/


















































































































































Текст
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА СИБИРСКОЕ ОТДЕЛЕНИЕ
ИНСТИТУТ ЦИТОЛОГИИ И ГЕНЕТИКИ
компьютерный шиз структуры,
«лжиии и эволиии
ГЕНЕТИЧЕСКИХ ШРШШШ
ПРОБЛЕМЫ ИНТЕЛЛЕКТУАЛИЗАЦИИ
НОВОСИБИРСК 1989
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА СИБИРСКОЕ ОТДЕЛЕНИЕ
ИНСТИТУТ ЦИТОЛОГИИ И ГЕНЕТИКИ
КОМПЬЮТЕРНЫЙ АНАЛИЗ СТРУКТУР
ФУНКЦИИ И ЭВОЛЮЦИИ
ГЕНЕТИЧЕСКИХ МАКРОМОЛЕКУЛ
ПРОБЛЕМЫ ИНТЕЛЛЕКТУАЛИЗАЦИИ
Сборник научных трудов
ответственный редактор д.б.я. Н.А.Колчанов
НОВОСИБИРСК 1989
2, ri В
S, ' . lunar1
Academy of Sciences of the USSR
Siberian Department
Institute of Cytology and Genetics
С f- c-vU" ' ~ д S,
I О"' ч ^ ¦"'"-Ч ¦"
e, . <~ - о ь
f
COMPUTER ANALYSIS of STRUCTURE.
FUNCTION AND EVOLUTION
of GENETIC MACROMOLECULES
.INTELLECTUALISATION PROBLEMS
Issue of scientific works
Editor Dr. N.A. Kolchanov
3,
I'fc ' 1 it ' Ко
II. 0 '" ;ь л <
J ?. ,,'iir
функциокаль^шх сайтов в .гюявдуклести.лннх послр.доБа--
тельностях ,..,.,,,.,.,.•,,,.,.,,.,,. ,,,,,,,,,,,,,,, у,-1
12. Д,Н.Бенюх, й.П.Пояомарешсо, Н.А.Колчаноз, Ю.А.Орлов.
Компьютерная система для исследования ЛНК(ГНК)-взаимодей
ствуюдих белков. ,,.,....,,,.,,,,..,,,,,,,,,.,,-,,,. ,а4Э
.Витяев, Н.Л.Подколодный. От экспертных систем а
создающим теории предметных областей...... ?р,;
.Е
Novosibirsk 1989
CONTENT
J. N.A.Kolchanov. Editors foreword 5
3. Solovyev V.V., Salichova Л.К., Rogozin I.B. System for
functional diagnostic of genetic texts 12
3. Shindyalov I.N., Kllkunova I.N. Computer system for
Investigation of molecular evolution on the basis of
homologlcal sequences analysis 41
4. Kapltonov y.V. Computer system for Investigating the
properties of mobile genetic elements 65
5. Hotfozin I.B., Kolchanov N.A., Solovyov V.V., Sredneva H.E.
A computer system helps through the analysis of a
polynucleotlde context for the role it plays In the
appearing of mutations 90
в. Solovyev V.V., Salamov A.A., Sallkhova Л.Х. Computer
aystest for Investigating structural organization of globu-
globular proteins 1. search for lnformstivs characteristics and
their usage for calculating the secondary structure ..111
7. Solovyev V.V., Sallkhova A.K., Salamov A.A. Computer
system for investigating structural organization of
globular proteins 2. Analysis of regularities and
calcullatlng Method of a helycal proteins tertiary
structure 131
8, Kel A.E,, Ischenko I.V., Omelyanchuk L.V. Computer
modelling of the translation process. 155
8. Ponoaarenko M.P., Shindyalov I.II., Kolchanov N.A. Expert
system for predicting topological structures of globular
protein on the basis of amino acid sequences...........168
10. Ponomarenko M.P., Orlov J.L. Expert system of fast
estimation of.biopolimers characteristics utility for
their classification 197
11. Kel A.E., Ponomarenko M.P. , Orlov J.L., ffischenko Т.П.,
Kolchanov N.A. The computer system for functional sites
analysis in polinucleotlde sequences 221
12. Benukh D.N., Ponomarenko M.P., Kolchanov H.A.,Orlov J.L.
Computer system analysis of DNA(RNA)-interactlng
proteins 243
13. Vltyaev E.E., Podcolodny N.L. From expert system to
systems producting subject field theories 264
Предисловие редактора
В последнее десятилетие молекулярная биология и генетика
переживают буршй период развития, связанный с появлением выс-
высокоэффективных методов исследования генетических макромолекул
( ДНК, РНК и белков), включая быстрые методы определения пер-
первичных структур, высокоразрешающие методы определения простра-
пространственных структур и разнообразные технологии работы с реком-
бинантной ДНК. Все это приводит к "взрывообразному" росту
количества молекулярно-генетической информации, которая с
большой скоростью накапливается в специализированных банках
данных [1].
Указанное обстоятельство стимулировало разработку компью-
компьютерных методов анализа молекулярно-генетической информации.
Эти метода, реализованные в виде пакетов программ, использу-
используются для интерпретации результатов молекулярно-генеткческиу
экспериментов, В настоящшее время создан ряд таких пакетов,
способных выполнять рутинный анализ отдельных аминокислотных и
полияукдеогидных последовательностей [2].
Вместе с тем. разработка таких компьютерных средств не
сможет приблизить нас к углубленному пониманию {а тем более -
решению) фундаментальных проблем молекулярной биологии и гене-
генетики. Для этого, на наш взгляд, необходимо создание высокопро-
высокопроизводительных "наукоемких" компьютерных систем для теоретичес-
теоретического исследования ключевых проблем молекулярной биологии и ге-
генетики.
Наиболее эффективный путь решения этой задачи состоит з
использовании идей Новой информационной технологии, ориентиро-
ориентированной на создание интеллекту авизированных компьютерных
систем, т.е. таких систем, которые способны самостоятельно
¦или при ограниченном общении с человеком! решать р. реальной
проблемной среде поставленные перед ними задачи [3].
Практическая реализация проекта "ГЕНОМ ЧЕЛОВЕКА", направ-
направленного на полную расшифровку этого генома [4J, имеющего
размер Ю9 нуклеотидов и содержащего до юо 000 генов,
свидетельствует о том, что уже в самое ближайшее время даже
для первичной обработки огромных массивов молекулярно-
генетических данных ( накопление, верификация, сравнительный
— 5 —
анализ, систематизация, распознавание генов и функциональных
сайтов) потребуются специализированные интеллектуализиро-
вавяые компьютерные системы, способные осуществлять
значительную часть этой обработки с минимальным участием
экспертов - биологов или даже в автоматическом режиме.
Отметим три проблемы, возникающих при разработке
интеллектуализированных компьютерных систем в молекулярной
биологии и генетике.
1. Формализация гигантских об"емов неформализованных
знаний из соответствующей предметной области, которые не могут
быть заложены в ЭВМ без предварительного приведения их к
специфическому "машинному" представлению в формате
стандартных структур данных. Заметим, что первым шагом в этом
направлении является создание специализированных баз данных,
хранящих хорошо формализованную информацию из соответствующих
содержательных областей молекулярной биологии и генетики.
2. Формализация знаний, навыков и опыта экспертов
специалистов в области молекулярной биологии и генетики, а
также в области анализа данных (стратегии применения методов
анализа данных, распознавания образов, оптимального
планирования экспериментов, математического моделирования;
теоретические знания и интуитивные представления о принципах
организации, функционарования и эволюции исследуемых классов
генетических макромолекул).
3. Разработка специализированных компьютерных систем для
автоматического производства, накопления и применения формали-
формализованных знаний о конкретной содержательной области молекуляр-
молекулярной биологии и генетики на основе анализа информации, содержа-
содержащейся в базах данных.
Настоящий сборник преимущественно состоит из работ, выпо-
выполненных в Лаборатории теоретичесокй молекулярной генетики
Теоретического отдела ИЦиГ СО АН СССР. При формировании
сборника мы избегали включения в его состав работ, посвященных
описанию пакетов программ, выполняющих рутинные функции
анализа молекулярно-генетических данных. Вместо этого акцент
был сделан на описание таких компьютерных систем, которые
ориентированы на глубокое исследование содержательных
проблем в различных областях молекулярной биологии и генетики
- 6 -
(таких как теория структурно-функциональной организации
генетических макромолекул, теория мутационного процесса,
теория мобильных генетических элементов, теория молекулярной
эволюции).
Компьютерные системы, описанные в настоящем сборнике,
находятся на различных стадиях интеллектуализации. Значите-
Значительная часть из них исходно разрабатывалась как пакеты
программ для исследования содержательных молекулярно
генетических проблем, впоследствии дополнявшиеся
специализированными базами данных. В настоящее время эти
комплексы содержат от десятков до сотен программных модулей с
разнообразными функциями.
К этой группе относятся : 1 > компьютерная система для
функциональной диагностики генетических текстов, разработанная
В.В. Соловьевым, А.К. Салиховой и И.Б. Рогозиным; 2) компью-
компьютерная система для исследования молекулярных механизмов
мутационного и рекомбинационного процесса, разработанная И.Б.
Рогозиным и соавторами; 3) компьютерная система, разработанная
В.В. Соловьевым, А.К. Салиховой и А.А. Саламовым для
исследования вторичной и третичной структуры белков; 4)
описанная в работе В.В. Капитонова компьютерная система для
исследования свойств мобильных генетических элементов и ряд
других.
Как правило, интеллектуализация затрагивает отдельные
блоки этих систем и диктуется необходимостью повышения их
эффективности и производительности. Например, И.Н. Шиндяловым
и И.Н.Кликуновой разработана компьютерная система для
исследования закономерностей молекулярной эволюции, включающая
программный модуль "Имитационное моделирование эволюции
семейств гомологичных макромолекул", который содержит
формальную модель предметной области, в которой работают
остальные программы этой системы. Фактически, указанный
программный модуль обеспечивает интеллектуализацию системы за
счет универсальной модели эволюционнного процесса.
Вторая группа работ представлена статьями, в которых
описаны компьютерные системы исследования структурно
функциональной организации генетических макромолекул с явно
выраженными элементами интеллектуализации. Работа М.П.
- 7 -
Editor's Foreword
Пономаренко и соавт. посвящена описанию демонстрационного
прототипа экспертной системы для предсказания топологической
структуры белков на основе их аминокислотных последовательнос-
последовательностей. В работе М.П. Пономаренко и Ю.Л. Орлова дано описание
демонстрационного прототипа экспертной системы для быстрой
оценки полезности использования произвольных характеристик
биополимеров дли их классификации. Отличительной особенностью
этой системы является использование нечетких эмпирических
исчислений в рамках теории аддитивной полезности Сэвиджа [5].
В статье А.Э. Келя и соавт. изложены принципы организации
интеллектуализированной компьютерной системы для исследования
функциональных сайтов в полинуклеотидных последовательностях.
Статья Д.Н. Бенюха и соавт. посвящена описанию интеллелекту-
ализированной компьютерной системы для исследования
ДНК(РНК)-взаимодействующих белков, основой которой является
библиотека программ (демонов) для точного распознавания
структурно-функциональных детерминант в аминокислотных
последовательностях белков.
Завершает сборник статья Е.Е. Витяева и Н.Л. Подколодного
в которой рассмотрен вопрос о построении формальной теории
предметной области на основе экспертных систем второго
поколения.
Заведующий лабораторией теоретической
молекулярной генетики теоретического
отдела ИЦиГ СО АН СССР д.б.н. Н.А. Колчанов
Литература
[1] EMBL/NIH Workshop "Future databases for molecular
biology", EMBL, Heidelberg, 25-27, February, 1987
[2] Rawlings C.J. Sowtware directory for molecular bio-
biologists, Macmillan Publ., Ltd., 1986
[3] Кузин Е.С., Ройтман А.И., Фоминых И.Б., Хахалин Г.К.
Интеллектуализация ЭВМ, М.: Высшая школа, 1989
[4] P.Berg. // Science, 1987. v.237 p.1411 ,
[5] Фишберн П. Теория полезности для принятия решений.
М.: Советское радио, 1980
- 8 -
In recent years molecular biology and genetics have expe-
experienced period of Impetuous development due to highly effi-
efficient methods of Investigating genetic macromolecules ( DMA,
RNA and proteins ). It leads to an exploslon-llke increase of
the quantaty of molecular and genetic information which Is ra-
rapidly accumulated In specialized data banks.
These circumstances stimulated working out computer me-
methods which realized as program packages are used for experi-
experimental molecular genetic data Interpreting. At present a num-
number of such packages exist and provide simple forms of analy-
analysis of amlno acid and polynucleotlde sequences.
At the same tine creation of these simple computer sys-
systems can not bring us nearer understanding of molecular biolo-
biology and genetics fundamental problems. We consider that in
order to Improve this situation specialized "science-
consuming" computer systems for theoretical study of key pro-
problems of molecular biology and genetics should be developed,
In which highly-efficient methods of data analysis and new in-
information techniques based on the ideas of artificial Intel-
Intellect and expert systems conception would be used.
The present book consists mainly the works carried out by
the scientists of the Laboratory of theoretical molecular ge-
genetics of the Institute of Cytology and Genetics of the Sibe-
Siberian Branch of the USSR Academy of Sciences. While collecting
papers we Ignored those devoted to a description of packages
of programs performing routine procedures of molecular-genetic
data analysis. We accented, Instead, on a description of com-
computer systems oriented to profound examination of problem*
rich in content from different fields of molecular biology and
genetics such as theory of structural and functional organiza-
organization of genetic macromolecules, theory of mobile genetic ele-
elements, theory of molecular evolution and so on.
The papers presented In the book can be classified into
three groups. The first group contains the papers with desc-
description of complicated packages of programs which have been
elaborated In the laboratory for many years. As a rule they
- 9 -
are complex computer systems of tens or hundreds program- molecular biology an
„.. . -- -- - <*""»ci.ion which is th/»
mlng modules including as a necessary element a specialized B"" one ln our field of investigation
data base containing definite types of molecular and genetic
Information. This group Includes the papers of: V.V.Solovyev
et al. - description of system for functional diagnostics of
genetic texts; I.B.Rogozln et al. - description of computer
eyetem for investigating polynucleotlde context role ln emer-
emergence of mutations and recombinations; I.N.Shlndyalov and
I.M. Kllkunova - description of computer system for studlng
the regularities of molecular evolution; V.V. Kapltonov - the
prlnclplles of computer system organization for investigating
a structure, function and evolution of mobile genetic
elements. The two papers also belong to this group. One is by
V.V. Solovyev , A.K.Sallkhova and A.A. Salamov - description
of computr system for examining structural organization of
globular proteins, and the oter by A.E. Kel, I.V. Ischenko and
V.V. Omelyantchuk - description of a computer model for trans-
translation process.
The second group are the papers devoted to Intellectual1- ,
zed computer systems for studying structural organization and
evolution of genetic macromolecules, namely, those by:
M.P.Ponomarenko - a demonstratlonal prototype of expert system
for predicting the topologlcal structure of globular proteins
on the basis of their amlno acid sequences; M.P.Ponomarenko
and J.L.Orlov - description of a demonstratlonal prototype of
expert system for a quick estimations of significance of arbi-
arbitrary characteristics of blopolymers for their classification.
Included also are the papers of : A.E. Kel et al. - on the
principles of organization of the lntellectuallzed computer
system for Investigating functional sites ln polynucleotlde
sequences, and D.N.Benukh et al. - description of computer sy-
system for studlng of DNA(RNA)-lnteracting proteins ( also Imp-
Implemented with using lntellectualizatlon principles >.
Finally the third direction of investigation Is represen-
represented by the papers of E.E.Vltyaev, N.L.Podcolodny ln which the
problem of second generation expert system Is considered In
conclusion It should be noted that despite of the complexity
in elaborating the lntellectuallzed computer systems ln
- 10 -
СИСТЕМА ФУНКЦИОНАЛЬНОЙ ДИАГНОСТИКИ
ГЕНЕТИЧЕСКИХ ТЕКСТОВ
В.В. Соловьев, А.К. Салихова. И.Б. Рогозин.
Институт цитологии и генетики СО АН СССР, г.Новосибирск
I. Введение
Накопление данных о первичных структурах ДНК, РНК и белков
привело к появление Банков данных нуклеотидных и аминокислот-
аминокислотных последовательностей. Такие Банки содержат информацию о ты-
тысячах фрагментов геномов организмов различных видов и их бел-
белках. С каждым годом эта информация практически удваивается,
однако, одна из основных задач молекулярной биологии и генети-
генетики - выяснение функционального смысла этих фрагментов макромо-
макромолекул - далека от окончательного решения.
Кроме накопления этих данных постепенно расширяется фронт
экспериментальных исследований, охватывающий изучение структу-
структурно-функциональных характеристик отдельных генов и белков. В
экспериментах показана сложная блочно-комбинаторная структура
регуляторных районов генов, особенно генов эукариот.
Это означает, что актуальны исследования структурно-
функциональной организации определенного района ДНК с привле-
привлечением всех накопленных знаний, которые позволили бы сформули-
сформулировать потенциальные функциональные возможности этого района
ивыявить его структурные особенности. Такая стратегия помогает
планировать конкретные эксперименты, которые можно провести,
определяя функциональную роль генетического текста в данной
молекулярно-генетической системе.
В связи с этим нами разрабатывается система функциональной
диагностики генетических текстов, которая использует информа-
информацию из базы знаний о структуре и функциях генетических сигна-
сигналов, мощный пакет программ анализа последовательностей ДНК,
РНК и белков и пакет программ классификации данных. Такая сис-
система необходима для эффективного проведения генно-инженерных
работ по конструировании молекулярно генетических систем с за-
заданными свойствами, а также для интерпретации данных в теоре-
теоретических и экспериментальных работах с генетическими текстами.
Общая схема системы приведена на рис.1. На этой схеме в
двойной рамке отражены модули системы, которые уже действуют в
виде комплекса программ на IBM-PC. Остальные блоки находятся в
стадии разработки и реализованы лишь их простейшие варианты.
Интеллектуальный
интерфейс
общения
с пользователем
Модуль
вывода
решений
База знаний
функциональных
сигналов
знания
о стратегии
анализа
последовательностей
База данных
функциональных
структур *
Пакет программ
вявления структурно-
функциональных характе-
характеристик в заданных
по следовательностях
пополнение
Базы
выявленными
характерис-
характеристиками
Интерфейс связи
с Базами данных
нуклеотидных и
аминокислотных
по следовате льно с тей
Рис. I. Общая схема системы функциональной диагностики
генетических текстов.
2.1. Описание реферата базы данных по функциональным сайтам.
База функциональных сайтов включает данные, полученные ре-
реферированием научной литературы, структура реферата дает пред-
представление о содержании базы, принципах сбора и описания инфор-
информации.
Предметом реферирования является один или несколько сходных
(по функциям; районов генетических макромолекул - ДНК, РНК и
белков. Каждая из этих последовательностей может содержать вы-
выявленные функциональные сайты, повторы или элементы вторичной
структуры и другие значимые характеристики, которые заносятся
- 13 -
в реферат и в дальнейшем в базу данных на ЭВМ.
Разработанный нами формат реферата максимально учитывает
' современные тенденции в этой области-* при описании особеннос-
особенностей функциональных районов используются многие принципы, пред-
предложенные в новом едином формате баз данных нуклеотидных после-
последовательностей (GenBank/EMBL/DDPJ). Кроме того, учтен опыт со-
создания базы знаний по функциональным сайтам в ИМГ АН СССР
(разработчики D. А. Каламбет и С.А. Сприжицкий).
2.2. Краткое описание полей.
В реферате отражаются структурно-функциональные особенности
генетических макромолекул, последовательность каждой из кото-
которых рассматривается введенной в направлении от 5'- к 3'- (вво-
(вводится одна нить ДНК или РНК) или от и- к с-концу для аминокис-
аминокислотной последовательности. Нумерация начинается с единицы, ко-
которая соответствует первому элементу вводимой последовательно-
последовательности.
Каждое поле реферата имеет свое название и формат записи
данных. Текстовые данные, по которым может быть осуществлен
поиск в базе функциональных районов, будем называть ключами.
Рассмотрим по порядку следования поля реферата.
1. Data.- дата заполнения и последней модификации реферата-
Пример.- 20.11.88/modiГу=22.11.89
2. identifier.- идентификатор записи Сключ длины до 10 символов,'.
Идентификатор записи должен быть уникальным в БД, начинать-
начинаться с буквы латинского алфавита и не содержать пробелов. В
идентификатор рекомендуется включать следующие составные час-
части; первые буквы латинского названия 'хозяина?' (или таксономи-
таксономической единицы;, в которых функционирует данный район; симво-
символы, отражающие функцию района C-6 символов); символы, отлича-
отличающие описываемые последовательности от других районов сходного
типа B-5 символов).
Пример: TAHTPII1TR (ТА - Trltlcum aestivum,
НТ - Mitochondrlal DNA, Pill - RNA polimerase III
promoter, TR - tRNA).
3. Signal name: название функционального сигнала
(ключ длины до 32 символов).
В базе данных существует библиотека общцх данных о функцио-
функциональных сигналах и их словарь. Эта информация включает тип мо-
молекулы, на которой функционирует определенный сигнал; процесс,
который он регулирует; общие сведения о молекулах, которые
участвуют в реализации функции сигнала,и процессы, на которые
он влияет.
Пример: PROMOTER UNA POLYHERASE III PLANT MITOCHONDRIAL tHHA
4. Host organism: организм, в клетках которого функционирует
данный сайт (ключи длиной до 64 символов;.
Если описываемые последовательности относятся к различным
видам, то их необходимо перечислить в порядке, соответствующем
порядку записанных последовательностей в разделе реферата
SEQUENCE.
Примеры: Trltlcum aestivum
1 Canis famillarls
2 Canis lupus
5. Sequence source: источник последовательности данного сайта
гключи длиной до 64 символов;.
Это поле предназначено для уточнения источника последовате-
последовательности. В нем отражается извлечение молекулы из органелл, ви-
вирусов, другого организма, искусственный синтез. Кроме того, в
этом поле можно указать номер соответствующей хромосомы орга-
организма хозяина.
Примеры: ARTIFICIAL, MITOCHONDRIAE, CHROMOSOME 5
1 ADENOVIRUS TYPE 2
2 ADENOVIRUS TYPE 5
6. Precursor site: идентификатор записи, описывающей сайт-
предшественник для мутантного сайта или
района (ключ длины до (С символов;.
Данное поле присутствует только в записях, описывающих му-
тантные сайты. Отличия мутантного сайта от "нормы" можно опи-
описать в таблице особенностей.
7. Keywords: ключевые слова или группы слов, характеризующие
функциональный район (ключи длины до 64 символов;.
Пример: PLANT,tRNA, promoter RNA polimerase III, mltochondrlae
в. cement: краткое описание особенностей функционирования
данного сайта fтекстовые поля длиной до 10 строк;.
В комментарий следует включать информацию, которая не опи-
описана в других полях.
- 15 -
Пример: No consensus eukaryoyic nuclear RNA polymerase II
promoter, no consensus -10 and —35 region as are found upstre-
upstream of some chloroplast tRNA genes. A pur Ine—rich motif is
found upstream of all the wheat mitochondrial tRNA genes,
the first 9 n. of wich are Identical except at one position
with the yeast mitichondrial promoter: ATATAAGTA 121.
Если комментарии относятся к отдельным описываемым района*!,
то их нужно нумеровать в соответствующем порядке.
9, References: краткие библиографические ссылки ( Первый автор,
журнал.том.-страницы (год)
Пример: 1. Suboch G.M., NAR 13:456-46711986)
Номера ссылок могут указываться в любом месте реферата в
квадратных скобках.
10. Feat-.зге table: Таблица особенностей-
Таблица особенностей предназначена для описания главным
образом следущих характеристик функциональных районов:
1. Структурной организации: наличия повторов, вторичной или
третичной структуры;
2. Функциональной организации: взаимодействие с другими
молекулами, функциональная роль отдельных фрагментов,
эффективность их функционирования;
3. Эволюционных особенностей: локализации мутаций, их вли-
влияния на функцию-
Принципы описания этих характеристик в основном соответст-
соответствуют новому формату баз данных нуклеотидных последовательнос-
последовательностей (GenBank/EMBL/DDPJ), однако есть отличия, связанные с опи-
описанием в одном реферате группы функциональных районов-
Описание структурно-функциональных характеристик осуществ-
осуществляется с помощью трех элементов;
1. Feature key - ключ характеризующий тип особенности
(имеется словарь таких ключей);
2. Location - локализация особенности в последовательности;
3. Qualifier - дополнительные сведения об особенности
(имеется словарь типов таких сведений).
Пример: Feature key '.Location/Qualifiers
районов, то она указывается один раз, как показано в этом при-
примере- При различном расположении указывается локализация осо-
особенности в каждом районе.
Пример:
HISC_SIGNAU''/UBLE=5'-bo;'/FINCTI0N="»'!*n<""!°-
-'/NOTE=**son'e homology sites are near it"
_1!44. .55/
,'/Ц0ТЕ="ГЛе tlrst 9 n. are aligned with the yeast
,'mltochondrtal promoter ATATAAGTA 121"
_2!24..35
J3120. .31
_4',79. .90
5! 65..76 „
TRNA
J 151. .122/note="location in alignment sequences"_
Если локализация особенности одинакова для всех описываемых
Отметим, что дополнительные сведения, касающиеся всех учас-
участков, описываемых данным ключевым словом, даются вначале, а
сведения, относящиеся к конкретному району - после указания его
локализации.
Если в реферируемой работе приводится выравнивание последо-
последовательностей участков с данной особенностью и построена их ко-
нсенсусная последовательность, то после ключа "asequence" мож-
можно привести эти данные. Это обязательно в двух случаях: когда
участки имеют разную локализацию или если последовательности
рассматриваемых функциональных районов не выравнены.
Пример;
ASEOUENCE !б> -Ьох
t ! ATATATGAAAAG
2!ATGTAAGAAGAA
3!CTACCGGАЛААв
, 4! ACTTAAGAACGA ;
. 5/ AGTAAAGAAGAG
В строке ключа "ASEOUENCE" должна стоять метка, присвоенная
ранее этим участкам квалификатором "LABLE"-
Консенсусная последовательность описывается ключем
"CONSENSUS". ' который также включаетсяя с соответствующей
меткой.
Пример:
CONSENSUS •'5' -ь°х
1NNHNAAGAANRR
- 16 -
- 17 -
Влияние на экспрессию или относительную эффективность опре-
определенного фрагмента можно задать с помощью ключа "efficiency".
Квалификаторы "condition" и "relative" описывают условия экс-
эксперимента, в котором получены эти данные, и величины относите-
относительной эффективности функционирования. Если эфективность изме-
измерена относительно какой-либо другой последовательности, то ее
яужно указать с помощью квалификатора "note".
Пример.-
EFFICIENCY— '• 5'—box/condition^" concurrent expression in vitro"
l,'high/relative=1.0/note="most strong expression"
2,'hlgh/relatlve=0.8
3!low/relative=O.1
jt:mldlle/relatlve=O.S_
_s:low/relatlve=O.2
После описания таблицы особенностей в реферате представлены
последовательности функциональных районов. Если они выравнены,
ТО ДЛЯ ПОЛЯ Sequence указавается квалификатор "alignment",
значение которого означает "пределы" выравнивания внутри райо-
районов. При различной локализации выравненных частей функциональ-
функциональных районов данные квалификаторы следует пронумеровать.-
SEQUENCE' * • 'ALIGNMENT^ • • 60
2 /ALIGNMENT^5¦-84¦
Пример:
11 Sequence. /A_INGHENT=1• -60
10 20 30 40 SO 60
1.GGTTAAAAAAAAAAAAAAAAAA—GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG
2.TT-AAAAAAAAAATAAAAAAAAAAAAAGGGGGGGGGGGGGGGGGGGGGGGG—GGGGGAA
2.3. Пример реферата функциональных районов.
Data.- 20. Ji.se/noDIfy=22. 11 .89
Identifier: ТЛМТР1ПТ
Signal паше: PROMOTER RNA POLYMERASE III
Host organism: TR1TICUM AESTIVUM
Sequence source: M1T0CH0NDR1AE
Definition: J. Gene tRNA ASP
2. Gene tRNA PRO
3. Gene tRNA SER/f(rffE="antlcodon GCT"
4. Gene tRNA TYR
5. Gene tRNA SER/n(y^^"anttcodon TGA"
Keywords: PLANT, tRNA,promoter RNA polimerase III, mitochondriae
Coaaent: Wo consensus eukaryoyic nuclear RNA polymerase II
promoter, no consensus —10 and —35 region as are found upstre-
upstream of some chloroplast tRNA genes. A purine-rich motif Is
found upstream of all the wheat mito— chondrlal tRNA genes,
the first 9 n. of wich are identical except at one position
with the yeast mltichondrial promoter: ATATAAGTA [21.
References: 1. Jouce P.B.M., Plant Mol.Biol. 10:251-262A988)
2. TabakH.F., CRC Crit.Rev.Biochem. 14:297-317A983)
3. Borsuk P., NAR 14:7508A986)
Feature key ', Location/Qualifiers
TRNA-
_,' 151. . 122/ц(у%?="location in alignment sequences"_
MISC-SIGNAL—¦'/LABLE=s'-bo*/FUNCTION="u'!*'!OII''!*'
,'/КОТЕ=°ЯОЯ1е "omology sites are near It
!44..SS/
^/le first 9 n. are aligned with the yeast
;mltochondrlal promoter ATATAAGTA 121"
ASEOUENCE
_2i24. .35
_3!20..31
_4!79..90
_5165. .76
/5'—box
1!atatatgaaaag_
21atgtaagaagaa_
3ictaccggaaaag_
4!acttaagaacga_
5:agtaaagaagag_
CONSENSUS ¦'5' -b°x
INNNNAAGAANRR
EFFICIENCY— ''5'-i>ox/coNDITION-"concur'"en' expression in vitro"
l/A/g/i/RELATIVE^1 -O/nQ^^="most strong expression"
— 2! h i g/i/RELATI VE=° •в
3! 1 (""/RELATIVE^ • J
- 19 -
_ ТЯ _
Coatinue
- Г ?";¦-/¦ ?Т7ГТТТП
¦ . ГГГТИЛАТГГГП
утгТТТТЛАЛЛТТ
-) .GTTTAAAAAATTT
' 5. ТТТТТТТТТТТТТ
'гл. Структура организации данных.
Важной задачей при создании баз данных является простота и
удобство как заполнения, так и доступа к имеющейся информации.
Фундаментом для её решения является правильно выбранная на на
чальном этапе разработки структура организации данных. При
этом должны быть выделены основные типы информации, обеспечена
возможность хранения полных сведений по каждому типу и быстро
го получения разнообразных выборок.
- 20 -
с,"
- Op и
- ЛЦ1 "I 1-1.
ва и '
- Fpa-к
n 1KB
j к а т а л о
i консенсусов стр
I- тип структуры
|- тип сигнала
- последовательно
сть
Рис, 2, Схема организации базы функциональных сайтов.
Описываемая здесь база данных по функциональным сигналам
имеет структуру близкую к реляционным базам данных. (Рис, 2'.
Вся информация хранится в нескольких каталогах, что позволит в
будущем расширять базу за счет образования новых каталогов дли
новых типов информации, Например,-о мутациях в сигналах. Еди-
Единицей хранения информации в каждом каталоге является запись.
Для каждого каталога основные поля записей приведены на рисун-
рисунке.
2.5. Режимы работы с базой.
Для работы с базой данных функциональных сайтов, помимо
стандартных режимов записи и поиска, разрабатывается режим
анализа хранящихся данных. При работе в этом режиме можно бу-
будет подготовить данные в необходимом формате и воспользоваться
любой программой из библиотеки программ анализа, которая пос-
постоянно пополняется. Ряд данных, полученных в результате такого
- 21 -
анализа, будет заноситься в качестве новой информации в саму
базу. Основные возможности для работы в диалоговом режиме при-
приведены ниже.
ЗАПИСЬ - внесение новой информации
- дополнение/изменение информации базы
- запись результатов работы программ анализа
ЧТЕНИЕ - каталогов
- данных по ключам
АНАЛИЗ - поиск по ключам
- формирование выборок по ключам разной
сложности
- построение консенсуса, частотной матрицы и
обработка данных другими программами
пакета "Контакст"
ПОДГОТОВКА ДАННЫХ
для специализированных программ и пакета
- выборка по ключам
- переход к определенному формату
представления данных
Таким образом рассмотренная структура базы данных, позво-
позволяет накапливать информацию о функциональных сайтах и подгота-
подготавливать ев для анализа с помощью специализированных пакетов
программ, описанных далее.
3. Поиск информативных характеристик, определявших
структуру функциональных районов.
Нуклеотидные последовательности функциональных районов оп-
определяют особенности опознания и функционирования генетических
сигналов. Наряду с такими легко конструируемыми и явно значи-
значимыми характеристиками как консенсусные последовательности, су-
существенными для функционирования часто являются энергетические
и геометрические параметры двойной спирали ДНК, наличие разли-
различных прямых повторов и элементов вторичной структуры, характер
нуклеотидного или динуклеотидного состава. Для задания и поис-
поиска таких характеристи мы использовали развитое программное
обеспечение разработанного нами пакета анализа генетических
текстов "Контекст" [1,2]. Данные, полученные из анализа после-
последовательностей, оцифровывались специальной программой
- 22 -
"Feature" и подвергались в дальнейшем дискриминантному анали-
анализу. Общая схема поиска информативных характеристик генетичес-
генетических текстов (нуклеотидных и аминокислотных последовательнос-
последовательностей) приведена на рис 3.
ПРОГРАММЫ
формирования выборок генетических текстов,
выполняющих сходные функции
РАСЧЕТ
характеристик генетических текстов с помощью
программ пакета "Контекст" (для каждой
последовательности из выборки)
ОЦИФРОВКА
характеристик генетических текстов с помощью
программы "Feature"
ДИСКРИМИНАНТНЫЙ АНАЛИЗ
выборок генетических текстов на основе
полученных характеристик и выявление информативных
характеристик, разделяющих выборки
ПОСТРОЕНИЕ МОДЕЛИ ФУНКЦИОНАЛЬНОГО СИГНАЛА
и функционала, осуществляющего его поиск
в заданных последовательностях
Рис.3. Блок схема системы поиска информативных характеристик,
определяющих функционирование генетических сигналов.
3.1. Возможности пакета программ "Контекст" для расчета
характеристик генетических текстов.
Пакет программ "Контекст" позволяет производить всесторон-
всесторонний анализ последовательностей ДНК, РНК и белков, включающий
- 23 -
как выявляло внутренних характернее и-- нуглеотидных и ;. ки-
кислотам?; "оолеловательнпстэй, так и быстрс" сравнишь ;г воя-
вояками данных. Этот пакет часто использовался для интерпретации
полученных экспериментальных данных, а также для плакирования
ряда биохимических экспериментов [3,4]. Пакет позволяет прово-
проводить комплексный анализ нуклеотидных к аминокислотных последо-
последовательностей и включает следующие группы программ:
Программы ввода, редактирования и анализа последовательностей.
Имя
программы
SEQ
PLAYS
GOMOL
REPEAT
ТАВЫ
GOMOLA
ТАВА
TRACT
PISTR
Функции
программы
Зедактор последовательности. Предназначен для
ввода, проверки и редактирования последовательно-
последовательностей ДНК, РНК и белков. Для повышения надежности
работы программа периодически осуществляет запись
последовательности в буферный файл.
Проведение различных манипуляций с последователь-
последовательностями. С её помощью можно производить объеди-
объединение произвольного набора последовательностей,
редактирование, печать последовательностей и т.д.
Осуществляет быстрый поиск групп несовершенных
повторов в последовательности, содержащей тысячи
нуклеотидов. Производит статистические оценки
ожидаемого числа повторов, границы 95% довери-
доверительного интервала и выявляет группы неслучайных
повторов.
Поиск неслучайных повторов оптимальной длины.
Анализ и компактный вывод результатов программы
"GOMOL" И "REPEAT".
Выполняет операции,сходные с программой "GOMOL",
но для аминокислотной последовательности
Анализ и компактный вывод результатов программы
"GOMOLA".
Определяет частотный состав трактов от 2 до 6 в
составе набора нуклеотидных последовательностей.
Выявляет статистически достоверные участки консер
вативные в группе последовательностей
Строит распределение трактов по длине последова-
последовательности.
- 24 -
SCREEN
7.ОШ
REPBAN
DESIGN
ruiacTOK
.!'. 'I. 3,0,.
aj-шт i.
¦ЛТЪ ОЬ
¦али;
ЗДУ
¦,1ЬНъ
¦"НОС Г Я
JiOK •
яо три
, детей у
'. в 1'рех ; .-.«•
открытой ры.
.ктеристик нуклботидны.^
аких как частоты мояо-, -¦:
гии формирования двойной
.1'ме> ДНК; угол локальной закру
о гомЪ"логичных~районов в группе~последог
.вестей. _____
¦жрованио случайных последовательностей
•чшх типов. .
^т ожидаемого числа и 9^% ловерительногс
цтервала для несовершенных повторов.
истрый поиск по Банку данных нуклеотидных пос-
>довательиостей "БЛНП" фрагментов гомологичных
аределенной последовательности, йспольоует
етод контекстного анализа L5J. .
истрый поиск по банку данных аминокислотных пос-
.леловательностей фрагментов,гомологичных опреде-
определенной последовательность. Использует метод
контекстного анализа [5].
Быстрый поиск по БДНП гомологичных последователь-
ностей на основе статистики по олигонуклеотидам.
Быстрый поиск по БД фрагментов;гомологичных корот
кой последовательности без статоценок.
Поиск неслучайных гомологии между исследуемой
последовательностью и списком последовательностей
из Банка данных нукдеотидных последовательностей.
Вывод последовательности в различных форматах с
указанием функциональных сайтов, кодирующих
областей и т.п.
Программы анализа функциональных сайтов.
поиск фрагментов неслучайной гомологии с консенсу-
консенсусом в нуклеотидных последовательностях
поиск фрагментов неслучайной гомологии с консенсу-
консенсусом в аминокислотных последовательностях
- 25 -
NABOR
WESMAT
WESISK
WESISD
WESOPT
PEROB
PERAS
PERISK
PERISM
PERISD
PEROPT
STATTR
программа формирования обучающих выборок в
заданном формате
программа формирования частотной матрицы w, распе-
распечатка матрицы и анализ отдельных сайтов из выборки
программа поиска сайтов в последовательностях ДНК
и РНК на основе частотной матрицы w.
программа поиска сайтов с делениями и вставками на
основе частотной матрицы.
программа выравнивания (оптимизации) выборки
сайтов на основе использования частотной матрицы W
программа формирования весовой матрицы w на основе
обучающих выборок по алгоритму перцептрон.
распечатка матрицы W и анализ отдельных сайтов из
обучающей выборки.
программа поиска сайтов в последовательностях ДНК
и РНК на основе матрицы w по методу перцептрон.
программа поиска сайтов на основе нескольких
матриц W.
программа поиска сайтов с делециями и вставками на
основе матрицы w.
программа выравнивания (оптимизации) выборки
сайтов на основе использования матрицы W.
выявление консервативных зон в наборе последова-
последовательностей на основе результатов программы TRAKT.
Программы построения выравнивания последовательностей и
вторичной структуры РНК.
FOLD
FOLDT
FOLDN
FOLDS
RP
ALIGN
расчет вторичной структуры длинных РНК на основе
результатов работы программы "REPEAT"
расчет вторичной стуктуры на основе совершенных
инвертированных повторов
расчет вторичной структуры РНК на основе
модифицированного алгоритма Нуссинов
расчет вторичной структуры РНК на основе
моделирования её самоорганизации
объединение массивов с повторами
выравнивание последовательностей на основе резуль-
результатов работы программ REPEAT или REPBAN.
Многие программы пакета включают статистические оценки
достоверности выявленных результатов анализа
последовательностей, разработанные в Теоретическом отделе ИЦиГ
СО АН СССР [5], что выгодно отличает их от ряда часто
используемых программ анализа генетических макромолекул. Все ¦
программы пакета являются оригинальными, оригинальны также
большинство алгоритмов, такие как быстрое сравнение с базами
данных, анализ функциональных сайтов.
3.2. Принципы и режимы оцифровки характеристик
генетических текстов.
С помощью описанных программ осуществляется расчет характе-
характеристик генетических текстов. При этом наиболее часто использу-
используются следующие функциональные возможности пакета "Контекст":
PLAYS
GOMOL
СОМРО
TRACT
WEIGHT
PERCEP
поиск участков Z-ДНК
поиск политрактов
поиск неслучайных прямых повторов
поиск неслучайных инвертированных повторов
поиск неслучайных симметричных повторов
расчет частот моно-, ди- и тринуклеотидов
расчет энергии фрагмента ДНК или РНК
поиск неслучайных трактов длиной до б-ти
нуклеотидов
поиск функциональных участков с помощью
весовой матрицы
поиск функциональных участков с помощью
матрицы алгоритма перцептрон •
Использование более сложных характеристик планируется при
дальнейшем развитии системы. Найденные характеристики, для
оценки их значимости на основе дискриминантного анализа, под-
подвергаются оцифровке, то есть перекодировке в числовые величины
Программой "Feature".
3.2.1. Оцифровка характеристик структур
(или участков генетических текстов)
Основные характеристики можно разделить на два класса: пер-
первый .Отражает локализацию структур в последовательности. Второй
описывает особенности, характеризуемые определенными числовыми
- 26-
- 27 -
величинами (называемыми весами), которые позволяют отличать
эти структуры друг от друга. Введено два типа описания струк-
структур: I) учет средних параметров всей совокупности структур; 2)
учет параметров линь для структуры с максимальным весом. Для
оцифровки структур использовались следующие характеристики по-
последовательности:
1) число структур в последовательности;
2) значение веса для структуры с максимальным весом;
3) локализация структуры с максимальным весом;
4) расстояние от заданной позиции структуры с
максимальным весом;
5) среднее значение параметра, характеризующего
локализацию структур;
6) среднее значение веса для всех структур;
7) среднее значение расстояния всех структур от
заданной позиции;
При этом средние значения рассчитываются либо для всей за-
заданной последовательности, либо может быть задана длина участ-
участка усреднения. В последнем случае появляются две дополнитель-
дополнительные характеристики:
8) локализация участка с максимальным средним весом
структур;
9) значение среднего веса структур на этом участке.
Типичными представителями структур являются участки Z-ДНК,
политракты, повторы и функциональные сайты. Каждая из этих
структур имеет свои особенности, отражением которых являются
специфические рекимы оцифровки. Так, например, массив повторов
перед оцифровкой можно "отфильтровать", выбрав лишь определен-
определенные классы повторов. Для этого при анализе повторов использу-
используются следущие рекимы:
а) задание минимальной длины повторов;
б) задание минимального числа несовпадений между
участками повторов:
в) задание минимального % гомологии между участками
повторов.
Для повторов применялось четыре типа весов, характеризующих
эти структуры:
a) w = ь-к. где Ь - длина участка повтора.
- 28 -
а К -число различий между участками повтора;
б) w = 1/s, где s - ожидаемое число повторов (длины ъ
с к различиями) в случайных последовательностях;
в) w = d, где d - расстояние между участками повторов;
г) w = (L-K)/d -вес, отражающий вероятность
взаимодействия между комплементарными участками
повторов в двухцепочечной ДНК.
При анализе функциональных сайтов использовались следующие
веса:
а) w = п (n-число несовпадений с консенсусом);
б) w = 1/s, {S - ожидаемое число сайтов в последова-
последовательности );
в) w = r (R - расстояние между функциональными блоками
для сложных сайтов);
г) w = s (s - число несовпадений между определенным блоком
и его консенсусом).
Кроме того, для учета отклонений характеристик структур от
определенных значений, добавлены следующие режимы оцифровки:
Ю) отклонение локализации структуры от оптимального;
11) отклонение веса структуры от оптимального.
Эти режимы могут учитывать, например, такие характеристики
промоторов прокариот, как их практически строгое расположение
перед точкой инициации транскрипции, а также существенное
ухудшение эффективности их функционирования при изменении рас-
расстояния между -10 и-Э5 боксами.
3.3. Дискриминанткый анализ и построение функционала
для поиска функционального сигнала
в заданных генетических текстах.
Оцифрованные характеристики записываются в файлы прямого
доступа, которые далее обрабатываются программами
дискриминантного анализа. Блок-схема этой части системы
приведена на рис.4.
- 29 -
Считывание значений оцифрованных характеристик
генетических текстов для различных выборок
I
Дискриминантный анализ разделения
различных пар выборок функциональных районов
I
Вычисление векторов средних
и ковариационных матриц наборов
характеристик для определенных выборок
Вычисление расстояний Махалонобиса D
между выборками
I
Отбор наиболее информативных характеристик
для разделения различных пар выборок
по критерию Фишера
т
Запись векторов Фишера и векторов средних
информативных признаков, используемых для
построения функционала поиска генетических
сигналов
Рис. 4. Блок-схема комплекса программ выявления
информативных характеристик, и построения
функционала поиска генетических сигналов.
Рассмотрим произвольную пару выборок нуклеотидных или ами-
аминокислотных последовательностей, которые характеризуются раз-
различными функциональными свойствами. Например, последователь-
последовательности одной из выборок содержат генетические сигналы опреде-
определенного типа, а в другой они отсутствуют. Задача поиска харак-
характеристик, которые позволяют идентифицировать генетические сиг-
сигналы, решается методом дискриминантного анализа [6]. Пусть
Хцг - наблюдаемое значение J-ro признака для t-ой последова-
последовательности в J-той выборке, где j=-\,..., p ; i=i,2 ; t=i,..,,
л(. Здесь р - число признаков и л? - число последовательностей
в /-той выборке. Введем линейную дискриминантную функцию как
линейно независимую комбинацию всех признаков :
- 30 -
rlt~
A)
В качестве меры разделения выборок примем расстояние
Махалонобиса D между двумя выборками:
~'
где вектор Х{=
= (Xr&)'S,~' (Х,-Хг)
V;i=i,2
B)
*»= }*
a S» - объединенная ковариационная матрица
S» = 1 22 ?
С помощью D ищем набор признаков, давдий лучщее разделе-
разделение. Такие наборы искались независимо для каадой пары выборок
Изложим, кратко алгоритм выбора информативных признаков, i")
Анализируется разделение между выборками тип. которое дает
каждый признак а отдельности из всех признаков выбираем приз-
признак с наибольшим D . 2) Выбранный признак рассматриваем совме-
совместно с каждым из оставшихся для отбора. Выявляется пара приз-
признаков, даодая лучшее разделение выборок. 3) Аналогично добав-
добавляем, по одному признаку к уже выбранным к признакам до тех
пор, пока выполняется условие:
F = (л, + л2 - к - 2)
Я! {
¦»
где Fe(Pitu2) _ р-критерий Фишера на заданном уровне
значимости «, а
Ч
(л,
- 2
4. Пример применения системы для поиска характеристик,
. . отличающих "сильные- и -слабые- промоторы прокариот.
Промоторы прокариот являются одним из наиболее изученных
генетических сигналов, который определяет эффективность тран-
транскрипции и точку старта синтеза РНК. На основании тщательного
- 31 -
изучения нескольких сот нуклеотидных последовательностей
промоторов и их мутационных вариантов построен консенсус
промоторов E.coli и ее фагов, состоящий из -Ю и -35 блоков, а
также нескольких преимущественно встречающихся нуклеотидов
вблизи старта транскрипции (рис. б) [7]. Показана хорошая
корреляция мевду сходством нуклеотидной последовательности
определенного промотора с консенсусом и функциональной
активностью промотора. Поэтому предложено оценивать "силу"
промотора по соответствию с полученной усредненной структурой
[8-И].
Однако имеется обширный ряд данных, которые свидетельствуют
о том, что на активность промоторов, кроме структуры рассмот-
рассмотренных стандартных блоков и оптимальных расстояний между ними,
существенно влияют характеристики нуклеотидных последователь-
последовательностей.окружающих консенсус (см. обзор. [I]). Появляются также
прямые экспериментальные данные о том, что изменение "силы"
•a
v
О 40
a
V
D
20-
tcTTGACal t
t tg TAtAaT
-50 -40 -30 -20
номер позиции
-10
Рис. 5. Вверху представлен консенсус промоторов E.coii.
На гистограмме по оси ординат отложена частота
встречаемости наиболее представленного' нуклеотида [7].
промотора болеэ чем на порядок может зависеть от нуклеотид-
- 32 -
ных последовательностей, расположенных как до -35 блока, так и
после старта транскрипции [2]. Поскольку явных общих блоков в
этих районах для разных генов не наблюдается, это означает,
что более сложные характеристики определяют отмеченные эффек-
эффекты. Поиск таких характеристик представляется важным как для
выяснения молекулярных механизмов функционирования промоторов,
так и для учета их при конструировании генетических систем с
заданными свойствами в генно-инженерных экспериментах.
4.1. Выборки нуклеотидных последовательностей промоторов.
В данном случае демонстрация работы системы функциональ-
функциональной диагностики генетических текстов проведена на трех
выборках (А, В и С) промоторов различной степени активности.
Формирование выборок промоторных районов проводилось с учетом
данных работы К.в. Артемьева и соавт. [13], которые при
анализе 188 промоторов E.coli и бактериофагов, разбили их на
группы по эффективности транскрипции. Выборка А, содержала
наиболее сильные, В - средние, с - более слабые промоторы. Эти
выборки описаны в таблице I. Для анализа были взяты районы
протяженностью 150 н.п., в том числе юо н.п. до и 50 н.п.-
после точки инициации транскрипции. Нуклеотидные
последовательности промоторных участков около 80 генов
прокариот были взяты из гейдельбергской базы данных 1985-1987
„р., а также из ряда литературных источников.
4.2. Набор характеристик нуклеотидных последовательностей,
использованный при анализе промоторов •
В данной работе ставилась задача определения таких харак-
характеристик нуклеотидных последовательностей, которыми различают-
различаются, выборки А, В, С. Рассматривались следующие характеристики,
рассчитываемые с помощью пакета "Контекст".
' I. Энергия участка двойной спирали ДНК расчиты-^валась
программой "СОМРО" (начало отсчета в -100):
В, - энергия участка в районе 1 -50 ;
/%.' - энергия участка в районе 51-100;
,ВЭ. - энергия участка в районе 101-150,
'VII. Насыщенность, промоторов неслучайными прямыми повторами
.CDJ-или инвертиврованными повторами (I) с менее чем ю-ю раз-
линиями между их участками определялась с помощью программы
"GOMOL":
Dj - ЧИСЛО ПРЯМЫХ ПОВТОРОВ;
D^ - максимальное значение веса для всех повторов.
Для характеристики использовались следующие значения веса:
при U 1 W={ l-k )№;
при i= 2 w=i/s, где s - ожидаемое число повторов;
при i= 3 w=-in s.
bl3 - среднее значение веса для повторов»
Ii> 1г' 1з ~ аналогичные величины для инвертированных повторов,
III. Содержание в промоторных районах сайтов гомологичных
консенсусной последовательности, поиск которых осуществляется
с помощью программы "SITE":
Sj - число сайтов^
s^ - максимальное величина веса сайтов;
s^ - средняя величина веса сайтов,
Для характеристик сайтов используются следующие значения весов:
при i= 2 w= 1/s, где s - ожидаемое число сайтов;
при 1= 3 W= -in S;
S4 - среднее расстояние от точки инициации транскрипции,
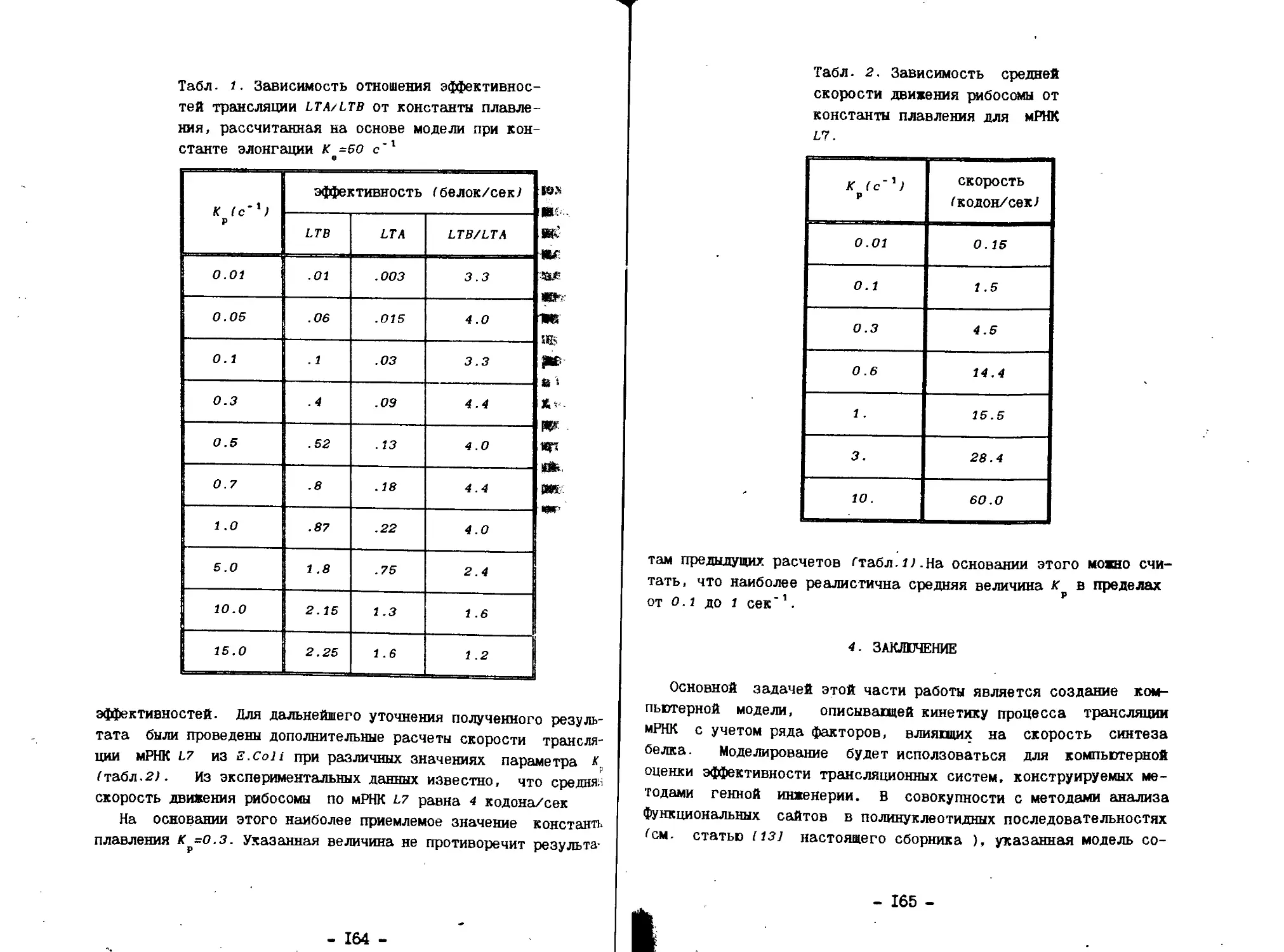
4.3- Факторы, разделявшие выборки промоторов
Средние значения характеристик и расстояние Махалонобиса
для 15 признаков приведены в таблице 2. Рассмотрим признаки,
которые разделяют выборки А и С - очень сильных и слабых
промоторов. Наилучшее разделение при использовании одной
характеристики дает признак s* . который отражает совершенство
консенсусной последовательности. Важность наличия консенсуса
для силы промотора отмечалась и ранее. Дальнейшее добавление
признаков, достоверно увеличивающее разделение выборок,
проходило в следующем порядке:
сстояние Махалонобиса
- это характеристика
консенсусов;
- число палиндромов в последовательностях;
отражавшая наличие нескольких
Таблица
I. Список
гены
сильных
промоторов
ТТ А1
Т7 А2
Т7 A3
Т7 АО
E.coli
E.coli
E.coli
E.coli
E.coli
E.coli
E.coli
E.coli
газ r
• LAMBDA
DO
RRN
RRN
RRN
RRN
RRN
RRN
RRN
RRN
PL
A
A
E
В
D
D
X
X
P1
P2
P1
P2
P1
P2
P1
P2
использованных
гены
средних
npOmuiuiJvjD
X174 В
X174 D
X174 A
G4 В
G4 С
FD 2
FD 8
FD 10
газ a
E.coli LPP
E.coli CMP A
E.coli UVRB
E.amylop. LPP
S.marcea. LPP
промоторов.
гены
слабых
птюмототют
PD 4
PD 2'
LAMDA PRM
PR'
PE(PRE)
E.coli ЬАС
E.coli RPL
)
KA
E.coli RPLJL-RPOBC
E.coli ARA -"-
E.coli ARA
E.coli ARA
E.coli REC
E.coli ARO
E.coli TRP
E.coli HIS
E.coli TUP
E.coli TUP
E.coli TRP
E.coli PHO
1 E.coli STR
TN3 BLA *
T7 С
BAD
С P1
С Р2
A
H1
В P1
В Р2
R
A
- 35 -
-34 -
Ej и Еэ - энергии плавления (или формирования двойной спирали
ДНК в районах -100 - -50 и I - 50.
Таким образом, сильные промоторные зоны, кроме наличия бо-
более совершенной консенсусной последовательности, характеризу-
характеризуются большей насыщенностью палиндромами, более легкоплавкими
нуклеотидными последовательностями в 5'-районах от промотора и
более "жесткой" двойной спиралью в районе за точкой инициации
транскрипции (см. величины средних значений в таб.2).
Достоверное увеличение набора признаков для разделения вы-
выборок А и В (очень сильные и средние промоторы) происходило в
следующем порядке:
Iрасстояние Махалонобиса
*з
0.99
К
3.17
Ег
5.62
7.02
Здесь,также как и в случае сравнения выборок А и С, главную
роль играет наличие совершенного консенсуса, что отражают ха-
характеристики (Sg и Sj ). Кроме того, более слабые промоторы
имеют более легкоплавкую зону -50 - О. Еще одной выявленной
характеристикой является наличие прямых повторов в районе бо-
более сильных промоторов.
Анализ рассматриваемых признаков для выборок В и С выявил
лишь один достоверный различающий их признак s|,
характеризующий более совершенный консенсус в выборке В.
Таким образом, наиболее важной характеристикой промоторов
прокариот является соответствие их нуклеотидной
последовательности консенсусу промоторов. Однако, как видно из
проведенного анализа, сила промотора может существенно
модулироваться такими факторами, как GC - богатость района
после точки инициации транскрипции (вклад этого параметра
достаточно высок, как видно из таблицы 2). Кроме того, сила
ггромотора зависит от наличия прямых повторов и палиндромов
на участке -100 - +50, окружающих точку инициации
транскрипции.
Также следует отметить, что, поскольку достоверное отличие
выборок А от С и А от В зависело не только от характеристики
максимально сходного с консенсусом фрагмента, но и от средних
характеристик таких фрагментов, то более сильные промоторы
отличаются наличием дополнительных фрагментов последователь-
последовательностей (кроме основного) сходных с консенсусом. Такие фрагмен-
фрагменты, содержащие несовершенные копии (мотивы) промоторных элеме-
о
V.
«t
о. cd
К О
{С ^
Н гв
ф а
а.
о S
а
X
ru го
ед
OJ CU
ед
со
cu го
со
ед
го го
- го
_
го го
го ги
о
— ги
о
^_
Гг>
LU
ги
ш
ш
о
ю
-о
Ц">
о*.
и>
о*.
"О
си
о
го
О
т
ii
г—
о
о
ги
иг>
IT-
ю
о
ги
—
го
fU
—
си
си
г—
—*
ю
ги
и>
о
j 94
си
о
ги
о
г-
о
ю
го
го
1--
1-ГО
ги
ю
ги
U/
Г1]
1»
го
о
ги
ги
о
ю
о
о
со
ю
*г
о
ю
w~
ю
^_
ю
ст.
го
CU
ч
CVJ
~
си
ги
о
с?.
СЛ
о
о
о
ю
о
о
о
о
о
о
о
—
о
си
о
СР.
ГУ
6
-3-
о
го
о
*
ги
о
су
го
о
ю
о
ГГ.
ст.
о
ю
о
о
Си
rJ
со
•ol
го
о
о
о
о
г—
nj
о
о
о
.41
о
о
о
о
г"
*
со
о
nj
о
о
nj
о
го
о
-
го
го
о
OJ
"~
о
о
о
о
о
о
ги
о
о
си
о
CXJ
о
•ol
¦ol
ст.
го
о
.99
о
со
го
о
го
о
J—
.01
г~
си
о
о
го .
о
.01
о
СО
о
**
о
о
Си
- 37 -
- 36 -
нтов, являясь сайтами связывания РНК- полимеразы, могу г повы-
повышать концентрацию этого фермента в промоторной зоне, тем са
мм, усиливая ее транскрипционную активность.
В заключение, следует отметить, что хотя с помощью
описываемой системы выявлены существенные характеристики,
которые можно интерпретировать как в теоретической работе, так
и использовать в генно-инженерных экспериментах, мы
рассмотрели далеко не все характеристики последовательностей
которые возможно получить на основе пакета "Контекст". Болев
детальный анализ промоторных районов на существенно
расширенных выборках этих районов будет опубликован в
следующих работах.
5. Заключение
Разработавшая нами система функциональной диагностики ге-
генетических текстов имеет широкие перспективы дальнейшего раз-
развития. Массовое секвенирование фрагментов генома и активные
экспериментальные исследования их функциональных особенности?
генерируют все больший объем информации, который невозможно
охватить в целом без определенного ее упорядочения и накопле-
накопления в базе данных. Поэтому развитие такой базы является весьма
актуальным не только для практического использования как исто-
источника сведений, но и для теоретического анализа на основе ком-
пьтерных систем. Особо сложная структурная организация функци
оналышх районов генов эукариот, многомодульное комбинаторна
строение их генетических сигналов позволяют считать необходи
мым дальнейшее развитие и расширение спектра возможностей ком
пьютерного анализа этих данных. Безусловно, кратко изложенна
в работе система является лишь начальным этапом этого пути, 1
настоящее время ее развитие происходит в направлении все боль
шей интеллектуализации режимов работы с данными и программами
Накопленный опыт анализа генетических текстов включается в си
стему в виде знаний о стратегиях анализа, которые различны дл
разных задач исследований.
Усложнение стратегии анализа увеличивается при переходе с
исследования одной последовательности к их выборкам, как э1
отражено на схеме.
В заключении следует отметить участие Тимошевской Е.А.
Селедцова И.А. в исследованиях, изложенных в §4, а так)
одна
после
- сравнение с БД
- выявление внут-
внутренней структуры
(повторы, вто-
вторичная структура
рамки и т.п.)
- поиск потенциа-
потенциальных функциона-
функциональных сайтов
| несколько
довательнос
- сравнение друг
с другом
- выравнивание
- анализ консерва-
консервативности локали- н
зации функциона-
функциональных сайтов
выборка
т и
- построение кон-
консенсуса, часто-
частотных матриц
- анализ консер-
ь вативных и ва-
вариабельных уча-
участков , их сопо-
сопоставление с фу-
функциональными
участками
- поиск значимых
характеристик
- классификация
- построение
деревьев
выразить благодарность Н.А.Колчанову за конструктивное обсуж-
обсуждение результатов работы.
ЛИТЕРАТУРА
•[1}. В.В.Соловьев, И.Б.Рогозин // препринт ИЦиГ СОАН СССР.
. Новосибирск. 1986. 70 с.
[2]. В.В.Соловьев и др. Введение в теорию генетических
текстов. Новосибирск. МГУ. 1987. 90 с.
[33. В.А.Потапов и др. // ДАН СССР. 1988. т.299. с. 1250-1255.
[4]. В.В.Капитонов и др. // Генетика. 1987. т.XXIII.
C.2II2-2II9.
' [5]. Н.А.Колчанов, В.В.Соловьев, А.А.Жарких. // Итоги науки и
техники ВИНИТИ АН СССР. Молекулярная биология. 1985.
т.21. с.6-37.
[6]. Болч Б., Хуань К. Многомерные статистические методы для
экономики. М,: Статистика. 1979. с.219-228,
t7]. Hawley D.K., Mellure W. // NAR 1983- v.11. p.223?-2556.
18]. Milligan M.E. et al. // MAR 1984. v.12. p.789-800.
19]. Staden R. // MAR. 1984, v.12. p.505-519.
[10]. Миронов А.А. ' Александров Н.Н. // Молекулярная биология.-
198?.- т.21. с.242-249.
tti]. McClure W.R. // Ann.Rev.Biochem.- 1985.- Vol.34.- p.171-204.
It?]. Higgins C.F. Genetic Engeneering. NY,1986. p.1-59
[133. Артемьев К.В., Васильев Г.В., Гуревич А.И. //
Биоорганическая химия. 1983. т.9. с. 1544-1557.
- 3S -
- 39 -
A SYSTEM TOR GENETIC TEXTS FUNCTIONAL DIAGNOSTICS.
V.V.Solovyov, A.K.Sallkhova, I.B.Rogozln.
Institute of Cytology and Genetics, Siberian Branch
of the Academy of Sciences of the USSR, Novosibirsk,
A computer system for analysis and search of genetic
macromolecules DNA, RNA and proteins functional characteris-
characteristics is described. This systea la created on IBM-PC and
consists from the next set of modules: Data Bass of functional
signals and functional structures, program package for genetic
texts analysis "CONTEXT", a system for eearehing informative
characteristics and Intellectual interface interacting with
user (Fig.l).
Section 2 is devoted for describing principles ana
formats of presentation data Rbout genetic xacromolecalee fun-
functional plcularitles, and also for Data Вата structure and re-
Blffis of working (Fig.2}.
Xa section 3 a system for searching lnfforaistlve characte-
characteristics is described (Fig.3). It consists Ггош packag:,
"CONTEXT" (part 3.1) lit consists fro» more than 30 programs}
program "Feature* for evaluating ьТ searched genetic text cha
ranteristice (part 3.2) and a sodsle for discriminant analysis
of sets of sequences fragments (part 3.3). Tha result of thi:«
system working are as reveal of regularities in functions'
structural genetic texts organisation, зо as functional, wht^>
permits to reveal certain genetic signals in target seqrsrenct-..
Section 4 presents sit example of using this system to s*.-
slysis of characteristics, determining activity of procaryulfз
genes promoter regions. It is shown that strong promoter re?! ¦
ens are characterised (except well—(ttuded coincidence to
consensus structure) by йоге siEpiyaelting ША regions in
-100 - +50 zone.
КОМПЬЮТЕРНАЯ СИСТЕМА ДЛЯ ИССЛЕДОВАНИЯ ЗАКОНОМЕРНОСТЕЙ
МОЛЕКУЛЯРНОЙ ЭВОЛЮЦИИ НА ОСНОВЕ СРАВНИТЕЛЬНОГО АНАЛИЗА
ГОМОЛОГИЧНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ.
И.Н. Шиндялов, И.Н. Кликунова
Институт цитологии и генетики СО АН СССР, Новосибирск, 680090
1. ВВЕДЕНИЕ
Определим информационную область, исследуемую в рамках
компьютерной системы, развиваемой в данной работе. Она
содержит совокупность сведений о генах, имеющих эволюционное
родство и характеризующихся гомологией первичной структуры.
Всю информационную область можно подразделить на четыре
класса, объединяющих достаточно однородные данные.
I. Нуклеотидные последовательности, соответствущие
кодирующим частям генов, интронам, спейсерам, молекулам РНК.
II. Описания гомологичного соответствия позиций
последовательностей (разные варианты выравнивания).
III. Структурно-функциональные характеристики белков и
РНК, кодируемых генами: пространственные структуры,
локализация функциональных районов, проекция
структурно-функциональных особенностей на первичную структуру;
IV. Системные характеристики кодируемых макромолекул:
геномные, тканевые, организменные, онтогенетические,
таксономические, эволюционные.
Рассматриваемая компьютерная система включает базу данных
и комплекс программ для анализа содержащейся в ней информации.
Отдельные программы представляют собой блоки, из которых могут
конструироваться программные средства для решения конкретных
прикладных задач.
На предлагаемой информационной области возможна
постановка ряда задач, обусловленных прежде всего неполнотой
описания имеющихся семейств генетических макромолекул.
Так, если мы располагаем информацией о
последовательностях макромолекул (I класс данных), то
представляет интерес получение информации II, III и IV
классов. Нетрудно также представить ситуации с различными
- 41
- 40 -
комбинациями исходных данных и возникающими в этой связи
задачами, направленными на реконструкцию более или менее
полного описания семейства макромолекул (I-IV классы данных).
Рассмотрим типичные задачи анализа гомологичных
последовательностей.
. 1. Задача выравнивания.
По первичным структурам генетических макромолекул
восстановить гомологичное соответствие позиций.
2. Задача таксономии.
По первичным структурам гомологичных макромолекул
восстановить взаимосвязь таксонов.
S. Задача исследования закономерностей эволюции.
По первичным структурам выравненных гомологичны
макромолекул и - о привлечением данных о ь.
структурно-функциональной организации, восстановит
последовательность ключевых событий их эволюционист;
возникновения,
4. Задача структурно-функциональной характеристики.
По первичным структурам выравненных гомологичк ¦
макромолекул реконструировать особенности ил
структурно-функциональной организации.
Для решения указанных задач нами разработана компьютерная
система исследования закономерностей молекулярной эволюции,
описанная ниже, включающая базу данных гомологичны/
нуклеотидных последовательностей и комплекс программ для
исследования закономерностей молекулярной эволюции на основе
сравнительного анализа гомологичных последовательностей.
2. БАЗА ДАННЫХ ГОМОЛОГИЧНЫХ НУКЛЕОТВДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Описание гомологичных последовательностей дается i ля
групп родственных макромолекул, объединенных в семейства.
Описание семейства включает следующие типы информации:
1. Последовательности членов семейства.
2. Выравнивание последовательностей (варианты).
3. Реферат для сзмействе и д,пя отдельных
последовательностей.
Реферат- для семейства включает следующие .информационные
поля:
IDE - идентификатор ;;еме*;гва;
FAM - название- семействе:
дои - список организмов, для которых з оанке имеется
последовательное? и;
HUM - число генов данного семейства й банки;
LEH - длинй л* г л Uj ii с ~-ча
SEQ - сгисс ^.r ifi'tc о, t Д1 "^t-w ч.^; •
DAT - дат 1 1 trii > fji ли ira, рефврата;
KOD -ФИО Г! г, ^ л . Asg!C
CLA - icrav о гек t. 6^i^cl „ f i емейства;
GRO - КЛ<з<"си с \ 1 ' i Щ.Л ii
FUH - ФУ1--СЦ ^ I
EXP - ОС" --f i 1 I и- ,r i
8EL - спиг! v if ч^. ' ' _ "*<
HSE - гомс т) j »¦ не имевшими
СОР - взаимс л i ¦- «с «I :го гзьа с ат.:ут^мя
белкэ^ ^ *
COG - взаим^ f-"" ft fi oi
гену, t , iri "i
REH - регуяч!» )Г ud ( ,
REG~- влиляис n wo ' " \ ! trtv*
гена и % -7л 1- г > -1 л ч
этого п' л и i r 1 О \а
НОМ.- степень гомологии внутри семейства и "лежду отде^(ькыми
группами;
ST8 - ключи;
PSE - харктеристика псевдогенов;
D - посттрансляционная модификация, деградация;
данного гека;
-¦ э едяцяи даыесте
.у гена или фунжшю
-42 -
RAT - скорость эволюции семейства:
В'№ - характеристика внутренних повторов;
1ST - информация об нитронах;
¦SIG - информация о сигнальной последовательности;
REF - список литературных источников;
СОН - общий комментария к семейству;
KEY - дополнительные ключевые слова;
Помимо реферата для семейства в целок1, для казкл
последовательности семейства имеется отдельный рефера:
содержащий описание особенностей данной последовательности, -
присущих всем последовательностям семейства.
Реферат для отдельной последовательности вк.лочг
следующие информационные поля:
IDE - идентификатор нуклеотидной последовательности;
FAN - название семейства;
GEN - положение данного гена в классификации (C1.AJ:
SOU - организм - хозяин данного гена;
len - длина последовательности гена;
DAT - дата ввода и последнего редактирования реферата;
KOD - Ф.И.О. референта и редактора;
Блок полей "особенности".
SkS - характеристика источника, из которого выделен гея;
INF - ключевые слова о последовательности;
НЕТ - гетерологическая экспрессия гена, особенности;
OVL - информация приводится, если введенная последовательное
кодирует более одного белкового продукта;
VAH - таблица вариантов нуклеотидной последовательности;
SIN - функциональная разметка нуклеотидных
последовательностей;
SIP - функциональная разметка белковых продуктов данного геы
PSE - харктеристика псевдогенов;
REF - список ссылок по реферату;
REV - список ссылок по последовательностям;
KEY - дополнительные ключевые слова;
- 44 -
3. КОМПЛЕКС ПРОГРАШ ДДГ ИССЛЕДОВАНИЯ ЗАКОНОМЕРНОСТЕЙ
молекулярной эволпник на c-jhobe сравнительного анализ*
гомологичных последоватежнхтг;?. ,
Этот комплекс в настоящее время содокаит ? ос
программ.
гу геупг
3.1 ПОСТРОЕНИЕ ФИЛОГЕНЕТИЧЕСКИХ ДЕРВЬЬЕЕ Ш ОСНОВЕ АНАЛИЗА И
ОБЪЕДИНЕНИЯ ЭЛЕМЕНТАРНЫХ ТОПОЛОГИЙ,
На вход программы подается набор выравненных гомологичных
последовательностей. На выходе получается гопо.яогчя
филогенетического дерева, которая указывает эволюционные
взаимосвязи между последовательностями :>5ис. 1}
Метод построения филогенетически*, деревьев ка осязай
анализа и объединения элементарных тэгологий, яеляицийоя о шона
из вариантов реализации принципа совместимое.ги, теодетй'геакк
рассмотренного в работе ?13, состоит и? трех Злаков;
С1. Блок пос-гюешя списка достоверкж. "четздрг •?"
(топологий для четырех последовательностей;,
С2. 'Блок построения дерева не, основе обг-эдинйГ-ия
достоверных "чвтверо:-см.
СЗ. Блок анализа м редактирования деревьев.
В ходе работы программы пеа/ыцоБательк,': применяются бмо^т
С1, С2, CS.
Рассмотрим более подробно функции первчислезшых Ялажов
(см. Рис. 1).
БЛОК С1. ПОСТРОЕНИЕ СПИСКА ДОСТОВЕРШХ "ЧЕТВЕРОК".
На вход блока подается набор выравненных гомологичных
последовательностей и задается порог' по достоверности fq. Для
построения спиыса "четверок" анализируются все возможные
"четверки" и выбираются те из них, для которых возможно
достоверное восстановление топологии дерева (без корня). Для
четырех после дователъностей возможно, три альтернативные
топологии, из которых выбирается одна. При этом оценивается
достоверность однозначности выбора.
Для оценки достоверности используется следующий подход
f^! (Рис. 1). Пусть Nj, N2, 1ц - числа совместимых позиций по
;.
;
4
i
2
1
i
'3CDEF04IJKL
;ea3«aaataac
t*fttgtaattta
1-121-111-33
s Ч /
V1 ,2
S,.2 ,
^ Чз
F.0.042
r"l
ac
?at.a«gccta
acgttcggctac
gtatctatattg
1
2
5
ABCDEPOKUKL
ttaigtaattta
acgataggccia
8 « с g
tL ? ?
я -о з
>~~'С 5
н,-з" г
~ ^ ^
n,.c ,
> <.,
?.0.037
¦i
4
1
г
i
5
ABCDEFGHI.'KL
tc«ap^%ataac
ttatgtaa?t;ta
scgttcggetac
a,»o 4
¦> 45
Uj.2* 2
/"" 4^
Ц..В
" 'Q
Г.0,411
->
J
1
3
1
4
ABCD
tcaa
EKHUKL
gaaataac
scgatagyccts
acgt
NS3"
tcggctec
1 4
in
2
3
2
3
4
2
5
ABCTEKb
ttatgtaa
acg&tags
acgttcg?
••¦j.j ^
,> ч
?»o.031
трем группам, соответствующим топологиям . i,
максимальное из Ы^, Mg, N3; ш - номер
НяН-+Н„-«-К3 - полное число анализируемых
ш
2, 8;
этой топологии;
позиций. Тогда
вероятность того, что топология ш соответствует реальной для
рассматриваемой четверки последовательностей составляет;
1к
си
Далее "четверки" упорядрчиваются по достоверности и из полного
списка "четверок" выбираются наиболее достоверные с помощью
порога по достоверности ?«,
БЛОК С2. ОБЪЕДИНЕНИЕ ДОСТОВЕРНЫХ "ЧЕТВЕРОК".
На вход блока С2 подается список отобранных достоверных,
"четверок", упорядоченных по величине достоверности. На выходе
программы получатся варианты топологии дерева. Объединение
осуществляется на основе последовательного добавления,
"четверок" к строящемуся дереву с эвристическим выбором
очередной добавляемой "четверки".
Рис. i. Схема построения филогенетического дерева, с помош
метода основанного на анализе и объединении элементарк;
топологий,
(а) Исходный набор из пяти гомологичных нуклеотихда
последовательностей. Цифрами указаны номо-
последовательностей, буквами - гомологичные позиции.
(б) Рассматриваются все возможные наборы из четыре
последовательностей и выделяются позиции, в котор-
наблюдается два варианта нуклеотидов, каждый из котор»
представлен в двух из четырех сравниваем
последовательностей. Группы совместимых позиций обозначен
цифрами.
(в) Каждой группе совместимых позиций сопоставляется от
из трех возможных топологий и для каждой топологи
подсчитывавтся число совместимых позиций (Nj, Н2, Н31. Из тр' "
топологий выбирается одна, характеризующаяся максимальным л
(помечена *} и для нее оценивается вероятность Р, соответствия
этой топологии реальному дереву эволюции.
(г) С помощью порога Р0=0.05 для вероятности
соответствия выбранных топологий реальному дереву эволюши
отбираются только те топологии, которые имеют вероятность
P<Pq, т.е. соответствует реальному дереву с вероятностью «
ниже Pq.
(д) На основе достоверных элементарных тополо<мй
конструируется общее дерево эволюции путем ^х
последовательного объединения ("сшивания").
- 46 -
БЛОК СЗ. АНАЛИЗ И РЕДАКТИРОВАНИЕ ПОЛУЧЕННЫХ ДЕРЕВЬЕВ,
Блок предназначен для манипуляций с вариантами деревьев з
диалоговом режиме и позволяет учесть дополнительные зн&ния, не
использованные при непосредственном построении дерена.
Таким образом, рассмотренная программа позволяет на
основе данных о родственных последовательностях дли
конкретного семейства получать новую информацию об
эволюционных взаимосвязях между ними.
3.2. ОЦЕНКА ИСТИННОГО ЧИСЛА ФИКСИРОВАВШИХСЯ СИНОНИМИЧНЫХ
И НЕСИНОНИМИЧНЫХ ЗАМЕН ДЛЯ ПАР ГЕНОВ.
Программа позволяет получать новую информацию о числе
синонимичных и несинонимичных мутаций, фиксировавшихся в ходе
Дивергенции двух нуклеотидных последовательностей, кодирующих
белки.
На вход подаются две выравненных последовательности. На
выходе получаются истинные числа синонимичных и несинонимичных
мутаций К8и ка, фиксировавшихся в ходе эволюции этих
- 47 -
лОр1"!/<риьакн;ге на
Л '
v до ^( сии» 4MMH4iui< i»< н, нсрмироеанное на чису
№ИЧНЫХ I ""«
¦yOi рамма ^!д(„^в-ллв. ло.л-ыь ^...сло мутационных событий,
гам числе с учетом повторных л обратных мутаций, которые и
видны при прямом сравнении последовательностей.
основным звенсж программы является матрица состветотви
видимых fp и Si и истинных (А и В; чисел различий (транзиций
трансверсий, соответственно; '.Рис.Ь}. Эта матрица получена
помощью имитационного моделирования эволюцг
последовательностей с различными числами транзиций
'граисверсий (см. 8,8),
В ходе работы (программы делается следующее. •
1, На основе сравнения последовательностей находят'
числа транзиций и трансперсий (?, и в,), классифицированные :'
вырожденности сайтов 1, т.е. по числу возникающих одинакова
колонов при мутировании данного сайта.
2. На основании матриц соответствия величины ?% и ч
преобразуются в истинные числа транзиций и трансверсий А,
. 0.
0.
«2-
* 0.
0.
0.
0.
0.
20
22
24
26
28
30
32
34
0
А -
0
0
0
0
0
0
0
0
.20
0
22
0
транзиций
.33
.35
.36
.38
.40
.40
.42
.43
0.
0.
0.
0.
0.
0.
0.
0.
41
43
45
47
48
51
S3
54
0.
0.
0.
0.
0.
0.
0.
0.
24
48
52
52
56
58
62
65
67
0
0
0
0
0
0
0
0
0
Р
.26
.56
.61
.87
.68
.72
.76
.77
.81
0
0.
0.
0.
0.
0.
0.
0.
0.
28
69
73
76
78
83
86
88
94
0
0
0
0
0
0
0
1
1
.30
.79
.84
.86
.91
.94
.97
.00
.01
0.32
0.89
0.81
0.98
1.01
1.02
1.06
1.07
1.09
0.
1.
1.
1.
1.
1.
1.
1.
1.
34
00
02
06
09
10
12
14
14
0.
1.
1.
1.
1.
J.
1.
1.
1.
зе
06
10
13
13
15
17
18
18
0.20 0.22 0.24 0.26 0.26 0.30 0.32 0.34 С.36
0.20
0.22
0.24
0.26
0.28
0.30
0.32
0.34
В -
0
с
0
0
0
/0
G
0
трансверсии
.26
.30
.34
.38
.43
.51
.56
.66
0
0
0
0
0
0
0
0
.26
.30
.35
.40
.44
.50
.56
.66
0.
0.
0.
0.
0.
0.
0.
0.
27
30
35
39
45
50
58
66
0.
0.
0.
0.
0.
0.
0.
0.
27
30
34
38
44
50
57
64
0.26
0.30
0.34
0.39
0.43
0.50
0.57
0.66
0.
0.
0.
0.
0.
0.
0.
0.
26
30
34
39
44
50
58
66
0.
0.
0.
0.
0.
0.
0.
0.
26
30
34
3S
44
50
57
вв
0.
0.
0.
0.
с.
0.
0.
0.
26
30
34
39
44
50
58
ее
0
о
п
0
о
о
0
0
.26
.30
.34
.39
.44
.50
.57
.67
Рис. 2. Фрагмент матриц соответствия видимого (Р и в)
и истинного (Айв) числа различий (траязиций и трансвер-
иий ) на позицию нуклеотидной последовательности.
8. На основе А, и В, находятся числа синонимичных
несинонимичных мутаций К и к с помощью формул ЕЗЗ:
I*2A2+N41C4.
а"
AVVg
B)
и В} - истиннее
Здесь: M,t - число сайтов вырожденности 1;
числа транзиций и трансверсий, соответственно; К^А.+В,,
Таким образом, программа позволяет получать новую
информацию о числе истинных событий фиксаций мутаций, имевших
место в ходе эволюции пары последовательностей от общего
предка (классифицированную на синонимичные и несинонимичные
замены).
- 48 -
B.S. АНАЛИЗ СЕМЕЙСТВ ГЕНОВ ПО СООТНОШЕНИЮ ФИКСИРОВАВШИХСЯ
СЩЦМИМЧНЫХ И НЕСИНОНИМИЧНЫХ ЗАМЕН.
Программа предназначена для анализа и характеристики
ceiietCTB генов с точки зрения соотношения синонимичных и
нвсинонимичных замен для набора гомологичных
последовательностей в целом. Она позволяет получить следующий
набо
набор информативных параметров: н, вп д~
U, Вс
Здесь н«
ИН-
среднее по
семейству отношение чисел несинонимичных и синонимичных замен;
- 49 -
o.ao|-
0.40 t
Рис.3. Зависимость H{K ), где н=к /к , к, - число
несинонимичных куклеотидкых замен, нормированное на число
несинонямичных сайтов, К,- число синонимичных замен,
нормированное на число синонимичных сайтов 1гуклеотидной
последовательности. Обозначения (в скобках даны параметры
линейной аппроксимации зависимости для некоторых семейств
гомологичных генов):
# - интерферон а человека (BQ=0.8ii, B1=-i.7i7)
| - гемагглютинин вируса гриппа (BQ==0.?36, В^-0.226)
f - а-гемоглобин (BQ=0.256, В^-0.022)
Ш - гистон Н4 (BQ=0.101, Bj=-0.020)
BQ
- 50 -
- отношение числа несинонимичных замен к
числу синонимичных для пары последовательностей J, и J2;
JiJ2
- числа синонимичных и
замен при дивергенции последовательностей J
Н - число последовательностей в семействе;
не синонимичных
и J2;
во,
параметры линейной аппроксимации
H-8lEB+B0
распределения пар генов семейства в плоскости (Н, кв), где
»%:
д_ , До - 98* доверительные интервалы для величин в„, в,.
Величины Кв и Ка определялись с помощью программы 3.2
оценки истинного числа фиксировавшихся синонимичных к
несинонимичных замен для пар генов.
На Рис.8 приведена зависимость Н(Кв) и ее линейная
аппроксимация для ряда белковых семйств. Различные семейства
демонстрируют весьма разнообразное поведение Н(КаК что,
вероятно, свидетельствует о специфическом характере
эволюционного процесса для них. Можно видеть, что наблюдаются
как отрицательные, так и близкие к нулю углы наклона.
Равенство нулю угла наклона означает постоянство отношения
И=Ха/Кв для всех пар нуклеотйдных последовательностей данного
семейства. Это может свидетельствовать о сохранении
функциональной нагруженное™ белков данного семейства.
Возможными объяснениеми отрицательного угла наклона могут быть
изменение функциональной нагруженное™, либо, неравномерный
Характер распределения фиксирующихся несинонимичных замен по
последовательности, приводящий к недооценке числа
нвеинонимичных замен К„ используемым методом и,
соответственно, к уменьшению величины н=к /к .
a S
8.4. АНАЛИЗ ИЗМЕНЕНИЯ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ АМИНОКИСЛОТ.
:*'; Программа предназначена для анализа и характеристики
ермейств с точки зрения изменений физико-химических свойств
ЧРИ мутациях, наблюдавшихся в ходе эволюции
последовательностей этих семейств.
Для анализа испольовались четыре наиболее существенных
- 51 -
характеристики аминокислот 14 3: размер боковой цепи (Gtb
яолярность (GgJ, изоэлектричеосая точка (G3^ гидрофобное?*
(O^J. Каждая аминокислотная замена характеризовалась величиной
iOjj - Суммарного изменения физико-химических характеристик
при замене аминокислоты типа i на тип j в к-ой позиции
последовательности:
Здесь
G* - значение характеристики G для
аминокислоты типа l C4J.
Рассматривая (Рис.4) распределение фиксировавшихся
актаций, выявленных при парном сравнении последовательностей
для различных семейств, отметим наблюдающееся разделение
белков на два класса.
1. С преимущественно консервативным характером мутацте
т.е. с преобладанием замен с малым uG (например, гистон Н4
р-глобин).
2. G преимущественно неконсервативным характером мутаций,
т.е. с одинаковой представленностью всех классов по (л-.
(например, гемагглютюшн, казеин).
Можно предположить, что изменения физико-химически1
свойств при заменах отражают функциональные особенности белкор
и характер эволюционных изменений их аминокислотны
последовательностей. Принадлежность к классу 1 может быт:
обусловлена консервативным характером эволюции белка.
Что же касается класса 2, то здесь возможны две ситуации
Первая - неконсервативные белки. Примером такого белка
очевидно, является казеин. Неконсервативный характе ¦;
фксирующихся замен для него сочетается с высокой частоте-
несинонимичных мутаций (большое н).
Вторая возможная ситуация - белки, у которых наряду i--1
сравнительно консервативными участками существуют небольш; >з
зоны, где преимущественно фиксируются замены, приводящие
большим изменениям физико-химических свойств аминокислот. Л'^
этих белков, несмотря на небольшие значения Н (ч"о
.1) B, С31 D! A) B) C) D)
Рис.4. Распределение частот встречаемости замен аминокислот
(*) в зависимости от разницы физико-химических свойств
*°i.j • По оси ординат - средние для семейства частоты замен,
по оси абсцисс - четыре интервала изменения физико-химических
свойств:
B) ДО, . =73+144, C) Д6, , =145*217,
te
l I
, =218*284. Единицы измерения «г, . - условные.
- 53 -
- 52
свидетельствует об их функциональной кагруженногтн:
наблюдаются аехонсервативяые замены с боль^лик значениями ас;
Тшсая картина наблюдается для нзйраминидбза, гемаггдатидашг
иммуноглобулина, интерферона. Способностью фиксироват
лесинонимичные мутации могут обладать эбласти антигеннк
f )*t«r > азк и г'емагглютиш-ffit:
лммуяоглсбу.лмиов, активяк
f ( зариабельны/ участков може
ером зволюцик этих белков
-ч !зико--хкммчесхих свойств
^о^. 'гэмагглзсгпишн, ыэйрамкнидао;
1 ° ь, слад
с л чь ь i
т"ит тос-ог fEOfeieiu ускользание ст 'лммушгогс отве?
f «( организма, а в белжах имьг/нкей систе?-
ч» г-ш, интерферон? - эе оперативно;
и l Ei иешЖ) в соответствии с антигенном ожруженз;
«г и 1 то есть, адаптивно эволюционируют беда:
к №ь ¦" системах.; иепосредстЕенно взаимодейству щи/.
3.8 L
3.6
з.г
з.о
2.6
J. -ту бужи
гке?он
2.4
3 5, АНАЛИЗ СООТКОЦЕНИЯ СИНОНИМИЧНЫХ И НЕСИНОНИМИЧНЫХ САЙТОг-
ЯУКЯЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЯХ,
Программа предназначена для анализа и характеристик
семейств с тсчяи зрения соотношения синонимичных
яесинонимичных сайтов.
Для данного гена рассматриваются все возможные заме
одного нухлеотида з каждой позиции, Если длк
последовательности I, то число таких замен 31 (в как,:;
иувлеотидной позиции возможно три замены}. Определим велкчк
Ig - число не синонимичных нуклеотидных замен из 31 возможшз-
¦л величин-.' й - число синонимичных нуклеотидных замен из
возможных. Тогда показатель, оценивающий соотношел
не синонимичных и синонимичных сайтов, определяется еле дум
образом-;
и '- VHc
Из особенностей генетического кода следует, что
54 -
15,4
Рис.5. Зависимость отношения числа потенциально косиновишгашх
1|- числу потенциально синонимичных сайтов нутслэотщщо^
Й^ледовател-ьностк u=8a/8e от н^/к,,.. Приведены средние
дай 32 семейств белков. Наблюдается полоаительная коррелждая
«Дичинами к и и. Коэф^.циент корреляции 0 50, достоверность 9Ш-
аминокислотном и колонном составе белка отношение
несинонимичных и синонимичных сайтов должео составлять
*<К8. Для реальных семейств генов эта величина колеблется в
Жапазоне от 2.74 до 3.88 СРис 5).
ч/- Наблюдается положительная корреляция между потенциально
( и ] и реализованным ( к ) отношением числа
к числу синонимичных замен. То есть, d белв-.sx с
частотами (|мксируицихся не синонимичных замен (более
Рионально нагруженнюли J наблюдается меньшее число
ЧрИнонимичных сайтов. Отметим, что пониженная частота
пцихся несинонимичных замен Kg не обусловлена меньшим
несинонимичных сайтов н , поскольку к нормировано на
сайтов. То есть, взаимосвязь и и н обеспечивается
Ят^вием отбора. Это позволяет сделать внвод о том, что в
¦М4-.
щюцессе эволюции формируется и поддерживается аминокислотный
я кодошшй состав, обеспечивающий минимальное чхсле
ае синонимичных сайтов для белков, характеризующихся большоз,
»1рос1да.ональной нагруженностью. Для последующего эволюционного
процесса это, фактически, означает адаптацию системы к
возникающим мутациям. Наблюдаемая оптимизация представляет
оэбой пример стабилизргрупщего отбора на молекулярном уровне.
3.8. ШИТАВДОННОЕ МОДЕЛИРОВАНИЕ ЭВОЛЮЦИИ СЕМЕЙСТВ ГОМОЛОГИЧНЫ).
ЖСЛЕаОВАТйЛЬНОСТЕЙ.
Программа предназначена для моделирования различных
эволюционных процессов, типичных для семейств гомологичных
последовательностей.
На вход программы подаются следующие исходные данные:
I - длина последовательности;
я - последовательность или способ ее генерации;
"л - временная схема дуплидирования последовательностей;
я - временная схема мутирования последовательностей;
к - логическая схема динамики вариабельности позиций;
? - схема имитационных экспериментов для решемж
конкретной задачи пользователя.
Е работах Фитча ?5, 61 для описания эволюции белковьи.
молекул предложена коварионная модель, взятая нами за основ,!
тгри моделировании динамики вариабельных позиций в белках.
Зое позиции аминокислотной последовательности беях.-
разбиваются на два класса: A) консервативные - для которы;
несинонимичные мутации запрещены; B) коварионы - для которых
яесинонимичные мутации разрешены. Предполагается, что при
йршссации мутаций происходит переход позиций из одного класса г,
другой с сохранением общего числа позиций каждого класса.
Для описания динамики вариабельности позиций используюты;
следующие параметры:
1, к - число коварионов;
2, Список позиций-коварионов (*);
3, р - скорость изменения состава коварионов. Пре-
Префиксации в последовательности каждой новой мутации список
зеоварионов обновляется на рк позиций.
На выходе программы получается модельное эволюционнс-j
- 56 -
дерево и набор "современных" последовательностей.
В ходе работы программы реализуется мутации и дупликации
последовательностей в соответствии со схемами N, и, к и
производятся многократные имитационные эксперименты в
.соответствии со схемой Р.
Описанная программа монет использоваться для решения
различных конкретных задач.
Рассмотрим некоторые примеры, иллюстрирующие возможности
рассматриваемой программы,
- ЗАДАЧА 1„ ГЕНЕРАЦИЯ МАТРИЦЫ ПРЕОБРАЗОВАНИЯ ВИДИМЫХ ЧИСЕЛ
РАЗЛИЧИИ ДЛЯ ПАРЫ ПОСЛЕДОВАТЕЛЬНОСТЕЙ В ВЕЛИЧИНЫ ИСТИННЫХ
-Ф1СЕЛ РАЗЛИЧИИ (для программы 8.1).
Воспроизводился процесс накопления мутаций в ходе
дивергенции двух нуклеотидных последовательностей длины 100.
Вся область изменения величин Р и б (видимых чисел транзииий и
-4*рансверсий на сайт) разбивалась на класса и на каждом шаге
"Накапливались значения А и В, соответствующие заданным Р и о.
;|Дзоизводилось большое число шагов моделирования с различными
;.#»данными Р и Q, обеспечивающее не менее 30 попаданий в каадый
¦3f Таким образом получались матрицы преобразования видимнх
¦ЯИсел транзиций и трансверсий для пары последователмостей в
'/величины истинных чисел транзиций и трансверсий (Рис. 2),
'$? задача 2. постгоение калибровочной шкалы для оценки
эщаптивности эволюции на основе анализа характера
Распределения пар генов по соотношению синонимичных и
^^синонимичных мутаций.
Ц' Рассматривались два варианта модели, отражающие
ичапциеся режимы эволюции: A) с сохранением локализации
арионов (адаптивная эволюция); B) с изменяющейся
кализацией коварионов (нейтральная эволюция). На рис. 6
едены калибровочные кривые, полученные для адаптивного и
рального режимов эволюции. Можно видеть, что для первого
ианта модели наблюдается отрацателышй наклон построенной
исимости, в то время как для второго варианта модели
юдается отсутствие наклона.
- 57 -
O.I
0.05
-0.05
-O.I
-0.15
- о.;
-0.2!
Табл.1. Отношение H=Ka/Ks и параметры линейной аппроксимации
нейршющдаза
-1
интерферон «*
человека
•во
0.1 0.2 0.3
0.S 0.6 0.7 o.fl
Рис.6. Зависимость характеристик линейной аппроксимаци
н=В1Кв+в0 КРИВОЙ H(KS' для м°Дельных последовательностей
различными параметрами эволюции.Обозначения: ¦ - режк'
эволюции 1 (р=о.о), • - режим эволюции 2 (р=о.1). Отдельны
точки соответствуют различным величинам к, характеризующие
число коварионов в модельной последовательности. Приведены
также данные для некоторых реальных семейств белков.
н=в1Кв+В0 для исследуемых семейств генов. Приведены
95»
доверительные интервалы для величин BQ и в1# Режим 1
соответстствует эволюции с постоянной локализацией ковариовов,
режим 2-е изменяющейся их локализацией в белке.
Семейство
генов
«
Интерферон а челов
Интерферон а быка
Интерферон а мыши
Казеин «s
Гистон НЗ мыши
Гистон нз
ГИСТОН Н2Ь
Гистон Н4
Э -Гемоглобин
а -Гемоглобин
Иммуногл. Схкролик
Лютропин
Фосфорилаза
Glu-амидотрансфер.
Na-уретическ пепт.
Эшсефалин
Инсулин
Пролактин
а - ТубуЛИН МЫШИ
Э - Тубулин
Рибосомалышй бел.
Цитохром с
Нейраминидаза
Гемагглютинин
Т-рецептор,У„-цепь
Нейраминидаза,
" - конец
гРанулин
н
0.44
0.29
0.34
0.69
0.01
0.05
0.10
0.07
0.27
0.23
0.21
0.26
0.11
0.11
0.13
0.18
0.09
0.31
0.02
0.12
В0
0.81
0.71
0.57
0.91
0.02
0.06
0.05
0.10
0.25
0.28
0.11
0.69
0.15
0.17
0.22
0.16
0.13
0.38
0.04
0.22
0.08-О.03
0.09-0.04
0.14
0.32
0.52
0.55
0.13-
0.19
0.79
0.46
0.61
-0.12
довер
во
и
0.67
0.48
0.31
0.11
-0.02
0.03
0.02
0.05
0.20
0.18
-0.03
-0.37
0.00
-0.09
0.14
-0.40
-0.73
0.22
0.01
0.06
-0.26
-0.16
0.10
0.60
0.33
0.52
-0.24
ИНТ.
(95*)
0.94
0.95
0.82
1.71
0.07
0.10
0.09
0.14
0.31
0.38
0.25
1.76
0.29
0.44
0.30
0.72
1.00
0.53
0.06
0.37
0.20
0.07
0.28
0.99
0.58
0.70
0.00
В1
-1.71
-4.50
-1.02
-0.40
-0.02
-0.00
0.03
-0.02
0.02
-0.03
0.32
-0.75
-0.07
-0.04
-0.15
0.01
-0.05
-0.09
-0.09
-0.04
0.09
0.08
-0.12
-0.22
0.03
-0.03
0.13
довер
ИНТ.
В. (95*)
-2.32
-6.93
-2.13
-1.78
-0.28
-0.02
Q.00
-0.04
0.02
-0.12
-0.03
-2.56
-0.39
-0.23
-0.29
-0.35
-1.11
-0.29
-0.04
-0.12
-0.08
0.01
-0.33
-0.31
-0.03
-0.08
0.07
-1.11
-2.07
0.08
0.98
0.23
0.03
0.05
0.00
0.07
0.04
0.67
1.50
0.24
0.14
-0.02
0.38
1.00
0.10
0.00
0.02
0.27
0.15
0.08
-0.13
0.10
0.01
0.19
ре-
режим
эво-
эволюции
1
1
1
?
2
2
2
2
2
2
?
2
2
2
?
2
2
2
2
2
2
2
1
1
2
?
2
- 58-
- 59 -
Оценка режима эволюции реальных семейств генов с помощью
построенной калибровочной шкалы осуществляется путем отнесения
режима эволюции семейства к одной из трех ситуаций:
1. Эволюция семейства адаптивна;
2. Эволюция семейства нейтральна;
3. Оценить характер адаптивности эволюции семейства
не представляется возможным.
В табл. 1 и на рис. 6 приведены результаты оценки режимов
эволюции для ряда белковых семейств. Исходя из характера
наклона зависимости H(KS'' определяемого значением
коэффициента линейной аппроксимации Bj, можно видеть, что гены
глобиновых, гистоновых и др. семейств эволюционирует в рамках
режима с изменяющейся локализацией коварионов, в то же время,
для генов гемагглютинина и нейраминидазы свойственен режим
эволюции с постоянной локализацией коварионов.
Коварионы с постоянной локализацией в генах оболочечных
белков вируса гриппа могут быть соотнесены с участками,
кодирующими антигенные детерминанты соответствующего белка
Вирус гриппа "ускользает" от действия иммунной системь
организма хозяина за счет фиксации мутаций в областях
антигенных детерминант оболочечных белков, что приводит н
резкому изменению их конформации и, тем самым, предотвращает
связывание имевшихся ранее вариантов антител со вновь
возникшими мутантными штаммами вируса гриппа. Для гистонов,
гемоглобинов и других белков, характеризующихся режимом 2,
таких строго локализованных коварионов, вероятно, нет.
Заметим, что для большинства проанализированных семейсть
характерен режим 2.
4. ЗАКЛЮЧЕНИЕ.
Описанная компьютерная система иллюстрируют традиционные
технологии, используемые для исследования закономерностей
молекулярной эволюции. Дальнейшее развитие указанной системы
состоит в создании интеллектуальных программных средств,
характеризующихся существенно большей гибкостью решения задач,
их более широким спектром и большим участием конечного
пользователя в выборе направления поиска решения.
Описанные программы и база данных гомологичных
нуклеотидных последовательностей представляют собой элементы
из которых может строиться интеллектуальная программная
система для исследования закономерностей молекулярной
эволюции. Среди рассмотренных программ следует особо отметить
программу "Имитационное моделирование эволюции семейств
гомологичных последовательностей" (см. п. 3.6.). Эта программа
содержит формальную модель предметной области, в которой
работают остальные программы, описанные в п. 8.1-3.в.
Программа 3.6 позволяет находить взаимосвязь между внутренними
параметрами эволюционного процесса и наблюдаемыми величинами,
например, между истинными и видимыми числами транзиций и
трансверсий - (A,B)»{P,Q); между параметрами динамики
коварионов и соотношением чисел синонимичных и несинонимичных
мутаций - (к,р)»(н,Кя).
Таким образом программа 3.6 обеспечивает
интеллекуализацию описанной компьютерной системы за счет
универсальной модели эволюционного процесса.
Существенной компонентой, необходимой для качественного
улучшения существующего комплекса программ в направлении его
интеллектуализации, является база знаний, содержащая сведения
об успешных применениях различных расчетных схем для решения
задач исследования закономерностей молекулярной эволюции и
хранящая биологически значимые результаты этого анализа.
В настоящем комплексе можно выделить совокупность
характеристик, которую можно рассматривать как исходный формат
для формирования такой базы знаний.
1. Совокупность полей базы данных гомологичных
последовательностей.
2. Топологии филогенетических деревьев, построенные
программой 3.1 с характеристикой достоверности и списками
противоречивых "четверок".
3. кя, ка - числа синонимичных и несинонимичных мутаций,
нормированные на сайт (см. п. 3.2).
4. н, BQ, Bj, дв0, йВ1 - характеристики семейств с точки
зрения распределения синонимичных и несинонимичных мутаций для
набора гомологичных последовательностей (см. п. 3.3).
5. ug - характеристика семейств с точки зрения изменений
Физико-химических свойств при мутациях (см. п. 3.4).
- 60 -
- 61 -
6. u - характеристика семейств с точки зрения соотношения
синонимичных и несинонимичн'- сайтоов (см. п. 3.5).
7. k, p - параметры динамики коварионов, оцененные с
помощью программы 3.6.
8. Режим эволюции семейств, оцененный с помощью программы
8.6 (адаптивный или нейтральный).
Создаваемая интеллектуальная система должна существенно
расширить спектр возможных постановок задач с целью достижения
наиболее полной структурно-функциональной и эволюционной
характеристики белковых семейств в рамках выбранной предметной
области.
Разработанные программные средства ориентированы на
использование компьютера типа IBM PC. Объем базы данных по
гомологичным последовательностям: 50 семейств, около 10
последовательностей на семейство.
ЛИТЕРАТУРА
II] Омельянчук Л.В., Колчанов Н.А. // В кн.: Алгоритмический
анализ структурной информации (Вычислительные системы,
ВЫП.112). Новосибирск. ИМ СО АН СССР. 1986. с. 46-55.
[2] Шиндялов И.Н. Исследование структурной организации и
эволюции глобулярных белков на основе моделирования с
помощью ЭВМ. Автореф. канд. дис. Новосибирск. ИЦиГ СО АН
СССР. 1988.
131 И W.-H., Wu C.-J., Luo С.-С. // Mol. Blol. Evol. 1985.
V.2. p.ISO.
[4] Bogardt R., Jones B.H.,Dwulet F., Garner W.H..Lehman L,D.
Gurd F.R.N. // J.MoX.Evol. 1980. V.15. p.197.
[5] Fitch W.M. // J.Mol.Evol. 1971. V.I. p.84.
[6] Fitch V.M., Markovltz E. // Biochemical Genetics. 1970. V.4.
p.579.
- 62 -
SUMMARY,
COMPUTER SYSTEM FOR THE INVESTIGATION OF MOLECULAR EVOLUTION
ON THE BASIS OF HOMOLOGICAL SEQUENCES ANALYSIS.
I.N.Shindyalov, I.N.Kllkunova
Institute of Cytology and Genetics, Siberian Branch, the
Academy of Sciences, Novosibirsk, 630090
The paper consists of four sections. Section 1 (INTRODU-
(INTRODUCTION) contains main classes of data that the described system
deals with: (I) nucleotlde sequences; (II) alignment of se-
sequences; (III) structural and functional characteristics of
proteins and RNA; (IV) systemic properties for macromoleoulea
(genonlc, tissue, organlsaal, ontogenetlc, taxonomlc, evolu-
evolutionary). Then typical problems of analysis of homologous
sequences are considered: A) alignment problem; B) taxonomy
problem; C) problem of evolution description; D) problem of
structural and functional characterization of macromolecules.
The computer system under consideration Includes data
base and a set of programs to analyze the Information
contained in it. Section 2 describes the DATA BASE OF
HOMOLOGOUS SEQUENCES for genes families, alignment versions
and a synopsis on the family and formats of fields used for
family description. In section 3 ve present a set of programs
for studying the molecular evolution based on comparative ana-
analysis of homologleal sequences. At present this set contains a
seven main groups of programs described belov.
Section 3.1 - CONSTRUCTING PHILOGENETIC TREES BASED ON
ANALYSIS AND JOINING ELEMENTARY TOPOLOGIES.
Section 3.2 - ESTIMATION OF THE NUMBER OF FIXED SYNONY-
SYNONYMOUS AND NON-SYNONYMOUS SUBSTITUTIONS FOR PAIRS OF GENES.
Section 3.3 - ANALYSIS OF FAMILIES OF GENES BY THE RATIO
OF FIXED SYNONYMOUS AND NON-SYNONYMOUS SUBSTITUTIONS.
Section 3.4 - ANALYSIS OF VARIATION OF AMINO ACID PHYSI-
PHYSICAL AND CHEMICAL PROPERTIES.
Section 3.5 - ANALYSIS OF THE RATIO OF SYNONYMOUS AND
NON-SYNONYMOUS SITES IN NUCLEOTIDE SEQUENCES.
Section 3.6 - IMITATION SIMULATION OF EVOLUTION OF HOMO-
- 63 -
LOGOUS SEQUENCE FAMILIES.
Section 4 (CONCLUSION) contains a description of a set of
characteristics vhlch can be considered as starting fornat for
the development of knowledge base in designing an Intellectual
computer system:
1. Fields of the data base of homologous sequences.
2. Topologies of philogenetlc trees produced by program
3.1 with the slgnlficancy estimates and lists of contradictory
elementary topologies.
3. К , К are the numbers of synonlmous and non-
¦ynonymous mutations related to a numbers of correspondings
¦ltes (see Sect. 3.2).
4. H, Bo, В., ДВ0, ABj are the characteristics of famlli-
•¦ from viewpoint of distribution of synonymous and non-
¦ynonymous mutations for a set of homologous sequences (see
Sect 3.3).
5. UG Is the characteristic of families from viewpoint of
variations of physical and chemical properties under mutations
(¦ее Sect. 3.4).
6. U is the characteristic of families from viewpoint of
the ratio of synonymous and non-synonymoue eltee (see sect.
3.5).
7. k,p are the parameters of dynamics of covarions evalu-
evaluated by program 3.6.
8. Mode of families evolution estimated by program 3.6
(adaptive or neutral).
The Intellectual system created should essentially make a
range of possible settings of problems wider to achieve the
most complete structural, functional and evolutionary charact-
characteristics for protein families within the subject field chosen.
The above software elaborated are aimed at IBM PC compat-
compatible computers.
КОМПЬЮТЕРНАЯ СИСТЕМА ИССЛЕДОВАНИЯ СВОЙСТВ
ПЕРЕМЕЩАЩИХСЯ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Капитонов В.В.
Институт цитологии и генетики СО АН СССР
1. Введение
В настоящее время установлено A-3), что значительную
долю генома эукариот составляют мобильные генетические эле-
элементы и повторяющиеся последовательности, способные к
внутригеномному распространению за счет транспозиции и реко-
мбинационных процессов, которые в дальнейшем будем называть
для общности перемещающимися последовательностями (ПП). От-
Отдельные классы ПП могут составлять до 5% генома эукариот, а
суммарная доля ПП достигает до 50$ [31. Поэтому при секвени-
ровании новых фрагментов генома чаще всего в их составе на-
находятся ПП. По причине наличия в ПП функциональных сайтов
различной природы и в связи с активным участием ПП в транс-
транспозициях и рекомбинационных процессах, приводящих к перест-
перестройкам генома, подобные элементы могут являться существенным
фактором эволюции и функционирования геномов, влияющих на
приспособленность организмов.
Это объясняет актуальность исследования структурно-фун-
хциональной организации и эволюции ПП.
Наблюдаемый экспоненциальный рост объема эксперимента-
экспериментальных данных по ПП делает необходимым применение новых тех-
технологий, основанных на более полном использовании ЭВМ (с
тенденцией к интеллектуализации программного обеспечения
Г41). Описанию подобных методов исследования свойств ПП, ко-
которые объединены в рамках комплекса программ на ЭВМ IBM PC
(см. Рис.1), и поевящена представленная работа.
2. МЕТОДЫ ОЦЕНКИ ПАРАМЕТРОВ ТРАНСПОЗИЦИИ ПП
Важнейшими характеристиками ПП являются частоты их тра-
транспозиции х и встраивания ц в геноме. Для теоретических оце-
оценок значений х и р нами предложен ряд методов.
-65 -
- 64 -
МЕТОДЫ ОПЕНКИ ПАРАМЕТРОВ
ТРАНСПОЗИЦИИ И ДИНАМИКИ
- Математические модели;
- Имитационные модели;
- Филогенетический анализ.
МЕТОДЫ АНАЛИЗА ОБМЕННЫХ
ПРОЦЕССОВ С УЧАСТИЕМ ПП
- эффективность протекания
обменных процессов;
- частота неравного
кроссинговера;
- эффективности блочной
конверсии.
БАЗА
ДАННЫХ
структурно-
фу нк ци ональная
организация;
динамические
свойства ПП;
эволюция ПП.
ОЦЕНКА ВЛИЯНИЯ ПП НА
ПРИСПОСОБЛЕННОСТЬ
ПОПУЛЯЦИИ
Рис. 1 Программный комплекс для исследования
свойств мобильных элементов и повторяющихся
последовательностей.
Предполагается, что перемещения ПП внутри генома проис-
происходят по схеме: транскрипция —» обратная транскрипция »
встраивание к-ДНК в геном.
2.1 Математическое моделирование эволюционной
динамики изменения количества ПП в геноме.
Метод основан на аналитическом описании зависимости
среднего внутригеномного числа ПП от времени. Рассмотрим его
на примере моделирования внутригеномной эволюции повторов
Alu.
Будем считать, то численность Alu в геноме определяется
процессами размножения и выщепления. Можно предположить сле-
следующее: а)повтор, образовав свое копию, сам сохраняется в
геноме; б)элемент, выщепившийся из генома, исключается из
общегеномного пула повторов; в)процессы размножения и выщеп-
выщепления повторов независимы; г)суммарное количество повторов в
геноме не может превосходить некоторой пороговой величины Mj
вероятность встраивания повторов уменьшается с ростом их чи-
- 66 -
ела геноме. Другие лимитирующие факторы протекания указанных
процессов отсутствуют; д)все повторы идентичны по их способ-
способности к размножению и выщеплению; е)процессы встраивания и
выщепления - реакции первого порядка по числу повторов в ге-
геноме N с константами х и м, соответственно.
Тогда изменение числа повторов со временем можно опи-
описать следующим уравнением:
dN _ , М - N
dt
= X
N -
A
где член (м-Ю/м учитывает вероятность встраивания повтора в
геном при условии, что в геноме уже имеется N из М
максимально допустимого числа повторов.
Для стационарного состояния при dN/dt = 0 имеет место:
М - N
м
B)
где N - количество повторов в стационарном состоянии.
Для оценки частоты выщепления ц воспользуемся такой
особенностью ПП, как наличие коротких фланкирующих прямых
повторов, возникающих в момент встраивания ПП в геном в ре-
результате дупликации фрагмента генома в месте встраивания.
Предполагается, что: 1) фланкирующие повторы функционально
ненагрукены и эволюционируют со скоростью фиксации нейтраль-
нейтральных мутаций v=5-10~9 замен на позицию за год; 2)повторные и
обратные мутации маловероятны и ими можно пренебречь.
Тогда, по формуле T=k/2Lv можно оценить время т, проше-
прошедшее с момента внедрения ПП в геном до момента его экспери-
экспериментального исследования, где к -количество нуклеотидных за-
замен в двух фланкирующих повторах длины L, окружающих ПП.
Легко показать [5 ], что среднее значение времени т
(которое фактически является средним временем нахождения ПП
в сайте его встраивания) связано с константой выщепления со-
соотношением
T=1//J . C)
Итак, анализируя фланкирующие прямые повторы, можно на-
найти среднее время т, а следовательно и частоту выщепления ц.
С помощью такого подхода могут быть также получены зна-
значения параметров х и М. Для повторов Alu нами получены след-
lO ^ »* ^ о in° \^q in ' luin ' f -j /ГОД)
упцие оценки: 9-10" s M
- 67 -
2-10? х«310 7, (J*10 7
Этот метод эффективен в качестве начального приближения,
когда детальные механизмы эволюции ПП не известны, и имеется
лишь ограниченный экспериментальный материал.
2.2 Имитационное моделирование эволюции ПП.
Наиболее типична ситуация, когда нухлеотидные последова-
вательности секвенированы лищь для малой доли ПП определен-
определенного класса(например, для Alu такая доля составляет менее
0.01*). Ясно, что apriory эволюционные характеристики такого
класса ПП, полученные из анализа ограниченной выборки,нельзя
распространять на всю совокупность ПП данзого класса без до-
дополнительных статистических оценок.
Для исследования таких усредненных характеристик, как
степень внутривидовой дивергенции, частоты перемещения (вст-
(встраивание и выщепление) ПП, удобно использовать метод иммита-
ционного моделирования на ЭВМ.
Рассмотрим имитационную модель эволюции ПП типа Alu.
Модель основана на следующих предположениях.
1)эволюция ПП определяется тремя процессами: встраивани-
встраиванием (вероятноять х), выщеплением (вероятность ц), мутировани-
мутированием (вероятность v);
2)число ПП не может превышать порогового значения (м);
3)процесс распространения ПП по геному описывается диффе-
дифференциальным уравнением A).
Моделирование эволюции ПП с реальными значениямии параме-
параметров х, ц, м и v затруднительно в связи с необходимостью ис-
использования больших объемов оперативной памяти ЭВМ и значи-
значительным временем счета. Поэтому, указанные параметры, а так
же продолжительность периода эволюции Т и число повторов Alu
в геноме N следует изменить при моделировании следующим об-
образом. Значения х, » и v возрастают на 2 порядка, а Т, Мим
уменьшаются на 2 порядка.
Корректность таких изменений параметров модели вытекает
из следующего простого преобразования уравнения A):
- (Хо)
(М/а) - (N/oQ
М/а
Ш/а) -
где а - некоторое положительное число (для Alu а=ю2).
Предполагалось, что в начальный момент эволюции в гено-
геноме имеется лишь несколько ПП. Каждый повтор Alu имел нуклео-
тидную последовательность длиной L=285 п.о. В качестве исхо-
исходного варианта Alu взята усредненная последовательность реа-
реальных повторов.
Согласно схеме перемещения повторов Alu в геноме пред-
предполагалось, что для размножения Alu необходимо наличие собс-
собственного промотора (РНК-полимеразы III) в первом мономере
Alu. При этом размер и расположение промотора соответствова-
соответствовали его локализации в реальных последовательностях. Считалось,
что мутация в любой позиции этого сайта приводит к потере
способности Alu к размножению.
Вначале моделируется накопление ПП в геноме за время
эволюции т. При этом через заданный интервал х осуществляет-
осуществляется процедура имитации эволюции, заключающаяся в моделирова-
моделировании процессов репликативной транспозиции, выщепления и мути-
мутирования с заданными частотами х, ц и У, соответственно. При
мутировании фиксируются только те замены, которые попадают в
промоторный участок. В целом, осуществляется к = Т/г тактов
имитационного эволюционного процесса.
Каждый повтор характеризуется пятью параметрами: i-но-
мер, присваевыемый в порядке встраивания повторов в геном;
х-время возникновения повтора(номер такта); у-номер повтора,
из которого образовался данный повтор; z - признак состояния
промотора повтора (z=0, если промотор функционален; г=1,если
он содержит мутацию); w-признак наличия повтора в модельном
геноме (w=o, если повтор сохраняется в модельном геноме,
w=i, если он выщепился из генома).
В результате полного цикла моделирования за время Т бу-
будет получена симуляционная матрица размерности Ш*5), где N-
общее число повторов, возникших с самого начала работы моде-
модели.
В итоге мы имеем возможность имитировать процесс опре-
определения характеристик и свойств Alu на основе анализа огра-
ограниченной экспериментальной выборки повторов Alu.
Действительно, для любых s повторов, случайно выбранных
из всех Ы повторов, полученных в результате моделирования,
можно определить среднюю степень дивергенции и другие дина-
динамические характеристики, если восстановлены нуклеотидные по-
последовательности этих S повторов в соответствии с их филоге-
- 68 -
- 69 -
нией. Тогда, проводя оценки динамических характеристик для
для большого числа выборок размера s, можно решить вопрос о
состоятельности оценок таких характеристик, полученных по
экспериментальной выборке этого размера.
Так как в первых трех столбцах симуляционной матрицы
(соответствующих параметрам i, x, у) фактически содержатся
филогенетические отношения для всех N элементов, возникших
за время т, т.е. эволюционное дерево G(N) с N висячими
вершинами, то задача сводится к построение поддерева G(S
для s повторов, случайно выбранных из всех сохранившихся l
модельном геноме N элементов [6].
Зная модельные временные интервалы между вершинами по-
полученного древа и скорость мутирования, можно в соответстви;
с филогенией ПП восстановить нуклеотидные последовательнос-
последовательности каждого из s ПП.
Процесс эволюции повторов Alu моделировался при одних
тех же параметрах Т = 8105 лет, х = Ю^лет, S = 55, v = 2.:
—7
10 замен на позицию в год. При этом значения М, х и
варьировались в следующих диапозонах: 6108 < М < 3-Ю4
4 6
10 < X < 10,
у
10~6<
< 2 10 т Рассматривались тольк.
такие комбинации значений хин, при которых число повторе
после т лет составляло не менее 5-Ю8. Для оценки степей
дивергенции повторов в модели анализировалось большое чис
разных случайных выборок, содержащих последовательности
повторов Alu модельного генома и полученные выборочные
значения степени дивергенции усреднялись.
Оказалось [ 6 ], что существует одна локальная область
значений параметров хин, для которых средняя степень
дивергенции по модельной выборке наиболее близка к
оценке, полученной из анализа 55 реальных повторов Alu (Г --
0.28). Найденная область значений хин такова:
10< х < 2.5-Ю, 6-10~8< м < 1.2-10 .
Максимальное значение средней степени дивергенции по
модельной выборке составляет D = 0.15 (при х = 2.5 10~7, н -
8 10 ). Такая степень дивергенции является максимально
достижимой в нашей модели.
Отметим, что значения х и м, при которых в модели дос-
достигается максимальная степень дивергенции, наиболее близкая
к реально наблюдаемой, хорошо согласуются с оценками, полу-
полученными в разделе 2.1.
С помощью данной модели можно исследовать вопрос о ко-
количестве секвенируемых ПП, необходимых для достаточно точно-
точного определения динамических характеристик этого класса ПП.
для этого изучалась зависимость ¦ средней степени дивергенции
от размера выборки ПП. Оказалось (рис.2), что со 100 и более
ПП точность оценки частот х и \> меняется незначительно.
D
0.2 -
0.15-
0.1 -
0.05-
0
5 10
Рис.8
50
100
Зависимость средней степени дивергенции D от
размера модельной выборки. К - размер выборки.
ПП
2.3 Метод филогенетического анализа ПП
Нами был разработан метод оценки частот транспозиции
основанный на использовании филогенетического анализа.
Пусть имеется набор ПП, для каждого из которых извест-
известна нуклеотидная последовательность. По этим ПП можно пост-
построить филогенетическое древо. На рисунке 3 приведено древо
для пяти ПП.
Длины ребер т', т^, т*, т^ и т^ можно рассматривать
пропорциональными временам нахождения ПП 1, 2, 3, 4 и 5,
соответственно, в сайтах инсерции. Поэтому среднее значение
этих длин f' можно рассматривать как величину, обратнопро-
порциональную частоте выщепления ПП из генома \х.
Рассмотрим расстояния между вершинами т*, т* и т^. Яс-
Ясно, что, например расстояние т^ пропорционально длительнос-
длительности временного интервала между такими двумя событиями, как
репликативная транспозиция ПП 1, приведшая к возникновению
- 70 -
-71 -
Рис.3 Филогенетическое древо для пяти ПП. 1, 2, 3, 4, 5-
- висячие вершины, соответствующие ПП;
т - длины ребер.
ПП 5, и следущая по очередности репликативная транспозиции
ПП 1, в результате которой образовался ПП номер 3. Чем бо-
больше частота репликативной транспозиции х, тем меньший вре-
временной интервал должен разделять последовательные транспози-
транспозиции одной и той же ПП. Поэтому величину т2, являющуюся сред-
средним значением длин т*, т^ и т*, можно считать обратнопропор-
циональной частоте транспозиции х.
Обычно порядок транспозиций не известен, поэтому струк-
структура филогенетического древа, вообще говоря, прямо не связа-
ана с порядком транспозиций. В таком случае удобно использо-
использовать интегральные характеристики филогенетических деревьев,
связанных с частотами перемещений ПП.
Одной из таких характеристик является величина, знач!
ние которой равно сумме всех ребер древа. Легко показать, ч1
для древа, построенного по к ПП, в нем имеется 2(к-1) реба:
длиной ii(i=i, г,..., 2(K-D), и справедлива формула
к т
1
1.1
C)
где т'= ? т'./к - величина, пропорциональная 1/м.
Второй интегральной характеристикой является величина,
равная сумме всех возможных расстояний Ъ1 между любыми двумя
висячими вершинами, каждая из которых соответствует ПП. В
- 72 -
общем случае, для дерева, построенного по к ПП, справедливо
соотношение
с|
I Lt = off + (Л3 , D)
где о и Э - коэффициенты.
Принимая во внимание, что значения коэффициентов а, р
гут сложным образом зависеть от конкретной структуры дерева
я, вообще говоря, не известны, можно использовать описанную
выше имитационную модель B.2) для оценки значений коэффици-
коэффициентов о, э и проверки возможности определения скоростей реп-
репликативной транспозиции и выщепления.
Для этого моделируется возникновение большого количест-
количества ПП за длительное время. При этом скорости транспозиции и
зыщепления задаются заранее. После окончания моделирования
случайным образом формируются выборки из К ПП, то есть из
тех ПП, которые сохранились в геноме за весь цикл моделиро-
моделирования. Для любой такой выборки можно восстановить филогене-
филогенетическое древо по матрице И, так как имеется информация о
том, какой повтор от какого повтора и когда произошел.
Таким образом, восстановив филогенетическое древо для К
ПП данной выборки, мы можем определить по формуле (8) значе-
значение величины т1, а следовательно значение константы ц. Срав-
Сравнивая полученное значение с заданным в модели, можно опреде-
определить точность рассматриваемого метода. Оказалось {табл. 1),
что для оценки величины м достаточно проанализировать небо-
небольшое количество ПП. При этом относительная ошибка составля-
составляет менее 50$ (с тенденцией некоторого завышения истинного
значения м).
Определив величину f' и анализируя ту же выборку разме-
размером к, можно определить значение суммы в левой части D).
Рассматривая большое количество аналогичных выборок то-
того же размера и предполагая справедливым для каждой выборки
Условие D), можно методом наименьших квадратов {считая зна-
значение f известным и равным 1/х) определить коэффициенты о, р
и их зависимость от размера выборки к (табл. 2).
- 73 -
Таблица 1 Зависимость относительных ошибок &ц и б\ опреде-
определения частот выщепления м и ют размера выборки к.
к
10
20
40
50
Таблица 2.
к
5
10
15
20
Sf (%)
70
50
45
45
бо
45
42
41
Зависимость коэффициентов а и Э от
размеров выборки к
а
22
95
220
390
со.
7
91
280
510
Для определения зависимости эффективности данного мето-
метода от значений частот хим. используемых в модели, эти зна-
значения мы изменяли в интервалах:
10 7< X <
10 5 И
10 7< М <
Из табл.3 видна слабая зависимость точности определения
частот транспрозиции, а и р от значений хим.
Таблица 3, Зависимость коэффициентов <*, р и ошибок оценки
б 6
X
2-Ю
2-Ю
2-Ю
2-Ю
частот
ч
Ю-5
ю-6
1.5-10
2-10
транспозиции бх и 6^ от х и ц.
2
2
1.5
1
60
66
59
55
6Х
(*)
65
69
65
60
ос
92
94
96 '
96
Э
90
91
89 .
90,
Для проверки работоспособности метода мы использовали^
набор филогенетических деревьев для Alu повторов [7 ]. Ока-
Оказалось, что полученные оценки достаточно близки к оценкам,
полученным методами 2.1 и 2.2.
3. МЕТОДЫ ОЦЕНКИ ЭФФЕКТИВНОСТИ ПРОТЕКАНИЯ
ОБМЕННЫХ ПРОЦЕССОВ МЕЖДУ ПОВТОРАМИ
Кластером будем называть такое расположение ПП в гено-
геноме, когда расстояние между любыми двумя соседними ПП пример-
примерно равно длине ПП.
- 74 -
Под обменными процессами подразумеваются процессы типа
конверсии, неравного кроссинговера, приводящие к перераспре-
перераспределению молекулярно-генетического материала в геноме.
8.1 МЕТОД ПСЕВДОКЛАСТЕРОВ
3 качестве меры эффективности протекания обменных
процессов с участием кластеризованных ПП удобно использовать
зависимость средней степени дивергенции Ф(х) между любыми
двумя ПП кластера ст внутригено*шого расстояния х между ними.
Требуется оценить: случаен ли вид этой зависимости к
и связана ли эта зависимость с протеканием обменных
процессов в кластере? Метод псездокластеров, позволяющий от-
ответить на эти вопросы, реализуется в виде пяти этапов.
За счет случайных перестановок ПП друг с другом форми-
формируется новая выборка кластеров того же размера, что и исхо-
исходная. Она состоит из псевдокластеров, каждый из которых об-
образован ПП, входящими на самом деле в различные реальные ис-
Рис.4 Формирование выборки псевдокластеров путем
случайных перестановок повторов в реальных
кластерах, а, в, с, d - реальные кластеры;
A',B',c-,D'- псевдокластеры;
A-1О)-номера ПП.
ходные кластеры. Если зависимость * (х), построенная по ре-
реальной выборке, связана с эффективностьс протекания обменных
эмых обменных процессов, то аналогичная зависимость, постро-
построенная по выборке псевдокластеров, уже не связана с обменными
процессами, так как рекомбинационное взаимодействие удален-
удаленных друг от друга ПП маловероятно.
- 75 -
Сравнение реальной и случайной выборок
По выборкам реальных кластеров и псевдокластеров строят
ся графики зависимости функции Фр(х) и $с(х).
В качестве меры различия между реальной и случайной
зависимостями Ф^х) и Фс(х) используется среднеквадратичное
отклонение с
Распределение среднеквадратичного отклонения
Генерируя большое число (М) выборок псевдокластеров,
находим среднеквадратичное отклонение для каждой из М выбо-
выборок от выборки реальных кластеров. В результате можно пост-
построить функцию распределения среднеквадратичного отклонения с
между реальной и случайной зависимостями Ф(х) (рис. 5).
Критические значения отклонения
Так как неизвестно, сколько выборок псевдокластеров не-
необходимо сформировать, чтобы свойства распределения среднек-
среднеквадратичного отклонения е были верно определены, использует-
используется следующий прием.
Для распределения е, построенного по первым N выборкам
псевдокластеров, находятся критические значения с^и eR2 та-
такие, чтобы они ограничивали слева и справа интервалы, в каж-
каждый из которых попадает 2-3* от общего числа всех наблюдае-
наблюдаемых значений величины с
_ Рис.5 Распределение среднеквадратичного отклонения
между реальной и случайной зависимостями Ф(х)
е - величина среднеквадратичного отклонения;
N- количество наблюдаемых значений е;
?к1 и ек2 ~ критические значения.
Затем генерируется И псевдокластерных выборок и строит-
строится распределение отклонений для суммарного множества выборок
псевдокластеров размером 2И. Для этого распределения вычис-
вычисляются новые критические значения е^ и е^. Если критичес-
критические значения сходятся, то распределение среднеквадратичных
отклонений считается заданным. Условие сходимости имеет вид:
- 76 -
к K2 i2
Если оно не выполняется, то генерируется новая выборка
из и псевдокластеров, и такай итерационная процедура продол-
продолжается до выполнения условия.
Проверка статистической гипотезы
Исходная выборка реальных кластеров случайно разделяет-
разделяется пополам так, что часть кластеров образуют одну выборку, а
оставшиеся - другую.
Для каждой из двух таких выборок определяются зависимо-
зависимости Ф^х) и Ф2(х). Сравнивая эти зависимости описанным выше
способом, находим среднеквадратичное отклонение е .
многократно разбивая исходную выборку реальных класте-
кластеров на две части, можно получить множество значений е .
Если в реальных кластерах обменные процессы с участием
ПП протекают с низкой частотой, то следует ожидать, что эле-
элементы множества е имеют закон распределения аналогичный за-
закону распределения, полученному при сравнении псевдокласте-
псевдокластеров с реальными кластерами.
Пусть статистическая гипотеза. HQ заключается в том, что
что выраженных обменных процессов между элементами внутри
хластера не имеется, то есть величина отклонения с распре-
распределена также, как и величина отклонения е. Если из всего
множества значений отклонения с только к элементов попало в
хритическую область (рис.5), то можно найти вероятность та-
такого события при условии справедливости гипотезы HQ.
Действительно, считая, что вероятность Р попадания
значений с в критическую область, при условии справедливос-
справедливости гипотезы HQ (и в силу определения критической области),
будет равна 0.05, а такке считая, что количество попаданий
в критическую область подчиняется биномиальному распределе-
распределению, вероятность попадания в критическую область к раз будет
равна
Р(к) = С' Рк A - Р)"
E)
где м-количество элементов множества е .
Если Р(к)<0.05, то гипотеза HQ должна быть отвергнута с
уровнем значимости 0.95, то есть в этом случае в кластерах
ПП имеют место обменные процессы типа неравного кроссингове-
Ра и генной конверсии.
- 77 -
Выявление обменных процессов в кластерах повторов Alu
Анализ одинаково ориентированных повторов показал, чт
с ввероятностью 85* можно утверждать об отсутствии система
тического воздейстивия обменных процессов на эволюцию одно
направленных повторов Alu внутри кластеров.
Анализ противоположно ориентированных повторов, показ
что распределение величин отклонения е .полученное при ера
внении частей реальнойвыборки друг с другом, сдвинуто в ст
рону малых значений среднеквадратичного отклонения е .
В соответствии с формулой E) такое событие имеет вероятн ¦
сть около 0.001. Таким образом, в ходе эволвции в кластерах
Alu протекают выраженные обменные процессы между противоп -
ложно ориентированными повторами.
3.2. ОЦЕНКА ЧАСТОТЫ НЕРАВНОГО КРОССИНГОВЕРА
ПО РАСПРЕДЕЛЕНИЮ ФЛАНКИРУЮЩИХ ПОВТОРОВ.
Рассмотрим метод оценки частоты неравного кроссингов»;».
(НК), протекапцего с участием ПП, на основе анализа свойств
коротких фланкирующих повторов.
Качественное описание модели
в районе встраивания ПП обычно происходит удвоение ко
роткого участка, и образуются идентичные фланкирующие (ФП)
повторы (рис.6). В митозе и мейозе в результате НК между
(D
(D
(И)
Рис.6 Неравный кроссинговер между одинаковыми кластерами;
1, г, з - номера ПП ; А, В, с - фланкирующие повто-
повторы; 1/г и г/1 - ПП, являющиеся результатом
неравного кроссинговера между ПП i и г.
кластеризованными ПП картина расположения фланкирующих уча-
участков может изменяться.
При неправильном спаривании между сестринскими хромати-
дами A) (рис.6) образуется новое распределение ФП. в первом
новом кластере (I) вокруг ПП 1/2 возникли разные ФП (А и В),
а во втором кластере (II) фланки ПП 2/1 (А и В) не совпадают
но их можно обнаружить вокруг других ПП этого кластера. По-
Подобная картина наблюдается и при взаимодействии гомологичных
хромосом с разными кластерами, однако в зтом случае располо-
расположение ФП может меняться без сдвига между хромосомами.
Итак, по расположению ФП в кластерах ПП можно оценить
приблизительно определить частоту кроссинговера. Для зтого
разработан метод, основанный на имитационном моделировании
кроссинговера между кластерами ПП в митозе и мейозе.
Предпологалось следующее:
1) ПП в кластерах распределены равномерно; г) в начальный
момент в геномах всех особей популяции кластеры идентичны и
каждый ПП кластера окружен ФП (идентичными для данного ПП но
отличными от ФП, окаймляющих другие ПП); 3) наличие незави-
независимых транспозиций ПП, когда каждый ПП кластера имеет опре-
определенную вероятность выщепления из генома; также возможно
встраивание в кластер новых ПП; 4) число потенциальных сайт-
сайтов встраивания ПП в кластере ограничено; Ь) популяция состо-
состоит из N диплоидных особей.
На ЭВМ имитировалось большое количество шагов кроссин-
кроссинговера описанным ниже способом.
3.2-1. Моделирование митотического кроссинговера
Рассматривается неравный кроссинговер между сестрински-
сестринскими хроматидами в каждой особи популяции (обмен между двумя
идентичными кластерами). Вероятность НК предполагалась об-
обратно пропорциональной величине сдвига. Тогда вероятность
того, что в результате кроссинговера образуется новый клас-
кластер, состоящий из к ПП, при условии, что до кроссинговера
кластер состоял из L ПП, может быть взята в виде
р = 4-и- и- -*-i
F).
В случае делеции при НК к=ь-п, где n-количество делети-
ровавшихся ПП в кластере. В случае дупликации к=ь+п, где
^-количество дуплицировавшихся ПП. Очевидно, что величина
сдвига при НК п может изменяться от 1 до (L-1).
Случай, когда п=о, соответствует тому, что НК не проис-
происходит, а происходит гомологичный обмен. Вероятность такого
события будет p=i/l F). Итак, в соответствии с F) на отде-
отдельном шаге моделирования случайно формируется величина сдви-
- 78 -
- 79 -
га при НК. Затем случайным образом определяется место обмена
в данном акте кроссинговера, и после этого с равной вероят
ностью производится выбор либо кластера с делецией, либо
кластера с дупликацией. Размер популяции остается неизменным
3.2.2. Моделирование мейотического кроссинговера
Моделируется неравный кроссинговер между гомологичными
хромосомами. Для этого случайным образом проводится равнове-
равновероятный выбор двух особей популяции, каждая из которых соде-
содержит свой кластер ПП.
Результат взаимодействия таких кластеров друг с другое
определяется вероятностным образом по формуле для вероятное
ти Qij)c того, что при кроссинговере между кластером, состоя-
состоящим из к ПП, и кластером, состоящим из j ПП, образуется но-
новый кластер, состоящий из i ПП [8]:
где
2/C + к)
2(.1 + к)
1|Ь (8)
при четной сумме j + k
при нечетной сумме j + к.
U + к)с- 1
Таким образом на данном шаге случайным образом задается
размер сдвига при кроссинговере по формуле (8). Затем, слу-
случайным образом, определяется направление сдвига и место ре-
комбинационного обмена, что приводит в перестановкам ФП.
Изменение расположения ФП и их сходство описывается
тремя параметрами, усредненными по всей популяции: к1 -коли-
-количество ПП в кластере, каждый из которых окружен идентичными
ФП, умноженное на два; Е„ - количество одиночных ФП. которые
в кластере присутствуют в единственном числе; к„- количество
всех остальных ФП в кластере.
В модели учтены процессы независимого выщепления и вст-
встраивания ПП. Временной шаг в модели соответствует среднему
интервалу времени между последовательными актами НК. Поэтому
вероятность транспозиции и выщепления на каждом шаге модели-
моделирования оказывается связанной с частотой НК.
В результате моделирования можно получить зависимость
параметров к
R-j от времени, которую легко использо-
вать для оценки частоты неравного кроссинговера
Оказалось, что в предположении постоянства частоты
не-
неравного кроссинговера », время существования кластера одноз-
однозначно определяется соотношением параметров i^/N, K2/N, E-/N,
где ы-суммарное количество ФП, лишь начиная с некоторого
критического момента, когда значения параметров становятся
установившимися. В среднем, равновесие наступает за 20 - 40
актов НК, поэтому однозначно определить частоту НК можно
лишь для тех кластеров, которые длительно эволюционируют.
Оказалось, что при достаточно большой величине отноше-
частоты транспозиции х к частоте НК v ( \/v > ю^) значение
частоты неравного кроссинговера оценить невозможно. Наиболее
оптимальна для оценки значения частоты НК при известной час-
частоте транспозиции следующая область изменения значений \/к:
10~2< x/i> < 102 .
В этом случае удается построить калибровочные зависи-
зависимости трех параметров модели от отношения хд>.
Данный метод был применен для оценки частоты НК с учас-
участием кластеризованных повторов Alu.
о
Оказалось, что в большинстве случаев х/к°<10 , то есть
можно считать, что частота НК повторов Alu имеет величину
а
порядка 10 за год.
3.3. ИССЛЕДОВАНИЕ БЛОЧНОЙ КОНВЕРСИИ В КЛАСТЕРАХ ПП
При исследовании эффективности генной конверсии между
ПП обычно используют эволюционное расстояние Кимуры [9].
Однако эта величина не позволяет оценить эффективность блоч-
блочной конверсии (БГК), когда в результате взаимодействия пов-
повторяющихся участков генома формируются короткие гетеродупле-
ксы ДНК с их последующей репарацией и коррекцией, приводящи-
приводящими к "передаче" короткого фрагмента (около 15 н.п.) с одного
ПП на другой. В качестве меры, позволяющей выявить последст-
последствия БГК с участием ПП, можно принять величину, пропорциона-
пропорциональную количеству участков совершенной гомологии, выявляемых
при сравнении ПП.
Будем предпологать, что чем ближе в пределах одной хро-
хромосомы расположены ПП, тем с большей эффективностью будет
протекать БГК. Поэтому удобно выборку исследуемых ПП соста-
составить из элементов, входящих в различные кластеры.
- 81 -
кластер 1
кластер 2
Рис.7 Перекрестная гомология. 1, 2 7 - ПП;
п1 ,- эффективное число участков совер-
совершенной гомологии между ПП i и J-
Одним из факторов, припятствующих выявлению БГК, яв.1:.
ется общая гомология ПП, обусловленная высокой степенью i
родства. Для ослабления влияния этого фактора рассматри
лись только такие пары ПП, локализованных в одном класте
которые филогенетически оказывались ближе к ПП из друге:
кластера, чем друг к другу. Это определялось при построек^
филогенетического древа методом совместимости для четы]s
последовательностей [10]). На рис.7 ПП 1 и г ближе к ПП -и
5, чем друг к другу. Поэтому четверка таких ПП включается в
анализ. Пусть nt г- эффективное число участков совершенной
гомологии (длина которых больше порогового значения 1) между
последовательностями ПП 1 и 2, расположенными в одном класте-
кластере, определяется следующим образом. Если при сравнении этих
ПП находятся два участка совершенной гомологии размером pi
(R1>i) И Е2 (E2>i), TO
,
$2/1 .
В качестве меры отличия БГК между элементами внутри
кластера от БГК между элементами, локализованными в удален-
удаленных друг от друга кластерах, рассматривались величины К, г и
к" 2, определяемые следующим образом (рис.7) :
К, г-
1,2 ,
1,2
1, г
П1,г+ П
где
П1,4+ П1,5+
П2,5
1 , 2
н
- 82 -
Рассматриваются всевозможные четверки, составленные из
пары ПП одного кластера и из пары ПП другого кластера. Про-
Процедура проводится для всех пар кластеров выборки. В резуль-
результате получаем распределения значений кс и к" Затем находим
аналогичные распределения ксл для псевдокластерных выборок
(раздел 3.1). Если к и ксд распределены по разным законам,
то БГК протекает с одной эффективностью между ПП, расположе-
расположенными в одном и том же кластере, и с другой - между ПП, рас-
расположенными в разных кластерах. Для сравнения распределений
К и К достаточно использовать непараметрический критерий
уилкоксона.
Если К и ксл распределены одинаково, то следует оцени-
оценить возможность того, что вид распределений к связан не с БГК
а с матрицей сходства анализируемых ПП. Для этого достаточно
получить выборку, состоящую из случайных нуклеотидных после-
последовательностей того же размера и с тем же частотным составом
что и в ПП, с матрицей сходства, идентичной матрице сходства
исходной реальной выборки. При этом из рассмотрения необхо-
необходимо исключать высококонсервативные участки функциональных
сайтов в реальных ПП.
Сравнивая количество участков совершенной гомологии в
реальной и случайной выборках, можно решить, не является ли
наблюдаемая блочная гомология только следствием высокой сте-
степени родства разных копий мобильных элементов.
Метод использовался для исследования блочной генной ко-
конверсии между кластеризованными повторами Alu. Исходная вы-
выборка состояла из 41 повтора Alu, расположенных в 9 класте-
кластерах. Минимальная длина участков совершенной гомологии равня-
равнялась 10 н.п..
Оказалось, что имеются основания предполагать наличие
блочной конверсии между Alu, эффективность которой не зави
сит от того, насколько близко они расположены друг от друга.
4. ОЦЕНКА ВЛИЯНИЯ ПП НА ПРИСПОСОБЛЕННОСТЬ ПОПУЛЯЦИИ
Разработана модель, учитывавшая повреждающие влияния
инсерций ПП в кодирующие и регуляторные районы генома.
В модель заложены следующие предположения: рассматрива
ется диплоидная панмиксическая популяция численностью N; ка-
- 83 -
ждай ПП имеет независимую от других элементов вероятност
вшепления из генома »; каждая ПП способна к независимой pF
пликативной транспозиции с вероятностью \; в геноме имеете
конечное число потенциальных сайтов встраивания ПП М; все и
М сайтов распределены равномерно; в геноме имеется к локусов
обладающих такими свойствами, что, можно выделить только два
аллельных состояния: первое - в локус встроился ПП, и второе
- ПП в зтом локусе нет: если особь популяции гомозиготна по
первому аллелю, то считается, что она имеет нулевую приспо-
приспособленность; приспособленность гетерозиготы и гомозиготы по
по второму аллелю предполагалась одинаковой; все к локусов
считались независимыми, поэтому можно было пренебречь нерав-
неравновесностью по сцеплению и считать общую приспособленность
приспособленность по всем к локусам мультипликативной. л
Рассмотрим один из к локусов. Пусть х-частота аллеля А1
Тогда частота аллеля А2 будет равна A-х). Можно показать,
что средняя приспособленность популяции w по данному локусу
удовлетворяет условию
* = 1 - X2.
(9)
Если воспользоваться диффузионным приближением [15], то
можно найти функцию плотности вероятности Ф(х) того, что ч&
стота аллеля А1 находится в интервале от х до х+Sx. Для зто
го достаточно определить среднее значение изменения частот;
за единицу времени Mgx и дисперсию изменения частоты х Ygx.
Оказывается, что
М*,= -мх + t>A-:
xd-x)/BW>dW/dx,
x= x(x-D/BN),
Ф(х) =
где С определяется условием нормировки j$(x)dx - 1,
\н/(м-Ю, если
1, если
< м-п
i м-п
п число
Так как х = / Ф(х)х<1х, то подставляя зто выражение i
формулу (9), можно найти среднюю приспособленность по локусу
В качестве меры выживаемости популяции удобно использо-
использовать генетический груз L [9 ]:
I, = (W - W)/W
max max
где w - оптимальное значение приспособленности. Если рас-
смотреть все к локусов совместно (считая справедливым пред-
- 84 -
положение о мультипликативности), то получается, что
L = 1 - A-Х2)К.
Используя ранее оцененные параметры распространения
повторов Alu в геноме [ 9 ], с помощью данной модели можно
вычислить генетический груз.
Оказалось, что при достаточно реальных значениях частот
перемещения повторов Alu, оценке имеющегося количества
повторов в геноме и числе потенциальных сайтов внедрения
Alu, генетический груз находится в интервале от 0.1 до 0.6.
Обычно считается, что у млекопитающих генетический груз не
бывает выше 0.5 [ 9], поэтому полученный результат означает,
что перемещения Alu, в целом, не нейтральны, и в ходе
эволюции повторов Alu должны возникать механизмы компенсации
их повреждающего влияния. Это могут быть блокировка
транспозиции давно внедрившихся в геном повторов, блокировка
внедрения повторов в кодирующие районы и т.п.
5. БАЗА ДАННЫХ ПО СТРУКТУРНО-ФУНКЦИОНАЛЬНОЙ
ОРГАНИЗАЦИИ И ЭВОЛЮЦИИ ПЕРЕМЕЩАЮЩИХСЯ
ПОСЛЕДОВАТЕЛЬНОСТЕЙ.
Для эффективной работы всего комплекса программ необхо-
необходимо наличие базы данных, содержащей как результаты экспери-
экспериментальных исследований свойств мобильных элементов, так и
информацию, возникшую в ходе исследований в рамках самого
комплекса программ.
Рассмотрим кратко формат базы данных в виде множества
информационных полей.
i-Ссылка на начало записи последовательности; 2- длина
последовательности; 3-локализация в геноме(номер хромосомы,
относительный адрес элемента); 4-ссылка на две последовате-
•Рыюсти фланкирующих участков, возникающих при интеграции
ПП; 5-ссылка на последовательность участка интеграции (раз-
(размер участка, сиквенс, локализация точки встраивания); 6.-
ссылки на промоторы полимеразы II (локализация промоторов,
Для каждого промотора локализация начал и концов транскрип-
т°в; 7.-аналогично для промоторов полимеразы III; 8.- ссыл-
ки на значения частот встраивания и выщепления (с привязкой
сайтам транспозиции); 9.-локализация кодирующих районов
- 85 -
со ссылками на функции белка и его свойства); 10.-ссылки на
различные функциональные сайты; 11.- ссылки на рекомбина-
ционные свойства (локализация двух взаимодействующих элем-
элементов, локализация обменов и их частота); 12.- ссылки на
изменения генома, возникающие при выщеплении ПП; 18.-чис-
18.-число копий в геноме; 14.-ссылки на внуклеточные факторы, вли-
влияющие на транспозицию ПП; 15.-название организма; 16.-ссылки
на математические модели динамики (вид уравнений, значения
коэффициентов и параметров модели); 17.-ссылки на результаты
имитационного моделирования (значения параметров, оценка
точности их определения); 18.-ссылки на результаты анализа
обменных процессов в кластерах (вид используемых функцио-
функциональных зависимостей и оценки степени неслучайности выявля-
выявляемых эффектов); 19.-ссылки на классифицирующие методы, пра-
правила и признаки классификации; 20.-описание влияния инсер-
ций и выяеплений ПП на приспособленность организмов.
5. ЗАКЛЮЧЕНИЕ
Основные перспективы развития и применения данного ком-
комплекса программ связаны прежде всего с проведением комплекс-
комплексных исследований свойств перемещающихся последовательностей,
а также с задачей автоматизации подобных исследований.
Во-первых, для этого необходима разработка методов
классификации семейств ПП по нуклеотидным последовательнос-
последовательностям (что является особенно актуальным для экспериментаторов)
на основе экспертной системы.
Во-вторых, необходимо уметь решать более сложную задачу
классификации методов математического и имитационного моде-
моделирования на основе выявления закономерностей (по нуклеотид-
нуклеотидным последовательностям; по экспериментальным данным о числе
копий ПП в геноме, о частотах транспозиции, о локализации ПП
и т.п.), определяющих применимость данных методов к теорети-
теоретическому исследованию свойств конкретных семейств ПП. Поэтому
база данных в перспективе должна включать в себя как опыт
моделирования различными исследователями, так и результату
моделирования, полученные с помощью комплекса программ, свя-
связанного с базой.
- 86 -
Автор выражает глубокую благодарность Колчанову Н.А. и
щахмурадову И.А. за активное участие в постановке задач мо-
моделирования свойств перемещающихся последовательностей, в
обсуждении результатов, и в разработке компьютерных алго-
алгоритмов, а также Митюговой Л.В. за участие в расчетах на ЭВМ.
ЛИТЕРАТУРА
[ 1] Хесин Р.Б. Непостоянство генома. М.: Наука. 1964. 472 с.
[ 21 Flnnegan D.J. // Int.Rev. of Cytol. 1985. v.93. p.281
[ 3] Rogers J.H. // Int.Rev. of Cytol. 1985. v.93. p.187
[ 4] Уотермен д. Руководство по экспертным системам. М.:
Мир. 1989. 470 с.
( 51 Шахмурадов И.А., Капитонов В.В., Колчанов Н.А. // Гене-
Генетика. 1989. т.XXV. с,1682
? В] Капитонов В.В., Шахмурадов И.А., Колчанов Н.А. // Гене-
Генетика. 1989. Т.XXV. с.1111
[ 7] Колчанов Н.А., Шахмурадов И.А., Капитонов В.В. // Мол.
биология. 1989. т.28. с.526
[ 8] Takahata N. // Genet.Res. 1981. v.38. p.97
[ 91 Кимура М. Молекулярная эволюция: теория нейтральности.
М.: Мир. 1985. 894 с.
[101 Омельянчук Л.В., Колчанов Н.А. // В кн.: Алгоритмичес-
Алгоритмический анализ структурной информации (Вычислительные сис-
системы, вып.112). Новосибирск. ИМ СО АН СССР. 1986. с.46-
55.
- 87 -
SUMMARY
Computer system for Investigating the
properties of mobile genetic elements
Kapltonov V.V.
Institute of Cytology and Genetics, Academy of Sciences of
the USSR, Siberian Division, Novosibirsk
At present it has been established that eukariotic ge-
genomes contain a considerable number of mobile genetic ele-
elements (MGE).
Such sequences are an essential factor of evolution and fun-
functioning of genomes affecting organism fitness. The present
paper is devoted to description of computer methods for stu-
studying MGE properties aggregated within a complex of programs
on IBM PC computer.
Section 2 describes the methods for estimating MGE tra-
transposition parameters. Firstly, there is a method for a math-
mathematical simulation of evolutionary dynamics of MGE number
in genome, using experimental information on flanking direct
short repeats. As a result one can obtain estimates
of both the replicative transposition frequency x and
that of MGE excision from the genome it. Secondly (Section
2.2), there is a method for imitation simulation of MGE evolu-
evolution in the genome permitting one apart from obtaining the
estimates of frequencies X and и to obtain those of accurate
determination of x and м depending on experimental data vo-
volume. Finally, in Section 2.3 we describe a method for eva-
evaluating transposition frequency values using philogenetic
analysis.
All three methods provide identical estimates and are
complementary.
In Section 3.1 pseudoclusters method is described which
makes is possible to estimate efficiency of exchange processes
(those of gene conversion and unequal crossingover) with ta-
taking part of clustered MGE. This method made it possible to
show that in clusters of Alu repeats the exchange processes
_ 88 -
can proceed between the oppositly-oriented repeats. Section
3,2 contains a description of a method for estimating the fre-
frequency of unequal crossingover, based on analysis (using imi-
imitation simulation) of redistributing direct short flanking re-
repeats in MGE clusters.
In Section 3.3 we present a method for revealing the ef-
effect of gene conversion. We managed to show their manifesta-
manifestation for Alu repeats in the human genome.
Section 4 describes a method for evaluating the in-
influence of MGE transposition on fitness, based on calculation
of genetic load value in the population.
Finally, Section 5 provides data base for structural-fun-
structural-functional MGE organization, that is designed for a joint detai-
detailed analysis of MGE properties by various methods within the
frameworks of a common complex of program.
- 89 -
КОМПЬЮТЕРНАЯ СИСТЕМА ДЛЯ ИССЛЕДОВАНИЯ РОЛИ ПОЛИНУКЛЕОТИДНОГ
КОНТЕКСТА В ВОЗНИКНОВЕНИЕ МУТАЦИЙ
И.Б.Рогозин, Н.А.Колчанов, В.В.Соловьев, Н.Е.Среднева
Институт цитологии и генетики СО АН СССР, г. Новосибирск
I. ВВЕДЕНИЕ
Представление о пассивной роли ДНК как мишени дг
возникновения мутаций сменяется пониманием большого значеж
полинуклеотидной последовательности в определении тип
мутации, их локализации и частоты. Экспериментальные дате-!
показывают, что в определенных участках ДНК - "горячих
точках, мутации возникают существенно чаще, чем в друг^
участках. Это свидетельствует о важной роли полинуклеотидног
контекста (конкретных вариантов полинуклеотиднн
последовательностей) в возникновении мутаций 11-6].
Появление эффективных методов секвенирования привело
быстрому накоплению данных о локализации мутаций
последовательностях ДНК. Однако, механизмы влиянм
полинуклеотидного контекста на возникновение мутаций остаютс
малоисследованными. В связи с этим важное значена
приобретает разработка компьютерных методов для анализ
особенностей полинуклеотидного контекста, влияющих :-г-
возникновение мутаций. Ниже дано описание разрабатываемой нам.
компьютерной системы для исследования роли этих особенностей.
2. СОДЕРЖАТЕЛЬНАЯ ФОРМУЛИРОВКА ПРОБЛЕМЫ
Большинство мутационных и рекомбинационных событий в ДН-:
можно отнести к следующим классам [71:
а) точковые мутации - множественные или одиночные замен;
нуклеотидов, делеции или вставки небольших участков ДНК;
б) общие рекомбинации - обмен между гомологичными фрагментам
ДНК;
в) сайт-специфические рекомбинации, на основе которых могу"
возникать инверсии, транслокации, делеции или вставкл
фрагментов ДНК большой протяженности, инсерции (делеции!
мобильных элементов, вирусов, фагов;
В работе описана часть компьютерной системы, связаная с
исследованием роли полинуклеотидного контекста в возникновении
одиночных и множественных нуклеотидных замен. Рассмотрим
возможные механизмы их возникновения.
Таутомеризация_нуклеотидов_181
Нуклеотиды, помимо наиболее вероятной конформации (при
которой они образуют комплементарные пары А-Т и G-C), способны
к таутомеризации и переходу в такие конформации, при которых
они могут формировать неканонические комплементарные пары.
Если в момент репликации в родительской цепи ДНК произойдет
переход нуклеотида в таутомерную конформацию, то в дочерней
цепи ДНК возникнет некомплементарная пара, т.е.
предмутационное состояние, которое может в дальнейшем породить
замену нуклеотида в данной позиции.
Дислокация_матричной_цепи_[62
При репликации ДНК, в результате сдвига матричной цепи
относительно праймерной и присоединения следующего вуклеотида
к праймерной цепи, в ДНК образуется некомплементарная пара,
которая может привести к возникновению мутации в
последовательности (рис.1)[51.
-Т-Т-С-А-А-Т-
Праймерная: -Т-Т-С ' -Т-Т-с-А
III -> Ml! -> I I I : I I
Матричная: -a-a-g-g-t-a- -а-а g-t-a- -a-a-g-g-t-a-
Рис.1. Схема возникновения предмутационного состояния
(отмечено двоеточием) в соответствии с моделью дислокации
матричной цепи [51.
_ер_сия_Х92
Это нереципрокный перенос информации от одной нити ДНК к
другой. Генная конверсия может возникать при формировании
несовершенного гетеродуплекса из нитей ДНК, принадлежащих двум
гомологичным удаленным участкам генома. Последующая репарация
корректирует одну нить ДНК по другой, обеспечивая перенос
генетической информации от одного участка ДНК к другому.
- 90 -
- 91 -
проявлявшийся в возникновении группы мутаций ( рис. 6. ).
Репар^ционная_корр^|сция_несовер^ешшх_гетеродхпле1ссов_ДНК11х.
Эти гетеродуплексы возникают при неправильном спаривал
близко расположенных взаимокомплементарных нитей коротк;
прямых повторов или комплементарных палиндромов в ДНК.
Т G Т G
Т G T G
т»а репарация т*а
G*C > G»C
Т G ШПИЛЬКИ Т*А
G*C G*C
5'...-A-A«T-C-...3* 5"...-А«Т-С-...3'
Рис. 2. Репарационная коррекция несовершенной
возникшей по участку комплементарного палиндрома.
шпильк:
(индуцированныймутагенез _)
В основе мишенного мутагенеза лежат прямые взаимодействия
мутагенных факторов с ДНК, приводящие к химической модификации
оснований. При немишенном мутагенезе происходит химическая
модификация пула нуклеотидов или модификация ферментов
репликации и репарации ДНК.
Мутации, как результату некоего процесса, предшествуе',
предмутационное состояние, т. е. повреждение структуры ШЖ,
которое с определенной вероятностью может привести к
образованию мутации. Становление мутаций - сложный
многостадийный процесс. Это затрудняет идентификации
механизмов влияния полинуклеотидного контекста на
возникновение мутаций.
3. ОПИСАНИЕ ПРИНЦИПИАЛЬНЫХ ОСОБЕННОСТЕЙ СИСТЕМЫ
3.1. База данных мутационных событий.
Содержит описание мутаций в молекулах ДНК с известными
полинуклеотидными последовательностями. Используемый формат
данных (рис.3) включает ряд информационных полей, содержащих
Идентификатор последовательности: LAC3SAC
Тип последовательности: ДНК
Авторы: T.A.Kunkel
Название: The autational specificity of DMA polymerasee-a
and - T during In vitro DNA synthesis
Ссылка: The J.of Biо1.Chemistry
Классификация об'екта: In vitro
Ключевые слова: eucarluotlc DNA polymers*»; point mutation,
mi epaiг
Тип мутаций: single base substitution, deletion
Предполагаемый механизм возникновения мутаций: mi ainser-
ainsertion of nucleotldes, nodel of dislocation mutagenesls
Способ возникновения мутаций: spontaneous mutations
Описание особенностей эксперимента: The base substitutions
errors to be analysed were produced during in vitro DNA
synthesis of the lacZa gene In M13mp2 DNA by purified DMA
polymerases -a and -?, In reactions containing all four
dNTP substrates at aquimolar concentrations. The aasey mea-
measures errors which result In a decrease in «-complementati-
«-complementation, identified as К1Sap2 plaques having light blue color.
Разметка функциональных зон:
pept 127.- 0. lacZa gene
Локализация мутаций:
substitution number of
position transition mutations
19
28
28
0 -> T
С -> T
С -> G
1
7
2
И Т.Л.
последовательность:
GCGCAACGCAATTAATGTGAQTTAG и Т.Д.
Рис.3. Пример описания последовательности НО] в банке
мутационных событий. В дальнейшем, на примере этой последо-
последовательности будут иллюстрироваться метода, представленные
в настоящей работе.
- 92 -
- 93 -
библиографическую информацию, нуклеотидную последовательность,
локализацию мутаций, вид нуклеотидных замен, качественную
характеристику мутаций { спонтанные, индуцированные и т.д. )
для индуцированных мутаций - название мутагенного фактора.
В настоящее время сотрудниками ИЦиГ СО АН СССР Колчановьа
Н.А, Циановым Г.Л., Мазиным А.В., Омельяячуком Л.В.
Рогозиным И.Б., Среднрвой Н.Е. разрабатывается более полным
формат данных.
3.2. Комплекс программ анализа полинуклеотидного
контекста.
Эти программы предназначены для выявления особенностей
полинуклеотидного контекста, связанных с возникновением
мутаций. Входной информацией для них являются полинуклеотидные
последовательности с указанием локализации и вида
наблюдавшихся в них мутаций. При анализе контекста
используется расширенное описание нуклеотидов в 15-ти
буквенном алфавите (см.таб. 1 на стр.2 25).
3.2.1. Анализ распределения мутаций по типам
нуклеотидных позиций.
Анализируется вектор FD) (где F(l) - частота мутирования
нуклеотида а, КB) - Т, FC) - G, FD) - С). Для анализа
используется критерий, основанный на биномиальном
распределении. Пусть общее число мутаций равно п, число
мутаций нуклеотида типа 1 равно kj. Причем, частота
мутирования нуклеотида F{i )=kj/n больше 0.25 (ожидаемой при
равномерном распределении мутаций по типам нуклеотидов ).
Вероятность такого события по случайным причина равна
P(k)« I Pln,J,0.26> » ? C-J«@.25)J(l-<0.25Mn~J A)
J-k J-k n
Малое значение (Р < О.01) свидетельствует о том, что для
исследуемого мутационного процесса характерно неслучайно
частое мутирование нуклеотида типа 1. При анализе
распределения мутаций по типам нуклеотидов может
использоваться также расширенный алфавит нуклеотидов
(см.таб. 1 на стр.гН). Пример использования этого подхода
приведен в таблице 1.
Таблица 1. Частота возникновения мутаций в нуклеотидах
определенного типа в последовательности lacbsac.
Тип нуклеотида
Частота мутирования
Plk)
А
0.05
-
Т
0.06
-
G
0.37
4*Ю-3
С
0.52
S
0.88
<10
8.2.2. Анализ мутационных переходов
Полезную информацию о молекулярных механизмах мутагенеза
может дать анализ мутационных переходов l -> J (где 1
исходный тип нуклеотида, J - тип нуклеотида, возникшего в
результате мутации). Анализируется матрица частот мутационных
переходов ТDх4) (где T(i,j) - частота мутационных переходов
из нуклеотидов 1-го в нуклеотиды J-ro типа). Пусть kj, - число
переходов из 1-го в J-й нуклеотид, п - общее число мутаций.
Пусть частота мутационного перехода T(i,J)=k1j/n превышает
1/12, ожидаемую при равномерном распределении мутаций по
возможным типам переходов. Для оценки неслучайности превышения
наблюдаемого числа переходов над ожидаемым используется
критерий A) биномиального распределения Pfkj,).
Малое значение P(kj,)<O.OI показывает, что для исследуемых
мутаций характерен неслучайно частый переход из 1-го в J-й тип
нуклеотида. Так, анализ мутаций в гене LACBSAC выявил
следующие неслучайные типы переходов: переходов с->т выявлено
57, ожидаем 9, P(kct)<IO ; переходов G->Y выявлено 40,
ожидаем 19, P'kgy' * Ю~5. При анализе мутационных переходов
может использоваться расширенный алфавит нуклеотидов.
8.2.3. Выявление горячих точек мутирования.
Мы использовали подход, приведенный в работе [ill.
Распределение мутаций по нуклеотидным позициям оценивалось
- 94 -
- 95-
исходя иа распределения Пуассона. Пусть среднее количеств
мутаций в позициях определенного типа равно и. Для групп
позиций, в которых произошло i мутаций , рассчитываете
ожидаемое число таких позиций в последовательности по формуле
R(i)-8«(i»1/i!)«e"'i, (S
где S - общее число позиций определенного типа в исследуемо
последовательности. Если наблюдаемое число таких сайте
превышает их ожидаемое число, то указанная группа являете
"горячими точками" мутирования.
3.2.4. Оценка кластеризации мутаций.
Для оценки ожидаемого расстояния между случай: :
разбросанными мутациями мы использовали предложенную в работ-
1121 оценку. Доля мутаций, находящихся на расстоянии не боле;
г оснований друг от друга, вычисляется по формуле:
г 1-1
К(г) ¦ ? Г * A - Г) , <3
1=1
где t - средняя частота сайтов мутирования в пересчете на ода
нуклеотид (вычисленная для определенной последовательности '
Вели наблюдаемые расстояния между сайтами возникновекы
мутаций отличаются от ожидаемых •(в соответствии с критерии
я2 ), то считается, что в последовательности наблюдаете!
кластеризация сайтов мутирования.
8.2.5. Анализ модели репарационной коррекции.
Для проверки различных гипотез о механизмах возникновени:
мутаций в молекулах ДНК нами разработан метод статвесов. Это'
метод является эффективным "инструментом" для изучения рол-
полинуклеотидного контекста в процессах возникновения мутаций
На основе выбранной модели механизма возникновения мутаци:
строится функция «(х^, *2 *~ц'• отражающая зависимости
локализации мутационных событий от факторов (х,
полинуклеотидного контекста, использующихся в модели. такими
факторами могут быть выявленные в последовательности структура
(повторы различных типов, сайты, политракты и т.д.) , которые,
как предполагается в рассматриваемой модели, обеспечивает
процессы возникновения мутаций или косвенно, влияют на них. Для
1-ой мутации в исследуемой последовательности вычисляется
- 96 -
значение построенной функции "мал =w(xi" «2'*-1'xnJ"
Полученные веса усредняются по всем мутациям, в результате
чего получается статвес группы мутаций ^¦г1еал ¦ Затем на ЭВМ с
помощью генератора случайных чисел моделируются случайные
мутации в исследуемой последовательности, расположение которых
a priori не зависит от рассматриваемых факторов контекста. Для
исследуемой последовательности строится значение Wc ч при
случайном "разбрасывании" 1000 групп мутаций. Каждая группа
содержит число случайных мутаций, равное наблюдаемому. На
основе распределения
*СЛуЧ
оценивается
вероятность
у
Р'*гчь<,„ <w^m/U ' группе случайных мутаций иметь значение
wpeaJ]iили большее. Если вероятность Р(?реал_<?случ_ )< О(где
q - малое число ), то выявляется достоверная связь данных
мутаций с рассматриваемыми факторами в последовательности.
Аналогично исследуются конкретные (индивидуальные) мутации,
когда вместо группы мутаций рассматривается только одна.
Модель репарационной коррекции предполагает наличие в
районе мутирования прямого повтора или комплементарного
палиндрома. Весовая функция имеет вид: W»(L-K)/R, где L
длина повтора, репарационная коррекция которого могла бы
вызвать возникновении данной мутации, К - число несовпадений
между участками повтора, R - расстояние между участками
повтора. Для каждой мутации учитывался повтор с максимальным
статвесом. Пример результатов анализа приведен на рисунке 4.
* *
• *
т - с
Т С ->
G - С
С - G
G - С
51...- G - С -...3'
G « С
Рисунок 4. Предполагаемый механизм возникновения Мутации Т ->
G в 188 позиции гена lacbsac, описанного на рисунке 3.
P(W
- 97 -
3.2.6. Анализ модели дислокации матричной цепи в ходе
репликации.
Эта модель предполагает наличие в районе мутирования
политракта ( рис.1). Связь политрактов с позициями мутаций
оценивается с помощью метода статвесов ( с использованием
статвеса V = L, где L - длина политракта). Пример анализа
приведен на рис.5 для мутации Т -> С в 49 позиции
последовательности гена LACBSAC, описанного в реферативной
карте. В данном случае выявлена связь положения этой мутации с
политрактом ТТТ, указывающая на возможность ее возникновения
на основе дислокации матричной цепи в ходе репликации, как это
показано на рисунке 5.
В'... СТТТ ...3'
Б'...ССТТ...З'
Рисунок б. Возможный способ возникновения мутации Т->с
(позиция * 49) в гене LACBSAC на основе дислокации матричной
.цепи в ходе репликации P(W <w )=0.08.
3.2.7. Анализ модели генной конверсии.
В настоящее время эта схема возникновения мутаций
исследуется на основе двух программ. Первая из них анализирует
характер распределения кластера мутаций на основе сравнения
предполагаемых донорных и акцепторных последовательностей с
использованием метода статвесов. Статвес для группы мутаций
(см.рис.6) вычисляется следующим образом: W-L2-LI, где L2 и
LI - правая и левая границы расположения кластера в
полинуклеотидной последовательности. Результаты, полученные на
основе этой программы, показаны на рисунке 6, где приведен
пример выявления генной конверсии между vx геном и псевдогеном
цыпленка. Анализ показал, что вероятность наблюдать такой
кластер мутаций по случайным причинам - Р(ИТ^„„ <?„тг„ )<О.О1,
(Jo сХЛ * \jJljf Ч *
что явно свидетельствует в пользу генной конверсии, как
возможного механизма возникновения мутаций в этой
последовательности. Вторая программа выявляет наличие
- 98 -
консервативных районов, характерных для генной конверсии, при
сравнении группы последовательностей, в которых возникли
мутации. Результаты, полученные на основе этой программы,
приведены на рисунке 9.
Li L2
1 AAATCCGGCTCCACAGCCACATTAACCATCACTGQOGTCCGAGCCGACGACAAT
2 CTCtecgTAtTcecaCAeaeattaaccatcaetggggtecAageegaOgaTQaG
„т,- та - A G TO G
3 СТС
ТА Т
СА
Рисунок 6. Пример выявления генной конверсии между v геном
A) и псевдогеном B) из]; внизу приведены нуклеотидные
заменыC); р(«случ.<«реал# )<О.О1. Позиции предполагаемого
конверсионного тракта Ll=66, L2=122.
3.2.8. Анализ консенсусов сайтов мутирования
Участки полинуклеотидного контекста, окружающие сайты
мутирования, могут быть представлены в виде набора выравненных
последовательностей. Для этого набора последовательностей
может быть осуществлен поиск консенсуса ( в расширенном 15-ти
буквенном алфавите ). Например, при построении статистического
консенсуса для набора горячих точек в каждой позиции набора
выравненных последовательностей поочередно для всех 15-ти
типов нуклеотидов оценивается их встречаемость.
Пусть в исследуемом наборе из п последовательностей (на
рисунке 7 п=14) в j-ой позиции рассматривается основание типа
1. Оно встретилось в этой позиции kj раз. Вероятность такого
события оценивается из биномиального распределения
п
Р(к.)= ? p(n,l,F(i)), где F(i) - частота основания 1 в
исследуемой последовательности (в которой произошли мутации).
Для позиции J выбирается такой тип основания, для которого
характерно минимальное значение P(kj), при условии, что
p(V<O.OI.
Статистический консенсус может быть построен также для
Динуклеотидов. Рассматриваются две соседних позиции J и J+1 (
яуклеотидн рассматриваются в 15-ти буквенном алфавите ) . Для
- 99 -
определенного динуклеотида (U,12) подсчитывается число ku 12
его встречаемости в J-ой и У+1)-ой позиции. Частота
встречаемости динуклеотида A1,12) в исследуемой
последовательности F(u,i2). Из соотношение
п
р(км ,_)= т, P(n,l,F<li,i2)) вычисляется вероятности
1 *11,12
наблюдать к^ 1а пар типа A1,12) по случайным причина*
(см.формулу (I)). Как и выше, отбирается пара с минимальным
PUU 12), если Plkjj 121 * О-01 (см- Рис- ?Ь Аналогично
статистический консенсус строится для тринуклеотидов, а также
для комбинаций нуклеотидов в позициях, разделенных
пространственно. Полученная информация в дальнейшем может
подвергаться дополнительной статистической обработке.
Позиция
28
48
53
159
165
192
213
217
226
232
233
234
243
250
Последовательность сайта
GCT С ACT
AGG С ТТТ
ТТА С ACT
ТТЛ С АСС
CGT С GTG
АСС С ААС
ОСА С АТС
АТС С ССС
ТТС G ССА
GCT G GCG
CTG G CGT
TOG С GTA
AGC G AAG
AOG С CCG
Число мутаций
9"
4
6
6
4
5
4
4
6
4
9
4
3
4
динуклеотид - wk с R
PU) = 0.0015 0.0003
Рисунок 7. Анализ динуклеотидов в выборке горячих точе-
мутирования в гене LACBSAC. Выявленные консервативны
динуклеотида подчеркнуты в последовательностях выборки. ,
8.2.9. Анализ связи мутаций с консенсусом
С помощью метода статвесов нами оценивается неслучайность
связи мутаций в исследуемой последовательности с консенсусом
( полученным ранее для мутаций в зтой или другой
последовательности ). Если для определенной позиции J
характерно наличие данного консенсуса (см.рис.8), то ей
присваивается статвес, равный количеству мутаций в этой
позиции:
V = м. ( 4 ).
4. ИССЛЕДОВАНИЕ РОЛИ ПОЛИНУКЛЕОТИДНОГО КОНТЕКСТА В
ВОЗНИКНОВЕНИИ СОМАТИЧЕСКИХ МУТАЦИЙ В ИММУНОГЛОБУЛИНОВЫХ ГЕНАХ
Рассмотрим возможности описанного выше комплекса программ
на примере исследования соматических мутаций в
иммуноглобулиновых генах. Соматические мутации в v-генах в
большинстве представлены нуклеотидными заменами. Они возникают
с высокой частотой в V-районах (V-сегменты генов
иммуноглобулинов и прилегающие к ним фланкирующие районы) и ею
наблюдаются в других районах. Молекулярные механизма этого
явления интенсивно исследуются, но многие особенности остаются
неясными.
Для анализа была взята выборка из 14 V-генов, в которых
выявлено 164 соматические мутации (таб.2) (см.обзор [141).
При исследовании модели репарационной коррекции было показано,
что в II 14 v ) меньше 0.26.
случайным причинам в
соответствии с критерием биномиального распределения равна
3.7*1р~ . столь низкое значение вероятности является весом»!
аргументом в пользу этого механизма возникновения соматических
мутаций. Однако, анализ индивидуальных мутаций показал, что
этот механизм не объясняет возникновение всех наблюдаемых в
этих генах соматических мутаций [14].
Анализ контекста, окружающего точки, для которых
наблюдалось наибольшее число соматических мутаций в
последовательностях VHI3 и VHI4, выявил новую закономерность.
рр
что в II из 14 v-генов значение Р(?„-„„ <?
Вероятность такого события по
_ 100 -
- 101 -
Таблица 2. Результата анализа v-генов иммуноглобулинов.
Краткое
наименование
VN1
VM2
VM3
VM4
VN6
VN6
VM7
VMS
VH9
VM10
VH11
VH12
VH13
VM4
Число
мутаций
3
6
7
4
13
8
10
13
10
10
9
10
42
24
P<W < *сл>
для модели
репарацион-
репарационной коррек-
коррекции
О.П
0.13
0.07
0.12 '
0.07
0.18
0.35
0.46
0.13
0.01
0.08
0.63
0.24
0.21
P<W < Усл)
для встреча-
встречаемости кон-
консенсуса RSY
О.ОБ
0.10
0.16
0.34
0.35
0.04
0.13
0.30
0.44
0.01
0.01
0. II
0.01
0.01
PlW < *сл>
для модели
дислокации
матричной
цепи
-
О.П
-
-
0.29
-
0.52
0.36
0.49
0.27
0.47
0.17
0.29
0.87
Примечание: черточка обозначает отсутствие мутаций,
возникновение которых могло объясняться исследуемым
механизмом.
Подавляющая часть горячих точек мутирования имела консенсус
RSY, при этом, сайт мутирования характеризовался наличием
нуклеотида S. Результаты анализа значимости консенсуса RSY в
14 v-генах приведены в таблице 2 с использованием статвеса,
вычисленного из соотношения D) (рис.8). Можно видеть, что для
ю из 14 v-генов значение Р<?_,„п <и„т„, ) меньше 0.25.
реал. олуН•
Вероятность такого события, вычисленная по биномиальному
распределению, равна 3.4*10 . Столь низкое значение
вероятности показывает, что контекст rsy связан с
возникновением соматических мутаций в исследованных генах
Интересно, что анализ RSY сайта для мутаций в
последовательности с-«ус гена tie](встроенного в кластер
иммуноглобулиновых генов) также выявил явную связь мутаций с
этим контекстом,( P(w
реал
<?случ.)<0-01»-
7В
5*.•.gGGTtcaocttcACTgattactaoatgAGCTtggg...3'
3 2 4 1 1 331
рисунок 8. Часть последовательности v-гена, в котором
наблюдались соматические мутации (их число указано цифрами под
позицией в которой они наблюдались)[16]. Большими буквами
выделены сайты мутирования, в которых обнаружен консенсус BSY.
Влияние соседних оснований на частоту возникновения
мутаций отмечалось и ранее для прокариотических ДНК-полимераз.
Авторы [17] предполагают, что соседние основания слева и
справа могут иметь большое значение для стабилизации
неканонической пары, возникающей в ходе репликации. Похожие
закономерности выявлены при анализе спонтанных мутаций,
возникающих при трансфекции вектора pZI89 в клетки обезьяны.
Для всех мутантных позиций оказалось характерным наличие слева
вполне определенных нуклеотидов. Консенсус имеет вид: YC, где
нуклеотид С - позиция мутирования. Предполагается, что зти
мутации возникли в ходе репарации трансфецированной ДНК,
поврежденной клеточными нуклеазами [17].
Анализ механизма дислокации праймерной цепи не выявил его
связи с возникновением мутаций в исследованных v-генах
( табл. 2).
Анализ механизма межгенной конверсии проводился для
последовательности Ух-гена цыпленка. Ранее, данные в пользу
межгенной конверсии были обнаружены при сравнении нуклеотидной
последовательности этого гена и 25 его псевдогенов [13].
Проведенный анализ областей выявил следующую картину
Распределения соматических мутаций в этом Ух-гене длиной 303
н-п- Предполагаемые области конверсии образовывали пять
основных групп сгущения соматических мутаций. В составе
- 102 -
- 103 -
районов, разделяющих группы сгущения , (спейсеров) неслучайнс
часто встречается тракт TCAS (Рис.9). Этот тракт встретился г
последовательности спейсеров б раз (суммарная длина спейсерог.
- 108 нуклеотидов). Всего таких трактов найдено 6. Вероятность
того, что в отрезок Р=108/303 попало 5 трактов из 6, ь
соответствии с биномиальным распределением - 0.02 (формула
A)). Такое малое значение вероятности показывает, что данных
тракт может играть роль определенного сигнала при генной
конверсии Ух-гена и его псевдогенов.
позиция последовательность спейсера
начала спейсера
2 GCGCTGAC TCAG CCG
62 AGA TCAC CTGCTCCGGQG
116 CCTGGCAGTOCCCCTG TCAC T
162 ACATCCCT TCAC GATTCTCCGGTTCC
201 САСАТТААССА TCAC TOGGGTCC
консенсус tcas
Рисунок 9. Результаты анализа спейсерных районов V^-гена
цыпленка [13]. Анализ выявил консервативный тракт tcas,
который, как мы предполагаем, может быть сигналом генной
конверсии.
Можно предположить следующую схему возникновения
соматических мутаций в генах иммуноглобулинов (см. [2]). Роль
инициирующего фактора в возникновениии соматических мутаций
играет нарушение нормальной структуры хроматина в V-райоках
генов иммуноглобулинов, происходящее1 в ходе их перестройки,
Освобождение ДНК от нуклеосом приводит к ее локальному
плавлению и формированию гетеродуплексов и шпилечных структур,
которые могут подвергаться репарационной коррекции. С другой
стороны, такой район в значительной степени экспонирован для
клеточных нуклеаз, которые могут повреждать ДНК V-районов.
Дальнейшая репарация с ошибками (особенно в RSY сайте)
приводит к появлению точковых замен. Для межгенной конверсия
также необходимо освобождение ДНК от нуклеосом. Относительные
вклады и возможность взаимосвязи этих трех механизмов требуют
дальнейшего анализа.
5.ПУТИ ИНТЕЛЛЕКТУАЛИЗАЦИИ СИСТЕМЫ.
Итак, в нашей работе создан весьма сложный комплекс
программ для анализа роли полинуклеотидного контекста в
возникновении точковых нуклеотидных замен. В настоящее время
этот комплекс расширяется в двух направлениях. Во-первых,
разрабатываются программы для выявления дополнительных
структурных особенностей ДНК, связанных с возникновением
мутаций ( энергия В-формы ДНК, углы Калладина-Дикерсона,
наличие z-формы ДНК, политрактов, различных типов повторов и
т.д.).
Во-вторых, разработан комплекс методов для исследования
таких типов мутаций, как делеции, дупликации [18].
Одновременно с этим ведется разработка более полной ¦ версии
банка данных, включающей, помимо нуклеотидных замен, большие
массивы информации по делениям, дупликациям, рекомбинациям и
т.д.
Сложность и разнообразие функций, выполняемых описанным
комплексом программ, и необходимость анализа обширных массивов
информации, находящейся в базе данных, остро поставили вопрос
интеллектуализации системы анализа мутационных и
рекомбинационных событий, которая в настоящее время находится
на стадии разработки демонстрационного прототипа. Помимо двух
описанных выше блоков ( комплекса программ для анализа
полинуклеотидного контекста и базы данных) эта система
содержит: базу знаний о роли полинуклеотидного контекста в
возникновении мутаций, интерфейс пользователя и программу,
обеспечивающую управление вычислительным процессом,
диспетчер.
5.1.База знаний.
Она предназначена для хранения формализованных знаний о
взаимосвязях между особенностями полинуклеотидного контекста и
- 104 -
- 105 -
мутациями.
В базе знаний содержатся результаты анализа
последовательностей из банка данных мутаций и рекомбинаций.
Для каждой проанализированной ранее последовательности
содержится следупцая информация:
1) характеристика распределения мутаций по нуклеотидным
позициям определенных типов;
2) характеристика мутационных переходов. Для каждого перехода
X -> Y (X - исходный нуклеотид, Y - возникший в результате
мутации) приведена его частота;
3) данные об особенностях контекста, связанных с
возникновением мутаций;
4) данные о механизмах возникновения мутаций;
5) информация из банка данных о исследуемой
последовательности, мутагене, организме и т.д;
в) система классификации результатов анализа. Все
проанализированные последовательности, информация о
результатах анализа которых хранится в базе знаний, разбиты на
ряд групп. Каждая группа включает последовательности, мутации
в которых возникли по сходным механизмам (при анализе выявлены
похожие особенности контекста).
5.2. Диспетчер.
Диспетчер обеспечивает управление работой системы.
Принципы его функционирования основаны на нашем практическом
опыте анализа роли полинуклеотидного контекста в возникновении
мутаций. Диспетчер выполняет три основные функции:
1) управление комплексом программ анализа полинуклеотидного
контекста;
2) оценка сходства характеристик полинуклеотидного контекста
для исследуемой группы мутаций с аналогичными
характеристиками, накопленными в базе знаний;
3) анализ ситуации, возникапцей в ходе работы системы.
- 106.-
5.3. Интерфейс с исследователем
Выполняет следующие функции:
1) ввод последовательности из банка данных мутационных событий
или с клавиатуры самим исследователем;
2) объяснение результатов анализа: мотивировка принятия той
или иной гипотезы о возможном молекулярном механизме
возникновении исследуемых мутаций или обоснование
неслучайности связи определенного контекста с возникновением
мутаций;
3) возможность вмешательства исследователя в работу системы на
любом этапе ее функционирования с целью прекращения
исследования или изменение его направления по желанию
пользователя.
ЛИТЕРАТУРА
СИ RipJey L.S. // Proc.Natl.Acad.Scl.USA 1982, V.79, p.4126
[2] Kolchanov N.A., Solovyov V.V., Rogozln I.B. // FEBs Lett.
1Э87, V.214, p.87
[3] Burns P.A., Gordon A.J.E., Gllcknan B.V. // J.Mol.Blol.
1987, V.194, p.385
[4] Loeb L.A., Kunkel T.A. // Aimu.Rev.Blochem. 1982, V.61,
p. 429
[5] Drake J.V., Baltz R.H. // Annu.Rev.Blochem. 1976, V.46,
p.11
[6] Салганик Р.И., Мазин А.В., Дианов Г.Л., Овчинникова Л.П.
// Генетика 1984, Т.20, с.1244
t7] Льиш Б. Гены. М.: Мир, 1987, 398 с.
(8] Watson J.D., Crick F.H.C. // Nature 19S3, V.171, p.982
E9] Meselson И., Redding С // Proc.Natl.Acad.Scl.USA 1975,
V.72, p.358
UO] Kunkel T.A. // The J.of Blol.Chea. 1985, V.280, p.12866
[11] Topsl M.D., Eadle J.S., Conrad M. /' The J.of Blol. Chea.
1986, V.261, p.9879
Gearhart P.J., Bogenhagen D.F. // Proc.Nail.Acad.Sol.USA
1983, V.80, p.3439
- 107 -
113] Reynaud C.-A., Anquez V., Grlmal H., Velll J.-C. // Cell
1987, V.48, p.379
[14] Рогозин И.Б., Соловьев В.В., Колчанов Н.А. Контекстная
предетерминированяость мутационного процесса
( соматические, спонтанные и индуцированные точковые
мутации) Новосибирск: ИЦиГ СО АН СССР. 1988. (препринт).
[15] Вегек С, Griffiths G.H., Mllstaln С. // Nature 1985
V.318, р.412
[161 Rabbits Т.Н., Наш1уп Р.Н., Baer R. // Nature 1983, V.306,
р.760
[171 Hauser J., Levlne A.8., Dixon К. // The EHBO J. 1987,
V.B, p.63
[181 Рогозин И.Б., Кель А.Э. // Тез.III Всес.совещании
"Теоретические исследования и банки данных по молекулярной
биологии и генетике". Новосибирск, 1988, с.51
SUMMARY
A COMPUTER SYSTEM HELPS THROUGH THE ANALYSIS OF A
POLYNUCLEOTIDE CONTEXT FOR THE ROLE IT PLAYS IN THE
APPEARING OF MUTATIONS
I.B.Rogozln, N.A.Kolchanov, V.V.SoXovyov, N.E.Sredneva
Institute of Cytology and Genetics, USSR Acadeay of Sciences,
Siberian Department, Novosibirsk
That the polynucleotlde context of specified variants
of the polynucleotlde sequences is of profound importance as
for the appearing of mutations, has become a fact by no*.
However, the molecular mechanisms by means of which the
polynucleotlde context comes to be an influence on the
appearing of mutations are what still hangs In the balance.
The efficacious methods of sequencing led to the fast
plling-up of data on the mutation localization in the
sequences. We have worked out в computer system which proved
to promote analysing the features of в polynucleotyde
context that Influences the appearing of mutations. At
present it consists of two substructures, namely The
Hutattonal Events Data Bank and The Complex of Methods To
Analyse The Correlation Between The Context Features And The
Appearing Of Singular and Multiple Substitutions.
The Mutatlonal Events Data Bank Is a description of
DMA-localized mutations, the DNA polynucleotlde sequences
known.
The Complex of Analysing Methods comprises of a number
of programmes with the following purposes:
1. Analysing various characteristics of the point
nutations, including the mutation distribution as for the
types of nucleotide positions, the mutatlonal transitions
etc,
2. Analysing the statistical non-randomness of the
correlation between the mutation localization and various
features of the context (direct repeats, complementary
palindromes, tracts, consensus of various sorts etc.9.
3. Constructing the consensus of "hot-spots" of
mutations.
On the base of the actual methods, we also study the
correlation between the somatic mutations in the Ig genes
and the features of the context.
Some mechanisms of autagenesis are shown to. contribute
to the appearing of the mutations. Firstly, a row of somatic
mutations showed a statistically reliable correlation with
the repeats, which Is In line with the model of reparatlonal
correction of the haterodupllces forming up throughout the
segments of direct repeats and complementary palindromes.
Secondly, reliability was revealed for the correlation
between a number of somatic mutations with the consensus
RSY- Finally, a conservative signal sequence, containing the
consensus TCAS, was detected at the gene conversion, the
V^-gene of the chlken taken as a demo.
Nowadays our complex forks In Its development. Firstly,
- 108 -
- 109 -
the nimber of context features (the energy of the B-form of
DMA, Calladlne-Dickerson's angles, polytracte, z-form of DNA
and so forth) is neveretoppingly upplng. Secondly, the work
at в complex of methods for studying various mutatlonal and
recomblnatlonal events Is In headway. On the base of merging
The Mutatlonal Events Data Bank and The Complex of Analysing
Methods, we are also developplng an lntellectualized system
for analysing mutatlonal and reeombinational events.
Besides, the system Is supposed to Include a Database, an
Interface with the user and a control program to run the
analysis.
- no -
КОМПЬЮТЕРНАЯ СИСТЕМА ДЛЯ ИССЛЕДОВАНИЯ
СТРУКТУРНОЙ ОРГАНИЗАЦИИ ГЛОБУЛЯРНЫХ БЕЛКОВ
I. Поиск информативных характеристик и их
использование при расчете вторичной структуры
В.В. Соловьев, А.А.Саламов, А.К. Салихова.
Институт Цитологии и Генетики СОАН СССР, Новосибирск 630090.
I.Введение
Определение пространственной структуры белков по аминоки-
аминокислотным последовательностям - одна из центральных задач моле-
молекулярной биофизики. Традиционные подходы, применяемые к расче-
расчету структуры небольших органических молекул, неэффективны для
решения этой задачи в связи со следующими ососбенностями: а)
огромным числом переменных, описывающих атомную структуру бел-
белка (vlo ); б) неопределенностью минимизируемого энергетическо-
энергетического функционала (например, диэлектрическая постоянная меняется
от 2 до 80 в разных участках белковой структуры); в) многоэкс-
тремальностьв минимизируемого функционала (причем нативная
структура может не соответствовать самому глубокому минимуму);.
г) трудностью учета взаимодействий с множеством молекул воды в
процессе сворачивания.
В связи с этим представляются перспективными подходы,
учитывающие процесс самоорганизации белка: структуры, образую-
образующиеся на ранних стадиях сворачивания, предполагаются достаточ-
достаточно консервативными, а более сложная структура формируется из
них, как из готовых блоков. В настоящей серии работ рассмотре-
рассмотрена разработанная нами компьютерная система для исследования
закономерностей строения и расчета вторичной и третичной стру-
структур глобулярных белков, реализующая этот подход.
Общая блок-схема системы приведена на рис.1. На первом
этапе работы производится анализ базы данных пространственных
структур белков ( Brookhaven Protein Data Bank) И базы данных
вторичных структур белков. Знания, полученные при этом анали-
анализе, используются для разработки методов расчета вторичной и
третичной структуры глобулярных белков.
- III -
БАЗА ДАНИЮ
вторичных структур
БАЗА ДАНШ
пространственных структур
ПАКЕТ ПРОГРАММ
формирования характерис-
характеристик вторичных структур
ПАКЕТ ПРОГРА»
анализа пространственных
структур белков
ПАКЕТ ПРОГРАММ
дискриминантногс анализ;
вторичных структур
ЗНАНИЯ
Закономерности
формирования
вторичной структуры
Закономерности
укладки
а-спиралей
ПРОГРАММА
расчета вторичное
структуры белка
ПАКЕТ ПРОГРАММ
моделирования укладки «-спиральных белков
Рис. I. Общая блок-схема компьютерной системы для
исследования структурной организации глобулярных
белков.
2. Расчет вторичной структуры глобулярных
белков по их аминокислотной последовательности.
На сегодняшний день существует четыре основных подхода к
разработке методов определения вторичной структуры (ВС) глобу-
глобулярных белков по известной первичной структуре. Первый основан
на поиске стереохимических закономерностей [I], второй исполь-
использует физические модели формирования ВС [2,3], третий базирует-
- 112 -
,я на поиске статистических закономерностей в белках с извест-
известной ВС [4-7] и четвертый учитывает информацию о структуре го-
мелегичяых белков [8-Ю]. Последние два подхода кажутся пер-
перспективными, так как число белков с известной структурой стие-
мительно растет. Однако конкретная реализация таких методов,
как правило, не дает хороших результатов [II], что.
по-видимому, связано с использованием упрощенных способов ста-
статистического анализа. С другой стороны, эти методы, как прави-
правило, учитывают лишь определенные характеристики аминокислотных
последовательностей, в связи с чем расчет ВС заданного белка
одновременно несколькими методами часто дает более надежные
результаты [12]. Поэтому актуальной является разработка нового
подхода к определению ВС белков, учитывающего все лучиие каче-
качества реализованных ранее методов, именно этому требованию от-
отвечает развиваемый нами подход, использующий дискриминантный
анализ, что позволяет интегрировать в методе расчета ВС самые
разнообразные характеристики.
2.1 Поиск наиболее информативных характеристик.
Изложенный метод является дальнейшим развитием предложен-
предложенного нами ранее метода определения ВС белков, основанного на
использовании данных дискриминантного анализа при выборе кон-
формации определенного участка полипептидной цепи [13]. В ос-
основу метода положены следующие известные принципы: 1) совмест-
совместный учет различных типов взаимодействий, формирующих и стаби-
стабилизирующих ВС белков; 2) учет наиболее полного набора индиви-
индивидуальных физико-химических свойств аминокислот и анализ харак-
характера их проявления в белках с известной пространственной стру-
структурой; з) конформационное состояние полипептидной цепи должно
удовлетворять требованиям компактности белковой глобулы и эк-
ранировакности гидрофобного ядра от растворителя.
В число взаимодействий, определяющих ВС, включены локаль-
локальные, средние и дальние. Локальные взаимодействия характеризуют
конформационнные свойства отдельного аминокислотного остатка.
Для их учета рассматривались такие свойства аминокислот, как
энергия транспорта по Тэнфорду, полярность, объем боковой
группы, поперечное сечение остатка, заряд и т. п. На конформа-
- ИЗ -
ционнное состояние остатка влияют особенности его окружения.
Для учета этого i -му остатку полипептидной цепи приписывались
значения физико-химических характеристик, полученные усредне-
усреднением по всем остаткам, лежащим на участке И-к ; i+k ). В сос-
состав учитываемых характеристик входили также коэффициенты
Чоу-Фасмана [5], описывапцие склонности остатков к формирова-
формированию ос-спиралей, р-структур, поворотов и неупорядоченных кон-
формаций, а также параметры склонностей дублетов аминокислот-
аминокислотных остатков, разделенных * остатками (*=0,1,2,3), к опреде-
определенному типу ВС, вычисленные нами по базе данных.
Учет дальних взаимодействий основан на том, что значите-
значительное число гидрофобных групп должно быть погружено в гидрофо-
гидрофобное ядро, а гидрофильные группы должны преимущественно нахо-
находиться на поверхности белка. При оценке склонности определен-
определенного участка полипептидной цепи к формированию а-спирали про-
проверялась возможность образования им гидрофобного кластера, ко-
который в геометрии а-спирали определялся как поверхность, выре-
вырезаемая центральным двухгранным углом ~120 Г вдоль которой
группируется максимальное число гидрофобных остатков. Для ко-
количественной оценки рассчитывались такие характеристики, как
число аминокислотных остатков в кластере, средняя гидрофобная
энергия кластера, суммарная энергия кластера и аналогичные ха-
характеристики для стороны, противоположной кластеру.
В крнформации р-структуры определенный участок полипеп-
полипептидной цепи образует две противоположные стороны (гидрофобную
и полярную). Тогда в участке, для которого оценивается склон-
склонность к формированию р-структуры, остатки разной четности при-
приписываем к разным сторонам и вычисляем средние значения Тэн-
фордовской энергии в и V на соответствующих сторонах.
Кроме того, роль дальних взаимодействий в формировании и
стабилизации ВС отражает ряд интегральных характеристик амино-
аминокислотных остатков, учитывающих их взаимодействие с окружением
в глобулярных белках. Это координационное число, или число
атомных контактов данной аминокислоты с ее окружением в бел-
белках; среднее расстояние до центра масс белка; доля поверхнос-
поверхности, доступная воде и т. п.
Таким образом, каждой аминокислотной позиции соответству-
соответствует набор значений признаков, характеризующих дальние, средние
- 114 -
и локальные взаимодействия. Выделяем следующие пары альтерна-
альтернативных классов ВС: а-спираль - неспираль («-«); р-структура
- не Э-структура (/з - Д); поворот - неповорот (t -~ t) ;
«-конец - не w-конец (н - ы\; с-кокец - не с-кокец (с - с)'.
Тогда задача поиска закономерной организации ВС может быть
сформулирована следующим образом: каждую аминокислоту рассма-
рассматриваемого белка необходимо отнести к одному из двух альтерна-
альтернативных состояний каждой пары конформационных классов. В такой
постановке задача решается методами дискриминантного анализа
[14].
Рассмотрим произвольную пару классов. Пусть xi]t - наблю-
наблюдаемое значение у-го признака для t-ro наблюдения в i-том
классе, где ;=и Р . 1 = иг , t = \ „,. здесь Р - число
признаков и л, - число наблюдений в i-том классе. Введем ли-
линейную дискриминантную функцию как линейно независимую комби-
комбинацию всех признаков :
A)
В качестве меры разделения классов примем расстояние
Махалонобиса D между двумя классами:
D?= (Xr ХгI S/' (Х»-Хг) B)
где
вектор Х(= (хи,х1г,... ,xip); i = i,2 и *„=
a S, - объединенная ковариационная матрица
С помощью D ищем набор признаков, дающий лучшее разделе-
разделение. Такие наборы искались независимо для каждой пары классов.
Изложим кратко алгоритм выбора признаков. I) Анализируется
Разделение между классами I и II, которое дает каждый признак
вотдельности; из всех признаков выбираем признак с наибольшим
D . 2) Выбранный признак рассматриваем совместно с каждым из
- 115 -
оставшихся для отбора пары признаков, дающей лучшее разделе-
разделение. 3) Аналогично добавляем по одному признаку к уже выбрани-
ым к признакам до тех пор, пока выполняется условие [14]:
р = (л, + пг - к - 2)
(
)
- * - 2)
1 + го
Формирование
различных
признаков для
участков вто-
вторичной струк.
(гидрофоб, мо-
моменты, энергия
гидрофобных
кластеров,шаб-
кластеров,шаблоны для пово-
поворотов и т.д.)
База данных
физ.-химических
характеристик
аминокислот
1
Вычисление зна-
значений признаков
для различных
классов ВС
База данных
вторичных
структур
белкоэ
Дискриминантныв анализ разделения
различных пар классов ВС
Вычисление векторов средних
и ковариационных матриц наборов
характеристик для определенных классов
_L
Вычисление расстояния Махалонооиса
кекду классами
Отбор наиболее информативных характеристик
для разделения различных пар классов
по критерии Фишера
Запись векторов Фишера и векторов средних
информативных признаков, используемых для
предгаид'угар яг.
Рис.
2. Блок-схема алгоритма поиска наиболее
информативных характеристик.
- 116 -
где Fa(H!)
значимости «, а
- F-критерий Фишера на заданном уровне
{л, + л2) {л,
- 2
Блок-схема алгоритма поиска наиболее информативных харак-
характеристик приведена на рис. 2. Метод реализован на СМ-4 и IBM-
PC. База данных, используемая в работе, содержит 73 белка с
известной ВС. Полный список всех отобранных характеристик при-
приведен в табл. I. Наборы характеристик и значения коэффициентов
дискриминантной функции для различных пар классов приведены в
табл. 2 - 8.
При анализе и-Л и с-с использованы следупцие параметры :
длина концевой области a-спирали равна 4-м, Р-структуры - 8-м
аминокислотам.
По выбранным характеристикам можно построить кривые, ха-
рактеризупцие склонность определенного участка заданного белка
находиться в «-спирали, р-структуре и р-повороте. После выбора
классифицирующих признаков для отнесения t -ой аминокислоты
конкретного белка к одному из классов можно использовать сле-
дущее правило классификации: если Д г р, то данная аминокис-
аминокислота относится к классу I, в противном случае-к классу II, где
д =
I - У
C)
,jr2{,... ,*
/ = 1,2. Здесь
¦ а р - некоторый порог; it = (*, t,jr2{,... ,*pt), ,2 десь
через Р = S, (Xr - Хр) обозначен вектор Фишера, а к" =
= 1/г р (X, + Хг).
Хотя больвая часть остатков может быть правильно клас-
классифицирована таким образом, это не гарантирует выделения
дискретных протяженных участков одного типа ВС, какими являют-
являются a-спирали и /J-структуры. Например, структура ap«pap в райо-
районе a-спирали правильно будет предсказывать состояние 5ОЖ оста-
остатков. Однако она явно является неприемлемой, как результат
предсказания a-спирали. Поэтому следупцим этапом построения
метода расчета ВС белков являлась разработка подхода, позволя-
позволяющего по этим кривым выделять локализацию ВС.
- 117 -
Табл. I Список
разделение мешу
отобранных характеристик, датам наилучшее
парами альтернативных классов.
Обозначение
F,
« = а
• ¦ в
F,,
пй
1.1.2.3.'
•• «.».!.
ЕГ
HI
н,
Нг
Н,
Н.
Нг
п5
Р.г
Характеристика
КонФодаационные параметры Чоу-Фасмана [,1^Д:
Параметры склонности к
в-спирали
Э-структуре
Э-поворотам
Попоэициоьянй параметр сезонности
к Э-поворотам [16)
Параметры склонности к
внутренней части
*f-KOHliy
с-концу
участку, смежному с tf-кошшм
участку, смешому с с-концом
структуры s [17]
Параметры склонности для дуплетов
разделенных *-1 остатками, во вторичнур
структуру типа s, (Вычислялись на
базе данных из 73 белков).
Гидрофобные свойства аминокислот
Энергия Тэнфорда (изменение свободной
энергии боковых цепей при переносе из
воды в этанол при 25 С ) [18]
Индекс гилропатии [19]
Характеристики гидрофобных взаимодействий
в а-спиралях.
Гидрофобный момент «-спирали [20]
Мера гидрофобноети:
Энергия Тэнфорда
Индекс гилропатии
Гидрофобная энергия кластера
Средняя гидрофобная анергия кластера
Длина кластера
Средняя анергия полярного сектора
Разность средних энергий гидрофобного клас-
кластера и полярного сектора
Характеристики гидрофобных свойств
Гидрофобный момент Э-структуры
Энергия гидрофобной стороны
Консенсусы для поворотов [51]
расовая матрица пешептрона для
остатков вокруг поворота
- 118 -
Табл.2. Характеристики, отобранные для разделения классов
(о - не <х)$
1
2
3
4
5
6
7
8
Характе-
го«
F.
Н,
Нг
Нэг
(Д,
s?C
Коэ».
дискри-
минант-
ной
функции
-21.5
34.8
0.2
-1.1
0.3
-0.2
-0.6
-0.3
1
1
-5
0
3
3
1
-1
Среднее
значение
и диспепсия
по струк-
структуре
.32
.06
.69
.74
.03
.64
.08
.86
± 0
± 0
± 2
± 0
± 1
± 1
± 0.
± 0.
20
07
45
39
26
54
47
33
по неструк-
туие
0.88
0.96
-2.64
0.50
2.56
2.46
0.77
-1.73
*
±
±
1
±
±
±
0
0
4
0
1
1
0
0.
21
08
15
34
23
24
4?
45
Расстояние
Махало
б отде
льности
4.42
1.56
0.58
0.49
0. 14
0.34
0.52
0.09
обиса
сум
марное
4.42
6.13
6.84
7,06.
7.13
7. 13
".г:
-.12
Табл.3. Характеристики, отобранные для разделения классов
О - не Э) 7
1
2
3
4
Характе-
Характеристика
'?:
н,
ТоэИГ
дискри-
минант-
ной
Функции
-16.6
26.0
-0.1
0.3
Среднее значение
и дисперсия
по струк-
структуре
1.44 ± 0.29
1.09 ± 0.08
2.87 ± 1.50
-0.68 ± 0.40
по неструк-
туое
0.92 * 0.24
0.99 ± 0.09
2.54 ± 1.?8
-0.45 ± 0.34
Расстояние
Махалоногнгя
в отде
4.40
1 .26
0.06
0 42
сум-
4.40
-•93
5 °7
Табл.4. Характеристики, отобранные для разделения классов
3
Характе-
Характеристика
Га 'го
1 К ho
—5=—1
дискри-
дискриминант -
ной
функции!
-4.3
-3.8
-3.5
1.4
1
1
1
1
Среднее значение
и диспеосия
по струк-
тусе
•95 ± 0.72
.14 ± 0.17
•03 t 0.09
•17 t 0.26
по неструк-
1.08 1
0.99 ±
0.99 t
0.99 ±
¦' -II-
0
0
0.
0.
46
19
10
22
Расст
Махало
в отде
3.48
о.бг
0.13
0.64
ояние
вобиса
сум-
3.48
3.9S
4.12
4.16
- 119 -
Табл.5. характеристики, отобранные для разделения классов
(С - не с ) <
Табл.8. Характеристики, отобранные для разделения классов
(» - не t L
1
2
3
4
Характе-
Характеристика
FDce
<Fr>.o
-F/
( F* >ro
1 Ко»».'
дискри-
дискриминант-
ной
ЛУНКШШ
-5.9
-4.5
5.3
-4.1
Среднее значение
и диспепсия
по струк-
структур
2.12 ± 0.76
1.13 ± 0.18
1.17 ± 0.19
1.07 ± 0.13
по неструк-
неструктуре
1.06 ± 0.48
0.98 ± 0.18
0.97 ± 0.21
1.00 t 0.14
Расстс
Махалот
в отде
льности
4.63
0.65
0.90
0.26
яние
обиса
сум-
суммарное
4.63
5.26
5.69
6.21
Табл.6. Характеристики, отобранные для разделения классов
(».- не и.ь
1
2
3
4
Характе-
Характеристика
( Fp")ro
( F/ho
Коэфф.
дискри-
минант-
ной
функции
-2.8
-3.1
-3.5
-4.3
Среднее значение
и дисперсия
по струк-
структуре
1.73 * 0.96
1.19 * 0.28
1.09 * 0.15
1.06 ± 0.14
по неструк-
неструктуре
1.04 * 0.50
0.96 ± 0.27
0.98 ± 0.16
0.99 ± 0.14
Расстс
Махало;
в отде
льности
1.68
0.70
0.44
0.21
>яние
ГСбиса
сум-
суммарное
1.68
3.39
2.88
2.95
Табл.7. Характеристики, отобранные для разделения классов
«V
1
2
3
4
- не с. K
Характе-
Характеристика
( Fp">,o
( Fj >ro
п
Коэфф.
дискри-
дискриминант -
ной
Функции
-2.7
-3.0
-4.2
1.4
Среднее значение
и дисперсия
по струк-
1.72 ± 0.67
1.19 * 0.27
1.13 ± 0.16
1.07 * 0.13
по неструк-
неструктуре
1.04 ± 0.52
0.96 ± 0.27
0.98 ± 0.16
0.99 ± 0.15
Расст
Махало)
в отде
ЛЬНОСТИ
1.66
0.71
0.69
0.28
эяние
тобиса
сум-
суммарное
1.66
2.37
2.99
3.03
- 120 -
1
г
)
4
5
6
7
8
Характе-
( f,h
( F, >ю
Р)?
^
F?
( Ш>«
коафф.
дискри-
минант-
ной
-6.4
0.2
0.2
-5.2
-0.9
-6.4
-0.8
2.9
1
1
1
-1
0
0
1
-0
Среднее
значение
по струк-
структуре
.34
.07
.03
.87
.49
.65
.10
.70
t
±
t
±
t
t
±
*
0.30
0.13
0.14
0.93
0.50
0.48
0.15
1.01
по неструк-
туре
0.66 ±
0.95 *
0.94 *
-2.47 ±
0.17 *
0.Э5 ±
0.94 *
-0.01 *
0.26
0.12
0.13
0.66
0.38
0.48
0.15
1.13
Расстояние
Махало]
в отде
льности
2.64
1.06
0.52
0.44
0.51
0.41
1.17
0.41
хобиса
сум-
2.64
3.55
4.03
4.51
4.61
5.00
5.08
5.13
2.2 Выявление дискретных вторичных структур.
Анализ информативных характеристик, отобранных для разде-
разделения различных ВС белков, показывает, что как для а-спиралей,
так и для Э-структур их и- и с-концы сформированы аминокисло-
аминокислотами со специфическими свойствами, отличными от внутренних
участков этих структур. Это свидетельствует в пользу того, что
и- и с-кокцевые участки играют важную самостоятельную роль при
формировании а-спиралей и р-структур. Поэтому предполагается,
что определенная конформация фрагмента белка (т.е. локализация
а-спиралей или р-структур) определяется совместным действием
трех элементов: «-концевого, внутреннего и с-концевого
участка.
В связи с этим для автоматического выявления дискретных
структур {а-спирали, Э-структуры) использовалось три интегра-
льных признака: Д„, Д(л , и Дс (где например Д(п вычисляется
как усредненное значение Д из C) для участка аминокислотной
последовательности с центром в /-той позиции). Для этих трех
признаков производился дискриминантный анализ обучающей выбор-
ки для классов "а-спираль - неспираль" и "Э-структура - не
^-структура", причем Ду, Д(„, Дй-рассчитывались для «-конца,
внутреннего участка и с-конца соответственно. Значения Дт для
массов (а - не а)9 и (р- не РO приведены в табл. 9.
При расчете вторичной структуры (например а-спирали) для
каждого участка длиной 9, начиная с «-конца данного белка,
- 121 -
Табл.9. Наборы интегральных характеристик
а) классы ( о - не о )9
1
2
3
Характе-
Характеристика
»U
»С
*»
дискри-
шнант-
ЯОЙ
функции
-1.1
-0.4
-0.4
Среднее значение
и дисперсия
по струк-
структуре
4.14 * 1.98
0.84 * 2.08
0.68 * 2.44
по яеструк-
туре
-2.38 ± 2.09
-2.23 * 1.53
-3.34 х 1.79
Расстояние
Махалонобиса
в отде
льности
9.75
4.96
3.94
сум-
суммарное
9.75
10.24
10.56
б) классы ( Э - не Э V
1
2
3
Характе-
Характеристика
*и
*с
*»
Коэфф.
лискри-
минакт-
ной
Дпгншгии
-1.3
-0.3
-0.2
Среднее значение
и дисперсия
по струк-
структуре
1.51 i 2.62
2.65 * 2.31
2.79 * 1.82
по неструк-
туте
-2.98 ± 1.67
-0.03 ± 1.76
-0.20 * 1.78
Расстояние
Махалонобиса
в отде
льности
6.90
2.80
2.27
сум-
суммарное
6.90
7.21
7.2В
вычислялось значение Д, аналогичное описанному выше для отде-
отдельной аминокислотной позиции. Причем признаки д. и Дс вычисля-
вычислялись по краям участка, а Д,„- по центральной области. Если
этот участок имел Д < р (где ? -некоторый порог), то он исклю-
исключался из расчета и рассматривался следупций фрагмент белка,
смещенный вправо на одну позиции. При д > р участок расширялся
в обе стороны за счет последовательного вклочения в него по
Одной позиции с одного из краев. На этих участках рассчитыва-
рассчитывались да и фрагмент белка с максимальной да считался потенциа-
потенциальной а-спиралью. Дальнейший поиск осуществлялся за с-кокцом
этой спирали. Аналогичная процедура выполнялась и для
Э-структур. Блок-схема алгоритма расчета вторичной структуры
белков приведена на рис.3.
Таким образом, мы получали набор потенциально возможных
а-спиралей и э-структур. Если некоторые а-спирали и
Э-структуры "конкурировали11 друг с другом , то выбирались те,
которые имеет большее значение д.
Рассмотренный метод дает предсказание вторичной структуры
(а, Р, с) при учете лишь признаков отдельных аминокислот с
точностыэ 57%. Учет специфических дипептидов увеличивает точ-
точность расчета до 63%. Данные тестирования метода на обучая^8
- 122 -
Расчет для «вж-
дов позиции ин-
форм. признаков
для классов
¦ поворот -
- неповорот
Аминокислотная последовательность
Расчет для каж-
каждого фрагмента
признаков
и »
Расчет для
дого фрагмента
признаков
Возможное рас-
расширение фраг-
фрагмента и выбор
участка с мак-
Выбор структур с максимальным »
в местах перекрытий
Учет возможны! ограничений на длину
и способы взаимной упаковки ВС
Конечное предсказание
Рис. 3. Блок-схема алгоритма расчета вторичной структуры
белков.
и контрольных выборках приведены в табл. 10-13. Точность пред-
предсказания не уступает одному из лучших зарубежных методов [6],
проверенному на столь же представительной выборке белков.
Отметим однако существенные отличия. Во-первых, наш метод
позволяет локализовать дискретные структуры. Во-вторых, точ-
точность локализации протяженных а-спиралей и р-структур достига-
ет 90% (табл.II,12). Отметим, что окончательная локализация
- 123 -
Табл.10.Суммарные результаты предсказаний на обучавшей
выборке из 73 белков.
Общее число
остатков
Число пред-
предсказанных
остатков
Предсказание
по позициях
Предсказание
ио структурам
Вероятность
правильного
предсказания
а-спирали
3814
3769
68.2 «
78.9 *
66.2 %
Э-струк-
туры
3124
2899
56.4 *
65.9 *
58.4 *
Суммарное предсказание
по 4 состояниям (а.э.с.г)
Суммарное предсказание
ПО 3 СОСТОЯНИЯМ (а.Э.С)
нерегу-
нерегулярные
участки
5009
5279
78.4 *
ИЗ НИХ
Э-повороты
2043
2143
54.3 *
59.9 *
69
.6 *
Табл.11.Зависимость точности предсказания а-спиралей
от их длины L
Предсказание
по позициям
по структурам
короткие
L < 8
50.9 *
56.7 *
средние
7 < L < 13
69.8 *
83.2 *
длинные
L > 12
79.5 *
96.8 *
Табл.12. Зависимость точности предсказания р-структур о:
Предсказание
по позициям
по структурам
короткие
L < 6
39.3 *
41.3 %
средние
5 < L < 9
59.5 *
76.8 *
длинные
L > 8
66.1 %
92.1 %
- 124 -
Табл.13. Результаты предсказаний по трем состояниям для
контрольной выборки
а-класс
1.Цитохром с (рис)
г.Цитохром с
{g.moUtchtanam)
З.Калмодулин
4.Тропонин С (индис)
5.Гормон роста соматропин
(свинья)
6.R0P белок
(плазмида Colet)
Р-класс
7.Иммуноглобулин G Fc
(человек)
8.Кислая протеинаэа
(эндотиапепсин)
Э.Актиноксантин
а + f класс
ю.Цитохром с,
@. vulgartsl
и.Тимидилат синтеза
[L. casst )
12.Ял-супероксид дисмутаэа
{B.slearothermophl/us)
о / Э класс
13.Роланеза
ы.Аспартат аминотранс^ераза
{G.gtllus)
15.Каталаэа
(В.taurus/
1б.Фосфоглицерат кинаэа
17.Рибулозо-бифосфат карбо-
КСИЛаэа [К.гиЬгит)
Среднее по всем белкам
Предсказание (*)
64.3
73.4
78.4
75.9
60.7
73.0
58.9
53.7
68.5
56.3
62.7
57.6
59.4
54.6
58.2
62.7
60.9
63.51
а-спиралей и р-структур, по-видимому, определяется при форми-
формировании белковой глобулы и зависит от тонких стереохимических
взаимодействий в третичной структуре белка. Поэтому, кроме по-
позиционного критерия точности расчета ВС, мы использовали и
показатель, отражающий число правильно предсказанных ВС. При
этом а-спирали и р-структуры считались правильно определенны-
определенными, вели не менее двух их остатков были правильно предсказаны.
Высокая точность расчета протяженных ВС имеет важное значение,
так как именно они определят1 третичнув укладку белка и их
правильная локализация необходима для большинства разрабатыва-
разрабатываемых методов расчета пространственной структуры белков, один
которых описан в настоящем сборнике [22].
- 125 -
Разработанная нами система выявления информативных харак
теристик аминокислотных последовательностей имеет широкие пер
спективы развития. Во-первых, в настоящее время рассмотрений
процесс приобретения знаний о закономерностях определяющих фо-
формирование ВС, применен к поиску антигенных детерминант бел
жов. Во-вторых аналогичный подход.разрабатывается для анализ;
сложной картины строения функциональных районов генов эукари
от. И в третьих, подходпредложенный нами для выделения диск
ретных целостных структур на основе криоых, характеризующие
отдельные элементы этих структур, представляется плодотворным
для дальнейшего развития компьютерных систем анализа и опреде
яения структуры и функции биополимеров. Такие системы в буду -
чем должны стать основой экспертных систем, с расширением их
базы знаний, введением в них интеллектуального интерфейса об-
общения с исследователем, который имел бы возможность модифици-
модифицировать режимы работы алгоритмов (например, привлечением к ре-
решению задачи известных экспериментальных данных).
Литература.
A1 LimV.I. // J.Mol.Blol. 1974. v.88. p.873
[2] Ptitsyn О.В. ,Finkelsteiii A.V.//Biopolymers 1983.v.22.p.15
[31 ZlmB B.H., Bragg J.K. // J. Chem. Phys. 1959. v.31. p.526
[4] Chou P., Fasman G. // Biochemistry 1974. v.13. p.222
[51 Gamier J., Oeguthorpe D., Robeon B. // J.Mol.Blol. 1978
v.120. p.97
[в] Glbrat J.F., Gamier J., Robson B. // J. Mol. Biol. 1987
v.198. p.425
[7] Qian H., Sejnowekl T.J. // J.Mol.Biol. 1988 v.202 p.865
[8] Levin J., Robson В., Gamier J., // FEBS Letters 19S6
v.205, p.303
[9} Nlshikawa K., Ool T. // BBA 1986 v.871, p.45
[103 Zvelebil M.J., Barton G., Taylor W.R., Stenberg M.J.E.
// J.Mol.Biol. 1987 v.195, p.957
till Kabsch V., Sander С // FEBS Letters 1983 v.155, p.179.
[121 Шульц Г., Ширмер Р. Принципы структурной организации
белков. М.: Мир, 1982. о.151
- 126 -
[131 Соловьев В.В., Капитонов В.В., Колчанов Н.А. // В кн.:
Теоретические исследования и банки данных по молекулярной
биологии и генетике (под ред. Ратнера В.А.). Новосибирск, ИЦиГ
СОАН СССР. 1986. С.138-147
[14] Болч Б., Хуанъ К.. Многомерные статистические методы для
экономики. М.: Статистика, 1979. с.219 - 228
[15] Chou P., Fasman G. // Ann. Rev. Blochem. 1978 v.47.p.251
[161 Chou P.Y., Fasman G.D. // J.Mol.Biol. 1977 v.115. p.135
[171 Argos P., Palau J. // Int. J. Peptlde protein res. 1982.
v.19 p.380
118] Rozakl Y., Tanf.ord С // J. Biol .Chem. 1971 v.246 p.2221
[191 Kyte J. and Doolittle R.F. // J.Mol.Biol.1982.v.157.p.106
B01 Elsenberg D. , Veiss R.M. and Terwllllger T. // PMAS
of USA 1984 v.8i p.140
[2U Coen F., Abarbanel R.I., Kuntz R., Fletterlck. //
>* Biochemistry 1983 v.22 p.4894
t22] Соловьев В.В., Салихова А-К., Саламов А.А. //
(настоящий сборник) Новосибирск: ИЦиГ СО АН СССР, 1989.
с. 12-40.
- 127 -
SUMMARY
COMPUTER SYSTEM FOR INVESTIGATIKG
STRUCTURAL ORGANISATION OF GLOBULAR PROTEINS
1. SEARCH FOR INFORMATIVE CHARACTERISTICS AND THEIR
USAGE FOR CALCULATING THE SECONDARY STRUCTURE.
Solovyev V.V., Salamov A.A., Sallkhova A.K.
Institute of Cytology and Genetics, Siberian Branch,
the USSR Academy of Sciences, Novosibirsk
Determination of a special structure of proteins
according to aalno acid sequences Is one of the key problems
of molecular biophysics. The present paper Is the first one of
the papers devoted to a computer system elaborated by us for
investigating the regularities of construction and calculating
secondary and tertiary structures of globular proteins. This
system takes account of a step-by-step proteins
••lf-organlzatlon process.
Section 1 Is an Introduction to the entire series and
describes shortly a general scheme of the whole system
(Flg.i).
Section 2 is devoted to a method of calculating the
secondary structure (SS) of globular proteins by their ami no
•eld sequence based on using data of discriminant analysis In
choosing conformation of a particular segment of polypeptlde
chain. A problem of searching for SS regular organisation has
been formulated here In the following way: each amlno acid or
fragment of the protein considered with the related set of
signs values characterizing different types of interactions,
should be attached to one of the two alternative SS classes.
(As pairs of alternative classes there have been taken a-hell*
- not-hellx la - a), Э-structure - not-p-structure (p - ?'•
tern - not-turn (t - t), N-end - not-N-end (N - Й), C-end -
not C-end (C - CM.
Section 2.1 deals with a search for the most informative
characteristics influencing a division of alternative classes-
- 128 -
The number of signs to be analyzed included both the
frequently used characteristics (various physical-chemical
parameters, hydrophoblc moments of a-hellces and ^-structure»
and so on) and a number of others (such as, the number of
amlno acids in hydrophoblc cluster, hydrophoblc cluster enegry
of a a-helix, energy of a hydrophoblc side of ^-structure and
so on). Sampling of Informative characteristics was performed
by Fisher's F-factor [141. Makhalonobis's distance was taken
as a measure for dividing Into classes. Fig. 2 Is a scheme of
an algorithm of search for the most informative
characteristics. The method has been realised on SM-4 and
IBM-PS computers. A complete list of all the chosen
characteristics Is given In Table 1. Sets of characteristics
and coefficients values of the discriminant function for
different pairs of classes are presented In Tables 2-8.
To attribute the t-th amlno acid of a particular protein
to one of the classes one may use the following rule of
classification: if Д г Р, then a given amino acid belongs to
class I, in the opposite case to class II, where Л = px-Y (I),
and P is certain threshold; x.= (x..,*_.,...,x .) Is the signs
1,2,...,p, calculated for t-th amlno acid. Here Fisher'•
vector Is designated by psS^lX^-XJ, and Y*= 1/2 p (Xj+X.,);
X; and X2 are the vectors, being mean for classes I and II,
end Sg'1 is the inverse united covarlon matrix.
Section 2.2. Is devoted to automatic revealing the
discrete secondary structures (a-hellxces and 0-structures).
For this a model of SS formation has been proposed that uses
the three Integral signs: 4wl д , and & (where, for example,
л,„1в clculated as the average value Д from (I) for a region
of amino acid sequence with the centre In the i-th position).
With these signs taken Into account a discriminant analysis of
teaching sampling was performed for classes of a "a-helix -
not-hellx" and "p-structure - not-p-structure", with Дм, Д1|(,
Ac Delng calculated for N-end, Internal region and C-end,
respectively. The values Д for classes (a - not-a) and ip -
not-PO are shown In Table 9.
Shown In Flg.3 is the cheme of an algorithm for
calculating the secondary structure of proteins. The method
- 129 -
had been considered to predict the three states of SS (a, p,
c) with the 63 % accuracy. Data on testing the method on
teaching and control samplings are shown in Tables 10-13,
Prediction accuracy competes with the best foreign methods
[в]. One may note, however, some essential distinctions.
Firstly, the methods permits one to localise discrete
structures. Secondly, the accuracy of revealing «xstended
o-hellces and p-structures is 90 К (Tables 11,12).
- 130 -
КОМПЬЮТЕРНАЯ СИСТЕМА ДЛЯ ИССЛЕДОВАНИЯ
СТРУКТУРНОЙ ОРГАНИЗАЦИИ ГЛОБУЛЯРНЫХ БЕЛКОВ
2. Анализ закономерностей и метод расчета
третичной структуры а-спиральных белков.
В.В. Соловьев, А.К. Салихова, А.А. Саламов
Институт цитологии и генетики СО АН СССР, г.Новосибирск
I.ВВЕДЕНИЕ
Для расчета пространственной структуры белковой молекулы
с учетом процесса самоорганизации недостаточно знания её вто-
вторичной структуры. Уникальность и быстрота сборки третичной
структуры белков обеспечивается некоторыми механизмами, точное
знание которых в настоящее время отсутствует.
В связи с этим нами разработана система для исследования
закономерностей пространственной организации глобулярных бел-
белков на основе компьютерного анализа рентгеноструктурных данных
(рис. I).
| БАЗА ДАННЫХ
j Broofchaven
i Data Base
ПАКЕТ ПРОГРАММ АНАЛИЗА
-гидрофобных моментов ««-спиралей
-соотношения типов вторичных структур
взаимного расположения пар спиралей
-плотности контактов меащу спиралями
-доли контактов разных типов
3 Н А Н И Я о закономерностях
строения пространственной структуры белка
МЕХАНИЗМ УКЛАДКИ
МЕТОД
РАСЧЕТА
третичной структуры ««-спирального
глобулярного белка
Рис. I. Схема компьютерной системы анализа и
расчета пространственной структуры белка
- 131 -
С помоиыэ программ этой системы проанализировано более 20-ти
а-спиральных белков из Базы данных пространственных структур
белков и получены знания о закономерностях встраивания
а-спиралей в глобулу, взаимной укладке пар а-спиралей,
значимости вкладов контактов разных типов, плотности контактов
между а-спиралями. Эти знания служат основой для разработки
гипотезы о механизме самоорганизации, на основе которой
предлагается метод расчета третичной структуры, в настоящей
работе описан разработанный нами пакет программ расчета
пространственной структуры для а-спиральных белков.
2. Пакет программ анализа пространственной структуры ;
а-спиральных глобулярных белков. ]
В данном разделе изложены методы и результаты анализа
строения а-спиральных глобулярных белков. Полученные законо-
закономерности важны для моделирования формирования пространственной
структуры этих белков.
2.1. Анализ особенностей встраивания
а-спиралей в глобулярное ядро.
Для исследования влияния взаимной упаковки отдельных
а-спиралей на формирование глобулярного белка с гидрофобным
ядпом и гидрофильной оболочкой, вводится понятие гидрофобного
момента, который отражает ассиметрию распределения неполярных
(гидрофобных) остатков всего белка или его части [1,21.
Гидрофобный момент а-спирали определяется как сумма
моментов отдельных аминокислот ( L длина а-спирали ):
L L
й = /__ Si - / Gi * °l
i = l 1=1
Здесь гидрофобный момент аминокислоты mt есть вектор, направ-
направленный по перпендикуляру, опущенному из центра масс боковой
группы на ось а-спирали ( в, единичный вектор в этом направле-
направлении). Величина ij равняется энергии гидрофобных взаимодействий
G^ для данной аминокислоты. Обычно используются значения гид
рофобной энергии Тэнфорда [31. Явный физический смысл имеет
направление момента, которое при совпадении с направлением
встраивания спирали в глобулу обеспечивает максимальную гжра-
- 132 -
нировку гидрофобных боковых групп аминокислот. Если гидрофоб-
гидрофобные и полярные аминокислоты распределены равномерно, момент
Таблица I. Гидрофобный момент всех а-спиралей на примере
нескольких белков.
название белка
гемоглобин
лошади
всего
лизоцим
бактериофага
всего
парвальбумин
карпа
всего
миоглобин
кашалота
всего
цитохром с551
всего
номер
спирали
2
3
4
5
6
6
I
2
3
4
5
6
7
7
I
2
3
4
4
I
2
3
4
5
6
6
I
2
3
4
4
длина
спирали
в ак
14
15
20
10
18
22
99
12
21
II
13
II
10
13
91
9
10
9
10
38
16
15
21
9
19
23
103
9
10
II
14
44
5.76
5.77
2.49
2.90
14.99
8.83
15.64
6.05
6.II
6.00
6.82
7.46
6.34
5.84
6.18
5.70
5.41
6.58
5.90
7.73
12.07
6.17
3.80
3.44
4.95
8.95
3.92
3.36
5.05
2.35
6.26
8.18
проекция
к центру '
белка в %
58
94
(-) 54
51
72
48
47
99
92
(-) 67
60
(-) 5
62
58
(-) 6
23
52
48
99
64
66
31
62
19
67
48
(-) 29
*)
Приведена величина компоненты гидрофобного момента в
процентах, взвешенная к количеству аминокислот в а-спирали.
Знак (-) означает что момент направлен от центра белка.
- 133 -
а-спирали близок к нулю: увеличение величины момента соответ-
соответствует выделению гидрофобного и гидрофильного кластеров на
поверхности а-спирали.
Сумма гидрофобных моментов «-спиралей, входящих в белок,
составляет гидрофобный момент всего белка. Принимая во вниман-
внимание существование гидрофобного ядра в глобуле, можно предполо-
предположить, что величина суммарного момента невелика, так как момен-
моменты разных а-спиралей должны быть направлены к центру белка.
Эти предположения были проверены при анализе 17
а-спиральных глобулярных белков из банка третичных структур
белков [41. Примеры величин гидрофобных моментов для каждой
а-спирали и суммарные моменты для нескольких белков приведены
в таблице I. В ней же приведены доли проекции момента отдель-
отдельных а-спиралей к центру белка, взвешенные на количество ами-
аминокислот в а-спиралях. Видно, что для большинства «-спиралей
значительная составляющая момента направлена к центру болка. в
22-х проанализированных белках только 16 % а-спиралей имеют
гидрофобный момент, направленный от центра, а для 64 * - про
екция момента к центру белка составляет более 50 * величины
полного гидрофобного момента а-спирали.
Для проверки направленности реальных гидрофобных моментов
к центру белка, мы сравнивали реальный гидрофобный момент все
го белка со случайным, который определялся как сумма гидрофоо
ных моментов а-спиралей, имеющих реальные величины и случайные
ориентации в плоскости, перпендикулярной осям «-спиралей. Реа-
Реальный суммарный момент в большинстве случаев меньше случайного
(таблица 2). Ситуации, для которых случайный гидрофобный мо
мент оказался меньше реального (например, леггемоглобин), мо
гут возникать у белков, функционирующих и составе мультимерних
комплексов и имеющих группы гидрофобных остатков на своих по
нерхностнх в местах контакта с другими суб'единицами.
Исследовании направления гидрофобных моментов вторичннх
структур и соотношения между суммарными реальными и случайными
моментами для нескольких белков проведены также в работе [.
"Анторами получены важные результаты о направлении моментов
<> спиралей к центру и значительном превышении случайного сум
марного момента но сравнении с реальным. Однако, в С!;] направ
Л(;ни(! гидрофобного момента «-спирали определилось из гоомитри
- 134 -
ческих соображений для идеальной спирали. Поэтому, разли«-чия
между суммарными реальным и случайным моментами оказались в
анализируемых авторами белках несколько выше рассчитанных нами.
Таблица 2. Суммарный гидрофобный момент 17-ти а-спиральных
белков для случаев реальной ориентации и случай-
случайного расположения моментов а-спиралей.
название
белка
гемоглобин
лошади («0
гемоглобин
лошади (р)
гемоглобин
. человека («0
гемоглобин
человека^
человека^)
деоксигемоглобин
человека (?)
леггемоглобин
лизоцим
лизоцим
миоглобин
человека
миоглобин
парвальбумин
карпа
релаксин
ферроцитохром С2
цитохром С
цитохром С
цитохром C55I
% ак
в спиралях
70.21
53.42
75.89
74.66
75.89
62.59
75.82
55.49
29.36
61.44
67.32
18.35
40.00
22.32
34.62
34.62
42.68
реальный
момент
15.64
17.98
20.63
5.12
20.63
17.52
34.17
6.18
5.35
3.92
19.02
7.73
3.68
6.80
8.32
7.40
8.18
случайный
момент
18.92
19.39
24.03
24.80
23.17
25.15
29.69
18.18
6.89
18.58
21.74
11.73
4.41
5.07
8.43
9.39
9.44
Таким образом, гидрофобный момент "грубо" характеризует
направление встраивания а-спиралей в гидрофобное ядро белка.
?.2. Взаимная укладка пар а-спиралей.
Предполагается, что одним из ранних этапов самоорганизации
после формирования вторичной структуры является взаимная укла-
- 135 -
юса соседних пар а-спиралей и образование, так называемой,
"сверхвторичной структуры" из двух или трех спиралей [5-81.
Мы исследовали закономерности попарного взаимодействия
а-спиралей, связанные с определенным расположением гидрофобных
групп на их поверхностях.
Для характеристики наличия или отсутствия контакта между
двумя спиралями было введено понятие плотности контакта:
».. = -4- * V ш.
"и
где Bj, - количество контактов между 1 и J атомами, принадлежа-
нами разным а-спиралям, D - длина более короткой из спиралей.
Учитывались контакты только между парами а-спиралей, для кото-
которых плотность контакта была выше 0.5, что соответствует сущее
твованию как минимум Юти контактов между атомами разных
а-спиралей.
' • • ••
•
-/*> «о -« -«о -"» -к> -<•
т
¦т
т
т
к
ю
и
to
9
го *о
. ..
tO ID 'ОО ЧО >*° ttO liP
Рис.
Зависимость угля между осями и спиралей от угла мпжду
их гидрофофбными моментами лля 80 >«- спиралей из 14 ти
белков.
Характер взаимного расположения двух контактирупцих
«- спиралей описывается углом между их осями о, а взаимное рас
положение гидрофобных кластеров на их поверхностях можно оха
рактеризонать углом между их гидрофобными моментами Ф. I'рафик
зависимости между этими двумя величинами ( рис. ','.) и гистог
раммы распределения о и Ф (рис;. Яа.б) попиоляют сделать неко
- 136 -
торые заключения о наиболее предпочтительных взаимных располо-
расположениях а-спиралей в белках.
9 +
а )
-160 -120 -80 -40
18
14
10
6
2
О
б )
60
120 160
Рис. За. Распределение по
углам между осям
а-спиралей.
Зб. Распределение по
углам между
гидрофобными
моментами спиралей
100 ' 140 ' 180
Так, например, чаще всего встречаются пары а-спиралей, оси
которых направлены перпендикулярно или актипараллельно друг
другу. Для классификации взаимной ориентации гидрофобных мо-
моментов были выделены четыре возможных ориентации отдельного
момента и семь типов его взаимного расположения (таб. 3).
Анализ а-спиральных белков показал, что а-спирали тяготеют
к контакту гидрофобными поверхностями (таб. 3). Характер этих
контактов таков, что взаимодействующие а-спирали образуют ком-
комплекс, имеющий общие гидрофобную и гидрофильную поверхности,
контактируя друг с другом краями гидрофобных кластеров (таб.
31. Такое расположение а-спиралей было названо [71 "полярным",
в отличие от "неполярного", когда гидрофобные моменты направ-
направлены в основном друг к другу (таб. 3).
- 137 -
Таким образом, вслед за [91 на более широкой выборке бел-
белков определено наиболее распространенное взаимное расположение
гидрофобных кластеров контактирующих а-спиралей с образованием
единых гидрофобной и гидрофильной поверхностей. Этот вывод яв-
является существенным для развития представлений о пространст-
пространственной организации белков.
Таблица 3. Количество различных типов взаимного расположения
моментов для параллельного (|| ) и перпендикулярного
( 1 ) расположения осей а-спиралей.
а-спирали показаны с торца и изображены схематично
прямоугольниками. Гидрофобные кластеры заштрихованы.
СИ ИИ
СИ СИ
¦и си
итого
оси
оси И 3
оси
оси 11 5
оси
оси
II
оси
оси
оси
оси
оси
т
30 / V.5
10
10
12
55
"полярное"
расположение
а-спиралей
"неполярнос"
расположение
«-спиралей
Исследовался также вопрос о количестве взаимных контактов
между а-спиралями. Результаты итого анализа приведены на рис
4. Из гистограммы видно, что основная масса «-спиралей копта
ктирует с двумя другими «-спиралями. Цля 46% ¦«-спиралей, име
пцих контакты с 3-мя - 5-и другими «-спиралями, всегда можно
выделить две или одну спираль,, плотность контакта с, которыми
значительно выше, чем с остальными. Отметим, что в 83 % олуча
ев контакты возникают между соседними (п прицелах олижайших
- 138 -
трех) по аминокислотной последовательности а-спиралями.
20
15
10
число
контактов'с 1-й
Рис. 4
2-мя
3-мя
4-мя
5-d а-спиралями
it-
Ik
It
Количество контактов у а-спиралей в II белках.
Заштрихованная область.соответствует спиралям,
у которых есть по два значимых контакта с другими
( 62 % ). Остальные 38 % а-спиралей имеют по
одному значимому контакту.
Рассматривались только белки, имеющие более
двух а-спиралей.
Полученные данные показывают, что, как правило, определен-
определенная а-спираль тесно взаимодействует с двумя другими. Это также
свидетельствует против модели полностью автономной укладки пар
спиралей на ранних этапах процесса самоорганизации и позволяет
предположить, что формирование нативной конформации, в том чи-
числе» попарные взаимодействия,происходят, видимо, после эта-
nas на котором все а-спирали образуют квазисферическую глобу-
глобулу,' контакты гидрофобными поверхностями экранируют их от воды.
2.3. Анализ атом-атомных взаимодействий.
Исходя из гидрофобной структуры белкового ядра,нередко
пренебрегают вкладом электростатических контактов. Так как
гидрофобные взаимодействия играют суще, ственнуг роль в процессе
самоорганизации, значимость электростатических контактов можно
грубо оценить при сравнении количества гидрофобных и электро-
электростатических контактов в глобулярном белке.
Мы рассмотривали контакты между всеми атомами, входящими
ь аминокислоты а-спиралей (кроме атомов водорода). Предполага-
Предполагалось, чт» атомы кислорода и азота поляризованы или Шгсут за-
рял, а атомы углерода нейтральны. Наличие или отсутствие кон-
контакта определялось геометрически. Считалось, что контакт есть,
- 139 -
fc |Г> Ю t- CO
Е- «d «d « «d
ж as ж x ж
*> О О О
g
! о
! t« E-
ssss
*d о О о
4) И S Е- i
X 4" «5 4i <
О О Р* 1
о о
к к
О О
о о
1 i
11
Е- й a a n u
«d л д в О
i ч аз
~ I 4» (О ~
: * 9 >:
о а
Е- X
л а
О i о Е-
О t* ti >- 4> >* 4»
a ? S а я а а
rt cu С
« о « о
I О К О №
• « ю ю m
13 i Д О К О О О
о ¦ч- so
Ьй - (X) ? Ьй
1~
о
со
"""
О)
<
с
4)
(Л
СО
X
Gin
GIU
с
¦п
о.
a
t_
4*
°"
О
О.
_
(О
о
4»
_J
>
<
90S
о
г-
ст.
со
OJ
ON
ю
го
со
г-
tO
сг-
OJ
со
о
OJ
155
ю
го
о
CU
о
ю
го
о
о
о
OJ
о
OJ
о
tO
о
о
0.09
0.14
10
о
CD
nj
О
.09
о
to
о
g
.09
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
8
о
8
О
о
о
о
о
го
О
о
о
о
а>
о
о
СО
о
о
0.06
0.05
го
О
О
О
о
о
о
to
о
о
о
о
со
о
о
го
о
о
о
го
о
to
о
о
to
о
о
ю
о
о
ч>
о
о
о
о
о
OJ
о
ю
о
о
.46
о
о
о
о
0. 18
0. 1 1
ю
о
о
о
.09
о
о
о
.93
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
OJ
го
ru
о
о
о
ю
о
о
о
о
о
0.05
0.04
о
о
о
о
о
о
о
ГО
о
о
о
о
о
о
о
OJ
о
о
nj
о
о
о
о
о
о
о
о
ю
о
о
ID
о
о
о
о
о
60.
о
о
о
to
О-
о
о
ю
ю
OJ
о
—
о.ег
г.31
о
о
ON
ю
-'
Г—
о
ON
CD
о
ГО
ГО
Г-
го
со
to
со
«г
ю
ю
о
\О
OJ
.во
.67
OJ
о*
ru
OJ
—
ю
о
г—
F
о
о
го
о
г-
о
On
¦ч-
о
0.62
0.49
ГО
о
о
го
о
о
о
CU
о
о*
ю
о
ч-
о
ю
о
о
о
со
о
t—
ю
о
го
СО
о
о
о
го
о
г-
о
0.43
0.4в
о
го
о
CU
ю
о
о
ю
о
ГО
СО
—
OJ
о
о
-
о
ги
-
СО
ч-
о
го
о
го
СО
OJ
о
го
о
СО
о
о
о
0.09
о
о
z^
о
о
о
zt
о
<»¦
о
о
о
о
о
OJ
о
о
On
о
о
о
о.
-
.59
о
го
о
о
ю
nj
о
_j *-.* tri ?4i 1Л %s *c m
- 140 -
если расстояние меаду полярными атомами не превышает в а для
гидрофобного контакта и 4 X - для электростатического.
С помощью программы "Контакт" проанализирована информация
по всем имеющимся «-спиральным белкам в Банке данных. Рассмот-
Рассмотрены различные типы контактов между неполярными и полярными
атомами, принадлежащими разным а-спиралям, находящимся как в
основной цепи, так и в боковой группе ( таблица 4 ).
Представлены данные о среднем числе контактов различных
типов для всех 20 аминокислот. Видно, что основное число внут-
рибелковых контактов приходится на контакты между атомами гид-
гидрофобных боковых групп, а также на контакты между этими боко-
боковыми группами и неполярными атомами основной цепи белка (см.
отроки 5-6 таблицы 4). Полярные (или заряженные) атомы в 40 I
случаев контактируют между собой, а в остальных - с полярными
атомами основной цепи белка {см. строки 1-2 таблицы 4).
Поскольку парциальные заряды на полярных атомах боковых
групп (лизина, аргинина, глутаминовой и аспарагиново!
кислот)обычно в несколоко раз выше, чем для атомов основное
цепи НО), то электростатические контакты между ними должны
давать значительный вклад в стабилизацию белковой конфармации.
Исследование атом-атомных взаимодействий в «-спиральных белках
с известной пространственной структурой позволяет сделать вы-
вывод о значительном количестве О») электростатических контак-
контактов внутри структуры белка. Вклад одного гидрофобного контакта
дает выигрыш энергии - 0.5 ккал/моль. а одного электростатиче-
электростатического до 4 ккал/моль. В связи с этим проведенный анализ под-
подтверждает необходимость учета этого типа взаимодействий при
расчете энергии определенных конформации белка.
3- Моделирование процесса самоорганизации t4,..
3.1. Выбор основной схемы процесса.
Ранее предложено несколько схем поэтапного расчета струк-
структуры а -спиральных белков t5.II]. Они базируются на первона-
первоначальном расчете низкоэнергетичных конформации пар соседних
«-спиралей, и их последующей взаимной укладке. На этом пути
достигнуты неплохие результаты. Так, в работе [5] по известной
схеме взаимодействия пар «-спиралей в достаточно ограниченном
конформационном пространстве изменения параметров взаимной
- 141 -
т
ориентации а-спиралей рассчитаны низкоэнергетичкые конформации
глобулярного ядра миоглобина со среднеквадратичным отклонением
от нативной~з а.
Однако, как показано в нашей работе, при взаимодействии
двух а-спиралей как правило образуется структура, имеющая об-
щую гидрофильную и гидрофобную поверхности.
В естественной для белков среде, водном окружении,
¦полярное" расположение а-спиралей должно быть энергетически
невыгодным, поскольку обширная гидрофобная поверхность оказы-
оказывается в контакте с водой. Поэтому, несмотря на то, что перво-
первоначальная автономная попарная укладка является привлекательным
подходом для теоретического расчета [5,12-14], по-видимому, она
не соответствует реальному пути процесса самоорганизации.
В работе [201 по_экспериментальному изучению процесса де-
натурации белков.показано, что на одной из начальных стадий
денатурации молекула белка проходит через состояние "расплав-
"расплавленной глобулы", характеризующееся близостью к нативной форме
по компактности и вторичной структуре. Авторы предположили,
что белок проходит через такое состояние и при сворачивании.
В данной работе с учетом этих данных предложен следующий
механизм формирования нативной структуры глобулярного белка:
1. первоначально все а-спирали, взаимодействуя гидро-
гидрофобными кластерами, образуют глобулу, в которой
экранируется большинство гидрофобных остатков;
2. на втором этапе за счет перемещений отдельных
а-спиралей на гидрофобной подложке, образуемой
остальными а-спиралями^формируется околонативная
компактная конформация белка;
3. путем локальных трансформаций эта структура пере
ходит в нативную.
3.2. Задание белковой конформации.
Поиск низкоэнергетичных конформации в нашей работе основан
на упрощенном представлении структуры белка и использовании
порогового энергетического функционала, аналогичного применя-
применяемым в других исследованиях по расчетам третичной структуры
[5,12-161.
В каждой вторичной структуре относительное расположение
- 142 -
i
составляющих ее элементов (аминокислот) строго фиксировано в
пространстве относительно друг друга. Используется упрощенное
геометрическое представление всех элементов белка.
При расчете энергии взаимодействия между разными
а-спиралями учитываются гидрофобные, электростатические и
ван-дер- ваальсовские взаимодействия.
При расчета энергии взаимодействия учитывается только по-
попарное взаимодействие между а-спиралями.
Задается начальное расположение а-спиралей в виде квази-
квазисферической глобулы, внутрь кототюй обращены гидрофобные клас-
кластеры а-спкралей.
Дальнейший: поиск низкоэнергетичных конформации осуществля-
осуществляется путем локальных перемещений этих спиралей.
На каждом этапе отбрасываются стерически запрещенные и
энергетически невыгодные конформацик.
8.3. Построение начального расположения а-спиралей.
В работе использовалось упрощенное представление геометри-
геометрического строения а-спирали: боковые группы аминокислот аппрок-
аппроксимировались сферами с центрами в Ср-атомах8 Атомы основной
цепи а-спирали представлены цилиндром радиусом 3.7ACI7I. Рас-
Рассматривались а-спирали с идеальной геометрией. Координаты
Сд-атомов находились по формулам [181:
XI - 3.17 * OOSCA«U0O«(I-i?+19))
YI - 3.17 s SINU«A00«CI-l)+19))
zi = i.5 * п-i;
где А = 3.14/18C., I - номер аминокислоты в а-спирали.
Укладка а-спиралей вокруг квазишарового ядра (то есть та-
таким образом, что длина, ширина и толщина глобулы примерно рав-
равны) является наиболее типичной для а-спиралышх глобулярных
белков.
В работе [191 доказано, что при описании белка квазишаро-
квазишаровому ядру можно сопоставить квазисферический многогранник.
Вершинами аппроксимирующего многогранника являются концы
«-спиралей, ребрами - соединяющие их участки полипептидной це-
цепи и оси а-спиралей.
В этой работе также показано, что каждому данному числу
спиралей соответствует единственный выпуклый многогранник
- 143 -
(для обеспечения контактов полярных остатков с водой) и огра-
ограниченное количество возможных расположений а-спиралей на нем.
Многогранники этого типа использовались нами для выбора нача-
начального расположения.
При выборе начального расположения для каждой а-спирали
задаются вершины многогранника, между которыми она расположе-
расположена. Однако, такое расположение обладает весьма большим числом
степеней свободы относительно поворотов спирали вокруг своей
оси. Поэтому, учитывая результаты анализа принципов встраива-
встраивания а-спиралей в глобулу, описанные выше, мы выбирали началь-
начальное расположение а-спиралей таким образом, чтобы их гидрофоб-
гидрофобные моменты были направлены к центру белка (см. рис.5).
Рис.5. Начальное расположение а-спиралей в домене 1 папаина. ¦
Для одной из них показан выбор системы отсчета и
ориентации встраивания.
Критерием для сравнения начального расположения с нативной
структурой может служить среднеквадратичное отклонение взаим-
взаимного расположения атомов в сравниваемых структурах 11511
'"- 144 -
<dy - d,
где
расстояние между атомами в рассчитанной структуре, а
l - в реальной. Ясно, что при сравнении построенного началь-
начальt
ного расположения с нативной конформацией среднеквадратичное -
отклонение должно существенно зависеть от выбора характерного '
размера многогранника - длины ребра А. Существует некоторое L'
оптимальное расположенное а-спиралей, наиболее близкое к нати- -^
вному. Ему соответствует длина ребра А • при увеличении дли- t
ны ребра спирали удаляются друг от друга и от реального рас-
расположения*, при уменьшении ребра среднеквадратичное отклонение
должно возрастать, а спирали оказываются в стерически запре- .
ценных конформациях.
В работе [19] показано, что длина ребра многогранника, ап- '-
проксимирующего белокj должна быть порядка двух диаметров '•
а-спирали. Радиус цилиндра, аппроксимирующего атомы основной .
цепи а-спиралирз.7 А. Поэтому диаметр а-спирали можно выбрать к
Ml--.'
i
Рис. В
Зависимость среднеквадратичного отклонения начального
приближения (RMS) от величины ребра многогранника.
Заштрихованная область соответствует задаваемым апри-
априорно параметрам. I) домен I папаина*, 2) цитохром С<
- 145 -
7 A, a величина ребра многогранника, следовательно, равна 14А.
Для проверки правильности такого задания длины ребра много-
многогранника построена зависимость среднеквадратичного отклонения
расположения а-спиралей от нативной конформации щм различных
длинах реберомногогранника (рис. 6J. Можно видеть, что размер
ребра4-15 А соответствует отклонению 5 А.(что является хоро-
хорошим начальным приближением для дальнейшего расчета структуры
белка]. Построенная таким образом структура не удовлетворяет
стерическим ван-дер-ваальсовсвим ограничениям и поэтому должна
подвергаться дальнейшей оптимизации.
8.4 Поиск низкоэнергетичных конформации.•
При расчете третичной структуры бэлковых молекул использо-
использовался пороговый энергетический функционал, учитывающий гидро-
гидрофобные взаимодействия, электростатические контакты между заря-
заряженными полярными и боковыми группами и ограничения на плот-
плотность упаковки аминокислот, налагаемые ван-дер-ваальсовскими
взаимодействиями. Для пары взаимодействующих а-спиралей функ-
функционал подробно описан в [51:
Е- '4
Энергия взаимодействия всего белка определялась в аддитив-
аддитивном приближении,как сумма энергий взаимодействия пар спиралей:
S = , Е. ,
21
где Bj, - энергии взаимодействия i и J а-спиралей.
При расчете третичной структуры осуществлялось задание
случайных расположений а-спиралей в заданной, области минимиза-
минимизации с помощью датчика случайных чисел. Выбиралась достаточно
большая область минимизации, охватывающая большую часть возмо-
возможных конформационных состояний данной белковой молекулы, учи-
учитывающая, что а-спирали связаны между собой небольшими участ-
участками полипептидной цепи. Параметры области минимизации, опре-
определяющие отклонение в обе стороны от начального расположения,
приведены ниже:
- 146 -
Чтение информации о вторичной структуре
Построение многогранника
Выбор начального расположения а-спирали
Сравнение начального расположения с
нативной структурой
Выбор случайного расположения а-спиралей
вблизи начального расположения в заданной
области минимизации
довлетворяются
стерические ограничения
да
, Расчет энергии полученной конформации
да
нет
Сравнение полученной конформации с
нативной по среднеквадратичному отклонению
и запись конформации
Рис. 7. Блок-схема программы расчета пространственной
структуры'а-спиральных глобулярных белков.
- 147 -
DX ж 7 Ae
DY = 10 A
DZ<i2A
WC = 1.57
СДВИГ ПО ОСИ X
СДВИГ ПО ОСИ Y
СДВИГ ПО ОСИ Z
угол вращения вокруг оси X
ограничивает угол разворота а-спирали во-
вокруг оси Z так, чтобы гидрофобный момент все время имел поло-
положительную составляющую к центру белка.
WY ¦ 3.14 угол вращения вокруг оси Y
\П = з.14 угол вращения вокруг оси Z
Блок-схема алгоритма расчета третичной структуры белковой
молекулы приведена на рис. 7. На получаемые конформаций нала-
налагались как энергетические, так и геометрические ограничения:
а-спирали не должны пересекаться (минимальное допустимое рас-
расстояние между осями а-спиралей - 3.5 А); концы а-спиралей, со-
соединенных участками полипептидной цепи, не могут расходиться
более чем на Nx2.5 A (N - число аминокислот между концами
а-спиралей).
На рис.8 приведено распределение по энергиям всех конфор-
конформаций, полученных при расчете структуры трехспирального домена
папаина. Самый левый большой столбец отражает количество кон-
конформаций, запрещенных по ограничениям, налагаемым
ван-дер-ваальсовскими взаимодействиями', остальные соответству-
соответствуют количеству конформаций с определенным значением энергетиче-
количество
89 954 ^конформаций
16
14
12
10
8
6
4
-24
Рис. 8.
-22 -20 -18 -16 -14
-12
I О
Количество низкоэнергетичных коцформаций
домена I папаина ( 3 а-спирали ).
Приведены среднеквадратичные откломоиия от
нативной структуры.
- 148 -
ского функционала. Видно, что число низкоэнергетичных конфор-
конформаций (с энергией меньше -20 ккал/моль) очень мало, 0.01 % от
всех 90 тыс. просмотренных структур.
Полученные результаты позволяют предполагать, что выбран-
выбранная схема укладки а-спиралей оказалась весьма продуктивной для
поиска малого набора низкоэнергетичных околонативных конформа-
конформаций. Распределение полученных конформаций по среднеквадратич-
среднеквадратичным отклонениям от нативной, приведенное на рис. 9, показыва-
показывает, что структуры с отклонением 6-9 А существенно удалены от
о
среднего RMS, равного 15 А.
Хотя, отобранные низкоэнергетичные конформаций содержат не-
несколько структур,не соответствующих нативной, следует учесть,
что основная проблема, возникающая при расчете белковых струк-
структур - перебор гигантского конформационного пространства всех
возможных вариантов укладки, оказывается практически решена.
По-видимому, более точный учет геометрии белковой структуры
(при восстановлении атомной структуры аминокислот) позволит
выбрать одну из нескольких полученных конформаций для последу-
шей оптимизации, которая может обеспечить более высокую
точность расчета.
Аналогичные расчеты проводились для цитохрома С D
а-спирали) и миоглобина F а-спиралей). В этих случаях также
выявлялось небольшое количество низкоэнергетичных конформаций,
среди которых были структуры с RMS, существенно отклоняющимися
от среднего. Однако эти расчеты для 90 тыс. просмотренных
структур оказались недостаточными для выявления околонативных
структур. По-видимому, с увеличением количества спиралей резко
возрастает число испытаний, требуемых для полного просмотра
конформационного пространства и выявления всех низкоэнергетич-
низкоэнергетичных конформаций.
Дальнейшее развитие этого подхода состоит в реализации
третьего этапа - перехода в нативное состояние путем локальных
трансформаций. Для этого необходимо провести тщательный анализ
полученного набора конформаций и осуществить поиск низкоэнер-
низкоэнергетичных структур вблизи каждой из них при небольшом разбросе
параметров, используя алгоритм восстановления атомной структу-
структуры белка по упрощенной геометрии. После чего возможна миними-
минимизация энергии и получение третичной структуры белка с высокой
- 149 -
точностью.
240
230
220
210
200
190
180 -
170 ¦
160
150
140
130
120
ПО
100
90
80
70
60
ьо
40
30
Z0
10
0
количество
конформаций
,—.
1—
2 4 6 f
1С
—|
_j 1 i |
12 14
—
-
i J j
16 18 2С
]
)
1
22
]
1
2
1>Лг
¦ +¦—ч-
4 28 28
Рис.9. Гистограмма количества разрешенных конформаций домена I
папаина для каждого среднеквадратичного отклонения от
нативной конформаций. Всего 2748 стерически разрешенных
конформаций.
Заключение.
Таким образом, в настоящей работе описан первый вариант
компьютерной системы, позволяющей как анализировать закономер-
закономерности строения белковой молекулы, так и проводить расчеты тре-
третичной структуры для класса а-спнралышх глобулярных белков
или а-спиральных доменов.
- 150 -
В работе частично использована идеология экспертных сис-
ем: разработана подсистема анализа закономерностей структурн-
рй организации глобулярных белков из Базы данных пространст-
пространственных структур и подсистема использования полученных знаний
формулировке модели и алгоритма расчета третичной структу-
Предполагается дальнейшее развитие системы в направлении
асширения её возможностей экспертной оценки третичной струк-
структуры заданного белка по его аминокислотной последовательности,
этой целью предполагается распространить подсистему анализа
Базы данных на все классы белков, а также дополнить алгоритми-
алгоритмический расчет структуры интерактивным режимом работы, исполь-
используя машинную графику. В таком режиме исследователь будет иметь
пополнительную возможность осуцествлять укладку модельного бе-
кового фрагмента с помощью графического редактора.
Разработанная система актуальна для теоретической и экспе-
экспериментальной работы с белковыми молекулами, особенно в связи с
развитием методов белковой инженерии при конструировании бел-
белков с новыми или измененными функциями.
Авторы выражают благодарность Н.А.Колчанову за конструк-
|тивное обсуждение результатов этой работы, которая явилась
развитием подходов к расчету структуры белковав разработке ко-
которых он принимал активное участие.
ЛИТЕРАТУРА
(П. Elsenberg D., Weiss R.M., Tervllliger Т.е. // Nature. 1982.
v. 299. p. 371-374.
[2J. Elsenberg D., Weiss R.M., TerwllllngerT.C., Vllcox V. //
FARADAY SYMP.CHEM.SOC. 1982. v. 17. p. 109-120.
?31. Kozakl Y., Tanford С // J.Blol.Chem. 1971. v. 246. p. 2211.
?41. PROTEIH DATA BAMK.- BROOKHAVEH HATIOHAL LABORATORY.
?61. Solovyov V.V., Kolchanov U.A. A // J.Theor.Blol. 1984. v.110.
P. 67-91.
|e61. Chothia C. , Levitt M. , Richardson D. // PKAS of the KSA.
1977. v. 74. P. 4130-4134.
If?]. Efinov A.V. // J. Mol. Biol. 1979. v.134. p. 23-40.
IB). LimV.I., Efinov A.V. //F5BS LETTERS. 1977. v.78. P.279-283.
-15: -
[91. Qoldanberg D., Crelghton T.//BIOPOLYMERS. 1985.v.24.p.167.
[10). Matthew J.B. //Ann.Rev.Blophys.Chem.l985.v.l4.p. 387-417.
(II I. Ptltayn O.B., Flnkelsteln A.V. //Int.J.of Quantum Che*.
1979. V. 16. P. 407-418.
[12). Levitt M. // J.Mol.Blol. 1976. v. 104. P. 59-107.
[13). Varsel A., Levitt M.//J.Mol.Blol.1976. v. 106. P. 421-437.
[14). Rashln A.A. Myoglobln as a model of protein folding.
Blomolecular structure, conformation, function and
solution.ed.R Srlnlvasan. v.2. Physico-chemical
and theoretic studies. 1981. Pergamon Press.
[15). Cohen F.B., Richmond T.J., Richards F.M. // J.Mol.Blol.
1979. v. 132. P 275-288.
[16). Yeas M., Jacobean J.W.//J.Theor.Blol.l979. v.77.P.263-305.
[17). Richmond T.J., Richards F.M.//J.Mol.Blol.1978.v.119. P.537.
[181. Волысенитейн М.Б. Биофизика.- м.: Наука, 1981.
119). Мурзин А.Г., Финкельштейн А.В. // Биофизика. 1983.т.28.
С.905-910.
[20]. Долгих Д.А., Абатуров Л.В., Бражников Е.В. // Докл.Акад.
наук СССР. 1988. т. 272. с. I48I-I484.
-152 -
SUMMARY
COMPUTES SYSTEM for INVESTIGATING
STRUCTURAL ORGANISATION Of GLOBULAR PROTEINS
2. Analysis of regularity and calculating method
of «-helix proteins tertiary structure.
V.V. Solovyov, A.K. Sallkhova, A.A. Salamov
Institute of Cytology and Genetics, Siberian Branch,
Academy of Sciences of th USSR, Novosibirsk
Protein secondary structure knowledge Is unsufflclent for
following the process of self-organization calculation of Its
tertiary structure. That Is why, a system for analysing terti-
tertiary organisation regularities using X-ray data had been creat-
1. These regularities are the base for developing a mechanism
bf self-organization and for building a tertiary structure ca-
calculating method (scheme on Flg.l).
Section 2 Is devoted to program package of analysis of
l-hellx globular protein tertiary structure from Brookheaven
pata Base and their results.
Part 2.1 is about investigation of an a-helix Insertion
Into protein globule. An idea of hydrophobic moment (HM) is
used [1]. HM values for «-helices of some proteins are shown
In Table 1. Only 16 % of a-helices have HM directed from the
kroteln center, and HM vectors are mainly directed to the cen-
center for 64 % of «-helices. As the majority of a-helices HM are
oriented to protein center, HM can be considered as a factor
^roughly" characterizing principles of Insertion for a-helix.
Packing «-helices In pairs Is the theme of 2-.2. Correla-
Correlation of lnter-axls angle и from inter-hydrophobic moment angle
for «-helices pair Is shown on Fig.2 and their histograms -
bn Fig. 3a,b. Analysis of contacting u-hellces gives that
l-helices are going to contact by their hydrophobic areas
[Table 3 black sides are hydrophobic). In 83 % cases «-helices
have contacts with the three nearest by ami no acid sequence
(r-hellces (Fig.4).
So as not all investigators take electrostatic interacti-
interactions Into account, we have checked its value B.3). Data on qu-
¦ntaty of electrostatic contacts between atoms of main and sl-
Se chains A-4 strings of Table 4) gives the value of 9 * for
{contacts of that type.
- 153 -
Section 3 is devoted to tertiary structure calculation
method which coincides to decided on the base of previous ana-
analysis and some data, as [20], mechanism or self-organisation:
1. Firstly, all a-hellces by hydrophobic Interactions form a
globular structure, hldrophoblc amino acids are hidden.
2. On the second stage by moving single a-hellces on hydropho-
hydrophobic area of others a neai—native compact protein structure
is formed.
3. At the end, this structure by local transformations reach
Its native structure.
Search on low-energy conformation Is based on simplified
structure of protein and using energy function (which Includes
Van-der-Vaals, hydrogen, hydrophobic and electrostatic Intera-
Interactions) as in other works [5, 12-16]. That's dlscribed In 3.2.
How to constract begining structure is described In 3.3.
a-hellces and parts of sequence between them lay down on the
polyhedron edge [19], so, that their hydrophobic clusters are
oriented to center (Fig.5). In [19] is shown that an edge
length in such case have to be as large as two helix diameters
(about 14 A). Checking that Information, a relation between
root mean square (RMS) from native structure and edge length
was calculated (Fig.6) for A) papaln domen 1 C a-helices)
and B) cytochrome С D a-helices). A region of a priori set-
ted parameters is underlined. Note the beginning structure has
rather small RMS from native, but is stereo-forbidden.
For calculating of tertiary structure C.4) on each step
a new localization of all a-helices is choosed randomly in
rather large conformattonal space of parameters [5] (Fig.7
with algorithm). Energy and RMS distributions for 90000
examined structures of papaln domen 1 are shown on Fig 8 and 9
consequently. One can see a small number of low-energy
conformations @.01 % of all) and among them there are
near-native.
So, the method is good enough to choose a small number of
low-energy conformations on which calculations of third stage
can be continued. The perspectives are in realisation the
third stage by taking Into account geometry of amino acids,
better functional and possibility of expert evaluations.
-154-
КОМПЬПГЕРНОЕ МОДЕЛИРОВАНИЕ ПРОЦЕССА ТРАНСЛЯЦИИ
А.Э.Кель, И.В.Ищенко, Л.ВОмельянчук
Институт цитологии и генетики СО АН СССР, г.Новосибирск
1. ВВЕДЕНИЕ
В последние годы возрастает интерес к теоретическому иссле-
исследованию процесса трансляции на основе компьютерного моделиро-
моделирования. Это обусловленно не только тем, что трансляция наряду с
репликацией и транскрипцией относится к числу фундаменталь-
фундаментальных генетических процессов, но также и требованиями генно -
инженерных исследований, направленных на разработку методов
конструирования искусственных молекулярно-генетических систем
с заданными свойствами.
Большинство работ, посвященных теоретическому исследованию
трансляции, были направлены на компьютерное моделирование про-
процесса элонгации 11-3]. При этом они не учитывали явным образом
неоднородности полинуклеотидного контекста и его влияния на
движение рибосом вдоль мРНК. Исключение составляют работы
14,5]. В работе 14] предложена компьютерная модель процесса
трансляции, учитывающая тормозящее влияние шпилек мРНК на про-
процесс элонгации. При этом предполагалось, что рибосома, подо-
подошедшая к стабильной шпильке, останавливается в этом участке до
тех пор, пока часть двойной спирали, включающая в себя очеред-
очередной транслируемый кодон, не расплавится за счет тепловых флук-
флуктуации (т.е. рассматривалось пассивное плавление-шпильки за
счет тепловых флуктуации без усиления этого процесса рибосо-
рибосомой; . В работе 15] проведен теоретический анализ влияния вто-
вторичной структуры РНК в области сопряжения цистронов на относи-
относительную эффективность их трансляции.
В рамках нашей работы внимание сосредоточено на моделирова-
моделировании процесса трансляции с детальным описанием влияния вторич-
вторичной структуры мРНК на элонгацию рибосом. Построенная имитаци-
имитационная модель позволяет оценивать эффективность процесса тран-
трансляции мРНК с учетом параметров ее вторичной структуры I энер-
энергетическая стабильность шпилек, количество комплементарных пар
в них, конхретное расположение шпилек в мРНК, время их
- 155 -
плавления и т.д.;.
2. МОДЕЛЬ ПРОЦЕССА ТРАНСЛЯЦИИ мРНК С УЧЕТОМ ВЛИЯНИЯ
ВТОРИЧНОЙ СТРУКТУРЫ
2 Л ОПИСАНИЕ ВТОРИЧНОЙ СТРУКТУРЫ мРНК
Следуя is], потенциальной вторичной структурой мРНК назовем
полный набор всех возможных совершенных спиралей (Sp), т.е.
спиралей, не содержащих нарушений комплементарности. Реально в
мРНК в каждый данный момент времени формируется лишь часть по-
потенциально возможных спиралей <Sp*>.
Состояние спирали определяется следующим набором параметров
(рис.:П: тремя структурными параметрами ( начало спирали ni ,
?0- энергия петли
L- энергия спирали
с с
с с
с с
A-U
A-U
A-U
A-U
A-U
A-U
A-U
A-U
кН1-»Д-1/«-И2
CCAUCCAACUC ACUUACC...
Рисунок 1. Параметры, характеризующие состояние шпильки
Энергия шпильки ffL= el+ gq.
конец спирали N2 , длина спирали L 1 ,¦ двумя энергетическими
I энергия ^стэкинг-взаимодействий спирали, содержащих L комп-
комплементарных пар,- энергия шпилечной петли, замыкавшей спи-
спираль gq); одним кинетическим параметром ( время нуклеации спи-
спирали т j.
тс является оценкой времени образования первой комплемен-
- 156 -
'арной пары спирали при сближении концов шпилечной петли на
участке мРНК при отсутствии стерических затруднений ^наличие
уже реализованной конкурирующей спирали или экранирование ри-
рибосомой потенциально возможного участка нуклеации;.
Теоретическое рассмотрение кинетики формирования шпилечной
петли РНК приводит к следующему соотношению, связывающему Кв-
константу скорости нуклеации и величину gq:
Кс= 1/Kexp(-GQ/RT), A)
где к - константа поворотной изомеризации вокруг единичной
связи в РНК, оцениваемая величиной порядка 10™с~г[7]. Заме-
Заметим, что тс= 1/к^.
Формирование шпильки длиной L проходит через последова-
последовательность промежуточных состояний (рис.2). Т.о. спираль можно
представить как набор микросостояний с соответствующими равно-
равновесными вероятностями реализации vr Переход между состояниями
выражается следующей кинетической схемой.-
с
с
А
А
А
А
А
А
С
С
С
и к^
и ^^
и к~
и
и
и
С
С
А
А
А
А
А
С
С
С
A-U
и
и
и
и
и
С
С
А
А
А
А
С
С
С
A-U
A-U -—>
и
и
и
и
С
С С
С С
A-U
. . . j^ A-U
A-U
A-U
A-U
A-U
Рисунок 2. Последовательность состояний
формировании шпильки
при
Можно показать, что для указанной схемы в равновесии веро-
вероятность каждого состояния выражается формулой;
Ы = А /П+А +. . .+А.
i i \ 1
- 157 -
.+А,;
L
12)
Здесь А, = expl-G/RT) и с, - энергия состояния spj, вычисляе-
вычисляемая из соотношения: С1=?1+с?о.
В нашей модели рассматривалась вторичная структура мРНК,
образованная набором шпилек, т.е. совершенных.спиралей, зам-
замкнутых шпилечными петлями. Рассматривалось равновесное состо-
состояние шпилек в соответствии с B).
2.2 МОДЕЛИРОВАНИЕ ЭЛЕМЕНТАРНЫХ АКТОВ ТРАНСЛЯЦИИ
Движение рибосомы вдоль мРНК во время белкового синтеза
рассматривается как стохастический пошаговый процесс. Каждый
шаг может быть отнесен к одному из 4 типов элементарных актов
в зависимости от положения рибосомы на мРНК и состояния ее
вторичной структуры. Это - инициация, свободная элонгация,
терминация и элонгация, сопровождающаяся плавлением шпильки
рибосомой.
Инициация, элонгация (в отсутствии тормозящего влияния вто-
вторичной структуры; и терминация описываются следувдими кинети-
кинетическими схемами.- к
in
для инициация R(O) —> rid,
к
для элонгация Rfj) —* Rtj+i),
для терминация R(N) —» R(O).
Здесь *ln- константа инициации, к - константа терминации;
R(O)-состояние свободной рибосомы, не ассоциированной с мРНК;
RIJ)-состояние, в котором рибосома транслирует У-ft кодон;
W-длина транслируемой части мРНК в кодонах.
Формула для вероятности перехода, соответствующая приведен-
приведенным выше кинетическим схемам, выглядит следующим образом.-
PU) = i-expf-kt) 13)
Здесь к - соответствующая константа перехода '*1п» *. или KJ ¦
Элонгация, сопровождающаяся плавлением шпильки рибосоме
допускает три варианта состояния кодона, который экраниров
шпилькой (щс.з). При переходе рибосомы с у-го на (j+il~& к
- 158 -
i С
I C
У 'с
I
I
f- (J+U-& кодон
' г
У-й кодон
A-U
A-U
A-U
A-U
A-U
A-U
С
С
A
...CUC
", вариант
С
С
С
п=3
САС. .
I
С С
с с
с с
A-U
A-U
A-U
г A-U
A-U п=2
L A U
. г С САС.
\\
. .cue
вариант //
с с
с с
с с
A-U
л-и
A-U
г A-U
A U п=1
I A U
. . г- С САС.
к
...сие
вариант ///
Рис.з. Возможные состояния (j+i1-го кодона в спирали, вызванные
тепловыми флуктуациями ее концов.
дон плавится л комплементарных пар. В зависимости от варианта
экранирования кодона л принимает значения от 1 до з. Этот
переход описывается следующей кинетической схемой.-
к к к
р р р
R (J) —> . . .—> RJJ) —> R(j+i)
п О
Здесь /г - константа скорости плавления рибосомой одной
комплементарной пары, & RJJ) - состояние комплекса 'рибосома-
у-й кодон" с л нуклеотидами (J+i1-го кодона, входящими в сос-
состав комплементарных пар. Уравнения, соответствующие приведен-
приведенной кинетической схеме, разрешаются относительно вероятности
перехода с у-го кодона на J+i кодон при различных значениях л
в виде функции я Ш . Время * начинает отсчитываться с момен-
момента трансляции рибосомой У-го кодона.
Вариант /. Первые три шага процесса соответствуют последо-
последовательному плавлению трех связей. Последний шаг - трансляция
уже неструктурированного кодона с константой элонгации Ке. Ве-
Вероятность перехода определяется формулой:
)-exp(-K-t)
p
- 159 -
Здесь k=(K/(Kt-KJ> ,
в=(кг-зкк +
-к )
р •
С=ККBК -К )/(К -К )г,
р • р • р •
D=K2K /IK -К ).
р • р •
Вариант //. Для трансляции (j+D-ro кодона необходимо рас-
расплавить 2 связи. Вероятность перехода определяется формулой:
J-exD(-Kt) (SI
Здесь К=1К /(К -KJ>Z.
В=КBК -К )/(К -К )*,
• р • р *
С=КК /(К -К ) .
р * р •
Вариант ///¦ Для трансляции (j+i1-го кодона необходимо рас-
расплавить одну связь• Вероятность перехода определяется формулой.-
Р lt)=l-k-exp(-K-t)+B-expl-K>t)
F)
Здесь k=K/(Kp-KJ,
В=К /(К -К ) .
• р •
Следует заметить» что зависимость элонгационного процесса
от прохождения модулирующих кодонов можно учитывать в опреде-
определении константы элонгации К^, уменьшая ее для трансляции ред-
редких кодонов.
2.3 ИМИТАЦИОННОЕ МОДЕЛИРОВАНИЕ ПРОЦЕССА ТРАНСЛЯЦИИ
Процедура моделирования
Предлагаемая модель относится к типу имитационных моделей и
описывает непосредственное движение группы рибосом вдоль мат-
матрицы с помощью пошаговой стохастической процедуры. Каждый эле-
элементарный шаг движения отдельной рибосомы задается соответст-
- 160 -
вушим распределением вероятности перехода P(t). с помощью
счетчика случайных чисел выбрасывается псевдослучайное число I
в интервале [0,1]. Затем по соответствующей функции P(t), за-
задаваемой одним из соотношений 13) - F), решается уравнение
5 = P(tl и определяется время соответствующего перехода t для
2-й рибосомы. Вычисляя t( для всех рибосом, расположенных на
мРНК и упорядочивая их по величине, получаем временной ряд
(t .), задающий очередность перехода рибосом на очередной ко-
дон Далее осуществляется выбор рибосомы с наименьшим време-
временем перехода и производится реализация последующего состоя-
состояния соответствующего варианта перехода. Как указывалось выше,
различается четыре варианта перехода; инициация, элонгация,
терминация и плавление;. Т.о. на каждом этапе расчета мы имеем
мРНК с заданным положением рибосом. В процессе расчета
запоминаются необходимые параметры движения рибосом fвремя
прохождения по мРНК, количество сошедших рибосом, количество
рибосом, одновременно находящихся на мРНЮ. После того как
количество оттранслировавшихся рибосом достигнет заданной
величины, расчеты прекращаются.
Входные данные модели
Первую группу данных образует набор <sP> шпилек вторичной
структуры исследуемой мРНК, описываемых параметрами представ-
представленными в пункте 2.1. Расчет вторичной структуры проводился
отдельной программой на основе поиска непрерывных комплемен-
комплементарных участков последовательности мРНК с выбрасыванием шпи-
шпилек, имеющих энергию меньше наперед заданного порога.
Вторая группа образуется совокупностью управлясцих парамет-
параметров, значения которых определяют динамический 'портрет"
трансляционной системы. Имеется 4 управляющих параметра: кон-
константа инициации х, , константа элонгации к , константа тер-
in в
минации Kt и константа плавления К ¦
р
Исходя из анализа литературных данных выбран следующий ин-
интервал изменения константы элонгации ю < к%< юо (с'х).
Выбор константы терминации основан на предположении, что
фаза терминации не является лимитирующей и поэтому константа
терминации должна быть, выбрана больше максимамального значения
константы элонгации: Kt> too (c~ ). Другими словами, скорость
"стока" рибосом с матрицы не должна быть меньше скорости
"скольжения" по ней рибосом.
Изменение скорости инициации меняет населенность матрицу
рибосомами и то среднее расстояние между ними, в рамках ко-
которого может происходить образование и разрушение шпилек вто-
вторичной структуры. Имеются ограниченные экспериментальные дан-
данные, согласно которым, населенность бактериальных матриц ри-
рибосомами поддерживается примерно равной 0.5 [8,91. Это значит,
что примерно 50% всей мРНК покрыто рибосомами. Предполагая,что
синтез белка происходит с максимально возможной эффективнос-
эффективностью, которая в свою очередь прямо пропорциональна населеннос-
населенности, будем выбирать такую константу инициации *1п, чтобы заве-
заведомо достигалась максимальная населенность. Проведенные расче-
расчеты показали, что максимальная населенность достигается при
константе инициации Kin> ю с. В дальнейших расчетах при
идентификации константы плавления нами было выбрано значение
к =ю с.
t п
Константа плавления является тем параметром, который ха-
характеризует степень влияния вторичной структуры на процесс
трансляции. Процедура определения этого параметра описана ни-
ниже. В качестве третьей группы параметров модели рассматрива-
рассматривались длина мРНК Lm и линейный размер рибосомы lr (т.е. коли-
количество кодонов мРНК, покрываемых одной рибосомой, принятое
равным 26 но]).
Выходные данные модели
После окончания компьютерного эксперимента происходит обра-
обработка полученных результатов и формируются выходные данные;
среднее время элонгации, средняя населенность, скорость синте-
синтеза f эффективности.
Населенность Н является характеристикой заполненности мРНК
рибосомами и выражается формулой Н = <Nr>-Lr/ L^, где <Иг> -
среднее количество рибосом на мРНК.
Эффективность трансляции ег Гили что одно и то же - ско-
скорость "схода" готовых белков с мРНК ) задается формулой
ЕГ ж <Нг>/<Те>, где <Тг> - среднее значение времени элонгации
- 162 -
усреднение времен прохождения отдельных рибосом по мРШ.
3. ИДЕНТИФИКАЦИЯ ПАРАМЕТРА ПЛАВЛЕНИЯ ШПИЛЬКИ РИБОСОМОЙ
Рассмотрим пример использования модели для оценки константы
плавления вторичной структуры мРНК рибосомой. Метод оценки
этой константы заключается в следующем. Многие прокариотичес-
кие гены имеют преимущественно полицистронную природу ^т.е. на
них синтезируются непрерывные молекулы мРНК, кодирующие два
или более белкам. Следуя гипотезе о влиянии вторичной структу-
структуры на трансляцию, естественно предположить, что цистроны с
различной вторичной структурой будут транслироваться с разной
эффективностью.
В частности, в работах [11,12] исследовалась эффективность
синтеза белка двумя сцепленными цистронами LTA и ltb в клетках
Е.СоИ. Оказалось, что мРНК LTB транслируется в s раза интен-
интенсивнее, чем мРНК lta. Прежде всего эффект влияния вторичной
структуры должен проявится в различной скорости синтеза бел-
белков, транслируемых с мРНК этих цистронов.
Для идентификации константы плавления К проведены расчеты
эффективности трансляции мРНК, кодируемых цистронами LTA и
LTB, при различных значениях константы плавления К . Рассчиты-
Рассчитывая эффективности трансляции при различных значениях этой кон-
константы для двух мРНК, мы вычисляли отношение этих эффективнос-
тей для фиксированных значений константы К . Для идентификации
константы к выбирался такой интервал ее изменения, при кото-
котором отношение эффективностей трансляции указанных мРНК прибли-
приближалось к 5, что соответствовало экспериментальным данным.
Накладывая дополнительно условие о соответствии средних
скоростей трансляции модельных мРНК и реальных, можно значи-
значительно сузить интервал возможных значений константы плавления.
Данные расчетов приведены в таблице 1.
Из таблицы 1 видно, что область максимального различия между
эффиктивностями трансляции мРНК lta и ltb принадлежит диапазо-
диапазону значений константы плавления о.01 - ю с'1. Дальнейшее уве-
увеличение константы плавления приводит к уменьшению отношения
- 163 -
Табл. l. Зависимость отношения эффективнос-
тей трансляции lta/ltb от константы плавле-
плавления, рассчитанная на основе модели при кон-
константе элонгации к =50 с'*
К (с'Л)
р
О.О1
О.О5
0.1
0.3
0.5
0.7
1.0
5.0
10.0
IS.О
эффективность
LTB
.01
.06
.1
.4
.52
.8
.87
1.8
2.15
2.25
LTA
.003
.015
.03
.09
.13
.18
.22
.75
1.3
1.6
fбелок/секУ
LT8/LTA
3.3
4.0
3.3
4.4
4.0
4.4
4.0
2.4
1.6
1 .2
тс
Ж
Ш 5
яр
т.
эффективностей- Для дальнейшего уточнения полученного резуль-
результата были проведены дополнительные расчеты скорости трансля-
трансляции мРНК L7 из г.Сол при различных значениях параметра к
(тэ.бл-2). Из экспериментальных данных известно, что средняя
скорость движения рибосомы по мРНК п равна 4 кодона/сек
На основании этого наиболее приемлемое значение константь
плавления К =о.з. Указанная величина не противоречит результа-
- 164 -
Табл. 2. Зависимость средней
скорости движения рибосомы от
константы плавления для мРНК
иг.
К (с''1)
р
О.О1
0.1
0.3
0.6
1.
3.
10.
скорость
^кодон/сек^
0.15
1.5
4.5
14.4
15.5
28.4
60.0
там предыдущих расчетов ("табл. 1).На основании этого можно счи-
считать, что наиболее реалистична средняя величина К в пределах
от 0.1 до 1 сек"'.
4. ЗАКЛЮЧЕНИЕ
Основной задачей этой части работы является создание ком-
компьютерной модели, описывающей кинетику процесса трансляции
мРНК с учетом ряда факторов, влияющих на скорость синтеза
белка. Моделирование будет исползоваться для компьютерной
оценки эффективности трансляционных систем, конструируемых ме-
методами генной инженерии. В совокупности с методами анализа
функциональных сайтов в полинуклеотидных последовательностях
^см. статью ИЗ] настоящего сборника ), указанная модель со-
I
- 165 -
ставит основу разработки экспертной системы компьютерного
конструирования систем трансляции с заданными свойствами.
ЛИТЕРАТУРА
11) MacDoneld C.T.,Glbbs J.H. 8 Pipkin A.C.//Biopollmers 1968.
v.6. p.l
12) Gordon J?.// J.Theor.Btol. 1969. v. 22. p. 515
13) HacDonald C.T.,Glbbs J.H. 8 Pipkin A.C.//Biopoltmers,1967,
V.7,p.707.
14) Hetjne в., Nilsson L. 8 Blomberg G. // J.Theor.Btol. 1977.
v.68. p.321-329
16) Лихонвай В.А., Шамин В.В-,Корженевский С.К.//Сборник тези-
тезисов /// всесоюзного совещания " Теоретические исследова-
исследования и банки данных по молекулярной биологии и генетике ".
НОВОСИбИрСК, 1988. О,-143
16) Миронов А.А., Дьяконова Л.П., Кистер А.Э.// Мол.биол.1984.
Т.18. С.1686-1693
17) Волькенштейн М.В. Биофизика. М..- Наука, 1988, с. 66
18) Palmiter R.D.// J.Biol.Chem. 1973. v. 248. p. 2095
19) Harnkao B. A. 8 Miller 0. L.//Annu .Rev .Biochem. 1973. v. 42.
p. 379-396
110) Спирин А-С Молекулярная биология. М..Высш.ок.,1986,с.136
111) Yamamoto Т., Suyama A., Mori N.,Yokota T. S Vada A.//FEBS.
1985. v.181. p. 377-380
112) Yamamoto T. 8 Yokota T.//J.Bacteriol. 1981. v.145
1131 Кель А.Э., Пономаренко М.П., Орлов Ю.Л., Мищенко Т. М.,
Колчанов Н.А. // ( настоящий сборник ), 221-242.
- 166 -
SUMMARY
THE COMPUTER MODELINO OP TRAMSLATIOM
Kel A.E., Ischenko I.W., OMlyJanchok L.W.
The work presents a computer model, describing the kinetics
of шКНА translation process regarding the peculiarities of Its
secondary structure. The secondary bBNA structure Is «ade out of
¦utually complementary areas of this molecule, forming the so
called hairpins spiral structures preventing rloosome movement
¦long mRHA.
An lmmltatlonal computer model, which allows to estimate the
effectiveness of an aRNA translation process regarding parameters
of Its secondary structure (energy stability of a hairpins, nmber
of complementary pairs In them and locallratlon of a hairpins) Is
suggested. The movement of rlbosomes along mBNA Is considered to
be a stochastic step-by-step process.
The quantitative Identification of such a model parameter as
a constant of halrplne melting by rlbosome Is carried out.
It appeared that the value of this parameter lies within
3,1-1 sec'1. The model describes the kinetics of translation
rocess In good accordance with the present experimental date and
be used for assessing the efficiency of the concrete mMA
ranelatlon process (natural as well as artificial, appearing Jn
Re-engineering construction, molecular-genetlcal systems).
- 167 -
ЭКСПЕРТНАЯ СИСТЕМА ДЛЯ ПРЕДСКАЗАНИЯ ТОПОЛОГИЧЕСКИХ СТРУКТУР
ГЛОБУЛЯРНЫХ БЕЛКОВ ПО ИХ АНИНОКИСЛОТНЬМ ПОСЛЕДОВАТЕЛЬНОСТЯМ
М.П.Поноиаренко, И.Н.Пкщдялов, Н.А.Колчанов
институт цитологии и генетики СО АН СССР, г.Новосибирск
1. Введение
Предсказание пространственной структуры белка по его ами-
аминокислотной последовательности - одна из центральных проблем
молекулярной биологии. Можно выделить [1] три основных подхода
е ее решению:
1) моделирование процесса самоорганизации молекулы белка с по-
помощью минимизации псевдоэнергетических функционалов 12, 3);
2) поэтапное предсказание уровней структурной организации бел-
белка М, 5, 6} на основе статистических методов;
3) комбинаторный перебор конформаций белковой молекулы и их
оценка на основе систем логических правил A, 7, 8).
Исследования с помощью этих подходов ведутся более 20 лет.
За это время получен ряд важных результатов (см., например об-
обзор (9)), однако окончательное решение проблемы предсказания
пространственной структуры по аминокислотной последовательности
белка до сих пор не найдено.
Представляется, что наиболее существенными факторами, пре-
препятствующими решению этой проблемы, являются сложность структу-
структурной организации глобулярных белков и недостаточная мощность
современных ЭВМ для точного расчета низкоэнергетических конфор-
конформаций белковых молекул.
В результате развития компьютерной технологии появились но-
новые, эвристические подходы к решению проблем, для которых зат-
затруднено применение традиционных алгоритмических методов. Одним
из таких подходов является технология экспертных систем НО).
Впервые использование экспертных систем для объединения тради-
традиционных методов анализа первичных структур белков (предсказание
их вторичной структуры, активных центров и т.п.) было предложе-
- 168 -
в A1, 12). Мы предлагаем другой путь применения этой техно-
технологии, состоящий в создании специализированной экспертной сис-
системы для автоматического производства, накопления и применения
хорошо формализованных знаний о взаимосвязях между первичными и
третичными структурами белков: методов распознавания особеннос-
особенностей пространственного строения белковых молекул по значенное
физико-химических и статистических характеристик их аминокисло-
аминокислотных последовательностей.
Важной характеристикой пространственного строения глобуляр-
глобулярных белков является их топологическая структура - взаимное рас-
расположение в пространстве ("справа", "слева", "сверху",
"снизу") различных локально упорядоченных участков (а-спиралей,
р-нитей и р-поворотов) A3). Она задается набором дискретных
признаков [1) (рис.1). Белки, топологические структуры которых
характеризуются одинаковыми значениями дискретных признаков,
образуют топологический класс 113]. Например, флаводоксинн.
нуклеотид-связывающие домены протеиназы К и каталаз относятся к
топологическому классу "флаводоксиноподобвых белков" (рис.1).
Настоящая работа посвящена описанию специализированной
экспертной системы для автоматического производства, накопления
и применения компьютерных методов распознавания топологических
классов белков по значениям физико-химических и статистических
характеристик их аминокислотных последовательностей. Приноси
работы системы состоит в автоматическом генерировании и последу-
последующей проверке различных гипотез о возможных взаимосвязях между
топологическими и первичными структурами белковых молекул. Дли
этой цели нами предложен оригинальный формат представления соде-
содержательных молекулярно-биологических знаний в терминах компьюте-
компьютерных методов распознавания структурно-функциональных детерминант
биополимеров по их первичным структурам. Важная особенность
предлагаемого метода автоматического производствазнаний состоит
в использовании механизма "обратной связи" между процессами по-
порождения и проверки гипотез: "новые" гипотезы генерируются не
посредством их полного перебора в рамках формата знаний, а на
основе тщательного логического анализа результатов проверки уже
рассмотренных гипотез. Таким образом, после каждого акта "выд-
"выдвижения и проверки" гипотезы происходит "самообучение" эксперт-
I
- 169 -
MKrnVSOTOBTKXMULUKeilBSOKDVmBTSOTnDILLHBDIIJ
LOCSAHeDBTLSBSBRFFIJUilSTKISOKKTALraSTavaDaKniBSfB
«aeOYOCVTVBTPLIVQIZPDKABQDCIBPOKKIAJI
l""»""l'".|.i..| n ).... I.... I ¦ ... t .... I
A •*
fi - fi
тип топологии
ЧИСЛО /j-нитвТ
ОРИЕНТАЦИИ j3-HKTER
ЗАКРУЧЕННОСГЬ ПЕРЕШНЕК МЕЖДУ хз-НИТЯМИ
ПОРЯДОК /3-НИТЕЙ В >3-ДИСТВ
ПАРАЛЛЕЛЬНАЯ
ПРАВАЯ
64312
Рис.1 Структурная организация флаводоксина из Clostridiun UP:
первичная (А), вторичная (Б), третичная (В,ПЗ)) струк-
структура. Топологическая структура представлена графически
(Г) и с помощью набора дискретных признаков (Д).
- 170 -
ной системы: она получает информацию, необходимую для определе-
определения наиболее перспективных направлений работы и исключения ту-
тупиковых, ложных путей поиска. Для оптимизации своей работы экс-
экспертная система использует два типа знаний:
(а) априорные знания (заложенные в нее разработчиками);
(б) апостериорные знания ("выведение" ею самостоятельно посред-
посредством генерации и проверки соответствующих гипотез).
Важно отметить, что именно благодаря активному использованию
экспертной системой указанных знаний ей удается успещно рещать
сложную и неалгоритмизуемую задачу производства новых (априори
неизвестных) знаний о взаимосвязях между первичными и топологи-
топологическими структурами белков, формализовать, накапливать и отоб-
отображать их в удобной и понятной для эксперта-биолога форме.
2. Формат знаний и принцип работы экспертной системы
Каждая конкретная предметная область изучает свой круг
объектов и явлений материального мира и решает возникающие при
этом содержательные задачи. Поэтому, единого для всех предмет-
вых областей формата представления знаний не существует. С
другой стороны, в конкретной предметной области есть неформали-
неформализованные знания экспертов, которые не могут быть заложены в ЭВМ
без предварительного приведения их к специфическому "машинному"
представлению в виде стандартных структур данных [141. Таким
образом, одной из центральных проблем, решаемых при создании
конкретной экспертной системы, является разработка формата ком-
компьютерного представления знаний о выбранной предметной области
40. 14, 15).
В рамках настоящей работы содержательные знания о взаимо-
взаимосвязях между топологическими структурами и аминокислотными по-
последовательностями белков представляются посредством следующей
"четверки" информационных полей:
« (Т, Т2). (Y
здесь:
- 171 -
•V ¦•
D »
<J<
- два топологических класса ^ и Т2 белковых молекул,
в различии которых но характеристикам аминокислотных
последовательностей состоит настоящее знание;
,Y_) - конкретный набор из п характеристик аминокислот-
вых последовательностей, по значениям которых мо-
можно отличить белки с топологией Т, от белков с
топологией Т2;
.- конкретный метод распознавания образов,
димо подставить значения характеристик
в который необхо-
(Y, Yn). для
того, чтобы различить белки с топологией Т1 от белков с
топологией Т„
D - текстовое описание рассматриваемого знания (кем, когда,как
и на каком материале оно получено и проверено, точность,
достоверность и т.п.).
Указанный формат представления знаний о взаимосвязях меж-
между топологическими структурами и аминокислотными последователь-
последовательностями белков обеспечивает их хранение в банках данных любого
типа (сетевых, иерархических или реляционных), так как для его
кодирования используются только стандартные структуры данных:
(l^.Tg) - два числа (коды в экспертной системе);
(Y1,...,Yn) - массив чисел (кодов в экспертной системе);
W - натуральное число (код в экспертной системе);
D - текстовый массив.
Важная особенность содержательных знаний, представленных в
терминах указанного формата - возможность их автоматического
применения с помощью специализированной программы-интерпрета-
программы-интерпретатора, способной выполнять только две элементарные операции:
- вычислять значение характеристики аминокислотной последовате-
последовательности по ее заданному коду, принятому в экспертной системе;
- подставлять вычисленное значение характеристики аминокислотной
последовательности в метод распознавания образов по его за-
заданному коду, принятому в экспертной системе.
Кроме того, предложенный формат позволяет организовать ав-
автоматический процесс порождения гипотез (т.е. еще не проверен-
проверенных знаний) о возможных взаимосвязях между топологическими
структурами и аминокислотными последовательностями белковых
- 172 -
молекул с помощью программы-генератора. которая Должна уметь
выполнять одну элементарную операцию:
- генерировать наборы кодов характеристик аминокислотных после-
последовательностей и значение кода метода распознавания образов
(принятые в экспертной системе).
Наконец еще одним, важным с практической точки зрении,
свойством предлагаемого формата является то, что сформулирован-
сформулированная на его основе гипотеза (т.е. формально заданный, во не
проверенный набор характеристик и метод распознавания образов)
может быть легко проверена на экспериментальных данных. При
этом, степень достоверности гипотезы может быть оценена колите-
ственно на основе точных статистических методов обработки резу-
результатов предсказания. Эта работа может быть осуществлена такие
тоетой программой-анализатором, способной выполнять три элеме-
арные операции:
оптимизировать (означивать) метод предсказания с помощью обу-
обучающих выборок экспериментальных данных;
- осуществлять с помощью программы-интерпретатора предсказание
топологий для контрольной выборки экспериментальных данных;
накапливать и статистически обрабатывать результаты предска-
предсказания топологий на контрольной выборке экспериментальных
данных.
многократное последовательное применение указанных программ
генератора и анализатора позволяет организовать автоматический
процесс производства знаний о взаимосвязях между топологически-
топологическими и первичными структурами белков посредством порождения соот-
соответствующих гипотез и их проверки A0,14,15) на эксперименталь-
экспериментальных данных (наборах аминокислотных последовательностей белков,
представляющих два заданных топологических класса Т., и Т2).
Кроме того, известно A0,14,15), что использование описанно-
описанного выше процесса автоматического производства знаний допускает
высокую степень его интеллектуализации посредством включения
"обратной связи" между проверкой и генерацией гипотез:
новые гипотезы порождаются на основе логического анализа резу-
результатов проверки ранее сгенерированных. При этом интеллектуа-
интеллектуальность указанного процесса достигается за счет использования
системой знаний двух типов:
- 173 -
(а) теоретических званий и практических навыков относительно
применения методов распознавания образов, анализа данных,
методов оптимального планирования экспериментов, физико-
химических и статистических свойствах аминокислотных оста-
остатков, способов их размещения в полипептидных цепях белков
и т.п. (эти знания априорно заложены разработчиками систе-
системы в программы генератор, анализатор и интерпретатор);
(О) знания о статистически достоверных взаимосвязях между ами-
аминокислотными последовательностями и топологиями рассматри-
рассматриваемых белков, предсказывающие способности проверенных на-
наборов характеристик аминокислотных последовательностей (эти
знания априорно не заложены в систему, а "выводятся" систе-
системой самостоятельно посредством генерации и проверки соотве-
тствуюаих гипотез и используются программой-генератором для
поиска наиболее перспективных направлений работы).
Таким образом, использование принципа "порождения и провер-
проверка гипотез с обратной связью" позволяет построить "машину логи-
логического вывода" новых (т.е. априори неизвестных) знаний на ос-
основе компьютерного анализа экспериментальных данных 116).
3. Описание экспертной системы
Предлагаемая в настоящей работе экспертная система предс-
представляет собой интеллектуальное программное средство, предназна-
предназначенное для автоматического производства формализованных знаний
о взаимосвязях между топологическими и первичными структурами
белков, выявляемых с помощью обучающих данных.
Входными данными экспертной системы являются аминокислот-
аминокислотные последовательности белков и их топологическая классификация.
В топологической классификации белков указывается два типа топо-
топологий (ниже они, для удобства, будут именоваться "искомой" и
"другой").
Выходные данные экспертной системы - это либо корректный
метод распознавания искомой топологии, либо вывод экспертной
системы о ее неспособности построить такой метод на основе пре-
представленных ей экспериментальных данных.
- 174 -
Метод распознавания искомой топологии включает:
(Y1,...,Yn) - набор из п характеристик аминокислотных после-
последовательностей (т.е. их кодов);
(I)
\. ЛИНЕЙНЫЙ ДИСКРИМИНАНТ ФИШЕРА A7):
п
Ф (Y.B) = Bq +У Bj-Yj . где:
IBq,...,^) - вектор коэффициентов.
\. ПРЕДСКАЗЫВАПЦЕЕ ПРАВИЛО A7]:
ЕСЛИ РАССМАТРИВАЕМЫЙ БЕЛОК имеет такие ЗНАЧЕНИЯ характери-
характеристик (Y1,...,Yn) аминокислотной последовательности,
при которых ЗНАЧЕНИЕ дискриминанта Ф (Y.B) > О,
ТО РАССМАТРИВАЕМЫЙ БЕЛОК имеет ИСКОМУЮ топологию,
,
ИНАЧЕ РАССМАТРИВАЕМЫЙ БЕЛОК имеет ДРУГУЮ топологию.
4. Архитектура экспертной системы
Функциональная схема предлагаемой экспертной системы при-
приведена на рис.2. Она состоит из трех функциональных блоков:
- БЛОК ВВОДА/ВЫВОДА ДАННЫХ;
- БЛОК ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ (гипотез);
- БЛОК СТАТИСТИЧЕСКОГО АНАЛИЗА МЕТОДОВ ПРЕДСКАЗАНИЯ (гипотез).
—I-
БЛОК
ВВОДА/ВЫВОДА
ДАННЫХ
«-6-
БЛОК
ГЕНЕРАЦИИ
МЕТОДОВ
ПРЕДСКАЗАНИЯ
(гипотез)
БЛОК
СТАТИСТИЧЕСКОГО
АНАЛИЗА
МЕТОДОВ
ПРЕДСКАЗАНИЯ
Рис.2 Функциональная схема экспертной системы.
- 175 -
•ункциональные связи между блоками показаны на рис.2 стрелками.
Связь I - ПОЛЬЗОВАТЕЛЬ вводит в экспертную систему ВХОДНЫЕ
ДАННЫЕ (топологии и первичные структуры белков);
Связи 2 и 3 - ВХОДНЫЕ ДАННЫЕ преобразуются к внутреннему предс-
представлению в экспертной системе и пересылаются в
БЛОКИ ГЕНЕРАЦИИ И СТАТИСТИЧЕСКОГО АНАЛИЗА МЕТОДОВ
ПРЕДСКАЗАНИЯ;
Связь 4 - очередной сгенерированный МЕТОД ПРЕДСКАЗАНИЯ пе-
передается в БЛОК СТАТИСТИЧЕСКОГО АНАЛИЗА МЕТОДОВ
ПРЕДСКАЗАНИЯ;
Связь 5 - результат анализа МЕТОДА ПРЕДСКАЗАНИЯ возвраща-
возвращается В БЛОК ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ;
Сшюи 6 И 7 - ПОЛЬЗОВАТЕЛЬ получает ВЫХОДНЫЕ ДАННЫЕ экспертной
системы;
Следует выделить Связь-5. Она реализует ОБРАТНУЮ СВЯЗЬ в
процессе поиска метода предсказания топологий: по ней БЛОК ГЕ-
ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ получает количественную оценку
предсказывающей способности только что сгенерированного им но-
нового метода предсказания топологий. Далее, этот блок активно
использует полученную оценку для конструирования следующих
вариантов метода предсказания: сопоставляет ее с аналогичными
оценками других, ранее сгенерированных и проверенных методов, с
помощью заложенных в него (априорных) знаний, выявляет взаимо-
взаимосвязи между наборами характеристик аминокислотных последовате-
последовательностей и предсказывающими способностями соответствующих мето-
методов и на этой основе планирует направление своей дальнейшей ра-
работы на наиболее перспективных путях поиска. Таким образом,
наличие "обратной" связи-5 (см. рис.2) между процессами генера-
генерации и проверки гипотез является принципиальной конструктивной
особенностью предлагаемой экспертной системы, делающей ее ин-
интеллектуальным программным средством.
Рассмотрим работу функциональных блоков экспертной систе-
системы более подробно.
- 176 -
5. Блок ввода/вывода данных
Главным назначении этого БЛОКА является преобразование
аминокислотных последовательностей белков (ВХОДНЫЕ ДАННЫЕ) во
внутреннее представление экспертной системы. Для каждой исполь-
используемой аминокислотной последовательности вычисляется набор из
8640 дискретных спектральных характеристик, описывающих разли-
различные способы размещения в ней остатков с определенными физико-
химическими и статистическими свойствами.
Для получения характеристик этих аминокислотных последова-
последовательностей использовались следующие 15 свойств остатков;
1) Энергия переноса из ВОДЫ в СПИРТ 118);
2) Энергия перехода из КЛУБКА в ГЛОБУЛУ A9);
3) Энергия Тэнфорда . B0);
Л) Индекс гидропатии 121];
5) Гидрофобность B2);
6) Полярность B2);
7) Громоздкость B3);
8) Координационное число [231;
9) Объем боковой группы 122);
10) Молекулярная масса I 9);
11) Расстояние до центра масс [ 9);
12) Содержание в белках E.coli (в %) I 9);
13) а-Спиральный коэффициент Чоу-Фасмана [24);
14) р-Структурный коэффициент Чоу-Фасмана [24);
15) Неструктурный коэффициент Чоу-Фасмана [24).
Различные способы размещения аминокислотных остатков в полипеп-
полипептидных цепях моделировались с помощью 18 базисов дискретных
спектральных преобразований, стандартные 125) графические изоб-
изображения которых приведены на рис.3. Более подробные описания
свойств остатков и базисов дискретных спектральных преобразова-
преобразований даны в работе 126]. Над каждой используемой аминокислотной
последовательностью производится три эвристических операции.
1) Аминокислотная последовательность РАЗБИВАЕТСЯ на 32
РАВНЫХ (с точностью до одного остатка) интервала;
- 177 -
Рис. З. Базисы дискретных спектральных преобразований, исполь-
использованные в настоящей работе.
- 178 -
X U)
F(
2) Значения свойств остатков на этих интервалах УСРЕДНЯЮТСЯ.
Получаются вектора средних значений свойств остатков на
интервалах разбиения IX Ш) (q - номер свойства остатков
от 1 до 15; 3 - номер интервала от 1 до 32);
3) Полученные векторы средних IX (J)] ПРЕОБРАЗУЮТСЯ в диск-
дискретные спектры (Y„у(в)' (? - номер базиса спектрального
преобразования от 1 до 18; в - номер базисного вектора-
строки от 1 до 32) по формуле 125]:
lYqy(s)J = [XqU)] [PVC,8))T1 B)
здесь:
q,v,s - номера свойств остатков, базисов спектральных преобра-
преобразований и базисных векторов, соответственно.
J - номера интервалов усреднения, от 1 до 32.
- вектор средних значений свойств остатков на интервглах;
) - невырожденная матрица размером 32x32: базис дискрет-
дискретного спектрального преобразования 125);
Уау(в) - дискретный спектр вектора средних IX-Ш' в базисе ве-
векторов-строк матрицы FT(s,j).
В результате применения трех указанных операций к аминоки-
аминокислотной последовательности получется 8640 дискретных спектраль-
спектральных характеристик. Они отражающих 18 различных механизмов (по
числу базисов спектральных преобразований) размещения в полипе-
полипептидных цепях белков 15 различных свойств остатков по 32 интер-
интервалам равной длины A8x15x32 = 8640). Формируя из этих характе-
характеристик различные наборы и подставляя их в методы распознавания
образов, БЛОК ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ строит гипотезы О
возможных взаимосвязях между топологическими и первичными стру-
структурами белков.
Кроме описанной выше функции, БЛОК ВВОДА/ВЫВОДА ДАННЫХ вы-
выполняет еще две:
- вводит в экспертную систему топологическую классификацию исс-
исследуемых аминокислотных последовательностей белков, преобра-
преобразует ее во внутренне "машинное" представление и пересылает в
БЛОК СТАТИСТИЧЕСКОГО АНАЛИЗА МЕТОДОВ;
- выводит ПОЛЬЗОВАТЕЛЮ выходные данные экспертной системы.
- 179 -
6. Блок генерации методов предсказания (гипотез)
Главным назначением этого БЛОКА является "генерация" разли-
¦»_х гипотез о возможных взаимосвязях между топологическими и
первичными структурами белков. Каждая такая гипотеза включает:
а) набор характеристик аминокислотных последовательностей (т.е.
список их кодов, принятых в экспертной системе);
в) метод распознавания образов (его код в экспертной системе),
в который необходимо подставить эти характеристики для того,
чтобы различить белки двух топологических классов, предложе-
предложенных системе в качестве входных данных.
Таким образом, формат представления гипотез БЛОКОМ совпадает
с форматом представления знаний (си. раздел 2). Отличие же ги
потезы от знания состоит в том, что она еще не проверена с по-
шащью реальных экспериментальных данных (т.е. гипотеза - пра-
вильчая, с точки зрения формата знаний, последовательность ко-
кодов экспертной системы). Именно такие абстрактные "гипотезы"
и генерирует данный БЛОК.
Важной особенность БЛОКА ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ
является то, что в процессе формулировки гипотез он не делает
полного их перебора (это невозможно, так как общее число вычи-
вычисляемых ею характеристик равно 8640, а в наборы для методов
предсказания входит, как правило, от I до 10). Нами заложено
в БЛОК 27 эвристических и логических правил, с помощью которых
планируется направление поиска статистически достоверных зако-
закономерностей. Каждое из этих правил реализовано в виде процедур
на языке FORTRAN-IV. Рассмотрим некоторые из них.
Один из вопросов, решаемым БЛОКОМ в каждом акте генерации
гипотез: "сколько характеристик должно быть в генерируемом
наборе ?". На разных этапах работы экспертная система исполь-
использует для решения этого вопроса различные правила. Так например,
ПРАВИЛО-20 устанавливает максимальное число характеристик в ме-
методах предсказание топологий:
- 180 -
ПРАВИЛО 20
Установить максимальное число характеристик в метода
предсказания топологий: „ ,
N = -1=2- - 1,
здесь: 3
N - МАКСИМАЛЬНОЕ число характеристик в методе предсказания;
К - число аминокислотных последовательностей, введенных в
экспертную систему в качестве ВХОДНЫХ ДАННЫХ.
Это правило учитывает закон дихотомий [37] и отражает очевж-
лый факт, что невозможно статистически достоверно описать вебо-
ьшое число объектов с помощью большого числа параметров.
Другая проблема, решаемая БЛОКОМ также в каждом акте генерв-
гш гипотез - "в каком порядке рассматривать характеристики (вя
х наборы) при генерации новой гипотезы ?". Этот вопрос решает-
к на всех этапах работы экспертной системы с помощью ПРАВИЛА-4:
ПРАВИЛО 4
При генерации методов предсказания (гипотез) характе-
характеристики (или их наборы) рассматривать в порядке УБЫВАНИЯ
количественных значений оценок их предсказывающих спо-
способностей, вычисляемых БЛОКОМ СТАТИСТИЧЕСКОГО АНАЛИЗА МЕ-
МЕТОДОВ ПРЕДСКАЗАНИЯ с помощью ПРАВИЛА-40 (см. раздел 7).
Это правило является выражением принципа "нетерпеливости"
из теории принятия решений 1281: сначала рассматривается все
амое лучшее, потом - то, что несколько хуже.
Важной особенностью работы БЛОКА ГЕНЕРАЦИИ является так-
также то, что он не "конценрирует" свое внимание на совершенство-
совершенствовании какого-либо одного метода предсказания. Напротив, поиск
статистически достоверных закономерностей ведется параллельно
в десятках направлений. При этом БЛОК ГЕНЕРАЦИИ работает следу-
следующим образом: (а) с помощью определенного планирующего ПРАВИЛА
формируются различные подмножества характеристик аминокислотных
последовательностей (например, подмножества характеристик, вы-
вычисляемых с помощью одинаковых физико-химических свойств остат-
- 181-
ков); (б) затем на сформированных подмножествах характеристик
строятся методы распознавания топологий, дающие достоверные ре-
результаты предсказания; (в) все характеристики, не вошедшие
в построенные методы предсказания, исключаются из дальнейшего
рассмотрения; (г) выбирается новое планирующее ПРАВИЛО для раз-
разбиения еще не исключенных из рассмотрения характеристик на но-
новую систему подмножеств и процесс поиска повторяется. Использо-
Использование такого механизма работы позволяет БЛОКУ ГЕНЕРАЦИИ избе-
избегать тупиковых направлений поиска. Рассмотрим, в качестве при-
примера одно из планирующих правил БЛОКА ГЕНЕРАЦИИ:
ПРАВИЛО I
Сформировать 8640 подмножеств характеристик: по
одной характеристике в каждом подмножестве. На каждом
из этих подмножествах построить по одному пороговому
методу предсказания топологий. С помощью ПРАВИЛА-40
вычислить предсказывающие способности построенных ме-
методов. Использовать их в качестве оценок индивидуаль-
индивидуальных предсказывающих способностей характеристик амино-
аминокислотных последовательностей.
Это правило обеспечивает выполнение принципа "максимальной
объективности" 110]: никакая характеристика не может быть иск-
исключена из рассмотрения без каких-либо на то оснований.
Всего в БЛОК ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ (гипотез) заложе-
заложено 27 таких правил, которые объединены в пять стратегий поиска
статистически достоверных взаимосвязей между топологическими
и первичными структурами белков. Эти стратегии применяются БЛО-
БЛОКОМ последовательно одна за другой. Ипользование этой системы
эвристических правил обеспечивает интеллектуальность функиони-
рования БЛОКА ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКАЗАНИЯ (гипотез).
7. Блок статистического анализа уеуодов предсказания
Блок предназначен для статистического анализа методов предс-
предсказания топологий белков по их аминокислотным последователыю-
- 182 -
:тям. Технология анализа методов предсказания аналогична эмпи-
яческиы логическим исчислениям CUHA-методов 129} ИСКУССТВЕН-
ИСКУССТВЕННОГО ИНТЕЛЛЕКТА. Принцип работы БЛОКА состоит в циклической
гроверке статистических свойств результатов работы методов пре-
чсказания с помощью реальных экспериментальных данных.
Используемые экспериментальные данные (первичные структуры
елков с известной топологической классификацией) случайным об~
азом разбиваются на независимые непересекающиеся) ОБУЧАЮЩУЮ в
КОНТРОЛЬНУЮ выборки равного объема. С помощью обучающей выборка
роизводится оптимизация свободных параметров исследуемого метс-
а предсказания. С помощью контрольной выборки - оценка его ра-
оты на независимых от обучения данных.
Для оптимизации работы БЛОКА в него также заложены эмпири-
еские правила, с помощью которых он быстро и объективно оцени-
ает предсказывающие способности исследуемого метода (гипоте-
(гипотезы). Рассмотрим в качестве примеров некоторые их них.
ПРАВИЛО 35
Оценить с помощью критерия Фишера для линейного диск-
дискриминанта A71 информативность набора характеристик из
рассматриваемого метода предсказания.
Это правило реализует использование пошагового дискриминан-
тного анализа A7) в работе экспертной системы.
Оценить
Пусть:
Na - число
Njj - число
Nc - число
Njj - число
ПРАВИЛО 38
адекватность результатов
ПРАВИЛЬНЫХ предсказаний
ОШИБОЧНЫХ предсказаний
ОШИБОЧНЫХ предсказаний
ПРАВИЛЬНЫХ предсказаний
ЕСЛИ: NQ + Nc > Na
предсказания топологий:
для ИСКОМЫХ
для ИСКОМЫХ
для ДРУГИХ
ДЛЯ ДРУГИХ
+ % + «с +
TO предсказание АДЕКВАТНО;
ИНАЧЕ предсказание НЕАДЕКВАТНО.
топологий;
топологий;
топологий;
топологий.
Это правило реализует анализ "а-импликативности" [29] рас-
рассматриваемых методов предсказания топологий.
- 183 -
ПРАВИЛО 40
Оценить достоверность результатов предсказания топо-
топологий на контрольных данных:
Пусть:
Na - число ПРАВИЛЬНЫХ предсказаний для ИСКОМЫХ топологий;
Nb - число ОШБОЧНЫХ предсказаний для ИСКОМЫХ топологий;
Nc - число ОШШОЧНЫХ предсказаний для ДРУГИХ топологий;
Ng - число ПРАВИЛЬНЫХ предсказаний для ДРУГИХ топологий.
Тогда
Вычислить количестенную оценку предсказывающей спо-
способности метода предсказания, по формуле:
R = - t In A),
здесь:
1 - значение точного критерия Фишера-Ирвина [30].
Это правило реализует анализ рассматриваемых методов предс-
предсказания топологий с точки зрения точных статистических критери-
критериев оценки соответствия результатов предсказания топологий реа-
реальным экспериментальным данным [30). Кроме того, в качестве ко-
количественной оценки предсказывающей способности метода исполь-
используется информационная интерпретация [31} вероятности 1.
ПРАВИЛО 42
ЕСЛИ во всех испытаниях метод предсказания топологий
давал АДЕКВАТНЫЕ и ДОСТОВЕРНЫЕ результаты,
был СБАЛАНСИРОВАН и ИНФОРМАТИВЕН,
ТО его использование статистически ОБОСНОВАНО;
ИНАЧЕ его использование статистически НЕ ОБОСНОВАНО.
Это правило подводит итог статистического анализа метода
предсказания топологий (гипотезы). Оно отражает эмпирический
опыт разработчиков системы: "для объективной оценки предсказы-
предсказывающей способности метода необходимо его проверять на различных
комбинациях обучапцих и контрольных данных, исследуя при этом
- 184 -
азличные особенности получаемых результатов". Полученный с
этого правила результат статистического анализа мето-
предсказания возвращается в БЛОК ГЕНЕРАЦИИ МЕТОДОВ ПРЕДСКА-
(гипотез). На этом работа БЛОКА заканчивается и начинает-
новый цикл "генерации и проверю'" методов предсказания топо-
(гипотез).
Всего в БЛОК заложено 15 различных эвристических правил,
пользование которых позволяет проанализировать широкий спектр
гатистических закономерностей работы методов предсказания то-
яогий белков по их аминокислотным последовательностям. Это
озволяет быстро и эффективно исключать из рассмотрения беспер-
пективные направления поиска статистически достоверных взаимо-
ей между топологическими и первичными структурами белков.
8. Реализация экспертной системы
Описанная выше экспертная система для автомаческого произ-
производства новых знаний "о взаимосвязях между первичными и тополо-
топологическими структурами белков" реализована в лаборатории теоре-
гической молекулярной генетики ИЦиГ СО АН СССР [26).
Экспертная система выполнена на языке FORTRAN-IV для мини-
СМ-4. Она состоит из двух программ:
SPEKTR - БЛОК ВВОДА/ВЫВОДА ДАННЫХ;
POISK - БЛОКИ ГЕНЕРАЦИИ И СТАТИСТИЧЕСКОГО АНАЛИЗА
МЕТОДОВ ПРЕДСКАЗАНИЯ (гипотез).
Программа SPEKTR осуществляет преобразование одной аминокис-
последовательности в численные значения 8640 дискретных
Вентральных характеристик не более, чем за 10 минут.
Программа POISK строит один корректный метод предсказания
пологий глобулярных белков по их аминокислотным последовате-
гтям (или делает вывод о том, что она не может этого сде-
гь) не более, чем за одни сутки.
- 185 -
9. Применение экспертной системы для предсказания
топологических структур белков
С помощью описанной выше экспертной системы нами были исс-
исследованы 55 аминокислотных последовательностей а/р-доменов гло-
глобулярных белков пяти топологических классов. Описание этих пос-
последовательностей дано в таблице I. Графическое изображение их
топологических структур приведено на рис.4.
С помощью указанных данных (первичные и топологические стру-
структуры белков) и описанной выше экспертной системы нами были
исследованы пять типов топологий а/р-доменов глобулярных бел-
белков. Для каждой из пяти указаных топологий искались такие
особенности строения их аминокислотных последовательностей, ко-
которые отличают их от других типов топологий а/р-доменов. Соот-
Соответственно, для каждого типа топологий были сформированы обуча-
обучающие данные для работы экспертной системы и на их основе постро-
построены пять методов распознавания этих топологий (по одному методу
для каждого исследуемого топологического класса).
' В табл.2 приведен один из построенных экспертной системой
методов, ориентированный на распознавание "аминоацил-тРНК-снн-
тетазоподобных" топологий (рис. 4в). Этот метод содержит пять
характеристик аминокислотных последовательностей, значения ко-
которых можно вычислить по формуле B) с помощью информации, со-
соответствующей значениям индексов "q", "v" и "в" из табл.2. В
последней колонке табл.2 даны значения коэффициентов "В^" для
линейного дискриминанта Фишера (I), для которого выше приведе-
приведено предсказывающее правило. Для четырех других типов исследуе-
исследуемых топологий с помощью экспертной системы построены аналогич-
аналогичные методы распознавания.
Описание результатов, полученных на независимых от обучения
контрольных данных с помощью пяти найденных экспертной системой
методов предсказания топологий, приведено в табл.3. Всего осу-
осуществлено 88 предсказаний способов упаковки аминокислотных пос-
последовательностей в топологии а/р-доменов пяти типов. В 62 из
НИХ были ПРАВИЛЬНЫЕ результаты G0%); в 26 - ОШИБОЧНЫЕ C0%).
Достоверность полученных результатов по точному критерию Фише-
Фишера-Ирвина 130] составила более 99% (табл.3, строка "ВСЕГО").
- 106 -
FXN
ADK
Ш
Рис,4 Топологические структуры а/р-доменов пяти типов,
использованных в качестве ИСХОДНЫХ ДАННЫХ для работы
экспертной системы (графическое представление [13]).
ОБОЗНАЧЕНИЯ:
FXN - флаводоксиноподобная топология (А);
ADK - аденилэт киназоподобная топология (Б);
TS - аминоацил-тРНК-синтетазоподобная топология (В);
RHO - роданезоподобная топология (Г);
DH - дегидрогеназоподобная топология (Д);
окружность - а-спираль; треугольник - р-нить;
стрелка - направление хода полипептидной цепи.
- 187 -
Таблица i
Описание выборки аминокислотных последовательностей
известными топологическими структурами а/р-доменов, котор
использовалась в качестве ИСХОДНЫХ ДАННЫХ для работы настоящее
экспертной системы.
тип
топо-
топологии
FXN
АПК
TS
RHO
Ш
ВСЕГО
ЧИСЛО
аминокислотных
последовательностей
»1
8
22
6
3
16
55
N2
6
12
4
2
9
33
*3
2
10
2
1
7
22
источник
данных о
топологии
N4
6
3
2
3
4
18
N5
2
19
4
0
12
47
ОБОЗНАЧЕНИЯ:
N1 - общее число аминокислотных последовательностей;
Ng - число последовательностей в ОБУЧАЮЩЕЙ выборке экспертной
системы;
N3 - число последовательностей в НЕЗАВИСИМОЙ выборке при испы
тании методов предсказания, построенных экспертной систе
ной.
Нд - число последовательностей с известной пространственной
структурой [321;
Ng - число последовательностей, привлеченных по гомологии.
- 188 -
Таблица 2.
Метод предсказания аминоацил-тРНК-синтетазо подобных
пологий по аминокислотным последовательностям белков,
внный экспертной системой (пояснения в тексте).
1
I
1
¦
.
0
1
2
3
4
1 5
СВОЙСТВО
аминокислотных
остатков
(qi)
-
молекулярная
масса [ 13
молекулярная
масса [11
молекулярная
масса 11)
молекулярная
масса [11
объем боковой
группы 114]
номер
базиса
дискретных
спектральных
преобразований
(см. рис.4 )
-
рис.4(з)
рис.4(а)
рис.4(а)
рис.4(а)
рис.4(з)
номер
компоненты
дискретного
спектра
(•)
-
21
23
30
13
21
коэффициент
в ЛИНЕЙНОМ
ДИСКНаИНАНТЕ
ФИШЕРА
(формула (I))
11.9
-15.0
41.8
44.3
• 80.3
-36.0
(*) - номера компонент указаны в соответствии с рис.3. Нумера-
Нумерация базисных векторов осуществляется "сверху в низ" от I
(самый верхний на рисунке) до 32 (самый нижний).
- 189 -
Таблица 3.
Тестирование методов предсказания топологий по аминокис-
аминокислотным последовательностям белков, построенных экспертной
системой, с помощью НЕЗАВИСИМЫХ данных.
тип
топологической
структуры
(ИСКОМЫЙ)
FXN
ADK
TS
RHO
DH
ВСЕГО
НЕЗАВИСИМЫЕ
данные
L1
2
10
2
1
7
22
h
15
6
15
15
15
66
h
17
16
17
16
22
88
предсказание для НЕЗАВИСИМА
данных
2
6
1
1
5
15
h
0
4
1
0
2
7
L6
7
1
4
5
2
19
h
8
5
11
10
13
47
Ч
58
68
70
68
81
70
74
88
48
62
99
99
ОБОЗНАЧЕНИЯ:
L1 - число последовательностей с ИСКОМОЙ топологией;
1<2 - число последовательностей с ДРУГОЙ топологией;
Lg - ОБЩЕЕ число аминокислотных последовательностей в
НЕЗАВИСИМЫХ ДАННЫХ;
1>д - число ПРАВИЛЬНО предсказанных ИСКОМЫХ топологий;
L5 - число ОШИБОЧНО предсказанных ДРУГИХ топологий;
Lg - число ОШИБОЧНО предсказанных ИСКОМЫХ топологий;
I«f - число ПРАВИЛЬНО предсказанных ДРУГИХ топологий;
Lg - суммарная ДОЛЯ правильных предсказаний;
Lg - ДОСТОВЕРНОСТЬ по точному критерию Фишера-Ирвина [30].
- 190 -
Таким образом, можно сделать вывод, что предложенная в
настоящей работе экспертная система действительно способна в
автоматическом режиме строить на основе обучающих данных новые
методы распознавания топологий по аминокислотным последователь-
последовательностям белков (точность предсказания 70*, достоверность 99*).
10. Заключение
Важно отметить, что производительность экспертной системы
составляет I новый метод распознавания топологий в сутки, что
в сотни раз выше, чем у эксперта-Оиолога, использующего тради-
традиционные методы эвристического поиска. Основой такой высокой
эффективности экспертной системы являются заложенные в нее
экспертные правила (знания) о способах генерации и проверки
методов предсказания топологий (гипотез о возможных взаимосвя-
взаимосвязях между топологическими и первичными структурами белков).
В настоящее время в систему заложено 42 таких экспертных прави-
правила. Именно они концентрируют внимание экспертной системы на
наиболее перспективных направлениях поиска и позволяют исклю-
исключать из рассмотрения ложные, тупиковые пути.
Эффективное использование экспертной системой заложенных в
нее эмпирических и теоретических знаний, позволяющих ей успешно
решать сложные и неалгоритмизуемые задачи, наглядно свидетельс-
свидетельствуют о ее компетентности и интеллектуальности.
Дальнейшее развитие предлагаемой экспертной системы будет
вестись в следующих направлениях:
- совершенствование априорных теоретических и эмпирических зна-
знаний, заложенных в экспертную систему, посредством уточнения
"старых" и добавления "новых" логических правил;
- расширение компетентности экспертной системы на "новые" клас-
классы содержательных молекулярно-биологических задач (в частнос-
частности, распознавание других типов доменов глобулярных белков);
- интеграция системы с Банками Данных и Пакетами прикладных
программ по молекулярной биологии и генетике.
Представляется, что развитие базы знаний экспертной системы,
расширение области ее компетентности на широкий крут биологичес-
- 191-
ких задач сделают ее высокоэффективным интеллектуальным програм-
нш средством анализа первичных структур биополимеров.
лихдгАТУРД
t 1] Cohen F.E., Steinberg M.J.E., Taylor W.R. // J.Mol.Biol.,
1982, v.156, 821-862.
С 2] Isogai Y., Neroethy G., Scheraga H.A. // PNAS USA, 1977,
V.74, « 2, 414-418.
I 3) Levitt И. // J.Mol.Biol., 1983, v.170, 729-764.
I 4) Chou F., Pasman G. // J.Mol.Biol., 1977, v.115, 135-175.
I 51 Лим В.И. и др. // Молекулярная биология, 1978, т.12, А.1,
206-232.
[ 6) Solovyov f.f., Kolchanoy N.A. // J.Theor.Biol, 1984, у.110,
67-91.
I 7] Галактионов С.Г., Родионов М.А. // Биофизика, 1980, т.25,
385-392.
[ 81 Busetta В., Barrens Y. // Biochem. Biophys. Acta, 1983»
v.709, 73-83.
[ 91 Шульц Г., Ширмер Р. Принципы структурной организации
белков, М.: Мир, 1982.
1101 Попов Э.В. Экспертные системы, М.: Наука, 1987.
111) Haiech J., Sallaotin J. // BilchUnie, 1985, у.67, 555-560,
112) Fiehleigh R.V., Robson В., Garnier J., Finn P. // FEBS
Letters, 1987, vr214, 219-225.
113) Richardson J.S. // Advances in protein chemistry, 1981,
v.34, 167-339,
114] Поспелов Г.С, Предисловие к русскому изданию, // (В кн.
Минский М. Фреймы для представления знаний. М.: Энергия,
1979, 3-5).
[151 Хейес-Рот Ф., Уотерман Д., Ленат д. Построение экспертных
систем. М.: Мир, 1987.
116) Уотерман Д. Руководство по экспертным системам. М.: Мир,
1989.
117) Волч Б., Хуань К.Дж. Многомерные статистические методы
для экономики. М.: Статистика, 1979.
Nozakl Y., Tanlord C.J. // J.Biol.Chem., 1971, v. 246,
2211-2217.
|{191 Rose G.D. et al. // Science, 1985, v.229, 834-838.
1B0] Tanlord C.J. // J.Amer.Chem.Soc, 1962, v.84, 4240-4247.
|f211 Kute J., Doolittle R.F. // J.Mol.Biol., 1982, v. 84,
105-132.
|122] Bogardt R.A. et al. // J.Mol.Evol., 1980, V. 15, 197-218.
fl23) Prabhakaran M., Ponnuswamy P.K. // J.Theor.Biol., 1979,
V.80, 485-504.
j[243 Chou P.Y., Fasman G.D. // Biochemistry, 1974, v.13,
211-222.
125) Elliot D.E., Rao K.R. // Fast transforms: Algorihtme,
Analyses, Applications. ACADBIIC PRESS, New York, 1982.
B63 Экспертная система для предсказания топологических струк-
структур доменов глобулярных белков. Новосибирск: ИЦиГ СО АН
СССР. 1989. (препринт), части 1,2,3. 165 с.
B73 Дуда Р., Харт П. Распознавание образов и анализ сцен. М.:
Мир, 1976.
[283 Фишберн П. Теория полезности для принятия решений. М.:
Наука, 1978.
1293 Гаек П., Гавранек Т. Автоматическое образование гипотез:
математические основы общей теории. М.: Наука, 1984.
[303 Леман Э. Проверка статистических гипотез. М.: Наука,
1979.
[313 Алгоритмы восстановления зависимостей, (ред. Вапник В.Н.),
М.: Наука, 1984, 124.
[323 BROOKHAVEN PROTEIN DATA BANK. Brookhaven National Labora-
Laboratory, USA, 1985.
- 192 -
- 193 -
SUMMARY
EXPERT SYSTEM FOR PREDICTING TOPOLOGICAL STRUCTURE
OF GLOBULAR PROTEIN ON THE BASIS OF AMINO ACID SEQUENCE
Ponomarenko II.P., Shindyalov I.N., Kolchanov N.A.
Institute of Cytology fc Genetics, Siberian Branch,
the USSR Academy of Sciences, Novosibirsk, USSR, 630090
The present paper deals with a description of a specialized
expert system for automatic production, accumulation and appli-
application of computer methods for recognizing protein topological
classes on the baeis of physical and chemical properties and
statistical characteristics of their amlno acid sequences. Des-
Description of protein topological structure 1в presented in fig.1.
Principle of system functioning consists in automatic gene-
generating and further test of various hypotheses on interdepen-
interdependence between topological and primary structures of protein
molecules. An original format was suggested here for presenting
knowledge in terms of computer methods for recognition of stru-
structural and functional determinants of biopolymere by their pri-
primary structures (sect.2).
An Important feature of the proposed method for automatic
production of knowledge consists in using a mechanism of "feed-
"feedback" between generation processes and testing the hypotheses.
New hypotheses are generated not by their complete sampling
within the knowledge format but using a logical analysis of
various hypotheses already tested. So, each act or
"generating and testing" a hypothesis is followed by "self-
teaching" of expert system: it receive» information needed for
determining the most perspective direction of work and exclu-
excluding deadlock, false ways of searching. To optimize its work
the two types of knowledge are used in expert system:
- 194 -
a) a priori knowledge (put into it by the elaborators) on theo-
theoretical methods and practical experience in application of
pattern recognition methods, data analysis, method of optimal
planning of experiments, physical, chemical and statistical
properties of amlno acid residues, description of their loca-
location in protein polypeptide chains;
b) knowledge on statistically significant interrelations betwe-
between amlno acid sequences and topologies of proteins and
predicting abilities of the tested characteristics of amlno
acid sequences.
Thus, using the principle of "generating and testing the
hypotheses with feedback" makes it possible to design "a dlvice
of logical deduction" of new (i.e. unknown a priori) knowledge
on the relationship between the topological structures of prote-
proteins and their amlno acid sequences.
The output of expert system is:
1) particular method for recognition of a definite topological
structures of proteins.
OR
2) conclusion of expert system on its inability to devise
such method based on experimental data presented.
Method of recognition produced by the expert system (sect.3)
is Fisher's linear discriminant A) with the known coefficients
Bo, B1, ..., Вц determined during the expert system running.
The linear discriminant contains a set of parameters Y1,..., Yn,
presenting the characteristics of amlno acid sequences, whose
form was also selected during the expert system functioning.
Section Л presents a description of expert system architec-
architecture with short outline of its main program units and functio-
functional connections between them.
Section 5 deals with characteristics of input data of ex-
expert system (amino acid sequences, topological structures of
proteins, physical and chemical properties of amino acid residu-
residues, bases of discrete spectral transformations, used for arran-
arrangement of continuous curves for variations of physical and che-
chemical properties along amino acid sequences in discrete spect-
spectrum Y (s) of vectors of the mean XQC) in basis of vector-
4 - 195 - Ч
lines of matrix ?v(s,J)).
Section 6 presents the program unit of generating methods
for predicting topological structures of proteins by their am:
no acid sequences. Examples of solving rules used by expert
system in its work are given.
Section 7 gives description of the program unit for statis-
statistical analysis of methods for predicting topologies. Examples
of the rules characterizing the methods are discussed in terms
of adequacy, balance, accurate predictions of protein topologi-
topological structures.
In section 8 we present a short description of conmon chara-
characteristics for expert system.
Section 9 provides the expert aystem application to elabo-
elaborating methods for predicting topological structures of a/0-
domalns of globular proteins according to their amlno acid se-
sequences.
The expert system has been shown to be able to devise pre-
predicting methods showing for control sample (five classes of
protein topological structures) the 70% accuracy, with signifi-
significance p < 0.01.
ЭКСПЕРТНАЯ СИСТЕМА БЫСТРОЙ ОЦЕНКИ ПОЛЕЗНОСТИ ИСПОЛЬЗОВАНИЯ
ПРОИЗВОЛЬНЫХ ХАРАКТЕРИСТИК БИОПОЛИМЕРОВ ДЛЯ ИХ КЛАССИФИКАЦИИ
М.П.Пономаренко, Б.Л.Орлов
Институт цитологии и генетики СО АН СССР, г.Новосибирск
I. Введение
Характерной особенностью современной молекулярной биологии
а генетики является интенсивный процесс их компьютеризации. Од-
Одним из существенных факторов, способствующих широкому внедрению
ЗЕМ в практику научных и технологических исследований по биоло-
биологии, стало накопление здесь огромных массивов экспериментальных
данных: расшифрованных фрагментов геномов различных организмов,
молекул РНК и белков. Объем указанной информации в настоящее
время достигает нескольких десятков мегабайт и удваивается пра-
практически ежегодно. Таким образом, уже в самое ближайшее время
для первичной обработки расшифрованных молекулярно-генетических
данных (накопление, верификация, хранение, сравнительный анализ,
систематизация, распознавание генов и функциональных сайтов)
потребуются специализированные интеллектуальные компьютерные
системы, способные осуществлять значительную часть этой обрабо-
обработки с минимальным участием экспертов-биологов, или даже в авто-
автоматическом режиме. Это обстоятельство стимулирует поиск новых
высокоточных информационно-компьютерных технологий автоматичес-
автоматического анализа первичных структур биополимеров.
Важной особенностью молекулярной биологии и генетики являет-
является разнообразие изучаемых объектов и явлений, гигантские объемы
неформализованных эмпирических знаний, которые не могут быть
заложены в ЭВМ без предварительного приведения их к специфичес-
специфическому, "машинному" представлению в формате стандартных структур
данных и/или в виде процедур анализа данных til. Таким образом,
одной из центральных проблем компьютеризации молекулярной биоло-
биологии и генетики является формализация накопленных здесь эмпириче-
эмпирических знаний с целью их последующего автоматического использова-
использования с помощью ЭВМ для решения широкого круга практических задач.
- 196 -
- 197 -
Одним из способов формализации эмпирических знаний является
выражение ах в терминах количественных соотношения между разли-
различными характеристиками объектов и явлений. Поэтому возникает
проблема выявления таких характеристик исследуемых объектов и
явлений, между значениями которых наблвдавтся статистически
достоверные количественные взаимосвязи. Учитывая высокую слож-
сложность и огромное разнообразие объектов и явлений, изучаемых
молекулярной биологией и генетикой (первичные, вторичные, топо-
топологические и пространственные структуры белков, полинуклеотид-
вые последовательности ДНК и РВК, регуляторные и функциональ-
функциональные сайты биополимеров и т.п.), можно предположить, что опре-
определяющую роль в формализации эмпирических знаний может сыграть
возможность автоматического поиска указанных высокаинформатив-
ннх количественных характеристик.
Настоящая работа посвящена описанию экспертной системы для
быстрой автоматической оценки полезности использования задан-
заданных характеристик для классификации биологических макромолекул,
пряпущ работы экспертной системы состоит в детальном анализе
широкого спектра статистических свойств каждой рассматриваемой
характеристики. Отличительной конструктивной особенностью сис-
системы является широкое использование нечетких эмпирических исчи-
исчислений в рамках теории аддитивной полезности Сэвнджа [21. Имен-
Именно это обстоятельство позволяет экспертной системе использовать
в процессе рассмотрения каждой предложенной ей характеристики
теоретические и эвристические знания следующих типов:
а) априорные знания по теории распознавания образов и матема-
математической статистике (заложенные в нее в виде библиотеки
подпрограмм на языке PORTRAN-77 для ПЭВМ IBM PC);
б) апостериорные знания о статистических свойствах рассматрива-
рассматриваемой характеристики, приобретаемые системой в процессе рабо-
работы и выраженные в количественной форме аддитивной полезности.
Важно отметить, что благодаря активному использованию указанных
знаний экспертная система успешно решает сложную и неалгоритми-
зируемую задачу быстрой оценки полезности использования задан-
заданных характеристик биополимеров для их классификации.
- 188 -
2. Описание экспертной системы
Предлагаемая в настоящей работе экспертная система представ-
представляет собой интеллектуальное программное средство, предназначен-
предназначенное для быстрого автоматического оценивания полезности предло-
предложенной характеристики биополимеров для заданного способа их
классификации. Определим задачу работы системы более подробно.
Рассматривается конечная выборка первичных структур биопо-
биополимеров (белков, участков ДНК или РНК). Предполагается, что все
рассматриваемые биополимеры объединены в два различных класса
(например, классы изофункциональных белков). Далее, предполага-
предполагается, что каждый из рассматриваемых биополимеров однозначно
характеризуется некоторым набором "наблюдаемых" параметров. Чи-
Число и природа этих параметров могут быть произвольными: физиче-
физические, стереохимические, статистические свойства аминокислотных
остатков и нуклеотидов, порядок их размещения в первичных стру-
структурах биополимеров, частоты использования олигопептидов или
олигонуклеотидов и т.п..
Далее, предполагается, что указанные параметры биополимеров
могут принимать различные значения. Однако, возможность появ-
появления конкретных значений этих параметров для биополимеров из
одного класса может быть охарактеризована определенной часто-
частотой. Следовательно, область значений рассматриваемых параметров
на множестве биополимеров определенного класса можно считать
вероятностным пространством. Тогда, любое отображение указан-
указанного вероятностного пространства на множество вещественных чи-
чисел - вещественнозначная случайная функция.
Таким образом, исследуемые с помощью предлагвмой экспертной
системы характеристики биополимеров суть описанные выше вещест-
веннозначные случайные функций. Соответственно, задача эксперт-
экспертной системы состоит в быстром автоматическом оценивании полез-
полезности использования предложенной ей характеристики биополимеров
для заданного способа их классификации.
Входные данные экспертной системы:
(Pj) 1=1>п - значения предложенной характеристики для биополи-
биополимеров одного из двух заданных классов (для опре-
- 199 -
деленности: биополимеров класса "I");
- число биополимеров в рассматриваемой выборке класса
,m
- значения этой же характеристики для биополимеров
другого из двух заданных классов (класса "II");
- число биополимеров в рассматриваемой выборке класса "II"
Выходные данные экспертной системы:
R - численное значение полезности использования предложенной
характеристики (F) для заданного способа разбиения рассма-
рассматриваемой выборки биополимеров на два класса: "I" и "II".
Отличительной конструктивной особенностью предлагаемой экс-
экспертной системы является использование нечетких эмпирических
исчислений в рамках теории аддитивной полезности Сэвиджа 12]. В
частности, ныходной параметр R принимает значения из интервала
вещественных чисел [-1, 1), где интерпретируется следующим об-
разом;
а) -1 ^ R < О - характеристика неприемлема для классификации;
б) 0 < R < 0,5 - характеристика может использоваться для клас-
классификации биополимеров только в сочетании с
другими, более "полезными" характеристиками;
в) 0,5 < R < I - характеристика сама по себе может быть испо-
использована для предложенной классификации био-
биополимеров.
Важной особенностью является то, что для вычисления числен-
численного значения полезности R предложенной для анализа характерис-
характеристики экспертная система использует эмпирические знания двух ти-
типов: „ ^
а) априорные знания по теории распознавания образов (необходи-
(необходимые и достаточные условия применимости различных методов
распознавания образов и кластер-анализа) и математической
статистике (точные критерии проверки статистических гипотез,
оценивание достаточных статистик распределений и т.п.). Эти
знания заложены в экспертную систему в виде подпрограмм на
языке FORTRAN-TT ГОШ IBM PC;
- 200 -
б) апостериорные знания о статистических свойствах рассматрива-
рассматриваемой характеристики биополимеров (типы распределения и обла-
области значений этой характеристики для классов "I" и "II";
предсказывающие способности при подстановке в различные ме-
методы распознавания образов и кластер-анализа; наличие "зану-
мленностей" по масштабу и "положению среднего значения" и
т.п.). Эти знания не заложены в экспертную систему априори,
а выводятся ею в процессе исследования статистических свойств
рассматриваемой характеристики.
Следует отметить, что активное использование экспертной си-
системой самостоятельно "добываемых" ею апостериорных знаний о
статистических свойствах рассматриваемой характеристики позво-
позволяет ей целенаправлено применять заложенные в нее точные апри-
априорные знания. Существенно, что с помощью априорных знаний экс-
экспертная система получает эмпирические оценки полезности рассма-
рассматриваемой характеристики с точки зрения различных частных аспе-
аспектов математической статистики, распознавания образов и кластер-
анализа. Все такие "частные" полезности измеряются количестве-
количественно, причем области их численных значений и способы интерпрета-
интерпретации тождественно совпадают с областью значений и способу интер-
интерпретации полезности R рассматриваемой характеристики (см. выле).
Именно это обстоятельство позволяет использовать в рассуж-
рассуждениях экспертной системы нечеткие исчисления теории аддитивной
полезности Сэвиджа [2]. В результате, система не делает жестких
"категоричных" суждений о рассматриваемых характеристиках био-
биополимеров, а дает объективную оценку их классифицирующих спосо-
способностей, основанную на разностороннем анализе их статистических
свойств. Следовательно, работа экспертной системы существенным
образом основана на последовательном использовании одного из
теоретических принципов ИСКУССТВЕННОГО ИНТЕЛЛЕКТА [3] - принци-
принципе "максимальной объективности": всякое решение принимается
только при наличии большого числа его различных обоснований. В
результате, характеристики биополимеров, рекомендованные экспе-
экспертной системой для заданного способа их классификации, обладают
высокими предсказывающими способностями, имея при этом различ-
различные (порой, даже взаимоисключающие) статистические свойства.
- 201-
Ииишп это обстоятельство н позволяет считать предлагаемую экс-
экспертную систему интеллектуальвш программаш средством.
3. Архитектура экспертной системы
функциональная схема экспертной системы приведена на рис.1.
Она состоит из семи функциональных блоков:
- БЛОК ВВОДА/ВЫВОДА ШЯОРМАЦИИ;
- БЛОК КОНТРОЛЯ ОБЛАСТИ ЗНАЧЕНИЙ ХАРАКТЕРИСТИКИ;
- БЛОК КОНТРОЛЯ ТИПА РАСПРЕДЕЛЕНИЯ ЗНАЧЕНИЙ ХАРАКТЕРИСТИКИ;
- БЛОК КОНТРОЛЯ ПРЕДСКАЗЫВАИЩ СПОСОБНОСТЕЙ ХАРАКТЕРИСТИКИ;
- БЛОК ВЫЧИСЛЕНИЯ ПОЛЕЗНОСТИ ХАРАКТЕРИСТИКИ;
- БЛОК УПРАВЛЕНИЯ ЗНАНИЯМИ;
- ВАЗА ЗНАНИЙ.
Между этими блоками экспертной системы существует 18 функциона-
функциональных связей. Они показаны на рис.1 стрелками.
Экспертная системе работает следующим образом. Сначала БЛОК
ВВОДА/ВЫВОДА ИНФОРМАЦИИ получает от пользователя входные данные
для работы экспертной системы и, с помощью БЛОКА УПРАВЛЕНИЯ
ЗНАНИЯМИ, заносит их в БАЗУ ЗНАНИЙ (связи I и 17). Затем, пос-
последовательно один за другим работают три БЛОКА КОНТРОЛЯ статис-
статистических свойств характеристики биополимеров (связи 2, 6 и 10).
Они используют в процессе исследования характеристики различные
апостериорные знания ("уже установленные") о ее статистических
свойствах (связи 3, 7, II и 18) и , в свою очередь, заносят та-
такие звания ("новые") в БАЗУ ЗНАНИЙ (связи 4, 8, 12 и 17). Кроме
того, каждый из трех указанных БЛОКОВ КОНТРОЛЯ может самостоя-
самостоятельно прекратить анализ рассматриваемой характеристики, если
она явно непригодна для классификации биополимеров (например,
если области значений характеристики для разных классов полнос-
полностью совпадают). Это делается посредством связей 5, 9 и 13. Пос-
Последним в экспертной системе работает БЛОК ВЫЧИСЛЕНИЯ ПОЛЕЗНОСТИ
ХАРАКТЕРИСТИКИ (связь 14). Он анализирует результаты статисти-
статистического анализа рассматриваемой характеристики, проведенного
тремя БЛОКАМИ КОНТРОЛЯ (связи 15 и 18). Эти результаты предста-
представляют собой количественные значения оценок полезностей рассмат-
- 202-
БЛОК
ВВОДА/ВЫВОДА
ИНФОРМАЦИИ
16
5
«
9
13
16
2
БЛОК КОНТРОЛЯ
ОБЛАСТИ ЗНАЧЕНИЙ
ХАРАКТЕРИСТИКИ
6
БЛОК КОНТРОЛЯ
ТИПА РАСПРЕДЕЛЕ-
РАСПРЕДЕЛЕНИЯ ЗНАЧЕНИЙ
ХАРАКТЕРИСТИКИ
10
БЛОК КОНТРОЛЯ
ПРЕДСКАЗЫВАШЩ
СПОСОБНОСТЕЙ
ХАРАКТЕРИСТИКИ
14
БЛОК ВЫЧИСЛЕНИЯ
ПОЛЕЗНОСТИ
ХАРАКТЕРИСТИКИ
I
3
4
7
8
. — 17 » п
БЛОК
УПРАВЛЕНИЯ
ЗНАНИЯМИ
а »
12
15
».
18
«
БАЗА
ЗНАНИЙ
(классная доска)
Рис.1 Функциональная схема экспертной системы.
- 203 -
риваемой характеристики с точек зрения областей и типов распре-
распределения ее значений на различных классах биополимеров, а такие
с точки зрения ее предсказывающих способностей (см. части 4, 5
и 6). На их основе вычисляется количественное значение оценки
полезности R использования рассматриваемой характеристики для
классификации биополимеров (см. выходные данные). Затем пара-
параметр R возвращается пользователю (связь 16), и работа эксперт-
экспертной системы заканчивается.
Следует отметить, что БАЗА ЗНАНИЙ, БЛОК УПРАВЛЕНИЯ ЗНАНИЯМИ
и функциональные связи 17 и 18 между ними - существенные кон-
конструктивные особенности экспертной системы. Именно с их помо-
помощью осуществляется универсальный механизм "наследования" знаний
о статистических свойствах рассматриваемой характеристики био-
биополимеров, получаемых различными БЛОКАМИ экспертной системы. В
результате - происходит оптимизация ее работы: каждый БЛОК кон-
концентрирует свое внимание на наиболее существенных, с точки зре-
зрения "ранее установленных" знаний, аспектах анализа предложенной
' характеристики и "игнорирует" рассмотрение тех ее особенностей,
которые становятся в процессе анализа "второстепенными". Кроме
того, к моменту завершения анализа предложенной характеристики
биополимеров экспертная система накапливает большое число коли-
количественных оценок полезности ее использования для классификации
биополимеров. Важно подчеркнуть, что несмотря на то, что ука-
указанные оценки полезности характеристики отражают различные ее
свойства, все они имеют одну и ту же область значений; 1-1; 1)
с единым способом ее интерпретации (см. раздел 2). Именно это
обстоятельство позволяет применить в работе экспертной системы
теорию аддитивной полезности Сэвиджа [2), дающую простой и гиб-
гибкий способ разностороннего статистического анализа характерис-
характеристик, в результате, экспертная система демонстрирует элементы
интеллектуального поведения: рекомендуемые ею для классификации
биополимеров характеристики обладают высокими предсказывающими
способностями, имея при этом различные (порой даже взаимоисклю-
взаимоисключающие) статистические свойства.
Рассмотрим работу основных функциональных БЛОКОВ экспертной
системы более подробно.
- 204 -
4. Блок контроля области значений характеристики
Главное назначение БЛОКА - оценивание полезности характери-
характеристики биополимеров для заданного способа их классификации с то-
точки зрения областей ее значений на двух альтернативных классах
"I" и "II" (см. раздел 2). Принцип работы БЛОКА состоит в:
а) оценивании областей значений рассматриваемой характеристика
на заданных классах биополимеров "I" и "II";
б) оценивании степени перекрывания указанных областей;
в) вычислении количественного значения полезности рассматрива-
рассматриваемой характеристики биополимеров с точки зрения спепени пе-
перекрывания областей ее значения для классов "I" и "II";
г) принятии решения о целесообразности использования предложен-
предложенной характеристики для классификации биополимеров или прек-
прекращении ее дальнейшего анализа.
Для выполнения указанных процедур статистического анализа исс-
исследуемой характеристики биополимеров в экспертную систему зало-
заложено 18 эвристических правил (априорные знания, см. выше). Рас-
Рассмотрим, в качестве примера, одно из них:
ПРАВИЛО 10
п - суммарное число биополимеров класса "I";
m - суммарное число биополимеров класса "II";
п - число биополимеров класса "I", попавших в
область значений для класса "II";
m - число биополимеров класса "II", попавших
в область значений для класса "I";
Тогда:
RI0 = 1 -
m
где:
RT0- значение полезности характеристики с точки зрения
суммарного числа нестрого классифицируемых с ее
помощью биополимеров.
Это правило является эвристическим. Оно отражает эмпиричес-
эмпирический опыт разработчиков системы: "чем меньше биополимеров одного
класса имеют значения рассматриваемой характеристики, попадаю-
попадающие в область ее значений для другого класса, тем в большей
степени эта характеристика подходит для классификации".
- '•'Об -
5. Блок контроля типа распределения значений характеристики
Данный БЛОК работает после БЛОКА КОНТРОЛЯ ОБЛАСТИ ЗНАЧЕНИЙ
ХАРАКТЕРИСТИКИ. Таким образом, он осуществляет статистический
анализ только тех характеристик биополимеров, для которых сте-
степень перекрывания областей значений на классах "I" и "II" приз-
признана достаточно малой (т.е. допускающей возможность классифика-
классификации биополимеров). Отметим, что обнаружение указанного свойства
характеристики на ограниченной выборке биополимеров еще не гара-
гарантирует воспроизведение его на выборках большего объема. Одной
из важных особенностей такой характеристики, существенно повы-
аавщей вероятность воспроизведения ее статистических свойств,
является близость распределений значений этой характеристики на
классах биополимеров "I" и "II" к нормальному распределению.
Кроме того, нормальность распределений значений рассматриваемой
характеристики на заданных классах биополимеров "I" и "II" яв-
является необходимым и достаточным условием применимости аналити-
аналитических методов оптимизации "свободных" параметров большинства
методов распознавания образов и кластер-анализа [4, 53, тради-
традиционно используемых для автоматической классификации объектов.
Принцип работы данного БЛОКА состоит в проверке четырех
различных статистических гипотез о степени близости распределе-
распределений значений исследуемой характеристики на классах биополимеров
"I" и "II". Для этой цели в экспертную систему заложено восемь
эмпирических и теоретических правил, реализованных в виде про-
процедур на языке FORTRAN-77 для ПЭВМ IBM PC. В частности, норма-
нормальность распределения значений исследуемой характеристики на
на заданном классе биополимеров проверяется с помощью критерия
Пирсона [6] для статистики %2 (ПРАВИЛО 24).
6. Блок контроля предсказывающих способностей характеристики
Этот БЛОК экспертной системы работает после БЛОКОВ КОНТРО-
КОНТРОЛЯ ОБЛАСТЕЙ И ТИПА РАСПРЕДЕЛЕНИЯ ЗНАЧЕНИЙ ХАРАКТЕРИСТИКИ. Та-
Таким- Образом, он анализирует только такие характеристики биопо-
биополимеров, области значений которых на классах "I" и "II" пересе-
- 206 -
каются достаточно слабо (так что можно осуществлять классифика-
классификацию биополимеров) и, что не менее важно, для которых указанное
свойство непересечения в достаточной степени воспроизводимо
благодаря нормальности их распределения и на классе "I", и на
классе "II". Следовательно, к началу работы данного БЛОКА экс-
экспертная система уже подробно исследовала статистические свойс-
свойства рассматриваемой характеристики биополимеров. Соответствен-
Соответственно, целью работы этого БЛОКА является выяснение практической
ценности использования исследуемой характеристики для заданно-
заданного способа классификации биополимеров.
Принцип работы БЛОКА состоит в циклическом выполнении сле-
следующей последовательности эвристических процедур:
а) каждая из предложенных экспертной системе выборок биополиме-
биополимеров классов "I" и "II" (см. раздел 2) разбиваются на две под-
выборки равного объема (назовем их для определенности "обуча-
ающими" и "контрольными" выборками соответствующих классов);
б) с помощью "обучающих" подвыборок биополимеров классов " и
"II" находится пороговое значение характеристики, оптималь-
оптимальное с точки зрения их заданной классификации;
в) с помощью найденного на "обучающих данных" порогового значе-
значения для заданной классификации биополимеров и (еще неисполь-
неиспользованных) "контрольных" подвыборок биополимеров классов "I"
и "II" осуществляется "контрольная" (независимая от "обуче-
"обучения") классификация биополимеров. Получается результат кон-
контрольной классификации:
И&- число ВЕРНО классифицированных полимеров класса "I",
Nb- число НЕВЕРНО классифицированных полимеров класса "I",
NQ- число НЕВЕРНО классифицированных полимеров класса "II",
Nd- число ВЕРНО классифицированных полимеров класса 1";
г) полученные результаты "контрольной" классификации биополиме-
биополимеров анализируются с помощью статистических и эвристических
критериев:
- 207 -
- суммарной доли ПРАВИЛЬНО классифицированных биополимеров;
- доли ПРАВИЛЬНО классифицированных биополимеров класса "I";
- доли ПРАВИЛЬНО классифицированных биополимеров класса 1";
- ДОСТОВЕРНОСТИ по точному критерию Фишера-Ирвина F1;
- ДОСТОВЕРНОСТИ по критерию статистики % 16).
д) на основании проведенного с помощью указанных пяти статисти-
статистических критериев анализа результатов "контрольной" классифи-
классификации биополимеров вычисляется, соответственно, пять различ-
различных оценок полезности рассматриваемой характеристики (по од-
одной оценке для каждого из пяти статистических критериев).
Указанные процедуры выполняются циклически одна за другой 10
раз. В результате осуществляется 10 независимых "контрольных"
испытаний рассматриваемой характеристики на различных комбина-
комбинациях "обучающих" и "контрольных" данных (процедура "а"). В хо-
ходе этих испытаний исследуются (процедура "г") получаемые с ее
помощью (процедура "в") классификации "контрольных" подвыборок
биополимеров. Важно подчеркнуть, что указанные "контрольные"
подвыборки биополимеров являются в каждом из этих 10 испытаний
независимыми от "обучающих" подвыборок биополимеров (процедура
"а"), с помощью которых подбираются оптимальные границы между
классами (процедура "б").
Для осуществления описанного выше сценария статистического
анализа классифицирующих способностей исследуемой характеристи-
характеристики в БЛОК заложено 23 теоретических и эвристических правила
(априорные знания, раздел 2). Рассмотрим, в качестве примера,
одно из этих правил. *
ПРАВИЛО 48
ЕСЛИ во всех 10 "контрольных" испытаниях характеристики,
получаемые с ее помощью классификации "контрольных"
подвыборок биополимеров (независимых от "обучающих"
подвыборок !!!) обладают следующими свойствами:
а) суммарная доля ПРАВИЛЬНО классифицированных био-
биополимеров больше 70%;
б) доля ПРАВИЛЬНО классифицированных биополимеров
в классе "I" больше 50%;
- 208 -
в) доля ПРАВИЛЬНО классифицированных биополимеров
в классе "II" больше 50Х;
г) достоверность по точному критерию Фишера-Ирвина
F3 больше 75Х;
д) достоверность по критерию %2 16] больше 90Х;
ТО рассматриваемая характеристика биополимеров реко-
рекомендуется для их заданной классификации;
ИНАЧЕ рассматриваемая характеристика биополимеров беспо-
бесполезна для осуществления их заданной классификации.
Это правило является эвристическим. Оно выражает эмпиричес-
эмпирический опыт разработчиков системы: "целесообразность использования
характеристики биополимеров для их классификации может быть
объективно оценена только в процессе решения реальных практиче-
ких задач".
В заключение следует особо подчеркнуть, что БЛОК КОНТРОЛЯ
ПРЕДСКАЗЫВАП1Щ СПОСОБНОСТЕЙ ХАРАКТЕРИСТИКИ - принципиально ва-
важный конструктивный элемент предлагаемой экспертной системы.
Наиболее ценным с практической точки зрения свойством этого
БЛОКА является то, что он рекомендует использовать для класси-
классификации биополимеров только те их характеристики, для которых:
а) на независимых от "обучения", "контрольных" данных показана
высокая точность (более 10%) и достоверность (более 9ОХ)
получаемых с их помощью классификаций биополимеров;
б) указанные свойства точности и достоверности классификаций
биополимеров воспроизводятся (не менее 10 раз подряд) на не-
независимых комбинациях "обучающих" а "контрольных" выборок,
построенных посредством случайных разбиений исследуемого ма-
массива надежных экспериментальных данных.
Эти требования накладывают весьма жесткие ограничения на стати-
статистические свойства характеристик, рекомендуемых для классифика-
классификации биополимеров. Однако, использование таких характеристик от-
открывает принципиально новые подходы к решению содержательных
биологических задач. В частности, появляется возможность пост-
построения методов высокоточной классификации биополимеров, привле-
привлекая для их оптимизации ("обучения") все известные эксперимента-
- 209 -
льние данные; достоверность и воспроизводимость результатов их
работы на независимых от "обучения" данных гарантируется при
этом тем тщательным статистическим анализом предсказывающих
способностей характеристик биополимеров, который осуществляет
ся этим БЛОКОМ экспертной системы (см., например, ПРАВИЛО 48).
7. Блок вычисления полезности характеристики
Этот БЛОК работает только с теми характеристиками, которые
рекомендованы ПРАВИЛОМ 48 (см. раздел 6) для осуществления за-
заданной классификации биополимеров. Следует отметить, что среди
предложенных экспертной системе характеристик биополимеров мо-
может быть много таких, которые обладают большими классифицирую-
классифицирующими способностями (например, характеристики для экспертной
системы может предлагать квалифицированный эксперт-биолог).
Поэтому, важно для каждой найденной характеристики иметь коли-
количественную оценку ее полезности при проведении заданной класси-
классификации биополимеров. Соответственно, назначение этого БЛОКА
экспертной системы состоит в вычислении для каждой рассматрива-
рассматриваемой им характеристики биополимеров эмпирической оценки полез-
полезности для заданного способа их классификации. Для этой цели
используется следующее эвристическое правило:
ПРАВИЛО 50
Пусть: R1a - полезность характеристики с '
степени пересечения областей
для биополимеров классов "I"
точки зрения
ее значений
и"П";
Тогда:
здесь:
It,fi - полезность характеристики с точки зрения
степени нормальности распределений ее зна-
значений для биополимеров классов "I" и "II";
R.q - полезность характеристики с точки зрения
ее классифицирующих способностей для биопо-
биополимеров классов "I" и "II";
«50= -?-
(R
18
3-R49)
- окончательная оценка полезности использова-
использования раой характеристики для за-
заданного
нчательная оценка полезн
рекомендованной характеристики для за-
заного способа классификации биополимеров.
Это правило является эвристисгаш. Оно выражает эмпиржчес
опыт разработчиков системы. Наибольший вклад в численное значе-
значение полезности Rg0 вносит количественная оценка ее классяфщщ-
рующих способностей R49, полученная в результате многократных
независимых испытаний этой характеристики для классификации
различных "обучающих" и "контрольных" выборок биополимеров. Та-
Таким образом, ПРАВИЛО 50 выражает очевидное эмпирическое сообра-
соображение: "практическая ценность для классификации объектов любой
их характеристики главным образом определяется тем, насколько
точной и содержательной получается построенная с ее поионыв
классификация реальных данных".
Кроме того, существенный вклад в полезность RgQ рассматри-
рассматриваемой характеристики вносят оценки полезности с точки зрения
степени нормальности распрелений и степени пересечения областей
ее значений для биополимеров классов "I" и "II" (Rg6 и R18 ,
соответственно). Таким образом с помощью ПРАВИЛА 50 учитывает-
учитывается выборочная природа входных данных экспертной системы, на ос-
основании статистического анализа которых она и делает вывод о
степени целесообразности использования той или иной характерис-
характеристики биополимеров для заданного способа их классификации. Оче-
Очевидно, нормальность распределений и низкая степень пересечения
областей значений исследуемой характеристики биополимеров на
заданных их классах существенно повышают вероятность воспроиз-
воспроизведения ее классифицирующих способностей на независимых данных.
Вычисленная БЛОКОМ количественная оценка полезности Rg0
рассматриваемой характеристики биополимеров для заданного спо-
способа их классификации - выходные данные экспертной системы.
Способ ее интерпретации приведен в разделе 2. После вычисления
этого параметра работа экспертной системы заканчивается.
- 211 -
-310 -
8. Реализация экспертной системы
Описанная в настоящей работе экспертная система для быст-
быстрой оценки полезности использования произвольных характеристик
биополимеров для заданного способа их классификации реализована
в лаборатории теоретической молекулярной генетики ИЦиГ СО АН
СССР. Она содержит 50 теоретических и эмпирических правил. Экс-
Экспертная система реализована на языке FORTRAN-?? ПЭВМ IBM PC.
С помощью модельных данных осуществлена калибровка шкалы
намерения полезности характеристик биополимеров с точки зрения
их использования для задач классификации. Исследована зависи-
зависимость вычисляемой экспертной системой полезности R от таких ва-
важных особенностей поведения рассматриваемой характеристики био-
биополимеров как (а) расстояние между ее средними значениями на
классах "I" и "II"; (б) соотношение между дисперсиями значений
этой характеристики на альтернативных классах биополимеров;
а так же (в) от объемов выборок биополимеров из альтернативных
классов "I" и "II". Представляется, что адекватное понимание
сложных взаимосвязей между указанными особенностями поведения
характеристик на альтернативных классах биополимеров и их клас-
классифицирующими способностями является необходимым условием для
построения содержательных с биологической точки зрения класси-
классификаций регуляторных и функциональных сайтов ДНК, молекул РНК
и белков. Именно эти обстоятельства и обусловили проведение
детальной калибровки шкалы полезности произвольных характерис-
характеристик биополимеров с точки зрения их классификации. Рассмотрим
в качестве примеров некоторые из указанных взаимосвязей, уста-
установленные в ходе калибровки полезности R, вычисляемой эксперт-
экспертной системой.
На рис.2 показана зависимость численного значения парамет-
параметра R от расстояния d между средними значениями характеристики
на двух альтернативных классах биополимеров, в качестве модель-
модельных данных для получения приведенной на рис.2 зависимости R(d),
нами использовались пары выборок вещественных чисел, значения
которых имели плотности распределения N (в0, 1) и N (so+d, 1),
соответственно (здесь, и далее, N (в,о) - стандартное обозначе-
обозначение плотности нормального распределения со средним значением в
1.0
0.5 -^
0.0
- 0.5 -h
- 1.0 *•••
R (d)
1 I I I I
Рис. 2 Пример зависимости полезности R от расстояния d между
средними значениями характеристики на двух альтернати-
альтернативных классах, измеренного в масштабе дисперсий ее зна-
значений на этих классах (пояснения в тексте).
и дисперсией о). Таким образом, рис.2 показывает зависимость
полезности R от расстояния d между сремнгат значениями характе-
характеристики на двух альтернативных классах, измеренного в масштабе
дисперсий ее значений на этих классах. Видно, что полезность R
указанных выборок может быть проинтерпретирована (см. раздел 2)
следующим образом: при d меньшем 0.5 выборки неразличимы (т.е.
характеристику с подобными распределениями значений на биополи-
биополимерах классов "I" и "II" бесполезно использовать для их класси-
классификации). При значениях d от 0.5 до 1.5 эти выборки уже можно
различить, однако для более надежной их классификации следует
привлечь дополнительные сведения. Если же значения d больше 1.5,
то выборки могут быть различимы только на основе анализа подоб-
подобной характеристики. Далее, при d большем 3 полезность R дости-
достигает своего максимального значения для выборок с нормальным за-
законом распределения значений характеристики. В предлагаемой эк-
экспертной системе в качестве максимального значения полезности
- 212 -
- 213 -
1.0 t-
0.5 f-
0.0
- 0.5 f-
- 1.0 -L
¦ ¦¦¦¦¦¦ ¦ i I i i i i « i i i i | i ' i ' » v
S 2 3 4
с*********************
Рис. 3 Пример зависимости полезности R от соотношения v между
дисперсиями значений характеристики на двух альтернати-
альтернативных классах при фиксированном расстоянии между ее сре-
средними значениями на этих классах (пояснения в тексте).
характеристики, плотности распределения значений которой на
классах "I" и "II" являются нормальными, выбрана величина 0,9.
Это обусловлено тем, что могут быть еще "более полезные" харак-
характеристики: например, характеристики, являющиеся специфическими
константами в пределах каждого из альтернативных классов биопо-
биополимеров (плотность распределения - 0(х)-функция Дирихле).
На рис.3 показана зависимость численного значения парамет-
параметра R от соотношения v между дисперсиями значений характеристики
на двух альтернативных классах биополимеров. В качестве модель-
модельных данных для получения приведенной на рис.3 зависимости R(u),
нами использовались пары выборок вещественных чисел, значения
которых имели плотности распределения N (О, 1) и N A.5, v),
соответственно. Таким образом, рис.3 показывает зависимость
полезности R от соотношения v между дисперсиями значении харак-
характеристики на двух альтернативных классах при фиксированном зна-
значении расстояния между ее средними значениями на этих классах.
- 214 -
Значения указанного расстояния и дисперсии в первой выборке
взяты равными 1.5 и I, соответственно, вследствие того, что они
(см. выше) являются критическими для зависимости полезности R
от расстояния d между средними значениями характеристики на
двух альтернативных классах (измеренного в масштабе ее диспер-
дисперсий на этих классах). Видно, что вычисляемая экспертной систе-
системой оценка полезности R так же правдоподобно описывает степень
разделимости этих выборок. При v меньшем 1.5 они почти полнос-
полностью различимы. При значениях и от 1.5 до 3 указанные выборки
еще можно различить, однако для более надежной классификации в
этом случае следует привлекать дополнительные сведения. Далее,
при v большем 3 рассматриваемые выборки разделить уже практи-
практически невозможно (т.е. бесполезно привлекать для классификации
биополимеров те их характеристики, значения которых имеют на
двух альтернативных классах подобные плотности распределения).
На рис.4 показана зависимость численного значения парамет-
параметра R от объемов п выборок биополимеров из двух альтернативных
классов. В качестве модельных данных для получения приведенной
на рис.4 зависимости R(n), нами использовались пары выборок ве-
вещественных чисел, значения которых имели плотности распределе-
распределения N (О, 1) и N A.5, I), т.е. критические (см. выше) с точки
зрения индивидуального использования характеристики биополиме-
биополимеров для их классификации. Таким образом, рис.4 показывает зави-
зависимость оценки полезности R характеристики биополимеров от объ-
объема известных экспериментальных данных, привлекаемых для иссле-
исследования ее классифицирующих способностей. Очевидно, что необхо-
необходимый для объективного анализа объем данных существенно зависит
от статистических свойств рассматриваемой характеристики биопо-
биополимеров. Объем же таких данных, в свою очередь, в значительной
степени определяет степень адекватности получаемой на их основе
оценки полезности привлечения характеристики биополимеров для
.41 классификации. Из рис.4 видно, что можно выделить три облас-
области значений п, в которых поведение вычисляемой экспертной сис-
системой полезности R существенно различается. При объемах п выбо-
выборок меньших 10 биополимеров полезность R использования произво-
произвольной характеристики для их классификации не может быть оценена
объективно. При объемах п таких выборок в диапазоне от 10 до 20
-215 -
1.0 *-
0.5
0.0
- 0.5 f-
- 1.0 ****
R (n)
20
I i ¦ i i I I—I—I—I—I—I—I—I—!—»¦ П
40
60
80
Рис. 4 Пример зависимости полезности R от объема п выборок био-
биополимеров, привлекаемых для исследования классифицирую-
классифицирующих способностей их произвольных характеристик (поясне-
(пояснения в тексте)..
биополимеров оценка R оказывается явно заниженной (т.е. выборки
нерепрезентативны). При п большем 20 полезность R практически
достигает максимального значения и перестает зависеть от объе-
объемов выборок классов "I" и "II". Таким образом, можно сделать
вывод, что экспертная система позволяет достаточно объективно
оценивать полезность использования характеристик биополимеров
с точки зрения их классификации (R слабо зависит от объемов вы-
выборок при п большем 20). Следует также отметить, что для этой
цели она может использовать выборки биополимеров весьма неболь-
небольшого объема: оптимальный объем выборок - 20-30 биополимеров
в каждом исследуемом классе. Эта особенность экспертной системы
является приципиально важной для молекулярной биологии и генети-
генетики, в которых огромное разнообразие изучаемых ими объектов и
явлений характеризуются, как правило, лишь фрагментарными и
разрозненными экспериментальными данными.
- 216 -
Оценена эффективность работы экспертной системы при обра-
обработке экспериментальных данных 17, 8). Ее быстродействие для
ПЭВМ IBM PC/AT-286 составило в среднем более 30 характеристик
биополимеров за 1 секунду. Показано, что методы функциональной
классификации белков по их аминокислотным последовательностям,
построенные с помощью рекомендованных экспертной системой хара-
характеристик их первичных структур, обладают точностью не менее
95% на независимых данных G). Кроме того, в результате статис-
статистического анализа списков характеристик ругуляторных сайтов ДНК,
найденных с помощью экспертной системы, предложен ряд новых мо-
молекулярных механизмов их функционирования [8].
Таким образом, можно сделать вывод, что предлагаемая в нас-
настоящей работе экспертная система для быстрой оценки полезности
характеристик биополимеров для их классификации - высокоэффек-
высокоэффективное интеллектуальное программное средство молекулярной био-
биологии и генетики, позволяющее решать широкий круг содержатель-
содержательных практических задач.
ЛИТЕРАТУРА
I I] Уотермен Д. Руководство по экспертным системам. М.: Мир.
1989.
[ 2] Фишберн П. Теория полезности для принятия решений. М.:
Наука. 1978.
[ 3] Попов Э.В. Экспертные системы. М.: Наука. 1987.
[ 4] Дуда Р., Харт П., Распознавание образов и анализ сцен,
U., Мир, 1976.
[ 51 Патрик Э. Основы теории распознавания образов. М.: -
Советское радио. 1980.
[ 6] Леман Э. Проверка статистических гипотез. М.: Наука,
1979.
[ ?] Бенюх Д.Н., Пономаренко М.П., Колчанов Н.А., Орлов Ю.Л.
// (настоящий сборник). 1989. с. 243 - 263 .
[ 8] Кель А.Э., Пономаренко М.П., Орлов Ю.Л., Мищенко Т.М.,
Колчанов Н.А. // (настоящий сборник). 1989. с. 221 -242 .
- 217 -
3IS -
SUMMARY
EXPERT SYSTEM OF FAST ESTIMATION OF
BIOPOLYMERS CHARACTERISTICS UTILITY FOR THEIR CLASSIFICATION
Ponomarenko M.P., Orlov Yu.L.
Institute of Cytology and Genetics, Siberian Branch,
the USSR Academy of Science, Novosibirsk, USSR, 630090
The work deals with the description of the expert system
of fast estimation of blopolymers arbitrary characteristics
utility for their classification.
The principle of its vork lies in a detailed analysis of
a wide spectrum of statistical properties of each characteris-
characteristic In question.
Input data of the expert system:
( Pj ) 1=1,...n - values of the propoused characteristics
for blopolymers of one of the two given
classes ( for definition : blopolymers of
class "I" );
( P, ) 1=1,...ra - values of the same characteristics for
blopolymeres of the other one of two given
classes ( class "I" );
n,m - sampling volumes of class "I" and "I", correspondingly.
Output data of the expert system :
R - numerical value of the utility of the proposed characte-
characteristic { P ) for the given way of dividing of blopolymers
Into two classes : "I" and "II"
The parameter R takes values from Interval [-1,1] of
real numbers, where It is Interpreted in the following way :
a) -1. < R < 0. - characteristic is unacceptable for classi-
classification;
b) 0. < R < 0.5 - characteristic may be used for classifica-
classification of blopolymers, but only in combina-
combination with other characteristics;
c) 0.5 < R < 1. - characteristic may be used by Itself for
classification of blopolymers.
- 218 -
To obtain the R numerical value of the proposed characte-
characteristics the system used knowledge of two types :
a) a priori knowledge on the theory of pattern recognition
( necessary and sufficient conditions of application of me-
methods of pattern recognition and of cluster-analysis ) and on
mathematical statistics ( exact criteria of statistic hypothe-
hypothesis control, and so on ). This knowledge Is given to expert
system by the creators.
b) a posteriori knowledge about the statistical properties of
the discussed blopolymer characteristics ( types of distribu-
distribution and spectrum of its values for classes "I" and "II" and
so on ).
This knowledge Is not put into the expert system, but is
"output" by it Independently In the process of characteristics
investigation^
Active use by the expert system of the independently "ob-
"obtained" knowledge about statistical properties of the descri-
described characteristics allows It to apply exact a priori knowled-
knowledge put by creators purposefully.
It Is noteworthy that with the help of a priori knowledge
the expert systemgets empiric estimations of the utility of
the characteristics under consideration from the point of view
of different particular aspects of mathematical statistics,
pattern recognition and cluster analysis. Moreover, all such
"particular" cases of utility have the same spectrum of values
and way of their Interpretation. This allows the expert system
to use the empiric calculus of additive utility theory. As a
result, the system does not make "categoric" opinions on
blopolymers characteristics, evaluating objectively their cla-
classifying abilities, based on the all-round analysis of their
statistical properties. The work of the expert system is based
on the using of the principle of "maximal objectivity" of
Artlflcal Intelligence : "any decision is taken only at the
presence of sufficient number of Its different substantia-
substantiations».
Expert system architecture is described In part 3. The
logical scheme of the system Is given In Flg.l. In parts 4,5,6
- 219 -
and 7 one can find short description of Its separate functio-
functional blocks. Examples of some a priori rules, put Into expert
system by creators are given. The whole number of such rules
la 50.
In part 8 the realisation of the expert system Is descri-
described.
The examples of the calibration scale of characteristics
are given In Fig. 2,3 and 4.
The effectiveness of expert system at the real experimen-
experimental data treatment Is estimated [7,8]. Its speed for IBM
PC/AT-286 was more than 30 characteristics per second. It Is
shown that methods of fuctlonal classification of proteins
based on characteristics, recommended by the expert system,
demonstrate on the Independent data the exactness not less
than 95V [7]. As a result of the regulatory DNA sites charac-
characteristics study, found by expert system, some new molecular
mechanisms of their work are suggested [8].
- 220
КОМПЬЮТЕРНАЯ СИСТЕМА АНАЛИЗА ФУНКЦИОНАЛЬНЫХ САЙТОВ
В ПОЛИНУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЯХ
Кель А.Э., Поиомаренко М.П-, Орлов ЮЛ., Мищенко Т.М-,
Колчанов Н-А.
Институт цитологии и генетики СО АН СССР, г.Новосибирск
1.ВВЕДЕНИЕ
Задача анализа структуры функциональных сайтов в поли-
нуклеотидных последовательностях давно стоит перед иссле-
исследователями. В основном это связано с потребность!) поиска раз-
различных функциональных сигналов в последовательностях с неиз-
неизвестными функциями [1,2]. Анализ структуры функциональных
сайтов проводится также с целью выявления механизмов их функ-
функционирования [3,4].
К настоящему времени имеется большое разнообразие подхо-
подходов и методов анализа функциональных сайтов и их поиска в
полинуклеотидкых последовательностях [1-5]. Однако, они имеют
ряд принципиальных ограничений на точность и эффективность
работы.
Целью направления, в рамках которого выполнялась работа,
является создание компьютерной системы анализа и поиска функ-
функциональных сайтов с использованием так называемой "Новой
информационной технологии" [в]- Эта технология основана на
принципах накопления и использования формализованных знаний о
структуре и функциях исследуемых объектов. Отличительной осо-
особенностью данного подхода является максимальное ориентирование
всей системы на потребности конечного пользователя - молеку-
молекулярного биолога.
В настоящей статье приводится описание первой части этой
системы, ориентированной на поиск закономерностей в распреде-
распределениях олигонуклеотидов и повторов различного типа в функцио-
функциональных сайтах. На основе обучающей выборки функциональных
сайтов в процессе работы системы производится вычисление боль-
больного количества (более 2-х. миллионов) характеристик, отража-
отражающих насыщеность определенными олигонуклеотидами и повторами
функциональных сайтов. Информативность каждой характеристики
-221 -
оценивается с помощью комплекса программ статистического ана-
анализа- Из всего множества характеристик выбирается набор инфор-
информационно значимых. На их основе возможен поиск в нуклеотидных
последовательностях функциональных сайтов определенного типа.
Существенно, что в предлагаемом подходе производится
автоматический выбор определенного набора информационно
значимых характеристик (из очень широкого набора) без задания
жесткого априорного предпочтения тем или иным характеристикам
и без формирования жесткой модели исследуемого функционального
сайта.
Такой подход имеет принципиальное отличие от подходов,
применявшихся ранее для исследования и поиска функциональных
сайтов, в которых модель функционального сайта, как правило,
задается исследователем a priori. В качестве примера можно
отметить описание функционального сайта в виде консенсуса, как
набора независимых позиций (см. рис. г).
ТТССС
отесс
атесс
итесс
АС АСС
АСЛСА
АСАСА
GGTAC
AGGGU
Consensus:
s
s. t
E.c
G4 :
аз :
t - 1 .
ФВО :
ф82 ;
X :
УРП :
Oil
TACTGCAAA
ТССТССТЛА
TCCTGCTAA
TCCTGCTAA
СЛАГЛСЛАС
AAAGACAAA
AAAGACACT
ТТAT AC AC A
ТТ AT AC AC Л
ас-
GCC
асе
асе
А- А
АТА
AT A
ACT
ACT
—
С-AAAAG
CAAAAAG
СА\АААА
CAAAAAG
CAAGA\C
ACAAAAG
ACAAAAG
CAAAAAC
С ДАЛЛАС
eaAAAAG
•GA-CTA
AGAG-T1
GGAGCTT
¦GAGCTT
ACT A-TГ
ACAATAT
AAAATAG
TGAAC-¦
тал ас-¦
Рисунок 3.
Модель функционального сайта при построении
конценсуса, как набора независимых позиций (на
примере OKI- облостей фагов, и оактерий)
1121.
2. ОПИСАНИЕ СИСТЕМЫ ПОИСКА ИНФОРМАЦИОННО ЗНАЧИМЫХ
ХАРАКТЕРИСТИК В ФУНКЦИОНАЛЬНЫХ САЙТАХ
Предлагаемая система поиска закономерностей ь ра<;преде
лениях олигонуклеотидов и повторенностей в функциональных гай
тах состоит из нескольких программных блоков (см- рис <.')••
г) Блок управлени моделями представлений и стратегиями
поиска; \.
2) Блоки расчета характеристик;
- 222 -
3) Блок статистического анализа;
4) Блок первичной интерпретации результатов.
Блок управления
моделями представ-
представлений и стратегиями
поиска
У
МОДЕЛЬ 1
ра оче т
повторен-
повторенностей
Стратегия
поиска 1
Стратегия
поиска 2
1
МОДЕЛЬ Z
расчет
частот оли-
олигонуклеотидов
Стратегия
поиска 1
1
N
Стратегия
поиска 2
МОДЕЛЬ 3
рзечет
частот
четверок
Стратегия
поиска 1
—¦
-1 i I
Блок
статисти-
статистической
проверки
1
Блок первичной
интерпретации
результатов
Запись в файл
информационно-значимых
характеристик
Рисунок 2
Блок-схема системы поиска закономерностей в
последовательностях функциональных сайтов.
На вход в систему подается две выборки данных.- выборка
сайтов определенного типа (выборка 1) и последовательности, не
относящиеся к функциональному сайту данного типа (выборка 2).
На выходе получаем набор информационно значимых
характеристик.
2.1 БЛОК УПРАВЛЕНИЯ МОДЕЛЯМИ ПРЕДСТАВЛЕНИЙ И
СТРАТЕГИЯМИ ПОИСКА
Предлагаемая система позволяет производить поиск инфор-
информационно значимых характеристик в различных моделях представ-
представления последовательностей функциональных сайтов и с использо-
-223-
вакием различных стратегий поиска- Моделью представления будем
называем конкретный способ описания информации, заключенной в
последовательностях функциональных сайтов. Под стратегией
поиска будем понимать конкретный набор процедур преобразования
этой информации в набор численных параметров, характеризующих
функциональные сайты (называемых в дальнейшем характерис-
характеристиками).
В предлагаемой версии системы реализованы следующие модели
представления (рис.2).
Модель 1. Выявление повторенностей различных типов в нуклео-
тидных последовательностях.
Модель 2. Выявление частот встречаемости олигонуклеотидов раз-
различной длины.
Модель 3. Выявление частот встречаемости подпоследовательнос-
подпоследовательностей длины 4 с предпочтительным расположением опреде-
определенных групп нуклеотидов в различных позициях
четверки.
В каждой модели представления поочередно реализуются
несколько стратегий поиска и отбора информационно значимых
характеристик, качественное описание которых дано в разделе
2.4
Блок управления осуществляет поэтапное переключение раз-
различных моделей представления, передает результаты расчета
характеристик в блок статистического анализа и на основе этого
анализа производит подключение той или иной стратегии поиска.
2.2 БЛОК СТАТИСТИЧЕСКОГО АНАЛИЗА
Блок предназначен для быстрого статистического оцени-
оценивания полезности заданной характеристики функциональных сай-
сайтов для их классификации.
Входные данные блока ¦.
р (П - значения предложенной характеристики для функциональ-
функциональных сайтов одного класса ;
р (J) - значения этой же характеристики для последовательнос-
последовательностей, не относящихся к фукциональным сайтам данного
класса. > • . :
1*1, ...,п и j=i,...,т , где пит- объемы выборок.
-224-
Выходные данные блока таковы.
R - численное значение полезности использования предложенной
характеристики для разделения двух представленых выборок.
Параметр R принимает значения из интервала [-7,1] и
интерпретируется следующим образом:
а) к е М, О] - характеристика неприемлема для классификации;
б) « ? (О, 0.51 - характеристика может быть использована для
классификации функциональных сайтов только в
сочетании с другими характеристиками ;
в) я е Ю.в, 17 - характеристика сама по себе может быть ис-
использована для классификации функциональных
сайтов.
Каждая характеристика функциональных сайтов, поступившая в
блок статистического анализа, оценивается с трех различных
точек зрения (подробнее см. в статье [7] данного сборника).
1) Оценивается полезность R^ применения рассматриваемой
характеристики для классификации функциональных сайтов с
точки зрения степени перекрывания областей ее значений на
заданных классах. Для этой цели в блок заложено 18 различ-
различных логических, статистических и эвристических правил.
2) Оценивается степень близости распределений значений рас-
рассматриваемой характеристики на заданных классах к нормальному
распределению (коэффициент полезности R2) ¦ Этот коэффициент
гарантирует высокую вероятность воспроизводимости статисти-
статистических закономерностей на независимых данных.
5) Оценивается статистическая достоверность классификации
функциональных сайтов на основе рассматриваемой характеристики
(коэффициент полезности R3). Вычисление этого коэффициента
осуществляется посредством многократной классификации случайно
сформированных подвыборок из представленой выборки сайтов на
основе критериев Фишера-Ирвина, "хи-квадрат", А- и
р-импликативности [8], суммарной доли правильно классифициро-
классифицированных сайтов.
Количественная оценка полезности использования рассмат-
рассматриваемой характеристики для функциональной классификации
сайтов вычисляется по следующей формуле;
-225-
Таким образом, наибольший вклад в численное значение
полезности я вносит оценка йз классифицирующих способностей
рассматриваемой характеристики функциональных сайтов. Кроме
'того, параметр й отражает такие важные статистические особен-
особенности исследуемой характеристики, как степень перекрывания
областей ее значений на разных классах функциональных сайтов
и близость распределения этих значений к нормальному распре-
распределению. В результате блок не дает жестких, "категорич-
"категоричных" суждений о рассматриваемых характеристиках функцио-
функциональных сайтов, а делает объективную разностороннюю оценку
их классифицирующих способностей, основанную на тщательном
анализе большого числа статистических свойств. Поэтому ха-
характеристики, рекомендуемые этим блоком для классификации
функциональных сайтов, обладают высокими предсказывающими
способностями, имея при этом различные (порой даже взаимо-
взаимоисключающие) статистические свойства-
После вычисления параметра ft работа блока заканчива-
заканчивается-
2.3 БЛОКИ РАСЧЕТА ХАРАКТЕРИСТИК
Блоки расчета характеристик являются независимыми прог-
программными модулями, каждый из которых реализует одну модель
представления последовательностей функциональных сайтов.
2.3.1 БЛОК АНАЛИЗА РАЗЛИЧНЫХ ТИПОВ ПОВТОРЁННОСТЕЙ
В данном блоке вычисляются характеристики, отражающие
насыщенность последовательностей функциональных сайтов повто-
ренностями четырех типов (см. рис.з)( с учетом их локализации
и нуклеотидного состава фрагментов этих повторенностей). Выде-
ляется два направления вычисления сходства в последовательнос-
последовательностях: прямое (варианты а и d на рисунке 3) и обратное (вари-
(варианты ь и с на рисунке з).
-226-
а ) б' > з-
J ~ * " 5'
*> ";:,., :::::::::::::::::: = %
с) 5.:. ,.. .. з-
а> 3< 5'
Рисунок з. Повторенности четырех типов, анализ которых
производится в последовательностях. Стрелками
показаны участки последовательностей, по которым
производилось сравнение, а) прямые повтореннос-
повторенности; ь ) симметричные; с) инвертированные; d) пря-
прямые комплементарные
На первом этапе характеристики расчитываются по следующим
формулам:
'¦¦$:
F,(k)=
) (Пх) -Пх+к) I х (р(а >-р(а I ж Ц(а ,а )
L xx*k хх+к
(Пх)-Пх+к) I х (Р(а
B)
Х.Г» 1
Гг(к)=
».V»i
(Г(х)-Г(к-х) I
(f(x)-f(k-xlI x (pta l-p(a II
C)
Здесь использованы следующие обозначения.
1) F^{k) и ^г(к) - значения характеристик для прямого
и обратного направления.
Я) х - номер позиции в функциональном сайте.
О) к - параметр, отражающий расстояние между началами
повторенностей.
4) я - код нуклеотида, стоящего в позиции
х. (см. табл. 1)
5) г(х) - функция, отражающая вес фрагмента повторенности,
расположенного в позиции х . Рассматривалось 12 вариантов
- 227 -
весовых функций г(х) (рис 4). С помощью этих функций задастся
различные предпочтения разным участкам последовательности
функционального сайта.
6) р{а) - функция, отражашая вес фрагмента повтореннос-
ти, содержащего код а . Рассматривалось и вариантов весовых
функций р(а). с помощью данных функций задаются разные пред-
предпочтения фрагментам повторенностей, содержащим те или иные
нуклеотиды-
7) б{а,ь) - функция, отражающая вес отношения между кода-
кодами нуклеотидов а и ь . В частности, если рассматриваются
прямые повторенное™ то б(а,ь)=1 при а ¦ ь и б(а,ь)=о при а *
ь. В данном весе может так же учитываться конкретный состав
нуклеотидов а и ь (или другие особенности отношений между ни-
ними). В системе рассматривалось в различных функций веса отно-
отношений между кодами, отражающих как прямое совпадение, так и
комплементарное взаимодействие¦
Таблица 1. Кодировка is групп нуклеотидов,
позициях анализируемых четверок.
встречающихся в
одно-
букв, код
А
т
G
С
R
Y
И
К
V
S
в
V
н
D
N
обозначение
А
т .
G
с
G/A
Т/С
А/С
G/T
Л/Т
в/С
-А
-Т
-G
-С
Ж
группа
нуклеотидов
А
т
G
с
G ИЛИ Л
т или с
Л ИЛИ С
G ИЛИ Т
А ИЛИ Т
G ИЛИ С
Т ИЛИ в ИЛИ С
А ИЛИ 0 ИЛИ С
Л ИЛИ Т ИЛИ С
л или г или в
любой
интерпретация
аденин
тимедин
гуанин
цитозин
пурины (большие)
пиримидины (малые)
амино (полож.заряд)
кето (отриц.заряд)
слабые взаимодейств.
сильные взаимодейст.
-228-
1)
S)
2)
.,
3)
4)
П—, „,-П-,
10)
I Ц ...rJ I
Рисунок -1. Варианты весовых функций, задающих различные пред-
предпочтения разным участкам последовательности функци-
функциональных сайтов.
Полученные таким образом характеристики передаются в блок
статистического анализа. После статистической проверки (по
заданному порогу) бракуются информационно незначимые характе-
характеристики. Оставшиеся характеристики передаются на второй этап
поиска информационно значимых характеристик.
На втором этапе работы данного блока производится следую-
следующие процедуры анализа информации.
1) Усреднение полученных характеристик для близких значений
к - расстояния между фрагментами повторенностей по формулам:
Здесь о^(к) и Qz(k) - значения характеристик для прямого
и обратного направления, а ы(к) - весовая функция, задающая
предпочтения различным расстояниям между повторенностями.
Вид функций и(к) аналогичен функциям, представленым на
рисунке 4.
2) Нахождение максимальных и минимальных величин
характеристик для близких значений к. При этом значения
характеристик вычисляются с использованием весовых функций
о(к).
На выходе данного блока получаем набор наиболее информа-
- 229 -
ционно значимых характеристик, отражающих различие двух пред-
ставленых выборок по параметрам повторенностей внутренних
фрагментов.
2.3.2 БЛОК АНАЛИЗА ЧАСТОТ ВСТРЕЧАЕМОСТИ
ОЛИГОНУКЛЕОТИДОВ
В данном блоке на первом этапе производится расчет частот
встречаемости моно-, ди-, три-, и тетрануклеотидных фрагментов
в последовательностях. Значения каждой характеристики пред-
представляют собой частоты встречаемости олигонуклеотида опреде-
определенного типа в данных последовательностях-
Полученные характеристики передаются в блок статистичес-
статистической проверки. На этом этапе анализа задается нежесткий порог
браковки информационно незначимых характеристик. Оставшиеся
характеристики передаются на следующую стратегию поиска.
На втором этапе производится оценка обедненности олиго-
нуклеотидами различных типов в б'- и з1- направлениях от опор-
опорных олигонуклеотидов, выявленых на первом этапе.
Полученные таким образом характеристики отражают насыщен-
насыщенность олигонуклеотидами различных типое протяженных участков
последовательностей функциональных сайтов. Схема получаемых в
данной модели характеристик представлена на рисунке 5.
Рисунок 5. Схема характеристик, получаемых в модели г.
И- олигонуклеотид, выявлений на первом этапе.
Щ| - область, обедненная определенными олигонуклеотидами в 5'
направлении. ЦЦ - область, обедненная определенными олигонук-
олигонуклеотидами в з7- направлении
2.3.3 БЛОК АНАЛИЗА ЧАСТОТ ВСТРЕЧАЕМОСТИ
ПОДПОСЛЕДОВАТЕЛЬНОСТЕЙ ДЛИНЫ 4
В данном блоке производится анализ частот встречаемости
подпоследовательностей длины 4 с предпочтительным располо
жением определенных групп нуклеотидов в различных позициях
четверки. Рассматривалось 15 групп нуклеотидов, встречающися в
позициях анализируемых четверок (см.табл.2)-
В данном блоке реализована одна стратегия поиска. Исходно
- 230 -
в последовательностях находятся все четверки различных
типов • Расчет значений характеристик, соответсвующих опреде-
определенным четверкам, производится по следующей формуле.-
V
/¦(*)
D)
Здесь »к - значение к -той характеристики ,-
и. - количество четверок t -того типа в
последовательности;
г{х) - весовые функции, определяющие вес четверок,
локализованных в позиции х.
В данном блоке реализовано 12 вариантов весовых функций
(рис-4), с помощью которых оценивается предпочтительное распо-
расположение информационно значимых четверок в последовательностях
функциональных сайтов-
2.4 КАЧЕСТВЕННАЯ ХАРАКТЕРИСТИКА СТРАТЕГИЙ АНАЛИЗА
ДАННЫХ
Каждая из рассмотренных моделей расчета характеристик
имеет одну или несколько стратегий поиска информационно
значимых характеристик.
Стратегии задается набором из л процедур ГРA>,Р<2),-",
f" ); i-тая процедура в конкретной реализации может выполнять
одно из к(" возможных однотипных преобразований информации и
передавать результат работы следующей процедуре, как показано
на схеме представленой ниже.
Р
где 51E2,...?п - индексы выполняемых процедур E,= 1, К).
На первом шаге задаются начальные значения индексов
?, = '.?,=*. ...?=?; выполняются последовательно все л ггроце-
дур и производится вычисление характеристики н(?,.?2.. .?п). На
следующем шаге значения индексов изменяются и система вновь
переходит к выполнению заданных процедур. Таким образом расчи-
расчитываются поочередно новые характеристики, каждая из которых
соответствует набору индексов f?,<?2 ••• Zn> , где i.s 5, s
- 231 -
к'" , то есть производится поиск информационно значимых ха-
характеристик в пространстве, задаваемом набором процедур
> р<
2.5 БЛОК ПЕРВИЧНОЙ ИНТЕРПРЕТАЦИИ РЕЗУЛЬТАТОВ
Данный блок представляет собой набор программ, которые
облегчают обработку и интерпретацию полученых результатов.
После завершения работы всей системы поиска закономернос-
закономерностей, найденые информационно значимые характеристики записыва-
записываются в промежуточные файлы в виде наборов индексов, использу-
используемых в различных стратегиях поиска. В блоке первичной интер-
интерпретации результатов данные характеристики перекодируются в
представление, доступное для пользователя.
В зависимости от решаемой задачи пользователь может с
помощью предлагаемых программ посмотреть конкретнуг локализа-
локализацию найденных информационно значимых детерминант в последова-
последовательностях функциональных сайтов; провести расчет вероятностей
возникновения по случайным причинам тех или иных свойств выяв-
ных детерминант.
Данный блок первичной интерпретации результатов оказы-
оказывает значительную помощь при генерировании и проверке различ-
различных гипотез о механизмах функционирования исследуемых функци-
функциональных сайтов.
3. РЕЗУЛЬТАТЫ АНАЛИЗА ВЫБОРОК ФУНКЦИОНАЛЬНЫХ САЙТОВ
ПРОКАРИОТИЧЕСКИХ ГЕНОВ
Для демонстрации работоспособности разработанной системы
мы провели предварительный анализ двух выборок функциональных
сайтов прокариотических генов; 1) промоторов E.coii некоторых
фагов и плазмид, 2) сайтов инициации трансляции мРНК E.coii.
Первая выборка содержала 40, а вторая J6 последова-
последовательностей. Для выборки промоторов брались участки последова-
последовательностей от -too до точки инициации транскрипции. Последова-
Последовательности сайтов инициации трансляции содержали по бо нуклео-
тидов к 5'- и з'- концу мРНК от инициаторного кодона-
- 232 -
3.1 АНАЛИЗ ЧАСТОТ ВСТРЕЧАЕМОСТИ МОНО- И ОЛИГОНУКЛЕОТИДОВ
В ВЫБОРКЕ ПРОМОТОРОВ
В качестве выборки последовательностей, не относящихся к
промоторам, использовался набор случайных последовательностей
с частотами нуклеотидов, такими же как и в выборке промоторов.
Анализ выявил наличие предпочтительного использования
динуклеотида аа в промоторах по сравнению со случайными пос-
последовательностями. Полезность данной характеристики оказалась
не большой (р=0.35). то есть, эта характеристика хотя и
является информативной, номожет быть использована только в
сочетании с другими характеристиками.
Поэтому для уточнения полученой закономерности был прове-
проведен анализ частот олигонуклеотидов в 5' из* направлениях от
каждой пары ал в последовательности.
Было выявлено 10 информационно значимых характеристик
(табл.2), соответствующих олигонуклеотидам, присутствующим с
низкой частотой в 5'- и 3'- направлениях от пар аа в промото-
промоторах. В большинстве случаев это были тугоплавкие олигонуклеоти-
ды (см. табл. г). По-видимому, данные характеристики отражают
наличие в промоторных зонах специфических легкоплавких
участков.
Таблица 2. Информационно значимые характеристики, соответст-
соответствующие олигонуклеотидам, присутствующих с низкой
частотой
олигонуклеотиды,
с низкой частотой
в 5'- направлении
от па аа
AGG
лес
AG
ТС А
GTC
СТА
CGC
олигонуклеотиды.
с низкой частотой
в 3'- направлении
от пар аа
TG
АС
AGG
3.2 АНАЛИЗ ПОВТОРЕННОСТЕЙ РАЗЛИЧНОГО ТИПА В ПРОМОТОРАХ
Некоторые результаты этого анализа приведены на рисунке 6.
233-
Шли выявлены информационно значимые характеристики, отражающие
повторенное™ всех < типов для различных расстояний между
повторенностями.
Наличие прямых и инвертированных повторов в структуре
промоторов было показано ранее различными авторами
[9,10]. Повторы четвертого типа (см.рис. бг.), которые можно
назвать прямыми комплементарными повторами, выявлены нами
впервые.
Рисунок 6. Наиболее информационно значимые повторенное™,
в последовательностях промоторов. Стрелками
показаны участки последовательностей, по которым
производилось сравнение. Цифрами показаны
расстояния между началами повторенностей.
а) прямые повторенное™; ь) симметричные;
с) инвертированные; d) прямые комплементарные.
3.3 АНАЛИЗ ЧАСТОТ ВСТРЕЧАЕМОСТИ ПОДПОСЛЕДОВАТЕЛЬНОСТЕЙ
ДЛИНЫ 4
Эта модель представления фукциональных сайтов проанализи-
проанализирована нами наиболее подробно. Исследовались промоторы и сайты
инициации трансляции. Во обоих случаях в качестве альтерна-
альтернативной выборки использовались наборы случайных последователь-
последовательностей с теми же частотами нуклеотидов, что и в реальных функ-
функциональных сайтах. Некоторые рэзультаты анализа приведены в
таблице з (а и ь).
- 2М-
Таблица з. Примеры информационно значимых подпоследователь-
подпоследовательностей длины 4, выявленных в функциональных сайтах
Г а)
G/A
G/A
Ж
С/А
—С
А/Т
Ы
А/С
А/С
С
четверки
нуклеотидов
А/Т
А/Т
С/А
А/Т
G/A
А/С
Ж
ж
А/С
А/С
А/С
А/Т
А/С
А/Т
-С
промоторы
л
я
А/С
А
А/С
А/С
четверки
однобук.
(из табл
R V Н
R V Н
N R V
R W М
D R W
V М D
сайты инициации
А
G/A
А
А/С
С
G/A
Н N А
П N R
СНА
В
коде
. 1)
А
N
Н
А
Н
М
номер
функции
(ИЗ рИС.2)
г
2
2
3
4
7
трансляции
н
с
R
1
2
9
полез-
полезность
.578
.640
.700
.676
.545
.587
.567
.636
.602
В промоторах в результате анализа был выявлен набор ин-
информационно значимых четверок (табл. за). Интересно отметить,
что в большинстве выявленных четверок содержится следупцее со-
сочетание нуклеотидов в трех последовательных позициях: RVH, где
R=(A или G) - пурины (большие по размерам), v=(a или Т)
легкоплавкие, я=(Л или С) - аминоподобные куклеотиды (несут
избыточный положительный заряд [11]). Это сочетание встречалось
в 90 % выявленых четверок.
Результаты анализа функциональных сайтов инициации
трансляции представлены в таблице зь. Почти все выявленные
четверки с коэффициентом полезности р > 0..5 оказались
насыщены нуклеотидами, несущими положительные заряды.
4. ЗАКЛЮЧЕНИЕ
4.1 ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Проведенный анализ показал работоспособность представ-
леной системы. На её основе проанализированы две выборки фун-
функциональных сайтов прокариотических организмов и выявлены
определенные закономерности в их структуре.
Во-первых, в промоторах некоторых прокариотических орга-
организмов найдены информационно значимые характеристики, связан-
- 235 -
ные с наличием повторенностей различного типа (прямые, инвер
тированные, симметричные и прямые комплементарные повтореннос-
ти). Известно, что прямые и инвертированные повторы могут
играть определенную роль в процессе функционирования промото-
промоторов [9,10], а именно - в процессе образования открытого комп-
комплекса ( структуры, содержащей протяженный расплавлений участок
ДНК в районе точки инициации транскрипции). Инвертированные
повторы часто встречаются в различных функциональных сайтах,
и могут служить для распознавания этих участков димерными
белками [12]. Функциональное значение симметричных повторов
может быть связано с формированием так называемой я-формы ДНК.
[13]. Необходимо отметить, что симметричные и прямые
комплементарные повторы не могут возникать с помощью блочных
перетасовок фрагментов ДНК (дупликаций или инверсий), и видимс
возникают конвергентным путем.
В промоторах также выявлены информационно значимые тройки
нуклеотидов. Мы предполагаем, что такие тройки нуклеотидов
могут влиять на процесс плавления ДНК в районе промотора.
Гипотетический механизм такого плавления под действием белка,
несущего положительно заряженные группы, представлен на рисун-
рисунке 8. Как показал анализ на основе весовых функций, кончен
трация таких троек растет в направлении к точке инициации
транскрипции (рис. 9).
Рисунок 8. Гипотетический механизм плавления ДНК в районе
промотора под действием белка, несущего положи-
положительно заряженные группы
-236-
промотор А2 фага Т7
-30 -80 -70 -60 -SO -40
CCAATCGACACCGGGGTCAACCGGATAAGTAGACAGCCTGATAAGTCGCACGAAAAACAG
-30 -20 - 10
GTATTGACААСATGAAGTAACATGCAGTAAGATАСАААТС
промотор RRN X Р2 E.coli
-ЭО -80 -70 -60 -SO -40
CGGCGGATGTGAATCACTTCACACAAACAGCCGGTTCGGTTGAAGAGAAAAATCCTGAAA
-30 -20 - 10
TTCAGGGTTGACTCTGAAAGAGGAAAGCGTAATATACGCC
промотор PR фага х
-90 -80 -70 -60 -50 -40
TTTCTTTTTTGTGCTCATACGTTAAATCTATCACCGCAAGGGATAAATATCTAACACCGT
-30 -20 - 10
GCGTGTTGACTATTTTACCTCTGGCGGTGATAATGGTTGC
Рисунок э. Примеры расположения информационно значимых четверок
RVMN в трех промоторах
Таким образом, промоторы насыщены информационно значимыми
тринуклеотидными фрагментами, которые могут влиять на их функ-
функционирование. Существенно,что эти фрагменты не локализованы
в строго опеределенных местах функциональных сайтов, а диспер-
диспергированы по ним (см. рис. э).
Можно предполагать, что функционирование промоторов осу-
осуществляется на основе двух групп факторов: облигатных и
факультативных. К облигатным, то есть присутсвупцим в обяза-
обязательном порядке, можно отнести консервативные блоки в последо-
последовательностях функциональных сайтов (например, -10 и -35 блок в
промоторах E.coli ) . Возможно, что именно облигатные
структуры обеспечивают базовый уровень активности функциона-
функциональных сайтов.
К факультативным факторам (присутсвупцих в варьируемых
- 237-
количествах и локализованных в различных местах) можно, напри-
например, отнести выявленные нами олигонуклеотилные фрагменты.
Можно предполагать, что роль таких факторов заключается в
модуляции работы промоторов, что обеспечивает их функциональ-
функциональную специфичность, соответствующую определенному уровню генной
активности.
4.2 СИСТЕМА ПОИСКА ИНФОРМАЦИОННО ЗНАЧИМЫХ ХАРАКТЕРИСТИК,
КАК ОСНОВА ЭКСПЕРТНОЙ СИСТЕМЫ АНАЛИЗА ФУНКЦИОНАЛЬНЫХ
САЙТОВ
Разработанная система поиска закономерностей в функцио-
функциональных сайтах является подсистемой разрабатываемой экспертной
системы анализа функциональных сайтов в нуклеотидных последо-
последовательностях. Обшая схема разрабатываемой экспетной системы
приведена на рисунке ю. Уже созданные блоки показаны толстыми
линиями, а разрабатываемые - пунктирными линиями.
Разрабатываемая экспертная система предназначена для
генерации и накопления знаний по функциональным сайтам и ис-
использования накопленых знаний для поиска функциональных сайтов
в неизвестных последовательностях.
^интеллектуальный"'
1 интерфейс '
I (проектирование
стратегий поиска
СИСТЕМА ПОИСКА
ЗАКОНОМЕРНОСТЕЙ
в функциональных
сайтах
2)автоматизиро-
I ванный поиск |
. функц. сайтов. .
. 3)поиск функц. .
I сайтов в визу- I
альнон режиме
1 альн
I БАЗА ЗНАНИЙ |
( по функциональ-!
, ним сайтам
, СИСТЕМА ПОИСКА ФУНКЦ. ,
, САЙТОВ В ПОСЛЕДОВА- ,
, ТЕЛЬНОСТЯХ j
Рисунок ю. Общая схема создаваемой экспертной системы анализа
функциональных сайтов в нуклеотидных последова-
последовательностях
Описанная нами система поиска закономерностей является
первой версией блока генерации знаний экспертной системы. Прк
- 238 -
анализе выборок функциональных сайтов с помощью этой системы
выявляются наборы информационно значимых характеристик, на
основе которых в блоке построения методов предсказания могут
строится методы предсказания функциональных сайтов. Наиболее
эффективные методы предсказания должны накапливаться в Базе
знаний по структурной организации функциональных сайтов-
Накопление в Базе знаний методов предсказания даст воз-
возможность быстро и эффективно производить поиск и анализ функ-
функциональных сайтов различных типов в неизвестных последователь-
последовательностях. Поиск функциональных сайтов будет осуществлятся в двух
режимах: в автоматическом и в режиме активного диалога с поль-
пользователем.
Создаваемая экспертная система будет содержать так назы-
называемый интеллектуальный интерфейс - систему "общения" пользо-
пользователя с экспертной системой (см.рис. 10). В нем будет предус-
предусмотрена возможность проектирования стратегий поиска закономер-
закономерностей. Пользователь по своему желанию сможет подключать те
или иные стратегии поиска, изменять вид весовых функций, то
есть практически осуществлять программирование процесса поиска
закономерностей.
ЛИТЕРАТУРА
[1] Staden R. // Nucl.Acids Res., 1984, V.12, p.S0S-Si9.
[2] Высоцкая Г.С, Гусев В.Д., Куличков В.А. // в сб."Теорети-
сб."Теоретические исследования и банки данных по молекулярной биоло-
биологии и генетике.", 1986, с.54-58.
[3] Milligan M.E., Hawley D.K., Entriken R., McClure W.R. //
Nucl.Acids Res., 1984, V.12, p.789-800.
[4] Шахмурадов И.А., Колчанов Н.А., Соловьёв В.В., Ратнер В.А.
// Генетика, 1986, т.ххп, с.357-367.
[5] Sherer G.F.E., Walktnshaw M.D., Arnott S., Когте D.J. //
Nucl.Acids Res., 1980, V.8, p.3895-3907.
[6] Уотермен Д. Руководство по экспертным системам: Пер. с
англ. // М.,Мир, 1989, 388 с.
[7] Пономаренко М.П., Орлов Ю.Л. Экспертная система быстрой
оценки полезности использования произвольных характерис-
характеристик биополимеров для их классификации. // в сб.Компьютер-
- 239 -
ные методы анализа генетических макромолекул. Проблемы
интеллектуализации., Новосибирск: ИЦиГ СО АН СССР, 1989.
[8] Гаек П..Гавранек Т, Автоматическое образование гипотез:
математические основы общей теории. // М., Наука, 1978
[9] Артемьев Н.В., Васильев Г.В., Гуревич А.И. //
Биоорганическая химия, 1983, т.9, с.1544-1557.
[10] Кель А.Э., Колчанов Н.А. // в сб. Теоретические
исследования и банки данных по моекулярной биологии и
генетике., Новосибирск: ИЦиГ СО АН СССР, 1986, с.87-90.
[11] Зенгер В. Принципы структурной организации нуклеиновых
кислот. Пер. с англ. // М..Мир, 1987, 584 с.
[12] Льган Б. Гены. // М., Мир, 1987.
[13] Hlrkin et al . // Nature, V.330, N. 6147, 1987, p.495-497.
SUMMARY
THE COMPUTER SYSTEM OF FUNCTIONAL SITES" ANALYSIS IM
POLINUCLEOTIDE SEQUENCES
Kel A.E., Ponomarenko M.P., Orlov Yu.L., Mlschenko T.M.
Kolchanov N.A.
The present paper Is devoted to description of computer
system for functional sites analysis In pollnucleotlde sequen-
sequences. This system search for different types of characteristics
In ollgonucleotides and repeats distribution In functional
sites. Significance of reveald characteristics is estimated by
the complex of statistic analysis programmes. Out of a great
number of characteristics the set of most valuable ones Is
chosen. On their basis the search for functional sites of a
certain type in nucleotlda sequences is possible.
The system consists of several programmed blocks. 1) Block
of controlling the process of characteristics search . 2)
Block of characteristics calculations. 3) Block of statistical
analysis. 4) Block of the Initial interpretation of results.
in Fig.2 block-scheme of the whole system Is presented.
In the present system three strategies of valuable chara-
characteristics search are realized:
1) Analysis of of different types of repeat in nucleotide
sequences. 2) Analysis of ollgonucleotides frequency of
different length. 3) Analysis preferable localization of
separate nucleotide groups in different positions of four
nucleotide length subsequences.
Statistical analysis Is carried out by calculating the
utility of a given characteristics at classification of
functional sites of the present type. The description of the
block of statistic analysis Is given in section 2.
Characteristics value shoving the presense of repeats Is
calculated by the formulas B} and C). Characteristics value
of shoving ollgonucleotide frequencies are calculated by for-
formulas D) and E).
Tvo functional sites samplings are analysed by the vorked
out system: Dpromoters of E.coll. some phages and plasmids,
2) Initiation translation sites. In these functional sites
- 241 -
informatively-valuable repetitions and ollgonucleotldes cl
certain type dlsperged on the whole sequency length we г
found. In Fig. 8 hypotetlc mechanism of the toim
ГОШШ. in г i.y . a tijrv .
oligonucleotlde participation In promotors functioning С in
process of the open complex forming) Is shown.
The worked out system of searching for regularities
functional sites is a subsystem of an expert system
functional sites analysis in nucleotlde sequences. The gem
scheme of this expert system is given in Fig.5.0.
the
- 242 -
КОМПЬЮТЕРНАЯ СИСТЕМА ДЛЯ ИЗУЧЕНИЯ ДНК(РНК)-
ВЗАИМОДЕЙСТВУЮЩИХ БЕЛКОВ.
Бенюх Д.Н., Пономаренко М.П,, Колчанов Н.А., Орлов С.Л.
Институт Цитологии и Генетики СО АН СССР,г.Новосибирск.
1. ВВЕДЕНИЕ
Молекулы ДНК и РНК играют в клетках живых организмов
исключительно важную информационную и структурную роль. Их
функционирование во многом определяется взаимодействием с
различными классами белковых молекул ( ДНК-, РНК- связывающими
бедками ). Поэтому понятен возрастающий интерес к проблеме
белок-нуклеиновых взаимдействий и к изучению белков,
взаимодействующих с ДНК(РНК). Отметим две наиболее важные
задачи, возникающие в этой области:
1) разработка методов выявления белков, взаимодействующих
с ДНК(РНК), на основе анализа их аминокислотных
последовательностей;
2) выявлекие в аминокислотных последовательностях белков
структурно-функциональных детерминант, обеспечивающих
выполнение указанных функций.
Структурно-фунуциональной детерминантой или просто
детерминантой будем называть участок первичной структуры
белка, обладающий специфическим составом аминокислотных
остатков, характеризующийся определенной локализацией в
первичной структуре и имеющий отношение к' определенной
биологической функции белка. Функциональный домен белка
(минимальная структурная единица, способная выполнять
определенную биологическую функцию) может состоять из
нескольких структурно-функциональных детерминант. Таким
образом, аминокислотным последовательностям белков определен-
определенного функционального семейства может быть поставлен в
соответствие набор структурно-функциональных детерминант,
обрузующий своеобразный структурно-функциональный "портрет"
данного семейства. Для такого описания семейства необходимо
накопление всей доступной информации о нем в базе знаний. В
свою очередь, это требует разработки библиотеки программ для
высокоточного распознавания структурно-функциональных детер-
детерминант. Эти программы используют информацию базы знаний и
способны получать новые данные о структурно-функциональной
организции как белков данного функционального класса, так и
новых, ранее не изученных семейств белков.
В настоящей работе дано описание компьютерной системы,
основанной на изложенных выше принципах, и предназначенной для
изучения белков, взаимодействующих с ДНК(РНК). Эта система
содержит базу знаний и комплекс программ, распознающих
структурно-функциональные детерминанты.
2. БАЗА ЗНАНИЙ ДЛЯ ФУНКЦИОНАЛЬНОГО СЕМЕЙСТВА БЕЛКОВ
При построении базы знаний (БЗ) для конкретных семейств
белков нами использовалась технология интегрированных баз
знаний. Необходимость ее применения диктовалась несколькими
требованиями:
1) простотой реализации базы знаний для конкретного
семейства белков;
2) пригодностью для быстрого тиражирования типовой БЗ;
3) уникальностью информации о каждом семействе белков.
Логическая схема типовой базы знаний для функционального
семейства белков приведена на рис. I. При наличии достаточно
полной информации о функциональном семействе в литературных
источниках, поля базы знаний, "несущие" информацию о наличии и
расположении структурно-функциональных детерминант, заполня-
заполняются посредством реферирования соответствующей литературы. В
качестве второго источника информации используются инстру-
инструментальные программные средства продукции знаний, например,
экспертная система для структурно-функциональной классификации
аминокислотных последовательностей [ I]. Наконец, в качестве
эффективного источника информации о структурно-функциональных
детерминантах при формировании баз знаний для новых семейств
белков может использоваться библиотека программных средств,
предназначенных для высокоточного распознавания структурно-
функциональных детерминант в исследовавшихся ранее семействах
белков. В этом смысле, создаваемая нами компьютерная система
- 244 -
Описание семейства
Описание белка
Семейство
белков
Название
Процесс
|
| Функция
j
j Клеточная
I структура
! Сайты связы-
I вания на ДНК
Белок
Название
Организм
Культура
клеток
(ткань)
Структурная
организация
Детерминанта
Название
Описание
Метод распо-
распознавания
Источник
информации
Первичная
структура
белка
Структурная
организация
Словарь
названий
Словарь
организмов
Словарь
культур
(тканей)
Список
источников
Список амино-
аминокислотных
последова-
последовательностей
Аминокислот-
Аминокислотная последо-
последовательность
Детерминанта
Длина
Начало
Конец
Рис.1.Логическая схема типовой базы знаний для функционального
семейства белков. Обозначения:
[CJ - Название списка информации. ? - Информационное поле.
Отношения: —»- -"один к одному"; —>-» "один ко многим",
- 245
обладает способностью к "самообучению".
База зданий для семейства белков содержит ряд "тради-
"традиционных" информационных полей:
1) название семейства;
2) название функции белков данного семейства;
3) название процесса, в котором участвуют белки данного
семейства;
4) название клеточной структуры, в которой обнаружены эти
белки;
5) название участков ДНК, с которыми взаимодействует
белок;
6) аминокислотная последовательность и ее длина;
7) название белка;
8) название организма, из которого выделен белок;
9) название культуры клеток (ткани), из которой выделен
белок;
Ю) ссылка на источник информации.
Кроме того, база знаний включает "нетрадиционные" поля,
описывающие каждую структурно-функциональную детерминанту:
1) краткое название структурно-функциональной
детерминанты;
2) полное описание этой детерминанты в текстовом виде;
3) локализацию детерминанты на аминокислотной последо-
последовательности (начало и конец).
Например, база знаний для семейства гистонов Н1 имеет
следующие варианты заполнения полей 1) и 2), описывающих
структурно- функциональные детерминанты II, III, VI и VII (см.
рис. 2).
Детерминанта II. Поле I: "N-концевой домен"; поле 2:
"N-концевой домен взаимодействует с ДНК; длина - 5-40 амино-
аминокислотных остатков; богат положительно заряженными аминокис-
аминокислотными остатками - Р, к, а также, Р и V; вариабельный".
Детерминанта III. Поле I: "Центральный гидрофобный'
домен"; поле 2: "Центральный гидрофобный домен формирует
глобулу белка, его длина 60 90 аминокислотных остатков;
аминокислоты Рим отсутствуют; консервативный »
гидрофобный; имеет конафиатившм; качало (G/H) P (P/V) Y и
повторы длиной 10-1е) аминокислотных остатков".
- 246 -
ГГ
11
\т
ГШ~|
ГШ]
Рис.2. Схема расположения структурно-функциональных
детерминант гистонов Ш. Обозначения:
НН - аминокислотная последовательность гистона Н1;
полная последовательность гистона Н1;
N-концевой домен;
центральный гидрофобный домен;
с-концевой домен;
гидрофобная область N-концевого домена;
область перехода N-концевого домена в центральный
гидрофобный домен;
консервативное начало центрального гидрофобного домена;
I -
II -
III -
IV -
v -
VI -
VII -
VIII - повторы центрального гидрофобного домена;
IX - вариабельная область центрального гидрофобного домена;
X - область перехода центрального гидрофобного домена в
С-концевой домен;
XI - повторы 1-го типа с-концевого домена;
XII - повторы 2-го типа С-концевого домена.
Детерминанта VI. Поле I: "Граница между N-концевым и
гидрофобным доменами"; поле 2: "Граница между Ы-концевым и
гидрофобным доменами характеризуется резкой сменой физико-
химических свойств аминокислотной последовательности и
консервативным разделяющим участком (G/H) P (P/v) Y ".
Детерминанта VII. Поле I: "Консервативное начало
гидрофобного домена"; поле 2: "Начало гидрофобного домена имеет
консервативный консенсус - (G/H) P (P/V) Y".
Помимо этого, в базу знаний заносится информация о
методах распознавания структурно-функциональных детерминант,
построенных с использованием перечисленной выше информации:
1) название метода распознавания структурно-функци-
структурно-функциональной детерминанты;
- 247 -
2; описание характеристик и способа применения этого
метода распознавания в текстовом виде.
Рассмотрим в качестве примера "портрет" семейства
гистонов Н1 в терминах структурно-функциональных детерминант.
Схема расположения структурно-функциональных детерминант
гистонов Н1 приведена на рис.2. Детерминанты 1-1V и vl-Xll
найдены и размечены на основе литературных данных [2,3,4,5].
Детерминанта v (гидрофобная область ы-концевого заряженного
домена) обнаружена и локализована при помощи экспертной
системы [ 1].
3. БИБЛИОТЕКА ПРОГРАММ ДЛЯ РАСПОЗНАВАНИЯ СТРУКТУРНО-
ФУНКЦИОНАЛЬНЫХ ДЕТЕРМИНАНТ
Разрабатываемая компьютерная система ориентирована на
создание таких методов распознавания структурно-функциональных
детерминант, которые позволяют выявлять эти детерминанты во
всех без исключения последовательностях белков данного
функционального семейства. Учитывалось, что реальная
локализация структурно-функциональных детерминант в каждой
конкретной последовательности семейства может варьировать на
определенном интервале. Предпологалось, что границы
варьирования не превышают 10% от длины детерминанты.
Построение распознающей программы для структурно-
функциональной детерминанты определенного типа осуществляется
в четыре этапа.
3.1 ГЕНЕРАЦИЯ МОДЕЛЕЙ ОПИСАНИЯ СТРУКТУРНО-ФУНКЦИОНАЛЬНЫХ
ДЕТЕРМИНАНТ
Построение распознающей программы для определенной
структурно-функциональной детерминанты начинается с визуаль-
визуального анализа участка аминокислотной последовательности, на
который проецируется данная структурно-функциональная детер-
детерминанта. Этот анализ проводится для выявления качественной
картины встречаемости и распределения по длине указанного
участка каждого из 20 типов аминокислотных остатков. Такая
процедура проделывается для каждой аминокислотной последова-
- 248 -
тельности из базы знаний. После этого, для всего семейства
белков строится перекодировка W из двадцатибуквенного алфавита
в частотную шкалу, отражающая в условных единицах качественную
картину специфичности аминокислотного состава данной
детерминанты. Пример таких перекодировок для ы-концевого
домена гистонов Н1 приводится на рис.3. Далее, в единицах этой
перекодировки строится профиль распределения аминокислотных
остатков вдоль участка детерминанты. Затем производится выбор
способа сглаживания G и функции предпочтения Т(Ь) для данного
профиля, которые качественно отражают распределение специфи-
специфических аминокислотных остатков по длине детерминанты. Примеры
способов сглаживания приведены на рис. 4, графического пред-
представления функций предпочтения - на рис. 5.
Таким способом описывается качественная картина состава и
распределения аминокислотных остатков на изучаемом участке
аминокислотной последовательности. Заметим, что подобная
качественная картина, являющаяся исходной информацией для
построения распознающих программ, может быть построена любым
исследователем, имеющим лишь самое общее представление о
структуре и функции белков изучаемого семейства и овладевшем
несложными навыками работы с компьютерной системой.
Поскольку предлагаемый способ описания детерминанты носит
качественный характер, для получения оптимального варианта
распознающей программы требуется перебор значительного
количества перекодировок, способов сглаживания и функций
предпочтения. Каждая комбинация указанных компонент фактически
задает модель исследуемой структурно-функциональной детер-
детерминанты. Действительно, конкретный вариант перекодировки
качественно отражает задаваемое исследователем предпочтение
функциональной значимости одних аминокислотных остатков над
другими. Выбор способа сглаживания определенным образом
учитывает характер локальных взаимодействий между остатками.
Наконец, значимость одних районов структурно-функциональных
детерминант перед другими задается функцией предпочтения.
Для поиска оптимального варианта необходим перебор всех
трех перечисленных выше компонент. Например, можно получить
до Ю вариантов перекодировок, до 5 вариантов способов
-249 -
а) f 2. { a,p.к }
1, { G.S.T.R }
W = ^ 0, { V,L,M,D,N,E,Q
i -2, { I,C.F,Y,W,H }
б) f 3. { К }
2, { A.P.R }
1, { V.L.M.S.T.G }
W = ^ -1, { I.D.N.E.Q }
-2. { F.Y.H }
I -3, { C.W }
в)
W =
г)
2,
1,
О,
-2,
W =
4,
3,
2,
1 ,
i
' 0.
-1 ,
-2,
I -э.
А,К }
P,S,T,R,G )
V,L,M,D,N,E,Q
I.CF.Y.W.H }
К }
R )
А }
; p,s,t,g }
; v.l.m >
: н }
[ I.C.F.Y.W >
С D.N.E.Q )
Рис.3. Примеры перекодировок аминокислотных проследовательнос-
тей. Приведено 4 перекодировки для ы-концевого домена
гистонов Н1. w - значение перекодировки.
a)
= 1.L
• = {
max
б) G
В) G. =
.,. Cj. Cltl ) , С
mln ( Ci.,, Clf Clt1 ) . С
1= 1 ,L/2
i= L/2.L
lt1
= 1 ,1
Рис.4. Примеры формул для сглаживания: а) - отсутствие сгла-
сглаживания; б) - максимум из трех значений перекодировки
до средины участка и минимум из трех - после; в)
усреднение по трем значениям ттерекодировки. Обозначе-
Обозначения: ь - длина участка последовательности; wt - значе-
значение профиля в i-й позиции;
профиля в i-й позиции.
- 250 -
- значение сглаженого
а)
л
б)
1
-1
в)
1
г)
1
1
Рис.5. Варианты графического представления функций предпочте-
предпочтения, а) и б) отражают предпочтение для одних амино-
аминокислотных остатков до средины участка и для других -
после; в) и г) задают большие веса остаткам в центре
участка. Длина участка нормирована на единицу.
сглаживания и до ю вариантов функций предпочтения. Это
осуществляется программой-генератором (схему фрейма для
программы-генератора см. на рис.6), на выходе из которой
получится ю * 5 * ю = 500 значений выходного параметра,
каждое из который характеризует определенную модель
представления структурно-функциональной детерминанты. Взвешен-
Взвешенное среднее для участка последовательности вычисляется по
формуле
. _ A Y*
где
G « 1
ъ± - длина детерминанты в i-й последовательности, Yj =
(L. ) - значение профиля в j-й позиции участка после
сглаживания и домножения на функцию предпочтения. Вычисление
-251 -
набор участков
последовательностей
набор
перекодировок
набор способов
сглаживания
—>—
—
\
Блок
переко-
перекодировки
1
Т
Блок
сглажи-
сглаживания
Блок выдачи
результата
\
Блок
вычис-
вычисления
харак-
терис-
теристики
X
\ \
набор функций
предпочтения
Блок
усред-
усреднения
Блок
пере-
перебора
I - поле фрейма, предназначенное для заполнения.
I | - функциональный блок фрейма.
Рис.6. Схема заполненного фрейма программы-генератора,
вычисляющей параметр X (для модели структурно-функци-
структурно-функциональной детерминанты). Схема дана в обозначениях [ 6].
характеристики X производится после заполнения фрейма для
программы-генератора.
3.2 АНАЛИЗ ПОЛЕЗНОСТИ ХАРАКТЕРИСТИКИ
В целях выявления характеристик для идентификации
структурно-функциональных детерминант с наибольшей
информационной значимостью, используется описанная программа-
генератор, которая формирует две выборки характеристик X. Одна
выборка {х^} вычисляется для участков аминокислотных
последовательностей белков данного семейства, содержащих
исследуемую структурно-функциональную детерминанту. Вторая
выборка {Х2> вычисляется для участков случайно отобранных
аминокислотных последовательностей из банка данных pip \ 7],
не принадлежащих к данному семейству. Анализ этих двух выборок
- 252 -
характеристик проводится с помощью экспертной системы для
быстрой оценки полезности [8]. Экспертная система разбивает
выборки характеристик {х,} и {х„} на независимые подвыборки
для обучения и контроля. Таким образом, все полученные
комбинации перекодировок, способов сглаживания и функций
предпочтения проходят тест на точность и достоверность
предсказания на независимых от обучения контрольных данных.
Поэтому вся информация о структурно-функциональных детер-
детерминантах базы знаний может быть использована для построения
методов распознавания.
В результате работы экспертной системы отбирается 5-10
выборок характеристик {X,} и {Х2>, которые разделяются с
наибльшей полезностью R. Фактически величина R характеризует
полезность рассматриваемого варианта модели ( комбинации
перекодировоки, способа сглаживания и функции предпочтения )
для целей распознавания структурно-функциональных детерминант,
так'как выборки характеристик {х;} и {х,} получены при помощи
данной модели. Одно из наилучших сочетаний перекодировки,
способа сглаживания и функции предпочтения с полезностью R
0,88, полученное при построении методов распознавания
N-концевого заряженного домена гистонов Н1, приведено на
рис.?. Кроме того, для отобранных характеристик {Х%} и (Х2},
вычисляется г, как среднее от {Х^} и {Х2>. Z представляет
собой порог, разделяющий выборки {X.J и {Х2>.
а)
w =
б)
f 2, { А,Р,К } В)
1, { G.S.T.R }
*> 0, { V,L,M,D,N,E,Q }
, -2, { I.C.P.Y.W.H }
Рис.7. Пример компонент одной из лучших моделей для
Ы-концевого домена гистонов Ш.
описания
-253-
3.3 ЗАПОЛНЕНИЕ ТИПОВЫХ ФРЕЙМОВ ДЛЯ РАСПОЗНАВАНИЯ СТРУКТУРНО -
ФУНКЦИОНАЛЬНЫХ ДЕТЕРМИНАНТ
Разработанная нами схема типового фрейма для программы
распознавания структурно-функциональных детерминант в
аминокислотных последовательностях белков приведена на
рис.8. Фрейм заполняются информацией, полученной на предыдущем
этапе: сочетанием перекодировки, способа сглаживания, функции
предпочтения и значением порога. Предлагаемая схема отличается
от схемы на рис.7 отсутствием "Блока перебора",
осуществляпцего в программе-генераторе перебор всех возможных
сочетаний компонентов модели структурно-функциональной
детерминанты. Кроме того, фрейм для распознающей программы
имеет "Блок принятия решения", который осуществляет сравнение
участок
последовательностей
перекодировока
г
Блок
переко-
перекодировки
способ
сглаживания
Блок
сглажи-
сглаживания
Блок выдачи
результата
функция
усреднения
Блок
усред-
усреднения
пороговое
значение
' |
Блок принятия
решения
- поле фрейма, предназначенное для заполнения.
- функциональный блок фрейма.
Рис. 8. Схема заполненного типового фрейма программы, для
распознавания структурно- функциональной детерминанты.
- 254-
с пороговым значением и вычисление "веса" исследуемого
участка. "Вес" участка пропорционален полезности R для данной
модели представления.
Последним шагом создания распознающей программы является
ее тестирование на базе данных. В таблице 1 приведены
результаты сканирования по последовательностям базы знаний
для гистонов Н1 с помощью программы, распознающей Ы-концевой
домен этих гистонов. При этом ставились следующие задания:
1) выяснить, во всех ли белках данного семейства можно
идентифицировать исследуемую структурно-функциональную
детерминанту;
2) установить точность локализации выявленной структурно-
функциональной детерминанты относительно ее локализации в БЗ.
Указанный домен был найден во всех последовательностях
БЗ. Точность позиционирования составила | L-L*|*1OO/ L = 92$.
Табл.1. Результаты позиционирования программы, распознающей N-
концевой домен гистонов Н1.
Номер
в базе
знаний
1
2
" 3
4
5
6
7
8
9
10
11
12
13
14
15
16
сред-
среднее
Последняя позиция N-концевого домена
По базе знаний
5
5
34
38
31
38
39
26
26
36
37
33
35
21
30
33
Ь= 29,1
По результатам сканирования
6
10
37
39
31
43
43
27
30
6
9
38
40
23
19
38
L*= 26,8
- 255-
4. ПРИМЕР ИСПОЛЬЗОВАНИЯ СИСТЕМЫ ДЛЯ ИЗУЧЕНИЯ ДНК(РНК)-
ЮАИМОДЕЙСТВУЩИХ БЕЛКОВ
Помимо описанной выше программы для распознавания N-
концевого домена, нами была разработана программа выявляющая
область перехода N-концевого домена в центральный гидрофобный
домен. Эта программа характеризуется высокой точностью
локализации указанной детерминанты.
На основе описанной выше программы распознавания N-
концевого домена гистонов Н1 и программы, распознающей область
перехода N-концевого домена в центральный гидрофобный домен и
построенной на основе предлагаемого нами подхода, была
разработана программа способная выявлять комплексную
детерминанту, состоящую из двух указанных структурно -
функциональных детерминант. С помощью этой программы было
проведено сканирование Банка PIR, целью которого было уста-
установление специфичности указанной детерминанты для:
а) гистонов Н1;
б) гистонов всех имеющихся в банке семейств { Н1, н?а,
Н2В, НЗ, Н4, Н5 );
в) всех ДНК(РНК) взаимодействующих белков банка PIR.
Результаты анализа приведены в Табл.2.
Всего программой было отобрано 157 аминокислотных
последовательностей, содержащих описанную выше детерминанту
{ из 6210 последовательностей Банка Pin ). Все отобранные
последовательности можно разделить на 3 группы.
Относительно первой группы (* 1 - 7 в Табл.2) достоверно
известно, что данные белки взаимодействуют с ДНК(РНК) [ 8].
1) База данных PIR содержит 19 последовательностей
гистонов Н1, из которых 16 было отобрано нами в базу знаний
для построения методов распознавания. Оставшиеся 8
последовательности не были использованы в базе знаний, так как
представляют собой фрагменты полных последовательностей
гистонов Н1. Из них 2 последовательности длиной менее 40
аминокислотных остатков не приринимались во внимание при
анализе баз данных, так как минимальная длина последова-
последовательностей при сканировании была равна 50-ти аминокислотным
остаткам. Что же касается 3-й последовательности, то в ней
-256-
Таол.2. Результаты сканирования по
распознающей комплексную
тексте)
базе данных PIR программы,
детерминанту.(пояснения в
Гру-
Группа
1
11
111
№
1
2
3
4
6
7
8
9
10
1 1
12
13
14
15
16
Названия семейств
и белков
His tone H1
His tone H2A, Н2В, НЗ
Н4, Н5
Nonhistone
chromosomal proteins
Eibosomal proteins
Initiatione fator IF-3
Н1з tidyl-tPNA-syntheta^e
Sigma ia protein
Virus coat proteins
Balbiani-ring-b
protein
Kinase-related
transforming protein
(ros )
Protein synthesis
inhibitor 11
Progesteron receptor
env polypotein
Hipothe-tical protein
Genome polyprotein
Othe proteins
Последовательностей
отобрано
17
59
4
15
1
1
1
14
1
о
1
1
1
2
2
35
всего в
банке
19
89
7
171
1
1
1
122
1
2
2
2
1
2
2
5787
Р
<10'10
ю-5
0.025
0.025
0.025
ю-6
0.025
0.0012
0.049
0.049
0.025
0.0012
0.0012
-
была распознана комплексная структурно-функциональная
детерминанта, характерная для гистонов Н1.
2) Из г= 89 последовательностей, принадлежащих другим
семействам гистонов 59 было отобрано, как содержащие комплекс-
комплексную детерминанту. Вероятность отбора к- 59 последовательностей
-257-
гистонов из 89 по случайным причинам, при условии, что всего
отобрано ,п= 157 последовательностей из d= 6210, можно оценить
из биномиального распределения
Р = 1
с* р"{1-р)п"\
где р = г / d.
Таким образом, р < 100. Этот результат позволяет сделать вывод
о наличии в большинстве последовательностей гистонов
комплексной детерминанты, состоящей из N-концевого домена и
области перехода N-концевого домена в центральный гидрофобный
домен, что соответствует литературным данным [2,3,4,5].
8) Банк PIR содержит 7 негистоновых хромосомных белков.
Из них в 4-х при сканировании была выявлена комплексная детер-
детерминанта, что позволяет сделать вывод о гистоноподобной
структуре ДНК- связывающего домена негистоновых хромосомных
белков. Вероятность такого события по случайным причинам
Р =• 10~5.
4) В Банке PIR содержится 1?1 последовательность
рибосомалышх белков. Из них было отобрано 15 последователь-
последовательностей. Вероятность такого события по случайным причинам
Р =» 10~5.
5) Остальные белки, о которых достоверно известно, что
они взаимодействует с ДНК(РНК) представлены фактором инициации
трансляции ( IF-3 ), гистидил-тРНК-синтетазой и сигма 1а субъ-
субъединицей РНК-полимеразы. Вероятности отбора этих после-
последовательностей по случайным причинам приведены в Таблице 2,
они тоже малы.
Вторая группа - белки, о которых мы не располагаем
информацией, позволяющей отнести их к ДНК (РНК)-
взаимодействупцим белкам из за неполноты сведений об этих
белках в базе данных PIR (J* 8 - 15 в Табл.2) . Тем не менее,
интересно, что в 14-ти белках оболочек вирусов из 122 имеющих-
имеющихся в базе, выявлена указанная комплексная детерминанта. Веро-
Вероятность отбора этих последовательностей по случайным причинам
Р « Ю~6. Этот результат, по нашему мнению, может свидетель-
свидетельствовать в пользу гипотезы о гистоноподбной структуре
N-жонцевого домена белков оболочек вирусов, которые участвуют
в упаковке вирусной ДНК(РНК).
-258-
Третью группу отобранных последовательностей представляет
белки, не взаимодействующие с ДНК (РНК) (Jf 16 в Табл. 2).
Таким образом, благодаря применению описанного в работе
подхода для представления знаний, достигнута высокая точность
распознавания комплексной детерминанты гистонов Н1, состоящей
из N-концевого домена и области перехода N-концевого домена в
центральный гидрофобный домен. Существенно также, что
предложенная технология позволяет выявлять белки, принад-
принадлежащие к группе ДНК(РНК)-взимодействуицих.
5.ЗАКЛЮЧЕНИЕ
Полученные результаты позволяют сделать следующие выводы:
1. Предложен новый подход для построения методов
классификации функциональных семейств белков. Этот подход
основан на применении концептуального представления экспертных
знаний [ 9] в молекулярной биологии. Отличительной чертой этого
подхода является отображение знаний в виде списков инфомации и
взаимодействующих с ними программ для представления и
обработки информации.
2. Другой отличительной чертой предложенного подхода
является применение качественных экспертных оценок для
формализации замеченных закономерностей строения аминокис-
аминокислотных последовательностей. Причем, эти оценки позволяют
получать количественные характеристики аминокислотных после-
последовательностей белков, эффективные для целей распознавания
структурно-функциональных детерминант.
3. В настоящей работе выявлены некоторые особенности
структурно-функциональной организации ДНК(РНК)-взаимодейству-
ДНК(РНК)-взаимодействующих белков.
4. Достигнута высокая точность распознавания структурных
особенностей аминокислотных последовательностей белков, что
позволяет считать эту технологию перспективной для решения
задачи функциональной разметки аминокислотной последователь-
последовательности. Это является особенно важным в связи с перспективой
полного секвенирования генома человека и некоторых
животных.Решение этой задачи потребует высокой точности
распознавания, так как предстоит осуществить фукциональную
-259-
идентификацию ~ 100 ООО генов и кодируемых ими белков.
Предлагаемая система для изучения ЛНК(РНЮ впаимо
действующих белков к настоящему времени реализована в виде
демонстрационного прототипа. Дальнейшее разитие системы будет
идти по еле душим направлениям.
1) Увеличение количества функциональных семейств белков в
базе знаний ( с перспективой охвата всех семейств ДНК(РНК)
взаимодействующих белков ).
2) Значительное увеличение количества распознающих
программ.
3) Реализация гибкого сценария распознавания структурно-
функциональных детерминант в диалоговом режиме.
4) Создание диалоговой автоматической системы для ведения
баз знаний и способов формализации знаний экспертов-биологов.
5) Создание диалоговой автоматической системы для
генерации характеристик аминокислотной последовательности
белков и анализа лучших вариантов.
R) Создание автоматической системы для проектирования и
реализации методов распознавания структурно- функциональных
детерминант белков.
7) Создание специализированной интеллектуальной системы,
позволяющей эксперту-биологу вести исследование аминокислотных
последовательностей ЛНК(РНК) взаимодействующих белков с целью
установления возможных молекулярных механизмов белок-
нуклеиновых взаимодействий и закономерностей . организации
соответствующих глобулярных белков.
-260-
"ЛИТЕРАТУРА"
[ 1] Стрелец-В.Б., Колчанов Н.А. Классификация аминокислот-
аминокислотных последовательностей на основе экспертных оценок по
физико-химическим и статистическим параметрам. // В сб.
Теоретические исследования и банки данных по молекулярной
биологии и генетике. Новосибирск. 1988. с 54-55.
[ 2] D.Kmiecik et al // Eur.J.Biochem., V150, 1985. PP359-3T0,
Primary structure of the two variants of a sperm-specific
his tone H1 from the annelid Platynereis dumerilii.
[ 3] Y.Ohe, H.Hayashi, K.Iwai // J.Biochem., V100, 1986,
PP359-368, Human spleen histone H1. Isolation and amino-
acid sequence of a main variant, H1B.
[ 4] K.D.Kole, R.G.York, W.S.Kistler // Biochimica at Biophy-
sica Acta, V869. 1986, pp223-229, Sequence of amino ter-
minalhalf of rat testis-spesific histone variant Hit.
[ 5] J.R.Vanfleteren, S.M.Van Bun, J.J.Ban Beeumen // Bio-
cheir.. J. ,V255, pp647-652, 1988, The primary structure of
the major isoform (H1.1 ) of histone H1 from the nematode
Caenorhabditis elegans.
[ 6] Уотермен Д. Руководство по экспертным системам. М.: Мир.
1989.
[ 7] PROTEINE SEQUENSE DATABASE of the Proteine Idetificatione
Resours (PIR), National Biomedical Reseach Foundation,
Gorgtown University Medical Center, Washington D.C., USA,
1987.
[ 8] Пономаренко М.П., Орлов Ю.Л. // (настоящий сборник).
1989. с.
[ 9] Лыоин Б. Гены. М.: Мир. 1987.
[10] Шенк Р. Концептуальная обработка информации. М.: Энергия.
1980.
-261-
SUMMARY
Compater system for analysis of DtfA(RNA)-ii»teracting proteins
Benukh D.N., Ротюваrente M.P., Kolchanov Я.А., Orlov Yu.L.
In this paper demonertatlng prototype of the Intellectua-
llxmd computer system for studying the proteinae interacting
w»«h the DNACHHAS Is presented.
Section 1 of paper is the Sntroductione to a problem.
There ara arguments in favour of it, that are firstly, working
JSKt of computer methods for identification of proteins Inter-
Interesting »hith the DNA(BHA) and, secondly, revealing the pecu-
peculiarities of proteins araino asid sequences alowing them to
perform function described above. In addition, definition for
щ structural-functional determinant is given as an area of the
primary structure of protein having s specific composltione of
ealno asid residues. Each structural-functional determinant is
related to a particuler protein function.
Sectione 2 is devoted to description of the knowlege base
of the computer eyatem. The knowlege base contains information
on certain functional families of proteins and corresponding
structural-functional determinants. Besides, the knowlege base
is fed with Information on methods of each determinant recog-
recognition. Fig. i is a scheme for location of structural-
functional determinants of histone HI. Fig. 2 is a logical
diagram of a typical knowlege base for protein functional
family.
Section 3 describes an approach for devising highly accu-
accurate methods of recognition of protein structural-functional
determinants. The technology of constructing a particular
recognition method starts whith generating a model for repre-
representing certan structural-functional determinant, followed by
a statistical analysis of usfulness of the model prodused for
a given determinant recognition. At the next stage the best
models are used to create programs recognizing a given struc-
structural-functional determinant. The final stage is testing of
-262 -
the obtained progissss. Figures 3, 4 end S ere examples of сеяв-
ponsnte for the aodel representing structural-functional
determinant. Fig. 8, 7 апй 8 Illustrate the scl»«aee of fraaee
for programs of structural-functional determinant recognition.
Tables 1 and 2 show the results of testing this progress,
In section 4 we discuss the results obtained in scanning
the PI8 data base by programs of racognizing K-end domain cf
histone HI and transition area of N-end domain to s central
hydrophobic domain. The results of this experi»ent are shorn
In Table 3.
Final section presents prospect for developing the compu-
computer system to study the proteins interacting «hith t3i« ША
ШНА). The described approach is assumed to be used for desig-
designing an intellectualized system which permits ?*i expert-
biologist to investigate amino aeld sequences of 01А$Ш(А}~
interacting proteins for finding possible molecular mechanises
of protein-nucleic interactions and regularities of
ment of the related globular proteins.
- 263 -
01 ЭКСПЕРТНЫХ СИСТЕМ К СИСТЕМАМ, СОЗДАНИЮ*
ТЕОРИИ ПРЕДМЕТНЫХ ОБЛАСТЕЙ
Витяев Е.Е, Подколодный Н.Л.
Институт математики СО АН СССР, ВЦ СО АН СССР,
Новосибирск 630090
1. ВВЕДЕНИЕ
Экспертными системами называют интеллектуальные программ-
программные системы, моделирующие процесс принятия решений челове-
человеком-экспертом. Специфика приложений экспертных систем по срав-
сравнению с другими системами искусственного интеллекта состоит в
следующем [16]: "Во-первых, экспертные системы применяются для
решения только трудных (не "игрушечных") практических задач.
Во-вторых, по качеству и эффективности решения экспертных
систем не уступают решениям эксперта-человека. В-третьих, ре-
решения экспертных систем обладают "прозрачностью", т.е. могут
быть объяснены пользователю на качественном уровне ... Это ка-
качество экспертных систем обеспечивается их способностью рас-
рассуждать о своих знаниях и умозаключениях. В-четвертых, экс-
экспертные системы способны пополнять свои знания в ходе диалога
с экспертом".
Разработка экспертных систем (ЭС) привела к многочислен-
многочисленным исследованиям строения человеческого знания и разработке
машинных методов "извлечения" знаний. Эти исследования показа-
показали, что эксперт имеет огромное число правил вида "если ,
то ..." вместе с другими, глубинными структурами знания. Пра-
Правила являются только верхней частью экспертных знаний - "по-
"поверхностными" знаниями. Их дополняют "глубинные" знания, вклю-
включающие теорию предметной области и знания о способе решения
проблем.
Теория предметной области может быть представлена сетью
структурных признаков, отношениями в сети могут быть, напри-
например, отношения причинно-следственной связи, отношения подчи-
подчиненности, отношения ассоциации и. т.д. Структурные признаки мо-
могут включать все родовидовые конкретизации или разбиения
признака.
Знания о способе решения проблем включают общие стратегии
управления применением знаний в конкретных ситуациях и приня-
- 264 -
тием решений. Стратегии имеют дело с организацией и ограниче-
ограничением поиска, избеганием вопросов или наблюдений, выводом по
аналогии, работают с неопределенной, противоречивой и неполной
информацией и т.д.
Экспертные системы первого поколения, как правило, сос-
состоят из набора правил и содержат только "поверхностные" зна
ния. Экспертные системы второго поколения имеют явное преде
тавление "глубинных" знаний, они содержат компоненты, представ
ляющие теорию предметной области и компоненту знаний о спосо-
способах решения проблем. В них, как правило, присутствует также
компонента, содержащая "поверхностные" знания, получающиеся в
результате применения "глубинных" знаний в конкретных ситуа-
ситуациях. "Вывод адекватных правил из "глубинных" знаний не триви-
тривиальная проблема, требующая обучающие стратегии, которые в боль
шей степени еще являются объектом исследований"[26].Существуют
экспертные системы, использующие только "глубинные" знания.
"Глубинные" знания точнее отражают структуру человеческого
знания. Известно, что человеческое восприятие целестно, а
восприятие и анализ ситуации происходит от целого к частному.
Целое в экспертных системах представляется теорией предметной
области. Эта теория должна с одной стороны охватывать все воз-
возможные случаи, а с другой стороны, быть достаточно общей и не
содержать ненужных деталей. Знания о способе решения проблем
являются знаниями-статегиями подведения частного под общее с
целью найти место этого частного в общей целостной картине те-
теории предметной области и затем сделать вывод относительно ка-
каких-либо свойств этого частного в соответствии с теорией пред-
предметной области, с этой целью иногда используются аксиомы вида:
VPi,PzeP; S4,S2eS: Преобразование iPl,Px) & Решение (P2,S2) &
Получить (S2,S4) => Решение (P^S^,
где Преобразование (Pj.Pj) - преобразование проблемы Pt в Р2;
Решение (P-^S^ - S± решение проблемы Р (i=i,2);
Получить (S2,S4) - получить решение St из S2.
Каковы с нашей точки зрения дальнейшие перспективы разви-
развития экспертных систем ?
Результаты, полученные в философской логике, теории изме-
измерений и аксиоматической теории принятия решений показывают,
что эмпирическая (экспериментально проверяемая) теория пред-
265 -
метной области представляет собой систему величин, связанных
между собой законами. Функциональный вид законов и набор мате-
математических операций, который имеет смысл осуществлять над чис-
числовыми значениями величин, определяются на лоияко-операцио-
нальном уровне эмпирическими системами величин и законов к со-
соответствующими системами аксиом. Эмпирические системы - алгеб-
алгебраические системы, в которых основное множество значений вели-
величины (значений величин) и множество отношений, операций к
констант интерпретируемы в системе понятий рассматриваемой
предметной области. Эмпирические системы удовлетворяют соот-
соответствующей системе аксиом S [14,15,18-20,24,25].
Величины и законы тесно связаны. Функциональный вид зако-
законов прост потоку, что числовые представления величин и закона,
связывающего эти величины, как правило, выводятся из одной к
той же системы аксиом [24]-
Эти результаты указывают наиболее строгий путь построения
эмпирических теорий в различных предметных областях. Для этого
все величины, знания и данные должны быть переведены на теоре-
теоретико-модельный уровень и исследованы аксиоматически.
Целостность знания и умение подводить частное под общее
может обеспечиваться следующим принципом "естественной" клас-
классификации знаний [8]: объекты, события и явления одного класса
описываются одинаковыми закономерностями, разных классов -
разными.
Полнота знаний должна обеспечиваться широким применением
методов индуктивного синтеза знаний по данным, примерам и сце-
сценариям рассуждения [4,26].
Следует заметить, что все упомянутые методы и подходы
применимы и к объективным (априорным) и к субъективным (экс-
(экспертным) знаниям. В последнем случае эксперт рассматривается
как своеобразный прибор.
В целом, упомянутые результаты, подходы и методы позволя-
позволяют существенно уточнить понимание "глубинных" знаний и
по-новому поставить цель создания экспертных систем. Конечной
целью создания экспертных систем является с нашей точки
зрения создание экспертных систем, создающих теории предметных
областей.
В соответствии с этой целью в данной работе предлагается
- 266 -
проект инструментальной научно-исследовательской экспертной
системы построения эмпирических теорий. Данный проект оснсвы-
ваэтся на упомянутых выше результатах, подходах и методах.
2. КРАТКОЕ ОПИСАНИЕ СИСТЕШ.
Главной особенностью системы являются развитые средства
построения базы знаний.Система предоставляет возможности ввода
и редактирования экспертных и априорных знаний в виде произ-
произвольной совокупности высказываний в языке первого порядка. В
диалоге с экспертом это множество высказываний приводится к
совокупности правил вида "если .... то ..." [22].
Формирование системы правил выполняется методом индуктив-
индуктивного синтеза ПРОЛОГ-программ на основании имеющихся в БД или
предъявляемых экспертом истинных или ложных примеров, либо
примеров правильного рассуждения [26].
Другой подход, использованный в системе - формирование
вероятностных правил методом индуктивного синтеза ПРОЛОГ-прог-
ПРОЛОГ-программ по выборкам из данных. Такие правила имеют статус законо-
закономерностей и находятся проверкой определенных статистических
критериев [4].
Система выявляет и устраняет "противоречия" в теории на
разных этапах ее построения. Проверка неполноты теории прово-
проводится в диалоге с экспертом путем выявления невозможности вы-
вывести некоторый указанный экспертом факт или утверждение [26].
Система позволяет осуществлять вывод из полученной тео-
теории систем аксиом теории измерений и аксиоматической теории
принятия решений для определения "истинных" шкал величин и за-
законов, а также вывод систем аксиом конструктивной теории моде-
моделей для получения конструктивных числовых представлений
"структурных" величин и законов [7].
В результате такого подхода конструируется ЭС, позволяю-
позволяющая решать любые задачи, выводимые из построенной теории, в
частности, это могут быть задачи предсказания ( прогнозирова-
прогнозирования, диагностики, заполнения пропусков и т.д. ), интерпретации
во введенной системе понятий всех полученных результатов ( об-
обнаруженных закономерностей, разнообразных следствий, процедур
шкалирования величин и законов и т.д. ), планирования экспери-
эксперимента ( управление поиском недостающей информации).
Далее излагаются теоретические предпосылки и основные ал-
- 267 -
горитмы используемые при разработке инструментальной ЭС.
3. ПОСТРОЕНИЕ ЭМПИРИЧЕСКИХ ТЕОРИЙ.
3.1. Качественная, логическая, количественная и конструк-
конструктивная эмпирические теории строятся последовательно. Циклы их
формирования обозначены на рис.1 соответственно одинарной,
двойной и тройной пунктирными линиями. В соответствии с упомя-
упомянутыми выше результатами переход теории из качественного сос-
состояния в количественное должен осуществляться через логическую
эмпирическую теорию, осуществляющую аксиоматический анализ
предметной области. Переход от качественной теории к логи-
логической осуществляется выделением из величин, данных и знаний
их логико-операциональной составляющей - всей эмпирической ин-
информации, выразимой в логике первого порядка и интерпретируе-
интерпретируемой в системе понятий качественной теории. Переход от логичес-
логической эмпирической теории к количественной осуществляется ис-
использованием теории измерений и аксиоматической теории приня-
принятия решений. Переход от логической эмпирической теории к конс-
конструктивной эмпирической теории осуществляется применением
теории конструктивных моделей НО]. Количественная и конструк-
конструктивная эмпирические теории дополняют друг друга и отличаются
выбором действительных или натуральных чисел для получения
числовых представлений. Совместное размещение всех эмпиричес-
эмпирических теорий в БД, при котором они бы дополняли друг друга и
составляли совместно результирующую эмпирическую теорию пред-
предметной области, можно осуществить на основе семантического
программирования и многосортных алгебр,реализуемых реляционной
базой данных [17].
Результирующая эмпирическая теория характеризуется тем,
что в ней есть реляционная база данных, база знаний,
функциональные и конструктивные зависимости и есть
вопросно-ответная система, основанная на логическом
программировании. Все данные, знания и результаты интерпрети-
интерпретируемы в системе понятий предметной области. Интерпретация
доступна конечному пользователю.
3.2. Качественная теория является исходным пунктом пост-
построения эмпирической теории. Она дает описание рассматриваемой
предметной области и тех знании о предметной области, которые
имеются к началу работы. Допускаются мало разработанные и
- 268 -
чисто интуитивные теории. Минимальные требованияк качественной
теории состоят в том,чтобы, во-первых, существовала система
понятий, в которой она формулируется и интерпретируется и,
во-вторых, существовали свойства, признаки, величины и
соответствующие измерительные процедуры, интерпретируемые в
этих понятиях. Система понятий, свойства, величины,данные,
априорные знания (если они есть) и модели должны храниться в
интеллектуальном банке данных (БД). Построение такой системы
в диалоге с пользователем составляет задачу 1.1. (Табл. 1 ).
Построенные далее логическая, количественная и конструктивная
эмпирические теории интерпретируются в системе понятий
качественной теории и заносятся в БД.
3.3. Начальное состояние логической эмпирической теории,
получается из качественной теории переводом на теоретико-мо-
теоретико-модельный уровень ее логико-операционной составляющей. В соот-
соответствии с методологическим принципом теории измерений (свойс-
(свойства определяются отношениями ) из всех свойств, величин, и
Признаков качественной теории пользователь выделяет множество
V интерпретируемых в системе понятий отношений и операций. Вы-
Выделение множества V проводится по методике [1] в диалоге с
пользователем (задача 2л.). Результаты измерительных процедур
и данные качественной теории представляются частично опреде-
определенными многосортными алгебрами В1 = < А д-; V >. Это может
оыть сделано простыми программами преобразования данных (зада-
(задача 2.2.). Априорные знания качественной теории представляются
системой аксиом S. Из всех эмпирически осмысленных аксиом
можно, как правило, удалить кванторы существования,вводя в них
интерпретируемые (в системе понятий качественной теории) опе-
операции над объектами (скулемовские функции). В результате можно
получить систему аксиом S, включающую только универсальные
формулы. Можно показать, что множество универсальных формул
логически эквивалентно множеству формул вида
»!_, ... xt (Afi& ... &А|^ => А|о) A)
Aj,kt Ak - атомарные формулы
1, если атомарная формула берется без отрицания,
о, если атомарная формула берется с отрицанием.
Алгоритм перевода универсальных формул в формулы A) опи-
F; [?2]. Такой путь преобразования системы аксиом в сово-
купность формул A) составляет задачу 2.3.6. В последнее время
формулы A) часто используются при задании базы знаний в экс-
экспертных системах [16]. Поэтому некоторые априорные знания ка-
качественной теории могут быть сразу заданы формулами A) (зада-
(задача г.З.а). Дальнейшее построение логической эмпирической
теории происходит за счет усиления системы аксиом S методом
обнаружения закономерностей [4]. Применяя к имеющимся данным Ш
этот метод с некоторым уровнем доверия а получим множество
закономерностей Ра в виде совокупности формул A) (задача
2.4.). В [5] доказано, что этим методом можно обнаружить любую
закономерность, выраженную универсальной формулой. Доказатель-
Доказательство проведено для предикатной сигнатуры, но его легко
распространить на общий случай. Ставя различные эксперименты
поанализу зависимостей между различными величинами и
обрабатывая получающиеся данные, можно пополнить систему
аксиом S различными множествами закономерностей Fa к получить
в результате теорию Т предметной области как совокупность
формул A). Эту теорию назовем логической эмпирической
теорией.
Если независимо определить атомарные формулы и их отрица-
отрицания и включить формулы к & -iA в теорию Т, то формулы можно
представить как хорновы дизъюнкты, а теорию Т как программу в
логическом программировании. Это обеспечивает эффективное
использование теории и получение из нее различных следствий
(задача 2.5.). К теории как к программе в языке ПРОЛОГ можно
обращаться с любыми вопросами вида А &А2...&А,, и автоматически
получать ответ. Теория может использоваться для получения
предсказаний неизвестных значений признаков методом [4]
(задача 2.7.).
Все результаты логической эмпирической теории нужно ин-
интерпретировать в системе понятий качественной теории. Интерп-
Интерпретируемость результатов следует из интерпретируемости отноше-
отношений и операций, интерпретируемости формул и следствий из них,
а также из интерпретируемости результатов классификации и
предсказания (см. соответствующие методы).
3.4. Количественная эмпирическая теория строится на осно-
основании результатов теории измерений и теории принятия решений о
числовых представлениях величин, законов и функций полезности.
270 -
Если в теории содержится какая-либо система аксиом теории из-
измерений или теории принятия решений, то используя соответству-
вдие процедуры шкалирования [14] (задача 3.1. Ь можно получить
числовые представления величин, функциональных зависимостей и
функций полезности. Выводимость системы аксиом из теории Т
может быть установлена логическим выводом (задача 2.5.), либо
методом обнаружения закономерностей, проверкой на данных SR
(задача 2.4.). Шкалирование может быть осуществлено либо по
данным методом решения систем линейных неравенств (задача
г,4.), либо в диалоге с экспертом, как это делается,
например, в [14] (задача 3-2.а.), либо планированием экспери-
эксперимента (задача 3-2.6.), в случае величин, измеряемых приборами.
Функциональные зависимости и функции полезности получаются из
систем аксиом с одновременным построением числовых предс-
представлений, входящих в них величин. Это позволяет простыми функ-
функциональными зависимостями описывать целые классы функций. В
теории измерений найдены системы аксиом для многих
физических величин и фундаментальных физических законов [241.
Результаты функциональной теории измерений [20] показывают,
что подобную систему величин, связанных между собой
фундаментальными законами, можно получить и во многих других
областях. Многие системы аксиом, описывающие функциональные
зависимости и функции полезности, содержат только отноше-
отношения линейного порядка для одной из взаимосвязанных величин.
Это позволяет применять их в различных предметных областях.
Числовые представления величин, функциональных зависимостей и
функций полезности, получаемые из систем аксиом адекватны и
интерпретируемы в системе понятий качественной теории [18].
3-5. Третий уровень построения эмпирической теории состо-
состоит в построении количественной и конструктивной эмпирической
теории. В.теории измерений [24.18,19] и теории принятия реше-
решений [14,19] нельзя получить числовые представления некоторых
величин и закономерных связей в силу ограниченности, использу-
используемого в них понятия числового представления. Величины и зако-
закономерные связи, описываемые частичными порядками, толерантнос-
тями, решетками и т.д. не могут быть сильным гомоморфизмом
вложены в поле вещественных чисел. Для числового представления
таких величин и закономерных связей можно использовать конс-
- 271 -
КАЧЕСТВЕННАЯ ЭМПИРИЧЕСКАЯ
ТЕОРИЯ
ЗАДАНИЕ ПРЕДМЕТНОЙ ОБЛАСТИ!
Система понятий,
признаки, величины,
измерительные процедуры,
данные,
априорные знания.
Результирующие
положения,
знания,
модели.
А
I 1
\/
БАЗА ДАННЫХ
ЛОГИЧЕСКАЯ ЭМПИРИЧЕСКАЯ ТЕОРИЯ!
отношения, операции,
измерительные процедуры,
частично-определенные модели
( данные ),
системы аксиом
( априорные знания ).
закономерности: ?<
"естественная"
классификация знаний,
логическое программирование
( знания, вопросно-ответная
система),
предсказание.
\
\i/
\
I КОЛИЧЕСТВЕННАЯ ЭМПИРИ-
! ЧЕСКАЯ ТЕОРИЯ
1 ( вещественные числа )
!числовые значения
!величин, данные,
!измерительные процедуры
I функции зависимости.
! функциональные зависи-
! мости.
! КОНСТРУКТИВНАЯ
! ЭМПИРИЧЕСКАЯ
! ТЕОРИЯ
!(натуральные числа,
! рациональные числа)
! эффект, выч.
! числовые значения
! величин, данные,
1 измерительные процед
! функции зависимости.
! функциональные зави-
зависимости ( общерекур-
!сивные функции ).
V.
V
Рис. 1 Схема построения эмпирической теории.
— построение качественной теории предметной области
=== построение логической теории предметной области
=г= построение количественной теории предметной области
- 272 -
труктивные числовые представления. Значениями величин в этом
случае являются натуральные, рациональные или другие эффекти-
эффективно вычислимые числа (например, какие-либо коды). Ближе всего
к понятию числового представления теории измерений находится
понятие конструктивного числового представления [7], основан-
основанное на конструктивизации эмпирических систем [ю]. Требование
конструктивности процесса шкалирования приводит к еще одной
формализации конструктивного числового представления.
Конструктивные числовые представления интерпретируются в
системе понятий качественной теории.
4. АЛГОРИТМ ИНДУКТИВНОГО СИНТЕЗА ЗНАНИЙ.
В общем случае подходы развиваемые в данной работе,
по-видимому, можно обобщить на системы вывода, управляемые об-
образцами [23]. Это широкий класс систем включающий системы, ос-
основанные на правилах, системы вывода на сетях, фреймах и дру-
другие. Такие системы работают с множеством модулей ( источников
знаний ), каждый из которых содержит образец, определяющий ус-
условия применимости модуля в зависимости от текущей ситуации.
Анализируя текущую ситуацию интерпретатор на каждом шаге рабо-
работы системы определяет набор модулей подходящих для обработки в
этой ситуации. Далее по определенным правилам разрешается
конфликт между конкурирующими модулями, выбирается один модуль
и передается на выполнение. Для нас наиболее вазгаымсвойством
таких систем является структурная независимость модулей
знаний, существенно облегчающая модификацию базы знаний.
Следует также отметить, что знания о решении проблем в
этих системах отделены от теории предметной области. Это дела-
делает систему более гибкой и позволяет по мере роста базы знаний
применять различные стратегии поиска решений.
Нам удобно при дальнейшем рассмотрении под модулем пони-
понимать некоторое правило, имеющее две части: предусловие, прове-
проверяемое на данных из БД и действие, модифицирующее БД. Система
основанная на правилах является частным случаем систем вывода,
управляемых образцами.
Существенным требованием является монотонность системы.
Это означает, что результат работы системы не зависит от по-
порядка использования модулей.
Введем формальное описание вычислительного акта, связан-
73 -
ного с выполнением модуля р в виде <р,х,у>
где р - имя модуля,
i - входные параметры,
у - выходные параметры.
Мы не будем уточнять смысл обработки и детально
описыватьмодуль. Будем считать, что интепретатор при обращении
к модулю требует предварительного выполнения совокупности
модулей р1,р2,... рк (обратный вывод).
Конечное (возможно пустое) упорядоченное множество троек
(<р1,х1,у1>,<р2,х2,у2>, ... , <pk,xk,yk>) назовем, следуя [26]
верхним уровнем трассировки модуля р при входе х и выходе у.
Если множество пустое, то это означет, что модуль р при
входе i получает на выходе у не требуя обращения к другим
модулям.
Это означает, что для модуля р при входе х явно ( прямой
вызов ) или косвенно ( интепретатор создает ситуацию, в
которой возникает возможность вызова модуля р) требуется
выполнение модуля р1 с входом х1. И если р1 при входе х1
возвращает у1 ,то вызывается р2 с входом х2 и если р2 при входе
х2 возвращает у2, то то требуется вызвать рк при входе хк
и если рк возвращает ук, то модуль р возвращает у.
Интепретацией И назовем конечное множество троек <р,х,у>,
где р - имя процедуры с п входными и га выходными переменными.
Выход у для модуля р в И называется корректным, если
<р,х,у>€М или у=о, если <р,х,у>Л1. Иначе, некорректным.
Вывод называется частично корректным, если корень любого
полного вывода входит в М. Вывод называется полным, если любая
тройка из И является корнем некоторого полного дерева вывода.
Вывод полностью корректный, если он частично корректный,
полный и всегда заканчивается.
Идея алгоритма индуктивного синтеза БЗ состоит в следую-
следующем. Начиная с некоторой (возможно пустой) БЗ системе последо-
последовательно предъявляются факты с известными оценками истинности
ищутся ошибки разного вида. Ьй корроктируется так, чтобы
система адекватно раоотала на предъянляемых известных фактах.
При коррекции Бй выполняете лиоо удаление ошибочного
правила из БЗ, лиоо синтьи ноьип, нршшш и его проверка (в
случае, когда при вьюоде некоторая промежуточная цель не раз-
разрешается ( отсутствуют правила)).
Перебор претендентов на новое правило в процессе синтеза
упорядочен по степени сложности правила.
Алгоритм индуктивного синтеза БЗ [26]:
1. Если фактов нет, то на 7. иначе читать факты с оценкой
истинности.
г. Если факт со значением истина, то на 3, иначе ъ.
3. Если система не находит подтверждения истинности
факта, то на 4, иначе 1.
4. Используя алгоритм диагностики неполноты знаний, найти
цель А , не разрешенную при выводе. Искать не помеченное
правило В, которое разрешает А. Добавить В в БЗ. Перейти на 2.
5. Если система доказывает истинность факта, то на 6,
иначе 1.
6. Используя алгоритм поиска некорректных правил, найти
правило, приводящее к появлению ошибки. Убрать его из БЗ.
Пометить правило. Перейти на 2.
7. Выдать БЗ.
8. Конец.
Опишем кратко алгоритм поиска некорректного правила. Мы
рассматриваем следующие типы ошибок: ЭС выдает ошибочное зак-
заключение; заключение, полученное ЭС верно, но не полно; система
не выдает никаких сообщений или выдает сообщение о
зацикливании.
Возможны другие варианты ошибочной работы системы, напри-
например, система выдает верный результат, но использованные при
этом рассуждения неверны; заключение верное, использованное
рассуждение верное, но можно получить этот же результат другим
способом, не представленным в системе. Эти варианты
покрываются первыми двумя типами ошибок, если в качестве ре-
результата работы системы рассматривать поиск логической цепоч-
цепочки, приводящей к решению.
Если некоторое правило из верхнего уровня трассировки р
сработало неверно,то неверно сработало само правило р. Будем
считать, что если правило р сработало верно, то верно
сработали правила (р1 ,р2,...рк).
используя это свойство, можно целенаправлен© локализовать
75 -
ошибочное правило в дереве вывода. Это будет неверно
сработавшее правило, имеющее самый высокий уровень. Поиск
этого правила можно осуществлять методом деления пополам.
5. "ЕСТЕСТВЕННАЯ" КЛАССИФИКАЦИЯ БАЗЫ ЗНАНИЙ.
"Естественная" классификация базы знаний проводится в со-
соответствии с принципом [8]:"объекты (события, явления) одного
класса описываются одинаковыми закономерностями ( знаниями ),а
объекты разных классов - разными группами закономерностей".
Этот принцип является конкретизацией критерия "естественности"
классификации Уэвелла : "чем больше общих утверждений об
объектах дает возможность сделать классификация, тем она
естественней".
В системе предполагается наличие двух алгоритмов класси-
классификации для детерминированных (истинных не модели данных ) и
вероятностных закономерностей, детерминированный случай явля-
является предельным для вероятностного.
Рассмотрим детерминированный случай. Будем предполагать,
что SR - алгебраическая система. Предположим, что детермини-
детерминированные закономерности не содержат отрицаний и следовательно,
могут рассматриваться как правила ПРОЛОГ-программы Рг.
Множество фактов ПРОЛОГ-программы определим как множество Ф
всех атомов, истинных в 01. Алгоритм классификации должен быть
в этом случае алгоритмом структуризации ПРОЛОГ-программы
Рг(Ф).
Классом назовем любое множество атомов К, истинных в И
замкнутое относительно прямого вывода по правилам программы
Рг(Ф). Рассмотрим программы Рг(Ф' ),Ф'<=Ф. Минимальной Эрбрано-
вой моделью К(Рг(Ф*)) программы Рг(Ф') называется множество
атомов, замкнутое относительно прямого вывода по фактам Ф'.
Классы К являются минимальными Эрбрановыми моделями программ
Рг(К) и минимальные Эрбрановы модели являются классами, так
как замкнуты относительно прямого вывода. В отличие от классов
вминимальных Эрбрановых моделях К(Рг(Ф')) явно указывается
множество атомов Ф', порождающих класс К.
Замкнутость относительно прямого вывода является "естест-
"естественным" требованием класса, так как по информации о принадлеж-
принадлежности объекта к некоторому классу (либо по признакам объекта,
определяющим эту принадлежность) мы всегда можем сделать вывод
- 276 -
о других признаках данного объекта или других закономерно
связанных с ним объектов.
Каждой программе Рг(Ф') поставим в соответсвие множество
правил Р(Рг(Ф')), применяющихся в процессе прямого вывода по
фактам Ф'. Если факты относятся к объектам, образующих в
некотором смысле класс, то в соответствии с принципом
"естественной" классификации к этому классу объектов будут
применяться только правила, являющиеся детерминированными
закономерностями для объектов этого класса. Определим
программу Р(Ф')=Р11Ф'. Минимальная Эрбранова модель этой
программы совпадает с минимальной Эрбрановой моделью
программы Рг(Ф'), так как из последней удалены правила, не
применяющиеся в процессе вывода.
Программа Р(Ф') является описанием класса К(Р(Ф')). Факты
Ф1 дают "минимальное" описание свойств объектов класса
К(Р(Ф')), к которыму они относятся. Класс К(Р(Ф')) . дает
полное описание свойств объектов класса. Правила Р дают набор
детерминированных закономерностей, описывающих объекты класса.
Новый объект будет подпадать под определение класса, если
он удовлетворяет свойствам Ф* и закономерностям из Р. После
этого программой Р(Ф') могут быть получены все возможные
следствия о других его свойствах.
6. ПРИЛОЖЕНИЕ. Таблица 1.
Классы решаемых задач
1. Формирование качественной теории.
1.1.Внесение в базу данных системы понятий, исходных данных,
признаков, величин, априорных знаний.
г. Формирование логической эмпирической теории.
2.1. Выделение отношений и операций.
2.2. Перевод данных на логичекий уровень.
2.3. Перевод априорных знаний в логическую форму:
а) заданием их формулами A);
б) заданием их универсальными формулами, введением операций
для кванторов существования и переводом их в формулы вида A).
2.4. Обнаружение закономерностей и внесение их в БД:
а) для фиксированной формулы A);
б) для формулA),генерируемых в порядке возрастания сложности.
2.5. Логический вывод.
2.6. Классификация объектов методом [8].
2.7. Предсказание методом [4].
3. Формирование количественной и конструктивной эмпирических
теорий.
3.1. Определение шкал величин и функциональных зависимостей
проверкой выполнимости систем аксиом теории измерений
или теории принятия решений:
- удаление из аксиом кванторов существования и введение
операций;
- приведение универсальных формул к виду A);
- проверка формулы на реальных данных решением задачи 2.4.
3.2. Построение шкал и функциональных зависимостей:
а) по данным;
б) в диалоге с экспертом или планированием эксперимента.
3.3. Определение конструктивных числовых представлений величин
и законов проверкой выполнимости систем аксиом (см.задачу 3.1.).
3.4. Построение конструктивных числовых представлений и
"конструктивных" законов:
а) по данным;
б) в диалоге с экспертом или планированием эксперимента.
ЛИТЕРАТУРА
[1] ВИТЯЕВ Е.Е. Анализ данных с применением языка эмпири-
эмпирических систем .-Автореф. дис ... канд. техн.наук.-Новоси-
Зирск,1982.-16 о.
[2] ВИТЯЕВ Е.Е. Числовое алгебраическое, и конструктивное
представление одной физической структуры.-В кн.: Логико-мате-
Логико-математические основы МОЗ ( Вычислительные системы, вып. 107). Но-
Новосибирск, 1985, с. 40-51.
[3] ВИТЯЕВ Е.Е. Закономерности в языках эмпирических сис-
систем и законы классической физики. В кн.: Эмпирическое
предсказание и распознавание образов ( Вычислительные системы,
вып. 79). Новосибирск, 1979, с. 45-56.
[4] ВИТЯЕВ Е.Е. Метод обнаружения закономерностей и метод
предсказания. В кн.: Эмпирическое предсказание и распознавание
образов ( Вычислительные системы, вып. 67). Новосибирск, 1976,
с. 54-68.
[5] ВИТЯЕВ Е.Е. Обнаружение закономерностей, выраженных
- 278 -
универсальными формулами. В кн.: Эмпирическое предсказание и
распознавание образов ( Вычислительные системы, вып. 79). Но-
Новосибирск, 1979, с. 57-59-
[6] ВИТЯЕВ Е.Е. Закономерности в языках эмпирических сис-
систем. В кн. Эмпирическое предсказание и распознавание образов
( Вычислительные системы, вып. 76). Новосибирск, 1978, с.
3-14.
[71 ВИТЯЕВ Е.Е. Конструктивное числовое представление ве-
величин. В кн. Методы анализа данных (Вычислительные системы,
вып. 111). Новосибирск, 1985, с.23-32.
[8] ВИТЯЕВ Е.Е. Классификация как выделение группы объек-
объектов, удовлетворяющих разным множествам согласованных законо-
закономерностей.- В кн.: Анализ разнотипных данных ( Вычислительные
системы, вып. 99). Новосибирск, 1983, с. 44-50:
[91 Дискретная математика и математические вопросы кибер-
кибернетики, т.1/ Под ред. СВ. Яблонского, О.Б. Лупанова.-М.:Нау-
Лупанова.-М.:Наука, 1974. -311С
[10] ЕРШОВ Ю.Л. Проблемы разрешимости и конструктивные мо-
модели. -М.: Наука, 1980.-415с
[11] ЗАГОРУЙКО Н.Г., САМОХВАЛОВ К.Ф., СВИРИДЕНКО Д.И.
Логика эмпирических исследований.-Новосибирск, 1978. -66с.
[12] ЗАГОРУйКО Н.Г. Методы обнаружения закономернос-
закономерностей. -М.: Знание, 1981.- б2о.
[131 КАРНАП Р. Философские основания физики.-М.: Прогресс,
1971.-387С.
[14] КИНИ Р.Л., РАйФА X. Принятие решений при многих кри-
критериях: предпочтения и замещения.-М.:Радио и связь,1981.-560с.
[15] КОЗЕЛЕЦКИй Ю. Психологическая теория решений.-М.:
Прогресс,1979.-5озс.
[16] ПОПОВ Э.В. Экспертные системы.-М.:Радио и связь.-1988.
[17] СВИРИДЕНКО Д.И. Проектирование J-программ. Постанов-
Постановка проблемы. В.кн.: Методы анализа данных (Вычислительные сис-
системы, вып. 111). Новосибирск, 1985, с.108-127.
[18] ПФАНЦАГЛЬ И. Теория измерений.-Мир,1976.-248с.
[19] ФИШБЕРН П.С. Теория полезности для принятия решений.
М.:Наука,1978.-352о.
[20] ANDERSON M.H. Algebraic Rules in Psyhologioal
ment.-Amer.Scientist,1979 ,v.67,p-555-563.
279 -
[21] CLAEK K.L. MoCABB F.G. PROLOGrA Language for
Implementing Expert Systems.-Machine Intelligence, 1980,v.10.
[22] CLOCKSIN W.P., MELLISH C.S. Programming in
Prolog.-New York, 1981.-280c.
[23] HAYES-ROTH P., WATERMAN D.A. Principles of Pattern
Directed Inference Systems.-1978.-p.577-601.
[24] Foundations of measurement.Vol.1./Krantz D.H.,Zuoe R.
D., Suppes P., Tversky A.-NY and London: Academic press,1971.-
577p.
[25] KRANTZ D.H..TVERSKY A. Conjoint measurement analysis of
composition rules in psychology.-Psyohol.Rew.,1971,v.78,p.151-169.
[26] SHAPIRO E.Y. Algorithmic Program Debagging.-MIT
Press, 1982.-210 p.
[27] STEELS L. Second Generation Expert Systems.//Future
generation computer systems.1985.v1D).
- 280 -
SUMMARY
Fro» Expert Systems to Those Developing Theories
of Subject Domains.
Vityaev E. E. , Podkolodny N. L.
The present paper deals with expert systems of the second
generation characterized by the formula:
Domain Theory + Problem Solving Knowledge = Heuristic Rules.
Tendencies of furthere expert systems development are
considered. The results of philosophical logic, measurement
theory and axiomatic theory of decision making are noted to
give more precise definition of the first component - Domain
Theory, and the idea of "natural" classification of knowledge
makes more precise definition of the second one. The above
results show that the aim of constructing expert systems is to
produce systems developing theories of subject domains. There
is a short description of project for instrumental expert
system of developing empirical ( experimentally proved )
theories in various subject domains. The project is based on
the following main results and methods. The results obtained
in philosophical logic, measurement theory and axiomatic
theory of making decisions show that the empirical theory of
subject domain is a system of quantities connected ( with each
other ) by laws. The quantities and laws are closely connected
with each other. A functional form of the laws is simple
because the numerical representation of quantities embodied in
the law and the functional form of the law are often produced
from the same system of axioms. The functional form of the
laws and a set of mathematical operations having sense for
numeric values of a certain quantity are determined at a
theoretical-model level bv systems of axioms and empirical
systems ( algebraic systems in wich the main set of quantity
( quantities) values and a set of relationships, operations
and constants are interpreted in terms of subject domain). For
determination feasibility of particular system of axioms on a
parti ally-determined empirical system a method of detecting
the regularities in the first order language has been
elaborated.
Thus, the empirical theory of subject domain should
contain the three complementary empirical theories -
juaiitative, logical, arid quantitative ones. Transition of
theory from a qualitative state to quantitative one is posible
mly through logical empirical theory. Logical theory performs
axiomatic analysis of subject domain. Transition from logical
theory to quantitative one takes place by means of the results
sf both the measurement theory and axiomat;c theory of
decision making. The qualitative empirical theorv is a
description of subject domain and that knowledge ( if any )
wich are already available in this subject domain. Description
of subject domain is setting a system of notions and class of
objects to be studied as well as quantities, data and
measuring devices ( including an expert ) interpreted in the
system of notions. Making more precise definition of the
component "Problem Solving Knowledge" is performed by the
methods of "natural" classification of knowledge based on the
principle: objects, events, phenomena of the same class are
described bv identical regularities, of different class by
different ones. The original method of "natural"
classification of objects is considered in the paper.
Подписано к печати 7.12.89 г. МН 10536
Формат бумаги 60 х 90 I/I6. Печ.л. 17,6. Уч.-изд.л. 12,i
Тираж 600. Заказ 333. Цена 80 коп.
Ротапринт Института цитологии и генетики СО АН СССР
630090, Новосибирск, пр. Академика Лаврентьева, 10