/








































































































Текст
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ ИМ. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ*
АНАЛИТИЧЕСКАЯ ХИМИЯ СУРЬМЫ
А. А. Немодрук
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1978
УДК 543 : 546.86
Серия «Аналитическая химия элементов»
Главный редактор
член-корреспондент АН СССР Ю. А. Золотов
Редакционная коллегия:
И. П. Алимарин, Ю. И. Беляев, А. И. Бусев, М. П. Волынец, А. Н. Ермаков, В. М. Иванов, А. В. Карякин, И. М. Кузьмин, С. Б. Саввин, Н. М. Ростоцкая (ученый секретарь)
Редактор тома «Аналитическая химия сурьмы» профессор П. К. Агасян
Адрес редколлегии: 117334, Москва, Воробьевское шоссе, 47а Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
20506-457
Н 055(02)-78 156'78’ кн- 2
© Издательство «Наука», 1978 г.
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов» — составит около 50 томов. Потребность в подобного рода издании назрела давно. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом возникло настоящее издание — серия «Аналитическая химия элементов», которое осуществляется впервые. Издание серии было начато по инициативе академика А. П. Виноградова, который с 1958 по 1975 г. был ее главным редактором.
Аналитическая химия любого элемента и его различных со-едйнений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы, как физические, так и физико-химические и химические, излагаются применительно для количественного определения данного элемента начиная с анализа сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
3
Монографии содержат обширную библиографию, доведенную^ до последних лет; они рассчитаны на широкий круг химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического-элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно по мере их подготовки. Вышли в свег монографии, посвященные торию, таллию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, бериллию, редкоземельным элементам и иттрию, никелю, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, селену и теллуру, алюминию, нептунию, трансплутониевым элементам, платиновым металлам, радию, кремнию, германию, магнию, рению, золоту, марганцу, кадмию, ртути, кальцию, фосфору, литию, олову, серебру, цинку, рубидию и цезию, вольфраму, мышьяку, сере, плутонию, азоту, барию, стронцию.
Готовятся к печати монографии по аналитической химии углерода, хрома, меди, ванадия.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Сурьма — один из давно известных и довольно часто используемых элементов. Она входит в состав многих сплавов цветных металлов, типографских шрифтов, подшипниковых сплавов. Сурьма и ее соединения используются в резиновой, красильной, спичечной, стекольной, фармацевтической, аккумуляторной, приборостроительной и в ряде других отраслей промышленного производства. Сурьма применяется при изготовлении солнечных батарей, инфракрасных детекторов, ферромагнитных приборов, огнестойких соединений, сурьмяных электродов для рН-метров. Особенно важной областью потребления сурьмы является полупроводниковая промышленность. В ряде случаев требуется сурьма очень высокой чистоты. В то же время содержание сурьмы в земной коре очень мало и не превышает 4-10~5%. В связи с этим аналитическая химия сурьмы характеризуется очень большим разнообразием методов ее отделения и определения, широким диапазоном определяемых концентраций и большим разнообразием анализируемых материалов. Особенно быстро аналитическая химия сурьмы развивалась за последние 25 лет в связи с прогрессом полупроводниковой промышленности. За это время возник и успешно развивался ряд новых разделов аналитической химии сурьмы, в том числе такие, как аналитическая химия сурьмы высокой чистоты и ее соединений, методы определения очень малых количеств сурьмы в различных материалах и т. п.
Общее быстрое развитие аналитической химии оказывало влияние на аналитическую химию сурьмы — она пополнялась новыми высокочувствительными, высокопроизводительными и быстрыми физическими и физико-химическими методами. Опубликовано большое число работ по аналитической химии сурьмы. Однако специальные руководства или монографии по аналитической химии сурьмы в мировой научной литературе фактически отсутствуют. Работы по аналитической химии сурьмы опубликованы во многих, часто малодоступных изданиях, сборниках, диссертациях, патентных описаниях и т. п. на различных языках народов нашей планеты. Поэтому возникла необходимость в монографии, в которой были бы рассмотрены и критически оценены различные методы определения, отделения и обнаружения сурьмы, а также сделаны рекомендации по применению наиболее подходящих
5
методов анализа в каждом конкретном случае в зависимости от анализируемого материала, содержания в нем сурьмы, требующейся точности, производительности и ряда других факторов.
Для восполнения этого пробела написана настоящая монография. В ней сделана попытка объективно рассмотреть и оценить все методы определения, отделения и обнаружения сурьмы и методы определения примесей в сурьме высокой чистоты и ее соединениях. Особое внимание уделено современным, наиболее надежным, быстрым, высокопроизводительным инструментальным методам, а также методам, характеризующимся высокой чувствительностью и точностью. В книге особенно подробно изложены новые и наиболее перспективные методы определения сурьмы в разнообразных промышленных и природных материалах, в том числе такие физические и физико-химические методы, как спектральные и химико-спектральные, рентгенофлуоресцентные, атомно-абсорбционной и атомно-эмиссионной спектрофотометрии, фотометрические и экстракционно-фотометрические и т. д. Рассматриваются также перспективы дальнейшего развития отдельных методов.
В книге сделана сравнительная оценка различных методов, указаны их достоинства, недостатки и ограничения. Приводятся такие важнейшие их характеристики, как чувствительность и точность.
В монографии нашли отражение работы, опубликованные в отечественной и зарубежной литературе до 1975 г. включительно и частично за 1976 г.
Автор выражает глубокую благодарность кандидату химических наук Е. Я. Нейману за помощь в написании раздела «Электрохимические методы», кандидату технических наук М. П. Бурмистрову за просмотр и обсуждение раздела «Спектральные методы» и Л. В. Морейской за просмотр и обсуждение разделов «Атомно-абсорбционная спектрофотометрия» и «Фотометрия пламени». Особенно глубоко автор признателен К. Г. Дмитриеву за большую помощь в подборе необходимой литературы, а также доктору химических наук Н. М. Кузьмину и кандидату химических наук В. И. Фадеевой за прочтение рукописи и ценные замечания.
Автор
Глава I
ОБЩИЕ СВЕДЕНИЯ О СУРЬМЕ
ПОЛОЖЕНИЕ СУРЬМЫ В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ
Сурьма принадлежит к пятой группе периодической системы химических элементов Д. И. Менделеева и входит в подгруппу мышьяка. Атомная масса сурьмы равна 121,75 [213, 992], атомный номер 51. Строение электронной оболочки: Is2, 2s2, 2р6, 3s2, Зр6, 3d10, 4s2, 4р6, 4d10, 5s2, 5р3.
Природная сурьма состоит из двух стабильных изотопов: 121Sb(57,25%) и 123Sb(42,75%). Важнейшими искусственно полученными радиоактивными изотопами сурьмы являются: 122Sb, i22mgb и 124Sb (табл. 1). В настоящее время известно около 25 искусственно полученных радиоактивных изотопов сурьмы, в том числе изотопы с атомной массой 116 (тип превращения: 0+, электронный захват, = 15,5 мин. и тип превращения (3+, Г,/, = = 60 мин.), 117 (р+, электронный захват, 71/, = 2,5 часа), 118 (электронный захват, Т,/с — 5,1 часа), 120 (0+, электронный захват, = 16,6 мин.), 124m (изомерный переход, Ту, = = 1,3 мин. и изомерный переход, = 21 мин.), 125 (Р~, 2,7 года), 126 (р~, Ту, = 9 час., р~, Ti/t = 28 дней), 127 (р", Т./( = = 93 часа), 129 (р~, Т,/г = 4,2 часа), 131 (£', Т,/2 = 23,1 мин.)
НАХОЖДЕНИЕ СУРЬМЫ В ПРИРОДЕ
Содержание сурьмы в земной коре очень мало и составляет 4-10_6% [130]. Однако, несмотря на это, сурьма — один из доступных металлов вследствие наличия ее руд и минералов. Кроме того, сурьма содержится в виде примесей в рудах многих других металлов, при переработке которых ее выделяют в качестве побочного продукта.
В природе наиболее часто встречаются соединения трехвалентной положительно заряженной сурьмы (сульфиды, тиосоли, антимониты, триоксид), затем трехвалентной отрицательно заряженной (антимониды). Соединения пятивалентной сурьмы в природе встречаются очень редко.
7
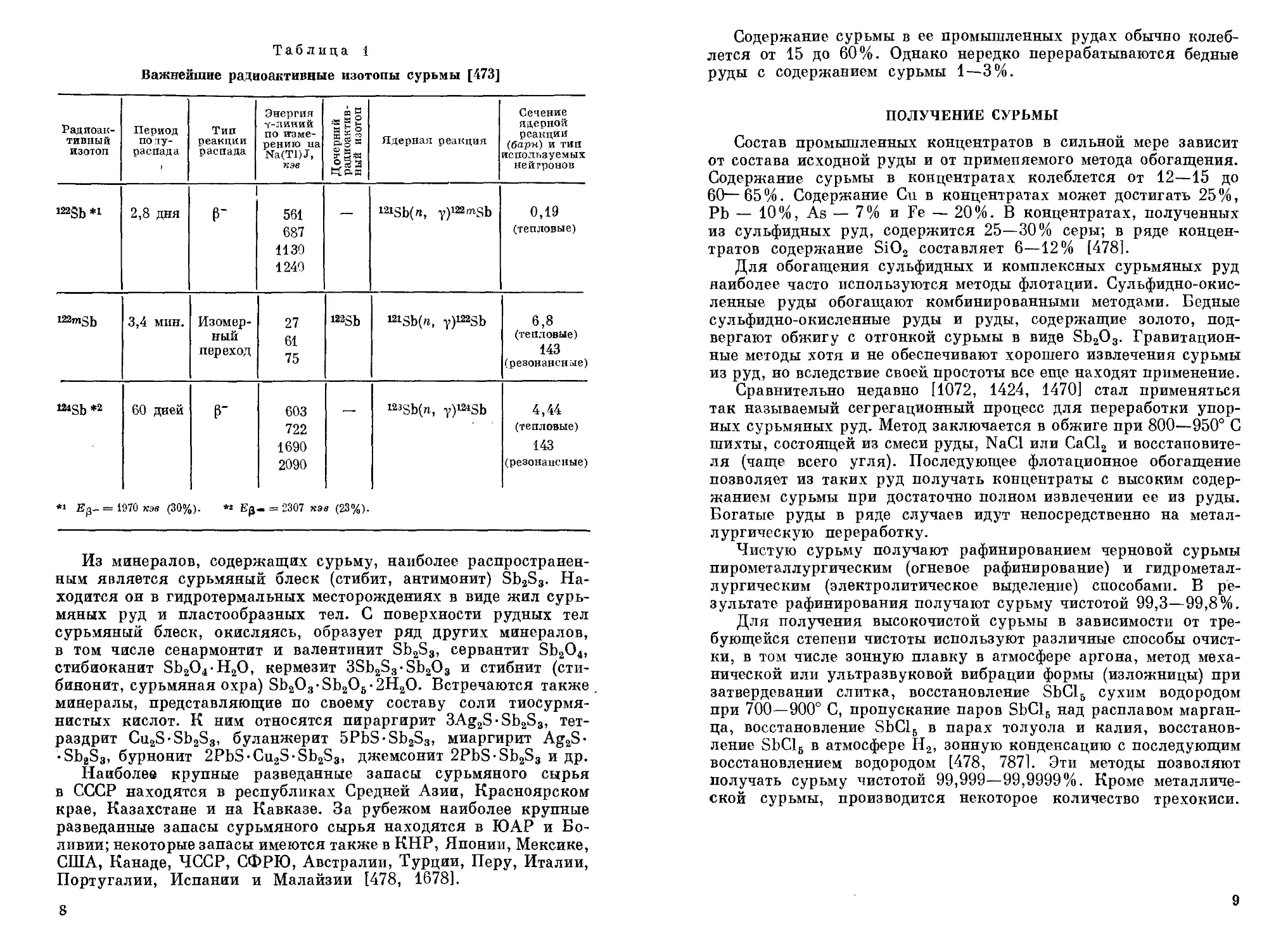
Таблица 1
Важнейшие радиоактивные изотопы сурьмы [473]
Радиоактивный изотоп Период по лу-распада 1 Тип реакции распада Энергия у-линий по измерению на Na(Tl)J, кэв Дочерний радиоактивный изотоп Ядерная реакция Сечение ядерной реакции (барн) и тип используемых нейтронов
122gb *1 2,8 дня Г 561 687 ИЗО 1249 — 121Sb(n, y)122mSb 0,19 (тепловые)
122mSb 3,4 мин. Изомерный переход 27 61 75 122Sb 121Sb(n, V)122Sb 6,8 (тепловые) 143 (резонансные)
124gb *2 60 дней г 603 722 1690 2090 — 123Sb(n, y)i2*Sb 4,44 (тепловые) 143 (резонансные)
*1 Eg- = 1970 кэв (30%). *2 Ер- = 2307 кэв (23%).
Из минералов, содержащих сурьму, наиболее распространенным является сурьмяный блеск (стибит, антимонит) Sb2S3. Находится он в гидротермальных месторождениях в виде жил сурьмяных руд и пластообразных тел. С поверхности рудных тел сурьмяный блеск, окисляясь, образует ряд других минералов, в том числе сенармонтит и валентинит Sb2S3, сервантит Sb2O4, стибиоканит Sb2O4-H2O, кермезит 3Sb2S3-Sb2O3 и стибнит (сти-бинонит, сурьмяная охра) Sb2O3-Sb2O6-2H2O. Встречаются также минералы, представляющие по своему составу соли тиосурмя-нистых кислот. К ним относятся пираргирит 3Ag2S-Sb2S3, тетраэдрит Cu2S-Sb2S3, буланжерит 5PbS-Sb2S3, миаргирит Ag2S-•Sb2S3, бурнонит 2PbS-Cu2S-Sb2S3, джемсонит 2PbS-Sb2S3 и др.
Наиболее крупные разведанные запасы сурьмяного сырья в СССР находятся в республиках Средней Азии, Красноярском крае, Казахстане и на Кавказе. За рубежом наиболее крупные разведанные запасы сурьмяного сырья находятся в ЮАР и Боливии; некоторые запасы имеются также в КНР, Японии, Мексике, США, Канаде, ЧССР, СФРЮ, Австралии, Турции, Перу, Италии, Португалии, Испании и Малайзии [478, 1678].
8
Содержание сурьмы в ее промышленных рудах обычно колеблется от 15 до 60%. Однако нередко перерабатываются бедные руды с содержанием сурьмы 1—3%.
ПОЛУЧЕНИЕ СУРЬМЫ
Состав промышленных концентратов в сильной мере зависит от состава исходной руды и от применяемого метода обогащения. Содержание сурьмы в концентратах колеблется от 12—15 до 60—65%. Содержание Си в концентратах может достигать 25%, РЬ — 10%, As — 7% и Fe — 20%. В концентратах, полученных из сульфидных руд, содержится 25—30% серы; в ряде концентратов содержание SiO2 составляет 6—12% [478].
Для обогащения сульфидных и комплексных сурьмяных руд наиболее часто используются методы флотации. Сульфидно-окисленные руды обогащают комбинированными методами. Бедные сульфидно-окисленные руды и руды, содержащие золото, подвергают обжигу с отгонкой сурьмы в виде Sb2O3. Гравитационные методы хотя и не обеспечивают хорошего извлечения сурьмы из руд, но вследствие своей простоты все еще находят применение.
Сравнительно недавно [1072, 1424, 1470] стал применяться так называемый сегрегационный процесс для переработки упорных сурьмяных руд. Метод заключается в обжиге при 800—950° С шихты, состоящей из смеси руды, NaCl или СаС12 и восстановителя (чаще всего угля). Последующее флотационное обогащение позволяет из таких руд получать концентраты с высоким содержанием сурьмы при достаточно полном извлечении ее из руды. Богатые руды в ряде случаев идут непосредственно на металлургическую переработку.
Чистую сурьму получают рафинированием черновой сурьмы пирометаллургическим (огневое рафинирование) и гидрометаллургическим (электролитическое выделение) способами. В результате рафинирования получают сурьму чистотой 99,3—99,8%.
Для получения высокочистой сурьмы в зависимости от требующейся степени чистоты используют различные способы очистки, в том числе зонную плавку в атмосфере аргона, метод механической или ультразвуковой вибрации формы (изложницы) при затвердевании слитка, восстановление SbCl5 сухим водородом при 700—900° С, пропускание паров SbCl6 над расплавом марганца, восстановление SbCl6 в парах толуола и калия, восстановление SbCl6 в атмосфере Н2, зонную конденсацию с последующим восстановлением водородом [478, 7871. Эти методы позволяют получать сурьму чистотой 99,999—99,9999%. Кроме металлической сурьмы, производится некоторое количество трехокиси.
9
ПРИМЕНЕНИЕ СУРЬМЫ
Сурьма находит широкое применение в современной науке и технике. В настоящее время важнейшим потребителем высокочистой сурьмы является полупроводниковая промышленность. Сурьму с содержанием примесей применяют как до-
норную добавку при’производстве германиевых полупроводников. Сурьма высокой чистоты служит исходным материалом для получения антимонидов Al, Ga и In. Антимонид индия используется для производства датчиков Холла, в счетно-решающих устройствах, в качестве ^фильтра и детектора ИК-излучения и т. п. Антимониды А1 и Ga используются при изготовлении высокочастотных диодов и триодов.
Сурьма входит в состав различных сплавов специального назначения. Например, она входит в состав так называемого твердого свинца (5—15%), используемого для приготовления аккумуляторных пластин, труб и листового проката для химической промышленности, для производства защитных оболочек электрических и телеграфных кабелей, для получения типографских и подшипниковых сплавов. Чистая сурьма используется для приготовления сурьмяных электродов для pH-метров и других приборов. Радиоактивный изотоп 124Sb используется в качестве источника нейтронов и у-излучения.
Трехокись сурьмы применяют в качестве ингредиента в производстве термостойких синтетических смол и полимеров. Различные соединения сурьмы служат исходным материалом в производстве ряда медицинских препаратов. Соединения сурьмы используются в текстильной промышленности в качестве протрав и при получении невозгораемых тканей, в стекольной промышленности при изготовлении оптических стекол, в качестве пигмента при изготовлении некоторых красок и эмалей, в резиновой и спичечной промышленности, в производстве ряда химических реактивов, в качестве люминесцентного покрытия при изготовлении ламп дневного света.
Глава II
ХИМИКО-АНАЛИТИЧЕСКАЯ ХА РАКТЕРИСТИКА СУРЬМЫ
И ЕЕ СОЕДИНЕНИЙ
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА СУРЬМЫ
Сурьма — металл блестящего серовато-белого цвета. Из жидкого состояния застывает в кристаллическом виде. Кроме кристаллической формы, известны три аморфные формы — желтая, черная и взрывчатая сурьма. В обычных условиях устойчива только кристаллическая сурьма. Основные физико-химические свойства кристаллической сурьмы представлены ниже.
Плотность при 25°С , г/см3
Т. пл., °C
Т. кип., °C
Электропроводность при 0° С, ол»-1 • ел»-1
Твердость по Моосу
по Бринелю, кг!мм2\
Теплота плавления, ккал/г-ато.и
Теплота испарения, ккал/г-атом
Уд. теплоемкость при ?0° С, кал/г-град Теплопроводность при 0° С, кал/см-сек-град
Уд. электросопротивление при 20° С, мком-см
Уд. электромагнитная восприимчивость Поперечное сечение захвата тепловых нейтронов, барн
Энергия ионизации, ав/г-атом
Sb° —»Sb+
Sb+ -»Sb2+
Sb2+ -» Sb3+
Sbs+ -4 Sb«+
Sb4+ -» Sb5+
Атомный радйус, A
Радиус иона, A
Sbs”
Sb6+
Sb3+
Потенциал ионизации, ов
6,678
630,5
1440
2,56-Ю-4
3,0—3,5 32,5—34,0
9,5
49,6 0,04987 0,045
43,045
6,6-10-*
2,6.
8,639' 16,5 25,3 44,02
55,4
1,40
2,77 0,62 0,92 0,48
11
При комнатной температуре на воздухе сурьма не окисляется, но ее пары легко сгорают с образованием Sb2O3. Желтая сурьма образуется при пропускании О2 или воздуха в жидкий SbH3 при — 90° С. При — 50° С желтая сурьма переходит в кристаллическую сурьму. Черная сурьма образуется при быстром охлаждении паров сурьмы. При охлаждении до ~400° С она переходит в кристаллическую сурьму.
Взрывчатая сурьма получается при электролизе раствора SbCl3 (17—33% SbCl3) в НС1 (пл. 1,12) при плотности тока в пределах 0,043—0,2 а/дм2. Полученная при этом сурьма переходит в кристаллическую сурьму со взрывом, вызываемым механическим воздействием (трение, царапание, удар, соприкосновение нагретым металлом).
Элементная сурьма нерастворима ни в воде, ни в органических растворителях, практически нерастворима в растворах HF, разб. H2SO4 и растворах щелочей; растворяется в смеси HNO3 и НС1 и смеси HNO3 и винной кислоты. При действии конц. HNO3 сурьма переходит в нерастворимую HSbO2, очень медленно растворяется в конц. НС1 и H2SO4 с образованием соответственно HSbCl4 и Sb2(SO4)3.
СОЕДИНЕНИЯ СУРЬМЫ
Неорганические соединения сурьмы
Соединения с кислородом. С кислородом сурьма образует четыре соединения: Sb2O3,Sb2O4, Sb6O12 и Sb2O5. Трехокись сурьмы Sb2O3 образуется при нагревании сурьмы на воздухе. Она бесцветна, плотность колеблется от 5,1 до 5,8 г/см2 в зависимости от происхождения или способа получения. Температура плавления 656° С, кипения 1550° С. Давление пара при 505° С 0,08 мм рт. ст., при 656° С — 8,5 мм рт. ст. Растворимость в воде составляет 0,016 г/л при 1,5° С и 0,010 г/л при 100° С; хорошо растворяется в растворах НС1 и HF; в разб. HNO3 растворяется мало, а в конц. HNO3 окисляется с образованием малорастворимой HSbO3; в кипящей конц. H2SO4 растворяется хорошо, но при охлаждении выпадает осадок малорастворимого сульфата. Растворимость в щелочных растворах невелика (в 0,005—0,1 М NaOH растворяется 4,4-10-3 молъ/л). Трехокись сурьмы немного растворяется в этаноле, несколько лучше — в глицерине, а также в растворах винной, бензойной, молочной, уксусной и в ряде других органических кислот.
Из растворов галогенидов при подщелачивании или из растворов щелочных антимонидов при подкислении выделяется гидроокись сурьмы — Sb(OH3). В большинстве случаев она выделяется частично обезвоженной и ее состав соответствует формуле SbO(OH) или HSbO2. Радикал SbO, называемый антимонилом, входит в состав ряда солей, играя в них роль одно-12
валентного металла. Метасурьмянистая кислота HSbO2 очень •слабая кислота (К = 10-11). Из ее солей наиболее известен малорастворимый NaSbO2, выделяющийся обычно в виде кристаллогидрата NaSbO2-3H2O.
Тетраокись сурьмы — Sb2O4 получается при разложении Sb2O5 нагреванием; при 750—800° С или окислением Sb2O3 кислородом воздуха при 410—450° С. Тетраокись сурьмы — белый кристаллический порошок, желтеющий при нагревании; плотность 7,5 г/см2-, при температуре выше 900® С заметно разлагается, а при 1030° С полностью переходит в Sb2O3; мало растворяется в воде (3,8-10~6 г/л) и в разбавленных кислотах.
Пятиокись сурьмы — Sb2O5 светло-желтого цвета, существует в двух формах — кристаллической (пл. 7,86 г/'сж3) и аморфной (пл. 3,76 г/<ш3); устойчива ниже 357° С, при более высоких температурах переходит через Sb6O12 в Sb2O4 и далее — в Sb2O3. Пятиокись мало растворяется в воде, в растворах щелочей растворяется с образованием антимонатов щелочных металлов, хорошо растворяется в соляной кислоте с образованием гидроксохлоридных комплексов сурьмы (V) и HSbCl6. С водой Sb2O6 образует ряд гидратов, содержащих от 1 до 6 молекул воды на одну молекулу Sb2O5 [935], однако определенные гидратные •формы нехарактерны и белый аморфный осадок (a:Sb2O5-г/Н2О) изменяет свой состав в зависимости от условий. Соли сурьмяной кислоты (антимонаты) производятся обычно от гексагидрокси-сурьмяной кислоты H[Sb(OH)6], отвечающей гидратированной мета-форме HSbO3-3H2O (К = 4-Ю-6).
Известны также антимонаты, соответствующие солям ряда полимерных сурьмяных кислот, в частности пиросурьмяной кислоте H4Sb2O7. Кислая калиевая соль пиросурьмяной кислоты K2H2Sb2O7-5H2O реагирует с ионами натрия с образованием нерастворимого пироантимоната, вследствие чего используется для качественного обнаружения натрия. Все соединения Sb(V) обладают окислительными свойствами, которые проявляются только в кислых растворах. Сурьма(У) способна частично окислять даже НС1 до С12.
Соединения с галогенами. Галогениды сурьмы(Ш) типа SbX3 известны для всех галогенов. Из галогенидов Sb(V) устойчивы -только SbCl6 и SbF6. Некоторые физико-химические свойства галогенидов сурьмы представлены в табл. 2. Трифторид сурьмы получается при растворении сульфата или хлорида Sb(III) во фтористоводородной кислоте. Трихлорид сурь-м ы гигроскопичен и расплывается на воздухе. Хлориды щелочных и щелочноземельных металлов и NH4C1 при достаточной их концентрации предотвращают гидролиз SbCl3 вследствие образования более прочных анионных хлоридных комплексов Sb(III). Трибромид сурьмы более гигроскопичен, чем трихлорид. Конц. HNO3 разлагает его с одновременным окислением Sb(III) до Sb(V). Трииодид сурьмы на воздухе устойчив даже
13
Таблица 2
Некоторые свойства галогенидов сурьмы
Галогенид внешний вид Плотность, г/см* ТПЛ. °C гкип> °с дн°гм, ккал/жоль 8 (в воде)
SbF3 Бесцветные кристаллы 4,38 292 319 —217,2 444,7 0/100 г воды
SbCl3 То же 3,14 73,4 221 —91,34 Не растворяется
SbBr3 » 4,15 97,0 288 —62,1 То же
SbJ3 Кристаллы* 4,85 170 401 —23,0
SbF6 Бесцветная жидкость 2,99 8,3 149,5 — Растворяется
SbCl5 Светло-желтые кристаллы 2,33 3 140 (разл.) —104,87 Разлагается
SbJ6 Темно-бурые кристаллы — 79 400,6 — »
* Ромбические — желтые, моноклинные—желто-зеленые, тригональные — красные.
при нагревании, он не гигроскопичен; азотной кислотой разла-гается с образованием Sb2O3 и J2.
Пентафторид сурьмы в сухом воздухе испаряется без разложения, во влажном воздухе образует гидрат SbFs-2H2O. Может быть легко получен при взаимодействии SbCl6 с газообразным HF. Пентахлорид сурьмы дымит на воздухе вследствие взаимодействия с содержащейся в нем влагой, при температуре выше 106° С разлагается на SbCl3 и С12; образуется при слабом нагревании сурьмы с хлором или хлорированием SbCl3; водой гидролизуется до сурьмяной кислоты; с хлоридами одновалентных металлов образует комплексные хлориды типа Me[SbCl6]. Известны также тетрахлорид и тетрабромид сурьмы [935].
Тригалогениды сурьмы (SbCl3, SbBr3, SbJ3) хорошо растворяются в инертных органических растворителях (бензол, толуол, СС14, СНС13, С2Н4С12), что широко используется для отделения и концентрирования сурьмы.
Соединения с серой. Известны три модификации Sb2S3: серочерная кристаллическая и две аморфные. Наиболее устойчива кристаллическая форма. Аморфная оранжевая форма образуется при действии H2S на растворы галогенидов сурьмы; температура ее плавления 548° С, температура кипения 990° С. Т р и с у л ь-фид сурьмы нерастворим в воде и разбавленных минеральных кислотах. Водород при 360° С восстанавливает Sb2S3 до Sb и H2S. Хлор, бром и иод взаимодействуют с Sb2S3 с образованием соответствующих галогенидов сурьмы и серы. Трисульфид сурьмы 14
на воздухе горит голубоватым пламенем с образованием смеси различных окислов сурьмы и SO2. Конц. HNO3 реагирует с Sb2S3, образуя различные продукты окисления, в том числе сурьмяную и серную кислоты и свободную серу. Растворы гидроокисей щелочных металлов и щелочных сульфидов растворяют Sb2S3 с образованием солей сурьмянистой и тиосурьмянистой кислот.
Известны соли ортотиосурьмянистой (H3SbS3), метатиосурь-мянистой (HSbS2), пиротиосурьмянистой (H4Sb2S6), ортотиотет-расурьмянистой (H6Sb4S9), метатиотетрасурьмянистой (H2Sb4O7), ортотиооктасурьмянистой (H10Sb8S17) и метатиооктасурьмянистой (H2Sb8S13) кислот.
Тетрасульфид сурьмы Sb2S4 образуется при нагревании Na3SbS4 с соляной кислотой.
Пентасульфид сурьмы — аморфное вещество ярко-оранжевого цвета; плотность 4,12 г/см\ при нагревании до 170° С разлагается на Sb2S3 и S, горит на воздухе с образованием Sb2O3 и SO2; в воде не растворяется, но легко растворяется в растворах щелочей и щелочных сульфидов с образованием смесей соответствующих антимонатов и тиоантимонатов в первом случае и тиоантимонатов — во втором.
Соединения с селеном и теллуром. Сурьма(Ш) с селеном образует ряд соединений, в том числе Sb2Se3, Sb3Se4, Sb4Ses и SbSe. Имеется указание на образование Sb2Ses [1151]. Известны соединения Sb с Se, в состав которых входят также галогениды (SbSeBr, SbSeJ) и сера (Sb2Se2S3). Описан трителлурид Sb2Te3 [1526].
Соединения с водородом. Известны два соединения сурьмы с водородом: дигидрид H2Sb2 и сурьмянистый водород (стибин) SbH3. Дигидрид сурьмы — твердое вещество, образуется только при взаимодействии антимонидов натрия или калия с водой. В аналитической химии сурьмы H2Sb2 не используется, в то время как стибин находит широкое применение в методах отделения сурьмы и в методах ее качественного и количественного определения.
Стибин— бесцветный газ с удушливым запахом, немного напоминающим запах H2S. Он в 4,3 раза тяжелее воздуха. Температура плавления его — 88,5° С, кипения — 17° С. В одном объеме воды растворяется 5 объемов SbH3; с водой не взаимодействует, хорошо растворяется в органических растворителях: в одном объеме этанола растворяется до 15 объемов SbH3, а в одном объеме СС14 или CS2 растворяется ~ 250 объемов SbH3 При комнатной температуре стибин медленно разлагается, а при температуре выше 150° С очень быстро распадается на Н2 и Sb, выделяющуюся на холодной поверхности в виде зеркала металлической сурьмы.
Стибин обладает сильными восстановительными свойствами и быстро окисляется даже слабыми восстановителями, разлагается концентрированными кислотами и щелочами. Образуется стибин при восстановлении растворимых соединений сурьмы цинком
15
в соляной кислоте, борогидридом натрия и при действии разбавленных кислот на антимониды ряда металлов (например, Mg3Sb2). Стибин образуется также при обработке бескислородными кислотами сплавов, содержащих сурьму.
По физиологическому действию стибин аналогичен арсину, он поражает центральную нервную систему и кровь. Признаки отравления стибином: головная боль, тошнота, замедленное дыхание, слабый пульс, усиленное мочеиспускание. Токсичность стибина несколько больше токсичности арсина 1752].
Соединения с металлами (антимониды). Антимониды образуются сплавлением сурьмы с металлами. Наибольшее практическое значение нашли антимониды элементов третьей группы периодической системы (AlSb, GaSb и InSb) благодаря их полупроводниковым свойствам [6].
Оксигалогениды. Для Sb(III) и Sb(V) известны оксигалогениды которые образуются из соответствующих галогенидов в результате их частичного гидролиза или из Sb2O3 и Sb2Os вследствие неполной замены атомов О атомами галогенов при обработке галогеноводородными кислотами. Известны антимонилфто-р и д SbOF, антимонипхлорид SbOCl, антимонил-бромид SbOBr, анти м о нилиодид SbOJ, а также более сложные продукты гидролиза соответствующих галогенидов, в. том числе Sb8O7F10, Sb4O3(OH)3Cl3, Sb4OsCl2, Sb4OsBr2) Sb4OsJ2,
Сульфогалогениды. Аналогично оксигалогенидам сурьма образует сульфогалогениды (тиогалогениды), в том числе SbSCl, Sb4S8Cl2, SbsS6Cl3, Sb8SuCl2, SbSBr, SbSJ. Для Sb(V) сульфогалогениды вследствие их большой склонности к гидролизу и малой устойчивости изучены недостаточно.
Оксисульфиды. Сурьма(Ш) образует ряд оксисупьфидов, в том числе Sb2OS2, Sb4OSs. Известны также оксисульфиды Sb(V) (например, Sb2O4S), а также оксисульфиды, содержащие одновременно Sb(III) и Sb(V) (например, Sb2O2S2).
Сульфаты. Для Sb(III) известен ряд кислых и основных сульфатов, а также средний сульфат Sb2(SO4)3. Все они легко гидролизуются.
Органические соединения сурьмы
Сурьмаорганические соединения — один из наиболее изученных классов металлоорганических соединений [357]. Сурьмаорганические соединения образуют как Sb(III), так и Sb(V). Соединения, содержащие Sb(III), рассматриваются как производные стибина. Наибольшее применение находят ароматические сурьмаорганические соединения типа R3Sb, R3SbX2 и RSb(OH)2O, где R — ароматический радикал и X — галоген. Как для Sb(III), так и Sb(V) известно большое число образуемых ими сурьмаор-ганических соединений, в том числе RSbX2, R2SbX, R3Sb, RSb = =SbR, RSbO и (R2Sb)2O, RSbX4, R2SbX3, R3SbX2, R4SbX, RsSb,. RSb(OH)2O, R2Sb(OH)O и R3SbO.
16
Соединения типа R4Sb.X легко диссоциируют в водных растворах на соответствующие ионы. Из соединений этого типа соли тетрафенилстибония используются в аналитической химии наиболее часто [938]. Катион Ph4Sb + образует с анионами металлгалогенидных кислот и некоторыми другими тяжелыми анионами малорастворимые в водных растворах ионные ассоциаты, растворяющиеся в неполярных органических растворителях, что используется для экстракционного отделения следовых количеств ряда металлов.
При действии трех молекул C6HsMgBr на молекулу SbCls образуется трифенилстибин (C6Hs)3Sb (пластинки, т. пл. 53° С). При избытке SbCl3 образуются C6HsSbCl2 (т. пл. 62° С) и (C6H5)2SbCl (т. пл. 68° С). Фенилстибиндихлорид при взаимодействии с хлором образует C6HsSbCl4, который легко гидролизуется с образованием фенилстибиновой (фенилсурьмяной) кислоты C6H5SbO(OH)2. При хлорировании (C6H5)2SbCl получается (C6Hs)2SbCl3, который при обработке горячим разбавленным раствором NaOH дает (CeH5)2SbO-• (ONa). Дифенилстибиновая кислота (C6H6)2SbO-• (ОН) реагирует с соляной кислотой с образованием (CeH5)2SbCl3 и (C6H5)2Sb(OH)Cl2. Трифенилстибин при взаимодействии с хлором образует (C6Hs)3SbCl2 (иглы, т. пл. 143° С). Гидролизом (C6H5)3SbCl2 или окислением (C6H5)3Sb перманганатом калия получают (С6Н5)3-• Sb(OH)2 (листочки, т. пл. 212° С).
Стибинобензол C6H5Sb=SbC6H5 получают восстановлением CeH5SbO(ONa)2 гидросульфитом. При взаимодействии солей диазония с растворами HSbO3 в щелочной среде протекает-реакция, аналогичная реакции Барта для мышьяковистых соединений, с образованием соответствующих арилпроизводных сурьмянистой кислоты. При окислении кислородом воздуха смесей? арилгидразинов с SbCl3 в присутствии СиС12 образуются соответствующие первичные и вторичные сурьмаорганические соединения. Эти же соединения могут быть получены исходя из SbCls.
Известен ряд гетероциклических сурьмаорганических соединений, в которых Sb является гетероатомом (например, с т и-бакридин). В органических соединениях Sb теряет свойства, характерные для нее в неорганических соединениях. Для определения Sb в сурьмаорганических соединениях их, как правило,, предварительно минерализуют.
17 '
Глава III
КАЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СУРЬМЫ
ФИЗИЧЕСКИЕ МЕТОДЫ
В связи с быстрым развитием и внедрением в практику физических методов количественного анализа они могут использоваться также для быстрого обнаружения сурьмы.
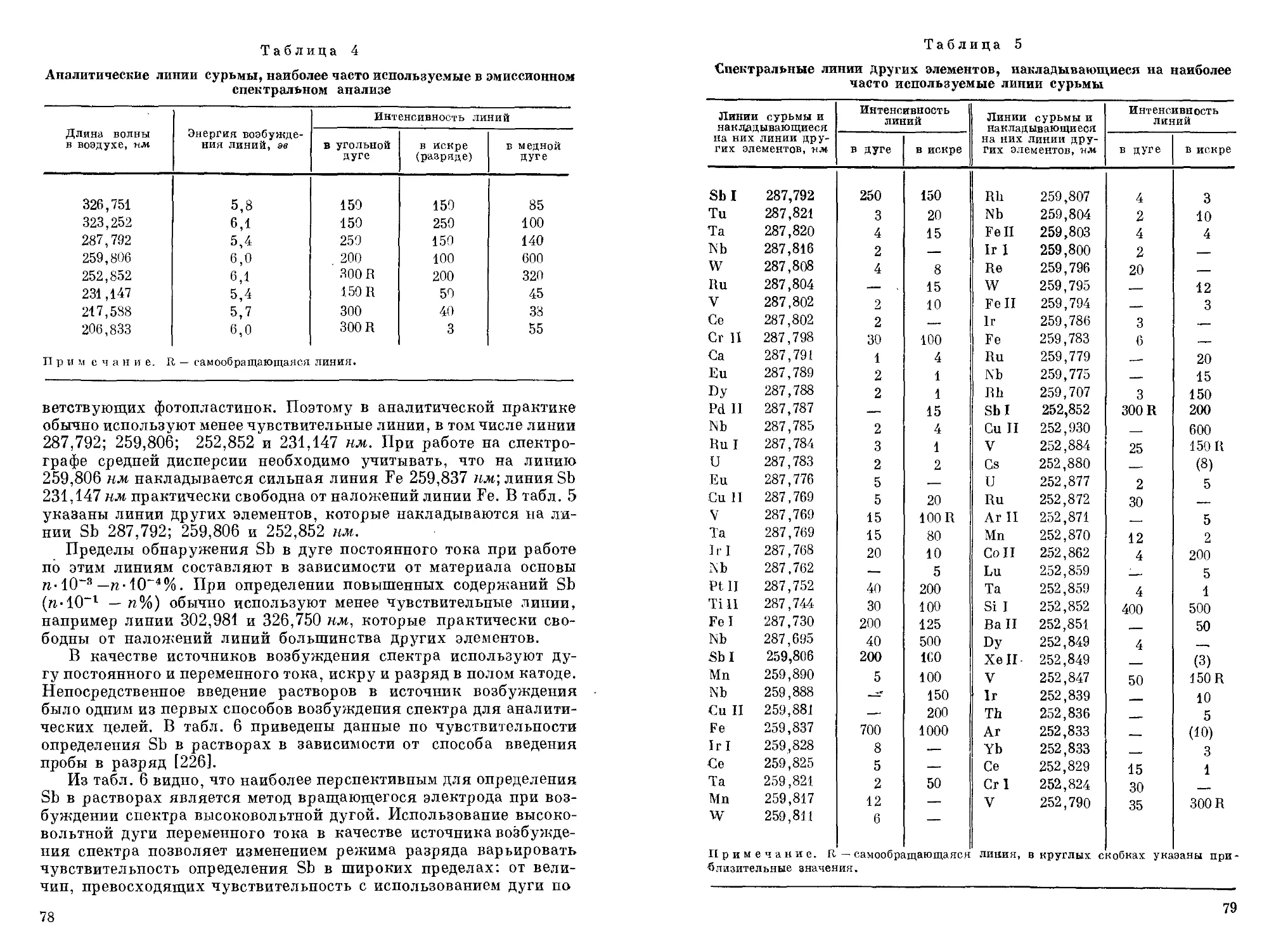
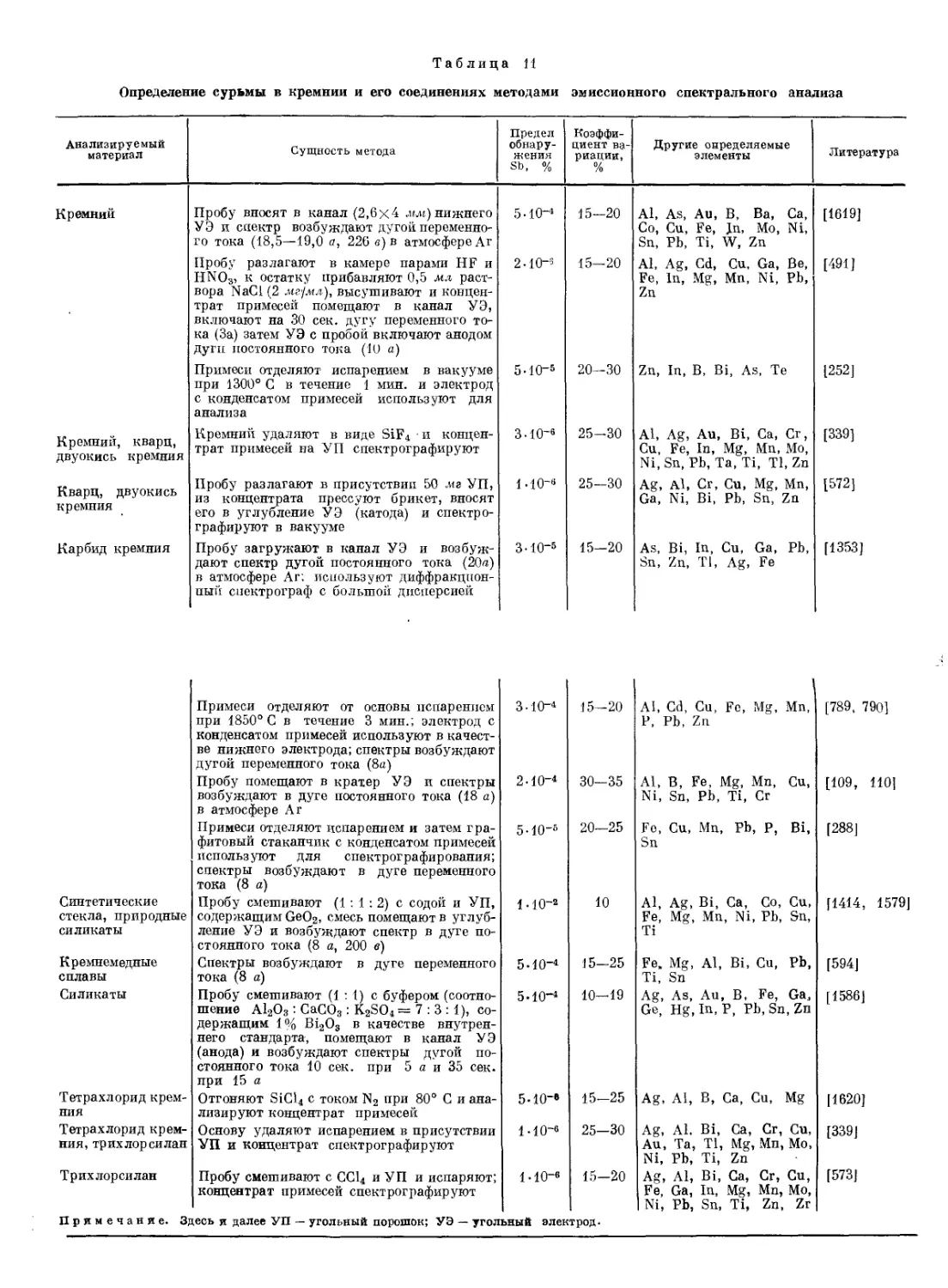
Спектральные методы. Для обнаружения Sb можно воспользоваться методом эмиссионного спектрального анализа, позволяющего без разложения исследуемого материала быстро обнаруживать Sb по ее линиям в спектре с высокой чувствительностью и одновременно получать информацию о наличии ряда других элементов.
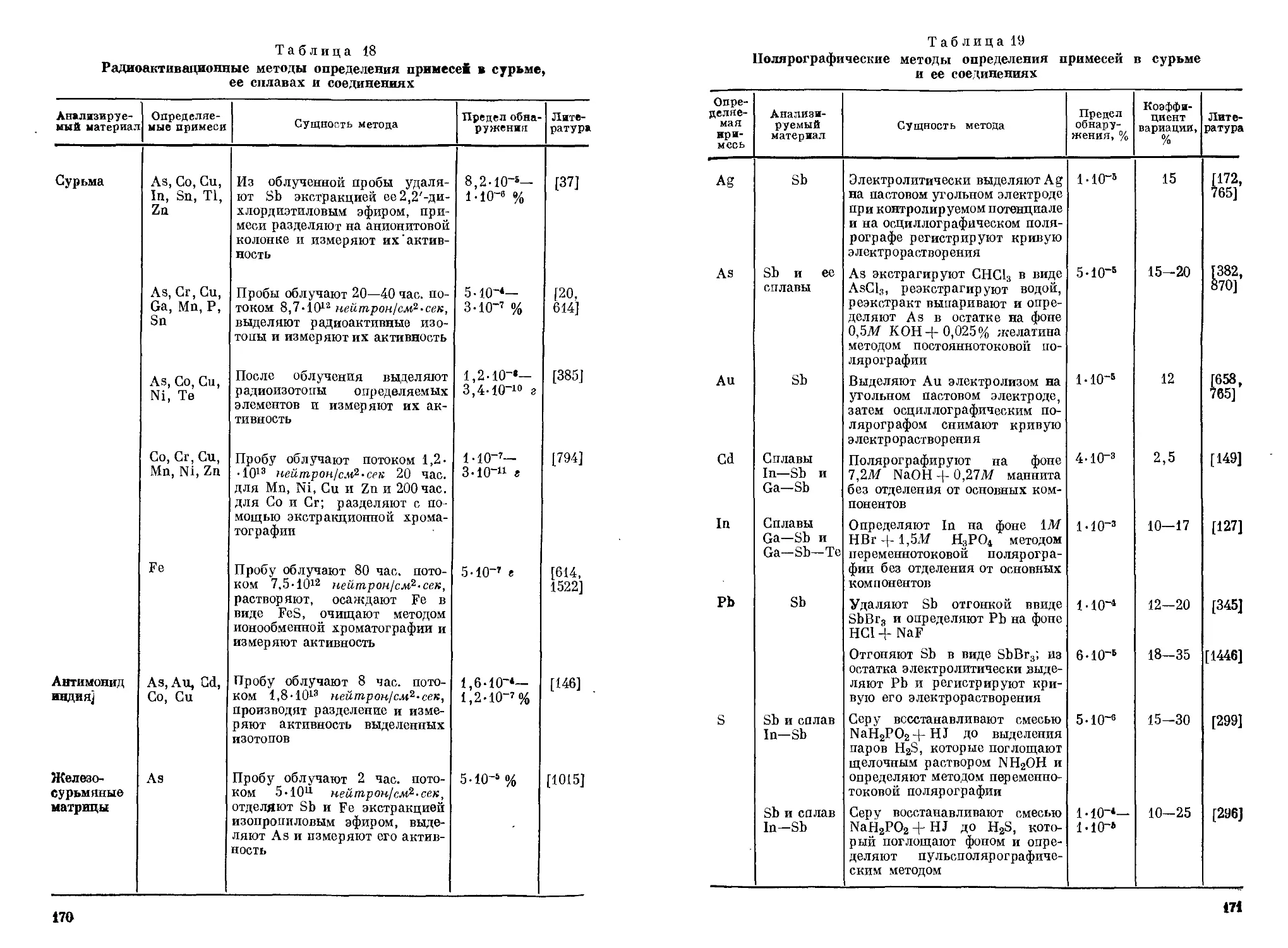
Эмиссионным спектральным методом Sb можно легко обнаруживать не только в сплавах, минералах, рудах и других твердых материалах, но также и в газах. Описан [943] метод качественного и количественного определения Sb одновременно с As, основанный на восстановлении их до гидридов, пропускании последних через электрический разряд и регистрации излучения Sb и As при 228,8 и 252,8 нм соответственно.
Анализ проводят в приборе (рис. 1), состоящем из реакционного сосуда 1, содержащего 25 мл 1%-ного раствора Na[BHJ, закрывающегося тефлоновой пробкой 2 с проходящей через нее трубкой 3 для ввода газа-носителя (Не) и отводной трубкой 4. Выделяющиеся газы током Не (200 мл/мин) выносятся через отводную трубку 4 в осушительную трубку 5, заполненную безводным CaSO4, и поступают в детектор 6, в котором под действием электрического разряда (800—900 в, 15 вт/ем) стибин распадается на Н2 и Sb; излучение атомов Sb выделяется из спектра монохроматором, и соответствующий ему фототок регистрируется показывающим или регистрирующим прибором. Перед анализом через прибор пропускают Не для удаления азота, линия эмиссии которого (244,80 нм) несколько накладывается на аналитическую линию Sb (252,8 нм).
Анализируемый раствор (объемом 'С 10 мл) с pH 7—11, содержащий от 5 иг до 2 мкг Sb, вводят шприцем непосредственно в раствор Na[BHJ через отросток 7, закрытый каучуковым или полиэтиленовым колпачком 8. Продолжительность анализа 3—5 мин. Обнаружению Sb мешают 100-кратные количества Ag, Си и Аз.
18
Сурьма может быть легко и быстро обнаружена также рентгенофлуоресцентным методом, методами атомно-абсорбционного и атомно-люминесцентного анализа.
Люминесценция кристаллофосфоров. Описан [691, 692] метод, обнаружения Sb по люминесценции кристаллофосфоров, образующихся при выпаривании анализируемого раствора с СаО или K4[Fe(CN)6]; в качестве источника УФ-излучения используют ртутно-кварцевую лампу ПРК-4. В первом случае в присутствии Sb кристаллофосфоры излучают желто-зеленое излучение, во
во-эмиссиоввого спектрального анализа
1 — реакционный сосуд; 2 — тефлоновая пробка; з — трубка для ввода газа-носителя; 4 — отводная трубка; 5 — осушительная трубка; 6— детектор; 7— отросток для ввода проб;
8 — каучуковый или полиэтиленовый колпачок
втором — желтое; предел обнаружения Sb составляет соответственно 1>10-6 и 1«10-3 мкг. Так как обнаружению Sb этим методом мешают многие элементы, то, как правило, Sb необходимо предварительно отделять.
Окрашивание пламени. При внесении соединений Sb в бесцветное пламя оно окрашивается в бледно-голубой цвет [581].
ХИМИЧЕСКИЕ МЕТОДЫ
Обнаружение сероводородом. Сурьма входит в V аналитическую группу катионов, для которых характерна способность осаждаться сероводородом из кислых растворов.
При осаждении Sb(V), наряду с образованием Sb2S6, образуется также Sb2S3 за счет частичного восстановления Sb(V) доЗЬ(Ш) сероводородом. Восстановительная способность H2S уменьшается с понижением температуры и увеличением кислотности. Более полному образованию Sb2S8 способствует также пропускание более сильного тока H2S.
Концентрированная НС1 растворяет сульфиды Sb при нагревании, что отличает их от сульфидов As. При нагревании оранжевого осадка сульфидов Sb(III) и Sb(V) с конц. HNO3 выделяется HSbO3 в виде белого аморфного осадка. Образование
сульфидов Sb при действии H2S в кислых растворах используется в систематическом ходе анализа для отделения катионов V аналитической группы [13, 16, 581, 707, 773].
Вместо H2S, характеризующегося высокой токсичностью, неоднократно предлагались другие серусодержащие реагенты [581], в том числе Na2S [707], тиоформамид [864], (NH4)2S2+jc [609], Na2S2O3 [558], CH3C(S)SNH4 [15591 ,P2S5 [1449]. Действие этих реагентов основано на их разложении с образованием H2S.
Стремление заменить токсичный H2S привело к разработке ряда бессероводородных схем качественного анализа. В одной из них [1444] предусматривается использование в качестве группового реагента на Sb(III), Bi(III), Sn(IV), Fe(III), Cr(III) и Al(III) ацетата натрия, осаждающего их в виде характерных осадков. Для идентификации отдельных элементов осадки рассматривают под микроскопом. В других схемах в качестве группового реагента используют комплексон III [868], Na2S2O3 [859], 8-оксихинолин и (C2H5)2NC(S)SNa [973].
Обнаружение гидролизом. При сильном разбавлении растворов Sb(III) водой выпадает белый осадок SbOCl, который при нагревании с конц. растворами НС1 или NH4C1 снова растворяется; SbOCl, в отличие от BiOCl, растворяется также в растворах винной кислоты с образованием SbOHC4H4O8 [895]. Растворы SbCl5 при разбавлении водой выделяют белый осадок SbO2Cl, который растворяется при нагревании с НС1.
Обнаружение действием щелочей. Гидроокиси щелочных металлов и аммиака из растворов солей Sb(III) и Sb(V) выделяют белые осадки соответственно HSbO2 и H3SbO4, растворяющиеся в избытке NaOH или КОН, но не растворяющиеся при добавлении карбонатов щелочных металлов и NH4OH. As(III) и As(V) при действии NaOH или КОН це образуют осадков, а осадок Sn(OH)4, в отличие от осадков гидроокисей Sb(III) и Sb(V), не растворяется в НС1.
Реакция с тиосульфатом. Тиосульфат натрия в слабокислых растворах при нагревании реагирует с Sb(III) и Sb(V) с образованием красно-оранжевого осадка Sb2OS2 [8, 13, 867, 869, 921, 1310]. Для уменьшения мешающего влияния других ионов металлов рекомендуется их маскировать ЭДТА.
В пробирку помещают 0,5 мл анализируемого раствора, содержащего 10 мкг Sb, добавляют 2 М NaOH до pH 2,0—2,5, 2 мл 5%-ного раствора ЭДТА (если после установления pH 2,0—2,5 выпадает осадок, то он растворяется при введении ЭДТА), несколько кристалликов Na2SaO3, и смесь нагревают до кипения. В присутствии Sb выпадает красно-оранжевый осадок.
Реакция с нитратом серебра. В присутствии аммиака Sb(III) восстанавливает ионы Ag+ до металла.
Для проведения реакции к 0,1—0,5%-ному раствору AgNO3 прибавляют равный объем конц. NH4OH, вводят предварительно подщелоченный анали-20
зируемый раствор и немного подогревают. В присутствии Sb(III) выделяется темный осадок серебра. Восстановители и окислители мешают [581].
Реакция с иод идами. В солянокислых растворах Sb(V) можно обнаружить по окислению ею иодид-ионов до иода, сообщающего раствору бурую окраску. Чувствительность реакции можно значительно повысить добавлением нескольких капель раствора крахмала (в этом случае реакционная смесь приобретает синюю окраску) или экстрагировать выделившийся иод небольшим количеством СНС13, окрашивающегося в фиолетовый цвет. Обнаружению Sb(V) мешают окислители [5811.
Восстановление металлами. Все металлы, стоящие в ряду напряжений левее Sb, в слабокислых растворах восстанавливают Sb(III) и Sb(V) до металлической сурьмы, выделяющейся в виде черного губчатого осадка. Вследствие более высокой избирательности реакции в качестве металла-восстановителя рекомендуется пользоваться оловом [13, 317, 734]. Предел обнаружения сурьмы 10 мкг. Наряду с оловом можно использовать Zn, Fe, Al и Mg. В щелочных растворах Zn и А1 восстанавливают Sb(III) и Sb(V) до металла, в то время как мышьяк восстанавливается до арсина и обнаружению Sb не мешает [1291].
Описана капельная реакция обнаружения Sb на медной пластинке с использованием алюминия [1334].
На медную пластинку^наносят анализируемый солянокислый раствор и прижимают к пластинке внутри раствора хорошо очищенную алюминиевую проволоку. В присутствии Sb образуется бархатно-черное пятно. Равные количества Sn не мешают; в присутствии больших количеств Sn выделившийся серебристый осадок металлического олова превращает черное пятно Sb в серое.
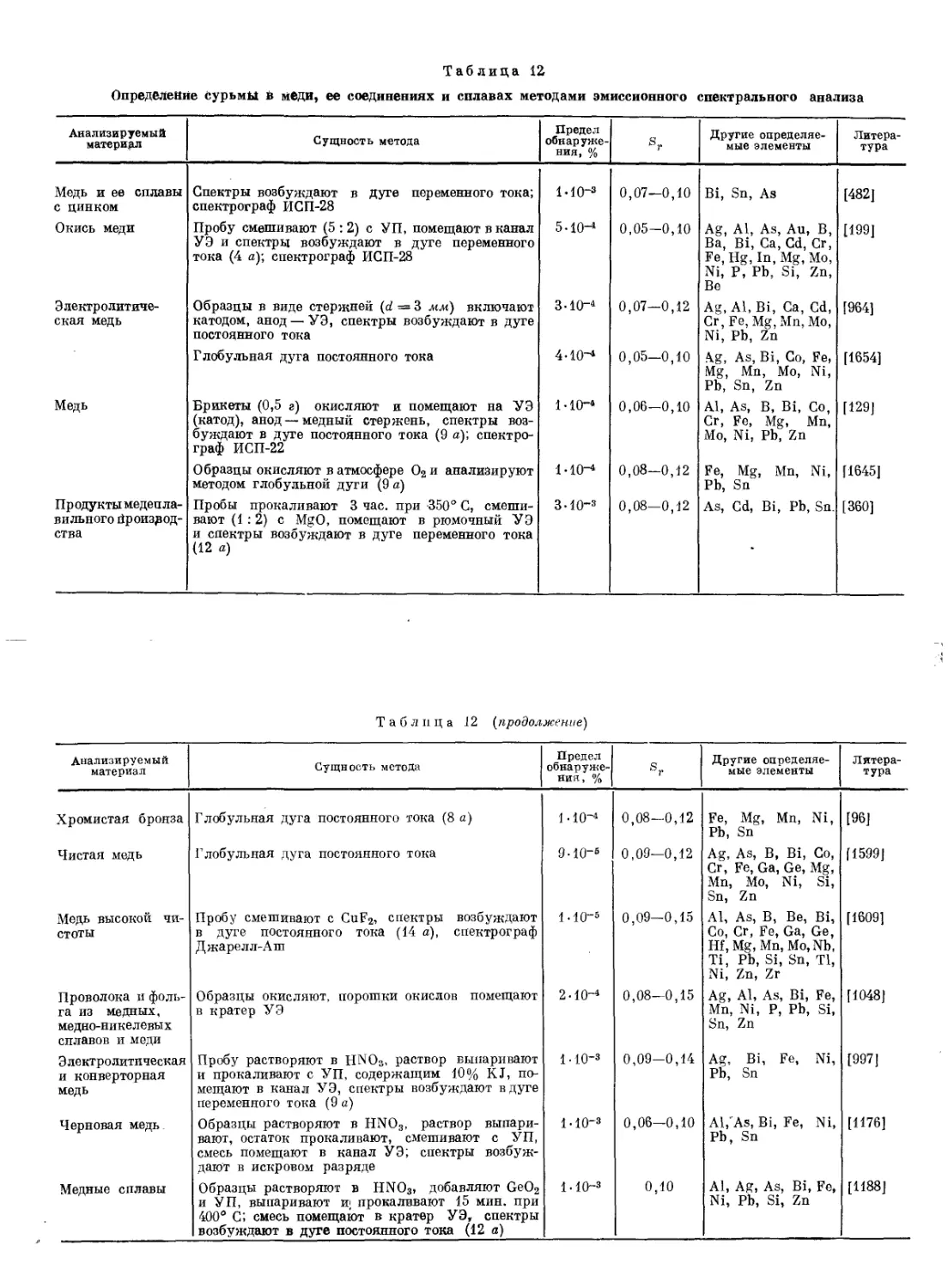
Реакция Марша [1284, 1285, 1320]. Эта реакция основана на восстановлении соединений Sb до SbH3, который при нагревании без доступа воздуха разлагается на Н2 и сурьму, отлагающуюся на холодной поверхности прибора в виде блестящего темного налета (зеркала).
Реакцию проводят в приборе Марша (рис. 2), состоящем из небольшой колбы 1 с пробкой с двумя отверстиями; через одно из них вставлена капельная воронка 2, доходящая до дна колбы, а через’другое — газоотводная трубка 3, соединенная с трубкой 4, заполненной безводным СаС12, и далее с трубкой 5 из тугоплавкого стекла. Трубка 5 имеет сужение 6 и узкий оттянутый конец 7.
В колбу вносят цинк, приливают 10—20%-ную HaSOi и ждут 15 мин. (до полного вытеснения воздуха из прибора), после чего водород зажигают у оттянутого конца трубки /in одновременно пламенем газовой горелки нагревают участок трубки 5 перед сужением 6. Если в течение 15—20 мин. в узкой части трубки не образуется металлическое зеркало (что указывает на то, что
1 Если водород зажечь, не дождавшись удаления воздуха, то произойдет взрыв образовавшегося гремучего газа.
21
Рис. 2. Прибор Марша
I — реакционная колба;
2 — капельная воронка;
з — газоотводная трубка;
4 — хлоркальциевая трубка;
5 — трубка иа тугоплавкого стекла;
6 — сужение;
7 — оттянутый конец трубки
используемые цинк и кислота не содержат Sb и As), то через воронку 2 осторожно, чтобы не впустить воздух в прибор, вводят исследуемый раствор. В присутствии Sb в узкой части трубки вблизи нагреваемого места, а иногда и впереди сужения образуется блестящий черный налет. Только As дает подобный налет. Для отличия Sb от As зеркало смачивают раствором NaClO или NaBrO, которые растворяют зеркало мышьяка, не изменяя зеркала сурьмы. Метод позволяет обнаруживать до 1 мкг Sb.
Экстракционно-иодидный метод [939, 1627]. Для быстрого обнаружения Sb предложен метод, основанный на образовании и экстракции HSbJ4.
К анализируемому раствору, 3,5—4,0 М по НС1, содержащему 10 мкг Sb, прибавляют равный объем насыщенного раствора K.J и 1—2 капли диэтилового эфира. В присутствии Sb эфирный слой окрашивается в желтый, оранжевый или красно-оранжевый цвет (в зависимости от содержания Sb). Если в исходном исследуемом растворе содержалась Sb(V) или вещества, окисляющие 1~ до 12, то перед экстракцией HSbJ4 прибавляют 1—2 капли 10%-ного раствора NazSOg для восстановления образовавшегося иода до иодид-ионов.
Образование ионных ассоциатов анионными галогенидными комплексами сурьмы(У) с катионами основных красителей. Эта группа реакций характеризуется очень высокой избирательностью и лежит в основе большинства фотометрических методов количественного определения сурьмы. Поскольку Sb(III) не образует подобных ассоциатов, то ее предварительно окисляют до Sb(V). Так как Sb(V) при хранении в растворах склонна к образованию полимерных ионов, не образующих анионных гексагалогенидных комплексов, то, как правило, ее предварительно восстанавливают до Sb(III) и затем снова окисляют до Sb(V).
Наиболее простым и удобным представляется метод с применением родамина С [317, 920, 1057].
Для обнаружения Sb на фарфоровую пластинку помещают каплю исследуемого раствора. Если присутствует Sb(V), то сначала в отдельной порции 22
раствора (0,5—1 мл) восстанавливают ее прибавлением одной капли 15%-ного раствора хлорида олова(П), и затем каплю восстановленного раствора наносят на фарфоровую пластинку, прибавляют 1—2 кристаллика NaNO2 для окисления Sb(IH) до Sb(V) и каплю конц. НС1, Через 5 мин. прибавляют каплю насыщенного раствора мочевины для устранения избытка HNO2 и затем вводят каплю 1 - IO'6 М раствора родамина С. В присутствии Sb краснорозовая окраска родамина С вследствие образования ионного ассоциата переходит в фиолетовую. Эта реакция позволяет обнаруживать до 0,5 мкг Sb [317]. Обнаружению Sb мешают Hg, Bi, Au, W и Mo, в присутствии которых наблюдается аналогичное окрашивание раствора.
Вместо родамина С можно пользоваться также метиловым фиолетовым [13, 370, 371, 7071.
Реакцию проводят в пробирке точно так же, прибавляют каплю 1 М NaNO2 и 3 капли конц. НС1. Через 1—2 мин. добавляют каплю насыщенного раствора мочевины, смесь разбавляют водой до 3 мл я вводят 3—4 капли 0,06%-ного раствора метилового фиолетового. В присутствии > 0,5 мкг Sb появляется тонкая суспензия кристалликов гексахлоростибата метилового фиолетового, имеющая в проходящем свете фиолетовую окраску. В отсутствие Sb раствор окрашен в зеленовато-желтый цвет. Присутствие до 1000-кратных количеств Sn, Fe, Си, Zn и многих других элементов обнаружению Sb не мешает.
Для обнаружения Sb в медных сплавах разработан бесстружко-вый метод с применением метилового фиолетового в качестве реагента [753, 938].
На поверхность исследуемого сплава наносят несколько капель HNO3. Через несколько минут HNO3 удаляют, протравленное пятно промывают водой, сушат фильтровальной бумагой и наносят две капли HNO3. После прекращения реакции раствор с осадком переносят в мерный цилиндр, лунку промывают 5—6 раз водой и 2—3 каплями НС1 (1 : 1). Раствор в цилиндре разбавляют до 1 мл, прибавляют 1,5' мл конц. НС1, вводят по каплям 10%-ный раствор хлорида олова(П) в НС1 (1 : 1) до исчезновения зеленой окраски и сверх этого еще 1—2 капли. Затем приливают 10%-ный раствор NaNO2 до возвращения зеленой окраски,"выдерживают 3—5 мин. и вводят раствор мочевины до прекращения выделения пузырьков N2, разбавляют водой до 7—8 мл, добавляют 2—3 капли 0,2%-ного раствора метилового фиолетового, хорошо перемешивают и экстрагируют гексахлоростибат метилового фиолетового 0,5мл толуола. В присутствии Sb толуольный слой окрашивается в синий цвет. Предел обнаружения Sb в медных сплавах 0,009%,
Для обнаружения Sb рекомендовано также применение малахитового зеленого [367]. В этом случае образуемый им ионный ассоциат экстрагируют толуолом. В присутствии >0,1 мкг Sb толуольный слой окрашивается в синий цвет.
Реакция с органическими реагентами. Сурьма(Ш) взаимодействует с многими серу- и кислородсодержащими органическими реагентами с образованием комплексов, окраска которых отличается от окраски самих реагентов. Наиболее чувствительные
23
цветные реакции Sb(III) дает с димеркаптотиопиронами [723]г 6-(2-хинолилазо)-3,4-диметилфенолом [623], 2-(хинолилазо)-п-крезолом [621], 2,3,7-триоксифлуоронами [506], пиридилазосоединениями [596, 1195], галлеином [107, 758], дифенилкарбазидом [215], лг-(меркаптоацетамидо)фенолом [1434] и тиолактамами [1009]. Однако эти реагенты недостаточно избирательны.
Заслуживает упоминания реакция с меркурофлуоресцеином [1663].
Исследуемый раствор (0,01 мл) с концентрацией Sb > 0,01 мкг!мл наносят на фильтровальную бумагу и после высушивания опрыскивают раствором, содержащим меркурофлуоресцеин и 8-оксихинолин (последний необходим для уменьшения мешающего влияния других элементов). В присутствии Sb(III) пятно в УФ-свете становится белым на черном фоне. Обнаружению Sb этим методом мешают As(III), Fe(III) и Ni(II).
Обнаружение сурьмы с применением импрегнированной фильтровальной бумаги. Достоинством реакций на фильтровальной бумаге, импрегнированной селективными реагентами, является малая продолжительность и высокая чувствительность. В качестве примера можно привести реакцию, проводимую на бумаге, пропитанной 4-оксибензотиазолом [1060]. При нанесении на эту бумагу капли исследуемого раствора, содержащего >0,1 мкг Sb, и облучении УФ-светом с длиной волны 365,0 нм появляется характерная желто-зеленая флуоресценция. Однако вследствие мешающего влияния многих элементов необходимо Sb предварительно отделять.
Для обнаружения Sb рекомендуется [1237] использовать бумагу, пропитанную Fe[Fe(CN)el. При нанесении капли раствора, содержащего > 0,5 мкг Sb(III), на полоске этой бумаги появляется голубое окрашивание, обусловленное восстановлением Fe(III) до Ге(П)и образованием берлинской лазури. Обнаружению-Sb мешают вещества, восстанавливающие Fe(III) до Fe(II).
Бумага, пропитанная фенилбензогидроксамовой кислотой, рекомендована для разделения и обнаружения многих ионов металлов, в том числе и Sb(III) [1527].
Для отделения Sb(III) от других ионов для последующего ее обнаружения применяют хроматографию анионных хлоридных комплексов металлов на бумаге, пропитанной три-н-октиламином в качестве жидкого ионообменника [1631].
Для быстрого обнаружения Sb в присутствии As и Sn рекомендуется метод с применением бумаги, импрегнированной раствором, содержащим 20 г Na2S2O3 и 2 г KJ в 100 мл.
После нанесения анализируемого раствора на полоску бумаги ее погружают в расплавленный парафин (80—90° С) на 1—2 мин. Затем бумагу извлекают и по охлаждении наблюдают окрашенные кольцевые зоны иодидов Sb,. As и Sn. Предел обнаружения — 2 мкг Sb в 0,005 мл раствора [399].
24
Методы бумажной хроматографии. Для обнаружения Sb(V) в присутствии As(V) и Sn(IV) исследуемый раствор хроматографируют на бумаге Ватман № 1, № 4 или Шлейхер и Шюлль с применением 150 мл смеси (1:1:1) «-бутанола, ацетилацетона и воды. Значения Rf составляют: 0 для Sb(V), 0,51 для Sb(III) и 0,65 для Sn(IV) [1089]. Для обнаружения Sb(V) на хроматограмме могут быть использованы KJ, дитизон, родизонат натрия, тиоацетамид, H2S, 8-оксихинолин, кверцетин, ализарин, рубеа-новодородная кислота и ряд других реагентов.
В одном из методов [356] Sb(III), As(III) и Sn(IV) разделяют на фильтровальной бумаге Ватман № 1 с использованием смеси (8 : 1,5 : 2,5) СН3СООН, ацетилацетона и воды. После высушивания хроматограммы опрыскивают раствором дитизона в СНС13 (0,5 мг/мл), Sb(III) образует оранжевое пятно, As — желтое и Sn — красное. Предел обнаружения 0,1 мкг Sb.
Описан [1581] метод разделения и обнаружения 23 ионов металлов, в том числе Sb, с использованием хлороформного раствора диэтилдитиокарбамината диэтиламмония.
Для разделения Sb(V), As(V), Sn(IV) и Hg(II) в качестве проявителя предложено использовать н-бутанол, насыщенный 3,4 М НС1 [1419]. Значение Rt для Sb(V) равно 0,34. Сурьму обнаруживают опрыскиванием хроматограммы 0,2%-ным раствором KJ.
Для разделения и обнаружения Sb(III), As(III) и Sn(II) предложен метод радиальной хроматографии на бумажных фильтрах с применением н-бутанола, насыщенного 3,5М НС1 [751]. Смесь (7 : 3) циклогексанола с НСООН оказалась эффективной для отделения Sb от других ионов металлов с целью последующего обнаружения Sb [1454].
Для обнаружения Sb(III) и Sb(V) в их смесях предложено [467] использовать распределительную хроматографию с применением н-бутанола в качестве проявителя. Значения Rj для Sb(III) и Sb(V) равны соответственно 0,64 и 0,97. Хроматограмму помещают в камеру, содержащую H2S. Образование соответствующих пятен сульфидов указывает на присутствие Sb(III) и Sb(V). Предел обнаружения 0,02 мкг{мл Sb(III) и 0,24 мкг/мл Sb(V).
Описан [998] метод обнаружения Sb в органических соединениях, включающий минерализацию исследуемого вещества в запаянной трубке, экстракцию Sb органическими растворителями и хроматографирование экстракта, содержащего Sb, на бумажном фильтре с применением смеси (4 : 3 : 3 : 2 : 3 : 0,6) конц. НС1, СН3СООН, н-бутанола, н-пропанола, этанола и ацетилацетона в качестве проявителя.
Тонкослойная хроматография. Для обнаружения Sb предложено использовать хроматографию на слоях крахмала с применением растворов (NH4)2S2+JC [966], на слоях целлюлозы — с использованием смесей спиртов или кетонов с кислотами [1281, 1541] и на слоях силикагеля — с использованием смеси (20 : 1) н-бутанола с IM НС1 [1411], а также хроматографирование ана
25
лизируемой смеси ионов в виде пирролидиндитиокарбаминатов смесью (2 : 1) бензола с СНС13 с последующим обнаружением сурьмы раствором CuSO4 [1516].
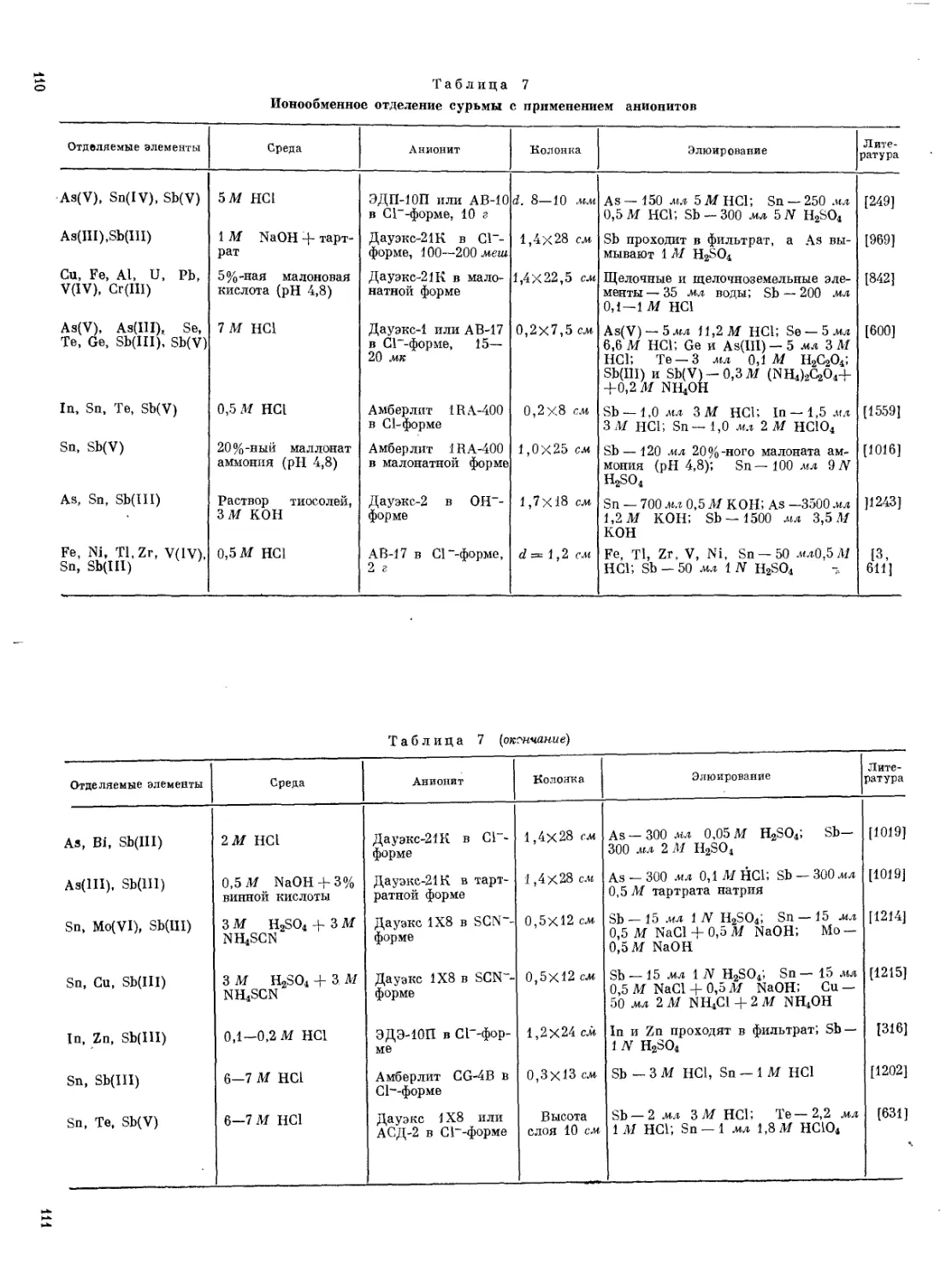
Адсорбционная хроматография. Для обнаружения Sb(III) в присутствии As и Sn предложен метод адсорбционной хроматографии.
Анализируемый раствор (5 капель) вносят в колонку (d = 0,6 ч- 0,7 см, h — 4 см}, заполненную А12О3; колонку промывают НС1 (1:4) и пропускают H2S. В течение 1 мин. образуется верхняя светло-желтая зона As2S3, затем оранжевая зонаЯЬгЯзи нижняя буро-коричневая 3OHaSnS2. Предел обнаружения Sb 7 .10~6 г-ион!л [556].
Методы кольцевой бани. Для разделения и обнаружения Sb(III) As(III) и Sn(II) используют метод кольцевой бани, в котором указанные элементы разделяют на бумаге Ватман № 1 в виде тартратных комплексов с применением 60 %-ного этанола в качестве проявителя [1524]. Описана [973, 1100] схема качественного анализа 20 катионов в одной капле раствора с использованием метода кольцевой бани. С применением соответствующих растворителей на бумажном фильтре получают 8 колец. В шестом кольце, содержащем Sb, Ash Bi, разделяют эти элементы на зоны в виде диэтилдитиокарбаминатов с применением ацетилацетона в качестве проявителя. В другой схеме качественного анализа 26 катионов [917] их разделяют с помощью групповых реагентов (HaS, НС1) на три группы, затем методом кольцевой бани каждую группу катионов разделяют на отдельные элементы и идентифицируют. Метод обеспечивает четкое разделение и идентификацию всех катионов при! их содержании в капле > 2 мкг.
Для обнаружения Sb в биологических и других материалах рекомендуют метод кольцевой бани, позволяющий обнаруживать до 1-10~? г-ион/л Sb [1644]. Для этого из анализируемого раствора объемом 5 лл выделяют Sb вместе с Си, Cd, Hg, Pb и Bi осаждением сероводородом. Из осадка сульфидов сурьму отделяют обработкой раствором (NH4)2S2+K и выделяют на бумажном фильтре методом кольцевой бани.
Метод кольцевой бани используют для обнаружения Sb в смеси с ионами других металлов на бумажном фильтре [1643]. Метод включает осаждение и переосаждение сульфидов с применением газообразных реагентов (H2S, NH3, Вг2) и (NH4)2S2+a;.
Метод кольцевой бани с использованием (NH4)2CS3b качестве группового реагента, быстро осаждающего Sb и ионы многих элементов при минимальном избытке самого реагента, рекомендован [1197] для обнаружения Sb в сложных смесях. Из образовавшегося пятна сульфидов 2%-ный раствор (NH4)2CS3 количественно вымывает Sb(III) и Sb(V), а также Se и Си в кольцевую зону, где они обнаруживаются после обработки раствором IM НС1.
Метод кольцевой бани успешно применяют для обнаружения Sb в воздухе [1642].
26
Анализируемый воздух пропускают через 4 М раствор H2SO4, содержащий 10% K.J, и экстрагируют SbJ3 бензолом. Экстракт наносят на кружок фильтровальной бумаги, бензол испаряют и вымывают SbJ3 смесью (2 : 1) бензола с этанолом. Для обнаружения Sb ее зону погружают в кипящий 5%-ный раствор 12-молибдофосфорной кислоты. Метод позволяет обнаруживать до 0,008 мкг Sb в пробе.
С целью повышения избирательности обнаружения Sb методом кольцевой бани рекомендуется использовать ЭДТА в качестве маскирующего реагента [1525].
Капельные реакции. По одной из них [1054—1056] для обнаружения Sb в органических и неорганических соединениях поступают следующим образом.
В микропробирку помещают немного исследуемого вещества, содержащего 10 мкг Sb, добавляют 10-кратное количество NH4Br, перемешивают и нагревают на небольшом пламени до образования сублимата в верхней части пробирки. Сублимат снимают ватным тампоном, смоченным раствором 15 М NaOH, содержащим5% Hg(CN)3. В присутствии Sb образуетсяSbBr3, который реагирует в щелочной среде с Hg(CN)2 с выделением черного осадка ртути, вследствие чего на ватном тампоне на месте сублимата появляется черное пятно. Обнаружению Sb мешает As, дающий аналогичную реакцию.
Микрокристаллоскопические реакции. По одному из методов [740] Sb(III) обнаруживают по образованию непрозрачных кристаллов рейнекеата Sb(III) в форме гантелей, сросшихся в друзы. В других случаях поступают следующим оброзом.
Для обнаружения Sb каплю анализируемого раствора, содержащего ^0,1 мкг Sb, ’помещают на предметное стекло микроскопа и с одной стороны капли вводят в нее кристаллик K.J, а с другой стороны — кристаллик RbCl или CsCl. В присутствии Sb образуются оранжевые шестиугольные кристаллы Bb2[SbJ8] mtnJCsJSbJJ соответственно [13, 114, 1418, 1420].
Полярографические методы. Для обнаруживания Sb рекомендованы [1037, 16231 полярографические методы. По одному из них [1037] Sb обнаруживают методом осциллографической полярографии на фоне IM НС1 по катодному току восстановления Sb(III) при —0,1 в1. Метод позволяет обнаруживать до 5* 10-5 г-ион/л Sb; As и Sn не мешают.
В другом методе [1623] Sb обнаруживают на фоне смеси 1 М НС1 и 1М винной кислоты по волне восстановления Sb(III) при —0,19 в. Для устранения мешающего влияния других элементов Sb предварительно отделяют в виде SbH3. Предел обнаружения зависит от чувствительности полярографа и составляет 2—10 мкг Sb.
1 Здесь и далее потенциалы указаны относительно нас. к. э.
27
Глава IV
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СУРЬМЫ
ХИМИЧЕСКИЕ МЕТОДЫ
Из химических методов определения Sb наибольшее значение1 имеют титриметрические методы. Гравиметрические методы по точности сопоставимы с титриметрическими, но характеризуются значительно большей продолжительностью и трудоемкостью. Вследствие этого в настоящее время они имеют ограниченное применение и вытесняются титриметрическими и различными физико-химическими и физическими методами.
Г равиметрические методы
Среди гравиметрических методов относительно большое значение имеют методы определения Sb в виде Sb2S3, Sb2O4 [1501 и металлической сурьмы [849, 1025, 1052, 1307].
Определение в виде Sb2S3. Один из первых методов определения Sb в виде Sb2S3 предложен Кларком [985]. Хенц [1145], затем Фортманн и Метцль [1626] усовершенствовали этот метод. Выделившийся осадок может содержать сульфиды Sb(III) и Sb(V), а также серу. При прокаливании в инертной атмосфере при 280— 300° С Sb2S5 полностью разлагается на Sb2S3 и серу, которая при этом улетучивается.
Для определения Sb в виде сульфида лучшим является метод, описанный в работе [150].
Анализируемый раствор, 2,5 М по НС1, нагревают до кипения и немедленно осаждают Sb быстрым током H2S при 90—100° С. После того как появится красный осадок Sb2S3, содержимое реакционной колбы периодически перемешивают, избегая размазывания осадка по стенкам колбы. Когда осадок потемнеет, ток H2S несколько ослабляют. Продолжительность осаждения обычно не превышает 30—35 мин. и заканчивается, когда весь осадок почернеет. Раствор с осадком разбавляют равным объемом воды, перемешивают, снова нагревают и продолжают пропускать H2S. После коагуляции осадка и охлаждения его отфильтровывают на предварительно прокаленный при 280— 300° С и взвешенный фильтрующий тигель, промывают несколько раз водой для удаления маточного раствора, а затем этанолом, высушивают при 100— 28
130° С в течение 2 час. в атмосфере СО2, после чего прокаливают еще 2 часа при 280—300° С и после охлаждения взвешивают. Относительное стандартное-отклонение результатов определения сурьмы Sr — 0,002 -е 0,003. Определению мешают все элементы, осаждающиеся H2S в условиях осаждения сурьмы.
Фортманн и Метцль [1626] при определении Sb этим методом в присутствии олова проводили осаждение, добавляя Н3РО4, маскирующую Sn. Саруди [1496] несколько улучшил этот метод.
В соответствии с разработанной им методикой выделяют смесь сульфидов Sb и Sn, растворяют их в НС1 (1 : 1), в полученный раствор прибавляют H2SO4 и выпаривают почти досуха. К остатку прибавляют конц. НС1, винную кислоту и Н3РО4, смесь нагревают до кипения, колбу с раствором погружают в кипящую воду и осаждают Sb пропусканием быстрого тока H2S. Выделившийся осадок отфильтровывают, промывают, прокаливают при 280—300° С в инертной атмосфере и взвешивают в виде Sb2S3. Метод позволяет определять Sb в присутствии значительно превосходящих количеств олова.
В работе [1569] также используется взвешивание в виде Sb2S3, но предварительно Sb переводят в растворимые тиосоли и последующим добавлением НС1 выделяют ее в виде нерастворимых сульфидов, которые отфильтровывают, высушивают, прокаливают в инертной атмосфере до Sb2S3.
Ряд методов предложен для гравиметрического определения Sb в виде Sb2S3 без применения H2S. Рай [1464] для этой цели рекомендует кипячение раствора, насыщенного NH4SCN. Хотя некоторые авторы [1350] рекомендуют этот метод как лучший вариант гравиметрического определения Sb в виде Sb2S3, однако для него характерна тенденция давать несколько заниженные результаты [1102]. Тиоацетамид [1064] и тиоформамид [1078] в тех же условиях также могут быть использованы для определения Sb в виде Sb2S3 (Sr = 0,002). Тритиокарбонаты щелочных металлов, предложенные для этой цели [1079], неудобны в связи с их малой устойчивостью при хранении.
Определение в виде Sb2S5. Описан [1513] метод определения Sb, в котором предварительно окисляют Sb(III) до Sb(V) и осаждают ее в условиях, исключающих образование Sb2S3 (высокая концентрация НС1, быстрый ток H2S). Осадок Sb2S5 высушивают при 105° С. Вследствие дополнительной операции окисления Sb(III) до Sb(V) и устранения избытка окислителя этот метод не нашел практического применения.
Определение в виде Sb2O4. Вместо взвешивания Sb в виде Sb2S3, требующего прокаливания выделенного осадка в инертной атмосфере, можно растворить влажный осадок в HNO3 и после выпаривания раствора досуха остаток прокалить и взвесить в виде Sb2O4 [894]. При прокаливании осадка следует тщательно защищать его от восстановительных газов. Вместо растворения влажного осадка Sb2S3, возможно разложение его обработкой раствором NH4OH и Н2О2, как это рекомендовано для GeS2 [1196].
29
В этом случае влажный осадок растворяют в 10 М NH4OH, приливают 25 мл 3%-ной Н2О2 и оставляют на холоду, после чего выпаривают на водяной бане досуха. Остаток смачивают несколькими каплями H2SO4 (1:1), выпаривают досуха, прокаливают при 800—850° С и взвешивают в виде Sb2O4.
Погрешность определения Sb такая же, как и в методе, основанном на взвешивании в виде Sb2S3, но он проще в выполнении, поскольку не требует инертной атмосферы при прокаливании осадка.
Интересным вариантом метода, основанного на использовании Sb2O4 в качестве весовой формы, является метод, предложенный Намоисом [1369] и впоследствии улучшенный другими авторами [1641]. Метод заключается в выделении Sb в элементном виде оловом (взятом в виде стержня), отделении выделившейся Sb, которую смачивают раствором HNO3 или смешивают с NH4NO3 и прокаливают на воздухе для переведения ее в Sb2O4. Однако в связи с трудностью количественного отделения выделенной элементной Sb от стержня метод дает плохо воспроизводимые результаты.
Другие методы, основанные на использовании неорганических реагентов. Ряд авторов [825, 1118, 1479,1603] в качестве весовой формы для определения Sb предлагают NaSbO3. Сурьму осаждают из щелочного раствора в виде NaH2SbO4. При прокаливании осадка при 450—700° С количественно образуется NaSbOg. Однако на стадии промывания (для удаления избытка NaOH) осадок частично растворяется и получаются заниженные результаты [1641].
Определение с применением органических реагентов. Методы определения Sb с применением органических реагентов имеют некоторое преимущество (по сравнению с определением в виде Sb2S3 или Sb2O4), заключающееся в отсутствии необходимости использовать H2S, и, вследствие большей молекулярной массы выделяемых соединений, позволяют определять меньшие количества Sb
Файгль [1053] рекомендует осаждать Sb 5-кратяым количеством пирогаллола.
Осадок отстаивают 2'часа, отфильтровывают, промывают]водой, высуши» вают при 110° С и взвешивают. Метод позволяет определять до 0,05 мг Sb в присутствии Аз и Sn [1350].
Уилсон [1649] в качестве реагента для гравиметрического определения Sb предложил использовать пропилгаллат.
К раствору,J 0,5 М по НС1 и содержащему 15—50 мг Sb, прибавляют 25 мл 10%-ного раствора реагента, и смесь оставляют на ночь. Выделившийся осадок отфильтровывают, промывают несколько раз раствором 0,5 М НС1 и 1—2 раза этанолом, высушивают при 110° С и взвешивают в виде Ci0HuO6(SbO). Мышьяк определению Sb не мешает.
30
Галловая кислота осаждает Sb(III) в виде комплекса, пригодного для ее гравиметрического определения [1112]. Осадок отфильтровывают и высушивают при 114—163° С [1350]. Для гравиметрического определения Sb рекомендованы также 9-м е т и л-2,3,7- триоксифлуорон [1639], т а н н и н [1364, 1576], галлеин [1363], мирецитин [1364], пирамидон [1076] и ряд других реагентов, но вследствие малой избирательности они не нашли применения.
Пиртя [1428] предложил определять Sb в виде 8-оксихи-нолината. Однако было показано [1103], что этот метод характеризуется малой точностью. Авторупересмотрел свой первоначальный вариант метода и значительно улучшил его [1429].
Для определения Sb к 15—20 мл анализируемого солянокислого раствора прибавляют 0,5—1,5 г винной кислоты, 15—20 мл 4%-ного уксуснокислого раствора 8-оксихинолина, нагревают до 60—65° С и постепенно прибавляют 10%-ный раствор аммиака; при pH 1,5 начинает выпадать желтый осадок. Раствор аммиака добавляют до pH 6,0—7,5 и смесь выдерживают 30—40 мин. Выделившийся осадок отфильтровывают, промывают 0,04%-ным раствором 8-оксихинолина до отсутствия иона хлора, высушивают при 105—110° С и взвешивают в виде Sb(C9HeON)3.
Кардвелл [967] предложил другой вариант определения Sb 8-оксихинолином, в котором ее выделяют в виде SbO(C9HeON) (C9H7ON)2.
Купферон [1425, 1426] позволяет определять Sb(III} в присутствии Sb(V) и As.
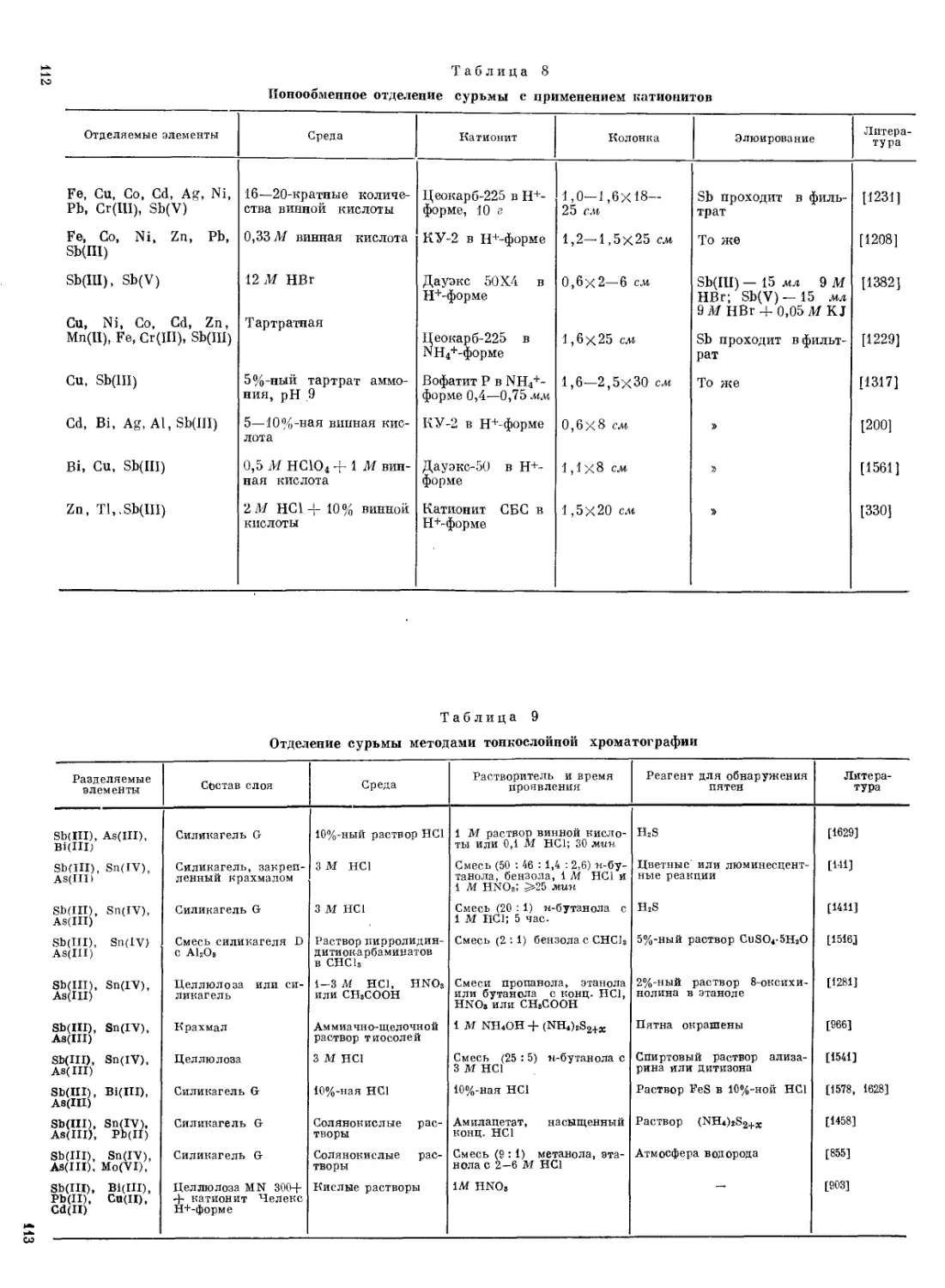
Описан [925] гравиметрический метод определения Sb(III), основанный на применении диэтилдитиофосфорной кислоты в качестве осадителя. Кроме диэтилдитиофосфорной кислоты из серусодержащих. органических реагентов для гравиметрического определения Sb(III) предложены: ф е и и л т и о-гидантоновая кислота [1275] и P-а минонафта-лид тиогликолевой кислоты [901, 902]. Вследствие малой растворимости образующихся осадков эти реагенты позволяют определять очень малые количества Sb (до 0,05 мг). Однако, многие элементы мешают.
Ряд гравиметрических методов основан на образовании нерастворимых в воде ионных ассоциатов, образуемых анионными комплексами Sb(V) с катионами органических оснований. Описан [98] метод, основанный на осаждении Sb(V) в виде гексахлороантимоната диантипирил метания. Этот метод позволяет определять Sb в присутствии As, Zn, Си и небольших количеств Bi. В другом методе [866] Sb(V) выделяют в виде гексафтороантимоната тетрафенил-арсония и гексафтороантимоната нитрония.
Для гравиметрического определения Sb(V) предложен метод,, основанный на выделении ее в виде нерастворимого в воде ионного>
31<
ассоциата, образуемого анионом SbCle с катионом тпраис-д и х л о-р о-б нс-этилендиаминкобальта (III) [898].
Анализируемый раствор (~ 50 мл), содержащий 1—50 мг Sb, насыщают хлором и прибавляют 10 мл 2%-ного раствора реагента в 2 М НС1. Через 2 часа образовавшийся осадок отфильтровывают, промывают водой и высушивают при 100—110° С.
Благодаря небольшому фактору пересчета массы осадка на Sb (0,2098), метод характеризуется высокой чувствительностью. Определению Sb не мешают большие количества As, Си, Zn, Cd, Fe, Hg, Sn, Bi и Pb.
Весьма удобным оказался метод, основанный на способности Sb(III) образовывать анионные иодидные комплексы, дающие нерастворимые в воде ионные ассоциаты с крупными органическими катионами. Преимущество таких методов заключается в том, что они пригодны для определения Sb независимо от степени ее окисления, так как присутствующие в растворе ионы J- восстанавливают Sb(V) до Sb(III). В качестве органического основания рекомендован 1,2- диморфолилэтан, который с Sb(III) в присутствии J- в зависимости от условий образует два нерастворимых в воде соединения: [C10H20O2N2H2] [SbJ5] и [C10H20O2N2H2] [SbJJ2 [873]. Последнее соединение использовано для гравиметрического определения Sb.
Методы, основанные на выделении сурьмы в элементном виде. Описан [1582] метод, основанный на осаждении Sb в элементном виде солями Ст(П). Осадок Sb отфильтровывают, промывают водой, этанолом, высушивают при 110° С и взвешивают. Определению Sb этим методом мешают Pt, Hg, Bi, Au, Se и Те, восстанавливающиеся солями Сг(П) до элементного состояния. Вместо Сг(П) можно использовать V(II).
Более эффективным является электролиз при контролируемом потенциале [279, 849—852]. Этот метод удобен тем, что не требует отделения полученной в результате электролиза Sb фильтрованием, так как ее взвешивают вместе с катодом, на котором она выделяется. Кроме того, с применением электролиза с контролируемым потенциалом в ряде случаев возможно последовательное определение нескольких элементов из одного раствора. Так, например, в работе [279] показана возможность последовательного определения в одном растворе Си и Sb, а также Sb и Sn, а в работах [279, 850, 851] — Си, Pb, Sn и Sb. Метод применен для определения указанных элементов в бронзах и латунях. Погрешность определения каждого элемента ~ 0,1%.
Гравиметрическими методами Sb определяют в бронзах и латунях [849—851], сплавах на основе свинца [852].
32
Титриметрические методы
Титриметрические методы определения Sb по сравнению с гравиметрическими находят значительно большее применение. Это объясняется тем, что они намного быстрее и проще позволяют определять меньшие количества Sb и характеризуются такой же или даже лучшей точностью [150, 335, 597, 774, 962]. Большинство титриметрических методов основано на титровании Sb(III) до Sb(V) растворами окислителей. Значительно меньше используются методы, основанные на титровании Sb(V) растворами восстановителей, а также методы комплексонометрического, осадительного и кислотно-основного титрования.
Оксидиметрическое титрование
Броматометрический метод. Из оксидиметрических методов наиболее распространенным является броматометрический метод, в котором Sb(III) окисляется до Sb(V) броматом калия в присутствии КВт, катализирующего окисление Sb(III) до Sb(V). При титровании в кислой среде выделяется свободный бром, который и является активным окислителем для Sb(III).
Броматометрическое определение Sb впервые осуществил Гюори [1133]. Затем этот метод неоднократно совершенствовался [7, 963, 1273, 1403, 1404]. В зависимости от содержания Sb в титруемом растворе в качестве титранта используют 0,01 — 0,5 N КВтО3. Растворы КВтО3 стандартизуют обычно по H3AsO3.
Титрование Sb(III) проводят, как правило, в соляно- или сернокислых растворах. По данным работы [7], более точные результаты получают при титровании в сернокислой среде. Следует иметь в виду, что HSO3NO, как образующаяся при разложении навески смесью HNO3 и H2SO4, так и содержащаяся в некоторых сортах H2SO4 в качестве примеси, сильно мешает [919]. Для ее разложения раствор необходимо разбавить водой и снова упарить до появления белого дыма H2SO4. Оптимальная концентрация НС1 или H2SO4 находится в пределах 2—5 N.
Для установления конечной точки титрования предложен ряд индикаторов, среди которых наиболее часто используют метиловый оранжевый. В конечной точке окраска раствора исчезает вследствие появления избытка брома, два атома которого присоединяются к индикатору по азогруппе с образованием бесцветного К,№-дибромпроизводного. Кроме метилового оранжевого, довольно часто используется метиловый красный [650, 759]. Рекомендован [928] также тропеолин 0, который в конечной точке в результате бромирования (атомы Вт замещают атомы Н в положениях 3 и 4) меняет коричнево-оранжевую окраску на зелено-желтую; предложены и дают хорошие результаты п-этоксихризои-дин, нафтоловый сине-черный, бриллиантовый понсо 5R, фуксин, а-нафтофлавон [1508]. В качестве индикаторов для броматомет-
2 А. А. Немодрук
33
рического титрования могут использоваться некоторые оксазовы [1491] и тиазины*[1490], которые необратимо окисляются избытком брома, изменяя при этом свою окраску. Наиболее чувствительными оказались тионин, метиленовый голубой, азур А и тио-нолин.
Богнар считает более удобными обратимые индикаторы, в качестве которых рекомендует пиридилазорезорцин [931], который в конечной точке оранжевую окраску меняет на зеленовато-желтую, и нитробензол азо резорцин [930], оранжевая окраска которого в точке эквивалентности переходит в лимонно-желтую.
Интересными индикаторами оказались фосфорномолибдено-ванадиевая [424] и кремнемолибденованадиевая [1564] кислоты. В присутствии Sb(III) они обратимо восстанавливаются до соответствующих синих форм, которые в конечной точке избытком КВгО3 снова окисляются до исходной окисленной формы с переходом синей окраски в желто-оранжевую. В качестве обратимого индикатора для броматометрического титрования Sb(III) предложено [1568] использовать известную в иодометрии иодокрах-мальную реакцию. Потенциометрическое установление конечной точки [450, 759, 1439] также дает хорошие результаты (в оптимальных условиях ошибка находится в пределах 0,15—0,20 отн. %). В связи с тем что при комнатной температуре Sb(III) окисляется недостаточно быстро, титрование броматом калия следует проводить при 60—70° С.
Для определения Sb броматометрическим титрованием с применением метилового оранжевого в качестве индикатора может быть рекомендована следующая методика.
К анализируемому раствору приливают 40 мл конц. НС1, 10 мл 10%-ного раствора сульфита натрия для восстановления Sb(V) до Sb(III) и кипятят несколько минут для полного удаления SO2 и As (в виде AsCl3). Затем раствор разбавляют водой до ~ 150 мл, прибавляют 1 г КВг, 2 капли 0,1%-ного раствора метилового оранжевого, нагревают до 70° С и титруют раствором 0,04— 0,1 А КВгО3 до исчезновения оранжевой окраски индикатора. В тех же условиях проводят холостой опыт.
Определению мешают все ионы, окисляемые броматом в кислой среде. Поэтому в их присутствии Sb необходимо предварительно отделять. Однако в ряде случаев отделения можно избежать. Так, например, при анализе тетраэдрита анализируемый раствор обрабатывают хлоридом титана(Ш), затем избыток Ti(III) и ряд других ионов, в том числе As(III), окисляют ванадатом аммония и титруют Sb(III) броматом калия [1413].
При определении Sb в растворах, содержащих As, его удобно удалять в виде AsCl3 нагреванием раствора при достаточной концентрации НС1 [663, 821, 1088, 1507]. В ряде случаев возможно определение Sb и некоторых других элементов без их отделения. Так, например, сплавы, содержащие Sb, Sn, Pb и Ст, а также-сплавы, содержащие Sb, Sn, Pb и Си, анализируют методом, 34
включающим броматометрическое титрование Sb(III), йодометрическое — Sn(II) и комплексонометрическое — остальных элементов [1404].
Броматометрическим титрованием определяют Sb в свинцовых [1088, 1553], свинцово-оловянных [1245], свинцово-оловянно-сурьмяных [262, 1030], медно-кадмиевых [846], алюминиево-сурьмяно-галлиевых [202, 7601, цинково-кадмиево-сурьмяных [1274], цинково-сурьмяно-теллуровых [6501, сурьмяно-оловянно-свинцово-хромовых [14041, полупроводниковых [452] и типографских сплавах [821], оловянных бронзах [12441, катализаторах [376], ртутно-сурьмянистых рудах [597], олове [1244], платиновых металлах [400], антимоните и арсените скандия [337], цилиндрите [538], тетраэдрите [14131, гальванических золото-сурьмяных ваннах [899], цинке [1244], гипергенных металлах [653], свинцовых рудах и продуктах их переработки [4841, органических соединениях [1665].
Перманганатометрия. По своему значению перманганатометрическое титрование Sb(III) идет вслед за броматометрическим. Его преимущество по сравнению с броматометрическим титрованием состоит в том, что реакция между Sb(HI) и МиО7 идет значительно быстрее. Однако титрование перманганатом требует соблюдения более строгих условий, в частности концентрация HCI в титруемом растворе должна быть 1,2—3 М. Одновременно в растворе желательно присутствие 10% H2SO4. Титрование необходимо проводить при 5—10° С [150]. В этих условиях расход КМпО4 настолько соответствует стехиометрическому, что можно пользоваться теоретическим титром раствора КМпО4. Титрованию Sb(III) мешают все элементы, взаимодействующие с МпО4, в том числе V (IV), Fe(II), As(III), Sn(II), Se(IV), Te(IV). Таким образом, в отношении избирательности перманганатометрическое титрование несколько уступает броматометрическому. Fe(III), присутствуя в значительных количествах, частично восстанавливается до Fe(II) при обработке сульфитом натрия в сернокислых растворах с целью переведения Sb(V) в Sb(III) и завышает результаты.
Индикатором при титровании Sb(III) перманганатом в большинстве случаев служит сам титрант, избыток которого окрашивает раствор в розовый цвет. Иногда в качестве индикатора используют метиленовый голубой [719] и метиловый оранжевый [6481, в присутствии которых раствор титруют до исчезновения окраски индикатора. Рекомендуется также потенциометрическое установление конечной точки [1173, 1346, 1646].
Для определения Sb может быть рекомендована следующая методика [150, стр. 296].
Анализируемый растворе концентрацией НС1 1,2—1,5 М и H2SO4 1,7— 2,0 М, содержащий 10—250 мг Sb(III) ине содержащий As(III), V(IV), Fe(II), SO|- и других окисляющихся перманганатом веществ, охлаждают до 5—10° С
2*
35
и титруют раствором 0,03—0,1 N КМпО4 до появления слабо-розовой окраски, не исчезающей в течение 5 сек.
Если предварительно к анализируемому раствору добавить 1—2 мл 1 М MnS04, то раствор можно титровать и при комнатной температуре. Если сурьма не полностью находится в виде Sb(III), то анализируемый раствор сначала обрабатывают сульфитом натрия, как и при броматометрическом титровании.
Перманганатометрическое титрование Sb(III) по точности дает примерно такие же результаты, как и броматометрическое, но вследствие того, что перманганатометрическому титрованию Sb(III) мешает большое число элементов, оно реже используется.
Перманганатометрическим титрованием определяют Sb в алю-миниево-магниевых сплавах [719], арсениде-фосфиде и арсениде-антимониде марганца [1646], медных сплавах [1346], теллуриде висмута и термоэлектрических материалах на его основе [988], сплавах на основе олова и свинца [1304], электролитах серебрения [775], баббитах [1223], щелочно-калиевых растворах [648].
Описано [560] раздельное определение Sb2S3 и Sb2S5 в их смесях с применением КМпО4 в качестве титранта.
Имеется указание [1173] на возможность перманганатометрического титрования Sb(III) в щелочной среде.
К 0,5—10 мл анализируемого раствора, содержащего от 3 до 60 мг Sb(III), приливают IM NaOH до его концентрации 0,1М, добавляют 0,1 г Н2ТеО4 и титруют 0,0002 N КМпО4.
Вследствие меньшей точности (ошибка 3—5%) перманганатометрическое титрование в щелочной среде не имеет преимуществ перед титрованием в кислых растворах.
Иодометрия. Метод основан на титровании Sb(III) до Sb(V) раствором иода в нейтральной или слабощелочной среде в присутствии тартрат-иона, необходимого для удержания сурьмы в растворе. Используются два варианта йодометрического титрования Sb(III). По одному из них, более часто используемому, Sb(III) непосредственно титруют раствором иода; по другому — избыток иода оттитровывают раствором Na2S2O3 [163, 889, 1621].
В качестве индикатора наиболее часто используют крахмал [889, 1184, 1226] и потенциометрическое установление конечной точки [194, 616, 1612, 1621]. Рекомендованы также высокочастотный [1448] и амперометрический [616] методы установления конечной точки.
При визуальном установлении конечной точки к анализируемому раствору « 100 мл),\ содержащему 5—200 мг Sb, прибавляют 10 мл 10%,-ного раствора сульфита натрия, разбавляют водой до 90—100 мл, подкисляют соляной кислотой до кислой реакции, нагревают до кипения и кипятят до полного удаления запаха SO2. Затем вводят 2 мл 10%-ного раствора винной кислоты, избыток NaHCO3 (до pH ~ 8,3), 5 мл 0,5%-ного раствора крахмала и титруют 36
0,02—0,1 N раствором иода до появления неисчезающей синей окраски. Одновременно выполняют холостой опыт. Ошибка определенияЗЬ 0,2—0,3%.
Наиболее часто для титрования используют водные растворы иода, содержащие KJ. Описано [1448] титрование Sb(III) метанольным раствором иода. При определении Sb(III) титрованием в неводных средах [615, 616] используют растворы иода в различных органических растворителях, в том числе в метаноле, этаноле, пропаноле, ацетонитриле. Хотя титрование в неводных средах менее удобно по сравнению с титрованием в водных растворах, однако оно позволяет определять при совместном присутствии ряд таких элементов, которые в водных растворах раздельно титровать не удается. Так, в работах [615, 616] показана возможность определения Sb(III), As(III) и Sn(II) в их смесях йодометрическим титрованием в различных неводных растворителях (метанол, этанол, ацетонитрил, пропанол и их смеси с СН3СООН) с применением потенциометрической и амперометрической индикации конечной точки. Разработанные методики позволяют определять 1—15 мкг'/экв Sb(III), As(III) и Sn(II) в их смесях с хорошей точностью. Показано [616], что амперометрическая индикация конечной точки титрования позволяет получать более точные результаты по сравнению с потенциометрической, а биамперометрическая индикация обеспечивает получение наиболее точных результатов.
Йодометрическим титрованием определяют Sb в рудах [1184], чугуне [1226], сплавах Ga—Sb—Se [395] и Sn—Pb—Sb [889], резине [163].
Титрование Sb(III) другими окислителями. Кроме КВгО3, КМпО4 и иода, для титрования Sb(III) в качестве титранта используют Ce(SO4)2, которым титруют Sb(III) в сернокислом растворе с использованием дифениламина в качестве индикатора [1114].
Рао [1466] показал, что Sb(III) может быть оттитрована раствором NaVO3 с применением JC1 как катализатора. Установлена возможность определения Sb(III) титрованием растворами NH4VO3 в среде конц. НС1 при комнатной температуре с потенциометрической индикацией конечной точки [510]. Определению Sb(III) не мешают многие ионы, за исключением Fe. Ошибка определения в пределах 0,4—1,1%.
Чихалик [981а] показал, что JC1 количественно взаимодействует с Sb(III), окисляя ее в нейтральной и слабощелочных средах до Sb(V). Для удержания Sb(V) в растворе титрование проводят в присутствии тартратов. Конечную точку титрования устанавливают потенциометрически. Ошибка определения Sb 0,3%.
В качестве титранта рекомендован также JG13 [1422, 1533]. Лучшие результаты получены при титровании в среде СН3СООН [1422].
Для определения Sb(III) используется ряд соединений, содержащих активный хлор, в том числе NaC10[1049, 1050, 1224]
37
и хлорамин Т [913, 914, 1341]. С применением NaClO титрование проводят в среде 0,8—1 М NaOH при 80—90° С с потенциометрической индикацией конечной точки. Титрование растворами хлорамина Т проводят при комнатной температуре в среде 2—6 М НС1 с применением JC1 в качестве катализатора. Кроме потенциометрической индикации конечной точки, рекомендовано использование визуальных индикаторов, из которых метаниловый желтый найден наиболее эффективным [1341], а также люминесцентных индикаторов [1050]. Из последних рекомендован люминол, люми-несцирующий в присутствии избытка С1О~.
Хлорит натрия использован для титрования Sb(III) в растворах НС1 [1108]. Описано титрование Sb(III) хлоратом калия [880], при проведении титрования в 6—7 М НС1 в присутствии КВг, ускоряющего окисление Sb(III) до Sb(V), с использованием нафтолового сине-черного в качестве индикатора ошибка определения Sb(III) составляет 0,20—0,25%.
Описано [886, 929, 979] титрование Sb(III) растворами N-бром-сукцинимида. В растворах 2,5—3,0 М НС1 TV-бромсукцинимид быстро окисляет Sb(III) до Sb(V), восстанавливаясь при этом до сукцинимида. Конечную точку титрования устанавливают как потенциометрически, так и визуально с применением фенилрозин-дулина или розиндона как обратимых индикаторов [929]. Рекомендовано также использовать метиловый красный [886]. Титрование Sb(III) N-бромсукцинимидом по точности сопоставимо с броматометрическим титрованием.
В качестве реагентов для титрования Sb(III) рекомендованы также Mn2(SO4)3 [1311, 15351, Co2(SO4)3 [1340], Pb(CH3COO)4 [904], K3[Fe(CN)e] [1232], K2MnO4 [881] и надбензойная кислота [1534]. Однако методы с их применением значительно уступают броматометрическому и перманганатометрическому методам по точности получаемых результатов; кроме того, сами реагенты менее доступны и многие из них неустойчивы при хранении.
Редуктометрическое титрование
Редуктометрические методы определения Sb по практическому. значению намного уступают оксидиметрическим методам. Наряду с прямым титрованием Sb(V) растворами восстановителей, в ряде случаев раствор восстановителя вводят в избытке, который затем оттитровывают подходящим окислителем.
Описаны [1230] методы определения Sb(V), включающие добавление KJ и титрование гипосульфитом иода, выделившегося в эквивалентном количестве содержанию Sb(V). Концентрация НС1 в пределах 6—7 М является оптимальной. Этим методом определяют сурьму в антимонатах, типографическом металле [1230] и сульфосоединениях сурьмы [51].
Для определения Sb(V) рекомендован SnCl2 [510, 511, 1014]. Хорошие результаты получены при титровании в среде 7,0—7,5 М
38
НС1 при 70—80° С с потенциометрической индикацией конечной точки. Этим методом можно последовательно титровать Sb(V) и Cu(II). Метод применен для определения Sb в баббитах [5101.
Применение более сильных восстановителей, в том числе солей Ti(III) [16811, Сг(П) [776] и V(II) [1811, менее удобно ввиду их малой устойчивости. Титрование SbCl5 растворами TiCl3 проводят в среде этилцеллозольва с потенциометрической индикацией конечной точки. Хорошие результаты получены при использовании алюминиевого электрода или электрода из нержавеющей стали в паре с нас. к.э. Возможно также визуальное установление конца титрования по обесцвечиванию раствора.
Прямое титрование Sb(V) раствором Сг(П) приводит к восстановлению ее до металла [7761. VC12 используют для определения Sb(V) и Т1(Ш) при их совместном присутствии. Сначала в среде 2 М НС1 титруют Т1(Ш), затем в среде 10 М НС1 титруют Sb(V). Конечную точку титрования устанавливают потенциометрически. Титрование Sb(V) этими восстановителями мало избирательно, так как при этом титруются все окислители, в том числе Fe(III), Cu(I) и Cu(II), Hg(I) и Hg(II), Ag(I), Bi(III), Mo(VI), Re(VII), Se(IV) и Se(VI), Te(IV) и Te(VI), Sn(IV) и U(VI). Кроме того, для получения правильных результатов необходимо титрование проводить в инертной атмосфере, а титруемый раствор предварительно освобождать от О2.
В качестве титранта для Sb(V) предложен 2,4-дитиобиурет [699]. Этот реагент в растворах НС1 (>3 М) восстанавливает Sb(V) до Sb(III). С его применением предложен амперометрический вариант титрования Sb(V). Титрование проводят по анодному току реагента при наложении внешней э.д.с. 0,55 в; в качестве индикаторного электрода применяют платиновый или графитовый электрод. Амперометрическому титрованию Sb(V) в 6 М НС1 не мешают большие количества Pb, Bi, Те, Ni, Со, Mn(II), Zn, Al, Cd, In, Sn, Hg, Ag. Мешающее влияние Fe(III) устраняется введением H3PO4, a Se(lV) — добавлением 50-кратных количеств Cu(II).
Комплексонометрическое титрование
Комплексонометрические методы описаны как для Sb(III) [978, 1567], так и для Sb(V) [1680]. В качестве индикаторов для комплексонометрического титрования Sb(IlI) рекомендованы 2-(2-тиазолилазо)-4-метоксифенол и 2-(2-тиазолилазо)-5-метокси-фенол [978], которые в точке эквивалентности изменяют зеленоголубую окраску на желтую. Прямое комплексонометрическое титрование Sb(V) возможно в среде диметилформамида с кондуктометрическим установлением конечной точки [1680]. Вследствие мешающего влияния многих ионов металлов комплексонометрические методы определения Sb не нашли применения.
Предложен [817] косвенный комплексонометрический метод определения Sb в ее двойных сплавах с Zn, In, Bi и Pb, основан
39
ный на связывании всех компонентов раствора в комплексонаты, титровании избытка ЭДТА раствором CuSO4 в присутствии 1-(2-пиридилазо)резорцина в качестве индикатора при pH 2—3, последующем повышении pH раствора до 5,0 с одновременным нагреванием до кипения и титровании тем же раствором сульфата меди ЭДТА, выделившегося в количестве, эквивалентном содержанию Sb.
Другие титриметрические методы
Описан [565] ускоренный метод определения Sb(III) в присутствии As, основанный на титровании кислоты, образующейся в количестве трех эквивалентов на 1 г-ион Sb(III) при смешении раствора SbCl3 с тартратом натрия. Образующуюся кислоту титруют раствором NaOH по фенолфталеину. В другую порцию раствора вводят раствор K4[Fe(CN)e], отфильтровывают выделившийся осадок Sb4[Fe(CN)e]3 и титруют в фильтрате свободную кислоту. По разности двух титрований рассчитывают содержание Sb. Вследствие большой трудоемкости, малой точности и мешающего влияния многих катионов этот метод не рекомендуется для практического использования.
Хоросани [1225] установил, что реакция 2SbCl3 + 6КОН-> -> Sb2O3 + 6КС1 + ЗН2О протекает количественно не только в присутствии тартрата, но и в его отсутствии.
Описан [618] косвенный метод определения Sb(III), основанный на осаждении ее анилином в виде Sb2O3, растворении осадка в титрованном растворе KJ и последующем меркурометричес-ком титровании избытка J~c применением дифенилкарбазона в качестве индикатора. Определению Sb мешают РЬ и Ag.
Свойство SbJ3 проявлять себя в среде метилэтилкетона как кислота использовано для разработки автоматического титрования ее изопропанольным раствором КОН с потенциометрическим установлением конечной точки [363]. Метод позволяет дифференцировано титровать Sb(III), As(III) и Bi(III) в их смесях.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
Фотометрические методы
В- настоящее время фотометрические методы определения Sb являются наиболее распространенными. Большим достоинством их является высокая чувствительность, малая продолжительность и простота выполнения. Немаловажным фактором, обусловливающим их широкое применение, следует считать наличие почти во всех химико-аналитических лабораториях необходимого недорогостоящего оборудования для фотометрического анализа.
40
Определение сурьмы с применением неорганических реагентов
Из методов, основанных на использовании неорганических реагентов, наиболее часто применяется иодидный метод. Значительно меньше применяется метод, основанный на образовании восстановленной фосфорносурьмяномолибденовой гетерололикислоты, а также бромидный.
Иодидный метод. Метод основан на образовании при достаточном избытке J- в растворах H2SO4 растворимого комплекса SbJ4, обладающего желтой окраской. На кривой светопоглощенил этого комплекса имеются два максимума: один, более интенсивный, при 330 нм (е = 3,2 • 104) и другой, менее интенсивный, при 425 нм (е = 4,0-103). Имея в виду более высокую чувствительность, в ряде работ [158,1045,1602, 1682] рекомендуется оптическую плотность растворов измерять при 330 нм. Однако в связи с большим мешающим влиянием других элементов оптическую плотность чаще измеряют в области второго максимума [480, 890, 1032, 1329, 1394], Определению Sb иодидным методом наиболее сильно мешает Bi, образующий желтый комплекс BiJ4, поглощающий свет практически во всем диапазоне поглощения SbJ4 и особенно в области максимума при 330 нм. Определению Sb иодидным методом при измерении оптической плотности при 330 нм также сильно мешают РЬ и Hg. При измерении оптической плотности при 425 нм допустимо присутствие 10-кратных количеств РЬ, 4-кратных — W, 5-кратных — Sn, 20-кратных — Си, 30-кратных — Мо [1045]; не мешают до 10—20 мкг/мл As, Hg и Fe. Мешают окислители. Таллий осаждается в виде T1J, который отделяют фильтрованием или центрифугированием [1329]. Для устранения мешающего влияния желтой окраски иона J3, образующегося вследствие восстановления Sb(V) до Sb(III), а также при взаимодействии J~ с окислителями и с кислородом воздуха, в реакционную смесь вводят аскорбиновую кислоту. В некоторых случаях используют NaH2PO2 [1394, 1682], тиомочевину [139, 229, 340, 400, 480, 1185], Na2S2O3 или Na2SO3 [469].
Для переведения всей сурьмы в SbJ4 требуется большой избыток KJ. При концентрации KJ менее 5% Sb(III) переходит в SbJ4 не полностью; оптимальная концентрация KJ в фотомет-рируемом растворе находится в пределах 5—8%. Вместо KJ можно использовать NaJ или NH4J. Концентрация H2SO4 должна быть в пределах 2,2—5,0 N, при концентрации 5N, вследствие довольно быстрого окисления J~ серной кислотой до J2, получаются завышенные результаты. С целью упрощения методики определения Sb часто в качестве реагента используют раствор KJ, содержащий аскорбиновую кислоту. Иногда наряду с аскорбиновой кислотой рекомендуется вводить тиомочевину [557, 1183], эффективно маскирующую медь.
4f
Иодидный метод характеризуется достаточно высокой избирательностью (при измерении оптической плотности при 425 нм) и при использовании подходящих маскирующих реагентов позволяет определять Sb в алюминиевых сплавах [843], чугуне [1185], нелегированных сталях [512], медно-оловянных сплавах [1436], сплавах Sb с Ан, а также в олове, свинце и меди [1043].
Для определения Sb к анализируемому раствору, содержащему 0,2—1 ме Sb (при измерении оптической плотности при 425 нм или на фотоэлектроколориметре с синим светофильтром) или 0,01—0,1 мг Sb (при измерении оптической плотности при 330 нм), добавляют 10 мл H2SO4 (1 : 1), 5 мл 2%-ного раствора', аскорбиновой кислоты, 10 мл 40%-ного раствора KJ, разбавляют водой до метки и через 5 мин. измеряют светопоглощение в кювете с I = 1 см относительно раствора холостого опыта. Ошибка определения Sb составляет 2-3%.
Для определения Sb в присутствии Bi сначала определяют Bi в растворе, содержащем 1% K.J; в этих условиях Sb практически не образуете J- цветной реакции. Затем в другой аликвотной части раствора при концентрации KJ 5—8% определяют суммарное светопоглощение Sb и Bi. Из полученного значения оптической плотности вычитают оптическую плотность, найденную для Bi, и по разности оптических плотностей находят содержание Sb.
Иодидный метод применяется для определения Sb в различных материалах, в том числе в алюминии и его сплавах и солях [843, 1294], бронзах [139, 340], ванадате натрия [1294], галлии и его окислах [1294], германии [500], железе [1294], чугуне [22, 951, 1185, 1477], нелегированных [1431] и легированных сталях [918], ферросплавах [690], железных рудах [735, 1277], золоте [735, 1682] и его сплавах [1043], кобальте, магнии и марганце и их хлоридах [1294], меди и ее сплавах [340], сплавах меди с оловом [544, 1043, 1153], мышьяке, его солях и окислах [539, 1294], тонких пленках сплава мышьяка с сурьмой [1602], олове [994], сплавах олова с сурьмой [405, 1043], оловянных бронзах [1266], органических соединениях [1665], пищевых продуктах [1331], полупроводниковых сплавах [452], припоях [512, 1032, 1279], свинце [890], свинцово-цинковых рудах [1183], смазочных материалах [1394], сурьмяных рудах и продуктах их переработки [480], нитратах тория и урана, хлориде хрома и цинке [1294], электролитах для получения свинцово-оловянного сплава [557], аккумуляторном скрапе [1279] и ваннах лужения [409]. Для определения SbH3 в смесях газов.также используется иодидный метод, включающий поглощение и окисление SbH3 до Sb(HI) [1086].
Для определения больших содержаний Sb (20—50%) в ее рудах, концентратах и продуктах переработки рекомендован метод дифференциальной спектрофотометрии, основанный на использовании светопоглощения SbJ4. Ошибка определения Sb 1% [540].
Метод восстановленной фосфорносурьмяномолибденовой гетерополикислоты. Метод основан на взаимодействии Sb(IHj 42
с МоОд в присутствии Р04 и аскорбиновой кислоты в качестве восстановителя с образованием восстановленной фосфорносурьмя-номолибденовой гетерополикислоты, обладающей голубой окраской (при 725 нм е = 7-103) [1132]. Сурьма и фосфор в состав комплекса входят в отношении 1:1.
Для определения Sb этим методом анализируемый раствор разбавляют водой до 80 мл, подщелачивают 0,5 М раствором NaOH по фенолфталеину до слабой розовой окраски, прибавляют 0,5 мл раствора фосфатно-молибдатного реагента (содержащего 50 мл смеси в отношении 6 : 1 0,1 М Na2MoO4 и 0,05 М КН2РО4 и 0,12 М по H2SO4), 2 мл 5%-ного раствора аскорбиновой кислоты, разбавляют водой до 100 мл и через^ 15 мин. измеряют оптическую плотность при 725 нм в кювете с I = 1 см.
Определению Sb этим методом мешают Ba, Си, Ag, Pb, As, V и W; допустимо присутствие до 100 мкг/мл Mg, Sr, Zn, Cd, Al, Ti, В галогенидов, NO3, Cl O3, до 50 мкг/мл Co, Ni, SiO3- Ошибка определения Sb<3%. Метод использован для определения Sb в силикатах [1138]. В качестве восстановителя применен Na2SO3. Для устранения мешающего влияния Fe(III) прибавляют комплексон III.
Метод сурьмяномолибденовой сини. Установлено [1328], что при взаимодействии Sb(III) с МоО4 с применением аскорбиновой кислоты в качестве восстановителя при pH 1,4 образуется соединение голубого цвета (Xmav = 725 нм, е = 1,1 -104).
Метод, основанный на образовании сурьмой(У) окрашенного бромидного комплекса. Сурьма(У) в растворах НВг образует желтого цвета бромидный комплекс (Атах = 366 нм). При концентрации НВг > 28% оптическая плотность растворов не зависит от концентрации НВг [1388]. Закон Бера соблюдается при содержании Sb(V) 2—50 мкг/мл. Определению Sb этим методом мешают многие элементы.
Косвенный метод определения сурьмы по образованию надтитановой кислоты [1074]. Метод основан на образовании желтого пероксидного комплекса Ti(IV) при взаимодействии H2TiFe с Н2О2 в присутствии Sb(III) вследствие разложения H2TiFe и образования фторидного комплекса сурьмы — H2SbF6. В качестве реагента используют раствор, 2,5 М по НС1 и 0,1 М по Ti(IV) с молярным соотношением Na2TiFe : NaF : Н2О2 = 1:4:1. Образование эквивалентного количества пероксидного комплекса Ti протекает практически мгновенно. Определению Sb этим методом мешают элементы, поглощающие свет в области поглощения пероксидного комплекса Ti, а также элементы, образующие прочные фторидные комплексы, в том числе Al, Zr, Fe, Si и В.
Косвенный метод определения сурьмы по иодокрахмальной реакции [469]. Метод включает экстракцию Sb бензолом в виде SbJ3 из раствора, 10 А по H2SO4, реэкстракцию SbJ3 из органической фазы водой, окисление в водном растворе J- бромом до JO3,
43
удаление избытка Вг2, добавление KJ, реагирующего с J03 с выделением иода, взаимодействующего с крахмалом с образованием синей окраски, интенсивность которой прямо пропорциональна содержанию Sb. По этому методу одному атому Sb соответствует 18 атомов иода, вследствие чего молярный коэффициент погашения в расчете на Sb составляет ~ 3,2 -105.
Определение сурьмы с применением органических реагентов
Методы определения Sb с применением органических реагентов многочисленны и разнообразны. Наиболее многочисленную группу составляют методы, основанные на образовании интенсивно окрашенных ионных ассоциатов анионом SbClg с катионами основных красителей, экстрагирующихся органическими растворителями. Большую группу фотометрических методов составляют также методы, основанные на цветных реакциях Sb(III) с серу-содержащими и кислородсодержащими органическими реагентами.
Методы, основанные на образовании ионных ассоциатов анионом SbClg с катионами основных красителей и их экстракции. Эта группа методов наиболее часто используется при определении малых содержаний Sb в различных материалах. Чувствительность определения Sb зависит от используемого основного красителя и в общем случае она тем выше, чем выше молярный коэффициент погашения красителя. Что же касается селективности, то она часто имеет обратный порядок [327, 329]. Однако даже с применением одного и того же основного красителя чувствительность и особенно селективность определения могут существенно меняться. Это происходит потому, что для Sb(III) и других элементов экстрагирующиеся ионные ассоциаты с основными красителями образуются при различных концентрациях ионов С1_ и Н+; при недостаточной концентрации ионов Н+ возможно образование частично гидролизованных хлоридных комплексов Sb(III), в том числе Sb(OH)C15, Sb(OH)2C14 и т. д., которые образуют с катионами основных красителей неэкстрагирующиеся ионные ассоциаты. При малой концентрации С1~ не образуются необходимые для получения ионных ассоциатов анионы SbClg- /
Сильное влияние оказывает природа органического растворителя. Так, например, при изготовлении бензолом ионного ассоциата аниона SbClg с катионом бриллиантового зеленого при концентрации НС1 < 2 М не мешаетFe(III), помешает Ga(III), а при снижении концентрации НС1 < 1 М он оказывает незначительное влияние. Т1 и Ан мешают определению Sb при концентрации НС1 1—12 М. При использовании в качестве экстрагента хлорбензола многие элементы сильно мешают при всех концентрациях НС1 [329]. Применение органических растворителей с более высокой диэлектрической проницаемостью расширяет интервал кислот
44
ности, в котором Sb хорошо экстрагируется. Однако если использовать органические растворители с более высокой диэлектрической проницаемостью (например, хлороформ, амилацетат), экстрагирующие Sb с более высокими коэффициентами распределения, то при этом снижается селективность определения Sb.
Замена части НС1 на H2SO4 не повышает селективности извлечения Sb. Оптимальная кислотность водного раствора зависит как от используемого основного красителя, так и от органического растворителя. Максимальная экстракция ионных ассоциатов SbCle с основными красителями, способными образовывать двухзарядные катионы (метиловый зеленый, иодный зеленый), по данным работы [327], приходится на меньшую концентрацию кислоты по сравнению с красителями, образующими однозарядные катионы. Это происходит потому, что красители, способные образовывать двухзарядные катионы, при повышении концентрации кислоты образуют эти двухзарядные катионы, которые с однозарядным анионом SbCG образуют неэкстрагирующийся ионный ассоциат. Однако при использовании полярных растворителей, таких, как хлороформ, дихлорэтан или смесь (5 : 1) бензола с нитробензолом, ионные ассоциаты также хорошо экстрагируются.
Чувствительность и селективность зависят не только от концентрации ионов Н+ и Sb(V), но и от концентрации и природы основного красителя, природы органического растворителя, устойчивости и коэффициента распределения образующегося ионного ассоциата, концентрации С1~, продолжительности экстракции, соотношения объемов водной и органической фаз [45, 71, 430—433, 435, 436, 438-441, 712, 858].
В ряде работ проведено сравнительное изучение различных основных красителей для экстракционно-фотометрического определения Sb [71—73, 327, 329, 514, 521, 798]. Установлено, что даже красители, относящиеся к одному классу и с близкой структурой, сильно отличаются по экстракционной способности. Так, например, бриллиантовый зеленый и кристаллический фиолетовый, принадлежащие к классу трифенилметановых красителей, по своей структуре и рефрактометрическим радиусам отличаются незначительно (3,64 и 3,60 А соответственно), в то же время экстракционная способность бриллиантового зеленого намного выше [327]. Общая основность красителей также не определяет их экстракционные свойства. Что касается трифенилметановых красителей, то их способность экстрагировать Sb хорошо объясняется предложенным рядом авторов [430—433, 435, 436, 438—441] механизмом ассоциации и экстракции, согласно которому образование ассоциата происходит по участку катиона красителя с наибольшей плотностью положительного заряда. Данные работы [327] подтверждают этот вывод; величина зарядов на граничных группах, несущих максимальный положительный заряд, возрастает в ряду: кристаллический фиолетовый < малахитовый зе
45
леный < бриллиантовый зеленый, и в этом же ряду растет их способность экстрагировать Sb.
Многие основные красители, в том числе и трифенилметановые, способны к агрегации, степень которой зависит от их концентрации [31, 203, 358, 415]. На агрегацию красителей оказывают существенное влияние строение самого красителя, природа растворителя и температура [413, 414]. Наиболее сильно красители агрегируются в водных растворах; с уменьшением диэлектрической проницаемости растворителя агрегация ослабевает, и для спиртовых растворов она уже мало характерна [31, 32, 203, 358, 413—415].
В оптимальных условиях экстракции Sb(V) с применением кристаллического фиолетового (при его исходной концентрации в водной фазе 1,66 • 10~4 М) краситель, находящийся в этих условиях в виде двух форм — мономерной (Хтах — 591 нм) и димерной (Хтах = 540 нм), образует с SbCls ионный ассоциат, бензольные экстракты которого также характеризуются двумя максимумами поглощения — при 610 и 550 нм [327]. Некоторое смещение максимумов поглощения объясняется явлением сольватохромии [361]. Однако при извлечении ионного ассоциата растворителями с более высокой диэлектрической проницаемостью, чем у бензола (хлорбензол, хлороформ, дихлорэтан и т. п.), и смесями бензола с высокополярными растворителями в спектрах экстрактов наблюдается только один максимум, принадлежащий мономерной форме красителя, т. е. наблюдается явление, обратное установленному для самих красителей. Таким образом ведут себя и другие красители, в том числе метиловый фиолетовый, бриллиантовый зеленый, малахитовый зеленый. Получение экстрактов с одним максимумом существенно увеличивает оптическую плотность экстракта. Таким образом, добавление к бензолу нитробензола, дихлорэтана и других высокополярных растворителей или использование только этих растворителей приводит к дезагрегации красителей, входящих в состав ионных ассоциатов. Растворители с диэлектрической постоянной 10 (нитробензол, спирты, нитрилы, альдегиды и т. п.) в качестве экстрагентов для экстракционно-фотометрического определения Sb(V) непригодны, так как сильно извлекают солянокислые соли самих красителей. Для экстракции ионных ассоциатов, образуемых SbCls с катионами трифенилметановых красителей, рекомендуется применять растворители с диэлектрической проницаемостью в пределах 4,8— 10,0 [327]. Эти растворители (хлорбензол, смеси бензола с нитробензолом или с дихлорэтаном) экстрагируют Sb(V) полнее, и получаемые экстракты характеризуются значительно большими молярными коэффициентами погашения. Добавление к бензолу циклогексанона и других кетонов, наоборот, уменьшает оптическую плотность экстрактов. Это объясняется тем, что кетоны хорошо извлекают Sb в виде HSbCls, присоединяясь к ней с образованием соответствующих неокрашенных сольватов [393].
46
Перед определением Sb ее необходимо полностью перевести в Sb(V). Однако поскольку растворы иногда содержат Sb(IV), которая очень медленно окисляется до Sb(V), то в этих случаях вначале всю Sb восстанавливают до Sb(III) сульфитом натрия, а избыток SO2 удаляют кипячением и затем окисляют Sb(III) до Sb(V). Иногда в качестве восстановителя применяют SnCl2 [393], что менее удобно. В качестве окислителей наиболее часто используют Ce(SO4)2 или NaNO2. Окисление Sb(III) сульфатом Ce(IV) проводят на холоду в среде ~6 М НС1. Избыток Ce(IV) восстанавливают добавлением NH2OH-HC1, который не восстанавливает Sb(V). Гидроксиламин восстанавливает также хлор, образующийся при взаимодействии Ce(IV) с НС1. При использовании NaNO2 для окисления Sb(III) избыток его устраняют добавлением мочевины [1125]. Экстракцию обычно проводят при комнатной температуре. Предварительное охлаждение до 0° С несколько повышает эффективность экстракции [1067].
Для маскирования ионов, мешающих экстракционно-фотометрическому определению Sb, весьма эффективным оказался гексаметафосфат натрия, образующий прочные комплексы со многими ионами металлов и не взаимодействующий с Sb(V). Его применение позволяет определять Sb в различных материалах без ее предварительного отделения. Таким образом определяют Sb в сталях [1351], двуокиси титана [1083, 1467], почвенных вытяжках и осадочных породах [1550], почвах и минералах [1549].
Перед измерением оптической плотности экстрактов рекомендуется [1067, 1221, 1549] фильтровать их через бумажные фильтры для удаления мути, обусловленной наличием в экстракте эмульгированной воды. В связи с тем что основные красители и их соединения с Sb(V) при этом частично сорбируются бумажными фильтрами, фильтрования через бумажные фильтры следует избегать. Для устранения мути лучше экстракты центрифугировать или отстаивать. Хорошие результаты дает также высушивание экстрактов безводным Na2SO4 [1467] или добавление небольших количеств этанола [469], существенно повышающего растворимость воды в неполярных и малополярных органических растворителях.
Ниже приводятся некоторые данные по экстракционно-фотометрическому определению Sb различными основными красителями.
Бриллиантовый зеленый. Экстракционно-фотометрическое определение Sb бриллиантовым зеленым характеризуется самой высокой чувствительностью [327]. Оптимальная концентрация НС1 при экстракции неполярными и малополярными растворителями (бензол, толуол, о-ксилол, хлорбензол, анизол, хлорбутан) составляет ЗМ, а при экстракции хлороформом — 7М. Некоторые данные по экстракционно-фотометрическому определению Sb с применением бриллиантового зеленого приведены в табл. 3.
При использовании бензола в качестве растворителя Sb можно определять в присутствии Fe, если ионный ассоциат экстраги
47
ровать из раствора с концентрацией HG1 < 2М. При концентрации HCI < 1 М Ga оказывает незначительное влияние, в то время как Т1 и Ан мешают во всем интервале кислотности. При использовании хлорбензола определению Sb сильно мешают посторонние элементы во всем возможном интервале концентраций HG1 (1—12М). Однако поскольку при определении малых содержаний Sb в различных материалах ее предварительно выделяют, то несколько меньшая селективность бриллиантового зеленого по сравнению с некоторыми другими основными красителями не имеет существенного значения.
Поскольку бриллиантовый зеленый, выпускаемый промышленностью, содержит ряд примесей, его рекомендуется предварительно очищать [1221].
G применением бриллиантового зеленого оптическую плотность экстрактов измеряют при 635 [1221], 640 [1067, 1666] или 650 нм [956]. Как показано в табл. 3, максимум светопоглощения ионного ассоциата зависит от используемого органического растворителя.
Экстракционно-фотометрическим методом с применением бриллиантового зеленого определяют Sb в железе, чугуне, сталях и сплавах на основе железа [408, 1074, 1351], индиевых сплавах [661, 662], кадмии и его солях [568], меди и ее сплавах [393, 408, 649, 686], минералах [1549], мышьяке [364], никелевых сплавах [686], оловянных рудах и продуктах их обогащения [1063], осадочных породах [1550], почвах [1549, 1550], продуктах свин-цово-цинкового производства [626], сточных водах заводов цветной металлургии [784], титане и его окислах [1083, 1467], фармацевтических препаратах [1467], феррохроме и хроме [393], цинке [769], его сплавах с галлием [661], цинковых электролитах [757].
Кристаллический фиолетовый. Кристаллический фиолетовый по практическому применению для экстракционно-фотометрического определения Sb почти не уступает бриллиантовому зеленому. Чувствительность определения Sb с его применением несколько ниже, но избирательность несколько выше [329].
Положение максимума светопоглощения гексахлорстибата кристаллического фиолетового в его экстрактах зависит от при-( роды органического растворителя: при использовании бензола он находится при 610, толуола — при 608, смеси (4 : 1) бензола с нитробензолом — при 600, хлорбензола — при 598, смеси (1 : 1) бензола с дихлорэтаном — при 596 и хлороформа— при 592 нм; условный молярный коэффициент погашения составляет соответственно 9,3-104; 8,4-Ю4; 11,6-104; 11,2-Ю4; 11,8-Ю4 и 11,5-Ю4 [327]. Оптимальная концентрация НС1 в водной фазе зависит от природы используемого органического растворителя и колеблется от 2 (бензол, толуол) до 4М (хлороформ). Извлечение Sb в оптимальных условиях равным объемом органического растворителя также зависит от его природы и составляет 90% для толуола, 95,9% для бензола, 98,7% для хлорбензола, 99% для хлороформа и 99,5% для смесей бензола с нитробензолом и с дихлорэтаном.
48
Таблица 3
Экстракционно-фотометрическое определение сурьмы с применением бриллиантового зеленого [327]
Органический растворитель 1снсъ М *’ \пах комплекса, 'нм ЕК0МПЛ’Ю ’ Извлечение Sb, % *2
Бензол 3 644 1,08 97,5
Толуол 3 644 1,06 97,1
о-Ксилол 3 644 1,05 96,2
Хлороформ 7 634 1,17 98,0
Хлорбензол 3 640 1,20 93,3
Анизол 3 640 1,15 93,0
Хлорбутан 3 632 1,19 95,0
** Оптимальная концентрация. *2 Извлечение равным объемом экстрагента.
Имеются данные [791] о высокой эффективности в качестве экстрагента трихлорэтилена.
G применением кристаллического фиолетового Sb определяют в висмуте [454], вольфрамовых концентратах [179], двуокиси германия [624], железе, железных рудах и сталях [70, 845, 1412],. кадмии [470], меди, медных концентратах и сплавах [94, 190, 642, 685, 686], минеральном сырье [476], никеле и его сплавах [686, 695], олове, его рудах и концентратах [596], природных водах [666], свинце [1046], ферровольфраме [632], феррониобии [786], ферротитане [632].
Метиловый фиолетовый. Этот краситель, также принадлежащий к группе трифенилметановых, образует с SbCl6 ионный ассоциат, экстрагирующийся органическими растворителями. Чувствительность экстракционно-фотометрического определения Sb с его применением ниже, чем с применением бриллиантового зеленого и кристаллического фиолетового; при использовании бензола е = 5,4-104 при АП1ах = 608 нм (2 М HG1); для СНС13 е = = 8,1 • 104, Хтах = 590 нм (4 М НС1) [327]. Несмотря на указанный недостаток, метиловый фиолетовый довольно часто используется для определения Sb в различных материалах. С его применением определяют Sb в алюминии [254], жаропрочных сплавах [497], железе, чугуне, сталях, железных рудах и ферросплавах [84, 444, 975, 1406], кадмии [456], меди и ее сплавах [93, 341, 359, 489, 490], молибдене и ферромолибдене [401, 645, 655], никеле и его сплавах [502], оловянных рудах и продуктах их переработки [596], припоях [277], рении [645], свинце [1105, 1106], таллии [320], титане [498], хроме и его сплавах [502, 545], цинке, цинковых сплавах, электролитах и растворах цинкового производства [332, 456, 700], тонких напыленных слоях стибнита [63].
49
Другие трифенилметановые красители. Исследовано применение ряда других основных трифенилметановых красителей для экстракционно-фотометрического определения Sb, в том числе иодного зеленого [327, 329], малахитового зеленого [327, 329, 1212], метилового зеленого [327, 329, 711], нильского голубого [35], основного бирюзового [788], основного синего К [515] фуксина [35, 519]. В связи с более низкой чувствительностью и меньшей доступностью по сравнению с бриллиантовым зеленым, кристаллическим фиолетовым и метиловым фиолетовым они не находят широкого применения.
Родаминовые красители. По своей практической значимости для экстракционно-фотометрического определения Sb родамины идут вслед за трифенилметановыми красителями. Из родаминов наиболее часто используется родамин С (иногда неправильно называемый родамином В или Б). Катион родамина С с анионом SbCl6 образует практически нерастворимый в воде ионный ассоциат, хорошо экстрагирующийся многими органическими растворителями (бензол, толуол, ксилол, СС14, СНС13, изопропиловый эфир и др.), в то же время они не извлекают родамина С. Поскольку растворимость ионного ассоциата в ароматических углеводородах, а также в СНС13 и СС14 ограниченна (например, в бензоле в расчете на Sb она не превышает 2 мкг/мл), то в экстракционнофотометрических методах определения Sb с родамином С чаще используют изопропиловый эфир [1171, 1233, 1390, 1580, 1637, 1683]. В этом растворителе максимум оптической плотности ионного ассоциата находится при 552 нм (е = 9,7-104). Другие кислородсодержащие растворители менее удобны, так как частично экстрагируют также и родамин С. Положение максимума светопогло-щения экстрактов ионного ассоциата несколько зависит от природы применяемого растворителя; в случае бензола он находится при 565 нм.
Экстракционно-фотометрическое определение Sb родамином С изучалось многими исследователями [326, 476, 956, 1013, 1125, 1169, 1302, 1390, 1460, 1550]. Для окисления Sb(III) до Sb(V) лучшим реагентом является Ce(SO4)2 в среде 6 М НС1. С его применением окисление проходит быстро и количественно без нагревания. Избыток Ce(IV) восстанавливают гидроксиламином. При использовании в качестве окислителя NaNO2 его избыток устраняют мочевиной [1125]. Определению Sb с применением родамина С мешают Fe, Ga, In TI и Au, образующие с родамином С одинаково окрашенные экстрагирующиеся ионные ассоциаты. Для устранения их мешающего влияния Sb предварительно отделяют или выделяют мешающие элементы. Так, например, Ан легко можно выделить восстановлением сульфитом до металла, a Ga и Fe — экстракцией их хлоридных комплексов перед окислением Sb(III) до Sb(V). Небольшие количества Fe(III) можно маскировать Н3Р04. Описан [1634, 1647] метод, в соответствии с которым Sb(V) в виде хлоридного комплекса выделяют экстракцией пзо-
50
пропиловым эфиром из 1,5—2М НС1 и бесцветный экстракт обрабатывают водным раствором родамина С, в результате чего в органической фазе образуется гексахлоростибат родамина С, интенсивность окраски которого пропорциональна содержанию Sb. Fe в этих условиях не экстрагируется и определению Sb не мешает.
Использование родамина С позволяет определять Sb в германии [1295], горных породах [1186, 1401, 1502, 1634], железе, чугуне и сталях [1091, 1233, 1577, 1661], картоне [1637], кремнии [1295], меди и ее сплавах [1039, 1580], минеральном сырье [325], мышьяке и его окислах [1544], никеле и его сплавах [1085], олове [995, 1091], органических соединениях [1241, 1665], платине, родии и их сплавах [393, 1648], почвах [1186, 1634], природных водах [1438, 1634], свинце [995, 996,1292, 1293, 1471, 1580, 1683], серебре и его сплавах [980], силикатных породах [1401], теллуре [1171], титане и его окислах [1179], цинке и цинковых электролитах [1139, 1482, 1615].
Из родаминовых красителей в качестве реагентов для экстракционно-фотометрического определения Sb применяются также родамин Ж [326], родамин 6Ж [263, 326, 518], родамин ЗС (этил-родамин С) [476], пиронин Ж [470] и бутилродамин С [326, 372, 1467]. Так как указанные родамины в условиях экстракции соответствующих ионных ассоциатов сами частично переходят в органическую фазу, они применяются довольно редко, хотя с применением бутилродамина С достигается очень высокая чувствительность определения Sb (е = 1,18-10s) [326].
Тиониновые (тиазиновые) красители. Из тиониновых красителей для экстракционно-фотометрического определения Sb исследованы метиленовый голубой [325], диметилтионин (азур I) [713] и триметилтионин (азур II) [713, 799]. Они образуют с анионом SbCl6 ионные ассоциаты, хорошо экстрагирующиеся хлороформом, хлорбензолом, бромбензолом, дихлорэтаном, трихлорэтиленом и их смесями, а также смесями бензола с дихлорэтаном и с нитробензолом. Методы с применением тиониновых красителей характеризуются высокой чувствительностью (е = 8-104-г--4-9-104). Мешающее влияние других элементов несколько больше, чем с применением трифенилметановых красителей и родамина С (мешают Au, Tl, Hg, Ga, Ti, J-, SON-, NO3). Недостатком тиониновых красителей является также то, что они сами заметно экстрагируются из солянокислых растворов. В связи с этим заслуживает внимания экстракционно-фотометрическое определение Sb триметилтионином из растворов H2SO4 в виде ионного ассоциата, образуемого комплексным анионом Sb(SO4)3 с катионом красителя [799]. При использовании в качестве экстрагента смеси (2 : 1) дихлорэтана с трихлорэтиленом достигается практически количественное извлечение Sb (Xmax = 660 нм, е = 8,6-104). Основным преимуществом экстракции из растворов H2SO4 является более
5i
высокая селективность определения Sb, а также то, что сам краситель при этом не экстрагируется.
Сафраниновые красители. Из сафранинов для экстракционно-фотометрического определения Sb описано применение сафранина Т (толусафранин, сафранин Ж) [584, 585, 952]. В среде 3,5 М НС1 анион SbCl6 образует с катионом красителя экстрагирующийся бензолом ионный ассоциат. Однако по чувствительности и селективности определения Sb сафранины уступают бриллиантовому зеленому, кристаллическому фиолетовому и родамину С и поэтому редко применяются.
Антипириновые красители. Несмотря на то что красители этой группы в качестве органических реагентов пока мало изучены, однако вследствие высокой чувствительности определения ряда элементов они довольно широко используются в аналитической практике. Для определения Sb в виде ионных ассоциатов с анионом SbCle изучен ряд антипириновых красителей. Лучшим из них оказался 4,4-бис- (Н-метилбензиламинофенил)антипирилкарбинол [100]. Образуемый им ионный ассоциат экстрагируется бензолом (Хтах = 575 нм, е = 1,3-105). Описано определение Sb (0.,00075— 0,002 %) в меди [99] и теллуре О2-10-5 %) [100] с использованием этого реагента.
Другие основные красители. Из других основных красителей взаимодействующих с анионом SbCl6 с образованием экстрагирующихся ионных ассоциатов, заслуживает внимания янус зеленый, представляющий собой продукт азосочетания диазотированного сафранина Т с NjN-диметиланилином. Образуемый им ионный ассоциат экстрагируется смесями бензола (а также толуола, ксилола) с ацетоном из 1,8—2,0 М НС1. Экстракционнофотометрическое определение Sb с применением януса зеленого характеризуется высокой чувствительностью (Хтах = 600 нм е = 1,18-10в) [186].
Имеются указания [324] на высокую эффективность красителя катионного синего К, позволяющего определять Sb в сильно разбавленных растворах ()>1.10_8 г/л). Образуемый им ионный ассоциат с анионом SbCle экстрагируют амилацетатом.
Исследован ряд других основных красителей различных классов и показано, что некоторые из них, в том числе 1,4-диметил-1,2, 4-триазолиний(3-азо-4>диметиланилин и 6-метокси-З-метилбенз-тиазол(азо>4-М-метилдифениламин, позволяют определять Sb с хорошей чувствительностью (е = 6,2-104 н- 6,9-104) [185, 583, 619].
Методы, основанные на образовании ионных ассоциатов анионными комплексами сурьмы(Ш) с катионами основных красителей. Эта группа экстракционно-фотометрических методов несколько проще, чем выше рассмотренная, так как не требует предварительного окисления Sb(III) до Sb(V) и удаления избытка окислителя. Однако по селективности определения Sb эти методы ус
52
тупают методам, основанным на образовании ионных ассоциатов анионом SbCle с катионами основных красителей, и поэтому мало используются.
В качестве реагентов для экстракционно-фотометрического определения Sb(III) предложены: антипирин [180] и ряд его производных, в том числе диантипирилметан [10, 24, 41], дианти-пирилметилметан [24, 10], диантипирилпропилметан [24, 10], диантипирилфенилметан [10], бцс-(4-диметиламинофенил)-(1,3-дифенил-4-хлорпиразолон-5-ил)карбинол [217], родамин С [1390]. Названные реагенты образуют с анионом Sb J4 окрашенные ионные ассоциаты, экстрагирующиеся многими органическими растворителями (СНС13, CeHe). Определению Sb этими методами мешает ряд элементов, в том числе Pb, Bi, Си, Tl, Ag, Hg, Pd, Pt, Os и Ir, дающие в условиях определения Sb анионные иодидные комплексы, также образующие ионные ассоциаты с указанными красителями.
Для повышения избирательности экстракционно-фотометрического метода, основанного на образовании окрашенного ионного ассоциата анионом SbJ4 с катионом диантипирилметана при определении Sb в морской воде, предложено сначала экстрагировать ее 0,01 М раствором диантипирилметана в СНС13 из раствора, 0,6 М по NaCl и 0,2—3,1 М по НС1, а затем экстракт обрабатывать смесью 10 мл 10 %-ного раствора аскорбиновой кислоты и 8 мл 10 %-ного раствора KJ в 0,5 НС1. При этом Sb(V) в экстракте восстанавливается до Sb(III), которая образует окрашенный ионный ассоциат состава [(DAM)H]Sb J4 [41]. К рассматриваемой группе методов следует также отнести методы, в которых Sb(III) сначала экстрагируют толуолом или другим подходящим органическим растворителем в виде SbJ3 или SbBr3, а затем к экстракту прибавляют раствор основного красителя и измеряют оптическую плотность образовавшегося в органической фазе окрашенного ионного ассоциата [688, 741]. Кроме иодидных и бромидных комплексов Sb(lll), для образования ионных ассоциатов могут использоваться также роданидные комплексы [1390], но с их использованием селективность определения Sb существенно снижается.
Пиридин-иодидный метод [150, 281, 522, 975, 993]. Этот метод основан на образовании ассоциата анионом SbJ4 с катионом пиридина, обладающего желтой окраской. Максимальная интенсивность окраски достигается при концентрации H2SO4 3— 4 М и концентрации KJ ~ 1%. Хлорид-ионы ослабляют окраску, а при больших содержаниях полностью препятствуют образованию окрашенного ионного ассоциата. Слишком большие количества пиридина также ослабляют окраску. Поскольку образующийся окрашенный ионный ассоциат практически нерастворим в воде, то для стабилизации окраски добавляют защитные коллоиды (желатин, гуммиарабик). As и Sn (несколько десятых миллиграмма) определению Sb не мешают. Мешают Bi, Со, Ni и Zn, образующие
53
осадки в условиях определения Sb. Вследствие низкой чувствительности и селективности пиридин-иодидный метод мало используется.
Методы, основанные на образовании сурьмой(Ш) окрашенных внутрикомплексных соединений. Сурьма(ПТ) способна одинаково легко образовывать внутрикомплексные соединения как с кислородсодержащими, так и с серусодержащими органическими реагентами. В связи с этим для Sb(III) предложено большое число органических реагентов, образующих с ней внутрикомплексные соединения, пригодные для ее фотометрического определения. Общим недостатком этой группы фотометрических методов является значительно меньшая селективность определения Sb по сравнению с методами, основанными на образовании ионных ассоциатов анионом SbClQ с катионами основных красителей.
Среди кислородсодержащих реагентов для определения Sb(III) наибольшее значение имеют 9-замещенные флуорона (2,3,7-триок-си-6-флуорона). Эти реагенты характеризуются высокой чувствительностью определения Sb(III) и образуют с ней нерастворимые внутрикомплексные соединения, в состав которых Sb и реагент входят в отношении 1 : 1. Эти соединения плохо экстрагируются органическими растворителями, поэтому реакцию проводят в водных или водно-зтанольных растворах и окрашенный золь стабилизируют защитными коллоидами (желатин, гуммиарабик, поливиниловый спирт, крахмал).
Из 9-замещенных флуорона наиболее часто используют 9-фенилфлуорон [22, 64, 65, 178, 495, 563, 627, 1084, 1218]. Фенил-флуорон включен в рациональный ассортимент реагентов для фотометрического определения Sb [496]. Оптическую плотность золей комплекса измеряют при 530 нм (е = 3,42-104) или 555 нм (е = 5,63-104) [1084]. Поскольку определению Sb с применением фенилфлуоронов мешают многие элементы, ее предварительно отделяют. Рекомендована [65] экстракция Sb в виде пиридин-иодид-ного комплекса.
Для этого к анализируемому раствору (15 мл) прибавляют 5 мл конц. H2SO4, 0,5 г винной кислоты, 0,1 г аскорбиновой кислоты и по охлаждении вводят 1 мл 10%-ного раствора KJ, 0,6 мл 10%-ного раствора пиридина и экстрагируют Sb 5 мл диэтилового эфира. Экстракцию повторяют еще 2 раза-(по 3 мл) и из полученного экстракта Sb реэкстрагируют (2 раза по 1,5 мл 1,5 N H2SO4). К реэкстракту прибавляют 0,3 мл 1%-ного раствора желатина, 0,3м.л этанольного растворафенилфлуорона, выдерживают 30 мин. и измеряют оптическую плотность при 530 нм в кювете с I = 1 см относительно раствора холостого опыта. Ошибка определения Sb составляет 5—8%.
С применением фенилфлуорона Sb определяют в ванадии и его окислах [563], в воздухе помещений сурьмяного производства [1218], галлии [64, 65], железе, чугуне, стали и аккумуляторной железной катодной массе [22, 495], индии [65], мышьяке [1084],
54
рудах [178], свинце, его сплавах и активной свинцовой аккумуляторной массе [627, 1084], таллии [64, 65], цинке [64].
Кроме 9-фенилфлуорона, для определения Sb рекомендованы 9-метилфлуорон (ХЭф = 530 нм, е = 4,05-104) [872, 1106, 1249, 1335], 9-пропилфлуорон (Хэ$ = 540 нм, е = 5,72-104) [1626, 1626 а], 9-(4-нитрофенил)-2,3,7-триокси-6-флуорон (п-нитрофенил-флуорон) [506], салицилфлуорон [522].
Заслуживает внимания экстракционно-фотометрический вариант определения Sb с применением флуоронов [522]. Наиболее эффективным здесь оказался салицилфлуорон, который совместно с антипирином в присутствии анионов сильных кислот (С1_, Вг~, J", NOg, SCN~, С1О4) образуете Sb(III) разнолигандное комплексное соединение, хорошо экстрагирующееся СНС13. В состав комплекса Sb, салицилфлуорон, антипирин и анион сильной кислоты входят в соотношении 1 : 1 : 3 : 1. При использовании в качестве анионов J~, SGN- и СЮ4 молярные коэффициенты погашения при = 510 нм составляют соответственно (8,7 ± 0,4) -104, (8,9 ± 0,4)-104 и (9,1 ± 0,2)-104.
Подобно флуоронам с Sb(III) реагируют кверцетин (pH— ~ 4,5, ХЭф = 420 нм, е = 1,2-104) [993], 3,5,7,4'-тетраоксифлавон (pH 0,85-1,10, ХЭф = 410ч-420 нм, е = 1,09-104) [1090], галлеин (pH 3—4, Хэф = 520 -г- 525 нм, е ~ 1,1-104) [107, 567], 2,3,7,8-тетраоксифеназин (pH 10, ХЭф = 538 нм, е = 4,0-104) [1416], 5,6- и 6,7-диокси-1,4-нафтохинон (pH ~ 1,0) [1327], 3,4-диоксиазобензол (pH 2, ХЭф = 490 нм, е = 2,1-104) [281], пирокатехиновый фиолетовый (pH 0,9—1,1, ХЭф = 560 нм, е — 1,33-• 104) [517, 918], пирогалловый красный (pH 4,5, ЛЭф = 420 нм, е = 4,545-103) [520], бромпирогалловый красный (pH 6,6—6,8, %Эф = 560 нм, е = 3,9-10*) [918, 977].
Кроме органических реагентов, содержащих в качестве реакционных групп гидроксильные и карбонильные группы, для определения Sb предложен ряд таких, в молекуле которых, наряду с кислородсодержащими группами, присутствуют амино-, алкиламино-, диалкиламино-, азо- и другие азотсодержащие группы. К таким реагентам относятся 4-(2-пиридилазо)резорцин (е = 1,2-•104), 5-(2-пиридилазо)-2-моноэтиламино-м-крезол (е = 3,82-• 104) и его 5-бром- (е = 5,6-104) и 3,5-дибромзамещенное (е = = 4,4-104) [185, 741, 742], 2-(2-пиридилазо)-5-диэтил-з4-амино-фенол и его 5-бром- и 3,5-дибромзамещенные [183, 184, 742], 6-(2-хинолилазо)-3,4-диметилфенол [620], 1-(2-хинолилазо)-2-нафтол (е = 1,24-104) [622], 2-(хинолилазо)-м-крезол (е = 9-103) [621], 8-оксихинолин и некоторые его замещенные [1178], N-метилана-базин-а'-азо-диметиламинофенол (Хтах = 600 нм, е= 3-104) [1], N-метиланабазин(а'-азо-4>,м-аминофенол (%1МХ = 570 нм, е = 5-•104) [705], 4-(2-М-метиланабазиназо)резорцин (%1Пах =
= 540 нм, X = 1,06-104) [2, 704], М-метиланабазин(а-азо>ди-
зтиламинофенол, К-метиланабазин(а'-азо>-«-гептилрезорцин
55
и пиридилазо-и-гептилрезорцин [1,2, 704—706]. Вследствие малой избирательности определения Sb эти реагенты не нашли практического применения.
Простейшим из серусодержащих реагентов является тиомочевина, образующая с Sb(III) комплекс желтого цвета [1389]. Для полного в связывания Sb в комплекс необходима высокая концентрация тиомочевины, которая при проведении определения в среде 47V H2SO4 должна быть >5% и в среде 4АГ НС1 >15% [540, 1436]. Описан [42] вариант, в котором определение рекомендуется проводить в водно-ацетоновой среде с концентрацией H2SO4 1,3 N. В этих условиях чувствительность цветной реакции несколько выше (е ~ 1,2-103).
Для определения Sb этим методом к анализируемому раствору (1—5 мг Sb) прибавляют 4 г тиомочевины, 0,9—1,0 мл конц. H2SO4, разбавляют ацетоном до 25 мл и измеряют оптическую плотность при 423 нм. Ошибка определения Sb составляет 1,2—1,9%. Определению Sb сильно мешает Bi; несколько меньше мешают другие элементы, тиомочевинные комплексы которых поглощают в области поглощения комплекса Sb.
Также малочувствительным является диэтилдитиокарбаминат натрия, рекомендованный для экстракционно-фотометрического определения Sb [924]. Несколько большей чувствительностью (е ~ 6-103 при 350—380 нм) характеризуется пирролидиндитио-карбаминат натрия [1255, 1256]. Для маскирования мешающих элементов используют лимонную кислоту, ЭДТА и KCN. Комплекс экстрагируют СНС13 или СС14. С использованием пирро-лидиндитиокарбамината натрия определяют Sb в железе, чугуне и нелегированных сталях [1255, 1258] и цинке [1257].
8-Меркаптохинолин рекомендован для экстракционно-фотометрического определения Sb в виде комплекса SbJ2A, где А —анион 8-меркаптохинолина (Хтах = 390 нм, е = 6,10-103) [1551].
В качестве реагентов для экстракционно-фотометрического определения Sb исследованы некоторые азопроизводные бензтиазола, в том числе 5-(2-тиазолилазо)-2-моноэтиламино-и-крезол (Хтах — = 580 нм, е = 3,0-104) [182], 6-метокси-З-метилбензтиазол (азо> 4-Н-метилдифениламин (Хгаах = 610 нм, е = 4,7 -104) [328]. Изучены также толуол-3,4-дитиол и некоторые другие реагенты, содержащие о-дитиольную группировку [1077], фенилдитиоуксус-ная кислота [30] и тиокапролактам [1532]. Однако вследствие малой избирательности определения Sb и низкой чувствительности эти реагенты не нашли применения.
Методы, основанные на образовании сурьм ой(У) окрашенных внутрикомплексных соединений. Сурьма(У) также способна, образовать с рядом органических реагентов внутрикомплексные соединения, пригодные для ее фотометрического определения. Высокой чувствительностью характеризуются цветные реакции Sb(V) с пирокатехиновым фиолетовым [103, 513], с которым она
56
при pH 2 реагирует в отношении 1:1 (е = 6,93-104, Хтах = = 528 нм).
Аналогично Sb(V) реагирует с пирогалловым красным и бром-пирогалловым красным [516], образуя при pH 2—3 окрашенные комплексы (Хтах = 520 нм, е = 0,98-104 и 1,29-104 соответственно).
Изучены [698] цветные реакции Sb(V) с 22 производными димеркаптотиопирона. Наиболее чувствительные цветные реакции дают диэтил- и метилфенилтиопирон. Для последнего комплекса при %тах = 360 нм е = (2,21 ± 0,05)-104. Для стабилизации окраски образующегося малорастворимого комплекса используют защитные коллоиды. Определению Sb(V) сильно мешают As, Hg, Си.
Цветные реакции Sb(V) с хелатообразующими органическими реагентами изучены недостаточно.
Косвенные фотометрические методы определения сурьмы. Из косвенных фотометрических методов определения Sb следует отметить метод, основанный на использовании в качестве реагента 1,10-фенантролината Fe(III), который взаимодействует с Sb(III) с образованием интенсивно окрашенного фенантролината Fe(II). Оптическую плотность измеряют при 530 нм (е = 2,22-104). Определению Sb мешают восстановители и окислители. Метод применен для определения Sb в трехокисях As и Bi [1396].
Методы, основанные на реакции стибина с диэтилдитиокарба-минатом серебра. Фотометрический метод определения As, основанный на цветной реакции AsH3 с диэтилдитиокарбаминатом серебра в пиридине, введенный в аналитическую практику в 1952 г. [1618], интенсивно изучался и в настоящее время является одним из наиболее эффективных методов определения следовых количеств мышьяка [535]. Однако определению As этим методом мешали Ge и, особенно, Sb, восстанавливающиеся в условиях определения As соответственно до GeH4 и SbH3, которые ззаимодействуют с диэтилдитиокарбаминатом серебра с образованием подобно окрашенных продуктов, но с меньшей интенсивностью окраски. Подробное изучение влияния Sb при определении As и взаимодействия SbH3 с диэтилдитиокарбаминатом серебра позволили найти условия, в которых цветная реакция достигает высокой чувствительности.
Авторы работы [1165] разработали метод, позволяющий определять SbH3 как в отсутствие, так и в присутствии As. Механизм цветной реакции SbH3 с диэтилдитиокарбаминатом серебра совпадает с таковым для AsH3 [926, 927, 1165] и состоит в образовании интенсивно окрашенного золя Ag. При этом одна молекула SbH3 выделяет 6 атомов Ag, a Sb3- окисляется до Sb3+ с последующим образованием диэтилдитиокарбамината. Вместо раствора диэтил-дитиокарбамината серебра в пиридине предложено [1165] использовать хлороформный раствор этого реагента, 0,1 М по пиридину. Вместо пиридина могут использоваться некоторые другие амины, в том числе этаноламин, этилендиамин, ди-«-пропиламин и три
57
этаноламин. Хотя кривые светопоглощения окрашенного продукта, образующегося с применением указанных аминов, довольно сходны по форме, но положение на них максимума светопоглощения и особенно интенсивность окраски существенно зависят от природы амина. При уменьшении концентрации амина оптическая плотность раствора остается постоянной только до некоторого предела, после чего она резко уменьшается. Эта предельная концентрация амина зависит от его основности: чем ниже основность амина, тем выше его предельная концентрация. Соотношение между предельной концентрацией амина и его основностью может быть представлено выражением: 1g с = 0,62 рКь — 4,9. Примерно к такому же выводу пришли Боде и Хахман [927], исследовавшие в качестве аминов пиперазин, эфедрин, 1,10-фенантролин.
Для определения Sb с применением цветной реакции SbH3 с диэтилдитиокарбаминатом серебра рекомендуется следующая методика [1165].
Пробу (1—40 мкг Sb) растворяют в HNO3. К раствору прибавляют избыток конц. НС1 и нагревают до прекращения выделения окислов азота. Остаток переносят в реакционную колбу емкостью 25 мл, прибавляют 5 мл 10%-ного раствора KJ, 4 капли 10%-ного раствора SnCl2-2H2O в НС1 (1 : 1), 10 мл конц. НС1 и ~ 3 г гранул Zn. Колбу тотчас же соединяют с насадкой с пористой стеклянной перегородкой или пробкой из стеклянной ваты. Над перегородкой имеется слой ваты, пропитанной РЬ(СН3СОО)2 для поглощения H2S. Выделяющиеся из реакционной колбы газы, пройдя этот слой ваты, через трубку (d = 2 -j- 3 мм) поступают в цилиндр или пробирку с 5 мл хлороформного раствора, содержащего 0,25% диэтилдитиокарбамината серебра и 1% этаноламина. Через 30 мин. измеряют оптическую плотность поглотительного раствора при 525 нм и по аналогично построенному калибровочному графику находят содержание Sb. Метод позволяет определять также Sb и As при их совместном присутствии. Для этого в одной аликвотной части раствора проводят анализ, как указано выше, и измеряют оптическую плотность, соответствующую суммарному содержанию Sb и As. Из другой аликвотной части раствора сначала удаляют As в виде AsCl3 упариванием раствора при 130° Сив остатке определяют Sb. Метод позволяет определять Sb в ряде материалов с ее содержанием до 1-10-4%.!
На примере анализа медного порошка показано, что при содержании Sb 4-10-3% ошибка «^7,5%. Стибин предложено также поглощать хлороформным раствором диэтилдитиокарбамината серебра, содержащим 1,10-фенантролин [1670]. Хотя этот метод несколько уступает по чувствительности экстракционно-фотометрическим методам с применением основных красителей, но уже в настоящее время превосходит их по воспроизводимости результатов. Замена цинка, используемого для получения SbH3, боро-гидридом натрия позволит существенно снизить значение холостого опыта и тем самым повысить чувствительность метода.
58
Люминесцентные методы
Люминесцентные методы определения Sb основаны на образовании соединений как с неорганическими, так и с органическими реагентами, обладающих характерным свечением в определенных условиях при возбуждении светом подходящей длины волны. Наиболее часто используется способность Sb(III) образовывать люминесцирующие комплексы с простейшими неорганическими соединениями (НВт, НС1). Интенсивность свечения этих комплексов значительно усиливается с понижением температуры, что использовано для разработки высокочувствительных методов определения сурьмы.
Наибольшая чувствительность определения Sb низкотемпературным люминесцентным методом достигается при использовании бромидных комплексов Sb(III) [664]. Спектр люминесценции Sb(III) в растворах НВт при —196° С представляет собой широкую полосу с максимумом при 640 нм. Люминесценция возбуждается светом в диапазоне 250—370 нм.
Сурьма(У) с НВт не образует люминесцирующих комплексов, однако в растворах НВт с концентрацией 8,6 М она количественно восстанавливается до Sb(IH). Таким образом, при определении в среде 8,6 М НВт не имеет значения исходная степень окисления Sb. Вследствие этого рекомендуется люминесцентное определение Sb проводить в среде 8,6 М НВт. Определению Sb при возбуждении люминесценции светом ртутно-кварцевой лампы со светофильтром УФС-4 и регистрации люминесценции при 600—640 нм не мешают 1000-кратные количества Li, Na, К, Mg, Са, Sr, Ba, Be, Zn, Cd, Al, Cr, Mn, Ni, а также NO3, SOf, POf, 100-кратные — Tl, Pb, Те и Bi и 10-кратные — Fe. На цвет люминесценции оказывает влияние присутствие ряда органических примесей, изменяющих его от красного до белого. Поэтому при определении малых содержаний Sb (1 • 10~5—1 • 10-7%) рекомендуется ее предварительно отделять экстракцией бензолом в виде SbJ3. С учетом указанных особенностей влияния посторонних веществ на люминесценцию Sb в замороженных растворах НВт рекомендуется следующая методика [664].
При определении Sb в жидкостях в каждую из двух платиновых чашек помещают пробу (10—50 мл), добавляют по 0,2 мл конц. H2SO4 и HNO3 и в одну из чашек вводят 0,05 мкг Sb. Растворы выпаривают на водяной бане, к остаткам прибавляют по 5 мл H2SO4 и по 0,2 мл 10%-ного раствора KJ, после чего растворы облучают светом лампы СВД-120А в течение 10 мин. в кварцевых пробирках для восстановления Sb(V) до Sb(III). Выделившийся при этом иод восстанавливают добавлением 0,1 мл 10%-ного раствора Na2S2O3 • •5Н2О. Из полученных растворов Sb экстрагируют бензолом (порциями по 1 мл) и затем реэкстрагируют водой. К реэкстрактам прибавляют по 0,1 мл конц. HNO3 и выпаривают в платиновых чашках на водяной бане. Остатки растворяют в 8,6 М НВг и измеряют интенсивность люминесценции при —196° С в области 600—640 нм, возбуждая ее ртутно-кварцевой лампой
59
СВ Д-120А со светофильтром УФС-4. Метод применен для анализа травильных растворов, воды, плавиковой, соляной, фосфорной и азотной кислот, этанола гипофосфита натрия, нитратов кобальта, никеля и алюминия с содержанием Предел обнаружения Sb — 1 нг!мл.
Описано также определение Sb по люминесценции ее бромидных комплексов в замороженных растворах в ряде других материалов, в том числе в соляной кислоте и тетрахлориде германия [77—79], растворах НВг [1242], галогенидах щелочных и щелочноземельных металлов [665], полупроводниковых материалах [647] и окислах щелочноземельных металлов [1657].
Способность Sb(III) в замороженных растворах НС1 образовывать люминесцирующие комплексы использована для прямого ее определения в соляной кислоте [79]. Оптимальная концентрация НС1 равна 4Д7. Люминесценцию возбуждают линией 310 нм, максимум люминесценции находится при 615 нм. Вследствие меньшей чувствительности определения Sb по люминесценции в замороженных растворах НС1, чем в растворах НВг, этот метод не нашел практического применения даже для определения Sb в соляной кислоте, из которой Sb обычно предварительно экстрагируют и определяют затем в среде 8,6 М НВг.
Люминесцентные методы определения Sb с применением органических реагентов основаны на образовании ими люминесци-рующих комплексов с Sb(III), которые образуют кислородсодержащие органические реагенты, в том числе бензоин [75], морин [40, 797] и триоксифлавон [1062]. Сурьма(У) образует люминесцирующие гексахлоростибаты сафранина Т [474] и этилродами-на С [475].
Несмотря на большую чувствительность определения Sb люминесцентными методами по сравнению с фотометрическими, они не нашли широкого применения вследствие большей продолжительности и трудоемкости, а также необходимости иметь специальное оборудование и помещение.
Хемилюминесцентные методы
Для определения Sb подробно изучена [338] реакция Sb(V) с люминолом, сопровождающаяся люминесцентным свечением, интенсивность которого пропорциональна концентрации Sb. Для окисления Sb(III) до Sb(V) могут использоваться NaNO2, КМпО4, КВгО3, Ce(SO4)2, Н2О2. Имеет значение порядок добавления реагентов; максимальная интенсивность хемилюминесценции достигается, если к раствору гидроокиси щелочного металла прибавляют раствор люминола, а затем анализируемый раствор, содержащий Sb в виде SbCle. Для определения Sb хемилюминесцентным методом предложена следующая методика [338].
К анализируемому раствору, 6—8 М по НС1, прибавляют несколько капель 10%-ного раствора NaNO2 и оставляют на 1—2 мин. для окисления 60
Sb(III) до Sb(V). Затем вводят насыщенный раствор мочевины для удаления HNO2, смесь разбавляют водой до концентрации НС1 0,6—1 М и сразу заполняют этим раствором микрогнезда кюветы, содержащие по 1—2 капли 0,02 М раствора люминола и 0,5 мл раствора NaOH такой концентрации, чтобы после введения анализируемого раствора значение pH смеси было в пределах 9—10. Добавление анализируемого раствора производят при темно-красном свете в микрогнездо кюветы, накрытой фотопластинкой. Через 20—30 сек. пластинку снимают и обрабатывают соответствующим образом. Интенсивность почернения пятен измеряют на денситометре. Проводят по три параллельных определения. Учитывают также результаты холостого опыта. Метод характеризуется высокой чувствительностью (0,05 мкг!мл). Присутствие до 50-кратных количеств Zn и 200-кратных — Cd и Sn определению Sb не мешает. Fe и Си даже в малых количествах гасят хемилюминесценцию.
Хемилюминесцентный метод применен для определения Sb в цинке и сульфата цинка без ее предварительного отделения [255]. Реакцию проводят при pH 11—12. Для устранения мешающего влияния следовых количеств Fe и Си вводят 1 каплю 0,1 М раствора сульфосалицилата натрия. При использовании навески анализируемого материала 0,1 г метод позволяет определять до 6-10~4% Sb в цинке 'и до 1,2 • 10-4% Sb в сульфате цинка с ошибкой ~8%.
Для определения Sb в солях алюминия и цинка хемилюминесцентным методом ее предварительно отделяют экстракцией в виде пиридин-иодидного комплекса. Этот вариант метода позволяет определять до 0,1 мкг Sb в пробе [256].
Хемилюминесцентные методы определения Sb трудоемки и не имеют практического значения.
Полярографические методы
Полярографические методы определения Sb характеризуются низкими пределами обнаружения, составляющими в ряде случаев 1-10-7—1-10~9%. Они позволяют одновременно с Sb определять РЬ [1216], As [207, 1096, 1216, 1405], Sn [1611], In [1071], Bi [416, 419 , 607 , 608, 1029, 1672], Си [530], Au [529], Bi и Си [459], Bi и As [1689], Bi, Pb, Se и Те [532], In, Cd и Sn [453], As и Sn [453, 1097, 1584], Bi, Pb и Sn [418], Cu, Fe и Bi [122]. В связи с этим полярографические методы определения Sb, особенно для малых ее содержаний, находят широкое практическое применение [86, 147, 269, 286, 368, 527, 569, 725, 1377].
Электрохимические превращения Sb можно представить следующей схемой:
+2е~ +3е- +зе~
sb5+Sb3+:-------- sb° ;-- sb3~. (i)
-2e- -зе_ -3e~
B зависимости от полярографического фона и от материала применяемого электрода на полярограммах Sb можно регистри
61
ровать токи разряда-ионизации, соответствующие любому из процессов, включенных в схему (1).
Полярографическими методами Sb обычно определяют с применением ртутных капающих [147, 368, 453, 569, 1584] и стационарных электродов [116, 134, 569, 1430], а также твердых электродов, изготовленных преимущественно из различных материалов на основе графита (спектрального графита, пропитанного эпоксидными смолами [88], смесью полиэтилена с парафином [605], угольной пасты [1348, 1439], стеклоуглерода [891, 1105, 1220].
Рис. 3. Полярограммы восстановления Sb(III) на фоне 1М НС1, полученные методами классической (7), переменнотоковой (2) и осциллографической (3) полярографии cSb(III)*^Oe З'Ион/л: 1 — 10; 2 — 0,5j 5—1,0 (осциллполярограмма полу чека при v =0,5 ejcen)
Определение Sb с применением ртутных электродов основано на использовании двух электродных процессов: разряда сольватированных или комплексных ионов Sb(III)
| SbLx]s~x + Зе~ —» Sb (амальгама) + х L~ (2)
и анодном растворении (ионизации) Sb из амальгамы с образованием сольватированных или комплексных ионов Sb(III)
Sb (амальгама) -|- х L~ — Зе~ —»[ЗЬЬж]8-зс. (3)
Здесь L- — однозарядный анион, однако замена однозарядного аниона многозарядным анионом не меняет существа обоих электродных процессов.
Последовательное осуществление процессов (2) и (3) составляет цикл разряда-ионизации Sb на стационарных ртутных электродах, лежащий в основе инверсионной вольтамперометрии [86, 1478], иногда называемой также амальгамной полярографией с накоплением [362, 368, 693, 725]. Процесс (2) описывает предварительное концентрирование Sb с образованием амальгамы, а процесс (3)— анодное растворение ее из полученной амальгамы.
Электрохимическое восстановление Sb(III) до элементной Sb [реакция (2)] исследовано методами классической, осциллографической, переменнотоковой и импульсной полярографии. На рис. 3 представлены типичные полярограммы восстановления Sb(III), полученные указанными методами. Наибольшую чувствительность
«2
определения Sb обеспечивает переменнотоковая полярография, наименьшую — классическая (рис. 3).
Для определения Sb с применением ртутного капающего электрода используют различные полярографические методы. Наиболее часто используется метод классической полярографии [368, 402, 608, 680, 1096, 1097, 1135, 1156, 1607, 1611]. Используются также другие методы, в том числе методы осциллографической [291, 292, 294, 295, 312, 451, 644, 764, 1037, 1352, 1370, 1395, 1478, 1604], переменнотоковой [116—119, 122, 124, 451, 453, 503, 720, 763, 1159, 1174, 1175, 1180, 1216, 1400, 1574], квадратноволновой [1192, 1373, 1395, 1400], импульсной [122, 343, 453, 1584] и тонкослойной [254, 721] полярографии.
В качестве фоновых электролитов в большинстве случаев рекомендуются растворы НС1 различной концентрации, в том числе 0,5М [694] <131 [295, 333, 344, 721, 1352, 1370, 1478], 2М [1395, 1574], ЗМ [95, 720, 822, 1175,1400], 4Л/ [204], Ш [453, 1530, 1546, 1604, 1664] и 8М [1635]. При этом потенциал полуволны или пика заметно смещается в сторону более отрицательных значений (от —0,15 в в 0,5 М НС1 до —0,36 в). Иногда в качестве фоновых электролитов используют растворы Н8РО4 [1216, 1672], H2SO4 [292, 876], смеси НС1 с фосфорной [1574], серной [95, 492, 822, 1174, 1175, 1605, 1606, 1672] или винной кислотой [1135], смеси Н2С2О4 с (NH4)2C2O4 [1097], СН3СООН, с CHsCOONa [312], лимонной кислоты с цитратом кальция [1096], H2SO4 с MnSO4 [1605, 1607], H2SO4 с KSCN [1180], NaF с СН8СООН [402]. В ряде случаев используют щелочные фоны. Для удержания в растворе всех компонентов пробы используют добавки реагентов, образующих в этих условиях растворимые комплексы. Наиболее часто применяют маннит [207, 607, 1029]; рекомендованы также глицерин [420], комплексон III [1345], тартрат калия [644], KCN [1623].
Сурьму полярографически можно определять по реакции (2) на фоне электролитов, в которых Sb(III) находится и разряжается на электроде в виде комплексных ионов. Сурьма может электро-восстанавливаться и в виде аква-ионов, однако, в связи с повышенной склонностью Sb(III) к гидролизу с образованием полярографически неактивных гидролизованных форм Sb(III), определение необходимо проводить в растворах с высокой концентрацией H2SO4 [292, 492, 1605—1607] или Н3РО4 [1672]. Отсутствие токов восстановления Sb(III) в слабокислых, нейтральных и слабощелочных растворах используется в ряде случаев для устранения ее мешающего влияния определению некоторых других элементов. Так, например, для определения Bi в присутствии Sb предложено использовать в качестве фона 1М NH4C1, содержащий 20% лимонной кислоты [721]. На этом фоне возможно определение микроколичеств Bi в сурьме без предварительного разделения. Способность Sb образовывать прочные комплексы с цитрат- и тартрат-ионами широко используется в полярографии для повышения разрешающей способности метода при определении Sb(III) в присут-
63;
ствии Bi(III) и Cu(lf). Эти ионы разряжаются в большинстве фонов в той же области потенциалов, что и Sb(III).
В ряде работ дается оценка мешающего влияния Bi(III) [368, 1037, 1174, 1400, 1478, 1530, 1574] и Cu(II) [569, 763, 1156, 1584, 1604] определению Sb(III) с применением различных полярографических фонов. Для классической полярографии допустимо трехкратное количество Bi(III) по отношению к Sb(III) при использовании солянокислых фонов, а в случае полярографии переменного тока оно может быть увеличено до 100-кратного [118]. Методом классической полярографии Sb(III) на фоне Н2С2О4 можно определять в присутствии 10-кратных количеств Сн(П) [1097]. Такое же количество Сн(П) допустимо при определении Sb(III) методом импульсной полярографии на фоне малоновой кислоты [1584]. С применением солянокислых фонов допустимо присутствие 3—4-кратных количеств Cu(II) по отношению к Sb(III). Определению Sb мешают также большие содержания РЬ [876, 1096, 1546]. На фоне щелочных растворов, содержащих маннит, возможно полярографическое определение Sb(III) в присутствии Sb(V) [206, 207, 607].
Применение неводных растворителей для полярографического определения Sb(III), как правило, мало эффективно [403, 1135]. Однако использование 50%-ных растворов диметилформа-мида, содержащих 5-метокси-8-оксихинолин в качестве комплексующего реагента, позволяет определять Sb в манганиновых и типографических сплавах, содержащих 10-кратные количества Си, за счет резкого сдвига волны восстановления Sb(III) в более электроотрицательную область потенциалов (ф»/г = —0,95 в) [122, 124].
При полярографическом определении Sb(III) с окислительным . растворением анализируемого материала (растворение с применением HNOS, НС1 + Н2О2, НС1 + HNOS и т. п.) возможно частичное, а иногда и полное превращение Sb(III) в Sb(V). В этом случае результат полярографического определения Sb будет занижен. Для устранения ошибки определения Sb, обусловленной частичным или полным нахождением ее в виде Sb(V), последнюю предварительно восстанавливают солями гидразина [118, 569], Na2SO8 [95, 644, 1029, 1544, 1545] или Na2S2O8 [492, 1584, 1664, аскорбиновой кислотой [204].
Полярографические методы с применением ртутного капающего электрода широко применяются для определения Sb в различных промышленных и природных материалах, в том числе в железе, чугуне и сталях [503, 823, 1037, 1216, 1264, 1309, 1478, 1574], полупроводниковых материалах [123, 343, 344, 451, 680, 720, 721, 1071], свинце и его сплавах [130, 142, 144, 148, 154, 220, 230, 246], рудах и концентратах [204, 1036, 1635], цицке и его солях [67, 416, 418, 420], цинковых электролитах [417], титане и его соединениях [822, 823, 1174, 1548], меди [1672], олове [1201], молибдене [644], кадмии [1584], цирконии и его сплавах [823], типографских сплавах [763, 820], ферромарганце [1352], манга-64
ниновых сплавах [763], арсениде галлия [1192], минеральном сырье [118, 373], селен-теллуровых минералах [736], стекле и силикатных материалах [492, 1395], вольфрамате кальция [1287], шламах медных анодов [1175], сульфидно-щелочных растворах [160], отработанных щелоках [1546], гидросульфите натрия [295], пенополиуретане [95], растительных материалах [1370], водах [1604], жидком аммиакате иодида аммония [1163].
Инверсионная вольтамперометрия. Пределы обнаружения Sb(III) при использовании ртутных капающих электродов методами осциллографической [291] и переменнотоковой [116, 118]
полярографии находятся на уровне п-10~7 г-ион1л. Однако в ряде случаев указанные пределы обнаружения Sb часто недостаточны для контроля ее содержания в материалах высокой чистоты и в материалах, используемых в электронной технике, ядерной физике и т. д. Для снижения пределов обнаружения Sb в последнее время успешно используется метод инверсионной вольтамперометрии [86, 233, 241, 533, 725,1377]. При определении Sb этим методом на полярограммах регистрируются анодные поляризационные кривые с характерными максимумами тока (рис. 4). Высота пиков
Рис. 4. Анодные поляризационные кривые амальгамы сурьмы на фоне IM НС1 cSb(III)’^®* г~ион/л> 1 “— 2; 2 — з 8; <₽э = —0,5 в (нас. к. а.); ю= 0,125 e/сек; Га — 3 мин.
ионизации пропорциональна концентрации Sb(III) в растворе.
Различные вольтамперометрические характеристики Sb(III) в ряде индифферентных электролитов исследованы в работах [233, 703]. Распределение в объеме ртутной капли сурь-
мы, выделяющейся в результате пред-электролиза, изучено в работах [247, 248]. Вследствие предвари-
тельного электролитического концентрирования предел обнаружения Sb удалось понизить до п-10~9 г-ион/л при использовании постояннотоковой полярографии и до 5>10~10 г-ион/л — переменнотоковой [116, 569].
При определении Sb методом инверсионной вольтамперометрии наиболее часто используют солянокислые фоны с концентрацией НС1 от 0,01Д7 [133, 293] до 4Л7 [243—245]. Для удержания Sb и
других элементов в растворе при использовании полярографических фонов с малой концентрацией НС1 используют добавки различных маскирующих реагентов, в том числе винной [67, 223, 290] и лимонной [290, 526] кислот. При определении Sb в кремнии в качестве фонового электролита рекомендована 0,2M HF [271]. Иногда используют смеси H2SO4 с НС1 [526], а при определении
з А. А. Немодрук
65
Sb в олове в качестве фона рекомендован 2,5М раствор H2SO4 в 50%-ном этаноле [221].
Показана возможность определения Sb методом инверсионной вольтамперометрии непосредственно в экстрактах, полученных после ее отделения экстракцией органическими растворителями [309—311, 1162]. Сурьму можно определять в среде некоторых неводных растворителей (например, в среде диметилсульфоксида) и ряда жидких кремнийорганических соединений, в которых можно одновременно определять также Ti и Nb [1503]. Рекомендовано применение смешанных растворителей [311, 875].
В зависимости от анализируемого материала, используемого фонового электролита, применяемого электрода и других условий предэлектролиз проводят при потенциале от —0,3 в [654] до —1,2 в [105]. Потенциал максимума анодного тока (в зависимости от состава фонового электролита) находится в пределах от —0,05 в [293] до -0,25 в [67, 243].
Методом переменнотоковой полярографии со стационарным ртутным электродом установлено [120], что при проведении пред-электролиза при потенциалах отрицательнее —1,0 в (ртутное дно) наблюдается резкое уменьшение тока ионизации Sb из амальгамы, что объясняется потерей части Sb вследствие образования SbH8, а также торможением разряда комплексного аниона SbCl4 на отрицательно заряженной поверхности электрода. Показано [161, 220, 531], что определяющим в данном случае является потеря Sb в виде SbH8, образующимся при потенциалах отрицательнее —0,8 в. Следовательно, для устранения образования SbHa и связанного с этим занижения результатов определения сурьмы необходимо проводить предэлектролиз при потенциалах не ниже -0,8 в.
На ионизацию Sb из амальгамы могут оказывать влияние электроосажденные совместно с ней в стадии предэлектролиза металлы с близкими потенциалами ионизации (Bi, Си) [239, 1378], а также интерметаллические соединения Sb, образующиеся при совместном электролизе с некоторыми металлами [240, 412]. Сурьма может образовывать интерметаллические соединения также и с материалом подложки в тех случаях, когда ртутные капельные или пленочные электроды получают с использованием, металлических подложек [289, 1219].
Первое из указанных затруднений устраняется предварительным отделением Sb от мешающих элементов выбором соответствующего потенциала предэлектролиза и полярографического фона. Второй источник помех (образование интерметаллических соединений с материалом подложки) может быть устранен путем использования стационарных ртутных электродов без металлических контактов.
Предварительное электрохимическое осаждение Sb может быть проведено не только на ртутных электродах, но и на поверхности твердых индифферентных электродов [86, 88]. Использование 66
твердых, преимущественно графитовых, электродов позволяет исключить применение металлической ртути.
При определении Sb методом инверсионной вольтамперометрии с применением твердых электродов возникают специфические осложнения, связанные с кристаллизацией ее на твердой электродной поверхности, что может отразиться на форме и величине регистрируемого аналитического сигнала. Кроме того, на поверхности твердых электродов, как правило, более сильно проявляется взаимодействие Sb с другими совместно электроосаждаемыми элементами [628] с образованием как интерметаллических [1498], так и химических [174, 531] соединений. На стадии предэлектролиза на твердых (платиновых и графитовых) электродах создаются условия для совместного осаждения с сурьмой Си [530], Se [531], Те [1498], Ан [529], Ag и Ni [1672].
Для определения Sb методом инверсионной вольтамперометрии весьма перспективно применение ртутно-графитовых электродов [270, 463—465, 525, 526, 533, 605, 628, 1065]. В отличие от стационарных ртутных электродов, для получения ртутно-графитового электрода не требуется каких-либо дополнительных операций, поскольку электрод образуется в процессе электролиза анализируемого раствора, в который вводится определенное количество соли Hg(II) [1065]. Применение ртутно-графитового электрода по сравнению с графитовым позволяет понизить предел обнаружения Sb практически на порядок и исключить образование интерметаллических и химических соединений Sb с другими элементами и тем самым устранить их мешающее влияние на определение Sb.
Ртутно-графитовый электрод имеет большое сходство с ртутным пленочным электродом, который нашел широкое применение в инверсионной вольтамперометрии [270]. Показано [525], что минимальное количество ртути, необходимое для определения Sb с применением ртутно-графитового электрода, равно 12-кратному по отношению к Sb. Малые количества ртути не оказывают заметного влияния на ионизацию электроосажденной Sb. При содержаниях Sb и Hg, близких к эквивалентным, образуются твердые растворы. Для устранения указанных осложнений и правильного определения Sb с применением ртутно-графитовых электродов рекомендуется вводить в раствор не менее чем 100-кратные количества Hg(II).
Проведенное сравнительное изучение условий определения Sb с использованием ртутного капельного, ртутного пленочного и ртутно-графитового электродов [891] показало, что для определения Sb на уровне п-10-7 — п-10-8 г-ион/л лучшим является ртутный капельный электрод. При определении Sb в присутствии Си, Bi, Se и Те применение ртутно-графитового электрода позволяет одновременно определять также указанные элементы [532, 1120].
Для определения Sb методом инверсионной вольтамперометрии может быть использована реакция электрохимического окисления Sb(III) до Sb(V), протекающая в растворах НС1 при потенциале
3*
67
Рис. 5. Поляризационные кривые ионизации гексахлоростибата родамина С, полученного электролизом в 1Л/ НС11 -10~? М родамина С
cSb(HI)' 10*а-ион/л: 1 — 0,5; 2 — 1,2;
3 — 2,0; (рэ = 0,8 в (нас. к. э);
v = 0.017 в/сек', Тг» = 5 мин.
0,80 ч- 0,85 в, поскольку графитовые электроды позволяют использовать область потенциалов положительнее потенциала ионизации Hg. При электроокислении Sb(III) до Sb(V) в растворах НС1, содержащих родамин С [87, 89] или малахитовый зеленый [940], на поверхности графитового электрода образуется пленка малорастворимого гексахлоростибата родамина С или малахитового зеленого, которые при последующей катодной поляризации электрода с осадком разрушаются с восстановлением Sb(V) до Sb(III). На полярограммах при потенциале — 0,35 в регистрируется пик тока электровосстановления (рис, 5) гексахлоростибата, высота которого пропорциональна содержанию Sb в растворе [85, 311, 891].
Для этой же цели предложено [536, 1605] использовать ряд других основных красителей. Предел обнаружения Sb этими методами составляет — 5 • 10—7 г-ион/л, что несколько хуже, чем при ее определении методом инверсионной вольтамперометрии с применением ртутных и ртутно-графитовых электродов; но так как подавляющее большинство других ионов в этих условиях электрохимически неактивно, то рассмотренные методы часто позволяют определять Sb в различных материалах без предварительного ее отделения.
Методы инверсионной вольтамперометрии находят широкое применение для определения Sb в различных материалах, в том числе в чугунах, железе и сталях [1348, 1575], меди и медных сплавах [87, 116, 526, 569, 1348, 1575,1585], олове [221, 222, 224, 225, 242, 318, 526], алюминии [131, 132, 731, 1503], галлии и его солях [243, 245, 293, 303], арсениде галлия [243, 245, 246, 303, 586], кад-.
мии и его солях [302, 318, 737], германии, тетрахлориде и тетрабромиде германия [105, 134], кремнии, двуокиси кремния, тетрахлориде и тетрабромиде кремния и трихлорсиланах [105, 133, 271, 310, 1503], цинке и цинковых сплавах [67, 737], серебре [605, 731], свинце [833], теллуре [116], мышьяке [303], хроме и его солях [940], барии [125], ртути [528], висмуте [1348], никеле и никелевых сплавах [590], припоях [1348], полиметаллических рудах и продуктах цветной металлургии [116], растворах гидрометаллургического производства [138, 319, 1545], шламах [1175], ниобии и тантале и их соединениях [223, 290], химических реактивах и препаратах [105], криолите [245, 586], материалах, используемых в электронной
68
технике [224, 654], сульфиде цинка [245, 586], сточных [232, 464] и природных [465, 1105] водах, фторидах магния и стронция [245, 586], растворах хлорида натрия [309], спирте [311].
Кулонометрические методы
Кулонометрические методы характеризуются очень высокой чувствительностью, а также хорошей воспроизводимостью и правильностью [257, 362]. Ошибка определения Sb находится в пределах 0,1—1% [351, 915]. Для определения Sb используются оба варианта кулонометрии — кулонометрия с контролируемым потенциалом и кулонометрическое титрование.
В варианте кулонометрии с контролируемым потенциалом для определения Sb используют два способа, основанных на восстановлении Sb на ртутном электроде [1041]. По первому способу Sb(V) предварительно восстанавливают гидразином до Sb(III), которую кулонометрически восстанавливают до металлической Sb на фоне IM НС1 + 0,4 М винной кислоты при потенциале —0,28 в. Метод предусматривает предварительную очистку фона электролизом при 0,0 в. При выполнении определения по второму способу электролитом служит смесь 6 М НС1 -ф- 0Л М винной кислоты, предварительно очищенная электролизом при 0,35 в. Далее определение проводят последовательно, восстанавливая Sb(V) и Sb(III) при —0,21 и —0,35 в соответственно. Количество Sb(III) находят по разности между найденным содержанием Sb(III) при восстановлении ее при —0,35 в и содержанием Sb(V). Определению Sb не мешают As(III) и РЬ(П); мешает Си.
Разновидностью кулонометрического определения Sb(III) при контролируемом потенциале является применение кулонометрического детектора [1583]. Действие детектора основано на электрохимическом окислении Sb(III) до Sb(V) на поверхности платинового электрода при 0,8 в. Этот метод использован для определения полноты отделения Sb методом ионнообменной хроматографии. Определению Sb этим методом мешают Ti, Се, Fe, Hg и Bi.
Значительно чаще для определения Sb используется кулонометрическое титрование. Описано определение Sb(III) кулонометрическим титрованием ионами МпО4, которые злектрогенерируются в 0,025 М MnSO4, 4 —7 TV по H2SO4 [350, 352]. Метод позволяет определять до 0,08 мкг Sb(III). При содержании Sb(III) 10 мкг применяют кулонометрическое титрование электрогенерированны-ми бромом [946, 1472], иодом [617, 915, 1287, 1651], гипогалогени-тами [1283] и бихроматом [351]. Конечную точку при кулонометрическом титровании Sb(III) наиболее часто устанавливают амперометрически [350, 1031, 1287] и биамперометрически [617, 946,1472]. Используются также потенциометрический, фотометрический и визуальный методы [1287]. В последнем случае в качестве индикатора применяют крахмал, образующий с избытком иода синее окрашивание.
69
Кулонометрическими методами определяют Sb в сплавах свинца и олова [617, 1041], органических соединениях и лекарственных препаратах [1410], стеклах [1651], сере и селене [3501, иодиде сурьмы [1559]. Кулонометрическое титрование использовано для ускоренного автоматического контроля содержания Sb в растворах [1474].
Амперометрическое титрование
Определение Sb амперометрическим титрованием обычно проводят, используя реакцию окисления Sb(III) до Sb(V) с платиновыми индикаторными электродами. Наиболее правильные и воспроизводимые результаты могут быть достигнуты при титровании Sb(III) на фоне 2М НС1 растворами КВгО3 или К JO3 при потенциале 0,4 в (меркуриодидный электрод) [675, 676]. Возможно также титрование на фоне — 2 М H2SO4, но в этом случае необходимо добавлять некоторое количество КС1 или NaCl. Кривые титрования состоят из двух отрезков: одного — горизонтального и второго — круто поднимающегося, соответствующего резкому подъему тока после достижения конечной точки, обусловленному тем, что избыток ВгО3 окисляет Вг“ или J- соответственно до Вг2 или J2, которые дают ток восстановления на платиновом индикаторном электроде. Титрованием броматом или йодатом калия можно определять от 5 до 800 мкг Sb(III) с ошибкой — 0,5 мкг при объеме титруемого раствора 25 мл и — 1 мкг при объеме 250 мл [1252]. Титрование проводят при 0,3 в с платиновым вращающимся электродом. Можно проводить титрование также с. применением двух индикаторных электродов при 0,015 в [278]. Применение КВгО3 или KJO3 в качестве титрантов позволяет определять Sb(III) в присутствии ряда элементов, в том числе Cu(II), Bi(III), Pb(II), Fe(III), Zn(II), Cd(II), Ni(II) [278, 944, 1028, 1087]. As(III) титруется вместе с Sb(III). Однако если проводить титрование в бикарбонатной среде с применением в качестве титранта К2СгО4, то в этих условиях Sb(III) окисляется полностью за 20—30 сек., тогда как As(III) реагирует с СгО4 значительно медленнее. Это различие использовано для раздельного определения Sb и As [17, 236].
Сурьму (III) можно определять амперометрическим титрованием с использованием в качестве титранта растворов солей Hg(II) на фоне 2М КОН с добавлением комплексона III. Индикацию ведут по току восстановления Hg(II) без наложения внешнего напряжения [137]. В присутствии Sn(II) наблюдается анодный ток, обусловленный окислением Sn(II) до Sn(IV) ртутью (II). Вслёдст-вие этого во время титрования анодный ток понижается, что может быть использовано для раздельного титрования Sn(II) и Sb(III) в их смесях. По окончании титрования Sn(II) ток остается постоянным до тех пор, пока титруется Sb(III), а после того, как оттитру-ется Sb(III) (вторая точка эквивалентности), ток возрастает [137]. As(III) титруется вместе с Sb(III). В некоторых случаях для ампе
70
рометрического определения Sb(III) ее предварительно окисляют добавлением раствора К2Сг2О7, избыток которого затем оттитровы-вают раствором соли Мора [214].
В качестве титранта предложен также JC1 [214]. С его применением Sb(III) титруют с использованием вращающегося платинового микроэлектрода. По окончании титрования Sb(III) хлорид иода восстанавливается на платиновом микроэлектроде на фоне СаС12, НС1, CHgCOONa или NaHCO3, образуя четкую волну восстановления при потенциалах от +0,2 до —0,2 в, высота которой пропорциональна концентрации JC1 в растворе. Лучшие результаты получены при титровании Sb (I II) на фоне 0,1М тартрата натрия, подкисленного НС1 при 0,1 в.
Хлорамин Т образует необратимую каталитическую волну восстановления на платиновом электроде как в кислых, так и в слабощелочных фонах (NaHCO3). При переходе от кислых растворов к щелочным волна восстановления хлорамина Т смещается в область отрицательных потенциалов (от 0,0 до —0,45 в). Однако высота волны восстановления хлорамина Т в обоих случаях пропорциональна его концентрации. Вследствие этого по окончании титрования Sb(III) ток резко возрастает, что обеспечивает установление четкой конечной точки и получение хорошо воспроизводимых результатов [750].
Гипобромитом натрия титруют Sb(III) на фоне NaHCO3 (pH 8,2) с применением вращающегося платинового микроэлектроДа; при этом ВгО~ дает волну восстановления при 0,3 в. В качестве титранта для амперометрического титрования Sb(III) предложен также Ce(IV) [1674]. Сурьму(Ш) можно определять амперометрическим титрованием с двумя поляризованными платиновыми электродами растворами Вг2 в СН3СООН [1421].
Кроме титрантов-окислителей, для амперометрического определения Sb рекомендовано применение ряда реагентов, образующих с ней нерастворимые или комплексные соединения. Тионалид образует прочное нерастворимое соединение с Sb(III), что использовано для ее амперометрического определения [724]. Метод позволяет определять Sb(III) в присутствии 5-кратных количеств Си и As, 200-кратных — РЬ и Fe. Показана возможность использования карбоксиметилдитиокарбамината кальция для амперометрического титрования Sb(III) [566], образующего с ней в кислой срёде (от pH 4 до 3 A" H2SO4) прочное растворимое соединение. Точку эквивалентности устанавливают по току анодного окисления реагента. Различие в устойчивости карбоксиметилдитиокарбаминатов Sb(III) и Bi(III) использовано для раздельного их определения при взаимном присутствии.
Хорошими реагентами для амперометрического титрования Sb оказались некоторые производные димеркаптотиопирона, образующие с ней нерастворимые соединения [696, 697]. Так, амилдимеркаптотиопирон (H2R) образует с Sb(III) соединение состава Sb(HR)3, нерастворимое в сильных кислотах. Титрование ведут
71
на фоне 3 N НС1 при 0,7 в с графитовым электродом по анодному току окисления избытка реагента. При определении 10—600 мкг Sb в растворе объемом 40 мл получены вполне удовлетворительные результаты [697]. Определению Sb(III) этим методом мешают Сп(П) и As(III), которые титруются вместе с Sb(III). Однако Sn и РЬ не мешают. Исследовано 12 производных димеркаптотиопирона, образующих с Sb(III) нерастворимые в кислотах осадки, в качестве реагентов для амперометрического титрования Sb(III) [696]. Лучшие результаты получены с применением 2,6-димеркапто-3,5-диэтил-1,4-тиопирона. Титрование ведут с применением графитового микроанода по анодному току окисления реагента при 0,7 в. Титрование Sb возможно на фоне H2SO4 и НС1 в широких пределах их концентраций. Точка эквивалентности фиксируется при соотношении Sb: реагент = 1:3. Поскольку Se(IV) и Te(IV) образуют более прочные соединения, чем Sb(III), то это различие использовано для раздельного определения Sb(lII) в ее бинарных смесях с Se(IV) и Te(IV). Ошибка определения каждого элемента 3%. Метод позволяет определять Sb в растворах с ее концентрацией 2,5 мкг/мл.
Методы амперометрического титрования Sb(III), основанные на образовании прочных нерастворимых и комплексных соединений с органическими реагентами, характеризуются низкими пределами обнаружения (до 10 мкг Sb в пробе), однако по точности (ошибка — 3%) уступают амперометрическому титрованию броматом калия (ошибка —0,1%).
Амперометрическим титрованием Sb определяют в сурьмяно-свинцовых и сурьмяно-свинцово-оловянных сплавах [944], полупроводниковых материалах, содержащих In, A], Se и Те [696], свинцово-оловянных припоях [697], свинцовых рудах и пылях свинцового производства и в водных растворах, полученных после выщелачивания этих пылей [236], типографских [566] и медноцинковых сплавах [1087].
Радиоактивационные методы
Радиоактивационные методы определения сурьмы основаны на регистрации излучения радиоактивных изотопов, образующихся в результате протекания соответствующих ядерных реакций с участием нерадиоактивных изотопов Sb, содержащихся в природной смеси. Подробно теория активационного анализа и применяемая аппаратура описаны в работах [159, 374, 485, 738, 839, 1371, 1423, 1509]. Активационные методы определения Sb характеризуются очень высокой чувствительностью и позволяют одновременно с Sb определять ряд других элементов. Для анализа, как правило, требуется небольшое количество анализируемого материала, которое в ряде случаев может быть 1 мкг. Существенным недостатком активационных методов является необходимость наличия малодоступного источника активации и тщательного соблюдения специ
72
альных условий работы с радиоактивными веществами, обладающими чрезвычайно высокой токсичностью.
Из всех активационных методов для определения Sb наиболее часто используются нейтронно-активационные методы. Для определения Sb анализируемый материал облучают потоком от 1011 [1203] до 1014 нейтрон/см? • сек [488]. При этом в природной смеси изотопов Sb, содержащей 57,25% 121Sb и 42,75% 128Sb, протекают следующие ядерные реакции:
изомерный переход
121Sb («, v) 122mSb —--—- 122Sb (Т1/2 = 2,8 дня),
(J 1уа=3,4 мин.) '*
128Sb (п, у) i2*Sb (Т1/2 = 60 дней).
Оба образующихся при этом радиоактивных изотопа, 122Sb и 124Sb, используются для определения Sb (некоторые их характеристики приведены в табл. 1).
При облучении пробы потоком 1014 нейтрон!см2-сек в течение 24 час. метод позволяет определять до 1 нг Sb [1131]. При определеиии Sb в висмуте облучением пробы (2 г) потоком 4Л011 нейтрон!см2-сек в течение 15 дней с последующим измерением у-активности изотопа 12,Sbno методу у, у-совпаде-ний предел обнаружения составляет 0,1 иг Sb [932]. При использовании быстрых нейтронов пробу перед облучением, кроме алюминиевой фольги или полиэтиленовой пленки, завертывают в кадмиевую фольгу [1109] или помещают в кадмиевый контейнер [949].
Продолжительность облучения пробы зависит от природы анализируемого материала, интенсивности используемого потока нейтронов и содержания Sb и может колебаться от нескольких долей минуты [1296, 1409] до 1 — 2 недель [945, 1002, 1239]. После облучения пробе дают остыть необходимое время для разложения короткоживущих изотопов, затем обычно протравляют ее для удаления поверхностных загрязнений.
Наиболее быстрыми, производительными и простыми являются недеструктивные инструментальные методы, основанные на у-спектрометрическом определении Sb в облученных образцах без выделения образовавшихся ее радиоактивных изотопов. Однако чувствительность определения Sb этими методами на 1—2 порядка ниже по сравнению с методами с выделением и радиохимической очисткой радиоактивных изотопов Sb. Кроме того, методы недеструктивного анализа непригодны для определения Sb в материалах с низким ее содержанием в присутствии элементов, которые образуют по реакции (н, у) изотопы с близкими энергиями у-квантов и периодами полураспада к энергиям у-квантов и периодам полураспада радиоактивных изотопов 122Sb и 124Sb. Тем не менее недеструктивный вариант вследствие малой трудоемкости и высокой производительности используется для определения Sb во многих материалах.
.73
Спектрометрия по у, ^-совпадениям использована для определения Sb в свинце [835] с применением сцинтилляционного гамма-спектрометра.
Образец (1—3 г) и стандарты помещают в кварцевые контейнеры и облучают в течение 2—10 дней в реакторе потоком 1,4-1012 нейтрон!см2-сек. Облученные пробы и стандарты выдерживают 4—5 дней и измеряют активность изотопа 124Sb по у-пикам 0,603 и 1,692 Мэе. Предел обнаружения 1-10-3 мкг Sb; при содержании Sb 1,5-10_®% ошибка составляет 20%, а при ее содержании 2,1-10-5% ошибка снижается до 10%.
Для определения Sb, Na, Си и As в полупроводниковом кремнии [1217] применен Се(Ы)-детектор в сочетании с 4096-канальным у-спектрометром и ЭВМ. Эта система проводит регистрацию, сглаживание и вычитание у-спектров и выдает полученные результаты в виде отпечатанных таблиц и у-спектра.
Предел обнаружения Sb недеструктивным активационным методом в сильной мере зависит от природы анализируемого материала и выбранной методики (интенсивность потока нейтронов, продолжительность облучения и остывания, величина навески, характеристика радиоизмерительной аппаратуры) и колеблется от 3-Ю-2 [836] до 2-10-9% [625]; абсолютное количество Sb, которое может быть определено, составляет 1-10-9г [625, 1061], а иногда даже 5 • 10-11 г [1239].Ошибка определения Sb, в зависимости от условий и содержания Sb, может колебаться от 0,7 [466] до 20—25% [989]. В ряде работ [427, 932, 1217, 1468, 1505, 1506, 1652] для достижения большей точности результатов анализа и учета влияния других элементов рекомендуется получаемые данные обрабатывать с помощью ЭВМ по соответствующим программам.
Недеструктивный активационный метод применяется для определения Sb в алюминии [841, 1688] и его сплавах [945], нитриде алюминия [421], аскорбиновой кислоте [1630], асфальте [982], висмуте [830, 1204, 1239] и его сплавах с сурьмой [48, 313], воздушной пыли [884, 13131, галените [21], германии [633, 1384, 1385], горных породах [230, 427,541, 949, 1061, 1289], графите [106, 1207], железе, чугуне и стали [135, 884, 1128, 1129, 1556, 1652], индии [1271J, карбиде кремния [468], кремнии [212, 762, 932, 950, 989, 1217, 1361], тетрахлориде кремния [1462] и эпитаксиальных слоях кремния [580], медй (1002], морских [642, 1427] и природных водах [4, 1040], нефти и нефтепродуктах [991, 1517], олове [1305], полифенолах [983], почвах [1528], растительных материалах [1316, 1528], рудах [466, 1270], свинце [835 -837, 1205, 1505, 1506], стандартных образцах металлов [1316], теллуре [5], титане [68], хроматографической бумаге [1409], циркалое [1099], эммитерных сплавах [625], трифенилах [8771 и фториде лития [331]. Благодаря высокой чувствительности и вследствие того, что для анализа, как правило, требуется небольшое количество анализируемого материала, эти методы часто используются в криминалистической практике [884, 892, 12961. Имеются указания [965] ®б использова-
74
иии нейтронно-активационного метода для проточного контроля состава производственных растворов на содержание в них Sb, As, Си, F, Au, Na, Ti, Cl, Al, Вг, Co, Eu, Hf, Zn, Мп, Ag и V.
Кроме тепловых нейтронов, для активации могут использоваться также быстрые нейтроны без замедления. В этом случае протекает ядерная реакция с образованием радиоактивного изотопа ’20Sb : 121Sb (п, 2 п) 120Sb (Т>/г 16,6 мин.).
Как уже указывалось выше, педеструктивный активационный метод не всегда может быть применен, а в тех случаях, где его применение возможно, ошибка определения Sb, как правило, выше, чем в методах с выделением и радиохимической очисткой образовавшихся изотопов Sb. Для выделения Sb из облученного материала могут использоваться все методы, описанные в главе V.
Для количественного выделения радиоактивных изотопов Sb в качестве носителя наиболее часто применяют природную смесь стабильных изотопов Sb, добавляя ее в виде растворимого соединения к облученной пробе в достаточных количествах (5 —50 мг).
Это обеспечивает практически полное выделение даже субмик-рограммовых количеств радиоактивных изотопов Sb. Однако полное выделение их не обязательно, если определить их выход. Для его определения находят количество выделенной аналитической формы (взвешиванием, титрованием или другим подходящим методом) и оценивают его по отношению к тому количеству, которое могло быть получено в случае полного выделения введенного носителя. В ряде работ [1092, 1312, 1660] описаны методы разделения элементов, рекомендуемые для применения в активационном анализе.
Активационные методы определения Sb не требуют использования реактивов особой чистоты и для них значение холостого опыта не имеет смысла. Загрязнения, которые могут иметь место на поверхности анализируемого материала, устраняются протравливанием после облучения пробы. Это преимущество активационных методов, наряду с высокой чувствительностью и надежностью, обеспечило им широкое применение, особенно в анализе материалов высокой чистоты и в тонких научных исследованиях.
Активационные методы с выделением и радиохимической очисткой образовавшихся изотопов Sb используются для ее определения в алюминии [639—641, 912, 1235, 1247, 1376, 848] и трехокиси алюминия [639], боре и нитриде бора [426], бериллии [523], ванадии и пятиокиси ванадия [145], висмуте [1204, 1659, 1660], вольфраме [144], галлии [1375] и арсениде галлия [640, 824, 825, 831, 1375], германии [610, 639, 640], горных породах [74, 449, 1276, 1554], железе, стали и чугуне Г987, 1033, 1113, ИЗО, 1280, 1590, 1653], железных метеоритах [1539], золоте [1676], индии [828, 829] и арсениде индия [115], каменных метеоритах [1136, 1234, 1236,1515], кремнии [38, 39, 275,282, 455,639, 640, 861, 1035, 1144, 1355, 1473, 1492, 1540, 1687], двуокиси кремния и кварце [282—285, 487, 639, 640], карбиде кремния [38, 276, 639, 640],
75
трихлорсилане [1342, 1461], трихлорметилсилане [38], тетрахлориде кремния [403, 640], стекле [1107], литии и его соединениях [50, 524], магнии [874] и его сплавах [1597], меди [1034, 1286,1675], мышьяке [39], никеле Г1000, 1001], олове Г1035], осмии [708], палладии [152], платине [1236] и ее сплавах с родием [488], свинце [144, 488, 907, 1206] и его сплавах [1457], селене [250, 772, 874, 1104], сере [771], серебре [1676], таллии [19, 604], теллуре [251, 772], титане и двуокиси титана [1380, 1381], углероде и графите [639, 640, 1203, 1330], в черном порохе [844], фосфоре [826, 885, 1093], цинке [827,1272, 1488] и его солях [1012] и цирконии [1070], в различных биологических материалах [1312, 1415, 1510], пищевых продуктах [1415], удобрениях [1402], органических материалах [1238, 1407, 1510], трифениле [1407], пластмассах [1034], нефтях и нефтепродуктах [1402, 1408], продуктах деления [1124], эпитаксиальных пленках кремния [162, 578], электролитах [561], реактивах [642, 1501], азотной и соляной кислотах [1339, 1517], перекиси водорода [1339], дистиллированной и деионизованной воде [639], ледниковом льде [1636], сточных, поверхностных и морских водах [642, 947, 1501]. Эти методы находят также применение в криминалистике [1487] и при изучении старинных произведений искусства [1299].
Иногда используется предварительное концентрирование Sb при ее активационном определении. Так, например, при определении Sb в морской воде предложено [1359] предварительно выделять ее хроматографическим методом с применением читозана в качестве сорбента.
Кроме нейтронно-активационных методов, для определения Sb используются также фотоно-активационные (у-активационные) методы [355, 356, 375, 865, 1263]. В работе [375] обсуждены возможности у-активационного определения отдельных элементов при помощи бетатрона с внутрикамерным облучением; предел обнаружения Sb составляет ~1 мкг. у-Активационный анализ для определения Sb имеет значительно меньшее значение, чем нейтронно-активационный. Наиболее перспективными областями его применения является массовый анализ проб на отдельные элементы со сравнительно высоким их содержанием и материалов с относительно простым составом. Инструментальный у-активационный анализ используется для определения Sb в воздухе [865], в сурь-мяно-циркониевом [356] и сурьмяно-фосфатном [355] ионообмен-никах.
Радиометрические методы
Из радиометрических методов определения Sb заслуживают внимания методы, основанные на измерении интенсивности обратно-рассеянного |3-из л учения, позволяющие производить анализ без разрушения образца. Этот метод применен для определения Sb в припоях [1263], фармацевтических препаратах [1497], цветных металлах [1194].
76
К радиометрическим методам определения Sb относится также метод изотопного разбавления с применением радиоактивных изотопов Sb [14, 18, 846]. Сочетание этого метода со субстехиометрической экстракцией успешно используется для определения Sb в различных материалах. В работе [253] Sb экстрагируют из раствора НС1 (1 : 9) толуолом в виде ионного ассоциата SbClg с катионом метилового фиолетового, взятом в субстехиометрическом количестве по отношению к Sb. Сурьму определяют также субстехиометрической экстракцией в виде купфероната Sb(III) [1686]. Для повышения избирательности определения сначала Sb(III) окисляют до Sb(V) и экстрагируют мешающие элементы в виде купферо-натов, затем восстанавливают Sb(V) до Sb(III) и в тех же условиях повторяют экстракцию. Метод субстехиометрической экстракции Sb(III) в виде купфероната применен для ее определения в висмуте и сурьмяно-висмутовых сплавах [770].Субстехиометрическая экстракция дихлорэтаном или хлороформом сурьмы в виде гексахлоростибата тетрафениларсония применена для определения ее в горных породах активационным методом. Использование субстехиометрической экстракции позволяет определять Sb без дополнительных химических операций.
ФИЗИЧЕСКИЕ МЕТОДЫ
Эмиссионный спектральный анализ
Эмиссионный спектральный анализ в настоящее время является одним из наиболее широко используемых методов определения малых содержаний Sb в металлах и их сплавах, горных породах, рудах, веществах высокой чистоты, полупроводниковых и многих других материалах [227, 287, 314, 369, 380, 398, 442, 635, 637, 681—683, 807]. Теоретические основы эмиссионного спектрального анализа изложены в ряде руководств и монографий [209, 226, 349, 709, 936]. Основными преимуществами эмиссионного спектрального анализа являются универсальность, высокая чувствительность и вполне удовлетворительная точность. Большая производительность и экономичность делают его незаменимым при массовых анализах однотипных проб, особенно с использованием современных приборов с фотоэлектрической регистрацией спектров [501, 710]. К числу достоинств спектрального метода следует также отнести в большинстве случаев малое количество вещества, необходимое для проведения анализа, составляющее иногда сотые доли грамма.
Эмиссионный спектр Sb состоит из нескольких тысяч линий [228, 748]. Некоторые характеристики аналитических линий Sb приведены в табл. 4. Наиболее чувствительные линии Sb расположены в УФ-области спектра: 206,833 и 217,588 нм. Однако их использование обычно затруднено в связи с отсутствием соот-
77
Таблица 4
Аналитические линии сурьмы, наиболее часто используемые в эмиссионном спектральном анализе
Длина волны в воздухе, нм Энергия возбуждения линий, эв Интенсивность линий
в угольной дуге в искре (разряде) в медной дуге
326,751 5,8 159 150 85
323,252 6,1 150 250 100
287,792 5,4 259 159 140
259,806 6,0 200 100 600
252,852 6,1 300 R 200 320
231,147 5,4 150 R 59 45
217,588 5,7 300 40 38
206,833 6,0 300 R 3 55
Примечание. В — самообращающаяся ЛИНИЯ.
ветствующих фотопластинок. Поэтому в аналитической практике обычно используют менее чувствительные линии, в том числе линии 287,792; 259,806; 252,852 и 231,147 нм. При работе на спектрографе средней дисперсии необходимо учитывать, что на линию 259,806 нм накладывается сильная линия Fe 259,837 нм', линия Sb 231,147 нм практически свободна от наложений линии Fe. В табл. 5 указаны линии других элементов, которые накладываются на линии Sb 287,792; 259,806 и 252,852 нм.
Пределы обнаружения Sb в дуге постоянного тока при работе по этим линиям составляют в зависимости от материала основы п-10~8 — п-10~4%. При определении повышенных содержаний Sb (п-10-1 — п%) обычно используют менее чувствительные линии, например линии 302,981 и 326,750 нм, которые практически свободны от наложений линий большинства других элементов.
В качестве источников возбуждения спектра используют дугу постоянного и переменного тока, искру и разряд в полом катоде. Непосредственное введение растворов в источник возбуждения было одним из первых способов возбуждения спектра для аналитических целей. В табл. 6 приведены данные по чувствительности определения Sb в растворах в зависимости от способа введения пробы в разряд [226].
Из табл. 6 видно, что наиболее перспективным для определения Sb в растворах является метод вращающегося электрода при возбуждении спектра высоковольтной дугой. Использование высоковольтной дуги переменного тока в качестве источника возбуждения спектра позволяет изменением режима разряда варьировать чувствительность определения Sb в широких пределах: от величин, превосходящих чувствительность с использованием дуги по
78
Таблица 5
Спектральные линии других элементов, накладывающиеся на наиболее часто используемые линии сурьмы
Линии сурьмы и накладывающиеся на них линии других элементов, нм Интенсивность линий Линии сурьмы и накладывающиеся на них линии других элементов, нм Интенсивность линий
в дуге в искре в дуге в искре
SbI 287,792 250 150 Rh 259,807 4 3
Tu 287,821 3 20 Nb 259,804 2 10
Та 287,820 4 15 Fell 259,803 4 4
Nb 287,816 2 — Ir I 259,800 2
W 287,808 4 8 Re 259,796 20
Ru 287,804 — 15 W 259,795 12
V 287,802 2 10 Fe II 259,794 3
Се 287,802 2 — Ir 259,786 3
Ст 11 287,798 30 100 Fe 259,783 6 ——
Са 287,791 1 4 Ru 259,779 20
Ей 287,789 2 1 Nb 259,775 15
Dy 287,788 2 1 Rh 259,707 3 150
Pd II 287,787 — 15 SbI 252,852 300 R 200
Nb 287,785 2 4 Си II 252,930 600
Ru I 287,784 3 1 V 252,884 25 150 R
U 287,783 2 2 Cs 252,880 (8)
Eu 287,776 5 — U 252,877 2 5
Си 11 287,769 5 20 Ru 252,872 30
V 287,769 15 100 R Ar II 252,871 5
Ta 287,769 15 80 Mn 252,870 12 2
Ir I 287,768 20 10 Coll 252,862 4 200
Nb 287,762 — 5 Lu 252,859 5
Pt II 287,752 40 200 Ta 252,859 4 1
Till 287,744 30 100 Si I 252,852 400 500
Fel 287,730 200 125 Ball 252,851 50
Nb 287,695 40 500 Dy 252,849 4
SbI 259,806 200 1С0 Xell 252,849 (3)
Mn 259,890 5 100 V 252,847 50 150 R
Nb 259,888 150 Ir 252,839 10
Си II 259,881 —— 200 Th 252,836 -- 5
Fe 259,837 700 1000 Ar 252,833 (Ю)
Ir I 259,828 8 — Yb 252,833 _ 3
Ce 259,825 5 — Ce 252,829 15 1
Ta 259,821 2 50 CrI 252,824 30
Mn 259,817 12 — V 252,790 35 300 R
W 259,811 6 —
Примечание. R — самообращающаяся линия, в круглых скобках указаны приблизительные значения.
79
Таблица 6
Нижние границы определяемых содержаний сурьмы в растворах по линии 259,806 нм
Метод анализа Способ возбуждения сн-10<, % Метод анализа Способ возбуждения Сн-10*. %
Вращающийся а 1,0 Вращающийся а 2,5
электрод б 1,0 ДИСК б 25
Медная искра а 5 в 5
б 10 Пористый элект- а 10
а 8 род б 25
Фильгуратор а 2,5
Примечание, а — высоковольтная дуга; б — конденсированная искра, искровой режим; в — конденсированная искра, дуговой режим.
стоянного тока [718], до весьма низких значений, при которых можно определять высокие концентрации Sb без предварительного разбавления. Наклон градуировочного графика увеличивается по мере уменьшения тока [334], поэтому используемый диапазон тока обычно находится в пределах 1—5 а. В этих условиях получают достаточно большие наклоны градуировочных графиков. Кроме того, для высоковольтных дуг снижается реабсорбция [601].
Установлено [630], что разряд в полом катоде в аксиальном магнитном поле может быть успешно использован в качестве источника возбуждения при определении Sb и элементов с низкими потенциалами возбуждения. Предел обнаружения Sb с применением полого катода с наложением магнитного поля на порядок лучше, чем без магнитного поля.
При определении Sb в твердом материале наиболее низкие пределы обнаружения достигаются в разряде в полом катоде или наиболее широко распространенной угольной дуге постоянного тока при помещении навески анализируемого материала в полость электрода. Снижению пределов обнаружения Sb в различных материалах способствует правильный выбор осветительной системы, спектрографа и фотоэмульсии. От осветительной системы как части спектральной установки требуется, чтобы она не ограничивала максимально возможный световой поток через апертуру спектрографа. Этому требованию отвечает из простых схем только прямое отображение источника на щель спектрографа (однолинзовая система освещения). Спектрограф должен одновременно обеспечить: высокое отношение интенсивностей линии и фона и достаточно высокий для фотографической регистрации уровень освещенности в фокальной плоскости камеры. Таким образом, он должен обладать как можно большей разрешающей силой (до предела, заданного естественной шириной линии) при достаточно
80
большой светосиле. Из отечественных приборов этим требованиям в полной мере отвечает спектрограф типа СТЭ-1.
Прямое определение Sb в сочетании с рядом других элементов производится в самых разнообразных материалах, в том числе в алюминии [54, 55, 1134], бериллии и его соединениях [305, 1297], боре [778, 1117] и фосфиде бора [26], ванадии и его окислах [234, 491, 1117], висмуте [809, 909, 1134], вольфраме и его соединениях [195, 739, 795, 1265], вольфрамовых рудах [1480], германии и его соединениях [559, 634, 905], горных породах [386, 730, 1182, 1240, 1336, 1443, 1599], графите и углероде [235, 397, 612], жаропрочных и тугоплавких сплавах [176, 177, 379, 1278, 1593], железе [425, 1134, 1441], железных рудах и минералах [198, 386, 636, 971, 1336], сталях [176, 546, 1278, 1441, 1593] и чугуне [61, 274, 546, 1250], золоте [404, 754, 909, 10951 и его сплавах [196, 389,390, 1167], индии [1168, 1308] и сплавах на его основе [814, 815, 1267], иттрии и его окислах [234, 272], алюмоиттриевом гранате [82], кадмии [598, 599, 1134] и кадмиевых сплавах [819], кобальте [60, 153, 1134], кремнии [252, 1619], кварце [154], карбиде кремния [109, 110, 288, 789, 790, 1353], кремниево-медных сплавах [594], силикатах [1586], технических стеклах [612, 1579], меди [129, 482, 964, 997, 1176, 1599, 1609, 1645, 1654], медных сплавах [96, 482, 1048, 1188, 1457,1463, 1566], окиси меди [199], продуктах медеплавильного производства [360] и медных электролитах [1298, 1600], молибдене и его соединениях [104, 237, 308, 795, 1325, 1347, 1443], мышьяке [472, 1134], никеле и никелевых сплавах [486], ниобии и его окислах [49, 972], олове [582, 744, 782, 812, 900, 1684] и его сплавах [1210, 1494, 1495], полупроводниковых материалах [668, 678, 806, 1298, 1684], припоях [210, 1101], свинце [481, 534, 908, 1154, 1155,1193, 1543,1655], свинцовых сплавах [126, 871], рудах [53, 667, 806, 1143] и пылях [811], РЗЭ и их окислах [234, 353], селене [154, 155, 499, 747, 818, 1134], селениде ртути [715], сере [189, 1134], серебре [388, 390, 391, 909, 1598], хлориде серебра [1362], стеклоуглероде [397], сульфидных рудах [638], тантале [237], теллуре [156, 591, 592, 1134, 1613], теллуровом баббите [1656] и теллуриде свинца [342], типографских сплавах [323], титане и двуокиси титана [288, 306, 1262], тории и его окислах [272], уране [1447], окислах урана [878, 1182, 1240] и урановых рудах [1443], ферросплавах [792, 793], фосфоритах [879], хроме [555, 729, 792] и его окислах [54, 55, 571], цинке [976] и цинковых рудах и минералах [1142], цирконии [679] и двуокиси циркония [1368], производственных растворах [205, 882, 1290, 1323, 1324, 1483], сточных и природных водах [429], азотной, серной, соляной, уксусной, фтористоводородной и бромистоводородной кислотах [111, 121, 407, 552, 574, 1008], воздушной пыли [12].
Прямой спектральный анализ широко применяется в экспрессных и автоматических методах контроля производства, в геологических, геохимических и гидрохимических исследованиях, для
81
контроля окружающей среды, в криминалистике и т. п. Используя различную упругость паров основного соединения и избыточного компонента, спектральный метод позволяет определять сверхстехиометрические количества одного из компонентов. Так, например, определяют незначительные сверхстехиометрические количества Sb в антимониде индия [629].
Однако в ряде случаев чувствительность прямого эмиссионного спектрального анализа бывает недостаточной, в частности для контроля производства веществ высокой чистоты. В таких случаях проводят предварительное концентрирование Sb. Наиболее простыми, удобными и быстрыми методами концентрирования примесей Sb являются физические методы, в частности методы отгонки (дистилляции) Sb в вакууме, на воздухе и в токе газа-носителя. Однако такие методы применимы только к материалам, основу которых составляют элементы и их соединения, причем их летучесть значительно ниже летучести Sb. Применение концентрирования методами дистилляции примесей требует тонкого измельчения анализируемого материала, поскольку скорость диффузии отгоняемых примесей в твердой фазе мала. Тонкоизмельченную пробу нагревают током большой силы в графитовом стаканчике, зажатом между графитовыми щеками охлаждаемых водой медных электродов. Пары выделяющихся примесей конденсируются на охлаждаемой графитовой или металлической капсуле, которая затем используется в качестве электрода дуги или искры при последующем спектральном определении Sb и ряда других выделившихся вместе с ней примесей.
Применение предварительного концентрирования Sb путем ее отгонки с целью достижения более низких пределов ее обнаружения методом эмиссионного спектрального анализа рекомендовано для определения Sb в чистой Fe2O3 [198], карбиде кремния [288, 789, 790], кремнии [252] и кварце [553], двуокиси титана [288], трехокиси вольфрама [195] и вольфраме после его окисления до трехокиси нагреванием при 1800 °C [795], молибдене и трехокиси молибдена [27, 795, 796, 1443], тантале [237], ниобии и тугоплавких сплавах на основе ниобия, вольфрама и молибдена [379].
Однако методы концентрирования отгонкой неприменимы для определения Sb в материалах, основу которых составляют элементы и соединения, недостаточно отличающиеся по летучести от Sb. Кроме того, метод отгонки несколько сужает число одновременно определяемых элементов, ограничивая их только теми, которые испаряются при выбранных условиях. Поэтому часто, особенно в случае анализа веществ высокой чистоты и полупроводниковых материалов, производят предварительное концентрирование Sb.
Выбор метода концентрирования зависит от природы анализируемого материала, а также от природы и числа определяемых примесей и’ряда других факторов. В связи с этим все методы предварительного концентрирования Sb и совместно с ней определяе-82
мых других примесей можно разделить на две группы: методы, основанные на отделении основы, и методы, основанные на выделении примесей. Воспроизводимость определения Sb с предварительным концентрированием, как правило, находится в пределах 15—35% [383, 727, 761, 780J. Предел обнаружения Sb с использованием концентрирования регулируется величиной навески и аппаратурными возможностями и ограничивается чистотой используемых реактивов, посуды и электродов, а также чистотой рабочей атмосферы и достигает п-10-7% [62, 383, 727] и даже ниже; обычно он находится в пределах п-10-6 — п-10~6 % [58, 80, 669, 717, 780, 1007, 1157, 1158, 1260].
Методы концентрирования, основанные на отделении основы. Эта группа методов, как правило, позволяет определять не только Sb, но и многие другие примеси, так как удаляется только основа. Из всех методов концентрирования, основанных на удалении основы, наиболее часто используется экстракция. Таким образом определяют Sb в уране, который селективно экстрагируют из 5— 6 М HNO3 трибутилфосфатом, и в оставшемся водном растворе после его выпаривания наряду с Sb определяют еще 23 элемента [493]. При определении Sb в сталях и чугуне Fe предварительно экстрагируют диэтиловым эфиром из раствора НС1, в водный раствор вносят угольный порошок и соль Bi (внутренний стандарт), выпаривают досуха и спектрографируют [546]. При определении Sb в ниобии и его соединениях Nb экстрагируют 60%-ным раствором ТБФ в бензоле из ЮМ H2SO4 [378]. Для определения Sb в селене рекомендовано удалять Se экстракцией раствором дитизона в СС14 [12001, а при определении Sb в золоте последнее отделяют экстракцией 2,2'-дихлордиэтиловым эфиром из 6Л/ НС1 [547]. Для определения Sb в кадмии его отделяют экстракцией СНС13 в виде пиридин-иодидного комплекса [727]. При определении Sb в трихлориде мышьяка последний удаляют экстракцией из растворов НС1 ж-ксилолом [669] или бензолом [384].
Вторыми по практической значимости являются методы концентрирования, включающие удаление основы селективной отгонкой или испарением. Таким методом концентрируют Sb и другие примеси для их последующего спектрального определения в германии, пленках германия и двуокиси германия, отгоняя Ge в виде GeCl4 после растворения пробы в смеси НС1 и HNO3 [112, ИЗ, 460—463], а также в самом тетрахлориде германия [ИЗ]. В кремнии, двуокиси кремния и кварце концентрируют примеси путем обработки пробы смесью HF и H2SO4, вследствие чего удаляется Si в виде SiF4 [339, 572]; в тетрахлориде кремния и трихлорсилане примеси концентрируют выпариванием основы [339, 573, 1620]. Выпаривание является наиболее простым способом концентрирования Sb и других примесей при их определении в водах и водных растворах с малым содержанием минеральных солей, а также в соляной, азотной, серной, уксусной, хлорной и в других кислотах. Иногда для определения примесей в растворах их упа-
83
ривают до малого объема и концентрат наносят на тонкие угольные диски и высушивают, а затем эти диски используют в качестве электродов [656]. При определении Sb и других примесей в мышьяке пробу растворяют в смеси НС1 с Вг2 и отгоняют As в виде AsCl3 и АаВг3 [669, 670] или окисляют мышьяк до As2O3, который затем удаляют возгонкой [472]. Селен рекомендуется удалять возгонкой в виде SeO2 [157]. Цинк испаряют при 500° С в вакууме [780, 781]. Аналогично может быть удален Т1 [781], J [860, 1098, 1629]. Примеси Sb и других элементов в легко летучих веществах, таких, как органические растворители и т. п., рекомендуется концентрировать путем удаления основы выпариванием [577]. В ряде случаев для предотвращения потерь Sb и некоторых примесей в виде летучих комплексов с этими органическими веществами выпаривание проводят в присутствии Н2С2О4, образующей с Sb и многими другими элементами более прочные нелетучие комплексы [554].
Методы концентрирования примесей, включающие удаление основы путем ее полного или частичного осаждения, также часто используются в спектральном анализе. Таким путем концентрируют Sb и другие примеси в свинце, его сплавах и соединениях. В одном из методов пробу растворяют в HNO3, раствор упаривают до малого объема; выпавший при этом Pb(NO3)2 отделяют, а маточный раствор упаривают досуха, прокаливают, взвешивают (для определения коэффициента обогащения, который необходим для расчета содержания Sb и других примесей в анализируемом материале) и спектрографируют [726]. В другом методе пробу свинца растворяют в HNO3 и РЬ осаждают в виде хлорида [1260] или сульфата [80]. Th рекомендуется выделять осаждением в виде оксалата или пероксида [307], Те — в виде Н2ТеО3 [728]. Описан метод концентрирования Sb и других примесей в меди, включающий выделение основной части Си электролизом. Электролит отделяют, выпаривают досуха, сухой остаток прокаливают, взвешивают и спектрографируют [43].
Методы концентрирования, основанные на выделении сурьмы и других примесей. Среди этой группы методов наибольшее значение имеют экстракционные методы. Выбор метода экстракционного концентрирования в значительной мере определяется природой анализируемого материала — он должен обеспечивать экстракцию Sb и других, подлежащих определению примесей, оставляя при этом основу в водной фазе.
При выделении Sb из Sr и Ва используют экстракцию Sb и примесей других металлов в виде диэтилдитиокарбаминатов хлороформом из водных растворов при pH 5—6 [550]. Экстракция Sb и других тяжелых металлов в виде диэтилдитиокарбаминатов использована для концентрирования их при определений в воде особой чистоты [62]. В некоторых случаях для расширения числа определяемых примесей, кроме диэтилдитиокарбамината натрия, используют реагенты, которые дополнительно экстрагируют дру-84
гие примеси, не образующие диэтилдитиокарбаминатов. Так, для выделения Sb и других примесей из солей щелочных металлов их экстрагируют СНС13 при pH 5,5—6,5 с применением в качестве реагентов диэтилдитиокарбамината натрия и 8-оксихинолина [761]. Этими реагентами экстрагируются также Al, Mg, РЗЭ. При определении Sb и других тяжелых металлов в лечебных минеральных водах экстракцию проводят хлороформом с применением диэтилдитиокарбамината натрия и дитизона при pH 3—9 [957].
Для концентрирования Sb при ее определении в цинке и цинковых [59] и никелевых электролитах [58] ее экстрагируют раствором диэтилдитиофосфорной кислоты в СС14. При определении Sb в алюминии концентрирование проводят экстракцией хлороформным раствором диантипирилметана из 2,5—3,0 М НС1 [218]. Из хрома Sb также экстрагируют с применением диантипирилметана из раствора, 1М по NH4SCN и 0,5—8М по H2SO4, в виде гек-сароданостибата диантипирилметания. При определении Sb и некоторых других элементов в солях фосфорной кислоты предложено экстрагировать их расплавленным 8-оксихинолином при 80—85°С и pH 5—8 [428]. Для концентрирования Sb и других примесей в солях Li, Rb и Cs рекомендовано экстракцию проводить алифатическими монокарбоновыми кислотами [733].
Довольно простым и удобным методом концентрирования Sb является соосаждение. Из носителей (коллекторов) наиболее часто используются гидроокиси Fe, Al, Sn, Ti, Zr и др. Но по эффективности лучшим коллектором для выделения Sb является гидратированная двуокись марганца, которая наиболее полно соосаждает Sb. Ее обычно получают введением в раствор соли Мп(П) и КМпО4, который окисляет Мн(П) до Mn(IV) и сам при этом восстанавливается до Mn(IV). Соосаждение Sb с МпО2 использовано для концентрирования Sb при определении ее в свинце [1038]. В ряде случаев в качестве коллектора используют CuS. Для этого в солянокислый раствор пробы добавляют соль меди(П) и пропускают H2S. Осадок отделяют, высушивают и озоляют при 600° С. Медь одновременно служит внутренним стандартом. Таким образом определяют Sb в хроме [101], хромовом ангидриде 1777], никеле и его сплавах [108], железе [1254], вольфраме, ниобии и тантале [984].
При определении Sb в Zn, Cd, Ga, In и ряде других металлов и их соединениях иногда в качестве коллектора используют гидроокись, получаемую частичным осаждением основы. Так, для концентрирования Sb в кадмии прибавляют NH4OH до получения небольшого осадка (pH 6,0—7,5) [716], который затем отделяют, высушивают, прокаливают, взвешивают и спектрографируют. Аналогично соосаждают Sb с Zn(OH)2 при определении ее в цинке 1716] и с гидроокисями Ga, In и Те при определении Sb в Ga, In и Те [551]. Однако эти коллекторы значительно уступают МнО2. При определении Sb в Bi в качестве коллектора предложено использовать Sn(OH)4 [477].
85
В ряде случаев используются методы ионного обмена. С их применением производят предварительное концентрирование Sb в геологических материалах [840], сталях [336], мышьяке и галлии [562] и минеральных водах [958]. Трудностей, связанных с количественным элюированием Sb и других определяемых примесей со смолы, можно избежать, если смолу озолить [336].
Сурьму можно выделить также методом цементации. Так, при определении Sb в кадмии ее выделяют из раствора пробы в HNO3 цементацией на цинковом порошке при pH 6,0 в присутствии NH2OH при 80° С [1007]. Вместе с Sb выделяются и другие металлы, стоящие правее Zn в ряду напряжений.
Способность Sb восстанавливаться до летучего SbH3 также используется в спектральном анализе как метод концентрирования. Описан [1588] прибор для восстановления сурьмы до SbH3, последний, проходя через нагретую стеклянную трубку, разлагается с выделением сурьмы, которую используют для спектрографирова-ния. В качестве восстановителя применяют цинк в среде НС1. Применение Na[BHJ в качестве восстановителя по сравнению с цинком позволяет существенно снизить значение холостого опыта и достигнуть предела обнаружения Sb 0,5 нг [943].
Рептгеноспектральпые методы
Рентгеноспектральные методы анализа являются весьма перспективными и в последнее время все чаще используются для определения Sb вследствие высокой экспрессности и хорошей точности. Они пригодны как для определения малых содержаний Sb при использовании больших количеств анализируемого материала, так и для очень малых количеств материала при больших содержаниях Sb. Как и в эмиссионном спектральном анализе, рентгеноспектральные методы позволяют определять Sb одновременно с рядом других элементов. Рентгеновский локальный анализ при помощи электронного зонда позволяет анализировать пробы объемом до 1 мкм3. Он удобен для исследования однородности распределения Sb по объему анализируемого образца, позволяет выявлять включения с аномальными концентрациями как Sb, так и других элементов в Sb и ее сплавах.
Основы рентгеноспектрального анализа и применяемая аппаратура описаны в ряде монографий, руководств и обзоров [28 29, 83, 442а, 687, 738, 813].
Характеристический эмиссионный рентгеновский спектр Sb возбуждается при бомбардировке образца потоком электронов. Он состоит из небольшого числа линий К-, L- и M-серий, интенсивность которых зависит от режима работы рентгеновской.трубки. Линия Sb Ка (0,470 А) наиболее чувствительна и используется для определения Sb в материалах с малым ее содержанием. Линия La (3,439 А) слабее примерно в 25 раз, но имеет очень малый фон [1514]. Значительно большее применение для определения Sb
86
нашли методы, в которых для возбуждения характеристических рентгеновских спектров Sb используют излучения, обладающие высокой энергией, в том числе излучения многих радиоактивных изотопов [1617].
Интенсивность аналитической линии Sb в рентгеновском спектре зависит от ее концентрации, природы основы, природы и концентрации других элементов, толщины слоя анализируемого материала, состояния его поверхности, степени измельчения и плотности упаковки, геометрии расположения анализируемого образца и используемой аппаратуры и практически не зависит от степени окисления Sb и химической природы соединений, в виде которых она представлена.
Абсолютное количество Sb, которое может быть определено в оптимальных условиях, составляет 1—2 мкг [1658], а иногда и нескольких долей микрограмма. Однако точность определения малых количеств Sb значительно снижается. При благоприятных условиях относительное стандартное отклонение составляет 0,002— 0,003. Рентгеноспектральный метод используют для определения Sb в монолитных образцах, порошкообразных материалах и водных и органических растворах.
При прямом рентгеноспектральном определении Sb для градуировки приборов необходимо применять стандартные образцы, которые по составу, состоянию поверхности и плотности должны соответствовать образцам анализируемого материала. Поскольку трудно иметь стандартные образцы, точно соответствующие анализируемому материалу, то для учета влияния других элементов, содержание которых в анализируемом материале отличается от их содержания в стандартных образцах, используют различные способы, в том числе эмпирически найденные, а также теоретически рассчитанные поправочные коэффициенты. Для быстрого учета этих коэффициентов используют ЭВМ, работающие по специально составленным программам [1570]. Соединение рентгеноспектрального прибора с ЭВМ позволяет определять Sb одновременно с рядом других элементов в течение 5—6 мин.
Прямым рентгеноспектральным методом Sb определяют в различных материалах, в том числе в сталях [1126, 1164, 1268, 1624], свинце [1379], свинцовом блеске [1531], сплавах свинца с сурьмой и свинца с оловом, а также олова с сурьмой [1148—1150], сплавах сурьмы с висмутом [1211], теллуром [1211], свинцом и медью [1570], свинцом и оловом [1148—1150, 1288], сурьмяных рудах, концентратах и продуктах сурьмяного производства [542, 990], вольфраме, его рудах, концентратах и сплавах [1306], типографских сплавах [1481, 1482], порошковом железе [1306].
В тех случаях, когда чувствительность прямого определения Sb рентгеноспектральным методом оказывается недостаточной, проводят предварительное концентрирование. Обычно при этом Вводят определенное количество другого подходящего элемента, который служит внутренним стандартом. Так, например, опреде
87
ляют Sb в растворах, содержащих также Sn, Se, As и Ge. Для концентрирования Sb и указанных элементов их соосаждают с CuS, образующимся при введении в раствор 100 мг Си (в виде СиС12 или CuSO4) и пропускании H2S [1658]. Концентрат высушивают и анализируют, используя медь в качестве внутреннего стандарта. Метод позволяет определять до 2 мкг Sb в растворе объемом до 500 мл. При определении 100 мкг Sb стандартное отклонение составляет 0,025; для 5—10 мкг Sb оно увеличивается до 0,1.
Экстракция также позволяет проводить групповое концентрирование примесей для последующего их определения. Описаны [1177] два варианта экстракционного концентрирования^ По одному из них выделяют Sb совместно с V, Cr, Мп, Fe, Со/ Ni, Си, Zn, As, Se, Pd, Ag, Cd, In, Те, Hg, T1 и Bi экстракцией CHC13 в виде диэтилдитиокарбаминатов из водного тартратно-ацетатно-го раствора при pH 5—6. В другом варианте Sb экстрагируют СНС13 совместно с Ti, V, Fe, Zr, Mo, Sn и Hf в виде купферро-натов.
Экстракты по каплям наносят на определенную ограниченную площадь фильтровальной бумаги, давая каждой капле полностью испариться. После полного испарения экстракта возбуждают спектры излучепием'рептгсновской трубки с вольфрамовой или хромовой мишенью (50 кв, 30 ма) и регистрируют линии Sb и других элементов сцинтилляционным или пропорциональным счетчиком в течение 100 сек. Одновременно проводят холостой опыт с использованием тех же экстрагентов. Предел обнаружения Sb ~ 1 мкг.
Атомно-абсорбционная спектрофотометрия
Атомно-абсорбционная спектрофотометрия — относительно новый метод химического анализа. Первые работы по его применению опубликованы в 1955 г. [856, 1633]. Вследствие высокой чувствительности и селективности, простоты выполнения и малой продолжительности анализа этот метод в настоящее время широко применяется для определения многих элементов, в том числе Sb [265, 659, 709, 863, 1011, 1024, 1303, 1315, 1538, 1558, 1632]. Метод основан на способности свободных атомов каждого элемента поглощать излучение только определенной резонансной частоты. Вводя анализируемый раствор в пламя горелки или используя другой атомизатор, переводят большую часть элементов, находящихся в растворе в виде химических соединений, в свободные атомы. Условия атомизации подбирают так, чтобы определяемый элемент возможно большей частью переходил в свободные невозбужденные атомы. Кроме растворов, в последнее время в атомно-абсорбционной спектрофотометрии успешно применяется вариант с использованием твердых образцов. Благодаря импульсному характеру испарения и отсутствия разбавления анализируемого материала, чувствительность определения элементов в этом варианте существенно повышается. Поглощение резонансного излучения атомным
88
паром пробы зависит от концентрации в нем атомов определяемого элемента или в конечном итоге от его концентрации в анализируемой пробе.
Подробно теоретические основы и практическое применение метода атомно-абсорбционной спектрофотометрии рассматриваются в ряде монографий [448, 659, 709, 863, 1024, 1047, 1161, 1442, 1465, 1475, 1486, 1650, 1662] и обзоров [91, 265, 857].
В спектре поглощения Sb наиболее чувствительными являются линии 217,59; 206,83 и 231,15 нм. Соотношение их пределов обнаружения равно 0,5 : 0,6 : 1 [1321]. Однако в основном используются только линии 217,59 [267, 268, 387, 1122, 1248, 1354, 1391, 1511, 1673] и 231,15 нм [387, 1248, 1354, 1391]. Хотя абсорбционный сигнал последней линии несколько ниже, чем линии 217,59 нм, но влияние фона в этом случае несколько меньше, и вследствие этого получаемые результаты характеризуются лучшей воспроизводимостью [1534]. В связи с тем что в области линии Sb 206,83 нм прозрачность пламени для указанной аналитической линии значительно снижается, эта линия не имеет практического значения. При использовании пламенных атомизаторов наиболее высокая чувствительность определения Sb достигается с применением воздушно-ацетиленового пламени; в случае пламени смеси N2O с СгН2 чувствительность несколько ниже, а для пламени смеси воздуха с природным газом — значительно ниже. Другие пламена для определения Sb используются довольно редко [266, 1023, 1392]. Исключение составляет только случай, когда Sb вводят в пламя в виде SbH3: здесь наиболее эффективным является пламя смеси Н2 с воздухом.
Чувствительность определения Sb с применением пламенных атомизаторов в сильной мере зависит от совершенства используемого прибора, окислительно-восстановительных свойств пламени, высоты просвечиваемой зоны, геометрии горелки и ряда других факторов. Указывается [1391], что при использовании воздушно-ацетиленового пламени и спектрофотометра Тектрон АА1000 и просвечивания пламени светом лампы с сурьмяным полым катодом на расстоянии 1,5—2 мм от края горелки чувствительность определения Sb в расчете на 1% поглощения света для линии 231,15 нм составляет 1,3..мкг/.чл и для линии 217,58 нм — 0,64 мкг1мл. Мостин и Куннингем [1354] считают, что при прохождении пучка света от лампы с сурьмянным полым катодом через воздушно-аце-тиленовое пламя на расстоянии 4—10 мм выше уровня горелки достигается наиболее высокая чувствительность определения Sb (спектрофотометр Перкин-Элмер 303, ток полого катода 20 ма, ширина щели монохроматора 1 мм, скорость распыления анализируемого раствора 3,5 мл/мин), которая составляет (на 1% поглощения света) 1,4 мкг!мл для линии 217,58 нм и 2,0 мкг/мл — для линии 231,15 нм.
Одним из наиболее эффективных методов концентрирования Sb для последующего ее определения методом атомно-абсорбци
89
онной спектрофотометрии является экстракция. Она удобно сочетается с определением Sb в получаемых экстрактах путем непосредственного их распыления в пламя. Кроме повышения чувствительности за счет концентрирования, она повышается также вследствие замены воды органическими растворителями, в среде которых чувствительность определения, как правило, несколько выше. Наиболее часто в качестве органического растворителя используют метилизобутилкетон, который из солянокислых растворов экстрагирует как Sb(V) [1321, 1387, 1469, 1667], так и Sb(III) [1141, 1392]. Сурьма(У) извлекается практически полностью, в то время как Sb(III) — только на ~50% [1387]. Для количественного извлечения Sb(III) из водных растворов ее экстрагируют после добавления KJ или NH4J метилизобутилкетоном, содержащим 5% трифенилфосфиноксида [954, 1594] или 0,2 М три-н-октил-амин [684].
Предложено также экстрагировать Sb раствором диэтилдитиокарбамината аммония в ксилоле [1248] или пирролидиндитиокар-бамината аммония в метилизобутилкетоне [974]. При использовании для экстракции Sb растворов ТБФ в углеводородах (например, в толуоле) атомное поглощение Sb значительно ниже (в ~4 раза) по сравнению с ее атомным поглощением при использовании метилизобутилкетоновых экстрактов [1667].
При определении Sb в почвах, горных породах и рудах рекомендуется предварительно концентрировать Sb отгонкой ее из смеси тонкоизмельченного анализируемого материала с NH4J [1391, 1511]. Сублимирующийся при этом SbJ3 отлагается на охлаждаемой поверхности, где его растворяют в определенном объеме 2—ЗМНС1, и полученный раствор вводят в воздушно-ацетиле-новое пламя.
Описан [1638] комбинированный метод определения Sb в геологических материалах, включающий предварительное ее отделение отгонкой в виде SbJ3 из смеси анализируемого материала с NHJ, растворение возгона в 10%-ной НС1, экстракцию Sb из полученного раствора 4%-ным раствором триоктилфосфинокиси в метилизобутилкетоне и распыление полученного экстракта в воздушно-ацетиленовое пламя. При определении Sb в никеле рекомендовано предварительно концентрировать ее соосаждением с МпО2 [955]. Некоторое повышение чувствительности определения Sb и ряда других элементов (до50%) достигается за счет применения диафрагм на конденсорных линзах [1147].
Одним из наиболее эффективных методов повышения чувствительности определения Sb атомно-абсорбционным методом является выделение ее в виде SbH3 и непосредственное введение его в пламя. Для переведения сурьмы в SbH3 в качестве восстановителя наиболее часто используют цинк в присутствии SnCl2 в сильнокислых растворах. Лучшим вариантом является метод с предварительным сбором образующегося SbH3 и последующим введением полученных газов в пламя смеси Н2 + Аг, свободно горящей 90
в воздухе [1432, 1669]. Вместо SnCl2 при восстановлении цинком может использоваться TiCl3, в присутствии которого также достигается полное восстановление сурьмы до SbH3 [1432]. С применением таблеток из порошка цинка продолжительность восстановления сурьмы до SbH3 несколько сокращается [1671].
Поллок и Уэст [1432, 1433] показали, что применение магния с добавлением небольших количеств TiCl3 позволяет существенно снизить значение холостого опыта. С использованием спектрофотометра Перкин-Элмер 403 возможно определение до 0,0012 мкг Sb в пробе. При содержании Sb в анализируемой пробе 0,1—0,6 мкг ошибка определения составляет ~ 3%.
Еще лучшим восстановителем является Na[BHJ, применение которого позволило снизить предел обнаружения Sb до нескольких нанограммов в пробе [1057—1059, 1367, 1499].
Для определения Sb в виде SbH3, как правило, используется пламя смеси Н2 с Аг, свободно горящее в воздухе [1140, 1209, 1499, 1587]. При использовании пламени смеси Н2 с Аг и воздухом и сборе выделяющегося SbH3 в специальный сосуд, позволяющий затем газовую смесь равномерно вводить в пламя, предел обнаружения составляет 5 иг Sb в пробе [1499].
Стибин, получаемый добавлением подкисленного анализируемого раствора к двойному объему 1%-ного раствора Na[BH4], пропускают через кварцевую трубку длиной 17 см, находящуюся в пламени стехиометрической смеси С2Н2 с воздухом, получаемого с применением щелевой горелки. За счет нагревания трубки SbH3 атомизируется с образованием невозбужденных атомов Sb. Через трубку пропускают свет лампы с полым сурьмяным катодом и измеряют атомное поглощение Sb. Метод не требует коррекции фона, как в случае генерации SbH3 цинком с применением пламени смеси Н2 с Аг и воздухом. Чувствительность определения Sb (на 1% поглощения света) при использовании линии 217,6 нм составляет 0,061 нг/мл. При концентрации Sb 0,02 мкг/мл коэффициент вариации составляет 4,9% [1592]. Определению Sb с применением Na[BH4] для генерирования стибина не мешают As, Sn, Si, Al, Мп, Ca, Mg, К, Na. Заметное влияние оказывают Сг, Си, Ni, Pb, Fe и HNO3. При содержании Sb в пробе от 0,05 до 0,5 мкг калибровочный график прямолинеен. При определении 0,3 мкг Sb при п — 15 коэффициент вариации составляет 5,2% [1367].
Методы с использованием графитовой кюветы. Как указывалось выше, в настоящее время используется ряд вариантов метода, позволяющих анализировать непосредственно сухие вещества. Наибольшее распространение из них получил метод графитовой кюветы [406, 448, 1140, 1290].
В кювету вносят навеску анализируемого материала. В случае анализа растворов их также вносят в[ кювету и выпаривают досуха. Сухую пробу испаряют в инертной атмосфере (Ar, N2) при 2000—2600° С за счет электрокон-тактного нагрева или мощной дуги постоянного тока (20—40 а), зажигаемой
61
между кюветой и электродом. Вследствие этого проба испаряется в течение 0,3—0,4 сек. и поглощение света образовавшимся атомным паром непрерывно регистрируют с помощью соответствующей аппаратуры.
Обычно используют интегральный метод регистрации (измеряют площадь под кривой, построенной в координатах поглощение света—время полного испарения пробы). Этот сигнал мало зависит от колебаний температуры кюветы, режима нагрева и ряда других факторов. В качестве аналитического сигнала возможно также использование пика поглощения при работе с приборами, имеющими приспособление для экстремальной настройки на сигнал. В этом случае для получения удовлетворительных результатов требуется тщательное соблюдение постоянства условий проведения анализа. Точность определения Sb с применением графитовой кюветы ниже, чем при использовании растворов, вводимых с постоянной скоростью в пламя. В оптимальных условиях коэффициент вариации составляет 4—12%, [1322], но абсолютная чувствительность этого метода исключительно велика (10~12— 1(ГН г Sb).
Исследованы [1433] некоторые приемы снижения предела обнаружения Sb с применением графитовой кюветы. Показано, что применение шариковой безэлектродной ВЧ-лампы имеет ряд преимуществ по сравнению с лампой с полым катодом.
Одной из разновидностей непламенной атомно-абсорбционной спектрофотометрии является метод с использованием графитового стержня в качестве атомизатора [1673]. Для определения Sb этим методом ее предварительно отделяют экстракцией метил-изобутилкетоном из раствора, 6 М по НС1.
Аликвотную часть экстракта вносят в канал (d = 1,5 мм) графитового стержня, растворитель испаряют, остаток озоляют (30 сек. при ~180 °C) и атомизируют (3 сек. при 2200° С). Поглощение’ света атомами Sb измеряют при 217,6 нм. Этот вариант метода позволяет определять до 10~10 г Sb с коэффициентом вариации ~ 3%.
Для определения Sb в остатках от выстрелов описан [1111] метод с использованием танталового ленточного атомизатора.
Налет от выстрела снимают с поверхности 5%-ной HNO3, раствор высушивают и остаток озоляют в низкотемпературном пламени. Золу растворяют в 1 М HNO3, раствор наносят на танталовую ленту атомизатора, сушат 45 сек. при 100° С, озоляют 30 сек. при 450° С, атомизируют 2 сек. при 2500° С и измеряют поглощение света линией 217,6 нм. Предел обнаружения Sb составляет 0,2 нг в пробе; коэффициент вариации ~ 8,5%.
Комбинированные методы. Описан также вариант атомноабсорбционного определения Sb, сочетающий распыление анализируемого раствора в пламя смеси Н2 с воздухом комбинированной горелки-распылителя при использовании кюветы для подогревания отходящих газов пламени [1338, 1484, 1485]. Кювету (Z = 45 см, d = 0,9 -т- 1,5 см) нагревают в электрической печи. 92
В этом случае достигается большая длина поглощающего слоя, а нагревание отходящих газов пламени до оптимальной температуры атомизации обеспечивает практически переведение всей Sb в атомный пар. В связи с этим чувствительность определения Sb значительно повышается.
В другом подобном методе [1610] предложено использовать кварцевую трубку (Z = 70 см, d = 0,9 см), помещенную в электрическую печь при 1200° С. С одной стороны трубки на расстоянии 3 мм от ее края размещена кольцевая горелка для пламени смеси Н2 с воздухом, а с другой — всасывающий патрубок вентилятора. При включении вентилятора пламя втягивается в трубку, в которой измеряется атомное поглощение Sb. Проба поступает в трубку в виде сухого аэрозоля за счет распыления нагретым до 110° С воздухом.
Описан [1213] автоматический вариант атомно-абсорбционного определения Sb в природных водах, основанный на образовании SbH3, пропускании его через трубчатую печь в качестве атомизатора и измерении атомного поглощения Sb. Метод позволяет анализировать до 40 проб в час. Предел обнаружения Sb составляет 0,5 мкг!л.
Методом атомно-абсорбционной спектрофотометрии определяют Sb в различных материалах, в том числе в алюминии и его сплавах [954, 1469], геологических материалах, минеральном сырье и горных породах [97, 732, 863, 954, 1338, 1391, 1485, 1638], железных рудах, железе, чугуне, стали и ферросплавах [888, 954, 1069, 1140, 1141, 1601], меди и медных сплавах [1392, 1534, 1673], мышьяке и его сплавах [1534], никеле, никелевых сплавах и соединениях [954, 955, 1594], олове и его сплавах [1354], оловянно-свинцовых припоях [1166], свинце, его сплавах и солях [267, 268, 1354, 1450], галенитах [1387], сплавах редких и цветных металлов [1140, 1321], полупроводниковых материалах [265, 1122], рудах [97, 15Ц, 1601, 1638], почвах [1391, 1594, 1638], силикатных материалах, -керамике и стеклах [652, 1587), чистых веществах [315], солях щелочных и щелочноземельных металлов [387], природных и сточных водах [1123, 1209, 1213, 1367], плутонии [1622], солях цинка и кадмия [387], синтетических волокнах [1321], пищевых продуктах [1367], пистолетных пулях [948], добавках к нефтепродуктам [1563], химических реактивах и препаратах [264—266, 268, 387].
Ввиду высокой чувствительности метод атомно-абсорбционной спектрофотометрии используется в криминалистических исследованиях, в том числе для определения Sb в копоти, остающейся возле огнестрельных ран, сделанных с близкого расстояния [1111, 1469]. Малая продолжительность и простота выполнения определения Sb методом атомно-абсорбционной спектрофотометрии обеспечивают его использование в автоматическом контроле содержания Sb в ряде материалов [1123, 1213, 1500].
93
Атомно-флуоресцентная спектрофотометрия
Наряду с атомно-абсорбционной спектрофотометрией для определения Sb применяется также метод атомно-флуоресцентной спектрофотометрии как в пламенном [1017, 1018, 1075, 1251, 1392, 1591], так и в непламенном варианте [1322]. Метод основан на переводе анализируемого материала в атомный пар, переходе атомов определяемого элемента иэ невоэбужденного в более высокое энергетическое состояние в результате поглощения резонансного излучения внешнего источника возбуждения, последующем переходе возбужденных атомов в исходное невозбужденное состояние, сопровождающимся характерным для каждого элемента флуоресцентным излучением, интенсивность которого в определенных пределах пропорциональна концентрации атомов определяемого элемента в атомном паре. Атомную флуоресценцию измеряют перпендикулярно к пучку внешнего источника возбуждения, проходящего через атомный пар.
В качестве атомизаторов для Sb наиболее часто используют пламена. Изучена [1251] возможность атомно-флуоресцентного определения Sb в различных пламенах с применением в качестве источника света высокоинтенсивной лампы с полым катодом и атомно-абсорбционного спектрофотометра Вариан-Тектрон АА4, видоизмененного для атомно-флуоресцентных измерений. Исследованы пламена смесей: Н2 — воздух, Н2 — Аг, Н2 — О2 — Аг и С2Н2 — воздух. Наиболее эффектным оказалось пламя смеси Н2 с Аг (диффузное) с расходом 0,95 л!мин Н2 и 5,5 л!мин Аг. Когда тушение флуоресценции мало, наибольшей чувствительностью характеризуются резонансные линии Sb 206,83; 217,58 и 231,15 нм, по которым пределы обнаружения Sb найдены равными соответственно 0,1, 0,03 и 0,1 мкг!мл. В пламени смеси Н2 с О2 и Аг (1,15 л!мин Н2, 0,2 л!мин О2 и 5,5 л!мин воздуха) пределы обнаружения Sb по тем же линиям несколько хуже (соответственно 0,1, 0,05 и 0,15 мкг/мл).
Изучены [1018] атомно-флуоресцентные характеристики Sb в пламени смеси пропана с воздухом с использованием излучения безэлектродной разрядной лампы с парами Sb, питаемой ВЧ-ге-нератором. Наиболее интенсивная флуоресценция отмечена также для линий 206,83; 217,58 и 231,15 нм. При использовании линии 217,58 нм предел обнаружения Sb составляет 0,05 мкг/мл; калибровочный график прямолинеен для растворов с концентрацией Sb 0,1—120 мкг!мл. Определению Sb в указанных условиях не мешают 100-кратные количества Cd, Со, Си, Fe, Hg, К, Mg, Мп, Мо, Na, Pb, Zn, а также AsOl”, РО^, SO^ и NO3.
В атомно-флуоресцентной спектрофотометрии Sb в качестве источников возбуждения часто используют безэлектродные лампы [1017, 1075, 1591, 1608]. Описана [1075] двухэлементная безэлек-тродная разрядная лампа в виде трубки как источник линейчатого спектра для атомно-флуоресцентного определения Sb и As. Для
94
повышения чувствительности определения Sb с применением безэлектродных ламп с УВЧ-возбуждением рекомендуется [15911 применение электрической модуляции. При использовании атомно-абсорбционного спектрофотометра Саусерн Аналитикал А-300, в котором изменены положение источников света и конструкция наконечника горелки, работающей на смеси С2Н2 с воздухом, а также для ограничения потока света от первичных конусов пламени применена маска, предел обнаружения Sb по линии 231,4 нм с использованием безэлектродных УВЧ-ламп с электрической модуляцией составил 0,04 мкг/мл/, это в ~ 5 раз меньше, чем без модуляции.
Массманном [1322] проведено сравнение атомно-абсорбционного и атомно-флуоресцентного методов с применением графитовой кюветы и установлено, что предел обнаружения Sb атомно-флуоресцентным методом несколько выше, чем атомно-абсорбционным. Коэффициент вариации в обоих случаях одинаков.
Для повышения чувствительности определения Sb атомнофлуоресцентным методом используется предварительное концентрирование ее экстракцией метилизобутилкетоном [1392]. Введение в атомизатор Sb в виде SbH3 также позволяет значительно повысить чувствительность ее определения. Описан [1608] высокочувствительный бездисперсионный атомно-флуоресцентный метод определения Sb с предварительным ее выделением в виде SbH3 с применением Na[BHJ в качестве восстановителя и пламени смеси Н2 с воздухом. Метод позволяет определять до 3 на Sb в пробе.
Атомно-эмиссионная спектрофотометрия
Один иэ вариантов метода атомно-эмиссионной спектрофотометрии, рекомендованный для быстрого качественного обнаружения Sb [943], пригоден также для ее количественного определения-Метод позволяет определять до 0,5 нг Sb в пробе. Методика количественного определения Sb совпадает с методикой, описанной для ее качественного обнаружения (см. главу III) с той разницей, что для количественного определения записывают кривую интенсивности излучения линии Sb 252,5 нм, и по площади, ограниченной этой кривой, с помощью калибровочного графика находят содержание Sb. Схема используемого прибора, разрядный детектор и оптическое измерительное устройство описаны в работе [941]. Коэффициент вариации составляет ~ 5%. Вместо Не в качестве газа-носителя может использоваться Аг [942]. Метод рекомендован для определения Sb в природных и сточных водах.
Описан [1573] комбинированный метод атомно-эмиссионного определения Sb, основанный на предварительном выделении ее в виде (C6H6)3Sb, газохроматографическом отделении от (C6H6)3As и введении его в атомно-эмиссионный детектор с применением УВЧ-плазмы в качестве атомизатора [1571, 1572].
95
Пробу (0,2—1 г) минерализуют нагреванием с HN03 и НС104 и из полученного раствора выделяют Sb вместе с As соосаждением с тионалидом (последний вводят в виде 2%-ного раствора в ацетоне) [1437]. Реакционную смесь кипятят 5 мин. и оставляют на 10 мин. Затем кипятят 30 мин. для удаления ацетона. После охлаждения осадок отфильтровывают на стеклянный фильтр со •слоем тионалида, высушивают и обрабатывают 1,5 М раствором Mg(CeH5)Br в диэтиловом эфире. Избыток Mg(C6H5)Br разлагают водой и образовавшиеся (CeH5)3Sb и (C6H5)3As экстрагируют диэтиловым эфиром, бензолом или толуолом. Аликвотную часть экстракта вводят в газохроматографическую колонку (d = 0,5 мм), заполненную Газхромом Q 80/100 с нанесенными на него 4% FFAP. Температура колонки 350° С, газ-носитель — Аг (100 мл/мин). Выходящую из колонки газовую смесь вводят в атомно-эмиссионный детектор с УВЧ-генератором (мощностью 30 вт) и регистрируют эмиссию линий Sb 259,8 нм и As 228,8 нм. Предел обнаружения для твердых материалов составляет 7,5 •10~6% Sb, для растворов и жидкостей — 0,125 нг/мл Sb . Коэффициент вариации находится в пределах 2,6—7,1%. Калибровочный график (в координатах интенсивность эмиссии — содержание Sb) линейный в пределах 0,01 — 1 мкг Sb.
Метод применен для анализа различных растительных и биологических материалов, каменного угля и летучей золы.
Молекулярный эмиссионный анализ
В последнее время для определения ряда элементов предложен метод молекулярного эмиссионного анализа с применением полого стержня [896, 897, 1099, 1557]. Определение Sb этим методом основано на ее свойстве давать характеристическое молекулярное излучение SbO при 355 нм, возникающее в маленькой полости на конце стержня при введении в полость SbH3 и нахождении полости в диффузном пламени смеси Н2 с N2 при пропускании через полость Оа [897, 1099]. Калибровочный график линеен при концентрации Sb от2-10~4 до 3-10-3%. Стандартное отклонение составляет 1,4-10-5 % Sb. Предел обнаружения 5-10~5% Sb. Определению 5 мкг Sb этим методом мешают только Ag и Си, если присутствуют в равных количествах.
Другие методы
Среди других методов определения Sb заслуживают внимания термоэлектрические методы, методы бумажной, тонкослойной и газовой хроматографии и кинетические методы.
Термоэлектрический метод рекомендован для определения Sb в свинцово-сурьмяных сплавах [347, 348]. Метод основан на измерении зависимости термоэлектродвижущей силы от концентрации Sb в сплаве. Могут использоваться три варианта контактирования анализируемого сплава с элементом сравнения: 1) пайка оловом, 2) контакт с помощью механического зажима и 3) контакт через
96
расплавленный металл. В последнем варианте образец анализируемого сплава и элемент сравнения одними концами погружают в расплавленный сплав Вуда. В качестве элемента сравнения используют висмут. Термоэлектродвижущая сила свинцово-сурьмяных сплавов прямолинейно убывает с увеличением содержания Sb. При содержании Sb в сплаве 4,7—8,5%стандартное отклонение не превышает ±0,18%.
Описано применение метода бумажной хроматографии для определения Sb в стекле [1146]. Используют фильтровальную бумагу Ватман № 1, импрегнированную 8-оксихинолином. Хроматограмму проявляют смесью бутанола с NH4OH. Содержание Sb находят по площади окрашенной зоны Sb. При определении 10— 100 мкг Sb ошибка составляет 5—7%.
Методы газовой хроматографии используются для анализа смесей SbCl3 с летучими хлоридами других элементов [1521], смесей SbH3 с летучими гидридами [1261] и смесей SbH3 с различными токсичными газами [1493]. Для определения SbCl3 в смесях с AsCl3 и GeCl4 в качестве неподвижной фазы используют полимеры на основе политетрафторэтилена [1521]. Для определения SbH3 в смеси с SnH4 используют колонку (I = 1 м, d = 4 мм), заполненную твердым носителем силь-о-сель, содержащим 25% силиконовой жидкости 702; температура колонки 36° С. Применение трикрезилфосфата в качестве неподвижной фазы дает возможность отделить SbH3 от GeH4, AsH3 и SnH4 [1261]. Продолжительность анализа ~5 мин.
Описан [1493] метод анализа многокомпонентных смесей, содержащих SbH3, Н2, Вг2, СО, СО2, Cla, С12О, НС1, HCN, HF, N2O5, NO2, фосген, окись этилена и моноэтаноламин. Метод газовой хроматографии применен для определения SbH3 в газовой фазе в процессе электроразрядного легирования при использовании электродов, содержащих Sb [66].
Тонкослойная хроматография. Описано [1160] определение Sb(III) в присутствии As(III) и Sn(IV).
Мышьяк предварительно отделяют в виде хлорида экстракцией бензолом из 6 М НС1. Из оставшейся водной фазы при pH 4,0 экстрагируют Sb и Sn бензолом в виде дитизонатов и хроматографируют на пластинках со слоем (0,2 мм) целлюлозы MN 300. Пластинки предварительно высушивают 5 мин. при 100° С, затем опрыскивают бензолом и сушат на воздухе. На пластинку наносят 20 мкл полученного экстракта и проявляют хроматограмму бензолом до перемещения фронта растворителя на 0,5—1 см. После этого хроматограмму высушивают и проявляют смесью (4 : 1) гексана с бензоломдо продвижения фронта растворителя на 10 см. Значения Rf сурьмы и олова составляют соответственно 0,51 и 0,10. По площади окрашенного пятна Sb находят ее содержание.
Для определения Sb в присутствии As и Bi методом тонкослойной хроматографии на 6 пластинок (15 X 15 см) со слоем силикагеля G, предварительно высушенных при 105° С в течение 30 мин., наносят на стартовую линию по
4 А. А. Немодрук
97
20 мкл анализируемого раствора (5 проб) и стандартного раствора Sb; пластинки погружают в воду на 0,5 см и хроматографируют 15 мин., после чего их высушивают при 105° С и повторяют хроматографирование в перпендикулярном направлении. Хроматограммы опрыскивают раствором FeS в 10%-ной НС1. Пятна Sb снимают с пластинок вместе с сорбентом, обрабатывают 25%-ным раствором винной кислоты, разбавляют этим же раствором до 20 мл, добавляют 2 г NaHCO3 и титруют Sb(III) 0,1 N раствором иода в присутствии крахмала в качестве индикатора. При определении 1—4 мг Sb ошибка % 7% [1628].
Жидкие мембранные электроды на основе ионных ассоциатов, образуемых анионом SbClg с катионами основных красителей (севроновым красным L и CL, флавиндулином О, феназиндули-ном О), предложено использовать для определения Sb(V) и Sb(III) [1066]. Эти электроды более селективны к Sb(V), чем к Sb(III). Потенциал электрода пропорционален концентрации Sb(V) в пределах 10-8—10~2 г-ион!л.
Кинетическое окислительно-восстановительное титрование Sb(lII). Реакционную смесь титруют раствором окислителя (KBrO3, J2, Ce(SO4)a, KaCraO7, KJO3) в строго определенных условиях (pH, температура, объем) при постоянной скорости подачи титранта с потенциометрическим, фотометрическим или визуальным (индикаторы ксиленоловый оранжевый, ферроин) установлением конечной точки. По продолжительности титрования, которое прямо пропорционально содержанию Sb, находят ее содержание. Метод позволяет определять Sb в растворах с ее концентрацией 8- 10~s—1,2-Ю“4 г-ион!л с ошибкой 2—5% [953, 1326].
Показана возможность раздельного определения Sb и As в их смесях [1595, 1596]. Метод основан на добавлении титранта и регистрации потенциала электродов во времени и анализе кривых, построенных в координатах потенциал—время. На этой кривой в присутствии обоих компонентов имеется два прямолинейных участка с различным наклоном, соответствующих титрованию As и Sb.
Масс-спектральный метод применен для определения Sb и одновременно с ней до 72 других примесей в Si, Ge и графите [766],
Глава V
МЕТОДЫ ОТДЕЛЕНИЯ СУРЬМЫ ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
Для отделения Sb от других элементов используется ряд методов. Выбор наиболее подходящего зависит от анализируемого материала и содержания в нем Sb, от выбранного метода определения Sb и от требований, предъявляемых к анализу в целом (продолжительность, точность и др-).
МЕТОДЫ ОСАЖДЕНИЯ
Осаждение в виде сульфидов. Долгое время важнейшими методами отделения Sb были методы, основанные на использовании свойств ее сульфидов [150]. Осаждение Sb в виде сульфидов в кислых растворах позволяет отделять ее от элементов, не входящих в группу H2S, а последующей обработкой выделенного осадка щелочным раствором отделять ее от элементов группы меди. Однако ряд элементов (As, Au, Pt, Sn, Ir, Ge, Se, Те и Mo) образует при этом растворимые соединения и вместе с Sb переходит в раствор.
Сурьму можно отделить от As осаждением его H2S из раствора, 9 М по НС1, при 15—20° С, пропусканием быстрого тока H2S. Для полной коагуляции осадка As2S5 смесь выдерживают 1 час при комнатной температуре, осадок отфильтровывают и промывают 8—9 М НС1, насыщенной H2S.
Для отделения Sb от Sn и Ge ее осаждают в виде сульфида из раствора, 0,2 М по НС1 и 0,4 М по HF (HF образует с Sn и Ge растворимые фторидные комплексы) [1301]. От олова Sb может быть также отделена осаждением сероводородом из солянокислых растворов в присутствии щавелевой или винной кислоты, необходимой для удержания Sn в растворе.
Поскольку работа с H2S неприятна вследствие его токсичности, то вместо него часто используют реагенты, которые в процессе осаждения постепенно разлагаются с выделением H2S, в том числе тиоацетамид [280, 394], тиоформашид [864], тиоацетат аммония [1529], тиомочевину [1542], Na2S2O3 [558, 859], P2S6 [1449], Na2S [707], (NH4)2S2+X [609]. Однако вследствие малой кон
4*
99
центрации H2S, ввиду медленного разложения ряда из них, количественное образование сульфидов сурьмы часто не достигается; в других случаях (Na2S, P2S5, полисульфиды аммония) разложение идет быстро и в связи с этим практически нет никакой разницы от осаждения самим H2S.
Осаждение органическими серу содержащими реагентами. С применением 2-меркапто-5-аминофенил-1,3,4-тиодиазола в качестве осадителя Sb можно отделить от большинства тяжелых металлов [1435]. Среди органических серусодержащих соединений найдены также такие, которые позволяют отделять Sb(III) от Sb(V). Показано [1679], что из среды конц. НС1 с применением 8-тиооксина осаждается Sb(V), а затем разбавленной НС1 осаждается Sb(III).
Осаждение другими органическими реагентами. С применением ализарина S сурьма может быть отделена от А1, Ст, Мп, Со, Ni, As и Cd в виде нерастворимого ализарината [34, 755]. Сурьму можно отделить от большинства элементов осаждением из галогенид-ных растворов добавлением водно-спиртового раствора дианти-нирилметана, образующего нерастворимые ионные ассоциаты с анионными галогенидными комплексами Sb. Наиболее полно Sb осаждается из иодидных растворов (количественное осаждение Sb в этом случае обеспечивается при ее концентрации 5-• 10~3 мкг!мл) [593].
Осаждение в виде металлической сурьмы. От Sn, Cd и ряда других элементов Sb можно отделить осаждением в виде металла в среде 0,4 М НС1 восстановлением железным порошком. Вместе с Sb осаждаются Си, Bi и частично РЬ и As [1362]. Для выделения Sb в элементном виде в качестве восстановителя применяют также другие металлы, в том числе губчатый свинец [714], кадмий в виде порошка [660] и алюминий в виде опилок [587]. С применением губчатого свинца одновременно с Sb выделяются Си и Bi. При выделении Sb с использованием порошка кадмия цементацию проводят в среде 6 М НС1 при нагревании. Из растворов с концентрацией Sb > 1,5 г-ион/л она выделяется количественно. С применением алюминия можно количественно выделять Sb, проводя цементацию при 60° С в 3%-ном растворе тартрата натрия. В этих условиях As(III) не выделяется. Однако в присутствии даже небольших количеств As(III) сурьма выделяется уже не полностью; присутствие равных или больших количеств As подавляет цементацию Sb. В 0,5 М НС1 происходит количественная цементация Sb, в то время как As остается в растворе. Если же в растворе присутствует Си, то алюминий восстанавливает As до арсина [587]. При определении Sb в галлии и сплавах индия с галлием и индия с цинком выделяют Sb цементацией ее на оловянном электроде из раствора, 0,5 М по НС1 [662].
100
МЕТОДЫ СООСАЖДЕНИЯ
Для отделения малых количеств Sb методы осаждения с применением коллекторов (носителей) используются довольно часто особенно в случае анализа веществ высокой чистоты, полупроводниковых материалов, реактивов и т. п.
Соосаждение сурьмы с гидроокисями металлов. Наиболее эффективным коллектором для выделения микроколичеств Sb является гидратированная МпО2, которая полнее других соосажда-ет Sb и поэтому наиболее часто используется [35, 46, 689, 955, 1152, 1293, 1302, 1397, 1438, 1471]. Гидратированная МпО2 образуется при введении в раствор КМпО4 и избытка соли Ми(П). Иногда соосаждение Sb проводят с МпО2, получаемой при взаимодействии КМиО4 с этанолом [1438]. Сурьму соосаждают из горячих растворов при pH 1—7. Для повышения селективности отделения Sb соосаждение рекомендуется проводить при pH 1,0—1,2 [1471]. Соосаждение с МиО2 позволяет количественно выделять до 1 мкг Sb из 100 мл раствора.
Для отделения микроколичеств Sb от Bi соосаждение проводят из раствора, 1,2 М по HNO3 [1397]. Вместе с Sb соосаждает-ся Sn. Соосаждению Sb мешает F" и ряд других веществ, маскирующих Sb [46]. Вслед за МпО2 по эффективности соосаждать Sb следует Fe(OH)3, затем А1(ОН)3. Сурьма количественно соосажда-ется с Fe(OH)3 из растворов с pH 6—9 [1073]. Для отделения микроколичеств Sb при ее определении в никеле и медно-никелевых сплавах в качестве соосадителя рекомендуется H2SnO3 [986]. При определении Sb в железе в качестве коллектора используют Сг(ОН)3 [1399], а при выделении ее из хрома — МнО2 или H2SnO3, в то время как из аммиачно-щелочных растворов — Fe(OH)3 [689].
Для выделения Sb из щелочных растворов эффективным коллектором является Mg(OH)2 [346, 1073], с которым при pH 10 она соосаждается практически количественно. Этот коллектор удобен для отделения Sb от металлов, образующих в указанных условиях растворимые соединения (Zn, Al, Сг, Be, Pb, Si, Sn, W). Микроколичества Sb могут быть выделены соосаждением из 0,5—1 М НС1 или H2SO4 с CuS или MoS3 [1293, 1401]. Для отделения Sb от Sn соосаждение проводят в присутствии Н2С2О4, а для предотвращения осаждения W и V — в присутствии винной кислоты.
Соосаждение сурьмы с тионалидом. Для выделения Sb из ферровольфрама используют тионалид [392]. При смешении ацетонового раствора тионалида с анализируемым водным раствором он выделяется в виде тонкодисперсного осадка и захватывает образующийся при этом тионалидат сурьмы. Соосаждение Sb с тионалидом позволяет выделять ее практически количественно (93— 95%) из растворов с ее содержанием до 0,05 мкг/'л. Вместе с Sb выделяется As. При выделении Sb из алюминия соосаждением с тионалидом в среде 0,5 М НС1 в раствор вводят небольшое количество Sn в качестве коллектора [254]. Микроколичества Sb могут
101
быть выделены в элементном виде с использованием Те в качестве носителя [100]. Этот метод особенно удобен для определения Sb в теллуре. Сначала Sb выделяют, соосаждая ее с частью Н2ТеО3, затем полученный осадок растворяют и выделяют Sb соосаждени-ем с элементным Те.
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Для отделения Sb довольно часто используется экстракция. Малая продолжительность и высокая избирательность экстракционных методов отделения Sb обеспечивают им широкое применение. В ряде случаев экстракционное отделение Sb непосредственно сочетается с ее определением в полученном экстракте.
Экстракция неорганических комплексов
Экстракция в виде хлоридов. В солянокислых растворах Sb(III) и Sb(V) находятся в виде различных соединений и ионов в зависимости от концентрации НС1 и присутствия хлоридов других металлов. Сурьма(Ш) в растворах с низкой концентрацией НС1 существует в виде гидролизованных форм, содержащих хлорид-ион; при более высоких концентрациях НС1 — в виде SbCl3, SbCl4, SbCl| и SbCl| . Сурьма(У) в растворах НС1 образует ряд гидроксохлоридных комплексов с различным содержанием ионов ОН- и С1_. Равновесие между отдельными формами Sb(V) в растворах, как правило, устанавливается очень медленно (в течение нескольких часов и даже суток) [1383]. Поэтому для экстракции, особенно Sb(V), имеет значение не только концентрация НС1 и хлоридов других металлов, но и предыстория приготовления водного раствора. Для экстракции Sb(III) и Sb(V) из растворов НС1 используется много различных органических растворителей. Углеводороды и их галоидзамещенные плохо экстрагируют Sb. Однако с их применением можно, например, отделить Sb(III) от As(III). Для этого экстрагируют As(III) бензолом из 6—10 М HG1. В этих условиях Sb(III) остается в водной фазе [893].
Из спиртов для экстракции Sb из солянокислых растворов наиболее часто пользуются изоамиловым спиртом [191, 193, 422, 602, 646]. При экстракции из растворов, в которых наступило гидролитическое равновесие различных форм Sb(V), экстракционное равновесие устанавливается быстро лишь при концентрации НС1 ;> > 9 М; в растворах с меньшей концентрацией НС1 оно устанавливается в течение нескольких часов и даже суток [193]. Экстракция Sb (HI) изоамиловым спиртом возрастает с увеличением концентрации НС1 до 6 М, затем снова уменьшается. Уменьшение экстракции из растворов с концентрацией НС1 6 М объясняется тем, что в этих условиях Sb(III) находится преимущественно в виде кислот H2[SbCl5] и H3[SbCl6], которые плохо экстрагируются [602, 646].
102
Из солянокислых растворов Sb(III) экстрагируется ио гидратно-сольватному механизму в виде комплекса [Н+ (H2O)m"S„’ • SbCl4] [273]. В присутствии LiCl, наряду с указанными соединениями, экстрагируется также соль лития [Li+(H2O)a:-SJ/-SbCl4]. Из концентрированных растворов LiCl даже при низких концентрациях НС1 Sb(V) экстрагируется изоамиловым спиртом на 100%, независимо от того, сколько времени выдерживались растворы перед экстракцией [602]. Изоамиловый спирт экстрагирует Sb(V) в виде комплексных кислот H[SbCl6] и H[SbCl5(OH)], катионная часть которых гидратирована и сольватирована несколькими молекулами изоамилового спирта [192, 273]. Экстракция Sb(V) изоамиловым спиртом из растворов НС1 использована для выделения 125Sb без носителя из облученного олова [191].
Простые эфиры из растворов НС1 хорошо экстрагируют Sb(V) и хуже — Sb(III). Диэтиловый эфир хорошо экстрагирует Sb(V) из растворов НС1 и тем лучше, чем выше концентрация НС1. Однако при концентрациях НС1 6 М растворимость диэтилового эфира в водной фазе значительно возрастает и поэтому обычно Sb(V) экстрагируют диэтиловым эфиром из 5—6 М НС1 [165]. Извлечение составляет 80—82%. Сурьма(У) экстрагируется диизопропиловым эфиром из 1—10 М НС1 [1044, 1233, 1512, 1615, 1634, 1647]. Максимум экстракции Sb (~ 99,5%) наблюдается из растворов 5—7 М НС1. Однако при этом экстрагируется также Fe(III). Поэтому в присутствии Fe(III) рекомендуется экстрагировать Sb(V) из раствора, 2 М по НС1 (извлекается 94% Sb), поскольку в этих условиях Fe(III) не экстрагируется. Большое различие в экстрагируемости Sb(V) и Sb(III) с применением диизопропилового эфира использовано для их разделения [10441. 2,2'-Дихлордиэтиловый эфир (хлорекс) избирательно экстрагирует Sb(III) из 11 М НС1, вследствие чего находит широкое применение [37, 102, 136, 259, 260, 800]. При экстракции простыми эфирами Sb(V) экстрагируется в виде кислоты H[SbCle], а также в виде частично гидролизованных продуктов, в том числе H[SbOCl4] и H[SbCl5(OH)]. Соотношение между отдельными экстрагируемыми формами в сильной мере зависит от концентрации НС1 и общей концентрации хлорид-ионов.
Кетоны экстрагируют Sb(V) и Sb(III) из растворов НС1 подобно спиртам [192]. Наиболее часто используется метилизобутилке-тон. Максимум экстракции Sb(V) с его применением достигается при концентрации НС1 8М и составляет ~ 100% [1121]. Необходимая концентрация НС1 может быть значительно снижена (до 0,5 М) при проведении экстракции в присутствии 9—11 г-экв/л H2SO4 или НС1О4; в этих условиях извлечение Sb(V) составляет 97—99%. Из растворов НС1 Sb(III) извлекается независимо от концентрации НС1 не более чем на 70%. Из раствора, 7,5 М по НС1, вместе с Sb(III) практически полностью экстрагируются ме-тилизобутилкетоном Ga, Fe(III) и Au(III), Ge(IV) — на 93%, Mo
103
(VI) - на 92%, In - на 67%, Sn(IV) - на 63%, W(VI) - на 61%, As(V) - на 16%, Zn-на 10%, Hf - на 9%, К - на 8%, РЬ - на 5%, Cu(II) - на 4%, Na, Со и Сг(Ш) - на 1%; Bi(III), Mn(II), Та, Ni, La, Nd, Sc, Рг и Ba не экстрагируются [1208]. Экстракционная способность алифатических кетонов уменьшается с увеличением молекулярного веса [1081].
С применением сложных эфиров (этилацетат, изоамилацетат) максимальное извлечение (~ 70%) Sb(III) достигается при экстракции из 2—3 М НС1 [52, 446, 1565]. Для Sb(V) оптимальная концентрация НС1 составляет при использовании этилацетата 7М [52] и при использовании изоамилацетата >8 М [1565]; при этом Sb(V) извлекается соответственно на 90 и 95%. Наиболее полно Sb(V) отделяется от Sn(IV) при экстракции ее из 7 М НС1 (фактор разделения 95) [1253]. Среди сложных эфиров особенно подробно исследована экстракция Sb(III) три-н-бутилфосфатом (ТБФ) [258, 259, 603, 646, 1026, 1172, 1227, 1228, 1476, 1547, 1666]. Максимальное извлечение Sb наблюдается при концентрации НС1 2—5 М [646, 1026, 1666]. Указание на то, что максимум экстракции Sb(III) приходится на растворы с концентрацией НС1 8 М [1172], ошибочно. Механизм экстракции Sb(III) трибутилфосфатом из растворов HCI исследовался рядом авторов [259, 603, 1227, 1476, 1547]. Установлено, что Sb извлекается в виде соединений ЗЬС13-ТБФ, ЗЬС13-2ТБФ и HSbCl4. Соотношение между этими формами в сильной мере зависит от концентрации НС1, общей концентрации С1—, температуры и избытка ТБФ. Из растворов с концентрацией HCI > 5М Sb(III) преимущественно экстрагируется в виде HSbCl4 [259].
Введение в водную фазу хлоридов щелочных, щелочноземельных и ряда других металлов повышает извлечение Sb(III). Особенно эффективными оказались LiCl, MgCl2 и А1С13. Согласно [1666], Sb(III) экстрагируется в виде Н[ЗЬС14-2ТБФ]. Из концентрированных растворов LiCl Sb(III) экстрагируется в виде соединения 2ЫС1-ЗЬС13-2ТБФ-п Н2О [1228]. Состав экстрагирующихся комплексов при экстракции Sb(III) из растворов LiCl зависит от концентрации LiCl и HCI в водной фазе и концентрации ТБФ в органической фазе и его избытка, а также от температуры. Коэффициент распределения Sb(III) из растворов LiCl значительно выше, чем из растворов НС1 той же концентрации. Экстракция Sb(III) трибутилфосфатом из растворов НС1 в присутствии LiCl использована для фотометрического определения Sb с бриллиантовым зеленым [1666]. Сурьма(У) из растворов НС1 хорошо экстрагируется ТБФ и тем лучше, чем выше концентрация НС1 [646, 1026]. Максимальный коэффициент распределения Sb(V), достигает ~ 104 [1026]. При экстракции Sb(V) из растворов с концентрацией НС1 9 М соотношение Sb : С1~ в экстрактах равно 1 : : 5, а при концентрации НС1 < 9 М оно равно 1 : 3. Эти данные позволили сделать заключение [603], что Sb(V) экстрагируется ТБФ соответственно в виде SbCl5 и SbOCl3. Кроме того, при высо-104
ких концентрациях HCI Sb(V) экстрагируется также в виде комплексной кислоты H[SbCle] подобно тому, как она извлекается рядом других кислородсодержащих экстрагентов [193, 602, 646]., Возможна также частичная экстракция Sb(V) в виде H[SbCl5(OH)] и H[SbOCl4]. При экстракции Sb(V), частично или полностью экстрагируются As, Ga, Hg, Ge, Tl, Bi, Fe, Sn.
Нитрометан, в отличие от ТБФ, характеризуется большой селективностью извлечения Sb. Он хорошо экстрагирует как Sb(HI), так и Sb(V). Высокая избирательность экстракции Sb(HI) нитрометаном из растворов НС1 использована для концентрирования микропримесей при определении их спектральным методом в трихлориде сурьмы [381].
Экстракция в виде бромидов. Полярность связи Sb(III) и Sb(V) с Вт- меньше полярности связи с С1_. Вследствие этого коэффициенты распределения бромидных комплексов Sb(III) и Sb(V) выше, чем хлоридных. Так, если хлоридные комплексы Sb(III) и Sb(V), плохо экстрагируются углеводородами и их галогенозамещенными, то бромидные комплексы этими растворителями экстрагируются хорошо. Например, Sb(III) хорошо извлекается бензолом из 6—16 М H2SO4 (содержащей 0,005 М НВт) в виде SbBr3 [1127]. Оптимальная концентрация H2SO4 и НВг составляет соответственно 10 и 0,03 М. В этих условиях при концентрации Sb(III) 8,2-10~9—9,85-10-3 г-ион!л она экстрагируется на 98,1—99,3%. Одновременно с Sb(III) экстрагируются As(III) (99,4%), Ge (98,2%), Se (95,6%), Hg(II) (74,1%), Bi (10%), Fe(HI) (5,5%), Nb (0,9%), Pa (0,9%) и Mg (0,6%). Экстракцию Sb(III) CC14 в виде SbBrs рекомендуется проводить из раствора, содержащего 5% КВг, при концентрации H2SO4 8 М. В этих условиях вместе с Sb(HI) экстрагируются Ge, Sn, As, Se и Hg [1560].
Спирты извлекают бромидные комплексы Sb с большими коэффициентами распределения, чем углеводороды и их хлорзаме-щенные. При концентрации Sb(III) 4-10~4 г-ион!л она количественно экстрагируется из раствора, 4 М по H2SO4, содержащего 0,1—2 г-ион/л Вт- [422].
Простые эфиры хорошо экстрагируют Sb(V) (95% из 5 М НВт) и хуже — Sb(HI) (37,4% из 2 М НВти 14,9% из 4М НВт). Экстракцию Sb(V) диэтиловым эфиром используют для концентрирования микропримесей при их спектральном определении в антимсниде индия [549]. Равный объем 2,2'-дихлордиэтилового эфира из 5 М НВт извлекает Sb(V) на 89,7% [548].
Экстракция в виде иодидов. Независимо от исходной степени окисления из иодидных растворов Sb экстрагируется в виде Sb(IH). Вследствие большей устойчивости иодидных комплексов Sb хорошо экстрагируется из менее кислых растворов, чем в случае бромидных и особенно хлоридных растворов. Из раствора, 5 М по H2SO4, содержащего 0,005—0,05 М KJ, Sb(III) хорошо экстрагируется бензолом и толуолом в виде SbJs [743, 1127, 1401, 1459]. При концентрации KJ 0,01 М равным объемом бен
105
зола Sb извлекается на 99,8% [1127]. Поел добавления KJ реакционную смесь необходимо выдерживать 10 мин. и затем добавлять экстрагент и встряхивать 2 мин. В этих условиях вместе с Sb извлекается 99,0% Sn, 85,1% Se, 51,2% Hg и 44,9% As. Для отделения Sb от Sn ее экстрагируют из 4 М HJ; коэффициент разделения составляет 1,58-104, а при повышении концентрации HJ до 6 М он возрастает до 2,4-104 [1253]. Спирты также хорошо экстрагируют Sb из иодидных растворов, но несколько менее избирательно [1080].
Экстракция Sb диэтиловым эфиром в виде пиридиниодидного комплекса используется для ее отделения при фотометрическом определении в In, Ga и Т1 [64, 65]. 2,2'-Дихлордиэтиловый эфир хорошо экстрагирует Sb из иодидных растворов, но несколько хуже, чем диэтиловый эфир [548]. При экстракции Sb метилиэобу-тилкетоном извлечение ее практически не зависит от концентрации H2SO4 при постоянной концентрации KJ и составляет 99,4% при концентрации KJ 0,5 М. При увеличении концентрации KJ свыше 0,5 М коэффициент распределения Sb несколько уменьшается [1120].
Экстракция в виде фторидов. Из растворов HF как Sb(III), так и Sb(V) экстрагируется плохо, вследствие чего экстракция в виде фторидных комплексов не имеет практического значения [260, 261].
Экстракция в виде роданидов. Для экстракции Sb в виде роданидных комплексов требуется высокая концентрация SCN~ [548]. Кроме того, имеют место нечеткое разделение фаз, а также низкие коэффициенты распределения.
Экстракция внутрикомплексных соединений
Серусодержащие реагенты. Из реагентов, образующих с Sb экстрагирующиеся внутрикомплексные соединения, наибольшее значение имеют серусодержащие реагенты и прежде всего диэтилдитиокарбаминат и его аналоги. Он реагирует с Sb(III) в отношении 3:1с образованием внутрикомплексного соединения, экстрагирующегося многими органическими растворителями. Экстракция протекает в широких пределах концентрации ионов Н+ — от сильнокислых растворов (107V H2SO4 или НС1) до pH 9 [923]. Экстракцией из сильнокислых растворов Sb(III) отделяется от многих элементов [65, 924, 1257, 1258, 1398]. Поскольку диэтил-дитиокарбаминат не образует с Sb(V) экстрагирующихся комплексов, то это различие используется для их раздельного определения в морской воде [1110], а также для отделения Sb от Bi и Си. Сначала окисляют Sb(III) до Sb(V) и экстрагируют Bi и Си в виде диэтилдитиокарбаминатов, затем в водной фазе восстанавливают Sb (V) доЗЬ(Ш) и экстрагируют диэтилдитиокарбаминат Sb(III) [1665]. Поскольку экстрагирующиеся диэтилдитиокарбаминаты образуют многие элементы, то диэтилдитиокарбаминат натрия
106
часто используют для группового выделения примесей [173]. Ди-этилдитиокарбаминат натрия обычно добавляют к водному раствору и здтем экстрагируют образовавшиеся диэтилдитиокарбамина-ты металлов. Однако в кислых растворах диэтилдитиокарбаминат переходит в диэтилдитиокарбаминовую кислоту, разлагающуюся с выделением CS2. В ряде случаев используют диэтилдитиокарба-минаты ряда тяжелых металлов, в том числе Pb, Zn и др. Так, например, для выделения Sb из цинка и его солей Sb экстрагируют хлороформным раствором диэтилдитиокарбамината цинка. Вследствие значительно большей устойчивости диэтилдитиокарбамината Sb(III) она вытесняет Zn из его комплекса и переходит в органическую фазу [1398].
В последнее время для экстракции металлов в виде диэтилди-тиокарбаминатов используют диэтилдитиокарбаминат диэтиламмо-ния, хорошо растворимый в органических растворителях. С его применением при экстракции Sb допустима более высокая кислотность водной фазы, чем с применением диэтилдитиокарбамината натрия [469]. В ряде случаев для экстракции Sb вместо водных растворов диэтилдитиокарбамината натрия используют растворы диэтилдитиокарбаминовой кислоты в СНС13 или СС14. Экстракция с применением хлороформного раствора диэтилдитиокарбаминовой кислоты применяется для выделения Sb иэ In, Ga и Т1 для ее последующего фотометрического определения [65]. Другие замещенные дитиокарбаминаты, в том числе дибенэил-, пирролидин-, пи-разолин-, глицин-, гидразин- и фенилгидразиндитиокарбаминат натрия, взаимодействуют с Sb(III) аналогично диэтилдитиокар-баминату. Природа заместителя мало отражается на их устойчивости при хранении в водных растворах. Этилксантогенат калия также реагирует с Sb(III) с образованием экстрагирующегося комплекса. При экстракции СС14 из растворов, 1,8—2,5 М по НС1, содержащих 0,25% этилксантогената калия, Sb отделяется от Bi и As [970]. Экстракцию Sb хлороформом в виде купфероната используют для отделения ее от макроколичеств РЬ [1005]. Исследована экстракция Sb(III) в виде внутрикомплексных соединений с другими серусодержащими реагентами, в том числе с а-ди-тионафтойной кислотой [148], 4-меркапторезорцином [960] и ди-этилдитиофосфорной кислотой [816]. £5
Аликфосфорные кислоты. Моно- и ди-(2-этилгексил)ортофос-форная и 2-этилгексилпирофосфорная кислоты в виде 1М растворов в гептане хорошо экстрагируют Sb(V) из растворов НС1, НВт, H2SO4, HNO3 и НС1О4 [411]. Кривые зависимости экстракции Sb(V) от природы экстрагента и минеральной кислоты во многом сходны и их форма зависит в основном от концентрации ионов Н+. Исключение представляют лишь НС1 и НВг, иэ концентрированных растворов которых Sb(V) экстрагируется по другому механизму. Из 9—11 М НС1 Sb(V) экстрагируется в виде кислоты H[SbCl6], а из 8 М НС1 — в виде H[SbCle] и H[Sb(OH)Cl5], сольватированных экстрагентом. При экстракции Sb(V) этими
107
экстрагентами из разбавленных растворов указанных кислот она извлекается по ионнообменному механизму в виде внутрикомплекс-ных соединений. Сурьма(Ш) алкилфосфорными кислотами экстрагируется хорошо как из растворов H2SO4, HNO3 и НС1О4, так и из галогеноводородных кислот. Если при экстракции Sb(III) из растворов H2SO4, HNO3 и НС1О4 одинаковой концентрации существенных различий не наблюдается, то при экстракции из галогеноводородных кислот коэффициенты распределения Sb(III) уменьшаются в ряду: НС1 НВг HJ HF [1282]. Изучена экстракция Sb(III) ди-(н-бутил)фосфорной кислотой [1137].
Экстракция алкилфосфорными кислотами использована для разделения Sb, In и Ga [1282] и для определения Sb в In и Bi [167]. Экстракцией из растворов H2SO4, HNO3 и НС1О4 0,25 М растворами моно- или ди-(2-этилгексил)фосфорной и 2-этилгексил-пирофосфорной кислот в инертных растворителях Sb(III) может быть отделена практически от любых количеств Zn, Cd, Си, Fe(II), Со, Ni, As, Сг и Mn [410]. Экстракцией Sb(III) ди-(2-этилгексил)-фосфорной кислотой из слабокислых или нейтральных растворов она может быть отделена от Sb(V) [847].
Другие реагенты. Из других реагентов, используемых для экстракции Sb, следует упомянуть N-бензоилфенилгидроксил-амин, 0,1 М хлороформные растворы которого экстрагируют Sb(III) из 5 М H2SO4 [613, 1300]. Экстракцией из 2 М НС11 %-ным раствором дифенилкарбазида в амиловом или изоамиловом спирте отделяют Sb(III) от Се, Y, Zr, Cd, U, Ва и Sr [783].
Экстракция ионных ассоциатов
Экстракция ионных ассоциатов, образуемых анионом SbCl6 с катионами основных красителей, широко используется в экстракционно-фотометрических методах определения микроколичеств Sb. Для отделения Sb(V) экстракцией в виде ионных ассоциатов иногда вместо основных красителей используют бесцветные или слабоокрашенные органические основания. Так, для выделения Sb из хрома Sb(V) экстрагируют 0,1 М раствором трибензиламина в СНС13 в среде 7—10 М НС1 [471]. Для экстракции Sb(V) в виде ионных ассоциатов используют растворы N-додецил-триалкилметиламинов в ксилоле, количественная экстракция достигается при концентрации НС1 в водной фазе >6М [1365]. 0,06 М раствор тридодециламина в ксилоле экстрагирует Sb(V) из конц. НС1 и из 1,5 М НС1, содержащей А1С13 (2,5 М), с коэффициентами распределения 100; при этом в значительных количествах также экстрагируются Fe(III), Ра, Zn, Ag и Hg [854].
Для отделения Sb(V) от Bi(III) рекомендуется [2161 экстрагировать ее из 8 М НС1 растворами диантипирилметана в инертных растворителях.
108
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Ионообменная хроматография. Для отделения Sb чаще используют аниониты, которые сорбируют как Sb(III), так и Sb(V) в виде анионных комплексов. Сорбированные анионитом элементы, в том числе и Sb, избирательно элюируют растворами подходящих реагентов (табл. 7). С применением катионитов, как правило, в ионообменной колонке поглощаются сопутствующие элементы, a Sb проходит в фильтрат (табл. 8). Для предупреждения сорбции Sb катионитом в раствор вводят винную кислоту или ее соли или другие маскирующие вещества.
Адсорбционная хроматография. Эффективным методом разделения является адсорбционная хроматография. Так, Ge и Sb могут быть разделены па колонке 0,5 X 45 см (с капилляром внизу, длиной 15 см), заполненной 2 г целлюлозы. Анализируемый раствор пропускают через колонку, на которой сорбируются оба элемента, затем элюируют Ge пиридином, a Sb — бензолом [1098]. Целлюлоза и некоторые ее замещенные, особенно фосфатцеллюло-за, прочно сорбируют Sb(III) из смеси (2 : 98) 90%-ной HNO3 с диэтиловым эфиром, которая затем легко вымывается с колонки 0,5—5 М водным или эфирным раствором НС1 [1358]. При этом Sb полностью отделяется от As(V), Hg(II), Au(III), U(VI), щелочных и щелочноземельных металлов, которые проходят через колонку. Fe(II), Fe(III), Mn, Ag, Zn, Ст, Co и Си сорбируются целлюлозой вместе с Sb, но избирательно вымываются с колонки растворами NH4SCN в смеси (1 : 4) метанола с диэтиловым эфиром.
Для выделения радиоактивных изотопов Sb из облученного урана применен Zrs(PO4)4 [860], который, в отличие от органических сорбентов, устойчив к радиоактивному излучению.
Тонкослойная хроматография. Этот метод разделения наиболее часто используется для отделения Sb от As, Sn и Bi (табл. 9). В качестве тонкого слоя чаще других используется силикагель [140]. В некоторых случаях используют его смеси с А12О3 [1516] и с крахмалом [141]. Применяется также целлюлоза [1281, 1541]. Поведение Sb и наиболее часто сопутствующих ей элементов (As, Sn) на тонких слоях целлюлозы сходно с поведением в методе бумажной хроматографии [1685].
Разделение органических соединений Sb(III) и Sb(V) на слоях А12О3 наиболее эффективно с применением в качестве растворителей смеси (90 : 10) бензола с этанолом и смеси (95 : 5) этилацетата с этанолом [219].
Бумажная хроматография. По своим возможностям методы бумажной хроматографии сходны с методами тонкослойной хроматографии. Основное различие заключается только в том, что в методах бумажной хроматографии сама бумага одновременно является тонким слоем и подложкой. Методы бумажной хроматографии позволяют отделять микро- и ультрамикроколичества Sb от элементов с близкими химическими свойствами. Эти методы ха-
109
Таблица 7
Ионообменное отделение сурьмы с применением анионитов
Отделяемые элементы Среда Анионит Колонка Элюирование Литература
As(V), Sn(IV), Sb(V) 5M HC1 ЭДП-10П или АВ-10 в С1“-форме, 10 г d. 8—10 мм As—150 ли 5МНС1; Sn —250 мл 0,5 М НС1; Sb — 300 ж 5.V H2SO4 [249]
As(III),Sb(III) 1 M NaOH тартрат Дауэкс-21К в С1“-форме, 100—200 меш 1,4x28 см Sb проходит в фильтрат, a As вымывают 1 М H2SO4 [969]
Cu, Fe, Al, U, Pb, V(IV), Cr(III) 5 %-пая малоновая кислота (pH 4,8) Дауэкс-21К в мало-натной форме 1,4x22,5 см Щелочные и щелочноземельные элементы— 35 мл воды; Sb — 200 мл 0,1—1 А/ НС1 [842]
As(V), As(IlI), Se, Те, Ge, Sb(III), Sb(V) 7М НС1 Дауэкс-1 или АВ-17 в С1 “-форме, 15— 20 мк 0,2x7,5 см As(V) — 5 мл 11,2 М НС1; Se — 5 мл 6,6 М HCl; Ge и As(IH) — 5 мл 3 М НС1; Те —3 мл 0,1 М Н2С2О4; Sb(IlI) и Sb(V)-0,3M (NH4)2C2O4+ +0,2 М NH4OH [600]
In, Sn, Те, Sb(V) 0,5,17 HCl Амберлит 1RA-400 в Cl-форме 0,2x8 см Sb — 1,0 мл ЗМ HCl; In—1,5 мл ЗМ HCl; Sn — 1,0 мл 2 М НС1О4 [1559]
Sn, Sb(V) 20%-ный маллонат аммония (pH 4,8) Амберлит 1RA-400 в малонатной форме 1,0x25 см Sb — 120 мл 20%-ного малоната аммония (pH 4,8); Sn—100 мл 9 А H2SO4 [1016]
As, Sn, Sb(III) Раствор тиосолей, ЗМ КОН Дауэкс-2 в ОН“-форме 1,7x18 см Sn — 700 мл 0,5 Af КОН; As —3500 мл 1,2 М КОН; Sb — 1500 мл 3,5 М кон ]1243]
Fe, Ni, Tl,Zr, V(1V), Sn, Sb(III) 0,5 М НС1 АВ-17 в С1 “-форме, 2 г <7=1,2 см Fe, Tl, Zr, V, Ni, Sn—50 млО,5М HCl; Sb — 50 мл 1 N H2SO4 [3, 611]
Таблица 7 (окончание)
Отделяемые элементы Среда Анионит Колонка Элюирование Литература
As, Bi, Sb(III) 2M HCl Дауэкс-21К в С1“-форме 1,4x28 см As — 300 мл 0,05 М H2SO4; Sb— 300 мл 2 М H2SO4 [1019]
As(HI), Sb(lII) 0,5 M NaOH + 3% винной кислоты Дауэкс-21К в тартратной форме 1,4x28 см As — 300 мл 0,1 М HCl; Sb — 300 мл 0,5 М тартрата натрия [1019]
Sn, Mo(VI), Sb(III) 3 M H2SO4 + 3 M nh4scn Дауакс 1X8 в SCN~-форме 0,5x12 см Sb — 15 мл 1 N H2SO4; Sn — 15 мл 0,5 М NaCl + 0,5 М NaOH; Мо — 0,5 М NaOH [1214]
Sn, Cu, Sb(III) 3 M H2SO4 + 3 м nh4scn Дауакс 1X8 в SCN“-форме 0,5x12 см Sb — 15 мл 1 N H2SO4; Sn — 15 мл 0,5 М NaCl + 0,5 М NaOH; Cu — 50 мл 2 М NH4C1 + 2M NH4OH [1215]
In, Zn, Sb(IlI) 0,1— 0,2 M HCl ЭДЭ-10П в С1"-фор-ме 1,2x24 см In и Zn проходят в фильтрат; Sb — 1 N H2SO4 [316]
Sn, Sb(III) 6-7 M HCl Амберлит CG-4B в С1~-форме 0,3x13 см Sb — 3 M HCl, Sn — 1 M HCl [1202]
Sn, Те, Sb(V) 6—IM HCl Дауэкс 1Х8 или АСД-2 в С1“-форме Высота слоя 10 см Sb — 2 мл 5M HCl; Те—2,2 мл IM HCl; Sn —1 мл 1,8 M HC1O4 [631]
Таблица 8
Ионообменное отделение сурьмы с применением катионитов
Отделяемые элементы Среда Катионит Колонка Элюирование Литература
Fe, Си, Со, Cd, Ag, Ni, Pb, Сг(Ш), Sb(V) 16—20-кратные количества винной кислоты Цеокарб-225 в Н+-форме, 10 г 1,0-1,6x18— 25 см Sb проходит в фильтрат [1231]
Fe, Co, Ni, Zn, Pb, Sb(III) 0,33 М винная кислота КУ-2 в Н+-форме 1,2—1,5x25 см То же [1208]
Sb(III), Sb(V) Си, Ni, Co, Cd, Zn, Mn(II), Fe, Cr(III), Sb(III) 12 М НВг Тартратная Дауэкс 50X4 в Н+-форме Цеокарб-225 в NH4+H£opMe 0,6x2—6 см 1,6x25 см Sb(IU) — 15 мл 9 (И НВг; Sb(V) — 15 мл 9 М НВг 4- 0,05 MKJ Sb проходит в фильтрат [1382] [1229]
Си, Sb(lII) 5%-ный тартрат аммония, pH 9 Вофатит Р в NH4+-форме 0,4—0,75 мм 1,6—2,5x30 см То же [1317]
Cd, Bi, Ag, Al, Sb(III) 5—10 %-на я винная кислота КУ-2 в Н+-форме 0,6x8 см [200]
Bi, Си, Sb(III) 0,5 М НС1О4 + 1 М винная кислота Дауэкс-50 в Н+-форме 1,1x8 см [1561]
Zn, Tl,,Sb(III) 2М НС1 -|- 10% винной кислоты Катионит СБС в Н+-форме 1,5 X 20 см » [330]
Таблица 9
Отделение сурьмы методами тонкослойной хроматографии
Разделяемые элементы Состав слоя Среда Растворитель и время проявления Реагент для обнаружения пятен Литература
Sb(III), As(III), Bi(III) Силикагель G- 10%-ный раствор НС1 1 М раствор винной кислоты или 0,1 М НС1; 30 мин H2S [1629]
Sb(III), Sn(IV), AS(III> Силикагель, закрепленный крахмалом 3 М НС1 Смесь (50 : 46 : 1,4 :2,6) «-бутанола, бензола, 1 М НС1 и 1 М HNO»; >25 мин Цветные' или люминесцентные реакции [141]
Sb(III), Sn(IV), As(III) Силикагель G- 3 М НС1 Смесь (20 : 1) м-бутанола с 1 М НС1; 5 час. H2S [14И]
Sb(III), Sn(IV) As(III) Смесь силикагеля D с А12О8 Раствор пирролидин-дитиокарбаминатов в СНС13 Смесь (2 : 1) бензола с CHC1S 5%-ный раствор CuSO4'5H2O [1516]
Sb(III), Sn(IV), As(III) Целлюлоза или силикагель 1-3 М HCI, hno8 или СНгСООН Смеси пропанола, этанола или бутанола с конц. НС1, HNOs или СНаСООН 2%-ный раствор 8-оксихи-нолина в этаноле [1281]
Sb(HI), Sn(IV), As(III) Крахмал Аммиачно-щелочной раствор тиосолей 1 М NHiOH + (NH4)2S2+X Пятна окрашены [966]
Sb(III), Sn(IV), As(III) Целлюлоза 3 М НС1 Смесь (25 : 5) н-бутанола с 3 М НС1 Спиртовый раствор ализарина или дитизона [1541]
Sb(IH), Bi(III), As(III) Силикагель G 10%-ная НС1 10%-ная НС1 Раствор FeS в 10%-ной НС1 [1578, 1628]
Sb(III), Sn(IV), As(III), Pb(II) Силикагель G Солянокислые растворы Амилацетат, насыщенный конц. НС1 Раствор (NH4)sS2+x [1458]
Sb(HI), Sn(IV), As(III), Mo(VI), Силикагель G- Солянокислые растворы Смесь (9 : 1) метанола, этанола с 2—6 М НС1 Атмосфера водорода [855]
и*. Sb(III), Bi(III), Pb(II), Cu(II), Cd(II) Целлюлоза MN 300-}- 4- катионит Челекс Й+-форме Кислые растворы IM HNOs — [903]
05
рактеризуются очень высокой чувствительностью, достигающей нескольких нанограммов Sb [1589]. Имеется ряд модификаций метода бумажной хроматографии, заключающихся в использовании бумаги, импрегнированной различными веществами. Например, для разделения Sb(I II), As(III) nSn(IV) предложено использовать бумагу Ватман № 1, импрегнированную Fe[Fe(CN)6] [1237]. С применением бумаги, импрегнированной Ti(WO4)2, Sb отделяется от многих ионов [1452]. На бумаге Ватман № 1, импрегнированной Sn(WO4)2, отделяют Sb от Ga, Fe, Cd [1456], а на бумаге, импрегнированной Sn3(PO4)4,— от As(III), Au(III), Pd(II), Pt(IV) и Hg(II) [1451]. На бумаге, импрегнированной SrCrO4, Sb отделяется от Sn(IV), Ti(IV), РЗЭ [1455]. Хорошие результаты получены при использовании ионообменных бумаг. Так, с применением бумаги, пропитанной фенилбензогидроксамовой кислотой, можно отделить Sb(III) от As(III), Bi(III), Fe(III), Pb(II), Cu(II), Ni(II), Co(II) [1527]. Эффективной оказалась бумага, пропитанная сильнокислотным катионитом Амберлит SA-2 [1527], а также три-н-ок-тиламином [1631] и сильноосновным анионитом SB-2 [1527].
Методом бумажной хроматографии разделяют Sb(IH) и Sb(V) [467, 1181, 1356, 1451, 1453]. Для повышения эффективности разделения часто используют различные комплексообразующие вещества. Использование роданидных комплексов позволяет разделять Sb(III), As(III), Bi(III) и Pb(II) [1308]. Хроматографирование в виде тартратных и оксалатных комплексов обеспечивает отделение Sb(III) от многих элементов, в том числе от As и Sn [1523, 1524, 1536]. В качестве растворителей для хроматографирования наиболее часто используются растворы НС1 и ее смеси с органическими растворителями (табл. 10).
Отделение Sb методами бумажной хроматографии часто сочетается с ее непосредственным качественным обнаружением на полученных хроматограммах (см. главу III), а также с количественным ее определением по площади окрашенных пятен, получаемых после обработки хроматограмм подходящими реагентами. Среди этих реагентов используются H2S, растворы FeS в соляной кислоте [467, 1168], растворы К J [1419, 1589], дитизона в СНС13 [887, 1519, 1589], 12-молибдофосфорной кислоты [1455]. Для обнаружения Sb(Vj на хроматограммах эффективным реагентом является смесь (1 : 1) 0,05%-ного раствора родамина С в 2—6 НС1 и 20%-ного раствора КВг [1337]. Интересным оказалось сочетание разделения методом бумажной хроматографии в активационном анализе с последующим определением злементов в зонах по их активности [922].
Экстракционная хроматография. В этом методе удачно сочетаются достоинства экстракции и хроматографии. Разделение основано на различии в распределении разделяемых веществ между двумя несмешивающимися жидкостями, одна из которых подвижна, а другая фиксирована на носителе. Теоретические основы и практическое значение экстракционной хроматографии рассмот-114
Таблица 10
Отделение сурьмы методами бумажной хроматографии
Отделяемые ионы Бумага Растворитель Литература
Sb(III), As(IU), Sn(IV) в 0,96 М тартрате Ватман № 1 Этанол 60%-ный [1524, 1536]
Sb(V), As(V), Sn(lV), Hg(II) To же н-Бутанол, насыщенный 3,4 М НС1; Я/ Sb — 0,34 [1419]
Sb(III), As(IH), Sn(IV) В Смесь (8 :1,5 : 2,5) СНзСООН, ацетилацетона и воды [887]
Sb(III), Ag, Cd, Sr, Ba, Cu, Co » Смесь (420 : 30 : 150) этанола, конц. НС1 и воды [922]
Sb(III), Sn(ll) в Смесь (1:1) н-пропанола и IM НС1 [1372]
Sb(V), Sn(IV) в То же [1372]
Sb(V), As(V) » » [1372]
Sb(III), Ga(III), Fe(III), Fe(ll), Cd(ll) Бумага, импрегни-рованная Sn(WO4)a Смесь (3:1:1) н-бутанола, диоксана и 10 М НС1 [1456]
Sb(lH), Sn(Il), Ti(lV), РЗЭ Ватман № 1, им-прегнированный SrCrO4 0,2 М Sr(NO3)2 (pH 6) [1455|
Sb(III), Bi(IlI), As(III), Pb(II); роданидные комплексы Ватман № 1 Смесь (40 : 20 : 10) ацетона, воды и конц. НС1 [1308]
Sb(III), As(III), Sn(IV) То же н-Бутанол, насыщенный 3 М НС1; Rf Sb— 0,34; As —0,83; Sn —0,99 [1589]
Sb(III), As(V), Sn(II) В 15%-ный раствор (NHACOg в 1 М NH4OH; Rf Sb — 0,26; Sn —0,09; As —0,84 [1168]
Sb(lll), Sb(V) Бумага SS н-Бутанол, насыщенн ый 1 M НС1; Rf Sb(III) — 0,64; Sb(V) —0,97 [467]
Sb(IIl), As(III), Sn(ll) Ватман № 1 60%-ный этанол [916]
Sb(V), As(V), Sn(IV) Ватман № 1 или № 4 или Шлейхер и Шюлль Смесь (1:1:1) н-бутанола, ацетилацетона и воды; Rf Sb—0; As —0,51 и Sn — 0,65 [1089]
рены в работах [1051, 1319]. В качестве неподвижных фаз (экстрагентов) для отделения Sb используются три-(н-октил)фосфинок-сид [968], ди-(2-этилгексил)фосфорная кислота [1027], аламин 336 [834], ТБФ [15, 56]. Для отделения Sb методом экстракционной хроматографии пригодны практически все вещества и их смеси, используемые для экстракции Sb.
415
Для отделения Sb(III) от As предложено [968] использовать колонку, заполненную порошком целлюлозы, обработанной три-(н-октил)фосфиноксидом. Для выделения Sb из смеси продуктов деления U и Np используют колонку с порошком политрифтормоно-хлорэтилена, смоченного ТБФ или ди-(2-этилгексил)фосфорной кислотой [1027]. Система аламин-336 — растворы НС1 оказалась эффективной для отделения Sb(V) от Sn(IV) [834].
ОТГОНКА В ВИДЕ ЛЕГКОЛЕТУЧИХ СОЕДИНЕНИЙ
Отгонка в виде трихлорида. Отгонка Sb в виде SbCl3 является эффективным методом ее отделения. Он характеризуется высокой селективностью и в соответствующих условиях Sb может быть отделена от всех элементов, в том числе и от As и Sn [36, 1022, 1113].
Разделение As, Sb и Sn отгонкой в виде хлоридов исследовано с применением их радиоактивных изотопов при определении в железе [1022], чугуне и сталях [1113]. Установлено, что при использовании навески 2 г и при отгонке AsCl3 при 135° С одновременно с ним отгоняется до 5—10% Sb [1113]. Снижение температуры отгонки с 135 до 112° С хотя и значительно уменьшает переход Sb в дистиллят, но полностью его не устраняет, и загрязнение дистиллята сурьмой всегда имеет место при отделении малых количеств As от больших количеств Sb. В этих случаях необходимо полученный дистиллят подвергнуть вторичной перегонке в тех же условиях. Остаток присоединяют к основному раствору, содержащему Sb и Sn, для последующего отделения Sb. При отделении малых количеств Sb от больших количеств Sn оно также частично переходит в дистиллят, содержащий Sb. Для полного отделения Sb от Sn в таких случаях требуется повторная отгонка [150]. Введение СаС12 в раствор перед отгонкой (до 3,0—3,4 М) позволяет полностью отделять As от Sb из растворов, 2,4—5,0 М по НС1 [36].
Поскольку As(V) не образует летучего хлорида, его восстанавливают добавлением гидразина. Иногда используют серный цвет. Соли Fe(II) и Сн(1) также восстанавливают As(V) и Sb(V), но сами образуют осадки в перегонной колбе, что приводит к возникновению толчков, особенно при отгонке Sb, которую проводят при 155— 165° С- Сурьма отделяется от Ge отгонкой последнего в виде GeCl4 при кипячении солянокислого раствора и пропускании С12. Вместо С12 можно использовать и другие окислители, в том числе КМпО4 и К2Ст2О7.
Отгонка в виде трибромида. Вследствие меньшей летучести SbBr3 по сравнению с SbCl3 (см. табл. 2) отделение Sb отгонкой в виде SbBr3 используется редко. Однако в случае выделения Sb из бромидных растворов наиболее простой представляется непосредственная отгонка SbBr3 [1305].
Отгонка в виде стибина. В настоящее время отгонка в виде SbH3 является одним из наиболее эффективных методов отделения 116
микро- и ультрамикроколичеств Sb и используется в различных методах ее определения. В качестве восстановителей используют цинк в присутствии небольших количеств SnCl2 или TiCl3. Применение вместо гранулированного цинка таблеток цинковой пыли позволяет значительно сократить продолжительность выделения SbH3 [1671]. Амальгамированный цинк в среде >9М НС1 также рекомендован в качестве восстановителя [277]. Магний, будучи более чистым в отношении примеси Sb, более удобен для выделения микроколичеств Sb [1432, 1433]. Однако лучшим является Na[BH4], который свободен от загрязнений Sb и довольно быстро переводит ее в SbH3 [1367, 1499]. Выделяющийся SbH3, как правило, непосредственно используют для определения или обнаружения Sb. Для использования всего образовавшегося SbH3 в последующем методе его определения через сосуд, в котором происходит восстановление, пропускают газ-носитель (воздух, Н2, N2 и др.).
Описано электрохимическое восстановление Sb до SbH3 [984]. Для определения Sb в органических веществах предложен метод разложения их нагреванием с магнием, в результате чего вся сурьма переходит в Mg3Sb3, который затем разлагают серной кислотой, а выделяющийся SbH3 током N2 вытесняют в поглотительную склянку, содержащую раствор окислителя [1191].
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Электролиз и электродиализ при контролируемом потенциале позволяет избирательно выделять из раствора либо Sb, либо другие элементы.
Электроосаждение наиболее часто используется при определении микроколичеств Sb методами инверсионной вольтамперометрии (см. главу IV). Миллиграммовые количества Sb осаждают при контролируемом потенциале в виде элементной Sb для ее гравиметрического определения [47, 279, 849—852]. Из лимоннокислого раствора Sb можно отделить от Bi и Sn [1025]. Описан [89] метод отделения, основанный на электроокислении Sb(III) до Sb(V) на графитовом электроде при потенциале 0,8 в в растворах НС1 в присутствии родамина С, образующего на электроде с Sb(V) осадок гексахлоростибата родамина С, используемый для последующего определения Sb методом инверсионной вольтамперометрии. Для выделения радиоактивной Sb, а также Cd, Pd и Ag из смеси продуктов деления рекомендован метод внутреннего электролиза в среде 5 М NaCl с использованием ячеек с разделенными катодным и анодным пространствами [1616].
Электрофорез. Сурьму этим методом обычно отделяют в присутствии веществ, обеспечивающих удержание Sb в растворе. Кроме того, применение комплексообразующих веществ расширяет также возможности разделения. Одним из комплексообразующих реагентов, наиболее часто используемых при отделении Sb
117
от других элементов, является винная кислота. С ее применением на полосах (Z = 75 см ) бумаги Итон-Дайхман 301 или 950 при напряжении 500 в при концентрации винной кислоты 0,5 М разделяются Sb(III), Sn(IV), и Te(IV) [1520]. При отделении Sb от 1г, Os, Pd, Pt, Си, Fe, Те и Se хорошие результаты получены при использовании 0,1 М растворов лимонной, винной, а-изоокси-масляной кислот или глицина [1333]. С использованием бумаги Ватман № 1 при градиенте потенциала 15 в/см в присутствии 0,05 М Н-(2-оксиэтил)иминодиуксусной кислоты разделяются Sb(III), As(V), Ge(IV), Te(lV). Au(III), Se(lV), Re(VII), W(VI) и Mo(VI); Sb(III), Ge(IV), As(V), Bi(III), V(V), Ta(V), Nb(V) и Cr(Vl); Sb(III), Te(IV), Mn(II), Cr(III) [1198, 1199].
Исследована [1332] подвижность комплексов Sb(III), Sn(lV) Hg(II), Pb(II), Bi(III), Ag(I), Gu(II), Zn(II), Mn(II), Ni(II), Co(II) и Fe(III) с ЭДТА и нитрилотриуксусной кислотой при высоковольтном электрофорезе (10 000 в) при pH 3—8. Изменение концентрации указанных веществ в пределах 0,01—0,05 М по-разному влияет на подвижность и размер пятен указанных элементов. Нитрилотриуксусная кислота оказалась более эффективной. Иногда вместо введения комплексообразующих реагентов в анализируемый раствор используют бумагу, импрегнированную этими реагентами [1116].
ДРУГИЕ МЕТОДЫ
Метод кольцевой бани применен для отделения Sb при ее определении в крови [1644]. Метод позволяет отделять Sb от Bi, Fe, Pb, Hg, Tl, Cu, Cd и Zn из растворов с ее содержанием до 10~7 г-ион!л.
Метод зонной плавки успешно использован для отделения Sb от Bi [933].
Глава VI
ОПРЕДЕЛЕНИЕ СУРЬМЫ
В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
АНАЛИЗ РУД, МИНЕРАЛОВ, ГОРНЫХ ПОРОД И ПОЧВ
Методы разложения. Поскольку ряд соединений Sb, особенно хлориды, легко летучи (см. табл. 2), то при разложении анализируемого материала необходимо принимать меры к устранению возможных ее потерь. Следует избегать кипячения растворов, содержащих НС1, или, если нельзя исключить эту операцию, проводить ее с применением обратного холодильника. Способ разложения анализируемого материала в сильной мере зависит от его природы и требований последующего метода определения Sb.
Руды, минералы, горные породы и почвы можно разлагать нагреванием с H2SO4, NaHSO4, Na2S2O7 и K2S2O7 или сплавлять с гидроокисями щелочных металлов. Для разложения кварца и силикатных материалов наиболее простым методом является обработка смесью HF и H2SO4 [492] и смесью HF с HNO3 [1338]. Для разложения сульфидных минералов и руд для последующего определения Sb методом атомно-абсорбционной спектрофотометрии рекомендуется применять смесь HNO3 и НС1. Сульфидные руды, содержащие Sb и другие элементы, образующие летучие хлориды, разлагают нагреванием в колбе Кьельдаля со смесью (NH4)2SO4, K2SO4, H2SO4 и серного цвета, необходимого для восстановления Sb(V) до Sb(III). В ряде случаев разложение анализируемого материала сочетают с отделением Sb. Так, например, поступают при определении Sb в каменных метеоритах радиоактивационным методом [1324]. Некоторые из методов разложения анализируемых материалов приводятся ниже при рассмотрении методов их анализа. Ряд методов разложения горных пород, руд и минералов описан в руководствах по их анализу [150, 1190].
Методы определения. В настоящее время в химическом анализе имеется тенденция к использованию инструментальных методов, характеризующихся высокой производительностью и экспрес-сностью. Особенно перспективными представляются методы, не требующие разложения анализируемого материала. Среди них наибольшее применение находят прямые спектральные методы.
119
По одному из них [1447] определение Sb а также Sn и РЬ в горных породах и рудах проводят следующим образом.
Пробу смешивают (3 : 1) с буфером, состоящим из 25 г угля, 25 г порошка гранита и 25 г серы, предварительно пропитанным раствором Bi(NO3)3 (внутренний стандарт). 35 мг полученной смеси помещают в кратер (d = 3 мм, h = = 5 жж) нижнего угольного электрода и сжигают в дуге переменного тока (12—16 а, 3 мин.). Используют спектрограф СТЭ-1 с решеткой 600 штрих 1мм. Эталоны готовят введением Sb2O5, SnO2 и РЬО2 в основу, состоящую из гранита, ультрабазальта и кварца, и смешивают с тем же буфером. Калибровочный график строят в координатах Л У — 1g с. При содержании Sb 0,0001 — 0,03% ошибка составляет 15—20%.
В другом методе [354] для определения Sb и As в горных породах поступают следующим образом.
Пробу разбавляют равным количеством смеси, состоящей из NH4C1, серы и угольного порошка в соотношении (3 : 1 : 6), и загружают в полость стального камерного электрода (h = 40 мм, d = 16 мм, толщина стенок 2 мм). В крышке электрода имеется сопло в виде угольного стержня (d = 6 мм, канал d = 1,1 ч- 1,4 мм). После нагрева электрода пламенем (1 мин.) зажигают дугу переменного тока (14 а, 3 мин.) между соплом и верхним угольным электродом. Используют спектрограф ИСП-30. Предел обнаружения Sb 4,8-10~s%.
Описано спектральное определение Sb, Sn, TI, Ge и Zn в горных породах [1504]. В геологических материалах определяют Sb одновременно с As, Cd и Zn. При содержании Sb 0,7—1% и навеске 50 мг ошибка определения <^15% [971]. Описан спектральный метод определения Sb и других 10 элементов в горных породах с использованием ZnS в качестве буфера и внутреннего стандарта [1288].
Для определения Sb, Ag, Си, Sn, Pb, Mn, В, Hg, Zn, W, Mo и As в глинистых осадках эталоны готовят введением окислов определяемых элементов (1-10-4—1 • 10-1%) в искусственную основу, состоящую из SiO2, А12О3, Fe2O3, MgO, CaO, Na2O и K2O, которую прокаливают при 900° С.
Пробы и эталоны разбавляют угольным порошком в 2 раза и помещают в канал угольного электрода. Спектры возбуждают в течение 30 сек. в прерывистой дуге (10 гц , 5 а). Затем, не прерывая экспозиции, делают переход на непрерывную дугу (50 гц, 5 а) в течение 15 сек. (что необходимо для возбуждения спектров В, Мо и W). Спектры (обе экспозиции) фотографируют на спектрографе PGS-2 на одно и то же место фотопластинки. Линию In 293,2 нм используют в качестве внутреннего стандарта для легколетучих элементов, а линию Pd 311,4 нм — для труднолетучих. Предел обнаружения Sb 1.10~2%; погрешность 7,1 —15,3% [1187].
Описано [879] определение следовых количеств элементов, в том числе Sb, в фосфатных горных породах с пределом обнаружения Sb и большинства других элементов в интервале 10-5—10-4% (Sr =0,1-4- 0,2).
120
Для определения следовых количеств Sb и 15 других элементов в кварце и карбонате кальция используют свободно горящую дугу и дугу, стабилизированную смесью газов (О2 + Аг или О3 + Не), с применением в качестве флюсов Li2GO3 или LiCl [937]. Использование стабилизированной дуги позволяет повысить чувствительность определения Sb и ряда других элементов в 2—10 раз.
Для быстрого определения Sb, а также Sn в рудах и продуктах их обогащения предложен рентгенофлуоресцентный метод [543]. С использованием источника излучения, энергетический спектр которого содержит 5% излучения с энергией 53 кэв, 3% излучения с энергией 84 кэв и 92% тормозного у-излучения с энергией до 800 кэв, и в качестве приемников флуоресцентного излучения Sb (26,4 кэв) и Sn (25,3 кэв) сцинтилляционного счетчика с кристаллом NaJ(Tl) метод позволяет определять Sb и Sn при их содержании до 0,1%.
Для определения Sb в горных породах, минералах, рудах, метеоритах и почвах предложен ряд вариантов активационного анализа. В некоторых случаях предусматривается определение только Sb > 2-10‘>5% (230, 1289]. В большинстве методик наряду с Sb определяют ряд других элементов. Так, например, в силикатных породах, кроме Sb, определяют Та, Th, Hf, U, Rb, Gs, Sr, Ba, Sm, Tb и Yb [949].
Описано [1061] активационное определение в горных породах Sb и 14 других элементов. С использованием 2&2Cf в качестве источника нейтронов предложен полевой метод анализа минералов, горных и осадочных пород, позволяющий определять Sb и 19 других элементов без разложения проб [1515]. G применением полупроводниковых детекторов в сочетании с ЭВМ недеструктивным вариантом активационного анализа определяют Sb и 30 других элементов в горных породах, рудах и минералах [427]. Однако, когда требуется более высокая чувствительность, проводится разложение облученного материала и выделение Sb.
Для определения Sb в ее рудах и концентратах наиболее точные результаты получают методом броматометрического титрования. При определении Sb в ртутно-сурьмянистых рудах или огарках, содержащих до 10% флюорита, поступают следующим образом.
К пробе (1 г) в конической колбе добавляют 0,5—1 г Н3ВО3, 2—3 г K2SO4 или Na2SO4, 1/4 часть беззольного фильтра (с? — 12 см), 30 мл конц. H2SO4 и нагревают до начала выделения дыма H2SO4. Затем колбу закрывают воронкой и продолжают нагревание до обесцвечивания ( ~ 3 час.). Остаток немного охлаждают, приливают 1 мл раствора клетчатки (для его приготовления 2 фильтра смачивают 150 мл конц. H2SO4 и размешивают до полного растворения) и вновь нагревают до обесцвечивания. К охлажденному остатку приливают 30 мл воды, кипятят 2—3 мин. и после охлаждения приливают 20 мл конц. НС1, 60—70 мл воды, нагревают до 60—70° С, добавляют 0,1—0,2 г КВт и титруют 0,025 N КВгО3 в присутствии метилового оранжевого [597].
121
При определении Sb в сульфидных рудах и минералах пробу разлагают нагреванием с HNO, или смесью H,S04 с Н902 [1507, 1508].
Для отделения As к 2 мл полученного сернокислого раствора прибавляют 5 мл воды, 10 мл 20%-пой НС1, 0,2 г КВт, немного N2H4.H2SO4 и отгоняют AsCl3 до получения 5 мл дистиллята. При содержании As < 0,8 мг к остатку вновь добавляют 5 мл 20%-ной НС1 и отгоняют еще 5 мл дистиллята. Если As присутствует в больших количествах (20—200 мг), то отгонку повторяют до 7 раз. В полученном дистилляте определяют As титриметрическим методом, а в оставшемся растворе титруют Sb броматом калия. Мешающее влияние Fe(III) устраняют добавлением Н3РО4, а РЬ и Hg осаждают в виде бромидов.
Описан ускоренный вариант броматометрического определения Sb в ее рудах и концентратах [1625]. Для определения Sb в рудах рекомендовано также йодометрическое титрование [1184].
Малые содержания Sb в горных породах, почвах и минералах наиболее часто определяют экстракционно-фотометрическими методами с применением бриллиантового зеленого [596, 1150,1549], кристаллического фиолетового [70, 476] и родамина С [171, 1186, 1401, 1502, 1634].
Для определения Sb в почвах и горных породах с применением бриллиантового зеленого раствор, полученный после растворения пробы, помещают в делительную воронку, устанавливают концентрацию НС1 — 6 М, добавляют 0,5 мл 5%-ного раствора NaNO3 и встряхивают для окисления Sb(III) до Sb(V). Избыток HNO3 устраняют добавлением мочевины. Затем добавляют 25 мл 10%-ного раствора (NaPO3)6 и экстрагируют гексахлоростибат бриллиантового зеленого 5 мл толуола. Экстракт фильтруют и измеряют его оптическую плотность при 640 нм. Метод позволяет определять до 1.10-4% Sb (Sr ~ ~ 0,15). Определению Sb этим методом мешают только Т1 и Au [1549].
Несколько видоизмененный вариант этого метода рекомендован для определения Sb в полевых условиях [1550]. Для достижения более высокой чувствительности в ряде случаев используют предварительное концентрирование Sb, в том числе соосаждением с GuS [1401] или отгонкой в виде SbH3 [171]. Таким путем возможно определение до 0,1—0,2 мкг Sb в пробе или до 2-Ю~5% Sb' (Sr = 0,15-1-0,25).
Малые количества Sb в горных породах, рудах, минералах и почвах определяют полярографическими методами [204, 373, 492, 1036].
По одному из них [492], рекомендованному для определения Sb и As в кварце и силикатных породах, пробу разлагают смесью HF и H2SO4 и отгоняют хлориды Sb и As. Полярографирование проводят на фоне 1 N H2SO4, содержащей КС1 (0,5 М) и метиленовый голубой (1 -10~5 Му, Sb и As образуют на этом фоне четкие волны соответственно с = —0,42 и —0,68 в. Метод позволяет определять до 1.10-4% Sb (Sr ~ 0,1).
122
Используются также методы атомно-абсорбционной спектрофотометрии.
По одному из них [1391] пробу (0,25—1,2 г) перемешивают в пробирке с 0,5 г NH4J. Пробирку вводят в пламя газовой горелки и возгоняют Sb в виде SbJ8) который оседает на холодной части пробирки. По охлаждении в пробирку вводят 20 дал 2М НС1, тщательно перемешивают и после отстаивания прозрачный раствор распыляют в пламя смеси воздуха с С2Н2. Атомное поглощение Sb измеряют при 231,8 или 217,58 нм. Предел обнаружения Sb 1-10-8% (Аг < 0,04).
Для определения микроколичеств Sb и многих других элементов в почвах, минералах, горных породах, метеоритах и различных видах минерального сырья рекомендован ряд активационных методов с выделением образовавшихся радиоизотопов [74, 449, 541, 848, 911, 1136, 1234, 1247, 1276, 1515], характеризующихся очень высокой чувствительностью (п-10~5—п-10~6%) и удовлетворительной точностью (Sr =0,05-4-0,10).
АНАЛИЗ МЕТАЛЛОВ, СПЛАВОВ, ПОЛУПРОВОДНИКОВЫХ И ДРУГИХ (МАТЕРИАЛОВ
При разложении металлов, сплавов, полупроводниковых и других материалов для последующего определения в них Sb необходимо соблюдать те же предосторожности по предотвращению возможных потерь Sb, которые указаны при рассмотрении методов разложения горных пород, минералов, руд и почв. Выбор метода разложения зависит от природы анализируемого материала и от последующего метода отделения и определения Sb.
Алюминий, его сплавы и соединения лучше всего растворять в смеси (3 : 1) HG1 и HNO3. Возможно растворение в соляной кислоте в отсутствие нагревания или при небольшом нагревании. Применяют также смесь H2SO4 и HNO3 [131, 132]. После упаривания раствора для удаления HNO3 основная масса алюминия выпадает в осадок в виде сульфата, который отфильтровывают, а фильтрат используют для последующего анализа. Растворение алюминия в растворах гидроокисей щелочных металлов применять не рекомендуется [1344], так как при этом часть Sb теряется вследствие образования SbH3.
Описан ряд методов определения Sb и ряда других примесей в алюминии, не требующих разложения пробы и выделения Sb, в том числе методы эмиссионного спектрального [54, 55, 1134] и активационного [841, 1688] анализа. Активационные методы рекомендованы также для определения Sb в алюминиевых сплавах 1945] и нитриде алюминия [421].
По одному из методов [1688] для определения Sb (а также As, Cr, Cu, Ga, Ge, Мп, Sm) пробу и стандарты облучают 24 часа, выдерживают 10 час., протравляют смесью НС1 и HNO3, промывают водой и ацетоном и, проведя
123
соответствующие измерения у-активности, находят содержание указанных элементов. Предел обнаружения Sb составляет 1-10~5%.
Для определения Sb в алюминии используются также методы, включающие разложение анализируемого материала. Одним из таких методов является экстракционно-фотометрический, включающий растворение навески в 20%-ном растворе NaOH, подкисление раствором HG1, отделение Sb осаждением тионалидом, отделение осадка и озоление при невысокой температуре. Остаток сплавляют с NaOH, плав выщелачивают водой и заканчивают определение Sb в виде гек'сахлоростибата метилового фиолетового. При использовании пробы весом 10 г предел обнаружения Sb оставляет 2-10~5% (Sr = 0,10-4- 0,15) [254]. Более простым представляется метод атомно-абсорбционной спектрофотометрии [954].
Пробу (1—10 г) растворяют в смеси НС1 и HNO3, избыток HNO3 удаляют нагреванием с НСООН и раствор выпаривают досуха. Остаток растворяют в НС1(1 : 1), добавляют 4 г аскорбиновой кислоты, 15 мл 10%-ноп НС1, содержащей 30% KJ и 10% аскорбиновой кислоты, разбавляют до 50л(л и экстрагируют SbJ3 10 мл 5%-ного раствора трифосфиноксида в метилизобутилкетоне. Экстракт распыляют в воздушно-ацетиленовое пламя и измеряют атомное поглощение Sb, используя в качестве источника излучения лампу с полым сурьмяным катодом. Метод позволяет определять до 1-10~5% Sb. При содержании Sb 1,38-10“3% стандартное отклонение составляет 2,7-10~4%.
Предложен непламенный вариант определения Sb > 1-Ю~10з (Sr = 0,05 -4- 0,10) в алюминии и его сплавах методом атомно-абсорбционной спектрофотометрии, включающий предварительное экстракционное отделение Sb [1673].
Для определения малых количеств Sb в алюминии рекомендован химико-спектральный метод [218], включающий экстракционное концентрирование определяемых примесей с применением диантипирилметана. Экстракт выпаривают на угольном порошке и спектрографируют. Высокой чувствительностью определения Sb (до 10‘5%, Sr ~ 0,15) в алюминии характеризуется полярографический метод [131, 132], основанный па предварительном электролитическом концентрировании Sb на ртутном электроде с последующей анодной поляризацией электрода при непрерывно меняющемся до нуля потенциале. Для определения Sb в алюминии, и его сплавах предложен ряд вариантов активационного метода, включающих выделение Sb из облученного материала [848, 912, 945, 1235, 1247, 1376]. Методы характеризуются очень высокой чувствительностью (до 1-10~6 %) и вполне удовлетворительной точностью (Sr ~ 0,1).
В сплаве AMG (94,5—96% А1, 0,3—0,7% Mg, 3,5—4,4% Sb и <0,4% Fe) предложено определять Sb перманганатометрическим титрованием [719].
В катализаторах на основе А12О3 (~ 85% А12О3, 5% V2O5, 5% TiO2) Sb определяют методом, включающим растворение пробы
124
В H2SO4 (1 : 1), выделение Sb осаждением в виде сульфида с применением тиоацетамида в качестве реагента, растворение осадка в H2SO4 (1 : 1), окисление Sb(III) до Sb(V) сульфатом Ge(IV) и экстракционно-фотометрическое определение в виде гексахлоростибата родамина С [883].
Для определения Sb[>8-10~8% и одновременно Cd и Bi в барии предложен осциллополярографический метод с предварительным электролитическим выделением определяемых элементов на ртутном электроде [125].
В бериллии и окиси бериллия Sb и ряд других примесей определяют спектральными методами [305, 1297].
По одному из них [305] навеску (0,2 г Be или 0,56 г ВеО) растворяют в 10 мл HCI (1 : 1) и 1 мл HNO3. Раствор упаривают до сиропа и 3 раза обрабатывают конц. HNO3. Затем добавляют конц. NH4OH до осаждения Ве(ОН)2, смесь высушивают, прокаливают сначала при 300° С и далее при 500° С для получения стекловидной негигроскопичной окиси бериллия. Спектры возбуждают в дуге постоянного тока (12а) при испарении 60 мг полученной окиси бериллия из канала (4x6 мм) нижнего электрода. Спектрограф ИСП-22, экспозиция 2 мин., щель 10 лк. Метод позволяет определять Sb и 25 других примесей при их содержании 10-3 — 10~4% (Sr ~ 0,2).
Разработан активационный метод определения Sb (а также As, Au, Ba, Cd, Gu, Mn, Mo, Ni и Sn) в бериллии, включающий экстракцию Sb хлороформом из облученного образца в виде диэтилдитиокарбамината [523].
Описан спектральный метод, позволяющий определять Sb, а также Go, Sn, Si, Al, Mg, Pb, Gr, Mn, Fe, Ni, Bi, Cd, Gu и Zn в боре [778, 779]. I
Эталоны и пробы смешивают (1 : 1) с графитовым порошком, содержащим 5% Ge (внутренний стандарт), помещают в отверстие угольного электрода и возбуждают спектры дугой постоянного тока (14 а). Метод позволяет определять указанные примеси в пределах га-10-2 — га.10-1% с ошибкой 9—40%.
В другом спектральном методе [1117] содержание Sb > 10-6% в боре определяют с использованием плазменного генератора в атмосфере аргона. Для определения Sbi>l-10~6% в боре, борном ангидриде и борной кислоте предложен химико-спектральный метод [81], включающий концентрирование определяемых примесей обработкой пробы HNO3 и HF. Кроме Sb, метод позволяет определять еще 23 элемента.
Описан [26] спектральный метод определения Sb и других 24 примесей в фосфиде бора.
Пробу смешивают (2 : 1) с NaCl и спектры возбуждают в дуге переменного тока (18 а). Предел обнаружения 1-10~4— 1-10~2%.
Для повышения чувствительности определения Sb и других легколетучих примесей в фосфиде бора рекомендуется применять
125
метод испарения; в этом случае предел обнаружения повышается примерно на порядок.
Для определения Sb (а также Au, Na и Вт) в нитриде бора рекомендован [246] активационный метод без разложения пробы. При использовании 180 мг пробы и облучении в течение 200 час. потоком 1,8-1013 нейтрон/см? сек предел обнаружения Sb составляет 1,6-10'8 г.
Сурьму О 3-10-4%) и ряд других примесей в пятиокиси ванадия предложено определять спектральным методом с испарением в воздухе и использованием разрядной трубки с полым катодом [494]. Фотометрический метод с предварительной экстракцией Sb в виде пиридин-иодидного комплекса и последующим фотометри-рованием в виде фенилфлуороната позволяет определять в пятиокиси ванадия до 5>10~5% Sb [563]. Активационный метод определения Sb в пятиокиси ванадия, включающий выделение Sb из облученного материала, характеризуется высокой чувствительностью (1-Ю'8—1-Ю'9 г) и удовлетворительной точностью (Sr — = 0,1-4- 0,2) [145].
Сурьму в висмуте определяют экстракционно-фотометрически [454, 657, 906], полярографическим [1348], спектрографическим [477, 809, 1117] и активационным [830, 1204, 1239, 1659] методами. Поскольку висмут не мешает экстракционно-фотометрическому определению Sb с применением кристаллического фиолетового [454] и родамина С [657], то ее непосредственно экстрагируют в виде окрашенных ионных ассоциатов из раствора, полученного растворением пробы, и измеряют оптическую плотность экстракта. В полярографическом методе [1348] сначала выделяют Bi с помощью катионнообменной смолы и в оставшемся растворе определяют Sb О 1 • 10~5 %) методом инверсионной вольтамперометрии с применением графитового пастового электрода. Один из спектральных методов определения Sb (а также Pb, Ag и Си) предусматривает использование литых образцов висмута (d — 9 мм) [809]. Спектры возбуждают искровым разрядом от генератора ИГ-2 (1,1 а, 0,55 мгн, 0,01 мкс/>), спектрограф ИСП-22, экспозиция 50 сек. При содержании Sb н-10”2 — н-10~3% ошибка определения 5—10%. По другому спектральному методу Sb из висмута предварительно выделяют соосаждением с H2SnO3. При использовании навески массой 5 г предел обнаружения достигает 1 • 10“4 % Sb (Sr ~ 0,1) [477]. Большинство активационных методов позволяет определять в висмуте Sb без растворения пробы с высокой чувствительностью (до 10'8%) [830, 1204, 1239]. Методы, включающие растворение облученного образца и выделение Sb, используются редко [1659].
Для определения Sb в ее сплавах с висмутом рекомендован метод [1226], в котором Bi маскируют комплексоном III и титруют Sb(III) раствором иода. Ошибка определения Sb 0,5%.
В теллуриде висмута, легированного сурьмой, и в ряде других подобных термоэлектрических материалов Sb определяют пер-126
манганатометрическим методом после отделения ее в виде сульфида [988].
В вольфраме, трехокиси вольфрама, вольфрамовых концентратах и рудах Sb наиболее часто определяют спектральными методами [104, 196, 378, 379, 739, 795, 1265, 1480]. Из них чаще используются методы, включающие предварительное отделение Sb испарением из анализируемой пробы [195, 379, 795]. Вольфрам предварительно переводят в трехокись. Высокая чувствительность определения Sb (1-10-4—1-10~6%) и удовлетворительная точность (Sr = 0,15-4-0,25) обеспечивают им широкое применение. Методы прямого спектрального определения Sb менее чувствительны (10“2—10~3%) и применяются для анализа вольфрамовых руд [1480], окислов и препаратов вольфрама [739, 1265]. В одном из методов [104] предусмотрена возможность прямого спектрального анализа порошкообразного вольфрама. Химико-спектральные методы, вследствие большой продолжительности и трудоемкости, для анализа вольфрама используются редко [378]. Для определения Sb в вольфрамовых концентратах предложен экстракционнофотометрический метод [179], в соответствии с которым пробу сплавляют с K2S2O7 и из сернокислого раствора выделяют Sb экстракцией амилацетатом. Экстракт упаривают до небольшого объема, остаток минерализуют нагреванием с HNO3, растворяют в небольшом количестве H2SO4 (1 : 1), прибавляют HG1 и экстрагируют Sb толуолом в виде гексахлоростибата кристаллического фиолетового.
Для определения Sb > 2,5 -10-6 % в вольфрамате кальция используют полярографический метод [785] включающий отделение Sb соосаждением с GdS из виннокислого раствора, растворение концентрата и определение Sb в полученном растворе.
В галлии, его сплавах и арсениде галлия Sb определяют экстракционно-фотометрическим [63, 64, 65, 661, 662], полярографическим [246, 293, 586], активационным [640, 825, 1375] и спектральным [629] методами. По одному из них [63, 64] для определения Sb > 5-10'6% (Sr = 0,08) в галлии ее отделяют экстракцией хлороформным раствором диэтилдитиокарбаминовой кислоты, затем отделяют ее от Sn и Мо экстракцией эфиром в виде пиридин-иодидного комплекса и фотометрируют в виде комплекса с фенил-флуороном. По другому методу [661, 662] Sb выделяют цементацией на оловянном электроде и определяют с применением бриллиантового зеленого.
Методы инверсионной вольтамперометрии используется для определения Sb > 4-10-6% (Sr = 0,16) в арсениде галлия [245, 246, 303, 586] и нитрате галлия [293] непосредственно в растворе, полученном после растворения пробы. Определению Sb этим методом мешают 3-кратные количества Си и 6-кратные — Bi. Для определения Sb > 1-10“5% (Sr — 0,1 -4-0,2) рекомендован также метод квадратно-волновой полярографии [1192]. Активационный метод [640, 825, 1375], включающий выделение Sb из облученного
127
материала, пригоден для определения 3-10-6% Sb в галлии и арсениде галлия.
В германии и двуокиси германия Sb определяют спектральным, фотометрическим, полярографическим и активационным методами. Из спектральных методов наиболее удобны методы, позволяющие определять Sb n-10-4% (Sr = 0,15 —е— 0,25) без разложения анализируемого материала [559, 564, 634, 905].
По одному из них [905] пробу (5 мг} загружают в конический канал (глубиной 3 мм) угольного электрода (d = 5 мм), покрывают слоем угольного порошка и полностью испаряют в дуге постоянного тока (12 а).
В другом методе [634] для определения Sb 2,5- 1(Г8 г (Sr = = 0,10), а также Ag, Ga, In, Bi и As в германии спектры возбуждают в газоразрядной трубке с полым угольным катодом. Для повышения чувствительности определения Sb и других примесей этим методом [634] отгоняют Ge в виде GeCl4. При использовании навески 1 г предел обнаружения Sb составляет ~ 1 • 10-7 % (5Г ~ ~ 0,25). В других химико-спектральных методах [112, ИЗ, 304, 461, 462, 756] для определения Sb в германии и двуокиси германия пробу растворяют в смеси HG1 и HNO3, отгоняют Ge в виде GeGl4 и анализуруют полученный концентрат. Предел обнаружения Sb ЬЮ-М-Ю'6 % (Sr= 0,12-н0,2).
Для определения Sb^l-10"7 г (5Г ~ 0,15) в пленках германия, нанесенных на стеклянную подложку, поступают следующим образом.
Пленку растворяют в смеси HNO3 и НС1, к раствору прибавляют 19 мг угольного порошка и при 60—75° С отгоняют GeCl4. Затем раствор выпаривают досуха. К остатку приливают 4 капли 1%-ного раствора NaCl и снова выпаривают досуха. Остаток помещают в кратер угольного электрода и возбуждают спектр в дуге постоянного тока (10 а). Используют спектрограф И СП-28 [460].
Для определения Sb ;> 1-10~4% (Sr = 0,10 -н 0,15) в двуокиси германия пробу растворяют в конц. HG1 при кипячении и пропускают через раствор С12 [624]. При этом Ge отгоняется в виде GeGl4. В полученном концентрате Sb определяют экстракционнофотометрическим методом с применением кристаллического фиолетового.
Высокой чувствительностью определения Sb (2-10-7 %, Sr = = 0,10-4-0,25) в германии и тетрахлориде германия характеризуется полярографический метод, включающий отгонку Ge в виде GeGl4, концентрирование Sb на электроде в форме висящей ртутной капли и регистрацию пика анодного растворения Sb при —0,105 в на фоне 0,2 М HG1 [134]. При определении Sb (а также Pb, Bi и Gd) в GeBr4 рекомендован метод, включающий испарение GeBr4, электролитическое выделение определяемых примесей и регистрацию пиков их анодного растворения [105]. При использовании
128
навески 10 г предел обнаружения Sb достигает 1-10”7% (Nr = 0,13).
Сурьму (> 1 • 10~7%) и ряд других примесей в германии и двуокиси германия определяют недеструктивными активационными методами [633, 640, 838, 1384]. Активационные методы, включающие выделение Sb из облученного германия [38, 610, 639], обладают еще большей чувствительностью (1-10“9 г Sb в пробе).
Для определения Sb в железе, сталях и чугуне наиболее часто используются спектральные методы. По одному из них [425] для определения Sb > 1-10~2% (Sr = 0,08-н 0,10) и 14 других примесей в железе пробу прессуют в брикет (d = 6 мм, масса 500 мг), который помещают на графитовую подставку, используя ее в качестве анода, и возбуждают спектр дугой постоянного тока (7 а), определяя при этом Sb, Cd, Pb, Sn и Zn. Затем спектрографируют малолетучие примеси (Al, Ni, Со, Сг, Si, Mg, Мп, Cu, Со, Be) с использованием расплавленной пробы в качестве катода (спектрограф ИСП-22). Для повышения чувствительности определения Sb и других элементов используют добавки носителей [1440]. Применение магнитного поля позволяет в тех же условиях понизить предел обнаружения Sb еще в 1,7 раза [1441].
При определении Sb 6• 10“4% в железе, сталях и жаропрочных сплавах рекомендованы спектральные методы, основанные на фракционной дистилляции [176, 1278]. Описан также ряд других простых вариантов спектрального определения Sb в железе, чугуне и сталях [274. 1250, 1441, 1593], феррохроме, феррониобии, феррованадии, ферромолибдене и в жаростойких сплавах на основе железа [54, 793]. Метод испарения примесей с конденсацией на угольной капсюле, служащей в дальнейшем электродом, позволяет снизить предел обнаружения Sb в железе, сталях, чугуне и трехокиси железа до п-10~4—-п-10-5% [198]. Химикоспектральные методы определения Sb в железе и сталях, включающие концентрирование Sb соосаждением с GuS [336, 1274] или отделение Fe экстракцией диэтиловым эфиром [546], характеризуются примерно такой же чувствительностью, как и метод испарения.
Для определения Sb >. 1% (Sr 0,01) в порошковом железе рекомендован рентгенофлуоресцентный метод [1020, 1021]. Этот метод часто используется для определения Sb и ряда других примесей в сталях [1126, 1164, 1283, 1624]. Для определения 0,0001 — 0,5% Sb, а также Ti, V, Сг, Мп, Go, Ni, Cu, W, As, Mo, Sn и РЬ в малоуглеродистых сталях образцы готовят в виде отполированных с одной стороны цилиндров, которые при измерении вращаются. Используют линии Ка, (кроме W, РЬ и Со, для которых используют линии Lai, LPl и соответственно). Место измерения фона находят по спектрограммам, записанным с дискриминатором и без него. Время измерения, необходимое для определения Sb, а также As и РЬ, с ошибкой 1% при их концентрации > 1 • 10~4% составляет 400 сек.
Як
5 А. А. Немодрук
129
Спектрофотометрические методы позволяют быстро и с высокой чувствительностью определять Sb в железе, сталях, чугуне, железных рудах и сплавах на основе железа. В ряде случаев фотометрическими методами можно определять Sb непосредственно в растворе, полученном после растворения пробы. Так определяют Sb в сером чугуне [1185], нелегированных [1431] и легированных [918] сталях методом, основанным на образовании и измерении окраски SbJ;. Лучшим вариантом этого метода, пригодным для определения Sb 0,001—0,025% (Sr = 0,02 0,07) в железе,
чугуне и сталях, является вариант, описанный в работе [918].
Экстракционно-фотометрические методы, включающие экстракцию окрашенного ионного ассоциата, образуемого SbClg с катионом кристаллического фиолетового [845, 1412] или бриллиантового зеленого [1351], также быстро позволяют определять Sb 4-10-4% (Sr = 0,06) в железе, сталях, чугуне и других продуктах черной металлургии. Предложен ряд методов [1082, 1233] определения Sb в сталях, в соответствии с которыми Sb экстрагируют в виде HSbCle кислородсодержащими органическими растворителями, а затем отмывают экстракт от мешающих элементов и контактируют его с водным раствором основного красителя, образующего в органической фазе интенсивно окрашенный ионный ассоциат. В методе [1577] Sb вначале отделяют экстракцией бензолом в виде иодида, реэкстрагируют в водную фазу и определяют экстракционно-фотометрическим методом с применением родамина С.
Иногда Sb предварительно отделяют отгонкой в виде SbCl3 и определяют с помощью метилового фиолетового [975] или выделяют ее экстракцией раствором пиро-2-этилгексилфосфорной кислоты в гексане и после реэкстракции Bi 2%-ным раствором KJ реэкстрагируют Sb 6 М HG1 и определяют с применением бриллиантового зеленого [408].
Определение Sb в чугуне и нелегированных сталях экстракционно-фотометрическим методом в виде пирролидиндитиокарбами-ната [1255, 1258] также требует ее отделения и характеризуется примерно на порядок меньшей чувствительностью по сравнению с описанными выше методами.
Для определения Sb в феррониобии рекомендован [786] экстракционно-фотометрический метод с применением кристаллического фиолетового, позволяющий определять Sb непосредственно в растворе, полученном после разложения пробы. При определении Sb этим методом в ферровольфраме и ферротитане ее предварительно выделяют в виде Sb2S3 соосаждением с HgS [632].
Среди полярографических методов имеется ряд таких, которые позволяют определять Sb в железе, чугуне и сталях без ее отделения [832, 1216, 1478]. Однако чувствительность и точность определения Sb могут быть существенно улучшены путем концентрирования Sb и особенно при использовании метода инверсионной 130
вольтамперометрии [1348, 1575]. Этим методом возможно определение до 1-10-6% Sb (Sr = 0,10 0,15).
Методом инверсионной вольтамперометрии определяют Sb > > 1-10-4% в ферросплавах и жаропрочных сплавах на основе железа без ее отделения [90].
В ферромарганце Sb 5-10“3% определяют методом осциллополярографии [1352].
Для определения Sb в железе, сталях и железных рудах простыми и быстрыми являются методы атомно-абсорбционной спектрофотометрии [954, 1141, 1387, 1601); простейший вариант — непосредственный анализ раствора, полученного после растворения пробы. При использовании воздушно-ацетиленового пламени возможно определение Sb при ее концентрации 2—20 мкг/мл (Sr = — 0,03 -г- 0,05) [1601]. В непламенном варианте возможно определение до 10“12 г Sb. Методы атомно-абсорбционной спектрофотометрии с экстракционным отделением Sb в виде HSbGl6 с применением метилизобутилкетона в качестве экстрагента и воздушно-ацетиленового пламени [954, 1141, 1387] характеризуются высокой чувствительностью (1 • 10—6%). В одном из таких методов [954] Sb экстрагируют 5%-ным раствором трифенилфосфиноксида в метилизобутилкетоне и экстракт распыляют в воздушно-ацети-леновое пламя.
Для определения Sb в железе, сталях, чугуне, железных метеоритах и других материалах на основе железа предложен ряд активационных методов, не требующих выделения Sb из облученного материала [135, 1556, 1632]. По одному из них [1652] при определении 2,5-10-4% Sb ошибка ~ 4,8%. В ряде других [987, 1033, 1113, ИЗО, 1222, 1280, 1539, 1590, 1653] Sb выделяют из облученного материала. Предел обнаружения Sb этими методами достигает 1 • 10“6 —1 • 10“7% (Sr = 0,07 н- 0,12).
Для определения Sb > 1 • 10-5% в золоте и его сплавах наиболее часто используют спектральные методы без разложения пробы [389, 404, 754, 1095, 1386]. В соответствии с методом [196] пробу золота растворяют в царской водке и раствор наносят на торец угольного электрода. Метод позволяет определять Sb 1-10“4% (Sr = 0,06 -т- 0,13) и одновременно с ней еще 22 примеси. Для определения Sb, а также Al, As, В, Ge, Ga и Sn в золотых сплавах поступают следующим образом.
Пробу (0,01 г) растворяют в царской водке и раствор (1 мл) вводят в аналитический промежуток вращающимся (10 об/мин) графитовым диском. Спектр возбуждают высоковольтной искрой. При содержании примесей 0,1 — 2% погрешность составляет 5,8—10,7% [1167].
Фотометрические методы, основанные на измерении оптической плотности SbJ4 [1043, 1682] или пиридин-иодидного комплекса Sb [735], позволяют быстро (~ 1 час.) и с хорошей точностью = 0,05-4-0,10) определять Sb > 3,5-10-4% в золоте и его сплавах.
5*
131
Для определения Sb в золотосодержащих отходах припоев предложен активационный метод без выделения Sb из облученного материала [1518]. В другом методе [1676] Sb определяют после выделения ее из облученного материала.
В индии чаще всего Sb определяют экстракционно-фотометрическими [64, 65, 661, 662] и спектральными [682, 814, 815, 905, 1189, 1267] методами. В одном из фотометрических методов [668, 806] Sb отделяют от основы экстракцией хлороформным растьо-ром диэтилдитиокарбаминовой кислоты. Метод позволяет определять до 5-10s% Sb (Sr = 0,1 -4- 0,2). Несколько проще и менее трудоемким является другой фотометрический метод [661, 662], включающий выделение Sb цементацией на оловянном стержне, растворение выделенной Sb, экстракцию ее в виде гексахлорости-бата бриллиантового зеленого и измерение оптической плотности экстракта. Метод рекомендован для определения Sb 5-10~6% (Sr ~ 0,10) в индии и его сплавах с цинком и галлием.
Методы прямого спектрального анализа позволяют быстро определять Sb и ряд других элементов в индии и его сплавах с галлием, со свинцом и висмутом, со свинцом и серебром [814, 815, 905, 1119]. Однако предел обнаружения Sb в этих методах составляет 10~а —10”3%, что в ряде случаев недостаточно. Поэтому, когда требуется более высокая чувствительность, применяют методы, включающие предварительное концентрирование Sb [682].
Для определения Sb в сплавах In—Ан—Са, In—Sb—Au—Ga и Pb—In—Bi—Sb—Au—Ga, используемых в полупроводниковых приборах, разработан спектральный метод с использованием проб в виде шариков (> 40 мк) или таблеток (d = 40 мк, h — = 100 мк) [678]. Сурьму (а также Au, Ga, Ni, Sn и Bi) в микрослитках многокомпонентных сплавов на основе индия определяют недеструктивным активационным методом [1271]. Используют 3—200 мкг анализируемого материала. Одновременно облучают 20—30 образцов потоком 1,2-1013 нейтрон/см2-сек в течение 20 час. Предел обнаружения Sb при использовании сцинтилляционных детекторов составляет 10-10 г, с применением Ge (Li)-детекторов — 10-9 г.
При определении Sb > 3-10-7% (Sr = 0,1 -4-0,2) в индии высокой чистоты и арсениде индия рекомендовав^ активационные методы, включающие выделение Sb из облученного материала [115, 639, 828, 829]. Для определения Sb > 1 • 10~4 % (Sr = 0,10 ц- 0,15) в сплавах индия с цинком предложен метод пере-мепнотоковой полярографии [1104]. Полярографирование проводят на фоне ЗМ НС1. Равные количества Си мешают. Метод применен также для определения Sb в тонких слоях полупроводниковых сплавов In—Sb—Те [451]. В этих же сплавах Sb определяют прямым броматометрическим титрованием, а также фотометрическим методом в виде иодида [451].
Сурьму и ряд других примесей в иоде определяют химико-спектральными методами [589, 745, 746]. По одному методу [745,
132
746] Sb концентрируют путем возгонки иода, смешанного с угольным порошком. При использовании навески 12 г предел обнаружения Sb составляет 6-10~’%. В другом методе [589] Sb концентрируют путем сублимации иода в камере, защищающей от попадания в концентрат внешних загрязнений.
Концентрат растворяют в HNO3 (1 : 1) и наносят на плоские торцы обработанных полистиролом угольных электродов В-3; используют спектрограф средней дисперсии. Предел обнаружения Sb составляет 3 - IO-7 % (S'r = 0,25 -s-ч- 0,35).
В трехокиси иттрия Sb > 1-10"5% (5Г = 0,15-4— 0,20) определяют спектральным методом, включающим отгонку их в виде фторидов из канала электрода с применением фторопласта в качестве фторирующего агента. Спектр возбуждают в угольной дуге. Спектрограф ДФС-13 [272].
Для определения Sb в кадмии наиболее часто применяются методы спектрального анализа, позволяющие определять Sb как без концентрирования [598, 599], так и с предварительным концентрированием [716, 717, 727, 1007]. Метод [598, 599] спектрального определения Sb > 1-10~4% (Sr < 0,2), а также Си, Ag, Bi, Со, Ni, Pb, TI, Sn и Zn в кадмии основан на испарении пробы в виде королька из анода угольного электрода. В ряде спектральных методов Sb и другие примеси в кадмии концентрируют цементацией на небольшом количестве цинкового порошка [1007], соосаждепием с МпО2 [707], отделением основной массы кадмия экстракцией СНС13 в виде пиридин-иодидного комплекса [727] или соосаждением примесей с небольшой частью основы в виде гидроокиси [716]. Предел обнаружения Sb 1 • 10~5—5-10~’% (5, = 0,2 4-0,3). Для определения Sb 5-10'®% (Sr = 0,10-4— 4- 0,20) в кадмии предложен ряд экстракционно-фотометрических методов с использованием в качестве реагентов метилового фиолетового [456] и кристаллического фиолетового [443, 470, 657].
В кадмии и его солях Sb > 1-10“’% предложено определять методами инверсионной вольтамперометрии [302, 318]. Метод импульсной полярографии, включающий выделение Sb соосаждением с МпО2, позволяет определять до 4-10-7% Sb (Sr = 0,12-4--4-0,25) [1668].
В кальции Sb определяют активационным методом с выделением Sb из облученного материала [1675].
В кобальте Sb определяют спектральными методами с использованием литых электродов [60, 153].
Для определения Sb в кремнии, кварце, карбиде кремния, стеклах, тетрахлориде кремния и трихлорсилане наиболее часто применяют методы спектрального анализа (табл. 11). Для определения Sb в кремнии и его соединениях высокой чистоты широко используются также активационные методы. Особенно удобны те из них, которые позволяют определять Sb без ее выделения [212, 468, 762, 932, 950. 989, 1144, 1361, 1366, 1540). Ме-
133
Таблица 11
Определение сурьмы в кремнии и его соединениях методами эмиссионного спектрального анализа
Анализируемый материал Сущность метода Предел обнаружения Sb, % Коэффициент вариации, % Другие определяемые элементы Литература
Кремний Пробу вносят в канал (2,6x4 .юг) нижнего УЭ и спектр возбуждают дугой переменного тока (18,5—19,0 а, 226 в) в атмосфере Аг 5-10-4 15—20 А1, Со, Sn, As, Au, B, Cu, Fe, Jn, Pb, Ti, W, Ba, Ca, Mo, Ni, Zn [1619]
Пробу разлагают в камере парами HF и HN03, к остатку прибавляют 0,5 мл раствора NaCl (2 мг-/мл), высушивают и концентрат примесей помещают в канал УЭ, включают на 30 сек. дугу переменного тока (За) затем УЭ с пробой включают анодом дуги постоянного тока (10 а) 2-Ю-5 15—20 Al, Fe, Zn Ag, Cd, Cu, In, Mg, Mn, Ga, Be, Ni, Pb, [491]
Примеси отделяют испарением в вакууме при 1300° С в течение 1 мин. и электрод с конденсатом примесей используют для анализа 5-Ю-5 20-30 Zn, In, B, Bi, As, Те [252]
Кремний, двуокись кварц, кремния Кремний удаляют в виде SiF4 и концентрат примесей на УП спектрографируют 3-10~6 25-30 Al, Ag, Au, Bi, Ca, Cr, Cu, Fe, In, Mg, Mn, Mo, Ni, Sn, Pb, Ta, Ti, Tl, Zn [339]
Кварц, двуокись кремния Пробу разлагают в присутствии 50 мг УП, из концентрата прессуют брикет, вносят его в углубление УЭ (катода) и спектрографируют в вакууме 1.10-е 25—30 Ag, Al, Cr, Cu, Ga, Ni, Bi, Pb, Mg, Mn, Sn, Zn [572]
Карбид кремния Пробу загружают в канал УЭ и возбуждают спектр дугой постоянного тока (20а) в атмосфере Аг; используют диффракцпон-пый спектрограф с большой дисперсией 3-Ю"5 15-20 As, Sn, Bi, In, Cu, Zn, Tl, Ag, Ga, Pb, Fe [1353]
Синтетические стекла, природные силикаты
Кремнемедные сплавы
Силикаты
Тетрахлорид кремния
Тетрахлорид кремния, трихлорсилан
Трихлорсилан
Примеси отделяют от основы испарением при 1850° С в течение 3 мин.; электрод с конденсатом примесей используют в качестве нижнего электрода; спектры возбуждают дугой переменного тока (8а)
Пробу помещают в кратер УЭ и спектры возбуждают в дуге постоянного тока (18 а) в атмосфере Аг
Примеси отделяют испарением и затем графитовый стаканчик с конденсатом примесей используют для спектрографирования; спектры возбуждают в дуге переменного тока (8 а)
Пробу смешивают (1:1:2) с содой и УП, содержащим GeO2, смесь помещают в углубление УЭ и возбуждают спектр в дуге постоянного тока (8 а, 200 в)
Спектры возбуждают в дуге переменного тока (8 а)
Пробу смешивают (1 :1) с буфером (соотношение А12О3 : СаСО3 : K2SO4 =7:3:1), содержащим 1% Bi2O3 в качестве внутреннего стандарта, помещают в канал У Э (анода) и возбуждают спектры дугой постоянного тока 10 сек. при 5 а и 35 сек. при 15 а
Отгоняют SiCl4 с током N2 при 80° С и анализируют концентрат примесей
Основу удаляют испарением в присутствии УП и концентрат спектрографируют
Пробу смешивают с СС14 и УП и испаряют; концентрат примесей спектрографируют
Примечание. Здесь и далее УП — угольный порошок; УЭ — угольный электрод.
ЗЛО-4 15—20 Al, Cd, Cu, Fe, Mg, Mn, P, Pb, Zn [789, 790]
2-10-4 30-35 Al, B, Fe, Mg, Mn, Cu, Ni, Sn, Pb, Ti, Cr [109, 110[
5-IO'5 20-25 Fe, Cu, Mn, Pb, P, Bi, Sn [288]
1•10-2 10 Al, Ag, Bi, Ca, Co, Cu, Fe, Mg, Mn, Ni, Pb, Sn, Ti [1414, 1579]
5-10-4 15-25 Fe» Mg, Al, Bi, Cu, Pb, Ti, Sn [594]
5-10-4 10—19 Ag, As, Au, B, Fe, Ga, Ge, Hg, In, P, Pb, Sn, Zn [1586]
5-10-» 15-25 Ag, Al, B, Ca, Cu, Mg [1620]
MO'6 25—30 Ag, Al. Bi, Ca, Cr, Cu, Au, Ta, Tl, Mg, Mn, Mo, Ni, Pb, Ti, Zn [339]
1•IO"6 15-20 Ag, Al, Bi, Ca, Cr, Cu, Fe, Ga, In, Mg, Mn, Mo, Ni, Pb, Sn, Ti, Zn, Zr [573]
тоды, включающие выделение Sb из облученного материала, применяются для ее определения в кремнии [38, 39, 275, 282, 455, 639, 861, 1355, 1540, 1687], кварце и двуокиси кремния [283— 285, 487], тетрахлориде кремния [1461], трихлорсилане [760, 999, 1342], трихлорметилсилане [38], карбиде кремния [38, 39, 276], стекле [1107] и эпитаксиальных пленках кремния [283— 285, 487]. Предел обнаружения Sb этими методами в кремнии достигает (4—5)-10-9% [39, 282] и даже 7-10~10% [1355], в тетрахлориде кремния — 1-10'8% [403] и в трихлорсилане — 8-10~8% [1342].
Полярографические методы используются реже, хотя более просты и во многих случаях обеспечивают высокую чувствительность определения Sb. Так, при определении Sb (а также Bi и Sn) в тетрахлорсилане методом инверсионной вольтамперометрии предел обнаружения достигает 4-10~6% (Sr = 0,1 —5- 0,2) [310]. В этом методе пробу разбавляют н-пропанолом до концентрации SiCl4 6,3 мол.“о и производят накопление указанных элементов.
Описан [491] полярографический метод определения Sb > > 1-10-1% и As в кварце и силикатах, включающий разложение пробы смесью HF и H2S04, отгонку Sb и As из остатка в виде хлоридов и полярографирование на фоне 1 N H2SO4, содержащей HG1 (0,5 М) и метиленовый голубой (1-1О-57И)
В технических, свинцовых и бариевых стеклах Sb > 5-10“5% (5,. = 0,1—0,2) определяют методом квадратно-волновой полярографии [1395], в соответствии с которым пробу (~ 100 мг) разлагают смесью HG1 и Н2С2О4 при нагревании до 155° G, избыток Н2С2О4 отгоняют и остаток растворяют в 2 М НС1, служащей одновременно полярографическим фоном. Для послойного определения Sb в эпитаксиальных структурах кремния применен метод инверсионной вольтамперометрии. Анализируют раствор, получаемый после электрохимического травления плавиковой кислотой, служащей также полярографическим фоном. Предел обнаружения Sb 5-10_1%, разрешение по глубине 2 мкм [271]. В стеклах Sb О 5-10~3% (Sr 0,01) определяют методом кулонометрического титрования электрогенерированным иодом [1651]. Высокой чувствительностью определения Sb (4-10-6%) в силикатах харак--теризуется метод непламенной атомно-абсорбционной спектрофотометрии [1587], включающий выделение Sb в виде SbH3 и введение его в атомизатор.
Сурьму ((> 4,3-10-5%) во фториде лития определяют активационными методами. По одному из них [50] Sb выделяют из облученного материала экстракцией хлороформом в виде 8-меркапто-хинолината и измеряют у-активность экстракта. В другом методе [331] пробу вместе с эталонами облучают потоком 4-1012 ней-трон/см2-сек в течение 15 час, затем дают остыть 75 час и измеряют ^-активность 12aSb 100-канальным сцинтилляционным у-спектро-метром. Предел обнаружения Sb — 8-10~5% (5r 20%).
136
Для определения Sb в нитрате лития предложен активационный метод [524], включающий выделение Sb из облученного материала двухкратным восстановлением до металла свинцом.
В магнии, его сплавах и соединениях Sb определяют различными методами. Активационные методы позволяют определять до !• 10_7 % Sb [874, 1597]. Для определения Sb во фториде магния предложен метод инверсионной вольтамперометрии [245].
Для определения Sb в меди, ее соединениях и сплавах наиболее часто используются спектральные методы (табл. 12). Экстракционно-фотометрическими методами с применением кристаллического фиолетового Sb определяют в черновой меди [649], медных концентратах [190], медно-цинковых сплавах [685], оловянных бронзах [94], медно-никелевых сплавах [686] с применением метилового фиолетового — в конверторной меди [359], безоловянных бронзах [93] и с применением родамина С — в медных сплавах [1580]. Эти методы позволяют определять Sb при ее содержании до 1-Ю'4—1-10~5% (Sr =0,08-4-0,15). Для определения Sb в меди и ее сплавах используются также другие фотометрические методы, в том числе иодидный — [139, 1068, 1266], иодидно-мочевинный [340] и тиомочевинный [139]. По чувствительности и избирательности эти методы значительно уступают экстракционно-фотометрическим.
Для определения Sb (0,01—0,15%) в анодной меди предложен полярографический метод с выделением Sb соосаждением с МпО2 [1672]. В меди высокой чистоты Sb определяют методом инверсионной вольтамперометрии [526]. Сурьму концентрируют соосаждением с Fe(OH)3. При содержании Sb 5-10~6—1•10-4 погрешность составляет 17—20%. Для определения 10~8—10~4% Sb в медных сплавах разработан метод вектор-полярографии [116].
Сурьму в бронзах и других медных сплавах определяют броматометрическим титрованием без ее отделения с погрешностью ~2% [959]. В оловянных бронзах и медных сплавах, содержащих олово, Sb предварительно выделяют соосаждением с Fe(OH)3 [1244]. Для определения Sb в ее сплавах с медью разработан метод амперометрического титрования раствором КВгО3 без ее отделения. Продолжительность титрования 5—10 мин., погрешность 2—3% [1087]. Титрованием раствором КВгО3 определяют Sb в медных сплавах [1346]. Гравиметрические методы, основанные на электролитическом выделении Sb при контролируемом потенциале, применены для ее определения в бронзах и латунях [849, 850, 852]. Коэффициент вариации 0,1—0,2%.
В молибдене и молибденовом ангидриде Sb наиболее часто определяют, спектральными методами. Большинство из них предусматривает концентрирование Sb методом испарения [27, 104, 197, 379, 795, 796]. Предел обнаружения Sb этими методами достигает 1-10~4—5-10~5/о (Sr = 0,1 -4- 0,2). Химическое концентрирование используется в редких случаях [308]. Для снижения пределов обнаружения Sb часто используют носители [1347J
137
Таблица 12
Определение сурьмы в меди, ее соединениях и сплавах методами эмиссионного спектрального анализа
Анализируемый материал Сущность метода Предел обнаружения, % sr Другие определяемые элементы Литература
Медь и ее сплавы с цинком Спектры возбуждают в дуге переменного тока; спектрограф ИСП-28 1-Ю-3 0,07—0,10 Bi, Sn, As [482J
Окись меди Пробу смешивают (5:2) с УП, помещают в канал УЭ и спектры возбуждают в дуге переменного тока (4 а); спектрограф ИСП-28 5-10-1 0,05-0,10 Ag, Al, As, Au, B, Ba, Bi, Ca, Cd, Cr, Fe, Hg, In, Mg, Mo, Ni, P, Pb, Si, Zn, Be [199]
Электролитическая медь Образцы в виде стержней (d = 3 мм) включают катодом, анод — УЭ, спектры возбуждают в дуге постоянного тока 3-10-4 0,07-0,12 Ag, Al, Bi, Ca, Cd, Cr, Fe, Mg, Mn, Mo, Ni, Pb, Zn [964]
Глобульная дуга постоянного тока 4-10-4 0,05—0,10 Ag, As, Bi, Co, Fe, Mg, Mn, Mo, Ni, Pb, Sn, Zn [1654]
Медь Брикеты (0,5 а) окисляют и помещают на УЭ (катод), анод — медный стержень, спектры возбуждают в дуге постоянного тока (9 а); спектрограф ИСП-22 1-10-4 0,06-0,10 Al, As, B, Bi, Co, Cr, Fe, Mg, Mn, Mo, Ni, Pb, Zn [129]
Образцы окисляют в атмосфере О2 и анализируют методом глобульной дуги (9 а) 1-10-4 0,08-0,12 Fe, Mg, Mn, Ni, Pb, Sn [1645]
Продукты медеплавильного производства Пробы прокаливают 3 час. при 350° С, смешивают (1:2) с MgO, помещают в рюмочный УЭ и спектры возбуждают в дуге переменного тока (12 а) 3-10-3 0,08-0,12 As, Cd, Bi, Pb, Sn. [360]
Таблица 12 (продолжение)
Анализируемый материал Сущность метода Предел обнаружения, % sr Другие определяемые элементы Литера тура
Хромистая бронза Глобульная дуга постоянного тока (8 а) 1 • IO’4 0,08—0,12 Fe, Mg, Mn, Ni, Pb, Sn [96]
Чистая медь Глобульная дуга постоянного тока 9-10-s 0,09—0,12 Ag, As, B, Bi, Co, Cr, Fe, Ga, Ge, Mg, Mn, Mo, Ni, Si, Sn, Zn [1599]
Медь высокой чистоты Пробу смешивают с CuF2, спектры возбуждают в дуге постоянного тока (14 а), спектрограф Джарелл-Аш 1-ю-5 0,09—0,15 Al, As, B, Be, Bi, Co, Cr, Fe, Ga, Ge, Hf, Mg, Mn, Mo, Nb, Ti, Pb, Si, Sn, TI, Ni, Zn, Zr [1609]
Проволока и фольга из медных, медно-никелевых сплавов и меди Образцы окисляют, порошки окислов помещают в кратер УЭ 2-10-4 0,08—0,15 Ag, Al, As, Bi, Fe, Mn, Ni, P, Pb, Si, Sn, Zn [1048]
Электролитическая и конверторная медь Пробу растворяют в HNO3, раствор выпаривают и прокаливают с УП, содержащим 10% KJ, помещают в канал УЭ, спектры возбуждают в дуге переменного тока (9 а) 1 • Ю-з 0,09—0,14 Ag, Bi, Fe, Ni, Pb, Sn [997]
Черновая медь Образцы растворяют в HNO3, раствор выпаривают, остаток прокаливают, смешивают с УП, смесь помещают в канал УЭ; спектры возбуждают в искровом разряде 1•IO'3 0,06-0,10 Al,'As, Bi, Fe, Ni, Pb, Sn [1176]
Медные сплавы Образцы растворяют в HNO3, добавляют GeO2 и УП, выпаривают и; прокаливают 15 мин. при 400° С; смесь помещают в кратер УЭ, спектры возбуждают в дуге постоянного тока (12 а) 1•IO-3 0,10 Al, Ag, As, Bi, Fe, Ni, Pb, Si, Zn [1188]
Таблица 12 (окончание
Литература [1463] [1417] о” 1 {1600] со
Другие определяемые элементы As, Bi, Cd, Fe, Pb, Sn Ag, Al, Bi, Fe, Ni,1 Pb, Zn As, Au, Bi, Cd, Fe, 1 Ni, Pb, Sn, Zn As, Bi, Fe, Ni Ag, Al, Au, Ba, Bi, Ca, Cd, Co, Cr, Fe, Mg, Mn, Mo, Ni, Pb, Pt, Sn, Те, Ti, V, Zn
to" 0,12 0,10 1 0,031—0,045 i 2 о 0,15—0,20
Предел обнаружения, % s-01-5 05 s £ 00 co I О СЧ 2
Сущность метода Образцы растворяют в HNO3, раствор выпари- вают, остаток прокаливают и смешивают (1 : 1) с УП; спектры возбуждают дугой постоянного тока । Образцы растворяют в смеси HCl и HNO3 и раствор вводят в разряд вращающимся угольным диском Образцы растворяют и раствор вводят на вращающийся дисковый УЭ; спектры возбуждают в искровом разряде Электролит вводят в разряд постоянного тока Медь вЬгделяют электролизом, электролит выпа- i ривают досуха, остаток прокаливают при 550° С; концентрат примесей возбуждают в дуге постоянного тока; спектрограф ИСП-28
Анализируемый материал Медные и медно- цинковые сплавы , i Бронзы 1 Медные сплавы Медный электролит Медь, ее соедине- ния и сплавы
и реагенты, участвующие в термохимических реакциях [749]. Хорошая чувствительность определения Sb (1-10-4%) достигается также с применением разрядной трубки с полым катодом [1325]. Методы прямого спектрального определения Sb в молибдене и его соединениях вследствие малой чувствительности (~ 10-3%) используются редко. Экстракционно-фотометрический метод с применением метилового фиолетового позволяет определять в молибдене и молибденитах до 5-10_6% [401], а метод осциллополярографи-ческой полярографии — до 1-10~4% Sb [644].
В мышьяке и его соединениях Sb наиболее часто определяют химико-спектральными методами. В одном из них [4721 Sb концентрируют путем испарения As2O3. Спектры получают при полном испарении проб концентратов в дуге переменного тока (16 а). Предел обнаружения Sb 1-10~б% (Sr = 0,20-^0,25). В другом методе [669] для определения Sb 1*10-8% (5Г ~ 0,3) ее концентрируют отгонкой основной массы AsCl3. При определении Sb в трихлориде мышьяка в качестве метода концентрирования используют экстракцию AsCl3 бензолом или лг-ксилолом из среды 10—12 М НС1. Оставшуюся водную фазу выпаривают с угольным порошком, содержащим 2,5% NaCl, и концентрат спектрографируют (ИСП-30, дуга постоянного тока 14 а). Предел обнаружения Sb 1-10~5% [384]. При использовании разряда в полом катоде предел обнаружения Sb снижается до 2-10-6% [383]. Для определения Sb > 1 • 10~4 % (Sr = 0,10-ь 0,15) в мышьяке описан также прямой метод [472], в соответствии с которым пробу вносят в канал угольного электрода и спектрографируют (ИСП-28, дуга переменного тока 16 а). Сурьму (> 1-10-4%) в мышьяке и трехокиси мышьяка определяют экстракционно-фотометрическим методом с применением родамина С [1544]. Метод включает отделение Аз экстракцией бензолом в виде АзС13. В другом методе [1084] Sb определяют с применением фенилфлуорона. Масс-спектраль-ный метод применен для определения Sb и ряда других примесей в пленках арсенида галлия [57].
Для определения Sb в органических диарсенидах пробу спекают с ZnO и серой. Мышьяк, образующий растворимые соединения, удаляют выщелачиванием раствором соды, и в остатке определяют Sb фотометрическим методом в виде иодидного комплекса [539]. Метод инверсионной вольтамперометрии позволяет определять до 1-10~8 з Sb в пробе [303].
В никеле и его сплавах Sb 2-10-4% определяют спектральным методом без предварительного отделения [108]. В другом методе [486] предусмотрено два варианта определения Sb. По одному варианту анализируют металлические образцы (дуга переменного тока 7—12 а, спектрограф ИСП-28 или ДФС-13); предел обнаружения Sb 1 -10~3% (Sr = 0,1 -д- 0,2). По другому варианту пробу переводят в окислы; спектры возбуждают дугой переменного тока 6а. При спектральном определении Sb > 2,5- 10~в% в никелевых электролитах применено групповое концентриро
141
вание Sb, As и Bi экстракцией их СС14 в виде диэтилдитиофосфатов [58]. Экстракционно-фотометрические методы определения Sb в никеле и хромо-никелевых сплавах с применением кристаллического фиолетового [695], родамина С [1085] и метилового фиолетового [502] позволяют быстро (20—40 мин.) определять Sb при содержании О !• 10-3%. Для определения Sb (1-10~9 г) в никеле описан активационный метод [1000, 1001], включающий выделение Sb из облученного материала. Методом инверсионной вольтамперометрии определяют Sb(>l-10“4% в хромо-никелевых сплавах [5901. Для определения Sb > 1 • 10~4% в никеле разработаны методы атомно-абсорбционной спектрофотометрии. По одному из них [955] Sb выделяют соосаждением с МпО2, концентрат растворяют и раствор распыляют в воздушно-ацетиленовое пламя. По другому методу [954] Sb экстрагируют 5%-ным раствором триоктилфосфи-ноксида в метилизобутилкетоне в виде иодида и экстракт распыляют в воздушно-ацетиленовое пламя. При содержании Sb 1,38-10~3% стандартное отклонение составляет 2,7•10^4%.
Сурьму в ниобии и пятиокиси ниобия наиболее часто определяют методами спектрального анализа. Ниобий предварительно переводят в пятиокись. Прямые методы [49, 972] позволяют определять до 1 • 10-4% Sb. Предварительное отделение Sb методом испарения снижает предел обнаружения Sb до 1 • 10-5% [379]. Метод, включающий концентрирование Sb соосаждением с CuS [643], и метод, в котором удаляют Nb экстракцией 60%-ным раствором ТБФ в бензоле в среде 10 М H2SO4 [378], также характеризуются высокой чувствительностью п-10-5% (Sr=0,15-r--г- 0,20). Метод инверсионной вольтамперометрии применен для определения Sb 5-10~4% (Sr 0,26) в ниобатах щелочных металлов и пятиокиси ниобия [290]. Предварительное выделение Sb экстракцией в виде диэтилдитиокарбамината позволяет снизить предел обнаружения Sb до 1’10_6% [223].
Для определения Sb в олове наиболее часто используются спектральные методы [782, 812, 900, 1684]. При проведении анализа с возбуждением спектров в дуге постоянного тока в атмосфере Аг, при большой скорости испарения предел обнаружения Sb составляет 5-10"5% [1684]. Описано [812] определение Sb с использованием литых и порошкообразных образцов. Наряду с Sb метод предусматривает определение еще 22 элементов с пределом обнаружения 5-10-3—1-10~5%. При строгой стандартизации условий возбуждения спектров и режима обработки фотопластинок при использовании аналитической пары линий Sb 252,852— Sn 245, 523 нм можно работать по твердому графику [582]. Описано [1495] определение Sb О 1 • 10~3% в олове и его соединениях с применением квантометра с возбуждением спектров как в низковольтной дуге (940 в), таки в высоковольтной искре (14000 в). Ряд спектральных методов предложен для определения Sb в различных сплавах, содержащих олово, в том числе в свинцово-оло-вянно-сурьмяных [1210] и антифрикционных [1494], а также в оло
142
ве лудильных ванн [744] и в электролитах для осаждения свинцово-оловянных сплавов [557].
Для определения Sb в оловянных сплавах наиболее удобными и точными (Sr = 0,0015 -4- 0,0025) являются титриметрические методы. Броматометрическим титрованием определяют Sb в баббитах (0,5—15% Sb) [934], лвинцово-оловянных сплавах (до 14% Sb) [1245], оловянных бронзах, припоях и подшипниковых [1244] и типографских сплавах [И, 1010]. В свинцово-оловянных сплавах предложено определять Sb также методом броматометрического титрования после ее выделения электролизом при контролируемом потенциале, что обеспечивает большую точность определения (Sr ~ 0,001) [852, 853].
Перманганатометрическим титрованием определяют Sb в белом металле [1304]. Биамперометрическим титрованием с применением КВгО3 в качестве титранта определяют Sb в свинцово-оловянносурьмянистых сплавах [944]. Амперометрическое титрование с использованием амилдимеркаптотиопирона в качестве титранта применено для определения Sb в свинцово-оловянных сплавах [697]. Разработан ряд экстракционно-фотометрических методов определения Sb в олове и свинцово-оловянных сплавах, в том числе с применением родамина С (> 1 • 10-3% Sb) [995], иодидным и тиомочевинным методами (> 1-10~2% Sb) [512]. Для определения Sb в олове рекомендован ряд методов инверсионной вольтамперометрии как без отделения Sb >> 5-10~5% (Sr = 0,10 -4--4- 0,15) [221, 224], так и с отделением ее экстракцией этилацетатом [507] или диизопропиловым эфиром [222, 225], а также отгонкой Sn в виде SnBr4 [507]. Нижняя граница определяемых содержаний Sb этими методами достигает 7-10-6—1-10_5% (Sr= 0,15 -4- 0,25).
Прямой полярографический метод применен для определения Sb 1-10~2% (5\ ~ 0,05) в черновом олове [1201]. Методами атомно-абсорбционной спектрофотометрии с использованием воздушно-ацетиленового пламени определяют Sb (3—17%, Sr = = 0,03 -t- 0,05) в полиграфических сплавах [1122] и в свинцово-оловянных припоях [1166]. Для определения Sb в олове рекомендован активационный метод с выделением Sb из облученного образца [1305].
В осмии Sb 0,1 мкг (Sr — 0,1 -4- 0,3) определяют активационным методом, включающим выделение ее из облученного образца [708].
В палладии Sb (6,4-10~7 г, Sr = 0,10-4-0,25) определяют активационным методом после выделения ее из облученного образца экстракцией 2,2'-дихлордиэтиловым эфиром [152].
Для определения Sb 1 • 10~3% в платине используют спектральный метод, позволяющий определять еще 18 других примесей [389а]. В сплавах платины с родием Sb (0,001—0,01%) определяют экстракционно-фотометрическим методом с применением родамина С после ее хроматографического отделения [482] или экстракции изопропиловым эфиром из 7,7 М НС1 [1648]. В продук-
143
тах, содержащих платиновые металлы, Sb определяют в виде ио-дидно-тиомочевинного комплекса после ее выделения соосажде-нием с Fe(OH)3 [400]. В платине и платино-родиевых сплавах Sb определяют активационными методами, включающими предварительное выделение ее из облученного образца [488, 1238].
Для определения Sb > 1 • 1Q»3% в окислах РЗЭ рекомендован спектральный метод [353].
Пробы смешивают (1 : 1) с угольным порошком, содержащим 4% NaF, и смесь помещают в канал угольного электрода. Спектры возбуждают в дуге постоянного тока (15 а) и регистрируют с помощью спектрографа ИСП-28.
В окислах РЗЭ и иттрия для определения Sb к пробе прибавляют фторопласт-3. Спектры возбуждают в дуговом разряде [234].
Для определения Sb 1 10~4% в рении предложен экстракционно-фотометрический метод с применением метилового фиоле тового [645]. В другом методе [507] пробу растворяют, Sb выделяют экстракцией диэтиловым эфиром в виде ионного ассоциата, образуемого анионом SM4~ с катионом пиридиния, и из экстракта реэкстрагируют Sb раствором H2SO4. В реэкстракте Sb определяют по светопоглощению фемилфлуороната Sb (III).
В ртути Sb > 3 • 10~7% определяют полярографическим методом, основанным на анодной поляризации электрода большой поверхности из анализируемой ртути на фоне iM КОН [528]. Полярографический метод использован также для определения Sb в сульфидно-щелочных растворах ртутно-сурьмяного производства [160]. Фон - 0,25 М NaOH + 0,25 М Ka2S.
В свинце, его сплавах, окислах, рудах и продуктах их переработки наиболее часто Sb определяют методами спектрального анализа (табл. 13). В указанных материалах Sb определяют также фотометрическими методами. Так, Sb 5 • 10~3% в свинцовой аккумуляторной массе определяют без ее отделения с применением фенилфлуорона [425, 627]. Для определения Sb в свинце предложен метод, включающий отделение его в виде PbSO4 и определение Sb с фенилфлуороном [1084]. В большинстве случаев при определении Sb в свинце предусматривается отделение ее соосаж-дением с МпО2 и последующим определением Sb в полученном концентрате экстракционно-фотометрическим методом в виде гексахлоростибата родамина С [1293, 1580, 1683] или метилового фиолетового [1006].
При определении Sb в отдельных свинцовых шариках для растворения шарика используют конц. H2SO4, к раствору добавляют 9.17 HCI, окисляют Sb(III) до Sb(V) нитритом натрия и экстрагируют Sb в виде гексахлоростибата кристаллического фиолетового [1046]. Предел обнаружения Sb в свинце экстракционнофотометрическими методами составляет от 5 • 10-3% [1293] до 2 • 10-4% [1580]. Для определения Sb 1 • 10-2% в свинце предложено также экстрагировать ее диизопропиловым эфиром из
Таблица 13 Определение сурьмы в свинце, его сплавах и соединениях методами эмиссионного спектрального анализа Литература ш —, ш со со Я оо ш 12 10 S? 2 м о о со 05 Ц uZ. ZL
Другие определяемые элементы й 2 й сл < < Л ।—। “3 — -Г аГ s-« Н о SJ < о Д’ тг> г- ф Ф гл с <л ca fe и ь"> и ю и Й д ® Й < < < □ N
Коэффициент вариации, % 7 „ 1 S 2 1 S 1 in Щ
Предел обнаружения, % ,о т Y 7 7 7 ? 7 о 2 ь ° 2 s 3 04 СО СО СМ
Сущность метода 1 100 мг пробы в виде диска (</=3,2 мм, h = 1,2 лея) помещают в углубление УЭ и полностью испаряют в дуге постоянного тока (12 а) Пробу в виде цилиндра вносят в отверстие УЭ ц спектры возбуждают в дуге переменного тока (8 а) Используют литые электроды (проба — анод); спектры возбуждают в дуге переменного тока (8—9 а) Используют жидкие электроды; спектры возбуждают в искре (13 кв, 0,01 мкф, 0,5 мгн\, спектрограф ИСП-22 Используют литые электроды, спектры возбуждают в | высоковольтной искре; спектрограф ИСП-22 Используют литые электроды, спектры возбуждают в дуге постоянного тока (7 а) Пробу растворяют в НМОз, свинец отделяют в виде PbSOn раствор выпаривают при 100—110° С и соли помещают в углубление УЭ (катода), спектры возбуждают в дуге постоянного тока (8 а) Пробу растворяют, свинец отделяют в виде РЬС12, фильтрат выпаривают досуха при 100-120° С,остаток дважды выпаривают с HNO3 и анализируют на кварцевом спектрографе с полым катодом
’ Анализируемый материал й S' Ф и S и S и 5 и
145
144
Таблица 13 (продолжение)
Анализируемый материал Сущность метода Предел обнаружения, % Коэффициент вариации, % Другие определяемые элементы Литература
Свинец отделяют в виде Pb(NO3)2 из конц. HNO3, раствор выпаривают досуха и остаток спектрографируют 5-Ю-6 15—25 Na, Са, Ag, Cd, In, Tl, Fe, Co, Zn, As, Cu, Bi [726]
Сурьму выделяют соосаждением с МпО2, осадок высушивают при 130° С, смешивают (1 :1) с УП, таблетируют и сжигают в дуге переменного тока; спектрограф ИСП-22 1-Ю-3 12 Sn [1038]
Свинец и его соединения Образцы растворяют в HNO3, свинец отделяют в виде PbSO4, фильтрат выпаривают, остаток смешивают (5:1) с УП, спектры возбуждают в дуге постоянного тока (13 а); спектрограф ИСП-22 1.10-5 20 21 элемент [80]
Свинцово-сурьмянистые сплавы Используют литые электроды, спектры возбуждают в конденсированной искре (12 кв, 1950 пф), 0,08 мгн) и регистрируют спектрографом большой дисперсии 0,3—15 1,8-6,9 Ag, Cd, Cu, Fe, Zn 1871]
Сурьмянистый свинец Используют литые электроды, спектры возбуждают в искре (8 кв, 3 мкф, 0,08 мгн) и регистрируют на спектрографе Q-24 3,28— 9,73 5 As, Cu, Fe, Zn [481]
Теллуристый свинец Используют литые электроды, спектры возбуждают в искре (2а, 0,01 мкф, 0,15 мгн) 9-Ю"3 6,8 Те [126]
Свинцовая пыль Эталоны и пробы смешивают (1 : 1) с УП, содержащим 5% Bi(NO3)3 5-Ю-2 6 As, Cd, Sn, Те 18П]
Свинцовые припои Пробу переводят в окислы, которые испаряют из кратера (3x4 УЭ (анода), спектры возбуждают дугой постоянного тока; спектрограф ИСП-28 12 Al, As, Bi, Cu, Fe, Sn, Zn [2Ю].
Таблица 13 (окончание)
Анализируемый материал Сущность метода Придел обнаружения, % Коэффициент вариации, % Другие определяемые элементы Литература
Пробу переводят в окислы и испаряют из канала УЭ, спектры возбуждают в дуге постоянного тока 1-10-» 12—20 Ag, As, Bi, Cd, Cu, Zn [1101]
Типографские сплавы Используют литые электроды, спектры возбуждают в конденсированной искре (0,005 мкф, 0,01 мкн, 0,8 а) 1-Ю-3 15-20 Cu, Pb, Sn [323]
Аккумуляторный лом, концентраты и хвосты свинцового производства Порошкообразные пробы возбуждают в дуге переменного тока (9,5 а); спектрограф ИСП-28 1-10-2 3-5 Al, As, Cu, Fe, Sn, Zn [201]
Сырье и полупродукты свинцового производства Порошкообразную пробу испаряют из канала УЭ, спектры регистрируют на кварцевом спектрографе средней дисперсии 1-10-2 7 As, Sn [806]
Галенит Пробу смешивают (2:3) с УП, помещают в канал (3x3 aiai) УЭ (анода) и спектры возбуждают в дуге постоянного тока 1-10-3 20-25 Bi, Cd [677]
Сульфид свинца Пробу смешивают с УП, помещают в углубление УЭ (анода) и спектры возбуждают в дуге постоянного тока (12 а) 5-Ю-4 5—15 Ag, As, Bi, Cu, Ni [53]
раствора НС1 и заканчивать определение измерением оптической плотности раствора иодидного комплекса [890].
Описан спектрофотометрический метод определения Sb в свинцово-цинковых рудах в виде иодидно-тиомочевинного комплекса [1183]. При определении Sb в продуктах свинцово-цинкового и кадмиевого производства сначала Sb отделяют экстракцией 0,5 М раствором ди-(2-этилгексил)фосфорной кислоты в гексане или октане из серно- или азотнокислого раствора, затем ее реэк-страгируют 9М НС1 и в реэкстракте определяют в виде гексахлоростибата бриллиантового зеленого [626]. Для определения Sb в свинце и его сплавах предложен косвенный фотометрический метод, включающий отделение ее осаждением в виде ионного ассоциата, образуемого SbGl6 с катионом дихлорэтилендиаминко-бальта, растворение осадка в растворе NaOH, добавление Н2О2 и комплексона III и измерение интенсивности образующейся фиолетовой окраски [1343]. При содержании Sb 0,01 -0,1% коэффициент вариации 2—3%.
Полярографическим методом определяют Sb в свинце без ее отделения с использованием 6,2 М HG1 в качестве фона [1664]. В сурьмянистом свинце определяют Sb с использованием в качестве фона 6,0 М НС]. Основную массу свинца предварительно отделяют осаждением в виде РЬС]2. При полярографическом определении Sb в рафинированном свинце в качестве фона используют 17V H2SO4. Свинец предварительно удаляют в виде PbSO4 [876]. При содержании Sb 2,5 • 10-3% коэффициент вариации ~ 5%. При использовании в качестве полярографического фона аммиачно-щелочного раствора ЭДТА сурьму определяют в свинце без его отделения [1345]. Разработаны полярографические методы [1343, 1606] одновременного определения Sb, Bi и As в свинце. С использованием в качестве фона IN H2SO4 -|- 0,17V HNO3 в присутствии 0,001 % метиленового голубого возможно определение до 2 • 10-4% Sb [1343]. Метод инверсионной вольтамперометрии позволяет определять в свинце до 1 • 10-5% Sb [833].
Сурьму в свинце определяют активационными методами как в инструментальном варианте [836, 837, 1205, 1506] с пределом обнаружения 4 • 10-6%, так и методами, включающими ее выделение из облученного материала [144, 837, 907, 911, 1206]. Этими методами возможно определение до 1 • 10“6% Sb (Sr = 0,06) [911]. Методы атомпо-абсорбционной спектрофотометрии [267, 268] при использовании пламени смеси С2Н2 с воздухом и измерении поглощения света линии 217,6 нм позволяют определять до 3 • 10~3% Sb (Sr = 0,05 +- 0,06).
В свинце [1379], свинцовом блеске [1531] и типографских сплавах [1481, 1482] Sb 1 • 10~3% определяют методами рентгенофлуоресцентного анализа по линии Sb Ка. В методе [1379] в качестве источника излучения для возбуждения спектров используют 241 Ат. Сурьму в типографских [1553], антифрикционных [1088], свинцово-сурьмянистых и свинцово-оловянно-сурьмянистых спла-148
вах [944] определяют методом броматометрического [1088, 1553] и биамперометрического [944J титрования без ее отделения.
В селене и его соединениях Sb наиболее часто определяют спектральными методами (табл. 14). Используются также активационные методы. Высокая чувствительность (1 • 10-6 — 1 • 10~7%)и возможность одновременного определения большого числа примесей (до 15 —20 элементов) обеспечивают им применение не только для анализа технического продукта [772, 11091, но и для анализа селена высокой чистоты [151, 250, 772, 1104, 1677]. Для определения Sb (0,06 —3 мкг) в селене предложен [350J метод кулонометрического титрования. Матрицу предварительно удаляют возгонкой, остаток растворяют в H2SO4 и титруют Sb(III) на фоне MnSO4 + 7AH2SO4 электрогенерированными ионами МпО4~.
В сере Sb > 1 • 10-4% (Аг = 0,05-4-0,10) предложено определять спектральным методом [189], заключающимся во вдувании порошкообразной пробы в виде узкой вертикальной струйки в разряд между горизонтально расположенными угольными электродами. Спектры возбуждают в дуге постоянного тока (20 а) и регистрируют с помощью спектрографа ИСП-30. Описан также активационный метод определения Sb до 9 • 10“8% (Sr = 0,10-4— 0,25) и 13 других примесей в сере [771]. Пробы и эталоны облучают потоком 8,7 • 101г нейтрон/см2 сек в течение 30 час. выделяют Sb и измеряют ее активность. Сурьму в сере (0,06—3 мкг Sb, Sr = =0,005 -j- 0,05) предложено [350] определять кулонометрическим титрованием электрогенерированными ионами МпО4" после удаления серы сжиганием.
В серебре и его сплавах Sb определяют спектральными методами с использованием проб в виде стержней. Спектры возбуждают в дуге переменного тока (12 а) [390]. В другом варианте [391] спектры возбуждают в глобульной дуге. Ошибка определения в обоих случаях 10—20%. С использованием электродов из анализируемого материала сурьму определяют с использованием возбуждения спектров в конденсированной искре (Sr ~ 0,05) [909] или в дуге постоянного тока 8 a (Sr ~ 0,15) [1598]. Определение Sb (5 • 10~°—2,5 • 10"3 %, Sr ~ 0,11) и 14 других примесей в хлориде серебра выполняют следующим образом.
Пробу смешивают с угольным порошком в соотношении (1 : 1). 40 мг смеси помещают в канал угольного электрода (анода). Спектры возбуждают в дуговом разряде и фотографируют на дифракционном спектрографе с дисперсией 0,25 нм/мм [1362].
Метод инверсионной вольтамперометрии с предварительным отделением Sb экстракцией этилацетатом позволяет определять в серебре до 5 • 10-6% Sb (5Г = 0,10 н- 0,15) [731]. Описан [1676] активационный метод определения Sb в серебре, включающий выделение Ag из облученной пробы осаждением в виде AgG] и дальнейшую очистку сурьмы до радиохимической чистоты. В различ-
149
Таблица 14. Определение сурьмы в селене и его соединениях методом эмиссионного спектрального анализа
Литература [155] [154] [499] [747] [818] [157] [1200] [715] [715]
Другие определяемые элементы о-? 4 ДЙ о Рн с? щ н О О О г - ~._Г - fi Г ф S..-'S.2 i g S a* v z: i § § as ° -'5 -55 —T с? ф fee ® fee. Дю ю o -£ of fe£^p ьр be
Коэффициент вариации, % in Ю ю ю ca lo ю to ю -ч-i СЧ1 О<1 •'г-* м co co 1 1 1 1 1 1 1 II оог-юогосм О О to тч CO T-f co
Предел обнаружения, % 7T? 1 ? 1 T ? T T О О О T о о о о о У Т У 2Т 1 У У Tv — I о У •*-< “У ю
Сущность метода Пробу помещают в канал нижнего УЭ, а в канал верхнего УЭ вносят УП, содержащий 1% NaCl; спектры возбуждают в дуговом разряде (8 а) Пробу вносят в канал У Эи сплавляют, добавляют еще пробы и снова сплавляют до заполнения канала Пробу помещают в канал УЭ; дуга постоянного тока (10 а); спектрограф Q-24 В пробу вводят 1% NaCl, спектры возбуждают дугой переменного (8—19 а) или постоянного тока (15 а); спектрограф ИСП-22 или ДФС-8 Пробу помещают в кратер УЭ и расплавляют, снова дополняют и расплавляют до заполнения кратера; дуга переменного тока (10 а); спектрограф ИСП-22 Пробу окисляют HN03 до SeO2 и отгоняют SeO2 при 315° С, остаток прокаливают при 400—450° С, прибавляют раствор NaCl, высушивают и помещают в кратер УЭ; дуга переменного тока (12 а) Пробу растворяют в HNO3, к раствору добавляют 6 М НС1 п отделяют Se экстракцией раствором дитизона в СС14; водный раствор выпаривают, остаток растворяют в 200 мкл 6 М НС1; 50 мкл этого раствора наносят на смесь 9,1 мг УП и 0,9 мг NaCl, помещают в кратер УЭ и высушивают; дуга постоянного тока (13 а)', спектрограф е дифракционной решеткой 600 штрих]мм Пробу смешивают (1 :10) с УП; дуга переменного тока; спектрограф ИСП-28 Возгоняют HgSe и концентрат примесей наносят на торец УЭ! дуга переменного тока; спектрограф ИСП-28
Анализируемый материал И S | я S ф ф н к ч ч >> 3 ф ф ь Q CJJ Q О,
ных серебряных сплавах определяют Sb экстракционно-фотометрическим методом с применением родамина С после отделения Sb соосаждением с МпО2. При содержании Sb 0,0041—0,31% ошибка определения 4—5% [980]. Для определения Sb в электролитах серебрения ее предварительно отделяют экстракцией раствором ди-(2-этилгексил)пирофосфоргтой кислоты в гептане, затем реэк-страгируют и титруют 0,1 N раствором КМпО4 [775].
В таллии Sb > 5 • 10“s% определяют фотометрическим методом с фенилфлуороном, отделяя ее от Т1 экстракцией раствором диэтилдитиокарбаминовой кислоты в СНС13, а затем от Sn и Мо* — экстракцией диэтиловым эфиром в виде пиридин-иодидного комплекса [64, 65]. Активационные методы, включающие выделение Sb из облученной пробы, позволяют наряду с Sb определять в таллии и другие элементы (As, Со, Cu, Fe, Ga, In, Те и Sn [604]; As, Cu, Mn, Zn [19]).
Для определения Sb в тантале применен химико-спектральный метод, включающий выделение ее соосаждением с CuS и прокаливание осадка при 500° С [643]. Спектральный анализ тантала по методу испарения позволяет определять Sb (в также Bi, Cd, Pb и Sn) с пределом обнаружения 1 • 10“4% (Sr = 0,08 -4- 0,11) [237]. В танталатах щелочных и щелочноземельных металлов Sb ф? 1 • 10“6 % (Sr = 0,10-4— 0,15) определяют без отделения методом инверсионной вольтамперометрии на фоне 0,2М НС1 + 0,1М винной кислоты [223].
В теллуре Sb определяют спектральными [156, 591, 592, 1613] и химико-спектральными [728] методами. Предел обнаружения Sb спектральными методами составляет 1 10“3—1 • 10“4% (Sr = = 0,1-4-0,2), химико-спектральными — 1 • 10“6% (Sr = 0,15 -4--4- 0,30). В теллуриде свинца Sb, а также Ag, Al, Bi, Cu, Fe, Mg, Si, Sn) определяют спектральным методом с пределом обнаружения 5 • 10“5% (Sr ~ 0,18) [342]. Для определения Sb в теллуристом баббите рекомендован химико-спектральный метод, позволяющий определять также As, Bi, Sn, Те и Zn [1656]. Экстракционно-фотометрический метод, включающий концентрирование Sb путем отгонки теллура в виде бромида, экстракцию Sb раствором диэтидитиокарбамината диэтиламмония в СНС13, выпаривание экстракта и обработку остатка смесью HNO3, НС1О4 и H2SO4 и экстракцию Sb в виде гексахлоростибата родамина С, позволяет определять в теллуре до 5 • 10“s %Sb (Sr — 0,15-4- 0,20) [1171]. При использовании инструментального активационного метода с применением стоканального 7-спектрометра при навеске 10 мг возможно определение в теллуре до 5-10“7% Sb (Sr ~ 0,15) [5]. Методы, включающие выделение Sb, позволяют определять до 9 • 10“8—1-10“7% Sb (Sr = 0,15 -н 0,30) [251, 772].
В титане и двуокиси титана Sb О 1 • 10"4% (Sr — 0,05-4— -4-0,15) определяют экстракционно-фотометрическими методами с применением метилового фиолетового [498], бриллиантового зеленого [1083, 14671 и родамина С [1179]. Для определения Sb
151
и 20 других примесей спектральным методом [306] в титане пред" варительно переводят титан в окислы.
При определении Sb в двуокиси титана пробу смешивают (4 : 1) с AgCl; 60 мг смеси помещают в отверстие угольного электрода (анода) и спектры регистрируют с помощью спектрографа ИСП-22.
В другом методе [1262] для определения Sb и других 13 примесей в двуокиси титана пробу разбавляют смесью ZnO с угольным порошком и вводят в канал угольного электрода; спектры воз-бу’ящают в дуге постоянного тока (10 а) и регистрируют кварцевым спектрографом большой дисперсии (0,3 нм/мм). При содержании Sd 0,001 -5% коэффициент вариации составляет 3—20%. В титане и двуокиси титана Sb > 5-10~6% (Sr = 0,03-4-0,12) определяют методом инструментального активационного анализа [68]. Методы с выделением Sb из облученной пробы используют для ее определения в порошках двуокиси титана, рутиле и анатазе [230] и в отдельных кристаллах двуокиси титана [1380]. Полярографическими методами определяют Sb (а также Си) в двуокиси титана, рутиле, анатазе и бруките [1548]. В двуокиси титана Sb (0,01 — 0,2%) определяют на фоне 1М раствора HCI [822]. Метод переменнотоковой полярографии рекомендован для определения Sb в растворах сульфата титана, содержащих до 345 г/л Ti (IV) и до 217 г/л Fe [1174].
Для определения Sb (до 5 10-5%) в тории и его соединениях предложен химико-спектральный метод, включающий отделение тория в виде оксалата или пероксида [307].
В углероде, угольном порошке и графите Sb определяют активационными [106, 1203, 1207, 1330] и спектральными методами [397, 612]. Инструментальными активационными методами определяют Sb > 5 10~9% в пирографите [106] и в аморфном угле, природном и искусственном графите (> 7 • 10~8%) [1207]. В ряде активационных методов предусматривается выделение Sb из облученной пробы. В одном из них [1203] при определении Sb в аморфном угле и графите пробу после облучения озоляют при 400° С в атмосфере О2. В другом методе [11301 при определении Sb в ядерном графите облученную пробу минерализуют нагреванием с HNO3 в колбе с обратным холодильником. Предел обнаружения Sb 1 • 10'8% (£,. = 0,15 -н 0,25). Для определения Sb в графите предложен спектральный метод, в соответствии с которым пробу смешивают с NaF, содержащим Bi (внутренний стандарт) [612]. Описаны [397] методы спектрального, атомно-абсорбционного и флуоресцентного определения Sb(10-7—10%) в графитовом порошке и стеклоуглероде.
Для определения Sb в уране, его окислах и рудах наиболее часто используются спектральные методы. Уран предварительно переводят в окислы. Большинство спектральных методов включает дистилляцию Sb с носителями, в качестве которых используют AgCl [1443, 1447], смесь (4 : 1) AgCl с AgF [1240] и смесь
152
(3 : 1) Li2GO3 c NH4G1 [1182]. Предел обнаружения Sb 1 • 10-5— 2 • 10~5% (S,. = 0,07 -4- 0,17) [1447]. Прямой спектральный метод определения Sb в материалах на урановой основе без использования носителей позволяет определять до 1 • IO-3 Sb % [878]. Для спектрального определения Sb > 2 • 10-4%, а также других 20 примрсей в уране предложен химико-спектральный метод, в котором уран предварительно выделяют экстракцией ТБФ [493].
В фосфоре Sb определяют активационными методами, включающими выделение Sb из облученной пробы с пределом обнаружения 3 • 10-в% [826], 4,5 • 1О'7о/о 1885] и 1 • 10“7% [1093].
Для определения Sb в хроме, его сплавах и окислах предложен ряд спектральных методов. В одном из них [729] для определения Sb О 5 • 10~4% в хроме используют фракционную дистилляцию: Sb и другие примеси из камеры испарителя поступают непосредственно в плазму дуги переменного тока (10а).
По методу [555] пробу смешивают (5 : 1) с угольным порошком. 100 мг смеси помещают в графитовый стаканчик, который устанавливают между щеками испарителя, нагревают до 1600° С и выдерживают при этой температуре 2 мин., принимая конденсат на торец угольного электрода. Предел обнаружения Sb 1 • 10“3% (Sr = 0,09 ч- 0,14).
Для определения меньших содержаний Sb проводят предварительное ее концентрирование соосаждением с GuS. При содержании Sb 1 • 10-г —1 • 10“4% Sr = 0,10-4— 0,19 [101]. Ряд спектральных методов [571, 777] предложен для определения Sb в окислах хрома. По одному методу [777] Sb 1 • 10~4 (Sr 0,3) выделяют соосаждением с GuS, осадок растворяют в смеси HG1 с HNO3 и анализируют в виде раствора. В случае анализа хромового ангидрида Cr(VI) предварительно восстанавливают до Сг(Ш). В хроме и его сплавах Sb определяют также экстракционно-фотометрическим методом с применением метилового фиолетового [545].
Для определения Sb в цинке, его сплавах и соединениях лучшими являются экстракционно-фотометрические методы, характеризующиеся высокой чувствительностью (1 • 10~7% [1489] или 0,05 мкг/мл [456]). В качестве реагентов используют родамин С [332, 1139, 1489, 1615], кристаллический фиолетовый [701], бриллиантовый зеленый [769] и метиловый фиолетовый [456].
При определении Sb 5-10~5% (.S’r = 0,10) в цинке спектральными методами его переводят в окись путем растворения в HNO3, выпаривания раствора досуха и прокаливания при 500° С. Остаток растирают, вводят в канал угольного электрода и возбуждают спектры в дуге переменного тока (4 а) [976].
Для достижения большей чувствительности проводят концентрирование путем вакуумной сублимации цинка при 560° С (предел обнаружения Sb 1 • 10~5% (8Г = 0,30 -н 0,35) [780], экстракцией Sb и некоторых примесей СС14 в виде диэтилдитиокарбами-
153
натов [59] или соосаждением с небольшим количеством Zn(OH)2, получаемой добавлением NH4OH к раствору пробы до pH ~ 6 (предел обнаружения Sb 10“5%, Sr = 0,2 н- 0,3) [716].
Для определения Sb 1-10~б% в цинковой обманке пробу смешивают с активированным углем (1 : 9); 40 мг полученной смеси вносят в углубление угольного электрода, покрывают слоем активированного угля и возбуждают спектры в дуге постоянного тока (8 а) [1142].
Полярографические методы позволяют одновременно определять в цинке Sb и Bi [417, 420] и Sb и Sn [67]. В полупроводниковых индий-цинковых сплавах Sb определяют методом тонкослойной хроматографии с использованием очень малых объемов растворов (5 —10 мкл); при содержании Sb ~2% ошибка определения <^5% [721]. Активационные методы, включающие выделение Sb из облученной пробы, используются для ее определения в цинке [827, 1272, 1488] с пределом обнаружения до 1-10-7 мкг в пробе (5Г ~ 0,25) [1272] или до 1-10~6% (5Г = 0,1 ч- 0,2) [827]. В электролитных растворах сульфата цинка активационный метод с выделением Sb из облученной пробы позволяет определять до 2-10"3 мкг/мл Sb [1012].
В цирконии, его сплавах и двуокиси циркония Sb определяют спектральными [679, 1368] и активационными методами [1070, 1555]. При определении Sb спектральными методами в цирконии последний путем прокаливания переводят в двуокись, которую смешивают с AgGl, и смесь вводят в канал угольного электрода; спектры возбуждают в дуге постоянного тока (12 а) и регистрируют спектрографом ИСП-22. Предел обнаружения Sb 1-10~3% [679]. В химико-спектральном варианте этого метода цирконий сначала удаляют в виде миндалята, водный раствор выпаривают с добавлением PbSO4 и заканчивают анализ как и в прямом варианте. Предел обнаружения Sb ~ 1-10~4% (5Г ~ 0,2). Метод с применением фракционного испарения (носитель AgCl) позволяет определять в двуокиси циркония до 1-10-4% Sb [1368]. Инструментальный активационный метод применен для определения Sb в циркалое [1555]. Метод с выделением Sb из облученного материала использован для ее определения в цирконии [1070].
Для определения Sb в щелочных и щелочноземельных метал- . лах, их окислах и солях наиболее часто используются химикоспектральные методы. Сурьму и другие примеси концентрируют экстракцией в виде диэтилдитиокарбаминатов [550, 761], комплексов с алифатическими монокарбоновыми кислотами [733], диэтилдитиокарбаминатов и 8-оксихинолинатов экстракцией при pH 3,7 и затем в виде дитизонатов при pH 9,0 [1115], а также соосаждением с CdS [575, 576] и осаждением в виде диэтилдитиокарбаминатов и сульфидов (образующихся за счет введения тибацетами-да). Предел обнаружения Sb в указанных материалах составляет 5-10“6—1-10~4% (Sr = 0,2-1-0,3). Люминесцентные методы рекомендованы для определения Sb в иодиде натрия [665] и окиси
154
кальция [1657]. В методе [665] Sb > 3-10-7% сначала отделяют экстракцией диэтиловым эфиром в виде пиридин-иодидного комплекса, затем! реэкстрагируют водой, реэкстракт выпаривают в присутствии HNO3, остаток растворяют в 0,5 мл конц. НВг и измеряют люминесценцию при 640 нм при температуре жидкого азота.
АНАЛИЗ ОРГАНИЧЕСКИХ ВЕЩЕСТВ, РАСТИТЕЛЬНЫХ И ЖИВОТНЫХ ПРОДУКТОВ
В растительных и биологических материалах определяют Sb активационными методами без ее выделения из облученной пробы. Так определяют Sb в хроматографической бумаге [1409], полифе-нилах [983], нефти и продуктах ее переработки [982, 991, 1517], листьях деревьев [13161, аскорбиновой кислоте [1630]. Предел обнаружения Sb этими методами достигает 1 -10—7—1 -10—8 % (Sr = = 0,10 -г- 0,15). Иногда при определении Sb в органических материалах ее выделяют из облученного материала. Наиболее часто для этой цели используют методы мокрой минерализации, в том числе нагревание со смесью H2SO4 и HNO3 [877, 1407] или же с этой же смесью с добавлением Na2SO4 [163], смесью HNO3 и Н2О2 [1510]. Применяют также простое сжигание в условиях, исключающих потери Sb [1408]. Из полученного остатка Sb выделяют экстракцией, осаждением или соосаждением и другими методами. Так определяют Sb в пищевых продуктах, органических и биологических материалах [877, 1415, 1510], нефти и продуктах ее переработки [1408], резине [163]. Предел обнаружения Sb достигает 1 -10-9—1 • 10"11 г, или 1 • 10-8—1-10-9%.
Спектральным методом определяют Sb в растительных и биологических материалах после их минерализации [1573].
В одном из методов [577] пробу смешивают с SrSO4 и минерализуют нагреванием со смесью H2SO4 и HNO3. Спектры возбуждают в дуге постоянного тока между угольными электродами.^ Предел обнаружения Sb ~ 1-10~6% (S, = 0,15 -е 0,20).
В органических растворителях определяют Sb^l’10-6% и другие 22 элемента спектральным методом после удаления растворителя испарением в присутствии Н2С2О4, необходимой для устранения потерь определяемых элементов [554].
Для определения Sb в лавсане 20—30 г пробы озоляют при 400—450° С в течение 5—6 час., золу разбавляют 10- или 100-кратным количеством смеси СпО и угольного порошка (1 : 9), смесь помещают в канал угольного электрода и спектры возбуждают в дуге переменного тока (18—20 а).
При определении Sb 3-10-6% в тиомочевине ее концентрируют на смеси угольного порошка и GdS с применением тиоацетамида и диэтилдитиокарбамината натрия в качестве осадителей. При определении Sb 5- 10-г% (5Г ~ 0,05) в смазочных материалах пробу минерализуют нагреванием со смесью H2SO4 и HNO3. В ми-
155
нерализате определяют Sb в виде иодидного комплекса [1394]. Для определения Sb 1 • 10~5% в картоне, используемом для изготовления тары для пищевых продуктов, из минерализата экстрагируют Sb диизопропиловым эфиром и фотометрируют в виде гексахлоростибата родамина С [1637].
В этаноле и его водных растворах Sb 1-10-в% определяют методом инверсионной вольтамперометрии без ее отделения [311]. Для определения Sb в растительных материалах рекомендован метод катодно-лучевой полярографии [1370]. При определении Sb в пенополиуретане ее предварительно извлекают конц. H„SO4 и затем полярографируют на фоне ЗМ HG1 [95].
АНАЛИЗ РЕАКТИВОВ, РАСТВОРОВ, ВОЗДУХА, ПРИРОДНЫХ И СТОЧНЫХ ВОД
В минеральных кислотах Sb наиболее часто определяют спект- • ральными методами [111, 552, 574, 1008], позволяющими, кроме Sb, одновременно определять ряд других примесей. Так, например, в методе [111] в соляной, азотной и фтористоводородной кислотах одновременно с Sb 1 • 10~7% определяют еще 18 элементов.
Пробу (50 г) выпаривают при 100° С с 20 мг угольного порошка, добавляют 0,5 мл 0,2%-ного раствора NaCl и снова выпаривают. Концентрат помещают в кратер (d = 4,5 мм, h = 4 мм) угольного электрода (анода). Спектры возбуждают дугой постоянного тока (10 а); спектрограф ИСП-22 или ИСП-28.
В другом методе [574], также включающем выпаривание анализируемой кислоты с угольным порошком, концентрат анализируют с применением газоразрядной трубки с полым катодом. Метод позволяет определять до 3-10~7% Sb (Sr = 0,15) в уксусной, соляной, серной и фтористоводородной кислотах. В методе [552] Sb и 14 элементов-примесей определяют в броме, азотной, соляной, бромистоводородной и фтористоводородной кислотах путем измерения относительной интенсивности линий элементов-примесей и элемента сравнения — кобальта в дуговом спектре сухого остатка, полученного после выпаривания пробы с угольным порошком. В качестве усиливающей добавки используют NaCl. При определении Sb в качестве аналитической пары линий используют линии Sb 259, 806 — Со 252,136 нм. Предел обнаружения Sb при навеске анализируемого вещества 10 и 50 г составляет соответственно 1-10~6 и 2 • 10“7 %. Для определения Sb и других 16 примесей в соляной и азотной кислотах предложен метод [1008].
Пробу (50 мл) упаривают до 0,5 мл, и концентрат наносят на торец уголь ного электрода, пропитанного хлороформным раствором органического стекла. В качестве внутреннего стандарта используют золото. Спектры возбуждают в дуге переменного тока (7 а), Спектрограф (J-24.
Методом инверсионной вольтамперометрии предложено определять Sb 1-10“6% в соляной и азотной кислотах, деионизован
156
ной воде и растворах аммиака [224]. Определение выполняют на фоне 0,3 М HG1. Для определения Sb в бромистоводородной кислоте рекомендован [77, 1242] метод, основанный на измерении люминесценции SbBr3 при температуре жидкого азота. Метод позволяет определять до 2,5 нг Sb в пробе объемом 0,5 мл. В соляной и азотной кислотах, а также в перекиси водорода Sb предложено [13391 определять активационным методом.
В гидросульфите натрия Sb, а также Zn и Bi определяют методом осциллополярографии с пределом обнаружения —10“4% (5Г = 0,1 ч- 0,2) [295].
Метод инверсионной полярографии [232] применен для определения Sb 1 • 10~s%, а также Cd, Си и Pb в растворах и сточных водах свинцово-цинкового производства; полярографическим методом определяют Sb 1 —60 г/л в растворах после гидрометаллургической выработки медно-сурьмянистых концентратов и промывных водах [1546].
В природных и сточных водах, а также в питьевой воде Sb определяют методами активационного анализа с предварительным концентрированием соосаждением с Fe(OH)3 [642], ионным обменом [1359] или испарением в замороженном состоянии (для избежания возможных потерь), используя для этого пробы объемом до 100 мл [1427]. Предложен также ряд методов без концентрирования Sb [4, 1040, 1636]. Для определения Sb в ледниках поступают следующим образом.
Пробу льда растаивают и 57 мл полученной воды помещают в 9—12 сосудов, содержащих по 3,5 мл конц. HNO3, и облучают 60 мин. вместе со стандартами па вращающемся стенде потоком 1,8-1012 нейтрон/см2-сек. Облученные пробы объединяют, прибавляют по 10 мг носителей определяемых элементов (Sb, As, Cu, Hg, Mn; для Cd — 20 мг) и проводят разделение и радиохимическую очистку. Активность выделенных радиоизотопов измеряют с помощью сцинтилляционного "/-спектрометра с кристаллом NaJ(Tl). Метод позволяет определять до 0,3 нг Sb в пробе или до 5-10~10% [1636].
Методы атомпо-абсорбционной спектрофотометрии также часто используются для определения Sb в природных и сточных водах. В большинстве методов Sb восстанавливают с помощью NaBH4 и образующийся SbH3 вводят в трубчатую печь [1123, 1213] или в водородно-воздушное пламя, защищенное аргоном [1209]. Предел обнаружения Sb 0,5 мкг/л. При содержании в пробе 0,3 мкг Sb Sr = = 0,052. Описано также применение атомно-абсорбционной спектрофотометрии для определения Sb в морской воде, рапе и растворах солей, включающей предварительное хроматографическое концентрирование Sb [1359].
Для определения Sb в морской воде предложен метод инверсионной вольтамперометрии [1105].
Через пробу воды пропускают газообразный НС1 до его концентрации 4 М п проводят электролиз 10 мин. при —0,5 в, затем регистрируют поляро
157
грамму до —0,2 в и по высоте пика находят суммарное содержание Sb и Bi. В другой такой же пробе проводят электролиз в среде 1 М НС1 при — 0,4 в; при этом выделяется только Bi, который определяют по его пику ионизации. Содержание Sb находят по разности.
В методе [465] для устранения мешающего влияния Bi, Fe и Си их отделяют экстракцией хлороформным раствором диэтилди-тиокарбамината цинка, предварительно окислив Sb(III) до Sb(V) бромной водой. Из оставшейся водной фазы экстрагируют Sb с применением диэтилдитиокарбамината натрия, из экстракта Sb реэкстрагируют соляной кислотой и определяют методом инверсионной вольтамперометрии с применением ртутно-графитового электрода. Предел обнаружения Sb 10 нг/л.
Метод катодно-лучевой полярографии [1604] предназначен для определения Sb > 0,1 мкг! л в речной и озерной воде. Недостатком его является необходимость длительного упаривания пробы (объем 200 мл) до 0,5 мл без кипячения.
Экстракционно-фотометрические методы используются для определения Sb в природных [41, 1438] и сточных водах предприятий цветной металлургии [784]. Для определения Sb >0,1 мкг/л в речных, озерных и морских водах предложен метод [1438], включающий концентрирование ее соосаждением с МпО2, последующее отделение Sb от соосажденных с ней элементов экстракцией мети-лизобутилкетоном, реэкстракцию и определение в реэкстракте с применением родамина С. При содержании в пробе 1дгкг Sb коэффициент вариации составляет ~2%. Для определения Sb > > 10 мкг/л в сточных водах предприятий цветной металлургии предложен экстракционно-фотометрический метод [784] с применением бриллиантового зеленого. Для раздельного определения Sb(III) и Sb(V) в природных и сточных водах предложен метод [666] с применением кристаллического фиолетового. Из одной пробы воды экстрагируют Sb(V) в виде гексахлоростибата кристаллического фиолетового, а затем в другой такой же пробе окисляют Sb(III) до Sb(V) и экстрагируют таким же образом всю сурьму. По разности находят содержание Sb(III).
При определении Sb > 1,2-1СГ6% в природных и сточных водах спектральным методом пробу выпаривают до получения сухого остатка, который затем смешивают (1 : 2) с буферной смесью, содержащей 50% (NH4)2HPO4, по 16,7% CaSO4-2H2O и MgSO4-2H2O, 16,6% Na2SO4 и 0,035% GeO2 (внутренний стандарт). Спектры возбуждают в дуге переменного тока (16 а) [429].
В методе [62] Sb > 2-10“7% концентрируют экстракцией хлороформом в виде диэтилдитиокарбамината, экстракт выпаривают с угольным порошком и анализируют. Для определения Sb > > 0,5 мкг/л предложен спектральный метод [943], включающий восстановление сурьмы с помощью NaBH4 до SbH3, подачу образовавшегося SbH3 током Не в дуговой разряд и измерение интенсивности линии Sb 252,5 нм.
158
В атмосферной пыли Sb наиболее часто определяют экстракционно-фотометрическими методами [175]. Методы включают фильтрование определенного объема анализируемого воздуха через плотные фильтры, их минерализацию и анализ минерализата. Для определения SbH3 > 1-10-4% в воздухе рекомендован метод [1662], в котором анализируемый воздух пропускают (100 мл/мин) через 50 мл 3 М раствора НС104, содержащего 72 г/л NaJ и 10 г/л J2. В полученном растворе определяют сурьму в виде SbJ7 по оптической плотности при 425 нм. Для анализа атмосферной пыли предложен спектральный метод, позволяющий наряду с Sb определять еще 12 элементов с пределом обнаружения 2-Ю-7 — 1-10”6 г [12]. При определении Sb в воздушной пыли активационными методами [884, 1313, 1314] фильтры не озоляют, а непосредственно подвергают облучению потоком нейтронов, а затем снимают у-спектры через различные промежутки времени и рассчитывают содержание Sb и других элементов.
Глава VII
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В СУРЬМЕ И ЕЕ СОЕДИНЕНИЯХ
Для определения примесей в сурьме и ее соединениях используются спектральные, фотометрические, полярографические, атомно-абсорбционные, люминесцентные и многие другие методы. Однако наибольшее значение имеют спектральные методы, позволяющие одновременно определять большое число примесей [479, 682, 801]. Ошибка определения примесей прямыми спектральными методами зависит от их содержания, анализируемого материала, используемой аппаратуры и ряда других факторов и колеблется от 3 —5 до 30 —50%; чаще всего она находится в пределах 10 —20%. Некоторые характеристики прямых спектральных методов определения примесей в сурьме и ее соединениях приведены в табл. 15.
Однако с развитием полупроводниковой промышленности и промышленности чистых веществ потребовалось определять значительно меньшие содержания примесей в сурьме и ее соединениях, чем те, которые можно определять прямыми спектральными методами. В связи с этим стали использоваться химико-спектральные методы, включающие предварительное концентрирование определяемых примесей. В большинстве случаев это достигается удалением основы различными методами, а также экстракцией Sb, в том числе экстракцией бутилацетатом [187, 446, 447, 671] и 2,2'-ди-хлордиэтиловым эфиром [102, 800, 803] из растворов HG1 и ди-(2-этилгексил)фосфорной кислотой [802], 2,2'-дихлордиэтиловым [805] и диэтиловым эфиром [549] из растворов НВг, отгонкой в виде SbBr3 [25, 457, 458] и SbCl3 [50а, 187], ионным обменом [767, 803, 804) и направленной кристаллизацией [808]; двухступенчатым концентрированием, включающим метод направленной кристаллизации и экстракции бутилацетатом [382]. Химико-спектральные методы характеризуются в среднем на 1—2 порядка более высокой чувствительностью по сравнению с прямыми спектральными методами. Краткие характеристики химико-спектральных методов определения примесей в сурьме и ее соединениях приведены в табл. 16. Эти методы, включающие концентрирование примесей путем их выделения из анализируемого материала (например, зонная плавка [606]), используются редко.
160
Таблица 15
Прямые спектральные методы определения примесей в сурьме и ее соединениях
Определяемые примеси Условия определения Предел обнаружения, % Литература
Ag, Al, As, В, Bi, Со, Cu, Fe, Ga, Ln, Mn, Ni, Pb, Sn, Zn Смесь (4:1) пробы с УП; испарение из углубления анода, дуга постоянного тока (9 а) 3-Ю-3-6-Ю-6 [69, 672]
Ag, As, Bi, Co, Cu, Mn, Na, Ni, Pb, Sn, Zn Дуга переменного тока, проба в виде брикетов, в случае анализа Sb2O3 дуга постоянного тока (8 «) 0,3-4-10-* [810]
Al, Bi, Cu, Ge, Fe, Mg, Si, Ti Дуга постоянного тока (10 а), испарение из анода смеси пробы с УП 1-10-2—5-IO"6 [800]
Ag, Al, As, Bi, Co, Cu, Fe, Mn, Ni, Pb, Sn, Zn Смесь пробы с УП; дуга переменного тока МО-3—2- 10~в [674]
As, Bi, Co, Cu, Fe, Mn, Na, Ni, Pb, Zn Дуга переменного тока; испарение проб из кратера УЭ 3-Ю-3—1-Ю-4 [188]
Ag, Al, As, Bi, Ca, Co, Cr, Cu, Fe, In, Mg, Mn, Na, Ni, Pb, Sn, Те, Zn Искровое возбуждение спектров с использованием брикетов; эталоны и пробы смешивают (4:1) с УП 6-Ю-4— 1 -10~6 [606]
Ag, As, Bi, Cu, Fe, Pb Искровое возбуждение спектров, эталоны готовят сплавлением 0,6-3-10-з [9]
Ag, As, Bi, Cu, Pb Искровое возбуждение спектров, эталоны готовят сплавлением 1-2-Ю-з [702]
Ag, As, Bi, Cu, Fe, Ni, Pb, Sn Искра между образцом и УЭ, эталоны готовят сплавлением 0,6-1-Ю-з [1042]
Bi, Cu Искровое возбуждение спектров, те же условия для определения Bi и Си в сурьмянистом свинце 1-10-2—5-10-4 [231]
Ag, Co, Cu, Fe, Ga, In, Mn, Pb, Sn Смесь 40 мг пробы и 10 мг УП испаряют из канала УЭ (анода) 6-Ю-4—6-10-е [671]
Al, Ca, Cu, Fe, Mg, Pb, Si Дуга постоянного тока (8 а), испарение пробы из анода МО-4—МО"6 [722]
Ag, As, Au, Bi, Cd, Co, Cu, Fe, Mn, Ni, Pb Искровое возбуждение спектра ОД-МО"3 [673]
6 А. А. Немодрук
161
Таблица 15 (окончание)
Определяемые примеси Условия определения Предел обнаружения, % Литература
Bi, In, Pb, Sn, Те Искровое возбуждение спектра и брикетирование проб и эталонов при определении Bi, Pb и Sn 1,0—5-Ю-3 [810J
15 примесей Растертые пробы сурьмы смешивают с УП; дуга постоянного тока (15 а) 1 -10~5—6-10-’ [51]
Ag, Bi, Cd, Cu, Pb, Sn Метод фракционной дистилляции с медленной разверткой спектров для антимонида индия 1-Ю-4—8- 10~в [238]
As, Bi, Cu, Fe, Pb, Zn Сурьму переводят в Sb2O3, смешивают с УП; дуга постоянного тока 1,0—1-Ю-2 [1246]
Ag, Al, As, B, Bi, Ca, Cd, Cu, Fe, In, Mg, Mn, Ni, Pb, Sn, Zn Порошкообразную пробу сурьмы смешивают (10:1) с УП; дуга постоянного тока МО-3— 1-Ю-6 [1357]
Ag, Al, As, Bi, Cu, Fe, Pb, Si, Sn Порошкообразную пробу сурьмы смешивают (2; 1) с УП; дуга постоянного тока (9 а) 3-10-3-2-IO"6 [1094]
As, Mo, P, Si, Sn, Те, Ti Порошкообразную пробу антимонида алюминия смешивают с УП 3-Ю-3—МО"5 [768]
Для определения примесей в сурьме и ее соединениях также широко используются спектрофотометрические методы. Они позволяют определять практически все элементы, в том числе такие, которые в настоящее время спектральными методами определить невозможно (например, S, С1).
По чувствительности определения примесей в сурьме и ее соединениях спектрофотометрические методы сопоставимы со спектральными, а по точности несколько превосходят их (коэффициент вариации 5—20%). Некоторые характеристики спектрофотометрических методов определения примесей в сурьме и ее соединениях представлены в табл. 17.
Для определения примесей в сурьме, ее сплавах и соединениях часто используются активационные методы. Некоторые характеристики активационных методов определения примесей в сурьме и ее соединениях приведены в табл. 18.
Для определения Se и Те в сурьме и ее соединениях применен люминесцентный метод при низкой температуре [445]. В качестве люминесцентных реагентов используют 2,3-диаминонафталин и
162
Таблица 16
Химико-спектральные методы определения примесей в сурьме и ее соединениях
Анализируемый материал Определяемые примеси Условия определения Предел обнаружения, % Литература
Сурьма высокой чистоты Ag, Al, As, Bi, Ca, Cd, Co, Cr, Cu, In, Mg, Mn, Ni, Pb, Те, Zn Экстракция основы 2,2'-дихлордиэтиловым эфиром из 11 М НС1 1-10~4—2-10-’ [800]
Ag, Al, Bi, Ca, Cd, Co, Cr, Cu, In, Mg, Mn, Ni, Pb, Ti, Zn Отделение основы экстракцией бутилацетатом из 8—11 М НС1 1-10“5—МО"8 [187, 446, 447]
Ag, Al, Au, Ba, Be, Ca, Cd, Co, Cr, Cu, Fe, Ga, In, La, Mg, Mn, Ni, Pb, Pt, TI, Zn Отделение основы отгонкой в виде SbBr3 i.io-«—i-io~7 [25, 457, 458]
Ag, Bi, Cu, Fe, Mn, Ni, Pb Отделение основы отгонкой из растворов НС1 1.10-5—МО-’ [187]
Ag, Al, Bi, Cd, Cu, Fe, Mg, Ni, Pb Отгонка основы из раствора НС1 + Вг2 и упаривание дистиллята с УП Ю-s-ю-7 [50а]
Ag, Al, Bi, Cd, Co, Cr, Cu, In, Mg, Mn, Ni, Pb, Zn Двухступенчатое концентрирование примесей, направленная кристаллизация и отделение Sb экстракцией 1-10~6—2-10-8 [322]
Bi, Ca, Cd, Co, Cr, Fe, Ga, In, Mg, Mn, Ni, Pb, TI, Zn Катионообменное концентрирование примесей из раствора 0,1 М по HF и 0,05 М по H2SO4 1.10-8—2.10-7 [767,804]
11 примесей Пробы смешивают с УП и хлорируют для удаления Sb; концентрат анализируют 1-10-5—1-10~7 [92J
Трихло-РВД сурьмы Al, Ag, Bi, Ca, Co, Cr, Cu, Fe, Mg, Mn, Ni, Pb, Ti, Zn Fe экстрагируют диэтиловым эфиром; из водной фазы удаляют Sb экстракцией бутилацетатом; водную фазу выпаривают вместе с эфирным экстрактом на УП 1-10-8—1-10~6 [382]
6*
163
Таблица 16 (окончание)
Анализируемый материал Определяемые примеси Условия определения Предел обнаружения,% Литература
Пентахлорид сурьмы Ag, AL, Au, Bi, Cd, Cu, Fe, Mg, Mn, Ni, Pb, Tl Испаряют основу в присутствии УП в атмосфере N2 1,5-10~4 -• 4,8-10-8 [808]
Антимониды галлия и индия Ag, Al, Bi, Ca, Cd, Co, Cu, Mn, Ni, Pb, Zn Экстракция основы 2,2'-дихлордиэтило-вым эфиром; примеси собирают на 20 мг УП; дуга постоянного тока 2-10-4—8-10~7 [803]
Антимонид алюминия Ag, Al, Bi, Ca, Cd, Co, Cu, Fe, Mn, Ni, Pb, Zn Катионообменное концентрирование примесей; дуга постоянного тока 2-10~4—8-10~7 [803]
Антимонид галлия Ag, Al, Bi, Ca, Cd, Co, Cr, Cu, Mg, Mn, Ni, P, Pb, Zn Основу экстрагируют 2,2'-дих лор диэтиловым эфиром, водный раствор выпаривают с УП 5-10~4—Ы0~7 [102]
Антимонид индия Ag, Bi, Cd, Cu, Mn, Ni, Pb, Zn Sb п In отделяют экстракцией диэтиловым эфиром в среде 5М НВг 2-10~6-5.10~7 [549]
Ag, Al, Ba, Bi, Ca, Cd, Co, Cr, Cu, Mg, Mn, Ni, Pb, Pt, Те, Zn Sb и In отделяют экстракцией ди-(2-этилгексил)фосфор-ной кислотой в среде 7-8 М НВт 5-10-5—1 -10~7 [802]
Ag, Al, Be, Bi, Ca, Cd, Co, Cr, Cu, Mg, Mn, Ni, Pb, Zn Sb и In экстрагируют 2,2'-дихлор диэтиловым эфиром в среде 7-8 М НВг 5-IO"5—2-10-’ [805]
Антимонид алюминия Ca, Cd, Co, Cr, Cu, Bi, Fe, Ga, In, Mg, Mn, Ni, Pb, Ti, Zn Примеси выделяют на колонке с катионитом Дауэкс 50 WX8 из растворов HF и H2SO4 2-Ю-4—8-10-’ [768]
Трихлорид сурьмы Ag, Al, Bi, Ca, Co, Cr, Cu, Fe, Mg, Mn, Ni, Pb, Ti, Zn Экстрагируют железо диэтиловым эфиром; из водного раствора удаляют Sb экстракцией бутилацетатом и водную фазу вместе с эфирным экстрактом выпаривают на УП 3.10-4—i-10-s [382]
164
Таблица 17
Фотометрические методы определения примесей в сурьме и ее соединениях
Определяемый элемент Реагент Сущность метода Предел обнаружения, % Литература
Ag Дитизон Экстрагируют Ag в виде диэтилдитиокарбамината, реэк-страгируют 1 М HgSOj, снова экстрагируют в виде дитизона-та и измеряют оптическую плотность (ОП) экстракта 5-Ю-6 [862]
То же Экстрагируют Ag в виде дити-зоната в присутствии цитрата и измеряют ОП экстракта 1-10-5 [1003]
As Молибдат аммония Пробу растворяют в смеси НС1 4- Вг2, отгоняют As в виде AsClg, переводят в синюю мо-либдомышьяковую кислоту (СММК) и измеряют ОП 1 - IO"6 [1219, 1259]
То же As экстрагируют СС14 в виде AsCl3, реэкстрагируют водой, переводят в СММК и измеряют ОП 5-10-в [23, 698а, 862]
Экстрагируют AsClg хлороформом, реэкстрагируют аммиаком (1 : 20), переводят в СММК и измеряют ОП 5-Ю-6 [365, 504, 505]
Осаждают As в виде MgNH4AsO4 в присутствии тартрата (MgNH4PO4 коллектор) и определяют в виде СММК, которую экстрагируют изоамиловым спиртом 2-IO"5 [509]
Экстрагируют Sb 2,2'-дихлор-диэтиловым эфиром из 12 М НС1, в водном растворе переводят в СММК, которую экстрагируют изоамиловым спиртом 1-ю-4 [171]
Для определения As в трихлориде сурьмы пробу (0,5 г) смешивают с 10 мл смеси (1 :6) H2SO4 и НС1 и экстрагируют СС14; реэкстрагируют As водой и определяют в виде СММК 1-ю-5 [172]
» Экстрагируют As в виде AsJ3 из 6—13 М НС1 с помощью СС14, переводят в СММК и измеряют ОП 1-ю-5 [366]
165
Таблица 17 (продолжение)
Определяемый элемент Реагент Сущность метода Предел обнаружения, % Литература
Аз Молибдат аммония Пробу растворяют в смеси НС1 и HNOg и отделяют Sb экстракцией из 10—11 М НС1 2,2'-дихлордиэтиловым эфиром в присутствии Вг2;из водного раствора As экстрагируют СС14, реэкстрагируют водой и определяют в виде СММК МО"6 [1318]
То же Отгоняют As из раствора НС1 и определяют в виде СММК 5-10-» [1393]
Молибдатами реагент Экстрагируют As в виде AsCl3 бензолом, реэкстрагируют водой, переводят в СММК и измеряют ОП 2-Ю-4 [65]
Гипофосфит натрия В среде 6 М НС1 с помощью NaH2PO2 восстанавливают мышьяк до золя элементного As и сравнивают со шкалой стандартов 2-10-4 [321]
Au Дитизон Экстракционное титрование Au дитизоном; мешающее влияние Ag устраняют промывкой экстракта дитизоната раствором NaCl 1-10-4 [698а]
Bi Сульфид натрия Сурьму отделяют от висмута методом бумажной хроматографии —‘ [1552]
Дитизон Экстрагируют Bi в виде дитизоната из раствора с pH 9—10 в присутствии KCN и тартрата и измеряют ОП 5-10-6 [23, 862]
Ca Глиоксаль-бис-(2-ок-сианил) Экстрагируют Са 0,01%-ным раствором азо-азокси БН в смеси (1:4) ТБФ с СС14, реэкстрагируют 0,1 М НС1 и измеряют ОП комплекса Са с гли-оксаль-5ис-(2-оксианилом) 1-10-5 [651]
Cd Дитизон Экстрагируют Cd раствором дитизона в СС14 при pH 14, реэкстрагируют раствором с pH 2, снова экстрагируют в виде дитизоната при pH 12 и измеряют ОП экстракта 4-10-в [862]
Cl ^Нитрат серебра Определяют нефелометрически в виде AgCl; Sb удерживают в растворе в виде фторидного комплекса 5-Ю-5 [-166]
166
Таблица 17 (продолжение)
Определяемый элемент Реагент Сущность метода Предел обнаружения, % Литература
Со Дитизон Экстрагируют Со в виде а-нит-розо-^-нафтолата из слабокислого раствора, экстракт промывают раствором KCN и измеряют ОП 4-Ю-6 [862]
Ст Дифенил-карбазид Отделяют Sb отгонкой или экстракцией в виде SbCl3, окисляют Сг(Ш) до Cr(VI) и измеряют ОП продукта взаимодействия Cr(Vl) с дифенилкарба-зидом 1 - 10-5 [509]
Си Дитизон Экстрагируют Си раствором дитизона в СС14 при pH 14, реэкстрагируют раствором с pH 2, затем реэкстрагируют ее IM НС1 и определяют в виде дитизоната 8-IO'» [862]
Диэтилди-тиокарба-минат Си экстрагируют СНС13 в виде диэтилдитиокарбамината в присутствии ЭДТА и измеряют ОП экстракта 5-Ю-5 [1445]
Неокупро-ин Си экстрагируют СНС13 в виде комплекса с неокупроином и измеряют ОП экстракта 5-10~5 [[1170]
2,2'-Биции-хониновая кислота Пробу растворяют в НС1 + + HNO3, выпаривают с НВг, остаток растворяют в НС1 (1:1), добавляют NH2OH-HC1 и 2,2'-бицинхониновую кислоту и измеряют ОП 2,5-10-3 [588]
Fe 1,10-фенантролин Экстрагируют Fe эфиром из 6М НС1, реэкстрагируют раствором винной кислоты и фотометри-руют в виде 1,10-фенантроли-ната 1-Ю-5 [862]
То же Фотометрируют в тартратном растворе в виде 1,10-фенантро-лината 1-Ю-1 [1393]
а, а'-Дипи-ридил Экстрагируют крезолом комплекс Fe(H) с дипиридилом и измеряют ОП экстракта 2-10"» (при навеске 5г) [423]
Ga Бриллиантовый зеленый Экстрагируют Ga в виде ионного ассоциата GaCl4~ с катионом бриллиантового зеленого и измеряют ОП экстракта 5-Ю-2 [377]
167
Таблица 17 {продолжение)
Определяемый элемент Реагент Сущность метода Предел обнаружения, % Литература
Hg Дитизон Экстрагируют Hg из 2,0—2,5/V, H2SO4 раствором дитизона в СС14, реэкстрагируют раствором КМпО4, снова экстрагируют раствором дитизона в СС14 и измеряют ОП экстракта 6-КГ" [862]
Ni Диметил-глиоксим Экстрагируют Ni в виде диме-тилглиоксимата, реэкстрагируют раствором НС1, окисляют бромом до Ni(lll), снова экстрагируют в виде диметилгли-оксимата и измеряют ОП экстракта 8-10-’ [862]
То же Ni экстрагируют СНС13 в виде диметилглиоксима.та, реэкстрагируют IM НС1 и измеряют ОП диметилглиоксимата Ni МО-3 [434, 1004]
Pb Дитизон Экстрагируют РЬ в виде дити-зоната при pH 9—10 в присутствии тартрата и измеряют ОП экстракта 7-10-» [862]
Р Молибдат аммония Экстрагируют Sb из раствора НС1 эфиром, Р переводят в молибдофосфорную кислоту, экстрагируют ее эфиром, восстанавливают в экстракте с помощью SnC!2 до фосфорномо-либденовой сини и измеряют ОП [165]
s Ацетат свинца Отгоняют Sb из 6.17 НС1, водный раствор вы таривают досуха, серу отгоняют в виде H2S (восстановитель НВг Д- + NaH2P02); H2S поглощают раствором РЬ(СН3СОО)2 и измеряют ОП золя PbS 1-Ю-4 — 4-10—5 [168, 170, 1360]
Парарозанилин Пробу сурьмы сжигают при >1100° С, образующиеся пары SO2 поглощают раствором Na2[HgCl4] и определяют с парарозанилином 1-10-» [698а]
Se 3,3'-Диами-нобензидин Se соосаждают с As восстановлением раствором SnCI2; из концентрата экстрагируют Se в виде комплекса с 3,3'-диами-нобензидином и измеряют ОП 2-10~4 1169]
468
Таблица 17 (окончание)
Определяемый элемент Реагент Сущность метода Предел обнаружения. % Литература
Si Молибдат аммония Si переводят в молибдокрем-невую кислоту, восстанавливают SnCl2 до кремнемолибденовой сини, которую экстрагируют изоамиловым спиртом и измеряют ОП экстракта 1-ю-4 5-10~5 [508] [509]
Те Хлорид олова (11) Селен восстанавливают гидразином до элементного и отфильтровывают, затем восстанавливают теллур хлоридом олова (II), осадок теллура отфильтровывают, растворяют, снова осаждают и измеряют ОП взвеси теллура 2-Ю"4 [537]
Т1 Кристаллический фиолетовый Т1 экстрагируют СНС13 при pH 2—3 в виде комплекса с 1,5-ди-(1-бензилбензимидазо-лил-2)-3-метилформазаном, реэкстрагируют НС1, затем экстрагируют толуолом в виде тет-рахлороталлата кристаллического фиолетового 1-ю-4 [437]
То же Т1 экстрагируют СНС18 в виде тетрахлороталлата бриллиантового зеленого, экстракт испаряют, остаток обрабатывают Н2О2, снова экстрагируют Т1 толуолом в виде тетрахлороталлата кристаллического фиолетового 1 • 10-« [434]
3,3'-диаминобензидин, образующие растворимые в толуоле и циклогексане люминесцирующие соединения с Se и Те. Пределы обнаружения Se и Те в трихлориде сурьмы составляют соответственно 5-10-7 и 1-10~4%. В другом методе [135а] для определения Se в сурьме образующееся соединение Se с 3,3'-диаминобензидином экстрагируют толуолом и измеряют интенсивность флуоресценции экстракта. В этом случае предел обнаружения Se достигает 1 • 10-5%; погрешность -~20%. Разработан [509] флуориметриче-ский метод определения Al > 1 • 10_Б% в сурьме в виде комплекса с эриохром синечерным В при pH 5. Описан [1537] метод определения бора в сурьме и ее соединениях, основанный на измерении флуоресценции комплекса бора с дибензоилметаном. Предварительно бор отделяют отгонкой в виде борнометилового эфира. При определении содержания бора 10-6—10~7% погрешность состав-
169
Таблица 18
Радиоактивационные методы определения примесей в сурьме, ее сплавах и соединениях
Анализируемый материал Определяемые примеси Сущность метода Предел обнаружения Лите- ратура
Сурьма As, Со, Си, In, Sn, Tl, Zn Из облученной пробы удаляют Sb экстракцией ее2,2'-ди-хлордиэтиловым эфиром, примеси разделяют на анионитовой колонке и измеряют их’активность 8,2-10-®— 1-Ю-6 % [37]
As, Сг, Cu, Ga, Mn, P, Sn Пробы облучают 20—40 час. потоком 8,7‘iO12 нейтпрон!см2*сек, выделяют радиоактивные изотопы и измеряют их активность 5-10-*— 3-10-’ % [20, 614]
As, Co, Cu, Ni, Те После облучения выделяют радиоизотопы определяемых элементов и измеряют их активность 1,2-10-»- 3,4-10-1° г [385]
Co, Cr, Cu, Mn, Ni, Zn Пробу облучают потоком 1,2-•1013 нейтрон/см2-сек 20 час. для Mn, Ni, Си и Zn и 200 час. для Со и Сг; разделяют с помощью экстракционной хроматографии 1-ю-7— 3-10-11 г [794]
Fe Пробу облучают 80 час. потоком 7,5-1012 нейтрон/см2-сек, растворяют, осаждают Fe в виде FeS, очищают методом ионообменной хроматографии и измеряют активность 5-10-т г [614, 1522]
Антимонид индия] As, Au, Cd, Co, Cu Пробу облучают 8 час. потоком 1,8-1013 нейтрон/см2-сек, производят разделение и измеряют активность выделенных изотопов 1,6-10-*- 1,2-10-’% [146]
Железосурьмяные матрицы As Пробу облучают 2 час. потоком 5-10й нейтрон/см2 -сек, отделяют Sb и Fe экстракцией изопропиловым эфиром, выделяют As и измеряют его активность 5-10’5% [1015]
170
Таблица 19
Полярографические методы определения примесей в сурьме и ее соединениях
Определяемая нри-месь Анализируемый материал Сущность метода Предел обнаружения, % Коэффициент вариации, % Литература
Ag Sb Электролитически выделяют Ag на пастовом угольном электроде при контролируемом потенциале и на осциллографическом поля-рографе регистрируют кривую электр ораствор ения 1•10~5 15 [172, 765]
As Sb и ее сплавы As экстрагируют СНС13 в виде AsCl3, реэкстрагируют водой, реэкстракт выпаривают и определяют As в остатке на фоне 0,5М КОН4- 0,025% желатина методом постояннотоковой полярографии 5-Ю-5 15-20 [382, 870]
Au Sb Выделяют Au электролизом на угольном пастовом электроде, затем осциллографическим по-лярографом снимают кривую электрорастворения 1•10~5 12 [658, 765]
Cd Сплавы In—Sb и Ga—Sb Полярографируют на фоне 7,2М NaOH + 0,27М маннита без отделения от основных компонентов 4-Ю-3 2,5 [149]
In Сплавы Ga—Sb и Ga—Sb—Те Определяют In на фоне 1М НВг+ 1,5x1/ Н3РО4 методом переменнотоковой полярографии без отделения от основных компонентов 1-Ю-3 10—17 [127]
Pb Sb Удаляют Sb отгонкой ввиде SbBr8 и определяют РЬ на фоне НС1+ NaF Отгоняют Sb в виде SbBr3; из остатка электролитически выделяют РЬ и регистрируют кривую его электрорастворения 1-10"4 6-10~Б 12-20 18-35 [345] [1446]
s Sb и сплав In—Sb Серу восстанавливают смесью NaH2PO2+HJ до выделения паров H2S, которые поглощают щелочным раствором NH2OH и определяют методом переменнотоковой полярографии 5-Ю-® 15-30 [299]
Sb и сплав In-Sb Серу восстанавливают смесью NaH2PO2 + НJ до H2S, который поглощают фоном и определяют пульсполярографиче-ским методом 1-10-‘— 1-10-» 10—25 [296]
471
Таблица 19 (окончание)
Определяемая примесь Анализируемый материал Сущность метода Предел обнаружения, % Коэффициент вариации, % Литература
Se Sb Выделяют Se соосаждением с элементным As, осадок растворяют и определяют Se методом переменнотоковой полярографии после накопления на стационарном ртутном электроде 1•ю~® 35 [300, 301]
Sb и сплавы Ga—Sb и Ga—Sb-Те 0,5 г пробы растворяют в 1,0— 1,5 мл конц. НС1 и 0,3—0,4 мл HNO.,. Добавляют 1М Н3РО4 до 100 мл, вводят 0,8 мл 1%-ного раствора желатина и определяют Se методом переменнотоковой полярографии 5-10-’ 15-20 [128]
Sn Sb Из лимоннокислого раствора избирательно сорбируют Sn(IV) силикагелем, вымывают раствором НС1 и полярографируют на фоне IM HCI + ЗМ NH4Cl 5-Ю-» 15-30 [1562]
Те Sb Восстанавливают теллур до элементного гидроксиламином и Na2S2O3, в результате чего Те соосаждается с элементной S; осадок растворяют в смеси H2SO4 + Вг2 и определяют Те методом квадратноволновой или пульсполярографии 2-10~5 20 [297]
Sb и сплав In—Sb Определяют Те методом катодной вектор-полярографии с предварительным накоплением элементного Те на стационарном ртутном электроде 1-10~e 10—25 [296]
Zn Sb Удаляют Sb отгонкой в виде SbBr3 и определяют Zn на фоне IM KJ + Н3РО4 (pH 1—2) методом переменнотоковой полярографии 1•10~« 18-27 [570]
Sn Трихлорид и пентахлорид сурьмы Предварительно Sb отделяют экстракцией н-бутилацетатом в среде 9,5М НС1, затем соосаж-дают Sn с NH4BeP04 в присутствии ЭДТА; осадок растворяют и определяют Sn методом инверсионной вольтамперометрии на фоне 3,55/ НС1 1-10-® 15-30 [981]
«72
ляет 12%. Для определения Mg в трихлориде сурьмы предложен флуоресцентный метод [76] с использованием в качестве реагента бис-салицилальэтилендиамина и измерением интенсивности голубого свечения при 450 нм, возникающего при облучении раствора УФ-светом. Предел обнаружения Mg 1-10_Б%, погрешность 10—15%. В табл. 19 приведены характеристики полярографических методов определения примесей в Sb, ее сплавах и соединениях.
Щелочные и щелочноземельные элементы в сурьме и ее соединениях определяют методами фотометрии пламени. Так, в сурьме определяют Li, Na, К и Са в остатке после удаления основы отгонкой в виде хлорида в токе С12- Предел обнаружения Na, К и Са составляет 1-10~Б%, для Li — 3-10~6% [1374]. По другому методу [509] Са в сурьме определяют по молекулярной полосе СаО при 622 нм. Предел обнаружения 5-10~Б% Са, ошибка 3—5%.
Описано [396] определение Na, К и Са в сурьме методом фотометрии пламени без отделения основы (предел обнаружения 1-10':!%). Рекомендованы [164, 211] методы фотометрии пламени для определения Na в сурьме и продуктах ее производства также без отделения основы. Предел обнаружения 1 • 10~4 %, ошибка ~5 %.
Для определения Те в антимониде галлия применен метод атомно-абсорбционной спектрофотометрии [1614].
Пробу (40—50 мг) помещают в специальную камеру и пропускают хлор для удаления основных компонентов в виде летучих хлоридов. Остаток, содержащий Те, растворяют в 3 М растворе НС1, раствор распыляют в пламя и определяют Те по поглощению света линией 214,3 ил«. Предел обнаружения Те 20 нг/мл-, ошибка ~ 3%.
Для определения Ан и Ag в сурьме и продуктах ее производства предложены [143] экстракцирнно-атомно-абсорбционные методы. Золото избирательно экстрагируют раствором ди-н-бутилсульфида в бензоле, и экстракт распыляют в пламя. Для определения Ag сначала отделяют Sb и Fe экстракцией 2,2'-дихлордиэтиловым эфиром и анализируют водную фазу. Предел обнаружения Ан и Ag составляет соответственно 5 и 100 нг/мл.
Рентгенофлуоресцентный метод применен для определения Sn и As в сурьмяно-свинцовых сплавах [1063]. С использованием спектрометра XRD-6, трубки с вольфрамовым анодом, кристалла-анализатора LiF и амплитудного анализатора импульсов метод позволяет определять 0,15—0,75% Sn и 0,05—0,60% As. При содержании 0,260% Sn и 0,175 % As стандартное отклонение составляет соответственно 0,012 и 0,008%.
Для определения Н, О и N в сурьме предложен метод, основанный на их выделении при плавлении образца в графитовом тигле в вакууме без ванны [910]. Для определения 0 в сурьме предложен также метод [1269], основанный на восстановлении окисной фазы в токе Н2 и титровании образовавшейся при этом воды реактивом Фишера. Варьирование навески от 0,05 до 24 г позволяет определять от 5-10-4 до 9%О с ошибкой -~8%.
17.1
ЛИТЕРАТУРА
1. Абдишева А. В. Науч. тр. Ташкентского ун-та, вып. 379, 147 (1970).
2. Абдишева А. В., Талипов Ш. Т. Узб, хим. ж., № 1, 5 (1975).
3. Абдишева А. В., Талипов Ш. Т. Узб. хим. ж, № 6, 9 (1974).
4. Абдуллаев А. А., Хамидова Р., Хатамов Ш., Захидов А. Ш. Сб. «Активационный анализ горных пород и других объектов». Ташкент, «Фан», 1967, с. 185.
5. Абраров О. А., Имамов Т. X., Сабиров С. С. Сб. «Активационный анализ чистых материалов». Ташкент, «Фан», 1968, с. 66.
6. Абрикосов Н. X., Шелимова Л. Е. Полупроводниковые материалы на основе соединений AIVBVI. М., «Наука», 1975.
7. Авилов В. Б. Тр. Уральск, электромех. ин-та инж. ж.-д. трансп., вып. 2, 74 (1959).
8. Адамович Л. Л., Ружинская Р. И., Андрущенко Д. А. Укр. хим. ж., 27, 817 (1961).
9. Азриелян О. П. Изв. АН СССР. Сер. физ., 4, 20 (1940).
10. Айтова В. X., Живописцев В. П. Уч. зап. Пермского ун-та, № 324, 178 (1974).
11. А Плова Л. Е., Успенский Е. В. Полигр. произ-во, № 11, 14 (1957).
12. Александров Я. Я., Гуния Г. С., Гунченко А. И,, Туркин Ю. И. Тр. Геофиз. обсерв., 314, 104 (1974).
13. Алексеев В. Я. Курс качественного химического полумикроанализа. М.— Л., «Химия», 1976.
14. Алимарин И. П., Билимович Г. Н. Int. J. Appl. Radiat. and Isotop., 7, 169 (1960).
15. Алимарин И. И., Большова Т. А., Бахарева Г. А. Ж. аналит. химии, 28, 1300 (1973).
16. Алимарин И. Я., Петрикова М. Я. Качественный и количественный ультрахимический анализ. М., «Химия», 1974.
17. Алимарин И. Л., Терин С. И. Зав. лаб., 20, 18 (1954).
18. Алимарин И. П., Яковлев Ю. ВЖабин А . И. Сб. «Применение меченых атомов в аналитической химии». М., «Наука», 1965, с. 58.
19. Алимарин И. П., Яковлев Ю. В., Щулепников М. Я., Власов Д. А., Чернов Г. М., Сурков Ю. А . Ж. аналит. химии, 16, 213 (1961).
20. Алимарин И. П., Яковлев Ю. В., Щулепников М. Я., ЛережогинГ. Я. Сб. «Радиоактивные изотопы и ядерные излучения в народном хозяйстве СССР», т. 1. М., Гостоптехиздат, 1961, с. 293.
21. Аллабергенов Б. Р., Акбаров У., Лобанов Е. М., Кишанов И. Изв. АН УзССР. Сер. физ.-мат. н., № 3, 56 (1970).
22. Амшеева А. А. Изв. вузов. Химия и хим. технол., 13, 339 (1970).
23. Ангерман В., Бастиус X. Сб. «Получение и анализ веществ особой чистоты». М., «Металлургия», 1968, с. 209.
24. Анджапаридзе Д. И., Акимов В. К., Бусев А. И. Сообщ. АН ГрузССР, 66, 317 (1972).
25. Андреев А. С. Тр. Ленинградского политехи, ин-та, 201, 35 (1959).
26. Андреева И. Ю., Клер М. М Ж. аналит. химии, 20, 448 (1965).
27. Аникеева Я. Л. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, с. 179.
174
28. Аппаратура и методы рентгеновского анализа, вып. 3. Изд-во ЛГУ, 1968.
29. Аппаратура и методы рентгеновского анализа, вып. 5. Изд-во ЛГУ, 1969.
30. А пситА. А., Янсон Э. Ю. Уч. зап. Латвийского ун-та, 117, 39 (1970).
31. Арван X. Л. Изв. АН СССР. Сер. физ., 20, 443 (1956).
32. Арван X. Л., Зайцева Н. Е. Оптика и спектроскопия, 10, 272 (1961).
33. Арефьева Т. В., Позднякова А. А. Сб. научи, тр. Гос. НИИ цвет, мет., № 14, 67 (1958).
34. Арешидзе Т. Тр. Тбилисского ун-та, 80, 213 (1961).
35. Арстамян Ж. М. Молодой научн. работник естеств. наук, 2 (16), 65 (1972).
36. Артыкбаев Т., Дюсебеков Б., Ганиев Ш. У., Черниловская А. И. Докл. АН УзССР, № 1, 42 (1972).
37. Артюхин П. И., Гилъберт Э. Н., Пронин В. А. Ж. аналит. химии, 21, 504 (1966).
38. Артюхин А. И., Гильберт Э. Н., Пронин В. А. Тр. Комиссии по аналит. химии АН СССР, 16, 169 (1968).
39. Артюхин П. И., Гильберт Э. Н., Силъванович Ю. А., Зав. лаб., 33, 676 (1967).
40. Астафьева И. Н., Плотникова Р. Н., Щербов Д. П. Сб. «Исследования в области химических и физических методов анализа минерального сырья», вып. 3. Алма-Ата, «Наука», 1973, с. 52.
41. А фанасъев Ю. А., Рябинин А . И., Ажипа Л. Т., Романов А . С. Ж. аналит. химии, 30, 1830 (1975).
42. Ая-месМи М. Я., Грановская П. Б. Уч. зап. Азербайджанского ун-та. Сер. физ.-мат. и хим. н., № 2, 49 (1962).
43. Бабина Ф. Л., Барабаш А. Г., Пейзулаев Ш. И., Семенова Е. Ф. Ж. аналит. химии, 20, 501 (1965).
44. Бабко А. К., Данилова В. Н., Каплан М. Л. Укр. хим. ж., 32, 1009 (1966).
45. Бабко А. К., Ивашкович Е. М. Ж. аналит. химии, 27, 120 (1972).
46. Бабко А. К., ЛГтокало М./7. Зав. лаб., 21, 767 (1955).
47. Багдасаров К. Н., Коваленко П. Н. Уч. зап. Ростовского ун-та, 40 ИЗ (1958).
48. Бакин В. М., Кофтюк В. А., Ламбрев В. Г. Зав. лаб,, 38, 50 (1972).
49. Баленко Э. П., Лифшиц Е. В. Зав. лаб., 31, 690 (1965).
50. Банковский Ю. А., Веверис О. Э., Пелекис Л. В. Сб. «Нейтронно-активационный анализ». Рига, «Зинатне», 1966, с. 89.
50а. Баранова Л. Л., Бирюкова Е. Я., Горюшина В. Г., Солодовник С. М Сб. «Научные труды Гиредмета», вып. 22. М., «Металлургия», 1968, с. 158.
51. Баранова Л. Л., Назарова М. Г., Солодовник С. М. Зав. лаб., 36, 1074 (1970).
52. Барбанелъ Д. Г., ЧжанХуа-ли. Сб. «Методы количественного определения элементов». Изд-во ЛГУ, 1964, с. 96.
53. Баринов В. М., Айдаров Т. К. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 323.
54. Басова Е. П., Жарова Н. И., Шаевич А. Б., Шубина С. Б. Зав. лаб., 28, 1075 (1962).
55. Басова Е. П., Шаевич А. Б., Шубина С. Б. Сб. «Материалы III Уральского совещания по спектроскопии, 1960». Свердловск, Металлург-издат, 1962, с. 77.
56. Бахарева Г. А., Алимарин И. П., Большова Т. А., Нахаба Т. И. Ж. аналит. химии, 29, 58 (1974).
57. Бедреинов В. П., Украинский Ю. М., Фурсов В. 3., Хромова Н. В. Электронная техника. Научн.-техн. сб. «Управление качеством и стандартизация», вып. 5, 34 (1970).
58. Белоглазова А. Д., Крупнов В. К. Зав. лаб., 35, 451 (1969).
175
59. Белоглазова А. Д., Крупнов В. К., Сафаева Ф. 3. Зав. лаб., j 34, 1057 (1968).
60. Белокриницкая Е. Е., Бондаренко В. В., Витушкина И. Н., Герасимова М. С., Гинзбург В. М., Граменицкий И. Н., Лифшиц Д. М., Крыжная Ю. Ф. Сб. «Материалы I Уральского совещания по спектроскопии, 1956». Свердловск, Металлургиздат, 1958, с. 59.
61. Белявская В. К., Ковалев И.М. Вести. Харьковского политехи, ин-та, № 26 (74), 106 (1968).
62. Беляев В. П., Калиниченко В. Р. Кузьмин Н.М., Якименко Л. М. Зав, лаб., 28, 685 (1962).
63. Бесполенкова Е. К., Гринзайд Е. Л., Надеждина Л. С. Тр. Комиссии по аналит. химии АН СССР, 16, 240 (1968).
64. Бирюк Е. А. Зав. лаб., 30, 651 (1964).
65. Бирюк Е. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 176.
66. Бирюков В. М., Виноградов Б. Г., Пережогин Г. А., Холопова М. Д. Электронная техника. Научи.-техн. сб. «Управление качеством и стандартизация», вып. 5, 96 (1970).
67. Влащук В. П., Подольский В. А., Семенко А. С. Сб. тр. Донецкого НИИ черн. металлургии, вып. 26, 288 (1961).
68. Блинков Д. И., Лобанов Е. М. Тр. по химии и хим. технол. (Горький), вып. 3 (24), 108 (1969).
69. Блох И. М., Меламед М. Г., Ноткина М. А., Поляков С. П., Солодовник С. М. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, с. 136.
70. Блюм И. А. Сб. «Методы химического анализа минерального сырья», вып. 8. М., «Недра», 1965, с. 191.
71. Блюм И. А. Экстракционно-фотометрические методы анализа с применением основных красителей. М., «Наука», 1970.
72. Блюм И. А., Васильева А. ПСкринская Р. Н. Тр. Казахского НИИ минеральн. сырья, вып. 5, 260 (1961).
73. Блюм И. А ., Соловьян И. Т., Шебалкова Г. Н. Зав. лаб., 27, 950 (1961).
74. Бобров В. А., Салмин Ю. П. Труды VII Конференции молодых исследователей ВНИИ минерального сырья. Секция аналитики и технологии. М., изд. ВИМС, 1969, с. 13.
75. Вожеволънов Е. А. Тр. ВНИИ хим. реактивов и особо чист. хим. веществ, вып. 22, 60 (1958).
76. Вожеволънов Е. А., Серебрякова Г. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 244.
77. Вожеволънов Е. А., Соловьев Е. А. Сб. «Методы анализа химических реактивов и препаратов», вып. 16. М., «Химия», 1969, с. 170.
78. Вожеволънов Е. А., Соловьев Е. А. Сб. «Методы анализа химических реактивов и препаратов», вып. 11. М., изд. ИРЕА, 1956, с. 87.
79. Вожеволънов Е. А., Соловьев Е. А. Труды ИРЕА, вып. 30, 202 (1967).
80. Бондаренко Л. С, Пейзулаев Ш. ИКарабаш А. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 315.
81. Бондаренко Л. С., Сотникова Н. П., Пейзулаев Ш. И., Каратыгина Г. Ф., Варенцова Р. М. Там же, с. 483.
82. Бондаренко С. К., Мамот Ж. А., Прокопьева С. В. Зав. лаб., 36, 945 (1970).
83. Боровский И. Б. Физические основы рентгеноспектральных исследований. Изд-во МГУ, 1956.
84. Борчева Т. А., Николаева Т. Ф., Харламов И. П. Сб. «Определение микропримесей». М., изд. МДНТП, 1968, с. 58.
85. Брайнина X. 3. Ж. аналит. химии, 21, 529 (1966).
86. Брайнина X. 3. Инверсионная вольтамперометрия твердых фаз. М., «Химия», 1972.
87. Брайнина X. 3., Нейман Е. Я., Трухачева Л. Н. Зав. лаб., 37, 16 (1971).
88. Брайнина X. 3., Ройзенблат Е. М., Белявская В. Б., Кива И. К., Фомичева Т. И. Зав. лаб., 33, 326 (1967),
176
89. Брайнина X. 3., Сапожникова 9. Я. Ж. аналит. химии, 21, 807 (1966).
90. Брайнина X. 3., Чернышева А . В. Зав. лаб., 42, 640 (1976).
91. Брицке М. Э. Зав. лаб., 35, 1329 (1969).
92. Бродская Б. Д.у Ноткина М. Л., Меньшова Н. П. Ж. аналит. химии,. 27, 151 (1972).
93. Бронзы безоловянные. Методы химического анализа. ГОСТ 15027. 9-69.
94. Бронзы оловянные. Методы определения содержания сурьмы. ГОСТ 1953. 10—74.
95. Булыгин Б. М. Зав. лаб., 32, 675 (1966).
96. Бурмистров М. 77., Морейская Л. В. Зав. лаб., 37, 428 (1971).
97. Бурынина А. Г., Фабрикова Е. А . Сб. «Методы анализа редкометальных минералов, руд и горных пород», вып. 2. М., «Недра», 1971, с. 65.
98. Бусев А. И., Богданова Е. С. Ж. аналит. химии, 19, 1346 (1964).
99. Бусев А. И., Богданова Е. С., Типцова В. Г. Ж. аналит. химии, 20, 585 (1965).
100. Бусев А . И., Типцова В. Г., Богданова Е. С., Андрейчук А. М. Ж. аналит. химии, 20, 812 (1965).
101. Буянов Я. В., Разумова Г. Я., Сорокина Я. Я., Яковлев Я. Я. Сб. тр. ЦНИИ черн. металлургии, вып. 19, 65 (1960).
102. Буянова Л. М., Юделевич И. Г., Архипова В. А . Ж. аналит. химии, 24,.-435 (1969).
103. Быковцева Т. ТЦерковницкая Я. А. Зав. лаб., 30, 943 (1964).
104. Вайнштейн Э. Е., Беляев Ю. Я., Ахманова М. В. Тр. Комиссии ПО' аналит. химии АН СССР, 12, 236 (1960).
105. Вайнштейн Ю. Я., Гинзбург К. Я. Сб. «Методы анализа химических реактивов и препаратов», вып. 18. М., изд. ИРЕА, 1971, с. 145.
106. Валиева М. Г., Лобанов Е. М., Усманова М. М. Сб. «Активационный анализ», Ташкент, «Фан», 1971, с. 106.
107. Ван Куй. Хуасюэ сюэбао, 22, 66 (1956).
108. Ван Чжэнъ-шу, Ху Синъ-хуэй, ЮйБо. Цзиньшу сюэбао, 9, 98 (1966Б
109. Василевская Л. С., Кондрашина А. Я., Макарова Г. А ., ЯанаринаН.А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 71.
110. Василевская Л. С., Кондрашина А . Я., Макарова Г. А ., Панарина Я. А-Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, с. 128.
111. Василевская Л. С., Кондрашина А . Я., Муравенко В. Л. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 511.
112. Василевская Л. С., Яоткина М. А ., Садофъева С. А ., Кондрашина А. Я. Зав. лаб., 28, 678 (1962).
113. Василевская Л. С., Садофъева С. А., Омелъяновская О. Д.у Кондрат шина А. Я. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 119.
114. Васильев А . А., Шуб М. Е. Ж. прикл. химии, 6, 560 (1933).
115. Васильев Я. Я.у Разимова Г. Я., Шуба Я. Д. Радиохимия, 10, 596 (1968).
116. Васильева Л. Я., Лац Р. Г., Позднякова А. А. Ж. аналит. химии, 22 1827 (1967).
117. Васильева Л. Я., Юстус 3. Л. Научн. тр. Гос. НИИ цвет. мет. № 27, 40 (1967).
118. Васильева Л. Я., ЮстусЗ.Л.Научн.тр. Гос.НИИ цвет, мет., № 28,5 (1969).
119. Васильева Л. Я., Юстус 3. Л. Авт. свид. СССР 322711; Бюл. изобр., № 36 (1971).
120. Васильева Л. Н.у Юстус 3. Л. Электрохимия, 3, 953 (1967).
121. Василевская Л. С., Кондрашина А. Я., Муравенко В. Л. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 514.
122. В ассерштейн Ш. Е., Чикризова Е. Г .Ж. аналит. химии, 24, 496 (1969), 123. Вассерштейн Ш. Е., Чикризова Е. Г. Зав. лаб., 35, 922 (1969).
177
124. Вассерштейн Ш. Е., Чикризова Е. Г., Аропина И. В. Зав. лаб., 37, 18 (1974).
125. Вахобов А . В., Вахобова Р. У., Лукьянов Г. Л., Докабарова Н. К. Изв. АН ТаджССР. Сер. физ.-мат. и геол.-хим. и., № 3, 37 (1972).
126. Вершинина Ф. ИВихарев Л. И. Сб. научи, тр. Всес. н.-и. горноме-таллург. ин-та цвет, мет., № 9, 186 (1965).
127. Веселаго Л. И. Ж. аналит. химии, 25, 72 (1970).
128. Веселаго Л. И. Сб. «Методы химического анализа», М., изд. МДНТП, 1969, с. 176.
129. Ветошкин В. Ф. Сб. «Спектральный анализ в Цветной металлургии». М., Металлургиздат, 1960, с. 391.
130. Виноградов А. П. Геохимия, № 1, 6 (1956).
131. Виноградова Е. Н., Васильева Л. Н. Ж. аналит. химии, 17, 579 (1962).
132. Виноградова Е. Н., Васильева Л. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 296.
133. Виноградова Е. Н., Каменев А. И. Тр. Комиссии по аналит. химии АН СССР, 15, 175 (1965).
134. Виноградова Е. Н., Каменев А. И. Ж. аналит. химии, 20, 183 (1962).
135. Винокурова Л. А., Долгов 10. М., Другаченок М. А., Салама тов И. И., Храмченков А . И. Зав. лаб., 38, 51 (1972).
135а. Владимирова В. М., Кучмистая Г. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 139.
136. Власов В. С., Кузьмин Н. М. Зав. лаб., 39, 1048 (1973).
137. В о Плотников А. ПСангина О. А . Зав. лаб., 30, 18 (1964).
138. Волкова В. С., Раннее Г. Г. Сб. «Автоматический контроль и управление при обогащении и гидрометаллургии цветных металлов». Ташкент, «Фан», 1971, с. 51.
139. Володарская Р. С. Зав. лаб., 25, 143 (1959).
140. Волынец М. П. Тонкослойная хроматография в неорганическом анализе. М., «Наука», 1974.
141. Волынец М. П., Чулкина Л. С., Фомина Т. В. Зав. лаб., 35, 554 (1969).
142. Вторыгина И. Н. Сб. «Комплексное использование металлургического сырья Урала», ч. 1. Свердловск, изд. УПИ, 1967, с. 148.
143. Галанова А. П., Кудрявцева А. К., Пронин В. А., Юделевич И. Г., Валл Г. А., Гильберт Э. Н. Изв. СО АН СССР, № 7. Сер. хим. н., вып. 3, 89 (1973).
144. Ганиев А. Г., Назметдинов М. К., Рыбное В. В., Мухтаров Р. М. Сб. «Активационный анализ чистых материалов, Ташкент, «Фан», 1968, с. 61.
145. Ганиев А. Г., Назметдинов М. К., Сабиров С. Изв. АН УзССР. Сер. физ.-мат. н., № 1, 40 (1968).
146. Ганиев А . Г., Худайбергенов У., Мухтаров Р. М. Сб. «Активационный анализ чистых материалов». Ташкент, «Фан», 1968, с. 55.
147. Гейровский Я., Кута Й. Основы полярографии. М., «Мир», 1967.
148. Гертнер М. Д., Янсон Э. Ю., Поммерс В. Р. Уч. зап. Латвийского унта, 88, 59 (1967).
149. Герцева Н. С. Тр. Ин-та металлургии АН СССР, вып. 14, 251 (1963).
150. Гиллебранд В. Ф., Ленделъ Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., Госхимиздат, 1957.
151. Гильберт Э. Н., Пронин В. А. Изв. СО АН СССР, № 4. Сер. хим. и., вып. 2, 168 (1969).
152. Гильберт Э. Н., Пронин В. А ., Силъванович Ю. А ., Ганиев А . Г. Сб. «Активационный анализ чистых материалов». Ташкент, «Фан», 1968, с. 38.
153. Гинзбург В. Л. Физ. сб. Львовского ун-та, вып. 4 (9), 423 (1958).
154. Гинзбург В. Л., Глуховецкая Н. П. Зав. лаб., 26, 559 (1960).
155. Гинзбург В. Л., Глуховецкая Н. П., Лернер Л. А. Зав. лаб., 28, 682 (1962).
156. Гинзбург В. Л., Лернер Л. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 459.
178
157. Гинзбург В. МГлуховецкая И. ПДанилова И. Н. Ж. аналит. химии,. 17, 1096 (1962).
158. Гинзбург Л. Б., Шкробот И. Ф. Сб. «Научные труды Гинцветмета». М., Металлургиздат, 1961, с. 18.
159. Гинн В. Успехи в области нейтронно-активационного анализа. М., Атомиздат, 1967.
160. Гладышев В. И., Джумашев А. Ж. аналит. химии, 25, 189 (1970).
161. Гладышев В. П., Каплин А . А ., Наурызбаев М. К., Вейц Н. 77., Стромберг А. Г. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ 1973 с. 185
162. Глухарева Н. А., Шейнин Р. А. Зав. лаб., 34, 57 (1968).
163. Гордон Б. Е., Меламед д. А. Криминалистика и судебная экспертиза. Респ. межвед. сб. научи, и научи.-метод, работ., вып. 3, 316 (1966).
164. Городенцева Т.Б. Тр. ВНИИ стандарт, образцов и спектр, эталонов, 4, 171 (1968).
165. Горюшина В. Г., Арчакова Т. А . Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 238.
166. Горюшина В. Г., Арчакова Т. А. Там же, с. 240.
167. Горюшина В. Г., Бирюкова Е. Я. Ж. аналит. химии, 24, 580 (1969).
168. Горюшина В. Г., Бирюкова Е. Я. Зав. лаб., 31, 1303 (1965).
169. Горюшина В. Г., Бирюкова Е. Я. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 150.
170. Горюшина В. Г., Бирюкова Е. Я., Романова Е. В. Там же, с. 239.
171. Горюшина В. Г., Романова Е.В. Научи, тр. Гиредмета, 47, 56 (1973).
172. Горюшина В. Г., Романова Е. В., Разумова Л. С. Ж. аналит. химии,. 28, 601 (1973).
173. Грекова И. М., Назаренко В. А. Зав. лаб., 35, 537 (1969).
174. Гридаев В. Ф., Каплин А. А., Портнягина Э. О. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ, 1973, с. 239.
175. Гудзовский Г. А., Щербаков Г. Г. Гигиена и санитария, № 12, 50 (1963).
176. Гулютин А. В. Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, с. 298.
177. Гулютин А. В., Епифанов Ю.В. Авт свид. СССР 144637; Бюл. изобр., № 3 (1962).
178. Гуркина Т. В., Коновалова К. М. Тр. Казахского НИИ минеральн. сырья, вып. 5, 249 (1961).
179. Гурьев С. Д., Сараева Н. Ф. Сб. научи, тр. Гос. НИИ цвет, мет., № 18, 37 (1961).
180. Гусев С. И. Тр. Пермского мед. ин-та, вып. 31, 155 (1960).
181. Гусев С. И., Кетова Л. А. Ж. аналит. химии, 17, 137 (1962).
182. Гусев С. И., Курепа Г. А., Поплевина Л. В., Шаламова Г. Г., Шурова Л. М., Песис А. С. Ж- аналит. химии, 24, 1319 (1969).
183. Гусев С. И., Поплевина Л. В. Ж. аналит. химии, 23, 541 (1968).
184. Гусев С. И., Поплевина Л. В., Шаламова Г. Г. Уч. зап. Пермского’ун-та, № 178, 214 (1968).
185. Гусев С. И., Поплевина Л. В., Песис А. С. Ж. аналит. химии, 22, 731 (1957).
186. Гусейнов И. К., Абдулаева А . Б. Сб. «Исследования в области неорганической и физической химии». Баку, «Элм», 1971, с. 222.
187. Гуткина Р. И. Сб. «Материалы II Уральского совещания по спектроскопии. 1958». Свердловск, Металлургиздат, 1959, с. 134.
188. Гуткина Р. И., Гайдукова В. Г. Сб. «Материалы I Уральского совещания по спектроскопии, 1956». Свердловск, Металлургиздат, 1958, с. 81.
189. Давлетшин Э. Ю., Айдаров Т. К. Ж. прикл. спектроскоп., 1, 73 (1964).
190. Данчева Р., Белева-Наумова С., Георгиева Е. Бюл. научи.-техн, информ. Нипроруда, № 2, 47 (1970).
191. Дакар Г. М., Иофа Б. 3., Радиохимия, 4, 744 (1962).
192. Дакар Г. М., Иофа Б. 3. Радиохимия, 7, 25 (1965).
193. Дакар Г. М., Иофа Б. 3., Несмеянов А. И. Радиохимия, 5, 428 (1963).
179
194. Даукшас КНарушкевичус Л. Уч. зап. Вильнюсского ун-та. Сер. мат., физ. и хим. н., 7, 161 (1957).
195. Дегтярева О. Ф., Островская М. Ф. Ж. аналит. химии, 18, 245 (1963).
196. Дегтярева О. Ф., Островская М. Ф. Зав. лаб., 26, 564 (1960).
197. Дегтярева О. Ф., Островская М. Ф. Зав. лаб., 30, 174 (1964).
198. Дегтярева О. Ф., Федяева Н. В., Островская М. Ф. Зав. лаб., 27, 842 (1961).
199. Дегтярева О. Ф., Федяева Н. В., Островская М. Ф., Астахина Л. Г’ Зав. лаб., 27, 844 (1961).
200. Деменев Н. В., Драчевская Р. К., Усубакунов М. Сб. «Химия комплексных соединений редких и сопутствующих элементов». Фрунзе, «Илим», 1971, с. 66.
201. Демченко С. Д. Научн. тр. Всес. н.-и. и проект, ин-та вторичн. цвет, мет., № 1, 81 (1969).
202. Денисова Н. Е., Цветкова Е. В. Зав. лаб., 27, 656 (1961).
203. Деркачева Л. Д. Изв. АН СССР. Сер. физ., 20 410 (1956).
204. Джумашев А., Дистагиев Г. К. Тр. Киргизского ун-та. Сер. хим. н. Фрунзе, изд. Кирг. ун-та, 1968, с. 54.
205. Дмитриев К. Г. Зав. лаб., 15, 63 (1949).
206. Долаберидзе Л. Д., Каткамидзе Д. К. Бюл. Кавказского ин-та минералки. сырья, (2), 99 (1959).
207. Долаберидзе Л. Д., Каткамидзе Д. К., Бугианишвили В. К. Сб. научн.-техн, информ. Мин-ва геол, и охраны недр, № 1, 128 (1955).
208. Драчевская Р. К., Ильясова И. Ф., Долгашева И. В., Деменев И. В. Сб. «Химия и технология сурьмы». Фрунзе, «Илим», 1965, с. 9.
209. Ельяшевич М. А. Атомная и молекулярная спектроскопия. М., Физ-матгиз, 1962.
210. Емельянов С. К. Уч. зап. ЦНИИ оловянной пром-сти, № 1, 52 (1964). 211. Емельянов С. К. Уч. зап. ЦНИИ оловянной пром-сти № 1, 74 (1965). 212. Ерохина К. И., Лемберг И. X., Макашева И. Е., Маслов И. А., Обухов А. П. Зав. лаб., 25, 821 (1960).
213. Ж. аналит. химии, 26, 198 (1971).
214. Жданов А. К., Хадеев В. А., Кубракова А. И., Бондаренко Н. В. Узб. хим. ж., № 2, 44 (1961).
215. Желъвис А. И. Ж. аналит. химии, 12, 421 (1957).
216. Живописцев В. П. Тр. Комиссии по аналит. химии АН СССР, 17, 309 (1969).
217. Живописцев В. П., Линчина А. П. Сб. научн. тр. Пермского политехи, ин-та, № 120, 105 (1972).
218. Живописцев В. П., Петров Б. И., Селезнева Е. А., Сибиряков И. Ф. Тр. Комиссии по аналит. химии АН СССР, 16, 80 (1968).
219. Журавлев В. Е., Трофимова Н. И., Кашина Л. В. Тр. Естеств.-научн. ин-та при Пермском ун-те, 13, 179 (1972).
220. Заичко Л. Ф., Захаров М. С. Ж. аналит. химии, 21, 65 (1966).
221. Заичко Л. Ф., Захаров М. С., Мордвинова В. Д. Изв. Томского политехи, ин-та, вып. 120, 50 (1964).
222. Заичко Л. Ф., Захаров М. С., Шипкова Л. Г. Изв. Томск, политехи, ин-та, вып. 167, 105 (1967).
223. Заичко Л. Ф., Михайлова 3. С., Каплан А. А. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ, 1973, с. 189.
224. Заичко Л. Ф., Янкаускас В. Ф., Захаров М. С. Зав. лаб., 31, 265 (1965).
225. Заичко Л. Ф., Янкаускас В. Ф., Захаров М. С. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, с. 192.
226. Зайдель А. И. Основы спектрального анализа. М., «Наука», 1965.
227. Зайдель А . И., К алитеевский И. ИЛипис Л. В., Чайка М Л. Эмиссионный анализ атомных материалов. М.— Л., Физматгиз, 1960.
228. Зайдель А. И., Прокофьев В. К., Райский С. М., Славный В. А., Трейдер Е. Я. Таблицы спектральных линий. М., «Наука», 1969.
229. Зайковский Ф. В. Ж. аналит. химии, 9, 155 (1954).
180
230. Зайцев Е. И., Скакодуб В. А., Сотсков Ю. П. Ж. аналит. химии, 27, 2055 (1972).
231. Зан X. X. Изв. АН СССР. Сер. физ., 11, 299 (1947).
232. Зарецкий Л. С., Утенко В. С., Пидорич М. И. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ, 1973, с. 193.
233. Зарубина Р. Ф., Колпакова Н. А., Каплин А. А. Зав. лаб., 37, 11 (1971).
234. Захария И. Ф., Измайлова Д. Н., Стайков А. И. Тр. по химии и хим. технол. (Горький), вып. 3 (24), 77 (1969).
235. Захария Н. Ф., Назарова Т. Ф., Щегольков С. В. Методы спектрального анализа угольного или графитового порошка и аналитических концентратов на его основе. Одесса, изд. ИОНХ АН УССР, 1970; РЖХим, № 1769-70 Деи.
236. Захаров В. А., Сангина О. А., Драгавцева И. А. Зав. лаб. 26, 537 (1960).
237. Захаров Е. И., Липис Л. В., Петров К. И. Ж. аналит. химии, 14, 135 (1959).
238. Захаров М. С., Айдаров Т. К. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 201.
239. Захаров М. С., Заичко Л. Ф. Ж. аналит. химии, 19, 1256 (1964).
240. Захаров М. С., Заичко Л. Ф. Изв. Томск, политехи, ин-та, вып. 164, 183 (1967).
241. Захаров М. С., Заичко Л. Ф. Там же, с. 221.
242. Захаров М. С., Заичко Л. Ф., Шипкова Л. Г. Изв. Томск, политехи, ин-та, вып. 120, 50 (1964).
243. Захарова Э. А., Килина 3. Г. Изв. Томск, политехи, ин-та, вып. 164, 254 (1967).
244. Захарова Э. А., Килина 3. Г. Совещание по аналитической химии полупроводниковых веществ, 1965 г. Тезисы докладов. Кишинев, «Штиин-ца», 1965, с. 31.
245. Захарова Э. А., Килина 3. Г. Изв. Томск, политехи, ин-та, вып. 204, 154 (1971).
246. Захарова Э. А., Килина 3. Г., Катаев Г. А. Сб. «Арсенид галлия». Томск, изд-во ТГУ, 1968, с. 471.
247. Захарчук Н. Ф., Зебрева А. И. Сб. «Химия и химическая технология», вып. 1. Алма-Ата, «Наука», 1970, с. 41.
248. Захарчук Н. Ф., Зебрева А. И., Козловский М. Т. Ж. аналит. химии, 26, 246 (1971).
249. Зверева М. И., Дмитриева Е. А. Уч. зап. ЛГУ, № 297, 41 (1960).
250. Звягинцев О. Е., Шамаев В. И. Ж. аналит. химии, 14, 603 (1959).
251. Звягинцев О. Е., Шамаев В. И. Радиохимия, 1, 717 (1959).
252. Зилъберштейн X. И., Семов М. П. Ж. прикл. спектроскоп.,2, 16 (1965).
253. Зимаков И. Е., Рожавский Г. С. Тр. Комиссии по аналит. химии АН СССР, 9 (12), 231 (1958).
254. Зинченко В. А., Степанова С. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 294.
255. 31нчук В. К,, Комлев О. И. Шсник Льв1вск. ун-ту. Сер. ф!з., xim., мех.-мат., Льв1в, вид-во Льв1вск. ун-ту, 1968, с. 118.
256. 31нчук В. К., Комлев О. И., Лисак Я. Г. Шсник Льв1вск. ун-ту. Сер. Х1м., вып. 11, 43 (1969).
257. Зозуля А. П. Кулонометрический анализ. М., «Химия», 1968.
258. Золотов Ю. А., Голованов В. И. Ж. аналит. химии, 25, 610 (1970).
259. Золотов Ю. А., Голованов В. И. Взаимное влияние элементов при их экстракции из хлоридных растворов. М., «Наука», 1971.
260. Золотов Ю. А., Иофа В. 3., Чучалин Л. К. Экстракция галогенидных комплексов металлов. М., «Наука», 1973.
261. Золотов 10. А., Кузьмин Н. М. Экстракционное концентрирование. М., «Химия», 1971.
262. Золотухш В. К., Гаврилюк A. I. Bi сник Льв1вск. ун-ту. Сер. xiM., вып. 6, 82 (1963).
263. Иванкова А. И., Щербов Д. И. Сб. «Исследование и разработка фотометрических методов для определения микроколичеств элементов в ми
181
неральном сырье». Алма-Ата, изд. Казахского НИИ минер, сырья, 1967, с. 188.
264. Иванов Н. П. Сб. «Методы анализа химических реактивов и препаратов», вып. 10. М., изд. ИРЕА, 1965, с. 98.
265. Иванов И. П., Козырева И. А. Там же, с. 105.
266. Иванов И. П., Козырева, И. А. Тр. ВНИИ хим. реактивов и особо чистых хим. веществ, вып. 18, 301 (1966).
267. Иванов И. П., Михельсон Д. М. Тр. н.-и. и проекта, ин-та азота, пром-сти и продуктов орган, синтеза, вып. 5, ч. 2, 66 (1970).
268. Иванов И. П., Михельсон Д. М., Баранов С. В., Пафралиди Л. Г. Сб. «Методы анализа химических реактивов и препаратов», вып. 18. М., изд. ИРЕА, 1971, с. 103.
269. Игнатенко Е. Л., Игнатенко Е. Г. Сб. «Физико-химические методы контроля производства». Ростов, изд-во РГУ, 1975, с. 12.
270. Иголинский В. А., Стромберг А. Г. Методы анализа химических реактивов и препаратов, вып. 5—6. М., Госхимиздат, 1963, с. 29.
271. Идрисова Р. А., Лебедева О. В. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ, 1973, с. 253,
272. Измайлова Д. И., Захария И. Ф. Укр. хим. ж., 34, 109 (1968).
273. Иофа Б. 3.. Дакар Г. М. Радиохимия, 6, 411 (1964).
274. Иофф° А. Я., Зоммер Л. Р., Михайлова М. Е., Пылина В. Р. Зав. лаб., 39, 1087 (1973).
275. Исаева Е. А., Макашева И. Е., Маслов И. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 100.
276. Исаева Е. А., Макашева И. Е., Обухов А. И. Ж. аналит. химии, 18, 979 (1963).
277. Ишутченко Е. И., Елисеева В. М. Зав. лаб., 21, 791 (1955).
278. Иованович М., Драгоевич М. Гласник хем. друшт. Београд, 27, 95 (1962).
279. Иованович М., Костич С. Гласник хем. друшт. Београд, 22, 353 (1957).
280. Казакова. А. И., Гладышева К. Ф., Зиновьева Л. Д. Зав. лаб., 33, 427 (1967).
281. Казлаукас Р. М., Нарушкявичус Л. Р., Шкадаускас Ю. С. Научи, тр. вузов ЛитССР. Химия и хим. технол., № 12, 15 (1970).
282. Калинин А. И. Кузнецов Р. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 93.
283. Калинин А. И. Кузнецов Р. А., Моисеев В. В., Мурин А. И. Докл. АН СССР, 141, 98 (1961).
284. Калинин А. И., Кузнецов Р. А., Моисеев В. В. Радиохимия, 4, 575 (1962).
285. Калинин А. И., Кузнецов Р. А., Моисеев В. В. Сб. «Радиохимические методы определения микроэлементов». М.— Л., «Наука», 1965, с. 176.
286. Каменев А. И. Вести. МГУ. Химия, № 2, 79 (1964).
287. Калинин С. К., Файн Э. Е. Эмиссионный спектральный анализ минерального сырья. М., «Недра», 1969.
288. Калинников В. Т., Штейнберг А. И. Зав. лаб., 30, 178 (1964).
289. Каменев А. И., Виноградова Е. Н. Вести. МГУ. Химия, № 3, 76 (1965).
290. Каменев А. И., Виноградова Е. Н. Зав. лаб., 33, 929 (1967).
291. Каменев А. И., Виноградова Е. Н., Козинцева В. И. Вести. МГУ. Химия, № И, 573 (1970).
292. Каменев А. И., Виноградова Е, И., Тиришин А. Вести. МГУ. Химия, № 13, 57 (1972).
293. Каменев А. И., Виноградова Е. И., Финогенова 3. М. Вести. МГУ. Химия, № 14, 240 (1973).
294. Каменев А. И., Грановский Ю. В., Козинцева В. И. Вести. МГУ. Химия, № 12, 192 (1971).
295. Каменев А. И., Финогенова 3. М., Виноградова Е. И. Вести. МГУ. Химия, № 3, 342 (1970).
296. Каплан Б. Я., Ревякина Г. Н., Резакова X. С., Ширяева О. И. Тр. Комиссии по аналит. химии АН СССР, 16, 103 (1968).
182
297. Каплан Б. Я., Сороковская И. А., Ширяева О. А. Зап. лаб., 30, 659 (1964).
298. Каплан Б. Я., Сороковская И. А., Ширяева О. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, 191.
299. Каплан Б. Я., Ширяева О. А. Зав. лаб., 35, 401 (1969).
300. Каплан Б. Я., Ширяева О. А. Зав. лаб., 36, 1178 (1970).
301. Каплан Б. Я., Ширяева О. А. Научи, тр. Гиредмета, 71, 49 (1976).
302. Каплин А. А., Катюхин В. Е., Семеняк Т. И., Стромберг А. Г. Сб. «Методы анализа химических реактивов и препаратов», вып. 20. М., изд. ИРЕА, 1971, с. 71.
303. Каплин А. А., Михайлова 3. Г., Пичугина В. М., Стромберг А. Г. Электронная техника. Научн.-техн. сб. Управление качеством и стандартизация, вып. 5, 27 (1970).
304. Карабаш А. Г., Пейзулаев Ш. И., Морозова Г. Г., Смиренкина И. И. Тр. Комиссии по аналит. химии АН СССР, 12, 25 (1960).
305. Карабаш А. Г., Пейзулаев Ш. И., Слюсарева Р. Л., Липатова В. М. Там же, с. 331.
306. Карабаш А. Г., Пейзулаев Ш. И., Сотникова Н. П., Сазонова С. К. Там же, с. 108.
307. Карабаш А. Г., Пейзулаев Ш. И., Усачева В. П., Морозова Г. Г., Мешкова В. М., Лобанова В. Л. Ж. аналит. химии, 16, 217 (1961).
308. Карабаш А. Г., Самсонова 3. Н., Смирнова-Аверина Н. И., Пейзулаев Ш. И. Тр. Комиссии по аналит. химии АН СССР, 12, 255 (1960).
309. Карбаинов 10. А., Карбаинова С. Н. Сб. «Методы анализа химических реактивов и препаратов», вып. 20. М., изд. ИРЕА, 1971, с. 63.
310. Карбаинов Ю. А., Стромберг А. Г. Ж. аналит. химии, 19, 1341 (1964).
311. Карбаинова С. И., Карбаинов Ю. А., Стромберг А. Г. Сб. «Методы анализа химических реактивов и препаратов», вып. 20. М., изд. ИРЕА, 1971, с. 65.
312. Каргин Ю. М. Сб. «Теория и практика полярографического анализа». Кишинев, изд-во АН МолдССР, 1962, с. 243.
313. Карев В. Н., Доля П. Г., Тутубалин А. И., Коваленко Л. И. Зав. лаб., 35, 297 (1969).
314. Катченков С. М. Спектральный анализ горных пород, изд. 2. Л., «Недра», 1964.
315. Кацков Д. А., Львов Б. В. Ж. прикл. спектроскоп., 10, 382 (1969).
316. Кацман Б. X. Ж. аналит. химии, 23, 1234 (1968).
317. Кертман Л. Качественный химический полумикроанализ. М.— Л., Госхимиздат, 1949, с. 160.
318. Кива Н. К., Брайнина X. 3. Сборник научных трудов по химии. Свердловск, изд. Свердловского ин-та нар. хоз-ва, 1971, с. 4.
319. Ким Л. П., Раннее Г. Г., Табачников М. М. Авт. свид. СССР 365544; Бюл. изобр., № 18 (1972).
320. Ким Хен Пак, Ким Сек, Ким Кван. Пунсок Хвахак, 2, 92 (1963); РЖХим, 1964, 12Г69.
321. КипарисоваЛ. С., Толмачева Л. П. Бюл. научн.-техн. информ. ЦНИИ олова, сурьмы и ртути, № 4, 59 (1961).
322. Киргинцев А. П., Прохорова А. С., ЮЭелевич И. Г. Изв. СО АН СССР, № 7. Сер. хим. н., вып. 3, 43 (1969).
323. Киселева И. А. Полиграф, произ-во, № 6, 23 (1958).
324. Киш П. П., Онищенко 10. К. Авт. свид. СССР 272648; Бюл. изобр., № 19 (1970).
325. Киш П. П., Онищенко Ю. К. Ж. аналит. химии, 23, 1651 (1968).
326. Киш П. П., Онищенко Ю. К. Ж. аналит. химии, 26, 514 (1971).
327. Киш П. П., Онищенко Ю. К. Ж. аналит. химии, 29, 102 (1974).
328. Киш П. П., Онищенко IO. К. Зав. лаб., 36, 520 (1970).
329. Киш П. П., Онищенко Ю. К., Погойда И. И. Ж. аналит. химии, 28, 1746 (1973).
330. Климов А. П., Полищук В. А., Гаспарян Н. Р. Сб. научн.-техн. тр. НИИ металлургии Челябинского совнархоза, вып. 2, 168 (1960).
183
331. Клишане Д. А., Пелекис Л. Л. Сб. «Нейтронно-активационный анализ». Рига, «Зинатне», 1966, с. 44.
332. Коваленко П. Н., Моричева Н. И. Изв. вузов. Химия и хим. технол.
2, 322 (1959).
333. Колпакова Н. А., Зарубина Р. Ф. Зав. лаб., 37, 12 (1971).
334. Колбановский Ю. Я., Крижановская М. К. Зав. лаб., 22, 1334 (1956).
335. Колътгоф И. М., Белчер Р., Стенгер В. А., Матсуяма Дж. Объемный анализ, т. III. М., Госхимиздат, 1961.
336. Комаровский А. Г., Васильева Л. А., Тихомирова Н. А. Зав. лаб. 37, 179 (1971).
337. Комиссарова Л. Н., Бырько В. М., Меньков А. А., Гусева Н. И. Зав. лаб., 34, 410 (1968).
338. Комлев О. Й., 31нчук В. К. ЕИсник Льв1вск. ун-ту. Сер. х!м., вып. 9, 50 (1967).
339. Кондрашина А. И., Муравенко В. П., Васильевская Л. С., Садофъева С. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 74.
340. Коньков А. Г. Доклады научной конференции, 1957 г., т. 1. Сталинград, 1958, с. 161; РЖХим, 1960, 8917.
341. Коньков А. Г. Тр. Сталинградского с.-х. ин-та, 8, 59 (1960).
342. Конышева II. М. Зав. лаб., 34, 692 (1968).
343. Копанская Л. С. Труды III конференции молодых ученых Молдавии. Естеств.-техн. н., вып. 1. Кишинев, «Штиинца», 1964, с. 25.
344. Копанская Л. С., Ляликов Ю. С., Кириллова С. Г. Изв. АН МолдССР. Сер. биол. и хим. н., № 6, 19 (1968).
345. КопанскаяЛ. С., Цвигун Г. В. Сб. «Исследования по химии координационных соединений и физико-химическим методам анализа». Кишинев, «Штиинца», 1969, с. 87.
346. Коренман Я. И. Тр. по химии и хим. технол. (Горький), вып. 1, 152 (1963).
347. Корж П. Д., Гуляева Г. П. Сб. научн. тр. Магнитогорского горно-металлург. ин-та, вып. 23, 12 (1961).
348. Корж П. Д., Гуляева Г. Я., Шинятулин И. Н. Зав. лаб., 29, 289 (1963).
349. Королев Н. В., Рюхин В. М. Эмиссионный спектральный анализ. М., «Машиностроение», 1971.
350. Костромин А. И., Ахмадеев М. X. Уч. зап. Казанского ун-та, 124, 166 (1965).
351. Костромин А. И., Ахметов А. А. Ж. аналит. химии, 24, 503 (1969).
352. Костромин А. И., Флегонтов С. А. Зав. лаб., 27, 528 (1961).
353. Костюкова Е. С. Научн. тр. Иркутского НИИ редк. мет., вып. 12, 13 (1965).
354. Костюкова Е. С., Райхбаум Я. Д., Герасимов К. С., Климова А. К-Ежегодник ин-та геохимии СО АН СССР. Новосибирск, «Наука», 1972, с. 386.
355. Котельников Л. А., Кузнецов Р. А. Вести. ЛГУ, № 22, 137 (1970).
356. Котельников Л. А., Кузнецов Р. А. Радиохимия, 11, 256 (1969).
357. Кочешков К. А., Сколдинов А. П. Синтетические методы в области металлоорганических соединений сурьмы и висмута, вып. 8. М.— Л., Изд-во АН СССР, 1947.
358. Кравец Т. П., Песъкина А. Л., Жидкова 3. В. Изв. АН СССР. Сер. физ., 14, 943 (1950).
359. ' Красильникова Л. И., Долгорукова К. II. Сб. научн. тр. Всес. Н.-и. горнометаллург. ин-та цвет, мет., № 9, 22 (1965).
360. Краснобаева И., Ташкова А. Докл. Волг. АН, 17, 917 (1964).
<361. Краснов К. С., Шилова Г. В. Изв. вузов. Химия и хим. технол., 8, 915 (1965).
362. Крешков А. П. Основы аналитической химии. Физико-химические (инструментальные) методы анализа. М., «Химия», 1970, с. 164.
184
363. Крешков А. И., Милаев С. М., Хантургаев Г. А., Хантургаева Г. И. Ж. аналит. химии, 29, 98 (1974).
364. Кристаллева Л. Б. Тр. Томского ун-та, 154, 271 (1962).
365. Кристаллева Л. В., Кристаллев П. В. Тр. Комиссии по аналит. химии АН СССР, 14, 279 (1963).
366. Круглова М. Н. Тр. ВНИИ стандарта. образцов и спектр, эталонов, 3, 59 (1967).
367. Крылова А. II. Судебно-мед. экспертиза, 2, № 3, 31 (1959).
368. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М., Госхимиздат, 1959.
369. Куделя Е. С. Спектральный анализ металлов и сплавов. Киев, Гостех-издат, 1961.
370. Кузнецов В. И. Докл. АН СССР, 52, 231 (1946).
371. Кузнецов В. И. Ж. аналит. химии, 2, 179 (1947).
372. Кузнецов В. И., Большакова Л. И. Ж. аналит. химии, 15, 523 (1960).
373. Кузнецов О. А., Попова Н. А. Зав. лаб., 28, 1147 (1962).
374. Кузнецов Р. А. Активационный анализ. М., Атомиздат, 1967.
375. Кузнецов Р. А. Радиохимия, 13, 475 (1971).
376. Кузнецова А. С. Зав. лаб., 34, 410 (1968).
377. Кузнецова В. К. Тр. Уральского политехи, ин-та, № 163, 14 (1967).
378. Кузнецова Н. Н., Крауз Л. С. Ж. аналит. химии, 18, 1090 (1963).
379. Кузовлев И. А., Алексеенко Б. П., Захарова Т. И., Платонова Н. В. Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, с. 229.
380. Кузовлев И. А., Свердлина О. А., Ковыкова Н. В. Ж. прикл. спектроскоп., 16, 5 (1972).
381. Кузьмин Н. М. Докторская диссертация. М., ГЕОХИ АН СССР, 1973.
382. Кузьмин И. М., Захарова Т. И., Чугунова В. В. Сб. «Методы анализа химических реактивов и препаратов», вып. 17. М., изд. ИРЕА, 1970, с. 49.
383. Кузьмин Н. М., Попова Т. Д., Саратовская В. Л. Зав. лаб., 37, 658 (1971).
384. Кузьмин Н. М., Попова Т. Д., Кузовлев И. А., Соломатин В. С. Ж. аналит. химии, 24, 899 (1969).
385. Кулак А. И. Ж. аналит. химии, 12, 727 (1957).
386. Кульская О. А., Яцюк О. Ф. Сб. «Атомная спектроскопия и спектральный анализ». Киев, «Наукова думка», 1974, с. 107.
387. Кумина Д. М. Сб. «Методы анализа химических реактивов и препаратов», вып. 21. М., изд. ИРЕА, 1973, с. 119.
388. Куранов А. А. Изв. АН СССР. Сер. физ., 23, 1140 (1959).
389. Куранов А. А., Ручкина Н. П. Сб. «Материалы I Уральского совещания по спектроскопии, 1956». Свердловск, Металлургиздат, 1958, с. 105.
389а. Куранов А. А., Ручкина Н. П. Сб. Материалы III Уральского совещания по спектроскопии, 1960». Свердловск, Металлургиздат, 1962, с. 91.
390. Куранов А. А., Свиридова М. М. Сб. «Материалы I Уральского совещания по спектроскопии, 1956». Свердловск, Металлургиздат, 1958, с. 85.
391. Куранов А. А., Свиридова М. М. Сб. «Материалы II Уральского совещания по спектроскопии, 1958». Свердловск, Металлургиздат, 1959, с. 116.
392. Курбатова В. И., Калашникова Л. Я., Емашева Г. И., Никулина И. И., Феофанова В. В. Тр. ВНИИ стандарта, образцов и спектр, эталонов, 5, 111 (1969).
393. Курбатова В. И., Макагонова Л. Н., Жарикова В. К. Тр. ВНИИ стандарта, образцов и спектр, эталонов, 4, 108 (1968).
394. Курбатова В. И., Петрова Н. П., Никулина И. Н., Жаворонкова Е. Т., Зобнина Н. А., Жилкина С. П. Тр. ВНИИ стандарта, образцов и спектр, эталонов, 7, ИЗ (1971).
395. Куртова Л. В., Смарина Е. И., Волкова Н. А., Рощина А. В., Уч. зап. Московского ин-та тонкой хим. технол., 1, № 1, 41 (1970).
185
396. Кустас В. Л., Мамонтова Л. И., Шепота Н. Г., Юделевич И. ГЛ Сб. «Синтез, очистка и анализ неорганических материалов». Новосибирск, «Наука», 1971, с. 259.
397. Кутейников А. Ф., Егорова В. А., Конькова Е. С., Заварцева И. Д., Беляев Ю. И., Карякин А. В., Челинцев А. М. Ж. аналит. химии, 25, 1999 (1970).
398. Кюрегян С. К. Эмиссионный спектральный анализ нефтепродуктов. М„ «Химия», 1969.
399. Лабий Ю. М. Укр. хим. ж., 28, 641 (1962).
400. Лабковская Д. Б., Братчикова Н. И., Лазарева Г. А. Сб. «Анализ благородных металлов». М., «Металлургия», 1965, с. 211.
401. Лазарев А. И., Лазарева В. И. Зав. лаб., 25, 405 (1952).
402. Лазарев Д., Павлов Д. Химия и индустрия, 30, 166 (1958).
403. Ламбрев В. Г., Власов В. С., Мякинъкова Т. В. Ж. аналит. химии, 25, 2306 (1970).
404. Ланцев И. П., Фалъкова О. Б., Денисова Л. К. Тр. Центр, н.-и. горно-развед. ин-та цвет., редк. и благород. мет., вып. 97, 182 (1971).
405. Лапицкая Е. В., Горбенко Ф. П., Велъштейн Э. И. Зав. лаб., 34, 1446 (1968).
406. Лебедев Г. Г., Львов Б. В. Зав. лаб., 34, 1139 (1968).
407. Левин А. Б. Тр. Уральского политехи, ин-та, № 94, 106 (1960).
408. Левин И. С., Леонтьева Е. Г. Зав. лаб., 35, 1325 (1969).
409. Левин И. С., Леонтьева Е. Г. Изв. СО АН СССР, Сер. хим. н., № 7, вып. 3, 134 (1969).
410. Левин И. С., Шаталова А. А. Докл. АН СССР, 161, 1158 (1965).
411. Левин И. С., Шаталова А. А., Ворсина И. А. Изв. СО АН СССР, Сер. хим. н., № 14, вып. 6, 89 (1967).
412. Левицкая С. И., Зебрева А. И. Электрохимия, 2, 92 (1966).
413. Левшин Л. В., Баранова Е. Г. Изв. АН СССР. Сер. физ., 20, 424 (1956).
414. Левшин Л. В., Бочарова В. Г. Оптика и спектроскопия, 10, 627 (1961).
415. Левшин Л. В., Горшкова В. К. Там же, с. 637.
416. Лекторская Н. А., Коваленко П. Н. Научн. докл. высш, школы. Химия и хим. технол., № 1, 102 (1959).
417. Лекторская Н. А., Коваленко П. Н. Уч. зап. Ростовского ун-та, 40, 173 (1958).
418. Лекторская Н. А., Коваленко П, Н. Уч. зап. Ростовского ун-та, 41, 95 (1958).
419. Лекторская Н. А., Коваленко П, Н. Сб. «Технология покрытий металлов и методы контроля производства. Ростов, изд-во РГУ, 1962, с. 118.
420. Лекторская Н. А., Коваленко П. Н. Сб. «Электрохимические и оптические методы анализа». Ростов, изд-во РГУ, 1963, с. 74.
421. Леушкина Г. В., Лобанов Е. М., Дутов А. Г., Матвеева Н. П. Тр. по химии и хим. технол. (Горький), вып. 3 (24), 106 (1969).
422. Ли Гын, Хан Дон Дж. Бунсок хвахак, 8, 38 (1970); РЖХим, 1971, 13Г159.
423. Липшиц Б. М., Смирнова Г. К., Куликов Ф. С. Зав. лаб., 27, 1199 . (1961).
424. Литеану К., Шутек А. Ж. аналит. химии, 23, 445 (1968).
425. Лифшиц Е. В., Мосова Л. Н. Зав. лаб., 28, 1329 (1962).
426. Лобанов Г. М., Валиева М. Г., Усманова М. М. Сб. «Нейтронно-активационный анализ». Ташкент, «Фан», 1971, с. 72.
427. Лобанов Е. М., Левушкин Ю. А., Власюга С. П. Сб. «Активационный анализ в народном хозяйстве». Ташкент, «Фан», 1974, с. 57.
428. Лобанов Ф. И., Буяновская А. Г., Гибало И. М. Ж. аналит. химии, 26, 1655 (1971).
429. Логинова Л. Г. Ж. аналит. химии, 14, 217 (1959).
430. Ломоносов С. А. Ж. аналит. химии, 22, 1125 (1967).
431. Ломоносов С. А. Ж. аналит. химии, 28, 1645 (1973).
432. Ломоносов С, 4., Багреев В. В., Шуколюкова Н. И., Сорокин Г. X., Кузьмич М. К. Ж. аналит. химии, 29, 1261 (1974).
186
433. Ломоносов С. А., Звездин М. К., Инишев В. Д. Ж. аналит. химии, 24, 1115 (1969).
434. Ломоносов С. А., Мильштейн Ф. Я. Зав. лаб., 33, 14 (1967). Ломоносов С. Л., Николаев А. В. Докл. АН СССР, 167, 354 (1966).
436. Ломоносов С. А., Попов Э. И., Сорокин Г. X., Ройтман Л. И., Инишев В. Д„ Лисунова Р. П., Кондратов В. К., Шуколюкова Н. И., Про-шутинский В. И. Ж. аналит. химии, 28, 1653 (1973).
437. Ломоносов С. А., Рыбакова Ю. А., Подчайнова В. И., Веднягина Н. П. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, с. 263.
438. Л омоносов С. А., Сорокин Г. X., Попов Э. И., Шуколюкова Н. И.,
Носова И. П. Ж. аналит. химии, 28, 1877 (1973).
439. Ломоносов С. А., Сорокин Г. X., Шуколюкова Н. И. Там же, с. 1869.
440. Ломоносов С. А., Сорокин Г. X., Шуколюкова Н. И. Там же, с. 2085.
441. Ломоносов С. А., Шуколюкова Н. И., Чупахин О. И., Сорокин Г. X.,
Там же, с. 2091.
442. Лонцих С. В., Педлер В. В., Райхбаум Я. Д., Хохлов В. В. Спектральный анализ при поисках рудных месторождений. Л., «Недра», 1969.
442а. Лосев Н. Ф., С магу нов а А. Н., Ревенко А. Г., Павлинский Г. В., Тарасенко С. А., Розова О. Ф., Величко Ю. И., Зав. лаб., 43, 160 (1977).
443. Лурье Ю. Ю., Филиппова Н. А. Зав. лаб., 18, 30 (1952).
444. Лурье Ю. Ю., Филиппова Н. А. Зав. лаб., 19, 771 (1953).
445. Луфт Б. Д., Новиков В. Б., Хусид Л. Б., Шемет В. В. Ж. аналит. химии, 28, 536 (1973).
446. Лысенко В. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 242.
447. Лысенко В. И. Тр. Комиссии по аналит. химии АН СССР, 15, 195 (1965)*
448. Львов В. В. Атомно-абсорбционный спектральный анализ. М., «Наука», 1966.
449. Любимова Л. И., Салмин Ю. П., Сергеева Т. В., Сочеванов В. Г., Ши~ ряева М. Б. Сб. «Активационный анализ». Ташкент, «Фан», 1971, с. 130.
450. Ляликов Ю. С., Копанская Л. С. Изв. АН МолдССР, № 12 (90), 47 (1961).
451. Ляликов IO. С., Копанская Л. С. Укр. хим. ж., 30, 91 (1964).
452. Ляликов Ю. С., Копанская Л. С., Сафронкова Н. Я. Сб. «Физика». Л., Изд-во АН СССР, 1962, с. 26.
453. Ляликов Ю. С., Чернега Л. Н., Бодю В. И. Тр. Кишиневского политехи, ин-та, вып. 13, 167 (1967).
454. Ляшенко Т. В. Металлург, и хим. пром-сть Казахстана. Научн.-техн, сб., № 6 (22), 67 (1962).
455. Макашева И. Е., Маслов И. А., Обухов А. Н, Ж. аналит. химии, 15, 329 (1960).
456. Максай Л. И., Силаева А. А. Сб. тр. Всес. н.-и. горнометаллург. ин-та цвет, мет., № 7, 343 (1962).
457. Малахов В. В., Протопопова Н. П., Трухачева В. А., Юделевич И. Г. Изв. СО АН СССР, № 9. Сер. хим. н., вып. 4, 78 (1968).
458. Малахов В. В., Протопопова Н. П., Трухачева В. А., Юделевич И. Г. Тезисы докладов по аналитической химии полупроводниковых веществ. Кишинев, «Штиинца», 1965, с. 51.
459. Малинина Р. Д. Тр. ЦНИИ черн. металлург., вып. 31, 158 (1963).
460. Малкова О. П., Жукова А. Н., Рудневский Н. К. Ж. аналит. химии, * 19, 312 (1964).
461. Малкова О. П., Туманова А. Н., Рудневский Н. К. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, с. 122.
462. Малкова О. П., Туманова А. И., Рудневский Н. К. Там же, с. 132.
463. Мальков Е. М. Зав. лаб., 34, 504 (1968).
464. Мальков Е. М., Федосеева А. Г. Зав. лаб., 36, 912 (1970).
465. Мальков Е. М., Федосеева А. Г., Стромберг А. Г. Ж. аналит. химии, 25, 1748 (1970).
187
466. Мальцев В. К., Мелътцер Л. В., Калмыков А. А., Щумиловский Н. И-Авт. свид. СССР 162355; Бюл. изобр., № 9 (1964).
467. Манева Д. Науч. тр. Высш, ин-т хранит, и вкус, пром-сти (Пловдив), 14, 233 (1967).
468. Марунина Н. И., Сухов Г. В., Ионов В. И., Прокофьева И. К., Рейф-ман М. Б. Сб. «III Всесоюзная конференция по карбиду кремния» М., изд. ИОНХ АН СССР, 1970, с. 258.
469. Марченко 3. Фотометрическое определение элементов. М., «Мир», 1971.
470. Марченко П. В., Вдовенко М. Е., Набиванец Б. М., Оболончик И. В., Спиваковская Н. Е. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 388.
471. Марченко П.ВВоронина А. И. УК. аналит. химии, 23, 868 (1968).
472. Масленников В. М., Романова Л. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 228.
473. Маслов И. А., Лучинский В. А. Справочник по нейтронно-активационному анализу. Л., «Наука», 1971, с. 87.
474. Матвеец М. А., Щербов Д. П., Ахметова С. Д. Ж. аналит. химии, 29, 740 (1974).
475. Матвеец М, А., Щербов Д. П., Ахметова С. Д. Сб. «Исследования в области химических и физических методов анализа минерального сырья», вып. 3. Алма-Ата, изд. Казахского НИИ минер, сырья, 1973, с. 88.
476. Матвеец М. А., Щербов Д. П., Ахметова С. Д. Там же, с. 91.
477. Меламед Ш. Г., Солодовник С. М. Тр. Комиссии по аналит. химии АН СССР, 12, 172 (1960).
478. Меркин Э. И., Русакова Т. В., Мельников С. М., Кочергина Д. Г. Зарубежная практика переработки сурьмяных руд. М., Цветметинфор-мация, 1972.
479. Методы анализа веществ высокой чистоты. М., «Наука», 1965.
480. Микаелян К. Пром-сть Армении, № 11, 40 (1966),
481. Миланов Б., Иванова Д. Рудодоб. и металлургия, 21, № 4, 14 (1966).
482. Миленина Д. И. Уч. зап. Горьковского гос. пед. ин-та, вып. 98, 94 (1969).
483. Минасян К. Пром-сть Армении, № 11, 40 (1966).
484. Минасян И. В. Научн. тр. н.-и. гидрометаллург, ин-та (Ереван), вып. 5, 251 (1965).
485. Михеева Л. М., Михеев И. Б. Радиоактивные изотопы в аналитической химии. М., Госхимиздат, 1961.
486. Младенцева О. И., Горожанкина И. И., Сухенко К. А., Аксенова А. В. Тр. Комиссии по аналит. химии АН СССР, 12, 355 (I960).
487. Моисеев В. В., Кузнецов Р. А., Калинин А. И. Reinstoffe in Wissen-schaft und Technik. Berlin, Akademieverlag, 1963, S. 449.
488. Моисеев В. E., Тимофеева А. И. Тр. Ин-та физ. мет. Уральского научн. центра АН СССР, вып. 28, 87 (1971).
489. Морачевский Ю. В., Барбанелъ Д. Г. Уч. зап. ЛГУ, № 211 62 (1957).
490. Морачевский IO. В., Барбанелъ Д. Г. Там же, с. 76.
491. Морачевский Ю. В., Зилъберштейн X. И., Пирютко М. М., Никитина О. И. Ж. аналит. химии, 17, 614 (1962).
492. Морачевский Ю. В., Калинин А. И. Зав. лаб., 27, 272 (1961).
493. Мосеева 3. И., Инчук Г. П., Соколов А. Б., Карабаш А. Г., Пейзулаев Ш. И. Ж. аналит. химии, 29, 1589 (1974).
494. Музгин В. И., Золотавин В. Л., Гаврилов Ф.Ф., Балаев В. Н. Сб. «Спектральный анализ в геологии и геохимии». М., «Наука», 1967, с. 161.
495. Мустафин И. С., Иванова А. И. Изв. вузов. Химия и хим. технол., 5, 504 (1962).
496. Мустафин И. С., Иванова А. Н., Ковалева В. С., Муллина Р. Н. Ассортимент реактивов на сурьму. М., изд. НИИТЭХим, 1970.
497. Мухина 3. С., Жемчужная И. А., Котова Г. С. Ж. аналит. химии, 17, 170 (1962).
188
498. Мухина 3. С., Никитина Е. И. Ускоренные методы анализа титана и его сплавов. М., Оборонгиз, 1961, с. 50.
499. Мюнке М. Зав. лаб., 34, 165 (1968).
500. Нагибин В. С., Угнивенко М. Г. Тр. Ин-та металлургии АН СССР, 11, 221 (1962).
501. Нагибина И. М., Прокофьев В. К. Спектральные приборы и техника спектроскопии, изд. 2. М.— Л., «Машиностроение», 1967.
502. Надежина Л. С. Тр. Ленинградского политехи, ин-та, № 201, 120 (1959).
503. Надежина Л. С., Гринзайд Е. Л., Новаковская Э. Г. Ж. аналит. химии, 23, 86 (1968).
504. Назаренко В. А., Бирюк Е. А., Бык Г. И., Винковецкая С. Я., Лебедева Н. В., Суричан Т. А., Шустова Н. Б. Сб. научн. тр. Гиредмета, 12, 77 (1959).
505. Назаренко В. А., Бирюк Е. А., Шустова М. Б., Лебедева Н. В., Винковецкая С. Я., Суричан Т. А., Флянтикова Г. В. Сб. «Методы определения и анализ редких элементов». М., Изд-во АН СССР, 1961, с. 442.
506. Назаренко В. А., Лебедева Н. В. Ж. аналит. химии, 10, 289 (1955).
507. Назаренко В. А., Лебедева Н. В., Бирюк Е. А. Сб. научн. тр. Гиредмета, 12, 150 (1959).
508. Назаренко В. А., Флянтикова Г. В. Зав. лаб., 24, 663 (1958),
509. Назаренко В. А., Шустова М. Б., Флянтикова Г. В., Никонова М. П., Равицкая Р. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 234.
510. Парушкявичюс Л. Р., Даукшас К. Научн. тр. вузов ЛитССР. Химия и хим. технол., № 1, 11 (1961).
511. Парушкявичюс Л. Р., Даукшас К. Уч. зап. Вильнюс, ун-та. Сер мат., физ. и хим. н., 7, 161 (1957).
512. Парушкявичюс Л. Р., Казлаускас Р. М. Ж. аналит. химии, 20, 1252 (1965).
513. Нарушкявичюс Л. Р., Казлаускас Р. М. Научн. тр. вузов ЛитССР. Химия и технол., № 7, 9 (1965).
514. Парушкявичюс Л. Р., Казлаускас Р. М. Научн. тр. вузов ЛитССР. Химия и хим. технол.-, № 9, 47 (1968).
515. Нарушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас Ю. С. Ж. аналит. химии, 26, 922 (1971).
516. Нарушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас IO. С. Научн.. тр. вузовЛитССР. Химия и хим. технол., № 17, 39 (1975).
517. Парушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас Ю. С., Вашки-те Л. П. Научн. тр. вузов ЛитССР. Химия и хим. технол., № 15, 61 (1973).
518. Нарушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас IO. С., Вирбали-те Д. К. Научн. тр. вузовЛитССР. Химия и хим. технол., № 13, 59* (1971).
519. Нарушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас Ю. С., Вирбали-те Д. К., Бумялнс В. В. Научн. тр. вузов ЛитССР. Химия и хим. технол., К» 14, 49 (1972).
520. Нарушкявичюс Л. Р., Казлаускас Р. М., Шкадаускас Ю. С., Карито-найте Н. П. Там же, с. 95.
521. Нахарова А. А., Товстопят Е. С., Еременко В. Я. Гидрохим. материалы, № 51, 196 (1969).
522. НевскаяЕ.М., Антонович В. П., Назаренко В. А., Ж. аналит химии, 28, 1104 (1973).
523. НегинаВ. Р., Замятнина В. Н. Ж. аналит. Химии, 16, 209 (1961).
524. Негина В.Р., Замятнина В. Н., Преснякова М. А., Чикишева Л. А. Радиохимия, 3, 473 (1961).
525. Нейман Е. Я., Брайнина X. 3. Ж. аналит. химии, 30, 1073 (1975).
526. Нейман Е. Я., Долгополова Г. М., Трухачева Л. Н., Дмитриев К. Г. Зав. лаб., 36, 649 (1970).
527. Нейман Е. Я., Игнатов В. И. Ж. аналит. химии, 31, 970 (1976).
189-
528. Нейман Е. Я., Кузнец Э. Д., Белова Л. А. Зав. лаб., 35, 277 (1969). -529. Нейман Е. Я., Суменкова М. Ф. Ж. аналит. химии, 30, 1012 (1975). 530. Нейман Е. Я., Суменкова М. Ф. Там же, с. 1625.
531. Нейман Е. Я., Суменкова М. Ф., Немодрук А. А. Там же, с. 1012.
532. Нейман Е. Я., Трухачева Л. Н. Зав. лаб., 38, 1058 (1972).
533. Нейман Е. ЯТрухачева Л. Н., Долгополова Г. М. Сб. «Электрохимия и коррозия», вып. 31. М., «Металлургия», 1970, с. 206.
534. Неймарк Л. Э. Тр. Ин-та металлургии и обогащ. АН КазССР, 31, 72 (1968).
535. Немодрук А. А. Аналитическая химия мышьяка. М., «Наука», 1976.
536. Немодрук А, А. Ж. аналит. химии, 28, 138 (1973).
537. Непеина Л. А. Зав. лаб., 28, 166 (1962).
538. Нестерова Ю. С., Арапова Т. А. Тр. Ин-та геол. рудн. месторожд., петрогр., минералогии и геохимии АН СССР, вып. 81, 36 (1962).
539. Нестерова Ю. С., Арапова Т. А. Сб. «Химический анализ минералов и их химический состав». М., «Наука», 1964, с. 36.
540. Неудачина Э. Ф., Сентар 3. Научн. тр. Среднеаз. н.-и. и проектн. ин-та цвет, металлургии, № 8, 109 (1973).
541. Никаноров Г. С., Коцуба А. С., Коваленко В. В., Сильванович IO. А. Сб. «Активационный анализ благородных металлов». Ташкент, «Фан», 1970, с. ИЗ.
542. Никитин В. Н., Оболенцев Л. А., Вайда В. Г. Тр. Центр, н.- и. горно-развед. ин-та цвет.,редк. и благородн. мет., вып. 97, 187 (1971).
543. Никитин Ф. В. Зав. лаб., 31, 966 (1965).
544. Никитина Е. И. Зав. лаб., 14, 938 (1948).
545. Никитина Е. И. Тр. Комиссии по аналит. химии АН СССР, 12, 311 (1960).
546. Никитина Е. И., Иванова Н. К. Зав. лаб., 30, 46 (1964).
547. Николаев А. В., Юделевич И. Г., Буянова Л. М., Валл Г. А., Чучали-наЛ.С., Пещевицкий Б. И., Храпай В. П. Изв. СО АН СССР, №14. Сер. хим. н., вып. 6, 78 (1970).
548. Николаев А. В., Юделевич И. Г., Буянова Л. М., Чучалина Л. С., Середнякова Т. П. Там же, с. 83.
549. Ноткина М. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 203.
550. Ноткина М. А., Добкина Б. М. Зав. лаб., 26, 1126 (1960).
551. Ноткина М. А., Солодовник С. М., Баранова Л. Л., Лушина В. К., Романцова Т. И. Зав. лаб., 28, 176 (1962).
552. Ноткина М. А., Солодовник С. М., Василевская Л. С., Назарова Н. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 513.
553. Нюссик Я. М., Индиченко Л. Н., Дудыкина А. С. Сб. «Спектральный анализ в геологии и геохимии». М., «Наука», 1967, с. 156.
554. Овруцкий М. И., Фрегер С. В., Кечко В. Н., Белогуб Т. Н. Сб. «Методы анализа химических реактивов и препаратов», вып. 18. М., изд. ИРЕА, 1971, с. 63.
555. Олейник А. М. Тр. Уральск." н.-и. хим. ин-та, вып. 16, 186 (1967).
556. Олыианова К. М., Чмутов К. В. Ж. аналит. химии, 11, 94 (1956).
557. Опарина А. Ф., БруйчеЕ. С. Ж. прикл. химии, 45, 1368 (1972).
558. Орловский А. Ж. русск. физ.-хим. о-ва, 15, 32 (1883).
559. О Сан'Вон. Вести. АН КНДР, № 4, 12 (1962); РЖХим, 1963, 19Г21.
560. Оспанов X. К., Алимпева С. Д. Авт. свид. СССР 305406; Бюл. изобр., № 18 (1971).
561. Остапенко В. Ф., Шмонин Л. И., Черевко В. И., Энкер М. Б. Сб. «Ядерно-физические методы анализа веществ». М., Атомиздат, 1971, с. 255.
562. Отмахова 3. И., Чащина О. В., Катаев Г. А. Изв. СО АН СССР, № 9, Сер. хим. н., вып. 4, 84 (1967).
563. Отраслевые технические инструкции. Сурьма. Методы химического анализа. РЭТИ, № 174—58, № 175—58. М., изд. Гиредмет, 1958.
190
564. Отраслевые технические инструкции. Германий. Методы химического анализа. РЭТИ, № 222—61. М., изд. Гиредмет, 1961.
565. Павликова Г. Н. Изв. СО АН СССР, № 6. Сер. хим. н., вып. 1, 53 (1960).
566. Павличенко В. А., Усатенко Ю. И., Тулюпа Ф. М. Хим. технология. Респ. межвед. темат. научн.-техн. сб., вып. 3, 88 (1968).
567. Пан Шу-вэй, Лу Линъ-лянъ. Хуасюэ сюэбао, 23, 117 (1957).
568. Панталер Р. П., Лебедев Н. В., Семенова Л. Н., Колесова П. А. Тр. по химии и хим. технол. (Горький), вып. 3 (24), 140 (1969).
569. Пац Р. Г., Васильева Л. Н. Сб. «Методы анализа с использованием полярографии переменного тока». М., «Металлургия», 1967, с. 66.
570. Пац Р. Г., Лукашенкова Н. В., Шувалова Е. Д., Загладина Т. В, Зав. лаб., 34, 1173 (1968).
571. Певцов Г. А. Тр. Комисии по аналит. химии АН СССР, 12, 314 (1960).
572. Певцов Г. А., Красильщик В. 3., Скузоватова Т. П. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 81.
573. Певцов Г. А., Манова Т. Г. Там же, с. 88.
574. Певцов Г. А., Манова Т. Г. Там же, с. 515.
575. Певцов Г. А., Манова Т. Г., Рачинская Л. К., Герасимова Т. С. Сб. «Методы анализа химических реактивов и препаратов», вып. 15. М., изд. ИРЕА, 1968, с. 69.
576. Певцов Г. А., Манова Т. Г., Хасянова С. Ф. Там же, с. 64.
577. Пейзулаев Ш. И., Попова Л. К., Слюсарева Р. Л. Зав. лаб., 26, 552 (1960).
578. Пережогин Г. А. Ж. аналит. химии, 27, 2222 (1972).
579. Пережогин Г. А., Гаврилова Л. П. Ж. аналит. химии, 30, 725 (1975).
580. Пережогин Г. А., Мещеряков В. Г. Сб. «Активационной анализ». Ташкент, «Фан», 1971, с. 95.
581. Петрашенъ В. И. Качественный химический анализ. М. Госхимиздат, 1948, с. 359.
582. Петренко О. П., Иванова Г. В. Уч. зап. ЦНИИ оловян. пром-сти, № 1, 88 (1965).
583. Пилипенко А. Г Kim П. П., Онищенко Ю. К. Доповщ! АН УССР, 5, 249 (1971).
584. Пилипенко А. Т., Нгуен Монг Шинъ. Зав. лаб., 33, 1074 (1967).
585. Пилипенко А. Т., Нгуен Монг Шинъ. Укр. хим. ж., 34, 1286 (1968).
586. Пичугина В. М., Зарубина Р. Ф., Джабарова Н. К., Каплин А. А. Сб. «Труды молодых ученых Томского политехнического ин-та», вып. 1. Томск, изд. ТПИ, 1973, с. 68.
587. Подберезская Н.К, Тр. Казахского НИИ минеральн. сырья, вып. 2, 223 (1960).
588. Подчайнова В. Н., Малкина Т. Г., Студенская Л. С. Тр. ВНИИ стандарт. образцов и спектр, эталонов, 2, 18 (1965).
589. Поливанова Н. Г., Фраткин 3. Г. Зав. лаб., 33, 970 (1967).
590. Поленов Ю. Н., Габитова А.Ш., Павлова В. В. Исследования по электрохимическим методам анализа, вып. 3. Изд-во Казанского, ун-та, 34 (1970).
591. Полякова В. В., Федорова В. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 460.
592. Полякова В. В., Федорова В. В. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, с. 172.
593. Попелъ А. А., Скребкова Л. М. Уч. зап. Казанского ун-та, 117, 184 (1957).
594. Попков К. К Лельчук С. Л., Кудрявцева А. С. Пласт, массы, № 1, 39 (1960).
595. Поплевина Л. В. Тр. Пермского мед. ин-та, 108, 45 (1972).
596. Попов М. А., Кипариеова Л. С. Уч. зап. ЦНИИ оловянной пром-сти, № 1, 55 (1960).
597. Попов М. А., Третьякова Т. М. Бюл. научн.-техн. информ. ЦНИИ олова, сурьмы и ртути, № 1, 37 (1962).
191
598. Порхунова Н. А., Ларина Л. К., Бакалдина II. С. Сб. тр. Всес. и.-и горнометаллург. ин-та цветн. мет., № 7, 380 (1962).
599. Порхунова Н. А., Ларина Л. К., Беленкова Н. С. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 400.
600. Преображенский В. К., Москвин Л. II. Радиохимия, 3, 309 (1961).
601. Преображенский Н. Г. Изв. вузов. Физика, № 6, 57 (1961).
602. Привалова М. М., Рябчиков Д. И. Ж. пеоргап. химии, 5, 1605 (1960).
603. Привалова М. М., Рябчиков Д. И. Тр. Комиссии по аналит. химии АН СССР, 9, 301 (1958).
604. Пронин В. А., Гильберт Э. И., Артюхин П. И. Изв. СО АН СССР, № 3. Сер. хим. н., вып. 1, 88 (1966).
605. Пронин В. А., Горностаева Е. Д. Ж. аналит. химии, 30, 1139 (1975).
‘606. Прохорова С. А., Шелпакова И. Р., Юделевич И. Г. Уч. зап. ЦНИИ оловянной пром-сти, 2, 58 (1967).
607. Пятницкий И. В., Клибус А. X. Укр. хим. ж., 33, 1077 (1967).
608. Пятницкий И. В., Ружанская Р. П. Укр. хим. ж., 35, 203 (1969).
609. Равич А. Ж. русск. физ.-хим. о-ва, 37, 1303 (1905).
610. Разумова Т. И., Шуба И. Д Васильев И. Я. Радиохимия, 12, 133 (1970).
611. РайковиИ В. Гласник хем.друшт. Београд, 29, 17 (1964).
612. Pajuh С. Р. Гласник хем. друшт, Београд, 31, 177 (1966).
613. Раковский Э. Е., Петрухин О. М. Ж. аналит. химии, 18, 539 (1963).
614. Раковский Э. Е., Смахтин Л. А., Яковлев Ю. В. Зав. лаб., 26, 1199 (1960).
615. Рамадан А. А., Агасян П. К., Петров С. И. Ж. аналит. химии, 29, 1144 (1974).
616. Рамадан А. А., Агасян П. К., Петров С. И. Ж. аналит. химии, 30, 89, (1975).
617. Рамадан А. А., Агасян П. В., Петров С. И. Зав. лаб., 41, 641 (1975).
618. Рао А. Л., Дж., Пури В. К. Ж. аналит. химии, 26, 996 (1971).
619. Рахматуллаев К., Рахматуллаева М. А., Азизов Ш. Сб. «Химия редких и рассеянных металлов». Ташкент, «Фан», 1975, с. 30.
620. Рахматуллаев К., Рахматуллаева М. А., Рахимов X. Р., Талипов Ш.Т. Ж. аналит. химии, 25, 1132 (1970).
621. Рахматуллаев К., Рахматуллаева М. А., Рахимов Х.Р., Талипов III. Т. Узб. хим. ж., № 3, 23 (1970).
622. Рахматуллаев К., Рахматуллаева М. А., Талипов Ш. Т., Атаев А. Ж. аналит. химии, 27, 1793 (1972).
623. Рахматуллаев К., Рахматуллаева М. А., Талипов Ш. Т. Ж. аналит. химии, 25, 914 (1970).
624. Ржезач 3., Дитз Ю. Зав. лаб., 29, 1176 (1963).
625. Риекстиня Д. В., Пелекис Л. Я. Изв. АН ЛатвССР. Сер. физ. и хим. н., № 4, 24 (1971).
626. Родина Т. Ф., Левин И. С., Чепик М. Н., Леонтьева Е. Г. Зав. лаб., 39, 19 (1973).
627. Розенблюм Е. И., Постман М. Я. Зав. лаб., 29, 929 (1963).
628. Ройзенблат Е. М., Врайнина X. 3. Электрохимия, 5, 496 (1969).
629. Рудневский Н. К., Максимов Д. Е., Буракова Л. П. Ж. прикл. спектроскоп., 5, 384 (1966).
630. Рудневский Н. К., Максимов Д. Е., Шабанова Т. М. Труды Уральской конференции по спектроскопии. Свердловск, изд. Уральск, научн. центра АН СССР, 1971, с. 12.
631. Рыбаков В. Н., Стронский И. И. Ж. неорган. химии, 4, 2449 (1959).
632. Рыбина Т. Ф., Заюкова Н. Д., Пертова Л. Г. Сб. тр. ЦНИИ черн. металлургии, вып. 79, 68 (1972).
633. Рычков Р. С., Глухарева Н.А. Зав. лаб., 27, 1246, (1961).
634. Рудневский Н. К., Туманова А. Н., Максимов Д. Е., Ломзилова Л. В. Ж. прикл. спектроскоп., 11, 783 (1969).
635. Русанов А. Н. Основы количественного спектрального анализа руд и минералов. М., «Недра», 1971.
192
636. Русанов А. К., Алексеева В. М., Ильясова Н. В. Ж. аналит. химии, 16 284 (1961).
637. Русанов А. К., Алексеева В.М., Хитрое В. Г., Количественное спектральное определение редких и рассеянных элементов. М., Гос-геолтехиздат, 1960.
638. Русанов А. К., Сердобова Л. И. Ж. прикл. спектроскоп., 12, 596 (1970).
639. Рычков Р. С., Беркутова И.Д., Глухарева Н.А., Гофман А. К., Кузнецова А. Г., Смирнова Н. Б. Сб. «Радиоактивные изотопы и ядер-ные излучения в народном хозяйстве СССР», т. 1. М., Гостоптехиздат, 1961, с. 267.
640. Рычков Р. С., Глухарева Н. А. Зав. лаб., 27, 1246 (1961).
641. Рычков Р. С., Кузнецова Г. А., Глухарева Н.А., Беркутова И.Д., Гофман А. К., Майзелъ О. А. Методы определения и анализа редких элементов. М., изд-во АН СССР, 1961, с. 439.
642. Рябинин А. И., Романов А. С., Хатамов Ш., Кист А. А., Хамидова Р. Ж. аналит. химии, 27, 94 (1972.)
643. Рябчиков Д. И., Вайнштейн Э. Е., Борисова Л. В., Волынец М. И., Королев В. В., Куценко 10. И. Тр. Комиссии по аналит. химии АН СССР, 12, 82 (1960).
644. Рябчиков Д. И., Гохштейн Я. П., Борисова Л. В. Там же, с. 265.
645. Рябчиков Д. И., Лазарев А. И. Сб. «Рений».) М., Изд-во АН СССР, 1961, с. 267.
646. Рябчиков Д. И., Привалова М. М. Ж. неорган. химии, 3, 1964 (1958).
647. Саввина Л. П., Головина А. П., Соловьев Е. А. Вестн. МГУ. Химия, № 16, 451 (1975).
648. Саламатина В. В., Батюк А. Г. Изв. АН КиргССР, № 2, 54 (1974).
649. Сараева Н. Ф., Черенкова А. Т. Сб. научн. тр. гос. н.-и., ин-та цвет, мет., № 34, 33 (1971).
650. Сафронкова Н. Н., Ляликов Ю. С. Зав. лаб., 27, 21 (1961).
651. Сачко В. В., Горбенко Ф.П. Сб. «Методы анализа химических реактивов и препаратов», вып. 15. М., изд. ИРЕА, 1968, с. 107.
652. Седых Э. М. Стекло и керамика, № 4, 46 (1971).
653. Сендерова В. М. Химические анализы и формулы минералов». М., «Наука», 1969, с. 28.
654. Серженко Л. В., Горжанова А. А. Сб. «Успехи полярографии с накоплением». Томск, изд-во ТГУ, 1973, с. 252.
655. Силаева Е. В., Курбатова В. И. Зав. лаб., 28, 280 (1962).
656. Силакова В. Г., Макулов Н. А., Манова Т. Г., Божеволънов Е. А. Ж. аналит. химии, 29, 1508 (1974).
657. Синякова С. И. Кроль Ч. Я. Тр. Комиссии по аналит. химии АН СССР, 12, 206 (1960).
658. Синякова С. И., Чулкина Л. С. Ж. аналит. химии, 23, 841 (1968).
659. Славин У. Атомно-абсорбционная спектроскопия. Л., «Химия», 1971.
660. Соколова Л. Д. Коваленко П.Н., Богдасаров К. Я. Ж. аналит. химии, 19, 1196 (1964).
661. Солдатова Л. А., Килина 3. Г., Катаев Г. А. Там же, с 1267.
662. Солдатова Л. А., Килина 3. Г., Катаев Г. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 177.
663. Солнцев Н. И. Сб. научн. тр. гос. н.-и. ин-та цвет, мет., № 12, 36 (1956).
664. Соловьев Е. А., Софина Н. И. Зав. лаб., 40, 773 (1974).
665. Соловьев Е.А., Софина Н.И., Божеволънов Е. А. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты»", ч. 2. Харьков, изд. ВНИИ монокристаллов, 1971, с. 99.
666. Соловьева Л. Я., Арзамасцев Е. В. Сб. «Материалы конференции по итогам научных исследований». М., изд. Ин-та общ. и коммун, гигиены АМН СССР, 1965, с. 42.
667. Солодовник С. М. Изв. АН СССР. Сор. физ., 19, 182 (1955).
7 а. А. Немодрук
193
668. Солодовник С. М. Сб.. «Аналитическая химия редких металлов и полупроводниковых материалов». М., «Металлургия», 1970, с. 140.
669. Солодовник С. М., Горюшина В. Г., Арчакова Г. А., Бродская Б. Д., Мальцева В. П. Сб «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 230.
670. Солодовник С. М., Горюшина В. Г., Бродская В. Д., Назарова М. Г., Лушина В. К. Ж. аналит. химии, 22, 1054 (1967).
671. Солодовник С. М., Кондрашина А. И. Сб. научн. тр. Гиредмета, 3. 148, 1968.
762. Солодовник С. М., Кондрашина А. И., Баранова Л. Л. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 241.
673. Солодовник С. М., Русанов А. К. Изв. АН СССР. Сер. физ., 9, 635 (1945).
674. Солодовник С. М., Русанов А. К. Изв. АН СССР, 19, 179 (1955).
675. Сангина О. А. Амперометрическое титрование. М., «Химия», 1967, с. 307.
676. Сангина О. А. Амперометрическое титрование в анализе минерального сырья. М., Госгеолтехиздат, 1957, с. 156.
677. Сосновская Л. Н. Тр. Ин-та минералог., геохим. и кристаллохим. редких элементов, вып. 18, 186 (1963).
678. Сотников В. С., Королев Н. В., Шумова В. В., Морозова М. И. Ж. аналит. химии, 21, 754 (1966).
679. Сотникова Н. П., Романович Л. С., Пейзулаев Ш. И., Карабаш А. Г. Тр. Комиссии по аналит. химии АН СССР, 12, 151 (1960).
680. Софронкова Н. Н. Сб. «Спектральные и химические методы анализа материалов». М., «Металлургия», 1964, с. 121.
681. Спектральный анализ в геологии и геохимии. М., «Наука», 1967.
682. Спектральный анализ полупроводниковых соединений и их компонентов. М., Цветметинформация, 1973.
683. Спектральный анализ чистых веществ. Л., «Химия», 1971.
684. Спиваков В. Я., Лебедев В. И., Шкинев В. М., Кривенкова Н. П.г Плотникова Т. С., Харламов И. П., Золотов Ю.А. Ж. аналит. химии, 31, 757 (1976).
685. Сплавы медно-цинковые. Методы определения сурьмы. ГОСТ 1652. 6-71.
686. Сплавы никелевые и медно-никелевые. Метод определения сурьмы. ГОСТ 6689. 15—75.
687. Справочные таблицы по рентгеновской спектроскопии. М., Изд-во АН СССР, 1953.
688. Старобинец Г. Л., Рахманъко Е. М. Сб. «Методы определения и анализа редких элементов III — VI групп периодической системы элементов Д. И. Менделеева». Свердловск, изд. Уральск, научн. центра АН СССР, 1975, с. 77.
689. Степин В. В., Поносов В. И., Новикова Е. В. Тр. ВНИИ станд. образцов и спектр, эталонов. 2, 14 (1965).
690. Степин В. В., Силаева Е.В., Плисс А. М., Курбатова В. И., Крючкова Л. М., Поносов В. И. Анализ черных металлов, сплавов и марганцевых руд. М., Металлургия, 1964, с. 297.
691. Столяров К. П., Григорьев Н. Н. Ж. аналит. химии, 14, 71 (1959).
692. Столяров К. П., Григорьев Н. Н. Сб. «Методы люминесцентного анализа неорганических ионов». Минск, Изд-во АН БССР, 1960, с. 32.
693. Стромберг А. Г. Ж. Всес. хим. об-ва им. Д. И. Менделеева, 8, 575 (1963).
694. Стромберг А. Г., Стромберг Э. А., Зав. лаб., 27, 3 (1961).
695. Студенская Л. С., Круглова М. И., Долгорукова Г. С., Вербицкая В. А. Тр. ВНИИ станд. образцов и спектр, эталонов, 2, 29 (1965).
696. Сурмий А. М., Аришкевич А. М., Усатенко Ю. И. Тр. Комиссии по аналит. химии АН СССР, 16, 144 (1968).
697. Сурмий А. М., Усатенко Ю. И., Аришкевич А. М. Зав. лаб., 33, 937 (1967).
194
698. Сурмий А. М., Усатенко Ю. И., Аришкевич А. М. Вопр. химии и хим. технологии. Респ. межвед. тематич. научн.-техн. об., вып. 12, 68 (1969).
698а. Сурьма. Методы анализа. ГОСТ 1367. О—1367. 7—5976.
699. Сухоручкина А. С., Усатенко Ю. И., Лозина Л. И., Горизонто-ва Т. Н. Вопр. химии и хим. технологии. Респ. межвед. темат. научн.-техн. сб., вып. 32, 44 (1974).
700. Сущенко В. Г., Шкал Н. И., Козьмина А. И. Сб. тр. Всес. н.-и. горнометаллург. ин-та цвет, мет., № 13, 36 (1968).
701. Совет экономической взаимопомощи. Рекомендации по стандартизации, PC 361—65. Концентраты цинковые флотационные. Методы химического анализа. Определение содержания сурьмы. М., изд. СЭВ, 1965.
702. Таганов К. И. Изв. АН СССР. Сер. физ., 4, 20 (1940).
703. Тайбаев В. К. Тр. Ин-та хим. наук АН КазССР, 32, 35 (1971).
704. Талипов Ш. Т., Джиянбаева Р.Х., Абдишева А. В. Ж. аналит. химии, 27, 1550 (1972).
705. Талипов Ш. Т., Джиянбаева Р. X., Абдишева А. В. Зав. лаб., 37, 387 (1971).
706. Талипов Ш. Т., Джиянбаева Р. X., Абдишева А. В. Узб. хим. ж., № 3, 9 (1971).
707. Тананаев Н. А. Капельный метод. Качественный анализ неорганических соединений капельным методом. М.—Л., Госхимиздат, 1954, с. 114.
708. Таниев А. Г., Рыбное В. В., Ризаева Ш. Сб. «Анализ и технология благородных металлов». М., «Металлургия», 1971, с. 278.
709. Тарасевич Н. И., Семененко К. А., Хлыстова А. Д. Методы спектрального и химико-спектрального анализа. Изд-во МГУ, 1973.
710. Тарасов К. И. Спектральные приборы. Л., «Машиностроение», 1968. 711. Тараян В. М., Арстамян Ж. М. Арм. хим. Ж., 27, 557 (1974).
712. Тараян В. М., Арстамян Ж. М., Мапучарян Л. М. Сб. «Успехи аналитической химии». М., «Наука», 1974, с. 188.
713. Тараян В. М., Овсепян Е. Н., Экимян М. Г. Уч. зап. Ереванского ун-та. Естеств. н., № 1 (119), 73 (1972).
714. Теньковцев В. В. Зав. лаб., 21, 525 (1955).
715. Типцова В. Г., Дворцан А. Г., Малкина Э. И. Ж. аналит. химии, 23, 1863 (1968).
716. Типцова-Яковлева В. Г., Дворцан А. Г. Зав. лаб., 37, 676 (1971).
717. Типцова-Яковлева В. Г., Дворцан А. Г., Семенова И. В. Ж. аналит. химии, 25, 686 (1970).
718. Трапицын Н. Ф. Тр. по мех., мат., физ. Мин-ва нар. образов. КиргССР, вып. 23, 82 (1967).
719. Турчинский М. Л., Нейман Л. 3. Зав. лаб., 30, 673 (1964).
720. Тюрин Р. С. Сб. «Исследования по химии координационных соединений и физико-химическим методам анализа». Кишинев «Штиинца», 1969, с. 93.
721. Тюрин Р. С., Мадан Л. Г. Изв. МолдССР. Сер. биол. и хим. н., № 3, 66 (1970).
722. Ули Сюэбао, 15, 324 (1959); РЖХим, 1960, 8956.
723. Усатенко Ю. П., Аришкевич О. М. Допов1д1 АН УССР, № 4, 504 (1962).
724. Усатенко Ю. П. Заморская Т. В. Зав. лаб., 30, 240 (1964).
725. Успехи полярографии с накоплением. Томск, изд-во ТГУ, 1973.
726. Устимов А. МТембер Г. А. Зав. лаб., 35, 1440 (1969).
727. Устимов А. МТембер Г. А., Чалков II. Я. Зав. лаб., 37, 660 (1971).
728. Устимов А. М., Чалков Н. Я. Зав. лаб., 35, 177 (1969).
729. Устинова В. II., Свечникова Е. А., Коровина А. Г., Василевская Л. В. Тр. ВНИИ стандарт, образцов и спектр, эталонов, вып. 4, 128 (1968).
730. Усубакунов М. УВлешинский С. В., Какеева М. К., Вуркуталие-ва К. Изв. АН КиргССР, № 2, 47 (1971).
у*
195
731. Утенка В. С. Изв. Томск, политехи, ин-та, вып. 164, 257 (1967).
732. Фабрикова Е.А., Баум Л, И., Бурынина А. Г., Марьина И.А.Т Мушко О. Л. Сб. «Спектральный анализ в геологии». М., «Недра», 1971, с. 236.
733. Федяшина А. Ф., Юделевич И. Г., Гиндин Л. М., Строкина Т. Г. Изв. СО АН СССР, № 3. Сер. хим. н., вып. 1, 83 (1966).
734. Файнберг С. 10. Зав. лаб., 6, 36 (1937).
735. Феръянчич Ф.А. Тр. Центр, н.-и. горноразвед. ин-та, вып. 28, 147 (1959).
736. Феръянчич Ф.А. Тр. Центр, н.-и. горноразвед. ин-та, вып. 70 188 (1967).
737. Фигелъсон Ю.А., Яковлева В. Г. Ж. аналит. химии, 27, 89 (1972).
738. Физические методы анализа. М., «Мир», 1967.
739. Филимонов Л. Я., Макулов Я. А., Захарова З.А. Тр. Комиссии по аналит. химии АН СССР, 12, 227 (1960).
740. Фиников В. Г. Уч. зап. Саратовского ун-та, 42, 97 (1955).
741. Фомина А. И., Агринская Я. А. Сб. «Новые методы химического анализа материалов». М., изд. МДНТП, 1971, с. 34.
742. Фомина А. И., Агринская Я. А., Золотов 10. А., Серякова И. В. Ж. аналит. химии, 26, 2376 (1971).
743. Фомина А. И., Агринская Я. А., Петрашенъ В. И. Тр. Новочеркасского политехи, ин-та, 220, 113 (1969).
744. Фомина О. А., Смирнов Я. С. Сб. «Материалы I Уральского совещания по спектроскопии, 1956». Свердловск, Металлургиздат, 1958, с. 68.
745. Фраткин 3. Г., Волохова М. И., Лоливанова Я. Г. Зав. лаб., 27, 846 (1961).
746. Фраткин 3. Г., Волохова М. Я., Лоливанова Я. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 479.
747. Фраткин 3. Г., Поливанова Н. Г. Зав. лаб., 26, 1372 (1960).
748. Фриш С. Э. Оптические спектры атомов. М.— Л., Физматгиз, 1963.
749. Фришберг А. А. Ж. прикл. спектроскоп., 8, 326 (1968).
750. Хадеев В. А., Жданов А. К., Речкина Л. Г. Узб. хим. ж., № 6, 28 (1960).
751. Хаму тина Я. А., Танцырева Л., Сергеева А., Полыницина М.. Тр. Сибирского технол. ин-та, вып. 36, 87 (1963).
752. Херд Д. Введение в химию гидридов. М., Госхимиздат, 1955.
753. Ховякова Р.Ф., Ломоносов С. А. Тр. Уральского политехи, ин-та, вып. 96, 142 (1960).
754. Храпай В. Л. Доклады межвузовской научной конференции по спектроскопии и спектральному анализу. Томск, изд-во ТГУ 1960, с. 70.
755. Хухия В. Л., Арешидзе Т. В. Зав. лаб., 21, 785 (1955).
756. Особо чистые германий, двуокись германия и терахлорид германия. Химико-спектральный метод определения примесей. ЦМТУ 05—51—67. М., изд. Гиредмет, 1967.
757. Чепик М. Я., Христофоров В. С. Лаборант-аналитик свинцово-цинковых заводов. М., «Металлургия», 1975.
758. Черкесов А. И., Бусев А. И. Ж. аналит. химии, 12, 268 (1957).
759. Чернихов IO. А., Черкашина Т. В. Зав. лаб., 24, 1057 (1958).
760. Чернихов 10. А., Черкашина Т. В. Зав. лаб., 25, 26 (1959).
761. Чернихов Ю. А. Черкашина Т.В., Ноткина М.А., Петрова Е.И.,
Меньшова Н. П., Луговская В. И., Горянская Г. П. Ж. аналит. химии, 21, 714 (1966).
762. Чернова А. И., Марунина Я. И., Мельчук И. А., Богатиков Б. Ф. Сб. «Химия и химическая технология». М., «Химия»,. 1972, с. 296.
763. Чикрызова Е. Г., Аронина И. В., Вассерштейн Ш.Е. Зав. лаб., 37, 18 (1971).
764. Чикрызова Е. Г., Мерян В. Т. Ж. аналит. химии, 27, 2052 (1972).
765. Чулкина Л. С., Синякова С. И. Сб. «Определение микропримесеп», № 2. М., изд. МДНТП, 1968, с. 25.
196
766. Чупахин М. С. Главин Г. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 104.
767. Чучалина Л. С. Химико-спектральный метод анализа галлия, сурьмы и золота высокой чистоты на примеси с экстракцией основы (5,|3'-ди-хлордиэтиловым эфиром. Кандидатская диссертация. Новосибирск,. Ин-т неорган. химии СО АН СССР, 1972.
768. Чучалина Л. С., Юделевич И. Г., Буянова Л. М., Старшинова Н. П., Валл Г. А. Ж. аналит. химии, 24, 905 (1969).
769. Цинк. Методы химического анализа. ГОСТ 11684—66 — 11689—66.
770. Шамаев В. И. Ж. аналит. химии, 24, 894 (1969).
771. Шамаев В. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 436.
772. Шамаев В. И. Там же, с. 464.
773. Шапиро М. Я. Зав. лаб., 8, 968 (1939).
774. Шапиро Г. Методы аналитической химии. Количественный анализ’ неорганических соединений, ч. II. М., «Химия», 1969, с. 1018.
775. Шаталова А. А. Зав. лаб., 36, 277 (1970).
776. Шитько П. П. Ж. аналит. химии, 12, 201 (1957).
777. Шафран И. Г., Певцов Г. А., Взорова И. Ф. Тр. Комиссии по аналит. химии АН СССР, 12, 317 (1960).
778. Швангирадзе Р. Р., Мозговая Т. А., Щетинина Э. В. Ж- аналит. химии,’ 17, 94 (1962).
779. Швангирадзе Р. Р., Симонова Н. В. Зав. лаб., 24, 881 (1958).
780. Шварц Д. М., Капорский Л. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 377.
781. Шварц Д. М., Капорский Л. Н., Нилова И. С. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, с. 107.
782. Шварц Д. М., Портнова В. В. Физ. сб. Львовского ун-та, вып. 4 (9),’ 493 (1958).
783. Шведов В. П., Шилкина М. И., Зиновьева В. К. Радиохимия, 1, 103’ (1959).
784. Шевелева О. В., Вуйдакова Л. Я. Уч. зап. ЦНИИ оловянной пром-сти, № 1, 57 (1964).
785. Шитова Е. И., Скрипник Н. А-. Научн. тр. ВНИИ люминофоров и особочистых в-в, вып. 10, 131 (1974).
786. Шишкина И. И., Сахарова Я. Г., Фрейдензон Л. И. Тр. Уральского НИИ черн. мет., вып. 9,55 (1970).
787. Шиянов А. Г. Производство сурьмы. М., Металлургиздат, 1961.
788. Шкадаускас Ю. С., Нарушкявичус Л. Р., Казлаускас Р. М. Научн. тр. вузов ЛитССР. Химия и хим. технол., 13, 53 (1971).
789. Штейнберг А. Н. Сб. «Методы анализа веществ высокой чистоты». М-, «Наука», 1965, с. 69.
790. Штейнберг А . Н. Тр. Ин-та металлургии АН СССР, 11, 243 (1962).
791. Штудлар Л., Яноушек И. Collect. Czechosl. Chem. Communs, 27, 1965 (1960).
792. Шубина С. Б., Шаевич А. Б., Басова Е. П., Зав. лаб., 26, 1364 (I960).’
793. Шубина С. Б., Шаевич А. Б., Басова Е. П. Сб. «Некоторые вопросы эмиссии и молекулярной спектроскопии». Красноярск, изд. Ин-та физики АН СССР, 1960, с. 82.
794. Щулепников М. Н., Шманенкова Г. И., Щелкова В. П., Яковлев Ю. В': Ж. аналит. химии, 28, 608 (1973).
795. Щербаков В. Г., Аникеева Н. П. Бюл. Ин-та металлокерамики и спец, сплавов АН УССР, № 6, 114 (1961).
796. Щербаков В. Г., Аникеева Н. П., Игнатова А. Я., Мигала Т. Г-Сб. тр. ВНИИ твердых сплавов, № 3, 56 (1960).
797. Щербов Д. П., Астафьева И. Н., Плотникова Р. Н. Зав. лаб., 39, 564 (1973).
798. Щербов Д. П., Матвеец М. А., Ахматова С. Д. Сб. «Исследования в области химических и физических методов анализа минерального сырья, вып. 2. Алма-Ата, «Наука», 1972, с. 94.
197
799. Экимян М. Г. Уч. зап. Ереван, ун-та. Естеств. н., № 2 (123), 60 (1973). 800. Юделевич И. Г., Артюхин П. И., Чучалина Л. С., Протопопова Н. П., Скребкова Л. М., Гильберт Э. Н., Пронин В. А. Ж. аналит. химии, 21, 1457 (1966).
801. ЮделевичИ. Г., Буянова Л. М. Методы анализа полупроводниковых соединений типа AnlBv и исходных веществ для их синтеза. Новосибирск, изд. Ин-та неорг. химии СО АН СССР, 1971.
802. Юделевич И. Г., Буянова Л. М., Протопопова Н. П., Юдина П. Г. Ж. аналит. химии, 24, 1759 (1969).
803. Юделевич И. Г., Буянова Л. М., Чучалина Л. С., Левин И. С., Шел-пакова И. Р., Протопопова II. И., Шубарова В. П., Архипова В. А., Старшинова Н. П. Тр. по химии и хим. технол. (Горький) вып. 3 (24), 89 (1969).
804. Юделевич И. Г., Буянова Л. М., Чучалина Л. С., Шелпакова И. Р., Ануфриева Г. В. Тр. по химии и хим. технол. (Горький), вып. 4 (35), 78 (1973).
805. Юделевич И. Г., Буянова Л. М., Шабурова В. П. Сб. «Промышленность химических реактивов и особо чистых веществ». М., изд. ИРЕА, 1970, с. 37.
806. Юделевич И. Г., Вершинина Ф. И., Сосновская Т. И. Сб. научн. тр. Всес. н.-и. горнометаллург. ин-та цвет, мет., № 9, 181 (1965).
807. ЮделевичИ. Г., Буянова Л. М., Шелпакова И. Р. Химико-спектральный анализ веществ высокой чистоты и полупроводниковых материалов. М., «Химия», 1978.
808. Юделевич И. Г., Виргинцев А. Н., Прохорова С. А. Ж. аналит. химии, 24, 1090 (1969).
809. Юделевич И. Г., Ковалева В. Г. Сб. тр. Всес. н.-и. горнометаллург. ин-та цвет, мет., № 5, 202 (1959).
810. Юделевич И. Г., Палатбеков Ф. Н., Ковалева В. Г. Зав. лаб., 25ц 305 (1959).
811. Юделевич И. Г., Сосновская Т. И., Вершинина Ф. И. Сб. «Спектроскопия. Методы и применение». М., «Наука», 1964, с. 87.
812. Юделевич И. Г., Шелпакова И. Р., Суконкина Т. А., Прохорова С. А. Зав. лаб., 34, 1074 (1968).
813. Юинг Г. В. Инструментальные методы химического анализа. М., Атомиздат, 1963.
814. Юкшинская Л. А. Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, с. 302.
815. Юкшинская Л. А., Солоп Е. В. Электронная техника. Научн.-техн. сб. «Управление качеством и стандартизация», вып. 5, 106 (1972).
816. Юлдашева К. Т., Абдусалямова Я., Пишанов П. X., Ганиев А. Г. Сб. «Радиохимические методы получения радиоактивных изотопов». Ташкент», «Фан», 1974, с. 54.
817. Юрист И. М. Зав. лаб., 32, 1050 (1966).
818. Юркин 11., Сильвестров И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 457.
819. Юсупов Н. Д., Шаврин А. М. Уч. зап. Пермского ун-та, № 159, 216 (1966).
820. Яворовский А. А., Галибей Л. И. Тр. Укр. НИИ полиграф пром-сти, 4, 104 (1956).
821. Яворовский А. А., Шиманский В. Н. Там же, с. 127.
822. Яковлев П. Я., Малинина Р. Д. Тр. ЦНИИ черн. металлург., вып. 24, 136 (1962).
823. Яковлев П. Я., Малинина Р. Д., Разумова Г. П., Дымова М. К. Сб. «Теория и практика полярографического анализа». Кишинев, Изд-во АН МолдССР, 1962, с. 198.
824. Яковлев Ю. В. Доклады советской делегации на Международной конференции по мирному использованию атомной энергии. Женева, 1955 г. Исследования в области геологии, химии и металлургии. М., Изд-во АН СССР, 1955, с. 90.
198
825. Яковлев Ю- В., Догадкин Н. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 162.
826. Яковлев Ю. В., Догадкин. Н. Я. Там же, с. 258.
827. Яковлев Ю- В., Догадкин Н. Н. Там же, с. 380. ’
828. Яковлев Ю. В., Догадкин Я. Я. Radiochemical Analysis, v. 1, 1965, р. 187.
829. Яковлев Ю- В., Догадкин Н. Н., Щулепников М. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, с. 198.
830. Яковлев Ю. В., Кулак А. И., Рябухин В. А., Рычков, Р. С. Мирное использование атомной энергии. II Международная конференция в Женеве, 1958 г. (Р/2023). М., Атомиздат, 1959.
831. Яковлев Ю- В., Щулепников М. Н. Методы определения и анализа редких элементов. М., Изд-во АН СССР, 1961, с. 436. '
832. Яковлева В. Г., Фигелъсон Ю. А . Ж. аналит. химии, 27, 89 (1972).
833. Янкаускас В. Ф., Пичугина В. МКаплин А. А. Зав. лаб., 35, 1431 (1969).
834. A bdel-Rassoul А . А ., Aly Я. FZakareia N .L. anal. Chem., 272, 27 (1974).
835. Adams F., Hoste J. Nucleonics, 22, 55 (1964).
836. Adams F,t Hoste J. Taianta, 9, 827 (1962).
837. Adams F.t Hoste J. Taianta, 10, 1093 (1963).
838. Adams F., Maenhaut W-, Hoste J. Anal. chim. acta, 61, 127 (1972).
839. Advances in Activation Analysis, v. 1. London — New York, Acad'. Press 1969 •
840. Ahrens L. N., Edge R. A., Brooks R. R. S. Air. Ind. Chem., 15, N 6, 102 (1961).
841. Akerman K-, Brafman M., Wozniak W., Falkowska W-, Adamuszek W-, Golkowska A. In Buch: Reinstoffe in Wissonschaft und Technik, Teil 1. Berlin, Academieverlag, 1966, S. 509.
842. Akki S. B., Khopkar S. M. Z. anal. Chem., 255, 130 (1971).
843. Alar A. Modern Methods for the Analysis of Aluminium Alloys. London, Chapman Hall, 1949.
844. A Ibu-Yaron A ., A miel S. J. Radioanal. Chem., 11, 123 (1972).
845. Alderisio A., Gauzzo F. Ricerca sci., 36, 1025 (1966).
846. Alexis R. Notes inform. CEA, N 1341, 50 (1970); РЖХим, 1971, 4Г173.
847. Alian A., Haggag A. J. Radioanal. Chem., 20, 429 (1974).
848. Alian A., Shabana R.t Sanad W-, Allam B., Khalita K. Taianta, 15, 262 (1968).
849. A Ifonsi B. Anal. chim. acta, 19, 276 (1958).
850. Alfonsi B. Ibid., p. 389.
851. Alfonsi B. Ibid., p. 569.
852. Alfonsi B. Anal. chim. acta, 25, 274 (1961).
853. Alfonsi B. Ibid., p. 374.
854. Alian A., Sanad W. Taianta, 14, 659 (1967).
855. Altotta G., Silvetti D. P. An. Asoc. quim. argent., 59, 63 (1971).
856. Alkemade С. T. Y-, Milatz J. M. W. J. Opt. Soc. Amer., 45, 583 (1955).
857. Allan J. E. Spectrochim. acta, 18, 605 (1962).
858. All-Sibai A., Fogg A. Analyst, 98, 732 (1973). ;
859. Alvarez B. Rev. Fac. cienc. Univ. Oviedo, 5, 5 (1964).
860. Aly H. F., Marei S., Zakaria N. Mikrochim. acta, N 1, 1 (1973).
861. Amano H. J. Jap. Inst. Metals, 23, 621 (1959).
862. Angermanu W., Bastius H. In Buch: Reinstoffe in Wissenschaft und Technik. Berlin, Academieverlag, 1963, S. 499.
863. Angina E. A., Billings G. K. Atomic Absorption Spectrometry in Geology. New York, Elsevier, 1972. '
864. Antia M., Arora R., Bhatnagar R. Analyst, 86, 202 (1961).
865. Aras N. K., Zoller W-, Gordon G., Lutz G. Anal. Chem., 45, 1481 (1973).
866. Archer V. S-, Twelves R. B. Taianta, 15, 47 (1968).
867. Arribas J., Alvarez B. Inform, quim. anal., 16, 174 (1962).
868. Arribas J., Alvarez В. Inform, quim. anal., 17, 145 (1963).
869. Arribas J., Alvarez B. Rev. Fac. cienc. Univ. Oviedo, 3, N 2, 5 (1962).
199
870. Asaoka H. Jap. Analyst, 23, 1049 (1974).
871. Asensi A. A ., Sanpedro P. An. Real. soc. esp. fis. у quim., B56, 289 (1960).
872. Asmus E., Brandt K. Z. anal. Chem., 179, 175 (1961).
873. Asmus E., Ziesche D. Z. anal. Chom., 210, 177 (1965).
874. Atalla L. T., Lima F. Publ. JAE, N 167, 1 (1968); РЖХим, 1969, 19Г142.
875. Athavale V. T., Burangey S. P., Dhaneshwar R. G. Analyst, 97, 302 (1972).
876. Athavale V. T., Dhaneshwar R. G., Mehta M., Sundaresan M. Analyst,
86, 399 (1961).
877. Aubonin G. Radiochim. acta, 1, 117 (1963).
878. Avni R. Spectrochim. acta, B23, 619 (1968).
879. Avni R., Boukobra A. Appl. Spectrosc., 23, 483 (1969).
880. Badhakrishnamurty C. J. Indian Chem. Soc., 51, 1045 (1974).
881. Baef G., DaalderA. Z. anal. Chem., 167, 430 (1959).
882. Baer W. K., Hodge E. S. Appl. Spectrosc., 14, 141 (1960).
883. BahrH., JedasC. Chem. anal. (PRL), 19, 895 (1974).
884. Bando S., Imahashi T. Jap. Analyst, 20, 49 (1971).
885. Bando S., Kishi H. Jap. Analyst, 19, 17 (1970).
886. Barakat M. L., Chehab S. K. Analyst, 90, 50 (1965).
887. Barnabas T., Barnabas J. Naturwissenschaften, 44, 61 (1957).
888. Barnett W. B., Kerber J. D. Atom. Absorpt. Newslett., 13, 56 (1974).
889. Basiiiska H., Rychcik W. Chem. anal. (PRL), 14, 563 (1969).
890. Basset J., Jones J. Analyst, 91, 176 (1966).
891. Batley H., Florence T. М.1. Electroanal. Chem., 55, 23 (1974).
892. Baumgartner F., Stark H., Schontag A. Z. anal. Chem., 197, 424 (1963).
893. Beard H. C., Lyerly L. A. Anal. Chem., 33, 1781 (1961).
894. Beckett E. G. Chem. News, 102, 101 (1910).
895. Beisltein F. K., Blase O. W. Bull. Acad. Petersb., 1, 204 (1890).
896. Belcher R., Bogdanski S. L., Townshend A. Anal. chim. acta, 67, 1 (1973).
897. Belcher R., Bogdanski S.L., Ghonaim S.A., Townshend A. Anal. chim. acta, 72, 183 (1974).
898. Belcher R., Gibbons D. J. Chem. Soc., N 4, 775 (1952).
899. Bern J., Rubel S. Chem. anal. (PRL), 17, 1259 (1972).
900. Berecka M. Chem. anal. (PRL), 7, 223 (1962).
901. Berg R., Fahrenkamp E. S. Z. anal. Chem., 112, 161 (1938).
902. Berg R., Roebling W. Berichte, 68, 403 (1935)..
903. Berger J. A ., Meyniel G., Petit J. J. Chromatogr., 29, 190 (1967).
904. Berka A., Dvorak V., Nemec U., Zyka J. Anal. chim. acta, 23, 380 (I960)..
905. Bayer J., Rienacker D. In Buch: Reinstoffe in Wissenschaft und Technik Berlin, Academieverlag, 1963, S. 409.
906. Bianchini A., Alladio L. Fonderia ital., 10, 439 (1961).
907. Bianchini A., Lanjranco G. Chim. e ind., 48, 816 (1966).
908. Bianchini A., Zanaroli L. Metallurgia ital., 52, 315 (1960).
909. Bianchini A., Zanaroli L. Metallurgia ital., 50, 349 (1958).
910. Biermann R. Erzmetall, 27, 428 (1974).
911. Bishay T. Z. J. Radioanal. Chem., 10, 51 (1972).
912. Bishay T. Z. J. Radioanal. Chem., 13, 87 (1973).
913. Bishop E., Jennings V. J. Taianta, 9, 581 (1962).
914. Bishop E., Jennings V. J. Ibid., p. 593.
915. Biswas S. D., Dey A. К. J. prakt. Chem., 21, 147 (1963).
916. В iswas S. D., Dey A . К. Z. anal. Chem., 192, 376 (1963).
917. Biswas S. D., Munshi K., Dey A. Chim. anal., 48, 208 (1966).
918. Blazejak-Ditges D., Klingeleers II. Z. anal. Chem., 248, 18 (1969).
919. Bloch L. Chem. Weekbl., 51, 548 (1955).
920. Bluavides В. An. Fac. farm, у bioqium., 7, 518 (1956).
921. Blank P., Racine J. Chim. anal., 36, 272 (1954).
922. Bock-Wertmann W., Schuize W. Proceeding International- Conference Modern Trends Activation Analysis. Texas College Station, 1965, p. 120.
923. Bode H. Z. anal. Chem., 132, 182 (1954).
924. Bode H. Z. anal. Chem., 144, 165 (1955).
925. Bode H., Aruswald W. Z. anal. Chem., 193, 415 (1963).
’200
926. Bode В., Bachman К. Z. anal. Chem., 229, 261 (1967).
927. Bode H., Bachman K. Z. anal. Chem., 241, 18 (1968).
928. Bogn&r J. Neh6zipari. miisz. egyet. kozl., 32, 124 (1972).
929. Bogn&r J. Nehezipari miisz. egyet. kozl., sorozat, [2], 22, 67 (1975).
930. Bogn&r U. Ibid., p. 121.
931. Bogn&r J. Ibid., p. 125.
932. Bogn&r J., Quittner P., Szabo E. Magy. kem. folyoirat., 73, 346 (1967);.
933. Bollen N. J., Essen M. J., Smith W. M. Anal. chim. acta, 38, 279 (1967).
934. Boltz G., WiedmannB., Kurella W. Metall, 10, 821 (1956).
935. Bothorel P. Dans le livre: «Nouveau traite de chimie minerale», t. XI. Paris, Masson et Cle, 1958, p. 502.
936. Boumans P. W- J. M. Theory of Spectrochemical Exication. New York, Plenum Press, 1967. ;
937. Boumans P. W- J. M., Maessen F. J. Bol. geol. у minero, 80, N 5, 47
(1969). :
938. BowdA. J., Burns D. T., Fogg A. G. Taianta, 16, 719 (1969).
939. Bozsai I. Magy. kem. folyoirat., 61, 305 (1955).
940. Brainina Kh. Z., Tchernyshova A. V. Taianta, 21, 287 (1974).
941. Braman B. S. Anal. Chem., 43, 1462 (1971).
942. Braman R. S., Dynako A. Anal. Chem., 40, 95 (1968).
943. Braman R. S., Justen L. L., Foreback С. C. Anal. Chem., 44, 2195 (1972)'.
944. Brandshaw G. Analyst, 88, 599 (1963).
945. Brooksbank W.. Ledicotte G., Dean J. Anal. Chem., 50, 1785 (1958).
946. Broun R. A., Swift E. B. J. Amer. Chem. Soc., 71, 2717 (1949).
947. Brune D., Landstrom 0. Radiochim. acta, 5, 228 (1966).
948. Brunelle R. L., Boffman С. M., Snow К. B. Anal. Chem., 53, 470 (1970).
949. Brunfelt A. 0., Steinnes E. Anal. chim. acta, 48, 13 (1969).
950. Buchanan J. D., Fleishman D., Guinn V. P., Rush R. R. WESKON Techh. Pap., N 3, 13 (1962); РЖХим, 1964, 9Г149.
951. Buciewicz J., Wozniacka D., Wozniacki J. Pr. Inst, odlew., 9, N 4, 238 (1961).
952. Burgess C., Fogg A. G., Burrs D. T. Analyst, 98, 605 (1973).
953. Burgess A. E., Ottaway J. M. Analyst, 97, 357 (1972).
954. Burke К. E. Ibid., p. 19.
955. Burke К. E. Anal. Chem., 42, 1536 (1970).
956. Burke R. W., Menis O. Anal. Chem., 38, 1719 (1966).
957. BurrielM. F., Alvarez В. C. Inform, quim. anal., 15, 151 (1961).
958. BurrielM. F., Alvarez В. C. Inform, quim. anal., 16, 68 (1962).
959. Burriel-Marti F., Garate M. E. 31е Congress internationale chimie in-dustrielle. Liege, 1958, v. 1. Bruxelles, 1959, p. 81.
960. Buscarons F., Alsina J. Inform, quim. anal., 19, 12 (1965).
961. Buscarons F., Alsina J. An. Real. soc. esp. fis. у quim., B59, 101 (1963).
962. CaldasA. Anal. chim. acta, 45, 532 (1969).
963. Camozzo S. Chimica, 31, 415 (1955).
964. Camunas A., Berrero J. 20е Congress Groupement d’Avancement de Me-tode Spectroscopique, 1957. Paris, Meru, 1958, p. 133.
965. Can. Chem. Process., 52, N 12, 72 (1968).
966. Canio V.., Petrovic S., Bern A. Z. anal. Chem., 213, 251 (1965).
967. Cardwell T. J., Basouri F. G., MacConnell B.B., MacGeeR. J. Austral.
J. Chem., 21, 359 (1968).
968. Cerrai E., Testa C. Energ. nucl., 8, 510 (1961).
969. Chakravarty T. В. Sci. and Cult., 28, 392 (1972).
970. Chakravarty T. B., De A. K.Z. anal. Chem., 242, 152 (1968).
971. Champ W. В. Pap. Geol. Surv Can., N 1, part B, 2 (1970).
972. Chandola L. C., Koranjikar B. P. Govt India Atom. Energy Commis. (Rept), N-BARC-495 (1970); РЖХим, 1971, 5Г211.
973. Chatterjee R., Day A. K. Microchem. J., 12, 151 (1967).
974. Chau У. K., Lum-Shue-Chan K. Anal. chim. acta, 48, 434 (1969).
975. Chetkowski W. Pr. Inst, huth., 19, 109 (1966).
976. Chira A В inkits В., Voiculescu A .Rev. chim. (RPR), 15, 352 (1964).
201
977. Christopher D. H., West T. S. Taianta, 13, 507 (1966).
978. Chromy V., Sommer L. Taianta, 14, 393 (1967).
979. Chu R. C., Barron G. P., Baumgardner P. A. W. Anal. Chem., 44, 1476 (1972).
980. Chwastowska Skorko-Trzybulowa Z. Chem. anal. (PRL), 9, 123 (1964).
981. Cieszewski A., Lukaszewski Z. Chem. anal. (PRL), 21, 687 (1976).
981a. Cihalik J. Chem. listy, 49, 1167 (1955).
982. Cinffolotti L., Colombo U., Malvano R., Sironi C. Advances Organical Geochemistry Oxford—London—New York—Paris, Pergamon Press, 1964, p. 433.
983. Cinffolotti L., Fasolo G. B., Malvano R. Energ. nucl., 10, 381 (1963).
984. Ciuhandu G. Rev. chim. (RPR), 11, 530 (1960).
985. Clarke F. W. Amer. J. Sci., 49, 48 (1870).
986. Clarke G. Analyst, 53, 373 (1928).
987. Cleyrerque C., Deschamps N. J. Radioanal. Chem., 17, 139 (1973).
988. Cluley H., Proffit P. Analyst, 85, 815 (1960).
989. Cojocaru V., Cristu M., Dorcioman D., Badanoiu M. Stud, si cere. fiz. Acad. RPR, 11, 447 (1960).
990. Colanovic K. Colloque Spectroscopic International 16. Heidelberg, 1971. London, Soc. Anal. Chem., 1971, p. 374.
991. Colombo U., Sironi G., Fasolo G., Malvano R. Anal. Chem., 36, 802 (1964).
992. Comptes rendus XXV Conference Cortina d’Ampezzo, 30 June — 8 July, 1969. IUPAC, 1970.
993. Constantinescu D., Baiulescu G., Oteleanu R. Stud, si cere. chim. Acad. RPR, 6, 438 (1958).
994. Coppins W., Price J. W. Metallurgia, 46, 52 (1952).
995. Coppins W., Price J. W. Metallurgia, 53, 183 (1956).
996. Corbett J. A. Metallurgia, 65, 43 (1962).
997. Cordis V. Rev. chim. (RPR), 15, 35 (1964).
998. Cronzoulon P. Chim. anal., 50, 483 (1968).
‘999. Csada J., Erdeyul-Schneer A., Rausch H. Kozp. fiz. kut. inet. kozl., 16, 219 (1968).
1000. Csajka M-, Ordbgh M. Kozp. fiz. kut. int. kozl., 12, 335 (1964).
1001. Csajka M., Ordbgh M., Szabo E. Acta chim. Acad. Sci. hung., 51, 161 (1967).
1002. Cuppers M. Ann. chim., 9, 509 (1964).
1003. Cyrankowska M. Chem. anal. (PRL), 6, 649 (1961).
1004. Cyrankowska M. Chem: anal. (PRL), 15, 209 (1970).
1005. Cyrankowska M., Downarowicz J. Chem. anal. (PRL), 10, 67 (1965).
1006. Cyrankowska M., Downarowicz J. Chem. anal. (PRL), 12, 137 (1967).
1007. Czakow J. Ibid., p. 181.
1008. Czakow J., Zawadzki Z. Prz. elektron., 4, 93 (1963).
1009. Czerepko K., Siekorska-Tomicka H. Chem. anal. (PRL), 10, 245 (1966).
1010. Czichoii H. Poligrafika, 13, 8 (19611.
1011. Dalton E. F., Malonowski A. J. Atom. Absorpt. News lett., 10, 92 (1971).
1012. Dams R., Hoste J. Anal. chim. acta, 41, 205 (1968).
1013. Dartlet J. C., Monkman J. L. J. Assoc. Offic. Agric. Chem., 47, 630 (1964).
1014. Dauksas K., Naruskevicius L. Mokslo darbai. Vilniaus univ. Matem., fiz. ir chem. mokslu ser., 7, 161 (1957).
1015. Davas R., Hoste J. Acta chim. Acad. Sci. hung., 50, 111 (1966).
1016. Dowson J., Magee R. Mikrochim. acta, N 3, 325 (1958).
1017. Daynall R. M., Thompson K. S., West T- C. Taianta, 14, 551 (1967).
1018. Daynall R. M., Thompson K. S., West T. C. Ibid., p. 1151.
1019. De A. K., Chakrabarty T. Indian J. Chem., 7, 180 (1969).
1020. De Bruyne P. Fermentatio, N 1, 28 (1964).
1021. De Bruyne P-, Dept F. Metallurgie (Belg.), 3, N 4, 79 (1962).
1022. De Bruyne P., Hoste J. Bull. Soc. chim. belg., 70, 221 (1961).
202
1023. Dean J. A.,Adkins J. E. Analyst, 91, 709 (1966).
1024. Dean J. A., Rains T. C. Flame Emission and Atomic Absorption Spectrometry, v., 1, 2. New York, Dekker, 1971.
1025. Dean J. A., Reynolds S. Anal. chim. acta, 11, 390 (1954).
1026. Denig R., Trautmann W., Hermann G. J. Radioanal. Chem., 5, 223 (1970).
1027. Denig R., Trautmann W., Hermann G. J. Radioanal. Chem., 6, 331 (1970).
1028. Deshmukh G. S., Eshwar M. C. Curr. Sci. (India), 29, 388 (1960).
1029. Deshmukh G. S., Naik V. S. Z. anal. Chem., 263, 206 (1973).
1030. Deur-Siffar D., Bauman E. Kern, u ind. 11, 695 (1962).
1031. Devanathan M. A. V., Fernando Q., Peries P. Anal. chim. acta, 16, 292 (1957).
1032. Dhoud P. V., Khopkar S. M. Separ. Sci., 8, 379 (1974).
1033. Dibbs H. P., MacMaster С. H. Chem. and Ind., N 5, 217 (1965).
1034. Diebolt J. Bull, inform. ATEN, N 69, Suppl., 7 (1968).
1035. Doge H. G., Ehrlich G. Reinstoffe in Wissenschaft und Technik. Berlin, Academieverlag, 1965, S. 481.
1036. Dolezal J., Beran P. Chem. listy, 50, 1757 (1956).
1037. Dolezal J., Janacek K. Collect. Czech. Chem. Communs, 25, 885 (1960).
1038. Downarowicz J., Zagorski Z. Chem. anal. (PRL), 4, 445 (1955).
1039. Dozinel С. M. Z. anal. Chem., 157, 401 (1957).
1040. Draskovic R., Radoslavljevic R., Takvac T., Zaric M. Nauc.-tehn. pregl. INTDI, 21, N 9, 21 (1971); РЖХим, 1972, 14Г181.
1041. Dunlap L. B., Shults W. D. Anal. Chem., 34, 499 (1962).
1042. Dunn E. I., Gestaro I. P., Weisenstein W. Proc. Amer. Soc. Test. Mater., 59, 573 (1960).
1043. Dym A. Analyst, 88, 232 (1963).
1044. Edwards F. C., Voigt A. F. Anal. Chem., 25, 351 (1953).
1045. Elkind A., Gayer K., Boltz D. Ibid., p. 1744.
1046. Elvers H. Lab-Prax., 15, 21 (1963).
1047. Elwell W. T., Gidley J. A. F. Atomic Absorption Spectrophotometry, New York, Pergamon Press, 1966.
1048. Elwell W. T., Peake D. M. Analyst, 96, 465 (1971).
1049. Erdey L., Buzas L, Acta chim. Acad. Sci. hung., 6, 93 (1955).
1050. Erdey L., Buzas L., Ibid. p. 77.
1051. Extraction Chromatography. Budapest, Akad. Kiado, 1975.
1052. Facsko G., Golumbioschi F. Rev. roum. chim., 11, 207 (1966).
1053. Feigl F. Z. anal. Chem., 64, 41 (1924).
1054. Feigl F., Chan F. L. Chem.-Analyst, 56, 13 (1967).
1055. Feigl F., Chan F. L. Ibid., p. 14.
1056. Feigl F., Vinzenz A. Spot Tests in Inorganic Analysis. New York, Elsevier, 1972.
1057. Fegriwe E. Z. Z. anal. Chem., 70, 400 (1927).
1058. Fernandez F. J. Atom. Absorpt. Newslett., 12, 93 (1973).
1059. Fernandez F. J., Manning D. S. Atom. Absorpt Newslett., 10, 86 (1971).
1060. Fernando P. Anal. chim. acta, 12, 432 (1955).
1061. Filb^ R. N., Haller W. A., Shan K. R. J. Radioanal. Chem., 5, 277 (1970).
1062. Filler T. D. Anal. Chem., 43, 725 (1971).
Ю63. Fillmore C. L., Eckert A. C., Scholle J. V. Appl. Spectrosc., 23, 502 (1969).
1064. Flaschka H., Jakobljevich H. Anal. chim. acta, 4, 247 (1950).
1065. Florence T. M. J. Electroanal. Chem., 27, 273 (1970).
1066. Fogg A. G., Al-Sibaai A. A., Burgess C. Anal. Lett., 8, 129 (1975).
1067. Fogg A. G., Yillings J., Marriott D. R., Burns D. Analyst, 94, 768 (1969).
1068. Foglino M. Metallurgia ital., 52, 355 (1960).
1069. Foster P., Bozon H., Molins R. Analysis, 1, 64 (1972).
1070. Fournet L.. Albert Ph. C. r. Acad, sci., 254, 1076 (1962).
203
1071. Frank C. W., Meloan С. E. Anal. Chem., 39, 379 (1967).
1072. Freeman G. A., Bampacek C., Evans L. G. Mine J. Metals, 13, 370 (1961).
1073. Fujinaga T., Koyama M., Izutsu K., Himeno S., Kawashima M. J. Chem. Soc. Jap. Chem. and Ing. Chem., N 8, 1489 (1974).
1074. Fukamouchi H., Enohara R., Uehara M., Terui S., Tokimoto Y. Jap. Analyst, 8, 353 (1959).
1075. Fulton A., Thompson К. C-, West T. S. Anal. chim. acta, 51, 373 (1970).
1076. Fungairino L. V. An. Real. acad. farm., 16, 209 (1950).
1077. Gagliardi E., Durst L. Monatsh. Chem., 102, 308 (1971).
1078. Gagliardi E., Musil A., Reischel K. Z. anal. Chem., 137, 252 (1952).
1079. Gagliardi E., Pilz W. Z. anal. Chem., 136, 344 (1952).
1080. Gagliardi E., Tiimmler P. Talanta, 17, 93 (1970).
1081. Gagliardi E., Wdss H. P. Anal. chim. acta, 48, 107 (1969).
1082. Gallazzi C., Maneschi S. Metallnrgia ital., 58, 311 (1966).
1083. Galliford D. J., Yardley J. T. Analyst, 88, 653 (1963).
1084. Ganapathy J. S., Kadam B., Venkateswarlu Ch. Indian. J. Chem., 11, 385 (1973).
1085. Ganapathy J. Parasurama J. H., Venkateswarlu Ch. ISI Bull., 24, 449 (1972).
1086. Gann W. Z. anal. Chem., 221, 254 (1966).
1087. Garate M., Astudillo M., Burriel-Marti F. An. Real. soc. esp. fis. у quim., B56, 775 (1960).
1088. Garate M. E., Garate M. T. Chim. anal., 40, 77 (1858).
1089. Garcia Ch. A. Rev. Fac. farm, у bioquim., 29, 22 (1967).
1090. Garg B. S., Trikna К. C., Singh R. P. Talanta, 16, 462 (1969).
1091. Garnier H. Courr. norm., 33, 862 (1966).
1092. Gebauhr W. Radiochim. acta, 5, 8 (1966).
1093. Gebauhr W., Martin J. Int. J. Appl. Radiat. and Isotop., 4, 173 (1959).
1094. Gerbatsch R., Artus G. Monatsber. Dtsch. Akad. Wiss. Berlin, 5, 489 (1963).
1095. Gerbatsch R., Artus G. Z. anal. Chem., 233, 81 (1966).
1096. Geyer R., Geissler M. Z. anal. Chem., 187, 251 (1962).
1097. Geyer R., Geissler M. Z. anal. Chem., 201, 1 (1964).
1098. Ghe A. M. Ann. chim. (Ital.), 50, 1321 (1960). .
1099. Ghonaim S. A. Proc. Soc. Anal. Chem., 11, 138 (1974).
1100. Ghose A. K., Dey A. K. Analyst, 95, 698 (1970).
1101. Ghosh M. K., Debnath P. C., Dasgupta S., Kar B. Indian Ind., 9, N 12, 29 (1965).
1102. Gibbons D. Ind. Chem., 29, 183 (1953).
1103. Gibbons D. Ind. Chem., 30, 185 (1954).
1104. Gijbels R., Dams R. Anal. chim. acta, 62, 191 (1972).
1105. Gilbert T., Hume D. Anal. chim. acta, 65, 451 (1973).
1106. Gillis J., Hoste J., Claeys A. Anal. chim. acta, 1, 291 (1947).
1107. Gills T. E., Marlow W. F., Thompson B. A. Anal. Chem., 42, 1831 (1970).
1108. Glabisz U. Chem. anal. (PRL), 9, 139 (1964).
1109. Gobrecht H., Tausend A., Bratter P., Witters G. Solid State Communs, 4, 311 (1966).
1110. Gohda S. Bull. Chem. Soc. Jap., 48, 1213 (1975).
1111. Goleb J. A., Midkiff D. A. Appl. Spectrosc., 29, 44 (1975).
1112. Gomez J. O., Romero J. G. Tec. met. Barcelona, 4, 461 (1948).
1113. Gomiscek S. Nova proiz., 11, 273 (I960).
1114. Gopala R. G., Gandikota M., Viswanath S. G. Anal. chim. acta, 78, 501 (1975).
1115. Gorczynska K., Gluzinska M., Clecierska-Stoklosa D. Chem. anal. (PRL), 14, 591 (1969).
1116. Goshima F., Ishino F. Jap. Analyst, 24, 480 (1975).
1117. Gostkowska B., Ekiert H. Prz. elektron., 7, 229 (1966).
1118. Goto H. J. Chem. Soc. Jap., 55, 326 (1934).
204
1119. Goto H., Kakita Y. J. Chem. Soc. Jap., Pure Chem. Sec. 77, 739 (1956).
1120. Goto H-, Kakita Y. J. Chem. Soc. Jap., Pure Chem. Sec., 82, 1212 (1961).
1121. Goto H., Kakita Y. Sci. Repts Res. Inst. Tohoku Univ., All, 1 (1959)»
1122. Gouin J. U., Holt J. L., Miller R. E. Anal. Chem., 44, 1042 (1972).
1123. Goulden P. D., Brooksbank P. Anal. Chem., 46, 1431 (1974).
1124. Greendale A. E.,Love D. Anal. Chem., 35, 632 (1963).
1125. Greenhalgh R., Riley J. P. Anal. chim. acta, 27, 305 (1962).
1126. Griffiths J. M., Whitehead H. R. NPL Rep. Chem., N 16 (1972); Anal. Abstr., 25, 3474 (1972).
1127. Grimanis A., Hadzistelios J. Anal. chim. acta, 41, 15 (1968).
1128. Gruber E., Hecht F., Sorantin H. Mikrochim. acta, N 5, 794 (1971).
1129. Gruber E., Sorantin H. Mikrochim. acta, N 2, 262 (1971).
1130. Gruverman J. J., Henninger W. A. Anal. Chem., 34, 1680 (1962).
1131. Guinn V. P., Lukens H. R. Trace Analysis. Physical Analysis, New York, Interscience Publishers, 1965, p. 345.
1132. Guyon J. C., Matulis R. M. Chem.-Analyst., 56 22 (1967).
1133. Gyory S. L. anal. Chem., 32, 415 (1893).
1134. Haftka E. J. Mikrochim. acta, N 2, 331 (1965).
1135. Haight G. Anal. Chem., 26, 593 (1954).
1136. Hamaguchi H., Nakai T., Endo T. J. Chem. Soc. Jap., Pure Chem. Sec., 82, 1485 (1961).
1137. Handley T. H. Anal. Chem., 35, 991 (1963).
1138. Hara S. Bull. Osaka Ind. Res. Inst., 10, 41 (1959).
1139. Hasek Z. Hutn. listy, 17, 733 (1962).
1140. Headridge J. B. Lab. Pract., 23, 5 (1974).
1141. Headridge J. B., Smith D. R. Lab. Pract., 20, 312 (1971).
1142. Hegemann F., Kostyra H. Metall, 9, 849 (1955).
1143. Hegemann F., Sybel C. Ibid., p. 91.
1144. Heinen K., Larrabee G. Anal. Chem., 38, 1853 (1966).
1145. Henz F. Z. anorg. Chem., 37, 16 (1903).
1146. Heyndryckx M. P. Publ. groupem. avancem. meth, spectrogr., N 4, 373 (1960).
1147. Hildon M. A., Sully G. R. Anal. chim. acta, 53, 192 (1971).
1148. Hirokawa K. J. Jap. Inst. Metals, 24, 559 (1960).
1149. Hirokawa K. J. Sci. Repts Res. Inst. Tohoku Univ., A13, 271 (1961)»
1150. Hirokawa K. J. Ibid., p. 279.
1151. Hofacker A. Liebigs Ann., 107, 6 (1958).
1152. Holak W. Anal. Chem., 41, 1712 (1969).
1153. Holler A. C. Anal. Chem., 19, 353 (1947).
1154. Holynska B., Panek M. Magy. kem. folyoirat, 68, 511 (1962).
1155. Holynska B., Panek M. Chem. anal. (PRL), 7, 749 (1962).
1156. Honrigan H., Robinson J. Anal. chim. acta, 10, 281 (1954).
1157. Hoste J. Chem. Weekbl., 58, 106 (1962).
1158. Hoste J., Soete D. Z. anal. Chem., 245, 221 (1969).
1159. Hoste J., Soete D. Anal. chim. acta, 28, 281 (1963).
1160. Hranisavlijevic-Jakovliec M., Pejkovic-Tadic J. Mikrochim. acta, N 5— 6, 936 (1965).
1161. Hubbard D. P. Annual Reports on Analytical Atomic Spectroscopy, 1972,'v. 2. London, Soc. Anal. Chem., 1973.
1162. Hubbard D. P., Vernon F. Anal. Lett., 2, 657 (1969).
1163. Hubicki W., Jusiak S. Ann. Univ» M. Curie-Sklodowska, 1958 (1960), S. 97; РЖХим, 1962, 1Д112.
1164. Hughes H., Tunney A. A. US NTIS, Publ. Rept. N 226975/IGA, 1973; Chem. Abstr., 80, 152394.
1165. Hulanicki A., Glab S. Chem. anal. (PRL), 15, 1089 (1970).
1166. Hwang J. Y., Sandonato L. M. Anal. Chem., 42, 744 (1970).
1167. Hyman H. M. Appl. Spectrosc., 16, 129 (1962).
1168. Imai H. J. Chem. Soc. Jap., Pure Chem. Sec., 80, 761 (1959).
1169. Imai H. Jap. Analyst, 11, 806 (1962).
205
1170. Ishihara I., Koga M. Ibid., p. 246.
1171. Ishihara I., Koga M., Komuro H. Ibid., p. 1566.
1172. Ishimori T., Watanabe K., Nakamura E. Bull. Chem. Soc. Jap., 33, 636 (1960).
1173. Issa I., El Sherif J. Chem.-Analyst, 45, 78 (1956).
1174. Itsuki K., Nagao M. Jap. Analyst, 8, 824 (1959).
1175. Itsuki K., Nishino K. Jap. Analyst, 9, 886 (1960).
1176. Itsuki K., Suzuki F., Nishino K. J. Jap. Inst. Metals, 24, 718 (1961).
1177. Iwasaki K., Tanaka K., Takagi N. Jap. Analyst, 23, 1179 (1974).
1178. Izquierdo A., Lacort G. Inform, quim. anal., 24, 208 (1970).
1179. Jablonski W. L., Watson C. A. Analyst, 95, 131 (1970).
1180. Jacobsen E., Rojahn T. Anal. chim. acta, 54, 261 (1971).
1181. Janardhan P. B., Paul A. Indian J. Chem., 7, 68 (1969).
1182. Janda J., Schausberger J., Schroll E. Mikrochim. acta, N 1, 122 (1963).
1183. Jankovsky J. Hutn. listy, 20, 205 (1965).
1184. Jankovsky J. Z. anal. Chem., 201, 325 (1964).
1185. Janousek I. Hutn. listy, 19, 276 (1964).
1186. Jardine M. A. Pap. Geol. Surv. Can., N 29, 11 (1963).
1187. Jankovsky J. Acta geol. et geogr. Univ. com. Geol. N 15, 247 (1968).
1188. Jaycox E. K. Proc. Amer. Soc. Test. Mater. (1962), 62. 529 (1963).
1189. Jaycox E. K. Ibid., p. 537.
1190. Jeffery P. G. Chemical Methods of Rock Analysis, 2-nd ed. New York, Pergamon Press, 1975.
1191. Jenlk J. Collect. Czech. Chem. Communs, 23, 1056 (1958).
1192. Jennings V. J. Analyst, 87, 548 (1962).
1193. Jimenez-Seco J., Buriel F., Sistiaga J. An. Real. soc. esp. fis. у quim., B58, 669 (1962).
1194. Jirkowski R., Kressner R. Chem. listy, 62, 1335 (1968).
1195. Johar G. S. Curr. Sci. (India), 38, 543 (1969).
1196. Johnson E. B., Dennis L. M. J. Amer. Chem. Soc., 47, 790 (1925).
1197. Johri K. N. Singh K. Mikrochim. acta, N 1, 1 (1970).
1198. Jokl V., Majer J. Acta geol. et geogr. Univ. com. Geol., N 15, 77 (1968).
1199. Jokl Y., Undeutsch M., Majer J. J. Chromatogr., 26, 208 (1967).
1200. Joshi B. D., Bangia T. R., Dalvi A. Govt India Atom. Energy Conimtis. (Rept), N BAPC-522, 1970.
1201. Kadame P. Magy. kem. lapja, 15, 330 (1960).
1202. Kambara T., Suemitsu T. Radioisotopes, 21, 423 (1972).
1203. Kamemoto Y., Yamagushi S. Bull. Chem. Soc. Jap., 36, 132 (1963).
1204. Kamemoto Y., Yamagushi S. J. Chem. Soc. Jap., Pure Chem. Sec., 82 1653 (1961).
1205. Kamemoto Y., Yamagushi S. J. Chem. Soc. Jap., Pure Chem. Sec., 83, 887 (1962).
1206. Kamemoto Y., Yamagushi S. J. Chem. Soc. Jap., Pure Chem. Sec., 84, 823 (1963).
1207. Kamemoto Y., Yamagushi S. J. Chem. Soc. Jap., Pure Chem. Sec., 85, 572 (1963).
1208. Kammori O., Takahari T., Bando S. Jap. Analyst, 16, 826 (1967).
1209. Kan Kwok-Tai. Anal. Lett., 6, 603 (1973).
1210. Kashima D., Kubota M. J. Spectrosc. Soc. Jap., 16, 133 (1967).
1211. Katayama S., Kim Kjon Hun. Jap. Analyst, 20, 29 (1971).
1212. Katigar S. Naturwissenschaften, 49, 325 (1962).
1213. Kato K., Murano M. Jap. Analyst. 22, 312 (1973).
1214. Kawabuchi K. J. Chem. Soc. Jap., Pure Chem. Sec., 87, 262 (1966).
1215. Kawabuchi K., Kaya M., Ouchi Y. Jap. Analyst, 15, 543 (1966).
1216. Kawahaka M., Mochizuki H., Kaziyama R. Irikura K. Jap. Analyst, 11, 973 (1962).
1217. Keenan J. A., Larabee G. B. Chem. Instrum., 3, 125 (1971).
1218. Kemka R., Klucik J. Pracovni lekaf, 12, 74 (I960).
1219. Kemula Y., Gaius Z. Bull. Acad. Polon. Nauk., Ser. chem., 10, 7313 (1959).
206
1220. Kendall R. Anal. Lett., 5, 867 (1972).
1221. Kerr G. 0., Gregory G. R. E. C. Analyst, 94, 1036 (1969).
1222. Keunkmann II. Metall, 14, 905 (I960).
1223. Khalifa H. El-Rarbary J. Mikrochcm. J., 3, 137 (1968).
1224. Khatun S., Khundkar M. J. Indian Cliem. Soc., 34, 114 (1957).
1225. Khorasanl S. S. Anal. chim. acta, 24, 316 (1961).
1226. Khorasanl S. S. Pakistan J. Sci. and Ind. Res., 8, 142 (1965).
1227. Khorasanl S. S. Pakistan J. Sci. and Ind. Res., 6, 233 (1963).
1228. Khorasanl S. S., Hakim Q. A. Pakistan J. Sci. and Ind. Res., 9, 220
(1966).
1229. Khorasanl S. S., Khundkar M. II. Anal. chim. acta, 25, 292 (1961).
1230. Khorasanl S. 5., Khundkar M. II. Anal. chim. acta, 21, 24 (1959).
1231. Khorasanl S. S., Khundkar M. H. Ibid., p. 406.
1232. Kiboki M. Jap. Analyst, 10, 19 (1961).
1233. Kidman L., Waite C. R. Metallurgia, 66, 143 (1962).
1234. Kiesl W. Z. anal. Chem., 227, 13 (1967).
1235. Kiesl W., Hecht F., Sorentin H. Mikrochim. acta, N 6, 954 (1964).
1236. Kiesl W., Seitner H., Kluger F., Hecht F. Monatsh. Chem., 98, 972 (1967).
1237. Kielezewski W., Thiel J. Chem. anal. (PRL), 10, 271 (1965).
1238. Killick R. A., Morris D. F. Taianta, 9, 879 (1962).
1239. Kim J.. Hoste J. Anal. chim. acta, 33, 449 (1965).
1240. King H. G., Neff С. M. Appl. Spectrosc., 17, 51 (1963).
1241. Kinoshita Y. J. Pharm. Soc. Jap., 78, 315 (1958).
1242. Kirkbright G. F., Saw C. G., Thopmson J. V-, West T. S., Taianta, 16, 1081 (1969).
1243. Klement R., Kuhn A. Z. anal. Chem., 152, 146 (1956).
1244. Klinkmann A. Metall, 14, 1005 (1960).
1245. Klinkmann A. Metall, 18, 821 (1964).
1246. Knipe F. G. Spectrochim. acta, 1, 49 (1959).
1247. Kobayashi M., Nagatsuka S., Sawai T. Radioisotopes, 13, 26 (1964).
1248. Kobayashi S., Otobe S. Jap. Analyst, 21, 1648 (1972).
1249. Koch O. G. Z. anal. Chem., 265, 29 (1973).
1250. Koenig P., Schmitz K. Arch. Eisenhiittenw., 44, 469 (1973).
1251. Kolivoda D., Sichra V. Anal. chim. acta, 59, 477 (1972).
1252. Konopik N., Szlaczka K. Osterr. chem. Ztg, 52, 205 (1951).
1253. Kosaric N. Kem. i ind., 10, 324 (1961).
1254. Kotrbova M. Hutn. listy, 15, 802 (1960).
1255. Kovacs E., Guyer H. Chimia, 13, 164 (1959).
1256. Kovacs E., Guyer H. Metallurgia ital., 56, 365 (1964).
1257. Kovacs E., Guyer H. Z. anal. Chem., 186, 267 (1962).
1258. Kovacs E., Guyer H. Z. anal. Chem., 208, 255 (1965).
1259. Kowalczyk M. Chem. anal. (PRL), 9, 331 (1964).
1260. Krasnobaeva N., Alexandrov S. Acta chim. Acad. Sci. hung., 64, 1 (1970).
1261. Kraus M., Amiel S. Isr. Atom. Energy Commis. (Repts), №1168, 102 (1968).
1262. Krishna T. .S’., Dhall P. K., Rao A. S. Govt. India Atom. Energy Commis. (Ropts), № AEET (met.), 5 (1964).
1263. Krivaii F. Chem. prum., 15, 39 (1965).
1264. Kroupa M., Hutn. listy, 19, 665 (1964).
1265. Kucharzewski R. Reinstoffe in Wissenschaft und Technik. Berlin, Aca-demieverlag, 1963, S. 403.
1266. Kuhn M. Chim. anal., 40, 11 (1958).
1267. Kuzma Z. Prz. elektron., 5, 9 (1964).
1268. Laffolie H. Arch. Eisenhiittenw., 38, 535 (1967).
1269. Lahl W., Fischer W. Chem. anal. (PRL), 17, 669 (1972).
1270. Lamb J. F., Prussin S. G., Harris J. 4., Hollan J. M. Anal. Chem., 38, 813 (1966).
207
1271. Lambrev V. G., Nekrasov V. V., Akolaev G. G., Bodin N. N., Dneprov-sky I. C. J. Radioanal. Chem., 6, 431 (1970).
1272. Lanfranco G., Bianchini A. Chim. e ind., 50, 337 (1968).
1273. Langer E. Пат. ПНР, № 62585, 15.08.71; РЖХим, 1973, 5Г138Р.
1274. Lasek J., Cermak K., Huth, listy, 17, 735 (1962).
1275. Lassier A. C. r. Acad, sci., 176, 221 (1923).
1276. Laul J. C., Case D. R., Wechter M., Schmidt-Bieck F., Lipschutz M. E. J. Radioanal. Chem., 4, 241 (1970).
1277. Lavric T. Rud.-metal, zb. Bratislava, Posebna Stevilka, 1966, S. 575; РЖХим, 1968, 5Г139.
1278. Laiansky J., Belohlavek 0. Hutn. listy, 21, 272 (1966).
1279. Lenhardt K. Z. Erzbergbau und Metallhuttenw., 19, 139 (1966).
1280. Lesbats A., Tardy M. R. J. Radioanal. Chem., 17, 127 (1973).
1281. Lesigang-Buchtela M. Mikrochim. acta, N 3, 408 (1966).
1282. Levin I. S., Shatalova A. A., Azarenko T. G., Vorsina I. A., Burtovaya-Balakireva N. A., Rodina T. F. Taianta, 14, 801 (1967).
1283. Liberti A., Lazzari P. Ricerca Sci., 26, 826 (1956).
1284. Liebig A. Chem. Ztg, 36, 1465 (1912).
1285. Liebig А. Ъ. angew. Chem., 72, 416 (1905).
1286. Lihi F., Patch P., Sorantln H. Z. anal. Chem., 221, 176 (1966).
1287. Lingane J. J., Bard A. J. Anal. chim. acta, 16, 271 (1957).
1288. Litomisky J. Chem. anal. (PRL), 7, 409 (1962).
1289. Lombard S. M., Marlow K. W., Tammer-James T. Anal. chim. acta, 55, 13 (1971).
1290. Lowe R. M. Abstr. Spectrochim. acta, 24B, 191 (1969).
1291. Luis P., Sa A., Mascaro A. Mikrochim. acta, N 1, 1 (1969).
1292. Luke C. L. Anal. Chem., 25, 674 (1953).
1293. Luke C. L. Anal. Chem., 31, 1680 (1959).
1294. Luke C. L. Anal. chim. acta, 39, 447 (1967).
1295. Luke C. L., Campbell M. E. Anal. Chem., 25, 183 (1956).
1296. Lukens H. R., Guinn V. R. Trans. Amer. Nucl. Soc., 10, 66 (1967).
1297. Lund M. A., Smith D. Atom. Energy Res. Establ. [Rept], № AM73, 1961.
1298. Liischow H. M., Steil H. U. Z. anal. Chem., 245, 304 (1969).
1299. Lux F., Braunstein L. Z. anal. Chem., 221, 235 (1966).
1300. Lybe S. J., Shendrikar A. D. Anal. chim. acta, 36, 286 (1966).
1301. Mac Cay L. W. J. Amer. Chem. Soc., 31, 373 (1909).
1302. MacNulty B., Woollard L. Anal. Chim. acta, 13, 64 (1955).
1303. Madsen R. E. Atom. Absorpt. Newslett., 10, 57 (1971).
1304. Maekawa C.. Kato K. Jap. Analyst, 15, 159 (1966).
1305. Maenhaut W., Adams F., Hoste J. J. Radioanal. Chem., 6, 83 (1970).
1306. Major T., Grade P. Magy. kem. folyoirat., 74, 196 (1968).
1307. Majumdar A. K., Bhowal S. Anal. chim. acta, 36, 399 (1966).
1308. Majumdar A. K., Das M. K. J. Indian Chem. Soc., 44, 828 (1967).
1309. Malenthal E. J. Amer. Lab., 5, 25 (1973).
1310. Malewski B. Math, und naturwiss. Unters., 15, 218 (1962).
1311. Malik W-, Ajmal M. J. Electroanal. Chem., 6, 450 (1963).
1312. Malvanop R., Grosso P., Lanardi M. Anal. chim. acta, 41, 251 (1968).
1313. Mamuro T., Fujita A., Matsuda Y. Ann. Rept Radiat. Center Osaka Prefect., 10, 9 (1969).
1314. Mamuro T., Matsuda Y., Takeuti T., Fujita A. Radioisotopes, 20, 117 (1971).
1315. Manning D. C. Atom. Absorpt. Newslett., 10, 123 (1971).
1316. Mantel M., Albu-Jaron A., Amiel S. Isr. Atom. Energy Commis. (Repts), N 1190, 118 (1969).
1317. Marczenko Z. Chem. anal. (PRL), 2, 255 (1957).
1318. Marczenko Z., Mojski M., Skibe H. Chem. anal. (PRL), 17, 881 (1972).
1319. Markl P. Extraction und Extraktionschromatographie in der anorgani-schen Analytik. Frankfurt am Main, Akademische Verlagsgesellschaft, 1972.
1320. Marsch H. Edinb. New Philos. J., 229 (1886).
208
1321. Masauki У., Masami S., Tsugio T. Anal. chim. acta, 47, 121 (1969),
1322. Massmann H. Spectrochim. acta, B23, 215 (1968).
1323. Mathery M. Acta chim. Acad. Sci. hung., 30, 399 (1962).
1324. Mathery M. Chem. anal. (PRL), 7, 75 (1962).
1325. Matic J. S., Pesic D. S. Rev. roum. chim., 10, 733 (1965).
1326. Mattel a H. Taianta, 16, 1267 (1969).
1327. Matsumoto U., Nagase Y. J. Pharm. Soc. Jap., 81, 873 (1961).
1328. Matulis R. M., Guyon J» C. Anal. Chem., 37, 1391 (1965).
1329. Maurice M. J., Lingen R. L. Anal. chim. acta, 28, 91 (1963).
1330. May S., Pinte G. J. Radioanal. Chem., 3, 329 (1969).
1331. Mazur H. Rocz. Panst. zakl, hig., 11, 217 (1960).
1332. Meier H., Zimmerhakl E., Albrecht W., Bosche D., Hecker W., Menge P.r Ruckdeschel A., Unger E., Zeitler G. Mikrochim. acta, N 3, 553 (1970).
1333. Meier H., Zimmerhackl E., Albrecht W., Bosche D., Hecker W., Menge P., Unger E., Ruckdeschel A., Zeitler G. Mikrochim. acta, N 5, 1070 (1969).
1334. Menzel H., Siewert G. Mikrochim. acta, N 6, 814 (1958).
1335. Meyer S., Koch O. Z. anal. Chem., 179, 175 (1961).
1336. Mihalka J. Acta chim. Acad. Sci. hung., 30, 359 (1962).
1337. Miketukova V. J. Chromatogr., 24, 302 (1966).
1338. Miksovsky M., Rubeska I. Chem. listy, 68, 299 (1974).
1339. Minczewski J., Jaskolska H., Wodkiewicz L. Prz. elektron., 4, 520 (1963).
1340. Minczewki J., Pszonicka M. Chem. anal. (PRL), 10, 1357 (1965).
1341. Mitranescu M. Bull. Stiint. si tehn. Inst, politehn. Timisoara, 13, 339 (1968).
1342. Miyakawa Y., Kamemoto Y. Jap. Analyst, 83, 1029 (1962).
1343. Miyashita F., Miyatani G. J. Jap. Inst. Metals, 38, 182 (1974).
1344. Mizuike A., Kono T. Radioisotopes, 19, 609 (1970).
1345. Mladenovic S. Гласник хем. друшт. Београд, 30, 239 (1965).
1346. Mohr Е. Chem. Techn., 10, 34 (1958).
1347. Molenda К. Pr. Inst. mech. prec., 15, 47 (1967).
1348. Monien H., Bohn D., Jacob P. Angew. Chem., 83 , 921 (1971).
1349. Monien H., Jacob P. Z. anorg. Chem., 260, 195 (1972).
1350. Morandat J., Duval C. Anal. chim. acta, 4, 498 (1950).
1351. Morello B. Metallurgia ital.. 58, 317 (1966).
1352. Morris A. G. Analyst, 87, 478 (1962).
1353. Morrison G. H., Rupp R., Klecak G. Anal. Chem., 32, 933 (1960).
1354. Mostyn R. A., Cunningham A. F. Anal. Chem., 39, 433 (1967).
1355. Motozima K., Baito S., Nakayama R. Anal. Instrum., 6, 567 (1968).
1356. Munchow P. Chem. Ztg Chem. Appar., 94, 113 (1970).
1357. Murty P. S., Marathe S. M., Kapoor S. K., Saraswathy M. Z. anal. Chem., 268, 285 (1974).
1358. Muzzarelli R. A., Marcotrigiano G. Taianta, 14, 489 (1967).
1359. Muzzarelli R. A., Raith G., Tubertin O. J. Chromatogr., 47, 414 (1970',
1360. Nag6rski B. Chem. anal. (PRL), 20, 225 (1975).
1361. Nagy L. G. Acta chim. Acad. Sci. hung., 52, 365 (1967).
1362. Naik R. C., Machalo J. J. Covt India Atom. Energy Commis. (Repts), № BARC-539 (1971).
1363. Naito T. J. Pharm. Soc. Jap., 58, 690 (1938).
1364. Naito T. J. Pharm. Soc. Jap., 59, 303 (1939).
1365. Nakayama G. J. Chem. Soc. Jap., Pure Chem. Sec., 83, 1258 (1962).
1366. Nakai T., Yajima S., Fujil J., Okada M. Jap. Analyst, 8, 364 (1959).
1367. Nakanishi S., Nakayama T. New Nippon Elec. Techn. Rev., 10, 54 (1975).
1368. Nakazima T., Fukushima H. Jap. Analyst, 9, 81 (1960).
1369. Namois R. Ind. chim. min. Met., 5, 89 (1918).
1370. Nangiot P. J. Polarogr. Soc., 11, 45 (1965).
1371. Nasfutiu T. Radiochemical Methods of Analysis. Bucharest, Acad. RSR, 1973.
1372. Nasfutiu T. Rev. roum. chim., 12, 839 (1967).
1373. Nashiro M-, Sugisaki M. Jap. Analyst, 15, 356 (1966).
1374. Neeb К. H. Z. anal. Chem., 200, 278 (1964).
209
1375. Neeb К. H., Stockert Н., Braun В., Bleich Н. Р. Z. anal. Chem., 245, 233 (1969).
1376. Neeb К. H., Stockert Н., Gebauhr W. Z. anal. Chem., 219, 69 (1966).
1377. Neeb R. Inverse Polarographie und Voltametrie. Berlin, Akademie-verlag, 1969.
1378. Neeb R., Kiehnast J. Z. anal. Chem., 241, 142 (1968).
1379. Neef J., Adams F., Hoste J. Anal. chim. acta, 62, 71 (1972).
1380. Neirinckx R., Adams F., Hoste J. Anal. chim. acta, 48, 1 (1969).
1381. Neirinckx R., Adams F., Hoste J- Anal. chim. acta, 46, 165 (1969).
1382. Nelson F., Michelson D. C. J. Chromatogr., 25, 414 (1966).
1383. Neumann H. M., Remette R. W- J. Amer. Chem. Soc., 78, 1848 (1956).
1384. Neve R., Soete D., Hoste J. Anal. chim. acta, 40, 379 (1968).
1385. Neve R., Soete D., Hoste J. Z. anal. Chem., 245, 221 (1969).
1386. Newcombe H., MacBryde W. A. E., Bartlett J., Beamish F. E. Anal. Chem., 23, 1023 (1951).
1387. Ng W. K. Anal. chim. acta, 64, 292 (1972).
1388. Nielsch W., Boltz G. Mikrochim. acta, N 3—4, 313 (1954).
1389. Nielsch W., Boltz G. Z. anal. Chem., 143, 81 (1954).
1390. Nielsch W., Boltz G. Ibid., p. 264.
1391. Nicolas D. J. Anal. chim. acta, 55, 59 (1971).
1392. Norris J. D., West T. S. Anal. chim. acta, 71, 458 (1974).
1393. Norwitz G., Cohen J., Everett M. Anal. Chem., 32, 1132 (1960).
1394. Norwitz G., Galan M. Anal. chim. acta, 61, 413 (1972).
1395. Noshiro M., Sugasaki M. Jap. Analyst, 15, 356 (1966).
1396. Novdk J., Dobi&sova L. Пат. ЧССР, N 142881 (1971).
1397. Ogden D., Reynolds G. F. Analyst, 89, 538 (1964).
1398. Ogiolda K. Zesz. nauk (PRL), N 383, 123 (1973).
1399. Okoshi H. Trans. Nat. Res. Inst. Metals, 17, 201 (1975).
1400. Okoshi H., Sudo E. Jap. Analyst, 18, 1376 (1969).
1401. Onishi H., Sandell E. Anal. chim. acta, 11, 444 (1954).
1402. Orddgh M. Atomtechn. tajek., 11, 123 (1968).
1403. Ottaway J. M., Bishop E. Anal. chim. acta, 33, 153 (1965).
1404. Ottendorfer L. J. Metallurgia, 57, 105 (1958).
1405. Packman G., Reynolds G. Analyst, 81, 49 (1956).
1406. Palaldn L., Rdducanu G., Popescu S. Commun. inst. certari chim., 1957—1962, v. 1, S. 1, s. a. 389; РЖХим, 1965, 1Г123.
1407. Panly J., Girardi F. Bull. Soc. chim. France, N 2, 244 (1963).
1408. Patek P., Bildstein H. Z. anal. Chem., 231, 187 (1967).
1409. Patek P., Sorantin H. Anal. Chem., 39, 1458 (1967).
1410. Patriarche G. J. pharm. Belg., 15, 327 (1960).
1411. Patriarche G. J. pharm. Belg., 20, 336 (1965).
1412. Pavlikova E., Kvapil M., Weiss D. Rudy, 10, N 4, 13 (1962).
1413. Pavlikova E., Kvapil M., Weiss D. Rudy, 11, N 3, 9 (1963).
1414. Pawlowska H., Lachnik E. Szklo i ceram., 16, 161 (1965).
1415. Pendharkar M. S. J. Indian Acad. Forens. Sci., 4, 50 (1965).
1416. Peters J. Z. anal. Chem., 241, 167 (1968).
1417. Petit R. 19е Congress Groupement d’Avancement Metode Spectroscopi-que, 1956. Paris, Meru, 1956, p. 111.
1418. Pfeil E. Chem. Lab. und Betr., 5, 519 (1954).
1419. Pfeil E. Chem. Lab. und Betr., 6, 123 (1955).
1420. Pfeil E. Chem. Lab. und Betr., 26, 190 (1975).
1421. Piccardi G. Ann. chim. (Ital.), 52, 201 (1962).
1422. Piccardi G., Legittimo P. Anal. chim. acta, 31, 45 (1964).
1423. Pierce T. B. In book: «Selected Annual Reviews of Analitical Sciences», v. 1. London, Soc. Anal. Chem., 1971, p. 133.
1424. Pinkney E. T., Plint N. Trans. Inst. Min. and Met., 76, 114 (1967).
1425. Pinkus A., Martin F. J. Chem. Phys., 24, 137 (1927).
1426. Pinkus A., Martin F. Ibid., p. 182.
1427. Piper D. L., Gales G. G. Anal. chim. acta, 47, 560 (1969).
1428. Pirtea J. Z. anal. Chem., 118, 26 (1939).
210
1429. Pirtea J. Z. anal. Chem., 208, 411 (1965).
1430. Pjescic M. G., Susie M. V., Mlnic D. J. Electroanal. Chem., 57, 429 (1974).
1431. Ploum H. Arch. Eisenhiittenw., 33, 601 (1962).
1432. Pollock E. N., West S. J. Atom. Absorpt. Newslett., 11, 104 (1972).
1433. Pollock E. N., West S. J. Atom. Absorpt. Newslett., 12, 6 (1973).
1434. Poonia N. S. Mikrochim. acta, N 1, 211 (1969).
1435. Popper E., Crdciuneanu R., Florean E. Omagiu acad. prof. Raluca Ripan (Bucure^ti), 1966, p. 467.
1436. Popper E., Olteanu I., Popescu G. Rev. chim. (PRP), 7, 367 (1956).
1437. Portmann J. E., Riley J. P. Anal. chim. acta, 31, 509 (1964).
1438. Portmann J. E., Riley G. P. Anal. chim. acta, 35, 35 (1966).
1439. Prasad B. N., Prasad В. B., Singh D., Rao В. V. J. Sci. Res. Banaras Hindu Univ., 10, 146 (1959-1960).
1440. Pravceva C., Ganev P., Delijska A., Ninova V. Z. anal. Chem., 254, 16 (1971).
1441. Pravceva C., Ganev P., Ninova K, Delijska A. Z. anal. Chem., 255, 113 (1971).
1442. Price W. J. Analytical Atomic Absorption Spectrometry. London, Heyden and Son, 1972.
1443. Prod. Group. U.K. Atom. Energy Auth. Rept. N 220 CS (1963).
1444. Prokopov T. S. Proc. Iowa Acad. Sci., 77, 347 (1971).
1445. Provaznik J., Knizek M- Chem. listy, 55, 79 (1961).
1446. Provaznik J., Mojzis J. Ibid., p. 1299.
1447. Pszonicki L. Chem. anal. (PRL), 7, 947 (1962).
1448. Pungor E., Balasz L. Magy. kem. folyoirat, 67, 11 (1967).
1449. Purgotti A. Gazz. chim. ital., 42, 58 (1912).
1450. Quarrece T., Powell R., Cluliey H. Analyst, 98, 443 (1973).
1451. Qureshi M., Akhtar J., Mathur K. N. Anal. Chem., 39, 1766 (1967).
1452. Qureshi M., Husain W. Separ. Sci., 4, 197 (1969).
1453. Qureshi M., Khan M. Anal. Chem., 35, 2050 (1963).
1454. Qureshi M., Khan M. J. Indian Chem. Soc., 41, 673 (1974).
1455. Qureshi M., Khan M. Z. anal. Chem., 232, 194 (1967).
1456. Qureshi M., Mathur K. N., Israili A. H. Talanta, 16, 503 (1969).
1457. Qureshi I. H., Nagi F. I., Nasra M., Cheema M. N. Radioanal. Chem., 7, 221 (1971).
1458. Rai J., Kukreja V. P. Chromatographia, N 2, 404 (1969).
1459. Ramette R. W. Anal. Chem., 30, 1158 (1958).
1460. Ramette R. W., Sandell E. B. Anal. chim. acta, 13, 455 (1955).
1461. Rausch H., Csada G., Szabo E. Chem. zvest., 21, 592 (1967).
1462. Rausch H., Imrene C., Czabo E. Kozp. fiz. kut. intez. kozl., 15, 229 (1967).
1463. Ray B. N., Ditta R. K. .1. Technol., 7, 91 (1969).
1464. Ray H. N. J. Indian Chem. Soc., 17, 586 (1940).
1465. Ramirez-Munor J. Atomic Absorption Spectroscopy and Analysis by Atomic Absorption Flame Photometry. New York, Elsevier, 1968.
1466. Rao R. Z. Anal. Chem., 165, 195 (1959).
1467. Ratcliffe R. J., Stevens S. G. Manuf. Chem. and Aerosol News, 35, N 9, 83 (1964).
1468. Rayudi G. V. S., Tiefenbach B., Jervis R. E. Trans. Amer. Nucl. Soc., 11, 81 (1968).
1469. Renshaw G. D., Pounds C. A., Pearson E. F. Atom. Absorpt. Newslett., 12, 55 (1973).
1470. Rey M. R. W. Trans. Inst. Min. and Met., 76, 101 (1967).
1471. Reynolds G. F., Tyler F. S. Analyst, 89, 579 (1964).
1472. Richter H. L. Anal. Chem., 27, 1526 (1955).
1473. Robertson D. S. Nature (Engl.), 210, 1357 (1966).
1474. Robinson D. Proc. Soc. Anal. Chem., 2, 142 (1965).
1475. Robinson J. W. Atomic Absorption Spectroscopy. New York, Decker, 1966—1967.
211
1476. Roland G., Duyckaerts G. Anal. chim. acta, 54, 423 (1971).
1477. Rooney R. C. Analyst, 82, 619 (1957).
1478. Rooney R. C. BCURA J. 10, 446 (1962).
1479. Rose H. Handbuch der analitischen Chemie, Bd 6. Leipzig, Springer, 1867, S. 288.
1480. Rossmanith K., Hanna Z. Monatsh. Chem., 91, 238 (1960).
1481. Rossouw A. J. J. Afr. Ind. Chem., 14, N 3, 44 (1960).
1482. Rossouw A. J. Philips. Serv. Sci. and Ind., 7, 203, (1960).
1483. Rotger K. Z. anal. Chem., 227, 321 (1967).
1484. Rubeska I., Moldan B. Analyst, 93, 148 (1968).
1485. Rubeska I., Moldan B. Appl. Opt., 7, 1341 (1968).
1486. Rubeska I., Moldan B. Atomova Absorpcni Spectrofotometrie, Praha, SNTL, 1967.
1487. Ruch R. R., Guinn V. P., Pinker R. H. Nucl. Sci. and Eng., 20, 381 (1964).
1488. Rutkowski W. Chem. anal. (PRL), 14, 905 (1969).
1489. Rutkowski W. Ibid., p. 1099.
1490. Ruzicka E. Acta Univ, palack. Olomuc, 18, 241 (1965).
1491. Ruzicka E. Collect. Czech. Chem. Communs, 29, 2244 (1964).
1492. Ruzicka J., Stary J., Lemon A. Taianta, 11, 1151 (1964).
1493. Saltzman В. E. Anal. Chem., 33, 1100 (1961).
1494. Sampedro P., Asensi A. Rev. cienc. apl., 11, 193 (1957).
1495. Sana S. Jap. Analyst, 13, 351 (1964).
1496. Sarudi J. Magy. kem. folyoirat., 64, 149 (1958).
1497. Schiller P. Kernenergie, 6, 514 (1963).
1498. Schmidt E., Gygax H. R. Helv. chim. acta, 48, 1178 (1965).
1499. Schmidt F. J., Royer J. L. Anal. Lett., 6, 17 (1973).
1500. Schmidt F. J., Royer J. L., Muir S. M. Anal. Lett., 8, 123 (1975).
1501. Schneer-Erdeg A., Csada G. J., Rausch H., Szabo E. Ann. Univ. sci. Budapest, Sec. chim., 9, 77 (1967).
1502. Schnepfe M. M. Taianta, 20, 175 (1973).
1503. Schonberg G., Gutmann V., Nedbalek E. Z. anal. Chem., 186, 115 (1962).
1504. Schroll E., Weninger M. Mikrochim. acta, N 2, 378 (1965).
1505. Schubiger A., Wyttenbach A. Chem. Rdsch. (Schweiz), 25, 1273 (1972).
1506. Schubiger A., Wyttenbach A. Helv. chim. acta, 56, 648 (1973).
1507. Schulek E., Barcza L. Ann. Univ. sci. Budapest., Sec. chim., 2, 527 (1960).
1508. Schulek E., Barcza L. Magy. tud. akad. kem. tud. eszt. kozl., 11, 349 (1959).
1509. Schulze W. In book: «Advances Activation Analysis», v. 1. London— New York, Intercience Publ., 1969, p. 1.
1510. Schulte К. E„ Henke G., Tjan K. S. Z. anal. Chem., 252, 358 (1970).
1511. Schweinberg D. P., Hefferman B. J. Proc. Austral. Inst. Min. and Met., N 241, 87 (1972).
1512. Schweitzer G. K., Stroms L. E. Anal. chim. acta, 19, 154 (1958).
1513. Scott F. W. Chem.-Analyst, 24, 16 (1935).
1514. Scott R. B. Development Applied Spectroscopy, v. 1. Chicago, Academic-Press, 1962, p. 17.
1515. Seitner H., Kiesl W., Kluger F., Hecht F. J. Radioanal. Chem., 7, 235 (1971).
1516. Senf H. J. Mikrochim. acta, N 5, 954 (1968).
1517. Shah K. R., Filby R. H., Haller W. A. J. Radioanal. Chem., 5, 413 (1970).
1518. Shanks D. E., Broadhead K. G., Heady H. H. Int. J. Appl. Radiat. and Isotop., 21, 671 (1970).
1519. Sherma J., Cline C. W. Taianta, 10, 787 (1963).
1520. Sherma J., Evans G., Frame H., Strain H. Anal. Chem., 35, 224 (1963).
1521. Sie^S. T., Bleumer J. P. A., Rijnders G. W. A. Separ. Sci., 1, 41 (1966).
1522. Simkovd M., Kukula F., Steiskal R. Collect. Czech. Chem. Communs,
30, 3193 (1965).
212
1523. Singh E., Dey A. Mikrochim. acta, N 3, 366 (1961).
1524. Singh E., Dey A. .1. Indian Chem. Soc., 38, 323 (1961)
1525. Singh E., Dey A. Z. anal. Chem., 183, 248 (1961).
1526. Sharma 8. Curr. Sci. (India), 19, 114 (1950).
1527. Sherma J. Anal. chim. acta, 36, 138 (1966).
1528. Shibuya M. Proceeding 4th Conference Radioisotopes. Tokyo, Incorporated Press, 1962, p. 1156.
1529/Shiff R., Tarugi N. Z. anal. Chem., 34, 456 (1895).
1530. Sholes T. R., Deumedd К. H. Metallurgia, 71, 295 (1965).
1531. Siemes H. Z. Erzbergbau und Metallhiittenw., 15, 463 (1962).
1532. Sikorska-Tomicka H. Chem. anal. (PRL), 15, 795 (1970).
1533. Singh B., Kashyap G. Indian J. Appl. Chem., 22, 15 (1959).
1534. Singh B., Sahota S., Singh A. Z. anal. Chem., 169, 106 (1959).
1535. Singh B., Sahota S., Verma B. Res. Bull. Panjab Univ., 10, 261 (1960).
1536. Singh E., Dey A. Anal. chim. acta, 24, 444 и (1961).
1537. Skorko-Trzubula Z., Boguszewska Z. INTERAN (Prague, 1976), Plenary Leet., S. 1, 1976, p. 125.
1538. Slavin S., Sattur T. W. Atom. Absorpt. Newslett., 7, 99 (1968).
1539. Smales A. A., Mapper D., Fouche K. F. Res. Group. U. K. Atom. Energy Auth., № NERE 5254 (1967).
1540. Smales A. A., Mapper D., Wood A. J., Salmon L. Atom. Energy Res. Establ. [Rept], № C/R 2254 (1957).
1541. Solic L., Turina 8., Mazjanovic V. Z. anal. Chem., 258, 31 (1972).
1542. Sotoniewicz R. Rocz. Chem., 40, 38 (1966).
1543. Sosa J. P., Russo N. Le MIT, № 3, 35 (1970); РЖХим, 1971, 20Г100.
1544. Souza T. L. C., Kerbyson J. D. Anal. Chem., 40, 1146 (1968).
1545. Spalenka M. Chem. anal. (PRL), 4, 385 (1959).
1546. Spalenka M. Huth, listy, 13, 150 (1958).
1547. Specker H., Cremer M., Jackwerth E. Angew. Chem., 71, 492 (1959).
1548. Stabryn T. Hutn. listy, 14, 515 (1959).
1549. Stanton R. E., MacDonald A. Analyst, 87, 291 (1962).
1550. Stanton R. E., MacDonald A. Bull. Inst. Min. and Met., N 667, 1 (1962).
1551. Stara V. Taianta, 18, 228 (1971).
1552. Steffek M. Chem. listy, 61, 654 (1967).
1553. Steihler H. Pap. und Druck, 9, 241 (1960).
1554. Steinnes E. A. Analyst, 97, 241 (1972).
1555. Steinnes E. A. Kjeller Rept, N 142, 11 (1971); РЖХим, 1971, 19Г117.
1556. Steinnes E. Scand. J. Met., 1, 137 (1972).
1557. Stiles D. A. Proc. Soc. Anal. Chem., 11, 138 (1974).
1558. Stre&ko V., Martiny E. Atom. Absorpt. Newslett., 11, 14 (1972).
1559. Stronski J. Rocz. chem., 34, 709 (1960).
1560. Studlar K. Collect. Czech. Chem. Communs, 31, 1999 (1966).
1561. Sulcek Z., Boseova M., Dolezal J. Collect. Czech. Chem. Communs, 34, 787 (1969).
1562. Sulcek L., Dolezal J., Michal J., Sychra V. Taianta, 10, 3 (1963).
1563. Supp G. R. Atom. Absorpt. Newslett., 11, 122 (1972).
1564. Sutek A., Leb J. Stud. Univ. Babes-Bolyai. Ser. quim., 14, 67 (1969).
1565. Suzuki N. J. Chem. Soc. Jap., Pure Chem. Sec., 81, 437 (1960).
1566. Szabo Z. L., Szakacs O., Zimmer K. Taianta, 10, 521 (1963).
1567. Szekeres L., Kardas E., Szekeres G. L. Chem.-Analyst., 54, 116 (1965),-
1568. Szekeres L., Sugar E., Pap E. Z. anal. Chem., 159, 418 (1958).
1569. T aimni J., Agrwal R. Anal. chim. acta, 10, 312 (1954).
1570. Takauchi K. Rigaku Denki J., 7, 316 (1965).
1571. Talmi Y., Andren A. W. Anal. Chem., 46, 2122 (1974).
1572. Talmi Y. Anal. chim. acta, 74, 107 (1975).
1573. Talmi Y., Norvell V. E. Anal. Chem., 47, 1510 (1975).
1574. Tamba M. G., Vantini N. Analyst, 97, 542 (1972).
1575. Tamba M. G., Vantini N., Maneschi 8. J. Electroanal. Chem., 31, 193 (1971).
1576. Tamm H. Chem. News, 24, 221 (1871).
213
1577. Tanaka К. Jap. Analyst, 14, 1165 (1965).
1578. Tanaka M. Jap. Analyst, 12, 631 (1963).
1579. Tanaka T., Yamasaki K. Jap. Analyst, 18, 324 (1969).
1580. Tanaka Y., Kawahara M. Jap. Analyst, 10, 185 (1961).
1581. Tanaka Y., Tanaka Y. Jap. Analyst, 13, 623 (1964).
1582. Tandon J., Mehrotra R. Z. anal. Chem., 162, 33 (1958).
1583. Taylor L., Johnson D. Anal. Chem., 46, 262 (1974).
1584. Temmerman E., Verbeek F. Anal. chim. acta, 43, 263 (1968).
1585. Temmerman E., Verbeek F. Anal. chim. acta, 47, 546 (1972).
1586. Tennant W. C. Appl. Spectrosc., 21, 282 (1967).
1587. Terashima S. Jap. Analyst, 23, 1331 (1974).
1588. Tesarik B. Chem. prum., 21, 352 (1971).
1589. Tewari S. Z. anal. Chem., 180, 109 (1961).
1590. Thompson B., La Fleur P. Anal. Chem., 41, 852 (1969).
1591. Thompson К. C., Wildy P. C. Analyst, 95, 776 (1970).
1592. Thompson К. C., Thomerson D. R. Analyst, 99, 595 (1974).
1593. Thompson К. C. Analyst, 94, 958 (1969).
1594. Thompson К. C., Burke К. E. Analyst, 99, 469 (1974).
1595. Tockstein A., Tocksteinova D. Collect. Czech. Chem. Communs, 34, 1625 (1969).
1596. Tocksteinova D., Tockstein A. Ibid., p. 2169.
1597. Todd R., Cuthbert G., Dickinson R. Prod. Group. U.K. Atom. Energv Auth., № 337S (1962).
1598. Tolle H. Neue Hiitte, 14, 691 (1969).
1599. Tolle H. Z. anal. Chem., 240, 162 (1968).
1600. Tomings N. Appl. Spectrosc., 14, 72 (1960).
1601. Tomlianovic M., Grobenski Z. Atom. Absorpt. Newslett., 14, 52 (1975).
1602. Tomlinson L. Res. Group. U. K. Atom. Energy Auth., № AERE-П11886 (1967).
1603. Tomula E. S. Huyllingskrift tilligrad Ossian Aschau, 1920, 124.
1604. Toren P. E. Anal. Chem., 40, 1152 (1968).
1605. Tsuiki M. J. Electrochem. Soc. Jap., 28, 293 (1960).
1606. Tsuiki M. J. Electrochem. Soc. Jap., 29, 45 (1961).
1607. Tsuiki M., Kawase T. Res. Repts Fac. Eng. Gifu, № 10, 26 (1960).
1608. Tsuijii K., Kuga K., Sugaya I. Chem. Lett., № 7, 695 (1975).
1609. Tymchuk P., Mykytiuk A., Russell D. S. Appl. Spectrosc., 22, 268 (1968).
1610. Uchida T., lida C. Appl. Spectrosc., 29, 58 (1975).
1611. Ueda S., Fuzihara K., Yamamoto Y., Ueda K. J. Chem. Soc. Jap., Pure Chem. Sec., 92, 422 (1971).
1612. Ueno K., Tachikawa T. Jap. Analyst, 8, 572 (1959).
1613. Ujhidyne F. K., Szilvassyne V. Zs. Vespremi vegyipari egyet kozl., 9, 71 (1965).
1614. Uny G., Tardif J., Spitz J. Anal. chim. acta, 54, 91 (1971).
1615. Van Aman R. E., Hollibaugh F. D., Kanzelmeyer J. H. Anal. Chem., 31, 1783 (1959).
1616. Von Kote F., Catonne J. C. Radiochim. acta, 13, 145 (1970).
1617. Varga S. Chem. listy, 64, 1253 (1970).
1618. Vasak V., Sedivec V. Chem. listy, 46, 341 (1952).
1619. Vecsernyes L. Z. anal. Chem., 182, 429 (1961).
1620. Vecsernyes L. Tavkozlesi kut. intez. kozl., 7, 51 (1962).
1621. Verma M., Singh Y. J. Sci. and Ind. Res., BB, № 10, 709 (1954).
1622. Vienne J. Anal chim. acta, 55, 37 (1971).
1623. Vignoli L., Cristian B., Gonezo F. Chim. anal., 43, 439 (1961).
1624. Visapaa A. Kem. teoll., 24, 401 (1967).
1625. Vorlicek J., Dolezal J. Hutn. listy, 18, 55 (1963).
1626. Vortmann G., Metzl A. Z. anal. Chem., 44, 525 (1905).
1626a. Vrbsky J., Fogl J. Sb. Vyssej skoly chem. technol. v Praze, N 8, 183 (1972).
1627. Vиксе vic-Kovacevic V. Farm, glas., 9, N 2, 43 (1953).
214
1628. Vukcevic-Kovacevic V., Seremet S. Ball. sci. Cons. Acad. sci. et arts RSFY, A14, 377 (1969).
1629. Vukcevic-Kovacevic V., Seremet S. Sci. pharm., 36, 26 (1968).
1630. Wagy L., Mester L., Takacs G. Magy. kem. folyoirat., 73, 137 (1967).
1631. Waksmudzki A., Przeczlakowski S. Chem. anal. (PRL), 11, 159 (1966).
1632. Walker C. R., Vita 0. A., Sparks R. W. Anal. chim. acta, 47, 1 (1969).
1633. Walsh A. Spectrochim. acta, 7, 108 (1955).
1634. Ward F. N., Lakin H. W. Anal. Chem., 26, 1168 (1954).
1635. Weiss D. Chem. listy, 52, 1815 (1968).
1636. Weiss H. V., Bertine К. K. Anal. chim. acta, 65, 253 (1973).
1637. Weis W., Stivers E. Tappi, 44, A175 (1961).
1638. Welsch E. P., Chao T. T. Anal. chim. acta, 76, 65 (1975).
1639. Wenger P., Duckert R., Blankpain С. P. Helv. chim. acta, 20, 427 (1937).
1640. Werner 0. Z. anal. Chem., 250, 17 (1970).
1641- Wenger P., Paraud P. Ann. chim. anal., chim., appl., 5, 230 (1923).
1642. West P. W, Leacer A. Anal. Chem., 34, 555 (1962).
1643. Weisz H. Mikrochim. acta, № 3—4, 376 (1954).
1644. Weisz H., Tellgmann C. Z. anal. Chem., 220, 161 (1966).
1645. Werner O. Metall, 16, 1062 (1962).
1646. Whipple E. R., Ridgley D. H. Anal. chim. acta, 35, 499 (1966).
1647. White С. E., Rose H. J. Anal. Chem., 25, 351 (1953).
1648. Widtmann V. Hutn. listy, 25, 733 (1970).
1649. Wilson A. D., Lewis D. T. Analyst, 88, 585 (1963).
1650. Wilson’s Comprehensive Analytical Chemistry, v. 4. Instrumentation for Spectroscopy. Analytical Atomic Absorption and Fluorescence Spectroscopy. Diffuse Reflectance Spectroscopy. Amsterdam, Elsevier, 1975.
1651. Wise W. M., Williams J. P. Anal. Chem., 36, 1863 (1964).
1652. Wispelaere C., Op de Beeck J. P., Hoste J. Anal. chim. acta, 64, 321 (1973).
1653. Wispelaere C., Op de Beeck J. P., Hoste J. Anal. chim. acta, 70, 1 (1974).
1654. Witkowska S. Chem. anal. (PRL), 11, 783 (1966).
1655. Witkowska S., Hlasiewicz W. Chem. anal. (PRL), 7, 211 (1962).
1656. Witkowska S., Filasiewicz W. Rudy i metale niezel., 6, 26 (1961).
1657. Witzmann H., Piesche L. Reinstoffe in Wissenschaft und Technik. Berlin, Academieverlag, 1963, S. 439.
1658. Wlotzka F. Z. anal. Chem., 215, 81 (1966).
1659. Wodkiewicz L. Nukleonika, 8, 545 (1963).
1660. Wodkiewicz L. Nukleonika, 10, Suppl., 83 (1965).
1661. Wolbling H., Broh R. Giessereitechnik, 16, 415 (1970).
1662. Woodward C. Annual Reports on Analytical Atomic Absorption, 1973, v. 3. London, Soc. Anal. Chem., 1974.
1663. Wronski M., Kubalska B. Chem. anal. (PRL), 15, 901 (1970).
1664. WUlrich M., Hanner A. Techn. Mitt. PTT, 36, 507 (1958).
1665. Wyatt P. Analyst, 80, 368 (1955).
1666. Yadav A. A., Khopkar S. M. Bull. Chem. Soc. Jap., 44, 693 (1971).
1667. Yakagisawa M., Takeuchi T., Suzuki M. Anal. chim. acta, 60, 282 (1970).
1668. Yamamoto Y., Kumamaru T., Hayashi Y., Капке M. Anal. Lett., 5, 717 (1972).
1669. Yamamoto Y., Kumamaru T., Hayashi Y., Tsuino R. Ibid., p. 410.
1670. Yamamoto Y., Капке M., Mizukami Y. Chem. Lett., N 7, 535 (1972).
1671. Yamamoto Y., Kumamaru T., Hayashi Y., Tsuino R. Anal. Lett., 5, 419 (1972).
1672. Yamazakt Y. Repts. Govt Chem. Ind. Res. Inst. Tokyo, 53, 213 (1958).
1673. Yanagisawa M., Takeuchi T., Suzuki M. Anal. chim. acta, 64, 381 (1973).
1674. Yang N. S., Tien С. С., Kao H. H. Hua Hsiieh Pao, 25, 106(1959); Chem. Abstr., 54, 6387 (1960).
1675. Yazima S., Kamemoto Y., Shiba K. J. Chem. Soc. Jap., Pure Chem. Sec., 82, 38 (1961).
1676. Yazima S., Kamemoto Y., Shiba K., Onoda Y. Ibid., p. 194.
215
1677. Yazima S., Kamemoto Y., Skiba K., Onoda Y. Ibid., p. 343.
1678. Yearbook of the American Bureau of Metal Statistics. New York, Elsevier, 1970.
1679. Yoshimura Ch., Noguchi H., Hara H. Jap. Analyst, 13, 1249 (1964).
1680. Yvshimura Ch., Tamura K. J. Chem. Soc. Jap. Pure Chem. Sec., 87.
625 (1966).
1681, Yosimura T., Hara H. Jap. Analyst, 15, 139 (1966).
1682. Yurash B. Chem.-Analyst, 56, 28 (1967).
1683. Zanaroli L. Metallurgia ital., 58, 323 (1967).
1684. Zanaroli L. Metallurgia ital., 61, 385 (1969).
1685. Zeitmeisl M. J., Haworth D. T. J. Chromatogr., 30, 637 (1967).
1686. Zeman A., Stary J., Riiiicka J. Talanta, 10, 981 (1963).
1687. Zmijewska W. Chem. Anal. (PRL), 15, 1025 (1970).
1688. Zmijewska W., Kozminska D. Chem. anal. (PRL), 9, 469 (1964).
1689. Zunki M., Kawase T. Res. Repts Fac. Eng. Gifu Univ., N 10, 26 (1960).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азота определение в сурьме 173
Алюминия определение в сурьме 169
Антимониды 7, 16
Бора определение в сурьме 169
Бумажная хроматография 109
Висмута определение в сурьме 169
Водорода определение в сурьме 173
Галлия определение в сурьме 166, 167
Гидриды сурьмы 15
Железа определение в сурьме 167, 170
Жидкие мембранные электроды 98
Золота определение в сурьме 166
171
Изотопного разбавления метод 77
Изотопы сурьмы 7, 8
Индия определение в антимониде индия 171
Ионообменная хроматография 109
Кадмия определение в
сурьме 166
антимониде галлия 171
антимониде индия 171
Калия определение в сурьме 173
Кальция определение в сурьме 166, 173
Кислорода определение в сурьме 173
Кобальта определение в сурьме 173
Кремния определение в сурьме 169
Кулонометрическое титрование 69
Лития определение в сурьме 173
Магния определение в трихлориде сурьмы 173
Марша метод 21
Масс-спектральный метод 98
Меди определение в сурьме 167
Минералы сурьмы 8
Мышьяка определение в сурьме 166, 171, 173
Натрия определение в сурьме 168
Олова определение в сурьме 172, 173
Ртути определение в сурьме 168
Ртутно-графитовый электрод 67
Свинца определение в сурьме 168, 171
Селена определение в сурьме 162, 168, 172
Серы определение в сурьме 168, 171
Серебра определение в сурьме 165, 171, 173
Сплавов анализ алюминия с магнием, железом и сурьмой 123 висмута с сурьмой 126 индия с галлием, золотом и сурьмой 132
индия с цинком 132
свинца с оловом 144
Стибина
определение 159
физико-химические свойства 15
Сурьмаорганические соединения 16
Сурьмы
217
галогениды 13
гидриды 15
изотопы 7, 8
кристаллофосфоры 19
кислоты 13
минералы 8
обнаружение
бесстружковым методом 23
восстановлением металлами 21
галлеином 24
гидролизом 20
димеркантотиопиронами 24
дифенилкарбазидом 24 импрегнированными бумагами
24
иодидами 21
капельными реакциями 27
люминесценцией кристаллофос-
форов 19
метиловым фиолетовым 23
методами
адсорбционной хроматографии 26
бумажной хроматографии 25
кольцевой бани 26
полярографии 27
тонкослойной хроматографии
25
эмиссионного спектрального анализа 18
микрокристаллоскоиическими
реакциями 27
нитратом серебра 20
по реакции Марша 21
окрашиванию пламени 19
родамином С 22
сероводородом 19
серусодержащими реагентами 56
тиосульфатом 20
экстракцией
иодидов 22
ионных ассоциатов 22
оксигалогениды 16
определение в
алюминии 123
арсениде галлия 127
барии 125
бериллии 125
биологических материалах 155
боре 125
бромистоводородной кислоте 157
бронзах 137
ванадии 126
в виде
бромидного комплекса 43
иодида 41
пентасульфида 29
сурьмяномолибденовой сини
43
тетраоксида 29
трисульфида 28
фосфорносурьмяномолибдено-
вой гетерополикислоты 42
висмуте 126
воздушной пыли 159
вольфраме 127
галлии 127
германии 128
гидросульфите натрия 157
графите 152
деионизованной воде 156
железе 129
золоте 131
индии 132
иоде 132
иттрии 133
кадмии 133
кальции 133
карбиде кремния 133
кобальте 133
кремнии 133
литии 136
магнии 137
минералах 119
минеральных кислотах 156
молибдене 137
морской воде 157
мышьяке 141
мышьякорганических соединениях 141
никеле 141
ниобии 142
олове 142
органических растворителях 155
осмии 143
палладии 143
218
питьевой воде 157
платине 143
почвах 119, 121
природных водах 157
растительных материалах 155
редкоземельных элементах 144
рении 144
ртути 144
свинце 144
селене 149
сере 149
серебре 149
силикатах 134, 136
соляной кислоте 156
сталях 129
сточных водах 157
таллии 151
тантале 151
теллуре 151
тиомочевине 155
титане 151
тории 152
трихлорсилане 133
углероде 152
уране 152
ферросплавах 130
фосфоре 153
фтористоводородной кислоте 156
хроме 153
цирконии 154
щелочноземельных металлах 154
•определение
методом
активации фотонами 76
бумажной хроматографии 97
газовой хроматографии 97
инверсионной вольтамперометрии 65
распределительной хроматографии 109
термоэлектрометрии 96 тонкослойной хроматографии
97
но люминесценции комплексов с бензоином 60
бромидом 59
люминолом 60
морином 60
этилродамином С 60 титрованием броматометрическим 33 бромеукцинимидом 38 иодом 36 кинетическое 98 перманганатом калия 35 солями
ванадия(П) 39 ванадия(У) 37 олова(П) 39 титана(Ш) 39 хрома(П) 39 церия(1У) 37 трихлоридом иода 37 хлоридом иода 37 хлоридом натрия 38 фотометрическое
антипириновыми красителями 52
бриллиантовым зеленым 47 бутилродамином С 51 диметилтионином 51 диэтилдитиокарбаминатом натрия 56 серебра 57 иодидным зеленым 50 катионным синим К 52 косвенными методами 40, 57 кристаллическим фиолетовым 48
малахитовым зеленым 50 меркаптохинолином 56 метиленовым голубым 51 метиловым зеленым 50 метиловым фиолетовым 49 нильским голубым 50 оксихинолином 55 основным бирюзовым 50 основным синим К 50 пиридин-иодидным методом 53 пирокатехиновым фиолетовым 56
пиронином Ж 51
по иодокрахмальной реакции 43
по образованию надтитановой кислоты 43
219
родамином Ж 51
родамином 6 Ж 51
родамином С 50
родамином 3 С 51
сафранином Т 52 тиомочевиной 56 триметилтионином 51 толуол-3,4-дитиолом 56 флуоронами 54 янусом зеленым 52 отделение методом
зонной плавки 118
кольцевой бани 118 осаждением в виде
металлической сурьмы 100 ;
еульфидов 99
осаждением органическими реагентами 100
отгонкой в виде стибина 116 трибромида 116 трихлорида 116 соосаждением с
гидроокисями металлов 85, 101
сульфидом меди 101
теллуром 102
тионалидом 101 хроматографией
бумажной 109, 115 ионообменной 109 распределительной 109 тонкослойной 97, 113 экстракционной 114 экстракцией в виде
алкилдитиофосфатов 85 алкилфосфатов 85, 107 бромидов 105
внутрикомплексных соединений 84, 100, 106, 108
диэтилдитиокарбамината 84, 106 иодидов 105 ионных ассоциатов 108 роданидов 106 фторидов 106 хлоридов 102 электродиализом 117 электролизом 117 электроосаждением 117 электрофорезом 117 Сурьмы руды 9 соединения с водородом 15 галогенами 13 кислородом 12 металлами 16 селеном 15 серой 14 теллуром 15
строение электронной оболочки 7 сульфогалогениды 16 физико-химические свойства 11 Таллия определение в сурьме 169 Теллура определение в сурьме 162, 169, 172
Тонкослойная хроматография 109
Фосфора определение в сурьме 168
Хлора определение в сурьме 166 Хрома определение в сурьме 167 Хроматография ионообменная 109
Цинка определение в сурьме 172
Щелочноземельных металлов опре^ деление в сурьме 173
Экстракционная хроматография 114 Электролиз 117
ОГЛАВЛЕНИЕ
От редколлегии..................................................... 3
Предисловие........................................................ 5
Глава I
Общие сведения о сурьме............................................ 7
Положение сурьмы в периодической системе элементов................. 7
Нахождение сурьмы в природе........................................ 7
Получение сурьмы................................................... &
Применение сурьмы.................................................. 10
Глава II
Химико-аналитическая характеристика сурьмы и ее соединений...... 11
Физические и химические свойства сурьмы........................... 11
Соединения сурьмы................................................. 12
Неорганические соединения сурьмы............................. 12
Органические соединения сурьмы................................ 16
Глава III
Качественное определение сурьмы................................... 18
Физические методы................................................. 18
Химические методы................................................. 19
Глава IV
Количественное определение сурьмы................................. 28
Химические методы................................................. 28
Гравиметрические методы...................................... 28
Титриметрические методы...................................... 33
Физико-химические методы......................................... 40
Фотометрические методы....................................... 40
Люминесцентные методы........................................ 59
Хемилюминесцентные методы.................................... 60
Полярографические методы..................................... 61
Кулонометрические методы..................................... 69
Амперометрическое титрование................................. 70
Радиоактивационные методы................................... 72
Радиометрические методы...................................... 76
221
•Физические методы............................................... 77
Эмиссионный спектральный анализ............................. 77
Рентгеноспектральные методы................................. 86
Атомно-абсорбционная спектрофотометрия...................... 88
Атомно-флуоресцентная спектрофотометрия..................... 94
Атомно-эмиссионная спектрофотометрия........................ 95
Молекулярный эмиссионный анализ............................. 96
Другие методы........................:...................... 96
Глава V
Методы отделения сурьмы от сопутствующих элементов............... 99
Методы осаждения................................................. 99
Методы соосаждения................................................ 101
Экстракционные методы........................................... 102
Экстракция неорганических комплексов....................... 102
Экстракция внутрикомплексных соединений.................... 106
Экстракция ионных ассоциатов............................... 108
Хроматографические методы......................................... 109
Отгонка в виде легколетучих соединений............................ 116
Электрохимические методы.......................................... 117
Другие методы................................................... 118
Глава VI
Определение сурьмы в природных и промышленных объектах......... 119
Анализ руд, минералов, горных пород и почв........................ 119
Анализ металлов, сплавов, полупроводниковых и других материалов 123
Анализ органических веществ, растительных и животных продуктов 155
Анализ реактивов, растворов, воздуха, природных.и сточных вод . . 156
Глава VII
Определение примесей в сурьме и ее соединениях................ 160
Литература...................................................... 174
Предметный указатель............................................ 217
Аналитическая химия сурьмы (Серия «Аналитическая химия элементов»), А.А. Немодрук. М., «Наука», 1978, стр. 222.
В книге рассматриваются основные химические свойства сурьмы и ее соединений методы ее отделения от других элементов. Изложены теоретические основы различных методов определения и концентрирования сурьмы, в частности, большое внимание уделено-методам определения сурьмы в промышленных и природных объектах.
Книга рассчитана на широкий круг химиков-аналитиков научно-исследовательских институтов и заводских лабораторий, преподавателей, аспирантов и студентов вузов.
Таблиц 19. Иллюстраций 5. Библ. 1689 назв.
Александр Андреевич Немодрук
Аналитическая химия сурьмы
Серия: «Аналитическая химия элементов*
Утверждено к печати ордена Ленина
Институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор Л. В. Рузайкина
Редактор издательства Р.’А. Баранова Художественный редактор С. А. Литвак Технический редактор Л. Н. Золотухина Корректоры Л. В. Письмам, Р. А. Гютина
ИВ 7015
Сдано в набор 26.06.78
Подписано к печати 31.10.78. Т-18128 Формат бОхЭО1/». Бумага типографская № 2 Гарнитура обыкновенная. Печать высокая
Усл. печ. л. 14. Уч.-изд. л. 16,6 Тираж 2000 экз- Тип. зак. 761
Цена 2 р. 90 к.
Издательство «Наука»
117485, Москва, В-485, Профсоюзная ул., 94а
2-я типография издательства «Наука»
121099, Москва, Г-99, Шубинский пер., 10