения окисления и
фосфорилирования.
Третьим пунктом сопряжения является
заключительный участок дыхательной цепи,
осуществляющий передачу электронов от
цитохрома а3 на кислород.
Об эффективности окислительного
фосфорилирования обычно судят по величине
отношения этерифицированного фосфата к
поглощенному кислороду, то есть так
называемому коэффициенту Р: О.
Поскольку окисление НАДН сопряжено
с переносом электронов через три этапа
сопряжения, окисление любого субстрата,
поставляющего в дыхательную цепь свои
электроны при участии НАДН, приведет к
образованию трех молей АТФ на пару
электронов, перенесенных к кислороду: ""' "
При окислении таких субстратов, как
сукцинат, ацилы жирных кислот, а-глице-
рофосфат, они переносят свои электроны
в дыхательную цепь при участии флаво-
протеидов, в процесс переноса электронов
вовлекаются только два этапа сопряжения,
а потому окисление таких субстратов даст
отношение Р : О, равное двум.
Наконец, окисление такого вещества,
как аскорбиновая кислота, которая может
непосредственно восстанавливать цитохром
с в обход первых двух этапов сопряжения,
дает величину Р: О = 1. Механизм
окислительного фосфорилирования, по П.
Митчеллу, состоит в использовании разности
протонового потенциала по обе стороны
от внутренней мембраны митохондрий для
синтеза АТФ. Такие соединения, как 2,4-ди-
нитрофенол и трифторметоксикарбонил-
цианидфенилгидразон, способны замыкать
протонный ток через внутреннюю мембрану
митохондрий, разряжая митохондриальный
«конденсатор», и препятствовать фосфори-
лированию. Олигомицин и дициклокарбок-
силдиимид блокируют и
фосфорилирование, и перемещение протонов. Сведения о
ядах, разобщающих окисление и
фосфорилирование, включены в т. I (с. 169-170).
Гормональный контроль окислительного
фосфорилирования в значительной степени
определяется уровнем инсулина,
эндогенных опиатов и гормонов щитовидной
железы. Показаны их кратковременные, не
требующие ни синтеза белка, ни транскрипции,
а также отсроченные эффекты. Последние
опосредуются повышением скорости
образования специфических мРНК. Опиаты, и
256 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
особенно инсулин, максимально
увеличивают эффект фосфорилирующего окисления.
Активнейшими стимуляторами работы
митохондрий служат тироидные гормоны,
располагающие митохондриальными
рецепторами, особенно Т3.
В результате действия тироидных
регуляторов происходит следующее:
>* возрастание концентрации
окислительных ферментов на единицу массы
митохондрий (особенно заметное при
гипертирозе);
>* усиление транспорта АДФ в
митохондрии. Так, при гипертирозе Т3 связывается
с транслоказой АДФ в митохондриях,
что обусловливает стимулирующий
эффект АДФ на окислительное фосфо-
рилирование;
>* повышение активности оксидоредуктаз
митохондрий (СДГ, цитохромоксидазы);
>* избирательное повышение активности
митохондриальной а-глицерофосфат-
дегидрогеназы в несколько раз.
Кроме того, считают, что активация
теплоотдачи тироидными гормонами может
быть следствием стимуляции ими
активности Na-K-АТФ-азы всех клеток, что влечет
за собой усиление транспорта Na в печени,
почках, мышцах, мозге. Последующая
стимуляция выработки энергии происходит
из-за снижения индекса АТФ/АД Ф+ АМФ,
вследствие субстратной регуляции при
возрастании необходимости её образования.
Старые источники настаивают на
разобщающем действии тиротоксических
концентраций гормонов щитовидной железы
на окислительное фосфорилирование. Так,
было показано (P.P. Рачев, Н.Д. Ещенко,
1975), что при тиротоксикозе:
>* фосфорилирующее звено дыхательной
цепи между НАД и ФАД находится в
разобщенном состоянии;
>- фосфорилирующее звено между цитохро-
мом b и Cj «латентно разобщено» (легко
прекращает свое функционирование).
Однако более новыми данными
опровергается такая точка зрения, и в настоящее
время принято считать, что лишь
фармакологические дозы гормонов щитовидной железы
оказывают действительно разобщающий
эффект, что же касается тиротоксикоза — то
такие дозы даже при кризах не
достигаются, и активация теплового рассеяния при
нём вызвана в основном стимулированием
деятельности калий-натриевого насоса
(К. Зонненблик и соавт., 1984, А. Гайтон,
1995). Более подробно эти вопросы
затронуты в разделах «Энергетический метаболизм
и его нарушения» и «Патофизиология
щитовидной железы».
АНАБОЛИЗМ УГЛЕВОДОВ
И ЕГО НАРУШЕНИЯ
Как уже говорилось выше, главной запасной
формой углеводов у животных служит
гликоген (рис. 48, с. 228). Синтез гликогена (рис. 45,
реакции 3, 5) — основной путь анаболизма
моносахаридов. Гликогеногенез (гликогенез)
начинается с продукции белка гликогенина,
к тирозиновому остатку которого крепится
первый глюкозный остаток, включаемый в
состав полимера (см. также рис. 55).
Гликогеногенез, как всякий
анаболический процесс, требует энергии, которая
поставляется и в форме АТФ, и в форме УТФ
(уридинтрифосфата). Глюкозо-6-фосфат
превращается в глюкозо-1-фосфат
(реакция 3). Последний активируется уридиннук-
леотидами с образованием УДФ-глюкозы
(высоко реакционноспособная кофермен-
тная глюкоза). Следующую реакцию (5 на
рис.45) катализирует гликогенсинтетаза
(ГС). При этом УДФ-глюкоза
присоединяется к свободной гидроксильной группе
в положении 4 на уже существующей
молекуле гликогена. Сочетанное действие
гликогенсинтетазы и «ветвящего» фермента
приводит к образованию линейных цепей
мономеров глюкозы, соединенных 1-4
связями, и боковых ветвей с 1-6 связями в
точках ветвления (рис. 48). Гормональный
контроль гликогеногенеза включает ряд
звеньев. Инсулин оказывает на этот
анаболический путь двоякое действие:
> снижает распад гликогена, переводя фос-
форилазу (Фз) в неактивную форму;
> стимулирует синтез гликогена,
обеспечивая гликогеногенез субстратом
(глюкоза-6-фосфатом) через
активирование гексокиназы;
Глюкагон, как антагонист инсулина:
> стимулирует функцию киназы фосфори-
лазы (КФ), активирующей Фз;
> препятствует активации гликогенсин-
тетазы (ГС).
Адреналин вмешивается в процессы
обмена гликогена следующим образом:
> как антагонист инсулина — стимулирует
активность киназы фосфорилазы (КФ).
> стимулирует гликогенолиз; .
> инактивирует ГС.
Тироидные гормоны и парат-гормон
также значительно стимулируют гликогенолиз.
Синтез гликогена происходит в сытом
организме, при значительном доступе
экзогенной глюкозы в портальную кровь. В
голодном состоянии доминирует его распад,
глюкозо-б-фосфат гепатоцитов переносится
в ЭПР специальным ферментом-трансло-
казой, и микросомальная глюкозо-6-фос-
фатаза освобождает из глюкозо-6-фосфата
глюкозу, попадающую в кровь. В мышце
синтез гликогена доминирует в покоящемся
состоянии, поскольку покоящиеся миоциты
не экспрессируют глюкагоновых
рецепторов. В состоянии активной функции мышцы
экспрессируют рецепторы глюкагона, и
гликогенолиз приводит к энергетической
утилизации глюкозо-1-фосфата в реакциях
гликолиза (см. далее, рис. 55).
Другим важным анаболическим путём
обмена углеводов является глюконеогенез.
Функционально это процесс, обратный гликолизу,
приводящий к образованию глюкозы и
гликогена из углеводных и неуглеводных исходных
продуктов (рис. 54). Но глюконеогенез — не
простое обращение гликолитических реакций,
поскольку, как читатель помнит, три из девяти
шагов гликолиза энергетически необратимы
(глюкокиназная, фосфофруктокиназная и пи-
Патофизиология углеводного обмена 257
руваткиназная реакции). Хотя энергетические
сдвиги при большинстве реакций гликолиза
достаточно малы (реакции эти могут
протекать и в обратном направлении), имеются
три этапа, которые можно рассматривать как
необратимые процессы:
Обход этих энергетических барьеров
обеспечивают специальные ферменты глю-
конеогенеза:
(a) глюкозо-6-фосфатаза;
(b) фруктозо-1,6-дифосфатаза;
(c) спаренные ферменты пируваткарбо-
ксилаза и фосфоэнолпируваткарбоксики-
наза, а также малатдегидрогеназа.
Деятельность этой системы позволяет превратить
пируват обратно в фосфоенолпируват через
оксалоацетат и малат (рис. 54 справа).
Исходными эндогенными продуктами
глюконеогенеза у человека и высших
животных могут быть многие, но отнюдь не любые
метаболиты:
>- Лактат, возникший в результате
анаэробного гликолиза в самой печени, а
равно и лактат, приносимый с кровью
к гепатоцитам (например, от мышц).
Циркуляцию глюкозы и лактата между
мышцами и печенью иногда обозначают
как цикл Кори.
>■ Гликогенные аминокислоты (те, которые
при дезаминировании превращаются в
промежуточные продукты цикла Кребса
или в ПВК и, в конце концов, дают
фосфоенолпируват). В эту группу входят все
аминокислоты, кроме лейцина. Лейцин
превращается в ацетил-Ко А и может дать
начало ацетоацетату и кетоновым телам.
Еще раз отметим, что ни у человека, ни у
других животных нет метаболических
путей, превращающих ацетил-Ко А в пируват
или сукцинат.
То есть сам ацетил-Ко А, а значит, и атомы
углерода жирных кислот, и лейцин, у
человека субстратами глюконеогенеза не
являются. Ряд аминокислот при дезаминировании
дает и гликогенные, и кетогенные продукты
(изолейцин, лизин, фенилаланин, тирозин).
Скелетные мышцы могут освобождать
некоторые гликогенные аминокислоты для нужд
печёночного глюконеогенеза.
дисахариды
гликоген,
крахмал
полисахариды
клеточной стенки I
свободная \ I / другие
глюкоза^щ^ \ I / моносахариды
глюкозо-6-фосфат
гликоген
\
УДФ-глюкоза
Г1Ф
АТФ ||
глюкоза Г6Ф
УТФ
3-фосфоглицерат
рибулозо-1,5-
дифосфат
СО,
Фн
АТФ
У и
Г Ф6Ф
( v
Фн
ФДФ
фосфоенолпируват
ГЗФ(ДОАФ)
ЗФГ
2ФГ
оксалоацетат
фосфоенолпируват
,ГГФ
со2
^
цикл трикарбоновых
кислот
оксалоацетат
УРАД4
некоторые
аминокислоты
АДФ
митохондрия
пируват
малат
| НАДН
оксалоацетат
,/,
С02 АТФ
пируват
?
пируват
Рис. 54. Глюконеогенез. Слева — субстраты и продукты, справа — обходные этапы глюконео-
генеза, в сопоставлении с гликолизом. П Ф — глюкозо-1 -фосфат, Ф6Ф-фруктозо-6-фосфат,
Г6Ф -глюкозо-6-фосфат, ФДФ -фруктозодифосфат, ДОАФ — диоксиацетон-фосфат,
ЗФГ-3-фосфоглицерин, 2ФГ—- дифосфоглицерин, остальные сокращения по тексту
> Глицерин, который в глюконеогенез
поступает в основном из продуктов липолиза.
> Оксалоацетат, пируват, а также
промежуточные продукты цикла Кребса, кроме
цитрата. При флоридзиновой глюко-
зурии у экспериментальных животных
показано, что введенные трикарбоновые
кислоты цикла Кребса, из-за
недоступности экзогенной глюкозы, практически
количественно переходят в эндогенную
глюкозу в реакциях глюконеогенеза.
> У растений и микроорганизмов
имеются изоцитрат-лиаза и малат-синтетаза;
которые позволяют расширить круг
субстратов глюконеогенеза и включить в него
непосредственно жирные кислоты.
Гормональный контроль
глюконеогенеза может осуществляться несколькими
путями:
а) прямо ( через сигнальные воздействия
нагепатоциты);
б) косвенно (через субстраты,
поступающие из периферических тканей, от миоци-
тов, липоцитов);
в) комбинированным путём.
> Глюкагон активирует пируваткарбо-
ксилазу и фосфоэнолпируваткарбок-
сикиназу, угнетает синтез
полипептидных цепей на рибосомах, увеличивает
скорость протеолиза белков в печени,
стимулирует липолиз в жировой ткани
и в печени, как активируя гормончувс-
твительную липазу гепатоцитов, так и
стимулируя транспорт жирных кислот
в митохондрии.
> Катпехоламины активируют гликогено-
лиз в мио- и гепатоцитах через
стимуляцию фосфорилазы, что приводит к
активации гликолиза, повышению уровня
сахара крови и возрастанию содержания
молочной кислоты в мышцах.
> Глюкокортпикоиды активируют
освобождение гликогенных аминокислот
инсулинзависимымимезенхимальными
тканями и стимулируют ферменты
глюконеогенеза, но при этом практически
не стимулируются липолиз и гликоге-
нолиз.
Патофизиология углеводного обмена 259
>* Инсулин комбинированным путём
подавляет глюконеогенез.
Дефицит
фруктозо-1,6-дифосфатазы — аутосомно-рецессивная
наследственная болезнь, приводящая к невозможности
использовать в реакциях глюконеогенеза
лактат, глицерин и аланин. Поддержание
уровня глюкозы в крови у больных требует
постоянного притока экзогенной глюкозы.
Развивается лактат-ацидоз, что ведет к
гипердентиляции, сонливости вплоть до
комы и сопровождается кетозом и
гипогликемией.
Глюкокортикоиды усиливают активность
фруктозо-1,6-дифосфатазы, что приводит к
мощному глюконеогенезу и гипергликемии.
Одна из моделей сахарного диабета
основана на отравлении животных аллоксаном.
Аллоксан (уреид мезоксалевой кислоты,
блокатор HS-групп) не только сам
повреждает В-клетки панкреатических островков
и провоцирует против них аутоиммунную
агрессию, но и изменяет процессинг
фермента фруктозо-1,6-дифосфатазы в лизосомах
печени. Ключевой энзим глюконеогенеза у
животных с аллоксановым диабетом теряет
триптофановый остаток, что резко
увеличивает его чувствительность к глюкокортико-
идной и иной аллостерической стимуляции.
Таким образом, механизм аллоксанового
сахарного диабета комплексный. В
естественных условиях сходным с аллоксаном
образом может действовать мочевая кислота.
Поэтому гиперурикемия часто сочетается с
гипергликемией и СД (см. гл. 14).
ОСОБЕННОСТИ ОБМЕНА
УГЛЕВОДОВ В РАЗЛИЧНЫХ
ТКАНЯХ
Рассмотренные нами метаболические пути
углеводного обмена по-разному
осуществляются в разных условиях, поэтому
метаболизм углеводов в различных органах и
тканях неодинаков.
Мышцы — важный потребитель глюкозы,
средоточие самого большого абсолютного
260 А.Ш. Зайчик, Л.П.Чурилов Патохимия
запаса гликогена и источник субстратов для
глюконеогенеза.
Цель миоцита — наиболее эффективно
использовать глюкозу для образования
АТФ, необходимого для осуществления
мышечного сокращения. В состоянии покоя
часть глюкозы резервируется в гликоген.
Миоциты богаты ферментами гликолиза,
а большое количество митохондрий
обеспечивает в них эффективную работу цикла
Кребса и окислительного фосфорилирова-
ния. Накопление лактата в гиалоплазме
миоцита происходит лишь в условиях крайнего
утомления, когда упомянутые механизмы
становятся недостаточными.
Следующая особенность
миоцита—отсутствие ключевых ферментов глюконеогенеза.
Обмен углеводов в миоците, таким
образом, обеспечивает создание в покое запаса
гликогена, а в условиях интенсивной
работы — использование этих запасов, а
также поступающей глюкозы, при наивысшей
мощности — в основном в анаэробном
режиме. В покое лактат и другие субстраты
глюконеогенеза должны транспортироваться в
печень и сердце, где и реутилизироваться
(см. ниже). Существуют некоторые
принципиально важные особенности фосфорилиро-
вания поступающей в миоцит глюкозы:
>■ Гексокиназа миоцита сильно
угнетается продуктом собственной реакции
(отрицательная обратная связь) и не
подконтрольна инсулину. Следовательно,
мышечная гексокиназа фосфорилирует
глюкозу только до тех пор, пока глюкозо-
6-фосфат используется в гликолизе или
для образования гликогена.
>* В миоците отсутствует глюкозо-6-
фосфатаза (равно как нет ее и в других
внепеченочных тканях за исключением
почек и тонкой кишки). Вот почему
использование глюкозы в мышечной
и многих других тканях ограничено,
исключительно внутриклеточным
метаболизмом.
Основные энергетические потребности
разных типов мышц удовлетворяются
неидентичным спектром субстратов. Скелетные
чдокринно-метаболические нарушения)
мышцы используют больше углеводов, а
сердечная питается в основном за счет
окисления продуктов метаболизма жиров.
Миокард и скелетные мышцы отличаются
не только по спектру субстратов, из которых
черпают ацетил-КоА. Имеются и
отличительные особенности лактатдегидрогеназы
(ЛДГ) в этих разновидностях мышечной
ткани. Из-за этого между скелетной мышцей
и кардиомиоцитом существует
фундаментальное различие в использовании пирувата
и образовании лактата.
Так, кардиомиоциты щлеют такое
большое количество митохондрий и снабжаются
кровью столь обильно, что работают, по
сути, почти всегда в аэробных условиях.
Кардиомиоцит окисляет не только пиру-
ват, но и поступающий с кровью из других
органов лактат.
Напротив, в скелетной мышце в условиях
быстро наступающего утомления, при
меньшей мощности митохондриальной системы
и худшем кровоснабжении максимальна
активность ферментов гликолиза, а
способность митохондрий окислять образующийся
НАДФН оказывается недостаточной, и
пируват имеет тенденцию превращаться в
лактат. Образовавшийся лактат
диффундирует в печень, где используется для ресин-
теза глюкозы, или в сердце, где окисляется.
Эти метаболические различия обусловлены
различиями в генетической экспрессии
субъединиц изоферментов ЛДГ.
Мозг, по сравнению с другими
органами, особенно зависит от уровня
метаболизма углеводов. Гипогликемия
вызывает стресс и чувство голода, затем
прогрессирующее помрачение сознания
и, наконец, кому от глубокой тканевой
гипоксии. Так, следствием снижения
вдвое концентрации глюкозы в крови,
притекающей к мозгу, могут быть потеря
сознания и смерть через несколько минут.
Энергетическое обеспечение мозга
обусловлено прежде всего аэробными
механизмами, т. е., ещё более, чем к гипогликемии,
мозг чувствителен к гипоксемии,
вызывающей чувство удушья, одышку и, в конце
концов, гипоксическую кому. Основной
катаболизм глюкозы в мозге осуществляется
по пути гликолиза и далее — в цикле трикар-
боновых кислот. В дополнение — пентозный
цикл обеспечивает восстановительными
эквивалентами (НАДФН) синтез нейромеди-
аторов, аминокислот, липидов, гликолипидов
и компонентов нуклеиновых кислот. Гексоки-
наза мозга обнаруживает высокое сродство к
глюкозе и эффективно удерживает глюкозу
в клетке. Перенос глюкозы в клетки
мозга- неинсулинозависимый процесс. Нейроны
получают виноградный сахар всегда, лишь
бы он был в крови. Это делает приоритетным
снабжение ЦНС глюкозой в условиях стресса.
При очень высокой гипергликемии
увеличение содержания глюкозы в ЦНС может
приводить к набуханию мозга и участвует в
патогенезе гиперосмолярной комы (см. ниже
«Патофизиология сахарного диабета»).
Активность гликолитических ферментов
мозга очень высока. АТФ и цитрат
контролируют активность ФФК по принципу
отрицательной обратной связи; АМФ
обеспечивает положительный контур контроля.
Ферменты цикла Кребса высоко
активны, что предотвращает накопление в мозге
лактата. Пируватдегидрогеназная система
превращает большую часть пирувата в
ацетил-КоА. Небольшая доля ацетил-КоА
идет на образование ацетилхолина.
Некоторое количество ацетильных групп
сохраняется в резервной форме N-ацетиласпартата.
Синтез аспартата и глутамата до некоторой
степени обеспечивается промежуточными
продуктами цикла Кребса — кетокислотами.
Чрезмерное использование промежуточных
продуктов цикла лежит в основе нарушения
метаболизма углеводов в ЦНС. Так,
например, если а-кетокислоты используются для
фиксации аммиака, то истощение их запаса
приведет к токсическому воздействию
аммиака на клетки мозга.
Однако в условиях длительного голодания
мозг способен к метаболизму кетоновых тел (в
частности, р-оксибутирата) для обеспечения
цикла Кребса субстратами. В крайних
случаях происходит дезаминирование глутамата
и аспартата, а образовавшиеся кетокислоты
используются как субстраты цикла Кребса.
Патофизиология углеводного обмена 261
ГЛИКОГЕНОЗЫ
ИАГЛИКОГЕНОЗ
Гликогенозы — группа наследственных
нарушений накопления гликогена и/или его
утилизации, создающих недостаточность
на путях поддержания уровня глюкозы в
крови (печень) и/или генерации энергии
(мышцы). Первые случаи гликогенозов
описали еще в 1922 г. швейцарские врачи
Й. Парнес и Р.Вагнер. Именно один из
гликогенозов (IIIтипа) был первой
наследственной болезнью печени, для которой
супругам Кори удалось раскрыть природу
первичного энзиматического дефекта.
К.Ф. Кори и Г.Т. Кори предложили сам
термин «гликогеноз» и систему нумерации
этих болезней. По традиции мы указываем в
данной главе номера гликогенозов, но надо
иметь в виду, что в 90-е годы XX века от
номерной классификации стали отказываться,
так как в нумерации после позиции 6 было
очень много разночтений. В настоящее
время доминирует патогенетическое деление
гликогенозов на печеночные, мышечные и
смешанные формы. Следует отметить, что
при гликогенозах количество гликогена
в тканях не всегда изменено. Поражение
может ограничиваться качественными
аномалиями животного крахмала. Основные
гликогенозы представлены на рис. 55.
Печеночные гликогенозы поражают
метаболический путь, при котором происходит
утилизация гликогена для поддержания
уровня глюкозы в крови. В то же время
нарушение нормальных ферментативных
реакций в печени вызывает избыточное
отложение гликогена в этом органе.
К данной группе относят прежде всего
гликогеноз 1а типа (синонимы — гепато-
ренальный тип, болезнь фон Гирке,
рациональное название — дефект глюкозо-6-фос-
фатазы). Среди всех гликогенозов
данный встречается чаще всего а наследуется
аутосомно-рецессивно.
Недостаточность глюкозо-6-фосфатазы
вызывает накопление гликогена в гепато- и
нефроцитах, так как в избытке присутс-
Рис. 55. Пути обмена гликогена. Сокращения: ФЗ — фосфорилаза, КФ — киназа фосфорилазы,
ГС — гликогенсинтетаза (в скобках — неактивные формы этих ферментов); глю — глюкоза,
фру — фруктоза, Ф — фосфат, ЛДГ — лактатдегидрогеназа. Показаны путь гликогенолиза в
печени при гипогликемии и в скелетной мышце — при физической нагрузке
твует глюкозо-6-фосфат —
предшественник субстрата гликогенсинтетазы и
активатор D-формы этого фермента.
Неспособность организма больного превратить
глюкозо-6-фосфат в глюкозу ведёт к
недостатку глюкозы в крови. Это
обусловливает относительно низкие уровни инсулина.
Результатом является усиление липолиза,
гиперлипопротеидемия (ГЛП) I или V типа,
а также ацетонемия, метаболический ацидоз
и ацетонурия.
Для больных характерны низкие натоща-
ковые уровни глюкозы и редуцированный
подъем уровня сахара крови после инъекции
глюкагона или адреналина. Обычны
повышенные уровни лактата, пирувата, тригли-
церидов, холестерина и мочевой кислоты.
В молодом возрасте формируется подагра
и уролитиаз. Даже относительно
непродолжительное голодание ведет к гипогликемии.
В тяжелых случаях такая гипогликемия
может привести к судорогам и замедлению
роста из-за хронически низкого уровня
инсулина, что сдерживает анаболические
процессы.
Больные дети имеют короткое туловище,
большой живот и увеличенную печень. Уже
в период новорожденности наиболее
заметным признаком является гепатомегалия.
Гепатомегалия обусловлена как накоплением
гликогена (не только в цитоплазме, но и в
ядрах клеток), так и накоплением липидов
(стеатоз печени). Почки также увеличены
и содержат депозиты гликогена в каналь-
цевом эпителии, но селезенка нормальна.
Дети отстают в физическом развитии.
В постпубертатный период на первый план
выступает гиперурикемия с ее
клиническими осложнениями (см. раздел
«Патофизиология нуклеинового метаболизма»).
Гликогеноз lb представляет собой редкую
аутосомно-рецессивную генокопию
предыдущего расстройства, с той разницей, что
поражен ген транслоказы глюкозо-6-фосфата
в ЭПР. Поэтому расщепление этого эфира
глюкозы недостаточно, несмотря на высокую
активность самой глюкозо-6-фосфатазы.
Клиника болезни усугубляется, по
сравнению с классической формой фон Гирке,
Патофизиология углеводного обмена 263
еще и нейтропенией с рецидивирующими
бактериальными инфекциями.
Другой печеночный гликогеноз
известен как лимит-декстриноз или
гликогеноз III типа (синонимы — ограниченный
декстриноз, болезнь Форбса-Кори). Это
аутосомно-рецессивный дефект
фермента, катализирующего гидролиз 1-6
связей в молекуле гликогена (фамильярно
именуемая «сучкорубом» — debranching
enzyme — амило-1,6-глюкозидаза).
Данное нарушение весьма распространено и
составляет примерно 1/4 всех случаев гли-
когенозов, протекающих с гепатомегалией.
В клинической картине болезни Форбса-
Кори наиболее характерна гепатомегалия
как следствие накопления гликогена. Для
молекулы гликогена, выделяемой из печени
больных, типичны короткие наружные
ветви. Гликогенолиз возможен в
незначительных масштабах, так как фосфорилироваться
могут только молекулы глюкозы, взятые из
патологически коротких цепей.
Характерно, что у больных глюкагон
повышает уровень сахара только на фоне
потребления углеводов, но не натощак,
поскольку число расщепляемых ветвей
незначительно и недостаток глюкозы
развивается быстро. В крови, в отличие от I типа
гликогеноза, не отмечается лактат-ацидоза
и гиперурикемии, однако бывает ГЛП.
Данный гликогеноз отличается более
доброкачественным течением и не угрожает жизни
ребенка, хотя может привести к фиброзу
печени, умеренной задержке физического
развития и полового созревания. У
некоторых больных в подростковом и зрелом
возрасте отмечается умеренная миопатия,
поскольку понижена и мышечная
утилизация гликогена.
Следующий печёночный гликогеноз —
болезнь Андерсена, гликогеноз IVтипа,
представляет собой дефект ветвящего фермента
гликогена. Это крайне редкое аутосомно-
рецессивное поражение.
Недуг проявляется в раннем детстве, как
печеночная недостаточность (могут быть
желтуха, асцит). В то же время гипоглике-
мический синдром не выражен. Развитие
264 АЖ Зайчик, Л.П.Чурилов Патохимия
болезни сопровождается ранним циррозом
печени и приводит к смерти очень быстро.
Данный тип гликогеноза, в отличие от всех
вышеописанных, практически не поддается
лечению.
Дефицит ветвящего фермента можно
обнаружить в лейкоцитах, мышцах и фибро-
бластах, равно как и в ткани печени. В связи
с малым возрастом больных мышечные
симптомы — гипотония, слабость —
затушеваны печеночными. Содержание гликогена
в печени обычно нормально или почти
нормально, но структура этого гликогена —
патологическая. Животный крахмал имеет
очень длинные и редкие наружные ветви.
Печеночное поражение доминирует и
при гликогенозе Via типа {болезнь Херса,
недостаточное количество или отсутствие
гепатоцитарной фосфорилазы). Патологоа-
натомически имеет место накопление в гепа-
тоцитах гликогена нормальной структуры.
Больные подвержены гипогликемии, ГЛП
и умеренному ацидозу. Клиника сходна с
течением гликогеноза I, но симптоматика
выражена в меньшей степени. Сниженная
активность фермента обнаруживается и в
гепатоцитах, и в лейкоцитах. Болезнь Хер-
са — один из редчайших гликогенозов. Его
классическая форма аутосомно-рецессивна.
Под названием гликогеноза VIb типа (ранее
применялись синонимы — VIII тип, IX
тип) фигурирует очень близкое к болезни
Херса по клинике заболевание, связанное с
дефектом киназы фосфорилазы Ь. В отличие
от классической болезни Херса,
недостаточность киназы фосфорилазы b сцеплена
с Х-хромосомой: это единственная болезнь
данной группы, которая встречается только
у мальчиков
Мышечные гликогенозы характеризуются
доминированием нарушений в
энергоснабжении скелетной мускулатуры, которые
чаще всего выявляются при физической
нагрузке и находят выражение в таких
симптомах, как миальгия, миоглобинурия,
судороги, слабость, ферментемия
мышечными энзимами. В этой группе находятся
гликогеноз Vmuna — болезнь Мак-Ардля,
чдокринно-метаболические нарушения)
гликогеноз Vllmuna, а также
ненумерованные по классификации Кори: дефицит
мышечной фосфоглицеромутазы и дефект
М-субъединицы ЛДГ.
Болезнь Мак-Ардля — отсутствие
мышечной фосфорилазы. Вероятно, это
наиболее редко встречающийся гликогеноз.
При этой аутосомно-рецессивной форме
патологии поражаются только скелетные
мышцы, в которых полностью отсутствует
фосфорилаза. Активность фосфорилазы в
гепатоцитах — нормальна, гипогликемия
не обнаруживается. При тяжелой
физической нагрузке могут возникнуть судороги,
миоглобинурия, повышение уровня
сывороточной креатинфосфокиназы. Утомление
не сопровождается адекватной продукцией
лактата. Хотя болезнь не сцеплена с
полом, большинство больных — мужчины.
Симптомы болезни обычно выражены
незначительно, что лишний раз
подчеркивает значимость внемышечных источников
энергии для сокращения мышц (жирные
кислоты и др.).
Характерная черта гликогеноза VII типа -
очень низкий уровень фосфофруктокиназы
в скелетных мышцах. Он составляет всего
1-3% от нормального. Больные способны
переносить умеренные физические
нагрузки. Клиника болезни сходна с нетяжёлым
вариантом болезни Мак-Ардля, но
отличается умеренной гемолитической анемией
кризового характера, провоцируемой
окислителями (см. т. III).
Дефект мышечной фосфоглицеромутазы
по клинике практически неотличим от
болезни Мак-Ардля и описан у единственного
больного-мужчины. Дефект
М-субъединицы ЛДГ тоже представляет собой аналог
гликогеноза V типа, но при физических
усилиях больные развивают необычный
лабораторный симптом — пируватемию, так
как ПВК мышц не восстанавливается в лак-
тат. Болезнь описана у лиц обоего пола, но
наблюдений недостаточно для однозначных
выводов о типе наследования.
Несколько особняком в группе
гликогенозов стоит гликогеноз II типа. Это генера-
лизованный гликогеноз, поражающий все
гликогенсодержащие клетки.
Синонимические названия данной болезни — гликогенная
кардиомегалия, болезнь Помпе. Фактически
это лизосомальный тезаурисмоз, так как
первичный дефект при гликогенозе II
типа — отсутствие лизосомной а-1,4- глю-
козидазы. Заболевание встречается нередко
и составляет почти 10 % от всех гликогено-
зов. Среди болезней накопления гликогена
эта форма наиболее злокачественна и не
имеет эффективного лечения, а больные
умирают в грудном возрасте. Имеется
модель заболевания у крупного рогатого
скота австралийской породы. Для данного
гликогеноза доступен пренатальный метод
диагностики, путём амниоцентеза. Ведущий
признак заболевания — увеличение сердца в
грудном возрасте, вслед за чем развивается
тяжёлая сердечная недостаточность. До
некоторой степени поражается и печень, но
главные события развиваются в мышечной
ткани: нарастает слабость скелетных мышц
(как следствие значительного накопления
неутилизируемого гликогена в скелетной
мускулатуре), формируется расширение
сердца и наступает смерть в раннем
возрасте, в результате сердечной недостаточности.
Одним из необычных внешних проявлений
служит увеличение языка.
Считается, что отсутствие фермента и
секвестрация гликогена в лизосомах
сдвигают равновесие распада и синтеза гликогена
в сторону синтеза. Как следствие гликоген
накапливается в клетках. Гипогликемия
натощак не развивается; уровень сахара крови
поднимается в ответ на глюкагон. В отличие
от болезни фон Гирке печеночный глико-
генолиз при болезни Помпе совершенно
нормален.
В биоптатах мышц находят высокое
содержание гликогена, который электронно-мик-
роскопически выявляется внутри лизосом,
типична низкая активность а-глюкозидазы.
Сниженная активность фермента
обнаруживается и в лейкоцитах, что связано с
умеренным иммунодефицитом. Отмечается
повышение уровня креатинфосфокиназы в
сыворотке.
Патофизиология углеводного обмена 265
Агликогеноз или гликогеноз 0, по
номерной классификации, представляет собой
наследственный аутооомно-рецессивный
дефицит УДФ-глюкозо-гликогентранс-
феразы, то есть гликогенсинтетазы (ГС).
В тканях больных отсутствуют отложения
гликогена. Тем не менее болезнь совместима
с жизнью, хотя пораженные дети нуждаются
в частом кормлении, у них имеются
тенденция к утренней гипогликемии, кетоз со
рвотой и судорогами, ЗПМР.
ГЛИКОЗАМИНОГЛИКАНЫ
И МУКОПОЛИСАХАРИДОЗЫ
Важными продуктами, получаемыми из
моносахаридов в результате анаболических
реакций, являются гликозаминогликаны
(ГАГ), по старой терминологии — мукополи-
сахариды. Это гетеросополимеры гексуро-
новых кислот (глюкуроновой, идуроновой,
получаемых из соответствующих
моносахаридов) и других производных глюкозы к
галактозы — аминосахаров (галактозамин,
глюкозамин). Молекулы ГАГ— это цепи
повторяющихся димеров гексуроновой
кислоты и амииосахара. В некоторых из них
вместо гексуроновой кислоты может стоять
галактоза (например, в дерматансульфате).
Компоненты ГАГ часто (за исключением
гиалуроната) сульфатированы и всегда
ацетилированы. Отдельные ГАГ отличаются
композицией димеров, внутридимерными и
междимерными связями, степенью ацетили-
рования и сульфатирования Сахаров.
Все они синтезируются на белковой
матрице-затравке, к которой крепятся через
остаток ксилозы. Этот процесс происходит
в аппарате Гольджи фибробластов, хонд-
роцитов, остеобластов, гладкомышечных,
тучных и многих других секреторных
клеток. В синтезе ГАГ участвует большое
семейство высокосубстратспецифичных
энзимов — гликозилтрансфераз, нанизывающих
моносахариды, переносимые с нуклеотид-
сахаров, на олигосахаридный акцептор. Во
время синтеза, с участием сульфатаз и ак-
266 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Табллица 13. Разнообразие гликозаминогликанов
Название
Гиалуроновая кислота
Хондроитин-сульфат А
Хондроитин-сульфат С
Дерматан сульфат
Кератан сульфаты 1 и II
Гепарансульфат и
гепарин
Кислый компонент
D-глюкуроновая к-та
D-глюкуроновая к-та
D-глюкуроновая к-та
L-идуроновая к-та
D-галактоза
О-глюкуронил-2-суль-
фат, L-идуроновая к-та
Внутриди-
мерная связь
рьз
Р1-3
рьз
рьз
Q1-3
Р1-4
Р1-4
а1-3
Аминосахар
N-ацетил-О-глюкозамин
N-ацетил-О-галактоза-
мин-4-сульфат
N-ацетил-О-галактоза-
мин-6-сульфат
N-ацетил-О-галактоза-
мин-4-сульфат
М-ацетил-О-галактоза-
мин-6-сульфат
N-ацетил-О-галактоза-
мин-6-сульфат
Междимер-
ная связь
Р1-4
Р1-4
Р1-3
Р1-3
P1-3
al-4
гиалуроновая кислота
связующий белок
кератансульфат
хондроитинсульфат
коровий белок
тивной формы сернокислого
остатка — фосфоаденозин-
фосфосульфата (ФАФС)
идет сульфатирование ГАГ.
Глюкуроновая кислота эпи-
меризуется в идуроновую
с помощью эпимераз, уже в
готовой матрице.
Наиболее важные ГАГ
перечислены в таблице 13.
Х-П''л'л'.'.Ы.
'ЧЧЧ'РР
субъединицы
ьщшттт
Рис. 56. Структура протеогликанов и
гликозаминогликанов (по А. Уайту и соавт., 1981).
Пояснения в тексте
Комплексы ГАГ и белков,
на которых они
синтезируются, называются протео-
гликанами. В протеогликане
белок составляет ядро, а ГАГ
выступают из него,
наподобие щетины щёток. Цепи гиалуроновой
кислоты составляют остов всей конструкции,
нанизывая крепящие белки (рис. 56).
ГАГ и протеогликаны — это основа
аморфного вещества соединительной ткани и в
силу этого представлены в организме
чрезвычайно широко.
Гиалуроновая кислота присутствует во
многих органах и тканях, дерматансульфат-
в коже, пуповине, связках, сухожилиях и
клапанах сердца; хондроитинсульфат А - в
хряще, костях, роговице;
хондроитинсульфат С — в хряще, связках, сухожилиях
и пуповине. Наивысшая концентрация
кератансульфатов свойственна хрящу
межпозвоночных дисков и костям, а гепарин в
Патофизиология углеводного обмена 267
изобилии имеется в артериальных стенках,
лёгких, печени, мастоцитах. Прозрачные
среды глаза богаты ГАГ.
ГАГ и протеогликаны ответственны за
контактное ингибирование и иные мор-
фогенетические взаимодействия в тканях,
как полианионы, они связывают
кальций и структурную воду, определяя гид-
рофильность ткани и ее тургор. С ними
тесно взаимодействуют как волокнистые
протеины соединительной ткани, так и
инфильтрирующие ткань белки плазмы,
например, липопротеиды (см. выше
«Нарушения липидного обмена»), с которыми
ГАГ могут образовывать комплексы. Пато-
генность многих микроорганизмов связана
с наличием у них активной гиалуронидазы
и способностью пенетрации через аморфное
вещество соединительной ткани,
следовательно, целостность ГАГ можно
рассматривать как одно из условий неспецифического
иммунитета. ГАГ представляют фактор
внутренней эластичности тканей, а в составе
синовиальной жидкости выполняют роль
смазки. С возрастом в тканях уменьшается
доля гиалуроната и растет содержание ке-
ратансульфатов.
ГАГ и протеогликаны физиологически
находятся в процессе постоянного
самообновления. Так, для хондроитин-сульфата хряща
время полужизни не превышает 9 суток. Это
значит, что в лизосомах идёт процесс распада
ГАГ и протеогликанов, уравновешивающий
постоянный синтез этих соединений. С этой
целью используется ряд ферментов — катеп-
син D, субстрат-специфичные гликозидазы,
сульфатазы.
Синтез ГАГ и протеогликанов из
углеводов и аминокислот и их лизосомальный
распад подвержены сложной гормональной
регуляции. СТГ стимулирует синтез ГАГ
и протеогликанов, особенно гиалуроната.
Этот эффект опосредован соматомединами
А и С. Соматомедин А стимулирует суль-
фатирование протеогликанов. При
акромегалии имеется накопление ГАГ в ряде
органов. Андрогены увеличивают синтез
гиалуроната, особенно в клапанах сердца,
коже, мышцах и половых органах.
N-ацетил-
галактоза
Г
soA
идуроновая
кислота
1
N-ацетил-
глюкозамин
SOA
гепарансульфат
идуроновая
кислота
—г"
SO.
дерматансульфат
глюкуроновая
кислота
N-ацетил-
галактоза
-Г
so4
Рис.57. Механизмы некоторых наследственных мукополисахаридозов. Цифровые обозначения:
1 — синдромы Хюрлер и Шейе, 2 —- синдром Сан-Филиппо А, 3 — синдром Сан-Филиппо В,
4 — синдром Слая, 5 — синдром Хантера, 6 — синдром Морото-Лами. Вверху — гепарансульфат.
Внизу — дерматансульфат
268 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушений)
Название
МПСШ(б-ньХюрлер)
МПС£(б-ньУльри-
ха-Шейе)
МПСИ(б-ньХантера)
МПС Ilia (Сан-Филип-
по А)
МПС 1Mb (Сан-Филип-
поВ)
МПС 111с (Сан-Филип-
поС)
МПС IVd (Сан-Фи-
липпо D)
МПСМ(Моркио)
МПСУКМарото-
Лами)
МПСУИ(Слая)
Множественная суль-
фатазная
недостаточность
Сиалидоз
Галактосиалидоз
Фукозидоз
Маннозидоз
Аспартилглюкозами-
1нурия
Начало
6-12мес
Позднее
6-12месили в
юности (мягкая
форма)
1-6 г
1-6 г
1-6 г
1-6 г
Варьирует
2-4 г
1-2 г
1-4г
От врожденного
до ювенильного
1-12г
3-12 мес или
ювенильный
6-18 мес
После 20 лет
Первичный дефект
а-идуронидаза
а-идуронидаза
Идуронсульфат
сульфатаза
Гепаран -N-сульфа-
таза
N-ацетил-а-глюкоза-
минидаза
АКоА: а-глюкозами-
нид-М-трансфераза
N-ацетилглюкоза-
мин-6-сульфатаза
N-ацетилгалактоза-
мин-6-сульфатаза
Арилсульфатаза В
Р-глюкуронидаза
Арилсульфатазы АС
Нейраминидазы
Защитный гликопро-
теид
а-фукозидаза
а-маннозидаза
Аспартилглюкозами-
намидаза
Субстрат
накопления
гсдс
дс
дсгс
ГС
ГС
ГС
ГС
ксхсв
ДС
гсдсхс
ГАГ и сульфатиды
Сиалалолигосаха-
риды, КС
Тоже
Олигосахариды,
гликопептиды
гликолипиды, КС
Олигосахариды, КС
Аспартилглюкоза-
мин, гликопептиды
Особенности
Грубое лицо,
сердечные заболеванияя,
анкилозы
Тоже
Тоже
Лицо слегка
искажено. Гирсутизм
Лицо слегка
искажено. Гирсутизм
Лицо слегка
искажено. Гирсутизм
Лицо слегка
искажено. Гирсутизм.
Дисплазия отростков
позвонков, шейная
нестабильность,
компрессия спинного
мозга, аортальная
нед-ть
Лицо слегка
искажено, сердечные заб-я,
анкилозы
Лицо искажено,
сосудистые заб-я
Ихтиоз, метахрома-
тическая липодист-
рофия
Миоклонус, гарго-
иллизм
Ангиокератома,
множественный дизостоз
Ангиокератома,
потеря электролитов с
потом, грубое лицо
Глоссомегалия,
грубое лицо
Грубое лицо, амина-
цидурия
Патофизиология углеводного обмена 269
Табллица 14. Наследственные мукополисахаридозы и другие лизосомальные расстройства
углеводного обмена
цнс
ЗПМР
НетЗПМР
ЗПМР
Тяжелая ЗПМР
Тяжелая ЗПМР
Тяжелая ЗПМР
Тяжелая ЗПМР
ЗПМР
ЗПМР
ЗПМР
ЗПМР
ЗПМР
ЗПМР
ЗПМР
Гепатоспленоме-
галия
+++
++
+++
+
+
+
+
+
++
++
+
++ (при ранних
формах)
++ (при поздних
формах)
++
+++
—
Клиника поражений органов
Опорно-двигательный аппарат
++++
+++
++++
+
+
+
+
++++++ очень
тяжелое,
карликовость с коротким
туловищем
++++
++++
+++
++ (при ранних
формах)
++
++
++
++
Зрение слух
Помутнение роговицы
Тоже
Дегенерация сетчатки
—
—
—
—
Помутнение роговицы,
глухота
Помутнение роговицы
Помутнение роговицы
Дегенерция сетчатки
Вишневые пятна
сетчатки
Вишневые пятна
сетчатки, помутнение
роговицы
Катаракта, помутнение
роговицы
Катаракта
Кровь I
Зернистость
лимфоцитов |
Тоже
Тоже
Тоже
Тоже
Тоже
Тоже
ТЗН
Ярко выраженная
зернистость
лейкоцитов
Ярко выраженная
ТЗН
Вакуолизация и
зернистость лейкоцитов
Вакуолизация
лимфоцитов
Тоже
То же и пенистые
клетки
Вакуолизация
лимфоцитов, ТЗН
Вакуолизация
лимфоцитов
ТЗН — токсическая зернистость нейрофилов, остальные сокращения по тексту.
270 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Тироидные гормоны способствуют
распаду ГАГ, в связи с чем гипотироз отличается
их патологическим накоплением,
приводящим к утолщению кожи и повышению ее
гидрофильности, вплоть до отеков и даже
анасарки. Глюкокортикостероиды
способствуют реполимеризации ГАГ, но угнетают и
их распад, и синтез de novo.
Инсулин значительно активирует
образование ГАГ из глюкозы и галактозы. Однако
усиливается и деградация ГАГ. Таким
образом, инсулин — промотор самообновления
ГАГ. При СД, особенно инсулинопеничес-
ком, а также экспериментальном аллокса-
новом, скорость обновления ГАГ сильно
снижена. Нарушение обмена ГАГ при СД
участвует в патогенезе диабетических анги-
опатий и вторичного иммунодефицита (см.
«Патофизиология сахарного диабета»).
Большую группу лизосомальных
наследственных болезней составляют мукополиса-
харидозы. Это тезаурисмозы, при которых
в основе патологического накопления ГАГ
лежат дефекты лизосомальных ферментов,
предназначенных для деградации данных
соединений (см. также т. I, стр. 167).
Все мукополисахаридозы (МПС) — ау-
тосомно-рецессивны, за исключением
болезни Хантера (МПС II типа), которая
рецессивна и сцеплена с Х-хромосомой.
Для большинства МПС характерны
тяжелое поражение
опорно-двигательного аппарата, часто — карликовый рост
больных, деформированные конечности,
гротескные гримасы, застывшие на лице
(синдром «гаргоиллизма» по названию
гаргоилл — химерических существ в декоре
собора Парижской Богоматери). Имеется
висцеромегалия, большой живот,
характерны грубая кожа, избыточное
оволосение, часто наблюдаются болезни сердца
и лёгких, при многих МПС развивается
умственная отсталость, поражаются органы
чувств. Как правило, накапливаемые ГАГ
выделяются с мочой. Рис. 57 представляет
обобщенную структуру ГАГ, с указанием
связей, гидролиз которых нарушен при
различных конкретных МПС.
Нозологические особенности отдельных
МПС и близких к ним лизосомальных
нарушений углеводного и гликолипидного
обмена представлены также в табл. 14.
Дифференциальная диагностика МПС
иногда представляет большую важность,
так как некоторые из них не затрагивают
интеллектуальное развитие личности, а
другие обусловливают тяжёлое слабоумие.
Особенно трагичной является ситуация при
синдроме Моркио, носители которого имеют
глубокие нарушения опорно-двигательного
аппарата и функций внутренних органов,
обречены на раннюю гибель, но
интеллектуально сохранны и даже могут
проявлять выдающиеся способности (случай
А.деТулуз-Лотрека). В связи с целями
дифференциальной диагностики в данную
таблицу вместе с истинными мукополи-
сахаридозами (11 синдромов) включены
также имеющие сходные внешние признаки
нарушения обмена олигосахаридной
части гликопротеидов, метаболизм которых
тесно переплетается с ГАГ. Это сиалидоз,
маннозидоз, фукозидоз, галактосиалидоз.
Сведения о муколипидозах I и III типов
(I-клеточной болезни и псевдосиндроме
Хюрлер) включены в раздел «Нарушения
липидного обмена». В отличие от МПС
мукополисахаридурия не характерна для
этих МПС-подобных заболеваний. В то
же время их отличительной особенностью
служит отсутствующая при истинных МПС
крайне высокая активность сывороточных
кислых гидролаз.
Рассмотрение основных путей углеводного
метаболизма и частных нарушений его
отдельных этапов подготавливает нас к подробной
дискуссии, посвященной патофизиологии
главного нарушения регуляции углеводного
обмена, затрагивающего все виды
метаболизма. Речь пойдет о сахарном диабете (СД).
Глава 9
ПАТОФИЗИОЛОГИЯ
САХАРНОГО ДИАБЕТА
(Написано при участии Ю.И. Строева и В.И. Утехина)
«Диабет всегда начинается незаметно»
А.Т. Камерон, 1945
• Определение и классификация
• Инсулинзависимый сахарный диабет
• Этиология ИЗСД
• Патогенез ИЗСД
• Метаболические нарушения при сахарном
диабете
• Патогенез острых осложнений СД
• Синдром гипергликемии и гипергликемическая
кома
• Гипогликемическая кома и проблема
гипогликемии
ВВЕДЕНИЕ
Сахарный диабет (СД) известен со времен
глубокой древности. Его симптомы описаны
еще в папирусе Эберса (Египет, 1500 лет до
н. э.) В Аюрведах Индии (за 600 лет до н. э.)
описана болезнь «мадхумеха»:
1) диабет тощих, физически слабых,
беспокойных с шершавой кожей, с полифагией
и полидипсией и
2) диабет тучных, сонливых с мягкой и
жирной кожей и выраженной полифагией.
Первых лечили обильной пищей,
вторых — диетой и голоданием. СД как болезнь,
при которой происходит мочеизнурение
и «плоть растворяется», описал античный
медик Аретей Каппадокийский (81-133
отР.Х.).
Считается, что сахарный диабет не был
частой болезнью в ранних цивилизациях.
И до сих пор в примитивных сообществах
охотников и собирателей встречаются лишь
его единичные случаи. Изменения экологии
Патогенез хронических осложнений СД
Инсулиннезависимый сахарный диабет
Генетика ИНСД
Морфология и гормонообразующая функция
островков при ИНСД
Патогенез ИНСД и его фазы
Патогенез осложнений ИНСД
Инсулинорезистентность: виды и механизмы
Экспериментальные модели сахарного диабета
Патофизиологические основы лечения СД
человека в современном цивилизованном
обществе сделали сахарный диабет частым
заболеванием.
По данным медицинской статистики:
>* больные СД составляют более 5 %
населения (13 млн человек только в США);
>* среди пожилых (возрастная группа от
65 до 74 лет) СД болен почти каждый
пятый;
>* риск заболевания СД удваивается на
каждые 20 % избыточного веса;
>* СД — третья по частоте причина смерти
(уносящая более 300 тыс. человек в мире
ежегодно);
>- СД — ведущая причина слепоты в
развитых странах;
>- половина больных инсулинзависимым
СД умирает от хронической почечной
недостаточности (ХПН);
>- 3/4 больных инсулиннезависимым СД
умирают от осложнений атеросклероза;
272 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
>* у больных СД в 2 раза чаще, чем в
популяции, бывают болезни сердца и в 17 раз
чаще — болезни почек.
В последние 10 лет отмечается рост
заболеваемости СД во всех странах мира. В
России не менее 3-5 % населения больны
СД. В популяции больных СД 6-8 %
составляют дети в возрасте до 14 лет. Ежегодный
прирост числа детей, больных ИЗСД,
составляет до 6 %. До 13 % больных сахарным
диабетом детей — младше 5 лет.
По данным ВОЗ, лидирующее место по
заболеваемости ювенильным СД занимают
страны Скандинавии, и в частности,
Финляндия, где частота этого заболевания в
различных популяциях от 30 до 60 случаев
на 100 тыс. населения. За последние
тридцать лет заболеваемость ИЗСД удвоилась
на северо-западе США, в Финляндии и
Норвегии. Наименьшая встречаемость — в
Южной Корее и составляет около 0,5 на 100
тыс. населения.
ОПРЕДЕЛЕНИЕ
И КЛАССИФИКАЦИЯ
Идиопатический СД — это хроническое
мультигормональное расстройство всех видов
метаболизма, характеризующееся
нарастающей гипергликемией, глюкозурией, развитием
осложнений, в основе которых лежат
повреждения сосудов, а также нейропатия.
Первичный сахарный диабет — идиопати-
ческое расстройство механизмов инсулино-
вой регуляции метаболизма. Это
заболевание может быть вызвано либо деструкцией
В-клеток панкреатических островков и
абсолютной инсулиновой недостаточностью,
либо комбинацией первичной
резистентности тканей-мишеней к инсулину, а В-клеток —
к глюкозе, порождающей относительную
недостаточность инсулина. Поэтому
первичный СД, составляющий около 95 % всех
случаев СД, делится на 2 типа и несколько
подтипов, поименованных ниже:
>* Первичный сахарный диабет I типа
(синонимы: инсулинзависимый, гипоин-
сулинемический, юношеский, ювениль-
ный, ИЗСД) составляет 20% от общего
числа случаев первичного СД и делится
на следующие подтипы:
1а — обусловленный комбинацией
генетического и средового воздействия;
lb — первичный, генетически
обусловленный, со спонтанным аутоиммунным
процессом, без явных признаков экзогенной
провокации;
1с — с первичным поражением В-клеток
экзогенными диабетогенами (химическими,
вирусными).
Каждый из этих вариантов вначале, пока
количество инсулина достаточно, чтобы
предотвратить кетоацидоз, но уже не
достаточно, чтобы поддерживать уровень
глюкозы, может условно расцениваться в
клинике как «инсулиннезависимый», что не
меняет его патогенетической сути и
принадлежности к I типу.
>■ Первичный сахарный диабет II типа
(синонимы: инсулиннезависимый, гиперин-
сулинемический, взрослых, пожилого
возраста, тучных, ИНСД) составляет
почти 80 % от общего числа случаев
первичного СД.
Данный тип включает несколько особых
подтипов. Среди них:
Па — ИНСД у нетучных больных
ПЬ — ИНСД у тучных больных
Пс — ИНСД в юношеском возрасте — до
5 % больных («диабет взрослых в
юности» — ДВЮ; или в англоязычной
литературе: «maturity onset diabetes of the young» -
MODY, «мэйсоновский тип»).
Вторичный сахарный диабет, или
диабетические (гипергликемические) синдромы,
возникает как следствие по отношению к
другим болезням, поражающим
поджелудочную железу или систему регуляции
углеводного метаболизма. К этой группе относятся:
а) вторичный СД, вызванный
неаутоиммунной деструкцией панкреатических
В-клеток при поражении поджелудочной
железы (хронический панкреатит, рак,
гемохроматоз, кистозный фиброз, пан-
Патофизиология сахарного диабета 273
Таблица 15. Критерии ИЗСД и ИНСД
изсд
Абсолютная инсулиновая недостаточность
Аутоиммунный процесс против В-клеток
панкреатических островков
Отсутствие первичной инсулинорезистентности
Высокий риск кетоацидоза
Нет связи с тучностью
Конкордантность однояйцевых близнецов 30-50%
ИНСД
Относительная инсулиновая недостаточность
Отсутствие аутоиммунного процесса против В-клеток
панкреатических островков
Первичная инсулинорезистентность
Низкий риск кетоацидоза
Связь с тучностью
Конкордантность однояйцевых близнецов 90-100%
креатэктомия, травма поджелудочной
железы, синдром Рефсума),
b) вторичный диабет, вызванный
эндокринными расстройствами с
гиперпродукцией контринсулярных гормонов
(синдром Кушинга, акромегалия, фео-
хромоцитома, глюкагонома, гипертироз,
гиперплазия эпифиза, множественная
неопластическая полиэндокринопатия
MEN-1),
c) вторичный ятрогенный диабет при
применении медикаментов (кортикосте-
роиды, АКТГ, оральные контрацептивы,
пропранолол, антидепрессанты, феноти-
азины, некоторые мочегонные),
d) вторичный диабет при генетически
обусловленных синдромах (липодистро-
фии, гликогеноз I типа, миотоническая
дистрофия и атаксия-телеангиэктазия,
синдром Вернера, эльфизм, синдром
Рабсона- Менденхолла, наследственные
гипоталамические формы вторичного
ожирения — см. раздел «Нарушения
липидного обмена», хромосомные
аномалии Дауна, Шерешевского-Тернера и
Клайнфелтера, семейная атаксия Фриде-
рейха).
Термин «I тип» часто используют как
синоним для ИЗСД, а термин «II тип»
относят к любому ИНСД. Однако эти
градации совпадают не полностью, поскольку
некоторые больные СД II типа могут быть
полностью инсулинзависимыми и
склонными к кетоацидозу. Подгруппа На больных
ИНСД — нетучные субъекты, экспрессиру-
ющие антигены ГКГС, предрасполагающие
к аутоиммунному инсулиту, имеют и
признаки иммунного ответа на островковокле-
точные антигены и вместе с тем первичную
инсулинорезистентность.
Следует помнить, что термины «ИЗСД»
и «ИНСД» описывают клиническое
течение конкретного случая (склонный к
кетоацидозу и резистентный по отношению к
кетоацидозу соответственно), тогда как
термины «1тип» и «И тип» относятся к
патогенетическим механизмам болезни (результат
доминирования аутоиммунного механизма,
или результат реализации иных механизмов
патогенеза соответственно).
Ниже следует таблица 15,
обрисовывающая основные
дифференциально-диагностические и патогенетические различия
двух типов первичного СД, существование
которых впервые научно обосновал Э. Лан-
серо(1880).
Несмотря на разную этиологию и отличия
в ключевом звене патогенеза, оба типа СД
при длительном хроническом течении
вызывают нарушения метаболизма, приводящие
к вторичной ангиопатии и связанным с ней
осложнениям.
Кроме того, имеются данные (Р. Лесли,
Р. Эллиотт, 1994) о том, что ежегодно более
1 % больных ИНСД становятся типичными
носителями ИЗСД с характерными
иммунологическими и метаболическими
особенностями, что, скорее всего, свидетельствует
об отсутствии жесткой альтернативности
двух типов заболевания, а вероятный
механизм развития этого явления — наслоение
иммунопатологического процесса на
инсулинорезистентность. В этой связи любо-
274 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
пытны данные о том, что экспрессия одного
из основных аутоантигенов В-клеток у
больных с ИЗСД — глутаматдекарбо-
ксилазы — усиливается гипергликемией
(А. Россини, 1993).
Существование ИНСД,
сопровождаемого и не сопровождаемого ожирением,
учитывая последние данные о продукции
адипоцитами контринсулярного цитокина
ФНОа, представляется весьма важным.
Также имеются основания предполагать,
что значительная часть нетучных больных
с ИНСД — это лица, находящиеся в ранних
стадиях эволюции ИЗСД и все еще
имеющие достаточно инсулина, чтобы
предотвратить возникновение кетоацидоза.
Итак, инсулинзависимый и инсулинне-
зависимый СД имеют немало общих
патогенетических звеньев, и в настоящее время
нельзя отрицать существования смешанных
и переходных форм между этими двумя
вариантами первичного СД.
Некоторые авторы (Ф. Фелиг, 1985)
отдельно выделяют СД беременных, именуя
его «прегнодиабет». Причина прегнодиабе-
та— возрастание при беременности
продукции контринсулярных гормонов.
Классификация ВОЗ подразумевает
под диабетом беременных впервые
выявленную или начавшуюся только во время
беременности пониженную толерантность
к глюкозе. При этом подчеркивается, что в
данную группу не входят забеременевшие
больные СД женщины. После родов у
женщин с прегнодиабетом явный диабет может
не возникнуть, но может и развиться СД I
либо II типа. Это свидетельствует о том, что
этиологически прегнодиабет не является
самостоятельной формой.
ИНСУЛИНЗАВИСИМЫЙ
САХАРНЫЙ ДИАБЕТ
ИЗСД составляет в целом от 10 до 20
процентов всех случаев СД и особенно значим
в детской и подростковой медицине.
Этиология ИЗСД
Этиология ИЗСД мультифакториальна.
Большинство авторов считают, что ряд
экзогенных факторов, включая вирусы и
химические диабетогены, способен
спровоцировать цитолиз островковых В-клеток,
вызывая аутоиммунный процесс. Однако
подобная провокация аутоиммунного
цитолиза возможна только в организме
генетически предрасположенных индивидов с
наследственными особенностями регуляции
иммунного ответа. При этом не все
предрасположенные заболевают, что указывает на
роль экзогенных факторов. Взаимодействие
генов и среды подобного характера известно
как мультифакториальное наследование
или аддитивно-полигенное наследование
с пороговым эффектом и провоцирующей
ролью факторов среды (см. т. I, с. 134-135).
Характерной особенностью ИЗСД является
то, что внешнее провоцирующее
воздействие, как правило, имеет наиболее важное
значение в течение раннего и сравнительно
ограниченного периода онтогенеза. Именно
поэтому больные ИЗСД чаще заболевают
в молодом возрасте. Своеобразие
взаимодействия генов и среды при развитии ИЗСД
позволило Д. Реймонду охарактеризовать
эту болезнь как «мечту эпидемиолога», но
Р. Котран и соавт. награждают ее не менее
эмоциональным эпитетом «кошмар
генетика» (1989).
Генетические маркеры
повышенного риска при ИЗСД
Впервые на наследственную природу СД
указал еще Б. Наунин (1890), генетическую
теорию его этиологии много позже развил
Н.Э. Симпсон (1964). К группе риска по
ИЗСД должны быть отнесены в первую
очередь носители определенных генетических
маркеров, особенно контактировавшие с
вирусными и химическими диабетогенами.
Конкордантность монозиготных близнецов
по ИЗСД не превышает 30-54 %. В отличие
от некоторых форм ИНСД и диабета
взрослых в юности ИЗСД не менделирует. В
настоящее время насчитывают до 20
различных хромосомных участков, проявляющих
Патофизиология сахарного диабета 275
признаки положительной сцепленности с
заболеванием. У детей ближайших кровных
родственников больных ИЗСД частота
заболевания составляет около 6%.
Согласно современным оценкам,
исключительный вклад в мультифакториальную
предрасположенность вносит регион генов
ГКГС в коротком плече 6-й хромосомы
между локусами DR и Bf. При ИЗСД
часто встречаются гены, кодирующие белки
DR! з)4' которые per se видимо, прямо не
определяют подверженности ИЗСД. Но
гены DQ, наследуемые сцепленно с ними,
диктуют эту подверженность. Наличие
аспарагиновой кислоты в 57-м положении
(3-цепи белка DQ препятствует развитию
ИЗСД. Если же в этом положении стоит
любая другая аминокислота, подверженность
организма ИЗСД возрастает.
Полагают, что сцепление локусов белков
ГКГС 2-го класса и ИЗСД объясняется
иммунологическими функциями белков
ГКГС (см. т.1, с. 99-102, 410-413). Белки
ГКГС 2-го класса служат для связывания
пептидных фрагментов экзоцеллюлярных
антигенов при их презентации в иммунном
ответе. 57-й аминокислотный остаток
пептида, кодируемого геном HLA-DQ-bl*3.2,
пространственно находится как раз в том
участке молекулы белка ГКГС 2-го класса,
который связывает презентуемые
фрагменты антигенов. Считается, что при наличии
в этом положении не аспарагиновой, а иной
аминокислоты (в частности, у человека —
серина, аланина или валина) происходит
перекрестная презентация чужих и своих
эпитопов и провоцируется аутоиммунный
процесс.
Среди европеоидов почти 95 % больных,
страдающих ИЗСД, являются носителями
антигенов ГКГС DR3, DR4 и/или их комбинации.
Среднемировой популяционный процент
носителей этого гаплотипа не более 4 %, в
североевропейских регионах он
поднимается до 45 %. Как уже отмечалось выше, при
рассмотрении классификации СД по
наличию генетических маркеров и особенностям
картины болезни ИЗСД подразделяют на
подтипы 1а и lb.
При подтипе lb отмечается частое
присутствие в ГКГС-наборе антигенов DR3(D3)-
Bg-Aj; подтип Охарактеризуется наличием
комбинации DR4(D4)-B15-A2-Cw3. Любой из
указанных генов положительно
неравновесно сцеплен с ИЗСД, но чаще всего они
присутствуют именно в указанных сочетаниях.
Комбинация lb сопровождается
развитием ИЗСД на фоне системного
аутоиммунного органоспецифического поражения
ряда эндокринных желез (аутоиммунная
полиэндокринопатия). Проявления ауто-
иммунитета против островковых В-клеток
носят стойкий характер и отмечаются на
протяжении длительного периода болезни.
В то же время выраженный иммунный ответ
на исулин отсутствует. ИЗСД lb примерно
у 20 % больных бывает компонентом двух
различных плюриорганных аутоиммунных
синдромов.
Первый включает: болезнь Аддисона,
гипопаратироз, витилиго, пернициозную
анемию, гипотироз, хронический активный
гепатит, алопецию, синдром мальабсорбции
и кандидоз.
Второй характеризуется сочетанием с
болезнью Базедова-Грейвса и тяжелой
миастенией (но не сочетается с гипопаратирозом,
кандидозом и аутоиммунным гепатитом).
Остальные элементы 1-го и 2-го синдромов
совпадают.
При симптомокомплексе 1а системная
аутоиммунная полиэндокринопатия
отсутствует, а в патогенезе прослеживается
роль инфекции, аутоиммунитет против
островковых В-клеток носит преходящий
характер, а аутоиммунный ответ на инсулин
всегда сильно выражен.
Ассоциация ИЗСД с гаплотипами DR3
и DR4 изучена наиболее подробно, вместе
с тем при ИЗСД у европеоидов повышена
частота встречаемости и некоторых других
генов ГКГС: А10, DR7, DRw8, B18, DRj. У
монголоидов ИЗСД положительно
неравновесно сцеплен с ГКГС-антигенами В12 и DYT, a
также DRg, аналогичным DR3 европеоидов.
У негроидов наряду с этим имеется
положительная корреляция с наличием DR7 и DR9.
276 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
Районы минимального и максимального
распространения гаплотипов,
ассоциированных с ИЗСД, совпадают с максимальной
(Скандинавия) и минимальной (Корея)
пораженностью этой болезнью (см. также
т. I, с. 495-496).
В ответ на экзогенные стимулы гены
ГКГС в В-клетках панкреатических
островков экспрессируются в зависимости от
структуры, и это может определять их связь
с ИЗСД. Так, гены ГКГС 1-го класса
кодируют белки, участвующие в презентации
пептидов из состава собственных белков.
Это придает им центральную роль в
регуляции аутотолерантности и в устранении
клеток, поврежденных вирусами.
Предполагается, что дефект белков ГКГС 1-го класса
приводит к их избыточной или аномальной
экспрессии на В-клетках панкреатических
островков, обусловливая аутоаллергичес-
кий процесс. У мышей линии NOD
аутоиммунный диабет возникает спонтанно (см. в
разделе «Экспериментальные модели СД»).
У них обнаружены аномалии базальной
экспрессии антигенов ГКГС 1-го класса.
Считается, что вероятность заболеть
ИЗСД при наличии гена DR3 возрастает
в 5 раз, гена DR4 — в 7 раз, а у гетерозигот
DR3/DR4OHa выше в 14,3 раза (Дж. Бэтче-
лор, 1989).
За пределами генов ГКГС также
существуют гены, связанные со склонностью
заболеть ИЗСД. Это гены 5'-фланкирующей
области, соседствующей с геном инсулина.
Их вклад в мультифакториальное
сцепление оценивается приблизительно в 10%,
что ставит данный локус на второе место по
значимости среди регионов неравновесного
сцепления. Условное обозначение данного
хромосомного региона — IDDM2 или 11р15,
что отражает его расположение в
одиннадцатой хромосоме. В этой же хромосоме вблизи
рассматриваемого локуса находится ген ин-
сулиноподобного фактора роста 2. Инсули-
новому гену предшествует олигонуклеотид
из 14 пар азотистых оснований. Его
молекулярная масса у 99 % индивидов либо более
^-метаболические
1600 D, либо менее 600 D. Подавляющее
большинство больных ИЗСД имеют только
короткий вариант. Лица, имеющие длинный
вариант или оба, болеют ИЗСД значительно
реже. Любопытно, что вирусы могут иметь
последовательность, сходную с коротким
олигонуклеотидом. Аутоантитела
спонтанно диабетических мышей NOD распознают
общие эпитопы на молекулах инсулина и
антигена ретровирусов р73. Генов этой
области у больных ИЗСД могут быть связаны
с патогенезом болезни как через особенности
процессинга инсулина, так и через индукцию
перекрестного аутоиммунного ответа на
В-клетки у носителей ретровируса.
Остальные локусы сцепления вносят в
мультифакториальную
предрасположенность существенно меньший вклад. Среди
них:
>- IDDM3, расположенный в 15-й
хромосоме (15q);
>- IDDM4, сцепленный с хромосомой llq,
находится вблизи гена фактора роста
фибробластов;
>* IDDM5, представляющий регион
хромосомы 6q вблизи области ESR,
располагается по соседству с геном суперок-
сиддисмутазы — фермента, влияющего
на устойчивость островковых В-клеток
к альтерации свободными радикалами;
>• во второй и в десятой хромосомах
располагаются локусы D2S326 и D10S193,
проявляющие положительное
неравновесное сцепление с ИЗСД. Оба локуса
находятся вблизи генов фермента глута-
матдекарбоксилазы, являющегося одной
из мишеней аутоиммунной атаки при
ИЗСД;
>• в 14-й хромосоме локализуются гены
иммуноглобулинов. Оказалось, что у
больных ИЗСД имеются определенные
аллотипические особенности тяжелых
цепей IgG (Gm). У лиц с различными Gm
иммунный ответ на инсулин различается
по силе;
>- обнаружена связь между И 3 СД и
гетерозиготным фенотипом р-цепи Т-клеточ-
ного рецептора лимфоцитов.
Ограниченность разнообразия Т-клеточного
рецептора — общая черта аутоиммунных
эндокринопатий, особенно характерная
для пациентов с ИЗСД подтипа lb;
> гены фактора некроза опухолей ( Ф Н Оа)
также обладают полиморфизмом,
ассоциированным с ИЗСД. Фрагменты
рестрикции 5,5 kb и 10,5 kb связаны
с гаплотипами DR3 и DR4 у больных
ИЗСД;
> один из недавно обнаруженных локусов
сцепления с ИЗСД, находящийся в 18-й
хромосоме, возможно, связан с
антигенами групп крови системы Kidd;
> в семьях, где ИЗСД болен отец, число
больных детей в 4-5 раз больше, чем в
семьях, где больна мать. Эти факты
показывают, что в патогенезе заболевания
определенную роль могут играть какие-то
факторы, ограниченные полом;
> иммунологический конфликт между
матерью и плодом по антигенам
системы АВО и Rh+ увеличивает вероятность
развития у ребенка ИЗСД 1.
Даже индивид, обладающий аллелями,
сцепленными с высоким риском ИЗСД,
может прожить долгую жизнь и не заболеть
сахарным диабетом. Предрасположенность
создаёт лишь определенную высокую
вероятность заболевания. Для реализации этой
печальной возможности нужны события,
которые для индивидуального онтогенеза
случайны.
Провоцирующее событие.
Вирусные и химические
диабетогены
Диабетогенные факторы — это события,
любое из которых с определенной долей
вероятности может запустить развитие ИЗСД
у носителей генетических особенностей,
описанных в предыдущем разделе. Ни один
из факторов не порождает сахарного диабета
в 100% случаев и ни один из них не
достаточен для развития болезни в организме,
лишенном соответствующих особенностей
реактивности.
Патофизиология сахарного диабета 277
Таким образом, этиология ИЗСД может
быть гораздо адекватнее рассмотрена с
позиций кондиционально — конституциональной
доктрины, чем с точки зрения монокауза-
лизма (см. также об этом т. I, стр. 42-45,85).
Выделяют диабетогены инфекционные и
неинфекционные. Ведущее значение
имеют инфекционные вирусные диабетогены.
Впервые вирусную теорию СД
сформулировал К.У. Тэйлор (1970).
С тех пор как Я.В. Юн и соавторы (1978)
выделили из органов ребенка, погибшего
от ИЗСД, вирус Coxsackie B4,
вызывавший у подопытных животных инсулит и
гипергликемию, энтеровирусная инфекция
Коксэки рассматривается многими
авторами как главный кандидат на роль фактора,
повреждающего островковые В-клетки.
У данного серотипа вируса Коксэки
обнаружены общие полипептиды с ферментом
глутаматдекарбоксилазой, которая служит
одной из основных мишеней аутоаллергии
при ИЗСД.
Необходимо иметь в виду, что до 15%
больных с ИЗСД относятся к подтипу lb и
имеют системную аутоиммунную полиэн-
докринопатию, при которой специальной
инфекционной провокации не требуется.
В связи с этим роль инфекции четче
выявляется, если вычленить из общего числа
больных лиц с ИЗСД подтипа 1а.
Далее было показано, что и другие
вирусы способны спровоцировать повреждение
В-клеток панкреатических островков. Среди
них возбудители:
>• краснухи;
>* эпидемического паротита;
>* энтеровирусы (не Coxsackie);
>- реовирусы;
>* цитомегаловирусы.
Эпизодически сообщалось о диабето-
генности вирусов кори, осповакцины и
вируса Эпштейна-Барр. Считается вполне
доказанной связь между краснухой в
третьем триместре у беременных и ИЗСД у
рождённых ими детей: до 40 % детей,
имевших врождённую краснуху, в первые 30 лет
внеутробной жизни заболевают ИЗСД. Пик
278 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
заболеваемости в катамнезе таких детей
приходится на 13 лет.
Для экспериментальных животных
особенно диабетогенен вирус энцефаломио-
кардита мышей. При введении мышам его
М-варианта у 40 % животных
воспроизводится модель ИЗСД (см. ниже). При этом
иммунодепрессанты и отсутствие тимуса
препятствуют индукции заболевания.
С точки зрения представлений о
провоцирующей роли инфекции в генезе
аутоиммунного процесса аутоантитела против
поверхностных и ядерных антигенов
возникают как аутоантиидиотипы к вирусным
белкам, обеспечивающим фиксацию вируса
на В-клетках и в их геноме. Согласно
эффекту иммунологического зеркала, такие
антиидиотипы должны связывать те же
структуры, что и панкреатотропные вирусы
(см. т. I, стр. 116-117).
Большинство диабетогенных вирусов
оказывают своё действие через индукцию
аутоиммунного цитолиза островковых
В-клеток. Такая индукция присуща
течению инфекций у наследственно
предрасположенных субъектов. Лимфотропные
диабетогенные вирусы действуют как по-
ликлональные активаторы аутоиммунитета
(коревой вирус, вирус Эпштейна-Барр) или
инактивируют Т-супрессоры (реовирусы).
Вирусные инфекции, поражающие
поджелудочную железу, вызывают накопление ин-
терлейкинов и интерферонов, что запускает
аберрантную экспрессию антигенов ГКГС на
В-клетках островков и ведет к аутопрезен-
тации поверхностных антигенов В-клеток и
к последующему аутоиммунному цитолизу.
Особенно важно усиленное выделение в
островках, подверженных действию инфекции,
интерферона-у. Имеется предположение,
что персистирование «медленных» вирусов
в геноме В-клеток ведет к экспрессии на
клетках аномальных белков, являющихся
неоантигенами.
Наконец, возможна вирусная индукция
аутоиммунитета через вызванное вирусами
изменение соотношения субпопуляций
лимфоцитов. В этом случае аутоаллерги-
ческий процесс может быть следствием
вирус-индуцированного дефицита супрессо-
ров и/или избытка эффекторов (см. также о
механизмах аутоаллергии т. I, с.499-504).
В анамнезе многих больных ИЗСД нет и
намёка на какие-либо диабетогенные
инфекции. В то же время существует обширный
опыт моделирования СД, в том числе — ин-
сулинзависимого, с помощью различных
химических воздействий, Например, аллоксана
(см. «Экспериментальные модели сахарного
диабета»).
Это привело к формированию
представлений о том, что существуют и химические
диабетогены.
Сильным химическим диабетогеном
является средство для борьбы с
грызунами — вакор, а также нитрозамины и нит-
розомочевина. Эти агенты цитотоксичны
для В-клеток в условиях перфузии in situ.
Предполагается также, что начальное
повреждение панкреатических островков
химическими диабетогенами вызывает
интерлейкин-зависимую аберрантную
экспрессию DQ-DR-белков и стимулирует
аутоиммунный процесс.
Аллоксан, мочевая кислота, стрептозо-
тоцин, применяемые для моделирования
ИЗСД у животных, очевидно, способны не
только прямо вредить клеткам-источникам
инсулина, но и изменять антигенную
структуру В-клеток или вызывать возникновение
перекрестных антигенных детерминант, что
и провоцирует аутоиммунный процесс: у
«химически индуцированных» животных
возникает инсулит. Диабетогенный
эффект химических диабетогенов снимается
или ограничивается антилимфоцитарной
сывороткой и иммунодепрессантами;
воспроизводится адоптивным переносом
лимфоцитов больных животных — здоровым;
не моделируется у бестимусных мышей; а в
ряде случаев его выраженность достаточна
только при наличии антигенов ГКГС D3
и/или D4.
Средство для лечения пневмоцисто-
за — пентамидин — также вызывает
выраженную деструкцию В-клеток с участием
прямого и аутоиммунно-опосредованного
механизмов.
Однако существуют и такие
экспериментальные модели инсулинопенического СД,
которые не вовлекают аутоиммунные
механизмы: например, дитизоновая, связанная
с прямой токсичностью блокатора цинка
дитизона для островковых В-клеток.
В последнее время внимание
привлекают химические диабетогенные потенции
белка коровьего молока — бычьего
сывороточного альбумина (БСА).
Пептиды из состава БСА способны запускать
перекрестный иммунный ответ против
антигенов В-клеток у генетически
предрасположенных к аутоаллергии субъектов.
Эпидемиологически показана связь
между высокой частотой ИЗСД и высоким
потреблением молока. Примером такой
популяции может служить Скандинавия.
Среди аборигенов Австралии, японцев,
полинезийцев и некоторых групп
североамериканских индейцев, употребляющих очень
мало коровьего молока и вскармливающих
грудных детей исключительно
естественным путём, ИЗСД представляет большую
редкость.
Имеются данные, что БСА обладает:
1) антигенными детерминантами
секвенциального типа, перекрестно реагирующими
с человеческим инсулином, а также
2) общими антигенными
детерминантами с мембранным белком В-клеток ICA69
(синоним р69).
Поскольку в физиологических условиях
молекулы БСА разрушаются в ЖКТ до
такой степени, что сохранность конформа-
ционных и даже многих секвенциальных
детерминант представляется
проблематичной, альбумин молока как фактор риска
имеет большое значение в диете грудных и
особенно новорожденных детей, у которых
достаточно крупные пептиды могут
всасываться в кишечнике неповрежденными.
Показано, что искусственное
вскармливание значительно повышает риск
возникновения ИЗСД, а естественное (в течение, по
крайней мере, первых трёх-четырёх месяцев
жизни) предохраняет от развития болезни.
В человеческом альбумине отсутствует
эпитоп, ответственный за молекулярную
Патофизиология сахарного диабета 279
мимикрию с островковыми антигенами
(Г.Герстайн, 1994).
Превентивный эффект естественного
вскармливания можно частично связать и
с наличием в грудном молоке IgA и
лимфоцитов, повышающих противовирусный
иммунитет ребенка.
При наличии раннего контакта с
пептидами альбумина коровьего молока дети,
имеющие генетические особенности (и прежде
всего антигены ГКГС D3 и D4), очевидно,
не могут адекватно разграничить ауто- и
гетероантиген, что запускает перекрестный
иммунный ответ с участием
антиальбуминового хелпера и «антисвоего» эффектора.
Данные о диабетогенных свойствах БСА
нашли подтверждение в диетологических
экспериментах на мышах линий ВВ и NOD,
у которых торможение диабетогенеза
достигается при замене коровьего молока в диете
мышат на гидролизаты и L-аминокислоты.
Возможно, что термическая денатурация
альбумина коровьего молока способна
повлиять на его антигенно-перекрестные
свойства, и на кипячении молока может
быть основан способ предупреждения его
диабетогенного эффекта.
Ряд других компонентов диеты
оказывает влияние на вероятность заболевания
ИЗСД.
Нитрозамины содержатся в копченых
продуктах и способны провоцировать
деструкцию В-клеток. Сезонное употребление
копченых продуктов, содержащих
повышенные количества нитрозосоединений, связано
с подъемами заболеваемости ИЗСД у детей
в Исландии. Эффект воспроизводится при
кормлении мышей копченостями из
сезонной диеты исландцев.
Существует связь между развитием
абсолютной инсулиновой недостаточности
и употреблением продуктов, содержащих
пищевые цианиды. Более 2 000 растений
могут обогатить диету цианидами. Среди них
абрикосовые косточки, миндаль, бамбук,
тапиока и особенно потребляемая в
тропической Африке кассава, клубни которой служат
питанием для почти 400 млн людей. Корни
280 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
и листья кассавы содержат цианогенные
гликозиды линамарин и лотавстралин,
токсичные для ЦНС и небезопасные для
панкреатических островков. Существенному
повышению уровня цианидов и тиоцианатов
в крови способствует и курение (X. Реслинг,
П. Лундквист, 1992).
Так как хронический алкоголизм связан
с развитием вторичного гемохроматоза и
панкреатита, а также усиливает проявления
аутоиммунитета, вероятно, следует отнести
злоупотребление алкоголем к числу
пищевых факторов риска сахарного диабета.
Широко обсуждается роль основных
нутриентов в этиологии ИЗСД. Хотя
механизмы взаимосвязи далеко не ясны,
такая зависимость, вероятно, существует.
Так, избыток легкоусвояемых углеводов
усиливает как деструкцию В-клеток, так
и инсулинорезистентность (B.C. Генес,
СВ. Генес 1993). Один из возможных
механизмов — усиление секреции инсулина
и вызванное этим увеличение экспрессии
поверхностных аутоантигенов В-клеток.
Наряду с диабетогенами,
поступающими с пищей, имеются и антидиабетогены:
обогащение рациона гидролизатом казеина
снижает у мышей NOD частоту ИЗСД по
сравнению с мышами, в диету которых
входили обычные растительные белки.
К антидиабетогенам относят и
серосодержащие аминокислоты, дефицит которых
усиливает токсичность пищевых цианидов,
а также антиоксиданты и цинк. В
большинстве случаев механизмы их действия
связаны с неспецифическим торможением
альтерации (некроза). Для цинка эффект
может быть обусловлен его участием в
депонировании инсулина. Ингибитор АДФ-
рибозилирования витамин РР в больших
дозах тормозит процессы апоптоза и некроза
и даже используется для лечения ИЗСД.
Нехватка витамина F у мышей тормозит
развитие диабета, вызывая иммунодефицит.
Добавление полиненасыщенных жирных
кислот из морепродуктов подавляет синтез
IL-1 и TNF, известных как цитотоксические
и антиинсулярные агенты для В-клеток (см.
«Патофизиология витаминного обмена»,
«Патофизиология обмена
микроэлементов»).
Итак, роль провоцирующего события при
ИЗСД состоит в экзогенном запуске
аутоиммунного процесса в островках Лангерганса.
При определенных условиях — например,
системных аутоиммунных полиэндокринопа-
тиях, имеющихся примерно у 10 % больных с
подтипом ИЗСД lb, — провокация не является
необходимой. ИЗСД — заболевание с
длительным иммунологическим продромом и еще
более пролонгированным периодом полной
метаболической компенсации. Максимальный
интервал от начала аутоиммунного процесса
до начала интолерантности к глюкозе
составляет 3-4 года, а наиболее длительный разрыв
между первыми проявлениями снижения
способности вырабатывать инсулин и явной
метаболической декомпенсацией —11—12 лет.
Кривая заболеваемости ИЗСД наиболее резко
поднимается вверх в возрастные периоды от
новорожденности до 3 лет, и от 9 до 13 лет, а
после 14 лет — прогрессивно снижается.
Таким образом, провоцирующее событие
должно произойти в раннем онтогенезе: в начале
внеутробной жизни или даже до рождения.
После определенного возрастного периода
потенциальные возможности большинства
экзогенных диабетогенов спровоцировать
деструкцию В-клеток снижаются. Более того,
спровоцированный диабетогенными
факторами процесс не всегда «доходит» до уровня
явного диабета (рис. 58, отражающий стадии
развития процесса).
Патогенез ИЗСД
Ключевое звено патогенеза ИЗСД —
прогрессирующая гибель В-клеток
панкреатических островков. Это приводит к
изменению гетероклеточных взаимоотношений
в островках, инсулинопении и
относительному и абсолютному избытку контринсу-
лярных островковых гормонов. В результате
нарушается утилизация глюкозы и
возникают нарушения всех видов метаболизма.
Хронические нарушения метаболизма
порождают осложнения ИЗСД, главные из
которых связаны с микроангиопатией.
генетическая
предрасположенность
проявление прогрессирую- остаточные нетВ-клеток
аутоаллергии щая потеря аутоиммунные
против В-клеток, В-клеток, проявления нет С-пептида
нормальный
инсулиновый
ответ
аутоиммунные
проявления
уменьшаются,
уменьшение
ресурсов
инсулинового
терминальный
диабет
осложнений
(или их нет)
<10%
В-клеток,
имеется С-
пептид,
глюкоза
натощак
Рис. 58. ИЗСДI типа как стадийный хронический аутоиммунный процесс, (по Дж. Эйзенбарту,
1984). Пояснения — в тексте. I — момент провоцирующего действия диабетогена
Основной механизм гибели В-клеток —
спровоцированная вирусными и/или
химическими диабетогенами аутоиммунная
альтерация. Не исключается и роль подавления
пролиферации В-клеток антиклеточными
аутоантителами и медиаторами
аутоиммунного воспаления, что нарушает процессы
регенерации в островках. Впервые о роли
аутоантител (к инсулину) в патогенезе СД
задумался Ф. Депиш (1928), а наличие
лимфоцитарного инсулита при СД показал
Г. фон Мейенбург (1940).
Особенности клеточного состава
и взаимоотношений цитотипов
в панкреатическом островке
Поджелудочная железа человека содержит
более 1 млн панкреатических островков.
Основные цитотипы — это р, или В — (до 70 %),
а, или А — (около 20%) и 8, или D-клетки
(5-10%), остальные цитотипы встречаются
гораздо реже и не во всех островках: РР-
клетки (до 2%), D1 клетки, хромаффинные
клетки (менее 1 %). В дальнейшем изложении
клетки островков везде обозначаются
латинскими буквами, по рекомендации ВОЗ.
Несмотря на то что каждому виду
свойственны свои специфические особенности
Патофизиология сахарного диабета 281
топографии цитотипов в панкреатическом
островке, у всех изученных видов в
островках выявлены гетероклеточные области, в
которых А-, В- и D-клетки располагаются
в тесной близости — контактируют друг с
другом через самые разнообразные
варианты межклеточных контактов.
Гетероклеточные области (зоны)
представляют тем больший интерес, поскольку
известно, что афферентные капилляры и
нервы входят в островок именно в этих зонах.
С точки зрения возможных
межклеточных связей, которые могут осуществляться
как паракринно, так и через межклеточные
контакты (щелевые, плотные, десмосомаль-
ные, «взаимопроникающие»),
гетероклеточные области (зоны) могут функционировать
как «пейсмейкер» всего островка.
Это означает, что информация,
полученная извне (через кровь, через нервные
окончания), транслируется из этих зон к другим
островковым клеткам паракринно и через
межклеточные контакты, т.е. в
функциональном смысле панкреатические островки
представляют собой синцитий.
В свете современных представлений
панкреатический островок рассматривается как
система контроля внеклеточных концентра-
282 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
ций питательных веществ. Система
располагается на расстоянии от кишки, поскольку
концентрация веществ в просвете кишки не
может дать сведений о действительных
значениях концентраций питательных веществ
во внеклеточной жидкости. Она
организована в отдельные тяжи клеток внутри
плотного ретроперитонеального окружения, что
обеспечивает защиту от травмы. Хрупкие,
обильно васкуляризованные микроорганы,
если собрать их в единый крупный орган,
были бы более уязвимы.
Что касается сути паракринных
влияний (см. также т. I, с. 62-64), то они
представляются в данном случае гораздо
более существенными, чем, к примеру,
эндокринные, поскольку концентрации
аутакоидов, выделяемых в межклеточные
пространства и взаимодействующих со
своими рецепторами на клетках-мишенях
in situ, во много раз выше, чем
разбавляемые в системной циркуляции и
действующие эндокринно. В соответствии с
классическими представлениями Л. Орчи
и Р. Унгара (1975) (рис. 59) паракринные
влияния гормонов основных цитотипов в
гетероклеточных зонах могут быть сведены
к следующему:
>- паракринно секретируемый глюкагон
стимулирует образование и секрецию
инсулина островковыми В-клетками;
>■ паракринно секретируемый
глюкагон стимулирует образование и сек-
+
>
)
—
1 в
клетки
(инсулин)
II
1Г"
||
"II
А
клетки
(глюкагон)
•■
II
||
II
\
+
г
D 1
клетки
(соматостатин)\
Рис. 59. Схема паракринных взаимодействий островковых
клеток по Л. Орчи и Р. Унгару (1975). Связи со стрелкой на
конце — стимуляция секреции и пролиферации, связи с
черточками на конце — торможение секреции и пролиферации
рецию соматостатина островковыми
D-клетками;
>* паракринно секретируемый соматоста-
тин тормозит образование и секрецию
глюкагона островковыми А-клетками и
соматостатина — D-клетками.;
>• паракринно секретируемый соматоста-
тин тормозит образование и секрецию
инсулина островковыми В-клетками;
>■ паракринно секретируемый инсулин
тормозит образование и секрецию
глюкагона островковыми А-клетками.
Итог анализа подобных влияний может
быть сведен к следующему заключению:
>■ в физиологических условиях
синтетическая и функциональная активность
островковых А-клеток контролируется
одновременными тормозными
влияниями соматостатина и инсулина; контроль
эффективен, если он бигормонален.
Бигормональность контроля возможна
только при интактности структурной
организации гетероклеточных зон.
Морфофункциональная основа
представлений о патогенезе ИЗСД
В ответ на альтерацию островкового В-кле-
точного компонента в панкреатических
островках развивается инсулит. Гибель
островковых В-клеток, экссудативные изменения,
инфильтрация островков лимфоцитами,
макрофагами, а при неонатальном начале
ИЗСД — и эозинофилами, все это
обусловливает полное нарушение топографических
взаимоотношений цитотипов
внутри панкреатического
островка, извращение нейровас-
кулярных взаимоотношений,
повреждение межклеточных
контактов. De facto, при ИЗСД
гетероклеточные зоны
прекращают свое существование и
островки представляют собой
дезорганизованное скопление
А- и D-клеток, когда доля
А-клеток возрастает до 75 %,
а доля D-клеток — до 25%.
Количество В-клеток прогрес-
Патофизиология сахарного диабета 283
сивно сокращается и к моменту
формирования явного ИЗСД составляет менее 10%
первоначального. Бигормональное
торможение А-клеток утрачивается. Поэтому для
явного ИЗСД характерны повышенные
уровни глюкагона, концентрации которого
избыточны, по отношению к увеличенным
концентрациям глюкозы:
> Натощаковые уровни глюкагона
относительно выше натощаковых уровней
глюкозы и не подавляются дальнейшим
повышением концентрации глюкозы в
крови.
> Гипогликемия не вызывает подъема
концентрации глюкагона в плазме, что
лишний раз свидетельствует об
автономности функции А-клеток от
контролирующего действия уровня глюкозы крови.
Выявляются следующие
особенности функций островковых А-клеток при
ИЗСД:
> сохраняется ответ А-клеток на
свободные жирные кислоты (СЖК), то есть
возрастание уровня СЖК подавляет
выделение глюкагона;
> ответ А-клеток на аргинин усилен;
> высокие концентрации инсулина
частично восстанавливают ответ А-клеток и на
аргинин, и на глюкозу.
Итак, естественное тормозящее паракрин-
ное влияние инсулина на выработку
глюкагона А-клетками при ИЗСД снимается. В
результате соотношение глюкагон/инсулин
в крови больных ИЗСД по мере развития
болезни «стремится к бесконечности» (Дж.
Фостер, 1994).
Стадии развития ИЗСД
Благодаря усилиям Дж. Эйзенбарта во
второй половине 80-х годов XX века была
сформулирована концепция стадийности
течения ИЗСД, основанная на трактовке
данного недуга как хронического
аутоиммунного заболевания (рис. 58). На рисунке
отражены следующие стадии процесса:
/. Стадия генетической
предрасположенности.
Характеризуется наличием
генетических маркеров повышенного риска ИЗСД.
Может продолжаться от нескольких
месяцев до десятков лет (имеются
случаи ИЗСД у грудных детей и у глубоких
стариков). В эту стадию отсутствуют как
нарушения углеводного обмена (в покое и
при нагрузочных пробах), так и
иммунологические аномалии. У лиц с системной
наклонностью к органоспецифическим
аутоиммунным заболеваниям в эту стадию
могут выявляться признаки аутоиммунной
патологии других органов. Индивиды в эту
стадию не больны, но должны включаться
в группы высокого диспансерного риска
по ИЗСД.
П. Провоцирующее событие.
Как правило, сравнительно
кратковременная инфекция или воздействие химического
диабетогена. Может протекать субклиничес-
ки и выявляться только по данным
серологических проб, секвенс-анализа латентного
вирусного генома и при тщательном сборе
анамнеза. Может происходить в пренаталь-
ном и раннем постнатальном онтогенезе.
III. Стадия явных иммунологических
аномалий.
Длится от 2-3 мес до 2-3 лет и
заключается в развитии смешанной аутоиммунной
реакции против клеток панкреатических
островков, опосредованной как аутореак-
тивными CDs-лимфоцитами, так и
циркулирующими аутоантителами. Появляются
положительные тесты на клеточный и
гуморальный аутоиммунитет против антигенов
островковых клеток, в частности
собственного инсулина, глутаматдекарбоксилазы и
карбоксипептидазы Н. Морфологически
выявляется прогрессирующий инсулит
с лимфоцитарной инфильтрацией CD8/
С04-клетками, активацией макрофагов,
иногда — эозинофильными инфильтратами.
Ресурсы инсулиновой секреции всё еще
достаточны не только для поддержания
нормального натощакового уровня глюкозы в
крови, но и для непатологических сахарных
нагрузочных проб.
284 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
IV. Стадия латентного диабета.
Характеризуется прогрессирующим
снижением выработки инсулина при
нормальном уровне глюкозы натощак.
По длительности соответствует III стадии
и зависит от генетически
детерминированного резерва В-клеток. Выявляются
дополнительно положительные нагрузочные
пробы с глюкозой (интолерантность к глюкозе
по критериям Национального института
здоровья США).
О патологической нагрузочной пробе с
глюкозой можно говорить, если:
>- через 120 мин после приема взрослым
пациентом 75 г глюкозы ее уровень в
венозной крови превышает 200 мг/дл
(11,1ммоль/л) и в любой из сроков —
60, 90, 120 мин — также превышал
200мг/дл (11.1 ммоль/л) — т.е.
требуется двухкратная фиксация повышенного
уровня в течение 2 часов после нагрузки.
По критериям Американской
диабетической ассоциации:
Человек без СД имеет уровень глюкозы в
крови натощак менее 110 мг/дл. Больной с
явным СД имеет уровень глюкозы в крови
натощак 126 мг/дл или более. При
нарушенной толерантности к глюкозе {скрытый СД)
уровень глюкозы в крови натощак между
110 и 126 мг/дл. При пероральной пробе на
толерантность к глюкозе: у человека без СД
уровень глюкозы в крови становится ниже
140 мг/дл за 2 часа после приема раствора
глюкозы. У больного с явным СД уровень
глюкозы в крови 200 или более мг/дл
через 2 часа после приема. При нарушенной
толерантности к глюкозе (скрытый СД)
уровень глюкозы в крови между 140 и 200
мг/дл через 2 часа после приема раствора
глюкозы (АДА, 2003).
В то же время степень выраженности
аутоиммунного процесса в стадию латентного
ИЗСД постепенно ослабевает. Клинических
и лабораторных симптомов гипергликемии
вне нагрузочных проб нет.
V. Явный диабет.
Регистрируется с момента, когда к
симптомам, свойственным предыдущей стадии,
присоединяется гипергликемия натощак
(более 140 мг/дл — 7,8 ммоль/л в венозной
крови утром, до еды, зарегистрированные
не менее чем дважды в разные дни). Это
влечет за собой связанные с гипергликемией
яркие клинические симптомы. Внешне, с
точки зрения пациента, латентный диабет
становится явным внезапно, иногда на
протяжении суток. Толчком служат
стрессы, острые заболевания и иные события,
увеличивающие потребность в инсулине.
К этому моменту более чем у 1/2 больных
первоначальные аномалии аутоиммунитета
совершенно сглаживаются. Вот почему
длительное время природа ИЗСД как
результата долгого аутоаллергического процесса не
осознавалась медиками. Существовали
ложные представления о возможности развития
«острого первичного инсулинопенического
диабета», а значение стресса
(психоэмоциональной травмы) в генезе ИЗСД
преувеличивалось вплоть до отведения ему роли
основного причинного фактора.
Повышение уровня глюкозы натощак
индуцирует последние резервы В-клеток,
в связи с чем сразу после начала явного
диабета бывает «медовый месяц» —
кратковременный период сниженной потребности
в инсулинотерапии. С-пептид у больных
ИЗСД вначале присутствует, но по мере
развития болезни его выработка понижается
примерно на протяжении 2 лет. Секреция
инсулина и С-пептида в ответ на глюкозу
исчезает раньше, чем в ответ на неглюкоз-
ные стимулы.
VI. Терминальный диабет.
Данная стадия — эквивалент полной
гибели В-клеток, с высокой потребностью
в инсулинотерапии (не менее 60 ME в
сутки), а также с начальными
проявлениями микроангиопатии, отрицательными
или слабоположительными тестами на
аутоиммунитет и полным исчезновением
В-клеток, а значит — и способности
генерировать их уникальный продукт С-пептид.
Характерно, что иммунореактивный
инсулин внепанкреатического и экзогенного
происхождения при этом может продолжать
Патофизиология сахарного диабета 285
обнаруживаться в крови. Стадия полного
отсутствия В-клеток может быть достигнута
относительно быстро — за 3,5 года. Но чаще
это требует более длительных сроков. По
мере применения инсулине-терапии могут
появляться признаки вторичной иммуно-
зависимой инсулинорезистентности, дозы
инсулина возрастают до плато, а иногда
потребность в инсулине становится
колеблющейся, в зависимости от эндогенных
иммунопатологических причин, в частности
состояния антиидиотипического иммунного
ответа (лабильный диабет).
Поскольку инсулинотерапия не
исключает развития микроангиопатии, а
пересаженные В-клетки отторгаются из-за новой
аутоиммунной атаки, даже если взяты от
идентичного близнеца, перспективы
лечения ИЗСД связаны с иммуносупрессивной
терапией, которая, однако, эффективна
только в I-IV стадиях болезни. При позднем
начале иммунотерапии ее побочные
эффекты не уравновешиваются достигаемым
снижением потребности в инсулинотерапии.
Поэтому наиболее актуальной
стратегической задачей является раннее выявление
ИЗСД и групп риска и профилактическое
лечение. ИЗСД более не может
восприниматься системой здравоохранения как
исключительно эндокринное заболевание.
Вся система диспансеризации больных и
профилактики этого недуга должна
строиться с учётом современных данных об
иммунопатологической природе болезни.
Гормоны островков Лангерганса
при ИЗСД
Основной гормон В-клеток — инсулин
содержится в крови взрослых людей в
концентрации 0,4-0,8 нг/мл (10-20 мкЕд/
мл). В кровь поступает и его неактивный
предшественник проинсулин (до 5% по
отношению к инсулину), а также
соединительный фрагмент (рис. 86, с. 439),
отщепляемый при процессинге инсулина, — С-
пептид (0,9-3,5 нг/мл). Секреция инсулина
(рис. 60) и С-пептида в кровь стимулируется
глюкозой, холинергическими нервами и
аминокислотами (лейцин, аргинин),
некоторыми гормонами энтериновой системы и
препаратами сульфонилмочевины, причем
\
к+ \а/\аЛл к* чЛЛллЛ к+
Рис. 60. Секреция инсулина В-клетками (по Дж. Бэкстеру, Ф. Гринспэну, 1994).
Пояснения в тексте
286 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
1з
X S
ИНСД2
15
30
45 60 мин
ИЗСД-1
Рис.61. Ранняя и поздняя фазы инсулиново-
го ответа на глюкозную нагрузку в норме (в
середине), при ИЗСДI типа (внизу) и при ИНСД
II типа (вверху)
в нормальных условиях in vivo отмечаются
2 характерные пика гиперинсулинемии:
ранний и поздний (2-10 мин и 10-60 мин),
обусловленные выбросом депонированных
продуктов и активацией их синтеза de novo
соответственно (рис. 61). В. Кумар и соавт.
(1997) подчёркивают, что синтез
инсулина de novo активируют лишь глюкоза и
холинергические нервные воздействия на
островки. Аминокислоты, энтерогормоны,
а также производные сульфонилмочевины
сильно потенцируют стимулирующий
эффект глюкозы на инсулиновую секрецию
и активизируют выброс готового гормона.
Секреция инсулина стимулируется также
рядом других гормонов, в частности АКТГ.
Кроме того, показано, что повышение
секреции инсулина наступает до начала
всасывания пищевых субстратов из кишки,
что может указывать на нервный механизм
первой фазы инсулиновой секреции
(«мозговая фаза»).
При ИЗСД в IV-V фазе болезни
характерно угнетение реакции инсулина, а
затем и С-пептида, вначале — на нервные
стимулы и глюкозу, а потом на
аминокислоты, причем страдают обе фазы ответа
инсулина. Концентрации проинсулина
при ИЗСД не имеют характерных
изменений, зато могут возрастать при гипо-
калиемии, инсулиноме и наследственной
проинсулинемии.
Поскольку у всех леченых и у
большинства не леченых инсулином больных ИЗСД
в крови имеются аутоантитела и антитела к
инсулину, данные определения иммунореак-
тивного инсулина при ИЗСД практически,
всегда неадекватны и искажены в сторону
занижения. Поэтому
радиоиммунологическое определение (РИА) и иммунофермент-
ный анализ (ИФА) инсулина при ИЗСД
не могут служить средством диагностики и
не заменяют генетико-иммунологических
данных и тестов на толерантность к
глюкозе. В диагностическом и прогностическом
плане для уточнения стадии болезни
гораздо полезнее определение С-пептида. Для
ИЗСД типично содержание С-пептида ниже
300 пМ/л и его прогрессивное падение до
нуля в течение 2-3 лет болезни.
Инсулин устраняется из кровотока за
15 мин: часть его, провзаимодействовавшая
с клеточными рецепторами, подвергается
эндоцитозу, а остальной гормон деградирует
в печени и почках.
Основной гормон А-клеток — глюкагон
содержится в крови в норме в
концентрации 75-150 пг/мл, но лишь 40% этого
количества— активный гормон,
остальное — проглюкагон и внепанкреатическии
глюкагон из подчелюстной слюнной
железы, кишечника и желудка — «большой
глюкагон плазмы». Обе последние фракции
малоактивны. Секреция глюкагона в плазму
стабильна. Его освобождение активируется
аминокислотами (особенно аланином и
аргинином) и сильно тормозится глюкозой,
возрастает при значительной физической
нагрузке (вследствие наступающей амино-
ацидемии и нейрогенной стимуляции его
секреции).
При голодании содержание глюкагона
резко увеличивается, в основном за счет
замедления его клиренса. Уровень глюкагона
повышен при семейной гиперглюкагонемии
и опухолях-глюкагономах. При РИА и ИФА
следует иметь в виду, что прямые
определения не отражают биологической активности
глюкагона.
ИЗСД характеризуется умеренной ги-
перглюкагонемией, однако даже 4-кратное
возрастание содержания глюкагона per se,
при нормальном уровне инсулина, не
отражается на толерантности к глюкозе.
Поэтому многие авторы не считают определение
глюкагона у больных ИЗСД
информативным тестом.
Открытый А. Бразо в 1973 г. гипотала-
мический ингибитор секреции иммуноре-
активного соматотропина —
соматостатин — основной гормон D-клеток
панкреатических островков, вырабатывается и за
пределами поджелудочной железы: в кишке,
в ростральной части перивентрикулярного
ядра гипоталамуса и в других отделах ЦНС,
включая даже кору больших полушарий
(т.1, с. 64-65,97,391,537).
Показано, что освобождение иммуноре-
активного соматостатина в
панкреатическую вену происходит в ответ практически
на все факторы, связанные с приемом
пищи (высокая концентрация глюкозы,
аргинина, лейцина, свободных жирных
кислот). Смесь аминокислот, панкрето-
азимин/холецистокинин, секретин также
стимулируют его секрецию. Соматотропи-
новый ответ вызывают те же субстанции,
которые стимулируют секрецию инсулина
и глюкагона.
Гормон служит мультисистемным
ингибитором и тормозит: аппетит, моторику
желудка, двенадцатиперстной кишки и
жёлчного пузыря, всасывание глюкозы,
все пищеварительные функции слюнных
желез, желудка, тонкого кишечника, под-
Патофизиология сахарного диабета 287
желудочной железы, а также выработку
других островковых гормонов, продукцию
большинства гормонов энтериновой
системы и аденогипофиза и т.д. Соматостатин
способен, таким образом, пролонгировать
усвоение пищи и ее утилизацию тканями.
Ингибиторные эффекты соматостатина
простираются шире и захватывают
метаболические и физиологические процессы,
прямо не относящиеся к питанию. Он тормозит
функции тромбоцитов, снижает основной
обмен, ингибирует некоторые
электрофизиологические процессы в возбудимых тканях.
Ставится вопрос о взаимоотношениях
соматостатина и механизмов анабиоза (см. выше
раздел «Анабиоз и зимняя спячка»).
Показано, что интрапортальное введение
соматостатина в физиологических дозах
задерживает поступление в кровоток
питательных веществ из кишки. По мнению
Р. Унгара (1977), соматостатин
информационно обеспечивает афферентную часть
оси «кишка — островок — кишка»,
контролирующую приток питательных продуктов
из ЖКТ сверх обычного уровня, в ответ на
сигналы от энтериновых гормонов.
При оценке данных РИА и ИФА следует
иметь в виду, что пероральные
антидиабетические средства, применяемые у больных
с ИНСД (толбутамид и его аналоги),
являются сильными стимуляторами продукции
соматостатина, на чем частично и основан
их эффект, а также принимать во внимание
пульсовой характер секреции соматостатина
и крайнюю вариабельность его содержания в
крови. При ИЗСД содержание
соматостатина в крови стабильно растет по мере утраты
функций В-клеток.
Панкреатический полипептид — гормон
РР-клеток — вырабатывается в ответ на
глюкозу, белки и физическую нагрузку,
стимулирует инсулиногенез и контролирует
некоторые функции желудочно-кишечного
тракта; при ИЗСД его содержание в крови
несколько возрастает.
Кроме названных в островках
выделяется еще множество пептидных
регуляторов: галанин, опиоиды, кортиколиберин,
288 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
IL-1, панкреастатин, нейропептид Y, ко-
кальцигенин, АКТГ-подобный пептид,
гастрин-рилизинг полипептид, вещество
Р, ТТГ. Большинство этих агентов
освобождается из пептидэргических нервных
окончаний и неиросекреторных клеток
апудоцитарного происхождения, поэтому
их объединяют под названием
«панкреатические нейропептиды». Те же нейропепти-
ды выделяются и в гипоталамусе, где
участвуют в регуляции вегетативных функций
и нейроэндокринной передаче информации
клетками различного, преимущественно
мезенхимального происхождения (см.
также т. I, с. 536-537). В островках, как и в
иных органах, постоянно вырабатываются
производные арахидоновой кислоты цик-
ло- и липооксигеназного ряда (т. I, с.
359-361). Динамика всех этих регуляторов
при ИЗСД мало изучена.
Иммунопатологические
механизмы ИЗСД
При ИЗСД отмечаются многочисленные
проявления клеточного и гуморального
аутоиммунитета против антигенов В-клеток
и иных элементов панкреатических
островков, а также против некоторых неостров-
ковых антигенов. Аутоиммунная агрессия
при ИЗСД предшествует каким бы то ни
было нарушениям толерантности к глюкозе,
причем опережение может составлять до
10 лет, хотя чаще бывает в сроки от 2 до 36
месяцев перед установлением нарушений
углеводного обмена.
К началу явного диабета, в зависимости
от чувствительности применяемых методов,
у 80-100% пациентов обнаруживаются
проявления ГНТ и ГЗТ против островков.
При ИЗСД 1а на протяжении первых 2-5
лет болезни явления аутоиммунитета
смягчаются до выявляемости 20 -30 % (Л. Варди
и соавт., 1988). При ИЗСД lb проявления
аутоиммуноагрессии держатся долго на
стабильном уровне, и до половины таких
больных имеют высокоположительные
иммунотесты независимо от длительности
заболевания (Р.Вольпе, 1981, А. Пауэре,
Дж. Эйзенбарт, 1985).
Клеточный аутоиммунитет
при ИЗСД
Клеточные механизмы аутоиммунной
агрессии — главная причина деструкции В-клеток
в ходе ИЗСД. Заболевание поддается
адоптивному переносу: СЭв-положительными
Т-лимфоцитами больных грызунов можно
вызвать инсулит и интолерантность к
глюкозе у здоровых животных-реципиентов.
Адоптивный перенос возможен у животных,
представляющих разные генетические
модели ИЗСД (как крыс ВВ, так и мышей NOD,
см. ниже). Однако конкретные
иммунопатологические нарушения у этих двух моделей
ИЗСД могут быть различными: В В имеют
выраженную наследственную лимфопению
и дефицит цитотоксических лимфоцитов,
а мыши NOD, наоборот, избыток Т-
эффекторов при дефекте Т-супрессоров. Неона-
тальная тимэктомия предотвращает или
задерживает диабет у всех этих животных.
Плазмаферез и антитела к антиинсулино-
вым аутоантителам не дают лечебного
эффекта при ИЗСД. В то же время различные
способы иммунотерапии, затрагивающие
клеточное звено иммунитета, могут быть
эффективны.
Наиболее удобным для массового скри-
нингового тестирования механизмов ГЗТ
при ИЗСД является реакция торможения
миграции лейкоцитов (РТМЛ), которая
положительна у 55-65 % пациентов в пред-
гипергликемическую фазу ИЗСД и в его
разгаре, причем как в аутологичной, так и в
ксеногенной системах.
У больных ИЗСД имеются различные
признаки активации Т-системы
иммунитета, например повышение доли Т-клеток,
экспрессирующих рецепторы IL-2 и
антигены ГКГС 2-класса.
В иммунологическую и начальную гипер-
гликемическую фазу болезни отмечаются:
>• инсулит с характерной мононуклеарной
клеточной инфильтрацией,
протекающий на фоне изменения субпопуляци-
онных соотношений лимфоцитов;
Патофизиология сахарного диабета 289
> повышенный уровень лимфоцитарной
цитотоксичности в культурах
панкреатических В-клеток;
> высокая активность NK-клеток;
> повышенная активность цитотоксичес-
ких Т-лимфоцитов in vivo;
> повышенная активность АЗКЦ против
островковых клеток;
> усиленный ответ лимфоцитов на ФГА
(что, впрочем, характерно только для
компенсированного состояния обмена
глюкозы и сменяется депрессией при
декомпенсации);
> цитотоксические Т-лимфоциты больных
ИЗСД вызывают освобождение
инсулина из В-клеток поджелудочной железы in
vitro;
> описаны клоны Т-клеток от больных
ИЗСД, цитотоксичные in vitro для
клеток инсуломы;
> среди С04-положительных клеток у
больных ИЗСД преобладают CD45R+
лимфоциты, выполняющие функцию
индукторов / супрессоров и активирующие
киллерную CD8+ активность.
Гуморальный иммунитет
при ИЗСД
Гуморальный аутоиммунитет при ИЗСД
также изменён, и в настоящее время
считается доказанным участие в развитии этого
заболевания аутоаллергии типа ГНТ.
У здоровых лиц аутоантитела против
В-клеток встречаются не более чем в 0,5 %
случаев. Но их встречаемость в 10-30
раз больше у ближайших родственников
больных и в 60 раз выше у больных с орга-
носпецифическими аутоиммунными
заболеваниями без ИЗСД, при прегнодиабете
процент антителоположительных
сывороток достигает 10 %, при ИНСД — 20 %, а при
ИЗСД — 100% в III стадию болезни.
Появление аутоантител предшествует
морфологической картине инсулита при ИЗСД.
При ИЗСД регулярно
обнаруживаются аутоантитела против целого ряда
панкреатических и внепанкреатических
антигенов:
>■ поверхностных антигенов островковых
клеток (AISA);
>• цитоплазматических антигенов
островковых клеток (AICA);
>* ядерных антигенов островковых клеток
(AINA);
>- собственного инсулина (AIA);
>• ряд антител неорганоспецифического
и панкреатотропного неостровкового
характера;
>• органоспецифические непанкреатотроп-
ные аутоантитела.
Аутоантитела против поверхностных
антигенов островковых клеток (AISA)
выявляются у 75-90 % пациентов со свежим ИЗСД
методом непрямой иммунофлюоресценции
и локализуются на плазматических
мембранах островковых клеток. Они направлены
против невидоспецифических антигенов,
что позволяет с успехом диагностировать
их наличие не только на срезах
человеческой поджелудочной железы, но и в пробах
с сывороткой пациента и органами свиньи,
крысы, морской свинки, обезьяны, мыши.
Антигены-мишени AISA представляют
собой различные мембранные гликоко-
ньюгаты: гликопротеиды (gplOO, gp26,
gpl20), гликолипиды, моносиалганглио-
зиды (GT3 и GM2-1). Ганглиозиды
островков, являющиеся мишенями аутоантител,
выделены из панкреатической ткани, и
показано, что их метаболизм зависит от
глюкозы. Интересно, что и результаты
определения аутоантител к островкам зависят
от степени гипергликемии и
метаболической компенсации диабета. Практически
все подобные антигены содержат остатки
сиаловой кислоты и выделяются на
колонках с человеческими AISA. Обнаруженные
антитела к мембранным гликопротеидам
В-клеток в 88 % случаев принадлежат к IgG
HB25%-KlgM.
AISA часто бывают нетканеспецифичны-
ми. В связи со своей универсальной ролью
в структуре поверхностных клеточных ре-
290 АЖ Зайчик, Л.П.Чурилов Патохимия
цепторов ганглиозиды и гликоконъюгаты,
против которых они направлены, входят в
состав ПМ ряда эндокриноцитов, нейронов
и клеток апудоцитарного происхождения
(см. выше «Нарушения липидного обмена»).
В ряде случаев аутоантитела к
поверхности В-клеток у больных ИЗСД и
животных-моделей ИЗСД могут быть
направлены против белков, входящих в состав
секреторных гранул инсулина, в частности
карбоксипептидазы Яили против изоформы
транспортера глюкозы — GluT2, экспрессия
которого у крыс В В понижена (А. Россини
и соавт., 1993).
Доказано, что AICA — аутоантитела
к цитоплазме клеток островков также не-
видоспецифичны. Они регистрируются в
начале болезни методом непрямой иммуно-
флюоресценции или иммуноцитохимически
на нефиксированных срезах.
Антиген AICA, обнаруженный датским
иммунопатологом X. Бэккесковом и
соавторами (1988), — белок с молекулярной
массой 64 кД, был идентифицирован и
оказался ферментом глутаматдекарбок-
силазой (ГДК), ответственным за синтез
гамма-аминомасляной кислоты (ГАМК).
ГДК встречается и во многих других тканях
(стероидпродуцирующие клетки, нефро-
циты, гепатоциты). Особенно широко она
представлена в нейронах.
В островках синтез ГАМК под действием
местной ГДК может играть определенную
регуляторную роль, влияя на выработку
глюкагона и соматостатина. На основании
опытов с перфузией островков ГАМК
предположили, что, при отсутствии глюкозы,
этот метаболит может утилизироваться
В-клетками с энергетическими целями. ГДК
экспрессирована в В-клетках, но не в других
гормональноактивных элементах островка.
Описан синдром одеревенелости (англ.:
Stiff-man syndrome), при котором имеются
аутоантитела к ГДК, перекрестно
реагирующие с антигенами многих эндокринных
желез. AICA при данном синдроме
наблюдаются регулярно, а частота ИЗСД
приближается к 30 процентам.
)-метаболические нарушения)
Аутоантитела больных ИЗСД распознают
ГДК из В-клеток человека, крысы и свиньи.
Описаны две изоформы ГДК — GAD 65 и
GAD 67. GAD 65 широко представлена в
В-клетках. Считают, что это — наиболее
вероятный антиген — мишень при ИЗСД.
Попытки увязать различный риск ИЗСД
с различиями в наборе аутоиммунных
анти-ГДК антител, показали, что
аутоантитела GAD 65 гетерогенны и узнают либо
эпитоп Е1 (между 240 и 435
аминокислотой), либо Е2 (между аминокислотными
остатками 451 и 570). Цель подобной
детализации — создание такого
дифференциального иммунологического теста, который
разграничивал бы пациентов, имеющих
проявления аутоиммунитета к островкам,
позволяя выделить группу высокого и
группу умеренного риска ИЗСД.
Наибольшей прогностической
ценностью обладают, по-видимому, аутоантитела
к фрагменту ГДК, полученному трипси-
новым перевариванием, пептиду 37 кД.
Наличие таких аутоантител у детей очень
тесно коррелирует с последующим
развитием ИЗСД. AICA — это исключительно
IgG. Как аутоантитела AISA, так и AICA
встречаются у спонтанно-диабетических
крыс линии ВВ.
AIA — это аутоантитела к собственному
инсулину больных ИЗСД.
Долгое время считалось, что антитела
к инсулину возникают только в результате
инсулинотерапии, особенно если
применяемые инсулины ксеногенны. Однако
первичная аутоаллергия на собственный
инсулин — не редкость у больных ИЗСД,
никогда инсулинотерапии не получавших
(Й. Шехтер и соавт., 1982).
Инсулин — поверхностный антиген,
присутствующий в мембране В-клеток. При
ИЗСД он может быть объектом
аутоиммунной атаки. Клональность антиинсулиновых
аутоантител ограничена, и они связывают
инсулиновые эпитопы, отличные от тех,
которые взаимодействуют с антителами,
возникающими при инсулинотерапии. AIA
избирательно взаимодействуют с В-клетками.
Патофизиология сахарного диабета 291
Нередко в ходе развития ИЗСД Л1Л
отмечаются и аутоантитела к
проинсулину.
Изредка антиинсулиновые
аутоантитела обнаруживаются
даже у здоровых лиц, никогда не
получавших инсулина и, что
особенно важно, их наличие типично
для больных в ранних стадиях
ИЗСД.
AIA выявляют с помощью
коммерческих РИА и ИФА-наборов,
причем РИА считается
предпочтительным для больных,
подвергавшихся инсулинотерапии,
а ИФА— в случае обследования
до начала лечения инсулином.
AIA признаны важнейшим
маркером иммунологических
нарушений у детей в догипер-
гликемические стадии ИЗСД.
У взрослых же они не обладают
столь выраженной
прогностической ценностью. По данным
Диабетологического центра им.
Джослина (США), 100% детей до 5 лет,
заболевающих ИЗСД, имеют AIA в латентную
стадию болезни. По некоторым данным, AIА
более стабильны в ходе болезни, чем AISA,
и их содержание прямо коррелирует с
вероятностью развития некоторых осложнений
(Р. Бок, 1988).
Имеется генетическая модель ИЗСД у
животных, вызванная спонтанной
выработкой AIA ( крысы WBH/Kobe).
Инсулин как антиген может презенти-
роваться и процессироваться иммуноком-
петентными клетками, но весьма вероятно,
что сами островковые В-клетки, при условии
аберрантной экспрессии DR-белков, могут
представлять инсулиновый аутоантиген.
AIA проникают через плаценту, но могут
оказать у новорожденных лишь
кратковременный эффект.
Помимо AIA при ИЗСД наблюдаются
аутоантитела и к другим гормонам островков:
к панкреатическому полипептиду (у 80%),
глюкагону (15%), соматостатину (10%),
кальцитонину.
AISA к
кгликолипидам
AISA к карбоксипептидазе Н
В-клетка
Рис 62, Мишени аутоантител при ИЗСД I типа.
Сокращения в тексте
AINA — антиостровковые антиядерные
аутоантитела, впервые были найдены на
ранних стадиях ИЗСД у детей. Вопрос об их
органоспецифичности не выяснен. Хотя
значительная часть сывороток больных ИЗСД
и лиц из групп высокого риска содержит
AINA, многие диабетологи считают их лишь
сопутствующим феноменом.
Различные мишени аутоантител к В-клет-
кам и соответствующие иммуноглобулины
показаны на рис. 62.
Среди других аутоантител,
обнаруживаемых при ИЗСД, встречаются аутоантитела
кнеостровковой ткани поджелудочной
железы, в частности к цитокератину
панкреатических клеток (у новорожденных
с ИЗСД и их родственников). Более 30%
больных имеют аутоантитела к
иммуноглобулинам, отличающиеся от
ревматоидных факторов, а также аутоантитела
IgM против лимфоцитарных мембран Т- и
В-лимфоцитов.
Почти 40 % больных ИЗСД, леченных
инсулином, имеют аутоантитела к рецептору
этого гормона. Очевидно, что эти аутоанти-
292 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
инсулин
инсулин антитело
к инсулину
f щ\— инсулиновый
f f \ рецептор
) клетка-мишень
инсулина
Ш
Et>
эффект
блокирующее
антиидиотипическое
а
щ
/
аутоантитело
против антител
к инсулину
\^/J блокада (инсулино-
резистентность)
О
миметическое
доза инсулина
повышается
г имитация
эффекта
Рис 63. Антиидиотипический механизм лабильного сахарного диабета. Пояснения в тексте
доза инсулина
избыточна
тела могут возникать у больных ИЗСД по
антиидиотипическому механизму — в ответ
на антитела против инсулина, как попытка
компенсации избыточного аутоиммунитета.
Эти аутоантитела к рецептору инсулина
могут обладать in vivo блокирующей
активностью в отношении инсулинового
рецептора и вызывать инсулинорезистентность,
но возможны и инсулиномиметические
эффекты антител к рецептору в отношении
метаболического и морфогенетического
действия инсулина; это может составлять
основу патогенеза лабильного диабета с
колеблющейся потребностью в экзогенном
инсулине.
Аутоантиидиотипические антирецеп-
торные антитела, стимулирующие антиин-
сулиновый рецептор, найдены у больных
сахарным диабетом и показана их
способность вызывать гипогликемический эффект
(Р.Тарделла и соавт., 1983). Й. Шехтер и
соавт. получили антиидиотипические
антитела против антител к инсулину в
эксперименте и убедительно продемонстрировали,
что эти антиидиотипы обладают
преходящим инсулиномиметическим действием
на клетки-мишени в культуре адипоцитов
(1988). Поскольку данный механизм может
участвовать в патогенезе криптогенных
гипогликемии и формировании
лабильного течения диабета, он проиллюстрирован
специальной схемой (рис. 63).
Кроме антипанкреатической аутоаллер-
гии у больных ИЗСД и их родственников
имеет место выработка аутоантител ко
многим другим клеткам и органам: к
гипофизу, к корковому и мозговому веществу
надпочечников, к неорганоспецифическим
ядерным антигенам печени, белкам цитоске-
лета. Обнаружена продукция блокирующих
аутоантител к щитовидной железе. Всё это
существенно для характеристики плюриг-
ландулярной полиэндокринной аутоимму-
нопатии при ИЗСД подтипа lb и связано с
патогенезом осложнений болезни.
Кроме того, при ИЗСД выявлены аутоан-
титела к антигенам ГКГС (DQ региона) и к
гормонам тимуса, которые могут вносить
свой вклад в механизмы диабетического
вторичного иммунодефицита. Все это
свидетельствует о том, что при ИЗСД в целом
нарушена идиотип-антиидиотипическая
сеть комплементарных взаимодействий
антител и биорегуляторов.
Важными участниками аутоиммунного
процесса при ИЗСД могут быть аутоанти-
тела к собственному сывороточному
альбумину пациентов, а также антитела к бычьему
сывороточному альбумину (БСА). Эти
иммуноглобулины часто регистрируются
с помощью иммуноферментного и
радиоиммунологического методов в сыворотках
в догипергликемические стадии ИЗСД,
особенно у детей и у носителей антигена
ГКГС D3. Предполагается, что
антиальбуминовые иммуноглобулины способны
перекрестно реагировать с белком р69 из
состава В-клеток.
Количество циркулирующих иммунных
комплексов у пациентов с ИЗСД чаще всего
повышено.
При ИЗСД имеется увеличение общего
содержания В-лимфоцитов и В-лимфоцитов,
активно продуцирующих имуноглобулины.
Особенно интересны данные о
значительном повышении при ИЗСД количества
CDg-положительных В-лимфоцитов,
поскольку именно они присутствуют в избытке
при многих иных аутоиммунных
заболеваниях (аутоиммунный тироидит, системные
неорганоспецифические заболевания
соединительной ткани) и ответственны за синтез
так называемых естественных аутоантител,
принадлежащих к низкоаффинным IgM,
перекрестно распознающим многие свои и
экзогенные антигены. Эти
иммуноглобулины кодируются генами зародышевой линии
Vn3.n5. Подобные лимфоциты у здоровых
взрослых составляют 10-20%, а более
значительные их количества обнаруживаются
в норме лишь в раннем постнатальном
онтогенезе (т. I, с. 435).
Патофизиология сахарного диабета 293
Механизмы действия
аутоиммунных эффекторов
на островки
Инсулинопродуцирующие клетки
панкреатических островков атакуются цитотокси-
ческими Т-лимфоцитами вследствие
экспрессии на мембране островковых В-клеток
отсутствующих в норме DR-белков.
Причиной анормальной экспрессии может быть
действие вирусов, влияние интерферонов,
кахексина, интерлейкинов или других лим-
фо- и монокинов. Не исключена и
экспрессия неоантигенов-продуктов латентного
вирусного генома. Аномалии экспрессии генов
ГКГС 2-го класса предшествуют инсулиту и
касаются только инсулинпродуцирующих
В-клеток и эндотелиоцитов сосудов
поджелудочной железы.
Гуморальные механизмы могут вызывать
деструкцию островковых В-клеток путем
комплементзависимого клеточного лизиса
или антителоопосредованной клеточной
цитотоксичности. Так, при впервые
выявленном ИЗСД отмечаются признаки
активации комплемента по классическому и
альтернативному пути и
комплемент-опосредованный лизис В-клеток.
Аутоиммунные эффекторы приводят к накоплению в
островках IL-1. Доказано, что IL-1
снижает освобождение инсулина В-клетками,
уменьшает их чувствительность к глюкозе
и к неглюкозным стимулам и обладает
прямым повреждающим действием на
В-клетки. Следовательно, через IL-1
освобождение инсулина при ИЗСД может
быть заторможено раньше, чем наступит
деструкция В-клеток. Действие IL-1 в
большинстве случаев опосредовано липидными
модуляторами. При ИЗСД в островках
накапливаются фактор активации
тромбоцитов и простагландины, подавляющие
острую фазу инсулинового ответа на
глюкозу. Антагонисты циклооксигеназы,
например аспирин, облегчают течение ИЗСД.
Цитокины действуют на В-клетки более
сильно in vivo, чем in vitro; в связи с этим
существует даже мнение, что В-клетка, с ее
низкой цитотоксической резистентностью,
становится как бы «невинной жертвой»
294 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
цитокино-медиаторного удара,
происходящего в результате аутоиммунного процесса,
имеющего иные мишени.
ИЛ-1, ФНОа, лимфотоксин и интерфе-
рон-у, выделяемые при инсулите
лимфоцитами и макрофагами, обладают синэргич-
ным потенцирующим цитотоксическим,
антипролиферативным и антисекреторным
эффектом на островковые В-клетки.
Не случаен и тот факт, что в пораженных
островках, несмотря на клеточную
деструкцию и большое количество медиаторов
пролиферации, практически полностью
отсутствуют митозы. По аналогии с ат-
рофическими тиропатиями, при которых
обнаружены аутоантитела-блокаторы роста
железы, можно ожидать обнаружения
аутоиммунных агентов ИЗСД, выключающих
митотическую активность В-клеток.
Гуморальные эффекторы аутоаллергии,
вероятно, не исчерпывают своё действие
на В-клетки индукцией цитолитического
эффекта.
В последнее время стало известно, что
некоторые фракции IgG из сывороток
больных с ИЗСД стимулируют высвобождение
инсулина in vivo и in vitro без
сопутствующего цитолиза.
Обнаружены иммуноглобулины-блокато-
ры освобождения инсулина, не вызывающие
деструкции клеток-мишеней. Для антител
против компонентов глюкозного рецептора
и/или глюкозопереносящих белков
взаимодействие с аутоантигенами будет приводить
к функциональному блоку ответа В-клеток
на глюкозу без их разрушения. Аутоанти-
тела к ГДК могут, вероятно, вмешиваться в
паракринную регуляцию инсулиновой
секреции, а аутоантитела к карбоксипептидазе
Н — в секрецию инсулина.
При всем разнообразии последствий
взаимодействия гормонообразующих клеток
с аутоиммунными эффекторами, начиная
от лизиса и кончая стимуляцией или
торможением роста и гормонопродукции,
наибольшее значение при ИЗСД принадлежит
избирательной аутоиммунной деструкции:
показано, что к началу явного диабета масса
железы снижается в 2, масса островков —
в 3,3, а масса В-клеток — более чем в 850 раз
(Дж. Фостер, 1994).
Интегральные представления
сторонников аутоиммунной теории патогенеза ИЗСД
можно свести к следующей схеме (рис. 64)
(Т. Аткинсон, Н. Мак Ларен 1989):
1) индивид, генетически
предрасположенный к ИЗСД, подвергается воздействию
чужеродного антигена, обладающего
близким сходством с неким компонентом
В-клеток панкреатических островков;
2) на поверхности профессиональных
местных антиген-представляющих клеток,
презентирующих антиген, появляется
этот экзоцеллюлярный антиген в
сочетании с молекулами белков ГКГС
класса 1;
3) после узнавания комплекса CD4+ и CD8+
лимфоцитами возникает мощный и
продолжительный иммунный ответ против
антигена;
4) специфические к чужеродному антигену
цитотоксические лимфоциты и антитела
с током крови поступают в
панкреатические островки и атакуют В-клетки,
несущие чужеродный антиген.
Поврежденные В-клетки усиливают продукцию
белков ГКГС класса 1 и класса 2.
Появление большего количества белков класса
1 усиливает атаку цитотоксических
Т-лимфоцитов;
5) появление белков ГКГС класса 2 на
клетках, не относящихся к иммунной
системе, в норме должно возбуждать
супрессорную активность. При ИЗСД
это приводит к обратному результату:
вследствие генетического дефекта
иммунной системы супрессорный ответ
не реализуется, напротив, иммунный
ответ усиливается. Из-за аберрантной
экспрессии антигенов ГКГС на самих
В-клетках, молекулы их эндоцеллю-
лярного антигена, сходного с
экзогенным, могут оказаться в сочетании
с гликопротеидами ГКГС класса 2
(такими как DQ 3.2) на клеточной
поверхности — и тогда они презентуются
Патофизиология сахарного диабета 295
наследственная предрасположенность
(ГКГС DRy4 и другие аллели)
вирусы
химические
диабетогены
иммунный ответ против поврежденных В-клеток
+
аутоаллергия против собственных В-клеток
1 \
повреждения В-клеток
молекулярная мимикрия
аутопрезентация
У
деструкция В-клеток
экспрессия
неоантигенов
аберрантная экспрессия
антигенов ГКГС 2 класса
на В-клетках
абсолютная инс у липовая
недостаточность
свободные радикалы
медиаторы воспаления
Рис 64. Патогенез иммунопатологического процесса при ИЗСДI типа
CD4+ лимфоцитам таким образом, что
иммунные эффекторы «путают» их со
своими антигенами;
6) антитела против чужеродного антигена
и аутоантитела против своих, перекрес-
тнореагирующих компонентов могут
«метить» В-клетки с последующей
активацией системы комплемента или
привлечением NK-клеток и цитолизом.
IL-1, выделяемый активированными
макрофагами, способен самостоятельно
повреждать В-клетки и тормозить их
пролиферативную и секреторную
активность;
7) поврежденные В-клетки и
активированные макрофаги производят
значительное количество свободных радикалов,
неспецифически повреждающих новые
В-клетки, которые особо чувствительны
к их действию;
8) белки теплового шока, ганглиозиды и
проинсулин поврежденных В-клеток
могут появляться на их поверхности
или выделяться наружу. Макрофаги
поглощают их и представляют Т-хелпе-
рам как «чужеродные» неоантигены (в
норме не встречающиеся на поверхности
В-клеток) — это вызывает новый раунд
иммунной атаки.
Эти этапы иллюстрируются на рис. 64.
Таким образом: особенности
аутоиммунного поражения В-клеток состоят как
в том, что разрушение В-клеток ускоряется
вследствие их необычайно высокой
чувствительности к иммунной атаке, так и в
том, что сама иммунная атака оказывается
более выраженной, длительной и менее
избирательной, чем должно было бы быть
при нормальном иммунном ответе.
Повышенная чувствительность В-клеток связана
с несколькими обстоятельствами:
>- При повреждении на их поверхности
появляется избыточное количество
молекул ГКГС класса 1 и начинается
аберрантная экспрессия гликопротеидов
ГКГС класса 2, что облегчает аутоаллер-
гическое поражение.
>* Данные клетки особенно подвержены
повреждающему действию цитокинов, в
296 АЖ Зайчик, Л.П.Чурилов Патохимия
частности, IL-1 обладает прямым
токсическим действием на В-клетки, но менее
токсичен для других типов клеток.
>- В-клетки необычайно ранимы по
отношению к свободным радикалам,
которые высвобождаются поврежденными
клетками и макрофагами, так как их
антиоксидантные ресурсы невелики.
Эндокринный статус при ИЗСД
Изучение нейроэндокринной системы при
ИЗСД определяется следующими
соображениями:
1) Оценка содержания островковых
гормонов и их предшественников с целью
уточнения диагноза и прогноза.
2) Оценка содержания гормонов —
потенциальных регуляторов выработки
островковых гормонов с целью уточнения
патогенеза.
3) Оценка содержания гормонов желез,
пораженных системным аутоиммуным
процессом.
4) Оценка содержания факторов,
отражающих развитие осложнений.
В рамках данной книги, посвященной
патофизиологии метаболизма и его
эндокринной регуляции, уместна будет краткая
характеристика состояния эндокринной
системы при самом важном метаболическом
заболевании человека — СД.
Среди пептидных гормонов наиболее
значительным и многообразным
физиологическим действием на островковыи аппарат
обладают гормоны энтериновой системы и
нейропептиды. Хорошо известен факт более
сильной стимуляции секреции инсулина
при пероральном приеме глюкозы по
сравнению с внутривенным ( в классической
литературе— «инкретиновый эффект»).
Несомненно, гормоны энтериновой
системы служат физиологически
значимым регулятором инсулиновой секреции,
позволяющим при приеме пищи начать
стимулировать островки до нарастания
концентрации глюкозы в крови.
Наиболее сильным стимулятором
секреции инсулина среди гормонов энтериновой
(эндокринно-метаболические нарушения)
системы считается желудочный ингибиру-
ющий полипептид (GIP). Его содержание
в портальной крови растет после приема
пищи, он увеличивает секрецию инсулина
дозозависимым образом. GIP
стимулирует анаболизм, увеличивает скорость
кругооборота липопротеидов, потенцирует
липидмобилизующие эффекты и ингиби-
рует глюкагонзависимый липолиз. Для
проявления инсулинотропного действия
GIP необходима гипергликемия насыщения.
При избыточном весе существует
гиперчувствительность к GIP.
Холецистокинин (ССК) в
физиологических концентрациях стимулирует
выработку всех основных островковых гормонов,
эффект этот особенно заметен у
новорожденных и сопровождается торможением
аппетита и усилением выработки пролак-
тина и АКТГ.
Вазоактивный интестинальный
полипептид (VIP) — имеет рецепторы на В-клетках,
стимулирует выработку инсулина и может
действовать как индуктор дифференциров-
ки. Параллельно VIP увеличивает
продукцию лютеинизирующего гормона, сомато-
тропина и пролактина в аденогипофизе.
Секретин (SC) стимулирует выработку
инсулина in vivo и in vitro. ССК, VIP и SC
не увеличивают свое содержание в крови
при насыщении, что не позволяет считать
их столь же значимыми проводниками ин-
кретинового эффекта, как GIP.
Гастрин, участвующий в усилении
освобождения инсулина и глюкагона в ответ на
смешанную пищу и аминокислоты,
стимулирует выработку соматотропина.
Мотилин стимулирует выработку
соматотропина и обладает СТГ-подобным
действием на островки.
Галанин вызывает наибольший интерес
среди панкреатических нейропептидов.
Выделяется пептидэргическими нейронами
и непосредственно влияет на островковые
клеткц. Считается, что роль его особенно
велика при стрессорных воздействиях
на островки. Галанин при внутривенном
введении значительно повышает уровень
соматотропина, вероятно ингибируя обра-
зование соматостатина D-клетками, и
является одновременно стимулятором аппетита.
Интерес к этому пептиду диктуется тем, что
при экспериментальном введении собакам и
мышам галанин вызывает у них транзитор-
ную инсулинопению и гипергликемию.
ИЗСД у мышей сопровождается
понижением продукции энкефалинов в
панкреатических островках.
Нейропептид Y угнетает
стимулированный ответ глюкагона на аминокислоты и
одновременно повышает аппетит.
IL -1 в малых дозах, в ранние сроки после
введения и в условиях гипергликемии
стимулирует, а в противоположных ситуациях
понижает освобождение инсулина из
островков у крыс, не влияя на освобождение
глюкагона (это имеет физиологический
смысл в продроме инфекций и при
лихорадке).
В околоушных железах крыс обнаружены
неидентифицированные пептиды,
усиливающие образование инсулина в ответ на
глюкозу, причем эффекты паротидэктомии
особенно сказываются на концентрации
глюкозы у старых крыс.
Часть АКТГ — полипептид 19-39 имеет
рецепторы на В-клетках и иногда
рассматривается в качестве основного кандидата
на роль центрального гипофизарного
регулятора работы инсулинпродуцирующих
клеток.
Сомагпотпропин, как и АКТГ,
вырабатывается в ответ на гипогликемию, что, в свою
очередь, влияет на островки. Он снижает
метаболическое действие инсулина,
являясь контринсулярным гормоном, мощно
стимулирующим липолиз, кетогенез и
глюконеогенез.
Инсулин и система «соматотропин — ин-
сулиноподобные факторы роста» (ИФР) —
две родственные эндокринные системы,
пересекающиеся на многих уровнях.
ИФР-1 и его рецепторы имеют высокое
сходство и перекрестную реактивность с
инсулином и его рецептором. ИФР-1
определяет чувствительность островков к СТГ.
Существует связь между нарушениями
в системе «соматотропин — инсулино-
Патофизиология сахарного диабета 297
подобные факторы роста» и патогенезом
осложнений ИЗСД.
Плацентарный аналог СТГ — хорионичес-
кий гонадотропин (ХГ) и пролактин,
называемый иногда «малый СТГ», действуют на
островки подобно СТГ.
Лютеинизирующий гормон (Л Г) через
стимуляцию синтеза прогестерона
активизирует освобождение инсулина.
Тироидные гормоны: тироксин (Т4) и
трийодтиронин (Т3) увеличивают базаль-
ный и стимулированный синтез инсулина
и глюкагона, но снижают эффект инсулина.
При гипертирозе функция В-клеток
повышена, но и резистентность к инсулину
увеличена, что приводит к гиперинсулинемии
и гипер-С-пептидемии.
Гормоны липидной природы также
способны влиять на работу панкреатических
островков. Все производные циклооксиге-
назного пути метаболизма арахидоновой
кислоты, особенно простпагландины (Pg E2
А), снижают стимулированную глюкозой
секрецию инсулина и увеличивают базаль-
ную выработку глюкагона и соматостатина.
Особенно велико их воздействие на первую
фазу инсулиновой секреции. Продукты ли-
пооксигеназного пути, особенно, лейкотри-
ены (Lt) и 15-гидропероксиэйкозатетрае-
новые кислоты, стимулируют инсулиновый
ответ. Простагландины снимают влияние
GIP на секрецию инсулина.
Витамин D (через обмен кальция)
усиливает секрецию инсулина. Метаболизм этого
тканевого гормона частично находится под
контролем инсулина, который влияет на
этап повторного гидроксилирования
витамина D. В связи с этим при ИЗСД у детей
рахит отягощает течение болезни и
создается порочный круг, связанный с патогенезом
диабетической остеопатии.
Общеизвестно контринсулярное
действие глюкокортикоидов, конкурирующих с
инсулином за гексокиназу и мощно
стимулирующих глюконеогенез. Особняком стоит
гидрокортизон, тормозящий in vivo распад
инсулина.
Прогестерон уменьшает скорость
пролиферации В-клеток, но усиливает освобожде-
298 АЖ Зайчик, ЛЛ.Чурилов Патохимия
ние инсулина. Лндрогены снижают уровни
секреции инсулина и его эффекты.
Катехоламины (КА), действуя в
минимальных концентрациях, ингибируют
освобождение инсулина и снижают его эффект,
не влияя на содержание глюкагона, хотя и
служат его синэргистами, а также тормозят
образование соматостатина. В средних
концентрациях, действуя как нейромедиаторы,
КА еще сильнее тормозят секрецию
инсулина (рецепторно опосредованный а-эффект),
усиливают выработку глюкагона
(рецепторно опосредованный р-эффект) и повышают
уровень соматостатина (а/р-эффект). Таким
образом, нейротрансмиттерное действие
Р-адреномиметиков — инсулинотропное,
аа-адреномиметиков — антиинсулиновое.
Серотонин у человека увеличивает, а
дофамин снижает освобождение инсулина. При
этом дофамин ингибирует выброс
соматостатина и стимулирует освобождение
глюкагона, действуя через адренорецепторы.
Метаболические нарушения
при сахарном диабете
Особенности метаболизма у больных
обеими формами диабета имеют много
общего, так как в основном они обусловлены
недостаточным действием инсулина на
клетки-мишени (абсолютный дефицит
инсулина при ИЗСД или относительный
дефицит инсулина на почве инсулиноре-
зистентности, на фоне избытка глюкагона
и других контринсулярных регуляторов).
Поэтому метаболизм при СД
характеризуется рядом общих черт, которые
рассматриваются ниже.
Механизмы действия инсулина
на ткани-мишени
Зависимость различных клеток от инсулина
неодинакова. В отношении метаболизма
глюкозы центральная нервная система,
ткани надпочечника, гонад, глаза принадлежат
к «инсулиннезависимым» и поглощают
глюкозу из крови без участия транспортёров,
запускаемых инсулином. В то же время
скелетные мышцы, липоциты, соединитель-
>иннО'Метаболические нарушения)
ная ткань, включая ее специализированные
виды, клетки крови и иммунной системы,
являются высоко «инсулинзависимыми».
Положение печени, почек, сердца и
некоторых других органов на шкале инсулинзави-
симости промежуточно.
Ранние эффекты инсулина опосредованы
его действием на поверхностный инсулино-
вый рецептор (рис. 65); в то же время,
подобно другим пептидным биорегуляторам,
инсулин путем рецепторного эндоцитоза
проникает внутрь клетки и далее — в
клеточное ядро, и его отсроченные и хронические
эффекты, включая трофическое и морфоге-
нетическое действие, опосредованы прямым
взаимодействием гормон-рецепторного
комплекса с цис-регуляторными
элементами генома.
Инсулин связывается с а-субъединицей
мембранного рецептора; после чего следует
аутофосфорилирование р-субъединицы
рецептора, которая начинает проявлять
активность тирозиновой протеинкиназы.
Последняя фосфорилирует ряд клеточных
субстратов, давая старт ранним
метаболическим эффектам. Среди посредников
действия инсулина в клетке лучше всего
охарактеризован белок IRS-1,
фиксирующий на себе, после фосфорилирования,
ряд инсулинзависимых энзимов, включая
фосфатидилинозитол-тригидроксилкиназу
(PI 3 К), активируемую при этом и
запускающую фосфатидилинозитоловый каскад
посредников метаболических эффектов
инсулина. Активация этого фермента, а
также фосфолипазы С ведет к накоплению в
клетке диацилглицерина и инозитолтрифос-
фата. Вследствие этого запускается проте-
инкиназа С, открывающая входные
кальциевые каналы и способствующая активации
кальмодулина и фосфодиэстеразы. Это
приводит к понижению внутриклеточной
концентрации ц-АМФ и катаболических
процессов в клетке, в то же время кальмоду-
линовая активация гуанилатциклазы
обеспечивает через ц-ГМФ и соответствующие
протеинкиназы стимуляцию анаболизма.
Белок IRS-1 фиксирует и активирует и
ряд серин-треониновых киназ (raf, MAP,
Инсулиномиметические
антитела Г\
л
а субъединицы
Гормон (инсулин)
[Субстрат — Туг — РО-*|
(активный фермент)
Эффект
гормона
Рис 65. Рецептор инсулина — пример гормонального рецептора тирозинкиназного типа
S6), которые стимулируют активность
ферментов, обеспечивающих синтез гликогена,
липидов и белка, а также через так
называемые Ras-ассоциированные белки, ускоряет
перемещение молекул инсулинзависимого
транспортера глюкозы GluT4 в
плазматическую мембрану мио- и липоцитов.
В инсулиночувствительных клетках
существует мощная антисигнальная система,
способная уменьшать или блокировать
информационные последствия
взаимодействия инсулина с его поверхностным
рецептором. К ней относятся вырабатываемый
адипоцитами и клетками иммунной
системы ФНОа (кахексии), который, действуя
через специфические рецепторы, снижает
активность тирозиновой протеинкиназы,
особенно в мио- и липоцитах.
Блок рецепторной тирозиновой киназы
может обеспечиваться и другими
поверхностными и внутриклеточными энзима-
Патофизиология сахарного диабета 299
тически активными белками — РС-1 и
альфа-2-Н8-гликопротеином. Белок Rad
из Ras-суперсемейства ГТФ-аз ингибирует
перемещение транспортера глюкозы GluT4.
Эти механизмы важны для понимания
неоднородной природы инсулинорезистен-
тности.
Цитокины липоцитов (в частности, при
ответе острой фазы и при колебаниях
массы тела) сильно влияют на эффективность
действия инсулина. Адипонектин
увеличивает эффективность инсулина, особенно
его тормозное действие в гепатоцитах на
глюконеогенез и стимулирующее — на
захват глюкозы тканями. Кахексии, напротив,
тормозит эффекты инсулина на пострецеп-
торном уровне (см. выше гл. 7).
Углеводный метаболизм при СД
Плазматическая мембрана гепатоцита (в
отличие от мио- и липоцита) свободно про-
300 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
ницаема для нейтральной глюкозы. Вступит
или не вступит глюкоза на путь гликолиза,
определяется итогом «борьбы» между глю-
кокиназой и глюкозо-6-фосфатазой (см.
выше, предыдущий раздел). Контроль
инсулина над глюкокиназой (1) и над
поступлением свободных жирных кислот в клетку
(2) определяет активизацию гликолиза и
пентозофосфатного цикла.
Поджелудочная железа в норме ни при
какой метаболической ситуации не выключает
секрецию инсулина и глюкагона, посылая их
в кровь параллельно и меняя лишь
соотношение между ними
Эндокринный эквивалент
«диабетического статуса» организма проходит под знаком
относительного дефицита инсулина и
избытка глюкагона. Это обусловливает целый
набор изменений в углеводном метаболизме.
>- В гепатоцитах, миоцитах и липоцитах
низкие уровни инсулина приводят к
угнетению гексокиназной (глюкоки-
назной) реакции, что ведет к
снижению образования глюкозо-6-фосфата.
Низкие концентрации инсулина,
контролирующего активность ферментов
гликолиза и почти всех ферментов пен-
тозного цикла, а также малые
количества глюкозо-6-фосфата, обусловливают
угнетение гликолиза и пентозного цикла.
>• Вследствие активации фосфорилазы
и ингибирования гликогенсинтетазы в
гепатоцитах усиливается гликогенолиз.
Так как происходит снижение
активности глюкокиназы и активируется
глюкозо-6-фосфатаза, фосфорилирова-
ние глюкозы ухудшается, распад фос-
форилированной глюкозы возрастает,
а нейтральная молекула глюкозы легко
покидает гепатоцит.
>- Вследствие активации ферментов глю-
конеогенеза гепатоциты усиленно
производят глюкозу «de novo» (глюка-
гон активирует ключевые ферменты
глюконеогенеза и обеспечивает глю-
конеогенез субстратами — усиливает
липолиз, тормозит синтез белка на
рибосомах). Так как мембрана покоящейся
мышечной клетки не имеет рецепторов
к глюкагону, — мышца на глюкагон не
реагирует.
>• Нарушается транслокация транспортера
GluT4, обеспечивающего облегченную
диффузию глюкозы в мио- и липоциты,
и снижается поглощение глюкозы
мышцами и жировой тканью после еды.
>* Полноценная работа цикла Кребса
нарушается, и как следствие, возникает
снижение окислительного фосфори-
лирования и дефицит АТФ. Работа
цикла Кребса дефектна по следующим
причинам:
1) активность ферментов цикла снижена
как результат низких уровней инсулина
(половина ферментов цикла косвенно
подконтрольна инсулину);
2) субстраты цикла (оксалоацетат и а-
кетоглутарат), активно отвлекаются в
глюконеогенез, резко активированный
при абсолютно или относительно
повышенных уровнях глюкагона;
3) содержание пирувата снижено,
поскольку угнетен гликолиз;
4) анаплеротический путь восполнения
оксалоацетата в условиях возрастания
уровней ацетил-Ко А снижает свою
активность, поскольку снижены
концентрации пирувата.
Как результат всех указанных явлений
развивается гипергликемия. Сладкий вкус
сыворотки крови при СД установил М. Доб-
сон(1776).
Когда концентрация глюкозы
превысит почечный порог для глюкозы (160-
200 мг/дл, при нормальном уровне — 80-120
мг/дл или 3,5-5,5 ммоль/л), то начинается
глюкозурия. Сладкий вкус мочи как симптом
СД выделил Т. Уиллис (1674), позже У.
Кален (1769) отделил сахарное мочеизнурение
от «безвкусного». Глюкозурия вызывает
осмотический диурез. Больной испытывает
полиурию (до 8-12 л/сут). Именно она
зачастую расценивается как первый симптом
сахарного диабета. Дополнительным
механизмом полиурии может служить снижение
эффективности взаимодействия АДГ с его
Патофизиология сахарного диабета 301
рецептором из-за гликозили-
рования последнего.
Потеря воды и
электролитов (натрия, калия, магния и
фосфора) с мочой, не
компенсируемая их достаточным
поступлением в организм, может
приводить к дегидратации.
Следствием этого является
полидипсия. Происходит ге-
моконцентрация, может
наблюдаться падение ОЦК, что
обусловливает
недостаточность периферического
кровообращения.
Прогрессирование
нарушений кровообращения и осмо-
лярного равновесия в
сочетании с ионным дисбалансом и
тканевой гипоксией способны
привести к гиперосмолярной
коме (рис. 66).
ДЕФИЦИТ ИНСУЛИНА
ИЗБЫТОК ГЛЮКАГОНА
сниженная
утилизация
глюкозы
возросшая
продукция
глюкозы
ГИПЕРГЛИКЕМИЯ
I
ГЛЮКОЗУРИЯ
I
ОСМОТИЧЕСКИЙ
ДИУРЕЗ
ГИПЕРОСМОЛЯРНОСТЬ
U
~■ ДЕГИДРАТАЦИЯ —
КОМА-
• ДВ С-синдром
ШОК
-► СМЕРТЬ -*-
Основной обмен при ИЗСД
повышен. Неусвоение
углеводов ведет к «голоданию среди
изобилия»(М. Рубнер),
поэтому при ИЗСД во многом воспроизводится
эндокринно-метаболическая картина
второго периода голодания (см. раздел
«Патофизиология голодания» выше). С этим связана
полифагия, как характерный симптом
диабета, хотя механизмы повышения аппетита
могут быть, по крайней мере, при ИЗСД,
связаны с исхуданием и снижением
продукции ФНОа, подавляющего аппетит. Вполне
возможно, что в условиях дефицита
инсулина может снижаться продукция лептина,
представляющего аппетитподавляющий
сигнал для гипоталамуса (см.
«Патофизиология первичного ожирения»).
Липидный метаболизм
при диабете
Организм больного сахарным диабетом
удовлетворяет свои энергетические
потребности прежде всего за счет окисления
жиров. Дыхательный коэффициент стремится
к 0,7, карбонурический коэффициент (C/N
Рис 66. Патогенез гипергликемической комы. Пояснения в
тексте
в моче) повышается (см. выше
«Патофизиология энергетического метаболизма»). Такое
изменение карбонурического коэффициента
— следствие выделения в мочу огромных
количеств недоокисленного углерода (углерод
глюкозы, кетоновых тел) — феномен,
известный как «дизоксидативная карбонурия».
Основные звенья нарушения липидно-
го обмена при СД представлены на рис.
67 — внизу.
В здоровом организме инсулин
способствует отложению жира в жировой ткани
и снижению его утилизации. Механизмы
этого множественные и не сводятся только
к сбереганию жира путем усиления
катаболизма экзогенной глюкозы:
>* Инсулин усиливает синтез жирных
кислот из глюкозы в гепато- и липоци-
тах. Под его влиянием активируется
реакция карбоксилирования ацетил-КоА
с последующим образованием малонил-
КоА, удлиняющего молекулу СЖК,
мишенью гормона служит фермент
302 AM Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
ацетил-КоА-карбоксилаза(ацетил-КоА:
С02-лигаза).
Инсулин противодействует влияниям
всех липолитических гормонов
(адреналина, глюкагона, СТГ, глюкокортикои-
дов), а также создает избыток изоцитрата
и а-кетоглутарата — активаторов ацетил-
КоА-карбоксилазы.
Известно, что жирные кислоты
транспортируются из печени в жировую ткань
в составе липопротеидов очень низкой
плотности (ЛПОНП), секретируемых
печенью (см. «Нарушения липидного
обмена»). Инсулин усиливает активность
липопротеиновой липазы,
осуществляющей клиренс ЛПОНП с переходом
жирных кислот в адипоциты.
Инсулин ускоряет транспорт глюкозы в
адипоциты и ингибирует основной ли-
ДЕФИЦИТ ИНСУЛИНА
ИЗБЫТОК ГЛЮКАГОНА
снижение утилизации глюкозы
—— снижение липогенеза
I
мобилизация жира из депо
липемия
метаболический
ацидоз —
- повышение кетогенеза
и холестериногенеза
кетонемия и
холестеринемия
кетонурия
потеря Na
Рис 67. Липидный обмен при сахарном диабете. Поясне
ния в тексте
политический фермент клеток жировой
ткани — гормонзависимую липазу.
>* Под действием инсулина активация
гликолиза обеспечивает липогенез
пластически (а-глицерофосфатом), а
активация пентозного пути — энергетически
(поставкой НАДФН2).
При ИЗСД резко увеличена скорость ли-
полиза и содержание СЖК в крови. Жиры,
освобождаемые адипоцитами, при этом
ресинтезируются в печени, и поэтому обе
формы сахарного диабета приводят к стеа-
тозу печени. При ИЗСД ожирение печени
наблюдается, несмотря на общее исхудание,
и липиды могут составлять до 1/3 веса
печени. Поэтому сахарный диабет (особенно
ИНСД) часто сопровождается гиперлипо-
протеидемией IV типа (с накоплением в
крови ЛПОНП). Из-за снижения активности
липопротеинлипазы при ИЗСД
может быть гиперлипопротеи-
демия I типа (персистирование
хиломикронов) и сочетание I и
IV типов — гиперлипопротеиде-
мия V типа.
Вторичные гиперлипопроте-
идемии при СД способны
ускорить развитие атеросклероза
(IV тип) и панкреатита (I тип).
Механизмы взаимосвязи
атеросклероза и диабета
особенно важны для возникновения
осложнений ИНСД. При диабете
количество антиатерогенных
дренажных липопротеидов высокой
плотности снижено, а при ИЗСД
повышена концентрация высо-
котромбогенного липопротеида
(а). Подробнее эти аспекты
нарушения липидного обмена при
СД рассматриваются выше (см.
раздел «Нарушения липидного
метаболизма»).
В условиях преобладания
эффектов глюкагона и других ан-
тиинсулярных гормонов (СТГ,
глюкокортикоидов) изменения
в липидном обмене способствуют
Патофизиология сахарного диабета 303
усилению кетогенеза в печени и
торможению утилизации ацетоуксусной кислоты на
периферии. Это важный механизм
диабетического кетоацидоза (см. выше «Нарушения
межуточного обмена липидов»).
Белковый метаболизм
при диабете
Инсулин — важнейший стимулятор
анаболизма вообще — является и ключевым
анаболическим гормоном белкового обмена,
снижающим деградацию и стимулирующим
синтез белков. Механизмы анаболизирую-
щего действия инсулина включают:
> прямую стимуляцию сборки рибосом и
процесса трансляции (по типу
инициирующего фактора);
> усиление активного транспорта
аминокислот в клетки (особенно, валина,
лейцина, изолейцина) при
одновременном торможении
глюконеогенеза в печени и
освобождения аминокислот
из клеток, особенно
мышечных;
> трофические долговременные
эффекты, связанные с
усилением синтеза ДНК, РНК
и митотической активности
ряда клеток.
Эффекты инсулина на рост и
белковый синтез синэргичны с
действием соматотропина.
При сахарном диабете синтез
белка угнетается и усиливается
его катаболизм, что ведет к таким
симптомам, как гипераминоаци-
демия, отрицательный
азотистый баланс, усиление экскреции
мочевины (все эти феномены,
однако, не способны отменить
возрастание величины карбону-
рического коэффициента!).
Отчасти это обусловливает
имеющий место при диабете
вторичный иммунодефицит,
который, кроме того, связан и с
нарушением энергетики инсулин-
зависимых макрофагов и лимфоцитов,
проявлением чего служит, например, феномен
«ленивых фагоцитов».
Усиление распада белков при СД
особенно характерно для мышц. Этот
процесс сопровождается выходом калия и
других внутриклеточных ионов в кровь
с последующей экскрецией их с мочой, в
условиях полиурии. Гипокалиемия и потеря
мышечных белков обусловливают симптом
мышечной слабости.
Прогрессирующая потеря воды и
электролитов приводит в конце концов к
внутриклеточной дегидратации, которая способствует
катаболическим процессам и диффузии
электролитов во внеклеточную жидкость.
Пока продолжается выделение мочи,
существует опасность потери калия организмом
во все более угрожающих количествах, что
ДЕФИЦИТ ИНСУЛИНА
ИЗБЫТОК ГЛЮКАГОНА
снижение утилизации глюкозы
■ повышение распада
белка
аминацидемия
усиление поглощения
глюкогенных аминокислот
I
усиление
глюконеогенеза
i
повышение выведения
азота с мочой
( гиперазоту рия)
потеря калия и др.
" ионов клетками
i
дегидратация
клеток
потеря калия организмом
Рис 68. Белковый обмен при сахарном диабете
304 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
может привести к развитию опасных тахи-
аритмий. Основные звенья нарушения
белкового метаболизма при СД представлены
схематично на рис. 68.
Патогенез острых
осложнений СД
Крайняя степень метаболических
нарушений при сахарном диабете приводит к коме.
Кома (от греческого «глубокий сон») —
патологический процесс, характеризующийся
крайней степенью торможения центральной
нервной системы с отсутствием
рефлекторных реакций, включая и нарушение
жизненно важных висцеральных рефлексов.
Патохимической основой всякой комы
служит глубокая тканевая гипоксия
центральной нервной системы, приводящая к
деполяризации нейронов и угнетению их
возбудимости.
Поскольку к тканевой гипоксии мозга
могут привести различные механизмы, по
патогенезу выделяют различные виды ком.
Некоторые из них встречаются как острые
осложнения СД (табл. 15а).
Кетоацидоз
и кетоацидотическая кома
Кетоацидоз типичен для ИЗСД. Кетонурию
при СД открыл еще В. Петтерс (1857), а
чуть позже И. Каулич описал клинику
диабетического кетоацидоза (1860). Примерно
14-31 % детей, госпитализируемых в США
по поводу сахарного диабета, имеют
кетоацидоз. Для молодых больных СД
кетоацидоз — основная причина смерти (смертность
при кетоацидозе составляет от 0,5 до 15,4 %).
Однако пока образуется С-пептид и имеются
В-клетки, тяжелый кетоацидоз бывает редко,
так как для предотвращения быстрого липо-
лиза в адипоцитах достаточно минимальных
количеств инсулина. Таким образом,
выраженный кетоацидоз — характеристика
терминального декомпенсированного диабета.
Различают кетоацидоз и кетоацидотичес-
кое состояние.
Кетоацидоз — это патохимические сдвиги
во внутренней среде организма. Кетоаци-
дотическое состояние — это клинические
(особенно нервно-психические) нарушения,
соответствующие данному расстройству
кислотно-основного баланса. Крайним
выражением кетоацидотического состояния и
является диабетическая кетоацидотическая
кома. Некоторые черты кетоацидоза при
различных нарушениях уже
рассматривались выше, в разделе «Кетоз и стеатоз
печени». Здесь приведена
детализированная характеристика именно диабетической
формы кетоацидоза.
Ведущее звено патогенеза
кетоацидоза — особенность гормонального статуса
больного («нехватка инсулина — избыток
глюкагона»), а также преобладание в этих
условиях эффектов других контринсуляр-
ных гормонов — катехоламинов, глюко-
кортикоидов, соматотропина. Практически
кетоацидоз может возникать как следствие
повышения потребности в инсулине (при
переходе скрытого ИЗСД в явный), или при
физическом или эмоциональном стрессе
(у пациента с компенсированным до этого
ИЗСД). Диабетический кетоацидоз может
стать и результатом усиления действия
любых контринсулярных гормонов.
При диабетическом кетоацидозе
накопление кетоновых тел связано как с ускорением
их образования, так и с нарушением их
утилизации.
Выше, в разделе «Нарушения липидного
обмена» подробно рассмотрены механизмы
усиления продукции кетоновых тел при
ИЗСД, что делает излишним повторное их
описание.
Отметим, что одного усиления
образования ацетил-КоА недостаточно для индукции
кетоза.
При нормальном метаболизме
существуют множественные альтернативные
пути использования этого универсального
катаболита, о которых здесь уместно еще
раз напомнить:
>• Ацетил-КоА может вовлекаться в
реакцию изоцитратной конденсации с
оксалоацетатом и сгорать в цикле Кребса
что представляет основной путь его
нормальной утилизации.
Таблица 15а. Дифференциально-диагностические критерии различных видов комы при острых осложнениях СД
(по ЮМ. Строеву, ЛЛ. Чурилову, 2004)
Критерий
Анамнез
Развитие
Поведение
Предшествующие
жалобы
Температура тела
Кожа
Тургор кожи
Язык
Мышцы
Глазные яблоки
Артериальное
давление
Глюкоза в крови
Глюкоза в моче
Ацетонурия
Креатинин крови
Диабетическая
кетоацидотическая
кома Куссмауля
Диагноз СД известен или впервые
установлен
Постепенное, дни или часы
Заторможенность
Общая слабость, жажда, полиурия,
тошнота, боли в животе, рвота
Нормальная или повышена в связи
с сопутствующей инфекцией
Сухая, шершавая
Пониженный
Сухой, красный
Тонус ослаблен
Мягкие
Понижено
Высокая
Присутствует
Всегда
Норма или слегка повышен
Гипогликемическая
кома
Диагноз СД известен, получает
инсулин
Быстрое, минуты
Возбуждение
Головная боль, дрожание рук,
сильная потливость,
головокружение
Нормальная
Мокрая, особенно кожа лба
Достаточный
Нормальный
Тонус хороший, судороги
Нормальный тонус
Нормальное или пониженное
Пониженная
Может быть, если долго не
мочился
Отсутствует
Нормальный
Гиперосмолярная
(гипергликемичес-
кая) кома
Диагноз СД известен
или впервые
установлен
Постепенное, дни или
часы
Вялость, сознание
долго сохранено
Общая слабость,
вялость судороги
Повышенная
Сухая
Пониженный
Сухой, красный
Тонус ослаблен
Мягкие
Пониженное
Очень высокая
Присутствует
Отсутствует
Повышен
кома в исходе ХПН при диабети-
ческом гломерулосклерозе
Болеет СД давно
Медленное, дни
Заторможенность, безразличие
Общая слабость, головная боль,
отеки
Нормальная или пониженная
Сухая 1
Кожа отечная |
Нормальный 1
Мышечные подергивания
Нормальные
Чаще высокое
Нормальная
Чаще отсутствует
Отсутствует |
Очень высокий |
р
|з
|о
ISIS
р
§
|с
13>
1<э
306 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
>• Ацетил-КоА может участвовать в ресин-
тезе жирных кислот и жиров.
>* Ацетил-КоА может участвовать в стеро-
идогенезе через р-окси-р-метилглутарат
и мевалонат.
>- Наконец, ацетил-КоА может
использоваться для образования кетоновых тел.
Атомы углерода из состава ацетил-КоА у
человека и высших животных путём обмена
могут оказываться в составе продуктов,
вовлекаемых в глюконеогенез. Однако,
необходимо еще раз подчеркнуть: только
микроорганизмы и растения могут прямо вовлекать
ацетил-КоА в синтез гексоз, у человека и
млекопитающих прямой путь синтеза
глюкозы из ацетил-Ко А отсутствует.
При сахарном диабете все пути
использования избытка ацетил-КоА оказываются
неэффективными или блокированными, за
исключением тех, которые ведут к кетозу
или образованию холестерина (см.
«Нарушения липидного обмена» выше). Так как
кетоновые тела — умеренно сильные
органические кислоты, им свойственно связывать
значительные количества катионов натрия.
Это приводит к метаболическому ацидозу и
способствует потере воды и электролитов.
Увеличивается анионный просвет (см. ниже
«Патофизиология кислотно-основного
равновесия»), как следствие— нарастает
внеклеточная дегидратация, которая, с
одной стороны, ведет к внутриклеточной
дегидратации, а с другой к гемоконцентра-
ции и недостаточности периферического
кровообращения, вплоть до гипотензии,
способной, в свою очередь, привести к шоко-
подобному снижению почечного кровотока
и даже развитию анурии.
Выведение в мочу ионов натрия, фосфора,
хлора, калия, магния per se способно вызвать
аритмию и усугубить недостаточность
кровообращения.
Кетоз — главная причина
метаболического ацидоза при диабетической коме. Но
имеются и дополнительные механизмы
нарушения кислотно-основного баланса.
В обмен на натрий, теряемый с мочой, из
клеток в межклеточную жидкость
выводятся катионы водорода, усугубляя ацидоз.
Декомпенсированный ацидоз вызывает
дыхание Куссмауля — патологическую инспи-
раторную одышку с крайне редким, шумным
и глубоким дыханием, которая формируется
под водительством гаспинг-центра,
замещающего при глубокой гипоксии ЦНС
функции пневмотаксического центра.
При дыхании Куссмауля теряется С02и
наступает гипобикарбонатемия, а
следовательно, усугубляется ацидоз. При гипоксии
в ткани мозга накапливается избыток лак-
тата, из-за чего еще более нарастает ацидоз.
Ацидоз при диабетической коме вызывает
по типу порочного круга появление или
усугубление инсулинорезистентности, так как
инсулин в кислой среде имеет пониженное
сродство к своему рецептору.
Инсулинорезистентность при глубоком
ацидозе обусловлена еще и тем что:
1. Ацидоз угнетает гликолиз на этапе фос-
фофруктокиназы.
2. Имеет место высокий уровнень СЖКв
крови.
3. Создаются высокие концентрации
физиологических антагонистов инсулина,
многие из которых — катехоламины,
глюкокортикоиды, вазопрессин —
усиленно освобождаются в предагональном
состоянии.
Еще более усугубляет гипоксию
негативное влияние кетоацидоза на функцию
гемоглобина (НЬ). Образующийся в ходе
гликолиза 2,3-дифосфоглицерат (2,3-ДФГ)
является аллостерическим модулятором
гемоглобина. При добавлении 2,3-ДФГ к
раствору чистого НЬ сродство НЬ к
кислороду значительно снижается. Этот эффект
обусловлен тем, что 2,3-ДФГ сам
связывается с дезоксигемоглобином.
Когда концентрация 2,3-ДФГ в
эритроцитах падает, сродство НЬ к кислороду
возрастает, а способность его отдавать
кислород тканям уменьшается. Главный
фактор, регулирующий образование 2,3-ДФГ,
это доступность фосфата. При кетоацидозе
и сопутствующем ему дефиците фосфата
уровень 2,3-ДФГ в эритроцитах пада-
Патофизиология сахарного диабета 307
ет. Сродство гемоглобина к кислороду
возрастает, и поступление его в ткани
через капиллярные стенки падает.
Снабжение тканей кислородом, нарушенное
из-за недостаточности периферического
кровообращения, еще более ухудшается.
Кетоацидоз не может быть купирован
никакими ощелачивающими воздействиями,
без адекватной инсулинотерапии.
Таким образом, диабетическая (кетоне-
мическая, кетоацидотическая) кома — это
острое осложнение ИЗСД, обусловленное:
> влиянием кетоновых тел и тканевой
гипоксии на клетки ЦНС;
> обезвоживанием;
> сдвигом рН в сторону ацидоза.
Следует помнить, что в детском возрасте
диабетическая кома проявляется чаще и
быстрее. Резкий кетоз на фоне
дегидратации ведет к угнетению ферментных систем
мозга, происходит снижение утилизации
глюкозы нейронами, что усугубляет
развитие гипоксии в нейронах.
Нарастание катаболизма белков и их
усиленное превращение в углеводы
вызывает повышенное образование
аммиака и мочевины, как
результат — возникает гиперазотемия
и гиперазотурия, следствием
которых является усугубление
интоксикации и гипоксии
тканей мозга. Как уже отмечалось
в предыдущем разделе и в главе
«Патофизиология белкового
обмена», нейтрализация аммиака в
мозгу при СД затрудняется из-за
дефицита кетокислот,
использованных для глюконеогенеза.
Поэтому интоксикация
аммиаком отягощает течение тканевой
гипоксии ЦНС.
Гипоксия нейронов
непосредственно приводит к:
> расстройству дыхания;
> сосудистому коллапсу;
> снижению мышечного
тонуса;
>* нарушению высшей нервной
деятельности.
Основные звенья патогенеза диабетического
кетоацидоза отражены на рис. 69.
Клинические аспекты диабетической комы
характеризуются следующим. Кома
развивается постепенно (от 12-24 часов до
нескольких суток). В развитии комы выделяют
четыре стадии:
1 — легкое кетоацидотическое состояние;
2 — выраженное кетоацидотическое
состояние;
3 — тяжелое кетоацидотическое состояние;
4 — собственно кома:
а) поверхностная;
б) выраженная;
в)глубокая;
г) терминальная.
Каждая стадия процесса имеет типичные
симптомы.
1-стадия, или легкое
кетоацидотическое состояние. Для йее характерны
слабость, вялость, потеря аппетита, тошнота,
рвота, головные боли, головокружения,
ДЕФИЦИТ ИНСУЛИНА
ИЗБЫТОК ГЛЮКАГОНА
возросшая
мобилизация
жиров
сниженная
утилизация
глюкозы
возросшая
продукция
глюкозы
кетонемия
1
кетонурия
I— гипергликемия *
глюкозурия
снижение
рН
плазмы
(7.1)
потеря
KuNa'
КОМА
осмотический
диурез
гипербсмолярностъ
и
дегидратация
смерть-
-шок
Рис. 69. Патогенез диабетического кетоацидоза. Пояснения
в тексте
308 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
желудочно-кишечные боли, которые могут
имитировать острые заболевания брюшной
полости. В этот период резко усиливаются
полидипсия иполиурия. Уровень сахара
превышает 15,4ммоль/л. Значительно
возрастает глюкозурия. Появляется ацето-
нурия. Если легкие не справляются с
окислением кетонов, ощущается запах ацетона в
выдыхаемом воздухе.
2-стадия, или выраженное кетоацидоти-
ческое состояние. В эту стадию отмечаются
прогрессирующая сонливость и оглушение с
притуплением восприятия (обнубиляция).
3-стадия, или тяжелое кетоацидоти-
ческое состояние. Характеризуется
сопором: глубоким патологическим сном, при
котором, однако, ещё сохранены болевая
чувствительность, глотательный,
зрачковый, роговичный рефлексы. Сухожильные
рефлексы еще высокие. Разбудить больного
при сопоре можно, но лишь при помощи
сильных раздражителей.
4-стадиЯу или собственно кома.
Характеризуется:
>* полной потерей сознания;
>* дыханием Куссмауля (шумное,
глубокое, редкое, с удлиненным вдохом
и коротким выдохом; каждому вдоху
предшествует продолжительная пауза);
>* запах ацетона в выдыхаемом воздухе
(запах моченых яблок); присутствие
последнего симптома связывается с
обязательным существованием
сопутствующих или вторичных легочных
нарушений, например, синдрома дыхательных
расстройств, нередко сопровождающего
тяжелый кетоацидоз, так как здоровые
легкие нацело окисляют кетоновые
тела.
>■ При классической кетоацидотической
коме кожа больного сухая, холодная;
тонус глазных яблок резко снижен; зрачки
сужены; мускулатура вялая и
расслабленная; температура тела снижена; пульс
малый, частый; имеется гипотензия;
возникает олигурия вплоть до анурии;
присутствуют атония кишечника и
синдром переполнения атоничного желудка;
отмечаются признаки гипокалиемии (на
ЭКГ: снижение ST и зубца Т, удлинение
QT, появление высоких и заостренных
Р, а также — патологического зубца U).
В зависимости от преобладания тех или
иных симптомов выделяют следующие
варианты кетоацидотического состояния
(нередко имитирующие другие виды патологии):
> сердечно-сосудистый (преобладание
сердечной или сосудистой недостаточности,
коллапс);
>• желудочно-кишечный (клиническая
картина ложного аппендицита или
перитонита, псевдохолеры, аддисонова
криза);
>* почечный (картина острой почечной
недостаточности, на первом плане —
дизурия, гиперазотемия, протеинурия,
цилиндрурия; ацетонурия и глюкозурия
отсутствуют, вследствие резкого
снижения клубочковой фильтрации);
>* энцефалопатический (картина острого
нарушения мозгового кровообращения).
Картина крови при диабетическом кето-
ацидозе следующая:
>* нейтрофильный лейкоцитоз;
>• увеличение СОЭ;
>* возрастание количества эритроцитов и Hb.
Лабораторные клинико-биохимические
показатели (нормы см. в приложении А,
в конце книги) при тяжелом диабетическом
кетоацидозе очень характерны:
>- Содержание глюкозы в крови таких
пациентов обычно бывает выше 18ммоль/
л, может достигать и 100ммоль/л.
>* Осмоляльность крови повышена (ос-
моляльность плазмы в норме 280-310
мосм/кг) осмолярность крови
рассчитывается по формуле:
2х(К+ + Na+) ммоль/л +
+ гликемия ммоль/л.
>• Присутствует крайне выраженная кето-
немия — до 17 ммоль/л и более.
>• Повышено содержание: неэстерифици-
рованных жирных кислот, триглице-
ридов, креатинина, остаточного азота,
мочевины, холестерина, билирубина.
> Снижен уровень: резервной щелочности,
стандартного бикарбоната плазмы, рН до
7,2-6,8.
> Ряд патологических характеристик
имеет моча. Отмечаются: кетонурия,
высокая относительная плотность, кислая
реакция, резкая глюкозурия, протеину-
рия, цилиндрурия, микрогематурия.
При леченом кетоацидозе нередко
наблюдается такое типичное осложнение, как
отек мозга. Отек мозга — не вполне точный
термин, так как патофизиологически
отмечается внутриклеточная гипергидратация
(набухание) клеток мозга. Считается, что
гипергидратация возникает как следствие
лечения кетоацидоза на фоне начавшегося
улучшения биохимических параметров
крови. Механизмы этого осложнения,
обусловливающего до 1/3 смертельных исходов
кетоацидоза, особенно в педиатрической
практике, связаны с применением в лечении
гипотонических растворов и стремительным
падением осмоляльности внеклеточной
жидкости при быстром действии инсулина,
вводимого с лечебной целью в больших
дозах.
Нарушения кислотно-основного баланса
при сахарном диабете не сводятся только
к метаболическому кетоацидозу.
Значительную проблему — особенно в клинике
осложнений инсулиннезависимого
сахарного диабета — представляет лактат-ацидоз,
который может даже обусловить развитие
лактат-ацидотической комы, угрожающей
жизни пациента.
Ацидоз вследствие накопления
молочной кислоты встречается довольно часто,
даже у здоровых (см. ниже
«Патофизиология кислотно-щелочного равновесия»).
Кратковременный компенсированный
лактат-ацидоз возникает при усиленной
мышечной работе особенно у
нетренированных людей (как следствие относительного
недостатка кислорода).
Длительный лактат-ацидоз встречается
при тяжелых поражениях печени (токси-
Патофизиология сахарного диабета 309
ческие дистрофии, цирроз), при сердечной
недостаточности, при недостаточности
внешнего дыхания и при других формах
гипоксии. Но наиболее тяжелый лактат-ацидоз
возможен при декомпенсированном ИНСД
II типа, леченном бигуанидами — блокато-
рами глюконеогенеза.
Уровень лактата в крови — результат
динамического равновесия образования и
утилизации этого метаболита.
При молочнокислой форме
метаболического ацидоза уровень лактата в крови
увеличен более 5 ммоль/л и рН
артериальной крови составляет 7,25 и меньше (при
нормальном уровне лактата до 2 ммоль/л).
Хотя лактат образуют все ткани,
основное его количество образуется в мозгу,
эритроцитах, скелетных мышцах и коже.
Утилизируется же он главным образом в
печени и почках. В судебной медицине
лактат-ацидоз считается механизмом трупного
окоченения.
Выделяют А- и В-типы лактат-ацидоза
(по Р. Когену и X. Вудсу, 1976). Для типа А
характерны тканевая гипоксия, гипотензия.
Для типа В тканевая гипоксия не типична.
Тип В подразделяется на подтипы (Blf B2,
В3) в зависимости от этиологии (табл. 16.).
Диабетический лактат-ацидоз является
результатом гипоксии в организме
больного и некоторых эффектов, применяемых
для лечения инсулиннезависимого диабета
лекарств. Его развитию способствует
физическое переутомление больного сахарным
диабетом.
Гипоксическое состояние, которое
способствует лактат-ацидозу, как правило,
всегда имеет место при тяжелом сахарном
диабете.
Бигуаниды, особенно феноформин,
еще более осложняют ситуацию, угнетая
глюконеогенез и транспорт электронов по
дыхательной цепи.
Клинически у 80% больных ИНСД II
типа лактат-ацидоз начинает проявляться
дисфункцией желудочно-кишечного тракта,
сопровождается внезапно возникающими
310 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 16. Классификация и причины лактат-ацидозов
Тип А стканевой
гипоксией
Бигуаниды
Кардиогенныи шок
Эндотоксический шок
Тяжелая анемия
Левожелудочковая
недостаточность
Тип В, при разных
заболеваниях
Сахарный диабет
Почечная
недостаточность
Болезни печени
Инфекции
Лейкоз
Тип В2 лекарства и токсины
Бигуаниды
Алиментарная, парентеральная
фруктоза, сорбитол, ксилитол
Этанол
Салицилаты
Метанол
Тип ВЗ наследственные
формы
Гликогеноз 1 типа
Синдром Лея
Дефицит фруктозо-1 ,б-
дифосфата
Метилмалонилацидемия
Гипераланинемия
нарушением чувствительности, угнетением
сознания, гипотензией, гипотермией,
дыханием Куссмауля, сильным обезвоживанием
и циркуляторым коллапсом.
Диагноз следует подтвердить, определив
уровень лактата в плазме (>5ммоль/л).
Как правило, соотношение лактат/пиру-
ват — около 15; при этом артериальная кровь
имеет рН ниже 7,25 и уровень бикарбонатов
в плазме составляет менее 22 мэкв/л.
Анионный интервал (anionic gap), подробная
характеристика которого включена в раздел
«Патофизиология водно-электролитного
обмена», слишком широк и превышает
нормальный разброс, составляя 4—12 ммоль/л.
В отсутствие кетонурии возросший
анионный интервал у больного сахарным
диабетом почти наверняка указывает на
лактат-ацидоз. Лактат-ацидотическая кома
лечится крайне трудно.
Принципиально коррекция состояния
заключается в лечении шока, гипоксии
тканей и ацидоза.
Токсические метаболиты следует извлечь
из плазмы при помощи гемодиализа. Инсу-
линотерапия способствует освобождению от
лактат-ацидоза. В качестве буфера
используют метиленовую синь, хотя значимость ее
применения остается сомнительной.
Бикарбонат натрия вводят для противодействия*
ацидозу. Однако его следует применять с
большой осторожностью, ибо передозировка
бикарбоната приведет к развитию алкалоза.
Необходимую дозу бикарбоната вычисляют
следующим образом:
потребное количество бикарбоната
(ммоль) = 25 — (бикарбонат плазмы в
ммоль/л х вес тела в кг х 0,3).
Синдром гипергликемии
и гипергликемическая кома
Гипергликемия — повышение содержания
глюкозы в крови выше 120 мг/дл (6,1
мМ/л). Этиология гипергликемии может
быть различной. Выделяют следующие
виды гипергликемий:
>* Алиментарная — временное повышение
уровня глюкозы в крови при быстром
поступлении в организм избытка
легкоусвояемых углеводов. У здорового
человека прием 75 г глюкозы через рот
может вызвать подъём её концентрации
в крови выше верхней границы нормы.
Однако при этом уровень глюкозы не
превышает 200 мг/дл и нормализуется в
пределах 3 часов (см. выше — критерии
нагрузочной пробы на толерантность к
глюкозе). Через 2-2,5 часа может даже
наступить гипогликемическая фаза, в
результате физиологического ответа
инсулина.
>• Стрессорная — отражает действие ка-
техоламинов, глюкагона, глюкокорти-
коидов и вазопрессина и наступает при
действии различных стрессоров, в том
числе психоэмоционального характера.
Ранее отдельные виды стрессорной
гипергликемии характеризовались, как
«эмоциональная», «наркозная»,
«психогенная» и т.д.
> Судорожная — наступает при
конвульсиях мышц вследствие усиления в них
гликогенолиза. Примером
гипергликемии при судорожных состояниях
служит повышение уровня глюкозы в
крови при эпилептических припадках
и столбняке. Мышцы напрямую не
освобождают глюкозу в кровь, очевидно,
эта гипергликемия зависит от усиления
глюконеогенеза.
> Эндокринные — развиваются при
гиперпродукции контринсулярных гормонов.
Гипергликемией сопровождаются
синдром и болезнь Иценко-Кушинга и
другие формы гиперкортицизма, гиперти-
роз, гиперпаратеироз, феохромоцитома,
акромегалия и гипофизарный гигантизм.
Контринсулярные регуляторы могут
вызывать гипергликемию, действуя на
различные звенья углеводного обмена. Так,
глюкагон, катехоламины и тироидные
гормоны способствуют гликогенолизу,
СТГ тормозит синтез гликогена,
стимулирует распад инсулина и секрецию
глюкагона, глюкокортикоиды повышают
интенсивность глюконеогенеза.
> Выраженная гипергликемия характерна
для клиники глюкагономы. Глюкагоно-
ма — апудоцитарная опухоль клеток
островков Лангерганса или эктопической
локализации, секретирующая
избыточное количество глюкагона с развитием
синдрома, включающего вторичный
сахарный диабет, особый дерматит,
стоматит, анемию, похудание и диарею.
К настоящему времени описано около
200 случаев. Этиология неизвестна.
Играют роль мутагены, вызывающие
соматические мутации и клональный
неопластический процесс А-клеток.
Опухоль может быть проявлением
MEN-синдрома I типа (см. ниже гл. 13).
Она достигает диаметра от 4 до 20 см.
Вес — до 50-100 г. Располагается чаще в
хвосте поджелудочной железы, но могут
быть и эктопические локализации.
Нормальный уровень глюкагона в сыворотке
(50-200 нг/мл) при глюкагономе повы-
Патофизиология сахарного диабета 311
шается в сотни раз (до 96 нг/мл). При
семейной неопухолевой гиперглюкагонемии
он повышен умеренно. Глюкагон
стимулирует продукцию инсулина и гастри-
на, усиливает липолиз и гликогенолиз
(последний — только в печени), ускоряет
перистальтику. Возникает
гипергликемия на фоне гиперинсулинизма, без
тенденции к кетоацидозу как при ИЗСДI,
но и не с ожирением (как при ИНСД II
типа), а напротив — на фоне исхудания
и гипохолестеринемии. Типичное редкое
проявление глюкагономы - гипоальбу-
минемия и гипоаминацидемия в
результате усиления глюконеогенеза. Потеря
белка соматическим сектором ведет к
своеобразному глюкагонемическому
дерматиту - мигрирующей
неполитической эритеме. Она сопровождается
стоматитом. Бывают диарея, стеаторея
и железодефицитная анемия. Опухоль
бывает доброкачественной и нередко —
злокачественной. Детальнее см.
монографию «Эндокринология подростков»,
Ю.И.Строева и Л.П.Чурилова (2004).
>- При абсолютной и относительной ин-
сулиповой недостаточности
гипергликемия сопровождает явный СД любого
типа (что впервые установил в 1835 г.
Ф. Амброзини); она рассматривается
подробно в данном разделе.
Гипергликемия при повышении
содержания глюкозы выше 8 мМ/л, приводит
к глюкозурии. При этом теряются ценные
энергетические эквиваленты. Повышая
осмотическую активность плазмы и мочи,
гипергликемия ведет к полиурии, жажде и
полидипсии. Хроническая гипергликемия
стимулирует неэнзиматическое гликиро-
вание внеклеточных белков и усиливает
синтез полиоловых соединений в клетках.
Это вызывает тяжёлые осложнения,
рассмотренные ниже в разделе «Хронические
осложнения СД». Не любая гипергликемия,
а лишь очень высокое содержание глюкозы
в крови вызывает опасное острое нарушение
водно-солевого метаболизма и гиперглике-
мическую кому.
312 АЖ Зайчик, ЛЛ.Чурилов Патохимия
Гипергликемическая кома
Гипергликемическая (гиперосмолярная) кома
встречается реже, чем диабетическая (ке-
тоацидотическая). В большинстве случаев
она возникает у больных с ИНСД старше
пятидесяти лет и относительно редко
бывает в детском и юношеском возрасте. В 50 %
случаев она развивается у лиц с
нераспознанным до того или плохо леченным
диабетом, часто в связи со стрессом, травмой,
болезнью, резкой дегидратацией организма
(рвота, понос, ожоги, обморожения, крово-
потеря, обильное мочеотделение при
применении диуретиков).
Данное осложнение может быть
спровоцировано инфузией белковых и углеводных
растворов, лечением иммунодепрессантами
и дилантином (фенитоином), диализом.
Наиболее частая клиническая ситуация,
приводящая к гиперосмолярной коме, —
декомпенсированный ИНСД у пожилых
больных, которые не могут себя обслужить
и лишены надлежащего ухода.
В условиях кетоацидоза тошнота, рвота,
одышка и другие острые симптомы
приводят больного к врачу ранее, чем разовьется
крайняя степень гипергликемии. При
ИНСД кетоацидоз отсутствует, и
гипергликемия может нарастать до высоких уровней
без обращения к врачу. Пожилые одинокие
пациенты, страдающие ИНСД и
находящиеся дома, в случае инфекции, травмы
или иного состояния, обусловливающего
повышение потребности в инсулине, могут
оказываться в ситуации, когда
комбинируются крайне высокая гипергликемия (около
55ммоль/л) и относительная
беспомощность. В этой ситуации больные не могут
поддерживать осмотический гомеостаз
организма достаточно частым и обильным
питьём. Это и усугубляет водно-солевые
нарушения. В таких случаях развивается не-
кетогенная гиперосмолярная
гипергликемическая кома. Такая кома — не менее опасное
осложнение СД, чем кетоацидоз, дающее без
надлежащего неотложного лечения почти
50%-ный уровень смертности. Ключевое
звено ее патогенеза — гиперосмолярность.
Показатель осмоляльности внеклеточной
чдокринно-метаболические нарушения)
жидкости увеличивается до 500 мосмоль/кг.
Это результат глюкозурического
осмотического диуреза, без достаточной компенсации
потерь воды.
Патогенез гиперосмолярной комы,
отраженный на рис. 66, зависит от следующих
факторов:
1. Гипергликемия. К гиперосмолярной коме
приводит лишь быстро нарастающий и
достигающий очень высокого уровня
избыток глюкозы в крови: 30-200 мМ/л
(600-3600 мг/дл).
2. Гипернатриемия, гиперхлоремия.
Уменьшение выделения натрия и хлора с мочой
обусловлено повышением секреции
альдостерона (в ответ на дегидратацион-
ную гиповолемию), а также снижением
почечного кровотока.
3. ^Высокое содержание остаточного азота,
в том числе — мочевины (из-за
ограничения диуреза), повышение содержания
общего белка сыворотки тоже вносят
вклад в гиперосмолярность.
Гиперосмолярность приводит к резко
выраженной внутриклеточной дегидратации:
несмотря на то что глюкоза продолжает
входить в клетки мозга и её содержание в
них несколько повышается, концентрация
этого осмотически активного вещества в
межклеточной жидкости и плазме крови
остаётся намного большей, чем в интра-
целлюлярном отсеке. Нарушение водного
и электролитного равновесия в клетках
мозга влечет выраженную неврологическую
симптоматику и тканевую гипоксию ЦНС
с потерей сознания. Дегидратация ведет к
сгущению крови (отмечаются повышение
гематокрита, концентрации гемоглобина,
ложный лейкоцитоз, повышение
концентрации факторов свертывания). В связи с
этим для гиперосмолярной комы характерно
возникновение множественных тромбозов и
тромбоэмболия сосудов. У многих больных
формируется тромбо-геморрагический
синдром, следует закупорка почечных
клубочков и прогрессирует острая почечная
недостаточность. Развивается все более
выраженная олигурия и даже полная анурия.
Течение гиперосмолярной комы
характеризуется тем, что осложнение развивается
в течение нескольких дней, реже — в более
короткие сроки. Вначале отмечается поли-
урия, вслед за которой быстро возникают
другие симптомы:
> Поверхностное частое дыхание, типа
тахипноэ, без запаха ацетона.
> Двусторонний спонтанный нистагм и
мышечный гипертонус, снижение
сухожильных рефлексов.
> Затемнение сознания, вплоть до сопора
и комы.
Ключевую роль в лечении данного
осложнения играет инфузия гипотонических
растворов. Малые дозы инсулина
применяются для снижения гипергликемии. Это
дополняется антиацидотической терапией
и коррекцией калиевого баланса.
Гипогликемическая кома
и проблема гипогликемии
Гипогликемия — синдром, развивающийся
при понижении содержания глюкозы в
крови ниже 80 мг/дл (3,8 мМ/л). Причиной
данного синдрома могут быть понижение
поступления глюкозы в кровь из печени
и/или кишечника, повышение её
утилизации тканями и выведения из крови, а также
комбинация этих механизмов.
Различают следующие виды
гипогликемии:
> Инсулиновую — при передозировке
инсулина у диабетических больных и
при наличии инсуломы — чаще
доброкачественной инсулинпродуцирующей
опухоли панкреатической либо
эктопической локализации, которая производит
инсулин автономно от глюкозного
контролирующего сигнала.
Инсулома (инсулинома) - причина
органического гиперинсулинизма (болезнь
Харриса), встречается с частотой 1:900-
1000 аутопсий. Может быть компонентом
MEN-I синдрома (см. гл. 13). Как неопла-
зия, она вызывается соматическими
мутациями. Патоморфологически это узел
Патофизиология сахарного диабета 313
размером от 0,3 до 7 см, ее локализация
чаще в хвосте поджелудочной железы,
под капсулой, но может быть и
кишечной, печеночной, бронхолегочной и иной.
Инсулиномы обеспечивают в крови
избыток инсулина и его предшественников
(проинсулин, С-пептид) независимо от
уровня глюкозы. Болезнь течет с резким
снижением уровня глюкозы в крови и
недостаточным снабжением ею
головного мозга, что влечет ряд прекоматозных
расстройств ЦНС, возникающих в
определенной последовательности (кора
мозга — подкорковые структуры — средний
мозг — продолговатый мозг), вплоть до
классической гипогликемической комы.
В ответ на гипогликемию,
провоцируемую у таких пациентов физической
нагрузкой,, ночными голодными
паузами, предменструальными изменениями
уровня эстрогенов и другими факторами,
секретируются катехоламины, глюкагон
и кортизол, что проявляется тревогой,
внутренней дрожью, холодным потом,
тахикардией и слабостью, затем —
судорогами (см. ниже). Классическая
триада Уиппла при инсулиноме -
гипогликемия ниже 2,75 мМ/л, поведенческие
нарушения (утренняя оглушенность и
дезориентированность, потери сознания
натощак, сумеречные состояния
сознания, дурашливость, пританцовывания,
агрессивность и пр.), купирование этих
явлений сладостями или инфузией
глюкозы. Со временем прогрессируют
гипогликемические энцефалопатия и
полиневрит, а также вторичное поли-
фагическое ожирение, усугубляющее
гиперинсулинизм, ускоряется развитие
атеросклероза. Злокачественные
инсулиномы — редкость. Подробные
сведения об инсулиноме содержат новейшие
монографии Н.А. Майстренко (2004),
Ю.И.Строева и Л.П. Чурилова (2004).
>- От недостаточности контринсуляр-
ных гормонов. Ее наиболее типичное
проявление — тенденция к снижению
уровня глюкозы в промежутках между
приёмами пищи, обусловливающая
314 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
интолерантность к голоданию. Именно
так часто проявляется поначалу гипопи-
туитаризм. Ведь практически все гипо-
физарные гормоны и секреты зависящих
от них периферических желез
являются контринсулярными регуляторами.
Поведение человека, генерирующего
неконтролируемые вспышки
раздражения на голодный желудок, знакомо
каждому из нас. Такие индивиды
являются объектом добродушных насмешек
в юмористических рассказах о мужьях,
возвращающихся с работы в состоянии
дисфории. Однако, кроме
сакраментальной фразы о том, что «путь к сердцу
мужчины лежит через его желудок»,
появление подобных симптомов должно
подсказывать профессионалу медику,
что у пациента возможно латентное
понижение функциональных
возможностей гипоталамо-гипофизарно-над-
почечниковой системы. Не случайно к
подобному поведению склонны жертвы
хронического стресса. Гипогликемия
сопровождает гипокортицизм, гипоти-
роз, острую недостаточность функций
мозгового вещества надпочечников.
>• От недостаточного расщепления
гликогена — данная разновидность характерна
для ряда гликогенозов и для агликогено-
за (см. предыдущий раздел). К этой же
группе можно отнести гипогликемию
при печеночно-клеточной
недостаточности и лейцинозе (синдром Мак-Куори).
При алкогольном опьянении у лиц с
алкогольной гепатопатией могут
развиваться выраженные гипогликемии,
если прием алкоголя не сопровождается
адекватным приемом пищи
(«функциональный гиперинсулинизм»).
>• Алиментарная — сопровождает общее и
углеводное голодание. Сюда же можно
отнести и эндогенное углеводное
голодание — при кишечной и энзимопатической
мальабсорбции углеводов (см. выше).
Транзиторная гипогликемия
новорожденных представляет собой переходное
состояние смешанного генеза, связанное
как с алиментарным фактором, так и с
относительной незрелостью печеночных
механизмов мобилизации глюкозы.
>- Как следствие глюкозурии — возможна
при отравлении монойодацетатом и фло-
ридзином (см. выше). Справедливости
ради надо отметить, что эти яды
нарушают не только ренальную реабсорбцию, но
и кишечное всасывание глюкозы.
>* Аутоиммунные формы — связаны с инсу-
линомиметическим действием аутоанти-
тел к инсулиновым рецепторам (см. ниже).
В контексте данной главы наиболее
важным представляется синдром гипогликемии
при СД.
Гипогликемия может наступить у
больного ИЗСД при лечении инсулином.
Этому способствуют пропуск приема пищи,
передозировка инсулина или неожиданные
физические упражнения, провоцирующие
усиленное поглощение глюкозы мышцами.
Но весьма часто патогенез неожиданной
гипогликемии у прежде хорошо
скомпенсированного больного не столь прост.
>* При ИЗСД постепенно теряется
способность освобождать глюкагон и катехола-
мины в ответ на умеренное понижение
содержания глюкозы в крови. Из-за этого
компенсация гипогликемии запаздывает.
Причина такой особенности —
автономная нейропатия, нарушающая продукцию
контринсулярных гормонов. Вследствие
микроангиопатии сосудов портальной
системы гипофиза, с течением времени,
при ИЗСД формируется вторичный ги-
попитуитаризм, ослабляющий ответ на
гипогликемию. Так как ранние
симптомы гипогликемии (раздражительность,
дисфория, потливость) связаны с
гиперпродукцией адреналина, недостаточная
мощность контринсулярного ответа
может привести к отсутствию этих
предвестников и неожиданному наступлению
тяжелой гипогликемии при ИЗСД.
>* Другой механизм, способствующий ги-
погликемиям при ИЗСД, — это
функциональные последствия диабетического не-
фросклероза (феномен Зуброды-Дана).
Формирующаяся хроническая почечная
Патофизиология сахарного диабета 315
недостаточность удлиняет время
циркуляции инсулина и понижает почечный
порог для глюкозы, вызывая её потерю.
> Если ИЗСД сопровождается
аутоиммунной полиэндокринопатией, то
гипогликемии способствует аутоиммунная
недостаточность функций коры
надпочечников.
> Лишь недавно были оценены в полной
мере потенциальные возможности иди-
отип-антиидиотипических
взаимодействий при патокинезе диабета, леченного
инсулином. Инсулинотерапия вызывает
выработку антиинсулиновых антител,
что заставляет повышать дозу гормона.
Но антиинсулиновые антитела
провоцируют выработку аутоантиидиотипичес-
ких иммуноглобулинов против эпитопов
активного центра антител к инсулину.
Некоторые из этих антител второго
порядка воспроизводят взаимодействие
инсулина с его рецептором и служат как
бы «приблизительной
иммунологической копией» гормона. Активация
рецептора такими аутоантиидиотипическими
иммуноглобулинами может вызвать
эпизод гипогликемии у больного,
соблюдающего все рекомендации врача и
не нарушающего диету. На рис. 63 выше
представлена схема участия этих
механизмов в патогенезе так называемого
«лабильного диабета».
Гипогликемия, независимо от ее
причины, приводит к типовым последствиям.
Головной мозг не использует инсулинза-
висимые переносчики глюкозы и должен
получать из крови не менее 6 г глюкозы
в час.
Гипогликемия на уровне 50-70 мг/дл
(до 3 мМ/л) вызывает нервозность, тремор,
потливость, тревогу, чувство голода и
гиперсекрецию глюкагона и катехоламинов. Она
per se представляет классический стрессор.
Далее могут быть притупление
чувствительности и состояние, сопровождаемое
дезориентацией и галлюцинациями. Типичны
случаи ложной диагностики алкогольного
опьянения у таких больных. Если не
происходит нормализации уровня глюкозы,
то при содержании глюкозы 30-50 мг/дл
(3-2,5 мМ/л) ограничения в выработке
АТФ в нейронах ведут к снижению
активности калий-натриевого и
кальций-магниевого насосов, утрате ионных градиентов,
деполяризации клеток ЦНС, клоническим
судорогам (имеющим определённое
компенсаторное значение из-за гликогенолиза
в мышцах и усиления поставки лактата в
реакции глюконеогенеза). Наконец,
следует потеря сознания. При дальнейшем
прогрессировании энергодефицита мозга
развивается гипогликемическая кома.
Гипогликемию следует купировать глюкаго-
ном. Кроме того, пользуются и глюкозой (в
случае комы — вводимой внутривенно). По
мере углубления гипогликемии сознание
утрачивается не<;разу. Возможны ситуации,
когда гипогликемический больной
совершает сложные автоматические или связанные
с волевой установкой действия, не вполне
отдавая себе отчёт в содеянном. В практике
судебно-медицинской экспертизы
зафиксированы случаи, когда диабетические
больные в состоянии выраженной
гипогликемии совершали насильственные действия,
квалифицировавшиеся как хулиганство
или иное уголовное преступление, но
освобождались от уголовной ответственности,
как не полностью вменяемые в момент их
совершения.
Феномен Сомоджи (утренняя
гипергликемия после ночного эпизода гипогликемии)
чаще наблюдается у детей, чем у взрослых,
а объясняется выбросом контринсулярных
гормонов в ответ на гипогликемический
приступ.
Феномен Сомоджи дифференцируют с
так называемым «рассветным феноменом
или феноменом утренней зари» —
повышением уровня глюкозы и потребности в
инсулине в ранние утренние часы вследствие
ночного избытка соматотропина. В отличие
от феномена Сомоджи этому явлению не
предшествует ночная гипогликемия.
В силу общих звеньев патогенеза всех ком,
связанных с тканевой гипоксией нейронов,
316 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
их проявления во многом сходны. В данном
разделе приводим таблицу 15а,
характеризующую дифференциальные признаки
основных видов комы, возможных при СД.
Патогенез хронических
осложнении СД
Главные осложнения СД связаны с
диабетическими ангиопатиями. Считается, что
диабетические ангиопатии — основная
причина инвалидизации и смертности больных
сахарным диабетом.
Диабетические ангиопатии — понятие,
включающее:
>• микроангиопатии (поражение капилляров,
венул, артериол, прежде всего
затрагивающее их базальные мембраны) и
>- макроангиопатии (поражение крупных
артерий).
В течении сахарного диабета любого типа,
как правило, имеет место комбинированная
ангиопатия с преобладанием
микроангиопатии в молодом возрасте и макроангиопатии
после 30-40 лет. При этом макроангиопатия
проявляется в быстро прогрессирующем
атеросклерозе.
Тем не менее, это зависит не только от
возрастных особенностей реактивности.
Главным образом тип метаболических и
иммунологических нарушений влияет на
преобладание того или иного вида ангиопатии.
Для ИЗСД наиболее характерна микро-
ангиопатия. При ИНСД резко ускоряется
развитие атеросклероза крупных сосудов
(макроангиопатии) (рис. 70).
Вследствие осложнений диабета могут
маскироваться его основные проявления.
Развившийся на почве микроангиопатии
вторичный гипопитуитаризм уменьшает
потребность в инсулине, а в условиях
хронической почечной недостаточности (ХПН),
также вызванной сосудистыми
нарушениями, замедляется клиренс инсулина и может
повышаться порог глюкозурии. Как
результат, регистрируется «псевдоремиссия»
болезни (феномен Зуброды-Дана), так как
глюкоза исчезает из мочи без увеличения
дозы инсулина.
Микроангиопатические
осложнения
Главное осложнение ИЗСД — системная
микроангиопатия (МиАП). Основные
изменения происходят в базальной мембране
капилляров. В процесс вовлекаются все
капилляры, имеющие базальную мембрану
(универсальная капилляропатия).
Поражение сосудов микроциркуляторно-
го русла заключается в отложении белков
плазмы крови вдоль базальных мембран
микрососудов, утолщении и гиалинизации
базальных мембран с накоплением
материала, дающего положительную PAS
(ШИК)-реакцию (гистохимическую
реакцию с реактивом Шиффа для выявления
гликопротеидов и нейтральных гликозами-
ногликанов).
диабетическая
ангиопатия
микроангиопатия
макроангиопатия
нефропатия
ретинопатия
1 1
нейропатия
1 пангипо-
питуитаризм
ИБС
инсульт
1
периферический
артериосклероз и
атеросклероз
Рис. 70. Проявления ангиопатии при сахарном диабете. Обозначения: ИБС — ишемическая
болезнь сердца. Остальные — по тексту
Общим для микроангиопатий всех
локализаций являются:
> аневризматические изменения
капилляров;
> утолщение стенки артериол, капилляров
и венул за счет накопления в базальной
мембране сосудов PAS (ШИК)
положительных веществ. Отложения вдоль ба-
зальных мембран могут быть гомогенными
или слоистыми (стоит подчеркнуть, что,
несмотря на утолщение базальных
мембран, микрососуды у больных ИЗСД более
проницаемы для плазменных белков);
> пролиферация эндотелия и перицитов;
десквамация эндотелия в просвет
сосудов (пролиферация и десквамация
могут привести к облитерации просвета
сосудов);
> тучноклеточная реакция в периваскуляр-
ной ткани.
Капилляры и артериолы поражаются
чаще всего в особо предрасположенных
местах, к которым относятся:
> почечные клубочки;
> сетчатка глаза;
> дистальные отделы нижних
конечностей;
> кожа и скелетные мышцы.
Базальные мембраны могут поражаться и
в несосудистых структурах (капсула Боуме-
на-Шумлянского, канальцы почек,
периферические нервы, вследствие поражения vasa
nervorum, а также плацента).
Клинически микроангиопатия проявляет
себя через возникающие на ее почве нефро-
патию и ретинопатию, а также в какой-то
степени — и нейропатию. Интересно, что
минимальные изменения по типу
микроангиопатий обнаруживаются в микрососудах
у глубоких стариков, не имеющих СД.
Патогенетическая связь
микроангиопатий с сахарным диабетом убедительно
доказывается при пересадках почки: если
донорская почка с признаками
микроангиопатий от больного сахарным диабетом
пересажена здоровому реципиенту — МиАП
Патофизиология сахарного диабета 317
регрессирует, если здоровая почка
оказывается в организме реципиента с сахарным
диабетом — в ее сосудах развиваются
микроангиопатий.
«Диабетическая нефропатия» —
первично хронический процесс, собирательный
термин, включающий в себя, по сути,
диабетический гломерулосклероз и хронический
пиелонефрит (П. Киммельштиль, К. У ил-
сон, 1936).
Поначалу имеет место гипертрофия и
гиперфильтрация нефронов, затем —
микроальбуминурия при нормальной скорости
фильтрации и, наконец, прогрессирующий
гломерулосклероз с постепенным
развитием ХПН по гломерулярному типу (процесс
может сопровождаться нефротическим
синдромом или его элементами — макро-
протеинурическая стадия).
Сочетание СД, ретинопатии, протеину-
рии, артериальной гипертензии и
диабетического очагового гломерулосклероза
описано как синдром Киммельштиля-Уилсона.
При диабетическом поражении почек
обнаруживаются:
>- вовлечение клубочков (по типу
диффузного или, в случае синдрома
Киммельштиля-Уилсона, очагового
гломерулосклероза — с отложением компактных"
узлов PAS-положительного материала
по периферии клубочков, а также экссу-
дативными поражениями). Характерные
изменения почечного клубочка при
диабетической микроангиопатий
представлены на рис. 71;
>- гиалиновый артериолосклероз
(отложения гиалиновых масс в стенках почечных
артериол, сужающие их просвет и
обычно сопровождаемые гипертензией);
>* пиелонефрит и даже изредка
встречающийся некротический папиллит
(как результат восходящей инфекции
мочевыводящих путей, которая нередко
развивается при диабетическом
иммунодефиците).
Диабетическая ретинопатия —
поражение сосудов сетчатки с появлением на глаз-
318 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
-.. mjA
Рис. 71. Диабетическая микроангиопатия
почек. Накопление гиалиновых масс
(узелковая форма диабетического гломеруло-
склероза) в клубочке почки. Окр. Гематок-
син — эозин, об. х 120, препарат Ю.И. Строева
ном дне микроаневризм, а в дальнейшем —
при выделении ретиной ангиогенного
фактора — новообразованных неполноценных
сосудов, проникающих в стекловидное тело.
При развитии пролиферативного ретинита
типично появление кровоизлияний, что
может обусловить отслойку сетчатки и
слепоту, особенно если вовлекается желтое пятно.
ИЗСД — главная причина слепоты и одна
из ведущих системных причин ХПН (см.
«Практикум»).
Диабетическая нейропатия может
проявляться как мононейропатия с невритом
отдельных спинномозговых и черепномозго-
вых нервов, или автономная нейропатия — с
проявлениями нарушений нервной
регуляции вегетативных функций, в частности
тазовых органов и в особенности
желудочно-кишечного тракта.
Наиболее часто встречается
симметричная периферическая нейропатия с
поражением двигательной и чувствительной
иннервации нижних конечностей.
Поражается и ЦНС (спинной и головной мозг,
особенно — промежуточный).
Диабетическая нейропатия
характеризуется повреждением шванновских клеток,
дегенерацией миелина и аксонов. Возможные
механизмы нейропатии связаны с микро-
ангиопатией vasa nervorum и сосудов
портальной системы гипоталамуса-гипофиза.
Ишемический механизм неврита
представляется наиболее вероятным при мононей-
ропатиях.
Автономная нейропатия de facto
протекает на фоне диэнцефальных и
вегетативных нарушений, обусловленных ишемией
гипоталамо-гипофизарной области мозга.
Это провоцирует комплекс таких
симптомов, как импотенция, дисменорея, снижение
основного обмена, гипотензия, дисгидроз,
атония мочевого пузыря, расстройства
аппетита, терморегуляции, сна, перистальтики
ЖКТ.
Но микроангиопатические механизмы,
безусловно, не единственные факторы,
формирующие диабетические нейропатии.
Важную роль играет и их метаболический
механизм, связанный (в условиях хронической
диабетической гипергликемии) с усилением
сорбитологенеза. Многоатомный спирт сор-
битол обладает прямой токсичностью для
шванновских клеток, нарушая работу их
ионных насосов. Лекарства-ингибиторы
альдоредуктазы, снижая образование сорби-
тола и фруктозы, тормозят диабетическую
нейропатию и отчасти ретинопатию.
Определенное значение имеет нарушение
действия на нервную ткань инсулиноподобных
факторов роста, контролирующих миелини-
зацию. Нельзя исключить, что центральные
проявления нейропатии у больных ИЗСД
связаны с патогенностью гипертензии и
гипогликемии и последствиями
микроинсультов и кровоизлияний в мозг.
Автономная нейропатия с выраженными
гастроинтестинальными расстройствами
может быть связана с дефицитом вазо-
активного интестинального полинептида,
нарушения эрекции объясняют снижением
продукции окиси азота в микрососудах, а
также атеросклерозом артерий, приносящих
кровь к половым органам.
Микроангиопатии и периферическая
нейропатия, на фоне снижения регенерации и
иммунитета, в сочетании с ускоренным
атеросклерозом артерий нижних конечностей
(особенно у пожилых пациентов с ИНСД),
комбинируются в крайне неблагоприятную
«патогенную констелляцию», приводящую
к незаживлению и инфицированию
микроповреждений нижних конечностей. У
больных СД очень легко могут образовываться
персистирующие язвы нижних конечностей.
Одна из важных профилактических мер при
СД - избегать микротравм ног.
Микроангиопатии развиваются на
протяжении не менее 2 лет, но было показано,
что при достаточно чувствительных методах
диагностики начальные признаки
микроангиопатии можно обнаружить почти у 58 %
больных ИЗСД детей.
Доказано, что развитие микроангиопатии
напрямую зависит от длительности и степени
гипергликемии. Диабетический гломеруло-
склероз развивается у всех лиц со «стажем»
сахарного диабета более 25 лет (И. Белл,
1953). Интенсивная инсулинотерапия,
удерживая концентрацию глюкозы крови в
нормальных пределах, позволяет резко
затормозить развитие и/или прогрессирование
микрососудистых осложнений СД. Однако
патологические изменения продолжаются
некоторое время после нормализации уровня
глюкозы в крови (явление «гипергликеми-
ческой памяти»), что, возможно, связано
с медленным метаболизмом продуктов
неферментативного гликирования белков
или с иммунопатологическими процессами,
вовлекающими эти продукты.
Согласно иммунопатологической теории
патогенеза микроангиопатии, в их
патогенезе участвует иммунокомплексный
механизм. Иммунные комплексы, включающие
инсулин и другие белки, обнаружены в
стенках микрососудов. Установлена
положительная корреляция между уровнем
циркулирующих иммунных комплексов
и клиническими проявлениями
микроангиопатии. Ряд авторов нашли связь между
антиинсулиновыми аутоантителами и
диабетическим гломерулосклерозом.
Микроангиопатия возникает и у больных
ИНСД, и у больных ИЗСД, не лечившихся
инсулином.
Иммуногистопатологические
исследования показали, что в составе депозитов при
Патофизиология сахарного диабета 319
микроангиопатии имеются IgG, IgM,
компоненты системы комплемента, альбумин,
фибрин, фибронектин, коллаген, липопро-
теиды низкой плотности. В образовании
этих депозитов могут принимать участие
и характерные для многих пациентов с
ИЗСД аутоантитела к альбумину, а также
аутоантитела к иммуноглобулинам и
другим компонентам плазмы. Таким образом,
иммунокомплексный васкулит — основа
осложнений ИЗСД (И. Кохен и соавт.,
1987). Считается, что гликирование ряда
белков может менять их антигенные
свойства и, таким образом, некомпенсированная
гипергликемия-стимулирует
иммунокомплексный процесс. Установлено, что
при применении ксеногенных инсулинов
МиАП развивается быстрее, чем при
использовании генно-инженерного
человеческого.
В патогенезе микроангиопатии могут
принимать участие неиммунокомплексные
аутоиммунные механизмы, в частности ци-
тотоксические. Так, у большинства больных
ИЗСД выявляются гипокомплементемия и
аутоантитела к почечной ткани.
При диабетической микроангиопатии
закономерно обнаруживаются аутоантитела
к коллагену IV типа, входящему в состав
базальных мембран.
Некоторые факты свидетельствуют о
существенной роли тромбоцитов в
патогенезе микроангиопатии. Действие инсулина
на тромбоциты опосредуется недавно
открытым специфическим тромбоцитарным
инсулиновым рецептором.
Тромбоциты больных СД имеют
особенности:
>• повышена склонность к агрегации и
реакции освобождения;
>- повышена чувствительность к
агрегирующим веществам,
непосредственно снижаемая действием инсулина
(см.Ю.Строев, В. Файтельсон, 1976);
>• повышена продукция тромбоксанов и
простагландинов;
>• снижена выработка простациклина
эндотелием сосудов.
320 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-
Эти особенности связывают с
нарушениями обмена Са++ в тромбоцитах.
Тромбоциты при ИЗСД могут
фиксировать антиинсулиновые антитела и способны
активироваться иммунными комплексами.
Активация тромбоцитов приводит к
появлению в плазме и стенке сосудов медиаторов
пролиферации, усиливающих митогенез
гладкомышечных клеток и синтез ими
компонентов базальных мембран: коллагена,
гликозаминогликанов.
Уровень ростовых факторов в сыворотке
больных ИЗСД пропорционален степени
гипергликемии.
Наряду с иммунопатологической
многими аргументами подкрепляется и
метаболическая теория патогенеза микроанги-
опатий.
Метаболическая теория связывает
развитие поражения сосудов с нарушением
обмена гликозаминогликанов сосудистой стенки
и появлением в ней избытка продуктов
неподавляемого дефицитом инсулина пути
обмена глюкозы — сорбитолового (полиоло-
вого) цикла — соединений типа сорбитола.
Пусковым моментом патогенеза микро-
ангиопатий, по этой теории, являются
нарушения углеводного метаболизма. Реакции
цикла полиолов катализируют ферменты
альдоредуктаза и сорбитолдегидрогеназа.
В цикле полиолов происходит образование:
>- сорбита из глюкозы
(фермент-катализатор: альдоредуктаза))
>• фруктозы из сорбита
(фермент-катализатор: сорбитолдегидрогеназа).
Сорбитоловый путь резко активизируется
при гипергликемии в тех клетках и тканях,
которые способны поглощать глюкозу вне
зависимости от действия инсулина
(нервная ткань, ткани глаза, почки). Возникает
внутриклеточное накопление глюкозы и
сорбитола, а затем и фруктозы.
Накопление этих метаболитов повышает
в клетках и в таких структурах, как
хрусталик, осмотическое давление. Снижается
содержание миоинозитола, диацилглицерина,
скорость кругооборота инозитфосфатидов.
Из-за этого снижается активность
зависящей от них протеинкиназы С. Как следствие
уменьшается и активность калий-натриевой
АТФ-азы. Внутриклеточная
гипергидратация обусловливает гибель клеток ( именно
таков патогенез набухания и помутнения
хрусталика при диабетических катарактах).
Так как фосфатидилинозитоловый
посредник и протеинкиназа С участвуют в
передаче внутриклеточных сигналов от
инсулинового рецептора внутрь клеток,
предполагается, что нарушения обмена
полиолов при гипергликемии могут вторично
усиливать или вызывать пострецепторную
инсулинорезистентность. Этот
гипотетический механизм^ может играть некоторую
роль при формировании патогенетических
взаимосвязей между ИЗСД и ИНСД, через
персистирующую гипергликемию.
Миоинозитол и ингибиторы альдоре-
дуктазы замедляют развитие как нейро-
патии, так и катаракты при СД. Согласно
метаболической теории, полиоловые
соединения могут вызывать и ангиопатию.
Установлена митогенность сорбитола для
гладкомышечных клеток сосудов. Однако
нельзя забывать, что клетки сосудистой
стенки не относятся к тем элементам, в
которых преобладает неинсулинзависимый
транспорт глюкозы. Поэтому механическое
перенесение на сосуды патогенных
механизмов полиоловой нейропатии вряд ли
возможно.
Неферментативное гликозилирование
(по^ современной терминологии — гли-
кирование), как метаболическая основа
диабетической микроангиопатии, может
связать между собой основные положения
иммунопатологической и метаболической
теорий.
Ключевое значение имеет действие
гипергликемии на белки, с образованием
конечных продуктов необратимого гликирования.
Неферментативные реакции глюкозы с
белками известны, как реакции Мейяра, или
реакции побурения.
Цепочка этих реакций начинается со
взаимодействия альдегидной группы (СНО)
глюкозы и аминогруппы (NH2) белка, в ре-
Патофизиология сахарного диабета 321
зультате чего образуется основание Шиффа.
Оно нестабильно и перестраивается в более
стабильное (но еще способное к
обратимому превращению) соединение, называемое
«продукт Амадори».
Если в организме гликозилированный
белок (в первую очередь коллаген или иной
долгоживущий протеин сосудистой стенки)
не распадается в течение нескольких
месяцев или лет, некоторые из его продуктов
Амадори медленно дегидратируют и
перестраиваются с образованием производных
форм, содержащих глюкозу. Это — конечные
продукты необратимого глубокого
гликирования (рис. 72).
Конечные продукты необратимого
глубокого гликозилирования (КПНГГ)
оказывают разнообразные патогенные эффекты на
соединительную ткань и сосуды:
> вызывают сшивку между коллагеном
базальных мембран микрососудов и
белками плазмы крови, особенно в условиях
интенсивной фильтрации в гломерулах
почек. Именно такие изменения
типичны для микроангиопатий;
>* «пришивают» липопротеиды атеро-
генных классов к коллагену крупных
сосудов, препятствуя работе дренажных
механизмов и ускоряя атерогенез;
>• делают белки депозитов резистентными
к протеолизу;
>- препятствуют взаимодействию
коллагена базальных мембран с ламинином
и протеогликанами, сшивая молекулы
коллагена между собой. Базальные
мембраны при этом теряют протеогликаны и
повышают свою проницаемость;
>■ стимулируют выделение тканями эндоте-
лина-1, цидвдинов и других факторов,
усиливающих свертывание, тромбообразо-
вание и способствующих спазму сосудов;
>■ наконец, КПНГГ могут менять
антигенные свойства белков и провоцировать
иммунокомплексный процесс (см. ниже).
При ИЗСД обнаруживается уменьшение
соотношения сульфатированных гликозами-
ногликанов и гиалуроновой кислоты в
сосудистой стенке, снижение содержания проте-
огликанов (гепарансульфата) и увеличение
н-оо
(^o£-NH2 + Н-С-ОН
I
он-с-н
I
Н-С-ОН
I
Н-С-ОН
I
СН2ОН
D-глюкоза
Г
пирролы \
7 ?
H-C-N-^ело^)
н2о
I
-С
I
он-с
н-
он
-н
н
с-он
I
Н-С-ОН
I
СН2ОН
альдимин
(Шиффово основание)
N
*o"c4QC
н н
С-О
I
ОН-С-Н
I
Н-С-ОН
I
Н-С-ОН
I
СН2ОН
кетоамин
(продукт Амадори)
1
н-с=о
I
с=о
I
с-н2
Н-С-ОН
I
Н-С-ОН
I
СН2ОН
3-дезоксиглюказон
СН2ОН
I
Н-С-ОН
I
Н-С-ОН
+ он-с-н
I
с=о
H-C-N
I I
н н
1
Рис. 72. Формирование продуктов необратимого неэнзиматического гликирования при СД.
Пояснения в тексте
322 ALU. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
продукции коллагена IV типа и фибронек-
тина, в особенности в пораженных почках.
Последние изменения могут
воспроизводиться хронически высокими
концентрациями глюкозы в культуре эндотелиоцитов.
При ИЗСД отмечена активизация
образования гиалуроната из глюкозы, повышенная
активность N-ацетилглюкозаминидазы в
плазме и моче, связанная с ускоренным
развитием микроангиопатии. Считается,
что степень активации N-деацетилаз зависит
от генетически детерминированного набора
изозимов, а также уровня и длительности
гипергликемии. У носителей определенных
изозимов гипергликемия провоцирует
ускоренный распад сульфатированных гликоза-
миногликанов, что способствует развитию
микроангиопатии.
Структурную и функциональную
целостность базальной мембраны сосудов в
известной мере определяет обмен амино-
сахаров.
В ходе генетически детерминированного
синтеза компонентов базальной мембраны
сосудов на рибосомах эндотелиоцитов и
перицитов синтезируются
полипептидные цепи лизина и пролина, которые под
влиянием гидроксилазы превращаются в
гидроксилизин и гидроксипролин.
Молекула галактозы при участии галактозил-
трансферазы присоединяется к остатку
гидроксилизина.
Далее, при участии гликозилтрансферазы,
глюкоза переходит в состав гликопротеина
базальной мембраны, имеющего высокое
число дисахаридных частиц, сцепленных
с гидроксилизиновыми остатками. Эта
реакция заканчивается образованием
растворимых частиц, которые, связываясь между
собой, образуют саму структуру базальной
мембраны. Большую организующую роль
при этом отводят фибронектину (см. т. I,
с. 343-344).
При гипергликемии эти сложные
процессы нарушаются:
>* возрастает активность
гликозилтрансферазы и усиливается синтез гликопро-
теидов.
>■ повышается количество
гидроксилизина и гликопротеидов в базальных
мембранах, что вызывает последующее
утолщение базальных мембран;
>- возрастает синтез нейтральных ГАГ, а
значит, увеличивается и их содержание
в крови, и их отложение в сосудах;
>• происходит гликозилирование
сывороточных белков. Гликозилированный
альбумин угнетает пролиферацию
эндотелиоцитов и продукцию ими
коллагена;
>- могут гликозилироваться и клеточные
белки, в первую очередь рецепторы,
находящиеся на поверхности клеток, а это
вызывает нарушение регуляции
клеточных функций, угнетение пролиферации
и секреторной способности клеток. В
частности, нарушается способность
эндотелиоцитов продуцировать окись
азота, что может иметь значение в
патогенезе макроангиопатических
осложнений. Показано замедление перехода
проренина в ренин, из-за гликирования
проренина. Это может вносить вклад в
нарушение почечного микрокровотока
при СД.
Важность роли неферментативного гли-
козилирования белков в патогенезе
микроангиопатии подчеркивает хороший эффект
применения ингибиторов необратимого
гликозилирования (например, аминогуа-
нидина) у больных с осложнениями СД и
на животных — моделях ИЗСД.
Наилучшим интегральным показателем
кратковременного баланса гликемии служит
определение уровня фруктозамина плазмы
(по К. Шатонеф-Ж.-П. Тоберу, 1987). Чем
больше фруктозамина — тем хуже уровень
компенсации СД за последнюю неделю.
Аналогичный тест, дающий представление
о долговременной метаболической
компенсации — за период порядка 3 месяцев, —
уровень гликозилированного гемоглобина. Оба
показателя зависят от скорости прямого
гликирования белков и коррелируют с
темпами развития ретино- и нефропатии.
Гликозилированный НЬ обладает
повышенным сродством к кислороду и имеет
остатки глюкозы, соединенные с N-концевым
валином р-цепи гемоглобина А. Он условно
обозначается Hb Ale.
У здоровых его содержание составляет
4-6%, у больных СД возрастает в 2-3 раза.
Из-за этого в капиллярах затрудняется
отщепление кислорода от НЬ и переход
кислорода в ткани и развивается гипоксия.
Гипоксия сопровождается расширением
венозного отдела капилляров,
возрастанием их проницаемости. Изменяется тонус
сосудов, возникают мешотчатые
расширения стенок капилляров и микроаневризмы,
составляющие элементы морфологической
картины микроангиопатии.
Эндокринная теория развития МиАП
постулирует, что существенную роль в
развитии микроангиопатии играют вторичные
гормональные нарушения. В связи с этим
определение содержания ряда гормонов у
больных ИЗСД полезно в прогностическом
отношении для предсказания динамики
развития микроангиопатии. Еще в 60-е
годы XX века было отмечено, что
диабетическая микроангиопатия не поражает
гипофизарных карликов, страдающих
ИЗСД, и существенное значение в ее
развитии стали придавать гиперпродукции
СТГ. Гормон роста служит митогеном для
клеток сосудистой стенки. В эксперименте
на крысах показано, что СТГ отягощает
течение сахарного диабета и
микроангиопатии. Передозировка СТГ per se вызывает
васкулярные поражения, напоминающие
микроангиопатии. Выраженная гиперсома-
тотропинемия обнаруживается у больных
ИЗСД, но сам уровень СТГ не
коррелирует со степенью микроангиопатии. Зато
отчетливая корреляция имеется между
активностью соматомедина С плазмы и
диабетическими проявлениями
микроангиопатии, особенно ретинопатией.
Соматомедины, особенно ИФР-1, могут
вызывать гиперфильтрацию и гиперплаз-
моток в нефронах, свойственные начальной
стадии диабетической нефропатии.
Патофизиология сахарного диабета 323
При диабетической ретинопатии
концентрации соматомедина А в плазме
периферической крови снижены. Гипогликеми-
ческие средства, усиливающие выработку
соматостатина, активизируют и течение
микроангиопатии.
Таким образом, нарушения в системе
гормон роста-соматомедины определенно,
имеют патогенетическое значение при МиАП.
Обнаружено глюкозазависимое
повышение содержания предсердного натрий-
уретического "фактора (ПНФ) у больных
в начальной стадии диабетического гломе-
рулосклероза. Содержание ПНФ
положительно коррелирует с увеличением скорости
клубочковой фильтрации, клиренсом
альбумина, суточной экскрецией альбумина с
мочой — то есть показателями, повышение
которых характерно для ранней стадии
диабетического гломерулосклероза.
Многими авторами подтверждено, что
микроангиопатии при ИЗСД связаны с
характерной дисфункцией ренин-ангиотен-
зин-альдостероновой системы: нарушением
превращения проренина в ренин,
накоплением проренина (см. ниже), дефектом
действия ренина на мишени.
Описанные иммунопатологическая,
метаболическая и эндокринная теории
патогенеза микроангиопатии проявляют тенденцию
к слиянию (рис. 73). Весьма вероятно, что
гликозилирование способно изменить
антигенные свойства белков и полипептидов и
спровоцировать аутоимунный процесс.
Гликозилирование Fc-рецептора макрофагов
и Fc-фрагмента иммуноглобулинов может
замедлить клиренс иммунных комплексов
и вызвать их накопление.
Гликозилирование гормонов и их рецепторов становится
причиной гормональных нарушений. Не
исключено, что дефицит действия
соответствующих полипептидных регуляторов и
приводит к нарушениям метаболизма и
пролиферации в сосудах. Гликозилированные
белки подвергаются усиленному
фагоцитозу, в связи с чем, вероятно, в качестве
раннего признака микроангиопатии фигурируют
накопления PAS (ШИК)-положительных
324 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
аутоиммунный процесс
гипергликемия
инсулинотерапия
т
иммунные
ккомплексы
гликирование
белков плазмы и
сосудистой стенки
нарушение
активации и рецепции
пептидных гормонов
т
| пролиферативный васкулит |
Рис 73. Патогенез микроангиопатии при сахарном диабете,
Пояснения в тексте
гранул в мононуклеарах крови. Таким
образом, соответствующие иммунодиагнос-
тические тесты в сочетании с определением
уровня гликозилированного гемоглобина,
фруктозамина и циркулирующих иммунных
комплексов могут быть полезны в ранней
диагностике микроангиопатии при ИЗСД.
Среди других представлений о патогенезе
диабетического гломерулосклероза
интересна особая точка зрения Дж. Фостера
(1994) — сторонника теории гиперперфузии
и гиперфильтрации. Согласно этой теории,
осмотический диуретический эффект
глюкозы обусловливает гиперфильтрацию и
гиперфункцию нефронов, что в конечном
счете ведет к их износу и поражению.
По мере развития микроангиопатии
диагностическое значение приобретают
маркеры, связанные с активацией
биосинтеза компонентов межклеточного вещества
в стимулированных клетках сосудистой
стенки и процессом дезорганизации
соединительной ткани, в особенности — базаль-
ных мембран.
Так, по ходу развития микроангиопатии,
ее почечных и офтальмологических
осложнений увеличивается содержание в плазме
фибронектина, фибриногена, ламинина и
проколлагена III. Сходно изменяется
содержание фибронектина в моче, что позволяет
считать этот показатель
адекватным отражением степени
диабетического гломерулосклероза.
В ряде случаев имеется
возможность скринингового
радиоиммунологического и иммуно-
ферментого тестирования этих
показателей.
Традиционный тест ранней
диагностики диабетической
микроангиопатии — выявление
микроальбуминурии. Степень
этого нарушения коррелирует с
гипертензией, содержанием
гликозилированного гемоглобина
и антиинсулиновых аутоанти-
тел, диабетической ретино- и
нефропатией, длительностью
ИЗСД. Диагностически значимым
является повышение экскреции альбумина выше
30 мкг/мин. Альбумин может определяться
в моче радиоиммунологически.
Традиционный тест на выявление
диабетической ретинопатии — исследование
глазного дна, применяется также
флюоресцентная ангиография.
Нарушение функций периферических
нервов у больных ИЗСД может
оцениваться по порогу восприятия вибрации.
Радиоиммунологическая и иммунофер-
ментная оценка маркеров и ранних
проявлений осложнений ИЗСД может
содействовать раннему началу
профилактической терапии и по своей прогностической
значимости превосходит традиционные
методы. Показана высокая
чувствительность тепловидения в качестве метода
раннего выявления и контроля степени
развития микро- и макроангиопатии
сосудов конечностей при СД (Ю.И. Строев,
1980-1982).
Вторичные изменения
нейроэндокринной системы
при ИЗСД
Аномалии в концентрациях и
эффективности гормонов и нейромедиаторов при ИЗСД
могут быть результатом вторичных
нарушений в нейроэндокринной системе. Поэтому
при оценке результатов определения
гормонов у больных ИЗСД следует учитывать
некоторые типовые ситуации.
> Отклонения в концентрации гормонов
могут быть результатом системного
аутоиммунного процесса, охватывающего
при ИЗСД lb ряд органов и тканей. Это
относится к гормонам надпочечников,
гипофиза, щитовидной и паращитовид-
ных желез. У больного в этом случае
должны быть клинические проявления
аутоаллергии.
> Отклонения в концентрации гормонов
могут отражать поражение нейроэндок-
ринного аппарата вследствие развития
типовых осложнений ИЗСД. По мере
развития микроангиопатии
прогрессирующее нарушение функций почек и
гипоталамо-гипофизарной области мозга
ведет к закономерно формирующемуся
«медленному» пангипопитуитаризму и
изменению состояния системы ренин-
ангиотензин — альдостерон.
> При поздно выявленном, недостаточно
компенсированном или лабильном
диабете из-за длительной гипергликемии
происходит гликирование ряда белков,
включая гормоны, прогормоны и их
рецепторы. При этом обычно
имеются высокие интегральные показатели
гипергликемии: гликозилированный
гемоглобин выше 9,0 %, гиперфруктоза-
минемия. Данное явление может
сопровождаться неэффективностью действия
гормонов на мишени, задержкой проте-
олиза прогормонов или аутоиммунной
инактивацией гликозилированных
белков. Типичными примерами служат
характерные для ИЗСД замедление
перехода проренина в ренин и пониженная
активность соматомединов при
нормальном уровне СТГ.
> Отдельные особенности метаболического
статуса больных ИЗСД могут вызывать
соответствующую реакцию
определённых гормонов, например, гиперосмия
вызывает накопление АДГ, а кетоацидоз
тормозит выработку тестостерона.
Патофизиология сахарного диабета 325
ИНСУЛИННЕЗАВИСИМЫЙ
САХАРНЫЙ ДИАБЕТ (ИНСД)
Определение
ИНСД — гетерогенная по этиологии и
патогенезу группа заболеваний,
характеризующихся мультифакториальной
наследственной предрасположенностью,
относительной инсулиновой недостаточностью и
инсулинорезистентностью.
Дебатируется вопрос о том, какая из
двух выделенных патогенетических основ
ИНСД первична. У большей части больных
ИНСД сочетается с ожирением и
коррелирует с пожилым возрастом, что заставляет
предположить особую роль длительного
действия выявляющих его факторов или
существенное значение возрастной экспрессии
предрасполагающих генов.
Значительное количество больных ИНСД
кроме резистентности к инсулину и
ожирения имеет еще и характерную комбинацию
расстройств (рис. 35, выше):
>- гиперлипопротеидемию IV или V
типов;
>■ артериальную гипертензию;
>* ускоренный атеросклероз;
>• гиперурикемию;
>• микроальбуминурию;
>- ожирение андроидного типа;
>• стеатоз печени;
>- гиперкоагуляцию и недостаточность
фибринолиза;
>• избыток производства адипоцитами
некоторых провоспалительных контрин-
сулярных цитокинов (кахексии, ИЛ-6,
резистин) и дефицит производства ими
противовоспалительного инсулин-сен-
сибилизирующего цитокина (адипонек-
тина).
Частое сочетание этих расстройств
позволило некоторым авторам объединить
их в единый синдром — метаболический
Х-синдром (см. выше в разделах
«Нарушения липидного обмена» и «Патофизиология
326 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
нуклеинового обмена»). В настоящее время
достоверно неизвестно, вызваны ли все эти
нарушения одними и теми же механизмами.
Однако имеются косвенные данные, что при
всех перечисленных расстройствах
системно нарушена работа натрий-водородного
обменного механизма клеточных
мембран, известного в литературе как «Na+/
H+-antiport» (см. ниже «Патофизиология
водно-электролитного обмена» и гл. 14).
Данная точка зрения включает, по крайней
мере, часть случаев ИНСД в состав более
широкого системного метаболического дефекта.
По-видимому, при Х-синдроме большую
роль играет аддитивно-полигенный
характер наследования перечисленных аномалий,
с пороговым эффектом по таким
провоцирующим факторам внешней среды, как
переедание. Не исключено, что
предрасполагающие гены для данных расстройств частично
являются общими. Так как конкордантность
однояйцевых близнецов для ИНСД намного
выше, чем для ИЗСД, то уместно
предположить, что число генов, участвующих в
мультифакториальном эффекте, меньше.
Однако роль провоцирующих факторов
внешней среды в случае ИНСД может
оказаться гораздо более сложной.
В связи с этим для ИНСД гораздо труднее
решить вопрос о первичности или вторич-
ности нарушений, входящих в Х-синдром.
Возможно, что, с точки зрения кондици-
ональной логики, такой вопрос в случае
ИНСД вообще неправомерен.
Дело в том, что помимо Х-синдрома
известны и другие четко установленные или
предполагаемые причины инсулинорезис-
тентности и относительной инсулиновой
недостаточности:
>* недостаточный ответ В-клеток на глю-
козный сигнал без их деструкции;
>• неправильный синтез проинсулина, либо
нарушение его протеолиза;
>• избыток циркулирующих антагонистов
инсулина;
>• аномалии инсулиновых рецепторов;
>- блокада инсулиновых рецепторов;
>• пострецепторные блоки разного уровня
в клетках-мишенях.
Все эти дефекты, вероятно, генетически
детерминированы и по крайней мере
большинство из них — моногенно. Хотя каждый
из подобных механизмов встречается редко,
в сумме эта гетерогенная группа составляет
существенную часть больных с диагнозом
«ИНСД».
Генетика ИНСД
Семейный риск при ИНСД очень
значителен: до 40% сибсов и 1/3 потомства у
больных имеет интолерантность к глюкозе
(Дж.Фостер, 1994).
В отличие от ИЗСД для ИНСД выявлены
регионы, где проявляется эффект
родоначальника и последствия инбридинга
(остров Науру, где СД больны 83 % коренных
жителей и район расселения индейцев пима,
в Северной Америке, среди которых до 86 %
больны СД).
Несмотря на это, лишь для отдельных
моногенных менделирующих
разновидностей ИНСД установлен точный тип
наследования.
Так, при диабете взрослых в юности
(MODY подтип 2) имеется уже
упоминавшееся в разделе «Патофизиология
углеводного обмена» высокопенетрантное
аутосомно-доминантное наследование
дефекта глюкокиназы, ген которой находится
в коротком плече 7-й хромосомы.
Глюкокиназа — фермент,
обусловливающий чувствительность островковых
В-клеток и гепатоцитов к уровню глюкозы
крови. Имеются веские основания полагать,
что именно она представляет собой «глю-
козный сенсор», определяющий
чувствительность этих клеток к колебаниям уровня
глюкозы в крови. Как следствие данный
дефект вызывает относительную неотве-
чаемость В-клеток на глюкозный сигнал. У
носителей дефекта развивается умеренно
тяжёлый ранний инсулиннезависимый
диабет, однако данная аномалия у европеоидов
не наблюдается при классическом позднем
ИНСД, хотя такая корреляция отмечена у
негров США, японцев и креолов острова
Маврикий. Описано еще 5 подтипов MODY
с дефектами разных транскрипционных
факторов, делающими В-клетки
«ленивыми» (MODY 1,3-6).
Аномалии самой молекулы инсулина
могут возникать в результате мутации
его структурного гена, что обусловливает
возникновение биологически дефектных
молекул инсулина.
Так, описан случай, когда больной имел на-
тощаковую гипергликемию (> 11 ммоль/л)
и натощаковую гиперинсулинемию (около
100 мкЕД/мл), но проявлял нормальную
чувствительность к экзогенно вводимому
инсулину. Оказалось, что инсулин больного
имел замещение лейцина на фенилаланин в
биологически активном участке молекулы
инсулина: в позиции 24 р-цепи. Этот мутант-
ный инсулин имел низкую биологическую
активность и пониженную способность
связывания с клетками-мишенями.
Описаны единичные случаи ИНСД при
генетически обусловленном неполном
превращении проинсулина в инсулин.
Обычно это превращение происходит
путем протеолиза, в В-клеточной грануле.
В норме оно осуществляется почти
полностью: только около 5 % секреторного
продукта В-клеток представлено проинсулином.
В тех случаях, когда превращение
проинсулина в инсулин происходит не полностью,
избыток проинсулина секретируется в
кровоток. Естественно, что в этих условиях
для поддержания гомеостаза глюкозы
секреция В-клеток должна заметно возрасти,
поскольку биологическая активность
проинсулина намного меньше, чем инсулина.
Так как проинсулин в ходе иммуноконку-
рентного анализа дает перекрестную
реакцию с инсулином, радиоиммунологически
и иммуноферментно при этом выявляется
гиперинсулинемическое состояние.
Больные такой формой ИНСД имеют
дефект структурного гена проинсулина,
что обусловливает аминокислотную за-
Патофизиология сахарного диабета 327
мену в точке отщепления терминального
С-пептида, а это в свою очередь
препятствует процессу нормального превращения
проинсулина в инсулин и терминальный
С-пептид.
При ИНСД в наибольшей степени
нарушен неокислительный метаболизм глюкозы,
особеннр — синтез гликогена, подавленный
гораздо сильнее, чем захват глюкозы
клетками и ее окисление до С02 и воды. Этот
дефект наблюдается даже у нетучных нор-
могликемических родственников многих
больных ИНСД.
Здесь следует указать на возможную
взаимосвязь между маркерным для данного
синдрома аллелем гликогенсинтетазы А2 (30 %
частоты против 8 % в контрольной группе) и
высокой вероятностью развития ИНСД.
Различные авторы указывают на
возможное участие в патогенезе ИНСД генов
транспортеров глюкозы, в частности GluT2,
контролирующего проникновение глюкозы
в гепатоциты и панкреациты, или GluT4,
функционирующего в норме в липо- и ми-
оцитах.
Не исключена решающая роль
генетических аномалий возможного контринсулярно-
го фактора В-клеток человека — амшина,
известного также под названием амилоидо-
генного пептида. В норме амилин — пептид,
содержащий 37 аминокислот,
продуцируется, пакуется и секретируется в небольших
количествах, вместе с инсулином. У многих
пациентов с ИНСД избыток амилина
откладывается в синусоидных пространствах
вокруг В-клеток. При этом из него формируется
амилоид (см. выше «Диспротеинозы»).
Ведутся дискуссии о первичности отложений
амилина или деструкции В-клеток.
Показано, что амилин снижает ответ В-клеток на
глюкозу, в частности у свиней. Амилоидные
отложения в островках больных ИНСД
практически всегда содержат амилин. При
ИНСД его секреция нарастает параллельно
секреции инсулина. У больных инсулинне-
зависимым диабетом амилин находится в
избытке в плазме. В самом островке накоп-
328 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ление амилина может вызвать снижение
секреторных потенций В-клеток. Амилин
известен как «диабет-ассоциированный
пептид» или гомолог кокальцигенина, он
способствует гликогенолизу в мышцах.
У некоторых больных ИНСД
обнаружены особенности гена, контролирующего
процессинг рецептора инсулина
(хромосома 19) и связанные с этим аномалии
рецептора инсулина в его тирозинкиназной
части. В отличие от тучных недиабетических
пациентов тучные больные СД имеют
пониженную тирозинкиназную активность в
клетках-мишенях инсулина. Аномальный
пептид инсулинового рецептора может
служить маркером ИНСД в значительной (до
1/4) части случаев (B.C. Генес, С.Г. Генес,
1993).
СР. Кан (1995) придает наибольшее
значение при ИНСД генетике пострецеп-
торных блокирующих факторов, указывая,
что более, чем у 15% больных ИНСД (как,
впрочем, и у 5% здоровых испытуемых)
имеется генетическая вариация в структуре
вышеописанного внутриклеточного
инсулинового посредника IRS-1, дефекты которого
вызывают снижение толерантности к
глюкозе у трансгенных мышей.
Выделен и секвенирован мембранный
гликопротеин РС-1, ингибирующий
тирозинкиназную активность рецепторов
инсулина и в избытке экспрессированный
в фибробластах и скелетных мышцах
некоторых больных ИНСД. РС-1 способен
снижать инсулинзависимую пролиферацию
в культуре клеток карциномы молочной
железы при его трансфекции.
Некоторые пациенты с ИНСД имеют
избыточную экспрессию гена Rad,
относящегося к Ras/ГТФ-азному суперсемейству
и участвующему в работе системы
внутриклеточного опосредования гормональных
сигналов. Его продукт ингибирует
инсулинзависимую активацию транспортера
глюкозы GluT4.
У некоторых больных ИНСД описан
избыточный синтез а2-8Н-гликопротеина,
который клонирован, очищен и оказался
блокатором инсулинорецепторной
тирозинкиназной, но не тирозинаминотрансфераз-
ной активности.
Таким образом, множественные, чаще
всего пострецепторные дефекты, при
ИНСД могут вызывать первичную
резистентность В-клеток островков к глюкозе,
а клеток-мишеней инсулина — к этому
гормону.
Выявляющим генетические аномалии
экзогенным фактором служит при ИНСД
переедание, повышающее нагрузку на
систему инсулиновой регуляции метаболизма и
формирующее ожирение и дополнительную
инсулинорезистентность.
Морфология
и гормонообразующая функция
островков при ИНСД
Для ИНСД характерны определенные
особенности морфологии и функции островко-
вых клеток ( по Р. Унгару, Л. Орчи, 1978;
Ф.Фелигу, 1985):
>- относительная гиперглюкагонемия,
несмотря на натощаковую
гипергликемию;
>- возрастание уровней глюкозы
плазмы после введения глюкозы per os не
уменьшает уровней иммунореактивного
глюкагона плазмы;
>- избыточен ответ А-клеток на
аминокислоты (аргинин, аланин);
>- инсулин, даже в фармакологических
дозах, не изменяет аномального ответа
А-клеток на аминокислоты;
>• общий объем островковой ткани в
поджелудочной железе заметно увеличен;
>■ увеличение общего объема островковой
ткани происходит за счет возрастания в
островках числа инсулинпродуцирую-
щих В-клеток;
>• базальная секреция инсулина при ИНСД,
как минимум нормальна (У. У орд и со-
Патофизиология сахарного диабета 329
авт., 1985), а по некоторым данным —
увеличена;
> в ответ на глюкозную нагрузку ранняя
фаза стимулированной инсулинопродук-
ции повышена, поздняя в абсолютном
выражении характеризуется таким же,
как в норме или даже абсолютно
большим, приростом концентрации инсулина
в крови (рис. 61, с. 266). Сохранена и ин-
сулиновая реакция на аргинин. Однако
у подавляющего большинства больных
ИНСД секреция инсулина в позднюю
фазу ответа на глюкозу и аминокислоты
относительно недостаточна в расчете на
единицу прироста концентрации инсу-
линогенных субстратов. Это позволило
У. Уорду и соавторам, обнаружившим
данную закономерность, заключить о
наличии функционального дефекта инсу-
линового ответа В-клеток при ИНСД;
> соматостатин-продуцирующие D-клетки
резко уменьшены в количестве
(относительно выраженной гиперплазии и
гипертрофии В-клеток);
> гетероклеточные зоны панкреатических
островков нарушаются вследствие резко
возросшего представительства В-клеток
в островке;
> относительное количество D-клеток
оказывается настолько несущественным,
что не может оказывать адекватного
тормозного паракринного влияния на
глюкагон-продуцирующие А-клетки;
> имеет место снижение продукции
соматостатина и клинически
эффективны некоторые стимуляторы его
выработки;
> нередко отмечается амилиновый амило-
идоз и фиброз островков.
Патогенез ИНСД
и его фазы
Явление инсулинорезистентности или
сниженной эффективности инсулина при
СД открыл В. Фальта (1924) всего через 2
года после первого применения гормона в
клинике (К. Штольте, 1922). В настоящее
время представление о первичности
инсулинорезистентности в патогенезе ИНСД
поддерживается большинством специалистов.
Переедание, а возможно и другие
экзогенные факторы провоцируют при ИНСД
генетически детерминированную инсули-
норезистентность. Низкая эффективность
действия инсулина теоретически могла
бы быть скомпенсирована высокой
чувствительностью и эффективностью работы
В-клеток и гиперпродукцией гормона. Но
при ИНСД, как правило, имеется их
десенсибилизация к глюкозе, в основе которой
могут лежать разнообразные и иногда
независимые от инсулинорезистентности
причины.
Это ведет к характерной трехфазной
картине течения ИНСД (Дж. Фостер, 1994),
включающей следующие периоды:
>• стадия начальной
инсулинорезистентности и компенсации;
>• стадия выраженной
инсулинорезистентности и относительной инсулиповой
недостаточности; интолерантность к глюкозе;
>* стадия снижения инсулиновой секреции
и явного диабета.
В первой стадии имеется инсулиноре-
зистентность, вызывающая гиперфункцию
В-клеток и их гиперплазию. Это повышает
выработку инсулина и позволяет больному
иметь нормогликемию, т. е. в этой стадии
болезнь компенсирована.
Во вторую стадию инсулинорезистент-
ность усиливается. Важным
патогенетическим механизмом усиления
инсулинорезистентности, по мере развития
компенсированного ИНСД, служит формирующееся у
больных ожирение.
Известно, что тучность, особенно
гипертрофический и андроидный тип ожирения,
ведет к развитию инсулинорезистентности.
Менее значителен эффект
гиперпластического и бедренно-гиноидного типа тучности.
Поскольку подавляющее большинство
взрослых больных ИНСД приобретают
или изначально имеют избыточный вес,
инсулинорезистентность, обусловленная
330 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
тучностью, существенно влияет на
гипергликемию у этих больных.
Пионер инсулинотерапии Э.П. Джослин
(1928) писал: «Можно думать, что
диабет— наказание за чрезмерную полноту, и
чем больше ожирение, тем сильнее карает
природа».
При ИНСД кетоацидоз нехарактерен
именно по тем же причинам, по которым
имеется ожирение, — вследствие наличия
инсулина, который сдерживает липолиз и
создает условия для утилизации ацетил-КоА
на пути стероидогенеза и липогенеза.
В условиях стресса, при наличии даже
минимума инсулина, печень резистентна к
глюкагону, поэтому уровни малонил-КоА
остаются высокими, угнетая окисление
жирных кислот и перекрывая путь кетогенеза.
Метаболической платой за это служит
тенденция к ожирению и крайне высокой
гиперхолестеринемии. При ИЗСД гипер-
холестеринемия более умеренная, а при
ИНСД она очень выраженная.
По мере увеличения объема адипоцитов
плотность рецепторов инсулина на
поверхности жировых клеток уменьшается,
что может затруднять их кластеризацию и
сшивку, связанную с гормон-рецепторным
взаимодействием.
Обнаружен цитокин, выделяемый адипо-
цитами и обладающий способностью резко
снижать киназную активность рецептора
инсулина в мио- и липоцитах, — ФНОа
(фактор некроза опухолей- альфа, кахексии). По
мере нарастания ожирения продукция этого
цитокина значительно растет и становится
избыточной. Вероятный биологический
смысл этого — ограничить дальнейший
приток в адипоцит трансформируемого в
жиры материала, тем более что названный
цитокин является аппетит-подавляющим
регулятором, откуда его второе название —
кахексии. Но у лиц, склонных к ожирению,
кахексиновый механизм подавления
неэффективен. В то же время, ФНОа вызывает
дополнительную инсулинорезистентность,
а также цитотоксичен для В-клеток, что
прокладывает патогенетические пути для
перехода ИНСД во 2-ю и 3-ю фазы развития
(С.Р.Кан;1995).
Однако ожирение у больных ИНСД «не
отвечает» за всю инсулинорезистентность,
т.к. инсулинорезистентность у них
сильнее, чем могло бы быть обусловлено только
тучностью. Об этом ясно свидетельствует
сравнение эффективности инсулина при
действии на клетки-мишени тучных
испытуемых, страдающих СД и не имеющих
этого заболевания. При равном относительном
избытке массы тела в первом случае эффект
инсулина меньше, чем во втором.
Более того, многие нетучные больные с
ИНСД тоже инсулинорезистентны и имеют
пониженную киназную активность инсули-
новых рецепторов. Справедливо и обратное:
многие случаи инсулинорезистентности не
сопровождаются тучностью и СД.
Возможно, усиление инсулинорезистентности по
ходу развития ИНСД наступает из-за
адаптивного снижения экспрессии инсулиновых
рецепторов при «сытом» состоянии клеток
и гиперинсулинемии.
По мере развития ИНСД возрастает
уровень резистина и ИЛ-6 и снижается
продукция адипонектина, нарушается
эффективность лептина, что усиливает
инсулинорезистентность.
Описан механизм, способный обусловить
при развитии ИНСД обратную связь
между гиперсекрецией инсулина и усилением
периферической
инсулинорезистентности. Речь идет об амилиновом механизме.
Краткая характеристика амилина была уже
приведена в данном разделе выше.
Наконец, нельзя исключить возможность
усиления инсулинорезистентности из-за
нарушения идиотип-антиидиотипических
взаимодействий с участием иммунной
системы и инсулинового рецептора.
Так или иначе, но перечисленные
механизмы ответственны за прогрессирование
болезни.
2-я стадия ИНСД по мере
формирования будет проявляться пролонгированной
гипергликемией после еды и может быть
выявлена глюкозной нагрузкой.
Патофизиология сахарного диабета 331
В третьей стадии нет существенного
дальнейшего нарастания инсулинорезистентнос-
ти, но снижаются секреторные возможности
В-клеток. Это ведет к гипергликемии
натощак и явному диабету. Эта стадия может
быть вызвана эндогенными токсическими
влияниями на В-клетки или
манифестацией, под влиянием гипергликемии, скрытых
генетических дефектов В-клеток,
ускоряющих их гибель или старение.
Следует подчеркнуть, что проявления и
развитие болезни существенно тормозятся
при соблюдении диеты.
Вопрос о первичности инсулинорезистен-
тности при ИНСД нельзя считать
окончательно решенным. У. Уорд и соавторы (1985)
выступили с оригинальной стройной
концепцией патофизиологии ИНСД, с позиций
первичности функционального дефекта В-клеток
(«как гипотеза ленивых В-клеток»).
Согласно этой концепции, недостаточный
секреторный ответ В-клеток на глюкозу
первичен при ИНСД. Однако
развивающаяся вследствие этого гипергликемия в
начальной стадии болезни компенсирует этот
дефект, повышая стимуляцию В-клеток и
уровень инсулиновой секреции до нормы.
Сама по себе первичная
инсулинорезистентность без нарушений отвечаемости
В-клеток на глюкозу не может вызвать
ИНСД, так как секреторные резервы ин-
тактных В-клеток очень велики. Опыты с
удалением островков показывают, что 10%
массы интактных В-клеток достаточно для
поддержания нормогликемии без
ограничений экспериментальных животных или
больных с резекцией поджелудочной
железы в диете. По этой причине, указывают
У. Уорд и соавторы, громадное большинство
лиц, страдающих тучностью и
периферической инсулинорезистентностью, имеют
гиперинсулинемию, но не гипергликемию.
Если инсулинорезистентность
развивается на фоне ограничения секреторной
отвечаемости В-клеток, ее воздействие на судьбу
процесса меняется и становится решающим.
Уровень глюкозы плазмы поднимается еще
выше, чтобы скомпенсировать
инсулинорезистентность с помощью гиперинсули-
немии. Но гиперинсулинемия становится
элементом порочного круга, усиливая
периферическую инсулинорезистентность из-за
влияния избытка гормона на жировой обмен
и экспрессию инсулиновых рецепторов.
Возможно, адипоциты при
гипергликемии ведут себя согласно программе «сытого
состояния»: перестают выделять адипонек-
тин и выделяют избыток кахексина,
провоцирующего дополнительную
инсулинорезистентность. Кроме того, для стимуляции
дефектных В-клеток требуются уровни
глюкозы, превосходящие почечный порог. Из-за
глюкозурии плазменный уровень глюкозы
не может подниматься бесконечно. Он
достигает примерно 250 мг/дл, далее глюкоза
теряется, а секреторные резервы В-клеток не
могут быть мобилизованы, так как
дальнейшая стимуляция дефектных клеток
возможна только при больших уровнях глюкозы.
Таким образом, степень гипергликемии
у больного ИНСД определяется
взаимодействием сниженной чувствительности
В-клеток к глюкозе и периферической
инсулинорезистентности. Состояние
патологического равновесия достигается
при больших или меньших концентрациях
инсулина (в зависимости от преобладания
инсулинорезистентности или дефекта
В-клеток), но инсулина всегда
недостаточно по отношению к имеющемуся избытку
глюкозы в крови. Уровню декомпенсации
соответствует концентрация глюкозы, при
которой присутствует глюкозурия.
Следовательно, с точки зрения концепции У. Уор-
да, инсулинорезистентность приобретает
при ИНСД ключевое значение только в
контексте имеющегося латентного дефекта
В-клеток. Предполагается, что дефекты
В-клеток гетерогенны, не обязательно
связаны с патологией рецептора глюкозы
или ее переносчика в В-клетки и частично
обратимы.
Патогенез осложнений ИНСД
Среди осложнений ИНСД в данном разделе
будут рассмотрены только макроангиопатия
332 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
и инсулинорезистентность, поскольку
механизмы гиперосмолярной комы и микроан-
гиопатических осложнений уже
обсуждались. Следует помнить, что ни ускоренный
атеросклероз, ни инсулинорезистентность
не являются монополией ИНСД, а могут
наблюдаться и при ИЗСД.
Макроангиопатия
Макроангиопатия при СД — это
поражение крупных артерий. Макроангиопатия
наблюдатся при любом типе СД, но
особенно тяжёлый характер носит при ИНСД,
обусловливая 60 % смертельных случаев
при данном заболевании. Наиболее частой
причиной, непосредственно приводящей к
смерти больного ИНСД, является инфаркт
миокарда (37%). У женщин
репродуктивного возраста инфаркт миокарда
встречается гораздо реже, чем у мужчин, но СД
практически уравнивает частоту инфаркта
у больных разного пола. Гангрена нижних
конечностей поражает больных СД в 160
раз чаще, чем лиц без СД. Инсульт при СД
встречается намного чаще. Это косвенно
указывает на главный механизм макроанги-
опатии — ускоренный атеросклероз.
Классификация макроангиопатии
включает:
>• хроническую ишемическую болезнь
сердца,
>- нарушения мозгового кровообращения
(апоплексические и ишемические,
хроническую недостаточность мозгового
кровообращения),
>- облитерирующий атеросклероз артерий
нижних конечностей и облитерирующие
сосудистые поражения других
локализаций (см. рис. 70).
При макроангиопатии, рассматриваемой
как ранний распространенный атеросклероз,
патологические изменения происходят в
сосудах среднего и крупного калибра и в
сосудах vasa vasorum. В совокупности эти
нарушения приводят к дистрофическим
изменениям сосудистой стенки. Если для
микроангиопатии ключевая
патогенетическая роль гипергликемии убедительно
доказана, то в развитии макроангиопатии
гипергликемия, хотя и играет существенную
роль, но, по-видимому, не имеет
исключительного значения.
Атерогенез, по современным
представлениям, является адаптивным ответом
сосудистой стенки на анормальные в
количественном и качественном отношении
липопротеиды (см. «Нарушения липидного
обмена»). Этот адаптивный ответ протекает
с пролиферацией гладкомышечных и иных
клеток сосудистой стенки, их пенистой
трансформацией, синтезом избыточного
количества компонентов
межклеточного вещества, кальцинозом и вторичной
активацией тромбоцитов и механизмов
свертывания крови аномальными липопро-
теидами и пораженной сосудистой стенкой.
При СД атерогенез ускорен, а клинические
проявления осложнений атеросклероза
носят ранний характер. По образному
выражению Ю.И.Строева (1997), сочетание
диабетических макро- и микроангиопатии
с гипертензией «создает для больных
сахарным диабетом своеобразный злополучный
бермудский треугольник, который
становится для большинства из них поистине
фатальным». Механизмы ускорения
атеросклероза при СД множественны. Дело в том,
что СД усиливает и ускоряет действие таких
признанных факторов риска атеросклероза,
какими считаются гиперлипопротеидемия
игипертензия. Помимо этого,
гипергликемия и гиперинсулинемия сами по себе
принадлежат к числу факторов риска
атеросклероза.
Как уже отмечено в главе «Нарушения
липидного обмена», при СД имеется
гиперлипопротеидемия с накоплением липопро-
теидов низкой и очень низкой плотности
(ЛПНП и ЛПОНП), а при ИНСД - еще и
снижение уровня липопротеидов высокой
плотности (ЛПВП), выполняющих
дренажную антиатерогенную функцию.
Причины этого заключаются в
увеличении секреции ЛПОНП печенью, как при
интенсивной потере веса, так и при
прогрессирующем ожирении, так как это служит
способом затормозить развитие стеатоза
печени.
Центральную роль в механизмах гипер-
липопротеидемии при СД играет гликиро-
вание липопротеидов. Л ПНП, подвергнутые
неэнзиматическому гликированию, более
активно захватываются клетками
сосудистой стенки, особенно макрофагами, через
ацетил-ЛПНП рецептор, чем нативные
ЛПНП. Отложение избытка холестеринсо-
держащего материала в сосудистой стенке
ускоряется. В то же время гликирование
ЛПВП приводит к укорачиванию времени
их жизни и снижению концентрации,
соответственно тормозится дренаж холестерина
из сосудистой стенки. Добавим к этому
ускорение стероидогенеза при ИНСД и
повышение концентрации особо атерогенного
липопротеида (а), стимулирующего тромбо-
генез и коагуляцию при ИЗСД.
Выше уже отмечалось, что гликированные
молекулы волокнистых белков
соединительной ткани сосудистой стенки захватывают и
фиксируют ЛПОНП и ЛПНП более
активно, чем у индивидов без СД.
Способствуя задержке натрия в организме
и нарушая функции почек, СД ускоряет
развитие гипертензии. Если ИЗСД связан с
патогенезом почечной гипертензии, то ИНСД
входит в структуру так называемого
«метаболического Х-синдрома>>, уже
упоминавшегося выше в разделах «Нарушения липидного
обмена» и «Патофизиология нуклеинового
обмена». Напомним, что системный дефект
водород-натриевого противопереносчика
клеточных мембран обусловливает, с одной
стороны, эссенциальную гипертензию, а с
другой — инсулинорезистентность, гипер-
инсулинемию, гипертриглицеридемию и
гиперурикемию. Толерантность к глюкозе
при этом снижена (см. гл. 14).
По мнению Дж. Стайнера (1985), сама
глюкоза может иметь атерогенное
действие (см. с. 171). Дополнительным важным
фактором является тромбофилитический
синдром при СД.
Гиперинсулинемия — важный
самостоятельный фактор риска атеросклероза (см.
с.171-172).
Патофизиология сахарного диабета 333
Гипоксия и стресс, вызванный
колебаниями концентрации глюкозы в крови также
атерогенны.
Несомненно, значимо и ожирение,
наблюдаемое при ИНСД и являющееся
независимым от гиперлипопротеидемии
фактором риска атеросклероза.
Экзогенный сорбит, по Д. Стауту (1980), ускоряет
атерогенез.
Следует считаться и с ускорением при
гипергликемии эндогенного сорбитологенеза
в полиоловом цикле.
Генетическое предрасположение к
повреждению сосудистых стенок
подтверждается склонностью к развитию ангиопатий у лиц,
больных СД и имеющих антиген ГКГС В8.
По данным Ю.И. Строева (1999), при
ИНСД атеросклероз отмечается в 91,3%
случаев, ИБС — в 43,1%, а ангиопатия
конечностей — почти у 30 % больных.
Приводим рис.74, демонстрирующий данные
термографии нижних конечностей при СД
и интегральную картину нарушений липопр-
отеиднового метаболизма у больных СД.
Инсулинорезистентность:
виды и механизмы
Инсулинорезистентность — пониженная
эффективность действия инсулина часто
сопровождает СД, но может наблюдаться и
при других болезнях.
Под инсулинорезистентностью (ИР)
понимают состояние, когда для
предотвращения гипергликемии и кетоацидоза
необходимо 200 и более международных единиц
гормона ежесуточно.
Однако в зависимости от возраста и массы
тела цифры могут быть и иными. Поэтому
наиболее корректна клиническая оценка ИР
по количеству инсулина, потребного для
утилизации одного моля экзогенной глюкозы.
ИР может быть первичной,
существующей до начала инсулинотерапии, и
вторичной, вызванной реакцией на инсулиноте-
рапию.
При ИНСД всегда имеется первичная ИР.
При ИЗСД, несмотря на частое присутствие
29
24
Q здоровые лица
Щ больные, сахарным
диабетом
общие тршли- холесте- суммарные свобод, а-липо- $-липо- а-липо- $-лшю- пребета-липо-
липиды цериды рин $-липо- жирные протеиды протеиды протеиды протеиды протеиды
протеиды кислоты электрофорез диск-электрофорез в
на бумаге полиакриламидном геле
Рис. 74. Ангиопатии при СД. Внизу — показатели липидного обмена у здоровых лиц (белые
столбики) и больных СД (черные столбики). Вверху: а — термограмма нижних конечностей при ИНСД
II типа. Заметна «термоампутация» стопы на стороне преимущественного атеросклеротического
поражения, б — крупномасштабная термограмма стоп при диабетической МиАП нижних
конечностей. Справа — температурная шкала
Патофизиология сахарного диабета 335
до начала инсулинотерапии антител к
собственному инсулину, эти иммуноглобулины
не обусловливают сколько-нибудь
выраженной первичной ИР. Однако вторичная ИР
при ИЗСД встречается нередко, особенно
если используют ксеногенные инсулины.
Бычий инсулин отличается от
человеческого тремя, а свиной — одной аминокислотой,
китовый имеет более далёкую от
человеческого структуру (рис. 86, с. 439). Тем не
менее даже свиной инсулин, в отличие от
рекомбинантного человеческого, вызывает
достаточно выраженный иммунный ответ,
что способствует развитию вторичной ИР.
По локализации дефекта,
обусловливающего снижение эффективности гормона, ИР
делится на следующие формы:
> пререцепторную,
> рецепторную,
> пострецепторную и
> комбинированную, причем последний
вариант наиболее распространен.
ИР может подразделяться на варианты
и по причине, лежащей в её основе. Данная
классификация предусматривает
вычленение следующих форм ИР:
A. Аномалии секреторного продукта В-клеток
— аномалии молекулы инсулина
— неполное превращение проинсулина
в инсулин
Б. Циркулирующие антагонисты инсулина
— повышенные уровни контринсуляр-
ных гормонов (СТГ, кортизол, глюка-
гон, катехоламины, тироксин и др.)
— негормональные антагонисты
инсулина, а именно свободные жирные
кислоты (СЖК), амилин, кахексии и другие
— антиинсулиновые антитела
B. Дефекты тканей-мишеней
— дефекты инсулинового рецептора
— блокирующие или
десенсибилизирующие антитела против инсулинового
рецептора
— пострецепторные дефекты
При первичной ИР у больных ИНСД
преобладают пострецепторные дефекты.
При вторичной, наоборот, большую роль
играют аутоиммунные механизмы и
циркулирующие антагонисты инсулина.
Разнообразные пре- и пострецепторные
дефекты рассматривались раньше. В данном
разделе мы коснемся некоторых деталей,
связанных с циркулирующими
антагонистами инсулина.
>• Гормональные антагонисты включают
все известные контринсулярные
гормоны: кортизол, соматотропин, глюкагон,
катехоламины, тироидные гормоны.
>• Негоромональные антагонисты
инсулина — это СЖК, цитокины, амилин,
кахексии и антитела.
Известно, что СЖК нарушают
периферическую утилизацию глюкозы.
При инсулинорезистентности инсулин
не способен блокировать повышение уровня
СЖК в крови за счет их освобождения из ли-
поцитов. СЖК ингибируют захват глюкозы
печенью и способствуют глюконеогенезу,
усилению гипергликемии и ГЛП. Это
патогенетическое звено играет роль при
метаболическом синдроме и, в частности, при ИНСД.
Ген белка, связывающего СЖК при их
проникновении в клетки, более тесно сцеплен с
инсулинорезистентными состояниями, чем
ген рецептора самого инсулина.
Повышенная утилизация СЖК угнетает
гликолиз и снижает потребление глюкозы,
что de facto антагонистично по отношению к
метаболическим эффектам инсулина.
Антитела к инсулину образуются у всех
больных, получающих инсулин. Примерно
у каждого тысячного больного, леченного
инсулином, в течение первых 2 месяцев
терапии это приводит к накоплению в
высоких титрах IgG, блокирующих инсулин
и формирующих выраженную ИР, иногда
с аллергией к инсулину, выражающейся в
ГНТ, часто — анафилактического характера.
Однако, в большинстве случаев антитела не
являются причиной
инсулинорезистентности (хотя они и взаимодействуют с
инсулином при иммуноконкурентных методах его
определения и обусловливают «кажущуюся
гиперинсулинемию»).
336 АШ. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Дело в том, что, какова бы ни была
поначалу интенсивность выработки проти-
воинсулиновых антител, их количество у
большинства больных имеет тенденцию к
снижению через 100-200 дней лечения.
Связанный в комплекс с антителом
инсулин может освобождаться из комплекса и
быть причиной тяжелых гипогликемических
состояний. Вследствие идиотип-антиидио-
типических взаимодействий и вторичной
выработки анти-антител, титр антиинсулино-
вых иммуноглобулинов сильно колеблется.
В последние годы описан синдром инсу-
линорезистентности, обладатели которого
имели антитела против рецепторов
инсулина. Циркулирующие антитела
связываются с инсулиновым рецептором in vivo,
блокируют его и приводят к возникновению
инсулинорезистентности.
Антирецепторные антитела принадлежат
в основном к Ig G и связываются с
рецептором через Fab фрагмент. Они обнаружены, в
частности, при синдроме acanthosis nigricans
у афроамериканцев (М. Блечер, 1984).
Антирецепторные антитела, в принципе,
могут действовать двояко (см. рис. 63):
1) как эффективные имитаторы инсулина
(подобно тироидстимулирующим
иммуноглобулинам при диффузном
токсическом зобе);
2) как конкурентные блокаторы инсулина
(подобно антителам к ацетилхолиновым
рецепторам при генерализованной
миастении). Показано, что антиинсулиноре-
цепторные антитела нередко тормозят
метаболические эффекты инсулина, но
стимулируют его митогенное действие,
в частности в культуре клеток плаценты
HIR С-В.
Специфичностью к рецептору инсулина
будут обладать антитела против активного
центра антиинсулинового антитела (анти-
идиотипические антитела). Чаще они
имитируют действие инсулина. Показано, что
подобные антитела способны вызывать как
гипогликемии (см. выше «Гипогликемия как
синдром...»), так и инсулинорезистентность
(вследствие неполной комплементарности к
инсулиновому рецептору и быстрой
десенсибилизации последнего). Ранняя,
стимулирующая фаза действия подобных
иммуноглобулинов может сменяться стойкой
поздней — блокирующей (С. Р. Кан, Дж. Флайер,
1980; Й. Шехтер и соавт. 1986).
Мощными модуляторами и порой блока-
торами действия инсулина служат цитоки-
ны адипоцитов и оседлых макрофагов
жировой ткани — адипокины (П. Гавел, 2002).
При ИНСД и метаболическом синдроме
снижена продукция инсулинсенсибилизи-
рующего цитокина адипонектина, алептин,
призванный адаптировать организм к
снижению потребления энергии и усилению ее
расхода, хотя и вырабатывается в больших
количествах, но не эффективен. Адипоциты
висцерального жира и накапливающиеся в
жировой ткани в избытке макрофаги при
ИНСД и метаболическом синдроме
выделяют в повышенных количествах кахексии,
ИЛ-6 и резистин, которые усиливают
инсулинорезистентность, ингибитор
активатора плазминогена PAI-1, способствующий
тромбофилическому синдрому, и ангио-
тензиноген, потенцирующий артериальную
гипертензию. Эти особенности адипоцитов
связаны с их ранним гистогенезом и
наблюдаются чаще у лиц, испытавших
недоношенность или внутриутробную гипотрофию.
В связи с этим низкий вес при рождении
эпидемиологически связан с повышенным
риском ИНСД и метаболического синдрома
(Д. Жаке и соавт, 2000, см. гл. 14).
Дж. Фостер (1994) указывает на
разнообразие заболеваний, сопровождаемых
инсулинорезистентностью. Кроме идиопа-
тического СД, при котором ИР чаще всего,
имеет пострецепторную природу, снижение
эффективности действия инсулина на его
мишени наблюдается также и при:
>- ожирении;
>• ИР-синдроме типа А (дефект инсулино-
вого рецептора);
>- ИР-синдроме типа В (аутоантитела к
инсулиновому рецептору);
> местной и системной липоатрофии;
> гиперплазии шишковидной железы;
> синдроме Ольстрема;
> атаксии-телеангиоэктазии;
> синдроме Рабсона-Менденхолла;
> синдроме Вернера;
> синдромах Лоуренса-Муна и Барде-
Бидла;
> синдроме эльфа (синдром Донохью-
Ушинда);
> Thalassemia major;
> миотонической дистрофии;
> симптоматических формах вторичного
СД, вызванных гиперпродукцией конт-
ринсулярных гормонов (например,
акромегалии, гипофизарном гигантизме).
Некоторые из этих состояний уже были
охарактеризованы в разделе «Нарушения
липидного обмена», так как клинически они
принадлежат к формам вторичного
ожирения или к местным липоатрофиям.
Ниже приводятся дополнительные
сведения об углеводном обмене при этих и других
нарушениях.
ИР-синдром типа Л отмечается у женщин
с поликистозом яичников и гирсутизмом и
обусловлен мутациями инсулинового
рецептора и гиперандрогенизмом (см. ниже
«Нарушения эндокринной регуляции»).
ИР-синдром типа В также характерен
для женщин, но связан с усилением
аутоиммунного ответа против ДНК и против
инсулинового рецептора. Ряд симптомов
(сиалоаденит, артралгии, нефропатия)
приближает его к системным аутоиммунным
расстройствам с неорганоспецифическими
аутоантителами. Подобно другим
аутоиммунным заболеваниям, этот инсулино-
резистентный синдром присущ пожилым
пациенткам.
Приобретенная или врожденная липо-
атрофия или липоатрофический диабет
характеризуются местным или системным
исчезновением жировой ткани, гиперлипоп-
ротеидемией IV типа, ожирением печени,
гирсутизмом, умственной отсталостью и
Патофизиология сахарного диабета 337
поражением почек на фоне гипергликемии
без кетоацидоза. Предполагается
комбинированный рецепторно-пострецепторный
блок действия инсулина у таких больных,
эффективен адипонектин.
Гиперплазия эпифиза вызывает макро-
генитосомию, преждевременное половое
созревание, гирсутизм, раннее
прорезывание и деформацию зубов и утолщение
ногтей, ИР, гипергликемию и кетоацидоз
(см. ниже «Патофизиология шишковидной
железы»).
Синдром Рабсона-Менденхолла (дефект
рецептора инсулина в хромосоме 19)
напоминает редуцированную клиническую
картину эпифизарной гиперплазии, но без
самого увеличения шишковидной железы.
При синдроме Ольстрема имеются
проявления множественной резистентности
тканей-мишеней к гормонам, включая пит-
рессин-резистентный несахарный диабет,
первичный гипогонадизм и умеренную ИР.
Это сопровождается слепоглухотой.
Синдром Вернера (дефект в 8-й
хромосоме) характеризуется наследственной
аутосомно-рецессивной пострецепторной
ИР, сопровождаемой атрофией инсулин-
зависимых тканей, гипогонадизмом,
алопецией, катарактой, прочерней и высокой
частотой некоторых злокачественных
опухолей.
При атаксии-телеангиоэктазии, кроме
проявлений, вошедших в ее наименование,
ИР сопровождается иммунодефицитом.
Синдром эльфа или леприконизм —
редкая аномалия у девочек с ИР, липоатрофией,
утолщением кожи, нефрокальцинозом,
гирсутизмом и «лицом эльфа» (гипертелоризм,
экзофтальм). Все перечисленные
синдромы, как полагают, связаны с дефектами
рецепторов инсулина (хромосома 19). По
авторам нарушение известно как синдром
Донохью - Ушинда.
При Thalassemia major ne исключен
механизм инсулинорезистентности, связанный
с нарушением эритроцитарного транспорта
инсулина к тканям-мишеням.
При всех этих состояниях уровень
циркулирующего инсулина повышен, но явный
338 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
СД может и не наступать при достаточных
резервах В-клеток.
Описаны случаи аномально быстрого
протеолиза экзогенного инсулина в месте
его инъекции как основа ИР. Прежде это
именовали повышенной инсулиназной
активностью. Так как не было обнаружено
никакой специфической протеазы
инсулина, то такая формулировка имеет лишь
историческую ценность. Однако следует
учесть, что подобным больным помогают
инъекции вместе с инсулином
неспецифических блокаторов протеолиза.
Анализируя разнообразие синдромов,
сопровождаемых ИР, необходимо указать на
часто повторяемые стигмы ИР, при наличии
которых врач может предполагать
повышенную наклонность к ИР.
Они могут наблюдаться при разных
заболеваниях и включают:
>- гирсутизм,
>• липоатрофии,
>- ожирение,
>■ нарушения полового созревания,
>* acanthosis nigricans — особенно значимый
неспецифический кожный симптом ИР
типа А, проявляющийся в виде
бархатистых на ощупь пятен
гиперпигментации на заднелатеральной поверхности
шеи, животе, в подмышечной области и
реже — в других местах.
Экспериментальные модели
сахарного диабета
В античной древности Аретей определил
СД как болезнь, «при которой плоть
перетекает в мочу», считая причиной этого
явления ненормальную работу почек.
Экспериментальное изучение СД началось
гораздо позже.
Еще в середине XVII столетия Й.-К. Брун-
нер предпринял попытку удаления
поджелудочной железы и отметил после панкре-
атэктомии резкое исхудание у подопытных
животных (1688).
В 1776 г. английский доктор Дж. Добсон
обнаружил «путём выпаривания урины»,
что вещество, придающее моче больных
сладкий вкус, — это сахар.
Первая догадка о связи СД и состояния
поджелудочной железы принадлежит
британскому хирургу Т. Коули, описавшему
больного СД с множественными
панкреатическими камнями (1788). Чуть позже
главный хирург Королевской артиллерии
Дж. Ролло (1797) заметил, что степень
выделения сахара с мочой и тяжесть поли-
урии коррелируют с содержанием круп и
растительных продуктов в рационе больных.
Он пришел к заключению, что в болезни
повинен пищеварительный тракт, а не почки,
и отметил «запах гниющих фруктов» в
выдыхаемом больными воздухе. Всё это
направило усилия экспериментаторов на создание
моделей СД на основе нарушения функции
поджелудочной железы. Тем временем,
М. Шеврёль установил, что в моче больных,
а также в их крови имеется избыток глюкозы
(1815). Поэтому после открытий К. Бернара
касательно печеночной продукции глюкозы
из гликогена и ее секреции в кровь возник и
«непанкреатический» подход к
моделированию проявлений СД (1857). Сахарный укол
К. Бернара — моделирование преходящей,
очевидно, стрессорной гипергликемии и
глюкозурии путем пункции дна IV
мозгового желудочка. Нарушения обмена глюкозы
при этом развиваются у животных спустя
1-2 ч. и длятся от 4-6 ч. (у кроликов), до 1-2
суток (у собак). На основании этих опытов,
К. Бернар счёл СД нарушением нервной
регуляции секреторной функции печени,
тем более, что разрушение экзокринной
части поджелудочной железы (путём её па-
рафинизации) СД не воспроизводило. Тем
временем в 1869 г. студент-медик П. Лан-
герганс открыл в поджелудочной железе
островки. Однако «панкреатическая»
теория СД, предложенная А. Бушаром еще в
1875 г., возобладала только после появления
первой успешной модели СД, основанной на
панкреатэктомии.
Панкреатэктомия как способ вызвать
экспериментальный СД впервые успешно
была предпринята Й. фон Мерингом и
О. Минковским, а независимо от них и поч-
Патофизиология сахарного диабета 339
ти одновременно— Де Доминичи в 1889 г.
Тотальное удаление поджелудочной железы
практиковалось этими пионерами
экспериментальной диабетологии у собак. У
животных развивалась типичная картина ИЗСД
с истощением, кетоацидозом и гибелью.
О. Минковский и Д. Хидон показали, что
пересадка кусочка поджелудочной железы под
кожу собаки предохраняет панкреатэкто-
мированное животное от СД. Позже (1908)
Й. Форшбах продемонстрировал на данной
модели, что СД после панкреатэктомии
может быть купирован при парабиотической
связи кровеносной системы подопытной и
интактной собак. Так была установлена роль
нехватки неидентифицированного гормона
поджелудочной железы в патогенезе СД.
Воспроизведение панкреатэктомической
модели СД у крысы долгое время не
удавалось, поскольку диффузное распределение
панкреатической ткани по брыжейке не
позволяло удалить железу целиком. P.O. Скау в
1957 г. методически разрешил эту проблему
(тотально удалил поджелудочную железу) и
получил у крысы типичный СД. Данная
модификация модели Меринга-Минковского
развеяла миф о том, что крысы резистентны
к воспроизведению сахарного диабета при
помощи панкреатэктомии.
Догадку об эндокринной роли
поджелудочной железы высказывал еще учитель
П.Лангерганса — Р. Вирхов (1854). Но
только в 1898 г. отечественный патолог
А.И.Яроцкий предположил, что
регуляцией обмена глюкозы занимаются именно
островки Лангерганса. Русский учёный
Л.В.Соболев (1901) усовершенствовал
экспериментальную модель СД на собаках,
предложив оригинальную методику ауто-
лиза экзокринной части панкреатической
железы при сохранении островков
Лангерганса, путём перевязки выводного протока
железы. Разрушение ацинозных элементов
железы не приводило к СД, в отличие от
тотальной панкреатэктомии и удаления
островков у собак с атрофированной
экзокринной частью поджелудочной жедезы.
На этой основе Л.В. Соболев высказал ряд
основополагающих идей, определивших
в дальнейшем развитие
экспериментальной диабетологии. Так, он постулировал,
что вещество, нехватка которого повинна
в возникновении СД, вырабатывается
островками Лангерганса и рекомендовал
получать его путём перевязки выводного
протока железы новорожденных телят, у
которых экзокринная часть железы слабо
развита. Именно подобный метод
применили позже Ф. Г. Бантинг, К.Х. Бест и Дж.
Р. Маклеод для выделения из островков
инсулина (1921-1922)1. Данное открытие
было отмечено Нобелевской премией по
медицине 1923 г. Ранее этих авторов в 1904 г.
отечественный врач А. А. Кулябко впервые
попытался замещать гормональную
функцию у больных СД экстрактами островковой
ткани костистых рыб, а румынский
патофизиолог Николае Пэулеску (1916,1921) даже
получил водный экстракт из островковой
ткани, очистил препарат и купировал его
внутривенными инъекциями симптомы СДу
панкреатэктомированных собак. Он описал
динамику гипогликемизующего действия
препарата и запатентовал открытый гормон
под названием панкреин (1922). Ф. Бантинг
и соавторы цитировали в своей первой
публикации работы Н. Пэулеску, но не указали,
что панкреин купирует гипергликемию.
Работ Пэулеску почему-то не заметил
Нобелевский комитет. Термин «инсулин»
придумал еще до открытия самого гормона
И.деМейер(1909).
В 1922 г. Ф.М. Аллен на собаках с
резекцией 90 % поджелудочной железы
показал, что поражение и гибель
оставшихся В-клеток могут быть вызваны
перекармливанием углеводами. В 30-х годах XX
столетия аргентинский эндокринолог
Б.А.Усай моделировал СД, удаляя
поджелудочную железу у южноамериканских
жаб и у млекопитающих, и показал, что
проявления экспериментального СД
значительно ослабевают, вплоть до полного
1 Достоверно известен день — 30 октября 1920 г.,
когда выпускник медицинского факультета
Ф.Бантинг прочел статью Л.В.Соболева и
решил применить его метод.
340 ALU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
исчезновения, при удалении аденогипофиза
и надпочечников. Предварительная гипо-
физэктомия делает животных значительно
более резистентными к диабетогенному
эффекту резекции поджелудочной железы.
Эти опыты позволили установить факт
продукции контринсулярных гормонов в
аденогипофизе и надпочечниках и также
удостоились Нобелевской премии по
медицине (за 1947 год). Впоследствии выяснили,
что субтотальная резекция поджелудочной
железы ведет к латентному сахарному
диабету, который переходит у подопытного
животного из скрытого в явный при нагрузке
контринсулярными гормонами (тироидные,
соматотропин, глюкокортикоиды) или при
перекармливании углеводами. Более того,
Л.Ивенсу, А.Хэму и Р. Хейсту удалось
получить стойкий СД у собак путём
хронического введения экстрактов аденогипофиза
(1927-1939).
Дальнейший прогресс в понимании
функций островков и их нарушения при СД
связан с введением в экспериментальную
практику химических моделей СД. Первой
среди них появилась аллоксановая модель.
Дж. С. Данн и Н. Г. Мак Летчи
обнаружили в 1943 г., что аллоксан (уреид
мезоксалевой кислоты) избирательно
повреждает островковые В-клетки, вызывая их
некроз, и приводит к развитию СД у крыс,
мышей, кроликов и собак. Аллоксановая
модель позволила установить роль В-кле-
ток, открытых ещё в 1908 г. Б. Лэйном, в
продукции инсулина, а их повреждения — в
генезе ИЗСД. Так как аллоксан может
возникать в организме из мочевой кислоты
через диалуровую, а также в связи с частым
клиническим сочетанием СД и гиперури-
кемии, отечественный автор Я.А. Лазарис
модифицировал аллоксановую модель и
предпринял успешную попытку вызвать
СД у крыс токсическими дозами мочевой
кислоты (600-680 мг/кг массы тела), на
фоне дефицита пищевых серосодержащих
аминокислот (1957).
В дальнейшем ( X. Маске, 1957) для
химического моделирования ИЗСД
применили дитизон (дифенилтиокарбазон)
и оксином (8-оксихинолин). Это так
называемые щипковые» модели», поскольку
действующие в них химические вещества
образуют комплексы с цинком в
секреторных гранулах В-клеток. Это нарушает
накопление инсулина и его секрецию.
Закономерно сахарный диабет по данной
модели возникает лишь у кроликов, В-клетки
панкреатических островков которых богаты
цинком. У морских свинок и крыс данный
микроэлемент не играет столь важной роли
в инсулиновой секреции и не накапливается
в В-клетках, следовательно, они к «цинк-
блокирующему» экспериментальному СД
устойчивы.
Еще одна «химическая» модель СД
основана на применении стрептозотоцина —
антибиотика, вызывающего избирательное
повреждение В-клеток у крыс. Однако
у других животных диабетогенные и
летальные дозы данного антибиотика очень
близки что делает эту модель, практически,
малопригодной и не всегда легко
воспроизводимой.
Большое значение имело установление
того факта, что многие «химические»
модели на деле являются
химико-иммунологическими, так как аллоксан, мочевая
кислота и стрептозотоцин запускают против
В-клеток аутоиммунный процесс, который,
собственно, и губит источники инсулина.
Это стало ясно из экспериментов,
доказавших, что данные модели не воспроизводятся
у тимэктомированных иммунодефицитных
животных. Развитие экспериментального
СД, индуцированного химически, может
быть заторможено антилимфоцитарной
сывороткой и иммунодепрессантами. В ряде
случаев СД, полученный по этим моделям,
подвергается адоптивному переносу при
введении лимфоцитов больных
подопытных животных интактным. В то же время
некоторые химически индуцированные
модели СД, например «цинковые», имму-
нонезависимы.
Развитие представлений о вирусных диа-
бетогенах неотделимо от становления
вирусной модели ИЗСД. Заражение М-вариантом
вируса мышиного энцефаломиокардита в
Патофизиология сахарного диабета 341
40% случаев приводит у подопытных
мышей к развитию аутоиммунного инсулита,
оканчивающегося ИЗСД. Данная модель
также блокируется тимэктомией и имму-
нодепрессией.
Еще В. Наунин (1895) сформулировал
первую генетическую гипотезу о природе
СД. С обнаружением в 1974 г. Дж. Нерупом
и соавторами связи между СД и генами ГКГС
исследования в области генетического
моделирования СД особенно активизировались.
Животные чистых линий, развивающие
спонтанный СД, служат объектами
эффективного изучения как ИЗСД так и ИНСД.
Подобных генетических моделей СД
известно в настоящее время множество. Это
чистые линии китайских хомячков, мышей
ККА, мышей ОВ/ОВ и DB/DB, мышей АВ/
АВ, колючих мышей, новозеландских
мышей, песчаных крыс, крыс В В (BioBreeding),
мышей NOD (Non Obese Diabetic), крыс
WBH/Kobe.
У мышей NOD имеется генетическая
аномалия экспрессии антигенов ГКГС 1 -го
класса. Это приводит к облегченной провокации
аутоаллергии против В-клеток и развитию
ИЗСД. Животные характеризуются
избытком Т-эффекторов, действующих против
островковых антигенов а также дефицитом
супрессорных функций лимфоцитов.
Крысы ВВ, напротив, характеризуются
наследственной лимфопенией и дефицитом
цитотоксических лимфоцитов. Тем не менее
они развивают спонтанный аутоиммунный
инсулит, который, так же как в случае
мышей NOD, приостанавливается неонаталь-
ной тимэктомией или применением имму-
нодепрессантов, а в естественных условиях
ведет к неизбежному ИЗСД.
Крысы WBH/Кобе самопроизвольно
вырабатывают в высоких титрах аутоан-
титела к собственному инсулину, которые
повреждают В-клетки и формируют основу
для развития ИЗСД.
Мыши КО, дефектные по гену адипо-
нектина, страдают ИНСД с выраженной
инсулинорезистентностью.
У мышей линий ОВ и DB развивается
ожирение, инсулинорезистентность и ИНСД.
Они уже рассматривались в главе «Пато-
физиолология липидного обмена», так как
представляют экспериментальные модели
первичного ожирения, имеющие,
соответственно, наследственный дефект гена лептина
(ОВ) или лептинового рецептора (DB).
История моделирования СД показывает,
насколько существенным является
осмысление тех или иных экспериментальных
моделей болезни для прогресса в понимании
её механизмов (В.Г. Баранов, 1983).
Патофизиологические основы
лечения СД
Близкое родство патофизиологии и
экспериментальной терапии делает уместным
включение данного краткого раздела в очерк
о механизмах сахарного диабета.
Лечение ИЗСД основывается на
патогенетической замещающей терапии
инсулином. В настоящее время, после
публикации данных специального исследования
сравнительной эффективности разных
методов инсулинотерапии (так называемая
программа DCCT, 1993), доминирует точка
зрения, согласно которой для профилактики
осложнений ИЗСД очень важно
интенсивное лечение препаратами человеческого ре-
комбинантного инсулина с целью взможно
более полного предупреждения эпизодов
гипергликемии (tight control doctrine). В то
же время признается, что такое лечение не
исключает полностью возможность
возникновения гипергликемии, увеличивает
примерно в 3 раза, по сравнению с менее
строгими схемами, риск гипогликемии и
способствует нарастанию веса пациентов, в
том числе — до уровня выше оптимального.
Наряду с инсулинотерапией применяют
специальные средства профилактики ней-
ропатии и микроангиопатии — например,
аминогуанидин.
Сводка дднных о принципах лечения,
профилактики и реабилитации при СД
содержится в недавно вышедшей на русском
языке под нашей редакцией
мини-энциклопедии (Американская диабетическая
ассоциация, 2003).
342 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Адекватного способа этиотропного
лечения ИЗСД до сих пор нет. Гистосовмес-
тимые островки через определенное время
после трансплантации закономерно
подвергаются аутоиммунной атаке, что ведет к
их недостаточности. Гистонесовместимые
островки атакуются как трансплантат.
Технические системы, имитирующие В-клетки,
не замещают их многообразных функций.
Возможность использования
непанкреатических гибридных клеток с
имплантированным геном человеческого инсулина
только изучается. Применение инсулина
не предотвращает, а при ксеногенности
инсулина, возможно, и ускоряет развитие
микроангиопатий, приводит к вторичной
инсулинорезистентности, а иногда — ла-
билизации диабета, липоатрофиям,
анафилаксии. При адекватной инсулинотерапии
до 60% больных ИЗСД в течение многих
лет могут сохранять удовлетворительную
функцию почек, с минимальными
гистологическими признаками диабетического
гломерулосклероза и эпизодической
микроальбуминурией. Продолжительность их
жизни после установления диагноза может
превысить 40 лет. В то же время у большого
контингента больных (до 40 %) развивается
прогрессирующий диабетический гломеру-
лосклероз, который за 15-25 лет ведет ктер-
минальной ХПН. У этой группы пациентов
эффективная антигипертензивная терапия
может продлить данный срок до 30-35 лет.
Но эти результаты не могут
расцениваться как радикальный прогресс в борьбе
с сахарным диабетом.
Поэтому существует потребность в
разработке новых методов этиотропной и
патогенетической терапии ИЗСД, учитывающих
новые данные, касающиеся его этиологии
и патогенеза.
Большие надежды возлагаются на
иммунотерапию. Более полувека назад в
исследованиях школы А.А. Богомольца была
впервые продемонстрирована
эффективность серотерапии СД цитотоксическими
сыворотками (Г.П. Сахаров, Д.М.
Российский 1937, 1942). Очевидно, играла
роль антилимфоцитарная составляющая
действия цитотоксических сывороток.
Ныне как на животных моделях, так и в
клинике у людей, показана высокая
эффективность иммунодепрессантной терапии,
при условии, что она назначается в I—III
стадии болезни, т. е. не позже возникновения
первых клинических симптомов
гипергликемии. Даже в IV-V стадии, если еще имеется
продукция С-пептида, иммунодепрессант-
ная терапия позволяет существенно
уменьшить дозу инсулина, увеличить продукцию
С-пептида и добиться ремиссии.
Более эффективными оказались
средства, действующие на клеточный иммунитет
и формирование аутоиммунного ответа.
Антилимфоцитарная сыворотка позволяет
уменьшить дозу инсулина, но дает
гематологические осложнения.
Иммуносупрессия, направленная против
эффекторов ГНТ (плазмаферез, обменные
гемотрансфузии, антиидиотипические
антитела против антител к панкреатическим
антигенам), оказалась недостаточно
эффективной (Дж. С. Эйзенбарт, А. Пауэре;
1985). Традиционные иммунодепрессан-
ты — глюкокортикоиды — в данном случае
нежелательны, вследствие их выраженного
контринсулярного эффекта. Однако
имеются сообщения об успешном применении
«мягкой» разновидности кортикостероид-
ной терапии препаратом «прегнозолон»
(Р. Бок; 1986).
Широко проводятся клинические
испытания эффективности терапии ИЗСД
циклоспорином А и его аналогами — сандимуном,
циамексоном, FK-506. Дозы 5-10 мг/кг веса
в день при 2-3 месячном цикле циклоспо-
ринотерапии обеспечивают протективный
эффект во И-Ш стадиях ИЗСД и стойкую
ремиссию у 33-50 % больных, которых
начали лечить в начале IV стадии. При
длительно текущем явном диабете — конец
IV и V стадия — эффект циклоспориноте-
рапии невелик: стойкая ремиссия следует
не более чем у 9 % больных. Циклоспорин
А действует преимущественно на функции
Т-хелперов и поэтому не затрагивает тече-
ние уже установившегося аутоиммунного
процесса. Он понижает интенсивность
антипанкреатической ГЗТ и титр аутоантител
к инсулину, но мало влияет на выработку
аутоантител к другим поверхностным
антигенам островковых клеток. Данный
антибиотик нефротоксичен и гепатотоксичен,
он понижает функциональные возможности
гипоталамо-гипофизарной системы и
вызывает вторичный гипогонадизм, имеются
сообщения о повышении частоты лимфом
при терапии циклоспорином. Циклоспорин
сам по себе цитотоксичен для В-клеток и ин-
гибирует освобождение инсулина, особенно
при определенном гаплотипе ГКГС.
Вероятность побочных эффектов должна
учитываться. В связи с этим назначение
циклоспорина, безусловно, целесообразно только
во П-Ш стадии болезни, до начала явной
клиники СД. Однако как раз в эти периоды
сложно предсказать вероятность прогрес-
сирования ИЗСД. Циклоспоринотерапии
должен предшествовать эффективный отбор
соответствующей группы больных, а также
определенная деонтологическая работа.
Делаются попытки модифицировать
применение циклоспоринотерапии при ИЗСД,
чтобы избежать негативных побочных
эффектов. При сочетанном применении у
больных ИЗСД циклоспорина А и антител
к рецептору IL-2 (мАТ ART18) удается
обойтись субтерапевтической дозой
антибиотика. Перспективной представляется
обработка лимфоцитов больных
циклоспорином А и антителами к IL-2-рецептору in
vitro с последующей реинфузией клеток
больным. При этом минимализуется
побочное органотропное действие иммуно-
депрессанта.
Имеется опыт получения ремиссии от
применения азатиоприна с тимостимули-
ном, антител к DR-белкам и других имму-
носупрессорных воздействий у больных с
ИЗСД.
Разработан способ экспериментальной
терапии стрептозотоцинового диабета
умышей антителами к мышиным I-J, 1-Е и
I-A -детерминантам, причем
полиспецифическая сыворотка обладает терапевтическим,
Патофизиология сахарного диабета 343
а моноспецифическая aHTH-I-J-сыворотка —
диабетогенным эффектом.
При циклоспоринотерапии обязательным
является контроль содержания лекарства в
крови. Во всех случаях при разнообразной
иммунотерапии ИЗСД информативно
определение ряда иммунологических
показателей: продукции IL-2, индуцированной
фитогемагглютинином экспрессии
рецепторов IL-2, бласт-трансформации
лимфоцитов. Если нет передозировки
циклоспорина, показатели этих тестов не должны
снижаться.
Существуют и другие новаторские
подходы в терапии ИЗСД, основанные на
раскрытии его иммунопатологической
природы. Так, Дж. С. Эйзенбарт и соавт. (1989)
рекомендуют применять инсулинотерапию
в предгипергликемическую стадию процесса.
Человеческий инсулин вводится нормо-
гликемическим больным с эквивалентной
дозой глюкозы, в расчете на связывание
инсулином антиинсулиновой иммунореак-
тивности и десенсибилизирующий эффект,
а не с целью использования гормонального
действия инсулина. По мнению авторов,
эффект подобного вмешательства не уступает
циклоспоринотерапии.
Многие авторы отмечают, что цитолиз
В-клеток, вне зависимости от причины,
требует усиления поли-АДФ-рибозилиро-
вания в клеточных ядрах. Поэтому высокие
(до 3 граммов в день) дозы витамина РР,
являющегося ингибитором полиаденозин-
дифосфатрибозилирования, оказываются
эффективным средством, тормозящим
разрушение В-клеток. Применяется нико-
тинамид, с целью профилактики стеатоза
печени, сочетаемый с метилметионином.
Исследования более чем на 20 000 пациентов
подтвердили эффективность никотинамида
для профилактики ИЗСД в группах риска,
но, к сожалению, данный метод не действует,
если В-клетки уже погибли.
Исходя из имеющихся данных о роли
липидных медиаторов и тромбоцитов в па-
344 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
тогенезе ИЗСД и микроангиопатий, можно
предположить, что ценным
поддерживающим средством комплексной терапии
больных ИЗСД окажутся аспирин, антиаг-
реганты и гепарин.
ИНСД в настоящее время лечится с
помощью диеты и химических препаратов,
понижающих содержание глюкозы в крови
(производные сульфонилмочевины и бигу-
аниды). Так как некоторые из них
индуцируют продукцию соматостатина, Ф. Фелиг
предложил примененить сам соматостатин
для терапии СД, однако эффективность
этого метода оказалась весьма умеренной.
Следует отметить, что с
патофизиологической точки зрения, несмотря на
существование двух типов СД, остается незыблемым
классическое положение В.Г.Баранова
(1966): «Без инсулина нет терапии
сахарного диабета». Поскольку относительная
инсулиновая недостаточность свойственна
и ИНСД, сам по себе диагноз ИНСД не
должен восприниматься как противопоказание
к инсулинотерапии. При неэффективности
диетотерапии и лечения упомянутыми
антидиабетическими средствами при наличии
тяжелых осложнений инсулин назначают и
больным со вторым типом СД.
Так как основу ИНСД составляет
первичная ИР, ведутся поиски средств,
обходящих неэффективное рецепторное звено и
оказывающих инсулиноподобное действие,
несмотря на ИР (адипонектин?).
В 1991-1992 гг. Й. Шехтер и
соавторы обнаружили, что ванадий (особенно
в форме ванадила сульфата) способен
оказывать инсулиноподобный эффект на
клетки в обход инсулинового рецептора. В
настоящее время ведутся клинические
испытания препаратов ванадия для лечения
ИНСД (см. ниже «Патофизиология обмена
микроэлементов»). У. Уорд и соавт. (1985)
выступили с идеей стимуляции секреции
оставшихся В-клеток больных ИНСД
а-адренолитиками, блокаторами синтеза
простагландинов и антиопиатэргически-
ми средствами. В 2003 г. была показана
возможность дифференцировки
костномозговых стволовых клеток человека в ос-
тровковые инсулинобразующие клетки in
vitro (А. Янус, 2003), а затем в Бразилии
проведена успешная экспериментальная
терапия ИЗСД собственными
костномозговыми стволовыми клетками пациентов
(X. Вольтарелли и соавт., 2007).
Сахарный диабет — комплексная
междисциплинарная проблема современной
медицины. Так как данная «метаболическая
болезнь № 1» затрагивает без исключения
все виды обмена веществ, мы и посвятили
ей в этой книге отдельную главу
Глава 10
ПАТОФИЗИОЛОГИЯ
ВОДНО-ЭЛЕКТРОЛИТНОГО
ОБМЕНА
«Организм без внешней среды, поддерживающей его существование,
невозможен, поэтому в научное определение организма должна входить
и среда, влияющая на него»,
И.М. Сеченов, 1861
«Внутренняя среда... образуется циркулирующей жидкостью, которая
окружает и омывает все тканевые элементы... Организм как бы укрыт
в своего рода оранжерее».
К. Бернар, 1878
• Водно-солевой обмен, его регуляция и нарушения
• Нарушения осмотического гомеостаза
• Гомеостаз калия и его нарушения
• Патофизиология кислотно-основного равновесия
Все реакции обмена веществ
происходят на мембранных носителях, либо
в водных растворах. Активность энзимов,
чувствительность рецепторов и
электрофизиологические явления на мембранах клеток
зависят от водно-электролитного окружения.
В связи с этим исключительное значение
для метаболизма как средства поддержания
гомеостаза имеют вода и ионы.
В данном разделе книги рассматриваются
регуляция обмена воды и важнейших ионов, а
также её нарушения. При этом особое
внимание будет уделено типовым патологическим
процессам, связанным с
водно-электролитным балансом, в частности — отёку (см. также
т. I, с. 216-220 — о местных отёках).
По А.Г. Гинецинскому (1964), ионный
состав биогенных жидкостей сходен с морской
водой, так как жизнь зародилась в
первичном океане. По закону
электронейтральности, в каждой единице объема любой
биологической жидкости содержится равное
количество миллиэквивалентов катионов и
анионов. Но в разных компартментах
клетки и различных бассейнах организма это
количество может отличаться, в связи с чем
электролиты определяют ионные градиенты
и ионные токи в живых тканях. Основные
внутриклеточные ионы — натрий и хлорид,
а внутриклеточные — калий и фосфат.
Переход ионов между отсеками клетки и
бассейнами организма тесно связан с транспортом
воды и некоторых осмотически активных
неэлектролитов (глюкоза, мочевина и пр.).
Этот переход и соответствующие
градиенты — объект сложной генетической, нейро-
эндокринной и паракринной регуляции, где
важнейшая роль принадлежит почкам.
Поскольку кислотно-основной баланс
представляет собой динамическое
соотношение катионов водорода и щелочных
346 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
анионов, в этот раздел включено также
обсуждение патофизиологии
кислотно-основного равновесия.
Данная книга не является руководством
по клинической патофизиологии. В
связи с этим частные вопросы нарушения
водно-электролитных показателей при
различных заболеваниях включены в
Практикум и т. III «Механизмы болезней
и синдромов». В этом томе представлены
общие аспекты и освещены типовые
нарушения водно-электролитного равновесия,
а также отдельные примеры из сферы
частной патофизиологии. Обсуждение
тесно переплетённого обмена кальция,
магния, фосфора и фосфатов вынесено в
отдельный раздел, представленный ниже
в главе «Нарушения эндокринной
регуляции».
ВОДНО-СОЛЕВОЙ ОБМЕН,
ЕГО РЕГУЛЯЦИЯ
И НАРУШЕНИЯ
В нескольких нижеследующих разделах
представлены закономерности обмена воды,
натрия и калия, а также механизмы
нарушений этих процессов.
Вода, натрий и регуляция
осмотического гомеостаза
Вода по относительной массе является
основным веществом, из которого состоит
организм человека. В теле новорожденного
ребёнка ее до 80 %, у взрослых мужчин — 60 %,
а у женщин (за счет большего содержания
маловодянистых адипоцитов) — 55 %.
Колебания этого содержания в норме
укладываются в 15 %-ный коридор (М.У. Гроер, 1981).
Вода выполняет в организме огромное
количество функций. Сама живая материя
на Земле имеет свойства, приуроченные к
возможностям и характеристикам ее водной
основы. Потенции воды как растворителя
определили материал, из которого состоит
тело1; её теплоемкость задала потребности
температурной адаптации,
электропроводность водных растворов повлияла на
электрофизиологические свойства клеток,
текучесть — на способы транспорта веществ
в живых системах, возможности выделения
веществ в водной фазе предопределили
характер конечных продуктов метаболизма.
Уникальные свойства воды определяются
дипольным характером ее молекул и
сильной степенью их межмолекулярных
взаимодействий, проявляющихся в образовании
ими бесчисленных водородных связей.
Не только растворение в воде полярных и
нейтральных молекул, но и образование
амфипатическими веществами, типа
полярных липидов, мицелл в водных растворах
являются прямым следствием стремления
диполей воды образовать побольше
водородных связей. Огромное значение имеет
способность воды к диссоциации,
предопределяющая кислотно-основные свойства
водных растворов (см. ниже). Показатель
диссоциации чистой воды — концентрация
в ней водородных катионов, которая
близка к Ю-7. Её отрицательный десятичный
логарифм, то есть рН чистой воды равен
7,0 при температуре 25 °С (А. Ленинджер,
1976). Способность воды к радиолизу и
фотолизу предопределяет наличие в
клетках свободных кислородных радикалов, а
значит — прооксидантных и антиоксидан-
тных систем. Наконец, структурированный
характер воды позволяет некоторым
авторам (А. Бенвенист, 1987) в гипотетическом
порядке ставить вопрос о её
информационной роли в живых системах и о наличии у
водных растворов структурной памяти.
Вся вода организма разделена на
бассейны или сектора. Два главных бассейна — это
внеклеточная жидкость (относительный
объем которой с возрастом растёт) и
внутриклеточная жидкость (чей относительный
1 Хол Клемент в романе «Операция
„Тяготение"» справедливо замечает, что, если бы
растворителем на планете была не вода, а
серная кислота, жизнь развивалась бы не на
углеводородной, а на кремний-органической
основе.
Патофизиология водно-электролитного обмена 347
Таблица 17. Количество и состав жидкости в различных отсеках организма (по БД. Роузу, 1994)
Отсек
Внутрисосудистый
Интерстициальный
Внутриклеточный
Трансцеллюлярные
Желудочный сок
Панкреатический сок
Пот
Объем, л
(на 70 кг массы)
3
11-12
27
1
2-2,5 за сутки
1,5-2 за сутки
0,2 за сутки
Na+ мэкв/л
142
145
12
—
60
130
45
К+ мэкв/л
4,5
4,4
150
—
7
7
5
CI- мэкв/л
104
117
4
—
100
60
58
НСОз мэкв/л
24
27
12
—
0
100
0
РО|~ мэкв/л
2
2,3
40
—
—
—
Таблица 18. Суточный водный баланс взрослого человека
Поступление воды в организм
1,1 -1,4 л — с жидкой пищей, питьём
0,8-1,0 л — с твердой пищей
0,3 л — эндогенная метаболическая вода
—
ИТОГО: 2,2-2,7 л
Выделение воды из организма
1,2-1,5 л — через почки
0,1-0,3 л — с испражнениями
0,4 л — через лёгкие
0,5-0,6 — через кожу
ИТОГО: 2,2-2,7 л
Таблица 19. Резко отрицательный водный баланс при полном сухом голодании
Минимальный расход жидкости
Минимальное к-во мочи — 0,5 л
Минимальная потеря воды легкими и кожей — 0,9 л
ИТОГО: 1,4 л
Максимальная мобилизация эндогенной воды
Метаболическая вода — 0,2 л
Вода из депо — 0,5 л
ИТОГО: 0,7 л
объём с возрастом уменьшается),
разделённые плазматическими мембранами клеток.
В свою очередь внеклеточный бассейн
делится на три отсека:
- Интерстициальную жидкость,
окружающую клетки. Этот отсек, по мнению
М.Горн и соавт. (1999), включает и
лимфу1. Его относительный объем
значительно больше в раннем детстве.
- Внутрисосудиетую жидкость,
относительный объем которой мало меняется
с возрастом, представленную плазмой
крови.
Многие авторы, в том числе А.Г. Гинецинский
(1964) и А. Гайтон (1989), относят лимфу к
внутрисосудистой жидкости из-за ее
уникальной транспортной роли в отношении белков
и липопротеидов, а также функции переноса
лимфоцитов.
- Трансцеллюлярную жидкость (объем,
находящийся в серозных и иных
специализированных полостях тела, а также
в полых органах ЖКТ). Этот сектор у
взрослых относительно больше.
- При патологии часто встречается
своеобразная внутренняя секвестрация трансцел-
люлярной жидкости в обособленный
пул, выключенный из свободного обмена
(«третье пространство»), образуемый
через 1-2 дня после начала болезни. Быстрое
всасывание этой жидкости может
порождать для больного самостоятельную
опасность (Б.И. Мирошников и соавт., 1994).
Такими обособленными секвестратами
могут быть жидкость в ЖКТ при илеусе,
экссудат в серозных полостях и т.д.
Гистогематический барьер отделяет
внутрисосудистую жидкость от интерсти-
циальной, а трансцеллюлярная заключена в
348 ALU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
эпителиальные мембраны. Так как компарт-
менты разграничены барьерами, разделение
воды по секторам обеспечивает разный
состав жидкостей в различных отсеках, что, в
свою очередь, формирует градиенты ионов
и давлений, существенные для реализации
физиологических функций организма.
Табл. 17 иллюстрирует разнообразие
состава жидкости в этих бассейнах.
У здорового взрослого индивида
существует нулевой водный баланс. Суточный
баланс воды в организме в норме
характеризуется следующими цифрами (табл. 18).
Отрицательный водный баланс приводит
к дегидратации.
Она влечет за собой гиповолемию, цир-
куляторную недостаточность и тканевую
гипоксию.
Смерть при полном лишении воды
наступает у взрослого человека за 6-8 дней, так
как ежесуточный дефицит водного баланса
(отрицательный водный баланс) составляет
при этом 0,7 л за счет неизбежных потерь
воды (табл. 19).
Положительный водный баланс приводит
к гипергидратации. Ее конечным
результатом будут:
>- отёки, то есть накопление воды в
межклеточном секторе;
>* водянки, то есть накопление трансцеллю-
лярной воды, в основном — в серозных
полостях;
>- набухание клеток — внутриклеточная
гипергидратация.
При движении воды и растворенных в ней
веществ через клеточные мембраны,
капиллярные стенки и эпителиальные слои большое
значение имеют такие физико-химические
явления, как простая и облегчённая диффузия
по концентрационному градиенту, активный
транспорт против градиента, фильтрация.
Эти процессы кратко охарактеризованы в т. I
данного руководства (с. 216-220). В данном
разделе есть необходимость напомнить
характеристики такого процесса, как осмос.
Осмос — движение воды через
полупроницаемую мембрану из области с более низкой
концентрацией растворённого вещества в
область с более высокой его концентрацией.
С осмосом связаны осмотическое давление -
величина, равная тому гидростатическому
давлению, которое надо приложить, чтобы
прекратить осмотический ток воды, а также
онкотическое давление — фракция
осмотического давления, создаваемая нефильтруемыми
через капиллярную стенку белками.
Осмотические силы привлекают воду
туда, где повышена концентрация
осмотически активных веществ. Например,
выведение с мочой глюкозы, если при
гипергликемии она не реабсорбируется путем
активного транспорта, снижает почечную
реабсорбцию воды {осмотический диурез).
Меру способности раствора создавать
осмотическое давление, действуя на
движение воды, называют осмоляльностью. Она
выражается в миллиосмолях на килограмм
воды (мОсм/кг). Миллиосмоль — тысячная
часть осмоля, который равен осмоляльности
раствора, содержащего в килограмме воды,
число Авогадро частиц (6-Ю23).
Практически удобно измерять осмотическую
активность жидкостей организма в мосм/л.
В этом варианте величина называется ос-
молярностью. Так как биологические
жидкости весьма разведены, то осмоляльность
и осмолярность численно близки и почти
идентичны.
Осмотически активные субстанции
отличаются по молекулярному размеру,
наличию и величине заряда и другим
свойствам. Поэтому некоторые из них свободно
проходят через биологические мембраны.
Такова, например, мочевина.
Другие осмотически активные частицы
(белки, глюкоза, катион натрия) не могут
свободно и быстро преодолевать
плазматические мембраны клеток.
Первые называют неэффективными, а
вторые — эффективными осмотически
активными веществами.
Только эффективные осмотически
активные вещества сильно влияют на
перемещение воды между секторами или её
Патофизиология водно-электролитного обмена 349
удержание в пределах какого-то
из секторов.
Осмоляльность, создаваемая
этими веществами, является
эффективной, так как
обеспечивает движение воды.
Эффективная осмоляльность называется
тоничностью.
Тоничность зависит не только
от концентрации растворённых
веществ, но и от проницаемости
мембран для этих веществ.
Основными осмотически
активными эквивалентами во
внеклеточной жидкости служат
катион натрия и анион хлора,
меньшее значение имеет
бикарбонат-анион. Из
неэлектролитов важны глюкоза и мочевина.
Роль всех остальных
осмотически активных веществ вместе
взятых не покрывает и 3 %
суммарной осмоляльности (рис. 75,
отражающий катионный и анионный состав
внеклеточной жидкости).
Осмолярность внеклеточной жидкости
(плазмы) — Р(мОсм/л) — может быть
приблизительно определена по формуле:
Р(мОсм/л) = 2 (Na+ + К+) +[глюкоза] +
[мочевина] + 0,03 [белок],
где все параметры выражены в мМ/л, а
белок — в г/л.
Непосредственное измерение данной
величины возможно, например, криоскопи-
ческим методом. Реальное измерение даёт
величину большую, чем подсчет, поскольку
в формуле не учтён ряд минорных
осмотически активных компонентов.
Осмоляльность жидкостей тела составляет
приблизительно 292±12 мОсм/кг. Растворы,
имеющие ту же эффективную осмоляльность
или тоничность, называются
изотоническими. Примером служит хорошо всем знакомый
0,9% раствор хлорида натрия.
У гипотонических растворов тоничность
ниже, а у гипертонических — выше
диапазона осмоляльности жидкостей тела.
Н2О в % от веса тела
75
ионный состав
внеклеточной жидкости
1 внутри-
клеточно
35
внекле-
точно
40
в сосудах
60
внутри- I
клеточно
40
внекле-
точно
20 |
в сосудах
участвует
в метаболизме в дыхании
Na+
Ка+
Са++
Mg++
153+
катионы
142
153~ мЭкв/л
анионы
101
24
17
11
с\-
нсоН
Prot~ J
органы*
кислот
вв
[со7]+н2о:
н2со3:
HCOJ"
Prot~
+
+
н+
н+
Рис. 75. Диаграмма содержания воды в организме, ионного
состава внеклеточной жидкости (справа),
функционирования карбонат-бикарбонатного и белкового буферов
(внизу), По Э. Керпель-Фрониусу, 1975. Обозначения:
Prot — белки, ВВ — резервная щелочность, остальные
обозначения — по тексту.
Важнейшая закономерность,
определяющая распределение воды между секторами в
организме, заключается в том, что снижение
тоничности внеклеточной жидкости
приведёт к перемещению воды из экстрацел-
люлярной во внутриклеточную жидкость.
Объём клеток возрастёт. Так происходит,
например, при утолении жажды
дистиллированной водой.
При гипертоничности внеклеточной
жидкости вода переходит из клеток в экстрацел-
люлярный сектор. Объём клеток снижается.
Так происходит, например, при несахарном
диабете или выраженной гипергликемии.
Если развивается гиперосмолярность
какого-то сектора, но без его гипертоничности
(накопление неэффективных осмоэквива-
лентов), то дегидратации секторов не
происходит. Примерами последней ситуации
служат отравление метанолом и уремия.
В норме распределение воды и натрия
между секторами и их количество в
организме регулируются следующим образом.
Переход жидкости и солей между внутри-
сосудистой и интерстициальной жидкостью
350 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
подчиняется старлинговскому равновесию
гидростатических и осмотических сил по
обе стороны капиллярной стенки и зависит
от проницаемости сосудов, определяемой
местными гуморальными регуляторами,
например, аминами, кининами и эйкозано-
идами. Избыток осмотических эквивалентов
и жидкости возвращается в кровь по
лимфатическим сосудам. Локальное нарушение
этих равновесных процессов ведет к
возникновению местного отёка (воспалительного,
а^л§ргидес?кого, гемодинамического либо
лимфодинамического). Эти вопросы
подробно разбираются в т. I (с. 216-220) и здесь
повторно не затронуты.
Переход воды и солей между
внутриклеточным и внеклеточным бассейнами также
подчиняется равновесным закономерностям
по Э. Старлингу. Однако поскольку
плазматические мембраны не выдерживают даже
минимальных гидростатических
градиентов, процесс организован таким образом, что
перемещение зависит практически
исключительно от градиентов осмотических.
Существенные изменения объема клеток
нежелательны для их гомеостаза, а в таких
органах, как головной мозг, могут вызвать
летальные последствия из-за сдавливания
клеток в замкнутой полости. Поэтому объём
тканей поддерживается, по мере
возможности, постоянным, а гидростатические
напряжения на их границах — минимальными
(Т. Андреоли, 1987). Поскольку клеточные
мембраны большинства клеток свободно
проходимы для воды, это достигается при
условии изотоничности цитоплазмы и ин-
терстициальной жидкости. Плазматические
мембраны не являются вовсе
непроницаемыми для натрия и калия. В связи с этим
существует постоянная тенденция к выходу
последнего из клеток и входу в них первого —
по градиентам. К тому же из-за наличия в
клетке относительного избытка
высокомолекулярных, не покидающих её анионов белка
и других органических веществ у мембраны
клетки действует эффект Доннана (см.т.1,
с. 216), который обусловливает некоторое
превышение концентрации катионов внутри
клетки над их уровнем снаружи.
Если бы действие этих пассивных
факторов, перераспределяющих жидкость на
данной границе секторов, не
уравновешивалось, оно привело бы к внутриклеточной
гипергидратации и разрыву клеток.
Поэтому все клетки постоянно
затрачивают от 30 % и более своей свободной энергии
на работу активной транспортной системы,
противостоящей этим изменениям и
выводящей из интрацеллюлярного сектора натрий,
а также возвращающей «вытекший» из нее
калий. При недостаточности K+-Na+-ATOa-
зы наступает набухание клеток, что заметно
уже в ранних стадиях энергодефицита,
вызванного гипоксией (см. т. I, с. 181-183).
Таким образом, мембрана клетки
непроницаема для катионов калия и натрия не
анатомически, а лишь в функциональном
смысле, поскольку утечки
уравновешиваются активным транспортом.
Этот феномен позволяет поддерживать
изоосмолярность интрацеллюлярной и экс-
трацеллюлярной жидкости в норме.
Однако если тоничность экстрацеллю-
лярной жидкости существенно изменится,
это приведет к необходимости включить
дополнительные компенсирующие механизмы
для сохранения осмотического гомеостаза.
При гипотоничности экстрацеллюлярной
жидкости клетки расстаются с частью калия.
Активатором этого процесса выступает
минимальное повышение объёма клеток. Такая
компенсация небезразлична. Потеря калия
клетками изменяет их биоэлектрические
процессы. Эквивалентом этого служит на
уровне целостного организма то состояние
психической загруженности и притупление
реакций ЦНС, которое можно наблюдать у
пациента с «водным отравлением» — то есть
внеклеточной гипотоничностью.
Гиперкалиемия и повышение выхода
калия в почечный фильтрат тоже порождают
при этом определённые побочные эффекты
(см. ниже).
При гипертоничности внеклеточного
раствора наиболее опасно изменение объёма
клеток мозга, смещающее его в черепной
коробке. Чтобы противостоять съеживанию,
клетки, особенно мозговые, генерируют
Патофизиология водно-электролитного обмена 351
дополнительные эффективные
осмотически активные эквиваленты — «идиогенные
осмоли», включая продукты протеолиза,
аминокислоты и, возможно, ещё какие-то,
до сих пор не идентифицированные
точно, соединения. Данный механизм имеет
огромное значение при терапии таких
расстройств, и недоучёт его роли, в частности,
обусловливает большую частоту набухания
и отёка мозга в результате интенсивного
лечения гиперосмолярных ком у
пациентов с сахарным диабетом (см. «Нарушения
сахарной регуляции»).
На уровне целостного организма
осмотический гомеостаз поддерживается с
участием механизмов, отраженных на рис. 76 (см.
также ниже раздел «Нарушения
эндокринной регуляции»).
Сенсорами данной системы служат ос-
морецепторы, реагирующие на уменьшение
объёма клеток, в которых они расположены
(-2 % и ниже), барорецепторы,
откликающиеся на уменьшение эффективного
циркулирующего объёма (от -10 % и ниже), а также
центр жажды в гипоталамусе,
представляющий собой нейроны, чувствительные к
ангиотензину II (в меньшей степени — III)
и атриопептину.
Раздражение этих сенсоров ведет к
выбросу эффектора — вазопрессина (АДГ) гипо-
таламо-гипофизарным нейросекреторным
аппаратом, изменению питьевого поведения
и активации ренин-ангиотензин-альдосте-
роновой системы. Все это вместе вызывает
задержку натрия и воды в соотношении,
корректирующем отклонения от
осмотического равновесия.
В почках действие данного механизма
имеет 2 важных взаимозависимых аспекта.
Один касается разводящего сегмента неф-
рона — толстого восходящего колена петли
Генле. Другой — его концентрирующего
сегмента — собирательных трубочек.
Разводящий мочу сегмент улавливает из
нее большое количество катионов натрия и
переносит в интерстиций почки, который
делается гипертоничным по отношению к
первичной моче.
Но толстое колено петли Генле
практически непроницаемо для воды. Поэтому
моча теряет тоничность, между ней и интер-
стицием создаётся градиент. Если действует
АДГ (см. о механизмах его эффектов — ниже
раздел «Патофизиология гипоталамуса»),
то открывается возможность именно за счёт
этого градиента вызвать максимальный
антидиурез. Если же действие АДГ стремится
к минимуму — система будет приближаться
к максимальному водному диурезу и
достигать предельных разведений дефинитивной
мочи.
Обратные связи в системе
осуществляются атриопептином, который
вырабатывается в сердце и мозге при гиперволемии и
гипертензии и тормозит жажду и продукцию
АДГ, стимулируя натрийурез и диурез. Есть
сведения о наличии уабаиноподобного
стероида подкапсулярного слоя
надпочечников, антагониста альдостерона. Кроме
того, при достижении высокой степени ги-
пертоничности интерстициальной жидкости
мозгового вещества почки ее клетки
вырабатывают PgE2, ингибирующий действие АДГ
в нефроне и продукцию активатора ренин-
ангиотензин-альдостероновой системы.
Возможно, синергично действуют в качестве
понижающих регуляторов задержки натрия
и воды и почечные кинины.
Волюмостатические аспекты этих
компенсаторных реакций предусматривают
также активацию при гиповолемии симпато-
адреналового механизма, который, через
вегетативные нервы почек и гормоны
медуллярной части надпочечников, ограничивает,
за счёт действия на (3-адренорецепторы,
перфузию клубочков и способствует задержке
натрия и воды.
Нарушения
осмотического гомеостаза
Хотя 40 % натрия связано в минеральном
веществе костей, 2-5 % этого электролита,
присутствующие во внеклеточной
жидкости, играют в осмотическом равновесии
ключевую роль. Важно подчеркнуть, что
концентрация натрия в экстрацеллюляр-
плотное
пятно
эфферентная
артериола
почки
входные сигналы:
\NaCl, \К+
\ объема
\давления
\pgE2
клубочек
почек
конвертаза
z. glomerulosa
. обезвоживание
осмолярность
на 2% и более
ангиотензин-It-
J
+" I аминопептидаза
ангиотензин-Ш
ангиотензиназы
продукты протеолиза
t объема
внутрисосудистой
жидкости
на 10% и более
атриопептин
Рис. 76. Основные закономерности регуляции осмотического гомеостаза (справа). Сплошные линии — осморецепторная, штриховые —
барорецепторная регуляция, штрих-пунктирные линии — обратная отрицательная связь. Роль ренин-ангиотензин-альдостероновой
системы (слева). Р — кровяное давление^ — эффективный артериальный объём, остальные обозначения и пояснения — в тексте
Патофизиология водно-электролитного обмена 353
ной жидкости не отражает его количества
в организме, а определяется количеством
экстрацеллюлярной воды (Н.Г. Левински,
1994).
Дефицит и избыток натрия и воды в
организме формально может быть разделён
на 4 категории: первичный избыток натрия,
первичный избыток воды, первичный
натриевый дефицит и первичный водный
дефицит. Практически подобные
изолированные нарушения не типичны, происходят
комбинированные расстройства.
Первичный избыток натрия влечет
вторичный избыток воды и выражается в системных
отёках (внеклеточной гипергидратации).
Первичный дефицит натрия чаще всего
присоединяет дефицит воды, таким образом
развивается внеклеточная дегидратация
(объемный дефицит). Чистый или
преобладающий избыток воды выражается в
синдроме гипонатриемии, а относительная
или абсолютная нехватка воды даёт гипер-
натриемию.
Системные отеки
Системные отеки (см. также т. I, с. 220) — это
проявления внеклеточной гипергидратации,
возникшие вследствие действия общих
для всего организма факторов,
нарушивших интегральные механизмы регуляции
водно-солевого гомеостаза. Такие отёки
обнаруживаются во многих частях
организма (а принципиально их минимальные
проявления могут быть зарегистрированы
достаточно чувствительным способом в
любой точке внеклеточного бассейна!). Они
являются результатом общих соматических
заболеваний. Тотальный тяжелый отёк тела
называется анасаркой.
При любых системных отёках
существуют, независимо от их этиологии, общие
патогенетические механизмы, а именно —
положительные общий водный баланс и
натриевый баланс организма, гиперактивность
или повышенная эффективность действия
ренин-ангиотензин-альдостероновой
системы, частичное или полное исчезновение
нормального присасывающего
отрицательного интерстициального гидростатического
давления тканей (А.Гайтон, 1989, Ю.Бра-
унвальд, 1994).
Данная триада может стать результатом
действия разных первичных нарушений,
затем взаимно провоцирующих друг друга.
В связи с преобладанием в начале
процесса того или иного этиологического фактора
и ключевого патогенетического механизма,
выделяют несколько видов системных
отеков: сердечные, почечные
(преимущественно, нефритические и нефротические),
печёночно-цирротические, голодные,
эндокринные и идиопатические.
>• Сердечные отёки развиваются при
сердечной недостаточности на фоне других
ее проявлений. При застойной сердечной
недостаточности нарушение
систолического опорожнения и/или диастоли-
ческого расслабления миокарда ведут к
накоплению «непрокачанной» крови в
сердце, границы которого расширяются,
а также в венах. Эффективный
артериальный объем снижается, что запускает
стандартную волюмосберегающую и
задерживающую воду, а также натрий-
реакцию по механизмам, отраженным
на рис. 76 выше. В легких случаях это
может стимулировать
сократительную способность сердца, и достигается
компенсация, но по мере отягощения
первичного процесса, вызвавшего
сердечную недостаточность, компенсация
будет ограничивать выведение натрия
и воды и создавать избыточный объем
для насоса, который и так не
справляется с прокачкой крови из вен в артерии.
Активный характер задержки воды и
натрия при сердечных отёках и роль
почек и вегетативной нервной системы
в их развитии доказываются
экспериментами, которые позволяют снижать
тяжесть отёчного синдрома на модели
сердечной недостаточности у крыс путем
денервации почек, без прямого
воздействия на миокард. Роль гиперфункции
ангиотензин-альдостероновой системы
доказывается эффективностью блокато-
ров альдостерона и ингибиторов
продукции ангиотензина в терапии сердечных
354 АЖ Зайчик, Л.П.Чурилов Патохимия j
отёков. Дополнительными факторами
патогенеза сердечных отёков служат
увеличение венозного давления и
ацидоз, которые при застойной сердечной
недостаточности повсеместно облегчают
транссудацию плазмы через
капиллярные стенки. При недостаточности левого
желудочка развивается гемодинамичес-
кий отёк легких (см. Практикум).
Отметим неэффективность
контррегуляции с помощью атриопептина при
сердечной недостаточности. Уровень
продукции гормона повышен, но его действие на
рецепторы в почке и мозге по неизвестным
причинам малоэффективно.
>* Почечные отёки — развиваются как при
остром гломерулонефрите, так и при
нефротическом синдроме,
сопровождающем многие подострые и хронические
заболевания почек (см. т. III и
Практикум).
>* Нефритические отёки возникают при
анурии в связи с нарушением клубочко-
вой фильтрации на почве васкулита
почек, каким является острый диффузный
гломерулонефрит. Не выводятся вода и
натрий, возникает их общий избыток.
Проявляется компенсаторное значение
отёков, выводящих избыточный объём,
опасный для гемодинамики, за пределы
сосудистого русла. Дополнительным
фактором при нефритических отёках
является генерализованное повышение
сосудистой проницаемости, связанное
с тем, что острый гломерулонефрит
сопровождается системным иммуноком-
плексным процессом. Важно отметить,
что продукция ренина у больных
увеличена, а система ренин-ангиотензин-
альдостерон гиперактивна, несмотря
на отсутствие дефицита эффективного
артериального объёма, в силу прямой
стимуляции пораженных клубочков и
юкстагломерулярных структур.
>* Нефротический отек — компонент
нефротического синдрома с его
селективной протеинурией и выраженной
гипопротеинемией. Предполагается, что
idoKpuHHO-метаболические нарушения
решающую роль играют 2 фактора. Во-
первых, при нефротическом синдроме
часто первично уменьшена способность
нефронов экскретировать воду и натрий,
за счет их избыточно эффективной
проксимальной реабсорбции, что вызывается,
возможно, первичным усилением работы
её активного и пассивного механизмов
(И.Г. Каюков, 1997). У таких больных
нет активации ренин-ангиотензин-аль-
достероновой системы, им не помогают
инфузии альбумина и блокаторы альдо-
стерона. Во-вторых, у части больных
определенно играют роль механизмы,
связанные с утратой онкотически активного
плазменного белка. Гипоальбуминемия
ведет к первичному отёку и уменьшению
эффективного артериального объема, а
это вводит в действие ренин-ангиотен-
зин-альдостероновый механизм. У таких
пациентов выраженная гипопротеине-
мия, и имеются признаки вторичного
гиперальдостеронизма (Ю. Браунвальд,
1994). Дополнительно влияют
повышение сосудистой проницаемости на почве
системных иммунопатологических
процессов, понижение функции почек, по
мере развития нефросклероза и другие
факторы. Данные закономерности
охарактеризованы также выше в разделе
«Нарушения композиции белков плазмы
крови».
>* Печёночные отёки включают не только
местный асцит при портальной гипер-
тензии, но и системные проявления при
печёночной недостаточности.
Считается, что расширение артериовенозного
шунтирования при циррозе печени, а
также застой крови в системе воротной
вены могут уменьшать эффективный
артериальный объём и способствовать
развитию основного звена патогенеза
системных отёков — вторичного
гиперальдостеронизма.
Кроме того, имеет значение снижение
инактивации альдостерона больной
печенью, а также уменьшение печёночного
биосинтеза альбумина и других белков,
приводящее к гипопротеинемии и час-
Патофизиология водно-электролитного обмена 355
тичнои утрате онкотического давления
плазмы. Есть данные и о продукции в
печени повышенных количеств анги-
отензиногена при печеночных отёках
(Н.Г. Левински, 1987).
> Голодные отеки развиваются в
результате гипопротеинемии, которая
наступает при ограничении синтеза белка
в голодающем организме и усилении
его катаболизма. Это запускает ту же
последовательность событий, которая
характерна для части нефротических
гипопротеинемических отеков.
Голодание, особенно квашиоркор,
характеризуется вторичным гиперальдостеронизмом,
который усугубляется относительным
увеличением массы надпочечников,
теряющих, в отличие от большинства
других органов, очень незначительный
процент своего веса даже при
длительном эндогенном питании (см. выше
«Голодание»). Голодные отеки
отягощаются у лиц, пьющих подсоленную
воду, чтобы заглушить чувство голода.
Авитаминозы, приводящие по ходу
голодания к сердечной недостаточности
(например, бери-бери у голодающих),
резко усугубляют отёчный синдром (см.
ниже «Патофизиология витаминного
обмена»).
> Эндокринные отёки — системные отеки
в результате первичных заболеваний
каких-либо эндокринных желез. При
трактовке этой сборной группы надо
учесть, что при сахарном диабете I типа
системные отёки могут быть результатом
микроангиопатии и хронической
почечной недостаточности с нефротическим
синдромом, а при сахарном диабете II
типа — результатом атеросклероза и
его сердечно-сосудистых осложнений
(см. выше «Патофизиология сахарного
диабета»). За вычетом этих случаев
системные отеки могут развиваться
при следующих эндокринопатиях: ги-
перкортицизме, гиперальдостеронизме,
гипотирозе и изредка— при синдроме
неадекватно повышенной секреции ва-
зопрессина. Детальная характеристика
механизмов этих явлении дана ниже
в разделе «Нарушения эндокринной
регуляции». Отметим здесь лишь
важность при каждом из этих нарушений
гиперактивности ренин-ангиотензин-
альдостеронового механизма, пусть и
не играющего при многих эндокринных
отёках первичной роли.
>- Идиопатический системный отёк —
синдром, поражающий женщин и
сопровождаемый периодическими проявлениями
отечности и вздутием живота.
Манифестации отеков способствуют жара и
пребывание в вертикальном положении.
Предполагается, что синдром вызван
генерализованным повышением
капиллярной проницаемости и вторичной
активацией ренин-ангиотензин-альдосте-
роновой системы в ответ на уменьшение
эффективного артериального объёма.
У части больных синдром развивается в
ответ на продолжительное применение
диуретиков как форма
гиперкомпенсации, особенно на фоне их внезапной
отмены. К системным отекам может
также приводить назначение эстрогенов
и вазодилататоров.
Внеклеточная изоосмолярная
дегидратация (объемный дефицит)
Комбинированное уменьшение количества
внеклеточной воды и натрия ведет к
синдрому объемного дефицита. При нормальных
механизмах почечной задержки натрия даже
полностью бессолевая диета не ведет к
развитию объёмного дефицита, так как почки
резко ограничат его экскрецию, а запас
натрия в организме достаточен.
Поэтому синдром развивается при потере
натрия и воды помимо почек или при
нарушении почечной способности удерживать
воду и натрий (Н.Г. Левински, 1994).
Экстрареналъные потери объема
внеклеточной жидкости наблюдаются при
патологии ЖКТ (рвота, диарея, отсасывание
содержимого ЖКТ, наличие свищей органов
пищеварительной системы). Это часто
сопровождается гипокалиемией и
нарушениями кислотно-основного баланса (см. ниже).
356 ALU. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения
Другая важная причина, связанная с
потерей жидкости помимо почек, —
секвестрация её в третьем пространстве (см. выше).
Примерами могут служить перитонит,
быстрая эвакуация асцита, кровоизлияния
в трансцеллюлярное пространство.
Поскольку пот содержит натрий,
особенно много — при интенсивном потении и над-
почечниковой недостаточности, профузное
потение, так же как и плазморея через
обожжённую поверхность при ожоговой болезни,
может вызвать транскутанное уменьшение
объёма внеклеточной жидкости.
Потеря внеклеточной жидкости через
почки может происходить при отсутствии
первичного заболевания самих почек. Это
бывает при длительном и интенсивном
применении диуретиков, особенно — салурети-
ков, при гипокортицизме (включая и болезнь
Аддисона, и гипоальдостеронизм — см. ниже
«Нарушения эндокринной регуляции»);
а также при осмотическом диурезе, например
глюкозурическом (см. выше).
Кроме того, острая и хроническая
патология самих почек часто вызывает избыточные
ренальные потери натрия и воды.
Это относится к полиурической фазе
острой почечной недостаточности, когда
фильтрация частично восстановилась, но
первичная моча проходит по нефрону, не
Таблица 20. Типовые нарушения водного баланса и осмотического гомеостаза
(по М. Шёдлиху, 1978; Б.И. Мирошникову и соавт., 1994)
Тип
нарушения
Внутриклеточная
жидкость
Внеклеточная
жидкость
Жажда
Отёки
ЦВД
Нарушения
гемодинамики
ЦНС
Диурез
ЕгНЬ
Белок
Гематокрит
MEV
МСН
Причина
(самая частая
в
реаниматологии)
Основная
опасность
Гипоос-
молярная
дегидратация
Ocm-N
К-во-ft
Вода +
0см4
К-во4
Вода-
—
—
U
+
U
U
ft
ft
ft
ft
u
Малое
поступление Na+;
осмодиурез
Гиповолемия
Изоосмо-
лярная
дегидратация
Ocm-N
К-во-N
BoflaN
Осм-N
К-во-U
BoflaN
+
—
V
+
u
u
ft
N
ft
N
N
Секвестрация, потери
изЖКТ
Гиповолемия
Гипер-
осмолярная
дегидратация
Осм-N
К-во4
Вода-
Осм-ft
К-во4
Вода +
+
—
N
"
ft
U
ft
ft
ftN
u
ft
Малое
поступление
воды
Тканевая
гипоксия
Гипоос-
молярная
гипергидратация
Ocm-N
К-во-ft
Вода +
0см4
К-во-ft
Вода-
—
+
ft
+
м
U
U
U
N
ft
U
Избыток АДГ
Отек мозга
Изоосмо-
лярная
гипергидратация
Ocm-N
К-во-N
BoflaN
Осм-N
К-во-ft
ВодаЫ
—
+
ft
+
U
N
u
u
u
N
N
Избыточная
инфузия, оли-
гоанурия
Сердечная
недостаточность; отёк
лёгких
Гипер-
осмолярная
гипергидратация
Ocm-N
К-во4
Вода -
Осм-ft
К-во-ft
Вода +
+
+
ft
+
N
и
V
U
V
U
ft
Гипертонические
инфузии
Отёк легких
Патофизиология водно-электролитного обмена 357
восстановившему натриевый градиент,
который обеспечивает её концентрацию.
При хронической почечной
недостаточности из-за снижения количества
функционирующих нефронов развивается
неспособность ограничивать экскрецию натрия
и воды при их недостаточном поступлении.
Если такие пациенты ограничивают себя в
питье и соли, у них постепенно
уменьшается объём внеклеточной жидкости (см.
Практикум).
При диурезе после длительной
обструкции мочевыводящих путей, например после
прекращения почечной колики, также
развивается экстренное выведение большого
количества натрия и воды.
Независимо от причин синдром потери
внеклеточной жидкости проявляется
снижением тургора кожи и мягких тканей,
сухостью слизистых, гипотензией, повышением
гематокрита и гиперпротеинемией,
явлениями гемоконцентрации (табл. 20, ниже).
Патофизиология гипонатриемии
и гипернатриемии
Разведение внеклеточной жидкости
абсолютным или относительным, по сравнению
с дополнительным количеством соли,
избытком воды приводит к гипотоничности
внеклеточного сектора, переходу воды в
клетки, тенденции к их набуханию,
компенсаторной экскреции из клеток калия и
гипонатриемии.
В норме гипоосмоляльность
внеклеточной жидкости Должна вызывать разведение
мочи, водный диурез и компенсироваться.
Это требует подавления секреции АДГ,
достаточного притока натрия и воды в
толстое колено петли Генле и в собирательные
трубки и, наконец, хорошей функции этих
отделов нефрона.
Конечно, при быстром приеме очень
большого количества воды, не содержащей
достаточно натрия, например при первичной
психогенной полидипсии или ее
разновидности — маниакальном пристрастии к пиву,
пациент может исчерпать способность почек
к дилюции мочи и испытать истинное
«водное отравление» — с головной болью, вкусом
медной пуговицы во рту, распуханием
языка, психоэмоциональной «загруженностью»
и заторможенными реакциями и т.д.
Но гораздо чаще гипонатриемия бывает
результатом невыполнения условий
компенсации, перечисленных выше, и почки не
реализуют свою максимальную способность
разводить мочу.
Соответственно трём условиям
компенсации гипонатриемия развивается вследствие
действия трёх групп причин.
>* При неадекватной гиперсекреции АДГ
(синдром Пархона — см. подробную
характеристику ниже в разделе
«Нарушения эндокринной регуляции»).
>- При недостаточной доставке натрия
и воды в дистальный сегмент нефрона.
Это происходит при снижении скорости
клубочковой фильтрации и/или при
усилении проксимальной канальцевой
реабсорбции. В обоих случаях, даже при
отсутствии действия АДГ, в силу
минимальной спонтанной утечки воды из
просвета нефрона в собирательных трубках,
разведение мочи будет прогрессивно
ограничиваться, вплоть до выведения мочи,
гипертоничной по отношению к плазме.
Следовательно, внеклеточная жидкость
будет становиться гипотоничной, и
разовьётся гипонатриемия. Эта ситуация
может формироваться и при первичном
синдроме объемного дефицита, когда
доставка натрия и воды в дистальную часть
нефрона компенсаторно ограничивается
при уменьшении фильтрации. Как это ни
парадоксально, но гипонатриемию может
развить при действии данного механизма
даже пациент с системными отеками и
абсолютным избытком натрия и воды в
экстрацеллюлярном секторе. Ведь при
системных отёках снижен эффективный
артериальный объем, способствующий
доставке натрия и воды в дистальную
часть нефронов.
>* Местные нарушения транспорта натрия
петлёй Генле и воды — собирательными
трубками — даже при нормальной фун-
358 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
кции АДГ и доставке осмотических
эквивалентов в дистальную часть нефронов
также обусловливают гипонатриемию.
Более подробно механизмы различных
ренальных расстройств, приводящих
к гипонатриемии, будут обсуждаться в
т. III и Практикуме.
Гипернатриемия ведет к гипертоничности
внеклеточной жидкости. Она развивается
при повышении концентрации натрия во
внеклеточном секторе и сопровождается
выходом воды из клеток, тенденцией к
развитию внутриклеточной дегидратации и
внеклеточной гипергидратации, жаждой и
рядом других компенсаторных явлений (см.
табл. 20). При повышении осмоляльности
экстрацеллюлярной жидкости до 320-330
мОсм/кг наступают явные клинические
симптомы со стороны ЦНС, связанные с
обезвоживанием клеток мозга. При
осмоляльности 360-380 мОсм/кг развивается
кома (см. гл. 9).
Существуют 4 основные этиологические
группы факторов, вызывающих гиперна-
триемию.
- Неадетатныйприёмводъшневозможность
удовлетворить жажду. Это происходит у
пациентов, пребывающих без сознания, в
коме или в беспомощном состоянии. Сюда
же относится «синдром путника в
пустыне или в океане», когда в экстремальных
ситуациях люди, находящиеся в полном
сознании и активные, лишены доступа к
пресной воде. Существует и «эссещиалъ-
ная гипернатриемия», представляющая
собой пониженную чувствительность
осморецепторов и/или центра жажды,
иногда сопряженная с выраженным
притуплением способности ощущать жажду,
то есть воспринимать гиперосмолярность
внеклеточной жидкости.
- Осмотический диурез с потерей воды
без натрия, за счет иных эквивалентов,
обеспечивающих тоничность мочи (при
кетонурии, глюкозурии, применении
маннитола). Механизмы этого явления
обсуждались выше в разделе
«Патофизиология сахарного диабета».
- Избыточная потеря воды без натрия
почками (при гипофизарном и нефроген-
ном несахарном диабете — см. ниже
«Нарушения эндокринной регуляции»)
или экстраренальным путём (профузное
потение при низкой тоничности пота,
имеющее ключевое значение при
тепловом ударе). Последний механизм
наблюдается при смешанном и экзогенном
перегревании, в частности при тяжелой
физической нагрузке в условиях
высокой температуры и влажности воздуха
(см. т. I, с. 378).
- Комбинированные причины могут
вызывать гипернатриемию. Например, при
назначении глюкозы через зонд
больному с инсультом, который не может
сообщить о том, что испытывает жажду,
будут действовать совместно первый и
второй механизмы.
В заключение данного раздела приводим
таблицу, отражающую типы нарушений
водного баланса и осмотического гомеостаза, с
указанием их основных клинико-лаборатор-
ных признаков. Дегидратация и
гипергидратация разделены на три подвида каждая,
в соответствии с состоянием тоничности
(эффективной осмоляльности)
внеклеточной жидкости (см. табл. 20).
Помимо нарушений баланса ионов натрия,
бикарбоната и хлора, тесно связанных с
осмотическим гомеостазом и кислотно-щелочным
равновесием и рассматриваемых поэтому в
соответствующих разделах, существуют
клинически значимые отклонения в
концентрации других электролитов — калия, кальция,
магния и фосфата. Регуляция обмена трёх
последних ионов тесно взаимосвязана и
обсуждается ниже в разделе «Патофизиология
околощитовидных желёз...»
Ниже представлена характеристика
калиевого баланса и его нарушений.
ГОМЕОСТАЗ КАЛИЯ
И ЕГО НАРУШЕНИЯ
Калий — главный внутриклеточный
катион. На 98 % он находится в клетках, где его
Патофизиология водно-электролитного обмена 359
концентрация в 40 раз выше внеклеточной.
Ион удерживается там калий-натриевым
насосом, при участии ионов магния. Так
как калия очень мало снаружи, даже
минимальные колебания его внеклеточной
концентрации могут оказать выраженные
биологические эффекты — совершенно так
же, как это происходит в отношении
внутриклеточного кальция (см. ниже). Калий
играет гигантскую роль в осуществлении
биоэлектрической активности клеток и
поддержании нервно-мышечной
возбудимости и проводимости. Если нет изменений
кислотно-основного баланса, внеклеточная
и внутриклеточная концентрации калия
изменяются однонаправленно, в противном
случае возможны их разнонаправленные
изменения. Ацидоз способствует обмену
внутриклеточного калия на водородные
катионы и гиперкалиемии, а алкалоз,
наоборот, благоприятствует гипокалиемии, так
как водородные катионы покидают клетки,
в обмен на калий (см. ниже).
Инсулин и р-адренэргические влияния
способствуют переходу калия в клетки,
а-адреномиметики — обратному току
калия. Минералокортикоиды (как и большие
количества глюкокортикоидов) усиливают
экскрецию калия почками. Они влияют и
на распределение калия между
внутриклеточной и внеклеточной жидкостью, ускоряя
его переход в клетки. При гиперкалиемии,
а также в ответ на ангиотензин II и АКТГ
клубочковая зона коры надпочечников
усиливает секрецию альдостерона (см. ниже
«Нарушения эндокринной регуляции»).
Избыток калия, кроме того, стимулирует
его секрецию в нефронах. Эти механизмы
лежат в основе калиевого баланса. Однако
почка не может контролировать выведение
калия столь жёстко, как экскрецию натрия,
водорода и бикарбоната, поэтому потери
калия чаще всего продолжают происходить
даже при снижении его концентрации в
плазме. Содержание калия в плазме,
вообще говоря, не отражает его общих запасов
в организме. Однако запасы эти гораздо
меньше натриевых, поэтому бескалиевая
диета может повлиять на уровень калия в
плазме. Плазменная концентрация калия
составляет в норме довольно узкий диапазон
3,5-5 мэкв/л.
Поступление калия в организм зависит от
состава диеты. Богаты калием мясо, фрукты
и овощи. Особенно его много в абрикосах, в
частности, получаемых из них кураге (1717
мг/100г), урюке (890 мг/100г), а также
в изюме (774 мг/100г), черносливе (648
мг/100г), бобовых (около 1000 мг/100г)
артишоках, бананах, дыне, картофеле.
Последний, из-за большого абсолютного
удельного веса в суточном рационе и немалого
содержания калия (426 мг/100 г), служит
основным источником этого иона, которого
человеку нужно 2-4 г в сутки.
В калиевом балансе больных следует
учитывать содержание калия в некоторых
лекарствах, например пенициллиновых
антибиотиках.
Выведение калия идёт не только через
почки, но и с потом, а также через ЖКТ.
При понижении уровня калия в плазме
ниже 3,5 мэкв/л развивается гипокалиемия.
Этому способствует отрицательный
калиевый баланс. Этиология гипокалиемии
разнообразна и включает:
- Бескалиевую диету.
- Усиленную экскрецию калия почками,
не восполняемую поступлением
калия — при выраженной полиурии любого
генеза, синдроме Лиддля —
наследственной недостаточности реабсорбции калия,
при почечном канальцевом ацидозе I и
II типов (см. ниже), действии на почки
некоторых антибиотиков.
- Потерю богатого калием содержимого
ЖКТ, особенно желудочного сока.
- Гиперальдостеронизм и выраженный
гиперкортицизм.
- Разбавление экстрацеллюлярной
жидкости при инфузии растворами,
содержащими мало калия.
- Массированный переход калия в клетки
(действие инсулина и адреналина,
семейный периодический гипокалиемический
паралич, алкалоз).
360 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
Отрицательный азотистый баланс и гипо-
магниемия способствуют гипокалиемии.
Основные проявления гипокалиемии
связаны с нарушением электрических свойств
мембран возбудимых тканей.
Возникают усталость, депрессия и
безразличие к окружающему, мышечная слабость
и гипотонус мышц, спазмы мышц нижних
конечностей, гипорефлексия и адинамия,
тошнота, рвота, атония кишечника, запор,
парестезии, повышается чувствительность
сердца к наперстянке, может быть сердечная
недостаточность.
На ЭКГ отмечается характерная картина:
снижение сегмента ST, уплощение зубца Т,
появление зубца U, широкой T-U волны.
Развиваются желудочковые тахиаритмии,
в тяжелых случаях — вплоть до трепетания
и мерцания. Это часто сопровождается
признаками метаболического алкалоза (см.
ниже).
Гиперкалиемия развивается при
повышении содержания калия во внеклеточной
жидкости до 5,5 мэкв/л*и выше.
Ее причины могут быть следующими:
- увеличение поступления калия в
организм, выше возможностей экскреции;
Это бывает, например, при
однообразной картофельной диете, при инфузии
избытка калийсодержащих препаратов;
- ограничение выведения калия почками
(острая почечная недостаточность в
олигоанурическую фазу, тяжелая
хроническая почечная недостаточность и
некоторые тубулопатии, гипоальдостеронизм
и гипокортицизм, действие спиронолак-
тона и некоторых других диуретиков);
- массированный выход калия из клеток
(цитолиз, гемолиз, синдром
длительного сдавливания, тромбофилитический
синдром и ДВС-синдром, внутренние
кровотечения, инсулинодефицитные
состояния, гиперкалиемический
периодический паралич, интоксикация
блокаторами натрий-калиевого насоса,
в частности уабаином и сердечными
гликозидами).
Гиперкалиемии способствует гиперосмо-
ляльность внеклеточной жидкости, а также
ацидоз.
Проявления гиперкалиемии связаны с
ролью калия в биоэлектрических процессах.
В целом её клинические симптомы менее
характерны, чем манифестация
гипокалиемии, а изменения на ЭКГ весьма патогно-
моничны.
Могут быть парестезии,
раздражительность, беспокойство, спазмы и колики в
животе, диарея, слабость в ногах, парезы
мышц. Отмечаются брадиаритмии. В
тяжелых случаях возможна остановка сердца.
Изменений на ЭКГ очень типичны. Зубец Т
становится высоким и узким, заострённым.
Интервал PQ увеличивается, ST
претерпевает депрессию, QRS расширяется, Р может
отсутствовать. Вместе с тем в отличие от
других расстройств, которые на ЭКГ могут
сопровождаться пикообразным высоким
зубцом Т, при гиперкалиемии интервал QT
не удлинён.
Экспериментальная модель
гиперкалиемии создаётся у животных путем ад-
реналэктомии и перевода на питьё воды с
добавкой хлорида натрия.
В связи с тем, что различные катионы и
анионы представляют кислые и щелочные
эквиваленты или обмениваются с протонами
и бикарбонатом, нарушения электролитного
баланса неразрывно связаны с
патофизиологией кислотно-щелочного равновесия.
ПАТОФИЗИОЛОГИЯ
кислотно-основного
РАВНОВЕСИЯ
Кислотно-основное (кислотно-щелочное)
равновесие или баланс (КЩР) в организме
прецизионно регулируется, а его
нарушения порой обусловливают возникновение
тяжёлой патологии. Прежде чем изложить
основные механизмы этих нарушений и
закономерности их компенсации, необходимо
ввести основные понятия, используемые в
этой сфере патохимии.
Патофизиология водно-электролитного обмена 361
Основные понятия, связанные
с регуляцией КЩР
и ее нарушениями
Кислота — это любое вещество, которое
может отдавать протон (Н+) при химических
реакциях и диссоциации, а щелочь — это
вещество, которое может связываться с
протоном (определение Л. Брёнстеда-Т. Ло-
ури)1. Кислоты постоянно возникают в
ходе метаболизма. Каждый день тело
человека продуцирует 20 М угольной кислоты и
80мМ нелетучих кислот. Это эквивалентно
добавке в жидкую среду организма
ежесуточно более чем 1 л концентрированной
серной кислоты.
Тем не менее организм поддерживает
внеклеточную концентрацию Н+ в очень
узком пределе — 40 ± 5 нМ/л. Столь узкий
диапазон имеет принципиальное
значение, так как большинство биохимических
жизненно важных процессов требует для
своего оптимального течения именно этих
условий.
К. Сёренсон (1909) предложил
применять отрицательные десятичные логарифмы
концентраций ионов водорода, выраженных
в грамм-ионах на литр. Этот показатель
называется рН, по первым буквам латинских
слов potentia hydrogeni — «сила водорода».
Диапазон нормального рН плазмы
крови — от 7, 35 до 7,45. Внутриклеточный рН
измерить сложнее, но в большинстве клеток
он слабокислый, так рН саркоплазмы мышц
составляет в покое около 6,9, а внутри
эритроцитов он равен 7,19±0,02. В отдельных
внутриклеточных компартментах рН
сильно варьирует — например, в работающей
лизосоме он около 2,0, а межмембранное
пространство митохондрий вообще самое
«кислое» место в клетке, где постоянно
возобновляется необходимый для запасания
1 В теоретической химии применяется более
строгое определение Т. Льюиса, по которому
кислотой является любой потенциальный
акцептор электронной пары, а основанием — любой
потенциальный её донор. В этом определении Н2
фигурирует как основание, а главной кислотой
организма будет молекулярный кислород.
энергии протоновый градиент. Клетки
простаты имеют самый низкий рН цитоплазмы
(менее 4,9), а в остеобластах рН достигает
8,5.
Кажется, что уровень самого показателя
рН в норме колеблется незначительно, но
это соответствует изменениям
концентрации катионов водорода в пределах 12% от
медианы, что не так уж мало. Более
значительные изменения рН крови в сторону
повышения или понижения концентрации
протонов связаны с патологическими
процессами. Колебания рН внеклеточной
жидкости от 6,8 до 7,8 вызывают глубокие
нарушения жизнедеятельности, но еще
совместимы с жизнью. За этими пределами
наступает смерть.
Любую образуемую при обмене веществ
кислоту можно рассматривать как
находящуюся в равновесии между ее
диссоциированной — (А-) и недиссоциированной
формами — (НА):
Н+ + А<-»НА.
Отношение концентрации свободного
протона, свободного аниона и связанной
пары протон-анион можно выразить как
константу диссоциации (Ка),
соответствующую средней точке титрования конкретного
буфера щелочью или кислотой:
Ка-[Н+][А-]:[НА],
рассчитывая уравнение для [Н+],
получаем:
[Н+] = Ка[НА]:[А].
Используя отрицательный десятичный
логарифм для каждой стороны уравнения,
имеем:
-lg[H+] = -lgKa-lg[HA]:[A-].
Отрицательный десятичный логарифм
концентрации протонов — (-lg [H+]) — это
и есть рН, a (-lg Ка) можно обозначить как
рКа — константу диссоциации.
Сменив знаки, получаем следующее
уравнение:
pH = pKa + lg[A]:[HA].
362 А.Ш. Зайчик, Л.П.Чурилов Патохимия
Это соотношение известно как уравнение
Хендерсона-Хассельбалха.
Оно позволяет:
1 — рассчитать рН кислотно-основной
системы по данным молярного отношения
кислоты и щелочи,
2 — вычислить величины константы
диссоциации (Ка),
3 — определить молярное отношение
кислоты и щелочи, когда известны значения
рН и рКа.
Поддержание этих констант (рН и рКа)
осуществляется автоматическими
механизмами связывания кислых или щелочных
эквивалентов во всех метаболических процессах
организма. Наиболее значимы и достаточно
хорошо известны буферные, дыхательные и
почечные механизмы поддержания кислотно-
основного равновесия. Прежде чем перейти к
их характеристикам, необходимо оговорить
некоторые ключевые понятия.
При нарушении КЩР развиваются
ацидозы и алкалозы.
Алкалоз — аномальное накопление
бикарбонат-аниона в организме или потеря
кислот.
Ацидоз — аномальное накопление кислот
в организме или потеря оснований.
Метаболический ацидоз (алкалоз) —
нарушение КЩР вследствие первичных
изменений уровня бикарбонат-аниона.
Дыхательный ацидоз (алкалоз) —
нарушение КЩР из-за первичных изменений
уровня парциального напряжения С02 в
артериальной крови.
Ацидемия — снижение рН артериальной
крови ниже 7,40.
Алкалемия — повышение рН
артериальной крови выше 7,40.
Введение двух последних терминов
призвано отразить тот факт, что сама по себе
величина рН артериальной крови даёт лишь
качественную, но не количественную оценку
сдвига КЩР, а нарушения
кислотно-основного баланса компенсируются с участием
не только плазмы, но и внутриклеточной
жидкости (Т. Андреоли, 1987; М.Горн и
соавт., 1999).
чдокринно-метаболические нарушения
Декомпенсированные ацидоз и алкалоз -
выход рН артериальной крови за границы
нормы (7,35-7,45) в соответствующую
сторону, при истощении буферной емкости
системы и неэффективной компенсации.
Коррекция КЩР — приспособительная
реакция со стороны органа, нарушение
функции которого вызвало первичное смещение
КЩР (например, лёгких — при дыхательных
ацидозе и алкалозе).
Компенсация КЩР — приспособительная
реакция со стороны органа, «неповинного»
в первичном смещении КЩР (например,
лёгких при метаболическом ацидозе и
алкалозе).
Ниже рассматриваются основные
закономерности срочной и долгосрочной
регуляции внеклеточного рН.
Буферные системы организма
Солевые и органические компоненты
биологических жидкостей поглощают и
освобождают протоны для стабилизации рН.
Но если добавлять кислоту или щелочь in
vitro в сосуд, где находится 5,5 л донорской
крови, то её способность поддерживать рН
в нормальном диапазоне очень скоро
исчерпается. Однако in vivo реактивация системы
обеспечивается почками и легкими.
Регуляторами, непосредственно
обеспечивающими быструю неполную
компенсацию сдвигов рН крови, являются буферные
системы жидких сред организма, а именно
крови и тканей. Деятельность лёгких,
почек, печени и кожи компенсирует сдвиги
рН более медленно, но зато полностью.
Буферы реагируют на изменения рН
немедленно, в течение 1 секунды. Буфер — это
раствор, содержащий слабую кислоту и ее
соль, образованную сильным основанием
(сопряженная кислотно-основная пара по
А. Ленинджеру, 1976). Такая система
стремится противостоять изменению рН после
добавления либо кислоты, либо щелочи.
Существует множество потенциальных
буферов, которые присутствуют как внутри,
так и вне клеток организма человека, во всех
его жидкостях.
Патофизиология водно-электролитного обмена 363
Основным и наиболее лабильным
внеклеточным буфером служит карбонат-би-
карбонатный.
Он состоит из угольной кислоты и
бикарбоната (гидрокарбоната) натрия, а его
константой является соотношение концентрации
кислоты и ее кислой соли:
[H] = K{H2C03/NaHC03}.
В норме оно составляет 1/20, при
константе ассоциации бикарбонатного буфера — 6,1.
Примерно 40 % всех высвобождаемых ионов
водорода нейтрализует внеклеточная
буферная система угольной кислоты и
гидрокарбоната натрия.
С учетом конкретных характеристик
данного буфера в норме уравнение Хен-
дерсона-Хассельбалха выглядит как
функциональная взаимосвязь
концентрации протонов во внеклеточной жидкости,
содержания в ней бикарбонат-аниона и
парциального напряжения углекислого газа
артериальной крови.
Концентрация С02, растворенного в крови, в
800 раз больше, чем концентрация НС03
(диссоциированной формы угольной кислоты).
Поэтому принято считать, что С02 находится в
динамическом равновесии с Нf. Концентрацию
Н2С03, Н+ и НСОз можно описать уравнением
Хендерсона-Хассельбалха, характерным для
главной буферной системы:
рН = рК + log [НС03]:[Н2С03].
В норме можно утверждать, что
количество Н+ прямо пропорционально зависит от
парциального давления С02:
рН = рК + log[HC03"]:a x рС02,
где a — константа растворимости С02 в воде
(0,03), а рС02 = 40 мм Hg. Заменяя члены
уравнения на реальные показатели,
характерные для здоровых людей, и зная, что
концентрация бикарбоната в плазме крови
довольно велика и составляет 23,9 мМ/л,
получаем соотношение, объясняющее
почему нормальный уровень рН внеклеточной
жидкости составляет именно 7,4:
рН = 6,1+log [23,9:1,2] = 7,4.
Для практических целей это соотношение
удобнее выразить как уравнение
действующих масс:
[Н+] (нМ/л) = 23,9хрС02 (мм Hg):
:[НС03] (мМ/л).
В таком виде оно пригодно для расчетов,
поскольку [НС03~ ] И рС02 количественно
измеряют в обычной клинико-лабораторной
практике. Отношения между [Н+] и рН в
физиологическом диапазоне отражены в
таблице (например, рН = 7,0 соответствует
концентрации ионов водорода в 100 мМ/л).
Закономерности функционирования
данного буфера приводились выше на рис. 75
(с. 349).
Фосфатный буфер, кислым и основным
компонентами которого служат,
соответственно, однозамещённый и двузамещённый
фосфаты натрия, облегчает экскрецию
протонов в почечных канальцах. Это важный
внутриклеточный буфер. Его константа
определяется соотношением:
[Н] = К {NaH2P04 /Na2HP04}.
Во внеклеточной жидкости соотношение
компонентов этого буфера составляет 1: 4,
а в моче, при рН 5,4, оно может достигать
25:1. Значение Ка для этого буфера равно
6,8. Именно за счёт деятельности данного
буфера обеспечивается большая часть
титруемой кислотности мочи. Фосфатный
буфер позволяет экскретировать кислые
эквиваленты в почках без значительной
потери бикарбонат-аниона.
Наибольшей общей емкостью, в силу
изобилия своих компонентов, обладает
белковый, и особенно — его разновидность — ге-
моглобиновый буфер (рис. 75):
[Н] = К {H-protein/Na-Protein}
[Н] = К {НЬ02/НЬ}.
Эти буферы — преимущественно
внутриклеточные, основанные на амфотер-
ности белков и обратимости окисления
гемоглобина. Константа Ка белкового
364 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения
буфера — 7,4. Белковая буферная
активность присутствует и в плазме. Кроме того,
внутриклеточные буферы могут принимать
участие в нейтрализации Н+,
образованного вне клеток, например, в силу явления
трансминерализации, когда при закислении
экстрацеллюлярной жидкости К+ выходит
из клеток в обмен на Н+ и Na+. До 60%
избытка кислых эквивалентов связывают
внутриклеточные буферные системы.
Проникающий в эритроциты С02 связывается с
гемоглобином, что восстанавливает
доступность бикарбоната:
ННв+ + НС03- <-> Нв° + Н2С03 <г>
<-» Н20 + С02.
Фактически и продукт дезаминирования
аминокислот и аминов — аммиак можно
рассматривать как важный компонент
системы NH4+/NH3. Дело в том, что аммиак,
выделяемый в ответ на ацидоз клетками
почек, связывает ион Н+. Катион аммония
отсутствует в клубочковом фильтрате, но
его кислые эквиваленты могут добавляться
к титруемой кислотности мочи,
обеспеченной кислыми фосфатами.
Итак, после взаимодействия протона с
буферной системой он оказывается в
состоянии связывания или нейтрализации с
последующим выведением его из
организма. Таким образом, компоненты буферных
систем расходуются, а стало быть, требуют
восполнения своего состава. Следовательно,
буферы — только первая линия защиты
постоянства рН внутренней среды организма.
Собственное их действие может удерживать
рН внеклеточной жидкости в биологически
допустимых пределах, несмотря на добавку
до 16 мэкв кислоты или до 29 мэкв щелочи
на литр плазмы.
Следует помнить, что в ответ на любой
сдвиг КЩР реагируют сразу все буферы,
а не один, причём по принципу изогидрии
это происходит одномоментно, с
изменением баланса компонентов каждого из них
(А. Гайтон, 1989). Восполнение состава
буферных систем требует участия легких,
почек и других органов и тканей.
Летучие кислые эквиваленты
и роль легких в КЩР
Количество и типы кислот, которые
непрерывно образуются в процессе
жизнедеятельности организма, различаются. Так,
в результате окисления субстратов в сутки
образуется от 10000 до 22000 миллимолей
С02. Углекислый газ — летучий эквивалент
угольной кислоты, что может быть
выражено равновесным уравнением карбонат-би-
карбонатного буфера:
Н+ + НС03- = Н2С03 - Н20 + С02.
Диоксид углерода — единственный
прямой гуморальный стимулятор
дыхательного центра, хорошо растворим и в воде, и
в липидах. Он проникает из митохондрий
в цитоплазму, а из неё — во внеклеточную
жидкость. Поскольку рС02в капиллярах
меньше, газ свободно проходит в плазму.
В плазме крови нет карбоангидразы и
С02 не образует Н2С03, а связывается с
плазматическими белками и формирует
соединения типа карбаминовой кислоты
(Я. Мусил и соавт., 1981). Протоны,
образующиеся при их диссоциации, связываются
фосфатом. Частично, С02 проникает в
эритроциты и там гидратируется карбо-
ангидразой. Карбоангидраза эритроцитов
превращает значительную часть угольной
кислоты в НС03_, который быстро
переносится из эритроцитов в плазму крови,
в обмен на хлорид-анион. Это явление
известно как феномен Хамбургера. В
тканях эритроциты, наоборот, отдают хлорид
в обмен на бикарбонат. Анионообменный
белок, ответственный за противоперенос
хлорида и бикарбоната, участвует не только
в транспорте С02 в лёгкие, но и в
поддержании КЩР почками, а также другими
клетками. Его биохимическое
наименование — белок 3-й полосы. Интересно, что
именно при возрастной деградации этого
белка образуется неорганоспецифический
антиген стареющих клеток, по которому
иммунная система распознаёт и подвергает
аутофагоцитозу состарившиеся клеточные
элементы (см. т. I, с. 150-152).
Патофизиология водно-электролитного обмена 365
Небольшая часть С02 в красных
кровяных клетках обратимо связывается
гемоглобином в карбаминогемоглобин, без
помощи энзимов. Протоны, полученные в
эритроците от диссоциации угольной
кислоты и карбаминогемоглобина, нейтрализуют
фосфат-анионы и гемоглобиновый буфер.
В связи с наличием всех этих
приспособлений транспорт С02 из клеток к альвеолам
не вызывает по пути существенных
изменений КЩР в экстрацеллюлярной жидкости.
Гиперкапния стимулирует, а гипокапния
тормозит вентиляцию. Газ выводится из
организма лёгкими, причём в естественных
условиях, у здорового человека на уровне
моря метаболическая продукция и
выведение С02 уравновешиваются при
концентрации углекислого газа в плазме 1,2 мМ/л, что
соответствует парциальному давлению
этого газа в 40 мм Hg. Контроль за основными
газовыми показателями осуществляется и
рефлекторно, через афферентные сигналы
с хеморецепторов чувствительных к С02.
Расположены хеморецепторы диффузно
по сосудам всего организма и
сконцентрированы в продолговатом мозге,
аортальном и каротидных синусах. По мнению Т.
Андреоли (1987), наибольшее значение
имеет чувствительнсть хеморецепторов
ЦНС к рН (рС02) цереброспинальной
жидкости. В связи с тем, что колебания рН
в цереброспинальной жидкости обычно
отстают от колебаний этого показателя
в плазме крови по амплитуде и по фазе,
легочная компенсация при острых
метаболических ацидозах и алкалозах запаздывает
с началом на 1-Зч.
Здоровые легкие могут нивелировать на
50-70% нарушение кислотно-основного
баланса за счет респираторной активности.
Повышение концентрации Н+ выше верхней
границы нормы приводит к увеличению
вентиляции легких в 4-5 раз, в то время как её
понижение уменьшает вентиляцию легких
на 50-75%.
Легочные механизмы поддержания
кислотно-щелочного равновесия
высокоэффективны. Лёгкие выступают эффектором
бикарбонатного буфера, сдвигая его
равновесие вправо. С учетом этого пределы
доступной регуляции расширяются до
возможности удержать рН плазмы в
совместимых с жизнью границах при добавке
до 23 мэкв кислоты или до 80 мэкв щелочи
на литр плазмы.
>- Подсчитано, что при экстремальных
физических нагрузках количество угольной
кислоты может возрастать в 20 раз. Но
и при этом регуляция выделения С02
достигается изменениями скорости и
объема вентиляции легких. Высокая
эффективность удаления кислоты из
организма объясняется высокой
способностью углекислоты к диффузии,
растворению в воде и практически
абсолютной способностью её к диссоциации
а также упомянутыми значительными
резервными возможностями легких.
Срыв функций легких в поддержании
КЩР ведет к респираторным нарушениям
постоянства рН, первичными изменениями
при которых служат те или иные
отклонения парциального напряжения углекислого
газа.
Нелетучие метаболические
кислоты и роль почек
в КЩР
Количество вырабатываемых в
организме нелетучих кислот составляет около
1 ммоль/кг массы тела в сутки. Нелетучие
кислые эквиваленты, при достаточном
уровне аэробного окисления углеводов и
белков, главным образом возникают путём
деградации серосодержащих аминокислот
метионина и цистеина, в форме серной
кислоты. Поэтому именно белковая пища
увеличивает кислотную нагрузку на систему
поддержания КЩР, хотя хороший
антрекот на вкус совсем не кислый. Напротив,
лимон — кислый на вкус, однако
содержащийся в нем цитрат — ощелачивающий
анион. При недоокислении энергетических
субстратов и гипоксии образуется много
лактата и кетоновых тел. Их накоплению
препятствует инсулин. Распад нуклеиновых
кислот прибавляет к нелетучим кислотам
Збб АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
мочевую, а катаболизм органических
фосфористых соединений приводит к
освобождению фосфат-аниона, что эквивалентно
генерации фосфорной кислоты.
Потребляемые продукты могут поставлять в организм
экзогенные кислые эквиваленты (источники
катионов, например — лизин) и щелочные
эквиваленты (например, анионный цитрат).
Прямой прием существенных количеств
сильных кислот и оснований через рот
бывает только при отравлениях, в частности
суицидных. Метаболические нелетучие
кислоты могли бы нейтрализовать всю
буферную ёмкость организма (составляющую
порядка 15 мМ/кг) за 10-20 суток. Но этого
не происходит, так как почки секретиру-
ют протоны, в чистом виде или в составе
катиона аммония — в мочу, регенерируя
бикарбонат НСОз и буферную емкость
соответствующих систем организма. На 90 %
эти процессы происходят в проксимальных
канальцах нефронов, на 5% — по ходу
петли Генле и еще на 5 % — в дистальных
канальцах и собирательных трубках. В
проксимальных канальцах действует механизм
протон-натриевого противопереносчика, а
в дистальных частях нефрона — АТФ-зави-
симый протонный насос.
Регуляторные механизмы поддержания
КЩР организма почками реализуются
значительно медленнее, чем реакции,
зависящие от легких. Активация канальцевой
секреции катиона водорода, как следствие
повышения парциального давления
углекислого газа, происходит через несколько
минут, а альдостероновый путь активации
противоградиентной секреции протона в
дистальных отделах нефрона развивается в
течение часов и суток. Ранее существовало
упрощённое представление о наличии
отдельных парциальных функций ацидогенеза и
аммониогенеза у почек, дидактически
разделявшихся при обсуждении этого вопроса
с фильтрацией, реабсорбцией и секрецией.
Такое разделение вряд ли можно признать
удачным. Следует согласиться с мнением
большинства клиницистов и физиологов,
что основные процессы, регулирующие
кислотно-основное состояние, базируются
на клубочковой фильтрации, канальцевой
(проксимальной и дистальной) секреции и
реабсорбции. Это и есть процессы,
обеспечивающие условно выделяемые ацидогенез
и аммониогенез. Аммониогенез и ацидогенез
усиливаются при нагрузке кислотами,
первый имеет особое значение при попадании
в организм сильных кислот. Конечно же,
действие этих механизмов неотрывно от
концентрационно-дилюционной функции
нефрона (см. также подробную
характеристику парциальных функций нефрона в
Практикуме и т. III).
Секреция протонов обеспечивает реаб-
сорбцию бикарбонатов и эквивалентного
количества, уравнивающего электрические
заряды катиона натрия. С мочой экскре-
тируется в норме лишь 2-5 из более чем
4 000 мМ фильтруемого за сутки
бикарбоната (С.И.Рябов, Ю.Н. Наточин, 1997).
Секреция протонов освобождает в клетках
канальцевого эпителия бикарбонат-анионы.
Эти бикарбонаты, вместе с фильтрующимся
в мочу натрием, реабсорбируются в кровь.
Протоны в первичной моче (4000 мМ в
сутки) нейтрализует профильтрованный из
плазмы бикарбонат. Но его потеря
уравновешивается бикарбонатом, генерированным
в клетках канальцев при секреции протона
и реабсорбированным оттуда в кровь. Часть
секретированных протонов (80 мМ в сутки)
нейтрализуется в моче не бикарбонатом, а
другими буферами — фосфатным и
аммониевым. Катион водорода в моче связывается
в виде аммония и в виде двузамещенного
фосфата (см. предыдущий раздел):
H+ + NH3 = NH4+,
н++нро4- = н2ро4-.
При этом сберегается
профильтрованный клубочком бикарбонат, что позволяет
почкам его реабсорбировать и добавить
к щелочным резервам крови. Из общего
количества в 80 мМ протонов, не
связывающих бикарбонат мочи, до 1/3 нейтрализует
фосфатный буфер. Еще 2/3
нейтрализуются путём переноса в состав иона аммония.
Аммоний и кислый фосфат обеспечивают
Патофизиология водно-электролитного обмена 367
общую кислотность дефинитивной
мочи, в которой, даже при
отравлении сильными кислотами,
практически не появляются свободные
кислоты. Однако таким косвенным
путем рН мочи можно снизить при
адаптации к увеличению
кислотности внутренней среды организма до
4,8(Н.Г.Левински, 1994).
Фактически почки восполняют
потери бикарбонатного иона при
функционировании лёгких,
поскольку первые метаболизируют глютамин,
посылаемый печенью, с образованием
бикарбоната и аммиака, связывающего Н+ и
секретируемого в мочу (В.Ю. Шанин, 1998).
Итоги работы этой системы характеризует
таблица 21.
Нарушение ацидо- и аммониогенеза
в почках при почечной недостаточности
развивается, по отношению к другим
парциальным почечным функциям, относительно
рано, а восстанавливается при обратимой
острой недостаточности функций почек
сравнительно поздно. Недостаточность
этих процессов приводит к выделительному
почечному ацидозу, когда кислые
эквиваленты не экскретируются из организма, а
бикарбонат не реабсорбируется должным
образом (П.В. Горизонтов, 1981).
Главные регуляторы почечной функции
по поддержанию КЩР — рС02 и
концентрация в плазме бикарбоната. Ацидоз
усиливает секрецию протонов, аммония, а также
реабсорбцию и генерацию бикарбонатов.
В частности, в 10 раз может увеличиться
выведение аммония в мочу. Алкалоз инги-
бирует вышеназванные процессы.
В последнее время установлено, что почки
имеют также бикарбонат-хлоридный проти-
вопереносчик (анионообменный белок 3-й
полосы — см. выше). Он функционирует в
дистальных отделах нефрона и позволяет
секретировать в мочу бикарбонат. Данный
механизм (своего рода «алкалигенез»)
усиливается при алкалозе. В дополнение
к этому, при повышении уровня
щелочных эквивалентов в плазме, фильтрация
Таблица 21. Выделение кислот почкой и фильтрация
бикарбоната (поДжЛ. Шейману, 1997)
Кислые и основные эквиваленты
Фильтруемый HCOi
Канальцевая реабсорбция НСО;
Выделение бикарбоната с мочой
Выделение титруемых кислот с мочой
Выделение аммония с мочой
Общая экскреция кислот
миллимоль/сутки
4150
4145
5
55
30
80 (55+30-5)
бикарбоната перестаёт уравновешиваться
его канальцевой реабсорбцией, что
ускоряет компенсацию. По этой причине даже
потребление соды в больших количествах
с пищей и напитками вызывает при
здоровых почках лишь минимальные сдвиги в
уровне бикарбоната, хотя моча значительно
ощелачивается, обогащаясь ионами
бикарбоната и однозамещенного фосфата натрия.
Ее рН может повышаться до 8,2 (А. Уайт
и соавт., 1981; Н.Г. Левински, 1994).
Множество дополнительных регулятор-
ных механизмов, кроме состояния карбо-
нат-бикарбонатного буфера, связанных
с другими параметрами водно-солевого
обмена, влияют на почечные функции по
поддержанию КЩР.
Это и объём внеклеточной жидкости, и
доступность хлорид-аниона, и имеющееся в
наличии количество калия. Ангиотензин II
при снижении объема внеклеточной
жидкости, среди прочих эффектов, усиливает
реабсорбцию бикарбоната почками. Кроме
того, в этих условиях снижается общий
объём клубочковой фильтрации и выход
бикарбоната в первичную мочу. Альдостерон
стимулирует в дистальной части нефрона
реабсорбцию натрия и секрецию протонов,
за счет активации протонового насоса и
увеличения электроотрицательности
содержимого просвета канальцев. Дефицит
хлоридов понижает экскрецию бикарбоната
и усиливает реабсорбцию натрия.
Выраженная гипокалиемия и общая ги-
покалия всегда увеличивают реабсорбцию
бикарбоната, что способствует развитию
метаболического алкалоза. С другой
стороны, гиперкалиемия снижает почечный
368 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
Таблица 22. Факторы регуляции почечного транспорта протонов
и бикарбонат-анионов (поДж. А. Шейману, 1997)
Проксимальный каналец
рС02
Фильтруемая нагрузка бикарбонатом
Карбоангидраза
Ангиотензин II
Паратирокринин
К+ сыворотки, HPOJ- сыворотки
Дистальный каналец
и собирательная трубка
Градиент рН
Электрическая разность
потенциалов
рС02
Альдостерон
Экскреция аммония
Доступность хлорида
аммониогенез и вносит вклад в
формирование ацидоза.
Некоторые из факторов, регулирующих
почечные функции по поддержанию КЩР,
в контексте общих изменений
водно-электролитного метаболизма перечислены в
табл. 22.
Дополнительные факторы
регуляции КЩР
Ряд физиологических процессов, за
пределами почек и лёгких, принимают участие
в поддержании КЩР, во взаимодействии
с вышеописанными основными
эффекторами.
Печень вырабатывает глютамин,
используемый почками для аммониогенеза,
и метаболизирует лактат и кетокислоты,
закисляющие внеклеточную жидкость.
Поэтому печёночная недостаточность ведет к
метаболическому ацидозу.
Скелет, как депо кальция и фосфатов,
может вмешиваться в компенсаторные
процессы под контролем парат-гормона и
кальцитонина (см. «Нарушения
эндокринной регуляции»), при длительных
отклонениях от равновесия в кислую сторону. В
этих условиях возрастает остеолиз и выход
в экстрацеллюлярную жидкость и
первичную мочу фосфатов и кальция. Кальций и
фосфат-анион реагируют во внеклеточной
жидкости с угольной кислотой, образуя
анион основного фосфата и анион
бикарбоната. Последние два аниона нейтрализуют
катионы кислот. В мочу при этом поступают
кальции, натрии,
анионы кислот и основной
фосфат-анион.
Последний подкисляется
обычным почечным
механизмом ацидоге-
неза до аниона кислого
фосфата. Кальций и
кислый фосфат
теряются с дефинитивной
мочой, а натрий и
бикарбонат реабсорби-
руются. Потеря 1 М
фосфата кальция
позволяет убрать из организма 4 эквивалента
кислоты. Механизм поддержания КЩР с
участием скелета высокопроизводителен.
Однако его работа оплачивается опасной
деминерализацией костей и зубов в
условиях адаптации к ацидозу (см. гл. 13, раздел
«Остеопороз»).
Все другие клетки принципиально также
способны участвовать в регуляции КЩР.
При избытке бикарбоната в экстрацеллю-
лярном бассейне они захватывают катион
натрия, в обмен на катионы водорода и
калия, экскретируемые из клеток.
Протоны нейтрализуют лишний бикарбонат, а
образовавшаяся углекислота диссоциирует
и С02 удаляется при вентиляции лёгких.
Калиевый катион и бикарбонат-анион
фильтруются в мочу.
В условиях понижения рН
внеклеточной жидкости эти равновесные процессы
принимают обратный ход. Клетки отдают
ион натрия в обмен на протоны и К+. Это
пополняет ресурсы свободного бикарбоната
во внеклеточном секторе. Снижение
фильтрации калия облегчает подкисление мочи
почками (см. выше). Работа данных
механизмов эффективна, но может вызывать
потерю калия или его избыток в плазме, что
влечет патологические последствия (выше
см. «Гомеостаз калия и его нарушения»).
Органы ЖКТ играют определённую роль
в поддержании КЩР в норме и могут быть
основным звеном нарушения КЩР при
патологии. Слизистая желудка экскретирует
в трансцеллюлярный сектор ежесуточно
Патофизиология водно-электролитного обмена 369
150 мМ протонов. При этом генерируется
и поступает во внеклеточную жидкость
организма эквивалентное количество
бикарбонат-аниона. Поджелудочная железа
и энтероциты секретируют бикарбонат в
просвет ЖКТ и нейтрализуют с его
помощью протоны. Не связавшиеся с
протонами бикарбонат-анионы реабсорбируются
кишечником. Соответственно, при потерях
желудочного содержимого усиливается
образование и реабсорбция бикарбоната и
его концентрация во внеклеточном секторе.
При потерях кишечного содержимого
реабсорбция бикарбоната из ЖКТ снижается.
Микрофлора ЖКТ образует при брожении
органические кислоты, которые могут
способствовать закислению внеклеточной
жидкости, — см. ниже (В.Ю. Шанин, 1998).
Натрий-водородный противопереносчик
(он же белок NHE), участвующий в
почечных процессах ацидогенеза и в обмене
катионов между клетками и внеклеточной
жидкостью, — один из важнейших универсально
распространённых белков организма. Это
регулятор внутриклеточного рН. Имеется
целое семейство таких белков. Известно не
менее пяти их разновидностей (Э.Г. Улум-
беков, Ю.А. Челышев, 1997):
> NHE1 — имеется на всех клетках в
базолатеральных отделах их мембран,
кодируется геном SLC9A1. Именно
дефект этого гена и данного белка ведет,
с течением времени при действии ряда
экзогенных риск-факторов к развитию
гипертонической болезни (эссещиалъной
гипертензии). Дефект делает клетки
склонными к задержке натрия и кальция
и экскреции протонов. Инсулин и
факторы роста активируют его деятельность
(см. Практикум). На страницах этой
книги многократно упоминался (см. выше
«Патофизиология нуклеинового обмена,
«Нарушения липидного обмена»,
«Патофизиология сахарного диабета») и еще
будет затронут (см. ниже «Нарушения
обмена микроэлементов» и гл. 14) так
называемый метаболический Х-синдром.
Считается, что развитие всех его
проявлений — гипертензии, СД, ожирения,
гиперурикемии, атеросклероза (см. рис.
35, с. 170) — так или иначе связано с
дефектом именно данного противопере-
носчика.
>- NHE2 — функционирует в канальцах
почек и слизистой кишечника.
>- NHE3— характерен для апикальных
отделов плазматических мембран эпителия
кишечника и почек.
>• NHE4 — локализован в клетках
слизистой желудка, участвует в механизмах
секреции НС1 желудочного сока.
>- NHE5 — регулятор рН и объема клеток
в ЦНС, участвует в механизмах защиты
от набухания и отёка мозга.
Способы оценки и параметры
КЩР
Имеются разные подходы к оценке КЩР.
Все они оперируют рядом важных
терминов (ацидоз, алкалоз, ацидемия, алкалемия,
декомпенсированные ацидоз и алкалоз,
коррекция и компенсация КЩР), расшифровка
которых дана выше. Наиболее быстро
показатели КЩР определяются микроионо-
метрическим методом на приборе, впервые
предложенном П. Аструпом (1957).
Независимо от применения того или иного метода
оценки КЩР ключевое внимание уделяется
нескольким важнейшим показателям:
>■ рН — отрицательный десятичный
логарифм концентрации водородных ионов,
выраженной в грамм-ионах на литр, в
биологических жидкостях организма.
Значение рН, близкое к норме, не всегда
исключает существование нарушений
КЩР, которые могут быть латентными
(компенсированными). Величина рН
более 7,4 свидетельствует об алкалемии,
арН ниже этого уровня — об ацидемии.
>• рС02 —парциальное давление С02 в
плазме крови в мм Hg. Это давление
углекислого газа в газовой смеси, находящейся в
равновесии с плазмой артериальной крови
при температуре 38 °С. Данный показатель
непосредственно отражает дыхательный
компонент регуляции КЩР. Гипервен-
370 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения
тиляция выводит его за нижнюю границу
нормального интервала, что ведет к
респираторному алкалозу. Гиповентиляция
ведет к гиперкапнии, парциальное
напряжение диоксида углерода становится выше
верхней границы нормального диапазона
рС02, что характерно для дыхательного
ацидоза. Однако изменения рС02 могут
быть и следствием компенсации со
стороны лёгких метаболических нарушений
КЩР. Чтобы отличить эти ситуации друг
от друга, требуется уделить внимание рН
и[НС03].
>• р02 — парциальное давление 02 в плазме
крови в мм Hg; определяется
применительно к тем же условиям, что и
предыдущий показатель. Если данная величина
в пределах нормы, то она не оказывает
существенного влияния на КЩР. Но
выраженная гипоксемия, то есть её
снижение ниже нижней границы нормы,
вызывает усиление анаэробного
гликолиза и продукцию лактата, что ведет к
метаболическому ацидозу. Гипоксемия
может способствовать гипервентиляции
и дыхательному алкалозу.
Оба газовых показателя могут
измеряться отдельно, в артериальной, венозной и
капиллярной крови, но для точной оценки
КЩР применяется определение в крови
артериальной.
>• [HCOj] — концентрация бикарбонат-
аниона в плазме. Данный показатель
может быть подсчитан для разных условий.
Наиболее важен:
>- Стандартный бикарбонат плазмы (СБ,
SB) — содержание иона
бикарбоната в плазме, полностью насыщенной
кислородом крови, уравновешенной
при 38 °С с газовой смесью, в которой
рСО2=40 мм Hg. Это главный
показатель, непосредственно отражающий
почечный компонент регуляции КЩР.
При метаболическом ацидозе, или при
компенсации дыхательного алкалоза
он может падать ниже нижней границы
нормы. При метаболическом алкалозе
(и при компенсации дыхательного
ацидоза) — повышаться выше верхней черты
диапазона, указанного ниже.
Таким образом, основанием для
суждения о наличии и характере ацидоза либо
алкалоза служат прежде всего показатели,
отраженные в табл. 23.
Ряд других показателей КЩР не имеет
универсального значения и определяется,
либо не определяется в зависимости от
выбора метода оценки состояния КЩР.
С начала 40-х годов XX века при
определении характера нарушений КЩР чаще
всего применялся метод расчёта буферных
оснований и избытка/дефицита оснований.
Согласно этому методу, необходим подсчет,
в дополнение к основным показателям —
рС02, [НСОз] и рН артериальной крови еще
и следующих индексов:
>* Буферные основания — БО или ВВ (buffer
base) — сумма всех анионов цельной
крови, принадлежащих к
вышеназванным буферным системам (фактически
SB + анионный запас белкового буфера,
см. выше рис. 75). Показатель БО рассчи-
Таблица 23. Величины основных показателей КЩР в крови
(по ИЗ. Клявзунику, 1978; М. Горн и соавт., 1999)
Параметры
РН
рС02 мм Нд
р02 мм Нд
НСОз мэкв/л
N (среднее
значение в
артериальной крови)
7,4
40
95
24
Диапазон нормы
в артериальной
крови
7,35-7,45
35-45
80-95
22-26
Диапазон нормы в
венозной крови
7,33-7,43
41-51
35-49
24-28
Диапазон нормы
в капиллярной
крови
7,35-7,43
34-45
80-50
21-25
Патофизиология водно-электролитного обмена 371
тывается для полного насыщения крови
кислородом при 38°С и рСО2=40 мм Hg.
Он зависит от уровня гемоглобина в
крови и в норме, при 140 г/л гемоглобина
составляет 41,7± 0,043 мэкв/л.
> Дефицит оснований — ДО (или BD —
base deficit) — разница между
фактической и должной величиной БО при
метаболическом ацидозе. Определяется
как количество оснований, которое
необходимо добавить к крови, чтобы довести
её рН до нормы при рС02 = 40 мм Hg, при
температуре 38° С и полном насыщении
крови кислородом.
> Избыток оснований — ИО (он же -BE
или base excess) — разница между
фактической и должной величинами БО
при метаболическом алкалозе. В норме,
условно говоря, не существует ни
дефицита, ни избытка оснований.
Фактически это выражается в том, что разница
должного и фактического Б О
находится в нормальных условиях в пределах
± 2,3 мэкв/л (по М. Горн и соавт. — ± 2
мэкв/л). Показатель отражает в общих
чертах количество неиспользованных
буферов крови, в основном — бикарбо-
натного и гемоглобинового. Выход этого
показателя из коридора нормы типичен
для метаболических нарушений КЩР.
Аномально высокие положительные
значения характерны для алкалоза (ИО);
в то время как аномально низкие
отрицательные характеризуют ацидоз (ДО).
В рамках описываемого метода оценки
нарушений КЩР диагностический алгоритм,
рекомендуемый М. Горн и соавт. (1999),
предусматривает следующие шаги:
1. Определить, отклоняется ли рН от
среднего нормального. Если он отклоняется
от 7,4 констатируется ацидоз (при его
понижении) или алкалоз (при
повышении).
2. Установить, выходит ли рН за границы
интервала нормы. Если он в нормальных
пределах — нарушение
компенсированное, если в критических пределах —
нарушение декомпенсированное.
3. Оценить рС02. Определить, в каком
направлении и на какую величину этот
показатель отклоняется от нормы (40 мм
Hg). Вообще говоря, если рС02
повышается рН должен снижаться (ацидоз).
Если рС02 понижается рН должен
повышаться (алкалоз).
4. Оценить содержание [НСОз].
Определить, в каком направлении и на какую
величину он отклоняется от 24 мэкв/л.
Вообще говоря, снижение [НСОз]
должно снижать и рН (ацидоз); повышение
[НСОз"] — повышать рН (алкалоз).
5. Определить, какой из параметров — рС02
или [НСОз"] — более соответствует по
направлению изменений сдвигу рН.
Например, если сдвиг рН свидетельствует
об ацидозе — имеется ли ацидотическое
повышение рС02 или ацидотическое
снижение [НСОз], или же °ба эти изменения?
Показатель, который по направлению
изменений соответствует динамике рН
и больше отклоняется от средней
нормальной величины, как раз и «повинен»
в нарушении КЩР. Тогда изменения
другого показателя, не соответствующие
по направлению логике изменения рН,
надо расценить как компенсаторные.
Если первичным оказалось изменение
[НСОз] ~~ нарушение КЩР по этиологии
метаболическое, если первичен сдвиг рС02
— оно респираторное. Если изменены, в
соответствии со сдвигом рН, оба
показателя — речь идёт о смешанном дыхательно-
метаболическом расстройстве.
Например:
рН=7,2
pC02=19MMHg
...IHCO3]-.7 мэкв/л
Диагноз: метаболический ацидоз с
частичной дыхательной компенсацией.
рН = 7,50
рС02= 20 мм Hg
...J.MCPil=.llM3KB/ji
Диагноз: дыхательный алкалоз с почечной
компенсацией.
372 АШ. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
В дополнение к этим основным индексам
и правилам их элементарной оценки
следует иметь в виду значительную ценность
такого клинического параметра, как
величина анионного зазора (в некоторых
текстах — анионного дефицита или пробела).
Анионный дефицит имеет важное значение
при дифференциации разных подвидов
метаболического ацидоза и в связи с этим
подробно рассматривается ниже, в
следующем разделе.
Иногда для характеристики патологии
КЩР имеет значение определение рНмочи.
Это широко варьирующий в норме (4,6-8,0)
показатель, который имеет тенденцию к
понижению при ацидозах и к повышению —
при любых алкалозах.
В 1963 г. У.Б. Шварц и А.С.Релмэн
показали, что существует более
предпочтительный метод наглядной оценки сдвигов
КЩР, не нуждающийся в определении ИО,
ДОиБО.
Это метод «доверительных полос» (рис. 77),
в основе которого лежит определение
положения данного случая на так называемой
номограмме Х.У. Дэвенпорта.
Если изобразить систему координат, где
по оси абсцисс откладываются значения
рС02в мм Hg, а по оси ординат — значения
[HCOj] в мэкв/л, и на ней провести
изобары, соответствующие разным значениям
рН, рассчитанным из уравнения Хендерсо-
на-Хассельбалха для получающихся
комбинаций этих параметров, то в центре всей
системы окажется «точка нормы» (НВ) —
лежащая на пересечении изобары рН=7,4
с ординатой 24 мэкв/л и абсциссой 40 мм
Hg. На рис. 77 вверху слева — номограмма,
отражающая направления начальных
патологических изменений от точки нормы при
респираторном и метаболическом алкалозе,
либо ацидозе, что соответствует четырём
основным квадрантам. Вверху справа
показано, в каком направлении компенсаторные
механизмы будут сдвигать начальные
патологические изменения рН, рС02и [HCOj]
при четырёх основных нарушениях КЩР.
Наконец, внизу слева номограмма
модифицирована с нанесением на неё полос
доверия — серых интервалов в ± 2
стандартных отклонения для компенсаторных
сдвигов этих трёх параметров у нормальных
и больных индивидов (в ответ на первичные
расстройства, указанные на графике
буквами). В таком интервале будут находиться
95 % индивидов, дающих компенсаторный
ответ на то или иное расстройство. Внизу
справа та же номограмма модифицирована
с учетом метода ДО/ИО. Вместо [HCOj] по
одной из осей отложены величины ДО и ИО
(см. подпись к рисунку).
Оценка случая производится путем
определения, входит ли данное сочетание
параметров в полосу доверия.
Например:
рН=7,16
рС02=71 мм Hg
[НСО3] = 30 мэкв/л.
Точка, соответствующая данному случаю
(на рис. 77 слева внизу — Z) находится вне
полосы доверия хронического дыхательного
алкалоза, значит, подобное сочетание
параметров КЩР не является простым
следствием почечной компенсации дыхательных
нарушений, а отражает комбинированное
расстройство, скорее всего — наложение на
хронический дыхательный алкалоз
метаболического ацидоза, что, конечно же, может
быть верифицировано только с учётом
клинических симптомов и анамнеза пациента.
Принципиально важно, что
компенсаторные сдвиги, работая «против» первичной
тенденции изменения рН, не исправляют
его ненормального значения. Не имея
номограммы, компенсаторные сдвиги при
первичных нарушениях КЩР можно оценить с
помощью формул из таблицы 24 (см. также
Практикум).
Этиология и патогенез
нарушений КЩР
Как уже отмечалось выше, к нарушениям
кислотно-основного состояния относятся
ацидоз и алкалоз. Ацидозы и алкалозы
делятся на экзогенные (вызванные
отравлениями кислотой или щелочью), а также
эндогенные (связанные с нарушениями ме-
первичное нарушение
компенсаторный ответ
20 40 60 -5 5 15
РаСОг ммНд — избыток кислоты, мМ
+ избыток оснований, мМ
Рис. 77. Номограммы для оценки нарушений КЩР методом доверительных полос. Вверху
слева — первичные нарушения (МАлк — метаболический алкалоз; РАлк — дыхательный алкалоз,
МАц — метаболический ацидоз, РАц — дыхательный ацидоз, О — острый, П —
продолжительный (хронический), НВ — нормальная точка); вверху справа — направления компенсации при
этих нарушениях. Внизу слева — доверительные полосы 95% интервалов компенсаторных
ответов (серые). Прерывистые линии — везде изобары рН плазмы.Обозначения те же.
По оси абсцисс везде парциальное напряжение С02 в артериальной крови, по оси ординат —
концентрация бикарбоната в мМ/л (по Н.Г. Левински, 1994).
Внизу справа — та же номограмма при методе дефицита (ДО) и избытка (ИО) оснований. По оси
абсцисс — дефицит и избыток оснований в мМ, по оси ординат рС02 в торр, сплошные
диагональные линии — изобары рН плазмы (По Я. Мусилу и соавт., 1981)
Пояснения в тексте
374 АЖ Зайчик, Л.П.Чурилов Патохимия^
ханизмов КЩР). Эндогенные расстройства
подразделяются на дыхательные (газовые)
и метаболические (негазовые), а также на
компенсированные и декомпенсированные.
Некоторые источники особо рассматривают
выделительные ацидоз и алкалоз (от
усиленной потери кислых, либо щелочных
эквивалентов или от недостаточной способности
почек к закислению мочи) в виде отдельной
категории, но наиболее часто, их включают в
разряд метаболических. В настоящее время
также принято включать ацидозы и
алкалозы от избыточного введения экзогенных
кислот и оснований в категорию
метаболических (Н.А. Уварова, 1994).
Дыхательный ацидоз возникает как
следствие гиповентиляции, что приводит к
гиперкапнии (повышение рС02 больше 40
мм рт. ст.), а следовательно к увеличению
концентрации катионов водорода и
снижению рН ниже 7,40. В обычных условиях
выделение из организма углекислоты равно
её продукции. Нарушение этого баланса
возможно вследствие повышенного
образования углекислоты или неспособности
дыхательной системы вывести её из организма.
Нарушение вентиляции лёгких может
протекать остро или хронически,
следовательно, дыхательный ацидоз тоже может
быть острым или хроническим. В ответ на
гиперкапнию буферные системы
повышают концентрацию бикарбоната в плазме,
примерно на 1мМ/л на каждые 10 мм Hg
прироста рС02. Почки усиливают
экскрецию протонов и реабсорбцию бикарбоната.
За счёт этого концентрация бикарбоната
в плазме растет до 3,5 мМ/л на каждые
10 мм Hg прироста рС02. В тех случаях,
когда буферные системы не в состоянии
компенсировать избыток С02, развивается
состояние декомпенсации, сопровождаемое
снижением рН. Так формируется газовый
ацидоз. При очень значительной
гиперкапнии и ацидозе снижается эффективность
действия катехоламинов на их рецепторы и
растёт тонус блуждающего нерва. Это ведет
к торможению компенсаторных реакций со
стороны сердечно-сосудистой системы.
шно-метаболические нарушения
Клинические формы патологии,
сопровождаемые дыхательным ацидозом,
многочисленны. Наиболее часто встречается
дыхательная недостаточность при острых
респираторных заболеваниях и синдромах.
При хронических заболеваниях лёгких с
нарушением альвеолярной вентиляции
хроническая гипокапния развивается не
всегда. В случае пневмофиброза и отёка
лёгких гипоксемия может в такой
степени стимулировать вентиляцию, что этого
оказывается достаточно для выведения
С02, с его большой диффузионной
способностью, несмотря на нарушения. Поэтому
имеется гипокапния. Дыхательный ацидоз
в этих случаях возможен при утомлении
дыхательных мышц и очень тяжёлом
течении первичного заболевания. К
наиболее частым причинам респираторного
ацидоза относятся пневмония, бронхит,
эмфизема, ларингоспазм, бронхоспазм,
пневмоторакс, гидроторакс и гематоракс,
аспирация, асфиксия, муковисцидоз, ги-
повентиляционный пиквикский синдром
при ожирении.
К выраженной дыхательной
недостаточности приводят патологические процессы и
заболевания, связанные с угнетением ЦНС
(дыхательного центра) или с
периферическими нервно-мышечными расстройствами.
В этой группе патологии лидируют травмы,
отеки и опухоли головного мозга. Большую
группу случаев представляют отравления,
нарушающие центральную стимуляцию
вентиляции, например барбитуровое и
опиатное. Известное значение в развитии
дыхательной недостаточности в клинике
имеют токсические эффекты лекарств,
нарушающих периферические или центральные
механизмы вентиляции: анестетиков, седа-
тивных препаратов, наркотиков, алкоголя,
миорелаксантов и др.
В отдельную группу патологии можно
выделить такие заболевания, как
полиомиелит, генерализованная миастения, гипофос-
фатемия, демиелинизирующие спинальные
поражения, при которых нарушается работа
дыхательных мышц. Краткую
характеристику основных признаков газового ацидоза
Патофизиология водно-электролитного обмена 375
можно найти в табл. 26 ниже (см. также
Практикум).
Дыхательный алкалоз является
результатом повышения альвеолярной вентиляции.
Развивается гипокапния. В порядке
компенсации тканевые буферы освобождают
протоны, концентрация бикарбоната в плазме
из-за этого снижается примерно на 2 мэкв/л
на каждые 10 мм Hg падения уровня рС02.
В клетках усиливается гликолиз и растет
концентрация лактата и пирувата в крови.
Возникают предпосылки для формирования
метаболического ацидоза. Наступает гипо-
фосфатемия, так как фосфаты используются
в клетке в ходе гликолиза.
Ответ почек на гипокапнию и алкалоз
замедлен, поэтому при остром дыхательном
алкалозе он не успевает развиться, но при
хроническом — подключается. В силу этого
хронический дыхательный алкалоз
характеризуется намного более значительным
снижением уровня бикарбоната в плазме — до
0,45 мэкв/л на каждые lOMMHg падения
уровня рС02что обеспечивается возросшей
почечной экскрецией этого аниона.
Важной особенностью водно-электролитных
нарушений при газовом алкалозе является
тенденция к гипокальциемии, связанная с
компенсаторным обменом ионами между
плазмой и скелетом. Усиливается и
выведение с мочой калия.
Этиология респираторного
ощелачивания связана прежде всего с причинами
гипервентиляционного синдрома. Любая
гипоксемия с р02 ниже 60 мм Hg и
сохранённой способностью к вентиляции
лёгких опасна, с точки зрения развития
респираторного алкалоза. Здесь и острая
гипоксия различного генеза (например,
при тромбоэмболии лёгочной артерии и
горной болезни). Хронические заболевания
с гипоксемией также вызывают
гипервентиляцию и гипокапнию (пневмофиброз,
бронхиальная астма, застойная сердечная
недостаточность). Гипервентиляция может
быть следствием хронической гемической
гипоксии при тяжелой анемии.
Стимуляция дыхательного центра при стрессе,
лихорадке, токсико-септическом шоке,
отравлении салицилатами, опухолях мозга,
черепно-мозговой травме и энцефалите,
длительно персистирующее закисление
цереброспиальной жидкости после
коррекции метаболического ацидоза, могут
обусловить дыхательный алкалоз. Тенденция к
дыхательному алкалозу имеется у
беременных. Наконец, данное расстройство может
быть ятрогенным, при неверно выбранном
режиме искусственной вентиляции лёгких
(см. табл. 26 и Практикум).
Метаболический ацидоз развивается
вследствие первичной гипобикарбонатемии.
Это происходит при накоплении в крови
органических кислот, как промежуточных
метаболитов обмена веществ, вследствие
их избыточного образования. Первичные
нарушения могут касаться также потери
оснований через ЖКТ или блока экскреции
кислот почками. Метаболический ацидоз
достоверно определяется при уровне
бикарбоната менее 24 мэкв/л и рН менее 7,40.
Снижение рН стимулирует дыхание, как
срочный компенсаторный механизм, что
уменьшает уровень парциального давления
углекислого газа в артериальной крови
максимально на 10-15 мм Hg. Создается
предпосылка к наслоению респираторного
алкалоза.
Снижение уровня бикарбоната на 1 мэкв/л
при остром метаболическом ацидозе даёт
компенсаторный ответ, обеспечивающий
уменьшение рС02 артериальной крови на
1,2 мм Hg (см. рис. 75 выше).
В компенсации участвуют обмен ионами
между плазмой и клетками, в частности,
эритроцитами, а также между
внеклеточной жидкостью и скелетом (см. выше).
Результатом является увеличение в плазме
содержания калия, кальция и натрия. При
хроническом течении процесса
прогрессирует деминерализация костей.
Наиболее адекватным механизмом
избавления организма от кислот является
экскреция их почками. Подкисление мочи в ответ
на метаболический ацидоз начинается
быстро. Падение рН мочи вначале обусловлено
376 АЖ Зайчик, Л.П.Чурилов Патохимия
увеличением экскреции аниона кислого
фосфата. Через несколько дней на полную
мощность включается механизм усиленного
аммониогенеза. Однако степень кислотной
нагрузки на организм может быть разной.
Почки способны десятикратно увеличить
экскрецию кислых эквивалентов. Но рН
мочи не может понижаться бесконечно. При
чрезвычайно высоких кислотных нагрузках
механизмы элиминации кислот бывают
недостаточными. Декомпенсированный
метаболический ацидоз сопровождается комой
(при рН ниже 7,2), дыханием Куссмауля и
тахиаритмиями (экстрасистолия, в тяжелых
случаях — фибрилляция желудочков).
Истинный «чистый» метаболический
ацидоз встречается довольно редко, обычно
это смешанное расстройство
кислотно-основного состояния.
Заболевания, в патогенезе которых
встречается метаболический ацидоз, очень
многочисленны. Уместно разделить
метаболический ацидоз на группы по этиологии
и величине анионного просвета (см. выше
данный раздел и раздел «Патофизиология
сахарного диабета»).
Анионный просвет (дефицит, интервал,
зазор, сокращенно — А) — искусственно
выводимое соотношение между концентрацией
катиона натрия и суммой концентраций
анионов хлорида и бикарбоната в плазме,
удобное для оценки происхождения
метаболического ацидоза в клинике. Он
определяется по нижеследующей формуле:
A--[Na+]-{[Cl-] + [HC03-]}.
По закону электронейтральности
количество катионов и анионов в плазме
эквивалентно. Поэтому анионный дефицит
отражает концентрацию неизмеряемых анионов.
В норме его величина 12 ±4 мэкв/л. Анионы
органических кислот, сульфаты,
производимые при метаболизме аминокислот,
анионные эквиваленты белка формируют
эту величину.
Метаболический ацидоз всегда сопряжен
с уменьшением содержания бикарбонат-
аниона. Но ситуация с другими анионами
отличается при разной этиологии этого
чдокринно-метаболические нарушения
расстройства. В большинстве случаев
метаболический ацидоз вызван причинами,
способствующими поступлению во
внеклеточную жидкость НС1. Правда, в прямой форме
это бывает только при экзогенном ацидозе.
Аналогичная ситуация сопровождает
экзогенное введение хлоридов (аммония,
аргинина и других катионных аминокислот),
усиленное парентеральное питание.
Но такие же изменения буферных
систем развиваются и при потере избыточных
количеств бикарбоната через ЖКТ (поносы,
свищи кишечника и вирзунгова протока,
хирургическое удаление части кишечника,
илеус, восстановительный период после
дыхательного алкалоза). Бикарбонаты
теряются и с мочой (наследственная врожденная
бикарбонатурия, нефропатия беременных,
почечный канальцевый ацидоз
проксимального и дистального типа, гипоальдостеро-
низм, передозировка калийсберегающих
диуретиков, ингибиторов карбоангидразы).
Механизмы и типы почечного гиперхлоре-
мического канальцевого ацидоза подробно
обсуждаются в т. III данной серии. Здесь
приводится их краткая характеристика
в виде табл. 25. Тип III в данной таблице
отсутствует, так как под ним в настоящее
время понимают не что иное, как развитие
почечного канальцевого ацидоза I типа у
детей.
Наконец, принципиально сходной
является ситуация при быстром разведении
крови, когда концентрация хлорида
стабилизируется быстрее, чем бикарбоната.
Во всех этих случаях формируется гипер-
хлоремический метаболический ацидоз.
Уровень хлоридов растет адекватно понижению
бикарбоната, и в итоге анионный просвет
остаётся в пределах нормы.
Сравнительно небольшое количество
состояний сопровождается таким
метаболическим ацидозом, при котором
убывающий бикарбонат-анион замещается не
хлорид-анионом, а иными анионами (лак-
татом, кетокислотами, сульфатом и т.д.).
Эти ситуации легко дифференцируются, по
увеличению анионного просвета, как
метаболический ацидоз с возрастанием анионно-
Патофизиология водно-электролитного обмена 377
Таблица 25. Гиперхлоремический почечный канальцевый ацидоз
(по СИ. Рябову, Ю.В. Наточину; 1997)
Характеристики
Патогенез
рН мочи при ацидемии
Бикарбонат плазмы
Экскретируемая фракция
бикарбоната при нормо-
бикарбонатемии
Диагностические пробы
Калий плазмы
Потребность в
бикарбонат-анионе (мэкв/кг в
сутки)
Сопутствующие
заболевания
Тип 1 (дистальный)
Иподкисления мочи в
дистальном сегменте
нефрона
5,3
< 10мэкв/л
Взрослые —3% Дети
(«тип III») —до 10%
Нагрузка бикарбонатом
или хлоридом аммония
U
1-2 (взрослые) 4-14 («тип
III» — дети)
Нефрокальциноз, уро-
литиаз
Тип II (проксимальный)
иреабсорбции
бикарбоната в проксимальном
сегменте нефрона
разный
14-20мэкв/л
15-20%
Нагрузка бикарбонатом
NwinU
10-15
Рахит, остеомаляция
Тип IV
Гипоальдостеронизм или
резистентность к мине-
ралокортикоидам
5/3
> 15 мэкв/л
Уровень альдостерона
плазмы
U
1-3, при коррекции
гиперкалиемии не
требуется
Нет
го дефицита. К ним относится прежде всего
кетоацидоз (при декомпенсации сахарного
диабета — см. выше «Патофизиология
сахарного диабета», голодании — см. выше
«Голодание», алкогольном опьянении,
нарушении обмена разветвлённых аминокислот,
гиперурикемии и других причинах — см.
выше «Кетоз и стеатоз печени» и гл. 9).
Большое значение имеет лактат-ацидоз
(при гипоксии, шоке, отравлении угарным
газом, циркуляторной недостаточности,
дефектах глюкозо-6-фосфатазы, фруктозо-
1,6-дифосфатазы, пируваткарбоксилазы и
пируватдегидрогеназы, при гипераланине-
мии и других метаболических
расстройствах).
Нередко бывает очень тяжелым лактат-
ацидоз при сахарном диабете II типа, когда
ему способствуют недостаточно активная
утилизация пирувата, применение
фруктозы и лечение бигуанидами, особенно, фено-
формином — см. выше «Патофизиология
углеводного обмена», «Патофизиология
сахарного диабета».
Дополнительно к изложенным выше
в этих главах механизмам и вариантам
этиологии лактат-ацидоза можно указать
на его закономерное возникновение у
тяжелых больных со злокачественными
неопластическими процессами. В малиг-
низированных клетках вследствие наличия
определённых онкобелков и изменения
митохондрий отсутствует эффект Пасте-
ра и гликолиз не подавляется в аэробных
условиях, поэтому они продуцируют
избыток лактата (см. т. III). Лактат-ацидозу
способствует срыв глюконеогенеза при
печёночной недостаточности различного
происхождения и при гиповитаминозе Bt
(см. ниже «Патофизиология витаминного
обмена»). В.Ю.Шанин (1998) указывает
на возможность развития метаболического
лактат-ацидоза с увеличением анионного
дефицита при усиленном всасывании в
кишечнике органических кислот.
Бродильная диспепсия, как разновидность
дисбактериоза, может вызывать продукцию
из клетчатки плохо метаболизируемого
D-изомера лактата.
Таким образом, ацидоз вследствие
накопления молочной кислоты встречается
довольно часто. Механизмы его
формирования при нарушении равновесия между
образованием и утилизацией лактата
освещались выше, в разделе «Патофизиология
сахарного диабета».
378 АЖ Зайчик, Л.П.Чурилов Патохимия
Напомним, что все разнообразие лактат-
ацидозов (по Р.Д. Когену и Г.Ф.Вудсу,
1976) классифицируется на тип А (от
гипоксии) и тип В (негипоксический, от
первичного нарушения глюконеогенеза
и повышенного освобождения лактата
печенью). Краткая сравнительная
характеристика этих разновидностей
молочнокислого ацидоза приводилась выше в табл.
16 (глава 9).
Еще раз подчеркнем, что диабетический
лактат-ацидоз протекает всегда с
увеличением показателя анионного промежутка
(интервала).
В отсутствие кетонурии возросший
анионный промежуток у больного сахарным
диабетом почти наверняка указывает
именно на лактатацидоз.
Некоторые рекомендации по
патофизиологическим основам лечения лактат-
ацидоза при сахарном диабете включены в
главу 9 выше.
Нарушения по типу метаболического
ацидоза с повышенным анионным зазором
характерны и для ряда отравлений,
поражающих почки и печень (метанолом, толуолом,
этиленгликолем, паральдегидом, салици-
латами), а также для уремии. Токсиканты,
перечисленные выше, не только сами
превращаются в органические кислоты —
муравьиную, глиоксалевую, щавелевую, — но
и блокируют метаболизм лактата и других
естественных органических кислот.
Уремия приводит к нарушению аммонио-
генеза и ацидогенеза.
Характеристики метаболического
ацидоза можно найти в табл. 23 ниже.
Метаболический алкалоз — состояние,
характеризующееся повышением уровня
бикарбоната и снижением концентраций
катиона водорода и хлорид-аниона во
внеклеточной жидкости. В основе этого лежат
либо потеря протонов (вследствие потери
хлорид-аниона или непосредственно), либо
нагрузка экзогенным бикарбонатом. При
метаболическом алкалозе возникает
компенсаторная гиповентиляция и гиперкапния до
рСО2=50-60 мм Hg, но возможности этого
чдокринно-метаболические нарушения
механизма ограничиваются создающейся
гипоксемией.
Этиология данного процесса может быть
связана с потерей ионов Н+, наступающей
вследствие:
>• потери желудочного содержимого при
рвоте и отсасывании через зонд,
врождённой хлоридорее;
>■ потери протонов почками при действии
петлевых диуретиков и тиазидов, при
первичном и вторичном гиперальдосте-
ронизме и выраженном гиперкортициз-
ме (см. ниже «Нарушения эндокринной
регуляции»), после хронической гипер-
капнии, при действии больших доз кар-
бенициллина и других пенициллиновых
антибиотиков, которые выводятся с
мочой как нереабсорбируемые анионы,
при гиперкальциемии и кальциурии,
гипомагниемии;
>• перемещения протонов в клетку из-за ги-
покалиемии и при углеводной нагрузке.
Альтернативный вариант этиологии и
патогенеза метаболического алкалоза зависит
от задержки избытка бикарбонат-аниона.
Это наблюдается:
>* вследствие ятрогенного назначения его
больших доз, а также передозировки его
предшественников;
>* при массивной гемотрансфузии;
>• при молочо-щелочном синдроме (см.
ниже «Патофизиология паращитовид-
ных желёз...»);
>• при применении ряда диуретиков;
>• при тяжёлом осложнённом муковисци-
дозе.
Причины и механизмы данного синдрома
дифференцируются в зависимости от того,
является ли потеря хлоридов и гипохлоре-
мия первичным патогенетическим звеном
процесса, или же первичные нарушения
касаются других ионов, а дефицит хлоридов
создаётся вторично.
В первом случае метаболический
алкалоз хлоридчувствителен, то есть
корректируется после инфузии хлорида натрия
и протекает с уровнем хлоридов в моче
Патофизиология водно-электролитного обмена 379
менее 10 мэкв/л. Объём внеклеточной
жидкости при таком алкалозе имеет
тенденцию к снижению. Это ограничивает
возможности почек по компенсаторной
экскреции бикарбоната, так как
приоритетными являются нейроэндокринные
стереотипы, направленные на борьбу с
гиповолемией, а они требуют задержки
воды и ионов.
Во втором случае процесс не
корректируется хлоридами и развивается при
уровне хлоридов мочи > 20 мэкв/л. Объём
внеклеточной жидкости в таких случаях
нормальный или повышенный.
Хлоридчувствительные формы
метаболического алкалоза разнообразны. К ним
относятся алкалоз при действии пеницил-
линовых антибиотиков, диуретиков, а
также метаболический алкалоз при быстром
восстановлении параметров КЩР после
дыхательного ацидоза (Дж.А. Шейман,
1997), алкалоз при потере желудочного
содержимого (В.Ю. Шанин, 1998).
Остальные вышеназванные формы
метаболического алкалоза принадлежат к
хлориднечувствительным. Многие формы
такого алкалоза протекают на фоне гипе-
ральдостеронизма и нарушений функций
ренин-ангиотензин-альдостероновой
системы. Это ведёт к задержке воды и натрия,
усиленной экскреции калия и протонов и к
гипокалиемии, ограничивающей
эффективность клубочковой фильтрации, а значит и
экскреции бикарбоната почками.
Дополнительно последствия различных форм
гиперальдостеронизма и гиперкортицизма
рассматриваются ниже в разделе
«Нарушения эндокринной регуляции».
Своеобразной формой метаболического
хлориднечувствительного алкалоза
характеризуется синдром Барттера (ортостати-
ческая гипотензия, снижение объёма вне-
Таблица 26. Основные признаки типовых нарушений КЩР
(по М. Горн и соавт., 1999; с изменениями и дополнениями)
Синдром
Острый дыхательный ацидоз
Хронический
компенсированный дыхательный ацидоз
Острый дыхательный алкалоз
Хронический дыхательный
алкалоз
Острый метаболический
ацидоз
Хронический метаболический
|ацидоз
Острый метаболический
алкалоз
Хронический метаболический
|алкалоз
РН
U
V
ft
ft
u
u
ft
ft
PaC02
ft
ft
ft
U
ft (компенсаторно)
-U (менее
значительно, чем при остром)
ft (сильно — почти
до 60 мм Нд)
ft (компенсаторно)
нсо3
Без изменений
ft (компенсаторно)
Без изменений
(снижение только при
длительном
процессе и адекватной
функции почек)
U (компенсаторно)
ft
У
ft
ft
Основные симптомы
Тахикардия, одышка,
головная боль,
беспокойство, потливость, цианоз,
аритмии, гипотензия,
летаргия, кома
Одышка, летаргия, кома
Парестезии,
головокружение
Нет особых симптомов
Тахипноэ, гипотензия,
влажная холодная кожа,
кома, дыхание Куссмауля.
Аритмии |
Слабость, анорексия,
общее недомогание
Миастения, гипорефлек-
сия, аритмии, апатия,
ступор
Без особых симптомов
380 А.Ш. Зайчик, Л.П.Чурилов Патохимия
клеточной жидкости, гиповолемия, гипер-
натриурия, гипокалиемия, метаболический
алкалоз — см. также т. I, с. 156). Механизмы
данного нарушения связывают с патологией
простагландиновой регуляции (избыточное
действие PgE2 на рениновую продукцию и
почечные функции).
Основные характеристики
метаболического алкалоза представлены в табл. 26,
как и краткие сведения о других типовых
нарушениях КЩР.
Следует иметь в виду, что у больного
могут одновременно развиваться 2 и более
разных нарушения КЩР.
При этом формируются сочетанные
картины показателей КЩР и симптомов.
Смешанные нарушения, обусловленные
сочетанием двух типов ацидоза или двух
типов алкалоза, приводят к более
существенным отклонениям рН, чем комбинации
алкалозов с ацидозами. В последних
случаях характер отклонения рН определяет
преобладающее из разноименных
нарушений.
Смешанный метаболический и
дыхательный ацидоз характерен для острой
сердечно-лёгочной недостаточности и для
тяжелого отёка лёгких. Циркуляторная
гипоксия вызывает гликолитический ответ
тканей и лактат-ацидоз, а дыхательная — ги-
перкапнию.
Смешанный респираторно-дыхательный
алкалоз может наступить при рвотах
беременных, так как потеря хлоридов и
желудочного кислого содержимого наслаивается
на характерный для нормального течения
беременности гипокапнический фон.
Метаболический ацидоз комбинируется
с газовым алкалозом при отравлении сали-
цилатами, этиленгликолем, сепсисе. В
последнем случае циркуляторное расстройство
ответственно за метаболический ацидоз, а
чдокринно-метаболические нарушения
гипервентиляция при лихорадке — за
газовый алкалоз.
Обратная комбинация метаболического
алкалоза и газового ацидоза свойственна
хроническим лёгочным больным при
лечении диуретиками, стероидами, седативными
средствами или дренировании у них
содержимого желудка.
При хронических неспецифических
заболеваниях лёгких комбинируются
хронический респираторный ацидоз и эпизоды
острого респираторного ацидоза при обострениях.
Метаболический кетоацидоз при диабете
из-за сильной кетонемической рвоты может
комбинироваться с метаболическим же
алкалозом. Возможны и тройные
комбинированные расстройства КЩР. Большое
значение в распознавании скрытого
метаболического ацидоза, наслаивающегося на
другие расстройства КЩР, имеет анализ
анионного промежутка, который при этом
всегда сильно увеличивается.
При алкалемии и ацидемии включаются
описанные выше механизмы компенсации,
основанные на обмене ионами между
различными секторами жидкости организма (см. ранее
в этой главе об эффекте Хамбургера и т.д.).
Поэтому при интерпретации состояния КЩР
еще более сложные комбинации возникают,
когда учитываются сдвиги рН во
внутриклеточной жидкости. Так, при гиперальдостеро-
низме (и крайней степени гиперкортицизма)
происходят ионные сдвиги, способствующие
внеклеточному метаболическому алкалозу и
внутриклеточному ацидозу.
Существование сложных
комбинированных нарушений КЩР подтверждает, что
диагностика в медицине не может быть
полностью формализована и основана только на
алгоритмической оценке лабораторных
данных, сколь бы ни был велик объем знаний
о конкретных патохимических процессах.
Решающим тут остаётся учёт анамнеза и
клинических симптомов.
Глава 11
ПАТОФИЗИОЛОГИЯ
ВИТАМИННОГО ОБМЕНА
«Невозможно обеспечить жизнь белками,
жирами, сахаром, солями и водой...»
Н.И.Лунин (1880)
«О значении неорганических солей для питания животных»
• Общие свойства витаминов и история идей в витаминологии
• Нарушения обмена энзимовитаминов
• Нарушения обмена редокс-витаминов
• Нарушения обмена гормоновитаминов
• Заключение
Витаминами называются
низкомолекулярные органические соединения
различного, относительно простого строения,
не относящиеся к аминокислотам, которые
человек или животные должны получать
извне полностью или частично (по Р.В. Ча-
говцу, Е.В. Лахно, 1968; П.Е. Калмыкову,
М.Н. Логаткину, 1974). Витамины не
являются для организма поставщиками энергии
и не имеют существенного пластического
значения. Однако, они либо играют
каталитическую роль, входя в состав ферментов
(энзимовитамины — как правило,
водорастворимые вещества), либо выполняют
сигнальные функции экзогенных прогормонов
и гормонов (гормоновитамины — как
правило, жирорастворимые вещества).
ОБЩИЕ СВОЙСТВА
ВИТАМИНОВ И ИСТОРИЯ
ИДЕЙ В ВИТАМИНОЛОГИИ
О болезнетворности качественно
неполноценной пищи эмпирически врачи
догадывались издавна. Болезнь бери-бери описана в
древнекитайском каноне медицины 2 500 лет
тому назад. Клиническая картина
авитаминоза А была известна медикам античной
Греции, а Гиппократ лечил куриную слепоту
печенью. Проявления цинги хорошо знали
еще участники VIII Крестового похода,
как об этом свидетельствовал летописец
Ж. де Жуанвиль, впервые ее подробно
описавший (1270). Корабельные врачи, начиная
с эпохи великих географических открытий,
когда появилось само староголландское
слово scorbec — «язвы во рту», впоследствии
трансформированное в скорбут,
пристально изучали цингу. Ещё бы — ведь Васко
да Гама (1469-1524), например, потерял
от этого недуга 65 % своей команды. Сэр
Ричард Хокинс (1593), а много позже и
другие английские адмиралы XVIII века,
по результатам исторического
исследования шотландского доктора Дж. Линда
(20-26 мая 1747 на борту корабля «Салис-
бери»), приказами вводили профилактику
цинги на флоте лимонным соком, а русский
флотоводец Ф.Ф. Ушаков (1745-1817) во
время зимовок моряков применял с той же
382 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
целью свежее мясо, чем предвосхитил
современные данные о роли пищевого белка для
определения потребности в аскорбиновой
кислоте. Рыбий жир применял для терапии
рахита еще Т. Сиденхэм в XVII столетии.
В1810 г. итальянский врач Дж.-Б. Марзари
постулировал, что причиной пеллагры
является неполноценность кукурузной диеты.
Возникновение научной витаминологии
произошло в XIX веке и связано с
именами французского патолога Ф. Мажанди
41816), русского врача Н.И. Лунина (1880)
и главного санитарного инспектора флота
Японской Империи адмирала К.Такаки
(1885,1887).
Ф. Мажанди первым установил, что
рост молодых собак не может
поддерживаться нормально, «если они получают
только основные поддерживающие жизнь
вещества — сахар, маслообразные и альбу-
моидные вещества». Н.И. Лунин углубил
эти исследования, и ему удалось в
экспериментах на мышах показать, что животные,
получающие извлечённые из натурального
молока казеин, жиры и лактозу, воду и
минеральные соли в адекватных количествах,
болеют и чахнут, несмотря на это. В то же
время свежее молоко «содержит ещё и
другие вещества, незаменимые для питания» и
спасает подопытных мышей.
К. Такаки на значительном
статистическом материале показал, что причиной
бери-бери (по-японски: как'ке), которая
поражала на японских кораблях в
несколько'раз больше моряков, чем на английских,
плавающих в тех же широтах, служило раз-
личие в рационе. Адмирал успешно снизил
заболеваемость бери-бери, обогатив диету
матросов молоком, овощами и мясом.
Догадку о том, что целое семейство болезней,
включая цингу, рахит и «язву роговицы»,
представляет собой дефициты различных
незаменимых экзогенных химических
веществ, высказывал еще в 1842 г. Дж. Бадд в
своей работе «Расстройства, происходящие
от неполноценного питания».
Однако химическая природа витаминов
и их подлинная роль в обмене веществ,
так же как патогенез авитаминозов, стали
выясняться только в XX веке. Именно эта
область медицинского знания собрала
самый большой, после иммунологии, урожай
Нобелевских премий, причём большинство
из них — по химии (см. табл. В в
приложении).
В 1886 г. по направлению голландского
правительства авторитетный
патологоанатом К. Пикельринг и известный невролог
К. Винклер прибыли на остров Яву, где
свирепствовали массовые заболевания бери-
бери. В помощники им колониальной
администрацией был выделен молодой местный
военврач X. Эйкман. Учёные из метрополии
придерживались инфекционной теории
заболевания и, не найдя ей заслуживающих
внимания подтверждений, уехали. X.
Эйкман подошел к проблеме
патофизиологически: остался в Индонезии и в ходе
десятилетней работы создал модель бери-бери
на курицах, доказав, что симптомы болезни
зависят от отсутствия пищевого фактора,
представленного в рисовых отрубях (1897).
Нобелевской премии за своё открытие он
дождался только через 32 года. Несмотря на
эти достижения, химии витаминов не
существовало. Поэтому авитаминозы оставались
крупной социально-экономической и
медицинской проблемой. В Русско-японскую
войну бери-бери вывела из строя больше
японских солдат, чем российская армия
и флот вместе взятые (1/6 часть личного
состава). В свою очередь, в царской армии
в 1-ю Мировую войну более полумиллиона
военнослужащих страдали цингой — что во
много раз превысило, например,
заболеваемость желудочно-кишечными
инфекциями.
Первый шаг к созданию теории
незаменимых пищевых факторов и к их
химической идентификации сделал преподаватель
физиологической химии Ф. Дж. Хопкинс
(1906,1910).
Он провёл серию опытов по исключению
из диеты экспериментальных животных
тех или иных натуральных ингредиентов, с
их заменой на химически очищенные
эквиваленты, что привело сначала к открытию
незаменимых аминокислот и обоснованию
Патофизиология витаминного обмена 383
положения о неодинаковой пищевой
ценности разных белков, а затем к констатации
факта, что развитие бери-бери, скорбута и
рахита также зависит от отсутствия
натуральных незаменимых компонентов, но
неаминокислотного характера,
дополнительных по отношению к основным пищевым
ингредиентам.
Практически одновременно польский
биохимик К. Функ выделил из рисовых
отрубей азотсодержащее вещество,
излечивающее экспериментальную бери-бери
(1911), кристаллизовал его и назвал
«витамин» — амин жизни. Ему же принадлежит
введение термина «авитаминоз» для
обозначения расстройств, вызванных дефицитом
витаминов в организме. В 1911-1912 гг.
Ф. Хопкинс и К. Функ выступили с теорией
авитаминозного происхождения скорбута,
рахита, пеллагры и бери-бери. В 1909 г.
немецкий ученый У. Степп обнаружил,
что спиртоэфирная смесь экстрагирует из
чёрного хлеба жирорастворимое вещество,
необходимое для роста мышей, и назвал его
фактор роста А. Позже Э.В.Мак-Коллюм и
соавт. (1913) показали гетерогенность
жирорастворимых незаменимых факторов и
существование первого жирорастворимого
витамина — незаменимого ростостимулиру-
ющего фактора А сливочного масла (1913). К
1922 г. Т.Б. Осборн и Л.Б. Миндел выделили
этот фактор. Что же касается функовского
«витамина В», то именно он (тиамин) стал
первым, выделенным в чистом виде (А. Вин-
даус, 1932) и искусственно синтезированным
(Р. Уильяме, Дж. Клайн, 1936) витамином.
Последующие исторические вехи в
развитии учения о витаминах связаны с идеей
Х.фон Эйлер-Хельпина и П. Каррера о
провитаминах — естественных
предшественниках, метаболизируемых в активные
витамины в организме. На этой основе ими
была доказана роль каротинов, как
провитаминов А, а затем осуществлён синтез
витамина А (1929-1933). Наряду с этим на
примере авидина яичного белка и биотина
желтка яиц была создана концепция
антивитаминов — специфических антагонистов
незаменимых пищевых факторов (М.А. Бо-
ас, 1926). В 20-40-е годы были открыты,
выделены и структурированы практически
все основные витамины, кроме
сложнейшего по строению — кобаламина. Эта задача
потребовала применения рентгенострук-
турного анализа и была решена только в
1948-1956 гг. (Д.Кроуфут-Ходжкин). В
эти годы сложилась историческая
номенклатура витаминов (по С. Дремонду), в
которой не должно удивлять отсутствие
некоторых цифровых индексов при букве В.
Обозначавшиеся ими витамины оказались
в дальнейшем формами уже известных или
артефактами.
В 1921 г. известный русский химик
Н.Д. Зелинский высказал ценнейшую
гипотезу, что витамины метаболически
необходимы потому, что связаны со структурой
и функцией ферментов.
Впоследствии эта догадка нашла
блестящее подтверждение в отношении многих
водорастворимых витаминов, в частности
в работах, доказавших коферментную роль
витаминов В2, В6 (П.Каррер, Р. Кун 1930-
1939), Bj (P.A. Питере, 1936), никотиновой
кислоты (О. Варбург, 1935) и т.п.
Как справедливо замечают П.Е.
Калмыков и М.Н. Логаткин, многие
водорастворимые витамины в коферментной форме по
строению представляют собой нуклеотиды
или нуклеотидоподобные вещества. Так,
например, рибофлавин входит в состав
мононуклеотидного (ФМН) и флавинаде-
ниндинуклеотида (ФАД); то же относится
и к ниацину (НАД, НАДФ и НМН - ни-
котинамидмононуклеотид); половина
молекулы КоА, содержащего пантотеновую
кислоту, также является нуклеотидом;
пиридоксаминфосфат можно рассматривать
как комплекс пиримидинового основания и
фосфорной кислоты, пирофосфатная форма
тиамина — это, по сути, пиримидин-нуклео-
тид-фосфат, пуриновые остатки и пентозы
имеются у кобамидного кофермента и т.д.
Данная закономерность не случайна — так
как структура белков кодируется полину-
клеотидами, то между информационными
последовательностями первых и последних
существует сродство, в силу чего природа и
384 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
избрала на роль коферментных «вставок» в
каталитические белки именно нуклеотиды и
подобные им молекулы. Схематично можно
представить витаминсодержащие кофер-
менты энзимовитаминов, как:
N - УВ - P(S),
где N — азотсодержащая часть (азотистое
основание или близкая к нему структура),
У В — рибоза и рибозопроизводные, Р —
фосфат или пирофосфат. Иногда в витаминном
коферменте стоит вместо фосфата сера (S).
Дальнейшее развитие патофизиологии
витаминного обмена связано с осознанием того
факта, что недостаток метаболического
действия витамина может быть связан не только
с прямым дефицитом витамина или его
предшественника в употребляемой пище
(первичные или экзогенные гипо(а)витаминозы),
но и с эндогенными причинами (вторичные
гипо(а)витаминозы).
К последним принципиально относятся:
>• нарушение всасывания витаминов в
ЖКТ;
>• недостаточный синтез витаминов эу-
бактериальной микрофлорой ЖКТ и
клетками организма (для тех витаминов,
которые частично формируются из
провитаминов в организме);
>* «обкрадывание» организма в отношении
витаминов паразитами и сапрофитами
>• поступление в организм пищевых или
лекарственных антивитаминов;
>* нарушение активации или усвоения
абсорбированного витамина, в результате
внутренних болезней (печени, почек,
реже — других органов);
>* энзимопатии, приводящие, к
метаболической неффективности витамина;
>■ повышенные потери витамина;
>* относительная недостаточность при
повышении, в результате нагрузок,
беременности, кормления грудью и других
ситуаций, требующих адаптации,
потребности в витаминах.
Жирорастворимые витамины
накапливаются в организме, причём их депо включают
печень и жировую ткань. Водорастворимые
витамины в существенных количествах не
депонируются, а при избытке выводятся.
Обнаружение этого факта привело к
появлению представлений о гипервитамино-
зах — патогенных последствиях избытка
витаминов, главным образом
жирорастворимых. Появление доступных синтетических
препаратов витаминов и период «витамино-
логического романтизма» в практической
медицине и в общественном сознании в
изобилии поставили материал для изучения
клиники и механизмов гипервитаминозов
(в частности, А и Д), в основном ятрогенных
или вызванных самолечением. По этому
поводу В.Герберт и С. Барнетт замечают, что
«нет нужды ставить дюжину
регулировщиков вместо одного на одном и том же
перекрёстке. От этого автомобильное движение
не улучшится, скорее наоборот. Достаточно
их просто заменять» (1981).
Следует отметить, что картина
авитаминозов характеризуется яркими симптомами
и чётко очерченными проявлениями. Это
возможно только при полном отсутствии
поступления некумулируемых и несинте-
зируемых эндогенно витаминов
(например, аскорбиновой кислоты — при цинге).
Если витамин присутствует в питании, но
в уменьшенном количестве, поступает, но
не усваивается полностью, если он
способен запасаться или частично
синтезируется — клиническая картина бывает менее
яркой и специфичной. Тогда-то и говорят о
гиповитаминозе, который можно трактовать
и как начальную стадию авитаминоза. В 60-е
годы XX века сформировалось понимание
энзимовитаминной недостаточности как ко-
ферментной недостаточности. Однако затем
внимание патологов привлекли наблюдения,
свидетельствующие, что во многих случаях
при типичных симптомах гиповитаминоза
чистая витаминотерапия оказывается
малоэффективной, зато помогает коррекция
белкового рациона или введение тех или
иных микроэлементов (см. следующий
раздел). И наоборот, на фоне дефицита белка
даже малые дозы ряда витаминов, например
ниацина, могут быть токсическими. Обмен
Патофизиология витаминного обмена 385
многих витаминов гормонозависим. Так,
гипофизэктомия увеличивает содержание
НАД в печени, тироксин необходим для
введения пантотеновои кислоты в коэнзим А,
а парат-гормон — для гидроксилирования
провитамина D.
С установлением факта присутствия
микроэлементов в структуре многих вита-
минзависимых коферментов (например,
кинурениназа содержит и Вби Mg2+) стало
ясно, что симптомы витаминной
недостаточности зависят от неэффективности всего
комплекса апофермент-активированный
витамин-ион, а не просто от нехватки ко-
фермента. При этом ионы микроэлементов
могут играть роль источника и транспортёра
электронов или стерически сближать
активные группы фермента. Таким образом, стало
ясно, что фактически при гиповитаминозах
зачастую имеется дефицит апофермент-
витамин-минерального комплекса. Кроме
того, было отмечено взаимодействие самих
витаминов, например фолата и витаминов-
метильных донаторов (Т. Терруан, 1969).
Синергистами являются витамины — анти-
оксиданты, например Е и А. Из-за подобного
взаимодействия нескольких витаминов и
микроэлементов в одной технологической
цепочке (например, витаминов Blf B2, PP,
В15 и микроэлементов железа и меди при
осуществлении биологического окисления)
одни и те же симптомы могут
«перекрываться» в клинической картине разных болезней
питания. Так, поражение высокоаэробных
эпителиальных клеток кожи (с ее
производными) и слизистых рта, глотки и пищевода,
в виде дерматита, изменений волос и ногтей,
глоссита, хейлита, эзофагита и др. присуще
клинике нехватки нескольких разных
витаминов и дефицита микроэлементов, все из
которых участвуют в биологическом
окислении (см. ниже «Патофизиология обмена
микроэлементов»). Синергизм в отношении
многих эффектов присущ аскорбиновой
кислоте и тиамину. В то же время витамины
могут проявлять и антагонизм. Например,
нафосфорилирование тиамин и пиридоксин
влияют противоположно. Антагонистичны
некоторые эффекты ретинола и филлохи-
нонов, пиридоксина и токоферолов.
Существует и прямой химический антагонизм,
например витамина С и цианкобаламина,
не позволяющий назначать их в одном
шприце. Витамины-донаторы метильных
групп (кобаламин) и их акцепторы (ниацин)
противоположно действуют на образование
липопротеидов печенью.
Благодаря подобным успехам в
понимании взаимодействий незаменимых факторов
питания и природы авитаминозов в 70-80-е
годы витаминологи перешли от терапии
моновитаминами к применению сначала
поливитаминов, а затем коферментных
форм витаминов и витамин-минеральных
комплексов. В развитых странах
авитаминозы стали редкостью, а гиповитаминозы, в
США, например, по признанию В.
Герберта (1980), часто встречались лишь у лиц с
эндогенной недостаточностью витаминов
(алкоголики, пациенты с мальабсорбцией)
и представителей бедных слоев населения,
особенно у малообеспеченных пожилых
людей и беременных женщин.
Но появилась и стала отрицательно
влиять на общественное здоровье иная
проблема, которую В. Герберт и С. Барнетт (1981)
метко охарактеризовали, как «nutritional
cultism» — культ здорового питания или
«пищевой культуризм». Имеются в виду
усилия пищевой индустрии, парамедиков и
части врачей по продаже и популяризации
витаминно-минеральных,антиоксидантных,
травяных и иных «оздоровляющих»
пищевых добавок, а также общественная
атмосфера своеобразной модной истерии вокруг
этих продуктов и их мегадоз, сложившаяся
в ряде стран в 80-е годы XX века. В
настоящее время, встречаясь с законодательными
ограничениями за рубежом, волна
агрессивной рекламы и армия «суперсэйлсменов»
докатились и до России, где всё это пока в
новинку. Поэтому медику-профессионалу
будет нелишне ознакомиться с книгами
на эту тему, чтобы приобрести иммунитет
к такому явлению, которое называется
«food quackery» и давно известно нашим
зарубежным коллегам (см. В. Герберт, 1980;
В. Герберт, С. Барнетт, 1981). Наиболее
386 АЖ Зайчик, Л.П.Чурилов Патохимия(
впечатляющие факты из этой области
содержит история лаэтрила (амигдалина).
Этот цианогенный гликозид был выделен
Э.Т.Кребсом из абрикосовых косточек
и усиленно внедрялся в США и других
странах в 60-80-е годы, как «витамин В17»,
стимулирующий противораковый
иммунитет. Клиническая эффективность нового
витамина не была доказана в исследованиях
методом плацебо, более того, у него
обнаружилась мутагенная активность. Когда
произошло несколько смертельных случаев
цианистого отравления, от приема больших доз
амигдалина, это повлекло судебные иски, и
империя лаэтрила, с ежегодным объёмом
продаж 1000000000 американских
долларов, развалилась. Другая подобная история
связана с пангамовой кислотой —
«витамином В15». Это вещество, выделенное «отцом
амигдалина» Э.Т.Кребсом (1943) из того
же источника, было впервые синтезировано
в СССР И.Н. Горкиной и В.Н. Букиным как
Б-глюконо-6-бис-( 1 -метилэтил )-аминоаце-
тат, и авторы получили на него патент США
в 1975 г. Уже в 1978 г., вследствие удачной
рекламы, витамин В15 был самым
покупаемым аптечным продуктом в Нью-Йорке.
Считалось, что он увеличивает скорость
окислительных процессов и может
служить метальным донатором. Впоследствии
оказалось, что клиническая эффективность
пангамовой кислоты переоценивалась, и не
всегда может быть подтверждена в
исследованиях с применением двойного
слепого контроля, а метальные группы в этом
соединении не так лабильны, как в других
донаторах. Как следствие «пангамовый бум»
пошёл на убыль. В Канаде, а затем и в США
применение препарата было даже
запрещено. Так как у него не найдено вредного
действия, в нашей стране он не исключён из
фармакопеи. И всё же пангамовая кислота
не является незаменимой, к тому же она не
служит коферментом. Поэтому нет
оснований относить ее к витаминам.
Однако период увлечения мегадозами
витаминов оставил и позитивный след в области
наших представлений о болезнях питания.
Речь идёт о том, что в результате усилий
шно-метаболические нарушения
двукратного нобелевского лауреата (по химии
и физике) Л. Полинга, автора знаменитой
книги «Витамин С и простуда» (1970) по
пропаганде больших доз аскорбиновой кислоты,
произошли некоторые сдвиги в понимании
самой роли витаминов. Относительно
собственно мегадоз витамина С говорится ниже
в специальном разделе. Здесь же, касаясь
истории идей в витаминологии, важно
оценить подход, который сам Л. Полинг назвал
«ортомолекулярной медициной». Согласно
этой концепции, рекомендованный
минимальный прием витамина — это лишь доза,
предотвращающая явный гиповитаминоз. Но
увеличение потребления витамина до ортомо-
лекулярного или оптимального количества,
хотя и не является необходимым, чтобы
избежать клинического расстройства здоровья,
может привести к оптимизации метаболизма,
сделать практически здорового человека более
здоровым. Критики Л. Полинга упрекали
его в неклиническом подходе, механицизме,
заангажированности (его Институт орто-
молекулярной медицины финансировался
крупнейшим производителем витамина С
компанией «Хоффман-Лярош»),
справедливо указывая, что гипердозы водорастворимых
витаминов не усваиваются, а
жирорастворимых — опасны. Ряд авторов подчёркивал, что
Полинг сделал данный вопрос «пресловутым»
и объективно служил интересам того самого
«food quackery» (В.Герберт, 1980).
Однако прошли годы, сам великий
учёный, по трагической воле судьбы, пал в
1994 г. жертвой того недуга, панацеей от
которого он считал витамин С, — рака,
а рациональное зерно идей Л. Полинга
всё-таки получило адекватную оценку как
подход к концептуальному прорыву в
витаминологии.
Вот что пишет по этому поводу Дж.Р. Смай-
тис (1998): «Роль Полинга в медицине пора
переоценить. Этот тот случай, к которому
применима знаменитая эпитафия на
могильном камне в Томбстоуне, Аризона:
«Здесь лежит тело Джорджа Томпсона,
Повешенного за убийство в 1882.
Он был прав, а мы не правы,
Но мы его вздернули — мир праху его».
Дело в том, что для современных целей
деление витаминов на коферментные и
прогормональные недостаточно. И ранее
появлялись сомнения в адекватности
такой простой классификации.
Например, не были очевидны коферментные
функции аскорбиновой кислоты, которая,
в отличие от других водорастворимых
витаминов, не претерпевает в организме
активирующей трансформации в нуклео-
тид-подобный кофермент, а содействует
работе энзимов в неизменённой форме.
Жирорастворимые витамины, кроме D и
F, лишь с большой натяжкой подходили
под категорию прогормонов. И только
после открытия и изучения роли
эндогенных окислителей и контролирующих их
антиоксидантных факторов в организме
(см. подробную характеристику в т. I
данного руководства, с. 193-196) стало
ясно, что большая группа водо- и
жирорастворимых витаминов взаимодействует
в организме как антиоксидантная система.
Следовательно, с современных позиций
можно говорить, что существуют энзимо-
витамины (Bt, В2, РР, В6, В12, пантотеновая
кислота, Н, фолиевая кислота и др.), гор-
моновитамины (A, D2, D3), а также витами-
ж-антиоксиданты или редокс-витамины
(А, Е, С, липоевая кислота).
Впрочем, данные функции могут
сочетаться в одном витамине (например, кофер-
ментная и антиоксидантная). В отношении
последней группы и приходится оценить
предвидение Л. Полинга — повышение доз
аскорбиновой кислоты выше
определённого, гарантирующего от цинги уровня, вряд
ли, усилит ее коферментную функцию,
но, очевидно, способно упрочить ресурсы
антиоксидантных систем организма. В
отношении витаминов, обладающих ан-
тиоксидантной функцией, конечно, также
справедливы и предупреждения о риске
гипервитаминозов (например, А и Е). Но
как раз по отношению к ним концепция
оптимальной дозы вместо минимальной
имеет, на наш взгляд, определенные права
гражданства. В заключение данного раздела
хотелось бы обратить внимание читателя
Патофизиология витаминного обмена 387
на то, что практически все витамины, так
или иначе, связаны с регуляцией процессов
окисления — в качестве коферментов
окислительно-восстановительных реакций, как
редокс-системы и «ловушки» электронов.
К 70-м годам XX века, вследствие
расширительного толкования понятия «витамин»,
классификации насчитывали их более 20
(М.И.Смирнов, 1974; К.С. Петровский,
1975). При этом часть таких веществ не
отвечала тому или иному критерию (а то и
нескольким критериям)
вышеприведённого определения истинных витаминов. Так,
холин образуется в достаточном
количестве при наличии метионина и адекватном
обеспечении белком, а также выполняет
и пластические функции как компонент
фосфолипидов (см. выше «Нарушения
липидного обмена»). По тем же критериям
и отсутствию коэнзиматической функции
можно вывести из числа витаминов орото-
вую кислоту (см. выше «Патофизиология
нуклеинового метаболизма»).
Ненасыщенные жирные кислоты {«витамин F») — это
пластические компоненты ряда липидов,
в связи с чем потребность в них лежит
отнюдь не в «витаминном» микродиапазоне
суточных доз. «Витамин U» — метилме-
тионин — de facto является производным
незаменимой аминокислоты и отделялся
от этой группы эссенциальных нутриентов
искусственно (см. выше «Патофизиология
белкового обмена»). Циклический
многоатомный спирт инозит, хотя и незаменим
для животных, выполняет пластическую
роль в инозитфосфатидах и в нём имеется
суточная потребность на уровне 1-1,5 г/с.
Карнитин (витамин Вт) синтезируется в
организме людей. Парааминобензойная
кислота незаменима, но служит составной
частью другого витамина — фолацина.
Наконец, важным критерием витаминного
характера той или иной субстанции служит
наличие соответствующей клинической
картины гиповитаминоза при ее отсутствии
в организме. Поэтому современный список
витаминов насчитывает у большинства
авторов только 13 наименований (Food and
Nutrition Board, National Research Council
388 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения
Таблица 27 Витамины человека
Витамин
А,
А2
D2
D3
Е
к,
к2
В,
в2
в6
РР(В5)
В3
вс,в9
н
|В12
с
р
-
Синоним
Ретинол
Дегидроретинол
Эргокальциферол
Холекальциферол
а- (J- у-токоферолы
Филлохинон
Фарнохинон
Тиамин
Рибофлавин
Пиридоксин
Ниацин
Пантотеновая1 кислота
Фолацин
Биотин
Кобаламин
Аскорбиновая кислота,
дегидро-аскорбиновая
кислота
Биофлавоноиды,полифенолы
Липоевая кислота
Группа
Антиоксиданты (Ж)
Гормоновитамины (Ж)
Антиоксиданты (Ж)
Гормоновитамины, энзи-
мовитамины (Ж)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Энзимовитамины (В)
Антиоксиданты,
энзимовитамины (В)
Антиоксиданты (В)
Энзимовитамины,
антиоксиданты (В)
Кем и когда выделен
Осборн, Миндел; 1922
Виндаус, 1931
Ивенс, Эмерсон, 1921
Дойзи, 1939
Янсен, Виндаус, 1926
Кун, Вейланд, Каррер, 1934
Кун, 1939
Хубер, 1867 Элвехьем, Вули, 1937 —
идентификация как витамина РР
Уильяме, 1933
Митчелл, Снелл, Уильяме, 1941
Кегель, 1935
Райке, Смит„1948
Сент-Дьёрдьи, 1927
Сент-Дьердьи, 1936
Рид, Гунсалюс, 1953
NAS Recommended daily allowances, 1980;
В. Кумар и соавт., 1997).
За некоторыми «развенчанными»
субстанциями до сих пор сохранено название
«витаминоподобные вещества». Однако
они не рассматриваются в данном разделе.
Интересующегося ими читателя отсылаем
к более детальным специальным
руководствам (М.И.Смирнов, 1974; Л. Страйер,
1994). Ниже обсуждается патология обмена
16 веществ, которые авторы относят к
витаминам человека, руководствуясь
вышеназванными критериями. Ряд из них могут в
существенных количествах синтезироваться
микрофлорой кишечника — тиамин, ниацин,
фолацин, кобаламин, витамин К,
пантотеновая кислота, липоевая кислота, пиридоксин,
рибофлавин. Впрочем, последний активно
поглощается Lactobacilli. Роль этого
синтеза не следует переоценивать, так как не
вполне выяснена возможность всасывания
названных бактериальных продуктов из
дистальных отделов толстого кишечника. К
тому же синтез одних витаминов возможен
лишь при наличии других (так, витамин К
не образуется эубактериальной флорой без
витаминов биотина и фолацина).
Клетки человека способны ограниченно
вырабатывать витамин D3 и ниацин, но для
этого требуется ряд условий (см. ниже).
Таблица 27 содержит перечисление
витаминов.
НАРУШЕНИЯ ОБМЕНА
ЭНЗИМОВИТАМИНОВ
Проявления и механизмы гиповитаминозов
по различным энзимовитаминам настолько
взаимосвязаны и перекрываются, что это
дало право некоторым авторам трактовать
их недостаточность как принципиально
поливитаминную. Однако для большинства
из них описаны и специфические
авитаминозы. В нижеследующем разделе, кроме
их этиологии и патогенеза, мы уделили
внимание практически важному вопросу о
пищевых источниках витаминов. К
сожалению, профессионалу медику не приходится
удовлетворяться расхожей мифологемой,
что «богатым источником витаминов
являются исключительно фрукты и овощи». Как
мы увидим, это далеко не в каждом случае
так. Чтобы не повторяться, заранее
отметим высочайшее содержание большинства
энзимовитаминов группы В в пекарских и,
особенно в пивных дрожжах.
Недостаточность тиамина
(болезнь бери-бери)
Название данного заболевания происходит
от индийского beri — ножные оковы, что
выражает особенности неуверенной
шатающейся походки больных.
В тиамине нуждаются все животные,
за исключением жвачных. В связи с этим
существуют модели данного заболевания, в
частности модель X. Эйкмана на курицах.
В ветеринарии известен аналог бери-бери у
голубей и лисиц — паралич Частека.
Бери-бери издавна распространена в
странах южной, юго-восточной и восточной
Азии как расстройство питания, связанное
с однообразным потреблением
полированного риса. Эпизодически она встречается
по всему миру, в частности, и в развитых
странах — например, у алкоголиков при
синдроме Вернике-Корсакова. По некоторым
данным, каждый четвёртый
госпитализированный алкоголик имеет тиаминовую
недостаточность. Фактически лёгкие формы
бери-бери весьма распространены в качестве
«болезней цивилизации» повсеместно, так
как увеличение потребления
рафинированных углеводистых продуктов и сахара,
характерное для рациона городских жителей
многих регионов, повышает потребность в
Патофизиология витаминного обмена 389
тиамине. Парентеральное введение
глюкозы, не сбалансированное дополнительным
тиамином, приводит к его недостаточности.
Так как тиамин при всасывании должен
превратиться в пирофосфат («кокарбок-
силазу»), который и служит его активной
формой, существенная недостаточность
этого витамина развивается при мальабсор-
бционных синдромах. Тиамин разрушается
при продолжительной варке и теряется при
рафинировании зернопродуктов. Поэтому
в этиологии данного гиповитаминоза
велика роль характера приготовления пищи.
Кофе ингибирует фосфорилирующее
всасывание В„ а рыба и морепродукты, столь
несчастливо сочетавшиеся с
полированным рисом в питании матросов адмирала
К.Такаки, обладают высокой активностью
тиаминазы, разрушающей витамин. По
некоторым данным, тиаминаза принадлежит
бактериям, имеющимся в морепродуктах,
а тиаминазная болезнь поражает
несколько процентов сельского населения в ряде
районов Японии (В.Б. Спиричев, 1966).
Взрослому человеку необходимо не менее
1,4-2,4 мг витамина Bt в день, в зависимости
от калоража диеты и количества углеводов.
Богатейшие тиамином продукты (здесь и
далее — в мг/100 г) — свинина (0,8), овсянка
(0,6), греча (0,5), печень (0,37), чёрный хлеб
(0,26), яйца (0,14).
Имеются сухая и влажная форма болезни
бери-бери. В последнее время формой тиа-
миновой недостаточности считают и
синдром Вернике-Корсакова. Некоторые авторы
указывают на то, что формы могут сменять
друг друга как стадии — при прогрессиро-
вании тяжести заболевания — от сухой к
влажной, а далее — и к влажной с
элементами синдрома Вернике-Корсакова.
Сухая форма имеет симптоматику
распространенного дегенеративного демиели-
низирующего полиневрита с
преимущественным, но не исключительным поражением
соматических нервов нижних конечностей.
Наблюдаются миальгия, атрофия мышц,
миастения, потеря в весе. Возникает арефлек-
сия, расстройство чувствительности, у птиц
характерен опистотонус. В дальнейшем
390 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения
NH2
N ^^с— NH2— N —С —СНз
j сн СН х^'с-СНг-СНзОН
СНз X S
в/
асоон
-
N N
РР (ниацин)
aCONH2
СНз
Н3С
СНз
НОНС— СНОН—СНОН—СН2ОН
СНз
I
N N
СО
NH
N
В2
С
II
О
НОН2С—С—СНОН —С—N—СН2—СН2—С
I
V
он сн2он ctZ
°V>VCh2oh 01Ч^<£сн2он
пиридоксол пиридоксаль
пн CH2NH2
\£Г7
пиридоксамин
В6
N N
СНз ОН
Вз (пантотеновая кислота)
СООН
I
(СН2)4
СН —СН—NH
ОН
/
S
\
\
/
СН2—С NH
Н (биотии)
с=о
О Н
ОН
t^cH2"NH~0"H
птеридин + парааминобензоат
птероевая кислота
глютамил
птероилглютамат
Вс (фолацин)
развивается фобический невроз, снижается
интеллект. При влажной форме развивается
острая сердечная недостаточность и отёки
маскируют (но не подменяют) мышечную
атрофию. При этом признаки миокардио-
дистрофии (дряблый бледный миокард,
расширение всех камер сердца) сочетаются
с расширением периферических сосудов и
усиленным шунтированием крови, что ведет
к изотонической перегрузке сердца. При
синдроме Вернике-Корсакова происходят
кровоизлияния в сосцевидные тельца, пери-
вентрикулярную область зрительного бугра,
передний мозжечок и дно 4-го желудочка.
В результате отмечаются выраженные
нервно-психические нарушения:
(офтальмоплегия, нистагм, атаксия, апатия, дезори-
Рис 78. Химическая структура
водорастворимых энзимовитаминов В, В2,
В3, PR Вб, Вс. Пояснения в тексте
ентация, спутанность сознания, безразличие
т.е. энцефалопатия Вернике, к которой
присоединяются при прогрессировании
болезни ретроградная амнезия,
неспособность усваивать новую информацию и
болтливость — корсаковский психоз). Первым
мысль о пищевом дефиците какого-то
«тонкого химического соединения» как главной
причине бери-бери высказал еще в 1804 г. на
Цейлоне врач Т. Кристи. Но в дальнейшем
поколения медиков, будучи под
впечатлением от массовости вспышек этой болезни в
организованных коллективах в тропиках (у
солдат, моряков, заключенных), считали ее
инфекцией. Чтобы доказать дефицитарную
природу недуга, понадобились не только
многолетние эксперименты X. Эйкмана на
Патофизиология витаминного обмена 391
курах, описанные выше, но и
эпидемиологическое исследование инспектора
пенитенциарных заведений А. Вордермана в тюрьмах
Индонезии, использовавших разные пайки
заключенных (1897). Сам Эйкман вначале
истолковал все эти данные в духе старой
инфекционной теории и думал, что
крахмал очищенного риса служит бактериям
сырьем для производства нейротоксина, а
рисовые отруби его инактивируют. Однако,
его преемник Г. Грийнс (1901) доказал, что
ключевую роль играет именно отсутствие
нестойкого водорастворимого незаменимого
вещества. Позже Г. Фрэзер и Э.Т. Стэнтон в
Малайзии показали, что загадочное
вещество экстрагируется спиртом и вместе с ним в
алкоголь переходит лечебная активность
рисовых отрубей при бери-бери (1909). После
создания модели бери-бери на голубях было
доказано, что птиц излечивают дрожжи
(Г.Шауманн, 1911). И, наконец, К.Функ
извлек из рисовых отрубей кристаллы
неочищенного витамина, способные
радикально предупреждать и лечить бери-бери
(1911), а через 15 лет был получен чистый
тиамин (см. табл. 27).
Метаболическая роль витамина Bj —
тиамина (см. формулу на рис. 78) хорошо
изучена. Это источник коферментов для
энзимов, расщепляющих межуглеррдные связи.
Он входит в состав пируватдекарбоксилаз-
ного комплекса, одного из ключевых
ферментов пентозного шунта — транскетолазы,
а также а-кетоглутаратдегидрогеназы.
При нарушении его обмена прежде всего
возникает расстройство окислительного
декарбоксилирования пирувата и вообще
а-кетокислот, и частично блокируется
метаболизм углеводов. Именно поэтому
течение гиповитаминоза Вх ухудшается
углеводистой диетой. По этой же причине
у больных бери-бери имеется понижение
дыхательного коэффициента, дизоксида-
тивная карбонурия, накопление и
выведение продуктов недоокисления пирувата,
которые и действуют токсически на ЦНС.
Возникает метаболический ацидоз и
энергодефицит, сказывающийся на работе
градиентных насосов клеток, в том числе
нервных, мышечных, сердечных. Так как
не метаболизируются до конца углеводы,
организм вынужден использовать больше
белков и жира, что ведет к исхуданию и
мышечной атрофии, а у детей — к задержке
физического развития. Азотистый баланс
становится резко отрицательным.
Нарушается синтез жирных кислот и переход
углеводов в жиры. Имеется торможение
переаминирования аминокислот. Страдает
из-за трудностей с образованием АКоА из
пирувата ацетилирование холина, и
возникает нарушение обмена ацетилхолина, что
в ЖКТ приводит к запорам. Большое
значение для развития симптомов бери-бери
имеет некоэнзимная функция тиаминпи-
рофосфата в нервной ткани: известно, что
его дефосфорилирование происходит при
генерации потенциалов действия (А. фон
Мюральт, 1962). Дефицит образования
тиаминпирофосфата проявляется
некротической энцефалопатией и при
наследственном митохондриальном синдроме Лея (см.
т. I, стр. 173-174). Ранним лабораторным
показателем тиаминового дефицита
служит снижение активности транскетолазы
эритроцитов, а клиническим — боли в
икроножных мышцах.
По водорастворимому тиамину не бывает
настоящего гипервитаминоза, однако
парентеральное введение его большой дозы может
вызвать анафилактоидный шок вследствие
способности данного витамина в
определенных дозах неспецифически дегранулировать
тучные клетки.
Недостаточность рибофлавина
Витамин В2был выделен в 1933 г. Р.
Куном. Для получения 1г лактофлавина
понадобилось почти пять с половиной тонн
обезжиренного молока. Вот уж поистине,
как у В.В. Маяковского — «В грамм
добыча — в год труды!» Двумя годами позже
структуру витамина определил П. Каррер
(рис.78).
Рибофлавин поступает в организм в
составе яиц (0,69), сыра (0,43), молока (0,19),
гречи (0,24), печени и мяса (0,13),
необходим взрослому в суточной дозе 1,9-3,0 мг
392 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
и всасывается в верхней части ЖКТ.
Гиповитаминозу В2 способствуют хранение
продуктов на свету, фототерапия, алкоголизм и
мальабсорбция. Хлорпромазин, адриамицин
и антидепрессанты ингибируют флавоки-
назу и препятствуют фосфорилирующему
всасыванию рибофлавина.
Арибофлавиноз (описан у человека
У. Себреллом и Р. Батлером в 1939 г.)
проявляется нарушением процессов
биологического окисления, поскольку данный
витамин входит в состав флавиновых дегид-
рогеназ. Клинически, следовательно, будут
страдать высокоаэробные эпителии кожи и
полости рта. Развиваются хейлоз (трещины
на губах), ангулярный стоматит и глоссит
(фуксиновый язык), себоррейный дерматит
носогубного треугольника, а также
мошонки, ушей и шеи, интерстициальный кератит
(васкуляризация роговицы), блефарит и
конъюнктивит. Снижается детоксикаци-
онный потенциал печёночных оксидаз в
отношении ряда лекарств. Нарушается
темновая адаптация и ухудшается цветовое
зрение. Довольно часто, особенно у детей,
этот гиповитаминоз сопровождается нор-
мохромной гипорегенераторной анемией и
лейкопенией. Проявления арибофлавиноза
усугубляются жировой и углеводной
нагрузкой. Интересно, что устойчивость к малярии
растет при арибофлавинозе, несмотря на
все эти неприятности. Очевидно,
плазмодии страдают от дефицита витамина еще
сильнее, чем клетки человека. Возможно,
нарушается выживание инфицированных
эритроцитов.
Патогенез проявлений арибофлавиноза
связан с участием В2 в окислительном фос-
форилировании, работе сукцинатдегидро-
геназы, моноаминоксидазы и митохондри-
альных оксидаз жирных кислот. Витамин
участвует в функции эритроцитарной
глутатионредуктазы, предохраняющей
красные кровяные клетки от аутоокисления.
Активность последнего фермента считается
чувствительным лабораторным тестом на
арибофлавиноз, как и снижение
экскреции его флюоресцентных производных с
мочой.
Недостаточность ниацина
(пеллагра) и его избыток
Дефицит витамина РР (ниацина)1
проявляется пеллагроидом, а в тяжелых
случаях — пеллагрой. Название болезни восходит
к итальянскому «pelle agra — шершавая
кожа», а первое научное описание дано
врачом испанского короля Филиппа V,
К.Казалем (1735). Исторически она была
известна как сезонное весеннее заболевание,
характерное для малообеспеченного
сельского населения южноевропейских районов
(Италия, Испания, Румыния, Молдавия,
Западная Грузия), массовое везде, где в
питании большую роль играла кукуруза.
Пеллагра отмечалась и в США, но
практически неизвестна у коренного населения
Центральной Америки — родоначальников
культуры маиса. Особенности экологии
питания индейцев предусматривают
эмпирически сложившееся известкование
кукурузных лепёшек, способствующее
повышению их ниациновой ценности. Само
название «витамин РР» дано по первым
буквам характеристики «pellagra preventive»
Дж. Гольдбергером, нашедшим в дрожжах
фактор, излечивающий подопытных
животных от болезни-аналога пеллагры (1926).
Ранее, доказав на себе невозможность
передачи пеллагры от человека к человеку
(1916), этот военный врач опроверг
доминировавшую тогда инфекционную теорию
этиологии данного недуга. Гольдбергер
путем опытов на 12 заключенных в штате
Миссисипи показал, что пеллагру вызывает
кукурузно-злаковая диета, лишенная
мясомолочных продуктов (1915).
Витамин РР требуется взрослому в
суточной дозе 14-25 мг, он устойчив к
термообработке. Синтез никотиновой
кислоты возможен из триптофана в кишечнике
(бактериями) и в самих клетках организма, в
присутствии витамина В6.60 мг триптофана
1 В русской литературе иногда обозначается, как
витамин В5, а в американской — как В3, что
может создавать путаницу, так как в европейских
текстах обозначение В3 закреплено за пантоте-
новой кислотой.
равноценны 1 мг витамина РР (ниациновый
эквивалент). Белки кукурузы и сорго
бедны триптофаном и содержат неусвояемый
связанный ниацин. Считается, что имеет
значение и высокое лейцин-изолейциновое
соотношение в белках этих культур, ибо
лейцин ингибирует конверсию триптофана
в витамин PP. Продукты, поставляющие
много витамина РР — это печень (15), мясо
(от 2,3 до 4,5 — больше всего, в баранине),
рыба (2,2 — больше всего в лососёвых). Из
растительной пищи относительно много
РР имеется в бобовых (2-2,6), грече (4,2),
чёрном хлебе (3,1), но его усвояемость из
растительных продуктов намного ниже.
Причина пеллагры может быть связана
с мальабсорбцией, алкоголизмом,
длительным приемом изониазида и цитостатиков,
а также белковым голоданием. Считается,
что дефицит вышеописанных витаминов
группы В способствует манифестации
пеллагры. В составе ниацин-зависимых
ферментов часто имеются
микроэлементы — цинк, магний, марганец, молибден и
кобальт. Поэтому многие авторы
подчёркивают роль полидефицита незаменимых
факторов питания в развитии этой болезни.
Вторичная пеллагра характерна из-за
нарушения доступности триптофана при болезни
Хартнупа и карциноидном синдроме (см.
выше — «Нарушения межуточного обмена
аминокислот...»)
Дж. Гольдбергер и соавт. описали у
собак спонтанный аналог пеллагры («чёрный
язык») (1928) и получили модель болезни
на крысах.
Никотиновая кислота и никотинамид,
объединенные под названием ниацин
(см. рис. 78), играют роль коферментов в
НАД-зависимых дегидрогеназах
(участниках тканевого дыхания, метаболизма
углеводов и аминокислот), НАДФ-зависимых
ферментах (пентозного шунта и синтеза
липидов), НМН-зависимых энзимах (ал-
когольдегидрогеназа, малик-фермент). Не
менее (а для клиники гиповитаминоза — по-
видимому, более) важна роль НАД как
субстрата поли-АДФ-рибозилирования.
Данный процесс (см. т. I, с. 144-145) участвует
Физиология витаминного обмена 393
в сшивке хромосомных разрывов и в работе
репаразной системы, а также имеет (при
нехватке НАД) ключевое значение в
механизме некробиоза и апоптоза клеток,
особенно — высокоаэробных. Поэтому при
пеллагре происходит ускоренная гибель ряда
клеток, не уравновешенная их пролиферацией.
В связи с торможением процессов
апоптоза и некробиоза в островках Лангерганса
высокие дозы никотинамида (3 г/сутки) были с
успехом использованы для замедления
скорости развития инсулинзависимого диабета у
лиц с доказанным наличием аутоиммунного
инсулита (Х.П. Чейз и соавт., 1990). Редокс-
витамины (см. ниже) проявляли синергизм
с высокими дозами никотинамида в
антидиабетическом действии.
Клиническая картина обычно включает
«синдром трёх Д»:
>- Дерматит носит двусторонне-симмет-
ричный характер и локализуется на
открытых участках кожи (например,
«ожерелье Казаля» — на шее). Это
эритема, связанная с облучением,затем
присоединяются гиперкератоз,
отшелушивание, трещины, гиперпигментация
и вторичная инфекция. Поражаются и
слизистые — имеется уретрит, вагинит,
проктит.
>- Диарея вызвана атрофией эпителия
желудка и кишечника и в типичном случае
представляет понос, вначале — без слизи
и крови. Имеется и поражение
начальных отделов ЖКТ -- гипертрофический
бороздчатый глоссит с отёком и
малиновой слизистой, афтозный стоматит с
выраженной гииерсаливацией,эзофагит.
Возможны эрозии и язвы ЖКТ. Именно
пеллагроидный компонент вызывает
сходную картину поражения ЖКТ при
полном длительном голодании (см.
соответствующий раздел выше). Гаст-
роинтестинальный синдром замыкает в
патогенезе пеллагры порочный круг, так
как происходит срыв всасывания ниаци-
на и триптофана, а также других нутри-
ентов. Именно дегидратация вследствие
мальабсорбции приводит к быстрой
394 АЖ Зайчик, Л.П.Чурилов Патохимия
смерти при острой форме пеллагры (за
2-3 недели).
>- Демещия представляет результат
хронического рецидивирующего поражения
ЦНС. При пеллагре психо-неврологи-
ческие проявления обильны и включают
неврастенический синдром, сильнейшие
нервно-мышечные и головные боли и
парестезии в форме жжения (описаны
случаи психоза и самоубийств больных
на этой почве!). Психоз протекает с
дезориентацией, бредом,
галлюцинациями. В ЦНС отмечается гибель
нейронов и дегенерация проводящих путей,
в спинном мозге картина напоминает
фуникулярный миелоз при дефиците
кобаламина. Психотические проявления
дефицита витамина РР навели
сторонников ортомолекулярной медицины на
мысль использовать витамин, в терапии
шизофрении, но клиническая
эффективность такого метода не была доказана.
>• Другие проявления болезни могут
включать анемию, миастению, миокар-
диодистрофию.
Метаболизм витамина РР в печени связан
с его необратимым метилированием. Как
метальный акцептор, никотинамид участвует
в регуляции печёночного синтеза ЛПОНП.
Прием ниацина снижает продукцию этих
липопротеидов, а также сильно уменьшает
скорость деградации Л ПВП, что
используется при профилактике и лечении некоторых
гиперлипопротеинемий (см. «Нарушения
липидного обмена»). Но большие дозы
витамина РР при длительном приеме
способствуют обратимому ожирению печени, так
как этот орган перестает нормально
выделять в кровь липиды. Метальные донаторы
(холин, метионин, бетаин) препятствуют
развитию стеатоза печени при совместном
с витамином РР использовании.
Чувствительным тестом на
гиповитаминоз РР служит определение пониженной
экскреции М^метилникотинамида с
мочой.
Еще одна особенность высоких доз
витамина РР, наблюдаемая у никотиновой
чдокринно-метаболические нарушения
кислоты, но не никотинамида, — это её гис-
тамин-либерирующий эффект.
Никотиновая кислота способна вызвать освобождение
гистамина из мастоцитов неспецифически,
без сенсибилизации, по типу аллергоидной
реакции. Это может сопровождаться зудом,
крапивницей, кожной гиперемией и жаром,
гиперацидным состоянием и даже
коллапсом (см. т. I, с. 451).
Дело в том, что при дозе более 50 мг на
прием организм большинства людей не
успевает быстро метаболизировать
никотиновую кислоту в амид. Впрочем, именно
данный аспект действия никотиновой кислоты
используется как лечебный, например при
синдроме Рейно. Таким образом, в
применении к этому витамину справедлива фраза
В. Герберта (1980): «Много витамина — это
не всегда хорошо. Много — это иногда
лучше, иногда хуже, но всегда — дороже».
Дефицит и избыток витамина В6
Витамин Вб — это пиридоксин, пиридоксаль
и пиридоксамин. Его структура (рис. 78)
определена Р. Куном (1939).
Потребность в данном витамине у
взрослых — 1,5-2,8 мг/сутки. Он устойчив к
термообработке, но теряется при
консервировании и фоточувствителен. Многие виды
продуктов богаты В6, в связи с чем чисто
диетический, изолированный дефицит
этого витамина у человека, вне
экспериментальных условий, не описан. Больше всего
пиридоксаля и пиридоксамина в печени,
почках, мясе (от 4,0 до 0,8). Из
растительных продуктов, где имеется пиридоксин,
лидируют ячмень (до 2,3), кукуруза (1,0),
соя (0,9).
Гиповитаминоз В6 возможен при
использовании лекарств — антагонистов
витамина: антитуберкулёзных средств
(изониазид, фтивазид, тубазид, циклосе-
рин); пеницилламина, используемого для
лечения гепатолентикулярной дегенерации
и цистинурии, L-ДОФА, а также эстрогенов,
в том числе — противозачаточных средств.
Значительно повышена потребность в
витамине при беременности, в связи с чем
гиповитаминоз В6даже считают (наряду с
дефицитом витамина Е) патогенетическим
фактором раннего гестоза. Стресс,
лихорадка, гипертироз и любые другие
метаболические ситуации, сопряжённые с ускорением
катаболизма белка, требуют повышенных
количеств Вб. Всасывание витамина
страдает при выраженной мальабсорбции, а распад
усилен при алкоголизме.
После всасывания витамин
активируется в печени в пиридоксальфосфат, при
участии рибофлавиновых ферментов путем
фосфорилирования. Витамин В6 — главный
компонент ферментов переаминирования и
дезаминирования, а также некоторых син-
тетаз и рацемаз аминокислот. Процессы,
определяющие физиологический состав смеси
аминокислот в крови и балансирующие его
при разном наборе аминокислот пищи,
зависят от этих реакций. Поэтому при
гиповитаминозе по данному витамину прежде всего
страдает обмен белка и наблюдается гипера-
миноацидемия, аминацидурия, оксалурия,
вызванная нарушением обмена глиоксалевой
кислоты, и отрицательный азотистый баланс,
а потребность в нём зависит от количеств
и качества белка в диете (см. выше
«Патофизиология белкового обмена»). Витамин
участвует не только в работе карбоксилаз, но
и в преобразовании арахидоновой кислоты
в линолевую, что делает его существенным
для обмена липидов. При дефиците как В6,
так и витамина F, у подопытных крыс
отмечается акродиния — себорейный дерматит с
поражением ушей, кончиков лапок, хвоста,
кожи вокруг рта. Себорейный дерматит
отмечен при данном гиповитаминозе и у людей.
Дерматит при пиридоксиновом дефиците
отличается от поражения кожи при нехватке
ненасыщенных жирных кислот наличием не
только шелушения, эритемы и пигментации,
но и отёка.
Пиридоксину присуща липотропная
активность, вероятно связанная с участием в
обмене метионина. Вместе с тем витамин В6
способствует синтезу антилипотропного ни-
ацина из триптофана и участвует в синтезе
сфинголипидов. На обезьянах
продемонстрировано ускорение атеросклероза при
Патофизиология витаминного обмена 395
дефиците пиридоксина, а у людей, больных
атеросклерозом, зафиксировано
увеличение потребности в витамине В6. Фермент
8-аминолевулинатсинтетаза, участвующий
в начальных этапах синтеза гема, использует
В6 как кофактор.
У алкоголиков, из-за ускорения
разрушения В6, вследствие влияния ацетальдегида на
активность его оксидазы, отмечается
нарушение утилизации железа костным мозгом,
сидеробластическая анемия, гемохроматоз.
Частично анемия связана и с понижением
скорости абсорбции кобаламина,
характерным для дефицита пиридоксина.
Нарушение переаминирования при
данном гиповитаминозе способно отразиться на
синтезе белка в быстро пролиферирующих
тканях, например костном мозге: отмечается
лейкопения. У детей при дефиците этого
фактора питания задерживается рост.
Витамин В6, в условиях нарушения белкового
обмена, находится в сложных
взаимоотношениях со своими апоферментами. При его
дефиците возможен избыток апофермен-
тов, синтез которых затем, при коррекции
гиповитаминоза, активируется витамином.
Пиридоксин — единственный витамин, при
дефиците которого наблюдаются эпилеп-
тиформные судороги (Ю.В. Букин, 1966).
Дело в том, что данный витамин необходим
для активности глутаматдекарбоксилазы,
обеспечивающей синтез тормозного
медиатора ГАМК, и для утилизации триптофана и
синтеза серотонина, при нарушении которых
образуются метаболиты типа ксантуреновой
кислоты. Эти аспекты биологической роли
В6 связаны с патогенезом судорог,
гипервозбудимости и повышенной чувствительности
к шуму у крыс и новорожденных детей с
дефицитом пиридоксина. Ксантуреновая
кислота препятствует инсулиногенезу, что
может обусловить развитие диабетоподоб-
ных состояний.
Неспецифические проявления
гиповитаминоза В6 включают хейлоз, глоссит и
другие, сходные с вышеописанными для
остальных витаминов группы В, симптомы.
Ранним лабораторным признаком ви-
таминодефицита служит в данном случае
396 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
пониженная экскреция 4-пиридоксиновой
кислоты с мочой. Понижается и активность
эритроцитарных трансаминаз.
При употреблении сверхвысоких доз
витамина В6 — от 2 г/сутки — развивается
сенсорная нейропатия с онемением кожи,
особенно вокруг рта, нарушением
вибрационной чувствительности. Эти проявления
трактуются как острый гипервитаминоз В6.
Недостаточность пантотеновой
кислоты
Апантотеноз получен только в
эксперименте у животных и проявляется судорогами,
парезами, дерматитом, эрозиями ЖКТ, ги-
полипидемией и стеатозом печени. Важной
чертой расстройства является
недостаточность стероидогенных органов —
происходит геморрагический некроз надпочечников.
У человека большую роль дефициту именно
данного витамина придают в патогенезе
педиолалъгии (эритромелалгии) —
заболевания, связанного с поражением малых
артерий дистальных отделов нижних
конечностей. Жжение в стопах считается
характерным субъективным проявлением
гипопантотеиоза. Суточная потребность в
пантотеновой кислоте (рис. 78) у
взрослого — около 10-15 мг. Больше всего витамина
в пчелином маточном молочке (50), печени
(9-30, особенно — в бараньей), в яйцах (до
2,7), в рыбе (лососёвые) и в хлебе (0,8).
Кулинарная обработка разрушает витамин
в значительной степени, при варке кислота
переходит в бульон. Более 3,4 мг витамина в
сутки синтезирует, при отсутствии дисбак-
териоза, микрофлора кишечника.
Метаболическая роль данного витамина
обширна, так как он входит в универсальный
катаболит — ацетилкоэнзим А, и в ацилпере-
носящий белок (а также его аналог — муль-
тиферментный комплекс, синтезирующий
жирные кислоты животных клеток).
Особенно велико участие пантотеновой кислоты
в процессах ацетилирования, утилизации
пирувата, синтезе липидов и стероидов.
Всего почти 80 ферментов зависят от
пантотеновой кислоты, в связи с чем она и
получила название от слова, обозначающего
по-гречески «вездесущий».
Недостаточность биотина
Витамин Н (биотин — см. рис. 78) впервые
был выделен из яичного желтка.
Интереснейшей природной особенностью этого
витамина оказалось наличие в яичном белке
его высокоспецифичного лиганда — гли-
копротеида аеиЪина, который оказался
способен к аффинному самопроизвольному
необратимому связыванию витамина Н со
значительным уменьшением энтальпии, то
есть в термодинамически выгодном режиме.
Авидин (70 кД) содержит 4 субъединицы,
и каждая связывает биотин. Система ави-
дин-биотин оказалась первой
биохимически охарактеризованной парой, которая
могла служить моделью избирательного
комплементарного взаимодействия. Ее
изучение оказало влияние не только на
витаминологию, но и на становление
микрокалориметрии и понимание
комплементарных взаимодействий вообще— в том
числе иммунологических (У.Бойд; 1969).
Был даже создан способ биохимического
определения различных веществ на основе
использования принципа их конкуренции
за авидин с биотинилированными
аналогами — предшественник иммуноконкурент-
ного анализа.
Человек нуждается в очень малых
количествах витамина Н — 0,15-0,20 мг/сутки.
Изучение гиповитаминоза Н затруднялось
тем, что из-за широкого распространения
биотина в природных продуктах (печень -
0,25; соя — 0,06; желтки яиц — 0,03, грибы
и цветная капуста до 0,02, рис — 0,012) в
естественных условиях это состояние у
людей в чистом виде не наблюдается.
Детям, почему-то убеждённым, что полезны
только не очень вкусные продукты, приятно
бывает узнавать, что высоким содержанием
биотина отличаются шоколад, арахис и
грибы. Дрожжи и здесь, как и для
витаминов группы В, оказываются среди лидеров.
К тому же биотин синтезируется кишечной
микрофлорой, и человек выделяет его с ка-
лом больше, чем потребляет. Для усвоения
пищевого витамина Н биотинидаза
поджелудочного сока должна освободить его от
белка. Гиповитаминоз Н развивается при
комплексном нарушении поступления
витаминов, например мальабсорбции, в условиях
дисбактериоза. Данному гиповитаминозу
способствует употребление в пищу сырых
яичных белков. В эксперименте показано,
что 12 белков, съедаемых в день, снижают
всасывание витамина Н до такого уровня,
что через несколько суток развивается
Н-гиповитаминоз. Имеются случаи
гиповитаминоза Н при парентеральном питании. У
крыс, цыплят, а также оленей картина биоти-
новой недостаточности включает остановку
роста, дерматит, облысение, депигментацию
производных кожи. У людей выражен
чешуйчатый себорейный дерматит носогубно-
го треугольника и волосистой части головы.
Это сопровождается конъюнктивитом,
гиперестезией, миальгией, анемией, атаксией и
часто — дислипопротеинемией. Считается,
что дефицит витамина Н — важное звено
патогенеза десквамативной эритродермии
Лейнера у новорожденных, когда имеется
понос и характерная себорея. При данной
болезни обнаружена недостаточность
панкреатической биотинидазы.
Витамин служит коферментом карбок-
силаз, включая такие важные энзимы, как
пируваткарбоксилаза, ацетил-КоА-кар-
боксилаза, пропионил-КоА-карбоксилаза.
В связи с этим он необходим для
адекватного синтеза жирных кислот и стеринов,
а также для образования оксалоацетата,
«втаскивающего» активные одноуглеродные
фрагменты в цикл Кребса.
Недостаточность фолацина
(фолиевой кислоты)
Существование данного витамина было
предсказано отечественным автором
В.В.Ефремовым (1926), открывшим ме-
галобластическую анемию беременных,
которую хорошо излечивала диетотерапия
печенью и дрожжами. Позже Л. Уиллз и
соавт. обнаружили, что сходная пищевая
Патофизиология витаминного обмена 397
цитопения у обезьян излечивается теми
же продуктами (1937), а Ф. Дей назвал
гипотетический незаменимый фактор,
ответственный за ее предупреждение,
«витамин М» — от англ. «monkey». Другое
традиционное наименование данного
витамина — Вс — связано с тем, что его свойства
изучались на цыплятах (chicken). Иногда
данный витамин обозначают как В9.
Фолиевую кислоту (фолацин) впервые
выделили из листьев (folia) шпината П.
Митчелл и соавт. (1941-1944), им же удалось
установить, что данное вещество является
птероилглутаминовой кислотой и содержит
остатки 2-амино-4-окси-6-метилптеридина,
глутаминовой и, что особенно интересно,
парааминобензойной кислот. Последняя
кислота известна как незаменимый фактор
для многих микроорганизмов. Глутамино-
вых остатков в молекуле витамина может
быть различное количество. Фолиевая
кислота представлена у животных в основном
гептаглутаминатами с у-глутамильными
связями, в бактериях — триглутаминатами.
Кишечная микрофлора человека
синтезирует некоторое количество фолацина при
наличии парааминобензойной кислоты (рис.
78). Фолаты содержатся главным образом в
растительной пище — бобах (160 мкг/100г),
петрушке (117), спарже и брокколи (более
100), шпинате (53). Но было бы ошибкой
полагать, что в животных продуктах их нет
(в печени -160, почках — 45, мясе —10).
Фолатами богаты и грибы. Лидируют по
содержанию фолата, как и в отношении
иных витаминов группы В, дрожжи.
Фолацин термически крайне неустойчив.
Кроме того, для его усвоения из природных
продуктов необходим гидролиз полиглута-
матов конъюгазами гликокаликса тонкого
кишечника до моноглутамата. Бобы, хотя и
содержат много полиглутаматов, считаются
плохим источником фолацина, так как в них
имеется термоактивируемый ингибитор
конъюгаз. В норме усваивается не более
30-40 % природных полиглутаматов. При
спру их усвоение резко падает. Полагают,
что при некоторых формах данного
заболевания может иметь место первичный дефект
398 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения
гидролаз, необходимых для мембранного
пищеварения предшественников
абсорбируемого фолацина. Порочным кругом в
развитии спру считается вторичное
нарушение регенерации кишечного эпителия при
отсутствии всасывания фолацина, которое
дополнительно ограничивает поступление
фолиевой кислоты в организм.
Различные формы спру, особенно тропические,
рассматриваются как состояния, клиника
которых главным образом, определяется
фолациновым гиповитаминозом.
Всасывание фолацина снижается у
алкоголиков под действием этанола, а также
при приеме ряда лекарств (фенитоина,
барбитуратов). Кислые пищевые продукты
понижают абсорбцию фолатов. В целом по
причине крайней неустойчивости к
кулинарной обработке и плохого всасывания
сбалансированный рацион содержит не более
2/3 от суточной потребности в фолацине,
которая составляет у взрослого человека до
400мкг/сут. Остальное дополняет
микрофлора кишечника. Беременность, кормление
грудью и другие состояния напряжённой
адаптации характеризуются очень
значительным повышением потребности в данном
витамине и соответствующим возрастанием
степени риска гиповитаминоза.
Установлено, что физиологическое развитие плода
протекает с такой высокой потребностью в
фолацине и кобаламине, что их
относительный дефицит в фетальный период
неизбежен, что и обусловливает фетальный мега-
лобластический эритропоэз (см. т. III). Тем
не менее абсолютный дефицит фолацина у
беременных связан со значительным
нарастанием частоты пороков развития нервной
трубки у потомства (М. Селлер, Н. Невин,
1984). Повышены потери фолатов у лиц с
экзоэритроцитарными гемолитическими
анемиями, когда внутриэритроцитарная
фолиевая кислота оказывается в плазме
и выводится с мочой, а также у больных
шелушащимися кожными заболеваниями.
Гемобластозы и злокачественные опухоли
тоже сильно увеличивают потребности в
фолацине. Наконец, фолацин теряется при
гемодиализе.
Запасы фолатов в организме
сосредоточены в печени и почках и приближаются к
5-20 мг, что оттягивает начало клинически
явного гиповитаминоза на несколько
месяцев после момента, когда поступление
фолацина становится неадекватным.
Заболевания печени повышают скорость
развития фолатной недостаточности. Фолацин в
крови переносится специальным
транспортером в виде №-метилтетрагидрофолата.
При поступлении в клетку данная форма
витамина превращается в деметилирован-
ную при участии витамина В12. Если этого
не происходит, клетки будут легко терять
фолат, так как полиглутаминированию и
сохранению в интрацеллюлярном отсеке
подвергается только тетрагидрофолат.
Именно поэтому при гиповитаминозе В12
развивается и нарушение использования
клетками фолацина (см. ниже).
Метаболическая роль фолацина крайне
важна (см. т. III гл. 1 и Практикум). Витамин
активен в форме тетрагидрофолата, который
возникает из дигидрофолата при действии
дигидрофолатредуктазы. Наследственный
дефект этого фермента ведет к стойкой
мегалобластической анемии.
Тетрагидрофолат — это кофермент, содействующий
включению одноуглеродных фрагментов
(метильной, формильной, метиленовой
групп) из серина и дериватов гистидина в
различные соединения. Важнейшим
аспектом этой функции является участие фолат-
зависимых ферментов в синтезе пуринов,
а также пиримидинового азотистого
основания тимина (см. выше «Патофизиология
нуклеинового метаболизма»). Фермент
тимидилатсинтетаза превращает дезокси-
уридил-монофосфат в дезокситимидил-
монофосфат. При этом тетрагидрофолат
окисляется в дигидрофолат и нуждается в
восстановлении. Если имеется
гиповитаминоз по фолацину, уридилаты не
превращаются в тимидилаты. Синтез РНК при этом
существенно не страдает. Но в ДНК тими-
новые нуклеотиды перестают включаться и
частично заменяются на уридиновые.
Образуется аномальная легко фрагментируемая
ДНК. Кроме того, синтез ДНК резко замед-
Патофизиология витаминного обмена 399
к вторичной гомоцистеинурии и нехватке
незаменимой аминокислоты метионина, что
способствует атеросклерозу, тромбозам и
стеатозу печени, понижает функцию
надпочечников (К.С. Мак-Кулли, 1969).
Лабораторные признаки фолациново-
го дефицита включают также снижение
уровня фолацина плазмы (в норме 6-15
нг/мл) и фолацина эритроцитов (в норме
150-600 нг/мл эритроцитарной массы).
Если причиной фолацинового дефицита
служат гемобластозы и печеночная
недостаточность, то уровень фолатов в плазме часто
нормален, несмотря на дефицит фолацина
в тканях.
Важным
дифференциально-диагностическим признаком, присутствующим (хотя
и не всегда) при кобаламиновом дефиците
и практически отсутствующим при фолаци-
новой недостаточности, служит демиелини-
зирующая полинейропатия.
Известно множество антивитаминов
фолиевой и парааминобензойной кислот,
которые широко применяются в
химиотерапии. Они могут вызывать клинически
выраженный фолациновый гиповитаминоз. Это
прежде всего цитостатики (аминоптерин,
аметоптерин, и особенно — метотрексат).
Данные лекарства ингибируют дигидро-
фолатредуктазу. Более слабое
аналогичное действие оказывают триметоприм,
пентамидин, пириметамин, триамтерен.
Сульфаниламиды тормозят у бактерий
синтез фолацина из парааминобензойной
кислоты, на чем основан их лечебный
эффект. Но это же свойство создает риск
дисбактериоза и фолациновой
недостаточности при сульфаниламидной терапии. Для
преодоления токсичности антагонистов
тетрагидрофолат-редуктазы пользуются
фолиновой кислотой (лейковорином).
ляется. Это ведет к нарушению клеточного
цикла быстро пролиферирующих клеток, в
частности гемопоэтических и
эпителиальных. Данные звенья патогенеза и
формируют картину фолат-дефицитных состояний.
Из-за замедления клеточного цикла эритро-
идные предшественники реже делятся и
дают меньше красных кровяных клеток. Они
дольше пребывают в интерфазе, синтезируя
гемоглобин. Формируется ядерно-цитоп-
лазматическая асинхрония. Получаются
мегалобласты и мегалоциты, имеющие
повышенный цветовой показатель, но
пониженный срок жизни. Нестабильная ДНК
формирует структуры типа колец Кабо и
телец Жолли. Развивается гипорегенераторная
гиперхромная мегалобластическая анемия.
Подобные же нарушения миелопоэза ведут к
тромбоцитопении и лейкопении со сдвигом
ядерной формулы гранулоцитов вправо и
гиперсегментацией их ядер. Устойчивость
мегалоцитов к гемолизу понижена, в связи
с чем мегалобластические состояния могут
даже сопровождаться гипербилирубине-
мией, хотя усиленный гемолиз и не служит
главным механизмом данных анемий.
Нарущается и пролиферация эпителия,
что проявляется хейлозом, гунтеровским
глосситом (сухой красный «лакированный
язык»), эзофагитом, конъюнктивитом, ат-
рофическими или эрозивными гастритом и
энтеритом. Вследствие этого бывает ахлор-
гидрия, поносы (реже — запоры), стеаторея.
Происходит задержка роста, ухудшение
заживления ран, развивается
иммунодефицит, возможно оживление хронических
инфекций и субфебрилитет. Фолин-зависи-
мые ферменты участвуют в синтезе серина
и валина. Тетрагидрофолат необходим и
для синтеза (путём метилирования гомо-
цистеина) метионина, а также для
катаболизма гистидина. При нехватке фолацина
накапливается промежуточный продукт
катаболизма последнего — форминоглута-
миновая кислота, определение которой в
моче обладает диагностическим значением
для дифференцировки между фолациновым
и кобаламиновым дефицитами. Дефицит
фолацина (и кобаломина) может приводить
Гиповитаминоз В12
Еще в XIX столетии Т. Аддисон и А. Бирмер
описали злокачественную анемию с
увеличением диаметра незрелых красных
кровяных клеток, сопровождаемую ахилическим
гастритом с атрофией слизистой желудка. В
400 А.Ш. Зайчик, Л.П,Чурилов Патохимия (эндокринно-метаболические нарушения
XX столетии Дж. X. Уиппл показал, что
введение печени в рацион собак с мегалобласти-
ческими анемиями приводит к стимуляции
кроветворения, а затем Ж.Р. Мино (в
англоязычной транскрипции Дж. Майнот,
канадский врач, сам одним из первых спасенный
от диабетической комы экспериментальной
инсулинотерапией) и У.П. Мерфи (1926)
добились излечения мегалобластических
состояний у человека большими
количествами печени. Однако, диетотерапия печенью
оставалась неэффективной при анемии
Аддисона-Бирмера. В связи с этим, ученик
Ж.Р. Мино, У.Б.Кэстл (1928),
предположил, что ее развитие зависит от внешнего
фактора пищи и внутреннего, выделяемого
слизистой желудка. В 1948 г. Э.Л. Смитом
и соавт. был выделен в кристаллической
форме агент, ответственный за лечебный
эффект печени при мегалобластической
анемии (внешний фактор Кэстла или, как
часто транскрибируют, Кае л а), чуть позже
Г.А. Баркер охарактеризовал его кофер-
ментную форму — кобаламин. Вещество
получило название витамин В12. В анион-
замещённой форме цианкобаламина он стал
использоваться в фармакотерапии. Но
только в пятидесятых годах Д. Кроуфут-Ходж-
кин удалось расшифровать крайне сложную
химическую структуру витамина (рис. 79),
используя рентгеноструктурный анализ
(1955). Витамин оказался кобальтсодержа-
щим геминоподобным соединением
(молекулярной массой 1356 Д). Позже выяснили
и природу внутреннего антианемического
фактора Касла. Это был гексозаминсодер-
жащий мукопротеин обкладочных клеток
слизистой желудка, образующий с внешним
фактором комплекс, защищающий его
разрушения в кишечнике. Лишь в составе
такого комплекса В12 эффективно всасывается в
тонком кишечнике с помощью специального
рецептора. В Скандинавии описано редкое
аутосомно-рецессивное наследственное
заболевание — болезнь Имерслунда-Грэсбека,
при котором сильно ограничено кишечное
всасывание витамина В12 из-за дефекта
данного кишечного рецептора и имеется проте-
инурия. Описаны и единичные наблюдения
мегалобластической анемии, связанной с
наследственным дефектом самого
внутреннего фактора Касла.
В 90-е годы XX века, при решающем
вкладе австралийского патолога Б.Х. Тоха,
было установлено, что злокачественная
анемия Аддисона-Бирмера — аутоиммунное
расстройство, вызванное аутоантителами
к компонентам водородного насоса и гас-
тринового рецептора обкладочных клеток
слизистой желудка, а также, у ряда
больных — аутоантителами против внутреннего
фактора Касла (см. т. I, с. 469, т. III, гл. 1 и
Практикум). Именно поэтому пероральная
терапия печенью у таких больных не давала
эффекта. Однако позже оказалось, что при
сверхвысоких концентрациях витамина В12
в кишечнике и суточной дозе от 200 мкг
он всасывается и без внутреннего фактора
Касла.
Кобаламин — уникальное соединение,
не образуемое ни животными, ни
высшими растениями. Лишь микроорганизмы
способны к созданию его молекул. В связи
с этим высоким содержанием витамина
В12 отличаются весьма неаппетитные
субстанции — осадок сточных вод (50 мкг/г),
речной ил (3 мкг/г), отходы производства
антибиотиков и навоз (0,1 мкг/г). Копрофа-
гия у животных играет определенную роль
в поставке этого витамина.
Микрофлора слепой кишки человека
синтезирует витамин В12при наличии
витамина PP. Высшие растения накапливают
сравнительно немного витамина, а
животные — гораздо больше. Ценными
источниками витамина являются печень (90—150
мкг/ЮОг), почки (20-50), рыба (11-15,
особенно, пикша и сельдь), мясо (2-8).
Человеку необходимо в день не более 2-3 мкг
витамина В12. Витамин устойчив к
термообработке пищи, но разлагается на свету.
Достоверно доказана этиологическая роль
строгого вегетарианства в возникновении
гиповитаминоза В12. К этиологии данного
нарушения самое прямое отношение имеют
и гельминтозы, так как многие
паразитические черви избирательно абсорбируют
витамин В12. Особенно этим отличаются
:€1^ТСНз
о=р-с V^4^~CH3
о но
сн
СНз
хромофорная
часть
нуклеотидная
часть
Рис 79. Строение водорастворимого энзимовитамина В12 Пояснения в тексте
402 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
широкий лентец и анкилостома.
Всасывание витамина страдает при энтеритах,
в том числе спру, резекции кишечника и
действии таких лекарств, как неомицин,
парааминосалициловая кислота и колхицин.
После всасывания витамин поступает в
портальный кровоток в комплексе с транс-
кобаламином II (35 кД) и проникает в гепа-
тоциты, причём, по некоторым данным, и в
этом процессе участвуют гастромукопроте-
ин или его фрагменты. В печени витамин
депонируется, причём в настолько
существенных количествах, что даже после тотальной
резекции желудка первые признаки явной
мегалобластической анемии отмечаются
при обычной диете лишь через 3-4 года.
По-видимому, в печени депонируемый
витамин прочно связан с транскобаламином!
(121 кД). Этот же комплекс проникает в
кровь и переносится плазмой. При аутосом-
но-рецессивном наследственном дефиците
транскобаламина II развиваются мегало-
бластическая анемия, лейкопения и
иммунодефицит (см.т.1, с. 510). При нехватке
транскобаламина Г гематологических
аномалий не отмечено. Существует и транскобала-
мин III, как полагают участвующий вместе
с остальными транспортерами витамина в
предупреждении потерь кобаламина.
Активными формами витамина В12 в
организме человека служат метилкобаламин и
аденозилкобаламин (см. т. III, гл. 1).
Установлено лишь 2 метаболические
реакции, в которых напрямую участвуют
эти коферменты. Первый необходим для
метилирования гомоцистеина в метионин.
Торможение этой реакции при
гиповитаминозе В12 имеет весьма фатальные для
метаболизма последствия. Нехватка метионина
затрудняет образование холина, а значит —
фосфолипидов, и экскрецию липопротеидов
печенью. Избыток гомоцистеина может
способствовать развитию атеросклероза
(см. выше). Но главное, перестает демети-
лироваться №-метилтетрагидрофолиевая
кислота и нарушается ее превращение в
тетрагидрофолат, удерживающийся внутри
клеток. Поэтому данный метаболит теряется
из клеток и, несмотря на достаточное
количество фолата и тетрагидрофолата в крови,
ткани испытывают гиповитаминоз по фо-
лацину. Вот почему основой клинической
картины гиповитаминоза В12, как и при фо-
лациновой недостаточности, служат
проявления мегалобластического кроветворения.
По этой же причине фолацин и кобаламин
являются синергистами, а большие дозы
фолацина эффективны при
гиповитаминозе В12. Так как фолацин необходим в
синтезе пиримидиновых и пуриновых нуклеоти-
дов не только гемопоэтическим клеткам, а
всем клеткам организма, в первую очередь
быстро пролиферирующим, то проявления
авитаминоза В12, как и нехватки фолиевой
кислоты, сопровождаются характерными
изменениями эпителия ЖКТ и
торможением его пролиферации. Нарушение
реэпителизации ведет к эрозиям и на фоне
лейкопении — к язвенно-некротическому
синдрому, поражающему слизистые рта,
пищевода, желудка, но особенно
кишечника (см. выше).
Второй из активных коферментов —
аденозилкобаламин — катализирует реакцию
превращения метилмалонил-КоА в сук-
цинил-КоА. При срыве этого процесса
накапливается избыток метилмалоновой
кислоты, выделяемой с мочой, что отличает
гиповитаминоз В12от фолациновой
недостаточности. Метилмалоновая и пропионовая
кислоты, которые находятся в организме
в избыточных количествах, переходят в
жирные кислоты с нефизиологическим
нечётным числом углеродных атомов в
скелете. Эти метаболиты, включаясь в липиды
нейронов, вызывают жировую дистрофию
нервных клеток и демиелинизацию нервных
волокон (П.Ф. Вэгли, 1948).
Дополнительное значение в патогенезе неврологических
нарушений при данном авитаминозе имеет
нарушение образования ацетилхолина из-за
холиновой недостаточности.
В последнее время установлено, что
разрушению кобаламина в организме
способствует закись азота. При однократном
наркозе закисью азота не развивается
дефицита этого витамина, но пролонгированные
курсы, например при лечении столбняка,
могут приводить к мегалобластической
анемии. При курении ускоряется инактивация
витамина В12, причём возможно даже
клиническое проявление недостаточного действия
данного витамина у заядлых курильщиков.
НАРУШЕНИЯ ОБМЕНА
РЕДОКС-ВИТАМИНОВ
В данный раздел включены сведения о
расстройствах метаболизма, связанных с
витаминами С, Е, А и липоевой кислотой, а
также полифенолами и биофлавоноидами
(«витамин Р»). При этом надо учитывать,
что у данных витаминов имеются и кофер-
ментные (липоат) либо, гормоноподобные
(витамин А) функции.
Все эти витамины участвуют в регуляции
редокс-состояния клеток, представляют
собой в определённых формах
естественные антиоксиданты — то есть соединения,
располагающие лабильными атомами
водорода, несущими неспаренный электрон.
Соединяясь с активными кислородными
радикалами (АКР) и липоперекисями,
антиоксиданты нейтрализуют их с
образованием стабильных несвободнорадикальных
продуктов (см. т. I).
В этом качестве редокс-витамины
взаимодействуют с антиоксидантами,
вырабатываемыми самим организмом: глутатионом, цис-
теином, тиоредоксином, положительными
глобулинами острой фазы и другими тиол-
содержащими соединениями, металлозави-
симыми и селенозависимыми ферментами —
разрушителями активных кислородных
радикалов, мочевой кислотой и билирубином.
De facto все эти участники метаболизма
представляют собой единую антиоксидан-
тную систему клеток, поскольку способны
восстанавливать друг друга и позволяют
осуществлять функцию нейтрализации
свободных кислородных радикалов как
в цитозоле (водорастворимые
компоненты — например, витамины С и Р), так и
на мембранах клеток (жирорастворимые
витамины А и Е).
Патофизиология витаминного обмена 403
Ярким примером взаимодействия редокс-
витаминов служит их кооперация в системе
глутатиона. Глутатион, образуемый с
участием аминокислоты цистеина, нейтрализует
АКР и должен сам после этого
восстанавливаться. Реактиватор глутатиондисульфида,
фермент глутатионредуктаза, зависит от
НАДФН, а значит, нуждается в витамине
PP. При восстановлении глутатиона
источником восстановительных эквивалентов
служит витамин С. Аскорбиновая кислота
может использоваться и для реактивации
витамина Е, а также каротиноидов; кроме
того, она замедляет окисление липопроте-
идов переносящих эти жирорастворимые
витамины. Биофлавоноиды существенно
уменьшают дозы аскорбиновой кислоты,
необходимые для достижения этих
эффектов. Прооксиданты, образуемые при
воспалении и повреждении клеток, в частности
деструкции пуринов, включают в ядрах
эксперссию гена, контролирующего фактор
транскрипции NF-kB. Данный сигнал
запускает каскад генетических событий,
приводящий к синтезу и активации цитокинов
и прогрессированию воспаления. Вместе с
тем он индуцирует синтез ферментов
антиокислительного действия — каталазы, глу-
татион-пероксидазы, супероксиддисмутазы
и антиокислительных острофазных белков.
Потенциал этого направления регуляции
зависит от доступности и запаса редокс-
витаминов.
Таким образом, редокс-витамины
проявляют отчётливый синэргизм между собой
и с рядом пищевых веществ, которые не
являются полностью незаменимыми (цис-
теин, изопреноиды, хиноны). Химические
формулы редокс-витаминов представлены
на рис. 80.
Цинга и действие мегадоз
витамина С
Цинга (скорбут, детская форма фигурирует
также под эпонимом болезнь Мёллера-Бар-
лоу) — это один из самых старых и массовых
авитаминозов, известных человечеству. Его
первые медицинские описания относят к
404 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
с=о
он—с I
ое-й
I
НС
I
он—сн
о
J
-н2
с=о
c=ol
+н2
C-OI
НС '
I
он—сн
I
сн2он
он
сн2он
С (аскорбиновая
ОН
СН2
0:
кислота)
Р (кверцетин)
СНз
CH
I
сн2
сн2
сн2
сн2
соон
липоевая кислота
9Нз 9Нз
'<£-(СН2)з—CH—(СН2)3—CH—(СН2)3
<СН3
Е (токоферол а-форма)
CH
СНз СНз
Рис 80. Химихеская структура редокс-витаминов С,Р,Е, липое
вой кислоты. Пояснения в тексте
XIII веку. Антискорбутные продукты
питания также были эмпирически открыты
давно: в 1671 г. Венет указал на
норвежский опыт лечения болезни ягодами и
на антицинготные свойства цитрусовых
фруктов. В 1750 г. русский географ,
первопроходец Камчатки, СП. Крашенинников
описал применение сланника-кедровника
от цинги народами Севера. В XVIII веке
Россия экспортировала хвою в качестве
антискорбутного средства. Но годы шли, а
цинга оставалась массовой
медико-социальной проблемой, связанной с сезонными
особенностями питания городской бедноты
и с жизнедеятельностью человека в
экстремальных условиях, так как ее этиология и
патогенез были неясными (см. выше). Вот
какой исторический факт описан в
документах, хранящихся в семейном архиве
автора. В самом начале 30-х годов прошлого
века, будучи врачом в г. Анадыре на
Чукотке, молодой выпускник медицинского
факультета Иркутского университета П.В.
Чурилов столкнулся с массовой
заболеваемостью цингой среди рабочих
рыбокомбината. Знакомство с рассказами Джека
Лондона выручило начинающего врача. Он
вспомнил, как золотоискатели
Аляски в одном из рассказов
американского писателя
обсуждают противоскорбутные
свойства
сланника-кедровника. Кедровник в изобилии рос
в тундре. Доктор приготовил
его спиртовой экстракт.
Излишне говорить, что в такой
форме профилактическое
средство приобрело у рабочих
большую популярность. В
результате вскоре на комбинате
не осталось ни одного
цинготного больного. Интересно
не только то, что порой из
художественных источников
можно почерпнуть дельный
научный совет.
Поразительно, что к этому, сравнительно
недавнему времени, хотя уже
существовала модель цинги
на морских свинках А. Хольста-Фрейлиха
(1907), уже делались попытки экстракции
антицинготного начала из капустного сока
(Н.Е. Бессонов, 1921), да и сам витамин С
(«гексуроновая кислота») уже был выделен
А. Сент-Дьердьи (1928) из коры
надпочечников и из паприки, систематических
представлений о его роли при цинге и научной
основы для специфической профилактики
этого недуга еще не было! Только в 1932 г.
А. Сент-Дьердьи и И.Л. Свирбели
идентифицировали гексуроновую кислоту как
фактор, излечивающий цингу, и переименовали
данное вещество в аскорбиновую кислоту.
А ведь цитированное выше исследование
Дж. Линда, показавшее, что не кислота как
таковая, а лишь компоненты лимонного сока
защищают от цинги было опубликовано еще
в 1753 г.! Более того, уже в 1850 г. А. Брай-
сон показал, что чистая лимонная кислота
не обладает противоцинготным действием,
а значит, от цинги, защищает другой
компонент цитрусовых. Но такой авторитет
витаминологии, как Э.Мак-Коллюм в
1917 г., неверно истолковав результаты
опытов на крысах по моделированию скорбута,
приписал смерть животных бактериальной
аутоинтоксикации и усомнился в
авитаминозной природе цинги. К тому же попытки
выделения витамина С из лимонного сока
наталкивались на сложности, связанные
с нестойкостью витамина в растительных
тканях при консервации. Еще ученые
викторианской Англии справедливо
сомневались в антискорбутных свойствах долго
хранимого лимонного сока и указывали, что
свежее мясо и кровь арктических животных
предохраняют от цинги коренных
обитателей Севера куда лучше (Ф.Дж. Джексон,
1899). Именно взяв в качестве источника
свежее животное сырьё — кору
надпочечников коров, — А. Сент-Дьердьи и добился
успеха, хотя искал не витамин, а катализатор
окислительно-восстановительных реакций
у млекопитающих.
Аскорбиновая кислота (рис. 80)
синтезируется всеми растениями и подавляющим
большинством животных из глюкозы.
Кроме Homo sapiens лишь обезьяны и
морская свинка не способны ее вырабатывать
и должны получать извне. Минимальная
потребность взрослого человека в витамине
С оценивается в 50-100 мг/сутки, но так как
она сильно повышается при разнообразных
состояниях, связанных со стрессами и
напряжённой адаптацией, то многие
источники рекомендуют более значительные дозы.
В природе 3 формы данного витамина —
восстановленная форма — L-аскорбиновая
кислота, наиболее активная в витаминном
отношении; продукт ее обратимого
окисления — дегидроаскорбиновая кислота,
способная восстанавливаться, а также
запасная растительная форма витамина
С — аскорбиген. Последняя связанная форма
наиболее устойчива к окислению и
сохраняет примерно треть витаминной активности.
Не менее 70% аскорбиновой кислоты в
растениях находится в запасной форме,
структура которой, как отмечено М.Н. Ло-
гаткиным и П.Е. Калмыковым (1974), имеет
общие черты с классической нуклеотидо-
подобной формулой, приведенной выше
для энзимовитаминов. В кислых растворах
аскорбиновая кислота и аскорбиген хорошо
сохраняются и термостойки. Открытие про-
Патофизиология витаминного обмена 405
стого способа предохранения аскорбигена в
зеленых кормах методом закисления (1928)
принесло Нобелевскую премию по химии
финскому учёному А. И. Виртанену.
Аскорбиновая кислота активно
всасывается в тощей кишке, и формы
гиповитаминоза С, вызванные нарушением ее абсорбции,
неизвестны.
В организме человека витамин С не
депонируется. Одномоментно в теле содержится
до 5 граммов аскорбиновой кислоты, причем
феноменально богаты ею надпочечники,
где, по подсчётам П. Хорнсби и А. Кри-
велло (1983), чистый витамин составляет
до 1 % сырого веса органа! Немало данного
витамина и в эритроцитах. Интересно, что
курение несколько понижает содержание
аскорбиновой кислоты в крови, хотя еще
никто не приобрёл скорбута только из-за
этой пагубной привычки.
Наиболее ценные пищевые источники
витамина С — растительные продукты
(черноплодная рябина — 2 000 мг/100 г,
хвоя— 1400, квашеная капуста — 1100,
шиповник — 1000, черная смородина — 300,
лимонник китайский — 250). Прекрасным
источником аскорбиновой кислоты могут
служить брокколи, киви, фейхоа, красный
и зеленый перец. Из животных продуктов,
существенные количества аскорбиновой
кислоты имеются в надпочечных железах,
сердце и печени, а вот молоко, например,
крайне бедно витамином С (1 мг/дл).
Метаболическая роль витамина С
определяется прежде всего тем, что аскорбиновая
и дегидроаскорбиновая кислота составляют
окислительно-восстановительную систему.
В этом качестве витамин является
необходимым для многих обменных реакций, в
частности особенно для процессов гидрок-
силирования и амидирования. Витамин С
участвует в гидроксилировании параокси-
фенилпировиноградной и параоксифени-
луксусной кислот, образовании серотонина
путём гидроксилирования триптамина.
Наиболее значительным аспектом гидрок-
силирующей роли аскорбиновой кислоты
является ее участие в метаболизме
коллагена.
406 АЖ Зайчик, ЛЛ.Чурилов Патохимия
Аскорбиновая кислота активирует из
неактивных предшественников пролил-
гидроксилазу и лизилгидроксилазу. Эти
ферменты обеспечивают гидроксили-
рование проколлагена. Отсутствие
нормального гидроксилирования не дает
формирующимся коллагеновым
молекулам возможности приобрести стабильную
спиралевидную конфигурацию, поэтому
затрудняется образование перекрестных
связей в коллагеновых фибриллах,
необходимое для образования тройных спиралей
тропоколлагена. В таком недоделанном
виде коллагеновые фибриллы плохо
секретируются фибробластами. Секрети-
рованные тропоколлагеновые молекулы
без должного гидроксилирования своих
пролиновых остатков проявляют
повышенную растворимость, не имеют
достаточной механической прочности и легко
перевариваются коллагеназами. Имеются
и сведения о дополнительном снижении
скорости синтеза коллагеновых пептидов
при гиповитаминозе С.
Поражается сильнее всего наиболее
насыщенный гидроксипролиновыми
остатками коллаген кровеносных сосудов, в
том числе их базальных мембран. Именно
поэтому цинга характеризуется
геморрагическим синдромом в форме вазопатии
(см. т. I, стр.251). Геморрагическая сыпь на
коже, кроме типичных экхимозов и пете-
хий, сопровождается перифолликулярным
гиперкератозом с характерными папулами,
имеющими кровяной венчик.
Так как ослабевает прикрепление
надкостницы к костям и фиксация зубов в манди-
булярных и максиллярных лунках,
появляются поднадкостничные кровоизлияния,
периостит и геморрагические пародонтит
с гингивитом. Могут быть субпериосталь-
ные переломы по типу «зелёной веточки».
Внутричерепные кровоизлияния способны
при тяжелом скорбуте вызвать смерть
больного. При дефиците аскорбиновой кислоты
снижается всасывание железа, а главное, его
отщепление от ферритина что в комбинации
с кровоточивостью обусловливает
появление гипохромной анемии.
чдокринно-метаболические нарушения
Если цинга разыгрывается в растущем
организме, при детском скорбуте —
болезни Мёллера-Барлоу, — возникают особые
нарушения остеогенеза: образование кости
на месте хряща задерживается, а хрящ
подвергается разрастанию и минерализации
(Т. Барлоу, 1885). Формируются ряды
хондроцитов, наступает их временная
минерализация. Но из-за замедленного
образования остеоидного матрикса в остеобластах,
тормозятся процессы рассасывания хряща.
Это приводит к расширению эпифизарных
зон и к образованию выступающих в мета-
физарную часть костномозговой полости
хрящевых выростов. Под воздействием
механических нагрузок появляется
деформация грудной клетки (корытовидное
западение грудины и выступающие чётко-
образные концы рёбер), а также варусное
искривление длинных трубчатых костей
нижних конечностей. В отличие от
рахитических цинготные чётки болезненны на
ощупь. Характерны и болезненные
поднадкостничные гематомы вдоль длинных
костей конечностей.
Доказана роль аскорбиновой кислоты в
активации гексокиназы, что существенно
для обеспечения нормального
проникновения глюкозы в клетки и отложения
глюкозы в печени. Дополнительное значение в
патогенезе системного поражения
соединительной ткани при гиповитаминозе С имеет
нарушение формирования гиалуроновой
кислоты. Витамин С участвует в синтезе и
метаболизме гормонов щитовидной железы.
Иммунодефицит, отмечаемый при цинге,
в настоящее время объясняют снижением
продукции защитных белков нейтрофилов.
Немаловажное значение имеет и нарушение
фибропластического компонента
воспаления — еще корабельный врач XVII века
Эпсом описал у цинготных больных
ослабление рубцов и вскрытие старых ран.
Так как витамин С известен в двух
формах, он может быть как прооксидантом
(в частности, восстанавливать железо,
усиливающее свободнорадикальные реакции),
так и антиоксидантом. Важно подчеркнуть,
что его антиоксидантные эффекты сильнее
проявляются при больших дозах, а проок-
сидантные — при малых. Кроме того,
направленность действия витамина С может
определяться доступностью его синергистов
и обеспеченностью железом (П. Хорнсби,
Дж. Кривелло, 1983; Л. Полинг, 1972; Дж.Р.
Смайтис, 1998).
Как уже упоминалось выше, в качестве
антиоксиданта витамин С непосредственно
нейтрализует АКР в водной фазе и
участвует в реактивации витамина Е, главного
антиоксиданта клеточных мембран.
Аскорбиновая кислота снижает скорость
окисления ЛПОНП, что способствует транспорту
витаминов Е и А и торможению процессов
атерогенеза. Ее антиатерогенный эффект,
как опосредованный через витамин Е, так и
собственный, был показан в ряде
эпидемиологических исследований (С. Тодд и соавт,
1995; Дг Пэндей и соавт., 1996). Однако
другие исследования обнаружили у
витамина С лишь токоферол-опосредованное
антиатерогенное действие (М. Стэмпфер,
Э. Римм; 1996).
Так как АКР мутагенны, то можно
полагать, что витамин С, как и его партнёры по
антиоксидантной системе, способны
повышать резистентность к неопластическим
процессам. По данным ряда исследований,
аскорбиновая кислота снижает риск
возникновения рака желудка и кишечника, так как
нейтрализует канцерогенные нитрозамины
пищи (Дж. Смайтис, 1998). Имелись
сообщения о протективной роли мегадоз
аскорбиновой кислоты при раке лёгких (Л. Йонг
и соавт., 1997).
Витамин С — участник окислительно-
восстановительных реакций,
приводящих к образованию кортикостероидов и
участвующих в метаболизме некоторых
катехоламинов. Поэтому обеспеченность
витамином очень важна для адекватной
работы надпочечников и мозга.
Существуют данные, документирующие активацию
коры надпочечников и снижение тяжести
аллергических и аутоиммунных
заболеваний при введении мегадоз витамина С.
Показано увеличение стрессоустойчивости
и функциональных возможностей надпо-
Патофизиология витаминного обмена 407
чечных желез, в частности у лиц с болезнью
Аддисона, понижение риска возникновения
болезней нарушенной адаптации при
действии больших доз витамина С (Д. Дас и
соавт., 1997).
В головном мозге при действии глута-
мата на NMDA-рецепторы в
возбуждающих синапсах происходит значительное
образование АКР, потенциально опасное
для нейронов. Нейтрализатором этих
эффектов выступает витамин С, который
секретируется в эти синапсы при обратном
захвате глутамата. Снижение антиокси-
дантных резервов и увеличение перекис-
ного повреждения нейронов показано при
болезни Альцхаймера (когда нейроны
содержат дефектный антиокислительный
белок пресенилин-1); при инсультах и
болезни Паркинсона, хорее Гентингтона
(когда увеличена продукция АКР в мозге),
синдроме Дауна (когда имеется
повышенная продукция перекиси водорода в
мозге на почве хромосомной аномалии);
при шизофрении II типа (когда увеличено
превращение антиоксидантного дофамина
в токсическую хиноновую форму — дофа-
минохром). При всех этих заболеваниях
прием больших доз витамина С имеет
значение антириск-фактора (Дж.
Смайтис, 1998).
В связи с тем, что образование АКР
является побочным эффектом окисления
пуринов при альтерации тканей, а также
происходит параллельно работе фермента
простагландин-Н-синтетазы, на
протяжении воспаления, мегадозы витамина С
могут оказывать противовоспалительный и
тканесберегающий эффект при инфекциях
и повреждении тканей. Это и служило
основой для рекомендации Л. Полинга (1973)
использовать мегадозы витамина С для
предупреждения простуды. Правда, позже было
показано, что эффект аскорбиновой
кислоты — симптоматический, а не этиотропный,
и ее эффективность подтвердилась лишь
как лечебного, но не превентивного средства
(Х.Хемиля, 3. Херман; 1996). Однако
удалось установить, что большие дозы
витамина стимулируют бактерицидную активность
408 АЖ Зайчик, Л.П.Чурилов Патохимия
и миграционную способность нейтрофилов,
что даже послужило для лечения синдрома
Чедиака-Хигаши (см. т. I, стр. 160,264).
Таким образом, адекватная поставка
витамина С очень существенна для снижения
риска развития и/или степени тяжести
многих распространенных и опасных
заболеваний. Неудивительно, что интегра-
тивные эпидемиологические исследования
Й.Энстрёма (1993) и Л. Тули и соавт.
(1995) выявили отрицательную корреляцию
между приемом и сывороточным уровнем
аскорбиновой кислоты, с одной стороны, и
уровнем смертности в различных
популяциях американцев с другой.
Применение мегадоз витамина С не
свободно от побочных эффектов и может быть
патогенным, особенно для индивидов с
некоторыми особенностями реактивности.
Основной метаболит аскорбиновой
кислоты — щавелевая кислота, известная как
компонент наиболее распространенных
почечных конкрементов. Как показано А. Чал-
мерсом и соавт. (1986), лица, наклонные
к оксалурии, имеют сниженную скорость
всасывания витамина С в кишечнике,
образуют из него больше оксалатов и мегадозы
витамина способны у них ухудшить течение
мочекаменной болезни. По мнению А. Дип-
лока (1995), у обычных индивидов, без
наклонности к оксалурии, мегадозы витамина
С не приводят к существенному повышению
выведения оксалатов с мочой, так как аскор-
бинат выводится в нативной форме.
Как уже указывалось, в присутствии
избытка свободного железа аскорбиновая
кислота оказывает прооксидантный эффект.
Поэтому не рекомендуется применять ее
мегадозы, если уровень ферритина
сыворотки превышает 120 мкг/л (В. Герберт,
1993-1995). Следует учесть, что не только
относительно редкие в большинстве
популяций талассемии и сидеробластические
состояния, но и гемохроматоз, частота
которого в европеоидных популяциях
очень велика, характеризуются избытком
свободного железа (см. ниже
«Патофизиология метаболизма железа»). Считается,
что во Франции первичный гемохроматоз
^метаболические нарушения
Таблица 28. Извращение данных лабораторных
анализов при приеме мегадоз витамина С
Ложно-положительные и завышенные
результаты
Глюкоза
Мочевая кислота
Креатинин
Щелочная фосфатаза
Фосфат
Парацетамол
Ложно-отрицательные
и заниженные
результаты
Билирубин
лдг
С02
Калий
КОМТ
МАО
Холестерин
Креатинфосфокиназа
ДОФА-р-гидроксилаза
Проба Грегерсена
вовлекает более 1 % населения, а в США
гетерозиготами по его гену является не
менее 12,5%, а больных более миллиона! С
учетом этого довольно большой группе
пациентов не показаны мегадозы витамина С,
несмотря на их очевидную биологическую
активность.
Мегадозы витамина С вызывают кризо-
вый гемолиз у лиц с дефицитом глюкоза-6-
фосфатдегидрогеназы, которых в мире более
60 миллионов. Установлено, что мегадозы
витамина С способствуют освобождению и
активации мышьяковистых соединений из
состава пищевых продуктов и повышают
токсичность мышьяка.
Для практических целей также важно
помнить, что мегадозы витамина С
извращают результаты ряда лабораторных тестов
(табл. 28).
С учетом вышеназванных ограничений
и индивидуальных особенностей ряд
авторов в новейших источниках (Дж. Смайтис,
1998) рекомендует увеличение ежедневного
приема витамина С здоровыми взрослыми
людьми до 500 мг. В то же время другие
(В.Герберт, 1980) настаивали на
ненужности увеличения минимальных
гигиенически рекомендованных доз (60 мг) и даже
подчеркивали негативные эффекты такого
использования аскорбиновой кислоты.
Выводы из сопоставления фактов о пользе
и вреде мегадоз витамина С, приведённых
выше, читатель может сделать сам.
Патофизиология витаминного обмена 409
Биофлавоноиды и полифенолы
«Оливки, лимоны, чеснок на
столе — не будет коротким
твой век на Земле».
(Староитальянская поговорка)
«По яблоку на день —
и доктор не надобен».
(Староанглийская поговорка)
Данная группа незаменимых пищевых
факторов, иногда обозначаемая как витамин
Р (от лат. permeabilitas — проницаемость),
была открыта А. Сент-Дьёрдьи (1936),
который показал, что натуральные лимоны и
паприка излечивают проявления вазопатии
у лиц, на которых не действует
синтетическая чистая аскорбиновая кислота.
Соединения данной группы содержат
ядро бензапирона, соединенное с гидро-
ксилами фенолов. Последние могут быть
метилированы и соединены с различными
остатками Сахаров (рамнозы, глюкозы и
т.д.). Известно более 600 веществ этой
группы (рис.80 представляет типичный
пример — кверцетин).
Большинство из подобных веществ
водорастворимо, но имеются и жирорастворимые
формы (в частности, в оливках и зеленом
чае). Растения вырабатывают витамин Р при
участии никель- и молибденсодержащих
энзимов. Многие растительные продукты (но
не животные ткани) имеют в своем составе
значительные количества витамина Р.
Знакомство с их перечнем убеждает в ценности
многих традиций и рецептов народной
кулинарии и медицины. Что касается био-
флавоноидов, гречиха содержит рутин,
чайный лист (особенно зелёный) — катехины,
мирецетин и кемпферол, цитрусовые — гес-
перидины (500 мг/100г), рис — госсипол,
ромашка — апигенин, яблоки — кверцетин,
бобовые, особенно соя и чечевица, — ге-
нистрин, свёкла — антоцианы, в частности,
высокометилированные бетаин и бетанин.
Полифенолы изобилуют в травах: куркума
длинная, входящая в состав индийской
пряности карри, насыщена куркумином,
тимьян содержит карвакуол и тимол,
розмарин и многие другие растения — феруловую
кислоту. Чернильный орешек содержит
галловую кислоту, образующую комплекс с
витамином С, — галоаскорбин. Полифенолы
имеются в большом количестве в шоколаде,
кофе, какао, белом вине.
Свежие оливки, оливковое масло (и,
особенно вода после технологического
промывания оливок при его отжиме) содержат
гидрокситирозол. В данных продуктах
витамин Р находится в сочетании с
полиненасыщенными жирными кислотами, что,
возможно, усиливает их эффект.
Много витамина Р алоэ (алоэмодин),
ревень (реин), шиповник (680мг/100г),
гибискус и кора чилийского дерева болдо
(болдин). Очень богаты витамином Р арония
(2000 мг/дл), голубика (300), логанова ягода
и брусника (600), признанные источники
этого витамина — смородина, особенно
чёрная (1000), и клюква (330), а также льняное
семя и софора японская. В составе чеснока
витамин Р сочетается с серосодержащими
антиоксидантными аминокислотами и
фитонцидами, обладающими выраженным
бактерицидным действием на
этиологический агент язвенной болезни — Helicobacter
pylori (Дж. П. Сивам и соавт., 1997).
Диетологи высоко оценивают красные
вина как источник комплекса биофлаво-
ноидов (кверцетин, рутин, катехин). В них
содержится в 10 раз больше витамина Р,
чем даже в таком богатом источнике, как
зеленый чай. Вдобавок красное вино
содержит каротиноид резерватол (см. ниже)
и мочевую кислоту, проявляющую
синергизм с редокс-витаминами. Однако эти
позитивные эффекты не отменяют вредного
действия содержащегося в данном продукте
алкоголя.
Минимальная суточная потребность в
витамине Р составляет для взрослых
ориентировочно 50-100 мг/сутки, хотя
сбалансированная диета даёт более полуграмма
биофлавоноидов и полифенолов в день.
Риск гиповитаминоза Р создается у лиц,
не употребляющих растительной пищи и
находящихся на упрощенной диете.
410 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
Первые проявления экспериментального
гиповитаминоза Р включают боли в ногах и
плечах, утомляемость, затем
присоединяются проявления вазопатии с образованием
петехий в местах механического давления
на кожу.
Биологическое действие витамина Р ранее
сводили только к капилляроукрепляющему
эффекту. На деле оно оказалось много шире.
Биофлавоноиды и полифенолы — мощные
антиоксиданты, реактивирующие суль-
фгидрильные соединения и витамин С,
а также глутатион и в известной степени
токоферолы. Первооткрыватель витамина
Р, венгерский биохимик А. Сент-Дьердьи,
отметил, что для эффекта данного витамина
необходимы были хотя бы минимальные
количества аскорбиновой кислоты, активность
которой значительно потенцировалась
биофлавоноидами и полифенолами. Кроме
эффектов, опосредованных витамином С,
следует указать, что витамин Р снижает
скорость окисления ЛПОНП, уменьшает
активность гиалуронидазы, предотвращает
переход адреналина в токсичный адрено-
хром.
Широко известно седативное и
гипотензивное действие многих богатых витамином
Р растений, из которых в России наиболее
популярна черноплодная рябина (арония).
Долгое время механизмы этого эффекта,
используемого в народной медицине,
оставались загадочными.
В настоящее время установлено, что
биофлавоноиды аронии и других растений,
например, Passiflora coerulea и Matricaria
recutitis (такие как: кверцетин, госсипол,
хризин, апигенин), способны связываться с
бензодиазепиновым рецептором в мозге.
Действие многих из них в очищенном
виде в десятки раз сильнее эффекта
элениума (Дж. Медина и соавт., 1997).
Именно этот аспект неантиоксидантного
действия витамина Р обусловливает его се-
дативный, гипотензивный, обезболивающий
эффекты. Влияя на бензодиазепиновый
рецептор тромбоцитов и уменьшая продукцию
тромбоксанов, витамин Р снижает тромбо-
генный потенциал кровяных пластинок, что,
наряду с предохранением липопротеидов от
окисления, существенно для его антиатеро-
генного действия.
По причинам, уже рассмотренным выше, в
разделе, посвященном витамину С,
биофлавоноиды изучались на предмет их
антиканцерогенной активности. Было установлено,
что витамин Р нейтрализует канцерогенные
для ЖКТ нитрозамины. Очень
значительный резонанс имело исследование П. Кне-
кта и соавт. (1997), показавших, что именно
биофлавоноиды яблок были фактором, от
которого зависело предупреждение
развития рака (лёгких и других локализаций) в
группе из 10000 финнов. Аналогичные
исследования существуют в отношении
чеснока, сои, зеленого чая, оливок — и снижения
риска раковых опухолей ЖКТ и
мочеполовой системы (Дж. Смайтис, 1998).
Информация о профилактической и
лечебной эффективности различных видов
еды, содержащей биофлавоноиды и
полифенолы, столь изобильна в современной
литературе, что это можно объяснить только
удовлетворением, получаемым авторами,
когда пишут о полезных вещах, которые к
тому же еще и вкусны.
Гипервитаминоз Р^неизвестен, как и
токсические эффекты биофлавоноидов.
Впрочем, В.Кумар и соавторы (1997)
указывают, что чеснок предохраняет не только
от язвенной болезни, атеросклероза и других
недугов, но и еще надёжнее — от поцелуев!
Гиповитаминоз
и гипервитаминоз Е
Витамин Е — это смесь из 4 токоферолов (от
греческого tocos phero — «потомство
несущий») и 4 токотриенолов. На рис. 80 дана
структура а-токоферола. Иногда в состав
данного витамина зачисляют и эфирные
производные вышеназванных соединений.
Витамин жирорастворим и был открыт
Г.М. Ивенсом и К.С. Бишопом в 1922 г. как
фактор сливочного масла и салата,
предупреждающий бесплодие. Как витамины
наиболее активны а-р-у-токоферолы,
соотношение эффективности которых выражается
как 100:40:8, наиболее сильные антиокси-
дантные потенции присущи 8-токоферолу.
В день взрослому нужно не менее 10-30 мг
токоферолов и токотриенолов. Потребность
в витамине сильно увеличивается при
мышечной работе и почти в полтора раза
больше у мужчин, нежели у женщин.
К счастью, витамин Е представлен во
многих продуктах, как растительных, так и
животных. Очень богаты витамином
растительное масло (60-120 мг/дл, особенно об-
лепиховое, соевое и кукурузное, но в гораздо
меньшей степени — 5мг/дл — оливковое).
Следовательно, витамина Е достаточно
много содержит и такой прозаический продукт,
как маргарин. Также превосходным
источником витамина Е признаны пшеничные
и кукурузные зародыши (проростки — до
25 мг/дл). Немало витамина в бобовых
(5), яйцах (3), морской рыбе (до 2,5). Надо,
однако, отметить, что в собственно рыбьем
жире обнаружены лишь следы
производных токола; витамин распределён в
клеточных мембранах тканей рыб.
Витамином Е богата и зелень тёмных тонов,
особенно шпинат, кресс-салат и люцерна,
она же — альфальфа.
Авокадо, батат и арахис поставляют
витамин Е на стол любителей более
экзотических продуктов. Диетический гиповитаминоз
Е — редкость. Но возможно нарушение
всасывания этого витамина при стеаторее
и непереносимости жиров, а также других
формах мальабсорбции. Малым резервом
токоферолов и плохим их всасыванием
отличаются недоношенные дети и дети с
внутриутробной гипотрофией, у которых
к тому же на фоне часто необходимой ок-
сигенотерапии снижена антиоксидантная
резистентность. Антивитаминами Е
являются пиридин, сульфаниламиды и хлорор-
ганические соединения.
В кишечнике витамин Е включается в
хиломикроны, затем всасывается в лимфу
и обменивается в крови с ЛПНП и другими
липопротеидами. Он распределен по тканям
повсеместно и присутствует в клеточных
мембранах, причём больше всего витамина
содержат жировая ткань, ЦНС, мышцы и
Патофизиология витаминного обмена 411
печень. Гиповитаминоз Е возможен при
наследственной абеталипопротеидемии, так
как нарушен его транспорт.
Еще в ранний период исследования
витаминной активности токоферолов было
установлено, что они предотвращают про-
горкание масла. Причиной этого является
антиоксидантная способность витамина Е,
захватывающего неспаренные электроны
АКР и не участвующего в дальнейшей цепи
аутоокислений. Витамин тормозит пере-
кисное окисление ненасыщенных жирных
кислот (образование липоперекисей). При
гиповитаминозе Е, в буквальном смысле
происходит «прогоркание жиров» в
клеточных мембранах. Витамин Е необходим и для
восстановления других жирорастворимых
редокс-витаминов в частности витамина
А и убихинона, известного как коэнзим Q.
Последнее соединение — важный участник
митохондриального процесса тканевого
дыхания, поскольку от него зависит передача
протонов с флавиновых ферментов,
содержащих витамин В2, на цитохромы. Иногда
убихинон называют витамином Q10. Хотя
это и не вполне корректно, так как
коэнзим Q может синтезироваться в организме
животных из тирозина и неизвестны
симптомы авитаминоза Q, очевидна витамино-
подобная роль данного редокс-соединения,
зависящего от витамина Е (П.Е. Калмыков,
М.Н. Логаткин, 1974). Ферменты фосфоли-
пид-глутатионпероксидаза и глутатион-пе-
роксидаза, участвующие в обеспечении
прерывания цепи образования липоперекисей,
содержат селен. Поэтому, можно сказать,
что витамин Е и данный микроэлемент
взаимодействуют с эндогенными
соединениями организма при осуществлении своих
антиоксидантных функций. Витамин С, как
уже отмечалось, способен восстанавливать
редокс-активность этой системы. Поэтому
все вышеперечисленные факторы
действуют комплексно и, в частности, симптомы
дефицита витамина Е и селена очень сходны
(см. т. I, с. 193-196).
В первую очередь срыв антиоксидантных
и витаминсберегающих функций
токоферолов ведет к повреждению клеток, имеющих
412 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
наибольшую мембранную поверхность,
высокую напряжённость процессов окисления
и активно вырабатывающих АКР-
мышечных волокон и нейронов. Могут страдать
также быстропролиферирующие клетки
(сперматогенный эпителий, гепатоциты и
эпителий нефронов, зародышевые ткани),
так как в состоянии высокой митотической
активности у них понижены антиоксидант-
ные резервы и идет интенсивный мембра-
ногенез. Гиповитаминоз Е сопровождается
тканевой гипоксией в органах с высокой
потребностью в кислороде.
Типичное проявление нехватки
токоферолов — выраженная миодистрофия,
охватывающая как скелетные (особенно
диафрагмальную), так и гладкие мышцы,
а также миокард. Происходит деструкция
митохондрий, фрагментация мышечных
волокон, отложение пигмента, сходного
с цероидом и представляющего
результат пероксидации липидов, появляются
микронекрозы. Ранним постоянным
признаком служит нарушение образования
креатинфосфата и креатинина, экскреция
которого с мочой снижается, при
параллельном появлении креатинурии.
Возникают гипотонус и слабость мышц, вплоть
до паралича задних конечностей у
экспериментальных гиповитаминозных крыс.
Характерно, что миодистрофия, как и все
основные проявления Е-гиповитаминоза,
имеет тем более тяжелый характер, чем
выше в диете витаминодефицитных
животных содержание полиненасыщенных
жирных кислот. Следовательно, именно с
нарушением их обмена связаны
наблюдаемые симптомы.
Со стороны ЦНС отмечаются демиели-
низация и глиоз в спинном мозге. Особенно
характерна дегенерация аксонов в задних
канатиках, присутствует отложение липо-
фусциновых пигментов в нервных ганглиях,
поражается и миелиновая оболочка
чувствительных нервов. Это ведет к атаксии,
гипорефлексии, дизартрии, гипоэстезии.
Вследствие пероксидации липидов и
вторичного нарушения обмена витамина А в
сетчатке возможна ее дегенерация.
чдокринно-метаболические нарушения
У крыс и других подопытных животных
гиповитаминоз Е вызывает азооспермию,
затем атрофию тестикул и стерильность
самцов. У самок сохраняется способность
беременеть, но к моменту имплантации
зародыш погибает. У человека не доказано
аналогичное поддерживающее фертиль-
ность действие витамина Е и нет сведений
о его стимулирующем влиянии на
сперматогенез. Предполагается, однако, роль
гиповитаминоза Е в патогенезе раннего токсикоза
беременных и у пациенток, наклонных к
спонтанным абортам.
Известно снижение резистентности
эритроцитов к гемолизу, наступающее
при гиповитаминозе Е. Из-за перекисной
трансформации эритроцитарной мембраны
красные кровяные клетки могут
приобретать шиповатый вид (акантоцитоз). Однако
у взрослых людей усиленный гемолиз,
предупреждаемый токоферолами и
провоцируемый диалуровой кислотой, показан
лишь in vitro. У обезьян, а такаже у
недоношенных и подверженных мальабсорбции
новорожденных, отмечали острую
гемолитическую анемию, купируемую витамином
Е (см. т. III).
При гиповитаминозе Е имеется
отложение цероидоподобного пигмента в клетках
слизистой кишечника, что сопровождается
их ускоренной гибелью. Присутствуют не-
кробиотические изменения в гепатоцитах
и канальцевом эпителии почек, возможны
гепатонекрозы и нефротический синдром.
Витамин Е определённо замедляет
скорость окисления атерогенных липопро-
теидов, что препятствует их отложению
в сосудистой стенке. Имеются данные о
снижении адгезивно-агрегирующих свойств
кровяных пластинок при действии этого
витамина. Сообщалось о его протромбин-свя-
зывающей активности. (В.И. Добрынина,
1976; Дж.Смайтис, 1998). Все это, вкупе с
многочисленными эпидемиологическими
данными, позволяет считать витамин Е
антиатерогенным.
В отношении предполагаемого
антиканцерогенного эффекта токоферолов данные
эпидемиологических исследований проти-
Патофизиология витаминного обмена 413
воречивы, от подтверждения протективной
роли в отношении рака желудка (Г.У. Коме-
ток и соавт., 1991) и до обнаружения прямо
противоположной тенденции к учащению
рака легких в финской популяции среди
тех, кто получал дополнительно витамин Е
(Группа АВТС, 1994). Последнее
исследование имело очень большой негативный для
пропагандистов антиоксидантных пищевых
добавок резонанс в медицинской печати.
Витамин Е кумулируется в организме,
обладает токсичностью и способен вызвать
гипервитаминоз. При гипервитаминозе Е
отмечается тромбоцитопатия и гипокоагу-
ляция (последняя связана с нарушением
всасывания витамина К), ослабление темно-
вого зрения из-за антагонизма с витамином
А, диспептические явления, гипогликемия.
Некоторые симптомы прямо
противоположны лечебным эффектам витамина — так,
избыток витамина Е приводит к появлению
чувства слабости, повышенной мышечной
утомляемости, головной боли и... ослаблению
сексуальной активности (В. Герберт, 1980).
Не исключено, что все это продиктовано
конкуренцией больших доз а-токоферола, чья
антиокислительная активность минимальна, с
основным антиоксидантом — у-токоферолом.
Повышенные дозы витамина Е могут, на фоне
высокого уровня андрогенов, привести к
мышечным судорогам, что следует учитывать при
попытках использования Е-витаминизации
в повышении функциональной готовности у
спортсменов.
Недостаточность липоевой
кислоты
Липоевая или тиоктовая кислота
(рациональное название — 6,8-дитиооктоно-
вая кислота — см. рис. 80) известна как
водорастворимый витамин, в двух
формах — а-липоевой кислоты и а-липоамида
(Л.Рид, И. Гунсалюс; 1953). В организме
биологически активной жирорастворимой
формой витамина служит его дигидроли-
поил-лизил.
Данный витамин является коферментом
энзима дигидролипоил-трансацетилазы при
окислительном декарбоксилировании пиру-
вата и других а-кетокислот, входит в состав
коэнзима А. Этот витамин, легко
проникающий через гематоэнцефалический барьер,
считается также тиоловым антиоксидантом,
сберегающим токоферолы и аскорбиновую
кислоту. Липоат необратимо связывает
тяжелые металлы в водорастворимые
комплексы, выводимые почками. Он обладает
липотропными средствами. Активизация
использования глюкозы и липидов в
реакциях окисления, наступающая при действии
липоевой кислоты, приводит к
восстановлению запасов гликогена в печени, понижению
уровня глюкозы и липопротеидов в крови.
Потребность в липоевой кислоте точно не
определена, лечебные дозы приняты на
уровне 4-25мг/сутки. Витамин содержат
и животные, и растительные продукты (в
мкг/кг — печень, сердце и почки — более
1000; говядина — 725; молоко — около 900;
рнс— 220; капуста — 115). Весьма богаты
тиоктовой кислотой темно-зеленые листья
овощей, особенно шпината и брокколи.
Дефицит липоевой кислоты ведет к так
называемому «пирувизму». Это
проявляется повышением содержания пирувата и
других кетокислот в крови, метаболическим
ацидозом, полиневритом, мышечными
спазмами. Наблюдается миокардиодистрофия.
Может развиваться ожирение печени.
Показана протективная роль липоевой кислоты в
отношении диабетических полинейропатии
и ангиопатии (Л. Паркер и соавт., 1995;
Д. Циглер и соавт., 1997).
Липоевая кислота — пример витамина,
сочетающего роль кофермента и редокс-
функции.
НАРУШЕНИЯ ОБМЕНА
ГОРМОНОВИТАМИНОВ
Жирорастворимые витамины A, D, К —
накапливаются в организме и способны
проникать в клеточные ядра и оказывать
воздействие на экспрессию генов, биосинтез
белков и активность ферментов, клеточную
414 ALU. Зайчик, ЛЛ. Чу рилов Патохимия (эндокринно-метаболические нарушения
дифференцировку, подобно гормонам.
Поэтому их можно рассматривать как
экзогенные прогормоны. Эти витамины сочетают
свои некоферментные гормоноподобные
функции с другими, например антиокси-
дантными и регулирующими состояние
клеточных мембран.
Витамин А и каротиноиды:
патофизиология дефицита
и избытка
Гемералопия (куриная слепота) —
нарушение темнового и сумеречного зрения, а
также кератомаляция — помутнение и
размягчение роговицы — были известны еще
античным медикам, лечившим их бараньей
печенкой. В XIX веке научные исследования
этих синдромов были начаты рядом врачей:
Ш. Маккензи (1857), показавшим связь
диеты и частоты кератомаляции у младенцев;
Д.Ливингстоном (1857), обнаружившим
связь между питанием и глазными болезнями
у участников своей знаменитой африканской
экспедиции; Г. Лобо (1860), описавшим
«эпидемическую бразильскую офтальмо-
патию» у детей скудно питающихся рабов
и вылечившим ее с помощью рыбьего жира;
М.Мори, обнаружившим в Японии (1896)
болезнь «икэн» (ксероз, кератомаляция,
диспепсия), также успешно лечимую рыбьим
жиром. Значительные вспышки гемералопии
были описаны у русских крестьян и их детей
в период Великого поста, причем болезнь
облегчалась конопляным маслом и
излечивалась рыбой (А. Савельев, 1862).
Научные исследования данного
заболевания особенно быстро прогрессировали в
начале XX века, когда был выделен
ростовой жирорастворимый фактор А чёрного
хлеба и сливочного масла (У.Степп, 1909,
Э.В.Мак-Коллюм и соавт.; 1913). Еще в
1909 г. швейцарский офтальмолог П. Кнапп
(1909) вылечил экспериментальную ке-
ратомаляцию у крыс, вызванную бедной
животными жирами диетой, с помощью
рыбьего жира. Огромную роль в
витаминологии, и в частности в обнаружении
витамина А, сыграла экспериментальная
методика раздельного злакового питания
крыс, примененная американскими
учеными Э. Мак-Коллюмом и М. Дэвис для
изыскания незаменимых компонентов
диеты. В 1913-1914 гг. им удалось показать, что
после 8-14 недель на диете, лишенной иных
животных жиров кроме свиного, крысы
перестают расти и заболевают «офтальмией»,
а добавка яичного желтка или сливочного
(но не растительного) масла рост
восстанавливает. В 1919 г. в детском доме в Дании
главный врач К. Блох заметил, что одна из
двух групп детей страдала от куриной
слепоты и тяжелой ксерофтальмии, а другая нет.
Он сравнил листы питания и обнаружил,
что употребление сливок предохранило
детей от болезни. Будучи знаком с
экспериментальными работами Э. Мак-Каллюма
и соавторов, этот педиатр назначил детям
рыбий жир и за неделю устранил симптомы
болезни, позже он отметил ускорение роста
воспитанников. Толчком к изучению
жирорастворимого антиксерофтальмического
фактора послужили массовые заболевания
ксерофтальмией в Германии, где население
в период Первой мировой войны, особенно
морской блокады 1916-1918 гг., сильно
страдало от нехватки животных жиров в
диете. В конце концов была установлена
структура выделенного витамина А (Т.Б. Осборн,
Л.Б.Миндел; 1922) а затем он был
синтезирован благодаря усилиям X. фон Эйлер-
Хельпина и П. Каррера. Ими же была
доказана роль каротиноидов как провитаминов
А (1929-1933).
Витамином А называют группу
натуральных и синтетических изопреноидов (см.
рис.81, где в качестве примера фигурирует
витамин Aj), включая ретинол (присущий
морским рыбам), ретинол-2 (выделенный от
пресноводных рыб)), их метаболиты — ре-
тиналь, ретиноевую кислоту. Провитамины
А— это а-р-у-каротины, криптоксантин и
некоторые другие. Витамин А является и
гормоноподобным соединением,
регулирующим экспрессию генома ряда клеток,
и редокс-витамином — одновременно. Ряд
каротиноидов, не имея собственно гормо-
ноподобной активности, не относятся к
Патофизиология витаминного обмена 415
н2с
н2с
он
витамину А, но являются антиокси-
дантами, притом более сильными,
чем витамин А. К ним принадлежат
ликопен, зиксантин, лютеин,
ксантофилл, резерватол, геранилы. Более
слабой антиоксидантной
активностью обладают такие естественные
изопреноиды, как лимонен, карвон,
ментол.
Интерес представляет
распределение витамина А и провитаминов А
в живой природе. Готовый витамин
А встречается исключительно в
тканях животных. Наиболее богат им
рыбий жир, в особенности — акулий
и палтусовый, в меньшей
степени — тресковый (19 мг/дл). Печень
животных содержит чуть меньшие
количества витамина А (4-14),
немало его в яйцах и сливочном
масле (0,6), сметане и сырах (0,3).
Считают, что животные получают
витамин из каротиноидов
растительной пищи, а рыбы — также из
зоопланктона. Полярные
млекопитающие накапливают рекордные
количества витамина А в печени,
питаясь рыбой. Каротиноиды —
исключительно продукт растительного
синтеза, обнаруженный в растениях за 100
лет до выделения витамина А (Г.В.Ф. Ва-
кенродер, 1831). Структура каротинов была
расшифрована в 1930-1933 гг.
нобелевскими лауреатами по химии П. Каррером и
Р. Куном. Этими провитаминами богаты
красно-оранжевые овощи и плоды, а также
зелень. По содержанию р-каротина, самого
активного провитамина А, лидируют
пальмовое масло (80), облепиховое масло (40), соя
(10), петрушка (8,5), морковь (7,2), курага
(5), томаты (3). Немало его содержат батат,
мускусная дыня, фенхель. Другие
каротиноиды преобладают в помидорах (ликопен),
кормовой и огородной капусте, брокколи,
луке-порее и черемше (зиксантин, лютеин).
Таким образом, пути накопления
витамина А в пищевых цепях представляют одну из
характерных взаимосвязей различных
организмов в экоценозах. Потребность взрос-
■Нз
СНз СНз
Ч-У' СНз С]
|С—СН=СН—С=СН—СН=СН—С
II
сн
сн2
СНз I
Л/ СН2ОН
Рис 81. Химическая структура гормонови-
таминов A, D, К. Пояснения в тексте
СН=СН—СН—СН—СНз
I I
СНз СНз
СНз
СН—СН2—СН2—СН2—СН—СНз
СНз
СНз
СН2—СН=С—(СН2)з
СНз—СН
(сн2)3
СНз—СН
(СН2)з
СНз—СН
СНз
лого человека в витамине А — 1,5 мг/сутки
(то есть 5000 условных международных
единиц — ME). При обычном питании на
3/4 потребность в витамине А
удовлетворяется за счет биотрансформации каротинов
в ретиноиды. Принято считать, что 0,6 мкг
Р-каротина соответствуют 1 ME витамина А.
Так как витамин А неустойчив к окислению,
вероятность его нехватки возрастает при
дефиците витамина Е, предохраняющего
витамин А от разрушения.
Каротины и ретинол всасываются в
тонком кишечнике, причём последний — в 2
раза быстрее. Известно, что усваивается не
более 1/6 общего количества р-каротина.
Всасывние идет вместе с эмульсией жиров,
особенно ему способствуют токоферолы и
ненасыщенные жирные кислоты. При стеа-
торее возникает гиповитаминоз А.
В энтероцитах каротиноиды
превращаются в витамин А. В меньшей степени этот
416 АЖ Зайчик, ЛЛ.Чурилов Патохимия
процесс имеет место и в печени.
Превращение катализирует железосодержащий
фермент р-каротин-15-15'-диоксигеназа.
Его активность стимулируется гормонами
щитовидной железы. Вот почему при гипо-
тирозе нарушается переход каротиноидов в
витамин А, провитамины накапливаются в
организме и вызывают каротинемическую
псевдожелтуху. В норме из каротиноидов
образуется ретиналь, восстанавливаемый
далее в ретинол при участии ниацин-за-
висимых дегидрогеназ. Ретинол образует
эфиры с пальмитиновой и иными жирными
кислотами и транспортируется хиломик-
ронами. Купфферовские клетки печени
депонируют эфиры ретинола, причем запас
у взрослого может быть достаточен на 2-3
года. Вследствие этого гиповитаминоз А при
отсутствии ретиноидов и каротиноидов в
диете поражает, как правило,
новорожденных, не имеющих запаса витамина. Ретинол-
эстераза освобождает витамин А, который
переносится в крови транстиретином (см.
раздел «Нарушения композиции белков
плазмы» выше). Освобождение ретинола
печенью — цинкзависимый процесс. При
печеночной недостаточности возможен
вторичный гиповитаминоз А. Транстире-
тин поглощается пигментными клетками
сетчатки и эпителиальными клетками. В
клетках витамин А связывается с
клеточным ретинолсвязывающим белком. Потеря
ретинолсвязывающего белка при протеину-
рии может способствовать гиповитаминозу
А. В эпителиях и других тканях эмбриона
имеется большое количество специального
внутриклеточного белка, связывающего
ретиноевую кислоту.
Метаболические функции витамина А в
сетчатке обеспечиваются ретинолом и ре-
тиналем, а в остальных органах зависят от
ретиноевой кислоты. Впрочем, одна кислота
не может обеспечить условий для
нормального развития зародышей.
Витамин А играет ключевую роль в
фоточувствительности сетчатки глаза,
потенцирует дифференцировку эпителиальных
клеток, в том числе особенно слизистых и
кожи, усиливает иммунитет, способствует
чдокринно-метаболические нарушения
синтезу хондроитинсульфатов и росту тела
(две последних, функции выражены у
молодых животных и детей). Ретиноиды и ка-
ротиноиды, в дополнение к этим функциям,
являются мембранными антиоксидантами и
фотопротекторами.
В сетчатке витамин А участвует в
обеспечении темновой адаптации, и в первую
очередь его дефицит сказывается на функции
палочек, наружные сегменты которых при
хронической нехватке витамина необратимо
дегенерируют. Альдегидная форма
витамина А — 11-цис-ретиналь взаимодействует в
клетках-палочках сетчатки с белком опси-
ном и формирует самый
фоточувствительный зрительный пигмент родопсин.
Родопсин имеет сложную структуру. Ретиналь
связан с лизиновыми остатками опсина по
типу шиффова основания. Кроме витамина
А в составе родопсина присутствуют
углеводный компонент, связанный с опсином
через аспарагиновую кислоту, а также фос-
фатиды (фосфорилхолин и фосфатидилсе-
рин). На свету ретиналь перемещается от
фосфолипидной к белковой части
комплекса. Под действием фотонов в
фоторецепторах родопсин распадается с освобождением
опсина и трансформы ретиналя, который
НАД-зависимая дегидрогеназа
восстанавливает до транс-ретинола, причем имеются
его необратимые потери. При распаде
родопсина изменяются электрические свойства
мембран палочек и генерируется потенциал
действия, передаваемый по нервным
волокнам в ЦНС, создавая светоощущение.
В темноте изомераза переводит
транс-форму в цис-ретинол, который ферментативно
окисляется до цис-ретиналя, опять-таки с
участием НАД. В этом процессе важна роль
цинксодержащей алкогольдегидрогеназы.
Постоянное осуществление описанного
цикла ведет к потерям ретиналя, которые
должны восполняться из запасов ретинола.
При дефиците ретинола родопсин не может
восстановиться в темноте.
Три различных йодопсина колбочек,
ответственных за цветовое зрение, также
содержат небольшое количество витамина
А. Однако только тяжелый дефицит вита-
Патофизиология витаминного обмена 417
мина приводит к сужению полей зрения и
сказывается на цветоощущении.
Следовательно, лишь при выраженном авитаминозе
колбочки не остаются интактными.
При гиповитаминозе А в сетчатке
наблюдается деструкция палочек. Сужаются ней-
роэпителиальный и гранулярноклеточный
слои сетчатки.
В результате наступает нарушение
сумеречного и ночного зрения (гемералопия и
никталопия). Тест на темновую адаптацию,
дающий результат более 45 секунд за 3
минуты, считается патологическим и
используется для выявления гиповитаминоза А.
Прежде чем перейти к характеристике
других аспектов патогенеза амитаминоза
А, необходимо резюмировать некоторые
факты, приведенные выше. Освобождение
ретинола из печени и его превращения в
сетчатке зависят от обеспеченности
дефицитными в организме алкоголиков цинком
и ниацином. В силу зависимости всасывания
ретиноидов от абсорбции липидов в
кишечнике поступление витамина А в организм
снижается при алкогольной мальабсорбции.
По этим причинам и из-за участия алкоголь-
дегидрогеназы в механизмах превращений
витамина в сетчатке алкоголики являются
второй по значению группой риска в
отношении гиповитаминоза А, после грудных
детей из малообеспеченных семей.
Не менее важным, чем роль ретинола
в биохимии зрения, является участие
витамина А в дифференцировке
эпителиальных клеток, особенно в эпидермисе и
железистых эпителиях, вырабатывающих
слизистый секрет. Ретиноевая кислота
предупреждает метаплазию железистого
эпителия в плоский ороговевающий. Она
делает это как гормон, посредством
регуляции экспрессии генов ряда рецепторов
факторов роста. Возможно также, что при
дефиците витамина А провоцируется
процесс апоптоза в эпителиальных клетках.
Гормоноподобный эффект ретиноидов
ассоциируют со взаимодействием витамина
А и белка-регулятора транскрипции,
связанного с онкогеном erbA. Данный
рецептор участвует и в опосредовании действия
ряда стероидных гормонов. Известно, что
витамин А в виде ретинол-фосфоманно-
зидов присутствует в мембранах клеток и
участвует в регуляции обмена мембранных
гликопротеидов.
Так или иначе, железистый эпителий
самой разной локализации при авитаминозе А
претерпевает кератинизацию. Фактически
гиповитаминоз провоцирует роговую
дистрофию (см. раздел «Диспротеинозы»).
Ороговение эпителия протоков слезных
желёз ведет ко ксерофтальмии.
Наблюдается атрофия слезных желез. Высыхание
(ксероз) конъюнктивы и роговицы ведет к
воспалению и кератомаляции. Размягчение
роговицы может закончиться образованием
ее язвы. Ранним признаком служат опалес-
цирующие беловатые очаги гиперкератоза
на склерах — бляшки Бито. Метаплазия
слизистых оболочек дыхательных путей
ведёт к значительному снижению местной
противоинфекционной резистентности,
вплоть до развития ларинготрахеобронхи-
та и пневмонии. Метаплазия наблюдается
в выводных протоках пищеварительных
желез, в пищеводе и других отделах ЖКТ.
Гиперкератоз в полости рта приводит к
образованию налета на языке. В слюнных
железах формируются ретенционные
кисты. Ухудшение пищеварения ведет к
порочному кругу, так как всасывание
жирорастворимых витаминов нарушается при
расстройстве пищеварительных функций
еще больше. Изменяется эпителий нефро-
нов и мочевыводящих путей. Возникают
циститы и пиелиты. Кератинизация и слу-
щивание могут привести к цилиндрурии и
даже послужить основой для
формирования нефроуролитиаза. Роговая дистрофия
и атрофия желез поражают и кожу. Имеется
фолликулярный папулёзный гиперкератоз
(симптом «жабьей кожи»), трещины кожи,
угри, кисты сальных желез, возможно
оживление грибковой и бактериальной
инфекции. Наиболее выраженные поражения
кожи бывают, если имеется сочетанный
гиповитаминоз А и В.
Описанные изменения сильно снижают
эффективность барьерной функции покро-
418 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
вов тела и, безусловно, отчасти
ответственны за отмечаемый при гиповитаминозе А
вторичный иммунодефицит.
Но витамин А, особенно в форме его
макрофагального метаболита 14-гидрокси-
ретинола, обладает и собственными
иммуностимулирующими функциями. В частности,
есть сведения об ускорении пролиферации
лимфоцитов и активизации фагоцитоза
под действием ретиноидов.
Эпидемиологические данные свидетельствуют, что
коррекция содержания витамина А в диете
детей тропических регионов подчас ведет к
понижению смертности от инфекций почти
на треть. При ответе острой фазы цитокины
вызывают снижение продукции ретинол-
связывающего белка печенью и его уровень
в крови снижается, что может уменьшать
биологическую доступность витамина А.
Витамин Айв большей степени ка-
ротиноиды обладают антиоксидантным
эффектом. Они способны тормозить
процессы перекисного окисления липидов,
хотя и менее эффективно, чем токоферолы.
Антиоксидантный эффект каротиноидов
способствует предупреждению катаракт
(Ч.Эбрамс и соавт., 1991). Каротинои-
ды лютеин, зиксантин и кантаксантин, в
отличие от р-каротина, накапливаются в
сетчатке и необходимы для ее функции.
Очень значительные количества
каротиноидов, главным образом р-каротина,
аккумулирует яичник и особенно желтое
тело беременности. Синтез
прогестерона и других стероидов предусматривает
активные перекисные процессы, в то же
время, каротиноиды призваны защитить
яйцеклетки от мутагенного действия
перекисей. Тяжелый авитаминоз А ведет к
стерильности животных.
Резерватол, изобильно представленный
в красном вине, арахисе и траве Polygonum
cuspidatum, является наиболее мощным по
своему биологическому действию кароти-
ноидным антиоксидантом и используется в
ряде восточных народных лекарств.
Уровень каротиноидов, особенно
ликопена и р-каротина, снижается в крови
пациентов с хроническими
гемолитическими анемиями, гемохроматозом, сахарным
диабетом, ВИЧ-инфекцией, — при
которых усилена, продукция прооксидантов и
некробиотические процессы (В. Пэрфитт
и соавт., 1994). Многие источники, в
частности Дж. Р. Смайтис (1998), рекомендуют
ежедневный обязательный прием ликопена
и р-каротина — по 10 мг каждого.
Антиоксидантные и частично дифферен-
цировочные эффекты ретиноидов и
каротиноидов в эпителиях послужили основой для
предположений об их возможном
противораковом и антинеопластическом действии.
Это, однако, не получило однозначного
подтверждения.
Ликопен, компонент помидоров и в
меньшей степени абрикосов и гуавы,
отличается от всех каротиноидов наиболее
выраженным тропизмом к жировой ткани и
липидам. Именно у него имеется
значительный антиоксидантный эффект в отношении
липопротеидов, способность вызывать
некоторое понижение тромбогенного потенциала
кровяных пластинок, а также отмечена связь
его повышенного содержания в крови и в
диете с понижением риска рака
поджелудочной железы. У р-каротина не отмечено этих
эффектов, зато зарегистрировано снижение
риска рака молочной железы при употреб-
леннии его повышенных количеств.
Каротиноиды снижают чувствительность клеток
молочной железы к эстрогенам. Ретиноиды
не показали какой-либо профилактической
противораковой активности в двойных
слепых исследованиях. Более того, Дж. Оуменн
и соавт. (1996) даже отметили в
эпидемиологическом исследовании повышение частоты
рака у заядлых курильщиков, получавших
витамин А и р-каротин, по сравнению с теми
из них, кто обходился без этих витаминных
добавок. Однако транс-ретиноевая кислота
оказалась эффективна в лечении одной из
форм острого миелобластного лейкоза — М3
(промиелоцитарного), протекающего с
транслокацией t(15,17) и экспрессией
неопластическим клоном химерного белка -
результата слияния рецептора ретиноевой
кислоты RAR-a и онкопротеина PML (см.
т. III, гл. 3).
Патофизиология витаминного обмена 419
Показана роль витамина А в
предупреждении рецидивов рака печени.
Установлено также, что 13-цис-ретиноевая кислота
снижает тяжесть течения и вероятность
рецидивов при некоторых
неонкологических кожных болезнях (псориаз, акне,
лейкоплакия).
Раннее название витамина А — витамин
роста. Имелось в виду подчеркнуть, что он
необходим для роста и развития зародыша.
Вероятно, отчасти это связано с дифферен-
цировочной ролью ретиноевой кислоты.
Но одна кислота не способна поддержать
эмбриогенез у крыс, следовательно, важны
и иные аспекты метаболического действия
ретиноидов: антиоксидантные эффекты,
взаимодействие с витамином Е, а возможно,
и обнаруженная у эфиров ретинола
способность стимулировать транскрипцию
некоторых белков, в частности в клетках печени.
Ключевое значение в росторегулирующей
активности витамина А может иметь
метаболизм хондроитинсульфатов. При дефиците
витамина А развивается нехватка фосфоаде-
нозилфосфосульфата (ФАФС). Причиной
этого служит увеличение распада ФАФС
под действием лизосомальных сульфатаз,
подавляемых ретиноидами. ФАФС
необходим для синтеза хондроитинсульфатов
костной и других видов соединительной ткани.
При авитаминозе А нарушается рост костей.
Так как мозг продолжает расти, ранний
авитаминоз А у животных ведет к повышению
внутричерепного давления, что осложняет
пагубное действие данного гиповитаминоза
на зрение ещё и возможным повреждением
зрительных нервов.
Метаболическая роль витамина А
позволяет считать его как гормоноподобным
соединением, так и редокс-витамином.
Организм не способен эффективно
выводить избыток витамина А. Поэтому
известны как острый, так и хронический гиперви-
таминоз А. Описания гипервитаминоза А
сохранились в фольклоре эскимосов, у
которых существовал запрет на употребление
в пищу печени полярных млекопитающих.
В 1897 г. от неизвестной причины погибли
участники шведской полярной экспедиции
С. Андрэ. Изучив их дневниковые записи,
Ф.Ф. Талызин (1970) предположил, что
причиной острого расстройства здоровья у
полярников могло быть употребление
печени убитого ими белого медведя. Впрочем,
альтернативная версия предполагает острый
трихинеллёз.
Гипервитаминоз А бывает результатом
передозировок при лечении, однако только
суточные дозы выше 50 000 ME при
продолжительном введении или одноразовый
прием порядка 1-6 млн ME причиняют
отравление. В 70-е годы XX века в США
приобрёл скандальную известность педиатр
Линдон X. Смит, который в своих книгах
(«Кормите детей правильно», 1970)
рассматривал гиперактивность и другие
отклонения в поведении детей как результат гипо-
витаминозов и усиленно пропагандировал
в популярной-форме лечение отклонений
в поведении высокими дозами витамина А
(месячные курсы по 50 000 ME в день). Так
как от выполнения этих советов ряд
пациентов пострадал, доктор был в 1973 г. лишен
лицензии. Известны и эпизоды случайных
отравлений, когда рыбий жир используется
вместо кулинарного растительного масла.
При отравлении рыбьим жиром
симптоматика гипервитаминоза А и D
комбинируется. Массовое подобное отравление
наблюдалось в шестидесятых годах в XX веке
в Ленинградской области после крушения
железнодорожного состава,
перевозившего этот продукт. Ценнейшие наблюдения
больных были описаны И.М. Воронцовым
и другими специалистами Педиатрического
института.
Гипервитаминоз А вызывает периосталь-
ный гиперостоз. Это сопровождается болью
и отеком вдоль длинных трубчатых костей,
у взрослых — кальцинозом связок.
Острое отравление сопровождается головной
болью, тошнотой и слабостью, ступором,
отёком соска зрительного нерва —
вследствие ликворной гипертензии и сдавления
черепно-мозговых нервов в отверстиях
костей головы. Картина напоминает
проявления внутричерепного объёмного
процесса. Может повышаться температура.
420 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения
При хроническом отравлении нарушается
пищеварение, пропадает аппетит,
отмечается потеря в весе. Снижается образование
секрета сальных желез и развивается сухой
дерматит, выпадают волосы, могут быть
проявления геморрагического синдрома. У
крыс и взрослых пациентов кости при гипер-
витаминозе становятся ломкими, в культуре
ткани высокие дозы ретиноидов тормозят
остеогенез и усиливают хондролитические
процессы. Это делает витамин А в высоких
дозах эмбриотоксичным. Синтетические
ретиноиды считаются тератогенами и не
должны употребляться беременными
женщинами.
Гипервитаминоз А сопровождается не-
кробиотическими изменениями и некрозом
гепатоцитов и почечного канальцевого
эпителия, возможно развитие фиброза печени.
Употребление избытка каротиноидов менее
опасно, так как они не обладают острой
токсичностью. Их трансформация в витамин
А— регулируемый процесс и тормозится
при угрозе гипервитаминоза. Однако и при
этом может проявляться гиперкаротинемия
с псевдожелтухой. Псевдожелтуха наиболее
заметна на ладонях и подошвах и в отличие
от билирубиновой желтухи не вовлекает
склеры.
Решая вопрос о безопасности этих
провитаминов, надо принять во внимание
цитированные выше данные о повышении
риска развития рака и сердечных
приступов при приеме избытка каротиноидов,
на фоне хронических интоксикаций
алкоголем и табачным дымом. Избыток
Р-каротина потенцирует действие гепато-
тропных ядов, нарушает усвоение других
важных каротиноидов — зиксантина и лю-
теина. Избыток кантаксантина вызывает
его обратимую кристаллизацию в клетках
сетчатки глаза.
Все это свидетельствует о том, что
ретиноиды и каротиноиды не должны быть
предметом «пищевого культуризма» и
любительского самолечения, так как это
мощные биологически активные вещества
с широким спектром действия.
Витамин D, рахит
и гипервитаминоз D
«И потекут простые будни:
Рахит, прививки, санпросвет.
Я стану старым, стану нудным
Ах, где мои 17 лет!»
(Е.В. Ноткин, Л.П. Чурилов
«Песня выпускников»)
Автор, гордящийся дипломом
врача-педиатра, с особым чувством приступает к
изложению раздела, посвященного этиологии и
патогенезу нарушений метаболизма «самого
педиатрического» витамина.
Наиболее древний объект, несущий
признаки рахита, — мумия священной
обезьяны, чей возраст 4 000 лет, описанная Лор-
те-Гейяром. Очевидно, у обезьяны было
генетическое нарушение обмена витамина
D, так как вообще в Египте и других древних
очагах цивилизации рахит был чрезвычайно
редок. Ископаемые доисторические
скелеты и останки древних египтян, нубийцев,
греков, индийцев, китайцев, инков и
вавилонян не имеют признаков перенесенного
рахита. Очевидно, сказывалось тропическое
и субтропическое расположение древних
центров культуры и натуральная диета
наших предков. Античный врач Соранус
(98-138 гг.) упоминает о размягчении
костей, но не ставит это в связь с диетой. В то же
время картины средневековых художников
и мастеров эпохи Возрождения, например
старых голландцев, часто изображают
явных рахитиков. Классическим считается
внешний вид рахитического младенца на
картине итальянского мастера Караваджо
(1569-1609) «Спящий амур» — здесь и
рахитические чётки, и утолщение эпифизов,
и деформация грудной клетки. На грустные
профессиональные размышления наводят
медика известные строчки Д. Кедрина:
«Эти гордые лбы винчианских мадонн/ Я
встречал не однажды у русских крестьянок/
У рязанских молодок согбенных трудом...»
Впрочем, рахит — недуг индустриального
общества и стал известен в Европе как
«английская болезнь», так как приобрел
повальное распространение в городах
Британии в XVII-XIX веках и был впервые
описан английским студентом Д. Уистлером
(1645) и доктором Ф. Глиссоном (1650).
Название rickets происходит от английского
ricks — копны, скирды, бугры. Ф. Ганземанн
рассматривает экологию рахита как «домес-
тикационной болезни», указывая, что он не
встречался в преиндустриальную эпоху и
является болезнью горожан, проводящих
жизнь в помещениях. Ему принадлежит
меткое наблюдение, что рахит поражает диких
животных в неволе, но не при свободном
содержании (1914).
Медицинская география рахита,
описанная С. Маффеи еще в 1844 г, представляет
определенный интерес. Эта болезнь
встречается в умеренных широтах, между 40 и 60
градусами северной широты, в тропиках и
Арктике встречается только на фоне общего
недоедания, не наблюдается в высокогорных
районах (выше 1000 м над уровнем моря).
По классическим данным, рахит встречается
у негров чаще, чем у проживающих на той
же территории белых, в городах, особенно
крупных промышленных центрах, чаще,
чем в сельской местности. Наиболее тяжело
поражаются дети малообеспеченных
родителей (Л.Р. Перельман, 1938). В 1909 г.
Х.Г. Шморль находил рахит в 96% аутопсий
у немецких детей, умерших в возрасте до
18 месяцев. Еще в 40-х годах XX века более
чем у половины детей в больницах США,
по данным аутопсий, отмечались признаки
нетяжелого рахита. В постиндустриальную
эпоху наступили, конечно же, определенные
изменения в эпидемиологии рахита,
связанные с универсализацией питания населения,
переходом к коттеджному расселению
горожан, а также с массовой медицинской
профилактикой этого заболевания. Рахит, как
результат диетических дефектов, перестал
в развитых странах быть массовым. Вместе
с тем появилась проблема патогенных
последствий гипервитаминоза D.
Профилактика рахита стала возможна
только после открытия витамина D. В конце
XIX столетия наиболее прогрессивной
концепцией этиологии и патогенеза рахита была
Патофизиология витаминного обмена 421
световая теория Т.А. Пальма, обратившего
внимание на сезонность болезни (максимум
заболеваемости — зимой) и особенности ее
географии. Пальм, не обнаруживший
болезни в Японии и в ряде южных стран, считал,
что солнечные лучи обладают
антирахитическим действием (1890). Это обострило
итерес к исследованию биологической
активности ультрафиолетовых лучей, и врач с
туманных Фарерских островов Н.Р. Финсен
был удостоен Нобелевской премии за
обнаружение в 1893 г. их противотуберкулезного
действия. Рахит всегда был «английской
болезнью». Неудивительно, что его лечение
рыбьим жиром впервые описал доктор Дар-
би из Манчестера (1789). Позже А. Труссо
во Франции назвал рахит болезнью
недостаточного питания и инсоляции и
рекомендовал рыбий жир для его лечения (1861). В
викторианской Англии педиатр У.Б. Чиддл
(1888), обнаружив рахит даже в местности с
жесткой водой и у детей из обеспеченных
семей, находившихся на искусственном
вскармливании, предположил, что он связан с
дефицитом незаменимого фактора,
имеющегося в грудном молоке, а Дж. Бленд-Саттон
(1889) наблюдал рахит у львят, лишенных
материнского молока, и корректировал их
развитие назначением рыбьего жира и
молотых костей. Э. Мелланби (1919) получил
первую экспериментальную модель рахита
у щенков, содержавшихся в помещении, на
диете с ограничением потребления молока
до 200 мл в день, и показал, что
продукты, богатые ранее открытым витамином
А, предупреждали недуг. Он заключил,
что антирахитический фактор — либо сам
витамин А, либо другое жирорастворимое
начало, сходное с ним по распределению в
пищевых продуктах. Это вызвало яростное
неприятие у шотландских врачей во главе с
Л. Финдлеем, который ранее (1908)
показал ключевую роль содержания в закрытом
помещении при рахите щенков, тем более
что англичанин Мелланби демонстрировал
рахитогенный эффект диеты, основанной на
овсянке, национальном блюде Шотландии
(К. Раджакумар, 2003). Затем в Австрии
Г. Чик и О. Хульдчинский продемонстри-
422 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения
ровали с применением рентгенографии,
что как рыбий жир, так и ультрафиолетовое
облучение лечат рахит у детей (1919). Э.В.
Мак-Коллюм в 1918-1919 гг. получил
сходное заболевание на своей уже
упоминавшейся крысиной модели, однако, не будучи
врачом, не имел клинических знаний,
позволяющих распознать в экспериментально
полученной болезни картину рахита. Он
показал своих крыс педиатру Дж. Хаулэнду,
который тут же установил диагноз. Затем
Мак-Каллюм и соавторы (1922) доказали,
что антирахитический фактор отличен
от витамина А, так как не разрушается в
условиях деструкции последнего. Позже
А.Ф. Хесс и X. Штинбок показали, что
эксприментальный рахит лечится наиболее
быстро ультрафиолетовыми лучами
диапазона 280-302 нм. Более того, оказалось, что
облучение дрожжей, растительного масла,
муки ультрафиолетом придает им
антирахитические свойства (1924). В 1900-1928
гг. А. Виндаус, занимаясь химией
стероидов, обнаружил, что в дрожжах содержится
холестериноподобное вещество эргостерин,
при облучении ультрафиолетом
приобретающее антирахитические свойства.
Производное эргостерина, излечивающее
рахит, было им идентифицировано и
получило название витамин D2. Впоследствии,
при облучении 7-дегидрохолестерина,
из кожи был получен витамин D3— xo-
лекальциферол. Рассматривавшийся А.
Виндаусом Dt оказался смесью стеринов,
и ныне это обозначение не употребляется
(рис. 81). Активностью витамина D
обладает и восстановленное производное другого
продукта облучения эргостерола — дигид-
ротахистерин. В растительных источниках
имеются 22,23-дегидроэргостерин и 7-де-
гидроситостерин, которые при облучении
переходят, соответственно, в витамин D4
(75 % от активности кальциферолов) и D5
(33 % от активности кальциферолов).
За 1 ME витамина D принято 0,025 мкг
холе(эрго)кальциферола. Суточная
потребность в витамине у детей выше, чем у
взрослых, и составляет 500 ME (12,5 мкг),
тогда как у беременных женщин — 400, а
у взрослых — 100 ME. Не случайно, рахит
поражает детей в возрасте 2-24 месяцев,
хотя известны и его поздние формы.
Впрочем, ряд источников, например
справочник Американской медицинской
ассоциации, рекомендует и для взрослых
дозы 200-400 ME в день. Особенно
обоснованным кажется некоторое увеличение
потребности у старых людей, ведущих
домашний образ жизни или прикованных к
постели и не подвергающихся инсоляции.
Эти контингенты подвержены старческой
остеомаляции, имеющей много общего
с рахитом и чувствительной к терапии
витамином D.
Большинство компонентов обычного
меню без специальной обработки содержит
весьма мало витамина D. Ислючительны-
ми источниками являются рыбий жир и
печень глубоководных морских рыб
(палтуса - 100000 ME на 100 г, тунца - 30 000,
трески — до 1500), мясо рыб (палтус — до
4 000, другие рыбы — от 150 до 1500).
Источники витамина у рыб загадочны. Их ткани не
подвергаются в толще воды сколько-нибудь
значительному ультрафиолетовому
облучению. Планктон, которым они питаются,
витамина D не содержит. Авторам неизвестно,
как ихтиологи решают эту проблему, хотя
в медицине еще доктор Дарби лечил рахит
«Oleum Jecoris Aselli». Печень наземных
животных почему-то содержит гораздо меньше
витамина D, хотя они и гуляют на солнышке
(говяжья — 100 МЕ/100 г). В яичном
желтке летом до 300 ME на 100 г, в сливочном
масле — до 100, сыре — до 200, молоке — до
4 МЕ/100 г продукта, причём во всех этих
случаях зимой содержание витамина в
естественных продуктах значительно (в 2-3
раза) понижается.
В обычных условиях до 80 %
необходимого количества витамина D организм может
синтезировать в коже из
7-дегидрохолестерина, провитамина D3. Провитамин
фотохимически переходит в превитамин, который
претерпевает температурно-зависимую
изомеризацию в стабильный витамин. Для
процесса необходим солнечный свет,
причём наиболее активна его УФ-В фракция,
Патофизиология витаминного обмена 423
то есть ультрафиолетовые лучи с длиной
волны 290-320 нм. Процесс проходит в
макрофагальных клетках собственно дермы
и, возможно, в базальном слое эпидермиса и
адипоцитах подкожно-жировой клетчатки.
Он ослабевает с возрастом, при кахексии и
при потемнении кожи. Гиперкератоз
вследствие авитаминоза А экранирует собственно
дерму и способствует развитию
гиповитаминоза D. Смог и обычное стекло лишают
солнечный свет антирахитической активности.
Понижается фотохимическая активность и
если солнце стоит низко над горизонтом.
Таким образом, диета, бедная кальциферолом,
доместикация и эколого-климатические
особенности могут вызывать экзогенный
гиповитаминоз D.
Витамин проникает в кровеносные
сосуды кожи и переносится с участием витамин-
D-связывающего белка в печень, где гидрок-
силируется в 25-м положении, превращаясь
в 25-гидроксивитамин D (кальцифедиол).
Последующее повторное гидроксилирова-
ние в эпителии проксимальных извитых
канальцев почек, под влиянием
зависимой от парат-гормона митохондриальной
оц-гидроксилазы, превращает этот
метаболит в наиболее активный 1,25-дигидрокси-
витамин D (кальцитриол). Почечное гид-
роксилирование стимулируется не только
парат-гормоном в ответ на гипокальциемию,
но и гипофосфатемией — непосредственно.
При избытке дигидроксилированного
витамина механизм отрицательной обратной
связи ингибирует его дальнейшую
активацию. При снижении уровня моногидрокси-
лированного витамина почечное гидрокси-
лирование увеличивается. Анаболические
гормоны СТГ и пролактин способны
усиливать продукцию кальцитриола в почках.
Кроме кальцитриола у человека образуются
альтернативные дигидроксильные формы
витамина (24,25 или 25,26-дигидроксивита-
мин D), которые значительно менее
активны. Их вырабатывают почки, кости, хрящ и
тонкий кишечник. Помимо почек, ниболее
активный 1,25-кальцитриол формируется
только плацентой и, возможно, некоторыми
лимфоцитами.
Гидроксилирование в печени и почках
необходимо и для активации пищевого
витамина D, который всасывается с липидами
в тонком кишечнике (главным образом
Bjejunum) и попадает сначала в состав хи-
ломикронов. Впоследствии он переносится
в печень и жировую ткань в комплексе с
D-связывающим а{ -глобулином. Снижение
уровня этого транспортного белка
характерно для нефротического синдрома и
печёночной недостаточности, а повышение — для
беременности и гиперэстрогенизма.
Основное депо витамина, как и в случае ретинола,
находится в адипоцитах.
Нарушение переваривания и всасывания
липидов (стеаторея) приводит к
вторичному гиповитаминозу D. Наиболее тяжелая
остеомаляция развивается при целиакии и
обструкции желчевыводящих путей (холес-
тазе). В последнем случае страдает не только
всасывание, но и печёночное
гидроксилирование витамина.
Менее тяжелое нарушение всасывания
витамина D развивается у пациентов с
резецированным по Бильрот-П желудком и
при панкреатической недостаточности. Во
всех этих случаях надо иметь в виду, что
при выраженной мальабсорбции
нарушается не только всасывание витамина D, но
и абсорбция в кишечнике самих кальция
и фосфора, так что даже абсорбированный
или синтезированный в коже кальциферол
не будет максимально эффективным.
Рахит и остеомаляция эндогенного типа
могут быть результатом неадекватного
метаболизма витамина D. При этом его
содержание в диете и всасывание достаточны, но
нарушена активация или усилен распад.
Разрушение активного витамина D идёт
в печени под действием системы микросо-
мальных оксидаз со смешанной функцией,
зависящих от цитохрома Р-450 (см. т. I,
стр. 162-163). Ряд лекарств (фенитоин,
фенобарбитал, рифампицин, карбамазепин)
индуцируют активность этих энзимов и
способствуют созданию эндогенного дефицита
витамина D.
Первое гидроксилирование витамина D
нарушается при печёночной недостаточ-
424 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения
ности. Второе гидроксилирование сильно
страдает при хронической почечной
недостаточности. В этих условиях остеомаляция
связана не только с эндогенным дефицитом
активного витамина D, но и еще как
минимум с тремя патогенетическими
механизмами:
>• при нефросклерозе замедлено выведение
метаболитов парат-гормона, которые
накапливаются в крови и усиливают
остеопороз, в то же время, если имеется
протеинурия, то может теряться вита-
MHH-D-связывающий белок;
>* в организме при ХПН аккумулируется
экзогенный алюминий (из пищи и
принимаемых многими почечными
больными антацидов), который
откладывается в костях и конкурирует с кальцием,
нарушая ход минерализации (см. ниже
«Патофизиология обмена
микроэлементов»);
>* наконец, антациды, принимаемые из-за
уремического гастрита, препятствуют
всасыванию фосфатов.
Известно наследственное рахитоподоб-
ное заболевание — витамин-D-зависимый
наследственный рахит I типа, при
котором имеется дефект именно почечной
оц-гидроксилазы. Рецессивный мутантный
ген локализован в 12-й хромосоме. Болезнь
проявляется задержкой физического
развития, тяжелыми ранними рахитическими
изменениями скелета, гипоплазией эмали
зубов, вторичным гиперпаратирозом, ги-
покальцифосфатемией, высоким уровнем
сывороточной щелочной фосфатазы, ами-
ноацидурией. Возможна коррекция
метаболизма при пожизненном заместительном
введении малых доз кальцитриола —
препарата дигидроксилированного витамина D.
Большие дозы обычного витамина D тоже
имеют терапевтический эффект.
Данный синдром не следует смешимать
с другим заболеванием — витамин-D-
зависимым наследственным рахитом II
типа, когда гидроксилирование проходит
нормально, но имеется дефект тканевых
рецепторов дигидроксивитамина D,
снижающий чувствительность клеток-мишеней.
Клинически это заболевание сходно с
первым, отличительными чертами являются
алопеция, milia, и эпидермальные кисты,
а также мышечная слабость. Его тяжесть
варьирует, по-видимому, из-за
возможности наличия в 12-й хромосоме у различных
больных нескольких мутантных генокопий,
и оно также отвечает на терапию большими
дозами витамина D.
Образование дигидроксивитамина D в
почечном эпителии проксимальных
извитых канальцев и реабсорбция фосфата
почками тормозятся неидентифицированными
пептидными факторами, синтезируемыми
при экспрессии онкогенов в различных ме-
зенхимальных опухолях. Под воздействием
этих факторов развивается такое паранеоп-
ластическое явление, как остеомаляция, при
которой также помогает кальцитриол.
В дальнейшем обзоре механизмов
нарушений обмена кальциферола мы коснемся
только тех аспектов минерального
метаболизма, которые непосредственно связаны с
рахитом и гипервитаминозом D. Более
детальное рассмотрение этиологии и
патогенеза остеомаляции как синдрома содержится в
разделе «Патофизиология паращитовидных
желез...» и в разделе «Остеопороз...» ниже.
Витамин D, вместе со своими
метаболитами и гормонами ряда желёз внутренней
секреции, фактически представляет собой
сложную сбалансированную эндокринную
систему (см. ниже рис. 131, 132, гл. 13).
Главными мишенями этого стероидного
гормона служат почки, ЖКТ и кости. Его
рецепторами снабжены также
околощитовидные железы, гипофиз, мозг,
активированные лимфоциты, макрофаги, тимус, кожа
и многие опухолевые клоны. Мышцы имеют
рецепторы только к кальцифедиолу.
Витамин D обладает следующими
метаболическими функциями, позволяющими
ему быть главным звеном гормональной
регуляции обмена кальция и фосфора и
участвовать в обеспечении некоторых
других процесов:
>- Кальцитриол увеличивает
проницаемость плазматической мембраны кле-
Патофизиология витаминного обмена 425
ток кишечного эпителия для кальция,
вследствие чего тот входит в энтероци-
ты по электрохимическому градиенту.
Кальцитриол увеличивает активный
транспорт кальция в митохондрии эн-
тероцитов, предупреждая его цитоплаз-
матический избыток, чреватый
некробиозом (т. I, с. 184-185). Наконец,
кальцитриол индуцирует на генетическом
уровне синтез кальцийсвязывающего
белка-переносчика, обеспечивающего
дальнейшее выкачивание кальция из
энтероцитов в кровь (Д. Бикл и соавт.,
1981). Независимо от кальция витамин
D стимулирует абсорбцию фосфата в
кишечнике.
> Роль витамина D в костях противоречива
и изучена менее детально. Известно тем
не менее, что его присутствие
необходимо in vivo и in vitro для отложения
солей кальция во вновь сформированном
остеоидном матриксе. Без витамина D
остеоид продолжает синтезироваться,
но задерживается его минерализация
и сильно страдает образование кости
на месте хряща, а также нарушается и
периостальная оссификация. В отличие
от нормы метаэпифизарные хрящевые
пластинки в рахитических костях
расширены и имеют нечёткие границы, под
ними имеется избыточное количество
остеоида, который не полностью
минерализован. Отсутствует нормальная зона
предварительной оссификации хряща,
поэтому пролиферативные хрящевые
клетки на границе эпифиза и диафиза
утрачивают регулярное палисадное
расположение. Остеоидное вещество и
костный мозг врастают в хрящ
беспорядочно. Наблюдается гиперплазия хряща
и остеоидного матрикса, который не
подвергается нормальному
рассасыванию, эти массы образуют выступающие
бугорки. В кости претерпевают
гиперплазию фибробласты и кровеносные
сосуды, возможно, в ответ на деформации и
микроповреждения. Рахитические кости
податливы и легко подвергаются
механическим деформациям, они медленнее
растут, что обусловливает внешние
проявления рахита со стороны скелета.
Установлено несколько аспектов
действия витамина D при окостенении. Повышая
всасывание ионизированного кальция и его
уровень в крови, витамин D препятствует
гиперпаратирозу, при котором парат-гормон
мог бы обеспечить остеомаляцию. Витамин
D способствует в кости и хряще окислению
пирувата в цитрат, а парат-гормон
препятствует переходу цитрата в изоцитрат и цис-
аконититат. Это позволяет им синергично
обеспечивать высокий уровень цитрата,
необходимого для образования
нерастворимых кальциевых солей и минерализации
(В.И.Добрынина, 1976).
Кальцитриол способствует дифференци-
ровке моноцитов и макрофагов в
остеокласты, без которые задерживается резорбция
хряща и избыточного остеоида, что
блокирует энхондральное окостенение. Он также
снижает синтез остеобластами коллагена
I типа, способствуя резорбции избытка
неминерализованного остеоида. Кальцифеди-
ол, в свою очередь, обладает способностью
индуцировать биосинтез минорных белков
костного матрикса — остеокальцина, фос-
фосиалопротеинов и остеонектина, нужных
для связывания кальция и коллагеновых
белков кости. Резорбцию остеоида кальци-
федиол не стимулирует.
Обеспечивая за счет действия на
кишечник и почки (см. ниже) высокий уровень
фосфата в плазме, витамин D поддерживает
концентрации фосфата и кальция на уровне
выше точки насыщения.
Если они (или хотя бы один только
уровень фосфата) снизятся — процесс
минерализации тормозится.
При рахите из-за нехватки активного
витамина D в первую очередь понижается
всасывание кальция и его освобождение
из кости. Это ведет к стимуляции функции
околощитовидных желез и избыточной
продукции парат-гормона. Есть также данные,
что стимуляция рецепторов витамина D в
самих паращитовидных железах в норме
сдерживает продукцию этого гормона.
Так или иначе, рахит — наиболее частая
426 АЖ Зайчик, Л.П.Чурилов Патохимия
в педиатрии ситуация, сопровождаемая
развитием вторичного гиперпаратироза.
Гиперпаратироз позволяет компенсировать
уровень ионизированного кальция в крови
и несколько усилить почечное гидроксили-
рование витамина D. Но при общем
дефиците провитамина или витамина D этого
оказывается недостаточно. Гиперпаратироз
способствует вымыванию кальция из
костей и потере фосфата с мочой. Развивается
фосфатурия и гипофосфатемия. Поскольку
уровень фосфата упал ниже точки
насыщения, а также ниже минимума, необходимого
для адекватного фосфорилирования
компонентов остеоида, минерализация последнего
оказывается невозможной. Если кости
активно растут, генез остеоида продолжается.
При дефиците витамина D, без адекватной
резорбции хряща и остеоида, формируется
рахитическая структура костей, наступают,
по мере действия механических нагрузок,
связанных с дыханием и развивающейся
моторикой ребенка, их характерные
деформации. При тяжелом длительном рахите и
образование остеоидного матрикса страдает,
а рост костей тормозится. Таким образом,
ранний рахит у детей 2-24 месяцев в
развернутой стадии проявляется следующими
симптомами, в основном отражающими
нарушение энхондрального остеогенеза:
>- Craniotabes — то есть размягчение костей
черепа, при котором уплощаются
затылочные кости.
>- Периостальные наслоения остеоида в
области лобных и теменных бугров («лоб
Сократа», «квадратная голова»), при
выздоровлении окостеневающие.
>• Утолщения эпифизов длинных
трубчатых костей («рахитические браслеты») и
«рахитические чётки» на грудных концах
рёбер. В отличие от скорбутных чёток
они не болезненны.
>- Деформации грудной клетки, связанные
с дыхательными движениями, в
частности «борозда Гаррисона» по линии
прикрепления диафрагмы, а также
«куриная грудь» с выступающей грудиной
и сдавленными с боков ребрами.
чдокринно-метаболические нарушения
>- По мере развития моторики ребенка,
начинающего ползать и ходить,
отмечаются искривления нижних конечностей
(genua vara и реже genua valga,
плоскостопие, саблевидные голени). Возможны
деформации таза («плоский таз»).
>- Задерживается закрытие родничков и
прорезывание зубов. Облегчается
возникновение их кариеса.
>- Имеются деформации лицевого черепа,
в частности высокое готическое нёбо,
седловидный нос.
>• Позвоночник претерпевает кифоз в
нижнегрудном отделе, а по мере
развития навыков ходьбы это дополняется
поясничным лордозом и сколиозом.
При позднем рахите у старших
дошкольников и подростков комбинируются
проявления нарушений энхондрального и периос-
тального окостенения. Типичны боли в
костях и суставах, размягчения костей черепа
не бывает, но симптомы со стороны нижних
конечностей и позвоночника присутствуют.
Если рост костей в длину к моменту
развития гиповитаминоза был уже завершен,
формируется остеомаляция — взрослая форма
витамин-Б-зависимого дизостеогенеза,
когда преобладают нарушения пермостального
окостенения. Она поражает пожилых
пациентов, особенно лежачих больных, а также
беременных и кормящих женщин. При
остеомаляции остеоид вырабатывается, но
не минерализуется. Это следует отличать от
остеопороза, когда снижается и образование
остеоида и его минерализация (см. гл. 13).
Впрочем, показано, что витамин D у
пожилых эффективен и при остеомаляции и при
многих формах остеопороза. По некоторым
наблюдениям, до четверти пожилых
пациентов больниц с различными диагнозами в той
или иной степени страдают от остеомаляции
и остеопороза. Считается, что к возрастной
остеомаляции предрасположены носители
определённых аллельных вариантов
рецептора кальцитриола.
Следует иметь в виду возможность
нарушения минерализации остеоида из-за
местных и системных причин, не связанных
с дефицитом активного витамина D. Это
приводит к картине «D-резистентного
рахита». Одним из подобных состояний является
наследственная аутосомно-рецессивная
гипофосфатазия. При дефекте щелочной
фосфатазы у больных замедляется гидролиз
пирофосфата, который в костях является
ингибитором минерализации. У детей
развивается рахитоподобное заболевание
скелета, у взрослых часты патологические
переломы. Повышается экскреция с мочой
фосфатидилэтаноламина. В отличие от
истинного рахита, при котором уровень
щелочной фосфатазы сыворотки повышен,
имеется резкое снижение её активности.
Приобретенные нарушения кристаллизации
фосфата кальция востеоиде и угнетение
жизнедеятельности остеобластов вызывает
лекарство этидронат, высокие дозы которого
провоцируют остеомаляцию.
> В почках витамин D усиливает реабсор-
бцию кальция в дистальных извитых
канальцах. Эта его функция в основном
опосредована через эффект парат-гормо-
на. Возможно, он играет пермиссивную
роль, контролируя рецепцию парат-
гормона почкой. По отдельным,
неподтверждённым данным, независимо от
парат-гормона витамин D способствует
также снижению экскреции фосфата и
аминокислот с мочой. Почечные этапы
метаболизма и действия витамина D
тесно связаны с патогенезом рахито-
подобных заболеваний, при которых
причиной остеомаляции являются
первичные дефекты ренальных витамин-D-
ассоциированных механизмов. Прежде
всего это тубулопатии, сопровождаемые
изолированной либо комбинированной
потерей фосфат-анионов и гипофос-
фатемией. При семейной сцепленной с
Х-хромосомой гипофосфатемической
остеомаляции имеется изолированный
дефект реабсорбции фосфатов почками
и у некоторых больных понижение
кишечного всасывания кальция. Это, как и
вышеназванный дефект щелочной
фосфатазы, ведет к рахиту, резистентному к
терапии витамином D. При нормальном
Патофизиология витаминного обмена 427
уровне кальцитриола в плазме имеется
сильная гипофосфатемия, концентрация
кальция бывает на нижней границе
нормы или субнормальна. Данный дефект
весьма распространен и ответственен за
большинство случаев рахита,
резистентного к витамину D. В лечении, наряду с
большими дозами витамина, используют
нагрузку фосфатами.
Сочетанная с другими дефектами
реабсорбции фосфатурия характерна для
синдрома Де Тони-Дебре-Фанкони. При
полном синдроме (см. также выше «Ами-
ноацидурия») отмечаются фосфатурия,
бикарбонатурия, аминоацидурия и глюко-
зурия. Развивается почечный канальцевый
ацидоз, который (как и хронические
метаболические формы ацидоза) дополнительно
препятствует нормальной минерализации
остеоида. У больных формируется
остеомаляция. Синдром может присутствовать
в структуре других наследственных
аномалий транспортных белков (болезни
Коновалова-Вильсона, цистиноза, синдрома
Лоу — см. выше «Нарушения транспорта
аминокислот...»). Его картина зачастую
сопровождает парапротеинемии с выделением
белков Бенс-Джонса.
Таким образом, нормальное действие
витамина D не может быть обеспечено не
только при нехватке пищевого кальция, но и
при гипофосфатемических состояниях (см.
также «Патофизиология паращитовидных
желёз...»).
Подытоживая имеющиеся сведения о
патохимических аспектах рахита,
подчеркнём, что истинный дефицит витамина D,
в отличие от других причин остеомаляции,
упоминавшихся выше, характеризуется
следующими лабораторными данными:
гипофосфатемией, гипокальциемией (в
тяжелых случаях) или нормокальциемией,
высокой активностью щелочной фосфатазы
сыворотки, гипоцитратемией, гиперпара-
тирозом, фосфатурией и гипокальциурией.
Содержание кальцифедиола снижено,
кальцитриола — может быть нормально
или субнормально. Кальций-фосфатный
428 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
коэффициент сыворотки, в норме равный
2, при рахите сильно увеличен.
>* Роль витамина D не исчерпавается
регуляцией остеогенеза. Еще классики
педиатрии — Н.Н. Филатов и М.С. Маслов,
а также основатели микробиологии, в
частности Р. Кох, отмечали, что рахит
и туберкулёз «идут рука об руку» и
взаимно отягощают друг друга. Но только
недавно была выяснена конкретная регу-
ляторная роль кальцитриола в иммунной
системе. Как известно, микобактерии
туберкулёза, как и отдельные грибки,
крайне устойчивы к бактерицидному
действию макрофагальных
кислородных радикалов из-за наличия у них в
клеточных стенках антиоксидантных
фенол-гликолипидов. Оказалось, что
стимулированные цитокинами
макрофаги человека синтезируют в больших
количествах кальцитриол, так как в них
индуцируется активность сц-гидроксилазы.
Кальцитриол усиливает в макрофагах
активность НАДФ-зависимой нитро-
ксидсинтазы и позволяет в завершающей
фазе фагоцитоза оросить микобактерии
туберкулёза активными
азотсодержащими радикалами, к которым они
неустойчивы. Это резко усиливает способность
макрофагов к завершению фагоцитоза
и ограничивает персистирование
возбудителей в фагоцитах, что важно для
прекращения распространения инфекции
по организму. Таким образом, получено
прямое доказательство роли витамина
D в противоинфекционном, в частности
противотуберкулёзном и
противогрибковом, иммунитете. У грызунов в этих же
целях используется метаболит другого
витамина — фолиевой кислоты — тет-
рагидроптерин (см. также т. I, стр. 3).
>* Имеются данные об анаболизирующем
эффекте кальцифедиола на скелетные
мышцы и даже гипотеза об
ограничительной роли витамина D в росте и развитии
мозга, «габариты» которого должны
соответствовать размерам черепной
полости при закрытии родничков. Однако
эти положения пока не подкреплены
достаточными доказательствами. Впрочем,
ещё классики педиатрии, в частности,
А.Ф. Тур в своей монографии «Рахит»
(1966), подчеркивали, что рахит — не
болезнь опорно-двигательного аппарата,
а системное эндокринно-метаболическое
расстройство, и указывали в этой связи,
что дебют рахита у грудных детей
сопровождается как раз не костной, а нервно-
мышечной и кожной симптоматикой:
ребенок становится раздражительным,
беспокойным, плохо спит, испытывает
дизгидроз и мышечный гипотонус.
Происходит облысение затылка. Тяжелый
рахит может сопровождаться
симптомами, вызванными системной гипокаль-
циемией, в частности тетанией.
Ухудшение вентиляции легких и снижение
иммунитета при рахите способствуют
развитию бронхолёгочных заболеваний.
Нет сомнения, что по мере расширения
наших знаний о метаболической роли
витамина D вне костей классическая точка
зрения, не ограничивающая патогенез
рахита проблемой окостенения, будет
подтверждаться.
Практическая педиатрия в XX веке
одержала победу над массовым рахитом. В
развитых странах добавление кальциферола в
молоко и питательные смеси,
индивидуальная профилактика с помощью
ультрафиолетового облучения или витамина D привели
к тому, что в обеспеченных слоях населения
экзогенный рахит стал встречаться у детей
реже, чем эндогенный. Но педиатры и
терапевты столкнулись с проблемой
противоположного знака, порожденной неосторожным
использованием новых возможностей
медицины. Имеется в виду гипервитаминоз D,
включая его отдалённые последствия.
Гипервитаминоз наблюдается при
продолжительной передозировке его
препаратов или одноразовом приеме токсической
дозы витамина. Этому способствуют
искусственное вскармливание, создающее
избыток кальция, а также неумеренное
комбинирование витаминного и
ультрафиолетового метода профилактики рахита,
дефицит других витаминов, в частности А,
Патофизиология витаминного обмена 429
сердце). Отмечается понижение активности
щитовидной железы и гонад.
Установлено прооксидантное действие
избытка витамина D на клеточные
мембраны и на липопротеиды, что в сочетании
с повреждением эндотелия и активацией
макрофагов может способствовать
появлению липоперекисей и развитию
атеросклероза. Даже нетяжелый гипервитаминоз D,
перенесенный в детстве, считается одним
из факторов риска атеросклероза в
последующие годы. При тяжелом отравлении у
грудных детей отмечен артериосклероз и
надклапанный стеноз аорты. Поражение
сосудов при гипервитаминозе D
усиливается при дефиците кобаламина и фолацина
из-за сопутствующих нарушений обмена
гомоцистеина (см. выше).
Разрушение белковой костной матрицы
в условиях гипопаратироза тормозится,
прекращается рост скелета в длину,
утолщаются диафизы и особенно эпифизы.
Раннее зарастание родничков ведет к
формированию микроцефалии и к повышению
внутричерепного давления. Это может
сопровождаться сонливостью и апатией.
Тяжелый острый гипервитаминоз D, при
случайном приеме крайне больших доз
рыбьего жира и собственно кальциферола,
иногда обусловливает смертельные исходы
в результате почечной недостаточности,
сдавления мозга, ацидоза и гиперкальци-
емических аритмий. Кроме того, большие
дозы витамина D тератогенны.
Таким образом, использование гормоно-
витамина кальциферола требует от доктора
такой же осторожности и продуманности,
как и применение других гормонов.
Е, Bj, С и Р. Гипервитаминоз у детей может
быть острым и хроническим. Его картина
проявляется чаще всего, начиная с 3-4
месяцев жизни, но это не значит, что он не
имеет отдаленных последствий. У ряда
индивидов даже не очень значительная
передозировка витамина вызывает тяжелый
гипервитаминоз. Это связано с собственным
синтезом кальцитриола активированными
макрофагами. При саркоидозе и в меньшей
степени других хронических гранулёматоз-
ных ГЗТ происходит усиленная конверсия
витамина D в его активнейшую форму, так
как каждая гранулёма содержит цитокин-
активированные макрофаги. Большое
значение имеет и усиление абсорбции витамина
D, кальция, фосфора, а также аффинитета
тканей к витамину D при выздоровлении
от мальабсорбционных синдромов.
Ранними признаками передозировки витамина
являются тошнота, головная боль, потеря
аппетита и веса, полиурия, жажда и
полидипсия. Могут быть запоры, гипертензия,
мышечная ригидность.
Проявления гипервитаминоза связаны с
усилением всасывания кальция и фосфора,
гиперкальциемией и гиперфосфатемией,
кальциурией, вторичным гипопаратирозом.
Снижается активность щелочной фосфа-
тазы. В щелочной среде в почках и других
тканях фосфаты и кальций образуют
труднорастворимые соли. Происходит
образование почечных конкрементов, кальциноз
почек и сосудов. Экспресс-методом гипер-
кальциурия хорошо выявляется по
немедленному интенсивному помутнению мочи
при добавлении к ней половинного
количества реактива Х.У. Сулковича1. В норме
помутнение бывает легким и развивается с
задержкой в несколько секунд, а при гипо-
кальциурии моча не мутнеет. Кальциноз при
отравлении витамином D наблюдается и в
других внутренних органах (печени, лёгких,
1 Реактив Х.У. Сулковича: в 50 мл
дистиллированной воды растворяют по 2,5 г щавелевой
кислоты и оксалата аммония, добавляют 5 мл
ледяной уксусной кислоты и доводят объем
раствора до 150 мл.
Нарушения обмена витамина К
В 1929 г. датский биохимик X. Дам в
экспериментах на цыплятах установил, что диета,
сбалансированная по витаминам группы
В экстрактом из дрожжей и содержащая
достаточно рыбьего жира (поставляющего
витамины А и D), всё же не спасает птиц от
кровоточивости. Фактор, предохраняющий
от коагулопатии, содержался в конопляных
430 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения
семенах. X. Дам назвал его витамином К (от
датского написания слова «коагуляция»).
Оказалось, что витамин К, отсутствующий
в свежей рыбной муке, появляется там при
гниении (Э.А. Дойзи, 1939). Возникла
подтвердившаяся впоследствии догадка, что
его могут вырабатывать бактерии. Формулу
витамина К установили в 1939 г.
швейцарский биохимик П. Каррер и Э.А. Дойзи
(рис. 81). Витамин оказался производным
2-метил-1,4-нафтохинона, имел две формы
(растительный фитил-нафтохинон — фил-
лохинон или витамин Ки бактериальный
дифарнезил-нафтохинон — менахинон или
витамин К2) и вскоре был синтезирован.
Дойзи и Дам стали нобелевскими
лауреатами в 1943 г. по медицине.
Витамин К — жирорастворимое
вещество. В СССР М.М. Шемякиным и
А.В. Палладиным впервые был получен его
синтетический водорастворимый аналог,
удобный для парентерального применения
(метилнафтохинон бисульфит или вика-
сол). Биологические свойства витамина К
позволяют считать его гормоновитамином,
индуцирующим синтез ряда белков (см. т. I,
раздел «Тромбоз»). Однако он является
и редокс-витамином, поскольку обладает
свойством принимать и отдавать протоны
и электроны, по типу превращения хинона
в гидрохинон и обратно. Установлена и
коферментная роль витамина К,
например, в менадион-редуктазе митохондрий
растительных клеток и печени. Можно
сказать, что данный незаменимый фактор
питания представляет собой ярчайшую
иллюстрацию относительности
разграничения витаминов на энзимо-, гормоно-
и редокс-витамины. По этой причине мы
рассматриваем его в завершение данного
раздела книги.
Потребность в витамине К у взрослого
в обычных условиях на смешанной диете
удовлетворяется, по-видимому, на 100%
микрофлорой верхних отделов толстого
кишечника. Эубактериальная флора, в
основном Е. Coli, синтезирует до 1,5 мг
витамина в сутки, в основном из
распространенного в обычной диете менадиона. В то же
время оценочная потребность взрослого не
превышает 0,2-0,3 мг/день, а минимальная
определена всего в 1 мкг/кг массы тела в
сутки. Так как беременные женщины
поставляют витамин растущему плоду,
потребности которого могут быть велики из-за
участия витамина К в синтезе ряда белков,
считается, что при беременности симбиоти-
ческий синтез витамина ниже потребности
в нем, и рекомендуют его прием в пределах
2-5 мг/день. У детей первых дней жизни
отсутствует поставка витамина
симбионтами. Потребность составляет не менее 1-12
микрограммов в сутки.
Хорошими источниками витамина К
являются листья зеленых овощей, плоды и
ягоды (шпинат — 4 мг/100 г; помидоры — 0,4;
земляника — 0,12; цветная капуста — 0,06).
Подчеркнем, что им богаты и некоторые
животные продукты (свиная печень — 0,6;
свинина — 0,15; говядина -0,1). Очень много
витамина в люцерне и крапиве.
Этиология гиповитаминоза К связана не с
его пищевым дефицитом, который возможен
только при полном голодании или в
экспериментальных условиях. Реально нехватка
витамина возникает:
>• При нарушении всасывания липидов
и стеаторее (особенно при ахолии).
Важно отметить, что диффузные
поражения печени и холестаз, часто
вызывающие стеаторею, способны
нарушить и витамин-К-зависимые
процессы в этом органе. С другой стороны,
холелитиаз может служить поводом
для хирургических операций,
предъявляющих повышенные требования к
системе гемостаза. Поэтому введение
викасола перед такими операциями
патофизиологически оправдано. В
норме 80% витамина Кх всасывается
в лимфу тонкого кишечника вместе с
липидами. Витамин К2 всасывается и в
толстом кишечнике, причём, в отличие
от Kj, его абсорбция неэнергозависима
и не требует участия специального
переносчика. В лимфе витамин
переносится липопротеидами, в основном,
хиломикроновой фракции.
Патофизиология витаминного обмена 431
> При дисбактериозах, в частности,
вследствие перорального лечения
сульфаниламидами и антибиотиками широкого
спектра действия, угнетающими симби-
онтную флору кишечника. Продукты,
богатые витамином, а также его
препараты рекомендуют для профилактики
гиповитаминоза при дисбактериозе.
> При отравлении антивитаминами К.
Свойства антивитамина К у дикумарина
впервые были открыты классиком
отечественной фармакологии М.Д. Маш-
ковским. Кумариновые антикоагулянты
ингибируют восстановительную
реактивацию витамина К из его неактивной
эпоксидной формы. Экологически
небезынтересно отметить, что и витамин
К, и его антивитамины присутствуют
в одних и тех же пищевых источниках
травоядных — например, в клевере.,
Кумариновые антикоагулянты, в
частности варфарин, до сих пор остаются в
арсенале медицины для лечения
гиперкоагуляции, например при синдроме
диссеминированноговнутрисосудистого
свертывания и тромбоэмболии
лёгочной артерии. Возможна, следовательно,
их передозировка. Надо считаться и
с широким использованием аналогов
варфарина, вакора и более современного
и сильнодействующего бродифакоума
в практике дератизации — в качестве
крысиных ядов. В связи с этим острый
гиповитаминоз возможен при
профессиональных и случайных отравлениях и
попытках самоубийства. Кумариновые
производные способны инактивировать
фермент эпоксидредуктазу на разный
срок. С точки зрения дератизации
крысиный яд тем эффективнее, чем дольше
животное пребывает в состоянии
дефицита факторов коагуляции после его
применения. Ведь для гибели крыса,
подверженная эффекту антивитаминов,
должна еще успеть получить
серьёзное случайное повреждение, чреватое
кровопотерей. В этом отношении вне
конкуренции стоит бродифакоум,
однократный прием которого выключает
эпоксидредуктазу на несколько месяцев.
Антагонистами действия витамина К
являются и некоторые другие лекарства,
действующие не через эпоксидредуктазу.
Среди них — гидантоин, а также цефа-
лоспорины 3-го поколения (моксалак-
там, цефоперазон, цефамандол), которые
не только вызывают дисбактериоз, но
и прямо конкурируют с витамином за
карбоксилазы.
>• Важнейшим проявлением
гиповитаминоза К является геморрагическая болезнь
новорожденных. Данное заболевание, под
названием melaena neonatorum, было
известно еще средневековым врачам,
а возможно — и задолго до нашей эры.
Книга Левит содержит рекомендацию
древним иудеям — не проводить циркум-
цизию у младенцев ранее 8-го дня после
рождения, что далеко не случайно
совпадает с типичными сроками окончания
периода наивысшего риска
гиповитаминоза К. При геморрагической болезни
новорожденных, на 2-4-й день жизни,
иногда — позже, возникают мелена
(черный дегтеобразный стул, окружённый
на пеленке красным ободком), пурпура,
реже гематемезис. Имеется выраженная
кефалогематома. Могут быть
кровотечения из пуповинного остатка, у
девочек — метроррагия, в более тяжёлых
случаях бывают кровоизлияния в мозг,
печень, легкие, надпочечники.
Патогенез расстройства связан с низким
содержанием у новорожденных витамин-
К-зависимых факторов коагуляции. Он не
превышает даже в норме 60 % от уровня
взрослых и закономерно снижается в первые
2-3 дня после рождения (Н.П. Шабалов,
1984). Так как симбионтная флора в
кишечнике устанавливается только по мере начала
кормления, она не поставляет витамина К в
организм в первую неделю постнатальной
адаптации. К тому же запас витамина К в
печени к моменту рождения невелик,
особенно у недоношенных и гипотрофичных
детей. Ни женское, ни коровье молоко не
являются богатыми источниками витамина К
(1,5 мкг/л и 6 мкг/л, соответственно). К тому
432 ALU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
же родовой стресс обязательно приводит к
появлению на слизистых оболочках ЖКТ
эрозий, а иногда и язв (см. т. I, гл. 18).
Поэтому системная недостаточность коагуляции
проявляется чаще всего именно меленой
и гематемезисом. Грудное вскармливание
обеспечивает ребенка частично
материнскими факторами свёртывания и снижает
вероятность и тяжесть геморрагической
болезни. Профилактика этого нарушения
осуществляется с посмощью введения
дополнительного витамина К в диету
беременных и викасола новорожденным (см. т.
III, гл. 4).
Биологическая роль витамина К, а
значит, и патогенез гиповитаминоза К связаны
прежде всего с тем, что данный витамин —
обязательный кофактор микросомального
печёночного фермента у-карбоксилазы.
Она превращает глутамиловые остатки
в ряде белков в ди-у-карбоксиглутаматы.
Этот процесс— посттрансляционный, но
предшествует секреции преобразуемых
белков. Подвергаемые у-карбоксилированию
белки — это факторы свёртывания II, VII,
IX, X (а возможно — и V). Кроме того, анти-
тромботические белки С и S также проходят
эту обработку. По некоторым данным, у-
карбоксилированию подлежат и некоторые
неколлагеновые белки костей, в частности
остеокальцин, выделяемый остеобластами
и составляющий в костях до 1 % белкового
содержания (см. ниже «Патофизиология па-
ращитовидных желёз...» и «Остеопороз...»).
Есть сведения и об участии витамина К в
посттрансляционной модификации
ферментов поджелудочного и желудочного сока, а
также альбумина. Данные об участии в
синтезе панкреатических ферментов особенно
интересны, в связи с имеющимися фактами
высокой диабетогенности некоторых
препаратов-антивитаминов К, в частности вакора.
Возможно, витамин К играет еще не
известную роль в островках Лангерганса или в их
предохранении от разрушения.
Входящие в белки у-карбоксильные
остатки находятся там блоками по 10-12
аминокислот. Подобный домен позволяет
этим протеинам связывать IV фактор
свёртывания — ионизированный кальций. Через
кальциевый мостик они прикрепляются к
полианионным поверхностям, что и даёт
возможность им выполнять как прокоагу-
лянтные (И, VII, IX, X, V), так и антикоагу-
лянтные (С, S) функции.
Таким образом, коагулопатия при
дефиците витамина К является комплексной и
охватывает как образование протромбиназы,
так и тромбина. Удлиняются время
свёртывания крови, частичное тромбопластиновое
и протромбиновое время. Тип
кровоточивости — гематомный. Ранним проявлением
служит снижение степени карбоксилирования
фактора VII и белка С, а затем — и остальных
вышеназванных протеинов. Лабораторным
признаком, который, правда, характерен и
для печёночной недостаточности, является
понижение протромбинового индекса ниже
70%. При выраженном гиповитаминозе он
может падать ниже 35 %. При истинном
гиповитаминозе К, в отличие от коагулопатий,
связанных с печеночной недостаточностью
и гепаринизацией, наблюдается
накопление в плазме некарбоксилированных
предшественников факторов свёртывания,
в частности протромбина (так называемые
PIVKA — proteins in vitamin К absence).
Установлена также необходимость
витамина К для оптимального прохождения
кальцификации остеоидной матрицы.
Антагонисты витамина К не применяют у
беременных, так как они вызывают нарушения
формирования скелета у эмбриона.
Имеются основания полагать, что вита-
мин-К-зависимые кальцийсвязывающие
белки могут участвовать в почечной реаб-
сорбции этого двухвалентного катиона.
В ходе своей коферментной функции
витамин К окисляется в эпоксид и инак-
тивируется. Как уже упоминалось выше,
эпоксидредуктаза восстанавливает его
активность, а кумариновые антивитамины
нарушают этот процесс.
В растениях и у бактерий витамин К
определённо является компонентом редокс-
системы и участвует в тканевом дыхании.
Менадион-редуктаза, способная
осуществлять перенос электронов от НАДН-зависи-
мых дегидрогеназ на цитохромы, имеется и
в печени животных. Считается, что участие
витамина К в процессах окислительного
фосфорилирования требует кооперации с
рибофлавином, аскорбиновой кислотой,
магнием и другими редокс-факторами.
Есть наблюдения о зависимости
активности креатинфосфокиназы, гексокиназы и
мышечной АТФ-азы у животных от уровня
обеспеченности витамином К. Антиокси-
дантный витамин Е в больших дозах инги-
бирует всасывание и действие витамина К,
которому он родствен химически (Е — бен-
зохинон, а К — нафтохинон). Точная роль и
место витамина К в редокс-системах клеток
изучены намного хуже, чем его участие в
модификации карбоксилируемых белков.
Но известно, что большие дозы витамина К
могут вызывать гипервитаминоз, основным
проявлением которого служит изменение
редокс-систем красных кровяных
клеток. Эритроциты могут подвергаться при
К-гипервитаминозе усиленному
образованию метгемоглобина и гемолизу. По этой
причине у новорожденных, получавших до
и после рождения значительные дозы
витамина К, может продляться и отягощаться
гемолитическая желтуха (см. т. III, гл. 4).
В последнее время появляются данные, что
антивитамины К могут разобщать
окисление и фосфорилирование на участках
до и после цитохромов в митохондриях
экспериментальных животных. Это может
подтвердить положение о наличии у данного
витамина функций, сопрягающих окисление
и фосфорилирование, что, возможно, делает
его участником энергообеспечения синтеза
наиболее быстрообмениваемых белков.
ЗАКЛЮЧЕНИЕ
Заканчивая рассмотрение
патофизиологических основ витаминологии, надо отметить,
что сложность экспериментального
моделирования гиповитаминозов состоит в том,
что продолжительность жизни подопытных
животных с авитаминозом лимитируется
наиболее опасным метаболическим рас-
Патофизиология витаминного обмена 433
строиством, возникающим в отсутствие
витамина. При этом, проявления, не менее
характерные для выпадения метаболической
роли того или иного витамина, но
требующие для своего развития большего времени,
могут просто не успеть развиться. Поэтому
не исключено, что механизмы и полная
картина многих гиповитаминозов еще
далеки от окончательного выяснения. Вместе
с тем нам кажется оправданным
предвидение А.М.Уголева (1987), полагавшего,
что какие-либо неизвестные незаменимые
факторы питания могут поставляться
человеку эубиотической микрофлорой, причем
потребность в них столь мала, что картина
их нехватки не описана, и само их
существование еще долго не будет очевидным.
Внимательное изучение витаминологии
показывает, что нет естественных продуктов
питания, которые были бы воплощённой
панацеей или, наоборот, несли ярлык
изначально «плохих». Например, кукуруза
бедна ниацином, но в изобилии содержит
витамин Е. Поэтому призывы типа «больше
фруктов — и все проблемы будут решены!»,
«зеленый чай — рецепт долголетия!»,
«сахар — белая смерть!», «витамин С убережет
вас от последствий курения!» и т.д. имеют
орнаментальный характер и принадлежат
не профессиональному, а паралогическому
мышлению. Трепетное отношение людей
к вопросам диеты эксплуатируется
пропагандистами «здоровых» пищевых добавок
и приносит большие деньги. Но в области
науки о питании не все кажущиеся
закономерности представляют практическую
ценность. Вспоминается фраза из трактата
о вреде огурцов, опубликованного в
знаменитом сборнике «Физики шутят» о том, что
90 % всех солдат ели огурцы.
Главное, что кажется неоспоримым,
принимая во внимание вышеизложенный
материал, это то, что хорошая диета есть
прежде всего диета разнообразная.
Для медика - экспериментатора
изучение патофизиологии витаминного
обмена — хорошая школа, показывающая, как в
медицине проходят путь от эмпирических
наблюдений к доказанному научному
434 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения
знанию. Интересующихся этими
вопросами читателей привлечет недавний обзор
К.Дж. Карпентера «Краткая история науки
о питании (в 4-х частях)» (2003).
В витаминологии, также как в
эндокринологии и учении о цитокинах, вполне
справедлив и очень актуален открытый
Л.Р.Перельманом (см. гл. 13 ниже)
принцип пермиссивного действия
биорегуляторов. Витамины действуют комплексно,
вместе с микроэлементами и
незаменимыми аминокислотами, для осуществления
той или иной метаболической цепи или
трансформации, зачастую, необходимо
параллельное или последовательное участие
нескольких незаменимых веществ. Их
соотношения могут, по-видимому, определять
относительные скорости альтернативных
метаболических процессов, влияя на
конечный результат.
Выше уже были представлены некоторые
примеры: так, в составе пируват-декарбок-
силазного комплекса находятся несколько
витаминных коферментов, превращения
незаменимого метионина требует участия фо-
лацина и кобаламина, липотропное действие
на гепатоциты присуще этим витаминам, а
также селену и метионину, регуляция
обмена кератина требует наличия эссенциальных
жирных кислот и ретинола, причем дефицит
любого из них оборачивается
гиперкератозом. Наконец, редокс-потенциал клеток
определяется поставками витаминов Е, К, С
и ряда микроэлементов. На разном пермис-
сивном фоне одна и та же доза какого-либо
витамина может действовать неодинаково.
Это означает, что в диетологии требуется
комплексный подход при коррекции
поставки незаменимых ингредиентов пищи.
Практически, любой из витаминов влияет
на окислительно-восстановительные
процессы, пусть не все они служат коэнзимами
оксидорелуктаз.
В свете недавнего открытия специального
гормона печени - гепцидина,
контролирующего метаболизм такого незаменимого
фактора пищи, как железо (см. гл. 11 ниже),
нет сомнений, что применение принципа
пермиссивности в витаминологии будет
расширяться. Регуляторная роль паратиро-
кринина в отношении метаболизма
витамина D известна давно (см. гл. 13 ниже). Мы
полагаем, что в дальнейшем будут открыты
и гормоны, регулирующие обмен других
незаменимых факторов питания, включая
витамины.
Глава 12
ПАТОФИЗИОЛОГИЯ ОБМЕНА
МИКРОЭЛЕМЕНТОВ
«Этот камень рычал когда-то,
Этот плющ летал в облаках!»
Н.С. Гумилёв. «Начало. Книга I. Дракон. Песнь I»
• Роль и место микроэлементов в физиологии и патологии обмена веществ
• Патофизиология метаболизма железа
• Патофизиология обмена других микроэлементов-металлов
• Патофизиология обмена важнейших микроэлементов-неметаллов
Основатель современной экологии
В.И.Вернадский подчёркивал
единство элементарного состава живых существ
и неживой среды, сочетанный характер их
эволюции (1921). Он ввел представления
о биогенной миграции атомов — переходе
элементов из организма в организм и их
распространении по планете, в силу
действия биогеоценотических связей,
объединяющих живых участников экологического
сообщества и его минеральную основу.
Развивая положения биогеохимии, другой
наш соотечественник ученик
Вернадского— А.П.Виноградов (1932) разработал
учение о биогеохимических провинциях и
влиянии локальных особенностей
минерального состава почвы, воды и продуктов
питания на патологию.
Эти положения особенно актуальны в
медицинской географии, гигиене и
патофизиологии.
роль и место
МИКРОЭЛЕМЕНТОВ
В ФИЗИОЛОГИИ
И ПАТОЛОГИИ ОБМЕНА
ВЕЩЕСТВ
Химический состав живых клеток более
чем на 90 % представлен всего четырьмя
элементами — углеродом, водородом,
кислородом и азотом. По сути дела,
крупнейшие разделы данной книги, посвященные
патологии обмена углеводов, липидов,
белков и нуклеиновых кислот, касались
как раз метаболизма этих элементов,
которые их и составляют. Еще более 9 % веса
тела представлено элементами, которые
находились в изобилии в воде первичного
океана, где зародилась жизнь, это: натрий,
хлор, фосфор, кальций, магний, калий и сера.
436 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Металлы этой группы слабо связываются
с отрицательно заряженными лигандами и
существуют в виде катионов, относительно
легко переносимых через клеточные
мембраны. Их клетки используют для создания
электрических биопотенциалов и биотоков,
а также в качестве «спусковых крючков»,
опосредующих передачу сигналов.
Патология метаболизма натрия, хлора,
калия обсуждалась в разделе
«Патофизиология водно-электролитного обмена», что
касается кальция, фосфора и магния —
детальный обзор на эту тему вынесен в
раздел «Патофизиология околощитовидных
желез...» ниже.
Роль серы, которой взрослый человек
потребляет в сутки не менее одного грамма,
в жизнедеятельности организма выяснена
недостаточно. Какие-либо специфические
расстройства, вызванные ее дефицитом,
пока не описаны. Однако выше шла речь о
том, что сера — важный структурный
компонент аминокислот метионина и цистина,
а дисульфидные связи представлены во
многих белках, включая гормоны (кальци-
тонин, инсулин). Сера имеется и в составе
витаминов (тиамин, биотин, липоат).
Богатейшими источниками серы служат сыры
(содержание 263 мг/100г), яйца и мясо (230
мг/100г).
При нормальном метаболизме,
дожигающем органические кислоты до С02 и Н20,
именно серосодержащие аминокислоты
служат главным источником нелетучих кислот
в организме, поскольку производство из них
серной кислоты (сульфат-аниона)
освобождает кислые валентности. О патофизиологии
обмена серосодержащих аминокислот речь
уже шла выше, в разделах, посвященных
белковому обмену и кислотно-щелочному
равновесию.
Таким образом, обсуждение в данной
книге к настоящему моменту коснулось всех
тех элементов, которые составляют более
99 % массы тела.
Однако оставшиеся, суммарная доля
которых в организме чуть менее 1 %,
чрезвычайно важны для здоровья. Это и есть
микроэлементы. Именно расстройства их
метаболизма ответственны за многие
нарушения, которые либо универсально
распространены (например, железодефицитная
анемия или кариес зубов), либо крайне
тяжелы (например, микседема, эксфолиа-
тивный акродерматит или селензависимая
миокардиодистрофия).
Понятие «микроэлементы» (ср. англ.:
«trace metals» — металлы, присутствующие
в следовом количестве), до известной
степени условно. К микроэлементам относят
металлические и неметаллические
элементарные составляющие организма,
содержание каждой из которых не превышает 1 мкг
на грамм веса живой ткани. По Р.А. Хен-
кину (1976), это элементы, суммарное
содержание которых в теле человека весом
70 кг составляет менее 4 граммов. Так как
натуральная еда содержит, как правило,
достаточные и сбалансированные самой
природой количества микроэлементов, в
течение многих лет медицина сталкивалась
только с эндемическими проявлениями их
дефицита и дисбаланса.
Исключение представлял обмен железа,
поскольку повсеместная широчайшая
распространенность его нарушений — давно
известный факт. В последнее время из-за
расширения применения практики полного
парентерального питания частота
встречаемости расстройств обмена микроэлементов
значительно увеличилась.
В данной главе даётся характеристика
патофизиологии нарушений, связанных
с наиболее важными микроэлементами.
Железодефицитные анемии — тема для
отдельного подробного обсуждения в
разделе «Патофизиология системы крови и
кроветворных органов» в т. III. Однако
железодефицит, как более широкое дизме-
таболическое состояние, рассматривается в
данной главе. Нарушения метаболизма йода
в пределах его главного потребителя —
щитовидной железы — описаны в главе
«Нарушение эндокринной регуляции», однако
ниже приводится краткая характеристика
роли йода в метаболизме.
В физиологических условиях
микроэлементы-металлы и неметаллы образуют
Патофизиология обмена микроэлементов 437
прочные комплексы с белками организма.
В составе этих комплексов они и
выполняют свою биологическую роль. Большинство
из них входит в состав активных центров
различных ферментов, в качестве активных
компонентов, ответственных за химический
катализ. Некоторые (цинк, йод)
обнаруживаются в составе прогормонов и даже активных
гормонов. Общеизвестна роль
микроэлементов в составе транспортных белков
(например, железа в гемоглобине). Микроэлементы
входят в состав редокс-систем, существенных
для продукции и инактивации кислородных
радикалов в организме (см. т. I, с. 193-196 и
данную книгу, раздел «Нарушения обмена
редокс-витаминов»). С учетом этих аспектов
по крайней мере 15 микроэлементов
считаются незаменимыми или, вероятно,
незаменимыми для человека. Последнее означает,
что их незаменимость показана только для
животных. К. Тэсмен-Джоунс (1987)
включает в эту группу 5 неметаллов — йод, селен,
фтор, кремний и, возможно, мышьяк, а также
10 металлов — железо, медь, цинк, кобальт,
марганец, никель, хром, ванадий, молибден
и, вероятно, олово.
Микроэлементы в патофизиологии
важны как источник заболеваний, вызванных
их дефицитом, дисбалансом или прямой
токсичностью. Последний аспект относится
не только к 15 вышеназванным, но и к
широкому кругу других микроэлементов, из
числа тех шестидесяти с небольшим, которые
вообще обнаружены в организме.
Дефицит микроэлементов, как и дефицит
витаминов, может быть экзогенным
(связанным с неадекватным приёмом их в пищу) и
эндогенным.
Экзогенный дефицит может быть
эндемическим региональным (как, например,
селеновый — в Восточной Финляндии и в
китайской провинции Ганьсу). Он может
развиваться у населения бедных стран при
недоедании (железодефицитные анемии
при квашиоркоре), а у пациентов из богатых
стран — при парентеральном
несбалансированном питании.
Эндогенная недостаточность
незаменимого микроэлемента наступает по разным
причинам. Среди них: расстройства
всасывания из-за желудочно-кишечных нарушений,
наследственных дефектов
белков-транспортёров, а также при наличии компонентов
пищи, препятствующих абсорбции
микроэлемента. Так, например, хронический
энтерит нарушает всасывание железа, при
болезни Имерслунда-Грэсбека отсутствует
возможность абсорбции кобальтсодержа-
щего витамина В12, при наследственном
дефиците металлотионеина не всасываются
ни цинк, ни медь, а фитаты и волокна
растительных продуктов, изобилующих в
питании населения некоторых районов Ирана и
Египта, не дают возможности использовать
цинк, попадающий в кишечник.
Развитию дефицитных состояний
способствуют потери микроэлементов с
мочой— в полиурическую фазу почечной
недостаточности, с пищеварительными
соками — при наличии фистул, с
отшелушивающейся кожей, выпадающими волосами,
и особенно при потере крови. Последнее
очевидно на примере железа.
Дисбаланс в обеспечении
микроэлементами состоит в том, что при значительном
превышении содержания некоторых
микроэлементов над оптимумом возникают
явления дефицита других микроэлементов.
Это может быть обусловлено конкуренцией
между элементами при всасывании и реаб-
сорбции, а также при включении в белки.
Так, избыток цинка или молибдена
способствует дефициту меди.
Токсические эффекты микроэлементов
настолько разнообразны и так зависят от
формы, в которой элемент попадает в
организм, и количества ядовитого вещества,
что должны и будут обсуждаться отдельно
в т. III данной серии, при рассмотрении
механизмов наиболее распространенных
отравлений.
В данном разделе мы лишь отметим, что
микроэлементы как яды часто хронически
накапливаются в организме, поступая в
него постепенно и в малых дозах (свинец),
но могут, при относительно больших дозах,
438 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
послужить и причиной острых
интоксикаций (мышьяк, таллий, ртуть). Механизмы
их токсичности, при всём разнообразии,
включают ингибирование ферментов путём
связывания с аминокислотами их
активных центров (например, свинец блокирует
8-аминолевулинат-синтетазу, с чем связано
развитие под его влиянием сидероахре-
стической анемии, а мышьяк — пируват-
киназу). Большое значение может иметь
повреждение микроэлементами структуры
нуклеиновых кислот, нарушение синтеза
белка (тяжелые металлы), фосфорилиро-
вания (фтор), мембранной проницаемости
(ртуть). Нельзя забывать о гаптенной
активности некоторых микроэлементов,
провоцирующих при попадании в легкие, в
эпидермис и кожу тяжелые гиперергические
реакции замедленного типа (никелевый и
йодный контактный дерматит, бериллиевый
аллергический альвеолит и дерматит). Ряд
токсических эффектов металлов
обусловлен нарушением продукции гормонов. Так,
свинец угнетает синтез тироидных
гормонов, тропных гормонов аденогипофиза,
надпочечниковых и гонадных стероидов
(Р.А.Хенкин, 1976).
Токсичность микроэлементов,
попадающих в тело человека в ультрамикромалых
дозах, наиболее значима для здоровья
пациентов, не способных экскретировать их
из организма.
Красноречивым примером служит
алюминий. Этот металл изобилует в земной
коре, являясь там одним из самых
распространённых. Но живые клетки избегают
алюминия. При хронической почечной
недостаточности минимальные количества
алюминия, попадающие в организм как при
гемодиализе и употреблении антацидов, так
и иными путями, накапливаются в нём.
Алюминий легко абсорбируется в ЖКТ и
аккумулируется в мозге, костях и эритроне.
Это ведет к инвалидизирующим
расстройствам, включая неврологические
нарушения (апатию, потерю памяти, деменцию,
дезориентировку в пространстве, мышечные
подёргивания и судороги), поражение
опорно-двигательного аппарата (остеомаляцию,
резистентную к лечению витамином D;
патологические переломы, миальгии и
миастению), а также анемию. Остеологические
аспекты отравления алюминием
рассмотрены также ниже в разделе «Патофизиология
паращитовидных желез...»
Время новых технологий породило
дополнительные возможности для контакта
организма с токсичными
микроэлементами — и это относится не только к сфере
профессиональной патологии. Достаточно
вспомнить о таких примерах, как
токсический энцефалит у производителей свинцовых
аккумуляторов, марганцевый паркинсонизм
у сварщиков и металлистов, бериллиоз у
изготовителей катодных трубок, отравления
у потребителей морепродуктов,
выловленных в водах, загрязненных промышленной
ртутью, йодный аутоиммунный тироидит у
пациентов, лечившихся от аритмии амио-
дароном, и т.д.
Наконец, особый вид нарушений обмена
микроэлементов известен в
радиоэкологической медицине. Он связан с адресной
инкорпорацией радиоактивных изотопов
биологически необходимых
микроэлементов. Это ведет к накоплению излучателей
прямо в клетках, осуществляющих те или
иные важнейшие функции. По сравнению
с метаболически безразличными или
быстро метаболизируемыми радиоактивными
веществами подобные задерживаемые в
организме источники мутагенного действия
могут быть особенно опасны. Так, например,
стронций и цезий задерживаются в костях.
Как следствие радиоактивные изотопы
цезия и стронция, попавшие в атмосферу при
ядерных испытаниях и авариях,
накапливаются в опорно-двигательном аппарате
человека, проникая в организм через пищевые
цепи естественных биогеоценозов. Доказано,
что этот процесс происходит особенно
быстро в биогеоценозах с короткими пищевыми
цепями, например в тундре, где биогенная
миграция атомов из почвы кратчайшим
путём, через ягель и оленя, приводит к
человеку. Так, кривые накопления
радиоактивного стронция у оленеводов Скандинавии и
Кольского Севера в 50-60-е годы XX века
Патофизиология обмена микроэлементов 439
точно отражали погодовую интенсивность
ядерных испытаний в Арктике.
По данным нанопатологии, частицы
бария, титана и других металлов из состава
рентгеноконтраста и даже считавшихся
инертными в биологическом отношении
протезов могут формировать в организме
наноэмболы и вызывать поражения печени
и других органов.
из самых распростаненных наследственных
болезней в европейских популяциях.
Существование среди нарушений обмена
железа как самой распространенной из де-
фицитарных болезней человека — железо-
дефицитной анемии, так и одной из самых
частых наследственных — гемохроматоза,
поразительный пример, подчёркивающий
огромную роль этого металла в функциях
клеток.
ПАТОФИЗИОЛОГИЯ
МЕТАБОЛИЗМА ЖЕЛЕЗА
(написано совместно с В.И. Утехиным)
Железо, содержание которого составляет на
грамм живой ткани от 55 (у мужчин) до 40 (у
женщин) микрограммов, не подпадает под
формальный критерий микроэлементов и
занимает промежуточное положение между
ними и той вышеназванной группой
элементов, содержание которых составляет до 10 %
веса тела. Однако, поскольку оно выполняет
свои функции в связанной с белками форме, а
не в ионизированном виде, его
метаболическая роль вполне соответствует
традиционному пониманию свойств микроэлементов.
Дефицит железа и его крайняя степень,
выражающаяся в железодефицитной анемии,
чрезвычайно распространены и в среднем
поражают не менее 12% населения и около
20% женщин. В беднейших странах этот
показатель превышает 50 % населения. В
группах высокого риска (беременные, кормящие,
новорожденные, недоношенные, подростки,
престарелые, лица с геморрагическими
заболеваниями, малоимущие слои населения)
частота дефицита железа повсеместно еще
выше например среди беременных — около
50 %. В различных странах, в том числе
характеризующихся высоким жизненным уровнем
населения, снижение запасов железа было
найдено при скрининг-обследованиях у 25 %
обследованных детей, 30 % подростков, но
лишь у 3 % мужчин (см. т. III, гл. 1).
Первичный гемохроматоз, связанный с
избытком железа в организме, считается одной
В данной книге вкратце рассматриваются
только метаболизм железа и железодефи-
цит, а также гемохроматоз — как патология
обмена веществ.
Роль железа в организме определяется
тем, что оно легко и обратимо окисляется и
восстанавливается.
Железо широко распространено в водах и
почвах земли и, быть может, именно поэтому
включено в многочисленные белки,
существенные для жизни растений и животных.
Перечислим важнейшие
железосодержащие белки:
>* гемопротеины (гемоглобин, миоглобин,
цитохромы, цитохромоксидаза, гомоген-
тизиноксидаза, пероксидаза, миелопе-
роксидаза, катал аза);
>- железофлавопротеины, в том числе —
цитохром-С-редуктаза, сукцинатдегид-
рогеназа, пролиноксидаза, НАДФ-де-
гидрогеназа, ацил-КоА-дегидрогеназа,
ксантиноксидаза и др;
>• белки, содержащие железо различных
молекулярных конфигураций, — транс-
феррин, ферритин, гемосидерин, мобил-
феррин, лактоферрин и др.
Роль железа в энергетическом
метаболизме красноречиво иллюстрирует только
один факт: около половины ферментов или
кофакторов цикла Кребса или содержат этот
металл, или нуждаются в его присутствии.
Последние работы свидетельствуют о том,
что железо необходимо и вне ферментов
и транспортных белков: для
формирования в мозге D2 — рецепторов (рецепторы
дофамина). Отсутствие или недостаток
дофаминергических рецепторов нарушают
440 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболическиенарушения)
Фракция железа
Гемоглобин
Миоглобин
Геминовые энзимы
Трансферрин
Ферритин и гемосидерин
ИТОГО:
Таблица 29. Железо в организме человека
Абсолютная величина, мг
Мужчины, на 70 кг
2670
350
8
6
800
3834
Женщины, на 50 кг
1500
220
7
5
320
2052
Относительная величина, %
Мужчины
69,6
9,1
0,2
0,2
20,9
100
Женщины
73,0
10,6
0,3
0,3
15,8
100
нормальное функционирование и развитие
дофаминергических нейронов.
Распределение железа в мозге отражает
локализацию окончаний нейронов,
производящих у-аминомасляную кислоту (ГАМК).
Предполагается, что низкий уровень железа
мешает деградации ГАМК или нарушает
функционирование нейронов,
производящих дофамин. Уже одно это показывает
глобальность последствий железодефицита
для организма. Железодефицит не сводится к
своему гематологическому проявлению, а
затрагивает функции всех клеток, особенно — в
высоко аэробных тканях. Не случайно
многие его проявления выражаются в аномалиях
поведения и психических симптомах.
Распределение железа в организме
характеризуется следующими цифрами (табл. 29).
Из данной таблицы видно, что запасы
железа у мужчин значительно превышают
таковые у женщин. Превышение достигает
100, а по другим данным, даже 200 %.
Отсюда ясно, почему железодефицит поражает
намного чаще женщин, в то время как ге-
мохроматоз, связанный с избытком железа,
наблюдается в основном у мужчин.
В связи с половыми различиями в обмене
железа следует отметить, что менструальные
кровотечения у женщин легко могут стать
причиной железодефицита. Показано, что
средняя кровопотеря в ходе менструации
составляет около 40 мл/цикл. А10 % женщин
теряют 80 мл/цикл, что эквивалентно потере
30 мг железа. Поскольку абсорбция 1 мг
железа/день требует 10-20 мг железа в пище,
понятно, почему при среднем содержании
железа в диете около 10 мг/день баланс
железа у обильно менструирующих шаток.
Железо присутствует в пищевых
продуктах в различных формах: в окисной (Fe3+) и
закисной (Fe2+). Наиболее хорошо
усваивается и быстрейшим, прямым способом
всасывается окисное железо в составе гемина
из животных продуктов. Таким образом,
диеты, богатые мясом, сводят вероятность
экзогенного железодефицита к минимуму.
Печень животных содержит 12-8 мг/100 г
железа, почки и яйца — около 6, мясо — 2
мг/100 г. Из растительных продуктов
лидируют овсянка (4,2), грибы (3,9), персики
и гранаты (от 3 до 4 мг/100 г). Но в
растительных, особенно зерновых продуктах, до
60 % железа находится в трудно усвояемой
форме, связанной с фитиновой кислотой.
Окисное негеминовое ферри-железо, в том
числе, из состава минеральных компонентов
диеты, должно освободиться из
органических комплексов и превратиться в закисное
ферро-железо, чтобы всосаться в клетки
слизистой кишечника. Потребность
взрослого человека в железе — 5 мг на 1 ОООккал
или 15мг/сутки. Но только 5-10 %
пищевого железа всасывается, то есть 1 -1,5 мг. При
дефиците железа всасывание растёт и,
например, у беременных достигает 4 мг/день.
Соляная кислота желудка и витамин С
способствуют переходу пищевого железа во
всасываемую, некомплексную ферро-форму.
Ускорение транспорта химуса,
гиповитаминоз С, избыток цинка и меди, ахилия и агас-
трическое состояние, энтериты повышают
риск железодефицита.
Патофизиология обмена микроэлементов 441
Всасывание железа в основном идёт в
верхней части тонкого кишечника. Желудок,
подвздошная и толстые кишки участвуют
в этом процессе в малой степени. Железо
гемина свободно проходит через клетки
интестинальной слизистой, внутри гем
деградирует под действием специальных
ферментов, а железо присоединяется к пулу
внутриклеточного белка ферритина. Еще в
1937 г. Р. Мак-Кене и Э. Видоусон
показали, что способность человека выводить
избыток железа из организма ограничена, и
предсказали, что всасывание железа должно
строго регулироваться и соответствовать
динамике потребностей в нем. П.Ф. Хан и
соавторы в 1943 г. зарегистрировали
усиление абсорбции радиоактивного железа
у собак с постгеморрагической анемией
после недельного латентного периода с
момента кровопотери. С тех пор классические
представления связывали всасывание
свободного закисного железа с рециркуляцией
кишечного трансферрина и с трансферри-
новым рецептором. Современные данные
М.Э.Конрада, Дж.Н.Умбрайта и др.
(1993-1998) показали, что это гораздо более
сложный процесс. Всасывание негеминового
ферри-железа требует связывания с
кишечным муцином, а затем - фиксации комплекса
на §ъ-интегрине люменальной мембраны
энтероцитов. Затем комплекс связывается с
мобилферрином — цитоплазматическим
белком энтероцитов, гомологом кальретикулина
(56кД). Другая «эстафетная команда», в
которую включены ферроредуктаза щеточной
каймы энтероцитов, восстанавливающая
ферри-железо в ферро-форму, а также ли-
ганд ферро-железа и других двухвалентных
катионов (кадмия, кобальта, свинца, меди,
цинка) — белок DMT-I, — обеспечивает
транспорт закисного железа в энтероциты.
Белок SFT - стимулятор транспорта
железа — обеспечивает индукцию DMT-I и
секрецию мобилферрина при недостатке железа в
организме. Обе команды передают железо на
апоферритин. В кровь из энтероцитов дорогу
железу открывает комплексообразование
с базолатеральным транспортером железа,
гефестином, который гомологичен медь-пе-
реносящему белку церулоплазмину, в этом
участвует также комплекс трансферринового
рецептора и ферропортина (см. ниже). Есть
основания полагать, что эти транспортные
системы действуют и в других ядерных
клетках организма, потребляющих железо.
Центральная регуляция обмена железа
связана с печенью и ее эндокринной
функцией. Главным регулятором метаболизма
железа в организме служит гормон гепа-
тоцитов гепцидин. Гепцидин
синтезируется в печени, его продукция усиливается
цитокинами И Л-1,6 и 8 при воспалении,
острофазном ответе и при перегрузке
железом. Именно гепцидин - главный сигнал,
вызывающий гипоферремию и гипоцинк-
емию при ответе острой фазы (см. т. I,
гл. «Преиммунный ответ. Лихорадка...»).
Гиперпродукция гепцидина участвует в
патогенезе гипохромной анемии длительно
и часто болеющих детей.
Гемоювелин, мембранный белок,
кодируемый в первой хромосоме и являющийся
ко-рецептором фактора морфогенеза костей,
стимулирует печеночную продукцию
гепцидина, а его растворимые фрагменты
подавляют гепцидинообразование. Мишень
гепцидина — белок ферропортин, экскретирующий
железо из клеток, которые его накапливают.
Ферропортин способствует переходу железа
из энтероцитов в кровь. Гепцидин подавляет
его экспрессию. Также гепцидин снижает
экспрессию вышеописанного транспортера
DMT-I, что уменьшает всасывание железа
(Т. Ганц и соавт., 2001,2003).
Доказано, что дефицит гепцидина (в
результате мутаций его гена, а также генов
ферропортина и гемоювелина)
способствует перегрузке железом и гемохроматозу,
а избыток данного печеночного гормона
(например, при его автономной продукции
некоторыми опухолями печени) ведет к
железодефициту и ответственен за
появление микроцитарной паранеопластической
анемии у онкологических больных (см.
т. III, гл. 3).
Таким образом, современное понимание
метаболизма железа основано на представ-
442 АШ. Зайчик, Л.П.Чурилов Патохимия
лении о его специфической гормональной
регуляции с учетом принципа обратной
связи. Это ведет к трактовке ряда форм
железодефицита и избытка железа как
эндокринных нарушений (см. ниже). Из
состава кишечного ферритина железо может
медленно переходить в трансферрин, этот
процесс активизируется при компенсации
железодефицита, когда трансферрина в
энтероцитах становится больше, а
ферритина — меньше. При избытке железа
развиваются обратные тенденции, и ферритиновая
фракция теряется со слущивающимися
энтероцитами. Таким образом, ось гепци-
дин — ферропортин и система абсорбции
железа могут модулировать интенсивность
этого процесса в широких пределах,
усиливая и при необходимости ограничивая его
поступление, при этом свободное
токсическое железо в энтероците не появляется.
Количество хранимого в организме
железа хорошо коррелирует с уровнями
железа сыворотки ( у женщин 80-95 мкг/дл
или 10,7-21,5 мкмоль/л, у мужчин 120—
125 мкг/дл или 14,3 — 26 мкмоль/л).
В физиологических условиях содержание
железа в сыворотке колеблется в течение
дня: оно снижается к вечеру, достигая
минимума около 21 час, и повышается,
достигая своего максимума, между 7 и 10 час
утра. Содержание сывороточного железа
снижено при острых или хронических
воспалительных процессах, при опухолях, при
острых инфарктах миокарда. Лихорадка и
ответ острой фазы всегда сопровождаются
гипоферремией, которая необходима для
их защитного эффекта. Понижая доставку
железа в ткани, организм достигает
торможения размножения железозависимых
бактерий и ограничения интенсивности
альтеративно-аутоокислительных
процессов (см. т. I, с. 28,370).
При выраженном дефиците железа,
доходящем до стадии анемии, содержание железа
в сыворотке понижается, однако
нормальные уровни железа сыворотки не исключают
латентного железодефицита.
Несмотря на давно известную врачам
эмпирическую эффективность сульфата
(эндокринно-метаболические нарушения)
железа при хлорозе (Г. Фёдиш, 1839),
до конца XIX века ученые сомневались в
возможности встраивать неорганическое
железо пищи в биополимеры организма, а
такие авторитеты физиологии, как Г. Бунге
(1885) и Э. Гамбургер (1878-79), отрицали
это, ссылаясь на то, что экспериментальные
животные выводят с фекалиями столько
же железа, сколько получают с пищей, хотя
эти данные были всего лишь отражением
нейтрального баланса железа, не
исключающими его обмена. Однако Г. Стокмен (1893,
1895) доказал, что неорганическое железо
усваивавается в ЖКТ и эффективно лечит
гипохромную анемию при пероральном
применении и инъекциях, он же
обнаружил, что из-за неточности аналитических
методов того времени медики повсеместно
заблуждались, переоценивая содержание
этого металла в растительной пище.
Усилиями этого автора было доказано, что при
гемолизе железо реутилизируется.
Показатели обеспеченности организма
железом связаны с его
белками-транспортёрами. В крови железо переносит
трансферрин, гликопротеид (80 кД) с
электрофоретическими свойствами р-гло-
булина, связывающий 2 атома металла в
ферри-форме каждой своей молекулой при
участии НСОд. Принципиально все клетки,
но в наибольшей степени эритробласты и
гепатоциты, способны к его экспрессии и
продукции. Сродство у этой удивительной
молекулы, продукта дупликации древнего
гена в хромосоме 3, к железу столь огромно,
что при его нормальной выработке в литре
крови присутствует теоретически лишь 1
атом свободного железа. Трансферрин —
отрицательный глобулин острой фазы, его
синтез при лихорадке понижают интерлей-
кины(ИЛ-1иИЛ-6).
Трансферрин переносит железо во многие
ткани, в том числе в основном в костный
мозг, и захватывается его клетками,
причем как эритроидными, так и иными. Этот
процесс опосредуется трансферриновым
рецептором первого типа, наибольшей
экспрессией которого отличаются эритро-
идные, другие миелоидные, плацентарные
Патофизиология обмена микроэлементов 443
и все быстропролиферирующие клетки
(включая неопластические). Этот
рецептор — димерный трансмембранный гли-
копротеин (190 кД), он может отрываться от
поверхности клетки и циркулировать,
связывая трансферрин в крови. Эритропоэтин
увеличивает его экспрессию. Трансферрин-
рецепторный комплекс поступает в
клетки-потребители железа эндоцитотически,
затем происходит закисление содержимого
эндосом, распад комплекса и рециркуляция
рецептора и трансферрина. Помимо данного
рецептора в печени имеется
низкоаффинный трансферриновый рецептор 2-го типа,
участвующий в захвате железа гепатоцита-
ми и важный для обратной связи в системе
саморегуляции продукции гормона гепциди-
на. В эритроидных клетках железо делится
между митохондриями, где включается в
гем, и белком ферритином, в миелоидных
существенная его часть попадает в защитный
белок лактоферрин, а в макрофагах
включается в основном в ферритин. Апоферритин
содержит полость, где после образования из
него ферритина хранится кристалл гидрата
закиси железа, содержащий более 4 000
атомов этого металла. Имеется целое семейство
тканеспецифических ферритинов. Феррити-
новая форма хранения железа обеспечивает
его депонирование, реутилизацию, а также
в небольшой мере — циркуляцию, так как
некоторое количество бедного железом
апоферритина имеется в плазме крови. Из
ферритиновой формы железо может
эффективно мобилизоваться. Ферритиновые
гранулы имеются в наибольшем числе в
макрофагах костного мозга, селезёнки и
печени и в сидеробластах— миелоидных
клетках костного мозга. При нарастающем
дефиците железа их количество понижается,
вплоть до исчезновения. После утилизации
гема и гемоглобина, освобожденного при
гемолизе, при участии гемопексина, гап-
тоглобина, механизмов аутофагоцитоза и
эндоцитоза, гемоксидазы фагоцитов железо
снова попадает в макрофаги, в «ферритино-
вый футляр». Из ферритина железо
мобилизуется быстро, и процесс его либерации
регулируется. Синтез ферритина в макро-ч
фагах стимулируется железом. С течением
времени и особенно при избытке железа и
при усиленном протекании в тканях сво-
боднорадикальных процессов (где железо
участвует в реакции Фентона, см. т. I, с. 190,
195), ферритин превращается в гемосидерин.
Гемосидерин — малорастворимый комплекс
кристаллов железа, не покрытый белковым
слоем, содержащий относительно немного
денатурированного ферритинового белка,
а также липиды.
Он обнаруживается в макрофагальных
и других клетках. Железо мобилизуется
из него, но очень медленно, и процесс
этот, по-видимому, не подлежит строгой
регуляции. Уровень ферритина сыворотки
рассматривается как характеристика запаса
железа в организме. Разработаны иммуно-
конкурентные методы его определения. По
разным авторам в норме цифры ферритина
сыворотки колеблются у мужчин в пределах
94-149 нг/мл, у женщин 34-76 нг/мл. При
выраженном железодефиците эти цифры
падают ниже Юнг/мл.
Известно, что низкие уровни ферритина
сыворотки отражают сниженные уровни
запаса ретикулоэндотелиального железа.
Высокие уровни ферритина сыворотки
наблюдаются у больных с патологией печени,
особенно при остром некрозе гепатоцитов,
у больных с малигнизирующимися
опухолями и с лейкозами. Больные с острыми и
хроническими воспалительными
заболеваниями тоже могут иметь повышенные
уровни ферритина, равно как и больные с
идиопатическими или вторичными
формами повышения уровня железа.
Избыток гемосидерина наблюдается при
усиленном разрушении эритроцитов и при
перегрузке организма железом.
Наиболее важные объективные критерии
состояния метаболизма железа связаны с
трансферрином.
Железосвязывающая способность
сыворотки (ЖСС) — это практически важный
показатель метаболизма железа, мера
количества трансферрина в циркулирующей
крови. Данная величина отражает
количество железа, которое может связаться с
444 А.Ш. Зайчик, Л.П.Чурилов Патохимия
трансферрином. В норме в 100 мл сыворотки
имеется достаточное количество трансфер-
рина, чтобы связать 250-450 микрограммов
железа. Поскольку в сыворотке в норме
содержится в среднем около 100
микрограммов железа/100 мл, трансферрин,
следовательно, насыщен железом в среднем на 1/3
при сбалансированном обмене железа.
«Ненасыщенную» или «латентную» же-
лезосвязыеаюищю способность [«unsaturated
ion-binding capacity» — ШВС] легко
измерить или при помощи радиоактивного
железа или спектрофотометрически. Сумма
ШВС и железосвязывающей способности
сыворотки составляет общую железосвя-
зывающую способность [«total iron-binding
capacity» — TIBC]. В норме Т1ВС
колеблется в пределах 30,6-84,6 мкмоль/л. Вычитая
количество железа сыворотки из общей
железосвязывающей способности, легко
узнать ненасыщенную (латентную) желе-
зосвязывающую способность:
TIBC- [железо сыворотки]=ШВС,
ШВС+ЖСС =Т1ВС.
При выраженном дефиците железа ШВС
и TIBC часто повышены. В норме
трансферрин насышен на 20-50 процентов. При
ферродефиците нередко обнаруживается
насыщение трансферрина на 15 и менее
процентов.
Суммируя, следует отметить, что при
выраженном, доходящем до анемии желе-
зодефиците:
>• увеличивается TIBC;
>* значительно увеличивается ШВС;
>- резко снижен процент насыщения
трансферрина.
Специального пути выделения железа из
организма в норме не существует. В ионной
форме оно не присутствует. Трансферрина
почки в мочу не пропускают. До 1 мг этого
металла теряется в день со слущивающими-
ся и отшелушивающимися клетками кожи и
слизистых. У мужчин этим потери железа в
норме и ограничиваются. Менструирующие
женщины, как уже говорилось выше, теря-
чдокринно-метаболические нарушения)
ют железо в гораздо большем количестве
(15-40 мг в месяц) с кровью.
Железодефицит — состояние, при
котором содержание железа в организме меньше,
чем в норме. Это процесс, развивающийся
через несколько последовательных фаз.
Этиология железодефицита всегда
связана с хроническим превышением расхода
(потерь) железа над его доставкой, то есть
отрицательным балансом железа. Это
может быть вызвано рядом причин, среди
которых наиболее важны:
- Неадекватное поступление железа с
пищей. У детей первого года жизни, чаще
всего железодефицит возникает именно
как следствие диеты, не содержащей
адекватного количества железа.
Новорожденному требуется около 160 мг
железа в день, недоношенному, лишенному
депо железа — 240 мг. У детей старших
возрастных групп к фактору диеты
могут присоединиться и другие.
Длительно и часто болеющие дети, в силу
подавления продукции трансферрина
при ответе острой фазы и расходования
железа для нужд дефензиновой
системы лейкоцитов, особенно подвержены
железодефициту. У взрослых,
благодаря меньшей суточной потребности
в железе (всосаться должно не более
1-2 мг железа в день), чисто пищевой
железодефицит бывает намного реже.
- Мальабсорбция железа — это редкое
явление, если только оно не связано с
агастрическим состоянием или
комплексными мальабсорбционными
синдромами. Известно, что у половины больных
после субтотальной резекции желудка
через несколько лет возникает железо-
дефицитная анемия. Многие из таких
больных имеют нарушение реабсорбции
пищевого железа, частично вследствие
ускоренного гастро-еюнального
транзита, частично вследствие быстрой
эвакуации пищи из двенадцатиперстной
кишки.
Патофизиология обмена микроэлементов 445
Хроническая кровопотеря. У взрослых
мужчин и женщин после менопаузы
основной причиной железодефицита
являются кровотечения из ЖКТ,
которые могут спровоцировать язвенная
болезнь, диафрагмальная грыжа, опухоли,
гастриты (алкогольные или вследствие
лечения салицилатами, стероидами, ин-
дометацином). У детей кровотечения из
ЖКТ тоже могут играть роль в развитии
дефицита железа, особенно при
анафилактических реакциях на свежее молоко,
гельминтозах и протозоонозах
кишечника. Наконец, нарушения в системе
гемостаза также могут вести к кровотечениям
из ЖКТ. Повторные эпизоды
кровохарканья от различных причин способны
привести к развитию дефицита железа.
Так, выраженным железодефицитом и
анемией характеризуются идиопатичес-
кий гемосидероз и синдром Гудпасчера
(прогрессирующий гломерулонефрит с
внутрилегочными кровоизлияниями, на
почве аутоантител к коллагену базаль-
ных мембран). Полименорея, менорра-
гия и метроррагии — типичные причины
дефицита железа у женщин. У мужчин
причиной нехватки железа тоже иногда
бывают кровотечения из мочеполового
тракта.
Расход железа на нужды эмбриона или
плода, входе беременности, а также
расход железа при лактации. У
вынашивающих потомство женщин средняя
потеря железа определяется тем его
количеством, которое идет на эритро-
поэз плода. В ходе родов имеет место
кровопотеря, отторжение последа и
утрата порядка 150-200 мг железа. После
родов, при лактации, будет использовано
еще около 900 мг. Все это вместе
эквивалентно кровопотере в два литра.
Поскольку большинство женщин начинают
беременность без особых запасов железа,
неудивительно, что дополнительные
потребности часто приводят к его
дефициту и даже анемии. Фактически 80-100
процентов женщин имеют истощение
запасов железа при беременности и
лактации, а потому беременная должна
получать железо дополнительно.
- Потеря железа вследствие внутрисо-
судистого гемолиза с гемоглобинури-
ей. В связи с этим дефицитом железа
осложняются некоторые хронические
гемолитические анемии.
- Нарушение включения железа в синтез
гема («ложные» железодефицитные
состояния — например, при врожденной
атрансферринемии — болезни Хейлмейе-
ра} порфириях, обсуждавшемся выше в
разделе «Патофизиология витаминного
обмена» гиповитаминозе В6,
сатурнизме). Эти формы патологии описываются
в т. III, при рассмотрении собственно
железодефицитных анемий.
- Комбинации указанных факторов. Соче-
танные причины, по-видимому, как раз
и лежат в основе большинства случаев
нехватки железа у взрослых.
1. Термин «истощение запасов железа»
обозначает раннюю стадию
железодефицита, когда:
>* запас железа уменьшен или
отсутствует, но:
>* концентрация железа в сыворотке в
норме;
>* гемоглобин в норме;
>- гематокрит в норме.
2. Термин «железодефицит без анемии»
относится к более серьезной ступени,
характеризующейся:
>• снижением или отсутствием запаса
железа;
>• низким содержанием железа в
сыворотке;
>- низким насыщением трансферрина,
Патогенез железодефицита отражает
прогрессирующее использование и истощение
депо железа, а также нарушения функций
железозависимых ферментов, рецепторов
и других белков.
Последовательных стадий в этом
процессе можно выделить три.
2.
446 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
но:
>■ без наличия истинной анемии.
3. Термин «железодефицитная анемия»
подразумевает еще более выраженную
степень железодефицита, когда
регистрируются:
>* низкая концентрация железа в
сыворотке;
>- низкая концентрация трансферри-
на;
>■ низкая концентрация гемоглобина в
единице объема крови;
>* низкое значение гематокрита;
>- гипохромия и микроцитоз
эритроцитов, кольцевидные эритроциты,
нередко — тромбоцитоз.
Железодефицитная анемия — конечная
стадия отрицательного баланса железа.
Дефициту железа свойственны так
называемые сидеропенические
негематологические симптомы, многие из которых
проявляются раньше наступления анемии.
Это — выраженные изменения кожи,
ногтей, волос, слизистых оболочек рта
(сухость и трещины кожи на руках и ногах,
уплощенные и вогнутые ломкие ногти, что
известно как «койлонихия», ангулярный
стоматит). Поражается слизистая верхних
дыхательных путей, пищевода, желудка,
имеется мышечная слабость, которая не
соответствует тяжести анемии,
присутствуют извращения вкуса, меняется поведение
больных.
Механизм этих проявлений связан с
дефицитом железозависимых рецепторов
и ферментов, в частности оксидаз,
участвующих в образовании поперечных сшивок в
коллагеновых белках.
Характерный признак железодефицита —
мышечная слабость — наблюдается у
большинства больных. Объясняют ее не только
анемией, но и дефицитом
железосодержащего фермента а-глицерофосфатоксидазы,
нарушениями обмена миоглобина. С
мышечной слабостью связывают и нередко
встречающееся у страдающих железодефи-
цитом девочек ночное недержание мочи.
Часто больные не способны удержать мочу
при кашле, смехе, не могут остановить
начавшееся мочеиспускание.
Железо — компонент цитохромов,
поэтому железодефицит обусловливает
поражение наиболее чувствительной к
кислородному голоданию эпителиальной ткани. Этим
картина нехватки железа напоминает
проявления дефицита витаминов группы В,
участвующих в биологическом окислении.
Железодефицит так же, как соответствующие
гиповитаминозы, проявляется стоматитом,
глосситом, эзофагитом, неинфекционным
мембранознымларингофаринготрахеитом,.
Последнее обусловливает при дефиците
железа весьма дезориентирующее врача
проявление — гиперемию небных дужек и задней
стенки глотки, охриплость голоса, дисфа-
гию— но без инфекции. Эти проявления
известны как синдром Пламмера-Уинсона
и снимаются при коррекции обмена железа.
Эзофагит при железодефиците повышает
риск развития опухолей пищевода. В
развитии таких симптомов имеет значение не
только тканевая гипоксия клеток слизистой,
вызывающая их ускоренную гибель. Очень
важны нарушения образования коллагена
при дефиците железа, которые
способствуют отслойке пораженного эпителия от ба-
зальных мембран и образованию паутинопо-
добных наслоений и мембран в пораженных
глотке, гортани и пищеводе.
При железодефиците страдают и более
глубокие отделы ЖКТ.
Как следствие снижения активности
многих ферропротеинов, уменьшается
способность париетальных клеток к секреции
соляной кислоты. Нарушение желудочной
секреции, иногда прогрессирует вплоть до
ахилии. У половины больных
обнаруживается атрофический гастрит. По
современным данным, внутриклеточное железо
способствует пролиферации клеток, что
и объясняет его развитие. Это приводит к
порочному кругу в патогенезе болезни, так
как падение кислотности желудочного сока
, затрудняет всасывание железа.
Таким образом, при ферродефиците
атрофии подвергаются слизистые с активно
пролиферирующими клетками (вовлекают-
Патофизиология обмена микроэлементов 447
ся язык, пищевод, желудок, тонкая кишка,
происходит кератинизация буккального
эпителия). У детей обнаруживаются
признаки нарушения кишечного всасывания
жиров, ксилозы и ряда других субстратов.
Страдают при нехватке железа и функции
нервной системы. Появляются парасте-
зии. Нарушается работа гипоталамуса, где
имеется значительное представительство
рецепторов железа. Извращение вкуса (pica
chlorotica) обнаруживается как у взрослых,
так и у подростков. Больные часто едят мел,
зубной порошок, уголь, глину, песок.
Особенно обращает на себя внимание
потребление льда (погофагия), сырой крупы, теста,
сырого мясного фарша. Бывает пристрастие
к запаху керосина, мазута, бензина, ацетона,
гуталина, выхлопных газов машин, резины
и даже... мочи.
Дети с дефицитом железа испытывают
трудности в учёбе, не способны к
длительной концентрации внимания, порой гипер-
активны.
Причина этих извращений вкуса и
аномалий поведения полностью не выяснена,
однако они проходят после назначения
препаратов железа и рецидивируют при
обострении железодефицита.
Изменения в количестве и активности
дофаминовых рецепторов и продукции ГАМ К
при дефиците железа, о которых шла речь
выше, вероятнее всего, ответственны за эти
психические проявления данного
нарушения обмена веществ.
Тот факт, что дефицит железа
вызывает столь обширные изменения функций
различных клеток, не означает, что
«железо— это железное здоровье», как иногда
ошибочно думают пациенты. Избыток
железа не менее патогенен, чем недостаток,
так как железо — мощный модулятор ауто-
окислительных процессов и в связи с тем,
что у организма нет эффективных путей
выведения его избытка.
Острое отравление железом особенно
опасно для детей, имеющих малые антиок-
сидантные ресурсы. У них приём 600-700 мг
железа в таблетках через рот может привести
к смерти. Механизмы отравления включают
некротический гастроэнтерит, тканевую
гипоксию, метаболический ацидоз, шок и кому
(К.А. Райе, Д.В. Санти; 1999).
Хроническая интоксикация избытком
железа приводит к развитию гемохроматоза,
впервые описанного клинически в 1865 г.
А.Труссо как «бронзовый диабет» и
впервые объясненного патогенетически
накоплением железа в организме в 1889 г. К. фон
Реклингхаузеном. Возможно при
перегрузках железом и развитие гемосидероза.
Гемохроматоз отличается патологически
повышенной или неподавляемой
абсорбцией железа из ЖКТ. Больше всего ускорено
выведение железа из энтероцитов в кровь.
Регуляторные белки IRP-I и IRP-2 в
норме могут взаимодействовать с железо-
чувствительным участком и-РНК транс-
ферринового рецептора и предохранять
ее от деградации. При избытке железа они
фиксируют его и прекращают
препятствовать деградации данной РНК, что ведет
к уменьшению экспрессии рецептора и
снижению поступления металла в клетки.
Эти же регуляторные белки способствуют
деградации и-РНК ферритина. Поэтому у
здоровых людей при избытке железа
экспрессия ферритина, наоборот, возрастает,
реципрокно по отношению к трансферри-
новому рецептору.
При гемохроматозе нарушается
способность к ингибированию всасывания и
освобождения в кровь железа его избытком. По
современным данным, гемохроматоз —
наследственная эндокринопатия. Около 60
поколений назад мутации, сберегающие в
организме железо, могли иметь адаптивную
ценность для населения северо-запада
Европы, жившего впроголодь. Это обусловило
их закрепление в популяциях, особенно — у
кельтских этносов. В современных условиях
богатой железом диеты адаптационное
значение этих генов утратилось, и они
обусловили высокую частоту гемохроматоза.
У человека имеется 4 генокопии
наследственного гемохроматоза. При наиболее
распространенном 1-м подтипе имеется
аутосомно-рецессивная мутация C282Y гена
448 А.Ш. Зайчик, Л.П.Чурилов Патохимия
HFE в 6-й хромосоме, болезнь проявляется
после 40 лет. При редком 2а-подтипе
имеется более ранняя манифестация болезни,
что соответствует ювенильному гемохро-
матозу. Мутация HFE2 в 1-й хромосоме
вовлекает белок гемоювелин, который
перестает стимулировать продукцию гепцидина.
Снижение гепцидинообразования ведет к
гиперэкспрессии ферропортина и DMT-I
и перегрузке железом. При редчайшем
подтипе 26 имеется дефект гена самого
гормона гепцидина НАМР в хромосоме 19.
Так как гепцидин — еще и фунгицидный
белок, у больных этим видом гемохроматоза
несколько снижается и противогрибковый
иммунитет. При 3-м подтипе
гемохроматоза установлена мутация печеночного
рецептора трансферрина. Как результат
обрывается обратная связь, и высокий
уровень железа перестает стимулировать
продукцию гепцидина. И, наконец, подтип
4 связан с единственной аутосомно-доми-
нантной генокопией болезни (ген SLC11АЗ/
ferroportin 1 в хромосоме 2). При этом имеет
место дефицит ферропортина, вызывающий
периферическую гепцидин-резистентность
при сохранном центральном звене
гормональной регуляции метаболизма железа.
Избыток железа связывает трансферрин
и ферритин, заполняя его валентности.
Деградация ферритина ведет к появлению
гемосидерина с его закисным железом.
При гемохроматозе железо, которое
особенно цитотоксично в его закисной форме,
откладывается во внутренних органах. Это
ведет к гибели паренхиматозных клеток и
их замещению на соединительную ткань.
Результатом служит органная
недостаточность.
Первичный гемохроматоз —
наследственно-обусловленное заболевание, с аутосом-
но-рецессивными дефектными генами и
пороговым эффектом, проявления которого
зависят от степени нагрузки экзогенным
железом. Оно наиболее часто поражает
европеоидов, являясь для них одним из
характернейших наследственных заболеваний
вообще. На первом месте по частоте болезни
стоит Франция, особенно — Бретань, где но-
тнно-метаболические нарушения)
сителями гена, предрасполагающего к
первичному гемохроматозу, являются до 13%
населения. Частота гомозиготного фенотипа
по дефектному гену в мире от 1/30 до 1/600,
гетерозигот от 5 до 13 %, а клинически
выраженных случаев — от 0,2 до 0,02 % населения.
Возможно, такая распространенность алле-
ля гемохроматоза связана с селективным
преимуществом — резистентностью к же-
лезодефициту, которая им обеспечивается.
Первичный гемохроматоз очень сильно
сцеплен с носительством алеля ГКГС А3 и
в меньшей степени с аллелями В7 и В14, так
как ген заболевания лежит в коротком плече
6-й хромосомы. Первичный гемохроматоз
развивается весьма медленно. За многие
годы количество железа в организме
больного может увеличиться с 2 до 40 граммов.
Проявлению болезни способствует нагрузка
железом, в том числе — при приеме
алкогольных напитков. Коньяки, портвейны,
красные вина, сидр содержат относительно
много железа. У народа банту в Южной
Африке описана пищевая перегрузка железом,
связанная с обычаем пить из железных
сосудов перебродившее в них домашнее пиво
и закусывать кровью животных. Гемосидероз
банту имеет генетическую основу и
поражает тех представителей африканских
этносов, у которых имеются «чувствительные»
аллельные варианты гена ферропортина,
заинтересованного в патогенезе редкого 4-го
подтипа гемохроматоза (см. выше).
Так как у мужчин выше потребление
железа и меньше возможностей для его экскреции,
более 90 % больных с перегрузкой железом
принадлежат к сильному полу, несмотря на
то, что ген гемохроматоза с полом не сцеплен.
Поражение гипофиза при гемохроматозе
проявляется пангипопитуитаризмом,
поджелудочной железы — вторичным сахарным
диабетом, миокарда— сердечной недостаточностью,
печени — циррозом, тестикул — импотенцией.
Имеются спленомегалия, сероватая
гиперпигментация кожи, связанная как с отложением
гемосидерина, так и с наступающей у
некоторых пациентов раньше гипопитуитаризма
первичной недостаточностью пораженной
гемохроматозом коры надпочечников. Увели-
Патофизиология обмена микроэлементов 449
чивается риск рака печени. Могут быть артрит
и полинейропатия.
Вторичный гемохроматоз развивается
при первичных анемиях с неэффективным
эритропоэзом и при хроническом
усиленном гемолизе, например при талассемиях.
При вторичном гемохроматозе
генетическая предрасположенность тоже имеется, но
выражена слабее. Он развивается
относительно быстрее первичного («ювенильно»)
и дает клиническую картину уже во 2-м
десятилетии жизни.
Поражаются сильнее всего печень и
сердце. Имеются задержка физического
развития, фиброз печени и сердечная
недостаточность.
Гемохроматоз — одно из немногих
заболеваний в современной медицине, где,
несмотря на все успехи фармакотерапии,
наиболее эффективным патогенетическим
средством лечения остаются, как и в средние
века, кровопускания (А.Г. Мотульски, 1987).
В отличие от гемохроматоза, гемосиде-
роз— относительно доброкачественная
форма перегрузки железом, при которой оно
накапливается в основном в макрофагаль-
ных клетках. Паренхима внутренних
органов, в частности печени, поражается редко.
Зато ретикулоэндотелиальные элементы
насыщены железом. Гемосидероз не
сопряжен с ускоренным всасыванием железа и
не даёт органной недостаточности. К нему
приводят многократные гемотрансфузии и
кровоизлияния.
ПАТОФИЗИОЛОГИЯ ОБМЕНА
ДРУГИХ МИКРОЭЛЕМЕНТОВ-
МЕТАЛЛОВ
До появления эмиссионной спектроскопии
(1929) сколько-нибудь прецизионные
исследования обмена микроэлементов у
человека и их содержания в рационах были
крайне затруднительны. Методологический
прогресс позволил оценить роль многих
металлов в норме и при патологии.
Цинк — незаменимый элемент в питании
человека. Потребность взрослого человека
в этом металле составляет 15-25 мг/сутки,
из которых, возможно, всасывается всего
около 5 мг. Цинком богаты бобовые, кунжут,
арахис, отруби (до 10 мг/100г), но во всех
этих продуктах он, к сожалению, связан с
фитиновой кислотой и малоусвояем.
Высоким содержанием цинка известны
моллюски, грибы, яичный желток, печень, мясо
(10-30 мг/100 г). Всасывание цинка идет в
тонком кишечнике, вероятно, с участием
транспортного белка, входящего в
поджелудочный сок, а также упоминавшегося в
разделе о метаболизме железа белка DMT-I
и тормозится фитатом, медью, фосфатом,
кальцием, волокнами клетчатки. В то же
время некоторые аминокислоты и пептиды
ускоряют его абсорбцию. Таким образом,
однообразное растительное питание, с
преобладанием неферментированных злаков
и дефицита белка ведет к гипоцинкозу.
Известны его эндемические очаги на Среднем
Востоке, где у сельского населения Ирана
и Египта выявлена цинково-белковая
недостаточность. Гипоцинкозу способствуют
энтериты, прием алкоголя, а также
увеличенные потери цинка из красных кровяных
клеток при хронически повышенном
гемолизе. Именно вторичная нехватка цинка
нарушает рост и физическое развитие детей,
больных серповидноклеточной анемией.
Цинка в теле человека содержится от 0,5
до 3 г, поэтому по формальному критерию
он, как и железо, не вполне отвечает
параметрам микроэлемента.
В крови находится лишь всего 1 %
цинка организма, остальное сосредоточено в
тканях. На 70 % цинк сыворотки (а его там
около 12-20 мкМ/л) связан с альбумином.
Гипоцинкемия вызывается интерлейкинами
и другими цитокинами острой фазы (ИЛ-2,
ИЛ-6, ФНО-а) через гепцидин и
сопровождает всякую лихорадку. Кроме того, гипо-
цинкемии способствует гипопротеинемия,
так как при ней увеличиваются потери этого
микроэлемента с мочой. Гипергликемия при
нагрузке глюкозой сопровождается гипо-
цинкемией. Цинк экскретируется главным
450 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
образом, с поджелудочным соком и далее — с
фекалиями. Из тканей и органов наиболее
богаты цинком простата, сперма,
эритроциты, печень, мозг и кожа с ее производными.
Гипоцинкоз впервые получен у крыс
У.Р. Тоддом и соавторами (1934), затем
обнаружен в ветеринарии в виде паракера-
тоза свиней (X. Такер, У. Сэлмон, 1955) и
цыплят, причем заболеванию способствуют
фитаты пищи, снижающие биодоступность
цинка (Б. О'Делл, 1969). Как показали обс-
следования находящихся на однообразной
растительной диете египетских и турецких
подростков из бедных семей, проведенные
в 1963-1967 гг. А. Прасад и соавторами,
гипоцинкоз у человека проявляется
задержкой физического развития, низкорослостью,
гипогонадотропным гипогонадизмом,
половым инфантилизмом, бесплодием,
иммунодефицитом (с паразитарными, грибковыми
и бактериальными поражениями),
характерным гиперкератотическим дерматитом на
локтях, коленях и в других участках кожи.
Очаги дерматита возникают вокруг
естественных отверстий. Дети с гипоцинкозом
едят глину, что имеет адаптивный характер:
показано, что глина задерживает выведение
цинка из ЖКТ.
У подопытных крыс, лишенных
цинка, возникает паракератоз — синдром,
характеризующийся анорексией, рвотой,
выпадением волос, поражением кожи и
слизистых, кератитом, задержкой роста.
Прерывается беременность, выкидыш имеет
множественные эмбриопатии. Нарушается
толерантность крыс к внутривенно
введённой глюкозе.
У человека имеется аутосомно-рецессив-
ный наследственный синдром, вызванный
дефектом кишечной абсорбции цинка — эн-
теропатический акродерматит,
дебютирующий при переходе на искусственное
вскармливание, когда в организм перестает
поступать материнский цинксвязывающий
протеин. Болезнь характеризуется
смешанным иммунодефицитом, отшелушивающим
дерматитом дистальных отделов
конечностей, головы и кожи вокруг естественных
отверстий («перипорит»), хроническим
энтеритом, асперматогенезом,
неврологическими нарушениями (см. т. I, с. 514).
Замедляется заживление ран. Острый
дефицит цинка вызывает раздражительность,
затем — депрессию. Характерна агейзия -
утрата вкусовых ощущений. Может быть
ухудшение сумеречного зрения, как при
авитаминозе А (см. выше «Патофизиология
витаминного обмена»).
Механизм этих нарушений связан с
биологической ролью цинка в организме. Всего
в организме насчитывается от 100 до 200
разных цинксодержащих металлопротеи-
нов. Цинк — компонент активных центров
РНК-полимераз, карбоангидразы, алкоголь-
дегидрогеназы, факторов транскрипции,
ферментов — оксидоредуктаз, трансфераз,
лиаз, гидролаз, изомераз, цитозольной
формы супероксиддисмутазы. Он необходим
для восстановления ретинола в сетчатке.
Данный металл непосредственно входит в
структуру активного гормона вилочковои
железы тимулина (см. ниже
«Патофизиология тимуса») и участвует в упаковке
инсулина, гонадотропинов и других гормонов,
причём блокатор цинка дитизон даже вызывает
инсулиновую недостаточность (см. выше
«Патофизиология сахарного диабета»). При
сахарном диабете повышен уровень цинка в
крови и его суточная экскреция.
Данный металл является также
минеральным антиоксидантом.
Цинк — липотропный фактор для печени,
участвующий вместе с витамином В6 в
образовании ненасыщенных жирных кислот,
синтезируемых в организме. Его дефицит
ускоряет развитие стеатоза печени и
атеросклероза. Рецептор глюкокортикоидов
является цинксодержащим белком. Цинк
участвует в формировании нормальной
вкусовой чувствительности.
Избыток цинка может вызывать острые и
хронические отравления.
Характерно отравление при вдыхании
паров цинка сварщиками, сопровождаемое
подъемом температуры, ознобом,
гиперсаливацией, головной болью, лейкоцитозом
и кашлем. При хроническом воздействии
цинковая интоксикация вызывает ане-
Патофизиология обмена микроэлементов 451
мию, нарушения функций цитоскелета
клеток крови, что выражается в
торможении хемотаксиса, фагоцитоза, пиноцитоза
и замедлении агрегации тромбоцитов.
Неврологические нарушения включают
сонливость. Если цинк поступает
ингаляционным путём — возможен пневмофиброз,
при оральном пути поступления бывают
тошнота, рвота, язвы желудка и панкреатит
(К. Г. Фалчук, 1994). При гемодиализе
избыток цинка может попасть в организм
парентеральным путём
Медь — требуется человеку ежесуточно в
дозе не менее 2-6 мг. Богатейший источник
меди — печень животных (15-74мг/кг),
затем идут мясо (3,7-5,4) и рыба (2,5-5).
Из растительной пищи меди много в
бобовых (3-6 мг/кг). Металл всасывается
в основном в желудке и верхней части
двенадцатиперстной кишки. В сыворотке
медь связывается временно альбумином,
переносится в печень и там вступает в связь
с белком церулоплазмином. В теле человека
примерно 0,25 г меди.
Печень — депо меди и средоточие многих
медьзависимых ферментов, в основном —
класса оксидоредуктаз. Избыток меди она
экскретирует с желчью, в форме особого
гликопротеидного комплекса. Именно из
печени медь перераспределяется между
органами и тканями (максимальные
количества идут в мозг, сердце, почки, надпочечники,
кожу, кроветворные клетки) при посредстве
церулоплазмина, который к тому же сам
является ферментом — ферроксидазой.
Церулоплазмин окисляет фибрин, катехола-
мины, витамин С, имеет большое значение
в функции аминооксидаз соединительной
ткани, в первую очередь — лизил-оксидазы,
при образовании поперечных сшивок в
соединительно-тканных волокнистых белках,
участвуя в продукции десмозина и изоде-
смозина. Этот протеин, раствор которого
имеет голубоватый цвет, обладает
свойствами модулятора аутоокисления, так как
способствует переходу закисного железа в
окисное в реакции Фентона (см. т. I, с. 190).
Кроме того, медь содержится в цитозоль-
ной форме фермента-прерывателя цепных
свободно-радикальных процессов, каким
является супероксиддисмутаза. Поэтому
вполне понятно огромное значение
церулоплазмина как положительного глобулина
острофазного преиммунного ответа. От
концентрации церулоплазмина весьма зависит
сывороточный уровень меди, так как в норме
90% из 13-22 мкМ/л, содержащихся в
сыворотке, связано именно с этим
транспортёром. Церулоплазмин связывает 6 атомов
меди на одну молекулу. Его ген находится в
3-й хромосоме, а биосинтез стимулируется
регуляторным белком, аналогичным IRP.
Экспрессии церулоплазмина способствуют
некоторые цитокины преиммунного ответа,
особенно — у-интерферон. Белок экспресси-
рован не только в печени, но в астроцитах,
мозговых оболочках, нейронах, на
сетчатке, в плаценте и других клетках. Гомолог
церулоплазмина — гефестин — имеется на
базолатеральной мембране энтероцитов
(см. выше о железе). Церулоплазмин -
связующее звено метаболизма железа и меди.
Он участвует в доставке этих ионов плоду
и регулирует их баланс в ЦНС. Показано,
что церулоплазмин способствует оттоку
железа в астроциты и защищает нейроны
от прооксидантного токсического действия
его избытка. Он обладает собственным
деполяризующим действием на нейроны,
способствует их агрегации при диффе-
ренцировке мозга и , что особенно важно,
окисляет серотонин и норадреналин.
Поскольку церулоплазмин аффинно связывает
ЛСД, считается, что ускорение окисления
норадреналина и подавление окисления
серотонина, обусловливающее психотоми-
метический эффект этого галлюциногена,
может быть церулоплазмин-зависимо
(В.Н. Зайцев и соавт., 1999).
Церулоплазмин уменьшает активность NO-синтазы и
конкурирует с некоторыми
антикоагулянтами. Ответ острой фазы сопровождается
гиперкупремией. Повышение сывороточного
уровня меди сопровождает также цирроз
печени, инфаркт миокарда, гемохроматоз,
коллагенозы и тиротоксикоз. Напротив,
при дефектах продукции церулоплазмина
452 АЖ Зайчик, ЛЛ.Чурилов Патохимия
в сыворотке имеется гипокупремия, иногда
несмотря на наличие и даже избыток меди
в тканях. Гипокупремией характеризуются
гипопротеинемические состояния,
особенно нефротический синдром и голодание,
мальабсорбционный синдром, а также
важные наследственные аномалии обмена
меди— болезнь Вильсона-Коновалова и
болезнь Менкеса (см. ниже).
Наиболее изучена кроветворная роль
меди. Это второй, после железа, по
значению гемопоэтический микроэлемент. Медь
необходима для эритропоэза и гранулопоэ-
за, она участвует в стимуляции созревания
ретикулоцитов, путем активации цитохро-
моксидазы всех, в том числе — гемопоэти-
ческих клеток, а также модуляции захвата
железа трансферрином, что необходимо для
включения железа в гем.
Экспериментальная медь-дефицитная гипохромная анемия
была впервые получена у крыс Э.Б. Хартом
исоавт.(1928).
Церулоплазмин является стимулятором
миелопоэза, увеличивает адгезивные
свойства лейкоцитов, служит гемопротектором
при ответе острой фазы и тормозит адгезию
тромбоцитов (Л.В. Кривохижина и соавт.,
2004).
Очень важна, по-видимому, и регуляция
медь-зависимых окислительных процессов в
соединительной ткани и коже. Эти аспекты
использования меди влияют на правильное
образование эластина сосудов и опорно-
двигательного аппарата, способствуют
кератинизации и синтезу меланина в коже,
пигментации волос. Витамин С и медь
имеют в костях общие точки приложения при
действии пролиноксидаз (см. выше:
«Патофизиология витаминного обмена»).
Медь связана с функциями нейроэндо-
кринной системы. Этот металл — кофермент
дофамин-р-гидроксилазы, участвующей в
синтезе нейромедиаторов. Описано высокое
физиологическое содержание меди в
подкорковых ядрах стрио-паллидарной
системы, пермиссивный эффект на освобождение
и действие инсулина, участие в функциях
тирозиназы, а значит — в окислении кате-
холаминов, связь с регуляцией менструаль-
чдокринно-метаболические нарушения)
ного цикла и гипоталамической
терморегуляцией. У овец дефицит меди — причина
тяжелой спинальной демиелинизирующей
нейропатии, известной как эндемическая
«вертячка» — «swayback» в бедных медью
районах Австралии и США (У.М. Нил и
соавт., 1931). Медь in vitro непосредственно
стимулирует освобождение ряда тропных
гормонов гипофиза. В коре надпочечников
имеется медьзависимый фермент А5-3-ке-
тостероид-изомераза, участвующий в
синтезе прогестерона из прегненолона.
Пищевой недостаток меди у человека
наблюдали при парентеральном питании и
у младенцев на молочном вскармливании,
не получавших своевременно прикорма и
коррекции по микроэлементам.
Проявления синдрома нехватки меди
разнообразны. Имеется микроцитарная нор-
мохромная анемия. Замечено, что на фоне
медного дефицита менее активно действует
витамин В12, что может приводить к макро-
цитарным анемиям. Отмечается остеопатия
с изменениями в костях, напоминающими
болезнь Мёллера-Барлоу, то есть дефицит
витамина С. У крыс формируется рахитопо-
добный синдром.
При дефиците меди тормозится рост,
нарушаются процессы ороговения в коже,
может возникать депигментация кожи и её
производных, особенно — волос.
Дегенерация эластина приводит к расслаивающим
аневризмам аорты, отмечается вазопатия.
Бывают расстройства терморегуляции с
тенденцией к гипотермии, умственные
расстройства, спинальные нарушения.
У подопытных крыс при медь-дефици-
тарных состояниях возникает анэструс и
прерывается беременность.
Наиболее частое наследственное
нарушение метаболизма меди (1/30000) было
описано впервые еще К. Вестфалем (1893),
затем в 1912 г. С.А.К. Вильсоном, а в
нашей стране — Н.В. Коноваловым (1948) и
известно как гепатолентикулярная
дегенерация (болезнь Коновалова-Вильсона). При
данной болезни в печени имеется аутосом-
но-рецессивный дефект лизосомального
белка (WND), ответственного за экскрецию
Патофизиология обмена микроэлементов 453
меди с желчью. Этот ген (WND)
локализован в длинном плече хромосомы 13. Медь
в печени связывается с шаперонинами
(HAHl/Atoxl и др.), которые
распределяют ее по отсекам гепатоцита. HAHl/Atoxl
передает ее на фермент АТФ-азу Р-типа
(АТР7В) в транс-часть аппарата Гольджи.
Этот фермент и встраивает ионы меди в це-
рулоплазмин, обеспечивает рециркуляцию
апоцерулоплазмина. Здоровые доношенные
дети рождаются с низкой концентрацией
церулоплазмина и медными запасами,
сосредоточенными в печени. В течение
первых месяцев внеутробной жизни
концентрация церулоплазмина в сыворотке
растет и происходит перераспределение
меди. При дефектах генов АТР7А и АТР7В
в 13-й хромосоме нарушается функция
АТФ-зависимых медь-транспортирующих
белков. Это может затрагивать кишечник
и другие органы (дефект АТР7А, что ведет
к общему дефициту меди, дестабилизации
эластин-коллагеновых структур и болезни
«курчавости Менкеса»), либо в ЭПР
клеток печени, экстрапирамидной системы и
плаценты (дефект АТР7В, основа болезни
Коновалова-Вильсона). При болезни
Коновалова-Вильсона и уровень сывороточного
церулоплазмина остаётся низким.
Устанавливается положительный медный баланс,
экскреция поступающей в организм меди
затруднена. Гепатоциты подвергаются
токсическому действию меди, развивают жировое
перерождение и гибнут, причём
формируется фиброз и цирроз печени, её хроническая
недостаточность. При разрушении гепато-
цитов медь эпизодически высвобождается
из печени и откладывается в других органах
и тканях, что обусловливает приступы
гемолитической анемии, разнообразные, в
основном моторные неврологические
нарушения, связанные как с отложением меди
в ЦНС, так и с недостаточностью функций
печени (хлопающий тремор). В десцемето-
вой мембране лимба роговицы появляются
патогномоничные для данного заболевания
золотисто-коричневые или зеленоватые
кольца Кайзера-Фляйгиера. Это может
сопровождаться катарактой, артритами,
холелитиазом и уролитиазом, синдромом
Фанкони (см. выше: «Аминоацидурия»).
Часто бывают гиперхолестеринемия и
инсулинорезистентность. Избыток меди в
сосудистой стенке способствует развитию
атеросклероза.
При болезни Менкеса (или «курчавости
Менкеса») происходит еще более сложное
нарушение обмена меди, рецессивного,
сцепленного с Х-хромосомой характера.
Дефицит меди возникает в печени, мозге,
сосудах и сыворотке крови, при этом в
кишечнике, почках и фибробластах кожи
её содержание нормально или повышено.
Снижено количество церулоплазмина и
имеется дефицит активности тканевых
аминооксидаз. Нарушается образование
перекрёстных связей в коллагене и эластине.
Развивается картина, похожая на
экспериментальный дефицит меди у животных, но
без признаков анемии. Доминирует
нарушение стабильности соединительно-тканных
структур. Это выражается в аневризмах,
эмфиземе, остеопорозе, иногда следует разрыв
сердца. Волосы больных имеют курчавый
вид (Дж.Х. Менкес, 1962). Таким образом,
если болезнь Коновалова-Вильсона
характеризуется гипоцерулоплазминемией на
фоне некоторых симптомов медного
отравления, болезнь Менкеса сочетает нехватку
церулоплазмина в сыворотке с симптомами
медного дефицита в ряде тканей.
Противоположное состояние — гиперцеру-
лоплазминемия — свойственно
аутоиммунному тироидиту, особенно при отклонениях от
эутироидного состояния. Наиболее высокий
уровень церулоплазмина отмечен при гипер-
тирозе (А.Ш. Зайчик и соавт., 2005).
Кобальт является ключевым элементом
кобамидного кофермента (витамина В12).
В этом качестве он необходим метилен- и
метилтрансферазам и участвует в
превращениях метионина и гомоцистеина (см. выше
«Патофизиология витаминного обмена»).
В сутки человеку необходимо 0,1 мкг
кобальта, при общем содержании в организме
около 14 мг (К. Фалчук, 1994). Кобаламин
вырабатывают только микроорганизмы. Ос-
454 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
тальные участники экоценозов получают его
по пищевым цепям. В моллюсках,
организме морских животных и растений, печени
(13 мкг/дл), почках, а также иле содержится
достаточное количество биогенного
кобальта. Из растительных продуктов кобальт
присутствует в красновато-фиолетовых овощах,
фруктах и ягодах — свёкле (12 мкг/дл),
землянике и клубнике (7-9 мкг/дл), гранатах,
а также в овсяной крупе.
Металл всасывается в тонком кишечнике,
в составе витамина В12. Кобальт, при
хорошем обеспечении медью и железом,
стимулирует эритропоэз, а при его отсутствии в
эндемических районах (Австралия) у жвачных
животных, которых снабжают кобаламином
микроорганизмы рубца, наступает витамин-
В12-дефицитная мегалобластическая гипер-
хромная анемия (Э.Дж. Андервуд, 1937).
Больше всего кобальта в сердце. Известно
также активирующее действие кобальта на
фосфатазы костей и энтероцитов. Найден
кобальт и в панкреатических островках, что
позволяет предполагать связь между этим
микроэлементом и инсулиновой секрецией.
Так как гомоцистинурия — фактор риска
атеросклероза, а метионин — липотропный
фактор, становится понятным, почему
скорость развития и тяжесть атеросклероза и
некоторых гиперлипопротеинемий
коррелируют со степенью кобальтовой
недостаточности (В.М. Сахарчук, 1978).
Кобальт в высоких дозах вызывает
отравление, происходит взаимодействие избытка
кобальта с липоевой кислотой. Это
затрагивает реакции декарбоксилирования, что
приводит в сердце к нарушению метаболизма
жирных кислот и пирувата. Формируется
миокардиодистрофия и энергодинамическая
сердечная недостаточность. Данное
заболевание было характерно для любителей
баночного пива в те годы, когда, в силу гигиенической
безграмотности, производители добавляли
в него хлорид кобальта для стабилизации
пены. Кобальт нарушает захват йода в
щитовидной железе и способствует зобу.
Марганец представляет собой
микроэлемент, общее содержание которого в
организме чрезвычайно мало — до 0,02 г. В сутки
человеку необходимо 2-5 мг этого металла.
Марганцем богаты в основном растительные
продукты. Листовые овощи содержат его
10-20 мг/кг, зерновые — 1-15мг/кг. Но
настоящим концентратором марганца
является чай. Чайный лист далеко опережает по
данному показателю все пищевые продукты,
накапливая до 200 мг/кг марганца! В то же
время печень животных, мясо и рыба имеют
невысокое содержание марганца (2-3 мг/кг).
Дефицит марганца в естественной
обстановке у человека не описан: даже чайно-хлебная
диета содержит его достаточно.
Марганец всасывается в тонком
кишечнике на 40-50%, в основном на
протяжении двенадцатиперстной кишки, и
связывается специальным
переносчиком — pj-глобулиновым белком
трансманганином. Металл накапливается в печени.
Высоким потреблением марганца
характеризуются также ЦНС, гонады, миокард
и скелет, почки и поджелудочная железа.
Экскреция микроэлемента идёт через
жёлчь и панкреатический сок, в малой
степени — через почки.
Гиперманганемия характерна дляинфаркта
миокарда и цитолиза в печени. При
судорожных заболеваниях отмечена гипоманганемия.
Марганец входит в состав небольшого
количества металлопротеинов-ферментов, а
также активирует многие энзимы, не входя
в их состав. Среди них — оксидоредуктазы,
гидролазы и лигазы. Наиболее важна роль
марганца в составе или при активации ксан-
тиноксидазы,амино-ацил-т-РНК-синтетаз,
диаминоксидазы, пируват-карбоксилазы
(где он входит в активный центр), фосфатаз.
Митохондриальный изофермент суперок-
сид-дисмутазы также является марганец-
зависимым.
Благодаря этому данный металл
участвует в окислительном фосфорилировании,
окислении жирных кислот, метаболизме
пуринов, холестерина и гликозаминогли-
канов, антиоксидантной защите клеток. Это
липотропный фактор, особенно активный
при дефиците холина, а также антиатеро-
генный микроэлемент.
Патофизиология обмена микроэлементов 455
Марганцевый дефицит у человека,
описанный при нарушении его всасывания,
характеризуется задержкой роста, гипогона-
дизмом, аномалиями скелета с замедлением
оссификации и снижением щёлочно-фосфа-
тазной активности, нарушением
метаболизма жиров, похудением, дерматитом, рвотой,
кетозом, гипохолестеринемией. Нарушается
также рост и пигментация волос. Характерно
резкое понижение продукции протромбина,
без дефицита витамина К.
Модель этого заболевания получена в
эксперименте на курицах, свиньях и
крысах. Марганцевая недостаточность у куриц
проявляется в форме пероза («болезнь
соскользнувших сухожилий»)-
искривления костей конечностей и гиперплазии их
эпифизов, причём сухожилия икроножных
мышц характерным образом соскальзывают
со своих мыщелков (Х.С. Вильгус и соавт.,
1937). У крыс марганцевый дефицит
снижает толерантность к глюкозе и приводит
к дегенерации В-клеток островков Лангер-
ганса. На безмарганцевой диете животные
растут, но самцы страдают бесплодием от
дегенерации семенников, а самки не
могут вскармливать потомство (Э.С. Орент,
Э.В.Мак-Каллюм, 1931).
Избыток марганца токсичен. Он нарушает
всасывание железа и конкурирует с медью при
обеспечении кроветворения, вызывая анемию.
В костях при гиперманганизме формируются
рахитоподобные изменения («марганцевый
рахит», известный как эндемическая болезнь
скота). Избыток марганца связан с
некоторым эндемическим учащением подагры. У
шахтеров известно профессиональное
заболевание, связанное с хронической ингаляцией
марганцевой пыли. Оно проявляется в виде
паркинсоноподобного синдрома, энцефали-
топодобных нарушений, астено-апатических
состояний. Характерно появление анорексии,
импотенции, судорог в нижних конечностях,
дизартрии. Могут быть даже симптомы
психозов — насильственные смех и плач. Фоном
этих нарушений служит пневмокониоз.
Интересно, что значительным избытком марганца
характеризуются естественные воды и почва
петербургского региона.
Молибден, которого в теле человека
примерно 70 мг, должен потребляться в
ежесуточных количествах 0,15-0,5 мг. Это
ключевой компонент активного центра
ксантиноксидазы, важный участник пури-
нового метаболизма (см. выше
«Патофизиология нуклеинового обмена»), от него
зависит также активность еще нескольких
оксидоредуктаз.
Экспериментальный недостаток
молибдена в питании крыс снижает образование
мочевой кислоты в печени и кишечнике.
При этом молибден может быть заменен
вольфрамом. Наблюдения указывают на
учащение у населения рака пищевода при
геохимическом дефиците молибдена. Области
с избытком молибдена характеризуются
высокой поражённостью подагрой и гипер-
урикемией (Армянское нагорье). Избыток
молибдена в экспериментальных условиях
вызывает стерильность у самцов крыс.
Хром присутствует в организме в общем
количестве около 0,6 мг, а потребляется в
сутки примерно 5-200 мкг этого микроэлемента.
Наиболее богата хромом (как и
молибденом) печень животных. Установлено, что
дефицит хрома нарушает рецепцию
инсулина и снижает толерантность к глюкозе. Это
относится только к трехвалентному хрому
(К. Шварц, В. Мерц, 1957). Возможно, ион
способствует, подобно ванадию (см. ниже),
экспрессии транспортёров глюкозы.
Избыток хрома приводит к поражению почек,
при ингаляции повышает риск рака легких,
как гаптен хром может вызвать контактный
дерматит.
Ванадий — микроэлемент, важность
которого для растений и животных давно
известна в агрохимии и ветеринарии.
Однако, так как в теле человека не более 100 мкг
ванадия, лишь недавно стала раскрываться
его, вполне возможно, уникальная роль в
человеческом организме. Х.А. Шредер и
И.И. Баласса (1961) установили его
значение в липидном обмене. Ванадий
накапливается в печени и жировой ткани, подавляет
холестериногенез, усиливает катаболизм
липидов и тормозит развитие
экспериментального атеросклероза. В клинических
456 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
условиях атеросклероз у пациентов
протекает с гипованадиемией: Противоположное
отклонение концентрации ванадия в
сыворотке — гиперванадиемия — отмечено при
маниакально-депрессивном психозе.
И. Шехтер и соавт. (1987) обнаружили,
что ванадил-сульфат (но не другие
сульфаты) обладает выраженным инсулиноподоб-
ным действием на липоциты in vitro и in
vivo. Кванадилу оказались чувствительны
даже клетки, резистентные к действию
инсулина. Интереснейшее свойство ванадия
«обходить» барьер инсулинорезистентности
нашло применение в терапии СД (см. выше
«Патофизиология сахарного диабета»).
Вероятно, оно связано с действием ванадила
на систему пострецепторных передатчиков
сигнала в клетках. Известно, что ванадий —
стимулятор аденилатциклазы, ингибитор
Na+-K+-ATO-a3bi, фосфофруктокиназы,
тирозиндефосфорилаз и аденилаткиназы.
Ванадий несколько уменьшает
выраженность отёчного синдрома при голодании. У
крыс без ванадия нарушается рост.
Дебатируется вопрос о связи нарушений
обмена ванадия с неоднократно
упоминавшимся на страницах этой книги
метаболическим Х-синдромом (гл. 14), с его
дефектом натрий-водородного мембранного
переносчика.
Никель присутствует в сыворотке крови
здоровых индивидов в концентрации до 0,01
мкг/дл, хотя его общее содержание в теле и
суточная потребность в нём не определены.
Пищевые источники никеля — морские
продукты, печень, сельскохозяйственные
растения содержат много никеля в эндемических
никелевых районах (Хибины). Больше всего
никеля в организме содержат гипоталамус,
гипофиз, печень, поджелудочная железа,
почки и легкие. В химии никель известен
своими каталитическими возможностями.
Предполагается, что и в живых клетках он
может входить в активные центры
биокатализаторов-энзимов.
Никель действительно обнаружен в
оксидоредуктазах, в частности — уреазе
бактерий, в гидролитических ферментах
человека. За ним признают
вспомогательную роль в обеспечении гемопоэза,
сходную с действием кобальта. В ядре никель,
способный взаимодействовать с
хроматином, по некоторым данным, участвует
в стабилизации структуры РНК. В крови
никель образует комплексы с альбумином,
с а2-микроглобулином и с гистамином.
Считается, что никель играет важную
роль при образовании липопротеидов.
Никель усиливает включение углеродной
метки из глюкозы в липиды и гликоген.
При экспериментальном атеросклерозе его
содержание снижается, но при клиническом
атеросклерозе данные разных авторов об
уровне никеля в сыворотке резко
противоречивы (Л.Р. Ноздрюхина, 1977).
Никель обладает выраженным пролак-
тостатическим действием, опосредованным
через гипоталамус, и влияет на
освобождение других гипофизарных гормонов
(Р.А.Хенкин, 1976).
Дефицит никеля не описан ни в
клинической, ни в экспериментальной форме.
Зато хорошо известны формы патологии,
связанные с избытком никеля. Это
контактный дерматит на никель как гаптен (см. т. I,
с. 482-483), а также некроз паренхимы
печени при оральном пути воздействия никеля.
При ингаляции никелевой пыли возможна
пневмония. Пораженность карциномами
дыхательных путей и, вероятно, почек
увеличивается при профессиональном и
бытовом контакте с никелевой пылью. Избыток
никеля сильно угнетает жизнедеятельность
многих растений. У подопытных животных
избыток никеля угнетает сперматогенез и
вазывает гипогонадизм и стерильность.
Стронций играет определённую роль в
процессах оссификации. До 0,024% веса
костной золы приходится на этот
микроэлемент. Дефицит кальция способствует
задержке стронция в костях. Избыток
стронция в диете подопытных животных
вызывает витамин-О-резистентный
стронциевый рахит. Стронциевый рахит описан в
Туркмении как эндемическая болезнь скота.
Так называемая уровская болезнь (болезнь
Кашина-Бека) в эндемичных районах
Прибайкалья в бассейне р. Урова поражает
Патофизиология обмена микроэлементов 457
людей и напоминает стронциевый рахит, с
которым, очевидно, имеет общие
этиологические факторы. Радиоактивный стронций,
кумулируясь в костях, способствует
развитию опухолей.
Кадмий даже при однократном введении
вызывает у подопытных млекопитающих
некроз тестикул и дегенеративные изменения
в яичниках. Микродозы кадмия поступают
в питьевую воду из старых водопроводных
труб. Считается, что избыток кадмия — один
из числа «мягких» факторов риска
атеросклероза.
Существует экспериментальная модель
энергодинамической сердечной
недостаточности на основе миокардиодистрофии,
вызванной кадмиевым отравлением (см.
Практикум).
Многие другие металлические
микроэлементы, например, олово, входящее в состав
гастрина, существенны для растений и
позвоночных животных, но их роль в патологии
человека не изучена.
ПАТОФИЗИОЛОГИЯ
ОБМЕНА ВАЖНЕЙШИХ
МИКРОЭЛЕМЕНТОВ-
НЕМЕТАЛЛОВ
Безусловно, самым важным участником
метаболизма среди всех микроэлементов
этой группы считается йод.
Его роль в метаболизме определяется
участием в структуре тироидных гормонов, а
также использованием йодидов и оксийоди-
дов макрофагами и полиморфонуклеарами
в качестве естественных бактерицидных
агентов. Само открытие йода Б. Куртуа в
1811 г. связано с его извлечением из золы
водорослей и губок, применявшейся на
Востоке древними китайцами за 1500 лет
до нашей эры, а на Западе — еще испанским
врачом XIII века Кассамидой для лечения
зоба. Первооткрыватель йода
предположил, что именно с этим элементом связано
противозобное действие золы водорослей.
В 1850 г. А. Шатен установил обратную
связь между содержанием йода в воде и
частотой эндемического зоба в Альпах. Однако
уже А. Хирш (1885) указал, что йод помогает
не при всяком зобе, и подчеркнул опасности,
связанные с йодотерапией. Тем не менее в
1895 г. Т.Э. Кохер начал систематическое
лечение препаратами йода больных
эндемическим зобом в Швейцарии, а в
эндемичных по дефициту природного йода районах
Огайо (США) Д.Мэрии и П. Кимбелл в
1917-1920 гг. в большом проспективном
эпидемиологическом исследовании
показали эффективность профилактики зоба у
детей йодированной солью.
Основные аспекты метаболизма йода
же освещаются ниже (см. «Нарушения
эндокринной регуляции»). Для целей данной
главы следует отметить, что минимальная
суточная потребность в йоде составляет у
взрослых 100-150 мкг, йодный оптимум —
150-300 мкг в сутки, а нормальное
содержание в крови — 5-12 мкг/дл. В период
полового созревания наивысшие потребности в
йоде. Пищевым источником йода могут
служить многие растительные и животные
продукты, но содержание в них этого галогена
широко колеблется и в приморских районах
в 5-10 раз превышает уровень,
характерный для тех же видов еды, произведённых
в горной местности, на удалении от морей.
Поэтому реальное поступление йода в
организм с пищей варьирует от менее чем 20
й до 700 мкг в день. Особенно богаты йодом
рыбий жир (770 мкг/дл), морские водоросли
и моллюски. В эндемических по дефициту
йода районах проводится профилактическое
йодирование поваренной соли.
Йодид всасывается в ионной форме
I", процесс происходит быстро, требуя до
получаса. От 5 до 30 % йода захватывает
щитовидная железа, небольшую часть —
лейкоциты, остальное выводится с мочой.
Интермедиарный обмен йода и его
нарушения будут рассмотрены при обсуждении
патофизиологии тироидной функции.
Напомним о патогенности избытка йода.
Контакт с йодом как гаптеном может вы-
458 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
звать дерматит («йодизм»), избыток йода
может способствовать аутоиммунному
тироидиту и нарушениям функции
щитовидной железы («йод-базедовизм»). Йод
как радионуклид избирательно поражает
щитовидную железу.
Чрезмерное поступление йода в
щитовидную железу ингибирует органификацию
и спаривание йодтирозиновых остатков
тироглобулина (эффект Вольфа-Чайкова).
У мышей линии NOD, получавших
йодированную воду, избыток йода провоцирует
аутоиммунный тироидит (Роуз Н.Р. и соавт.,
1999). У лиц, леченных от аритмий амиода-
роном (кордароном), содержащим в одной
таблетке годовую потребность человека в
йоде, нередко возникают аутоиммунный
тироидит или базедова болезнь (Э. Ро-
ти, Л.И.Браверманн, 2000). По мнению
Н. Амино (2002), избыток потребляемого
йода способствует развитию аутоиммунных
заболеваний щитовидной железы,
увеличивая иммуногенность тироглобулина и
вызывая аномальную экспрессию антигенов
ГКГС II класса на тироцитах.
Следовательно, для предрасположенных к аутоаллергии
индивидов прием избыточных количеств
йода с пищей и с различными витаминно-
минеральными добавками или лекарствами
опасен. Высокая распространенность
аутоиммунных тиропатий заставляет крайне
осторожно подходить к поголовному
использованию населением йодированной
соли и любых содержащих йод препаратов,
которые нужны только при реально
доказанном геохимическом йодном дефиците
(Ю.И. Строев, Л.П. Чурилов, 2005).
Фтор — микроэлемент огромного
биологического значения.
Анион фтора влияет на активность адени-
латциклазы, поэтому концентрации фтора
в клетках поддерживаются на очень малом
уровне (в мышцах — 2 мг/кг). В силу этого
практически весь фтор в организме
сосредоточен внеклеточно, в костях (до 490 мг/кг), в
зубах (до 560 мг/кг). Зубная эмаль —
наиболее насыщенная данным галогеном ткань
организма (0,02 % ее веса представлено именно
этим элементом). Ее поверхностный слой,
самое твёрдое вещество тела человека,
содержит максимальное количество фтора — в
нём 0,7 % фтор-апатита. Всего в эмали зуба
до 4 % апатитов составляет именно фтор-
апатит — Са10(РО4)б(Р)2. Его кристаллы
уложены особенно плотно, для микробов он
наименее преодолим, поэтому без фтора эмаль,
обедненная этим минорным компонентом,
более подвержена кариесу. Интересно, что
рекордное содержание фтора обнаружено в
эпифизарном «песке» и в ткани самого
эпифиза (см. гл. 13). Подробнее о роли фтора в
патофизиологии стоматологических
заболеваний - см. специальное учебное пособие
(Л.П. Чурилов, 2006).
Фтор в костях и зубах способствует
минерализации. Но фториды усиливают эти
процессы лишь при адекватном количестве
кальция. Избыток фтора при недостатке
кальция минерализацию костей тормозит.
Для фтора вообще характерен очень узкий
диапазон оптимального биологического
действия. Как недостаток, так и избыток
этого элемента патогенны.
Взрослому в сутки необходимо 1,5-4 мг
фтора. До 2/3 этого количества всасывается
из питьевой воды, а остальное из пищи.
Если содержание фтора в воде между 0,9
и 1,5 мг/дл — развитие зубов происходит
нормально, а поражённость кариесом мала.
При понижении содержания фтора растёт
заболеваемость кариесом зубов. Эмаль и
дентин кариозных зубов дефторированы.
Особенно кариогенной является вода,
содержащая менее 0,5 мг/дл фтора. В
прошлом, до введения фторирования воды и
фторсодержащих зубных паст, в
Великобритании, например, в районах, где питьевая
вода содержит лишь следы фторида (Уэльс),
поражённость кариесом доходила до 98%
населения.
Известно и нарушение минерализации
остеоида при нехватке фтора.
В то же время, если в воде более 1,2 мг/дл
фторида, у людей, употребляющих такую
воду с детства, весь период развития зубов,
наблюдается особое поражение организма,
проявляющееся, в частности, дистрофией
зубной эмали, не связанной с кариесом, -
Патофизиология обмена микроэлементов 459
эндемический флюороз. Крапчатая эмаль
содержит фторидов в 16 раз больше нормы.
На такой эмали имеются растущие
беловато-желтые пятнышки. Она трескается, и
края зубов становятся хрупкими.
Флюороз обусловлен тем, что избыток
фтора провоцирует апоптоз энамелоблас-
тов.
Крапчатость зубной эмали при флюорозе
сочетается со слабостью мышц, вялостью,
потерей в весе, анемией, ломкостью костей,
кальцификацией сухожилий, образованием
остеофитов («системный флюороз»).
Описан и профессиональный флюороз
при контакте с фторидами в алюмииево-
магниевом производстве, а также при
изготовлении фторсодержащих минеральных
удобрений. Если избыток фтора
наблюдается при уже сформировавшихся постоянных
зубах, то эмаль не меняется, но системные
проявления болезни имеются.
Острая интоксикация содержащимся в
некоторых инсектицидах фторидом, как
блокатором аденилатциклазы, приводит
к тошноте, желудочным коликам, рвоте
и поносу. Наблюдаются гипокальциемия
и судороги, возможен коллапс, остановка
дыхания и смерть.
Селен — незаменимый микроэлемент,
значение которого связано с его ключевой
ролью в антиоксидантных системах клеток.
Уровень селена в крови поддерживается в
пределах 1,9-3,17 мкМ/л.
Потребность в селене — 50-200 мкг в
сутки, причём она зависит от доступности
других антиоксидантов (витамина Е) и
микроэлементов (например, молибдена). Селен
содержится в повышенных количествах в
морепродуктах и чесноке.
В ряде работ конца 30-х-начала 40-х
годов прошлого столетия, в частности
проведенных К. Шварцем в голодающей
нацистской Германии, было установлено,
что у человека и животных бедная белком
и витаминами диета в сочетании с
интоксикациями ведет к стеатозу и некрозу
гепатоцитов и циррозу печени. В поисках
незаменимых ингредиентов пищи,
предупреждающих алиментарный цирроз, было
установлено, что большую роль играют
витамин Е и серосодержащие аминокислоты,
в частности — метионин. Однако оказалось,
что не менее важен загадочный
низкомолекулярный «третий фактор», почти в 500
раз более активный, чем токоферол. Более
чем через 20 лет поисков он был
идентифицирован К. Шварцем и К. Фолтцем как
селен (1957). Впоследствии ветеринарами
в эндемичных по дефициту селена районах
Новой Зеландии и Скандинавии была
показана возможность профилактики и лечения
болезни белых мышц и других недугов
домашних животных селеном.
Селен — мощный антиоксидант,
компонент глютатион-пероксидазы, фосфо-
липид-глютатион-пероксидазы, других
оксидоредуктаз и некоторых трансфераз
(см. также т. I, с. 193-196). Фибробласты
и многие другие человеческие клетки
не растут в культуре без добавок селена.
Провинция Ганьсу в КНР— эндемический
район селеновой недостаточности,
известна существованием местной кэшаньской
болезни — особой формы многоочаговой
некротической миокардиодистрафии. В
ветеринарии аналогичные эндемические
синдромы описаны у свиней и жвачных
животных. Самое низкое в мире
геохимическое содержание селена (и самая низкая
в Европе продолжительность предстоящей
жизни сорокалетних мужчин) отмечено в
Финской Карелии (Л.А. и Н.С. Гавриловы,
1991). Швеция, Дания и Новая Зеландия,
отличающиеся геохимическим дефицитом
селена тоже находятся среди лидеров по
поражённости атеросклерозом и
некоторыми новообразованиями. Обнаружена
связь между селеновой недостаточностью
у беременных и развитием муковисцидоза
у плода (Д. Уоллак, 1997). Нехватка селена
вызывает аритмии, миопатии, азооспермию,
некрозы печени, угри, а в тяжелых случаях
— дилатационную кардиомегалию и
застойную сердечную недостаточность. Селен
увеличивает противовирусную и
противоопухолевую резистентность у животных,
связывает токсические тяжелые металлы
460 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
(прежде всего кадмий и ртуть). Дж.Р. Смай-
тис (1998) приводит данные о дефиците се-
лензависимых ферментов и эффективности
добавок селена в качестве поддерживающего
лечения, облегчающего состояние больных
при бронхиальной астме, респираторном
дистресс-синдроме, сахарном диабете, раке
молочной железы, ВИЧ-инфекции.
В то же время избыток селена способен
замещать серу в метионине и цистеине.
Это происходит у растений,
произрастающих на солончаках, богатых данным
микроэлементом. Прием в пищу избытка
селена, в частности таких растений,
вызывает острое отравление с симптомами
алопеции, дистрофии ногтей, чесночным
запахом изо рта, эмоциональной
лабильностью, часто переходящей в апатию.
Эндемический селиноз описан в Юте и других
юго-западных и центральных засушливых
районах США при превышении
рекомендованной суточной нормы потребления
селена в 5-6 раз.
Кремний входит в состав костей,
сухожилий, кожи, артериальных стенок и тканей
глаза. Данный микроэлемент необходим для
нормального раннего роста и
минерализации костей. Он тормозит развитие
экспериментального атеросклероза. Предполагают,
что все эти эффекты зависят от его участия
в образовании перекрёстных сшивок кол-
лагеновых молекул. Ингаляция частиц,
содержащих двуокись кремния, активирует
лёгочные макрофаги и ведет к
формированию гранулём и силикозу легких.
Патофизиология обмена
микроэлементов находится в фазе активного развития.
Многие важные её проблемы, например
истинная роль селена в антиоксидантной
защите, ванадия и хрома в диабетологии,
стали выясняться лишь в последние годы.
Это сопровождается ростом массовой
популярности полиминеральных добавок как
средства «оптимизации питания». В связи
с этим повторим мысль, звучавшую выше
в разделе «Патофизиология витаминного
обмена». Ни витамины, ни микроэлементы
сами по себе не являются неким
«количественным эквивалентом здоровья». Об этом
красноречиво свидетельствуют такие
факты, как связь между йодными добавками и
аутоиммунным тироидитом, обсуждавшаяся
выше токсичность избытка селена, меди,
марганца и фтора и т.д.
По-настоящему здоровым, в том
числе—в аспекте микроэлементов, является
не искусственно усиленное, а разнообразное
естественное питание.
Глава 13
НАРУШЕНИЯ ЭНДОКРИННОЙ
РЕГУЛЯЦИИ
«Функциональное единство оранизма не является следствием простой
механической сборки его частей... Организм содержит две системы связи...
медленную почтовую систему химических посланий и быструю}
телеграфную — нервных».
Дж. Бернал. «Наука в истории»
История идей в эндокринологии и её основные принципы
Метаболизм гормонов и общие механизмы эндокринных нарушений
Патология гипоталамо-гипофизарного нейросекреторного аппарата
Патофизиология шишковидной железы
Патофизиология аденогипофиза
Гормон роста и патофизиология ростовых и анаболических процессов
Тиротропный гормон и нарушения тироидной функции
Патофизиология надпочечных желез и их регуляции
Патофизиология гонадотропной функции и гонад
Патофизиология околощитовидных желёз и кальций-фосфорно-магниевого метаболизма
Остеопороз и эндокринно-метаболические заболевания костей
Заключение
Для мобилизации отдельных
составляющих реактивности — реактонов —
организму требуются эндогенные сигналы — ак-
тоны. В роли эндогенных актонов
выступают различные биорегуляторы — гормоны,
нейротрансмиттеры, аутакоиды, ионы,
субстраты, антитела (см. т. I, с. 114-115).
В данном разделе речь пойдет о нарушениях
гормональной регуляции метаболизма и
жизнедеятельности клеток.
Гормоны — это органические сигнальные
молекулы беспроводникового системного
действия. Они распознаются рецепторами
и влияют на экспрессию генов и активность
ферментов в клетках-мишенях на удалении
от места своей продукции.
В отличие от них нейротрансмиттеры
(нейромедиаторы), также имеющие
органическую сигнальную природу, действуют
лишь топически, в пределах того синапса, в
который освобождаются проводником.
Тканевые аутакоиды (например
медиаторы воспаления) — это органические
сигнальные молекулы беспроводникового
зонального действия, вырабатываемые, влияющие
на рецепторы и инактивируемые в пределах
ограниченного участка органа или ткани.
Типичными биорегуляторами данной
группы являются эйкозаноиды, которые,
несмотря на явное участие в химической регуляции
многих процессов, рассматриваются вне
данного раздела (см. т. I, с. 155-159, 359-361).
Приведённые градации относительны,
так как одна и та же молекула в разных
ситуациях может выступать в любой из
этих трёх ипостасей, примером чего служат
катехоламины, в частности норадреналин
(см. также т. I, с. 346).
Нередко гормоны распространяются
лишь в пределах ограниченной циркуля-
торной сети: так происходит с гипоталами-
ческими либеринами и статинами, а также
462 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
с многими гормонами ЖКТ, которые
предохраняются от системного распространения,
так как инкретируются в портальные
системы, снабжённые собирающими сосудами
между двумя капиллярными сетями (см.
с. 503, рис. 96). Такой гормон попадает в
кровь в пределах первой капиллярной сети
(например, в гипоталамусе, клубочках почек
или непарных органах брюшной полости) и
аффинно связывается рецепторами клеток,
снабжаемых вторичной капиллярной сетью
(например, в гипофизе, в районе петли Генле
или в печени).
Гормоны не являются ни катализаторами,
как энзимы, ни коферментами, как витамины,
ни пластико-энергетическим сырьем, как
нутриенты. Э. Старлинг образно называл их
вестниками. Их роль — информационная,
поскольку, в зависимости от своего
взаимодействия с рецепторами и от состояния
клеток-мишеней в контексте других сигнальных
эффектов, они включают и выключают на
генетическом уровне определенные клеточные
программы или модулируют эпигенетически
эффективность их осуществления.
В отношении обмена веществ, клеточной
пролиферации, роста тканей,
долговременных адаптивных реакций гормоны служат
основными регуляторами,
определяющими направленность и эффективность этих
процессов. Нейротрансмиттеры в своих
воздействиях на эти стороны
жизнедеятельности или зависят от регулирующих
эффектов гормонов, или даже опосредуют
свои влияния через гормоносекретирующие
клетки. Ценными особенностями
гормональной регуляции являются ее
беспороговый принцип и большое разнообразие
пермиссивных взаимодействий гормонов,
исключающее господство стереотипии.
В то же время проводниковая нервная
регуляция, осуществляемая на основе
пороговых стереотипных ответов, играет
приоритетную роль в управлении
движением, в сборе афферентной информации,
в регуляции быстрых типовых реакций на
различные стимулы.
Гормональная и нервная регуляция, а
также иммунная регуляция клеточных
функций неразрывно связаны между собой.
Примерами могут служить необратимое
нарушение развития ЦНС при врождённых
формах гипотироза (синдром Фаггё), а также
наличие секреторной иннервации у многих
эндокринных желёз (эпифиза, мозгового
вещества надпочечников, щитовидной железы,
островков Лангерганса). Высшим
проявлением нейроэндокринного единства является
существование феномена нейросекреции
(см. ниже). Эволюционное происхождение
и взаимоотношения нервной и эндокринной
регуляции в контексте концепции единого
иммунонейроэндокринного аппарата
подробно обсуждены в т. I данного руководства
(с. 61-65) и в недавно вышедшем учебном
пособии (Е.А. Корнева, 2003).
ИСТОРИЯ ИДЕЙ
в эндокринологии
И ЕЁ ОСНОВНЫЕ ПРИНЦИПЫ
Концепция гормональной регуляции
метаболизма сформировалась на основе
длительной эволюции ряда основополагающих
идей, о развитии и современном состоянии
которых необходимо напомнить перед
обсуждением механизмов конкретных эн-
докринопатий.
Истоки эндокринологии при желании
можно проследить, начиная с воззрений
античных медиков, в частности Гиппократа
и Птолемея Диоскорида, которые считали
интеграцию организма результатом
надлежащего смешения разнородных
гуморальных начал, а индивидуальность —
следствием уникальной комбинации «жизненных
соков». Парацельс продолжил эту линию,
проповедуя концепцию «Архея» —
своеобразного центрального процессора,
управляющего органами с помощью алхимических
субстанций.
Позже доктрина гуморальной регуляции
наложила определяющий отпечаток на
взгляды Ж.-Б. Ван Гельмонта,
исходившего из существования «археев органов».
Интересно, что он признавал в организме
регуляторную роль газообразных веществ
(и ввел само понятие «газ»), что неожиданно
перекликается с таким новейшим
открытием, как медиаторная функция окиси азота
(Р.Ф. Фуршготти соавт., 1998). Р.Декарт
в самом начале XVII века подчеркивая, что
рефлекторный принцип роднит животных,
человека и машину, полагал именно
шишковидную железу вместилищем главного
«немашинного» компонента — души,
управляющей телом, то есть интуитивно угадал
ключевую роль нестереотипной надмеха-
нической регуляции в организме. В XVIII
столетии Э. Бургхааве продолжил развитие
и пропаганду ятрохимической доктрины.
Тем не менее значение проводниковых
и механических связей было ясно оценено
гораздо раньше, чем роль связей
гуморальных, в связи с анатомической очевидностью
первых и незримым характером последних.
Еще Герофил Халкидонский, Алкмеон
и Эразистрат (перед 300 г. до н. э.)
распознали регуляторную роль мозга и нервов.
Поэтому в науке о живом в XVIII и начале
XIX столетия безраздельно господствовал
взгляд, согласно которому «нервная система
и есть само животное, остальные системы не
более, чем слуги» (П. Кювье). Предельное
развитие этой теории приводило к
невесёлому заключению, что «человек —
высокопросвещённая машина... какой-нибудь
пустяк, волокно незначащее могло бы
сделать дураками Эразма или Фонтенеля»
(Ж.-О. де Ламетри).
Тем удивительнее, что именно
современник и соотечественник цитированных
выше авторов выступил с догадкой,
противоречившей основному умонастроению
медиков той эпохи и на столетие
опередившей своё время. Речь идёт о
французском враче Теофиле Бордю (см. также т.
I, стр. 53-56). В своей книге под скромным
названием «О медицинском анализе крови:
исследование о хронических заболеваниях»,
опубликованной в 1775 г., признавая роль
нервной регуляции, а также постулируя,
что специальные отделы мозга управляют
разными органами и частями тела с
помощью импульсов, переданных через нервы,
Нарушения эндокринной регуляции 463
Т. Бордю высказывает и первую научную
формулировку принципа гормональной
регуляции. Он пишет: «Каждый орган служит
фабрикой и лабораторией специфического
гуморального агента, который, по
приготовлении и приобретении индивидуально
присущих ему свойств, возвращается в
кровь. Кровь обладает специфическими
свойствами, приобретенными в органах,
через которые она проходит. Каждый орган
посылает в нее свою эманацию... Таким
образом, кровь несет в своем потоке
экстракты всех органов, необходимые для жизни
целостного организма, и обладает
количественными и качественными
характеристиками, не поддающимися экспериментальному
определению химиков».
Особенно замечательно, что Бордю, в
духе представлений нашего времени,
отводил каждому органу (а не избранным
железам) определенную внутрисекреторную
регуляторную функцию.
В начале XIX века гуморальную доктрину
в области патологии развил К. фон Роки-
танский (1849), считавший химические
вещества интеграторами
жизнедеятельности тела как целого и настаивавший на
существовании «безлокальных» расстройств
регуляции.
К1844 году развитие анатомии
обеспечило Й. Мюллера достаточным количеством
фактов, чтобы сделать обобщение о делении
желез на занятые внешней и внутренней
секрецией. Последние, по мнению немецкого
учёного, всегда лишены выводных протоков
и посылают секрет в кровь и/или лимфу.
В разряд их, по Мюллеру, попали не только
щитовидная железа или надпочечники, но и
селезёнка, лимфоузлы, печень и т. д.
Характерно, что классики той эпохи не выделяли
гормоны как сигналы, а рассматривали
любое химическое вещество, которым «железа»
обогащает кровь, как «инкрет». Например,
по К. Бернару, выделение печенью глюкозы
в кровь под действием симпатических
нервов — есть её внутренняя секреция (1857).
Постепенно накапливались клинические
и патологоанатомические наблюдения,
позволившие поставить определённые
464 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
симптомокомплексы в связь с видимыми
поражениями тех или иных «беспротоковых»
желёз. Историческое значение здесь имели
наблюдения Т. Коули (1785),
зарегистрировавшего калькулёз поджелудочной железы
как причину сахарного диабета, Т. Аддисона
(1849), описавшего бронзовую болезнь как
результат туберкулёзного поражения
надпочечников, Дж. Флаяни (1802), К.Х. Парри
(1825), Р.Дж. Грейвза (1835) и, наконец,
К.А. фон Базедова (1840), установивших
системные нозологические последствия
гиперплазии и предполагаемой
гиперфункции щитовидной железы. Личный хирург
британского короля (и предполагаемый
фигурант дела Джека Потрошителя) У .О. Галл
показал, что синдром микседемы
вызывается атрофией или аплазией щитовидной
железы (1873-1874). П. Мари, а позже
Э. Бриссо и Г. Мейге (1886,1895)
установили связь опухолей гипофиза с акромегалией
и гигантизмом.
Еще в те годы из разрозненных данных
аутопсий начала складываться картина
общей этиологии эндокринных заболеваний,
которая к настоящему времени обогатилась
и усложнилась (см. ниже).
Вторым важным источником аргументов
в поддержку концепции гуморальной
регуляции были экспериментальные данные по
экстирпации и пересадке «беспротоковых
желез» и применению их экстрактов и го-
могенатов.
По понятным причинам первыми
внимание экспериментаторов, хорошо
осведомленных о последствиях оскопления,
привлекли тестикулы. Еще Дж. Хантер
успешно пересаживал их (1780), а А. А. Бер-
тольд установил, что такая трансплантация
маскулинизирует петуха-кастрата (1849).
Ш. Броун-Секар обнаружил, что
билатеральная адреналэктомия вызывает
смертельный коллапс у собак, но приписал этот
результат сопутствующей травме нервных
сплетений (1857). Й. фон Меринг и О. Мин-
ковский воспроизвели сахарный диабет
у собак путём полной панкреатэктомии
(1889). П.М. Шифф убедился, что
тотальная тироидэктомия приводит к
смертельным судорогам у собак, но не вызывает этого
синдрома у кроликов (1884). Чуть позже
Э. Глей объяснил эти различия, открыв
околощитовидные железы, удаление которых и
было причиной судорог у собак, и которые
у кроликов локализованы на расстоянии от
щитовидной железы и щадятся при тироид-
эктомии (1891). Наконец, Ф. Хофмайстеру
(1894) удалось воспроизвести микседему с
помощью тироидэктомии и показать, что
утрата щитовидной железы сказывается на
неполовозрелых животных сильнее, чем на
взрослых. Р. Бетанкур и С.А. Серрано
добились улучшения у пациентки с микседемой,
подсадив ей под кожу долю щитовидной
железы овцы (1890). Это подтолкнуло на
смелый клинический эксперимент доктора
Г.Р. Мюррея, начавшего лечение тяжёлой
микседемы у 46-летней больной
подкожными инъекциями экстракта овечьих
щитовидных желез (1891). Опыт увенчался полным
успехом, и пациентка дожила до 79 лет при
поддерживающей терапии и полном
исчезновении симптомов болезни, пережив
своего исцелителя. Наконец, Ж. Такамине
(1901) удалось химически охарактеризовать
действующее начало обнаруженного^еще
М. Вюльпианом (1856) секрета хромаффин-
ного вещества медуллярной части
надпочечников и установить, что это амин —
адреналин. Все эти факты создали основу для
важнейшего теоретического обобщения:
«... специфические растворимые продукты
выходят в кровь и оказывают действие на
клетки других составных частей тела.
Имеется взаимосвязь между разными клетками
организма, осуществляемая путями,
отличными от используемых нервной системой»
(Ш. Броун-Секар, Ж. д'Арсонваль, 1891).
В результате опыта, произведённого
Э.Г. Старлингом и У.М. Бейлиссом (1904),
было показано, что гуморальная субстанция,
инкретируемая слизистой оболочкой
двенадцатиперстной кишки после попадания
в нее пищи, стимулирует панкреатическую
секрецию даже при полной денервации
этих образований. Почти одновременно
Дж. Эдкинс открыл, что экстракт клеток
антрального отдела слизистой желудка
Нарушения эндокринной регуляции 465
стимулирует секрецию соляной кислоты и
пепсина клетками его пилорической части,
и назвал действующее начало «гастрином».
Интерпретируя эти новаторские результаты,
Э.Г. Старлинг в 1905 г. и ввел само понятие
«гормон» (от греческого «хорцеко» —
пришпориваю), определив его как «вещество,
вырабатываемое в одной части организма
и переносимое кровью или лимфой к
какой-либо другой части, функция которой
вследствие этого изменяется». Чуть позже
Н. Пенде ввел термины «эндокринология»
и «эндокринные железы» (1909)— и
концепция гормональной регуляции после 230-
летнего существования добилась наконец
полноправного положения в клинической и
экспериментальной медицине. Как ни
парадоксально, это произошло на фоне, казалось
бы, безраздельного триумфа нервизма, когда
работы критически отнёсшегося к данным
о секретине и гастрине И.П. Павлова по
нервной регуляции пищеварения были
только что увенчаны Нобелевской премией
(1904).
Вслед за этим, в контексте классического
представления К.' Бернара о постоянстве
внутренней среды и о примате нервной
системы в регуляции его химических аспектов
(1856), встал вопрос о регуляторных
взаимоотношениях различных эндокринных
желез и о неироэндокриннои взаимосвязи.
Поначалу идеи эндокринологов сводились
к констатации антагонизма или
синергизма между разными железами внутренней
секреции и отделами вегетативной нервной
системы.
Большое значение имели полученные на
модели гипофизэктомии данные Дж. Аско-
ли и Т. Леньяни (1912), а также Б. Аллена
и Ф.Смита - Дж. Грезера (1916, 1920)
о стимулирующем действии гипофиза на
рост организма, пролиферацию и функцию
периферических эндокринных желёз. Среди
инкреторных органов появился кандидат на
роль центрального регулятора — нижний
мозговой придаток. Идея о центральной
регуляторной роли гипофиза — одна из
главных в медицине первой трети XX столетия.
Вдохновлённый ею в годы занятий
медициной, доктор М.А. Булгаков впоследствии
напишет в «Собачьем сердце» — повести,
весь сюжет которой вращается вокруг ксено-
генной трансплантации нижнего мозгового
придатка: «Гипофиз — закрытая камера,
определяющая человеческое данное лицо.
Это — в миниатюре — сам мозг... Этих
гормонов в гипофизе... господи!» (1925).
В попытках обнаружить системность в
«эндокринной системе» сначала X. Эппин-
гер и соавторы, а затем Б. Ашнер (1912)
гипофиз
щитовидная железа
PNS
поджелудочная железа
околощитовидная железа
яичник
-*
^^^™"™~
торможение
^-
i -^
SNS
хромаффинная система
Рис. 82. Взаимодействие желёз внутренней секреции и вегетативной нервной системы согласно
представлениям X. Эппингера-Б. Ашнера (1912). PNS — парасимпатическая нервная система;
SNS — симпатическая нервная система
466 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
построили первые схемы взаимоотношении
эндокринных органов и нервной системы,
выглядевшие примерно как на рис. 82.
Однако ряд авторов, и в первую очередь
первооткрыватель липидной природы
гормонов коры надпочечников А. А. Богомолец
(1908), справедливо указывали на
теоретическую недостаточность таких построений,
подчёркивая, что близость или
противоположность того или иного конкретного
эффекта двух гормонов еще не равнозначна
синергизму, либо антагонизму взаимных
влияний соответствующих желёз. А.А.
Богомолец прозорливо указал на возможность
продукции разных гормонов одной железой
и на множественность эффектов гормонов.
Немедленно это было доказано Г. Ивенсом
и Дж. Лонгом, получившим множественные
эффекты от введения аденогипофизарной
вытяжки крысам (1921). Оценивая
совокупность эндокринологических знаний
того времени, А.А. Богомолец писал:
«Эндокринная система менее всякой другой
физиологической системы соответствует
понятию системы вообще» (1932). Особое
значение он придавал полученным его
учеником Л.Р. Перельманом (1924) данным
о пермиссивном действии гормонов (см.
т.I, стр.55). Л.Р.Перельман показал, что
предварительная кастрация у котов и
самцов-собак снимает судорожный синдром,
вызванный удалением паращитовидных
желёз, несмотря на то что уровень кальция
в крови кастрированных паратиропривных
животных остаётся низким. Таким образом
«конечный результат опыта определяется
физиологическим состоянием
периферического органа... Состояние периферии
оказывается решающим моментом в
реакции её на гормональные и нервные
влияния» (А.А. Богомолец, 1937). Через 14 лет
Д. Ингл и Дж. Энгел пришли к аналогичной
идее (1951). Ныне представление о
пермиссивном действии одного гормона на
эффекты другого — один из краеугольных камней
эндокринологии. Эффект одного
биорегулятора может меняться под действием
другого — на пострецепторном уровне, на эффек-
торном уровне или путём влияния одних
гормонов на экспрессию рецепторов других.
Нарушения пермиссивного действия
гормонов способны приводить к патологии. Так,
глюкокортикоиды контролируют
экспрессию катехоламинового рецептора и пермис-
сивно влияют на концентрации цикло- АМФ
в клетках, облегчая действие катехоламинов
и на пострецепторном уровне. Поэтому в
условиях гипокортицизма адреналин не
оказывает должного гликогенолитического
действия, и болезнь Аддисона протекает с
тенденцией к гипогликемии. В то же время
гиперкортицизм усиливает гипертензивное
действие катехоламинов, что имеет значение
в патогенезе многих форм повышения
артериального кровяного давления.
Пермиссивность означает, что в общей
эндокринной оркестровке, помимо сумма-
ции, прямого антагонизма или синергизма,
имеет место важнейшее явление, когда
гормон действует не просто в качестве
комплементарного рецептору сигнала-триггера,
но и выполняет роль символа, включающего
пакет плейотропных эффектов, создающих
сцену или контекст для интерпретации
других сигналов. Логика нервного регулятора
бинарна и дискретна, но «химический
компьютер» эндокринной системы оперирует
более многозначными символами, чем
просто «ноль и единица» — то есть сигнал и его
отсутствие! В области доминирования
нервной регуляции «работают» усредняющие
формы контроля, и применимы широкие,
ступенчатые регуляторные характеристики
типа «симпатический тонус» и
«парасимпатический тонус». Но эндокринная
регуляция индивидуализует своё воздействие
на конкретную мишень в определенный
момент ее онтогенеза и поэтому достигает
большой многогранности. Огромную роль
для перехода к новому уровню
представлений о нейроэндокринном взаимодействии и
о сочетании стереотипии и
индивидуализации в нейроэндокринной регуляции имели
работы У. Кеннона и Г. Селье (1915,1936),
приведшие к открытию стресса, а также
идентификация адренокортикальных
стероидных гормонов (Т. Рейхштейн, Э. Кендалл
и соавт.; 1936-1948). Эти аспекты истории
идей в эндокринологии отражены подробно
в т. I данного руководства (с. 55, 522-530).
Истинные нейроэндокринные
взаимоотношения стали выясняться только по мере
установления контролирующей роли
гипоталамуса по отношению к гипофизу.
Б. Ашнер, Ж. Камю и Г. Русси(1912,1913)
заподозрили, что именно повреждение
области подбугорья, прилегающей к гипофизу,
частично обусловливает метаболические
эффекты, приписываемые гипофизэктомии.
В 30-х годах, когда различными авторами
уже были разделены разные гормональные
начала аденогипофиза, супруги Б. и Э. Шер-
рер создали представления о нейросекре-
ции и доказали, что гипоталамус выделяет
несколько белковых гормонов. Г. Харрису
и Д. Джекобсону удалось доказать, что
гормоны гипоталамуса достигают
аденогипофиза по портальной системе и
управляют функциями последнего (1937-1952).
М. Саффран (1955) ввел для этих
регуляторов обозначение «рилизинг-факторы»
(ср. современный термин — либерины). В
последующие три десятилетия многие из
них, а также гипоталамические
тормозные гормоны — «ингибитинг- факторы»
(ср. современное — статины), которых
оказалось меньше, чем рилизингов, были
очищены и идентифицированы (Р. Гий-
мен, Э.В.Шалли, 1977). Выяснилось, что
нанограммовые количества посылаемых в
портальный кровоток либеринов и стати-
нов управляют функцией (и в известных
пределах — функциональной дифферен-
цировкой) аденогипофизарных клеток,
которые выделяют в системный кровоток
микрограммовые количества тропных
гормонов регулирующих пролиферацию,
дифференцировку и секрецию клеток ряда
периферических желез внутренней
секреции. В то же время В. Дю Виньо выяснил
нонапептидную природу гипоталамических
гормонов, посылаемых через нейрогипофиз
прямо в системный кровоток (1954) — см.
также т. I, с. 57. Сформировалось
представление о высшем нейроэндокринном
интеграторе и передатчике — гипоталамо-
гипофизарной системе (современный тер-
Нарушения эндокринной регуляции 467
мин — гипоталамо-гипофизарный нейросек-
реторный аппарат, см. т. I, с. 530-545).
Еще М.М. Завадовский (1933)
выдвигал концепцию «плюс-минус
взаимодействий» — то есть отрицательных обратных
связей как основы взаимоотношений
эндокринных желёз. Развивая эти поиски,
А. Минз создал представления о
«гипофизарно-тироидной оси», объединяющей эти
железы (1943). Однако явственно теория
обратных связей как формы
взаимодействия гипоталамо-гипофизарного нейро-
секреторного аппарата и периферических
эндокринных желез, подконтрольных
гипофизу, оформилась только после
создания и популяризации основ кибернетики
(Н. Винер, 1948). Практически немедленно
после выхода в свет винеровской
«Кибернетики: науки об управлении в животном и
машине» Р. Хоскинз (1949) объединил идеи
обратной связи и гипофизарно-тироидной
оси в своей концепции «тиростата», а позже
Г. фон Эйлер и Б. Хольмгрен
экспериментально доказали реальность тироксинового
игибирования секреции аденогипофизарно-
го ТТГ (1957). В дальнейшем представления
о сервоконтроле гормональйых функций
стали одной из основ эндокринологии.
В настоящее время твёрдо установлено,
что принцип обратной связи, когда
регулируемый параметр оказывает обратное влияние
на продукцию гормонального
регулирующего сигнала, служит убиквитарной основой
самоуправления в эндокринной системе.
Регулируемым параметром может быть
концентрация гормона в крови.
Повышение уровня гормона периферической
эндокринной железы может понижать
секрецию тропного гормона
аденогипофиза или либерина гипоталамуса {длинная
петля обратной связи). Возможны
подобные соотношения между концентрациями
тропного гормона и рилизинг-фактора
(короткая петля обратной связи). Наконец,
сам тропный гормон или либерин понижают
собственную продукцию (ультракороткая
петля обратной связи). В роли
регулируемого показателя может выступать и
содержание какого-либо метаболита, подавляющее
468 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
по принципу сервомеханизма продукцию
гормона, повышающего такой показатель.
Пример — взаимоотношения концентрации
кальция и секреции парат-гормона, уровня
глюкозы и гормонов островков Лангерганса,
калий-натриевого соотношения в плазме —
и секреции альдостерона, осмоляльности
внеклеточной жидкости — и продукции
вазопрессина.
Гормоно-рецепторное взаимодействие
двусторонне. В течение первой половины
XX столетия внимание эндокринологов
было приковано к изменениям количества
гормонов как причине заболеваний. Но в
1942 г. Ф. Олбрайт описал псевдогипопа-
ратироз, связанный с неотвечаемостью
тканей на парат-гормон, вырабатываемый в
более чем достаточных количествах. Таким
образом, был заложен первый камень в
основание «эндоэндокринологии» или учения
о патологических последствиях рецептор-
ных и пострецепторных дефектов клеток-
мишеней. Впоследствии были открыты
рецепторные и пострецепторные формы
тканевой резистентности практически ко
всем известным гормонам. Развитие
представлений о рецепторах гормонов позволило
распространить принцип сервоконтроля и
на гормоно-рецепторные взаимоотношения.
Так, при гиперинсулинизме, вызванном
перееданием, по механизму сервоконтроля
подавляется экспрессия инсулиновых
рецепторов, и могут формироваться инсули-
норезистентность и сахарный диабет тучных
(см. гл. 9,14).
Наряду с отрицательными обратными
связями в отдельных случаях во
взаимоотношениях центральных и периферических
эндокринных образований были
обнаружены проявления положительной обратной
связи. Так, при овуляции эстрогены,
синтезируемые под влиянием гонадотропинов,
способствуют пиковому повышению
концентрации Л Г и ФСГ.
Представления об обратных связях в
ауторегуляции эндокринной системы имеют
большое значение для диагностики и
лечения эндокринопатии. В соответствии с ними
эндокринные заболевания можно разделить
на первичные эндокринопатии и вторичные
эндокринопатии.
При первичных поражениях
патологический процесс, вызвавший нарушения
гормональной регуляции (аутоиммунное
повреждение, воспаление, опухоль,
генетический дефект) локализован в самой железе.
При вторичных эндокринопатиях начальное
поражение (которое также может
принадлежать к одной из перечисленных абзацем
выше категорий) находится в пределах гипо-
таламо-гипофизарного нейросекреторного
аппарата, а дисфункция периферической
эндокринной железы является следствием
этого процесса. Принцип обратной связи
накладывает отпечаток и на основные
подходы к лечению эндокринных болезней
и на их диагностику. В области лечения
приходится считаться с тем, что более или
менее длительное назначение экзогенного
гормона периферической железы подавляет
продукцию гипофизарных и гипоталами-
ческих стимуляторов её деятельности.
Поэтому резкое прекращение заместительной
терапии провоцирует синдром отмены
(кортикостероиды при гипокортицизме).
Более того, назначение
гормонального'регулятора вызывает ответ, понижающий в
той или иной мере чувствительность
организма к нему (см. раздел «Патофизиология
сахарного диабета» — о явлении вторичной
инсулинорезистентности). Первым
сформулировал это в виде «принципа обратной
чувствительности» Дж. Б. Коллип:
«Индивидуальная чувствительность к вводимым
гормональным препаратам обратно
пропорциональна содержанию или продукции
гормона в собственной железе» (1934).
Еще более важные последствия диктует
феномен обратной связи для
дифференциальной диагностики эндокринопатии.
Очевидно, что первичные и вторичные
эндокринопатии очень часто могут быть
разграничены по противоположным
изменениям концентраций тропных гормонов
(или иногда — гипоталамических факторов)
в крови. Так, при первичном гипотирозе,
вызванном йодной недостаточностью, гипо-
таламо-гипофизарный аппарат, повинуясь
Нарушения эндокринной регуляции 469
принципу обратной связи, отвечает на
низкое содержание тироксина и трийодтирони-
на в крови повышением продукции ТТГ. В
то же время вторичный гипотироз,
вызванный гипопитуитаризмом, протекает при
низкой концентрации как тироидных, так и
тиротропного гормонов. При болезни фон
Базедова (первичный гипертироз и
гиперплазия щитовидной железы аутоиммунного
происхождения) содержание ТТГ в крови
понижено, в то время как при вторичном
базедовизме, спровоцированном опухолями
гипоталамо-гипофизарной локализации,
вырабатывающими стимуляторы тироид-
ной функции, повышены и уровень ТТГ, и
уровень тироидных гормонов. Классическая
болезнь Аддисона — первичная хроническая
недостаточность коры надпочечников —
вызвана инфекционным, либо аутоиммунным
адреналитом, а изредка — атрофией коры
надпочечников аутоиммунного генеза.
Поэтому продукция проопиомеланокортина
(ПОМК) — прогормона, содержащего
центральный стимулятор адренокортикоцитов,
АКТГ, — компенсаторно возрастает. Так как
продуктом протеолиза
проопиомеланокортина является и МСГ, кожа больных
приобретает бронзовый оттенок и наблюдается
характерный «симптом грязных локтей».
Если гипокортицизм имеет центральное
происхождение и относится к вторичным
эндокринопатиям, то в силу первичного
гипопитуитаризма продукция
проопиомеланокортина и его дериватов, наоборот,
понижается и гиперпигментации кожи не
наблюдается, напротив — возможен даже
обратный процесс («белый аддисонизм»).
Легко заметить, что это создает возмож-
h
«» о
о w
г 2
a. «d
S
О
С
8
I
первичные
эндокринопатии
периферических
желез
гипопитуитаризм
норма
периферическая
гормонорезистентность
либо
автономная секреция
гипофизарных гормонов
(например, опухолью)
автономная секреция
периферических желез
(например, опухолью)
>-
N высокая
концентрация гормона периферической железы
Рис 83. Роль принципа обратной связи в распознавании первичных и вторичных эндокринопатии
Пояснения в тексте.
470 АШ. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
ность для точной дифференциальной
диагностики, если эндокринолог располагает
прецизионными данными о концентрациях
гормонов в крови (рис. 83).
К сожалению, до конца 60-х годов XX
века это «если» было весьма существенным
ограничением в эндокринологии. Насколько
приблизительны были представления об
истинном содержании гормонов при тех или
иных состояниях, можно судить по тому
обстоятельству, что классик эндокринологии
30-х годов Г. Цондек устанавливал факт
беременности, используя биотест, по
потемнению спинки лягушек при добавлении
в воду женской мочи.
Неудивительно, что природа многих
эндокринопатий из-за этого длительное
время представала в извращённом свете.
Так, болезнь фон Базедова считалась
расстройством центрального происхождения,
так как имелись отдельные больные, у
которых подобный симптомокомплекс
наблюдали при опухолях области турецкого
седла. Проверить, как же обстоит дело с
секрецией ТТГ у большинства больных,
не было никакой возможности, хотя, по
современным данным, гипертироз носит
вторичный характер не более чем в 5 %
случаев (И. Чопра, Д.Соломон, 1983).
Революционизирующую роль в эндокринологии
сыграла разработка иммуноконкурентных
методов определения концентраций
биологически активных веществ, сначала — на
основе радиоактивной метки (радиоимму-
ноассей — РИА), а затем — с применением
других, более безопасных методов мечения
гормонов и антител (иммуноферментный
анализ — ИФА, иммунохемилюминесцен-
тный анализ — ИХЛА). Родоначальниками
этих методов были С.Берзон и Р. Ялоу
(1968), искавшие способ установления
причин инсулинорезистентности при сахарном
диабете.
Принцип подобной методики отображен
на рис. 84 и состоит в конкуренции между
известным количеством меченого
биорегулятора и неизвестным количеством на-
тивного биорегулятора за антитела к нему.
Чем больше проба биологической жидкости,
полученная от обследуемого индивида,
содержит гормона, тем меньше меченого
гормона включается в иммунный комплекс.
Если затем осадить иммунные комплексы и
измерить количество метки в надосадке, то
можно прецизионно оценить содержание
гормона в биологической пробе,
предварительно построив калибровочную кривую
на основании измерений со стандартами,
содержащими фиксированные количества
гормона.
Немедленно после введения РИА общие
представления об эндокринной патологии
пришлось корректировать. В одном из
исследований, посвященных измерению
концентрации паратгормона, авторы
метода РИА, в качестве контрольных проб,
от субъектов с заведомо интактной, как
им казалось, паратироидной функцией,
использовали кровь больных раком лёгких.
Каково же было их изумление, когда у
пациентов с овсяноклеточным раком лёгких
концентрации иммунореактивного
паратгормона оказались необычайно высоки.
Таким образом, была конкретно доказана
возможность существования внежелезистых
эндокринопатий — нарушений продукции
и/или действия гормонов, не связанных
с первичными поражениями ни самих
эндокринных желёз, ни регулирующих их
деятельность приборов. К заключению о
возможности существования подобных
расстройств теоретически пришёл еще Г.
Цондек, писавший, что: «Функциональные или
анатомические изменения эндокринных
желёз не всегда должны рассматриваться
как первичная причина заболеваний. Во
многих случаях эти изменения являются
лишь реакцией на болезненные процессы,
локализованные в других органах» (1935).
В вышеприведённом примере продуцентами
пептида, родственного паратгормону
оказались опухолевые клетки-апудоциты.
Внежелезистыми могут оказаться
эндокринопатий, вызванные эктопической
продукцией гормонов, за пределами
основной образующей их железы. Кроме того, к
внежелезистым принадлежат и нарушения
эффективности действия гормонов, вы-
__/ — антитела к гормону
х / — меченый гормон
——/ — нативный гормон
инкубация \ 1 '
-< + >-
>
V
1
X
Рис. 84. Принцип иммуноконкурентных методов определения гормонов. Пояснения в тексте
званные их связыванием и инактивацией
циркулирующими антагонистами,
нарушениями рецепции гормона в тканях-мишенях
и пострецепторными дефектами таргетных
клеток. Примеры подобных ситуаций даны
в разделе «Патофизиология сахарного
диабета» при обсуждении разнообразия его
инсулинонезависимого типа и механизмов
инсулинорезистентности. Эндокринные
расстройства могут возникать и при
аномальной скорости разрушения гормона.
Так, замедление клиренса альдостерона и
половых стероидов в печени при
хронической печеночной недостаточности, например
вследствие цирроза, ведет к вторичным
внежелезистым гиперальдостеронизму,
гиперэстрогенизму — с соответствующей
Нарушения эндокринной регуляции 471
симптоматикой (отёки, гинекомастия и
т.д.).
Становление точных методов измерения
гормональных концентраций, сделавшее
эндокринологию «зрячей», вновь привлекло
внимание к старой эндокринологической
идее, которую А.А. Богомолец (1937)
формулировал следующим образом: «...все
органы и клетки организма обладают
эндокринной функцией». ВI томе данного
руководства (с. 62-66, рис. 8) читатель может
найти подробную характеристику отличий
между разными способами химической
сигнализации в организме (панокрин-
ным, паракринным, специализированным
паракринным, наружным и внутренним
аутокринным, юкстакринным, нейромеди-
472 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
аторным, нейроэндокринным и, наконец,
собственно эндокринным). В настоящее
время очевидно, что какой-либо (или сразу
несколько) из этих способов гуморальной
регуляции присущи практически каждой
клетке. Эндокринология, пройдя навеянную
нервизмом стадию, когда за определёнными
клетками или органами признавали
исключительную способность выделять гормоны,
вернулась к точке зрения своего
основоположника Т. Бордю и подтвердила, что в
организме нет монополии на гуморальную
регуляцию, а клетки и управляют с
помощью химических сигналов, и управляются
ими. Филогенетически и гормоны, и их
рецепторы возникают очень рано, многие
из них имеются у одноклеточных задолго
до появления эндокринных желёз и нервной
системы (принцип эволюционного оппор^
тунизма или структурно-функциональных
блоков — по Р. Гиймену и A.M. Уголеву,
1984). Принципиально генетические
программы гормонообразования имеются в
каждой клетке.
Конечно, собственно выделение гормонов
системного или дистантного действия в
кровь и лимфу (эндокринная сигнализация)
присуще не любой клетке, но и не
сосредоточено лишь в особых железах, как полагал
Й. Мюллер. Само по себе становление этой
идеи оказалось возможным вследствие
появления точных иммуноконкурентных
и иммуногистохимических методов
обнаружения гормонов и зависело от открытия
диффузной (дисперсной) эндокринной
системы.
Отход от традиционной догмы,
приурочивавшей продукцию каждого гормона к
одному анатомически определимому
образованию, совершился на основе прогресса тех
исследований, которые привели к открытию
секретина и гастрина (см. выше). Э.Г. Стар-
линг предсказал, что гормонов ЖКТ вскоре
будет открыто множество. Обнаружение
гормоносодержащих экстрактов различных
пищеварительных органов, обладающих
подобными свойствами, не заставило себя
ждать. Были найдены (по их эффектам)
холецистокинин-панкреатозимин (А. Айви
и Э. Олдберг, 1927, А. Харпер и Г. Рэйпер,
1943), вещество Р (Г. фон Эйлер и Дж. Гед-
дам, 1931), обнаружена инсулинотропная
активность пептидов двенадцатиперстной
кишки (Ж. Ля Барр, Ж. Ле Дрю, 1935).
Однако только развитие химии пептидов
позволило в 60-70-х годах XX века
выделить гастроинтестинальные пептидные
гормоны в чистом виде. Тем временем в 1938 г.
гистолог Ф. Фейртер обнаружил множество
разнообразных светлых эпителиальных
клеток с признаками гормональной секреции
в слизистых оболочках органов
пищеварения, сформулировал понятие о диффузных
эндокринных эпителиальных органах и
ввел представление о паракринном
(действующем на непосредственных соседей)
химическом сигнале. Подобные клетки
были обнаружены позже yi вне ЖКТ — в
слизистых оболочках бронхов, щитовидной
железе, почках, островках Лангерганса и
других органах. К этой же категории были
причислены и пинеалоциты эпифиза.
Обобщая эти данные, А.Дж.Э.Пирс (1968)
выдвинул концепцию APUD-системы и назвал
подобные диффузные эндокриноциты «апу-
доцитами» (от англ. amine precursor uptake
and decarboxylation — захват и декарбокси-
лирование аминных предшественников),
имея в виду, что эти клетки способны к
синтезу гормонов — производных аминокислот.
Впоследствии оказалось, что им присуща и
продукция аминов, и выработка регулятор-
ных пептидов, причём часто вопреки догме
«одна клетка — один гормон» — в пределах
одного клеточного типа. Так, В-клетки
островков Лангерганса выделяют и
инсулин, и ГАМ К. Все эти клетки имеют черты
эндокриноцитов и паранейронов, обладают
маркерами: хромогранином А, ферментами
декарбоксилазой и нейрон-специфической
енолазой, а также, по существу, являются
нейроэндокринными (Ю.А. Малашхия
и соавт., 1990). Первоначально А. Пирс
постулировал, что все они расселяются по
организму из нейроэктодермы нервного
гребешка, но позже А. Эндрю доказала, что
диффузные эндокриноциты могут иметь
также эндодермальное и мезодермальное
происхождение (1981). Окончательное
формирование концепции диффузной гас-
троэнтеропанкреатической эндокринной
системы произошло к середине 80-х годов
в работах Ч. Вейля (1985). Хотя в
настоящее время данная концепция не свободна
от противоречий с новыми фактами (не
все апудоциты могут производить
пептидные гормоны, многие из них не связаны
эмбрионально с нейроэктодермой, ряд гор-
монообразующих эктопических опухолей
не имеет апудоцитарных характеристик),
не приходится отрицать, что она сыграла
в развитии современной эндокринологии
важнейшую роль.
Принципиальное значение имело
неожиданное наблюдение, что практически
все пептидные гормоны, первоначально
найденные в ЖКТ (гастрин, вещество Р,
холецистокинин, ВИП, инсулин, глюка-
гон и другие), содержатся также и в ЦНС.
И, напротив, некоторые первично открытые
в ЦНС нейропептиды (соматостатин, ней-
ротензин, эндогенные опиаты и пр.) позже
были найдены как инкреторные продукты
диффузных эндокриноцитов кишечника и
островков Лангерганса. Более того,
выяснилось, что эти пептиды, будучи гормонами
в ЖКТ, служат нейротрансмиттерами в
пептидэргических системах мозга или
выделяются в безимпульсном режиме после
антидромного распространения по
афферентным нервным проводникам теми же
нейронами, которые оперируют и аминными
нейромедиаторами (Г.Дж. Докрей, 1978;
Дж.М. Поляк, С.М.Блум, 1977). Стали
понятны механизмы эктопических
эндокринных синдромов, наподобие описанного
выше овсяноклеточного рака лёгких, когда
гормоны в избытке вырабатываются
опухолями из апудоцитов (апудомами). В ЖКТ
(в основном аппендиксе и подвздошной
кишке) подобные опухоли — карциноиды
встречаются особенно часто в силу того, что
в пищеварительных органах найдено не
менее 15 типов диффузных нейроэндокринных
клеток (табл. 30). Отметим, что из
перечисленных в ней регуляторов лишь глюкозоза-
висимый полипептид пока не зафиксирован
Нарушения эндокринной регуляции 473
в ЦНС в качестве пептидэргического ней-
ротрансмиттера — остальные же обладают
«кишечно-мозговым» дуализмом.
Это относится не только к пептидам. По
производству индоламинового гормона
мелатонина диффузные эндокриноциты
ЖКТ опережают эпифиз (И.М. Кветной,
B.C. Хавинсон, 2001).
В настоящее время считают, что одни и
те же пептиды служат в мозге глобальным
средством химического кодирования тех
форм нейронной активности, которые
связаны с отдельными древнейшими
функциями (половым и пищевым поведением,
поддержанием водно-электролитного баланса,
позитивным и негативным подкреплением,
памятью), а в ЖКТ используются локально
как паракринные аутокоиды или гормоны.
Более того, по У. Кэннону и А. Розенблю-
ту (1951), антидромное распространение
и секреция афферентными нервами таких
нейропептидов служит реальной основой
явления «нервной трофики», суть которого
долго оставалась неясной, несмотря на то
что оно привлекало внимание неврологов
и эндокринологов со времен Ф. Мажанди
(1824) и до работ такого активного
исследователя этого феномена, каким был
А.Д.Сперанский (1935). А.Дж.Э.Пирс
прямо заключил, что « апудоциты
продуцируют пептиды, действующие и как гормоны,
и как нейромедиаторы. Они действуют как
эффекторы третьего звена, поддерживая
или модулируя функции соматических и
вегетативных нейронов, фактически
служа трофическими субстанциями как для
нейронов, так и для соматических клеток»
(1977). Всё это рассматривают как прямой
результат эволюционного оппортунизма по
Р. Гиймену — то есть приспособления архе-
типических блоков к новым функциям. Как
указал Г.Кобата (1982), гормоны —
древняя форма коммуникации клеток, поэтому
диффузная эндокринная система, вероятно,
возникла задолго до компактных желёз
внутренней секреции.
С открытием дисперсной эндокринной
системы стали предметными вопросы эн-
474 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 30. Гормоны гастроэнтеропанкреатической части диффузной
эндокринной системы. Сокращения: Ж—желудок, ТК—тонкий кишечник, ПЖ—поджелудочная
железа, ТлК—толстый кишечник, остальные сокращения — как в тексте
Тип клеток
р
DI
ЕС
ECL
G
1
к
LFG
Мо
N
Р
S
А
В
PP(F)
Локализация
Желудок (Ж), тонкий
кишечник (ТК),
поджелудочная железа (ПЖ)
Ж,ТК,ПЖ, толстая
кишка (ТлК)
Ж/ГК,ТлК
Ж
Ж,ТК
ТК
ТК
ТК,ТлК
ТК
ТК
Ж/ГК
ТК
пж,ж
ПЖ
пждк
Вырабатываемые
биорегуляторы
Соматостатин
ВИП
Серотонин
Серотонин, гнетами н,
дофамин
Гастрин, опиаты
Холецистокинин
Глюкозозависимый
инсулинотропный
пептид
Глюкагоноподобный
пептид 1, другие
дериваты глюка гона
Мотилин
Нейротензин
Бомбезиноподобный
пептид
Секретин
Глюкагон
Инсулин, ГАМ К,
амилин, С-пептид,
проинсулин
Панкреатический
полипептид
Эффекты в ЦНС
Понижение продукции СТГ, механизмы
анабиоза
Медленный сон, стимуляция продукции СТГ и
пролактина, подавление полового поведения
Усиление стресса, циркадный ритм кортико-
либерина, формирование положительных
эмоций, механизмы сна, мозговое
кровообращение
Дофамин — пролактостатин, модулятор
механизмов стресса, гистамин—- см. т. 1, с. 504-505
Гастрин — стимулятор продукции СТГ,
ингибитор продукции ТТГ и ЛГ, участник механизмов
долговременной памяти. Опиаты — см. т. 1,
С. 505-506,530-534,550-S53
Насыщение, подавление исследовательского
поведения, дремота, анальгезия, регуляция
быстрого и медленного сна, активатор сомато-
маммотрофной функции
Не найден в ЦНС
Участник механизма лептиновой регуляции
аппетита
Активатор голода и пробуждения
Индуктор пищевого поведения, гипотермии,
гипотензии, ингибитор продукции ТТГ, СТГ,
пролактина, блокирует груминг у крыс
Насыщение, антидиурез, гипотермия, груминг
у крыс
Не установлены |
Не установлены
Не установлены для инсулина. ГАМК —
широко распространенный тормозный медиатор
Гипотермия, подавление одышки
докринной функции сердца (атриальный
натрийуретический полипептид, кардио-
депрессорный полипептид), почек (ренин,
эритропоэтин, производные кальциферола),
печени (ангиотензиноген, производные
кальциферола, гепцидин) и других органов.
Оказалось, что многие диффузные эндо-
криноциты выделяют прогормоны-пред-
шественники, а активный гормон вовсе не
обязательно вырабатывается в конкретной
железе, а может даже формироваться вне
клеток, в крови. Примером служат ангио-
тензины II и III, предшественник которых
возникает в печени и диффузных эндокри-
ноцитах разной локализации, а активные
гормоны образуются прямо в плазме, за счёт
протеолитического эффекта почечного
ренина и лёгочной ангиотензин-конвертазы.
Наконец, можно сказать, разрушилась та
искусственная «берлинская стена», которая
делила непримиримых представителей
лагеря нервистов и лагеря гуморалистов,
сломавших немало копий в попытке выяснить
«кто главнее в организме?». Мозг оказался
de facto крупнейшей эндокринной железой,
а обе формы регуляции — двумя
сторонами нейроэндокринной сети, в которой их
функции не конкурируют, но делят сферы
преимущественной применимости и
взаимодействуют. Очень показательны в этом
свете некоторые данные, не исключающие
общего эмбрионального происхождения
гипоталамуса и всего гипофиза (У.Х. До-
хедэй, 1979).
Дальнейшее развитие эндокринологии
поставило вопрос о том, что иммунная
система, функции которой также основаны на
комплементарном взаимодействии
химических сигналов и рецепторов, не остаётся в
стороне от нейроэндокринного аппарата.
В 60-е годы прошлого века была раскрыта
эндокринная роль тимуса, а позже стало
ясно, что эта уникальная эндокринная
железа не только посылает в системный кровоток
типичные гормоны (тимозин и тимулин), но
и служит на протяжении раннего онтогенеза
местом паломничества лимфоидных
клеток-мишеней, которые, проходя через нее,
подвергаются действию множества местных
Нарушения эндокринной регуляции 475
паракринных и юкстакринных гуморальных
регуляторов-цитокинов, определяющих
пути их дифференцировки (Дж. Миллер,
1961 — подробно см. т. I, с. 397-399). Затем
стало ясно, что цитокины лимфоидных и
макрофагальных клеток способны изменять
функции гипоталамуса и гипофиза, а также
обладают гормоноподобным действием
на периферические эндокринные железы.
Более того, оказалось, что гормоны
(особенно пептидные) и их рецепторы часто
служат объектом аутоиммунного ответа, а
при патологии — мишенью аутоаллергии.
Из-за гомологии многих биорегуляторов и
рецепторов бактерий и высших организмов
подобная аутоаллергия, имеющая мишенью
эндокринную систему, служит частой
причиной первичных, вторичных, железистых
и внежелезистых эндокринопатий.
Аутоаллергия оказалась одним из самых частых и
важных механизмов эндокринных болезней.
Так, например, рецептор микроорганизма
Yersinia enterocolytica, подобный тиротро-
пиновому рецептору фолликулярных
клеток щитовидной железы, вызывает у
индивидов, предрасположенных к аутоаллергии,
перекрёстный аутоиммунный ответ,
поражающий тироциты и приводящий к болезни
фон Базедова (Л.Хариссон, 1984). Было
показано существование в норме, при
патологии и в экспериментальных условиях ан-
тиидиотипических антител, полностью или
частично имитирующих либо блокирующих
действие пептидных и аминокислотных
гормонов на клетки-мишени, появились данные
о гормоноподобном действии цитокинов
(см. т. I, с. 471 -473). Все эти факты убедили
современных эндокринологов, что
иммунная система, биорегуляторными сигналами
которой служат цитокины и антитела, в
норме и при патологии включается в ней-
роэндокринные взаимодействия, вследствие
чего сложилась концепция иммунонейроэн-
докринного коммуникативно-регуляторного
интегрирующего апарата (Е.А. Корнева,
1987, 2003; Х.О. Безедовский, 1989).
Появилась даже оригинальная точка зрения, что
некоторые лимфоциты представляют собой
своего рода циркулирующую разновидность
476 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
апудоцитов — теория неироиммунного диф-
ферона (В.В. Абрамов, 1986).
Большой путь от основополагающих
работ отечественных ученых И.Г. Савченко
(1891), Е.С. Лондона (1899) и СИ. Металь-
никова (1925) и до признания важной роли
нервной и прежде всего гипоталамической
регуляции иммунных процессов прошла
нейроиммунология (см. новейший обзор
Е.А. Корневой, 2007).
Подверглись эволюции и классические
представления о биохимических
механизмах продукции и действия гормонов, а также
общей этиологии и патогенезе эндокрино-
патий.
МЕТАБОЛИЗМ ГОРМОНОВ
И ОБЩИЕ МЕХАНИЗМЫ
ЭНДОКРИННЫХ НАРУШЕНИЙ
В настоящее время насчитывают около 100
гормонов млекопитающих. Химически они
подразделяются на три группы:
- пептиды и гликопротеиды,
- производные тирозина (возможно,
учитывая индоловый характер серотонина
и мелатонина, правильнее формулировка
«производные аминокислот»),
- дериваты холестерина.
Важно отметить, что одна и та же клетка
может вырабатывать одновременно или
на разных стадиях своего онтогенеза
разные гормоны одной химической группы, а
иногда — даже гормоны разных химических
групп (например, см. т. I, с. 546-547 — об
эскалаторной теории гистогенеза
надпочечников).
Биосинтез гормонов, по современным
данным, характеризуется следующими
особенностями.
Пептидные гормоны образуются по схеме
препрогормон — прогормон — активный
гормон, иллюстрируемой на рис. 85.
Вначале образуется гетерогенная ядерная
РНК, содержащая транскрипты экзонов и
интронов, затем при сплайсинге интроновые
копии удаляются, из экзоновых же
формируется м-РНК препрогормона, снабжённая
полиадениловым хвостом, длина которого
определяет срок ее жизни в клетке. На её
основе путём трансляции возникает
препрогормон, имеющий сигнальную
универсальную последовательность HaN-конце,
необходимую для переноса молекулы внутрь ШЭР.
Далее, в комплексе Гольджи, происходит
протеолитическое удаление этого
сигнального пептида, как правило, по связям между
аргининовыми или аргининовым и лизи-
новым остатками. Образуется прогормон
(например, проинсулин — см. рис.86, или
прокальцитонин), а иногда — сразу гормон
(например, пролактин или гормон роста).
Прогормон пакуется в секреторные
пузырьки и непосредственно в них, или в пределах
эндоплазматического ретиКулюма
происходит дальнейший протеолиз, с образованием
активного гормона. Так осуществляется,
например, вырезание С-пептида (рис. 86)
с формированием инсулина. Впрочем,
часто прогормон секретируется как таковой
вместе с готовым гормоном, а протеолиз и
вычленение активного гормона (гормонов)
идёт частично на периферии, причём по-
разному в различных тканях или на разных
стадиях гормонального ответа. Примером
может служить процессинг гипофизарного
проопиомеланокортина в надпочечниках
при стрессе. Некоторые пептидные гормоны
(ТТГ и все гонадотропины) перед секрецией
гликозилируются в аппарате Гольджи, что
удлиняет время их жизни в крови.
Пептидные гормоны, как содержащие одну,
так и имеющие две субъединицы, могут
кодироваться одним геном (инсулин, парат-
гормон) или двумя генами (хорионический
гонадотропин, ЛГ). Иногда один гормон
в разных органах может получаться из
различных предшественников, закодированных
неидентичными генами (соматостатин).
Часто, наоборот, один прогормон служит
предшественником нескольких гормонов,
получаемых из него путем
посттрансляционного протеолиза. Например, проопиоме-
ланокортин аденогипофиза служит сырьем
Нарушения эндокринной регуляции 477
ГЕН:
фланкирующая
ДНК
Пре-мРНК
процессию РНК
мРНК
препрогормои
для продукции АКТГ, р-ли-
потропина, а-МСГ, р-МСГ,
а-р-у-эндорфинов, энкефа-
лина и кортикотропинопо-
добного промежуточного
полипептида (см. т. I, стр.
542-543).
После транскрипции
одних и тех же генов
возможен альтернативный
сплайсинг гетерогенной
ядерной РНК с
образованием различных м-РНК,
приводящих в дальнейшем к
трансляции неидентичных
гормонов в разных тканях.
Так, С-клетки
щитовидной железы формируют
преимущественно кальци-
тонин, а ЦНС — пептид,
связанный с геном кальци-
тонина, — в обоих случаях,
на основе общего
транскрипта препрокальцитони-
на. Важной особенностью
характеризуется синтез
пептидных либеринов
гипоталамуса. Показано, что
образование многих из них
(тиролиберина, гонадо-
либерина, соматолибери-
на и пролактин-рилизинг
фактора) идёт в цитозоле
нейросекреторных клеток,
нерибосомальным путём.
Оно не чувствительно к
пуромицину и РНК-азе
и напоминает механизм
формирования глутатиона
и антибиотических
пептидов у микроорганизмов. Для осуществления
этих процессов служит набор АТФ-зависи-
мых цитоплазматических либерин-синтетаз
(Л.Гэррин и соавт., 1971).
Стероидные гормоны синтезируются по
совершенно иному пути. Сырьём служит
холестерин (в коре надпочечников и гонадах —
при образовании минералокортикоидов,
интрон
v
интрон интрон
транскрипция (ядро)
\
4 4%
интроновые копии
хллллллаллалХ^^а)
\ЛЛЛЛЛ/ ^ '
1 трансляция (рибосомы)
]
(ШЭР)
активный
гормон
фрагменты
(КОМПЛЕКС
ГОЛЬДЖИ)
секреторные пузырьки
кровь
Рис. 85. Общая схема образования полипептидных гормонов.
Поли-А — полиаденилат. Остальные обозначения — как в тексте
глюкокортикоидов, прогестинов, андрогенов
и эстрогенов), либо 7-дегидрохолестерин (в
коже, а затем — печени, стимулированных
макрофагах и почках, при образовании
кальциферола). Превращения стероидов состоят
в отщеплении алифатической боковой цепи,
гидроксилировании и дегидрировании, либо
ароматизации А-кольца. Читатель может
обратиться к схеме этих процессов (рис. 114,
стр. 548 в т. I данного руководства и рис. 112
478 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
56
55
54
53
52 51 50 49 48
47
46
45
44
<Лей}
-J3
59/
ТВал
60/
/Про/
.42
41
Алу^о
или).
ч39
ТГли)
\38
(Про,
.!* у
К^У
ш?
1(фен)
мой /♦
>-с А-цепь
2(Йлей)
оГвал)
34-/A-V
ТГлу>Ч
4ч-ТГлиЬ>
5^!!
-♦
соон
>(Вал1
ЛАсп
iTpe
^Т"Р/"27
26
37
Ml
(Ала)з4
С-петид ГГлуШ
JW32
W*31
(АлаТ
ПроГ29
28
Рис. 86. Структура проинсулина, инсулина и С-пептида. Обозначения аминокислот -
традиционные (см. с. 502).
ниже). Находящийся в составе липосом
холестерин и его дериваты претерпевают
многократный перенос в митохондрии и ГЭР,
причём наблюдается характерный контакт
и «мультипликация» мембранных структур,
участвующих в биосинтезе органоидов и
липосом. Так как пути стероидогенеза
предусматривают участие множества ферментов
(например, при продукции эстрогенов — не
менее шести), биосинтез стероидов зависит
от многих генов, включая как те, которые
кодируют ферменты, так и те, которые
кодируют внутриклеточные посредники
действия стимуляторов стероидогенеза —
ключевые белки (А. Дазо, 1983). Поэтому
существует много наследственных дефектов
стероидогенеза, тогда как число
наследственных дефектов продукции пептидных
гормонов невелико. Вместе с тем биосинтез
пептидных гормонов нередко происходит
эктопически, в опухолевых клетках, так
как требует растормаживания одной-двух
генетических программ. Однако биосинтез
стероидов в эктопических опухолях и
неэндокринных тканях не типичен. Стероиды
чаще секретируются в готовом виде.
Возможна и периферическая трансформация
прогормонов стероидного происхождения в
гормоны. Именно так расценивается судьба
витамина D в организме. Прогормон,
полученный при ультрафиолетовом облучении
в клетках кожи, или поступивший с пищей,
превращается в активную дигидроксиформу
гормона с участием печени, почек, а иногда и
активных макрофагов (см. выше
«Патофизиология витаминного обмена»).
Почки могут превращать прогестерон в
дезоксикортикостерон, что имеет значение
при развитии отёков у беременных.
Плацента переводит дегидроэпиандростерон
фетальной зоны коры надпочечников
плода в эстриол (см. т. I, с. 568-569). Эта
способность присуща и трофобластическим
опухолям. Липоциты превращают в эстриол
андрогены, что имеет значение в патогенезе
гиноидной формы ожирения (см. выше
«Нарушения липидного обмена»). Наконец,
большая часть андрогенной активности
принадлежит образуемому на периферии
5-дигидротестостерону, для которого
тестостерон гонад и сетчатой зоны надпочечников
фактически служит прогормоном.
Производные тирозина претерпевают при
биосинтезе катехоламинов и дофамина (в
мозговом веществе надпочечников, парааор-
тальных ганглиях плода и новорожденного,
апудоцитах) гидроксилирование и дека-
рбоксилирование в свободном состоянии
и пакуются в специальные секреторные
гранулы.
В щитовидной железе основные этапы
превращений тирозина на пути к тироидным
гормонам, наоборот, протекают при тирози-
новых остатках, связанных в составе особого
белка тироглобулина (см. рис. 109 ниже).
Тирозилы сначала йодируются в двух
положениях, давая монойодтирозин (МИТ)
и дийодтирозин (ДИТ); затем окисляются
и конденсируются между собой, формируя
йодтиронины — тетрайодтиронин и трийод-
тиронин. Йодтиронины отщепляются от
тироглобулина, который фолликулярные
тироциты захватывают из коллоида
щитовидной железы и секретируются в кровь в
виде тироксина (Т4) и трийодтиронина (Т3).
Т3 возникает из Т4 путём отщепления йода
от наружного фенольного кольца (дейоди-
рование). Т3 намного более активен и для
него фактически Т4 служит прогормоном.
Дейодирование идёт в клетках-мишенях
и представляет дополнительный уровень
регуляции эффективности тироидной
функции (см. ниже с. 552 и далее, и выше
«Голодание»).
Хранение и секреция гормонов в кровь
представляют важный этап их метаболизма,
объект многочисленных расстройств при
патологии.
Наиболее хорошо хранится неполярный
стероид кальциферол. Прекращение поступ-
Нарушения эндокринной регуляции 479
ления и биосинтеза его предшественника в
организме взрослого человека дает эффект
в виде остеопороза только через несколько
месяцев. Тироглобулин щитовидной железы
содержит двухнедельный запас тироидных
гормонов. Его лизис в ходе подострого
тироидита может вызывать выброс в кровь
порций гормонов и эпизоды гипертироза на
фоне общей, свойственной этому
заболеванию тенденции к ограничению
функциональных возможностей железы.
В-клетки островков Лангерганса имеют
инсулина не более чем на 5 дней секреции,
причём его правильная упаковка нарушается
блокаторами цинка, что позволило создать
дитизоновую модель сахарного диабета на
животных (см. «Патофизиология сахарного
диабета»). Другие пептидные гормоны
запасаются в еще меньших количествах.
Практически не запасаются остальные
стероидные гормоны в силу полярного
характера своих молекул. Семенники
содержат тестостерона не более чем 15-17 % от
суточной потребности, поэтому стероидогенез
в них характеризуется высокой постоянной
интенсивностью и легко нарушается при
острых поражениях яичек, например орхите.
Стероидные гормоны освобождаются в
кровь постоянно, на основе разницы в
концентрациях, и связываются с липопротеида-
ми и стероидпереносящими белками.
В отличие от них белковые гормоны и
производные тирозина, как правило,
поступают в кровь неравномерно. Секреция
катехоламинов мозговым веществом
надпочечников происходит активно, в ответ на
симпатический нервный сигнал. Тироидные
гормоны секретируются путем пиноцито-
за и протеолиза тироглобулина, которые
стимулируются как симпатическими
нервами, так и (в основном) гуморальными
сигналами — ТТГ и тиростимулирующими
иммуноглобулинами. Если печень и другие
неэндокринные клетки освобождают свои
белки в кровь тонически, мало реагируя на
специальные стимулы, то для секреции
пептидных гормонов, как правило, требуется
активный экзоцитоз секреторных пузырьков.
Этот процесс предусматривает триггерную
480 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
функцию нервной системы, метаболитов
или, чаще всего других гормонов, работу
цитоскелета, затрату макроэргов (в форме
ГТФ) и важную роль кальция. Именно так
секретируются гормон роста, глюкагон,
инсулин. Рис. 60 (см. выше гл. 9) дает
представление о сложности данного процесса на
примере секреции инсулина.
Секреция этого гормона В-клетками
островков Лангерганса стимулируется
глюкозой, некоторыми аминокислотами
(аргинин, лейцин), глюкагоноподобным
пептидом I и другими энтериновыми
гормонами, особенно желудочным ингибиру-
ющим полипептидом. Секреция инсулина
стимулируется также холинергическими
вагальными и р-адренергическими
симпатическими нервными окончаниями, па-
ракринным глюкагоном, АКТГ и СТГ. В то
же время она тормозится соматостатином,
а-адренергическими влияниями, избытком
самого инсулина, амилином, галанином и
цитокинами. До воздействия секреторных
стимулов выход катионов калия поляризует
мембрану В-клеток и препятствует
входному току ионов кальция через
потенциал-зависимые кальциевые каналы. Цитоплазма-
тический уровень ионизированного кальция
остаётся низким. При действии
секреторного триггера, например захвате глюкозы
В-клеткой, ситуация меняется. Глюкоза
метаболизируется, что ведет к генерации
АТФ, усилению работы К+-Ка+-зависимой
АТФ-азы плазматической мембраны,
снижению выходного калиевого тока и захвату
калия клеткой. Деполяризация приводит
к открытию потенциалозависимых
входных кальциевых каналов, Са2+ входит в
цитоплазму извне и из внутриклеточных
компартментов, где его судьба определяется
цикло-АМФ зависимыми механизмами,
активированными стимулятором инсулиновой
секреции. Кальций влияет на
микротрубочки и миозиновые филаменты и способствует
движению цитоскелета и перемещению
секреторных гранул с инсулином, который
подвергается экзоцитозу, с участием белков-
кальэлектринов, способствующих слиянию
плазматической мембраны и мембран
секреторных пузырьков (фьюзогенный эффект).
Секреция пептидных гормонов, как
правило, тесно сопряжена с их биосинтезом,
который она активирует. Например, глюкоза
вызывает быстрый пик выброса инсулина
в кровь через 10-15 мин, связанный в
основном с процессами, описанными выше,
но затем этот пик переходит в длительное
повышение секреции гормона, которое
достигает плато примерно через час и вызвано
активацией его биосинтеза de novo (рис. 61,
выше, гл. 9). По этим же причинам тропные
гормоны активируют и выброс, и синтез
гормонов клеток-мишеней например, АКТГ
уже через 3-12 мин вызывает выброс в кровь
кортикостероидов, а в более отдалённые
сроки (1-3 ч) под его влиянием развёртывается
картина активации стероидогенеза.
Некоторые гормоны освобождаются в
кровь постоянным, меняющимся по
интенсивности потоком (паратгормон, пролактин,
тироидные гормоны). Но для большинства
характерен импульсный режим секреции,
когда гормон поступает^ кровоток
дискретными порциями-толчками. Инсулин
сочетает пульсовой и постоянный режим
освобождения, но многие другие — АКТГ,
СТГ, гонадотропины, стероиды —
секретируются только импульсно. В последнее время
обнаружилось, что толчковой дискретный
характер секреции сам по себе важен для
действия гормона, а его нарушения
свойственны некоторым формам патологии. Так,
люлиберин стимулирует продукцию ЛГ в
аденогипофизе при пульсовом введении, но
те же дозы этого гипоталамического рили-
зинг-фактора тормозят продукцию Л Г, если
вводятся в непрерывном режиме. Утрата
импульсного характера секреции люлиберина,
при сохранении его продукции, закономерно
наблюдается у больных психогенной ано-
рексией-булимией (см. выше «Голодание»).
Микроимпульсное введение инсулина более
эффективно для контроля уровня глюкозы
в крови, чем непрерывная инъекция той же
общей дозы. Микроимпульсы поступления
СТГ в кровь совпадают с периодами
быстрого сна. В более долговременном плане ос-
вобождение гормонов в кровь подчиняется
определённым биоритмам. Под контролем
супрахиазматического ядра гипоталамуса,
серотонина и мелатонина эпифиза (см. т. I,
с.80-82 а также раздел «Патофизиология
шишковидной железы» в данном томе) в
зависимости от уровня общей освещённости
в видимом диапазоне спектра формируется
циркадный (околосуточный) ритм секреции
кортиколиберина, а следовательно — АКТГ
и глюкокортикоидов. По А. Шафарчи-
ку и соавт. (1983), у человека и дневных
млекопитающих акрофаза секреции гипо-
таламо-гипофизарных стимуляторов кор-
тикостероидогенеза приходится на 7-8 ч.
утра, а минимум — на поздний вечер (20 ч.)
Установлено, что данный ритм накладывает
отпечаток не только на стероидогенез, но и
на ход пролиферативных процессов в коре
надпочечников (Л.П. Чурилов, 1986).
Существуют ритмы с периодом, превышающим
сутки (инфрадианные), и более коротким,
чем циркадные (ультрадианные). Широко
известны колебания продукции гонадо-
тропинов и половых гормонов с
околомесячным периодом. Инфрадианной
периодизации подвержена активность секреции
тироидных гормонов. Суммарный результат
биоритмологических особенностей
гормонального фона определяет периодические
околомесячные и сезонные изменения
физической, психоэмоциональной и
интеллектуально-поведенческой активности
индивидов, что впервые описано
концепциями Г. Свободы-В. Флейса (1898) и
А.Л. Чижевского (1924).
Социально-поведенческая дизритмия является фактором
риска нейроэндокринных заболеваний.
Описаны «болезнь акклиматизации»,
вызванная быстрой сменой часовых
поясов, и учащение гипертензии у субъектов,
занятых ночной работой. В обоих случаях
отмечаются аномалии продукции гипота-
ламических, гипофизарных и надпочеч-
никовых гормонов, гиперфункция ренин-
ангиотензин-альдостероновой системы.
Характерно лечебно-профилактическое
действие эпифизарного гормона
мелатонина при болезни смены часовых поясов.
Нарушения эндокринной регуляции 481
Исчезновение нормального циркадного
ритма продукции АКТГ характерно для
болезни Иценко-Кушинга. Нет сомнений,
что биоритмологические подходы в
будущем значительно скажутся на развитии
эндокринологии(И.И. Дедов, В.И. Дедов,
1991). Например, уже сейчас данные о
содержании тироидных гормонов у женщин
должны корректироваться в зависимости
от того, на какой день месячого цикла
брали пробу для анализа (см. ниже — о
транспорте гормонов в крови).
После секреции гормоны попадают в
равновесную систему, где концентрации
связанного и свободного биорегулятора в
крови соотносятся между собой и зависят
от баланса скорости поступления и скорости
удаления гормона из крови, а также
количества и аффинности белка-переносчика.
Транспорт гормонов осуществляется с
током крови, лимфы и межклеточной
жидкости. У пептидов время полужизни в крови
порядка 3-7 мин, а у гликопротеидов может
быть более часа, но принципиально остается
весьма коротким у всех гормонов, что
позволяет эндокринной системе оперативнее
менять гормональный фон. Пептидные
гормоны распространяются в свободном
виде, но тироидные и стероидные, в силу
гидрофобности, требуют солюбилизато-
ров-переносчиков. Наиболее известные
транспортёры гормонов — транстиретин
(преальбумии), переносящий тироидные
гормоны и ретинол; тироксин-связываю-
щий глобулин, тестостерон-связывающий
глобулин, кортикостероид-связывающий
глобулин, альбумин, который
неспецифически фиксирует тироидные и многие
стероидные гормоны (см. «Нарушения белкового
состава плазмы крови» выше). Стероиды и
тироидные гормоны могут также
переноситься липопротеидами особенно высокой
плотности.
Эндокринологические руководства
традиционно числят нарушения транспорта
гормонов среди важных причин эндокрино-
патий, по аналогии с фармакологическими
данными о том, что связывание с белками
крови очень существенно для определения
482 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
биодоступности лекарств. Однако Дж. Уил-
сон (1994) справедливо указывает, что при
интактной обратной связи в системе
гипоталамус-гипофиз-периферические железы
сдвиги концентраций свободного активного
гормона, наступающие от изменения
связывания с плазменными белками, не будут
иметь стабильного характера, а эффективно
нейтрализуются компенсаторным
изменением его секреции. Так, эстрогены
увеличивают содержание тироксинсвязывающего
глобулина и делают биодоступность тироксина
меньшей. В ответ на это в эстральную фазу
месячного цикла у женщин гипоталамо-
гипофизарный нейросекреторный аппарат
увеличивает, по сервопринципу, продукцию
тиролиберина и ТТГ, щитовидная железа
добавляет в кровь тироидных гормонов — и
эффективная биодоступная концентрация
этих регуляторов остается нормальной.
В плазме наступает кажущееся увеличение
концентрации общего Т4, не означающее
наступления
функционально-метаболического гипертироза. Однако это небезразлично
при истолковании данных о концентрациях
тироидных гормонов в крови у женщин.
То же имеет место при беременности и
применении эстрогенных контрацептивов.
При нарушении нормальных механизмов
сервоконтроля гормональных концентраций
изменения уровня связывания гормонов в
крови становятся существенным
патогенетическим фактором эндокринопатий (см.
ниже). Так, даже небольшие дозы глюкокор-
тикоидов, вводимых с лечебной целью,
способны вызвать синдром Кушинга у пациента
с гипопротеинемическими состояниями
и снижением продукции кортикостероид-
связывающего белка (печеночной
недостаточностью, нефротическим синдромом,
микседемой).
Инактивация гормонов — важный этап
их метаболизма. Каждый, кому случалось
иметь дело с запавшей кнопкой дверного
звонка, помнит, как надоедливо звучит его
непрекращающийся сигнал. Химический
сигнал-гормон, как и любой другой, для
эффективного использования должен
своевременно прекращать своё действие — инак-
тивироваться. Уровень гормона зависит и от
его секреции, и от скорости инактивации:
ПК - СС/К,
где
ПК — плазменная концентрация
гормона,
СС — скорость его секреции,
К — метаболический клиренс гормона.
Гормоны инактивируются как в
клетках-мишенях после их проникновения
во внутриклеточное пространство, так и
в нетаргетных органах, главным образом
печени и почках. Иммунные комплексы с
участием гормонов подвергаются клиренсу
в фагоцитах. Основной путь инактивации
пептидных гормонов — протеолиз. Хотя
исторически сложились представления о
существовании для некоторых гормонов
специальных протеаз, на деле эти процессы в
основном осуществляются
неспецифическими протеолитическими ферментами клеток-
мишеней. Так, не обнаружено специальной
«инсулиназы», хотя условно этим понятием
оперируют многие эндокринологические
руководства. Вместе с тем для многих парак-
ринных пептидов, например интерлейкинов,
кининов, ангиотензинов имеются
специальные конвертазы (например, интерлейкин-
конвертаза ICE, ангиотензин-1-конвертаза,
калликреин).
Гормоны тирозинового происхождения
инактивируются специальными
ферментами. Катехоламины разрушаются моноами-
ноксидазой (МАО) и катехол-ортометил-
трансферазой (КОМТ) тканей-мишеней и
печени. Окислительное дезаминирование
и орто-метилирование этих гормонов
ведет к появлению и экскреции с мочой
таких продуктов, как ванилил-миндальная
кислота, норметанефрин и метанефрин,
концентрации которых измеряют в
диагностических целях. Блокада ферментов
катаболизма катехоламинов, например с
помощью лекарств, приводит к
значительному увеличению чувствительности тканей к
катехоламиновым сигналам (см. выше
«Нарушения межуточного обмена
аминокислот»). Тироидные гормоны дейодируются
Нарушения эндокринной регуляции 483
в тканях-мишенях и печени, дезаминиру-
ются и деконденсируются в печени, затем
окисляются, а в небольших количествах
рециркулируют с желчью.
Стероиды подвергаются в печени
восстановлению, для чего имеются
специальные энзимы, затем — неспецифическому
гидроксилированию и, наконец, образуют
гидрофильные парные соединения с
активными формами серной или глюкуроновой
кислот и экскретируются в желчь и мочу.
Из ЖКТ их дериваты могут реабсорбиро-
ваться, а содержание продуктов распада
стероидов в моче измеряется в
диагностических целях.
Как указывалось выше, в связи с
закономерностями гормонального транспорта сами
по себе колебания скорости деградации
гормона не приводят к эндокринной патологии,
если функционирует сервостатический
механизм гипоталамо-гипофизарного
контроля его эффективных концентраций. Может
быть, единственным примером такого рода
патологии служит инсулинорезистентность
к экзогенному гормону из-за его аномально
быстрого расщепления при подкожных
инъекциях.
Но при патологии обратные связи в
системе могут быть нарушены, и клиренс
гормонов становится важной детерминантой
их эффективной концентрации. Гипертироз
ускоряет катаболизм стероидов, делая
пациентов с болезнью фон Базедова
относительно «резистентными» к глюкокортикоидам.
Печеночная недостаточность замедляет
клиренс альдостерона, серотонина, эстрогенов,
кортизола и других гормонов, что
накладывает отпечаток на симптомы, наблюдаемые
у больных: импотенцию, гинекомастию,
отёки, пальмарную эритему, и сосудистые
«звёздочки» при циррозе печени объясняют
избытком соответствующих гормонов.
Как впервые было установлено У.Э. Нок-
сом (1951), гормоны опосредуют действие
на физиологические процессы через
активность энзимов. Позднее, из работ Б.О'Мэлли
и Р.ТШимке с соавторами (1965,1972),
стало ясно, что под влиянием гормонального
сигнала меняется и активность, и
количество молекул ферментов.
Достигая клеток-мишеней, гормоны
взаимодействуют с рецепторами. Основные
аспекты общих механизмов действия
гормонов в настоящее время представляются
следующим образом.
Первым этапом служит
комплементарное взаимодействие гормона с
внутриклеточным или мембранным поверхностным
гормональным рецептором. Специфичность
этих рецепторов высока, но не абсолютна.
Так, кортизол взаимодействует не только с
глюкокортикоидным, но и с минералокорти-
коидным рецептором, и наоборот — альдо-
стерон способен действовать на рецепторы
глюкокортикоидов. Инсулин часть своих
эффектов опосредует не через инсулиновый
рецептор, а через рецепторы инсулинопо-
добных ростовых факторов.
В общем случае взаимодействие
гормона с рецептором описывается уравнением
Скэтчарда, и зависимость между долей
связанного гормона и общим количеством
гормона в системе носит линейный характер
(рис. 87 с соответствующей формулой). Для
ib/f
Рис. 87. Типичный график Скэтчарда для
гормон-рецепторного взаимодействия. B/F —
соотношение связанного с рецептором и
свободного гормона; В — количество
связанного с рецептором гормона; 1/Ка — величина,
обратная равновесной константе диссоциации
гормон-рецепторного комплекса (Ка); RT —
общее количество рецепторов; F — количество
связанного гормона
484 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
гормонов, имеющих один класс рецепторов,
этот график представляет прямую. Однако
при наличии негативного кооперативного
взаимодействия между рецепторами, когда
связывание молекул гормона понижает
аффинность оставшихся свободными молекул
рецептора (инсулин), а также при
существовании нескольких классов рецепторов
одного гормона, с разным сродством (альдо-
стерон) скэтчардовский график становится
вогнутым.
До недавнего времени казалось
устоявшимся представление о том, что белковые и
катехоламиновые биорегуляторы действуют
на мишени, исключительно с поверхности —
не проникая внутрь клеток, а стероидные и
тироидные — только через внутриклеточные
рецепторы (А.Н. Юдаев и соавт., 1976).
Однако подобное разграничение, хотя и
диссоциация
гормонов
и рецепторов
проводни
'ковая последов
вательность
Рис 88. Перенос гормонов в клетку и в ядро посредством
рецепторного эндоцитоза. Г— полипептидный гормон,
Р — рецептор, пояснения в тексте
имеет известный смысл, не должно более
восприниматься как абсолютное или как
диктуемое исключительно гидрофильным
или гидрофобным характером молекул
биорегуляторов. Еще в 80-х годах XX века
было твёрдо установлено, что белковым
регуляторам не может приписываться только
дистантный механизм действия. Доказано,
что многие белковые гормоны и даже
антитела после их рецепции на плазматической
мембране таргетных клеток подвергаются
адсорбтивному эндоцитозу и, таким путём,
оказываются внутри клеток, а далее могут
транспортироваться в органоиды, включая
ядро (Г. Расмуссен, 1982), — см. рис. 88. Это
позволяет им иметь «раннюю волну»
эффектов, наблюдаемых на протяжении первых
минут после взаимодействия с клеткой и
опосредованную мембранным рецептором
и пострецепторными
посредниками, а также «позднюю волну»,
наступающую в сроки 2-4 ч и
позже, связанную с
внутриклеточным и внутриядерным
переносом белковых регуляторов и
адресованную непосредственно
генам. Доказан внутриядерный
перенос инсулина (А. Голдфайн
и соавт., 1982), хорионического
гонадотропина (К. Яскеляйнен
и соавт., 1983), люлиберина
(Ю. Шлессингер и соавт., 1983),
многих других пептидных
гормонов, а также антител (A.IH.
Зайчик, 1973; Д. Аларкон-Се-
говия, 1973).
В «позднюю волну»
эффектов белковых гормонов входят,
в частности, такие эффекты,
как морфогенетическое
действие гормонов, регуляция
пролиферации клеток-мишеней,
индукция синтеза ключевых
белков, контролирующих
каскады внутриклеточных событий,
например ключевых протеинов
стероидогенеза (А. Дазо, 1983).
У стероидов и тироидных
гормонов тоже имеется не только
аппарат Гольджи
внутриклеточный набор отсроченных
эффектов, но и определенные эпигенетические
ранние эффекты, связанные с их действием
на мембраны и мобилизацией
внутриклеточных посредников того же типа, что и у
белковых гормонов (И. Ненчи, 1981). Так,
например, тироидные гормоны именно
через поверхностные рецепторы
опосредуют свой усиливающий эффект на захват
клетками аминокислот и глюкозы, а также
прямо активируют мембранную АТФ-азу
плазмолеммы (Б.Г. Катцунг, 1998). Таким
образом, дистантный и внутриклеточный
механизм — не альтернативные виды
действия белковых, либо тироидных и
стероидных гормонов, а сотрудничающие
способы передачи соответственно раннего
и отсроченного гормональных эффектов
(Дж.Бэкстер, 1987).
Механизмы внутриклеточного
опосредования эффекта гормонов на поверхностные
рецепторы разнообразны и представлены
на рисунках 89-90. В качестве комментария
заметим, что различные внутриклеточные
посредники действия гормонов (циклонук-
леотиды ц-АМФ и ц-ГМФ, кальций и
связывающие его белки, фосфатидилинозит и его
производные, эйкозаноиды, пептиды) — это,
в большинстве случаев не альтернативные
мессенджеры, как полагали в 80-х годах XX
века, а взаимодействующие части целостной
системы. Тот или иной гормон, оперируя с
разными типами рецепторов, может в разной
степени затрагивать различные части этой
системы. Но целостный ансамбль
эффектов, вызываемых гормоном, всегда требует
участия нескольких интрацеллюлярных
посредников.
Механизмы действия гормонов на
внутриклеточные рецепторы представлены на
рис. 91. Гормон связывается с цитоплаз-
матическим рецепторным белком, чтобы
попасть в ядро. До прихода гормона ци-
топлазматический рецептор блокирован
белком-инактиватором. Для глюкокорти-
коидных рецепторов таким инактиватором
служит белок теплового шока БТШ-90.
Белки теплового шока используются в этом
качестве и другими цитоплазматически-
Нарушения эндокринной регуляции 485
ми гормональными рецепторами (см. т. I,
стр. 145-148). В клеточном ядре гормон
взаимодействует с цис-регуляторными
элементами хроматина, формируемыми
комплексами ДНК и негистоновых белков,
которые называют «ядерными
рецепторами». Тироидные гормоны располагают и ми-
тохондриальными рецепторами. Изменения
конформации цис-регуляторного элемента
хроматина способствуют ассоциации гена
с ДНК-зависимой РНК-полимеразой и его
экспрессии. И тироидные, и стероидные
гормоны имеют, кроме этого,
эпигенетические механизмы действия, опосредованные
через звенья, аналогичные мессенджерам
белковых гормонов. Внутриклеточные
рецепторы тироидных и стероидных
гормонов вместе с рецепторами витамина
А (см. «Патофизиология витаминного
обмена») принадлежат к классу белков-
регуляторов транскрипции и гомологичны
онкобелку-продукту онкогена erbA. Один
гормон может иметь несколько
разновидностей erbA-подобных рецепторов
(например, тироксин). Минералокортикоиды,
глюкокортикоиды, эстрогены, андрогены,
кальциферол и прогестины имеют разные
подтипы подобных рецепторов. Лиганды
некоторых аналогичных рецепторов всё
еще не найдены, что вселяет надежду на
обнаружение новых стероидных гормонов.
По некоторым данным (А. Уайт и соавт.,
1981; В. Шрайбер, 1982),
^охарактеризованные гипотетические гормоны-стероиды
вырабатывают тимус (тимостерин), а также,
возможно, субкапсулярный слой минерало-
кортикоидной зоны коры надпочечников
(кардиотропные или натрий-уретические
стероиды — антиминералокортикоидного
действия). Клонирование и секвенирование
новых рецепторов стероидного типа может
быть началом воспроизведения ситуации с
эндогенными опиатами, когда рецепторы
были открыты ранее своих естественных
лигандов.
Рецепторы гормонов на плазматической
мембране характеризуются известным
разнообразием. Они принадлежат по меньшей
мере к пяти типам, использующим посред-
Рис. 89. Циклонуклеотид-протеинкиназная часть опосредующего Рис. 90. Роль кальция и инозитфосфатидов в опосредовании гормо-
механизма гормонального сигнала. Обозначения — как в тексте нального сигнала. N — гуанилатсвязывающий белок, ДАГ — диацил-
глицерин, ФИТФ — фосфатидилинозитолтрифосфат, ГТФ — гуано-
зинтрифосфат, ГДФ — гуанозиндифосфат, R — рецепторная
субъединица
Нарушения эндокринной регуляции 487
Ядро
никовые механизмы по-разному
(рис. 89,90 и выше рис. 65).
>- Крупнейшая группа
поверхностных гормональных
рецепторов принадлежит к
семейству
ассоциированных с G-белками (ГТФ-
связывающими белками).
Прояснение механизмов их
функционирования было
начато первооткрывателем
циклонуклеотидных
вторичных посредников,
лауреатом Нобелевской премии
по медицине 1971 г. Э.У. Са-
зерлендом (младшим).
Современная концепция
функционирования G-белков
сложилась благодаря
работам А. Г. Гилмэна и М. Род-
белла, также удостоенных
этой премии за 1994 год.
В т. I данного руководства
имеется подробная
характеристика системы G-белков
и нарушений, связанных с
ними (с. 120-123, включая
схему на рис. 20). В данном разделе нет
необходимости в буквальном
повторении, но не будет лишним краткое напо-
мицание основных сторон работы этого
сложнейшего механизма. Рецепторы
этой группы имеют внеклеточный домен,
связывающий гормон или иной
биорегулятор, а также внутримембранную
часть, пронизывающую плазмолемму
несколько раз (откуда второе название
группы — серпантинные рецепторы),
и, наконец, — внутриклеточный домен,
способный связывать клеточный
адаптер — G-белок. Ассоциация лиганда и
рецептора ведёт к связыванию и гидролизу
G-белком ГТФ, что позволяет запустить
цепи из нескольких внутриклеточных
посредников, параллельно
обеспечивающих ансамбль ранних метаболических
изменений, диктуемых гормоном.
Одновременно активируется аденилатцикла-
за, причём образованная ц-АМФ обес-
I Интернализация I
Эффекты I
гормонов I
Стероидные
гормоны
Рис 91. Механизмы действия стероидных (С) и тироидных
(Т) гормонов. ЯР — ядерный рецептор, Р — гомолог онко-
белка егЬА
печивает активацию ряда протеинкиназ
класса А, фосфорилирующих энзимы и
распознающих белки клетки. Данный
циклонуклеотид также способствует
напрямую открытию натриевых ионных
каналов мембраны. Другая параллельно
развивающаяся линия эффектов, стар-
туемая G-белком, включает ферменты
инозитол-фосфатазы, известные под
условным именем «фосфолипаза С». Эти
энзимы прямо из инозитол-фосфатидов
мембраны изготовляют фосфатидил-
инозитол-4,5-дифосфат, переходящий в
трифосфоинозит (ИТФ) и диацилглице-
рин (ДАГ), - рис. 89-90.
ДАГ активирует протеинкиназы класса
С, в свою очередь фосфорилирующие ряд
функциональных белков клетки (рис. 90).
Третья линия событий, спровоцированных
активацией рецептора из данного семейства,
заключается в генерации из материала
мембраны арахидоновой кислоты и эйкозаноидов
(в чём участвуют фосфолипазы С и А2 и
488 А.Ш. Зайчик, Л.П.Чурилов Патохимия
ДАГ-липаза). Эйкозаноиды дополняют веер
гормонозависимых эффектов,
опосредованных G-белками. Наконец, под влиянием
ИТФ из внутриклеточных резервуаров
освобождается еще один важный участник
ансамбля посредников гормонального
эффекта — кальций. Кальций входит в
стимулированную гормоном клетку и извне,
чему способствует открытие, при участии
аденилатциклазных механизмов, ионных
каналов. Кальций не представляет собой
альтернативного независимого посредника
гормонального действия в клетке, но
взаимодействует с вышеназванными мессен-
джерами. Он распознаётся
внутриклеточным рецепторным белком кальмодулином
и некоторыми протеинами цитоскелета.
Цитоскелет под влиянием кальция
обеспечивает движение органоидов или клетки,
что вносит вклад в ансамбль гормональ-
нозависимых эффектов. Кальмодулин в
одних системах включает фосфодиэстеразу
и снижает эффективность аденилатцик-
лазной передачи (например, именно так
катехоламины вызывают расслабление
гладких мышц, сокращающихся под
влиянием ацетилхолина). Но в других системах
и, что особенно важно, при действии
тройных гормонов на периферические железы
внутренней секреции кальмодулиновый и
аденилатциклазный механизмы выступают
синергистами. К данной группе
гормональных рецепторов принадлежат рецепторы Л Г,
ФСГ, ТТГ, АКТГ, вазопрессина, глюкагона,
катехоламинов, паратгормона, соматостати-
на, кальцитонина, бомбезина, опиатных
пептидов, серотонина, мускариновый рецептор
ацетилхолина и рецепторы многих других
биорегуляторов. Здесь же — обонятельные
и зрительные рецепторы. При этом АКТГ,
адреналин, кальцитонин, ФСГ, глюкагон,
Л Г, паратгормон— все повышают уровень
ц-АМФ в клетках-мишенях, так же как и
ускоряют кругооборот инозит-фосфатидов.
АКТГ, вазопрессин, ТТГ и Л Г ещё и заметно
повышают цитоплазматическую
концентрацию ионизированного кальция. В то же
время соматостатин, оперируя через G-белки,
уровень ц-АМФ в таргетной клетке снижа-
чдокринно-метаболические нарушения)
ет. Это свидетельствует, что разные
биорегуляторы в различных клетках используют
вышеописанный комплекс посредников
неодинаково. Зарегистрированы при
патологии и получены в эксперименте антитела
к рецепторам этого класса, имитирующие и
блокирующие действие соответствующих
гормонов (ТТГ, АКТГ, гонадотропинов).
>* Другая группа гормональных рецепторов
сама представляет собой тирозиновые
протеинкиназы. Эти рецепторы не
оперируют с G-белками, как бы сокращая
вышеописанную посредниковую цепь
(см выше рис. 65, гл. 9). Каждый такой
рецептор — мембранный гликопротеид
из пары а-субъединиц снаружи и двух
Р-субъединиц, пронизывающих
мембрану. Наружные субъединицы связывают
гормон, а внутренние гидролизуют АТФ
и фосфорилируют себя и
внутриклеточные функциональные белки,
вызывающие гормонозависимый ответ клетки.
Показанный на рис. 65 рецептор — инсу-
линовый. Помимо важнейшего гормона
инсулина к этой группе принадлежат
рецепторы инсулиноподобных ростовых
факторов I и II, фактора роста
эпидермиса, тромбоцитарных ростовых факторов
и, возможно, фактора роста нервов. Как
видно из этого перечня, данный тип
рецепции очень важен в эндокринной
регуляции ростовых и анаболических
процессов. По некоторым данным,
подобные двухвалентные рецепторы при
активации должны попарно сшиваться
гормоном или антителом. Уровень
ц-АМФ снижается при действии
инсулина и ростовых факторов. Вместе с тем
подобная рецепция не исключает участия
компонентов посредниковой системы,
на которой основывается адаптерная
роль G-белков. Так, эпидермальный
ростовой фактор сочетает тирозин-
протеинкиназный механизм активации
клетки-мишени с повышением уровня
цитоплазматического кальция и
активацией инозит-фосфатидов, как и гормоны,
влияющие через G-белки. Тирозиновые
протеинкиназы кодируются многими
Нарушения эндокринной регуляции 489
онкогенами. Получены в эксперименте
и зарегистрированы при патологии как
имитирующие действие гормона, так и
блокирующие его антитела к рецепторам
данного класса (в частности, к инсули-
новому и к рецептору эпидермального
ростового фактора).
> Меньше известно о механизмах
опосредования внутриклеточных эффектов
пролактина, плацетарного лактогена и
гормона роста. Эти гормоны (а с ними —
ряд цитокинов и фактор роста нервов)
имеют рецепторы в виде
трансмембранной молекулы из одной субъединицы
гомологичного строения, которая не
является протеинкиназой и не вовлекает
G-белки.
Имеются данные Л.М. Удебин (1982),
что данный рецептор опосредует своё
действие в цитоплазме через пептидные
мессенджеры. По данным Д. Тёркингтона
и соавт., эти посредники в ядре приводят
к увеличению экспрессии генов протеин-
киназ (1973). Известно, что ряд эффектов
СТГ опосредован вторичной продукцией
в тканях инсулиноподобных ростовых
факторов, рецептируемых тирозиновыми
протеинкиназами. В пренатальном периоде
тканевым посредником действия СТГ
служит ИФР-П (соматомедин А), а после года
в большинстве тканей (кроме зубных
зачатков) — ИРФ-1 (соматомедин С). Вероятно,
их можно рассматривать как
внутриклеточные и паракринные тканевые вторичные
посредники, зависящие от действия СТГ на
его рецептор. Мутации рецептора СТГ
нарушают продукцию соматомединов (дефицит
соматомедина С при синдроме
карликовости Ларона). Есть сведения, что рецепторы
этого класса мобилизуют внутри клетки
также эйкозаноиды. Получены антитела к
рецепторам пролактина, имитирующие его
эффекты, и зарегистрированы аутоантитела
к рецептору СТГ, тормозящие рост детей
(Д. Миракян и соавт.; 1983).
> Уникальный характер имеет рецептор
атриопептинов (иредсердных натрий-
уретических факторов). Этот
гормональный рецептор сам является трансмем-
бранно расположенной гуанилатцик-
лазой. Взаимодействие атриопептина с
рецептором ведёт к накоплению ц-ГМФ
в клетке-мишени. Цикло-ГМФ
способствует активации внутриклеточных
сериновых и треониновых протеинкиназ,
фосфорилирующих ансамбль белков-
эффекторов. Неизвестны антитела,
которые бы влияли на данный рецептор
стимулирующим, либо блокирующим
образом. Однако при застойной
сердечной недостаточности отмечена
резистентность атриопептиновых рецепторов
к действию данного сердечного гормона,
не дающая возможности облегчить
отёчный синдром с помощью атриопептина
(В.Я.Багров, 1987). В последнее время
обнаружено, что N0 — липофильный
газообразный медиатор — также
активирует гуанилатциклазу в клетках-мишенях
(Б.Г. Катцунг, 1998).
>* Взаимодействие биорегуля тора с рецеп -
тором может приводить непосредственно
к открытою ионных каналов в мембране:
хлоридных (ГАМК, глицин и другие
возбуждающие аминокислоты), натриевых
и калиевых (ацетилхолин при действии
на никотии-холинергический рецептор).
Данные рецепторы могут блокироваться
аутоантителами (что лежит в основе па
тогенеза генерализованной миастении).
Так как агонисты рецепторов канального
типа формально не относятся к
гормонам, этот вид рецепции не
рассматривается в данной главе детально.
Как уже отмечалось, в рецепторной
эндокринологии сохраняют свою актуальность
основные принципы эндокринологии
гормональной: принцип обратной связи и
принцип пермиссивного эффекта. Количество
большинства гормональных рецепторов
на клеточной поверхности и в цитозоле
понижается при избытке гомологичного
гормона. Сродство многих рецепторов,
особенно тирозин-иротеинкиназного типа, к их
гормонам снижается после связывания
гормона. Одни гормоны гетерологично влияют
на экспрессию и аффинность рецепторов
490 А.Ш. Зайчик, Л.П.Чурилов Патохимия
других гормонов. Так, эстрогены повышают
экспрессию прогестиновых рецепторов.
Распределение гормональных рецепторов
в организме неравномерно. Тироидными
рецепторами располагает каждая клетка.
Глюкокортикоиды и катехоламины
действуют на очень широкий круг органов и
тканей. Но рецепторы глюкагона, например,
сосредоточены практически исключительно
в печени. Альдостерон имеет очень узкий
круг мишеней (почки, ЖКТ, слюнные и
потовые железы). Проникновение глюкозы
во многие жизненно важные органы (ЦНС,
диафрагмальная мышца, сердце, ткани глаза,
почка, надпочечник, гонады) не зависит от
инсулина и т.д. Спектр эффектов гормона
может ограничиваться не только
распространенностью его рецепторов, но и
эффекторов, а также нюансами его транспорта.
Из-за существования портальных циркуля -
торных систем многие гормоны практически
не попадают за пределы определённых зон.
Некоторые биорегуляторы действуют, по
сути дела, только паракринно (мюллеров
ингибирующий полипептид). В организме
ответ нейроэндокринной системы бывает
всегда плюригландулярным и вовлекает
различные влияния нескольких гормонов
на разные клетки. Примером может служить
интегральный ответ на гипогликемию:
глюкагон и адреналин побуждают печень к
гликогенолизу и тормозят гликогеногенез,
а также, вместе с кортизолом, усиливают в
этом органе глюконеогенез. Параллельно
в липоцитах СТГ кортизол и адреналин
стимулируют липолиз, причём глицерин
печень будет превращать в глюкозу, а
жирные кислоты послужат
альтернативным источником энергии. При этом захват
глюкозы липоцитами эти гормоны
останавливают. Адреналин способствует распаду
мышечного гликогена, кортизол ингиби-
рует потребление глюкозы в инсулинзави-
симых тканях и в ряде органов (например,
лимфоидных) сдвигает баланс распада и
синтеза белка в катаболическую сторону,
поставляя субстраты для глюконеогенеза
(см. также выше «Патофизиология
углеводного обмена»).
чдокринно-метаболические нарушения)
Всё это свидетельствует о большой
пластичности и об интегрированном
ансамблевом характере деятельности эндокринной
системы. Патология эндокринной системы
затрагивает все виды метаболизма, а также
рост, размножение, регенерацию, идивиду-
альное развитие организма. Классические
представления о патогенности недостатка
или избытка того или иного гормона
сложились еще на заре эндокринологии. В XX
веке к ним добавилось понимание того,
что тканевая резистентность или
гиперчувствительность к действию гормона может
вызвать картину болезни даже при его
адекватной продукции.
Современное представление о том, чем и
как могут быть вызваны эндокринопатии,
отражено на рис. 92.
Эндокринопатии всегда српровождаются
избыточным или недостаточным эффектом
какого-либо гормона на те или иные
мишени, что и порождает клиническую картину.
Известны, правда, и исключения. Так,
дефицит кальцитонина у человека бессимптомен,
так как кальций-фосфатный гомеостаз при
этом эффективно поддерживается
компенсаторными изменениями, связанными с его
антагонистом паратгормоном и
кальциферолом. Избыток тестостерона у мужчин и
прогестерона — у лиц обоего пола также не
дает клинических симптомов.
Недостаточное действие гормона может
быть следствием нескольких типовых
причин, так или иначе затрагивающих железу,
где он производится:
>• нарушение поставки гормона в кровь
эндокринной железой происходит при
деструкции, либо информационной
блокаде гормонообразующих клеток.
Причиной деструкции могут быть
различные процессы:
^> Негормонообразующие опухоли,
как это происходит, например, при
хромофобных (нуль-клеточных)
опухолях гипофиза, вызывающих
гипопитуитаризм. К этой же
этиологической группе может быть
отнесена гипофункция железы при
метастазах в нее неэндокринных
гипофункциональные
состояния
деструкция
блокада
(в т.ч. аутоиммунная)
блокада
ускорение
деградация
антитела-блокаторы
антагонисты
дефекты
повреждение
атрофия
гиперфункциональные
состояния
опухоль
гиперплазия
антитела-стимуляторы
эктопическая
продукция
ятрогения
блокада
деградация
антитела-имитаторы
агонисты
синергисты
повреждение
Рис 92. Возможные механизмы эндокринопатий.
торный аппарат. Пояснения в тексте
опухолей и инфильтрации лейкоз-
ными бластами.
^ Инфекция и последовавшее
воспаление с альтерацией железы (примером
служит острый тироидит, туберкулёз
надпочечников как причина болезни
Аддисона — и т.д.).
Нарушения эндокринной регуляции 491
i. ГГНСА — гипоталамо-гипофизный нейросекре-
^ Неинфекционное воспаление,
главным образом — аутоаллергической
природы (например, аутоиммунный
гипопаратироз, аутоиммунный гипо-
физит). Известны случаи деструкции
эндокринных органов при их
воспалении, вызванном травмой. Примером
492 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
служит случаи вторичного сахарного
диабета, развившегося у офицера
после боевого огнестрельного ранения в
брюшную полость,
сопровождавшегося обширной травмой поджелудочной
железы (Ю.И.Строев, Л.П. Чурилов,
2004).
^ Острое нарушение кровообращения,
приводящее к ишемическому некрозу
или тромбоэмболической апоплексии
эндокринного органа (примером
служит синдром Уотерхауза-Фри-
дериксена — острая надпочечниковая
недостаточность при двусторонней
тромбоэмболической апоплексии
мозгового вещества надпочечников,
следующая на фоне диссеминирован-
ного внутрисосудистого свёртывания
крови).
>- Генетически обусловленный дефект
синтеза гормона, приводящий к его
недостатку или ненормальной структуре (как
при наследственных формах гииотироза
и некоторых случаях инсулиннезависи-
мого сахарного диабета).
>- Дефицит компонентов, из которых
производится гормон (гипотироз при
геохимической недостаточности йода;
иммунодефицит при нехватке цинка,
входящего в состав тимулина;
гипофункция щитовидной железы и мозгового
вещества надпочечников при
наследственном нарушении окисления
тирозина, из которого строятся их гормоны).
>* Интоксикации, избирательно
нарушающие продукцию гормонов. Сюда
относятся как случаи ятрогенного
подавления продукции гормонов при
лечении и самолечении (например,
индукция гипокортицизма метирапоном,
гипотироза — мерказолилом), так и
влияния пищевых факторов. Роданиды
и цианиды репы, редьки, кабачковых,
краснокочанной капусты, маниоки и
тапиоки в больших дозах могут вызывать
понижение продукции тироидных
гормонов. Так, подсчитано, что килограмм
репы по антитироидному действию
приближается к половине таблетки
мерказолила.
>■ Важно отметить, что в некоторых
случаях блокада рецепторов тропных гормонов
на эндокриноцитах антирецепторными
аутоантителами ведет к
неэффективности стимуляции железы ее
физиологическими регуляторами и отсутствию
продукции гормона интактными в
других отношениях эндокриноцитами,
несмотря на метаболическую потребность
в нем. Так как многие тропные гормоны
(гормоны-стимуляторы гормонообра-
зования) служат для своих мишеней и
секреторными, и митогенными
сигналами (М. Павликовски, 1982), то такой
информационный блок ведет не просто
к недостаточности гормонов
регулируемой железы, но и к ее атрофии, за счёт
«апоптоза по умолчанию», без признаков
воспалительного или иного
деструктивного процесса. Примерами могут
служить атрофия коры надпочечников при
наличии блокирующих аутоантител к
рецепторам АКТГ (М. Давидай и соавт.,
1985), гипергонадотропный гипогона-
дизм из-за аутоаллергии против
рецепторов ФСГ (X. Диас и соавт., 1982) и
аутоиммунный атрофический гипотироз,
вызванный иммуноглобулинами,
блокирующими рецептор ТТГ (Ж.П. Десент,
Ж.Л. Вемо, 1982). Подробно о подобных
аутоиммунных блоках образования
гормона говорится в т. I (с. 471-473), а также
ниже при рассмотрении конкретных
эндокринопатий.
Во многих случаях недостаточное
действие того или иного гормона имеет
внежелезистое происхождение:
>- Недостаточный переход прогормона в
гормон вызывает симптомы дефицита
его активности, даже при не
нарушенном синтезе. Примерами могут служить
подавление перехода проренина в ренин
при сахарном диабете, связанное с
механизмами диабетической нефропатии, а
также витамин D-резистентные формы
рахита, рассмотренные в разделе, пос-
вященном нарушениям витаминного
обмена выше. При дефекте фермента
5а-редуктазы ткани-мишени не
превращают тестостерон в дигидротестостерон,
что ведет к частичному гипоандрогениз-
му, ибо собственная андрогенная
активность предшественника намного ниже,
чем дигидроформы этого стероида.
> Циркулирующие антагонисты гормона
могут снизить его эффективные
концентрации. В роли таких антагонистов
нередко выступают антитела к гормону
(вазопрессину — при неопухолевых
формах несахарного диабета, инсулину— при
вторичной инсулинорезистентности).
Неиммунологическими циркулирующими
антагонистами, вызывающими понижение
эффективности действия гормона, могут
быть другие гормоны (симптоматический
вторичный сахарный диабет при гипер-
кортицизме, феохромоцитоме), а также
ятрогенно введённые лекарства (гипоанд-
рогенизм и импотенция под влиянием
противоязвенного гистаминового блокатора
циметидина), или метаболиты (избыток
свободных жирных кислот снижает
эффективность действия инсулина).
> Как уже отмечалось выше, при
нормальном функционировании
сервомеханизмов обратной связи одно только
усиление связывания гормона с белком-
переносчиком, как и изолированное
ускорение деградации гормона, не могут
вызвать стабильного дефицита действия
гормона. Преходящие изменения будут
компенсироваться. Но в условиях
нарушенного механизма обратной связи как
увеличение степени связывания, так и
скорости разрушения гормона способны
отразиться на его эффективной
концентрации. Это хорошо проясняют следующие
примеры. Повышение содержания тесто-
стеронсвязывающего глобулина способно
вызвать снижение действия тестостерона
у женщин, поскольку у них нет механизма
обратной связи, контролирующего
уровень этого стероида. Высокий уровень
транстиретина и тиросвязывающего
Нарушения эндокринной регуляции 493
глобулина плазмы не ведет к гипотирозу
у здоровых людей, но важен для больных,
находящихся на экзогенном тироксине.
Поскольку тироидные гормоны ускоряют
катаболизм глюкокортикоидов, гиперти-
роз способен вызвать переход скрытого
гипокортицизма в явный или обусловить
резистентность к лечению кортикостеро-
идами, хотя сам по себе не ведет к гипо-
кортицизму, если только резервы коры
надпочечников и гипофиза адекватны.
>- Периферическая резистентность к
гормонам — важная внежелезистая причина
понижения эффективности их действия.
Такая резистентность часто связана с
рецепторными механизмами. Это может
подразумевать:
^ наследственный дефект
внутриклеточных рецепторов по типу
одиночных аминокислотных замен или
отсутствия их экспрессии, как это
наблюдается в отношении
рецепторов андрогенов при тестикулярной
феминизации — синдроме
Морриса; псевдогипоальдостеронизме,
наследственной резистентности к
кортизолу;
^> наследственный дефект мембранных
поверхностных рецепторов
(почечный питрессин-резистентный
несахарный диабет, синдром
карликовости Ларона, некоторые формы
тяжелого наследственного инсулино-
резистентного сахарного диабета);
^ блокаду рецепторов аутоантитела-
ми — как это отмечено при myasthenia
gravis— в отношении Н-холинерги-
ческих, а при acanthosis nigricans — в
отношении инсулиновых рецепторов;
*s> подавление экспрессии генов
рецепторов эпигенетическими
воздействиями — считается, что подобным
образом переедание и ожирение
могут способствовать развитию гипер-
инсулинемических форм сахарного
диабета.
Не менее важен пострецепторный
механизм гормонорезистентности. При псев-
494 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
догипопаратирозе имеет место дефект
сопряжения рецептора паратгормона и
рецепторов ряда других регуляторов с аде-
нилатциклазами, за счет наследственного
дефекта гуанилнуклеотидсвязывающего
регуляторного белка. Многие формы инсу-
линнезависимого сахарного диабета зависят
от наличия пострецепторных дефектов в
опосредовании действия инсулина на его
мишени (см. «Патофизиология сахарного
диабета»).
Иногда рецепторная нечувствительность
к гормону связана с неспособностью ткани
выработать под его влиянием тканевой
белковый гормон-посредник. Так, при
карликовости Ларона продукция СТГ адекватна,
но ткани из-за дефекта рецептора СТГ не
образуют под действием гормона роста
нужного количества одного из соматомеди-
нов — инсулиноподобного фактора роста I.
Важно подчеркнуть, что тканевая
резистентность бывает не только к пептидным или
тирозиновым гормонам, но и к стероидам.
Так, А. Вингерхёдс и соавт. описали
пациента с выраженым гиперкортицизмом, но
без признаков синдрома Иценко-Кушинга
(1976).
>- Наконец, если сами клетки-мишени
атрофированы или повреждены, они
могут не отвечать даже на сильный
гормональный сигнал. Глюкагон, например,
перестает вызывать гипергликемию при
тяжелой печёночной недостаточности.
В условиях истощения адипоциты
прекращают отвечать выработкой лептина
на инсулин и глюкагоноподобный
пептид — и перестает тормозиться аппетит.
Вазопрессин не эффективен при
выраженном нефросклерозе.
Избыточное действие гормона также
может иметь несколько типовых причин.
>■ Недостаточность физиологического
сервомеханизма, контролирующего
продукцию гормона по типу обратной
связи (например, снижение
чувствительности В-клеток островков Лангерганса
к глюкозе при некоторых формах инсу-
линнезависимого сахарного диабета или
клеток гипоталамуса, вырабатывающих
кортиколиберин, к глюкокортикоидам и
АКТГ — при болезни Иценко-Кушинга).
>- Обход физиологического механизма
обратной связи из-за появления
избытка стимуляторов гормонообразующих
клеток, не включённых в сервомеханизм.
Данная группа гиперфункциональных
эндокринопатий почти исключительно
имеет аутоиммунную природу и вызвана
иммуноглобулинами-агонистами
рецепторов тропных гормонов. Типичным
примером служит болезнь фон Базедова,
при которой гиперплазия и
гиперфункция щитовидной железы вызываются, в
обход гипоталамо-гипофизарной
регуляции, аутоантителами к ганглиозидной
и белковой частям рецепторов ТТГ.
Подобные аутоантитела могут вызвать
изолированную гиперфункцию железы,
но чаще обусловливают гиперплазию
и гиперфункцию. Хронические
неэндокринные заболевания могут создавать
неиммунологические сигналы,
стимулирующие продукцию того или иного
гормона, с развитием его вторичной
гиперпродукции. Примером служит
вторичный гиперальдостеронизм, за-
креплющий механизм системных отёков
при сердечной недостаточности. В роли
сигнала, побуждающего в конечном
итоге к гиперфункции ренин-ангиотензин-
альдостероновой системы, выступает
снижение объема сердечного выброса.
>• Утечка гормона из железы вследствие
ее деструкции (примером служат
эпизоды гипертироза при развитии лим-
фоцитарного подострого тироидита Де
Кервена).
>* Нерегулируемая гиперпродущия гормот,
как правило, относительно высоко
дифференцированной доброкачественной
эндокринной опухолью в пределах той
железы, которая служит его основным
источником (таков гипертироз при
токсической аденоме щитовидной железы,
персистирующая лакторея — аменорея
при пролактиноме аденогипофиза).
> Метаболический блок во
взаимопревращениях гормонов может вызвать не
только недостаток гормона, являющегося
конечным звеном цепи превращений, но
и избыток промежуточного гормонально
активного продукта. Характерным
примером служит врождённая гиперплазия
коры надпочечников, вызванная
дефицитом 21-гидроксилазы. При дефиците
этого фермента страдает биосинтез ми-
нералокортикоидов, а избыток
предшественников превращается в андрогены.
Поэтому гипоальдостеронизм и соль-
теряющий синдром могут сочетаться с
гиперандрогенизмом и вирилизацией
девочек, либо макрогенитосомией и
преждевременным половым созреванием
мальчиков.
Синдром избыточной эффективности
действия гормона также может иметь внеже-
лезистое происхождение. Основные формы
этого явления следующие:
> Эктопическая автономная от серво-
контроля продукция избытка гормонов
вне основных эндокринных желез. Этот
вариант эндокринной гиперфункции,
как правило, связан с образованием
гормонов, кодируемых одним-двумя
генами, в опухолевых, чаще
злокачественных, клетках апудоцитарной природы
(синтез АКТГ и кокальцигенина овся-
ноклеточными карциномами бронхов, а
также серотонина и энтериновых
пептидов — карциноидами ЖКТ, гормонооб-
разование в тимомах). Изредка опухоли
неапудоцитарнои природы могут давать
подобный эктопический синтез
(продукция тироидных гормонов в опухолях
яичника).
> Продукция избытка активного гормона
из прогормонов в неопухолевых
периферических тканях. Примером служат
гиперэстрогенизм при ожирении и при
печеночной патологии из-за избыточной
конверсии андростендиона в эстрогены
адипоцитами или гепатоцитами, а также
эндогенный гипервитаминоз D при сар-
коидозе из-за образования макрофагами
Нарушения эндокринной регуляции 495
гранулём избытка высокоактивной
формы кальциферола.
>- Ятрогенный избыток гормона или его
аналога может быть результатом лечения
или самолечения. Часто это относится
к избыточному приему адреномимети-
ков, тироидных гормонов, андрогенов
и кортикостероидов. Интересно, что
некоторые растительные пищевые
продукты содержат действенные аналоги
животных гормонов. Такова солодка,
имеющая в своём составе аналоги альдо-
стерона. Злоупотребление лакричными
конфетами и усиленное траволечение
солодкой вызывают картину, сходную с
первичным альдостеронизмом,
провоцируя артериальную гипертензию.
>• Аутоантитела к гормональным
рецепторам периферичеких тканей-мишеней
могут имитировать избыточное действие
соответствующих гормонов. Как
правило, они образуются по идиотип-антии-
диотипическому механизму, в ответ на
антитела к гормону. Инсулиномиме-
тические аутоантитела обнаружены у
больных «криптогенной» гипогликемией
и лабильным инсулинзависимым
сахарным диабетом, а также получены
экспериментально (см. выше рис. 63, гл. 9).
Связываясь с инсулиновым рецептором,
они временно воспроизводят часть
эффектов гормона, нарушая соотношение
между уровнем глюкозы и активностью
В-клеток (В. ДиМарио и соавт, 1977;
Й. Шехтер и соавт., 1982).
>* Тканевая гиперчувствительность к
гормонам встречается реже, чем
резистентность. Однако описаны почечная
гиперчувствительность к минералокор-
тикоидам (псевдогиперальдостеронизм с
низким уровнем ренина и альдостерона
и задержкой натрия), а также глюкокор-
тикоидная глаукома у больных, которые
особенно чувствительны к влиянию
кортикостероидов на внутриглазное
давление. Широко известна индукция
тироидными гормонами экспрессии
рецепторов катехоламинов в сердце и
496 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
—■" —,^—^—^^™^™^—^^—■' '
сосудах, участвующая в патогенезе ги-
пертензии и миокардиодистрофии при
гипертирозе.
>- Изолированные понижение связывания
гормона в крови и ускорение его
деградации, как правило, недостаточны для
индукции его избыточного действия в силу
нивелирующего эффекта нормальных
механизмов обратного контроля. Но
эндокринологи традиционно не оставляют
без внимания возможность появления
стабильного внежелезистого избытка
гормона, связанного с особенностями
его транспорта и катаболизма. Дело в
том, что при нарушениях нормальных
взаимоотношений тропных и
периферических гормонов или в сочетании
с действием других факторов данный
механизм приобретает известное
значение. Так, при голодании, печёночной
недостаточности и нефротическом
синдроме дефицит альдостеронсвязывающих
белков плазмы и недостаточный распад
альдостерона в печени вносят свой вклад
в развитие вторичного гиперальдосте-
ронизма и отёков, которые обусловлены
прежде всего, другим фактором —
понижением онкотического давления плазмы
(см. выше гл. 10).
Эндокринопатии бывают моноглан-
дулярными, то есть поражающими одну
эндокринную железу. Но встречаются и
плюригландулярные поражения. В случае
вовлечения в дисфункцию многих желез
необходимо учесть возможность системных
причин такого поражения.
Плюригландулярные синдромы бывают
гипоталамо-гипофизарными,
аутоиммунными, рецепторными и наследственными.
При центральной этиологии плюриглан-
дулярное поражение является вторичным
по отношению к пангипопитуитаризму (ти-
роидная, надпочечниковая недостаточность
и гипогонадизм).
При аутоиммунном плюригландулярном
синдроме аутоантитела поражают
несколько желёз внутренней секреции, имеющих
общие или сходные антигены. Известен
аутоиммунный синдром Шмидта (инсу-
чдокринно-метаболические нарушения)
линзависимый сахарный диабет,
аутоиммунный тироидит, гипопаратироз и гонад-
но-надпочечниковая недостаточность — см.
также раздел «Патофизиология сахарного
диабета»). При наличии аутоантител к
17а-гидроксилазе аутоаллергия поражает
стероидопродуцирующие клетки в коре
надпочечника и в гонадах. Аутоиммунные
плюригландулярные синдромы часто
сопровождаются неэндокринными
аутоиммунными расстройствами (атрофический гастрит,
витилиго), а также другими признаками
нарушенного имунитета (кандидоз).
Рецепторная плюригландулярная
аномалия развивается при дефекте гуанидин-свя-
зывающей части гормональных рецепторов
семейства G-белков. Наблюдается
неэффективность действия многих гормонов на
их мишени (псевдогипопаратироз, гипер-
гонадотропный гипогонадизм и первичный
гипотироз).
Описаны 3 различных плюригландуляр-
ных наследственных синдрома,
характеризующихся аномалией экспрессии онкогенов и
неопластическими процессами в разных гор-
монообразующих клетках (MEN-I, На, lib -
от англ. multiple endocrine neoplasiae).
Это аутосомно-доминантные растройства
с варьирующей пенетрантностью. Носитель
синдрома MEN наследует один из двух ал-
лельных генов дефектным, а во втором, на
протяжении онтогенеза, имеет место
соматическая мутация. Если она предоставляет
клеткам-мутантам селективные
преимущества, формируется доброкачественная или
злокачественная опухоль.
При MEN I имеется дефект гена под
аналогичным названием из 11-й хромосомы,
который служит антионкогеном. При других
типах MEN-синдрома генетический дефект,
вероятно, связан с протоонкогеном RET из
10-й хромосомы, онкобелок которого
является мембранной тирозин-протеинкиназой
со свойствами ростового рецептора.
Синдром MEN-1 (он же множественный
эндокринный аденоматоз или синдром Вер-
мера) включает доброкачественную аденому
паращитовидной железы, а также
доброкачественную либо злокачественную гормоно-
образующую опухоль островковых клеток
поджелудочной железы (в порядке
убывания частоты это может быть гастринома, ин-
сулинома, значительно реже — глюкагонома
или ВИПома). Гастриномы обусловливают
у таких больных тяжёлые язвы желудка
(синдром Цоллингера-Эллисона). Для MEN
I характерны также гипофизарные аденомы
(соматотропинома, пролактинома, реже —
кортикотропинома). Бывают карциноиды из
диффузных эндокриноцитов ЖКТ, липомы
и аденомы коры надпочечников.
Синдром MENU отличается присутствием
медуллярной карциномы С-клеток
щитовидной железы и доброкачественной либо
злокачественной феохромоцитомы.
Он имеет две разновидности. При MEN На
(синдроме Сиппля) добавляется гиперпара-
тироз, а при MEN lib синдроме Горлина (он
же — MEN III) гиперпаратироз нетипичен,
зато бывают невромы кожи и слизистых,
ганглионевромы и внешний вид
пациентов может напоминать синдром Марфана
(арахнодактилия, высокий рост, длинные
конечности).
Завершив рассмотрение общей
патологии эндокринной системы, мы должны
присоединиться к мнению А.Т. Камерона
(1945), полагавшего что: «Взаимодействие
различных эндокринных желёз является
примером изумительной корреляции
функций животного организма».
Следующие разделы посвящены
обсуждению частной патологии желез
внутренней секреции. Читатель должен учесть, что
материал, касающийся сахарного диабета
вынесен в отдельную главу 9. Ожирение
рассматривается в главе «Нарушения ли-
пидного обмена». Детальная характеристика
гипоталамо-гипофизарногонейросекретор-
ного аппарата и надпочечников,
содержащаяся в т. I данного руководства (с. 530-566),
в этой книге не будет повторена, хотя
эндокринологические аспекты патологии
гипоталамуса, гипофиза и надпочечников,
не связанные с темой стресса, включены
именно в данный том. Эндогенные опиаты
Нарушения эндокринной регуляции 497
как нейроэндокринные регуляторы
рассмотрены в т. I (с. 561-565,581-586).
ПАТОЛОГИЯ ГИПОТАЛАМО-
ГИПОФИЗАРНОГО
НЕЙРОСЕКРЕТОРНОГО
АППАРАТА
Архетипы поведения человека, связанные
с пищевыми, половыми, непроизвольными
двигательными формами активности,
эмоциями, положительным и отрицательным
подкреплением действий и поддержанием
температурного и метаболического гомеос-
таза, контролируются лимбической системой
(рис. 93,94). Лимбическая система включает
древнейшую часть коры — палеокортекс
(фронтоорбитальная кора, подмозолистая
извилина, поясная извилина, парагиппокам-
пальная извилина и крючок). Связанный
двусторонней коммуникацией с неокор-
тексом, палеокортекс поясом охватывает
субкортикальные лимбические
структуры — перегородку, параольфакторную
область, эпиталамус, передние ядра чертога,
часть базальных ганглиев, морской конёк,
миндалину. В сердцевине этого лимба
лежит гипоталамус — главный выходной
информационный канал лимбической
системы. Он связан с чертогом, куда стекается и
где переключается сенсорная информация,
касающаяся всех видов чувствительности.
Медиальный пучок переднего мозга,
пронизывающий его от палеокортекса через
подбугорье, к ретикулярной формации,
позволяет гипоталамусу иметь двустороннюю
нервную связь с центрами вегетативной
нервной системы в стволе мозга. Книзу
гипоталамус переходит в воронку и
соединяется ножкой с гипофизом через нервные
проводники и портальную систему сосудов.
Гипофиз служит главным «интерфейсом»
гипоталамуса во взаимодействии с
периферическими железами внутренней секреции.
В области подбугорья гематоэнцефаличе-
ский барьер разрешает доступ регуляторов
и метаболитов в ЦНС, чтобы обеспечить
заднегипоталамические ядра
(гипертензия, расширение зрачков,
дрожь, „центр теплопродукции",
пробуждение, симпатические центры)
дорзо-латеральные ядра
(вне разреза)
(- центры аппетита, жажды,
ангиотензиновые рецепторы)
вентромедиальное ж
(насыщение, соматолиберин,
нейросекреция)
дорзомедиальное ядро
(регуляция ЖКТ, КРФ)
перифорникальное ядро
(голод, гипертения, гнев, КРФ)
сосцевидное тело
(пищевые рефлексы)
аркуатное и
перивентрикулярное ядра
(либерины, статины, КРФ,
лептиновые рецепторы,
пищевое поведение, нейропептид Y)
интегративные
ядра .
паравентрикулярное ядро
(окситоцин, вазопрессин, КРФ)
установочные
точки
медиальная преоптическая
область (сокращения мочевого
пузыря, брадикардия, гипотензия,
сон, парасимпатические центры)
задняя преоптическая и
передняя гипоталамическая
область (термостат — „центр
теплоотдачи", потоотделение,
тиростатин)
перекрест
зрительных нервов
супраоптическое ядро
(вазопрессин, КРФ)
Рис, 93, Блок-схема функциональных зон и ядер гипоталамуса. Пояснения в тексте. Сокращения:
КРФ — кортиколиберин, АД — артериальное кровяное давление, ЖКТ — желудочно-кишечный
тракт
Рис. 94, Лимбическая система и
взаимодействующие с ней образования
(по А. Гайтону, 1989)
Извилина морского конька
Нарушения эндокринной регуляции 499
канал обратной связи между мозгом и
метаболизмом Хотя гипоталамус
обменивается сигналами с вышележащими
отделами ЦНС, его регуляторные образования
продолжают функционировать даже после
отделения от них, что говорит о решающей
роли гуморальных сигналов, переносимых
кровью, для работы его сервомеханизмов.
Гипоталамус вовлекается в реакции лим-
бической системой головного мозга и
обеспечивает эмоциональный и поведенческий
ответ лимбических структур, адекватный
рефлекторной и эндокринной гомеостазиру-
ющей реакции. Поэтому его можно считать
центром, обеспечивающим вегетативную
основу эмоционально-поведенческих реакций,
что крайне важно для обучения и даже для
специфически человеческих социальных
форм адаптации. Гипоталамус может быть
охарактеризован как орган биосоциальной
интеграции или аффективный дирижёр
поведения.
Немаловажно, что именно гипоталамус —
это отдел промежуточного мозга, занятый
контролем установочных точек гомеостаза.
Здесь имеются вегетативные нервные
центры, задающие термостатную установочную
точку (см. т. I, стр. 379-384), массостатную
установочную точку (см. раздел
«Нарушения липидного обмена»), осмостатную
установочную точку, а также баростатические
и другие центры поддержания балансовых
констант организма. Физиологически это
выражается в регуляции теплоотдачи и
теплопродукции, аппетита и насыщения,
жажды и диуреза, индукции противоположных
изменений кровяного давления.
Дирижирующие воздействия гипоталамуса на обмен
веществ осуществляются по принципу
компенсации отклонений метаболических
констант от установочных точек за счёт
координированного ответа эндокринной и
автономной нервной системы.
Таким образом, уникальное положение
гипоталамуса и нейросекреторные потенции
его нейронов делают этот небольшой отдел
мозга, составляющий всего 0,8 % его объема,
главным нейроэндокринным регулятором
(рис. 93).
Гипоталамо-гипофизарный нейросекре-
торный комплекс представляет собой высший
нейроэндокринный трансмиттер организма,
координирующий эндокринную регуляцию
обмена веществ с работой вегетативной
нервной системы и интегральными
эмоционально-поведенческими реакциями лимби-
ческой системы (К. Ледерис, У.Вил, 1978).
Он состоит из нескольких важных
отделов:
>• гипоталамуса, рассматриваемого как
отдел промежуточного мозга и центральное
звено лимбической системы (рис. 94);
>- нейрогипофиза (передняя часть —
срединное возвышение, задняя часть —
собственно задняя доля гипофиза);
>• аденогипофиза (передняя доля,
существующая у всех индивидов, и
промежуточная доля, сливающаяся у человека
с передней и как обособленное
образование представленная только у плода,
детей до 1 года и при беременности).
Как уже говорилось выше, ЦНС участвует
в регуляции метаболизма и подвержена,
в свою очередь, регуляторному влиянию
гормонов и метаболитов. Мозг располагает
для влияния на метаболизм несколькими
каналами.
Парагипофизарная нервная регуляция
осуществляется по пути гипоталамус —
медиальный пучок переднего мозга —
ретикулярная формация — вегетативные
симпатические и парасимпатические нервные
центры — черепно-мозговые и
спинномозговые нервы — эффекторные органы. Прямую
иннервацию, влияющую на метаболические
процессы, получают печень, жировая ткань,
ЖКТ, поджелудочная железа и островки
Лангерганса, паращитовидные железы,
щитовидная железа и мозговое вещество
надпочечников, кости.
Парагипофизарная гуморальная
регуляция использует ток
цереброспинальной жидкости и осуществляется по пути:
гипоталамус — цереброспинальная
жидкость — венулы сосудистого органа
концевой пластинки — системный кровоток.
500 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Цереброспинальный путь передачи
центральных гормональных влияний изучен
мало, но привлекает растущее внимание
исследователей в связи с возможностью
вмешательства в его деятельность при
процедуре ликворосорбции (Н.Г. Михайлова
и соавт., 1998). Гормоны гипоталамуса и
гипофиза могут цереброспинальным путем
достигать не только системного кровотока,
но и спинномозгового канала. С участием
системы цереброспинального ликворного
транспорта провоцируются их системные
эффекты, например усиление двигательной
и подавление пищевой активности кор-
тиколиберином. Часть гипоталамических
продуктов может попадать в системный
кровоток из портальной системы, используя
ветви верхнегипофизарной артерии.
Трансгипбфизарная гуморальная
регуляция осуществляется либеринами и статина-
ми мелкоклеточных нейронов гипоталамуса
через портальную систему в аденогипофизе,
а также включает секрецию
крупноклеточными нейронами гипоталамических нона-
пептидных гормонов в кровь в пределах
нейрогипофиза. Этим путем регулируется
деятельность коры надпочечников, гонад,
щитовидной железы, молочных желез, а
также координируются ростовые и
анаболические процессы, функции жировой ткани,
водно-солевой метаболизм и многие другие
процессы.
Патофизиологию этих
взаимодействующих образований целесообразно
рассмотреть отдельно.
Патофизиология гипоталамуса
Гипоталамус вместе с нейрогипофизом
развивается из выпячивания нейроэктодермы
дна промежуточного мозга. Он насыщен
нейросекреторными клетками.
Гипоталамические мелкоклеточные ней-
роныу имеющиеся в аркуатном ядре, пери-
вентрикулярной зоне, вентромедиальном и,
частично, паравентрикулярном и супраоп-
тическом ядрах, синтезируют (по крайней
мере, частично — нерибосомальным путём)
и выделяют стимуляторы секреции гипофи-
зарных тропных гормонов (либерины или,
при неидентифицированной структуре, —
рилизинг-факторы), а также ингибиторы
секреторной деятельности аденогипофи-
за — статины (при неидентифицированной
структуре — ингибитинг-факторы). Автор
идеи гипоталамической либерин-статиновой
регуляции М. Саффран первоначально
полагал, что для каждого аденогипофизарного
гормона имеется пара контрарегулирующих
факторов, но реальная регуляция оказалась
экономнее. Так как доминирующее влияние
гипоталамуса на секрецию пролактина в
гипофизе является тормозным, а на
продукцию остальных аденогипофизарных
регуляторов — стимулирующим, то
пересечение ножки гипофиза вызывает пангипопи-
туитаризм, но растормаживает и повышает
пролактиновую секрецию.,
Краткая характеристика производимых
в мелкоклеточных ядрах гипоталамических
биорегуляторов аденогипофиза содержится
в табл. 31.
Многие гормоны этой группы
синтезированы и доступны как лекарства (тиро-
либерин, гонадолиберин и др.). Особенно
перспективно применение длительно
действующего аналога соматостатина — октрео-
тида, который, в частности, эффективно
подавляет гормонообразование в самых
различных апудомах.
Нейросекреторные клетки гипоталамуса
помимо этого вырабатывают множество ней-
ропептидов паракринного местного действия
и содержат к ним рецепторы. Эти пептиды
очень важны в регуляции поведения и го-
меостаза. Нейропептид Y в аркуатном ядре
стимулирует аппетит и пищевое поведение,
его продукция чувствительна к лептину ади-
поцитов и к гормонам ЖКТ (см. «Липостат
и патофизиология ожирения»). Ангиотен-
зин II — пептид жажды в латеральных
гипоталамических ядрах. Холецистокинин
стимулирует насыщение. Нейротензин служит сомато-
маммотироидным ингибитором и индуктором
гипотермии и гипотензии. Опиатные пептиды
регулируют болевую чувствительность и осу-
Нарушения эндокринной регуляции 501
Таблица 31. Биорегуляторы мелкоклеточных нейросекреторных клеток гипоталамуса.
(*—пептиды, полностью изображенные на рис. 95)
Гормон
Тиролиберин
Кортиколиберин
Люлиберин
Соматолиберин (соматокринин)
Соматостатин
Пролактостатин (основной)
Пролактостатин-пептид
Меланостатин
М С Г-рилизинг фактор
Пролактин-рилизинг фактор
Структура
Пироглутамилгистидилпролина-
мид*
Пептид из 41 аминокислоты
Декапептид*
Пептид из 44 аминокислот
Пептид из 14 аминокислот, ЖКТ
производит прогормон из 28
аминокислот*
Дофамин
Пептид из 56 аминокислот
Пролиллейцилглицинамид*
Не идентифицирована
Не идентифицирована
Примечание
Одновременно — слабый пролакто-
либерин
См. т. I, стр. 503-505
Одновременно — фоллиберин
Главный стимулятор продукции СТГ
Одновременно — тиростатин,
слабый кортикостатин. Снижает
продукцию ряда гормонов ЖКТ
Переднее перивентрикулярное и
аркуатное ядра
Карбокси-конец предшественника
люлиберина
Карбокси-конец окситоцина
Стимулятор продукции МСГ
Предположительно ВИП
ществляют положительное эмоциональное
подкрепление поведения. Они модулируют
пищевое поведение, стимулируют продукцию
соматолиберина, МСГ-рилизинг-фактора,
при тормозя этом выработку пролактостати-
на, тиролиберина и гонадолиберина, а также
подавляют продукцию вазопрессина и корти-
колиберина, умеряя стресс.
Подробные данные об этих и других ги-
поталамических нейропептидах имеются
в т. I данного руководства, с. 537, табл. 37.
Структура важнейших из них представлена
на рис. 95.
В нейросекреторных элементах
гипоталамуса производятся и биогенные амины.
Выше уже шла речь об уникальной пролак-
тостатической роли дофамина. Этот амин,
кроме того, подавляет продукцию гонадо- и
тиролиберина, а также соматостатина. Серо-
тонин увеличивает продукцию практически
всех либеринов. Наоборот, ГАМК — тормоз-
ный медиатор гипоталамуса. Норадрена-
лин обладает усиливающим действием на
кортиколибериновую и тиролибериновую
активность. Ацетилхолин усиливает
кортиколибериновую и соматолибериновую
активность. Гистамин снижает продукцию
пролактостатина.
Сложные механизмы саморегуляции
гипоталамической секреции действуют так,
чтобы избегать одновременного усиления
продукции контрарегуляторных гипотала-
мических факторов. Реципрокные
взаимоотношения и сервомеханизмы в норме
практически исключают параллельное действие
стимуляторов, приводящих к активации
противоположных метаболических путей.
При патологии, однако, эти соотношения
могут нарушаться. Так, один из негативных
аспектов воздействия переохлажения
заключается в том, что при гипотермическом
стрессе активируется как продукция корти-
колиберина, так и выработка тиролиберина,
а конечные результаты усиленного действия
адренокортикальных и тироидных гормонов
на энергетический метаболизм во многом не
совпадают, особенно это касается
соотношения запасаемой и рассеиваемой энергии.
Гипоталамические либерины служат
и секреторными, и митогенными
стимуляторами для клеток аденогипофиза. Их
действие осуществляется через рецепторы,
ассоциированные с G-белками, путём
повышения в клетках мишенях концентраций
ц-АМФ и кальция, ускорения кругооборота
инозит-фосфатидов и мобилизации
соответствующих протеинкиназ. Они также
502 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Вещество Р
Нейротензин
Р-Эндорфин
АКТГ (кортикотропин)^
©©г
Аигиотеиэии
Окситоцим
ВДМММВД
Ala
Arg
Asn
Asp
Cys
Gin
Glu
Gly
His
lie
№ХЯ
Алании
Аргинин
Аспарагин
Аспаргиновая к-та
Цистеин
Глутамин
Глутаминовая к-та
Глицин
Гистидин
Изолейцин
(й©©@®®®©?
Leu
Lys
Met
Phe
Pro
Ser
Thr
Trp
Tyr
Val
5®ЙХЯ©®
Лейцин
Лизин
Метионин
Фенилиаланин
Пролин
Серии
Треонин
Триптофан
Тирозин
Валин
©
Вазоактивный кишечный полипептид
Q^JSJSJD^JDJS^JDJD^^^^^^JDJDJ^^J^J^^JDJu^^
Соматостатии
(ЗД£)
Холецистокиииноподобный пептид
&ш£&1&ш&ш
Рис. 95. Структура некоторых нейрогормо-
нов и нейропептидов, вырабатываемых в
гипоталамо-гипофизарном нейросекреторном
аппарате (по Ч. Наута и соавт., 1984)
проникают внутрь таргетных клеток и их
ядер и влияют на экспрессию генов и
синтез м-РНК, вызывая в известных пределах
заданных предыдущей дифференцировкой
секреторную переориентацию клеток адено-
гипофиза. Статины тоже опосредуют своё
действие, приводящее к понижению уровня
внутриклеточной ц-АМФ и кальция через
G-белки. Хроническая гиперсекреция либе-
ринов может вызывать не только
гиперфункцию, но и гиперплазию соответствующих
аденогипофизарных клеток-мишеней.
Гипоталамические гормоны попадают
в аденогипофиз по портальной системе
кровообращения, которая включает
капиллярную сеть в гипоталамусе,
принимающую эти регуляторы от мелкоклеточных
нейронов. В области этой капиллярной
сети повышенная проницаемость гемато-
энцефалического барьера даёт возможность
кровяным метаболическим и гормональным
сигналам достигать нейросекреторных ги-
поталамических клеток и модулировать их
секреторные ответы. Далее идут короткие и
длинные собирательные вены воронки
(срединного возвышения) числом до двадцати.
Наконец, имеется вторая капиллярная сеть,
диффузно распределённая в пределах всего
аденогипофиза, где и находятся таргетные
клетки (рис.96). Венозные коллекторы от
аденогипофиза ведут через нейрогипофиз
в гипофизарные вены и кавернозный синус,
а далее — в системную циркуляцию. Пор-
Нарушения эндокринной регуляции 503
первичная
капиллярная —^
сеть
нижнегипоф\
зарная артерия
тальная система гипофиза
обеспечивает в его
ножке абсолютно наивысший
уровень удельного
кровотока — 10 мл/г ткани в
минуту! В силу полной
достаточности коллатералей в
этом органе даже тромбоз
одной из гипофизарных
артерий не вызывает его
инфаркта. Клиническая
картина некоторых
нарушений функции гипофиза,
ранее приписывавшаяся
тромбозу гипофизарных
артерий, на деле является
следствием аутоиммунного
гипофизита — синдром Ши-
хена (Шиена).
В настоящее время
известно, что существуют
анастомозы, по которым
венозная кровь может поступать
обратно в медиобазальный
гипоталамус и в срединное
возвышение — из нейро-
гипофиза. Это позволяет
нонапептидам
крупноклеточных ядер гипоталамуса
принять участие в регуляции секреции
аденогйпофизарных гормонов и в серво-
контроле продукции гипоталамических
факторов. Характеристика портальной
системы гипоталамуса и гипофиза
приведена также в т. I (стр. 539). Отметим, что без
портальной системы либерины и статины
разводились бы в системной циркуляции и
аффинно извлекались оттуда рецепторами
негипофизарных клеток, что не позволяло
бы поддерживать их эффективные
концентрации в аденогипофизе.
Крупноклеточные секреторные нейроны
гипоталамуса сосредоточены в магноцеллю-
лярных частях супраоптического и паравен-
трикулярного ядер, а также в преоптической
области. Они вырабатывают рибосомаль-
ным путём количественно самые значимые
гипоталамические гормоны — нонапептиды,
с hi as та
optica
carotis
interna
верхнегипофи
зарные артерии
трабекулярная
артерия
^
портальные вены
вторичная
капиллярная сеть
гипофизарная
вена
Рис. 96. Гипоталамо-гипофизарный нейросекреторный аппарат,
его портальная система кровообращения (по В. Лингаппе и
соавт.,1997)
первыми обнаруженные и
охарактеризованные в подбугорье (Э. Шаррер, 1938, В/ Дю
Виньо, 1955). Супраоптическое ядро у
человека выделяет в основном аргинил-вазопрес-
син, главные функции которого — осморе-
гуляция, стимуляция стресса и повышение
кровяного давления, а паравентрикулярное
преимущественно окситоцин, известный как
утеротоник и галакторетик. У свиньи
выделяется эволюционно древний лизил-вазо-
прессин, отличающийся от человеческого
одной аминокислотой в 8-м положении, а
у низших позвоночных — вазотоцин
(имеющий в 3-й позиции не фенилаланин, как у
человека, а изолейцин), который обладает
смешанной вазопрессин-окситоциновой
активностью. По современным
представлениям, у человека понятия «вазопрессин»
и «антидиуретический гормон (АДГ)»
абсолютно равнозначны, хотя ранее и были
разночтения по этому вопросу (рис. 95).
504 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
В связи с этим вазопрессин в дальнейшем
изложении фигурирует и как АДГ.
Нонапептиды крупноклеточных
элементов гипоталамуса транслируются в
виде препрогормонов (21 кД), в структуре
которых находятся специальные
пептиды-переносчики нейрофизины (ЮкД).
Нейрофизин II образует с вазопрессином
комплекс — белок Х.Б. ван Дейка (рис. 97).
Нейрофизин I связывает и переносит окси-
тоцин. Следует подчеркнуть, что, вопреки
ранним представлениям, каждый отдельно
взятый нейрон синтезирует либо окситоцин,
либо вазопрессин, и в нейрогипофизе нет их
общего предшественника. Препрогормоны
гликозилируются в прогормоны (23 кД) и
пакуются в гранулы. Аксовазальным током
они транспортируются в терминали
аксонов, в заднюю долю гипофиза, а частично
— в медиальное возвышение и в III мозговой
желудочек. Поэтому пересечение ножки
гипофиза и его разрушение не лишают
организм вазопрессина и окситоцина, которые
продолжают попадать в аденогипофизарный
и системный кровоток и цереброспинальную
жидкость. Лишь повреждение переднего
гипоталамуса может привести к абсолютно
недостаточной продукции этих гормонов. В
терминалях аксонов часть гранул
представляет собой запасной пул гормонов, а часть —
готовый к кальцийзависимой секреции путём
экзоцитоза. Специальные протеазы
отщепляют от прогормонов нейрофизины II (10
кД) или I (12кД), а также дополнительные
пептиды — например, 39-аминокислотный
вазопрессин-ассоциированныйгликопроте-
ид — в аксонах супраоптического или мела-
ностатин — в аксонах паравентрикулярного
ядра. Все эти агенты попадают в кровь.
Общим стимулятором секреции обоих
нонапептидов является боль, а общим
ингибитором — этиловый спирт. Нонапептиды
закономерно секретируются в системный
кровоток при стрессе.
Регуляция вазопрессиногенеза, изученная
Ж. Вернеем, отражена на рис. 97 и
характеризуется следующими особенностями.
Продукция вазопрессина максимальна
ночью и минимальна в полдень. АДГ —
гормон, секреция которого наиболее сильно
стимулируется (по убывающей мощности)
гипотензией, гиповолемией и, наконец ги-
перосмоляльным состоянием внеклеточной
жидкости. Даже 2 %-ный прирост осмоляль-
ности плазмы от ее нормального уровня в
281,7 мосмоль/кг Н20 ведет к стимуляции
центральных гипоталамических осморе-
цепторов, синтезу и выбросу вазопрессина.
Гиповолемия рефлекторно, через рецепторы
растяжения левого предсердия и легочных
сосудов, стимулирует выброс и синтез АДГ
еще активнее. Этот механизм включается
при дыхании под положительным
давлением, длительном стоянии на одном месте и
быстрой вазодилятации. Напротив, гипер-
волемия, невесомость, ныряние под воду
и охлаждение тормозят этот механизм и
понижают продукцию АД Г.,Немаловажным
последствием этого служит усиленный
диурез, который желателен не во всех
подобных ситуациях.
Ряд стимулов, в частности гипотензия,
тошнота, рвота и курение очень сильно
стимулируют вазоирессиногенез, почти
не отражаясь на продукции окситоцина.
Система продукции вазопрессина имеет
феноменальный резерв мощности и в
экстремальных ситуациях, например при
шоке, способна нарастить его
концентрацию в крови более чем 1000 раз.
Повышение уровня вазопрессина от 2 пМ/л до
500пМ/л при стрессах — обычное дело.
При гиповолемическом шоке она
превышает 600 пМ/л. Медиатор жажды ангиотензин
II стимулирует, а кортикостероиды
тормозят продукцию вазопрессина. Порог жажды
несколько выше порога вазопрессинового
ответа (292 мосм/кг).
Установлено также стимулирующее
влияние на продукцию АДГ ацетилхолина через
Н-холинорецепторы и нейропептида Y.
Гипноз и гиперкапния подавляют
вазоирессиногенез и способствуют диурезу.
Метаболическое действие вазопрессина
обусловлено его влиянием на V2 и Vt —
рецепторы, причём для стимуляции первых
требуются существенно меньшие
концентрации гормона. V-рецепторы ассоцииро-
Нарушения эндокринной регуляции 505
ваны с G-белками, пострецепторный меха- (АП) существует целое семейство (около
низм передачи вазопрессинового сигнала 200, из которых у млекопитающих экспрес-
типичен для этой разновидности рецепторов сируются АП0-АП10). Устройство АП
и включает стимуляцию аденилатцикла- позволяет им образовывать гидрофобные
зы и протеинкиназ (преимущественно у гомотетрамеры, пропускающие молекулу
У2-рецептора),фосфатидил-инозитолового воды (а АП-3,7,9 и 10 — также мочевину
обмена (преимущественно у Угрецептора), и глицерин), но отталкивающие катионы
кальций-кальмодулинового взаимодействия и анионы. АП важны в патогенезе многих
и простагландинового синтеза в клетке-ми- нарушений водно-солевого обмена и ряда
шени (рис. 97,98, а также выше — 89,90). других болезней и синдромов: отека мозга
Спектр эффектов нонапептидов представ- (кетоацидоз блокирует экспрессию АП-1 в
лен в виде сравнительной табл. 32. Ниже нейронах — см. также выше гл. 9), глаукомы,
речь пойдет вначале о влиянии вазопрес- катаракты, кишечной мальабсорбции, нару-
сина. шений слюноотделения и потоотделения,
некоторых форм бронхиальной астмы.
Основной У2-опосредованный эффект АП-2 - посредник эффекта АДГ на поч-
вазопрессина — усиление реабсорбции воды ки — уникален для дистальных канальцев и
(но не электролитов!) в дистальных каналь- собирательных трубок. АП-2 тесно связан
цах и собирательных трубках. Вазопрессин- с патогенезом нарушений вазопрессиновой
зависимый механизм способен реабсорби- регуляции и полиурией. Задержка экспрессии
ровать до 13% всей первичной
мочи. Первую теорию действия
АДГ на почки создал профессор
Педиатрического института
А.Г. Гинецинский в 50-х годах
XX века. По его концепции, АДГ
увеличивает проницаемость
межклеточных промежутков
трубок для воды за счёт
стимуляции активности местной гиа-
луронидазы. В настоящее время
(Х.Валтин, 1992) известно, что
АДГ, через координированное
действие вышеназванных
посредников, способствует
активации цитоскелета эпителиоцитов
почек и фосфорилированию
мембранных белков, которые
собираются в особые кластеры
внутримембранных частиц на
люменальной стороне клетки
(рис. 98) и формируют канал-
переносчик для молекул воды
(эффект «аквапереносящего
белка» или аквапорина-2).
Первый аквапорин был
открыт в 1988 г. П. Эгром,
лауреатом Нобелевской премии по
медицине за 2003 г. Аквапоринов
боль, стресс, ацетил-
холин, никотин
рецепторы
растяжения
левого предсердия
и легочных сосудов]
барорегуляторы
каротидного синуса
и дуги аорты
нейрофизин II
системный кровоток
катаболический
эффект,
экспрессия
VIII I
фактора /
свертывания реабсорбция
воды
эндотелий
-*- сосуды
тромбоциты \
I сужение
Wp. тромбогенеза
Рис. 97. Регуляция секреции вазопрессина и его
эффекты. СОЯ — супраоптическое ядро, ПВЯ — паравентри-
кулярное ядро, V, и V2 — вазопрессиновые рецепторы,
ГАМК — гамма-аминомасляная кислота, vWF— фактор фон
Виллебранда
506 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
тесный
клеточный
контакт
просвет
канальца
(собирательной
трубки)
перитубулярная
жидкость
конгломераты
внутримембранных
частиц
Рис 98. Механизм действия АДГ на почки. Пояснения в тексте.
АП-2 в почках вызывает физиологическую
полиурию новорождённых (Ю.В. Наточин,
2001).
Как единственный гормон, способный
стимулировать почечную реабсорбцию воды без
задержки натрия, АДГ в экстремальных
ситуациях обеспечивает максимальную экономию
водных и волемических ресурсов организма.
Эволюционно это оправдывается тем, что
множество тревожных ситуаций
заканчивается кровотечением. Вазопрессиновый ответ
особенно важен при травмах, кровопотере
и дегидратации. По некоторым данным, ва-
зопрессин способствует и созданию
натриевого градиента петлёй Генле, что важно для
поддержания концентрирующей способности
нефронов. Он активизирует фильтрацию, но
не оказывает натрийуретического действия.
АДГ, как об этом ясно говорит его
второе название, служит важным
регулятором кровообращения,
особенно при
стрессе, коллапсе и шоке.
Вазопрессин (через
Vj -рецепторы)
является мощным вазоконс-
триктором для сосудов
кожи и мышц.
Предполагается его участие
в перераспределении
кровотока при стрессе
и в обеспечении
рабочей гиперемии
функционирующих мышц, а
также в централизации
кровообращения при
компенсированном
шоке. Очевидна роль
вазопрессина в
патогенезе гипертензии, в
том числе стрессорной.
Интересно, что
большие дозы этого
гормона способны сужать и
коронарные сосуды,
что даже послужило
для создания модели
коронарной
недостаточности на собаках (С.И.Теплов, 1962).
Не исключено, что именно вазопрессиновый
компонент стресса имеет наиболее тесную
связь с действием стресса, как фактора риска
сердечно-сосудистой патологии. Вазопрес-
синовые рецепторы в каротидных синусах
делают барорецепторы более
чувствительными к колебаниям системного
артериального давления, что отражает необходимость
более мощного использования барорефлек-
сов для регуляции кровяного давления в
экстремальных ситуациях. Сверхвысокие
концентрации АДГ, создающиеся в пред-
агональных состояниях, способны сильно
изменить и реологические свойства крови.
Кровяные пластинки и эндотелий распола-;
гают вазопрессиновым рецептором и
отвечают на экстремальную гипервазопрессине-
мию усилением тромбогенной активности.
Вазопрессин усиливает экспрессию фактора
фон Виллебранда эндотелием и освобож-
Нарушения эндокринной регуляции 507
Таблица 32. Эффекты нонапептидных гормонов гипоталамуса
Структуры и функции
Реабсорбция Н20 почками
Артериальное давление
Коронарные артерии
Стресс
Перистальтика кишечника
Сокращения матки
Выделение молока
Эйякуляция спермы
Кожные и мышечные сосуды
Метаболическое действие
Тромбоциты
Клеточный иммунитет
ЦНС
Эффект вазопрессина
Сильно тормозит
Повышает при высоких концентрациях
Сужает
Сильная КРФ-подобная активность
Стимулирует
Стимулирует
Слегка стимулирует
Слегка стимулирует
Сужает
Сильный активатор глюконеогенеза,
гликолиза, гликогенолиза и
окисления жирных кислот в печени. Слегка
увеличивает липогенез в адипоцитах.
Антикетогенное действие
Активатор адгезии, агрегации, тромбо-
генеза — в больших концентрациях
Не влияет
Усиливает память, обучаемость,
тревогу. Понижает реакцию на боль
Эффект окситоцина
0,5% от эффекта вазопрессина
Слегка понижает
Слегка расширяет
Слегка усиливает эффект КРФ
Не влияет
Мощный стимулятор
Основной стимулятор
Сильно стимулирует
Расширяет
Значительно стимулирует
липогенез в адипоцитах. Антикетогенное
действие
Не влияет
Активирует
Понижает реакцию на боль
дение печенью VIII фактора свёртывания
(через У2-рецепторы), тем способствуя
гемокоагуляции. Синдром диссеминиро -
ванного внутрисосудистого
свёртывания — тромбообразования, регулярно
сопровождается сверхвысокими уровнями АДГ.
Вазопрессин — гепатотропный
метаболический гормон, активатор ряда важных
обменных процессов. Он стимулирует у
голодных животных печёночный глюконеогенез,
у сытых — гликолиз, в обоих случаях
тормозит образование и ускоряет распад гликогена.
Под влиянием вазопрессина усиливается
зависящая от гормонов надпочечников
гипергликемия. Вазопрессин усиливает
захват и окисление печенью жирных
кислот и их этерификацию. Он предупреждает
кетогенный эффект глюкагона, продукция
которого при стрессе тоже увеличена. АДГ
обладает инсулиноподобным действием на
жировую ткань, стимулируя в ней липогенез,
захват глюкозы и «убирая» в адипоциты
избыток неэтерифицированных жирных
кислот плазмы. Поэтому под его влиянием
развивается длительное снижение уровня
неэтерифицированных жирных кислот
плазмы и, возможно, АДГ, в какой-то степени,
предохраняет при стрессе от диабетогенного
действия других стрессорных гормонов.
Вазопрессин действует и на ЦНС. В т.1,
стр. 539-540 подробно освещена его роль
как синергиста КРФ и активатора стресса.
АДГ связан с индукцией чувства тревоги и
понижает реакцию на боль. Этот нейрогор-
мон стимулирует память и влияет у крыс
на время сохранения реакций избегания
по отношению к повторным стрессорам.
Г. деВиед установил, что даже небольшие
количества вазопрессина радикально
улучшают обучаемость у крыс (1984).
Совокупность системных эффектов
вазопрессина позволяет расширительно
переоценить его роль в организме. Она, конечно
же, не исчерпывается осморегуляцией, а
может быть охарактеризована как
стимуляция противодействия организма развитию
экстремальных состояний.
Окситоцин как системный регулятор не
остаётся в тени своего более знаменитого
508 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
собрата. Как уже говорилось, боль служит
общим стимулятором, а этиловый спирт —
общим ингибитором продукции нонапеп-
тидов. В связи с этим алкоголь обладает
мочегонным, но никак не родостимулирую -
щим действием. Специфически и наиболее
мощно секрецию окситоцина активируют
нейроэндокринные рефлексы,
возбуждаемые растяжением матки и влагалища, а
также механическими воздействиями на
соски молочных желёз. У мужчин она
стимулируется при рефлексе эйякуляции. Эти
сигналы, а также эстрогены стимулируют
секрецию окситоцина, почти не затрагивая
вазопрессинообразование. Гиперосмоляль-
ность межклеточной жидкости усиливает
выброс окситоцина незначительно. Стресс
характеризуется окситоцин-либерирую-
щим действием. У окситоцина не отмечено
циркадного ритма секреции. У женщин к
моменту овуляции его уровень растет в 3-4
раза, а после нее падает. В родах он может
на короткое время возрасти в 30-50 раз, а
при сосании груди у кормящей матери — в
8-10.
Рецептор окситоцина принадлежит к той
же группе, что и вазопрессиновый. Однако
стимуляция окситоциновых рецепторов
снижает в клетках уровень ц-АМФ.
Возможно, более значительную роль в
опосредовании его эффектов играют внутриклеточный
кальций и открытие ионных каналов в
мембранах клеток-мишеней, по крайней мере,
известно его деполяризующее действие на
миоциты матки и миоэпителиоциты
грудных желёз.
Окситоцин усиливает сокращения матки
при идущих родах с момента растяжения
родовых путей, ускоряет течение родов и
отхождение последа, но запускать роды не
может. Пермиссивное синергичное действие
на маточный эффект окситоцина оказывают
эстрогены. При дефиците окситоцина роды
возможны, но удлинены. Для рефлекторной
экскреции молока из грудной железы
окситоцин необходим, при его отсутствии она
прекращается. Этими эффектами
определяется клиническое применение окситоцина
как лекарства.
Окситоцин обладает в почках слабым
вазопрессиноподобным эффектом, а кроме
того, стимулирует кровоток и фильтрацию
в кортикальных нефронах. Метаболически
он проявляет инсулиноподобное действие
на жировую ткань, стимулируя в ней липо-
генез, захват глюкозы и свободных жирных
кислот плазмы. Поэтому под его влиянием
развивается длительное снижение уровня
неэтерифицированных жирных кислот
плазмы, и, возможно, вместе с АДГ в какой-то
степени окситоцин предохраняет от кетоза
при стрессе. Окситоцин снижает ответ ЦНС
на боль, что может иметь физиологическое
значение при родах. Имеются
свидетельства, что данный гормон стимулирует
клеточный иммунитет (Е.А. Михайлова и
соавт., 1986).
Кроме вазопрессина и окситоцина, в
задней доле гипофиза человека обнаружен
родственный им нейропептид когерин,
стимулирующий in vitro и у животных в
экспериментальных условиях ритмические
перистальтические сокращения кишечника.
Остаётся неясным, играет ли он какую-то
физиологическую роль in vivo у людей и
освобождается ли в системный кровоток, а
также каково его взаимодействие с мотили-
ном энтериновой системы.
Описывая регуляцию продукции нанопеп-
тидов и их эффекты, мы уже коснулись
патологических последствий дефицита
окситоцина. Что касается вазопрессина, то в связи с
большой значимостью и относительной
распространенностью форм патологии,
связанных с нарушениями его продукции и
рецепции, они будут рассмотрены отдельно ниже.
Нарушения функций гипоталамуса
разнообразны и порой трудно отделимы от
гипофизарных нарушений. Ниже
описываются вначале общая этиология и патогенез
синдромов, свойственных поражениям тех
или иных частей гипоталамуса, а затем
некоторые отдельные нозологические единицы
применительно к нарушению продукции
отдельных гипоталамических регуляторов.
При этом ожирение, первичная и ряд
вторичных форм которого в настоящее время
считаются патологией гипоталамического
липостата, вынесено в главу «Нарушения
липидного обмена».
В связи с двойственной — нервной и
эндокринной — природой гипоталамуса
его поражения приводят к симптоматике,
которую и неврологи, и эндокринологи
рассматривают в контексте своей
специальности. Патофизиологически важно помнить
о том, что нейросекреция сосредоточена
именно в тех же клетках, которые участвуют
в рефлекторных цепях. Поэтому
применяемые в разных конкретных медицинских
специальностях термины не исключают
друг друга, а часто — синонимичны. Так,
диэнцефальные синдромы в невропатологии
и шпоталамические эндокринопатии в
эндокринологии — это две стороны одного и того
же патологического процесса. Обратимые,
начальные проявления гипоталамической
дисфункции могут условно обозначаться в
практике как «вегетативная дистопия» или
даже «астено-невротический синдром».
Гипоталамусу свойственны полярное
расположение центров, регулирующих
противоположные функции (например,
центры насыщения расположены вентро-
медиально, а голода — дорзолатерально;
парасимпатические ядра, а также центры сна
и теплопродукции находятся в переднем,
а симпатические центры пробуждения и
теплоотдачи — в заднем гипоталамусе и
т.д.). Это приводит к возникновению гомео-
статических и поведенческих расстройств
противоположного знака при разной
топографии очаговых поражений подбугорья.
Кроме того, билатеральные центры имеют
обширные перекрёстные нервные связи,
следовательно, для формирования
клинических расстройств необходима
двусторонняя альтерация гипоталамуса. Массивные
медиальные поражения часто
распространяются на близко лежащие симметричные
центры, а латеральные достигают
необходимых для этого размеров редко. Поэтому,
например, гипоталамическое ожирение
при двусторонней деструкции медиальных
Нарушения эндокринной регуляции 509
центров встречается гораздо чаще, чем ги-
поталамическая кахексия. При локализации
процесса в районе медиального возвышения
даже одиночный очаг небольших размеров
дает картину серьёзных нейроэндокринных
расстройств из-за нарушения функции
портальной передачи гормонов.
При хроническом развитии
гипоталамического процесса нижележащие нейроэн-
докринные центры могут компенсировать
многие его проявления. Острое выпадение
функций подбугорных образований
приводит к выраженным расстройствам. Поэтому
при хронических процессах (например,
опухолях) даже большой по объёму
местный очаг часто не сопровождается
отчётливыми симптомами. В то же время совсем
небольшие острые локальные повреждения
(травмы, ишемические и геморрагические
инсульты) чаще ярко симптоматичны.
Острые и хронические поражения подбугорья
нередко полярны и по знаку наступающих го-
меостатических расстройств. Гипергликемия
и гипертермия более типичны для острых, а
гипогликемия и гипотермия — для
хронических процессов. Острое тяжелое диффузное
повреждение подбугорья, случайное
нейрохирургическое или экспериментальное
разрушение гипоталамуса ведут к коме и
атональной недостаточности витальных
вегетативных функций, хроническое поражение
проявляется нарушениями когнитивной
сферы и метаболическими расстройствами.
(У. Наута, А. Фейертаг, 1984; Л. А. Фромен,
1987; Дж. Дэниеле, Дж. Мартин, 1994).
Общая этиология гипоталамопатий
характеризуется действием различных
факторов в разном возрасте.
У новорожденных они вызываются
родовой травмой, кровоизлияниями,
гидроцефалией, ядерной желтухой.
У детей проявляются семейные
наследственные синдромы (Лоуренса-Муна,
Барде-Бидла, Прадера-Вилли — см. стр. 216 и
далее). Могут приводить к диэнцефальному
поражению менингиты и энцефалиты.
Известен, например, вирусный энцефалит
Экономо с поражением гипоталамуса и
летаргией.
510 АЖ Зайчик, ЛЛ. Чурилов Патохимия
В зрелом возрасте большое значение
приобретают токсические поражения
гипоталамуса, в частности, в лидеры по частоте
выходит синдром Вернике-Корсакова (см.
«Патофизиология витаминного обмена»),
когда из-за хронической алкогольной
интоксикации возникают полигиповитаминоз,
демиелинизация, нейропатия и
своеобразное диэнцефальное расстройство с потерей
краткосрочной памяти. Гипоталамус — один
из самых радиочувствительных отделов
ЦНС. Известны гипоталамопатии у детей и
взрослых при ятрогенном лучевом
воздействии и профессиональном облучении.
У лиц любого возраста, в первую очередь
молодых, важной причиной
гипоталамопатии являются негормонообразующие и
гормонообразующие опухоли подбугорной и
соседствующей локализации (по убывающей
частоте: краниофарингиомы и их
разновидности — эпендимома, эпидермоидная киста;
астроцитомы, дисгерминомы — включая так
называемые «эктопические пинеаломы»,
ганглиоцитомы). Гипоталамус
повреждается крупными гипофизарными опухолями и
менингиомами, изредка может вовлекаться
в клеточную инфильтрацию при гемоблас-
тозах и саркоидозе. У лиц пожилого возраста
на первый план выдвигаются сосудистые
поражения гипоталамуса, связанные с
церебральным атеросклерозом и последствиями
гипертензии (Ф. Плам, В. ванУйтерт, 1978).
Следует отметить, что часто, при наличии
явного гипоталамического нейроэндокрин-
ного расстройства, в подбугорье не удаётся
визуализовать или обнаружить при аутопсии
внешних признаков какого-либо локального
процесса. Прежде такие ситуации трактовали
как «психогенные расстройства» —
психогенная дисменорея, нейрогенная анорексия и т. д.
Однако и они не лишены нейрохимической
основы и вызваны нарушением регуляции
образования гипоталамических гормонов.
Общий патогенез гипоталамопатии
характеризуется следующими особенностями,
которые комбинируются при разной
этиологии и топике поражений:
>* Синдромом нарушения пищевого гомео-
стаза. Гипоталамус содержит лептино-
idoKpuHHO-метаболические нарушения)
вые рецепторы и воспринимает, либо
выделяет множество регуляторов,
стимулирующих аппетит (нейропептид Y,
эндорфины и энкефалины, ГАМК,
дофамин) и вызывающих реакцию насыщения
(холецистокинин, ВИП, нейротензин,
тиролиберин, катехоламины, серотонин,
соматостатин). Опухоли вентромедиаль-
ного гипоталамуса вызывают ожирение,
полифагию и агрессивное поведение.
Нарушения продукции гипоталамотроп-
ного пептида липоцитов — лептина, а
также его рецепции в гипоталамусе
ведут к расстройству функционирования
гипоталамического липостата и
изменению установочной точки массы тела
индивида. Выше детально анализируется
патофизиология первичного и
гипоталамического вторичного ожирения
(см. «Нарушения липидного обмена»).
Описаны редкие случаи гипоталамиче-
ской анорексии и кахексии, связанной
с поражением центров аппетита. Выше
(в разделе «Голодание») обсуждаются
гипоталамические аспекты, механизмов
нейрогенной анорексии-булимии
(стойкое отсутствие импульсного режима
секреции гонадолиберина, повышение
продукции кахексина, вазопрессина
и кортиколиберина, снижение ответа
аденогипофиза на тиролиберин и люли-
берин, пойкилотермия).
>- Синдром расстройства терморегуляции.
Изменение установочной точки
температурного гомеостаза в гипоталамусе при
посредстве эндогенных пирогенов и про-
стагландиновых модуляторов
происходит при любой лихорадке (см. т. I, гл. 13).
Извращение работы гипоталамического
терморегулятора с активацией термоге-
неза имеет место при декомпенсирован-
ном перегревании и декомпенсирован-
ной гиперосмоляльной дегидратации.
Известны и другие гипоталамические
расстройства гомойотермности — па-
роксизмальная гипотермия-аритмия,
чувствительная к противосудорожным
препаратам, и гипертермия в ответ на
воздействие некоторых нейролепти-
Нарушения эндокринной регуляции 511
ков. Пойкилотермия наблюдается при
повреждении заднего гипоталамуса,
острая гипертермия — при диэнцефальной
травме и инсультах.
> Извращение сна и бодрствования при
гипоталамопатиях может выражаться в
ажитации и бессоннице — при
повреждении переднего подбугорья или сомнолен-
тных состояниях — у больных с
поражением заднего гипоталамуса. Обширные
острые повреждения гипоталамуса ведут
к коме.
> Расстройства
эмоционально-поведенческих и когнитивных функций включают
нарушение памяти (при поражении
медиального гипоталамуса и сосцевидных
телец) и даже слабоумие. Типична утрата
кратковременной памяти с сохранением
долговременной, например, при синдроме
Вернике-Корсакова. Описаны аномалии
эмоционально-мотивационной сферы:
безудержная гневливость и агрессия,
садомазохистское поведение,
дурашливость у гипоталамических больных в
связи с повреждениями центров ярости
(в латеральном гипоталамусе) и
наказаний (в перивентрикулярных ядрах) — см.
также т. I, стр. 531-533.
> Расстройства водно-солевого гомеостаза
характерны для поражения ангиотензи-
нергических регуляторов латерального
гипоталамуса и для нарушений вазопрес-
синогенеза. Последние рассматриваются
ниже отдельно. Помимо несахарного
диабета гипоталамические нарушения
питьевого поведения включают так
называемые церебральную гипонатриемию
и церебральную гипернатриемию.
Клинически эти нарушения расцениваются
как психогенная первичная полидипсия,
адипсия и гиподипсия. При первичной
«психогенной» полидипсии в отсутствие
нарушений вазопрессиногенеза больные
испытывают периодическую обсес-
сивную тягу к поглощению жидкости,
часто — охлаждённой. Развивается
гипоосмоляльная гипергидратация при
гипоосмоляльности мочи. В тяжёлых
случаях может быть набухание клеток и
симптомы «водного отравления» и
«ложного ожирения» (см. «Патофизиология
водно-электролитного обмена»). Это
состояние сходно с синдромом
избыточной продукции вазопрессина, однако при
гиперсекреции вазопрессина больные не
могут экскретировать разведённую мочу
(см. ниже). Вероятно, имеются
особенности чувствительности центров жажды
к ангиотензину и какие-то формы связи
ангиотензиновых и опиатэргическйх
воздействий в гипоталамусе. В отличие
от несахарного диабета осмоляльность
плазмы не превышает 290 мосм/кг, а
вазопрессин не помогает, но приводит
к ухудшению самочувствия. Первичная
гиподипсия характеризуется снижением
реактивности центра жажды или его
повреждением. Несмотря на
гипернатриемию, больные жажды не ощущают,
а функция почек у них нормальная.
>• Вегетативные расстройства также
часто имеют гипоталамическую природу,
ибо в головном мозге нейроны,
воздействующие специфически на функции
вегетативной нервной системы,
сконцентрированы в подбугорье. Часть из них
посылает непрерывающиеся проводники
к спинальным вегетативным центрам,
другие переключают свои моторные
выходы на стволовых структурах мозга.
Примерами поражения вегетативной
нервной системы при гипоталамопатиях
служат диэнцефальный эпилептический
синдром с пароксизмальными приступами
гиперактивности автономных нервов, а
также, в известной степени,
диабетическая автономная нейропатия, механизм
которой связан с ангиопатией сосудов
гипоталамо-гипофизарной системы (см.
«Патофизиология сахарного диабета»).
В подростковой медицине в качестве
обратимого возрастного расстройства
гипоталамических функций может
трактоваться вегето-сосудистая дистония
(Л.И. Левина, Л.В. Щеглова, 2006).
>■ Собственно нейроэндокринные
гипоталамические расстройства — до из-
512 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
вестнои степени условно очерченное
понятие, подразумевающее состояния с
определённой, выясненной аномалией в
продукции тех или иных гормонов под-
бугорья — либеринов, статинов и/или
нонапептидов. Среди них выделяются:
- гипоталамический дизгонадизм;
- гипоталамический гипотироз;
- гипоталамические нарушения сома-
томаммотропной функции;
- гипоталамо-адреналовые
дисфункции;
- аномалии вазопрессиновой
регуляции;
- смешанные нарушения.
В классической отечественной
литературе с её холистическим подходом
применительно к таким расстройствам иногда
употребляется расширительный эпитет
«церебральные», а с точки зрения
свойственного классическим западным
источникам редукционного, дифференциального
подхода предпочтителен термин
«гипоталамические». Следовательно, встретив
наименования «церебральное ожирение»,
«церебральная дисменорея», «церебральная
карликовость», читатель не должен думать
исключительно о коре больших
полушарий или воспринимать такие расстройства
как лишённые органической топической
основы.
Гипоталамический дизгонадизм известен
в нескольких формах. Преждевременное
половое созревание у мальчиков бывает
результатом термином района III желудочка,
инфильтрирующих гипоталамус. Эти
опухоли, прежде именовавшиеся «эктопическими
пинеаломами», секретируют хорионический
гонадотропин и а-фетопротеин; первый
обусловливает ускоренную
маскулинизацию, концентрация второго изменяется для
их диагностики. У девочек pubertatio praecox
может быть результатом гипоталамических
гамартом, вырабатывающих люлиберин.
Препубертатной формой гипоталами-
ческого гипогонадизма является синдром
Кальмана (ольфакторно-генитальная дис-
плазия). Аносмия сочетается с задержкой
полового созревания, аменореей и
бесплодием. У мужчин развивается первичная ев-
нухоидность. Могут быть цветовая слепота,
глухота, расщелины твердого нёба, верхней
челюсти и губы. Синдром трактуется как
дизэмбриогенетическое нарушение,
касающееся срединных структур мозга и черепа.
Он носит семейный характер и чаще
поражает мальчиков. Нарушается продукция
гонадолиберина, иногда— в сочетании с
тиролиберином. Вводимый таким больным
гонадолиберин оказывает лечебный эффект
(см. также с. 585).
Постпубертатный гипоталамический
гипогонадизм (также: психогеная,
церебральная аменорея или бесплодие) бывает
чаще у женщин. Синдром выражается во
вторичной гипоменорее или аменорее.
Отсутствует пульсовой микроритм секреции
гонадолиберина, и имеется ановуляция,
несмотря на нормальные функциональные
потенции гонадотрофов аденогипофиза и
клеток яичников. Секреция ЛГ в ответ на
гонадолиберин угнетена сильнее, чем ФСГ.
Нередко у больных имеется гиперпролакти-
немия гипоталамического происхождения,
связанная с нарушением сервомеханизмов
контроля гипоталамической дофаминовой
секреции эстрогенами. Нарушению
секреции гонадолиберина и утрате её пульсового
паттерна способствуют резкие изменения
веса и физическая нагрузка. Поэтому
синдром отмечается у балерин, спортсменок,
больных психогенной анорексией-булими-
ей, а также в виде мужского бесплодия — у
мужчин-спортсменов и у энтузиастов
быстрого похудения обоего пола.
Гипоталамический гипотироз — редкое
нарушение, при котором базальный
уровень ТТГ нормален или слегка понижен, но
ответ аденогипофиза на гипоталамическую
стимуляцию отсрочен и усилен. Это может
сочетаться с дефицитом продукции других
либеринов.
Гипоталамические нарушения регуляции
функций коры надпочечников наблюдаются
при тяжелых хронических стрессах, у
пациентов с реактивной депрессией и с аффек-
Нарушения эндокринной регуляции 513
тивными расстройствами. Секреция КРФ
у них перестаёт подавляться глюкокор-
тикоидами. Утрачивается суточный ритм
продукции КРФ и АКТГ. Немаловажно,
что аналогичные нарушения свойственны и
классической болезни Иценко-Кугиинга (см.
ниже), которая в определённом проценте
случаев вызвана именно кортиколиберин-
продуцирующими, автономными от серво-
регуляции опухолями гипоталамуса.
Гипоталамические причины могут
провоцировать нарушения регуляции сомато-
маммотрофной функции.
Идиопатическая гиперпролактинемия-
аменорея у женщин детородного возраста
при исключённой пролактиноме гипофиза
имеет гипоталамическое происхождение
и связана, как полагают, с ослаблением
дофаминергической супрессии пролакти-
ногенеза.
Психосоциальный нанизм описан впервые
у воспитанников сиротских домов
нацистской Германии (и в художественной
форме — Г.Грассом в «Жестяном барабане»).
В дальнейшем было установлено, что при
хронической эмоциональной депривации
и стрессе продукция соматостатина в
гипоталамусе может быть усилена, а реакция
гипофиза на соматолиберин уменьшена, что
приводит к торможению роста, даже при
полноценной диете. При благоприятном
психоэмоциональном окружении рост
восстанавливается. Очень близка по своим
проявлениям к данному расстройству
идиопатическая недостаточность секреции СТГ. При
этом расстройстве в гипофизе не отмечается
локальных патологических процессов,
большинство пациентов дают нормальный гипо-
физарный ответ на соматолиберин, однако
собственная гипоталамическая продукция
соматолиберина, тиролиберина, а иногда
и гонадолиберина понижена. Некоторые
гамартомы и ганглиоцитомы подбугорья
могут вырабатывать соматолиберин. С ними
связывают своеобразную патологическую
форму высокорослости — церебральный
гигантизм — редкий синдром ускорения
роста и полового созревания, с
расширением мозговых желудочков и аномальной
продукцией гипоталамических факторов, но
без признаков СТГ-продуцирующих аденом
гипофиза. Синдром обычно сопровождается
гиперкортицизмом, липодистрофией и гепа-
тоспленомегалией.
Комбинированные гипоталамические
нарушения, как правило, сочетают
дефицит нескольких гормональных функций в
связи с разрушением клеток-продуцентов
соответствующих либеринов. Чаще всего
их причиной служат расположенные над
турецким седлом негормонообразующие
опухоли — краниофарингиомы,
составляющие до 5 % всех внутричерепных
новообразований. При этом могут сочетаться
задержка полового развития, роста, гипотироз,
гиперпролактинемия и несахарный диабет.
Неэндокринные проявления включают гид-
роцефальный синдром и нарушения зрения,
связанные с травматизацией перекрестья
зрительных нервов.
Несахарный диабет
и другие нарушения
вазопрессиновых механизмов
Несахарное мочеизнурение (ср. латинское:
diabetes insipidus — буквально «безвкусный
диабет») — синдром гипотонической поли-
урии и полидипсии, вызванных
неспособностью удерживать воду и концентрировать
мочу из-за недостаточной эффективности
вазопрессинового механизма.
Несахарный диабет (НД) встречается у
0,5 % всех эндокринных больных, чаще
всего — подростков и молодых людей. Имеются
две главные этиологические разновидности
НД: центральная и периферическая.
Центральный НД — результат
нарушения синтеза и секреции вазопрессина. Он
обыкновенно вызван поражением
гипоталамуса или же гипофиза и гипоталамуса.
Центральный НД делится на 4 подтипа
(А.Мозес, Д. Стриттен, 1994). Рис. 99
изображает, как меняется осмоляльность
мочи при изменениях осмоляльности
плазмы крови у больных с разными формами
центрального НД. При первом подтипе (1)
нет секреции АДГ ни в ответ на
дегидратацию, ни после вливания гипертонического
514 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
осмоляльность
мочи,
Моем/кг
1400 1
1200 1
1000 Н
800 "1
600 -\
400
200 1
Осмоляльность
плазмы крови, Моем /кг
Рис. 99. Реакция осмоляльности мочи в ответ на изменения
осмоляльности плазмы крови в норме (N) и при различных
подтипах центрального несахарного диабета (1-4),
пояснения в тексте
раствора. Следовательно, у подобных
пациентов отсутствует активность вазопрес-
син-продуцирующих клеток или убиты
сами эти клетки, а осмоляльность мочи не
реагирует на сдвиги осмоляльности
плазмы. При втором подтипе (2) дегидратация
вызывает резкий прирост концентрации
АДГ в крови, но гиперосмоляльный раствор
не действует. Следовательно, пациенты с
дефектным центральным осморецептором
реагируют на гиповолемию, но не на
прогрессирующий прирост осмоляльности. У
них АДГ-продуцирующие клетки
функционируют. У носителей центрального НД
третьего подтипа повышена установочная
точка осмотического гомеостаза, снижена
чувствительность центрального осморе-
цептора и имеется замедленный ответ ва-
зопрессиногенеза на прирост осмоляльности
плазмы (3). Наконец, описаны пациенты
с центральным НД и сдвигом
кривой, изображённой на рис.
99, вправо. У них, очевидно,
нормален порог функции ос-
морецептора, но снижен объем
секреции вазопрессина,
приходящийся на единицу прироста
осмоляльности плазмы, т.е. ва-
зопрессиногенез относительно
недостаточен (4).
Важно отметить, что
пациенты со всеми этими подтипами
центрального НД, кроме
первого, могут получить
облегчение от фармакологической
стимуляции вазопрессиноге-
неза, в том числе при курении,
так как никотин активирует
АДГ-продуцирующие клетки.
Обычно для этбй цели
применяют N-холиномиметики и
клофибрат. Иногда
применяются антидиуретики (хлор-
пропамид). У этих пациентов
также усиливается вазопрес-
синогенез в ответ на тошноту
и рвоту. Клиническая тяжесть
первого подтипа — наивысшая.
При нём необходима
заместительная терапия (десмопрессин
в инъекциях, липрессин интраназально).
Центральный НД бывает наследственным
и приобретенным.
Наследственная форма центрального
аутосомно-рецессивного НД наблюдается
изолированно и в структуре синдрома
Вольфрама (в американских источниках —
синдром DIDMOAD). Расстройство включает,
помимо НД (DI), сахарный диабет (DM),
атрофию зрительного нерва (ОА),
глухоту (D) и вестибулярные нарушения. При
этом изначально уменьшено количество
АДГ-продуцирующих клеток
гипоталамуса. Имеется модель наследственного НД
на крысах (линии Брэттлборо), имеющих
дефицит вазопрессин-продуцирующих
клеток.
Этиология центрального приобретенного
НД включает все поражения гипоталамуса,
перечисленные в предыдущем разделе.
Примерно треть случаев приходится на опухоли,
треть — на гранулёмы, травмы и воспаления.
НД регулярно возникает у децеребрирован-
ных пациентов, жизнедеятельность тела
которых поддерживается искусственно.
До 1/3 пациентов с центральным НД
не имеют указанных выше первичных
процессов, и у них болезнь считается «иди-
опатической», что не значит
«беспричинной». Для развития симптомов болезни
необходимо поражение более чем 80%
АДГ-секретирующих элементов.
Особенно опасны процессы в области воронки,
поскольку здесь конвергируют аксоны
многих АДГ-продуцирующих клеток. По
классическим представлениям, главными
причинами служат местные опухоли (кра-
ниофарингиома), метастазы (рака молочной
железы), ксантомы (болезнь Хэнда-Шю-
лера-Крисчена, см. «Нарушения липид-
ного обмена»), гранулёмы (саркоидоз и
инфекционные гранулёматозы). Не только
механические (родовая), но и
электрические травмы головы могут провоцировать
НД. Во многих случаях «идиопатического»
НД истинной причиной является
аутоиммунный гипоталамический энцефалит (в
неврологии — «диэнцефалит») и/или
аутоиммунный гипофизит (А. Пупляр, 1982) с
лимфоидной инфильтрацией вазопрессин-
продуцирующих структур, аутоантителами
к нонапептидам и нонапептид-синтезирую-
щим нейронам и дегенерацией последних.
Именно такие формы НД провоцируются
вирусными инфекциями: гриппом,
энцефалитом. При родах усиление окситоциноге-
неза, по всей вероятности, тоже вызывает к
действию дополнительные аутоиммунные
механизмы контроля уровня окситоцина.
В связи с этим в структуре послеродового
аутоиммунного гипофизита — синдрома
Шихена (см. ниже) — как правило, имеется
иммунопатологический центральный НД.
Избыток минералокортикоидов
способствует задержке натрия и переустановке
точки регуляции гипоталамического ос-
мостата на более высокий уровень. Поэтому
могут появляться симптомы вторичного
Нарушения эндокринной регуляции 515
центрального НД. Центральные формы НД
не всегда являются необратимыми, а могут
разрешаться по мере регенерации и
нормализации состояния АДГ-продуцирующих
структур.
Периферический НД может быть не-
фрогенным. Нефрогенный НД связан с
неотвечаемостью почек на АДГ и протекает
при высоком уровне АДГ в крови. Он
может вызываться рецепторными или пост-
рецепторными дефектами У2-рецепторов
почек, либо поражением клеток-мишеней
вазопрессина. Важнейшая причина
почечного НД — нарушение из-за различных
рецепторных и пострецепторных дефектов
(всего их насчитывают до 25!) экспрессии
АП-2. Экспрессия АП-2 в почках нарушается
не только при мутациях, обусловливающих
наследственный почечный питрессин-резис-
тентный несахарный диабет, но также при
анаэробной инфекции под действием
токсина клостридий. Последнее приводит при этой
болезни к гипотонической полиурии.
Экспрессия АП-2 и других АП нарушается при
ртутном и никелевом отравлении, что важно
для развития сулемовой почки и никелевой
нефропатии. Простагландин Е2 и цитокины
острой фазы препятствуют, а кинины
способствуют экспрессии АП-2, что существенно
для развития шоковой почки.
Чаще встречаются 2 наследственных
почечных синдрома вазопрессин-резистент-
ного НД, представляющие собой генокопии.
Наследственный НД первого типа
рецессивен, сцеплен с районом q28 длинного плеча
Х-хромосомы и чаще поражает мальчиков,
хотя может проявляться и у девочек. У
большей части этих больных АДГ не вызывает
ц-АМФ зависимого ответа на стимуляцию
У2-рецепторов. Второй тип наследственного
почечного НД аутосомно-рецессивен. У этих
больных ц-АМФ-зависимый компонент
пострецепторного механизма срабатывает,
но отсутствует последующая активация
аквапорина-2. Литий и фториды блокируют
АДГ-рецептор, поэтому существуют
лекарственные формы почечного НД,
поражающие до 1/3 больных, леченных литием и
наркотизированных метоксифлюраном.
516 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Фторид натрия (тот самый ингредиент,
о наличии которого в зубной пасте с
торжеством оповещает реклама) позволяет
моделировать нефрогенный НД на собаках
и крысах. Антибиотик демеклоциклин
блокирует аденилатциклазу эпителия дис-
тальных канальцев и нарушает действие
АДГ. Рецепция АДГ в почках страдает
при гликировании рецептора у больных с
сахарным диабетом, а также при
системных иммунопатологических процессах,
когда возможно аутоиммунное поражение
АДГ-рецепторов. Действие вазопрессина
в почках становится менее эффективным
при гипокалиемии, гиперкальциемии,
обструкции мочевыводящих путей, почечных
заболеваниях, препятствующих созданию
гиперосмоляльности интерстиция почки
(фенацетиновая нефропатия), хотя все
эти состояния не относятся собственно к
НД (Т.Э. Андреоли, 1987; Б. Ривз, Т.Э.
Андреоли, 1992). Полиурия наблюдается и
без НД у больных с осмотическим диурезом
(например, при кетонурии и глюкозурии),
а также при утрате натриевого концентру -
ющего градиента петли Генле (полиуричес-
кая фаза ОПН). Ни та, ни другая ситуации не
относятся к нефрогенному НД и отличаются
от него гипертоничной или нормотоничной
мочой. При НД моча гипотонична (как
правило, при пробе С.С. Зимницкого удельный
вес до 1,005). При пробе с сухоядением,
небезопасной для больных НД, нормальные
испытуемые поднимают удельный вес мочи
до 1,020 за 12 ч. У больных НД он не
превышает 1,010. Нефрогенный НД не поддаётся
лечению АДГ. В полном соответствии с
принципом «Лечи подобное подобным»
иногда при нефрогенном НД с успехом
применяют... хлортиазидные диуретики (на фоне
ограничения потребления соли). Причина их
парадоксального действия на вазопрессин-
резистентные почки состоит в том, что они
вызывают салурию. Снижается скорость клу-
бочковой фильтрации, растет реабсорбция в
проксимальной части канальцев, не
зависящая от вазопрессина, уменьшается доставка
натрия к восходящей части петли Генле и как
следствие способность разводить мочу.
Особая аутоиммунная форма
периферического НД вызвана наличием аутоантител к
самому АДГ. Аутоантитела нарушают
транспорт гормона и вызывают его ускоренную
инактивацию в печени, почках и плаценте.
Д. Миракян и соавт. (1985) обнаружили
аутоантитела к вазопрессину почти у 40 %
детей с периферическими формами НД.
Аутоиммунитет к вазопрессину и вазопрес-
син-секретирующим структурам может
усиливаться при беременности. Кроме того,
плацента — источник мощной пептидазы
гипоталамических нонапептидов, которая
особенно активно расщепляет вазопрессин.
Поэтому беременность обостряет течение
НД, фактически используя и
периферический, и центральный механизмы.
Патогенез любых форм НД зависит от
конкретной этиологии болезни. Нередко НД
развивается столь остро, что пациент
способен указать день и час, с которого он считает
себя больным. Общими чертами патогенеза
являются нарушение водно-солевого
обмена и сопутствующие неврологические
расстройства, которые могут диктоваться
первичным интракраниальным процесом,
или же возникать на почве внутриклеточной
дегидратации.
Вследствие потери воды без натрия (а
сам вазопрессин, по некоторым данным,
даже обладает некоторым натрийурети-
ческим действием, утраченным при НД)
развиваются гипотоническая полиурия
(осмоляльность мочи до 200 мОсм/кг), ги-
постенурия (до 1,005), никтурия. Экскреция
мочи обычно возрастает до 3-6 л, но может
быть и до 18 л в сутки. Более обильная
полиурия не характерна для Н Д и может быть,
скорее, связана с психогенной полидипсией
или иными (осмотическими) механизмами
мочеизнурения. Умеренно растет
концентрация натрия в плазме, что вызывает
жажду. Это ведёт к полидипсии. Многие
авторы (Ф.Фелиг, 1985) указывают, что
лица с истинным НД особенно
предпочитают утолять жажду ледяной водой. Это
расценивается как отражение связи между
вазопрессиновыми и терморегуляторными
центрами в гипоталамусе. У детей НД может
Нарушения эндокринной регуляции 517
дебютировать ночным энурезом, иногда
с проявлениями гипертермии и потерей
в весе. Если больной имеет возможность
регулярно утолять жажаду, его состояние
компенсируется. Однако у беспомощных
пациентов, в бессознательном состоянии,
при недоступности воды, а также при
распространении гипоталамического процесса
на центры жажды и их разрушении течение
НД отягощается. Гиперосмоляльность
межклеточной жидкости прогрессирует и
может достичь 350 мОсм/кг воды. Нейроны
обезвоживаются и съёживаются. Это ведет к
обнубиляции сознания, прогрессирующему
ступору с пониженной реакцией на внешние
раздражители, в конце концов
летаргическое состояние может отягощаться вплоть
до гиперосмоляльной комы, очень сходной с
таковой при сахарном диабете, хотя и
протекающей без гипергликемии. Характерно,
что осмотически активные соединения не
вызывают этого нарушения, если могут
свободно проходить через мембрану клеток
(мочевина). Однако и хлорид натрия— при
НД, и глюкоза — при сахарном диабете —
свободой трансцеллюлярного проникновения не
обладают и «вытягивают» воду из нейронов и
иных клеток. Если гипернатриемия
прогрессирует медленно, нейроны успевают поднять
внутриклеточную концентрацию
собственных осмотически активных метаболитов
(«идиогенных осмолей»), удерживая воду
и смягчая дегидратацию. Но быстрое
повышение осмоляльности плазмы (например,
при остром НД у беспомощных пациентов)
особенно опасно (см. также гл. 9 выше).
Противоположным по характеру своего
патогенеза является синдром неадекватно-
избыточной продукции вазопрессина. В
европейской литературе он известен как синдром
Пархона (в честь выдающегося румынского
эндокринолога К.И. Пархона, первым
описавшего это заболевание в 1933 г. как гипер-
гидропексический синдром или несахарный
антидиабет). За океаном его предпочитают
называть синдромом Шварца-Барттера,
описавших это нарушение на 44 года позже
(У.Б.Шварц, Ф. Барттер; 1967).
Фигурирует также абревиатура SIADH (от англ.:
синдром неадекватной секреции АДГ).
В дальнейшем изложении используется
патогенетический термин «гипергидропек-
сический синдром».
Нарушение представляет собой
гипотоническую гипергидратацию организма из-за
автономной или нерегулируемой продукции
избытка АДГ.
Этиология гипергидропексического
синдрома прежде всего связана с вазопрессино-
мами. Часто это эктопические источники
АДГ. Вазопрессин может синтезироваться
в клетках мелкоклеточного бронхогенного
рака лёгких, в карциномах ЖКТ, мочевыво-
дящих путей, поджелудочной и
предстательной желёз, тимомах, саркоме Юинга и даже
лимфомах и лимфосаркомах (Дж. Серенсен
и соавт, 1995). Возможны и вазопрессиномы
гипоталамичеокой локализации. Помимо
опухолей гипоталамуса, гиперстимуляцию
вазопрессиновых нейронов могут вызвать
лекарства (хлорпропамид, клофибрат,
алкалоиды барвинка, фенотиазины, трицик-
лические антидепрессанты, карбамазепин).
Отравления никотином, дихлофосом и его
аналогами протекают с гиперсекрецией АДГ.
Наконец, ятрогенный гипергидропексичес-
кий синдром возикает от передозировки
препаратов самого вазопрессина.
Местные процессы в гипоталамусе и около
него способны провоцировать гиперсекрецию
АДГ путём раздражения клеток,
вырабатывающих вазопрессин, без их деструкции.
Поэтому гипергидропексический синдром может
наблюдаться при энцефалите, менингите,
местных абсцессах, гематомах и негормоно-
образущих опухолях, демиелинизирующих
нервных заболеваниях. Однако во всех этих
случаях он развивается реже, чем НД.
Значительное число случаев
гипергидропексического синдрома и вазопрессинемии
связано с первичной лёгочной патологией,
даже при отсутствии вазопрессином
(хронические инфекционные пневмопатии, об-
структивный синдром, острая дыхательная
недостаточность, вентиляция под
положительным давлением, гнойные заболевания
плевры и лёгких). Полагают, что при этом
518 ALU. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
усиливается неадекватная состоянию
водно-солевого обмена стимуляция вазопрес-
синогенеза через барорецепторы лёгких. По
неясным причинам синдром сопровождает
также перемежающуюся порфирию.
Так как центральный осморецептор и ос-
морецепторы печени работают при участии
АП, предполагается, что дефекты АП-1,4,8 и
9 в ЦНС и печени, вероятно, могут нарушить
работу контрольных механизмов вазопрес-
синогенеза и обусловить противоположные
по отношению к НД гипервазопрессинеми-
ческие состояния даже без вазопрессином
(И.В.Кабанов, Л.П. Чурилов, 2007).
Синдром неадекватной гиперпродукции
АДГ характеризуется следующими
патогенетическими звеньями. Из-за усиленной
задержки воды (без натрия) развивается
дилюция внеклеточной жидкости и гипона-
триемия. Из-за гиперволемии подавляется
компенсаторная продукция альдостерона.
В этих условиях гипергидратация организма
не сопровождается выраженными отёками, а
носит внутриклеточный характер, поскольку
вода перемещается в область более высокой
тоничности. К. Пархон, однако, описал и
больных с отёчным синдромом. Больные
могут иметь избыточный вес, причём в отличие
от истинного ожирения их масса тела весьма
лабильна. Моча концентрирована, больные
экскретируют переменные её количества,
и бывают периоды олигурии и полиурии с
ухудшением и ремиссией общих
симптомов. Гипервазопрессинемия может иметь
собственные эффекты внутри ЦНС, помимо
гипонатриемии. Больные тревожны,
мнительны, подвержены стрессам,
раздражительны. Картина собственно водного отравления
развивается уже при концентрации натрия
плазмы менее 120 мЭкв/л и включает
головную боль, головокружения, анорексию,
тошноту, характерный «вкус медной пуговицы»
во рту. По мере углубления гипонатриемии
и набухания клеток, в том числе— в ЦНС,
могут, при уровне натрия менее 110 мЭкв/л,
проявляться признаки дезориентации,
сонливости, слабости, психической
«загруженности» в острых тяжелых случаях следуют
аритмия, миоклонус, судороги и кома.
Гипонатриемия может иметь и иные
причины, независимые от секреции АДГ
(см. «Патофизиология
водно-электролитного обмена»), в частности, может быть
связана с избыточным потреблением воды,
превосходящим экскреторный максимум почек
(более 18 л в сутки), или же диктоваться
общей нехваткой натрия в организме и диете.
Характерно возникновение гипонатриемии
при нарушении механизмов поддержания
натриевого градиента в почках (полиуриче-
ская стадия ХПН). Ложная гипонатриемия
свойственна тяжелой гипергликемии, так
как внутриклеточная вода перемещается во
внеклеточный отсек и делает кажущуюся
концентрацию натрия меньше. Все эти
состояния надо дифференцировать с
классическим гипергидропексическим синдромом,
также как и гипонатриемию, связанную с
гипокортицизмом-гипоальдостеронизмом
или гипотирозом. В сложных случаях для
этого, дополнительно к определению
концентраций соответствующих гормонов,
служит тест с водной нагрузкой, поскольку при
истинном гипергидропексическом синдроме
больные экскретируют воду замедленно (см.
также гл. 10).
ПАТОФИЗИОЛОГИЯ
ШИШКОВИДНОЙ ЖЕЛЕЗЫ
«Введение мелатонина у
людей вызывает сонливость...
Несмотря на отдельные
наблюдения
противоположного характера, чтение
длинных учебников патологии
не стимулирует секрецию
мелатонина!»
(Р.А.ДеЛилли, 1989)
Шишковидная железа или эпифиз известна
более двадцати двух столетий, со времён
Герофила (III век до н.э.). Возможно,
именно эпифиз — тот объект
человеческого организма, который вызвал в своей
истории наибольшее количество научных
Нарушения эндокринной регуляции 519
споров. Первооткрыватель эпифиза
считал его контролёром интеллектуальной
деятельности человека. Название органу
триста лет спустя дал К. Гален, считавший
его лимфоузлом. А. Везалий (1543) описал
эпифиз анатомически и постулировал, что
шишковидное тело регулирует циркуляцию
цереброспинальной жидкости, так как орган
этот примыкает к задней части III мозгового
желудочка. Точку зрения К. Галена
разделял первый отечественный исследователь
эпифиза В. Юровский (1695), а
англичанин Д. Гибсон в те же годы развил теорию
А. Везалия и предположил, что эпифиз —
клапан разделения крови и лимфы. Более
возвышенное толкование роли эпифиза
принадлежит Р. Декарту, посчитавшему в
середине XVII столетия именно этот орган
весом 150-200 мг вместилищем
бессмертной души. Видный анатом Ж. Уинслоу
рассматривал этот орган как нервный
ганглий (1732), а Ф.Я. Генле через полтора
века увидел в нём железу (1879). Наконец,
догадку об эндокринной функции эпифиза
первым высказал на заре эндокринологии
Й.О.Л.Хюбнер(1898), который установил
связь между поражениями шишковидного
тела и преждевременным половым
созреванием. Дж.Б. Пелицци (1910) подтвердил
эти наблюдения, вследствие чего опухоли
эпифиза с ранним половым созреванием
получили название синдрома Пелицци
(«macrogenitosomia praecox»).
Русский учёный И.Ф. Цион в конце
XIX-го века начал экспериментальное
изучение шишковидного тела, вводя его
экстракты животным. Однако, в отличие от
нижнего мозгового придатка, экспериментальное
удаление верхнего — технически сложная,
травматичная операция. Поэтому, в
дальнейшем экспериментальное изучение роли
эпифиза сильно отстало от успехов в
физиологии гипофиза. К 1912 г. К. Фоа удалось в
опытах на цыплятах-петушках трёх-,
пятинедельного возраста показать, что эпифизэ-
ктомия ускоряет половое созревание. Позже
Й. Изава воспроизвел эффект ускорения пу-
бертации эпифизэктомией у курочек (1927).
Грегориу и Уррахиа продемонстрировали
признаки активации секреторной
деятельности и даже гиперплазии аденогипофиза
у эпифизэктомированных животных (цит.
по А.А. Богомольцу, 1937). В результате
сравнительно-анатомических исследований
С.С. Халатов высказал мнение о
рудиментарной природе эпифиза, восходящего к
париетальному «третьему глазу» низших
амфибий (1944). В дальнейшем в эпифизе
ряда животных, но не млекопитающих были
обнаружены светочувствительные клетки
(Е.И. Чазов, В.А. Исаченков, 1974). Так
как многие авторы потерпели неудачу при
попытке выделить эпифизарные гормоны,
сформировалась точка зрения, что эпифиз
функционирует лишь до определенного
возрастного периода, приблизительно — до
пубертата, задерживая половое созревание,
а далее инволюцирует или обызвествляется
(Н. Пенде, 1937, М.С. Маслов, 1960).
Однако прорыв в учении об эпифизе
как эндокринном органе, последовал,
когда А.Б. Лернер и соавт. (1958) выделили
и синтезировали гормон шишковидной
железы — мелатонин и доказали его
специфичность именно для этого органа (сейчас
известно и о его продукции в диффузных
эндокриноцитах), а также показали
присутствие как у детей, так и у взрослых (рис. 100).
Р. Дж. Вуртмэн, Дж. Аксельрод и Д.Э. Кел-
ли обнаружили суточную периодичность
эпифизарной секреции мелатонина с
ночным максимумом и показали, что свет
угнетает его продукцию и ускоряет половое
созревание и рост крыс, а эпифизэктомия
нивелирует эти эффекты (1968). В начале
70-х годов всё это привело к своего рода
«эпифизомании» в эндокринологии, когда
проблемы изучения шишковидного тела
привлекали не только научное, но, как в
начале века было с гипофизом, широкое
общественное внимание. Автор этих строк решил
поступить в мединститут в 1973 г., и одним
из ярких впечатлений, подтолкнувших к
такому решению, была... статья в газете
«Правда» — «Восхождение к эпифизу»,
знакомившая общественность с достижениями
лаборатории члена ЦК КПСС академика
Е.И. Чазова в этой области.
520 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Н серотонин
Н ацетилсеротонин
■сн2-снг-ш2"ачс"Г > OIV
трансфераза т^
О
II
CH2-CH2-NH-C-CH3
н т^ т^ н
Н NH
оксииндол ортометил-
трансфераза
О
CH2-CH2-N-C-CH3
и ii a
мелатонин
Рис 100. Гормоны эпифиза. Подчёркнуты биорегуляторы, секретируемые шишковидной железой
в кровь
В дальнейшем были выяснены механизмы
ритмического контроля за секрецией
гормонов эпифиза и их сдерживающее влияние на
гипоталамо-гипофизарныйнейросекретор-
ный аппарат. За эпифизом укрепился статус
«тесных башмаков гипофиза»
(Б.В.Алёшин, 1974). У шишковидной железы
обнаружились и другие гуморальные продукты,
влияющие на функции минералокортико-
идной зоны коры надпочечников, тимуса и
лимфоцитов, гонад (Дж. Фэррел и соавт.,
1959; К.И. Пархон, Шт. Милку, 1962). Так,
введение антиэпифизарной цитотокси-
ческой сыворотки стимулировало рост и
функции клубочковой зоны и тормозило
функции тимуса (А.М.Хелимский, 1969).
Был обнаружен широкий круг эффектов
мелатонина в области иммунитета и анти-
оксидантной резистентности,
непосредственно не связанных с его ритмической
гипоталаморегулирующей функцией (см.
ниже). В последующем мелатонин и
пептидные экстракты эпифиза («эпиталамин»)
стали применяться в клинической практике
(В.Х. Хавинсон и соавт., 1991). Однако
затем интерес к изучению эпифиза несколько
снизился, видимо, в связи с тем, что иные
гормоны шишковидного тела, кроме
мелатонина, так и не были получены в чистом виде.
По крайней мере, первоапрельский номер
канадского медицинского журнала за 1982
год вышел с изящной анонимной шуточной
статьёй-мистификацией о якобы открытом
гормоне шишковидного тела — вольволоне,
«регулирующем положение тела во сне».
Тем самым иронически подводился итог
«моде на эпифиз». Как следствие, например,
глава об эпифизе, присутствовавшая в
ранних изданиях роббинсовского руководства
по патологии, из последнего издания 1997 г.
уже была исключена. Мы все же полагаем
необходимым уделить в данном издании
должное внимание этой, в перспективе
далеко не исчерпанной, теме.
Современные представления о роли
эпифиза сводятся к следующему.
Шишковидное тело — орган, паренхима
которого состоит из светлых и тёмных пине-
алоцитов, относящихся к нейроэндокринной
APUD-системе. Это нейрогормональныи
передатчик, сопрягающий деятельность
гипоталамо-гипофизарного нейросекре-
торного комплекса, а также — трансгипо-
физарно и парагипофизарно — активность
периферических эндокринных желёз и
диффузных эндокриноцитов с природными
ритмическими процессами, прежде всего
Нарушения эндокринной регуляции 5 21
фотопериодическими явлениями. Эпифиз
вовлечен в контроль сезонных и суточных
ритмов половой активности и иных форм
гомеостазирующего поведения. Для многих
животных важно произвести на свет
потомство весной, так как это увеличит шансы
на его выживание. Поэтому и половая
активность большинства животных сезонна.
Освещение в видимом диапазоне тормозит
превращение серотонина в мелатонин, что
способствует активизации гонадотропной
деятельности гипоталамо-гипофизарного
нейросекреторного аппарата. В темновой
период продукция мелатонина усиливается
(так, в 3 часа ночи его концентрация в крови
превышает утреннюю в 3 раза). У хомяков
эпифиз активирует секрецию мелатонина,
если ежесуточный световой период
укорачивается до менее чем 11 часов. Световой
сигнал от сетчатки (рис. 101) поступает
по ретино-гипоталамическому тракту к
супрахиазматическому ядру гипоталамуса,
пейсмекеру эндогенных и акцептору
экологически обусловленных гормональных
биоритмов. Супрахиазматическое ядро
адресует свои сигналы мелкоклеточным
нейроэндокринным клеткам гипоталамуса,
и в первую очередь главному источнику
гонадолиберина — аркуатному ядру. Арку-
атное ядро модулирует выработку
гонадолиберина. Другие гипоталамические факторы
(кортиколиберин, пролактостатин, тиро-
либерин и меланоэргические регуляторы)
также меняют свою активность в зависи-
мелкоклеточные
ядра
эпифиз
спинной мозг
Рис 101. Фотопериодическая регуляция секреции эпифиза с участием ЦНС и гипоталамо-гипофи-
зарный нейросекреторный аппарат. ГТГ— гонадотропины, hv — электромагнитное излучение
522 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
мости от таких циклических воздействий.
Нисходящие тракты несут сигнал от гипо-
таламического пейсмекера в ретикулярную
формацию, что оказывает действие на цикл
«сон-бодрствование», и далее — в спинной
мозг, к вегетативным нейронам,
посылающим преганглионарные волокна в
верхнешейный симпатический ганглий (Р. Мур,
1978; В.Н. Анисимов, Р.Дж. Рейтер; 1990).
Постганглионарные норадренергические
проводники от этих ганглиев в составе
п. pinealis no ходу сосудов проникают в
эпифиз, где иннервируют пинеалоциты. Через
Р-адренорецептор и циклоаденилат-про-
теинкиназный механизм нервный сигнал
влияет на скорость захвата триптофана
пинеалоцитами и на активность ферментов
биосинтеза мелатонина (рис. 101),
особенно N-ацетилметилтрансферазы. В темноте
происходит усиленный захват триптофана
для продукции индолопроизводных и
переход серотонина в мелатонин, причем
завершающий этап — уникальная реакция,
контролируемая специфическим эпифи-
зарным ферментом гидроксииндол-орто-
метилтрансферазой. Важно отметить, что
эпифиз не просто воспроизводит ритмику
входных сигналов. Активность гидроксиин-
дол-ортометилтрансферазы в нём обладает
собственными ритмическими колебаниями.
Возможно, шишковидное тело
чувствительно и к электромагнитному излучению иных
диапазонов, выполняя функции
своеобразного биокомпаса (Е.С. Ром-Бугославская,
1986). Еще в 70-е годы XX века
предполагалось, что техногенное излучение различных
диапазонов может вносить возмущения в
работу эпифиза, и даже были попытки
связывать с этим действием явление акселерации
роста и развития детей. В последние годы
получены прямые свидетельства того, что
нарушение светового режима, избыточное и
круглосуточное освещение, микроволны,
радиоволны, электромагнитные поля и
ионизирующее излучение подавляют продукцию
мелатонина, способствуя увеличению риска
канцерогенеза в молочной и предстательной
железах (Д.Блэск и соавт., 1988; Б.Уилсон
и соавт., 1988).
Мелатонин и неидентифицированные
пептиды эпифиза обладают выраженным
прямым и опосредованным через серото-
нинэргические нейроны воздействием на
подбугорье, попадая туда через кровь и
цереброспинальный ликвор III желудочка,
откуда могут захватываться и
транспортироваться в гипоталамус при участии
танницитов.
Доказано снижение секреции гонадо-
либерина и гонадотропинов, меланоцито-
стимулирующего гормона, тиролиберина и
тиротропного гормона, кортиколиберина и
адренокортикотропного гормона под
влиянием мелатонина. Мелатонин и, возможно,
эпифизарные пептиды являются
индукторами синтеза опиатов в нейросекреторных
клетках. Пептиды эпифиза (но не мелано-
тонин) — тормозят продукцию СТГ и рост
крыс.
Парагипофизарные влияния эпифиза
могут быть адресованы ряду периферических
эндокринных желёз напрямую, поскольку
его продукты поступают в кровь большой
сильвиевой вены. Дж. Фэррел (1960),
обнаружив гиперплазию клубочковой зоны
коры надпочечников при подсадке
шишковидного тела, постулировал
существование «адреногломерулокортикотропина»
эпифиза, стимулирующего пролиферацию
и редуктазную активность клеток этой
зоны, усиливающего биосинтез минерало-
кортикоидов. Эти функции приписывали
1 -метокси-1,2,3,4-тетрагидро- р-карболину,
но не удалось показать факта его
физиологической продукции в эпифизе.
И мелатонину, и его дериватам — 5-ок-
ситриптофолам, а также особенно —
пептидам эпифиза, в частности, 8-аргининвазото-
цину присуще прямое антигонадное
действие на семенники и яичники (Шт. Милку и
соавт.; 1963; Д.У. Черзмен, 1970). В
последнее время установлено, что в зависимости
от пермиссивного фона и состояния гонад,
эпифизарные регуляторы варьируют своё
действие, и определенные их фракции могут
оказать и прогонадное влияние (Е.С. Ром-
Бугославская, 1986). Эпифизарные
факторы оказывают гипогликемизирующее
действие и облегчают течение аллоксано-
вого сахарного диабета. Однако сведения
о влиянии эпифиза на углеводный обмен
противоречивы (Шт. Милку, 1962). Кроме
вышеназванных экспериментальных
данных об экстрактах эпифиза, имеется
клинический синдром, при котором гиперплазия
шишковидного тела сочетается с сахарным
диабетом и инсулинорезистентностью. (см.
гл.9, синдром Рабсона-Менденхолла).
Отмечено контринсулярное действие
препаратов мелатонина. Руководствуясь
этими сведениями и данными о
чувствительности шишковидной железы к
электромагнитному излучению, детские
эндокринологи установили прямую
корреляцию между количеством времени,
которое дети проводят перед дисплеями
компьютеров, и степенью риска развития
ювенильного сахарного диабета.
Имеются ранние работы Н. Пенде (1954)
об антипаратироидном влиянии
экстрактов эпифиза, способствующих переходу
кальция из крови в ткани, что представляет
интерес, так как само шишковидное тело
очень часто кальцифицируется. Но из-за
противоречивости этих данных многими
авторами вмешательство эпифиза в
регуляцию уровня глюкозы и кальция отрицается
(В.И. Кандрор, 1991).
Мелатонин обладает снотворным и седа-
тивным эффектами, угнетает
биоэлектрическую активность мозга и, вероятно, при
посредстве опиатов оказывает анальгети-
ческое действие. Гипомелатонинемия и
снижение экскреции мелатонинопроизводных
с мочой отмечены при бессоннице, болезни
смены часовых поясов, депрессивных
синдромах и психозах.
Патология эпифиза включает
некоторые опухоли, гиперплазию, атрофию,
гипо(а)плазию, ишемический и
геморрагический некроз диспротеинозы, стеатоз,
а также кальцификацию этого органа. При
кальцификации кальцинаты щадят пине-
алоциты, поэтому считается, что функция
эпифиза, несмотря на обызвествление, долго
остается сохранной. Эпифизит с исходом в
глиоз или склероз может поражать шишко-
Нарушения эндокринной регуляции 523
видное тело при нейроинфекциях, сепсисе
и аутоиммунных процессах.
Аплазия (гипоплазия) шишковидной
железы носит врождённый характер или
следует под воздействием периэпифизарных
новообразований, сдавливающих эпифиз.
Она может протекать бессимптомно, но
иногда сопровождается преждевременным
половым созреванием.
Опухоли эпифиза составляют до 1 %
внутричерепных неоплазм. Наиболее часто (в
50 % случаев) это герминомы (также
именуемые тератомами или, при внеэпифизарной
локализации, например в подбугорье,
традиционно неточно обозначавшиеся как
«эктопические пинеаломы»).
Герминомы поражают почти
исключительно мужчин и состоят не из эпифизарных
элементов, а из зародышевых клеток, не
мигрировавших в гонады и переднее
средостение. Именно в средостении и половых
железах возникают аналогичные опухоли.
Герминомы часто прорастают в подбугорье
и соседние с эпифизом структуры, вызывая
ряд характерных симптомов— атрофию
зрительных нервов, несахарный диабет,
гипогонадизм, гидроцефалию из-за
блокады сильвиева водопровода. Давление на
верхние бугорки четверохолмия связывают
с симптомом Парино — сочетанным
параличом взора вверх. Давление на мозжечок
и ствол мозга проявляется характерной
широкой походкой. Так как герминомы в
некотором проценте случаев вырабатывают
хорионический гонадотропин, то они могут
обусловливать преждевременное половое
созревание и ускорение роста.
Данная причина не является
единственной основой для синдрома макрогенитосо-
мии при патологии эпифиза.
Негормонообразующие опухоли
эпифиза— главным образом астроцитомы и
глиобластомы (25 % от числа всех
онкологических поражений этого органа) — могут
нарушать продукцию мелатонина и/или
функцию гипоталамических гонадоли-
беринсдерживающих центров. Это также
ведет к pubertatio ргаесох. Гипотетически
пинеаломы эпифиза— то есть опухоли из
524 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рис 102. Преждевременное половое
созревание (Macrogenitosomia praecox — синдром
Пелицци) у мальчика 1 г. 10 мес. с
гидроцефалией и опухолью мозга. (По Лиссеру-Эсками-
лья, 1982)
его гормонообразующих клеток,
составляющие до 20 % спектра эпифизарных
новообразований, могут секретировать избыток
гонадостатических регуляторов и тормозить
половое созревание. На практике такие
случаи описаны в основном у взрослых обоего
пола, но редки и не всегда сопровождаются
гипермелатонинемией.
Макрогенитосомия (синдром Пелицци)
поражает гораздо чаще мальчиков (95%),
чем девочек. Клинически отмечается
раннее (до 10 лет) проявление комплекса
вторичных половых признаков (рис. 102),
увеличение и функциональное созревание
гонад. Гипергенитализм сопровождается
ускоренным ростом мускулатуры и
скелета, но из-за влияния андрогенов и раннего
закрытия эпифизарных хрящей высокого
роста не бывает. Может отмечаться
увеличение относительной длины туловища, по
сравнению с конечностями (антимарфано-
идный хабитус).
Прогноз определяется характером
опухоли, а также степенью и скоростью
нарастания гидроцефалии. Доброкачественные
пинеаломы, чаще встречаемые у взрослых,
растут медленно и не инфильтративны.
Пинеалобластомы (чаще поражающие
детей) по морфологии и поведению близки
к медуллобластомам (нейробластомам)
головного мозга, растут быстро, инфиль-
тративно и известны своими метастазами
(обычно в спинной мозг). При
неблагоприятном сочетании вышеупомянутых
факторов без лечения больные могут умирать
за 1-2 года, в доброкачественных случаях
опухоль развивается 7-10 лет, не давая
выраженных симптомов (Д.Я. Шурыгин, 1966;
Р.ДеЛилли, 1989).
По мнению ряда авторов, следует
выделять симптомокомплекс гиперпинеализ-
ма — половая инфантильность, евнухоид-
ный вид, высокий рост, длинные
конечности, ожирение, инсулинорезистентность
(Ш. Милку, 1962), однако существование
такой нозологической формы не является
общепризнанным.
Гормон эпифиза мелатонин оказался
не только блокатором активации половых
желез, но и активатором антиоксидантной
системы клеток, стимулятором
противоопухолевого иммунитета (см. также т. I,
с. 79-80). Установлена его способность
повышать активность Т-лимфоцитов и синтез
антител. Пептидные экстракты эпифиза
также стимулируют клеточный иммунитет.
Мелатонин ритмически влияет на секрецию
Нарушения эндокринной регуляции 525
опиатов, контролирующих ряд параметров
иммунологической реактивности.
Эпифиз осуществляет иммуномодулирующее
действие путем стимуляции продукции
эндогенных опиатов. Гормоны эпифиза
оказывают положительный эффект при
раке молочной железы. Удаление эпифиза
ускоряет развитие меланом и способствует
химическому канцерогенезу у подопытных
животных, а экстракты эпифиза обладают
некоторым противоопухолевым
действием.
Поскольку шишковидное тело инволюци-
рует с возрастом и несет определенные
биохронометрические функции, некоторые
авторы высказывали догадку о том, что именно
эпифиз имеет отношение к механизмам
старения (см. также т. I, с. 80). Эпифизарная
недостаточность может играть важную
роль в происхождении старческих психо-
нейроэндокринных и иммунологических
изменений (В.Н. Анисимов, Р.Дж. Рейтер
1990). С возрастом уменьшается секреция
эпифизарного мелатонина (Р.Л. Растинг,
1993). Как считает К.Э.Финч (1990),
обнаруживший, что гипоталамо-гипофизарная
система и управляемые ею гонады своими
гормональными сигналами взаимно
усиливают обоюдное старение, существует
возрастная тенденция повышения порога
чувствительности гипоталамуса к половым
гормонам, способствующая возникновению
гормонозависимых опухолей, в частности
рака молочной железы. Пептидные
экстракты эпифиза способны восстанавливать
ингибирующее действие эстрогенов на
гипоталамус. Наконец, эпифизэктомия
укорачивает, а мелатонин и пептидные гормоны
эпифиза удлиняют среднюю и
максимальную продолжительность жизни грызунов
(О.Мальм и др., 1955, В.Пьерпаоли и др.
1989, В.Н.Анисимов, Р.Дж. Рейтер, 1990).
Оживление интереса к эпифизу связано в
основном с данными исследований конца
ХХ-начала XXI века об удлинении жизни
у позвоночных и беспозвоночных животных
при введении мелатонина (В. Пьерпаоли,
Дж. Маэстрони и др.). Ряд авторов считают
эпифиз ритмическим координатором
работы дисперсной нейроэндокринной системы,
которая по общему производству
мелатонина превосходит саму шишковидную
железу. Найден мелатонин и в лейкоцитах,
а также мастоцитах, что связывают с его
ролью как иммунорегулятора
(В.Н.Анисимов, И.М.Кветной и соавт., 2000). В то
же время, имеются данные о лейкозогенном
действии избытка мелатонина как
эндогенного канцерогена (В.И. Романенко, 1983).
Недавно Д.Льюк (2001) установлено, что
эпифизарный «песок» состоит из апатита
с рекордным содержанием фтора, а
шишковидная железа превосходит все органы
по тропизму к этому микроэлементу. Более
того, имеются данные, что прием
повышенных количеств фтора угнетает продукцию
мелатонина и может способствовать
преждевременному половому созреванию и
укорочению жизненного цикла у
млекопитающих. Эти данные особенно важны ввиду
массового фторирования соли и зубной
пасты в целях профилактики кариеса зубов
в ряде регионов. Несомненно, шишковидное
тело — по-прежнему актуальный объект
медицинских исследований, связанных со
скоростью онтогенеза.
Следовательно, Рене Декарт, возможно,
был не так уж далек от истины, когда
поместил бессмертную душу именно в
шишковидную железу.
Таким образом, маленький орган,
укрытый большими полушариями головного
мозга, несмотря на весь прогресс,
достигнутый в изучении его физиологии и патологии,
еще таит, в ожидании усилий больших
полушарий Читателя, немало загадок.
ПАТОФИЗИОЛОГИЯ
АДЕНОГИПОФИЗА
Гипофиз известен со времён К.Галена
(IIвек н.э.), считавшего его фильтром для
образования носовой слизи. Впоследствии
нижнему мозговому придатку, или, как его
именовали в средние века, «головке розы»,
526 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
не раз приписывали экзотические функции.
А. Пикколомини в XVI веке
охарактеризовал его как пробку, удерживающую в мозге
жизненный дух. Виллизий, Г. Бургхааве и
С. делаБоэ видели в нём орган образования
цереброспинального ликвора или лимфы, а
Ф.Мажанди (1847) — лимфоузел. В
первой половине прошлого века он считался
целиком нервным образованием. Но
изначальную догадку о секреции нижним
мозговым придатком специфических продуктов
высказали еще в XVII столетии А. Литтре.
Секреторные признаки в клетках гипофиза
впервые отметил Й.Ф. Меккель.
Окончательно эндокринные функции признали за
гипофизом после открытия П. Мари связи,
между его аденомами и анарушениями роста
(1886). Русский гистолог П.И. Перемежко
описал аденогипофиз, нейрогипофиз и
гипофизарную щель (1867), а его польский
коллега В. Михалкович (1875) — сложный
эмбриогенез этого органа.
Напомним читателю (см. также т. I данного
руководства, с. 537-538), что нижний
мозговой придаток связан с гипоталамусом ножкой
и находится в фиброзной капсуле, в турецком
седле, углублении основной кости черепа.
Вес его не превышает 900 мг, тем не менее
этот микроорган ведает сложнейшими регу-
ляторными функциями в отношении обмена
веществ, функций периферических желёз,
роста и размножения. Он состоит из
эмбриологически разнородных частей — аденогипо-
физа, происходящего из кармана М.Х: Ратке,
выпячивания крыши ротоглоточной
эктодермы, и нейрогипофиза, опускающегося в
эмбриогенезе вниз и навстречу, в виде впячи-
вания дна диэнцефальной нейроэктодермы
третьего мозгового желудочка. Образуется
сложный орган, объединённый общей
сосудистой системой и капсулой и отделённый
сверху от основания мозга выростом твёрдой
мозговой оболочки — диафрагмой турецкого
седла (рис. 103). В целом надёжность защиты
гипофиза отражает его исключительную роль
в организме: «Ни один орган человеческого
тела не защищен так хорошо, не расположен
столь центрально и не спрятан столь
тщательно» (Х.Кушинг, 1930). Но диафрагма
относительно других частей турецкого седла
представляет более слабое место. При
прободении этой диафрагмы из-за внутричерепной
гипертензии орган может сдавливаться и
даже атрофироваться.
дно промежуточного
мозга
гипоталамус
7lf/£—\ каРман
(Г)/ Ратке
крыша
ротоглотки
туберальная
часть
воронка
нейрогипофиз
ротоглотка
срединное
возвышение
аденогипофиз
дистальная часть
Глоточные гипофизы"
dura
Т. arachnoidea
диафрагма седла
аденогипофиз
нормальная топография
основная кость
Т. arachnoidea ^"^
несостоятельная
диафрагма
седла
Т. arachnoidea аденогипофиГ
синдром пустого турецкого седла
Рис. ЮЗ. Эмбриогенез (вверху) и топография (внизу) гипофиза в норме и при синдроме «пустого»
турецкого седла (ЦСЖ — цереброспинальная жидкость)
Нарушения эндокринной регуляции 527
С этой особенностью связан
развивающийся при первичной несостоятельности
диафрагмы и тенденции к внутричерепной
гипертензии синдром «пустого»
(увеличенного) турецкого седла (К. Буш, 1951).
Его распространенность (обнаружен в 5 %
случаев аутопсий лиц, умерших при
явлениях гипофизарной патологии) заставляет
посвятить несколько строк данной аномалии
локализации гипофиза. Типичные носители
синдрома пустого турецкого седла — тучные,
много рожавшие женщины с повышенным
кровяным давлением. Они нередко
жалуются на головные боли, а 10% даже отмечают
ликворею из носа. На фоне упомянутых выше
анамнестических факторов, способствующих
ликворной гипертензии, дивертикул арах-
ноидной оболочки проникает в седло через
несостоятельную диафрагму. Аденогипофиз
уплощается и оттесняется в сторону. Ней-
рогипофиз чаще всего не смещается.
Топографические взаимоотношения изменены и
выглядят, как на рис. 103 справа.
Рентгенограмма черепа может выявить в полости
увеличенного седла цереброспинальный ликвор
(в арахноидном мешочке), что создает ложное
впечатление отсутствия гипофиза. Функция
гипофиза может совсем не пострадать. Чаще
отмечаются ее минимальные изменения:
гиперпролактинемия, связанная с травмой
ножки гипофиза, снижение стимулированной
секреции СТГ, дефекты полей зрения из-за
давления на перекрестье зрительных нервов.
Синдром дифференцируют от увеличения
турецкого седла при аденомах гипофиза.
Любопытной особенностью синдрома «пустого»
турецкого седля является высокая частота
аутоиммунитета к антигенам дистопиро-
ванного гипофиза — до 70% больных имеют
антигипофизарные аутоантитела.
Отметим, что по ходу эмбрионального
перемещения клеток будущего аденогипо-
физа, в основной кости и рядом с ней, могут
сохраняться остатки аденогипофизарной
ткани — «глоточные гипофизы», служащие
источником опухолей и эктопической
секреции гипофизарных гормонов.
Нейрогипофиз включает заднюю долю,
локализованное на границе с гипоталамусом
срединное возвышение и воронку (инфун-
дибулярную часть), соединяющую заднюю
долю со срединным возвышением.
Патофизиология нейрогипофиза касается
нарушения освобождения в кровь гипота-
ламических нонапептидов — вазопрессина
и окситоцина. Эти вопросы подробно
рассмотрены выше. В данном разделе мы
сосредоточим внимание только на
патофизиологии аденогипофиза. Будут изложены
те вопросы, которые не нашли отражения в
т. I настоящего руководства, где
характеристика гипофиза даётся лишь применительно
к его участию в стрессе (т. I, с. 537-544).
Аденогипофиз, составляющий до 75%
массы органа, включает переднюю долю с ее
дистальной и туберальной частью.
Последняя лежит выше диафрагмы турецкого седла.
Как вкратце уже упоминалось при
рассмотрении структуры гипоталамо-гипофизарного
комплекса, у плода и у детей до 5 лет, а также
при беременности может быть выделена
промежуточная доля с полостью (гипофизарной
щелью). В остальных случаях
промежуточная доля у человека сливается с передней
воедино и представлена рудиментарными
фолликулами и кистами Ратке,
содержащими коллоидно-слизистый секрет.
Аденогипофиз является местом
образования тропных и некоторых других белковых
гормонов, управляющих периферическими
эндокриными железами, ростовыми и
анаболическими процессами, обменом веществ
и размножением. Отечественный гистолог
А. Достоевский в 1884 г. установил наличие
в аденогипофизе трёх клеточных типов —
ацидофилов, базофилов и хромофобов, не
воспринимающих ни кислых, ни основных
красителей. С тех пор считалось, что хромо-
фобы — гормонально неактивные элементы,
из которых в ходе конечной дифференци-
ровки возникают остальные функционально
активные клетки. По мере развития
электронной микроскопии и иммуногистохимии,
в первую очередь благодаря исследованиям
М.Г. Фаркухар (1969, 1971), выяснилось,
что в аденогипофизе 6 клеточных типов:
>* Соматотрофы, относящиеся к ацидофи-
лам, локализованы в основном латераль-
528 АЖ Зайчик, Л.П. Чурилов Патохимия
но, вырабатывают гормон роста (СТГ) и
составляют около 40-50 % аденогипо-
физарных клеток. Их гранулы, размером
300-400 нм, окрашиваются красителем
оранжевым G.
>* Лактотрофы, коих до 20%, а при
беременности — более половины, также аци-
дофильны, находятся преимущественно
кнаружи от соматотрофов и формируют
пролактин. Их неправильной формы
гранулы заметно крупнее — 400-700 нм. Они
прокрашиваются эритрозином. При
лактации число лактотрофов тоже возрастает.
>• Тиротрофы (6%) многоугольны по
форме, базофильны и вырабатывают
ТТГ. Они Гомори-положительны, дают
ШИК-реакцию на гликопротеиды, а ти-
онин-альдегидный метод окрашивания
придаёт их гранулам (диаметром 140-
200 нм) сине-фиолетовый цвет. Больше
всего тиротрофов в переднемедиальной
части аденогипофиза. При первичном
гипотирозе их число резко возрастает.
>* Гонадотрофы (3-4 %), которые
выделяют оба гонадотропных гормона — люте-
инизирующий (ЛГ) и фолликулостиму-
лирующий (ФСГ), также базофильны,
ШИК- и Гомори-положительны, но
форма их округлая, а окраска тионин-
альдегидным методом придаёт их
гранулам (200-250 нм) пурпурно-фиолетовый
цвет. Они локализованы по
преимуществу в глубоких латеральных частях
железы. При кастрации гонадотрофов
становится намного больше, а
беременность снижает их количество, возможно,
в ответ на активацию гонадотропной
секреции плаценты.
>* Кортиколипотрофы, подробно
рассмотренные в т. I, составляют не менее
15-20%, базофильны, имеют красные
гранулы (при комбинированной окраске
ШИК- и тионин-альдегидным методом).
Диаметр гранул — 100-200 нм. Они экс-
прессируют ген проопиомеланокортина
(TIOMK) — общего пептидного
предшественника, содержащего среди своих
265 аминокислот идущие друг за другом
(эндокринно-метаболические нарушения)
полные последовательности нескольких
гормонов:
- у-меланоцитстимулирующего
гормона (у-МСГ),
- АКТГ (в структуре которого могут
быть выделены а-МСГ и CLIP или
кортикотропиноподобный
промежуточный полипептид),
- р-липотропина (из которого могут
образовываться, последовательно, у-ли-
потропин и его фрагмент — р-МСГ);
- р-эндорфина (аминокислотные
остатки с 61-го по 91-й), а-эндорфина
(остатки 61-76) и у-эндорфина (61-77);
- а также N-концевой гликопептид из
76 аминокислот, обладающий мито-
генным действием на адренокорти-
коциты клубочковой зоны
надпочечников.
И ПОМК, и его производные попадают
в кровь и цереброспинальную жидкость.
Процессинг ПОМК с образованием его
фрагментов частично идет на периферии
(например, в надпочечниках), а главным
образом — в аденогипофизе. ПОМК и его
дериваты подробно охарактеризованы в т. I
(гл. 18 и рис. 102). Скопления кортиколи-
потрофов имеются в медиальной мукоидной
части аденогипофиза, в pars tuberalis и на
границе с нейрогипофизом. Туберальные
кортиколипотрофы при первичном гипо-
кортицизме гиперплазируют, в отличие от
локализованных в задней части
аденогипофиза. Гиперкортицизм ведет в кортиколи-
потрофах к трансформации Крукса — когда
отмечаются гиалинизация их цитоплазмы и
исчезновение гранул.
>• Хромофобы (до 20 % клеток
аденогипофиза) имеют лишь скудные гранулы и не
содержат тропных гормонов. В
настоящее время признают, что среди них могут
быть не только незрелые, но и
истощившие свои секреторные возможности
клетки, а также звёздчатые клеточные
элементы с неясными, вероятно,
неэндокринными функциями. Не исключена
продукция в отдельных хромофобах ка-
ких-то гормональных начал — например,
факторов роста, эритропоэтина и т. д.
Секреторные клетки аденогипофиза
вырабатывают свои гормоны на
полисомах ШЭР, в упаковке гранул принимает
участие комплекс Гольджи, секреция идёт
путём экзоцитоза, а вне секреции гранулы
сливаются с лизосомами и разрушаются,
причём это может приводить к образованию
миелиновых фигур.
Говоря о гистологическом составе
аденогипофиза, надо учесть, что, по словам
М. Люсьена, Г. Ришара и Ж. Паризо (1925),
«гипофиз не обладает гистологической
структурой, совершенно постоянной.
Относительное число его различных элементов,
группировка их, морфологический характер
секреторных продуктов подвержены
значительным изменениям при нормальных
условиях, при разных проявлениях
физиологической активности индивидов».
Очевидны значительные изменения клеточного
состава железы при лактации, кастрации,
беременности, зимней спячке животных,
хроническом стрессе, а также с возрастом.
Сводку классических данных по этому
вопросу дает руководство Н.Б. Медведевой
(1946). В настоящее время вопрос ставится
еще радикальнее. Некоторые клетки
аденогипофиза принципиально производят
несколько гормонов (кортиколипоторфы)
путём альтернативного процессинга и/или
протеолиза общего предшественника. По
мнению А. Хэма и Д. Кормака (1979),
многие клетки аденогипофиза, по-видимому,
способны вырабатывать помимо основного
близкородственный ему гормон (не
исключено, по крайней мере — в ацидофильных
аденомах, существование «соматомаммот-
рофов», в которых отмечено присутствие
и СТГ, и пролактина, а также выработка
альтернативных гонадотропинов в одной
клетке). Выше уже отмечалось, что гипота-
ламические либерины и статины служат для
аденогипофизарных клеток не только
функциональными активаторами,
стимулирующими секрецию и биосинтез соответствующих
гормонов, но и регуляторами пролиферации.
Кроме того, биосинтез аденогипофизарных
Нарушения эндокринной регуляции 529
гормонов эффективно подавляется блокато-
ром транспортных РНК пуромицином, тогда
как блокатор транскрипции актиномицин
D не оказывает такого действия. Очевидна
большая роль долгоживущих РНК в
определении интенсивности и направленности
аденогипофизарной секреции (Ю. А. Панков,
1976). В связи с этим имеется точка зрения
(Дж.Саймингтон, 1969), что в каждый
отдельно взятый момент времени
клеточный спектр аденогипофиза и секреторная
ориентация отдельных клеток отражают не
только действие возобладавших в
процессе гистогенеза консервативных программ
клеточной дифференцировки. Известное
значение имеют вариабельные программы
дифференцировки, при действии которых
переключение секреторной ориентации может
достигаться под влиянием гипоталамических
регуляторов и их внутриклеточных
посредников, на посттранскрипционном уровне,
например, варьированием судьбы разных
долгоживущих транскриптов.
По крайней мере, можно считать
доказанным, что аденогипофиз выделяет больше
гормональных начал, чем имеет клеточных
типов.
Патофизиология аденогипофиза
приобрела большой объем сведений в результате
экспериментальной гипофизэктомии. Первым
произвёл эту операцию В. Хорсли в 1886 г.
Начальные попытки осуществлялись
травматическим транссфеноидальным методом.
Усовершенствование операции позволило
применять менее опасные трансназальный и
трансоральный методы, сделав возможным
серийное получение модели гипофизарной
недостаточности (Дж. Асколи, Т. Леньяни,
1912). Последствия экспериментальной
гипофизэктомии представляют собой
комбинированную недостаточность функций
ряда периферических желез (гонад, коры
надпочечников, щитовидной и молочной),
а также — что заметно у молодых
животных — остановку ростовых и уменьшение
анаболических процессов.
Животное прекращает расти, эпифи-
зарные швы преждевременно замыкаются,
отмечается персистенция тимуса, гипоплазия
530 А.Ш. Зайчик, Л.П.Чурилов Патохимия
гонад, надпочечников и щитовидной железы,
задержка пубертата, Развивается
прекращение астральных циклов или обрывается у
кормящих самок лактация. Животные
приобретают дистрофические изменения кожи,
волос и зубов, гипопластическую анемию,
очень характерно развитие иммунодефицита.
Ограничивается ретенция азота,
усиливается потеря воды и хлорида натрия, имеется
гипогликемия, понижается основной обмен,
может быть ожирение. В меру
сопутствующего повреждения гипоталамуса возможны
несахарный диабет, нарушения аппетита и
терморегуляции, потеря в весе. Животные в
исходе такой плюригландулярной
недостаточности, вызванной полной гипофизэктоми-
ей, погибают за несколько дней, если только
не располагают эктопическими источниками
аденогипофизарных гормонов.
В результате экспериментальных и
клинических наблюдений за результатами ги-
пофизэктомии, действием гипофизарных
экстрактов и продуктов, антигипофизарных
иммуноглобулинов, а особенно вследствие
успехов в биохимической и иммуногисто-
химической характеристике
аденогипофизарных регуляторов достоверно установлена
продукция аденогипофизом следующих
гормонов: гормона роста (СТГ), пролактина
(Прл), тиротропного гормона (ТТГ), адрено-
кортикотропного гормона (АКТГ), гонадотро-
пинов — фолликулостимулируюгцего (ФСГ) и
лютеинизируюгцего (Л Г) гормонов, липотро-
пинов (а-Л пТГ и р-Л пТГ), меланоцитостиму-
лирующих гормонов (а-МСГ, р-МСГ, у-МСГ),
а также эндогенных опиатов. Опиоидные
пептиды аденогипофиза многочисленны. Это а-,
р-, и у-эндорфины, пептиды динорфинового
семейства — динорфины А и В и неоэндор-
финыаир.
Мет-энкефалиноваяпоследовательность имеется в ПОМК, но энкефалины в
аденогипофизе не процессируются (подробно
см. т. I, с. 560-565). Установлена продукция
в пределах аденогипофиза эритропоэтина,
соматомедина Си основного фактора роста
фибробластов, но нет ясности относительно
их секреции в системный кровоток и
клеточных источников. Продукция других
регуляторов, в частности андрогенстимулирующего
(эндокринно-метаболические нарушения)
пептида Л.Н. Оделла-У. Д. Паркера (1980),
данной эндокринной железой расценивается,
как гипотетическая.
С общепатофизиологической точки
зрения заболевания аденогипофиза
представляют результат гипопитуитаризма или
гиперпитуитаризма.
Гипопитуитаризм чаще бывает
мозаичным или тотальным (пангипопитуитаризм),
то есть касается угнетения продукции
нескольких или всех гормонов нижнего
мозгового придатка.
Синдром Шихена, гипофизарный нанизм,
гипогонадотропный гипогонадизм
представляют собой примеры тотального и
парциального гипопитуитаризма.
Гиперпитуитаризм чаще, бывает
парциальным и в основном связан с утратой
чувствительности тех или иных
секреторных элементов аденогипофиза и/или
гипоталамуса к сервоингибирующим обратным
связям. Это оборачивается развитием
гиперплазии, вплоть до формирования гор-
монообразующих аденом гипофиза.
Аденомы гипофиза встречаются не менее
чем у 10 % нейрохирургических опухолевых
больных, и, возможно, значительная их
часть столь мала, что не диагностируется.
Макроаденомы (более сантиметра в
диаметре) имеют выраженную «объёмную»
симптоматику, связаную с воздействием
на хиазму зрительных нервов
(нарушения зрения), соседние гипоталамические
структуры, а иногда — и с повышением
внутричерепного давления. Микроаденомы
(диаметром до 10 мм) чаще ограничивают
свои проявления дизгормональными
явлениями. В 1/3 случаев аденомы гипофиза не
образуют гормонов, зато в 15 % формируют
их сразу несколько. Наиболее обычны про-
лактиномы, вызывающие персистирующую
лакторрею-аменорею женщин и бесплодие у
мужчин {синдромы Форбс-Олбрайта, Хиа-
ри-Фроммеля, Аргонза-дель Кастильо, Аль-
хумады-дель Кастильо) и присутствующие
в 30% случаев. Соматотропиномы (25%)
ответственны за гипофизарный гигантизм
и акромегалию. Лдренокортикотропиномы
(10%) обусловливают болезнь Иценко-Ку-
шжга, а после адреналэктомии — синдром
Нельсона. Опухоли из тиротрофов (1 %) и
гонадотрофов (2,5%), способные вызвать
вторичный гипертироз и гипергонадотроп-
ный гипергонадизм, исключительно редки.
Нам кажется удобным вначале уделить
внимание механизмам различных
нозологических форм пангипопитуитаризма. Затем,
по функционально-осевому принципу, мы
обсудим роль и спектр нарушений
продукции каждого из основных гипофизарных
гормонов, не отделяя от этого патологию
соответствующих периферических желёз.
Пангипопитуитаризм как
синдром и его основные формы
Недостаточность нескольких или всех
гормонов гипофиза, часто сочетающаяся с гипотала-
мической дисфункцией, встречается нередко
и имеет множество причин. Субклинические
легкие формы проявляются как латентная
гипоталамо-гипофизарная недостаточность.
Клинически выраженные формы
пангипопитуитаризма, связанные с определёнными
этиологическими факторами,
рассматриваются как отдельные нозологические единицы:
таковы описанный в 1914 г. М. Симмондсом
септико-эмболический некроз аденогипофиза
(синдром Симмондса) и очерченный в 1937 г.
Н.Л. Шихеном послеродовый
пангипопитуитаризм или синдром Шихена (Шиена),
считающийся самой частой формой
пангипопитуитаризма вообще.
В целом синдром может иметь гипота-
ламическое, гипофизарное и смешанное
происхождение.
Наиболее часто встречается в структуре
пангипопитуитаризма дефицит СТГ и гона-
дотропинов. Дефицит АКТГ и ТТГ, кроме
причин, описанных ниже, встречается редко.
Секреция пролактина при гипопитуита-
ризме аденогипофизарного происхождения
понижена, но при гипоталамических формах
повышается из-за снятия дофаминового
блока. Несахарный диабет характерен для
гипоталамических и смешанных форм гипо-
Нарушения эндокринной регуляции 5 31
питуитаризма, но не наблюдается при чисто
гипофизарных формах. Гипоталамические и
смешанные варианты пангипопитуитаризма
могут сопровождаться кахексией, но для чисто
гипофизарных, скорее, характерен
избыточный вес. В этом смысле введённый М.
Симмондсом термин «гипофизарная кахексия»
не вполне точен (см. историю этой неточности
выше в разделе «Истощение и кахексия»).
Тяжесть пангипопитуитаризма
варьирует от минимальной, манифестирующей
тенденции к гипогликемии натощак и
пониженной стрессоустойчивости, до
тяжелой, выражающейся в плюриглан-
дулярной недостаточности и коме. При
остром тяжёлом пангипопитуитаризме
гипопитуитарная кома сочетает
признаки гипотироидной и гипокортикоидной
(наблюдаются гипотермия, гипонатри-
емия, гипогликемия, судороги, адинамия
и ступор).
По этиологии пангипопитуитаризм
делится на врождённый и приобретённый.
Врождённые формы связаны с агенезией всего
нижнего мозгового придатка или только его
аденогипофизарной части. Приобретенные
случаи бывают результатом травматической
деструкции гипофиза, включая случайные и
хирургические травмы, базальное энцефа-
лоцеле, а также радиационные поражения.
Острые сосудистые катастрофы, включая
тромбозы и разрыв аневризм, поражают
гипофиз особенно часто при наличии синдрома
диссеминированного внутрисосудистого
свёртывания, при родах и на фоне сахарного
диабета. По мнению многих авторов, в
частности А. Пупляра (1982), главной причиной
приобретенного пангипопитуитаризма
служит аутоиммунный гипофизит, причём имеют
значение и циркулирующие аутоантитела,
илимфоцитарная инфильтрация органа.
Аутоиммунный гипофизит сочетается с другими
аутоиммунопатиями, например тироидитом
Хасимото и атрофическим гастритом.
В гипофизе бывает и острое гнойное, и
хроническое инфекционное гранулёматоз-
ное воспаление (саркоидоз, туберкулёз, люэс,
бруцеллез). Наконец, большой распростра-
532 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
ненностью в качестве причин пангипопитуи-
таризма отличаются негормонообразующие
неопластические процессы гипоталамуса
и самого гипофиза (краниофарингиомы,
герминомы, хордомы, глиомы, менингиомы
и большие аденомы, гистиоцитоз X,
метастазы других опухолей). Редкой причиной
являются диспротеинозы — амилоидоз и
гемохроматоз гипофиза. Описан пангипопи-
туитаризм у больных тезаурисмозами.
В любом случае для явной манифестации
пангипопитуитаризма необходима потеря
около 90 % ткани аденогипофиза.
Недостаточность различных гипофи-
зарных гормонов вносит вклад в патогенез
и картину пангипопитуитаризма и нередко
наступает в определённой
последовательности (СТГ-ГТГ-АКТГ-ТТГ).
Дефицит СТГ у детей вызывает
снижение ростовых темпов, а у взрослых иногда
манифестирует гипогликемиями натощак,
гипопластической анемией (возможно, и
из-за нехватки гипофизарного эритропоэти-
на), атрофией мышц а также образованием
периорбитальных морщин.
Из-за гипогонадизма пациентки
предъявляют жалобы на аменорею, пациенты
страдают понижением либидо, импотенцией,
снижением оволосения тела (выпадают
волосы на лобке и под мышками, а изредка
следует и тотальная алопеция), наблюдается
бесплодие. При послеродовом пангипопи-
туитаризме не устанавливается лактация,
и имеет место выраженная гипогалактия.
При гипоталамической форме, наоборот,
может быть лакторея. Изменения кожи,
делающие ее сухой и толстой, утомляемость,
интолерантность к холоду и снижение
эмоционально-интеллектуальной активности,
сонливость, запоры могут отражать дефицит
ТТГ. Уменьшение функциональных
возможностей коры надпочечников из-за
нехватки АКТГ приводит к понижению стрес-
соустойчивости, усугубляет утомляемость и
интолерантность к инсулину, проявляется
в похудении, снижении аппетита.
Больные имеют гипонатриемию и субъективно
ощущают вкусовую тягу «к соленому»,
хотя и не в такой степени, как при болезни
ндокринно-метаболические нарушения)
Аддисона (см. ниже). Кожа может светлеть
в силу уменьшения продукции МСГ. Из-за
дефицита опиатных пептидов характерна
депрессия. Ожирение может развиваться в
силу гипотироза и нехватки липотропных
гормонов. Но развитие гипокортицизма и
гипоталамические поражения могут вызвать
и глубокое исхудание.
Несахарный диабет не всегда
сопровождает пангипопитуитаризм. Может даже быть
параллельный синдром Пархона (см. выше).
Наконец, в зависимости от этиологии
картина дополняется местными симптомами,
например, признаками объёмного интракра-
ниального процесса.
Особенности некоторых форм
пангипопитуитаризма должны учитываться в
медицинской практике.
Синдром (болезнь) CuMMqndca
представляет собой гипоталамо-гипофизарную
недостаточность вследствие деструкции
аденогипофиза и части подбугорья. Оригинальное
наблюдение автора (1914) было у пациентки
46 лет с заболеванием, спровоцированным
родовым сепсисом и тромбо-эмболическим
некрозом гипофиза. М. Симмондс наблюдал
ее в гипопитуитарной коме, в терминальной
стадии после десятилетнего
пангипопитуитаризма. Возможны и случаи вследствие
.гнойных инфекций, гранулёматоза и т.д.
В клинической картине доминируют
явления вторичного гипокортицизма и дефицита
ТТГ и СТГ с анорексией, обезвоживанием,
обессоливанием, гипотермией, анемией и
исхуданием. Отмечается картина проге-
рии — преждевременного развития внешних
признаков старения, зависящая, по всей
вероятности, от сочетания обезвоживания,
гипотироза и гипогонадной алопеции.
Синдром Шихена — хронический
послеродовый пангипопитуитаризм.
Провоцирующим фактором, который даёт толчок началу
болезни, служит нарушение питуитарного
кровообращения. В большинстве случаев
это связано с синдромом диссеминиро-
ванного внутрисосудистого свёртывания,
шоком и коллапсом. Прежде синдром
Шихена был нередким осложнением после
родов из-за большой распространенности
Нарушения эндокринной регуляции 533
послеродовых кровотечений. Его развитию
благоприятствует гиперплазия аденогипо-
физа у беременных, которая не подкреплена
адекватным усилением кровоснабжения по
портальной системе. Однако в настоящеее
время акушерская частота синдрома Ши-
хена сократилась. Следует отметить, что
синдром может поражать и небеременных
женщин, и мужчин (например, при шоке,
или при гемагглютинации у больных серпо-
видноклеточной анемией). Циркуляторное
расстройство не вызывает при синдроме
Шихена острого тотального некроза
гипофиза. Запускается аутоиммунный процесс, и
развивающийся аутоиммунный гипофизит
довершает деструкцию гипофиза в подост-
ром и даже хроническом режиме. Аутоан-
титела к гипофизу регулярно выявляются
при синдроме Шихена и чаще всего при
классическом послеродовом поражении,
направлены против пролактотрофов.
Клиническая картина отличается
доминированием симптомов дефицита пролактина и
гонадотропинов. Имеются явные признаки
гипотироза, а манифестация гипокорти-
цизма менее значительна, чем при болезни
Симмондса. По крайней мере, кахексия
нехарактерна, и могут быть отёки.
Рассмотрев тотальную гипофункцию аде-
ногипофиза, перейдём теперь к обсуждению
парциальной дисфункции различных гипо-
физарных осей, включая как гипофункцио-
нальные, так и гиперфункциональные
центральные и периферические нарушения.
Гормон роста
и патофизиология ростовых
и анаболических процессов
«Карл Великий был сыном
Пипина Короткого. Легко
быть великим, имея такого
отца!»
(Ф.Д. Кривин, 1972)
Гормон роста (соматотропный гормон, со-
матотропин — СТГ) открыт Г.М. Ивенсом
и Дж.А. Лонгом (1921) и получен в чистом
виде через 35 лет. Он представляет собой
пептид из 191 аминокислоты с двумя сульф-
гидрильными мостиками и третичной
структурой в виде а-спирали. Из общего числа
160 аминокислот идентичны в СТГ и хори-
оническом гонадотропине (ХГ) плаценты и
термином человека. Таким образом, СТГ и
ХГ, гены которых находятся в 17-й
хромосоме, — близкие гомологи (83 % структуры,
рис. 104). Известную гомологию с ними
проявляет и про л актин (16% структуры), в
связи с чем у СТГ, кроме основной, имеется
слабая лактогенная активность. Более того,
ХГ имеет ростостимулирующую активность,
а в плаценте экспрессируется вариант СТГ
(В-СТГ). Гормон роста видоспецифичен.
У человека высоко активен, кроме
гомологичного, лишь гормон обезьян. Интересно,
что, по данным И. Резвани и соавт. (1973),
рецепторы СТГ у крыс способны
реагировать на гормон роста различных животных.
Гипофиз выделяет СТГ в виде мономера
и димера. Есть основания полагать, что
плазминоподобные ферменты в организме
должны активировать СТГ путём протеоли-
за, вычленяя из него наиболее действенную
часть. Тогда гипофизарный СТГ можно
рассматривать как прогормон.
Секреция СТГ сильно стимулируется
гипоталамическим соматолиберином (он
же — соматокринин), а также в меньшей
степени — тиролиберином и р-эндорфином,
допамином, серотонином, норадренали-
ном, ацетилхолином. Соматотропиногенез
тормозится соматостатином (нейроны
которого в подбугорье реципрокно связаны
с соматокрининовыми), адреналином и не-
этерифицированными жирными кислотами.
Гормон выделяется импульсно, причём в
сутки бывает 4-10 коротких периодов
высокой секреции, в том числе — при физических
усилиях, стрессе, гипогликемии, в моменты
медленноволновой фазы сна (вспомним
народное поверье: «Когда летишь во сне —
растёшь»), при приеме в пищу аминокислот
(особенно аргинина). Эстрогены, вазопрес-
син и глюкагон усиливают освобождение
СТГ, а прогестины тормозят.
В клинике тесты на трансгипоталами-
ческую провокацию выделения СТГ осу-
534 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рис. 104. Аминокислотная последовательность гормона роста человека и его гомология с
человеческим плацентарным лактогеном. СТГ представлен полностью, для плацентарного лактогена
даны отдельные, не совпадающие с гормоном роста аминокислотные остатки
(по Ю.А. Панкову, 1976)
ществляются с помощью аргинина и инсу-
линовой гипогликемии. СТГ секретируется с
нарастающей силой на протяжении детства,
его продукция резко увеличена у подростков,
снижается после 20 лет, остается стабильной
до 40, а затем еще более уменьшается. При
беременности и кормлении она резко
возрастает, причём в первом случае большая часть
СТГ— плацентарного происхождения. Время
полужизни СТГ в плазме — от 20 до 30 мин, а
катаболизм гормона преимущественно идёт в
печени и в какой-то степени в почках.
СТГ опосредует свое действие через
рецепторы соматомаммотрофного типа,
которые стоят особняком от других
ретрансляторов гормонального сигнала (см. выше)
и, вероятно, имеют пептидные
внутриклеточные посредники
Метаболическое действие СТГ
включает небольшое количество собственных и
широкий спектр пептид-опосредованных
эффектов этого гормона. Тканевыми ауто-
кринными и паракринными посредниками,
обязательными для реализации
большинства эффектов СТГ, служат инсулиноподоб-
ные факторы роста (соматомедины).
Соматомединов (см. также т. I) известно
до семи, все они действуют путём
понижения активности аденилатциклазного
механизма в различных клетках, но наибольшее
физиологическое значение имеют сомато-
медин С (инсулиноподобный фактор роста
I -ИРФ-1) и соматомедин А
(инсулиноподобный фатор роста II — ИРФ-П).
ИФР-1 — посредник всех основных
ростовых и метаболических эффектов СТГ в
постнатальный период. Это основной пеп-
Нарушения эндокринной регуляции 535
тид из 70 аминокислот, кодируемый в 12-й
хромосоме и наполовину гомологичный
проинсулину. ИФР-1 может образовываться
всеми мезодермальными, многими экто-
дермальными и некоторыми эндодермаль-
ными клетками, но наиболее активно
продуцируется и выделяется в системный
кровоток макрофагами печени, вследствие чего
может рассматриваться как печёночный
СТГ-зависимый гормон. Время полужизни
ИФР-1 в плазме составляет до получаса, но
он имеет белки-переносчики, продлевающие
срок его существования в крови в 6-18 раз.
Переносчик ИФР-1 третьего типа поступает
в плазму пропорционально уровню СТГ.
Секрецию ИФР-1 стимулирует и ХГ
плаценты. Кроме печени, ИРФ-I формируется
для паракринных и аутокринных нужд в
почках, костях, мышцах, ЖКТ и самом аде-
ногипофизе. ИФР-1 ингибирует продукцию
соматокринина, замыкая обратную связь
в системе центральной регуляции роста и
анаболизма. При ожирении уровень ИФР-1
возрастает, вызывая снижение продукции
СТГ. В то же время похудение и белковая
недостаточность, включая начальные
стадии алиментарной дистрофии, квашиоркор,
нейрогеную анорексию и другие подобные
состояния, понижают продукцию ИФР-1
и растормаживают биосинтез СТГ. Кто из
нас не помнит знаменитого родительского:
«Как ты похудел и вытянулся за время
болезни!» Есть основания полагать, что
это — не чисто кажущееся впечатление.
Интересные наблюдения связаны с тем, что
у подростков со сниженной массой тела, по
данным Ю.И.Строева (2005), рост часто,
наоборот, опережает средние должные
показатели. Можно полагать, что и здесь
сказывается данный соматомедин-зависи-
мый эффект.
Во время внутриутробного развития рост
тела не определяется СТГ. Это доказывается
нижеследующими наблюдениями: при
наследственном дефиците СТГ матери
рождают детей с дефицитом СТГ, но нормальной
длиной тела. Нормального размера и
младенцы у пигмеев-мбутси Заира и Бурунди,
хотя у них соматомедин С-опосредованный
эффект СТГ в постнатальном онтогенезе
нарушен в силу доминантной мутации.
Количество ИФР-1 у пигмеев снижено, хотя
уровень СТГ выше, чем у здоровых
европейцев, а средний рост — 135 см.
В качестве ростового фактора тканей
плода действует соматомедин А — ИФР-Н,
продукция которого требует минимального
уровня гормона роста, но к надфизиологи-
ческим концентрациям СТГ практически
нечувствительна. ИФР-П — нейтральный
пептид из 67 аминокислот, закодированный
в 11-й хромосоме и гомологичный ИФР-1
почти на 70%. Соматомедин А известен в
двух формах. Как паракринный и аутокрин-
ный регулятор он вырабатывается в тех же
клетках, что и ИФР-1. В
постэмбриональном периоде ростовое действие ИФР-Н
ослабевает на протяжении первого года
жизни и является актуальным только для
зубных зачатков. Интересно, что у пигмеев
относительный размер зубов крупнее, чем
у представителей других народов, так как
действие соматомедина А у них не
нарушено. Вместе с тем у больных
акромегалией и гипофизарным гигантизмом, когда
усилено ИФР-1-опосредованное действие
СТГ, характерным симптомом является
наличие больших межзубных
промежутков — диастемы, а относительный размер
зубов снижен.
Соматомедин В, объединяющий 4
разных кислых пептида, охарактеризованный
хуже других соматомединов, по-видимому,
участвует в СТГ-зависимом усилении роста
глиальных клеток. Так как СТГ сам
стимулирует продукцию инсулина в В-клетках,
формально инсулин тоже может быть
причислен к кругу посредников действия СТГ,
вместе с соматомединами, на рецепторы
которых он способен влиять (Дж. Уилсон,
1994).
Следует отметить, что соматомедины, в
свою очередь, опосредуют те присущие им
ростовые эффекты, которые связаны с
ускорением деления клеток, через стимуляцию
самого эффективного фермена
млекопитающих клеточной орнитиндекарбоксилазы,
генерирующей непосредственные актива-
536 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
торы пролиферации — полиамины (см. т. I,
гл.12).
СТГ непосредственно стимулирует рост
хондроцитов и сам оказывает острые кон-
тринсулярные эффекты на метаболизм —
снижает синтез липидов в адипоцитах и
поглощение глюкозы печенью и жировой
тканью, усиливает освобождение жирных
кислот в кровь. В то же время он
способствует синтезу инсулина и соматомединов,
что позволяет, в несколько отсроченном и
пролонгированном режиме, запустить через
эти посредники более широкий круг проли-
феративных процессов и, главное,
осуществить продлённое инсулиноподобное действие.
Например, СТГ и инсулин оба стимулируют
поглощение аминокислот рядом тканей.
Вышеописанное замечательное двухфазное
приспособление позволяет усиливать рост
и анаболизм и в то же время не
расплачиваться за это гипогликемией и снижением
катаболизма (В.Р. Лингаппа, 1997).
Из-за своеобразия взаимоотношений СТГ
и инсулиновой регуляции нарушения
продукции СТГ небезразличны для углеводного
метаболизма. При гипофизарном нанизме
больные подвержены гипогликемии между
приёмами пищи. При гигантизме и
акромегалии развивается инсулинорезистентность.
Гипергликемия в норме тормозит продукцию
СТГ. Но при инсулинозависимом сахарном
диабете концентрации СТГ парадоксально
повышены, как полагают, в связи с инсули-
ноподобным действием соматомединов и в
попытке оказать инсулинотропный эффект
на В-клетки. У гипофизарных карликов и
гигантов сахарный диабет протекает при
резко пониженной скорости развития микро-
ангиопатии, что свидетельствует об участии
СТГ и соматомединов в ее патогенезе. СТГ
ускоряет разрушение инсулина. Наконец,
гиперпродукция СТГ способствует синтезу
контринсулярного соматостатина, а при
депанкреатизации и на фоне инсулинозави-
симого сахарного диабета СТГ не оказывает
ростостимулирующего действия (см. также
«Патофизиология сахарного диабета»).
СТГ опосредует через соматомедины и
инсулин следующие эффекты: усиление
включения аминокислот, уридина, тимиди-
на и сульфата в клетки хряща и ускорение
синтеза белка, нуклеиновых кислот и
пролиферации хондроцитов и клеток всех других
тканей, индукция положительного азотистого
баланса, понижение производства мочевины,
усиление использования липидов, начальное
понижение и последующие нормализация
и повышение уровня глюкозы, свободных
жирных кислот и аминокислот в крови,
увеличение синтеза гликогена в мышцах и печени.
СТГ благоприятствует синтезу кетоновых тел,
в связи с чем сахарный диабет у лиц с сомато-
тропиномами осложняется кетоацидозом.
СТГ также влияет на метаболизм других
гормонов: ускоряет синтез ренина и альдо-
стерона, активацию кальциферола путём
его гидроксилирования, влекущую за собой
усиление всасывания и тканевого
использования кальция, а также конверсию Т4 в Т3.
Было бы искажением трактовать
ростовые эффекты СТГ как ограниченные лишь
соединительной тканью или
опорно-двигательным аппаратом. Он способствует росту
практически всех мягких тканей.
Достаточно указать на его лимфопролиферативное
(и иммуностимулирующее!) действие. СТГ
способствует также гипертрофии
миокарда и почек, эритропоэзу, заживлению ран
(рис. 105). Имеются наблюдения об
эффективном использовании его при лечении
инфаркта миокарда. Характеризуя эффекты
СТГ, А. Гайтон (1989) кратко и ёмко
формулирует, что система СТГ-соматомедины
стимулирует рост «всего, что может расти».
С момента окостенения метаэпифизарных
хрящей (примерно к 21-23 годам у юношей
и 23-25 — у девушек) кости более не растут
в длину, хотя могут продолжать утолщаться
при периостальном росте. Поэтому
патогенез и проявления нарушений, связанных
с СТГ, зависят от длительности и времени
начала дисфункции.
Нарушения функционирования системы
СТГ — соматомедины приводят к
различным расстройствам ростовых и
анаболических процессов.
При постпубертатном угнетении
продукции СТГ у взрослых лиц симптомов его
кора больших полушарий
медленный сон
ST-
А
»**
V*
ъействие_
'ИФР-Г
печень
X поджелудочная
железа
соматостатин
Г /\шпс
/ у \+
1е "" ^^
7?
инсулин
ин
таламус
гипофиз
ИФР-1
+
9-1
+ I
хондрогенез
остеогенез
гиперплазия, 1
гипертрофия
мягких тканей \
анаболическое
действие \
глюкостати-
ческий эффект
липидомобили- 1
зующий эффект
кетогенное
действие
эритропоэти-
ческий эффект
лактопоэти-
ческий эффект \
иммуностимули- 1
рующее действие \
Рис. 105. Регуляция системы СТГ— инсулиноподобные ростовые факторы и биологическое
действие СТГ. ИФР-1 — инсулиноподобный фактор роста I; SCr — соматокринин, SS — соматостатин,
ST — соматотрофы
538 ALU. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
недостаточности может вообще не быть.
Углублённое обследование взрослых лиц с
недостаточной продукцией гормона роста
выявило несколько сниженную
толерантность к физической нагрузке, тенденцию
к избыточной массе тела и понижению
соотношения массы мышц и жировой
ткани. Смертность от сердечно-сосудистых
заболеваний в этой группе увеличена, но
частота злокачественных новообразований
снижена.
Гипофизарный нанизм — первичная
форма торможения роста, связанная с
дефицитом продукции СТГ в аденогипофизе.
Ее частота невелика — не более чем 1/1000
лиц с задержкой роста, обращающихся
к врачу, являются истинными гипофи-
зарными карликами. В популяции частота
гипофизарного нанизма — от 1/20000 в
России до 1/4000 в Америке, Африке и на
Ближнем Востоке. До половины больных
имеют изолированную идиопатическую
недостаточность СТГ, часто вторичную по
отношению к дефициту соматокринина. До
35 % имеют раннюю сочетанную
недостаточность гипофизарных гормонов вследствие
опухолевого разрушения аденогипофиза,
аутоиммунного процесса, облучения. Около
15 % страдают от изолированного дефицита
СТГ, вызванного ранним воздействием тех
же причин. Считается, что соматотрофы
менее резистентны к ряду неблагоприятных
факторов, чем другие секреторные клетки
аденогипофиза. Большинство форм
гипофизарного нанизма — приобретенные. Имеется
модель наследственного гипофизарного
нанизма, созданная собаководами, в виде
карликовых пород декоративных собак.
Существует наследственный гипофизарный
нанизм и у человека, притом в аутосомно-
рецессивной (дефект гена СТГ) и в
сцепленной с Х-хромосомой разновидности. Особая
форма гипофизарного нанизма, сочетанного
с гипогонадотропным гипогонадизмом,
именуется «питуитарный инфантилизм» и
бывает вызвана краниофарингиомами или
аутоиммунным процессом, разрушающим
соответствующие клетки-регуляторы.
При гипофизарном нанизме рост
тормозится часто уже на втором-четвёртом годах
жизни, но в зависимости от этиологии, это
может произойти и позже. Если нет
сопутствующего дефицита других гипофизарных
гормонов, то задержка роста и весовой
прибавки носит пропорциональный характер
(однако голова может иметь слегка больший
относительный размер, а лоб — выступать),
а также сопровождается микроспланхнией и
не сопряжена с отклонениями в
психомоторном или половом развитии. Гипофизарный
нанизм нередко сочетается с некоторым
избытком жира и гипогликемиями натощак.
Высокий, пронзительный голос многих
лилипутов не должен восприниматься
как симптом гипогонадизма, а связан с
особенностями миниатюрной гортани как
резонатора. Н.А. Зарубина указывает на
такой характерный признак, как удлинённые
ресницы (1985). У гипофизарных карликов
ускорено старение кожи (геродерма), что
связано с замедлением её обновления и
иногда — вторичным гипотирозом. Но нет
достоверных данных о скорости общего
старения при гипофизарном нанизме.
Конечный рост таких пациентов может
быть в пределах 90-120 см. Критерием
карликовости считается рост, отстающий
от среднепопуляционного возрастного на
два-три сигмальных отклонения (как
правило, менее 130 см — у мужчин и 120 см — у
женщин). Имеются сообщения о
гипофизарных карликах ростом менее 70 см.
Нередко они совершают успешную
артистическую и цирковую карьеру. Самой
низкорослой жительницей Земли, среди
всех медицински освидетельствованных
гипофизарных карликов, считалась
мексиканская цирковая артистка Люсия Зарате
(1864-1890), чей рост был 51см при весе
2,3 кг. Микросомия подобных пациентов
принципиально не означает какой-либо
формы умственной либо социальной
неполноценности. По свидетельству
современников, один из самых миниатюрных
гипофизарных карликов был генералом
эфиопской армии. Авторы сожалеют о
невозможности опубликовать здесь фото этой
исторической личности в парадном мундире
или во время руководства боевыми
действиями. Впрочем, всем известны фото таких
крупных государственных и политических
деятелей, как Ясир Арафат (157 см), Давид
Бен Гурион (152 см.), Н.И. Ежов (152 см.),
Дэн Сяопин и королева Виктория (по 152
см.), карьере которых не помешала низко-
рослость. Гипофизарным карликом был
конкурент Т. А. Эдисона изобретатель сетей
переменного тока Ч.П. Стайнметц (122 см.).
Низкорослость не отняла у И. Канта и но-
белианцев М.Фридмана и Б. Мак-Клинток
(все по 152 см) возможности стать
гигантами познания.
Самым маленьким в истории жителем
Земли был страдавший прогерией
американец Кальвин Филипс (1791-1812), ростом
67 см. Из ныне живущих землян самый
маленький - непалец Кхагендра Тхара Магар.
В 2006 г. в 14 лет его рост составил 50,8 см
при весе 4,5 кг. Учитывая юный возраст
и современные методы лечения нанизма,
можно полагать, что у Кхагендры все
впереди - во всяком случае, австрийцу Адаму
Рейнеру в свое время удалось вырасти со
115 до 214 см и единственному побывать как
гипофизарным карликом, так и гигантом.
Задержка роста чаще всего имеет
причины, не связанные с дефицитом его гипота-
ламических и гипофизарных стимуляторов.
Поскольку рост — аддитивно-полигенный
признак с пороговым эффектом,
определяемым комплексом экзогенных факторов,
эти причины многочисленны — некоторые
руководства насчитывают до 56
разновидностей нанизма и гипостатуры. Образно
говоря, карликовость — гигантская проблема.
Поэтому эрудиция в вопросах нарушений
ростовых и анаболических процессов у
человека необходима доктору для успешной
дифференциальной диагностики.
Детальная характеристика различных
многообразных причин синдрома
замедления роста — предмет пособий по
эндокринологии. Однако в данном учебнике уместно
напомнить о механизмах некоторых из них,
а также о патофизиологической основе их
дифференциации.
Нарушения эндокринной регуляции 539
Самая частая причина отставания детей в
росте — конституциональная его задержка
(98%). Полигенность контроля роста
делает предсказания ненадёжными, но имеется
корреляция между ростом обоих родителей
и ребёнка. Питание, особенно в грудном
периоде раннего детства и в подростковом
периоде, небезразлично для реализации
генетически детерминированной потенции
роста. Рост может тормозиться при нехватке
белка в питании маленьких детей, при
дефиците незаменимых аминокислот, витамина
D, цинка и, возможно, других факторов.
Выше уже шла речь о психосоциальном
гипоталамическом нанизме (до 1 % случаев
задержки роста).
При раннем и врождённом гипотирозе
рост также значительно тормозится (гм-
потироидный папизм, частота которого
примерно вдвое-выше, чем гипофизарно-
го). Тироидные гормоны необходимы для
влияния ИФР-1 на хондроциты, усиливают
продукцию СТГ в ответ на различные
стимулы, активизируют амино-ацил-т-РНК-
синтетазы и процессы трансляции. Всё это
приводит к тому, что без них линейный рост
у животных практически полностью
останавливается. Гипотироидная карликовость
сопровождается нарушением
психомоторного развития детей и умственной
отсталостью, тем более выраженной, чем раньше
сформировалась нехватка тироидных
гормонов (см. ниже). Сами признаки гипотироза
могут присутствовать и у гипофизарных
карликов как следствие гипопитуитаризма,
но не бывают столь выражены. Дефицит
андрогенов в период полового созревания
может повлечь отсутствие пубертатного
ростового скачка, но не способен вызвать
нанизм. Макрогенитосомия (см. выше) и
другие причины раннего гипергонадизма
способствуют преждевременному
закрытию метаэпифизарных зон, вследствие чего
ребёнок быстро растет в раннем детском
возрасте, но рост его рано останавливается
и бывает ниже среднего. Выраженная
низкорослость бывает при дизгенезии гонад, в
частности, синдроме
Шерешевского-Тернера, причём уровень СТГ не понижен, а чувс-
540 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
твительность к нему уменьшена, что, однако,
преодолевается при применении больших
доз СТГ. Новые генетические данные
проливают свет на патогенез низкорослости
и высокорослости при аномалиях числа
гоносом. Идентифицирован ген SHOX
— гомеозисный ген низкорослости. По
одной копии находятся в X и Y хромосомах.
Низкорослыми получаются гемизиготные
генотипы, например ХО. Низкорослость при
синдроме Шерешевского - Тернера, мезомели -
ческой дисплазии Лангера, дисхондростеозе
Лери-Вейля — продиктована именно гапло-
недостаточностью гена SHOX.
Так как инсулин является соматомеди-
ном, при тяжелом раннем инсулинозависи-
мом диабете у детей бывает низкорослость, в
сочетании с такими признаками, как
инфантилизм и стеатоз печени, она входит в
диабетический синдром Мориака (см. гл. 9).
Весьма редко встречается, но
представляет теоретический интерес
нечувствительный к лечению СТГ синдром Ларона
(аутосомно-рецессивная карликовость при
высоком уровне СТГ, дефекте рецептора
СТГ, делающем невозможным адекватный
синтез ИФР-1, пониженном уровне ИФР-1
и II). Выше уже шла речь о аутосомно-до-
минантном дефиците ИФР-1 у африканских
пигмеев. Наконец, низкорослость с
диспропорциями и дизгенезией скелета,
искривлением конечностей может сопровождать
некоторые наследственные тезаурисмозы
(характеристику которых можно найти
в разделах «Патофизиология липидного
обмена» и «Патофизиология углеводного
обмена»), генетические остеохондродисп-
лазии, D-резистентные формы рахита (см.
«Нарушения витаминного обмена»).
Истинная гипофизарная карликовость
сопровождается понижением содержания
СТГ, а также ИФР-1 и третьего ИФР-пере-
носящего белка в плазме крови, реагирует
на терапию СТГ. Истинный гипофизарный
нанизм лечится в настоящее время реком-
бинантным СТГ. Рекомбинантный гормон
доступен с 1985 г. Гормон животных был
малоэффективен, поэтому длительно
существовала практика лечения гипофизарного
нанизма трупным СТГ. В 80-х годах
обнаружился повышенный риск прионовой
губчатой энцефалопатии (болезнь Крёйтц-
фелъда-Якоба, см.т. I, стр. 413),
передаваемой с препаратами трупного гипофиза.
Ятрогенная прионовая энцефалопатия была
найдена у многих успешно излеченных от
нанизма пациентов. Применение трупного
человеческого СТГ в ряде стран запрещено
с 1984 г. Подробно карликовость и
низкорослость рассмотрена в монографии
«Эндокринология подростков» Ю.И. Строева
и Л.П. Чурилова (2004).
Гипофизарный
гигантизм-акромегалия — это расстройство, возникающее из-за
хронической гиперпродукции гормона
роста. Как впервые показал К. Бенда (1900),
в 99 % случаев этиология болезни связана
с соматотропиномой — опухолью из эози-
нофильных клеток, вырабатывающихСТГ.
Соматотропиномы почти всегда
доброкачественны, хотя изредка проявляют инва-
зивность. Аденогипофизарная локализация
типична, хотя описаны эктопические
соматотропиномы — в парафарингеальной
области, синусах основной кости, ЖКТ, бронхах и
даже поджелудочной железе. В 1 % случаев
синдром гигантизма-акромегалии
обусловлен гиперпродукцией соматокринина
гипоталамуса и вторичной неопухолевой
гиперплазией соматотрофов. Частота
акромегалии невелика — порядка 5-7 случаев
на 100000 населения. Гигантизм
встречается примерно в 5 раз чаще. Интересно, что
среди гипофизарных гигантов соотношение
мужчин и женщин — примерно равное, но
среди пациентов с акромегалией женщины
значительно преобладают. Беременность
считается фактором риска для гиперплазии
эозинофильных клеток гипофиза.
Подавляющая часть больных представляет
спорадические случаи, хотя имеются семейные
формы этого синдрома.
Описана генетическая модель
гипофизарного гигантизма, полученная
собаководами. Наследственными гипофизарными
гигантами являются представители двух
пород: бульдоги и ирландские волкодавы.
Интересно, что особи породы карликовых
бульдогов — не гипофизарные карлики, а
нормальные родоначальники своих более
знаменитых гигантских сородичей. У всех
собак-моделей гигантизма и примерно у 40 %
пациентов с гигантизмом или акромегалией
на почве соматотропиномы в клетках
опухоли выявляется мутация а-субъединицы
белка Gs, вследствие которой этот элемент
пострецепторного ретранслятора сигнала
соматокринина утрачивает способность к
гидролизу ГТФ и «зависает» в хронически
активном состоянии. Аденилатциклазный
передаточный механизм пролонгированно
активируется и заставляет соматотрофы
вырабатывать избыток СТГ и ускоренно
пролиферировать. Мутация может быть
соматической и лишь изредка, при семейном
гигантизме, квалифицируется как
генеративная.
В остальных случаях природа
трансформации клеток соматотропиномы иная и пока
неясна. Предполагается, что в отдельных
случаях акромегалии-гигантизма с
нормальными концентрациями СТГ в крови может
иметь место гиперчувствительность тканей
к СТГ, избыток соматомадина С, но такие
наблюдения эксклюзивны.
Врожденный и ранний избыток СТГ при
наличии соматотропином ведёт к
ускоренному росту костей в длину и гипофизарному
гигантизму. Гигантским считается рост
более 200 см у мужчин и 190 см — у женщин
(или превышающий среднепопуляционное
значение на 2 и более сигмальных
отклонения). Существуют этносы, у которых
средний рост велик и процент лиц, формально
подпадающих под критерий гигантизма,
намного выше, чем у большинства народов.
Таковы, например тутси, населяющие
Руанду, Бурунди и Заир (средний рост —
185 см). Курьёзно, что тутси проживают
территориально там же, где и пигмеи. Об
удивительном соседстве племён гигантов и
карликов писал еще Геродот. Это косвенно
свидетельствует о примате генетических
факторов над экологическими в
определении роста. Зафиксированы случаи
сверхгигантского роста (более 2,5 м).
Нарушения эндокринной регуляции 541
По имеющимся в литературе данным, на
1993 г. среди достоверных, медицински
освидетельствованных случаев гигантизма
самым высокорослым из когда-либо живших
на Земле людей был наш соотечественник
Фёдор Махнов (1881-1912), чей рост
составлял 285 см (при весе 182 кг). Рост Джона
Миддлтона из Ливерпуля (1578-1623) был
282 см. Из хроник известны «ирландские
гиганты» — Корнелиус Мегрэт (1742-1768)
и Чарльз ОЪрайен (1761-1783), у скелетов
которых еще Дж. Хантер, поместивший
один из этих препаратов в музей
Королевского Хирургического Колледжа в Лондоне,
зафиксировал выраженное увеличение
турецкого седла и необычную толщину костей.
Читатель, несомненно, отметил, что век этих
гигантов был, увы, недолог. Аналогичный
экспонат (скелет императорского слуги
Буржуа, рост которого при жизни достигал
227 см) был включён в экспозицию
петербургской Кунсткамеры Петром Великим,
который, впрочем, и сам был гигантом не
только в историко-политическом, но и в
биологическом смысле (204 см).
Недостоверные исторические сведения имеются и
о людях, достигавших трёхметрового роста.
Имена многих гипофизарных гигантов
хранит история медицины и... баскетбола.
Из приведённых примеров ясно, что
сверхвысокий рост, также как и гипофизарный
нанизм, вполне совместим с социальной
активностью, физической и
интеллектуальной полноценностью. В настоящее время,
самым высокорослым представителем
человечества, по-видимому, считается
петербуржец Александр Алексеевич Сизоненко
(1959г.р.)-248см.
В дифференциально-диагностическом
плане стоит отметить, что высокий рост — не
всегда результат гипофизарной
гиперфункции. Он может быть последствием
удлинённого ростового периода. В этом случае
даже при нормальных концентрациях СТГ
высокорослость достигается путём
задержки полового созревания и окостенения
метаэпифизарных хрящей. Именно поэтому
многие носители раннего и врождённого
гипогонадизма обоего пола высокорослы.
542 ALU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Высокий рост характерен для синдрома
Клайнфелтера (46XXY), а также для евну-
хоидности при ранней кастрации. Выше уже
упоминалось об ускорении роста при
гипотрофии у подростков в связи с выпадением
ингибирующего влияния ИФР-I на
центральные ростовые регуляторы. Однако надо
подчеркнуть, что причины высокорослости
не так разнообразны, как карликовости, и
супергигантизм имеет исключительно ги-
персоматотропное происхождение.
Если гиперпродукция СТГ носит очень
выраженный характер, то уже при
незаконченном росте тела в длину появляются
признаки увеличения мягких тканей и
внутренних органов — акромегалия. При
гиперсекреции СТГ, начавшейся после
окостенения метаэпифизарных хрящей,
акромегалия может быть единственным
проявлением болезни, а рост акромегаликов
совсем не обязательно является гигантским.
Такая акромегалия дебютирует чаще в
среднем возрасте — 40-45 лет. Гипофизарный
гигантизм переходит в акромегалию, если
соматотропинома продолжает секрецию
после пубертата. И при гигантизме, и при
акромегалии секреция СТГ не подавляется
гипергликемией. В дальнейшем патогенез
акромегалии-гигантизма обсуждается
совместно как двух форм (а часто — стадий)
одного заболевания.
При данном синдроме усилено
метаболическое действие и увеличено содержание
в крови СТГ и ИФР-I. Кроме ускоренного
роста тела в длину, многие проявления
зависят от гиперплазии и гипертрофии
мягких тканей. Как правило, имеются глос-
сомегалия, увеличение гортани, диастема
(см. выше) — всё это меняет голос больных,
и он становится низким, звучит, как бы «из
глубины».
Черты лица грубеют, костные выступы
подчёркиваются, имеется прогнатизм, нос,
губы и уши увеличиваются, из-за
синусита и прогнатизма рот часто полуоткрыт,
отмечаются выраженные кожные складки,
периорбитальная гипертрофия мягких
тканей. Кожа маслянистая, влажная и тёплая.
Бывают гипертрихоз, акантоз, фиброзные
кожные полипы, кисты сальных желез.
Увеличиваются стопы и кисти, размеры
головы. Нередко бывает кифосколиоз.
Часто кожа гиперпигментирована. Все это
делает внешний вид больных столь
характерным, что, по выражению Дж. Дэниелса
и Дж. Мартина (1994), они «похожи друг
на друга больше, чем на членов собственных
семей». Читатели-любители киноискусства,
несомненно, помнят лицо выдающегося
французского комика Фернанделя.
Однако акромегалия-гигантизм, к
сожалению, порождает не только косметологи-
ческие, но и метаболические проблемы у
пациентов.
До 30 % носителей этого синдрома имеют
сниженное либидо, потенцию и дисменорею,
так как у них присутствует гиперпролак-
тинемия. Не менее 80 % интолерантны к
глюкозной нагрузке. Хотя большая часть
пациентов компенсирована в отношении ин-
сулиновой регуляции, 25 % развивает явный
сахарный диабет, нередко осложняемый
кетоацидозом. ИФР-I вызывает у пациентов
гиперплазию щитовидной железы. От 3 до
7 % имеют гипертироз, и гораздо большая
часть больных проявляет признаки
относительной избыточности тироидной функции,
вероятно, за счёт усиленной конверсии Т4 в
Т3 (повышение основного обмена,
потливость, непереносимость жары). У больных
ускорено развитие сердечно-сосудистых
заболеваний, имеется гипертензия при
низком уровне ренина, связанная с неальдо-
стероновой задержкой натрия и воды. Почки
характеризуются гипертрофией и
гиперфункцией, но, по-видимому, менее эффективны
в отношении прессорного натрийуреза.
Имеется кардиомегалия, осложняемая мио-
кардиодистрофией и застойной сердечной
недостаточностью по левожелудочковому
типу. Висцеромегалия повышает внутри-
брюшное давление и предрасполагает к
грыжам. Имеется дегенеративный остеоартрит,
связанный с гиперплазией хрящей, синдром
лучезапястного туннеля, парестезии.
Повышена частота лимфопролиферативных
заболеваний, полипоза кишечника и карцином
толстой кишки. При большом размере и ин-
Нарушения эндокринной регуляции 543
вазивном росте соматотропиномы гипофиза,
как и другие неоплазмы района турецкого
седла, дают неврологические расстройства.
Как правило, с самого начала заболевания
имеются головные боли. Более
значительные проявления включают ринорею (при
распространении опухоли вниз), дефекты
полей зрения (при сдавлении хиазмы
зрительных нервов), офтальмоплегию (при
прорастании в пещеристый синус),
гидроцефалию, изредка — височную эпилепсию
(при латеральном распространении).
Все эти нарушения метаболизма и
органных функций понижают среднюю
продолжительность жизни при гипофизарном
гигантизме-акромегалии.
Описаны парциальные формы
акромегалии и односторонний гемигигантизм,
когда ускоренный рост касается половины
тела или только отдельных органов (частей
тела) — головы, сердца и т. д. Существуют
спорадические и семейные формы этих
состояний, предположительно связанные
с региональными особенностями парак-
ринного опосредования активности СТГ
(М.И. Балаболкин, 1974). На рис. 106
представлено наблюдение Ю.И. Строева (1994),
касающееся больной с парциальной формой
акромегалии, — томограмма черепа с выра-
женно асимметричным ростом костей.
Существуют внешне напоминающие
акромегалию заболевания негипофизарной
природы, при которых имеется гиперплазия
мягких тканей конечностей и лица. Они,
видимо, отражают аномалии паракринной
соматомединовой регуляции ростовых
процессов и должны дифференцироваться
с акромегалией по нормальному уровню
СТГ и отсутствию характерных для
последней метаболических отклонений. Это
прежде всего синдром Бамбергера-Мари:
гипертрофическая пневмопатическая остео-
артропатия. Хронические нагноительные
заболевания лёгких и печени
провоцируют у наследственно предрасположенных
пациентов крайне выраженные симптомы
«барабанных палочек», «часовых стёкол»,
отёк и артропатию дистальных отделов
конечностей, гипергидроз.
56-1
в-1
50-»
У
/
Рис 106. Асимметричный рост костей черепа
при парциальной акромегалии у пациентки
Е.Д., 17 лет. Томограмма головы. Фото
предоставлено Ю.И. Строевым (1994)
При синдроме Турена-Соланта-Голе
(пахидермопериостоз) имеется массивное
утолщение кожи лица, кожи и костей
дистальных отделов конечностей, гипергидроз
и себоррея. Также наблюдаются симптомы
«барабанных палочек», «часовых стёкол» и
остеоартриты.
Этиология и патогенез этого
нарушения, которое бывает и спорадическим, и
семейным и поражает в основном мужчин,
неизвестны.
544 А.Ш. Зайчик, Л.П.Чурилов Патохимия{
Тиротропный гормон и
нарушения тироидной функции
Щитовидная железа легко подвергается
воздействиям неблагоприятных экологических
факторов как из-за своей поверхностной
локализации, так и в связи с присущими этому
органу метаболическими особенностями
(кумуляция йода и других микроэлементов,
интенсивное перекисное окисление). Особая
роль среди этих неблагоприятных факторов
отводится ионизирующим излучениям и
потенциально токсичным компонентам диеты.
Немаловажную роль играет и высокая
чувствительность данного органа к иммуности-
муляции. Вследствие всего этого в среднем
не менее 5,5-6% населения имеют те или
иные расстройства тироидной функции, и
их число быстро растёт. Поэтому в данном
томе будет представлена очень подробная
характеристика патологии щитовидной
железы. Чтобы представить себе значение
этих вопросов для практического
здравоохранения, достаточно упомянуть данные
Ю.И, Строева и соавт. (1998) о 17-кратном
увеличении частоты одного только диагноза
«аутоиммунный тироидит» у
госпитализированных подростков Санкт-Петербурга за
последние 10 лет.
Основным регулятором тироидной
функции является аденогипофиз.
Тиротропин (ТТГ) вырабатывается базо-
фильными тиротрофами аденогипофиза.
Структура ТТГ сложна, и его продукция
вне аденогипофиза маловероятна, но
имеются данные (А. Дольва и соавт., 1983) о
возможности эктопической продукцим ТТГ
в островках Лангерганса, особенно в неона-
тальный период.
Как обнаружено в 1971 г. Т. Ляо и
Дж. Пирсом, ТТГ — это гликопротеид с
молекулярной массой 28,3 кД, имеющий
211 аминокислотных остатков и состоящий
из двух нековалентно связанных
субъединиц— а- (13,6 кД, 2 углеводных остатка,
кодируется в 6-й хромосоме) и р- (14,7 кД,
1 углеводный остаток, кодируется в
хромосоме 1). Гормон богат серой: он содержит 5
чдокринно-метаболические нарушения)
дисульфидных мостиков в а-субъединице
и 6 — в р-субъединице. Интересно, что
а-субъединица ТТГ идентична в этом
гормоне и других гликопротеидных гормонах
аденогипофиза и плаценты- ФСГ, ЛГ,
ХГ, то есть гонадотропинах. Более того,
а-субъединица ТТГ практически невидоспе-
цифична, и у быка и овцы выглядит почти
как у человека. Однако р-субъединицы этих
гликопротеидных гормонов отличаются,
они разные у различных животных и несут
иммунологическую специфичность.
Именно в р-субъединице локализован участок,
связывающийся с рецептором ТТГ. Но
р-субъединица активируется или
приобретает нужную для воздействия на таргетные
клетки конформацию только в комплексе с
а-субъединицей. Также обстоит дело и для
гонадотропинов, причём их универсальная
а-субъединица может выступать
активатором специфических эффектов любой из
уникальных р-субъединиц, а, возможно,
защищает их при транспорте от разрушения
(рис. 107). Данный пример иллюстрирует
экономность информационных средств
эволюции: одна и та же полипептидная цепь
была использована для построения
дивергентного семейства гормонов с разными
функциями.
Выработка ТТГ в аденогипофизе
находится под позитивным контролем гипоталами-
ческого тиролиберина и негативным — со-
матостатина.
И гипоталамическое, и гипофизарное
звено регуляции тироидной функции
чувствительны к обратному контролю
сервомеханизма, основанного на понижении
продукции ТТГ при избытке тироидных
гормонов и самого ТТГ. При этом тироксин
и особенно трийодтиронин, в который
тироксин активно превращается в тиротрофах
гипофиза, действуют на ядерные
рецепторы тиротрофов, блокируя генетические
программы биосинтеза ТТГ.
Дополнительными ингибиторами базальной и
стимулированной секреции ТТГ служат дофамин
и глюкокортикоиды, в связи с чем еще
классик эндокринологии Н. Пенде говорил
об антагонизме питуитарно-тироидной и
Нарушения эндокринной регуляции 545
а-субъединица
(общая для ТТГ,
ГТГ и ХГ)
-ОН
РисЮ7. Гомология ТТГ и других гликопротеидных гормонов аденогипофиза и плаценты.
Структура субъединиц ТТГ (по Н.А. Юдаеву и соавт., 1976). У — углеводные остатки
питуитарно-супраренальной «осей».
Суточный ритм продукции ТТГ предусматривает
понижение его уровня после полуночи. При
сохранности гипоталамо-гипофизарной
связи острое и хроническое
переохлаждение вызывает усиление продукции ТТГ,
особенно у новорожденных, а стресс и
тепло — понижают продукцию этого гормона.
В связи с сильным влиянием обратных
связей на продукцию ТТГ концентрации ТТГ
и тироидных гормонов при практической
интерпретации результатов лабораторных
обследований должны всегда
сопоставляться друг с другом. Одни и те же абсолютные
цифры в контексте разных сочетаний могут
означать присутствие различных синдромов
(см. выше рис. 83).
При первичном гипотпирозе низкая
продукция тироидных гормонов вызывает
гипертрофию и гиперфункцию гипофизарных
тиротрофов. Концентрации ТТГ в крови
растут, причём, как правило, еще до
развития гипотироза. Выработка тиролиберина
увеличена, а тиростатических факторов —
снижена. Гипофиз так увеличивается, что
может ошибочно подозреваться его опухоль.
Параллельно активируется продукция
близких к ТТГ по структуре гонадотропи-
нов, снижается выработка СТГ и АКТГ, и
может возникать гиперпролактинемия, так
546 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
как выработка в гипоталамусе тиропро-
лактостатического медиатора —
дофамина — бывает понижена. В тяжелых случаях
при первичном гипотирозе вследствие этого
компенсаторного гипофизарного ответа
даже отмечаются нарушение полей зрения,
лакторея и ускорение полового созревания.
Однако при нормализации тироидной
функции все эти явления исчезают в силу их
обратимости.
При вторичном гипотирозе речь идет о
недостаточной стимуляции исходно
нормальной щитовидной железы со стороны
гипоталамуса или гипофиза. В типичном
случае концентрации ТТГ снижены или
нормальны, хотя низкий уровень тиро-
идных гормонов требует их повышения.
Многие пациенты, однако,
демонстрируют более сложную картину. У них при
низком содержании тироидных гормонов
концентрация ТТГ повышена, как при
первичном гипотирозе. Причиной этого
явления (наблюдаемого при так
называемом «синдроме болезненного эутироза»)
служит продукция в гипофизе
недостаточно активного ТТГ, в частности, не
подвергшегося адекватному гликозилированию.
Высокий уровень иммунореактивного ТТГ
при этом не означает усиления тиротроп-
ной стимуляции щитовидной железы.
Введение тиролиберина активизирует
гликозилирование ТТГ и повышает его
биологическую активность.
Гипертироз чаще всего не является
результатом гипоталамических или гипофи-
зарных нарушений, а происходит из-за
наличия негипофизарных иммунологических
стимуляторов щитовидной железы, либо
автономных от гипофизарного контроля
источников продукции тироидных гормонов
(см. ниже о болезни фон Базедова-Грейвса и
токсических аденомах щитовидной железы).
Соответственно, уровень продукции ТТГ и
тиролиберина при подобных формах гипер-
тироза, как правило, снижен.
Однако, примерно в 5 % случаев
гипертироз можно считать центральным. Причины
вторичного гипертироза следующие.
Автономные от периферического и ги-
поталамического контроля опухоли из ти-
ротрофов аденогипофиза могут насыщать
организм ТТГ и вызывать гипертироз. Как
правило, это макроаденомы. Они могут
параллельно продуцировать СТГ. В
опухолевых клетках изготавливается, наряду
с нормальной, дефектная а-субъединица
ТТГ, не вступающая в комплекс с р-субъеди-
ницей. Присутствие таких а-субъединиц в
крови является диагностическим признаком
ТТГ-образующих аденом гипофиза.
У некоторых больных с центральным
гипертирозом в гипофизе нет аденом, но их
тиротрофы резистентны к ингибирующему
действию тироидных гормонов. Поэтому
секреция ТТГ происходит на повышенном
уровне, и весь тиростат стабилизируется при
состоянии гипертироза.
Биологическое действие ТТГ, время
полужизни которого в крови около 1 ч, основано
на комплементарном связывании этого
гормона с поверхностным клеточным
рецептором, который является белково-ганглиозид-
ным комплексом, содержит 744
аминокислоты и принадлежит к семейству рецепторов,
ассоциированных с G-белками (см. выше).
Пострецепторные эффекты данного
транслятора предусматривают активацию аде-
нилатциклазы и ц-АМФ-протеинкиназных
механизмов в клетках-мишенях, а также
участие фосфатидил-инозитоловых и каль-
ций-кальмодулиновых посредников.
ТТГ располагает рецепторами и вне
тироидных клеток. Экстратироидные эффекты
этого гормона включают, в частности,
усиление липолиза в жировой ткани.
Однако главное метаболическое
действие ТТГ опосредовано практически
исключительно, через щитовидную железу.
Для щитовидной железы ТТГ служит не
только стимулятором захвата йода, синтеза
тироидных гормонов и их освобождения
в кровь. Доказано, что при исключении
влияния ТТГ на тироциты у
млекопитающих щитовидная железа инволюцирует,
её фолликулярный эпителий уплощается
и перестает обновляться. Эмбриональное
развитие железы требует действия ТТГ,
при его отсутствии может развиться
врождённый атироз. Таким образом, ТТГ — еще
и митогенный, и морфогенетический
гормон для тироцитов. Осуществляя эти
функции параллельно, ТТГ действует
через неодинаковые механизмы. Имеются
данные, что функция тироцитов
стимулируется с участием ганглиозидной части
рецептора ТТГ и протеинкиназы А, а их
рост — с участием белковой части этого
рецептора, протеинкиназы С и, возможно,
липидных посредников и процессов,
зависящих от интернализации комплекса ТТГ
и его рецептора.
Все это при патологии может приводить к
диссоциированным и мозаично-сочетанным
нарушениям функций щитовидной железы
и ее роста. Таким образом, щитовидная
железа — главная мишень ТТГ.
Щитовидная железа в силу своей
поверхностной локализации была известна,
видимо, еще древним египтянам, фрески
которых, по мнению Э. Рено, содержат
изображения божества Тота с признаками
зоба и гипотироза. Первое литературное
описание этого органа, дошедшее до нас,
принадлежит К. Галену. Название Glandula
thyroidea дал железе в 1656 г. Т. Вартон за
сходство со щитом. Достоверно известно,
что уже средневековые арабские врачи
знали о крайней степени гиперплазии
щитовидной железы — зобе — и успешно применяли
операцию струмэктомии — не позднее XI
века (Абул-Казим). Эндокринные
функции у железы первым заподозрил еще Т.У.
Кинг (1836), а доказал — путем
экспериментальной тироидэктомии и пересадки
железы — П.М.Шифф (1884). Наш
соотечественник Н.А. Бубнов (1884) первым
попытался выделить из коллоида железы
ее гормоны. А. Шатен в серединеХ1Х века
сформулировал гипотезу о йод-дефицитном
происхождении зоба (1850). Затем в работах
Т. Кохера была показана эффективность
йода при микседеме (1895), а усилиями
Э.Бауманна (1896) было установлено, что
Нарушения эндокринной регуляции 547
йод входит в состав приготовленного им
концентрата специфического
гормонального начала железы — йодотирина, а коллоид
пациентов с эндемическим зобом
содержит мало йода. Этапное значение имели
работы А. Магнус-Леви (1895),
разработавшего технику непрямой калориметрии,
начавшего объективную оценку функций
щитовидной железы in vivo и доказавшего
метаболическую роль её гормонов (см. выше
раздел «Энергетический обмен...»). Магнус-
Леви показал, что йодотирин воспроизводит
эффекты щитовидной железы на основной
обмен. В дальнейшем А. Освальд
выделил из коллоида йодсодержащий белок
тироглобулин (1899). Значимость йодно-
щитовидной темы в медицине начала XX
столетия подтвердил Нобелевский комитет,
удостоивший премии по медицине за 1909 г.
Т. Кохера. Позже Э.Кендалл (1919)
получил йодсодержащий гормон щитовидной
железы в кристаллической форме, дав ему
название тироксин, а К.Р. Хэрингтон с
соавторами (1926,1927) установил его строение
и синтезировал. Затем М. Гросс показал, что
в железе синтезируется и трийодтиронин,
гормональная активность которого выше.
Современные представления о
патофизиологии щитовидной железы можно
резюмировать следующим образом.
Щитовидная железа возникает на 4-й
неделе эмбрионального развития из
выпячивания вентральной стенки глотки между I и
II жаберными карманами. Из эндодермаль-
ного эпителия этого зачатка формируются
фолликулярные тироциты — Л-клетки
щитовидной железы, а также близкие к
ним клетки М. Аскенази (В-клетки). А и В
клетки трансформируются друг в друга и,
вероятно, имеют общие функции — синтез
тироксина и трийодтиронина. В-клетки
несколько крупнее и содержат биогенные
амины, в частности, серотонин. Предполагается,
что метаплазия В-клеток при тироидите
Хасимото и других тиропатиях приводит к
появлению оксифильных клеток К. Хюр-
тле, характерных для этих поражений (см.
ниже). Помимо этого щитовидная железа
548 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
имеет и третий клеточный тип, который
происходит из производных V пары жаберных
карманов — ультимобранхиальных телец,
врастающих в эмбриональную железу. Это
— парафолликулярные С-клетки,
источники кальцитонина и кокальцигенина (старое
название — полипептид, ассоциированный с
геном кальцитонина). Нарушения кальцито-
ниновой регуляции рассмотрены в разделе,
посвященном патологии паратироидно-
кальцитониновой регуляции
кальций-фосфорного обмена, о кокальцигенине известно,
что он при системном действии вызывает
гипотензию и тахикардию. Это убиквитар-
ный пептид, синтезируемый многими ней-
роэндокриноцитами разной локализации.
В бронхах он участвует в спазме гладких
мышц, в ЦНС считается активатором
симпатических центров гипоталамуса, подавляет
пищевое поведение и вызывает повышение
кровяного давления (Я.А. Рейд, 1998).
В тироидной патологии значение С-кле-
ток определяется тем, что это источники
медуллярного рака щитовидной железы.
При нарушении миграции эмбриональных
клеток щитовидной железы они остаются
по пути миграции или перемещаются в
средостение. По ходу трахеи, под языком
и в грудной клетке возможно образование
эктопической тироидной ткани и С-клеток,
которые могут инфицироваться, озлока-
чествляться или подвергаться гиперплазии,
давая загрудинный или язычный зоб.
В зрелой щитовидной железе паренхима
имеет псевдодольчатое строение и разделена
неполными перегородками, берущими
начало от капсулы органа. Псевдодольки состоят
из фолликулов, в центре которых
расположен коллоид — концентрат секретируемого
тироцитами тетрамерного гликопротеида
тироглобулина (660 кД), уникального тиро-
идного белка, богатого тирозином и йодом,
используемого для синтеза и запасания
гормонов щитовидной железы. Фолликулы
окружены базальной мембраной с
проживающими на ней А-клетками. Фолликулярный
эпителий ведет биосинтез гормонов, причём
цилиндрическая, кубическая или
уплощённая форма клеток отражает их более
или менее активную функцию. Биосинтез
требует постоянного поглощения йода из
крови, продукции и секреции
тироглобулина, эндоцитоза тироглобулина и отщепления
от него йодированных тирозинопроизвод-
ных. В железе более 30 млн фолликулов,
соединенных через отверстия в базальной
мембране и формирующих оплетённую
капиллярами сеть. Между фолликулами
имеются интерфолликулярные островки,
которые содержат фолликулярные и
парафолликулярные клетки и участвуют в
регенерации фолликулов.
Синтез тироидных гормонов
определяется следующими основными процессами
(рис. 108, 109). Характеризуя их этапы,
мы упомянем и некоторые их первичные
нарушения.
Клетки фолликулярного эпителия
постоянно синтезируют на рибосомах ШЭР, гли-
козилируют в ШЭР (маннозилирование) и
комплексе Гольджи (галактозилирование),
пакуют в секреторные пузырьки и выделяют
в коллоид тироглобулин.
Для образования гормонов щитовидной
железы необходим йод, содержащийся в
основном в морецродуктах. Значительные
территории, удалённые от морей, относятся
к геохимическим провинциям, где
потребность в йоде обеспечивается с известным
напряжением. В таких регионах
гиперплазия щитовидной железы и гипотироз
встречаются чаще. До 250 млн человек
населяет эту зону, куда преимущественно
входят высокогорные районы (Альпы,
Анды, Гималаи, Нагорный Дагестан, Саяны,
Папуа) и внутриконтинентальные
территории (район Великих Озёр, Полесье, часть
Сибири, Средней Азии). В разных регионах
фактическое суточное поступление йода
колеблется от 20 до 700 мкг. Оптимальное
обеспечение йодом требует его поставки
на уровне 150-300 мкг/сутки. Суточное
потребление йода на уровне менее 80 мкг
опасно, так как не позволяет поддержать
эутироидное состояние без стимуляции же-
i-моноиодтироаян
НО-/ У- СН2-
/=\ ?Н2 1+ У L 3 мои
-/ \-СН2-СН—СООН —с' j
hKD"
NH2
I
сн—соон
(МИТ)
L-тирозин
, , NH2
/ \ I
СНг-СН—СООН
L-3,5- дийодтирозин
(ДИТ)
2но-^ Vch2-ch-cooh—► но"\ У~°\ #
NH2
I
CH2-CH—COOH -
L-3,5- дийодтирозин
I I
NH2
Ь-3,5,3\5'-тетраиодтиронин I
Т4 + H2C-CH—COOH
OH l
I
серии
NH2
NH2
но_/ \-CH2-CH—COOH + НО -^ V-CH2-CH—
I
L-3-моноиодтирозин Ь-З.З-дииодтирозин
СООН-
СООН + L серии
L-3,5,3'
Рис. 108. Биосинтез тироидных гормонов и их предшественники
плазма
крови
аминокислоты
Рис. 109. Цитофизиологические основы продукции тироидных гормонов. ТГ — тироглобулин,
МИТ — монойодтирозин, ДИТ — дийодтирозин, ТЗ — трийодтиронин, Т4 — тироксин.
Пояснения в тексте
550 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
лезы ТТГ и формирования её гиперплазии
(см. также выше «Патофизиология обмена
микроэлементов»).
Пищевой йод в форме J" всасывается уже
в желудке и в верхней части кишечника
всего за полчаса. Тироциты захватывают
йод из плазмы крови и концентрируют его
в 25-50 раз, а при необходимости — и в
несколько сот. Работает энергозависимый
йодный насос, позволяющий поглощать йод
против градиента концентраций. Против
этого транспортера возникают аутоантитела,
которые участвуют в патогенезе
аутоиммунного тироидита и гипотироза (Н. Амино,
2002). Происходит противоперенос йодида
и натрия с участием специального
транспортного белка (65 кД), a K+/Na+-ATOa3a
нормализует натрий-калиевое соотношение,
затрачивая АТФ. В сутки железа поглощает
не менее 120 мкг йода. Так как эта система
не строго специфична, многие ионы служат
конкурентными антагонистами транспорта
йода. Транспорт йода блокируют тио-
цианаты, тиооксизолидоны и роданиды,
содержащиеся в некоторых продуктах,
обладающих антитироидным действием, или
«струмогенных» (здесь не только
экзотические для нас кассава, тапиока, маниока, но и
привычные для европейцев крестоцветные и
кабачковые: репа, цветная и краснокочанная
капуста, горчица, брюква, турнепс). Диета,
перенасыщенная этими ингредиентами, в
том числе — молоко вскармливавшихся ими
коров, способствует гипотирозу. Массовый
гипотироз на почве струмогенной диеты и
содержания углеводистых струмогенов в
воде отмечен во многих регионах Африки
(центральная часть, Мозамбик), Америки
(Колумбия, Чили), а также в других местах.
Формированию гиперплазии при наличии
струмогенов способствуют высокое
содержание в воде фтора и ее жесткость.
В этом смысле реформу Петра Великого,
после которой в России с её громадными
пространствами, принадлежащими к йод-
дефицитным зонам, репа уступила место
не содержащей струмогенов картошке, еще
предстоит оценить с точки зрения улучшения
эндокринологического здоровья населения!
чдокринно-метаболические нарушения)
Пертехнетат, содержащий
радиоактивный технеций, захватывается железой, но
не задерживается в коллоиде, что позволяет
использовать этот анион при сцинтилляци-
онном сканировании щитовидной железы.
Не менее 80 мкг йода в день расходуется на
синтез гормонов щитовидной железы. Йод,
не абсорбированный тироцитами, а также
попадающий обратно в кровь вследствие
утечки из железы (которая в норме
достигает 40-100 мкг/сутки и может повышаться
при патологии), частично утилизируется
макрофагами и нейтрофилами, которые
оказывают бактерицидное действие на объекты
фагоцитоза с помощью йодидов и оксийоди-
дов. Выведение йода из организма
осуществляется ЖКТ и почками, причём в ЖКТ йод
подвергается полной рабсорбции.
Следующий этап функционирования ти-
роцитов — органификация йода, то есть его
окисление — превращение из йодид-аниона
в связанную с тироглобулином форму. Этот
процесс происходит на щёточной кайме
тироцитов и катализируется тироидной пе-
роксидазой. Мембранно-связанная гем-со-
держащая пероксидаза щитовидной железы
бывает объектом аутоиммунитета, и титр ан-
титиропероксидазных аутоантител
коррелирует с тяжестью аутоиммунного тироидита
(см. ниже). Под ее воздействием йод
связывается с остатками тирозина в тироглобу-
лине коллоида. До четверти всех тирозилов
йодируются. Органификация приводит к
образованию как монойодтирозина (МИТ),
так и дийодтирозина (ДИТ). Последующий
этап — спаривание или конденсация йодти-
розиновых остатков с образованием тиро-
ниновых структур — ведет к образованию
на тироглобулиновой молекуле связанных
с ней тироксина (3,5,3,5-тетрайодтиронин)
и трийодтиронина (3,5,3-трийодтиронин).
Первый формируется путём конденсации
двух остатков ДИТ, а второй — из МИТ и
ДИТ. Уникальная третичная структура и
конформация тироглобулина могут быть
ключевыми в этом процессе. Аутоантитела
к тироглобулину часто присутствуют в
высоких титрах при различных тиропатиях.
Некоторые из них, по-видимому, служат
лишь свидетелями иммунопатологических
процессов, но относительно других имеется
предположение, что они способны нарушать
сложный конформационно-зависимый
процесс конденсации, идущий на тироглобули-
новой матрице. Так как конденсация тоже
катализируется пероксидазным
комплексом, блокаторы пероксидазы, в частности,
тиомочевина, служат мощными
ингибиторами функции щитовидной железы, действие
которых не снимается избытком йода в
отличие от перхлората и тиоцианатов. В
патофизиологии существует тиомочевинная
модель острого гипотироза и гипотироидной
комы на грызунах. Практически важно, что
очень высокое поступление йода в железу
ингибирует органификацию и спаривание
йодтирониновых остатков тироглобули-
на {эффект Вольфа-Чайкова). Поэтому
применение больших доз йода в течение
длительного времени может вызвать гипо-
тироз и зоб («йоднаямикседема»). Особенно
чувствительны к струмогенному действию
йода плод и лица с гипертирозом, леченные
радиоактивным йодом. Так как йод может
у предрасположенных к аутоиммунным
тиропатиям лиц провоцировать и болезнь
фон Базедова, ясно, что с точки зрения
патологии щитовидной железы применение
йодосодержащих лекарств (амиодарон,
йодид калия, витаминно-минеральные
композиции с йодом) должно
контролироваться и противопоказано определённому
кругу лиц.
На моль тироглобулина приходится в
норме 6-7 остатков МИТ, 4-5 молекул ДИТ, 4
моля тироксина и 0,2-0,3 — трийодтирони-
на. Чем хуже железа обеспечена йодом, тем
выше доля МИТ и трийодтиронина (Т3), что
считается компенсаторным явлением.
Под влиянием секреторных стимулов
тироциты захватывают обратно порции
йодированного тироглобулина. Это
осуществляется путём пиноцитоза. Коллоидная
капелька сливается с лизосомой. В фаголизо-
соме происходит протеолиз тироглобулина,
освобождаются тироксин и трийодтиронин,
и имеет место частичное дейодирование
тироксина в Т3, повышающее в секрете железы
Нарушения эндокринной регуляции 551
долю высокоактивного Т3. Отщепление
гормонов от тироглобулина подавляется
большими дозами йода, а также
препаратами лития, применяемыми при психических
заболеваниях. Дегалогеназа, требующая
НАДФ, освобождает йод из оставшихся
непревращёнными в гормоны ДИТ и МИТ
для повторного использования.
ТТГ способен активизировать захват и
транспорт йода железой, синтез
тироглобулина и его йодированных производных,
пиноцитоз коллоида и освобождение в кровь
Т3 и Т4.
Помимо эндемического йодного
дефицита и токсических поражений описываемых
сложных процессов химическими струмо-
генами пищевого и лекарственного
происхождения имеются наследственные дефекты
ответственных за функции тироцитов
белков, которые тоже приводят к первичному
гипотирозу и даже зобу. В этих случаях
гипотироз носит врождённый, часто —
семейный характер. В эпидемиологическом и
медико-географическом отношении важно
различать эндемические формы гипотироза
и зоба, связанные с региональной
особенностью экологии человека (геохимический
дефицит йода, традиционное употребление
струмогенов) и спорадические формы
гипофункции и гиперплазии железы вследствие
действия индивидуальных причинных
факторов и условий. Районы, где частота
зоба превышает 10% населения, считаются
эндемическими.
О роли аутоиммунных факторов в генезе
спорадического гипертироидного зоба речь
еще пойдёт ниже (см. болезнь фон Базедова).
Что же касается спорадического гипоти-
роидного или эутироидного зоба, вполне
возможно его возникновение на почве
наследственных дефектов тироцитов. При
близкородственном скрещивании, в малых
популяциях и изолятах, не исключено
накопление дефектных генов и учащение
встречаемости подобных нарушений. Такой
семейный, спорадический по своей сути,
синдром может ложно казаться
эндемическим.
552 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Четыре главных наследственных тиро-
патии — все аутосомно-рецессивного типа,
имеют следующую характеристику.
>- При дефекте мембранного
белка-транспортёра (65кД) возникает системная
неспособность желудка, слюнных желёз
и тироцитов улавливать и
концентрировать йодид и ряд других неорганических
анионов. Тест с захватом пертехнета-
та — отрицательный.
>* Отсутствие или дефицит тироидной
пероксидазы ведет к невозможности для
йодида отдать электрон, превратиться в
свободный йод и органифицироваться.
Часто при этом имеется нарушение слуха
(синдром Пендреда).
>• Дефицит дегалогеназы приводит к
нарушению реутилизации йода, остающегося
в МИТ и ДИТ. Это повышает
потребность в йоде и делает синтез тироидных
гормонов малоэффективным, хотя и
возможным.
>* Синдром, при котором не происходит
конденсация остатков МИТ и ДИТ в
тироглобулин-связанные Т3 и Т4,
ранее характеризовавшийся условно как
«дефицит конденсазы», оказался
гетерогенной группой из, по меньшей мере,
трёх разных наследственных нарушений
(аномальный тироглобулин, нарушение
протеолиза тироглобулина и нарушение
взаимодействия йода с тирозином).
Все эти нарушения в зависимости от
экспрессии генов и условий среды могут
вызвать различной тяжести функциональное
расстройство. Следовательно, зоб, который
возникает при каждом из этих нарушений,
может быть эутироидным (с
компенсированной функцией) или, наоборот, ярко
гипотироидным, с развитием кретинизма,
когда малая функциональная активность
тироцитов, даже стимулированных ТТГ, не
может быть компенсирована их числом.
После секреции в плазму большая часть
тироидных гормонов переходит в белково-
связанную, неактивную форму,
уравновешиваясь с малой активной свободной
фракцией. На 75 % тироидные гормоны
связываются с тироид-связывающим глобулином
(из фракции а-глобулинов). Большая часть
остального пула связана с транстиретином,
мигрирующим на электрофореграмме, как
преальбумин (см. выше «Нарушения
белкового состава плазмы крови»). Наконец,
небольшая часть связывается с
сывороточным альбумином. Уменьшение связывания
способствует более выраженному действию
гормонов, и наоборот. Тироид-связывающий
глобулин комплексирует Т4 намного более
активно, чемТ3, а транстиретин, вообще,
связывает только Т4. Поэтому лишь 0,02%
Т4 и целых 0,3 % Т3 остаётся свободным.
Соответственно, время полужизни Т4 — около
7 дней, а Т3 — только 30 часов.
Все белки, связывающие тироидные
гормоны, печёночного происхождения.
Поэтому плазменное связывание
тироидных гормонов понижается при печёночной
недостаточности, голодании. Андрогены и
большие дозы глюкокортикоидов
подавляют продукцию тироид-связывающего
глобулина, а эстрогены, клофибрат, тамоксифен,
перфеназин, наркотики героин и метадон,
а также, вероятно, эндогенные порфирины
при перемежающейся порфирии —
повышают. При нефротическом синдроме
связывающие эквиваленты теряются из плазмы,
аспарагиназа разрушает
тироид-связывающий глобулин, а салицилаты, фуросемид и
дилантин конкурентно вытесняют
тироидные гормоны из комплексов с белками. При
беременности экспрессия
тироид-связывающего глобулина растёт, а при акромегалии-
гигантизме понижается. Наконец, имеются
наследственные дефициты
тироид-связывающего глобулина и транстиретина, а также
наследственный избыточный синтез первого
из этих транспортёров тироидных
гормонов. При патологии в плазме отмечаются
аутоантитела к тироксину. Однако они не
влияют существенно на гормонозависимые
функции.
Свободные гормоны, достигая клеток-
мишеней, взаимодействуют с их
поверхностными и ядерными рецепторами. Т4 в
клетках практически нацело переходит
в Т3 в результате дейодирования. Моно-
дейодинация, катализируемая тканевой
дейодиназой, — важнейший этап
метаболизма тироидных гормонов, поскольку, по
современным представлениям, его можно
приравнять к переходу прогормона
(тироксина) в наиболее активный гормон (трийод-
тиронин), биологическое действие которого
в 3 с лишним раза сильнее. Этот процесс
регулируется в адаптивных целях. Только
13 % циркулирующего Т3 выделяется
непосредственно тироцитами, остальной гормон
возникает путём периферического
дейодирования Т4. Отщепление йода от наружного
кольца, в положении 5' — приводит к
появлению высокоактивного изомера Т3. Дейо-
дирование внутреннего кольца в положении
5 — порождает малоактивный обратный или
реверсивный Т3 (рТ3). В норме около 40 % Т4
метаболизируется этим путём.
Т4-5'-дейодиназы — микросомальные
сульфгидрильные ферменты печени и
почек, коры надпочечников и гонад (1тип),
а также ЦНС, гипофиза, бурого жира и
плаценты (IIтип). В этих органах в
основном и формируется Т3. В буром жире
активация II типа дейодиназы происходит1
рефлекторно, в ответ на симпатическую
стимуляцию липоцитов при холодовой
адаптации.
Описана активация Т4-5'-дейодиназы
II подтипа при гипотирозе, в частности,
сопровождающем аутоиммунный тирои-
дит, когда это считается компенсаторным
явлением. Дейодиназа I типа, в отличие от
II типа этого энзима, ингибируется про-
пилтиоурацилом и реагирует на гипотироз
понижением активности.
При голодании и практически любых
тяжелых болезнях образуется повышенное
количество рТ3, об адаптивной роли этого
явления см. выше раздел «Голодание».
Конверсия Т4 в Т3 ослаблена у плода,
новорожденных и престарелых. Она понижается при
голодании, тяжелом стрессе и под действием
некоторых медикаментов (йопагност и
другие йодоконтрастные препараты, амиодарон,
пропранолол, дексаметазон). Как полагают,
Нарушения эндокринной регуляции 553
в основе этого лежит торможение
активности дейодиназы I типа, а для йодсодержа-
щих лекарств — обоих типов. Дальнейший
катаболизм тироидных гормонов
предусматривает их последовательное дейодирование,
окисление углеродного скелета, а также
глюкуронидизацию и сульфатирование.
Метаболиты выводятся с желчью в ЖКТ и
частично реабсорбируются.
Некоторые эффекты тироидных
гормонов (ускорение захвата аминокислот,
активация калий-натриевой АТФ-азы)
детерминированы поверхностной рецепцией
гормонов, часть зависит от внутримито-
хондриального действия, но большинство
определяются влиянием на специфические
ядерные рецепторы тироидно-стероидного
семейства, гомологи онкобелка c-erbA,
воспринимающие, исключительно, Т3.
Имеется два типа тироидных внутриклеточных
рецепторов — TRa-рецепторы (подтипы оц
и а2), кодируемые 17-й хромосомой и
преобладающие в мозге, а также TRp рецепторы
(подтипы рг и р2)» закодированные в хромо
соме 3 и преобладающие в печени и почках.
Сердце содержит в равной мере оба типа
TR-рецепторов. При наследственном
дефекте Т3-связывающего домена TRpt-рецепто-
ров формируется аутосомно-рецессивный
синдром Рефетова — системная
резистентность к действию тироидных гормонов.
Спектр биологических эффектов
тироидных гормонов широк, так как их
рецепторы имеются у всех клеток (наименьшую
чувствительность к тироидным гормонам
проявляют спленоциты и семенники).
Часть эффектов опосредована через
регуляцию транскрипции генов, некоторые
зависят от влияния гормонов на судьбу
долгоживущих РНК и трансляцию, а
отдельные — осуществляются на
посттрансляционном уровне. Всего тироидные гормоны
активируют различным путём более 100
клеточных ферментов.
Некоторые эффекты тироидных
гормонов на клеточном уровне перечислены в
табл. 33.
554 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 33. Цитологические и биохимические эффекты тироидных гормонов
Мишени
Клеточная мембрана
KVNa+АТФаза
Рибосома
Митохондрия
Метаболизм углеводов
Метаболизм липидов
Метаболизм белков
Метаболизм нуклеиновых
кислот
Взаимоотношения с другими
гормонами
Пермиссивное действие на
эффекты катехоламинов
Витаминный обмен
Эффекты
Усиление поглощения аминокислот, глюкозы, калия; выведения натрия,
кальция, фосфора
Активация, увеличение хемиосмотических градиентов, прирост основного
обмена. Ускорение генерации биопотенциалов и реполяризации
Стимуляция связывания амино-ацил-тРНК, пептидил-синтетазной и транс-
локазнои реакции
Увеличение аэробного окисления, в токсических фармакологических дозах
(но не в физиологических!) разобщают окисление и фосфорилирование
Ускорение всасывания и окисления глюкозы и распада гликогена,
контринсулярное действие
Стимуляция липолиза, окисления жирных кислот, уменьшение стероидо-
генеза, индукция рецепторов Л ПНП
Усиление синтеза мочевины, отрицательный азотистый баланс, активация
биосинтеза дифференцировочных протеинов в ЦНС, скелете, гонадах и
других тканях. Индукция синтеза миозина в миокарде
Синтез ц-АМФ, активация начальных этапов синтеза пуринов и пиримиди-
нов, дифференцировочная активация синтеза ДНК и РНК. Синтез коротко-
живущих РНК. Индукция метаморфоза у головастиков
Стимуляция освобождения инсулина, глюкагона, соматостатина,
повышение печеночного распада стероидов и кортикостероидогенеза, снижение
синтеза катехоламинов в мозговом веществе надпочечников, тимотропное
действие на гормонообразование в вилочковой железе, подавление
секреции ТТГ и тиролиберина
Стимуляция транскрипции и экспрессии Р-адренорецепторов; ТТГ
усиливает экспрессию агадренорецепторов на самих тироцитах
Повышение потребности в большинстве витаминов
С точки зрения физиологии органов и
систем и клинических критериев оценки их
функции уместно указать основные
физиологические эффекты тироидных гормонов
(табл. 34).
Естественно, что при столь широком
круге эффектов отклонения в содержании
тироидных гормонов способны вызвать
разнообразную и яркую симптоматику.
Основные синдромы при заболеваниях
щитовидной железы отражают нарушения
ее функции, а также роста.
Как уже упоминалось выше, синдромы
дисфункции и дисплазии железы могут мозаично
сочетаться. Так, существуют гипотироидная,
эутироидная и гипертироидная
разновидности зоба. Вместе с тем при гормонообразующей
аденоме или атирозе функциональное
состояние железы всегда однозначно определено.
Большое место в спектре тиропатий
занимают воспаления железы — тироидиты,
рассматриваемые ниже.
Наконец, следует особо отметить
значительную роль аутоаллергических факторов
в развитии всех этих синдромов поражения
щитовидной железы.
Гипертироидные
и гипотироидные состояния
Избыточное действие тироидных гормонов
формирует гипертироз. Недостаточное
действие тироидных гормонов
характеризует гипотироз. Состояние адекватной
метаболическим и онтогенетическим нуждам
тироидной функции именуют эутироз.
Считается, что истинный гипотироз и
гипертироз сопровождаются сочетанными
однонаправленными изменениями
содержания Т3и Т4. Примерно у 10% тяжело-
Нарушения эндокринной регуляции 555
Таблица 34. Физиологическое действие тироидных гормонов на органы и системы
Органы и ткани
Сердце
Сосудистая система
Жировая ткань
Мышцы
ЦНС
ЖКТ
Легкие
Система крови и кроветворения
Кости
Почки
Другие эффекты
Эффекты
Положительный хронотропный и инотропный эффекты, за счёт увеличения
экспрессии и аффинности р-адренорецепторов и биосинтеза
высокоактивных в АТФ-азном отношении тяжёлых а-цепей миозина
Повышение систолического артериального кровяного давления и
пульсовой разницы (сенсибилизация к катехоламинам). Повышение ОЦК
Липолиз
Ускорение реакций, усиление катаболизма белка
Развитие головного мозга, синтез короткоживущих РНК и белков,
стимуляция обучаемости. Повышение возбудимости, лабильности
Повышение аппетита, ускорение перистальтики, ускорение всасывания
углеводов, стимуляция островков Лангерганса
Ускорение газообмена, одышка
Усиление эритропоэза, укорочение жизни эритроцитов
Индукция дифференцировки, роста. Потеря кальция
Увеличение кровотока, фильтрации, диуреза
Стимулируют окисление во всех органах, кроме зрелого головного мозга,
лимфоидной ткани, матки, яичек, аденогипофиза
больных людей без каких-либо первичных
нарушений функций щитовидной железы
и тиротропной активности гипофиза из-за
вышеописанных изменений конверсии Т4 в
Т3 и нарушений связывания и катаболизма
тироидных гормонов, сопряжённых с
укорочением времени их нахождения в крови,
возникает так называемый «синдром
болезненного эутироза». При этом состоянии,
дифференцируемом от истинного гипоти-
роза и эутироза, как правило содержание Т3
повышено, а Т4 — нормально или снижено.
Около 1 % пациентов с данным необычным
синдромом имеют высокий уровень Т4, в
основном, это лица, получавшие препараты
йода (см. также с. 503).
Гипертироз как синдром имеет
различную этиологию. Принципиально он может
происходить из-за повышения продукции
тироидных гормонов в тироцитах (в ответ на
иммунопатологические, гипоталамические
и гипофизарные стимулы, а также в
автономном режиме); из-за деструкции тироид-
ной железы и повышенного освобождения
запасённых гормонов (при тироидитах)
и, наконец, — из-за введения экзогенных
пищевых или лекарственных тироидных
гормонов. Субклинический гипертироз
характеризуется нормальным содержанием
тироидных гормонов при концентрации
сывороточного ТТГ менее 0,1 мЕд/л.
В таблице 35 причины гипертироза
фигурируют в порядке убывания их частоты.
В данном перечне причины в позициях
с 5-й по 10-ю характеризуются
исключительной редкостью, вплоть до уникальных
казуистических наблюдений, и суммарно
составляют менее 1 % всех случаев
гипертироза.
Гипотироз как синдром характеризуется
большим разнообразием этиологии. В 95 %
случаев поражение носит первичный
характер и касается тироцитов. Около 5 % случаев
вторичны и связаны с гипоталамо-гипо-
физарными дисфункциями. Эксклюзивно
редким является внежелезистыи гипотироз
при синдроме Рефетова (см. выше).
Первичный гипотироз бывает врождённым
(при наследственных дефектах тироцитов,
рассмотренных выше; при врожденном ати-
розе или дисгенезии железы; а также у
новорожденных детей от матерей, находящихся
под воздействием йода и тиростатиков). Но
556 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 35. Этиология гипертироза
1) Аутоиммунная стимуляция функции и роста желе-
зы (болезнь фон Базедова-Грейвса) — 85%
2) Автономная гиперфункция (токсический мульти-
нодулярный зоб — болезнь Пламмера; токсическая
фолликулярная аденома) — 5—7%
3) Деструкция железы при тироидитах (подострый
лимфоцитарный тироидит, дебют хронического ау-
тоиммунного тироидита Хасимото) — 5—7%
4) Йод-базедовизм (гипертироз у йоддефицитных
больных после лечения йодом)
5) Ятрогенный и пищевой гипертироз
б) Временный гипертироз новорожденных от мате-
рей с болезнью фон Базедова-Грейвса
7) Тиротропиномы гипофиза и других локализаций
8) Гиперпродукция тиролиберина гипоталамуса
(дефект сервомеханизмов)
9) Дисгерминомы и струма яичника (эктопическая
продукция гормонов)
10) Гормонообразующая карцинома щитовидной
железы, обычно метастатическая
большая часть случаев первичного гипоти-
роза — разнообразные приобретенные
поражения. О субклиническом гигютирозе
говорят, когда при нормальной концентрации
Т4 и Т3 уровень ТТГ превышает 2,0 мЕд/л.
Причины приобретенного первичного
гипотироза перечислены ниже в табл. 36.
Патогенез гипотироза и гипертироза
определяется нарушением эффектов, зависящих
от тироидных гормонов и компенсаторными
реакциями со стороны гипоталамо-гипофи-
зарного аппарата.
Основные проявления этих синдромов
в сравнительной форме представлены ниже
в табл. 37.
Среди конкретных заболеваний,
вызывающих гипертироз, наиболее
распространенной является болезнь фон Базедова
(термин, употребительный в континентальной
Европе, особенно — в Германии и странах
Восточной Европы; в англоязычных
странах — синоним: болезнь Грейвса, болезнь
Парри, в Италии — болезнь Флаяни).
Болезнь фон Базедова — системное муль-
тиорганное аутоиммунное расстройство,
основными проявлениями которого
являются диффузный токсический зоб (то
есть сочетание выраженных гипертироза и
гиперплазии щитовидной железы), а также
офтальмопатия и миокардиодистрофия.
И гипертироз, и гиперплазия железы, и
многие другие симптомы при болезни
Базедова имеют аутоиммунное происхождение.
Пораженность данной болезнью в
различных странах достигает 1-2% населения,
причем, как и при иных
иммунопатологических заболеваниях, большинство больных
составляют женщины (84 %) с максимумом
встречаемости в возрасте 15-40 лет.
Типично поражение носителей антигенов
ГКГС DR3 и В8, по крайней мере, среди
европеоидов. У монголоидов повышенный
риск болезни сцеплен с носительством
антигенов ГКГС Bw46 и В5, а среди негроидов
прослеживается ассоциация с наличием в
гаплотипе ГКГС антигена В17.
Этиология болезни точно не выяснена.
Имеются основания полагать, что
аутоиммунный процесс против компонентов
тироцитарного рецептора ТТГ возникает у
Таблица 36. Этиология первичного
приобретённого гипотироза
I. Деструкция тироцитов
1) Хронический аутоиммунный тироидит Хасимото
2) Идиопатическая микседема (возможно, субклини-
ческий вариант аутоиммунного тироидита)
3) Ятрогенные причины (радиотерапия и резекция
железы по поводу гипертироза, облучение шеи при
других диагнозах)
4) Подострый лимфоцитарный тироидит
(преходящий гипотироз)
5) Цистиноз
П. Гипофункция тироцитов без их разрушения
1) Эндемический йоддефицитный зоб
2) Эндемический зоб, вызванный струмогенами
диеты
3) Ятрогенный гипотироз при передозировке тиро-
статиков
4) Йодная микседема
Нарушения эндокринной регуляции 557
Таблица 37. Проявления гипертироза и гипотироза.
Органы и системы
Кожа и ее
производные
Глаза, лицо
Сердечно-сосудистая
система
Дыхательная система
ЖКТ
ЦНС
Опорно-двигательный
аппарат
Почки
Кровь и
кроветворение
Репродуктивная
система
Метаболизм
Гормоны
Гипертироз
Тёплая, тонкая, влажная, возрастные
изменения замедлены, потливость, тонкие
мягкие волосы, онихолиз, при болезни
фон Базедова — претибиальный отёк
Ретракция верхнего века, увеличение
глазной щели, периорбитальный отёк,
блеск глаз, редкое мигание,
гиперпигментация век. При болезни фон
Базедова возможен экзофтальм
Гипертензия, увеличение пульсовой
разницы, тахикардия, тахиаритмии,
сердечная недостаточность с высоким
минутным объемом. Усилена продукция
сердечного атриопептина
Одышка, снижение ЖёЛ
Повышение аппетита, ускорение
перистальтики, поносы
Гиперкинезия, психоэмоциональная
лабильность, бессонница, повышение
интеллекта, нервной возбудимости,
тревожность, ажитация, речь ускорена
Мышечная утомляемость, тремор,
гиперрефлексия, остеопороз, гипока-
лиемический парез
Полиурия, увеличение скорости
фильтрации
Ускорены эритропоэз и гемолиз,
лейкоцитоз, гипопротеинемия
Дисменорея, олигоменорея, снижение
фертильности, потенции. У лиц обоего
пола повышена конверсия андрогенов в
эстрогены. У мужчин — гинекомастия
Повышен основной обмен, снижена
толерантность к перегреванию, может
быть гипергликемия, имеется гипохо-
лестеринемия,гипотриглицеридемия,
отрицательный азотистый баланс,
увеличение потребности в витаминах
и железе, потеря в весе
При первичном гипертирозе —
понижение ТТГ; увеличены Т4, ТЗ, часто — и
захват йода тироцитами
Гипотироз
Холодная, толстая, отёчная. Ломкость
ногтей, сухие, толстые и ломкие волосы.
Ускорение возрастных изменений кожи
Птоз век, периорбитальный отёк,
выпадение волос наружной трети бровей,
обеднение мимики лица, увеличение языка.
Повышение гидрофильности и слизистый
отек кожи
Гипотензия, брадикардия, застойная
сердечная недостаточность с уменьшением
минутного объёма, снижение зубца Т,
вольтажа ЭКГ, выпот в перикарде.
Ослаблена продукция атриопептина. Ускорение
развития атеросклероза
Гиповентиляция, гиперкапния, выпот в
плевре
Анорексия, запоры, асцит
Заторможенность, снижение интеллекта,
сонливость, речь замедлена, хриплый голос,
депрессия
Мышечная утомляемость, ригидность,
гипорефлексия
Снижение скорости фильтрации и диуреза
Замедление эритропоэза, снижение
всасывания железа, анемия
Гиперменорея (снижен уровень тиреоста-
тина, который является и гонадостатином),
снижение либидо, бесплодие
Понижен основной обмен, снижена
толерантность к гипотермии; гиперхолестери-
немия, гипертриглицеридемия, повышение
чувствительности к инсулину, снижение
потребности в витаминах, положительный
азотистый баланс, прибавка в весе
При первичном гипотирозе — увеличен
ТТГ, снижены ТЗ, Т4, захват йода тироцитами
зависит от этиологии
558 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
генетически предрасположенных субьектов
вследствие провокации перекрёстной
иммунной реактивности. Экзогенный
провоцирующий антиген может быть
инфекционным. Л. Харрисон (1984) установил наличие
у Yersinia enterocolitica антигенов, подобных
ТТГ, а также анамнестический иерсиниоз
у многих лиц, заболевших впоследствии
базедовой болезнью. Ф. Надир и соавт.
(1983) показали возможность генерации
тиростимулирующих аутоантител к
рецептору ТТГ по идиотип-антиидиотипическому
механизму как иммуноглобулинов,
направленных против активных центров антител к
ТТГ (см. также т. I, с. 115-116). Возможно,
имеются и иные экзогенные инфекционные
и неинфекционные агенты, провоцирующие
аутоаллергическое поражение рецепторов
ТТГ, на основе генетической
предрасположенности. У части больных болезнь
возникает на фоне аутоиммунного плюри-
органного синдрома, включающего аутоал-
лергические поражения паращитовидной
железы, островков Лангерганса,
иммунопатологические орхит, оофорит, гипокорти-
цизм, а также аутоиммунный атрофический
гастрит (см. «Патофизиология сахарного
диабета» — касательно синдрома Шмидта). В
подобных случаях роль экзогенной
провокации минимальна, а на первый план выступает
дефицит лимфоцитарной супрессии.
Классическая литература прежних лет содержит
подкреплённое описаниями отдельных
наблюдений из практики мнение о большом
значении психоэмоционального стресса в
этиологии данного заболевания (В.Г.
Баранов, Н.Ф. Николаенко, 1966). Однако
современные источники трактуют роль стресса
лишь как выявляющую латентную болезнь
и не признают возможности существования
какой-либо чисто «психосоматической»
формы базедовой болезни, без первичной
аутоиммунной основы (Л. Вартофски, 1994,
П.РидЛарсен, 1987).
Патогенез болезни фон Базедова
определяется ключевым звеном — развитием
аутоиммунитета, направленного против
ряда антигенов щитовидной железы и иных
органов и тканей.
Основные эффекторы болезни — тиро-
стимулирующие аутоантитела. Они были
обнаружены еще в 1956 г. новозеландскими
исследователями Д. Эдамсом и X. Пьюр-
взом как «длительно действующий
стимулятор щитовидной железы». Поначалу было
ясно только, что стимулятор отличается по
срокам и, возможно, механизму действия
от ТТГ и содержится в глобулиновой
фракции плазмы крови больных. Затем
оказалось, что это — иммуноглобулины класса
IgG (Г. Крисе и соавт., 1965). Мишенью
этих аутоантител является рецептор ТТГ
(С. Мэнли и соавт., 1974), а еще точнее — его
ганглиозидная часть. Тироидстимулирую-
щие аутоантитела способствуют активации
ц-АМФ-зависимых и фосфатидил-инози-
толовых посредниковых механизмов в
клетках-мишенях, таким образом воспроизводя
действие ТТГ в «продлённом» варианте,
с более поздним пиком. Под их влиянием
активизируется захват йода, работа перок-
сидазы тироцитов, йодирование тироглобу-
лина, накопление коллоида и его пиноцитоз,
освобождение тироидных гормонов в кровь.
Аутоантитела данного вида воспроизводят
при их введении подопытным животным
картину гипертироза, но не все симптомы
болезни фон Базедова.
Наряду с тиростимулирующими ауто-
антителами в плазме крови больных
обнаружены аутоантитела, стимулирующие
рост щитовидной железы (пролиферацию
тироцитов). В настоящее время доказано,
что их специфичность не идентична ауто-
антителам, стимулирующим продукцию
тироидных гормонов. Наиболее вероятно,
их мишенью служит белковая часть
рецептора ТТГ (Д. Дониак и соавт., 1982;
В.М. Виерсинга и соавт., 1984). Подобные
аутоантитела найдены и у больных иными
формами гиперплазии щитовидной железы:
мультинодулярной гиперплазией, эутироид-
ным спорадическим зобом, а также, в низких
титрах — у клинически здоровых индивидов
(X. Шац и соавт., 1984; Й. Рисдаль и соавт.,
1984). Возможно, определенный уровень
их продукции вписывается в
физиологическую систему регуляции роста щитовид-
ной железы, а при патологии имеет место
эксцесс данного механизма. Так или иначе
гиперплазия и гиперфункция железы при
болезни фон Базедова возникают в ответ
на неидентичные сигналы. Параллельное
развитие зоба и гипертироза зависит от
совместной генерации цитостимулирующих
аутоантител двух разных специфичностей
(Х.А. Дрексхейдж и соавт., 1981). У лиц,
которые вырабатывают одну разновидность
аутоантител, скорее всего возникают иные
тиропатии — гиперплазия без тиротоксико-
за или, наоборот, гипертироз без зоба.
Спектр антитироидных аутоантител при
болезни фон Базедова не исчерпывается
этими двумя популяциями иммуноглобулинов.
Установлено, что у больных, особенно после
лечения радиоактивными препаратами
йода, возникают аутоантитела к рецептору
ТТГ, которые не стимулируют, а блокируют
гормонообразование. По-видимому, они
служат конкурентными антагонистами
ТТГ. Подобные иммуноглобулины могут
вызывать у больных базедовой болезнью
переход к гипотироидной фазе
заболевания. Ряд авторов обнаружили сходные
блокирующие аутоантитела у пациентов с
гипотирозом различной приобретенной
этиологии и считают, что они могут обусловить
гипофункцию железы, а их аналоги-бло-
каторы ростовых процессов — ее атрофию
(Ж. Десент, Ж. Вемо, 1982; М. Закария,
Дж. Маккензи, 1984). При болезни фон
Базедова часто имеются аутоантитела не ан-
тирецепторного характера — против тиропе-
роксидазы, против тироглобулина, ядерных
антигенов. Считается, что при этом
заболевании их роль — свидетельская. Возможно,
они связаны с патогенезом аутоиммунного
тироидита, который может сопровождать
болезнь фон Базедова (такая комбинация
иногда именуется «хаситоксикоз» — см.
ниже) или же осложнять леченую базедову
болезнь. Принципиально важно отметить,
что болезнь фон Базедова и аутоиммунный
тироидит — разные иммунопатологические
поражения железы. При болезни фон
Базедова доминируют антитироидные реакции
ГНТ, опосредованные циркулирующими
Нарушения эндокринной регуляции 559
аутоантителами. Важнейшей
отличительной чертой служит наличие аутоантител к
рецептору ТТГ. Так как тиростимулирую-
щие аутоантитела интернализуются вместе
с рецептором и не включают деструктивные
эффекторные механизмы, классическая
болезнь фон Базедова, по клиническим
критериям (отсутствие боли и других локальных
признаков воспаления) и по морфологии
биоптатов железы, представляет тироидную
гиперплазию и гиперфункцию, но не флогоз.
При аутоиммунном тироидите картина
отличается по всем этим позициям (см. ниже).
Доминирует ГЗТ против железы. Имеется
лимфоидная инфильтрация железы и
клинические, равно как и морфологические,
проявления её воспаления. Аутоантитела
носят не антирецепторныи характер, а
направлены против тиропероксидазы,
тироглобулина, других коллоидных и ци-
тозольных аутоантигенов. Соответственно,
и явления иммунологической стимуляции
роста и функции железы отсутствуют, а
имеется общее развитие процесса в
сторону прогрессирующей деструкции железы
и в перспективе — гипотироза (Р. Вольпе,
1984). Справедливости ради надо указать на
нередкое сочетание у одного больного,
одновременно, либо на разных этапах развития
болезни ГНТ и ГЗТ против аутоантигенов
щитовидной железы, что естественно с
общеиммунологических позиций (см. т. I,
с. 459). Именно это приводит к
формированию «хаситоксикоза», когда у пациентов
симптоматика и спектр аутоаллергии
оказываются смешанными (Д. Дониак, 1982).
В этом случае биоптаты железы могут
сочетать и признаки гиперфункциональной
гиперплазии, и лимфоцитарную
инфильтрацию, а полнокровие железы диктуется
и гиперфункцией, и тироидитом. В силу
возможности перехода у одного пациента
от тироидита Хасимото к болезни фон
Базедова (и наоборот), а также, изредка, — от
первичной приобретенной микседемы к ги-
пертироидным состояниям, Л. Вартофски
рекомендует рассматривать эти тиропатии
как близкие между собой, аутоиммунные
расстройства (1994).
560 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Большое клиническое значение имеет
тот факт, что при болезни фон Базедова
аутоаллергия не ограничивается интрати-
роидными мишенями, а затрагивает другие
органы и ткани.
Ещё первооткрыватели диффузного
токсического зоба указывали, что состояние
щитовидной железы не исчерпывает всех
основных признаков болезни (Дж. Флаяни,
1802; К. X. Парри, 1825; Р. Дж. Грейвз, 1835;
К.А. фон Базедов, 1840). Наконец,
Ф.Мёбиус сформулировал «мерзербургскую
триаду», согласно которой не только зоб,
но и экзофтальм, а также миокардиопатия
составляют важнейшие элементы болезни.
Последней он придавал столь большое
значение, что писал: «Больные болезнью
Базедова страдают сердцем и умирают от
сердца» (1846).
Поражение сердца при болезни фон
Базедова характеризуется тахикардией,
мерцательной аритмией, повышенным
проникновением кальция в кардиомио-
циты, снижением на них количества а- и
Рис. 110. Офтальмопатия и зоб при болезни фон
Больная А.К., 36 лет (по В. Фальта, 1913)
увеличением числа р-адренорецепторов, но
при сохраненном суточном ритме
чувствительности к катехоламинам. Со временем
развиваются кардиомегалия и сердечная
недостаточность.
Тиротоксическая инфильтративная
офтальмопатия — важный внешний синдром,
изучение которого обогатило представления
о болезни целым списком «именных»
симптомов, описанных классиками терапии
(рис.110).
Здесь, наряду с характерным недиплопи-
рующим экзофтальмом — то есть
выпячиванием глазных яблок вперёд — симптом
Дальримпля (широкое раскрытие глазных
щелей), симптом Штельвага (редкое
мигание), некоординированно быстрое
сокращение верхнего века и обнажение участка
склеры при взоре вверх (сицптом Кохера),
отставание верхнего века от радужки при
взоре вниз (симптом Грефе), яркий блеск
глаз (симптом Краузе), слабость
конвергенции (симптом Мёбиуса), гиперпигментация
век (симптом Еллинека), тремор закрытых
век (симптом Розенбаха),
неспособность наморщить лоб
(симптом Жофруа). Все эти
симптомы приведены здесь
не для того, чтобы напомнить
читателю о том, какой широкий
круг тем может быть затронут
на грядущем госэкзамене по
терапии, а по другой
причине. Давайте вместе удивимся,
как много ценных научных
наблюдений, с которых и
начинается патофизиология, может
сделать доктор без единого
научного прибора. Конечно
же, после введения в практику
эффективных тиростатических
средств тяжелая
тиротоксическая офтальмопатия стала
редкостью. Изучая механизмы
офтальмопатии, установили,
что ретроорбитальная
клетчатка и периорбитальные
ткани, включая веки и мышцы,
подвергаются отёку, и имеет
Базедова.
Нарушения эндокринной регуляции 561
место накопление гликозаминогликанов.
Функция глазодвигательных мышц,
например, мышцы Мюллера, поднимающей
верхнее веко, стимулируется, а при тяжелых
формах поражения происходят некробиоз и
деструкция мышечных волокон, фиброз,
нарушение смыкания век, и возможна потеря
зрения. Так как катехоламинолитические
лекарства облегчали офтальмопатию, ее
стали связывать с сенсибилизацией тканей
глаза к катехоламинам под действием ги-
пертироза.
Но попытки объяснить всю картину
болезни как следствие одного гипертироза
оставались неудачными. Было хорошо
известно, что степень экзофтальма и других
глазных симптомов не всегда коррелирует
с тяжестью гипертироза, зачастую не было
параллелизма в реакции составных
элементов триады Мебиуса на тиростатическое
лечение. Экзофтальм, например,
обнаруживали и при гипотирозе, и при
аутоиммунном тироидите. Одно время, на фоне
ошибочной трактовки болезни фон Базедова
как гипофизарной патологии даже
постулировали существование «гипофизарного
экзофтальмического фактора». Миокар-
диодистрофию у больных трактовали как
результат повышающего действия избытка
тироидных гормонов на чувствительность
миокарда к катехоламинам, а изменения в
сердце характеризовали весьма общим и
патофизиологически нейтральным термином
«обменно-дистрофические» (Г.А. Котова,
1991).
Наконец, такое проявление болезни, как
багрово-синюшный уплотнённый, брас-
летообразный, зудящий отек кожи вокруг
нижней трети голеней — «претибиалъная
микседема» или дермопатия вообще не
получало разумного объяснения ни с точки
зрения гипертироидной, ни в контексте ка-
техоламиновой версии (хотя и отмечалось у
5-10% больных).
Лишь после открытия
иммунопатологических основ патогенеза болезни фон
Базедова произошло важное изменение в
подходах к ее экстратироидным симптомам.
В настоящее время установлено, что все они
в решающей мере зависят от присутствия
аутоантител различной специфичности.
Как установлено А.У. Нэтаном и соавт.
(1983), миокардиодистрофия при болезни
фон Базедова связана не только с
сенсибилизацией миокарда к катехоламинам, но и с
присутствием особых аутоантител к кардио-
миоцитам, которые обладают
стимулирующим действием на миокард, способствуют
входному току кальция в его клетки.
При наличии выраженной офтальмопатии
и претибиального отёка сыворотка крови
больных содержит цитотоксические
аутоантитела и лимфоциты против антигена,
общего для тироцитов, а также фибробластов,
жировой и мышечной ткани. Аутоантитела
к ретроорбитальным липоцитам, фибро-
бластам и антигенам мышц, активирующие
аденилатциклазу клеток-мишеней,
вызывают периорбитальные изменения, отек век,
пролиферацию липоцитов и гипертрофию
мышечных волокон, экзофтальм и ещё ряд
симптомов офтальмопатии, претибиальный
отёк (К. Манн и соавт., 1979; С. Мэнли и
соавт., 1982). Эти изменения могут быть
пассивно перенесены иммуноглобулинами,
вводимыми экспериментальным животным
и лечатся иммунодепрессантами. Известное
значение в их развитии может иметь и
действие лимфоцитарных цитокинов. Интересно,
что тиротоксическую офтальмопатию
отягощает курение.
Более того, сами антитироидные
иммуноглобулины, вполне возможно,
неизбирательны в своих стимуляторных и иных
эффектах. Установлено, что иммунные
комплексы с участием аутоантител к тирогло-
булину оседают в тканях глаза и участвуют
в развитии инфильтративных изменений
(П.РидЛарсен, 1987).
У аутоантител к рецепторам ТТГ
щитовидной железы обнаружена способность
активировать не только гормонообразова-
ние в этой железе, но и стероидогенез в коре
надпочечников и гонадах (К. Трокудес и
соавт., 1979).
Таким образом, с современных позиций
болезнь фон Базедова рассматривается не
просто как эндокринопатия, но как системное
562 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
мультиорганное аутоиммунное
расстройство! Следует оценить с этой точки зрения и то
обстоятельство, что наиболее эффективные
тиростатики (мерказолил) проявляют и им-
мунодепрессивное действие.
Впрочем, новейшие данные показали,
что теорию экзофтальмического фактора
в архив сдавать рано. На роль этого начала
претендует липокин адипонектин,
известный своей защитной ролью в отношении
атеросклероза и метаболического
синдрома (см. гл. 14). По данным Т.П. Комбса и
соавт. (2004), удивительным результатом
гиперадипонектинемии у трансгенных
мышей является значительная гиперплазия
бурой жировой ткани, особенно — между
лопаток, а также появление двустороннего
экзофтальма из-за накопления ретроор-
битального жира. В этой связи уместно
предположить, что именно адипонектин или
адипонектин-миметические аутоантитела
как раз и являются тем легендарным «экзоф-
тальмическим фактором» при болезни фон
Базедова, о котором упоминали еще В.Г.
Баранов и В.В. Потин (1977). Всем известное
антиатерогенное действие болезни фон
Базедова и сам фенотип больных тиротоксикозом,
противоположный хабитусу больных
метаболическим синдромом, могут быть связаны
не только (и не столько) с гиперпродукцией
тироидных гормонов, сколько с возможной
гиперадипонектинемией или адипонектино-
подобным действием некоторых аутоантител
при данном системном аутоиммунном
заболевании. Конечно, это предположение еще
нуждается в проверке.
Другие аспекты патогенеза и проявлений
болезни фон Базедова следуют из
характеристики эффектов тироидных гормонов и
симптомов гипертироза, приведённых выше
в табл. 34,37.
Отметим, что наиболее опасное острое
осложнение болезни (с частотой 2-8%) — ти-
ротоксический криз. В ответ на инфекции,
травмы, хирургические вмешательства,
отмену антитироидных лекарств, лечение
радиоактивным йодом, тяжёлый стресс больной
базедовой болезнью средней или тяжелой
степени может развить синдром крайне
выраженного острого действия избытка
тироидных гормонов и катехоламинов. Отмечаются
гиперпиретическая лихорадка, тахикардия и
мерцательная аритмия, крайняя ажитация,
беспокойство вплоть до психоза, спутанное
сознание, тошнота, рвота, понос, полиурия,
прогрессирующая дегидратация и потеря
натрия, мышечная слабость, парезы, резкое
расширение кожных сосудов, гипертензия,
желтуха. Итогом может быть развитие комы,
коллапса, сердечной недостаточности и
смерть. Летальность, еще недавно
превышавшая 70 %, даже в условиях современного
лечения, составляет около 20-25 %. Этиология
и патогенез тиротоксических криза и комы
могут быть охарактеризованы следующим
образом (Дж. Хиггинс, 1985).
Этиологические факторы ведут к дополнительному
освобождению тироидных гормонов и
катехоламинов, которые пермиссивно влияют на
эффекты друг друга. Большое значение
придают резкому повышению доли свободных
гормонов в плазме и снижению тироид-свя-
зывающей способности её белков, возможно,
в силу появления циркулирующих блокато-
ров связывания (Л. Вартофски, 1994).
Не исключено и определённое значение
«резонансного» увеличения титра тирости-
мулирующих аутоантител, в силу
волнообразного характера идиотип-антиидиотипи-
ческих взаимодействий. Непосредственной
причиной нарушений сознания и работы
жизненно важных органов служат водно-
солевые расстройства, прежде всего потеря
натрия, избыток калия, обезвоживание.
Среди нозологических единиц,
связанных с гипотирозом, в особом рассмотрении
нуждаются ранняя и врождённая форма ги-
потироза с выраженным нарушением
психоинтеллектуального развития — кретинизм и
поздняя форма приобретенного гипотироза
у подростков и взрослых — микседема. Оба
синдрома протекают, как правило, с
компенсаторной гиперплазией щитовидной
железы и многими общими проявлениями
гипотироза (см. табл. 37, выше).
Кретинизм — термин, связанный со
старофранцузским «chretien», что лишено какого
Нарушения эндокринной регуляции 563
бы то ни было эмоционального негативизма
и буквально означает «христоподобный»,
«христианский».
Врождённая форма гипотироидного
кретинизма известна как синдром Фагге.
Столетие назад врожденный кретинизм был
весьма нередкой причиной нарушения
физического и психомоторного развития детей
в районах эндемической йодной
недостаточности (Швейцария и Тироль, Карпаты,
Тянь-Шань, Карачай, Сванетия, Верхняя
Волга, Кордильеры, Синьцзян, Индонезия).
Ныне его частота резко уменьшилась (около
1/5000) в связи с йодной профилактикой,
при этом возросла доля спорадических
случаев, основанных на наследственных
тироидных аномалиях (рис. 111).
Врожденный недостаток тироидных
гормонов тем сильнее нарушает развитие организма,
чем раньше сказывается. До формирования
у плода собственной тироидной секреции
большое значение имеет тироидная функция
у матери. Материнский гипотироз
способствует фетальному. При недостатке тироидных
гормонов страдает дифференцировка мозга,
нарушается не только генез короткоживущих
РНК, связанных с формированием энграмм
памяти, но и само образование синаптических
связей в формирующемся мозге.
Поэтому многие проявления синдрома
Фагге, в отличие от микседемы (которую
У. Галл называл «кретиноидное состояние
взрослых») необратимы или устойчивы к
лечению. Слабоумие при синдроме Фагге
глубокое, вплоть до степени имбецильнос-
ти или даже идиотии, с неспособностью к
обучению и самообслуживанию. К тому же
психоэмоциональная сфера при кретинизме
Фагге часто формируется с преобладанием
негативных эмоций, контакт с больными
достигается нелегко. В отличие от этого, при
приобретенном синдроме Галла (см. ниже)
речь идет об ухудшении функционирования
мозга, сформированного в основе своей
правильно. Поэтому кретинизм при мик-
седеме не столь глубок* умственная
активность понижена, но эти проявления хорошо
корректируются лечением, эмоциональный
фон у больных при кретинизме Галла более
Рис. 111. Случай спорадического
(врожденного) кретинизма (по В. Фальта, 1913)
позитивный. Помимо развития мозга, при
врожденной и ранней форме гипотироза
сильно страдает рост организма (см. выше
о гипотироидном нанизме). Нарушается
удлинение костей, костный возраст отстает от
паспортного, задерживается развитие зубов,
формируется челюстно-лицевая дисплазия
— западает переносица, лицо приобретает
характерное выражение (полуоткрытый
рот, широко посаженные глаза, грубые
черты, виден язык, мимика обеднена, волосы
редкие). Часто отмечают атонию мышц,
выступающий живот и пупочную грыжу.
Ранние проявления врожденного
гипотироза— продлённая желтуха новорожденных,
плохое сосание, запоры, сонливость, сухая
кожа. На острове Папуа и в Андах описана
особая форма врожденного гипотироидного
кретинизма с проявлением церебральных
параличей, косоглазия, глухоты.
564 ALU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Приобретённая гипотироидная кретино-
идность и микседема были впервые описаны
как синдром Галла. Нарушение поведения
и умственной активности таких больных
проявляется заторможенностью реакций,
медлительностью, апатией, снижением
интеллекта. Выше уже отмечалось, что эти
явления менее глубоки и обратимы в
отличие от синдрома Фагге. Часто при миксе-
деме у взрослых, у которых формирование
стереотипов социальной жизни,
профессиональной деятельности и семейного
поведения уже завершено, проявления кретинизма
вообще малозаметны. Наступившая крети-
ноидность может выражаться в неглубокой
дебильности, которая в рутинной
обстановке мало сказывается на адаптации больных.
Тем не менее даже начальная степень
гипотироза отражается на умственной
активности и интеллектуальной эффективности
индивида. П.Б. Ганнушкин подчёркивал,
что и в состоянии легчайшей начальной, так
называемой функциональной дебильности
пациент проявляет замедление мышления,
утрату чувства юмора, неспособность к
творческой неординарной деятельности (1926).
Учитывая большую распространенность
легких форм гипотироза, особенно среди
женщин после 40 лет, подобные явления,
редко связываемые обыденным сознанием
с эндокринной патологией, не должны
ускользать от внимания профессионала-
эндокринолога, особенно, когда речь идет
о развившемся за относительно недолгий
срок изменении привычного образа того или
иного индивида в глазах окружающих.
Микседема характеризуется плохим
аппетитом, запорами, интолерантностью к
холоду, ожирением и гиперхолестеринемиеи. При
дефиците тироидных гормонов уменьшается
продукция предсердного натрийуретичес-
кого полипептида. Создаётся тенденция к
задержке натрия и воды в организме.
Замедляется распад гликозаминогликанов. Их
гидрофильные скопления в коже, подкожном
жире, голосовых связках, языке, некоторых
внутренних органах задерживают в этих
тканях воду и способствуют слизистому
отёку и утолщению кожи, огрублению лица,
низкому хриплому тембру голоса (последний
симптом, безусловно, зависит и от
сдавливания возвратного гортанного нерва зобом).
Микседема приводит к увеличению языка с
отпечатками зубов, расширению границ
сердца и водянке серозных полостей. Отекают
слизистые щек, что сопровождается их
болезненными прикусываниями (Ю.И. Строев,
2002). Уровень Т3 и Т4 при гипотироидном
кретинизме и микседеме всегда понижен,
содержание ТТГ повышено, если речь не
идет о первичной гипоталамо-гипофизарной
недостаточности. В связи с аутоиммунным
характером многих случаев приобретённой
микседемы у больных могут присутствовать
антитироидные аутоантитела и лимфоциты.
В Мозамбике, на фоне высокого
содержания в диете населения пищевых цианидов,
являющихся струмогенами, и
полигиповитаминоза, описан синдром гипотироза с
проявлениями микседемы, полиневрита и
энцефалопатии — «конзо» (X. Рестлинг и
соавт., 1986).
Нелеченая микседема способна
прогрессировать до терминальной стадии микседе-
матозной комы. Этиология микседематозной
комы включает переохлаждение, инфекции,
травмы и действие лекарств, угнетающих
ЦНС. Как указывает Дж.Хиггинс (1985),
типичные случаи этой комы наблюдаются у
пожилых пациенток с гипотирозом в зимние
месяцы, на фоне респираторных инфекций
и приема седативных средств.
Это экстремальное состояние
выражается в прогрессирующей гипотермии
недостаточности кровообращения и ступоре
на фоне гиповентиляции и гиперкапнии.
Неадекватно стимулируется продукция
вазопрессина и возможна гипонатриемия,
провоцирующая судороги и набухание
мозга. Смертность при микседематозной коме
велика — от 15 до 50 %.
Нарушения ростовых процессов
в щитовидной железе
Нарушения ростовых процессов в данном
органе делятся на диффузные и очаговые.
Диффузные нарушения включают
гипоплазию железы вплоть до ее атрофии (при
Нарушения эндокринной регуляции 565
врожденной этиологии и полном отсутствии
тироидной ткани — атироз).
Гиперплазия железы в зависимости от
выраженности ее увеличения делится на
5 степеней тяжести (I — прощупывается
перешеек железы, II — железа заметна при
глотании, III — железа заметна при осмотре,
«толстая шея», IV — изменена
конфигурация шеи, не любая, а лишь данная и более
тяжёлая степени гиперплазии щитовидной
железы именуются «зоб», V — зоб огромных
размеров, выходящий за пределы шеи). При
пожизненном прогрессировании
эндемического зоба обнаруживалось увеличение
щитовидной железы до гигантских размеров
и веса — более 5 кг!
Очаговые нарушения роста («узлы»)
щитовидной железы чаще всего наблюдаются
при ее опухолях (токсическая аденома, рак)
и неопухолевой нодулярной гиперплазии.
Основной механизм гиперплазии железы
связан с хронической гиперпродукцией
ТТГ. Очень редко — это результат
опухолевой, автономной секреции тиротропина.
В подавляющем большинстве случаев речь
идёт о компенсаторном увеличении
выработки ТТГ в ответ на пониженную функцию
тироцитов. Причины понижения функций
тироцитов рассматривались в предыдущем
разделе. При эндемическом зобе это чаще
всего йодный дефицит и присутствие стру-
могенов в пище и воде. При спорадическом
зобе могут быть актуальны наследственные
и аутоиммунные причины гипофункции
тироцитов (см. выше).
Аутоантитела, стимулирующие рост
щитовидной железы, также могут быть
причиной спорадического зоба как в структуре
болезни фон Базедова, при сопутствующем
гипертирозе, так и изолированно, на фоне
эутироза. Содержание ТТГ при
иммунопатологических формах гиперплазии железы
понижено. При зобе поначалу гиперплазия
железы диффузна. Данная стадия
обратима при коррекции содержания тироидных
гормонов (простой диффузный зоб, он
же — коллоидный зоб). Но затем, по мере
накопления коллоида, из-за более быстрого
увеличения одних фолликулов и атрофии
других возможно развитие многоузловой
структуры гиперплазированной железы
(мультинодулярный зоб). Большое значение
в приобретении многоузловой структуры
имеет поликлональная неоднородность
тироцитов. Из-за неоднородности они
по-разному отвечают на одни и те же
количества митогенных стимуляторов, будь то
ТТГ или аутоантитела, стимулирующие их
рост. В подавляющем большинстве случаев
больные достигают эутироза, в связи с чем
простой и мультинодулярный зоб
клиницисты иногда характеризуют как «простой
нетоксический»у в противоположность зобу
при болезни фон Базедова (диффузный
токсический) и микседеме (гипотироидный).
Мультинодулярные формы зоба иногда
бывают гипертироидными, но степень
гипертироза меньше, чем при болезни
фон Базедова, а аутоантител сэкстрати-
роидной направленностью и выраженных
экстратироидных симптомов, как
правило, нет (Р. Де Лилли, 1989). В клонах
клеток, дающих начало микроузелкам при
мультинодулярной гиперплазии, может
экспрессироваться мутантный вариант
онкогена gsp. Онкобелок этого онкогена
способен вызвать хроническую активацию
ГТФ-связывающего протеина Gs.
Считается, что данный механизм может
содействовать автономной гиперсекреции некоторых
подобных узелков, нечувствительной к
уровню ТТГ. Это и приводит к гипертиро-
идному варианту мультинодулярного зоба.
Отметим, что у носителей
мультинодулярного зоба повышена частота рака и аденом
щитовидной железы (см. ниже), что тоже
может объясняться экспрессией онкогенов
(С. МакФи, Д. Бауэр, 1997).
В щитовидной железе могут возникать
местные и существовать метастатические
неопластические клоны.
В большинстве случав это
доброкачественные аденомы. Менее одного процента
всех узловых поражений железы составляют
злокачественные карциномы. Опухолевые
узловые поражения обычно одиночные.
«Холодные» узлы, то есть малоактивно
захватывающие йод, могут принадлежать
566 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
как к аденомам (90%), так и к карциномам
(10%), но «горячие» — то есть с
повышенным уровнем абсорбции йода — как правило,
относятся к доброкачественным.
Карциномы железы образуются de novo,
а не за счёт гипотетической малигнизации
аденом. Этиологию и патогенез рака
щитовидной железы связывают с радиационным
воздействием. Повышение частоты рака
данной локализации отмечено при ятроген-
ном1 и профессиональном облучении
головы и шеи, у лиц, пострадавших от атомных
бомбардировок в Японии, жертв ядерных
испытаний на Маршалловых островах и
аварии на Чернобыльской АЭС. Так, у
«хибакуся» — лиц, переживших атомную
бомбардировку, — в катамнезе частота тиро-
идных карцином составила на протяжении
40 лет 7-10%.
Опухолям щитовидной железы
способствует наличие тироидита Хасимото и ноду-
лярных форм зоба.
По своей структуре и происхождению
карциномы щитовидной железы,
составляющие около 1 % раковых поражений, делятся
на 4 формы.
- Папиллярный рак (75-85 %). Это
наиболее частая форма карциноматозного
тироидного поражения. Характерно
образование сосочков из атипичных
малигнизированных клеток, ядра
которых крупны и содержат настолько
нежный хроматин, что кажутся пустыми
и прозрачными. Патологоанатомы со
своеобразным мрачным юмором
именуют этот признак «глазок сиротки Энни»
(Д. Берне, В. Кумар, 1997). В
опухоли обнаруживают также
концентрические кальцификаты — псаммоматозные
тельца. Механизмы малигнизации при
данной форме рака связывают с
перемещением протоонкогена RET в хромосоме
10, кодирующего ростовой рецептор,
в связи с чем этот ген гиперэкспресси-
руется на новом месте и формируется
онкобелок РТС, характерный для клеток
папиллярного тироидного рака. Опухоль
не образует гормонов, метастазирует
чаще всего в региональные лимфоузлы,
реже — гематогенно, в лёгкие. Прогноз
более благоприятен, чем при иных
формах тироидного рака — с вероятностью
10-летнего выживания 85 %.
Фолликулярный рак (до 20%). Данная
форма поражает пациентов более
старшего возраста и учащается в районах с
эндемическим йодным дефицитом. Ткань
опухоли образует небольшие
фолликулы. В ней часто присутствуют в избытке
клетки К. Хюртле. Хорошо
дифференцированные опухоли этого класса могут
стимулироваться ТТГ и приводить к
гипертирозу. В то же время им
свойственны гораздо более частые гематогенные
метастазы, чем предыдущей форме рака.
Медуллярный рак (5%) представляет
собой нейроэндокринную апудому из
С-клеток щитовидной железы. Эти
опухоли секретируют кальцитонин, а
иногда — и другие гормоны (ВИП, серо-
тонин, соматостатин). Они вызваны
соматической мутацией или генеративной
мутацией протоонкогена RET. В случае
генеративной мутации медуллярный рак
у молодых больных и даже детей может
входить в структуру синдрома MEN На
или lib (см. выше). Но до 20% случаев
опухоли протекают изолированно, в
этом случае опухоль поражает пожилых
пациентов. Особенностью морфологии
опухоли часто является ее мультицен-
трический характер и отложение в ней
эндокринного амилоида (см. раздел
«Диспротеинозы»). Гиперсекреция
кальцитонина уравновешивается
системой регуляции кальциевого гомеостаза
и протекает без особых симптомов.
Гиперпродукция ВИП в опухоли может
приводить к поносам. Медуллярный рак
в структуре MEN-синдрома протекает
агрессивно, гематогенно метастазирует,
и более половины больных умирают в
течение 5 лет. Но изолированная
медуллярная карцинома имеет значительно
лучший прогноз.
Недифференцированные анапласти-
ческие карциномы щитовидной железы
Нарушения эндокринной регуляции 567
(менее5%), особенно их гигантокле-
точная разновидность, представляют
одну из опаснейших и самых агрессивно
растущих опухолей человека. Из-за
быстрой инфильтрации соседних тканей и
отдалённых метастазов смерь больного
наступает в пределах года с момента
диагноза.
В щитовидной железе возможно
развитие лимфом, сарком (которые, в отличие от
карцином, не экспрессируют кератин и не
имеют кератиновых промежуточных фила-
ментов), метастатических опухолей трахеи и
пищевода но все подобные случаи, за
исключением лимфом, эксклюзивно редки.
Доброкачественные аденомы
щитовидной железы представляют собой
одиночные, небольшие по размеру (обычно до 4
см в диаметре), гиперпластические узлы.
Большая их часть сохраняет
фолликулярную структуру (фолликулярные аденомы),
некоторые образуют пальцеобразные или
сосочковые скопления клеток (папиллярные
аденомы). Небольшой процент аденом
представлен клетками Хюртле-Лскенази.
Аденомы обязательно инкапсулированы, они не
инфильтрируют окружающую тироидную
ткань, а сдавливают её. Аденомы растут
медленно, достигая конечного стабильного
размера. Резкое увеличение и болезненность
аденоматозного узла представляет не малиг-
низацию, а результат внутриопухолевого
кровоизлияния.
В большинстве клетки аденом менее
функционально активны, чем нормальные
тироциты. Встречаются, однако, и редкие
токсические аденомы, способные активно
производить тироидные гормоны,
совершенно автономные или слабо зависящие от
ТТГ. Они могут вызывать гипертироз. В
щитовидной железе описаны нетироцитарные
доброкачественные опухоли (липомы,
гемангиомы, тератомы), а также различные
опухолеподобные кисты.
Тироидиты
Щитовидная железа часто является
ареной воспаления, так как локализована на
шее, в непосредственном соседстве с часто
инфицируемыми органами, богато крово-
снабжается и служит ареной биохимических
процессов с участием агрессивных
радикалов. Большое значение имеет участие
аутоиммунных механизмов в регуляции её роста
и функций, при определённых условиях
оборачивающееся аутоаллергией.
Острые, подострые и хронические
тироидиты неодинаковы по этиологии и
отличаются по патогенезу и последствиям.
Острый гнойный тироидит — гнойное
инфекционное воспаление щитовидной
железы. Это редкое заболевание, как
правило, имеет банальную этиологию и
вызывается стрептококками, стафилококками и
другими гноеродными микроорганизмами,
реже — сальмонеллами и энтеробактером.
При иммунодефицитных состояниях, в
частности, у больных ВИЧ-инфекцией,
описан острый тироидит, вызванный
условно патогенной микрофлорой. Причиной,
например, может служить Pneumocystis
carinii. При туберкулёзе также описан ка-
зеозный тироидит, а при сифилисе —
гуммозный. Щитовидная железа вовлекается
в саркоидоз. Есть наблюдения, касающиеся
грибковых тироидитов (Candida, Mucor,
Actinomyces). Поскольку патогенез болезни
представляет типичное острое воспаление
(т. I, с. 273-351), оно проявляется местными
краснотой, гипертермией, отёком, болью.
Гормональные функции железы, благодаря
действию гипоталамо-гипофизарных
сервомеханизмов, остаются в пределах нормы,
хотя бывают лихорадка, лейкоцитоз и
проявления ответа острой фазы.
Острый негнойный тироидит
представляет асептическое воспаление железы в
ответ на её механическую травму или
облучение, может сопровождаться преходящими
симптомами гипертироза.
Подострый тироидит (синонимы — гра-
нулёматозный тироидит, гигантоклеточныи
тироидит или тироидит Ф.Де Кервена,
1904) имеет вирусную этиологию.
Предполагается участие некоторых серотипов
вирусов кори и свинки (Э. Эйлан и со-
авт., 1957). По данным П. Чандрасомы и
568 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
К. Тэйлора (1998), есть много свидетельств
о провоцирующей роли энтеровирусов
Коксэки и ECHO, вируса Эпштейна-Барр,
эпизодически отмечается связь с ОРВИ
(аденовирусной, гриппозной и иной
этиологии). Тироидит Де Кервена может
начаться остро или протекать с самого начала
малосимптомно. После острой вирусной
респираторной инфекции пациентка
(болезнь чаще поражает женщин 30-50 лет,
носительниц антигена ГКГС В35) чувствует
слабость, недомогание, миальгии, а
поскольку происходит смешанная нейтрофильно-
мононуклеарная инфильтрация
щитовидной железы, также симптомы растяжения её
капсулы (боль с иррадиацией в ухо, челюсть
и даже в затылок). Лейкоцитоз
сопровождается крайне выраженным ускорением
СОЭ, что свидетельствует о значительной
интенсивности ответа острой фазы,
управляемого интерлейкинами. Железа может
увеличиваться, в том числе асимметрично,
и приобретать узловатую консистенцию. В
биоптате инфильтрат содержит нейтрофи-
лы и небольшое количество лимфоцитов
и макрофагов. Отмечаются деформация и
разрыв фолликулов, вокруг них могут быть
гранулёмы с гигантскими клетками. В крови
обнаруживаются в невысоких титрах ауто-
антитела к тироглобулину.
Порой для дифференциации с болезнью
фон Базедова требуется исследование
скорости захвата радиоактивного йода. Этот
показатель при подостром тироидите Де
Кервена понижен. Из-за образования в
железе лимфокинов, влияющих на тироциты,
а также из-за утечек при разрывах
фолликулов концентрация тироидных гормонов
в первые дни может повышаться, а затем
несколько снижается, что сопровождается
соответствующими компенсаторными
изменениями секреции ТТГ. Заболевание
самоограничивается в течение полутора-
трёх месяцев, не оставляя структурных и
функциональных дефектов, хотя возможны
рецидивы и развитие гипотироза.
Хронический тироидит всегда
представляет собой иммунопатологическое
воспаление щитовидной железы с длительным
волнообразным течением, чаще всего — без
тенденции к спонтанному
самоограничению.
Хронический тироидит известен в трёх
вариантах:
- хронический неспецифический лимфо-
цитарный тироидит с преходящим
гипертирозом (иногда характеризуется
как подострый лимфоцитарный
тироидит) — доброкачественое
иммунопатологическое поражение железы с ГЗТ
умеренной интенсивности, имеющее
тенденцию к медленному, малосимп-
томному течению. Болезнь поражает лиц
среднего возраста, в основном, женщин,
довольно нередко — после родов. Чаще
других болеют носительницы антигена
ГКГС DR5. Лимфоидная инфильтрация
железы незначительна и ведет к ее
некоторому безболезненому равномерному
увеличению. В железе нет лимфоидных
фолликулов и плазматических клеток.
Наблюдаются только малые лимфоциты.
В крови не обнаруживается
значительных титров антитироидных аутоанти-
тел. Содержание тироидных гормонов
в плазме транзиторно возрастает, что
ведёт к снижению уровня ТТГ и скорости
захвата радиоактивного йода. Затем, в
течение 2-8 месяцев, уровень тироидных
гормонов спонтанно нормализуется,
однако уровень стимуляции поглощения
йода при введении экзогенного ТТГ
остается на длительное время пониженным.
Бывает фаза транзиторного гипотироза.
Этиология болезни неизвестна,
предполагается роль неидентифицированных
вирусов. Клиническое значение болезни
определяется тем, что данная форма
тироидита вызывает преходящий ги-
пертироз и служит, вероятно, второй по
частоте, после болезни фон Базедова,
причиной этого синдрома.
- аутоиммунный хронический тироидит
Хасимото (получивший своё название
в честь наблюдательного японского
хирурга X. Хасимото (1912),
«вторгшегося» в пределы смежной медицинской
специальности) — наиболее часто ветре-
чающийся вид тироидита.
Заболеваемость им неуклонно увеличивается со
второй половины 60-х годов XX века и
составляет львиную долю всей тироид-
ной патологии. Чаще всего он поражает
женщин молодого и среднего возраста
(95 % случаев). Ф. Фелиг и соавт. (1985)
оценивают распространенность болезни,
включая её латентные формы, среди
женщин прямо-таки катастрофической — в
10% популяции! Нередко
аутоиммунный тироидит развивается у подростков,
как правило, на фоне
предшествующей ювенильной гиперплазии железы
(А.Ш. Зайчик и соавт., 1999), с которой
он связан патогенетически (И. Кобаяши,
1997). Дело в том, что усиление
пролиферации тироцитов в патологических и
нормальных условиях предусматривает
участие ростостимулирующих аутоан-
тител против белковых эпитопов
рецептора тиротропного гормона. Хотя дети и
подростки не составляют большинства
пораженных аутоиммунным тироиди-
том, он тем не менее служит главной
причиной гипотироза и зоба в педиатрии,
да и вообще — у населения районов с
геохимически достаточным поступлением
йода (Л.П. Чурилов и соавт., 2006).
Тироидит Хасимото — это
прогрессирующее аутоиммунное заболевание, которому
свойственны параллельное развитие ГНТ
и ГЗТ против антигенов щитовидной
железы (Л. Вайнер, 1997). Цитотоксические
лимфоциты и аутоантитела поддерживают
тенденцию к постепенному развитию
гипотироза по мере разрушения ткани железы.
Вначале состояние пациентов эутироидное,
но оно достигается за счёт активации
продукции ТТГ и захвата йода железой при ее
сниженном функциональном резерве. Затем
в течение длительного времени существует
латентный гипотироз с компенсаторным
возрастанием перехода Т4 в Т3 и снижением
уровня первого при нормальной
концентрации второго. Наконец, падают концентрации
обоих тироидных гормонов, и гипотироз
становится манифестным. Считается, что
не менее 90 % случаев приобретенного гипо-
Нарушения эндокринной регуляции 569
тироза, включая многие, квалифицируемые
как микседема, начинаются с явного или
субклинического аутоиммунного
хронического тироидита (Л. Вартофски, 1994).
В отличие от всех иных форм воспаления
данного органа при тироидите Хасимото
вследствие лимфоцитарной инфильтрации
железа может очень значительно
увеличиваться, имеет плотно-эластическую
консистенцию, что иногда образно характеризуется
клиницистами как «лимфоаденоидный
зоб». Истинная гиперплазия железы при
тироидите Хасимото не выражена.
Однако, лимфопролиферативные изменения
так характерны, что при биопсии в железе
иногда видны даже вторичные лимфоидные
фолликулы, как будто сама она становится
периферическим лимфоидным органом.
Наряду с малыми лимфоцитами
присутствуют плазматические клетки. Коллоидные
фолликулы уменьшены и немногочисленны.
Они содержат по периферии особые,
крупные, обладающие ярко-эозинофильной,
содержащей много митохондрий и лизосом,
цитоплазмой клетки Хюртле, результат
метаплазии фолликулярного эпителия,
вероятно, происходящие из В-клеток Аскенази
(см. выше). Эти клетки, иногда отмечаемые
и при других тиропатиях, формируют
гиперпластические узелки. По мере развития
болезни прогрессирует фиброз железы, не
выходящий за пределы её капсулы.
Тироциты экспрессируют при данной
болезни аберрантные антигены ГКГС II
класса, чего в норме не наблюдается. Этим
они «подставляют» под удар иммунной
системы сами себя, представляя собственные
пептиды лимфоцитам (Дж.-Ф. Боттаццо и
соавт., 1983). С Dg-положительные
цитотоксические лимфоциты в железе
сенсибилизированы против собственных тироглобулина
и микросомальных антигенов. Имеется
модель наследственного аутоиммунного
хронического тирюидита, спонтанно
развивающаяся у цыплят и гончих собак.
Получена модель приобретённого аутоиммунного
тироидита при воздействии радиоактивного
йода на морских свинок — по Дж.Тэйлору
и соавт. (1961).
570 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Йодная нагрузка — очевидный фактор
риска этой болезни. Йодированная вода
способствует ее развитию у генетичеки
предрасположенных мышей (Н. Роуз и
соавт., 1999). Подробнее см. монографию
Ю.И. Строева и Л.П. Чурилова (2004).
А. Чезарео и соавт. (1998) описали
семейную наследственную форму аутоиммунного
тироидита. Что касается его приобретённой
разновидности, то важную роль в этиологии
болезни в последнее время придают
генетической предрасположенности (в 3,2-3,7 раза
повышена вероятность развития данной
формы тироидита у носителей гаплотипов
ГКГС, включающих гены DR3 или DR5,
а также В8). Провоцирующим событием
у предрасположенных индивидов может
быть вирусная инфекция или применение
с лечебной целью йода, интерферонов
(особенно, а-ИФН и р-ИФН) и интерлейкина-2.
Патогенность этих цитокинов доказана при
изучении развития тироидита Хашимото в
катамнезе больных вирусным гепатитом С,
получавших курсы интерферонотерапии.
Упомянутые цитокины способствуют
повышению экспрессии антигенов ГКГС I
класса и аномальной экспрессии антигенов
ГКГС II класса на тироцитах. Тироидит
Хасимото может наблюдаться в структуре
системных аутоиммунных заболеваний с
неорганоспецифическими аутоантителами
(системная красная волчанка, ревматоидный
артрит, синдром Шёгрена). Больные
аутоиммунным тироидитом находятся в группе
повышенного риска развития В-клеточных
лимфом и папиллярного рака щитовидной
железы. При длительном существовании
тироидита Хасимото эти новообразования
поражают до 5 % больных. У больных
отмечается дефицит супрессорной функции
в отношении определённых клонов
лимфоцитов. Болезнь часто сочетается с иными
аутоиммунными эндокринопатиями и
аутоиммунными рецепторными заболеваниями
(среди них: инсулинозависимый сахарный
диабет, болезнь Аддисона — сочетание,
наблюдаемое в виде синдрома Шмидта;
пернициозная анемия, болезнь Верльгофа,
гипопаратироз).
Наиболее характерная
иммунопатологическая находка при данной форме
аутоиммунного тироидита — аутоантитела
к тироидной пероксидазе. Их титр
коррелирует с тяжестью заболевания и нарушением
гормональных функций железы.
Аутоантитела к тироглобулину, ядерным и иным
тироцитарным аутоантигенам также часто
обнаруживают у таких больных, но им
приписывают иные свойства. Если антиперок-
сидазные аутоантитела обладают цитоток-
сичностью и патогенетическим действием,
то антитироглобулиновые играют, по одним
данным, скорее свидетельскую роль, а по
другим — коррелируют с ходом ГЗТ и, возможно,
катализируют протеолиз тироглобулина
(К. Рот и соавт., 1997; С. Пол и соавт., 1997).
Могут присутствовать аутоантитела против
нетироглобулиновых белков коллоида.
При тироидите Хасимото развитие
гуморальных проявлений аутоиммунитета имеет
опережающий характер по отношению к
клеточным цитотоксическим механизмам
и гипотирозу. Поэтому существенные титры
аутоантител против тироглобулина и
тироидной пероксидазы обнаруживаются уже в
начале болезни. Например, у младших
подростков они фиксируются в крови на фоне
ювенильной гиперплазии органа, до
формирования клинически значимых проявлений
тироидита (Ю.И. Строев и соавт., 1998).
Аутоантитела к рецепторам ТТГ
наблюдаются при тироидите Хашимото, но чаще
всего бывают блокирующими, а не
стимулирующими — как при болезни фон Базедова,
способствуя развитию гипотироза.
Как уже отмечалось выше, при наличии,
в дополнение к перечисленным
иммунопатологическим эффекторам, стимулирующих
аутоантител к рецепторам ТТГ, больной
может развивать комбинированную картину
болезни фон Базедова и тироидита
Хашимото — «хаситоксикоз». При хаситоксикозе
содержание Т3 и Т4 повышено, а уровень ТТГ
и скорость захвата радиойода понижены.
Следует подчеркнуть, что аутоиммунитет
против щитовидной железы —
физиологическое явление. В низких титрах различные
антитироидные аутоантитела присутствуют
Нарушения эндокринной регуляции 571
у 50-90% клинически здоровых
индивидов, особенно — пожилых женщин. Для
тироидита Хасимото, как и для иных ауто-
иммуных тиропатий, характерны высокие
титры аутоантител, свидетельствующие не
просто о проявлении аутоиммунитета, но о
наличии аутоаллергии к щитовидной железе
(Ю.И. Строев, Л.П. Чурилов, 1999).
- Хронический фиброзно-инвазивный тиро-
идит Риделя (синоним: струма Риделя)
описан в 1896 г. (Б. Ридель). Это самый
редкий из хронических тироидитов.
Фиброз при этом заболевании охватывает
не только всю железу, но и
распространяется на соседние шейные структуры,
а нередко — это сочетается с фиброзом
языка и околоушных слюнных желёз,
ретробульбарным и ретроперитонеаль-
ным фиброзом (синдром Ормонда) и
фиброзом средостения. Тироидит Риделя,
видимо, является элементом в структуре
системного иммунопатологического
расстройства соединительной ткани. Болеют
чаще пожилые женщины. Железа
приобретает каменную плотность и неподвижна
при пальпации, относительно соседних
тканей. Пока сохраняются остатки ти-
роцитов, состояние больной может быть
компенсированным, эутироидным. В
исходе, в результате атрофии тироцитов,
развивается гипотироз. В механизмах
болезни предполагается участие ГЗТ и
ГНТ. У больных отмечается смешанная
нейтрофильно-мононуклеарная
инфильтрация железы, в крови регистрируются
различные антитироидные аутоантитела,
в железе — васкулит. Вероятно, большое
значение имеет выработка цитокинов,
содействующих фиброгенезу — тром-
боцитарного фактора 4, фактора роста
фибробластов и т.п. Струма Риделя
может вызвать стридор, сдавливая трахею.
Патофизиология надпочечных
желёз и их регуляции
Надпочечники — источник почти 60
стероидных, катехоламиновых и пептидных
гормонов. Это единственные производители
глюко- и минералокортикоидов, главные
продуценты андрогенов в женском
организме, важнейшие эффекторы стресса —
типового неспецифического нейроэндокринного
ответа организма на повреждение и угрозу
гомеостазу. Они участвуют также в поставке
прогестинов и эстрогенов, опиатов, в
конверсии тироидных гормонов. Все человеческая
жизнь — от появления на свет до
предсмертной агонии — проходит под знаком
высочайшей активности этих небольших органов,
суммарный вес которых всего лишь около
8 граммов. Не удивительно, что именно их
гормоны нашли самое широкое клинико-
фармакологическое применение.
Нарушения функций надпочечников
очень распространены. Их суммарная частота
превышает 1,5% населения, а в некоторых
регионах оказывается еще выше. Так, среди
эскимосов одна тйлько врожденная гиперплазия
коры надпочечников, редкая у представителей
других этносов, охватывает до 0,2 % всех детей.
Патология надпочечников включает не только
первичные приобретённые и наследственные
нарушения, но и вторичные эндокринопатии
вследствие расстройств регуляции роста и
функций этих органов.
Настоящий раздел, таким образом,
посвящен, в основном, этиологии и патогенезу
нарушений адренокортикотропной регуляции,
адренокортикальной и адреномедуллярной
функций.
Основные механизмы регуляции
функций надпочечников
В I томе данного руководства содержится
подробное описание истории изучения,
структуры и функции надпочечных желёз,
а также спектра производимых ими
гормонов (с. 514-543). Там же рассматриваются
вопросы центральной регуляции функций
надпочечников со стороны гипоталамо-ги-
пофизарного нейросекреторного аппарата и
вегетативной нервной системы, и дано
описание участия этих органов в стрессе (т. I, с.
492-513).
В данной книге эти вопросы повторно не
освещаются, ниже лишь напоминаются их
основные положения.
572 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Надпочечники — органы, корковое
вещество которых у взрослых состоит из
клубочковой (минералокортикоидной),
пучковой (глюкокортикоидной) и сетчатой
(андрогенопродуцирующей) зон. Мозговое
вещество — параганглий, состоящий из кате-
холаминопродуцирующиххромаффинных
клеток. Надпочечник растёт внутрь — с цен-
трипетальной миграцией клеток от капсулы
к мозговому веществу. При этом его клетки
видоизменяют свою функциональную
программу и дифференцировочные характерис-
т . ! под действием секреторно-митогенных
ciэмуляторов. Для клубочковой зоны
основными такими стимуляторами служат
ангиотензин II и АКТГ (дополнительную
роль играют калий-натриевое соотношение,
липотропин, МСГ и продукты эпифиза).
Для пучковой зоны эта роль безраздельно
принадлежит АКТГ. Клетки сетчатой зоны
находятся под сочетанным контролем АКТГ,
и — в более слабой степени — гонадотропи-
нов, пролактина и нейротензина. Секреция
мозгового вещества усиливается в ответ на
симпатический нервный сигнал.
Таким образом, главными гипоталамиче-
скими регуляторами надпочечников служат
вазопрессин, кортиколиберин и
ангиотензин, а аденогипофизарным — адренокорти-
котропин (АКТГ).
Адренокортикотропиновая
регуляция
Основной гипоталамо-гипофизарный путь
стимуляции коры надпочечников
предусматривает участие адренокортикотропина
(АКТГ), существование которого доказал
П.Э. Смит (1926), а выделил в чистом виде
П. Белл (1953).
Это хорошо изученный полипептид из 39
аминокислот, структура которого у человека
установлена Т.Н. Ли и соавт. (1961) и
представлена на рис. 95 (см. выше). Он не имеет
существенных видовых различий в своей
биологически эссенциальной части (с 1-го
по 24-й аминокислотный остаток), в связи
с чем АКТГ животного происхождения
действует у человека не менее, а иногда — более
эффективно, чем гомологичный. Все
видовые различия относятся к пептиду с 25-й по
39-ю аминокислоту, который для действия
на рецептор не обязателен. (Ю.А. Панков,
1976; Д.К. Клонофф, Дж.Х. Карам, 1998).
АКТГ и его взаимоотношения с ПОМК, а
также биологические эффекты и основные
закономерности секреции
охарактеризованы в т. I, с.512-514.
Секреция АКТГ стимулируется корти-
колиберином и вазопрессином, а также
ангиотензином II гипоталамуса, холи-
нергическими и серотонинергическими
нейронами подбугорья, а ингибируется
опиатами, ГАМК-эргическими нейронами,
и — наиболее сильно — глюкокортикостеро-
идами. Последние подавляют и продукцию
кортиколиберина. Эффект отрицательной
обратной связи носит двуфазный
характер — процессинг и секреция АКТГ
подавляется кортизолом через глюкокортикоидный
рецептор I типа остро, в первые 10 мин., а
затем наступает продлённая фаза
подавления продукции всего ПОМК, растянутая на
несколько часов и опосредованная через II
тип глюкокортикоидных рецепторов.
АКТГ — короткоживущий гормон, и уже
через 10 мин. его концентрация в плазме
снижается более чем наполовину. Однако в
кровь секретируется долгоживущий поли-
прогормон ПОМК, который может давать
АКТГ и другие пептиды в продлённом
режиме при его протеолизе в надпочечниках
и других органах.
Рецепторы АКТГ относятся к типичным
представителям семейства,
ассоциированного с G-белками. По Л.Д. Гэррину и
соавторам (1971), немедленный пострецепторный
механизм предусматривает активацию
аденилатциклазы и спектра протеинкиназ
А, фосфорилирующих белки-участники
стероидогенеза. Это позволяет достичь
увеличения выброса и синтеза стероидных
гормонов, обращаясь к трансляционным и
посттрансляционным механизмам, в
течение нескольких минут после воздействия
АКТГ на рецептор. По Р. Фариси и соавт.
(1981), важным моментом служит
активация фосфатидил-инозитоловых
посредников и протеинкиназы С, ответственных за
митогенное действие на клетки-мишени.
Наконец, А. Дазо и соавт. (1983) показали,
что АКТГ опосредует свои отсроченные
эффекты (проявляющиеся 2-4 ч спустя после
рецепции) через ключевые белки стероидо-
генеза, дерепрессирующие транскрипцию
определенных ферментов в адренокорти-
коцитах.
АКТГ очень сильно стимулирует
выработку глюкокортикоидов и андрогенов, а
также умеренно повышает продукцию ми-
нералокортикоидов, уступая в этом первые
роли ренин-ангиотензиновой системе.
Ренин-ангиотензиновая,
кининовая и атриопептиновая
регуляция
При рассмотрении стресса в т. I ренин-анги-
отензиновая регуляция упоминалась лишь
вскользь (с. 521). Подробная
характеристика ее роли в осмотическом гомеостаза и в
поддержании эффективного артериального
объёма была дана выше в разделе
«Патофизиология водно-электролитного обмена».
В связи с существенной ролью ангиотен-
зина II в стимуляции минералокортикоид-
ной секреции (и в небольшой степени —
также глюкокортикоидной) в данном разделе
необходима отсутствовавшая в I томе
характеристика ренин -ангиотензиновой системы.
При ознакомлении с этим материалом
читатель может пользоваться уже знакомым
рисунком 76, приведённым выше.
Центральным звеном этой системы
служит юкстагломерулярный аппарат почек,
включающий специализированный сегмент
дистального канальца — плотное пятно, а
также приносящую и выносящую артериолы
клубочков.
Ренин, гликопротеид из 274
аминокислот, он же — фермент аспартилпротеаза,
вырабатывается юкстагломерулярными
клетками, лежащими вблизи macula densa в
стенке афферентных и, в меньшей степени,
эфферентных артериол клубочков почек
(Дж.Бэкстер, 1987; Я.А. Рейд, 1998). Эти
клетки являются гранулярными миоэпи-
телиальными и, по некоторым
предположениям, тесно связаны с APUD-системой.
Нарушения эндокринной регуляции 573
Непосредственно они вырабатывают пре-
проренин, который переходит в проренин
(340 аминокислот), подвергающийся про-
теолизу, вследствие чего в этих клетках и их
паракринном окружении, а также в плазме
крови формируется ренин. Переход проре-
нина в ренин заторможен при инсулинза-
висимом сахарном диабете, что является
результатом гликирования прогормона и
приводит к нарушению почечного
кровотока, способствующему диабетической не-
фропатии (см. «Патофизиология сахарного
диабета»). Проренин продуцируется также в
печени и мозге, но неясно, переходит ли он в
ренин с той же эффективностью, как
почечный. Рениноподобные ферменты имеются в
надпочечниках, матке, кровеносных сосудах,
плаценте, сердце и слюнных железах. В
коре надпочечников — это компонент очень
активной местнЬй ренин-ангиотензиновой
системы, значительно влияющей на рост и
функцию адренокортикоцитов.
Скорость секреции ренина почками
служит главной детерминантой активности
всей ренин-ангиотензин-альдостероновой
системы. Ренин — короткоживущий
биорегулятор с периодом полураспада в крови
15-20 мин. Продукция ренина
стимулируется:
>• Снижением содержания натрия и
хлорида в первичной моче, которое
ощущается плотным пятном и приводит к
паракринной стимуляции его клетками
юкстагломерулярных продуцентов
ренина. Механизм этой рецепции зависит от
сочетанного натрий-калий-хлоридного
транспорта. Бессолевая диета усиливает
продукцию ренина. Повышение приема
калия понижает продукцию ренина в
связи с тем, что калий напрямую
усиливает секрецию минералокортикоидов.
>- Изменением растяжения барорецепторов
афферентных артериол клубочка в ответ
на понижение перфузирующего почки
кровяного давления, причём важна
определённая скорость этого понижения.
Сигнал также передаётся паракринно,
без участия нервов, или воспринимается
самими миоэпителиоцитами. Переход
574 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия
в вертикальное положение усиливает
продукцию ренина.
>* Симпатическими нервами и, в меньшей
степени, мозговым веществом
надпочечников — через норадреналин, прямо
стимулирующий ргадренорецепторы
' ' рениновых клеток. Норадреналин и
адреналин действуют и опосредованно,
через повышение перфузирующего
клубочек давления (внепочечные р-рецепто-
ры) и через снижение доставки хлорида
к плотному пятну, что зависит уже от
а-рецепторов катехоламинов.
>• Многие простагландины (в частности,
Е2) усиливают секрецию ренина и ведут
к гиперплазии ренин-продуцирующих
клеток (синдром Барттера).
>• Секреция ренина угнетается при
повышении содержания натрия и
хлоридов в фильтрате, солевой нагрузке,
повышении перфузирующего почки
давления, артериальных гипертензиях
(кроме катехоламиновой, что крайне
негативно влияет на возможности
компенсации при феохромоцитомах — см.
ниже); снижении симпатической
сигнализации. Денервация почек подавляет
продукцию ренина. Атриопептин и
и вазопрессин снижают ренинообразо-
вание. Оно угнетается также обратным
действием ангиотензина II на юкста-
гломерулярные клетки, что известно
как «короткая петля обратной связи»
- ренин-ангиотензин-альдостероновой
системы. Поскольку минералокорти-
коиды задерживают натрий и хлор, с
участием этих ионов связана «длинная
петля обратной связи», сдерживающая
продукцию ренина.
Ангиотензиноген — продуцируемый
печенью гликопротеидный препрогормон
(молекулярной массой 57 кД с более чем
400 аминокислотными остатками). Он
находится в а2-глобулиновой белковой
фракции плазмы. Синтез ангиотензиногена
стимулируют глюкокортикоиды, эстрогены,
тироидные гормоны и ангиотензин И.
Беременность, тиротоксикоз и гиперкортицизм
(эндокринно-метаболические нарушения)
протекают с выраженной гиперангиотензи-
ногенемией.
Под влиянием ренина 10 N-концевых
аминокислот ангиотензиногена
отщепляются, формируя ангиотензин I — прогормон,
лишенный собственной биологической
активности (см. выше рис. 95).
Ангиотензин I служит субстратом для
фермента дипептидил-карбоксипептидазы,
известного под условным названием «анги-
отензин-конвертаза» (см. рис. 76, с. 326).
Этот фермент эндотелиоцитов и плазмы,
имеющийся повсеместно, но наиболее
активный на сосудистом ложе лёгких —
важный участник протеолиза пептидных про-
гормонов и гормонов. Он отщепляет 2
аминокислоты с карбокси-конца декапептида
ангиотензина I, превращая его в октапептид-
ный гормон плазмы крови — ангиотензин II,
первый из известных гормонов, не имеющий
клетки-источника, а возникающий прямо
в плазме. Ангиотензин-конвертаза
расщепляет и инактивирует брадикинин, что
делает ренин-ангиотензиновую и хининовую
систему антагонистическими регуляторами
кровяного давления. Данный фермент
также участвует в инактивации вещества Р и
эндогенных опиатов. При неспецифических
хронических лёгочных заболеваниях его
активность может повышаться и понижаться.
Ингибиторы ангиотензинконвертазы
вошли в широкую клиническую практику как
средства против гипертензии.
Ангиотензин II — ключевой эффектор в
рениновой системе. Это гормон,
действующий через G-протеин-ассоциированные
рецепторы двух типов — ATI и АТ2. ATI
рецепторы преобладают в гладких миоцитах
сосудов и чувствительны к сульфгидриль-
ным восстановителям, АТ2 — преобладают
в коре надпочечников, почках, мозге и матке,
а от состояния сульфгидрильных групп не
зависят. Гормон опосредует ATI-действие
в сосудах через стимуляцию фосфатидил-
инозитолового посредника и кальций с
активацией протеинкиназы С.
ATI-зависимые эффекты ангиотензина II в мозге и
надпочечниках, а также, возможно, его АТ2-
эффекты связаны с ингибированием адени-
Нарушения эндокринной регуляции 575
латциклазы и протеинкиназ А (Ф. Бампас,
1991). Ангиотензин II — мощный вазоконс-
триктор (в десятки раз активнее норадре-
налина!). Не только сам гормон вызывает
острый прессорный эффект. Его действие
на гипоталамус способствует активации
симпатической нервной системы. В
параганглиях, в том числе мозговом веществе
надпочечников, он усиливает освобождение
катехоламинов. В сердце он действует как
положительный инотропный эффектор.
Все это продлевает его влияние и ведет к
артериальной гипертензии.
В почках ангиотензин II не только
подавляет продукцию ренина, замыкая обратную
связь, но и усиливает реабсорбцию натрия
в проксимальных канальцах. Он вызывает
сужение почечных сосудов.
Ангиотензин II в мозге стимулирует центр
жажды и является прямым дипсогеном —
провокатором питьевого поведения. Через
ангиотензинергические нейроны он
усиливает продукцию вазопрессина в гипоталамусе,
что способствует выработке АКТГ.
Ангиотензин II — тропный гормон мине-
ралокортикоидной зоны коры надпочечиков,
что впервые доказано Г. Дж. Нассдорфером
и соавт. (1982). Он усиливает продукцию
альдостерона, действуя на несколько
этапов — как ранних (конверсия холестерина в
прегненолон), так и поздних (превращение
кортикостерона в 18-гидроксикортикосте-
рон, рис. 112). В больших концентрациях
он стимулирует и глюкокортикостероидо-
генез.
Кроме того, ангиотензин II способствует
секреции кортикостероидов, а для клеток
клубочковой зоны служит митогеном.
Интересно, что и на гладкие миоциты сосудов
этот гормон оказывает митогенное действие,
а в кардиомиоцитах способствует
внутриклеточной гипертрофии.
Время жизни гормона в кровотоке
коротко — до 2 мин. При последовательном
протеолизе он дает ряд дериватов. Среди
них — продукт отщепления аспарагинового
остатка аминопептидазой — гептапептид
ангиотензин III. Он обладает слабой, сходной
с предшественником биологической
активностью, также стимулируя и минералокор-
тикоидный биосинтез, и чувство жажды.
Его уровень у человека составляет до 20 %
от концентрации ангиотензина II.
Семейство так называемых ангиотензи-
наЗу имеющихся на эндотелии сосудов всех
органов, кроме лёгких, довершает
инактивацию ангиотензинов. Некоторые из них
параллельно активируют антагонистически
действующие на гладкие миоциты кинины.
Работа ренин-ангиотензиновой системы in
vivo неотделима от регуляторных эффектов
других вазоактивных пептидов — хининов и
атриопептинов.
Кининовая система была рассмотрена в
т.1 данной книги (с. 348-350), к тому же
влияние кининов на функции
надпочечников незначительно и сводится к активации
освобождения катехоламинов из их
мозгового вещества.
Атриопептины существенно влияют на
минералокортикоидные реакции и
нуждаются в кратком описании.
Так называемые секреторные кардио-
миоциты предсердий, которые относят
кнейроэндокринным клеткам,
концентрируются в ушках предсердий, но
присутствуют и в желудочках. Эти клетки синтезируют
препрогормон из 151 аминокислоты, из
которого образуются атриопептин А (28
аминокислот) и атриопептин В (32
аминокислоты). Оба пептида имеют сходную
циклическую часть (17 аминокислот), а также
отходящие от нее короткую и длинную
ветви (рис.113). Атриопептины убиквитарны
и продуцируются также апудоцитарными
клетками лёгких и нейроэндокриноцитами
ЦНС и вегетативных ганглиев. В мозге, в
частности, преобладает атриопептин С (22
аминокислоты, цикл и одна нециклическая
ветвь). Тем не менее, во многих
источниках атриопептины (они же — предсердные
натрийуретические факторы)
рассматриваются как гормоны сердца, воплощение
его давно предполагавшихся и, наконец,
описанных эндокринных функций.
Секреция атриопептинов возрастает при
гиперволемии, растяжении предсердий, пе-
576 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
сн3соон
прегненолон- СНЗ t?-3^-оксистероид-
ч^>^, синтетаза С=0 дегидрогеназа
гладкая
НО
,V,^*4^
Гс^
гладкая
ацетат эидоплазматическая холестерин д5 прегненолон эндоплазматическая
сеть (микросомы) митохондрии сеть (микросомы)
f
21-гидроксилаза
т СН3
I _ 21-гидрок<
O^s^k^ гладкая °
СН2ОН
11а гидроксилаза
СН2ОН
прогестерон эндоплазматическая 11 дезоксикортикостерон митохондрии кортикостерон
I сеть (микросомы)
17а гидроксилаза
гладкая ^%У
эндоплазматическая
сеть (микросомы)
11а-гидроксилаза \ 2
О
HOv>,4iJPH
гладкая
эндопла
сеть (микросомы)
_ 17а окси митохондрии кортизол
17а оксипрогестерон эндоплазматическая ->
г , . дезоксикортикостерон
18 гидроксилаза
носн2он
18 дегидрогеназа
митохондрии
18-оксикортикостерон
митохондрии
СН2ОН
н&=°
альдостерон
Рис. 112 Цитофизиологические перемещения и биохимические превращения при биосинтезе
кортикостероидов (по В.И. Кандрору, 1991)
реходе в горизонтальное положение,
физических усилиях, солевой нагрузке. При этом
механизм стимуляции атриопептиногенеза
растяжением миокарда не требует участия
рефлексов (Ф. Нидлмен, 1987). Глюкокор-
тикоиды, альдостерон и вазопрессин
усиливают секрецию атриопептинов, их уровень
растет также при сердечной и почечной
недостаточности. Атриопептины сильно
подавляют функцию ренин-ангиотензин-аль-
достероновой системы, уменьшая секрецию
ренина и альдостерона, а также снижают
продукцию АДГ. Атриопептины действуют
на особые рецепторы трех типов — ANPA_C —
представляющие собой трансмембранную
гуанилатциклазу (см. выше) Стимуляция
этого энзима повышает уровень ц-ГМФ и
снижает количество кальция в цитозоле
клеток-мишеней. Затем гормоны
подвергаются эндоцитозу. В минералокортикоидной
зоне коры надпочечников представлены
атриопептиновые рецепторы, угнетающие
продукцию альдостерона непосредственно.
Атриопептины оказывают вазодилята-
торный и гипотензивный эффект, снижают
реабсорбцию и увеличивают фильтрацию
натрия в почках. Это позволяет им быть
основными эффекторами при реализации
важнейшего физиологического
механизма — прессорного натрийуреза, предохра-
Нарушения эндокринной регуляции 577
няющего от перехода острых гипертензий
в хронические. При повышении кровяного
давления, как установлено А. Гайтоном
(1989), почки усиливают экскрецию натрия
и воды, что обеспечивает мощную
долговременную стабилизирующую реакцию в
отношении артериального кровяного
давления (см. также т.1,с. 8-9). Данная реакция
требует одновременно усиления действия
атриопептинового и ослабления влияния
ренин-ангиотензинового звена пептидной
регуляции объёма внеклеточной жидкости.
Таким образом, данные гормоны
предохраняют от перегрузки организма жидкостью и
солью, препятствуют гипертензий и
развитию застойной сердечной недостаточности.
Эффект атриопептинов — не только
периферический. Обнаружены нейроны,
чувствительные к атриопептину, в
гипоталамусе— в прилегающей к дну III желудочка
передне-вентральной части перивентри-
кулярного ядра. Эта область гипоталамуса
связана с регуляцией кровяного давления и
водно-солевого гомеостаза (см. выше гл. 9).
Время полувыведения атриопептинов из
крови — 3-5 мин., сопоставимо с
аналогичным показателем ангиотензинов. Их
разрушает в основном нейтральная эндопептидаза.
Есть указания на продукцию в наружной
субкапсулярной части минералокорти-
коидной зоны коры надпочечников не-
идентифицированных натрийуретических
стероидов антиальдостеронового действия,
связанных с атриопептинами (Х.Э. ДеВар-
денер, Э.М. Кларксон, 1985). Ставится
вопрос о существовании естественных лигандов
уабаиновых и дигоксиновых рецепторов,
которые обладали бы натрийуретическим
действием за счет блокирования калий-
натриевого насоса. Этим гипотетическим
соединениям даже отводят важную роль в
сенсибилизации сосудов к прессорным
воздействиям при недостаточном прессорном
натрийурезе, свойственном эссенциальной
гипертензий (Ф.Нидлмен, 1987). Но их
строение и источник неизвестны, хотя они
отождествляются некоторыми авторами
с предполагаемыми антиальдостероновыми
нгы-
атриопептин- С
Рис. 713. Структура атриопептинов у человека,
(по Я.А. Рейду, 1998)
кортикостероидами. Если такие натрийу-
ретические вазоконстрикторные агенты
будут найдены, то речь пойдет еще об одном
модуляторе эффектов ренин-ангиотензин-
атриопептиновых взаимодействий.
Повышение активности ренин-ангиотен-
зин-альдостероновой системы и снижение
эффективности атриопептиновых контра-
регуляторных механизмов считаются
ключевыми в патогенезе всех видов системных
отёков (см. «Патофизиология
водно-электролитного обмена»). Эти расстройства
приводят к вторичному гиперальдостеронизму
(см. ниже). Нарушениям регуляции ренин-
ангиотензин-альдостероновых механизмов
и связанной с Ними атриопептиновой и
кининовой регуляции придают большое зна-
578 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
чение в патогенезе гипертензий и сердечной
недостаточности (данные вопросы будут
детально освещены в т. III «Механизмы
болезней и синдромов»).
Основные синдромы, связанные
с дисфункцией надпочечников
Закономерности кортикостероидогенеза
были раскрыты на основе классических
работ О. Хехтера и Г. Пинкуса в 50-60-х
годах XX века.
В дополнение к схеме стероидогенеза в
надпочечниках, приведённой в т. I (с. 517), в
данной книге на рис. 112, с. 576, дана краткая
цитофизиологическая схема их продукции,
отражающая локализацию и
ферментативное обеспечение основных этапов
биосинтеза кортикоидов.
Гормоны коры надпочечников
выполняют разные, но частично
перекрывающиеся биологические функции. Удаление
надпочечников смертельно, однако при
введении минералокортикоидов адрена-
лэктомированным животным они избегают
острой гибели и живут в условиях глубоких
метаболических расстройств и пониженной
стрессоустойчивости.
Минералокортикоиды, в основном — аль-
достерон и дезоксикортикостерон,
важнейшие регуляторы калий-натриевого гомеос-
таза и объема внеклеточной жидкости. Их
действие адресовано, главным образом,
почкам и тесно увязано с функцией ре-
нин-ангиотензиновой и атриопептиновой
систем. Минералокортикоиды выражен-
но действуют также на ЖКТ, потовые и
слюнные железы. Во всех клетках-мишенях
они адресуют свое отсроченное действие
цитозольному минералокортикоидному
рецептору (он же — глюкокортикоидный
рецептор I типа), который обеспечивает
их проникновение в ядро и дерепрессию
определённых генетических программ.
Рецептор связывает альдостерон в 100 раз
активнее, чем кортизол. Под действием
этих эффекторов клетки собирательных
трубок и соединительного сегмента нефро-
на начинают активно захватывать натрий и
обменивать его на калий и водород.
Следовательно, при наличии натрия в фильтрате
почек, пропорционально его доступности
альдостерон и его аналоги будут
осуществлять задержку натрия и воды,
способствовать экскреции калия и подкислению
мочи. Если натрия в первичной моче мало,
эффективность этих процессов
минимальна. Ренин и ангиотензин, делая натрий
доступным для обменного транспорта,
создают условия для эффективной альдосте-
роновой регуляции, а не просто повышают
концентрации альдостерона. Атриопептин
служит неконкурентным антагонистом
альдостерона. Потеря калия и водорода и
задержка натрия происходят под влиянием
гормонов в потовых и слюнных железах,
тонком и толстом кишечнике.
Большое значение имеет действие
минералокортикоидов за пределами почек.
Установлено, что у этих гормонов
имеются быстрые эффекты, не связанные ни
с цитозольным рецептором, ни с
модуляцией транскрипции. Как и предсказывает
принцип Г. Расмуссена (см. выше — о
механизмах действия гормонов), ранние
эффекты минералокортикоидов связаны с
их прямым действием на плазматические
мембраны клеток, на водород-натриевый
противопереносчик и фосфатидил-инози-
толовые посредники. Даже минимальные
концентрации альдостерона активируют на
протяжении 1-2 мин. данную транспортную
систему, что ведет к задержке ионов натрия
и потере протонов и ионов калия всеми
исследованными клетками, даже теми, где
ядерные механизмы действия
минералокортикоидов неактуальны (например,
лейкоцитами и миоцитами, мозгом и плацентой).
Эти эффекты влияют на объём клеток, их
чувствительность к другим гормонам и на
системный водно-солевой и
кислотно-основной обменн (см. гл. 10).
Расстройства секреции
минералокортикоидов известны в форме гипоальдосте-
ронизмаи гиперальдостеронизма. И тот и
другой синдромы могут быть результатом
надпочечниковых поражений (первичные
формы) и нарушений вненадпочечниковой
регуляции (вторичные формы).
Нарушения эндокринной регуляции 579
Ф
Глюкокортпикоиды (у
человека прежде всего кортизол
и в меньшей степени — кор-
тикостерон и кортизон) —
гормоны универсального
метаболического действия,
основные эффекторы стресса
и контринсулярные
регуляторы, сигналы, сдерживающие
воспаление и иммунный
ответ в умеренных,
контролируемых рамках нормергичес-
кого течения и
предупреждающие шок. В малых и средних
количествах они действуют
на глюкокортикоидный
рецептор II типа, а в больших
концентрациях оказывают
и некоторый минералокор-
тикоидный эффект — через
рецептор I типа.
Механизмы их действия
рассмотрены в т.1 (с. 522-
527). Подчеркнем, чтов
большинстве тканей, кроме
печени, они способствуют
катаболизму белков и липи-
дов, понижают
использование глюкозы в тех клетках,
которые имеют лишь инсу-
линзависимые глюкозные
транспортёры. Этим
определяется их диссимиляторное
действие. Одновременно в печени они
стимулируют синтез глюкозы и гликогена,
захват аминокислот, синтез ряда белков и
использование жирных кислот. В органах
с инсулиннезависимыми транспортёрами
глюкозы, например в ЦНС, сердце и диа-
фрагмальной мышце глюкокортикоиды
увеличивают потребление глюкозы (рис. 114).
Глюкокортикоиды — пермиссивные
регуляторы экспрессии рецепторов многих других
гормонов. Они влияют на скорость апоптоза
в лимфоидных и иных клетках.
При повышении продукции глюкокорти-
коидов развивается гиперкортпицизм. Он
может быть как первичным, надпочечниковым,
так и вторичным — при аномалии регуляции
глюкоза
(относительно^) I
Ф
лимфоидная ткань
№
соединительная
ткань
жировая ткань
-е-^^-о-
Рис. 114. Биологические эффекты глюкокортикоидов
(по Н.А. Юдаеву и соавт., 1976)
глюкокортикостероидогенеза. Так как АКТГ
и другие регуляторы совместно активируют
продукцию глюко- и минералокортикоидов,
нередко при гиперкортицизме имеются и
черты гиперальдостеронизма. Первичная
недостаточность функций коры
надпочечников или отсутствие должной стимуляции
со стороны ее регуляторов могут обусловить,
соответственно, первичный или вторичный
гипокортицизм.
В т. I данного руководства не
рассматривались закономерности метаболизма
кортикостероидов на этапах их транспорта и
инактивации, связанные с внежелезистыми
нарушениями глюко- и минералокортикоид-
ной регуляции. Эти пробелы восполняются
в данном разделе.
580 АЖ Зайчик, Я/7. Чурилов Патохимия (эндокринно-метаболические нарушения)
После попадания в кровь и глюко-, и
минералокортикоиды связываются белком
транскортином, а-глобулиновым протеином
печёночого происхождения (50 кД). Его
продукция усиливается при гиперэстрогенизме,
беременности, гипертирозе, сахарном
диабете. Существует наследственная аномалия,
проявляющаяся в аутосомно-рецессивном
усилении продукции транскортина. Все эти
состояния характеризуются повышенным
связыванием кортикостероидов. При
подобных ситуациях происходит уменьшение
фракции активного свободного кортизола,
что влечёт активацию продукции АКТГ.
Устанавливается нормокортицизм при
повышенном уровне неактивного связанного
кортизола.
Гипотироз, печёночная недостаточность
и нефротический синдром ведут к
понижению уровня транскортина. Он понижен при
болезни Рустицкого-Калера и семейном
наследственном дефиците этого глобулина.
Во всех этих случаях устанавливается за
счет действия центрального сервомеханизма
нормокортицизм с пониженным резервом
неактивного кортизола в крови.
Кортизол связывается транскортином
активнее других глюкокортикоидов и имеет
большее время иолувыведения из плазмы
крови — 1-1,5 ч против 45 мин. — у корти-
костерона. Только 10% плазменных
глюкокортикоидов свободны и метаболически
активны.
Минералокортикоиды связываются не
только транскортином, но и альбумином,
в обоих случаях — менее активно, чем
глюкокортикоиды. Поэтому более 30 % их
общего содержания в плазме
представлено свободными гормонами. Понижение
продукции альбумина и транскортина при
печёночной недостаточности и голодании,
их потеря при нефротическом синдроме
косвенно способствуют вторичному ги-
перальдостеронизму и системному
отёчному синдрому (см. также
«Патофизиология водно-электролитного обмена»).
Время полувыведения альдостерона —
20-30 мин. Как глюкокортикоиды, так
и минералокортикоиды инактивируют-
ся путём образования водорастворимых
парных соединений — глюкуронидов и
сульфатов — в печени, а затем экскретиру-
ются с мочой и желчью. Свободные кортико-
стероиды составляют не более 1 % суточной
экскреции. Печеночная недостаточность,
голодание, кахексия различной этиологии,
гипотироз замедляют их инактивацию,
что имеет значение для поддержания ги-
перальдостеронизма и относительного ги-
перкортицизма при этих состояниях.
Андрогены сетчатой зоны (андростендион,
дегидроэпиандростерон и его сульфат, а
также самый активный из них, хотя и сек-
ретируемый в наименьших количествах
андроген — тестостерон) участвуют в
регуляции половых функций, стрессе и
контроле анаболических процессов. У мужчин
их образование при стрессе существенно
для наиболее эффективной
психологической и метаболической адаптации, а также
уменьшения негативных последствий
дистресса. У лиц обоего пола они влияют на
формирование либидо. К тому же у
женщин надпочечники — это единственный
источник андрогенов, необходимых для
оптимального развития клитора, что связано
с возможностью испытывать оргазм. Выше
уже шла речь о механизмах стимуляции их
продукции. Они секретируются в свободном
виде и секс-связывающем глобулином и
связываются с альбумином. Конверсия этих
гормонов идет не только путем образования
парных соединений в печени, но и в тканях-
мишенях, где они метаболизируются в более
активные тестостерон и дегидротестостерон.
Плацента и жировая ткань, особенно
локализованная в гиноидных зонах, способны
превращать андрогены в эстрогены (см.
раздел «Нарушения липидного обмена»).
Нарушения секреции андрогенов могут
сопровождать расстройства продукции
других кортикостероидов (например,
тотальный гиперкортицизм при синдроме
Иценко-Кушинга). Однако возможны и
изолированные нарушения их секреции, в
частности, надпочечниковый гиперандро-
генизм при некоторых опухолях и
врождённой гиперплазии коры надпочечников. Эти
Нарушения эндокринной регуляции 581
нарушения известны под собирательным
названием «адреногенитальные синдромы».
Катехоламины мозгового вещества — нор-
адреналин и дофамин — очень
существенные компоненты обеспечения острого
ответа организма на повреждение, участники
регуляции гемодинамики. Кроме того, путем
процессирования проопиомеланокортина в
корковом и мозговом веществе
надпочечника образуются опиатные пептиды, прежде
всего энкефалины, что необходимо в фазу
физиологического выхода из стресса (т. I,
с. 530-535). Известны гормонообразующие
опухоли мозгового вещества
надпочечников, нарушающие баланс адреномедулляр-
ных гормонов.
Таким образом, всё разнообразие
патологии надпочечников можно условно
подразделить на состояния, характеризующиеся
гиперадренализмом и гипоадренализмом.
Оба синдрома могут быть тотальными, то
есть предусматривающими уменьшение
выработки гормонов всех функциональных
частей надпочечников, и парциальными —
сводящимися к избирательной
недостаточности функции какой-либо части. Термин
«тотальный гиперадренализм»
неупотребителен в европейских источниках, поскольку
фактически этот синдром представляет
собой, скорее, гиперкортицизм и относится к
гиперфункции только коры надпочечников.
Тотальный гипоадренализм, напротив,
известен и в виде гипокортицизма, и в форме,
затрагивающей как секрецию коркового, так
и мозгового вещества (апоплексия
надпочечников или синдром Уотерхауза-Фреде -
риксена). Отметим, что из-за универсальной
роли АКТГ как стимулятора адренокорти-
коцитов всех трёх зон гиперкортицизм не
является только гиперглюкокортицизмом.
Тотальный гиперкортицизм включает, как
правило, более или менее выраженные
симптомы гиперальдостеронизма и гиперан-
дрогенизма. В дальнейшем изложении
патофизиология этих синдромов и относящихся
к ним наиболее важных нозологических
единиц рассматривается последовательно.
Патофизиология гиперкортицизма
Синдром избыточного действия кортико-
стероидных гормонов или гиперкортицизм
возможен в четырёх основных ситуациях
(рис.115):
>- При избыточной продукции
стимуляторов коры надпочечников в гипотала-
мо-гипофизарном нейросекреторном
аппарате (вторичный гиперкортицизм).
>• При избыточной продукции адренокор-
тикальных стимуляторов вне гипотала-
мо-гипофизарного аппарата (синдром
эктопической продукции АКТГ).
>• При первичной гиперплазии и
гиперфункции клеток самой коры надпочечников
(первичный гиперкортицизм).
>* При экзогенном введении кортикостеро-
идов (ятрогенный гиперкортицизм).
Практически эти варианты синдрома
отличаются по уровню АКТГ в крови
(повышен — при вторичном и, особенно,
эктопическом и снижен — при ятрогенном
и, особенно, — при первичном гиперкорти-
цизме). Различить их помогают также тест с
подавлением продукции кортикостероидов
дексаметазоном (см. ниже) и симптомы,
зависящие от этиологии процесса.
Вторичный гиперкортицизм может
развиваться на почве формирования частично
автономной от сервоконтроля аденомы
гипофиза, вырабатывающей АКТГ. Эта
причина присутствует в 50-80% всех
наблюдений эндогенного гиперкортицизма.
Наиболее часто болеют женщины среднего
возраста, установлен повышенный риск у
много рожавших пациенток. Подобные ад-
ренокортикотропиномы гипофиза в девяти
случаях из десяти бывают базофильными
или хромофобными микроаденомами. Их
частота среди всех гипофизарных аденом
не превышает 10%. Описаны отдельные
наблюдения, когда аналогичную картину
вызывала неопухолевая гиперплазия кор-
тиколипотрофов аденогипофиза. Возможно,
в этих эпизодических случаях большую
роль играет первичная гиперпродукция
кортиколиберина. Гиперкортицизм на почве
582 АЖ Зайчик, Я/7. Чурилов Патохимия (эндокринно-метаболические нарушения)
эритема
„буйволиный
горбик"
геморраги
ческий синдром
истончение
конечностей
неопухолевая гиперплазия
и гиперфункция
(базофильная I хромофобная
микроад^нома I макроаденома
луно
образно^
лицо
нормальное
багровый турецкое
стрии J седло
MJ
/ избыток
\кортизола
, N1 +
гиперфункция коры надпочечников
свисающий]
живот
гирсутизм
замедленное
заживание
ран
остеопороз
компрессированные
„рыбьи" позвонки
микрокоду ля^ная неопухолевая
гиперплазия коры надпочечников
й пневморетро-перито-
неограмма при карциноме
правого и атрофии левого
надпочечников
коры
надпочечников
К^глюкостерома)
карцинома коры
надпочечников
Рис. 715. Механизмы и проявления гиперкортицизма. Синдром Иценко-Кушинга (по Ф. Неттеру
из CIBA Medical Illustrations Collection)
адренокортикотропиномы гипофиза в
России именуется болезнью Иценко-Кушинга.
Впервые двух таких больных описал
воронежский невропатолог Н.М. Иценко (1926),
приоритет которого неоспорим, но, к
сожалению, совершенно не известен на Западе,
где ссылаются на описание «гипофизарного
базофилизма» видным американским
нейрохирургом X. Кушингом (1932).
Болезнь Иценко-Кушинга — результат
мутации Gs белка в адренокортикотрофах,
при которой этот белок продлённо остаётся
Нарушения эндокринной регуляции 583
в стимулированном состоянии, как бы
реагируя на несуществующий кортиколибери-
новый сигнал. Подобная поломка
универсальна для различных гормонообразующих
аденом аденогипофиза и уже упоминалась
выше, при рассмотрении соматотропином.
Адренокортикотропинома гипофиза
не является полностью автономной от
сервоконтроля при производстве АКТГ.
Это используется в дифференциальной
диагностике — дексаметазон в большой
(но не в малой) дозе подавляет продукцию
глюкокортикоидов у больных с болезнью
Иценко-Кушинга более чем на 50 %, а лица,
имеющие синдром Иценко-Кушинга, то есть
гиперкортицизм эктопической, первичной
или ятрогенной этиологии, не реагируют
столь выраженной супрессией выработки
глюкокортикоидов при дексаметазоновой
пробе. Если же малая доза дексаметазона
эффективно подавляет кортикостероидо-
генез, то диагноз болезни Иценко-Кушинга
маловероятен, и приходится предполагать,
скорее, симптоматический транзиторный
гиперкортицизм (например, вызванный
депрессией, стрессами, юношеским диспи-
туитаризмом, хроническим алкоголизмом
и т.д.). На кортиколиберин больные с
болезнью Иценко-Кушинга чаще всего дают
преувеличенно сильный ответ. Примерно у
10% аденомы не отвечают на КРФ, так как
имеют рецепторный или пострецепторный
дефекты.
Адренокортикотропиномы — чаще
доброкачественные опухоли, но они растут более
агрессивно, чем другие аденомы гипофиза,
могут быть злокачественными и метаста-
зировать.
Так как не более 10% больных имеют
крупные опухоли, нейрохирургические
симптомы объёмного процесса при
болезни Иценко-Кушинга — большая редкость.
В связи с этим связь с аденомами гипофиза
при данной болезни долго не была
очевидной, и лечение сводилось в былые времена
к удалению или резекции надпочечников.
Если удалить надпочечники у пациента
с болезнью Иценко-Кушинга, не убрав
аденому гипофиза, то на фоне возникшего
ятрогенного гипокортицизма, в силу
увеличения стимуляции кортиколипотрофов
гипоталамическим кортиколиберином,
происходит бурное прогрессирование
адренокортикотропиномы. Она увеличивается и
насыщает организм своими секреторными
продуктами. У больного развивается
синдром Нельсона — появляются признаки
объёмного процесса в черепе и темнеет кожа, так
как опухоль вырабатывает ПОМК,
содержащий последовательность МСГ, а АКТГ в
высоких концентрациях проявляет МСГ-по-
добную активность. Признаков гиперкорти-
цизма нет из-за отсутствия надпочечников,
а симптомов болезни Аддисона обычно нет,
так как все больные после адреналэктомии
получают кортикостероиды.
Гиперпигментация кожи, сосков и
особенно участков, подвергаемых постоянному
трению, например, локтей и кожи под
браслетами и кольцами, может присутствовать
и при болезни Иценко-Кушинга, если
адренокортикотропинома секреторно очень
активна и велика. Болезнь Иценко-Кушинга
приводит к двусторонней гиперплазии коры
надпочечников. Утрачивается нормальный
суточный ритм продукции кортикостерои-
дов, притупляется стрессорная стимуляция
глюкокортикостероидогенеза. Картина
самого гиперкортицизма не отличается от
случаев, имеющих иную этиологию, и
рассматривается ниже.
В 15-30% случаев эндогенный
тотальный гиперкортицизм связан с первичным
поражением клеток коркового вещества
надпочечников. Такие поражения
именуют АКТГ-независимым синдромом
Иценко-Кушинга. Это чаще всего результат
гормонально активных опухолей из клеток
коры надпочечников. Опухоль,
вырабатывающая глюкокортикоиды, — глюкостеро-
ма — обусловливает классический синдром
Иценко-Кушинга при низкой продукции
АКТГ. Поражение бывает чаще всего уни-
латеральным, хотя как редкость описаны
и двусторонние глюкостеромы. Примерно
у 55 % больных опухоль доброкачественна,
но в остальных случаях проявляет признаки
584 АШ. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
злокачественного роста. Злокачественные
опухоли — глюкостеромы — относительно
чаще встречаются у мужчин, а
доброкачественные преобладают у женщин. Стеро-
идогенез в глюкостероме не подавляется
дексаметазоном. Разновидность
глюкостеромы, продуцирующая в дополнение к
глюкокортикоидам и минералокортикоидам
еще и существенные количества андрогенов,
называется глюкоандростерома. В этом
случае картина синдрома Иценко-Кушинга
дополняется у мальчиков преждевременным
половым созреванием, а у женщин —
вирилизацией. Андрогенные гормоны чаще
продуцируют карциномы. В аденомах
выработка андрогенов нехарактерна.
Существует первичная неопухолевая
микроподулярная гиперплазия коркового
вещества надпочечников, которая вызывает
АКТГ-независимый синдром
Иценко-Кушинга у подростков и молодых людей.
При этом значительно тормозится рост и
задерживается окостенение эпифизарных
хрящей. В отдельных случаях можно
говорить о семейном аутосомно-доминантном
характере этого редкого нарушения (сим-
птомокомплекс Карнея — помимо этого
поражения включает: голубые невусы, мик-
сомы кожи, грудных желёз и предсердий,
соматотропиному аденогипофиза, опухоли
нервов, яичек и других эндокринных желёз,
пигментные родинки).
Однако У.Дж. Райли и Н.К. Мак-
Ларен (1984), а также A.M. Невилль и
М.Дж. О'Хэйр (1984) указывали на наличие
при данном расстройстве аутоантител к коре
надпочечников и предполагают их ростости-
мулирующее и АКТГ-подобное действие, по
аналогии с соответствующей аутоиммунной
тиропатией (см. выше). Аутоантитела к
коре надпочечников обнаруживались при
синдроме Иценко-Кушинга и другими
авторами (Д. Петраньи, 1983; X. Андрада
и соавт., 1979). Учитывая
экспериментальные данные о возможности получения
стероидогенных и митогенных для ад-
ренокортикоцитов иммуноглобулинов с
помощью иммунизации ядерными адрено-
кортикальными антигенами — in vivo (A.
Ш. Зайчик, 1969-1988; Л.П. Чурилов, 1986)
и при иммунизации гибридом рецепторами
АКТГ — in vitro (X. Людденс и соавт., 1982),
предположение о возможности
аутоиммунной этиологии надпочечникового синдрома
Иценко-Кушинга выглядит достаточно
обоснованным.
С. Дж. МакФи в последнем издании
своей «Патофизиологии болезней» (1997)
прямо называет адренокортикостимулиру-
ющие аутоантитела основной причиной мик-
ронодулярной двусторонней гиперплазии
коры надпочечников — что авторы данной
книги могут только поддержать!
Еще более редкая форма
надпочечникового первичного гиперкортицизма
развивается при билатеральной крупноузловой
гиперплазии коры надпочечников. При этом
гиперплазируют островки интерреналовой
ткани, расположенные вокруг
надпочечников, крупные узлы, содержащие структуры,
характерные для всех зон коры, возникают
и в основных надпочечных железах.
Механизм этого расстройства связывают с
необычным АКТГ-подобным действием на
клоны аномальной интерреналовой ткани
ЖИП (желудочного ингибирующего
полипептида). В связи с этим, гиперкортицизм
у таких пациентов особенно характерен
после еды, при высоком уровне продукции
данного гормона энтериновой системы.
Концентрация АКТГ— низкая, ритм его
продукции утерян, дексаметазон, так же как
и при других надпочечниковых вариантах
гиперкортицизма, не подавляет продукцию
кортикостероидов.
Синдром эктопической автономной
секреции адренокортикальных стимуляторов
вызывает оставшиеся 5-15 % случаев
эндогенного гиперкортицизма. Данный феномен,
в отличие от классической болезни
Иценко-Кушинга, в 75% случаев отмечается у
мужчин. Почти всегда это — эктопическая
продукция АКТГ, однако описаны и
единичные случаи эктопической продукции
кортиколиберина. Источниками
эктопических стимуляторов коры надпочечников
бывают опухоли из клеток диффузной
Нарушения эндокринной регуляции 585
эндокринной системы (апудомы). Впервые
подобная АКТГ-продуцирующая опухоль
описана У.Х. Брауном в 1928 г. — в лёгких.
АКТГ вырабатывается чаще всего при ов-
сяноклеточном бронхогенном раке лёгких
(до половины всех случаев эктопической
продукции). В качестве эктопических
продуцентов этих гормонов по частоте
следующими идут: карциноиды ЖКТ,
медуллярный рак щитовидной железы, опухоли
островков Лангерганса, тимомы. Реже его
продукция исходит из опухолей слюнных
желез, феохромоцитомы и нейробластомы.
Как редкие и редчайшие описаны меланомы,
лимфосаркомы, раковые опухоли половых
органов, вырабатывающие АКТГ.
Эктопические опухоли могут вырабатывать все
дериваты проопиомеланокортина — липот-
ропины, опиаты, МСГ. Иногда у больных
обнаруживаются продукты необычного
процессинга ПОМК — «большой АКТГ» и
«средний АКТГ», биологическая активность
которых может быть понижена. Кортико-
либериновая секреция описана у опухолей
панкреатических островков, С-клеток
щитовидной железы и нефробластомы.
Имеется сообщение о медуллярной
карциноме щитовидной железы,
вырабатывавшей кортиколиберин и пролактолиберин,
а также кальцитонин. Важным отличием
гиперкортицизма, вызванного эктопической
секрецией КРФ, служит подавляемость
кортикостероидогенеза высокими дозами
дексаметазона — ведь АКТГ при этом
исходит из аденогипофиза. При гиперсекреции
эктопического АКТГ дексаметазонового
подавления не бывает. Некоторые АКТГ-
синтезирующие опухоли секретируют
также гастрин, серотонин, гонадотропины,
СТГ. Продукция АКТГ в овсяноклеточных
карциномах легких нередко сочетается с
выработкой АДГ, а также иногда — окситоцина.
Так как уровень АКТГ при эктопической
секреции очень высок и превышает
характерный для болезни Иценко-Кушинга в
несколько раз, этого оказывается достаточно
для проявления МСГ-подобного эффекта
и для заметной стимуляции секреции всех
зон коры надпочечников. Поэтому
отличительными особенностями гиперкортицизма
при эктопической продукции АКТГ служат
очень значительная гиперпигментация кожи
и чёткие симптомы гиперальдостеронизма и
гиперандрогенизма, накладывающиеся на
обычную доминирующую картину избытка
глюкокортикоидов — таковы гипокалиемия,
мышечная слабость. В связи со
злокачественным характером большинства опухолей,
обеспечивающих эктопическую секрецию
АКТГ и КРФ, у больных часто не бывает
характерного для других случаев
гиперкортицизма ожирения.
Ятрогенный гиперкортицизм возникает
при лечении глюкокортикоидами. Как
правило, это происходит не при
применении сверхвысоких доз в кратковременном
режиме — как у больных с экстремальными
состояниями, поскольку в таких случаях
потребность организма в глюкокортикоидах
очень велика.
В опасности оказываются пациенты,
получающие средние дозы гормонов (от 100
мг в день в пересчёте на кортизол) на
протяжении долгого времени — не менее двух
недель. Такая ситуация, например, создаётся
при лечении глюкокортикоидами
иммунопатологических и аутоиммунных болезней
(А. Голдфин, 1998).
Описав этиологию тотального
гиперкортицизма, коснемся теперь вопросов
патогенеза его проявлений.
При гиперкортицизме нарушаются все
виды метаболизма.
Липидный обмен у больных
характеризуется следующим.
Наиболее характерной чертой
гиперкортицизма, отмеченной у 94 % больных,
считается особая форма вторичного ожирения,
названная по характеру распределения жира
«центральной» (Дж.Б. Тиррель, 1987). Жир
накапливается на животе, в сальниках, в
брыжейке вокруг абдоминальных
внутренностей, на лице, в надключичной и задне-
шейной области, между лопаток. При этом
конечности остаются обычного диаметра
или даже, в половине случаев — истончают-
586 A.LU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ся (имеет значение как атрофия мышц,
охарактеризованная ниже, так и потеря жира).
Столь неравномерное распределение жира
отражает особенности глюкокортикоидных,
инсулиновых и андрогенных рецепторов
в различных адипоцитах (см. раздел
«Нарушения липидного обмена»). Основная
причина — избыток глюкокортикоидных
рецепторов в центрипетально
расположенных липоцитах (см. рис. 115). По Г. Крусосу
и соавт. (2002), глюкокортикоиды
способствуют липолизу в соматическом и липоге-
незу — в висцеральном жире.
В эпоху, когда доктора любили образные
выражения, эти проявления получили
яркие названия — лунообразное лицо, бычий
загривок, горб бизона и т.д., которые
мнемонически хороши, но малоупотребительны в
современной медицине с её подчёркнутым
вниманием к деонтологии. Больные имеют
гиперлипопротеинемию, как правило, II
типа (накопление ЛПНП, ЛПОНП, гипер-
холестеринемия, гипертриглицеридемия),
связанную с подавлением экспрессии апо-В
рецептора глюкокортикоидами и усилением
синтеза триглицеридов в печени.
Так как глюкокортикоиды — in vitro для
большинства тканей, кроме печени — ли-
политические гормоны, ожирение при ги-
перкортицизме, вообще говоря,
труднообъяснимо. Считается, что большое значение у
больных имеет полифагия, а также
вторичный гиперинсулинизм, обязательно
возникающий при гиперкортицизме, особенно,
АКТГ-зависимом, в ответ на гипергликемию
и прямое стимулирующее действие АКТГ
и, возможно, — гидрокортизона на остров-
ковые В-клетки.
Углеводный обмен при гиперкортицизме
характеризуется ослаблением эффектов
инсулина, стимуляцией продукции
эндогенной глюкозы из липидов и аминокислот.
Жировая ткань, мышцы и лимфоидные
органы уменьшают потребление глюкозы в
пользу ЦНС, миокарда и диафрагмы, гонад,
надпочечников и тканей глаза, куда глюкозу
переносят неинсулинозависимые
транспортёры. В печени усиливается не только
глюконеогенез, но и синтез гликогена, а
также секреция в кровь новообразованной
глюкозы. В результате формируются такие
симптомы, как гипергликемия, глюкозурия,
полиурия и полидипсия характеризующие
симптоматический стероидный диабет
примерно у 10 % больных с недостаточными
резервами В-клеток островков Лангерган-
са. Стероидный диабет — резистентен к
лечению инсулином, как II тип первичного
сахарного диабета, но в то же время
осложняется кетозом — как первичный сахарный
диабет I типа (см. также «Патофизиология
сахарного диабета»). Такое необычное
сочетание наблюдается в силу кетогенности
глюкокортикоидов. Микроангиопатия при
стероидном диабете нехарактерна; очевидно,
гиперкортицизм не благоприятствует её
иммунопатологическим механизмам.
В остальных случаях npji
гиперкортицизме ситуация стабилизируется путём
развития гиперинсулинемии. Явного
диабета не наблюдается, но пробы с глюкозной
нагрузкой могут выявить интолерантность.
К тому же гиперинсулинемия способствует
ожирению и атеросклерозу (см. выше и в
разделе «Патофизиолология липидного
обмена»).
Белковый обмен в целом изменяется в
сторону усиления катаболических
процессов в большинстве клеток. Это ведет к
отрицательному азотистому балансу. Но
в печени и ЦНС синтез белка не страдает,
а в отношении многих протеинов — даже
усиливается. Наиболее явно катаболическая
направленность белкового метаболизма при
данном синдроме сказывается в мышцах,
коже, соединительной ткани, костях, лим-
фоидных органах.
Мышечная слабость, отмечаемая в 60%
случаев, связана и с потерей белка
мускулатурой, и с тенденцией к гипокалиемии,
особенно — при выраженном минералокор-
тикоидном компоненте гиперкортицизма.
Особенно важно, что глюкокортикоиды
вызывают избирательную атрофию
быстрых мышечных волокон II типа, наиболее
зависящих от аэробного распада и поставок
глюкозы. Это выражается в слабости
проксимальных мышечных групп конечностей,
Нарушения эндокринной регуляции 587
их истончении (частота — 58% случаев).
Глюкокортикоиды угнетают коллагеноге-
нез в фибробластах. Это ведет к
истончению кожи (симптом папиросной бумаги),
вазопатии (36%), плохому заживлению
ран, а в сочетании с иммунодепрессивным
действием этих гормонов — провоцирует
кожные бактериальные и грибковые
инфекции. Растяжение истончённой кожи
при ожирении ведет к образованию
характерных стрий (52%), багрово-фиолетовый
цвет которых зависит от индуцированных
глюкокортикоидами гипертензии, плеторы,
эритроцитоза.
Так как глюкокортикоидами угнетается
синтез белков в костях и затрагивается
обмен витамина D, все больные страдают
остеопорозом, 58 % жалуется на боли в
спине, что сопровождается гиперкальциемией
и кальциурией (40%), приводя порой к
камнеобразованию (18%). На этом фоне
рост больных — при детском и ювенильном
гиперкортицизме — тормозится за счет
ряда аспектов действия кортикостероидов
(угнетение продукции СТГ, соматомединов
и тироидных гормонов, снижение синтеза
белка и гликозаминогликанов в хрящах).
Водно-солевой обмен у больных с гипер-
кортицизмом характеризуется тенденцией
к гипернатриемии, задержке воды, отёкам
(18%), гипокалиемии и ускоренной потере
кальция с мочой, у 72 % больных имеется
повышение артериального кровяного
давления. Как и другие водно-солевые
нарушения, гипертензия связана с минералокорти-
коидной гиперволемией и сенсибилизацией
сосудов к катехоламинам. Следует учесть,
что глюкокортикоиды сильно повышают
продукцию печёночного ангиотензиногена,
уровень ангиотензина тоже повышается,
хотя альдостерона и так вполне
достаточно. Это вносит дополнительный разлад в
регуляцию кровяного давления.
Глюкокортикоиды тормозят эффект атриопептинов
на пострецепторном уровне и усиливают
продукцию вазоконстриктора эндотелина.
Они понижают синтез моноксида азота.
Столь комплексная по своим механизмам
гипертензия бывает значительной и может
стать основной причиной сердечной
недостаточности, инсультов и смерти таких
больных. Часто повышается и
внутриглазное давление, вплоть до глаукомы.
Кислотно-основной баланс
характеризуется достаточно необычной ситуацией
метаболического алкалоза, продиктованного
усиленным выведением калия и протонов
с мочой. Степень нарушений значительна
при большом избытке гормонов и
выраженном минералокортикоидном эффекте (как
правило, этим отличаются глюкостеромы и
синдром эктопической продукции АКТГ).
Гиперкортицизм обладает многими
важными внешними и функциональными
признаками со своеобразным патогенезом.
Плетора делает лицо больных красным
(84%), а избыток андрогенов и кожные
инфекции, в силу глюкокортикоидного
иммунодефицита, способствуют появлению
на лице пустуло-папулёзного периораль-
ного дерматита и акнэ (40%). При избытке
андрогенов в структуре синдрома гиперкор-
тицизма у женщин появляется избыточное
оволосение тела — гирсутизм. Его частота
при гиперкортицизме у пациенток
приближается к 80 %. Это сопровождается дисмено-
реями (76 %). У лиц обоего пола
гиперкортицизм подавляет импульсный ритм секреции
гонадолиберина, что понижает ответ Л Г на
гонадолиберин. У женщин это способствует
аменорее, а у мужчин — ведет к снижению
либидо, импотенции, уменьшению тестикул.
Продукция андрогенов в яичках падает, что
не компенсируется слабыми надпочечнико-
выми андрогенами, даже если их количество
повышено. Поэтому при синдроме Иценко-
Кушинга женщины становятся несколько
более, а мужчины — менее маскулинными.
Из-за инсулинорезистентности появляются
очаги кожного acantosis nigricans — тёмные
бархатистые пятна в местах трения (см.
также «Патофизиология сахарного диабета»).
Для гиперкортицизма всегда характерны
лимфопения, эозинопения, нейтрофиль-
ный лейкоцитоз, отражающие индукцию
глюкокортикоидами миелопоэза, а также
588 А.Ш. Зайчик, Л.П.Чурилов Патохимия
хоуминга и апоптоза лимфоидных клеток,
разрушения эозинофилов.
Гиперкортицизм имеет поведенческие
эффекты: от эйфории — при остром действии
гормонов до депрессии — при хроническом
повышении их уровня. У многих больных
развиваются тревожность и бессонница,
особенно страдает фаза быстрого сна.
Своеобразная эмоциональная
лабильность, обидчивость и некоторая эгоцентрич-
ность больных с синдромом Иценко-Ку-
шинга легко узнаваемы (поведение
«Карлсона, который живёт на крыше»). В тяжелых
случаях описан кортикостероидный психоз
с галлюцинациями и маниями.
Парциальные формы гиперфункции
коркового вещества надпочечников
предусматривают резкое преобладание гиперсекреции
и эффектов какой-либо группы кортико-
стероидов. De facto, чисто парциальных
форм, практически, нет, так как и эффекты
гормонов, и действие стимуляторов их
продукции перекрываются. Выше уже шла
речь о глюкостеромах, создающих избыток
глюкокортикоидов. Хотя формально это
вызывает, казалось бы, парциальный
гиперкортицизм на практике у больных, как
правило, отмечаются и минералокортикоидные,
и андрогенные эффекты.
Тем не менее можно выделить состояния
с преобладанием гиперактивности тех или
иных зон коры надпочечников или
гиперсекреции какой-либо группы кортикоидов.
Среди них представляет интерес, в
первую очередь, гипералъдостеронизм.
Выделяют первичный альдостеронизм, вызванный
избыточной нерегулируемой секрецией
минералокортикоидов в коре
надпочечников, а также вторичный, то есть вызванный
чрезмерной ренин-ангиотензиновой
стимуляцией вариант альдостеронизма.
Первичный гиперальдостеронизм
известен как синдром Конна (Дж. Конн, 1955).
Его самая типичная причина — альдосте-
рома коры надпочечников,
вырабатывающая минералокортикоиды в автономном
чдокринно-метаболические нарушения)
режиме. Чаще всего это аденома, но описаны
и альдостерон-продуцирующие карциномы
коры надпочечников. Альдостеромы могут
иметь эктопическую локализацию (яичник,
кишечник, щитовидная железа). Так как
минералокортикоиды не ингибируют
продукцию АКТГ, альдостеромы не
сопровождаются, в отличие от глюкостером, атрофией
остальной коры надпочечников.
Разновидностью альдостером являются
глюкокортикоид-подавляемые минералокор-
тикоид-продуцирующие аденомы, в которых
сервомеханизм ограничения минералокорти-
коидной секреции не полностью отсутствует
и может быть индуцирован большими дозами
глюкокортикоидов. Такие
доброкачественные опухоли характерны для наследственного
первичного альдостеронизма Imuna. Во время
мейоза в гаметах, из которых развиваются
носители данной аномалии, происходит
неравномерный кроссинговер, и формируется
результат слияния участков двух разных
генов — гена альдостерон-синтетазы и гена 11-
(3-гидроксилазы. Гибридный химерный белок
CYPIIB1-CYPHB2 сохраняет активность
ключевого фермента биосинтеза альдосте- ■
рона, но приобретает сильную зависимость
от стимуляции АКТГ.
АКТГ как митоген оказывается для
клубочковой зоны таких больных
сверхэффективным. Глюкокортикоиды, снижая
продукцию АКТГ, тормозят экспрессию
данной аномалии.
Изредка бывает первичный
гиперальдостеронизм при неопухолевой билатеральной
гиперплазии клубочковой зоны коры
надпочечников. Природа этого явления
неизвестна, но, по аналогии с микронодулярнои
гиперплазией, предполагают важную роль
аутоиммунной стимуляции роста и
функций адреногломерулокортикоцитов. Такая
гиперплазия порождает либо неподавляе-
мую продукцию минералокортикоидных
гормонов (идиопатический первичный
гиперальдостеронизм), либо частично
подавляемую их продукцию (неопределённый
гиперальдостеронизм).
Псевдогиперальдостеронизм развивается
при употреблении в пищу избытка солодки
(лакрицы), которая содержит ингибитор
11-р-гидроксилазы — глицерризиниевую
кислоту. У лиц, жующих табак и
употребляющих леденцы с лакрицей, а также у
энтузиастов траволечения — при передозировке
солодки — превращение кортизола в
кортизон тормозится, избыток первого оказывает
альдостероноподобное действие.
Воспроизводятся признаки альдостеронизма (см.
ниже). Аналогичный ферментативный
дефект бывает при гипертензивной форме
врожденной наследственной гиперплазии
коры надпочечников, вызванной дефектом
11-р-гидроксилазы (см. характеристику
врождённых адренокортикальных
гиперплазии ниже). Псевдогиперальдостеронизм
также воспроизводится при синдроме Лид-
для — первичной гиперчувствительности к
альдостерону, когда при нормальном уровне
минералокортикоидов наступают гипока-
лиемия, гипертензия и гипоренинемия (см.
выше гл. 10). Экзогенный альдостеронизм
может быть результатом ятрогенного
введения минералокортикоидов, которые входят
в состав некоторых назальных капель и
аэрозолей.
При всех формах первичного
альдостеронизма и при псевдоальдостеронизме
активность ренина низкая. Продукция ренина
тормозится за счет активации рецепторов
растяжения избыточным объёмом
внеклеточной жидкости и повышенным давлением,
создающимися при превышении
адекватного уровня альдостерона.
Вторичный гипералъдостеронизм,
распространенный во много раз больше, чем
первичный, протекает при высоком уровне
ренина (что иногда обозначается звучным
термином ренинизм) и активной ренин-ан-
гиотензиновой стимуляции минералокор-
тикоидного стероидогенеза. Данная форма
альдостеронизма создается в нескольких
ситуациях (см. также раздел «Системные
отеки»):
>* При ишемии почек, вызванной
атеросклерозом и артериосклерозом бассейна
почечных артерий, что обычно связано с
Нарушения эндокринной регуляции 589
хроническими гипертензиями,
диабетическим гломерулосклерозом {синдром
Киммельстила-Уилсона) и т.д.
>• При снижении объёма внутрисосудистой
жидкости (застойная сердечная
недостаточность, нефротический синдром,
хроническая печёночная
недостаточность, голодание, другие гипопротеи-
немические состояния, передозировка
мочегонных и слабительных).
>* При гипонатриемии и усиленных
потерях натрия (хроническая почечная
недостаточность, почечный канальцевый
ацидоз).
>* При первичной гиперплазии юкстагло-
мерулярного аппарата (синдром Барт-
тера, избыток простагландина Е2).
>- При рениномах — опухолях, секретирую-
щих ренин автономно (юкстагломеруляр-
ной и, редко, овариальной локализации).
>* При беременности так как эстрогены
стимулируют продукцию ангиотензи-
ногена и ренина, а прогестины снижают
эффективность действия альдостерона
(«физиологический вторичный
альдостеронизм»).
При вторичном гиперальдостеронизме
в адреногломерулокортикоцитах
наблюдаются признаки гиперсекреции и
гипертрофии-гиперплазии, однако опухоли
или проявления нодулярной гиперплазии
отсутствуют. Уровень альдостерона при
вторичном альдостеронизме выше, чем при
первичном.
Патогенез и проявления гиперальдосте-
ронизма до известной степени типичны, но
варьируют в зависимости от этиологии.
При любом альдостеронизме минерало-
кортикоиды вызывают задержку натрия
и воды, а также потерю калия и катионов
водорода почками. Поначалу
стимулируется реабсорбция натрия в собирательных
трубках и обмен натрия на калий и протоны
в дистальных извитых канальцах.
Повышается обьём внеклеточной жидкости, и
растёт артериальное кровяное давление.
Гипертензия — главный симптом первично-
590 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
го альдостеронизма. От 8 до 12% больных
артериальной гипертензией имеют синдром
Конна. Повышается особенно диастоличес-
кое давление. Формируются гипертрофия и
дилятация сердца. Гипертензия
осложняется колебаниями давления ортостатического
характера, поскольку при потере калия
барорецепторы малочувствительны и не
обеспечивают адекватной постуральной
стабилизации давления.
По достижении и длительном удержании
определенного уровня гипертензии
включается и «наращивает обороты» механизм
прессорного натрийуреза, предохраняющий
от хронической гипертензии. Это означает,
что усиливается секреция атриопептинов,
что ведет к ускользанию собирательных
трубок из-под эффекта альдостерона.
Выделение натрия и воды увеличивается,
это позволяет пациентам с первичным
альдостеронизмом не иметь отёков. Но
в дистальных извитых канальцах атри-
опептиновые рецепторы не имеют столь
выраженного представительства, поэтому
там действие альдостерона продолжается в
полной мере. Это ведет к продолжению
потери калиевых и водородных катионов. Ги-
пернатриемия носит умеренный характер
из-за разведения натрия, но гипокалиемия
и выделительный метаболический алкалоз
прогрессируют.
Истощение ресурсов калия ведет к целому
ряду патологических последствий (см. выше
также «Патофизиология
водно-электролитного обмена»). При гипокалиемии
развиваются мышечная слабость, прострация.
Калийурия оборачивается увеличением
диуреза, особенно по ночам. Полиурия с
потерей калия исключает возникновение
отёков, но значительная калиевая деприва-
ция нарушает работу градиент-создающих
биоэлектрических систем организма. На
ЭКГ притупляется и сглаживается Т-зубец
и появляется выраженный U-зубец. Могут
быть тяжелые тахиаритмии, парестезии,
атония кишечника.
Длительная тяжёлая гипокалиемия
сказывается на работе почек («гипокалиемичес-
кая нефропатия»). Эффективность работы
АДГ-зависимого механизма реабсорбции
воды снижается, что усиливает полиурию
и никтурию, наблюдающиеся у таких
пациентов.
Так как входной ток калия сопровождает
утилизацию клетками глюкозы, тяжёлая
гипокалиемия понижает толерантность к
виноградному сахару. Это диабетогенное
действие независимо от других контрин-
сулярных эффектов кортикостероидов и
корректируется при нормализации
электролитного баланса.
Из-за перехода водородных
эквивалентов внутрь клеток и в мочу развивается
метаболический выделительный гипока-
лиемический гипохлоремический алкалоз.
В условиях алкалоза снижается уровень
ионизированного кальция в плазме, что
ведет к тетании и положительным симптомам
Труссо и Хвостека. При первичном гипер-
альдостероизме отёки наблюдаются лишь
после развития осложняющих его сердечной
и почечной недостаточности или при
наличии сопутствующих тромбофлебитов (не
более чем у 3 % пациентов).
При вторичном гиперальдостеронизме
важным звеном патогенеза является гипер-
ренинангиотензинемия. Как известно,
компоненты ренин-ангиотензинового каскада
понижают эффективность атриопептиновой
регуляции. Поэтому прессорно-натрий-
уретический механизм оказывается
значительно менее эффективным, а необходимое
для его работы подавление продукции
ренина — невозможным. Это приводит к
регулярному формированию системных
отёков и существенному абсолютному
избытку натрия у больных с различными
этиологическими формами вторичного
альдостеронизма. В ряде случаев
(печеночная недостаточность, гипопротеинемии)
некоторое дополнительное значение имеет
снижение скорости деградации или степени
белкового связывания минералокортико-
идов (см. выше также «Патофизиология
водно-электролитного обмена» и
характеристику механизмов отёков в т. I, с. 212-
213). Единственное исключение — синдром
Барттера, когда гиперальдостеронизм носит
вторичный характер, но выраженных отёков
и гипертензии не наблюдается.
Таким образом, данные формы альдосте-
ронизма различны этиологически,
патогенетически и клинически.
Гиперпродукция в надпочечниках половых
стероидов известна в разных формах и
сопровождает различные болезни и синдромы.
Наиболее характерными общими
проявлениями гиперандрогенизма служат гирсутизм,
олигоменорея, угри и вирилизация.
Традиционное собирательное название
для нарушений продукции половых
гормонов надпочечниками — адрено-генитальные
синдромы. Эта сборная группа включает
приобретенные и врождённые формы.
Приобретенные нарушения половых функций и
признаков на почве гиперпродукции адрено-
кортикальных половых гормонов связаны с
различными опухолями, вырабатывающими
секс-стероиды.
Выше уже отмечалась возможность
адренокортикального гиперандрогенизма
при гиперкортицизме вследствие болезни
и синдрома Иценко-Кушинга, в том числе
факт существования глюкоандростером.
Описаны андростеромы (1-3 % всех
опухолей надпочечников) и кортикоэстромы
надпочечников (отдельные наблюдения).
При андростеромах, которые у детей,
как правило, злокачественны, а у взрослых
нередко бывают и доброкачественными,
отмечается автономная гиперпродукция
андрогенов, связанная с соматической
мутацией (мутациями) генов стероидогенных
эзимов в опухолевом клоне. Андростеромы
достигают очень значительных размеров
(до 1,5 кг весом). У женщин андростерома
вызывает быструю вирилизацию
(гирсутизм, перераспределение жира по
мужскому типу, огрубение голоса, облысение,
атрофия молочных желез, олигоменорея и
аменорея, гипертрофия клитора, выделение
мускулатуры и повышение физической
работоспособности, изменение стереотипов
поло-ролевого поведения). У детей обоего
пола развивается преждевременное половое
созревание (у мальчиков — изосексуального,
Нарушения эндокринной регуляции 591
а у девочек — гетеросексуального типа). Рост
костей в длину рано останавливается из-за
опережающего костного возраста, больные
остаются невысокими. Это дополняется
местными объемными и
общеонкологическими симптомами, которые очень важны
для дифференциальной диагностики, так
как сходная картина может иметь
множество вненадпочечниковых и неопухолевых
причин (см. ниже «Патофизиология гона-
дотропной функции и гонад»). У взрослых
мужчин многие случаи небольших по
размеру андростером остаются, вероятно,
нераспознанными.
При кортикоэстромаХу которые,
наоборот, хорошо распознаются именно у мужчин,
имеет место продукция эстрогенов в мутант-
ном опухолевом клоне адренокортикальных
клеток. Эти оаухоли — злокачественные,
инвазивные, феминизирующие. У носителя
кортикоэстромы отмечаются гинекомастия,
часто — гипотрофия тестикул, феминизация
телосложения и стереотипов поведения.
Некоторое количество эстрогенов выделяют
многие глюкостеромы, андростеромы и
карциномы надпочечников, но это маскируется
секрецией андрогенов.
Большое значение имеет врождённая
форма адренокортикального
гиперандрогенизма, связанная с несколькими разными
аутосомно-рецессивными наследственными
дефектами ферментов стероидогенеза, при
которых возникший метаболический блок
благоприятствует синтезу андростероидов,
в ущерб продукции кортизола. Эти
наследственные заболевания входят в
собирательную группу адреногенитальных синдромов,
известных как «врождённая гиперплазия
коры надпочечников». Данные нарушения
более детально рассматриваются ниже, в
разделе, посвященном гипокортицизму, так
как при них могут доминировать явления
нехватки глюко- и/или минералокортико-
идов. Гиперандрогенизм при этих
расстройствах имеется не всегда. С выраженной
гиперпродукцией андрогенов надпочечниками
протекают лишь некоторые из них:
- Дефицит 3-$-ол-дегидрогеназы
(нарушается переход 55-прегненолона в
592 АШ. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
прогестерон, накапливается дегидро-
изоандростерон). Так как данное ан-
дрогенное соединение обладает лишь
незначительной активностью, то у
пациенток развивается нерезко выраженный
вирилизм; а у пациентов, из-за дефицита
активных андрогенов надпочечников и
тестикул, могут быть черты наружного
гермафродитизма, что выражается в
неполной маскулинизации гениталий.
Отмечаются врождённые крипторхизм
и гипоспадия.
- Дефицит 21-гидроксилазы приводит к
нарушению перехода прогестерона в
дезоксикортизол и 17а-прогестерона — в
11-дезоксикортикостерон. Болезнь
составляет до 95 % всех случаев
врожденной гиперплазии коры надпочечников
и известна в нескольких аллельных
вариантах, связанных с мутациями
гена CYP21 в хромосоме 6. В избытке
появляются основные андрогены
надпочечников, что ведет, даже при неполном
блоке, к ярко выраженному вирилизму
у женщин и pubertatio praecox у детей, у
мужчин увеличиваются оволосение тела
и размеры полового члена, возрастает
половое влечение, формируется макро-
генитосомия. При врожденном дефиците
данного фермента у девочек отмечается
псевдогермафродитизм из-за
маскулинного вида наружных гениталий.
- Дефицит 11-$-гидроксилазы приводит
к наиболее проксимальному блоку
стероидогенеза — на этапе между 11-
дезоксикортизолом (субстанция «S»
Рейхштейна) и кортизолом и между
11-дезоксикортикостероном и корти-
костероном. Результатом, среди других
симптомов, также служит вирилизация у
пациенток и pubertatio praecox у детей.
Так как истинный и ложный
гермафродитизм может иметь множество ненадпо-
чечниковых и неэндокринных причин, все
подобные случаи являются поводом для
определения хромосомного пола (см. т. I,
стр. 65-71) и других дифференциально-
диагностических иследований (см. ниже
«Патофизиология гонадотропной функции
и гонад»).
Патофизиология гипокортицизма
Гипокортицизм, то есть недостаточная
продукция и/или эффективность гормонов
коры надпочечников, известен в тотальной
и парциальных формах.
Тотальный гипокортицизм может быть
первичным (связанным с разрушением или
дисфункцией собственно коры
надпочечников) и вторичным (вызванным
последствиями хронического введения экзогенных
кортикостероидов, а также гипоталамиче-
скими и гипофизарными опухолями,
нарушающими продукцию кортиколиберина и
АКТГ). Первичный гипокортицизм намного
более распространён.
В основе первичного гипокортицизма
лежит первичная хроническая
недостаточность функций коркового вещества
надпочечников, в явной форме наблюдаемая
примерно с частотой 50 случаев на 1 млн
населения.
Это нарушение описано впервые
выдающимся клиницистом Т. Аддисоном (1855)
как бронзовая болезнь (по характерной
гиперпигментации, которая может
присутствовать у больных) и носит в настоящее
время наименование болезнь Аддисона.
Всего через полтора с небольшим десятилетия
клинику и развитие аддисоновой болезни
ярко описал в рассказе «Живые мощи»
классик русской литературы И.С.
Тургенев, который врачом, как известно, не был.
Однако, проявления данного недуга весьма
характерны и не ускользнули от внимания
профессионально наблюдательного
писателя.
В настоящее время известно, что
этиология болезни Аддисона чаще всего (в
80 % случаев) является аутоаллергической.
Деструкция коры надпочечников
происходит в результате избыточно интенсивного
аутоиммунного процесса, направленного
против антигенов адренокортикоцитов или
общих аутоантигенов коры надпочечников
и гонад (антигены стероидопродуциру-
ющих клеток). Наиболее часто мишенью
аутоиммунитета при болезни Аддисона
служат стероидогенные ферменты. В
первую очередь характерны аутоантитела к
21-стероидгидроксилазе (присутствуют у
60 % больных). Эти иммуноглобулины не
только блокируют фермент и вызывают
нарушение перехода прогестерона в дезок-
сикортикостерон. Они служат эффекторами
цитотоксического процесса, разрушающего
адренокортикоциты. В надпочечниках
обнаруживается иммунопатологическое
воспаление — аутоиммунный адреналит.
В надпочечниках, наряду с деструктивными
изменениями и фиброзом, имеется лимфо-
цитарная инфильтрация коры.
Нередко, особенно при раннем начале
болезни, обнаруживаются аутоантитела к
17-а-гидроксилазе. Так как данный
фермент присутствует в изобилии и в гонадах,
у больных регистрируются
взаимодействия иммуноглобулинов с цитоплазмой
стероид-продуцирующих клеток гонад и
надпочечников, и даже может быть гипер-
гонадотропный гипогонадизм, в дополнение
к клинике болезни Аддисона (К. Крон и
соавт., 1992).
Описаны и аутоантитела к рецепторам
АКТГ как причина болезни Аддисона
(Дж.Финк, Г. Билл; 1982). В этих случаях
иммуноглобулины взаимодействуют не
столько с цитоплазмой,сколько с
плазматическими мембранами клеток коры
надпочечников. Отмечается прекращение
пролиферации и атрофия клеток коры
надпочечников, но без признаков адреналита.
Имеется модель болезни Аддисона,
полученная индукцией у крыс аутоиммунного
адреналита введением соответствующей
специфической цитотоксической
антисыворотки (Т. Ирино, А. Гроллмен, 1968).
Болезнь Аддисона встречается
семикратно чаще у носителей гена ГКГС DR3,
который сцеплен и с другими аутоиммунопати-
ями (см. т. I, с. 98-104, 391-396, 465-467).
Отмечается также ее учащение при гаплоти-
пе ГКГС, содержащем аллель В8.
Не удивительно, что чаще всего её
проявления входят в структуру комбинированных
Нарушения эндокринной регуляции 593
аутоиммунных синдромов, поражающих и
другие эндокринные железы, а также
неэндокринные клетки.
Наиболее известен синдром Шмидта
(сочетание аддисоновой болезни,
аутоиммунного тироидита, инсулинзависимого
сахарного диабета). Это состояние
характеризуется более поздним началом
симптомов, сцеплением с гаплотипами ГКГС В8 и
DR3. Распространена и такая комбинация:
болезнь Аддисона, гипопаратироз,
хронический кожно-мышечный кандидоз. В этом
случае начало болезни раннее, имеется ауто-
сомно-рецессивное семейное наследование,
сцепления с упомянутыми аллелями ГКГС
не обнаружено. В общей сложности, до 80 %
больных с аддисоновой болезнью имеют
субклинические и явные аутоиммунные
тиропатии, 12-15 % — аутоиммунный
сахарный диабет, не менее 6 % — аутоиммунный
гипопаратироз (при частоте аутоантител к
паращитовидным железам до 26%). Не
менее четверти женщин, больных аддисоновой
болезнью, (те, у кого отмечены
аутоантитела к стероид-продуцирующим клеткам)
развивают аутоиммунный оофорит, а 5 %
мужчин — орхит, приводящие к первичному
гипергонадотропномугипогонадизму.
Кроме того, почти в половине случаев
присутствуют аутоантитела к клеткам
слизистой желудка, в 9 % мишенями являются
гастриновый рецептор или гастро-мукопро-
теин, что ведет к атрофическому гастриту и
аутоиммунной мегалобластической перни-
циозной анемии (анемия Аддисона-Бир-
мера). Другие аутоиммунные стигмы при
аддисоновой болезни включают частое
развитие витилиго, алопеции, хронического
активного гепатита, мальабсорбционных
синдромов — с аутоантителами
соответствующей специфичности.
Вторая по значению причина болезни
Аддисона (и первая, описанная самим Т. Ад-
дисоном) — инфекционные адреналиты,
в первую очередь — при туберкулёзе коры
надпочечников. Данная причина
ответственна примерно за 20 % случаев болезни.
Туберкулёзные бактерии попадают в кору
594 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
надпочечников при гематогенной диссе-
минации лёгочного, почечного или гаст-
роинтестинального туберкулёза. Многие
отечественные фтизиатры подчёркивают
значение так называемых «безлокальных
форм туберкулёза» — то есть ранней и
хронической туберкулёзной интоксикации,
которые в российской классификации
туберкулёза стоят отдельными позициями и
засчитываются в национальную статистику
по туберкулёзу. За рубежом эти формы в
классификацию и статистику не входят, так
как при них нет локального туберкулёзного
очага ни в одном органе (что, между прочим
даёт существенное кажущееся улучшение
эпидемиологических показателей по этим
странам). При туберкулёзной интоксикации
нет туберкулёзного адреналита. Однако
симптомы надпочечниковой
недостаточности столь очевидны, что
дифференцирование этого состояния с болезнью Аддисона
нетуберкулёзной этиологии является одной
из важных задач терапевта и фтизиатра
(О.И. Король и соавт., 1997). Считается,
что это связано со способностью
циркулирующих на ранних стадиях туберкулёза
цитокинов угнетать продукцию кортикос-
тероидов, однако эти аспекты патогенеза
туберкулёзной интоксикации пока изучены
недостаточно.
Туберкулёзный адреналит протекает
хронически и заканчивается казеозным
некрозом, а часто — и кальцификацией как
коркового, так и мозгового вещества надпочечников.
Кроме туберкулёза, в редких случаях
хронический деструктивный адреналит
вызывают другие инфекции, в первую
очередь — гистоплазмоз, кокцидиоз и другие
гранулёматозы (в том числе саркоидоз, для
которого инфекционный возбудитель не
установлен). У больных ВИЧ-инфекцией
и при других иммунодефицитных
состояниях типичны адреналиты, вызванные
малопатогенной для обычного человека
оппортунистической микрофлорой. Чаще
всего этиологическим агентом являются
цитомегаловирус (возможно
внутриутробное поражение надпочечников плода),
криптококки, Mycobacterim avium.
Поражения этими инфекциями могут быть
менее распространенными, щадя 30-50 %
ткани надпочечников или же (как при
многих случаях цитомегаловирусной
инфекции) ограничиваются мозговым
веществом.
Хронические инфекционные поражения
надпочечников вызывают развитие картины
аддисоновой болезни.
Однако, надо учитывать, что сепсис
любой этиологии (у детей — особенно часто
при молниеносной форме менингококковой
инфекции и генерализованной синегнойной
инфекции) провоцирует диссеминиро-
ванную внутрисосудистую коагуляцию и
тромбообразование, следствием чего может
быть острый тромбоз надпочечниковых
сосудов и апоплексия надпочечников. Это
ведет к двустороннему некрозу и острой
недостаточности коркового и мозгового
вещества надпочечников (синдром Уотерха-
уза-Фридериксена). У выживших больных
возможны кальцификация, фиброз
надпочечников и развитие симптомокомплекса
хронического гипокортицизма.
Инфаркт и апоплексия надпочечников
возможны и без инфекций, при ДВС-син-
дроме неинфекционной этиологии,
например, травмах и акушерских кровотечениях.
Редкие и казуистические формы
тотального гипокортицизма вызываются амилои-
дозом, гемохроматозом, облучением и
удалением надпочечных желез, метастатическими
негормонообразующими карциномами
(лёгких, грудной железы) и лимфомами
надпочечников.
Многие лекарства вызывают гипокорти-
цизм, блокируя ферменты стероидогенеза
(метирапон, аминоглутетимид, трилостан,
кетоконазол). Последний, как и некоторые
другие противогрибковые средства,
обладает антистероидогенной активностью для
надпочечников и гонад и применяется при
ВИЧ-инфекции против микозов, что
усиливает опасность ятрогенного гипокортицизма,
так как СПИД-ассоциированные инфекции
Нарушения эндокринной регуляции 595
и сами по себе приводят к адреналиту (см.
выше). Лекарство, применяемое при корти-
костеромах, — митотан — при передозировке
может вызвать гипокортицизм как цитоток-
сичное для адренокортикоцитов. Функция
надпочечников понижается при действии
рифампицина, опиатов, дилантина.
Наконец, имеются наследственные и
врожденные состояния с гипокортициз-
мом — врожденная гиперплазия коры
надпочечников, вызванная ферментативными
дефектами стероидогенеза, при которой
гипокортицизм имеет
мозаично-парциальный характер; а также врожденная
гипоплазия надпочечников, известная в двух
формах — анэнцефальной (когда недоразвит
и головной мозг), а также мегалоцитарной
наследственной, когда недоразвиты
только надпочечники, и немногие имеющиеся
адренокортикоциты имеют вид гигантских
клеток. Интерес представляет
наследственный аутосомно-рецессивный дефект
фермента десмолазы, осуществляющей
отщепление боковой цепи холестерина,
превращающее его в 85-прегненолон. При
этом страдает выработка всех стероидных
гормонов надпочечников, в адренокорти-
коцитах накапливаются липидные капли, и
развивается тотальный гипокортицизм. У
человека описан наследственный псевдоги-
покортицизм (синдром Виндерхёдса-Крусо-
са), связанный с пониженной
чувствительностью тканевых рецепторов к кортизолу.
Синдром характеризуется высоким уровнем
общего, свободного кортизола крови, АКТГ
в крови и дериватов кортизола в моче при
клинике умеренного гипокортицизма или
же без клинических признаков. Могут быть
умеренная артериальная гипертензия и
гипокалиемический алкалоз, так как АКТГ
повышает уровень дезоксикортикостерона.
Полная резистентность тканей к кортизолу
летальна (В. Шрейбер, 1987).
Патогенез основных проявлений болезни
Аддисона связан с дефицитом различных
кортикостероидов и избытком проопиоме-
ланокортина и его дериватов.
В течение длительного периода болезнь
находится в латентной фазе, когда
уровень кортикостероидов достаточен для
спокойного функционирования в обычной
обстановке, но истощенные ресурсы коры
надпочечников не позволяют адекватно
стимулировать их выработку при стрессе и
увеличении потребности в этих гормонах.
В латентную фазу состояние больных
компенсированное, но травма, операция или
сильные переживания могут вызвать криз
надпочечниковой недостаточности. Для
появления очевидных симптомов болезни
необходимо разрушение или
нефункциональное состояние не менее чем 90 % общей
массы адренокортикоцитов.
Если этот порог перейдён, то появляются
симптомы гипоальдостеронизма (прежде
всего потеря натрия и хлоридов,
обезвоживание, гиперкалиемия), а также глюкокортико-
идов (гипогликемия, слабость утомляемость,
сонливость, анорексия). Компенсаторно
повышается продукция АКТГ и его предшес-
твеника прополигормона ПОМК. Это ведёт
к гиперпигментации кожи и слизистых за
счет МСГ-подобного действия данных
регуляторов. Гиперпигментация выражатся не
только в диффузном потемнении покровов
тела. Обнаруживаются пигментные пятна и
сгущения оттенка в складках кожи, на местах
действия солнечных лучей. Темнеют соски,
ареолярные кружки, локти, колени,
ногтевые ложа, перианальная и перивагинальная
области. Характерны пигментные пятна на
слизистой щек, нёбе, дёснах.
Углеводный обмен при аддисоновой
болезни характеризуется тенденцией к быстрому
и стойкому понижению уровня глюкозы в
крови натощак. У детей возможны
спонтанные гипогликемии даже при своевременном
приеме пищи. Характерна плоская глике-
мическая кривая, высокий дыхательный
коэффициент натощак. В печени и мышцах
резко уменьшено количество гликогена.
Это вносит вклад в патогенез мышечной
слабости и адинамии, характерных для
данного заболевания. Гипогликемия при
аддисонизме может вызвать кому и быть,
смертельной.
596 A.UJ. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Нарушения белкового обмена
определяются дефицитом глюкокортикоидов, что
ведёт к снижению синтеза печёночных
протеинов и гипопротеинемии, а также
нехваткой андрогенов, тормозящей
анаболические процессы в мышцах. Мышечная
масса уменьшается. Вместе с тем снимается
катаболическое действие глюкокортикоидов
на обмен белка во многих других тканях
и органах, например, в лимфоидных. Для
аддисонизма характерны эозинофилия и
лимфоцитоз, так как срок жизни этих клеток
крови при гипокортицизме увеличивается.
Ограничение распада белка и нуклеиновых
кислот во многих тканях может привести к
тому, что выведение азота уменьшается.
Однако из-за нарушений водно-минерального
метаболизма, ограничивающих скорость
фильтрации (см. ниже), в крови может
быть некоторая ретенционная преренальная
азотемия. *
Нарушается и липидный обмен: при адди-
соновой болезни больные сильно худеют,
что связано не только с обезвоживанием
и потерей мышечной массы, но и с
уменьшением жировых запасов. В условиях
гипогликемии, под влиянием изменения глюка-
гон-инсулинового соотношения в пользу
глюкагона, в жировой ткани ослабевает
липогенез, и усиливается липолиз. К тому
же сильно понижен аппетит.
Водно-солевой обмен при первичной
болезни Аддисона изменен по типу гипоаль-
достеронизма. Многие больные испытывают
неодолимую тягу к потреблению поваренной
соли, фактически все они соль-зависимы.
При исключении потребления соли
наступает смерть. Имеется гиповолемия, снижается
скорость клубочковой фильтрации. При
водной нагрузке у больных усугубляется
гипонатриемия, и их почки оказываются не
в состоянии должным образом экскрети-
ровать воду. К тому же имеется вызванная
гипокортицизмом гипервазопрессинемия.
Таким образом, пациенты легко впадают во
внутриклеточную гипергидратацию с
набуханием клеток и «водным отравлением».
Нехватка хлоридов и натрия обусловливает
частые рвоты. Имеется выраженная гипер-
кальциемия.
Гипоосмоляльность плазмы и
внутриклеточная гипергидратация при болезни
Аддисона могут провоцироваться инфузиями
глюкозы. Не один больной с аддисоновой
болезнью был потерян при попытке
коррекции гипогликемии подобной инфузией, без
учёта необходимости повысить тоничность
внеклеточной жидкости. Глюкоза
захватывается клетками, освобождается вода,
которая дополнительно разводит и без того гипо-
натриемичную плазму. Следует набухание
клеток гипоталамуса, их неспецифическое
раздражение, активация термостатического
центра, лихорадка, коллапс и смерть.
Кислотно-основное равновесие в случае
болезни Аддисона сдвинуто в кислую
сторону. Ацидоз носит умеренный характер.
Уровень бикарбоната в плазме повышен.
Важные проявления болезни Аддисона
связаны с гипотензией, которая характерна
для 90% пациентов. Кровеносные сосуды
уменьшают свою чувствительность к кате-
холаминам, возможно и прямое
уменьшение продукции катехоламинов в мозговом
веществе надпочечников. Течение болезни
характеризуется частыми ортостатически-
ми коллапсами и приступами пониженного
кровяного давления. Повышается
проницаемость капилляров, происходит расширение
микрососудов.
Сердечно-сосудистая система в
условиях гиперкалиемии страдает от аритмий.
Характерна экстрасистромия. Гипотензия
и аритмии могут привести к смертельному
шоку в стрессорных ситуациях.
Выше уже шла речь о лимфоцитозе и эо-
зинофилии в крови при болезни Аддисона.
Картина дополняется гипорегенераторной
анемией в связи с отсутствием андрогенов
и глюкокортикоидов, стимулирующих
эритропоэз. Отмечаются нейтропения и
моноцитоз. Вместе с тем существенного
понижения противоинфекционного
иммунитета не бывает.
Гипокортицизм меняет функции ЦНС и
поведение больных. Характерны эмоцио-
Нарушения эндокринной регуляции 597
нальная лабильность, раздражительность,
отсутствие способности к длительной
концентрации внимания, часто
доминирует апатия. Многие пациенты необычайно
чувствительны к резким запахам, звукам,
вкусовым раздражителям. Если при ги-
перкортицизме ритм ЭЭГ учащен, то при
аддисоновой болезни появляются волны,
более медленные, чем а-ритм. Психическое
состояние при аддисоновой болезни ярко
описал один из пациентов французского
клинициста Ж. Де ля Фуа (1887): «Я
совершенно разбит, силы оставляют меня, я не
способен ни на малейшее усилие...». Всякое
движение внушало этому пациенту такой
страх, что он ложился, чтобы не производить
никаких усилий.
Механизмы и проявления вторичной
недостаточности коркового вещества
надпочечников несколько отличаются от
классической болезни Аддисона, и иногда такие
состояния называют «аддисонизмом».
Этиология вторичного аддисонизма
иногда связана с разрушением кортиколипотро-
фов аденогипофиза и/или кортиколибери-
новых нейронов гипоталамуса опухолями
и аутоиммунными процессами. Примером
может служить вторичный гипокортицизм
при синдроме Шихена (см. выше).
Но самой частой причиной является
экзогенная — симптомы гипокортицизма
становятся результатом быстрой отмены
хронической глюкокортикоидной терапии.
Уже применение кортикостероидов на
протяжении 6-7 дней ведет к
значительному угнетению продукции собственного
АКТГ. При многонедельном лечении доктор
вынужден учитывать это и, в случае
ожидания сильного стресса, например,
операции — повышать больному дозу экзогенных
кортикостероидов.
Для нормализации реактивности гипо-
таламо-гипофизарного нейросекреторного
аппарата после такой терапии требуется
не менее 2-3 месяцев, а к норме
продукция кортизола не возвращается в течение
6-9 месяцев. Поэтому дозы отменяемых
кортикостероидов снижают постепенно.
При быстром снижении симптомы
гипокортицизма и даже аддисонического криза
(см. ниже) проявляются и при нормальной
концентрации глюкокортикоидов в крови,
так как ткани адаптированы к их хронически
высокому уровню (А. Голдфин, 1998).
В развитии вторичного гипокортицизма
сменяются три стадии. На первой базальная
продукция и уровень кортизола нормальны,
но ответ на стресс резко понижен, так как
мала центральная стимуляция кортикосте-
роидогенеза. На второй секреция АКТГ и
КРФ еще более угнетаются, начинают
проявляться симптомы снижения базального
уровня кортизола, вне стресса. Наконец,
отсутствие АКТГ постепенно ведет к
вторичной атрофии коры надпочечников. Это
способствует переходу в состояние глубокой
базальной и стимулированной гипокорти-
золемии.
При вторичных формах гипокортицизма
содержание АКТГ понижено или нулевое.
Поэтому комплекс эффектов, связанных
с избытком ПОМК отсутствует, а
главное — нет гиперпигментации покровов тела.
Наоборот, кожа может светлеть («белый
аддисонизм»). Вторичная надпочечниковая
недостаточность часто ограничивается в
основном гипоглюкокортикоидными
проявлениями. Выраженный гипоальдостеронизм
и гипоандрогенизм при ней более редок.
Это облегчает состояние больных — гипо-
волемия и гиперкалиемия бывают нечасто.
Наблюдается гипонатриемия из-за
неспособности к эффективной экскреции воды,
но она не бывает столь глубокой. Менее
выражены сдвиги кислотно-щелочного
баланса. Не характерны выраженные
азотемия и креатининемия, которые типичны
при первичной болезни Аддисона. Однако
гипогликемия может быть ярко выражена,
так как первичное поражение состоит в
гипопитуитаризме.
Наивысшим проявлением
гипокортицизма (как первичного, так и вторичного)
является острый аддисонический (аддисо-
нов) криз. Это экстремальное состояние,
вызванное резким несоответствием между
598 A.LU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
потребностью в кортикостероидах и их
поставкой, провоцируется стрессорами. Так
как хроническая надпочечниковая
недостаточность имеет длительную латентную фазу,
аддисонов криз может быть дебютным
проявлением заболевания у больных, которые
до этого не были своевременно
диагностированы — например, в ответ на травмы или
медицинские вмешательства, проведённые
без учёта особенностей их надпочечниковой
функции. Первично острое течение
надпочечниковой недостаточности при
септическом синдроме Уотерхауза-Фридериксена,
апоплексии гипофиза, кровоизлиянии в
надпочечники — также даёт картину
подобного криза, отличающуюся отсутствием
предвестников.
Криз характеризуется быстрым
развитием глубоких гипонатриемии, внеклеточного
обезвоживания, гиповолемии, гиперкали-
емии, гиперкальциемии — на фоне
прогрессирующей гипогликемии. В то же время
клетки мозга и других органов набухают.
Возникают симптомы водного отравления.
Аддисонов криз проявляется высокой
лихорадкой, болями в суставах и мышцах,
прострацией, тошнотой, рвотой и поносом,
усугубляющими потери натрия и хлоридов.
Растяжение капсул абдоминальных органов
провоцирует такие боли, что наблюдается
картина «острого живота», и больной может
ошибочно трактоваться, как хирургический.
Кома, аритмия и гипотензия, резистентная
к катехоламинам, могут привести к
смертельному нарушению кровообращения и
дыхания. Картина комы, при наличии
выраженных желудочно-кишечных проявлений,
трудно отличима в финальных стадиях от
одного из вариантов кетоацидотической
комы, что может вызвать попытку лечения
инсулином и смерть (см. «Патофизиология
сахарного диабета»).
Синдром Уотерхауза-Фридериксена
представляет собой острую недостаточность
функций коркового и мозгового вещества
надпочечных желёз из-за их апоплексии.
Наиболее часто он развивается в ходе
диссеминированного внутрисосудистого
свёртывания-тромбообразования.
Причиной тромбозов, инфарктов и апоплексии
надпочечников служит активация тромбо-
генных свойств их эндотелия
циркулирующими цитокинами, при системном
действии медиаторов воспаления, характерном
для сепсиса и токсико-септического шока.
Большое значение в генезе синдрома имеют
генерализованные инфекции с
бактериемией и вирусемией. Наиболее часто синдром
осложняет менингококковый сепсис.
Отмечены его случаи при стрептококковой
и пневмококковой инфекции поражении
Haemophilus influenzae и даже в дебюте
тяжёлого полиомиелита. Механизм
поражения имеет много общего с феноменом Сана-
релли-Шварцмана. Эндотоксины бактерий
вызывают экспрессию дополнительных
рецепторов цитокинов и молекул клеточной
адгезии на поверхности эндотелия сосудов
ряда органов, в том числе — надпочечников.
При волнах бактериемии провоцируется
активация сторожевой полисистемы плазмы
и региональный или системный ДВС-синд-
ром (см. т. I, стр. 235).
В результате надпочечники могут
превращаться в буквальном смысле в заполненные
свернувшейся кровью мешочки.
Синдром встречается в любом возрасте,
хотя именно у детей является самой частой
причиной острой надпочечниковой
недостаточности. Неифекционные факторы,
способствующие синдрому — травма (в
том числе, родовая), тяжелые стрессы при
хирургических вмешательствах, ранениях,
ожоговой болезни, родах.
Проявления синдрома близки к тяжёлому
аддисоническому кризу, гипогликемия и
гипотензия обыкновенно бывают сильно
выражены. На эту картину наслаиваются
проявления тромбо-геморрагического
синдрома (например, петехии) и основного
заболевания (например, менингеальные
явления). Основные проявления синдрома
Уотерхауза-Фридериксена отражены на
рис. 115а.
Наряду с тотальной формой гипокор-
тицизма известны парциальные случаи
недостаточности продукции тех или иных
Коллапс,
выраженная
гипотензия
Менингококцемия,
менингококки в
цереброспинальной
жидкости или
мазке из зева
Характерная
температурная кривая
39'С
зб-с
Звездчатая
восходящая
геморрагическая
сыпь, цианоз,
прострация
Апоплексия
надпочечников
2 Дни
Рис 115а. Синдром Уотерхауза-Фридерикса (по Ф. Неттеру, CIBA Medical Illustrations Collection)
600 А.Ш. Зайчик, Л.П.Чурилов Патохимия
адренокортикостероидов. Часто они
мозаично сочетаются с гиперпродукцией других
кортикоидов. Подобные комбинированные
состояния нередко являются результатом
энзиматических блоков при врождённой
гиперплазии коры надпочечников, что
рассматривается ниже отдельно.
Однако возможны и изолированные
нарушения продукции отдельных классов
адренокортикальных гормонов, без
усиления синтеза других.
Первичный изолированный гипоальдо-
стеронизм может быть результатом
наследственного дефекта биосинтеза альдостерона
на этапе 18-гидроксилазыи 18-дегидрогена-
зы. Данная форма, в отличие от вариантов
врождённой гиперплазии, не
сопровождается адреногенитальным синдромом и
имеет тенденцию к спонтанной ремиссии.
Приобретенные изолированные нарушения
биосинтеза альдостерона могут возникать в
любом возрасте и быть следствием действия
блокирующих аутоантител к 18-дегидроге-
назе. Такой гипоальдостеронизм сочетается
с другими аутоиммунными эндокринопа-
тиями. Дефект биосинтеза альдостерона
вызывают некоторые препараты. При всех
формах первичного гипоальдостеронизма
уровень ренина в крови — высокий.
Вторичный гипоальдостеронизм
развивается от недостаточной функции ренин-анги-
отензиновой системы. При этом варианте
синдрома уровень ренина в крови низкий.
Существует несколько характерных
состояний, приводящих к гипоренинемическому
вторичному гипоальдостеронизму.
Почечный канальцевый ацидоз IV типа
(см. также раздел «Патофизиология
почек» в т. III и гл. 10 выше) поражает
в основном пожилых мужчин с
предшествующими подагрой, сахарным
диабетом, хроническим пиелонефритом. Он
является следствием прогрессирующего
нефросклероза, охватывающего юкста-
гломерулярный аппарат почек.
Продукция ренина понижается, страдает переход
проренина в ренин. Ренинпродуцирующие
клетки не отвечают на стимуляторы,
например, на АКТГ.
чдокринно-метаболические нарушения)
Послеоперационный гипоренинемичес-
кий гипоальдостеронизм развивается после
удаления альдостеромы.
Гипоальдостеронизм может быть ятро-
генным, в результате передозировки и
отмены экзогенных минералокортикоидов
(например, флудрокортизона) и лакрицы
(см. выше). В этих случаях он
развертывается после отмены лечения, в связи с
подавлением ренинообразования в предыдущую,
гиперальдостероновую фазу.
Проявления гипоальдостеронизма
включают гиперкалиемию, метаболический
ацидоз, гипотензию, гипонатриемию и
гиповолемию. В тяжелых случаях бывают
обмороки, брадикардия вплоть до блокад
и припадков Морганьи-Эдамса-Стокса с
судорогами и помрачением сознания.
Псевдогипоальдостеррнизмом
называется синдром резистентности почек и
других мишеней альдостерона к действию
минералокортикоидных гормонов. Это
обусловлено дефектом
минералокортикоидных рецепторов. На фоне проявлений
кажущегося гипоальдостеронизма уровень
альдостерона и ренина высок. При псевдо-
гипоальдостеронизме затруднено лечение
минералокортикоидами, и приходится
корректировать состояние больных введением
хлорида натрия.
Комбинированные проявления
парциального гипо-и
гиперкортицизма
Врождённая гиперплазия коры
надпочечников (или врождённый адреногенитальный
синдром, в дальнейшем — ВДКН) из-за
мозаичного и разнообразного сочетания
проявлений гипо- и гиперкортицизма
нуждается в отдельном описании.
Впервые данный синдром описан в 1865 г.
Л.де Кречио, но его наследственный
характер распознали лишь в середине
следующего столетия. Идея о ферментативном
блоке стероидогенеза как основе данного
расстройства принадлежит Ф. Барттеру и
соавторам (1951).
Существует целое семейство генных
мутаций, блокирующих те или иные этапы
стероидогенеза и вызывающих варианты
данного синдрома.
К ним относятся: дефекты 21-гидроксила-
зы, 1 ip-гидроксилазы, Зр-ол-дегидрогеназы,
17-гидроксилазы, 17-редуктазы, 5а-редукта-
зы, 20,22-десмолазы и 17,20-десмолазы. На
схеме (рис. 112, выше) показаны участки
путей стероидогенеза. где действуют эти
ферменты и локализуются данные блоки.
От 90 до 95% случаев ВДКН связаны с
различными мутациями, снижающими
активность 21 -гидроксилазы. Так как ген этого
фермента CYP21 в хромосоме 6 находится
по соседству с генами ГКГС, у заболевания
отмечается неравновесное сцепление с
некоторыми гаплотипами ГКГС (В5, В14> Bw47,
BW5i> Bw60, DR1).
В мире средняя частота классической 21-
гидроксилазной недостаточности составляет
от 1/5000 до 1/20000. Но у эскимосов она
очень велика — до 1/490 новорождённых.
Частота гетерозиготного носительства ауто-
сомно-рецессивных дефектов
21-гидроксилазы повсеместно весьма значительна — от
1/35 до 1/76, а у эскимосов — является
практически сплошной (Р.И. Четковски,
и соавт., 1984). Ряд форм ВДКН часто
поражает евреев — неклассический вариант
21-гидроксилазного дефекта встречается
у ашкенази с частотой до 3%, что в 10 раз
выше среднеевропейских цифр, дефицит
11-р-гидроксилазы нередко встречается у
марокканских евреев.
При различных мутациях гена CYP21 (де-
леция, инверсии) могут возникать разные
клинические варианты 21-гидроксилазной
недостаточности, известные как
классическая форма (в 75 % случаев — вирилизующая
и соль-теряющая, в 25 % случаев — только
вирилизующая), а также неклассическая
форма (отличающаяся менее тяжёлыми
проявлениями, в частности, поздним началом
вирильного синдрома).
Классическая недостаточность
21-гидроксилазы ведет к полному блоку превращений
17а-оксипрогестерона в 11-дезоксикортизол
и прогестерона — в 11-дезоксикортизол.
Избыток этих метаболитов превращается в
андростендион, что ведет к повышенной вы-
Нарушения эндокринной регуляции 601
работке надпочечниковых андрогенов. Уже
внутриутробно присутствует гиперандроге-
ния. В то же время эффективность синтеза
минералокортикоидов и глюкокортикоидов
страдает, что ведет к усилению функции
кортиколипотрофов аденогипофиза и
нарастанию уровня АКТГ, стимулирующего рост
коры надпочечников и андростероидогенез.
При выраженном дефиците альдостерона
гипертрофируется юкстагломерулярный
аппарат почек. Кора надпочечников резко
гиперплазирована за счет клубочковой
и сетчатой зон. Бывают микроузловая и
диффузная формы гиперплазии. Вид
органа напоминает кору больших полушарий
головного мозга. Вес одного органа может
достигать 60 г! Вадренокортикоцитах
изобилуют липидные включения и
присутствует липофусцин. В тестикулах мужчин
могут быть гиперплазия и даже опухоли,
а яичники у женщин проявляют признаки
вторичной гипотрофии из-за высокого
уровня андрогенов.
Клинически проявляются 2 синдрома —
гиперандрогенизм и гипокортицизм, причем
последний — преимущественно в форме
первичного гипоальдостеронизма.
С рождения отмечаются признаки
псевдогермафродитизма. У девочек вирилизуется
вид наружных гениталий (гипертрофия
клитора, смыкание половых губ по типу
мошонки, формирование урогенитально-
го синуса, напоминающего фаллоидную
уретру). Мальчики при рождении имеют
нормальные гениталии. Так как надпочеч-
никовые андрогены относятся к слабым и
при избытке конкурируют с тестостероном
за рецепторы, иногда и у мальчиков также
могут быть аномалии строения наружных
половых органов — гипоспадия и двусто-
ронный крипторхизм. В дальнейшем следует
pubertatio praecox, по гетеросексуальному
(у девочек) или изосексуальному (у
мальчиков) типам.
Неклассический вариант расстройства
приводит к рождению детей с внешне
нормальными половыми признаками, но
происходит постнатальная вирилизация и
гетеросексуальное преждевременное половое
602 ALU. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
созревание у девочек, а у мальчиков —
развивается преждевременное изосексуальное
половое созревание: рост ускорен, и рано
заканчивается окостенением метаэпифи-
зарных хрящей, костный возраст опережает
паспортный, отмечаются макрогенитосомия
и гипертрихоз.
От 65 до 75 % лиц с классическим
дефицитом 21-гидроксилазы страдают явным
врожденным гипокортицизмом-гипоаль-
достеронизмом. Отмечается врождённая
гиперпигментация кожи, со 2-5-й недели
внеутробной жизни характерен сольте-
ряющий синдром — слабость, полиурия,
гипотония мышц, гипотензия, потеря
веса, гиперкалиемия с характерной ЭКГ,
гипонатриемия, гипохлоремия, ацидоз,
срыгивания и фонтанирующая рвота,
позднее — тяга к солёной пище. При сольтеря-
ющей форме смертность на 1-2 году жизни
велика и приближается к 40 % (Е.В.
Плотникова, 1989).
У остальных гипокортицизм носит
скрытый характер и компенсируется гиперрени-
немией и гиперпродукцией АКТГ.
Стёртые и лёгкие формы ВДКН
служат основной причиной гирсутизма и
адренархе у девушек. Различные авторы,
обследуя пациенток с гирсутизмом,
выявили лабораторные признаки ВДКН у
значительных групп — от 1,5 до 30 % от всех
случаев гирсутизма (Р.И. Четковски, 1970)!
Очевидно, что гирсутизм — достаточный
повод для углублённого обследования на
наличие 21-гидроксилазной формы ВДКН
путем измерения сывороточного базаль-
ного и АКТГ-стимулированного уровня
17-гидроксипрогестерона. В отличие от
андростером, при ВДКН гиперпродукция
андрогенов чувствительна к подавлению де-
ксаметазоном. При введении этого стероида
понижается содержание андрогенов в крови
и экскреция их метаболитов — 17-кортико-
идов — с мочой.
Таблица 38. Редкие формы врожденной гиперплазии коры надпочечников
Дефектный
энзим
20,22-десмолаза
(сидром П ра дер-
Га ртн ера)
зр-ол-дегидро-
геназа
17-а-гидрокси-
лаза
11 Р-гидрокси-
лаза
Стероиды,
выделение
которых с
мочой растёт
Нет
Д5-Зр-стеро-
иды
Прегнандиол,
тетрагидро-
кортикостерон
Тетрагидроде-
зоксикорти-
зол, тетрагид-
родезоксикор-
тикостерон
Дефицитные
гормоны
Все стероидные
Глюкокортикоиды,
альдостерон
Андрогены,
эстрогены, кортизол,
альдостерон
Глюкокортикоиды,
альдостерон
Соединения,
появляющиеся в
избытке
Холестерин в коре
надпочечников
Дегидроизоан-
дростерон (слабый
андроген,
антагонист тестостерона)
Кортикостерон,
дезоксикортико-
стерон
Андрогены, 11-де-
зоксикортикосте-
рон
Примечания
Тотальный гипокортицизм,
гипогонадизм, врожденный
мужской
псевдогермафродитизм
Гипокортицизм и
гипогонадизм. Врожденный
наружный
псевдогермафродитизм у плодов обоего
пола
Половой инфантилизм у
женщин,
псевдогермафродитизм у мужчин; гипер-
тензия, задержка соли и
воды, гипокалиемический
алкалоз
Женский
псевдогермафродитизм, гипертензия,
задержка соли и воды,
гипокалиемический
алкалоз. Уступает по частоте
(1/100000) лишь дефектам
21-гидроксилазы
Нарушения эндокринной регуляции 603
Болезнь может быть распознана путем
антенатальной диагностики (амниоцентез,
анализ ДНК клеток биоптата ворсин
хориона). В случае пренатального диагноза
назначение дексаметазона матери
блокирует гиперпродукцию андрогенов и
позволяет избежать псевдогермафродитизма, в
дальнейшем гормонально-заместительная
терапия необходима в течение всей жизни.
Больные ВДКН входят в группу высокого
риска развития острой надпочечниковой
недостаточности.
Определённые особенностями
характеризуются остальные формы ВДКН,
затрагивающие кору надпочечников и
зависящие от других ферментативных дефектов
(табл. 38).
Некоторые разновидности нарушения
биосинтеза половых стероидов
проявляются в основном расстройством их конверсии
в периферических тканях. Они
рассматриваются ниже, в разделе, посвященном
нарушению гонадотропной и эндокринных
гонадных функций.
Патофизиология мозгового
вещества надпочечников
Основы структурно-функциональной
организации медуллярной части надпочечных
желёз уже рассматривались в т. I (с. 516,
520-521, 527-529). Вкратце напомним, что
мозговое вещество составляет около 10%
веса надпочечников, заключено в головке и
теле каждого надпочечника и отсутствует в
его хвостовой части.
Мозговое вещство надпочечников
представлено хромаффинной тканью, открытой
Н. Вюльпианом (1856), по способности
окрашиваться в зелёный цвет солями
железа. Хромаффинные клетки названы так
за своё сродство к солям хрома,
обусловленное присутствием катехоламинов. Хро-
маффинная ткань — часть функционально
единой и эмбриогенетически родственной
симпатохромаффинной системы организма,
включающей, помимо неё, симпатическую
нервную систему (Ф.Э. Крайер, 1987).
Симпатохромаффинная система участвует
в патогенезе многих заболеваний и
синдромов у человека (гипертонической болезни,
некоторых вторичных гипертензий, гипо- и
гипергликемий, аритмий, ортостатического
коллапса, гипотонической болезни).
У человека хромаффиноциты
наличествуют, кроме медуллярного слоя
надпочечников, и в других параганглиях.
Параганглии — небольшие органы, происходящие из
нервного гребня и расположенные паравер-
тебрально — от основания черепа до крестца,
а также в стенках мочевого пузыря. Они
находятся вблизи симпатических стволов
и в стенке крупных сосудов (примерами
являются крупнейшие из них — каротидные
тела, подключичные тела, яремные
клубочки и аорто-лёгочные тела). В виде отдельных
клеток хромаффиноциты лежат по ходу
вегетативных нервов и в автономных ганглиях.
У плода и детей первых 3-4 месяцев жизни
хромаффинная ткань особенно развита и
образует самые крупные парааортальные
ганглии — органы Цукеркандля,
локализованные в буром жире, близ места
ответвления нижней брыжеечной артерии.
Хромаффинная ткань мозгового вещества
организована в балки и гнездоподобные
скопления вокруг богатой сосудистой сети,
анастомозирующей с сосудами коры
надпочечников.
По этим сосудам часть артериальной
крови поступающей от ветвей почечных и
диафрагмальных артерий и аорты в
капилляры коркового вещества, затем переходит
в мозговое, но большая её доля проходит
по перфорирующим сосудам от капсулы
надпочечника — прямо в мозговое
вещество. Побывав в его синусоидах и приняв
продукты секреции мозгового вещества,
часть венозной крови может попадать
обратно, в синусоиды коркового вещества
через анастомозы. Предполагается, что это
создает дополнительные возможности для
координации продукции катехоламинов и
кортикостероидов, а градиент гормональных
начал, создаваемый в сосудах, участвует в
дифференцировке зон коры надпочечника
(П. Хорнсби, А.Кривелло, 1983).
604 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Секреторные функции хромаффинной
ткани надпочечников характеризуются
следующим. За пределами мозгового
вещества надпочечников синаптическими
передатчиками симпатических нейронов
служат норадреналин и дофамин. Мозговое
вещество надпочечников способно их
вырабатывать, особенно у плода, у которого
норадреналин — основной катехоламин
стресса (см. в т. I раздел «Онтогенетические
аспекты стресса»). Но резко
преобладающим катехоламином в секрете этого отдела
надпочечных желёз у взрослых является
адреналин (У.С.фон Эйлер, 1946). У крыс,
даже новорожденных, адреналиновые
клетки в мозговом веществе
надпочечников составляют 75 %. Это происходит из-за
большой активности в мозговом веществе
надпочечников (но не в других
параганглиях) фермента фенилэтаноламин-1^-метил-
трансферазы, превращающей норадреналин
в адреналин. Пути биосинтеза и
деградации катехоламинов в хромаффиноцитах
надпочечников представлены на рис. 116.
Ключевым ферментом при продукции
катехоламинов является тирозингидрокси-
лаза. Интересно, что обнаружены антитела
против этого энзима, значительно
активирующие его функцию (Дж. Хэйкок, Дж. Уэй-
майр, 1982). Это позволяет ставить вопрос о
возможности аутоиммунной гиперфункции
хромаффиноцитов.
Мозговое вещество надпочечников
вырабатывает и нейропептиды — нейротензин,
мет-энкефалин и р-эндорфин (Б. Ливетт
и соавт., 1981). ПОМК экспрессируется
в самих хромаффинных клетках и может
поступать сюда из гипофиза для протео-
литической обработки. В хромаффинных
клетках можно выявить также присутствие
нонапептидов, вещества Р, нейропептида Y,
динорфина, ВИП и бомбезина.
Регуляция деятельности мозгового
вещества, главным образом нервная, и
опосредована холинергическим симпатическим
сигналом. Нервный путь, активирующий
хромаффиноциты, начинается в
гипоталамусе и проходит через ретикулярную
формацию, спинной мозг, солнечное сплетение.
Секреторные ветви малого чревного нерва
иннервируют хромаффиноциты и
стимулируют их секрецию, причём доходящие
до мозгового вещества волокна являются
постганглионарными, так как само
мозговое вещество — не симпатический ганглий,
как неверно указано в некоторых старых
источниках, а именно параганглий.
Нейроны второго порядка в этом симпатическом
пути не сконцентрированы в отдельном
узле, а рассеяны, согласно направлению
ноЧ/1 н н
о
II
н с—он
I тирозин-
~ С — NH2 гидроксилаза
НО
тирозин
СНз
PNMT
сн3 ^ L II
ног^у^ п п ноГ
он он он
N-метиладреналин адреналин норадреналин
Рис. 116. Биосинтез катехоламинов в мозговом веществе надпочечников. ААД — декарбоксилаза
ароматических аминокислот, PNMT — фен ил-этанол амин-1Ч-метилтрансфераза
Нарушения эндокринной регуляции 605
их эмбриональной миграции, по всей его
протяжённости — от пограничного ствола
и до самого мозгового вещества (В.И.
Ильина, 1946). Достигающие надпочечников
парасимпатические ветви блуждающего и
диафрагмального нервов, по-видимому,
передают вазомоторные импульсы и не
являются секреторными. Гуморальные факторы
имеют для секреции катехоламинов
вспомогательное значение. Кортикостероиды
при длительном повышении их уровня в
оттекающей от коры надпочечников крови
могут повышать синтез фермента тирозин-
гидроксилазы и стимулировать тем самым
продукцию катехоламинов.
Стимулирующим действием на катехоламиногенез
обладает и пептид нейротензин,
выделяемый нервными окончаниями в мозговое
вещество.
До момента секреции адреналин хранится
в особых гранулах вместе с упаковочным
белком хромогранином и АТФ. Ацетилхо-
линовый сигнал обусловливает входной ток
кальция в цитоплазму хромаффиноцитов,
активацию сократительных белков цито-
скелета и экзоцитоз содержимого гранул
в кровь. Адреналин-продуцирующие и
норадреналин-продуцирующие клетки
различаются диаметром гранул (у последних
он меньше) и их плотностью (у последних
она выше). Отдельных дофаминоцитов в
мозговом веществе не найдено.
Во сне секреция катехоламинов
надпочечниками минимальна, содержание их в
крови невелико и при обычной
жизнедеятельности. Однако любая активация
симпатической нервной системы, особенно при
стрессе (травмы, опасность, гипогликемия,
боль, физическая нагрузка, гипертермия)
ведет к массированному катехоламиновому
секреторному ответу.
При сильном стрессе уровень
катехоламинов в крови возрастает в 4-5 раз. Родовой
стресс приводит к абсолютно максимальным
значениям концентрации катехоламинов
(см. в т. I раздел «Онтогенетические аспекты
стресса»). При максимальной стимуляции
в условиях тяжёлого стресса в крови
зарегистрированы концентрации адреналина — в
50, а норадреналина — в 20 раз выше нормы
(Ф.Хокер, 1988).
Катаболизм гормонов происходит в
основном в ходе их окисления и
метилирования, при обратном захвате хромаффин-
ными клетками, а также путем образования
парных соединений в печени. С мочой
выводится только 5 % дериватов катехоламинов.
Время полувыведения катехоламинов из
крови — 1-2 мин. Основными продуктами
распада этих гормонов вне ЦНС являются
ванилил-миндальная и гомованилиновая
кислоты.
В клетках-мишенях имеется 3 подтипа
Р-адреналовых рецепторов и 2 подтипа —
а-адренорецепторов (последние, в свою
очередь, представлены семью
разновидностями). Рассмотрение вопросов адренорецеп-
ции выходит за рамки данной книги и
приведено в современных-фармакологических
руководствах (см. Б. Катцунг, 1998). Ниже
представлены лишь данные, необходимые
для понимания контролируемых
гормонами медуллярного вещества надпочечников
эффектов. Ни единое природное вещество
не действует строго избирательно на один
вид адренорецепторов. Следует обратить
внимание на то, что адреналин повышает
работу сердца и расширяет артериолы
сердца, мозга и мышц, сужая сосуды кожи и
кишок. Норадреналин незначительно влияет
на сердце, но выраженно повышает общее
периферическое сопротивление за счет вазо-
констрикторного эффекта в большом круге.
Таким образом, оба гормона способствуют
систолической, а норадреналин — еще и
диастолической артериальной гипертензии
(В. Шрейбер, 1987).
Адреналин (он же эпинефрин) — главный
агонист р2-адренергических рецепторов,
а норадреналин влияет, в основном, на
Pj-адренорецепторы. На а-адренорецепторы
влияют оба главных катехоламина, причём
норадреналин — несколько сильнее на сц-
рецептор, а эпинефрин — на а2-рецептор.
В связи с этим их действие на органы и ткани
неидентично (см. ниже табл. 39, рис. 117, а
также табл.31 в т.1, на стр. 528). Катехол-
амины действуют на типичные G-протеин-
гипоталамус
кора
надпочечников
спинной
мозг
сн2о'
сн2о'
нор-
адреналин
артериаль- дечный основгГой
нов кровяное выб обмен
давление
метаболиты в моче (за сутки):
адреналин-
коньюгат
СН2о'
адреналин
Рис. 117. Регуляция адреномедуллярной секреции и биологические эффекты надпочечнико-
вых катехоламинов как основа проявлений феохромоцитомы (по Ф. Неттеру из CIBA Medical
llustrations Collection). Пояснения в тексте
Нарушения эндокринной регуляции 607
Таблица 39. Распределение и результаты стимуляции основных катехоламиновых рецепторов
Подтип
рецепторов
а1
a2
PI
Г
рз
pi
D2
Ткани и органы
Гладкие мышцы иннервируемых
сосудов, мочеполового тракта.
Дилататор зрачка. Пиломотроные
мышцы. Сердце, ЖКТ. Слюнные и
потовые, апокринные, аксиллярные,
ладонные и подошвенные железы
Гладкие мышцы ЖКТ. Постсина.-
птические в ЦНС. Тромбоциты.
Лимфоциты. Адренергические и
холинергические нервные
окончания. Гладкие мышцы некоторых
сосудов. Липоциты
Эндокриноциты
Сердце. Эндокриноциты.
Липоциты
Гладкие мышцы дыхательных путей,
матки, мочевого пузыря, сосудов.
Скелетные мышцы. Печень.
Эндокриноциты. Гладкие мышцы ЖКТ
Липоциты
Гладкие мышцы
Нервные окончания
Эффекты
Сокращение, вазоконстрикция (кожа). Эякуляция.
Сокращение матки и мочевого пузыря. Сокращение, дилатация,
адаптация к зрению вдаль. Пилоэрекция. Положительный
инотропный, хронотропный эффекты. Увеличение
тонуса сфинктеров, торможение перистальтики. Секреция
амилазы, пота
Расслабления различные, в зависимости от отдела.
Бодрость, тревога, страх. Агрегация. Хоуминг. Подавление
освобождения медиаторов. Сокращение,
вазоконстрикция. Подавление липолиза (в буром жире-липолиз,
калоригенез)
Подавление секреции инсулина, глюкагона, ренина,
стимуляция — соматостатина
Положительный инотропный, дромотропный,
хронотропный эффекты. Аритмогенный эффект. Снижение
продукции ренина. Липолиз
Расслабление, бронхо- и вазодилатация (в том числе —
здоровых коронаров). Усиление захвата К+ и гликогено-
лиза. Увеличение производства лактата, вазодилатация.
Стимуляция гликогенолиза, глюконеогенеза, выхода
в кровь катионов калия. Активация секреции ренина,
инсулина, глюкагона, соматостатина, эритропоэтина.
Расслабление
Стимуляция липолиза, калоригенез
Расширение сосудов почек и брыжейки, вазоконстрикция
других бассейнов
Модуляция освобождения медиаторов. В ЦНС — пролак-
тостатический эффект
ассоциированные рецепторы, в передаче их
эффектов в клетках большую роль играют
в случае р-рецепторов — аденилатциклаз-
но-протеинкиназный посредник, а в случае
a-рецепторов — фосфатидил-инозитоловый
мессенджер. В последнем варианте
активность аденилатциклазных механизмов не
стимулируется, а тормозится. Кальмодулин-
кальциевые модуляторы участвуют в
передаче сигнала от обоих типов рецепторов.
Дофамин влияет на D-рецепторы,
которых обнаружено не менее 4 подтипов
(Dj, D2, D4, D5). Эти рецепторы из того же
семейства, что и адренорецепторы.
Стимуляция Dx -рецепторов усиливает, а D2-penen-
торов — ослабляет работу аденилатциклазы
и соответствующих протеинкиназ А. Более
слабо этот катехоламин действует на р-ад-
ренорецепторы, а еще слабее — на а-адре-
норецепторы.
Регуляция выделения катехоламинов
мозгового вещества надпочечников и их
биологические эфекты как основа
проявлений патологии мозгового вещества
надпочечников представлены на рис. 117.
Спектр нарушений функций мозгового
вещества надпочечников включает прежде всего
состояния гиперфункции этого нейроэндок-
ринного органа. Среди них — гормонообразу-
ющие опухоли — феохромоцитома и другие,
приводящие к злокачественной гипертензии и
тяжелому нарушению обмена веществ.
608 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Возможна и гипофункция
медуллярного вещества надпочечников, для которой
характерны прежде всего гипотензивный
синдром и так называемая «автономная
нейропатия».
Хромаффиномы (в 90 % случаев —
доброкачественная — феохромоцитома, а в
10% — злокачественная — феохромоблас-
тома) — гормонообразующие опухоли
мозгового вещества надпочечников, других
параганглиев или симпатических ганглиев.
Нефункционирующие образования того
же генеза в параганглиях иногда называют
хемодектомами.
Хромаффиномы имеют частоту не менее
2 случаев на 1 млн населения и наиболее
типичны для лиц зрелого возраста (30-50
лет). Так как они могут оставаться
нераспознанными, некоторые авторы оценивают
их частоту намного выше — до 20 на 1 млн
населения (К.Н. Казеев, А.Н. Антонов,
1991).
Они обнаруживаются не менее, чем у
0,3-0,7 % больных с повышенным кровяным
давлением, являясь важной причиной
вторичной гипертензии. Не менее 1/10 случаев
являются семейными, аутосомно-дрминан-
тными. По образному выражению Р. Де
Лилли: «Феохромоцитома — зверюшка
общительная и любит компанию». Дело в том,
что феохромоцитома входит в структуру
пяти разных наследственных плюригланду-
лярных синдромов — по 40 % всех семейных
случаев относятся к синдромам MEN Па и
MEN lib (см. выше), а также 1 % приходится
на болезнь фон Реклингхаузена (семейный
нейрофиброматоз с феохромоцитомой),
и остальные — наблюдаются в структуре
синдрома фон Гиппеля-Линдау (частота
1/36000, аутосомно-рецессивный дефект в
хромосоме 3, проявления: гемангиобластома
ЦНС, чаще — мозжечка, феохромоцитома,
ангиомы сетчатки, карцинома почки, кисты
поджелудочной железы и почек, цистаде-
номы придатка яичка), а также синдрома
Штруге-Вебера (частота — 1/10000, имеет
спорадический характер, дефект
неизвестен, проявления: ангиоматоз мозговых
оболочек и кожи — в области иннервации
VII черепно-мозгового нерва, кальцифи-
кация участков мозга, задержка
умственного развития, судороги, хромаффиномы).
В случае MEN-синдромов этиология
опухоли связана с экспрессией мутаций
протоонкогена RET, локализованного в
хромосоме 10qll.2 и кодирующего рецептор-
ассоциированную тирозинкиназу. Мутации
различны — либо мутирует экстрацеллю-
лярный цистеинсодержащий домен белка
RET (при синдроме MEN На), либо
меняется треонин на метионин во
внутриклеточном домене того же белка (при MEN lib).
Изредка при этом наблюдается не опухоль,
а диффузная гиперплазия мозгового слоя
надпочечников (см. также выше «Общая
патология эндокринной системы»).
Остальные случаи хромаффином —
спорадические. С. Дж. МакФи (1997)
остроумно называет хромаффиному «десятичной»
опухолью, имея ввиду, что 1/10 случаев
имеет вненадпочечниковую природу, 1/10 —
расположена вне брюшной полости, в 1/10
наблюдений опухоль множественная в
1/10 — имеет двусторонний характер и, в
довершение всего, — по 10% случаев следуют
у детей, не сопровождаются гипертензией,
являются злокачественными.
Доброкачественные феохромоцитомы
невелики по размерам — весом до 100 г,
феохромобластомы могут достигать
громадной величины (более 2 кг). Тип строения
феохромоцитом ближе к ткани мозгового
вещества надпочечников и может быть трабе-
кулярным или альвеолярным, у некоторых
феохромоцитом и многих феохромобластом
он хаотический. В отличие от
феохромоцитом, феохромобластомы метастазируют в
региональные лимфоузлы, лёгкие, печень
и кости.
Большинство хромаффином секретируют
в основном норадреналин. Экстраадренало-
вые опухоли вообще адреналина не
вырабатывают. Некоторые адреналовые образуют
поровну адреналин и норадреналин. Реже
(обыкновенно, при MEN-синдромах)
встречается чисто адреналиновая секреция. Ред-
Нарушения эндокринной регуляции 609
кие злокачественные хромаффинобластомы
способны вырабатывать преимущественно,
дофамин что сопровождается экскрецией
больших количеств гомованилиновой
кислоты. Как апудоцитарные клетки хромаф-
финоциты способны и к продукции других
гормонов. Опухоли-хромаффиномы могут
вырабатывать существенные количества
энкефалинов, АКТГ, ВИП, серотонина,
эритропоэтина, интерлейкина-6 — что
вносит вклад в патогенез и симптоматику.
Маркерами хромаффином служат хромо-
гранин и нейрон-специфическая енолаза,
последнее обстоятельство позволяет
отнести их к апудомам.
Патогенез опухолей определяется
высвобождением гормонов и реакцией тканей
на них.
Более половины больных имеют
клиническую картину в виде частых (от
нескольких раз в неделю, до 25 раз в сутки)
катехоламиновых кризов. В значительном
проценте случаев, из-за меньшей
секреторной активности опухоли и
десенсибилизации тканей к катехоламинам, имеется
хроническое течение с редкими кризами.
Наконец, опухоль может быть асимптомной
(«немой»).
Ниже описана клиническая картина,
характерная для течения хромаффином
норадреналинового типа.
Пароксизмы при феохромоцитоме
выражаются в приступах гипертензии
(систолическое давление до 320 мм рт. ст!),
сердцебиения, тахиаритмии, жара, затем —
профузного потения («диафорез»). Это
может сопровождаться тревогой, страхом,
головными болями и болью в груди,
пояснице, животе. Зрачки расширены, бывают
диплопия и временное ухудшение зрения
из-за нарушения аккомодации и ишемии
сетчатки. Продолжительность такой ка-
техоламиновой атаки — от 5 мин. до суток
и более. Провоцируют ее механические
воздействия на опухоль, стресс,
физические усилия, повышение внутрибрюшного
давления, ингибиторы моноаминоксидазы
и ряд адренергических лекарств. При
локализации опухоли в стенке мочевого пузыря
описаны необычные кризы в результате
мочеиспусканий.
Гипергликемия, избыток жирных кислот
и калия отмечаются при таких приступах
в крови.
Кислотно-основное равновесие сдвинуто
в сторону метаболического ацидоза, имеется
гиперл актацидемия.
Гипертензия — основной симптом
болезни, его развитие обусловливает прогноз.
Она может быть постоянной и кризовой, а
объясняется усилением насосной
деятельности сердца и повышением
периферического сопротивления. Гиперренинемия
может затруднять работу прессорного
натрийуретического разгрузочного
механизма. Еще большую роль играют прессор-
ные центральные эффекты катехоламинов.
У больных бледное лицо и холодное
влажное рукопожатие — за счет
периферического вазоконстрикторного действия,
опосредованного через а-рецепторы. Характерны
обмороки и ортостатическая гипотензия,
так как снижение объема плазмы из-за
периферической вазоконстрикции ведет к
уменьшению возможностей компенсации
уровня системного артериального
кровяного давления при постуральных
изменениях. Головные боли из-за нарушения
церебрального кровообращения особенно
характерны при продукции в опухоли
серотонина.
Избыток катехоламинов повышает
потребность миокарда в кислороде, создаёт
тенденцию к относительной коронарной
недостаточности, что может вызвать даже
симптомы грудной жабы.
Развивается миокардиодистрофия,
может быть застойная сердечная
недостаточность.
Для функций ЖКТ характерны запоры,
может быть атония кишечника и кишечная
непроходимость.
Больные худеют, теряя жировые
запасы, причем избыток свободных жирных
кислот в крови дополнительно усиливает
риск аритмии и калоригенный эффект.
Основной обмен повышен. До 28 % случаев
протекают с лихорадкой на фоне кризов. В
610 АЖ Зайчик, ЛЛ.Чурилов Патохимия
развитии лихорадки велика роль не только
повышенного под действием катехоламинов
калоригенеза, но и гиперпродукции интер-
лейкина-6 — эндогенного пирогена. Как и
при гипертирозе, больные плохо переносят
жару. Толерантность к глюкозе
уменьшается, при малых функциональных ресурсах
В-клеток панкреатических островков может
быть вторичный сахарный диабет.
Отмечается задержка мочеиспускания из-за спазма
шейки мочевого пузыря.
Характерны (особенно — на высоте криза)
эритроцитоз (в случае продукции
опухолью эритропоэтина он крайне выражен),
лимфоцитоз, эозинопения, нейтрофильный
лейкоцитоз, ускорение СОЭ (особенно, при
гиперпродукции интерлейкина-6).
Повышается свёртываемость крови.
Симпатоадреналовый криз обычно
заканчивается фазой, когда доминируют явления
«противорегуляции» — падает артериальное
давление, краснеет лицо, замедляется пульс.
Пациент испытывает тошноту, рвоту,
гиперсаливацию, выделяется большое количество
мочи.
Продукция различных гормонов при
хромаффиномах может отклонять картину
болезни от данной, наиболее типичной.
Избыток энкефалинов создаёт
симптоматику так называемых «вагальных
кризов» — с падением давления и другими
симптомами, имитирующими действие
парасимпатической вегетативной нервной
системы (10%). Продукция ВИП
дополняет картину болезни диареей (6%).
Выработка ассоциированых с паратирокри-
нином пептидов ведет к гиперкальциемии
(4 %). АКТГ из хромаффином даёт картину
синдрома эктопической продукции адре-
нокортикотропина (см. выше). При этом,
в частности, наблюдается не ацидоз, а
гипокалиемический алкалоз.
Хромаффиномы адреналинового типа
отличаются приступами гипотензии и
красной, тёплой кожей из-за
периферической вазодилатации. При подобных
опухолях тип течения болезни может быть
интермиттирующим кризовым, с норма-
чдокринно-метаболические нарушения)
лизацией давления между приступами.
Адреналиновые хромаффиномы
характеризуются более кратковременными
кризами, в то же время, метаболические
сдвиги при высокой продукции
адреналина могут быть более выражены, чем в том
случае, если опухоль вырабатывает лишь
норадреналин.
Срок жизни больных хромаффиномами
укорочен из-за осложнений, даже если сама
опухоль и доброкачественна.
Наиболее типичные и тяжелые
осложнения могут включать фибрилляцию
желудочков, отек лёгких в связи с левоже-
лудочковой недостаточностью, инфаркт
миокарда или почки и особенно часто —
инсульт. Возможно развитие илеуса и
«острого живота», провоцирующего опасное для
больных хирургическое вмешательство,
появление кето- и лактат-ацидоза,
обострение латентного гипертироза. Часто
отмечаются проявления лейкоцитокластического
васкулита, тромбоэмболической болезни и
ДВС-синдрома. На высоте криза бывают
случаи острой почечной
недостаточности. Терминальное состояние больных с
феохромоцитомой, комбинирующее эти
синдромы, при преобладании острого
расстройства гемодинамики известно как
«катехоламиновый шок».
При диагностике гормонообразующих
хромаффином большое значение имеет
регистрация повышенного выделения с
мочой продуктов деградации
катехоламинов (рмГ рис. 117), не подавляемого кло-
нидином, то есть имеющего автономный
источник.
Кроме хромаффином, в мозговом
веществе надпочечников и в параганглиях
нередко бывают невробластомы. Это очень
злокачественные опухоли из нейробластов
нервного гребня. Они являются самыми
частыми солидными экстракраниальными
опухолями у детей до 5 лет и практически не
встречаются у непедиатрических пациентов.
Причиной опухоли служит амплификация
онкогена N-myc. Этот онкоген в норме
представлен одной копией в коротком плече
Нарушения эндокринной регуляции б 11
хромосомы 2. В результате его
амплификации появляется множество копий данного
онкогена, которые могут располагаться в
ядрах клеток опухоли экстрахромосомно, в
виде характерных сателлитов — двойных
малых хроматиновых телец. Копии гена
переносятся и в хромосому 13, где выглядят как
гомогенно окрашиваемый регион длинного
плеча. Часто в опухолевом клоне имеется
и аномалия хромосомы 1 (частичная моно-
сомия 1 из-за делеции фрагмента короткого
плеча этой хромосомы). Все эти характерные
признаки служат для цитогенетической
диагностики невробластом. В структуре опухоли
характерны розетки Гомера-Райта —
скопления примитивных нейробластов вокруг
центрально расположенной гомогенной массы
эозинофильных нервных волокон.
Данные опухоли могут вызвать некоторое
увеличение уровня циркулирующих катехол-
аминов и содержат нейрон-специфическую
енолазу. Но, в отличие от хромаффином,
они не обусловливают столь значительного
избытка катехоламинов и не выделяют хро-
могранина. Невробластомы
характеризуются инфильтративным агрессивным ростом,
метас,тазируют в кости черепа (метастаз
Гетчинсона), в частности, обусловливая
небазедов экзофтальм, а также в печень
(метастаз Пеппера) и ьпсЪстный мозг.
Неметастазирующие
доброкачественные ганглионевромы той же локализации
и происхождения, также могут повышать
уровень катехоламинов в крови и не
образуют хромогранина. Их маркером служит
белок S100. Данные опухоли поражают и
взрослых, и детей.
По некоторым данным, возможно одоб-
рокачествление невробластом, при котором
их клетки спонтанно созревают и
превращаются в шванноциты и многоядерные
гигантские ганглиоциты. Невробластома с
признаками созревания считается
переходной и квалифицируется как ганглионевроб-
ластома. В конечном итоге, после полного
созревания, возможна её трансформация в
ганглионеврому, что является чуть ли не
единственным примером подобного рода в
онкологии. v
Гипофункция мозгового вещества
надпочечников часто сочетается с гипокор-
тицизмом, если первичное поражение
надпочечников носит тотальный двусторонний
характер (см., например, выше — синдром
Уотерхауза-Фридериксена — при
апоплексии надпочечников).
Двусторонняя адреналэктомия не
приводит к симпатоадреналовой недостаточности,
если обеспечена заместительная терапия, и
симпатическая нервная система индивида
не имеет других дисфункций и аномалий.
Однако развитие адреномедуллярной
недостаточности возможно при
некоторых дополнительных условиях
(конституциональная ваготония, сахарный диабет,
недостаточность симпатической нервной
системы, аномалии обмена тирозина).
Основным проявлением
симпатоадреналовой недостаточности служит нарушение
адаптации системы кровообращения к
переходу тела в вертикальное
положение — ортостатический коллапс. В норме
стимуляция барорецепторов и
симпатических нервов обеспечивает при этом
более чем двукратный прирост продукции
норадреналина, что компенсирует
возможное изменение системного давления путём
увеличения периферического
сопротивления, работы сердца и выхода крови из
венозных депо. Ортостатический коллапс
может иметь три причины. При первичном
снижении объёма циркулирующей крови
(кровопотеря, дегидратация, тяжелая
анемия) и при периферической резистентности
к катехоламинам (гипокортицизм, действие
лекарств) коллапсы возникают несмотря на
полноценную реакцию симпатоадреналово-
го аппарата то есть на фоне гиперадренерги-
ческой реакции.
Первичная и вторичная недостаточность
адреномедуллярной функции создаёт ор-
тостатические коллаптоидные явления на
фоне гипоадренергической реакции.
Вторичная недостаточность мозгового
слоя надпочечников возникает при
повреждениях головного мозга (травмы, демиели-
низирующие энцефалопатии, инфекции,
действие фенотиазидов и других лекарств),
612 ALU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
при спинальных повреждениях (травмы,
спинная сухотка, сирингомиелия), а также
при нарушениях функции периферических
симпатических нервов (токсические, в том
числе — алкогольные нейропатии, амилои-
доз, порфирия и, наиболее часто —
диабетическая вегетативная нейропатия — см. выше
«Патофизиология сахарного диабета»).
При инсулинзависимом сахарном
диабете в результате хронической
гипергликемии, при участии механизмов глики-
рования белков и вследствие стимуляции
полиолового цикла, развиваются микроан-
гиопатия и полинейропатия. Одно из
проявлений этих осложнений — вегетативная
(автономная) нейропатия. Вследствие
вегетативной нейропатии и гипопитуита-
ризма, обусловленного ангиопатией
сосудистой системы гипоталамуса-гипофиза,
ЦНС больных утрачивает способность
к симпатической мобилизации в ответ
на гипогликемический сигнал и другие
стрессоры. Этому способствует
«привыкание» к эпизодическим гипогликемиям.
В результате развивается недостаточность
секреторного ответа мозгового вещества
надпочечников на гипогликемию, а также
дефицит глюкагона при гипогликемии, так
как стимуляция глюкагоновой реакции
зависит во многом от катехоламинов. Своего
рода порочный круг способствует
учащению гипогликемии и разбалансировке
состояния углеводного обмена (С.Э. Да-
гого-Джек и соавт., 1993). Особенно
большую роль играет дефицит продукции
адреналина.
Первичная недостаточность функций
симпатоадреналовой системы наблюдается
в нескольких формах.
Генерализованная симпатоадреналовая
недостаточность охватывает и
гипофункцию мозгового вещества надпочечников, и
гипотонус симпатических нервов. Больные
имеют пониженное «рабочее» артериальное
давление, брадикардию в покое, наклонны к
ортостатическим коллапсам, зачастую
страдают от метеоропатии, когда на их состоянии
сильно отражаются колебания
атмосферного давления. Классические источники даже
оперируют для подобных ситуаций
понятием «гипотоническая болезнь».
В современной литературе из всего
разнообразия данного синдрома вычленяется
первичная вегетативная дисфункция двух
разных типов (I и II), а также семейная диз-
автономия (Ф. Крайер, 1987).
При первичной вегетативной
дисфункции I типа (идиопатической ортостатиче-
ской гипотензии) нет каких-либо первичных
заболеваний ЦНС или полинейропатий. Но
у больных имеется низкий базальный
уровень секреции катехоламинов и тенденция к
гипотензии, сопровождаемая ортостатичес-
кими коллапсами. По-видимому, эти
пациенты и подразумеваются под историческим
термином «гипотоническая болезнь».
При первичной вегетативной
дисфункции II типа — синдроме Шая-Дрейджера
или множественной системной атрофии —
картина идиопатической ортостатической
гипотензии дополнена дегенеративным
заболеванием ЦНС с нарушенной дофа-
миноэргической регуляцией и
проявлениями, напоминающими паркинсонизм. У
больных нормальный базальный уровень
катехоламинов, но недостаточный
стимулированный.
При семейной дизавтономии — синдроме
Райли-Дея — имеется тяжелейшее
нарушение как симпатической, так и
парасимпатической регуляции включая функцию
мозгового вещества надпочечников.
Поэтому симпатоадреналовая недостаточность
не ограничивается ортостатически-гипо-
тензивным синдромом. Больные не могут
адекватно потеть и не переносят жару,
нарушена аккомодация зрения, имеется
недержание кала и мочи,
желудочно-кишечные расстройства.
При фенилкетонуриях (см. выше
«Патофизиология белкового обмена») нарушается
производство тирозина из фенилаланина.
Это способствует понижению
функциональных возможностей систем катехоламиново-
го синтеза. У больных могут быть тенденция
к гипотензии и другие проявления адрено-
медуллярной недостаточности.
Патофизиология гонадотропной
функции и гонад
«Пришла проблема Пола
— Румяная фефёла!»
Саша Чёрный, «Песня о поле», 1908
Пол человека детерминируется
наследственно. Развитие и поддержание комплекса
его признаков, как и функция размножения
в целом, зависят от эндокринных
регуляторов — гонадотропных гормонов и гормонов
гонад.
Эндокринные факторы не могут изменить
хромосомный пол человека, который
определяется в момент зачатия. Но гонадный
пол, развиваемый в ходе эмбриогенеза, а
также эндокринные, анатомо-физиологические
и психолого-поведенческие признаки пола,
формируемые как до, так и после рождения,
могут существенно видоизменяться под
воздействием гормональных расстройств
и воздействий. Это приводит индивида к
диссоциации между хромосомным полом,
с одной стороны, и полом эндокринным,
психосоматическим, наконец полом
паспортным, с другой.
Пол — важнейший социальный признак
человека. Поэтому проблемы пола и
психофизиологических основ поло-ролевого
поведения всегда интересовали не только
(и не столько) медиков, сколько широкую
общественность. В этом контексте ныне
часто обсуждаются проблемы равенства полов,
свободы определения пола и т.д. Заметим, что
хотя социально-правовое равенство полов
является необходимым условием свободного
развития общества, с медицинской точки
зрения свободы от генов, детерминирующих
развитие индивида, не бывает. Поэтому и
биологического равенства полов,
понимаемого как их одинаковость и эквипотентность, не
может быть. С этой точки зрения, индивиды,
у которых половая самоидентификация,
паспортный пол и хромосомный пол не
совпадают, воспринимаются медиками прежде
всего как пациенты.
Поэтому обсуждение этих сложных
проблем в данной книге ограничивается их
Нарушения эндокринной регуляции 613
медико-биологическими гранями. Раздел,
предлагаемый вниманию читателя, не
является полным описанием патофизиологии
заболеваний системы органов размножения.
Мы коснемся только эндокринопатий,
затрагивающих половую дифференцировку
и функцию гонад как желез внутренней
секреции. Более детальные сведения о
механизмах других болезней половых органов
можно будет почерпнуть в т. III данного
руководства.
Половая дифференцировка
В данном разделе рассматривается с
одинаковой степенью детализации не весь
целостный механизм определения и
поддержания пола, а лишь его эндокринологические
аспекты. Хромосомное определение пола и
его нарушения при гоносомных аберрациях
детально обсуждались ранее в т. I данного
руководства (с. 65-70,136).
До определённой стадии эмбриогенеза
каждый индивид представляет собой
соматического «андрогина» с
недифференцированным бипотентным гонадным зачатком
(гонадным валиком), содержащим
первичные половые хорды и мигрировавшие из
стенки желточного мешка примордиальные
половые клетки, способные к
альтернативной дифференцировке.
Эта структура формируется на 4-й
неделе внутриутробной жизни. Она содержит
корковое и мозговое вещество. На 7-8
неделе происходит дивергенция и зачатки
дают либо мужскую, либо женскую гонаду.
В мужском варианте преимущественное
развитие получают мозговые, а в женском —
корковые структуры половых валиков. При
этом хорды превращаются либо в семенные
канальцы, либо в овариальные фолликулы;
мезенхимные клетки формируют либо
клетки Лейдига, либо текальные и стромальные
элементы яичника, а примордиальные
половые клетки превращаются в сперматогонии,
или овогонии, соответственно. Каждый плод
располагает вольфовым (мезонефральным)
протоком, оформляющимся к 25-30-му
дню беременности, и мюллеровым (пара-
мезонефральным) протоком, формирую-
614 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
щимся к 44-48-му дню (А.Г. Кнорре, 1959,
Э.Г.Улумбеков, Ю.А. Челышев, 1997).
Вольфов проток может
дифференцироваться в эпидидимис, семявыносящий проток,
семенные пузырьки и эякуляторные
протоки. Мюллеров может дать фаллопиевы
трубы, матку и верхюю часть влагалища.
Наконец, имеются универсальные
зачатки наружных гениталий — урогенитальный
синус, мочеполовые бугорок, складки и
валики. Эти структуры можно преобразовать
либо в пенис и мошонку, либо в клитор и
половые губы.
Напомним, что мужской пол
развивается только при наличии Y-хромосомы, а
еще точнее — её SRY-гена, если правильно
реализуется определённая цепь эмбрио-
генетических событий. При отсутствии
Y-хромосомы действует спонтанная
тенденция к развитию всех бивалентных
эмбриональных зачатков по женскому типу,
которая не требует особых гормональных
активаторов. При отсутствии гонад у плода
вольфов проток регрессирует, а мюллеров
дифференцируется по женскому типу. При
кариотипе 45X0 (синдром Шерешевско-
го-Тернера) формируется яичник,
хотя последующее развитие в нем
фолликулов нарушено, и его
полноценность требует кариотипа 46ХХ.
Развитие мужского пола при
кариотипе, лишенном Y хромосомы,
зафиксировано только в
редчайших эксквизитных случаях
переноса SRY гена из Y, хромосомы — в
Х-хромосому из-за неправильного
кроссинговера при гаметогенезе. Таких
наблюдений около 50. При этом кариотип
46ХХ, а фенотип мужской. Больные низкого
роста, имеют нормальный пенис, маленькие
плотные яички, в которых, однако,
практически нет сперматозоидов. Уровень гонадо-
тропинов повышен, тестостерона — снижен,
эстрогенов — выше нормы.
Целостный ход биосинтеза мужских
половых гормонов отражен на рис. 118.
Критической в процессах развития
первичных мужских половых признаков
является 8-я неделя внутриутробного развития.
3$-гидроксистероид-
дегидрогеназа /изомераза
| прогестерон
Р45017а I 17а.-гидроксилаза
\17-ОН-прогестерон
17f 20-лиаза
| андростендиощ
| дигидротестостерон \
S
It
is
Si з
Рис 118. Биосинтез андрогенов и их превращения в
различных органах и тканях
Чтобы произошла полная реализация
программы маскулинизации, ген SRY из
семейства ДНК-регуляторных генов Sox,
находящийся в коротком плече
Y-хромосомы, близ центромера, вступает в действие
на 6-7 неделе эмбрионального развития,
запуская ряд генов, локализованных в других
хромосомах и обеспечивающих программу
синтеза тестостерона у плода, выработку
фермента А4-5а-редуктазы, необходимой
для конверсии этого андрогена в дегидротес-
тостерон (эти гены находятся в аутосомах)
и синтез пептида, ингибирующего развитие
мюллерова протока и тканевых
рецепторов андрогенов (эти гены присутствуют в
Х-хромосоме). Важное значение имеет ген
SRA1 семейства Sox, локализованный в
хромосоме 17, кодирующий дополнительные
факторы, опосредующие часть эффектов
гена SRY.
Белковые продукты, синтез которых
запускается геном SRY, представляют
собой «мужские антигены»,
используемые в иммунологическом тестировании
истинного пола. Мужской антиген H-Y, он
же — фактор,определяющий развитие тес-
тикул (ФРТ), служит иммунологическим
маркером мужского пола, отсутствует при
отсутствии Y хромосомы и при делеции ее
SRY-участка, в то же время при наличии
более чем одной Y-хромосомы его
концентрации кратно повышаются.
При мутациях в 17-й хромосоме и
отсутствии функций гена SRA1 действие ФРТ
не полностью эффективно. Происходит
развитие плодов с кариотипом 46XY по
женскому типу (реверсия пола) или развивается
врождённая камптомелическая дисплазия,
при которой большинство больных-мужчин
имеет женский фенотип. Взаимоотношения
этого гена с расположенным там же геном
хорионического гонадотропина не уточнены.
Процесс первичной мужской половой
дифференцировки занимает 8-14-ю недели
эмбриогенеза. До 50 дня гонады
индифферентны. Действие продуктов гена SRY (и,
возможно, SRA1) ведет на 7-8 неделе к диф-
ференцировке первых клеток Ф.Лейдига и
клеток Э. Сертоли в зачатке гонады. Клетки
Лейдига начинают продукцию фетального
тестостерона, их деятельность
стимулируется плацентарным хорионическим гона-
дотропином.
Фетальный тестостерон направляет
развитие зародыша по мужскому варианту.
Он способствует на 7-10 неделе развития
преобразованию вольфова протока в эпиди-
димис, семенные пузырьки, эякуляторные
протоки и семявыносящий проток. В тканях,
под воздействием экспрессируемой при
наличии гена SRY А4-5а-редуктазы, тес-
Нарушения эндокринной регуляции б 15
тостерон переходит в дигидротестостерон,
необходимый для преобразования общнго
зачатка наружных гениталий по мужскому
типу — в мошонку и пенис, а также для
развития простаты (рис. 118). Эти процессы
завершаются к 14 неделям беременности,
хотя нисхождение яичек в мошонку и рост
пениса продолжаются и в дальнейшем. В то
же время с начала восьмой недели первые
клетки Сертоли вырабатывают локальный
фетальный гормон — высокомолекулярный
гликопротеид, известный под названием
мюллеров тормозный фактор (антимюл-
леров ингибирующий пептид). Этот
регулятор обеспечивает быструю инволюцию
мюллерова протока, к началу 9-й недели
беременности, но продолжает выделяться
у плода мужского пола до момента родов и
даже в раннем постнатальном периоде. Его
гомолог выделяется яичками и в
последующем (см. ниже).
Итак, продукция тестостерона и антимюл-
лерова ингибирующего пептида в гонадах
плода предопределяет на втором-третьем
месяцах внутриутробного развития
формирование внутренних половых органов по
мужскому типу. Метаболит андрогенов 5а-
дигидротестостерон, неконверсируемыи в
эстрогены, контролирует маскулинный тип
формирования наружных гениталий на 3-4
месяце фетогенеза {соматический пол).
В то же время значительная часть
тестостерона у плода любого пола и в плаценте
метаболизируется в эстрогены. При
наличии Y-хромосомы и высокой продукции
тестостерона обеспечивается достаточное
содержание эстрогенных метаболитов,
подавляющее формирование обратной связи
между продукцией лютеинизирующего
гормона (ЛГ) и эстрогенов в развивающемся
мозге. Это ведет к установлению мужского,
нециклического типа гипоталамо-гипофи-
зарной регуляции гонадотропных функций
и обеспечивает на 4-6 месяце фетогенеза
мужской тип дифференцировки головного
мозга и гипоталамо-гйпофизарного нейро-
секреторного аппарата, что лежит в основе
нейропсихической маскулинизации в
последующей жизни.
616 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Созревание центров секреции гонадо-
тропинов идет под контролем эстрогенов,
полученных из тестостерона, а центров,
определяющих половое влечение — под
совместным контролем андрогенов и их
ароматических эстрогенных производных.
Центры, ответственные за поддержание
поведения, соответствующего избранной
половой роли, у мужчин организуются под
влиянием только андрогенов. Организация
трех этих мозговых центров идет
последовательно и частично перекрывается
(формирование «мозгового пола»).
Циклический тип секреции гонадотропи-
нов и высокая чувствительность механизма
обратной связи между концентрацией
эстрогенов и продукцией Л Г обнаружены
у истинно гомосексуальных мужчин, но
отсутствуют у гетеро- и бисексуалов. Таким
образом истинный первичный
гомосексуализм не должен трактоваться как чисто
психосоциальное явление, так как он
является болезненным нарушением полового
развития мозга (Г. Дёрнер и соавт., 1987).
В постнатальном онтогенезе, в период
полового созревания, активизация стерои-
догенной функции семенников, выходящих
из-под ингибирующего действия эпифиза,
ведет к приросту продукции
андрогенов — тестостерона и дигидротестостеро-
на — и к маскулинизации внешнего облика
подростков, сопровождаемому развитием
мужских вторичных половых признаков,
гиперплазией тестикул и наружных
гениталий, началом полноценного сперматогенеза,
который продолжается всю жизнь. По
крайней мере, зафиксированы случаи рождения
полноценных детей у глубочайших старцев,
в том числе лиц более чем 100-летнего
возраста. Хрестоматийный пример — рождение
философа Конфуция, отцу которого к тому
моменту перевалило далеко за 80.
Моноаминергические механизмы
участвуют в половой дифференцировке мозга.
Так, у лиц с парафилиями и
транссексуальными поведенческими нарушениями
установлено снижение скорости захвата
серотонина постсинаптическими нервными
окончаниями и тромбоцитами. Блокаторы
синтеза серотонина феминизируют половое
поведение самцов у грызунов, а ингибиторы
моноаминоксидазы, способствуя
накоплению серотонина и катехоламинов,
маскулинизируют поведение самок (И. Лехтинен,
1971; А.А. Ткаченко и соавт., 1995).
В норме половое созревание
начинается у мальчиков в 10-12 лет (рис. 119).
Это происходит благодаря первичным
изменениям в сервомеханизмах,
определяющих взаимоотношения тестикул и ги-
поталамо-гипофизарного аппарата. Перед
пубертатом гипоталамус и гипофиз очень
чувствительны к ингибирующему влиянию
тестостерона на продукцию гонадолиберина
и гонадотропинов. Начало пубертата
знаменуется понижением этой чувствительности.
Появляются ночные всплески гонадолибе-
риновой и лютропиновой секреции. Затем
они распространяются и на дневное время.
Наконец происходит переустановка
гипоталамуса на новый уровень чувствительности
к андрогенам. Секреция гонадолиберина и
чувствительность гонадотрофов к гонадоли-
берину возрастают. Это ведет к стимуляции
ЛГ продукции тестостерона в яичках и к
их гиперплазии. Андрогены обеспечивают
формирование признаков возмужания, в
основном — путём дифференцированной
стимуляции ростовых и анаболических
процессов. При этом до четверти всей задержки
азота, обусловленной андрогенами и
связанной у подростков с активизацией синтеза
нуклеиновых кислот и белков, приходится
на пролиферативно-гипертрофические
прцессы в гонадах и гениталиях.
Остальная задержка азота связана с
ростом скелета и мышечной массы, ростом
волос и гипертрофией сальных желез,
ростом и изменением конфигурации гортани и,
наконец, — активизацией эритропоэза.
Особенно чувствительны к андрогенам мышцы
плечевого пояса и груди, в связи с чем
маскулинизируются очертания фигуры. Весь
процесс пубертата занимает около 5 лет,
после чего дополнительная андрогенизация
у мужчин уже не усиливает выраженности
половых признаков. Правда, отдельные
проявления маскулинности— например,
высшие черепно-мозговые
центры включают („будят")
аденогипофиз
акнэ
волосы
под мышками
увеличение
грудных
желез
увеличение
матки
менструации
оволосение
лобка
ороговение
вагинального
эпителия
округление j
контуров
тела
прекращение
роста костей
в длину
начало
созревания
фолликулов,
овуляции и
лютеинизации
появление интер-
стициальных клеток,
стимуляция
пролиферации сперматогенного
эпителия ФСГ и
тестостероном
м-образная линия
роста волос
акнэ
оволосение лица
— развитие мышц
увеличение
гортани,
изменения
тембра голоса
некоторые
увеличение
грудных желез
волосы
на лобке
увеличение
пениса, простаты и
семенных пузырьков
прекращение роста
тела в длину
Рис 119. Нормальные половая дифференцировка и половое созревание у человека (по Ф. Неттеру
из CIBA Medical Illustrations Collection). Пояснения в тексте
618 А.Ш. Зайчик, ЛЛ. Чу рилов Патохимия (эндокринно-метаболические нарушения)
мышечная масса и степень оволосения
груди — могут нарастать на протяжении более
длительного срока.
В отсутствие гена SRY все
вышеописанные события не происходят, и формируется
соматический и психический женский пол,
причём примордиальная гонада
превращается в яичник. В диапазоне между 50-м
днём и 90-м днём беременности
формируются и успевают достичь стадии ооцитов
1 порядка женские гаметы. Они обрывают
своё развитие в профазе первого мейоти-
ческого деления и приобретают оболочку из
фолликулярных клеток, образуя приморди-
альные фолликулы, которых к 7-му месяцу
внутриутробной жизни насчитывается до
10 млн, а к рождению их число падает до
2 млн. Вольфов проток регрессирует к 101/2
неделям беременности, мюллеров
дифференцируется во внутренние гениталии, а
общий зачаток наружных гениталий после
11 недели, без влияния фетальных андроге-
нов, даёт наружные женские половые
органы. В дальнейшем развитии женских гамет
следует период покоя. За его время число
ооцитов сократится до 400 000.
В постнатальной жизни единственным
эндогенным источником андрогенов в
организме здоровых женщин служит кора
надпочечников. В период пубертата активизация
внутрисекреторной деятельности яичников,
продуцирующих женские половые стероиды
(эстрогены и прогестерон), осуществляемая
под контролем гипоталамуса и гипофиза, в
соответствии с заложенным в фетальный
период циклическим паттерном, приводит
к бурному развитию вторичных женских
половых признаков и началу месячных
(рис. 119). Период покоя в овогенезе
заканчивается, на пике содержания Л Г
завершается первое мейотическое деление,
образуются и циклически выходят в половые пути
полноценные яйцеклетки. Только немногим
более 400 из них будет использовано за
всю предстоящую жизнь половозрелой
женщины. Таким образом, если мужчина
при длящемся много десятилетий процессе
самообновления сперматогенного эпителия
тестикул имеет буквально миллиарды
шансов стать отцом, даже у женщин идеального
здоровья это число шансов ограничено всего
лишь сотнями.
Следует отметить, что большое
количество сперматозоидов уравновешивает
то обстоятельство, что каждый из них
намного более уязвим для неблагоприятных
влияний, чем отдельно взятая яйцеклетка.
Поэтому, частота женского и мужского
бесплодия, практически одинакова, несмотря
на разительные отличия в количественных
результатах ово- и сперматогенеза.
Отметим также, что яичник, в отличие от
яичка, очень плюрипотентная эндокринная
железа. Помимо половых стероидов,
различные клетки яичника являются источником
еще более чем 30 белковых гормонов и
паракринных регуляторов (Дж. Окленд и
соавт., 1992). В минорных количествах все
они обеспечивают нормальную регуляцию
женских половых функций. Однако, при
патологии именно яичник может служить
эктопическим источником избытка ряда
гормонов (см. ниже «Нарушение
эндокринных функций женских гонад»).
Асинхрония рассмотренных выше
процессов формирования половых признаков и их
зависимость от таких метаболических
факторов, как ароматизация половых стероидов
и их рецепция тканями, создают почву для
множества врождённых и приобретённых
рассогласований и нарушений соответствия
хромосомного, гонадного, нейроэндокрин-
ного, психического и соматического пола.
Нарушения половой
дифференцировки.
Гермафродитизм
Присутствие у индивида ткани как яичек,
так и яичников называется истинным
гермафродитизмом.
Истинный гермафродитизм издавна
вызывал пристальное внимание медиков,
поскольку идея «промежуточного существа»
глубоко укоренилась в культуре и
философии. О гермафродитах мы можем прочесть в
античных мифах, описывающих прототипа,
Нарушения эндокринной регуляции 619
давшего название этому синдрому — сына
Гермеса и Афродиты, слившегося в единое
существо с нимфой Сольмацией, а также
в философских трудах B.C. Соловьёва,
считавшего стремление к андрогинности
направлением эволюции человека.
Один из интереснейших случаев
истинного гермафродитизма, когда гермафродит
имел нормальных детей в браке, описан
выдающимся поэтом, доктором медицины
И.В.Гёте.
Но наиболее эксквизитное описание,
поистине уникального случая истинного
гермафродитизма, принадлежит Р. Вирхо-
ву и соавторам. Ими был описан истинный
гермафродит, при рождении названный
Катарина Гоман и воспитанный, как девочка. В
16 лет этот человек посчитал себя мужчиной
и начал половую жизнь с женщинами. В 20
лет у него появились менструации и
гинекомастия. Психофизиологическая ориентация
гермафродита изменилась, и он начал вести
половую жизнь с мужчинами как
нормальная женщина, но детей не имел. В 42 года
менструации прекратились. Тогда этот
замечательный исторический персонаж еще
раз изменил стереотип своего сексуального
поведения и, переменив имя Катарина на
Карл Гоман, женился и имел в браке
здорового сына. Обследовав Катарину-Карла,
Р. Вирхов обнаружил живые
сперматозоиды, а правильные менструации говорили
о наличии функционального яичника. Со
времён Вирхова подобных случаев больше
не описано, хотя имеются свидетельства о
более чем четырёх сотнях менее ярких
наблюдений истинного гермафродитизма.
У истинного гермафродита могут быть
один яичник и один семенник или овотести-
кула (овотестис) и яичник (в более чем 60 %
наблюдений). Реже бывают двусторонние
овотестикулы (более 20%), а самое редкое
сочетание — овотестикула и яичко (10%).
При этом обычно в разнополых гонадах или
разнополых частях овотестикул имеются
гаметы разных полов. Часть истинных
гермафродитов формирует гаметы лишь одного
пола. Не более половины из них имеют
зрелые яйцеклетки, а достаточное для мужской
фертильности количество полноценных
сперматозоидов образуют лишь отдельные
истинные гермафродиты.
Строение наружных половых органов у
истинных гермафродитов промежуточное,
около 7 % имеют наружные гениталии
женского типа, в 12 % наблюдений
присутствует пенис и уретра. Однако в 75 % случаев
истинные гермафродиты воспитываются и
осознают себя в социально-половом
отношении как мужчины (Дж. Императо-Мак-
Джинли, 1987).
При истинном гермафродитизме не
описано случаев самооплодотворения,
поскольку к периоду полового созревания
сохраняется формирование зрелых гамет
лишь одного типа, почти всегда —
яйцеклеток (Л.Л. Либерман, 1966). В пубертатный
период иногда происходит смешанная
вирилизация-феминизация. Чаще
преобладает влияние эстрогенов. До 80 % истинных
гермафродитов имеют гинекомастию, почти
50 % менструируют (при наличии уретры
и гипоспадии — в форме периодической
гематурии).
Истинные гермафродиты могут иметь
детей. Зафиксировано много случаев
беременности и успешных родов у истинных
гермафродитов, ведущих половую жизнь как
женщины, при условии удаления остатков
тестикул и коррекции наружных гениталий.
Отдельные истинные гермафродиты,
ведущие половую жизнь, как мужчины также
являются фертильными.
Истинные гермафродиты имеют
нормальный интеллект и физическое развитие, но
рост их часто — ниже среднего. Продукция
гонадотропинов не понижена, а часто —
увеличена.
Истинный гермафродитизм
предрасполагает к возникновению злокачественных
опухолей из половых клеток — гонадоблас-
том, которые поражают овотестикулу или
яичко в 3 % случаев.
Патогенез истинного гермафродитизма
связан с судьбой гена SRY.
Кариотип истинных гермафродитов — в
80 % случаев 46ХХ, в большей части остав-
620 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
шихся наблюдений — мозаичный 46XX/XY,
и лишь изредка — 46XY.
Большинство из больных имеют мозаи-
цизм с переносом в части клеток SRY-гена
на Х-хромосому или на аутосому. У
некоторых гермафродитов с мозаицизмом Н. Йос-
со и соавт. (1965) обнаружили признаки,
указывающие на двойное оплодотворение
при их зачатии.
Клетки из овариальной части гонадной
ткани больных при мозаицизме не имеют
антигена H-Y, а из тестикулярной части —
располагают этим маркером. Доза антигена
понижена из-за инактивации несущих
эктопический SRY-ген хромосом в части
соматических клеток, по случайному принципу.
Есть точка зрения, что истинный
гермафродитизм и синдром «кроссинговерных
мужчин 46ХХ», описанный в
предыдущем разделе, являются промежуточным и
крайним проявлениями одного и того же
заболевания (стр.614). По крайней мере,
при семейных случаях истинного
гермафродитизма описана родословная, где брат
и дядя пробанда — истинного
гермафродита — имели этот синдром.
Ложный гермафродитизм — синдром, при
котором наличествуют гонады одного пола
и половая принадлежность имеющихся
гонад соответствует кариотипу (46XY — при
мужском и 46ХХ — при женском
псевдогермафродитизме).
Однако формирование других половых
органов и вторичных половых признаков
характеризуется при данном синдроме
интерсексуальностью.
Мужской псевдогермафродитизм —
результат разнообразных нарушений в
эмбриогенезе гонад, продукции, метаболизме и
рецепции андрогенов. Он характеризуется
наличием парных семенников и неполной
маскулинизацией (могут быть гипоспадия,
микрофаллия, плохо развитая мошонка).
Женский псевдогермафродитизм —
наиболее часто является результатом появления
эндогенных или экзогенных андрогенов
в критический период от 8 до 12 недели
киематогенеза, когда плод с кариотипом
46ХХ чувствителен к их морфогенетиче-
скому влиянию. У больных имеются
яичники, но фенотип, как правило, мужской.
Это вызывается врождённой гиперплазией
коры надпочечников, арренобластомой во
время беременности у матери или приемом
матерью андрогенов и прогестинов в первом
триместре беременности.
Псевдогермафродитизм распространен
намного шире, чем гермафродитизм
истинный. Так как перемена
психолого-поведенческих половых стереотипов у детей
после 2 лет происходит с трудом, важной
задачей является диагностика истинного
пола и выбор желаемого пола с коррекцией
проявлений псевдогермафродитизма, по
возможности в раннем детском возрасте.
Этиология и патогенез мужского
псевдогермафродитизма
Основные причины мужского
псевдогермафродитизма сводятся к трём группам:
нарушению дифференцировки и развития
тестикул, дисфункции тестикул и
нарушению рецепции андрогенов.
Нарушение дифференцировки и развития
яичек может быть результатом различных
аномалий Y-хромосомы.
Полная делеция этой хромосомы во всех
или в части клеток ведёт к кариотипу ХО
или XO/XY. Возможна также делеция
короткого плеча этой хромосомы, при
сохранении самой Y-хромосомы — кариотип
XY(p-) или XY/ XY(p-) — при мозаицизме.
Если мутация носит ограниченный характер
и затрагивает изолированно гены,
контролирующие ключевые этапы в тестикулогенезе,
то кариотип может быть XY, без
хромосомных аномалий.
Клинически это проявляется в виде
нескольких форм врождённой дисгенезии
гонад — нарушения, при котором страдает
дифференцировка как гормонообразующей,
так и семяобразующей части тестикул.
При «чистой» дисгенезии гонад кариотип
46XY, но фенотипический пол — женский,
с первичной аменореей, высоким ростом,
евнухоидными чертами телосложения, без-
Нарушения эндокринной регуляции 621
волосыми подмышками и лобком. Яички
не закладываются вообще, производные
вольфова протока регрессируют, а мюлле-
рова — сохраняются. Реже бывают
рудиментарные остатки тестикулярной ткани,
дающие минимальные признаки
маскулинизации. Механизм нарушения основан на
отсутствии эмбрионального фактора диф-
ференцировки тестикул или его рецептора.
Мужской антиген присутствует в части
случаев и не выявляется у некоторых больных.
Описаны семейные родословные с
наследованием по Х-сцепленному рецессивному
и аутосомно-доминантному типам. Почти
треть пациентов страдает от гонадобластом
и дисгермином рудиментарных гонад.
Смешанная дисгенезия гонад отличается
мозаичным кариотипом 45XO/46XY, а
также присутствием яичка — с одной стороны,
и атрофичной гонады — с другой. Фенотип
больных — наиболее часто — женский,
напоминающий синдром Шерешевского-
Тернера, иногда — классически яркий,
нередко — неполный (см. т. I, стр. 136). Могут
быть и мозаичные случаи с промежуточным
строением гениталий и даже мужской
фенотип. На стороне яичка имеется семявынося-
щий проток, в то же время есть матка и часто
фаллопиевы трубы. Тестикула выделяет
андрогёны и обусловливает пубертатную
маскулинизацию, но в ней утолщены
стенки семенных канальцев, и образуется мало
сперматозоидов, вследствие чего больные
бесплодны.
Мозаицизм объясняется потерей Y-хромо-
сомы из-за ее анафазной задержки или
нерасхождения при митозе. Атрофия одной
гонады может зависеть от преобладания
в ней клеток с кариотипом 45X0. Хотя
сохранённая тестикула в постнатальном
периоде выделяет андрогёны, очевидно,
что в критический период маскулинизации
наружных гениталий ее функция была
недостаточна, поскольку они не
маскулинизировались. Задержка ингибирующего влияния
продуктов тестикулы между 8-й и 14-й
неделей развития вызывает персистирование
производных мюллерова протока.
Варьирование фенотипа может зависеть
от преобладания линий ХО или XY-клеток
в сохранной гонаде в критический период.
Лгонадизм — вариант дисгенезии гонад
при кариотипе 46XY и полном отсутствии
как гонад, так и производных обоих
протоков — вольфова и мюллерова. При этом
регрессия зародышевого яичка происходит
после выделения антимюллерова пептида,
но до начала продукции фетальных андро-
генов. В результате мюллеров проток
исчезает, а вольфов — не дифференцируется в
мужские гениталии и также регрессирует. В
классическом варианте внутренних половых
органов нет совсем, а наружные — женского
типа. В зависимости от точного момента
регрессии тестикулы, возможна феноти-
пическая вариация. Этиология синдрома
неизвестна, точно определено отсутствие
хромосомных аберраций.
Иногда нарушение дифференцировки
гонад при мужском псевдогермафродитизме
бывает частичным и касается лишь клеток
Лейдига, синтезирующих андрогёны, а
семенные канальцы развиваются нормально.
Фенотип таких больных женский,
иногда—с расщепленной недоразвитой
мошонкой. Мюллеровых производных нет, а из
вольфовых имеются семявыносящий проток
и придаток яичка.
У взрослых семенные канальцы
нормальны, но сперматогенез понижен. Клеток
Лейдига почти совсем нет. Уровень андрогенов
соответствует нормальному женскому. Они
имеют адренокортикальное происхождение.
Уровень ЛГ сильно повышен, но ни ЛГ,
ни хорионический гонадотропин не дают
прироста содержания андрогенов в крови.
Уровень ФСГ нормален. Очевидно, имеет
место отсутствие или торможение развития
клеток-предшественников лейдиговских.
Вероятнее всего, это связано с дефектом
рецепторов ЛГ/хорионического гонадо-
тропина на этих клетках. Видимо,
присутствующие у больных вольфовы структуры
требуют для дифференцировки очень
незначительного количества андрогенов. В то же
622 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
время мюллеровы структуры регрессируют,
так как антимюллеров ингибирующий
полипептид — продукт клеток Сертоли, а не
клеток Лейдига.
Во многих случаях мужского
псевдогермафродитизма дифференцированные тес-
тикулы перестают функционировать, что и
вызывает нарушение полового развития.
Это может быть проявлением
недостаточности антимюллерова ингибирующего
фактора. Чаще всего клинически пациент
выглядит как мужчина с парными
семенниками, полной дифференцировкой воль-
фовых производных и маскулинизацией
наружных половых органов. В юношеские
годы происходит пубертат по мужскому
типу. Однако имеется односторонний
крипторхизм, а на противоположной
стороне — паховая грыжа с семенником и мюл-
леровыми структурами в грыжевом мешке
(матка и фаллопиевы трубы). От 5 до 13%
пациентов имеют опухоли яичка. Описаны
семейные случаи с различными типами
наследования.
Считается, что нарушения как синтеза,
так и секреции, структуры, рецепции и,
наконец, своевременности выделения
антимюллерова фактора могут обусловливать
данную аномалию.
Значительно чаще дисфункциональный
мужской псевдогермафродитизм является
результатом различных нарушений
продукции андрогенов тестикулами и адренокорти-
коцитами. Это — невирилизующие формы
адреногенитального синдрома (врожденной
гиперплазии коры надпочечиков) у мальчиков
(см. выше). Девочки при этих энзимопатиях
рождаются нормальными, лишь при дефиците
3-р-гидроксистероиддегидрогеназы бывает в
последующем незначительный вирилизм. Все
нарушения аутосомно-рецессивны, многие
затрагивают и продукцию неполовых стероидов
в коре надпочечников (см. выше).
У пациентов с мужским хромосомным
полом тестикулы нормально
дифференцируются и мюллеровы производные
регрессируют, но наружные половые органы из-за
фетальной недостаточности тестостерона
сформированы по женскому типу, в виде
урогенитального синуса или (при дефиците
3-р-гидроксистероиддегидрогеназы) —
промежуточны.
Краткая характеристика этих форм
мужского ложного гермафродитизма дана
ниже в табл. 40. Дополнительные сведения
о данных аномалиях можно найти выше
(с. 554-556).
Третья принципиально возможная форма
мужского псевдогермафродитизма связана
с полной или частичной
нечувствительностью тканей-мишеней к андрогенам.
Самая важная ее разновидность — полная
андрогенорезистентность или синдром
Морриса (тестикулярная феминизация). Это
состояние уже описывалось в т. I данного
руководства, с. 69-70,125).
Вкратце добавим, что при данном
нарушении — мужской хромосомный пол
(46XY), нормально дифференцируются
семенники, находящиеся в пахах, а
мюллеровы производные отсутствуют. Несмотря
на нормальный или повышенный уровень
тестостерона, вольфовы производные не
развиваются, а наружные гениталии —
женские. Уровень ЛГ повышен, ФСГ —
нормален или повышен. Уровень эстрогенов на
нижней границе женской нормы.
Эстрогены имеют тестикулярное прЪисхождение.
Синдром связан с женской
психофизиологической ориентацией. Больные осознают
себя женщинами и воспитаны как девочки.
При половом созревании у них появляется
женский бюст. Синдром наследуется либо
с Х-хромосомой, либо аутосомно-доминан-
тно и ограничен полом. Патогенез связан с
отсутствием цитоплазматического
рецептора дигидротестостерона, либо с постре-
цепторным и ядерным дефектом ответа на
дигидротестостерон. До половой зрелости
яички нормальны. После полового
созревания клетки Лейдига гипертрофированы, но
сперматогенез угнетён.
Частичная форма резистентности к
андрогенам — синдром Рейфенштайна — отли-
Нарушения эндокринной регуляции 623
Таблица 40, Дефекты биосинтеза тестостерона при мужском псевдогермафродитизме
Дефектный
фермент
Холестерин-20-
22-десмолаза
3-р-гидрокси-
стероид-дегид-
рогеназа
17а-гидрокси-
лаза
17,20-десмолаза
17Р-гидрокси-
стероиддеги-
дрогеназа
Наружные
гениталии
Женские или
урогенитальный синус
Промежуточные
Женские или
урогенитальный синус
Женские или
урогенитальный синус
Женские или
урогенитальный синус
Корти-
зол
U
V
V
N
N
Альдостерон
U
U
ft (дезокси
кортикостерон,
кортикостерон,
гипертензия)
N
N
Андрогены
U
ft (дегидроэпи-
андростерон
периферического
происхождения) tt(17-OH-
прегненолон)
U
U
U (тестостерон)
ft (андростен-
дион)
Половое созревание
Не доживают
Гинекомастия при
низком уровне эстрогенов
Гинекомастия при
низком уровне эстрогенов
Задержано
Гинекомастия при
низком уровне тестостерона
и высоком —
эстрогенов. Происходит
активная периферическая
конверсия андростен-
диона в эстрон
чается меньшей степенью пубертатной
феминизации и более маскулинизированным
фенотипом, но принципиально аналогична
синдрому Морриса.
Как известно, во многих периферических
тканях — печени, сальных железах,
волосяных фолликулах и коже наружных половых
органов — действует фермент 5-а-редукта-
за, превращающая тестостерон в более
активный 5-а-дигидротестостерон (рис. 118).
Этот НАДФ-зависимый фермент также
способен восстанавливать двойную связь
в 4-5 положении в С-21 стероидах. При
наследственной аутосомно-рецессивной
аномалии данного энзима развивается
нарушение мужской половой дифференцировки,
ограниченное наружными гениталиями,
некоторыми вторичными половыми
признаками и простатой. Производные воль-
фова протока, развитие которых зависит от
концентрации тестостерона, оформляются
нормально. Мюллеров проток, в основном,
инволюцирует в срок. Однако
предстательная железа недоразвита, оволосение лица
и тела недостаточно, мужская линия роста
волос на лбу и висках не оформляется, и
акнэ в пубертатный период не характерны.
Имеется гипоспадия с открытием уретры в
промежности. Может быть урогенитальный
синус с отдельным отверстием вагины или
слепая рудиментарная вагина. В детстве
часто бывает крипторхизм, но в пубертате
яички нисходят в мошонку, которая растет
и пигментируется. Пенис увеличивается,
и гортань претерпевает маскулинизацию.
Упомянутые особенности зависят от
сохранения тестостерон-зависимых и
выпадения дигидротестостерон-зависимых
вторичных половых признаков. Хотя оба
андрогена имеют идентичный цитозоль-
ный рецептор, очевидно, его аффинитет к
дигидротестостерону в ряде структур выше,
или связывание рецептора с хроматином
более выражено при наличии на рецепторе
восстановленного андрогена. Уровень ЛГ
624 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
увеличен, клетки Лейдига гиперплазиро-
ваны, уровень ФСГ несколько повышен,
что может быть результатом повреждения
сперматогенеза при крипторхизме.
Известно, что для эффективного созревания
сперматозоидов существенны
температурные условия (завершающие этапы процеса
нарушаются при температуре выше 34 °С).
В мошонке температура ниже, чем в
брюшной полости и паховом канале. Именно
этим объясняют снижение сперматогенеза
при крипторхизме, в том числе и в случае
данной 5-а-редуктазной формы мужского
псевдогермафродитизма.
Патофизиология женского
псевдогермафродитизма
Для развития данного синдрома необходим
внутриутробный избыток андрогенов при
наличии у плода кариотипа 46ХХ.
Избыток андрогенов может создаваться
как самим плодом, так и возникать в
материнском организме.
Первая ситуация имеет место при вири-
лизующих формах врожденной гиперплазии
коры надпочечников у девочек. Вторая
бывает либо результатом секреторной
деятельности материнских опухолей -андростером
разной локализации, либо провоцируется
ятрогенно (экзогенно).
Вирилизующиё формы врожденной ад-
ренокортикальной гиперплазии уже были
рассмотрены выше, в главе посвященной
патофизиологии надпочечников. Мальчики с
этими синдромами (кроме недостаточности
3-р-гидроксистероиддегидрогеназы) имеют
при рождении нормальный половой
фенотип. У девочек выраженная вирилизация
наступает при дефицитах 21-гидроксилазы
и 11-р-гидроксилазы. Слабовирилизующим
действием обладает и дефект 3-р-гидрок-
систероиддегидрогеназы, описанный выше
в разделе о мужском
псевдогермафродитизме. Как уже отмечалось выше, дефицит
21-гидроксилазы, ген которой находится в
хромосоме 6, вместилище генов главного
комплекса гистосовместимости наиболее
часто наблюдается у носителей
определённого гаплотипа ГКГС — Bw47. Среди всех
этнических групп болезнь встречается чаще
всего у эскимосов Аляски (с. 554).
Из-за дефицита кортизола нарушения
сопровождаются избытком АКТГ,
нехваткой альдостерона и кортикостерона. Это
вызывает сольтеряющий синдром.
При дефекте 11-р-гидроксилазы, в связи с
наличием гипердезоксикортикостеронемии,
у больных развивается артериальная гипер-
тензия (см. выше).
Для всех синдромов этой группы
характерны промежуточный тип наружных половых
органов при рождении и последующая пос-
тнатальная вирилизация. Сильнее всего это
выражено при наиболее часто
регистрируемом 21-гидроксилазном дефиците. Так как
генотип женский, и тестикулярной ткани нет,
не вырабатывается антимюллеров пептид, и
мюллеровы структуры развиваются
(имеются фаллопиевы трубы и матка). Андрогенов
синтезируется достаточно, чтобы
маскулинизировать наружные гениталии и вызвать
вирилизацию вторичных половых
признаков, но недостаточно, чтобы индуцировать
развитие вольфовых производных. Больные
обоего пола в детстве растут быстро, но
ранняя оссификация метаэпифизарных хрящей
делает их в конечном итоге относительно
невысокими. У мальчиков гиперпродукция
андрогенов подавляет генез гонадотропинов,
поэтому при наличии ярких внешних
признаков возмужалости, а также эрекции и
нормального либидо, сперматогенез в тестикулах
малоэффективен, и могут быть азооспермия
и малые по размеру яички.
Если имеются гормонально-активные
андростеромы коры надпочечников, ар-
ренобластомы или лютеомы яичников, а
также при наличии у матери вирилизую-
щей формы врождённой гиперплазии коры
надпочечников, — материнский организм
вирилизует плод, обладающий кариотипом
46ХХ. Во всех случаях роль защитного
барьера может играть плацента, способная
ароматизировать значительные количества
андрогенов в эстрогены.
Назначение тестостерона и других
андрогенов беременным может вызвать вирилизацию
Нарушения эндокринной регуляции 625
плода женского пола, но такие случаи весьма
редки. Часто назначаемые при беременности
с целью её сохранения прогестины, по
некоторым данным, могут преобразовываться
в слабые андрогены, но их этиологическая
роль в развитии женского
псевдогермафродитизма не доказана. Вместе с тем убедительно
показано, что прогестерон, назначенный при
беременности, в высоких дозах способен
нарушать развитие тимуса у зародышей обоего
пола (Н.И. Чурилова, 1999) — см. ниже
«Патофизиология тимуса как эндокринной
железы». Диэтилстильбэстрол при
назначении беременным вызывает блокаду 3-р-гид-
роксистероиддегидрогеназы и парадоксально
усиливает влияние андрогенов на плод, хотя
сам является эстрогеном. Экзогенные
андрогены не способны вызвать дифференцировку
вольфовых производных у женского плода.
Их маскулинизирующий эффект отражается
только на строении наружных гениталий.
При агенезии мюллеровых структур на
почве аутосомно-доминантных мутаций
развивается полное отсутствие (или
гипоплазия) вагины и гипоплазия (отсутствие)
матки. Могут быть эктопии и дисгене-
зии почек и аномалии строения шейного
отдела позвоночника по типу синдрома
Клиппель-Фейля. Это состояние
сочетается с нормальным женским кариотипом и
функциональными яичниками и известно
как синдром Майера-Рокитанского-Кюсте-
ра-Хаузера. При хирургической коррекции
и восстановлении влагалища у пациенток
с нерезко выраженной гипоплазией матки
возможна даже беременность.
Рассмотрев нарушения половой диффе-
ренцировки, переходим теперь к
эндокринным аспектам регуляции функций гонад и
нарушениям этих процессов.
Гонадотропные функции
Гипоталамо-гипофизарныйнейросекретор-
ный комплекс при некотором
сдерживающем и циклообразующем влиянии эпифиза
(см. выше) осуществляет гонадотропные
функции у лиц обоего пола.
Аркуатное ядро гипоталамуса в импуль-
сно-пиковом режиме выделяет невидоспе-
цифический декапептид гонадолиберин
(рис. 120), стимулятор пролиферации и
секреторной активности гонадотрофов
аденогипофиза. Преимущественно люли-
бериновое или фоллибериновое действие
гонадолиберина на эти клетки зависит от
пермиссивного фонового содержания
различных половых стероидов и от скорости
нарастания концентрации гонадолиберина.
Самой важной детерминантой
селективной стимуляции освобождения ФСГ либо
ЛГ служит частота пульсации секреции
гонадолиберина. Длительная постоянная
секреция или инфузия гонадолиберина
может, наоборот, оказывать гонадостатический
эффект. Гонадолиберин имеется во многих
отделах головного мозга и обладает
поведенческими эффектами. Опиатные пептиды
тормозят, а норадреналин — стимулирует
его продукцию.
Гонадотрофы аденогипофиза,
составляющие в нем до 10% и часто соседствующие
с лактотрофами, образуют под действием
гонадолиберина гликопротеидные гормоны,
близкие по структуре — лютропин (Л Г) и
фоллитропин (ФСГ). Большинство
гонадотрофов вырабатывают оба гонадотропина,
некоторые специализированы. Гонадотро-
пины состоят из вырабатываемой в избытке
общей а-субъединицы, которую разделяют с
ТТГ и хорионическим гонадотропином (см.
с. 502), а также р-субъединицы, имеющей у
каждого из этих регуляторов свою
структуру (рис. 120). В гонадотрофах образуются
сначала прогормоны, затем они процессиру-
ются в гормоны и приобретают углеводные
компоненты путём посттрансляционной
модификации. В небольших количествах
нормальный аденогипофиз выделяет и хо-
рионический гонадотропин.
Л Г имеет у человека молекулярную массу
28,5 кД и содержит более 15% углеводов, а
ФСГ (34 кД) — 16%. Углеводные остатки
необходимы для поддержания
биологической и иммунологической специфичности
гонадотропинов и замедляют их захват и
распад в печени. Так как оба гормона пос-
626 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
5-о -(про)(^ NH2
гонадолиберин
(3 — субъединица лютропина человека
Рис. 120. Структура гонадолиберина (вверху) и р-субъединицы лютропина человека
(по Н.А. Юдаеву и соавт., 1976, Д.К. Клоноффу, Дж.К. Караму 1998)
тупают в кровь нерегулярными пиковыми
импульсами, а время полувыведения фол-
литропина больше, чем лютропина,
концентрации ЛГ в крови характеризуются в
течение суток крайней вариабельностью,
а ФСГ — несколько более постоянны.
ФСГ в яичках индуцирует сперматогенез,
а в яичниках — созревание фолликулов.
ЛГ— стимулятор развития интерстици-
альных клеток семенников и секреции
андрогенов, у самок он также стимулирует
разрыв фолликулов, образование жёлтого
тела и секрецию эстрогенов и
прогестерона. Биологическое действие ЛГ и ФСГ
на мужской и женский организм более
детально рассматривается в следующих
разделах.
Гонадотропиновая секреция
находится под сложным контролем комплекса
гормонов гонад. Подробно эти вопросы
затронуты ниже, при рассмотрении
эндокринных функций гонад. Но вкратце следует
отметить, что малые концентрации
эстрогенов снижают частоту секреторных пиков
гонадолиберина и чувствительность к нему
гонадотрофов. Однако длительное и
существенное повышение концентрации
эстрогенов, напротив, увеличивает освобождение
гонадолиберина и Л Г. Эта положительная
обратная связь устанавливается в конце
периода полового созревания. Прогестерон
в малых концентрациях снижает частоту
импульсов гонадолибериновой секреции,
но при значительном повышении уровня
прогестерона чувствительность
гонадотрофов к гонадолиберину и продукция ЛГ в
гипофизе растут.
Продукция гонадолиберина, ЛГ и ФСГ
подавляется тестостероном. Тестостерон сам
снижает частоту импульсов гонадолиберина
и Л Г, а также переходит в гипофизе в эстра-
диол, который уменьшает чувствительность
гонадотрофов к гонадолиберину. Выше уже
шла речь о том, что именно ослабление этого
сервомеханизма даёт толчок гормональным
изменениям при половом созревании.
Сильным тормозом секреции ФСГ (но
не Л Г) у лиц обоего пола служит продукт
клеток Сертоли и гранулёзных клеток
яичника — ингибин. Противоположное действие
оказывает другой пептид обратной связи тех
же клеток — активин.
Нарушения эндокринной регуляции 627
В ходе нормального полового цикла
женщины концентрации гонадотропинов
меняются сложно-согласованным образом
(см. рис. 125). У мужчин также имеются
ритмические вариации в их продукции.
Детальнее эти вопросы обсуждены ниже.
Постоянно высокий уровень
гонадотропинов в крови характерен для первичного
гипогонадизма и эктопической выработки
этих гормонов, а низкий — для вторичного
гипогонадизма.
Пролактин (лактотропный гормон, ПРЛ)
является аденогипофизарным
биорегулятором соматомаммотрофной группы. Этот
гормон можно считать и регулятором
половых функций, поскольку его мишенями
служат как экзокринная молочная железа,
так и гонады, и анаболическим регулятором,
близким к СТГ и плацентарному лактогену.
ПРЛ — древняя эволюционная находка и
имеется у амфибий и других животных, не
располагающих молочными железами. ПРЛ
животных активен у человека и после 25-го
аминокислотного остатка отличается от
человеческого очень мало (например, у
бычьего и овечьего гормона 95 % общих
аминокислот с человеческим, а у свиного — 98%!
Симпатичное животное с пятачком и здесь
доказывает свою биохимическую близость
к людям).
Гормон синтезируется лактотрофами
аденогипофиза, составляющими 15-20%
его массы. При беременности эта доля может
возрасти до 70 %. Как уже отмечалось выше,
в разделе, посвященном патофизиологии
гипоталамо-гипофизарного нейросекре-
торного аппарата, аркуатное и паравентри-
кулярное ядра гипоталамуса регулируют
продукцию пролактина в основном путём ее
сдерживания через дофамин, выполняющий
роль пролактостатина, путём воздействия на
02-рецепторы лактотрофов.
В связи с этим многие дофаминомимети-
ки являются блокаторами лактации.
Жженый ячмень и другие крупы, входящие в
состав эрзац-кофейных напитков, содержат
дофаминовые соединения, что позволяло
еще древним китайцам использовать эти
продукты для прекращения лактации у
женщин. По этой же причине современные
диетологи не рекомендуют употребление
эрзац-кофейных напитков при кормлении
грудью.
Стимулятором со свойствами пролак-
толиберина является гипоталамический и
гипофизарный аутокринный ВИП.
Существуют и другие кандидаты на роль пролакто-
либерина (см. ниже). При стрессе опиатные
пептиды стимулируют пролактиногенез,
что считается важным условием развития
стресса без дистресса, так как анаболизиру-
ющее действие пролактина и стимуляция
его малыми дозами андростероидогенеза
существенно активируют фазу
восстановления при стрессе и, возможно, позволяют
избежать стрессорного иммунодефицита (см.
т. I, стр. 490-543). С другой стороны, для
опиатных наркоманок характерна аменорея,
что также связано с возможным нарушением
опиатергической регуляции секреции ПРЛ.
Пролактолиберином является и тиролибе-
рин, хотя две эти функции не обязательно
изменяются однонаправленно.
Суточный ритм пролактиногенеза
характеризуется, как и у СТГ, ночным максимумом.
Ген прогормона ПРЛ находится в
хромосоме 6. Из прогормона в лактотрофах
процессируется собственно ПРЛ,
имеющий одну цепь из 198 аминокислот, три
дисульфидных связи и молекулярную
массу порядка 23 кД (рис. 121), который
выделяется в кровоток. Препролактин с
молекулярной массой около 100 кД в
минимальных количествах также секретируется в
кровь. Кроме лактотрофов аденогипофиза,
небольшое количество пролактина
выделяют Т-лимфоциты.
Рецептор пролактина относится к сома-
томаммотрофному типу и опосредует своё
действие через белковые внутриклеточные
и аутокринные посредники (см. выше).
Получены блокирующие и имитирующие
эффекты ПРЛ антитела к его рецептору, что
обосновывает гипотезу о возможности
аутоиммунной резистентности к ПРЛ или
аутоиммунной «псевдогиперпролактинемии».
У человека за пределами гонад, молочных и
628 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Рис. 121. Структура пролактина человека, дополнительно показаны некоторые отличия в
структуре бычьего пролактина — например, в положении 10 (по Н.А. Юдаеву и соавт., 1976)
сальных желез, клеток иммунонеироэндок-
ринного коммуникативно-регуляторного
аппарата не обнаружено значительного
количества рецепторов ПРЛ. У многих
животных рецепторы ПРЛ распространены в
организме более широко.
Пролактин — стимулятор роста и
лактации молочной железы. При половом
созревании усиленный рост железы в основном
регулируется эстрогенами, затем —
эстрогенами и прогестинами. Во время
беременности высокий уровень эстрогенов
стимулирует гиперплазию и гиперфункцию лактот-
рофов аденогипофиза у матери и у плода.
Плацентарный лактоген и, в меньшей мере,
пролактин готовят молочные железы матери
к грудному кормлению, но высокий уровень
эстрогенов пермиссивно ингибирует
действие ПРЛ на лактацию, поэтому продукция
молока наступает только после родов, на
фоне быстрого снижения выработки
эстрогенов. После родов, при начале грудного
вскармливания продукция пролактина
дополнительно стимулируется йри сосании
груди. Во время сосания прирост продукции
ПРЛ зависит от рефлекторно мобилизуемых
серотонинергических воздействий. Гормон
считается также стимулятором секреторной
активности жёлтого тела, откуда его старое
название — лютеотропный гормон, лютеот-
ропин. Эта функция ПРЛ имеет большое
значение при беременности. Гипофизэк-
томия у беременных самок не приводит,
однако, к прерыванию беременности, так как
функции ПРЛ матери дублируются
плацентарным лактогеном и ПРЛ плода.
Как анаболический регулятор пролактин
адресует своё действие иммунной системе.
Рецепторы ПРЛ обнаружены на макрофагах
и лимфоцитах, ПРЛ считается активатором
иммунитета (В.А. Романов, 1986).
Гиперпролактинемия у плода и матери
существенна для предохранения от
негативных эффектов родового стресса и для
иммуностимуляции.
У млекопитающих анаболические
эффекты ПРЛ включают синергизм с СТГ в
стимуляции роста, особенно — внутренних
Нарушения эндокринной регуляции 629
органов, с половыми гормонами — в
индукции вторичных внешних половых
признаков, эритропоэтическое действие, липогене-
тическое действие на жировую ткань. ПРЛ
способствует гипергликемии и оказывает
ренотропный эффект. ПРЛ стимулирует
также рост волос, развитие сальных желёз
и их секрецию. Действие ПРЛ на ЦНС у
всех позвоночных индуцирует стереотипы
материнского поведения.
ПРЛ — физиологический ингибитор
освобождения гонадолиберина, он снижает
продукцию ЛГиФСГ,ав больших дозах — и
половых стероидов. Поэтому после родов
на период грудного вскармливания, с
высоким уровнем ПРЛ в крови, обеспечивается
временный гипогонадизм и естественный
противозачаточный эффект. Очевидно, это
способствует регуляции частоты
беременностей и более полноценному раннему
уходу за потомством. Обнаружен адренокорти-
котропный эффект ПРЛ (Е.Е. Фомичева и
соавт., 2007).
Вместе с тем гиперпролактинемия как у
мужчин, так и у женщин часто служит
причиной вторичного гипогонадизма, аменореи
и бесплодия (см. ниже).
Особенности структуры и функций
тестикул как эндокринных желёз
Нарушения функции тестикул широко
распространены. До 6 % мужчин
бесплодны из-за той или иной формы и степени
тестикулярной недостаточности. Опухоли
семенников являются самой частой формой
злокачественных неоплазий у мужчин 20-
35 лет и третьей по значению
онкологической причиной смертности в этой возрастной
группе. Все эти нарушения часто сопряжены
с эндокринной дисфункцией яичек.
Мужские гонады были, вероятно,
первыми эндокринными железами, относительно
которых медики накопили богатый опыт
наблюдения последствий их экстирпации.
Кастрация, то есть прижизненое
удаление яичек или яичников, упоминается в
древнейших источниках. Она издавна
практиковалась достаточно широко: в
ветеринарии для придания животным спокойного,
терпеливого и выносливого характера, в
ритуальных и религиозных целях — для
достижения спасения души (секты скопцов),
в социальных целях — у людей, которым
поручался присмотр за гаремными
женщинами (евнухи), в виде кары за
изнасилование (что, по сообщениям прессы, и в XX веке
практиковалось пенитенциарной системой
некоторых стран). В период развития
научной медицины появились наблюдения
за результатами кастрации, предпринятой
хирургами по поводу опухолей гонад.
Естественно, на базе этого опыта,
особенно связанного с кастрацией мужчин,
предположение об эндокринной роли гонад,
в частности, яичек — возникло рано. Еще
в конце XVIII века К. Гуфеланд в книге
«Макробиотика» пророчески писал: «Но я
опять напоминаю, что мужское семя
назначено не только для отделения, но ещё более
для обратного поступления в кровь и для
собственного нашего укрепления. Coitus
nimius debilitat — medicus excitat». В 1849 г.
А. Бертольду удалось экспериментально
продемонстрировать наличие у тестикул
внутрисекреторной функции. В 30-х годах
минувшего века усилиями Нобелевских
лауреатов по химии А. Бутенандта и Л.С. Ру-
жички, а также Ж. Давида были выделены
мужские половые гормоны.
В настоящее время известно, что мужская
гонада — яичко (семенник) — служит как
органом сперматогенеза, так и источником
андрогенов, причём это основной орган,
секретирующий в кровь тестостерон и ди-
гидротестостерон. Так, первый на 95 % имеет
у мужчин тестикулярное происхождение.
Установлена также секреция нормальными
семенниками эстрадиола.
Гормоны яичка вырабатывают интер-
стициальные клетки Лейдига, а
сперматогенезом занимаются семенные канальцы,
где находятся клетки Сертоли. Яичко — и
источник, и одна из важных мишеней
андрогенов, поскольку последние имеют
рецепторы в клетках семенных канальцев и
нужны для сперматогенеза.
В клетках Лейдига из готового
холестерина липопротеидов плазмы крови и в мень-
630 А.Ш. Зайчик, Л.П.Чурилов Патохимия*
шей мере из холестерина, синтезированного
de novo, вырабатываются андрогены. Путь
андростероидогенеза в семеннике показан
выше на рис. 118. Он содержит 5 этапов,
катализируемых соответствующими
энзимами, причём лишь пятый этап, зависимый
от 17-р-гидроксистероид-дегидрогеназы,
практически уникален для семенника, а
остальные воспроизводятся и в коре
надпочечников (см. также т. I, с. 517). Ключевой
этап всего процесса — отщепление боковой
цепи холестерина с помощью цитохрома
P450scc и превращение холестерина в прегне-
нолон — находится под контролем Л Г аде-
ногипофиза. Пролактин, стимулирующий
в малых дозах продукцию тестостерона,
напротив, в высоких концентрациях подавляет
андростероидогенез. Кроме того, пролактин
нарушает конверсию тестостерона в более
активный дигидротестостерон.
Взрослый молодой мужчина секретирует
в кровь до 6000 мкг тестостерона в сутки,
причём запас этого стероида в тестикулах в
200 раз меньше, то есть кругооборот андро-
генов характеризуется огромной скоростью.
Дигидротестостерон образуется и в яичке, и
на периферии, во всех мишенях андрогенов
в количестве около 3 000 мкг в день
Интересно и практически важно, что
нормальный мужчина не лишён продукции
женских сексуальных начал.
Периферические ткани содержат аромата-
зу, превращающую у мужчин в норме до 0,3 %
суточной продукции тестостерона и более
1,5% суточной продукции дигидротестосте-
рона в женские половые гормоны — эстрон и
эстрадиол. Эстрадиол, кроме того, возикает и
в самой тестикуле. Таким образом, тестику-
лярный эстрадиол и эстрадиол, полученный
путём конверсии тестостерона и эстрона,
составляет у мужчин до 45 мкг в день. К тому
же периферические ткани вырабатывают в
сутки до 66 мкг эстрона. Особенно
значительная часть этого естественного эстрогенного
фона здорового мужчины вырабатывается
жировой тканью, в этом плане активен
также мозг. Эстрогенная функция липоцитов
повышена при ожирении (см. раздел
«Нарушения липидного обмена») и усиливается с
чдокринно-метаболические нарушения)
возрастом. В дополнение к тестикулярным и
адипоцитарным эстрогенам следы женских
половых стероидов имеются и в секрете
сетчатой зоны коры надпочечников. Эстрогены
мужчины — частичные синергисты и
частичные антагонисты эффектов андрогенов. Они
существенны для подавления продукции
ФСГ при реализации гипоталамо-питуитар-
но-гонадной обратной связи.
В крови гормоны яичек на 50 %
связываются со специфическим сексогормон-связы-
вающим глобулином. Только 3 %
тестостерона свободно, оставшееся количество
связано с альбумином. Последние две фракции
биологически доступны тканям.
Андрогены действуют на
внутриклеточный рецептор, кодируемый в длинном плече
Х-хромосомы. Превращение тестостерона
в дигидротестостерон имеет место прямо в
ядрах клеток-мишеней. И
дигидротестостерон, и тестостерон, который служит для
первого прогормоном, связываются с одним
и тем же рецепторным белком, но комплекс
с участием тестостерона наиболее активно
действует на экспрессию генов в одних
тканях (мышцы, вольфовы производные),
а дигидротестостероновый комплекс — в
других (предстательная и сальные железы,
волосяные фолликулы). Имеются и отличия
в репертуаре программ, запускаемых двумя
основными тестикулярными стероидами
(см. ниже). Некоторые эффекты андрогенов,
например, секреция жидкости семенными
пузырьками, не опосредованы генетически,
а зависят от прямой активации гормонами
выработки цикло-АМФ.
Спектр эффектов андрогенов включает
эмбриональную маскулинизацию развития
производных вольфова протока, контроль
органогенеза молочных желёз, вирилизацию
формирования наружных половых органов,
стимуляцию развития вторичных половых
признаков возмужалости в пубертатный
период. Андрогены способствуют развитию
либидо у лиц обоего пола, а также реализации
мужских психофизиологических стереотипов
полового поведения и мужской тенденции к
выбору активных оборонительных реакций
при стрессе (см. ниже рис. 122, с. 631).
Нарушения эндокринной регуляции 631
Это анаболизирующие
гормоны, способствующие
биосинтезу белка во многих
органах: гениталиях, ЦНС,
скелетных мышцах, скелете, почках
и миокарде, печени, слюнных,
потовых, сальных и молочных
железах. Под влиянием андро-
генов стимулируется синтез
не любых белков, а
связанных с дифференцировочными
программами упомянутых
органов в мужском организме.
Андрогены — стимуляторы
сперматогенеза и
секреторной деятельности простаты,
сальных желёз и семенных
пузырьков (где они не только
увеличивают выход семенной
жидкости, но и обогащают ее
фруктозой, необходимой для
движения сперматозоидов).
Для клеток вышеназванных
желез андрогены служат и
митогенными стимулами. Они
способствуют липолизу,
особенно в гиноидных зонах (см.
«Патофизиология липидного обмена»), в то же время
в органах, гиперплазирующихся под андро-
генным влиянием, эти гормоны усиливают
биосинтез фосфолипидов. Важный аспект
метаболической активности как мужских,
так и женских половых гормонов —
стимуляция окостенения эпифизов длинных
трубчатых костей — связан с продукцией
под их контролем белка остеокальцина и
является основным механизмом,
останавливающим рост конечностей в длину.
Андрогены повышают основной обмен, несколько
способствуют гипергликемии, уменьшают
потери немочевинного азота, ускоряют
катаболизм пуринов.
Важным аспектом их деятельности
служит стимуляция супрессорных функций
лимфоцитов. В вилочковой железе
андрогены способствуют инволютивным
изменениям тимоцитов и эпителия.
Следует отметить физиологическую роль
андрогенов в половой системе женского
II
& I з>
? I I в I
» N
§ II
У \13
Рис 122. Регуляция продукции тестикулярных андрогенов и
их биологические эффекты в пренатальном и постнатальном
периодах. Т — тестостерон, Э — эстрадиол, X — холестерин
организма — они, например, способствуют
нормальной скорости и репертуару
биосинтеза белков в матке и яйцеводах. Еще
раз напомним, что формирование либидо и
сила полового влечения различного знака
зависят у лиц обоего пола от андрогенов.
Именно андрогены — первичные
эротогенные гормоны любого взрослого индивида
(Д. Линде, Д.Гамбург, 1972), в то время
как знак эротических предпочтений зависит
от того поведенческого типа, который был
запрограммирован при дифференцировке
организма и особенно мозга.
Андрогены яичек в различные возрастные
периоды секретируются неодинаково.
Первый максимум их продукции достигается
во 2-м триместре внутриутробной жизни
плода. При этом плазменная концентрация
тестостерона поднимается до 3-4 нг/дл.
Уже в 3-м триместре содержание андрогенов
падает. После рождения на протяжении
первых 3-4 месяцев андростероидогенез вновь
активируется (плазменное содержание
тестостерона составляет до 2,5 нг/дл). Но со
632 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
2-й половины 1-го года жизни и до возраста
11-12 лет в секреторной деятельности яичек
наступает спад. С 12 до 17 лет продукция
тестостерона плавно и быстро нарастает,
достигая после пубертата максимума. До 60
лет она у большинства здоровых мужчин не
снижается, обеспечивая уровень
тестостерона плазмы около 5 нг/дл. В старости
происходит некоторое уменьшение плазменного
уровня тестостерона, особенно заметное на
восьмом десятке лет жизни, но и у
долгожителей он составляет более 2,5 нг/дл. В связи
с длительным характером активной
функции тестикул, даже люди очень преклонного
возраста могут сохранять фертильность.
Американец Джон Шелл (1788-1919) имел
26 детей, из них первого — в возрасте 19
лет, а последнего — в 126. Зафиксирован
уникальный случай рождения у другого
долгожителя трех сыновей в трех разных
столетиях.
Краткие сведения о семяобразующей
функции яичек и эндокринной роли клеток
Сертоли можно для целей данной главы
резюмировать следующим образом. Клетки
Сертоли находятся в семенных канальцах
в составе их сперматогенного эпителия,
выполняя функции своего рода нянек или
поддерживающих элементов для
находящихся тут же гамет и их предшественников.
Их рост и пролиферация стимулируются
андрогенами, без которых сперматогенез
невозможен. ФСГ аденогипофиза стимулирует
созревание сперматид. Этот аденогипофи-
зарный гормон также индуцирует в клетках
Сертоли выработку андроген-связывающего
белка, что позволяет накапливать андрогены
в просвете канальца и удерживать там их
критическую концентрацию, нужную для
сперматогенеза. ФСГ индуцирует в серто-
лиевых клетках продукцию трансферрина,
что необходимо для обеспечения
сперматогенного эпителия железом. По данным
Б.В.Покровского (1976), ФСГ
стимулирует экспрессию рецепторов Л Г на клетках
Лейдига, косвенно способствуя андростеро-
идогенезу. У половозрелого мужчины ЛГ и
ФСГ взаимозаменяемы при поддержании
сперматогенеза.
Клетки Сертоли сами не образуют
тестостерона, но рецептируют андрогены и
превращают тестостерон клеток Лейдига в
дигидроформу, а также в эстрадиол. Этот
процесс стимулируется в них с помощью
ФСГ. Эстрадиол клеток Сертоли паракрин-
но подавляет андростероидогенез в клетках
Лейдига.
Собственная эндокринная активность
клеток Сертоли связана с тем, что они
способны к выработке пептидных
гормонов — ингибина и активина.
Ингибин — сигнал обратной связи в гипо-
таламо-гипофизарно-гонадной оси, может
сильно подавлять продукцию гонадолибе-
рина и, несколько слабее, уменьшать
секрецию ФСГ, но не ЛГ. Это гликополипептид
массой около 10 кД, гомологичный феталь-
ному антимюллерову ингибирующему
полипептиду, который продуцируют плодные
предшественники клеток Сертоли.
Активин — другой полипептид клеток
Сертоли, реализующий механизм
положительной обратной связи с аденогипофи-
зом, стимулирует продукцию в последнем
ФСГ.
При нарушениях сперматогенеза
происходит усиление продукции ФСГ. Константа
спермообразования у взрослого
человека — 74 дня. Следовательно, острые
воздействия на сперматогенный эпителий могут не
сказываться на количестве сперматозоидов
более двух месяцев. Вышеизложенные
положения иллюстрируются рис. 122.
Эндокринные аспекты нарушения
функций гонад у мужчин
Нарушения тестикулярной эндокринной
функции могут быть условно разделены на
мужской гипогонадизм и гипергонадизм.
Возможно развитие их различных форм.
Гипогонадизм у мужчин — шестикуляр-
ная недостаточность, проявляется
нарушением сперматогенеза, гипоандрогенизмом,
часто — недоразвитием вторичных половых
признаков и, как правило, бесплодием.
Гипогонадизм может иметь первичное,
вторичное и внежелезистое происхождение. Из-за
нехватки андрогенов внешний вид больных
Нарушения эндокринной регуляции 633
54
похож на картину,
наблюдавшуюся после кастрации у евнухов
(«евнуходизм»), рис. 123.
Вторичный гипогонадизм
развивается из-за поражения
гипоталамо-гипофизарного
нейросекреторного аппарата и
бывает гипогонадотропным.
Он наблюдается либо как
врождённый, либо как
приобретённый синдром при
множестве разных нарушений.
Врождённые формы
вторичного гипогонадизма чаще всего
связаны с наследственными
дефектами. При синдроме
Кальмана (см. также выше, раздел,
посвященный патофизиологии
гипоталамо-гипофизарного
нейросекреторного аппарата)
имеется сцепленная с Х-хромо-
сомой рецессивная
недостаточность фактора, ответственного
за миграцию гонадолибери-
нергических нейронов из
обонятельного мозга в аркуатное
ядро. Недостаточность
продукции гонадолиберина и
гонадотропинов сочетает признаки
гипогонадизма с аносмией и
дефектами срединных
анатомических структур мозга и
черепа, глухотой и синдактилией.
Больные внешне евнухоидны
и имеют крипторхизм. Частота
этого нарушения находится в
пределах 1/10000-1/60000.
Описан наследственный ау-
тосомно-рецессивный дефект
р-субъединицы Л Г — синдром Паскуалини.
У больных евнухоидный вид, но продукция
ФСГ не нарушена, поэтому сперматогенез
идёт удовлетворительно, хотя из-за гипо-
андрогенизма отмечают олигозооспермию
и поэтому в большинстве случаев фертиль-
ность снижена.
Синдром Мэддока, описанный в виде
единичных случаев неизвестной этиологии,
это сочетание евнухоидизма и хронической
(Т*\
■
i
Рис. 123. Слева - евнухоидизм у подростка (по В. Фальта,
1913) вследствие рано развившегося гипогонадизма.
Справа — последствия кастрации, произведённой во взрослом
состоянии. 49-летний пациент, которому 12 лет назад
удалены по медицинским показаниям тестикулы. Гипогонадизм,
вторичное ожирение
вторичной адренокортикальнои
недостаточности. У пациентов отсутствует продукция
гонадотропинов и АКТГ. Так как клиника
синдрома часто развертывается после
пубертата, его отнесение к врожденной
разновидности вторичного гипогонадизма
вызывает сомнения.
Краниофарингиомы могут нарушать
изолированно продукцию гонадолиберина,
вызывая врождённый вторичный гипогонадизм.
634 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Приобретенный вторичный гипогонадизм
встречается нередко. К нему могут привести
различные факторы, угнетающие
продукцию гонадолиберина и гонадотропинов.
Среди них — тяжелые физические нагрузки
(гипогонадизм спортсменов), голодание,
стресс (см. соотвествующий раздел данной
книги и т. I), аутоиммунные поражения
гипоталамуса и гипофиза (см. выше). Нередко
такой гипогонадизм сочетается с другими
гипоталамо-гипофизарными синдромами и
симптомами — несахарным
мочеизнурением, гипергликемией, нарушениями
термостатической и липостатической регуляции.
Так как в аркуатном ядре имеются и
половые, и липостатические регуляторы, очень
часто гипогонадизм сочетается с гипотала-
мическим ожирением. Поэтому в клинике
существует термин «адипозогениталъная
дистрофия», обозначающий сочетание
приобретенного гипогонадизма и гипота-
ламического ожирения (см. выше в разделе
«Патофизиология липидного обмена»
рис.41, с. 201). Данное понятие обозначает
не нозологически специфическую болезнь,
а симптомокомплекс, который может иметь
различные (в том числе, опухолевые,
травматические, аутоиммунные, инфекционные,
комбинированные) причины, поражающие
дугообразное ядро.
Важной причиной приобретенного
вторичного гипогонадизма у мужчин, и у
женщин служит персистирующая гипер-
пролактинемия.
Гиперпролактинемия у лиц обоего пола
может быть результатом пролактином
гипофиза (реже — соматотропином и адрено-
кортикотропином), а также побочным
эффектом лечения нейролептиками, дофами-
нолитиками, антидепрессантами и другими
лекарствами. Гипоталамическая
гиперпролактинемия может быть следствием
разрушения дофаминергических регуляторов в
подбугорье. При травматическом обрыве
ножки гипофиза он продолжает
функционировать, причем пролактогенез
усиливается. При гиперпродукции тиролиберина
развивается сочетание гиперпролактинемии
и вторичного гипертироза.
У мужчин гиперпролактинемический
гипогонадизм проявляется гипоандрогениз-
мом. Клетки Лейдига гипофункциональны
и могут даже атрофироваться. Семенные
канальцы сохраняются. Пациенты теряют
половое влечение, имеют гинекомастию,
уменьшается сперматогенез. Однако
считают, что не более 15 % мужчин с гиперпро-
лактинемическим гипогонадизмом имеют
настолько выраженные расстройства, что
обращаются к врачу. Тем не менее до 8 %
случаев импотенции и до 5 % случаев
мужского бесплодия вызваны пролактиномами
или периферическими формами
гиперпролактинемии (см. ниже). Гораздо ярче
клиника женского гиперпролактинемического
синдрома. Поэтому более подробно
этиология и патогенез гиперпролактинемических
нарушений рассматриваются в следующем
разделе.
Первичный гипогонадизм у мужчин в
классическом виде наблюдается при
кастрации. Поэтому прежде чем представить
обзор его разнообразной этиологии,
опишем типовые
гормонально-метаболические и патофизиологические последствия
экстирпации тестикул. Многие симптомы
кастрации однотипны у мужчин и женщин
и воспроизводятся в соответствующем виде
при удалении яичников. Поэтому описание
этого синдрома в разделе, посвященном
нарушениям овариальных функций,
повторено не будет. Евнухоидный внешний вид
кастратов воспроизводится полностью или
частично во всех случаях, когда имеется
ранний гипоандрогенизм любой этиологии.
Следовательно, данное описание приложи-
мо не только к евнухоидизму, вызванному
кастрацией (см. рис. 123).
Симптомокомплекс кастрации
развивается при полном удалении гонад. Если
оставить даже небольшую часть ткани семенника
(1 %), компенсаторная гипергонадотропная
гиперфункция оставшейся ткани
предотвратит развитие гипоандрогенизма.
Последствия кастрации тем выраженней,
чем раньше она произведена. У лиц,
кастрированных в зрелом возрасте, нарушения
Нарушения эндокринной регуляции 635
могут быть минимальны и выражаться в
основном в стерилизации. Даже частота
бритья бороды после удаления семенников
у половозрелых пациентов длительное
время остается неизменной, так как многие
морфогенетические эффекты андрогенов
(и, стало быть — их отсутствия)
нуждаются в длительном временном периоде для
своего полного проявления. Более того, при
сформировавшихся стереотипах полового
поведения половозрелые кастраты могут
сохранять длительное время способность
к обычной половой жизни и рефлекторно
поддерживаемое либидо, хотя у них
имеется аспермия. В дальнейшем поздний
гипоандрогенизм выражается в
уменьшении оволосения тела, истончении волос на
голове, снижении либидо, эрекции, могут
быть приливы крови к лицу, как у женщин
в климактерический период. Уменьшается
мышечная сила, возникает геродерма —
постарение кожи, могут быть гинекомастия и
изменение распределения жира по
ганоидному типу.
При кастрации неполовозрелого
организма изменения касаются наиболее широкого
круга органов и систем.
При раннем гипоандрогенизме долго
продолжается рост конечностей в длину, а
развитие пубертатных особенностей таза и
плечевого пояса, костей черепа — наоборот,
задерживается.
Поэтому при евнухоидизме рост
больных высокий за счет изменения длины ног
(нижние конечности составляют не менее
половины длины тела +5 см). Размах рук
превышает длину тела более, чем на 5 см.
В то же время таз и плечи относительно
узкие, затылочная кость несколько уплощена.
Нередко бывает искривление позвоночника,
связанное с отставанием формирования
мышечного каркаса осанки при
гипоандрогенизме. Все это создает вид астеничный,
хотя евнухоиды физически чаще всего
полноценные люди (рис. 123 — слева). При
кастрации, произведенной во взрослом
состоянии, нередко бывает вторичное
ожирение (рис. 123 — справа). Развитие гениталий
«останавливается на том этапе, когда была
произведена кастрация» (Н.В. Медведева,
1947). Полового созревания не наступает,
а выраженность имевшихся вторичных
половых признаков уменьшается. Голос у рано
кастрированных мужчин из-за сохранения
детского строения гортани остается
высоким, горловым, лишен обертонов. Однако
гортань растёт, что создает иногда очень
своеобразный, уникальный вокальный
тембр. Это обстоятельство даже служило в
прошлом причиной кастрации певчих. До
сих пор любителей музыки волнует
драматическая история блистательного
итальянского певца XVIII-ro столетия Фаринелли,
(К. Броски) кастрата с детства, обладавшего
неповторимым голосом, близким к
своеобразному сопрано.
Затянувшийся инфантилизм сменяется
у кастратов ранней старостью, у многих
из них укорочен или отсутствует период
психосоматической зрелости. Из-за гипер-
липопротеинемии быстрее развивается
атеросклероз.
Тем не менее, вышеназванный
исторический пример, а также история средневекового
французского философа П. Абеляра и
хроникальные сведения о евнухах,
занимавших видные военные, политические и
государственные должности в Византии,
Оттоманской Порте и других странах
Востока, — подчёркивают, что при евнухоидизме,
связанном с первичным гипогонадизмом,
индивид остается умственно полноценной
личностью и может быть полноценным
социально, тем более, что происходит
сублимация половой энергии в иных формах
активности.
Метаболические последствия первичного
гипогонадизма и кастрации включают на-
клоность к ожирению (больше — у женщин),
снижению основного обмена, гиперхолес-
теринемии, креатининемии и креатинурии
(больше — у мужчин). Несмотря на контр-
инсулярный характер действия половых
стероидов на обмен глюкозы, кастрация не
ведет к гипогликемии.
Первичная тестикулярная
недостаточность разнообразна по этиологии и бывает
как врожденной, так и приобретенной.
636 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Врожденный характер первичный
мужской гипогонадизм носит при синдроме Клай-
нфельтера (47XXY, частота 1/400-1/500),
который проявляется в тестикулах дизгене-
зией семенных канальцев и андрогенной
недостаточностью. Сюда же относятся многие
случаи мужского псевдогермафродитизма.
Хромосомные нарушения,
обусловливающие первичную тестикулярную
недостаточность, рассмотрены в т. I данной книги
(с. 136), а врожденный мужской
псевдогермафродитизм подробно охарактеризован в
специальном разделе данной главы выше.
Некоторые формы врожденной тестику-
лярной недостаточности вызывают только
дефект сперматогенеза при отсутствии
гипоандрогенизма. Таковы варикоцеле и
крипторхизм (составляющие до 40 % всех
причин мужского бесплодия), а также ау-
тосомно-рецессивный синдром системной
неподвижности клеточных ресничек, при
котором азооспермия сочетается с
хроническими обструктивными бронхитами. Эти
состояния не являются эндокринопатиями
и подлежат рассмотрению в т. III. При
синдроме дель Кастильо имеется наследственная
аплазия герминативных клеток спермато-
генного эпителия, уровень сперматогенеза
резко понижен, а содержание ФСГ
повышено. При этом клетки Сертоли сохранены,
и гипоандрогенизма не наблюдается. Есть
предположение, что аналогичные состояния
могут быть результатом приобретенного
аутоиммунного поражения,
спровоцированного вирусной инфекцией.
Приобретенная первичная тестикулярная
недостаточность, кроме кастрации,
развивается в результате воспаления тестикул
и их придатков. Орхит и орхоэпидидимит
бывают инфекционными (наиболее
характерно их развитие при свинке), а также
аутоиммунными. Аутоиммунные орхиты
могут провоцироваться травмой яичек и
нарушением гематотестикулярного барьера
(модель такого заболевания была получена
на животных Э. Витебским). Провокатором
аутоиммунного орхита может выступить
и вирусная инфекция (эпидемический
паротит, вирус ECHO, арбовирусы). При
свинке от 7 до 12% пациентов мужского
пола в катамнезе развивают гипогонадизм
(В.Н. Тимченко, 1986). Известны
токсические причины аутоиммунного орхита.
Очень характерен аутоиммунный орхит
при хроническом алкоголизме, который, по
некоторым сведениям (Д. ван Тиль, 1977),
отмечается почти у 60 % таких пациентов и
представляет важную причину бесплодия
при алкоголизме, особенно — юношеском
и раннем. При многих системных болезнях
с иммунопатологическим компонентом
гипогонадизм также может быть
результатом аутоиммунных поражений яичек.
Гранулёматозные хронические инфекции
(особенно часто — проказа и туберкулёз)
также поражают яички.
Яички — один из самых радиопоражаемых
органов человека, так как, по закону Ж. Бер-
гонье-Л. Трибондо, радиочувствительность
ткани пропорциональна количеству в ней
незрелых, пролиферирующих элементов,
не обладающих выраженными антиокси-
дантными ресурсами. При дозах более 200
миллиГрей, как правило, наступает
нарушение сперматогенеза и андростероидогенеза,
повышаются уровни Л Г и ФСГ. Патогенез
этих нарушений связан с радиационным
орхитом и ишемией яичек, провоцируемой
их облучением. Сперматогенез и продукция
андрогенов нарушаются под действием ци-
тостатиков, спиронолактона, кетоконазола
и других лекарств.
При хронической печёночной
недостаточности, в частности, циррозе, а также при
хронической почечной недостаточности
у многих больных-мужчин отмечается
вторичный гипогонадизм. Считается, что
при поражении печени это связано с
усиленным переходом андростендиона, не
захватываемого печенью, в эстрогены во
внепеченочных клетках. Гиперэстрогенизм
тормозит продукцию Л Г, что нарушает
работу тестикул.
Больные циррозом печени и нефроскле-
розом имеют гиперпролактинемию. Ее
происхождение не вполне выяснено. Очевидно,
сказывается задержка клиренса ПРЛ. Гипер-
пролактинемия способствует гипогонадизму
и гинекомастии у таких мужчин.
Существование гипогонадизма при первичных парап-
легиях, связанных с поражениями спинного
мозга, свидетельствует об известной роли
вегетативной нервной системы в регуляции
тестикулярных функций. Большое значение
при этом имеют нарушения
кровообращения в денервированных яичках и аномалии
эрекции и эякуляции, которые, как и любая
остановка регулярной эвакуации спермы,
снижают сперматогенез. Однако показано,
что у спинальных больных страдает и
функция клеток Лейдига.
Несмотря на различия в этиологии и
патогенезе клиническая картина первичного
гипогонадизма у мужчин однотипна.
Если первичный гипогонадизм
развивается до полового созревания, формируется,
евнухоидность. У взрослых мужчин
первичная тестикулярная недостаточность, как и
при кастрации, сопровождается выпадением
волос на лице и теле, их истончением на
голове, уменьшается мышечная сила, стареет
кожа, могут нарушаться либидо и эрекции,
возникает олигоспермия. Впрочем, в
некоторых случаях (микоплазменный орхит,
лихорадочные заболевания, целиакия)
сперматогенез страдает сильно, а гипоанд-
рогенизм не возникает.
Третья важная группа причин гипоандро-
генизма у мужчин является внежелезистой
и связана с полным (синдром Морриса)
или частичным (синдром Рейфенштейна)
отсутствием тканевых рецепторов андроге-
нов. Поскольку эти нарушения носят
врождённый характер и относятся к мужскому
псевдогермафродитизму, они подробно
рассматриваются в соответствующем
разделе выше.
Гипергонадизм у мужчин выражается
прежде всего в тестикулярном гиперан-
дрогенизме. При раннем и врожденном
гипергонадизме это связано с синдромом
изосексуального преждевременного
полового созревания. Механизмы нормального
пубертата были описаны выше.
Нарушения эндокринной регуляции 637
Преждевременным считается появление
у мальчиков вторичных мужских половых
признаков и увеличение половых желёз в
возрасте ранее 10 лет. Преждевременное
изосексуальное половое созревание у
мальчиков встречается вдвое реже, чем у девочек.
Оно сопровождается ранним пубертатным
скачком роста с быстрой его остановкой из-
за преждевременного окостенения метаэпи-
физарных зон. Психическое развитие при
этом, как правило, соответствует возрасту,
то есть отстает от физического. Истинное
или полное преждевременное половое
созревание характеризуется активацией как
гормонообразующей, так и сперматогенной
функции тестикул. Ложное (неполное)
манифестирует гиперандрогенизмом и
маскулинизацией вторичных половых признаков,
но без появления сперматогенеза по
взрослому типу. При классическом полном изо-
сексуальном половом созревании
активирована вся ось гипоталамус-гипофиз-гонады,
поэтому в тестикуле гиперфункция
эндокринных элементов сочетается с диффузной
активизацией сперматогенеза. Однако при
многих случаях ложного неполного
преждевременного полового созревания, хотя
первичной активации гонадотропного
механизма нет, он может активироваться
вторично. Кроме того, при автономном, опухолевом
источнике андрогенов в яичке, сверхвысокая
местная концентрация андрогенов может
при низком уровне ФСГ вызвать локальную
активизацию сперматогенеза. Поэтому диф-
ференцировка ложного полового созревания
бывает затруднительна.
Синдром преждевременного полового
созревания может иметь и негонадные
причины. К нему приводят нарушения
функций шишковидной железы (в частности,
ее гамартомы, вырабатывающие хориони-
ческий гонадотропин — см. выше — синдром
Пелицци) и сетчатой зоны коры
надпочечников (см. выше — андростеромы и адрено-
генитальный синдром). При эпифизарных
причинах преждевременного возмужания
активируется и гонадотропная, и гонадная
функции — по истинному типу. Считается,
что возможны и неэпифизарные цереб-
638 АЖ Зайчик, Л.П.Чурилов Патохимия
ральные причины истинного повышения
секреции гонадолиберина и/или гонадо-
либериновой чувствительности аденоги-
пофиза, приводящие к истинному полному
преждевременному половому созреванию.
Среди них называют опухоли, инфекции и
повреждения мозга.
Следует, однако, подчеркнуть, что гона-
дотропин-секретирующие опухоли адено-
гипофиза, хотя они и встречаются нередко
и достигают больших размеров, как правило,
не вызывают клиники гипергонадизма.
Описаны лишь единичные случаи
увеличения тестикул и гиперандрогенизма в
результате опухолевой продукции
аденомами гипофиза, соответственно, ФСГ и
Л Г. В большинстве случаев секретируются
гонадотропины с дефектной структурой
или их отдельные субъединицы. Это может
быть связано с аномалией их гликозили-
рования или с наличием аутоантител к их
субъединицам. Так или иначе, гораздо чаще
гипергонадизма при гонадотропиномах
аденогипофиза развивается гипогонадизм.
Он отличается от первичного гипогона-
дизма высоким содержанием ингибина в
крови (в ответ на гиперпродукцию ФСГ).
Для первичного гипергонадотропного ги-
погонадизма характерен низкий уровень
концентрации ингибина.
При надпочечниковых вирилизующих
синдромах активируется андростероидоге-
нез вне тестикул. Яички остаются
небольших размеров, увеличения продукции го-
надотропинов и активации сперматогенеза
нет. Преждевременное половое созревание
чаще протекает по ложному типу. Изредка
в преждевременном половом созревании
повинна гепатобластома, секретирующая
хорионический гонадотропин.
Гонадные причины преждевременного
возмужания у мальчиков связаны с
доброкачественными (аденомы) и
злокачественными (карциномы) опухолями интерстициаль-
ных клеток семенника. Опухоли насыщают
организм андрогенами. Сперматогенез в
части пораженной тестикулы
стимулируется в соответствии с имеющимися
гормональными и морфологическими изменениями,
(эндокринно-метаболические нарушения)
фертильность также наступает в раннем
возрасте. Отличительной особенностью
опухолевого гипергонадизма является
одностороннее увеличение яичка.
Существуют аутосомная, ограниченная
мужским полом, и рецессивная, Х-сцеплен-
ная формы наследственной неопухолевой
гиперплазии клеток Лейдига. Возможно,
речь идет об их первичной
гиперчувствительности к ЛГ. При этом клиническая
картина соответствует очень раннему
истинному преждевременному половому
созреванию (в возрасте до 2 лет!) с двусторонним
увеличением яичек.
Преждевременное гетеросексуальное
половое созревание у мальчиков
вызывается при избыточном автономном синтезе
эстрогенов в организме. Чаще всего
источником служат аденомы и карциномы из
клеток Сертоли (реже — клеток Лейдига),
если они продуцируют аномально большие
количества женских половых гормонов. При
этом обычно развивается гиперэстрогенная
гинекомастия. Интересно, что эстрогены
могут образовываться самими опухолевыми
клетками или оставшейся здоровой частью
тестикул — под влиянием опухолевого хори-
онического гонадотропина. В первом случае
продукция собственных гонадотропинов и
тестостерона в неопухолевой части
семенников понижена. Имеется азооспермия. Во
втором случае, наоборот, — уровень
гонадотропинов в плазме высокий, продукция
тестостерона в неопухолевой части гонад
повышена, и азооспермия не характерна.
Также описаны и нетестикулярные
опухоли, вызывающие данный синдром
(например, локализованные в коре
надпочечников).
Вообще мужские гонады нередко
поражаются опухолями (2-6 случаев на 100000
мужского населения). К опухолям яичек
предрасполагает высокий уровень
гонадотропинов, в частности, при первичном ги-
погонадизме, некоторых формах мужского
псевдогермафродитизма и синдроме Кляйн-
фельтера. В 40 раз выше риск этих
новообразований при неопущении яичка в мошонку.
Нарушения эндокринной регуляции 639
Самые распространенные неоплазмы
тестикул (95% от общего числа) —
злокачественные семиномы (герминомы из
гаметогенных клеток эпителия), затем
идут дизэмбриогенетические опухоли.
Дизэмбриогенетические опухоли яичек —
эмбриокарциномы, хориокарциномы,
опухоли из желточного мешка, тератомы и
тератобластомы — отличаются продукцией
хорионического гонадотропина.
Специфическим маркером подобных опухолей
считается р-субъединица этого гормона.
Семиномы выделяют её не более, чем в 10 %
случаев, а другие вышеназванные
неоплазмы — в 90-100%. Ещё один маркер этих
опухолей — а-фетопротеин. Хорионический
гонадотропин опухолевого происхождения
влияет на продукцию половых стероидов
(см. выше), а а-фетопротеин обладает
стимулирующим действием на тромбоциты, в
частности, побуждая их к фрустрированно-
му фагоцитозу.
Около 5 % опухолей яичка представлены
неоплазмами из клеток Лейдига и клеток
Сертоли. Эти опухоли в 9/10 случаев
доброкачественны и образуют половые стероиды.
В зависимости от знака этих гормонов они
могут вызвать феминизацию или
ускоренную маскулинизацию у своих носителей.
Эндокринные функции женских
половых органов
Практически все органы женской половой
системы способны к эндокринной и парак-
ринной активности.
Яичники — женские гонады, органы
овогенеза и источник более чем 30 гормонов и
паракринных биорегуляторов, влияющих
на циклическое осуществление женских
половых функций. Без нормальной функции
яичников невозможна беременность.
Яичник содержит под капсулой корковый
слой, где расположены фолликулы, основная
герминативная и гормонообразующая часть
органа. Большая часть из них — незрелые
примордиальные фолликулы, имеющие
тонкий слой фолликулярных клеток -гранулёзу
— и окружённые базальной мембраной.
Часть фолликулов в яичнике половозрелой
женщины находится на разных стадиях
созревания и инволюции. Наиболее крупные
могут развивать толстую оболочку — теку.
Здесь же могут обнаруживаться желтое тело,
а также результат его инволюции — белые
тела. Клетки гранулёзы и внутренней части
теки, а также клетки желтого тела являются
стероидогенными. Они способны к выработке,
соответственно, эстрогенов, андрогенов и про-
гестинов. Но половыми стероидами гормоно-
образование в яичниках не исчерпывается.
Некоторое представление о
чрезвычайном богатстве внутрисекреторных
потенций яичников и о сложности овариальной
регуляции даёт нижеследующая таблица
(табл.41). Поистине, уникальное
разнообразие регуляторных факторов яичника
заставляет с уважением вспомнить
доктрину преформистов, размещавших в данном
органе «бесконечный ряд гомункулусов».
В рамках данного раздела
рассматриваются лишь основные эндокринные аспекты
функций яичников, а ниже представлено
обсуждение почти исключительно эндок-
ринно-метаболических аспектов
овариальной патологии.
Во время внутриутробного развития
эндокринная функция яичников не требуется
для проявления признаков женского пола.
Как уже описывалось выше, при отсутствии
гена SRY, эмбриональная гонада и мюлле-
ровы структуры развиваются в яичник и
внутренние гениталии спонтанно, а вольфов
ход регрессирует. В фетальном яичнике
закладывается до 7 млн овогониев,
развитие которых останавливается в профазе
мейоза, к рождению их остается до 1 млн,
к 10 годам — не более 0,4 млн. Небольшая
часть этого пула будет использоваться в
половозрелом организме на протяжени
всего фертильного периода. Таким образом,
овоцит до его созревания и оплодотворения
может жить десятки лет. Это способствует
кумуляции мутаций в овоцитах и делает
риск генетических дефектов плода у
пожилых женщин много большим.
640 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 41. Овариальные гормоны и аутакоиды
(поДж. Окленду, В.Лингаппе, 1997, модифицировано)
Биорегулятор
Андрогены
Эстрогены
Прогестины
Ингибин (фолликулостатин)
Активин
Антимюллеров ингибирующий
полипептид
Фоллистатин
Релаксин
Активатор и ингибитор мейоза
ооцитов
Фолликулярный регуляторный
белок (ингибитор секреции и роста
клеток гранулёзы)
Активатор плазминогена
Внеклеточные мембранные белки
Инсулиноподобный фактор роста
(ИФР)-1
Аналог эпидермального фактора
роста (ЭФР)
Трансформирующий фактор роста
(ТФР)-а
Основной фактор роста фиброб-
ластов
Трансформирующий фактор роста
(ТФР)-Р
Тромбоцитарный ростовой фактор
(ФРТ)
Фактор роста нервов
Проопиомеланокортин (ПОМК)
Энкефалин
Динорфин
Гонадолиберин
Окситоцин
Вазопрессин
Ренин
Ангиотензин II
Атриопептин
Источник
Тека, интерстициальные клетки
Гранулёза, желтое тело
Жёлтое тело
Гранулёза, тека, жёлтое тело
Гранулёза
Гранулёза, cumulus oophorus
Фолликулы
Жёлтое тело, тека, плацета, матка
Фолликулярная жидкость
Фолликулярная жидкость,
гранулёза, жёлтое тело
Гранулёза
Гранулёза, фолликулярная
жидкость
Гранулёза
Гранулёза, тека
Тека, интерстиций
Жёлтое тело
Гранулёза, тека, интерстиций
Гранулёза
Яичник
Жёлтое тело, интерстиций,
гранулёза
Яичник
Яичник
Гранулёза, фолликулярная
жидкость, другие структуры
Гранулёза, жёлтое тело
Яичник, фолликулярная жидкость
Фолликулярная жидкость, тека,
жёлтое тело
Фолликулярная жидкость
Фолликулярная жидкость, желтое
тело, яичник
Факторы, управляющие его
продукцией
ЛГ
ФСГ
Л Г, пролактин
ФСГ, ЭФР, ИФР 1, гонадолиберин,
ВИПДФР
ФСГ
ЛГ,ФСГ
ЛГ,ФСГ
ПРЛ,ЛГ,окситоцин, Рд
?
ФСГ, гонадолиберин
?
ФСГ, гонадолиберин
СТГ, ФСГ, ЛГ, ЭФР, ТФР, ФРТ,
эстрогены
Гонадотропины
ФСГ
7 1
ФСГ, ТФРа, фибронектин
? 1
7 1
7 1
7 1
7 1
7 1
ЛГ,ФСГ, Pg F2a
7 1
ЛГ,ФСГ
Ренин 1
7 1
Нарушения эндокринной регуляции 641
Окончание таблицы 41
Биорегулятор
Ингибитор и стимулятор
образования желтого тела
Ингибитор секреторных всплесков
гонадотропинов
Блокатор связывания ЛГ с
рецепторами
Нейропептиды (Y, кокальцигенин,
вещество Р, соматостатин, гистидил-
метиониновый пептид)
Вазоактивный кишечный пол и
пептид (ВИП)
Онкобелок c-mos
Эйкозаноиды, в том числе — про-
стагландины (Рд)
Источник
Фолликулярная жидкость
Гранулёза
Жёлтое тело
Нервные окончания
Нервные окончания
Ооциты
Повсеместно
Факторы, управляющие его
продукцией
7
7
7
?
7
Эмбриогенетические
Различные
Во втором триместре беременности
временная активация продукции
гонадотропинов обеспечивает созревание гона-
достата — системы гипоталамус-гипофиз-
яичник, связанной обратными связями в
функциональное целое. Затем продукция
гонадотропинов понижается.
В детстве секреция гонадотропинов у
девочек стабильно низкая. Незрелый яичник
вырабатывает минимальные количества
половых стероидов. В момент родов
большое значение имеют изменения в уровне
половых гормонов, связанные со стероидо-
генной активностью плодоплацентарного
комплекса. Плацента насыщает организм
плода эстрогенами. В первые дни после
родов их уровень падает. Эти изменения
обусловливают особое переходное
состояние новорожденных — гормональный
криз, или синкаиногенез. Он проявляется у
60-70 % новорожденных, чаще — девочек.
Симптомы синкаиногенеза, в период 3-6
дня жизни включают нагрубание молочных
желёз, угри, десквамативный вульвовагинит,
арборизацию носовой слизи, иногда отек
кожи наружных гениталий. Падение
уровня эстрогенов на 5-8 день жизни вызывает
бурную реакцию матки новорожденной
девочки, эндометрий отторгается, и наступает
метроррагия, длящаяся 1-2 дня. Продукция
гонадотропинов на фоне быстрого падения
уровня половых стероидов временно
возрастает. Все эти изменения иногда образно
характеризуют как «малый пубертат». И в этот
период, и в дальнейшем источники основной
части половых гормонов — неовариальные.
В последующем детстве у девочек половые
стероиды надпочечникового
происхождения играют большую роль для организма
(адренархе).
При половом созревании девочек, которое
начинается в 9-11 лет, происходит
установление импульсного характера секреции
гонадолиберина в гипоталамусе и
повышается чувствительность гонадотрофов к
гонадолиберину, а секреция гонадотропинов
также приобретает пульсирующий характер.
Эпизодические повышения гонадотропной
функции и стероидогенеза в гонадах у
девушек-подростков происходят во сне.
По достижении критической массы тела
в 48 кг или критической комбинации массы
жировой ткани, воды и общего веса тела
гипоталамус девочек делается менее
чувствительным к обратному ингибирующему
влиянию половых стероидов. У девушек с
ожирением это может происходить раньше, а
при пониженной массе тела — задерживаться.
Подобная связь анатомически закреплена
перекрыванием в дугообразном ядре центров
642 А.Ш. Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
продукции гонадолиберина и зон,
ответственных за эффект липостата (см. выше
«Патофизиолология липидного обмена»).
Увеличенная секреция овариальных
гормонов приводит к развитию полного
комплекса вторичных женских половых
признаков (рис. 119).
В дальнейшем у половозрелых женщин в
течение всего детородного периода
поддерживается циклический характер гонадо-
тропиновой и гонадной секреции,
описанный подробно ниже. В постменопаузальном
периоде из-за израсходования фолликулов
продукция овариальных гормонов резко
снижается, а аденогипофизарных гонадо-
тропинов — наоборот, значительно растет.
Обычно менопауза наступает в 50-51 год.
Гонадотропная функция гипоталамо-ги-
пофизарного нейросекреторного аппарата,
в норме характеризуется в половозрелом
женском организме следующим.
При низком пермиссивном уровне
эстрогенов пики гонадолибериновой секреции
следуют часто и гонадолиберин стимулирует
в аденогипофизе преимущественно
продукцию ФСГ. При высоком пермиссивном
уровне эстрогенов пики гонадолибериновой
секреции становятся реже, а в аденогипофизе
стимулируется в основном продукция ЛГ.
Гонадотропины оказывают своё действие
на яичник через рецепторы семейства,
связанного с G-белками, и циклонуклеотид-
протеинкиназные посредники.
Принципиально для яичника ФСГ
является стимулятором роста и пролиферации
клеток гранулёзы, экспрессии рецепторов
ЛГ, активатором конверсии андрогенов в
эстрогены (ароматазная реакция).
ЛГ для яичника служит стимулятором
андростероидогенеза во внутренней части
теки (в основном как и в тестикулах, на
этапе превращения холестерина в прегне-
нолон), а также способствует превращению
гранулёзных клеток в лютеиновые,
формируя желтое тело.
Стероидогенез в яичниках и биологическая
характеристика овариальных стероидов
обладают некоторыми важными особенностями.
В яичнике синтезируются андрогены
(клетками интерстиция и теки под
контролем Л Г), эстрогены (путем ароматизации
андрогенов в клетках гранулёзы, а также в
желтом теле — под контролем ФСГ) и про-
гестины (в клетках желтого тела, под
контролем Л Г). Пути биосинтеза этих гормонов
показаны на рис. 124. Как и адренокортико-
циты, овариальные клетки предпочитают в
качестве сырья для стероидогенеза готовый
ХН из ЛПНП плазмы крови, хотя могут
производить ХН и сами, из ацетата.
Главный овариальный андроген (С-19-
стероид) — андростендион. Его собственная
андрогенная активность мала. Он
преобразуется в эстрогены, в основном —
яичником, но также жировой тканью и другими
органами. Возможно и преобразование ан-
дростендиона в тестостерон. Превращение
андростендиона в тестостерон у женщин в
норме незначительно. Оно может
усиливаться при наличии андростером яичников
и при понижении ароматазной реакции в
яичниках. Это приводит к гирсутизму и
даже вирильности. (см. ниже). Метаболизм
и биологические эффекты андрогенов
рассматривались выше.
Эстрогены яичников (С-18-стероиды) —
это образуемый из андростендиона эстрон
(Et) и дериват тестостерона — эстрадиол
(Е2). Адипоциты и другие экстраовариаль-
ные ткани дают преимущественно Ej. У
женщин детородного возраста преобладает
Е2, так как доминирующий фолликул и
желтое тело синтезируют в основном именно
его. В постменопаузальный период, при
отсутствии функционирующих фолликулов,
большее значение приобретает Ej. Из Et и Е2
в почках, печени и других тканях может
формироваться эстриол (Е3), основной эстроген,
выводимый с мочой. При беременности пло-
до-плацентарный комплекс подключается к
продукции эстрогенов, поставляя, главным
образом, эстриол (Е3). Биологическая
активность Е^Ез/Ез соотносится примерно
как 1/10/3. Печень — главный орган
катаболизма и взаимопревращения эстрогенов.
Синтез оксипроизводных эстрогенов в
печени и других тканях стимулируется ти-
Нарушения эндокринной регуляции 643
РОИДНЫМИ ГОрМОНаМИ, а ПрОДуК- Фолликулярнаяфаза
\
лютенновая фаза
ция эстриола при гипертирозе,
наоборот, снижается. В печени
эстрогены подвергаются также
орто-метилированию до катёхол-
эстрогенов, которые выводятся с
мочой.
Интересно, что у жеребцов
основным местом продукции эстра-
диола, которым их моча
уникально богата, служат ... семенники,
содержающие эстрогенов намного
больше, чем яичники кобыл. Хотя
он
/ 7а-гидроксипрегненолон
НО
■ОН
НО"
НО*
НО"
16а-гидроксиэстрон
17$-эстрадиол
Рис 124. Пути стероидогенеза в яичниках.
Пояснения в тексте
В.В. Маяковский (1914) отмечал: «Деточка,
все мы, немного —: лошади!..», у приматов
продукция эстрогенов в семенниках
минимальна, а в надпочечниках — несравнимо
меньше, чем в яичниках, поставляющих у
женщин 95 % этих гормонов.
Эстрогены — гормоны-индукторы
считывания генетических программ,
ответственных за развитие вторичных женских
половых признаков. Они действуют через
образование комплекса в цитозоле клеток-
мишеней с рецепторным белком эстрофи-
лином I. Комплекс стероида и рецептора
(эстрофилин-П-58-стероидный комплекс)
поступает в ядра клеток, где
взаимодействует с цис-регуляторными элементами
хроматина. Возможно, в некоторых тканях,
в частности, матке внутриядерному
транспорту комплекса содействует а-фетопроте-
ин. Эстрогены осуществляют стимуляцию
роста в ряде органов, прежде всего, половых.
Они способствуют росту матки и протоков
молочных желез. В связи с этим гиперэс-
2-гидроксиэстрон
и другие метаболиты
2-гидроксиэстрадиол
и другие метаболиты
трогенизм является фактором риска для
развития миом матки, а также рака грудной
железы. Эстрогены стимулируют
окостенение метаэпифизарных хрящей и
останавливают рост тела в длину у девушек. При
нехватке эстрогенов отложение кальция
в костях нарушается, что лежит в основе
остеопороза, развивающегося у женщин в
постменопаузальный период. В то же время
в организме половозрелой женщины в
детородный период эстрогены способствуют
повышению содержания кальция в крови и
его отложению в длинных трубчатых костях,
вызывая потерю кальция тазовыми костями.
В сальных железах и кожных волосяных
фолликулах эстрогены тормозят
пролиферацию клеток и секрецию.
Внутриклеточным медиатором ростовых
эффектов эстрогенов во многих тканях
служат орнитиндекарбоксилаза и её
продукты — полиамины путресцин и спермидин
(см. т. I, с. 147,276,321).
Эстрогены обладают способностью
стимулировать биосинтез фосфолипидов,
644 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
особенно — в половых органах, но также и
в печени. С этим связаны их липотропный,
антиатерогенный и гиполипопротеинеми-
ческий эффекты.
Другие аспекты действия эстрогенов
рассмотрены ниже при описании овариально-
менструального цикла.
Так как именно андрогены служат в
нормальном женском организме основными
предшествениками эстрогенов, остаётся
лишь удивиться прозорливости 3.
Фрейда, который, задолго до открытия половых
стероидов (1921) писал: «...в каждом
человеке имеются мужские и женские элементы,
только, в соответствии с принадлежностью
к тому или иному полу, одни несоизмеримо
более развиты, чем другие, поскольку дело
касается гетеросексуальных лиц».
Единствеными реально значимыми
природными прогестинами (С-21-стероиды) у
человека служат прогестерон и 17-гидрок-
сипрогестерон. Их вырабатывают интер-
стициальные и текальные клетки яичника,
но, особенно — желтое тело (временная
эндокринная железа, формируемая
внутри яичника из остатков фолликула после
овуляции — см. ниже). В желтом теле
биосинтез прогестерона примерно в 30-40 раз
эффективнее, чем в других овариальных
структурах. Прогестерон вырабатывается
и в плаценте (см. ниже). Некоторые
женщины вырабатывают необычный прогес-
тин — 20-(3-оксипрегнандиол. Показано,
что, попадая через грудное молоко к
новорожденному, это соединение блокирует
образование диглюкуронида билирубина
печенью ребенка и вызывает продлённую
неонатальную гипербилирубинемию.
Транспорт прогестерона в крови
осуществляют транскортин и орозомукоид.
Прогестины, гормоны-индукторы
программ беременности, действуя через двуком-
понентный цитоплазматический А-В—
рецептор, обеспечивающий внутриядерное
проникновение гормона и его
взаимодействие с негистоновыми белками хроматина,
осуществляют комплекс изменений,
которые нужны для имплантации и гестации.
Они активируют секрецию желез
эндометрия, децидуальную реакцию, ингибируют
сокращения миометрия, тормозят на
центральном уровне окситоциновую секрецию в
ответ на растяжение влагалища. В грудных
железах прогестины способствуют
гиперплазии железистых клеток.
Прогестерон ускоряет дифференцировку
тимоцитов, обладает иммуносупрессорным
эффектом. Имеются наблюдения о связи
между прогестероном и уровнем продукции
белков острой фазы в печени. Прогестерон
также является термогенным гормоном,
повышая уровень температуры тела в лю-
теальную фазу овариально-меструального
цикла. Другие эффекты прогестерона
рассмотрены ниже.
Интегральная характеристика
ритмических эндокринных функций гипоталамо-ги-
пофизарно-овариального аппарата в норме
сводится к следующему.
В пубертате, под влиянием импульсно-
циклических колебаний секреторной
активности гонадотрофов, начинается
циклическое созревание и атрезия групп фолликулов
(от 3 до 30 за один раз) в яичнике и
соответствующие цикловые изменения в уровне
эстрогенов и прогестинов. Эти колебания
заставляют эстроген-зависимые ткани и
органы претерпевать созревание и фазовые
изменения. Проявлением этих
взаимодействий гипоталамуса, гипофиза, яичников и
матки служит овариально-менструальный
цикл, а установление первого такого периода
носит название менархе. В ходе
осуществления менструального цикла периодически
меняется и активность других эндокринных
желез — щитовидной, надпочечников, а
также продукция ряда белков в печени, функция
всех органов-мишеней овариальных
гормонов. Ритмичность деятельности яичника, а
также клеток-мишеней его гормонов связана
с необходимостью обеспечить оптимальные
условия, с одной стороны, для полового акта
и оплодотворения яйцеклетки, с другой -
для сохранения и развития зиготы.
Циклическая деятельность гипоталамо-
гипофизарно-гонадной оси и тканей-ми-
Нарушения эндокринной регуляции 645
эндометрий
шенеи овариальных гормонов
продолжается вплоть до возрастного уменьшения
гонадотропной и гонадной эндокринной
активности (климактерического периода).
Циклизм заключается в чередовании
фолликулярной фазы (когда преобладают
продукция андрогенов в теке яичника и их
ароматизация в эстрогены в гранулёзе) и лю-
теальной фазы когда остатки разорванного
при овуляции зрелого фолликула насыщают
организм прогестероном).
В начале очередного цикла яииник
(рис. 125) под влиянием го-
надолиберина усиливаются
секреция ФСГ и, с некоторым
отставанием по фазе, — также
ЛГ. Под влиянием ФСГ в
яичнике группа приморди-
альных фолликулов начинает
созревать и расти. Обычно
лишь один фолликул из
группы достигает пика созревания
и даёт начало зрелой
яйцеклетке, которая и выйдет в
половые пути женщины в
день овуляции, на границе
фолликулярной и лютеаль-
ной фаз. Л Г при участии ФСГ
стимулирует в яичнике также
синтез, превращения и
секрецию стероидных гормонов.
Клетки теки, побуждаемые
Л Г, вырабатывают андрогены,
и те диффундируют в клетки
гранулёзы, где превращаются
с участием фермента аромата-
зы, чувствительного к ФСГ, в
эстрогены (17-р-эстрадиол и,
в меньшей степени, эстрон,
эстриол). В результате
уровень эстрогенов нарастает в
течение всей фолликулярной
фазы. Под влиянием
эстрогенов эндометрий в матке
пролиферирует, утолщается
и приобретает прямые
железы, содержащие жидкий
секрет. Вагинальный
эпителий накапливает гликоген,
слущивается, и животный крахмал
переходит в лактат, обусловливая снижение рН
вагинального содержимого и предохраняя
половые пути от развития ряда
патогенных микроорганизмов. В самом яичнике
эстрогены, вместе с ИФР-1, стимулируют
пролиферацию фолликулярных клеток, что
еще больше увеличивает фолликул.
Первичные фолликулы переходят во
вторичные. Появляется доминантный фолликул,
11
примордиаль- созревающий овуляция
иый фолликул граафов пузырек
желтое тело
инволюцирующее
желтое тело
менструация
14
Дни
овариально-менструального цикла
Рис. 125. Ритмические гормональные и физиологические
изменения при нормальном овариально-менструальном цикле
(по А. Уайту и соавт., 1981; А.А. Пищулину, 1991)
646 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
опережающий в росте все прочие и
приобретающий толстую оболочку — теку.
Наружная тека имеет соединительно-тканную
структуру, а внутренняя тека — представлена
интерстициальными клетками, близкими к
клеткам Лейдига тестикул и образующими
андрогены. Эстрогены и ФСГ стимулируют
экспрессию рецепторов Л Г на клетках теки и
гранулезы. Это ведет к началу стимуляции с
помощью ЛГ продукции андрогенов в теке,
эстрогенов и, особенно — прогестерона в
гранулёзе в конце фолликулярной фазы.
Андрогены замедляют дальнейшую
пролиферацию клеток гранулезы. Прогестерон
стимулирует выброс ФСГ и его пик в середине
цикла. ФСГ усиливает транспорт жидкости
в полость фолликула. Вторичный фолликул
увеличивается в диаметре и превращается
в третичный (граафов пузырёк). Под
влиянием эстрогенов наступает срединный пик
секреции ЛГ.
В середине цикла (чаще на 11-13 день),
на фоне всплеска в секреции обоих гонадо-
тропинов, вызванного гиперэстрогенизмом
ипрогестеронемией, а также максимума
продукции яичниковых андрогенов,
доминантный созревший фолликул разрывается,
нарушая целостность стенки яичника.
Яйцеклетка выходит в яйцевод и далее
движется к эндометрию. Создается возможность её
оплодотворения.
Остаток фолликула под действием ЛГ
превращается в желтое тело. Клетки
желтого тела под действием Л Г и пролактина
синтезируют и выделяют большое количество
эстрогенов и особенно — прогестерона. При
этом уровень ФСГ и Л Г понижается, так как
прогестерон инактивирует положительный
механизм обратной связи, и секреция адено-
гипофиза временно находится лишь под ин-
гибирующим влиянием 17-р-эстрадиола.
Прогестин-эстрогеновая стимуляция
эндометрия приводит к превращению прямых
желёз в извитые, жидкий секрет
трансформируется в густой, начинается его активное
выделение (секреторная фаза развития
эндометрия). Прогестерон поддерживает
тонус шейки матки и делает миометрий тела
матки менее возбудимым.
При быстром ингибировании прогестина-
ми и эстрогенами секреции Л Г, желтое тело,
зависящее от его тропного действия, может
сохраняться лишь недолгое время, уровень
прогестерона достигает акрофазы на 8-9
день после овуляции, и corpus luteum инво-
люцирует в пределах 2-х недель. Это ведет
к стремительному падению концентраций
эстрогенов и прогестерона. В отсутствие
этих гормонов эндометрий не может больше
поддерживаться в секреторной фазе и
отторгается. Следует менструация, которая
начинается на фоне минимального содержания
прогестиновых и эстрогеновых стероидов
и знаменует окончание трёхфазного цикла,
средняя общая продолжительность которого
составляет около 28 суток. Сама
менструальная фаза цикла длится 3-5 дней.
Надо отметить, что эндометрий матки как
в небеременном состоянии, так и при
беременности является источником нескольких
гормонов и множества цитокинов. Здесь
вырабатываются пролактин, релаксин, ренин,
опиаты, кортиколиберин, кокальцигенин,
фибронектин, разнообразные факторы
роста, интерлейкины и эйкозаноиды.
На основе этих потенций при
формировании плаценты с участием плода беременная
женщина обретает новый мощный
эндокринный орган.
При этом в случае оплодотворения и
наступления беременности характер
эндокринной активности гонад меняется и
система выходит из циклов путем продления
прогестиновой фазы за счет образования
формирующейся плацентой хорионического
гонадотропина. Имплантация следует как
раз на пике лютеальной секреции
прогестерона. С 11-12 дня после оплодотворения
концентрация хорионического
гонадотропина удваивается каждые 2-3 дня. Секреция
хорионического гонадотропина, в отличие
от Л Г и ФСГ, не ингибируется прогестинами
и эстрогенами, поэтому он поддерживает в
сохраняющемся желтом теле беременности
длительную прогестероновую секрецию,
обеспечивающую с помощью высокого
уровня прогестинов нормальное протекание
беременности, вплоть до становления сте-
роидогенной функции самой плаценты, не
менее чем в течение 6-8 недель. К тому же
беременная матка перестает выделять про-
стагландины, оказывающие при обычных
циклах лютеолитический эффект.
Пик секреции хорионического гонадо-
тропина приходится на 80-й день
беременности.
Примерно с конца 11-й недели
беременности продукция хорионического гонадо-
тропина уменьшается, но беременность
развивается при стабильно высоком уровне
прогестерона благодаря плаценте, которая
работает вместе с плодом как единый стеро-
идогенный комплекс, превращая фетальные
андрогены — в эстрогены и вырабатывая
прогестины.
Плацента — активный
полигормональный эндокринный орган. Например, синтез
эстрогенов жёлтым телом и плацентой столь
мощен, что за 9 месяцев гестации у
женщины образуется больше эстрогенов, чем за
100 лет непрерывных овариально-менстру-
альных циклов.
Помимо описанного выше
хорионического гонадотропина и половых стероидов (в
основном, эстриола и прогестерона)
плацента вырабатывает еще ряд гормонов.
Хорионический соматомаммотропин (он
же — плацентарный лактоген) — гомолог
СТГ и ПРЛ, стимулирующий при
беременности развитие молочных желёз. Его уровень
в крови беременных растет с б-й недели и до
6-го месяца беременности. Данный гормон
в настоящее время считают основным
сигналом, обусловливающим метаболические
изменения в организме женщин,
характерные для беременности. Под его влиянием
происходит адаптация к метаболической
ситуации своего рода «ускоренного
голодания», создающейся при беременности. Этот
контринсулярный регулятор предохраняет
плод от гипогликемии. При беременности
быстрый отток субстратов
энергетического метаболизма к плоду и их утилизация
создают постоянную тенденцию к гипогли-
Нарушения эндокринной регуляции 647
кемии. Плацентарный лактоген усиливает
липолиз, повышает уровень свободных
жирных кислот в крови матери, а также
содержание глюкозы и, наконец, кетоновых
тел. Именно поэтому беременные женщины
предрасположены к развитию кетоацидоза.
Известна также связь беременности и
декомпенсации сахарного диабета (см. выше в
разделе «Патофизиология сахарного
диабета» — о прегнодиабете). Наряду с глюкозой,
благодаря действию этих
гормонально-метаболических механизмов, кетоновые тела
служат важным источником энергии для
плода и беременной матери (см. выше о ке-
тоацидозе — в разделе «Патофизиолология
липидного обмена»).
Пролактин (ПРЛ) — вырабатывается в
плаценте клетками синцитиотрофобласта.
Высокий уровень ПРЛ при беременности
поддерживают аденогипофиз матери и
плода, а также децидуальная ткань плаценты.
ПРЛ стимулирует иммунитет, функции
желтого тела беременности, готовит к
лактации молочные железы и участвует в
регуляции водно-солевого обмена у плода.
Вместе с плацентарным лактогеном он
участвует в вышеописанной
метаболической перестройке организма беременной
женщины.
Плацентарные аналоги СТГ и сам гормон
роста, образуемые плацентой могут
обусловить некоторое увеличение мягких тканей и
хрящевых органов при беременности
подобно тому, как гормон роста вызывает это при
акромегалии.
Релаксин (молекулярная масса около
6 кД), близкий к инсулину и ИФР, содержит
две полипептидных цепи (22 и 32
аминокислоты с двумя дисульфидными мостиками),
выделяется цитотрофобластом (и желтым
телом), служит гормоном подготовки к
родам. Релаксин оказывает при беременности
расслабляющее действие на матку, перед
родами расширяет её зев, размягчает шейку
и уменьшает плотность лонного и иных
тазовых сочленениий. Раннее введение
большой дозы релаксина вызывает
рассасывание плода у крыс, не нарушая продукции
прогестерона.
648 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ПОМК, его дериваты и кортиколиберин
также образуются в плаценте и имеют
отношение к развитию при беременности
некоторых поведенческих особенностей, а
также к относительному гиперальдостеро-
низму у беременных и индукции акта родов
(здесь особенно важен кортиколиберин).
Под их влиянием в конце беременности
синтез половых стероидов уменьшается, а
глюкокортикоидов — растёт.
Плацента выделяет также тиролиберин,
соматостатин, ингибин и активин,
различные факторы роста, цитокины, проренин и
специфические маркеры беременности —
Р-гликопротеид (имеющий, по некоторым
данным, иммуносупрессорные функции)
и плазменный белок А (с неизвестными
функциями).
Совокупность действующих на организм
и, в частности, — на ЦНС, сигнальных
факторов, вырабатываемых плацентой, играет
решающую роль в нейроэндокринном
формировании так называемой «доминанты
беременности».
Таким образом, эндокринные и парак-
ринные продукты женских половых органов
осуществляют регуляторные эффекты в
самом яичнике, обратную связь с гипота-
ламо-гипофизарным нейросекреторным
аппаратом и ЦНС, модулируя, в
частности, половое поведение женщины, а также
стимуляторные влияния на матку,
яйцеводы, влагалище, благоприятствующие
транспортировке гамет, оплодотворению
и имплантации, успешному протеканию
беременности.
Более детальное обсуждение акушерско-
гинекологических сторон этой темы можно
найти в соответствующих руководствах и в
т. III данной книги.
Нарушения эндокринных функций
женских гонад
и пролактиновой регуляции
Условно эндокринные дисфункции
яичников можно подразделить на препубертатные,
относящиеся к репродуктивному периоду, а
также к менопаузе и постменопаузе.
Нарушения полового созревания у
девушек могут выражаться в преждевременном
(как по изосексуальному, так и по
гетеросексуальному типам) или в задержанном
половом созревании, а также принимать
форму своевременного, но
гетеросексуального полового созревания, с элементами
вирилизации.
Иногда преждевременно развиваются
лишь отдельные вторичные половые
признаки. При гиперчувствительности клеток
грудных желёз к эстрогенам это выражается
в преждевременном телархе — увеличении
молочных желёз. При усилении андро-
геновой секреции в сетчатой зоне коры
надпочечников может быть изолированное
оволосение лобка и подмышек —
преждевременное пубархе или адренархе.
О преждевременности полового
созревания всего организма говорят, когда полный
набор его признаков появляется у девочек
8 лет и младше. Преждевременное
созревание может быть истинным (центральным, со
стимуляцией продукции гонадотропинов) и
ложным (периферическим, при неизменной
или пониженной гонадотропиновой
секреции и повышенном уровне половых
гормонов яичников или надпочечников).
При истинной разновидности
преждевременного изосексуального пубертата
причины гиперсекреции гонадотропинов
могут быть связаны с опухолями, другими
очаговыми процессами, энцефалитами и
менингитами, поражающими подбугорье.
Очень часто причиной служат неидентифи-
цируемые микроаденомы или повышенная
чувствительность аденогипофиза к гонадо-
либерину, развивающаяся на рецепторном
и пострецепторном уровне, поэтому 90 %
таких пациенток при рутинном клиническом
обследовании расцениваются как случаи
«идиопатического конституционального»
преждевременного пубертата.
Если преждевременная феминизация
девочек протекает на фоне множественной
фибродисплазии костей, асимметрии лица,
нарушений функций черепно-мозговых
нервов, появления кожных пятен цвета кофе
с молоком, это представляет собой синдром
Нарушения эндокринной регуляции 649
МакКьюна-Олбрайта. Причины
стимуляции гонадотропной функции могут быть
связаны в подобных случаях с аномалиями
формирования турецкого седла и срединных
образований черепа.
Другой симптомокомплекс, включающий
изосексуальную преждевременную
феминизацию, известен каксиндром
Сильвера-Расселла (помимо преждевременного пубертата
отмечаются клинодактилия, выраженная
задержка роста, вплоть до примордиаль-
ной карликовости, асимметрия фигуры,
уменьшенное деформированное турецкое
седло и треугольное лицо). Нередко
врождённый первичный гипотироз способствует
развитию ускоренного истинного полового
созревания. Тиролиберин, в избытке си-
тезируемый при этом, способен оказать
пролактолибериноподобный и гонадолибе-
риноподобный эффекты.
Ложное изосексуальное преждевременное
половое созревание девушек связано с эст-
рогенообразующими опухолями овариаль-
ного и надпочечникового происхождения.
К нему может привести также ятрогенное
действие лекарств, содержащих женские
половые стероиды и изредка — опухоли,
локализованные вне ЦНС и вырабатывающие
хорионический гонадотропин.
Практически все случаи
гетеросексуального преждевременного полового созревания
у девушек вызваны либо вирилизующей
формой врождённой гиперплазии коры
надпочечников (обычно 21-гидроксилазной
недостаточностью — см. выше), либо анд-
ростеромами яичника или надпочечника.
Задержка полового созревания у девочек
выражается в несвоевременно позднем
появлении вторичных половых признаков
(позже 13 лет) и в первичной аменорее к
возрасту 16 лет или старше (в норме
менструации начинаются не позднее, чем через
5 лет от возраста телархе).
Это может быть вызвано нарушением
формирования мюллеровых структур при
нормальной эндокринной и овогенной
функции гонад. Однако отмечаются и случаи
гипогонадизма как гипергонадотропного
(связанного с первичной овариальной
недостаточностью, например, с синдромом Шере-
шевского-Тёрнера), так и гипогонадотропно-
го (вызванного вторичной дисфункцией
яичников на почве гипоталамо-гипофизарной
патологии — например, синдрома Колмена).
Более подробно спектр, этиология и
патогенез таких нарушений рассмотрены ниже
в разделе, посвященном постпубертатным
гонадным дисфункциям и выше — в разделе
о псевдогермафродитизме.
При своевременном гетеросексуальном
половом созревании пубертат наступает в
срок, но характеризуется смешанной
картиной феминизации и вирилизации. К такому
развитию событий могут привести наиболее
часто синдром поликистозных яичников (см.
ниже) и, реже, нетяжёлые малосимптомные
формы вирилизующей адренокортикальной
гиперплазии (см. выше).
Наиболее широким спектром женских
гонадных эндокринных расстройств
характеризуется репродуктивный период. Такие
синдромы, как дисменорея и пременстру-
альная дисфория знакомы очень многим
женщинам.
Дисменорея — то есть болезненные
месячные, сопровождаемые тошнотой, головной
болью, диареей — поражает до 50 % женщин.
Патогенез первичной дисменореи связан с
избыточным образованием простагландинов
в эндометрии перед месячными и во время
менструации. Простагландины, особенно
PgF2a, действуя локально и системно,
вызывают усиленное сокращение и ишемию
матки, боль и другие симптомы.
О вторичной дисменорее говорят, когда
имеется провоцирующее ее первичое
заболевание. Наиболее тяжелая вторичная
дисменорея наблюдается при наличии в
брюшной полости эктопических островков
эндометрия, реагирующих на циклические
изменения эстрогенной и прогестиновой
активности.
Более 150 показателей
функционирования женского организма циклически
650 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
изменяются при осуществлении овари-
ально-менструального цикла (см. рис. 125
выше). Не удивительно, что так называемый
синдром предменструального
напряжения, отмечаемый почти у 5-10% женщин,
характеризуется большим разнообразием
проявлений, описание которых выходит за
рамки данного неклинического руководства.
Наиболее типична
эмоционально-поведенческая сторона этого симптомокомплекса,
выражающаяся в тревожости,
раздражительности, эмоциональной лабильности
таких пациенток. Многие проявления
синдрома предменструального напряжения
различаются, в зависимости от конституции
женщин. У очень женственных пациенток
с перевесом эстрогенов он выражается в
нервной напряженности и в нагрубании
груди.
У женщин с мальчишеским хабитусом и
перевесом прогестинов/андрогенов
симптомы включают депрессию и боли в животе и
пояснице. Нередки боли в молочных
железах —мастодиния.
По современным представлениям, в
основе синдрома лежат особенности
гормонального фона в конце лютеальной фазы
цикла — пониженая продукция эндогенных
опиатов и повышенная — пролактина,
повышение выработки вазопрессина, нарушения
в содержании а-МСГ. Следствием этого
являются метаболические изменения —
тенденция к гипогликемии, задержке натрия и
воды. У некоторых пациенток обнаружена
аутоаллергия к собственному прогестин-
связывающему белку, которая
формирует циклический иммунокомплексный, а
иногда — даже анафилактический процесс.
Описана, например, периодическая пре-
менструальная крапивница.
Несколько реже встречаются различные
серьёзные нарушения, обусловливающие у
половозрелых, прежде менструировавших
женщин прекращение месячных. Это
выражается во вторичной аменорее.
Из дальнейшего рассмотрения мы
исключаем физиологическую вторичную
аменорею при беременности и в менопаузе,
а также неэндокринные формы вторичной
аменореи — например, связанные с
атрофией эндометрия на почве его повторных травм
при выскабливаниях {синдром Эшермена).
Эндокринные формы вторичной
аменореи включают расстройства гипоталамо-
гипофизарного генеза {вторичная овари-
альная недостаточность) и яичникового
происхождения {первичная овариальная
недостаточность).
При вторичной овариальной
недостаточности, как правило, функционирующие
фолликулы имеются, но происходят анову-
ляторные циклы. Лечение может
восстановить овуляцию. Если причины овариальной
недостаточности имеют центральный
характер, хроническое отсутствие овуляции
протекает чаще на фоне низкого уровня
эстрогенов. При этом циклической
гиперплазии эндометрия нет, и характерным
признаком является отсутствие кровотечений
при назначении прогестинов. В некоторых
случаях (тиропатии, гиперпролактинемия),
вторичная овариальная недостаточность
протекает при нормальном или высоком
уровне эстрогенов. В этом случае рост
эндометрия продолжается, и он может
отторгаться в сроки, не совпадающие с ожидаемыми
сроками месячных, давая
дисфункциональные маточные кровотечения при отсутствии
овуляции. Назначение таким пациенткам
прогестерона провоцирует кровотечение.
Вторичная овариальная недостаточность
(гипогонадотропный женский гипогона-
дизм) может быть гипоталамической
этиологии.
Как тяжелый острый, так и
продолжительный хронический стресс могут нарушить
пульсовой ритм секреции гонадолиберина
в медиобазальном гипоталамусе. Поэтому
стрессогенный образ жизни способен
вызвать у пациенток вторичную аменорею
при нормальных яичниках. Экстремальные
соревновательно-тренировочные нагрузки
вызывают гонадолибериновую
недостаточность и вторичную надовариальную
аменорею у спортсменок.
Так как аркуатное ядро является
структурой, вовлеченной в лептиновую регуляцию
массы тела, продукция гонадолиберина и
месячные нарушаются при голодании и у
больных нейрогенной анорексией-булимией
и другими формами кахексии (см. выше).
Лекарственная гипоталамическая
вторичная овариальная недостаточность
может провоцироваться адренергическими,
дофаминэргическими и опиатэргическими
средствами, что нередко приводит к
вторичной аменорее у наркоманок.
Гипофизарная вторичная овариальная
недостаточность характерна для гипопи-
туитаризма (см. выше), в частности, его
послеродовой формы у женщин — синдрома
Шихена.
Нередко (в 10-40 % всех случаев
вторичной аменореи) она вызвана гиперпролак-
тинемическим синдромом. В этом случае
пролактин стимулирует в гипоталамусе
продукцию дофамина, который через
эндогенные опиаты блокирует выработку
гонадолиберина. Параллельно осуществляется и
прямое антиэстрогенное действие пролакти-
на на яичники.
Как уже отмечалось выше, в разделе,
описывающем патофизиологию тестикулярной
недостаточности, избыточная продукция
пролактина как у женщин, так и у мужчин
является достаточно частым нарушением
функций аденогипофиза и нуждается в
отдельной характеристике, так как её
проявления не исчерпываются гипогонадотропным
гипогонадизмом и аменореей.
Пролактин (см. выше) это аденогипофи-
зарный тропный гормон, периферическими
мишенями которого в основном служат эк-
зокринная молочная железа, а также, по
новым данным, — макрофаги и лимфоциты.
Таким образом, дефицит пролактина
проявляется гипогалактией и даже агалактией.
Эти симптомы типичны для дебюта
послеродовой гипофизарной недостаточности у
матерей. Имеются основания полагать что
пролактиновый дефицит вызывает
некоторое понижение иммунитета, особенно что
касается продукции секреторного
иммуноглобулина А. По крайней мере, накопление
Нарушения эндокринной регуляции 651
соответствующих лимфоцитов в лактирую-
щей молочной железе и секреция IgA в
молозиво и молоко, как и некоторые функции
макрофагов — пролактинзависимы. Тем не
менее у нелактирующих женщин и у мужчин
дефицит пролактина трудно обнаружить.
Напротив, избыток пролактина ярко
манифестирует у лиц обоего пола. До трети
всех пациенток с гиперпролактинемией
и подавляющее большинство пациентов
развивают этот синдром на почве гормоно-
образующей опухоли аденогипофиза — про-
лактиномы.
Пролактиномы — самые
распространённые гормонообразующие аденомы
гипофиза, они составляют более 30 % опухолей этой
подгруппы. Есть сведения, что микропро-
лактиномы имеются у 2-9 % людей, судя по
данным аутопсий случайно погибших лиц.
В 90 % случаев больные с микропролакти-
номами — женщины. Макропролактиномы
более редки, но встречаются чаще у мужчин.
Кроме пролактином, гиперпролактинемию
могут вызывать любые локальные процессы,
разрушающие ножку гипофиза, а также ан-
тидофаминергические лекарства. Опиаты и
эстрогены потенцируют эффекты
пролактина и стимулируют его секрецию. Последние
ускоряют рост лактотрофов, поэтому эстро-
генные контрацептивы считаются фактором
риска пролактином.
Избыток тиролиберина при первичном
гипотирозе также способен вызвать
усиленный пролактиногенез. Наконец, существуют
соматотропиномы и адренокортикотропи-
номы, секретирующие пролактин наряду
с основным гормоном. При хронической
почечной и печёночной недостаточности
гиперпролактинемия является внежелезис-
той и вызвана замедлением инактивации
данного гормона.
У женщин умеренная
гиперпролактинемия укорачивает лютеальную фазу ова-
риального цикла и ведет к нерегулярным
месячным и бесплодию. При выраженной
гиперпролактинемии может наблюдаться
клссический синдром персистирующейлак-
тореи -аменореи.
652 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Данная нозологическая единица
прежде считалась редким расстройством и
дробилась, в зависимости от наличия или
отсутствия выявленной аденомы гипофиза и
предшествовавших беремености и родов, на
несколько синдромов (синдром Форбс-Ол-
брайта, синдром Хиари-Фроммеля, синдром
Аргонза-дель Кастилъо, синдром Альхума-
ды-дель Кастилъо). Ныне эти состояния
объединили в один синдром и его частоту
оценивают, по меньшей мере, как 1/1000 — у
женщин (и 1/2 800 — у мужчин), причем
наиболее часто он диагностируется в расцвете
лет (в возрасте от 20 до 40).
Помимо упомянутых выше проявлений,
для синдрома бывают характерны гипе-
рандрогенизм (пролактин стимулирует
продукцию дегидроэпиандростерона в коре
надпочечников), ожирение с
перламутровыми стриями, наклонность к депрессивным
состояниям.
Все эти проявления не обязательны и
зависят от сопутствующих гипоталамо-ги-
пофизарных нарушений. Например, первое
описание данного синдрома вообще сделано
у женщины с галактореей и кахексией
(«молочная сухотка» ).
Для гиперпролактинемии характерна
склонность к эмоционально-личностным
расстройствам и даже к суицидам, что
связывают с изменением у больных уровня
биогенных аминов. Общая частота
гиперпролактинемии у населения доходит до
1,7 %, что делает гиперпролактинемический
синдром одной из актуальных проблем
эндокринологии. Каждый третий случай
женского бесплодия сопровождается ги-
перпролактинемией (Л.К. Дзеранова,
1999). Хроническая внутричерепная ги-
пертензия является фактором высокого
риска развития патологической
гиперпролактинемии в подростковом возрасте.
Стойкое повышение уровня пролактина
наблюдается почти у трети подростков с
головными болями, особенно - девушек.
Более подробно гиперпролактинемические
состояния описываются в недавно
вышедшем руководстве (Ю.И. Строев, Л.П.
Чурилов, 2004).
Редкие причины овариальной
недостаточности центрального генеза связаны с
синтезом аномальных а- или р-субъединиц
гонадотропинов или продукцией их
биологически неактивных форм с нарушенным
гликозилированием.
Если все вышеназванные причины
хронической гипоэстрогеновой ановуляции и
вторичной гипогонадотропной овариальной
недостаточности исключены,
предполагается наличие так называемой
изолированной гипогонадотропной гипофункции
яичников — синдрома, этиология которого
неясна, но, предположительно, связана с
наследственностью. Патогенез этого редкого
вида вторичного гипогонадизма
ассоциируется с недостаточной чувствительностью
гонадолибериновых нейронов гипоталамуса
к дофамину и, возможно, с избыточным
действием на гипоталамо-гипофизарный
нейросекреторный аппарат ингибина.
Еще предстоит оценить истинную
распространённость малоизученной внежелезистой
формы овариальной гипергонадотропной
недостаточности, связанной с аутоантитела-
ми к циркулирующим гонадотропинам.
Первичная овариальная
недостаточность — группа расстройств, для которых
характерны аменорея при гиперпродукции
гонадотропинов, а также поражения
женских гонад: дизгенезия, резистентность к
гонадотропным воздействиям, либо
деструктивные процессы в яичниках.
Генетические причины первичной гипо-
эстрогенной гипергонадотропной
недостаточности яичников включают, в первую
очередь, полное или частичное отсутствие
одной из Х-хромосом. При кариотипе
45X0 развивается синдром Шерешевско-
го-Тернера — дизгенезия яичников (см.
также т.1, с. 136). В 40% случаев возможны
также мозаичные кариотипы с отсутствием
Х-хромосомы в части соматических клеток.
При этом классический синдром Шерешев-
ского-Тернера связан с женским
хромосомным полом и половым недоразвитием.
Но некоторые из мозаичных пациентов
имеют клетки с кариотипом 45X0 и 46XY,
Нарушения эндокринной регуляции 653
представляя случаи мужского
псевдогермафродитизма с делецией Y-хромосомы в
части клеток (см. выше). У таких больных,
в отличие от женщин с синдромом 45X0,
крайне высок риск тестикулярных опухолей
в рудиментарных клетках яичек. Поэтому
кариотип всех лиц с гипергонадотропной
аменореей в возрасте до 30 лет должен
проверяться цитогенетически.
Синдром 45X0 — очень
распространенная аномалия гоносом (частота не менее
1/3000 женщин). Яичники представлены
соединительнотканными тяжами, больные
бесплодны, внутренние гениталии
рудиментарные женские, наружные —
инфантильные женские, развитие вторичных половых
признаков задержано. Имеют место
первичная аменорея с гипоэстрогенной ановуля-
цией, низкорослость, крыловидные кожные
складки на шее над плечами, бочкообразная
грудная клетка, стигмы дизэмбриогенеза
(например, готическое нёбо, эпикантус, не-
вусы, низко посаженные деформированные
ушные раковины), а также дисплазия костей
и суставов, коарктация аорты, наклонность
к лимфостазу. Коэффициент интеллекта
у больных может быть в пределах нормы,
ближе к её нижней границе. Характерны
гиперсоциальность и большая внушаемость.
Синдром истощенных яичников
(«преждевременный климакс») представляет
собой состояние, развивающееся при
раннем исчерпании запаса примордиальных
фолликулов. Вместо 50-51 года,
последние фолликулы расходуются до возраста
37-38 лет. Наступает гипоэстрогенизм. Он
манифестирует симптомами, сходными с
постменопаузальным периодом,
наступающим при естественном старении. Женщины
молодого возраста испытывают
климактерические симптомы: приливы крови к голове,
потливость, раздражительность, депрессию.
Имеется остеопороз. Возникают аменорея и
бесплодие.
Считается, что этиология и патогенез
данного расстройства связаны с утратой
слишком большого числа фолликулов на
почве инфекций, соматических мутаций и
аутоиммунных поражений оогониев,
причём эти факторы могут начать действовать
частично еще при внутриутробном развитии
яичника. Во многих случаях у больных
имеются сопутствующие плюригландулярные
аутоиммунные полиэндокринопатии, а
также аутоиммунная пернициозная анемия.
Первичная врождёная овариальная
недостаточность с аменореей характерна и для
некоторых наследственных дефектов стеро-
идогенеза. При дефиците 17а-гидроксилазы
она сочетается с сексуальным
инфантилизмом и избытком минералокортикоидов,
ведущим к гипертензии, дефицит 17,20-лиазы
характеризуется половым инфантилизмом.
Эти растройства подробно рассматривались
выше как причины адреногенитального
синдрома. При галактоземии образуются
метаболиты, токсичные для яичников, и
также формируется овариальная
недостаточность.
Редкая разновидность овариальной
недостаточности — синдром резистентного
яичника (синдром Сейведжа), при котором
развитие фолликулов останавливается
перед антральной стадией, а овуляция не
имеет места. При этом женские гонады
утрачивают чувствительность к ФСГ.
Имеются, очевидно, наследственные формы
этого нарушения, связанные с рецепторной,
либо пострецепторной аномалией. Однако
есть сведения и о синдроме резистентного
яичника аутоиммунной природы: в связи
с наличием блокирующих аутоантител к
рецепторам ФСГ (Л. Харрисон, 1983).
Приобретенная первичная овариальная
недостаточность может явиться следствием
хирургической экстирпации гонад
(кастрации) вследствие удаления опухолей и их
метастазов. Она также развивается при
облучении яичников, применении химиотера-
певтических цитостатических средств,
особенно — алкилирующих агентов. Причиной
овариальной недостаточности может быть
двусторонний оофорит (воспаление
яичников). Этиология оофорита — инфекционная
и аутоиммунная. Особенно характерно
хроническое течение процесса и развитие
гипогонадизма при иммунопатологических
оофоритах. Аутоиммунный оофорит бывает
654 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
частью плюригландулярных аутоиммунных
синдромов, сочетаясь с
иммунопатологическими поражениями щитовидной,
околощитовидных, поджелудочной желез, но
особенно часто — коры надпочечников.
Интересно, что овариальная недостаточность
характерна для носительниц синдромов Ди
Джорджи и Незелофа с аплазией тимуса и
иммунодефицитом.
Гиперандрогенизм у женщин, связанный
с продукцией избытка андрогенов в
надпочечниках, в частности, в опухолях-анд-
ростеромах, также вызывает овариальную
недостаточность и аменорею на фоне гипер-
эстрогенизма, так как избыток андрогенов
активно превращается в яичнике и других
тканях в эстрогены.
Важнейшей и самой распространенной
формой первичной овариальной
недостаточности, сопровождаемой хроническим
отсутствием овуляции при наличии гиперэст-
рогенизма, служит синдром поликистозных
яичников (описан впервые отечественным
автором С.К. Лесным в 1928 г., известен
также как синдром Штейна-Левенталя,
синдром склерокистозных яичников, синдром
овариальной гиперандрогении неопухолевого
генеза).
Синдром наблюдается у 3-6%
гинекологических пациенток. Классические
проявления включают ожирение, скрытый ин-
сулинонезависимый сахарный диабет, гир-
сутизм, аменорею с дисфункциональными,
внецикловыми маточными кровотечениями,
бесплодие, гипертензию. Гормональный
фон, помимо гиперинсулинемии,
характеризуется повышением уровня Л Г (с
выраженными пиками секреции), недостатком
ФСГ (со сглаженными пиками секреции),
гиперандрогенизмом, гиперэстрогенеми-
ей — за счёт эстрона, вырабатываемого при
периферической конверсии андрогенов, а не
эстрадиола — продукта яичниковой арома-
тазной реакции.
Сложная этиология и патогенез данного
синдрома представляют собой яркую
иллюстрацию полигормонального контроля
овариальных функций (рис. 126).
Механизмы этого синдрома могут служить
доказательством условности такого понятия, как
«гонадотропные гормоны», в том смысле,
ожирение >w
\сексосвязывающего глобулина
| белка связывающего ИФР-1
I трансформации
в
эстрон
4 риск рака матки,
I 1 \дисфункционалъные£^—
| гиперэстрогенемия \ * \ кровотечения
Рис. 126. Патогенез синдрома поликистозных яичников
что гормональные регуляторы, не
относимые классической эндокринологией ни к
половым, ни к гонадотропным, могут, тем
не менее, иметь для гонад и их патологии
важнейшее значение.
По современным данным (Н.А. Беляков
и соавт., 2004), ключевым звеном в
патогенезе этого гетерогенного синдрома в
большинстве случаев служит инсулинорезистен-
тность. Она порождает гиперинсулинизм и
ожирение. Гиперинсулинемия способствует
также гипертензии. В условиях избытка
инсулина печень понижает продукцию
сексосвязывающего глобулина и белка,
связывающего ИФР-1. Возрастают
биодоступность и уровень концентрации свободных
андрогенов, эстрогенов и ИФР-1 (см. также
«Патофизиология сахарного диабета»).
Андрогены усиливают инсулинорезис-
тентность и конверсируются на периферии
в эстрогены. Гиперэстрогенизм в момент,
не соответствующий ходу нормального
овариально-менструального цикла, ингиби-
рует продукцию ФСГ перед менструацией.
Пременструальный всплеск концентрации
ФСГ не происходит, что нарушает процесс
вовлечения новой группы фолликулов в
созревание.
Гиперэстрогенизм повышает у
пациенток с данным заболеванием риск развития
эндометриального рака матки и, по
некоторым данным, — рака молочных желёз.
В свою очередь, повышенная активность
(и продукция) ИФР-1 сильно
стимулируют рост теки и стромы яичника. Белочная
оболочка органа становится очень толстой,
что само по себе мешает ходу овуляции.
Кроме того, тека усиленно производит
андрогены, чему способствует пермиссив-
ный эффект ИФР-1 на её чувствительность
к ЛГ. Сам инсулин, при очень высоких
концентрациях, действуя на текальные
рецепторы ИФР-1, усиливает овариальный
андростероидогенез. Андрогены
способствуют в яичнике атрезии фолликулов,
дальнейшему росту стромы, нарушению
нормальных внутрияичниковых обратных
связей, обеспечивающих в норме выбор
доминирующего фолликула.
Нарушения эндокринной регуляции 655
Из-за ановуляции развивается дефицит
прогестерона. Нехватка прогестерона
нарушает пульсовую секрецию гонадолиберина.
Уровень ФСГ, а с ним и интенсивность
ароматазной реакции падают ещё ниже.
Это окончательно тормозит развитие
фолликулов.
Из-за их атрезии в гонадах формируются
кисты. Уровень ЛГ стабильно высок, но
лишен срединного пика, типичного для
нормальных циклов. В измененном яичнике
возрастает секреция ингибина,
закрепляющего аномальный характер гонадотропой
регуляции. Гиперандрогенизм вызывает
гирсутизм и акнэ. Отмечается гиперэндор-
финемия.
Интересно, что весь этот комплекс
изменений в гонадотропной регуляции
развивается и при других причинах гиперандроге-
низма (синдром Иценко-Кушинга,
врождённая вирилизующая адренокортикальная
гиперплазия, андростеромы). Своеобразное
беспиковое повышение продукции ЛГ и
дефицит ФСГ также ведут у таких больных
к вторичному поликистозу яичника. Может
отмечаться и пониженная чувствительность
тканей к инсулину (например, из-за гипер-
кортицизма).
Овариальная гиперандрогения может
иметь и иную, опухолевую природу. Чаще
всего опухоли из клеток теки и гранулёзы
вырабатывают эстрогены и прогестины и
не дают синдрома вирилизации. Вирили-
зующие опухоли яичников бывают редко
(0,1 % от всех неопластических поражений
этого органа). Считается, что большую
роль в их возникновении играют остатки
мужской эмбриональной гонады,
сохранившиеся в яичниках при скрытом мозаицизме
по половым хромосомам. Андробластомы
секретируют эстрогены и андрогены и
являются наиболее частыми среди опухолей этой
группы. Реже встречаются дизгерминомы,
продуцирующие, вдобавок к андрогенам,
ещё и хорионический гонадотропин.
Гиперандрогенизм вызывает у носительниц
опухолей аменорею, мужское лобное
облысение, гирсутизм, дефеминизацию.
656 А.Щ. Зайчик, Л.П.Чурилов Патохимия
Следует помнить и о возможности редких
карциноидных опухолей из диффузных
эндокринных клеток яичников,
вырабатывающих серотонин и тироидные гормоны
(«овариальная струма»).
Иногда, при нарушении фолликулогенеза,
происходит нормальный переход к лютеаль-
ной фазе цикла, формируется желтое тело,
наступают продукция прогестерона и
переход эндометрия к секреторному состоянию.
Однако, яйцеклетка не выходит из
фолликула. Все презумптивные признаки овуляции
наличествуют, менструация наступает, но
женщины бесплодны, а менструальное
отделяемое не содержит яйцеклетки. Этиология
и патогенез этого редкого нарушения,
именуемого синдром лютеинизации интактного
фолликула неизвестны. Возможно, речь
идет об устойчивости механизмов секреции
фолликулярной жидкости к действию гона-
дотропинов или о дефекте внутрифоллику-
лярной паракринной регуляции.
Большинство нарушений овариально-
менструального цикла касается
фолликулярной фазы и овуляции. Однако, известны
и изолированные нарушения его лютеаль-
ной фазы.
Их суть заключается в том, что секреция
прогестинов либо укорочена по времени
{недостаточность лютеальной фазы), либо
недостаточна по интенсивности
(неадекватность лютеальной фазы).
Изредка речь идёт о внежелезистной ре-
цепторной нечувствительности эндометрия
к прогестинам, вызванной генетическим
дефектом рецепторов прогестерона.
Акушерство и гинекология, а также
андрология и урология — большие и
сложные области медицины, и авторы не могут
взять на себя ответственность за более
развёрнутое изложение патофизиологии
акушерско-гинекологических и андро-уро-
логических расстройств, так как не обладают
соответствующей компетенцией в этих
разделах клинической медицины и не ставят
в данной книге иных задач кроме краткого
чдокринно-метаболические нарушения)
рассмотрения тех аспектов патологии,
которые наиболее тесно связаны с эндокринной
регуляцией и нарушениями обмена веществ.
Некоторые другие вопросы
патофизиологических основ акушерства и гинекологии, а
также андрологии и урологии войдут в том
III настоящей серии «Механизмы болезней
и синдромов».
НАРУШЕНИЯ ЭНДОКРИННЫХ
ФУНКЦИЙ ТИМУСА
Тимус был известен еще античным
анатомам, и Руфус Эфесский (100 до н.э.) считал
этот орган вместилищем храбрости и души.
Название железе дал К. Гален за сходство её
контура с листом тимьяна. В XX веке,
благодаря работам Дж. Миллера (1961), стала
ясна роль тимуса в иммунной системе.
Вилочковая железа, или тимус, является
одним из центральных лимфоидных органов
иммунной системы и в этом качестве служит
ареной дифференцировки и клональной
селекции Т-лимфоцитов. Поэтому патология
тимуса сопровождается
иммунопатологическими расстройствами — его гипоплазия
и аплазия свойственны многим врождённым
формам иммунодефицитов; нарушения
структуры и функций тимуса как иммуно-
компетентного органа, в частности, его
гиперплазия с появлением в нём В-лимфоцитов
и лимфоидных фолликулов типичны для
многих аутоаллергических заболеваний.
Вилочковая (в старой литературе —
зобная) железа развивается из передней кишки,
закладываясь на 4-й неделе киематогенеза,
раньше всех иммунных и эндокринных
органов в вентральной части III и дорзаль-
ной — IV жаберных карманов. Затем ее
развитие протекает в составе бранхиогенного
комплекса, вместе с зачатками
околощитовидных и щитовидных желёз. В ранний
период своего развития железа сохраняет
связь с глоткой посредством тимико-фа-
рингеального протока, который иногда не
полностью облитерируется. По его ходу на
шее могут сохраняться эктопические очаги
Нарушения эндокринной регуляции 657
ткани тимуса, присутствующие почти у
каждого третьего (С. Грей, Дж. Скенделакис,
1990). В свою очередь, в тимусе сохраняются
клетки, вырабатывающие паратгормон и
кальцитонин, а могут быть и целые,
включённые в вилочковую железу, паратироид-
ные тельца, что учитывается в эндокринной
хирургии и при определении источника
эктопического гормонообразования.
Эпителий тимуса, по современным
представлениям (Б. Ван Годекер, 1986), может
иметь смешанное экто-эндодермальное
происхождение (светлые эктодермальные
и тёмные эндодермальные клетки). Чисто
эпителиальным тимус остается лишь до 8-й
недели эмбриогенеза, затем там появляются
незрелые Т-лимфоциты-предшественники.
В строму органа врастают капилляры и она,
как и паренхима, заселяется мезенхималь-
ными макрофагальными клетками. После
12-й недели тимус представляет эпители-
ально-лимфоидно-мезенхимальный орган.
К рождению он состоит из 2-х долей,
покрытых соединительно-тканной капсулой и
содержащих разделённые перегородками
дольки. В последних различают корковое
и мозговое вещество. Корковое содержит
менее зрелые Т-лимфобласты и протимоци-
ты. Наружная субкапсулярная часть коры
тимуса богата светлыми эпителиоцитами,
лежащими под капсулой. Они содержат
множество вакуолей. Здесь же находятся много-
отростчатые крупные дендритные клетки и
клетки-няньки, охватывающие лимфоциты
своими отростками. И те и другие содержат
электронноплотные гранулы. Контактируя с
дендритными клетками и субкапсулярными
эпителиальными клетками, а также проникая
в клетки-няньки посредством эмпериполеза,
Т-лимфобласты претерпевают ранние стадии
дифференцировки по пути Т-лимфопоэза
под влиянием паракринных регуляторов,
выделяемых этими клетками и находящимися
в их вакуолях и гранулах.
Во внутренней части кортикального слоя
находятся тёмные эпителиоциты. В них
тоже имеются вакуоли с секреторным
содержимым. Ранние тимоциты претерпевают
здесь новые паракринные влияния, экспрес-
сируют Т-клеточный рецептор и проходят
негативную селекцию с устранением ауторе-
активных клонов, причём в этом участвуют
индукторы апоптоза и макрофаги.
Кортикальный слой отделён от системной
циркуляции гематотимическим барьером.
В мозговом слое находятся более
дифференцированные и зрелые Т-клетки,
мигрировавшие из коры тимуса. Они подвергаются
позитивной и негативной селекции, в связи
с чем не более 5 % выходит в циркуляцию, а
остальные апоптозируют и фагоцитируются
местными макрофагами. В медуллярном
слое имеются тёмные эпителиоциты,
содержащие секреторные вакуоли. Светлые
эпителиальные клетки формируют в этом
слое тельца Хассела (в классической
литературе — тельца Гассаля), в структуре
которых имеются как признаки секреторной
активности (центрально расположенный
коллоид в виде кист), так и апоптотической
инволюции. Многие авторы отрицают
секреторную роль телец Гассаля.
Важным элементом медуллярного слоя
являются разбросанные здесь миоэпителио-
циты (миоидные клетки). У них имеются
черты антигенной общности с мышечными
волокнами, в частности, миозин и
поверхностные рецепторы. В мозговом веществе
тимуса имеются также антигенпредстав-
ляющие интердигитирующие клетки.
В контакте с ними идут антигензависимые
этапы дифференцировки Т-лимфоцитов.
Тимус богато васкуляризован и иннерви-
рован вегетативными нервами, что служит
основой для гипотезы о парагипофизарных
нервных влияниях на его деятельность.
Артерии представлены в корковом, а вены — в
мозговом веществе. В периваскулярных
пространствах нормального тимуса
имеются отдельные В-лимфоциты (менее 1 %
всех лимфоидных клеток органа). Но в
паренхиме здорового тимуса их практически
нет. Интересно и важно, что часть нервных
холинергических окончаний, достигающих
вилочковой железы, оканчивается в ее
паренхиме на миоидных клетках, имеющих
Н-холинергический рецептор, аналогичный
мышечному.
658 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Детали организации коркового и
мозгового слоя тимуса отражены в т. I (с. 382-384).
Тимус как иммунокомпетентный орган,
включая патологические аспекты его
иммунологических функций, уже был подробно
рассмотрен в т. I данного руководства
(стр.382-384,481-483).
Настоящий раздел ограничивается
обсуждением достоверно известных современных
сведений о гормонах и некоторых паракрин-
ных факторах тимуса и роли нарушений
эндокринных функций вилочковой железы
при патологии.
Патология тимуса как органа во всех
деталях обсуждена не будет. Читатель
дополнительно адресуется к I тому данной книги
и недавней монографии В.П. Харченко и
соавторов «Болезни вилочковой железы»
(1998), трактующей, в частности,
неэндокринологические аспекты тимологии.
Эндокринные функции тимуса
постулировали задолго до обнаружения его
центральной роли в иммуногенезе, но
конкретные механизмы его внутрисекреторной
активности стали раскрываться гораздо
позже, чем активности иммунологической.
Полная ясность в этом вопросе не
достигнута и поныне.
Эндокринные потенции и продукция
истинных, системно действующих гормонов
вилочковой железы, принадлежат, в
первую очередь, клеткам эпителиальной части
тимуса. Однако тимические лимфоциты —
тимоциты, а также местные макрофаги и
фибробласты — способны вырабатывать
множество паракринных цитокинов, гормо-
ноподобная роль которых связана с тем, что
их клетки-мишени могут проходить через
зону действия этих цитокинов в тимусе при
миграции и кровообращении.
Внутрисекреторную активность
тимуса заподозрил еще в начале XX столетия
Й. Гаммар в своей знаменитой статье
«Полвека исследований тимуса» (1905). Им было
открыто явление возрастной инволюции
вилочковой железы. Тимус при рождении
весит 15 г, а к пубертату достигает
максимальной массы порядка 50 г (3. Кемилева, 1984).
Однако, если измерять массу паренхимы без
учёта жировой ткани, то максимум в 38 г
достигается уже в раннем детстве (1-5 лет),
а далее идет прогрессивное уменьшение ее
веса (М. Кендалл, 1981). В настоящее время
известно, что атрофия тимуса идет наиболее
быстро в период 25-40 лет (до 5% чистой
паренхиматозной массы в год). В старости
она замедляется, но расчет показывает, что
экстраполированный возраст
предполагаемой полной атрофии с заменой тимуса на
соединительную и жировую ткань приходится
на 120 лет. Механизмы возрастной атрофии
тимуса генетически запрограммированы и
управляются паракринными цитокинами,
возможно, оказывая влияние на развитие
старческого иммунодефицита (Дж. Сри
Рам, 1967; О.В. Зайратьянц, A.M. Берщан-
ская, 1998).
Тот же И. Гаммар (1923) открыл, что
инволюция тимуса ускоряется в эксперименте
экстрактами надпочечников. Тироидные
гормоны, вероятно, снижая доступность
свободных глюкокортикоидов (см. выше),
тормозят инволюцию тимуса.
Затем оказалось, что самые разные
воздействия достаточной силы
провоцируют инволютивные изменения в тимусе.
Й. Гаммар назвал это явление «акциден-
тальной инволюцией тимуса», имея в виду
не случайный характер самого феномена,
а случайность разнообразных стимулов,
вызывающих закономерный инволютивный
ответ вилочковой железы. Так как открытие
произошло до создания концепции стресса,
феномен не был увязан с действием
гормонов надпочечников на лимфоидные органы.
По современным данным, акцидентальная
инволюция — это нарастающий по
времени, динамический процесс гипофункции
и атрофии лимфоидных элементов тимуса
сначала в корковой, а затем — и в
медуллярной его зонах. Вместе с тем эпителиальные
и макрофагальные клетки микроокружения
тимоцитов по ходу акцидентальной
инволюции могут даже активизироваться. Ак-
цидентальную инволюцию расценивают как
явление, зависящее от апоптоза и эмиграции
лимфоцитов и связанное с кумулируемым
действием стрессов, прежде всего,
опосредованное глюкокортикоидами (В.П. Харченко
и соавт., 1998). При болезни Аддисона
инволюция тимуса сильно тормозится.
Уже после работ Тамара удалось показать,
что неонатальная тимэктомия (особенно в
первые 48 ч жизни) вызывает у животных
(в первую очередь, грызунов) задержку
роста, кахексию, деминерализацию костей и
повышенную электровозбудимость, которые
заканчиваются гибелью (А.А. Богомолец,
1937; М.С. Кахана, 1968). Даже из данного,
очень общего описания ясно, что
развивающийся при этом тотальный
иммунодефицит не объясняет всех эффектов тимэкто-
мии. Экстирпация железы в более поздние
сроки не сопровождается столь фатальной
картиной. X. Визель в начале XX столетия
выдвинул гипотезу, что тимус выделяет «ва-
готонизирующий» гормон. В дальнейшем
попытки выделить и охарактеризовать
гормональное начало тимуса традиционными
методами классической
экспериментальной эндокринологии (кормление железой,
введение ее экстрактов) много раз терпели
неудачу, хотя, с другой стороны, были
накоплены данные, что тимус — очень
чувствительная мишень гормональных воздействий
различных эндокринных желёз, с которыми
он двусторонне взаимодействует. Половые
гормоны стимулировали атрофию тимуса и
наоборот — экстракты вилочковой железы
задерживали половое созревание у
подопытных животных (Д.Я. Шурыгин, 1966).
Из вилочковой железы были получены
липидные экстракты с гипергликемизиру-
ющим действием (И. Потоп и соавт., 1960),
а введение водных экстрактов, наоборот,
усиливало действие инсулина и вызывало
гипогликемию (К. Пархон и соавт., 1928).
СТГ и тироидные гормоны способствовали
росту тимуса и частично нивелировали
эффекты неонатальной тимэктомии.
Введение молодым мышам антител к СТГ
вызывало гипофункцию тимуса,
прекращаемую введением гормона роста (А. Уайт и
соавт., 1981), что послужило основой для
гипотезы о контроле ряда функций тимуса,
как эндокринного органа со стороны СТГ.
Нарушения эндокринной регуляции 659
Антитимоцитарная сыворотка, в свою
очередь, вызывает дегрануляцию
ацидофильных клеток аденогипофиза (В.Э. Торбек
и соавт., 1979). Интересные данные были
получены И.В. Беляевой и А.Ш. Зайчиком,
продемонстрировавшими стероидогенные
эффекты IgG антисыворотки против
ядерных антигенов клеток тимуса (1986).
Патологоанатомические наблюдения
свидетельствовали о наличии гиперплазии
тимуса почти у 80 % больных генерализованной
миастенией. Изучение особенностей детей
с лимфатико-гипопластическим диатезом
поставило вопрос о связи между
гиперплазией тимуса, гипоплазией надпочечников,
параганглиев и щитовидной железы, с одной
стороны, и повышением частоты синдрома
внезапной младенческой смерти — с другой
(т. I, стр. 105-106). Но доминировала точка
зрения, что связь между тимусом и этими
формами патологии опосредуется прежде
всего изменениями в его иммунологических
функциях. В частности, М.У. Хесс показал,
что у неонатально тимэктомированных
животных задержка роста и кахектический
синдром не развиваются, если содержать их
в гнотобиотических стерильных условиях,
то есть решающую роль в генезе тимэктоми-
ческого синдрома играет иммунодефицит,
а не гормонодефицит (1968). В результате,
многие авторы к началу 70-х годов
разуверились в эндокринной природе тимуса, считая
её недоказанной (М. Юлес, И. Холло, 1963;
Н.Т.Шутова, 1966).
Позже методы иммуногистохимии и
молекулярной эндокринологии всё-таки
позволили идентифицировать некоторые
истинные гормоны тимуса. Оказалось, что
их главной мишенью служит иммунная
система. Было показано, что у неонатально
тимэктомированных животных подсадка
ткани тимуса в миллипоровой капсуле,
непреодолимой для клеток, а также введение
его концентрированных экстрактов,
позволяют предотвратить кахексию, несколько
ускорить рост и восстановить
иммунологическую реактивность. В настоящее время
выделено и охарактеризовано более 40
биорегуляторов пептидной, гликопротеидной
660 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
и стероидной природы, вырабатываемых
клетками тимуса. У многих из них
определена структура, а ряд внедрён в клиническую
практику в качестве иммуностимуляторов.
Как уже упоминалось, в основном они
продуцируются эпителиальными элементами
вилочковой железы — субкапсулярными
светлыми эпителиоцитами, свободными
дендритными клетками кортикального
слоя и также— тёмными эпителиоцитами
мозгового вещества. Кортикальные субкап-
сулярные эпителиоциты содержат вакуоли с
паракринными тимическими регуляторами,
а медуллярные — вакуоли, накапливающие
гормоны системного действия. При
введении экзогенных гормонов тимуса секреция и
продукция этих гормонов в эпителии тимуса
ингибируется по механизму обратной связи.
Наоборот, при низком уровне гормонов
тимуса в сыворотке крови удается наблюдать
признаки секреторной активации в эпите-
лиоцитах. Секреторная деятельность эпи-
телиоцитов тимуса ослабевает в условиях
дефицита цинка. (У. Савино и соавт., 1983).
Это связано с тем, что один из главных
системно действующих биорегуляторов,
выделяемых тимусом в кровь — тимулин (ранее
известный как тимический сывороточный
фактор) содержит металлический цинк в
своей структуре и нуждается в нём для
своей биологической активности (Ж.Ф. Бах,
1984). Насколько известно в настоящее
время, это — уникальный случай продукции
металлосодержащего гормона. Нехватка
цинка в питании (см. также
«Патофизиология обмена микроэлементов») приводит
к выраженному иммунодефициту. При
невсасывании цинка (энтеропатический
акродерматит) развивается
иммунологическая недостаточность (см. также т. I, с. 484).
Все эти явления отражают нарушение
продукции тимулина, связанное с цинковым
дефицитом так же, как эндемический гипо-
тироз связан с йодной недостаточностью.
Интересно, что и при нехватке цинка, и при
тимэктомии тормозится рост организма, и
снижается эффективность действия СТГ,
а инъекции некоторых тимусных гормонов
его восстанавливают.
Сывороточные концентрации
известных тимических гормонов максимальны
в детстве и снижаются после 40 лет. При
аплазии и гипоплазии тимуса доказано
понижение концентраций тимозинов и
тимулина. Помимо гормонов эпителиальных
клеток тимуса, макрофаги, фибробласты и
тимоциты вилочковой железы выделяют
множество цитокинов паракринного
действия (Р.С. Шулоф и соавт., 1987). В тимусе
имеются полигормонально активные ней-
роэндокринные апудоциты, выделяющие
различные гормоны диффузной эдокринной
системы. Маркеры апудоцитов несут клетки-
няньки. Эти элементы могут вырабатывать
вазопрессин и окситоцин, гормоны —
производные ПОМК и т.д. В.В.Абрамов в своей
концепции нейро-иммунного дифферона
высказал даже смелую мысль о том, что
некоторые лимфоцитоподобные клетки,
покидающие тимус, на деле являются
циркулирующими апудоцитами, и именно они
ответственны за известный феномен
продукции Т-лимфоцитами гормонов группы
ПОМК, а также нейропептидов (1986).
Из всего этого многообразия пермиссивно
взаимодействующих биорегуляторов не
более 8 субстанций действуют системно и могут
считаться истинными и специфическими
тимическими гормонами — в классическом
эндокринологическом смысле этого слова
(Ж.Ф. Бах и соавт., 1990). Таблица42 ниже
представляет современные данные об
основных гормонах и гормоноподобных
биорегуляторах тимического происхождения.
Обращают на себя внимание эффекты,
свидетельствующие о влиянии тимических
гормонов на нейроэндокринную систему,
особенно — стимуляция некоторыми из
них адренокортикальной функции (Д. Хи-
ли и соавт., 1983; А. Сивас и соавт., 1982).
Это может иметь большое значение при
осуществлении надпочечниковых
компонентов преиммунного ответа на
инфекцию, в частности, при провокации стресса
антигенными раздражителями наряду с
известным АКТГ-подобным действием ряда
монокинов и интерлейкинов, приводящим
Нарушения эндокринной регуляции 661
Таблица 42. Гормоны тимуса
Гормон
Тимопоэтин
Тимический
гормональный фактор
Тимический фактор X
Тимулин
агтимозин
а5-тимозин
а7-тимозин
Р4-тимозин
а-протимозин
тимостерин
Гомеостатический ти-
мусный гормон
Свойства
5562 Д, 49 аминокислот,
синтезирован. Фрагмент 32-36 — тимо-
пентин
3200 Д, термостабилен
4200 Д
857 Д, нонапептид, содержит цинк,
термостабилен, синтезирован
3108 Д, 28 аминокислот, N-конец
ацетилирован, гормон
синтезирован
Полипептид
2500 Д
4982 Д
Около 12500 Д
Липофильный, неполярный, сте-
роидоподобный. 400 Д структура
неизвестна
2000, термостабилен, гликопроте-
ид, структура не уточнена
Эффекты
Наиболее ранние этапы дифференцировки
Т-лимфоцитов, блокада нейромышечной
передачи, понижение интенсивности аутоаллерги-
ческих процессов
Активатор Т-лимфоцитов, восстанавливает их
функции
Стимулирует развитие лимфоцитоза, усиливает
ГЗТ
Стимулятор заключительных этапов
дифференцировки — лимфоцитов, способствует
образованию CD8+ — цитотоксических лимфоцитов.
Восстанавливает иммунологическую
реактивность Т-клеток in vitro
Поздние этапы Т-лимфопоэза, дифференцировка
С04+-лимфоцитов, индукция синтеза ИЛ-2 и
экспрессии его рецептора на Т-клетках. Стимуляция
роста бестимусных карликовых мышей
Поздние этапы Т-лимфопоэза, кортиколибе-
риноподобный эффект, АКТГ-подобное
действие, инсулиноподобное действие. Частичное
восстановление клеточного иммунитета при
аплазии тимуса
Поздние этапы дифференцировки Т-лимфоцитов,
образование CD8+ — супрессоров. Понижение
уровня триглицеридов плазмы
Ранние этапы дифференцировки Т-лимфоцитов
Предшественник ага2-тимозинов
Восстанавливает рост, содержание лимфоцитов
и реакцию на антигены при тимэктомии.
Оказывает гипергликемизирующий эффект
Синергист СТГ, антагонист ПТ, АКТГ и ТТГ
к активации стероидогенеза при иммунном
ответе. У. Пьерпаоли и соавт.( 1977), а также
Ю.А.Гриневич и В.Ф.Чеботарёв (1989)
считают, что именно тимус — через
собственные гормоны и цитокины Т-лимфоцитов, а
также регулируя активность аутоиммунных
клонов — оказывает решающие влияния при
координации функций нейроэндокринной и
иммунной систем.
Литература содержит сообщения и о
других биологически активных продуктах
тимуса. Например, Ф. Райе и соавт. (1980)
выделили из тимуса лейкогененол — аналог
тироидных гормонов и некий антитироид-
ный фактор.
Однако эти гормоны не включены в
приведённую таблицу, так как многие из
них химически не идентифицированы и в
ряде случаев негомогенны. Часто эти
факторы совпадают по эффектам с цитокинами
или комбинациями цитокинов, а также с
другими биорегуляторами могут быть их
фрагментами (например, тимопентин). Так
называемый «тимусный фактор плазмы»
человека оказался иденичен транстиретину.
Схема гормональных взаимоотношений
тимуса и других эндокринных органов, а также
структура одного из гормонов вилочковой
железы — al-тимозина — представлены на
рис. 127, с. 662.
662 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
гипоталамус
гонадолиберин
тиролиберин
соматостатин
кортиколиберин
аденогипофиз
соматотропин
тимус
тимулин,
тимозин,
тимопоэтин
\
лимфоидная
ткань,
селезенка
тимозин а/
-*-
• пролиферация; стимуляция роста
-<— — — — инволюция; торможение роста
Рис. 127. Гормоны тимуса и взаимоотношения вилочковой и других эндокринных желёз. По А.
Уайту и соавт. (1981), А.А. Пищулину (1991). Справа — структура тимозина а, по X. Барбуто (1998)
Многие гормоны тимуса выделены в
чистом виде и синтезированы. Некоторые
из них доступны для клинического
применения. Инъекции тимопоэтина успешно
применены для лечения ревматоидного
артрита. Тимулин проходит испытания в
качестве иммунорегулирующего
препарата при лечении хронического активного
гепатита, ВИЧ-инфекции и рака. Тимозин
(сц-фракция) показал обнадёживающие
результаты при клинических испытаниях
у онкологических пациентов и у больных
гепатитом (X. Барбуто и соавт., 1998).
Патология вилочковой железы включает
гипопластические, гиперпластические и
диспластические процессы.
Среди первых большое значение имеют
врождённые: аплазия тимуса — синдром Ди
Джорджи, гипоплазия тимуса — синдром
Незелофа; тимус гипоплазирован также при
ретикулярной дисгенезии, тяжелом соче-
танном иммунодефиците и атаксии-теле-
ангиоэктазии. Все эти состояния приводят
к первичной иммунологической недостато-
чости с полным выпадением Т-клеточных
и существенным нарушением В-клеточных
функций. Их этиология, патогенез и
проявления подробно охарактеризованы в т. I
(с. 475-484). В контексте данной главы
отметим лечебный эффект при этих
состояниях, оказываемый рядом гормонов тимуса
(см. табл. 42). Врождённая аплазия тимуса
Нарушения эндокринной регуляции 663
часто сочетается с аплазией паращитовид-
ных желез.
Приобретённая гипоплазия тимуса
характерна для гиперкортицизма, облучения,
отравления цитостатиками и голодания. Во
всех случаях имеет место вторичная
иммунологическая недостаточность (т. I, с. 485).
При многих иммунопатологических
заболеваниях — иммунокомплексных вас-
кулитах, системных аутоиммунопатиях с
неорганоспецифическимиаутоантителами,
аутоиммунных полиэндокринопатиях, ин-
сулинозависимом сахарном диабете —
происходит атрофия или гипоплазия истинной
паренхимы тимуса — его эпителиальных
клеток, сопровождаемая гиперплазией
лимфоидных элементов и образованием
В-клеточных фолликулов вне паренхимы
тимуса, в периваскулярных внутридоль-
ковых пространствах. При этом продукция
тимических гормонов снижается, то есть
имеет место тимическая недостаточность
и «ложная» гиперплазия органа — за счёт
лимфоидных элементов (В.П. Харченко и
соавт., 1998).
Интересно, что дело обстоит абсолютно
аналогично тироидиту Хасимото: вилочковая
железа, совершенно так же, как щитовидная
при аутоиммунном тироидите, формирует
лимфоидную имитацию истинной
гиперплазии, увеличивается за счёт лимфоидных
фолликулов, теряя фолликулы
эпителиальные. Такое впечатление, что тимус недаром
именовался ранее железой зобной!
В тимусе наблюдаются и
гиперпластические процессы, в том числе —
неопластического характера. К гиперпластическим
проявлениям патологии вилочковой железы
относят тимико-лимфатический статус,
опухоли, в том числе — тимомы и гиперплазию
паренхимы с лимфоидными фолликулами.
Тимико-лимфатический статус считается
выраженным проявлением лимфатико-
гипопластического диатеза, крайнего,
пограничного варианта конституции.
Состояние индивидов с тимико-лимфатическим
статусом характеризуется тимомегалией и
гипоплазией надпочечников и хромаффин-
ной ткани. Прежде считалось, что именно
тимомегалия приводит к повышению риска
стридора и синдрома внезапной смерти.
В настоящее время, неоднозначные связи
между тимомегалией и синдромом
внезапной младенческой смерти истолковываются
более сложно (см. подробное описание
данного диатеза в т. I данного руководства,
с. 104-106). Хотя некоторые аспекты учения
о тимико-лимфатическом статусе
представляют сейчас лишь исторический интерес,
тем не менее, нельзя отрицать, что носители
этого своеобразного состояния
реактивности имеют пониженную стрессоустойчивость,
вследствие тенденции к гипокортицизму и
симпато-адреналовой недостаточности. В то
же время, имеет право на существование и
точка зрения, согласно которой латентная
функциональная недостаточность
надпочечников, по крайней мере в части
подобных случаев, является первичной, нехватка
глюкокортикоидных влияний приводит к
гиперплазии лимфоидной составляющей
тимуса без выраженной гиперплазии его
эпителиальных элементов и, во всяком
случае, — с его эндокринной гипофункцией.
В этом случае развивающееся состояние
вилочковой железы уместно квалифицировать
как приобретённую тимомегалию на фоне
недостаточности тимических гормонов
(В.П. Харченко и соавт., 1998).
В тимусе могут развиваться различные
неопластические образования — лимфомы,
карциноиды, метастатические процессы,
герминомы. Тератомы считаются самыми
частыми опухолями тимической
локализации. С эндокринологической точки зрения
представляет большой интерес карциноид
тимуса. Эта апудоцитарная опухоль в
отличие от карциноидов ЖКТ, как правило,
не синтезирует серотонина и гистамина,
но может быть источником эктопической
продукции пептидных гормонов, наиболее
часто — АКТГ и вазопрессина. Тимус часто
вовлекается в процесс при
лимфогранулематозе и Т-клеточном лимфолейкозе.
Тимомы — опухоли из собственно
эпителиальных клеток тимуса. Тимомы — редкие
664 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
опухоли, но в переднем средостении
считаются одними из наиболее вероятных.
Тимомы в 90 % случаев
доброкачественны. Злокачественные тимомы проявляют
тенденцию к инвазивному росту и метаста-
зируют. Однако цитологические признаки
злокачественности присутствуют лишь у
некоторых инвазивно растущих тимом.
Такие опухоли именуют карциномами
тимуса. Тимомы содержат множество
лимфоцитов со свойствами созревающих и
зрелых Т-клеток. Опухолевый клон
представлен светлыми кортикальными эпите-
лиоцитами (до 79%, в том числе — в 5%
случаев — клетками-няньками), светлыми
медуллярными (16%) и изредка — тёмными
эпителиоцитами (В.П. Харченко и соавт.,
1998).
Важной особеностью тимом служит
наличие у больных, поражённых этими
опухолями, системных аутоиммунных проявлений.
Чаще всего — в 40-50 % случаев, особенно
при так называемых «лимфоидных
тимомах» (по терминологии П. Бернатца, 1961),
в которых много лимфоцитов, а
эпителиальные клетки немногочисленны и близки по
виду к обычным тимическим кортикальным
эпителиоцитам, — отмечаются
сопутствующие аутоантитела к Н-холинергическому
рецептору и генерализованная миастения.
Реже, как правило, — при так
называемых «веретеноклеточных тимомах» (по
Бернатцу), дальше отстоящих от нормы по
морфологическим признакам и
содержащих немного лимфоцитов и эпителиоциты
медуллярной дифференцировки,
обнаруживаются аутоантитела к клеткам крови,
гипогаммаглобулинемия и аутоиммунные
гемоцитопении (6-20% случаев).
Эпизодически тимомы сочетаются с системными
аутоиммунными поражениями
соединительной ткани и неорганоспецифическими
аутоантителами (бывают дерматомиозит — в
5% наблюдений, красная волчанка— в 2%,
ревматоидный артрит и склеродермия —
менее, чем в 1 %). Описаны сочетания тимомы
и аутоиммунных тироидита и гипопарати-
роза. Аутоиммунные поражения типичны
и для карцином тимуса.
По-видимому, нормальная отбраковка
аутореактивных клонов при тимомах
нарушена, что и вызывает подобные явления.
Возможны также нарушения
дифференцировки клонов цитотоксических
лимфоцитов, выполняющих супрессирующие
функции. При перечисленных выше и
других иммунопатологических
заболеваниях содержание тимических гормонов в
крови снижено, а при некоторых введение
этих гормонов терапевтически эффективно.
Однако прямые доказательства того, что
аутоиммунный синдром при тимомах связан с
эндокринной гипофункцией тимуса,
отсутствуют. Возможно, решающую роль играют
как раз юкстакринные и паракринные
взаимодействия. В опухолевых клетках тимом
обнаруживают иммуногистохимическими
методами как избыток гормонов вил очковой
железы, так и нормальное их содержание, и
даже отсутствие.
Гиперплазия тимуса наиболее часто
включает не просто его увеличение, а и
диспластические процессы, когда в тимусе
оказываются практически начисто
отсутствующие там в норме В-лимфоциты и даже
В-лимфоидные фолликулы. Это явление,
ранее упрощённо именовавшиеся «тими-
том», затем было охарактеризовано как
лимфофолликулярная гиперплазия вилоч-
ковой железы. Данная форма гиперплазии
тимуса в еще большей степени, чем тимомы,
сопряжена с аутоаллергическими
расстройствами.
По данным Р. Де Лилли (1989),
лимфофолликулярная гиперплазия тимуса
найдена в 80-85% случаев
генерализованной миастении, присутствует у многих
больных с другими аутоаллергическими
расстройствами: болезнью фон Базедова и
тироидитом Хасимото (40-50 %), болезнью
Аддисона, системными аутоиммунопатиями
с неорганоспецифическими
аутоантителами, иммунопатологическим гломеру-
лонефритом, аутоиммунным гепатитом и
циррозом печени.
При инфекционно-аллергических
поражениях — бронхиальной астме, ревма-
тизме — частота обнаружения лимфофол-
ликулярной гиперплазии тоже повышена.
Существуют разногласия по поводу того,
первичны или вторичны лимфоидные
изменения тимуса при аутоиммунных
заболеваниях. Очевидно, в зависимости от состояния
паренхимы тимуса и конкретного вида ауто-
иммунопатии вопрос этот может решаться
по-разному (Я. Мак-Кей, 1987).
Ранее большое значение в развитии
лимфоидных фолликулов в тимусе придавали
гипотетическому нарушению гематотими-
ческого барьера, якобы экспонирующему
скрытые забарьерные аутоантигены и
провоцирующему аутоиммунный тимит.
Но, как убедительно показано А.С.
Шевелёвым (1986), антигены тимуса и в норме
не отделены от иммунологического
распознавания, так как в нём, как и в ЦНС,
имеется локальная внутритимическая система
иммунокомпетентных клеток. Более того,
частичная резекция тимуса в клинике и в
экспериментальных условиях хотя и нарушает
соответствующий барьер, не провоцирует
аутоиммунных заболеваний и лимфофол-
ликулярной гиперплазии (В.П. Харченко
и соавт., 1998). К тому же, по последним
данным, лимфоидные фолликулы с
накоплением, пролиферацией и плазматизацией
В-лимфоцитов образуются не прямо в
паренхиме тимуса, а в пределах
расширяющихся внутридольковых периваскулярных
пространств, куда клетки фолликулов
мигрируют из крови (X. Кристин, 1989).
Подчеркнем, что истинная гиперплазия
тимуса, то есть разрастание его
эпителиальной части, не всегда наблюдается при
появлении лимфоидных фолликулов.
Выше уже шла речь о сочетании появления
лимфоидных фолликулов с гипоплазией
и атрофией истинной паренхимы, как это
происходит при тимической
недостаточности, связанной с некоторыми системными
иммунокомплексными и аутоиммунными
поражениями. В-лимфоидные фолликулы
находят и при аутопсии клинически
здоровых лиц без признаков аутоаллергических
болезней и гиперплазии паренхимы тимуса
(в 10% наблюдений).
Нарушения эндокринной регуляции 665
Весьма аргументирована точка зрения
В.П. Харченко и соавт. (1998), которые
указывают, что собственно гиперплазия
эпителиоцитов тимуса встречается гораздо
реже, чем лимфоидные фолликулы вилоч-
ковой железы и, по существу, гиперплазия с
лимфоидными фолликулами наблюдается в
тимусе только в двух случаях — при
генерализованной миастении I типа и при болезни
фон Базедова.
Характерно, что оба заболевания вызваны
антирецепторными аутоантителами. Кроме
того, принципиально важно, что только
при генерализованной миастении I типа и
при болезни фон Базедова продукция ти-
мических гормонов гиперплазированным
эпителием тимуса повышена. При болезни
фон Базедова гиперфункция тимуса как
эндокринной железы носит временный
характер, тесно связана с влиянием
избытка тироидных гормонов и, возможно,
неидентифицированных стимулирующих
аутоантител на тимические эпителиоциты.
Выше, в разделе, посвященном патологии
щитовидной железы, основные механизмы
этого политаргетного аутоиммунного
заболевания уже рассматривались.
Что касается генерализованной
миастении и, в частности, ее I типа, которому
свойственны гиперплазия и гиперфункция
эпителия тимуса, механизмы данного
заболевания обсуждаются ниже.
Генерализованная миастения (старые
названия — myasthenia gravis, тяжёлая
миастения) — аутоиммунное рецепторное
заболевание, связанное с нарушением
идиотип-антиидиотипической регуляции
продукции и действия тимопоэтина,
выражающееся в прогрессирующем параличе
скелетных мышц из-за блокады и
дегенерации Н-холинергических рецепторов под
воздействием аутоантител.
Генерализованная миастения известна с 1672 г. в описании
британского врача Т. Уиллиса.
Болезнь встречается нередко (1/10000)
и поражает в 2 раза чаще женщин. Главное
ее клиническое проявление — утомляемость
и прогрессирующая слабость скелетных
ббб АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
мышц, вовлекающая всё новые группы.
Обычно явные симптомы начинаются с
мускулов, имеющих моторные единицы малого
размера — глазных. Больные не могут
совершить в быстром темпе несколько миганий
подряд. Затем вовлекаются мышцы орофа-
рингеальной группы, шеи, проксимальных
отделов конечностей. В тяжелых случаях
страдают все мышечные группы.
Смертность составляет не менее 5-10%, причина
гибели — паралич дыхательных мышц.
Почти 90 % больных имеют аутоантитела
к постсинаптическому Н-холинорецептору
мышц. Эти иммуноглобулины относятся к
нескольким клонам неодинаковой
специфичности. Они могут блокировать рецептор,
провоцировать его разрушение с участием
комплемента и эндоцитоз, сшивая
рецепторы между собой и уплощая поверхность
постанаптической мембраны. В норме
освобождение ацетилхолина в моторных
бляшках снижается при повторных пресина-
птических разрядах, что создаёт экономию
медиатора и не снижает постсинаптический
ответ, ибо параллельно вступают в действие
дополнительные рецепторы. При
генерализованной миастении этот механизм не
обеспечен достаточным числом резервных
Н-холинорецепторов, поэтому паралич у
больного проявляется при повторяющейся
или длительной активности мышечной
группы (Д. Драхманн, 1987).
Болезнь моделируется на животных,
иммунизированных Н-холинорецептором,
и пассивно переносится животным
сывороткой и её IgG. Реже миастенические антитела
принадлежат к классу IgA и IgM.
У большинства пациентов заболевание
сочетается с другими аутоиммунными
расстройствами, наиболее часто тиропатиями
и иными эндокринопатиями, дерматомио-
зитом и другими болезнями с неорганоспе-
цифическими аутоантителами. Имеется
связь с определёнными антигенами ГКГС
(В8, DR3,4). Более того, тимэктомия,
которая при I типе миастении даёт лечебный
эффект, не избавляет от сопутствующих
аутоиммунопатий, которые после удаления
тимуса даже обостряются. При миастении
состояние тимуса ненормально
практически всегда. У 30 % больных имеются тимомы,
у подавляющего большинства отмечаются
в тимусе лимфоидные фолликулы. Это
может протекать на фоне различных проли-
феративных и функциональных изменений
эпителиальных клеток и различного уровня
секреции тимических гормонов.
Выделяют, согласно данным В.П. Хар-
ченко и соавт. (1998), 2 разновидности
прогрессирующей генерализованной
миастении.
При I типе имеется гиперсекреция
тимических гормонов и гиперплазия паренхимы
тимуса в сочетании с лимфоидно-фоллику-
лярной дисплазией. Клинически больные
с этой формой заболевания моложе,
подавляющее их большинство — женщины,
они имеют особенности ГКГС, упомянутые
выше, и высокие титры аутоантител при
их узком спектре. У больных поражаются
мышцы глаз, а дизартрия является
редкостью. Тимэктомия облегчает
состояние больных, хотя это не коррелирует со
снижением уровня тимических гормонов.
Вероятно, тимус играет роль органа, где
происходит сенсибилизация, а тимэктомия
оказывает иммуносупрессирующий
эффект, то есть патогенез болезни
опосредован иммунологически, а не напрямую —
избытком тимопоэтина.
При II типе содержание тимических
гормонов понижено, паренхима тимуса
атрофирована, несмотря на наличие лимфо-
идной фоликулярной дисплазии, нарушено
формирование телец Гассаля. Больные
обоего пола, возраст их — средний. Ассоциация
с гаплотипами ГКГС — иная (В2, В7, DR2).
Титр аутоантител ниже, а спектр — шире,
сильнее выражен дефицит Т-супрессоров.
Клинически часто встречается дизартрия, а
общая тяжесть миастении не столь велика,
как при I типе. Операция тимэктомии при
данном типе миастении неэффективна и
может даже усугубить болезнь.
Гормональная и иммунологическая
функции тимуса взаимозависимо затрагиваются
в патогенезе генерализованной миастении.
Гормон эпителиоцитов тимуса тимопоэтин
Нарушения эндокринной регуляции 667
ПАТОФИЗИОЛОГИЯ
околощитовидных
ЖЕЛЁЗ И КАЛЬЦИЙ-
ФОСФОРНО-МАГНИЕВОГО
способен сам связываться с Н-холинерги-
ческим рецептором, в частности, на
поверхности миоидных клеток тимуса. В
эксперименте он блокирует нервно-мышечную
передачу (Р. Лисяк, Р.Барчи; 1984).
Непосредственно в эпителиальных клетках
тимуса находятся пептиды, реагирующие
с сывороточными антихолинорецепторны-
ми аутоантителами больных миастенией.
Предполагается, что в основе патогенеза
болезни может лежать выработка в
аномально высоких титрах антиидиотипических
аутоантител против антител к тимопоэтину.
Эти иммуноглобулины, частично имитируя
активность тимопоэтина, связываются с
Н-холинорецептором и блокируют его,
запуская, в отличие от самого
тимопоэтина, процессы разрушения рецептора и его
интернализации. Толчком к смещению
идиотип-антиидиотипического равновесия
может быть инфекция. По крайней мере,
антигены, сходные с мишенью аутоантител,
обнаружены у некоторых дифтероидных
бацилл и микробов кишечной группы.
По-видимому, как и при других
аутоиммунных эндокринопатиях, мы имеем дело
в случае двух типов генерализованной
миастении с различным патогенезом болезни.
При I типе может играть ключевую роль
нарушение идиотип-антиидиотипической
сети с участием тимопоэтина, подобно
аналогичному процессу с участием ТТГ,
описанному при болезни фон Базедова. При
II типе может быть решающим дефицит
Т-супрессоров из-за нарушения их
созревания при атрофии эпителия тимуса, в связи с
чем происходит активация клонов
«естественных аутореактивных лимфоцитов» (см.
т. I с. 607, примечание 21). О лечении данного
заболевания см. Н.М. Жулёв и соавт., 2005.
В заключение раздела хотелось бы
подчеркнуть, что в ходе накопления данных
о механизмах эндокринных дисфункций
тимуса, по нашему мнению, еще не раз себя
проявит аналогия с гиперфункциональными
и гипофункциональными аутоиммунными
тиропатиями, учитывая близость
происхождения этих двух эндокринных желёз.
МЕТАБОЛИЗМА
Роль кальция в жизни организма настолько
велика, что неверно было бы просто сказать,
что кальциевый метаболизм, как и всякий
минеральный, регулируется клетками — и
этим всё исчерпывается. Ведь множество
интрацеллюлярных процессов, от митоза и
рождения клеток, до апоптоза и их гибели —
в свою очередь — регулируются кальцием,
при участии специфически распознающих
его белков (кальмодулина, кальэлектри-
нов, кальпаинов и т.д.). От кальция зависит
генерация потенциалов действия и
электромеханическое сопряжение, передача
гормонального сигнала и клеточная локомоция.
Кальций регулирует и скорость жизненно
важных внеклеточных процессов —
например, свёртывания крови.
Все клетки — от примитивных
одноклеточных организмов — до нейронов коры
больших полушарий человека жизненно
зависят от обмена кальция. По мнению
К. и Ч.Р.Клеемен (1981), это связано с тем
обстоятельством, что жизнь зародилась в
среде первичного океана, богатой кальцием.
Характерно, что паратироидный гормон
впервые обнаруживается у наземных животных,
переселившихся в среду, где кальций стал
менее доступен. Будучи важным регулятором,
ион кальция в то же время ядовит для клеток,
и значительное повышение его
внутриклеточной концентрации запускает механизмы
клеточной гибели, участвуя в некробиозе и
апоптозе (см. т. I, с. 174-196). Внутри клеток
концентрация кальция в 10000-100000 раз
меньше, нежели снаружи. Поэтому уровень
кальция вне и внутри клеток подлежит
прецизионному контролю, а при попадании в ци-
тозоль кальций эффективно секвестрируется
митохондриями и ЭПР.
Метаболизм кальция в организме тесно
переплетён с обменом фосфатов, связы-
668 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
вающих большую часть
внеклеточного кальция в виде кристаллов гидрокси-
апатита (эмпирическая формула
которого — Са10(РО4)6(ОН)2), в композитных
минерализованных структурах — костях.
В организме около 2 кг кальция и более 1 кг
фосфора. Это два его главных минеральных
компонента. Из данного количества 98%
кальция и 85 % фосфора связано в костях
и зубах.
По мнению Г. Кретцинджера (1978),
именно роль фосфата как ключевого
участника энергетического метаболизма главного
внутриклеточного аниона, концентрации
которого в клетках в 100 раз превышают
наружные, предопределила биологический
выбор кальция на роль убиквитарного
регулятора, как и необходимость поддерживать
на низком уровне внутриклеточный уровень
этого катиона. Коль скоро клетки стали
поддерживать кальциевый градиент, появилась
возможность использовать его модуляцию в
информационных целях.
Близкая физико-химическая аналогия
двух щелочно-земельных катионов — Са2+
и Mg2+ привела к тесному переплетению их
метаболизма. Магний — важный кофактор
некоторых аденилатциклаз, фосфатаз и
фосфорилаз, участник трансфосфорилиро-
вания, что связывает его судьбу в организме
и с фосфором. Большая часть магния (60%)
тоже депонирована в скелете. В силу
взаимозависимости обмена этих ионов, наличия
общей регуляции и комбинированных
нарушений патофизиология обмена магния также
рассматривается именно в данном разделе.
Регуляция кальциево-фосфорно-
магниевого гомеостаза
Вопросы метаболизма кальция, фосфатов
и их регуляции с участием кальциферола
уже затрагивались выше, при рассмотрении
рахита и гипервитаминоза D (см.
«Патофизиология витаминного обмена»). Судьба
кальция и фосфора в организме отражена
на рис. 128.
Содержание кальция в диете
нормируется и не должно быть менее 0,6 г за сутки.
Обычно у взрослых людей за сутки с пищей
поступает 0,6-1 г кальция, но у любителей
оздоравливающих пищевых добавок и ви-
таминно-минеральных композиций этот
показатель порой превышает 1,5 г. Кальций
плохо всасывается в ЖКТ. Всего 125-200
мг в день абсорбируют двенадцатиперстная
кишка и верхняя треть тощей. Интересно,
что одновременно определённое количество
данного иона (до 0,2 г в сутки) экскретиру-
ется в подвздошной кишке. Кальций
выводится также почками (до 0,3 г в сутки) и, в
малом количестве, потовыми железами (до
0,1 г/сутки). Менее 1 % всего кальция
находится в интерстициальной жидкости.
В плазме 40 % кальция связано с белками,
в основном с альбумином (связанная форма
кальция), 15 % — с кислыми органическими
анионами (комплексная форма кальция), а
остальной кальций свободен. Процент
связанного кальция (СвСа) может быть оценён
по эмпирической формуле:
СвСа( %) = 0,8А (г/л)+0,2Г(г/л)+3,
где А — концентрация в плазме альбумина,
а Г — глобулинов.
Количество общего кальция в плазме
понижается при гипоальбуминемии, но это
не оказывает влияия на содержание
катиона кальция. Содержание ионизированного
кальция в плазме находится в обратной
зависимости от рН и от концентрации
фосфат-аниона: гиперфосфатемия и
алкалоз способствуют появлению признаков
гипокальциемии, хотя уровень общего
кальция при этом не меняется. Ацидоз и
гипофосфатемия, наоборот, повышают
содержание ионизированного кальция в
плазме.
Кальций экскретируется почками в
количествах, составляющих примерно 0,15-0,3 г
в сутки, причем этот процесс лишь при
очень низких содержаниях кальция в диете
определяется поступлением данного иона
в организм. При нерезко сниженном,
нормальном и избыточном насыщении диеты
кальцием между скоростью экскреции
кальция с мочой и его содержанием в рационе
нет строгого параллелизма. Поэтому можно
сказать, что собственно почечные механиз-
Нарушения эндокринной регуляции 669
Са Р
|0,5-1,5г/0,6-2,0г
внутриклеточная
Са
0,25-0,5 г
-4-
0,1-0,2 г
фекалии
Рис 128. Биологическая локализация, обмен и судьба фосфора и кальция в организме.
Пояснения — в тексте
мы как сохранения кальция, так и
выведения его избытка не обладают большой
лабильностью. Они должны эффективно
взаимодействовать с кишечными
механизмами. Кальций реабсорбируется в почках в
дистальной части канальцев (15%) и в ещё
большей мере — в проксимальной части
(60 %) и петле Генле (25 %).
Уровень ионизированного кальция в
плазме регулируется взаимодействующими
гормонами паратирокринином и кальцито-
нином, а также витамином D. Под их
контролем приблизительно 0,5 г кальция в сутки
у взрослого индивида обменивается между
скелетом и плазмой крови.
Фосфор2, в отличие от кальция,
абсорбируется в ЖКТ, напротив, очень активно.
С пищей в среднем в день поступает около
1,2 г фосфора. Для диагностики нарушений
фосфорно-кальциевого обмена
концентрации фосфора в крови следует определять
натощак, так как, в отличие от уровня
кальция, они растут после еды.
В тощей кишке всасывается до 90%
суточного потребления фосфатов. Почки
экскретируют 15 % фильтрующихся
фосфатов с мочой, в равновесном с поступлением
этих ионов режиме. Фосфат может активно
секретироваться в канальцах. Реабсорбция
фосфата происходит на 9/10 — в
проксимальных канальцах, а на 1/10 — в более
дистальных частях нефрона.
В дополнение к 85 % фосфора,
депонированным, как уже отмечалось выше, в костях и
зубах, мягкие ткани содержат существенную
часть связанного фосфора и фосфат-аниона
(до 14%). Всего 1 % фосфора находится во
внеклеточной жидкости. Макроэргические
фосфатные соединения и фосфорилиро-
ванные активные метаболиты в норме не
могут свободно покидать клетки. Поэтому
только 12% фосфатов плазмы связано с
670 АШ. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
белками, остальные представлены
свободными фосфат-анионами. Уровень фосфора в
плазме зависит от факторов, регулирующих
обмен кальция. Но кальциевый гомеостаз
не является единственной детерминантой
фосфорного обмена. Кроме этого, судьба
фосфора определяется ходом
энергетического метаболизма в клетках. B.C. Ильин,
вообще предпочитал говорить не о
фосфорном, а об «углеводно-фосфорном обмене»,
имея в виду исключительную зависимость
судьбы фосфата от катаболизма углеводов
(1966). При активации синтеза гликогена
фосфаты переходят внутрь клеток. Поэтому
глюкоза, инсулин, сахаристая пища —
вызывают гипофосфатемию из-за
перемещения фосфат-анионов в клетки. Алкалоз,
особенно дыхательный, также провоцирует
гипофосфатемию, как полагают М.М. Горн
и соавт. (1999), в силу активации клеточного
гликолиза и образования
фосфоросодержащих метаболитов глюкозы. Дыхательный
ацидоз после торможения гликолиза лакта-
том, наоборот, приводит к выходу фосфата
из клетки и гиперфосфатемии. В силу этих
некальциевых факторов, влияющих на
уровень фосфора в плазме, концентрация
фосфатов имеет чёткий суточный ритм,
тогда как у ионизированного кальция такая
периодизация отсутствует. Низший уровень
фосфатов в плазме наблюдается утром, а
после полудня и ночью имеются 2 пика
(М.Ф. Холик и соавт., 1994).
Процессы депонирования кальция и
фосфора в костях и их абсорбции/экскреции в
кишечнике и почках взаимно
сбалансированы так, что концентрация этих ионов в
крови изменяется в весьма узких пределах
(8,8-10,4 мг/дл или 22-26 мМ/л кальция и
2,5-4,5 мг/дл или 9-13 мМ/л фосфата, см.
также приложение А).
Магний — преимущественно
внутриклеточный катион, четвертый по абсолютному
содержанию в организме (Л.Г. Смит, 1987).
Тело взрослого человека содержит около
25 г магния. В интрацеллюлярной
жидкости его концентрация в 8 раз выше, чем
в интерстициальной. Взрослому человеку
в день нужно не менее 3,5-4,5 мг магния,
чтобы не расходовать его костные резервы.
Богаты магнием зелень, где он выполняет
ключевую роль при фотосинтезе в составе
хлорофилла, морепродукты и мясо, орехи и
семечки, бобовые, бананы и цитрусовые,
шоколад, патока и кокосы. Впрочем, если этих
разносолов на столе нет, полезно помнить,
что очень богаты данным металлом маковые
зерна, а также самый обыкновенный... чай.
Магний всасывается в тонком
кишечнике при участии витамина D примерно на
40 % от его поступления с пищей. Избыток
фитиновой кислоты и жирных кислот, а
также алкоголь отрицательно влияют на его
абсорбцию. Высокие концентрации магния
в кишечном содержимом мешают
всасыванию кальция, но не наоборот. Магний
экскретируется почками, причём
эффективность его реабсорбции может достигать
95 %. Почки варьируют экскрецию магния в
равновесном, по отношению к поступлению
этого электролита, режиме, в широчайшем
диапазоне — от 1 до 250 мМ в день.
Алкоголь препятствует реабсорбции магния в
нефронах. Кальций и магний конкурируют
при реабсорбции. Магний — составная часть
минерального вещества костей, участник
работы трансфосфорилирующих ферментов
и амино-ацил-т-РНК-синтетаз,
обеспечивающих условия для трансляции белков.
В электрофизиологических процессах
определённое значение имеет роль магния как
антагониста кальция, проявляющаяся в их
различном влиянии на ЦНС.
Центральные органы, регулирующие каль-
ций-фосфорно-магниевый обмен — парати-
роидные железы.
В гистологии данные органы называются
околощитовидными или паратироидными.
Несмотря на это, авторы считают
возможным сохранить в данном разделе
традиционный для патофизиологической литературы
классический комбинированный термин
«паращитовидные железы».
Нижние паращитовидные железы
возникают из того же третьего глоточного
кармана, который даёт начало и тимусу
(см. выше), а верхние являются дериватами
четвёртого глоточного кармана. Таким обра-
Нарушения эндокринной регуляции 671
зом, у верхних и нижних полюсов каждой
из долей щитовидной железы, вне капсулы
последней, но под её фасцией в норме
обнаруживается по одной паращитовидной
железе.
Однако топография данного
эндокринного органа, быть может, наиболее изменчива
среди всех эндокринных желёз. У очень
значительной части людей (более 10%)
дополнительные паращитовидные тельца
обнаруживаются по всему ходу
эмбриональной миграции глоточных карманов: в том
числе, в тимусе, переднем средостении, близ
каротидных артерий. Они служат нередко
источником эктопических гормонообразу-
ющих опухолей.
Околощитовидные железы — наиболее
«молодое» органное открытие
эндокринологов. Верхние наружные околощитовидные
железы впервые описал шведский анатом
И.К. Сундстрём только в 1880 г.
Э. Глей, в 1891 г., интересуясь
последствиями тироидэктомии, показал, что у собак
удаление щитовидной железы (как
оказалось впоследствии, вместе с паращитовид-
ными) вызывает судороги и смерть. В 1893 г.
Ж. Николя описал у летучих мышей нижние
внутренние паращитовидные железы. Затем
Э. Глей установил, что у кролика, в отличие
от собаки, удаление щитовидной железы не
ведет к.тетании, так как у первого имеются
отдельно лежащие «дополнительные
щитовидные желёзки» (1892, 1897). И только
после опытов Вассале и Дженерали стало
ясно, что это самостоятельные нетироидные
органы, удаление которых воспроизводит
паратиропривную тетанию у животных.
Долгое время после этого в
эндокринологии удерживалось ложное представление
об антитоксической функции паращито-
видных желез в отношении попадающих
в организм бактериальных спазмогенных
токсинов. Острые модели тетании не
позволяли пролить свет на роль паращитовид-
ных желёз в регуляции метаболизма, так
как животные быстро погибали. Но в 1906
Н. Эрдгейму удалось создать модель
хронической несмертельной тетании на крысах,
располагающих небольшими добавочными
островками паратироиднои ткани, удаляя
у них основные паращитовидные железы.
В этих условиях, наблюдая за подопытными
животными он установил нарушение
минерализации костей и зубов при удалении и
частичное восстановление её — при подсадке
этих органов. У.Г. Мак-Коллюм и К. Фе-
гтлин (1909) на данной модели показали,
что паратироидэктомия понижает уровень
кальция в крови. Наконец, A.M. Хенсон
(1924) получил из желёз экстракт,
предотвращающий тетанию у паратироидэкто-
мированных собак и повышающий уровень
кальция в крови. Еще в 1910 г. Э. Сильвест-
ри показал, что кастрация кроликов-самцов
понижает у них мышечную возбудимость и
делает животных менее чувствительными
к тетаногенным ядам. Развивая эту
концепцию, Л.Р. Перельман (1925) предпринял
классическое исследование, в котором
продемонстрировал, что сама по себе кастрация,
в отличие от паратироидэктомии, не влияет
на уровень кальция в крови у котов и собак.
В то же время, наступление тетании при
удалении паратироидных желёз у котов и собак
зависит не только от уровня кальция, но и
от уровня половых гормонов. При низком
уровне половых гормонов после кастрации
тетания отсутствует, даже если кальций
крови снижается с 10,8 до 4,6 мг/дл.
Данное исследование, проведённое на
модели паратиропривной тетании, имело
значение, далеко выходящее за рамки
патофизиологии паращитовидных желез. Оно
впервые продемонстрировало возможность
пермиссивного действия одних гормонов на
эффекты других.
В последующем А. Хэму (1940) удалось
показать, что основные эффекты
гормонального начала паращитовидных желез связаны
с повышением содержания кальция в крови,
а гиперплазия железы индуцируется именно
гипокальциемией.
Паращитовидные железы состоят из
капсулы, стромы и недольчатой паренхимы,
в которой представлены мелкие главные
клетки двух подвидов: тёмные, содержащие
секреторные гранулы и, вероятно,
покоящиеся, и светлые — лишённые таких гранул
672 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
паратирокрин (паратгормом)
кальцитонин
Рис. 129.Структура паратгормона и кальцитонина человека (по Б. Катцунгу, 1998> А.Уайту, 1981)
и секреторно активные, последние также
богаты гликогеном. В железе имеются также
более крупные оксифильные клетки,
появляющиеся, очевидно, путём трансформации
главных в период пубертата и с возрастом
всё более многочисленные. Оксифильные
клетки рассматриваются как результат
инволюции главных. Функция оксифиль-
ных клеток точно не известна доныне. По
последним данным, паратгормон может
синтезироваться и в них.
Главные клетки обладают очень
развитым ГЭР, в оксифильных ГЭР представлен
слабее. Оксифильные клетки богаты, а
главные — относительно бедны митохондриями.
Интересная особенность нормальной
структуры паращитовидных желез — наличие в
каждой из них большого количества жира,
накапливаемого с возрастом (у пожилых —
до 70 % объема желез). При гиперплазии и
опухолях количество жира в
паращитовидных железах резко снижается.
Основной продукт паращитовидной
железы — паратирокринин (прежние
названия — паратирин или паратгормон).
Структуру паратироидного гормона
расшифровали в 1970 г. Х.Д. Найел и соавторы.
Его выделяют главные клетки.
Паратгормон — это пептидный регулятор
(рис. 129), состоящий из 84 аминокислот
(молекулярной массой чуть более 9,5 кД).
Паратгормон возникает из препрогор-
мона длиной в 131 аминокислотный
остаток (молекулярной массой около 12,5 кД,
синтезируется на полисомах) через стадию
прогормона (90 аминокислот, образуется
в ЭПР под действием клипазы), причем
его процессинг модулируется ферментом
фурином. Прогормон поступает за счет
энергозависимого механизма в комплекс
Гольджи, где протеолитический мембран-
но-связанный комплекс (триптическая
клипаза) вычленяет из него активный
гормон. Препрогормон кодируется в 11-й
хромосоме, а фурин — в 15-й. Оба экспрес-
сируются совместно. Весь процесс синтеза и
секреции (которая может происходить как в
виде экзоцитоза специальных гранул, так и
в безгранулярном режиме) занимает около
30 мин., причём 15 мин. тратится на
упаковку готового гормона в гранулы.
Паратироидная секреция активируется, в
основном, в ответ на снижение
концентрации ионизированного (свободного) кальция
в крови. Опосредованно гиперфосфатемия
также активирует паращитовидные железы,
Нарушения эндокринной регуляции 673
снижая концентрацию ионизированного
кальция.
Так же, как кальций, но значительно слабее,
на секрецию паратгормона влияет и магний.
Однако тяжелая длительная гипомагниемия
парадоксальным образом подавляет секрецию
паратгормона, так как магний необходим
самим паратироцитам для выделения их
гормонов (см. ниже). Главные клетки располагают
кальциевым сенсором — трансмембранным
гликопротеином, вмонтированным в их
плазматическую мембрану. Таким же сенсором
обладают, кроме паратироцитов, С-клетки
щитовидной железы и некоторые клетки мозга и
почек. Этот рецептор кодируется в хромосоме
3, при повышении уровня экстрацеллюляр-
ного ионизированного кальция он блокирует
экспрессию генов гормона паращитовидных
желез и ключевого фермента его активации.
В настоящее время доказано, что выработка
паратгормона, преимущественно
регулируется in vivo на посттранскрипционном уровне.
При повышении уровня иона кальция в крови
происходит стимуляция рецептора, активация
пострецепторного Gq-белка и нарастание
концентрации кальция в цитозоле, блокирующее
функцию главных клеток. Мутации данного
сенсора дают при гомозиготности тяжелый
наследственный неонатальный гиперпара-
тироз, а у гетерозигот — доброкачественную
семейную гипокальциурическую гиперкальци-
емию (см. ниже).
Кальциевый сенсор может модулировать
не только быстрый выброс из клеток
готового гормона. Установлено, что к кальцию
чувствительны протеазы, разрушающие
в норме около 90 % образующегося пара-
тирокринина. Таким образом, изменяя их
активность, кальциевый сигнал способен
влиять на долговременный пул гормона,
через скорость его разрушения. При
избытке кальция возможна практически полная
деградация паратгормона в главных клетках
под действием нейтральных
кальций-зависимых протеаз с секрецией его неактивных
С-концевых пептидов.
Клетки паращитовидной железы
вырабатывают также пептид, подобный паратиро-
кринину и закодированный в 12-й хромосоме
геном, произошедший, вероятно, от общего
с паратирокрининовым геном
предшественника.
Это убиквитарный пептид, к синтезу
которого способны и многие апудоцитарные
клетки, и неопластические клоны, а также
различные органы плода и взрослого —
сосуды, плацента, мозг, лёгкие, сердце, молочная
железа. Поэтому основная часть данного
паракринного регулятора производится за
пределами собственно паращитовидных
желёз. Именно паратирокринин-подобный
пептид, а не сам паратгормон, как
считали ранее, ответственен за большую часть
случаев эктопической продукции гипер-
кальциемических регуляторов. Данный
пептид имеет 141 аминокислоту, первые
30 из которых высокогомологичны
соответствующим аминокислотам паратгормона и
обеспечивают сходство их биологического
действия.
Так как его экспрессия — не редкость при
многих апудомах и иных неопластических
процессах, с избытком паратирокринин-
подобного пептида связывают остеопороз,
сопровождающий многие злокачественные
новообразования. В норме у взрослых
пептид не занят регуляцией кальциевого
обмена. Однако делеция гена паратирокри-
нин-подобного полипептида приводит к
тяжелой остеохондродисплазии и даже гибели
плодов крыс. Пептид необходим для роста
хондроцитов и задерживает минерализацию
хрящей. Большое значение имеет недавно
открытый факт, что у животных и человека
именно данный пептид обеспечивает
трансплацентарный перенос кальция к плоду,
захват кальция молочными железами и
насыщение им грудного молока. В женском
и особенно в коровьем молоке этого пептида
исключительно много. Возможно, он как-то
связан и с сокращениями матки.
Интересной особенностью данного
биорегулятора служит его способность
подавлять пролиферацию эпидермиса, причём
изучаются его потенциальные антипсориа-
тические свойства. В этой связи вспомним о
том, что молочные ванны и грудное молоко
674 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
эмпирически издавна применялись в
косметологии для улучшения вида и свойств
кожи. Есть сведения, что данный регулятор
необходим для развития волосяных
фолликулов и молочных желёз (Д.М. Шёбек,
Г.Дяе. Стрюлер, 1997).
Возможно, дефицит этого пептида связан
с патогенезом кожного кандидоза у больных
с гипофункцией паращитовидных желёз.
В дальнейшем изложении роль и функции
паратирокринин-подобного пептида больше
не обсуждаются.
Дополнительно стимулирующую роль в
паратироидной секреции могут играть
симпатические р-адренергические нервные
воздействия и гистаминергические влияния на
Н2-рецепторы. Таким образом, регуляция
паращитовидных желёз осуществляется,
насколько известно на данный момент, по
парагипофизарному принципу. Впрочем,
как и для гормонов, секреция которых
подлежит гипоталамической регуляции,
имеется циркадный ритм паращитовидной
активности, согласно которому акрофаза
секреции паратгормона наступает после
восьми часов вечера. Секреция
паратгормона ночью втрое выше, чем днём и на
протяжении всех суток имеет импульсный
характер. У человека не обнаружено гипо-
физарных регуляторов секреции
паращитовидных желёз, но у рыб, которые не имеют
отдельных паращитовидных телец, пролак-
тин гипофиза и другой аденогипофизарный
паратоподобный гиперкальциемический
гормон выполняют функции паратгормона.
Интересно, что и у человека паратгормон и
пролактин имеют общие эффекты —
например, оба стимулируют активацию витамина
D. Имеются предпосылки к существованию
гипоталамо-гипофизарной регуляции
функций паращитовидных желёз и у
человека. Ведь паращитовидные железы и
аденогипофиз близки по эмбриональному
происхождению.
Из гипоталамуса быка были получены
пептиды, введение которых вызывало
гипокальциемию у животных только при
наличии паращитовидно-щитовидного
комплекса — предполагаемые кандидаты на роль
центрального паратиростатина.
Благодаря вышеописанным механизмам,
паращитовидные железы могут
осуществлять срочные (выброс готового
паратгормона), отсроченные (синтез гормона de
novo) и отдалённые (гиперплазия) аспекты
реакции на гипокальциемию.
Активная форма витамина D — каль-
цитриол — подавляет экспрессию гена
паратгормона, реализуя дополнительную
обратную связь в данной системе. Этот
эффект не зависит от гиперкальциемии,
вызываемой кальцитриолом. Секреция
готового паратгормона ингибируется также
через а-адренорецепторы.
Время полувыведения паратгормона из
плазмы крови составляет 20-30 мин. и,
насколько известно, он не имеет существенной
связанной с белками плазмы фракции. Ин-
тактный парат-гармон подвергается проте-
олизу в паратироцитах и в плазме, причём он
расщепляется на короткий амино-концевой
пептид, который высокоактивен (вся
биологическая эффективность человеческого
паратгормона сосредоточена в его первых
34-х аминокислотах N-конца, а большая её
часть — в первых двух аминокислотах), и
более длинный неактивный карбокси-кон-
цевой пептид. По некоторым данным,
может образовываться также средний пептид.
Печень поглощает и разрушает нативный
паратирокринин, но не захватывает
средний и С-концевой пептиды — продукты его
деградации.
N-концевой пептид паратгормона имеет
очень короткий срок полувыведения из
циркуляции (до 10 мин.), так как инактиви-
руется клеткамимишенями путём эндоцито-
за, а также на 45 % экскретируется с мочой.
С-концевой пептид паратирокринина
длительное время циркулирует в крови и в
норме на 60 % выводится почками. При
почечной недостаточности экскреция С-концевого
пептида паратгормона особенно сильно
замедляется, он накапливается в крови и
создаёт ложное впечатление гиперпаратироза,
которое, однако, чаще всего не равнозначно
избытку биологически активного гормона.
Нарушения эндокринной регуляции 675
Дело в том, что многие иммунологические
методы определения паратгормона,
особенно разработанные давно, основываются на
применении антисывороток, опознающих
его средний пептид или С-конец. Такие
методы опеделяют неактивную форму
гормона в сумме с активной. При диагностике
нарушений, связанных с паратгормоном,
важно использовать методы, определяющие
содержание интактного гормона или же
применять двойное определение — с антителами
против как N-концевого, так и С-концевого
пептидов. Средний и С-концевой пептиды
паратирокринина обладают определённым
патофизиологическим действием и
расценивались, отчасти — преувеличенно, как одни
из важных «уремических токсинов». Им
приписывалось нейротоксическое и
антигонадное действие (К. Клеемен, Ч. Клеемен,
1981).
Парат-гормон (как и его аналог парати-
рокринин-подобный полипептид)
взаимодействует с гликопротеидным рецептором
на клетках-мишенях, принадлежащим к
семейству, ассоциированному с G-белками.
Рецептор кодируется в хромосоме 3,
имеет более 400 аминокислот и гомологичен
рецептору кальцитонина (см. выше). Пост-
рецепторная передача от этого рецептора
осуществляется с участием циклонуклео-
тид-протеинкиназного посредника, а также
фосфолипазы С, инозит-фосфатидов и
кальция. Дефект данного рецептора
приводит к наследственной остеодистрофии Ол-
брайта. Рассматриваемые здесь рецепторы
в изобилии представлены в костях и почках,
а в ЖКТ, по-видимому, большее значение
имеют не прямые, а кальцитриол-опосредо-
ванные эффекты парат-гормона.
Для понимания механизмов действия
паратгормона и патогенеза нарушений
кальций-фосфорного гомеостаза полезно
вспомнить основы гистофизиологии костной
ткани, которая служит главной мишенью
кальций-фосфорорегулирующих гормонов.
Подробнее эти вопросы рассматриваются
в т. III при изложении патофизиологии
частных заболеваний
опорно-двигательного аппарата. Ниже представлена лишь
минимальная необходимая информация по
этому вопросу.
Напомним, что кость состоит из так
называемых основных многоклеточных
единиц ремоделированиЯу ответственных за
локальные формообразование и местные
концентрации кальция и фосфора. В
составе таких единиц имеются мононуклеарные
потомки недифференцированных мезен-
химальных клеток — остеобласты. Они
синтезируют коллаген I типа, располагают
рецепторами паратгормона и ответственны
за отложение органического остеоида и его
последующую минерализацию. Маркером
их активности служит секретируемый ими
энзим — щелочная фосфатаза.
Минерализация обеспечивается при участии минорных
неколлагеновых кальций-связывающих
белков остеобластов, которые содержат
остатки а-карбоксиглютаминовой кислоты,
фиксирующие кальций. К ним относятся
остеокальцин и матриксный карбоксиглю-
тамил-содержащий белок. Карбоксиглю-
таминирование обоих белков зависит от
витамина К (см. выше «Патофизиология
витаминного обмена»). Остеокальцин
уникален для костей и зубов, и его уровень в
крови отражает скорость остеогенеза.
Параллельно через тромбоспондин, ос-
теонектин и остеопонтин эти фиксаторы
кальция (и магния) закрепляются на колла-
геновой матрице. Окружая себя
минерализованным остеоидом, остеобласты
превращаются в остеоциты, цитоплазма которых
образует отростки, через гаверсовы канальцы
остеоида связанные с соседними остеоцита-
ми. Остеоциты участвуют в локальной пери-
лакунарной деструкции кости и могут
влиять на быстрые колебания уровня кальция в
крови. Однако основную остеолитическую
функцию в единицах ремоделирования
кости выполняют потомки моноцитов —
гигантские многоядерные макрофаги костей —
остеокласты. Остеокласты перемещаются и
образуют в участках резорбируемой кости,
в особых лакунах Хаушипа (Дж. Хаушип,
1820), активный слой, прикрепляясь через
специальный адаптер — а\$Ъ~интегрин — к
676 АЖ Зайчик, Л.П.Чурилов Патохимия
остеопонтину. Они выделяют на своей
активной гофрированной каёмке коллагеназу
и маркерный фермент — кислую фосфатазу,
лизируя минерализованный остеоид и
растворяя кристаллы гидроксиапатита. Для
этого, с помощью специальных протонного
АТФазного насоса и карбоангидразы II типа
ими локально создаётся зона кислого рН=4
(М.Ф. Холик и соавт., 1994). Молодой
неминерализованный остеоид устойчив к
их действию. Повреждённая кость при
воспалении резорбируется ими и заменяется
остеобластами на новую. Молодые
остеокласты имеют рецепторы паратгормона и
кальцитонина, но на зрелых остаются лишь
последние. Нет у них и рецепторов
кальцитриола. Дифференцировка остеокластов
зависит от гранулоцитарно-моноцитарного
колониестимулирующего фактора, ИЛ-6 и
паратгормона.
Остеобласты и остеокласты
функционируют согласованно, что приводит к
обновлению всего кальция костей за период,
примерно, в 5-6 лет. Рост костей в длину
зависит от энхондрального образования
костной ткани на месте метаэпифизарного
хряща, а в ширину (толщину) — от пери-
остального окостенения (см. также выше
«Витамин D, рахит и гипервитаминоз D»).
Костная ткань находится под контролем
многих гормонов. Так, СТГ, пролактин,
инсулин и андрогены способствуют синтезу
остеоида. Глюкокортикоиды снижают в
костях синтез коллагена, а также, препятствуя
действию кальцитриола в кишечнике и
уменьшая почечную реабсорбцию кальция,
способствуют потере этого иона и остео-
порозу. Эстрогены способствуют синтезу
остеоида и отложению кальция в костях как
опосредованно через главные регуляторы
кальциевого обмена, так и непосредственно.
Мощными паракринными
стимуляторами остеогенеза служат различные факторы
роста (фибробластов, тромбоцитов, а также
трансформирующий и инсулиноподобный).
Резорбция кости стимулируется через про-
стагландины такими паракринными
регуляторами, как ИЛ-1, кахексии, лимфотоксин и
интерферон-у.
чдокринно-метаболические нарушения)
Но решающей остаётся регуляция с
помощью кальцитонина, кальцитриола и
паратгормона.
Паратгормон способен осуществлять в
организме следующие эффекты,
определяющие ход вышеописанных процессов (см.
также рис. 130 ниже):
>- стимуляцию второго гидроксилирования
витамина D в почках, превращающего
этот прогормон в активный гормон
1,25-дигидроксивитамин D (кальцитри-
ол, см. «Патофизиология витаминного
обмена»). Кальцитриол — не полный
синергист действия паратгормона. Он,
подобно паратгормону, стимулирует
нарастание содержания кальция и магния
в плазме, но, в отличие от паратирокри-
нина, задерживает и фосфаты.
>- активацию остеокластов, остеолиза
и освобождения кальция из костей
(Н.А. Барникот, 1948). Гормон
способствует появлению у молодых
остеокластов специфической
гофрированной каёмки, с помощью которой они
резорбируют костное вещество, а также,
в более отдалённые сроки, увеличивает
само количество остеокластов, ускоряя
их дифференцировку из моноцитов.
Гормон стимулирует перилакунарный
остеолиз глубокими остеоцитами. В
последнее время показано, что
активирующее действие гормона на зрелые
остеокласты носит непрямой характер.
Оно паракринно опосредовано цитоки-
нами, выделяемыми в ответ на гормон
в остеобластах и фибробластах (ИЛ-1,
кахексином и лимфотоксином, а также,
возможно, ИЛ-6 и гранулоцитарно-
моноцитарным колониестимулирую-
щим фактором). Параллельно этому
паратгормон через остеобластические
рецепторы стимулирует и остеогенез.
При высоких концентрациях гормона
преобладает стимуляция остеолиза, при
низких — остеогенеза. Периодические
курсовые воздействия небольших доз
паратгормона оказывают
анаболический эффект на костную ткань.
Нарушения эндокринной регуляции 677
В целом паратирокринин способствует
отрицательному костному балансу, то есть
соотношейию темпов остеогенеза и остеоли-
за, с преобладанием последнего, показателем
чего служат наблюдаемые при гиперпарати-
розе повышение выведения оксипролина
и сиаловых кислот с мочой. Кальцитриол
действует синергично с паратирокринином,
а 24,25-дигидроксивитамин D (секальци-
ферол) стимулирует остеогенез.
>* Паратирокринин уменьшает клиренс
кальция и магния в почках. Причина
этого — повышение эффективности
реабсорбции кальция (и магния) в
дистальных канальцах нефронов;
кальцитриол действует синергично. Следует
учесть, что в проксимальных канальцах
реабсорбция кальция под действием
паратирокринина снижается, хотя этот
эффект по абсолютной величине менее
значим, чем дистальная активация
обратного всасывания.
>- Усиление экскреции фосфата с мочой;
это сопровождается также понижением
реабсорбции сульфата, бикарбоната,
натрия, хлоридов и аминокислот. В
силу подобных эффектов паратгормон
способствует развитию выделительного
ацидоза (см. выше, гл. 10). Кальцитриол
выступает частичным антагонистом и
частичным синергистом
паратирокринина, задерживая и фосфат, и кальций.
>- Увеличение всасывания кальция
(магния) в ЖКТ. Этот эффект, по-видимому,
отчасти опосредован через кальцитриол,
который действует аналогично, но
вдобавок — способствует еще и абсорбции
фосфатов.
>- Паратгормон сильный положительный
инотропный регулятор, стимулирующий
сердечные сокращения. Он также
повышает кровяное давление и, в связи с этим,
клубочковую фильтрацию.
>* Паратгормон оказывает нерезко
выраженное контринсулярное действие на
углеводный обмен.
>- Имеются сообщения о его угнетающем
действии на сперматогенез, индукции
паратгормоном гиперлипопротеинемии
и провокации им кожного зуда. Но see
эти наблюдения относятся к
нефизиологически высоким дозам гормона.
У паратгормона существует
гормональный физиологический антагонист, реци-
прокно влияющий на кальцийфосфатный
метаболизм.
Гормон С-клеток щитовидной
железы — кальцитонин (ранее называвшийся
тирокальцитонином) был открыт в 1962 г.
Д.Коппом и соавторами, которые
полагали, что он производится там же, где
и паратгормон. Этим авторам удалось
заметить, что искусственно повышенная
концентрация кальция в крови снижается
быстрее, если щитовидно-паращитовидный
комплекс интактен, чем если он удалён.
Затем П.Ф.Хирш и соавт. (1963) доказали
тироидное происхождение кальцитонина.
У рыб, амфибий, ретилий и некоторых
птиц кальцитонин производят специальные
железы — ультимобранхиальные тельца, а у
млекопитающих их клетки погружаются в
щитовидную железу, то есть с ними
происходит примерно то же, что и с хромаффин-
ной тканью мозгового вещества, которая
оказывается внутри другой эндокринной
железы (А.А. Булатов, 1976). Наконец,
благодаря иммунофлюоресцентному анализу,
А.Г.Пирс и Г. Буссолати (1967) показали,
что источником гормона в щитовидной
железе служат именно происходящие из
нервного гребешка парафолликулярные
светлые клетки (С-клетки). Краткая
характеристика веретеновидных, крупных, арги-
рофильных клеток-источников
кальцитонина, составляющих в норме всего 0,1 % массы
железы и локализованных в медиальной
части ее боковых долей, уже была приведена
выше, в разделе, отражающем
патофизиологию тироидной функции.
Кальцитонин — пептид (молекулярной
массой 3421 Д) из 32-х аминокислот, из
которых 7 остатков на амино-конце замкнуты
дисульфидной связью в кольцо (см. рис.
129 выше).
Гормон синтезируется из прокальцитони-
на (15 кД). Соответствующий ген находится
678 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
в 11-й хромосоме и известен как ген каль-
цитонина/кальцитонин-ассоциированного
пептида-1 или «ген а». Транскрипция того
же гена а, который кодирует кальцитонин,
ведет при альтернативном процессинге
к синтезу кокальцигенина — пептида,
ассоциированного с геном кальцитонина (37
аминокислот). Нормальные С-клетки
выделяют практически только кальцитонин,
но опухолевые производят оба пептида.
Физиологическая продукция
кокальцигенина, в отличие от кальцитонина, присуща
многим нейросекреторным клеткам
диффузной эндокринной системы, в связи с
чем он обнаруживается в мозге, слизистой
бронхов и в других органах. Дело в том, что
в мозге и апудомах экспрессирован другой
ген 11-й хромосомы — ген р, транскрипт
которого даёт при процессинге только м-РНК
кокальцигенина, но не кальцитонина.
Считается, что пептид,
ассоциированный с кальцитониновым геном, может
выполнять паракринные функции. У него
обнаружен бронхоспастический эффект, а
также кардиотропное и нейротропное
действие, но в фармакологических дозах. Его
гормональная роль неизвестна. В последнее
время найден еще один пептид, кодируемый
геном, соседним с геном кальцитонина и
освобождаемый вместе с кальцитонином — ка-
такальцин (21 аминокислота). Он сходен
с кальцитонином по биологическому
действию. Предполагается, что все эти
регуляторы могут находиться с полигормональным
предшественником прокальцитонином в тех
же соотношениях что гормоны кортиколи-
потррфов с проопиомеланокортином.
С-клетки, представляющие классические
элементы APUD-системы (Б.В. Алёшин,
1981), располагают кальциевым сенсором,
основная роль в стимуляции выработки
кальцитонина принадлежит повышению
концентрации ионизированного кальция.
Кальцитонин секретируется в кровь,
причём время его полу выведения 2-15 минут.
В крови, особенно при гиперкальцитонине-
мии опухолевого генеза, обнаруживаются не
только мономер, но и различные олигомеры
кальцитонина.
Гормон воздействует на кальцитониновый
рецептор. В основном такие рецепторы
находятся в костях (остеокласты), почках (кайма
клеток коркового восходящего колена петли
Генле) и ЖКТ (желудок, кишечник).
Кальцитонин действует также в мозге и в иммунной
системе, предположительно, через рецепторы
вышеописанного родственного ему пептида.
Рецепторы кальцитонина, вместе с
рецепторами паратгормона, пептида,
ассоциированного с геном кальцитонина, составляют
особое подсемейство рецепторов, связанных
с G-белками. К ним примыкают сходные
рецепторы секретина, амилина, соматолибе-
рина, ВИП и Ж И П. Внутриклеточное
опосредование эффектов кальцитонина вовлекает
циклонуклеотид-протеинкиназный
посредник, инозит-фосфатиды и кальций.
Уровень кальцитонина у женщин меньше,
чем у мужчин, и сильно снижается в пос-
тменопаузальном периоде, что, возможно,
частично объясняет патогенез
климактерического остеопороза у женщин.
Эффекты кальцитонина сводятся к тому,
что этот регулятор:
>* Подавляет резорбцию костного вещества
остеокластами, а при хроническом
введении — и остеогенез остеобластами.
>• Подавляет реабсорбцию кальция и
фосфата, а также натрия, калия и магния в
почках.
>- Снижает секрецию гастрина и соляной
кислоты в желудке, трипсина и
амилазы — в экзокринной части
поджелудочной железы, повышает секрецию натрия,
калия, хлорида и воды в кишечнике.
Интересно, что пентагастрин стимулирует
секрецию кальцитонина так же сильно,
как гиперкальциемия. Следовательно,
существует ось С-клетки-желудок, где
имеется сервомеханизм обратной связи
концентраций гастрина и кальцитонина.
Часть гастроинтестинальных эффектов,
возможно, зависит от отмечавшейся
выше общности строения рецепторов
кальцитонина и некоторых энтериновых
гормонов.
>- Обладает выраженным прямым аналь-
гетическим действием на уровне
гипоталамуса и лимбической системы через
рецепторы кокальцигенина и, возможно,
амилина.
>* Возможно, тормозит активацию
макрофагов.
Общее направление этих эффектов делает
кальцитонин главным антигиперкальциеми-
ческим и гипофосфатемическим гормоном. У
многих животных он очень активен. Кальци-
тонины лосося и угря, несмотря на отличия в
антигенной специфичности, у человека в 100
раз более мощно действуют на обмен кальция
и фосфора, чем гомологичный
собственный кальцитонин. У людей, по-видимому,
кальцитонин менее важен, как регулятор
метаболизма этих ионов. По крайней мере,
при интактной паратироидной функции, ни
гиперкальцитонинемия ни тироидэктомия
у человека не сопровождаются сколько-
нибудь выраженными проявлениями
расстройств кальциевого обмена. Однако, при
медуллярных опухолях щитовидной железы,
продуцирующих много кальцитонина и
кокальцигенина, ремоделирование костной
ткани замедлено. Очевидно, при аномалиях
паратироидной функции состояние кальци-
тониновой регуляции у пациентов
приобретает большее значение. По крайней мере, как
фармакологический препарат, кальцитонин с
успехом используют при терапии остеопоро-
за, гиперпаратироза и болезни Педжета.
нарушения регуляции
кальций-фосфорно-
магниевого обмена
Типовые нарушения кальциево-фосфорного \
обмена проявляются в синдромах гипокаль-
циемии, гиперкальциемии, гипофосфатемии
и гиперфосфатемии.
Гипокальциемия часто сочетается с ги-
перфосфатемией, гиперкальциемия — с ги-
пофосфатемией. Однако, возможны и иные
сочетания (табл. 43).
Нарушения эндокринной регуляции 679
Гипокальциемия, то есть снижение
содержания ионизированного кальция в
сыворотке крови, как синдром, проявляется прежде
всего нарушениями
электрофизиологических процессов на клеточных мембранах, так
как ионы кальция участвуют в
формировании потенциалов действия. Характерны
повышение нервно-мышечной возбудимости и
тетания. При тетании пациент испытывает
периферические и периоральные
парестезии, имеется карпо-педальный спазм.
Порой развиваются судороги, бронхоспазм
и ларингоспазм. Судороги провоцируются
алкалозом, в том числе — дыхательным при
гипервентиляции, а также гипомагниемией
и гиперкалиемией. В то же время, ацидоз и
гипокалиемия неспецифически снижают
судорожную готовность. На ЭКГ
удлиняется интервал QT, возникают признаки
нарушения реполяризации миокарда и
могут проявляться аритмии и резистентность
миокарда к действию сердечных гликозидов.
Скрытая судорожная готовность у больного
оценивается при проверке ряда характерных
симптомов (рис. 131):
Симптом Труссо — судороги в области
кисти («рука акушера», «пишущая кисть»
через 2-3 мин. после перетягивания плеча
жгутом).
Симптом Хвостека — сокращение мышц
лица при перкуссии в месте выхода
лицевого нерва, кпереди от наружного слухового
прохода.
Симптом Гофмана — сокращение круглой
мышцы век и лобной мышцы при поколачи-
вании у наружного края глазницы.
Важно помнить, что симптоматика ги-
покальциемии в силу универсального
значения кальция не ограничивается этими
нарушениями. Типичны дисфагия,
нарушения регулярности кишечных отправлений,
могут быть рвоты из-за пилороспазма, боли
в животе отражают расстройство
перистальтики кишок.
Нередко неопределённые астенические
жалобы больных, их
тревожно-беспокойное поведение, бессоница, приступы тоски
и депрессии, даже проявления гипотиро-
за — отражают клинические последствия
680 AM Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Синдромы
Аденома
Гиперплазия
Эктопическая секреция
Вторичный гиперпаратироз
Остеолиз при метастазах в
кости
Саркоидоз
Гипервитаминоз D
Молочнощелочной синдром
Наел, гиперкальциемия —
гипокальциурия
Первичный гипопаратироз
Псевдогипопаратироз
Размер паращито-
видн. желез
Увеличена одна
Все увеличены
Все нормальны
Все увеличены
Все нормальны
Все нормальны
Все нормальны
Все нормальны
Слегка увеличены
все 4
Все нормальны
Все нормальны
Са2+ сыв.
ft
ft
ftft
ftUN
ft
ft
ft
ft
ft
V
U
PO|~ сыв.
ft
U
u
ftUN
ft
ft
ft
ft
ft
ft
»
Паратгормон сыв.
ft
ft
U (ft-пептид,
ассоциированный с геном парати-
реокринина)
ft
U
u
u
ft
u
N/ft
выраженои гипокальциемии и исчезают
после коррекции кальциевого обмена
(Ю.И. Строев и соавт., 1997).
Патогенез гипокальциемии всегда так
или иначе связан с абсолютной или
относительной недостаточностью паратироидной
регуляции. Гипокальциемия бывает острой
и хронической.
Острая гипокальциемия может
возникать при переливании цитратной крови,
действии ряда кальций-связывающих
лекарств (гепарин, ЭДТА, протамин), а также
медикаментов, нарушающих всасывание
кальция и магния в кишечнике и почках
(петлевые диуретики, аминогликозидные
антибиотики, цисплатина, амфотерицин
В), средств, влияющих на освобождение
кальция в костях (бифосфонаты, пликами-
цин). Она наблюдается в полиурическую
стадию острой почечной недостаточности,
при сепсисе (в связи с блокирующим
действием цитокинов на эффекты паратгормо-
на), а также в ходе острого панкреатита. В
последнем случае ее механизм не вполне
ясен. Скорее всего, имеет значение дефицит
всасывания кальция и магния при
панкреатической стеаторее, когда эти катионы
остаются в составе фекальных мыл. Есть
данные об образовании кальцийсодержащих
мылоподобных соединений в ретроперито-
неальной жировой клетчатке из-за некроза
поджелудочной железы и освобождения её
гидролаз. Сообщалось также о снижении
секреции паратирокринина и
резистентности к нему при нехватке каких-то неиденти-
фицированных факторов панкреатического
происхождения.
Транзиторная гипокальциемия
характерна для новорожденных в связи с
недостаточным содержанием кальция в женском
молоке и большой потребностью в этом катионе.
Массированное освобождение фосфатов
при рабдомиолизе, деструкции опухолевой
ткани также может быть связано с острой
гипокальциемией. При пересадке печени как
осложнение развивается гиперцитратемия,
так как трансплантат в течение некоторого
времени не способен эффективно метабо-
лизировать лимонную кислоту. Это также
ведёт к острой гипокальциемии. Острая
гипокальциемия опасна, если носит тяжелый
характер, но во многих случаях она является
преходящей и малосимптомной.
Хроническая гипокальциемия всегда
развертывается в характерную клиническую
картину, описанную выше. Наиболее часто к
хроническому дефициту кальция приводят
Таблица 43. Основные нарушения кальций-фосфорного метаболизма
Щелочная ф-за
ft
ft
N(ft)
N/ft
ft
N/ft
N
N
N
N
N
Ca2* мочи
ft
ft
ft
N/ft
ft
ft
ft
U
a
N/0
N/U
ц-АМФ
мочи
ft
ft
ft
ft
a
0
0
ft
ft
u
о
Примечание
Остеопатия. Уролитиаз. Метастатиче ский кальциноз. Язва желудка
Тоже
Онкологическая симптоматика. Остеопатия и уролитиаз менее
выражены
Симптомы первичного заб-я, напр., рахита
В кости и в метастазах образуются цитокины-активаторы остеолиза,
в опухоли — аналоги кальцитриола
Гранулёмы производят кальцитриол
Увеличен уровень витамина D
Сопутствующий алкалоз
Аутосомно-доминантное наследование дефекта Са-сенсора
Часто — аутоантитела к паращит. ж-зе; метастатич. кальциноз
Пострецепторный дефект G-белка, наел, остеодистрофия Ол-брайта
гипопаратироз и псевдогипопаратироз,
хроническая почечная недостаточность, рахит,
гипомагниемия (Дж.Т.Роттс, 1994).
Гипокалъциемия может подразделяться на
три основных типа. Гипокальциемический
синдром возникает:
>* При отсутствии или недостатке парати-
рокринина (гипопаратироз, абсолютная
паратирокрининовая недостаточность).
>* При нормальном или повышенном
количестве паратирокринина
(неэффективное действие паратирокринина
на мишени, периферическая
паратирокрининовая резистентность).
>- При быстрой потере внеклеточного
кальция с перекрытием
функциональных резервов паращитовидных желёз и
декомпенсацией, на фоне максимального
усиления секреции паратирокринина
(относительная паратирокрининовая
недостаточность).
Гипопаратироз может развиваться как
наследственное заболевание и
приобретённый синдром.
Наследственный гипопаратироз —
редкое заболевание с постепенным началом и
сопутствующими пороками развития. Он
встречается, в частности, в структуре синд-
]ения эндокринной регуляции 681
рома Ди Джорджи (см. т. I, с. 135,318,383) и
вызван хромосомной аномалией — делецией
участка короткого плеча хромосомы 22.
Эта форма сочетается с пороками сердца и
тяжёлым иммунодефицитом из-за аплазии
вилочковой железы и проявляется рано.
Описаны также изолированные формы
наследственного гипопаратироза — аутосомно-
доминантная, с аномальной структурой
паратирокринина; аутосомно-рецессивная — с
мутацией первого интрона гена гормона,
нарушающей его сплайсинг, а также
сцепленная с Х-хромосомой — с неизвестным
дефектом. Обнаружен семейный наследственный
полигландулярный аутоиммунный синдром,
при котором имеется раннее аутоиммунное
поражение паращитовидной, щитовидной,
половых и надпочечных желёз.
Для приобретенныхформ гипопаратироза
характерны более резкое начало и
отсутствие семейных и дизэмбриогенетических
проявлений.
В прошлом типичной формой
приобретенного гипопаратироза был синдром,
развивавшийся в результат неудачной
операции резекции щитовидной железы.
Теперь ятрогенный хирургический
гипопаратироз формируется после слишком
аденома
(чаще одиночная)
гиперплазия
главных клеток
гиперплазия
оксифильных
клеток (<1%)
карцинома
2%
приём больших
количеств Са и Р
(молоко) облегчает
симптомы болезни,
нормализует уровень
щелочной фосфатазы,
замедляет резорбцию
компенсаторное |
повышение активности |
остеобластов и повышение |
активности щелочной |
фосфатазы в сыворотке ■
перелом
костная
киста
остеобласт остеокласт
субпериостальная
резорбция
Рис 130. Механизмы развития гиперпаратироза (по Ф. Неттеру из CIBA Medical Illustrations
Collection). Са — ионы кальция, Р — ионы фосфата, А — гидоксиапатит (даллит),
N — нормальные параметры, Вит. — витамин, остальные сокращения и пояснения — в тексте
Нарушения эндокринной регуляции 683
радикальных операции по поводу
гиперпаратироза. Другая ятро-
генная причина —
дистанционное и эндогенное облучение при
радиотерапии и
радиодиагностике заболеваний органов шеи.
По мнению П. Буркхардта
(1982), одной из важнейших
причин приобретенного гипопарати-
роза надо считать аутоиммунный
паратироидит. Этот процесс
происходит на фоне параллельного
вовлечения в аутоаллергические
реакции других органов, что
выражается в сопутствующих
аутоиммунных тироидите, адреналите, ин-
сулите (иногда с исходом в ИЗСДI
типа) и недостаточность функций
соответствующих эндокринных
желёз. Может быть аутоиммунный
атрофический гастрит и перни-
циозная анемия. Кандидоз часто
сопровождает данный синдром,
который связывают с нарушением
функций Т-лимфоцитов.
Редкими и исключительными
причинами приобретенного гипо-
паратироза могут быть амилоидоз,
гемохроматоз, гемосидероз
паращитовидных желёз после
множественных переливаний крови.
Инфекционные паратироидиты,
травмы и апоплексия, как правило,
не вовлекают все железы, и гипопа-
ратироз при них не развивается.
Своеобразная форма секреторной
недостаточности паращитовидных желёз, без
деструкции паратироцитов, наблюдается
при тяжелой гипомагниемии (содержании
катиона магния менее 0,4 мМ/л при норме
0,8-1,2 мМ/л). Дело в том, что магний —
кофактор аденилатциклаз. Хотя этот ион
влияет на секрецию паратгормона аналогично
кальцию, и при умеренной гипомагниемии
паратироциты слегка стимулируются (см.
выше), при крайнем дефиците магния
вступает в действие иной механизм —
нарушаются ц-АМФ-зависимые процессы
секреции паратгормона и пострецепторной
Рис. 131. Положительный симптом Труссо у пациента с
гипопаратирозом и скрытой судорожной готовностью
(поВ.Фальта, 1932)
передачи паратирокрининового сигнала
в клетках-мишенях. Известна первичная
наследственная гипомагниемия из-за
дефекта абсорбции магния в ЖКТ, а также
вторичная — чаще всего связанная с
синдромом мальабсорбции, голоданием, почечной
недостаточностью, потерями магния при
лактации, его активной утилизацией при
остеогенезе или неадекватным по магнию
парентеральным питанием.
Самая распространенная причина
гипомагниемии — хронический алкоголизм,
особенно у недоедающих и асоциальных
пациентов. Как уже говорилось, при
алкоголизме всасывание магния страдает, а
выведение — усиливается. Способствует
684 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
гипомагниемии и гипокальциемии и
алкогольный панкреатит.
Интересно, что симптомы глубокой
гипомагниемии сами по себе напоминают белую
горячку. У пациента наблюдаются
беспокойство, парестезии, замедленное
мышление, утомляемость, бессонница и
галлюцинации. Затем следует помрачение сознания,
вплоть до прекомы. Имеются мышечная
дрожь, карпо-педальный спазм, очевидно,
связанный с индуцированной вторичной
гипокальциемией. Бывают паралитическая
кишечная непроходимость, вторичная
почечная гипокалиемия и связанные с ней
тахиаритмии, повышается чувствительность
сердца к гликозидотерапии.
Особенность картины гипопаратироза
и гипокальциемии при тяжелой
гипомагниемии — отсутствие избытка фосфатов
в крови, характерного для большинства
других случаев гипопаратироза. Дело в том,
что диета, лишенная магния, как правило,
дефицитна и по фосфору. Секреция и
уровень паратгормона восстанавливаются при
гипомагниемии немедленно после
парентерального введения магния.
Гипопаратироз различного генеза при
тяжелом хроническом течении вызывает
нарушение развития зубов, ухудшение
качества их эмали и ускоренный кариес.
В костях отмечаются проявления
остеосклероза и остеомаляции, в базальных ганглиях
головного мозга, связках, сухожилиях,
сосудах отмечаются петрификаты, что
может нарушать ликвородинамику и
приводить к внутричерепной гипертензии. Рано
обызвествляются реберные хрящи. При
начале в раннем возрасте гипопаратироз
ведет к низкорослости.
Очень типичны двусторонние катаракты.
Характерны нарушения роста волос, ногтей
и раннее постарение кожи. Эти проявления
могут отражать дефицит паратирокринин-
ассоциированного полипептида.
Периферическая резистентность к па-
ратирокринину может формироваться в
результате рецепторных и пострецептор-
ных дефектов в клетках-мишенях, а также
возникать как следствие гиповитаминоза
D (включая и формы рахита, резистентные
к экзогенному витамину D — см. выше).
Особый случай неэффективного действия
паратирокринина представляет хроническая
почечная недостаточность.
При витамин-Б-зависимых и почечных
нарушениях имеется и гипокальциемия,
и гипофосфатемия. Периферический пос-
трецепторный дефект тканей-мишеней
паратирокринина известен как псевдогипо-
паратироз и протекает с гипокальциемией
и гиперфосфатемией, как вышеописанные
истинные дефициты паратгормона.
Витамин-Б-зависимые формы
резистентности к паратгормону, сопровождаемые
вторичным увеличением его секреции, но
без адекватной компенсации нарушений
минерального метаболизма, уже
рассматривались выше в разделе «Патофизиология
витаминного обмена».
Хроническая почечная недостаточность
отражается на кальций-фосфорно-магние-
вом метаболизме из-за действия нескольких
факторов.
Во-первых, при нефросклерозе
уменьшается продукция кальцитриола почками.
Во-вторых, в организме больных
задерживается фосфат. И то, и другое ведут к
пониженной эффективности действия паратгормона.
Развивается вторичный гиперпаратироз.
Итогом является ренопривная остеодист-
рофия. Фосфат связывает кальций с
образованием у больных внескелетных очагов
кальциноза. К тому же из-за уремического
гипергастринемического гастрита больные
употребляют антациды, многие из которых
содержат дополнительные фосфаты и
алюминий, нарушающий при проникновении в
кости процесс минерализации.
Алюминий из данного и из случайных
пищевых источников накапливается в костях
больных хронической почечной
недостаточностью, почки которых плохо выводят
его из организма. Он снижает активность
остеобластов, и кости таких пациентов, даже
при терапии витамином D, не в состоянии
эффективно захватывать из крови кальций.
Формируются гиперкальциемия, болезнен-
Нарушения эндокринной регуляции 685
ная остеомаляция, патологические
переломы, имеют место и другие проявления
алюминиевой интоксикации — проксимальная
миопатия, деменция, явления, сходные с
болезнью Альцхаймера. Все это усугубляет
неэффективность действия паратгормона
(см. также «Патофизиология обмена
микроэлементов» выше).
Ранее в патогенезе остеодистрофии при
нефросклерозе придавали большое
значение длительной циркуляции в крови таких
больных фрагментов паратгормона, не
выводимых почками. В настоящее время
установлено, что собственная биологическая
активность этих фрагментов минимальна,
поэтому их токсическое действие
оспаривается. Однако не исключено, что такие
неактивные фрагменты могут, на каком-то этапе
метаболизма нативного паратирокринина,
конкурентно снижать его регуляторную
эффективность.
Псевдогипопаратироз — первая постре-
цепторная болезнь, описанная в
патофизиологии (см. выше — «Общая патология
эндокринной системы»).
При данном заболевании органы-мишени
не отвечают должным образом даже на
повышенные концентрации паратгормона (и
некоторых других гормонов, влияющих на
рецепторы, ассоциированные с G-белками).
Вместе с тем внеклеточный домен рецептора
интактен. Известны 3 формы псевдогипо-
паратироза.
При классической форме 1а
(остеодистрофии Олбрайта) имеется наследственный
дефект а-субъединицы гуанидинсвязыва-
ющего Gs.a белка, сопрягающего рецептор с
аденилатциклазой. Поэтому, паратгормон
(и некоторые другие гормоны — например,
аденогипофизарные) не способны повысить
содержание ц-АМФ и включить протеинки-
назы А в клетках.
Развиваются задержка роста, брахидак-
тилия, гипергонадотропный гипогонадизм
и очаги кальцификации вне скелета.
При псевдогипопаратирозе типа ip не
отмечается дефекта а-субъединицы белка
Gs.a, но продукция ц-АМФ нарушена из-за
дефицита другого, неидентифицированного
компонента G-белкового адаптера.
Клиническая картина отличается изолированной
резистентностью лишь к паратгормону, а
фенотип больных нормален.
При псевдогипопаратирозе типа II нет ни
дефицита а-субъединицы Gs.a, ни нарушений
в продукции ц-АМФ. Выработка циклонук-
леотида может даже быть повышена. Однако
пострецепторный механизм несостоятелен
в каком-то ином из своих компонентов.
Гормонорезистентность ограничивается
паратирокринином. Для больных особенно
характерна гиперфосфатемия. Данное
расстройство связывают с наследственными
D-резистентными формами рахита.
Один из самых курьёзных терминов в
арсенале эндокринологической
патофизиологии — это псевдопсевдогипопаратироз.
Вспоминается фраза поэта И.Г. Зыкова
«Если существуют муравьеды — должен
быть и муравьедоед!»
При псевдопсевдогипопаратирозе (4
приставки на один корень!) больные имеют
фенотип, свойственный остеодистрофии
Олбрайта, но уровень кальция у них
нормальный, так же, как и синтез ц-АМФ в
ответ на паратгормон. Так как эти
пациенты — родственники больных классической
формой псевдогипопаратироза,
развивающие впоследствии лёгкую гипокальциемию,
считается, что псевдопсевдогипопаратироз
наблюдается при гетерозиготности по гену
класического псевдогипопаратироза типа 1а
и неполной пенетрантности.
Относительная недостаточность
паратирокринина сопровождает состояния
с быстрой потерей кальция организмом.
Мощность паратгормональной регуляции
оказывается недостаточной для
обеспечения нормокальциемии, несмотря на полное
использование функционального резерва
паращитовидных желёз.
В этой группе преимущественно
ситуации, приводящие к острой гипокальциемии
и гиперфосфатемии.
Прежде всего это синдром массированного
цитолиза — практически во всех его видах.
При гепатолизе, гемолизе, применении ци-
686 AM Зайчик, Л.П.Чурилов Патохимия
тостатиков, вызывающих массовую гибель
неопластических клеток, при синдроме
длительного раздавливания, тяжелом
травматическом и ожоговом шоке — возникает
освобождение фосфата, калия и в меньшей
мере — магния из клеток. Ионизированный
кальций покидает кровь. Если, как это
нередко наблюдается, цитолиз
сопровождается острой почечной недостаточностью,
то гиперфосфатемическая гипокальциемия
может быть весьма значительной.
При эффективном лечении фиброзного
остеита, а также быстром заживлении
переломов развивается «синдром голодных
костей», и массированный отток кальция
в скелет может вызвать гипокальциемию,
поэтому необходимы поддерживающие
дозы кальция.
Гиперфосфатемия как синдром
рассматривается немедленно вслед за гипокальци-
емией, так как наблюдается практически
исключительно в четырёх клинических
ситуациях — и все они связаны с
понижением сывороточного уровня ионов кальция.
Гиперфосфатемией характеризуются:
>* гипопаратироз и псевдогипоцаратироз
(см. выше);
>- цитолитический синдром
(рассмотренный только что);
>* почечная недостаточность (см. том
III данного издания);
>- акромегалия и гигантизм (см. выше
«Гормон роста и регуляция ростовых и
анаболических процессов»).
Гиперфосфатемия способствует
метастатической кальцификации вне костного
скелета. Пороговой для этого процесса
является величина произведения
концентраций (мг/л) ионизированных кальция и
фосфата в сыворотке 700 и выше. Как уже
отмечалось, гиперфосфатемия — важный
фактор, приводящий к уремической осте-
одистрофии.
В тяжёлых случаях, например, у
уремических больных гиперфосфатемия вносит
вклад в развитие ацидоза, понижает
содержание ионов кальция в сыворотке.
чдокринно-метаболические нарушения)
Гиперкальциемия — повышение
содержания кальция в сыворотке крови, как
синдром встречается значительно чаще, чем
гипокальциемия.
Проявления выраженной гиперкальци-
емии включают анорексию, тошноту, рвоту,
запор, гипотонию мышц, утомляемость,
депрессию, иногда — сонливость.
Характерны жалобы на ухудшение памяти. Острая
тяжелая гиперкальциемия может быть
причиной комы. Гиперкальциемия нарушает
функции почек, вызывая резистентность к
АДГ и полиурию. Если она сопровождается
гиперфосфатемией или хотя бы протекает
без гипофосфатемии — у больных
провоцируется метастатическая кальцификация
сосудов, почек, роговицы, слизистой желудка
и соединительной ткани, особенно — вблизи
суставов за счет отложения фосфата кальция.
Гиперкальциемия может протекать на фоне
различного содержания фосфата в сыворотке,
но, как правило, сопровождается умеренной
гипермагниемией. На ЭКГ укорачивается
интервал Q-T, могут быть аритмии.
Обнаружив у пациента гиперкальциемию,
надо придать должное значение
дифференциальному диагнозу, так как одни причины
этого состояния крайне опасны
(злокачественные опухоли), а другие не представляют
сложности в лечении (например, иммобили-
зационная гиперкальциемия устраняется по
мере начала реабилитационных процедур у
лежачих больных).
Основные этиологические факторы ги-
перкальциемии могут быть сгруппированы
в 5 категорий:
>- Паратироидные причины включая
первичный гиперпаратироз.
>- Почечная недостаточность и связанные
с ней состояния (включая тяжелый
вторичный гиперпаратироз).
>- Опухолевые непаратироидные причины
включая неоплазмы, производящие
полипептид, ассоциированный с геном
паратирокринина и опухоли с костными
метастазами.
>• Факторы, связанные с избыточой
активностью витамина D (включая экзо-
Нарушения эндокринной регуляции 687
генный гипервитаминоз и состояния с
повышенной эндогенной продукцией и
активацией этого витамина).
>• Ускоренное ремоделирование костной
ткани
Первичный гиперпаратироз — заболевание,
вызванное продукцией и действием избытка
паратирокринина паращитовидных желёз.
Данное заболевание описано Ф. фон Реклин-
гаузеном (1891) и носит его имя (первая
болезнь фон Реклингаузена или фиброзно-кис-
тозная остеодистрофия). Сам автор считал
этот недуг хроническим воспалением. Позже
отечественный эндокринолог А.В. Русаков
(1924) доказал связь болезни с
гиперфункцией паращитовидных желез и описал ключевое
звено её патогенеза — образование в костях
гигантоклеточных разрастаний — эпулидов
или остеобластокластом, локализованных
в лакунах Хоушипа. Клетки эпулидов ведут
активную резорбцию кости и выработку
остеоида. Смена генераций костной ткани
резко ускоряется. При этом скорость
восстановления минерализованной кости отстаёт
от остеолиза и образования фиброзной
ткани. В кости формируются кисты,
заполненные мукоидом и коричневатой жидкостью.
Множественные кисты дают характерную
картину «мыльной пены» на
рентгенограмме. Недостаточно минерализованные кости
деформируются, могут быть патологические
переломы. Поднадкостничная локализация
кистозно-фиброзных изменений делает
кости пациентов крайне болезненными.
Отмечается истончение и исчезновение
кортикального слоя зубных альвеол, кости
черепа приобретают вид «солонки» из-за
множественных мелких очаговых дефектов
минерализации — пятнистого склероза,
рентгенологами описаны «таз типа карточного
сердца», бедренная кость «типа пастушьего
посоха», колоколообразная грудная клетка
и «рыбьи позвонки». Отмечаются кальци-
фикация хрящей, проявления подагры и
псевдоподагры (см. выше «Патофизиология
нуклеинового метаболизма»).
Болезнь не является редкой и поражает
не менее 0,1 % населения,
преимущественно — женщин среднего возраста. Если
учесть стертые субклинические формы,
частота болезни может доходить, по оценке
Дж.Т.Поттса (1994), до 1% популяции!
Таким образом, вместе с сахарным диабетом,
тироидитами и ожирением, данная форма
патологии может расцениваться как
принадлежащая к числу самых важных
эндокринных болезней, составляющих
буквально каждодневную заботу эндокринолога.
Описана редкая врожденная форма,
принадлежащая к структуре синдромов
множественных эндокринных неоплазий
типа I и На (см. выше о MEN-синдромах в
разделе «Общая патофизиология
эндокринной системы»).
При первичном гиперпаратирозе (см.
рис. 130) характерны гиперкальциемия, ги-
пофосфатемия, гиперкальциурия, высокая
активность остеокластической щелочной
фосфатазы в крови. Так как кальций влияет
на пострецепторную передачу вазопрес-
синового сигнала, при повышении его
уровня в первичной моче почки делаются
относительно резистентными к АДГ и
формируется полиурия, полидипсия и синдром
несахарного диабета. Для классического
течения болезни характерны метастатический
нефрокальциноз, уролитиаз, калькулезный
холецистопанкреатит, кальцификация
сосудов, слизистой желудка, роговицы. Имеется
гипертензия. Часто бывает язвенная болезнь
двенадцатиперстной кишки, желудка и
пищевода. В патогенезе последних
проявлений играет роль не только почечная
недостаточность и последствия кальциноза
сосудов и органов ЖКТ. Паратгормон сам
индуцирует гипертензию, а, возможно, и
продукцию избытка гастрина. По крайней
мере выше отмечалось, что его антагонист
кальцитонин является прямым
ингибитором продукции гастрина, пепсина и соляной
кислоты в желудке. С самого начала
отмечаются гипотонус и слабость мышц, а также
классические проявления гиперкальциемии
и гипермагниемии со стороны ЦНС. Бывают
гипертензия, зуд. Патогенез первой связан с
кардиотропным действием
паратирокринина, а последнего — с избытком паратирокри-
нин-подобного пептида.
688 А.Ш. Зайчик, Л.П.Чурилов Патохимия
В практике английских коллег
существует зарифмованная поговорка, описывающая
разнообразную клинику первичного гипер-
паратироза следующим образом «painful
bones, renal stones, abdominal groans and
psychic moans».
Паращитовидные железы (или хотя бы
одна из них) при первичном гиперпаратиро-
зе всегда первично гиперфункциональны и
гиперпластичны, и могут поражаться (в
порядке убывания частоты встречаемости):
>- аденомой из главных клеток (85%),
как правило, в одной из желез. Следует
учесть, что в 10% случаев аденомы
находятся в эктопически расположеных
дополнительных паращитовидных
железах, как правило, — в средостении.
Описаны случаи удаления всех четырёх
паратироидных желез без ликвидации
гиперпаратироза. Когда впоследствии
был обнаружен эктопический очаг
продукции паратгормона в средостении, его
удаление сделало пациентку кальций-
зависимой;
>• гиперплазией (10 %), как правило, — всех
паращитовидных желёз, хотя иногда
степень гиперплазии бывает в них крайне
неравномерной;
>* карциномой из главных клеток (2-5%,
одиночна, неагрессива но обладает инва-
зивным ростом, метастазирует в печень,
кости и лёгкие);
>• множественными аденомами (до 2%),
локализуются в нескольких железах.
Во всех этих ситуациях гиперпродукция
паратирокринина носит автономный от
кальциево-фосфорного контроля характер.
Гиперпластические процессы в железе
поликлональны, но аденомы и карциномы
моноклональны. При аденоме происходит
мутация, из-за которой ген-промотор
синтеза паратгормона в 11-й хромосоме
оказывается перед геном, кодирующим D-циклин,
белок-стимулятор деления клеток. Это и
ведет к ускоренной репликации главных
паратироцитов. В карциномах часто
отмечают утрату антионкогенов, в частности,
гена Rb.
чдокринно-метаболические нарушения)
Экстремальным проявлением болезни
служит гиперпаратироидный гиперкальци-
емический криз. Это тяжёлая дегидратация
вследствие высокой гиперкальциемии
(более 3,5 мМ/л или 14 мг/дл), гиперкаль-
циурии и резистентности почек к АДГ.
Провоцируются кризы переломами, приемом
антацидов и нарушением диеты. Остро
развиваются полиурия, рвота, гипертермия,
боли в костях, мышцах и животе,
имитирующие «острый живот». Обостряются
панкреатит, язвенная болезнь, и может наступить
острая почечная недостаточность. Уровень
фосфатов в крови повышается. Гипертен-
зия и аритмия способствуют сердечной
недостаточности и отёку лёгких. Состояние
больного быстро становится коматозным.
Кома имеет черты смешанной, уремически-
гиперкальциемической.Летальность
превышает 50 % (Дж. Хиггинс, 1985).
Помимо первичного, выделяют
вторичный и третичный гиперпаратирозы,
обсуждаемые ниже.
Вторичным называется гиперпаратироз
вследствие хронического усиления функции
и пролиферации паратироцитов в ответ на
первичное непаратироидное заболевание,
приводящее к гипокальциемии и/или ги-
перфосфатемии.
Наиболее часто вторичная гиперплазия и
гиперфункция паращитовидных желез
следует при рахите и других хронических
формах недостаточности витамина D (см. выше
«Патофизиология витаминного обмена»),
а также при хронической почечной
недостаточности. Вторичному гиперпаратирозу
способствует длительная нехватка кальция
в диете. При вторичном гиперпаратирозе
продукция паратгормона подконтрольна
уровням кальция и фосфора. Большая часть
случаев вторичного гиперпаратироза,
связанных с почечной недостаточностью,
протекает на фоне гиперфосфатемии, поэтому
для них весьма типичны очаги кальциноза
в мягких тканях, даже в коже (что известно
как кальцифилаксия).
Своеобразная форма вторичного
гиперпаратироза известна в космической медицине.
Нарушения эндокринной регуляции 689
Длительное воздействие невесомости на
организм, а возможно и быстрое
перемещение по орбите в электромагнитном поле
Земли, вызывают активизацию паратиро-
цитов, деминерализацию костного скелета,
гиперкальциемию и остеомаляцию. Этому
у космонавтов способствует относительная
гиподинамия. Физические упражнения в
космическом полёте призваны отчасти
затормозить эти процессы. Тем не менее агра-
витационная дисфункция паращитовидных
желез служит одним из факторов, пока что
ограничивающих длительность пребывания
человека в космосе.
Третичным гиперпаратирозом условно
называют состояние, связанное с развитием
автономно секретирующей паратгормон
аденомы паращитовидной железы в
условиях длительного предварительного
гиперфункционирования последней (первичное
заболевание вне паратироидной ткани
ведет к гиперфункции, та — к гиперплазии,
а последняя переходит в доброкачественную
опухоль).
Третичная форма синдрома, как и
первичная, связана с автономией источника
паратгормона от сервоконтроля.
Кроме гиперпаратироза, возможны и
иные причины гиперкальциемии, связанные
с дисфункцией паращитовидных желез, но
без их гиперплазии.
Лечение солями лития в психиатрии (при
маниакально-депрессивном психозе) ведет
иногда к понижению чувствительности па-
ратироцитов к кальциевому ингибированию
и гиперфункции этих клеток. Гиперплазия
и аденомы при этом, как правило,
отсутствуют.
Наследственный аутосомно-доминан-
тный дефект кальциевого сенсора у гете-
розигот приводит к семейной гипокальци-
урической гиперкальциемии. В отличие от
первичного гиперпаратироза, реабсорбция
кальция в почках не понижается, так как
ее торможение паратгормоном требует
наличия интактного почечного кальциевого
сенсора. Гиперкальциемия протекает на
фоне нормофосфатемии. Паращитовидные
железы нормальны или слегка гиперплази-
рованы.
У гомозигот тот же дефект
оборачивается тяжелым наследственным врождённым
первичным гиперпаратирозом, опасным для
жизни младенцев.
Опухолевые непаратироидные процессы
также могут вызывать гиперкальциемию.
Остеодистрофия у больных, страдающих
опухолями и гемобластозами, принадлежит
к числу наиболее значимых паранеопла-
стических явлений (Дж. Позднер, 1991).
Чаще всего деструкция костного вещества
сопровождает болезнь Рустицкого-Калера
(см. выше «Диспротеинозы»), лимфомы,
чешуеклеточный и эпидермоидный рак
лёгкого, опухоли молочной железы, почек.
Реже гиперкальциемией характеризуются
овсяноклеточный бронхогенный рак лёгкого
и другие опухоли с костными метастазами.
Данная форма гиперкальциемии — не есть
прямое следствие механического и
ферментативного «ломания» костной ткани
метастазами или опухолью как таковой.
Опухоли, не метастазирующие в кости,
вызывают этот синдром чаще, чем те, которые
имеют костные метастазы.
Несколько механизмов обеспечивают па-
ранеопластическую гиперкальциемию.
К ним относятся:
- продукция неопластическими клонами
пептида, ассоциированного с геном
паратирокринина (лимфомы,
чешуеклеточный и эпидермоидный рак лёгкого,
опухоли почек и др.);
- выработка опухолевым клоном кальцит-
риола (лимфомы);
- продукция опухолью, проросшей в
кость или её костными метастазами, а
также соседствующими с ними клетками
костного мозга — цитокинов-стимуля-
торов остеолиза и блокаторов действия
паратгормона (кахексии, лимфотоксин,
ИЛ-1, другие цитомедины, известные
под собирательным наименованием
«фактор активации остеокластов», а
690 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
также простагландины Е-ряда.
(метастазы овсяноклеточного рака и раковых
опухолей груди);
- хотя саркоидоз Бека не является
опухолевым процессом, макрофагальные
клетки его гранулём синтезируют
витамин D (см. выше «Патофизиология
витаминного обмена»), поэтому гипер-
кальциемия при саркоидозе примыкает
по механизмам к паранеопластической.
Последнее замечание позволяет
перейти к краткой характеристике следующей
этиологической группы: гиперкальциемии,
обусловленной нарушениями метаболизма
витамина D.
Сюда относятся как случаи, связанные
с D-гипервитаминозом, так и вызванные
ненормальным обменом обычных количеств
витамина D.
Гипервитаминоз D уже был
охарактеризован выше (см. «Патофизиология
витаминного обмена»), как и эндогенная
гиперкальцитриоловая гиперкальциемия
при саркоидозе.
В данном разделе добавим, что описан так
называемый синдром Уилъямса — врождённые
надклапанный стеноз аорты и «лицо эльфа»,
задержка психомоторного развития,
гиперкальциемия и наследственная
гиперчувствительность тканей к витамину D, спонтанно
уменьшающаяся после первого года жизни.
Усиленная резорбция кости и остеогенез
наступают и во многих других ситуациях,
когда при нормальном состоянии пара-
щитовидной регуляции и обеспеченности
витамином D в отсутствие неопластических
процессов баланс между выведением и
поступлением кальция в крови устанавливается
на несколько повышенном уровне.
Так, гипертироз, особенно
аутоиммунного происхождения, зачастую связан с
гиперкальциемией и ускорением остеолиза-
остеогенеза, с некоторым преобладанием
первого процесса. Дебатируется вопрос о
возможности аутоиммунной стимуляции
функций паращитовидной железы, по
аналогии со щитовидной, при болезни фон
Базедова.
Длительная иммобилизация и
применение диуретиков — тиазидов могут вызвать
гиперкальциемию. Поскольку витамин А
активирует остеокластическую функцию,
гиперкальциемия отмечается и при гипер-
витаминозе А (см. выше «Патофизиология
витаминного обмена»).
Наконец, гиперкальциемия может быть
экзогенной. Экзогенная форма
гиперкальциемии на фоне экзогенного алкалоза известна
как молочно-щелочной синдром (в тяжелой
острой форме — синдром Барнетта).
Синдром развивается при приеме в пищу
или с антацидной целью больших количеств
молока и при использовании язвенными
больными абсорбируемых антацидов
(например, карбоната натрия).
Считается, что нарушение характерно
только для индивидов, неспособных
подавлять кишечное всасывание кальция при его
избытке в содержимом кишечника.
Развивается гиперкальциемия, ведущая
к полиурии, потере натрия с мочой,
подавлению продукции паратирокринина и, как
следствие, задержке бикарбонат-аниона.
Формируется алкалоз, дополнительно
стимулирующий задержку кальция в
почках. Гиперкальциемия, в результате
осуществления этого порочного круга,
отягощается.
Начальные симптомы включают слабость,
раздражительность, мышечные боли и
апатию. Более тяжёлое течение приводит к
острой почечной недостаточности, гиперфос-
фатемии и эктопической кальцификации,
так как кальций-фосфорное произведение
концентраций в сыворотке превышает
критическую цифру 700 мг/л.
Состояния, связанные с
гиперкальциемией, нередко сопровождаются гипофосфате-
мией и изредка — гипермагниемией.
Гипермагниемия встречается
относительно нечасто, так как нормальные почки могут
варьировать экскрецию магния в 250 раз. Но,
при почечной недостаточности, проявления
гиперкальциемии и гипермагниемии
накладываются друг на друга.
Особенно этому способствует
использование почечными больными с уремической
гастропатией магний-содержащих анта-
цидов. Симптоматическая острая гипер-
магниемия возникает при использовании
сульфата магния как гипотензивного и
спазмолитического средства.
Магний в высоких концентрациях
оказывает седативный эффект на ЦНС, курарепо-
добное действие на нейро-мышечные
синапсы, вызывает брадиаритмии и гипотензию.
Гиперкалиемия и ацидоз усиливают, а
гиперкальциемия — ослабляет эффекты
избытка магния. Кальций — антагонист магния
при терапии тяжелой гипермагниемии.
Гипофосфатемия — может быть
результатом временного ухода фосфатов в клетки
и абсолютного дефицита фосфора в
организме.
Углеводистая диета и дыхательный
алкалоз способствуют переходу фосфатов внутрь
клеток. В обоих случаях, уровень фосфат-
анионов в крови снижается умеренно
(оставаясь выше 1 мг/дл).
Абсолютный, но неглубокий дефицит
фосфатов развивается при гиперпарати-
розе, различных формах рахита (см. выше
«Патофизиология витаминного обмена»),
гипомагниемии, дилюции внеклеточной
жидкости, гемодиализе.
Выраженная гипофосфатемия возможна
лишь при действии факторов,
существенно уменьшающих абсолютное количество
фосфатов. Это такие состояния как:
наследственная, сцепленная с Х-хромосомой
гипофосфатемия, хронический алкоголизм
и острый синдром похмелья,
диабетический кетоацидоз в фазу выздоровления
(см. выше «Патофизиология сахарного
диабета»), связывание фосфатов в
кишечнике, откармливание после голодания (см.
выше «Голодание»), Изредка фосфаты
захватываются агрессивными, активно
пролиферирующими неопластическими
Нарушения эндокринной регуляции 691
клонами (Дж.П. Кнохел, 1981).
Хроническая выраженная гипофосфатемия вызывает
расстройства энергетического обмена и
функций ряда клеток и органов.
Больной испытывает парестезии и
онемение пальцев, мышечные и сердечные боли,
ухудшается память, бывают судороги, может
помрачаться сознание.
Наблюдаются гипофункция и укорочение
жизни клеток крови, в которых возникает
нарушение ресинтеза АТФ и энергетического
метаболизма. Это ведет к гемолитической
анемии, иммунодефициту, а в условиях
эксперимента — и к нетяжелой тромбоцитопатии.
Характерны печеночная
недостаточность, метаболическая энцефалопатия с
разнообразными поведенческими и нервно-
психическими проявлениями. Имеется
мышечная слабость и в тяжёлых случаях,
даже рабдомиолиз.
В данном разделе мы рассмотрели только
некоторые нарушения кальций-фосфор-
магниевого метаболизма и его гормональной
регуляции. Так или иначе все они касались
костей и почек. Многие другие минеральные
факторы (алюминий, о котором шла речь
выше, фтор, роль которого
охарактеризована в разделе «Патофизиология
микроэлементов») также взаимодействуют с этой
сложной системой и влияют на гомеостаз
органов и тканей опорно-двигательного
аппарата и зубов.
Остеомаляция, то есть недостаточная
минерализация костного вещества, как и ос-
теопороз — то есть снижение массы костного
вещества, могут иметь много иных причин,
помимо освещаемых в разделе
«Патофизиология витаминного обмена» и в данном
разделе ниже.
Более подробная характеристика этих
процессов относится к III тому
данного издания — «Механизмы болезней и
синдромов».
692 АЖ Зайчик, Л.П.Чурилов Патохимия
ОСТЕОПОРОЗ
И МЕТАБОЛИЧЕСКИЕ
ЗАБОЛЕВАНИЯ КОСТЕЙ
(Написано при участии Ю.И. Строева)
Основными патологическими процессами,
затрагивающими белковый и минеральный
обмен в костной ткани, являются
остеопороз, остеосклероз, остеолиз, остеомаляция и
другие. На страницах данной книги при
рассмотрении ряда эндокринных заболеваний
(психогенной анорексии, гипопитуитаризма,
соматотропиномы, гиперпролактинеми -
ческого синдрома, глюкагономы, гипогона-
дизма, гипертироза, приобретенных форм
гипотироза и особенно — гиперкортицизма
и (гиперпаратироза) и при обсуждении
метаболической патологии (голодания, амино-
ацидопатий, рахита, болезни Менкеса) уже
шла речь о таком важном проявлении всех
вышеназванных болезней, как остеопороз.
Под остеопорозом, согласно
определению Л.Я. Рожинской, базирующемуся на
заключении группы экспертов ВОЗ (2000),
понимают «прогрессирующее снижение
костной массы в единице объема кости по
отношению к нормальному показателю у
лиц соответствующего пола и возраста, с
обязательным нарушением
микроархитектоники костной ткани, приводящим к
повышенной хрупкости костей и увеличению
риска их переломов от минимальной травмы
и даже без таковой». Как видим, решающую
роль придают снижению плотности костной
ткани и качественному критерию — такому
изменению ее микроархитектуры, которое
снижает механическую прочность костей
(Б.Л. Смолянский, В.Г. Лифляндский,
2006).
По Т. Хану (1999), в практическом
здравоохранении данный термин
применяется для обозначения любых поражений
костей, повышающих их хрупкость и риск
переломов.
Остеопороз — часть более общего
собирательного понятия «остеопения». Данный
термин употребляют и для обозначения
чдокринно-метаболические нарушения)
своего рода «предостеопороза», то есть
любых случаев, когда костная масса снижена,
а патологические переломы не возникают.
Предполагается, что при этом снижение
плотности костного вещества недостаточно
выражено или качественные изменения
микроструктуры костей не столь явные,
как при остеопорозе. При собственно ос-
теопорозе образование остеоида страдает
значительно больше, чем его
минерализация. Это отличает его остеомаляции, при
которой кости вообще размягчаются, то
есть имеется относительный дефицит
минерализации по отношению к количеству
образуемого остеоида (см. выше
характеристику основных форм гипофосфатемической
и гипокальциемической остеомаляции).
Классической остеомаляцией, как указано
выше в гл. 11, характеризуется дефицит
витамина D. Остеомаляция входит в круг
остеопенических расстройств, хотя и
отличается от остеопороза. При остеоатрофии
уменьшается общая масса костного
вещества, и происходит истончение костей, но без
нарушения костной плотности. Типичная
остеоатрофия свойственна здоровым людям
в глубокой старости, таким образом
физиологическую возрастную остеоатрофию
следует дифференцировать от остеопороза,
в том числе, старческого (Г. Тенчов, Г. Хад-
жибеков, 1962).
Остеопороз не исключает остеомаляции и
остеоатрофии и при определенных условиях
может с ними сочетаться. Так, общей
закономерностью является развитие вторичного
остеопороза при наличии заболеваний,
приводящих к остеомаляции. Дело в том,
что гипокальциемия, свойственная
тяжелой остеомаляции (см. выше), как правило,
провоцирует вторичный гиперпаратироз,
а стало быть, способствует остеопорозу.
Глубокая старость непременно ведет к
снижению продукции половых стероидов, что
дополняет картину остеоатрофии сениль-
ным остеопорозом.
Эпидемиология остеопороза
характеризуется следующим. В прошлом остеопороз
распознавался лишь в далеко зашедшей
стадии по своим осложнениям — патологи-
Нарушения эндокринной регуляции 693
ческим переломам. Развитие рентгенологии
и особенно — костной денситометрии,
основанной на лучевом, а в последующем —
ультразвуковом контроле плотности костной
ткани, позволило распознавать остеопороз
(и остеопению вообще) на более ранних
стадиях. Разработка биохимических и им-
муноконкурентных методов определения
маркеров повышенной резорбции кости и
костеобразования сделала раннюю
диагностику остеопороза доступной. Это и привело
к осознанию факта широчайшей
распространенности остеопороза и к появлению
представления о латентном асимптоматическом
остеопорозе или остеопении. Остеопороз
стал в наши дни самым частым
метаболическим заболеванием опорно-двигательного
аппарата в силу повсеместной гиподинамии,
патогенного действия хронического стресса,
а также в связи с широчайшим
употреблением лекарств, нарушающих обмен костной
ткани, например, нестероидных
анальгетиков и алюмосодержащих антацидов.
По частоте и медико-социальной
значимости среди болезней обмена остеопороз
следует сразу за нарушениями липидного
метаболизма, сахарным диабетом и железо-
дефицитными состояниями. В старших
возрастных группах он наблюдается примерно
у 50 % женщин, а остальные 50% имеют
высокий риск его развития. В США ежегодно
происходит 1,3 млн переломов, в том числе
500000 переломов тел позвонков и 247 000
переломов шейки бедра — в основном у лиц
с остеопенией и остеопорозом (Л. Мелтон,
1997). Смертность от причин, связанных с
патологическими переломами вследствие
остеопороза, гораздо выше, чем от
остальных переломов.
У 60 % женщин с впервые
выявленным гипотирозом имеется остеопения, в
том числе у 5% — остеопороз. По данным
М.Р.Некрасовой с соавт. (2003), в России у
72,7% женщин с эндогенным гипертирозом
выявляется остеопения, а у 19,2% —
остеопороз с преимущественным поражением
кортикальной костной ткани. Остеопороз
нередко является компонентом
наследственных синдромов. Так, он возникает при
синдроме Шерешевского-Тернера более
чем у половины подростков старше 18 лет
(Д.Гертнер, 1999). По данным группы
экспертов ВОЗ, среди населения мира после
50 лет остеопороз определяется у 13-18%
женщин и у 7-8% мужчин. Остеопения
встречается еще чаще: так, среди
обследованных москвичей старше 50 лет пораженность
остеопенией — 50%, а остеопорозом — 28%
(Б.Л. Смолянский, В.Г.Лифляндский,
2006).
Итак, изменения в методах диагностики
позволили оценить истинную
распространенность остеопороза и всю
медико-социальную значимость этого нарушения. На
протяжении многих лет остеопороз неверно
рассматривался как заболевание пожилых
людей или даже элемент физиологического
старения (!), Но, на деле, в развитии
остеопороза существенную роль играют
наследственные факторы и метаболические
нарушения, проявляющиеся с детства, поэтому
в настоящее время его рассматривают как
педиатрическую и подростковую, а не чисто
гериатрическую проблему (Ю.И. Строев,
Л.П. Чурилов, 2004).
Этиология и патогенез остеопороза в
последнее время подверлись интенсивному
изучению. Фактически имеется полигенная
наследственная предрасположенность к
ускоренному развитию остеопороза в виде
непрерывного спектра — от самого
незначительного (когда лишь пролонгированное
действие ряда эпигенетических факторов и
случайные сильные патогенные воздействия
могут со временем сформировать
остеопороз, который будет расценен в клинике
как «сенильный»), до самого значительного
(при сопровождаемом классическим ранним
первичным остеопорозом несовершенном
остеогенезе, когда имеется дефект сс-цепей
коллагена I типа, и генетический фактор
резко преобладает над эпигенетическими
влияниями). Между этими полюсами лежит
все разнообразие условно «первичных» и
«вторичных» случаев болезни. Совершенно
так же генетический дефект
at-антитрипсина позволяет пульмонологу говорить о гену-
инной эмфиземе, а на другом полюсе лежит
694 А.Ш. Зайчик, Л.П.Чурилов Патохимия
эмфизема сенильная, но обе — лишь две
ипостаси нарушения механо-химического
баланса между протеолитическими и анти-
протеолитическими влияниями на бронхо-
легочный аппарат (см. т. I, с. 125-126).
Остеопороз — полиэтиологическое
заболевание. Он представляет собой проблему
метаболического/баланса костной ткани (ос-
теостата) подобно тому, как ожирение —
проблема баланса липогенеза и липолиза в
ткани жировой ( массостата — см. гл. 7).
Остеопороз всегда развивается в
результате преобладания темпов резорбции костей
{остеолиза) над темпами их формирования
(остеогеиезом).
Первичный остеопороз (наиболее
распространенный, в особенности — его постме-
нопаузальный и старческий варианты — до
85 % всех случаев) имеет явную
конституционально-генетическую основу, являясь
полигенным заболеванием с пороговым
эффектом проявления, зависящим от
действия эпигенетических факторов — образа
жизни, питания, кальциевого баланса,
экологических факторов, интеркуррентных
болезней. Мультифакториальное действие
генов включает особенности генов
рецепторов половых стероидов, витамина D, генов
коллагена и коллаген-ассоциированных
белков, лептина и его рецепторов, минорных
витамин-К-зависимых костных протеинов,
гемоювелина и т.п. Связь между балансом
массы тела и костной массы — не чисто
внешняя, а глубинная: дело в том, что адипокин
лептин принимает участие в гипоталамичес-
кой регуляции состояния как массостата,
так иостеостата.
На мышах с нарушениями лептиновой
регуляции массы тела показано, что лептин
снижает скорость накопления костной
массы, а при его дефиците или
неэффективности его действия скорость остеогенеза выше
скорости остеолиза (Т.-А. Кок, Ж. Оверс,
2003). Так, у мышей линии Цукера (ob),
лишенных гена лептина (см. гл. 7), костная
масса повышена несмотря на имеющийся
гиперкортицизм и гипогонадизм, которые,
казалось бы, должны были вызвать у них
вторичный остеопороз. Она нормализуется
(эндокринно-метаболические нарушения)
при интрацеребральном введении таким
мышам лептина (К.М. Степпен и соавт.,
2000) У людей с дефектами продукции
лептина его введение стимулирует рост костей
в длину ( Дж. Матарезе и соавт., 2003).
У тучных подростков избыток эстрогенов
и неэффективность действия лептина на
его рецептор коррелируют с большей
костной массой (К.О. Клейн и соавт., 1998).
В аркуатном ядре гипоталамуса найдены
лептин-чувствительные симпатические
нейроны, контролирующие костную массу
по балансовому принципу — через
соотношение остеогенеза и остеолиза (Ф. Элефте-
риу с соавт., 2003). Другие цитокины тоже
участвуют в патогенезе остеопороза. ИЛ-1 и
кахексии способствуют выработке в костях
местными остеобластами ИЛ-6, а последний
привлекает и активирует остеокласты и
благоприятствует остеолизу. Этот эффект
тормозится эстрогенами и адипонектином,
что, вероятно, связано с патогенезом ряда
форм остеопороза, в частности,
постклимактерической и сенильной. Возможно,
адекватная секреция адипокинов и
трансформация стероидов в жировой ткани
важна и для предохранения от остеопороза
костной ткани. Костная ткань —
классическая инсулинозависимая ткань, объект энер-
го-субстратной депривации под влиянием
глюкокортикоидов. Не случайно описан
глюкокортикостероидный остеопороз у
лиц с гиперкортицизмом (см. выше гл. 13).
С уверенностью можно утверждать, что
хронический стресс — фактор риска
остеопороза, который, по Г.Селье, правильнее будет
причислять к болезням нарушенной
адаптации (см. т. I, с. 566-567).Пиковая костная
масса достигается в онтогенезе к 25 годам,
после чего степень и скорость проявления
остеопороза и родственных ему нарушений
зависят от темпов утраты костной массы и
от соотношения остеогенеза и остеолиза,
которое с возрастом смещается в сторону
последнего. Если пиковая костная масса к
25-30 годам невелика, то вероятность
последующего остеопороза резко повышена. На
величину пиковой костной массы влияют
генетический статус, уровень физической
Нарушения эндокринной регуляции 695
активности и характер питания, в первую
очередь, адекватность потребления кальция,
витамина D и белка (Л.А. Марченкова,
2003). Главными источниками биологичес-
—1си доступного кальция являются молочные
продукты и соки цитрусовых с добавкой
кальция (А.В. Мазурин, И.М. Воронцов,
1975). Потребление большого количества
белка, серосодержащих аминокислот и
органических кислот может способствовать
деминерализации кости, как и любые
формы пролонгированного ацидоза (С. Крэйн,
М. Холик, 2005), так как при этом костная
ткань отдает кальций, участвуя в
реактивации буферов (см. гл. 10). Факторами риска
остеопороза, по М.Т. МакДермотту (2001),
Л.Я. Рожинской (1999), являются:
семейный анамнез (переломы у матери),
некоторые фенотипические признаки (блондинки
с хрупким телосложением и голубыми
глазами, а также монголоиды, которые
поражаются тяжелее негроидов — в силу
большей пиковой костной массы у последних).
К факторам риска у женщин принадлежат
также позднее наступление менархе (после
15 лет), особенности акушерского анамнеза
(более 3 родов или отсутствие родов),
ранняя овариэктомия, ановуляция, олиго- и
аменорея. У лиц обоего пола условиями,
способствующими остеопорозу, являются
курение сигарет, избыточное употребление
алкоголя или кофеина, малоподвижный
образ жизни, низкое потребление кальция,
непереносимость молочных продуктов,
избыточное потребление мяса и рыбы,
длительный прием ряда лекарственных
препаратов (кортикостероиды, левотирок-
син, противосудорожные средства, гепарин,
антагонисты витамина К). Так, 5 дней
приема по 40 мг преднизолона приводят
к увеличению экскреции кальция с мочой
в 2 раза! Предрасполагают к остеопорозу
хронические неврологические, гепатоби-
лиарные болезни и мальабсорбция. Имеют
значение ряд экологических факторов.
Известно, что невская (вернее, ладожская) вода
считается чрезвычайно «мягкой».
Минерализация воды в водопроводной сети Санкт-
Петербурга редко достигает 0,8-1мг/л.
В.Е. Каземирский с соавт. (1988)
высказывают предположение о роли слишком
«мягкой» питьевой воды в возникновении
остеопороза у жителей Санкт-Петербурга.
Если генетические, нутрициологические
и иные факторы, действовавшие в раннем
онтогенезе, обусловили малую пиковую
костную массу, достигнутую к моменту
биологического взросления, а процессы,
ведущие к ее утрате, напротив, приобрели
высокую интенсивность рано, то
клинически значимый остеопороз формируется
также рано. Возможно, развитие первичного
остеопороза даже в подростковом периоде
(первичный ювенильный остеопороз). По
А.П. Крысь-Пугач и Т.А. Кинчая-Полищук
(2000), типичный подростковый
первичный остеопороз формируется после 14 лет.
Этиология его остается не ясной. Все его
проявления зачастую могут исчезать вскоре
после начала полового созревания. В более
ранних возрастных группах остеопороз
(порой даже врожденный) носит вторичный
характер по отношению к ранним первичным
заболеваниям, вызвавшим преобладание
резорбции костей над их формированием
(см. ниже классификацию). Ранние формы
первичного остеопороза могут составлять до
5-10% всех его случаев. Но в большинстве
случаев первичного остеопороза (около
90%) его развитие происходит
относительно медленно и достигает клинически
явных стадий лишь в старческом периоде
{первичный сенильный остеопороз), либо в
постменопаузу, когда ослабевает остеоген-
ное действие половых стероидов (первичный
постменопаузальный остеопороз).
Вторичный остеопороз — также
полиэтиологический синдром, при котором
аналогичные нарушения баланса кос-
теобразования и рассасывания костной
ткани развиваются вследствие первичных
эндокринных, генетических,
метаболических, иммунопатологических и иных
заболеваний, лекарственных интоксикаций и
ятрогенных причин и т.п. Остеопения и
остеопороз могут быть следствием
хронических заболеваний, тормозящих остеогенез
и стимулирующих остеолиз (эндокринных,
696 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
ревматических, гематологических,
гастроэнтерологических, пульмонологических,
нефрологических), результатом
алкоголизма или лекарственной терапии, прежде
всего, глюкокортикоидной. Явный гипого-
надизм всегда грозит развитием остеопоро-
за. Гормональная контрацепция вызывает
тенденцию к снижению костной массы в
результате возникающей гипоэстрогении,
особенно у курящих женщин в возрасте
старше 20 лет (Т. Канди с соавт., 1993).
Среди вторичных остеопорозов наиболее
часто встречается стероидный остеопороз
(например, у больных гормонозависимой
бронхиальной астмой). Заболевания,
вызывающие вторичный остеопороз,
способствуют и проявлению остеопороза первичного.
При наличии ранней стадии первичного
остеопороза и провоцирующих болезней
вторичный остеопороз развивается быстрее
и течет тяжелее.Остеопороз по патогенезу
разделяют на формы с высоким костным
обменом и низким костным обменом, что
верифицируется по маркерам костеобразо-
вания и резорбции кости.
Приводим (табл. 44) классификацию
остеопороза, принятую Российской
ассоциацией остеопороза в 1997 г., с некоторыми
нашими дополнениями и изменениями.
Заболевания и состояниия, перечисленные
в классификации, более или менее подробно
охарактеризованы в различных разделах
тт. I-III данного учебника и Практикуме.
Таблица 44
Классификация остеопороза.
А. Первичный остеопороз
1. Постменопаузальный остеопороз (I тип)
2. Сенильный остеопороз (II тип)
3. Ювенильный остеопороз
4. Идиопатический остеопороз
Б. Вторичный остеопороз
I. Заболевания эндокринной системы
1. Эндогенный гиперкортицизм (болезнь и
синдром Иценко-Кушинга)
2.Тиротоксикоз
3. Гипогонадизм
4. Гиперпаратироз
5. Сахарный диабет (инсулинозависимый)
6. Гипопитуитаризм, полигландулярная
эндокринная недостаточность
II. Ревматические заболевания
1. Ревматоидный артрит
2. Системная красная волчанка
3. Анкилозирующий спондилоартрит
III. Заболевания органов пищеварения
1. Состояние после резекции желудка
2. Мальабсорбция
3. Хронические заболевания печени
IV. Заболевания почек
1. Хроническая почечная недостаточность
2. Почечный канальцевый ацидоз
3. Синдром Фанкони
V. Заболевания крови
1. Миеломная болезнь
2. Талассемия
3. Системный мастоцитоз
4. Лейкозы и лимфомы
VI. Другие заболевания и состояния
1. Иммобилизация
2. Хронические обструктивные заболевания
легких
3. Алкоголизм
4. Нервная анорексия
5. Голодание и другие нарушения питания
6. Трансплантация органов
VII. Генетические нарушения
1. Несовершенный остеогенез
2. Синдром Марфана и другие фибриллинопатии
(системная дисплазия соединительной ткани
марфаноподобного типа)
3. Синдром Элерса-Данло (несовершенный
десмогенез)
4. Гомоцистинурия и лизинурия
5. Болезнь Менкеса
VIII. Медикаментозные воздействия
1. Кортикостероиды
2. Антиконвульсанты
3. Иммунодепрессанты
4. Агонисты гонадотропин-рилизинг гормона
5. Антациды, содержащие алюминий
б.Тироидные гормоны
Различают местный, распространенный
и генерализованный остеопороз. Местный
остеопороз наблюдается в области
костного очага заболевания. Распространенный
остеопороз может охватить кости целой
конечности. Генерализованный остеопороз
охватывает кости всего скелета и
отмечается при эндокринных заболеваниях,
гиповитаминозе D и К, а также в старости.
В пределах кости как органа остеопороз
бывает диффузный, очаговый, пятнистый
и смешанный.
Нарушения эндокринной регуляции 697
Умеренные физические нагрузки
кальцитонин
половые стероиды -
положительный кальциевый баланс 1
соматомедины, СТГ
витамин D
Маркеры:
— остеокальцин
— аминоконцевой пропептид
коллагена I типа
— щелочная фосфатаза
Клетки-участники:
— остеобласты
Рис 132. Патогенез остеопороза
Основные факторы риска остеопороза и
показатели баланса между остеогенезом и
остеолизом представлены на рисунке 132.
Особое внимание следует обратить на
объективные критерии (маркеры),
позволяющие оценить интенсивность остеогенеза и
резорбции костной ткани — двух процессов,
от соотношения которых зависит
формирование костной массы (Ю. Франке, Г. Рунге,
1995).
Важнейшими свидетелями этих
процессов считаются показатели остеобластичес-
кой и остеокластической активности.
1. Показатели остеобластической
активности (костного формирования).
- Остеокальцин сыворотки крови — некол-
лагеновый белок, продукт остеобластов,
содержащий полиглутамиловые остатки,
связывающие кальций и фиксирующие
данный белок к гидроксиапатиту костей.
Его значение определяется тем, что
остеокальцин:
> один из самых информативных
показателей формирования кости и
скорости ее ремоделирования;
> концентрация его в сыворотке
увеличивается в случаях,
сопровождающихся минерализацией кости.
- Аминоконцевой пропептид проколла-
гена Imuna, основной белок фибрилл
костного матрикса, образующийся при
формировании остеобластами коллагена
I типа из его предшественника.
t Маркеры:
— экскреция оксипролина
резорбция кости _ экскреция дезоксипиридинолина
— кислая фосфатаза
Клетки-участники:
— остеокласты
— глюкокортикоиды
I— отрицательный кальциевый баланс
генетические дефекты коллагена,
остеокальцина, фибриллина и др.
ТзД4
I— вазопрессин
— цитокины острофазного ответа
'— лептин
2. Показатели остеокластической
активности (костной резорбции).
> Оксипролин — циклическая
аминокислота, представленная в коллагене
костей в наибольших количествах
(см. о коллагеногенезе и его патологии
т. I, с. 342-345). Усиление
экскреции оксипролина с мочой отражает
активизацию процессов резорбции
костного вещества.
> Экскреция свободных пиридиновых
поперечно-сшитых соединений
коллагена.
> Экскреция дезоксипиридинолина
(ДПИД или Pyrilink D) и пиридино-
лина с мочой.
Данные компоненты зрелого коллагена
попадают в плазму крови и мочу только в
результате деятельности остеокластов.
> Тартрат-резистентная кислая
фосфатаза — один из ее изоферментов,
характерный для остеокластов. Во
время резорбции кости она
подвергается экзоцитозу и также присутствует
в крови.
Коварство остеопороза состоит в том, что
на ранних стадиях он протекает вообще без
специфических жалоб и физикальных
симптомов. Из-за неспецифических жалоб — на
сердце» на общую слабость, на боли в
конечностях, спине, груди и суставах - подобные
лица часто трактуются как страдающие
«нейроциркуляторной дистонией кардиаль-
698 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
ного типа». Ведущий симптом — ноющие
боли в спине и/или в нижних конечностях,
реже — в верхних. В поздних случаях
наблюдаются микропереломы и переломы
тел позвонков, проксимальных отделов
бедренных костей и дистального отдела
лучевой кости. Переломы любой локализации
возникают даже при незначительных
механических воздействиях на кость. Возможны
выраженные деформации грудной клетки
с последующим развитием хронического
торако-диафрагмального легочного сердца с
риском пневмоний. При гипокальциемии
может возникать тетания с ларингоспазмом или
бронхоспазмом (см. выше гл. 13). В ранней
диагностике остеопороза и остеопении и для
прогнозирования переломов используются
периферическая количественная
компьютерная томография костей, гамма- и
ультразвуковая денситометрия, радиографическая
абсорбциометрия (рентгеновская
денситометрия). Для точного определения костной
массы используют однофотонную абсорбци-
ометрию, двухфотонную абсорбциометрию,
компьютерное томографическое
сканирование, двухэнергетическую рентгеновскую
абсобциометрию (А.П. Калинин и соавт.,
2003). Самым информативным параметром
при этом служит так называемый Т-критерий,
рекомендованный группой экспертов ВОЗ в
1994 г. Т-критерий — это результат сравнения
костной массы пациента с таковой у здоровых
лиц. Он указывает, на сколько стандартных
отклонений (SD) минеральная плотность
кости у конкретного пациента ниже или выше
средних значений для здоровых (пик костной
массы). Так, Т-критерий «-2» свидетельствует
о том, что у пациента минеральная плотность
кости на 2 стандартных отклонения ниже
пика костной массы. По Т-критерию и судят,
есть ли у пациента остеопороз (Т.О. Чернова с
соавт., 2001; М.Т. МакДермотт, 2001), причем
градации таковы:
Т-критерий > -1 Норма.
Т-критерий между -1 и -2,5 .. Остеопения.
Т-критерий< -2,5 Остеопороз.
Другой параметр — Z-критерий —
выражается в числах стандартных отклонений
ниже или выше возрастной нормы (или
в процентах от возрастной нормы) и
показывает, соответствует ли костная масса
возрасту и могут ли другие, невозрастные
факторы влиять у данного пациента на
избыточную ее потерю. С учетом Z-кри-
терия традиционно деление остеопороза
на умеренный (потеря костной массы до
30 %, нет рентгенологических симптомов,
Z- критерий ниже возрастной медианы на
2,5-3 SD), выраженный (есть
рентгенологические признаки, Z-критерий снижен
на 3-4,5 SD) и резко выраженный (есть
жалобы на боли в костях и утомляемость,
имеется развернутая рентгеновская картина,
включая зоны Лоозера и патологические
переломы, Z-критерий снижен более, чем
на 4,5 SD). При диагностике остеопороза
измеряют уровень остеокальцина, амино-
концевого пропептида коллагена 1 типа в
крови и концентрацию оксипролина в моче.
Для правильной оценки резорбции кости
используется отношение концентраций
дезоксипиридинолина и креатинина в моче.
Количество тартрат-резистентной кислой
фосфатазы в плазме отражает скорость
остеопороза. Важно определение содержания в
сыворотке крови общего и ионизированного
кальция, неорганического фосфата, других
электролитов, паратирокринина, кортизола,
лептина, половых гормонов, тироидных
гормонов и ТТГ, а иногда — и кальцитонина.
Обязательно определение щелочной
фосфатазы, особенно изоферментов ее костной
фракции (Т. Хан, 1999). Костная щелочная
фосфатаза считается важным маркером
активности остеобластов (см. рис. 132).
Деструкцию мукополисахаридов костной
матрицы и уровень ответа острой фазы,
приводящего к продукции остеолитических
цитокинов, отражают сиаловые кислоты,
орозомукоид и С-реактивный белок. Важно
обнаружение повышенного уровня костной
щелочной фосфатазы и патологических
показателей маркеров остеобластической
активности и ответа острой фазы.
Прогноз и исходы остеопороза нередко
напрямую связаны с характером основной
патологии, его вызывающей. Например,
миеломная болезнь, системные заболевания
соединительной ткани, нервная анорексия
имеют плохой прогноз, а если остеопороз
вызван ятрогенным гиперкортицизмом то
прогноз при устранении причинного
фактора благоприятный. Впрочем, при любых
продолжительных остеопоротических
процессах полной реставрации костей не
происходит, лишь отдельные костные
перекладины утолщаются на фоне общей
деминерализации кости — эта стадия течения процесса
известна как гипертрофический остеопороз
(Г. Тенчов, Г. Хаджибеков, 1962).
Важным эндокринологическим средством
лечения остеопороза служит
рассматривавшийся в 13-й главе выше кальцитонин —
пептидный гормон С-клеток щитовидной
железы и дисперсной эндокринной системы.
Кроме торможения остеолиза и активности
макрофагов кальцитонин через кокальциге-
ниновые и амилиновые рецепторы в ЦНС
и опиатэргические механизмы головного
мозга оказывает также выраженное аналь-
гетическое действие. Кальцитонин лосося
(миакальцик, кальсинар, карил) во много
раз активнее человеческого и выпускается
в виде подкожных и внутримышечных
инъекций, а также в виде интраназального
аэрозоля. Лечение кальцитонином сочетают
с приемом витамина 03(холекальциферола)
и препаратов кальция. В целях подавления
увеличения резорбции кости в ночное
время препараты кальция лучше принимать в
вечерние часы (СМ. Брызгалина и соавт.,
2003). Передозировка витамина D может
привести к развитию нефрокальциноза и не-
фролитиаза. Дифосфонаты или бифосфона-
ты (этидронат натрия, алендронат натрия -
производные памидроновои и тилудроновои
кислот) подавляют резорбцию костной
ткани и вызывают вторичное снижение
активности остеобластов. Однако они способны
провоцировать вторичный гиперпаратироз
(С. Крэйн, М. Холик, 2005). Усиливают
синтез белка в костной ткани анаболические
стероиды (ретаболил, неробол). Но
неосторожное использование анаболических
стероидов может быть причиной задержки
роста у детей и подростков, а также других
Нарушения эндокринной регуляции 699
осложнений (Б. Катцунг, 1998).
Перспективным средством лечения некоторых форм
остеопороза может оказаться витамин К.
Подробно вопросы терапии остеопороза
рассмотрены в ряде недавних монографий
(см., напр., Б.Л. Смолянский, В.Г. Лиф-
ляндский, 2006).
Лечение и профилактика остеопороза —
актуальная задача современной медицины,
поскольку остеопороз — одно из главных
массовых неинфекционных социально
значимых метаболических заболеваний и
частый вторичный синдром при эндокри-
нопатиях и других метаболических
нарушениях.
ЗАКЛЮЧЕНИЕ
Для современной науки и культуры
характерно стремление выйти за рамки традиций,
норм и канонов, выработанных теми или
иными узкими областями знания или
подняться над «цеховым мышлением». В эпоху
постмодернизма научная мысль и
культурное новаторство стремятся к смешиванию
разнородных стилей и традиций, ученые и
деятели культуры тяготеют к
междисциплинарному и межкультурному подходу.
В медицине это ведет к слиянию усилий
представителей различных специальностей
в решении проблем, не укладывающихся в
формальное ложе тех или иных
номинальных специализаций.
В индустриальном обществе искания
ученых были сосредоточены на
материальной и энергетической стороне природных
и социальных явлений. В медицине в
центре внимания стояли те аспекты, которые
связаны с вещественно-энергетической
стороной жизнедеятельности организма. В
постиндустриальную эпоху повсеместно в
центр творческих исканий перемещаются
проблемы, связанные с информационной
стороной действительности. Медицина —
не исключение. Не следует воспринимать
информатику узко, как цеховую
принадлежность компьютерщиков. У живых клеток
— своя природная информатика. Мы видим
700 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
организм как погрешимую
информационную систему и начинаем осознавать, что
заболевания могут возникать в результате
нарушений сигнализации, рецепции сигналов,
их пострецепторной передачи, мимикрии
сигналов, архивирования, разархивирова-
ния, считывания и исполнения программ, по
которым живут клетки организма (см. т. I).
Это делает особенно актуальными и
широкими задачи и перспективы эндокринологии.
Ведь именно гормоны — главные системные
сигналы организма. Учитывая
универсальную способность всех клеток к продукции
гормонов и аутакоидов, наличие дисперсной
эндокринной системы и эндокринных
функций нейроцитов и иммунокомпетентных
клеток, можно утверждать, что эндокринные
звенья играют существенную роль в
патогенезе, а часто -ив этиологии практически
всех самых распространенных болезней. Так,
эндокринную природу имеют практически
все формы ожирения (см. гл. 7), различные
вторичные артериальные гипертензии - при
гиперкортицизме, гипералъдостеронизме,
гипертирозе, феохромоцитоме, гиперинсу-
линизме, вазопрессиноме, гиперпродукции
ренина (см. Практикум), а первичный
гемохроматоз и многие формы железоде-
фицита оказались аномалиями продукции
печеночного гормона гепцидина (гл. 12). В
настоящее время известно, что
эндокринные нарушения, не являясь причинными
факторами атеросклероза, служат важными
условиями, нередко определяющими
скорость его развития, тяжесть течения, исход;
влияющими на результат взаимодействия
многочисленных факторов риска с
организмом конкретного пациента. Эндокринный
контекст определяет вероятность тех или
иных патогенных событий в
сердечно-сосудистом континууме (Ю.И. Строев, Л.П.
Чурилов, 2004). Так, гипотироз резко
ускоряет атерогенез и даже позволяет получить
алиментарную модель атеросклероза у
хищных животных. Гипертироз, напротив,
тормозит развитие атеросклероза. Сахарный
диабет любого типа как и гиперинсулинизм
— служат факторами риска атеросклероза.
Сложные и неоднозначные влияния на
атерогенез оказывают половые гормоны.
Эстрогены способствуют продукции ан-
тиатерогенных ЛПВП, уровень которых
у мужчин существенно ниже. Андрогены
увеличивают уровень ЛПНП. Кастрация
у мужчин и у самцов подопытных
животных приводит к снижению коэффициента
атерогенности плазменных Л П. Эстрогены
способствуют ускорению окисления ХН
печенью, а андрогены тормозят эти
процессы (Д. Поповичи, В. Сэхляну, 1969).
Эстрогены способны тормозить образование
пенистых клеток в атеросклеротических
поражениях (П.-А. Бурасса и соавт. 1996)
и эффект этот опосредован
эстроген-зависимыми цитокинами Т-лимфоцитов (Р. Эль-
хаге и соавт., 2000). Широко известна роль
прогестин-содержащих контрацептивов в
качестве факторов риска
сердечно-сосудистых заболеваний. Внимание патологов,
изучающих связь тучности, сахарного диабета
и атеросклероза в рамках метаболического
синдрома (см. главу 14 ниже) в настоящее
время привлекают липокины и, более
всего — открытый Тошимаса Ямаучи и
соавторами (2001) новый адипоцитарный
гормон — адипонектин. Этот пептид
жировой ткани повышает чувствительность
клеток-мишеней к инсулину и тормозит
атерогенез у подопытных мышей (см. с. 718).
Таким образом, кардиологу не обойтись без
эндокринологических знаний.
Примеры эндокринных заболеваний
можно найти в практике гастроэнтерологии,
пульмонологии, гепатологии, нефрологии,
стоматологии, хирургии. Само поведение
человека трактуется как интегральный
результат реализации генетических программ,
зависящий от метаболизма, в особенности —
от его нейроэндокринной, прежде всего, ги-
поталамической регуляции. Следовательно,
и психоневрологические нарушения могут
зачастую иметь эндокринную подоплёку.
Примером могут служить фобические
неврозы при гипокальциемии (Л.П. Чурилов,
Ю.И. Строев, 2007).
Завершая изложение раздела о
нарушениях эндокринной регуляции, считаем
необходимым еще раз обратить внима-
Нарушения эндокринной регуляции 701
ние читателя на общие закономерности,
как лежащие в основе физиологических
принципов гуморальной регуляции, так и
определяющие формы и масштабы
развивающихся нарушений.
В принципе структурная и
функциональная организация различных звеньев
гуморальной регуляции соответствует
положениям основного закона материального
мира, действующего, по крайней мере, в
пределах нашей Галактики — закона отражения.
Комплементарностъ молекул — способ его
реализации в живом организме.
Однако всегда ли мы в состоянии
адекватно определить реальные механизмы
нарушений регуляции, пользуясь доступными
в настоящее время методами исследований?
Как правило, мы можем получить
информацию, соответствующую одномерному, а не
многомерному (объемному) отображению
происходящих процессов, а это лишает
исследователя возможности правильного
истолкования обнаруживаемых плейотропных
эффектов различных биорегуляторов.
Как следует из материалов, изложенных
в предшествующих разделах, многие
физиологически активные соединения обладают
разносторонним влиянием на таргетные
системы, однако, последствия подобных
воздействий далеко не всегда могут
контролироваться исследователями.
В основе многих заболеваний лежит
нарушение информационного разграничения
сфер влияния между системной нейроэн-
докринной регуляцией (гормоны и нейро-
трансмиттеры) и местными аутакоидными
механизмами аутокринного и паракринного
характера (цитокины и другие медиаторы
воспаления).
Как в здоровом организме, так и при
штатном, нормергическом успешном
функционировании типовых патологических
процессов эти управляющие программы не
конфликтуют между собой, так как корот-
коживущие аутакоидные сигналы
эффективны только на небольшом удалении от
источника, а их молекулярные носители не
достигают регуляторно значимых
концентраций в системном кровотоке.
С другой стороны, системные нервные
и гормональные сигналы не унифицируют
и не парализуют основанную на аутакоид-
ной регуляции оптимальную и топически
разнообразную работу местных защитных
механизмов, так как доступ гормонов и ней-
ротрансмиттеров в очаги нормергического
воспаления ограничен барьерными
изменениями микроциркуляции и
функциональным парезом вегетативных нервов. Данный
принцип впервые был сформулирован нами
как «правило системно-местного защитного
равновесия» (А.Ш. Зайчик, Л.П. Чурилов;
1999). При гиперергическом протекании
патологических процессов, например, при
различных видах шока и избыточном ответе
острой фазы он может остро нарушаться,
что снижает резистентность организма. На
наш взгляд, один из основных факторов,
обеспечивающих бесконфликтную
реализацию системных и местных защитных
программ — структурно-функциональная
целостность мезенхимальных тканей, с
одной стороны, составляющих арену местных
типовых патологических процессов, а с
другой - производящих уникальный репертуар
аутокоидов и гормонов - от медиаторов
воспаления до кортикостероидов.
Микроструктура мезенхимы влияет на поведение
ее клеток и в норме позволяет разграничить
информационные сферы влияния аутакои-
дов и гормонов и бесконфликтно сочетать
эти формы гуморальной регуляции.
Синдром Марфана и близкие к нему
марфаноидные фибриллинопатии могут
служить примерами хронического нарушения
системно-местного защитного равновесия,
когда аномалии самосборки ключевых
молекул активной мезенхимы приводят к
избыточному системному действию некоторых
аутакоидов и хроническим мультиорганным
поражениям (Ю.И. Строев, Л.П. Чурилов,
2007).
Ввиду сложности эндокринной
регуляции, несмотря на огромные возможности
диагностических исследований
(доступность определения концентраций
практически всех известных гормонов), осмысление
эндокринных нарушений остается нелёгкой
702 А.Ш. Зайчик, Л.П.Чурилов Патохимия
задачей, актуальной для врача любой
специальности. Тактика выбора лечебных
воздействий в практической
эндокринологии соответствует лишь Плоскостной, а не
объемной составляющей Вселенского закона
отражения. Известная вариабельность в
реализации последствий нарушенной функции
эндокринных желез, пермиссивность
эффектов информационных молекул — лишний
раз заставляют нас задуматься над
порочностью бытующих в гормональной терапии
принципов. При обнаружении более низких
(по сравнению с нормативами)
концентраций того или иного гормона в крови,
современный практикующий врач-эндокринолог
пытается компенсировать недостающие
количества данного соединения, а при более
высоких — снизить его избыток в
организме. Данный прием не учитывает основного
принципа реактивности организма — выбор
форм и масштабов реагирования
индивидуален. Классик отечественной патофизиологии
чдокринно-метаболические нарушения)
Л.Р. Перельман особо подчеркивал: «Для
каждого организма граница между
физиологической и патологической регуляцией
индивидуальна». В связи с этим, появление
в ближайшем будущем дополнительных
диагностических приемов, позволяющих
оценить мультифакториальность эффектов
использующихся методов медицинского
воздействия, будет являться основанием
для разработки индивидуальных регуля-
торных профилей и необходимых методов
терапии, что позволит нам приблизиться к
пониманию тех закономерностей, которые
соответствуют не плоскостному, а
объемному (трех- и более многомерному)
отображению явлений, лежащих в основе процессов
гуморальной регуляции.
В силу универсального значения
эндокринологических знаний для всех отраслей
медицины, обособившаяся в XX столетии,
эндокринология становится медицинской
наукой XXI века.
Глава 14
ПОНЯТИЕ О МЕТАБОЛИЧЕСКОМ
СИНДРОМЕ
(написано с участием Ю.И. Строева)
«Очень интересна наследственная связь между артериосклерозом,
подагрой, диабетом и, пожалуй, также конституциональной тучностью. Все эти
болезненные состояния, по-видимому, сродны друг другу, причем сущность
сродства их совершенно неясна».
Г. Штрюмпель «Учебник частной патологии и терапии
внутренних болезней», 1915
• История изучения
• Эпидемиология
• Определения
• Этиология
• Патогенез
На страницах данного тома уже
неоднократно шла речь о метаболическом
синдроме (МС), своеобразном
«перекрестке» патохимии, когда сочетаются нарушения
липидного, углеводного, нуклеинового,
белкового и водно-электролитного
обменов (см. выше гл. 6, 7, 9, 10). Эта проблема
в современной медицине — и своего рода
«перекрестокмнений», вызвавший только с
1988 г. по настоящее время около 5000
публикаций в мировой прессе (Ю.И. Строев
и соавт., 2007). Идут жаркие дискуссии об
оптимальных критериях синдрома, о роли
различных генетических особенностей в его
этиологии, о возможном общем
патогенетическом звене, порождающем сочетанные
метаболические нарушения, о
целесообразности использования данного диагноза в
целях оценки риска сердечно-сосудистых
заболеваний и о конституциональном
характере МС. С начала XXI века
«метаболический синдром» стал едва ли не самой
изучаемой формой патологии, интересующей
врачей практически всех специальностей:
терапевтов, эндокринологов, кардиологов,
нефрологов, офтальмологов, хирургов и др.
Метаболический синдром — основа
развития ИНСД, а также сердечно-сосудистых
заболеваний, которые на сегодня являются
ведущей причиной инвалидизации и
смертности населения развитых стран мира.
Представления о метаболическом
синдроме подошли в настоящее время к
кризису, связанному с «перепроизводством»
непрерывно обновляющейся информации о
его возможных проявлениях и отсутствием
общепринятой доказанной концепции его
патогенеза. В связи с этим есть
тенденция трактовать МС как «метаболический
симптомокомплекс» без общей этиологии
или, напротив (в связи с его широкой
распространенностью в популяции), видеть в
нем своего рода пограничный вариант
конституции индивидов, то есть своеобразный
«метаболический диатез». У теоретиков и
клиницистов нет единого понимания МС:
Н.А. Беляков справедливо указывает, что
само понятие «синдром» ограниченно
применимо в данном случае, и справедливее
было бы говорить о «хроническом мета-
704 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
болическом дисбалансе» (Н.А. Беляков и
соавт., 2005).
В третьем издании данного учебника
авторы решили дать читателю возможность
«постоять и подумать на этом перекрестке». По
дидактическим причинам глава, посвященная
МС, дана в заключение книги, после
рассмотрения отдельных патохимических нарушений,
которые могут сочетаться при МС.
История изучения МС
Ученые-медики уже давно обратили
внимание на связь между артериальной ги-
пертензией (АГ), сахарным диабетом (СД)
и атеросклерозом (АСК). Современные
представления о МС во многом основаны
на исследованиях СД в начале XX века. Еще
в 1922 г. классик отечественной терапии
Г.Ф. Ланг первым в мировой медицине
заметил частое сочетание СД, гипертонической
болезни (ГБ), АСК, ожирения и расстройств
жирового и пуринового обменов.
Сочетание АГ, гипергликемии и гиперурикемии
было описано в 1923 г. шведским врачом
Э. Кюлином. Частое сочетание гиперхолес-
теринемии, гиперурикемии, ожирения и
АГ находили и описывали в своих
публикациях классик отечественной терапии А.Л.
Мясников и патологоанатом Д.М. Гротэль
(1926). В 1931 г. в Вене выдающийся
австрийский эндокринолог В. Фальта и его
коллега Р. Боллер обратили внимание на
существование инсулин-чувствительной и
инсулин-резистентной форм СД. Вскоре
британский диабетолог Г.П.Химсуорт
предложил нагрузочный тест, при котором
обследуемым лицам глюкоза вводилась
перорально, а инсулин — внутривенно.
Таким образом, он установил, что у молодых
лиц худощавого телосложения
чувствительность к инсулину была такой же, как у лиц
без СД, в то время как у лиц среднего
возраста с избыточной массой тела к вводимому
внутривенно инсулину отмечалась
выраженная резистентность. Так сформировалось
представление о снижении
чувствительности к инсулину при тучности и о феномене
инсулинорезистентности. В 1947 г. Дж. Вог
обратил внимание на то, что ожирение
мужского типа (андроидное, абдоминальное,
«яблочное») чаще встречается у лиц с СД
2 типа и сердечно-сосудистыми
заболеваниями. Годом позже, в 1948 г., возможность
развития АГ на фоне избыточной массы тела
и гиперурикемии показал выдающийся
советский терапевт Е.М. Тареев. В монографии
«Гипертоническая болезнь» он прямо писал:
«Представление о гипертонике особенно
часто ассоциируется с ожирелым гиперсте-
ником, с нарушением белкового обмена, с
засорением крови продуктами неполного
метаморфоза — холестерином, мочевой
кислотой...». В 1960-1965гг.Р.ЯлоуиС.Берсон,
разрабатывая радиоиммунологическое
определение гормонов, показали, что ожирение
часто сочетается с гиперинсулинемией и ин-
сулинорезистентностью, и высказали идею
о том, что именно ожирение, протекающее
как с СД, так и без него, является причиной
инсулинорезистентности.
Бурный рост в мире заболеваемости СД,
ГБ и АСК во второй половине XX в. явился
причиной «рецидива» научного интереса к
проблеме взаимоотношения перечисленых
заболеваний. Дж. М. Ривен в начале 60-
х гг. высказал мысль о том, что в основе
АСК, СД, АГ и ожирения «может лежать
общий фундаментальный дефект» (1963).
А в 1966 г. французский врач Ж.-П. Камю,
«реанимировав», по сути, забытые труды
цитированных выше медиков, ввел в
практику медицины новое понятие —
«метаболический трисиндром», описав у одного
больного сочетание гиперлипидемии, СД
2 типа и подагры. Двумя годами позже
X. Менерт и X. Кульман (1968) выявили
подобные метаболические нарушения при
АГ и СД, предложив новый термин —
«синдром изобилия». Современный термин,
определяющий сочетание указанных выше
заболеваний — «метаболический
синдром», — ввели в 1980 г. в ГДР М. Ганефельд
и В. Леонгардт. Несмотря на то, что
некоторые звенья патогенеза этого синдрома были
давно установлены и изучены (в частности,
роль инсулинорезистентности и гиперинсу-
линемии), в теориях его происхождения до
сих пор остаются белые пятна. Это явилось
поводом к рождению очередного термина —
«синдром X» (Ривен Дж.М.,1988), который,
на наш взгляд, не совсем удачен, так как в
медицине уже существуют несколько
«синдромов X». Например, этот термин
применяют для обозначения одного из вариантов
ишемической болезни сердца.
Термин, предложенный Дж.М. Ривеном,
основывался на достигнутых к середине
80-х годов прошлого века более глубоких
знаниях в области клинической биохимии.
Поэтому «синдром X», по версии автора,
включал периферическую инсулинорезис-
тентность, гиперинсулинемию, пониженную
толерантность к глюкозе, дислипидемию (в
основном гипертриглицеридемию и гипоаль-
фахолестеринемию), а также артериальную
гипертензию.
Дж.М. Ривен специально подчеркивал,
что этот синдром может возникать у лиц с
нормальной массой тела, и поэтому даже не
включил ожирение в список его проявлений.
Учитывая, что перечисленные заболевания
по отдельности и вместе прогностически
неблагоприятно сказываются на
продолжительности последующей жизни, Н. Каплан
(1989) пошел дальше, придумав
мрачноватый термин — «смертельный квартет»,
который объединяет ожирение (особенно
верхней половины туловища — андроид-
ное) с низкой толерантностью к углеводам,
гипертриглицеридемией и артериальной
гипертензией. Сочетание «синдрома X» и
приступов апноэ во сне стали называть
«синдром Z» (А.И. Сергеев с соавт., 2005).
В 1991 г. австралиец П. Циммерт
предложил включить в синдром X центральное
ожирение и заменить его термином
«синдром инсулинорезистентности», а еще
лучше — вернуться к термину немецких
авторов «метаболический синдром». Но в
США продолжал широко употребляться
термин «синдром инсулинорезистентности», в
частности, по рекомендации Американского
общества эндокринологов-клиницистов.
В своей недавней статье Дж. М. Ривен
специально разделяет эти два понятия,
Понятие о метаболическом синдроме 705
указывая на то, что метаболический
синдром — всего лишь удобный клинический
инструмент в руках врачей, и его
прогностическая цель — выявить прежде всего лиц
с более высоким риском развития
сердечнососудистых заболеваний. Синдром же
инсулинорезистентности — это патогенетически
обоснованный термин, используемый для
обозначения закономерного комплекса
нарушений и связанных с ними клинических
исходов у лиц с инсулинорезистентностью и
компенсаторной гиперинсулинемией. Инсули-
норезистентность, в понимании Дж.М. Ри-
вена, не болезнь, а скорее — предболезнь,
то есть диатез, пограничная аномалия
конституции, которая (при наличии
гиперкалорийного питания и определенного образа
жизни) повышает вероятность развития
нарушений толерантности к глюкозе, дис-
липидемии, эндотелиальной дисфункции,
гемодинамических нарушений (включая
АГ), нарушений в системе фибринолиза и
коагуляции и т.д. В 1998 г. ВОЗ
рекомендовала к использованию именно европейский
термин «метаболический синдром», так
как результаты исследований показали,
что инсулинорезистентность не является
причиной всех компонентов
метаболического синдрома, и патогенез последнего не
сводится только к ней.
Эпидемиология МС
Распространенность МС в разных
популяциях колеблется от 5 до 30 %. Дж.М. Ривен
(1988,1999) считает, что на сегодня в мире
25 % лиц среднего возраста имеют
инсулинорезистентность и как следствие ее — МС.
Синдром несколько чаще встречается у
мужчин, чем у женщин. В США, по данным
Э.С. Форд с соавт. (2002), он с учетом
возраста присутствует в среднем у 23,7%
лиц, а в группе старше 60 лет — у 43,5%.
Однако МС не должен восприниматься как
мужское заболевание, хотя его и связывают
с гиперандрогенизмом и симпатикотонией
(А.И. Сергеев с соавт., 2005).
Установлено, что его частота среди женщин в Иране
706 А.Ш. Зайчик, Л.П.Чурилов Патохимия
(43 %), Омане (25 %) и Турции (40 %) выше,
чем среди мужчин (25, 20 и 28 %,
соответственно). У афроамериканцев
распространенность МС среди женщин более чем
наполовину выше, чем у мужчин, а среди
латиноамериканок — на четверть выше, чем
у мужчин того же этноса. Самой
пораженной этнической группой, по данным ВОЗ,
являются американские индейцы, среди
которых частота МС у женщин доходит до
58% и превышает ее у мужчин (44%). Не
случайно недавно вышедшая монография
Н.А. Белякова и соавт. (2005) озаглавлена:
«Метаболический синдром у женщин».
В Финляндии и Швеции МС
наблюдается у 10-15% лиц с нарушенной
толерантностью к глюкозе, у 42-64% лиц — с
гипергликемией натощак и у 78-84%
лиц - с ИНСД (Б. Изомаа с соавт., 2001).
Подробные данные о частоте МС в России
приводятся в монографии В.А. Алма-
зова с соавт. (1999) и Н.А. Беляковым и
СЮ. Чубриевой в недавно опубликованном
руководстве «Ожирение» (2003). Всего в
мире более 310 млн лиц с МС, и их
число, согласно существующим тенденциям,
превысит к середине текущего столетия
полмиллиарда. Вероятность развития МС
существенно повышается с возрастом.
Не исключено, что факторы риска,
связанные с инсулинорезистентностью, могут
воздействовать на предсуществующей
конституционально-генетической основе с
детства, например, при гипоталамическом
синдроме пубертатного периода (ожирении
с розовыми стриями) — самой частой
эндокринной патологии у подростков, которую
мы трактуем как возможную предтечу
классического МС (ЮМ. Строев, Л.П. Чурилов,
А.Ю. Бельгов, 2004).
Развитию МС способствуют как
неисправимые (генетические,
демографические — пол, возраст), так и исправимые
(употребление большого количества жирной
пищи, гиподинамия-гипокинезия, стрессы,
алкоголь, курение) факторы (С.А. Бутрова,
2000). Тесно связаны с формированием
МС количество и эндокринная активность
висцерального жира. Ориентировочным
(эндокринно-метаболические нарушения)
скрининговым показателем висцерального
ожирения служит окружность талии (И. Ле-
мье с соавт., 2000). Многие авторы (Д. Дан-
стен с соавт., 2004; М. Стерн с соавт., 2002;
Б.Исомаа с соавт., 2001) настаивают, что
МС значительно увеличивает риск развития
сердечно-сосудистых заболеваний (в 3 раза)
и риск смерти от них (в 2 раза), а также
повышает риск развития СД (в 5 раз).
Но в последнее время появились
исследования, показавшие, что риск, связанный
с МС, не превышает суммарного
дополнительного риска сердечно-сосудистой
патологии, порождаемого каждым из входящих в
него нарушений. На этом основании Б. Кан
с соавт. (2005) в специальном совместном
заключении Европейской и Американской
Диабетических Ассоциаций значимость
МС в прогнозировании риска развития
сердечно-сосудистых заболеваний ставят
под сомнение и критикуют даже саму
необходимость использования этого диагноза,
входящего в Международную
классификацию болезней, в широкой практике.
Критерии МС
В последние годы понятие «метаболический
синдром» все время расширяется. В него
теперь включают (или с его патогенезом
увязывают) не только нарушения углеводного
и липидного обменов, но и гиперурикемию,
микроальбуминурию, гипертрофию
миокарда, повышение содержания фибриногена в
крови, увеличение адгезивной и агрегаци-
онной способности тромбоцитов,
дисфункцию эндотелия со снижением продукции
NO, повышение концентраций некоторых
реагентов острофазного ответа, активности
ингибиторов тканевого активатора плазми-
ногена, активацию симпатической нервной
системы, гиперандрогенизм и аномалии
продукции некоторых регуляторных пептидов
адипоцитарного происхождения — липоки-
нов (см. выше гл. 6).
Ниже приводится сводная таблица
метаболических и функциональных показателей,
а также расстройств, связываемых разными
авторами с МС (таблица 45).
Понятие о метаболическом синдроме 707
Таблица 45. Лабораторно-диагностические признаки МС
Метаболические и
функциональные параметры
Нарушения углеводного
обмена
Нарушения липидного
обмена
Тромбогенные факторы
Концентрации реактантов
острофазного (преиммун-
ного) ответа и липокинов
в крови
Дисфункция сосудистой
стенки
Прочие метаболические
и функциональные
расстройства
Проявления метаболических и функциональных нарушений
Сниженная толерантность к глюкозе
или СД 2 типа (ИНСД)
Повышение содержания висцерального жира
(по данным МРТ)
Увеличение концентрации апо-В-ЛП
Снижение концентрации а по- А-1 -Л П
Появление ЛПНП и ЛПВП малой плотности
Повышение концентрации апопротеина С III
Гипертриглицеридемия
Повышение фибриногена
Повышение ингибитора активатора плазминогена I типа
Повышение вязкости крови
Повышение ИЛ-6
Повышение кахексина
Повышение резистина
Повышение С-реактивного белка
Повышение белка-стимулятора ацилирования
Гиперлептинемия
Снижение адипонектина
Микроальбуминурия
Повышение продукции несимметричного
изомера диметиларгинина
Снижение продукции N0
Увеличение экспрессии молекул клеточной адгезии
Артериальная гипертензия
Гиперурикемия
Повышенное образование свободных радикалов и
липоперекисей
Снижение прессорного натрийуреза и тенденция
к задержке в клетках катионов натрия и кальция
Гомоцистинемия
Неалкогольное ожирение печени
Синдром поликистозных яичников
Лейкоцитоз
Приступы обструктивного апноэ во сне
Контрактура Дюпюитрена
Нарушение продукции мелатонина
Наиболее авторитетные исследования
критериев МС принадлежали группе
экспертов ВОЗ (ВОЗ, 1999) и третьему докладу
Экспертной группы национального совета
США по определению, оценке и лечению
гиперхолестеринемий у взрослых (АТР
III, 2001). Эти определения не полностью
согласовались между собой. Эксперты ВОЗ
подчеркивали роль нарушений углеводного
обмена и инсулинорезистентности, эксперты
АТР — роль нарушения липидного
метаболизма и критерии состояния жировой ткани.
К тому же эти определения не были этнически
универсальными. Ввиду укрепления липо-
киновой теории патогенеза МС (см. ниже) и
по организационно-практическим причинам,
изложенным в недавнем критическом обзоре
(Р. Кан и соавт., 2005), Международной
диабетической федерации (МДФ, 2005) пришлось
выработать согласованный и приемлемый для
708 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
Таблица 46.Современные критерии МС
(ВОЗ —Доклад группы экспертов ВОЗ, 1999; ATP III — Третий доклад Экспертной группы
национального совета США по определению, оценке и лечению гиперхолестеринемий у взрослых, 2001;
МДФ — Согласованное общемировым консенсусом определение Международной диабетической
федерации, 2005)
ВОЗ (1999)
ATP III (2001)
МДФ (2005)
ГЛАВНЫЙ КРИТЕРИЙ: ИЗСД 2 типа
или повышенный уровень глюкозы
натощак, или интолерантность
к глюкозе, или инсулинорезис-
тентность, верифицированная
с помощью клэмп-теста Р.А. Де
Фронцо(1979)
ПЛЮС ЛЮБЫЕ 2 из следующих
признаков:
- соотношение окружностей талии
и бёдер >0,90 у мужчин и >0,85 у
женщин;
- триглицериды сыворотки более
1,7 мМ/л либо холестерин ЛПВП
ниже 0,9 мМ/л для мужчин и <1,0
для женщин;
- артериальное давление выше
140/90 мм рт. ст.;
- микроальбуминурия выше 20
мкг/мин либо соотношение аль-
бумин/креатинин мочи более 30
мг/г
ЛЮБЫЕ 3 и более из
следующих признаков:
— окружность талии более
102 см для мужчин и 88 см для
женщин;
— триглицериды сыворотки
более 1,7 мМ/л;
— артериальное давление
выше 130/85 мм рт.ст.;
— холестерин ЛПВП ниже 1,0
мМ/л для мужчин и <1,3 мМ/л
для женщин;
— глюкоза сыворотки крови
натощак более 5,6мМ/л
ГЛАВНЫЙ КРИТЕРИЙ: центральное
ожирение, подтвержденное у европеоидов
окружностью талии от 94 см для мужчин
и от 80 см для женщин (для иных рас
— поправки по специальной таблице, с
отсчетом от меньших окружностей)
ПЛЮС ЛЮБЫЕ 2 из следующих
критериев:
— триглицериды сыворотки от 1,7 мМ/л
или специальное лечение от гипертриг-
лицеридемии;
— холестерин ЛПВП ниже 1,03 мМ/л
для мужчин и <1,29 для женщин или
специальное лечение от гипоальфали-
попротеидемии; *
— артериальное давление
систолическое от 130 мм рт. ст. или диастолическое
— от 85 мм.рт.ст., либо специальное
лечение от артериальной гипертензии;
— глюкоза сыворотки крови натощак
от 5,6 мМ/л либо ранее установленный
диагноз СД 2 типа
применения в разных этнических популяциях
«общемировой консенсус по критериям МС».
Ниже, в таблице 46, даны для сравнения
все 3 перечня критериев МС.
Прочими метаболическими и
функциональными проявлениями МС в настоящее
время считаются:
Артериальная гипертензия
Гиперурикемия
Повышенное образование свободных
радикалов и липоперекисей
Снижение прессорного натрийуреза и
тенденция к задержке в клетках Na+ и Са++
Гомоцистинемия
Неалкогольное ожирение печени
Синдром поликистозных яичников
Контрактура Дюпюитрена
Лейкоцитоз
Приступы обструктивного апноэ во сне
Разумеется, не все эти параметры и
проявления обладают равной значимостью. Так,
согласно Н.А. Белякову и соавт., следует
выделять обменный, системный и
нозологический уровень структуры МС (2005),
отделяя при этом вторичные заболевания
и осложнения (такие, как ИБС и ХПН) от
СД, АГ, атеросклероза и синдрома
поликистозных яичников (см. гл. 7,8,9,13), которые
составляют первичную нозологическую
тетраду МС у женщин.
По мнению Дж. Ривена (1999),
метаболический синдром объединяет, в первую
очередь, следующие клинические и
метаболические расстройства:
>• абдоминальное ожирение (андроидное,
гипертрофическое, «яблочное»),
>- инсулинорезистентность и/или гиперин-
сулинемию,
>* нарушение толерантности к глюкозе
(ИНСД 2 типа),
>* артериальную гипертензию,
>• дислипидемию,
>• нарушение гемостаза,
>- гиперурикемию
>* микроальбуминурию.
Предложено считать метаболический
синдром полным, когда инсулинорези-
стентность проявляется всем комплексом
нарушений, или неполным, когда инсули-
норезистентность сочетается не со всеми
составляющими этого синдрома.
Нам импонирует определение МС по
Н.А. Белякову и соавт. (2005), которое
ниже и приводится:
«МС — это совокупность нарушений
гормональной регуляции углеводного,
жирового, белкового и других видов
обмена под действием внешних и внутренних
факторов с характерным развитием СД II
типа, гипертонической болезни, ожирения,
атеросклероза и последующих осложнений,
преимущественно, ишемического генеза».
Этиология МС
Существуют несколько важных
генетических предпосылок МС и ряд эколого-гигиени-
ческих факторов, играющих пороговую роль
для развития этого аддитивно-полигенного
синдрома. В каком-то смысле массовое
появление МС, ставшего проблемой
здравоохранения во второй половине XX века, можно
интерпретировать как реакцию человечества
на переход к относительно малоподвижному
и изобильному образу жизни. По
классической гипотезе Дж. В. Нила (1962), многие
тысячелетия эволюция человека проходила
в условиях давления естественного отбора в
пользу закрепления «генов бережливости»,
обеспечивавших повышенную живучесть и
экономное расходование энергетического
сырья в условиях дефицита еды и
подвижного образа жизни крестьян, охотников,
собирателей. К этому можно добавить, что
климатические факторы тропиков и
субтропиков, где сформировался человек,
способствовали закреплению аллелей, сберегающих
Понятие о метаболическом синдроме 709
натрий в условиях напряженной адаптации
к перегреванию.
Не следует забывать о «теории узкого
горла», принимаемой многими антропологами
и согласующейся с данными об
ограниченном полиморфизме митохондриальных
генов. Согласно ей, все ныне живущие
люди — потомки весьма ограниченной по
размеру группы кроманьонцев, выживших
в наиболее экстремальный период,
связанный с пищевым дефицитом (У.У. Хавеллз,
А.К.Уилсон, 1987; см. также т. I,
«Повреждение митохондрий»).
По Дж.В. Нилу, в новую эпоху именно
то, что обеспечивало популяционную
адаптацию к голоданию и перерасходу калорий,
стало бременем «цивилизованного»
человека. Эпидемиология МС свидетельствует о
том, что его «вспышки» возникают во 2-3-м
поколениях ийдивидов в сообществах,
испытывающих резкую ломку колониально-
аграрных стереотипов существования и
урбанизацию с повышением уровня жизни,
например, в ранее отсталых, но быстро
богатеющих в результате эксплуатации своих
природных ресурсов странах (тенденция
«From West — to the rest»). Известно, что
одним из новых центров пандемии МС
стали Ближний Восток и
страны-нувориши района Персидского залива (П. Сигал,
П.Циммерт,2005).
Несмотря на то, что единый генетический
маркер МС в рамках полиэтиологического
подхода к МС как
конституциональному диатезу еще не определен, ряд генов,
сцепленных с повышенной вероятностью
развития МС или обусловливающих
сниженную толерантность к пищевой нагрузке,
перечисляются ниже.
>- Генетические дефекты инсулинового
рецептора в хромосоме 19. Их известно
более 50, причем у одного индивида они
могут сочетаться, вызывая не только (и
не столько!) снижение количества
рецепторов на клетках, сколько снижение
их аффинитета к гормону (Ю.Г.Надь,
2001), нарушение экспрессии их
полипептидных цепей, уменьшение ти-
розин-протеин-киназной активности
710 АЖ Зайчик, ЛЛ. Чу рилов Патохимия (эндокринно-метаболические нарушения)
пострецепторного механизма (см. гл. 8,
13), ускоренную деградацию
рецепторов. Снижение эффективности
информационного действия инсулина ведет
к компенсаторной гиперинсулинемии
и/или гипергликемии, что способствует
развитию прочих компонентов МС (см.
ниже). По-видимому, мутации гена
инсулинового рецептора — не основная
причина инсулинорезистентности в
популяции (Дж. Хоун с соавт., 1994;
С.Тэйлор с соавт., 1992).
>• Генетический дефект хромосомы 15q,
обусловливающий пострецепторную
мультигормональную резистентность
при синдроме поликистозных яичников
(см. гл. 13). У пациенток сочетаются
инсулинорезистентность типа А с acanthosis
nigricans (см. выше гл. 9), гиперандроге-
низмом, гиперинсулинемией, гиперпро-
лактинемией и гиперлептинемией, и
развивается МС. Данное нарушение имеет
довольно высокую частоту в популяциях
(Ж. Мюллер-Д. Виланд с соавт., 1989).
>• Ген белка-переносчика длинноцепочеч-
ных свободных жирных кислот в
хромосоме 4. Данный ген экспрессирован в
тонком кишечнике и печени и его мутации,
широкораспространенные в популяциях,
связаны с инсулинорезистентностью,
повышением уровня СЖК в крови, их
ускоренным окислением и всасыванием,
а также риском развития МС (М. Про-
хазка с соавт., 1993; Л.И.Бейер с соавт.,
1995). Один из дефектов у крыс SHR,
воспроизводящих эссенциальную гипертен-
зию и другие компоненты МС человека,
находится как раз в хромосоме 4.
>• Возможно, что при МС, как и при
наследственной гиперлипопротеидемии
IV типа, имеется усиленное
превращение углеводов в жиры гепатоцитами,
что связано с малой чувствительностью
печеночной фосфофруктокиназы к
обратному ее ингибированию продуктами
этого синтеза.
>- Ген ($3-адренорецептора (см- гл. 13).
Э. Гарсиа-Руби с соавт. (1998), а
позже — А.В. Жукова с соавт. (1999) нашли,
что мутация Тгр64 Arg в гене рз-адреноре-
цептора может играть определенную роль
в формировании нарушения
чувствительности периферических тканей к
инсулину. При этом снижается активность
гормонозависимой липазы, и липолиз
требует компенсаторной
гиперактивности симпато-адреналового механизма,
которая достигается под воздействием
лептина и инсулина, но провоцирует АГ,
гиперлептинемию и гиперинсулинемию.
Сниженный липолитический эффект
катехоламинов на адипоциты —
характерная черта крыс SHR, на которых
получена генетическая модель неполного МС
(Т. Айтман с соавт., 1997).
>■ Инсулинорезистентность может
обусловливаться наследственным
изменением структуры глюкозопереносящего
белка GLUT 1 (А.Е. Понтироли с соавт.,
1996), а также уменьшением
концентрации глюкозного переносчика GLUT 4
(см. выше гл. 8, 9) в жировой ткани при
нормальном его содержании в скелетных
мышцах (Т.С. Цяо с соавт., 1997).
>• Получены данные о связи между
формированием инсулинорезистентности и
высокой активностью ренин-ангиотен-
зиновой системы. В качестве маркеров
предрасположенности рассматриваются
полиморфные гены-кандидаты,
продукты экспрессии которых могут влиять на
активность этой системы (см. гл. 13), в
частности, ген ангиотензин-конвертазы и
ген ангиотензиногена (М.И. Балаболкин
с соавт., 2001; Т. Катсуя с соавт., 1995).
Однако роль ренин-ангиотензиновой
системы в развитии артериальной гипер-
тензии при СД останется сомнительной
до тех пор, пока не появятся данные о
развитии АГ при длительной
гиперинсулинемии, свойственной
органическому гиперинсулинизму — инсулиноме
(см. гл. 9). Возможно, гипертензивное
действие оказывает не столько гиперин-
сулинемия, сколько сопутствующая ей
высокая продукция гормона амилина.
>• Значительную роль в формировании
МС у индивидов, предрасположенных к
нему, играет гиперактивность гипотала-
мо-гипофизарно-надпочечниковой
системы и симпато-адреналовых реакций.
Гиперкортизолемия и симпатикотония
создают предпосылки для усугубления
инсулинорезистентности, свойственной
МС, снижают выработку адипонектина
(см. ниже). Андроидное центрипеталь-
ное ожирение при МС похоже на таковое
при гиперкортицизме и ювенильном
диспитуитаризме, когда отмечаются АГ
и снижение чувствительности гипотала-
мо-гипофизарного аппарата к обратному
ингибирующему кортикостероидному
сигналу (Ю.И. Строев с соавт., 2003).
В этой связи важно, что у ряда больных
с МС имеются генетические дефекты в
5-м локусе гена глюкокортикоидного
рецептора, снижающие чувствительность
гипоталамо-гипофизарной системы
к гиперкортицизму (П. Бъёрнторп,
Р. Росмонд, 2000). Отмечены и
особенности полиморфизма генов рецепторов
дофамина и лептина, связанные при
МС с гиперактивностью
симпатической нервной системы (Н.А. Беляков,
С.Ю.Чубриева, 2003).
>* Мембранно-транспортная теория
патогенеза нарушений, формирующих
МС, придает ключевое значение в его
развитии генетическим особенностям
мембранных белков-противоперенос-
чиков ионов (см. выше гл. 6, 7, 9, 10).
Нарушение трансмембранного обмена
Na+-H+, оцениваемого по Na+- Li+-npo-
тивотранспорту, является генетическим
маркером, связанным с развитием ГБ.
При наличии сверхэкспрессивного гена
SRC9A1, управляющего
натрий-протонным противопереносчиком,
возникает внутриклеточное накопление Na+
и Са++, увеличение чувствительности
клеток к действию ангиотензина и но-
радреналина, формируется АГ. Инсулин
способствует экспрессии этого гена, а
крысы SHR, развивающие спонтанно
АГ и другие компоненты МС, обладают
Понятие о метаболическом синдроме 711
генетическим дефектом данного
белка. Очевидно, системные мембранные
нарушения присутствуют и при МС.
Так, при полном МС И.А. Романенко
с соавт. (2001) обнаружили предельно
низкие значения пластичности мембран
эритроцитов и наиболее крупные
размеры клеточных агрегатов. В 6,8 % случаев
популяция патологических эритроцитов
была представлена мишеневидными,
фрагментированными и
каплеобразными формами, а переходные формы
эритроцитов выявлялись в 46,9 %
случаев. При исследовании красной крови
у больных СД 2 типа Ю.И. Строевым
с соавт. (2001) выявлено статистически
высоко достоверное (р<0,001)
уменьшение объема эритроцитов при наличии
инсулинорезистентности и гиперинсу-
линемии, Что может иметь под собой
мембранно-генетические предпосылки.
>- Ген ADD 1/SREBP1 влияет одновременно
на дифференцировку фибробластов в ли-
поциты, экспрессию рецепторов ЛПНП
и на синтез жирных кислот и стероидов
в печени, что позволило К. Йокояме с
соавт. (1993) связать его полиморфизм с
предрасположенностью к МС. Продукт
гена опосредует влияние СЖК и
инсулина на липогенез.
>* Генетические расстройства, вызванные
полиморфизмом генов субстратов ин-
сулинового рецептора IRS-I и II, гли-
когенсинтетазы, глюкокиназы (см. гл.
8, 9), гормоночувствительной липазы,
липопротеиновой липазы, ферментов,
способствующих превращению
углеводов в липиды, а также гены, связанные
с преобладанием быстрых красных гли-
колитических мышечных волокон также
могут вносить вклад в формирование
предрасположенности к МС. Так, при
МС и ИЗСД 2 типа К.Ф. Петерсен с
соавт. (2004) обнаружили
наследственный митохондриальный дефект
окислительного фосфорилирования в мышцах,
способствующий снижению окисления
СЖК и жировой дистрофии мышечных
712 АЖ Зайчик, Я/7. Чу рилов Патохимия (эндокринно-метаболические нарушения)
волокон (см. также т. I, «Повреждение
митохондрий»).
>- Ген адипонектина в хромосоме 3q
связан с развитием МС и ИНСД 2 типа,
его мутации нарушают продукцию и
мультимеризацию данного коллагено-
подобного липокина, что способствует
инсулинорезистентности, стеатозу
печени, атеросклерозу и СД (Дж.П. Уайтхед
и соавт., 2005).
Очевидно, при формировании
аддитивно-полигенного наследования МС с
пороговым эффектом имеется пермиссивное
действие продуктов разных генов, сцепленных с
риском МС. Так, инсулинорезистентность
могла бы быть скомпенсированной, если
бы не генетические ограничения ответа
В-клеток островков Лангерганса на
гипергликемию; гиперинсулинемия не давала бы
АГ, если бы не наличие сверхэкспрессивных
аллелей натрий-водородного насоса; МС не
влиял бы столь тромбогенно на сосудистую
стенку, если бы не дефицит продукции
адипонектина и т.д.
В этиологии МС роль выявляющих
пороговых факторов играют особенности
взаимодействия предрасположенных к нему
индивидов со средой включая
гиперкалорийное, обогащенное жирами и
легкоусвояемыми углеводами питание, злоупотребление
алкоголем, курение сигарет, хронический
стресс, гиподинамию, дефицит пищевых
антиоксидантов (И.С. Либерман, Ю.И.
Строев, СБ. Иванова, 1993). Большое
значение среди эпигенетических предпосылок
развития МС придают внутриутробной
гипотрофии и недоношенности (Д. Баркер
с соавт., 1993). Считается, что эти факторы
могут сформировать у индивида
«бережливый фенотип», предрасполагающий в катам-
незе к инсулинорезистентности, АГ и
ожирению (Д. Жаке с соавт., 2000). Так как при
МС важную роль играют цитокины, в том
числе характерные для ответа острой фазы и
хронического воспаления (Дж. Сазерленд,
Б. МакКинли, 2004), не исключено, что в
будущем обнаружатся этиологические фло-
гогенные агенты, побуждающие липоциты
их вырабатывать, например, аутоантитела
или не идентифицированные пока
латентные инфекции.
Патогенез МС
Существующие представления о патогенезе
МС укладываются в рамки трех теорий.
Наиболее старая из них — глюкоцентрическая.
В конце 80-х гг. прошлого века ей на смену
пришла липоцентрическая теория. Наконец,
в настоящее время наиболее бурно
развиваются исследования в русле липокиновои
теории МС.
Совокупность нарушений, свойственных
МС, часто наблюдается у больных с
повышенным риском заболевания СД 2 типа или ранее
диагностированным СД 2 типа и предполагает
наличие ключевого патогенетического звена,
связанного с этой болезнью. Всеми
исследователями у лиц с МС обнаружена тканевая
резистентность к усвоению глюкозы (М.Т. Мак-
Дермотт, 1998). Поэтому глюкоцентрическая
концепция МС все еще принимается многими
авторами. По глюкоцентрической гипотезе, в
основе развития как СД 2 типа, так и МС
лежит единый патологический процесс —
инсулинорезистентность периферических тканей,
следствием чего является гиперинсулинемия.
Именно инсулинорезистентность и
сопутствующая ей гиперинсулинемия связывают
все метаболические расстройства,
наблюдающиеся при МС (Ю.В. Зимин с соавт.,
1999; Н.А. Беляков, СЮ. Чубриева, 2003).
Из-за инсулинорезистентности возникает
«додиабетическая» гипергликемия, которая,
однако, способствует гликированию белков
и соответствующим дизрегуляторным и
ангиопатическим изменениям (см. гл. 9).
Захват и окисление глюкозы мышцами
снижаются, недостаточно тормозится глюконе-
огенез в печени. Вместе с тем другие органы
и ткани могут оказаться селективно более
чувствительными к тем или иным эффектам
инсулина. Например, гиперинсулинемия будет
способствовать пролиферации сосудистых
гладкомышечных клеток, дисфункции
эндотелия, гиперактивации ионообменных
механизмов, задерживающих натрий и кальций,
развитию ожирения и гиперлептинемии. Это
подключает патогенетические механизмы
формирования остальных компонентом МС.
По липоцентрической теории, ключевую
роль в развитии метаболического
синдрома играет абдоминально-висцеральная
жировая ткань. Андроидное ожирение
рассматривается как «генератор» и один из
основных компонентов МС. По П. Бъёрн-
торпу (1990), причина всех нарушений
при МС — центральный тип ожирения.
По данным Я.В. Благосклонной с соавт.
(1998), у 88% больных независимо от
степени андроидного ожирения имеет место
инсулинорезистентность, в то время как при
гиноидном ожирении она выявляется лишь
у 32 % больных, причем в основном — при
ожирении III—IV ст.
Абдоминальный жир обладает высокой
метаболической активностью, является
богатым источником СЖК, а также секретиру-
ет большое количество цитокинов, особенно
ФНО-сс и лептина, которые поступают
отсюда непосредственно в портальную
систему и печень. Адипоцйты абдоминальной
области липолитически более
чувствительны, чем другие. В абдоминальных липоцитах
гораздо больше р-адренорецепторов с более
высокой активностью. В остальных
липоцитах преобладают ос-адренорецепторы.
Важно, что Рз-адренорецепторы липоци-
тов не регулируются катехоламинами по
принципу обратной связи, и липолиз в них
особенно активизируется по мере развития
свойственной МС симпатикотонии.
Важным звеном патогенеза МС, связанным
с метаболической ролью висцерального жира
и подчеркиваемым липоцентрической
теорией, служит избыток СЖК в крови. СЖК в
крови появляются в результате освобождения
их из липоцитов с помощью гормоночувстви-
тельной липазы и в связи с работой липопро-
теиновой липазы на эндотелии капилляров
легких, сердца и ряда внутренних органов (см.
гл. 7). Инсулинорезистентность не позволяет
инсулину эффективно ингибировать липолиз.
Понятие о метаболическом синдроме 713
Избыток СЖК снижает чувствительность
печени и других тканей к инсулину
вторично как в силу поставок альтернативного
субстрата окисления, так и из-за нарушений
в пострецепторной передаче инсулинового
сигнала, обусловливая порочный круг в
патогенезе МС. При избытке СЖК в гепато-
цитах формируется избыток ацил-КоА и его
производных типа церамидов, активируется
N-концевая киназа белка предранней реакции
протоонкогена c-jun (см. т. 1). Это нарушает
работу протеинкиназы С и пострецепторное
фософрилирование тирозина в субстратах
инсулинового рецептора IRS-I и И, особенно
у носителей их мутантных аллелей. Вместе
с тем инсулиночувствительность разных
пострецепторных метаболических путей в
одной и той же клетке оказывается
различной. Белок SREB-lc (белок, связывающий
стерол-чувствительный элемент, важный
участник опосредования эффекта инсулина на
липогенез) сохраняет чувствительность к
стимулирующему действию инсулина и СЖК, и
липогенез в печени возрастает. Этому могут
способствовать мутации SREB (см. выше).
В связи с этим, СЖК в печени
увеличивают продукцию ЛПОНП и обогащают
триглицеридами другие ЛП, а продукция
ЛПВП тормозится. Это способствует сте-
атозу печени и дислипемии, что приводит
к усилению активности липопротеиновой
липазы, дальнейшему росту концентрации
СЖК и повышению активности
свертывающей системы крови (повышение
уровня фибриногена, снижение факторов
фибринолиза). Следствием этого является
гипертриглицеридемия (Н.А. Беляков,
С.Ю.Чубриева,2003).
При избытке СЖК захват глюкозы в
мышцах снижается. Этим звеньям
патогенеза МС способствуют наследственные
особенности белка-переносчика СЖК и
дефекты митохондриального окисления СЖК
в мышцах у многих лиц с СД 2 типа (см.
выше). Все это вызывает компенсаторные
гиперинсулинемию и гиперлептинемию,
которые стимулируют симпатическую
нервную систему. Инсулин усиливает
экспрессию противопереносчика натрия/водорода,
714 ALU. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Си
Г_\С\\\
"ВюоиТГ
лпонп
Артериальная
IЛПВП гипертензия
ж малые плотные v^
Тлпнп
сосуды
Глюкоза.
t t \
СЖК ^инсулин СНС
j^Lty поджелудочная
(инсулин железа
В°со2
печень
жировая
ткань
|ЛПВП
Ml
Cm t
ВюоиТГ
малые плотные
ЛПНП
Глюкоза
фибриноген у ИАП-1
Тромбофилический
синдром
Гликоген
адинопектин
ТГ (внутримышечные
включения)
Рис 133. Патогенез метаболического синдрома (по Дж.М. Ривену и соавт., 2002). А — Ключевое
звено патогенеза, согласно липоцентрической теории. ТГ — триглицериды, СНС —
симпатическая нервная система, СЖК — свободные жирные кислоты. Б — Роль липокинов и провоспали-
тельных цитокинов, согласно липокиновой теории. ЦРБ — С-реактивный белок, ИАП-1 —
ингибитор активатора плазминогена I типа. Остальные обозначения по списку сокращений
способствующего задержке натрия и
кальция и развитию АГ (рис. 133, верх).
Таким образом, по липоцентрической
теории, главным звеном МС служит избыток
жира, превышающий возможности
липоцитов по его безопасному хранению. Это
вызывает «выплеск» липидов за пределы
малых липоцитов подкожного жира и
исчерпание возможностей их пролиферации.
Нарушается дифференцировка субкутанных
липоцитов. Жир вызывает их гипертрофию,
возрастание размера клеток, а затем, по ис-
черпании резервов депонирующей функции
подкожной жировой ткани, перемещается в
липоциты абдоминальные.
Упрощенно последствия этого можно
представить следующим образом.
Установлено, что число инсулиновых рецепторов
на одном эритроците — около 40, а на ге-
патоцитах и липоцитах — до 150-200 000.
Функционирование рецептора инсулина
описано выше в гл. 8,9.
При ожирении липоциты накапливают
жир и становятся во много раз больше по
размеру. В таком случае плотность
инсулиновых рецепторов на единицу поверхности
липоцита падает. Это по принципу обратной
связи и является сигналом к увеличению
продукции инсулина В-клетками
поджелудочной железы (в особенности, при
дефектных пострецепторных механизмах).
Следовательно, чем выше степень
ожирения, тем выраженнее гиперинсулинемия
(М.Н.Балаболкин, 1998), что
подтверждается и на практике (чувство голода,
потливость). Увеличение доли висцеральных
липоцитов в силу их метаболических и
сигнальных особенностей, перечисленных
выше, создает субстратную и цитокиновую
информационную «нагрузку» нагепатоциты
и способствует развитию стеатоза печени
и дислипемии. Возникает гиперлипопро-
теидёмия IV типа (см. гл. 7), происходит
усиленный обмен эфирами холестерина
между ЛПОНП и ЛПВП при помощи
белка-транспортёра эфиров холестерина. Из-за
этого время циркуляции и концентрации
ЛПВП сокращается, липопротеидные
фракции обогащаются триглицеридами, средний
размер частиц ЛПНП становится под
действием липопротеиновой липазы меньше, а
их атерогенный потенциал — выше.
Описанные дислипемия и гипергликемия
вкупе с гиперинсулинемией оказывают ате-
рогенное действие на сосуды, способствуют
АГ. Выше, в разделе «Местные формы утраты
жировых запасов», уже охарактеризованы
различные формы липодистрофий. При этих
заболеваниях подкожный жир утрачивается
частично или полностью, и организму
приходится «перебрасывать» липиды в висцераль-
Понятие о метаболическом синдроме 715
ные липоциты, печень и мышцы. Характерно
и патогенетически демонстративно, что при
этом возникают инсулинорезистентность,
стеатоз печени, гиперлипопротеидемия IV
типа, гиперкоагуляция, избыток провоспа-
лительных цитокинов в крови, а иногда — ги-
перандрогенизм и другие проявления МС,
даже без общего ожирения. Приобретенные
липодистрофий с неполным МС
развиваются у больных ВИЧ-инфекцией, получающих
лечение антипротеазными препаратами
(Э. Дэнфорт, 2000; А. Керр с соавт., 1998).
Многие подобные синдромы
сопровождаются продукцией различных аутоантител (в
том числе — к рецепторам инсулина) и могут
быть связаны с вялотекущим аутоиммунным
панникулитом, активирующим продукцию
липокинов. Патогенез и проявления
липодистрофий наводят на мысль об особой роли
в развитии МС жира, расположенного вне
подкожных адипоцитов.
Еще до открытия широкого спектра
липокинов М. Пулио с соавт. (1994)
подчеркивали, что для скрининга МС окружность
талии — важнейший антропометрический
признак. После обнаружения эндокринных
функций жировой ткани и особенно после
открытия гипоталамо-липоцитарной ней-
роэндокринной оси и роли лептина липо-
центрическая теория патогенеза МС
трансформировалась в липокиновую теорию. С
точки зрения липокиновой теории основные
составляющие МС формируют не столько
субстратно-энергетическая роль
продуктов липоцитов, сколько информационные
воздействия на организм липоцитарных
сигнальных молекул.
Выше уже неоднократно шла речь о
продукции адипоцитами цитокинов,
регулирующих метаболизм (см. гл. 7,9).
На сегодняшний день установлено, что
адипоциты служат важным источником
ряда сигнальных молекул паракринного
и эндокринного действия, участвующих в
патогенезе МС (рис. 133 Б), таких как:
>* лептин
>* ФНО-а (кахексии)
>■ интерлейкины-1,6и8(ИЛ-1,6,8)
716 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
>* ингибитор активатора плазминогена I
типа (ИАП-1)
>• белок-стимулятор ацилирования
>• ангиотензин II
>• резистин
>- адипсин
>* белок, родственный протеину агути
>- транформирующий фактор роста-р
(ТФР-Р)
>* адипофилин
>■ адипонектин
Лептин был уже подробно
охарактеризован и неоднократно упомянут выше
как иммунонейроэндокринный
регулятор, основной сигнал для гипоталамуса о
насыщении и стимулятор динамического
действия пищи (гл. 7, гл. 9, гл. 13, см. также
т. I). В контексте патогенеза МС добавим,
что гиперлептинемия всегда сопровождает
МС. У мышей линии Цукера (db) дефектен
рецептор лептина, и наблюдается картина
неполного МС. При ожирении у человека в
большинстве случаев имеется селективная
центральная резистентность к лептину, а
его периферическое действие
сохраняется и ввиду гиперлептинемии может даже
усиливаться (Н.А. Беляков с соавт., 2005).
При ожирении выявлена связь между
концентрацией лептина в крови, уровнем
инсулина и глюкозы, установлена способность
лептина нарушать связывание инсулина с
рецепторами в адипоцитах (П. Гавел с
соавт., 1996). Гиперлептинемия вносит вклад
в формирование инсулинорезистентности,
поскольку лептин ингибирует экспрессию
инсулиновой м-РНК и секрецию инсулина
(А.Л.Пеллет с соавт., 1997). В печени
лептин снижает тирозин-фосфорилирующее
действие инсулина на IRS-1 и тормозит
связывание этого важного пострецептор-
ного посредника инсулина с белком GRB-2,
участвующим в анаболических и ростовых
эффектах инсулина (Б. Кохен с соавт.,
1996).
Хроническая гиперинсулинемия
способствует гиперлептинемии, при этом лептин
может выступать в роли сигнала липоцитов
островковым В-клеткам о недостаточной
чувствительности жировой ткани к
инсулину (X. Ларссон с соавт., 1996). Лептин
напрямую и через стимуляцию секреции
СТГ усиливает процессы ангиогенеза, что
при МС может способствовать поражению
сосудов. Лептин оказывает стимулирующее
действие на симпатоадреналовые реакции и
при МС способствует АГ (Э.У. Шек с соавт.,
1997; М. Аизава-Абе с соавт., 2000). В свою
очередь его продукцию подавляют катехо-
ламины, что при МС может нарушаться.
Антагонист лептина — гормон желудка
грелин (аналог СТГ) — повышает
чувствительность тканей к инсулину, причем его
концентрации в крови понижаются при
ожирении и синдроме поликистозных
яичников, тесно связанном с МС (К. Шофль
с соавт., 2002). Наконец, данный цитокин
может способствовать секреции липоцита-
ми других провоспалительных цитокинов,
уровень которых при МС повышается.
Интересно, что лептин вырабатывается и
за пределами жировой ткани — в желудке,
мышцах, сердце, мозге и сосудах, состояние
которых при МС закономерно нарушается,
а лептиновый рецептор экспрессирован на
клетках сосудистой стенки и в миокарде.
Гиперлептинемию при МС связывают с
патогенезом гипертрофии миокарда,
хронической сердечной недостаточности и ан-
гиопатии, свойственных многим пациентам.
Избыточная продукция лептина служит
маркером воспаления и поражения жировой
ткани и ведет к сдвигу редокс-состояния
организма в прооксидантную сторону (с
гиперпродукцией перекисных соединений).
В то же время лептин — стимулятор
окисления СЖК в митохондриях и эндогенное
липотропное вещество, снижающее риск
стеатоза печени и жировой дистрофии
мышц (П. Шульце, Й. Кратч, 2005), при
применении у больных с липодистрофиями
он помогает преодолеть инсулинорезистен-
тностъ. По-видимому, при МС некоторые
эффекты лептина усилены, тогда как другие
из-за присутствия генетических дефектов,
отмеченных выше, невозможны. В целом
у лиц с МС уровень лептина коррелирует
со степенью риска сердечно-сосудистых
заболеваний и тесно связан с количеством
абдоминального жира.
Кахексии (ФНО-а) уже рассмотрен
детально в т. I как гуморальный агент
вторичной альтерации, цитокин-регулятор преим-
мунного и иммунного ответов, эндогенный
пироген, индуктор апоптоза и некробиоза.
Как медиатор паранеопластической ка-'
хексии он рассматривается при изложении
патофизиологии онкогематологических
заболеваний (см. т. III, гл 3).
В данном томе мы касались роли кахексина
в нейрогенной анорексии-булимии (см. гл. 3)
и при нарушениях депонирования липидов
(гл. 7), а также рассмотрели его в качестве
одного из медиаторов инсулинорезистентности
(гл. 9). В отношении патогенеза МС роль
кахексина несомненна, так как его продукция
существенно возрастает при инсулиноре-
зистентных формах ожирения и связана с
абдоминальным жиром, в особенности. Этот
цитокин тормозит тирозин-протеинкиназную
активность рецептора инсулина и экспрессию
глюкозного переносчика GLUT-4 в мышцах
и самих липоцитах (Дж. Паолиссо с соавт.,
1998; Й. Чэнь с соавт., 2002). Липоциты при
повреждении и перегрузке жиром секрети-
руют кахексии вместе с другими провоспали-
тельными липокинами — ИЛ-6, ИЛ-8и ИЛ-1,
что ведет к характерному для МС нарастанию
концентрации С-реактивного белка,
экспрессии на эндотелиоцитах молекул клеточной
адгезии (компонентом которых служит С-
реактивный белок) и сдвигу соотношения
гемостатических и антигемостатических
механизмов в тромбофилическую, прокоа-
гулянтную сторону. Кахексии способствует
секреции лептина и гиперлептинемии. У лиц
с МС гиперинсулинемия и резистентность
гипоталамуса к анорексигенным сигналам не
позволяют кахексину снизить потребление
пищи, но у лиц с осложненным МС этот
цитокин способствует формированию сердечной
недостаточности. При тяжелой хронической
сердечной недостаточности он участвует в
патогенезе феномена «сердечной кахексии».
Гиперсекреция жировой тканью провоспали-
тельных цитокинов — характерная черта МС.
Понятие о метаболическом синдроме 717
Возможно, это отражает вялотекущий
воспалительный процесс в жировой ткани типа
хронического малосимптомного панникулита,
связанный с дегенерацией перегруженных
жиром липоцитов (липотоксический эффект),
возможными аутоиммунными поражениями
этих клеток или же их еще не
идентифицированной хронической инфекцией. По СП. Вай-
сбергу с соавт. (2003), в гиперлипокинемии
при МС могут быть повинны не только
липоциты, но и макрофаги жировой ткани,
количество и активность которых возрастает.
Ингибитор активатора плазминогена I
типа (ИАП-1) необходим для поддержания
цитоархитектуры рыхлой соединительной,
а значит — и жировой ткани, так как он
способствует прикреплению липоцитов к
межклеточным структурам. При МС этот
цитокин секретируется адипоцитами в избытке,
и его системное действие снижает активность
антигемостатических механизмов сосудистой
стенки и плазмы (см. т. I). Это сочетается с
продукцией адипоцитами фибриногена и
других протромботических регуляторов и может
способствовать АГ и сердечно-сосудистым
осложнениям МС, а также диабетической ан-
гиопатии. Указанные нарушения продукции
ИАП-1 вызываются кахексином.
При МС жировой тканью в избытке
секретируется белок-стимулятор ацилиро-
вания. Он способствует захвату глюкозы
липоцитами и, возможно, гепатоцитами с
превращением ее в липиды. Он стимулирует
диацил-глицерол-ацилтрансферазу и,
наоборот, ингибирует гормоночувствительную
липазу (И. Мюррей с соавт., 1999).
Ангиотензиноген и ангиотензин II также
вырабатываются в адипоцитах, причем их
продукция при МС усиливается и у
носителей МС коррелирует с АГ. К тому же
свойственная МС гиперинсулинемия нарушает
трансмембранные ионообменные
механизмы, приводит к стимуляции Na+-H+-npoTH-
вотранспорта, что тесно связано с
ускорением входа в клетки кальция и с нарастанием
рН цитоплазмы. Это вызывает повышение
чувствительности гладкомышечных клеток
сосудов к ангиотензину и другим прессор-
ным агентам с ростом периферического
718 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
сосудистого сопротивления и давления
крови. При МС этому способствует сим-
патотонический эффект лептина.
Резистин — пептид, индуцируемый при
дифференцировке липоцитов из фиброб-
ластов. В зрелых адипоцитах его экспрессия
подавляется (К. Стэппен с соавт., 2002). При
МС его концентрация повышена. Резистин
вызывает инсулинорезистентность гепато-
цитов и мышечных клеток in vivo и in vitro,
но некоторые исследователи считают его не
патогенетическим фактором МС, а лишь
«свидетелем» этого процесса (О. Уккола, 2002).
Адипсин вырабатывается жировой
тканью при липолизе и, будучи центральным
антагонистом лептина, стимулирует центр
голода в гипоталамусе. У лиц с МС при
пищевом цикле «голод-насыщение» может
нарушаться нормальное чередование
выделения липоцитами лептина и адипсина
(Я.В. Благосклонная с соавт., 1998).
Белок, родственный протеину агути,
напоминает по структуре протеин грызунов
агути, который у них служит центральным
антагонистом а-МСГ и препятствует
развитию тёмной окраски шерсти, способствуя
синтезу феомеланина, придающего волосам
рыжеватый цвет. Гомолог агути-протеина у
людей блокирует в гипоталамусе рецепторы
а-МСГ 3-го и 4-го типов, что ведет к
торможению анорексигенного лептин-зависимого
эффекта а-МСГ, обусловливает лептино-
резистентность гипоталамуса, вызывает
полифагию и ожирение (см. гл.7). При МС
продукция этого белка в липоцитах
нарастает. Тяжелое ожирение с рыжим цветом
волос зафиксировано также у людей с
мутацией, нарушающей связывание антагониста
белка-гомолога агути а-МСГ с гипоталами-
ческими рецепторами (Дж. Манне с соавт.,
1995).
Адипофилин — белок-маркер накопления
адипоцитов разной локализации, его
уровень в крови повышен при МС и болезнях,
для которых МС служит фактором риска
(Н.А. Беляков с соавт., 2005).
Транформирующий фактор роста —
Р-противовоспалительный цитокин,
ингибитор коллагеназ и эластаз, медиатор
фиброплазии (подробно об этих его
качествах см. т. 1, гл. 12). Интересно, что данный
цитокин не только препятствует гемопоэти-
ческой трансформации костного мозга, но и
служит мощным индуктором тромбоцитар-
ных факторов роста, способствуя тем самым
ангиогенезу и атерогенезу. Продукция
данного цитокина стимулируется лептином, в
частности, в Т-лимфоцитах-хелперах 1-го
типа. Таким образом, при МС имеются
условия для его избыточной продукции и
участия в патогенезе. При ожирении у
мышей с неполным МС он вырабатывается в
избытке. Любопытно, что при МС зачастую
обнаруживается ранняя контрактура Дю-
пюитрена (дигитальный склероз), которая
может предшествовать явному СД 2 типа
и другим осложнениям МС (Ю.И. Строев,
Л.П. Чурилов, М.В. Цой, 2005).
Патогенез этого нарушения может быть
связан как раз с замедлением процессов
деградации коллагена и эластина под
действием ТФР-р. Интересно, что избыточное
действие ТФРР характерно для синдрома
Марфана и близких к нему фибриллино-
патий, при которых нарушается структура
кровеносных сосудов и развивается
деструкция стенки крупных артерий,
опосредованная провоспалительными факторами
макрофагов. Не исключено, что это служит
патогенетическим связующим звеном
между системной дисплазией соединительной
ткани (фибриллинопатиями) и
метаболическим синдромом.
Адипонектин — главный кандидат на
роль «защитника организма» от развития
МС. Выше мы уже упоминали о его ре-
гуляторной роли (гл. 7, 9). Адипонектин
секретируют практически только липоциты,
причем, в липоцитах висцерального (но не
подкожного) жира его продукция по мере
роста индекса массы тела сильно снижается.
При МС его концентрация также сильно
снижается обратно пропорционально доле
абдоминального жира. Данный цитокин
служит противовоспалительным агентом,
конкурентным антагонистом Clq-фактора
комплемента и кахектина, сдерживает
активацию макрофагов, сенсибилизирует ткани
Понятие о метаболическом синдроме 719
к действию инсулина, будучи, возможно, его
главным пермиссивным союзником. Через
рецептор 1-го типа адипонектин ускоряет
окисление глюкозы и СЖК в мышцах (в
печени — через рецептор 2-го типа), подавляет
глюконеогенез. Это — антиатерогенный,
липотропный для печени и антиваскулит-
ный для сосудов фактор, член семейства
коллагеноподобных белков. Он образует
мультимеры и циркулирует в крови в
разных формах, причем наиболее
высокомолекулярные его комплексы тропны к гепатоци-
там и сосудам. В миоцитах и эндотелиоцитах
имеется лиганд адипонектина — Т-кадгерин
(Дж.П. Уайтхед с соавт., 2005; Н.А.
Беляков с соавт., 2005). Адипонектин и
кахексии — реципрокные антагонисты, причем
действие тиазолидиндионов,
способствующих восстановлению чувствительности
тканей к инсулину, опосредовано именно
через индукцию первого. Продукция
адипонектина снижается под влиянием половых
стероидов как у мужчин, так и у женщин.
У новорождённых продукция адипонектина
очень высока. Адипонектин эффективен
при экспериментальной терапии у
животных — моделей ожирения и МС. Кахексии,
ИЛ-б, а также глюкокортикоиды и кате-
холамины, присутствующие при МС в
избытке, снижают экспрессию адипонектина.
Продукция адипонектина снижена у мышей
КО, не имеющих гена AGly-фрагмента
адипонектина, и эти мыши развивают модель
МС, а трансгенное введение дефектного
гена приносит им облегчение. Более того, у
трансгенных мышей, экспрессирующих
введенный им ген AGly-фрагмента, отмечалась
гиперадипонектинемия. Удивительным
результатом гиперадипонектинемии
является буйное разрастание бурой жировой
ткани, особенно — между лопаток, а также
появление двустороннего экзофтальма
из-за накопления ретроорбитального жира
(Т.П. Комбс с соавт., 2004).
В этой связи уместно предположить,
что адипонектин или адипонектин-миме-
тические аутоантитела как раз и являются
тем легендарным «экзофтальмическим
фактором» при болезни фон Базедова, о
котором упоминал еще В.Г. Баранов (1966)!
Всем известное антиатерогенное действие
болезни фон Базедова и сам «анти-МС»
фенотип больных тиротоксикозом могут
быть связаны не только (и не столько) с
гиперпродукцией тироидных гормонов (см.
гл. 13), сколько с возможной гиперадипо-
нектинемией или адипонектиноподобным
действием некоторых аутоантител при
данном системном аутоиммунном заболевании.
Это предположение нуждается в проверке,
однако уже ясно, что именно
адипонектин — основной кандидат на роль
патогенетического комплексного терапевтического
средства при МС.
В рамках любой из теорий патогенеза
МС большое значение имеют вторичные
причинно-следственные связи и порочные
круги, связанные со взаимным влиянием
его составляющих. Наиболее вероятна такая
последовательность обменных расстройств
при МС: развитие метаболического
синдрома начинается на фоне полигенных
наследственных дефектов и алиментарно-гиподина-
мического образа жизни с абдоминального
ожирения, приводящего к формированию
липотоксического повреждения адипоци-
тов, недостаточности их депонирующей
функции и дефицита адипонектина. Это
ведет к липокиновому ответу, описанному
выше, что и способствует дальнейшему
нанизыванию патогенетических звеньев
МС, в частности, инсулинорезистентности.
Затем возникает группа нарушений, которая
состоит из гиперинсулинемии, гипертриг-
лицеридемии, гипоальфахолестеринемии,
артериальной гипертензии и СД 2 типа, что
ускоряет атерогенез и формирует
сердечнососудистые и тромботические осложнения.
Существует концепция, согласно которой
факторы риска развития атеросклероза
являются следствием единого комплекса
эндокринно-метаболических нарушений,
связующим звеном которого является
снижение чувствительности тканей к инсулину
(Н.А. Беляков, СЮ. Чубриева, 2003).
Нарушения липидного обмена при
метаболическом синдроме имеют атерогенный
характер. Большое значение в развитии
720 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
атеросклероза играет повышенное
содержание в плазме крови продуктов перекисного
окисления липидов, при этом отчетливо
прослеживается значение генетической
предрасположенности (Е.Ф. Давиденко-
ва, И.С. Либерман, Ю.И. Строев с соавт.,
1995).
Гиперинсулинемия, гипераполипопроте-
идемия-В и высокая концентрация мелких
плотных частиц ЛПНП, объединенные в
«атерогенную метаболическую триаду»,
создают почву для развития атеросклеро-
тических поражений.
Ожирение является фактором высокого
риска развития ГБ, гиперлипидемии, АСК,
ИБС и СД 2 типа и, возможно, их
объединяет (Н.А. Беляков, СЮ. Чубриева, 2003).
Инсулинорезистентность и
гиперинсулинемия, типичные для андроидного ожирения,
являются факторами риска развития СД
2 типа. При этом важную роль играют
пониженная чувствительность инсулиновых
рецепторов на тканевом уровне и снижение
ответной продукции инсулина на
гипергликемию. Образно говоря, В-клетки островков
Лангерганса из-за годами продолжающейся
гиперинсулинемии, наконец, истощаются
(их масса падает, нарушается конверсия
проинсулина в инсулин), секреция
инсулина начинает снижаться, и в соответствии с
этим гипогликемия постепенно перерастает
в пониженную толерантность к углеводам
(скрытый или латентный диабет, по
старой терминологии), а затем — и в явный
сахарный диабет вплоть до потребности в
инсулине в ряде случаев.
Взаимотношения атеросклероза и СД
существенно меняются при АГ,
сопутствующей СД: так, по нашим наблюдениям,
частота инфаркта миокарда у больных СД
с АГ была в 3,5 раза, а инсультов — в 16,5
раза выше, чем при СД без АГ (Ю.И.
Строев, 1985, 1995; Ю.И. Строев, Л.П.
Чурилов, 2004)! Анализ причин летальности
при СД показал, что ведущей причиной
явился АСК разной локализации (34,3%),
второе место заняла ГБ в сочетании с АСК-
25,7% (Ю.И. Строев,1988). Предпринятое
Ю.И. Строевым гистохимическое изучение
топографии диабетического артериоло-
склероза в тех же органах, в которых был
в свое время был изучен гипертонический
артериолосклероз (Н.Н. Аничков с соавт.,
1948; А.М. Фишберг,1925; С.С. Вайль,1940;
К.Г. Волкова, 1958), показало, что при
СД и ГБ существует и морфологическое,
и определенное топографическое сходство
артериолосклероза с той лишь разницей,
что при СД изменения микроциркулятор-
ного русла первичны по отношению к АГ
(Ю.И. Строев,1978, 1998; А.А. Кедров,
Ю.И. Строев, 1983). Таким образом, СД и
АГ — два взаимоотягощающих компонента
развернутого осложненного МС,
поражающих множество органов-мишеней: сердце
и магистральные сосуды, почки, головной
мозг, микрососудистое русло практически
всех внутренних органов.
Гиперинсулинемия приводит к падению
содержания глюкозы в инсулиннезави-
симых клетках, прежде всего, в нейронах.
Этим объясняются приступы волчьего
аппетита, тяга к сладкому и поведенческие
особенности у лиц с подобным
функциональным гиперинсулинизмом. Наши
наблюдение пациентов с ожирением показывает,
что приступы голода у них нарастают во
второй половине суток, т.е. к ночи. Это не
удивительно, так как давно установлено,
что продукция инсулина находится под
влиянием блуждающего нерва, «царство»
которого, как известно, начинается именно
в это время и достигает максимума в
ночные часы. Именно таких лиц в вечернее и
в ночное время суток посещают приступы
гипогликемии с булимией, тахикардией,
проливными потами, раздражительностью
и нередко — с повышением артериального
давления. Поведение человека,
генерирующего неконтролируемые вспышки
раздражения на голодный желудок, знакомо
каждому из нас. Не случайно к подобному
поведению склонны жертвы хронического
стресса при наличии латентного гипопиту-
итаризма (см. гл. 13).
С этой точки зрения представляется, что
АГ — следствие патофизиологического
ответа организма на гипогликемию, которая
Понятие о метаболическом синдроме 721
сама по себе является сильнейшим стрессом.
Это сопровождается повышением
продукции и приведением в действие гормонов,
способных повышать уровень глюкозы, а
это, по существу, симпато-адреналовые и
связанные с ними регуляторы (глюкокор-
тикоиды, катехоламины, глюкагон и др.).
Ведь если не считать гипогликемизиру-
ющего действия ингибитора всасывания
глюкозы соматостатина, то единственным
гормоном, понижающим уровень глюкозы
в крови, остаётся инсулин. Но
практически все антагонисты инсулина способны
повышать артериальное давление, что при
гипогликемии как раз наблюдается. Таким
образом, гиперинсулинемия запускает целый
каскад реакций, приводящих к повышению
артериального давления:
>- повышает непосредственно и через
индукцию гиперлептинемии активность
симпатической нервной системы и
концентрацию катехоламинов в крови,
что приводит к увеличению сердечного
выброса и к спазму сосудов;
>* инсулин как митогенный фактор
усиливает пролиферацию гладкомышечных
клеток резистивных сосудов, суживает
их просвет и повышает общее
периферическое сопротивление;
>- гиперинсулинемия нарушает
трансмембранные ионообменные механизмы. Как
уже указывалось, она приводит к
стимуляции №+-Н+-противотранспорта в
клеточных мембранах, что тесно связано
с ускорением входа в клетки кальция и
с ростом рН цитоплазмы. Это вызывает
повышение чувствительности
гладкомышечных клеток сосудов к норадреналину
и ангиотензину с ростом
периферического сосудистого сопротивления и
кровяного давления. Избыток инсулина
не позволяет эффективно осуществлять
прессорный натрийурез и срывает работу
главного механизма, предохраняющего
организм от перехода острой АГ в
хроническую. Это повышает реабсорбцию
натрия в почках, что приводит к развитию
гиперволемии, возрастанию содержания
Na++ и Са++ в стенках сосудов и
способствует вазоконстрикции.
Словом, при МС возникает постоянный
повод к манифестации и стабилизации
артериальной гипертензии. Однако вопрос о
ведущих механизмах развития АГ при МС
остается открытым и требует дальнейших
исследований (М.В. Шестакова, 2001).
Можно представить цепочку формирования
МС и таким образом:
ожирение + извращение липокиновой
функции жировой ткани -» инсули-
норезистентность
—»гиперинсулинемия -> гипогликемический синдром -»
симпатикотония —> артериальная
гипертензия+тромбофилия + дислипопро-
теидемия —> атеросклероз.
В данную схему мы намеренно не
включили гиперурикемию. Нередко наблюдаемая
при МС гиперурикемия может быть
обусловлена повышением продукции мочевой
кислоты в связи с меньшей эффективностью
энергетического обмена и перерасходом
такого источника пуринов, как АТФ.
Продукция пуринов явно увеличена при ожирении
(см. гл. 6). Уровни триглицеридов и мочевой
кислоты в плазме проявляют сильную
положительную корреляцию (Г. Галлер, М. Гане-
фельд, 1979). Метаболическая связь между
ними, по X. Грейлингу (1969), объясняется
тем, что как синтез жиров, так и продукция
фосфорибозил-пирофосфатов требуют
НАДФН и зависят от интенсивности
функционирования пентозного шунта. При МС
гиперэкспрессия водород-натриевого проти-
вопереносчика, стимулируемая гиперинсу-
линемией, приводит к перерасходу
макроэргов и продукции дополнительных пуринов.
При подагре выяснена защитная роль
апопротеина В и содержащих его ЛПНП
и ЛПОНП. Адсорбируясь на поверхности
кристаллов уратов, апо-В препятствует
активации этими кристаллами лейкоцитов
и медиаторных систем воспаления. Таким
образом, дислипемия при гиперурикемии
может иметь компенсаторное значение.
При МС отмечено и снижение экскреции
722 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
мочевой кислоты почками, возможно,
связанное с гиперандрогенизмом и системной
эндотелиальной дисфункцией.
При МС гиперурикемия — вторичный
диабетогенный фактор из-за аллоксано-
подобного действия уратов, и некоторыми
авторами считается независимым фактором
риска АСК (см. гл. 6). Однако вопросы о
связи гиперурикемии (подагры) с СД и
АСК все еще дискутабельны. Несмотря на
широко распространенное мнение о диабе-
тогенности и атерогенности гиперурикемии
(Г. Галлер, М. Ганефельдт, 1979), до сих
пор не установлено явной корреляции
между уровнями мочевой кислоты, инсулина и
глюкозы в крови. Гиперурикемия —
далеко не постоянный компонент МС, хотя и
включалась основоположником концепции
МС Ж.-П. Камю (1966) в его структуру.
Вопреки обилию известных диабетогенных
и атерогенных факторов при подагре, СД
и ИБС сопутствуют ей не часто, и она не
может считаться неоспоримым фактором
риска этих заболеваний. К тому же сама
мочевая кислота — эндогенный антиокси-
дант (Н.Н.Коновалова с соавт., 2001). По-
видимому, из факторов риска, связанных
с нарушениями пуринового обмена, имеет
значение не концентрация самой мочевой
кислоты, а состояние участвующего в пури-
новом обмене фермента ксантиноксидазы,
которая в определенных условиях
продуцирует избыток свободных радикалов. В
последнее время появились данные о проок-
сидантном эффекте больших концентраций
мочевой кислоты (концепция уратов как
редокс-челнока (М.Р. Хейден, С. Тиаги,
2004). Имеются также данные о
блокировании синтеза N0 эндотелием при избытке
мочевой кислоты, что может быть связано с
уровнем эндогенного блокатора нитроксид-
синтазы асимметричного диметиларгинина.
Все это, конечно же, укрепляет
представления о гиперурикемии, как важном
компоненте МС. По-новому в последнее время
оценивается и роль хронического стресса в
этиологии МС. По Г.Т. Крусосу (2000) при
стрессе кортиколиберин, кортизол и
симпатические нервные сигналы способствуют
подавлению продукции соматолиберина
и гормона роста. Дефицит гормона роста
растормаживает гипертрофию и
гиперплазию висцеральных адипоцитов, причем
кортизол способствует переходу липидов в
висцеральный жир. Если при этом наступает
дистресс и снижается продукция половых
стероидов, то исчезает их сдерживающий
эффект на развитие висцерального
ожирения. Не случайно гиперкортицизм, как
при болезни Иценко-Кушинга, так и при
других заболеваниях, связанных с
перенапряженной и нарушенной стрессорной
адаптацией (юношеский диспитуитаризм)
имеет так много общих с классическим МС
признаков (см. гл. 13 выше). Эксперименты
Г.Т. Крусоса на макаках-циномольгусах
подтвердили, что хронический стресс с
исходом в стадию истощения моделирует
инсулинорезистентность и висцеральное
ожирение, характерные для МС. Это
позволяет включить МС в число «болезней
цивилизации», то есть заболеваний, для которых,
по Г. Селье, роль главного фактора риска
играет хронический стресс, превышающий
адаптационные ресурсы гипоталамо-гипо-
физарно-надпочечниковой системы.
Значение МС состоит в том, что при нем
пациенты даже с легкой непереносимостью
глюкозы подвержены повышенному риску
атеросклеротических заболеваний,
обусловленных сопутствующими нарушениями
артериального давления и обмена липидов
(МакДермотт М.Т., 1998). Коррекцию этих
нарушений следует производить
патогенетически, а именно — добиваясь снижения
резистентности к инсулину, нормализации
количества и сигнальной функции жировой
ткани в организме.
В данной главе учебника мы ставили
задачу познакомить читателя с этиологией и
патогенезом МС.
Вышедшие в последние годы
всеобъемлющие руководства и обзоры, посвященные МС
и ожирению (Н.А. Беляков, В. А. Мазуров,
2003; Н. А. Беляков и соавт., 2005, Ю.И.
Строев и соавт., 2007), содержат подробное
описание клиники, диагностики, лечения и
профилактики МС и его осложнений.
ПОСЛЕСЛОВИЕ
Главной темой данной книги, как и
предыдущего тома серии «Общая
патофизиология с основами иммунопатологии»,
остаётся повреждение и ответ на него. Метаболизм
как целостная система химических реакций,
в широком смысле, и представляет собой
такой генетически детерминированный ответ
организма на воздействие его химического
окружения. Имея наследственную основу,
метаболические процессы модифицируются
в соответствии с индивидуальным опытом
и экологическими факторами, например,
характером питания.
Основная особенность любых
компенсаторно-приспособительных реакций
организма включая и метаболические реакции
состоит в том, что они несовершенны.
Поскольку оптимальность их ограничена, они и
сами способны порождать вред. Вот почему
в известном смысле и данная, и предыдущая
книги серии посвящены парадоксам защиты.
Патофизиологическая интерпретация
конкретных биохимических процессов может,
таким образом, быть неоднозначной.
В связи с этим, на страницах данной
книги были представлены, по мере
возможности, различные концепции, отражающие
альтернативные точки зрения по поводу
патофизиологического смысла тех или иных
патохимических явлений.
Тяжкий путь через все предыдущие 722
страниц окончен, и читатель вот-вот
перевернёт последнюю из них. Благодаря
читателя за внимание, авторы подчёркивают,
что у них было благое намерение — в ходе
изложения, по возможности, читателю не
наскучить.
Авторы — специалисты в области
патофизиологии, и сознают пределы своей
компетентности. Но, метаболические
заболевания — широкая, междисциплинарная
проблема, актуальная для многих областей
медицины и требующая учёта данных
генетики, иммунологии, экологии и гигиены,
а не только биохимии или патологии. По
мере своих сил, в ходе изложения
патофизиологии обмена веществ мы стремились
знакомить читателя с её проблемами
широко, не игнорируя и тех аспектов, которые
724 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
изучаются студентами-медиками на других
кафедрах.
Не претендуя на всеобъемлющее
изложение вопросов патофизиологии обмена
веществ, мы попытались описать, в первую
очередь, те аспекты патохимии, которые
обычно не находят достаточно подробного
освещения в курсе частной
патофизиологии.
На страницах данной книги
неоднократно возникает вопрос о нормальных либо
изменённых концентрациях метаболитов
и содержании регуляторов в различных
биологических жидкостях. Для удобства
читателя данные о нормах
биохимических и эндокринологических показателей
вынесены в отдельное приложение 1-Аи
представлены в конце книги. При
знакомстве с приложением следует иметь в виду
относительный и ситуативный характер тех
или иных параметров функционирования
организма и взаимозависимость различных
нормативов.
В ходе изложения упоминались
концепции и факты, созданные или открытые
выдающимися химиками и биохимиками
прошлого. Для справки приводим приложение
2-В, где читатель найдёт данные о химиках
нобелевских лауреатах, удостоенных премии
за разработку вопросов, прямо или косвенно
повлиявших на становление представлений
о метаболизме и его нарушениях.
Йо многих случаях, физические рамки
учебника и полиграфические ограничения
не позволили в достаточной степени
насытить текст иллюстрациями, которые бы
облегчали его восприятие. С целью
компенсации этого недостатка авторы рекомендуют
обращаться к знаменитой схеме Д. Никол-
сона «Метаболические пути», имеющейся в
свободном доступе в сети Интернет (http://
www.iubmb-nicholson.org/chart.html).
В отличие от I тома данной серии в
данном издании мы сочли возможным
включить в список литературы лишь основные
источники, а читатель, интересующийся
более подробными библиографическими
данными, может почерпнуть их в списке
литературы тома I, поскольку многие ссылки
перекрываются.
Так как метаболизм регулируется, в
первую очередь, гормонами, в данном томе
были детально рассмотрены вопросы
патофизиологии эндокринной системы, а
расстройства функций многих других органов
и систем затронуты лишь в иллюстративном
плане, при обсуждении типовых нарушений
метаболизма.
В третьем томе данной серии —
«Механизмы развития болезней и синдромов» -
специально проанализированы этиология
и патогенез наиболее распространённых и
практически важных заболеваний,
поражающих другие органы и системы организма,
причём авторы оставляют за собой право,
по необходимости, возвращаться при этом
к описанию метаболических проявлений
данных форм патологии.
Приложение 1
Таблица А
Некоторые нормальные метаболические и эндокринологические показатели
(по К. Айзелбахеру и соавт., 1994; NBME, 1991; РДж. Элину и соавт. 1988; РЭ. Берману и соавт. 7 994;
ЮЛ. Лея, 1986; Ив. Пенчеву и соавт.; 1962, Ю.И. Строеву, Л.П. Чурилову, 2006;
докладу объединённого комитета экспертов ФАО-ВОЗ; 1974)
Параметр
Где определяется
Интервал нормы
Единицы
измерения
Биохимические и эндокринологические лабораторные параметры:
Адреналин
Ацетоуксусная к-та
Азот остаточный
Азот
АКТГ
Аланинаминотрансфераза
Альдостерон
а1-антитрипсин
Ангиотензин II
Андростендион
Аспартатаминотрансфераза
Аммоний
Аммоний
Аскорбиновая к-та
Билирубин общий
Билирубин прямой
Билирубин непрямой
Бикарбонат
Белок
Белок общий
Альбумин
Глобулины
Вазопрессин
Ванилил-миндальная к-та
Витамин А
Гастрин
Гемоглобин
Гемоглобин гликозилированный
Гематокрит
17-гидроксипрогестерон
моча
плазма
кровь/сыворотка, по
Кьельдалю
фекалии
плазма, 8-00
кровь
плазма, 8-00
сыворотка
плазма, 8-00
плазма
кровь
цельная венозная кровь
моча
сыворотка
сыворотка
сыворотка
сыворотка
сыворотка
моча
сыворотка
сыворотка
сыворотка
плазма при обычном
питьевом режиме
моча
сыворотка
сыворотка
кровь
кровь
кровь
плазма
ДО 275
до 100
0,2-4,5
1,2-1,6
До 18
0-0,58
до 220
0,8-2,1
10-30
Ж 3,5-7; М 3-5
0-0,58
$7-65
30-50
23-57
5,1-17
1,7-5,1
3,4-12
21-28
ДО 0,1
60-78
35-55
23-35
1,5-5,6
до 40
0,7-3,5
40-200
135-175 М; 120 160Ж
до 6% от общего НЬ
41-53 М; 36-46 Ж
М 0,2-9; Ж
(фолликулярная фаза) 0,6-3; лютеаль-
ная 1,5-10,6
нМ/сутки
мкМ/л
г/л
г/сутки
пМ/л
мкКат/л
пМ/л
г/л
нМ/л
нМ/л
мкКат/л
мкМ/л
мМ/сутки
мкМ/л
мкМ/л
мкМ/л
мкм/л
мМ/л
г/сутки
г/л
г/л
г/л
пМ/л
мкМ/сутки
мкМ/л
нг/л
г/л
%
объёмные %
нМ/л
726 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
Параметр
Глюкагон
Глюкоза натощак
Гаптоглобин
Дигидроэпиандростерон
11 -дезоксикортизол («S»)
Железо
Железо-связывающая способность,
общая (и насыщение)
Жир
Инсулин
Йод радиоактивный, захват
Кальций
Кальций ионизированный
Кальций
| Кальцитонин
| Кальцитриол
Калий
|Каротиноиды
| Катехоламины
| Кислотность титруемая
| Кортизол в 8-00
| Креатин
| Креатинин
| Креатинин
| Креатинфосфокиназа
| Лактатдегидрогеназа
| ЛПНП (холестерин)
ЛГ
| Магний
| Медь
| Мочевинный азот
| Молочная кислота
| Мочевая кислота
| Метилмалоновая кислота
| Натрий
Где определяется
плазма
сыворотка
сыворотка
плазма
плазма
сыворотка
сыворотка
стул, при наличии не
менее 50 г жира в суточном
рационе
сыворотка или плазма
натощак
за 24 ч щитовидной
железой
плазма
плазма
моча при потреблении
200 мг/день
плазма
плазма
сыворотка
сыворотка
моча, свободные
моча
плазма
моча
моча
сыворотка
сыворотка
сыворотка, метод Вакера
плазма
плазма
сыворотка
сыворотка
сыворотка
венозная плазма
сыворотка
моча
сыворотка
Интервал нормы
50-100
3,8-6,1 (в пробе на
толерантность до 7,8)
0,83-2,67
7-31
до 30
14-26М; 11-21 Ж
31-85 (насыщение
0,2-0,45)
ДО 6,0
43-186
5-30
2,2-2,6
1,1-1,4
ДО 3,8
до 50
40-160
3,5-5,0
0,9-5,6
до 590
20-40
138-635
М: до 380; Ж: до 760
8,8-14
53-106
Ж 0,17-1,17; М 0,42-1,50
60-100
3,36-4,11
Ж: (кроме овуляции)
5-25; овуляторный пик
25-100; постменопауза
выше 50. М: 5-20
0,8-1,2
11-22
1,2-3,0
0,6-1,7
0,18-0,48
0-3,5
135-147
Единицы
измерения |
нг/л
мМ/л
г/л
нМ/л |
нМ/л |
мкМ/л |
мкМ/л
г/сутки
пМ/л
% от дозы
мМ/л |
мМ/л
мМ/сутки
нг/л
пМ/л |
мМ/л |
мкМ/л |
нМ/сутки |
мЭкв/сутки
нМ/л
пМ/сутки
мМ/сутки
мкМ/л
мккат/л
между нар. Ед/л
мМ/л
мЕд/л
мМ/л
мкМ/л
мМ/л
мМ/л
мМ/л
мг/сутки
мМ/л
Приложения 727
Параметр
Норадреналин
Р-оксимасляная к-та
Окситоцин
Осмол ял ьн ость
Основной обмен
Прогестерон
Пиру ват
Пролактин
рн
р02
рС02
Свинец
Своб. жирные кислоты
СТГ
Тестостерон
Тироксин
Трийодтиронин
Трийодтиронин, абсорбция
Трийодтиронин реверсивный
ТТГ
Триглицериды
Ферритин
Фосфатаза кислая
Фосфатаза щелочная
Фосфор неорг.
Фолиевая кислота
ФСГ
Фибриноген
Хлорид-анион
Холестерин
Хорионический гонадотропин
Где определяется
плазма (ЭДТА, метабисуль-
фат)
плазма
плазма
плазма
при массе тела М66/Ж56
кг
плазма
венозная плазма
сыворотка
артериальная кровь
артериальная кровь
артериальная кровь
сыворотка
плазма
плазма после 100 г
глюкозы, принятой через рот.
плазма
сыворотка
плазма
на синтетической смоле
плазма
сыворотка, плазма
сыворотка
сыворотка
сыворотка
сыворотка
сыворотка
плазма/эритроциты
плазма
цитратная плазма
сыворотка
плазма
плазма, р-субъединица
Интервал нормы
104-548
до 300
1-4, овуляторный пик у
Ж 4-8
285-295
М: 1725/Ж: 1487
Ж: лютеальный пик выше
16. М, а также
неполовозрелые девочки, Ж в
преовуляторную фазу и в
постменопаузе до 6
60-170
2-15
7,35-7,45
75-105
35-45
ДО 1,0
до 180
ДО 5
Ж: до 3,5; М: 10-35; Дети:
0,17-0,7
64-155
U-2,9
25-35
0,15-0,61
0,4-4,0
оптимум 0,5-2,0
0,4-1,81
15-400 М; 10-200 Ж
0-0,90
0,5-2,0
1,0-1,5
3-25/100-425
Ж (кроме дня овуляции), М
5-20; Ж (овуляция) 12-30;
Ж(постменопауза) 12-30;
Дети: до 5
2-4
98-106
5,2-6,18
М и небеременные Ж: до
3
Единицы ]
измерения
пг/мл
мкМ/л
пМ/л
мОсм/кг
Ккал/сутки
нМ/л
мкМ/л
мкг/л
усл. Ед.
ммНд
ммНд
мкМ/л
мг/л
мкг/л
нМ/л
нМ/л
нМ/л
% ОТ ДОЗЫ
нМ/л
международных Ед/л
мМ/л
мкг/л
нкат/л
мккат/л
мМ/л
мкг/л
мЕд/л
г/л
мМ/л
мМ/л
мЕД/л
728 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
Параметр
Церулоплазмин
Цианкобаламин (В12)
Цинк
Эстрадиол
SB (стандартный бикарбонат)
BE или BD (дефицит или избыток
оснований)
ВВ (сумма оснований всех буферных
систем крови)
АВ (истинный бикарбонат крови)
Где определяется
сыворотка
сыворотка
сыворотка
плазма
микрометодом Аструпа
микрометодом Аструпа
микрометодом Аструпа
микрометодом Аструпа
Интервал нормы
270-370
74-516
11,5-18,5
Ж 70-220, макс, при
овуляции; М до 180
22-27
от -2,3 до +2,3
40-60
19-25
Единицы
измерения
мг/л 1
пМ/л
мкМ/л
пМ/л
мМ/л
мЭкв/л
мМ/л
мМ/л
Приложение 2
Таблица В
Нобелевские премии по химии, связанные с открытиями,
существенными для медицины и физиологии
Год
1901
1902
1903
1907
1909
1915
1926
1927
1928
1929
1930
1932
1937
1938
1939
1943
1945
1946
1947
1948
1953
1954
1955
1956
| 1957
Лауреаты
Я.Х. Вант-Гофф
Г.Э. Фишер
С. А. Аррениус
Э. Бюхнер
В. Оствальд
P.M. Вильшедтер
Т. Сведберг
Х.О. Виланд
А.О.Р. Виндаус
А. Харден, Х.К.А.С. фон Эйлер-Хёль-
пин
X. Фишер
И.Лэнгмюр
У.Н. Хеворс, П. Каррер
Р. Кун
А.Ф.Й. Бутенандт, Л. Ружичка
Д. де Хевеши
А.И. Виртанен
У.М. Стэнли, Дж.Х. Нортроп, Дж.Б.
Самнер
Р. Робинсон
А.В.К. Тизелиус
X. Стаудингер
Л.К. Полинг
В.ДюВиньо
С.Н. Хиншелвуд, Н.Н. Семёнов
А.Р.Тодд
Предмет(ы) исследований
Осмотическое давление растворов
Синтез Сахаров и нуклеотидов. Специфичность ферментов.
Принцип комплементарности в субстрат-ферментном
взаимодействии. Полипептидная природа белков |
Теория электролитической диссоциации |
Гликолиз. Спиртовое брожение в бесклеточных системах |
Фундаментальные принципы катализа и химического
равновесия. Доказательство каталитической функции ферментов |
Химия растительных пигментов и выделение хлорофилла |
Разделение молекул центрифугированием |
Жёлчные кислоты и их производные. Показал активацию
водорода в реакциях дегидрирования
Холестерин и витамин D
Ферментативные превращения Сахаров
Синтез тема, химия геминовых пигментов и хлорофилла
Поверхностные свойства амфифильных липидов в растворах
Структура и химия моносахаридов, витамина С, витамина А,
каротиноидов, витамина В2, флавинов
Химия каротиноидов и витаминов, выделение рибофлавина, его
роль во флавиновых ферментах, исследования витаминов А и В
Структура половых стероидов. Структура высших терпенов и
полиметиленов включая половые феромоны млекопитающих.
Биологическая роль изопреноидов
Метод меченых атомов
Консервация аскорбигена в растительных кормах
Получение ферментов и вирусных белков в кристаллической
форме
Установление структуры и синтез алкалоидов, в частности —
морфина
Электрофорез и адсорбционный анализ, установление состава
белков сыворотки крови
Химия макромолекул
Вторичная и третичная структура белков
Установление структуры витамина Н и нонапептидных гормонов,
первый синтез пептидных гормонов in vitro
Цепные свобод но радикальные реакции
Структура нуклеотидов и нуклеотид-содержащие коферменты
730 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
Год
1958
1961
1962
1964
1968
1969
1970
1972
1975
1977
1978
1980
1982
1988
1989
1991
1993
1997
2001
20Q2
2003
2004
2006
Лауреаты
Ф.Сэнгер
М. Кальвин
М.Ф. Перутц, Дж.К. Кендрью
Д. К. Ходжкин
Л. Онзагер
Д.Г.Р. Бартон, О. Хассель
Л.Ф.Лелуар
К.Ф. Анфинсен, С. Мур, А. Стайн
Дж.У. Корнфорт, В. Прелог
И.Р. Пригожий
П.Д. Митчелл
П. Берг, У. Джильберт, Ф. Сэнгер
А. Клуг
Й. Дизенхофер, Р. Хубер X. Михель
СОлтменД.Р.Чек
P.P. Эрнст
К.Б. Мюллис, М. Смит
П.Д. Бойер, Р.Дж. Уолкер, Й. Скау
УС. Ноулз, Р. Ноёри, К.Б. Шарплесс
Дж.Б. Фенн, К. Танака, К. Вютрих
П. Эгр, Р. Мак-Киннон
А. Чехановер, А. Гершко, И. Роуз
РД. Корнберг
Предмет(ы) исследований
Установление первичной структуры белков, в частности,
инсулина |
Усвоение С02 растениями, роль фосфоглицериновой кислоты
Структура гемоглобина |
Строение витамина В12
Реципрокные соотношения в термодинамике необратимых
процессов |
Концепция конформации макромолекул
Открытие роли уридиновых нуклеотидов в биосинтезе
углеводов. Демонстрация окисления жирных кислот в бесклеточной
системе
Строение и каталитическая активность рибонуклеазы. Связь
третичной структуры, конформации и функции белков
Стереохимия ферментативных реакций, железосодержащие
ферменты, биосинтез холестерина
Термодинамика неравновесных систем, теория диссипативных
структур
Хемиосмотический механизм окислительного фосфорилирова-
ния в митохондриях
Рекомбинантные ДНК, установление нуклеотидной
последовательности ДНК
Электронно-микроскопический кристаллографический анализ,
нуклеосомное строение нуклеопротеидов хроматина. Роль
гистонов
Пространственная структура фотосинтезирующих центров
РНК как катализатор (рибозимы)
Высокоразрешающая спектроскопия ядерного магнитного
резонанса
Полимеразная цепная реакция и олигонуклеотидный
сайт-направленный мутагенез
Ферментативный механизм синтеза АТФ и открытие K+-Na+-
АТФазы
Хиральный катализ, в том числе — для избирательного
синтеза оптических изомеров лекарств и биологически активных
молекул
Методы мягкой десорбции-ионизации в масс-спектрометрии
и спектроскопии ЯМР для структурного анализа белков в
растворах
Открытие аквапоринов и установление структуры калиевых
каналов в биомембранах
Убиквитин-опосредованная деградация белков в клетке
Молекулярный механизм транскрипции в эукариотических
клетках
СПИСОК ОСНОВНОЙ ЛИТЕРАТУРЫ
На русском языке и языках с кириллической графикой:
1. Абдуллаев Н.Х. // Пробл. эндокринол. и гормонотерапии.-1965.-К1.-с.Ю2.
2. Авроров П.П. // Обмен веществ и развитие энергии в организме при полном голодании. Дисс. СПб, 1900.
3. Адо А.Д., Новицкий В.В. (Ред.) Патологическая физиология. Томск, 1994, 466 с.
4. Алёшин Б.В. Гистофизиология гипоталамо-гипофизарной системы.М.:Медицина, 1971.
5. Алёшин Б.В. Система APUD (система ПОДАП). В кн.: Актуальные вопросы современной нейроэн-
докринологии. Нейробиологические аспекты. М.:Наука, 1981.-c.6-24.
6. Алмазов В.А., Благосклонная Я.В., Шляхто Е.В., Красильникова Е.И. Метаболический
сердечно-сосудистый синдром.-СПб.: СПбГМУ, 1999. - 202 с.
7. Альбицкий П.М. Односторонность и ошибочность современного физиологического учения о значении
продуктов обмена для организма и о деятельности выделительных органов. Необходимость нового
учения и основные начала его. Пгд: 1918.
8. Американская диабетическая ассоциация. Диабет от А до Я. Необходимые Вам знания о сахарном
диабете - в простом изложении. Пер с англ./ Под ред. А.С. Фокина, А.А. Фокина, Л.П. Чурилова, Ю.И.
Строева. СПб: ЭЛБИ, 2003. - 206 с.
9. Андреева М.П., Раскин A.M. Заболевания надпочечников. В кн.: Многотомное рук-во по внутренним
болезням. Т.7.-С.423-499.
10. Анисимов В.Н., Рейтер Р.Дж.-Вопр.онкол.-1990.-т.36.-К 3.-С.259-268.
11. Анисимов В.Н., Кветной И.М., Комаров Ф.И., Малиновская Н.К., Рапопорт СИ. Мелатонин в
физиологии и патологии желудочно-кишечного тракта. —М.: Советский спорт, 2000.
12. Аничков Н.Н. О патологических процессах, связанных с отложением в организме двоякопреломляющих
жиров. Труды О-ва Русск. Врачей, 1912.- с. 90.
13. Аничков Н.Н. Учебник патологической физиологии. 4-е изд., Л.: Биомедгиз, 1938.-432 с.
14. Аничков Н.Н., Волкова К.Г., Захарьевская М.А. Патологическая анатомия гипертонической болезни.
/ Труды IV сессии АМН СССР. - М.: Изд. АМН СССР, 1948. - С. 18-29.
15. Арешкина Л.Я. Витамин В-12 в животном организме. М.:Наука, 1976.-112 с.
16. Бабичев В.Н. Нейроэндокринология пола.-М.: Наука.-1981
17. Бадалян Л.О. Наследственные болезни. Ташкент: Медицина УзССР,1980.- 414 с.
18. Бадалян Л.О., Таболин В.А., Вельтищев Ю.Е. Наследственные болезни у детей. М.:Медицина,1971,
366 с.
19. Балаболкин М.И. // Акромегалия.-М.:Медицина.-1974.-128 с.
20. Балаболкин М.И. // Эндокринология.- 1985.-Т. 31.- N 5.- с. 48-55.
21. Балаболкин М.И., Мамаева Г.Г., Кравченко Т.В. Полиморфизм генов у больных с артериальной ги-
пертензией и сахарным диабетом инсулиннезависимого типа. / Актуальные проблемы современной
эндокринологии. Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С. 19.
22. Баранов В.Г. (ред.) Болезни эндокринной системы. Многотомное рук-во по внутренним болезням.
Л.: Медицина, т.7,-676 с.
23. Баранов В.Г. (Ред.). Руководство по клинической эндокринологии.-Л.:Медицина, 1977.
24. Баранов В.Г. (ред.) Экспериментальный сахарный диабет: роль в клинической диабетологии. Л.:Наука,
1983.-240 с.
25. Баранов В.Г., Потин В.В. Болезни щитовидной железы. / Руководство по клинической эндокринологии
под ред. акад. АМН СССР В.Г. Баранова. Л.: «Медицина», Л.О., 1977. - С. 348-441.
26. Барановский А.Ю., Ворохобина Н.В. ( ред.) Ожирение: клинические очерки. СПб.: Невский диалект,
2007.- 240 с.
27. Белицер В.А., Цыбакова Е.Т. // Биохимия.-1939.-T.4.-N 5.-С.516.
28. Бельгов А.Ю.,Строев Ю.И. Гормональный профиль юношеского ожирения. В сб.: Избранные вопросы
внутренней патологии подростков, (ред. Л.И. Левина), СПб: ППМИ, 1993.-c.99-103.
29. Беляков Н.А.(ред.), «Альтернативная медицина».-Архангельск-СПб: СЗКИ.-1994.-456 с.
30. Беляков Н.А., Мазуров В.И. (ред) Ожирение. Руководство для врачей.- СПб.: Издательский дом
СПбМАПО,2003,520с.
31. Беляков Н.А., Чубриева СЮ. Метаболический синдром. / Ожирение/ Под ред. Н.А. Белякова и В.И.
Мазурова. - СПб.: Издательский дом СПбМАПО, 2003. - С. 96-119.
732 А.Ш. Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
32. Беляков Н.А., Чубриева С.Ю., Глухов Н.В., Чубкин И.В. Роль лептина в патогенезе синдрома по-
ликистозных яичников и метаболического синдрома у женщин // Клиническая патофизиология,
2005.-T.2.-N1.-с. 73-80.
33. Бернар К.,Лекции по экспериментальной патологии.-М.,Л.:Биомедгиз,1937.-512 с.
34. Билибин А.Ф.Врач и больной. // Вестн. АМН CCCP.-1967.-N9.-C.55-61.
35. Блага Ф. // Арх. namn.-1963.-N 1.-C.13.
36. Благосклонная Я.В., Красильникова Е.И., Бабенко Ю.А. Ожирение и его потенциальная роль в
развитии метаболического синдрома. // Новые Санкт-Петербургские врачебные ведомости.— 1998. — №4.
- С. 43-48.
37. Богданова Т.С., Подвигина Т.Т. Кортикостероиды и энергетический обмен мозга. В сб.:. Гипофи-
зарно-адреналовая система и мозг.Л.:Наука, 107-134.
38. Богомолец А. А, К физиологии надпочечных желез, Супраренотоксины // Русский врач, 1909, т. 29.—С.
972-978.
39. Богомолец А. А. Аутолизотерапия (специфическая стимулирующая цито-токсинотерапия) //
Врачебное дело.—1936.—№ 1.—С. 1—6.
40. Богомолец А.А. (Ред.) Руководство по патологической физиологии . Тт. 1-3. М.: Биомедгиз, 1935-
1936.
41. Богомолец А.А. Избранные труды.-Киев: Изд-во АН УССР, т. 1-3,1958.
42. Богомолец А.А. К вопросу о микроскопическом строении и физиологическом значении надпочечных
желёз в здоровом и больном организме. Дисс. СПб., 1909
43. Богомолец А.А. К вопросу о микроскопическом строении надпочечников в связи с их отделительной
деятельностью. Одесса., 1905.
44. Богомолец А.А. Патология эндокринных желёз. В кн. Рук-во по патологической физиологии (ред.
А.А. Богомолец). Т. И. Биомедгиз: 1937.-С.419-532.
45. Бойд У. Основы иммунологии. М.:Мир, 1969,647 с.
46. Борисова Е.А. Кровоснабжение комплекса гипоталамус-гипофиз. В кн.: Нейроэндокринология.
Ч.1,Кн. 1-2; СПб: Изд-во РАН, 1994. С. 212-229.
47. Боткин СП. Курс клиники внутренних болезней и клинические лекции в 2 томах.-М.:Медгиз, 1950.
48. Браунштейн А.Е. // Вопр. питания.-1957.-t.16.-N 5.-С.18.
49. Браунштейн А.Е. В кн.: Пробл. Советской физиол. биохим. и фармакол. Т.2.-М.Д949.-С.707.
50. Брин Б.М. Учение об авитаминозах. В кн. Рук-во по патологической физиологии (ред. А.А.
Богомолец). Т.Н. Биомедгиз: 1937.-С.301-354.
51. Брызгалина СМ., Каширина Е.П., Королева Ю.Б. Проблемы в терапии остеопороза. / Тезисы Всеросс
научно-практ. конф. «Клиническая эндокринология — достижения и перспективы», посвящ. 80-летию
со дня рожд. Засл. деят. науки РСФСР, проф. Д.Я. Шурыгина. - СПб., 2003. — С 153-154.
52. Быков В.Л. Цитология и общая гистология. Функциональная морфология клеток и тканей человека.
СПб.: СОТИС, 1998,520 с.
53. Бутрова С.А. Метаболический синдром и роль абдоминального ожирения в его развитии (вопросы
патогенеза, проявления, современные подходы к лечению). / Ожирение. Метаболический синдром.
Сахарный диабет 2 типа. Под ред. академика РАМН И.И. Дедова. - М., 2000. - С. 38-43.
54. Вайль С.С. Изменения сосудов при гипертонической болезни. - Л., 1940.
55. Вапцаров И. и соавт. Диспротеинемии. София: Медицина и физкултура.-1978.-336 с.
56. Ёельтищев Ю.Е. Наследственные аномалии обмена витаминов. Сер. Итоги науки и техники. Генетика,
М.: ВИНИТИ, 1980.-т.5.-с. 123-148.
57. Вельтищев Ю.Е., Темин П.А. (ред.) Наследственные болезни нервной системы.— М.: Медицина,
1998.-496 с.
58. Вернадский В.И., Химическое строение биосферы Земли и ее окружение. Л.: Наука, 1965.-374 с.
59. Весёлкин П.Н., Ильин B.C. Патологическая физиология обмена энергии. В кн. Многотомное рук - во
по патологической физиологии. Т. II. М.:Медицина, 1966.-С.203-234.
60. Виленский Л.И. Клиника и терапия болезней расстройств питания. Иваново.-1947.-4 с
61. Винер Н. Кибернетика. М.: Советское радио.,1958.
62. Вирхов Р. Патология, основанная на теории ячеек (целлюлярная патология) в применении к
микроскопической анатомии нормальных и ненормальных тканей.-М., 1859,-472 с.
63. Вихерт A.M. и соавт. Кальциноз артерий. М.: Медицина, 1970.-152 с.
64. Войткевич А.А., Соболева Э.Л. Промежуточная доля гипофиза. Л.: Медицина, 1968,144 с.
Литература 733
65. Волкова К.Г. Артериальная гипертензия как фактор, усиливающий развитие атеросклероза. / Труды
XIV Всес. съезда терапевтов. — М., 1958. - С. 45-49.
66. Воловик А.Б. Алиментарная дистрофия у детей. В сб.: Работы ленинградских врачей за годы
Отечественной войны.-1943.-вып.5.-с. 15-21.
67. Воронин В.В. Руководство по патологической физиологии.-Тбилиси: Грузмедгиз, 1947-1948, ч.
1-2.-304с.
68. Воронцов И.М.,Кельмансон И.А., Цинзерлинг А.В. Синдром внезапной смерти грудных детей., СПб:
Изд-во специальной лит-ры, 1995,220 с.
69. Всемирная Организация Здравоохранения // Энергетические и белковые потребности. Доклад
специального объединённого комитета ФАО/ВОЗ Серия техн.докл.- N 522.- Женева, 1974.
70. Вундер П. А. Эндокринология пола.-М.: Наука, 1980.
71. Гаврилов Л.А., Гаврилова Н.С. Биология продолжительности жизни. М.: Наука, 1991.-280 с.
72. Гален Клавдий. О назначении частей человеческого тела. М., 1971.
73. Галл ер Г., Ганефельд М., Яросс В. Нарушения липидного обмена. Диагностика, клиника, терапия.
М.: Медицина, 1979.-327 с.
74. Ганнушкин П.Б. Клиника психопатий, их статика, динамика и систематика. М.,1933.
75. Гаршин В.Г. Туберкулёз при алиментарной дистрофии, сб.: Работы ленинградских врачей за годы
Отечественной войны.- 1943.-вып.5.-с. 121-127.
76. Гелынтейн Э.М. Некоторые итоги изучения клиники алиментарного истощения.В сб.: Труды
эвакогоспиталя ФЭП N 50,1943.- вып. 7.-С.2-25.
77. Генес B.C., Генес С.Г. // Патол. физиол. эксперим. терап.- 1993.- N 10.- с.253-304
78. Геодакян В. А. Эволюция логики детерминации полов и долголетие.// Природа.- 1983.-N 1 .-с.70-80.
79. Гераклит Эфесский // В кн.: Антология мировой философии в 4-х томах. М.:Мысль-т. 1 .-с.275-280.
80. Герасимов Г.А., Мельниченко Г.А., Фадеев В.В. Мифы отечественной тиреоидологии и
аутоиммунный тиреоидит. CONSILIUM-MEDICUM. [serial online] 2001 [cited 2002 Feb 10]; 3(11): [4 screens].
Available from: URL: www/media/consilium/01_ll/525.shtml.
81. Гертнер Д. Болезни костей и нарушения минерального обмена у детей. / Эндокринология. Под ред. Н.
Лавина. Пер. с англ. - М:Практика, 1999. — С. 480-516.
82. Гинецинский А.Г. Физиологические механизмы водно-солевого равновесия. М.;Л.: Наука.-1964.-
428с.
83. Гиппократ. Избранные книги.-М.:Биомедгиз, 1936.-511 с.
84. Гогин Е.Е. Гипертоническая болезнь. - М., 1997. - 400 с.
85. Голубева И.В. Гермафродитизм. Клиника, диагностика, лечение.-М.: Медицина.-1980.
86. Гольдгабер Д.Я. Молекулярные основы болезни Альцгеймера. Автореф. дисс. к.б.н. — М.: МГУ, 2003.
-48с.
87. Гордиенко В.М.,Козырицкий В.Г. Ультраструктура желёз эндокринной системы. Киев: Здоров'я,
1978.-288с.
88. Горизонтов П.Д. (Ред.). Гомеостаз. М.: Медицина, 1981,464 с.
89. Горкин В.З. Биохимия и биозимическая фармакология обмена некоторых моноаминов. В кн.: Орехович
В.Н. (Ред.). Молекулярные основы патологии. М.:Медицина., с.80-110.
90. Горн М.М. и соавт. Водно-электролитный и кислотно-основной баланс (краткое руководство).
СПб.-М.: Невский диалект -Бином, 1999.-320 с.
91. Гроллман А. Клиническая эндокринология и ее физиологическое значение. М.:Медицина.-1969.
92. Гротэль Д.М. О влиянии алиментарной дистрофии на коронарный артериосклероз, грудную жабу и
острый инфаркт миокарда. Учён. Записки 1-го Лен. Мед. Ин-та.- 1944.-С.28-34.
93. Гумилёв Н.С. Стихи и поэмы. Л.: Сов. Писатель, 1988.
94. Давидсон Дж. Биохимия нуклеиновых кислот. М.:Мир, 1976.
95. Давиденкова Е.Ф., Либерман И.С, Строев Ю.И. и др. Раннее выявление атерогенных сдвигов обмена
веществ и доклинических признаков поражений сердечно-сосудистой системы в семьях больных
атеросклерозом./ Липопротеиды и атеросклероз. Тезисы докл. симпозиума, поев. 110-летию со дня рожд.
акад. Н.Н. Аничкова, 21-23 ноября 1995 г - СПб., 1995. - С . 30.
96. Давыдовский И.В., Общая патология человека.- М.: Медицина.-1969.-611 с.
97. Дедов И.И., Дедов В.И. Биоритмы гормонов. - М.: «Медицина». - 1992. - 256 с.
98. Декарт Р. Избранные произведения.-М.:Госиздат, 1950.-710 с.
99. Дзеранова Л.К. Синдром гиперпролактинемии, успехи медикаментозной терапии. / Нейроэндокри-
нология. Под ред. Е.И. Маровой.- Ярославль, 1999. - С. 201-240.
734 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
100. Диво-93.// М.: АО Дива - Русская книга.-1993.
101. Дильман В.М. и соавт. Метаболическая регуляция иммунного гомеостаза. В кн. Нейро-гуморальная
регуляция иммунного гомеостаза. Л.:1986.-с.191-192.
102. Добрынна В.И. Биологическая химия. М.: Медицина, 1976.-504 с.
103. Дю ла Фуа Г. Руководство к внутренней патологии. -т.1.-СПб.:Практическая Медицина, 1899.-975 с.
104. Жукова А.В. Нефедова Ю.Б., Винник Т.А. и др. Анализ углеводного метаболизма у лиц, страдающих
гипертонической болезнью, в зависимости от TRP64ARG полиморфизма р3-адренорецептора. //
Артериальная гипертензия. - 1999. - № 5. - С. 19-20.
105. Жулев Н.М., Сайкова Л.А., Полякова Л.А., Дементьева Л.Н., Баранов В.В., Косачев В.Д., Жулев С.Н.,
Цинзерлинг Н.В. История изучения миастении на кафедре невропатологии им. С.Н. Давиденкова
СПб МАПО. Мат-лы I Российско-японского семинара по проблемам иммунонейроэндокринологии.
С.Петербург, 15-17 июня 2005 г.// Клиническая патофизиология. — 2005. — т. 2.-N 1. — с. 89.
106. Завадовский М.М. Усп. совр. биол.-1933.-т.2.-К 4-5.-c.86.
107. Зайко Н.Н., Быць Ю.В. (Ред.) Патологическая физиология. Киев: Логос, 1996.-648 с.
108. Зайчик А, Ш. Теоретическое и практическое значение учения о цитотоксинах // Пат. физ. и экспер.
тер.-1981.-Т. XXV.-№ 2.-C. 46-55.
109. Зайчик А.Ш., Васильев А.Г., Кравцова А.А., Утехин В.И., Чурилов Л.П. Иммуноглобулины к антигенам
хроматина как агенты иммуно-нейроэндокринного взаимодействия. Мат-лы I Российско-японского
семинара по проблемам иммунонейроэндокринологии. С.Петербург, 15-17 июня 2005 г.// Клиническая
патофизиология.— 2005. — т. 2. — № 1. — с. 86-87.
110. Зайчик А.Ш., Карев В.Е., Строев Ю.И., Утехин В.И. Цинзерлинг В.А., Чурилов Л.П. Иммунный ответ
как саногенный и патогенный фактор в развитии аутоаллергических и инфекционных заболеваний. В
кн.: Избранные лекции ведущих ученых Медицинского факультета СПбГУ, СПб: СПбГУ, 2005. — с.
12-27.
111. Зайчик А. Ш., Кравцова А. А., Трухманов М. С„ Чурилов Л. П, Физиологическое значение
специфических иммуноглобулинов в регуляции стероидогенеза в клетках коркового вещества надпочечников
// Физиологический журнал СССР им. И. М. Сеченова.- 1985.-Т. LXXI.-Nb 1,-С. 136-147.
112. Зайчик А, Ш., Перельман Л. Р. Цитотоксическая стимуляция коры надпочечников в норме и при
гипотиреозе // Цитотоксины в соврем, медицине.— Киев, 1966,—Т. 3.—С. 100—106.
113. Зайчик А.Ш. и соавт.- Иммунологическая регуляция клеточных функций.-Л.- ЛПМИ.- 1988.-
128с.
114. Зайчик А.Ш., Утехин В.И., Чурилов Л.П., Слободской Е.В. Патофизиология сахарного диабета. В кн.:
Нарушения иммунитета и метаболические расстройства. Ч.1.-СП6: Изд-во ППМИ, 1995, с. 86-187
115. Зайчик А.Ш., Чурилов Л.П. Исследование иммунологических процессов методом реакционной
микрокалориметрии прямого потока. В кн.: Общая и клиническая патофизиология.(ред. В.Ю. Шанин).-СПб.:
НПЦ Техноторф, 1999.- с. 39-47.
116. Зайчик А.Ш., Чурилов Л.П. Микрокалориметрия в медико-биологических исследованиях. // Успехи
соврем. Биол.-1982.-т. 93.-вып.3.-с.448-465.
117. Зайчик А.Ш., Чурилов Л.П. О некоторых семантических аспектах преподавания общей патологии.// В
сб.: Материалы научной конференции, посвященной 100-летию кафедры патофизиологии СПбГМУ
им. акад. И.П.Павлова 23-29 октября 1998 г.-СПб:СПбГМУ, 1998.- с. 60-64.
118. Зайчик А.Ш., Чурилов Л.П. Основы общей патологии. Ч. I. Основы общей патофизиологии. СПб:
ЭЛБИ-Спецлит, 1999.-624 с.
119. Зайчик А.Ш., Чурилов Л.П. Информационные аспекты развития патологических процессов. В сб.:
Чтения им. П.М. Альбицкого,СПб: СПбГМУ, 2000. - с. 16-34 .
120. Зайчик А.Ш., Чурилов Л.П. Патофизиология. Т. И. Основы патохимии. СПб: ЭЛБИ-Спецлит, 2000.
- 688 с.
121. Зайчик А.Ш., Чурилов Л.П. Патофизиология. Т. I Общая патофизиология. Изд. 2-е. СПб: ЭЛБИ, 2001
- 624 с.
122. Зайчик А.Ш., Чурилов Л.П. Патофизиология. Т. II Основы патохимии. Изд. 2-е СПб: ЭЛБИ-Спецлит,
2001.-688 с.
123. Зайчик А.Ш., Чурилов Л.П. Аутоиммунитет как система физиологической регуляции морфофункци-
ональных процессов. Клин, патофизиол. — 2002. — N 2. — с.8-17.
124. Зайчик А.Ш., Чурилов Л.П. Патофизиология Т. III. Механизмы развития болезней и синдромов. Вып.
1 Патофизиологические основы гематологии и онкологии. СПб: ЭЛБИ, 2002. — 624 с.
Литература 735
125. Зайчик А.Ш., Чурилов Л.П., Балашов Л.Д., Филатов СВ. Регуляция функций и роста клеток коры
надпочечников антинуклеарными иммуноглобулинами различной специфичности. Медицинский
академич. Журнал СЗО РАН РФ-2003. - т. 3. - с. 69-71
126. Зайчик А.Ш., Чурилов Л.П., Утехин В.И., Фокин А.С., Ирошникова Г.П., Беляева И.В. Введение в
экспериментальную патологию. Учебно-методическое пособие для студентов- медиков, СПб: ЭЛБИ
- СпбГУ, 2003. - 380 с, илл.
127. Зайчик А.Ш., Чурилов Л.П. Патофизиология. Т. I Общая патофизиология с основами
иммунопатологии. Изд. 3-е, дополненное и исправленное. СПб: ЭЛБИ, 2005. — 656 с.
128. Зарубина Н.А. Заболевания околощитовидных желёз. В кн.: Старкова Н.Т. (Ред.) . Клиническая
эндокринология. Руководство. М.: Медицина, 1991.-е. 164-187.
129. Зимин Ю.В. Инсулинорезистентность, гиперинсулинемия и артериальная гипертензия. //Кардиология.
- 1996.-№11.- С. 81-91.
130. Зимин Ю.В. Происхождение, диагностическая концепция и клиническое значение синдрома инсули-
норезистентности или метаболического синдрома X. // Кардиология. - 1998. - № 6. - С. 71-81.
131. Ибн Сина Абу Али. Канон врачебной науки.- Ташкент:Изд-во АН УзССР, 1954.-кн.1.-579 с.
132. Иванова Л. А., Канорский С.Г., Зингилевский К.Б., Ростовцева О.Н., Сай А.Н. / Актуальные проблемы
современной эндокринологии. Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С. 80.
133. Иверсен Л. Химия мозга. В кн.: Мозг. М.:Мир.,1984.-с.141-166.
134. Ильин B.C. Патологическая физиология углеводно-фосфорного обмена. В кн. Многотомное рук-во
по патологической физиологии. Т. И. М.:Медицина, 1966.-е. 270-311.
135. Иссерсон О.Д. К вопросу о летальности от алиментарной дистрофии в Ленинграде с ноября 1941 г. по
ноябрь 1942 г. Учён. Записки 1-го Лен. Мед. Ин-та.- 1944.-е. 96-100.
136. Иценко Н.М. // Юго-восточный вестник здравоохр.-1924.-Н 3-4.-с!136.
137. Кабанов И.В., Чурилов Л.П. Аквапорины и гипервазопрессинемия. Медицина XXI век. — 2007.
-№5(6). -с. 62-73.
138. Кавецкий Р.Е. Нарушения кислотно-щелочного равновесия. В кн. Многотомное рук-во по
патологической физиологии. Т. И. М.:Медицина, 1966.-С.438-454.
139. Кадыков Б.И. Голодание. В кн. Многотомное рук-во по патологической физиологии. Т. И. М.:Меди-
цина, 1966.-С.235-269.
140. Казека TJP. Метаболический синдром // Врачебный практикум: Новосибирск, 2002. — 50 с.
141. Каземирский В.Е., Медведев А.П., Данилова Н.А., Боровской Н.В. О вероятном влиянии минерального
состава питьевой воды на развитие остеопороза у подростков. / Матер. IV Междунар. конгресса «Эко-
лого-социальные вопросы защиты и охраны здоровья молодого поколения на пути в XXI век». — СПб:
Изд-во регионального отделения Российского Фонда «Здоровье человека», 1998. — С. 230-231.
142. Калинин А.П., Тишенина Р.С., Котова И.В., Титова Н.В. Двойная энергетическая абсорбциометрия и
биохимические маркеры костного метаболизма при первичном гиперпаратиреозе. / Тезисы Всеросс.
научно-практ. конф. «Клинические достижения и перспективы», посвящ. 80-летию со дня рожд. Засл.
деят. науки РСФСР, проф. Д.Я. Шурыгина. - СПб., 2003. - С 183-184.
143. Калицин Д.Ст. Биохимични аспекти на атеросклерозата. София:Медицина и физкултура, 1972,
155 с.
144. Калмыков П.Е., Логаткин М.Н. Современные представления о роли составных частей пищи. Л.:Ме-
дицина, 1974.
145. Кальве Э., Прат У. Микрокалориметрия. Применение в физической химии и биологии. М.: ИЛ,
1963.-447 с.
146. Калюжный И.Т., Калюжная Л.И.// Гемохроматоз: гиперпигментация кожи, пигментный цирроз печени,
«бронзовый» диабет. СПб: ЭЛБИ, 2003. — 48 с.
147. Камерон А.Т. Достижения современной эндокринологии. М.: ГИИЛ, 1948.-640 с.
148. Кандрор В.И. Заболевания надпочечников. Анатомия и физиология. В кн.: Старкова Н.Т. (Ред.).
Клиническая эндокринология. Руководство. М.: Медицина, 1991.-е. 278-296.
149. Кандрор В.И. Заболевания шишковидной железы. Заболевания вилочковой железы. В кн.: Старкова
Н.Т. (Ред.). Клиническая эндокринология. Руководство. М.: Медицина, 1991.-е. 366-377.
150. Капланский С.Я. Молекулярная патология обмена аминокислот. В кн.: Орехович В.Н. (Ред.).
Молекулярные основы патологии. М.:Медицина., с. 13-41.
151. Капланский С.Я. Патологическая физиология белкового обмена. В кн. Многотомное рук-во по
патологической физиологии. Т. И. М.:Медицина, 1966.-С.455-486.
152. Карлик Л.Н. Патологическая физиология. М.-Л.: Биомедгиз,1936,408 с.
736 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
153. Кассиль В.Г., Уголев A.M., Черниговский В.Н. Регуляция выбора и потребления пищи и обмен веществ.
// Усп. физиол. HayK.-1970.-T.l.-N 4.-C.64-97.
154. Катцунг Б. Г. (Ред.). Базисная и клиническая фармакология. М.,СПб.: Бином-Невский диалект, 1998.-
Тт. 1-2
155. Кахана М.С. Патофизиология эндокринной системы. М.:Медицина, 1968,314 с.
156. Каюков И.Г. Современные представления о патогенезе нефротического отёка. В кн.: Рябов СИ., На-
точин Ю.В. Функциональная нефрология.-Спб.: Лань, 1997.-С.108-121.
157. Кедров А.А., Строев Ю.И. О взаимодействии болезней и о механизмах влияния их друг на друга.//
Терапевтический архив. - 1983. - № 1. - С. 65-70.
158. Кемилева 3. Вилочковая железа. М,; Медицина, 1984,252 с.
159. Керпель-Фрониус Э. Педиатрия. Будапешт: Изд-во Академии наук Венгрии, 1975,622 с.
160. Кленов Э.Н. Некоторые показатели обмена веществ у больных с алиментарным истощением. Труды
эвакогоспиталя ФЭП N 50,1943.- вып. 3.-С.59-63.
161. Климов А.Н. Причины и условия развития атеросклероза. В кн.: Превентивная кардиология (ред.
Г.Н.Косицкий).-М.:Медицина, 1977.-С.260-321.
162. Климов А.Н., Никульчева Н.Г. Липопротеиды, дислипопротеинемии и атеросклероз. Л.: Медицина,
1984.- 168с.
163. Клиорин А.И. Заболевания органов пищеварения. // Пособие по педиатрии., Л.: ВМОЛАК, 1976.-
с.83-100.
164. Клотц И. Энергетика биохимических реакций.-М.:Мир.-1970.
165. Клявзуник И.З. Справочник по реаниматологии. Минск: Беларусь, 1978.-240 с.
166. Книга рекордов Гиннеса-1988.// М.: Советская Россия.-1989.
167. Кнорре А.Г. Гистоазбука. В кн.: Асклепий и музы, СПб.: ГПМА, 2000. - с. 217-220.
168. Кнорре А.Г. Краткий очерк эмбриологии человека. Л.:Медицина, 1967.-268 с.
169. Козловская Л.В., Мартынова М.А. Учебное пособие по клиническим лабораторным методам
исследования с элементами программирования). М.: Медицина, 1975.-С.246-272.
170. Коновалова Н.Н., Тишенина Р.С., Пархимович P.P., Мылов Н.М. Является ли подагра фактором риска
развития сахарного диабета и ишемической болезни сердца? / Актуальные проблемы современной
эндокринологии. Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С. 101.
171. Коренная В.В. Некоторые статистические данные о состоянии общественного здоровья в России в
90-е годы. В сб.: Мат. Научной конференции студентов СПбГПМА 29-30 апреля 1999 г., СПб: ППМИ,
с. 13.
172. Коркач В.И. Роль АКТГ и глюкокортикоидов в регуляции энергетического обмена. Киев: Здоров'я,
1979,152 с.
173. Корнева Е.А. Взаимодействие нервной и иммунной систем в норме и патологии. Л.:Наука, 1987,16 с.
174. Корнева Е.А. Введение в иммунофизиологию. СПб: ЭЛБИ, 2003.— 48 с.
175. Корнева Е.А. Основные этапы и тенденции развития иммунофизиологии. К 20-летию основания
международного научного общества по нейроиммуномодуляции. Медицина XXI век. - 2007.— № 6(7).—
с. 16-23.
176. Корниенко И.А. Возрастные изменения энергетического обмена и терморегуляции. М:Наука, 1979,
57 с.
177. Котова Г.А. Диффузный токсический зоб. В кн.: Старкова Н.Т. (Ред.). Клиническая эндокринология.
Руководство. М.: Медицина, 1991.-е. 116-134.
178. Котова СМ., Карпова Н.А., Максимцева И.М., Жорина О.М., Сохор А.Я. Остеопенический синдром
у детей и подростков. / Актуальные вопросы эндокринологии. Тезисы докл. Росс, конф., посвящ.
100-летию со дня рожд. акад. АМН СССР В.Г. Баранова 6-7 апреля 2000 г., Санкт-Петербург. - СПб.,
2000. - С. 225.
179. Кошечкин В. А. Биохимическая генетика нарушений метаболизма липидов плазмы крови. Сер. Итоги
науки и техники. Генетика, М.: ВИНИТИ, 1980.-т.5.-с. 85-122.
180. Краевский Н.А. Патологическая анатомия алиментарного истощения. Труды 1-й терап. конференции.
Горький.-1943.- с. 424-428.
181. Краснодембская К.Д. Промежуточная доля гипофиза. В кн.: Нейроэндокринология. 4.2 СПб: Изд-во
РАН, 1994. С.6-35.
182. Краснопольская К.Д. Биохимическая генетика болезней накопления гликозаминогликанов. Итоги
науки.-сер.Генетика человека.- 1980.-т.5.-с.42-84.
183. Кремер Ю.Н. Биохимия белкового питания. Рига, 1965.
Литература 737
184. Кривохижина Л.В. и соавт. Церулоплазмин - регулятор клеточного состава периферической крови.
В кн.: Дизрегуляционная патология органов и систем. Тезисы докл. III Российского конгресса по
патофизиологии. Москва 9-12 ноября 2004 г. М.: ГУНИИОПиП, 2004. - с. 70.
185. Крысь-Пугач А.П., Кинчая-Полищук Т.А. Остеопенический синдром и остеопороз у детей и подростков.
// Ортопедия, травматология и протезирование. - 2000. — № 2. - С. 35-38.
186. Крэйн С, Холик М. Метаболические болезни костей. В кн. Внутренние болезни по Тинсли Р. Харри-
сону.М.: Практика, т.6.— с. 2724.
187. Кэттайл Д., Арки Р.Патофизиология эндокринной системы.Спб.М. Невский Диалект—Изд-во БИ-
НОМ.2001.-336с.
188. Лабори А. Регуляция обменных процессов. М.: Медицина, 1970.-384 с.
189. Ламетри де Ж.-О. Человек-машина. Минск.: Литература, 1998,704 с.
190. Ланг Г.Ф. Гипертоническая болезнь. Л., 1950
191. Ланг Г.Ф. Клиника алиментарной дистрофии. Труды 1-й терап. конференции. Горький.-1943.- с.
406-424.
192. Ланг Г.Ф. О гипертонии. // Архив государственного клинического института для усовершенствования
врачей. - Л., 1922. - Т.1. - С. 16-66.
193. Лаптева Н.Н. Патофизиология белкового обмена. М.:ЦОЛИУВ.-1970.-335 с.
194. Левина Л.И. (Ред.) Подростковая медицина: руководство для врачей. СПб: Спецлит.-1999.- 700 с.
195. Левина Л.И. (Ред.) Подростковая медицина: руководство для врачей. СПб: Спецлит. — 1999. — 700 с.
196. Лейбсон Л. Г. Происхождение и эволюция эндокринной системы. В кн.: Эволюционная физиология,
ч. И, Л., Наука, 1983, с. 3-52.
197. Лейтес СМ. Патологическая физиология жирового обмена. В кн. Многотомное рук-во по
патологической физиологии. Т. И. М.:Медицина, 1966.-С.312-345.
198. Лейтин В.А. Свиридов Д.Д. В кн.: Стенка сосудов в атеро- и тромбогенезе.М.:Медицина, 1983.
с.155-175.
199. Ленинджер А. Биохимия. М.:Мир, 1976,958с.
200. Лесной С.К. // Акуш. и raH.-1968.-N 2.-C.184-191.
201. Линдеман В. К. Учебник общей патологии. Киев, 1910-1911.
202. Литвицкий П.Ф.(Ред.).Патофизиология.Курс лекций. М.:Медицина, 1996.-752 с.
203. Ловягина Т.Н., Баньковская Э.Б. // Ж. эволюц. биохим. физиол.-1970,-т.З.-285-304.
204. Логаткин М.Н. (ред.) Критерии адекватного питания. Л.: ЛПМИ, 1984,88 с.
205. Лондон Е.С. Физиология и патология пищеварения. Пгр.: Практическая медицина.-1916.-168 с.
206. Лопухин Ю.М. Холестериноз. М.:Наука, 1986
207. Мажанди Ф. Краткое основание физиологии. М.,Т.1.- 1830.
208. Мазурин А.В., Воронцов И.М. Справочник по детской диететике. - М: Медицина, 1975.
209. Мазуров В.И. Биохимия коллагеновых белков. М.:Медицина.-1974.-248 с.
210. Майстренко Н.А. (ред.) Органический гиперинсулинизм. СПб: ЭЛБИ, 2004. — 160 с.
211. МакДермотт М.Т. Секреты эндокринологии. 2-е изд-е исправ. и дополн. (пер. с англ.), М-СПб - Бином-
Невский диалект, 2001. — 464 с.
212. МакДермотт М.Т. Секреты эндокринологии./ Пер с англ. - М.: ЗАО «Издательство БИНОМ», 1998.
-416с.
213. Малахов Г.П. Очищение организма. Целительные силы. Т.1, СПб.: АО Комплект, 1995. — 360с.
214. Мальцев СВ. Причины и механизмы развития трофологической недостаточности у подростков/
Материалы II Национальной Ассамблеи. Охрана репродуктивного здоровья населения, 18-20 апреля
1997 г. - Москва, 1997. - С. 38-39.
215. Малашхия Ю.А., Манько В.М., Гургенидзе Т.В..- Иммунокомпетентные клетки спинномозговой
жидкости человека в норме и при заболеваниях нервной системы.-Тбилиси: Сакартвело.-1990.-
с.89-92.
216. Маслов М.С. Аномалии конституции (диатезы) в детском возрасте. В кн.: Многотомное рук-во по
педиатрии (ред. А.Ф.Тур). М.:Медгиз, Т. 1.-е. 471-526.
217. Медведева Н.Б. Патология углеводного обмена. Патология жирового и липоидного обмена. В кн.
Рук-во по патологической физиологии (ред. А.А. Богомолец). Т. II. Биомедгиз: 1937.-С.122-258.
218. Медведева Н.Б. Экспериментальная эндокринология. Киев: Изд-во АН УССР, 1946,744 с.
219. Меерсон Ф.З. (Ред.) Физиология адаптационных процессов. Сер. Рук-во по физиологии. М.:Наука,
1986,639 с.
220. Мечников И.И. Этюды о природе человека. М.: Научное слово, 1904.
738 А.Ш. Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
221. Мирошников Б.И. (Ред.). Экстренная абдоминальная хирургия. СПб.,1994, с. 134-147.
222. Митюшов М.И. Гипофизарно-адреналовая система и стресс. В сб.:. Гипофизарно-адреналовая система
и мозг.Л.:Наука, 192-202.
223. Михайлова Е. А. и соавт. Роль окситоцина в модуляции иммунных процессов. В кн. Нейро-гуморальная
регуляция иммунного гомеостаза. Л.:1986.-с.51.
224. Михайлова Н.Г. и соавт. Ликворосорбция у больных героиновой наркоманией. Ж. Невропатол. и
Психиатр, им. Корсакова.-1998.-N 6.- с. 32-34.
225. Москаленко Е.П. и соавт. Роль минералокортикоидного гормонопоэза в регуляции структуры и
функции лимфоидной системы. . В кн. Нейро-гуморальная регуляция иммунного гомеостаза.
Л.:1986.-с.52-53.
226. Мусил Я. Биохимия патологических процессов. М.:Медицина, 1985.
227. Мусил Я., Новакова О., Кунц К. Современная биохимия в схемах. М.:Мир, 1981.- 216 с.
228. Мясников А.Л. Гипертоническая болезнь и атеросклероз. М.:Медицина, 1965.- 616 с.
229. Мясников А.Л. Клиника алиментарной дистрофии. Л., 1945.
230. Мясников А.Л., Гротель Д.М., О содержании в крови холестерина и мочевой кислоты в связи с
конституцией и болезнями обмена веществ. Тр. X съезда терапевтов СССР. - М.; Л.: Госиздат, 1926.
-с.291-299.
231. Надь Ю.Г. Механизмы гиперинсулинемии при сахарном диабете. / Актуальные проблемы современной
эндокринологии. Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С. 142.
232. Наточин Ю.В. Клиническая физиология почек у детей. В кн.: Клиническая нефрология детского
возраста (Ред. А.В. Папаян, Н.Д. Савенкова).- СПб.: Сотис-1997.-е. 9-42.
233. Некрасова М.Р., Давыдова Л.И., Суплотова Л.А. Оценка минеральной плотцости костной ткани у
больных диффузным токсическим зобом. / Тезисы Всеросс. научно-практ. конф. «Клиническая
эндокринология - достижения и перспективы», посвящ. 80-летию со дня рожд. Засл. деят. науки РСФСР,
проф. Д.Я. Шурыгина. - СПб., 2003. - С. 209-210.
234. Ноздрюхина Л.Р. Биологическая роль микроэлементов. Новосибирск: Наука СО, 1977.
235. Омура Ю. Активация высшей нервной деятельности эндогенными веществами насыщения возрастает
в ходе потребления пищи // Мат-лы I Российско-японского семинара по проблемам иммунонейро-
эндокринологии. С.Петербург, 15-17 июня 2005 г.// Клиническая патофизиология. — 2005. — т.2.
-№ 1.-е. 82-83.
236. Орехович В.Н. (Ред.). Молекулярные основы патологии. М.:Медицина., 1966.-319 с.
237. Павлов И.П. (1911) О пищевом центре // ПСС.-М.,-Л.: Изд-во АН СССР, 1952.-т.6.-с. 9-27
238. Пальцев М.А., Пауков B.C., Улумбеков Э.Г. (ред.). Патология. Руководство. М. ГЭОТАР-МЕД, 2002.
-960 с.
239. Павлов И.П. Полное собрание трудов.-М.-Л.: АН СССР, 1940-1949.-тт. 1-5.
240. Павлов И.П., Ненцкий М., Ган М., Массен В.Н. (1892) Экковский свищ вен, нижней полой и воротной,
и его последствия для организма. // ПСС.-М.,-Л.: Изд-во АН СССР, 1952.-т.2.-кн. 1.-е. 210-238.
241. Панков Ю.А. Гормоны гипофиза. В кн.: Юдаев Н.А. (Ред.). Биохимия гормонов и гормональной
регуляции. М.: Наука.-1976.- с. 44- 93.
242. Паскаль Б. Мысли. М.: Художественная литература, 1974.
243. Пашутин В.В. Лекции общей патологии (патологической физиологии).-Казань, 1878, Ч.1.-427 с.
244. Певзнер Л. Основы биоэнергетики. М.:Мир.-312 с.
245. Пенчев Ив.(ред.) Ендокринно-обменна диагностика. София : Медицина и физкултура, 1962, с.
496-500.
246. Перельман Л.Р. К вопросу о функциональной взаимосвязи паращитовидных и мужских половых желёз.
Саратов: Саргублит, 1924,11с.
247. Перельман Л.Р. Лекции по патологической физиологии 1947-1948 учебного года, (в записи А.С.
Левитиной). Рукопись. Архив кафедры патофизиологии СПбГПМА.
248. Перельман Л.С. Патология водного и минерального обмена В кн. Рук-во по патологической
физиологии (ред. А.А. Богомолец). Т. И. Биомедгиз: 1937.-С.259-300.
249. Перельман Л.С. Патология питания. Патология окислительных процессов и дыхательный
коэффициент. Голодание. Патология белкового обмена. В кн. Рук-во по патологической физиологии (ред. А.А.
Богомолец). Т. И. Биомедгиз: 1937.-c.5-121.
250. Петровский К.С. Гигиена питания. М.: Медицина, 1975.-400 с.
Литература 739
251. Писарева СВ., Строев Ю.И., Чурилов Л.П. Лептин - нейроиммуноэндокринный регулятор обмена
веществ. Актуальные проблемы диагностики, лечения и профилактики заболеваний.Труды Мариин-
ской больницы, вып.Ш, СПб:: СПбГПМА, 2004. - с.112-120.
252. Пищулин А.А. Заболевания женских половых желёз. В кн.: Старкова Н.Т. (Ред.). Клиническая
эндокринология. Руководство. М.: Медицина, 1991.-е. 378-412.
253. Подвысоцкий В.В. Основы общей и экспериментальной патологии. Изд. 4-е., СПб: Изд-во ГЛ.Риккера,
1905.- 922 с.
254. Покровский А.А., Крыстев Л.П. Печень, лизосомы, питание. София: Изд-во Болгарской академии
наук, 1977,207 с.
255. Покровский А.А., Тутельян В.А. Лизосомы.-М:Наука, 1976,382 с.
256. Поленов А.Л. (ред..). Нейроэндокринология. Ч.1,Кн. 1-2; 4.2 СПб: Изд-во РАН, 1994.
257. Поленов А.Л., Константинова М.С, Гарлов П.Е. Гипоталамо-гипофизарный нейросекреторный
комплекс. В кн.: Нейроэндокринология. Ч.1,Кн. 1-2; СПб: Изд-во РАН, 1994.С. 139-187.
258. Попеску О. Синдромы в педиатрии. Бухарест:Мед.изд-во, 1977.-477 с.
259. Попов А.В., Кузнецов А.С, Лозовский В.Т. // Вопр. мед. химии,- 1978.-N 5.-е. 670-675.
260. Поповичи Д., Сэхляну В. Гормоны и сердечно-сосудистая патология. М.:Медицина, 1969.-392 с.
261. Пресман А.С. Электромагнитные поля в биосфере. М.: Знание, 1971.64 с.
262. Пригожий И.Р. Введение в термодинамику необратимых процессов.М.:ИЛ,1960.
263. Прутков Козьма. Сочинения. М.: Художественная литература, 1976.
264. Разбойников Св. Синдроми и рядко диагностицирани болести на двигателната система и на соеди-
нителната тъкан. / Синдроми и рядко диагностицирани болести.- София: Медицина и физкултура,
1967.-С. 439-541.
265. Рачев P.P., Ещенко Н.Д. Тиреоидные гормоны и субклеточные структуры. М.: Медицина, 1975.—
296 с.
266. Рожинская Л.Я. Системный остеопороз. Практическое руководство. - М., 2000.
267. Рожинская Л.Я., Марова Е.И., Сазонова Н.И., Чернова Т.О. Неинвазивная количественная оценка
массы кости в диагностике остеопороза эндокринного генеза. / Тезисы Всероссийской
научно-практической конференци «Клиническая эндокринология — достижения и перспективы», посвященная
80-летию со дня рождения Заслуженного деятеля науки РСФСР, профессора Д.Я. Шурыгина. - СПб.,
2003.-С.716.
268. Розенфельд Е.Л. Молекулярная патология обмена углеводов. В кн.: Орехович В.Н. (Ред.).
Молекулярные основы патологии. М.:Медицина., с.80-110.
269. Розенфельд Е.Л. Молекулярные основы некоторых наследственных энзимопатий. Итоги науки.-сер.
Генетика человека.-1980.-т.5.-с.З-41.
270. Рокитанский К., Руководство к общей патологической анатомии. М., 1849.
271. Романенко В.И. Мелатонин как возможный эндогенный лейкозогенный (бластомогенный) агент //
Гематол. трансфузиол. — 1983. — № 2. — С. 47-50.
272. Романенко И.А., Назаров СБ., Новожилов Е.А., Гринштейн В.Б. Реологические свойства эритроцитов
у родственников больных сахарным диабетом. / Актуальные проблемы современной эндокринологии.
Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С 167.
273. Романов В.А.Макрофаги очага воспаления в условиях эндогенной гиперпролактинемии. В кн. Ней-
ро-гуморальная регуляция иммунного гомеостаза. Л.:1986.-с.62.
274. Ром-Бугославская Е.С // Шишковидная железа./ БМЭ, изд. 3-е.- т. 27.-е. 1326-1341.
275. Роти Э., Браверманн Л.И. / Браверманн Л.И. (ред.). Болезни щитовидной железы (пер. с англ.). - М.:
«Медицина», 2000. - С. 401-17.
276. Рябов Г.А. (Ред.) Экстренная анестезиология. М.:Медицина, 1983,280 с.
277. Рябов СИ., Наточин Ю.В. Функциональная нефрология.-Спб.: Лань, 1997.-304 с.
278. Рязанцев Н.В. (1892) Пищеварительная работа и выделение азота в моче. // Арх. биол. наук.-1986.-
т.4.-вып.4.-с.391-410.
279. Сахарчук В.М. Патогенез атеросклероза. Роль микроэлементов. В кн.: И.М. Ганджа, Н.К. Фуркало ./
Атеросклероз.- Киев: Здоров'я, 1978.- с.106-115.
280. Свечников В. А. Болезнь голодания (алиментарная дистрофия). Л. изд-во 2-го Л МИ.- 1947.-103 с.
281. Свешникова Н.А. К вопросу о состоянии белкового обмена при алиментарном истощении. Сб. научн.
Работ ГИДУВа, 1942.-195-198.
282. Селье Г. Очерки об адаптационном синдроме.-М.:Медгиз, 1960.-254 с.
283. Семёнов Н.Н. Цепные реакции. Л.:Госхимтехиздат, 1934.
740 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
284. Сергеев А.И., Голиков А.П., Пель В.И., Белянская Е.В. Инсулинорезистентность и метаболический
синдром. СПб.: ВМедА, 2005. - 32 с.
285. Серов В.В., Ярыгин Н.Е., Пауков B.C. Патологическая анатомия.Атлас, М.:Медицина, 1986.-368 с.
286. Сиротинин Н. Н. Эволюция резистентности и реактивности организма. М., Медицина, 1981,236 с.
287. Скулачёв В.П. Аккумуляция энергии в клетке.- М.:Наука.-1972.
288. Слуцкий Л.И. Биохимия нормальной и патологически изменённой соединительной ткани. Л.:Меди-
цина.-1969.-376 с.
289. Смирнов М.И. (Ред.) Витамины. М.:Медицина, 1974.-491 с.
290. Смиттен Н.А., Шаляпина В.Г. Периферическая нейроэндокринная хромаффинная система
позвоночных. В кн.: Нейроэндокринология. Ч.1,Кн. 1-2; СПб: Изд-во РАН, 1994. С.362-394.
291. Соловьёв B.C. О смысле любви. В кн.: Сочинения в 2-х томах. 2-е изд., М., 1990.-т.2.-с.493-547.
292. Смолянский Б.Л., Лифляндский В.Г. Лечение остеопороза. СПб.: Издательский дом «Нева», 2006.
-256 с.
293. Соллертинская Т.Н. и соавт.// Роль дерморфина в регуляции процессов зимней спячки у
млекопитающих. Физиол. Ж. им. И.М. Сеченова, 1992. - т. 78. - № 4. - с.1-13.
294. Старкова Н.Т. (Ред.) . Клиническая эндокринология. Руководство. М.: Медицина, 1991.-512 с.
295. Стаут Р.У. Гормоны и атеросклероз.-М.: Медицина, 1985.-236 с.
296. Строев Ю.И. Болезни эндокринной системы. В кн. Подростковая медицина: рук-во для врачей. (Ред.
Л.И.Левина).Спб.:Спецлит,1999.-с.436-469.
297. Строев Ю.И. и др. Экология, щитовидная железа, тепловидение. В сб.: Прикладная оптика-98.
СПб.:1998,с.55-56.
298. Строев Ю.И., Либерман И.С. Общая характеристика ангиопатий при сахарном диабете. В кн.: Генетика
сахарного диабета. Л.: Медицина, 1988,-с. 21-35.
299. Строев Ю.И., Чурилов Л.П. Дисплазия соединительной ткани и эндокринная регуляция. В кн.: Каду-
рина Т.И. (ред.). Дисплазии соединительной ткани.,СПб.: Изд. дом МАПО, 2007 ( в печати).
300. Строев Ю.И, Чурилов Л.П., Йеми Расин и соавт. Гипотрофия юношей призывного возраста -
актуальная социальная проблема.// В кн.: Материалы ежегодной городской научно-практической
конференции с международным участием «Здоровье молодых», Санкт-Петербург, 25 ноября 2005 г. — СПб:
СПбГУ,с.16-17.
301. Строев Ю.И. О взаимоотношениях сахарного диабета и атеросклероза. / Наследственность и другие
факторы в патогенезе атеросклероза. Профилактика некоторых его осложнений. Сб. науч. тр. ЛПМИ
под ред. Р.С. Иванова. - Л.: Издательский отдел ЛПМИ, 1985. - С. 64-69.
302. Строев Ю.И. Характеристика артериальной гипертензии при сахарном диабете по данным
эндокринологического стационара. /Современные аспекты артериальных гипертензии. Матер. Всеросс. научной
конф. 19-21 декабря 1995 года. - СПб., 1996. - С. 180-181.
303. Строев Ю.И. Частота и формы артериальной гипертензии у больных сахарным диабетом. / Патология
внутренних органов при нейро-гормональных и обменных нарушениях. Сб. науч. тр. ЛПМИ под ред.
Ю.И. Строева. - Л.: Издательский отдел ЛПМИ, 1988. - С. 58-66.
304. Строев Ю.И., Вольфберг Е.Д., Попова А.Е., Самойлова Т.С. К ранней диагностике диабетических
микроангиопатий методом биопсии кожи. / Патология сердечно-сосудистой системы при нарушениях
нейро-гормональной регуляции. Сб. трудов ЛПМИ под ред. А.А. Кедрова. - Л., 1978. - С. 45-47.
305. Строев Ю.И., Зайчик А.Ш.,Чурилов Л.П и др. Функциональное состояние щитовидной железы у
подростков Санкт-Петербурга с ювенильным зобом. В сб.: Экология и здоровье детей - основа устойчивого
развития общества. СПб: 1997, с 196-198.
306. Строев Ю.И., Карповская Е.Б. Два случая микроаденомы гипофиза у девушек-подростков с нервной
анорексией. - Новые Санкт-Петербургские врачебные ведомости. - 1998. — № 1. - С. 59-61.
307. Строев Ю.И., Садов С.А., Шейх Зиарат А. Сахарный диабет и красная кровь. /Актуальные проблемы
современной эндокринологии. Матер. IV Всерос. конгресса эндокринологов. - СПб, 2001. - С. 196.
308. Строев Ю.И., Цой М.В., Чурилов Л.П., Шишкин А.Н. Классические и современные представления о
метаболическом синдроме.Часть I. Критерии, эпидемиология, этиология./ Вестник С.-Петербургского
Ун-та, сер. 11, Медицина. — 2007. — вып. 1. — с. 3-15
309. Строев Ю.И., Цой М.В., Чурилов Л.П., Шишкин А.Н. Классические и современные представления о
метаболическом синдроме. Часть II. Патогенез./ Вестник С.-Петербургского Ун-та, сер. 11, Медицина.
— 2007. (в печати).
310. Строев Ю.И., Чурилов Л.П. Бельгов А.Ю., Чернова Л.А.. Ожирение у подростков. Изд. 2-е испр. и
дополненное.СПб.: «ЭЛБИ-СП6», 2006. - 216 с.
Литература 741
311. Строев Ю.И., Чурилов Л.П. Йод и интеллектуальный потенциал России. Медицина XXI век. 2005; 1
(1): 14-21.
312. Строев Ю.И., Чурилов Л.П. Классические и современные представления об ожирении. В кн.:
Ожирение: клинические очерки, (п/ред. Барановского А.Ю., Ворохобиной Н.В.) СПб.: Диалект, 2007, гл.
3, с. 67-88.
313. Строев Ю.И., Чурилов Л.П. Плюсы и минусы очередной кампании поголовной йодной профилактики
в современной России. Актуальные проблемы диагностики, лечения и профилактики заболеваний..
. Труды Мариинской больницы, вып.Ш, СПб:: СПбГПМА, 2004. - с.101-112
314. Строев Ю.И., Чурилов Л.П. Эндокринные заболевания и атеросклероз. / Актуальные проблемы
диагностики, лечения и профилактики заболеваний. Труды Мариинской больницы. Выпуск III. - СПб.:
Издание СПбГПМА, 2004. - С. 91-101.
315. Строев Ю.И., Чурилов Л.П. Эндокринология подростков.(ред. А.Ш. Зайчик) СПб.:ЭЛБИ, 2004, 380
с. илл.
316. Строев Ю.И., Чурилов Л.П. О диагностическом значении определения тиротропного гормона: всегда
ли уровень тиротропного гормона служит объективным критерием функции щитовидной железы?
Медицина - XXI век, 2006. -№ 4(5), с. 58-66
317. Строев Ю.И., Чурилов Л.П., Бельгов А.Ю. О возможности трансформации гипоталамического
синдрома пубертатного периода в метаболический кардиоваскулярный Х- синдром. / Бюлл. НИИ
кардиологии имени В.А. Алмазова. Том И. - 2004. - № 1. - С. 229.
318. Строев Ю.И.Топографическая анатомия диабетической микроангиопатии. / Профилактика и лечение
сердечно-сосудистых заболеваний. Матер, научно-практич. конф. (в рамках «Недели здорового сердца»)
10-13 ноября 1998 г. - СПб., 1998. - С. 40-41.
319. Струков А.Н., Серов В.В. Патологическая анатомия. М.:Медицина, 1993,687 с.
320. Таболин В.А. и соавт. Урикопатии. В кн.: Справочник по детской диететике, (ред. И.М. Воронцов, А.В.
Мазурин), Л.: Медицина, 1977.- с. 186-187.
321. Талызин Ф.Ф. Ядовитые животные суши и моря. - М.: Знание, 1970. - 96 с.
322. Тареев Е.М. Гипертоническая болезнь. - М., 1948. - 21 с.
323. ТатоньЯ. Ожирение. Патофизиология, диагностика, лечение.-М.:Медицина, 1981.
324. Тенчов Г., Хаджибеков Г. Рентгеновское исследование при эндокринных заболеваниях. / Эндокринно-
обменная диагностика. Пер. с болг. - София: Медицина и физкультура, 1961. - С. 135-141.
325. Терещенко И.В. Лептин и его метаболизм. // Проблемы эндокринологии. - 2001. — Т. 47. — № 4.
-С. 40-45.
326. Тиц П.У. (ред) Клиническая оценка лабораторных тестов. - М., «Медицина». - 1986. - 480 с.
327. Тодоров Й. Клинические лабораторные исследования в педиатрии. София: Медицина и физкулту-
ра.-1961.
328. Тур А.Ф. Физиология и патология детей периода новорожденности.Л.,1955.
329. Тургенев И.С. Живые мощи. Собр. соч. В 5-ти томах .М.,1994.
330. Уайт А. и соавторы. Основы биохимии. Тт. 1-3, М.:Мир, 1981.
331. Уварова Н.А. Нарушения кислотно-основного состояния. В кн.: Адо А.Д., Новицкий В.В. (Ред.)
Патологическая физиология. Томск, 1994, с. 241-245
332. Уголев A.M. Естественные технологии биологических систем.- Л.:Наука.-1987.-317 с.
333. Уголев A.M. Теория адекватного питания и трофология.- Спб.:Наука.-1991.-272 с.
334. Уголев А.М. Энтериновая (кишечная гормональная) система. Л.:Наука.-1978.-315 с.
335. Улумбеков Э.Г., Челышев Ю.А. Гистология (введение в патологию). М.:ГЭОТАР, 1997.-950 с.
336. Фелиг Ф., Бекстер Дж.Д., Бродус А.Е., Нромен Л.А. (ред.)- Эндокринология и метаболизм,- М.-
Медицина.-т. 1-2.-1985.
337. Филаретов А.А., Данилова О.А. Кора надпочечника. В кн.: Нейроэндокринология. 4.2 СПб: Изд-во
РАН, 1994. С. 125-152.
338. Фойт К. Руководство по физиологии. СПб.: Изд-во Л. Германна, 1885.-тт. 1-4.
339. Фокин А.С. и соавт. Влияние функционального перенапряжения центральной нервной системы и
эндокринных нарушений на развитие экспериментального атеросклероза у собак. Бюлл. Эксперим.
Биол. Мед.-1975.-Ы 5.-С.28-35.
340. Франке Ю., Рунге Г. Остеопороз. Пер. с нем. - М: Медицина, 1995. - С. 117-121.
341. Фрейд 3. Лекции по введению в психоанализ.-М., 1922, тт. 1-2.
342. Халатов С.С. Об условиях отложения в организме анизотропных жиров и их свойствах. Тр. О-ва
русск. Врачей, 1912.-е. 1.
742 АЖ Зайчик, Л.П. Чурилов Патохимия (эндокринно-метаболические нарушения)
343. Халатов С.С. Учебник патологической физиологии.-М.-Л.:Медгиз, 1933.-531 с.
344. Халатов С.С. Холестериновая болезнь. М., 1946.
345. Хан Т. Метаболические болезни костей. / Эндокринология. Под ред. Н. Лавина. Пер. с англ. - М:
Практика, 1999. - С. 455-479.
346. Харрис Г. Основы биохимической генетики человека.-М.:Мир,1973.-226 с.
347. Харченко В.П. и соавт. Болезни вилочковой железы. М.: Триада-Х, 1998.-232 с.
348. Хелимский A.M. Эпифиз (шишковидная железа). М.: Медицина, 1969.
349. Хорст А. Молекулярные основы патогенеза болезней.-М.:Медицина, 1982.-454 с.
350. Хочачка П.,Сомеро Дж. Стратегия биохимической адаптации.-М.:Мир, 1977.-400 с.
351. Хэм А., Кормак Д. Гистология.-М.; Мир, 1982.-Т. 5.-С 49-223.
352. Цанев А. Затлъстяване. София : Медицина и физкултура.-1968.-188 с.
353. Цинзерлинг А.В.Динзерлинг В.А. Патологическая анатомия. СПб.:Сотис, 1996.
354. Цинзерлинг В.Д. // В кн.: Тр. 14-го Всесоюзного съезда терапевтов. М. Медгиз, 1958.- с.49-58
355. Цинзерлинг В.Д. Патология истощения в 1941-1942 гг. Труды 2-й терап. конференции. Вологда,
1943.-С.147-153.
356. Цыган В.Н. и соавт. Синдром хронической усталости и иммунной дисфункции. СПб: ВМедА.— 2001
357. Чазов Е.И., Исаченков В.А. Эпифиз: место и роль в системе нейроэндокринной регуляции.-М.:На-
ука.-1974.-280 с.
358. Чеботарёв Д.Ф., Фролькис В.В. Сердечно-сосудистая система при старении. Л.:Медицина, 1967.-
256 с.
359. Чернихова Е. А., Оганов B.C., Бакулин А.В. Костно-метаболические нарушения при нервной анорексии.
// Остеопороз и остеопатии. — 2000. — № 4. — с. 22-24.
360. Чернова Т.О., Игнатков В.Я., Григорян О.Р. Костная денситометрия в клинической практике. / Там
же. - С. 723.
361. Черноруцкий М.В. Алиментарная дистрофия с общеврачебной точки зрения. В сб.: Работы
ленинградских врачей за годы Отечественной войны.-1943.-вып.5.-с. 3-11.
362. Чижевский А.Л. Физические факторы исторического процесса. Калуга, 1924.
363. Чолаков В. Нобелевские премии. Учёные и открытия. М:Мир, 1986,369 с.
364. Чурилов Л. П. Митогенные эффекты специфических цитостимулирующих иммуноглобулинов //
Механизмы регуляции физиологических функций. Тез. докл. Ленингр. гор. конф. молод, ученых и
специалистов.—Л., 1985.—С. 112.
365. Чурилов Л. П. Цитогенетические механизмы стимулирующего действия специфических
иммуноглобулинов на стероидогенез в клетках коркового вещества надпочечников.—Л.: Дисс. к.м.н., 1986.
366. Чурилова Н.И. Развитие вилочковой железы в эмбриогенезе крыс при введении прогестерона. //
Морфология.-1994.-вып.7.->П2.-с.82-88.
367. Чурилов Л.П. (ред.) Механизмы развития стоматологических заболеваний. Клиническая
патофизиология для стоматологов. СПб.: ЭЛБИ, 2006,504 с. илл.
368. Чурилов Л.П. Новое о патогенезе ожирения. Мир медицины. — 2001. — № 3-4. — с. 80-82.
369. Чурилов Л.П. Обмен веществ, интеллект и лидерские качества. Мир медицины. — 2001. — № 1-2.
-с.50-51.
370. Чурилов Л.П., Строев Ю.И, Смирнов В.В., Муджикова О.М., Писарева СВ., Утехин В.И., Цой М.В.
Аутоиммунный тироидит - актуальная проблема современной эндокринологии. Вестник СПбГУ, сер.
Медицина, 2006. - № 1. - с. 3-25.
371. Чурилов Л.П., Строев Ю.И., Цой М.В. Прогностическое значение некоторых серологических и ультра-
сонографических признаков аутоиммунного тироидита Хасимото. / Тезисы Всероссийской научно-
практической конференци «Клиническая эндокринология — достижения и перспективы», посвященная
80-летию со дня рождения Заслуженного деятеля науки РСФСР, профессора Д.Я. Шурыгина. - СПб.,
2003.-С. 261-262.
372. Шабалов Н.П. Переходные (пограничные) состояния. В кн.: Справочник неонатолога. М.: Медицина,
1984, с. 84-94.
373. Шанин В.Ю. Клиническая патофизиология. СПб: Изд-во специальной литературы, 1998,750 с.
374. Шанин В.Ю. Типовые патологические процессы. СПб:Изд-во специальной литературы, 1996, 278 с.
375. Шевелев А. С„ Синявский О. А, Тимус как забарьерный орган // Иммунология.—1986.—№ 3.—С.
8-13.
376. Шейман Дж.А. Патофизиология почки. М.:Бином, 1997.-224 с.
Литература 743
377. Щестакова М.В. Сахарный диабет и артериальная гипертония: роль инсулинорезистентности. /
Актуальные проблемы современной эндокринологии. Матер. IV Всерос. конгресса эндокринологов.
-СПб, 2001.-С. 238.
378. Шредингер Э.Что такое жизнь с точки зрения физика.-М.:Атомиздат,1972,258 с.
379. Шрейбер В. Патофизиология желёз внутренней секреции. Прага: Авиценум, 1987. — 494 с.
380. Штрюмпель Г. Учебник частной патологии и терапии внутренних болезней, девятое русское издание,
Петроград, 1915; перевод 19-го немецкого издания.
381. Шурыгин Д.Я. Заболевания зобной (вилочковой) железы. В кн.: Многотомное рук-во по внутренним
болезням.Л.:Медицина, 1966-т. 7.-е. 533-545.
382. Шустов СБ., Халимов Ю.Ш. Функциональная и топическая диагностика в эндокринологии: Научно-
методическое издание. - СПб.: ЭЛБИ-СПб, 2001. - 239 с.
383. Шутова Н.Т. Патология холестеринового и липоидного обмена. В кн. Многотомное рук-во по
патологической физиологии. Т. II. М.:Медицина, 1966.-С.357-397.
384. Шутова Н.Т.,Черникова Е.Д. Патологическая физиология развивающегося организма.- Л:Медицина,
1974.-152 с.
385. Шхвацабая И.К., Метелица В.И., Бётиг 3. Эпидемиология сердечно-сосудистых заболеваний. М.:
Медицина, 1977.- 368 с.
386. Энгельгардт В.А. // Казанск. мед. ж.-1931.-т.27.-с.496.
387. Эрман М.В. Нефрология детского возраста в схемах и таблицах. СПб.: Спецлит, 1997.-414 с.
388. Эфроимсон В.П. Биосоциальные факторы повышенной умственной, активности человека. Препринт
ВИНИТИ в 2-х томах., М.: ВИНИТИ, 1982.
389. Эфроимсон В.П. К биохимической генетике интеллекта..// Природа-1976.-N 9.-С.62-73.
390. Юдаев Н.А. (Ред.). Биохимия гормонов и гормональной регуляции. М.: Наука.-1976.-380 с.
391. Яковлев В.А. и соавт. Препараты липоевой кислоты. В кн.: Клиническая фармакология (ред. В.В.
Закусов).-М.: Медицина, 1978.-С.367-368.
392. Ярилин А.А. и соавт. Иммунологические функции тимуса. Итоги науки.-сер.Иммунология. М.:ВИ-
НИТИ-1990.-188с.
На языках с латинской графикой:
393. Abderhalden, E. Weiterer Beitrag zur Frage nach der Verwertung von tief abgebautem Eiweiss im tierischen
Organismus. Z. Physiol. Chem.-1909. - 61:194-199.
394. Adams D.D.,Purves H.D.-Proc.Univ. Otago Med. School.- 1956.-v.34.-pp.l 1-12.
395. Addison Th. On the constitutional and local effects of diseases of the surrenal bodies. London., 1855.
396. Ahmed S.A. et al- Amer.J.Pathol.-1985.-v.l21.-pp.531-551.
397. Aitman TJ, Gotoda T, Evans AL, Imrie H, Heath KE, Trembling PM, Truman H, Wallace CA, Rahman A,
Dore C, et al. Quantitative trait loci for cellular defects in glucose and fatty acid metabolism in hypertensive
rats. Nat Genet 1997;16:197-201.
398. Aizawa-Abe M. et al. Pathophysiological role of leptin in obesity-related hypertension //J. Clin. Invest.-
2000.-v.l05.-p.l243-1252.
399. Alarcon-Segovia D„ Llorente L. Antibody penetration into living cells. III. Effect of antiribonucleoprotein
lgG on cell cycle of human peripheral blood mononuclear cells. // Clin. Immunol. Immunopathol., 1982.— V.
23.-N1.-P. 22-23.
400. Alberti KG, Zimmet PZ, Shaw J., for the IDF Epidemiology task force consensus group.The Lancet. — 2005.
-v.366.-pp.l059-1061.
401. Alberti KG, Zimmet PZ: Definition, diagnosis and classification of diabetes mellitus and its complications.
Part 1. Diagnosis and classification of diabetes mellitus, provisional report of a WHO consultation. Diabet
Med 1998,15:539-553.
402. Albright F. et al. // Endocrinologie.-1942.-v.30.-pp. 922-932.
403. Allen B.-Science.-1916.-v.44.-p.755.
404. Amino N, Miyai K, Kuro R, Tanizawa O, Azukizawa M, Takai S, Tanaka F, Nishi K, Kawashima M, Kumahara
Y. Transient postpartum hypothyroidism. Fourteen cases with autoimmune thyroiditis. Ann Intern Med
1977;(87):155.
405. Amino N. Hashimoto's thyroiditis. In: Thyroid and its diseases. 2002.— Ch. 7. pp.1-19. Available from: URL:
http://www.thyroidmanager.org/thyroidbook.html.
406. Andrada J.A. et al.// Arch. Pathol. Lab. Med.-1979.-v.l03.-pp.244-246.
744 АЖ Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
407. Andreoli Т.Е. The posterior pituitary. In: Wyngaarden J.B., L.H.Smith. Cecil's Textbook of Medicine. 18th
Ed. Philadelphia a.e.: W.B.Saunders, Vv 1-2.-1988. Pp. 1305-1313.
408. Andrew A. et al.-Gen.Compar.Endocrinol.-1982.-v.47.-pp.249-265.
409. Aretaeus of Cappodocia, 1837, Of the Causes and Signs of Acute and Chronic Disease (translated by T.
F.Reynolds), Pickering, London.
410. Ascoli G.,Legnani Т.- Muench.Med.Wochenschr.- 1912.-Bd.59.-s.518.
411. Atwater W.O., Benedict EG. Bull. Off. Exp. Stns U.S. Dept Agric.-v.109, Washington, 1902.
412. Aurbuch G.D. Polypeptide and amine hormone regulation of adenylate cyclase.// Ann. Rev. Physiol.-1982.-
v.44.-pp.653-666.
413. Aurex J., Cock T Leptin: cutting the fat off the bone. //The Lancet. - 2003. - Vol. 362. -P. 1572-74.
414. Bach J.-F. Physiology of the endocrine function of the thymic epithelium. // Progr. Immunol.—1984.—V.
5,-Int. Congr. Immunol.-Kyoto.-aug. 21-26.- N 183.-P. 1563-1570.
415. Baier LJ, Sacchettini JC, Knowler WC, Eads J, Paolisso G, Tataranni PA, Mochizuki H, Bennett PH,
Bogardus C, Prochazka M. An amino acid substitution in the human intestinal fatty acid binding protein
is associated with increased fatty acid binding, increased fat oxidation, and insulin resistance. J Clin Invest
1995;95:1281-1287.
416. Baumann, E., Uber das normale vorkommen von jods im thierkorper, Z. Physiol. Chem..- 1896.-21:319.
417. Baxter J.D. Disorders of the adrenal cortex. In: Wyngaarden J.В., L.H.Smith. Cecil's Textbook of Medicine.
18th Ed. Philadelphia a.e.: W.B.Saunders, Vv 1-2.-1988. Pp. 1340-1360.
418. Bayliss W.M., Starling E.H. Mechanism of pancreatic secretion. //J. Physiol.( London).-1902.-v.28.-p.325-
353.
419. Beaudet A. The glycogen storage diseases. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2099-2105.
420. Beaudet A.L. Lysosomal storage diseases. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2090-2099.
421. Beaumont J.L., Fredrickson D. et al. // Classification of hyperlipidemias and hyperlipoproteinemias., Bull.
WHO.-1970.-N43.-p.891.
422. Beaumont J.L., Beaumont V Autoimmune hyperlipidemia. // Atherosclerosis.-1977.-v.26.-N 4.-pp.405-
418.
423. Behrman R.E. (Ed.) Textbook of Pediatrics. Philadelphia, 1992.
424. Bernal J. Science in history. London: Watts, 1954.-736 pp.
425. Bernard С Lecons de Physiologie Experimental, Appliquee a la Medecine., Bailliere et Fils, Paris, 1856.
426. Berson S., Yalow R.S. Hurvey Lect.-1968.-v.62.-p.107.
427. Berthold, A. A., 1849, Transplantation der Hoden, Arch. Anat. Physiol. Wiss. Med. 16:42.
428. Bettencourt, R., and Serrano, S. A., 1890, Un cas de myxoedeme traite par la greffe hypodermique du corps
thyroid d'un mouton, Sem. Med. 10:204.
429. Biermann E.L. Atherosclerosis and other forms of artheriosclerosis. In: Harrison's Principles of Internal
Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 1106-1116.
430. Bjorntorp P. «Portal» adipose tissue as e generator of risk factors for cardiovascular disease and diabetes. //
Arteriosclerosis. - 1990. - Vol. 10. - P. 493-496.
431. Bjorntorp P., Rosmond R. The metabolic syndrome: a neuroendocrine disorder? // Brit. J. Nutr.-2000.-v.83.-
suppl.l.-pp. 49-57.
432. Blecher M. Receptors, antibodies and disease. // Clin. Chem., 1984.- V 30.-N 7.-P. 1137-1156.
433. Bock R.// Diabetes Forecast.- 1988.- 2.- 40-43.
434. Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1996;45:3-10.
435. Bordeu T, Analyse medicinale du Sang: Recherches sur les malade chronique, Ruault, Paris, 1775.
436. Bosi E. et al.// Diabetes.- 1991.- 40.- 977-983.
437. Bourassa P.-A. et al., // Proc. Natl Acad.Sci. USA. - 1996. - v. 93. - pp. 10022-27.
438. Bournaud C, Orgiassi J.J. J. Endocrinol. Invest. 2003; - (26) - Suppl. 2. - P. 49-56.
439. Braunwald E. Edema. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.:
McGraw-Hill, 1994. Pp. 183-186.
440. Bray G.A. Complications of Obesity.// Ann. Intern. Med.-1985.-v.103.-pp. 1053-1062.
441. Brown M.S., Goldstein J.L. The hyperlipoproteinemias and other disorders of lipid metabolism. In: Harrison's
Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2058-2069.
442. Brown TR, Zhao G, Palmer КС, Sundick RS: Thyroid injury, autoantigen availability, and the initiation of
autoimmune thyroiditis. Autoimmunity 1998; 27:1-12
Литература 745
443. Brown-Sequard Ch.E., d'ArsonvalJ.-Arch.Physiol.Norm.Pathol. (Paris).-1891.-t.23.-p.491.
444. Brown-Sequard, С E., 1856, Recherches experimeniales sur la physiologie et la pathologie des capsules sur-
renales, C. R. Acad. Sci. 43:422.
445. Bruck K.,Wunnenberg B. Non-shivering thermogenesis and its integration in the thermoregulatory system.
In: Терморегуляция, адаптация к холоду.Новосибирск: СО Наука, 1970, с.28-39.
446. Bryson, A. (1850) On the respective values of limejuice, citric acid, and the nitrate of potash, in the treatment
of scurvy. Med. Times Gaz. (Lond.) 21:212-214,435-436.
447. Bubnow, N. A., 1884, Beitrage zu der untersuchung der chemischen bestandtheil der schilddruse des menichen
und des rindes, Z. Physiol. Chem. 8:1.
448. Budd, G. (1842) Lectures on the disorders resulting from defective nutriment. Lond. Med. Gaz. 2:632-636,
712-716,743-749,906-915.
449. Bunge, G. (1885) Ueber die Assimilation des Eisens. Hoppe-Seyler Z. Physiol. Chem. 9:49-59.
450. Burckhardt P. Idiopathic hypoparathyroidism and autoimmunity.// Hormone Res.-1982.-v.16.-N 5.-pp.
304-307.
451. Burr, G. O. & Burr, M. M. (1930) On the nature and role of the fatty acids essential in nutrition. J. Biol.
Chem. 86:587-621.
452. Burstein M., Samaille J. Nouvelic methode de separation et de disage des lipoproteines de faible densite //
Ann. Biol. clinique.-1959.-t. 17.-p.23.
453. Barker, D.J., Hales, C.N., Fall, C.H., Osmond, C, Phipps, K., Clark, P.M.,. Type 2 (non-insulin-dependent)
diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Dia-
betologia, 1993.- 36,62-67.
454. Camus J.-P. Goutte, diabete, hyperlipemie une trisyndrome metaboliquey/ Rev. Rhumat. et Mai. Osteo-ar-
ticul., 1966.-t.10.-p.33.
455. Cannon W.B. Bodily changes in pain, hunger, fear and rage. N.Y.-London, 1934.
456. Carnegie P. R., Me Kay J. R. Evolution of studies on antireceptor antibodies and disease // Springer Semin.
Immunopathol,- 1982,- V. 5.- P.. 379- 388.
457. Carpenter K.J. A Short History of Nutritional Science: Part 1 (1785-1885). J. Nutr. - 2003. - 133:
638-645.
458. Carpenter K.J. A Short History of Nutritional Science: Part 2 (1885-1912). J. Nutr. - 2003. - 133:
975-984.
459. Carpenter K.J. A Short History of Nutritional Science: Part 3 (1912-1944).J. Nutr. - 2003. -133: 3023-
3032.
460. Carpenter KJ. A Short History of Nutritional Science: Part 4 (1945-1985) J. Nutr. - 2003. - 133: 3331-
3342.
461. Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin
resistance in patients receiving HIV protease inhibitors. AIDS 1998;12:F51-8.
462. Carr B.D., Wilson J.D. Disorders of the ovary and female reproductive tract. In: Harrison's Principles of
Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2017-2036.
463. Castle, W. B. & Townsend, W. С (1929) Observations on the etiologic relationship of achylia gastrica to
pernicious anemia. II. Am. J. Med. Sci. 178:762-777.
464. Cawley, T, 1788, A singular case of diabetes, consisting entirely in the quality of the urine; with an inquiry
into the different theories of that disease, Med. J. (London) 9:286.
465. Chandrasoma P., Taylor C.L. Concise Pathology, 3rd Ed. Stamford: Appleton & Lange, 1998,1041 pp.
466. Cheadle, W. B. (1888) A discussion on rickets. Br. Med. J. ii: 1145-1148.
467. Chen H. et al. // Cell. -1996. - v.12. - pp. 318-320.
468. Chevreuil, M. E., 1815, Note sur le sucre de diabete, Ann. Chim. (Pans) 95:319.
469. Christie, T. (1804) Letter on beriberi. In: An Essay on the Diseases Incident to Indian Seamen (Hunter, W.,
ed.) pp. 77-87. Honorable E. India Co., Calcutta.
470. Chrousos G.P. The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the
metabolic syndrome: neuro-endocrine and target tissue-related causes. Int J Obes Relat Metab Disord.-
2000.- 24.- Suppl. 2. - pp. 50-55.
471. Churilov L.P., .Stroev Yu.L, Mudzhikova O.M.,Pisareva S.V.,Sadov S.A. Anti-thyroid Autoimmunity in
Adolescent Hypothalamic Obesity: Simpson-Page' Syndrome as Immuno-neuroendocrine Disorder. In:
International symposium "Interaction of the Neurous and Immune Systems in Health and Disease" May 31st
-June 2nd, 2007. Abstracts. St. Petersburg: ElBi Publ., 2007. - pp.17-18.
472. Clerc G. Atherosclerosis as an immune disease? // Med. hypotheses.-1991.-V.36.-N 1.- pp.24-26.
746 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
473. CockT.-A.,AuwerxJ. Leptin: Cutting the fat off the bone. //Lancet- 2003. -N362. -p. 1572-1574.
474. Cohen A.S. Amyloidosis. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.:
McGraw-Hill, 1994. Pp. 1625-1630
475. Cohen В., Novick D., Rubinstein M. Modulation of insulin activity by leptin. Science. — 1996. — v. 274.-
pp.l 185-1188.
476. Collip, J. В., and Anderson, E. M., 1935, Studies on the thyrotropic hormone of the anterior pituitary, / Am.
Med. Assoc. 104:965.
477. Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L. Endogenous glucose production is inhibited by the
adipose-derived protein АсгрЗО. J Clin Invest 2001; 108:1875-81.
478. Combs TP, Pajvani U.B., Berg AH. A transgenic mice with a deletion in the collagenous domain of adiponectin
sisplays elevated circulating adiponectin and improved insulin sensitivity // Endocrinology. -2004. -v. 145.
-pp.367-385.
479. Comby J. L'uricemie chez les enfants. // Arch, de med. des enfants.-1901.-t.4.-Nl.-pp.l-20.
480. Cooley D.G. Iss Dich schlank: Ullstern, Frankfurt a. M.., Berlin, 1963.
481. Cotran R.S., Kumar V., Robbins S.L. Robbins' Pathologic Basis of Disease. 4th Ed. Philadelphia a.e.:
W.B.Saunders, 1989.
482. Cryer Ph.E. The adrenal medulla and the sympathetic nervous system. In: Wyngaarden J.B., L.H.Smith. Cecil's
Textbook of Medicine. 18th Ed. Philadelphia a.e.: W.B.Saunders, Vv 1-2.-1988. Pp. 1461-1467.
483. Cummings. D., Shwartz.W Genetics and Pathophysiology. //The Annual Review of Medicine.-2003.-
N54.-P.453-471
484. Cundy T, Cornish J., Roberts H., Tolder H., Reid I.R. //| Obst. & Gyn. - 1998. - Oct, 92 (4 Ptl). - p.
569-573.
485. Cushing H. Studies in intracranial Physiology and Surgery. London: Oxford Univ.Press, 1925.
486. Dam, H. (1935) The antihaemorrhagic vitamin of the chick. Nature, Lond. 135:652-653.
487. Danforth Jr E. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet 2000;26:13.
488. Daniels G.H., Martin J.B. Neuroendocrine regulation and diseases of anterior pituitary and
hypothalamus. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp.
1891-1918.
489. Daniels G.H., Martin J.B. Neuroendocrine regulation and diseases of the anterior pituitary and
hypothalamus. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp.
1891-1918.
490. Daw K., Powers A.C. // Diabetes.- 1995.- 44.- 216-220.
491. DeFronzo R.A., Tobin J., Anders R. Glucose clump technique- a method for quantifying insulin secretion and
resistance.// Amer. J. Physiol.-1979.-v.237.-pp.214-223/
492. DeGroot LJ. et al. Endocrinology, 3rd ed. Philadelphia:Saunders, 1995.
493. DeLillis R.A. The Endocrine System. In: Cotran R.S., Kumar V, Robbins S.L. Robbins' Pathologic Basis of
Disease. 4th Ed. Philadelphia a.e.: W.B.Saunders, 1989.-pp. 1205-1276.
494. Denke MA. Connections between obesity and dyslipidemia. Curr Opin Lipidol. 2001;12:625-628.
495. Dobson, M., 1776, Experiments and observations on the urine in a diabetes, Medical Observations and
Inquiries (London) 5:298.
496. Dockray, G. J„ 1978, Polypeptides in brain and gut: Cholecystokinin-like peptides, in Gastrointestinal
Hormones and Pathology o/ the Digestive System (M. Grossman, V. Speranza, N. Basso, and E. Lezoche, eds.),
pp. 263-269, Plenum Press, New York.
497. Doniach D. Les immunoglobulins stimulantes de croissance (TCI), peuvent-elles expliquer certains goitres
sporatiques thyroidiens? // Ann. endocrinol.— 1982.— v. 43.— N 6.— P. 543—547.
498. Donnan EG. The theory of membrane equilibris.- Chem.Rew.-1924.-col.l.-p. 73.
499. Drachman D. Myasthenia gravis. New EnglJ.Med.-1994.-v.330.-p.1797.
500. Drexhage H. A., Wiersinga W M„ Gaag R. D, van der, Thyroid growth stimulating immunoglobulins //
Aktuel. Endocrinol, und Stoffwechsel.- 1984.-V. 5.-N l.-P. 104-108.
501. Dunstan DW, Zimmet PZ, Welborn ТА et al. The rising prevalence of diabetes and impaired glucose tolerance.
The Australian Diabetes, Obesity and Lifestyle Study. Diabetes Care 2002;25:829-34
502. Durnin J.V.G.A., Passmore R. Energy, work and leisure. London: Heineman, 1967.
503. Dusantez-Fourt 1., Djiane J., Kelly P. A. et al. Differential biological activities between mono- and bivalent
fragment of anti-prolactin receptor antibodies // Endocrinology—1984.—V. 114.—N 3.—P. 1021—1027.
504. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415-28.
505. Edel H. // Z. arztl. Fortblid., 1963.-Bd.57.-S. 535-540
Литература 747
506. Edkins, J. S., 1905, On the chemical mechanism of gastric secretion, Proc. R. Sot. London Ser. В 76:376.
507. Eisenbarth G. S. Autoimmune Beta Cell Insufficiency-Diabetes Mellitus Type I // Triangle, 1984.-V. 23-N
34.-P. 111-124.
508. Elefteriou E, Takeda S., Lin X., Armstrong D., Karsenty G. Monosodium-glutamate sensitive hypotalamic
neurons contribute to the control of bone mass. // Endocrinology. - 2003. - N 144. - p. 3842-3847-
509. Elhage R. et al., // Endocrinology. - 2000/- v. 141. - N 1. - pp. 462-66.
510. Elin R. Reference intervals and laboratory values of clinical importance. In: Wyngaarden J.B., L.H.Smith.
Cecil's Textbook of Medicine. 18th Ed. Philadelphia a.e.: W.B.Saunders, 1988, v 2.-pp.2394-2404.
511. Endroczi E. Limbic System, Learning and Pituitary-Adrenal Function. Budapest: Academiai Kiado, 1972.
5i2. Engel EG. Recent Progress in Horm. Res. (Ed. G.Pinchus).-1951.-v.6.-p.277.
513. Eppinger H., Hess L. Die Vagotonic Berlin, 1910.
514. Estivariz P. E., lturriza F, McLean С et al. Stimulation of adrenal mitogenesis by N-terminal proopiocortin
Peptides // Nature.-1982.-V. 297.- N. 5865.-P. 419-422.
515. Euler U.S. von. Noradrenaline. Chemistry, Physiology, Pharmacology and Clinical Aspects. Springfield,
1956.
516. Evans, H. M., and Long, J. A., 1921, The effect of the anterior lobe administered intraperitoneally upon growth,
maturity, and oestrus cycles of the rat, Anat. Rec. 21:62.
517. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive
summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on
Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Adult Treatment Panel III).
JAMA 2001;285:2486-2497.
518. Falchuk K.H. Disturbances in trace element metabolism. In: Harrison's Principles of Internal Medicine. 13th
Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 481-484.
519. Falta W. Erkrankungen der Blutdruse. 2 Auflage., Berlin Springer Verlag, 1913.
520. Falta W., Boiler R. Insularer und insulinresistenter diabetes. Klin Wochenschr 10 (1931) 438 -443.
521. Farese R. V. The phosphatidate-phosphoinositide cycle: an intracellular messenger system in the action of
hormones and neurotransmitters // Metabolism,— 1983.—V. 32.—N 6.—P. 628—641.
522. Farid N. R., Briones-Urbina R., Baer E. Graves's disease—the thyroid stimulating antibody and
immunological networks. // Mol. Aspects of Med.- 1983.-6.- 5,- 355-457.
523. Farid N.R. , Linticum D.S.-Anti-ldiotypes, Receptors and Holecular Mimicry-1988.-Springer Ver-
lag.-N.Y.-l-317.
524. Farquhar M.G. Multiple pathways of exocytosis, endocytosis and membrane recycling: validation of Golgi
route. // Fed.Proc.-1983.-v.42.-N 8.- pp.2407-2413.
525. Fernel J. De naturali parte medicinae. Paris, 1542.
526. Feyrter F. Ueber Diffuse Endocrine Epitheliale Organe., Barth:Leipzig, 1938.
527. Findlay, L. (1908) The etiology of rickets: a clinical and experimental study. Brit. Med. J.ii:13—17.
528. Fink J. N., Beall G. M. Immunologic aspects of endocrine diseases //J. A. M. A-1982.-V. 248.-N 20.-P.
2692-2700.
529. Fishberg A.M. Anatomic findings in essential hypertension. // Arch. Intern. Med. - 1925. - Vol. 35., № 5.
- P. 650-668.
530. Flajani, G., 1802, Cllemone d'osservalioni e reflessiuni di chirurgia. Vol. 3, p. 370, Salomoni, Rome.
531. Ford E.S., Giles W.H., Dietz W.H. Prevalence of the metabolic syndrome amoung US adults: findings from the
third National Healthand Nutrition Examination Survey. //JAMA. - 2002. - Vol. 287 (3). - P. 356-359.
532. Foster D.W Anorexia nervosa and bulimia. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 452-455.
533. Foster D.W. The lipodystrophies and other rare disorders of adipose tissue In: Harrison's Principles of Internal
Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2131-2136.
534. Fox I.H. Metabolic basis for disorders of purine nucleotide degradation. // Metabolism.-1981.-v.30.-N
6.-pp.616-634.
535. Fredrickson D.S., Levy R.I. Familiar hyperlipoproteinemia. // In: Metabolic Basis of Inherited Disease / Ed.
J.B.Stanbury et al.-N.Y, 1972.-pp.l99-212.
536. Freissmuth M., Gillman A.G. G-Proteins and the regulation of second messenger system. In: Harrison's
Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994.Pp. 426-431.
537. Friedman L.S., Isselbacher KJ. Anorexia, nausea, vomiting, and indigestion. In: Harrison's Principles of
Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 208-213.
538. Friedman M. et al.-Amer. J.Med.-1968.-v.44.-p.525-537.
748 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
539. Frohman L.A. Heuroendocrine regulation and its disorders.The anterior pituitary. In: Wyngaarden J.B.,
L.H.Smith. Cecil's Textbook of Medicine. 18th Ed. Philadelphia a.e.: W.B.Saunders, Vv 1-2.-1988. Pp.
1280-1305.
540. Fujita H. Adrenal cortex // In: Functional morphology of endocrine glands / Kurosumi and Fujifa, eds,—
Stuttgart and Tokyo: George Thieme and Igaku Shoin.- 1975.-P. 300-342.
541. Funk, C. (1911) On the chemical nature of the substance which cures polyneuritis in birds, induced by a diet
of polished rice. J. Physiol. 43:395-400.
542. Fuster W et al. New Engl. J. Med.-1992.-v.326.-N 5.-pp.312-327.
543. Garrod A.E. Inborn errors of metabolism. Oxford: Univ.Press, 1909.
544. Garsia-Rubi E. et al. Trp64Arg variant of the 3-adrenoreceptor and insulin resistance in obese postmenopausal
women//J. Clin. Endocrinol.-1998.-v.83.-pp.4002-4005.
545. Gerstein H.C. // Diabetes Care.- 1994.- 17.- 1.- 13-19.
546. Giffin J.E., Wilson J.D. Disorders of the testes. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2006-2017.
547. Gley, E., 1891, Sur les fonclions du corps thyroide, С R. Soc. Biol. 3:841.
548. Goldberger, J. (1916) The transmissibilty of pellagra. Experimental attempts at transmission to the human
subject. Publ. Hlth Repts 31:3159-3162,3165-3167.
549. Goldstein J.L., Brown M.S the LDL Receptor focus and the genetics of familiar hypercholesterolemia.-Ann.
Rev.Gen.-1979v.13.-pp. 259-289.
550. Goldstein J.L., Brown M.S. Genetic aspects of disease. In: Harrison's Principles of Internal Medicine. 13th
Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 339-349.
551. Goodpaster В., Thaete E, Simoneau J. et al. Subcutaneosus a abdominal fat and thigh muscle composition
predict insulin sensitivity independently of visceral fat. // Diabetes. - 1997. - Vol. 46. - P. 1579-1585.
552. Graves, R. J., 1835, Clinical lectures, London Med. Surg. J. 7:516.
553. Greenspan ES. et al. Basic and Clinical Endocrinology. 5th ed. Stamford: Appleton & Lange, 1997.
554. Gull. W. O., On a cretinoid state supervening in adult life in women, Trans. Clin. Soc. London 1873-1874. -
7:180.
555. Guyton A.C. Textbook of Medical Physiology/ 7th Ed. Philadelphia a.e.: W.B.Saunders Co, 1986.
556. Hann, P. E, Bale, W E, Ross, J. E, Balfour, W. M. & Whipple, G. H. (1943) Radioactive iron absorption by
gastro-intestinal tract: influence of anemia, anoxia, and antecedent feeding distribution in growing dogs. J.
Exp. Med. 78:169-188.
557. Hales, C.N., Barker, D.J., Clark, P.M., Cox, L.J., Osmond, C, Winter, P.D., 1991. Fetal and infant growth and
impaired glucose tolerance at age of 64. British Medical Journal 303,1019-1022.
558. Hamburger, E. W. (1878-9) ITber die Aufnahme und Ausscheidung des Eisens. Z. Physiol. Chem. 2:
191-205.
559. Hammar J. Die normal-morphologische Thymus-forschung im letzten vier tei ljahrhundet. Analyse und
Synthese nebst einingen. Worten Zuden Funktionstrage// Leipzig.-1936.
560. Hanefeld M., Leonhardt W Das metabolische Syndrome // Dtsch. Gesundh. Wes. - 1980.- Vol. 36. - S.
545-551.
561. Hansemann D.R. Uber das konditionale Denken in der Medizin und seine Bedeutung fur die Praxis.-Ber-
lin.- 1912.
562. Harington, С R.,, Chemistry of thyroxine. 1. Isolation of thyroxine from the thyroid gland, BiochemJ.
1926.-20:293.
563. Harrison L.C.-Proc.Endocr.Soc.Austral.-1984.-v.27.-p.318.
564. Hart, E. В., McCollum E. V, Steenbock, H. & Humphrey, G. С (1911) Physiological effect on growth and
reproduction of rations balanced from restricted sources. University of Wisconsin Agricultural Experiment
Station Bull. 17:131-205.
565. Havel P.J, Kasin-KarakasS., Vueller W et al. Relationship plasma leptin to plasma insulin and adiposity in
normal weight and overweight women: effects of dietary fat content and sustained weight loss. //J. clin.
Endocrinol. - 1996. - Vol. 91. - P. 4406-4413.
566. Havel PJ. Control of energy homeostasis and insulin action by adipocyte hormones: leptin, acylation
stimulating protein, and adiponectin. Curr Opin Lipidol 2002;13:51-9.
567. Havel RJ. Classification of the hyperlipidemias. // Annu. Rev. Med.-1977.-v.28.-pp.l95-209.
568. Havrankova J. et al.-Proc. Natl Acad. Sci. USA .-1978.-v.75.-pp. 5737.
569. Hawker E- Brit.J.Hosp.Med.-1988.-v.39.-N4.-pp.280-287.
Литература 749
570. Hayden M. R, Tyagi S.C. Uric acid: A new look at an old risk marker for cardiovascular disease, metabolic
syndrome, and type 2 diabetes mellitus: The urate redox shuttle.// Nutr. & Metab. 2004,1:10
571. Haynes W.G. et. al. Receptor-mediated regional sympathetic nerves activation by leptin.//J. Clin. Invest-
1997.-v.l00.-pp.270-278.
572. Healy D. L., Hodgen G. D., Schulte H. M. et al. The thymus— adrenal connection: thymosin has corticotropin
releasing activity in Primates // Science- 1983.-V.222.-N4630.-P.1353-1355.
573. Hench Ph.S., Kendall E.S. et al. Proc. Staff. Meet. Mayo Clin.-1949.-v..24.-p.l81.
574. Herbert E. et al. Biosynthesis of ACTH and related peptides. In: Hormonal Proteins and Peptides.-1987.-
v.l3.-pp.59-87.
575. Herbert V. Nutrition Cultism. G.F. Stickley & Co: Philadelphia, 1980.-234 pp.
576. Herbert V., Barrett S. Vitamins and «Health» Foods: The Great American Hustle.- G.E Stickley & Co:
Philadelphia, 1980.-190 pp.
577. Higgins J.R. Thyroid and adrenal disorders in critically ill patients. Prog. Crit. Care Med.-1985.-v.2.-
pp.262-272.
578. Himsworth, H.P. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types.
Lancet. I (1936) 127-130.
579. Hintz R.L. Disorders of growth. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.:
McGraw-Hill, 1994. Pp. 1918-1921.
580. Holick M.E et al. Calcium, phosphorus and bone metabolism: calcium-regulating hormones. In: Harrison's
Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2137-2151.
581. Holmes O. Human acid-base physiology. London.: Chapman & Hall, 1993.
582. Hoist, A. & Fro'lich, T. (1907) Experimental studies relating to «ship beri-beri» and scurvy. II. On the
etiology of scurvy. J. Hyg. (Cambridge) 7:634-671.
583. Hornsby P.,Crivello J.F. // Mol. & Cell. Endocrinol.-1983.-v.30.-N l.-pp.l-20,123-147.
584. Hoskins, R. G„ 1949, The thyroid-pituitary apparatus as a servo (feed-back) mechanism, /Clin. Endocrtnol.
// 9:1429,
585. Hotamisligil G. S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alfa: direct
role in obesity-linked insulin resistance // Science. — 1993. — v. 259. — pp. 87-91.
586. Houssay, B. A., and Biasotti, A., 1931, The hypophysis, carbohydrate metabolism and diabetes.
Endocrinology 15:511.
587. Ingle D.J. Recent Progress in Horm. Res. (Ed. G.Pinchus).-1951.-v.6.-p.l59.
588. International Diabetes Federation. In: Promoting diabetes care, prevention and a cure worldwide.
Backgrounder 1. The IDP consensus worldwide definition of the metabolic syndrome. Pp. 1-7. Awailable from:
www.idf.org [ 7 screens, 2nd June 2005].
589. Isomaa В., Almgren P., Tuomi T. et al. Cardiovascular morbidity and mortality associated with the metabolic
syndrome.// Diabetes Care. - 2001. - Vol. 24(4). - P. 683-689.
590. Isselbacher K. et al (Eds). Appendix: laboratory values of Clinical Importance. In:Harrison's Principles of
Internal Medicine. 13th Ed., V2.-N.Y. a.e.: McGraw-Hill, 1994, pp. 2489-2496.
591. Isselbacher K. et al (Eds). Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.:
McGraw-Hill, 1994.
592. Jackson R., Gotto A.M. Hypothesis concerning membrane structure, cholesterol, and atherosclerosis.//
Atherosclerosis Rev.-1976.-v. 1 .-p. 1 -21.
593. James W.H.T. (Ed.) The Adrenal Gland. 2nd ed. Raven Press, 1992.
594. Jaquet, D., Gaboriau, A., Czernichow, P., Levy-Marchal, C.,. Insulin resistance early in adulthood in
subjects with intrauterine growth retardation. Journal of Clinical Endocrinology and Metabolism, 2000. — 85,
1401-1406.
595. Joslin S. The treatment of Diabetes. 4th Ed. Lea & Febiger: Philadelphia, 1928.
596. Kahn R., Buse J., Ferrannini E., Stern M. The metabolic syndrome: time for critical appraisal. Joint statement
of ADA and EASD. Diabetologia, 2005. - v. 48. - pp.1684-1699.
597. Kaplan N.M. The deadly quartet. Upper body obesity, glucose intolerance, hypertriglyceridemia, and hyperte-
sion. // Arch. Intern. Med. - 1989. - Vol. 149. - P. 1514-1520.
598. Katchalsky A. In: Chemical Dynamics of Macromolecules and its Cybernetics Significance., (Ed. S. Devons),
N.Y.: Columbia Univ. Press, 1969.
599. Katsuya Т., Horiuchi M., Chen y. et al. Relation between delenion polymorphism of angiotensin-convertin-
genzyme gene and insulin resistance, glucose intolerance, hyperinsulinemia, and dyslipidemia. // Arterioscler.
- Thromb.. Vase. Biol. - 1995. - Vol. 15, № 6. - P. 779-782.
750 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
600. Кау М. М, В., Goodman S. R., Sorensen К, Senescent cell antigen is immunologically related to band 3 //
Proc. Nat, Acad. Sci. USA.- 1983.- V, 80.- N 6,-P. 1631-1635.
601. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type
2 diabetes. Diabetes 2002; 51:2944-50.
602. Kendall M.D. (ed.) The Thymus Gland. London a.e.: Acad. Press, 1981,218 pp.
603. Kleeman K.E., Kleeman Ch.R. Parathyroid hormone and calcitonin. Annu. Rev. Physiol., 1982.-pp/
247-344.
604. Klein K.O., Larmore K.A., de Lancey E., Brown J.M., Considine R.V., Hassink S.G. Effect of obesity on
estradiol level, and its relationship to leptin, bone maturation, and bone mineral density in children. //J. Clin.
End. and Metab. - 1998. - V. 83. - N 10. - p. 3469-3475.
605. Knapp, P. (1909) Experimenteller Beitrag zur Ernahrung von Ratten mit ku" nstlicher Nahrung und zum
Zusammenhang von Erna" hrungssto" rungen mit Erkrankungen der Conjunctiva. Z. Exp. Pathol. Therap.
5:147-169.
606. Knochel J.P. Disorders of magnesium metabolism. In: Harrison's Principles of Internal Medicine. 13th Ed.,
Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2187-2190
607. Kocher Т.- Korrespondenzbl. Schweiz. Arzte.-1895.-Bd.25.-s.3.
608. Koopmans H.S. et al. Effect of cholecystokinin-pancreatozymin on hunger and thurst in mice.// Behav. Biol.
-1972.-v.7.-p.441-444.
609. Korbonits M. Leptin and thyroid - A puzzle with missing pieces. //Clin. Endocrinol.—1998. — Vol.49.—P.
569-572.
610. Kritchevsky D. Dietary fiber // Annu. Rev. Nutr.-1988.-v.8.-p.301-328.
611. Krohn K. et al. // Lancet.-1992.-N8796.-pp.770-773.
612. Kuhn, R., Reinemund, K., Weygand, F. & Stro" bele, R. (1935) U" ber die Synthese des Lactoflavins. Ber.
Deutsch. Chem. Gesells. 68:1765-1774.
613. Kumar P., Clark M. Clinical Medicine. 5th Ed. W.B. Saunders.Edinburgh a.e.,2002.-1446 pp.
614. Kumar V., Cotran R.S., Robbins S.L., Schoen F Robbins' Pathologic Basis of Disease. 5th Ed. Philadelphia
a.e.: W.B.Saunders, 1994.
615. Kylin E: Studies of the hypertension-hyperglycemia-hyperuricemia syndrome (Studien ueber das hypertonie-
hyperglykamie-hyperurikamiesyndrom.) Zentralblatt fuer Innere Medizin.-1923.- 44:105-127.
616. Lane M.A. // Amer. J. Anat.-1907.-v.7.-pp. 409-421.
617. Langerhans, P.„ Beitrage zur mikmskopischen Anatomie der Bauchspeichetdruse, Lange, Berlin, 1869.
618. Larsson H., Elmstahl S., Ahren B. // Deabetes. - 1996. - v.45. - pp.1580-1584.
619. Lederis K., Veale W. Current studies of hypothalamic function. Basel a.e.: S.Karger, 1978,218 pp.
620. Lehrke M., Lazar M.A. Inflamed about obesity. Nat Med 2004,10:126-127.
621. Leja J.J. Biezako klinisko analizu obligatas normas.Pec: Ilustracijas patologiskas fiziologijas lekcijam.RMI:
Riga, 1986, i. 82-84.
622. Lemieux I, Pascot A, Couillard C, et al. Hypertriglyceridemic waist: a marker of the atherogenic metabolic triad
(hyperinsulinemia; hyperapolipoprotein B; small, dense LDL) in men? Circulation. 2000; 102:179-184.
623. Leonhardt W. et al. // Diabetologia, 1972.-N 8.-pp.287-291.
624. Lerner A. et al. Structure of melatonin.-J. Amer. Chem. Soc.-1959.-v.81.-p.6084-6086.
625. LeRoith D. Evolutionary origin of the endocrine system.// J.Endocrinol.-1987.-V.112.-suppl.-p.27.
626. Leschner A.I. An Introduction to Behavioral Endocrinology. N.Y.-Oxford:Univ.Press,1978.
627. Levey G.S., Robinson A.G. Introduction to the general principles of hormone-receptor interactions. //
Metabolism.-1982.-v.31.-N7.-pp.639-645.
628. Levin K. // Clin.Chem.-1977.-v.23.-pp.929-937.
629. Levinsky N.G. Acidoses and alcaloses. In: Isselbacher K. et al (Eds). Harrison's Principles of Internal Medicine.
13th Ed., V2.-N.Y. a.e.: McGraw-Hill, 1994,
630. Levinsky N.G. Fluids and Elecrolytes. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y.
a.e.: McGraw-Hill, 1994. Pp. 242-252.
631. Li M.,Kane J. Effect of nicotine on the expression of leptin and forebrain leptin reseptors in the rat.//Brain
research.-2003-991(l-2) - P. 222-31.
632. Liberman Y.A.,Sculachov V.P. // Biochem.Biophys.Acta.-1970.-v.216.-p.30.
633. Lind, J. A Treatise of the Scurvy, p. 60. MillanEdinburgh, UK, 1753 (Reprinted 1953 by University of
Edinburgh Press).
Литература 751
634. Lingappa V.R. Disorders of the Hypothalamus and Pituitary Gland. In: McPhee S.J. et al.
Pathophysiology of Disease. An Introduction into Clinical Medicine. 2nd Ed. Stamford: Appleton & Lange, 1997, Pp.
449-469.
635. LiVoisi V.A.et al.(Eds). Pathology. 3rd Ed. Philadelphia a.e.:Harwal PubL, 1994.-508 p
636. Livshits G., Trofimov S.,Pantsulaia Т., Genetic variation of circulating leptin is involed in genetic variation
of hand bone size and geometry.//Osteoporosis international.-2003.-14(6).—P.476-83.
637. Luddens H. et al. Studies on the porcine adrenal ACTH-receptor. // Hoppe Seyler's Z. Physiol. Chem.-
1982.-v.363.-N9.-p.942.
638. Ludens J.H. et al. Rat adrenal cortex is a source of circulating ouabainelike compound. // Hypertension.-
1992.-v.l9.-N6.-Pt.2.-pp.721-724.
639. Luke J. Fluoride Deposition in the Aged Human Pineal Gland.// Caries Res 2001;35:125-128
640. Magendie, E (1816) Sur les proprie' te' s nutritives des substances qui ne contiennent pas d'azote. Ann.
Chim. (ser. 2) 3:66-77,408-410.
641. Magnus-Levy, A.,, Gaswechtel bei thyroidea, Z, Klin. Wochenschr. 1895.- 32:650.
642. Magnus-Levy, A„ 1907, Der stofFwechsei bei erkrankungen einer drusen ohne ausfuhrgang, in: Handbuch der
Pathologie des Stoffwechsels (C. von Noorden, ed.), pp. 311 -354, A Hirschwalt, Berlin.
643. Malmros H. // Lancet, 1969.-2.-479.
644. Manley W.W. et al. Springer's Seminars in Immunol.- 1982.-V.5.-N. 4.-pp.413-431.
645. ManneJ., Argeson A.C., Siracusa L.D. // PNAS USA.-1995.-v.92.-N 1 l.-pp.4721-4724.
646. Margulius H.S. Kallikreins and Kinins. Hypertension.-1995.-v.26.-p.221.
647. Marie, P., 1886, Sur deux cas d'acromegalie, Hypertrophic singuliere, non congenital des extremites super-
ieures, inferieurs et cephalique, Rev. Med. 6:297.
648. Marine, D. & Kimball, O. P. (1920) Prevention of simple goiter in man. Arch. Intern. Med. 25:661-672.
649. Mason J.B., Rosenberg I.H. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.:
McGraw-Hill, 1994. Pp. 437-440..
650. Mason J.B., Rosenberg I.H. Protein-Energy Malnutrition . In: Harrison's Principles of Internal Medicine.
13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 440-446.
651. Matarese.G., Sanna,V., Lechler.R et al. Leptin accelerates autoimmune diabetes in female NOD mice.
//Diabetes. -2002.--N51.-P.1356-1361.
652. Matey H. Genetyczne podstawy autoimmunizacji // Immunol, pol.— 1984.—V. 9.—N 1.—P. 65—72.
653. McCance, R. A. & Widdowson, E. M. (1937) Absorption and excretion of iron. Lancet ii: 680-684.
654. McCollum, E. V. & Davis, M. (1915) The nature of the dietary deficiencies of rice. J. Biol. Chem. 23:
181-230.
655. McPhee S.J. et al. Pathophysiology of Disease. An Introduction into Clinical Medicine. 2nd Ed. Stamford:
Appleton & Lange, 1997,604 pp.
656. Mehnert H., Kuhlmann H. Hypertonic und Diabetes mellitus. // Dtsch. Med. J. - 1968. - Vol. 19. - S.
567-571.
657. Mellanby, E. (1921) Experimental rickets. Med. Res. Council Spec. Rept. Ser. No. 61.
658. Melton LJ. 3rd. Osteoporosis: Magnitude of the problem: Worlwide and future. // 4th International
Symposium. June 4-7,1997. - Washington, 1997. - p. 23.
659. Miller J.F. Immunological Function of the Thymus.-Lancet.-1961.-v.2.-N 7205.-pp. 748-751.
660. Millingto J.,Chan J.,Van Donsen H. Sleep loss reduses diurnal rhythm amplitude of leptin in healthy men.//
Journal of Neuroendocrinology .-2003.-N15(9).-P.851-854.
661. Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorilation // Biochem.Biophys.
Res.Comm.-1965-v.41 .-p.445.
662. MohrM.,KohlerH.// Dt. Gesundt. Wesen, 1969.-Bd.24.-S.l 167-1171.
663. MonkebergJ.G.//Virch. Arch.-1903.-Bd.121.-S. 141-166.
664. Moore R.Y. // In: Yen S.S,C, Jaffe R.B. (Eds). Reproductive Endocrinology: Physiology, Pathophysiology
and Clinical Management.-Philadelphia: W.B. Saunders Publ.-1978.-pp.3-33.
665. Moses A.M., Streeten D.H.P. Disorders of the neurohypophysis. In: Harrison's Principles of Internal Medicine.
13th Ed., Vv 1-2.-N.Y a.e.: McGraw-Hill, 1994. Pp. 1921-1930
666. Mueller, J., 1844, Handbuch der Physiologie des Menschen fur Vorlesungen, Holschen, Coblenz.
667. Muller-Wieland D, Taub R, Tewari DS, Kriauciunas KM, Sethu S, Reddy K, Kahn CR. Insulin-receptor gene
and its expression in patients with insulin resistance. Diabetes 1989;38:31-38.
668. Murray I. et al. //J. Biol. Chem. - 1998. - v. 274. - pp. 36219-36225.
752 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
669. Nan-ping Wang. Unveiling the role of PPAR-6 in endothelial biology. // Chinese J. of Pathophysiology.
-June, 2006. - Vol. 22. - N 13 (Supplement). - P. 68.
670. Nathan A.W. et al. // Brit. J. Exp. Pathol.-1983.-v. 64.-N 5.-pp. 474-478.
671. NBME (National board of medical examiners).- Laboratory Values Tables. In: Self-test in the Part I Basic
Medical Sciences.Philadelphia, 1991.-pp4-6.
672. Neel, J.V. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress". American Journal of
Human Genetics 1962,14,353-362.
673. Nenci I. et al. The plasma membrane as an additional level of steroid-cell interaction.// J.Steroid. Bio-
chem.-1981.-v.l5.-pp.231-234.
674. Neville A.M., O'Hare M J. The Human Adrenal Cortex. Pathology and Biology. An Integrated
Approach.-Berlin a.e.: Springer Verlag, 1982,354 pp.
675. Novak K., Kaczmarec P., Mackowiak P. et al. Rat thyroid gland express the long form of leptin receptors,
and leptin stimulates the function of the gland in euthyroid non-fasted animals. //International Journal of
Molecular Medicine. - 2002. - N 9. - P. 31-34.
676. Nussdorfer G.G. et al.-Atti delPlnst.Venet.Sci. Lett. Arti.-1979-1980.-v.l38.-pp.l71-178.
677. O'Malley B.W. et al. // 5th Symp. Gene Transcript. In Reproductive Tissue, 1972.-p.17
678. Odeleye O., de Courten M., Pettitt D et al. Fasting hyperinsulinemiais a predictor of increased body weight
gain and obesity in Pima Indian children. // Diabetes. - 1997. - Vol. 46. - P. 1341-1345.
679. Olefsky J.M. Obesity. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-
Hill, 1994. Pp. 440-446.
680. Omura Y. The hypothalamus and feeding behavior. Shinkei Kenkyu No Shimpo. — 1966. — v. 10. — N 1. — pp.
84-97.
681. Orci L., Unger R.// Lancet- 1975.-1243-1244.
682. Ord, W. M,„ On myxoedema, a term proposed to be applied to an essential condition in the cretinoid affection
occasionally observed in middle-aged women, Med. Chir.-Trans. London 1878.- 61:57.
683. Osancova K., Hejda S.//Nutrit. Rep. Internat, 1972.-N 6.-pp. 191-198.
684. Osborne, T. & Mendel, L. B. Feeding experiments with isolated food substances. Carnegie Institution of
Washington, 1911. - Publ. No 156.
685. Pallett A.L., et al. Advancing age and insulin resistance: role of plasma tumor necrosis factor-alfa // Amer. J.
Physiol. - 1998. - v. 275. - pp. 294-299.
686. Palm, T. A. The geographical distribution and aetiology of rickets. Practitioner. — 1890. — 45: 270-279,
321-342.
687. Paltauf A. Ueber die Beziehungen der Thymus zum plotzlichen Tod.//Wien.Clin Wschr.-1889.-v.46.-pp.
877-881.
688. Paolisso G. et al. // Hypertension.-1999.-v.34.-pp. 1047-1053.
689. Parry, С. Н,„ Collections from the unpublished medical writings of the late Caleb lhllier Parry, Vol. 2,
Underwoods, London, 1825.
690. Pauling L.-Vitamin С and longevity- Agressologie.-1983.-v.24.-N7.-pp.317-319.
691. Pawlikowsky M. The link between secretion cind mitosis in endocrine glands Life Sci. —1982.— V. 30.—N
4.-P. 315-320.
692. Pearce A.G.E. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD
series and the embryologic, physiologic and pathologic inmlications of the concept. //J. Histochem. Cyto-
chem.-1969.-v.17.-N 5.-pp. 303-313.
693. Pende N. Konstitution und innere Sekretion.,1924.
694. Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin
resistance. Science 2003; 300:1140-42.
695. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-
resistant offspring of patients with type 2 diabetes. N Engl J Med 2004; 350:664-71.
696. Pevet P.-In: Human Reproduction. Proc. 3rd World Congress, 1981.-pp.544-549.
697. Pierpaolis W et al. Interdependence between neuroendocrine programming and the generation of immune
recognition in ontogeny. // Cell.Immunol.-1977.-v.29.-N l.-pp.l6-27.
698. Plum F. van Uitert R. // Non-endocrine diseases of Hypothalamus./ In: The hypothalamus (Eds: Reichlin
S. et al.).-N.Y.: Raven Press.-1978.-p.415.
699. Pontiroli A., Carpa E, Veglia F et al. Genetic contribution of polymorphism of the GLUT 1 and GLUT 4
genes to the susceptibiluty to type 2 (nob-insulin-dependent) diabetes mellitus in different population.//
Acta Diabetol. - 1996. - Vol. 33. - P. 193-197.
Литература 753
700. Posner J.B. Paraneoplastic Syndromes.-Neurol.Clin.-1991.- v. 9.- p. 919.
701. Potts J.T. Diseases of parathyroid gland and other hyper- and hypocalcemic disorders. In: Harrison's Principles
of Internal Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2151-2172.
702. Pouliot MC, Despres JP, Lemieux S, et al. Waist circumference and abdominal sagittal diameter: best simple
anthropometric indexes of abdominal visceral adipose tissue accumulation and related cardiovascular risk in
men and women. Am J Cardiol. 1994;73:460-468.
703. Pouplard A. Pituitary autoimmunity Hornrione Res.-1982.-V. 16.- P. 289-297.
704. Prasad, A. S., Miale, A., Farid, Z., Sandstead, H. H. & Schulert, A. (1963) Zinc metabolism in patients with
the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypogonadism. J. Lab. Clin.
Med. 61:537-549.
705. Prochazka M, Lillioja S, Tait JF, Knowler WC, Mott DM, Spraul M, Bennett PH, Bogardus С Linkage
of chromosomal markers on 4q with a putative gene determining maximal insulin action in Pima Indians.
Diabetes 1993;42:514-519.
706. Putnam F.W. The plasma proteins. N.Y.: Acad. Press, 1975
707. Rajakumar K. Vitamin D, Cod-Liver Oil, Sunlight, and Rickets: A Historical Perspective./ Pediatrics.-2003.-
V.112.-N.2.-,pp.el32-el35
708. Rasmussen H. Hormone action— 1981 In: Functional Regulation on cellular and Molecular Levels. —N. Y.
а. е.-1982.-P. 402-406.
709. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities: the role of insulin
resistance and the sympathoadrenal system. N Engl J Med 1996;334:374-381.
710. Reaven, G.M. Role of insulin resistance in human disease. Diabetes 37 (1988) 1595-1607.
711. Reaven,G.M. The Metabolic Syndrome or the Insulin Resistance Syndrome? Different Names, Different
Concepts, and Different Goals. Endocrinol Metab Clin N Amer. 2004; 33:'283-303.
712. Reichstein Т., von Euw J. Helvet. Chim. Acta, 1938.-v.21.-l 197.
713. Ries W. Fettsucht. Barth: Leipzig, 1970.
714. Riley W., Maclaren M, Autoimmune adrenal disease Pediat. Adolesc. Endocrinol. —1984. —V. 13,—P.
162-172.
715. Risdall J. E., Dahiberq P. 0., Westermark B. Influence of thyroid autoantibodies on thyroid cellular growth
•in vitro /J.Mol. and Cell. Endocrinol.- 1-V. 34.-N 3.-P. 215-219.
716. Rojdmark S.,Calissendorf J., Brismar K.Alcohol ingestion decreases both diurnal and noctural secretion of
leptin in healthy individuals. //Clin Endocrinolog. - 2001. - N 55. - P. 639-647
717. Rollo, J. An Account of Two Cases of the Diabetes Meltitus, Dilly, London, 1797.
718. Rose NR; Rasooly L; Saboori AM; Burek CL, Linking iodine with autoimmune thyroiditis.Environ Health
Perspect 1999 Oct; 107; Suppl 5:749-52.
719. Rosenberg I.H. Nutrition and nutritional requirements. In: Harrison's Principles of Internal Medicine. 13th
Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 437-440.
720. Rosenberg L.E. Inherited diseases of amino acid metabolism and storage. In: Harrison's Principles of Internal
Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2117-2125.
721. Rosenberg L.E.Short E.M. Inherited diseases of membrane transport. In: Harrison's Principles of Internal
Medicine. 13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2125-2131.
722. Rossini A.A. et al. // Diabetes Reviews.- 1993.- 1.- 43-75.
723. Roth C. et al. Autoimmune thyreoiditis in childhood: Epidemiology, clinical and laboratory findings in 61
patients.// Exptl. Clin. Endocrinol. Diab.-1997.- 105.-suppl.4.-pp.66-69
724. Rubner M. Die Quelle der tierischen Warme.// Zeit. f. Biol.-1894.-12.
725. Rubner M. Probleme des Wachstums und der Lebensdauer. Wien: Mitt. Ges. Innere Med. und Kinder-
heilk.l908.-Bd.7.- p.58-72.
726. Ryan KJ. -In: Maternal-Fetal Endocrinology.Philadelphia:W.B.Saunders, 1980, p 3.
727. Safiran, M-., Schally, A. V, and Bonfey, B. G, Stimulation of the release of corticolropin from the adenohy-
pophysis by a neurohypophysial factor, Endocrinology 1955.- 57:439.
728. Salas M.A. et al. / Interleukin-6 and ACTH act synergistically to stimulate the realease of corticosterone
from adrenal gland cells.// Clin. Exptl Immunol.-1990.-v. 79.-N 3.-pp.470-473.
729. Samson WK. et al. Gastrointestinal hormones: CNS location and sites of neuroendocrine action.// Endocrinol.
Exp.-1982.-v.16.-N 3-4, pp.177-189.
730. Sapolsky R. et al. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-realising factor.-Sci-
ence.-1987.-v.238.-N 4826.-pp. 522-524.
754 А.Ш. Зайчик, Л.П.Чурилов Патохимия (эндокринно-метаболические нарушения)
731. Savino W. et al. Thymic hormone-containing cells.... //J. Histochem. Cytochem.-1984.-v. 32.-p.942-
946.
732. Scharrer B.,Scharrer E. Neuroendocrinology. N.Y.-London: Columbia Univ. Press, 1963,289 pp.
733. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HE A novel serum protein similar to Clq, produced
exclusively in adipocytes. J Biol Chem 1995; 270:26746-26749.
734. Schimke R.T.// Bull.Soc. Chim. Biol.-1966.-v.48.-p.1009.
735. Scholl С et al. //J. Clin. Endocrinol. Metab.-2002.-v.87.-N 10.-pp.4607-4610.
736. Schulze P. Ch., Kratzsch J. Leptin as a new diagnostic tool in chronic heart failure. Clinica Chimica Acta
362(2005)1-11
737. Schwarz, K. & Foltz, С М. (1957) Selenium as an integral part of Factor 3 against dietary necrotic liver
degeneration. J. Am. Chem. Soc. 79:3292-3293.
738. Scriver С et al. The Metabolic and Molecular Basis of Inherited Disease. 7th Ed. McGraw-Hill, 1995.
739. Segal P., Zimmert P. The 1st International Congress on Prediabetes and the Metabolic Syndrome. Diabetes
Voice, 2005.-v.50.-N 2.-pp. 45-47.
740. Seguin, A. & Lavoisier, A. L. (1789) Premier me' moire sur la transpiration des animaux. Me' m. Acad. Sci.
Paris 601-661.
741. Shechter Y. et al. Mice immunized to insulin develop antibody to insulin receptor. //J. Cell. Biochem.-
1983.-v.21.-N2.-pp.l79-185.
742. Sheehan H.L. Post partum necrosis of the anterior pituitary. J. Pathol. Bacteriol, 1937.-v.45.-p.189.
743. Shek E.W., Brands M.W., Hall J.E. // Hypertension. - 1998. - v. 31. - pp. 409-414.
744. Shufeng Chen et al. Peroxysome proliferation - activator receptor-ycoactivator-la polimorphisms are as-
sociayed with the level of lipids in Chinese population. // Chinese J. of Pathophysiology. -June, 2006. - Vol.
22. - N 13 (Supplement). - P. 69-70
745. Shulof R.S. et al. Thymic pathology and biochemistry. Adv. Clin. Chem.-v.26.-p.203.
746. Simmonds M. Uber Kachexie hypophysaren Ursprungs, Dtsch.Med.Woch.-1916.-N7.
747. Smythies J.R. Every Person's Guide to Antioxydants. Rutgers Univ. Press: New Brunswick a.e., 1998.-
146 pp.
748. Sodemann W. A„ Sodemann Th. M. Pathologic physiology. Philadelphia: W.B.Saunders, 1985.
749. Soffer L.J., Dorfman R.J., Gabrilove J.L. The human adrenal gland. Philadelphia., 1961.
750. Steppan CM. et al. The hormone resistin links obesity to diabetes // Nature. — 2001. — N 409. — pp.
307-312
751. Steppan СМ., Crawford D.T., Chidsey-Frink K.L., Ke H., Swick A.G. Leptin is potent stimulator of bone
growth in ob/ob mice. // Regul. Pept. - 2000. - N 92. - p. 73-78.
752. Stern M, Williams K, Gonzalez-Villalpando С et al. Does the metabolic syndrome improve identification of
individuals at risk of type 2 diabetes and/or cardiovascular disease? Diabetes Care 2004;27(11):2676-81
753. Stockman, R. The treatment of chlorosis by iron and some other drugs. Br. Med. J.—1893. — i: 881-885,
942-944.
754. Stoleke H. Kongenitale Nebennierenrindenhyperplasie mit maximaler Virilisierung (penile urethra). Kasuistik
der Weltliteratur und ein eigener Beitrag// Ztschr. F. Kinderheilunde. — 1970. — N107(4). — pp. 343-365.
755. Stroev Ju.L, Churilov L.P. Autoimmune thyroiditis: interdisplinary approach. The 5th International
Congress of Pathophysiology. June 28-Jule 1, 2006. Beijing, China. (Part I, Oral presentation). / Chinese J. of
Pathophysiology. -June, 2006. - Vol. 22. - N 13 (Supplement). - P. 95.
756. Stroev Yu.L, Churilov L.P., Fedash V.V., Scripnic V.V, Mikhailov V.I. «Triunghiul Bermudelor» in diabetul
zaharat: microangio-patia, ateroscleroza, hipertensiunea arteriala (comparatii clinice si patologice). A IV-a
Conferinta Nationala de studii interdisciplinare in medicina interna (cu participare internationala). Rezumate.
- Bucuresti, 3-4 martie 2006. - Pp. 19-20.
757. Stroev Yu.L, Churilov L.P, Mudzhikova O.M. Behavioral Disorders in Autoimmune Endocrinopathies. In:
International symposium "Interaction of the Neurous and Immune Systems in Health and Disease" May 31st
-June 2nd, 2007. Abstracts. St. Petersburg: ElBi Publ, 2007.- pp. 86-87.
758. Strojev Ju. I., Tchourilov L.P., Staffel R. E. et al. Disorders of menstrual function in adolescent girls with
Hachimoto's autoimmune thyroiditis. / Abstracts of XXII World Congress on Pediatric and Adolescent
Gynecology. May 31-June 3 1998, Helsinki, Finland.
759. Stroyev Ju.L, Tchourilov L.P., Gadiliya M.D., Lytvinscayaju. O. Disorders of menstrual function in adolescent
girls with differens internal patology. / Abstracts of 7-th European Congress on Pediatric and Adolescent
Gynecology. Vienna- Austria, 12-15 March 1997.
760. Stryer L. Biochemistry. San Francisco: WH. Freeman et Co, 1994
Литература 755
761. Sutherland J, McKinnley В, Eckel RH. The Metabolic Syndrome and Inflammation. Metabolic Syndr Rel
Disord 2004; 2:82-104.
762. Suzuki T. Physiology of Adrenocortical Secretion. In: Front. Horm. Res.-1983.-v.l 1.-215 pp.
763. Sydenham T. Observationes medicae circa morborum acutorum historiam et curationem. Londini, 1676.
764. Szafarchyk A. et al. -J.Steroid. Res.-1983.-v.l9.-Nl.-pp.l09-1015.
765. Szent-Gyo'rgi, A. Observations on the function of the peroxidase systems and the chemistry of the adrenal
cortex. Description of a new carbohydrate derivative. Biochem. J. -1928.-22:1387-1409.
766. Szondy E., Horvath M., Merey L., Szekely 1., Lenguel E., Fust G., Gero S. Free and completed anti-lipoprotein
antibodies in vascular diseases // Atherosclerosis— 1983.— V. 49.—N 1.—P. 69—77.
767. Takaki, K.On the cause and prevention of kak'ke. Sei-I-Kwai Med. J.-1885.- 4 (suppl. 4): 29-37.
768. TakamineJ.- Amer. J.Pharm.-1901.-v.73.-p.523.
769. Talbot F.B. // Amer. J. Dis. Child.-1938.-v.55.-p.455.
770. Tardella L. et al. //J. Clin. Lab. Immunol.-1983.-v.l2.-N3.-pp.l59-165.
771. Taylor SI, Cama A, Accili D, Barbetti F, Quon MJ, de la Luz Sierra M, Suzuki Y, Roller E, Levy-Toledano R,
Wertheimer E, et al. Mutations in the insulin receptor gene. Endocr Rev 1992;13:566-595.
772. The Diabetes Control and Complications Trial Research Group. // New England J. of Medicine.- 1993.-
329.- 977-986.
773. Thiel, van D.H. et al.-Clin. Immunol. & Immunopathol.-1977.-v.8.-N2.-pp.311-317.
774. Toh B.-H., Gleeson P.A.-Immunol. Today.-1991.-v.12.-N 7.-pp.233-238.
775. Tsao T, Stenbit A., Li J. et al. Muscle-specific transgenic complimentation of GLUT 4-deficient mice. Effects
on glucose but not lipid metabolism. //J. Clin. Investr. - 1997. - Vol. 100. - P. 671-677.
776. Tucker, H. F. & Salmon, W. D. (1955) Parakeratosis or zinc deficiency in the pig. Proc. Soc. Exp. Biol. Med.
88:613-616.
777. Tunbridge WMG, Evered DC, Hall R, et al. The spectrum of thyroid disease in a community. The Whickham
Survey. Clin Endocrinol. 1977; 7:481-7.
778. Ukkola O. Resistin - a mediator of obesity-associated insulin resistance or an innocent bystander? // Eur.
J. Endocrinol. - 2002. - v. 147.- pp. 571-574.
779. Unger R., Orci L. // Diabetes,- 1977.- 28.- 3.- 241-244.
780. Vague J. La differenciation sexuelle, facteur determinant des formes de Pobesite. Presse Med. 1947; 30:
339-40.
781. Vardi P. et al.- // Diabetes Care.- 1987.- 10.- 5.- 645-654.
782. Volk B.W, Wellmann K.F. // In: Diabetic Panreas.- 1985.- 367-384.
783. Vollerthun R., Moller W. // Cell.Tisssue Res.-1980.-v.213.-p. 393.
784. Volpe R, Auto-immunity in the endocrine system.—Berlin e. a.: Springer Verlag.—1981.—167 p.
785. von Basedow, С A„ Exophthalmos durch hypetrophie des zell-gewebes in der augenhohle, Wochenschr.
Heilkunde 1840.-6:197.
786. von Mering, J Minkowski, O., Diabetes mellitus nach pankreas extirpation, Zentralbl. Klin. Med. 1889.-
10:393.
787. Vulpian M. - C.R.Acad. Sci.-1856.-v.43.-p.663.
788. Wallace D. Ref. after: Cooke R. Study: less food for longer life.// Newsday.-1997.-July, 25th, p.4.
789. Ward W et al. // In: Comparison of Type I and Type II Diabetes.- 1985.- 137-158.
790. Wartofsky L. Diseases of the thyroid. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv 1-2.-N.Y.
a.e.: McGraw-Hill, 1994. Pp. 1930-1953.
791. Wasson T. (Ed.) Nobel Prize Winners. An H.WWilson Biographical Dictionary, w. 1-2, N.Y.: H.W.Wilson
Co, 1987.
792. Watson S J. The endorphin family of opioid peptides: biochemistry, anatomy and physiology. In: Wyngaarden
J.B., L.H.Smith. Cecil's Textbook of Medicine. 18th Ed. Philadelphia a.e.: W.B.Saunders, Vv 1-2.-1988. Pp.
1268-1271.
793. Webster A.D.B. Metabolic defects in immunodeficiency diseases. // Clin.Exptl.Immunol.-1982.-v.49.-
pp.1-10.
794. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with
macrophage accumulation in adipose tissue. J Clin Invest 2003; 112:1796-808.
795. Whipple A.O. Hyperinsulinism in relation to pancreatic tumors // Surgery. — 1944. — v.16.— p. 89-305.
796. White A., Levine R.- History of Hormones.-In: Biol. Regulat. & Develop., N.Y.-London, 1982.-pp.l-24.
797. Whitehead J. P., Richards A. A., Hickman I. J., Macdonald G. A., Prins J. B. Adiponectin - a key adipokine in
the metabolic syndrome (Editorial) In: Diabetes, Obesity and Metabolism, 2005, March. Pp. 1—17
756 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
798. WHO. World Health Report - 1995. WHO: Geneva, 1998.
799. WHO. World Health Statistics Annual, 1970. WHO: Geneva, 1973.
800. Williams G.H., Dluhy R.G. Diseases of the adrenal cortex. In: Harrison's Principles of Internal Medicine.
13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 1853-1976.
801. Wilson J.D. Hormones and hormone action. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 1883-1884.
802. Wilson J.D. Vitamin deficiency and excess. In: Harrison's Principles of Internal Medicine. 13th Ed., Vv
1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 472-481.
803. Wilson J.D., Foster D.W Williams'Textbook of Endocrinology. 8th ed.,Philadelphia:Saunders, 1992.
804. Wilson J.D., Griffin J.E. Disorders of sexual differentiation. In: Harrison's Principles of Internal Medicine.
13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2039-2052.
805. Witebsky E., Rose N. H., Terplan K., Paihe J. N., Egen L. W. Chronic thyroiditis and autoimmunization //
JAMA.- 1957.- V. 161.- P. 1439-1470.
806. Wortmann R.L. Gout and other disorders of purine metabolism. In: Harrison's Principles of Internal Medicine.
13th Ed., Vv 1-2.-N.Y. a.e.: McGraw-Hill, 1994. Pp. 2079-2088.
807. Wurtman R.J., Axelrod J. Formation of melatonin and physiologic effects of melatonin.// Adv. Pharma-
col.-1968.-v.6.-pp.l41-151.
808. Wyngaarden J.B., L.H.Smith. Cecil's Textbook of Medicine. 18th Ed. Philadelphia a.e.: W.B.Saunders, Vv
1-2.-1988.
809. Yalow, R.S., and Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Invest. 39 (1960)
1157-1175.
810. Yalow, R.S., Glick, S.M., Roth, J., and Berson, S.A. Plasma insulin and growth hormone levels in obesity and
diabetes.'Ann. N. Y. Acad. Sci. 131 (1965) 357-373.
811. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Нага К et al. The fat-derived hormone adiponectin
reverses insulin resistance associated with both lipoatrophy and obesity. Nature Medicine. — 2001. — 8,
941-946.
812. Yokota T. et al. // Blood. - 2000. - v. 96. - pp.1723-1732.
813. Yokoyama C, Wang X., Briggs M. et al. // Cell. - 1993. - v. 75. - pp.187-197.
814. Youfei Guan. Metabolic nuclear receptors and the metabolic syndrome. // Chinese J. of Pathophysiology.
-June, 2006. - Vol. 22. - N 13 (Supplement). - P. 67.
815. Zaichik A.Sh., Churilov L.P.,Kravtzova A.A.,Utekhin V.J. Antibodies to nuclear antigens penetrate into
living endocrine cell nuclei and stimulate target cell growth and function. The 5th International Congress of
Pathophysiology. June 28-Jule 1,2006. Beijing, China./ Chinese J. of Pathophysiology. -June, 2006. - Vol.
22. - N 13 (Supplement). - P. 361-362.
816. Zaichik A.M., Churilov L.P., Kravzova A.A., Utekhin VJ. Anti-thyroid Autoimmunity in Adolescent
Hypothalamic Obesity: Simpson-Page' Syndrome as Immuno-neuroendocrine Disorder. In: International symposium
"Interaction of the Neurous and Immune Systems in Health and Disease" May 31st -June 2nd, 2007. Abstracts.
St. Petersburg: ElBi Publ., 2007. - pp. 88-89.
817. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L,Friedman JM. Positional cloning of the mouse obese
geneand its human homologue. Nature 1994;372:425-32.
818. Zilberfarb V.,Grosfield A.,Turban S. Hypoxia increases leptin expression in human Paz6 adipose cells.//Dia-
betologia .-2002.-45(4).-P. 527-523.
819. Zimmert P. Challenges in diabetes epidemiology. From West to the rest. Kelly West Lecture 1991, Diabetes
Care 15 (1992) 232-252.
820. Zunz K., Mering F. Inwifern bei influsst Nahrungszuhufr die thierischen Oxydationsprocesse? // Arch/ ges.
Physiol. Mensch. Thiere, 1883.-Bd.32.-S. 173-221.
i
Краткий предметный указатель
авитаминозы 34,355,382
агликогеноз 237,275,767
аденомы гипофиза 217,530,651
адипозо-генитальная дистрофия 216,218, рис.41
адипонектин 210,562,718
адрено-генитальные синдромы 591,637
адреномедуллярная недостаточность 611
азотистый баланс 51,61,80,360
аквапорины 505,515,730
акродиния 395
акромегалия 80,540-543,667
акцидентальная инволюция тимуса 658
алактазия 243
алиментарный маразм 34,45-47,63
алкалоз 310,362-364,375
алкаптонурия 86
алкогольная гепатопатия 123, 232
альбинизм 90-91
альдостерома 588,600
амилоидоз 121-123,532,683
аминоацидопатии 72,84,93,231
аминоацидурия 72-75,112
анальбуминемия 98,100
анасарка 39,270,353
андростерома 584,591,624
анорексия-булимия 31,48,54,170
антивитамины 384,399,431
антиоксиданты 99,280,387,403
апантотеноз 396
арибофлавиноз 392
артериолосклероз 176,317,720
асцит 47,263,354,557
атеросклероз 143,175-196,325,453
ахолия 155,430
ацидоз 360,380,427, 600
Болезнь Аддисона 275,356,592-593
- Альцхаймера 122,407,685,734
- Андерсена 263
- Барракера-Симмондса 224,226
- бронзовая 83,592
- Вольмана 151,173,197
- Деркума 222
- «икэн» 414
- Имерслунда-Грэсбека 400,437
- Иценко-КушингаЗН, 582-583
- «как'ке» 382
- Коновалова-Вильсона 74,452-453
- Крёйтцфельда-Якоба 121,540
- кэшаньская 437,459
- Маделунга 221
- Мак-Ардля 264
- Мёллера-Барлоу 406,452
- Менкеса 98,452-453,692
- острова Тэнжи (Тэнжир) 163
- Паркинсона 95,407
- Помпе 265
- Рустицкого-Калера 105,580,689
- Симмондса 222,531-533
- смены часовых поясов 481,523
- уровская 456
- фон Базедова-Грейвса 148,556-565,665
- фон Гирке 135,169,261,265
- фон Гиппеля-Лйндау 608
- фон Реклингхаузена 608
- Форбса-Кори 263
- Харриса313
- Хартнупа 72-73,393
- Хейлмейера 445
- Херса 264
- Хэнда-Шюлера-Кристиана 163
- ямайская рвотная 233
вазопрессинома 219
внежелезистые эндокринопатии 470-471,555
водный баланс 347-348,353
Врождённая гиперплазия КН 495,571,593,600
Галактоземия 243-245,653
Гемералопия 227,414,417
гемосидероз 37,447-449,683
Гемохроматоз 272,441,447-450,532,594
Гемоювелин 441,448,694
Генерализованная миастения 374,664-665
гепато-ренальный синдром 111
гепцидин 434,441,448,451,700
гермафродитизм 618-625
гиалиноз 113-114,123
гиперадренализм 581
гиперазотемия 109-110,307-308
гиперальдостеронизм 354-355,494,577
Гиперальфалипопротеинемия 174
Гипераминоацидемия 76,80-81.63
гипераминоацидурия 245
Гиперандрогенизм 337,591,637,654
гипервалинемия 87
гипервитаминозы 135,428-429,433,496,668
гипергидратация 211,309,348,511,518,596
Гипергликемия 311-312,319,331,449,536
Гипергонадизм 531,632,637-638
Гиперкалиемия 47,350,360,595
Гиперкальциемия 678-680, 686-687
гиперкератоз 124,393,417
гиперкортицизм 62,81,170,216,581-88,694,711
гиперкупремия 451
Гиперлипопротеидемии 54,143,163-164, 235
Гипермагниемия 690
Гиперманганемия 454
Гипернатриемия 312,358,517
758 АЖ Зайчик, ЛЛ. Чурилов Патохимия (эндокринно-метаболические нарушения)
гиперпаратироз 75,141-142,679-689,696
гиперпинеализм 524
Гиперпитуитаризм 530
Гиперплазия тимуса 664-665
гиперпролактинемические синдромы 634-652
гиперпротеинемия 100
Гипертензия 138,181-183,589-590
Гипертироз 18-19,83,148,273, 297,469,546,554
Гипертирозинемия 91
гипертоническая болезнь 182,704
гиперурикемия 46,125,131-137, 263, 721-722
Гиперфосфатемия 668,682,685-686
гиперхлоремия 312
гиперхолестеринемия 151,164-66,178-9,453
гиперцистинурия 72
гипоадренализм 581
гипоальдостеронизм 356,493, 600
гипоальфалипопротеидемия 174
гипобеталипопротеидемия 173
гипогликемия 87,263-265,313-315,530-532
гипогонадизм 210-11,216-20,632-38
гипокалиемия 303,359,380, 590, 679,684
гипокальциемия 100,459, 679-681, 684,686, 692
гипокортицизм 468-69,492-93,532,592-97,601
гипокупремия 452
гипомагниемия 673, 681-83
гипоманганемия 454
гипонатриемия 357,518,564,596-97,602
гипопаратироз 275,429,491,496,680-81,683-86
гипопитуитаризм 209,222,325,530-532,692
гипоплазия тимуса 138,662-63
гипопротеинемия 66,100-103,449,557
гипоталамопатии 509-10
гипотироз 18-19,82-83,91,170-71,512,545,555
гипотироидный нанизм 563
гипоурикемия 138-39
гипоферремия 104,441-42
гипофизарный гигантизм 311,530,540,542
- нанизм 80,216,538-41,563
- гипогонадизм 211,530
гипофосфатемия 47,426-27,668,691
гипохолестеринемия 151,311,455
гипоцинкоз 449-50
гистидинурия 72,93
гликогенозы 237,248,261-64
гликосфинголипидозы 144
глюкагонома 103,221, 273, 287,311,497
глюкоандростерома 584
глюкозурия 222,282
голодание 28,240,300,308-09
гомоцистинурия 87,90,454,696
гунтеровский глоссит 399
Диабетогенные факторы 277
диастема 542
дизгенезия гонад 539
динамическое действие пищи 19,51,53.203,211
дисбеталипопротеидемия 171,151
дисглобулинемия 100
дискератоз 124
дисменорея 318,510, 512,557,649
дисперсная эндокринная система 472,525, 699
диспротеинемия 100
диспротеинозы 105,112-115,327,532,566,689
диэнцефальные синдромы 509
дуоденопривная модель ожирения 205
дыхание Куссмауля 305-06,308, 310,376,379
дыхательный алкалоз 371-75, 379-80
- ацидоз 362,373-74,379, 670
Евнуходизм 633
Железодефицит 445-47, 700
Задержка полового созревания 649
зимняя спячка 22,56-8,81,287
зоб 551-52,556,560,565,569
Импринтинг геномный 200, 217
индиканурия 73
инсулиннезависимый сахарный диабет 271,325
инсулинома 121,185,209, 221,313
инсулинорезистентность 172, 210-211,271, 273,
306
истощение 222-223,531
ихтиоз 124,227,268
Йод-базедовизм 458,469,556
Каловая аутоинтоксикация 69
калорийный (калорический) коэффициент 26
калориметрия 22-24
калорический эквивалент 23,25-6
карбонурия 28,301
кастрация 629, 635, 671
кахексия 50,205.221-223,531,533
квашиоркор 34,38,45-47,63,68,101,355,437,535
кератомаляция 414
кетоацидоз 28,43,81,93,128, 228-31, 272-74,304,
380
койлонихия 446
кома 15-16,78,304-315,358,379,518,688
кортикоэстрома 591
коэффициент Рубнера 28,39,301
Креатинурия 112
креаторея 68
кретинизм 83,91,552,562-564
криоглобулинемия 105
кристаллические артриты 141-144
ксантоматоз 150-151,163-169
Лактат-ацидоз 263,310,377-78,380,610
лейциноз 86-7,314
лептин 20-1,40,45,49,51,53-4,198,210,650,694,
716
лиенторея 68
лизинурия 74,696
лимит-декстриноз 263
липоатрофии 223-24.337-38
липокины 236,336, 696,700
759
липоматоз 209,221-22
липостат (массостат) 53-54,197,202-15,509-10,
694
липотропные факторы 160,234-35
Макроангиопатия диабетическая 184,316,31 -334
макроглобулинемия Вальденстрёма 100
мерзебургская триада 560-61
метаболический алкалоз 373,378-80,590
- ацидоз 263,362,371,375-80,391,447,600
- блок 495,591
-синдром 172,703-719
микроангиопатия диабетическая 316-19,586
микседема 436,551,556,561-64,569
молочно-щелочной синдром 690
муковисцидоз 67,101,374,459
мукополисахаридозы 237,269-70
Нейрогенная анорексия 48,197, 202,210,510
нервная трофика 473
несахарное мочеизнурение 217-18,513-18,532
нефритические отёки 354
нефротические отёки 104,354
Ожирение 20,184,197-221,718-722
ортостатический коллапс 611
основной обмен 9-26,35,41,57,211-12,301,727
остеомаляция 377,423-24,426,685,691-92
остеопороз 426,432,479,557,643,653,678,692-99
Панкреатэктомия 338
панникулит 223-25,715,717
парапротеинемии 100,102,105, .225,427
педиолальгия (эритромелальгия) 396
пеллагра 66,392-93
первично рассеянное тепло 12
пермиссивное действие гормонов 466-67,508,554
пероз 455
печеночные отёки 354
пищевой культуризм 385
плазморея 356
плюригландулярные эндокринопатии 496,653
подагра 129-36,141-42,246,263,708,721
полиурия и полидипсия 74,305,308,429,516
преждевременный пубертат 337,512,523,638,649
принцип обратной связи 467-68,489
провитамины 414,416
пролактинома 216,218,497,634
протеинурия 102-03,121, 245,308,424
Рак щитовидной железы 565-66,585
Рахит 74-5,297,382-83,420-428,455-57,492,
681,692
ренин-ангиотензиновая система 40,573-77,588
ренинизм 589
рецепторные эндокринопатии 485,492-93,496
роговая дистрофия 124,152,417
Саркоидоз 135,429,510,515,531,567,594,680
сахарный диабет 181-84,217,230,271(гл.9), 696,
700
селиноз 460
сердечные отёки 353
симптомокомплекс Карнея 584
синдром MEN I 496-97
- MEN II 497,566,608
- Аргонза-дель Кастильо 218,530,652
- Бамбергера-Мари 543
- Барнетта 690
- Барттера 135,379,574,590
- болезненного эутироза 546
- Брутона 100
- Вернера 170,237,337
- Вольфрама 514,760
- Галла 563-64
- де Тони-Дебре-Фанкони 427
- ди Джорджи 654,662,681
- Донохью-Ушинда (эльфа) 337,690
- Карпентера 216,218,434
- Келли-Зигмиллера 133
- Киммелынтиля-Уилсона 317
- Клайнфелтера (Кляйнфелтера) 220,273,542
- Конна 588,590
- Корсакова-Вернике 389-90,510-11
- Ларона 489,493-94,540
-Лериша 177
- Леша-Найхена (Нихена) 133
-Лея 310,391
- Лиддля 359
- Лоу 74,427
- Лоуренса-Муна- Барде- Бидла 216-17,337,559
- Людера-Шелдона 75
- Майера-Рокитански - Кюстера-Хаузера 625
- МакКьюна-Олбрайта 649
-Марфана 497,696,701,718
-Мориака 184,540
- Морриса 623, 637
- Мэддока 633
- Незелофа 654,662
-Нельсона 531,583
- объемного дефицита 355,357
- одеревенелости 290
-Ольстрема 216-17,337
- Ормонда 571
- Пархона 216,219,357,517-18,532,761
- Паскуалини 633
- Пелицци 519,524,637
- Пендреда 552
- Пламмера-Уинсона 446
- Прадера-Вилли 216-17,509
- предменструального напряжения 650
- пустого турецкого седла 49,537
- путника в пустыне 358
- Рабсона-Менденхолла 273,337,523
- Райли-Дея 612
- Рейфенштейна 637
- Рейно 105,394
- Рефетова 553,555
760 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
- Рея 233,236
- Ригера 224
- Роули-Розенберга 75
- Сильвера 649
- Сиппля 497
- «социального едока» 50
- Стюарта-Морганьи-Мореля 217
- Сейведжа 653
- Турена-Соланта-Голе 543
- Уильямса 690
- Уотерхауза-Фридериксена 598-99,611
- Фагте 83,462,563-64
- Фанкони 73-4,102,138,453,696
- Форбс-Олбрайта 218,530
- Хиари-Фроммеля 218,652
- Цоллингера-Эллисона 67,497
- Чедиака (Шедьяка) -Хигаши 408
- Шая-Дрейджера 612
- Шварца-Барттера 517
- Шихена (Шиена) 216,515,530-33,597,651
- Шерешевского-Тернера 220,272,539-40, 621
- Шмидта 496,558,570,593
- Штейна-Левенталя 654-655
- Штруге-Вебера 608
- Эйнджелмена 217
- эктопической секреции АКТГ 585
- Элерса-Данло(са) 696
- Эшермена 650
системные отеки 104,353-55,589
ситостеринемия 151,173
стеатоз печени 46,143,146,152,160,168,228-34
стеаторея 44,155,173,311,399,423
Тезаурисмозы 270,540
тетания 671,679,698
тимико-лимфатический статус 663
тироидиты 554,567-71,663
тиротоксическая офтальмопатия 556,560
тирозиноз Медеса 91
тубулопатии 60,71,360,427
Уремия 109-10,134.349,378
уролитиаз 91,133,137,263,417,453,681,687
Фенилкетонурия 76,84,85,92,612
Феохромоцитома 95,311,574,606-08,700
Фибриллинопатии 696,701
Флюороз 459
Хаситоксикоз 559,570
Целиакия (спру) 59,68-9,101,155,397-98,402,
637
цинга (скорбут) 381,383,403-06,426
Экзофтальм 337,557,560-62,611,719
эндокринные отёки 355
эссенциальные аминокислоты 61,63-66
- жирные кислоты 152,387
Юношеский диспитуитаризм 219,706,722
Ятрогения 220,273,375,378,384,495,556,581,
695
Список используемых сокращений
Англоязычные сокращения
АА (amyloid associated) фибриллярный амило-
ид-ассоциированный белок
АВ (actual bicarbonate) истинный бикарбонат
крови
АСЕ (angiotensin converting enzyme) ангиотензин-
конвертаза
AD (amyloid dermatological) кожный амилоид
АЕ (amyloid endocrine) эндокринный амилоид
AF (amyloid fibrillary) фибриллярный компонент
амилоида
AIA (anti-insulin autoantibodies) анти-инсулино-
вые аутоантитела
AICA (anti-islet cytoplasmic autoantibodies)
аутоантитела к цитоплазме островковых клеток
AINA (anti-islet nuclear autoantibodies)
аутоантитела к ядрам островковых клеток
AISA (anti-islet surface autoantibodies)
аутоантитела к поверхностным антигенам островковых
клеток
AL (amyloid light chain) амилоид из лёгких цепей
иммуноглобулинов
APUD (amine precursor uptake and decarboxylation)
захват и декарбоксилирование аминных
предшественников
AS (amyloid senile) старческий амилоид
ATTR - транстиретиновый компонент амилоида
ВВ (buffer base) сумма оснований всех буферных
систем крови
BD (buffer deficit) дефицит оснований
BE (buffer excess) избыток оснований
С11 11 -стероидгидроксилаза
С17 17-стероидгидроксилаза
С21 21-стероидгидроксилаза
С D (cluster designation or cluster of differentiation)
обозначение скопления функциональных
поверхностных маркеров
C/N (carbogen/nitrogen) карбонурический
коэффициент
ССК (cholecystokinin) холецистокинин
CLIP (corticotropin-like intermediate polypeptide)
кортикоптропиноподобный промежуточный
полипептид
CR (complement receptor) рецептор фактора
комплемента
DCCTRG (diabetes close control trials research
group) исследовательская группа
клинического метода жесткого контроля степени
компенсации сахарного диабета
DIDMOAD (diabetes insipidus, diabetes mellitus,
optic atrophy, deafness) синдром Вольфрама
El (estrone) эстрон
E2 (estradiol) эстрадиол
E3 (estriol) эстриол
ECHO (enteric cytopathogenic human orphans)
энтеропатогенные человеческие вирусы-
сиротки
GIP (gastric inhibitory polypeptide) желудочный
ингибирующий полипептид
GluT (glucose transporter) транспортёр глюкозы
GluT4 (glucose transporter 4) инсулинозависимый
белок-транспортёр глюкозы 4 типа
Hb (hemoglobin) гемоглобин
HbAlc гликозилированный гемоглобин
HLA (human leukocytic antigens) человеческие
антигены гистосовместимости
ICE (Interleukin-1 converting enzyme) цитоп-
лазматические нейтральные цистеиновые
протеазы
IDDM (insulin dependent diabetes mellitus)
первичный инсулинзависимый сахарный диабет I
типа
IL-1 (interleukin-1) интерлейкин-1
IL-2 (interleukin-2) интерлейкин-2
LATS (long acting thyroid stimulator) длительно
действующий стимулятор щитовидной
железы
Lp(a) (lipoprotein (а)) липопротеид (а)
МСН (mean corpuscular hemoglobin) среднее
содержание гемоглобина на 1 эритроцит
MEV (mean erythrocyte volume) средний объём
эритроцита
МНС (major histocompatibility complex) главный
комплекс гистосовместимости
MODY(maturity oncet diabetes of the young) диабет
взрослых в юности
NADH (nicotine amide dinucleotide reduced)
восстановленная форма никотинамиддинук-
леотида
NK (natural (normal) killers) естественные
(нормальные) киллеры
NOD (non-obese diabetic mices) нетучные
диабетические (мыши)
PAS (periodic acid — Schiff) гистохимическая
окраска периодной кислотой — реактивом
Шиффа
762 АЖ Зайчик, Л.П. Чурилов Патохимия
Pg (prostaglandin) простагландин(ы)
PgI2 (prostacyclin) простациклин
PYY полипептид YY3-36
RMR (resting metabolic rate) основной обмен
SB (standard bicarbonate) стандартный
бикарбонат
SC (secretin) секретин
SD standard deviation, стандартное отклонение
SIADH (syndrome of inappropriately high)
vasopressin secretion) синдром Пархона
TIB С total iron binding capacity
TNF tumor necrosis factor
UIBC unsaturated iron binding capacity
VIII-vWF (von Willebrand's factor) фактор фон
Виллебранда
VIP vasoactive intestinal polypeptide
Русскоязычные сокращения
2,3-БФГ 2,3-бифосфоглицерат (дифосфоглице-
рат)
АГТГ адреногломерулокортикотропин
АД аутосомно-доминантный тип наследования
АДГ антидиуретический гормон
АЗКЦ антителозависимая клеточно-опосредован-
ная цитотоксичность
АИКАР аминоимидазол-карбоксамид-риботид
АКР активные кислородосодержащие радикалы
АКТГ адренокортикотропный гормон
ала аланин
АЛАТ аланин-аминотрансфераза
АМФ аденозинмонофосфат, аденозинмонофосфор-
ная кислота
АПУД захват и декарбоксилирование аминных
предшественников
АР аутосомно-рецессивный
арг аргинин
АСАТ аспартатаминотрансфераза
АС К атеросклероз
асн аспарагин
асп аспарагиновая кислота
АТФ аденозинтрифосфорная кислота
АФ РТ аденозинфосфорибозилтрансфераза
АХАТ ацил-холестерин-ацилтрансфераза
ББД белки Bence-Jones
БСА бычий сывороточный альбумин
БТШ белки теплового шока
вал валин
чдокринно-метаболические нарушения)
ВДКН врождённая дисплазия коркового вещества
надпочечников
ВИП вазоактивный интестинальный полипептид
ВИЧ вирус иммунодефицита человека
ВОЗ Всемирная организация здравоохранения
ГАМ К гамма-аминомасляная кислота
ГБ - гипертоническая болезнь
ГДК глутаматдекарбоксилаза
ГДФ гуанидиндифосфат
ГЗТ гиперчувствительность замедленного типа
гис гистидин
ГКГС главный комплекс гистосовместимости
ГКГС-1 белки первого класса главного комплекса
гистосовместимости
ГКГС-2 белки второго класса главного комплекса
гистосовместимости
Г-КСФгранулоцитарныйколониестимулирующий
фактор
Гл глицерин
ГЛ гормонозависимая липаза
гли глицин
глн глутамин
ГЛП гиперлипопротеинемия
глу глутаминовая кислота
ГЛЮ глюкоза
ГМК гладкомышечные клетки
ГМ-КСФ гранулоцитарно-моноцитарный колони-
естимулирующий фактор
ГНТ гиперчувствительность немедленного типа
ГПП-1 глюкагоноподобный пептид 1
ГС гепаран-сульфат
ГТГ гонадотропные гормоны
ГТФ гуанозинтрифосфорная кислота
ГФРТ гуанозинфосфорибозил трансфераза
ГЭР гладкий эндоплазматический ретикулюм
ДАГ диацилглицерин
ДВС диссеминированное внутрисосудистое
свёртывание
ДДТ 4,4-дихлор-дифенилтрихлорэтан
ДИТ дийодтирозин
ДК дыхательный коэффициент
ДНК дезоксирибонуклеиновая кислота
Д ОФ А диоксифенилаланин
ДПИД дикетопиридинолин
ДС дерматан-сульфат
ЖИП желудочный ингибирующий полипептид
ЖКТ желудочно-кишечный тракт
Жл желудок
ЖСС железосвязывающая способность
сыворотки.
ЗПМР задержка психомоторного развития
ИБС ишемическая болезнь сердца
ИД Г изоцитратдегидрогеназа
ИЛ интерлейкин(ы) — с соответствующими
номерами
иле изолейцин
ИМФ инозимонофосфат
ИР инсулинорезистентность
иРНК информационнная рибонуклеиновая
кислота
ИРФ инсулиноподобный ростовой фактор (то же,
что ИФР)
ИТФ инозинтрифосфорная кислота
ИФ ингибитинг-фактор
ИФА иммуноферментный анализ
ИФН интерферон
ИФНу- гамма- интерферон
ИФР инсулиноподобный фактор роста
ИХЛА иммунохемилюминесцентный анализ
КА катехоламины
кД килодальтон
КК калорический коэфффициент
КоА коэнзим А
КОМТкатехин-ортометилтрансфераза
КПНГГ конечный продукт необратимого
глубокого гликирования
КПТ карнитин-пальмитоилтрансфераза
КРИА коммуникативно-регуляторный интегратив-
ный аппарат
КрК креатиновый коэффициент
КРФ кортикотропин-рилизинг фактор
КС кератан-сульфат
КФ киназа фосфорилазы
к-ФРФ - кислый фактор роста фибробластов
КЩР кислотно-щелочное равновесие
КЭ калорический (калорийный) эквивалент
ЛГ лютеинизирующий гормон
ЛДГ лактатдегидрогеназа
лей лейцин
лиз лизин
ЛОР органы уха, горла, носа
ЛП липопротеид(ы)
ЛПВП липопротеиды высокой плотности
ЛПГ липотропный гормон
ЛПЛ липопротеиновая липаза
Список используемых сокращений 763
ЛПМИ Ленинградский педиатрический
медицинский институт
ЛПНП липопротеиды низкой плотности
ЛПОНП липопротеиды очень низкой плотности
ЛППП липопротеиды промежуточной плотности
Л ПС липополисахариды
ЛпТГ липотропный гормон (то же, что и ЛПГ)
ЛСД - диэтиламид лизергиновой кислоты
ЛХАТ лецитин-холестеринацилтрансфераза
МАО моноаминоксидаза
МАПО медицинская академия последипломного
образования
мг/дл миллиграммы на децилитр (мг%)
ME международные единицы
мет метионин
МиАП микроангиопатия
МИТ монойодтирозин
М-КСФ моноцитарный колониестимулирующий
фактор
М-КСФ моноциГарный колониестимулирующий
фактор
МО минутный объём
мОсм миллиосмоль
МПС мукополисахаридозы
МС метаболический синдром
МСГ меланоцитостимулирующий гормон
МХБ-I макрофагальный хемотактический
белок!
мЭкв миллиэквивалент
НА нейрогенная анорексия
НАД никотинамиддинуклеотид
НАДФН никотинамид-динуклеотидфосфат,
восстановленная форма
НАДФН никотинамиддинуклеотид-фосфат
НАСА Национальное управление США по
аэронавтике и исследованию космического
пространства
НД несахарный диабет
НМН никотинмононуклеотид
НЭЖК неэтерифицированные жирные кислоты
00 основной обмен
ОПН острая почечная недостаточность
ОРВИ острая респираторная вирусная инфекция
О ЦК объём циркулирующей крови
ПВЯ паравентрикулярное ядро
пг пикограмм
ПЖ поджелудочная железа
ПК пируваткиназа
ПМ плазматическая мембрана
764 АЖ Зайчик, ЛЛ.Чурилов Патохимия (эндокринно-метаболические нарушения)
пМ пикомоль
ПНФ предсердный натрийуретический фактор
(атриопептин)
ПНФ пуриннуклеозидфосфорилаза
ПОМ К проопиомеланокортин
ППСМСП площадь поперечного сечения мышц
середины плеча
ПРЛ, Прл пролактин
про пролин
РИА радиоиммунологический анализ
РМ реакционная микрокалориметрия
РГ|К рибонуклеиновая кислота
р02 парциальное напряжение кислорода
рС02 парциальное напряжение углекислого газа
рТЗ реверсивный (обратный) трийодтиронин
РТМЛ реакция торможения миграции лейкоцитов
РФ рилизинг-фактор
САИКАР сукцинил-амидоимидазол-карбоксамид-
риботид
СД сахарный диабет
СДГ сукцинатдегидрогеназа
СДД специфическое динамическое действие
(пищи)
сер серии
СЖК свободные (неэтерифицированные) жирные
кислоты
СКВ системная красная волчанка
СОЭ скорость оседания эритроцитов
СОЯ супраоптическое ядро
СТГ соматотропный гормон
Т тестостерон
ТЗ трийодтиронин
Т4 тироксин
ТГ триглицериды
ТИД теплопродукция, индуцированная диетой
тир тирозин
ТК тонкий кишечник
ТлК толстый кишечник
тре треонин
три триптофан
ТТГ тиротропный гормон
ТФР тромбоцитарный(е) фактор(ы) роста
ТФРа трансформирующий ростовой фактор а
УДФ уридиндифосфат
УФ-В ультрафиолетовые лучи диапазона 290-
320 нм
ФАД флавинадениндинуклеотид
ФАО (Food and agriculture organization)
Продовольственно-сельскохозяйственная
организация ООН
ФАФС фосфоаденозинфосфосульфат
ФГА фитогемагглюттинин
фен фенилаланин
Фз фосфорилаза
ФКУ фенилкетонурия
ФЛИП фосфолипиды
фМ фемтомоль
ФМН флавинмононуклеотид
ФНО фактор некроза опухолей
ФРПФ фосфорибозилпирофосфат
ФРТ фактор развития тестикул
ФРУ фруктоза
ФРФ фактор роста фибробластов
ФСГ фолликулостимулирующий гормон
ФФК фосфофруктокиназа
ХГ хорионический гонадотропин
ХКА холестериновый коэффициент атерогеннос-
ти
ХН холестерин
ХПН хроническая почечная недостаточность
ц-АМФ циклический аденозинмонофосфат
ЦВД центральное венозное давление
ц-ГМФ циклический гуанозинмонофосфат
цис цистин (дицистеин)
ЦНС центральная нервная система
ЦСЖ цереброспинальная жидкость
ШИК-РАБ-реакция гистохимическая окраска
реактивом Шиффа
ШЭР шероховатый эндоплазматический рети-
кулюм
ЩУК щавелевоуксусная кислота
ЩФ щелочная фосфатаза
Э эстрадиол
ЭДТА этилендиаминтетраацетат
ЭКГ электрокардиограмма
ЭПР эндоплазматический ретикулюм
ЭФР эпидермальный фактор роста
ЭХ эфиры холестерина
ЭЭГ электроэнцефалограмма
ЯР ядерный рецептор
Resume
This Manual is the second volume of the series (after vol. I published earlier in 1999, titled
"Fundamentals of General Pathology" and before next vol. Ill — " Mechanisms of Diseases and
Syndromes" — still to be published). The textbook is a comprehensive review in Pathophysiology
of Metabolic and Nutritional Diseases and in Endocrine Pathophysiology.
The book deals not only with pathophysiological meaning of biochemical disorders. It covers
also the task of brief medical synopsis of basic Biochemistry and introduces the readers into
Clinical Chemistry. 14 chapters contain the consequent analysis of the chemical aspects of
pathological processes. Both acquired and inherited disorders are discussed. The textbook pays
proper attention to the Pathophysiology of energetic metabolism, including resting metabolic
rate concept, calorimetry and microcalorimetry. The disorders of eating behavior and mechanisms
of starvation and inanition, as well as historical and modern views on obesity and overeating
are presented. The textbook deals with pathology of protein and nucleic acid metabolism,
with particular attention to gout and aminoacidopathies mechanisms. Later goes the detailed
discussion of carbohydrate metabolism disorders. Pathophysiology of diabetes mellitus is
specifically emphasized and arranged as separate chapter. The part, related to lipid metabolism,
besides general aspects, familiar disorders and lipoprotein turnover, contains extensive review
in natural history of atherosclerosis. Considerable part of the book is dedicated to the nutrition
diseases caused by inappropriate supply of different essential nutrients, with special charters
about vitamins, trace elements, antioxidants, essential amino acids and essential fatty acids.
The approach to this and other problems covered by the textbook is interdisciplinary. The
pathophysiological mechanisms and pathomorphological descriptions are intermingled with
profound excursion into the items of Hygiene, Preventive Medicine, Human Ecology, Medical
Geography, Pharmacology, Psychosomatic Medicine etc. The conclusive part of the book is the
largest chapter, covering the field of Endocrine Pathology. It has not only General Endocrinology
remind and basic endocrine pathology synopsis, but also several divisions dedicated to the
disorders of every concrete endocrine gland and to the disorders of APUD system as well. The
composition of material in endocrinological part of the textbook is non-traditional. The text is
arranged along the natural functional relationships, existing within endocrine system itself. Step
by step every particular endocrine "axis" with its derangement is described. Profound description
of pathophysiological basis for Obstetrics and Gynecology, as well as that for Andrology and
Urology can be found in this manual. The text represents most fruitful pathologic theories of
the past, including those which for a long time did not find sufficient illumination in medical
teaching literature, and current views on biochemical aspects of pathology. Most recent advances
of Human Medical Genetics related to the mechanisms of Endocrine and metabolic diseases also
are embedded into the textbook. The Manual has over 40 and more than 130 figures, including
original ones and also borrowed from rare classical sources of the past.
The manual can be especially interesting for Western specialists, because, together with
original and bright interpretation of common heritage, shared by world Pathophysiology, it
gives also the description of authentic views and concepts, achieved by Russian Pathology. This
contribution is not broadly known in Western countries due to previously existed informational
separation. Some chapters of the book are also available in interactive CD version. The textbook
may be recommended for medical, public health and pediatric school students. It can be useful
for postgraduate refreshing courses and for independent self-studies of medical doctors. It is
written with quite informal style. Bright language and good sense of humor displayed on its pages
may protect the readers from boredom. The book is of certain interest not only for medical and
research professionals and students, but also for large number of readers in endeavor to broaden
their erudition in medical and biological field (768 pp., 135 fig., 49 tables).
СОДЕРЖАНИЕ
ГЛАВА1
ОБЩИЕ АСПЕКТЫ ПАТОФИЗИОЛОГИИ ОБМЕНА ВЕЩЕСТВ И РЕГУЛЯЦИИ
МЕТАБОЛИЗМА 7
ГЛАВА2
ЭНЕРГЕТИЧЕСКИЙ МЕТАБОЛИЗМ И ЕГО НАРУШЕНИЯ 9
ОСНОВНОЙ ОБМЕН: ИСТОРИЧЕСКИЕ АСПЕКТЫ 9
ТЕРМОДИНАМИЧЕСКИЕ И БИОХИМИЧЕСКИЕ ОСНОВЫ БИОЭНЕРГЕТИКИ 10
УСЛОВИЯ ИЗМЕРЕНИЯ ОСНОВНОГО ОБМЕНА 17
МЕТОДЫ ОПРЕДЕЛЕНИЯ ОСНОВНОГО ОБМЕНА И ИХ ПРИНЦИПЫ.
КАЛОРИМЕТРИЯ . 22
КАЛОРИЧЕСКИЙ ЭКВИВАЛЕНТ КИСЛОРОДА 25
ДЫХАТЕЛЬНЫЙ КОЭФФИЦИЕНТ И КОЭФФИЦИЕНТ РУБНЕРА, ИХ
ИЗМЕНЕНИЯ ПРИ ПАТОЛОГИИ 27
ЗНАЧЕНИЕ ТЕРМОДИНАМИКИ В МЕДИЦИНЕ , 29
ГЛАВА3
ГОЛОДАНИЕ 31
ИСТОРИЯ ИЗУЧЕНИЯ ГОЛОДАНИЯ 31
ПРИЧИНЫ ГОЛОДАНИЯ 32
КЛАССИФИКАЦИЯ ГОЛОДАНИЯ 33
ПРОДОЛЖИТЕЛЬНОСТЬ ЖИЗНИ ПРИ ГОЛОДАНИИ 35
ПЕРЕРАСПРЕДЕЛЕНИЕ РЕСУРСОВ ПРИ ГОЛОДАНИИ 36
ПЕРИОДЫ ГОЛОДАНИЯ И ИХ ЭНДОКРИННО-МЕТАБОЛИЧЕСКАЯ
ХАРАКТЕРИСТИКА 38
КВАШИОРКОРИ АЛИМЕНТАРНЫЙ МАРАЗМ 46
НЕЙРОГЕННЫЕ АНОРЕКСИЯ И БУЛИМИЯ 48
ОТКАРМЛИВАНИЕ ПОСЛЕ ГОЛОДАНИЯ 51
ЛЕЧЕБНОЕ ГОЛОДАНИЕ 52
ЦИКЛ ГОЛОДАНИЕ-НАСЫЩЕНИЕ 53
ГЛАВА4
АНАБИОЗ И ЗИМНЯЯ СПЯЧКА 55
ГЛАВА5
ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО ОБМЕНА 59
ОБЩИЕ АСПЕКТЫ ПАТОЛОГИИ БЕЛКОВОГО ОБМЕНА 59
НАРУШЕНИЯ КОЛИЧЕСТВЕННОГО ПОСТУПЛЕНИЯ БЕЛКА В ОРГАНИЗМ .60
НАРУШЕНИЯ КАЧЕСТВЕННОГО СОСТАВА БЕЛКОВ 63
НАРУШЕНИЯ ПЕРЕВАРИВАНИЯ БЕЛКОВ. КИШЕЧНАЯ
АУТОИНТОКСИКАЦИЯ 66
НАРУШЕНИЯ ЧРЕЗМЕМБРАННОГО ТРАНСПОРТА АМИНОКИСЛОТ 71
АМИНОАЦИДУРИЯ 75
ПУТИ МЕТАБОЛИЗМА АМИНОКИСЛОТ 76
ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ БЕЛКОВОГО ОБМЕНА И ЕГО НАРУШЕНИЯ
ПРИ ЭНДОКРИНОПАТИЯХ 79
НАРУШЕНИЯ МЕЖУТОЧНОГО ОБМЕНА АМИНОКИСЛОТ И ИХ
ПРОИЗВОДНЫХ 83
НАРУШЕНИЯ КОМПОЗИЦИИ БЕЛКОВ ПЛАЗМЫ 96
НАРУШЕНИЯ КОНЕЧНЫХ ЭТАПОВ ОБМЕНА БЕЛКА 106
ДИСПРОТЕИНОЗЫ 112
ГЛАВА 6
ПАТОФИЗИОЛОГИЯ НУКЛЕИНОВОГО ОБМЕНА 125
ИНФОРМАЦИОННЫЕ И ПАТОХИМИЧЕСКИЕ АСПЕКТЫ НУКЛЕИНОВОГО
МЕТАБОЛИЗМА 125
ПУТИ ОБМЕНА ПУРИНОВ 126
НАРУШЕНИЯ ПУРИНОВОГО ОБМЕНА 129
ЭТИОЛОГИЯ ПОДАГРЫ 132
ПАТОГЕНЕЗ ПОДАГРЫ 135
ДРУГИЕ НАРУШЕНИЯ ПУРИНОВОГО ОБМЕНА 138
НАРУШЕНИЯ ОБМЕНА ПИРИМИДИНОВЫХ ОСНОВАНИЙ ...;139
КРИСТАЛЛИЧЕСКИЕ АРТРИТЫ 141
ГЛАВА 7
НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА 143
ВВЕДЕНИЕ ; 143
ЛИПИДЫ ОРГАНИЗМА И ПИЩИ. АЛИМЕНТАРНАЯ ЛИПИДНАЯ
НЕДОСТАТОЧНОСТЬ '. 144
НАРУШЕНИЯ ПЕРЕВАРИВАНИЯ И ВСАСЫВАНИЯ ЛИПИДОВ 153
ТРАНСПОРТ ЛИПИДОВ В ОРГАНИЗМЕ И ЕГО НАРУШЕНИЯ 155
ГИПЕРЛИПОПРОТЕИДЕМИИ И ДРУГИЕ ДИСЛИПОПРОТЕИДЕМИИ 164
АТЕРОСКЛЕРОЗ 175
НАРУШЕНИЯ ПРОЦЕССОВ НАКОПЛЕНИЯ И МОБИЛИЗАЦИИ ЛИПИДОВ .197
ЛИПОСТАТ И ПАТОФИЗИОЛОГИЯ ПЕРВИЧНОГО ОЖИРЕНИЯ 202
ИСТОЩЕНИЕ И КАХЕКСИЯ 222
ЛИЗОСОМАЛЬНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ ЛИПИДОВ 225
КЕТОЗ И СТЕАТОЗ ПЕЧЕНИ 228
ГЛАВА8
ПАТОФИЗИОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА 237
УГЛЕВОДЫ ПИЩИ И ОРГАНИЗМА. ПИЩЕВОЙ УГЛЕВОДНЫЙ
ДИСБАЛАНС 237
ПЕРЕВАРИВАНИЕ И ВСАСЫВАНИЕ УГЛЕВОДОВ. ПОСТУПЛЕНИЕ ГЛЮКОЗЫ
В КЛЕТКУ 239
НАРУШЕНИЯ ВСАСЫВАНИЯ И ПЕРВИЧНОГО ПРЕОБРАЗОВАНИЯ
УГЛЕВОДОВ 242
КАТАБОЛИЗМ УГЛЕВОДОВ И ЕГО НАРУШЕНИЯ 246
АНАБОЛИЗМ УГЛЕВОДОВ И ЕГО НАРУШЕНИЯ 256
ОСОБЕННОСТИ ОБМЕНА УГЛЕВОДОВ В РАЗЛИЧНЫХ ТКАНЯХ 259
ГЛИКОГЕНОЗЫИАГЛИКОГЕНОЗ 261
ГЛИКОЗАМИНОГЛИКАНЫ И МУКОПОЛИСАХАРИДОЗЫ 265
ГЛАВА9
ПАТОФИЗИОЛОГИЯ САХАРНОГО ДИАБЕТА 271
ВВЕДЕНИЕ 271
ОПРЕДЕЛЕНИЕ И КЛАССИФИКАЦИЯ 272
ИНСУЛИНЗАВИСИМЫЙ САХАРНЫЙ ДИАБЕТ 274
ИНСУЛИННЕЗАВИСИМЫЙ САХАРНЫЙ ДИАБЕТ (ИНСД) 325
ГЛАВА 10
ПАТОФИЗИОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА 345
ВОДНО-СОЛЕВОЙ ОБМЕН, ЕГО РЕГУЛЯЦИЯ И НАРУШЕНИЯ 346
ПАТОФИЗИОЛОГИЯ КИСЛОТНО-ОСНОВНОГО РАВНОВЕСИЯ 360
ГЛАВА 11
ПАТОФИЗИОЛОГИЯ ВИТАМИННОГО ОБМЕНА 381
ОБЩИЕ СВОЙСТВА ВИТАМИНОВ И ИСТОРИЯ ИДЕЙ
В ВИТАМИНОЛОГИИ 381
НАРУШЕНИЯ ОБМЕНА ЭНЗИМОВИТАМИНОВ 388
НАРУШЕНИЯ ОБМЕНА РЕДОКС-ВИТАМИНОВ 403
НАРУШЕНИЯ ОБМЕНА ГОРМОНОВИТАМИНОВ 413
ЗАКЛЮЧЕНИЕ 433
ГЛАВА 12
ПАТОФИЗИОЛОГИЯ ОБМЕНА МИКРОЭЛЕМЕНТОВ 435
РОЛЬ И МЕСТО МИКРОЭЛЕМЕНТОВ В ФИЗИОЛОГИИ И ПАТОЛОГИИ
ОБМЕНА ВЕЩЕСТВ 435
ПАТОФИЗИОЛОГИЯ МЕТАБОЛИЗМА ЖЕЛЕЗА 439
ПАТОФИЗИОЛОГИЯ ОБМЕНА ДРУГИХ МИКРОЭЛЕМЕНТОВ-МЕТАЛЛОВ..449
ПАТОФИЗИОЛОГИЯ ОБМЕНА ВАЖНЕЙШИХ МИКРОЭЛЕМЕНТОВ-
НЕМЕТАЛЛОВ 457
ГЛАВА 13
НАРУШЕНИЯ ЭНДОКРИННОЙ РЕГУЛЯЦИИ 461
ИСТОРИЯ ИДЕЙ В ЭНДОКРИНОЛОГИИ И ЕЁ ОСНОВНЫЕ ПРИНЦИПЫ 462
МЕТАБОЛИЗМ ГОРМОНОВ И ОБЩИЕ МЕХАНИЗМЫ ЭНДОКРИННЫХ
НАРУШЕНИЙ 476
ПАТОЛОГИЯ ГИПОТАЛАМО-ГИПОФИЗАРНОГО НЕЙРОСЕКРЕТОРНОГО
АППАРАТА 497
ПАТОФИЗИОЛОГИЯ ШИШКОВИДНОЙ ЖЕЛЕЗЫ 518
ПАТОФИЗИОЛОГИЯ АДЕНОГИПОФИЗА 525
НАРУШЕНИЯ ЭНДОКРИННЫХ ФУНКЦИЙ ТИМУСА 656
ПАТОФИЗИОЛОГИЯ ОКОЛОЩИТОВИДНЫХ ЖЕЛЁЗ И КАЛЬЦИЙ-
ФОСФОРНО-МАГНИЕВОГО МЕТАБОЛИЗМА 667
НАРУШЕНИЯ РЕГУЛЯЦИИ
КАЛЬЦИЙ-ФОСФОРНО-МАГНИЕВОГО ОБМЕНА 679
ОСТЕОПОРОЗ И МЕТАБОЛИЧЕСКИЕ ЗАБОЛЕВАНИЯ КОСТЕЙ 692
ЗАКЛЮЧЕНИЕ 699
ГЛАВА 14
ПОНЯТИЕ О МЕТАБОЛИЧЕСКОМ СИНДРОМЕ 703
ИСТОРИЯ ИЗУЧЕНИЯ МС 704
ЭПИДЕМИОЛОГИЯ МС 705
КРИТЕРИИ МС 706
ЭТИОЛОГИЯ МС 709
ПАТОГЕНЕЗ МС 712
ПОСЛЕСЛОВИЕ 723
СПИСОК ОСНОВНОЙ ЛИТЕРАТУРЫ 731
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 758
СПИСОК ИСПОЛЬЗУЕМЫХ СОКРАЩЕНИЙ 761
RESUME 765