































































































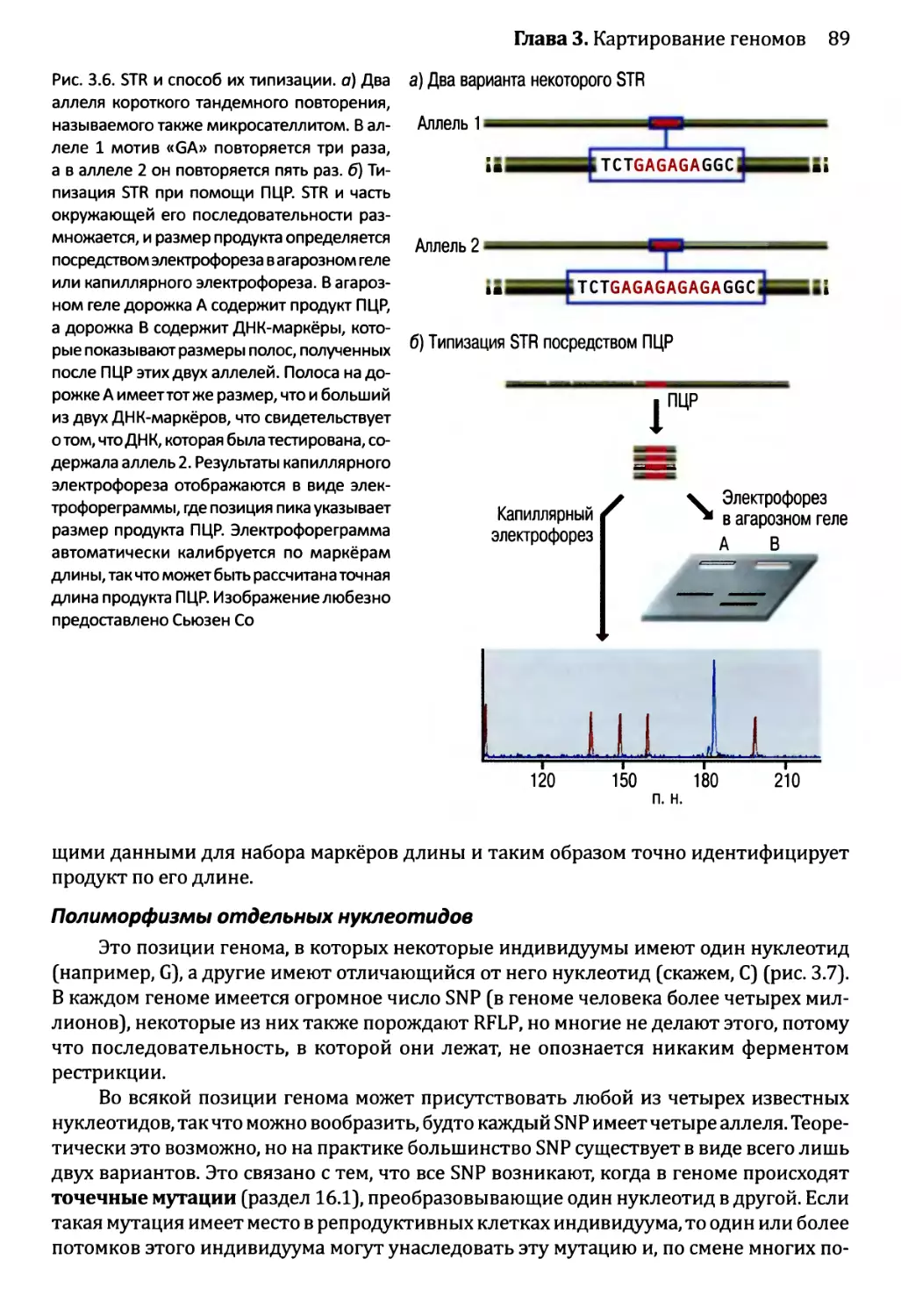






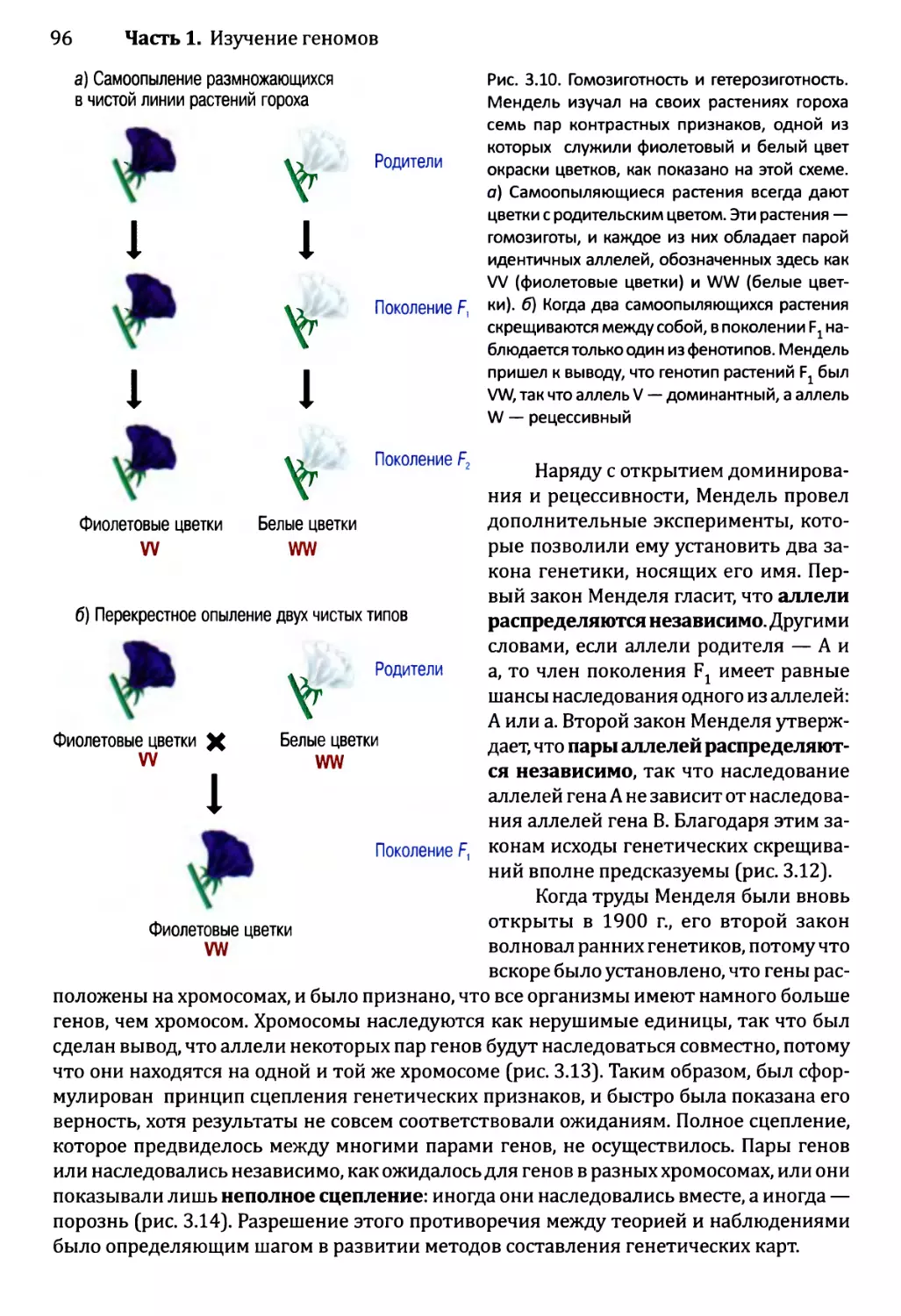
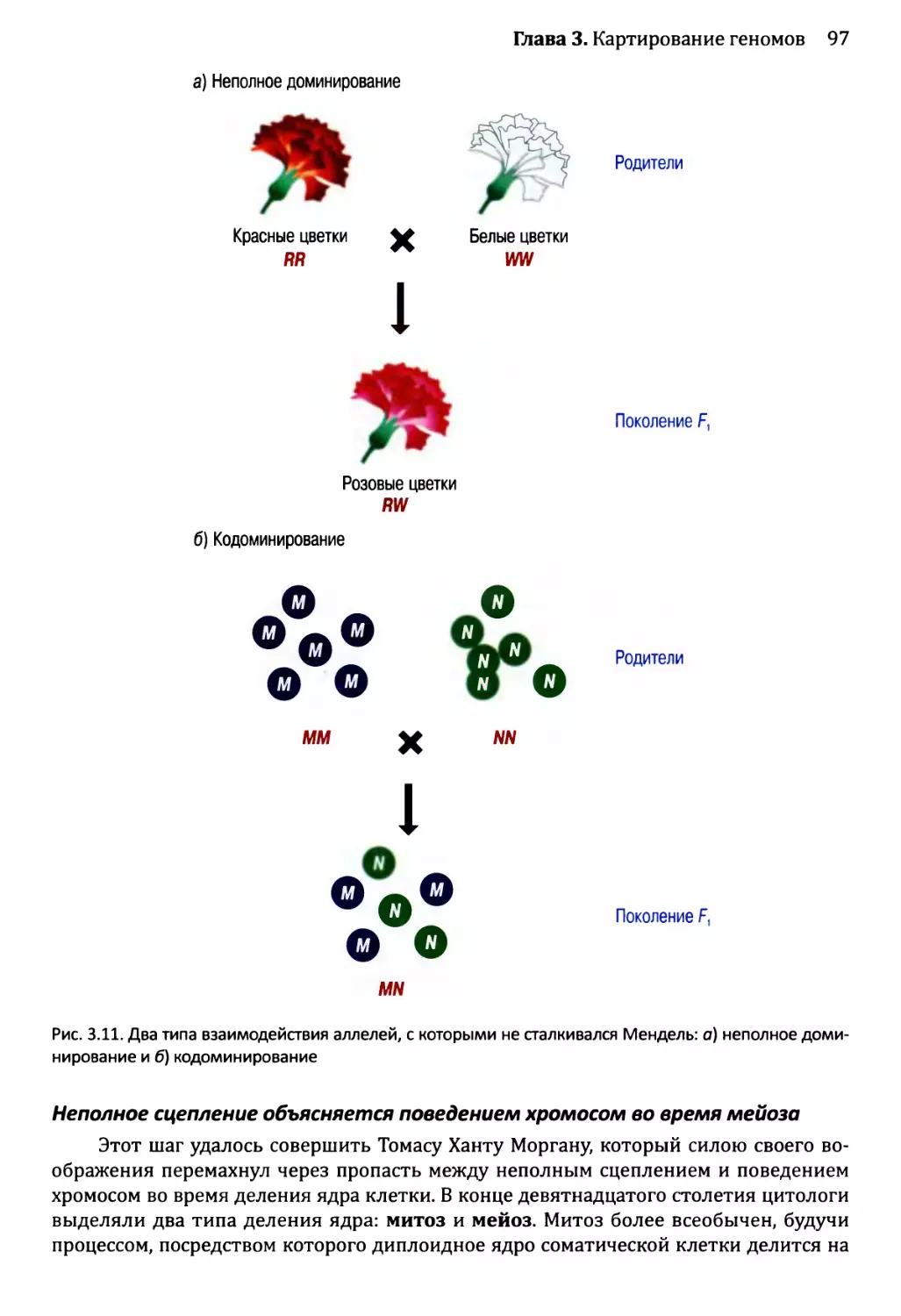



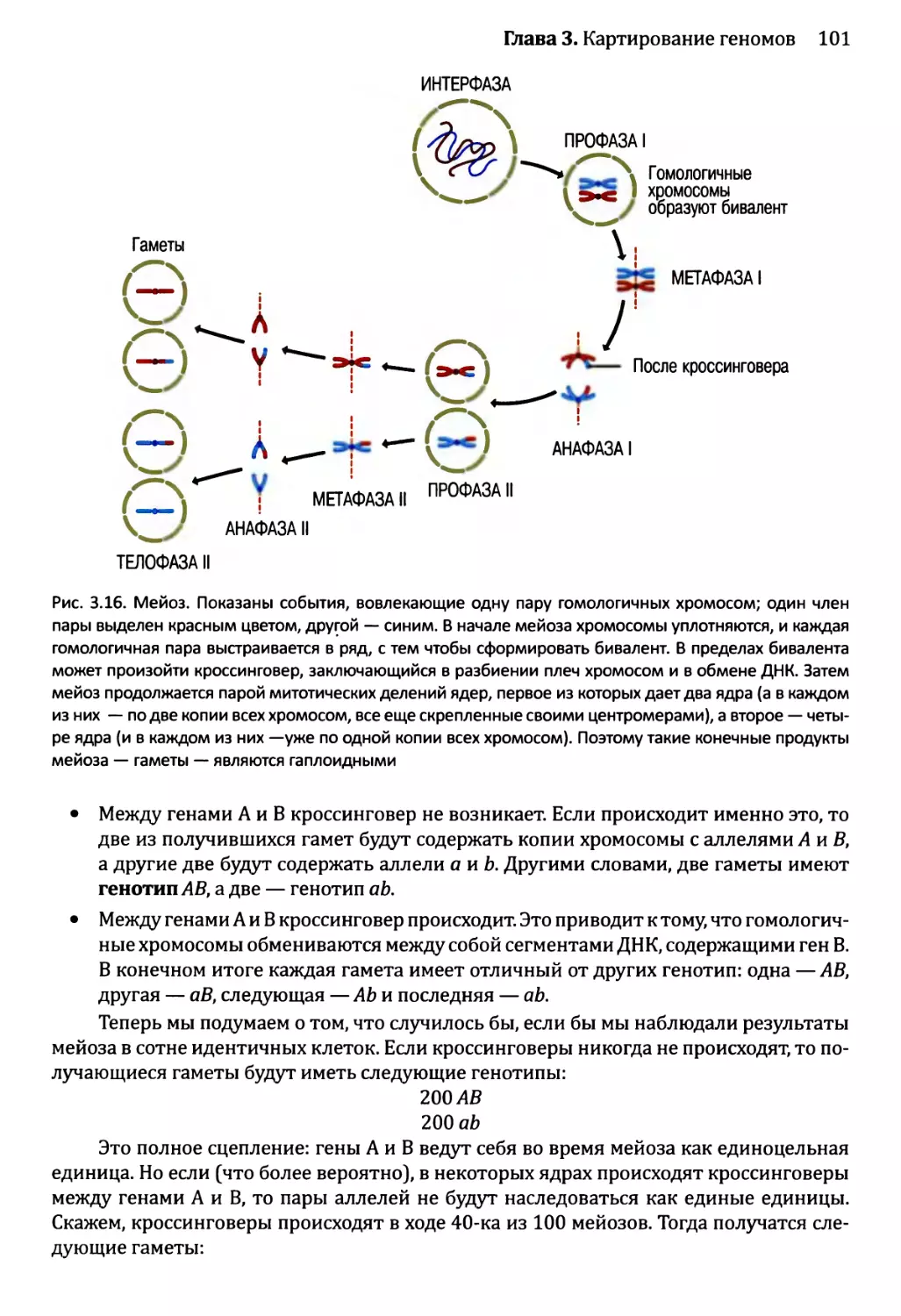



































































































































































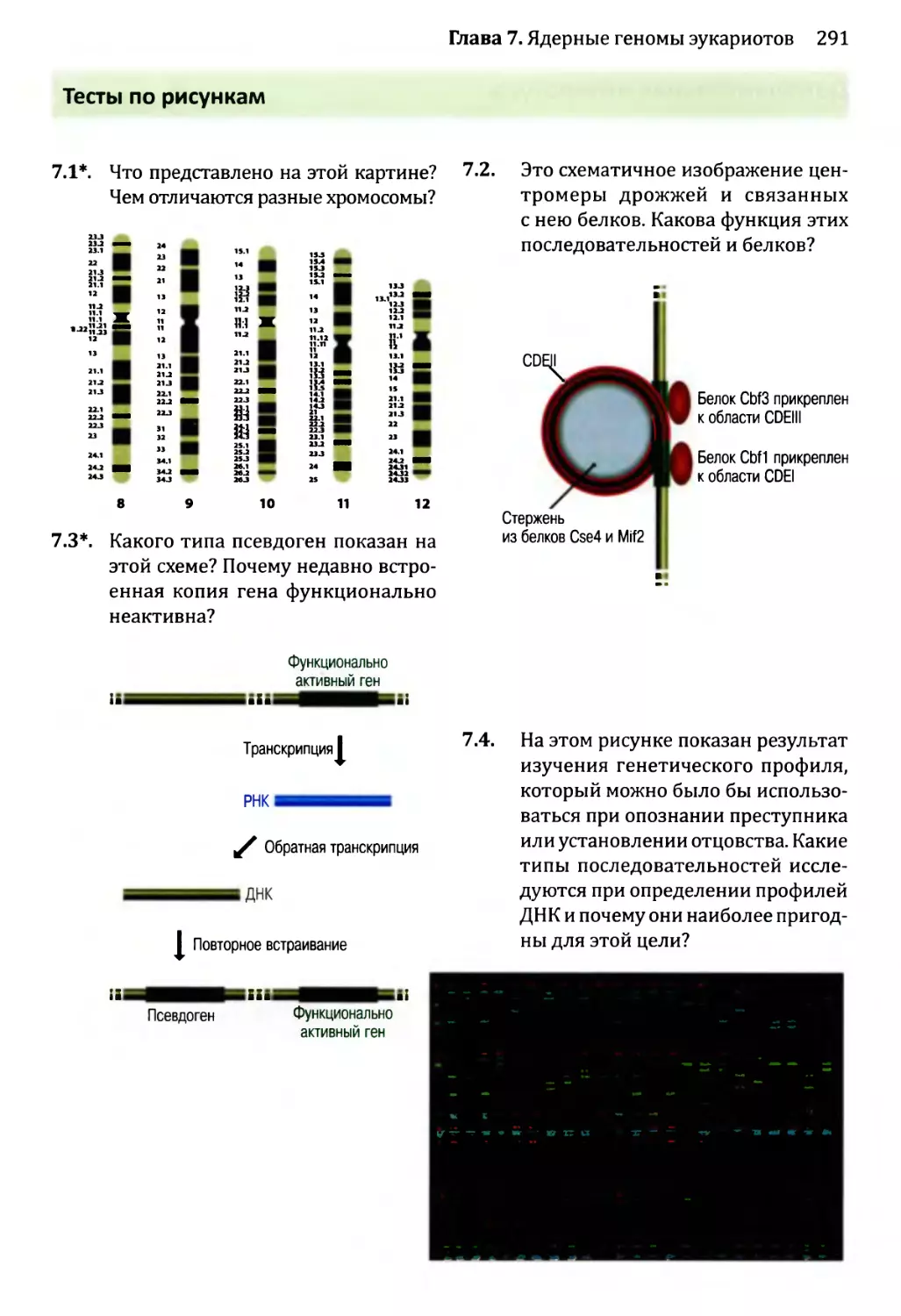




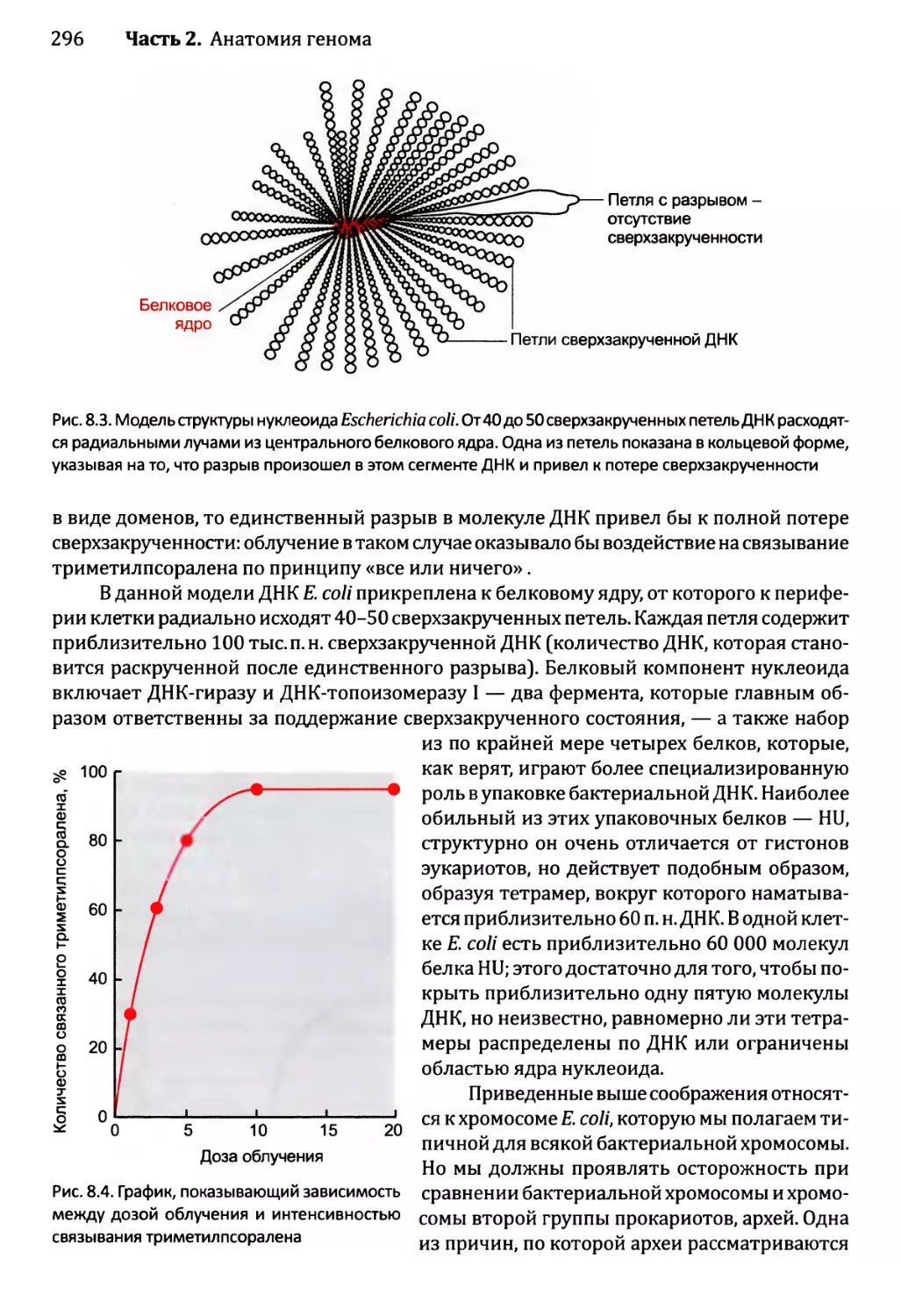









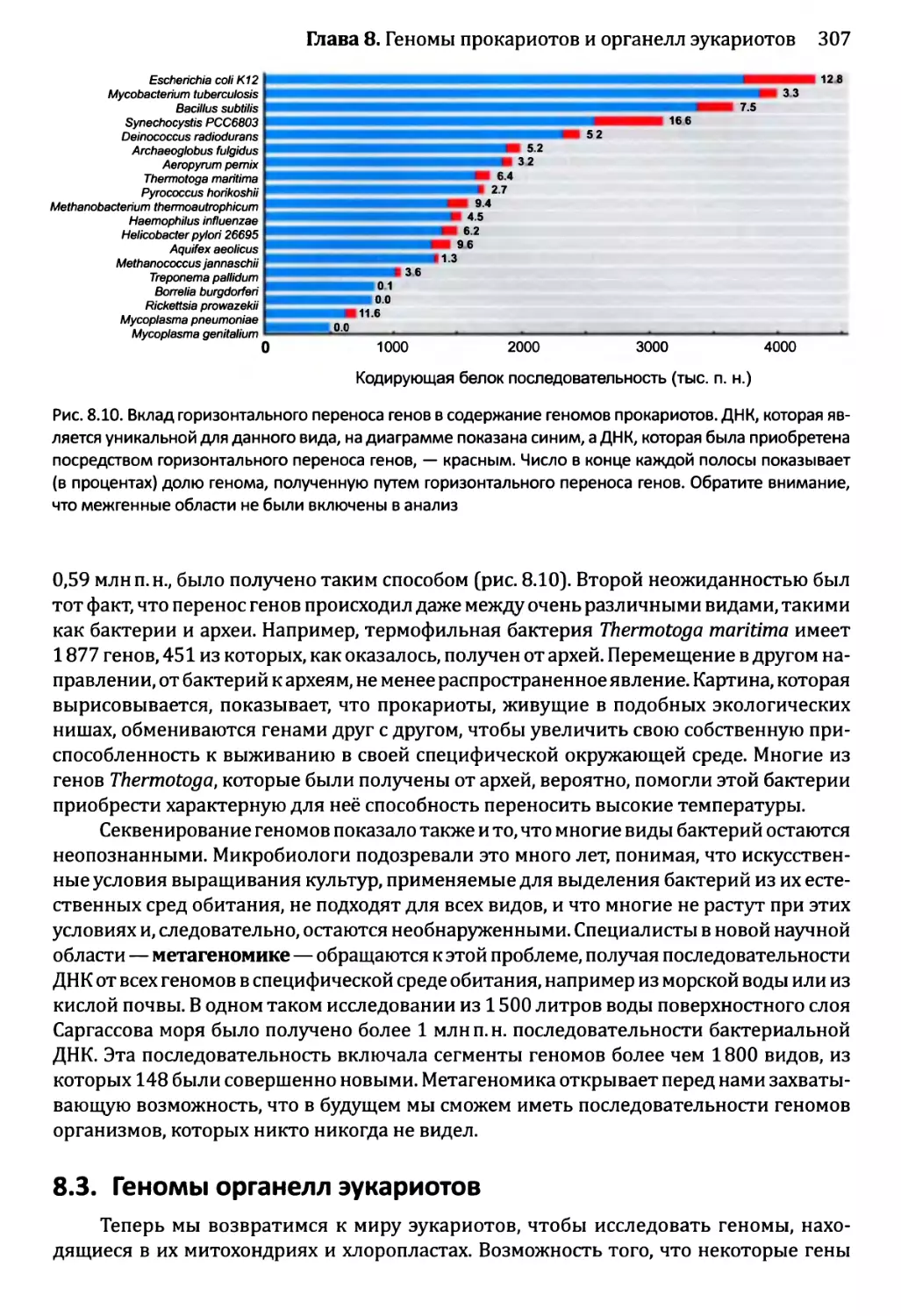


























































































































































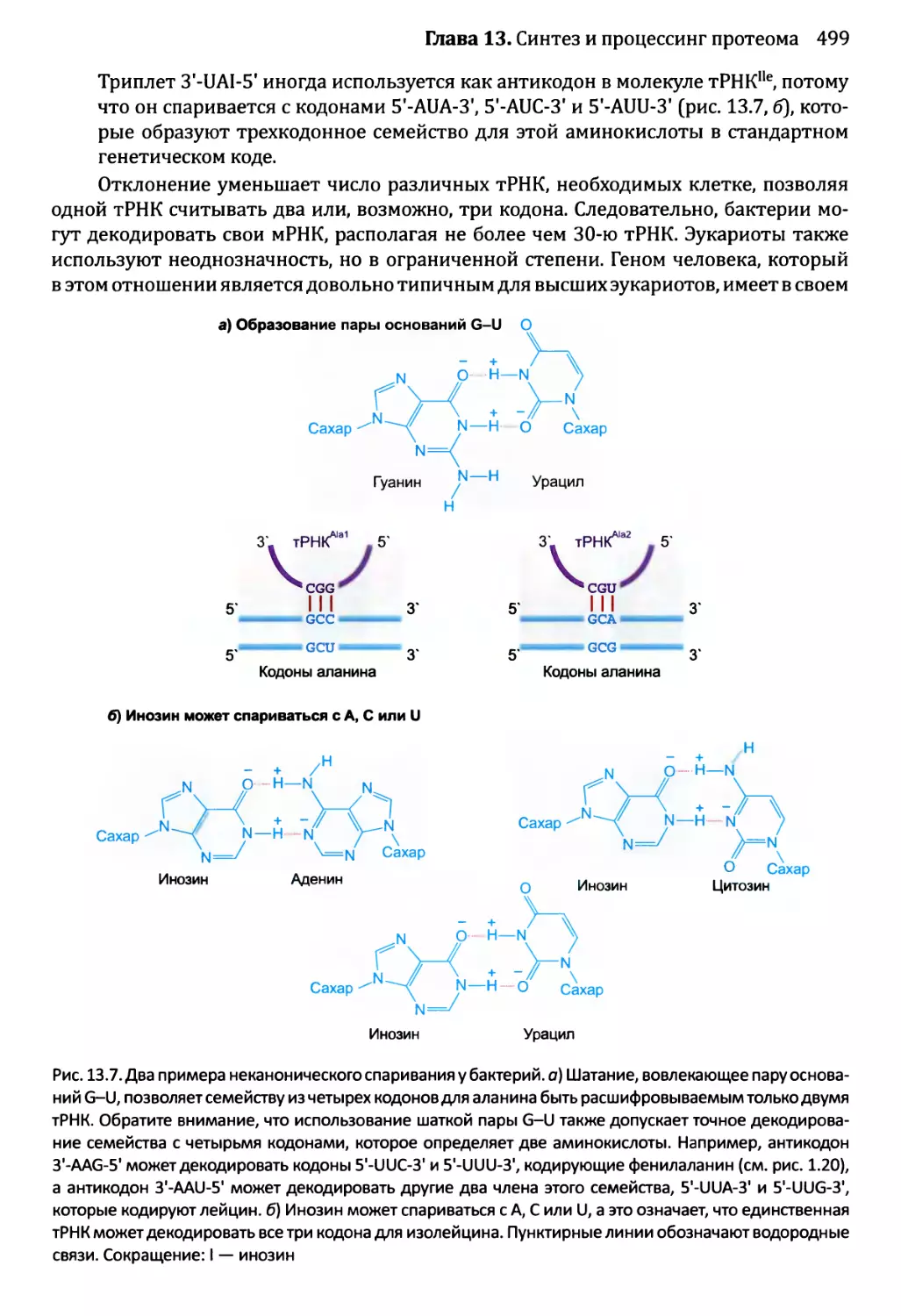














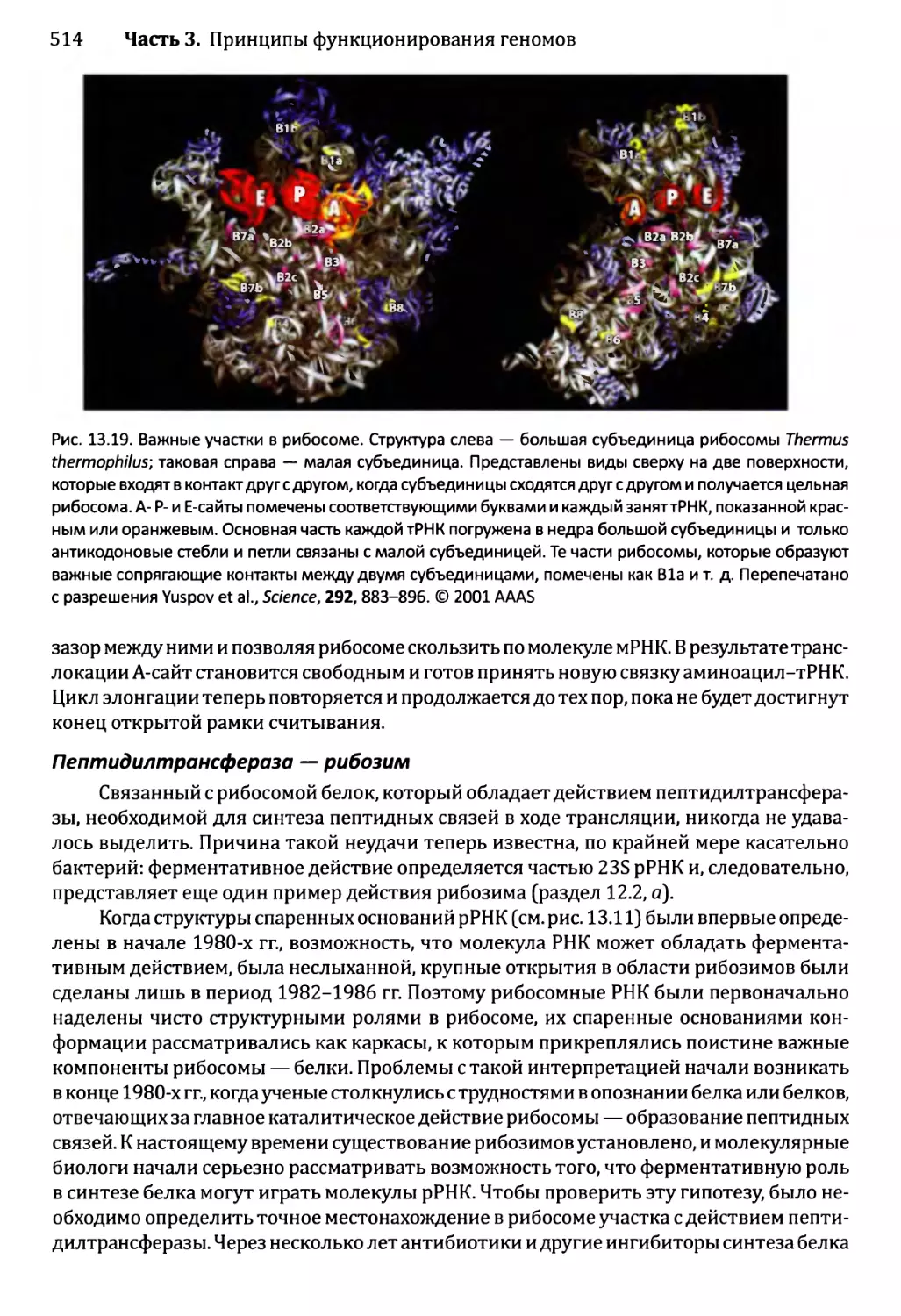
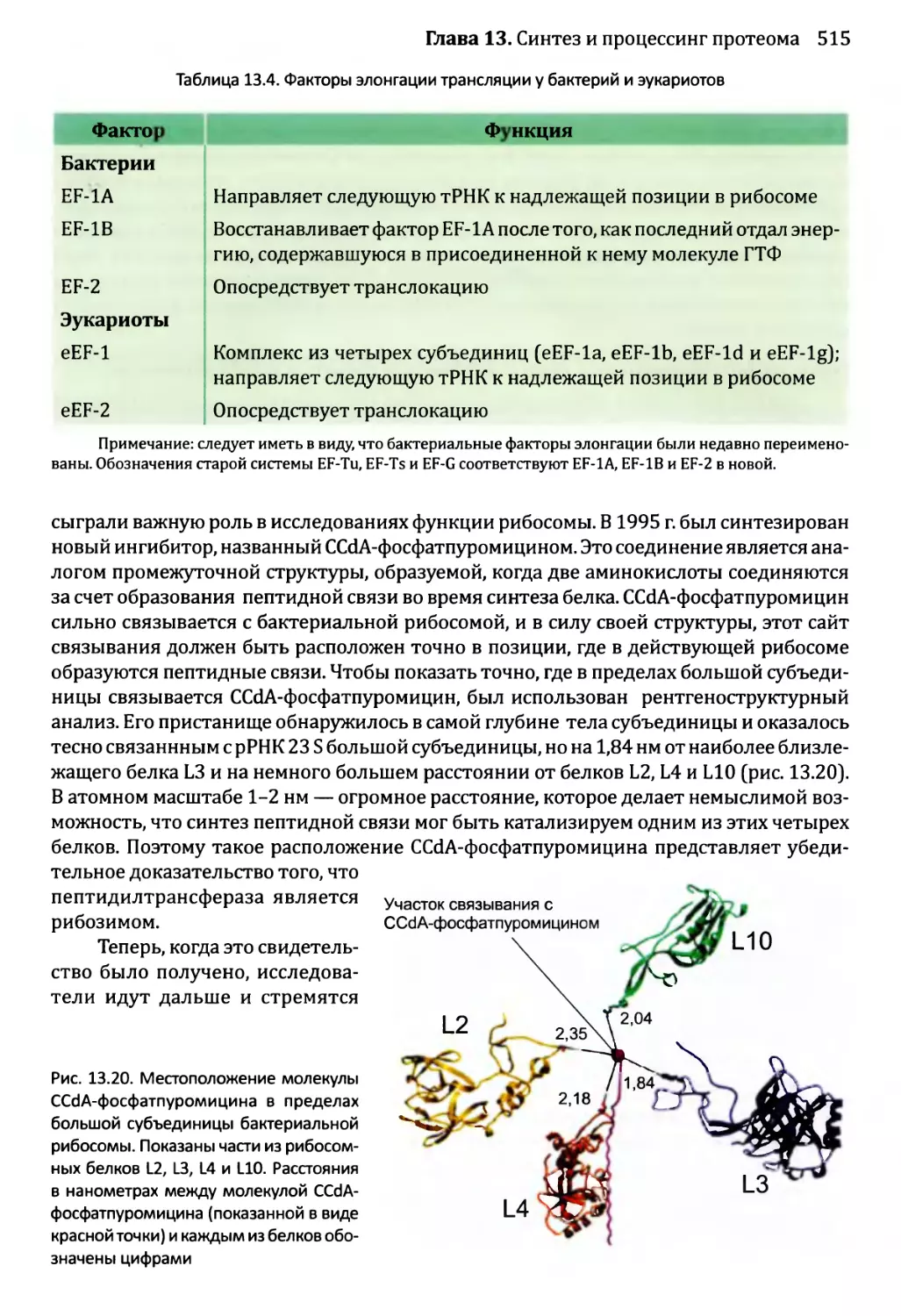








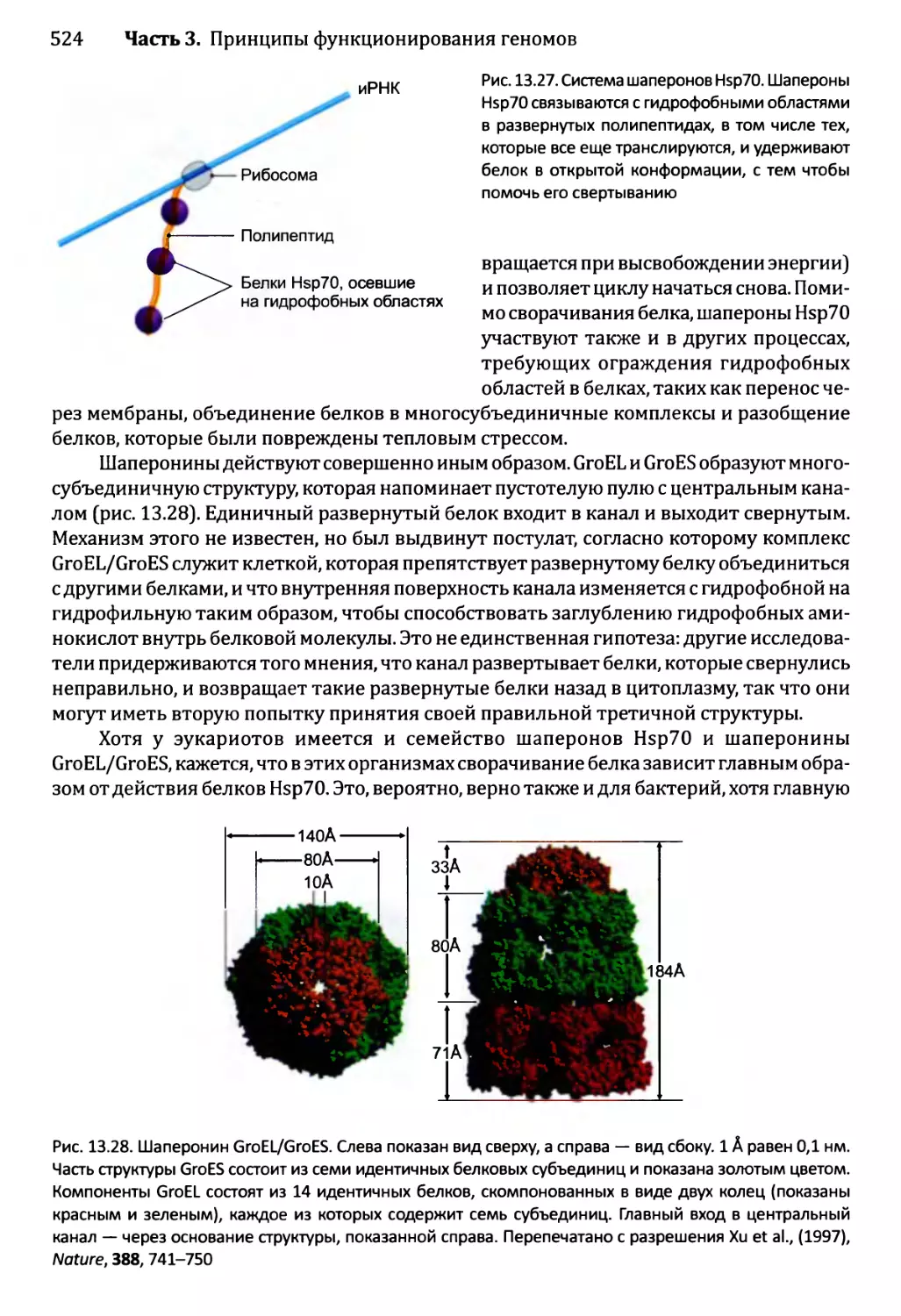






































































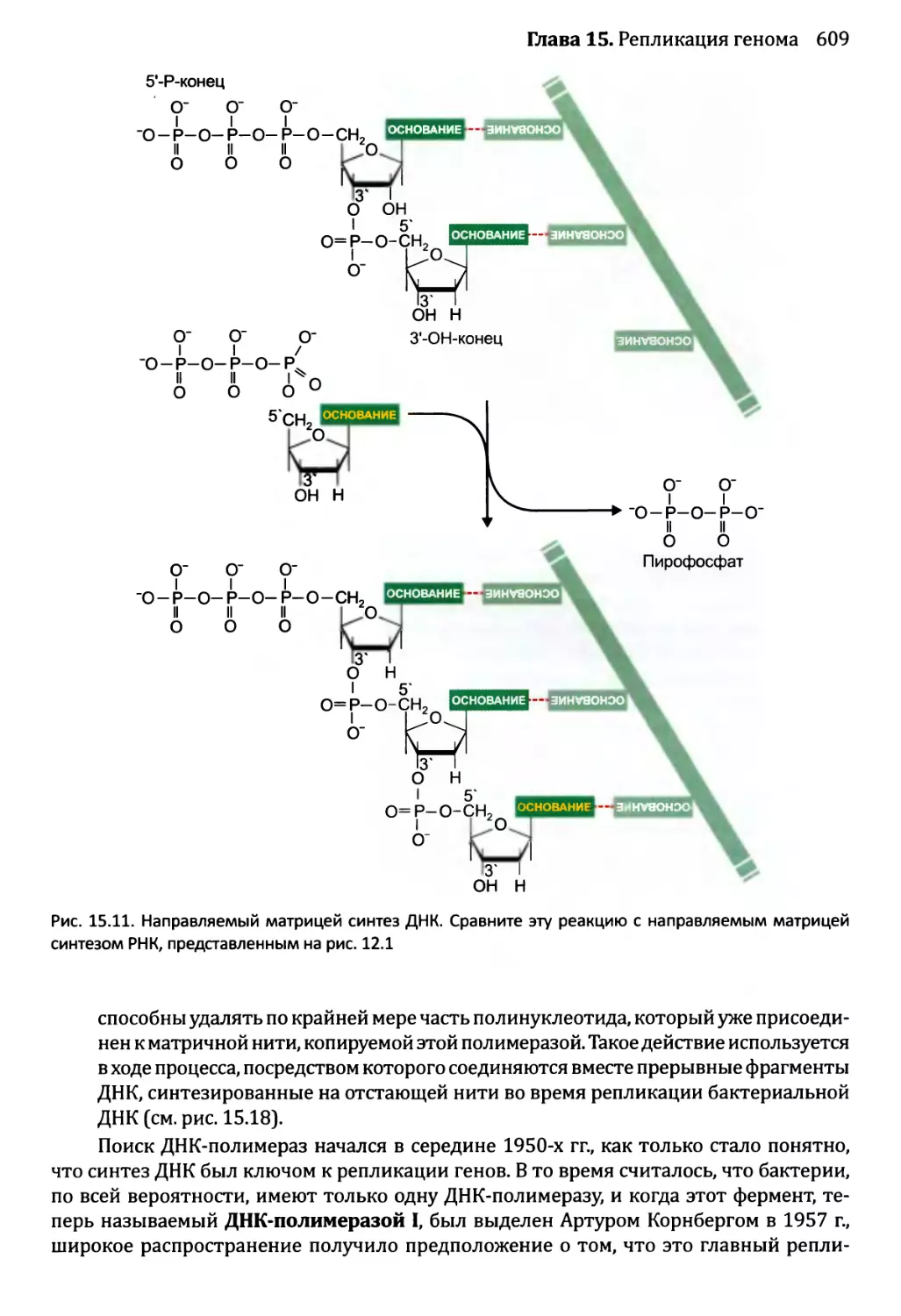



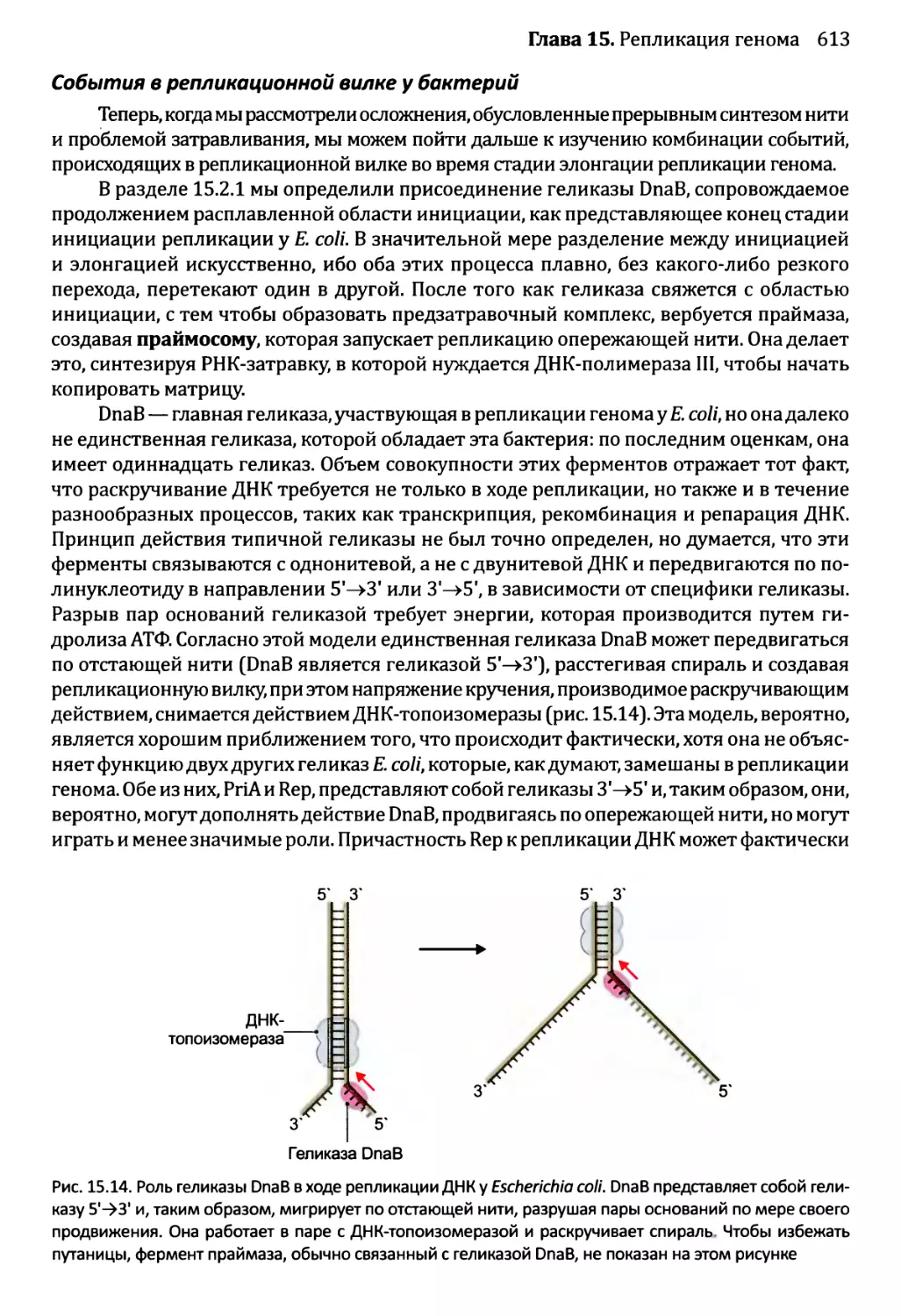
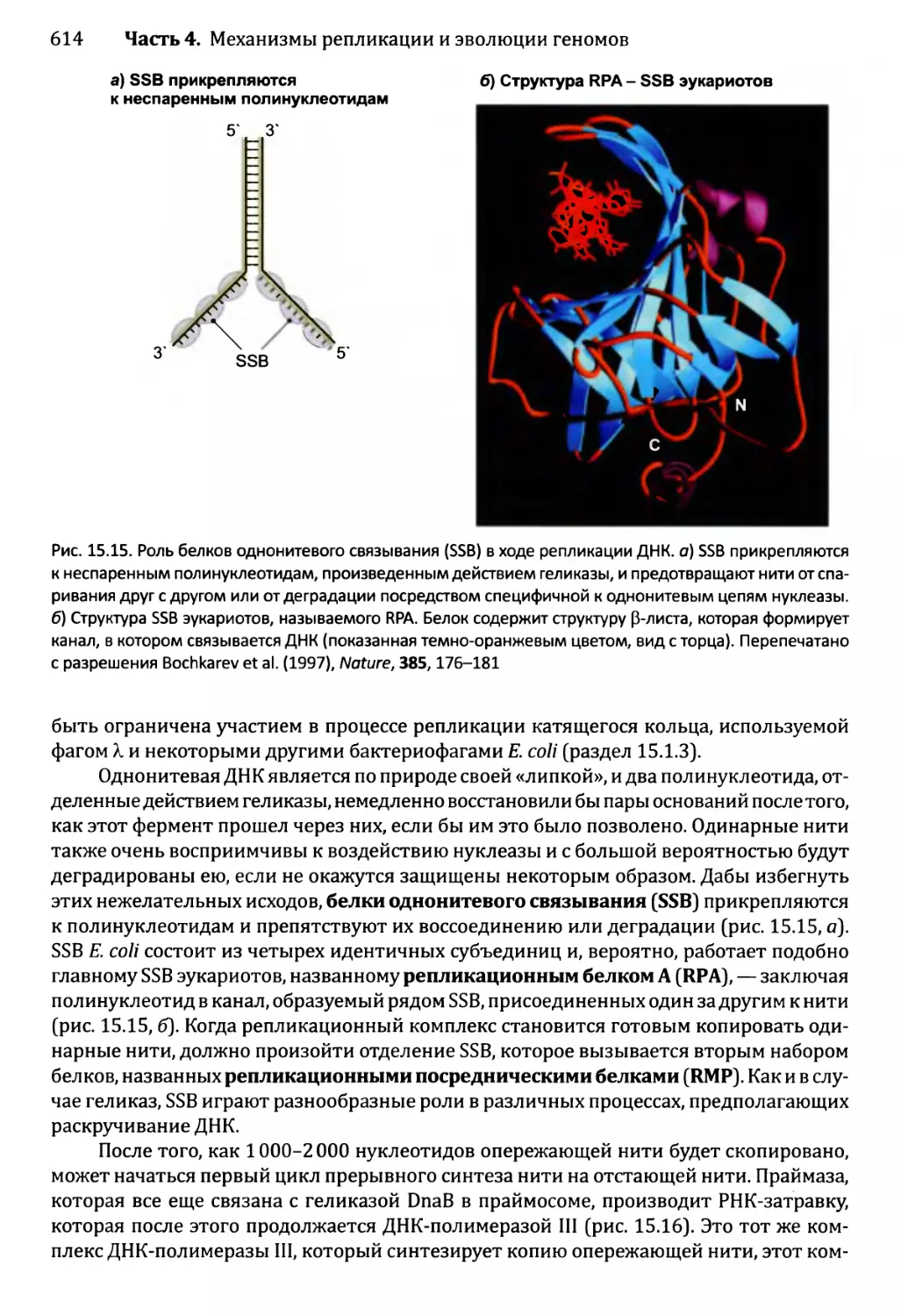
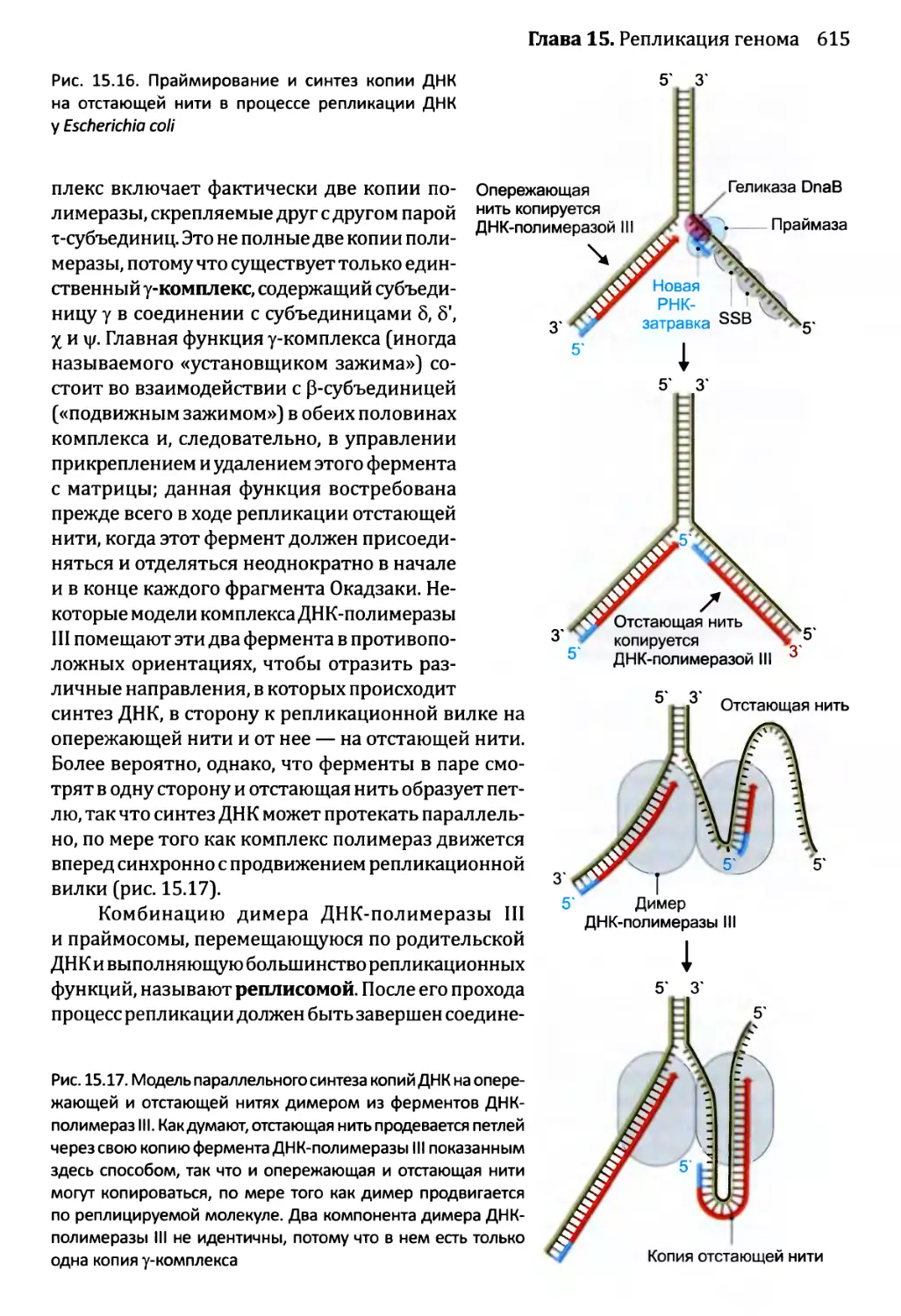
























































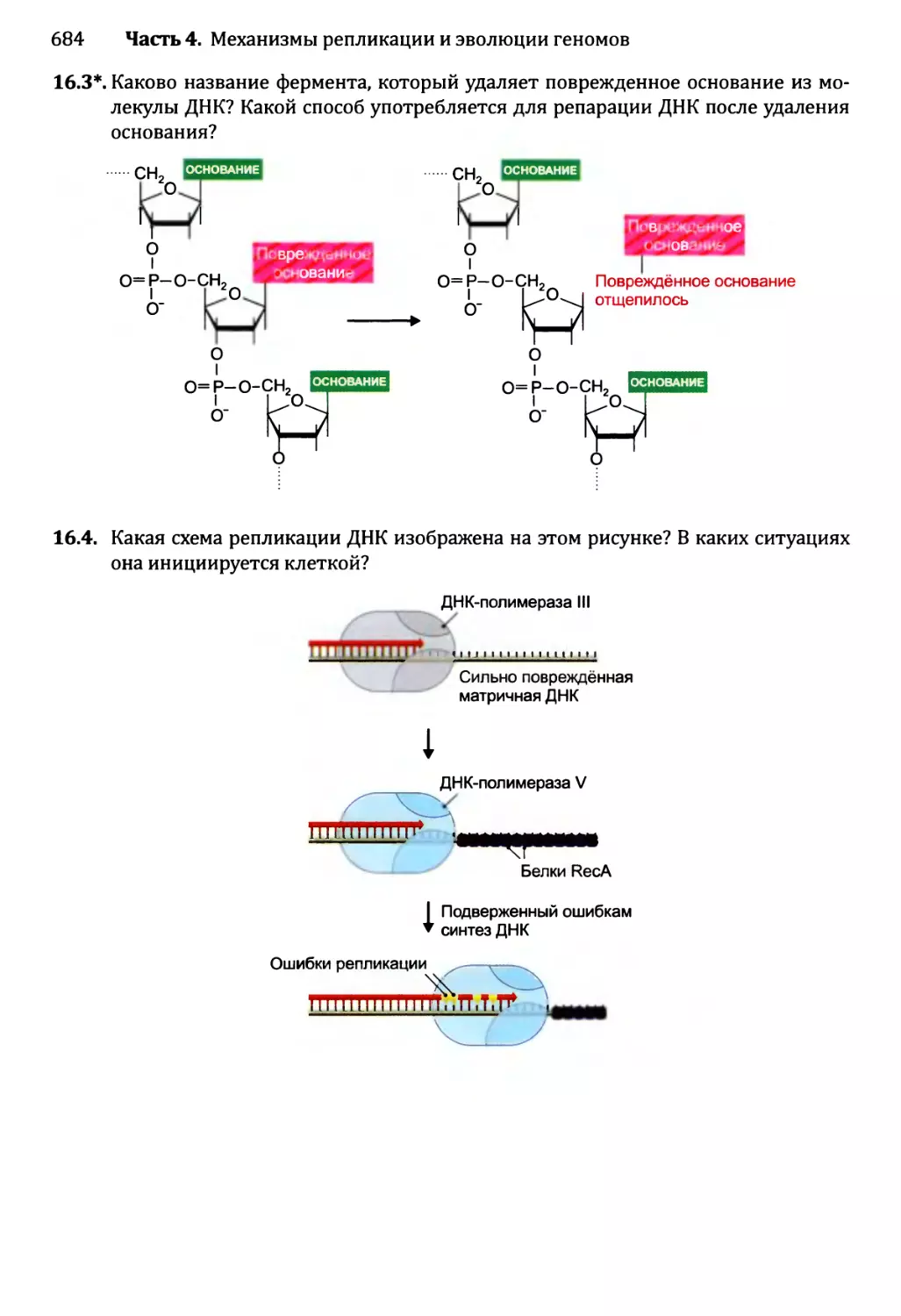



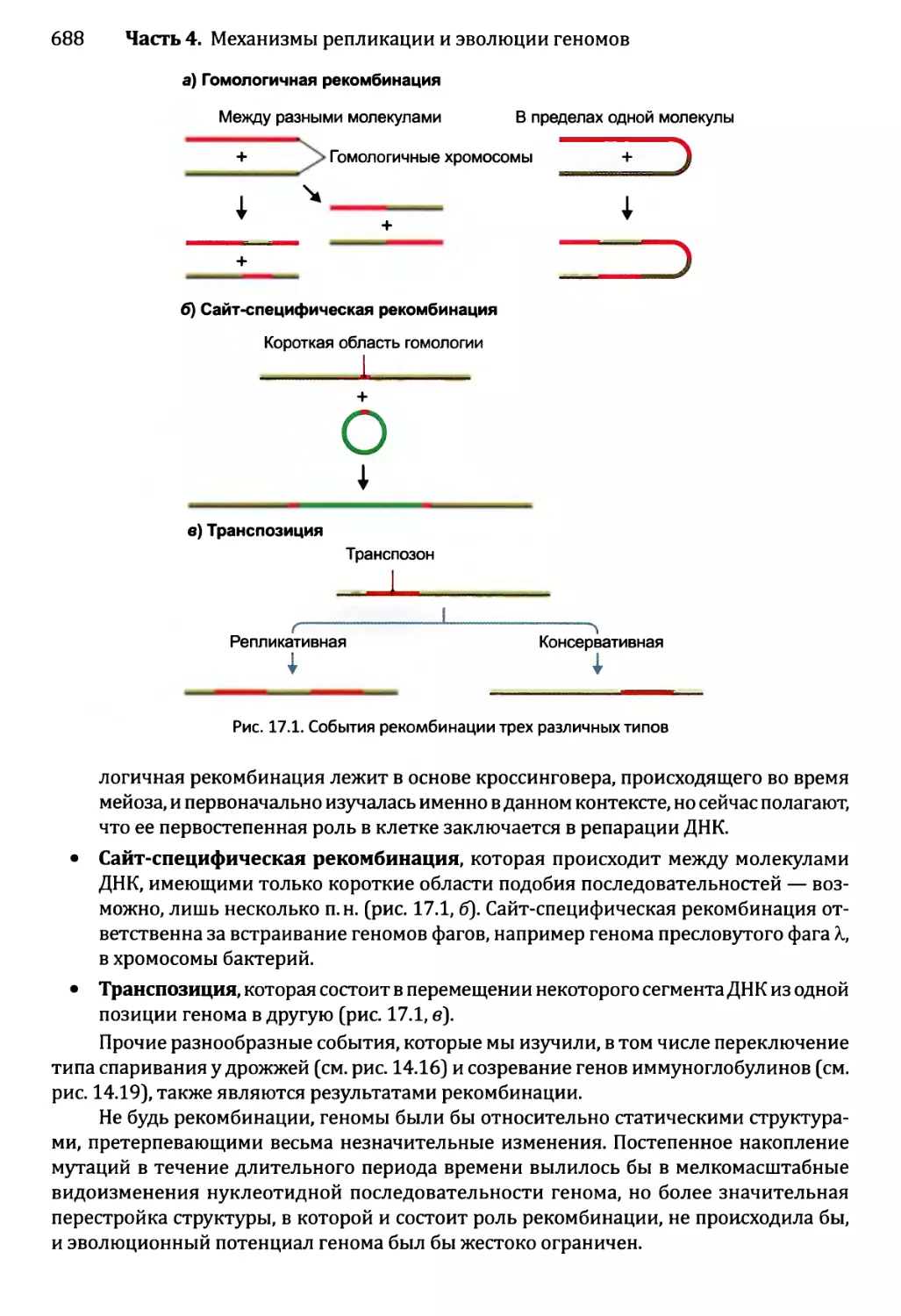

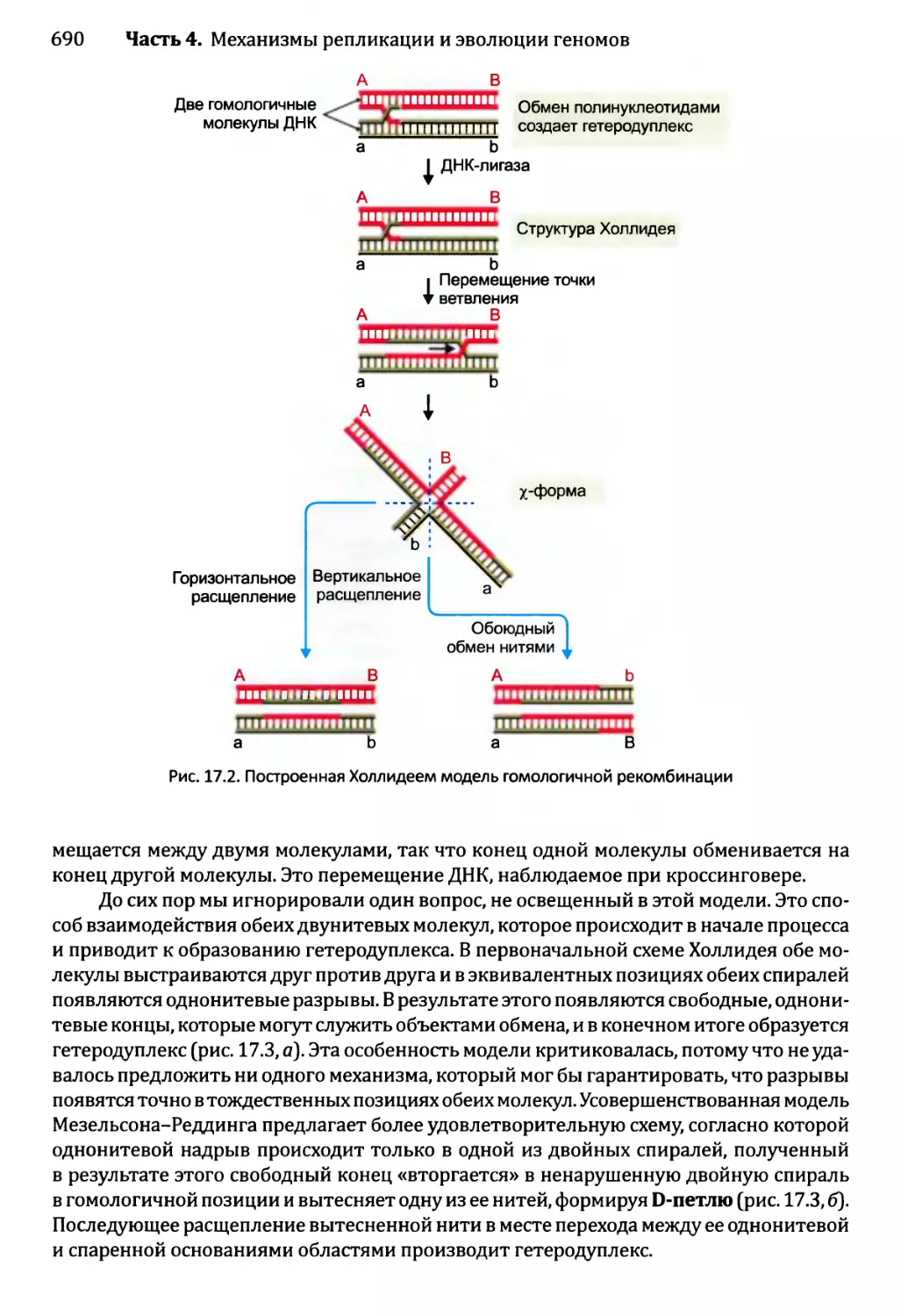







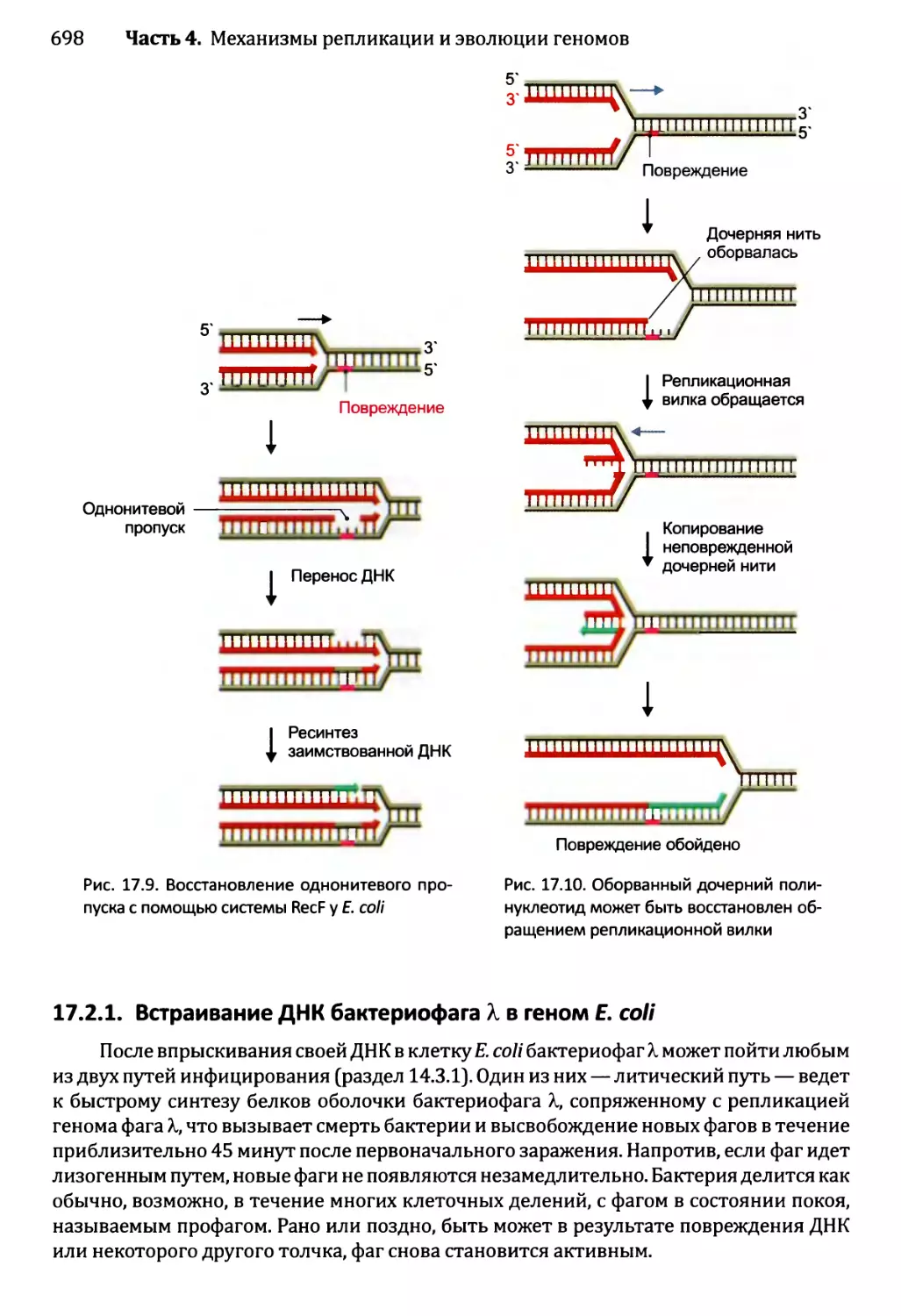




















































































































































































































Автор: Браун Т.А.
Теги: материальные основы жизни биохимия молекулярная биология биофизика микробиология генная инженерия переводная литература издательство геномы
ISBN: 0-8153-4138-5
Год: 2011




E.S75
Терри А. Браун
ГЕНОМЫ
Терри А. Браун
ГЕНОМЫ
Перевод с английского А. А. Светлова
Под редакцией д. б. н., проф. А. А. Миронова
Москва ♦ Ижевск
2011
УДК 577.2
ББК 28.070
Б 875
Издание осуществлено при финансовой поддержке Российского
фонда фундаментальных исследований по проекту № 08-04-07075
Браун Т. А.
Геномы / Пер. с англ. — М.-Ижевск: Институт компьютерных исследований, 2011. —
944 с.
Предлагаемая книга является первым наиболее полным и авторитетным руководством
по интенсивно развивающейся области науки — молекулярной генетике, аналогов которо-
му в мировой научной литературе нет. Издание охватывает молекулярную генетику от самых
основ до экпрессии генома и молекулярной филогении. Изложение сопровождается огромным
количеством цветных рисунков, в конце каждой главы приводятся задачи и вопросы, а также
библиографический список.
Книга предназначена для студентов, аспирантов и исследователей в области молекуляр-
ной биологии, генетики и биоинформатики, а также всех специалистов, работающих в смежных
с биологией и медициной областях.
ISBN 0-8153-4138-5 (англ.)
ISBN 978-5-4344-0002-2 (рус.)
© Garland Science Publishing, 2007
© Перевод на русский язык:
Ижевский институт компьютерных исследований, 2011
Перевод англоязычного издания Genomes 3rd ed./Т. A. Brown опубликован с разрешения издатель-
ства Garland, являющегося подразделением компании Taylor & Francis.
В данной книге содержится информация, полученная из авторитетных и весьма уважаемых ис-
точников. Перепечатанный в виде цитат материал приводится с разрешения авторов и указанием
источников. В книге представлен обширный список цитируемой литературы. Приложены все
усилия к тому, чтобы опубликованная информация была надежной, однако автор и издатель не
могут нести ответственность за достоверность всех приведенных материалов или последствия
их использования.
Все права защищены. Никакая часть данной книги не может быть перепечатана, воспроизведена,
передана или использована в какой бы то ни было форме электронными, механическими или
любыми иными средствами, которые известны в настоящее время или будут изобретены впо-
следствии, включая фотокопирование, запись на магнитный носитель, микросъемку, или при
помощи любой другой системы хранения и обработки информации, если на то нет письменного
разрешения издательств.
http://shop.rcd.ru
http://ics.org.ru
Оглавление
Предисловие..........................................................xi
Примечание читателю................................................xiii
Сокращения........................................................xviii
Часть 1, Изучение геномов.............................................1
Глава 1. Геномы, транскриптомы и протеомы.............................3
1.1. ДНК..........................................................6
1.1.1. Гены состоят из ДНК...................................6
1.1.2. Структура ДНК.........................................8
1.2. РНК и транскриптом..........................................16
1.2.1. Структура РНК........................................16
1.2.2. Виды и содержание РНК в клетке.......................18
1.2.3. Созревание РНК-предшественника.......................19
1.2.4. Транскриптом.........................................20
1.3. Белки и протеом.............................................21
1.3.1. Структура белка......................................21
1.3.2. Протеом..............................................25
Глава 2. Изучение ДНК...................................................37
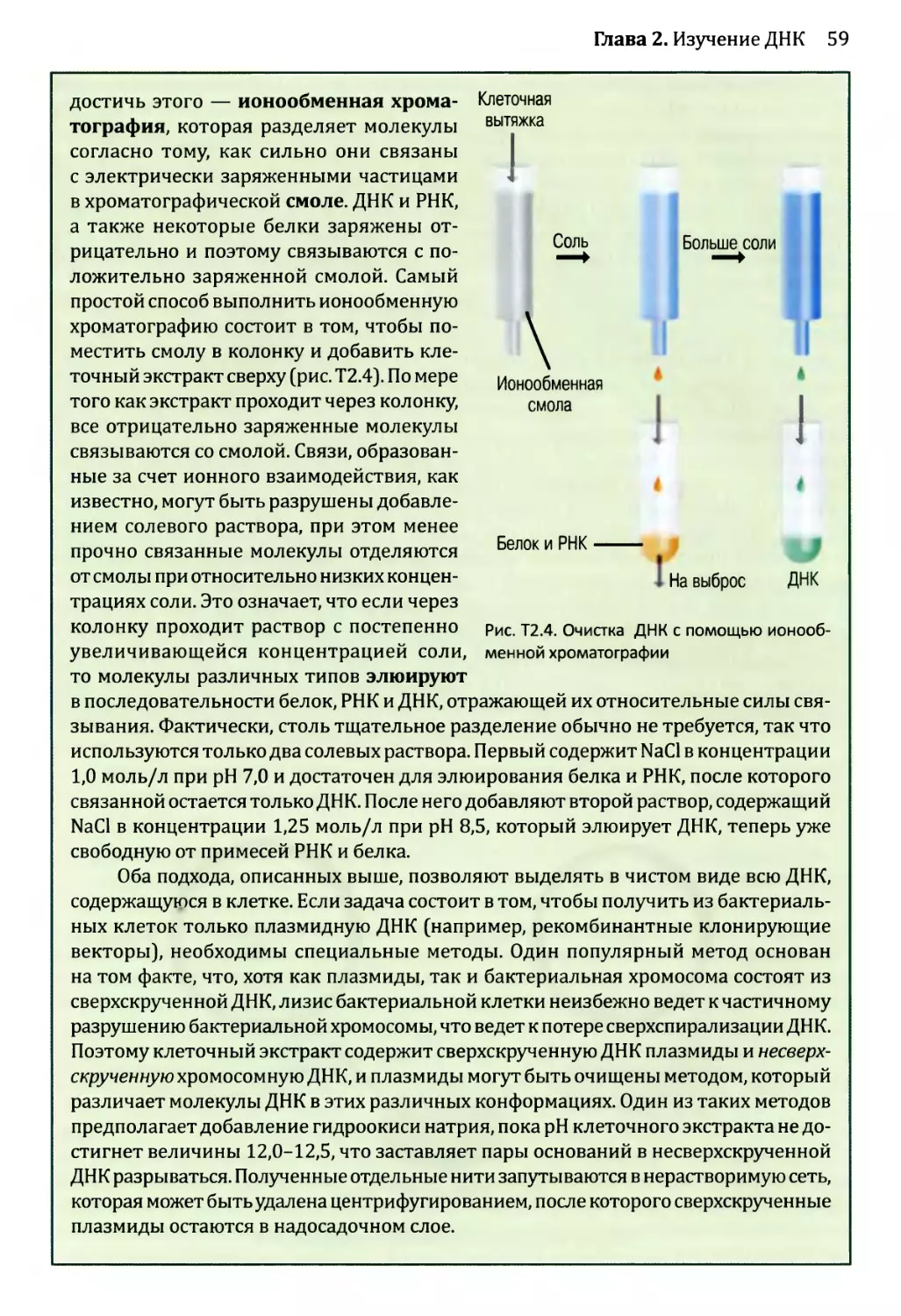
2.1. Ферменты для манипуляций с ДНК..............................39
2.1.1. ДНК-полимеразы.......................................40
2.1.2. Нуклеазы.............................................46
2.1.3. ДНК-лигазы...........................................51
2.1.4. Ферменты модификации концов..........................53
2.2. Клонирование ДНК............................................54
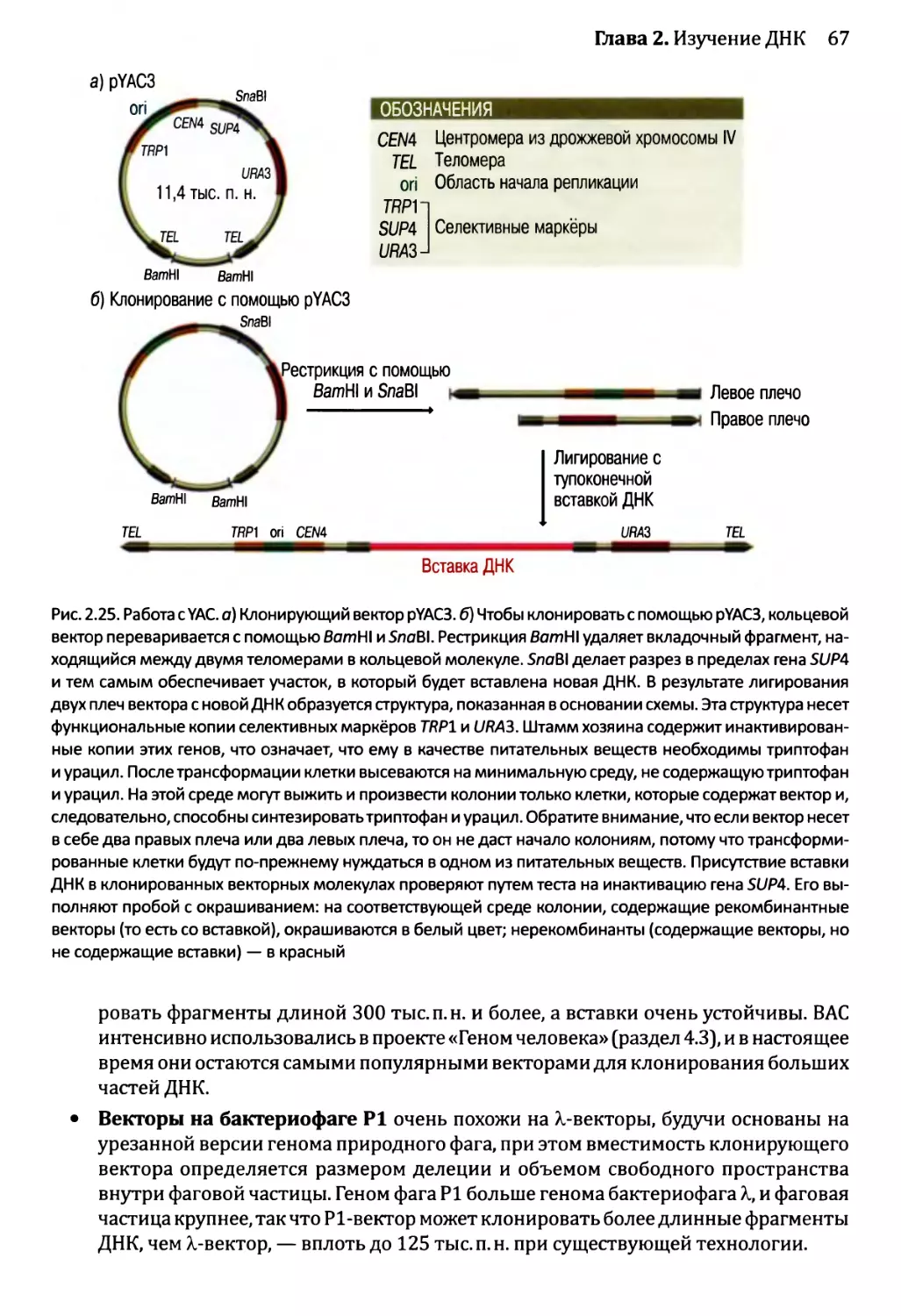
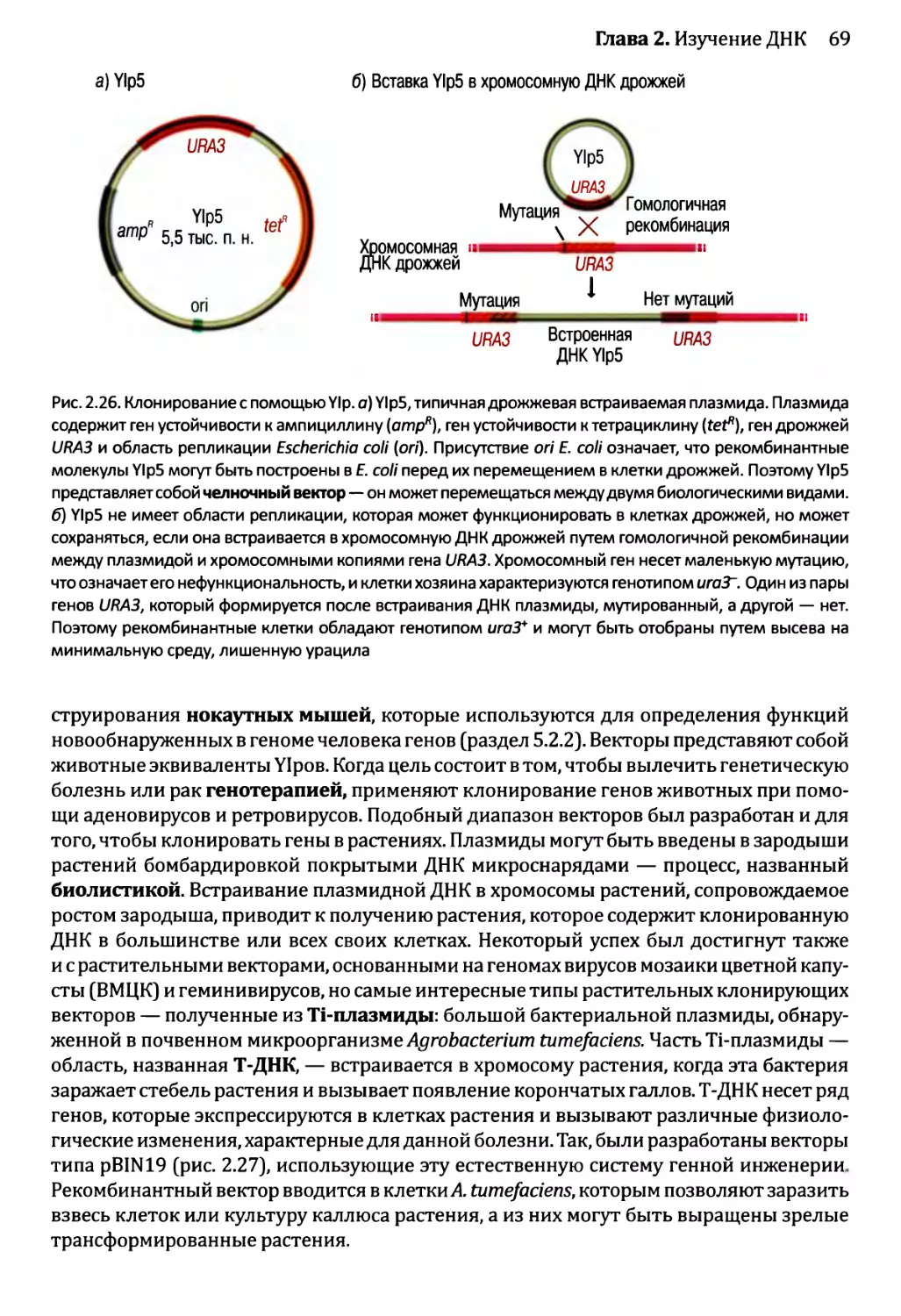
2.2.1. Клонирующие векторы и путь их применения.............56
2.3. Полимеразная цепная реакция (ПЦР)...........................70
2.3.1. Проведение ПЦР.......................................70
2.3.2. Применение ПЦР.......................................72
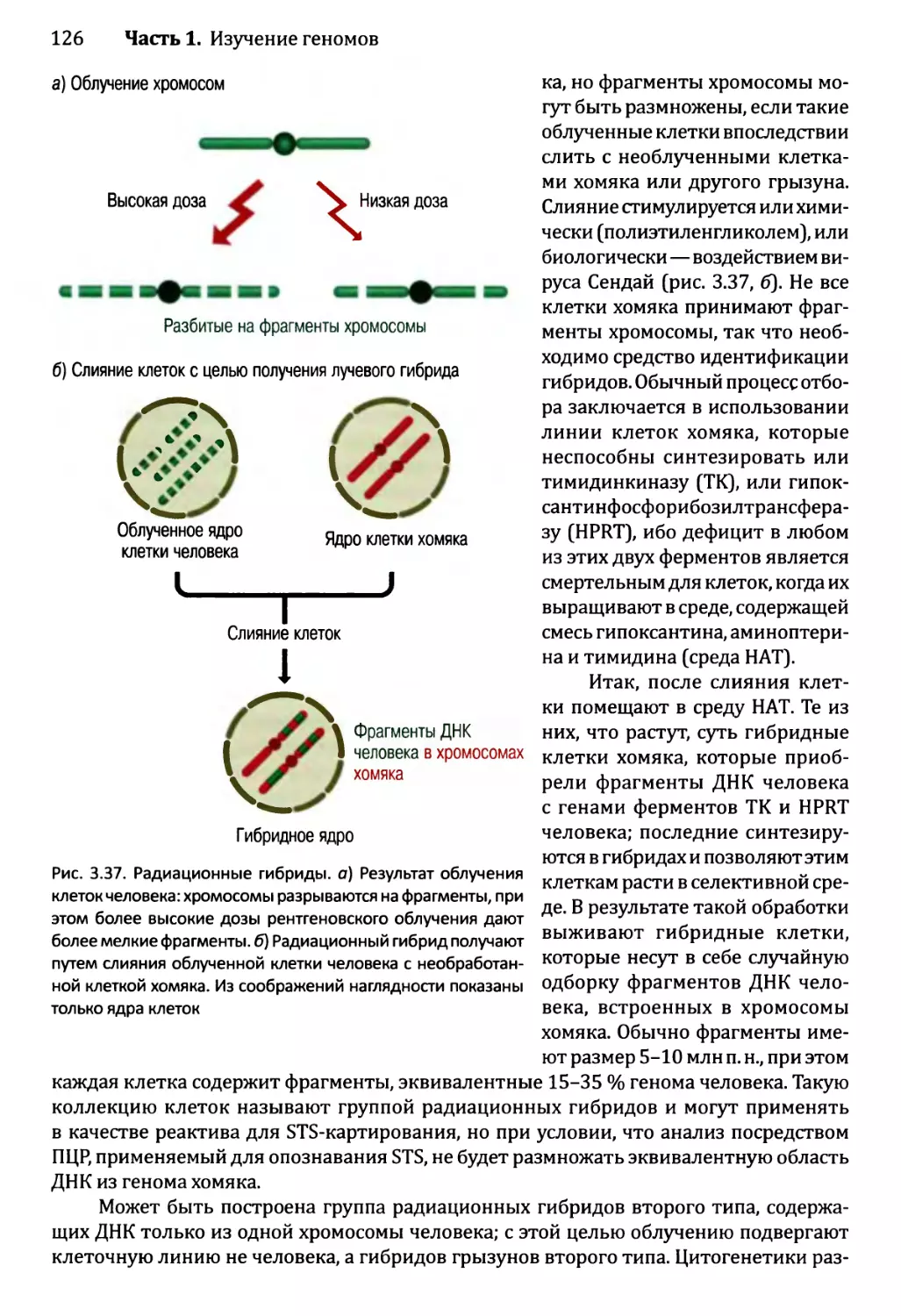
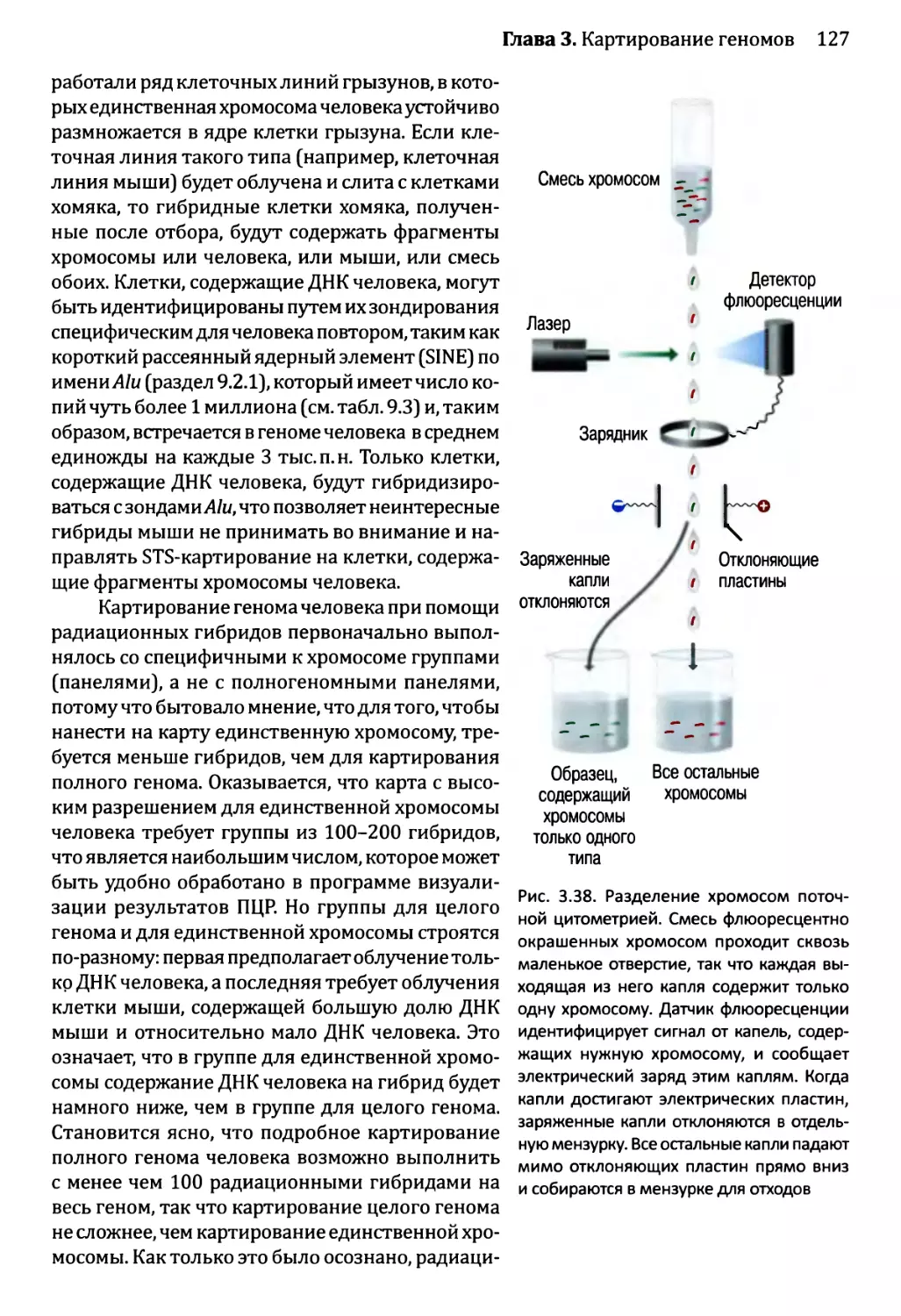
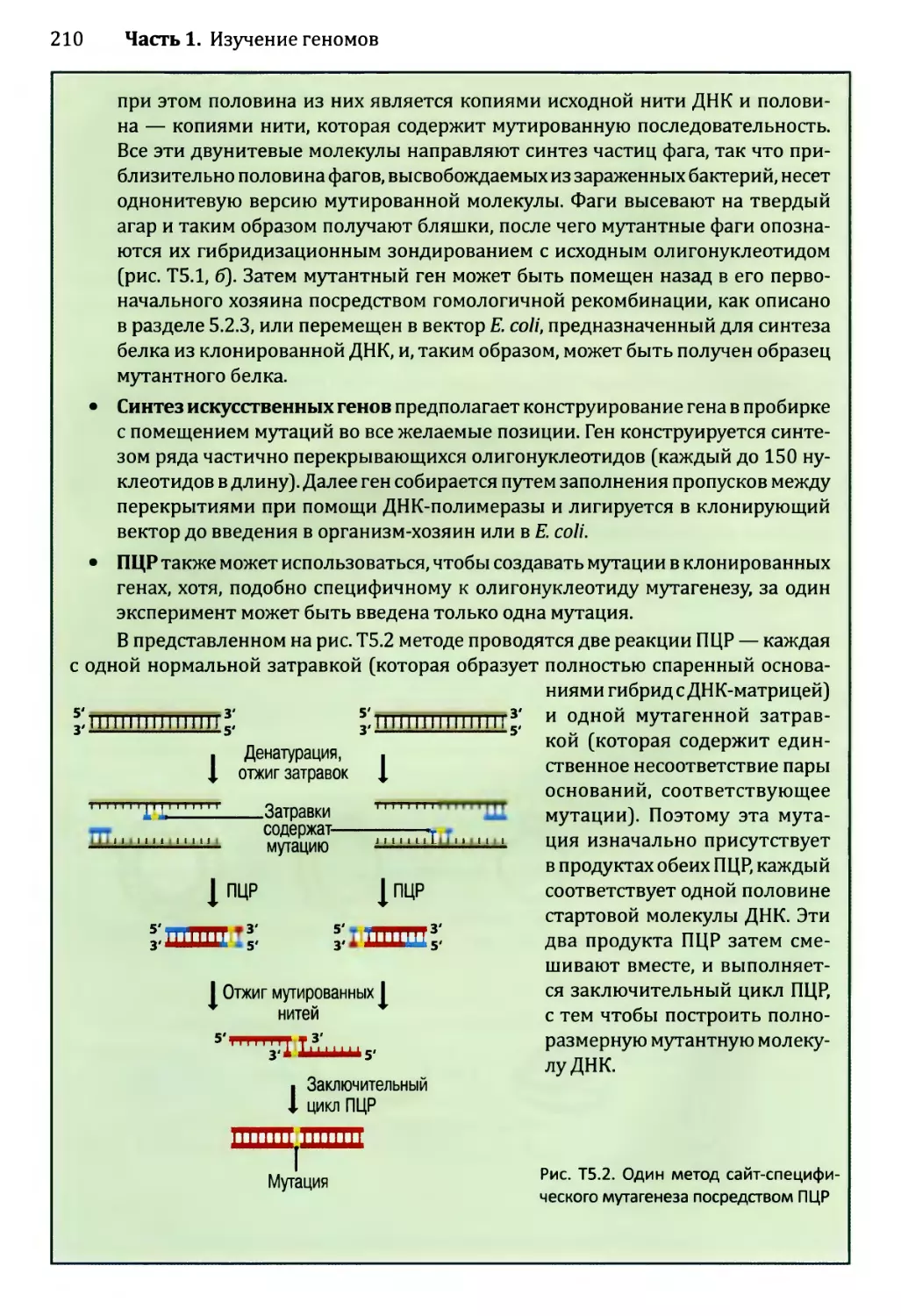
Глава 3. Картирование геномов...........................................81
3.1. Генетические и физические карты.............................83
3.2. Составление генетических карт...............................84
3.2.1. Первыми известными маркёрами служили гены............84
3.2.2. ДНК-маркёры для составления генетических карт........85
3.2.3. Анализ сцепления — основа составления генетических карт.95
3.2.4. Анализ сцепления генетических признаков у организмов
различного типа.....................................103
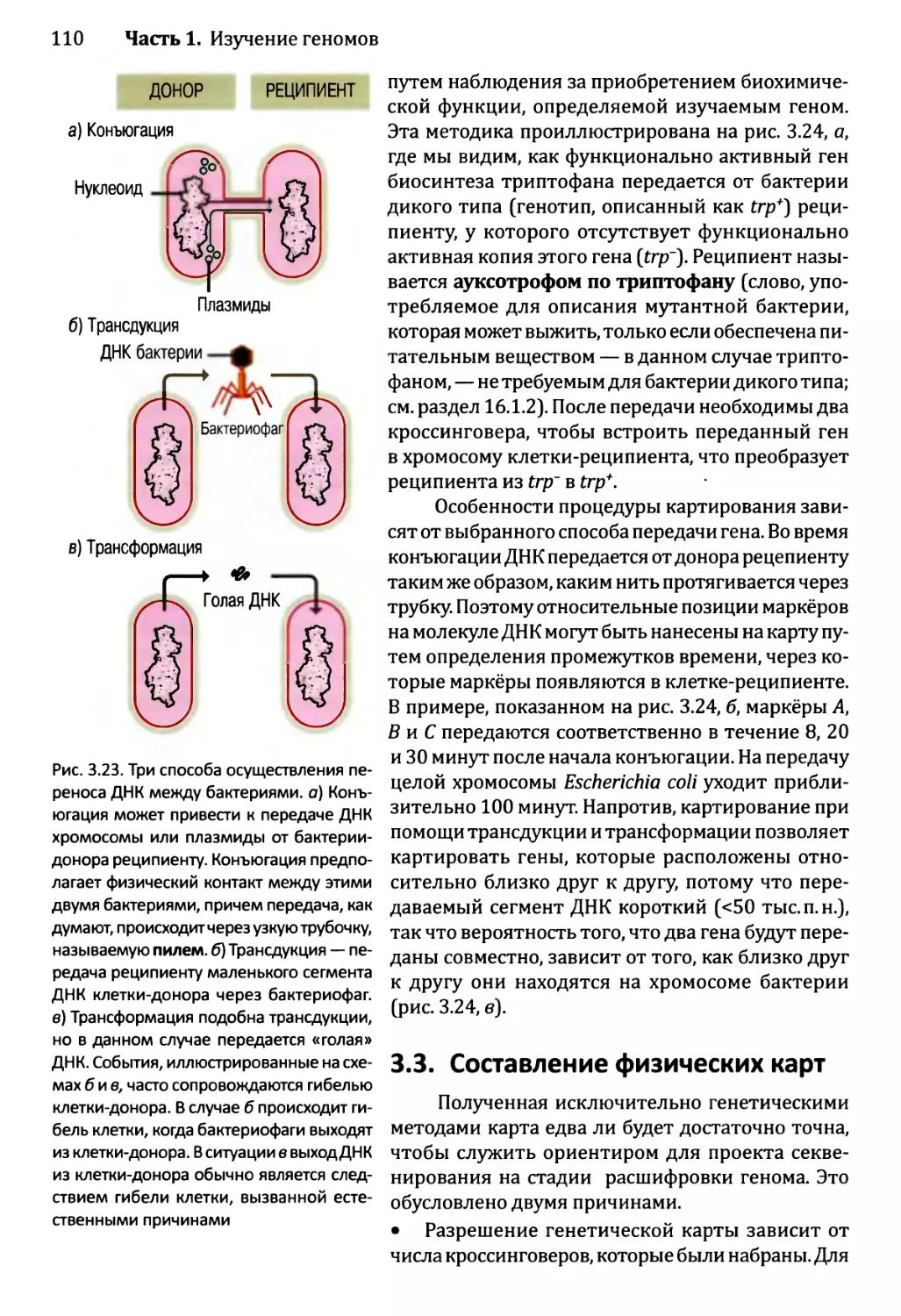
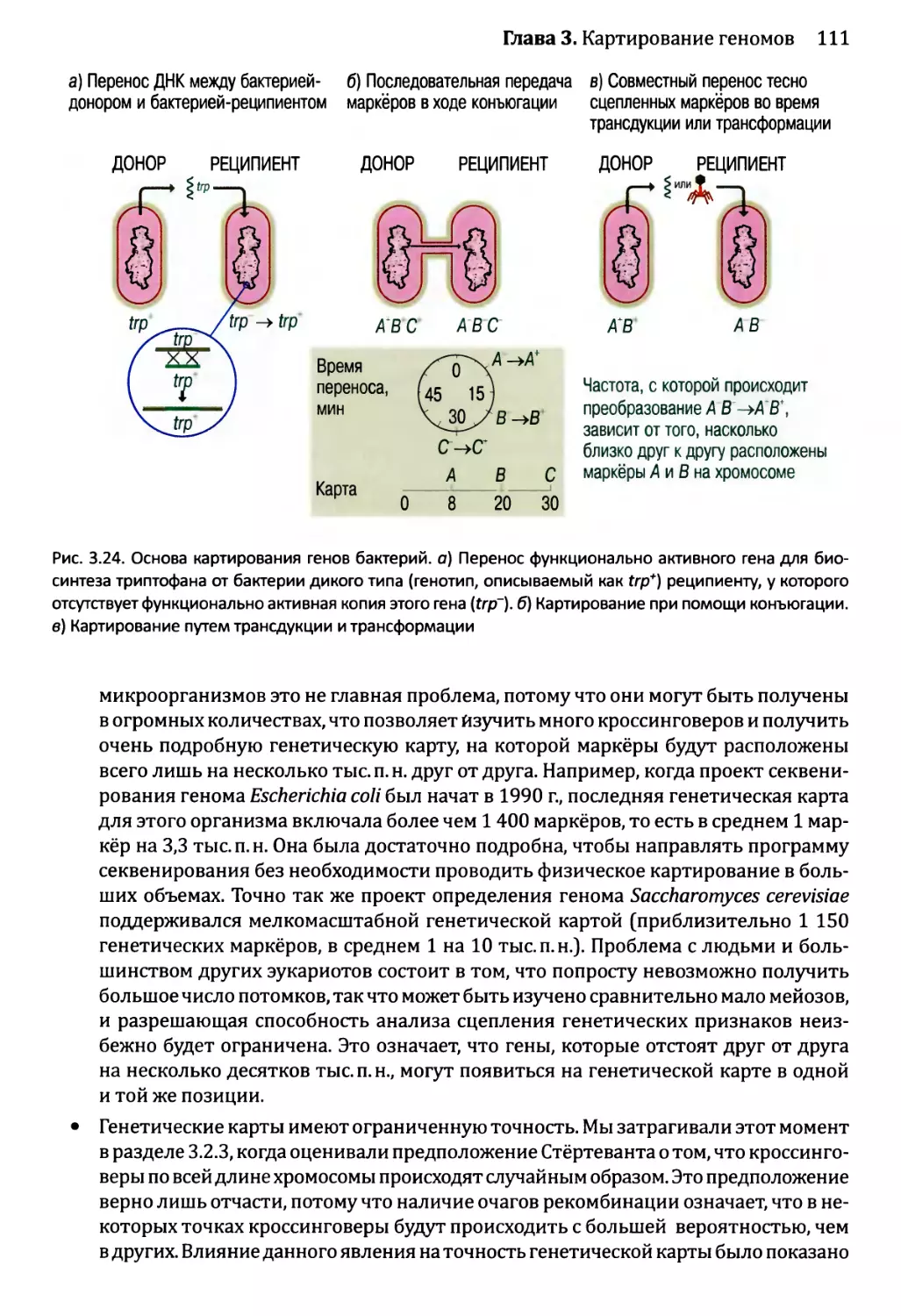
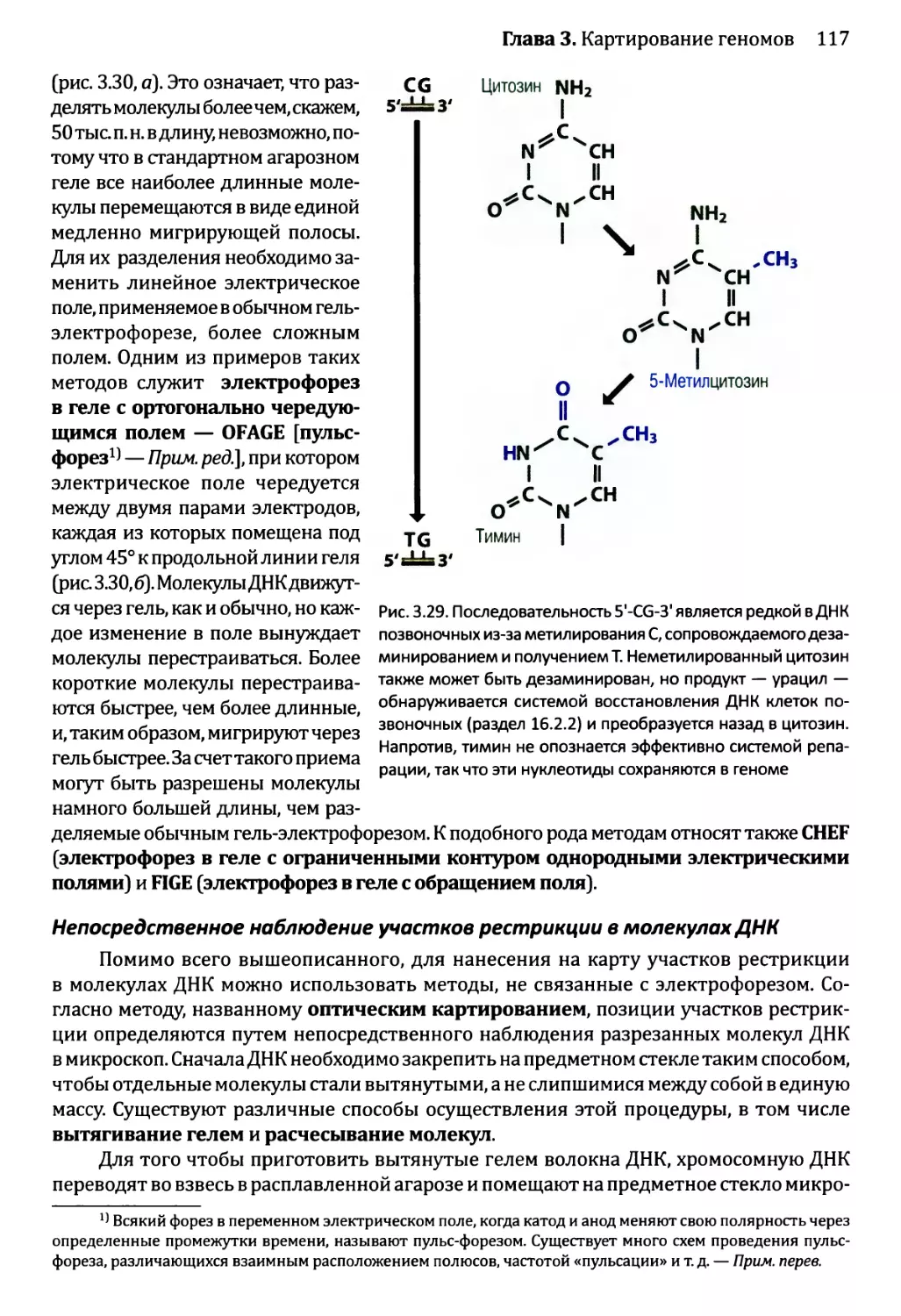
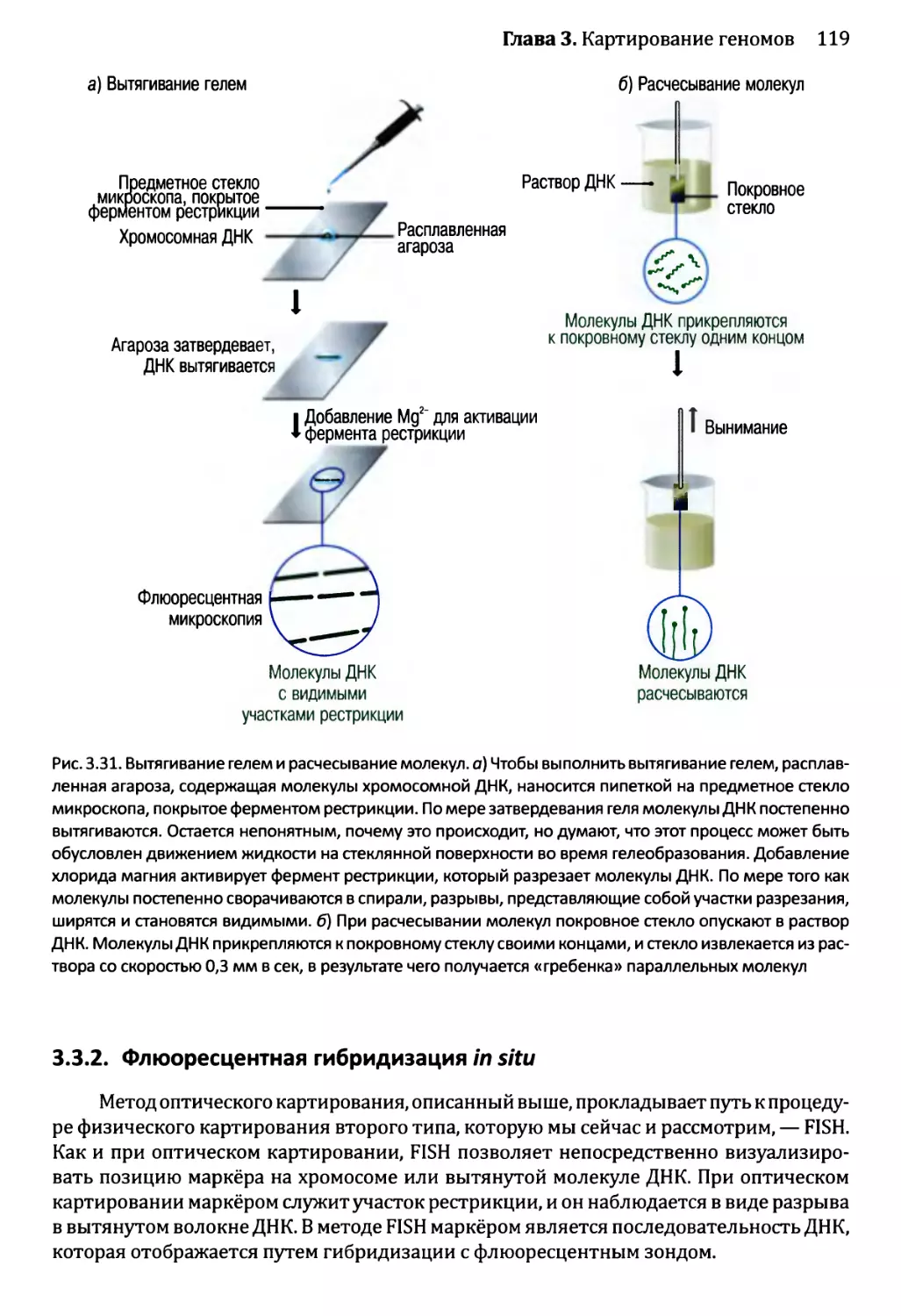
3.3. Составление физических карт................................110
3.3.1. Составление рестрикционных карт.....................112
3.3.2. Флюоресцентная гибридизация in situ.................119
3.3.3. Картирование с помощью меченых участков
последовательности..................................123
vi Оглавление
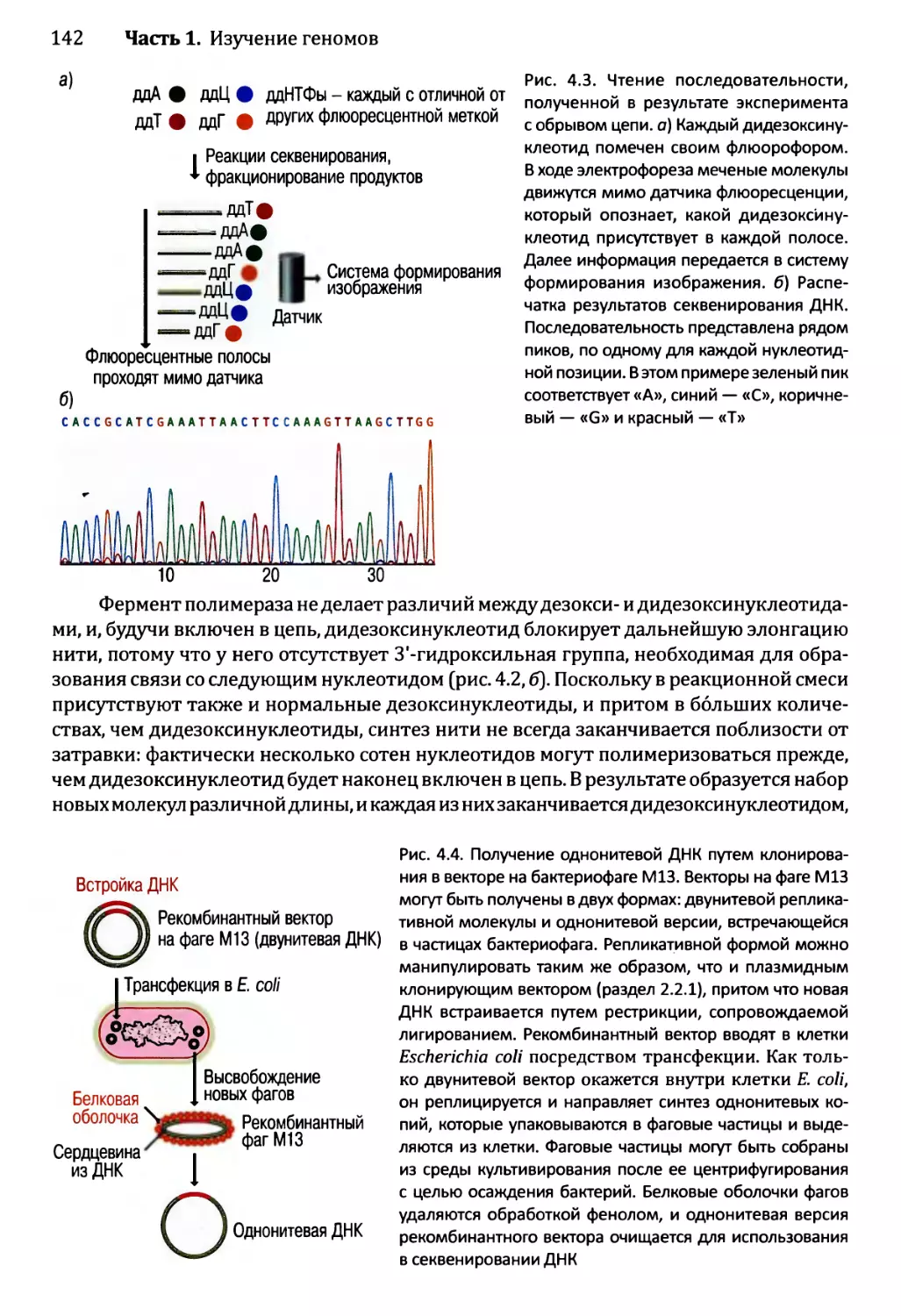
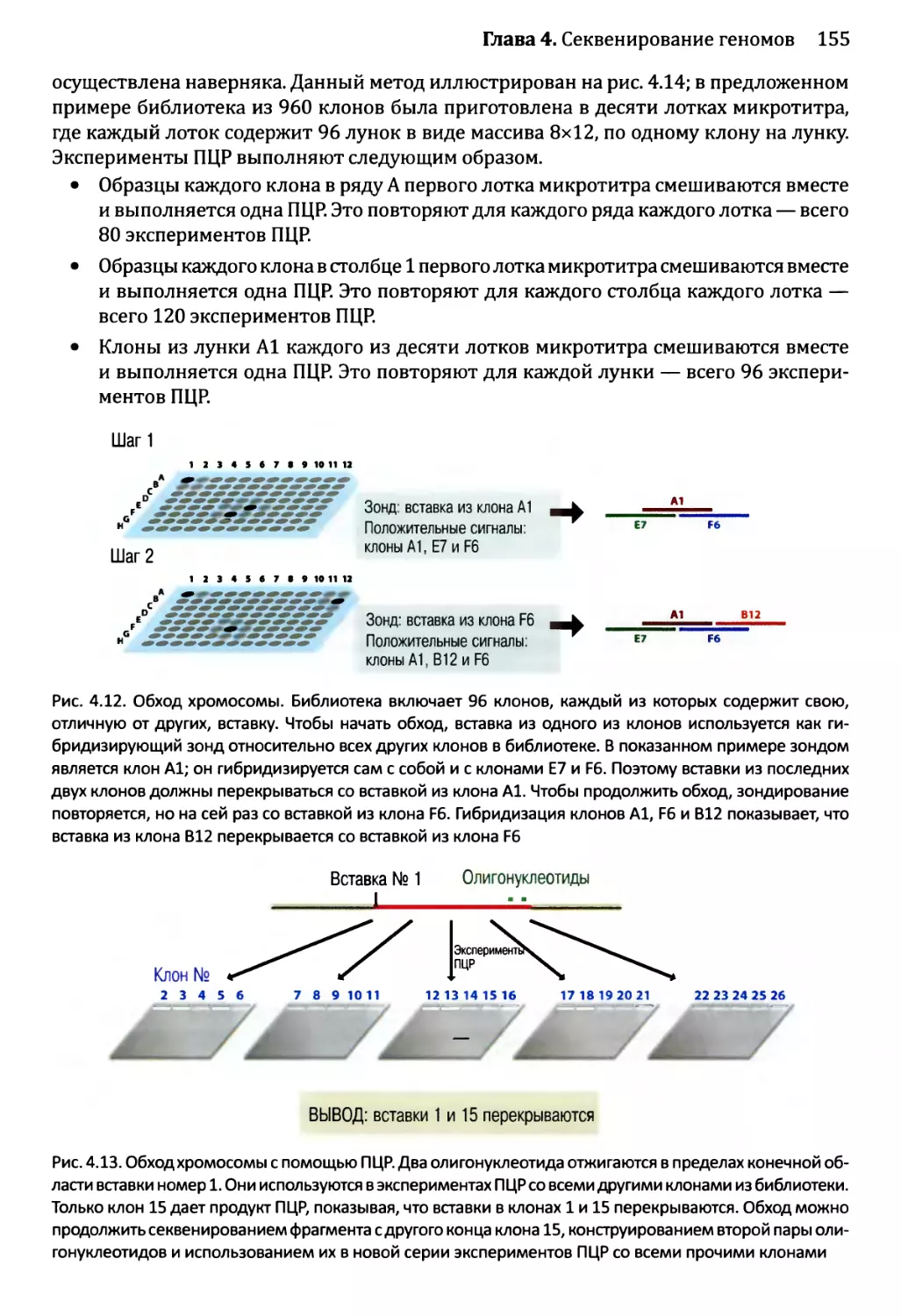
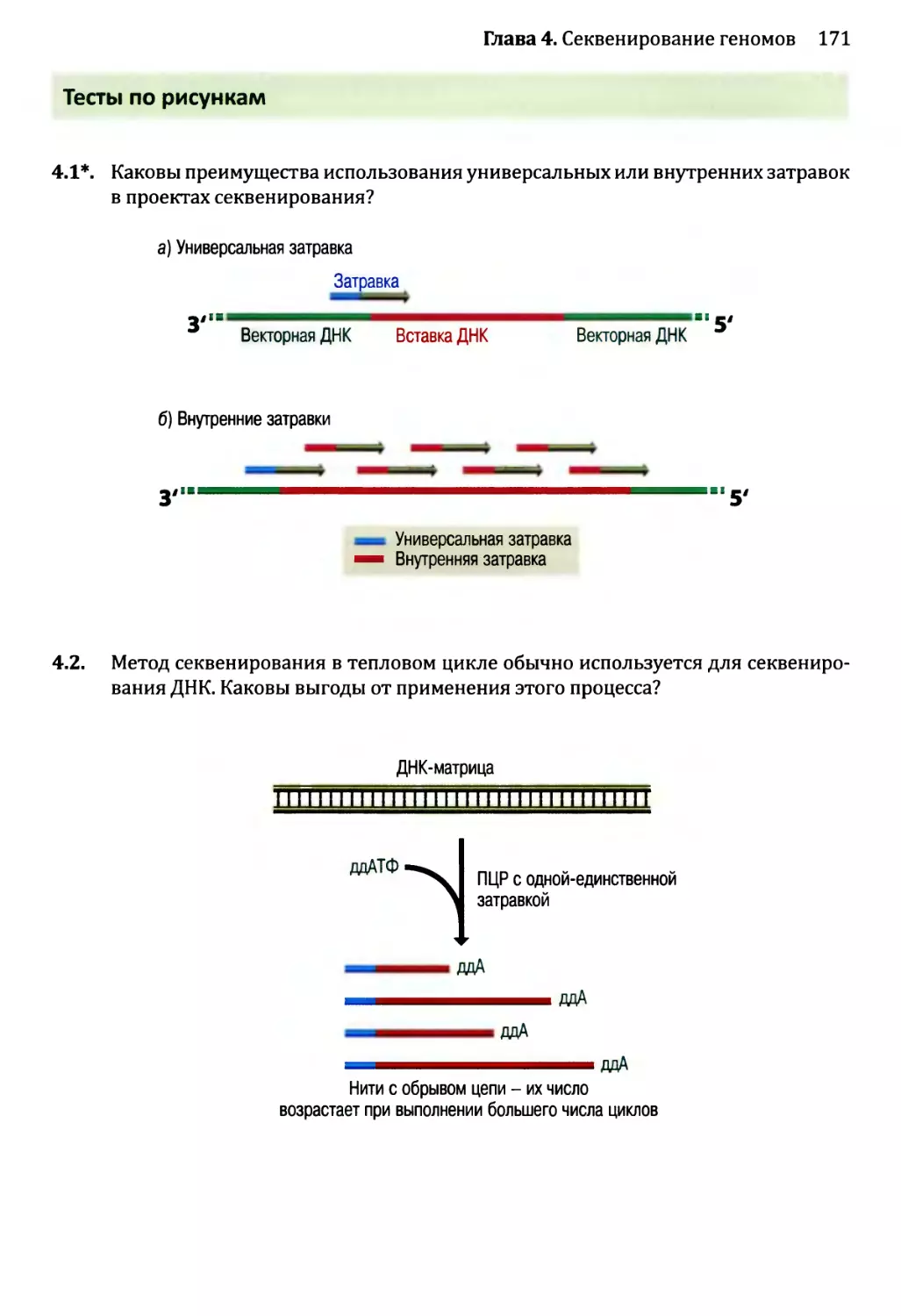
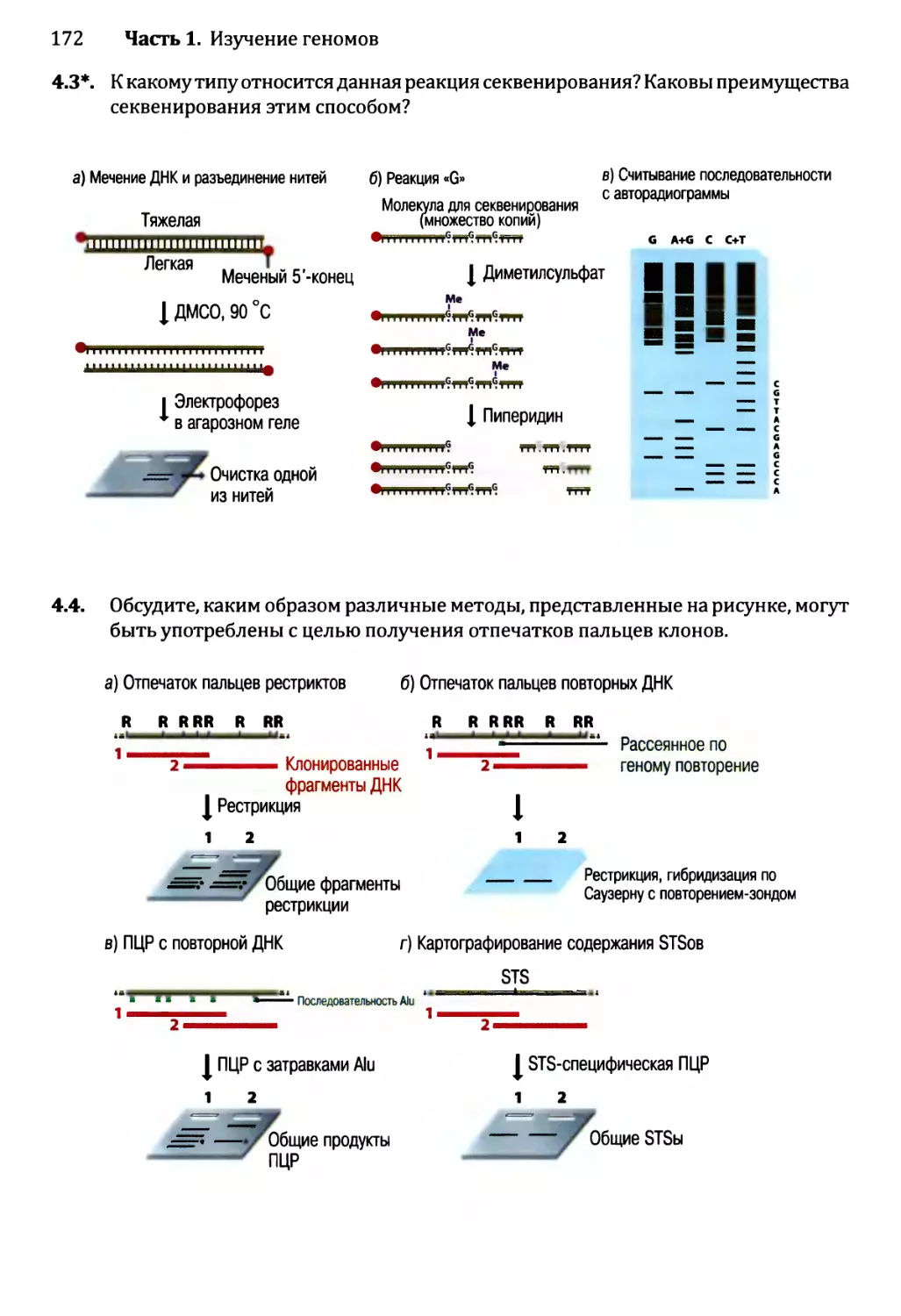
Глава 4. Секвенирование геномов.........................................138
4.1. Методология секвенирования ДНК.................................139
4.1.1. Секвенирование ДНК с обрывом цепи.......................139
4.1.2. Альтернативные методы секвенирования ДНК................146
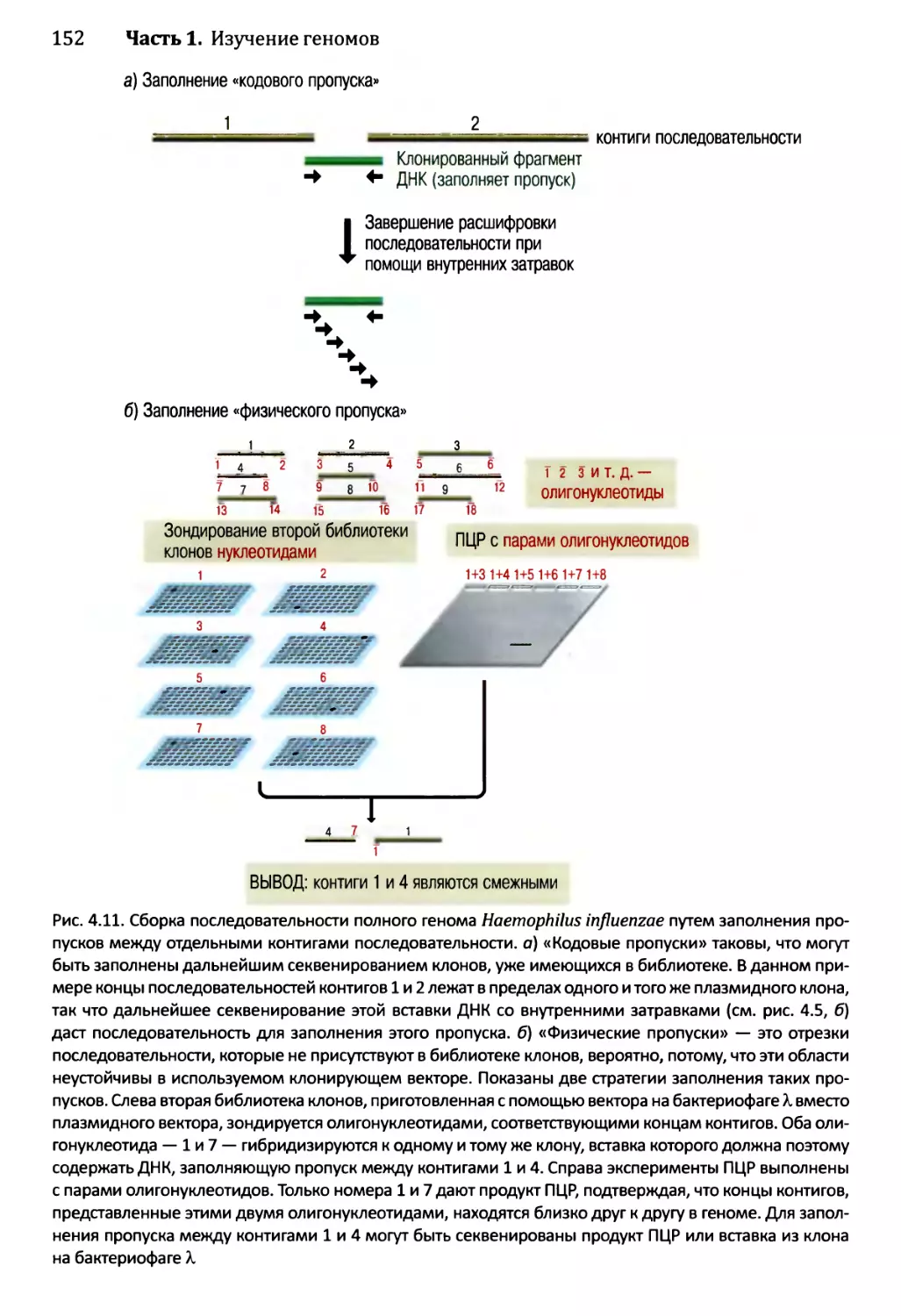
4.2. Сборка непрерывной последовательности ДНК......................150
4.2.1. Сборка последовательности методом дробовика.............150
4.2.2. Сборка последовательностей методом сборки контигов
из клонов...................................................153
4.2.3. Секвенирование методом дробовика для полных геномов...158
4.3. Проекты расшифровки генома человека............................161
4.3.1. Стадия картирования в проекте «Геном человека»..........161
4.3.2. Секвенирование генома человека..........................163
4.3.3. Будущее проектов по изучению генома человека............164
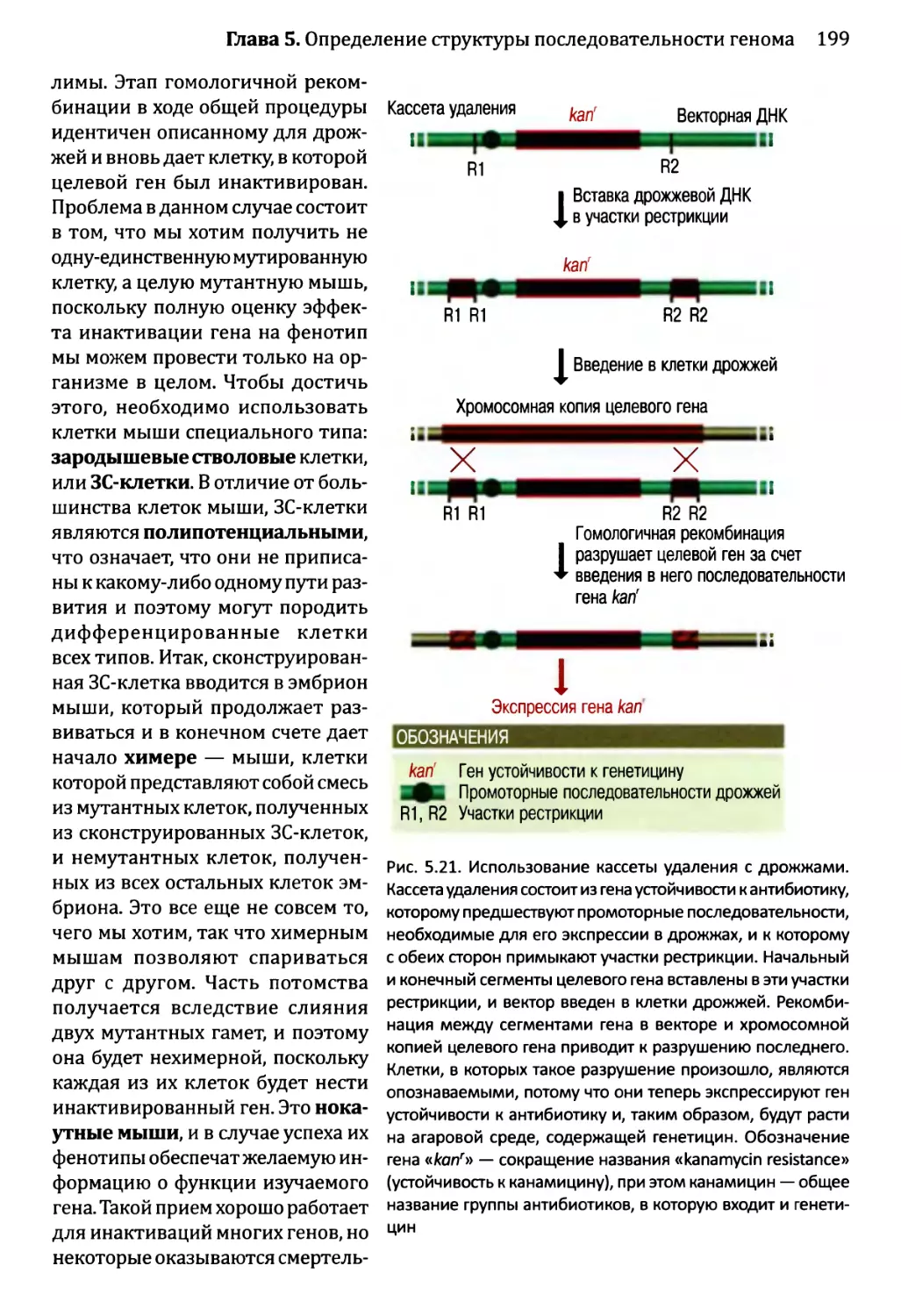
Глава 5. Определение структуры последовательности генома и функций,
ее составляющих.........................................................175
5.1. Определение местоположения генов в последовательности генома.176
5.1.1. Картирование генов с помощью анализа последовательности.176
5.1.2. Экспериментальные методы определения местоположения
генов...................................................185
5.2. Определение функций отдельных генов............................191
5.2.1. Компьютерный анализ функций гена........................192
5.2.2. Экспериментальное определение функций гена..............197
5.2.3. Более подробные исследования активности белка,
кодируемого неизвестным геном...........................205
5.3. Наглядный пример: аннотирование последовательности
генома Sacharomyces cerevisiae.................................208
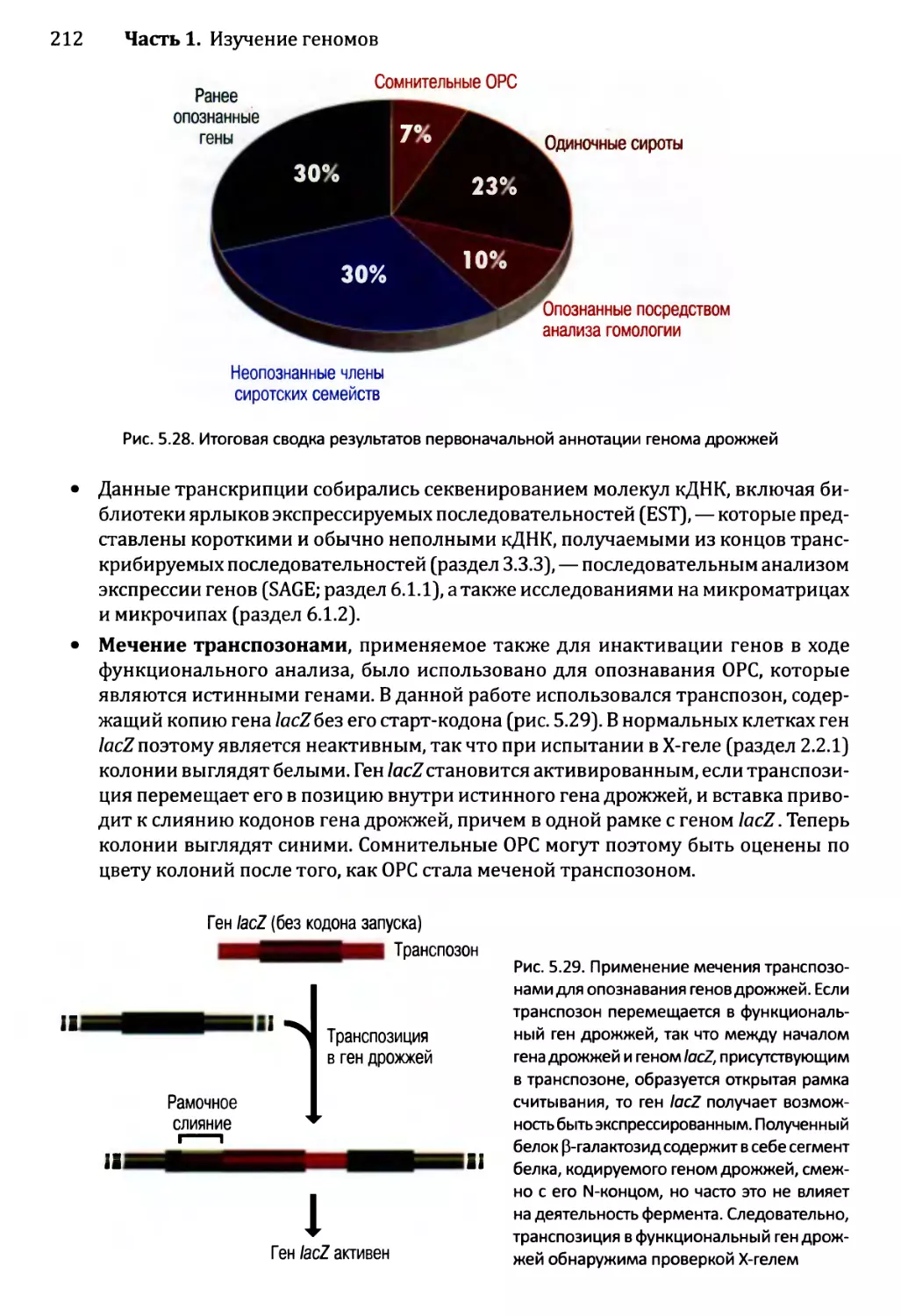
5.3.1. Аннотирование последовательности генома дрожжей.........208
5.3.2. Приписывание функций генам дрожжей......................213
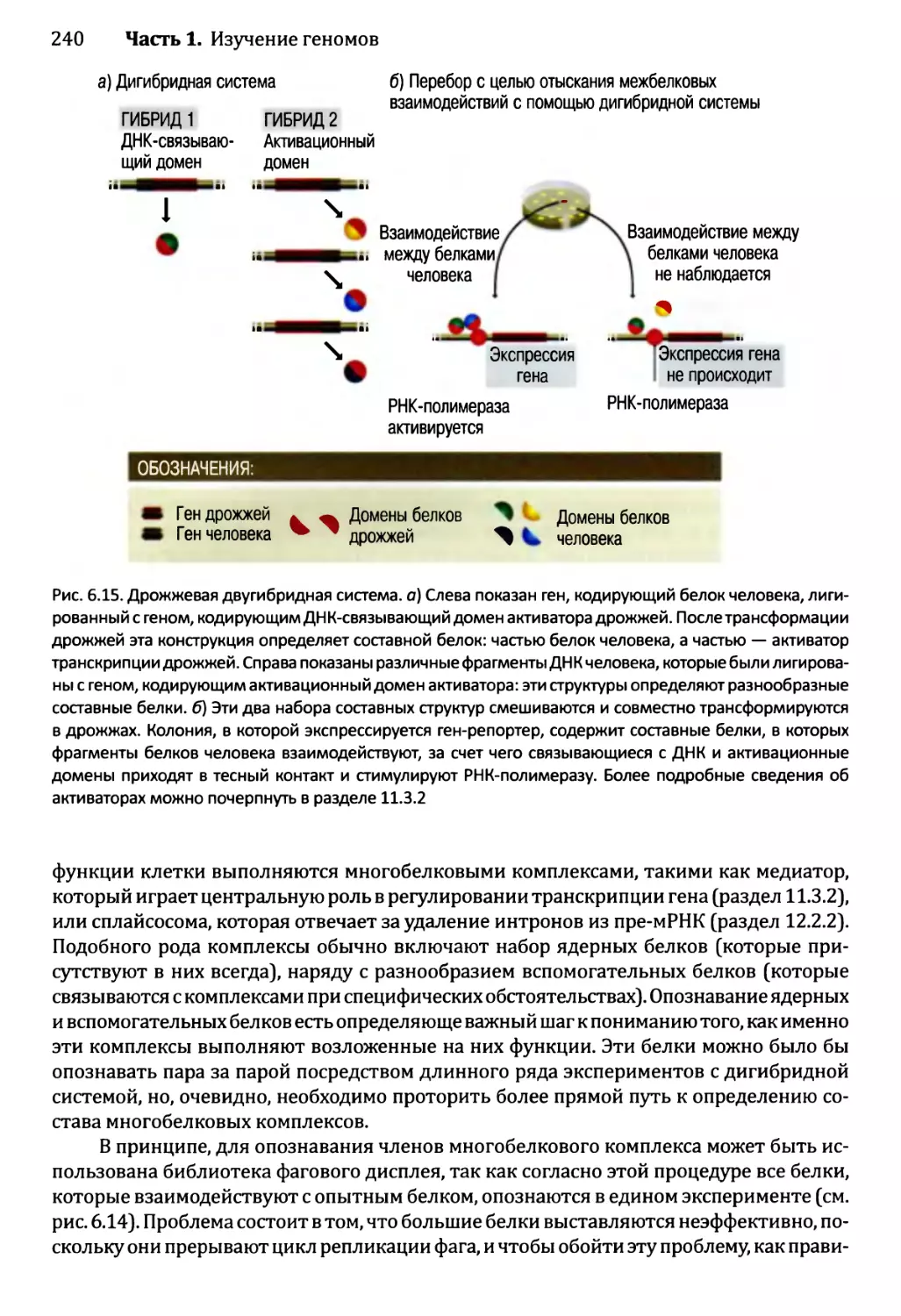
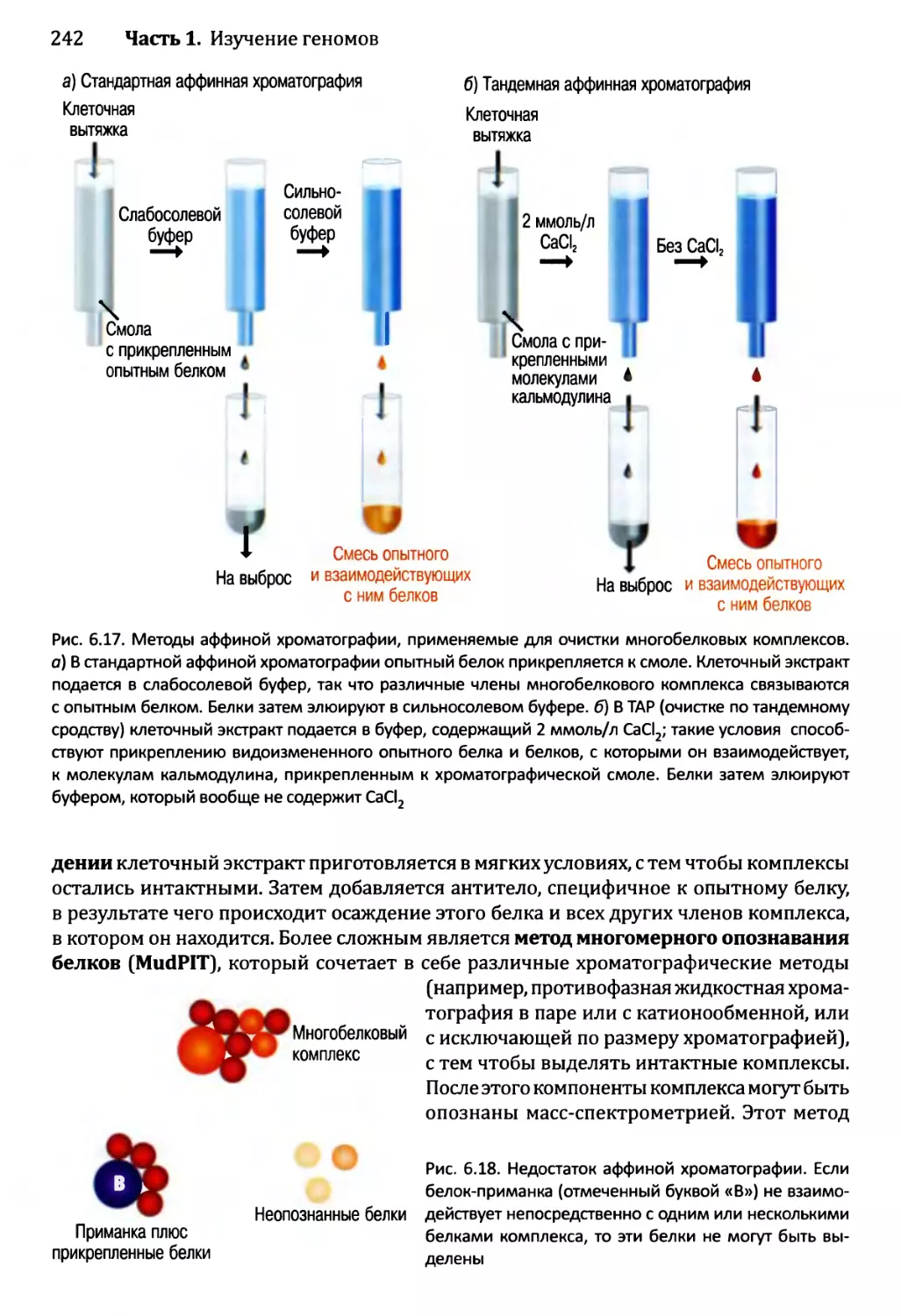
Глава 6. Постижение механизмов функционирования генома..................223
6.1. Изучение транскриптома.........................................224
6.1.1. Изучение транскриптома посредством анализа
последовательности......................................224
6.1.2. Изучение транскриптома с помощью анализа
на микроматрицах или чипах..............................225
6.2. Изучение протеома..............................................233
6.2.1. Определение профилей белков — методология для
опознавания белков в протеоме...........................233
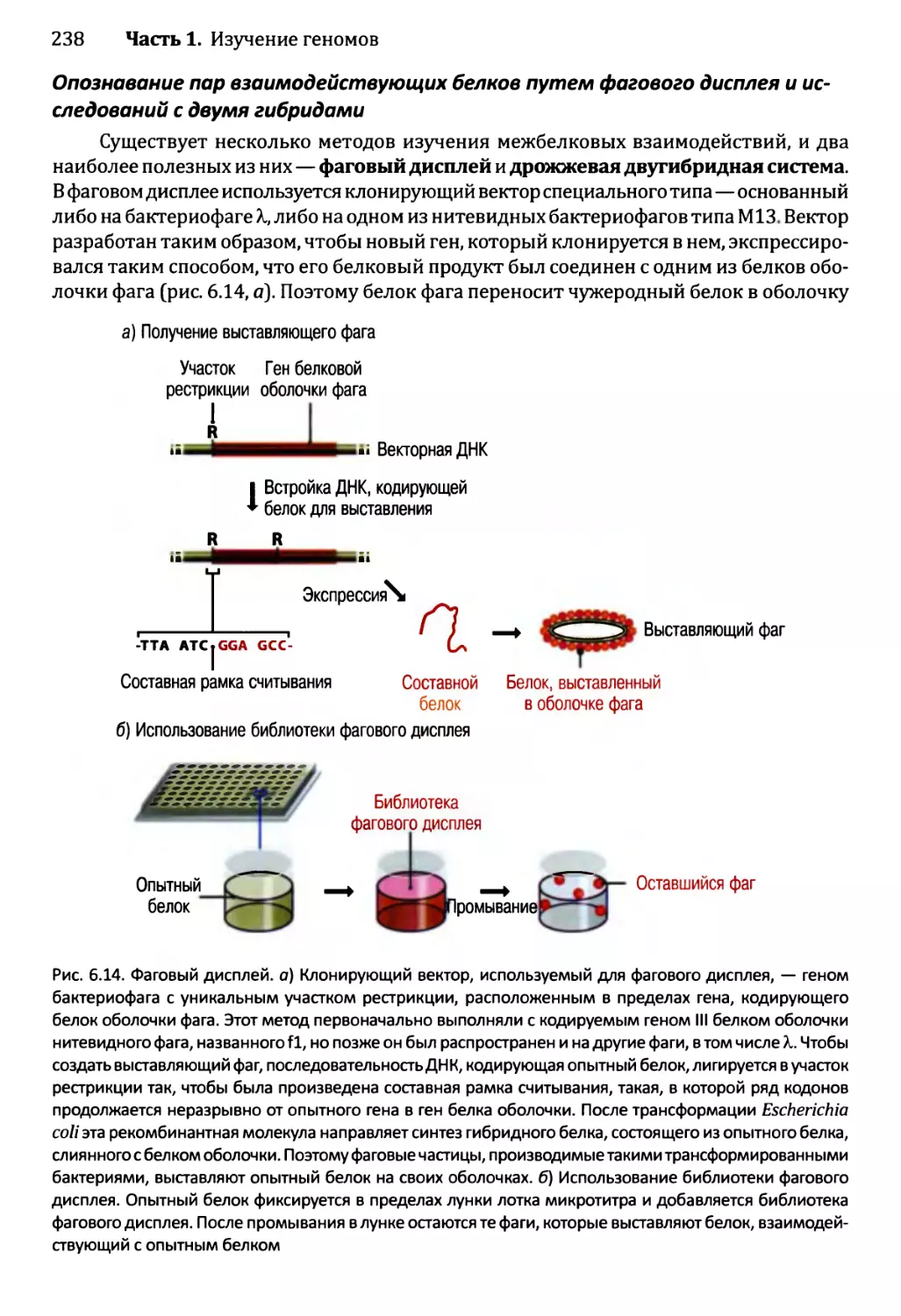
6.2.2. Опознавание межбелковых взаимодействий..................237
6.3. За пределами протеома..........................................245
6.3.1. Метаболом...............................................245
6.3.2. Постижение биологических систем.........................247
Часть 2. Анатомия генома..............................................257
Глава 7. Ядерные геномы эукариотов....................................259
7.1. Ядерные геномы содержатся в хромосомах.........................260
7.1.1. Упаковка ДНК в хромосомах...............................261
Оглавление vii
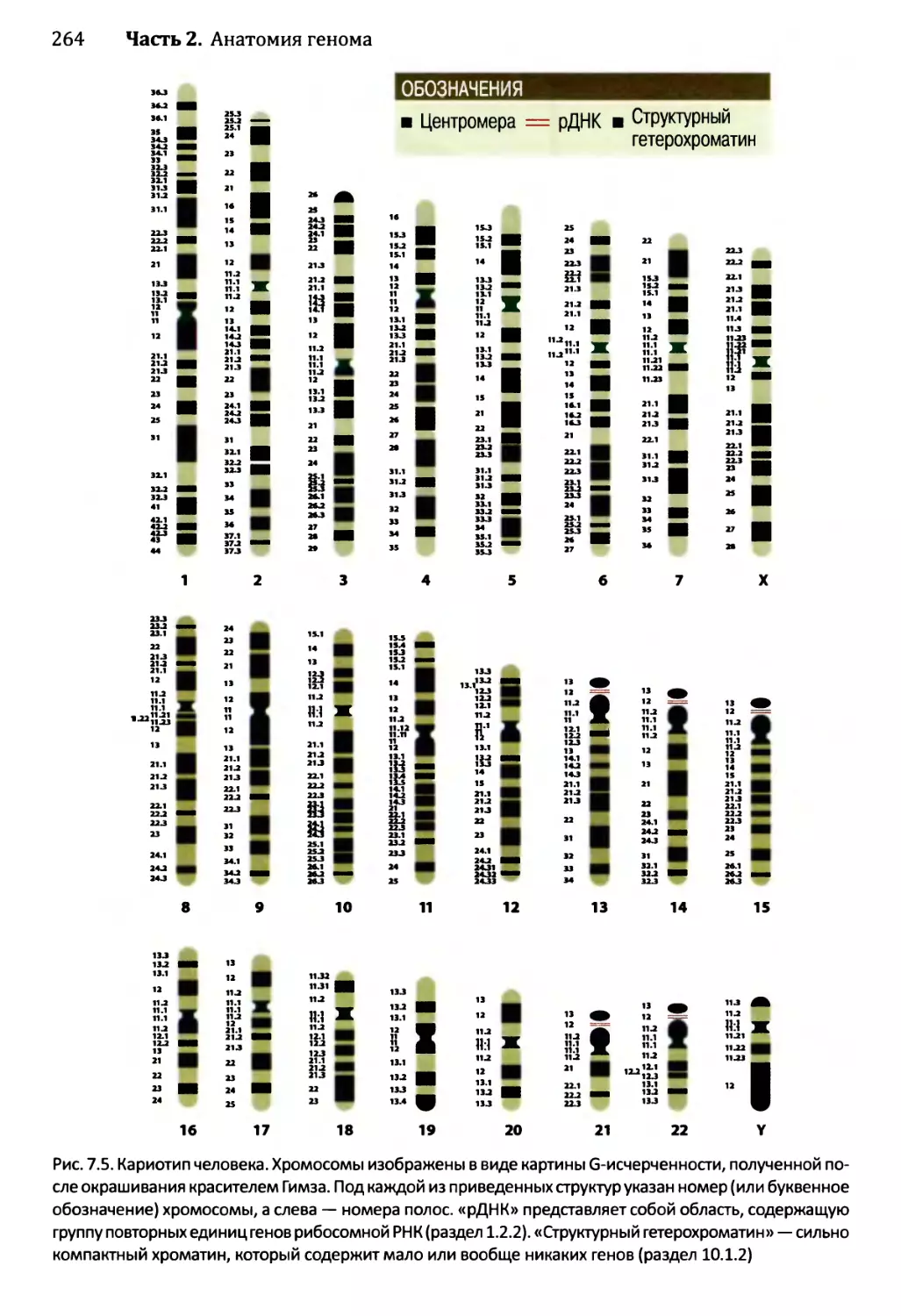
7.1.2. Особенности хромосом в период метафазы..............262
7.2. Генетические характеристики ядерных геномов эукариотов.....267
7.2.1. Где именно в ядерном геноме эукариотов находятся гены?.268
7.2.2. Каким образом организованы гены в ядерном геноме
эукариотов?.................................................269
7.2.3. Сколько генов находится в ядерном геноме эукариотов
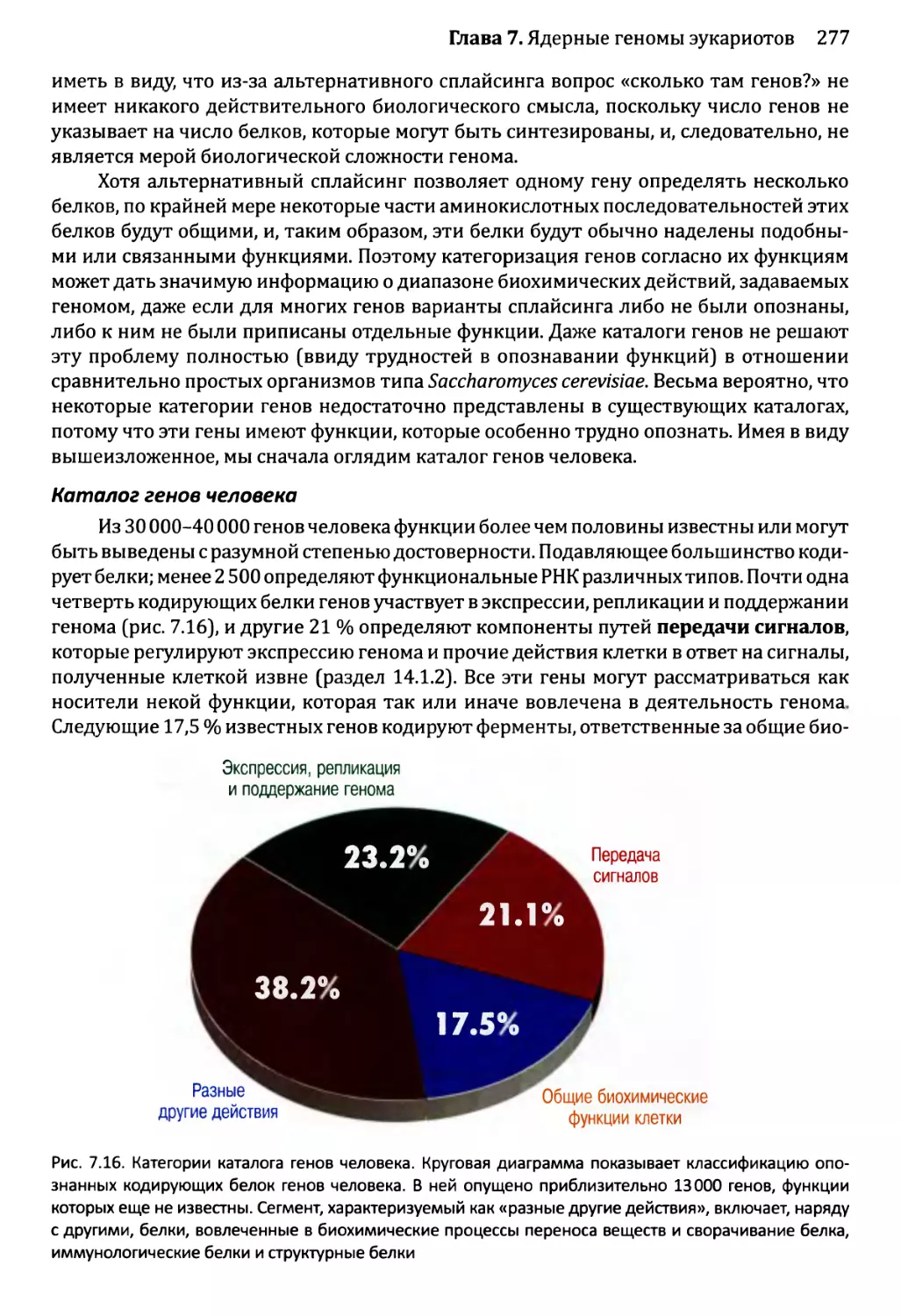
и каковы их функции?........................................276
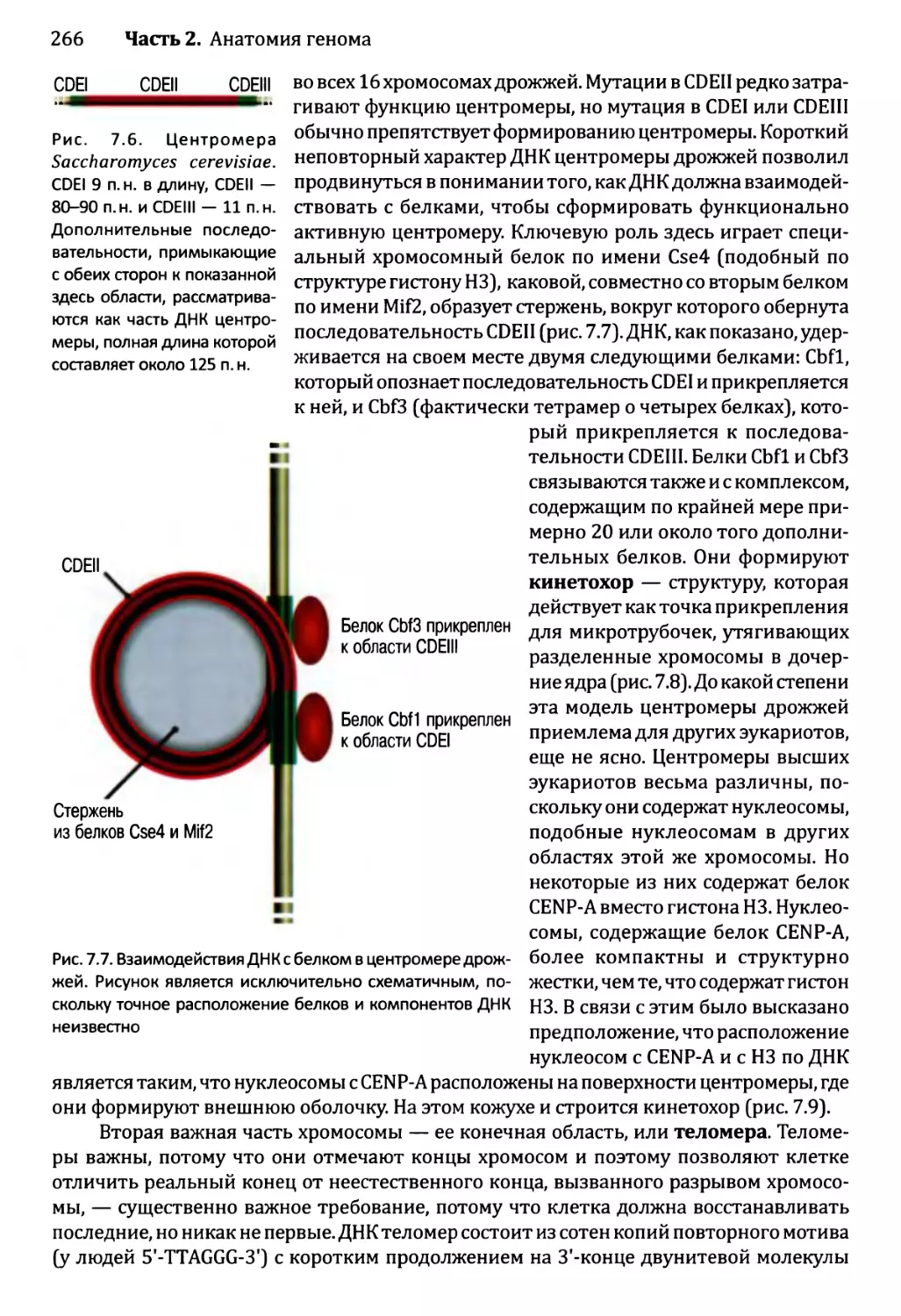
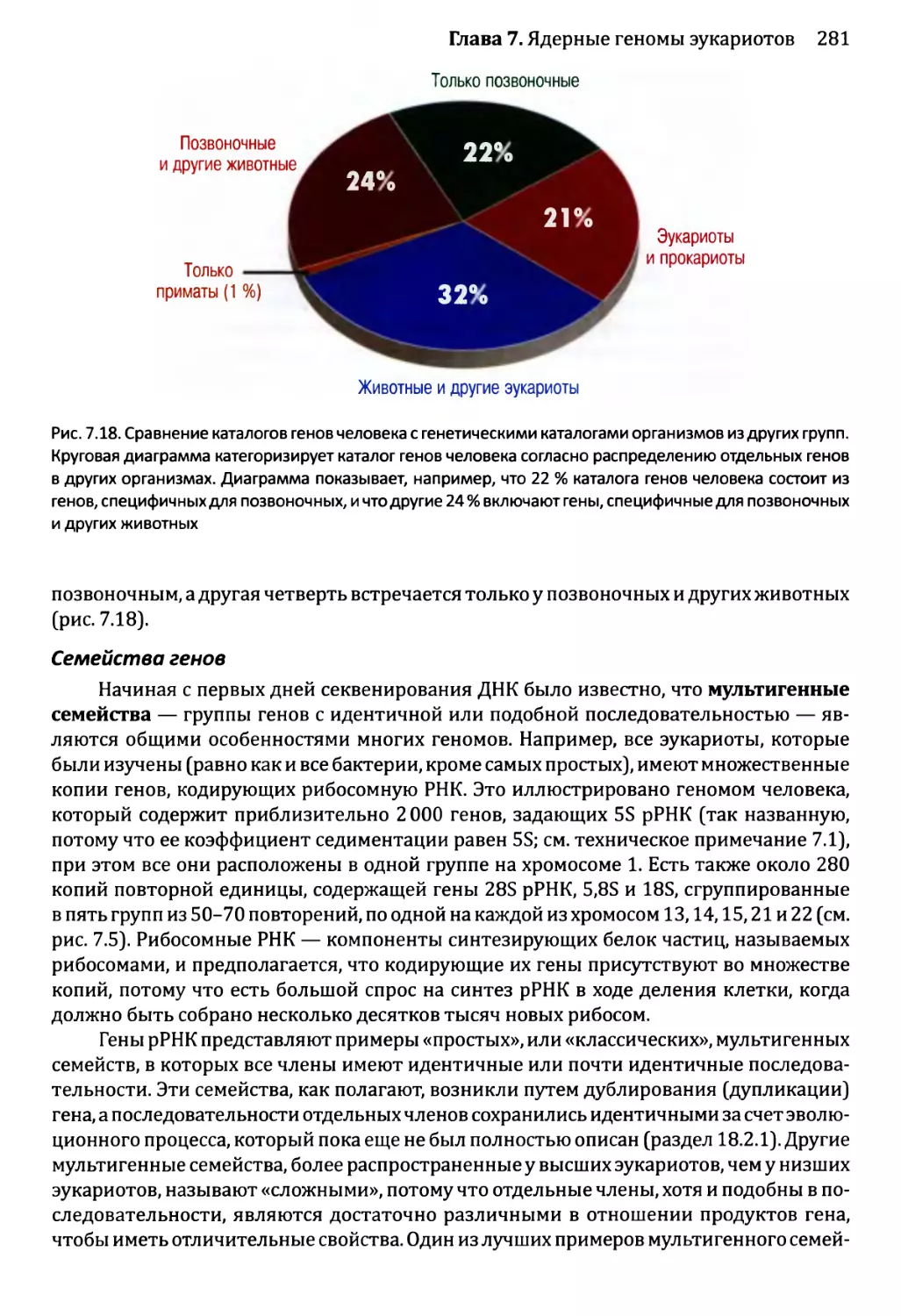
7.2.4. Содержание повторной ДНК в ядерных геномах эукариотов..284
7.2.5. Рассеянные повторы..................................286
Глава 8. Геномы прокариотов и органелл эукариотов...................293
8.1. Физические характеристики геномов прокариотов..............294
8.1.1. Хромосомы прокариотов...............................294
8.2. Генетические характеристики геномов прокариотов............299
8.2.1. Каким образом организованы гены в геноме прокариотов?..300
8.2.2. Сколько генов имеется в геноме прокариотов и каковы
их функции?.................................................303
8.2.3. Геномы прокариотов и понятие биологического вида....304
8.3. Геномы органелл эукариотов.................................307
8.3.1. Происхождение геномов органелл......................308
8.3.2. Физические характеристики геномов органелл..........309
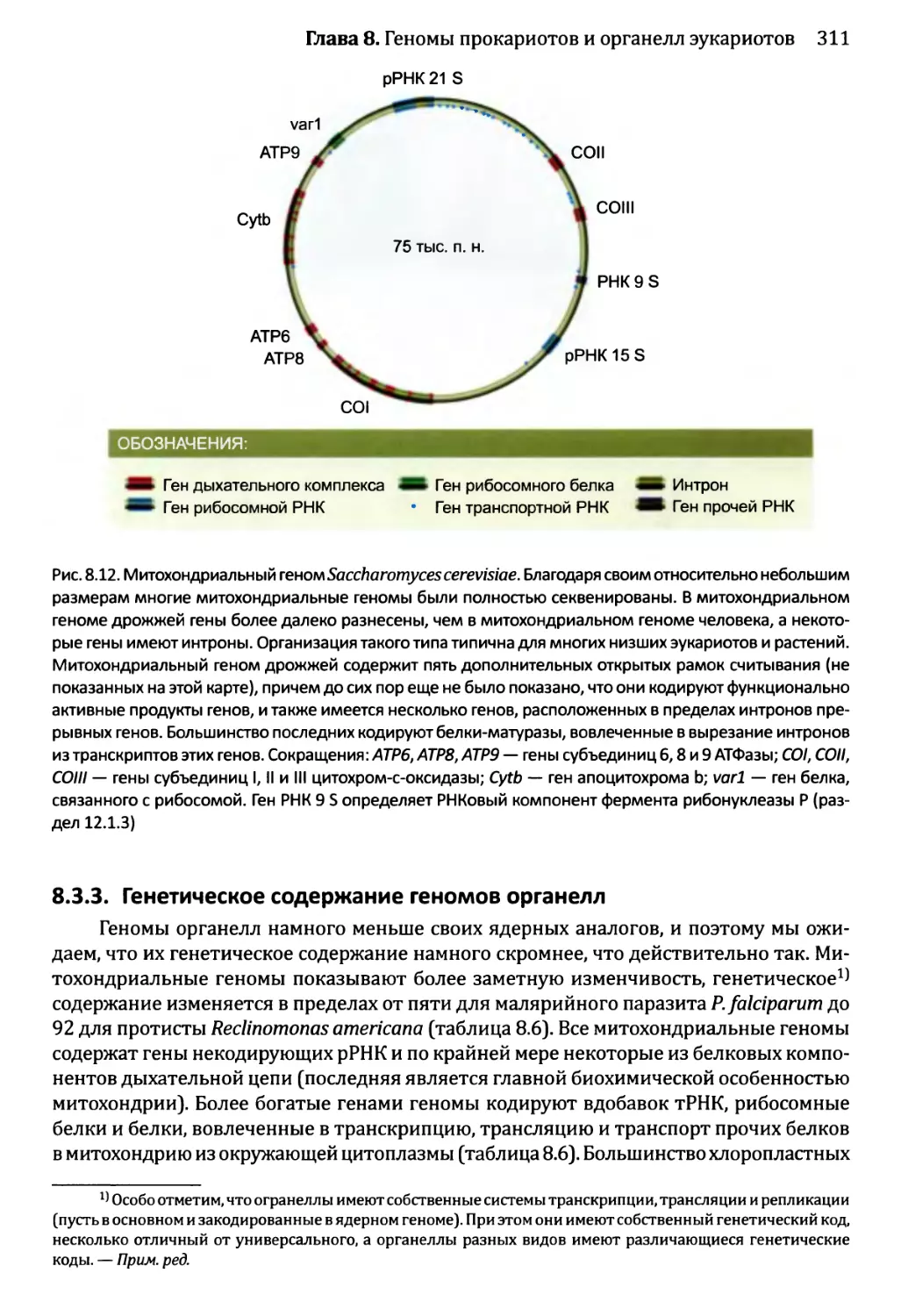
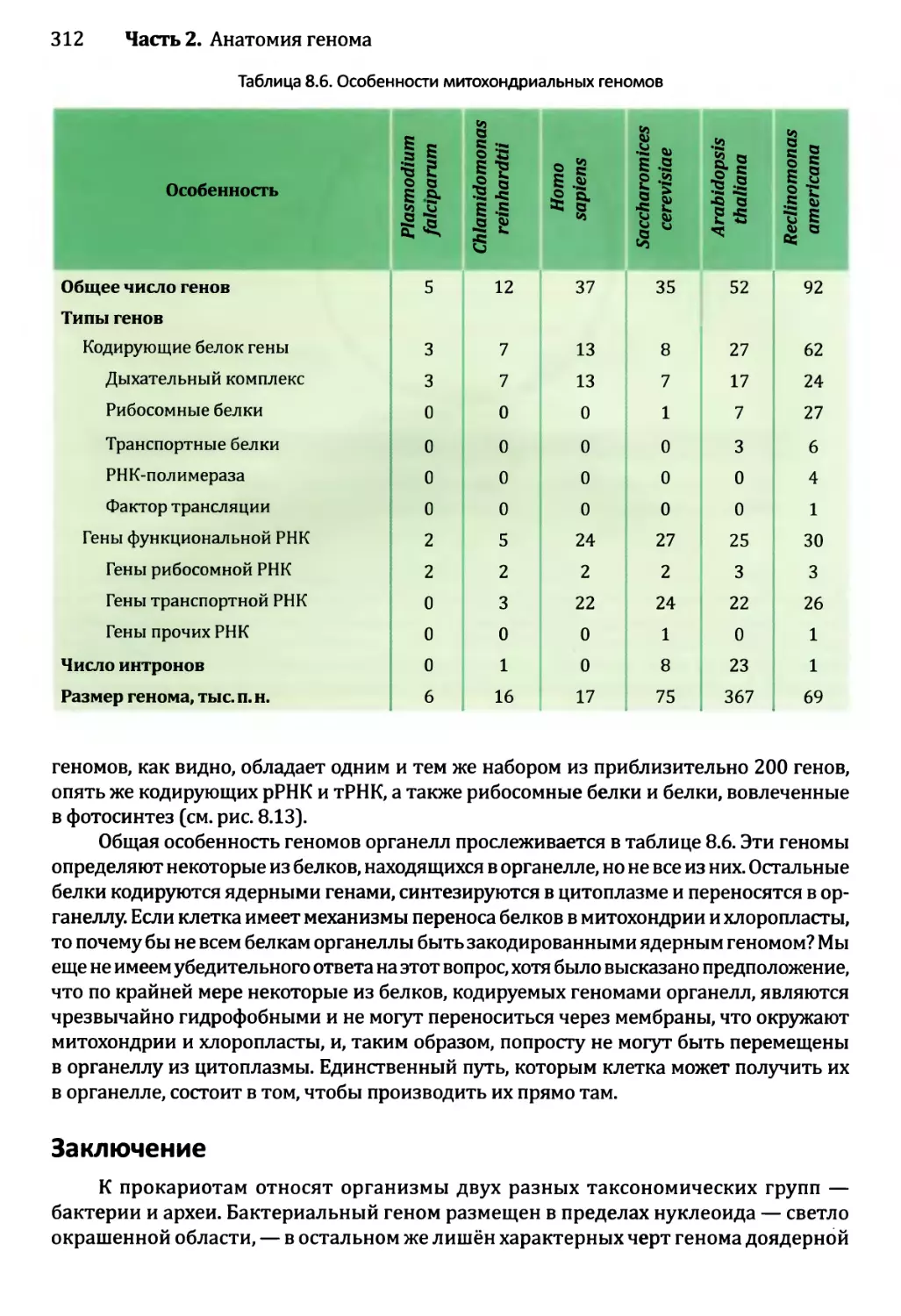
8.3.3. Генетическое содержание геномов органелл............311
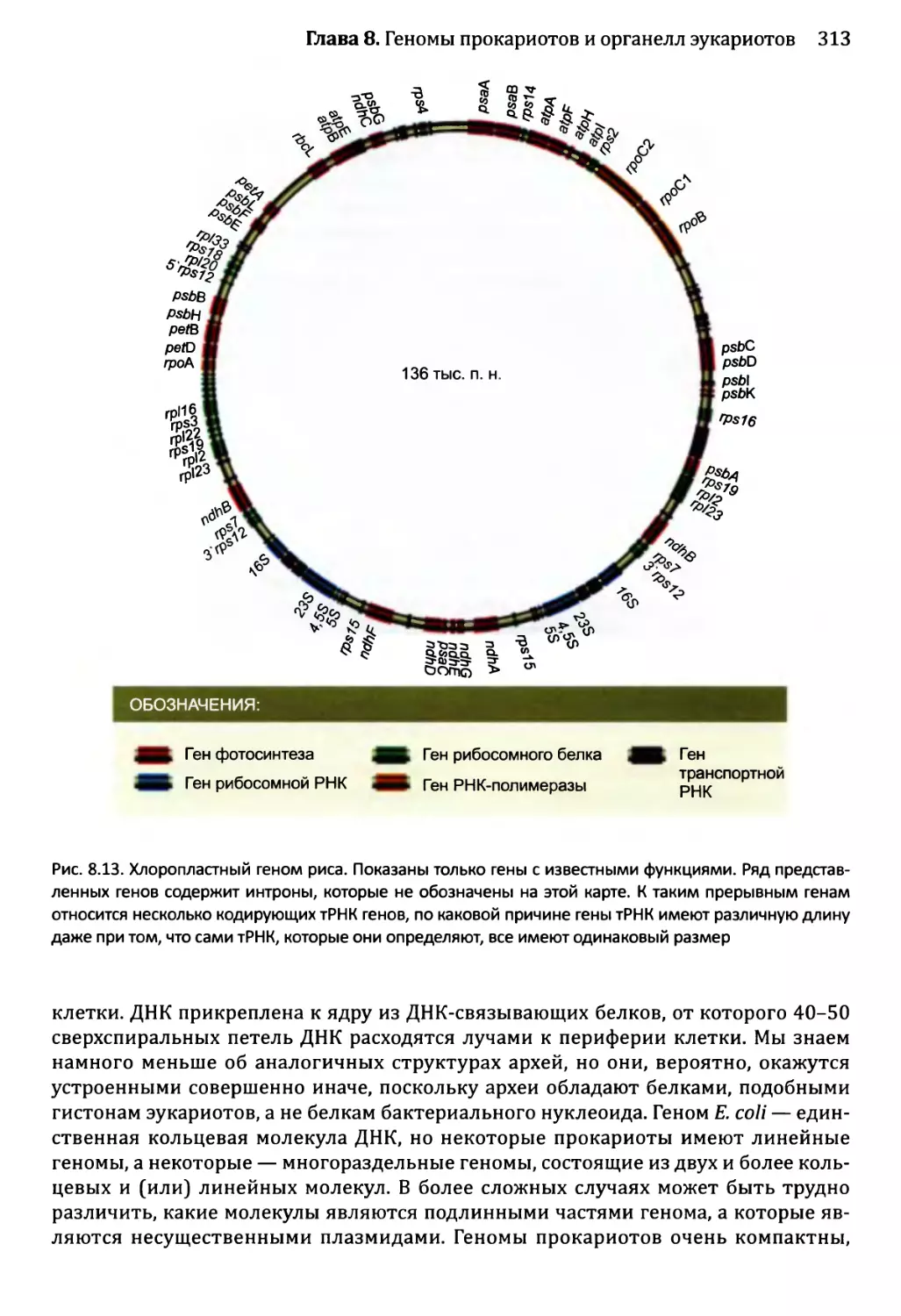
Глава 9. Геномы вирусов и мобильные элементы генома.................321
9.1. Геномы бактериофагов и вирусов эукариотов..................322
9.1.1. Геномы бактериофагов................................322
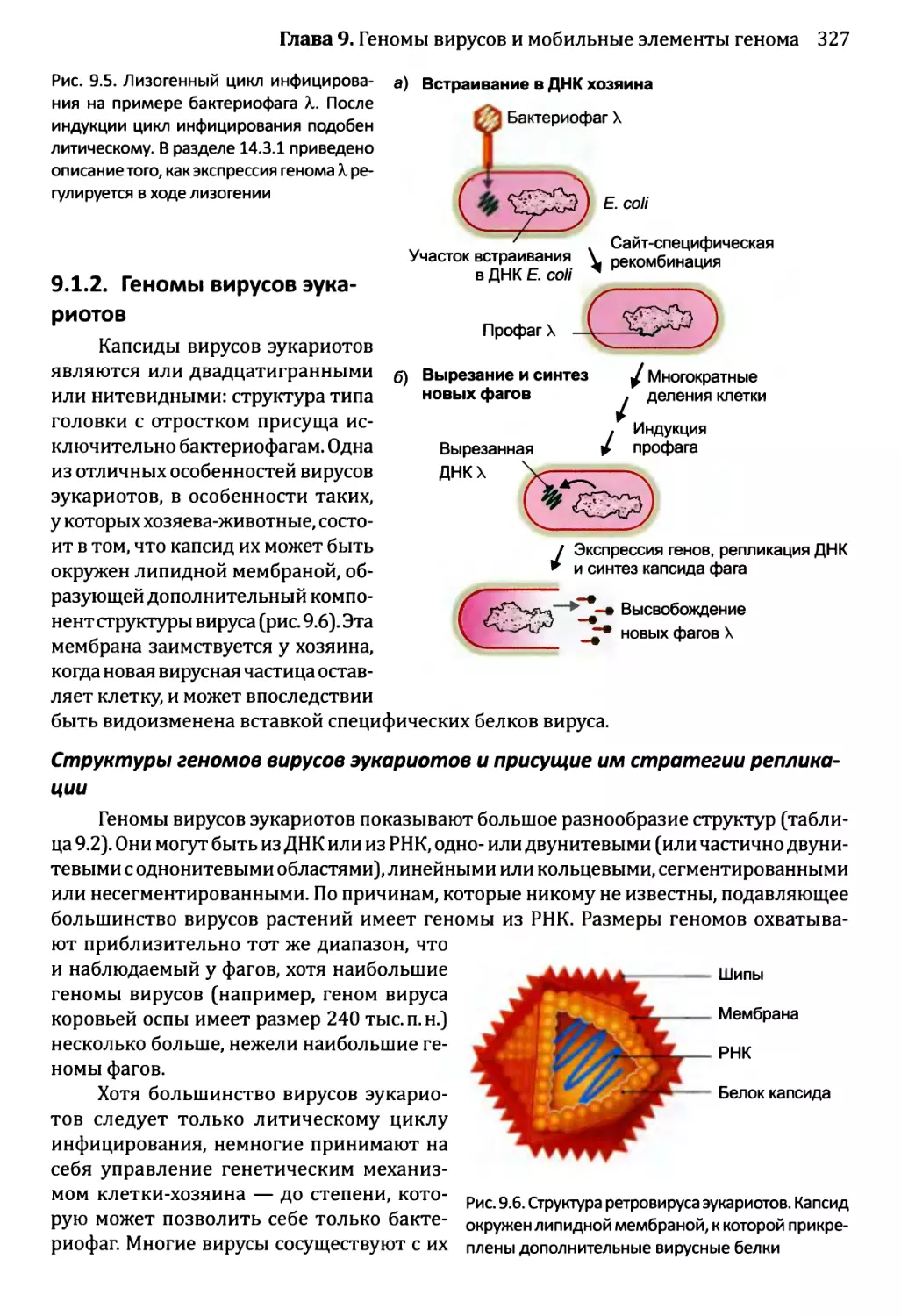
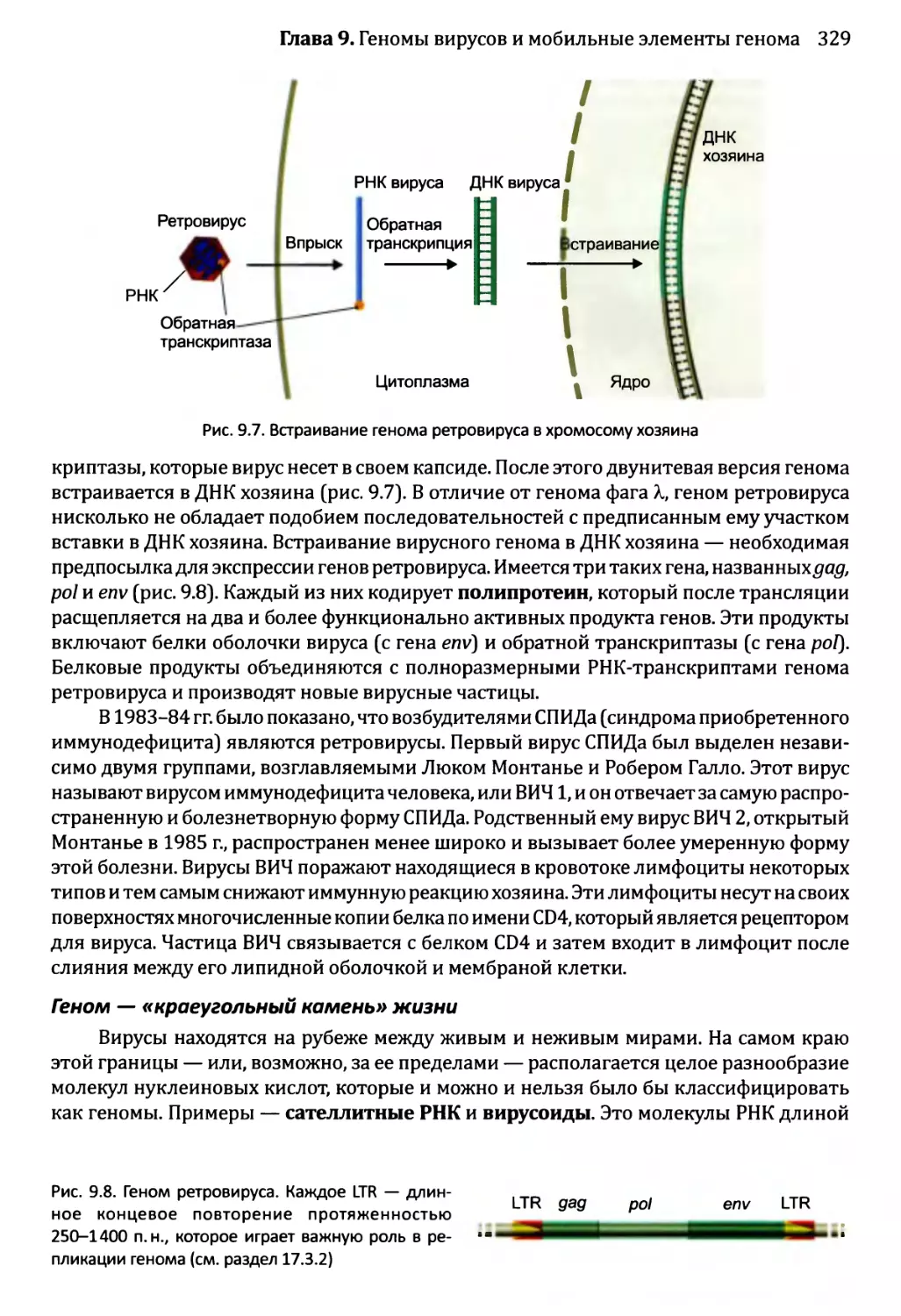
9.1.2. Геномы вирусов эукариотов...........................327
9.2. Мобильные элементы генома..................................331
9.2.1. Транспозиция через РНК-посредник....................332
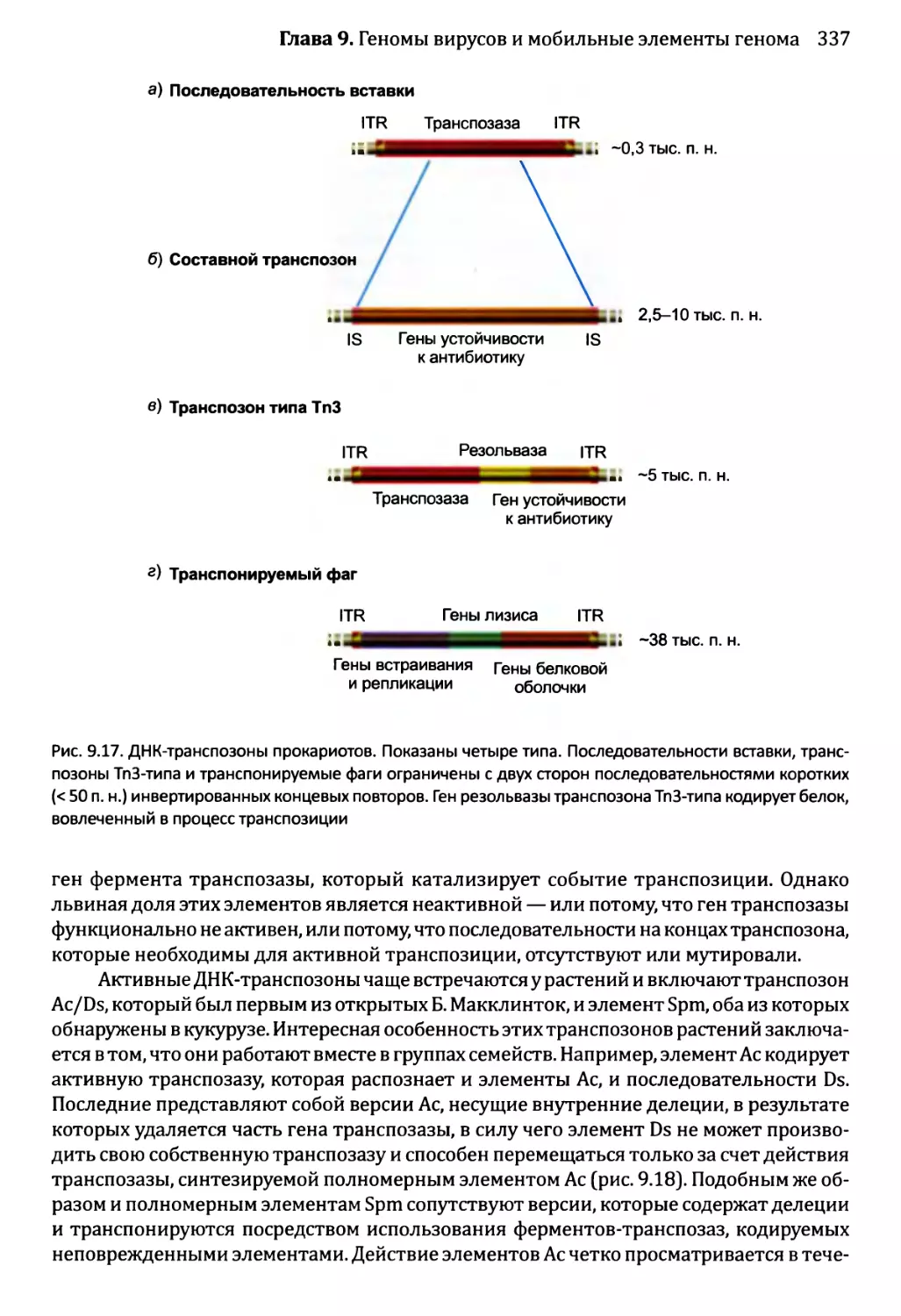
9.2.2. ДНК-транспозоны.....................................336
Часть 3. Принципы функционирования геномов..........................347
Глава 10. Доступ к геному...........................................349
10.1. Внутри ядра...............................................350
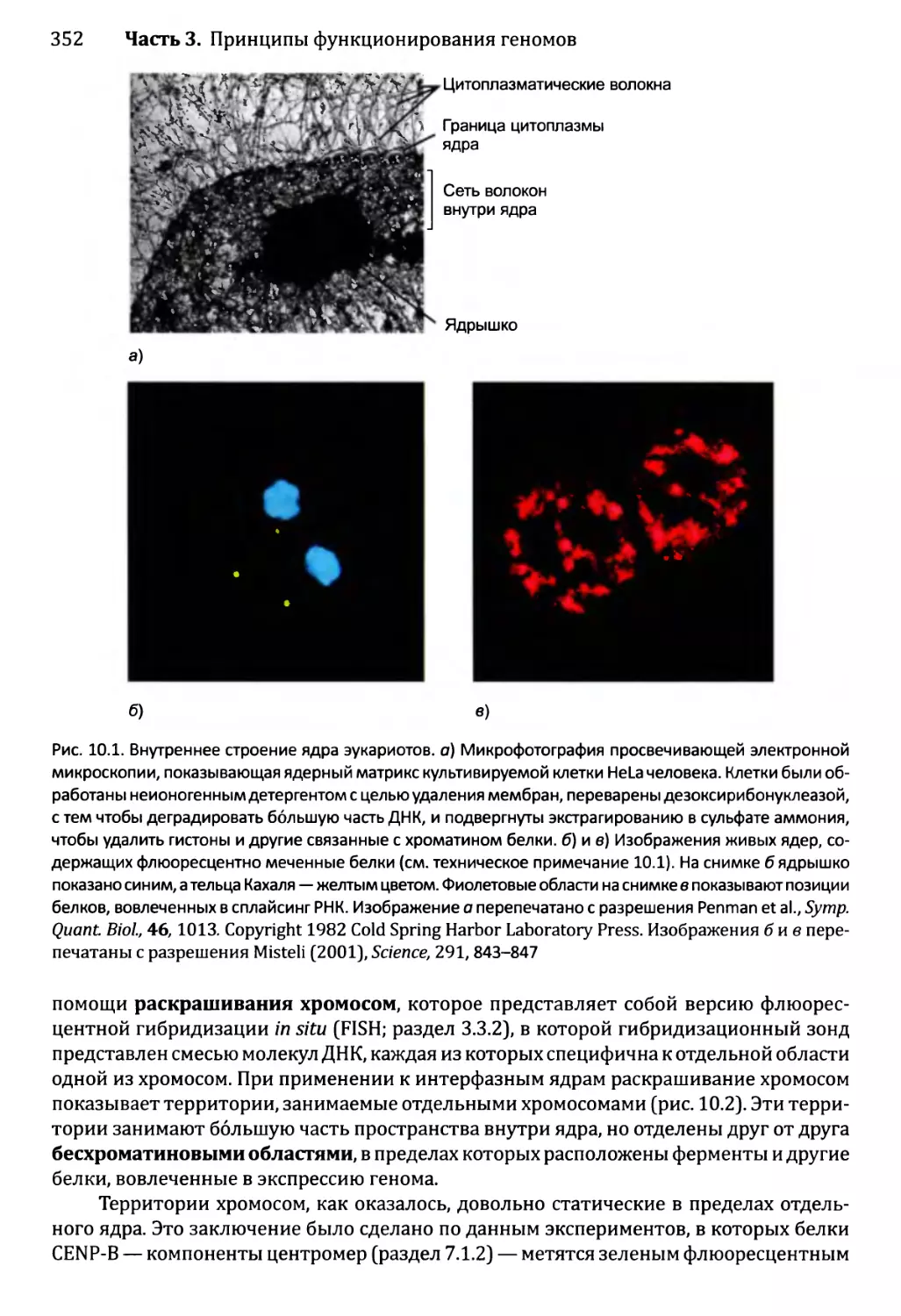
10.1.1. Внутреннее строение ядра эукариотов................350
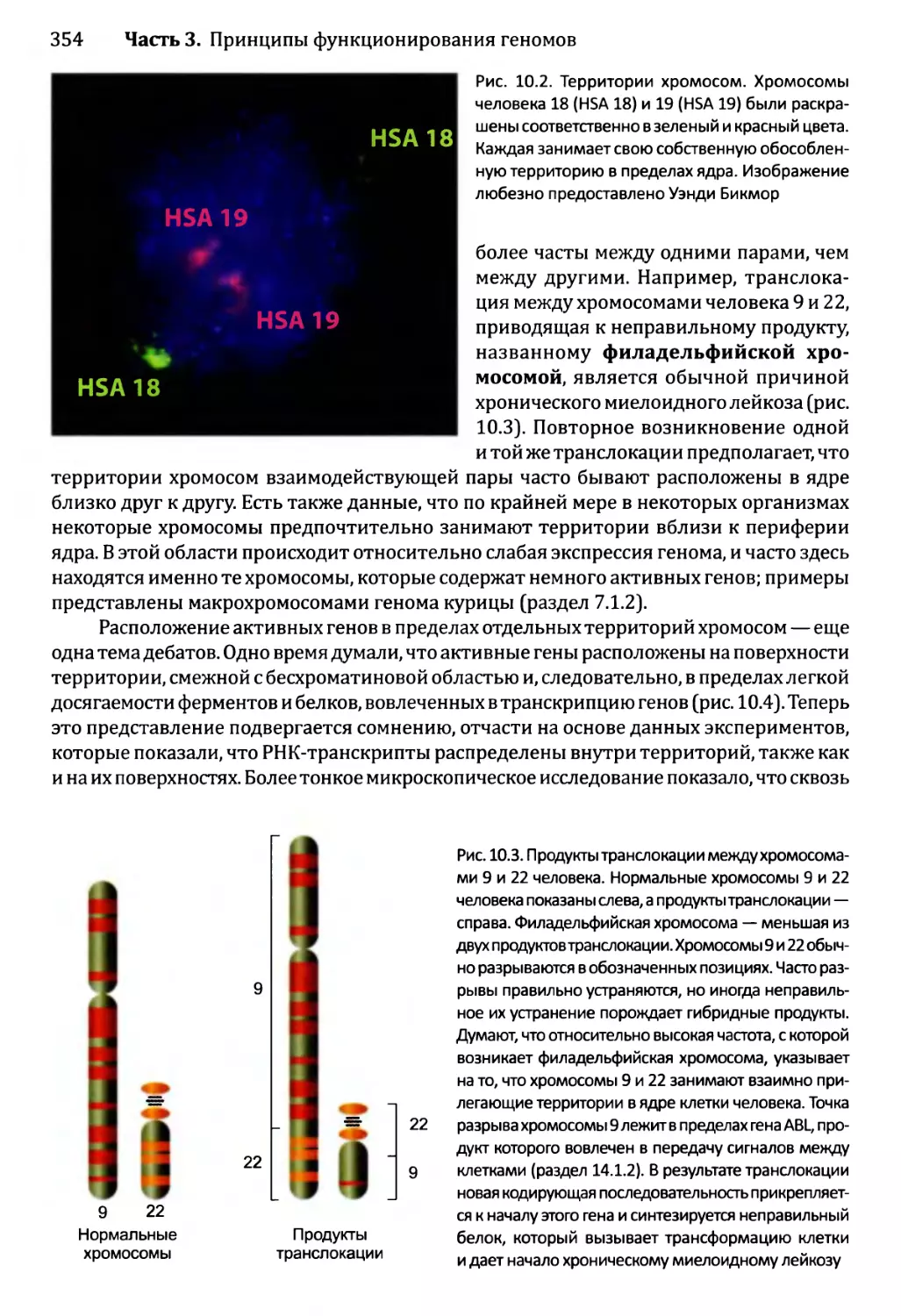
10.1.2. Хроматиновые домены................................355
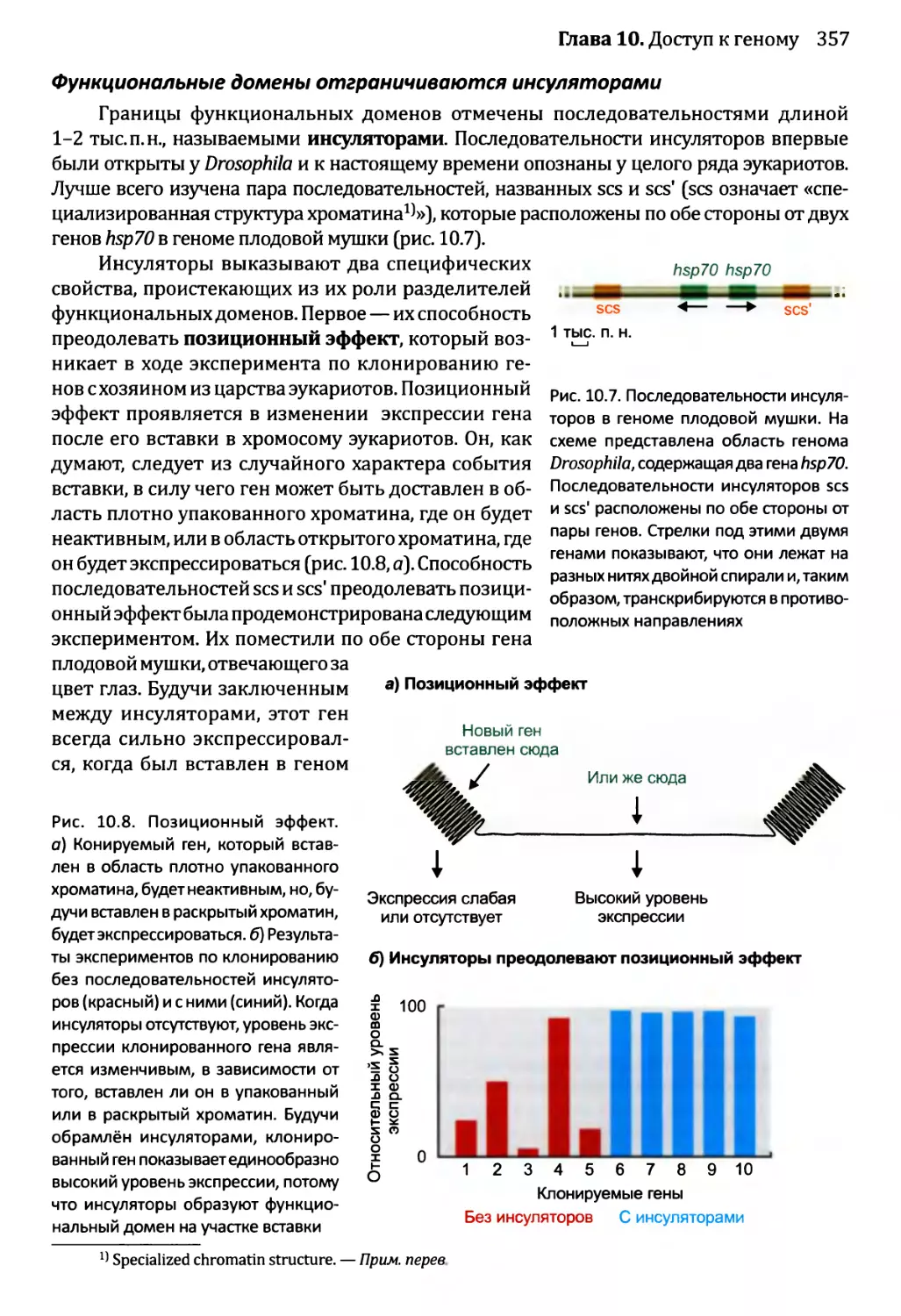
10.2. Модификации хроматина и экспрессия генома.................360
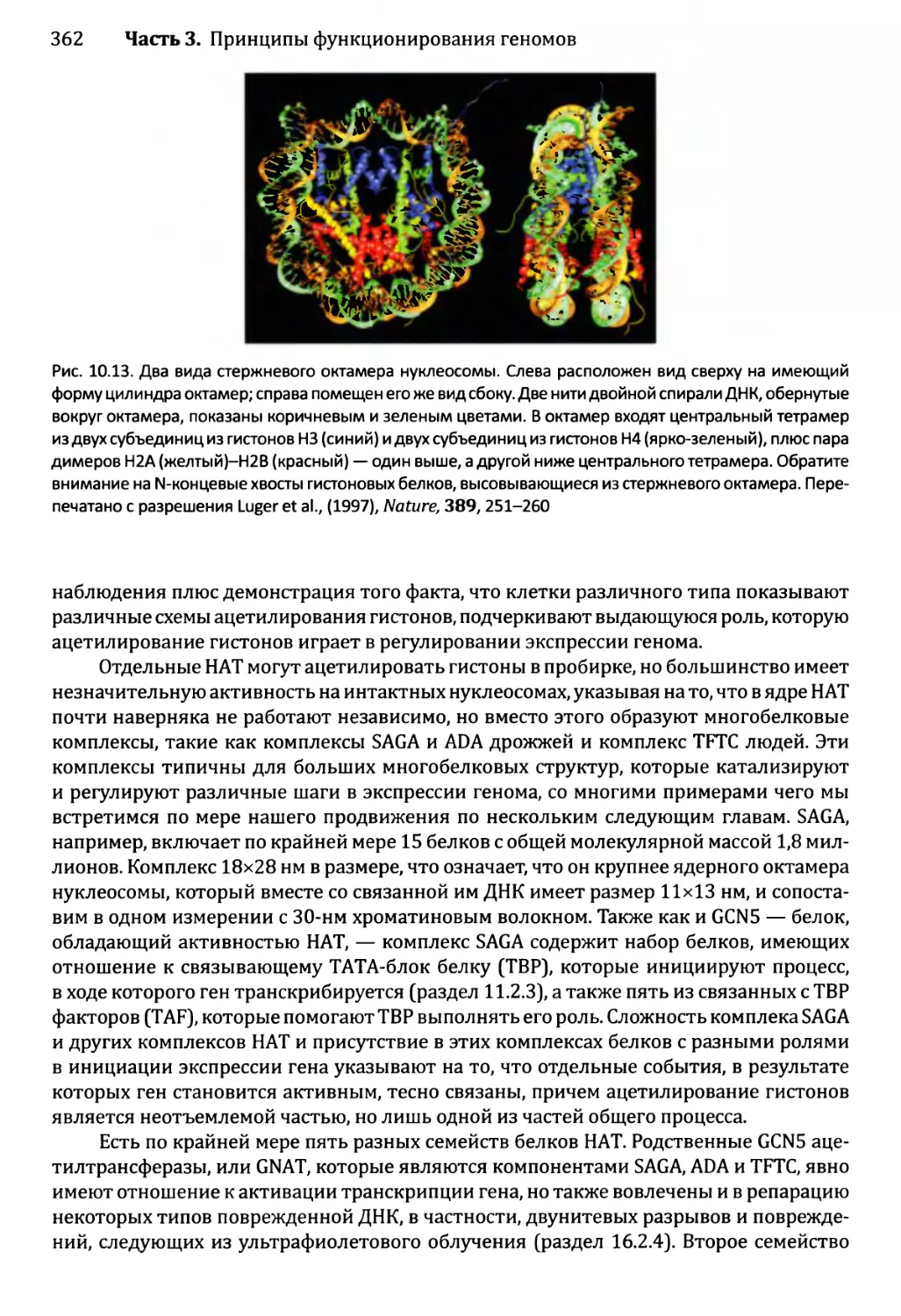
10.2.1. Химическая модификация гистонов....................361
10.2.2. Влияние перестройки нуклеосом на экспрессию генома....365
10.3. Модификация ДНК и экспрессия генома.......................367
10.3.1. Подавление генома путем метилирования ДНК..........367
Глава 11. Сборка комплекса инициации транскрипции...................379
11.1. ДНК-связывающие белки и их сайты связывания...............380
11.1.1. Особые характеристики ДНК-связывающих белков.......381
11.1.2. Определение позиций сайтов связывания с ДНК в геноме..389
11.1.3. Взаимодействие между ДНК и связывающимися с ней
белками....................................................394
viii
Оглавление
11.2. Взаимодействия белков с ДНК в ходе инициации транскрипции...398
11.2.1. РНК-полимеразы.......................................398
11.2.2. Последовательности, распознаваемые при инициации
транскрипции.................................................399
11.2.3. Сборка комплекса инициации транскрипции..............402
11.3. Регулирование инициации транскрипции........................407
11.3.1. Стратегии управления инициацией транскрипции у бактерий.408
11.3.2. Регуляция инициации транскрипции у эукариотов........413
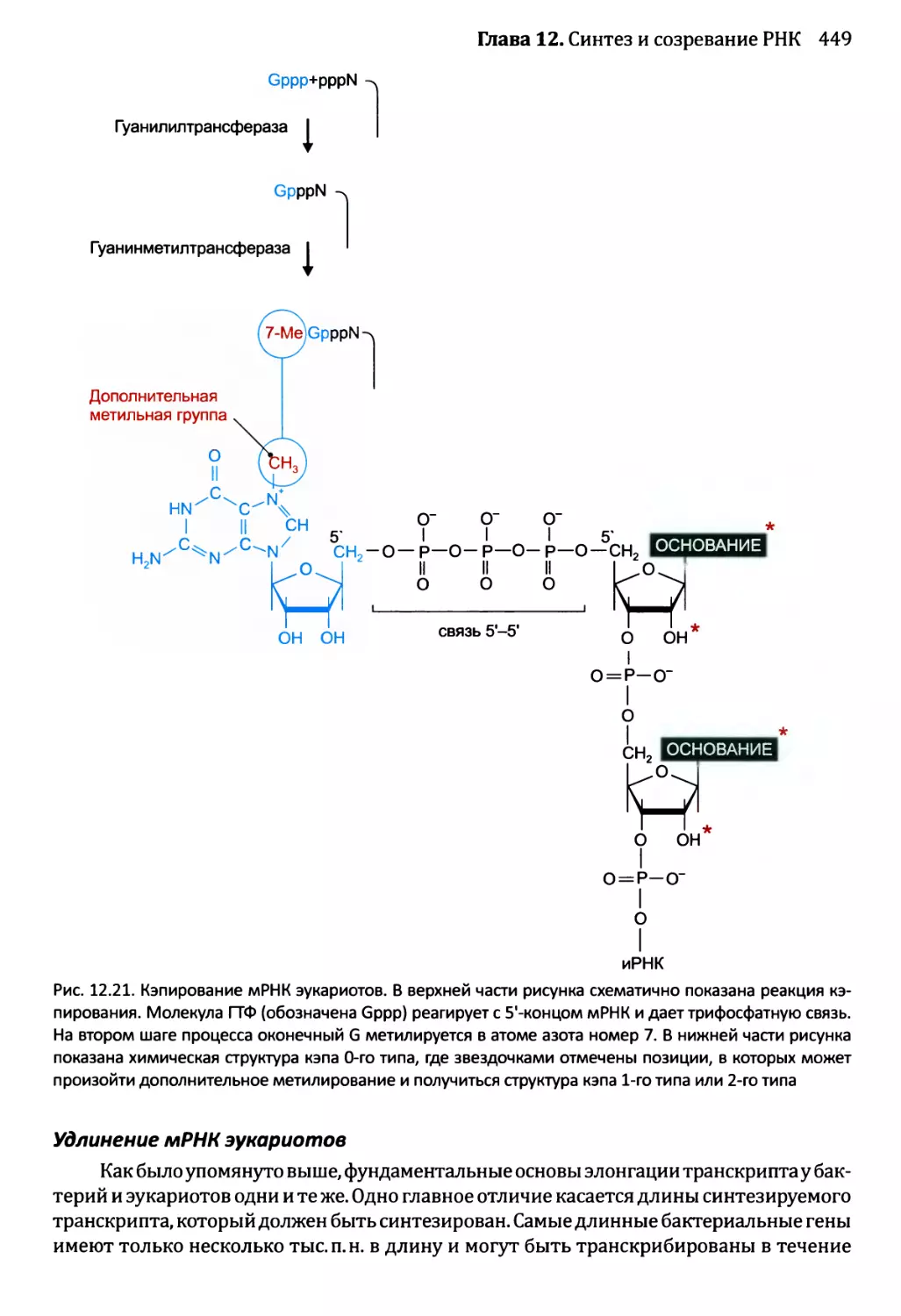
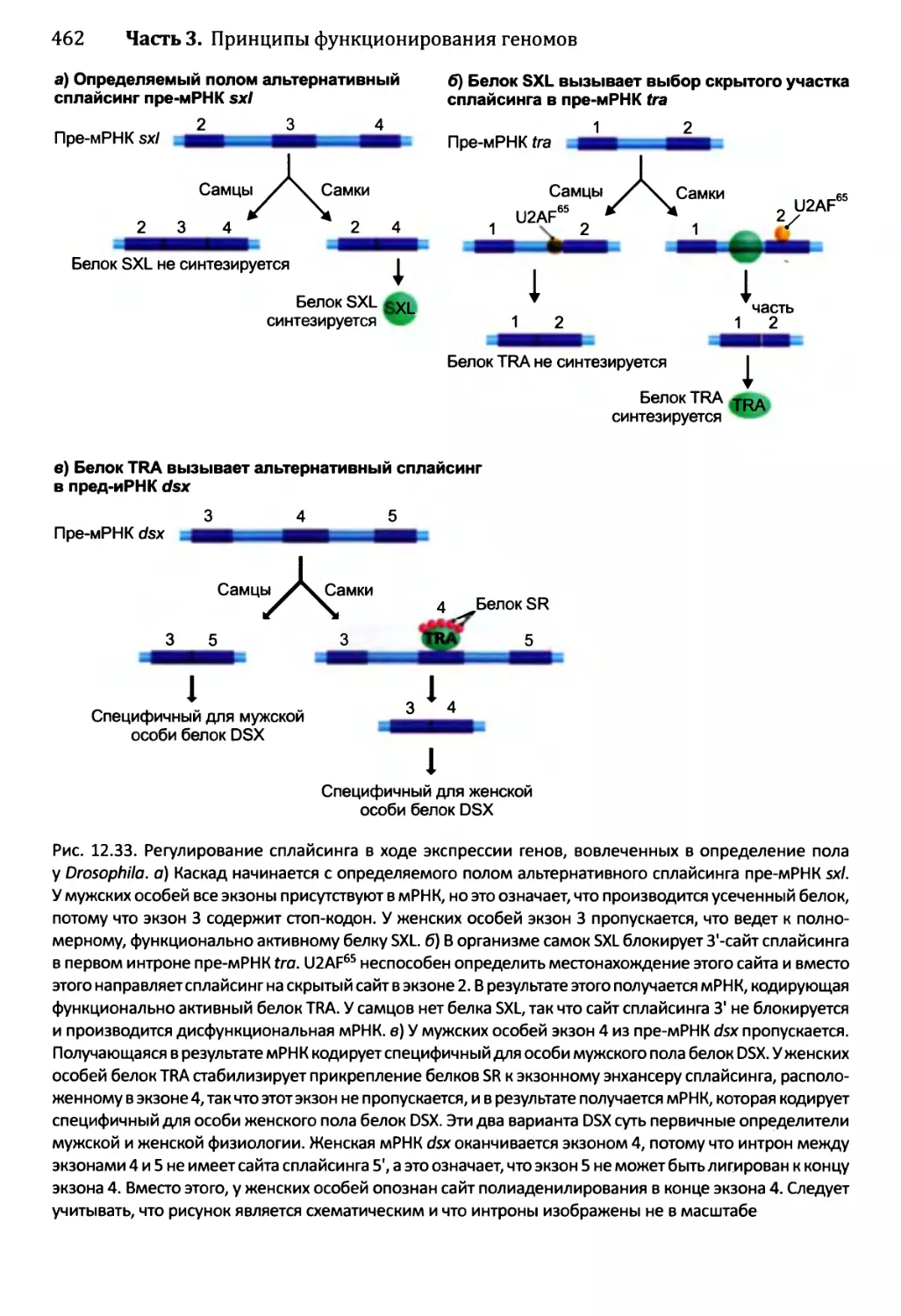
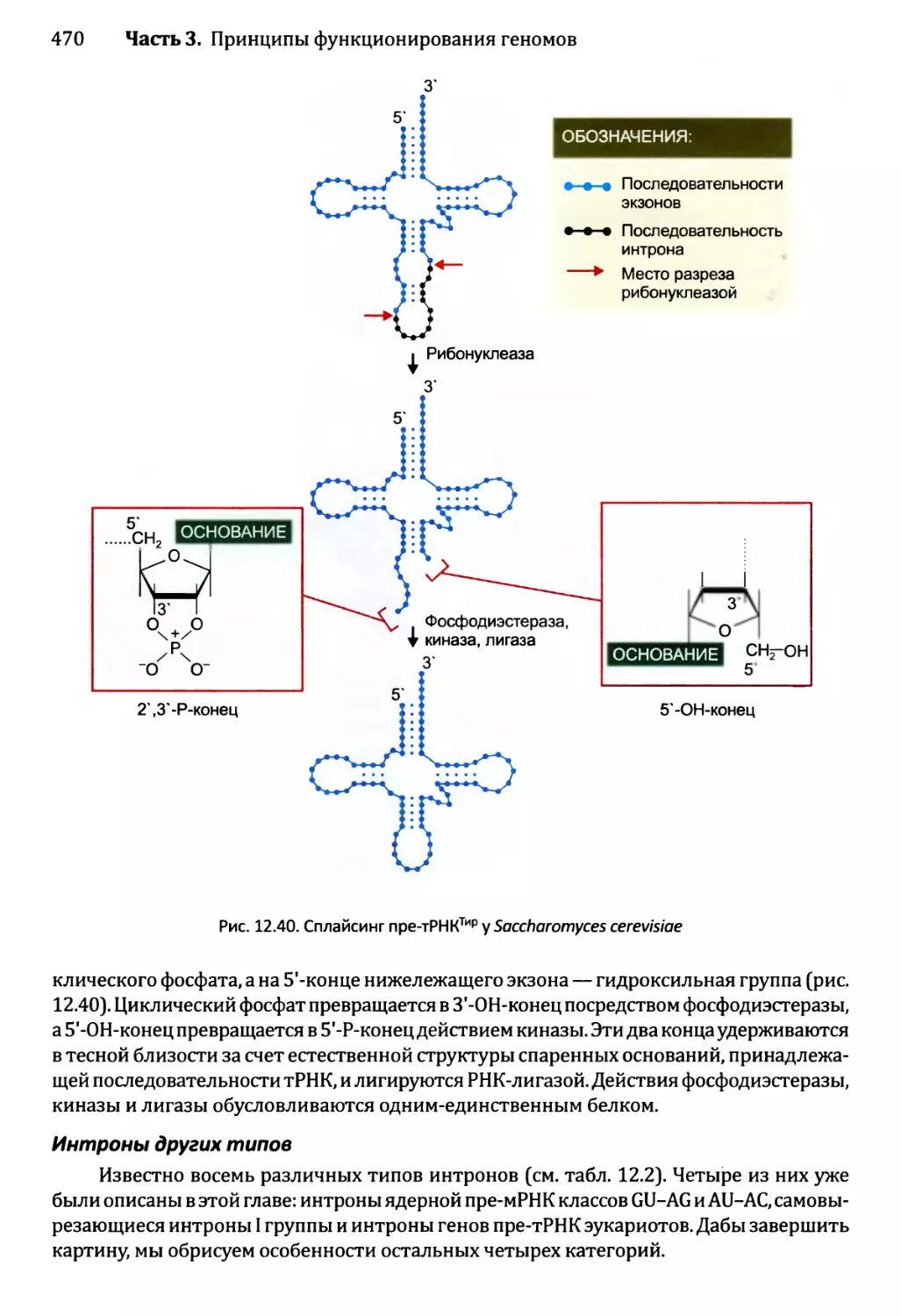
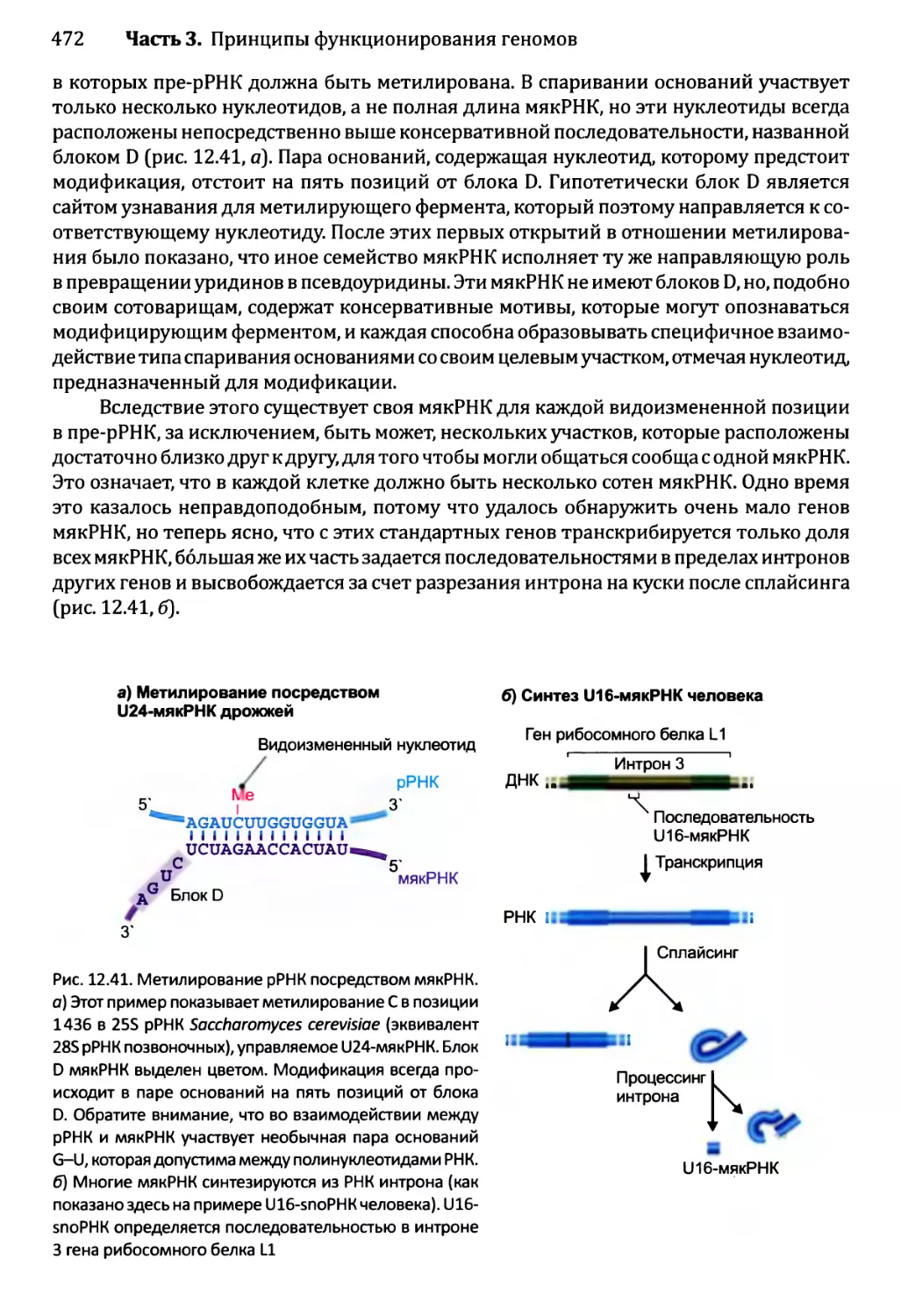
Глава 12. Синтез и созревание РНК.....................................429
12.1. Синтез и созревание РНК бактерий............................430
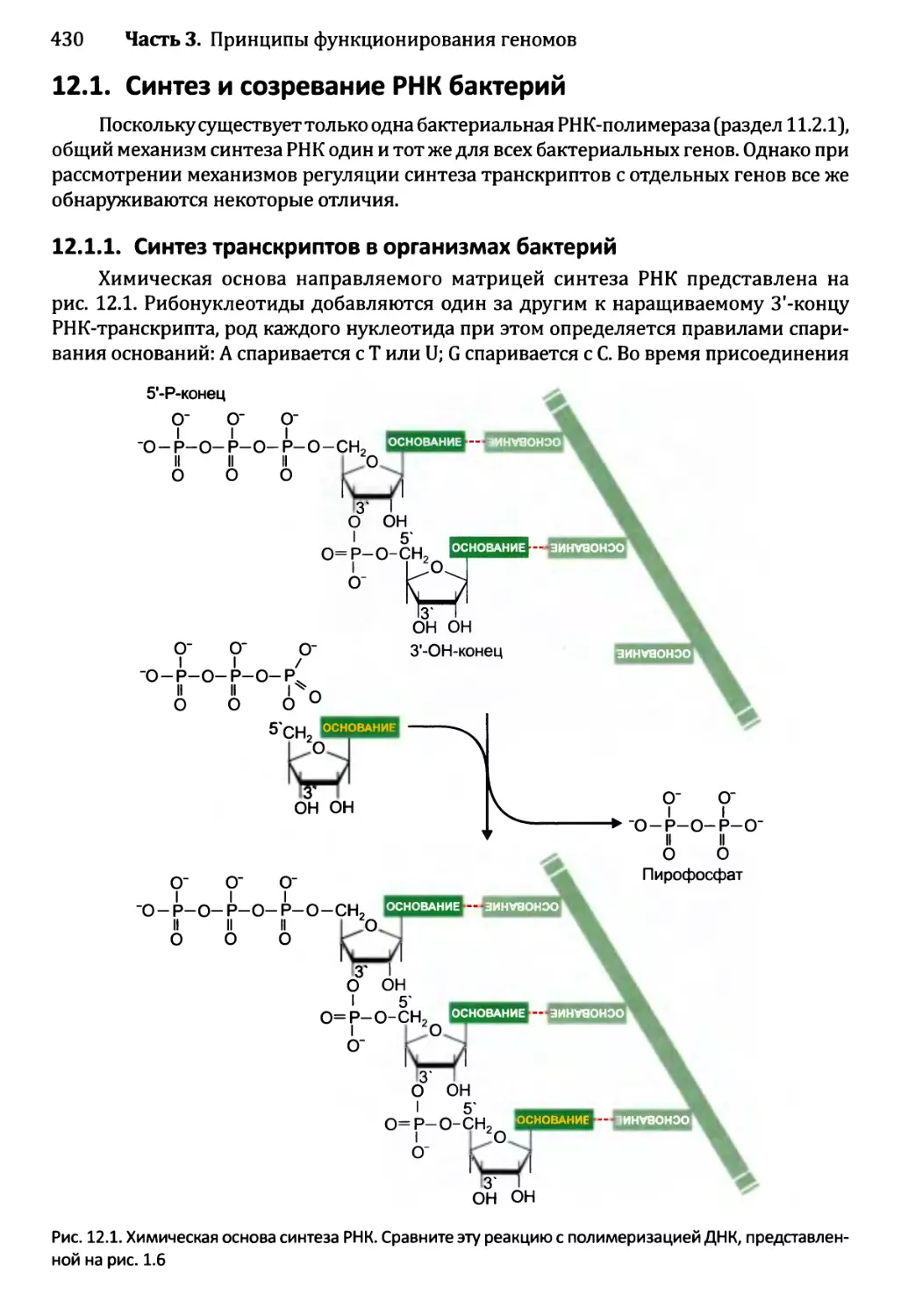
12.1.1. Синтез транскриптов в организмах бактерий............430
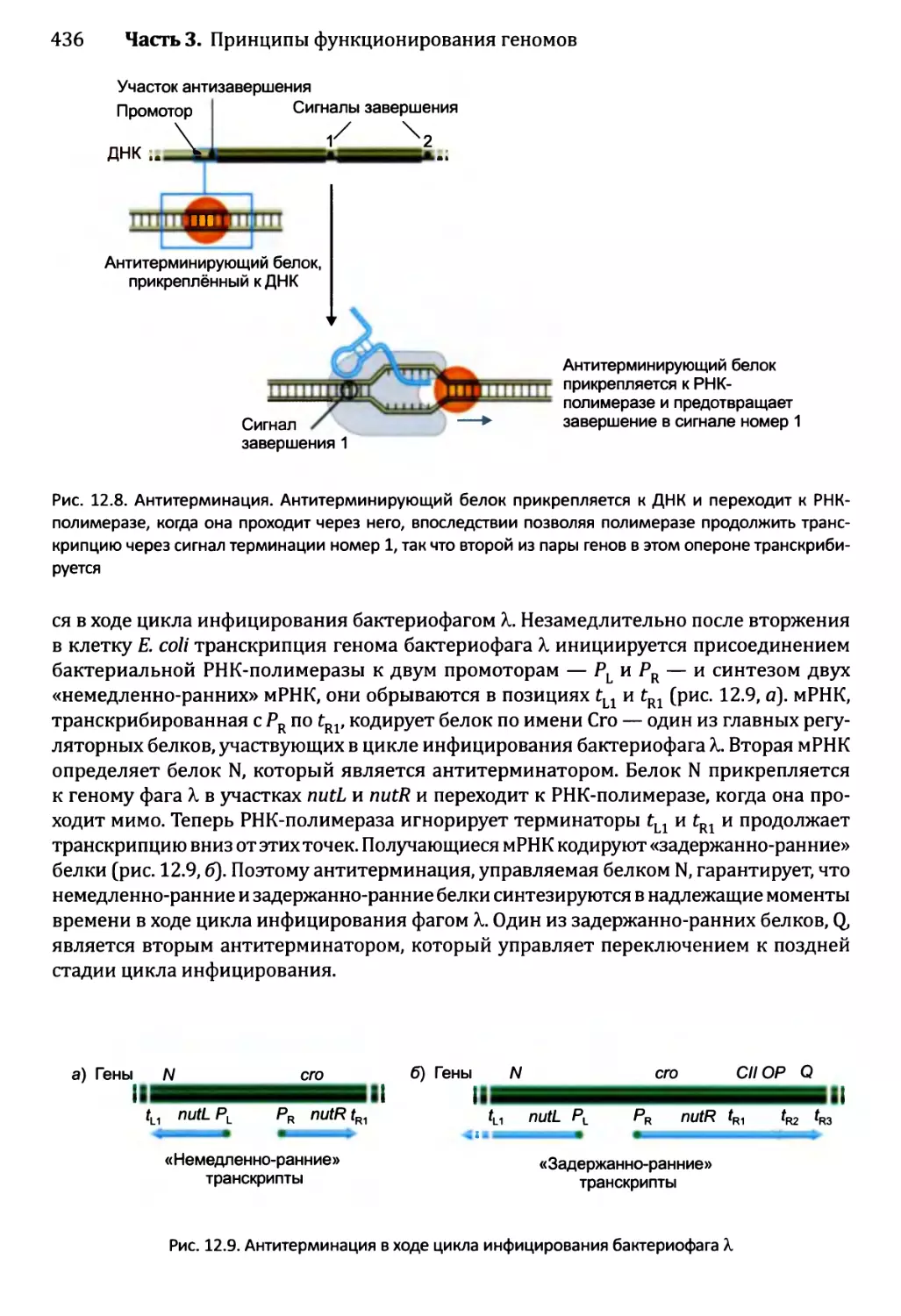
12.1.2. Управление выбором между элонгацией и терминацией....435
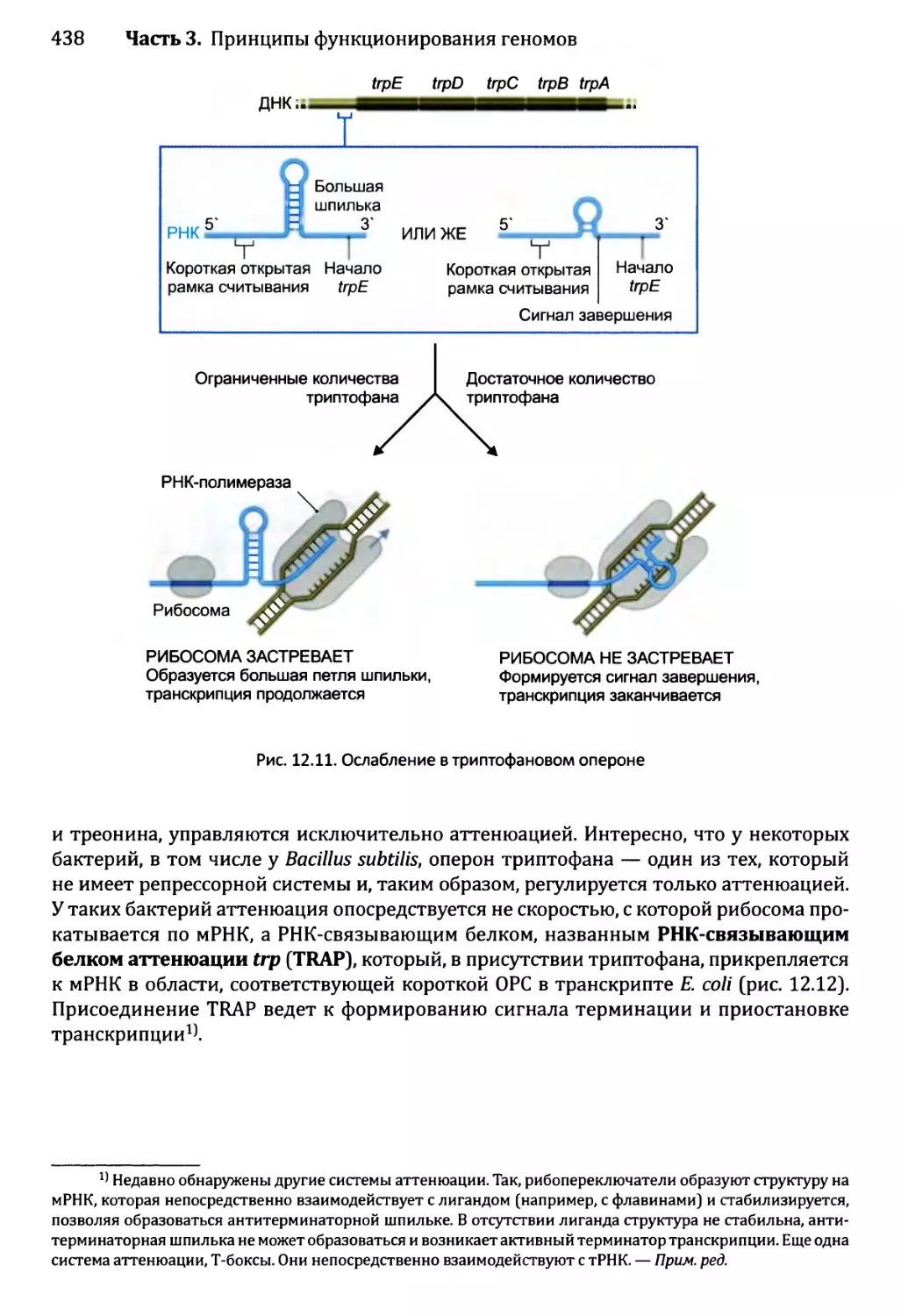
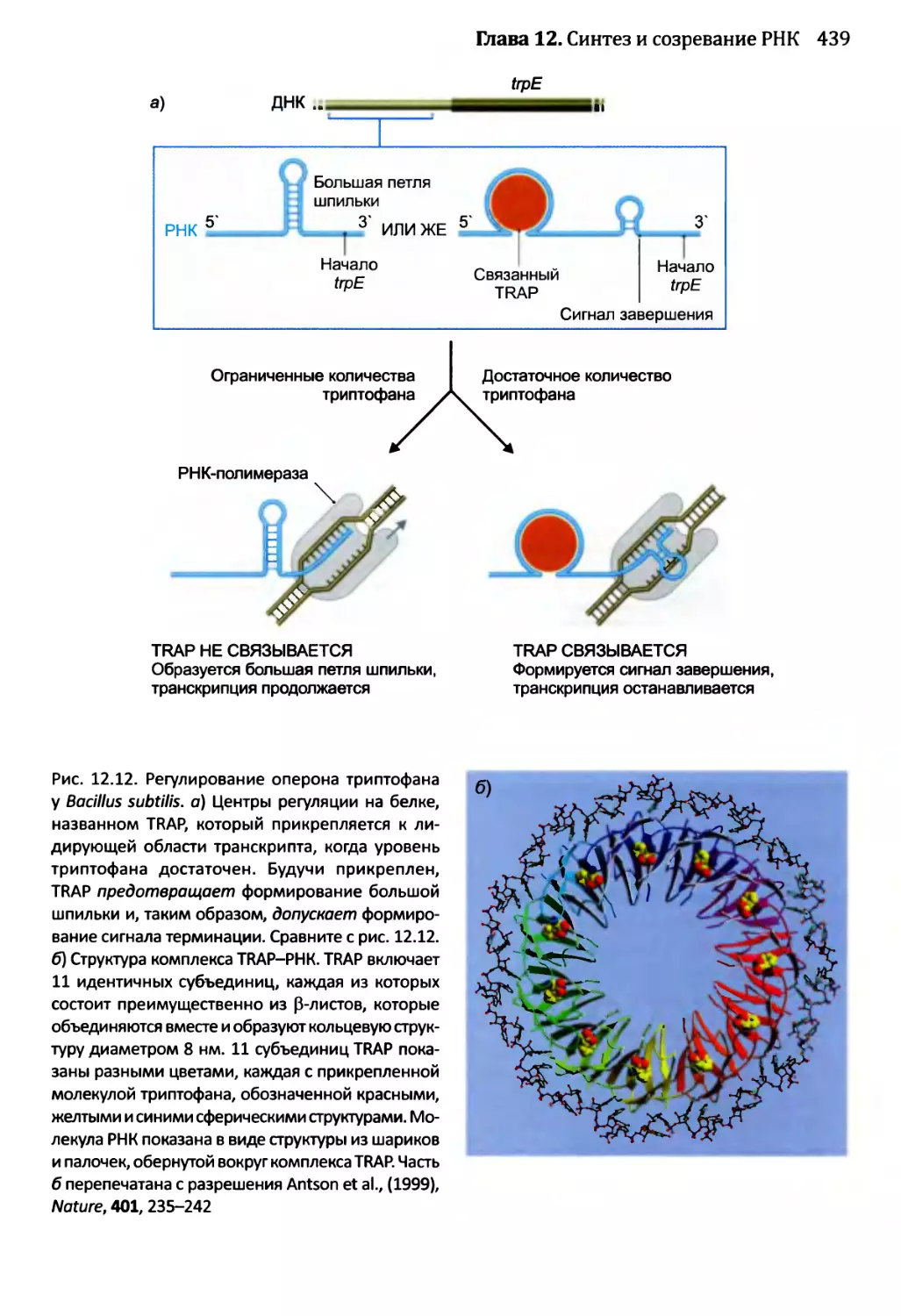
12.1.3. Созревание бактериальных РНК.........................441
12.1.4. Деградация бактериальных РНК.........................445
12.2. Синтез и созревание РНК эукариотов..........................447
12.2.1. Синтез мРНК эукариотов РНК-полимеразой II............447
12.2.2. Удаление интронов из ядерной пре-мРНК................454
12.2.3. Синтез функциональных РНК у эукариотов...............465
12.2.4. Сплайсинг пре-рРНК и пре-тРНК у эукариотов...........466
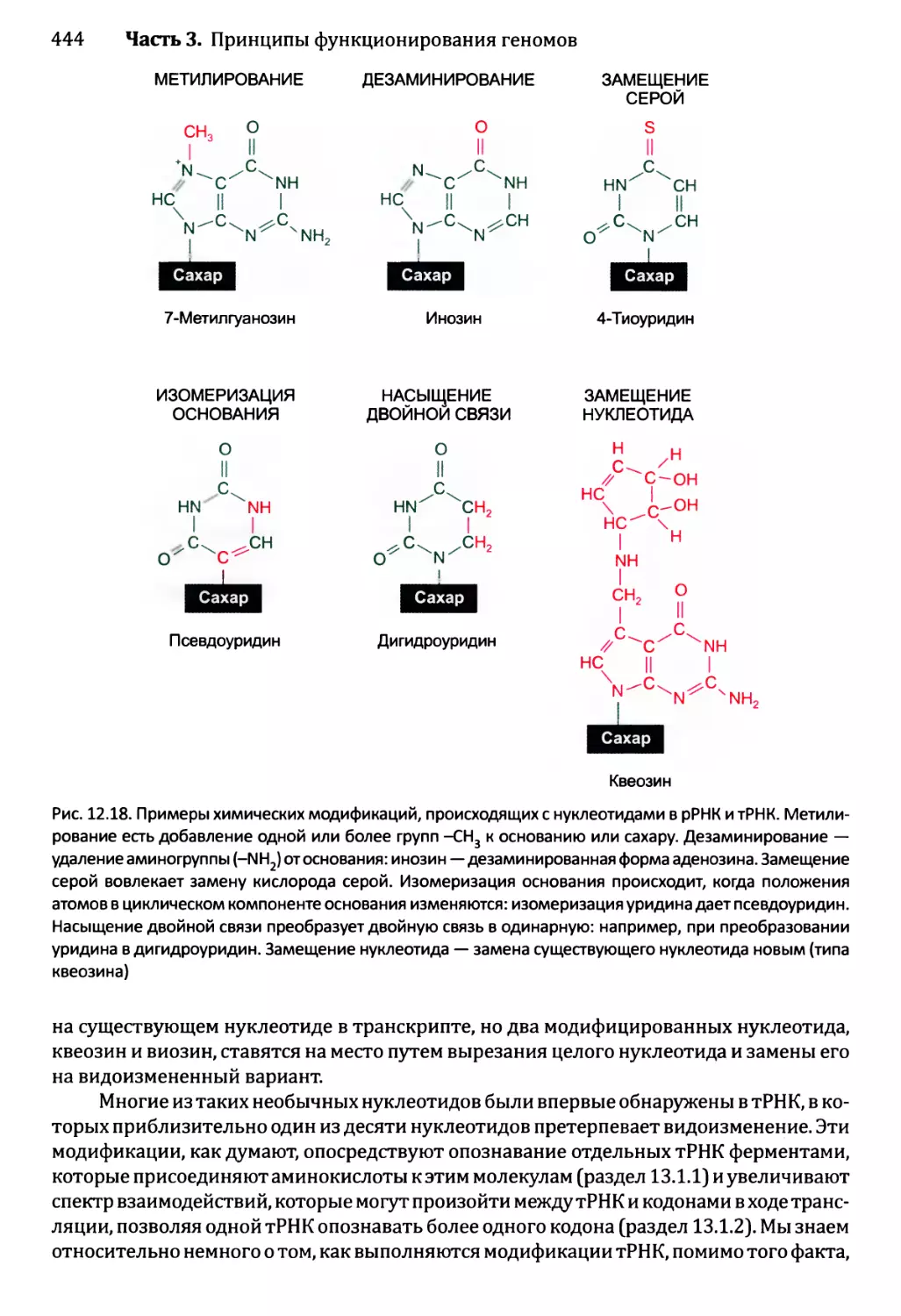
12.2.5. Химическая модификация РНК эукариотов................471
12.2.6. Деградация РНК эукариотов............................475
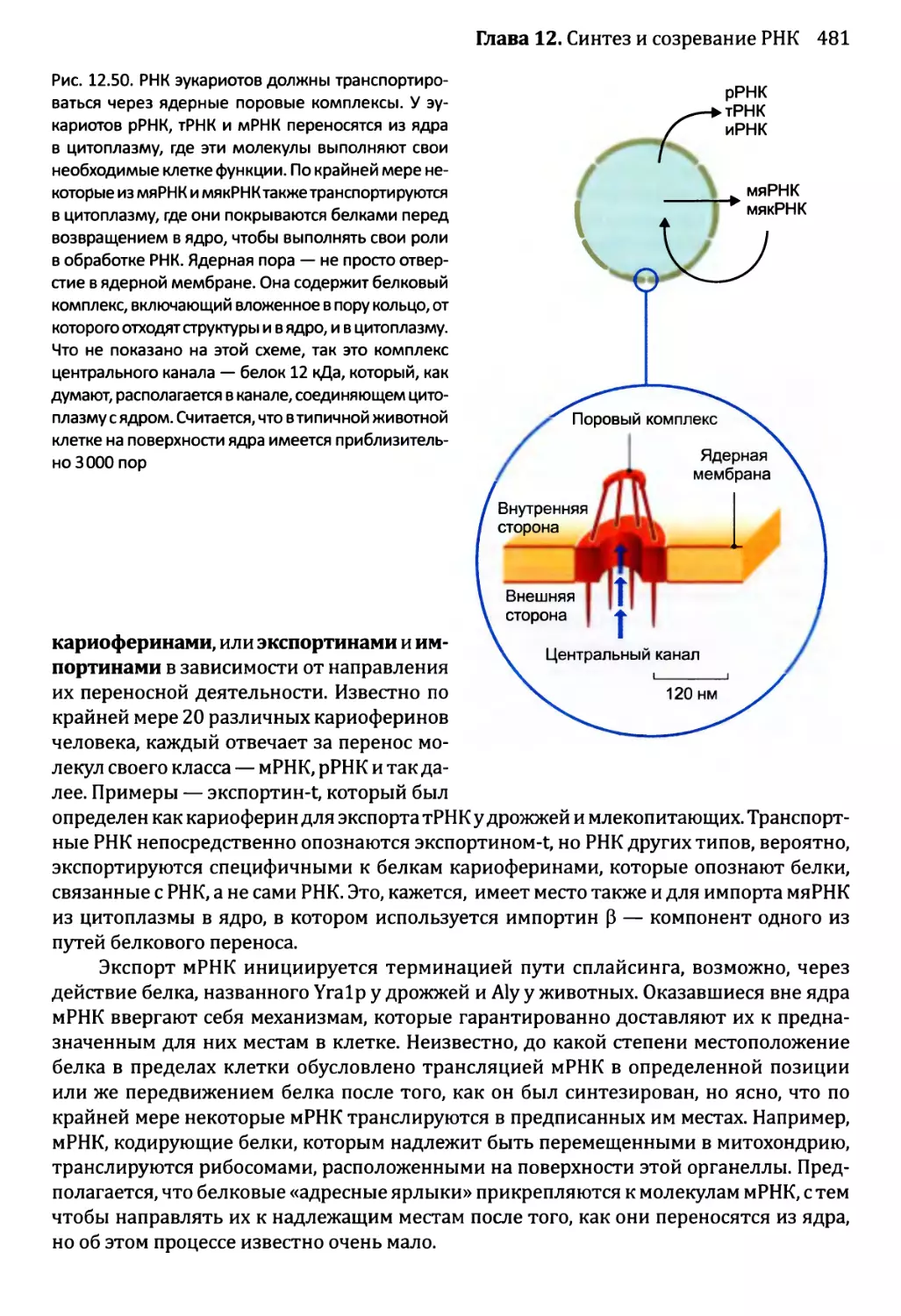
12.2.7. Перенос РНК в пределах клетки эукариотов.............480
Глава 13. Синтез и процессинг протеома................................492
13.1. Роль тРНК в синтезе белка...................................493
13.1.1. Аминоацилирование: присоединение аминокислот
к молекулам тРНК............................................493
13.1.2. Взаимодействия кодон-антикодон: прикрепление
молекул тРНК к мРНК.........................................498
13.2. Роль рибосомы в синтезе белка...............................501
13.2.1. Структура рибосомы...................................501
13.2.2. Инициация трансляции.................................504
13.2.3. Стадия элонгации в ходе трансляции...................512
13.2.4. Терминация трансляции................................518
13.2.5. Трансляция у архей...................................519
13.3. Посттрансляционный процессинг белков........................520
13.3.1. Сворачивание белка...................................521
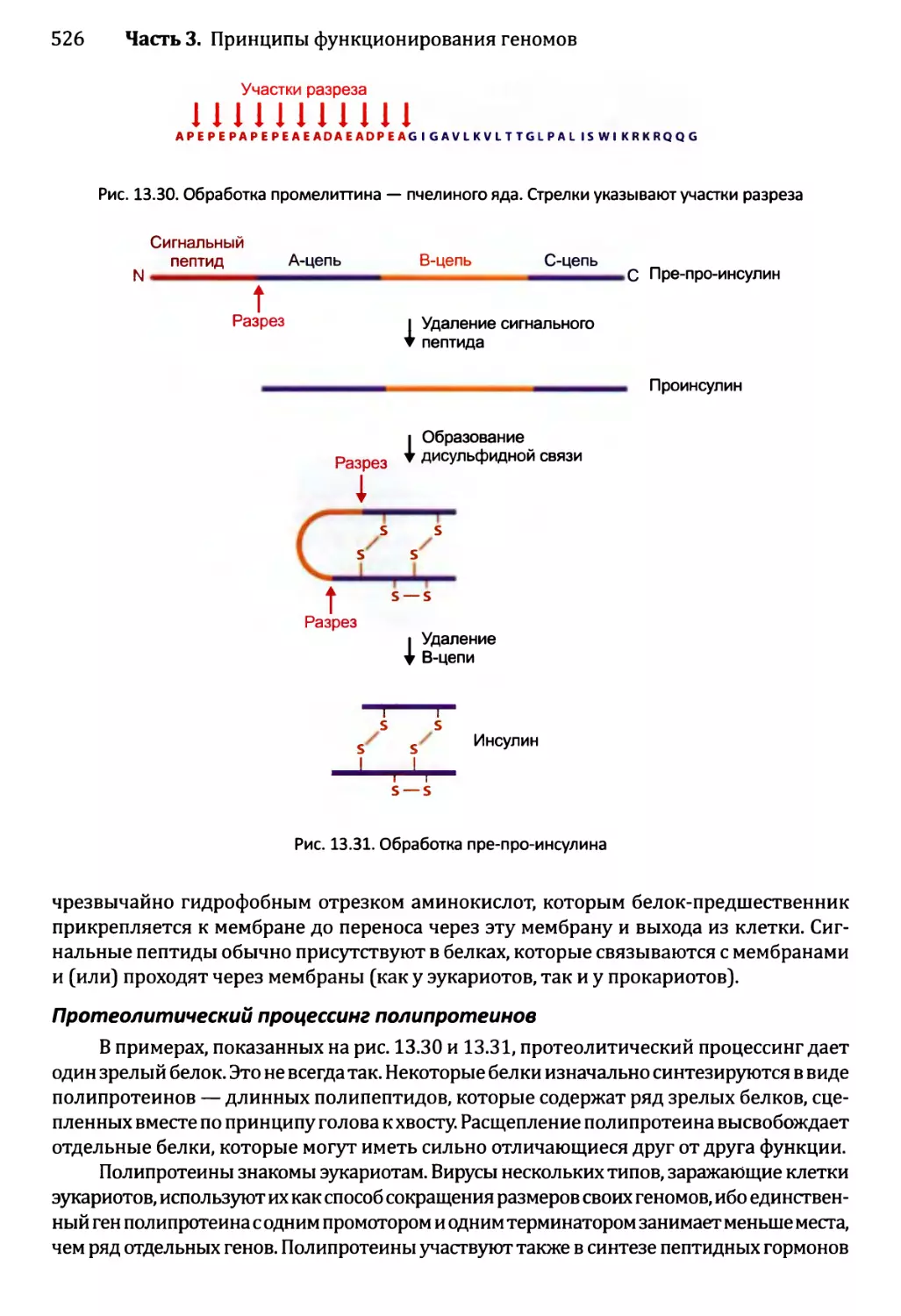
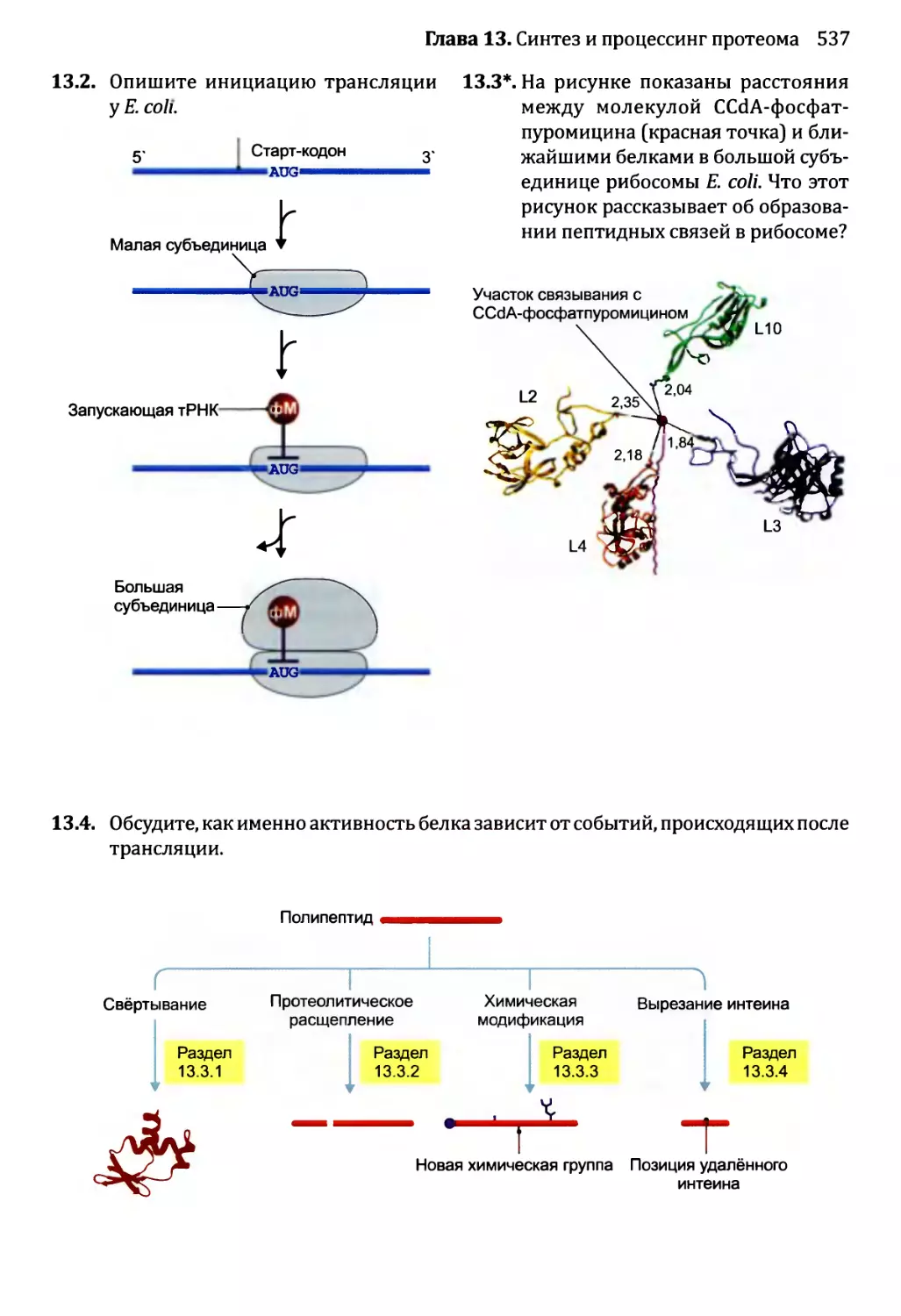
13.3.2. Обработка протеолитическим расщеплением..............525
13.3.3. Обработка химической модификацией....................527
13.3.4. Интеины..............................................529
13.4. Деградация белка............................................530
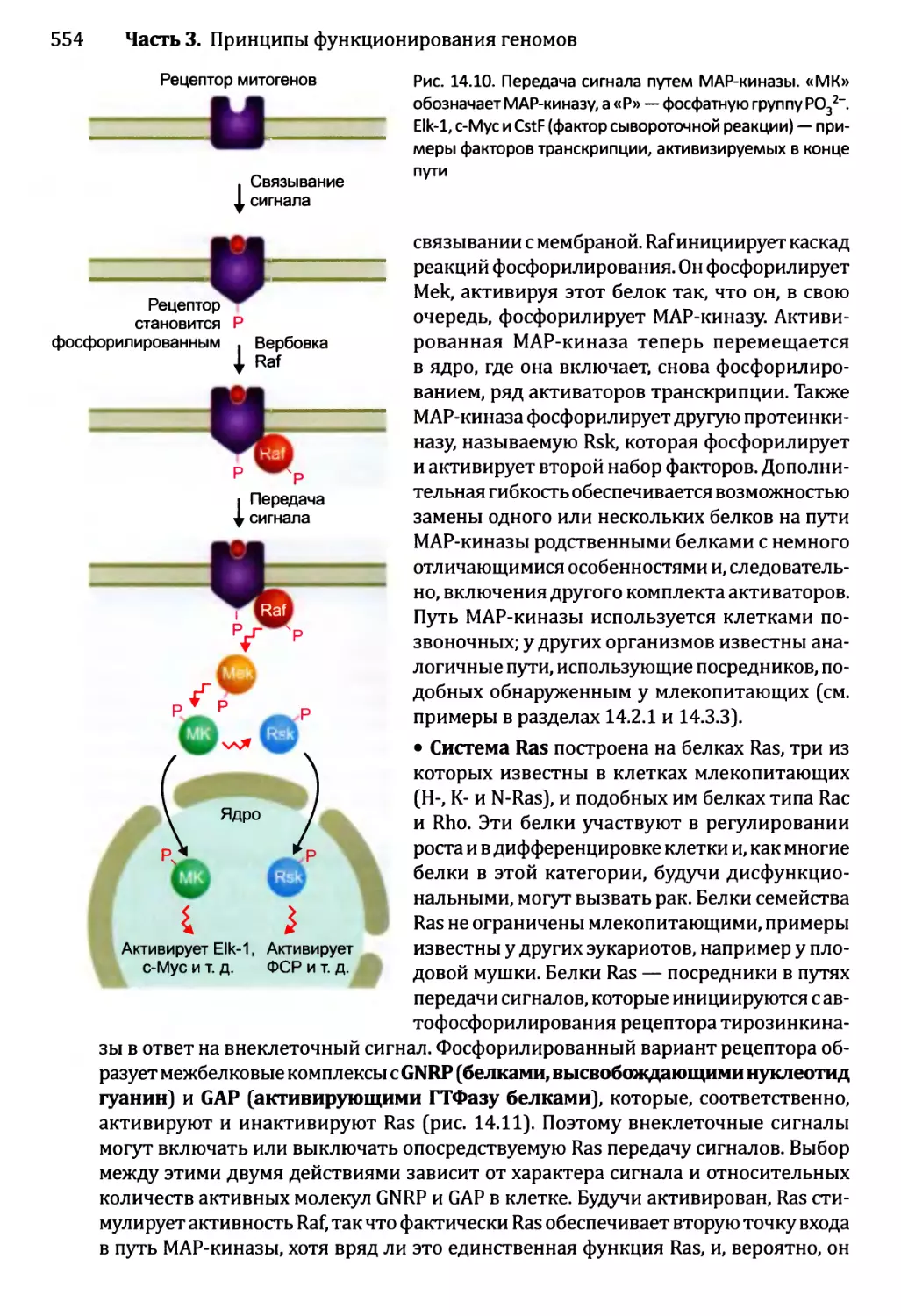
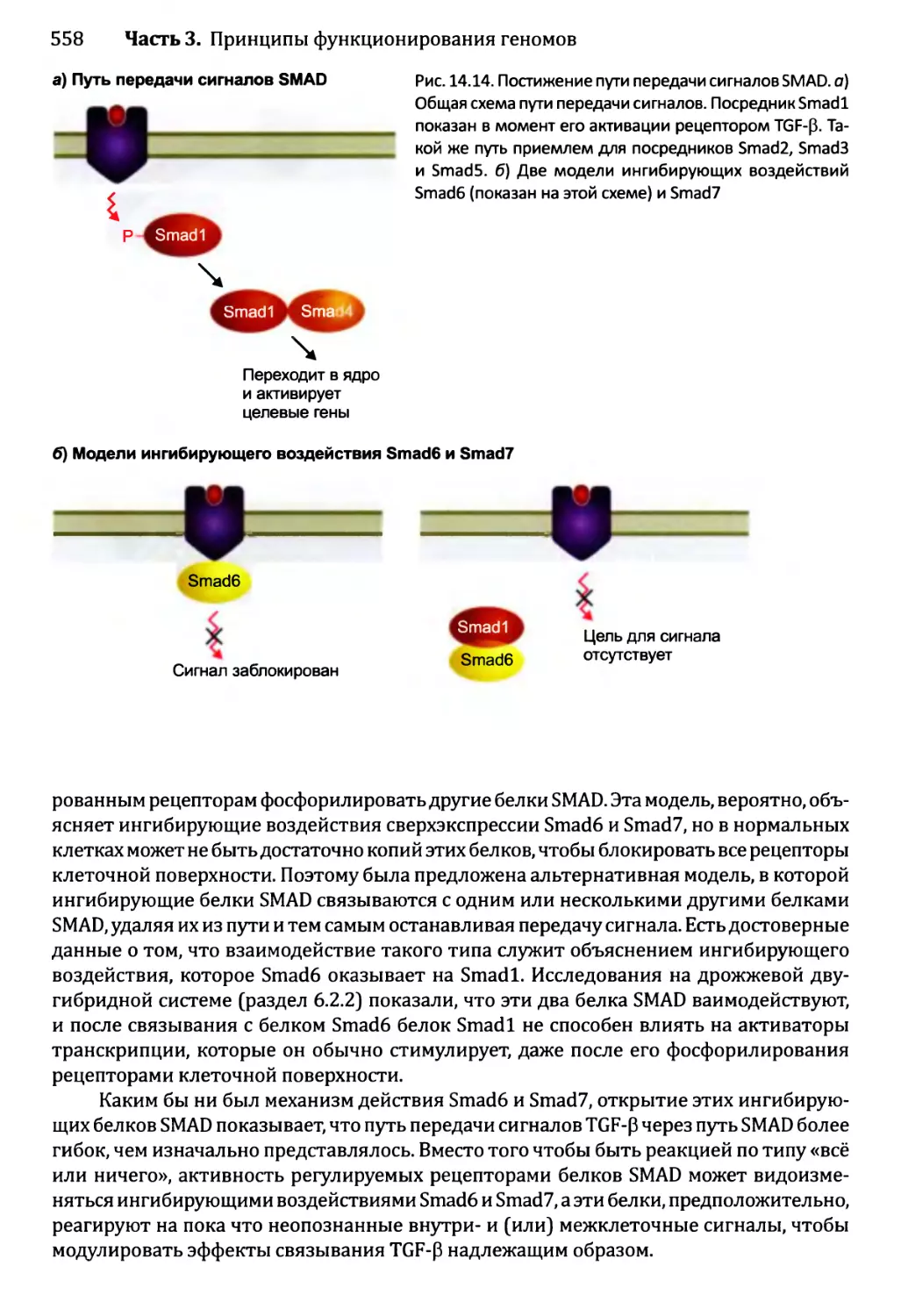
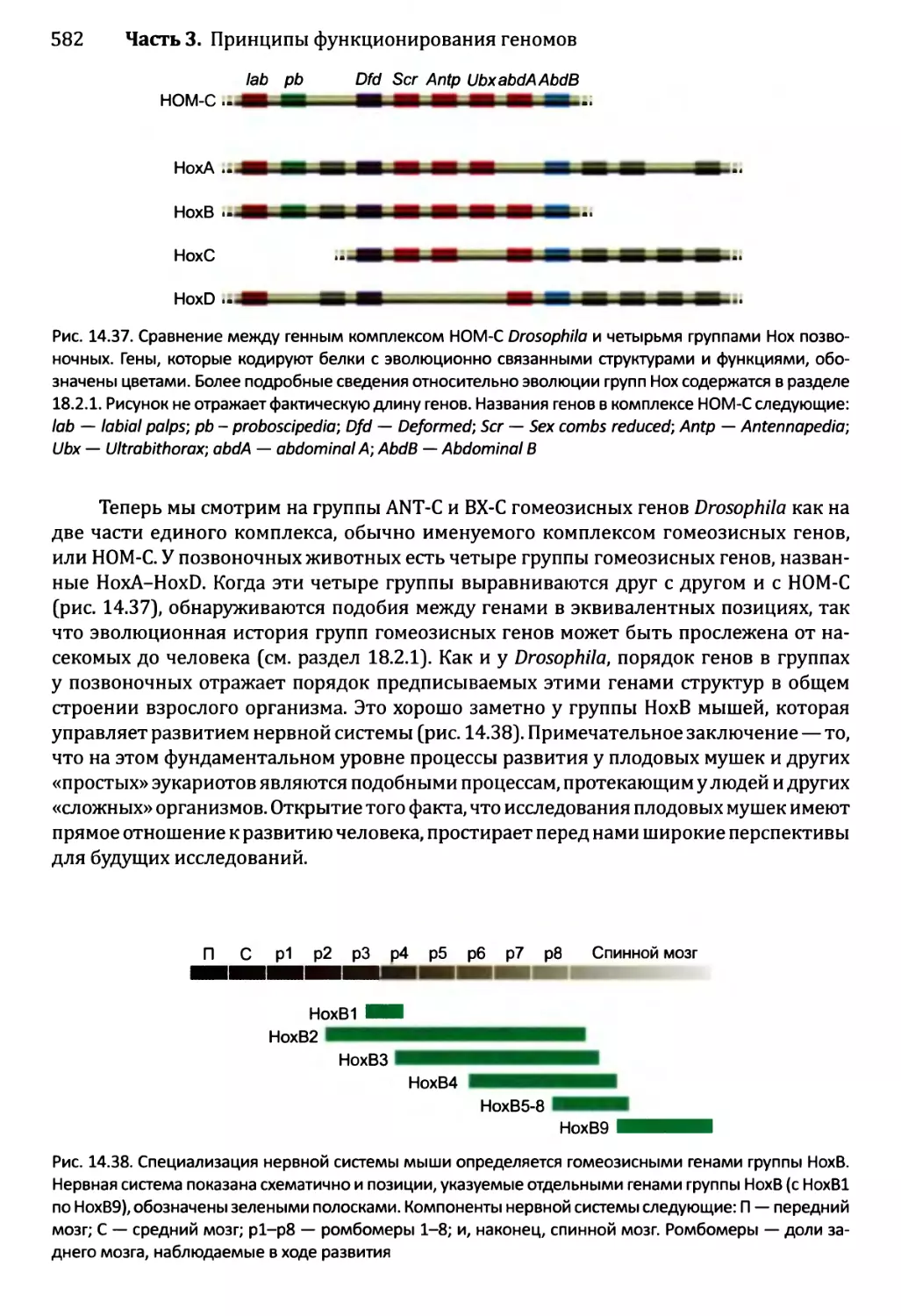
Глава 14. Регулирование активности генома.............................540
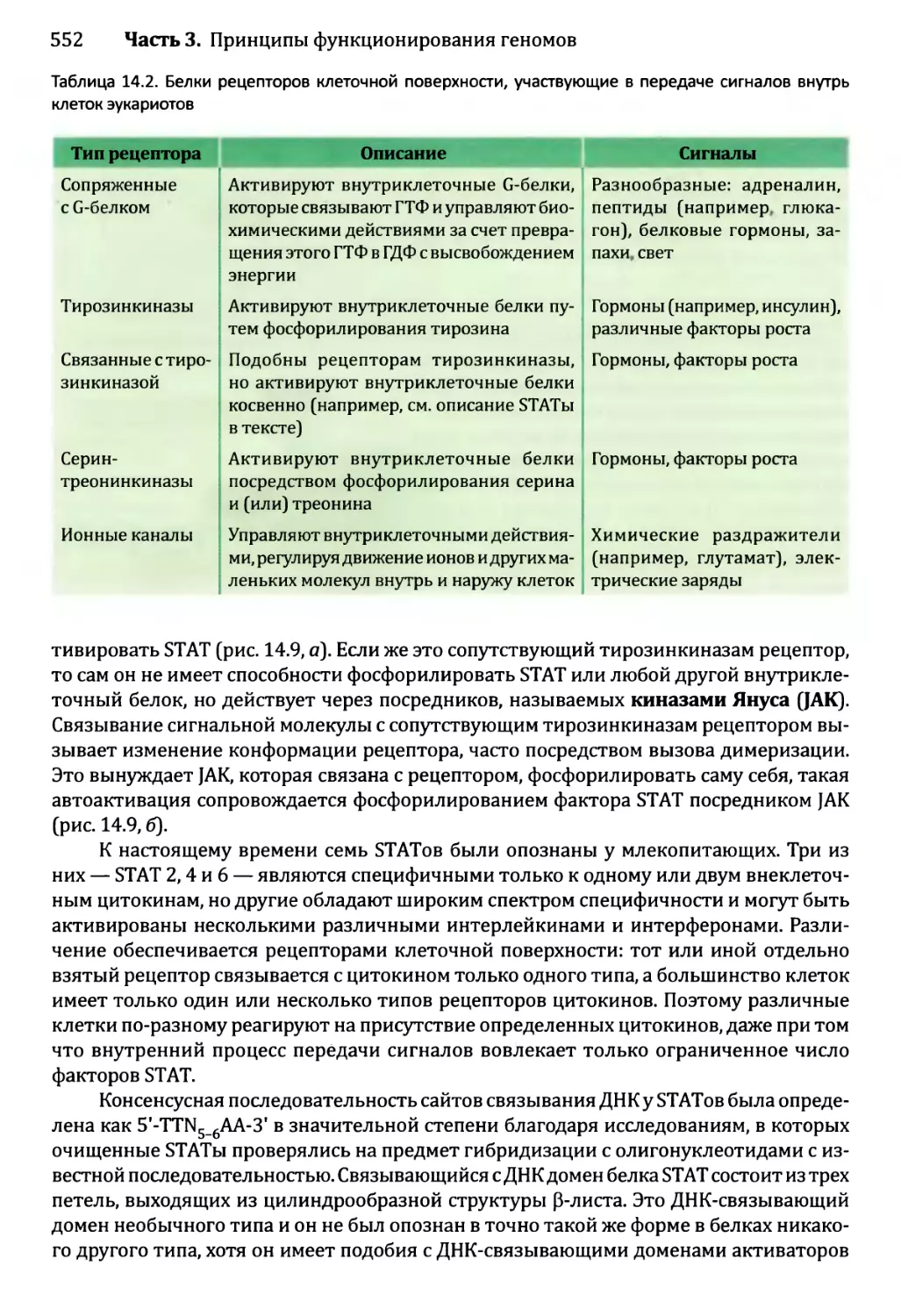
14.1. Непостоянные изменения в активности генома..................543
14.1.1.'. Передача сигнала посредством импорта внеклеточного
сигнального соединения......................................544
14.1.2', Передача сигналов опосредствуется рецепторами клеточной
” поверхности...........................................549
Оглавление ix
14.2. Постоянные и полупостоянные изменения в активности генома....559
14.2.1. Перестройки генома....................................559
14.2.2. Изменения в структуре хроматина.......................564
14.2.3. Регулирование генома петлями обратной связи..........566
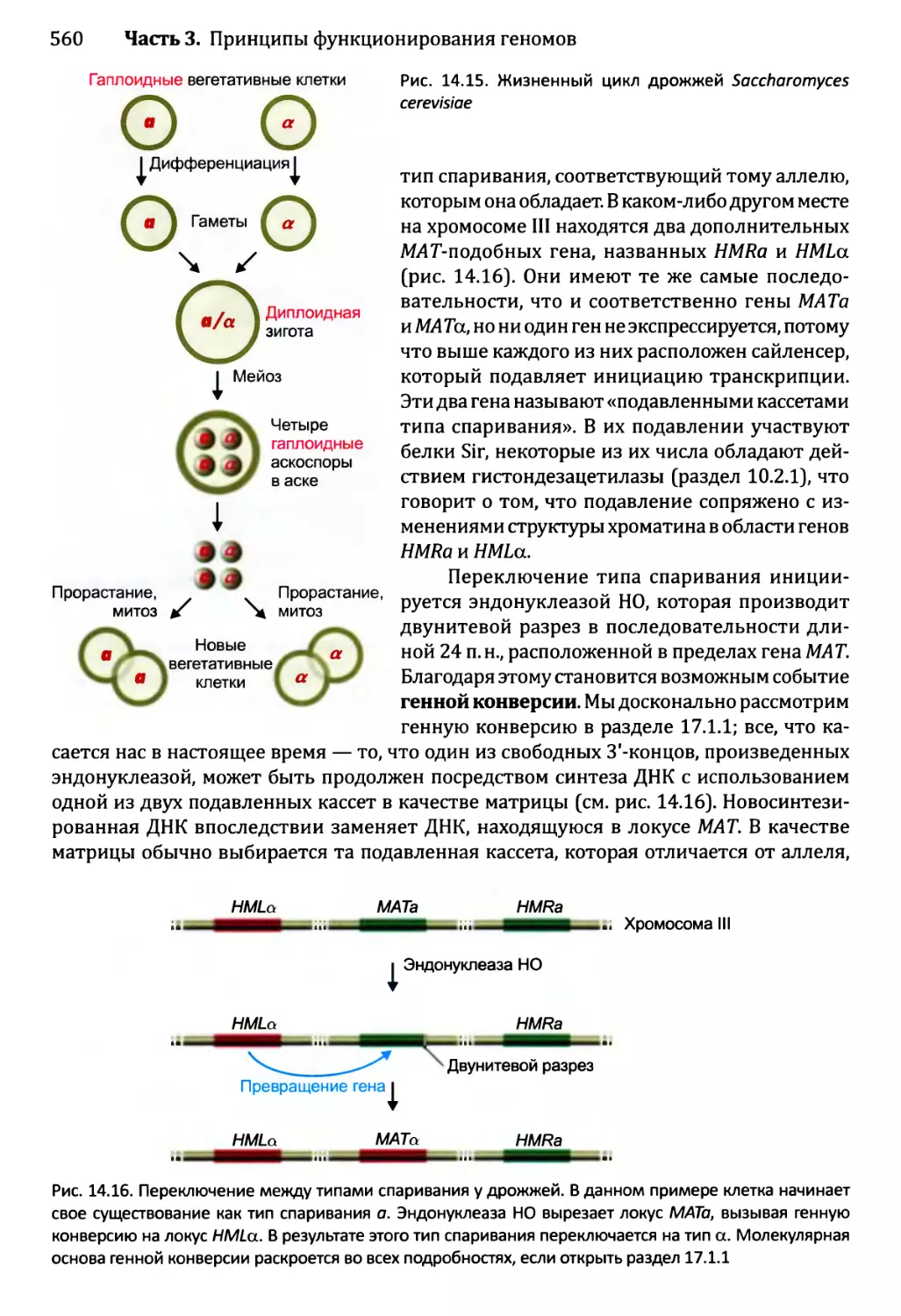
14.3. Регулирование активности генома в ходе развития организма...566
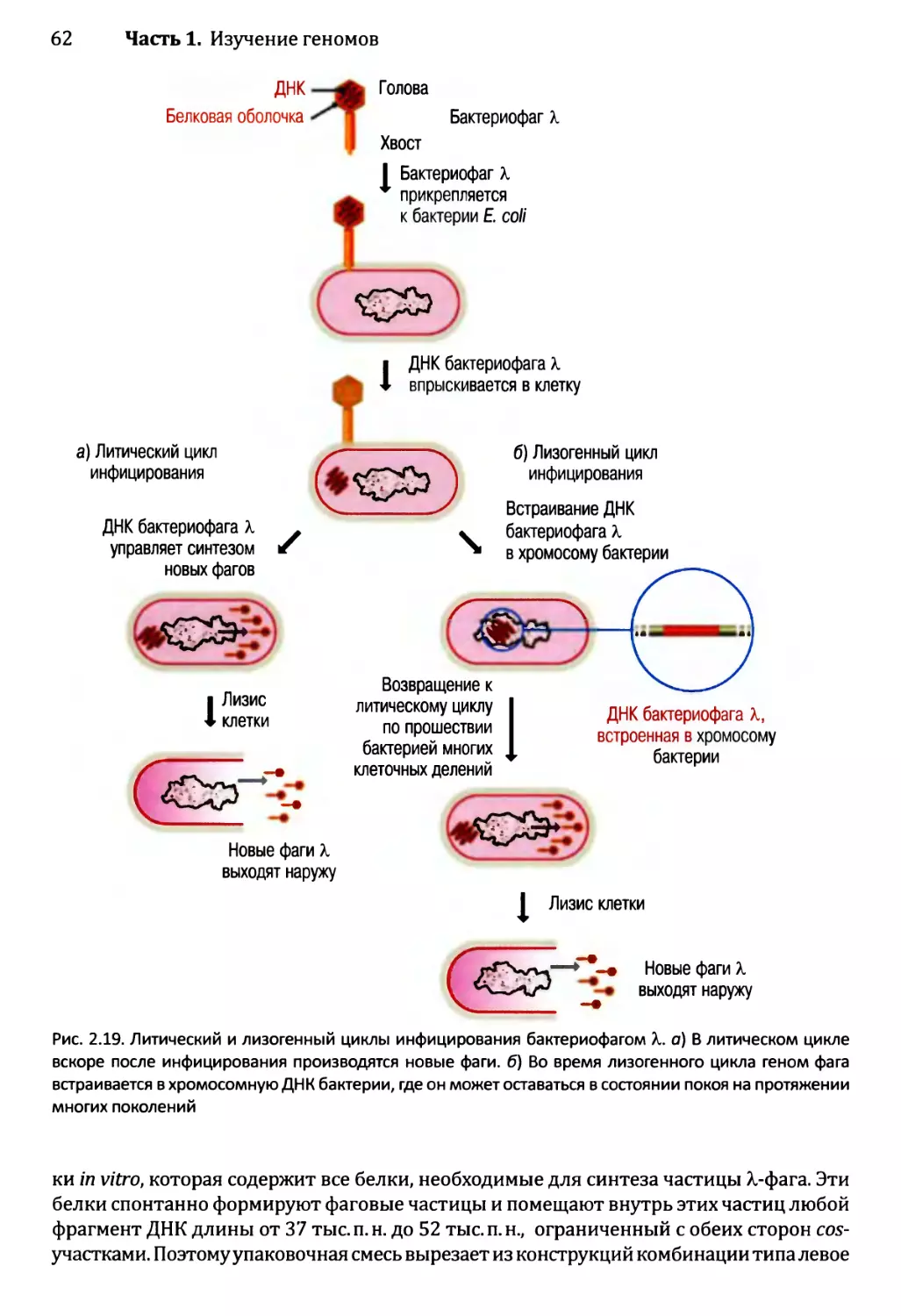
14.3 .1.ЪЛизогенный цикл бактериофага X.......................567
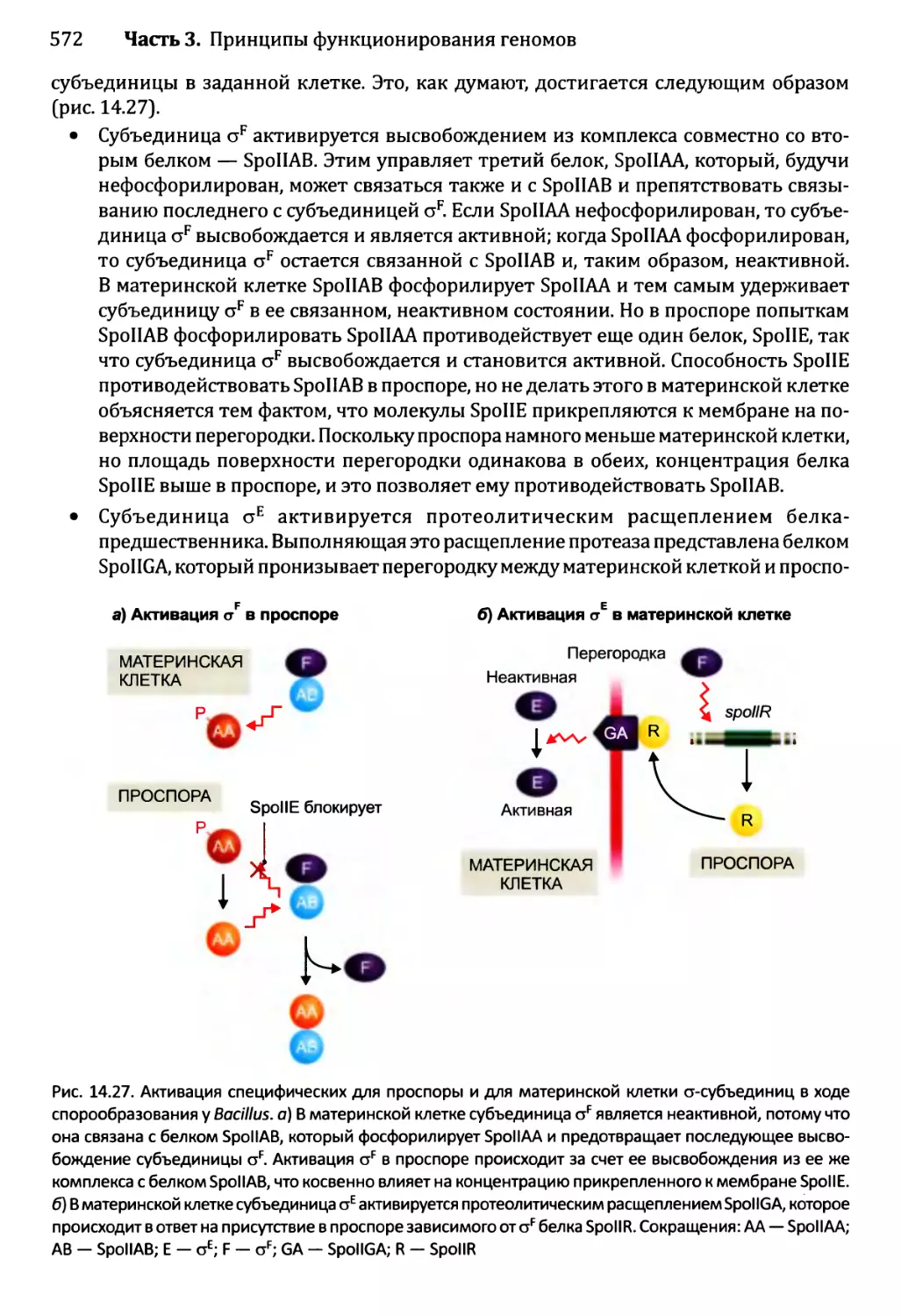
14.3Д Спорообразование у Bacillus...........................569
14.3. 3. Развитие яйцевода у Caenorhabditis elegans..........573
14.3) 4. Развитие Drosophila melanogaster....................577
Часть 4. Механизмы репликации и эволюции геномов......................593
Глава 15. Репликация генома...........................................595
15.1. Топологическая проблема.....................................597
15.1.1. Экспериментальное доказательство предложенной Уотсоном
и Криком схемы репликации ДНК...............................597
15.1.2. ДНК-топоизомеразы решают топологическую проблему
наделе.......................................................600
15.1.3. Вариации на тему полуконсервативности................602
15.2. Процесс репликации..........................................604
15.2.1. Инициация репликации генома..........................604
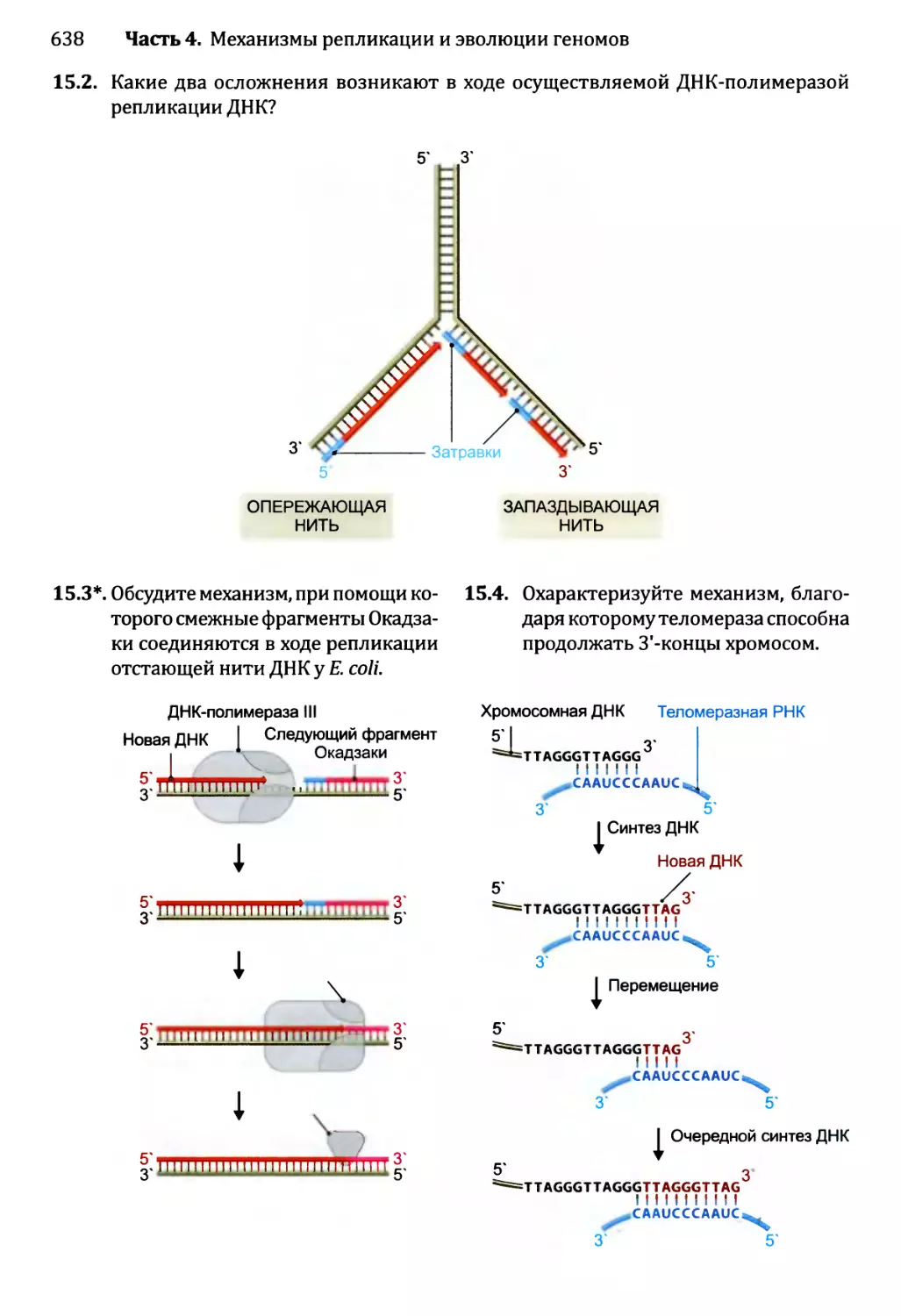
15.2.2. Стадия элонгации репликации..........................608
15.2.3. Терминация репликации................................619
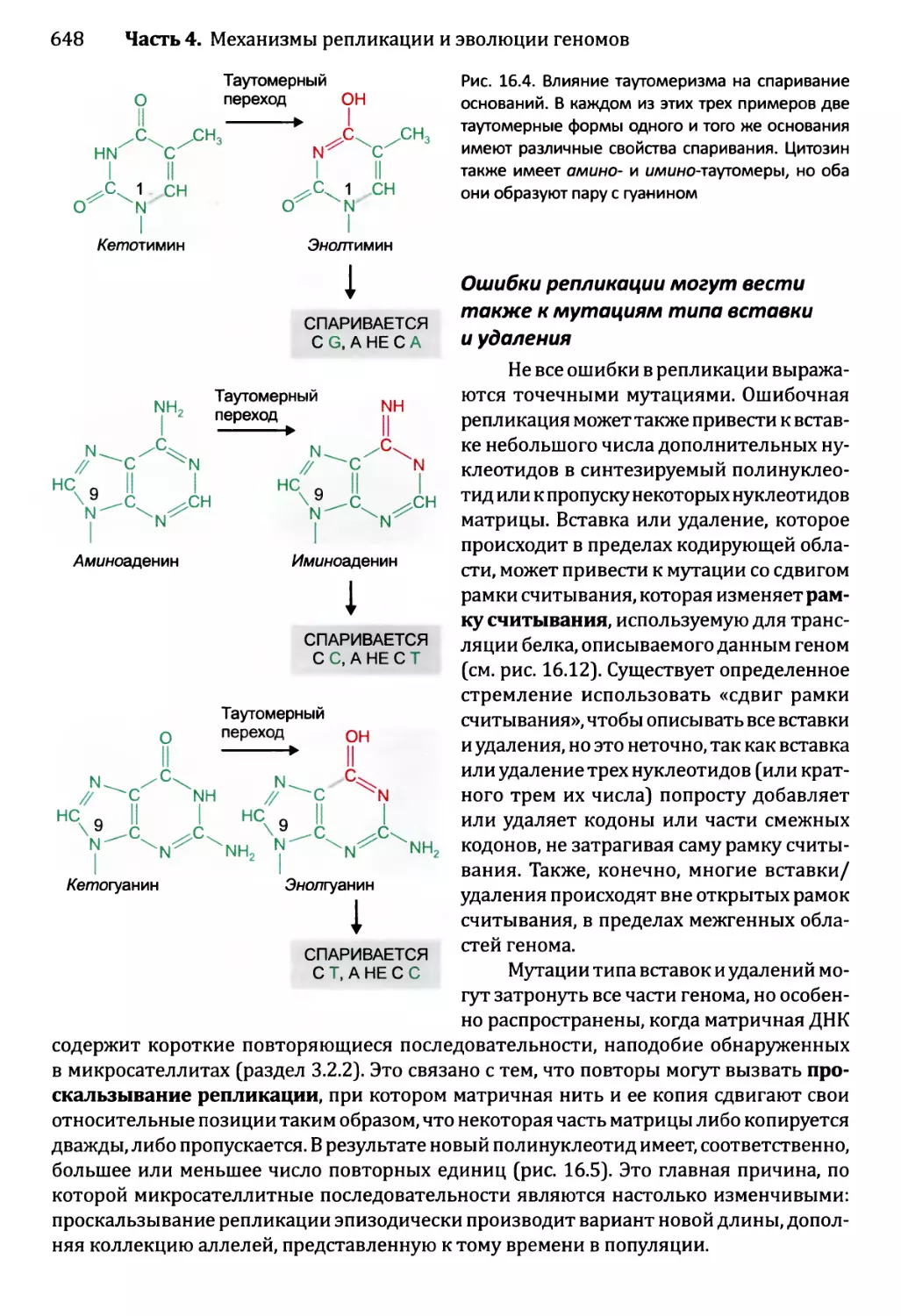
15.2.4. Поддержание концов линейной молекулы ДНК.............622
15.3. Регулирование репликации генома у эукариотов................627
15.3.1. Согласование репликации генома и деления клетки......627
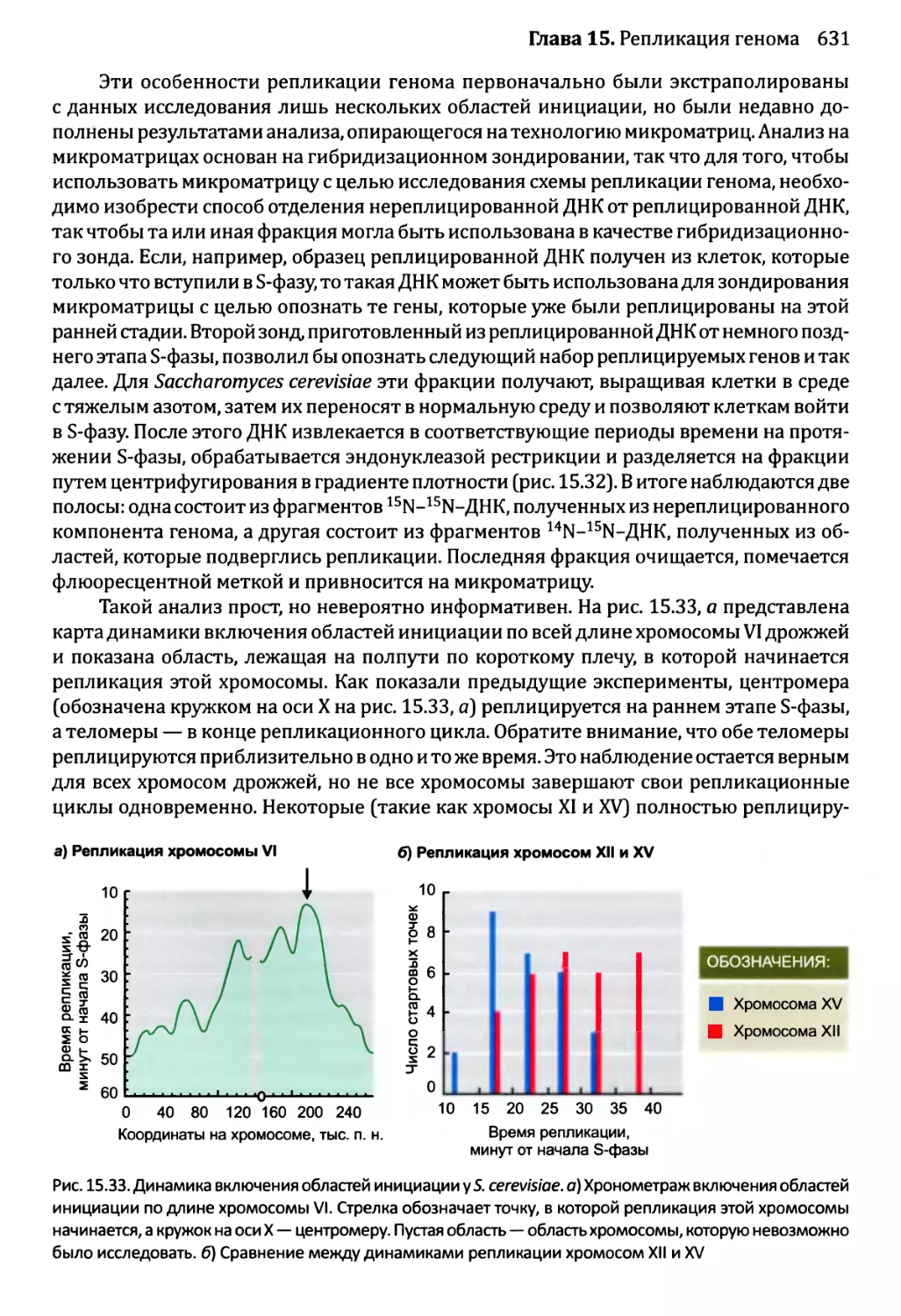
15.3.2. Управление во время S-фазы...........................630
Глава 16. Мутации и репарация ДНК.....................................641
16.1. Мутации.....................................................642
16.1.1. Причины мутаций......................................643
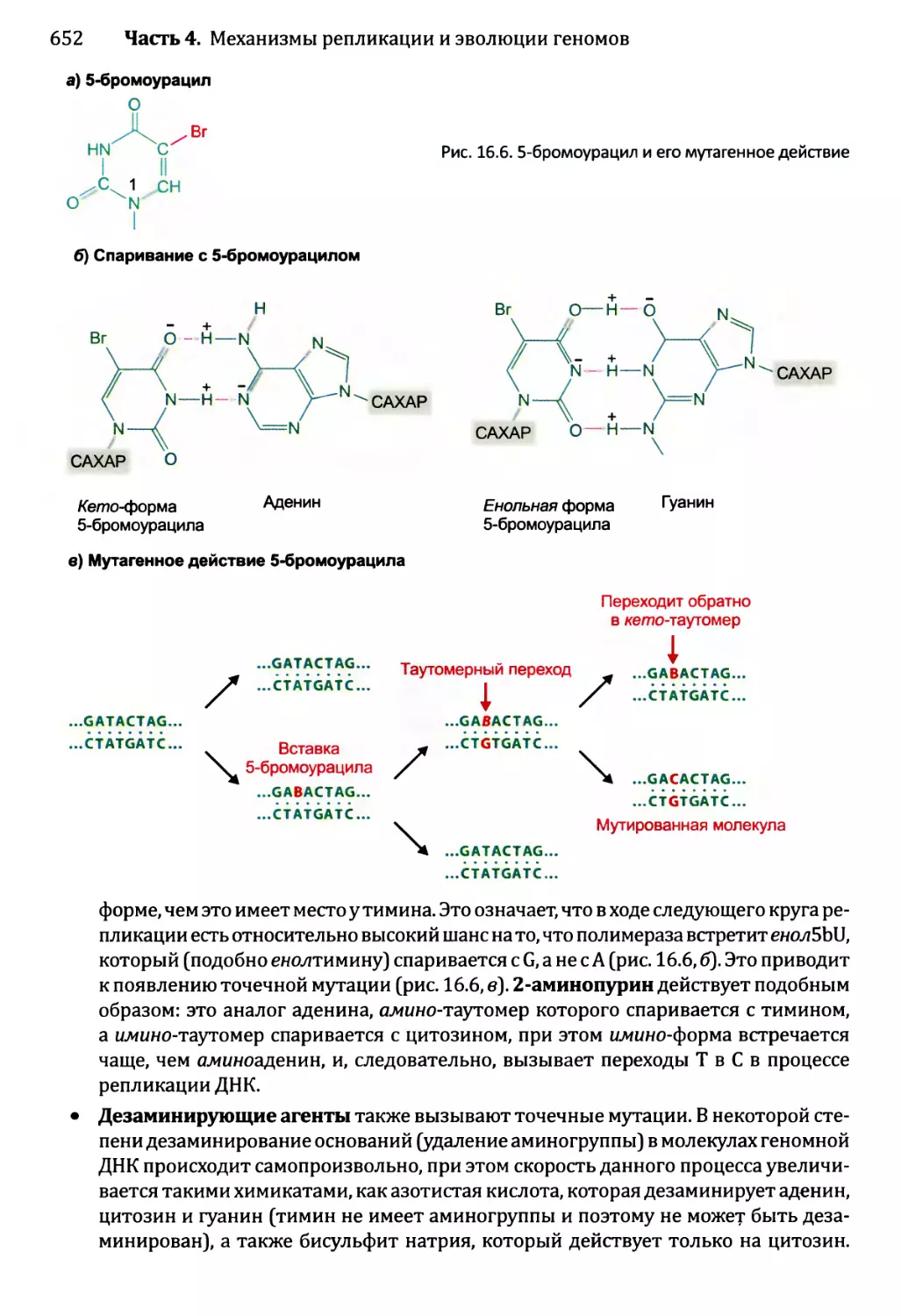
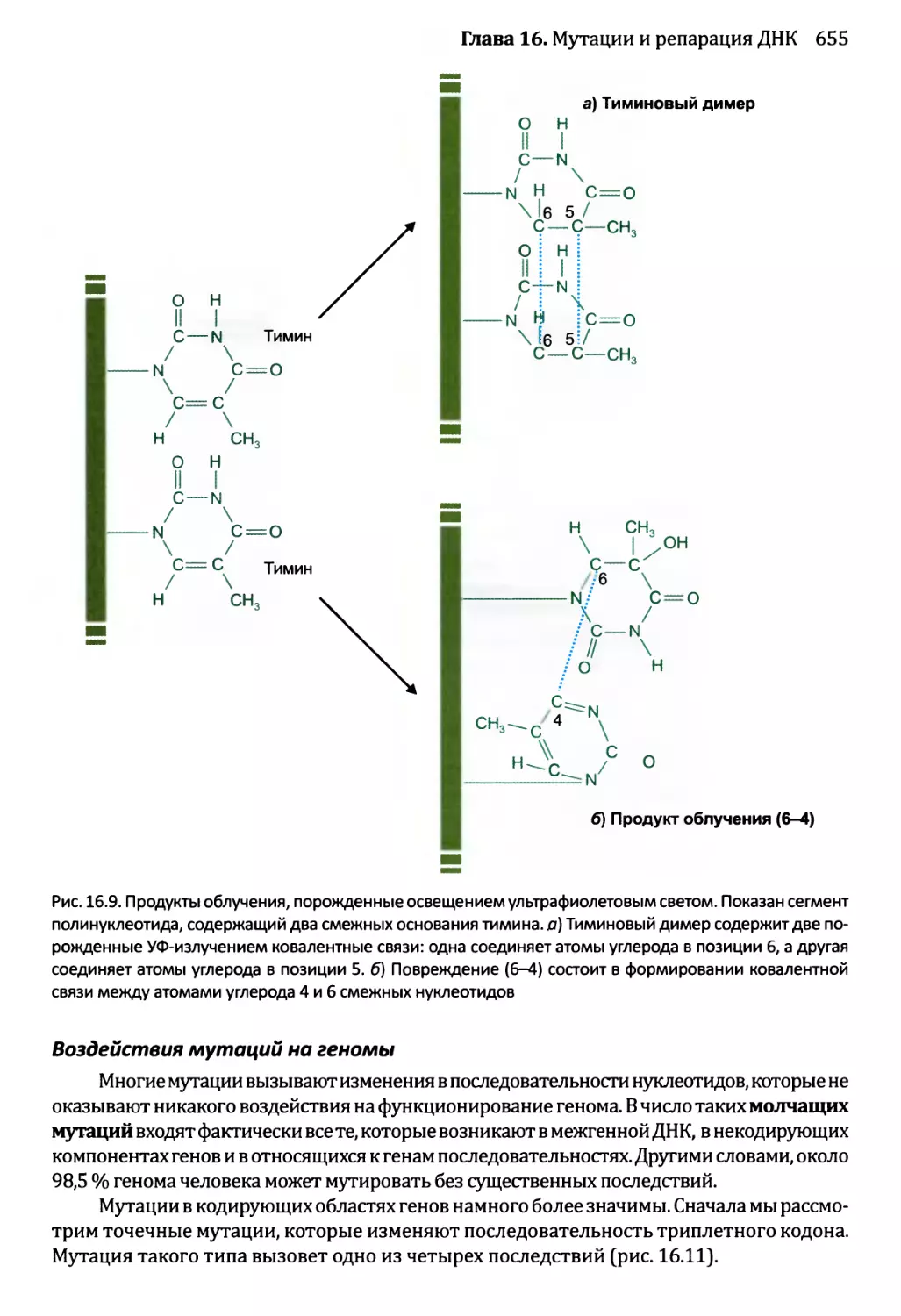
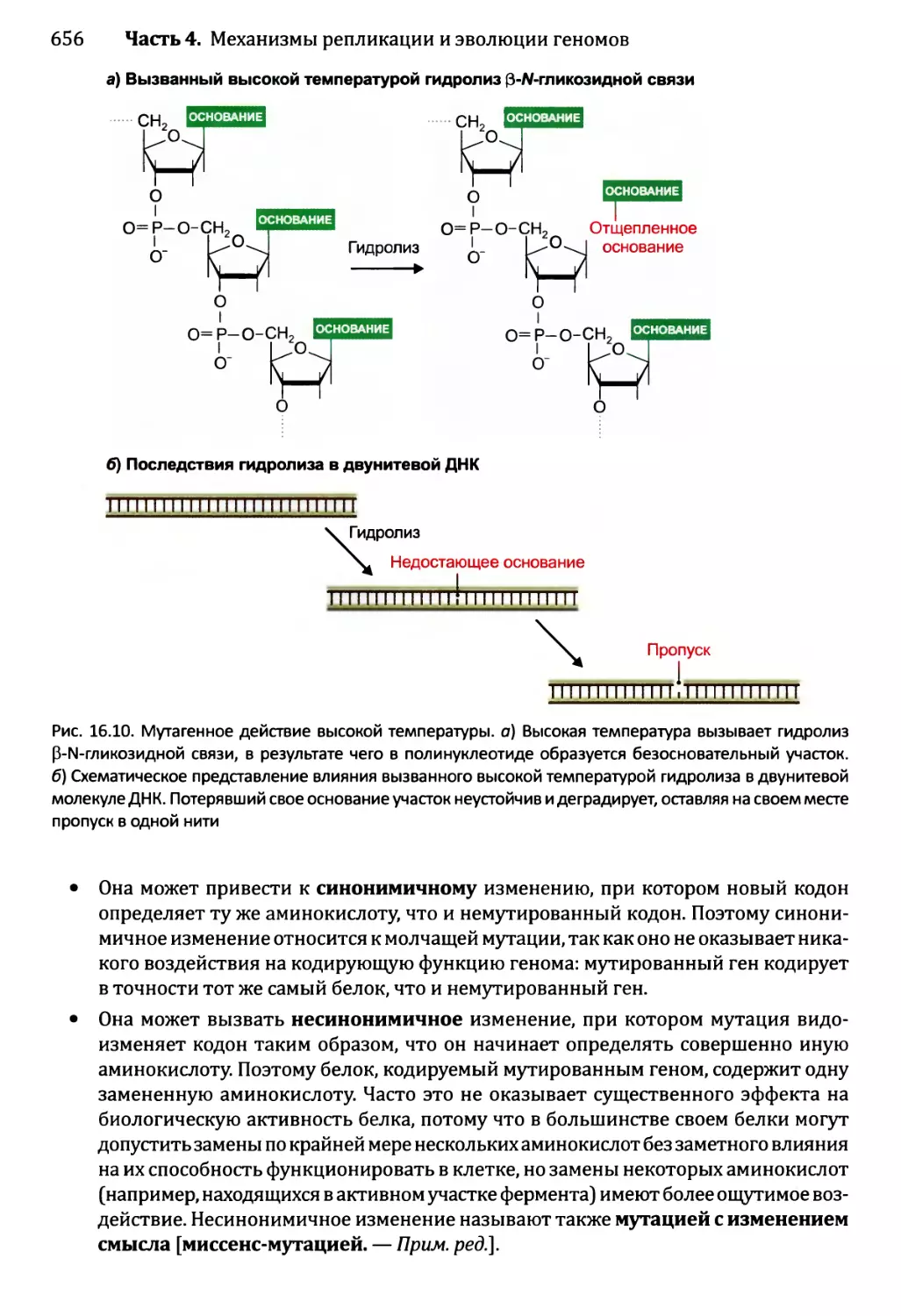
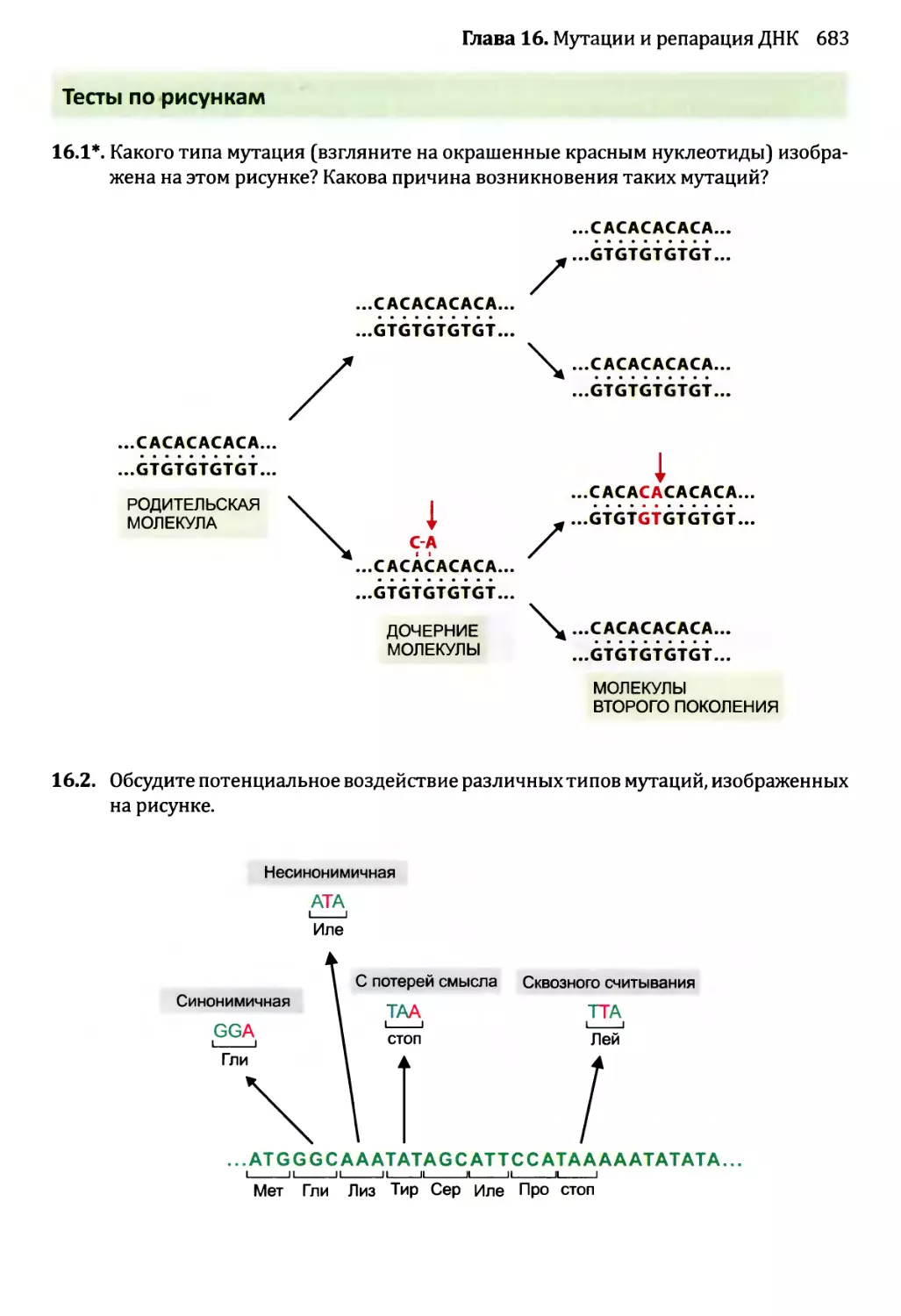
16.1.2. Воздействия мутаций..................................654
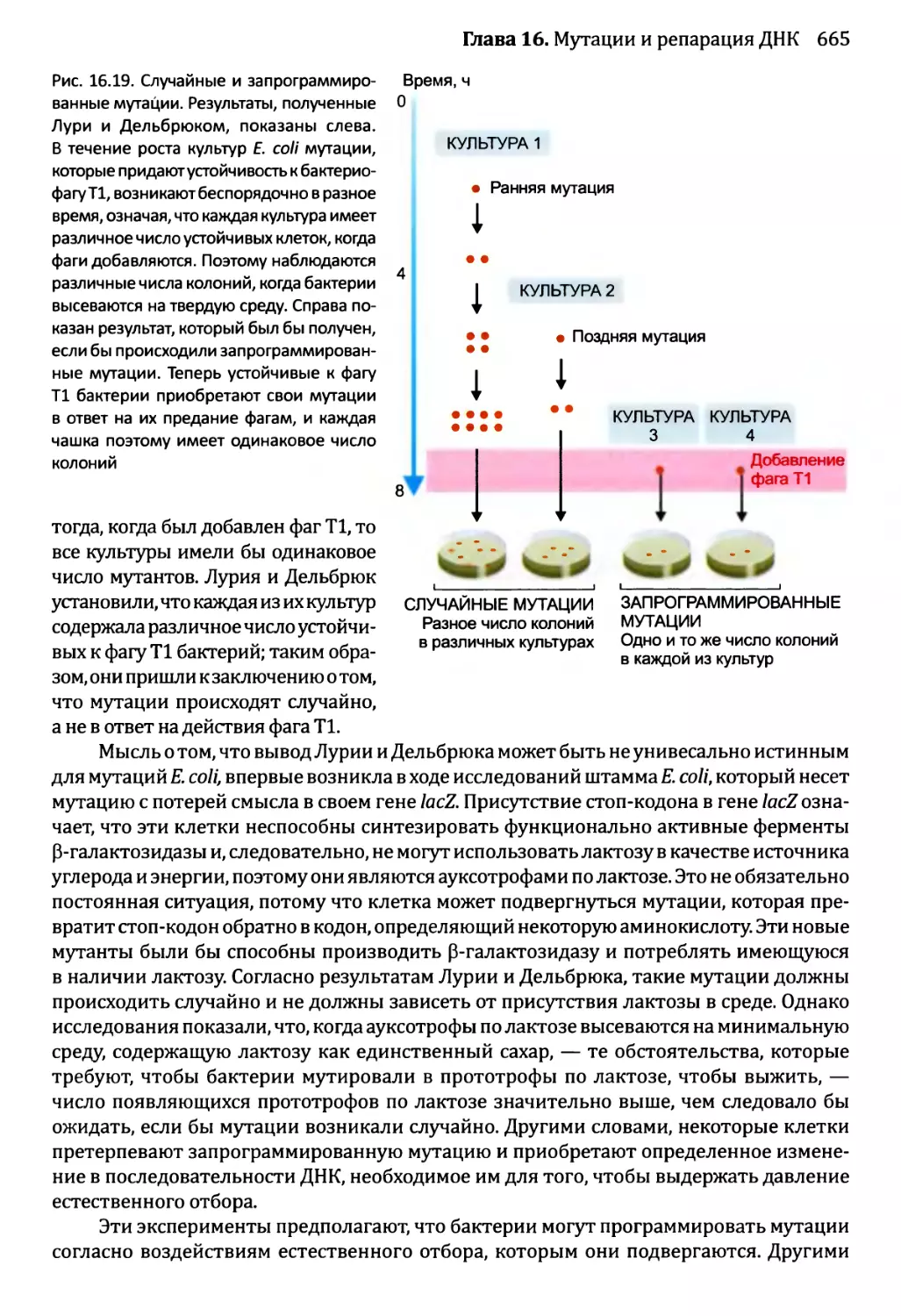
16.1.3. Сверхмутация и возможность запрограммированных мутаций....662
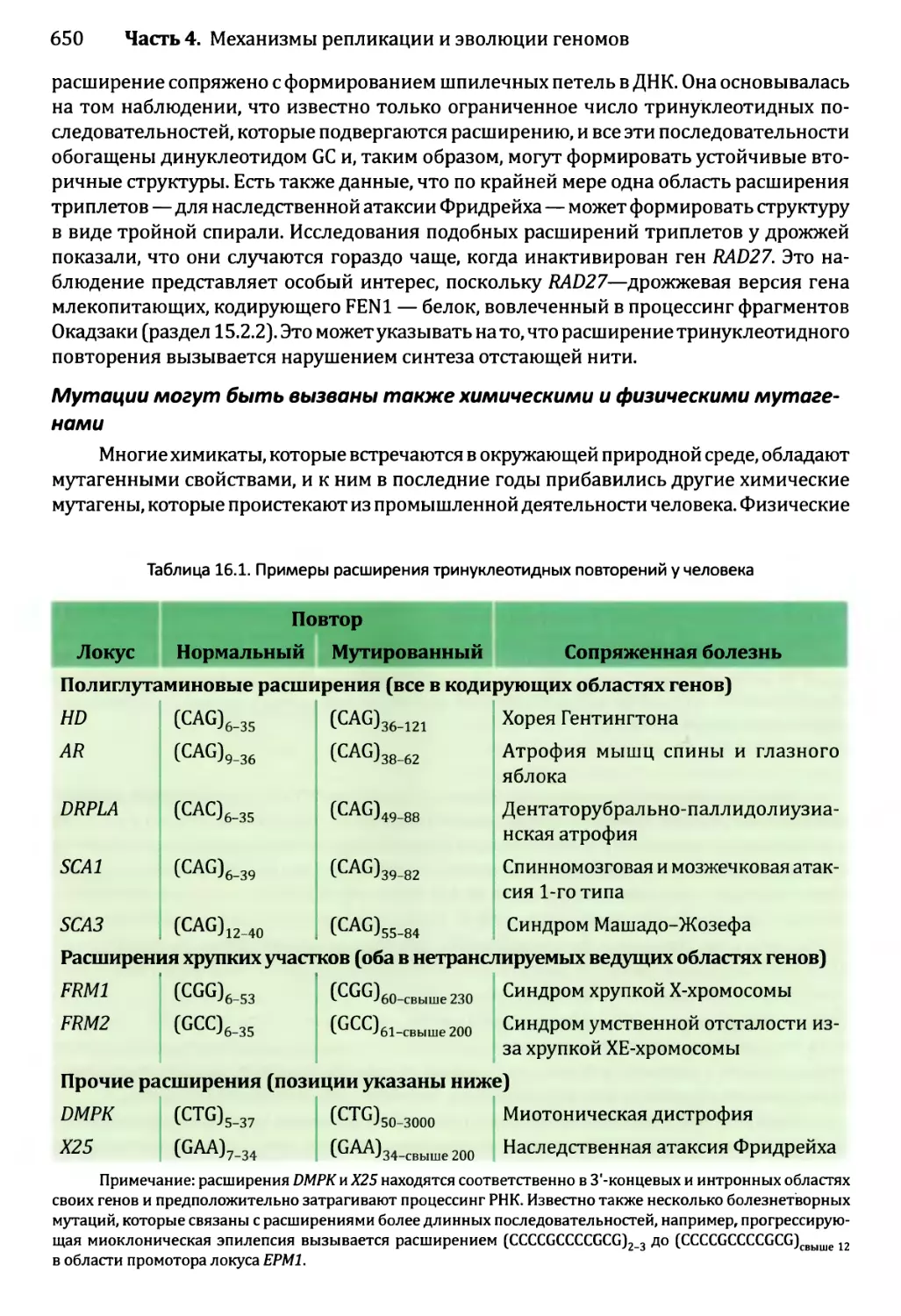
16.2. Репарация ДНК...............................................666
16.2.1. Системы прямого восстановления заполняют надрывы
и исправляют модификации нуклеотидов некоторых типов.........667
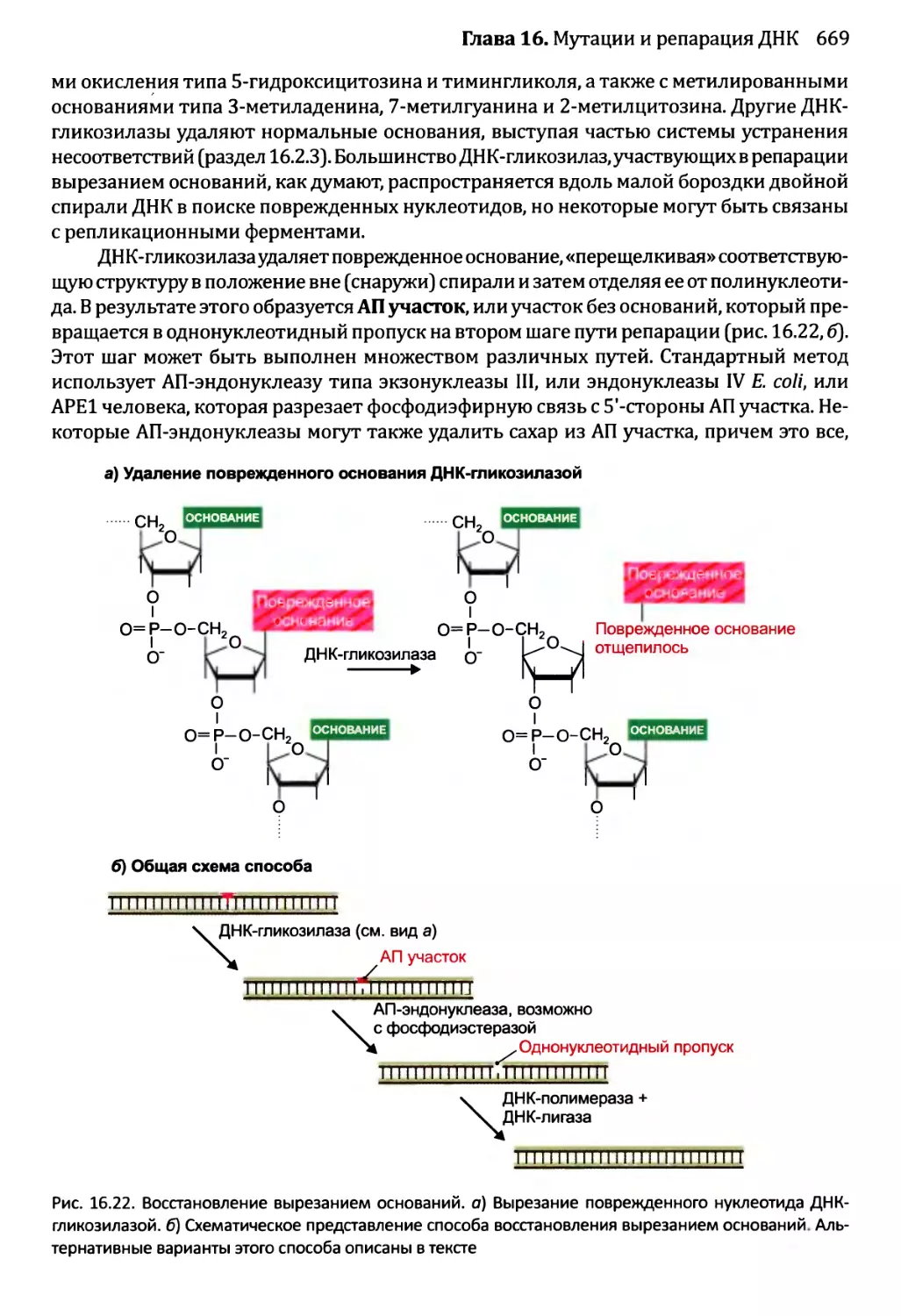
16.2.2. Восстановление вырезанием............................668
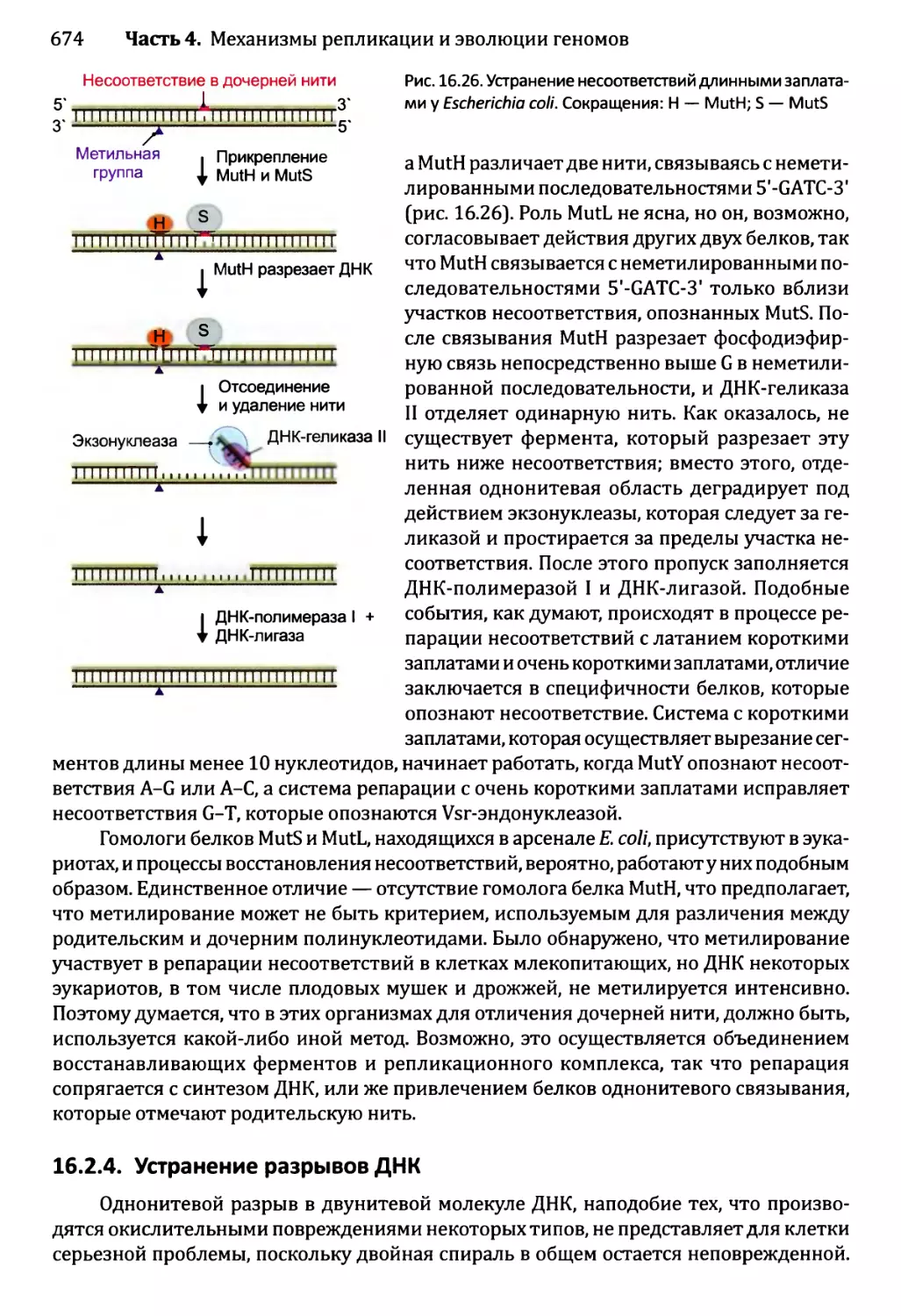
16.2.3. Устранение несоответствий: исправление ошибок репликации ...672
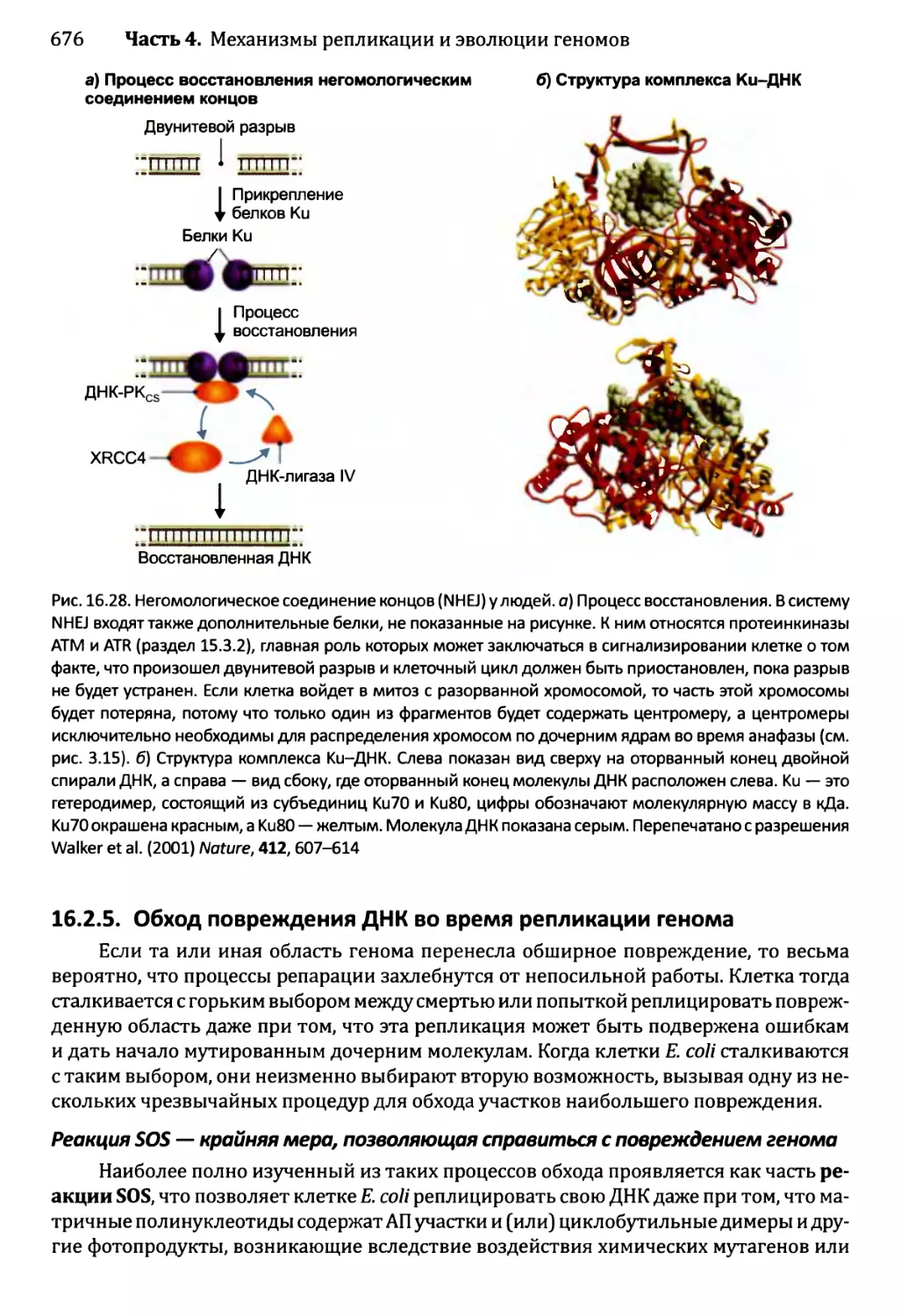
16.2.4. Устранение разрывов ДНК..............................674
16.2.5. Обход повреждения ДНК во время репликации генома.....676
16.2.6. Дефекты репарации ДНК лежат в основе многих болезней
человека, в том числе и появления раковых образований.......678
Глава 17. Рекомбинация................................................687
17.1. Гомологичная рекомбинация...................................689
17.1.1. Модели гомологичной рекомбинации.....................689
17.1.2. Биохимия гомологичной рекомбинации...................692
17.1.3. Гомологичная рекомбинация и репарация ДНК............696
X
Оглавление
17.2. Сайтспецифическая рекомбинация............................697
17.2.1. Встраивание ДНК бактериофага X в геном Е. coli.....698
17.2.2. Сайтспецифическая рекомбинация — помощь в генной
инженерии..................................................700
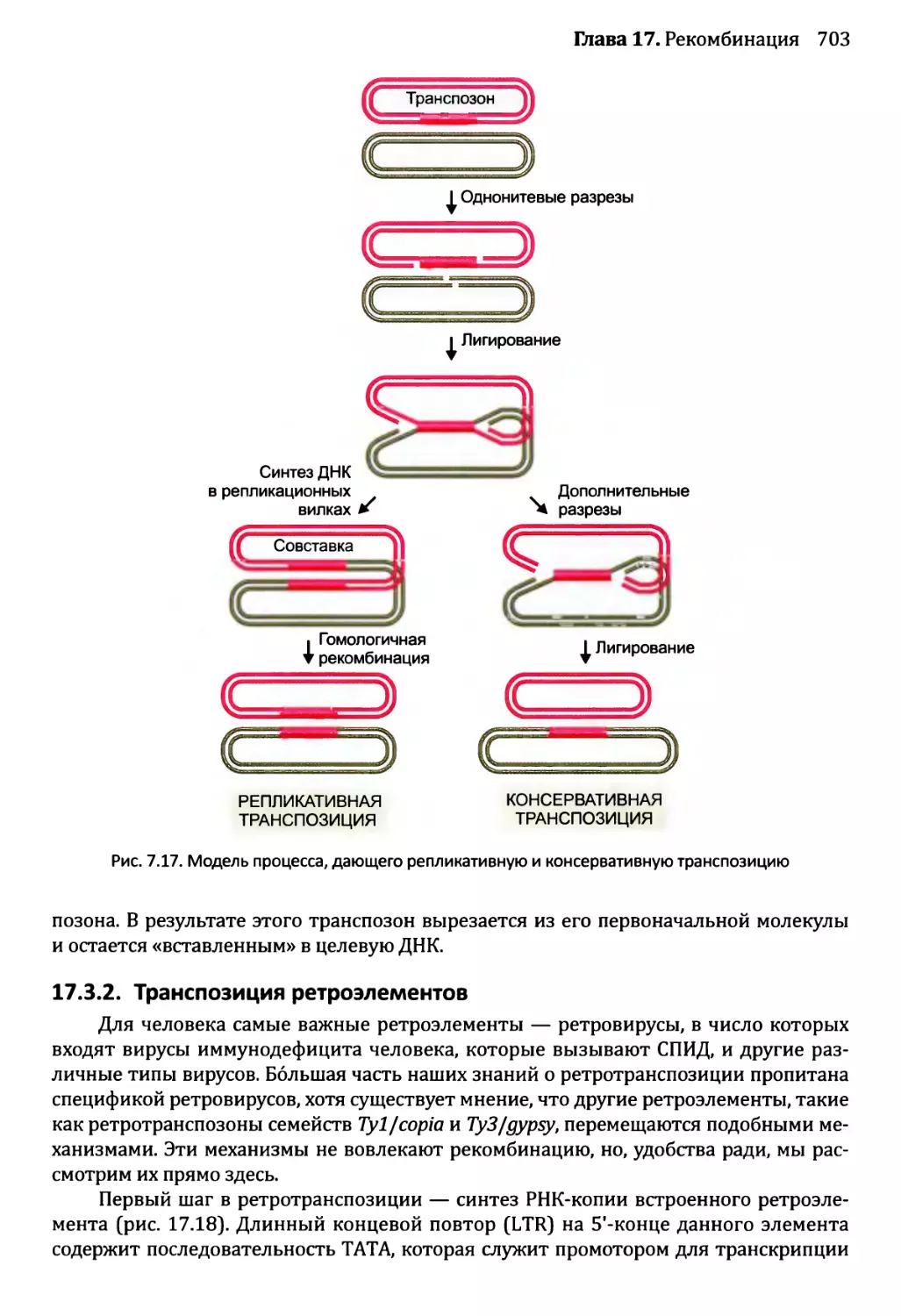
17.3. Транспозиция..............................................701
17.3.1. Репликативная и консервативная транспозиция
ДНК-транспозонов...........................................702
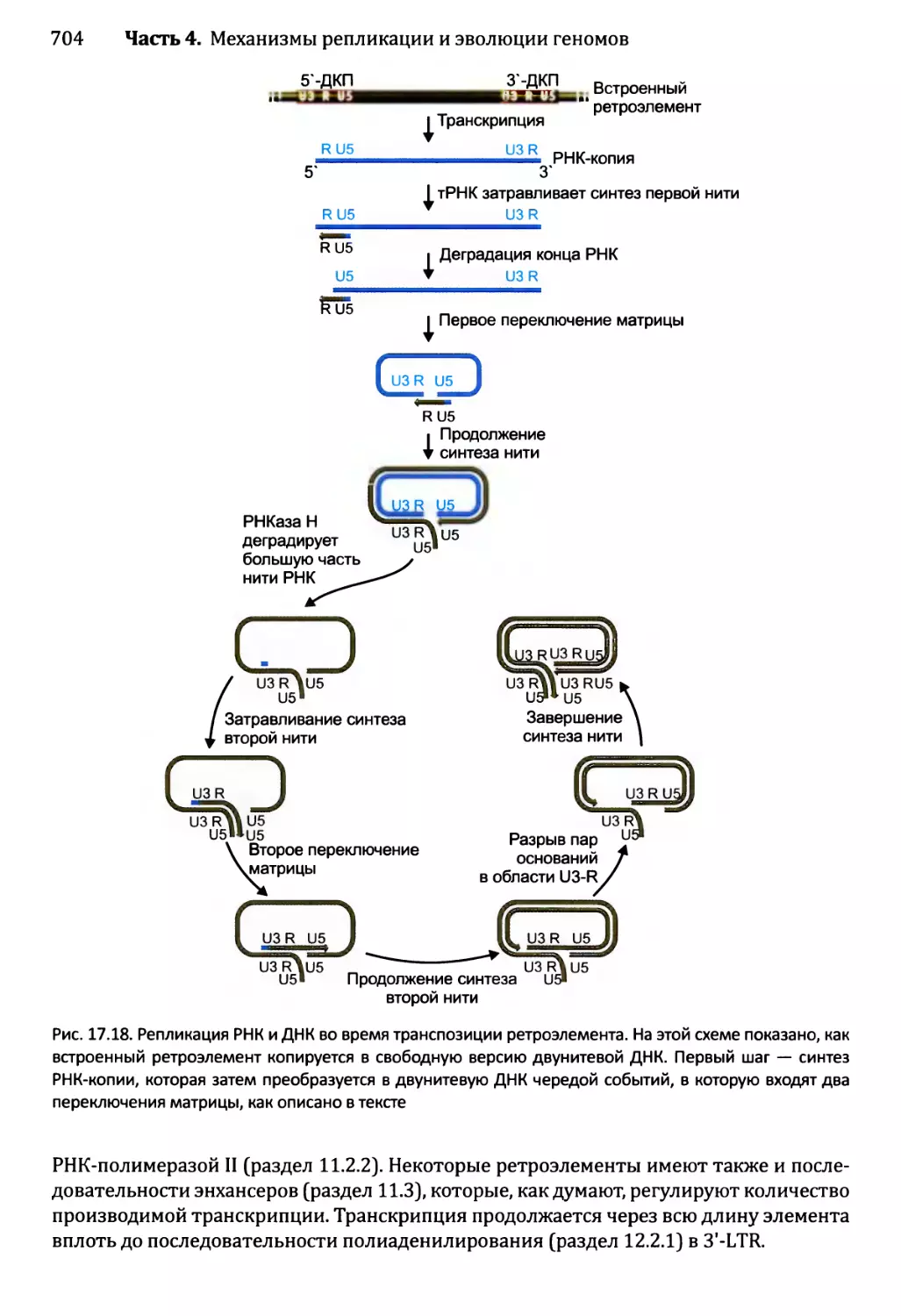
17.3.2. Транспозиция ретроэлементов........................703
17.3.3. Каким образом клетки минимизируют вредное влияние
транспозиции?..............................................706
Глава 18. Механизм эволюции геномов.................................713
18.1. Геномы: первые десять миллиардов лет......................714
18.1.1. Происхождение геномов..............................714
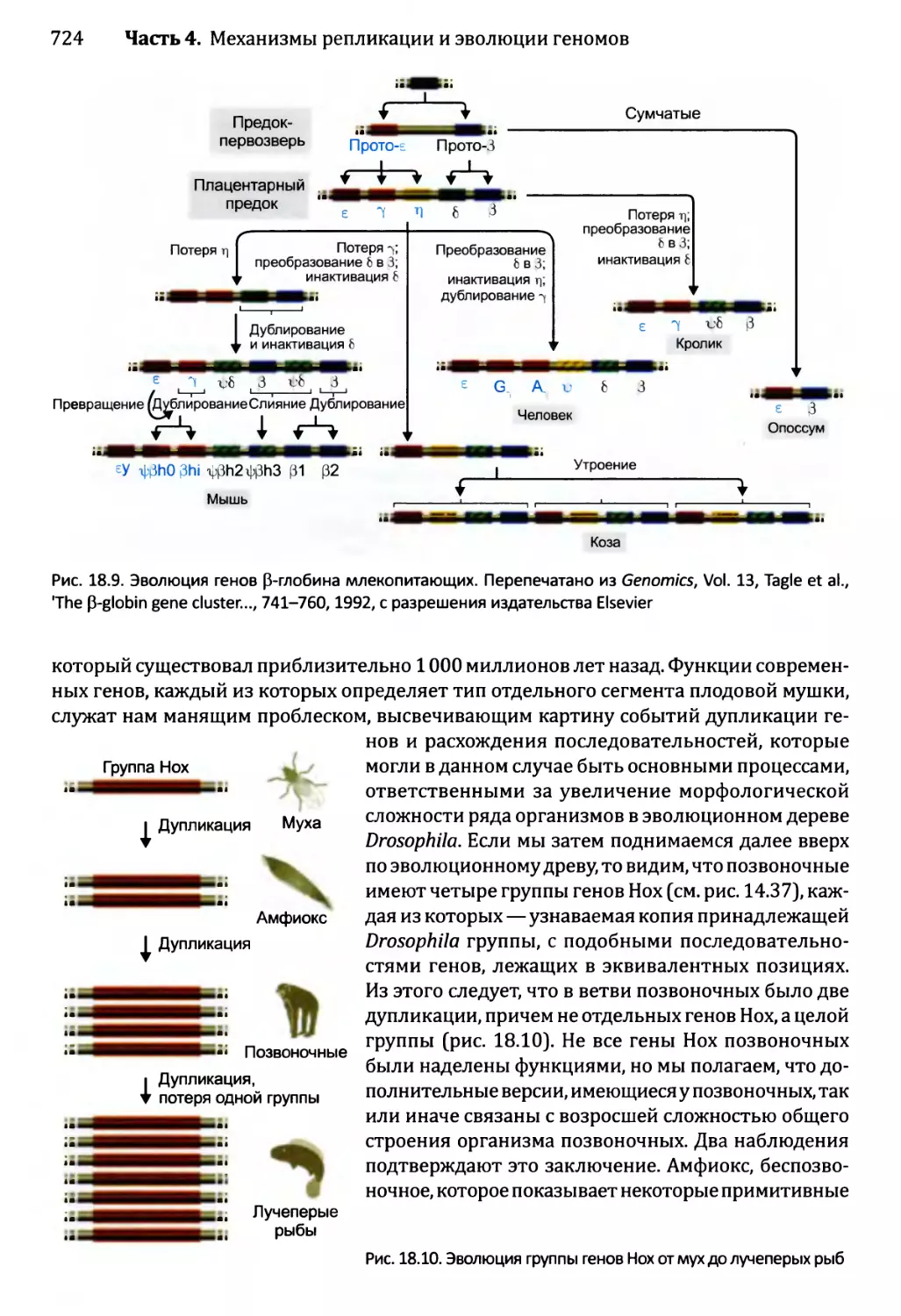
18.2. Приобретение новых генов..................................719
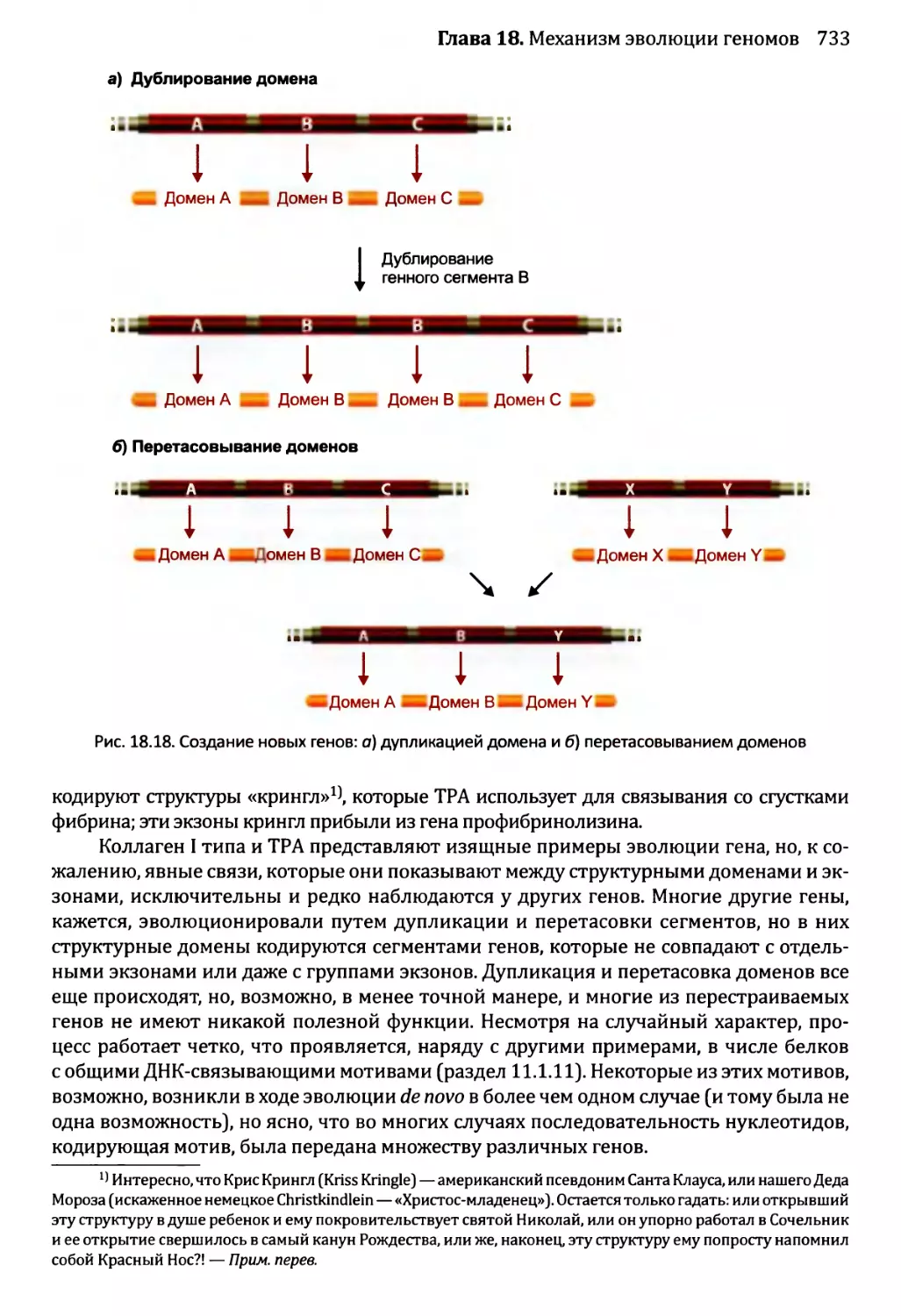
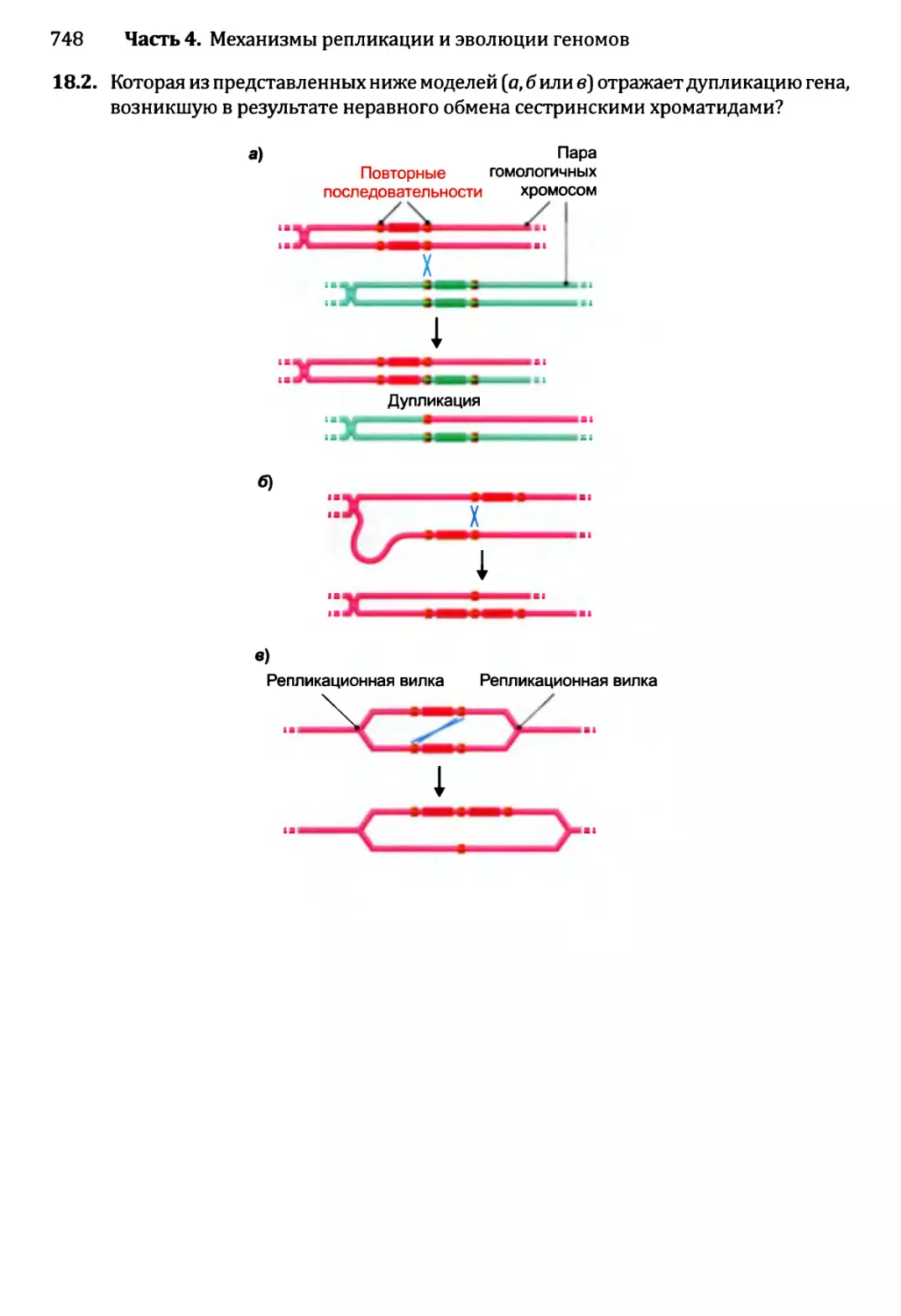
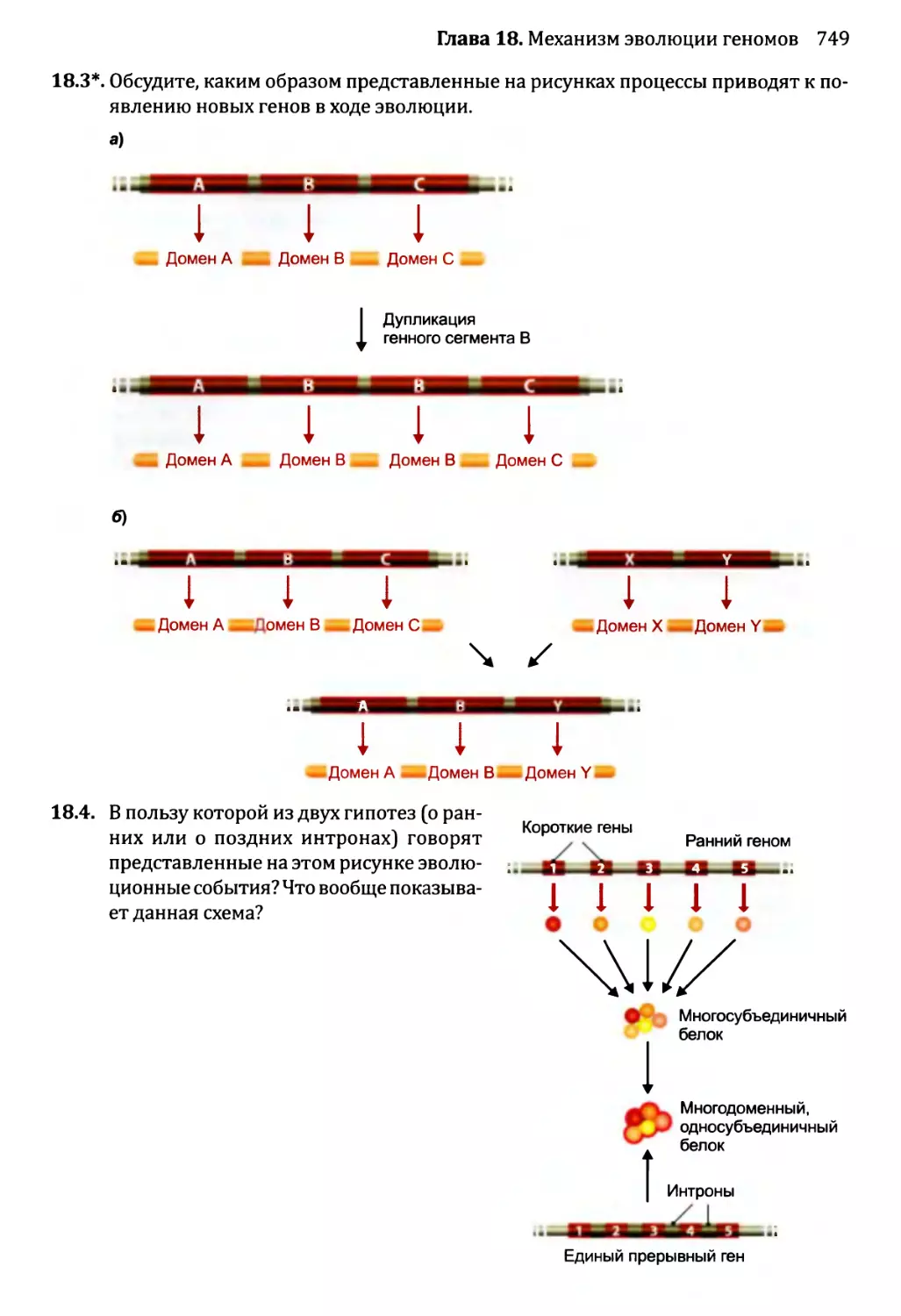
18.2.1. Приобретение новых генов в ходе событий дупликации.721
18.2.2. Приобретение новых генов от других видов...........735
18.3. Некодирующая ДНК и эволюция генома........................736
18.3.1. Мобильные генетические элементы и эволюция генома...737
18.3.2. Происхождение интронов.............................738
18.4. Геном человека: последние пять миллионов лет..............741
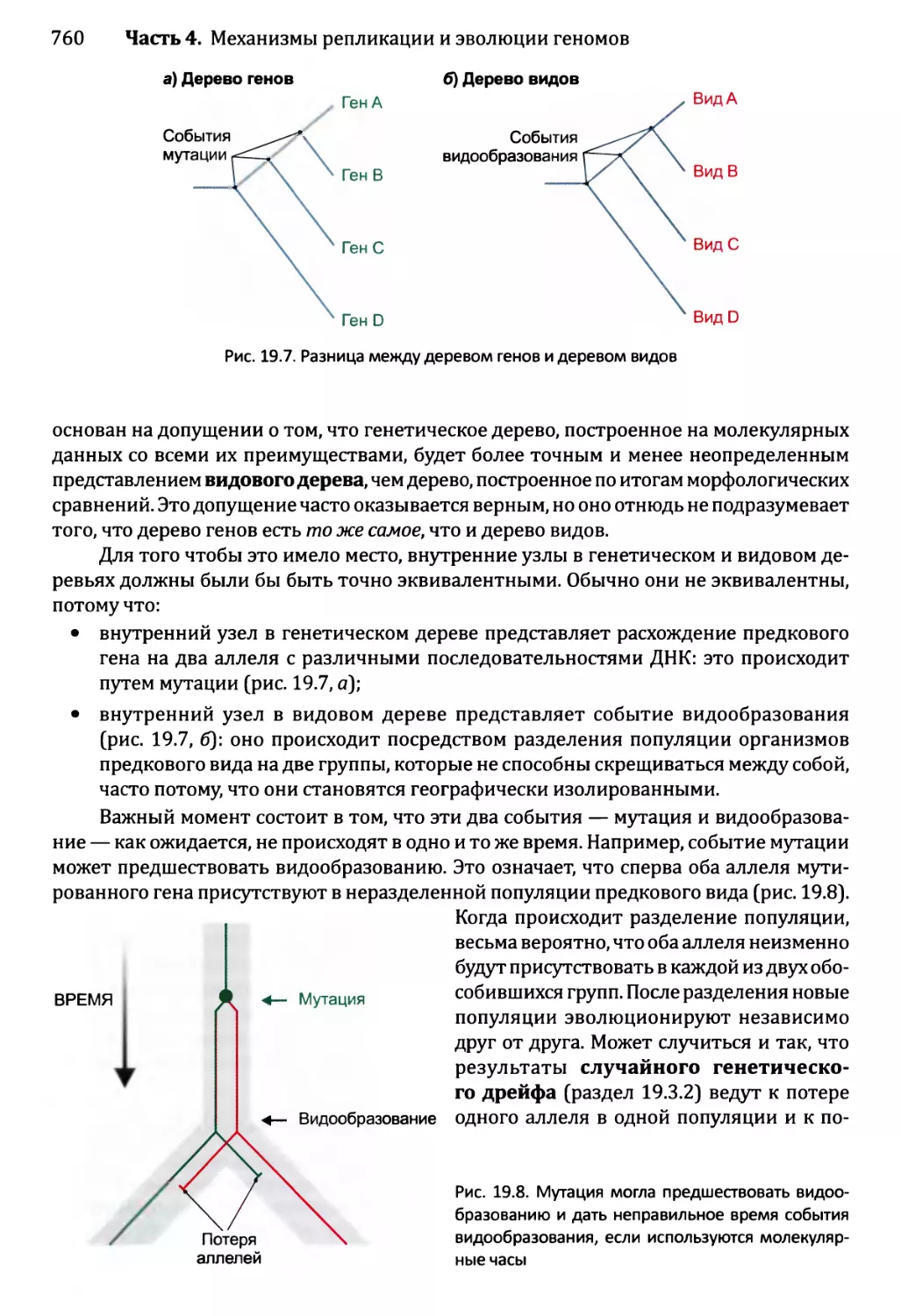
Глава 19. Молекулярная филогенетика.................................752
19.1. От классификации до молекулярной филогенетики.............753
19.1.1. Зарождение молекулярной филогенетики...............753
19.2. Восстановление филогенетических деревьев на основе ДНК....757
19.2.1. Основные характеристики построенных на основе ДНК
филогенетических деревьев..................................757
19.2.2. Восстановление дерева..............................761
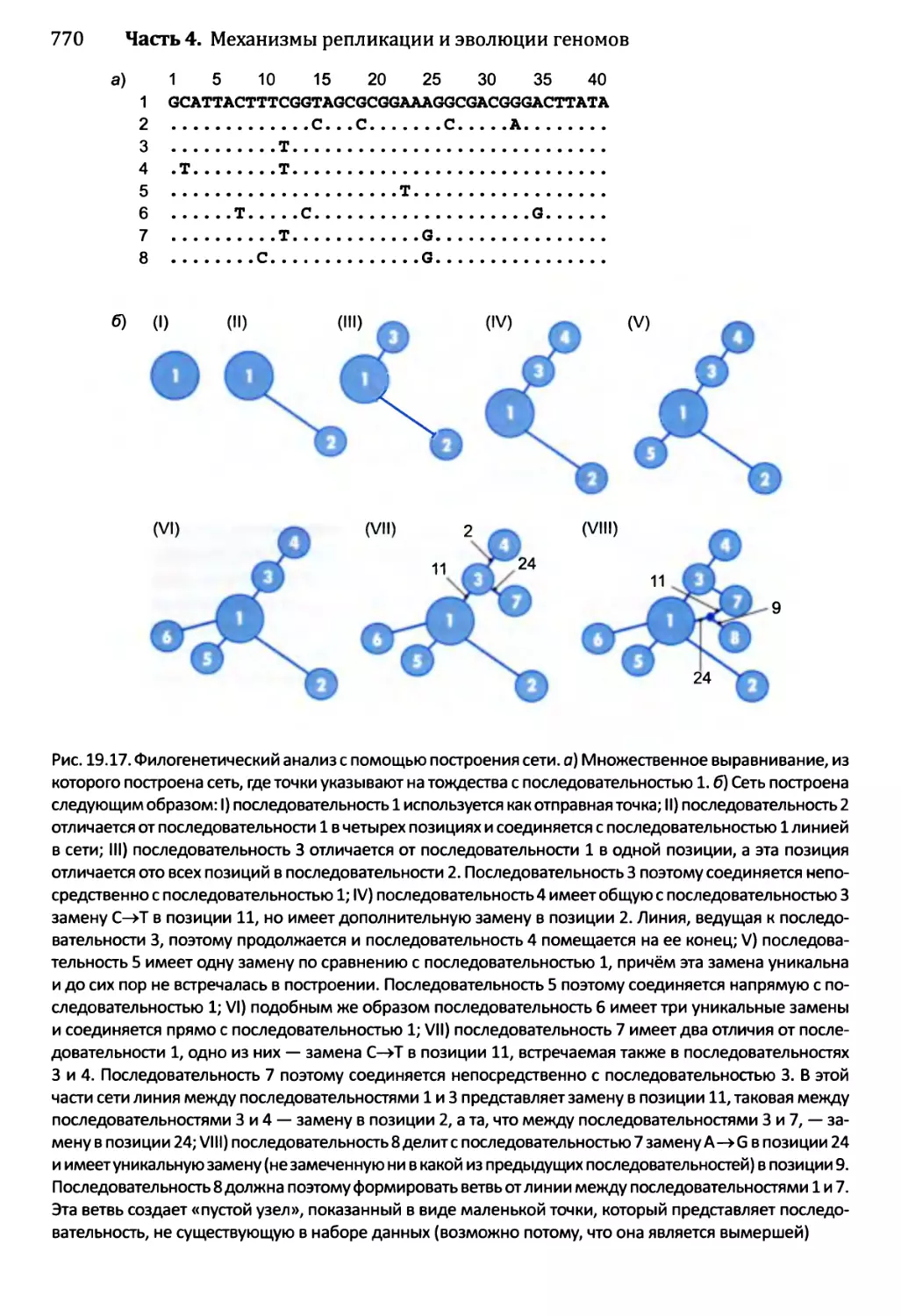
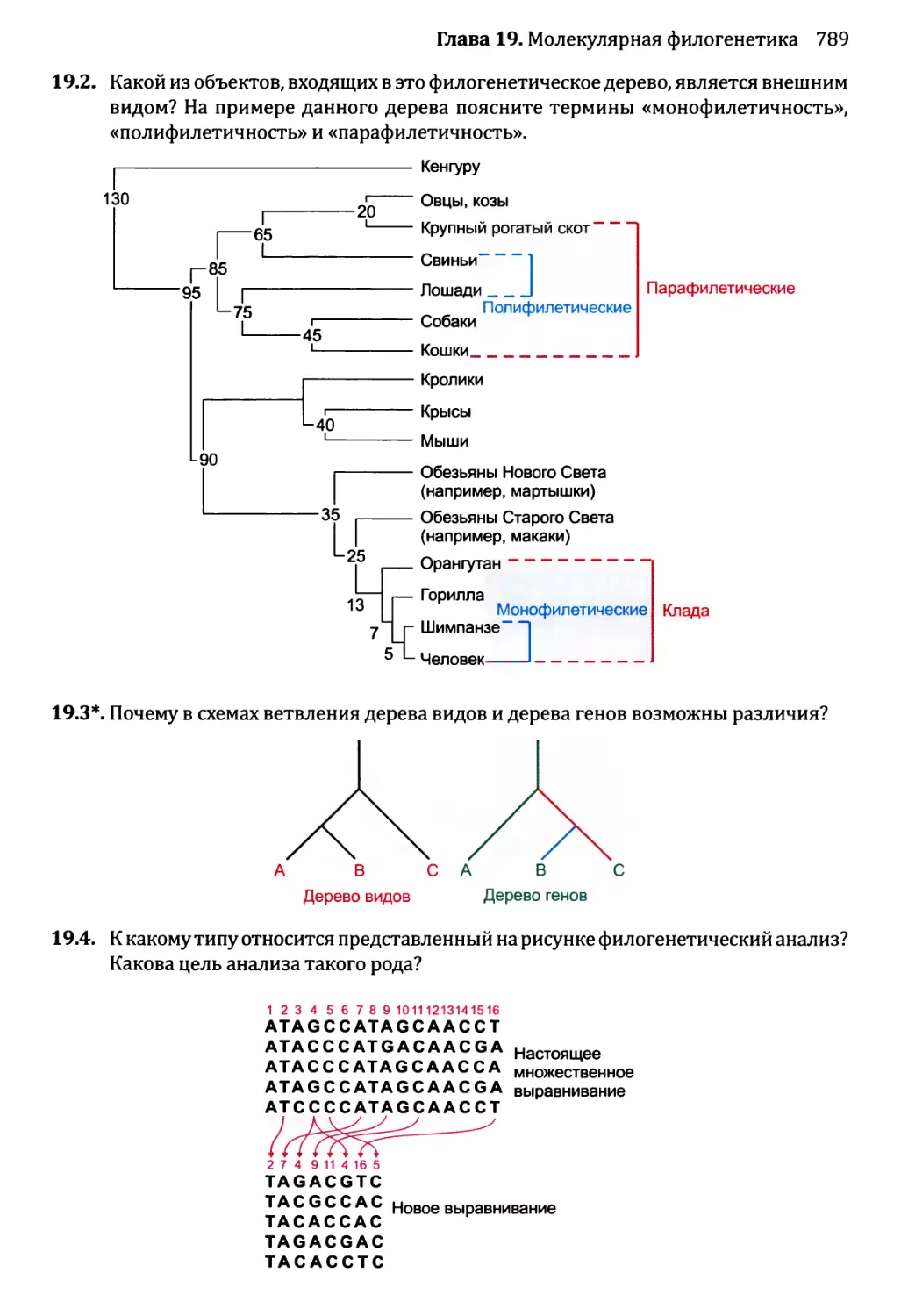
19.3. Возможности применения молекулярной филогенетики..........771
19.3.1. Примеры использования филогенетических деревьев....771
19.3.2. Молекулярная филогенетика как инструмент изучения
предыстории человека.......................................774
Приложение..........................................................792
Словарь терминов....................................................821
лагодарности........................................................871
Предметный указатель................................................874
Предисловие
Со времени выпуска второго издания книги «Геномы» прошло четыре года,
отмеченных удивительными открытиями.
Расшифрованные последовательности хромосом человека появлялись через
равные промежутки времени, и был завершен проект секвенирования генома
шимпанзе. Число эукариотов с частично или полностью установленными после-
довательностями геномов растет впечатляющими темпами, а новые генетические
последовательности прокариотов публикуются в виртуальном пространстве каж-
дую неделю. Экспериментальные методы изучения транскриптомов и протеомов
дают нам ключи к новому пониманию механизма экспрессии генома, а недавно
возникшая дисциплина — системная биология — связывает учение о геномах
с клеточной биохимией. Все эти достижения были включены в третье издание
книги «Геномы». В частности, материал, ранее умещавшийся в самостоятельную
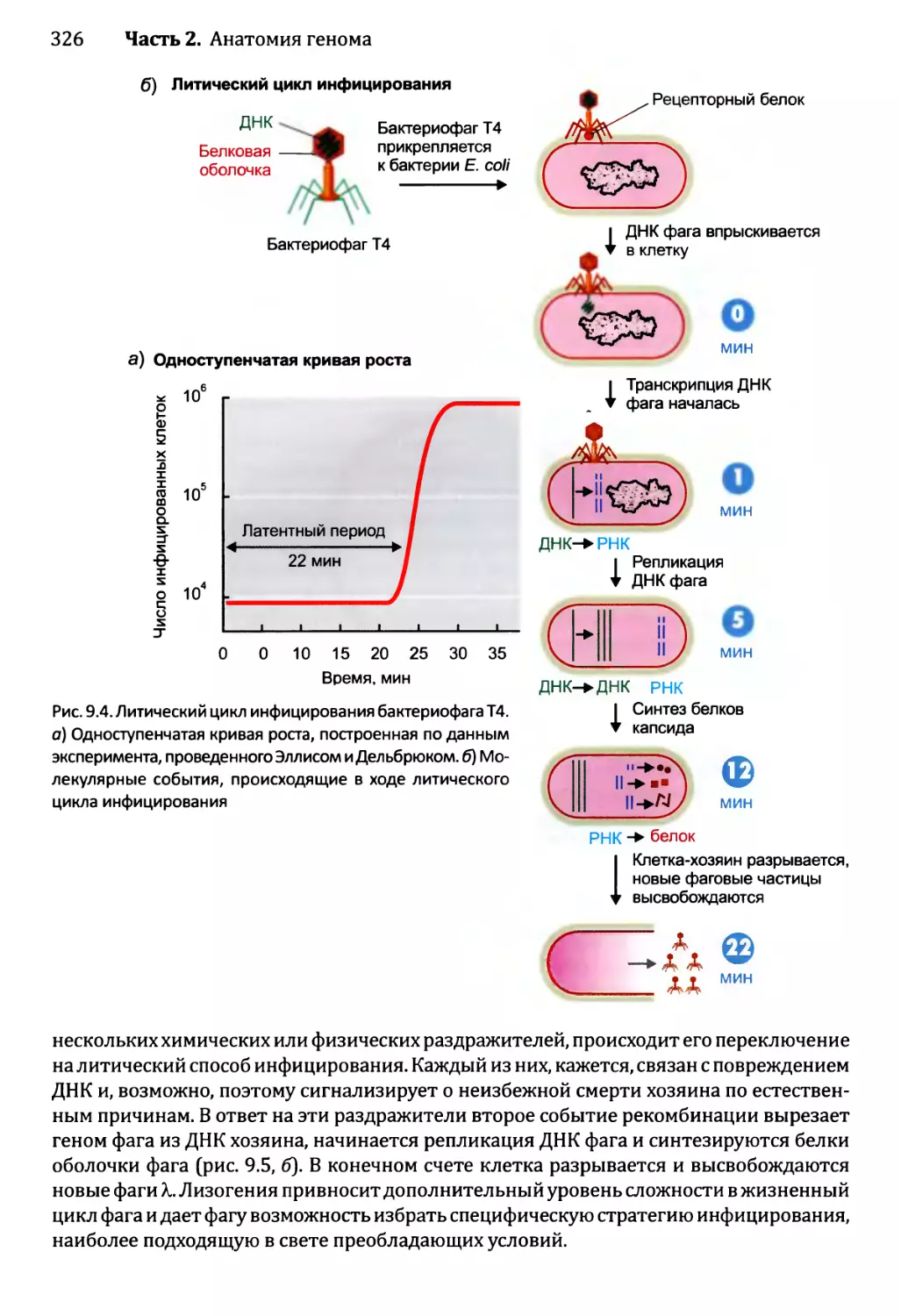
главу по анатомии генома, был расширен до трех глав, и, кроме того, я значительно
увеличил объем сведений о постгеномике, написав отдельные главы по анализу
последовательностей, а также по изучению транскриптомов и протеомов. Наряду
с этим, я воспользовался возможностью дать более глубокое описание процессов
экспрессии, репликации и рекомбинации генома.
Эти изменения привели к увеличению объема «Геномов», и, с тем чтобы
компенсировать этот «недостаток», я попытался сделать книгу более удобной для
читателя. Выделенные цветом поля теперь используются только для описания
технических методов, так что в целом текст книги меньше разбит на разнородные
части, что обусловливает более цельное изложение материала. Иллюстрации
были полностью переработаны с целью привнесения в рисунки большей ясности
и наглядности и придания книге более привлекательного внешнего вида. Списки
рекомендуемой дополнительной литературы и перечни задач в конце каждой
главы подверглись столь же всесторонней переоценке.
В ходе этого пересмотра я учитывал множество отзывов от ряда лекторов
и студентов из разных уголков мира. Этих людей буквально «так много, что их
невозможно упомянуть», так что я хотел бы сказать им общее «спасибо». Один
человек, для которого я приберег индивидуальную благодарность,— Даниела
Делнери из Манчестерского университета, комментарии которой касательно
глав по постгеномике и молекулярной эволюции были столь исчерпывающи-
ми, что я счел ненужным, за малым исключением, самостоятельно проводить
какие-либо изыскания в этих областях. Я чрезвычайно признателен Теду Ли
с биологического факультета Государственного университета Нью-Йорка за то,
что он взял на себя устрашающую (по крайней мере для меня) задачу написания
всеобъемлющих наборов вопросов и задач для каждой главы; эти задания для
закрепления материала значительно повысили качество книги. Помимо этого,
я благодарю Доминика Холдсуорта и Джеки Харбора из «Гарленд сайенс» за огром-
ное содействие в работе над третьим изданием книги «Геномы», а также Мэтью
Макклементса за выполненную им превосходную переработку иллюстративного
материала книги. Наконец, третье издание «Геномов» не появились бы на свет
xii Предисловие
без поддержки моей жены Кери. В разделе «Благодарности» к первому изданию
я написал: «Если вы находите эту книгу полезной, то вы должны благодарить
Кери, не меня, потому что именно она способствовала тому, что настоящая книга
была написана». И мне очень приятно, что один или два человека и в самом деле
откликнулись на мой призыв.
Т. А. Браун,
Манчестер
Примечание читателю
Я постарался сделать третье издание «Геномов» настолько удобным для читате-
ля, насколько это возможно. С этой целью я снабдил его различными методическими
средствами, одни из которых помогают читателю свободно оперировать материалом
книги, другие — делают ее эффективным учебным пособием.
Организация книги
«Геномы» разделены на четыре части:
Часть 1 — «Изучение геномов» — начинается с вводной главы, которая знакомит
читателя с геномами, транскриптомами и протеомами, после чего переходит к методам,
сосредоточенным на клонировании и ПЦР, которые использовались в догеномную эпоху
для исследования отдельных генов (глава 2). Затем описываются более специализи-
рованные методы, применяемые для изучения геномов, в том порядке, в котором они
использовались бы в типичном проекте расшифровки генома: методы построения
генетических и физических карт (глава 3); методология секвенирования ДНК и стра-
тегии, используемые для сборки непрерывной последовательности генома (глава 4);
методы опознавания генов в последовательности генома и определения функций этих
генов в клетке (глава 5) и, наконец, подходы к изучению транскриптомов и протеомов
(глава 6). Проект «Геном Человека» был мною положен в основу композиции 1-й части,
однако данный лейтмотив отнюдь не заглушает все прочие темы, и я постарался в пол-
ной мере охватить стратегии, которые использовались ранее и используются поныне
для изучения геномов других организмов.
Часть 2 — «Анатомия геномов» — рассматривает анатомию геномов различного
типа, встречающихся на нашей планете. Глава 7 освещает ядерные геномы эукариотов
и ставит главный акцент на геноме человека. Глава 8 рассматривает геномы прокариотов
и органелл эукариотов, причем последние включены сюда ввиду их прокариотического
происхождения. Наконец, глава 9 описывает геномы вирусов и мобильные генетические
элементы; они сгруппированы вместе, потому что мобильные элементы некоторых
типов имеют определенное отношение к геномам вирусов.
Часть 3 — «Механизм функционирования геномов» — охватывает материал,
суть которого в прошлом была неадекватно описана как: «ДНК переходит в РНК и, да-
лее, — в белок». Глава 10 обращается к приобретающему все большую значимость во-
просу о том, как структура хроматина влияет на экспрессию генома. Глава 11 описывает
сборку комплексов инициирования транскрипции у прокариотов и эукариотов и вклю-
чает детальное рассмотрение ДНК-связывающих белков, каковые играют центральные
роли на начальных стадиях экспрессии генома. Главы 12 и 13 сообщают подробности
о синтезе транскриптома и протеома, а глава 14 рассматривает регулирование актив-
ности генома. Сохранить объем главы 14 в разумных пределах трудно, поскольку к ре-
гулированию генома имеют отношение очень много различных тем, но я надеюсь, что,
используя конкретные примеры для иллюстрации общих вопросов, сумел достигнуть
удовлетворительного баланса между краткостью и широтой охвата.
xiv Примечание читателю
Часть 4 — «Принципы репликации и эволюции геномов» — связывает ре-
пликацию, мутацию и рекомбинацию ДНК с последовательной эволюцией геномов,
происходящей с течением времени. В главах 15-17 описаны молекулярные процессы,
отвечающие за репликацию, мутацию, репарацию и рекомбинацию ДНК, а в главе 18
рассмотрены пути, по которым эти процессы, как принято считать, сформировали
структуру и генетическое содержание геномов за время эволюции. Наконец, глава 19
посвящена использованию все более и более информативных сведений молекулярной
филогенетики для установления эволюционных отношений между последователь-
ностями ДНК.
Организация глав
Основные результаты
Каждая глава начинается с перечня «основных результатов». Они были сформули-
рованы очень тщательно и представляют собой не просто ряд конспектов фактического
содержания каждой главы, но, напротив, показывают уровень и качество знаний, кото-
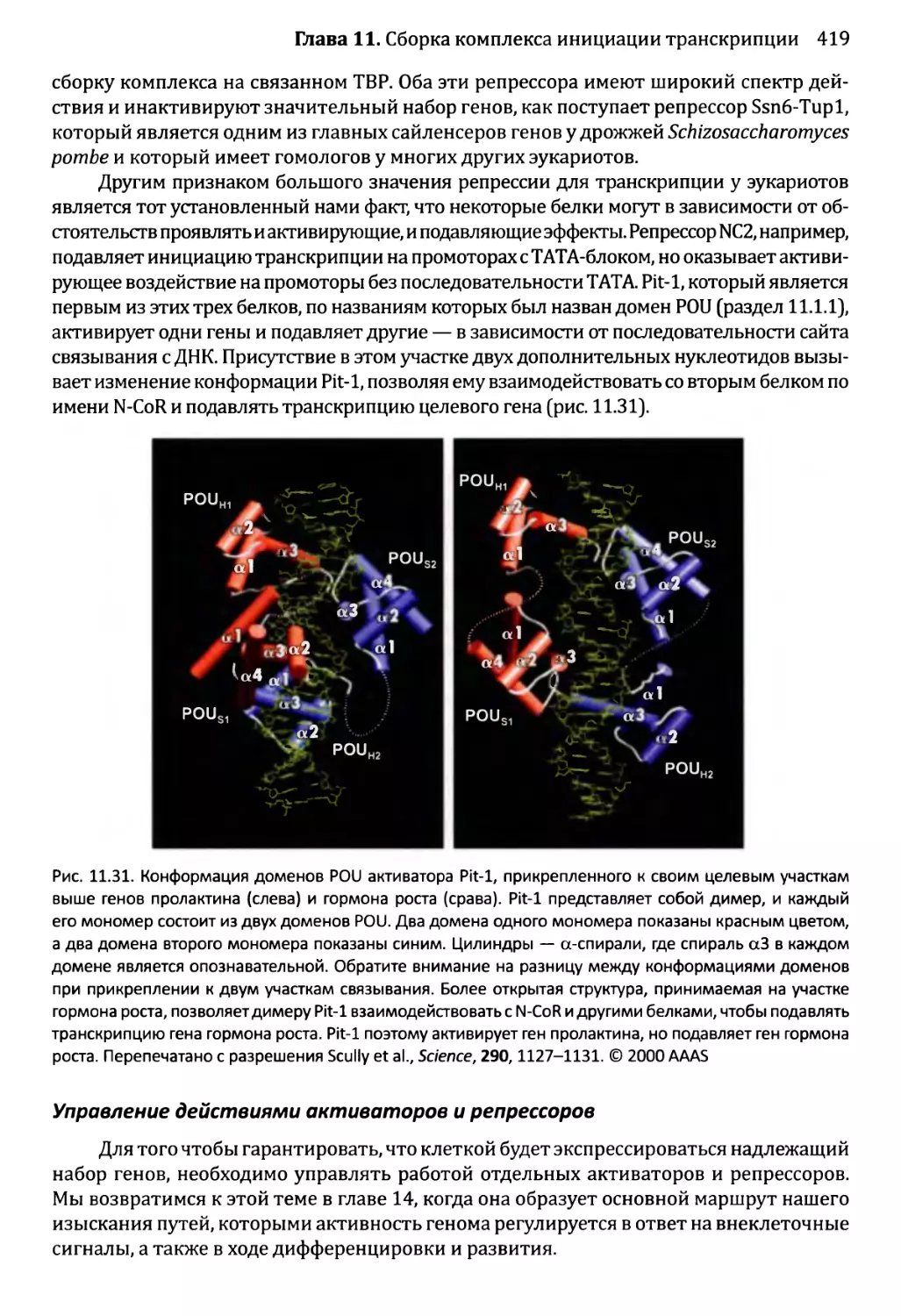
рые студент должен приобрести по прочтении главы. Поэтому «основные результаты»
перечисляют то, что студент должен уметь описать, нарисовать, обсудить, объяснить,
оценить, при этом каждый глагол подобран таким образом, чтобы в точности указать
действие умудренного знакомством с этой главой студента. Цель данного средства со-
стоит в том, чтобы избавить студентов от сомнений по поводу того, что они должны из-
влечь из каждой главы, а следовательно, и беспокойств о том, достаточно ли тщательно
они проработали материал.
Иллюстрации
Хорошая схема несомненно стоит тысячи слов, плохая может запутать читателя,
а излишняя попросту отвлекает. Поэтому я постарался сделать так, чтобы каждая
иллюстрация была необходима и служила своей цели, а не просто разбивала бы текст
и придавала бы книге привлекательный вид. Также я старался сделать рисунки воспро-
изводимыми, потому что, по моему мнению, оное делает их намного более полезными
в качестве учебного пособия для студента. Я никогда не понимал склонности некоторых
авторов превращать иллюстрации учебника в произведения искусства, потому что если
студент не может перерисовать ту или иную диаграмму, тогда это просто иллюстрация,
которая не помогает студенту усвоить информацию, каковую она призвана в себе нести.
Иллюстрации в «Геномах» настолько ясны, просты и незагромождены, насколько это
возможно.
Технические примечания
Основной текст дополнен и расширен рядом технических примечаний, заключен-
ных в отдельные рамки. Каждое техническое примечание представляет собой само-
стоятельное описание какого-либо метода или группы методов, важных в изучении
геномов. Технические примечания предназначены для чтения вкупе с основным текстом,
причем каждое из них расположено в том месте книги, где применение этого метода
упоминается в первый раз.
Примечание читателю xv
Вопросы, задачи и тесты по рисункам
В конце каждой главы приведены упражнения для самостоятельной работы че-
тырех различных типов:
• Вопросы закрытого типа охватывают ключевые пункты главы и проверяют пони-
мание студентом основ материала. Традиционалисты иногда ставят под сомнение
ценность вопросов закрытого типа из-за формальной оценки знаний, но не может
быть никакого сомнения в их ценности как средства повторения пройденного
материала: если студенты могут точно ответить на каждый из таких вопросов, то
они почти наверняка превосходно знают фактическое содержание главы.
• Вопросы открытого типа требуют самостоятельно сформулированного ответа
длиной 50-300 слов или иногда просят нарисовать диаграмму либо таблицу с по-
яснениями. Вопросы в полной мере охватывают содержание соответствующей
главы, сформулированы довольно близко к тексту, и ответы на большую часть из
них могут быть проверены путем простого сопоставления их с соответствующи-
ми частями текста. Студент может пользоваться вопросами открытого типа для
систематической работы над материалом главы или может выбирать отдельные
вопросы, с тем чтобы оценивать свою способность отвечать на вопросы по опреде-
ленным темам. Вопросы открытого типа могут быть использованы также и на
контрольных работах «с закрытой книгой».
• Изыскательские задачи требуют более развернутого ответа. Они изменяются
по характеру и трудности, самая простая из них требует немного больше усилий,
чем обыкновенный литературный обзор, при этом цель этих задач состоит в том,
чтобы студент продвинулся в своих изысканиях на несколько стадий от той точки,
от которой отправляются «Геномы». Иные задачи требуют, чтобы студент оценил
утверждение или гипотезу на основании своего понимания материала книги, по
возможности дополняя его чтением дополнительной литературы по предмету. Есть
надежда, что эти задачи в некоторой степени разовьют мышление и критическое
осмысление пройденного материала. Отдельные задачи довольно трудны, в не-
которых случаях до такой степени, что нет и не может быть однозначного ответа
на поставленный вопрос. Они предназначены для того, чтобы вызвать прения
и обсуждения, которые расширяют круг знаний участвующих в них студентов и по-
нуждают их тщательно обдумывать свои утверждения. За изыскательские задачи
можно приняться студентам, работающим индивидуально, или же, напротив, они
могут служить отправной точкой для обсуждения в группе.
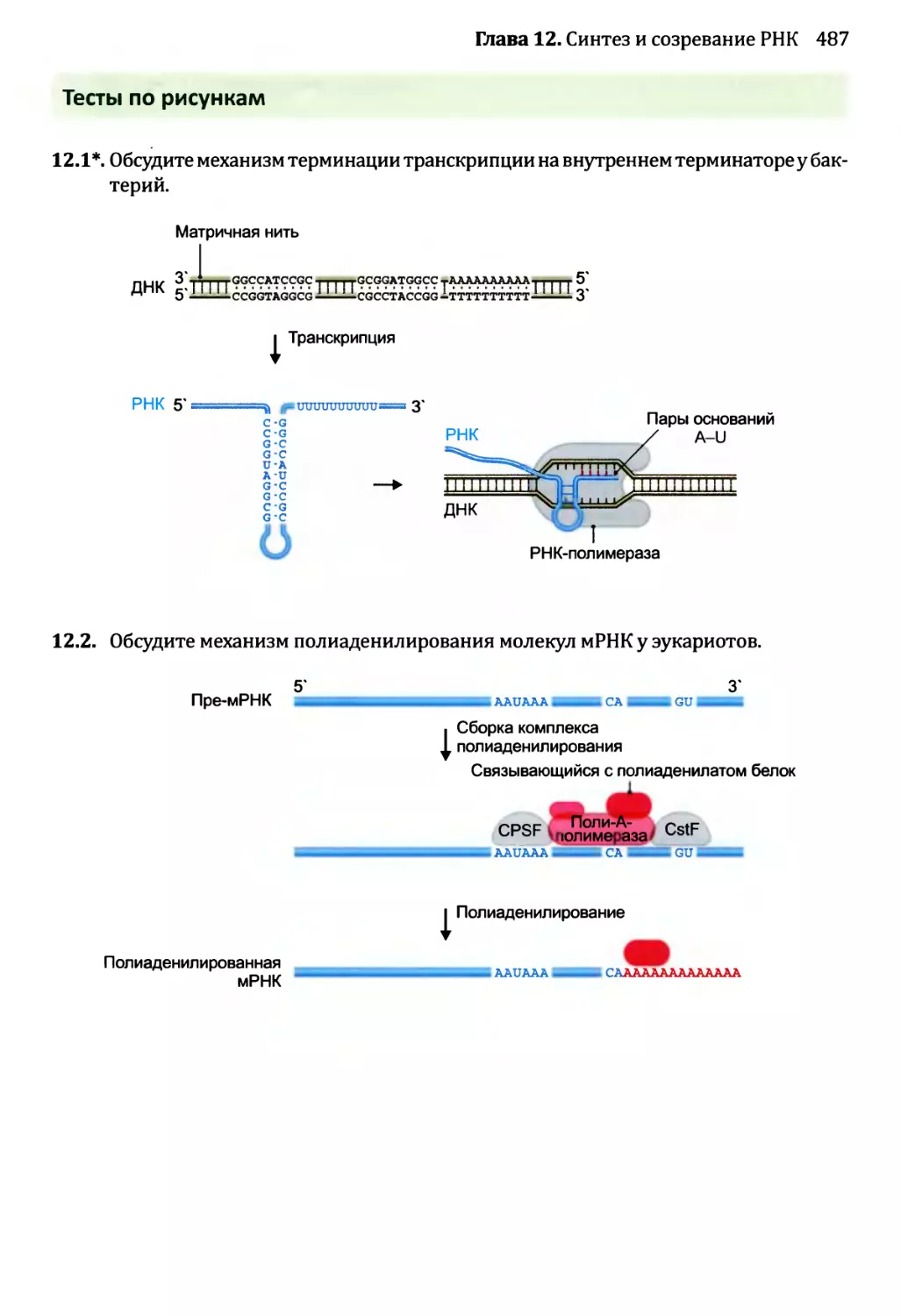
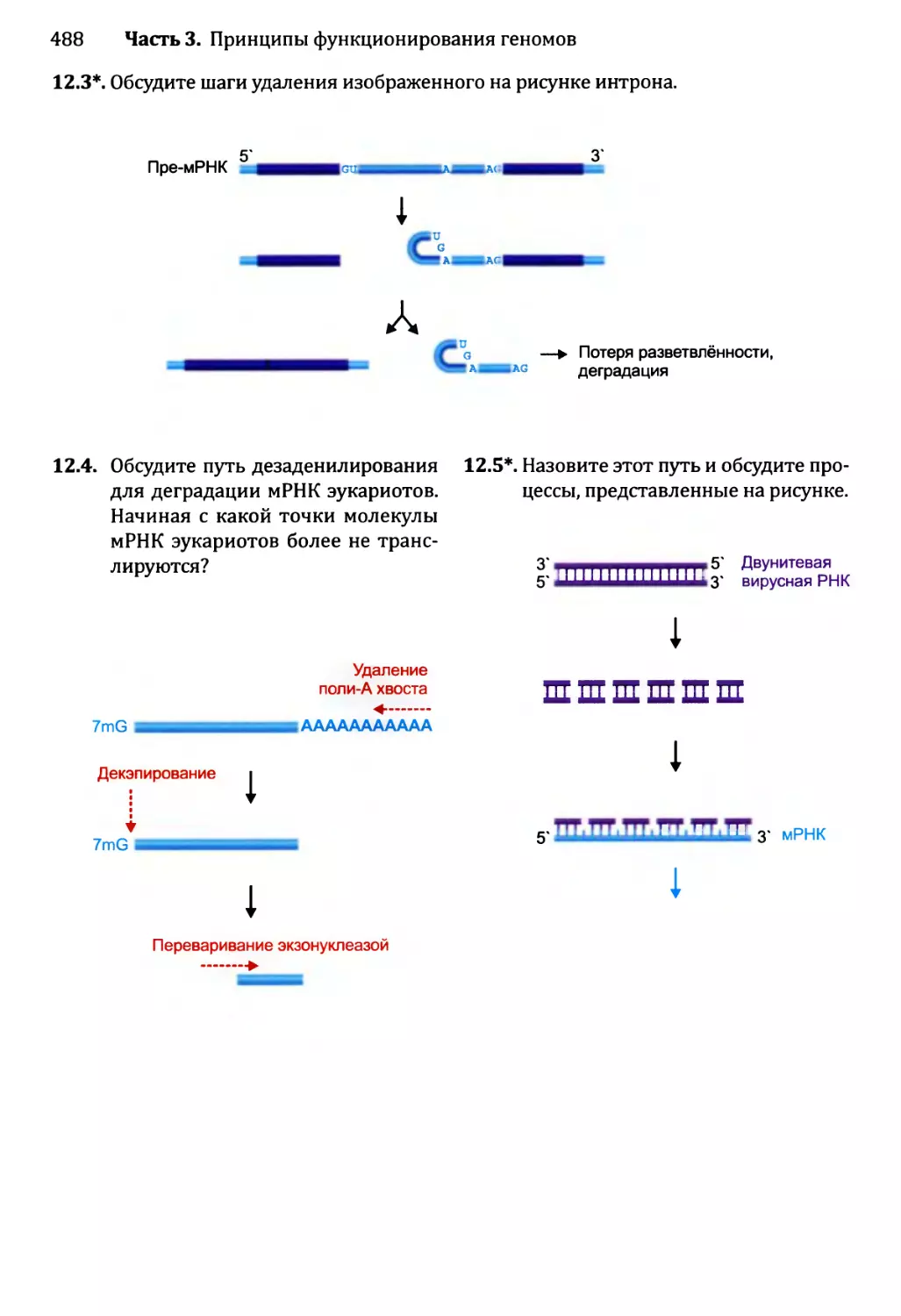
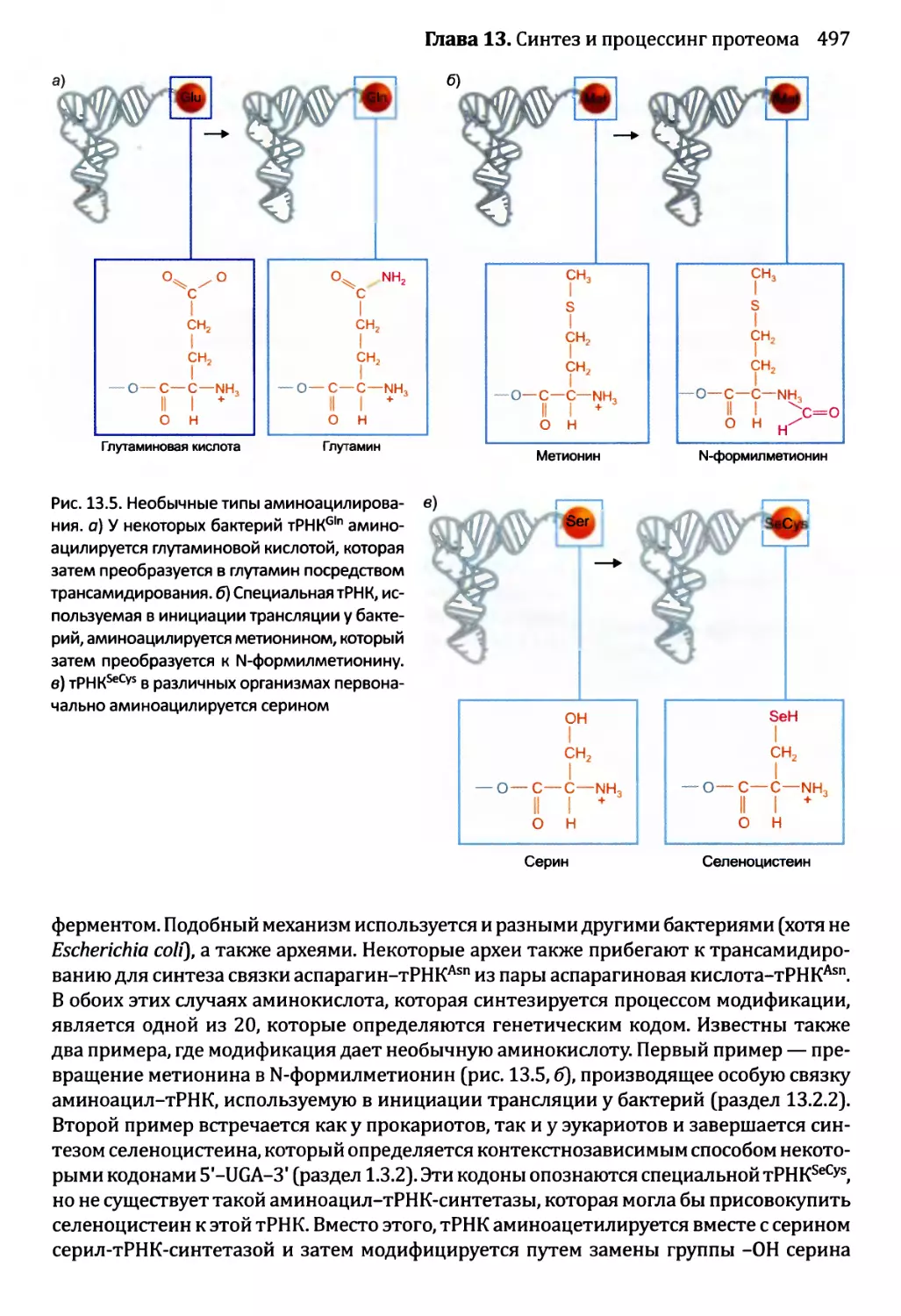
• Тесты по рисункам подобны вопросам открытого типа, но ставят отобранные
иллюстрации предстоящей главы в фокус упражнения. Эти тесты ценны в качестве
средства соединения фактической информации, полученной от чтения текста, со
структурами и процессами, которые иллюстрированы рисунками. Хорошая схема
действительно стоит тысячи слов, но только в том случае, если эта схема изучена
тщательно и полностью понята. Тесты по рисункам как раз помогают добиться
такого понимания.
Ответы на нечетные вопросы закрытого типа, открытого типа и тесты по рисункам
даны в приложении. По запросу, ответы на все вопросы будут высланы преподавателям,
приобретшим книгу, через Garland Science Classwire™. Для изыскательских задач вместо
ответов будет предоставлено руководство к решению.
xvi Примечание читателю
Дополнительная литература
Списки литературных источников в конце каждой главы включают те исследовател ь-
ские статьи, обзоры и книги, которые мне видятся как самые полезные источники дополни-
тельного материала. Мое намерение на всем протяжении «Геномов» состояло в том, чтобы
студенты могли использовать такие списки литературы для получения дополнительной
информации при написании развернутых докладов или диссертаций на определенные темы.
Поэтому я включал в них исследовател ьские статьи, но только в том случае, если был уверен
в том, что их содержание будет понятно большинству читателей книги, а не только особо
одарённым единицам. Акцент поставлен на доступные обзоры типа Перспектив «Science»,
Новостей и Обзоров «Nature» и статей в журналах «Trends»; одно из достоинств таких общих
статей состоит в соответствующем контексте и «каркасе» для той или иной части работы.
Большинство списков литературы разделено на секции, отражающие организацию инфор-
мации в главе, и в некоторых случаях я добавлял в конце сведений об источнике несколько
слов, характеризующих отличительную ценность каждого такого источника, с тем чтобы
помочь читателю принять решение, который из них стоит просмотреть в первую очередь.
Списки литературы для чтения далеко не всеисчерпывающие, и я поощряю читателей про-
вести некоторое время за просмотром полок своих собственных библиотек1) в поиске других
книг и статей. Зачастую бывает, что такой просмотр позволяет обнаружить интересные
вещи, о существовании которых и не подозреваешь!
Словарь терминов
Лично я весьма одобряю использование словарей терминов в качестве методи-
ческих средств и поэтому привел обширный глоссарий в третьем издании «Геномов».
Каждому термину, который выделен полужирным шрифтом в тексте книги, дано опреде-
ление в словаре терминов, наряду со множеством допол нительныхтерминов, с которыми
читатель мог бы встретиться при обращении к книгам или статьям из списков литера-
туры для чтения. Каждый термин из словаря терминов приведен также и в алфавитном
указателе, так что читатель может быстро найти путь к соответствующим страницам,
где интересующий его термин из словаря описан более подробно.
Для преподавателей
Ресурс Garland Science Classwire™, расположенный по адресу http://www.classwire.
com/garlandscience, предлагает учебные ресурсы и инструменты систематизации курса
для читающих его преподавателей. Он содержит изображения из третьего издания
«Геномов» в форматах JPEG и PowerPoint®. Вопросы закрытого типа, вопросы откры-
того типа, изыскательские задачи и тесты по рисункам, для которых в приложении не
дано никаких ответов или руководств, полезны для задания их в качестве домашней
работы и в качестве вопросов к экзамену. Ответы и руководства к этим упражнениям
будут высланы преподавателям по запросу через Classwire™. Преподаватели, кото-
рые приобрели настоящее издание «Геномов», могут дополнительно получить доступ
к ресурсам из других наших учебников. Classwire™ может послужить также гибким
и удобным в работе инструментом систематизации курса, который позволяет препо-
давателям создавать веб-сайты для своих занятий. Он предлагает такие средства, как
составитель программы курса, календарь курса, центр сообщений, планировщик курса,
виртуальный рабочий день и менеджер ресурсов. Для всего этого не требуется знания
программирования и владения тонкими техническими навыками. *
и Интернета. — Прим. ред.
Примечание читателю xvii
Перечень рецензентов
Автор и издатель выражают глубочайшую признательность следующим рецензен-
там за их кропотливый труд над совершенствованием этого издания.
Дин Деннер, Медицинская школа Университета Эмори
Даниела Делнери, Манчестерский университет
Юрий Дуброва, Лестерский университет
Барт Эгген, Гронингенский университет
Роберт Фоулер, Государственный университет Сан-Хосе
Адриан Хол, Университет Шеффилда Холама
Глин Дженкинс, Аберистуитский университет
Торстен Кристенсен, Орхусский университет
Майк Макперсон, Университет Лидса
Эндрю Рид, Манчестерский университет
Дарси Расселл, Бейкерский колледж
Амал Шервингтон, Университет Центрального Ланкашира
Роберт Слейтер, Хартфордширский университет
Клаас Сварт, Вагенингенский университет
Джон Тейлор, Ньюкаслский университет
Гёйдо ван ден Акервекен, Утрехтский университет
Васси Варе, Лехигский университет
Мэтью Аптон, Манчестерский университет
Сокращения
5-bU 5-бромоурацил CRM машина перестройки хромати-
А аденин; аланин на
ABF фактор связывания ARS CstF фактор стимуляции расщепле-
Ac/Ds мобильные элементы расте- ния
ний — активатор/диссоциа- СТАВ цетил триметил аммонийбромид
тор С-КД С-концевой домен
ala аланин cys цистеин
ANT-C комплекс Antennapedia D аспарагиновая кислота
arg аргинин DAG 1,2-диацилглицерин
ARMS амплификация рефракторной Dam ДНК-аденинметилаза
мутационной системы DAPI 4,6-диамино-2-фенилиндолди-
ARS автономно реплицирующаяся гидрохлорид
последовательность DASH динамическая аллелеспецифи-
asn аспарагин ческая гибридизация
А-сайт акцепторный сайт рибосомы DBS участок связывания двуните-
ASO аллелеспецифический олигону- войДНК
клеотид Dem ДНК-цитозинметилаза
asp аспарагиновая кислота Dfd Deformed
ВАС бактериальная искусственная DMSO диметилсульфоксид
хромосома Dnmt ДНК-метилтрансфераза
bis 1Ч,1Ч’-метиленбисакриламид DPE нижерасположенный промотор-
BLAST основное программное средство ный элемент
поиска локальных выравнива- DSB двунитевой разрыв
ний DSP1 белок 1 системы развития
Bx-C комплекс Bithorax спина-брюхо
C цистеин; цитозин dsRAD РНК-зависимая дезаминаза
CAP белок-катаболитный регулятор dsRBD домен связывания двунитевой
CASP CTD-связанный SR-подобный РНК
белок E глутаминовая кислота
СЕРН Центр изучения полиморфиз- Е-сайт сайт выхода из рибосомы
мов человека EDTA этилендиаминтетраацетат
CHEF электрофорез в геле с ограни- ченными контуром однородны- ми электрическими полями eEF EEO фактор элонгации у эукарио- тов величина электроэндосмоса
CJD болезнь Крейцфельда-Якоба EF фактор элонгации
Col колицин elF фактор инициации у эукарио-
CPSF фактор расщепления и специ- тов
фичности полиаденилирова- ния EMS этилметансульфонат
Сокращения xix
eRF фактор высвобождения (релиз- фактор) у эукариот ICAT закодированный изотопом яр- лык по сродству
ES эмбриональный ствол ICF иммунная недостаточность,
ESE энхансер сплайсинга неустойчивость центромеры
ESS сайленсер сплайсинга и лицевые аномалии
EST ярлык экспрессируемой после- IF фактор инициации
довательности 1g иммуноглобулин
F фертильность; фенилаланин IHF клеточный фактор встраивания
FEN эндонуклеаза откидной створки ile изолейцин
FIGE гель-электрофорез в обращае- Inr инициатор
мом поле Ins(l,4,5)P: 3 инозитол-1,4,5-трифосфат
FISH флюоресцентная гибридизация IPTG изопропилтиогалактозид
in situ IRES внутренний участок входа ри-
FRAP восстановление флюоресценции босомы
после фотообесцвечивания IS инсерционная последователь-
G глицин; гуанин ность
G1 фаза G1 митоза ITF клеточный фактор встраивания
G2 фаза G2 митоза ITR концевой инвертированный по-
GABA у-аминомасляная кислота втор
GAP белок, активирующий ГТФазу JAK киназа Януса
GFP зеленый флюоресцентный белок К лизин
gin глутамин L лейцин
glu глутаминовая кислота LCR управляющая локусом область
giy глицин leu лейцин
GNRP белок, высвобождающий ну- клеотид гуанин LINE длинный рассеянный ядерный элемент
GTF общий фактор транскрипции LTR длинный концевой повтор
H гистидин lys лизин
ГАТ гипоксантин + аминоптерин + M метионин; фаза митоза
тимидин MALDI-TOF времяпролетная масс-спектро-
HBS гетеродуплексный участок свя- зывания метрия с лазерной десорбцион- ной ионизацией матрицы
his гистидин MAP белки, ассоциированные с ми-
HLA лейкоцитарный антиген чело- века кротрубочками; активируемый митогеном белок
HMG группа ядерных негистоновых белков с высокой электрофоре- MAR область прикрепления к ма- триксу
тической подвижностью MeCP метил-СрС-связывающий бе-
HOM-C комплекс гомеотического гена лок
HPRT гипоксантинфосфорибозил- met метионин
трансфераза MGMT О6-метилгуанин-ДНК-метил-
HTH спираль-поворот-спираль трансфераза
I изолейцин mol моль
хх Сокращения
MudPIT метод многомерной идентифи-
кации белков
MULE Mutator-подобный транспони-
руемый элемент
N 2'-дезоксинуклеозид-5'-трифос-
фат; аспарагин
NHEJ негомологическое соединение
концов
NJ объединение соседей
NMD опосредствованный бессмыс-
ленным кодоном распад РНК
OFAGE электрофорез в геле с ортого-
нально чередующимся полем
OLA анализ методом лигирования
олигонуклеотидов
Отр белок внешней мембраны
ORC комплекс узнавания области
инициации
OTU оперативная таксономическая
единица
Р пролин
P-сайт пептидильный сайт рибосомы
РАС искусственная хромосома на
основе бактериофага Р1
PADP полиаденилатсвязывающий
белок
PAUP филогенетический анализ по
критерию бережливости
PCNA ядерный антиген пролифери-
рующих клеток
phe фенилаланин
PHYLIP программный пакет для фило-
генетического анализа
PIC предынициаторный комплекс
PNA (ПНК) пептидная нуклеиновая кисло-
та
pro пролин
PSE проксимальный элемент после-
довательности
PSI-BLAST позиционное итеративное основ-
ное программное средство поис-
ка локальных выравниваний
PtdIns(4,5)P2 фосфатидилинозитол-4,5-
бисфосфат
PTRF фактор высвобождения поли-
меразы I и транскрипта
Ри пурин
Ру пиримидин
Q глутамин
R аргинин; пурин
RACE быстрое размножение концов
кДНК
RBS участок связывания рибосомы
RC комплекс репликации
RF фактор высвобождения
RFC фактор репликации С
RFLP полиморфизм длины рестрик-
та
RHB домен гомологии Rel
RISC РНК-индуцируемый подавляю-
щий комплекс
RLF фактор лицензирования репли-
кации
RMP белок-посредник репликации
RPA белок репликации А
RRF фактор повторного использова-
ния рибосом
RTVL ретровирусоподобный эле-
мент
S серин; стадия синтеза
SAGE последовательный анализ экс-
прессии генов
SAP активируемый стрессом белок
SAR область прикрепления к карка-
су
SCAF SR-подобный CTD-связанный
фактор
SCS специализированная структура
хроматина
Se-cis селеноцистеин
ser серин
siPHK короткая интерферирующая
РНК
SINE короткий рассеянный ядерный
элемент
SMAD семейство, связанное с
SMA/MAD
Сокращения xxi
SNP полиморфизм отдельного ну- val
клеотида VNTR
SRF фактор сывороточной реак- ции W
SSB белок однонитевого связыва- X-gal
ния
SSLP полиморфизм длины последо- Y
вательности YAC
STAT передатчик сигнала и актива- тор транскрипции Yip
STR простой тандемный повтор
STS меченый участок последова- АДФ
тельности АМФ
Т треонин; тимин АП
TAF TBP-связанный фактор
ТАР очистка по тандемному срод- АТФ
ству АТФаза
TBP связывающийся с блоком ТАТА белок; связывающийся с тело- мерой белок БГЭ ВИО
ТЕМЭД Н,1Ч,1Ч',1Ч'-тетраметилэтиленди- ВИЧ
амин
TF фактор транскрипции ГДФ ГМФ
TGF трансформирующий фактор ро-
ста
thr Ti треонин опухолеродный ГТФ HDAC гяРНК
TIC TAF- и инициаторзависимый
кофактор ДНК
Tm температура плавления ДНКаза
Tn TOL транспозон толуол дНТФ
TPA активатор профибринолизина дсн дАТФ
ткани
TRAP trp РНК-связывающий белок аттенюации
ДЦТФ
тРНК транспортная РНК
trp триптофан ДДАТФ
tyr тирозин
U урацил ддЦТФ
UCE вышележащий элемент управ-
ления ддГТФ
UTR нетранслируемая область
валин
переменное число тандемных
повторений
аденин или тимин; триптофан
5-бромо-4-хлоро-3-индолил-
P-D-галактопиранозид
пиримидин; тирозин
дрожжевая искусственная хро-
мосома
дрожжевая интегрируемая
плазмида
аденозин-5'-дифосфат
аденозин-5'-монофосфат
апуриновый/апиримидино-
вый
аденозин-5'-трифосфат
аденозин-5’-трифосфатаза
бычья губчатая энцефалопатия
вирус иммунной недостаточно-
сти обезьян
вирус иммунной недостаточно-
сти человека
гуанозин-5'-дифосфат
гуанозин-5'-монофосфат
гуанозин-5'-трифосфат
гистондезацетилаза
гетерогенная ядерная РНК
дезоксирибонуклеиновая кис-
лота
дезоксирибонуклеаза
2'-дезоксинуклеозид-5'-трифос-
фат
додецил сульфат натрия
2'-дезоксиаденозин-5'-трифос-
фат
2’-дезоксицитидин-5'-трифос-
фат
2',3'-дидезоксиаденозин-5'-три-
фосфат
2’,3'-дидезоксицитидин-5'-три-
фосфат
2',3’-дидезоксигуанозин-5'-три-
фосфат
xxii
Сокращения
ДДНТФ 2',3'-дидезоксинуклеозид-5'- трифосфат
ддТТФ 2',3’-дидезокситимидин-5'-три- фосфат
дГТФ 2’-дезоксигуанозин-5'-трифос- фат
ЖХВР жидкостная хроматография вы- сокого разрешения
ккал килокалория
кДНК комплементарная ДНК
кДа килодальтон
ЛОШ логарифм отношения шансов
мкм микрометр
млрд п. н. миллиард пар нуклеотидов
млн п. н. миллионов пар нуклеотидов
миРНК микроРНК
мРНК матричная РНК
мцРНК малая цитоплазматическая РНК
млн л. миллионов лет
мякРНК малая ядрышковая РНК
мяРНК малая ядерная РНК
мяРНП малый ядерный рибонуклео- протеид
нг нанограмм
нм нанометр
НАД никотинамидадениндинуклео- тид
НАД-Н никотинамидадениндинуклео- тид восстановленный
ННРПОК наследственный неполипозный рак прямой и ободочной кишки
НТФ нуклеозид-5'-трифосфат
ОЗУ память с произвольной выбор-
кой
ОРС открытая рамка считывания
пг пикограмм
п. н. пара нуклеотидов
ПЦР полимеразная цепная реакция
ПНФаза полинуклеотидфосфорилаза
РНК рибонуклеиновая кислота
РНКи РНК-интерференция
РНКаза рибонуклеаза
РНП рибонуклеопротеид
рРНК рибосомная РНК
РТ-ПЦР ревертазная ПЦР
РПЭ-ПЦР ПЦР с рассеянными повторны-
ми элементами
слРНК сплайсированная лидерная РНК
СПИД синдром приобретенного имму-
нодефицита
ТК тимидинкиназа
тмРНК транспортно-матричная РНК
тыс. п. н. тысяч пар нуклеотидов
УФ ультрафиолетовый
УТФ уридин-5’-трифосфат
ЦТФ цитидин-5'-трифосфат
цАМФ циклический АМФ
цГМФ циклический ГМФ
ЭРВ эндогенный ретровирус
ЯМР ядерный магнитный резонанс
Часть 1
Изучение геномов
Часть 1 — «Изучение геномов» — описывает
методы и научные подходы, которые лежат
в основе наших знаний о геномах. Мы начинаем
со вводной главы, которая знакомит читателя
с геномами, транскриптомами и протеомами,
и затем, в главе 2, переходим к методам, ко-
торые сосредоточены на клонировании ДНК
и полимеразной цепной реакции и использу-
ются для изучения коротких фрагментов ДНК,
таких как отдельные гены. Глава 3 открывает
наше изложение геномики, описывая прин-
ципы построения генетических и физических
карт, а глава 4 показывает связь между карти-
рованием и секвенированием. По мере чтения
главы 4 читатель убедится в том, что хотя кар-
та может быть ценным помощником в сборке
длинной последовательности ДНК, картиро-
вание не всегда является необходимой пред-
посылкой к секвенированию генома. В главе
5 мы рассматриваем различные подходы, при-
меняемые для истолкования последователь-
ности генома, а в главе 6 мы изучаем методы,
употребляемые для исследования механизмов
функционирования генома путем управления
синтезом транскриптома и протеома и — по-
средством этого — определения биохимиче-
ских возможностей клетки.
Глава 1
Геномы, транскриптомы
и протеомы
Глава 2
Изучение ДНК
Глава 3
Картирование
геномов
Глава 4
Секвенирование
геномов
Глава 5
Определение структуры
последовательности генома
и функций её составляющих
Глава 6
Понимание механизма
функционирования генома
Глава 1
Геномы, транскриптомы
и протеомы
По прочтении главы 1 вы сможете:
• Давать определения терминам «геном», «транскриптом» и «протеом», а также
отвечать на вопрос о том, каким образом они взаимодействуют в процессе экс-
прессии генома.
• Описывать два эксперимента, по результатам которых молекулярные биологи
пришли к заключению о том, что гены находятся в ДНК, а также разъяснять
ограничения этих экспериментов.
• Давать подробное описание структуры полинуклеотидов и указывать харак-
терные химические различия между ДНК и РНК.
• Обсуждать данные, на основании которых Уотсон и Крик построили модель
структуры ДНК в виде двойной спирали, и описывать основные характеристики
этой структуры.
• Проводить различия между кодирующей и функциональной РНК и приводить
примеры РНК обоих типов.
• Вкратце описывать процессы синтеза и созревания РНК в клетке.
• Давать подробное описание различных уровней структуры белка и объяснять,
почему в основе разнообразия белков лежит разнообразие аминокислот.
• Описывать основные характеристики генетического кода.
• Объяснять, по какой причине функция белка зависит от последовательности
аминокислот.
• Перечислять главные роли белков в живых организмах и связывать разнообразие
первых с функцией генома.
Жизнь, какой мы ее видим, создается геномами мириад организмов, с которыми мы
делим нашу планету. Каждый из этих организмов обладает геномом, который содержит
биологическую информацию, необходимую для построения и поддержания организма
живущего в настоящий момент времени представителя данного вида. Большинство
геномов, в том числе геном человека и геномы всех остальных клеточных форм жизни,
построены из ДНК (дезоксирибонуклеиновой кислоты), однако некоторые вирусы име-
ют геномы из РНК (рибонуклеиновой кислоты). ДНК и РНК — полимерные молекулы,
состоящие из цепей мономерных звеньев, называемых нуклеотидами.
Геном человека, типичный для геномов всех многоклеточных животных, состоит
из двух различных частей (см. рис. 1.1):
4
Часть 1. Изучение геномов
Ядерный геном
Митохондриальный
геном
°р8°о
°ООО
Рис. 1.1. Ядерный и митохондриальный компоненты генома человека
• Ядерный геном включает приблизительно 3 200 000 000 нуклеотидов, распреде-
ленных по 24-м линейным молекулам ДНК (самая короткая — 50000000 нуклео-
тидов в длину, а самая длинная — 260 000 000), при этом каждая из них содержится
в отдельной хромосоме. Этот набор из 24 хромосом состоит из 22 аутосом и двух
половых хромосом: X и У. В целом на ядерный геном человека приходится при-
близительно 35000 генов1).
• Митохондриальный геном — кольцевая молекула ДНК длиной 16569 нуклео-
тидов, многочисленные копии которой расположены в производящих энергию
органеллах, называемых митохондриями. Митохондриальный геном человека
содержит всего лишь 37 генов.
Каждая из приблизительно 1013 клеток в теле взрослого человека имеет свою соб-
ственную копию (или копии) генома; единственные исключения составляют клетки не-
скольких особых типов — например, красные кровяные тельца, у которых в их полностью
дифференцированном состоянии нет ядра. В подавляющем большинстве клетки являются
диплоидными и, таким образом, содержат по две копии всех аутосом плюс две половые
хромосомы (XX у женщин и ХУу мужчин), то есть всего 46 хромосом. Такие клетки называют
соматическими, в отличие от половых клеток, или гамет, которые являются гаплоидны-
ми и содержат лишь 23 хромосомы — по одной из каждой пары аутосом и одну половую
хромосому. Клетки обоих типов заключают в себе около 8000 копий митохондриального
генома — приблизительно по 10 копий в каждой митохондрии.
Геном — хранилище биологической информации, но сам по себе он не способен
выдать эту информацию клетке. Использование биологической информации, содер-
жащейся в геноме, возможно лишь благодаря согласованным действиям ферментов
и других белков, которые участвуют в сложной серии биохимических реакций, назы- В
В последнем релизе — 24 тыс. генов. — Прим. ред.
Глава 1. Геномы, транскриптомы и протеомы 5
ГЕНОМ
Транскрипция
ТРАНСКРИПТОМ
РНК-копии активных генов, кодирующих белки
Трансляция
ПРОТЕОМ
Ассортимент белков клетки
Рис. 1.2. Геном, транскриптом и протеом
ваемой экспрессией генома (см. рис. 1.2). Первичным продуктом экспрессии генома
является транскриптом, представляющий собой совокупность молекул РНК, получен-
ных из тех кодирующих белок генов1), биологическая информация которых необходима
клетке в данный момент времени. Транскриптом поддерживается за счет процесса,
названного транскрипцией, в ходе которой отдельные гены копируются в молекулы
РНК. Вторичный продукт экспрессии генома — протеом — есть ассортимент белков
клетки, определяющий характер биохимических реакций, которые клетка способна
осуществлять. Белки, составляющие протеом, синтезируются посредством трансляции
отдельных молекул РНК, входящих в состав транскриптома.
Эта книга посвящена геномам и их экспрессии. В ней рассказывается о том, как геномы
изучаются (часть 1), как они организованы (часть 2), как функционируют (часть 3) и как
реплицируются и эволюционируют (часть 4). Возможность написать эту книгу появилась
лишь совсем недавно. Начиная с 1950-х гг. молекулярные биологи изучали отдельные гены
или маленькие группы генов и на основании результатов этих исследований создали бога-
тейшую научную теорию о механизмах функционирования генов. Однако методы, которые
позволили исследовать полные геномы, появились лишь в последние 10 лет. Отдельные
гены все еще интенсивно изучаются, но теперь такая информация об отдельных генах ин-
терпретируется в контексте генома как целого. Этот новый, более широкий ракурс не только
охватывает геномы, но и распространяется на всю биохимию и клеточную биологию. Сегодня
ученым уже недостаточно понять отдельные биохимические пути или внутриклеточные
процессы. Вызов незнанию теперь брошен системной биологией, которая пытается спле-
сти все эти пути и процессы в сети, могущие описывать общие законы функционирования
живых клеток и живых организмов.
Эта книга проведет вас через сокровищницу наших современных знаний о геномах
и покажет вам, что эта захватывающая область исследования образует основу нашего
непрестанно совершенствующегося понимания биологических систем. Однако сначала
мы должны обратить внимание на главные принципы молекулярной биологии и рассмо-
треть основные характеристики биологических молекул трех типов, входящих в состав
геномов и участвующих в их экспрессии: ДНК, РНК и белков.
В последнее время выясняется все большая роль некодирующих РНК, поэтому в транскриптом вклю-
чают все РНК, быть может за исключением рибосомных и транспортных, а не только белок-кодирующих. —
Прим. ред.
6
Часть 1. Изучение геномов
1.1. ДНК
ДНК была открыта в 1869 г. Иоганном Фридрихом Мишером, швейцарским биохи-
миком, работавшим в германском городе Тюбингене. Первые экстракты, которые Мишер
получил из белых кровяных телец человека, представляли собой неочищенные смеси
ДНК и хромосомных белков, но в следующем году он переехал в Швейцарию, город Базель
(в котором теперь располагается научно-исследовательский институт, названный его име-
нем), и приготовил чистый образец нуклеиновой кислоты из спермы лосося. Химические
анализы Мишера показали, что ДНК имеет кислотный характер и богата фосфором, а также
позволили ему предположить, что отдельные молекулы являются очень большими, хотя
лишь в 1930-х гг., когда к ДНК были применены биофизические методы исследования,
удалось окончательно установить огромную длину ее полимерных цепей.
1.1.1. Гены состоят из ДНК
Сегодня тот факт, что гены состоят из ДНК, настолько общеизвестен, что даже
трудно себе представить, что в течение первых 75-ти лет после ее открытия истинная
роль ДНК не подозревалась. Уже в 1903 г. У С. Саттон осознал, что структуры наследо-
вания генов соответствуют поведению хромосом во время деления клетки; именно эта
догадка привела к созданию хромосомной теории — предположению, что гены нахо-
дятся в хромосомах. Исследование клеток методами цитохимии (после окрашивания
их красителями, специфично связывающимися лишь с биохимическими веществами
одного типа) показало, что хромосомы состоят из ДНК и белка, наличествующих при-
близительно в равных количествах. После этого биологи пришли к выводу о том, что
должны существовать миллиарды различных генов и поэтому генетический материал
должен быть способен принимать множество различных форм. Но ДНК, казалось, не
удовлетворяла данному требованию, потому что в начале двадцатого века думали, что
все молекулы ДНК были одинаковыми. С другой стороны, было известно, и притом точ-
но, что белки представляют собой весьма изменчивые полимерные молекулы, причем
каждая образована уникальной комбинацией 20-ти химически различных аминокис-
лотных мономеров1) (раздел 1.3.1). На основании этих фактов ученые попросту приняли
гипотезу о том, что гены состоят из белка, а не из ДНК.
Ошибочное представление о структуреДНК продолжало пребывать в умах ученых,
но к концу 1930-х гг. стало известно, что ДНК, подобно белку, обладает огромной из-
менчивостью. Первоначальная гипотеза о том, что генетическим материалом является
белок, не утратила своей силы, но в конечном счете была ниспровергнута результатами
двух важных экспериментов:
• Освальд Эвери, Колин Маклеод и Маклин Маккарти показали, что ДНК есть актив-
ный компонент трансформирующего начала (экстракта бактериальных кле-
ток), которое, будучи смешано с безвредным штаммом Streptococcus pneumoniae,
преобразует эти бактерии в вирулентную форму, способную вызывать у мышей
пневмонию при введении в организм (см. рис. 1.3, а). В 1944 г., когда результаты
этого эксперимента были опубликованы, лишь несколько микробиологов призна-
ли, что наблюдаемая трансформация обусловлена передачей генов из экстракта
клеток в живые бактерии. Но как только это положение было принято, истинное
значение «эксперимента Эвери» стало ясным: гены бактерий должны были со-
стоять из ДНК.
На самом деле набор аминокислот не был точно известен вплоть до 60-х годов XX века. Например, два
цистеина, соединенные S-S связью, считались отдельной аминокислотой — цистином. — Прим. ред.
а) Трансформирующее начало
Глава 1. Геномы, транскриптомы и протеомы 7
б) Эксперимент Херши-Чейз
Мышь выживает
с . ДНК
Белковый капсид
Безвредные бактерии
Безвредные бактерии + Мышь умирает
трансформирующее начало.
обработанное протеазой
Безвредные бактерии + Мышь умирает
трансформирующее начало
Фаг, прикрепленный
| Перемешивание в мешалке
А
А СО
со А Фаги отсоединены
или рибонуклеазой
Безвредные бактерии + Мышь выживает
трансформирующее начало,
обработанное дезоксирибонуклеазой
। Центрифугирование
Бактериальный
осадок “
70 % 32Р
20 % 35S
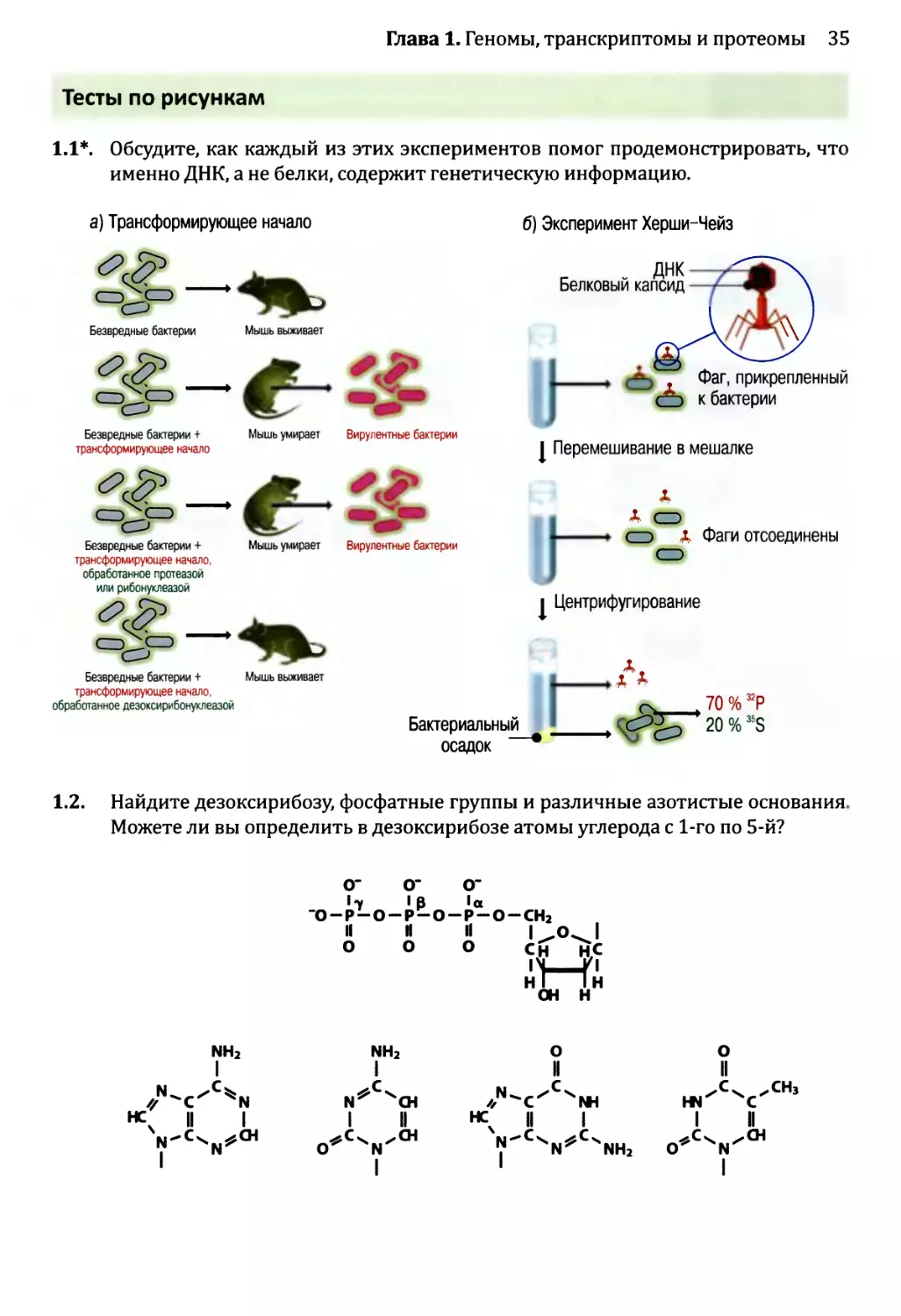
Рис. 1.3. Два эксперимента, которые позволили предположить, что гены состоят из ДНК:
а) Эвери с сотрудниками показал, что трансформирующее начало состоит из ДНК. В двух верхних строках
показана череда событий при введении мышам безвредных бактерий Streptococcus pneumoniae как с до-
бавлением трансформирующего начала (клеточного экстракта, полученного из вирулентного штамма
S. pneumoniae), так и без него. Если трансформирующее начало присутствует, то мышь умирает, потому
что гены в трансформирующем начале преобразуют безвредные бактерии в вирулентную форму, при-
чем эти вирулентные бактерии впоследствии извлекаются из легких мертвой мыши. Две нижние строки
показывают, что обработка протеазой или рибонуклеазой не оказывает никакого воздействия на транс-
формирующее начало и что трансформирующее начало инактивируется дезоксирибонуклеазой.
б) Эксперимент Херши-Чейз, в котором были использованы бактериофаги Т2. Каждый бактериофаг
содержит молекулу ДНК, заключенную в белковый капсид, к которому прикреплены «тело» и «ноги»,
позволяющие бактериофагу прикрепляться к поверхности бактерии и вводить свои гены в ее клетку.
ДНК бактериофагов была помечена 32Р, а белок — 35S. Спустя несколько минут после инфицирования
культура была перемешана, чтобы отделить пустые частицы фага от поверхности клеток. Затем культура
была центрифугирована, в результате чего бактерии вместе с генами фагов собрались в капле осадка на
дне пробирки, а более легкие частицы фагов остались плавать в суспензии. Херши и Чейз обнаружили,
что бактериальный осадок содержит большую часть помеченного 32Р компонента фагов (ДНК) и только
20 % материала, помеченного 35S (белка фагов). Во втором эксперименте Херши и Чейз показали, что
новые фаги, порожденные в конце цикла инфицирования, содержали менее 1 % белка из родительских
фагов. Более подробное описание цикла инфицирования бактериофагами приведено на рис. 2.19
• Альфред Херши и Марта Чейз применяли мечение радиоактивными изотопами,
с тем чтобы показать, что при заражении бактериальной культуры бактериофа-
гами (тип вируса) главным компонентом бактериофагов, который проникает
в клетки бактерий, является ДНК (см. рис. 1.3, б). Это было важнейшее наблюдение,
потому что к тому времени уже было известно, что в течение цикла инфицирова-
ния гены инфицирующих бактериофагов используются для управления синтезом
8
Часть 1. Изучение геномов
новых бактериофагов и что этот синтез происходит в клетках бактерий. Однако
если в клетки бактерий вводится только ДНК инфицирующих бактериофагов, то
из этого следует, что гены этих бактериофагов должны состоять из ДНК.
Хотя теперь нам и ясно, что эти два эксперимента предоставляют очевидные ре-
зультаты, которые говорят нам, что гены состоят из ДНК, биологов того времени было
не так легко убедить в этом. Оба эксперимента имеют ограничения, которые оставляют
скептикам возможность утверждать, что белок все же может быть генетическим мате-
риалом. Например, были сомнения о специфичности фермента дезоксирибонуклеазы,
которую Эвери с сотрудниками применяли для инактивации трансформирующего
начала. Этот результат — ядро доказательства того, что трансформирующим началом
является ДНК, — был бы ошибочным, если, как казалось возможным, фермент содержал
бы нижтожное количество (следы) загрязняющей протеазы и, следовательно, был бы
способен вызывать также деградацию белка. Ни один из экспериментов с бактериофага-
ми не позволяет сделать окончательного вывода, как подчеркнули Херши и Чейз, когда
они опубликовали полученные ими результаты: «Наши эксперименты ясно показывают,
что физическое разделение фага Т2 на генетическую и негенетическую составляющие
возможно... Химическое определение генетической составляющей, однако, должно
подождать до тех пор, пока на некоторые вопросы... не будет найден ответ». Огляды-
ваясь в прошлое, можно сказать, что эти два эксперимента важны не из-за того, что они
говорят нам, но потому, что они заставили биологов задуматься о том, что ДНК могла бы
быть генетическим материалом и поэтому заслуживает изучения. Именно результаты
этих экспериментов побудили Уотсона и Крика работать с ДНК, и, как мы позже увидим,
именно совершенное ими открытие двойной спирали — структуры ДНК — разрешило
приводивший ученых в замешательство вопрос о том, каким образом гены могут репли-
цироваться, и окончательно убедило научный мир в том, что гены состоят из ДНК.
1.1.2. Структура ДНК
Имена Джеймса Уотсона и Фрэнсиса Крика настолько тесно связаны с ДНК, что
легко забыть о том факте, что когда они начали свою совместную работу в октябре
1951 г., подробная структура полимерной цепи ДНК уже была известна. Их вклад со-
стоял не в том, что они определили структуру ДНК perse, но в том, что они показали, что
в живых клетках две цепи ДНК переплетаются и образуют двойную спираль. Поэтому
сначала мы должны ознакомиться с теми данными, которые были известны Уотсону
и Крику, когда они приступали к своей работе.
Нуклеотиды и полинуклеотиды
ДНК — линейный, неразветвленный полимер, мономерные звенья которого пред-
ставлены четырьмя химически различными нуклеотидами, могущими сочетаться между
собой в любом порядке и образовывать цепи длиной в сотни, тысячи или даже мил-
лионы единиц. Каждый нуклеотид в полимере ДНК состоит из трех компонентов (см.
рис. 1.4):
• 2'-дезоксирибозы, которая представляет собой пентозу, относящуюся к клас-
су сахаров, состоящих из пяти атомов углерода. Эти пять атомов углерода про-
нумерованы числами 1' (произносится «один-штрих»), 2' и так далее. Название
«2'-дезоксирибоза» указывает на то, что этот специфический сахар есть произво-
дная рибозы, в которой гидроксильная группа (-ОН), присоединенная к 2’-атому
углерода в молекуле рибозы, была заменена атомом водорода (-Н);
Глава 1. Геномы, транскриптомы и протеомы 9
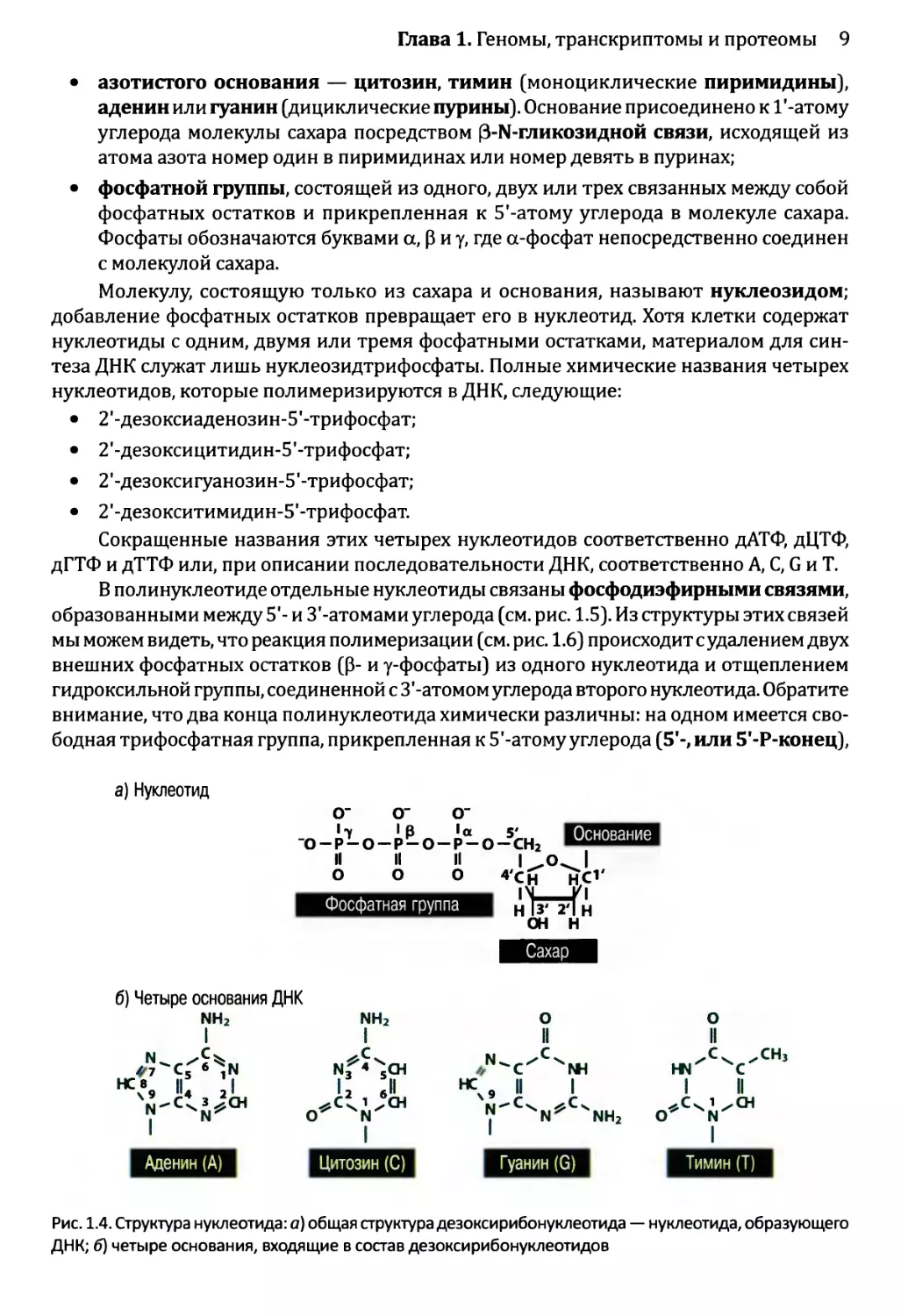
• азотистого основания — цитозин, тимин (моноциклические пиримидины),
аденин или гуанин (дициклические пурины). Основание присоединено к Г-атому
углерода молекулы сахара посредством (З-Ы-гликозидной связи, исходящей из
атома азота номер один в пиримидинах или номер девять в пуринах;
• фосфатной группы, состоящей из одного, двух или трех связанных между собой
фосфатных остатков и прикрепленная к 5'-атому углерода в молекуле сахара.
Фосфаты обозначаются буквами а, р и у, где а-фосфат непосредственно соединен
с молекулой сахара.
Молекулу, состоящую только из сахара и основания, называют нуклеозидом;
добавление фосфатных остатков превращает его в нуклеотид. Хотя клетки содержат
нуклеотиды с одним, двумя или тремя фосфатными остатками, материалом для син-
теза ДНК служат лишь нуклеозидтрифосфаты. Полные химические названия четырех
нуклеотидов, которые полимеризируются в ДНК, следующие:
• 2'-дезоксиаденозин-5'-трифосфат;
• 2’-дезоксицитидин-5’-трифосфат;
• 2'-дезоксигуанозин-5’-трифосфат;
• 2'-дезокситимидин-5’-трифосфат.
Сокращенные названия этих четырех нуклеотидов соответственно дАТФ, дЦТФ,
дГТФ и дТТФ или, при описании последовательности ДНК, соответственно А, С, G и Т.
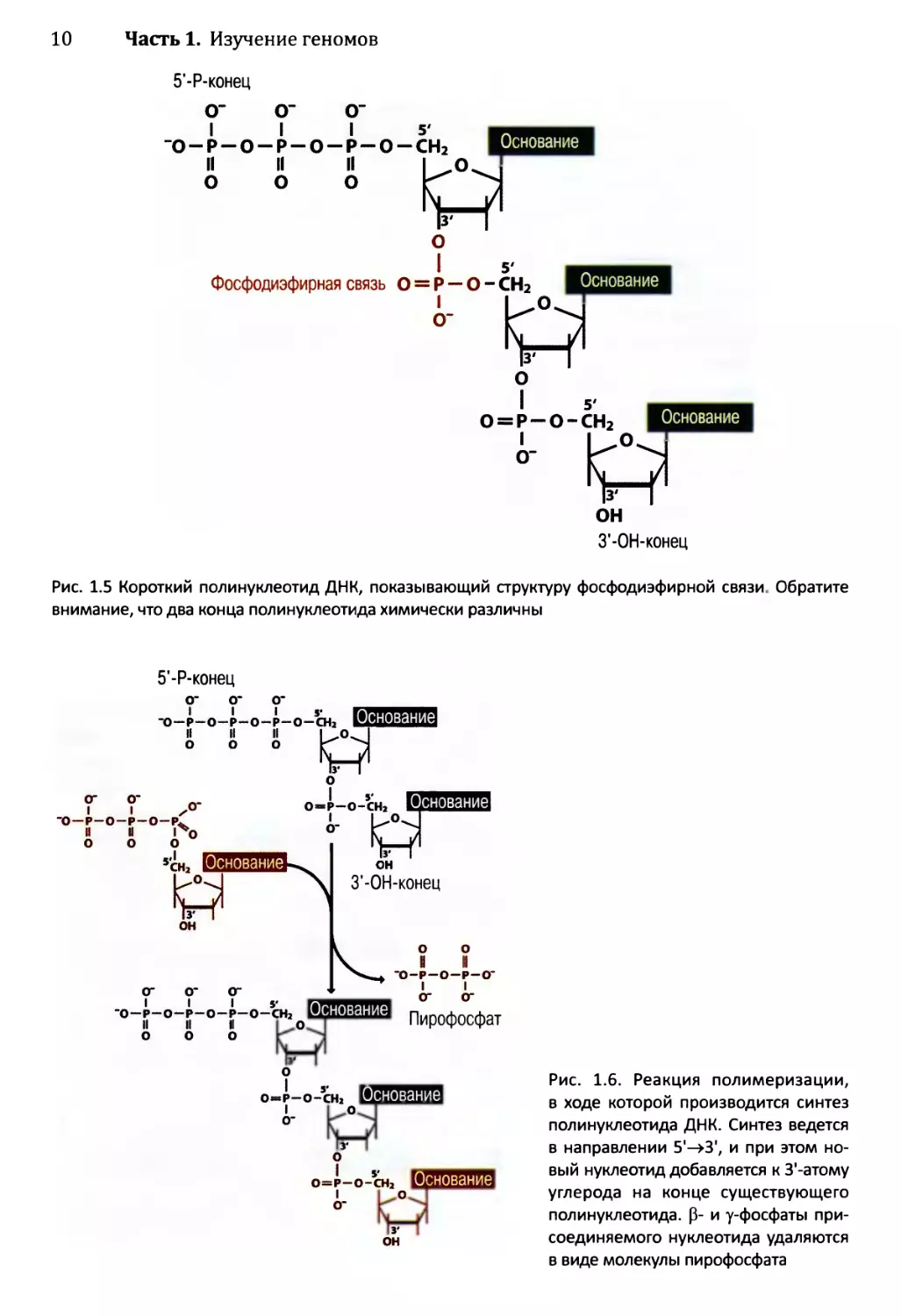
В полинуклеотиде отдельные нуклеотиды связаны фосфодиэфирными связями,
образованными между 5'- и З’-атомами углерода (см. рис. 1.5). Из структуры этих связей
мы можем видеть, что реакция полимеризации (см. рис. 1.6) происходит с удалением двух
внешних фосфатных остатков (Р- и у-фосфаты) из одного нуклеотида и отщеплением
гидроксильной группы, соединенной с З’-атомом углерода второго нуклеотида. Обратите
внимание, что два конца полинуклеотида химически различны: на одном имеется сво-
бодная трифосфатная группа, прикрепленная к 5'-атому углерода (5’-, или 5'-Р-конец),
а) Нуклеотид
О“ О"
л ~ 1Р
О“
, г- ’а 5*
O-P-O-P-O-P-O-CHz
Основание
о
о
0 4'Сн НС1
Фосфатная группа
он н
| Сахар
б) Четыре основания ДНК
nh2
Аденин (А)
nh2
I
Гуанин (G)
Тимин (Т)
Цитозин (С)
I
Рис. 1.4. Структура нуклеотида: а) общая структура дезокси рибонуклеотида — нуклеотида, образующего
ДНК; б) четыре основания, входящие в состав дезоксирибонуклеотидов
10
Часть 1. Изучение геномов
5'-Р-конец
О’ О’ О-
он
З'-ОН-конец
Рис. 1.5 Короткий полинуклеотид ДНК, показывающий структуру фосфодиэфирной связи. Обратите
внимание, что два конца полинуклеотида химически различны
5'-Р-конец
Рис. 1.6. Реакция полимеризации,
в ходе которой производится синтез
полинуклеотида ДНК. Синтез ведется
в направлении 5'->3', и при этом но-
вый нуклеотид добавляется к З'-атому
углерода на конце существующего
полинуклеотида. 0- и у-фосфаты при-
соединяемого нуклеотида удаляются
в виде молекулы пирофосфата
Глава 1. Геномы, транскриптомы и протеомы 11
а на другом — незамещенная гидроксильная группа, соединенная с З'-атомом углерода
(3'-, или З'-ОН-конец). Это означает, что полинуклеотид характеризуется химическим
направлением, обозначаемым как 5'—>3' (вниз на рис. 1.5) или как 3'—>5' (вверх на
рис. 1.5). Важное следствие полярности фосфодиэфирной связи заключается в том, что
химическая реакция, необходимая для продолжения полимера ДНК в направлении 5’—>3',
отличается от реакции, способной наращивать полимерную цепь в направлении 3'->5'.
Все природные ферменты ДНК-полимеразы способны осуществлять синтез только
в направлении 5'—>3', что значительно усложняет процесс, в ходе которого двунитевая
ДНК реплицируется (раздел 15.2).
Данные, подтвердившие гипотезу о двойной спирали
До 1950 г. различные эксперименты показывали, что молекулы клеточной ДНК со-
стоят из двух или нескольких полинуклеотидов, соединенных друг с другом некоторым
неизвестным образом. Возможность того, что раскрытие природы этого соединения
могло бы обеспечить понимание принципа работы генов, побудила Уотсона и Крика, по-
мимо других ученых, попытаться разрешить эту загадку структуры. По словам Уотсона,
приведенным в его книге «Двойная спираль», их работа была отчаянной гонкой про-
тив знаменитого американского биохимика Лайнуса Полинга, который первоначально
предложил неправильную модель тройной спирали, что дало Уотсону и Крику время,
необходимое им для завершения работы над моделью структуры двойной спирали.
Сейчас уже трудно отделить факт от вымысла, особенно относительно роли, сыгранной
Розалиной Франклин, чьи исследования с помощью рентгеноструктурного анализа
обеспечили большую часть экспериментальных данных, свидетельствующих в пользу
двойной спирали, и которая сама была очень близка к разгадке ее структуры. Но одно
совершенно ясно — открытие двойной спирали, совершенное Уотсоном и Криком в суб-
боту 7 марта 1953 г., было исключительным по своему масштабу научным прорывом
в биологии двадцатого века.
Для того чтобы вывести структуру двойной спирали, Уотсон и Крик использовали
информацию четырех категорий:
• Биофизические данные различных видов. Особенно важным было содержание
воды в волокнах ДНК, потому что это позволило определить плотность ДНК в во-
локне. Число нитей в спирали и интервал между нуклеотидами должны были со-
ответствовать плотности волокна. Модель тройной спирали Полинга опиралась на
неправильно измеренное значение плотности, которое привело к предположению,
что молекула ДНК упакована плотнее, чем это есть на самом деле.
• Рентгеновские дифрактограммы (см. техническое примечание 11.1), большая
часть которых была получена Розалиной Франклин, показали спиральный характер
структуры и обозначили некоторые из основных параметров этой спирали.
• Отношения оснований, установленные Эрвином Чаргаффом в Колумбийском
университете в Нью-Йорке. Чаргафф выполнил длинный ряд хроматографиче-
ских исследований образцов ДНК из различных источников и показал, что хотя
абсолютные значения количества оснований различны у разных организмов, ко-
личество аденина всегда совпадает с количеством тимина, а количество гуанина
равно количеству цитозина (см. рис. 1.7). Эти отношения оснований позволили
сформулировать правила спаривания оснований, которые явились ключом к от-
крытию структуры двойной спирали.
12
Часть 1. Изучение геномов
Бактерии
Escherichia coli
Клетки человека
Очистка ДНК
Рис. 1.7. Эксперименты по установлению отно-
шений оснований, выполненные Чаргаффом.
ДНК была извлечена из разных организмов
и обработана кислотой, с тем чтобы гидроли-
зировать фосфодиэфирные связи и высвобо-
дить отдельные нуклеотиды. Затем количество
нуклеотидов каждого вида было определено
с помощью хроматографии. Приведенные на
рисунке численные данные показывают неко-
торые из фактических результатов, полученных
Чаргаффом. Они указывают на то, что в преде-
лах погрешности эксперимента количество аде-
нина совпадает с количеством тимина, а коли-
чество гуанина равно количеству цитозина
Хроматография для определения
количества каждого нуклеотида
I 1
Отношение оснований Отношение оснований
А:Т 1,00 А:Т 1,09
G:C 1,00 G:C 0,99
• Моделирование — единственный серьезный метод исследования, который Уот-
сон и Крик выполнили самолично. Масштабные модели возможных структур ДНК
позволили им проверить свой вариант относительного расположения различных
атомов, дабы убедиться в том, что пары, которые образуют связи, не расположены
слишком далеко друг от друга, а также в том, что остальные атомы не находятся
столь близко, чтобы мешать друг другу.
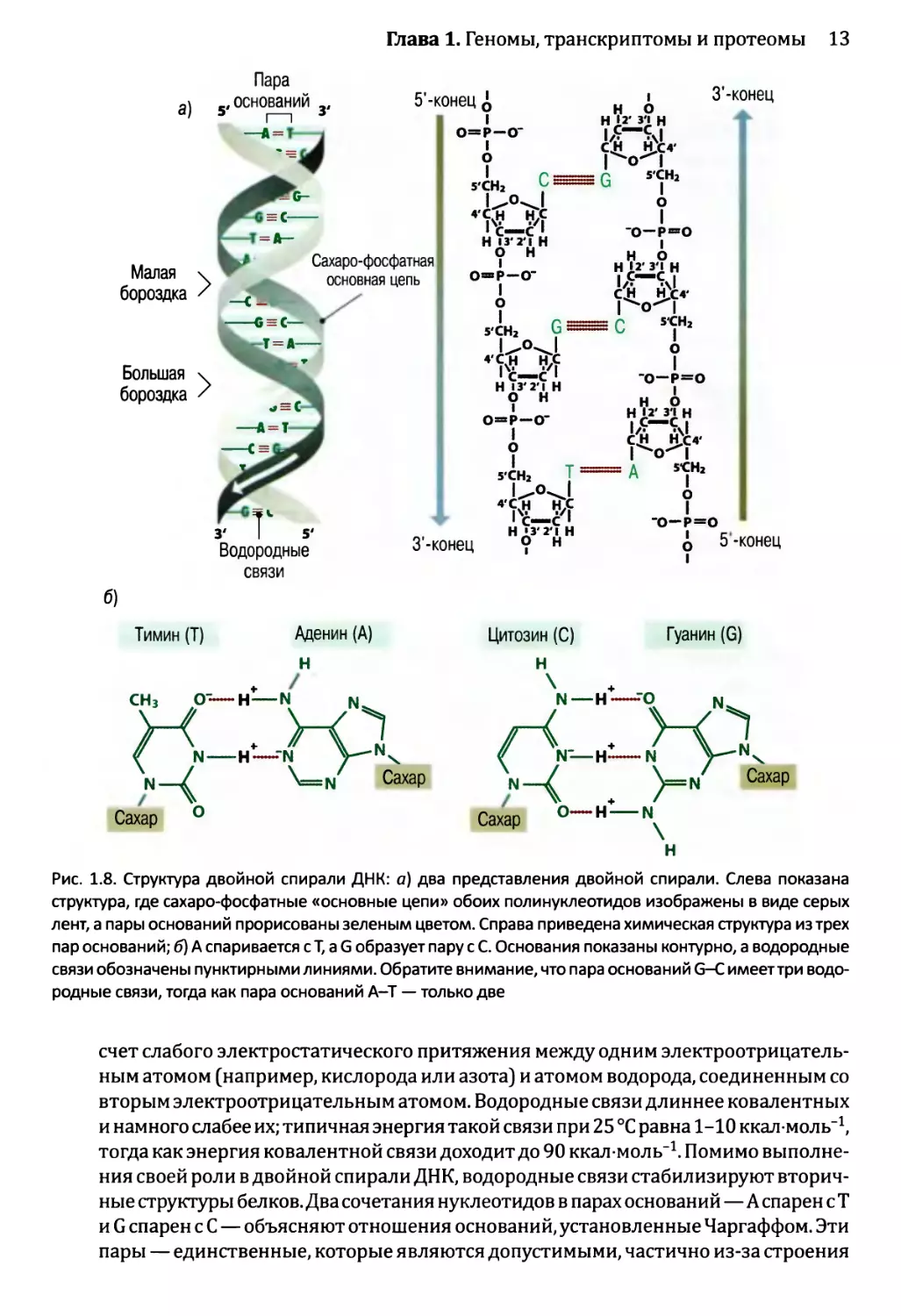
Основные характеристики двойной спирали
Двойная спираль правосторонняя, а это означает, что, если бы она была винтовой
лестницей и вы поднимались по ней вверх, то перила на внешней стороне лестницы
находились бы справа от вас. Две нити спирали бегут во встречных направлениях
(см. рис. 1.8, а). Спираль стабилизируется химическими взаимодействиями двух ти-
пов:
• Спаривание оснований между двумя нитями осуществляется путем образования
водородных связей между аденином на одной нити и тимином на другой нити или
же между цитозином и гуанином (см. рис. 1.8, б). Водородные связи возникают за
Глава 1. Геномы, транскриптомы и протеомы 13
связи
Тимин (Т) Аденин (А)
Цитозин (С) Гуанин (G)
Рис. 1.8. Структура двойной спирали ДНК: а) два представления двойной спирали. Слева показана
структура, где сахаро-фосфатные «основные цепи» обоих полинуклеотидов изображены в виде серых
лент, а пары оснований прорисованы зеленым цветом. Справа приведена химическая структура из трех
пар оснований; б) А спаривается сТ, a G образует пару с С. Основания показаны контурно, а водородные
связи обозначены пунктирными линиями. Обратите внимание, что пара оснований G-С имеет три водо-
родные связи, тогда как пара оснований А-Т — только две
счет слабого электростатического притяжения между одним электроотрицатель-
ным атомом (например, кислорода или азота) и атомом водорода, соединенным со
вторым электроотрицательным атомом. Водородные связи длиннее ковалентных
и намного слабее их; типичная энергия такой связи при 25 °C равна 1-10 ккал моль-1,
тогда как энергия ковалентной связи доходит до 90 ккал моль-1. Помимо выполне-
ния своей роли в двойной спирали ДНК, водородные связи стабилизируют вторич-
ные структуры белков. Два сочетания нуклеотидов в парах оснований—А спарен с Т
и G спарен с С — объясняют отношения оснований, установленные Чаргаффом. Эти
пары — единственные, которые являются допустимыми, частично из-за строения
14 Часть 1. Изучение геномов
азотистых оснований и относительных положений атомов, способных к образова-
нию водородных связей, и частично потому, что пара должна быть между пурином
и пиримидином: пара пурин-пурин была бы слишком большая, чтобы поместиться
внутри спирали, а пара пиримидин-пиримидин была бы слишком маленькой.
• Стекинг-взаимодействие, иногда называемое п-п-взаимодействием, обусловлено
гидрофобными взаимодействиями между смежными парами оснований и придает
двойной спирали дополнительную стабильность, как только нити соединились
спариванием оснований. Эти гидрофобные взаимодействия возникают потому, что
структура воды, образуемая за счет водородных связей, вталкивает гидрофобные
группы во внутренние части молекулы.
Для удержания двух полинуклеотидов вместе важно и спаривание оснований, и их
стекинг, но спаривание оснований имеет еще одно значение благодаря его биологиче-
ским следствиям. Ограничение, состоящее в том, что А может спариваться только с Т,
a G может образовывать пару только с С, означает, что в ходе репликации ДНК могут
производиться совершенные копии родительской молекулы посредством простого
способа, состоящего в использовании последовательностей уже существующих нитей
для считывания с них последовательности новых нитей. Это так называемый направ-
ляемый матрицей синтез ДНК, и эта система используется всеми клеточными ДНК-
полимеразами (раздел 15.2.2). Поэтому спаривание оснований позволяет молекулам
ДНК реплицироваться системой, которая является настолько простой и изящной, что,
как только структура двойной спирали была провозглашена Уотсоном и Криком, все
биологи окончательно убедились в том, что гены действительно состоят из ДНК.
Двойная спираль обладает гибкостью структуры
Двойную спираль, описанную Уотсоном и Криком и показанную на рис. 1.8, а,
называют В-формой ДНК. Ее характерные особенности заключаются в размерных па-
раметрах: диаметр спирали 2,37 нм, подъем 0,34 нм на пару оснований и шаг (то есть
высота полного витка спирали) 3,4 нм, что соответствует десяти парам оснований на
один виток. Полагают, что в живых клетках ДНК находится преимущественно в такой
В-форме, но теперь ясно, что молекулы геномной ДНК не полностью однородны по
структуре. Главным образом это связано с тем, что все нуклеотиды в спирали обладают
гибкостью, позволяющей им принимать слегка отличающиеся молекулярные формы.
Чтобы принимать эти разные конформации, относительные положения атомов в ну-
клеотиде должны немного изменяться. Для этого есть ряд возможностей, но самые
важные конформационные изменения включают вращение вокруг p-N-гликозидной
связи, изменение ориентации основания относительно сахарного звена и вращение
вокруг связи между 3'- и 4'-атомами углерода. Вращение обоих видов оказывает на
двойную спираль существенное влияние: изменение ориентации основания влияет
на относительное расположение двух полинуклеотидов, а вращение вокруг связи 34'
затрагивает конформацию сахаро-фосфатной основной цепи.
Поэтому вращения в пределах отдельных нуклеотидов ведут к значительным из-
менениям в общей структуре спирали. С 1950-х гг. было известно, что, когда волокна,
содержащие молекулы ДНК, подвергнуты воздействию различной относительной влаж-
ности, происходят изменения в размерных параметрах двойной спирали. Например,
видоизмененная версия двойной спирали, называемая A-формой (см. рис. 1.9), имеет
диаметр 2,55 нм, подъем 0,29 нм на пару оснований и шаг 3,2 нм, что соответствует 11
парам оснований на виток (см. табл. 1.1). К другим разновидностям относят В'-, С-, С’-,
Глава 1. Геномы, транскриптомы и протеомы 15
Таблица 1.1. Характерные параметры различных конформаций двойной спирали ДНК
Параметр В-ДНК А-ДНК Z-ДНК
Тип спирали Правосторонняя Правосторонняя Левосторонняя
Диаметр спирали, нм 2,37 2,55 1,84
Подъем на пару осно- ваний, нм 0,34 0,29 0,37
Высота полного вит- ка (шаг), нм 3,4 3,2 4,5
Число пар оснований на полный виток 10 11 12
Топология большой Широкая, Узкая, глубокая Плоская
бороздки глубокая
Топология малой Узкая, мелкая Широкая, мелкая Узкая, глубокая
бороздки
С"-, D-, Е- и Т-ДНК. Все они — правосторонние спирали, подобные В-форме. Возможна
также более существенная перестройка структуры, которая ведет к левосторонней
Z-ДНК (рис. 1.9), представляющей собой более тонкую версию двойной спирали и об-
ладающей диаметром всего 1,84 нм.
Линейные размеры различных форм двойной спирали не показывают, каковы
самые, по всей вероятности, существенные различия между ними. Они состоят не
в разности значений диаметра и шага, но в степени доступности внутренних областей
спирали с поверхности структуры. Как показано на рис. 1.8 и 1.9, ДНК В-формы не имеет
абсолютно гладкой поверхности: вместо этого вдоль спирали винтовыми линиями за-
кручиваются две бороздки. Одна из этих бороздок относительно широкая и глубокая
и называется большой бороздкой; другая — узкая и менее глубокая и называется ма-
лой бороздкой. А-ДНК также имеет две бороздки (рис. 1.9), но в данной конформации
по сравнению с В-ДНК большая бороздка еще глубже, а малая бороздка — более мелкая
и более широкая. Z-ДНК тоже отличается от остальных форм: одна бороздка фактически
не существует, а другая очень узкая и глубокая.
В ДНК всех форм часть внутренней поверхности по крайней мере одной из бороздок
образована химическими группами, прикрепленными к нуклеотидным основаниям.
В главе 11 мы увидим, что экспрессия биологической информации, содержащейся в ге-
номе, опосредствуется ДНК-связывающими белками, которые прикрепляются к двойной
спирали и регулируют активность генов, содержащихся в ней. Для того чтобы исполнить
свои функции, каждый ДНК-связывающий белок должен прикрепиться к ней в опреде-
ленном положении поблизости от гена, на чью активность он должен повлиять. Этой
цели может достичь (по крайней мере с некоторой степенью точности) белок, способ-
ный проникать в бороздку, в которой последовательность ДНК может «считываться»
без раскрытия спирали, сопровождаемого разрывом пар оснований. Важное следствие
этого условия заключается в том, что ДНК-связывающий белок, структура которого
позволяет ему распознавать определенную последовательность нуклеотидов в В-ДНК,
например, не мог бы быть способен распознать такую же последовательность, если
ДНК приняла бы иную конформацию. Как мы увидим в главе 11, конформационные
16
Часть 1. Изучение геномов
В-ДНК А-ДНК z-днк
Рис. 1.9. Структуры В-ДНК (слева), А-ДНК (в центре) и Z-ДНК (справа). Модели упаковки (сверху) и струк-
турные модели (снизу) отображают различные конформации молекул ДНК. Обратите внимание на
взаимные различия в диаметре спиралей, в числе пар оснований на полный виток и в топологии боль-
шой и малой бороздок у этих молекул. Перепечатано с разрешения KendrewJ. (Ed.), Encyclopaedia of
Molecular Biology. © 1994 Blackwell Publishing
изменения вдоль молекулы ДНК, наряду с другими структурными полиморфизма-
ми, обусловленными последовательностью нуклеотидов, могут играть важную роль
в предопределении специфичности взаимодействий между геномом и специфическими
ДНК-связывающими белками.
1.2. РНК и транскриптом
Первичный продукт экспрессии генома — транскриптом (см. рис. 1.2) — пред-
ставляет собой совокупность молекул РНК, полученных из тех кодирующих белок
генов, биологическая информация которых востребована клеткой в данный момент
времени. Молекулы РНК транскриптома, равно как и многие другие молекулы РНК,
транскрибированные с не кодирующих белки генов, синтезируются в ходе процесса,
называемого транскрипцией. В этом разделе мы окинем взглядом структуру РНК и за-
тем более подробно рассмотрим молекулы РНК различного типа, присутствующие
в живых клетках.
1.2.1. Структура РНК
РНК — полинуклеотид подобный ДНК, но с двумя важными химическими отличия-
ми (см. рис. 1.10). Во-первых, сахар в нуклеотиде РНК представлен рибозой, а во-вторых,
Глава 1. Геномы, транскриптомы и протеомы 17
Рис. 1.10. Химические различия между
ДНК и РНК: а) РНК содержит рибону-
клеотиды, в которых сахар представ-
лен рибозой, а не 2'-дезоксирибозой.
Отличие этих сахаров состоит в том, что
в молекуле рибозы к 2-атому углерода
прикреплена гидроксильная группа,
а не атом водорода; б) вместо тимина
РНК содержит пиримидин урацил
а) Рибонуклеотид
О" О" О"
Ills'
"О —Р —О —Р —О —Р —О —СН2
II II II | .0^1
о о о сн нс1'
WI
н
он он
б) Урацил
Основание
HN СН
СН
РНК содержит урацил вместо тимина. Таким образом, четыре нуклеотида, используемые
в качестве строительного материала для синтеза РНК, следующие:
• аденозин-5'-трифосфат;
• цитидин-5’-трифосфат;
• гуанозин-5’-трифосфат;
• уридин-5'-трифосфат.
В сокращенной форме названия этих нуклеотидов записываются соответственно
как АТФ, ЦТФ, ГТФ и УТФ или, в однобуквенной записи, — соответственно А, С, G и U.
Как и в случае ДНК, полинуклеотиды РНК образованы за счет фосфодиэфирных
связей 35 ’, но эти фосфодиэфирные связи менее устойчивы, чем таковые в полинуклео-
тиде ДНК, из-за косвенного влияния гидроксильной группы, прикрепленной к 2'-атому
углерода в молекуле сахара. Длина цепей РНК редко превышает несколько тысяч нуклео-
тидов, и хотя многие из них образуют внутримолекулярные пары оснований (например,
см. рис. 13.2), большинство из них одно-, а не двунитевые.
Ферменты, ответственные за транскрипцию ДНКвРНК, называют ДНК-зависимыми
РНК-полимеразами. Это название указывает на то, что ферментативная реакция, ко-
торую они катализируют, дает в результате полимеризацию РНК из рибонуклеотидов
и происходит по зависимой от (направляемой) ДНК схеме, а это означает, что последо-
вательность нуклеотидов в матричной ДНК определяет последовательность нуклеоти-
дов в синтезируемой РНК (см. рис. 1.11). Допустимо сокращать название фермента до
РНК-полимеразы, если контекст, в котором употребляется это название, гарантирует
малую вероятность спутать его с РНК-зависимыми РНК-полимеразами, которые
участвуют в репликации и экспрессии геномов некоторых вирусов. Химическая основа
направляемого матрицей синтеза РНК эквивалентна представленной на рис. 1.6 для
случая синтеза ДНК. Рибонуклеотиды добавляются один за другим к растущему З'-концу
РНК-транскрипта, при этом тип каждого нуклеотида определяется правилами спарива-
ния оснований: А образует пару с Т ил и U; G спаривается с С. Точно так же, как и в случае
18
Часть 1. Изучение геномов
ДНК 3'—-TACCCAACGCAATTC----------- 5'
AUGG-----
5' 3'
РНК
Рис. 1.11. Направляемый матрицей синтез
РНК. РНК-транскрипт синтезируется в на-
правлении 5'->3’, а ДНК при этом считыва-
ется в направлении 3'—>5\ причем после-
довательность транскрипта определяется
спариванием его оснований с основаниями
ДНК-матрицы
3Z—TACCCAACGCAATTC---------5"
AUGGGUUG —►
5' У
полимеризации ДНК, во время присоединения каждого нуклеотида р- и у-фосфаты от-
щепляются от подаваемого мономера, а гидроксильная группа отрывается от З'-атома
углерода в нуклеотиде на конце цепи.
1.2.2. Виды и содержание РНК в клетке
Типичная бактерия содержит 0,05-0,10 пг РНК, что составляет приблизительно 6 %
ее полной массы. Клетка млекопитающих, будучи намного крупнее, содержит больше
РНК (в целом — 20-30 пг), но эта величина представляет лишь 1 % полной массы клет-
ки. Наилучший способ понять содержание РНК в клетке — это разделить эти вещества
на категории и подкатегории в зависимости от выполняемой ими функции. Известно
несколько способов такой классификации, и наиболее содержательная схема показана
на рис. 1.12. Первичное разделение — между кодирующей РНК и некодирующей РНК.
Кодирующая РНК образует транскриптом и состоит из молекул только одного класса,
информационных, или матричных РНК (мРНК), которые представляют собой транс-
крипты кодирующих белки генов и, следовательно, транслируются в белки на второй
стадии экспрессии генома. Матричные РНК редко составляют более 4 % от общего коли-
Рис. 1.12. Виды и содержание РНК в клетке. На этой схеме представлены виды РНК, встречающиеся во
всех организмах, и ее категории, присутствующие только в клетках эукариотов
Глава 1. Геномы, транскриптомы и протеомы 19
чества РНК и, будучи недолговечны, деградируют вскоре после синтеза. Бактериальные
мРНК имеют периоды полураспада не более нескольких минут, а у эукариотов большая
часть мРНК деградирует в течение нескольких часов после синтеза. Столь быстрый обо-
рот означает, что состав транскриптома непостоянен и может быстро менять структуру
путем изменения скорости синтеза тех или иных мРНК.
РНК второго вида упоминается как «некодирующая», поскольку эти молекулы
не транслируются в белки. Однако лучшим названием будет «функциональная РНК»,
поскольку оно подчеркивает тот факт, что, хотя некодирующая РНК и не является
частью транскриптома, различные ее виды все же играют важные роли в клетке.
Известно несколько видов функциональной РНК, наиболее значимы из которых
следующие:
• Рибосомная РНК (рРНК) присутствует во всех организмах и обычно является
самой многочисленной РНК в клетке (в активно делящихся бактериях она состав-
ляет более 80 % от общего количества РНК). Ее молекулы являются компонентами
рибосом — структур, в которых осуществляется синтез белка (раздел 13.2).
• Транспортная РНК (тРНК) — маленькие молекулы, которые также вовлечены
в синтез белка и, подобно рРНК, встречаются во всех организмах. Функция тРНК
состоит в переносе аминокислот к рибосоме и обеспечении соединения аминокис-
лот в порядке, определяемом последовательностью нуклеотидов транслируемой
мРНК (раздел 13.1).
• Малая ядерная РНК (мяРНК, snPHK), также называемая U-PHK, потому что ее
молекулы богаты уридиннуклеотидами, находится в ядрах эукариотов. Эти моле-
кулы участвуют в сплайсинге (одном из ключевых шагов процесса созревания),
который преобразует первичные транскрипты кодирующих белки генов в моле-
кулы зрелой мРНК (раздел 12.2.2).
• Малая ядрышковая РНК (мякРНК, snoPHK) размещается в ядрышках клеток
эукариотов. Она играет центральную роль в химической модификации молекул
рРНК, направляя ферменты, которые осуществляют модификации, к определен-
ным нуклеотидам, где должны быть выполнены изменения, например добавления
метильной группы (раздел 12.2.5).
• МикроРНК (миРНК, miPHK) и короткая интерферирующая РНК (киРНК,
siPHK) — маленькие молекулы РНК, которые регулируют экспрессию отдельных
генов1) (раздел 12.2.6).
1.2.3. Созревание РНК-предшественника
Наряду со зрелой РНК, описанной выше, клетки содержат также молекулы РНК-
предшественника. РНК многих видов, особенно у эукариотов, первоначально синтези-
руется в виде молекулы РНК-предшественника, или пре-РНК, которая должна созреть,
прежде чем сможет выполнять свою функцию. В число различных этапов созревания
(события процессинга), полный свод которых вместе с подробным описанием приведен
в главе 12, входят следующие (см. рис. 1.13):
• Модификация концов происходит во время синтеза мРНК эукариотов. В итоге
большинство ее молекул получает необычный единичный нуклеотид, называемый
кэпом, на свой 5'-конец и поли-А хвост — на З'-конец.
На самом деле есть еще много типов РНК, задействованных в различных процессах. — Прим. ред.
20
Часть 1. Изучение геномов
Пред-РНК ------
(-------------------1--------
Модификация концов Сплайсинг
Раздел Разделы
12.2.1 12.2.2 и
112.2.4
Разрезание
Разделы
12.1.3 и
12.2.3
Химическая модификация
Разделы
12.1.3 и
12.2.5
I
Новая химическая
группа
Кэп Поли-А хвост Позиция
удаленного интрона
Рис. 1.13. Схематическое представление событий обработки РНК четырех типов. В организмах разных
биологических видов происходят различные (не все) события из данного набора
• Сплайсинг — удаление сегментов из внутренней части РНК-предшественника.
Многие гены, особенно эукариотов, имеют внутренние сегменты, которые не
несут никакой биологической информации. Их называют интронами, и в ходе
транскрипции гена они копируются наряду с содержащими информацию экзона-
ми. Интроны удаляются из пре-мРНК путем реакций разрезания и соединения.
Незрелая пре-мРНК образует фракцию ядерной РНК, называемую гетерогенной
ядерной РНК (гяРНК).
• События разрезания особенно важны в процессинге молекул рРНК и тРНК, многие
из которых первоначально синтезируются с единиц транскрипции, определяющих
сразу несколько молекул. Поэтому для того чтобы произвести зрелую РНК, цепи
пре-рРНК и пре-тРНК должны быть разрезаны на куски. Процессинг данного типа
происходит как у прокариотов, так и у эукариотов.
• Химическая модификация производится с рРНК, тРНК и мРНК. Молекулы рРНК
и тРНК всех организмов модифицируются путем добавления новых химических
групп, при этом эти группы присоединяются к определенным нуклеотидам в каж-
дой молекуле РНК. Химическая модификация мРНК, называемая редактированием
РНК, имеет место у многих эукариотов.
1.2.4. Транскриптом
Хотя транскриптом и составляет менее 4 % от общего количества РНК в клетке,
это наиболее значимый компонент, потому что в него входят молекулы кодирующей
РНК, которые используются на следующей стадии экспрессии генома. Важно обратить
внимание на то, что транскриптом никогда не синтезируется de novo. Каждая клетка
получает часть транскриптома своих материнских клеток при ее появлении на свет
в результате клеточного деления и поддерживает свой транскриптом на протяжении
всего периода жизни. Даже находящиеся в состоянии покоя клетки бактериальных спор
или семян растений имеют транскриптом, хотя трансляция этого транскриптома в белки
может быть полностью остановлена. Поэтому транскрипция отдельных кодирующих
белки генов не приводит к синтезу транскриптома, но вместо этого поддерживает транс-
криптом, заменяя деградировавшие молекулы мРНК, и вызывает изменения в составе
транскриптома через включение и выключение различных наборов генов.
Глава 1. Геномы, транскриптомы и протеомы 21
Даже у простейших организмов вроде бактерий и дрожжей многие гены активны
в любой момент времени. Поэтому транскриптомы очень сложны и содержат копии со-
тен, если не тысяч, различных молекул мРНК. Обычно молекулы мРНК того или иного
вида составляют лишь малую долю всей мРНК клетки, причем даже наиболее многочис-
ленная разновидность редко вносит более 1 % в общее количество мРНК. Исключения
представляют клетки, для которых характерна высокоспециализированная биохимия,
отраженная в транскриптомах, в которых преобладают мРНК одной или нескольких
разновидностей. Хороший пример — развивающиеся семена пшеницы: они в больших
количествах синтезируют белки-глиадины, которые накапливаются в дремлющем зерне
и обеспечивают источник аминокислот для прорастающих зародышей. В развивающихся
семенах молекулы мРНК, предназначенные для синтеза глиадинов, могут составлять
не менее 30 % транскриптома некоторых клеток.
1.3. Белки и протеом
Вторичный продукт экспрессии генома — протеом (см. рис. 1.2) — есть ассорти-
мент белков клетки, определяющий характер биохимических реакций, которые клетка
способна выполнять. Эти белки синтезируются путем трансляции молекул мРНК,
входящих в состав транскриптома.
1.3.1. Структура белка
Белок, подобно молекуле ДНК, представляет собой линейный, неразветвленный
полимер. Мономерные звенья белков называют аминокислотами (см. рис. 1.14), и об-
разующиеся из них полимеры, или полипептиды, редко бывают более 2 000 единиц
в длину. Как и в случае ДНК, основные характеристики структуры белка были опреде-
лены в первой половине двадцатого века, причем эта стадия биохимии белка достигла
высшей точки в 1940-х и в начале 1950-х гг., когда Полинг и Кори описали главные
конформации, или вторичные структуры, принимаемые полипептидами. В последние
годы интерес науки сфокусировался на вопросе о том, каким образом эти вторичные
структуры объединяются и образуют сложные, трехмерные формы белков.
Четыре уровня структуры белка
Структуры белков традиционно классифицируют на четыре уровня. Эти уровни
являются иерархическими, при этом белок строится поэтапно, и каждый уровень струк-
туры зависит от нижележащего:.
• Первичная структура белка образуется за счет объединения аминокислот в полипеп-
тид. Аминокислоты соединяются посредством пептидных связей, которые образуют-
СОО-
I
I
I
I
I
I
H3N— С — Н
Рис. 1.14. Общая структура аминокислот. Все аминокислоты име-
ют одинаковую общую структуру, включая центральный а-углерод, i
с которым соединены: атом водорода, карбоксильная группа, ами- 1
ногруппа и боковая группа, или радикал R. У аминокислот разных R
видов боковые группы различны (см. рис. 1.18)
I1 I2
H3N—С—СОО" + H3N—С—СОО"
н н
|*н20
R1 Н R2
I I I
H3N—С —С—- N—C—СОО“
N-конец | || | С-конец
НО н
I I
Пептидная связь
22 Часть 1. Изучение геномов
Рис. 1.15. В полипептидах аминокислоты соеди-
нены между собой пептидными связями. На ри-
сунке показана химическая реакция, в результате
которой две аминокислоты связываются вместе
пептидной связью. Такая реакция полимериза-
ции называется конденсацией, потому что в ре-
зультате ее протекания высвобождается вода
ся в ходе реакции полимеризации между
карбоксильной группой одной аминокис-
лоты и аминогруппой второй аминокис-
лоты (см. рис. 1.15). Обратите внимание,
что, как и в случае полинуклеотида, оба
конца полипептида химически различны: один несет на себе свободную аминогруппу
и называется амино-, NH2- или N-концом; другой имеет свободную карбоксильную
группу и называется карбоксильным, СООН- или С-концом. Поэтому направление
полипептида может быть выражено либо как N->C (на рис. 1.15—слева направо), либо
как C->N (на рис. 1.15 — справа налево).
• Ко вторичной структуре относятся различные конформации, которые может
принимать полипептид. Вторичные структуры двух главных типов — а-спираль
и [3-лист (см. рис. 1.16). Они стабилизируются, главным образом, водородными
связями, которые образуются между различными аминокислотами полипептида.
В большинстве своем полипептиды достаточно длинные и могут быть свернуты
в целый ряд вторичных структур, одна за другой по длине молекулы.
• Третичная структура обусловлена сворачиванием компонентов вторичной струк-
туры полипептида в трехмерную конфигурацию (см. рис. 1.17). Третичная структура
сохраняет свою устойчивость с помощью различных химических сил, в особенно-
сти — водородных связей между отдельными аминокислотами, электростатиче-
ских взаимодействий между боковыми группами заряженных аминокислот (см.
рис. 1.18) и гидрофобных сил, которые требуют, чтобы аминокислоты с неполяр-
ными («боящимися воды») боковыми группами были ограждены от воды путем
погружения их во внутренние области белковой молекулы. Кроме того, эту роль
могут выполнять ковалентные связи — дисульфидные мостики, — образуемые
между остатками аминокислоты цистеина, находящимися в различных позициях
последовательности полипептида.
• Четвертичная структура строится путем объединения двух и более полипепти-
дов (при этом каждый из них свернут в свою собственную третичную структуру)
в многосубъединичный белок. Не все белки образуют четвертичные структуры,
но это свойство присуще многим белкам со сложными функциями, в том числе не-
скольким, вовлеченным в экспрессию генома. Некоторые четвертичные структуры
сплачиваются дисульфидными мостиками между различными полипептидами,
что дает устойчивые многосубъединичные белки, которые не могут быть легко
разъединены на составные части. К другим четвертичным структурам относят-
ся более свободные объединения субъединиц, стабилизируемые водородными
связями и гидрофобными силами; это означает, что такие белки, в зависимости
Глава 1. Геномы, транскриптомы и протеомы 23
Рис. 1.16. Две главные единицы
вторичной структуры, обнаружи-
ваемые в белках: а) а-спираль
и б) 0-лист. Полипептидные цепи
изображены схематично. Боковые
группы не были показаны, с тем
чтобы не загромождать рисунок.
Устойчивость каждой из струк-
тур обеспечивается водородны-
ми (Н) связями между группами
С=О и N-H различных пептидных
связей. Представленная здесь
конформация 0-листа антипа-
ра ллельна, то есть две составляю-
щие его цепи бегут во встречных
направлениях. Существуют также
параллельные 0-листы
а) а-спираль
б) 0-лист
Водородная
N-конец
Рис. 1.17. Третичная структура
белка. Приведенная здесь во-
ображаемая структура белка
состоит из трех а-спиралей, по-
казанных в виде локонов, и четы-
рехнитевого 0-листа, обозначен-
ного стрелками
24 Часть 1. Изучение геномов
от функциональных требований клетки, могут возвращаться к составляющим их
полипептидам или изменять свой субъединичный состав.
В основе разнообразия белков лежит разнообразие аминокислот
Белки обладают богатым разнообразием функций. Это связано с тем, что аминокис-
лоты, из которых они построены, сами по себе химически разнообразны. Поэтому разные
последовательности аминокислот дают различные сочетания химических реакционных
способностей, причем эти комбинации определяют не только общую структуру собран-
ного из них белка, но также и расположение на поверхности его структуры химически
активных групп, которые обусловливают химические свойства белка.
Разнообразие аминокислот связано с природой боковых групп, потому что у всех
аминокислот именно эти части отличаются друг от друга и структура этих частей зна-
чительно варьирует. Белки состоят из набора 20-ти аминокислот (рис. 1.18; табл. 1.2).
Некоторые из них имеют боковые группы, которые представляют собой маленькие, отно-
сительно простые структуры, например, единственного атома водорода (в аминокислоте,
называемой глицином) или метильной группы (аланин). Другие боковые группы —
большие, сложные ароматические боковые цепи (фенилаланин, триптофан и тирозин).
В основном, этот набор представлен незаряженными аминокислотами, но две из них за-
ряжены отрицательно (аспарагиновая кислота и глутаминовая кислота), а три заряжены
положительно (аргинин, гистидин и лизин). Некоторые аминокислоты полярны (напри-
мер, глицин, серин и треонин), другие неполярны (например, аланин, лейцин и валин).
а) Неполярные боковые группы
СНз
I
Аланин
Н3С СНз
\н
I
Валин
НзС СНз
СН
I
СНз
I
Лейцин
СНз
I
СН,
I
НзС—СН
I
Изолейцин
НзС—СНз
/ \
HN хСНз
^С“
W ТОО-
СНз
I
S
I
СН,
I
СНз
I
Метионин
б) Полярные боковые группы
НО СНз SH
I I I
Н СНз НО—СН СНз
1111
Глицин Серин Треонин Цистеин
Прол ин Фенилаланин Триптофан
СНз СНз СНз
I I I
Тирозин Аспарагин Глутамин
в) Отрицательно заряженные боковые группы
V
к
I
Аспарагиновая
кислота
I
СНз
I
СН,
I
Глутаминовая
кислота
г) Положительно заряженные боковые группы
ЫНз
H3N i=N+Ha
I I
СНз NH
I I
ch, CH2 ^-7
I I HCZ I
СНз СНз N—C
I I H I
СНз CH3 СНз
I I I
Лизин Аргинин Гистидин
Рис. 1.18. Боковые группы аминокислот. Эти 20 аминокислот традиционно рассматриваются в качестве
определяемых генетическим кодом
Глава 1. Геномы, транскриптомы и протеомы 25
Рис. 1.19. Структуры селеноцистеина и пирро-
лизина. Части молекул, показанные коричне-
вым цветом, отражают различия между этими
аминокислотами и, соответственно, отличия от
цистеина и лизина
SeH
I
сн2
I
Селеноцистеин
Н
С—сн2
// \
N, X—СНз
\н н
I
с=о
I
NH
I
сн2
I
сн2
I
сн2
I
сн2
I
Пирролизин
Представленные на рис. 1.18 20 аминокислот традиционно рассматриваются в ка-
честве аминокислот, определяемых генетическим кодом (раздел 1.3.2). Именно эти ами-
нокислоты соединяются одна за другой при трансляции молекулы мРНК в белок. Однако
эти 20 аминокислот сами по себе не представляют предел химического разнообразия
белков. Разнообразие оказывается даже еще большим из-за двух факторов:
• по крайней мере две дополнительные аминокислоты — селеноцистеин и пирро-
лизин (рис. 1.19) — могут быть вставлены в полипептидную цепь в ходе синтеза
белка, причем их вставка обусловена модифицированным считыванием генети-
ческого кода (см. рис. 13.1.1);
• во время процессинга белка некоторые аминокислоты модифицируются добав-
лением новых химических групп (например, ацетилированием или фосфорили-
рованием) или присоединением крупных боковых цепей, состоящих их сахарных
звеньев (раздел 13.3.3).
Поэтому белки обладают огромным потенциалом химической изменчивости, одна
часть коего непосредственно определяется геномом, другая — проистекает из характера
процессинга белка.
1.3.2. Протеом
Протеом охватывает все белки, находящиеся в клетке в данное время. «Типичная»
клетка млекопитающих, например клетка печени (гепатоцит), как полагают, содержит
10 000-20 000 различных белков, всего около 8109 отдельных молекул, что составляет
приблизительно 0,5 нг белка или 18-20 % от общей массы клетки. Число копий отдель-
ных белков изменяется в чрезвычайно широких пределах, от менее чем 20 000 молекул
на клетку для белков самых редких типов до 100 миллионов копий для наиболее рас-
пространенных. Любой белок, число копий которого превышает 50 000 на одну клетку,
считают относительно избыточным, и в среднестатистической клетке млекопитающих
в эту категорию попадают около 2000 белков. При исследовании протеомов клеток
26
Часть 1. Изучение геномов
Таблица 1.2. Буквенные обозначения аминокислот
Аминокислота Обозначение
Трехбуквенное Однобуквенное
Аланин Ала А
Аргинин Apr R
Аспарагин Асн N
Аспарагиновая кислота Асп D
Валин Вал V
Гистидин Гис Н
Глицин Гл и G
Глутамин Глн Q
Глутаминовая кислота Глу Е
Изолейцин Иле I
Лейцин Лей L
Лизин Лиз К
Метионин Мет М
Пролин Про Р
Серин Сер S
Тирозин Тир Y
Треонин Тре Т
Триптофан Трп W
Фенилаланин Фен F
Цистеин Цис С
млекопитающих различных типов среди таких избыточных белков просматривается
очень мало различий; это предполагает, что большинство из них — вспомогательные
белки, которые исполняют обыденные биохимические отправления, происходящие во
всех клетках. Белки, которые придают клетке ее специализированную функцию, часто
оказываются весьма редкими, хотя встречаются исключения вроде огромных количеств
гемоглобина в красных кровяных тельцах.
Связь между транскриптомом и протеомом
Поток информации от ДНК к РНК, устанавливающийся в ходе транскрипции, не
представляет из себя никакой концептуальной трудности. Полинуклеотиды ДНК и РНК
имеют весьма подобные структуры, и мы можем легко понять, как РНК-копия гена
может быть получена путем направляемого матрицей синтеза, в котором учитываются
правила спаривания оснований, с которыми мы уже знакомы. Вторую стадию экспрессии
генома, в ходе которой молекулы мРНК транскриптома направляют синтез белков, по-
нять не так легко, если действовать аналогичным образом, то есть просто рассматривать
структуры молекул, которые в ней участвуют. В начале 1950-х гг., вскоре после того, как
была открыта структура двойной спирали ДНК, несколько молекулярных биологов по-
Глава 1. Геномы, транскриптомы и протеомы 27
пытались разработать схемы, согласно которым аминокислоты могли бы прикрепляться
к мРНК упорядоченным образом, но во всех этих моделях по крайней мере некоторые
из связей должны были быть короче или длиннее, чем допускали законы физической
химии, и посему каждая такая гипотеза незаметно уходила в небытие. В конечном счете,
в 1957 г., Фрэнсис Крик проложил путь через джунгли запутанных гипотез и предсказал
существование стыковочной молекулы-переходника, которая могла бы образовывать
мост между мРНК и синтезируемым полипептидом. Вскоре после этого стало ясно, что
такими стыковочными молекулами являются молекулы тРНК, и как только этот факт
был установлен, была быстро построена подробная модель механизма синтеза белков.
Мы исследуем этот процесс в разделе 13.1.
Другим моментом синтеза белка, интересовавшим молекулярных биологов
в 1950-х гг., была информационная проблема. Она относится ко второму важному
компоненту связи между транскриптомом и протеомом — генетическому коду, кото-
рый и определяет, как последовательность нуклеотидов мРНК транслируется в после-
довательность аминокислот белка. В 1950-х гг. было признано, что для описания всех
20-ти аминокислот, обнаруженных в белках, необходим триплетный генетический код —
такой, в котором каждое кодовое слово, или кодон, состояло бы из трех нуклеотидов.
Двухбуквенный код описывал бы только 42 = 16 кодонов, что недостаточно для кодиро-
вания всех 20-ти аминокислот, тогда как трехбуквенный код мог бы дать 43 = 64 кодона.
Генетический код был составлен в 1960-х гг. — частично путем анализа полипептидов,
являющихся результатом проводимой в бесклеточных системах трансляции искусствен-
ных молекул мРНК с известной или предсказуемой последовательностью, и частично
путем анализа на основе очищенных рибосом, в ходе которого определяли взаимное
соответствие последовательностей РНК и получаемых с них аминокислот. Когда эта
работа была закончена, стало ясно, что известные 64 кодона делятся на группы, и при
том члены каждой группы кодируют одну и ту же аминокислоту (рис. 1.20). Только
триптофан и метионин имеют лишь по одному кодону, все остальные аминокислоты
кодируются двумя, тремя, четырьмя или шестью кодонами. Эту особенность кода на-
зывают вырожденностью. Код имеет также четыре пунктуационных кодона, которые
указывают точки в пределах мРНК, в которых трансляция последовательности нуклео-
тидов должна начинаться и заканчиваться (рис. 1.21). Старт-кодоном обычно служит
триплет 5'-AUG-3', который определяет также метионин (так что большинство новых
синтезированных полипептидов начинается с метионина), хотя в нескольких мРНК для
этой цели употребляются другие кодоны, как то: 5'-GUG-3' и 5'-UUG-3’. Три стоп-кодона
представлены триплетами 5'-UAG-3', 5'-UAA-3' и 5'-UGA-3'.
Генетический код не универсален
Первоначально думалось, что генетический код должен быть один и тот же во всех
организмах. Довод состоял в том, что, будучи когда-то утвержден, код не мог бы более
изменяться, потому что предоставление нового значения любому отдельному кодону
привело бы к масштабным нарушениям аминокислотных последовательностей белков.
Это суждение представляется вполне обоснованным, так что кажется удивительным,
что в действительности генетический код не универсален. Код, показанный на рис. 1.20,
верен для подавляющего большинства генов в подавляющем большинстве организмов,
но отклонения от него широко распространены. В частности, митохондриальные геномы
часто используют нестандартный код (таблица 1.3). Это явление было впервые открыто
в 1979 г. группой Фредерика Сенгера в Кембридже, Великобритания, который обнару-
жил, что несколько митохондриальных мРНК человека содержат последовательность
28
Часть 1. Изучение геномов
иии иис Фен иси исс Ррп UAU UAC Тир UGU UGC Цис
UUA ПоГл UCA vcp UAA стпп UGA стоп
UUG леи UCG UAG Ь 1 UI1 UGG Трп
сии CCU CAU CGU
CUC Лей ССС Пгп САС Гис CGC Дпг
CUA ССА 1 ipu САА CGA “pi
CUG CCG CAG Глн CGG
AUU ACU AAU AGU
AUC Иле АСС Тпр ААС Асн AGC Сер
AUA АСА । рс ААА AGA
AUG Мет ACG AAG ЛИЗ AGG Apr
GUU GCU GAU GGU
GUC Do п GCC Ала GAC Асп GGC Гли
GUA DdJI GCA GAA Г п\/ GGA
GUG GCG GAG 1 Лу GGG
Рис. 1.20. Генетический код. Аминокислоты обозначены стандартными трехбуквенными обозначениями
(см. табл. 1.2)
5'-UGA-3', которая обычно кодирует завершение трансляции, во внутренних позициях,
где синтез белка, как следует ожидать, не должен останавливаться. Сравнения с ами-
нокислотными последовательностями белков, кодируемых этими мРНК, показали, что
в митохондриях человека триплет 5'-UGA-3' является кодоном триптофана и что данное
отклонение является лишь одним из четырех отклонений кода, наблюдаемых в этой
генетической системе. Митохондриальные гены в других организмах также показывают
отклонения кода, хотя по крайней мере одно из них — использование триплета 5'-CGG-3'
в качестве кодона триптофана в митохондриях растений, — вероятно, корректируется
в ходе редактирования РНК (раздел 12.2.5), прежде чем происходит трансляция.
Нестандартные коды известны также и для ядерных геномов низших эукариотов.
Зачастую модификация ограничена только в пределах маленькой группы организмов,
и нередко она вовлекает переназначение стоп-кодонов (таблица 1.3). В среде прокарио-
тов модификации менее распространены, но известен один пример у вида Mycoplasma.
ирнк
I I
Кодон начала Кодон окончания
(обычно AUG) (UAA, UAG или UGA)
Более важный тип видоизменения
кода — контекстнозависимое пере-
назначение кодонов, каковое происхо-
дит, когда белок, который должен быть
синтезирован, содержит селеноцистеин
или пирролизин. Белки, содержащие
пирролизин, редки и, вероятно, присут-
ствуют только в группе прокариотов,
называемой археями (глава 8), но селе-
нопротеины широко распространены во
многих организмах (например, фермент
Рис. 1.21. Позиции пунктуационных кодонов в мРНК глутатионпероксидаза, которая помога-
Глава 1. Геномы, транскриптомы и протеомы 29
Таблица 1.3. Примеры отклонений от стандартного генетического кода
Организм Кодон Должен кодировать Фактически кодирует
Митохондриальные геномы
Млекопитающие UGA стоп Трп
AGA, AGG Apr стоп
AUA Иле Мет
Drosophila UGA стоп Трп
AGA Apr Сер
AUA Иле Мет
Saccharomyces cerevisiae UGA стоп Трп
CUN Лей Тре
AUA Иле Мет
Грибы UGA стоп Трп
Кукуруза CGG Apr Трп
Ядерные геномы и геномы прокариотов
Различные протисты UAA, UAG стоп Глн
Candida cylindracea CUG Лей Сер
Micrococcus sp. AGA Apr стоп
AUA Иле стоп
Euplotes sp. UGA стоп Цис
Mycoplasma sp. UGA стоп Трп
CGG Apr стоп
Контекстнозависимые переназначения кодонов
Различные UGA стоп Селеноцистеин
Археи UAG стоп Пирролизин
Примечание: буква N означает любой нуклеотид.
ет защищать клетки людей и других млекопитающих от окислительного повреждения).
Селеноцистеин кодируется триплетом 5’-UGA-3', а пирролизин — кодоном 5'-UAG-3'.
Таким образом, эти кодоны имеют двойное значение, потому что в рассмотренных ор-
ганизмах они, как обычно, используются в качестве стоп-кодонов (таблица 1.3). Кодон
5’-UGA-3’, который определяет селеноцистеин, отличается от истинных стоп-кодонов
наличием структуры типа шпильки в мРНК, помещенной у прокариотов непосредственно
ниже кодона селеноцистеина, а у эукариотов — в З'-нетранслируемой области (то есть
в части мРНК после стоп-кодона). Опознавание кодона селеноцистеина требует взаи-
модействия между шпилечной структурой и специальным белком, который вовлечен
в трансляцию таких мРНК. По всей вероятности, аналогичная система работает и для
определения пирролизина.
30
Часть 1. Изучение геномов
Связь между протеомом и биохимией клетки
Биологическая информация, кодируемая геномом, находит свое заключительное
выражение в белке, биологические свойства которого определяются его свернутой
структурой и пространственным расположением химических групп на его поверхности.
Путем определения белков различных типов геном способен строить и поддерживать
протеом, полные биологические свойства которого формируют фундаментальное осно-
вание жизни. Протеом способен играть эту роль благодаря огромному разнообразию
белковых структур, которые могут быть сформированы, разнообразию, позволяющему
белкам выполнять целый спектр различных биологических функций. Эти функции
можно разбить на следующие категории.
• Биохимический катализ — это роль белков особого типа, называемых фермен-
тами. Ферментами катализируются центральные метаболические пути, которые
снабжают клетку энергией, равно как и процессы биосинтеза, в результате кото-
рых образуются нуклеиновые кислоты, белки, углеводы и жиры. Биохимический
катализ также ведет последовательную экспрессию генома через согласованные
действия ферментов наподобие РНК-полимеразы.
• Формирование структуры, которая на клеточном уровне определяется белками,
составляющими клеточный скелет, является первостепенной функцией также
и некоторых внеклеточных белков. Пример — коллаген, который является важным
компонентом костей и сухожилий.
• Движение — осуществляется сократительными белками, из которых самыми из-
вестными примерами являются актин и миозин в волокнах клеточного скелета.
• Транспорт материалов по всему телу — важная деятельность белков: например,
гемоглобин переносит кислород в кровотоке, а альбумин сыворотки переносит
жирные кислоты.
• Регулирование клеточных процессов — опосредствуется сигнальными белками
типа STAT (преобразователи сигналов и активаторы транскрипции, раздел 14.1.2)
и белками типа активаторов, которые связываются с геномом и влияют на уровни
экспрессии как отдельных генов, так и групп генов (раздел 11.3). Действия групп
клеток регулируются и координируются внеклеточными гормонами и цитокинами,
многие из которых — белки (например, инсулин — гормон, который управляет
уровнями сахара в крови, а интерлейкины — группа цитокинов, которые регули-
руют деление и дифференцировку клетки).
• Защита тела и отдельных клеток — функция ряда белков, в том числе антител
и белков, участвующих в свертывании крови.
• Функции хранения — выполняются белками типа ферритина, который действует
как склад железа в печени, и глиадинов, которые запасают аминокислоты в дрем-
лющих семенах пшеницы.
Такое разнообразие функций белка и придает протеому его способность преоб-
разовать проект, заложенный в геноме, в существенные особенности процесса жизни.
Заключение
Геном — хранилище биологической информации, которой наделен каждый орга-
низм, живущий на нашей планете. Подавляющее большинство геномов состоит из ДНК,
за малым исключением в лице тех вирусов, которые имеют геномы из РНК. Экспрессия
Глава 1. Геномы, транскриптомы и протеомы 31
генома — процесс, в ходе которого информация, содержащаяся в геноме, выражается
в клетке. Первичным продуктом экспрессии генома является транскриптом — совокуп-
ность молекул РНК, полученных из кодирующих белок генов, которые являются актив-
ными в клетке в определенное время. Вторичный продукт — протеом — представляет
собой ассортимент белков клетки, определяющих характер биохимических реакций,
которые клетка способна выполнять. Экспериментальные данные, показывающие, что
гены состоят из ДНК, впервые были получены между 1945-м и 1952-м гг., но именно
открытие структуры двойной спирали Уотсоном и Криком в 1953 г. убедило биологов
в том, что ДНК действительно является генетическим материалом. Полинуклеотид
ДНК — неразветвленный полимер, состоящий из многочисленных копий четырех хими-
чески различных нуклеотидов. В двойной спирали два полинуклеотида закручиваются
один вокруг другого, при этом находящиеся в нуклеотидах основания располагаются на
внутренней части молекулы. Полинуклеотиды соединены водородными связями между
основаниями, причем А всегда спаривается с Т, a G всегда образует пару с С. РНК также
представляет собой полинуклеотид, но, во-первых, отдельные его нуклеотиды отлича-
ются по структуре от встречающихся в ДНК нуклеотидов, а во-вторых, молекулы РНК
обычно однонитевые. Копированием генов в молекулы РНК занимаются ДНК-зависимые
РНК-полимеразы в ходе процесса, названного транскрипцией, в результате которого
синтезируется не только транскриптом, но также и ряд функциональных молекул
РНК, которые не кодируют белки, но тем не менее играют в клетке жизненно важные
роли. Многие РНК первоначально синтезируются в виде молекул-предшественников,
которые обрабатываются реакциями разрезания и соединения, а также химическими
модификациями, после чего получаются их зрелые формы. Белки также представляют
собой неразветвленные полимеры, но в них элементарными структурными единицами
являются аминокислоты, сцепленные между собой пептидными связями. Последова-
тельность аминокислот — первичная структура белка. Более высокие уровни струк-
туры — вторичная, третичная и четвертичная — формируются путем сворачивания
первичной структуры в трехмерные конформации и последующей ассоциации отдель-
ных полипептидов в многобелковые структуры. Белки функционально разнообразны,
потому что отдельные аминокислоты имеют различные химические свойства, что при
сочетании различными способами дает белки с широким диапазоном химических осо-
бенностей. Белки синтезируются посредством трансляции мРНК, в ходе которой правила
генетического кода определяют, какую аминокислоту будет кодировать каждый три-
плет нуклеотидов. Генетический код не универсален, его разновидности встречаются
в митохондриях и у низших эукариотов, а некоторые кодоны в отдельно взятом гене
могут иметь два разных значения.
32
Часть 1. Изучение геномов
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
1.1 *. Какое из следующих утверждений
о геноме организма является ЛОЖ-
НЫМ?
а. Геном содержит генетическую ин-
формацию для построения и под-
держания живого организма.
Ь. Геномы клеточных организмов
состоят из ДНК.
с. Геном способен экспрессировать
заложенную в нем информацию
без участия ферментов и белков.
d. Геномы эукариотов состоят из
ядерной и митохондриальной
ДНК.
1.2 . К соматическим клеткам относят
те, что:
а. Содержат гаплоидный набор хро-
мосом.
Ь. Порождают гаметы.
с. Не имеют митохондрий.
d. Содержат диплоидный набор хро-
мосом и составляют большинство
клеток человека.
1.3 *. Которое из следующихутверждений
описывает поток генетической ин-
формации в клетках?
а. ДНК транскрибируется в РНК,
которая затем транслируется
в белок.
Ь. ДНК транслируется в белок, ко-
торый затем транскрибируется
в РНК.
с. РНК транскрибируется в ДНК,
которая затем транслируется
в белок.
d. Белки транслируются в РНК, ко-
торая затем транскрибируется
в ДНК.
1.4 . В начале двадцатого столетия дума-
ли, что белки, в принципе, могут не-
сти генетическую информацию. На
основе каких из следующих данных
было сделано это заключение?
а. Хромосомы состоят из приблизи-
тельно равных количеств белка
и ДНК.
Ь. Было известно, что белки состоят
из 20-ти различных аминокислот,
тогда как ДНК состоит только из
4-х нуклеотидов.
с. Различные белки, как было извест-
но, имеют уникальные последова-
тельнослитогда как все молекулы
ДНК, как думали, имеют одну и ту
же последовательность.
d. Все вышеперечисленное.
1.5 *. Связями какого типа отдельные ну-
клеотиды молекулы ДНК соединены
друг с другом?
а. Гликозидными.
Ь. Пептидными.
с. Фосфодиэфирными.
d. Электростатическими.
1.6 . Какие из следующих методов Уотсон
и Крик активно использовали в раз-
гадке структуры ДНК?
а. Построение моделей молекул
ДНК с тем, чтобы убедиться в пра-
вильном взаимном расположении
атомов.
Ь. Рентгеноструктурный анализ мо-
лекул ДНК.
с. Хроматографические исследова-
ния с целью определить относи-
тельный состав нуклеотидов ДНК
из различных препаратов.
d. Генетические исследования, ко-
торые продемонстрировали, что
генетическим материалом явля-
ется ДНК.
Глава 1. Геномы, транскриптомы и протеомы 33
1.7 *. Эрвин Чаргафф изучал ДНК различ-
ных организмов и показал, что:
а. ДНК является генетическим ма-
териалом.
Ь. РНК транскрибируется с ДНК.
с. Количество аденина в данном
организме равно количеству ти-
мина (а количество гуанина — ко-
личеству цитозина).
d. Двойная спираль скрепляется
в единый тяж водородными свя-
зями между основаниями.
1.8 . Определение транскриптома клетки
формулируется как:
а. Все молекулы РНК, присутствую-
щие в клетке.
Ь. Кодирующие белок молекулы
РНК, присутствующие в клетке.
с. Молекулы рибосомной РНК, при-
сутствующие в клетке.
d. Молекулы транспортной РНК,
присутствующие в клетке.
1.9 *. Как ДНК-зависимые РНК-полиме-
разы выполняют синтез РНК?
а. Они используютДНК как матрицу
для полимеризации рибонуклео-
тидов.
Ь. Они используют белки как матри-
цу для полимеризации рибону-
клеотидов.
с. Они используют РНК как матрицу
для полимеризации рибонуклео-
тидов.
d. Им не требуется никакой матри-
цы для полимеризации рибону-
клеотидов.
1.10 . Функциональная РНК какого типа яв-
ляется главным компонентом структур,
необходимых для синтеза белка?
а. Матричная РНК.
Ь. Рибосомная РНК.
с. Малая ядерная РНК.
d. Транспортная РНК.
1.11 *. Определение протеома клетки зву-
чит как:
а. Все белки, которые клетка способ-
на синтезировать.
Ь. Все белки, присутствующие в клет-
ке на протяжении всей ее жизни.
с. Все белки, представленные в клет-
ке в данный момент времени.
d. Все белки, которые активно син-
тезируются в клетке в данный
момент времени.
1.12 . Какой уровень структуры белка
описывает свернутую конформацию
многосубъединичного белка?
а. Первичная структура.
Ь. Вторичная структура.
с. Третичная структура.
d. Четвертичная структура.
1.13 *. Ковалентные связи какого типа
соединяют остатки цистеина, рас-
положенные в различных местах
полипептида?
а. Дисульфидный мостик.
Ь. Водородная связь.
с. Пептидная связь.
d. Фосфодиэфирная связь.
1.14 . Считается, что в большинстве своем
избыточные белки в клетке явля-
ются вспомогательными белками.
Какова же их функция?
а. Они ответственны за специфиче-
ские функции клеток определен-
ных типов.
Ь. Они ответственны за регулиро-
вание экспрессии генома в клет-
ках.
с. Они ответственны за удаление от-
ходов метаболизма из клеток.
d. Они ответственны за общие био-
химические действия, которые
происходят во всех клетках.
1.15 *. Которое из следующих утверждений
относится к вырожденности генети-
ческого кода?
а. Каждый кодон может определять
более одной аминокислоты.
Ь. Большинство аминокислот имеет
более одного кодона.
с. Есть несколько старт-кодонов.
d. Стоп-кодоны могут кодировать
также и аминокислоты.
34
Часть 1. Изучение геномов
1.16 . Какая из следующих функций бел-
ков не относится к биологическим?
а. Биологический катализ.
Ь. Регулирование клеточных про-
цессов.
Вопросы открытого типа
1.1 *. Расположите во времени события:
открытие ДНК, открытие того факта,
что ДНК является генетическим ма-
териалом, открытие структуры ДНК
и расшифровка первого генома.
1.2 . Химические взаимодействия каких
двух типов стабилизируют двойную
спираль?
1.3 *. Почему специфичное спаривание
между А и Т, а также между G и С дает
гарантию высокой точности репли-
кации ДНК?
1.4 . Каковы два важных химических от-
личия между РНК и ДНК?
1.5 *. Почему молекул ы мРНК имеют корот-
кие периоды полураспада по сравне-
нию с другими молекулами РНК?
1.6 . Действительно ли мРНК претер-
певает трансляцию в той же самой
форме, в какой она синтезируется
с матрицы ДНК?
1.7 *. Испытывают ли когда-либо клетки
недостаток в транскриптоме? Обо-
снуйте ваш ответ.
Изыскательские задачи
1.1 *. В тексте (страница 11) есть утверж-
дение, что Уотсон и Крик открыли
структуру двойной спирали ДНК
в субботу 7 марта 1953 г. Обоснуйте
это утверждение.
1.2 . Подумайте, почему двойная спираль
незамедлительно приобрела всеоб-
щее признание как верная структура
ДНК.
с Перенос генетической информа-
ции.
d. Транспорт молекул в многокле-
точных организмах.
1.8. В чём состоит важная роль водо-
родных связей, электростатических
взаимодействий и гидрофобных сил
во вторичной, третичной и четвер-
тичной структуре белков?
1.9* . За счет чего белки могут иметь так
много разнообразных структур
и функций, когда все они синтези-
руются только из 20-ти аминокис-
лот?
1.10. В дополнение к наличию известных
20-ти аминокислот белки имеют до-
бавочное химическое разнообразие
из-за двух факторов. Каковы эти два
фактора и каково их значение?
1.11* . Каким образом кодон 5’-UGA-3'
может функционировать и как
стоп-кодон, и как кодон для видо-
измененной аминокислоты селено-
цистеина?
1.12. Как геном управляет биологической
активностью клетки?
1.3*. Какие эксперименты привели к откры-
тию генетического кода в 1960-е гг.?
1.4. На транскриптом и протеом смотрят
как соответственно на промежуточ-
ный и наконечный продукт экспрес-
сии генома. Оцените обоснованность
и ограничения этих терминов в ра-
курсе нашего понимания экспрессии
генома.
Глава 1. Геномы, транскриптомы и протеомы 35
Тесты по рисункам
1.1*. Обсудите, как каждый из этих экспериментов помог продемонстрировать, что
именно ДНК, а не белки, содержит генетическую информацию.
а) Трансформирующее начало
б) Эксперимент Херши-Чейз
Безвредные бактерии
Мышь выживает
Безвредные бактерии + Мышь умирает
трансформирующее начало
Безвредные бактерии + Мышь умирает
трансформирующее начало,
обработанное протеазой
с . ДНК
Белковый капсид
Фаг, прикрепленный
к бактерии
| Перемешивание в мешалке
А
A CD
CD А Фаги отсоединены
или рибонуклеазой
Безвредные бактерии + Мышь выживает
трансформирующее начало,
обработанное дезоксирибонуклеазой
। Центрифугирование
Бактериальный
осадок “
70 % 32Р
20 %
1.2. Найдите дезоксирибозу, фосфатные группы и различные азотистые основания.
Можете ли вы определить в дезоксирибозе атомы углерода с 1-го по 5-й?
О“
О-
О-
l-y I р 1а
“О-Р-О-Р-О-Р-О-
II II II
ООО
N
nh2
I
NH2
nh2
36
Часть 1. Изучение геномов
Дополнительная литература
Книги и статьи об открытии двойной спирали и других важных вехах в изу-
чении ДНК
Brock, T.D. (1990) The Emergence of Bacterial Genetics. Cold Spring Harbor Laboratory
Press, New York. A detailed history that puts into context the work on the transforming principle
and the Hershey-Chase experiment.
Judson, H.F. (1979) The Eighth Day of Creation. Jonathan Cape, London. A highly readable
account of the development of molecular biology up to the 1970s.
Kay,L. E. (1993) The Molecular Vision of Life. Oxford University Press, Oxford. Contains
a particularly informative explanation of why genes were once thought to be made of protein.
Lander, E.S. and Weinberg, R. A. (2000) Genomics: journey to the center of biology.
Science 287:1777-1782. A brief description of genetics and molecular biology from Mendel to
the human genome sequence.
Maddox, B. (2002) Rosalind Franklin: The DarkLadyofDNA. HarperCollins, London.
McCarty, M. (1985) The Transforming Principle: Discovering that Genes are Made ofDNA.
Norton, London.
Olby,R. (1974) The Path to the Double Helix. Macmillan, London A scholarly account of the
research that led to the discovery of the double helix.
Watson, J. D. (1968) The Double Helix. Atheneum, London. The most important discovery
of twentieth century biology, written as a soap opera.
Крик Ф. Безумный поиск. М.-Ижевск: РХД, 2004.
Глава 2
Изучение ДНК
По прочтении главы 2 вы сможете:
• Описывать операции, выполняемые в ходе клонирования ДНК и полимеразной
цепной реакции (ПЦР), и называть области применения и досадные ограничения
этих методов.
• Перечислять действия и охарактеризовывать основное назначение ферментов
различного типа, используемых в исследовании рекомбинантной ДНК.
• Идентифицировать важные особенности ДНК-полимераз и проводить разли-
чие между разными ДНК-полимеразами, применяемыми в исследованиях под
эгидой геномики.
• Описывать, с примерами, способ, которым эндонуклеазы рестрикции разрезают
ДНК, и объяснять, как изучают результаты переваривания рестриктазами.
• Проводить различие между лигированием тупых и липких концов и объяснять,
как эффективность лигирования тупых концов может быть увеличена.
• Описывать (достаточно подробно) ключевые особенности плазмидных клони-
рующих векторов и пояснять, как эти векторы используются в экспериментах
с клонированием.
• Охарактеризовывать применение векторов на бактериофаге X для клонирова-
ния ДНК.
• Приводить примеры векторов, используемых для клонирования длинных ку-
сков ДНК, и оценивать преимущества и ограничения векторов каждого из обо-
значенных типов.
• Озвучивать основные моменты клонирования ДНК у дрожжей, животных и рас-
тений.
• Описывать технологию проведения ПЦР, обращая особое внимание на значе-
ние затравок и температур, используемых во время воздействия тепловых
циклов.
Фактически все, что мы знаем о геномах и экспрессии генома, было открыто экс-
периментально: теоретические изыскания сыграли очень небольшую роль в этой или
любой другой области молекулярной и клеточной биологии1). В принципе, можно изу-
чать «факты» о геномах, не зная многого о том, как эти факты были получены, но если
мы хотим прийти к настоящему пониманию данного предмета, мы должны подробно
ознакомиться с методами и научными подходами, которые использовались для изучения
геномов. Следующие пять глав охватывают эти методы исследования. Сначала мы об-
ратимся к методам, основанным на клонировании ДНК и полимеразной цепной реакции.
Эти методы очень эффективны при работе с короткими сегментами ДНК, в том числе
с отдельными генами, и позволяют получать массу информации на этом уровне. В главе 3
Это не совсем верно — теоретические исследования геномов позволили сделать ряд важных и неожи-
данных открытий, которые впоследствии получили экспериментальное подтверждение. — Прим. ред.
38 Часть 1. Изучение геномов
мы перейдем к методам, разработанным для построения карт геномов, и увидим, как
методы генетического картирования, впервые развитые почти столетие назад, были
обогащены дополнительными методами для физического картирования геномов. Глава 4
устанавливает связь между картированием и секвенированием и показывает, что, хотя
карта и может служить ценным помощником при сборке длинной последовательности
ДНК, картирование не всегда является необходимой предпосылкой к секвенированию
генома. В главе 5 мы рассматриваем различные подходы, которые используются для ис-
толкования последовательности генома, а в главе 6 — исследуем методы, применяемые
для изучения экспрессии генома. По мере чтения главы 6 вы начнете осознавать, что
понимание того, как геном определяет биохимические возможности живой клетки, есть
одна из главных исследовательских задач в современной биологии.
Инструментарий методов, употребляемых молекулярными биологами для изуче-
ния молекул ДНК, был собран в течение 1970-х и 1980-х гг. До того времени единственным
путем, позволявшим изучать отдельные гены, была классическая генетика, имеющая
в своем арсенале процедуры, которые мы встретим в главе 3. Развитие более прямых
методов изучения ДНК стимулировалось крупными достижениями в области биохимиче-
ских исследований, благодаря чему в начале 1970-х гг. молекулярные биологи приобрели
ферменты, которые они теперь могли использовать для манипулирования молекулами
ДНК в пробирке. В естественном виде эти ферменты синтезируются в живых клетках
и вовлекаются в такие процессы, как репликация, репарация и рекомбинация ДНК,
которые мы встретим в главах 15,16 и 17. Для определения функций этих ферментов
многие из них были очищены, а реакции, которые они катализируют, — досконально
изучены. После этого молекулярные биологи приняли эти чистые ферменты на воору-
жение как инструменты для того, чтобы управлять молекулами ДНК предопределен-
ным образом — делать их копии, разрезать их на более короткие фрагменты и снова
соединять их вместе, но уже в комбинациях, не существующих в природе (рис. 2.1). Такие
манипуляции лежат в основе технологии рекомбинантной ДНК, в которой новые,
или «рекомбинантные», молекулы ДНК строятся из частей встречающихся в природе
хромосом и плазмид.
Методология рекомбинантной ДНК привела к развитию клонирования ДНК, или
клонирования генов, в котором короткие фрагменты ДНК, возможно, содержащие един-
ственный ген, встраиваются в хромосому плазмиды или вируса и затем реплицируются
в организме хозяина — бактерии или эукариота (рис. 2.2). В разделе 2.2 мы подробно
опишем, как выполянется клонирование гена и укажем причины, благодаря которым
этот метод произвел революцию в молекулярной биологии.
Гены
Копирование Разрезание Перестановка
Рис. 2.1. Примеры манипуляций, которые могут быть выполнены с молекулами ДНК
Глава 2. Изучение ДНК 39
Рис. 2.2. Клонирование ДНК. В этом примере предназначенный для
клонирования фрагмент ДНК встраивается в плазмидный вектор,
который затем последовательно реплицируется внутри бактерии-
хозяина
ДНК плазмиды
Встройка новой ДНК
Клонирование гена стало распространенным экс-
периментальным методом к концу 1970-х гг. Следующее
главное крупное техническое достижение появилось в се-
редине 1980-х гг., когда была изобретена полимеразная
цепная реакция (ПЦР). ПЦР представляет собой неслож-
ный метод: все, чего он достигает, — это многократное
копирование короткого сегмента молекулы ДНК (рис. 2.3),
однако эта реакция приобрела огромное значение для
многих областей биологического исследования, и не в по-
следнюю очередь — в изучении геномов. Подробно ПЦР
будет рассмотрена в разделе 2.3.
I
Новая ДНК
Репликация в бактериях
Бактериальная 1
культура \
Ген
Полимеразная цепная реакция (ПЦР)
Рис. 2.3. Полимеразная цепная реакция (ПЦР) применяется для получения копий отобранного сегмента
молекулы ДНК. В этом примере копируется один-единственный ген
2.1. Ферменты для манипуляций с ДНК
Технология рекомбинантной ДНК была одним из главных факторов, которые
внесли вклад в быстрый прогресс знания об экспрессии генов, произошедший в течение
1970-х и 1980-х гг. Основа технологии рекомбинантной ДНК — способность управлять
молекулами ДНК в пробирке. Это, в свою очередь, зависит от наличия очищенных
ферментов (их действия известны и поддаются управлению), каковые поэтому могут
быть использованы для привнесения заданных изменений в молекулы ДНК, которыми
манипулирует биолог. Ферменты, доступные молекулярному биологу, делятся на четыре
широкие категории.
• ДНК-полимеразы (раздел 2.1.1) представляют собой ферменты, которые синте-
зируют новые полинуклеотиды, комплементарные существующей матрице ДНК
или РНК (рис. 2.4, а).
• Нуклеазы (раздел 2.1.2) расщепляют молекулы ДНК, разрывая фосфодиэфирные
связи, соединяющие один нуклеотид со следующим (рис. 2.4, б).
40 Часть 1. Изучение геномов
• Лигазы (раздел 2.1.3) соединяют молекулы ДНК друге другом, синтезируя фосфо-
диэфирные связи между нуклеотидами или на концах двух разных молекул, или
на двух концах одной и той же молекулы (рис. 2.4, в).
• Ферменты модификации концов (раздел 2.1.4) производят изменения на концах
молекул ДНК, благодаря чему открывают в проекте экспериментов с лигированием
новое дополнительное измерение и выступают одним из средств мечения молекул
ДНК радиоактивными и другими маркёрами (техническое примечание 2.1).
2.1.1. ДНК-полимеразы
Многие из методов, применяемых для изучения ДНК, зависят от синтеза ДНК-копий
всех или части имеющихся в наличии молекул ДНК или РНК. Это требование существен-
но для ПЦР (раздел 2.3), секвенирования ДНК (раздел 4.1), мечения ДНК (техническое
примечание 2.1) и многих других процедур, занимающих центральное место в иссле-
дованиях молекулярной биологии. Фермент, который синтезирует ДНК, называют
ДНК-полимеразой, а тот, который копирует существующую молекулу ДНК или РНК,
называют направляемой матрицей ДНК-полимеразой (ДНК- или РНК-зависимой
ДНК полимеразой).
а) ДНК-полимеразы
Матрица ДНК Матрица РНК
.............. ДНК-копии
б) Нуклеазы
I—
Эндонуклеаза
I
Экзонуклеаза
Внутренние разрезы Нуклеотиды удаляются с концов
в) Лигазы
Одна молекула ДНК Две молекулы ДНК
Рис. 2.4. Действия: а) ДНК-полимераз, б) нуклеаз и в) лигаз, а) Деятельность ДНК-зависимой ДНК-полимеразы
показана слева, а деятельность РНК-зависимой ДНК-полимеразы — справа, б) Действия эндонуклеаз и экзо-
нуклеаз. в) Серая молекула ДНК лигируется сама с собой (слева) и со второй молекулой ДНК (справа)
Глава 2. Изучение ДНК 41
Техническое примечание 2.1: мечение ДНК
Прикрепление к молекулам ДНК радиоактивных, флюоресцентных или
иного типа маркёров
Мечение ДНК лежит в основе многих процедур молекулярной биологии, вклю-
чая гибридизацию по Саузерну (раздел 2.1.2), флюоресцентную гибридизацию
in situ (FISH; раздел 3.3.2) и секвенирование ДНК (раздел 4.1). Оно позволяет
определять местоположение специфической молекулы ДНК — на нитроцеллю-
лозной или нейлоновой мембране, в хромосоме или в геле — путем обнаруже-
ния сигнала, испускаемого тем или иным маркёром. В некоторых приложени-
ях используются также меченые молекулы РНК (техническое примечание 5.1).
Для того чтобы метить молекулы ДНК, часто используются радиоактивные
маркёры. Можно синтезировать нуклеотиды, в которых один из атомов фосфора
будет заменен на 32Р или на 33Р, либо один из атомов кислорода в фосфатной группе
будет заменен на 35S, либо же один или несколько атомов водорода будут заменены
на 3Н (см. рис. 1.4). Такие радиоактивные нуклеотиды также служат субстратом
для ДНК-полимераз и таким образом встраиваются в молекулу ДНК посредством
любой реакции синтеза нити, катализируемой ДНК-полимеразой. Кроме того, по-
меченные нуклеотиды или отдельные фосфатные группы могут быть прицеплены
к одному или обоим концам молекулы ДНК в ходе реакций, катализируемых Т4-
полинуклеотидкиназой или концевой дезоксинуклеотидилтрансферазой (раз-
дел 2.1.4). Радиоактивный сигнал может быть обнаружен счетом сцинтилляций, но
для большинства экспериментов молекулярной биологии необходима позиционная
информация, так что обнаружение сигнала осуществляется экспозицией чувстви-
тельной к рентгеновскому излучению пленки (авторадиография1^: см. пример на
рис. 2.11) или чувствительного к радиации фосфоресцирующего экрана (фосфорес-
центная визуализация). Выбор между различными радиоактивными метками за-
висит от требований процедуры. Изотоп 32Р дает высокую чувствительность, потому
что он обладает высокой энергией эмиссии, но хорошая чувствительность сопрово-
ждается низким разрешением из-за рассеивания сигнала. Изотопы низкой эмиссии
типа 35S или 3Н дают меньшую чувствительность, но зато большее разрешение.
Требования охраны здоровья и окружающей среды привели к тому, что в по-
следние годы радиоактивные маркёры стали менее популярными и для многих
процедур они теперь в значительной степени заменены нерадиоактивными альтер-
нативами. Наиболее полезные из них — флюоресцентные маркёры, которые явля-
ются основными компонентами методов типа FISH (раздел 3.3.2) и секвенирования
ДНК (раздел 4.1.1). Флюоресцентные метки с различными длинами волны эмиссии
(то есть, различных цветов) встраиваются в нуклеотиды или присоединяются не-
посредственно к молекулам ДНК и детектируются подходящей пленкой, флюорес-
Данный метод изредка встречается также и под именем радиоавтографии, но термин «автора-
диография» в большей мере отражает суть этого метода, поскольку принято располагать приставки,
обозначающие сущностные (радио) качества объекта при корне слова, а все указывающие обстоятель-
ственные (авто) признаки—дальше от него. По сути это метод регистрации распределения радиоактив-
ных веществ в исследуемом объекте. Пленка с чувствительной к радиоактивному излучению эмульсией
накладывается на поверхность (срез). Радиоактивные вещества как бы сами себя фотографируют (авто-
матически). Места почернения на пленке после проявления соответствуют локализации радиоактивных
частиц. Используется в биологии, медицине, технике. — Прим, перев.
42
Часть 1. Изучение геномов
центной микроскопией или датчиком флюоресценции. Способы нерадиоактивного
мечения других типов основаны на хемилюминесцентной эмиссии, но они имеют то
неудобство, что сигнал не генерируется непосредственно меткой, но вместо этого
должен быть «проявлен» обработкой меченой молекулы химикалиями. В наиболее
популярном из таких методов используется мечение ДНК ферментом щелочной фос-
фатазой, которая детектируется с применением производных диоксетана. которые
этим ферментом дефосфорилируются с возбуждением хемилюминесценции.
Механизм действия направляемой матрицей полимеразы
Направляемая матрицей ДНК-полимераза синтезирует новый полинуклеотид ДНК,
последовательность которого предписывается через правила спаривания оснований
нуклеотидной последовательностью копируемой молекулы ДНК или РНК (рис. 2.5).
Новый полинуклеотид всегда синтезируется в направлении 5’->3': ДНК-полимеразы,
которые производили бы ДНК в другом направлении, неизвестны в природе.
Важная особенность направляемого матрицей синтеза ДНК состоит в том, что
ДНК-полимераза неспособна использовать в качестве матрицы полностью однонитевую
молекулу. Чтобы начать синтез ДНК, этот фермент должен иметь под рукой короткую
двунитевую область на З'-конце, к которой (с уже ее З'-конца) он будет присоединять
новые нуклеотиды (рис. 2.6, а). Способ удовлетворения этой потребности в живых клет-
ках при репликации генома описан в главе 15. В пробирке реакция копирования ДНК
инициируется прикреплением к матрице короткого синтетического олигонуклеотида,
как правило, длины приблизительно 20 нуклеотидов, который и служит затравкой для
синтеза ДНК. На первый взгляд, потребность в затравке могла бы показаться нежеланным
осложнением, связанным с применением ДНК-полимераз в технологии рекомбинантной
ДНК, но это было бы глубочайшим заблуждением. Поскольку отжиг затравки к матрице
зависит от комплементарного спаривания оснований, позиция в пределах молекулы
матрицы, с которой начнется копирование ДНК, может быть задана за счет синтеза за-
травки с соответствующей последовательностью нуклеотидов (рис. 2.6, б). Благодаря
этому может быть скопирован короткий специфичный сегмент намного более длин-
ной молекулы матрицы, что намного ценнее, чем копирование случайного фрагмента,
которое лишь и было бы возможным, не будь необходимости в затравливании синтеза
ДНК (так что функция позиционирования с лихвой окупает все затраты, связанные
с затравливанием). В полной мере значение затравливания вы оцените, когда мы будем
рассматривать ПЦР в разделе 2.3.
Вторая общая особенность направ-
5 ляемых матрицей ДНК-полимераз — то,
ДНК 3'--TACCCAACGCAATTC
ATGG----►
5' 3'
Новая ДНК
3'--TACCCAACGCAATTC
ATGGGTTG----►
5' 3'
Рис. 2.5. Действие ДНК-зависимой ДНК-по-
лимеразы. Новые нуклеотиды прибавляются
к З'-концу растущего полинуклеотида, при этом
последовательность этого нового полинуклеоти-
да определяется последовательностью матрицы
5" ДНК. Сравните с процессом транскрипции (на-
правляемый ДНК синтез РНК), представленным
на рис. 1.11
Глава 2. Изучение ДНК 43
а) Для синтеза ДНК необходима затравка
yjLU I i I I I I L1 1 I I I g*
Затравка
уТ77. ....... .-XX.
ДНК-полимераза
-1L1I11J 1111 1 J LX
Синтез ДНК не происходит
Новая ДНК
। I I I I I1. .XXX
Синтез ДНК
б) Затравка определяет ту часть молекулы ДНК, которая будет скопирована
Затравка
5* У
. - 1XJ.JJX 1-XXIJJX1-XIJJJ J XIJ.1 .... .JJ, ... i ;; И I _г._,
Рис. 2.6. Роль затравки в направляемом матрицей синтезе ДНК. а) Для того чтобы начать синтез нового
полинуклеотида, ДНК-полимеразе необходима затравка, б) Последовательность этого олигонуклеотида
определяет позицию, в которой он прикрепляется к матрице ДНК и, следовательно, определяет об-
ласть матрицы, которая будет скопирована. Когда для получения новой ДНК in vitro используется ДНК-
полимераза, затравкой обычно служит короткий олигонуклеотид, полученный химическим синтезом.
Подробное описание процесса затравливания синтеза ДНК in vivo приведено в разделе 15.2.2
что многие из этих ферментов являются многофункциональными, то есть способными
расщеплять молекулы ДНК, равно как и синтезировать их. Это отражение способа, кото-
рым ДНК-полимеразы действуют в клетке во время репликации генома (раздел 15.2). На-
ряду со способностью осуществлять синтез ДНК в направлении 5'->3’, ДНК-полимеразы
могут также производить действия одной или обеих экзонуклеаз (рис. 2.7).
• Действие экзонуклеазы 3'^5' позволяет ферменту удалять нуклеотиды с З'-конца
нити, которая только что была синтезирована. Это действие называют коррек-
цией, потому что оно позволяет полимеразе исправлять ошибки путем удаления
нуклеотида, который был вставлен неправильно.
• Действие экзонуклеазы 5'->3' встречается реже, но присуще некоторым ДНК-
полимеразам, коим для выполнения своей природной функции необходима способ-
ность удалять в ходе репликации генома по крайней мере часть полинуклеотида,
уже присоединенного к матричной нити, которую такая полимераза копирует.
Виды ДНК-полимераз, используемых в научных исследованиях
Некоторые из направляемых матрицей ДНК-полимераз, применяемых в исследо-
ваниях молекулярной биологии (таблица 2.1), представляют собой варианты фермента
ДНК-полимеразы I Escherichia colt, который играет центральную роль в репликации
генома этой бактерии (раздел 15.2). Этот фермент, иногда называемый полимеразой
Корнберга в честь открывшего ее исследователя Артура Корнберга, обладает действием
экзонуклеаз 3'->5’ и 5'->3’, что ограничивает его применимость для манипуляции ДНК.
В основном, его применяют при мечении ДНК (техническое примечание 2.1).
44
Часть 1. Изучение геномов
Рис. 2.7. Синтез ДНК и действия экзонуклеаз, присущие ДНК-
пол имеразам. Все ДНК-полимеразы могут синтезировать
ДНК, а многие обладают также и свойствами одной или
обеих экзонуклеаз
Из действий двух таких экзонуклеаз именно
действие экзонуклеазы 5 >3' вызывает боль-
шинство проблем, когда для манипуляций моле-
кулами в пробирке используется ДНК-полимераза.
Это связано с тем, что фермент, обладающий та-
ким действием, способен удалять нуклеотиды
с 5'-концов полинуклеотидов, которые только что
были синтезированы (рис. 2.8). Маловероятно, что
полинуклеотиды будут расщеплены полностью,
потому что действие полимеразы обычно намного
сильнее, чем функция экзонуклеазы, но некоторые
методы не будут работать, если 5'-концы новых
полинуклеотидов будут укорочены каким-либо
образом. В частности, секвенирование ДНК бази-
руется на синтезе новых полинуклеотидов, начи-
нающихся одинаковыми для всех них 5'-концами,
которые были помечены затравкой, используемой
для инициации реакций секвенирования. Если же имеет место «откусывание» 5'-концов,
не важно, до какой степени, то определить правильную последовательность ДНК ста-
новится невозможно. Когда секвенирование ДНК начали претворять в жизнь в конце
1970-х гг., в нем использовалась видоизмененная версия фермента Корнберга, названная
полимеразой Клёнова. Полимераза Клёнова первоначально приготовлялась путем раз-
резания естественного фермента ДНК-полимеразы I Е. coli на два сегмента с помощью
протеазы. Один из этих фрагментов сохранял действия полимеразы и экзонуклеазы
3’—>5', но терял функцию экзонуклеазы 5’->3', присущую необработанному ферменту.
Этот фермент все еще часто называют фрагментом Клёнова в память об этом старом
а) Синтез ДНК в направлении 5' -> 3’
Затравка Новая ДНК
5*
lllllllllllllllltlllllll
Матрица
ДНК-полимераза
б) Действие экзонуклеазы 3’ 5’
Неверный нуклеотид
5>
3'i
> -
ДНК-полимераза обращает
свое направление
в) Действие экзонуклеазы 5' -> 3’
Замещенные
нуклеотиды
lllllllllllllIIIIIIIII
ТТТЩГЩ5<
методе его приготовления, но в настоящее время его почти всегда готовят из клеток
Е. coli, в которых ген полимеразы был спроектирован таким образом, чтобы конечный
фермент имел желательные свойства. Но фактически полимераза Клёнова теперь редко
используется в секвенировании и находит свое главное применение в мечении ДНК (см.
техническое примечание 2.1). Это связано с тем, что в 1980-е гг. был разработан фермент
по имени секвеназа (см. табл. 2.1), который имеет превосходящие свойства в отношении
секвенирования. В разделе 4.1.1 мы
Новая ДНК н Л
5 YTTTTTTTTT1!* 3 возвратимся к свойствам секвеназы
3' 5' и посмотрим, почему благодаря им
этот фермент является идеальным
Действие экзонуклеазы 5' -> 3’ для секвенирования.
Сегмент новой ДНК расщепляется
U5
Рис. 2.8. Благодаря действию экзонуклеазы
5'->3', ДНК-полимераза может расщеплять
5'-концы полинуклеотида, который только
что был синтезирован
Глава 2. Изучение ДНК 45
Таблица 2.1. Особенности направляемых матрицей ДНК-полимераз, применяемых в исследованиях
молекулярной биологии
Полимераза Описание Основное применение Перекрестная ссылка
ДНК-полимераза I Неизмененный фермент Е. coli Мечение ДНК Техническое примечание 2.1
Полимераза Клёнова Измененная версия ДНК- полимеразы IЕ coli Мечение ДНК Техническое примечание 2.1
Секвеназа Измененная версия ДНК- полимеразы I фага Т7 Секвенирование ДНК Раздел 4.1.1
Полимераза Taq ДНК-полимераза I Thermus aquaticus ПЦР Раздел 2.3
Обратная полимераза РНК-зависимая ДНК- Синтез кДНК Раздел 5.1.2
полимераза, полученная из
различных ретровирусов
Фермент ДНК-полимераза I Е. coli имеет оптимальную температуру реакции
37 °C, каковая является обычной температурой естественной окружающей среды
бактерии в кишечнике млекопитающих, например человека. Поэтому в пробирке
реакции с использованием полимеразы Корнберга, фрагмента Клёнова или секве-
назы проводятся при 37 °C и прерываются повышением температуры до 75 °C или
выше, что заставляет белок развертываться, или денатурировать, и терять свою
ферментативную активность. Этот режим совершенно адекватен для большинства
методов молекулярной биологии, но по причинам, которые станут ясными в разделе
2.3, ПЦР требует термостабильной ДНК-полимеразы — такой, которая способна
функционировать при температурах намного выше 37 °C. Подходящие ферменты
могут быть получены из бактерий типа Thermus aquaticus, которые живут в горя-
чих источниках при температурах до 95 °C и чей фермент ДНК-полимераза I имеет
оптимальную рабочую температуру 72 °C. Биохимическая основа термостабильности
белка до конца не ясна, но, по всей вероятности, она связана со структурными осо-
бенностями, которые уменьшают степень денатурации белка, которая происходит
при повышенных температурах.
Большое значение в исследованиях молекулярной биологии имеет ДНК-
полимераза еще одного типа. Это обратная транскриптаза (иногда называемая
ревертазой), которая представляет собой РНК-зависимую ДНК-полимеразу и,
таким образом, снимает копии ДНК с РНК-, а не с ДНК-матриц. Обратные транс-
криптазы вовлечены в циклы репликации ретровирусов (раздел 9.1.2), в том числе
вирусов иммунодефицита человека, которые вызывают синдром приобретенного
иммунодефицита, или СПИД; эти вирусы имеют геномы из РНК, которые копируются
в молекулы ДНК после заражения хозяина. В пробирке обратная транскриптаза может
быть использована для получения копий ДНК из молекул мРНК. Эти копии назы-
вают комплементарными ДНК (кДНК). Их синтез важен для некоторых способов
клонирования генов и методов, применяемых для картирования областей генома,
кодирующих заданные молекулы мРНК (раздел 5.1.2).
46
Часть 1. Изучение геномов
2.1.2. Нуклеазы
В технологии рекомбинантной ДНК нашел свое применение целый спектр нуклеаз
(таблица 2.2). Некоторые нуклеазы имеют широкий диапазон действий, но большинство
из них представлено или экзонуклеазами, удаляющими нуклеотиды с концов молекул
ДНК и (или) РНК, или эндонуклеазами, делающими разрезы во внутренних фосфодиэ-
фирных связях. Некоторые нуклеазы являются специфичными к ДНК, а некоторые —
к РНК, одни работают только с двунитевой ДНК, а другие — только с однонитевой ДНК,
иные же вовсе не разбирают молекул, на которые они воздействуют. В последующих
главах мы встретим примеры различных нуклеаз, когда будем рассматривать методы,
в которых они используются. Здесь мы подробно рассмотрим нуклеазы только одного
типа: эндонуклеазы рестрикции, которые играют центральную роль во всех аспектах
технологии рекомбинантной ДНК.
Эндонуклеазы рестрикции позволяют разрезать молекулы ДНК в определен-
ных позициях
Эндонуклеаза рестрикции — фермент, который связывается с молекулой ДНК
в области определенной последовательности и делает двунитевой разрез в этой по-
следовательности или около нее. Из-за специфичности последовательности позиции
разрезов в пределах молекулы ДНК могут быть предсказаны (при условии, что по-
следовательность ДНК известна); это позволяет вырезать определенные сегменты из
более крупной молекулы. Такая возможность лежит в основе клонирования генов и всех
других аспектов технологии рекомбинантной ДНК, в которой требуются фрагменты
ДНК с известной последовательностью.
Известно три типа эндонуклеаз рестрикции. В случае типов I и III нет никакой воз-
можности строгого управления позицией разреза относительно определенной последо-
вательности в молекуле ДНК, которая опознается ферментом. Поэтому такие ферменты
менее полезны, ибо последовательности получающихся фрагментов точно не известны.
Ферменты II типа не страдают этим недостатком, потому что разрез всегда произво-
дится в одном и том же месте — или в пределах распознаваемой последовательности
Таблица 2.2. Свойства важных нуклеаз, применяемых в исследованиях молекулярной биологии
Нуклеаза Описание Главное использование Перекрестная ссылка
Эндонуклеазы рестрикции Специфичные к по- следовательности ДНК-эндонуклеазы из многих источников Много применений Раздел 2.1.2
Нуклеаза S1 Эндонуклеаза, специ- фичная к однонитевой ДНК и РНК, из гриба Aspergillus oryzae Картирование транскриптов Раздел 5.1.2
Дезоксирибону- клеаза I Эндонуклеаза, спец- ифичная к двуните- вой ДНК и РНК, из Escherichia coli Нуклеазный футпринтинг Раздел 11.1.2
Глава 2. Изучение ДНК 47
(сайта узнавания), или очень близко к ней (рис. 2.9). Например, фермент II типа EcoRI
(выделенный из Е. coli) разрезает ДНК только в гексануклеотиде 5’-GAATTC-3'. Поэтому
переваривание ДНК ферментом II типа дает воспроизводимый набор фрагментов, по-
следовательности которых предсказуемы, если последовательность целевой молекулы
ДНК известна. Всего было выделено более 2500 ферментов II типа, и более 300 из них до-
ступно для использования в лаборатории. Многие ферменты имеют гексануклеотидные
целевые участки, но другие узнают более короткие или более длинные последователь-
ности (таблица 2.3). Известны также примеры ферментов с вырожденными сайтами
узнавания; это означает, что они разрезают ДНК во всех участках из семейства таких
связанных участков. Например, Hinfl (из Haemophilus influenzae) опознает последователь-
ность 5'-GANTC-3' (где N — любой нуклеотид) и, таким образом, производит разрезы
в последовательностях 5’-GAATC-3’, 5'-GATTC-3', 5’-GAGTC-3' и 5’-GACTC-3’. Большинство
ферментов делает разрезы в пределах сайта узнавания, но немногие типа BsrB\ произ-
водят разрез в определенной позиции вне этой последовательности.
Таблица 2.3. Некоторые примеры эндонуклеаз рестрикции
Фермент Последователь- ность узнавания (сайт узнавания) Тип концов Конечные последовательности
А1и\ 5'-AGCT-3' 3'-TCGA-5' Тупые 5'-AG СТ-3' З'-ТС GA-5'
Sou3A\ 5'-GATC-3' 3'-CTAG-5' Липкие, выдается 5' 5'- GATC-3' З'-CTAG -5'
Hinft 5'-GANTC-3' 3'-CTNAG-5' Липкие, выдается 5' 5'-G ANTC-3' З'-CTNA G-5'
BamHl 5'-GGATCC-3' 3'-CCTAGG-5' Липкие, выдается 5' 5'-G GATCC-3' З'-CCTAG G-5'
BsrBi 5'-CCGCTC-3' 3'-GGCGAG-5' Тупые 5'- NNNCCGCTC-3' 3'- NNNGGCGAG-5'
EcoBA 5'-GAATTC-3' 3'-CTTAAG-5' Липкие, выдается 5’ 5'-G AATTC-3' З'-CTTAA G-5'
Pstl 5'-CTGCAG-3' 3'-GACGTC-5' Липкие, выдается 3’ 5'-CTGCA G-3' З'-G ACGTC-5'
NotA 5'-GCGGCCGC-3' 3'-CGCCGGCG-5' Липкие, выдается 5’ 5'-GC GGCCGC-3' З'-CGCCGG CG-5'
Bgl\ 5'-GCCNNNNNGGC-3' 3'-CGGNNNNNCCG-5' Липкие, выдается 3' 5'-GCCNNNN NGGC-3' З'-CGGN NNNNCCG-5'
Сокращение: N—любой нуклеотид. Обратите внимание, что в подавляющем большинстве сайты
узнавания обладают симметрией инверсии: при чтении в направлении 5’->3' опознавательная
последовательность одинакова в обеих нитях.
48
Часть 1. Изучение геномов
Разрезы, производимые в различных позициях
1Ш
Опознавательная I Эндонуклеаза рестрикции I или III типа
последовательность ।
Эндонуклеаза рестрикции II типа
t
Все разрезы в одной и той же позиции
Рис. 2.9. Разрезы, производимые эндо-
нуклеазами рестрикции. В верхней части
схемы ДНК разрезана эндонуклеазой ре-
стрикции I или III типа. Разрезы сделаны
в немного различных позициях относи-
тельно последовательности узнавания,
так что получающиеся фрагменты имеют
различную длину. В нижней части схемы
показано действие фермента II типа. Все
молекулы разрезаны точно в одной и той
же позиции, что дало абсолютно идентич-
ные пары фрагментов
Ферменты рестрикции раз-
резают ДНК двумя различными
способами. Многие делают простой
двунитевой разрез, после которого
остаёеся тупой, или ровный, конец,
но другие разрезают две нити ДНК
в разных позициях, обычно отсто-
ящих друг от друга на два или на
четыре нуклеотида, так что полу-
чающиеся фрагменты ДНК имеют
на каждом конце короткие однонитевые выдающиеся куски. Их называют липкими,
или неровными, концами, потому что в результате спаривания оснований между ними
молекула ДНК может обратно склеиться воедино (рис. 2.10, а). Некоторые дающие
липкие концы рестриктазы оставляют 5'-выступы (например, 5аиЗА1 и Hinfl), тогда как
другие оставляют З’-выступы (например, PstI) (рис. 2.10, б). Одна особенность, которая
крайне важна в технологии рекомбинантной ДНК, состоит в том, что некоторые пары
ферментов рестрикции имеют различные сайты узнавания, но дают одни и те же липкие
концы; например, рестриктазы 5апЗА1 и ZtamHI обе дают липкий конец 5’-GATC-3’, хотя
сайт узнавания 5аиЗА1 состоит из четырех пар оснований, a ZtamHI опознает последо-
вательность из шести пар оснований (рис. 2.10, в).
Анализ результатов переваривания рестриктазами
После обработки ДНК эндонуклеазой рестрикции полученные фрагменты могут
быть исследованы электрофорезом в агарозном геле (техническое примечание 2.2)
с целью определения их размеров. Если исходная ДНК была относительно короткой
молекулой и после рестрикции было получено не более двадцати фрагментов, то обычно
возможно подобрать такую концентрацию агарозы, чтобы каждый фрагмент просма-
тривался в виде отдельной полосы в геле. Если начальная ДНК длинна и, таким образом,
дает много фрагментов после переваривания ферментом рестрикции, то независимо
от выбранной концентрации агарозы гель может попросту показать размытое пятно
ДНК, потому что там присутствуют фрагменты всевозможной длины, которые все сли-
ваются воедино. Это часто наблюдаемый результат, когда геномная ДНК разрезается
ферментом рестрикции.
Если последовательность исходной ДНК известна, то могут быть предсказаны
последовательности и, следовательно, размеры фрагментов, полученных после обра-
Глава 2. Изучение ДНК 49
Техническое примечание 2.2: электрофорез в агарозном геле
Разделение молекул ДНК различной длины
Гель-электрофорез — стандартный метод для разделения молекул ДНК раз-
личной длины. Он находит множество применений в анализе размеров фрагментов
ДНК и может употребляться также и для разделения молекул РНК (см. техническое
примечание 5.1).
Электрофорез заключается в движении заряженных молекул в электрическом
поле: отрицательно заряженные молекулы мигрируют к положительному электро-
ду, а положительно заряженные молекулы мигрируют к отрицательному электроду
Первоначально этот метод выполняли в водном растворе, в котором преобладаю-
щими факторами, влияющими на скорость миграции, были форма молекулы и ее
электрический заряд. Это не особенно пригодно для разделения ДНК, потому что
большинство молекул ДНК имеет одинаковую форму (линейную), и, хотя заряд
молекулы ДНК зависит от ее длины, различия в заряде недостаточны, чтобы обе-
спечить эффективное разделение. Ситуация меняется, когда электрофорез выпол-
няется в геле, потому что теперь форма и заряд молекулы отходят на второй план,
а критическим детерминантом скорости перемещения становится ее длина. Это
связано с тем, что гель представляет собой сеть пор, через которые молекулы ДНК
должны проходить, чтобы достигнуть положительного электрода. Более коротким
молекулам поры препятствуют меньше, чем более длинным молекулам, и поэтому
первые продвигаются через гель быстрее. По этой причине молекулы различной
длины образуют полосы в геле.
В молекулярной биологии используются гели двух типов: агарозные гели,
которые описаны здесь, и полиакриламидные гели, которые будут описаны в тех-
ническом примечании 4.1. Агароза — полисахарид, который образует гели с порами
в пределах от 100 до 300 нм в диаметре, причем размер пор зависит от концентрации
агарозы в геле. Поэтому концентрация геля определяет диапазон фрагментов ДНК,
которые могут быть разделены. Диапазон разделения зависит также от величины
электроэндосмоса (ЭЭО) агарозы, каковая служит мерой количества связанных
анионов сульфата и пирувата. Чем больше ЭЭО, тем медленнее скорость миграции
отрицательно заряженной молекулы, например ДНК.
Агарозный гель приготовляют следующим образом: размешивают необхо-
димое количество порошка агарозы в буферном растворе, нагревают его, чтобы
растворить агарозу, и затем выливают расплавленный гель на плексигласовую
пластину с лентой по бортам, предотвращающей расползание геля. Затем в гель
помещается решетка, чтобы оформить лунки для образцов. Далее гелю позволяют
схватиться и после этого выполняют электрофорез в геле, погруженном под буфер.
С тем чтобы следить за ходом электрофореза, к образцам ДНК перед их загрузкой до-
бавляют один или два красителя с известной скоростью перемещения. Полосы ДНК
визуализируются путем вымачивания геля в растворе бромистого этидия, при этом
данное соединение встраивается между парами оснований ДНК и флюоресцирует,
будучи активировано ультрафиолетовым излучением (рис. Т2.1). В зависимости
от концентрации агарозы в геле фрагменты от 100 п. н. до 50 тыс. п. н. в длину мо-
гут быть разделены на резкие полосы после электрофореза (рис. Т2.2). Например,
0,3%-й гель может использоваться для молекул от 5 тыс. п. н. до 50 тыс. п. н. в длину,
50
Часть 1. Изучение геномов
Лунка для образцов
I Вымачивание в 0,5 мкг/мл растворе
Д, бромистого этидия в течение 15 минут
Агарозный гель
Пленка-подложка из
прозрачного для УФ-
лучеи пластика
Рис. Т2.2. Диапазон размеров фрагментов, лежащий
в пределах разрешающей способности, зависит от
концентрации агарозы в геле. Электрофорез был
выполнен стремя различными концентрациями ага-
розы. Метки указывают размеры полос на левой
и правой дорожке. Фотография любезно предостав-
лена «BioWhittaker Molecular Applications»
Полосы ДНК
флюоресцируют
УФ УФ УФ
эис. Т2.1. Полосы ДНК в агарозном геле визуа-
лизируются путем окрашивания бромистым эти-
дием
а 5%-й гель — для молекул длины 100-500 п.н. В 4%-м или 5%-м агарозном геле
могут быть разделены фрагменты длиной менее 150 п. н., что позволяет различать
между собой полосы, представляющие молекулы, которые отличаются друг от друга
размером лишь только на один-единственный нуклеотид. Однако в случае более
крупных фрагментов разделять молекулы близкого размера удается не всегда, даже
в гелях с более низкой концентрацией агарозы.
ботки специфическим ферментом рестрикции. В таком случае полоса, соответствующая
желательному фрагменту (например, содержащему тот или иной ген), может быть
идентифицирована, вырезана из геля и очищена с выделением из нее ДНК. Даже если
его размер неизвестен, фрагмент, содержащий ген или какой-либо иной сегмент ДНК,
представляющий интерес, может быть идентифицирован методом так называемой
гибридизации по Саузерну при условии, что часть последовательности в пределах
этого фрагмента известна или может быть предсказана. Первый шаг — переместить
фрагменты рестрикции из агарозного геля на мембрану из нейлона или нитроцеллю-
лозы. Это делают, помещая мембрану на гель и позволяя буферу промочить ее, увлекая
ДНК из геля к мембране, где она становится связанной (рис. 2.11, а). В результате этого
процесса полосы ДНК становятся закрепленными на поверхности мембраны в относи-
тельных позициях, аналогичных таковым в геле.
Глава 2. Изучение ДНК 51
а) Тупые и липкие концы
IIIIIIIIIIIIIIIH
уШШ1( 1111UU 5* * iflllHii '"JIIILH 5'
Тупые концы Липкие концы
б) 5'- и З'-выступы
гШП^ПШ Г у
| BamHI | Pstl
гШП««е 5“ТС1ШП? vTWTGC>3' *свлшп1-
3^^—CCTAG5* G****»5 3 «G yACGTC*——— 5
в) Липкий конец, произведенный различными ферментами
5/ттттт^^ттттт у
3'1L1Ucctagg*11u 5'
| BamHI
5“тс«
йппжшп?
| Sau3AI
гШШста65. 5'GATCnnn?
Рис. 2.10. Результаты переваривания ДНК различными эндонуклеазами рестрикции, а) Тупые концы
и липкие концы. 6) Липкие концы различных типов: З'-выступы, производимые BamHI, и З'-выступы,
производимые Pst\. в) Одни и те же липкие концы, произведенные двумя различными эндо-
нуклеазами рестрикции: 5'-выступ с последовательностью 5'-GATC-3' производится и BamHI
(опознает 5'-CGATCC-3') и ЗаиЗМ (опознает 5'-САТС-3')
Следующий шаг — подготовка гибридизирующего зонда, который представляет
собой меченую молекулу ДНК, последовательность которой комплементарна последова-
тельности целевой ДНК, которую мы желаем детектировать. Зондом мог бы, например,
служить синтетический олигонуклеотид, последовательность которого соответствует
части интересующего нас гена. Поскольку зонд и целевые ДНК взаимно комплементарны,
они могут спариваться или гибридизироваться между собой, при этом позиция гибри-
дизированного зонда на мембране идентифицируется путем детектирования сигнала,
выделяемого меткой, прикрепленной к зонду. Для проведения гибридизации мембрана
помещается в стеклянную бутыль с меченым зондом и некоторым буфером, после чего
бутыль плавно вращается в течение нескольких часов, с тем чтобы зонд имел достаточ-
ную возможность гибридизировать свою целевую ДНК. Затем мембрану промывают,
чтобы удалить с ее поверхности все зонды, которые не гибридизировались, и, наконец,
детектируют сигнал от метки (см. техническое примечание 2.1). В представленном на
рис. 2.1, б примере зонд помечен радиоактивной меткой, а сигнал детектируется авто-
радиографией. Полоса, которая заметна на авторадиограмме, соответствует фрагменту
рестрикции, который гибридизируется с зондом и, значит, содержит искомый ген.
2.1.3. ДНК-лигазы
Фрагменты ДНК, которые были получены путем обработки эндонуклеазой рестрик-
ции, можно снова соединить вместе или прикрепить к новому фрагменту с помощью
52
Часть 1. Изучение геномов
а) Перенос ДНК из геля на мембрану
ДНК-маркеры Рестрикты ДНК
Агарозный гель
Гель
Фитиль
Подложка
Бумажные салфетки
Нейлоновая мембрана
Нейлоновая мембрана
б) Гибридизационный анализ
Зонд ДНК
Нейлоновая мембрана
Полосы гибридизации
Л* 3
Радиоавтограмма
2 3
Рис. 2.11. Гибридизация по Саузерну, а) Перемещение ДНК из геля на мембрану, б) Мембрана зондируется
меченно радиоактивно молекулой ДНК. На полученной авторадиограмме одна полоса гибридизации
заметна на дорожке 2 и две — на дорожке 3
ДНК-лигазы. Данная реакция требует энергии, которая обеспечивается добавлением
к реакционной смеси или АТФ, или никотинамидадениндинуклеотида (НАД) в зависи-
мости от типа применяемой лигазы.
Наиболее широко используемую ДНК-лигазу получают из клеток Е. coli, зараженных
бактериофагом Т4. Этот фермент участвует в репликации ДНК фага и кодируется его
геномом. В природе его роль заключается в синтезе фосфодиэфирных связей между не-
связанными нуклеотидами, принадлежащими какому-либо одному из полинуклеотидов
двунитевой молекулы (рис. 2.12, а). Чтобы соединить вместе два фрагмента рестрикции,
лигаза должна синтезировать две фосфодиэфирные связи, — по одной в каждой из
нитей (рис. 2.12, б). Это ни в коем разе не выходит за пределы возможностей фермента,
но реакция может произойти, только если концы, которые должны быть соединены,
случайно сойдутся достаточно близко друг с другом — лигаза не способна ухватиться за
них и свести их вместе. Если две молекулы ДНК имеют комплементарные липкие концы
и эти концы сошлись в результате случайных событий диффузии в лигирующей смеси,
то между двумя выступами могут образоваться переходные спаривания. Такие пары
оснований не особенно устойчивы, но они могут сохраняться достаточное для фермента
лигазы время, чтобы он мог прикрепиться к стыку и синтезировать фосфодиэфирные
связи, а тем самым — соединить концы вместе (рис. 2.12, в). Если же молекулы ДНК
имеют тупые концы, то они не могут спариться друг с другом даже временно, и процесс
лигирования становится намного менее эффективным, даже когда концентрация ДНК
высока и пары концов находятся в относительно тесной близости.
Большая эффективность лигирования липких концов способствовала развитию
методов преобразования тупых концов в липкие концы. В одном таком методе корот-
кие двунитевые молекулы, названные линкерами или адаптерами, прикрепляются
к тупым концам. Линкеры и адаптеры работают немного различными способами, но
и те и другие содержат сайт узнавания для эндонуклеазы рестрикции и, таким образом,
производят липкий конец после обработки соответствующим ферментом (рис. 2.13).
Глава 2. Изучение ДНК 53
Другой способ создания липкого конца — при-
крепление гомополимерного хвоста, которое
состоит в добавлении нуклеотидов одного за
другим к З'-концу нити тупого конца (рис. 2.14).
Вовлеченный в эти реакции фермент называют
концевой дезоксинуклеотидилтрансфера-
зой, которую мы встретим в следующем раз-
деле. Если реакционная смесь содержит ДНК,
фермент и только один из четырех стандартных
нуклеотидов, то новый отрезок полученной
однонитевой ДНК будет полностью состоять
только из нуклеотидов исключительно этого
вида. Это может быть, например, поли-G хвост,
который позволит молекуле спариться с други-
ми молекулами, несущими поли-С хвосты и соз-
данными таким же способом, но с добавлением
в реакционную смесь не дГТФ, а дЦТФ.
2.1.4. Ферменты модификации
концов
Одним из ферментов модификации кон-
цов является концевая дезоксинуклеотидил-
трансфераза (см. рис. 2.14), получаемая из
ткани вилочковой железы теленка. По сути,
она представляет собой независимую от ма-
трицы ДНК-полимеразу, потому что способна
синтезировать новый полинуклеотид ДНК без
спаривания поступающих нуклеотидов с суще-
ствующей нитью ДНК или РНК. Его главная
роль в технологии рекомбинантной ДНК за-
ключается в прикреплении гомополимерного
хвоста, как было описано выше.
Также часто используются два других
фермента модификации концов. Это щелоч-
ная фосфатаза и полинуклеотидкиназа Т4,
которые действуют в дополнение друг другу.
Щелочная фосфатаза, которую получают из
различных источников, включая Е. coli и ки-
шечную ткань теленка, удаляет фосфатные
группы с 5'-концов молекул ДНК, что препят-
ствует лигированию этих молекул друг с дру-
гом. Два конца, несущих 5’-фосфаты, могут быть
лигированы друг с другом, и обработанный
фосфатазой конец может лигироваться с не-
обработанным фосфатазой концом, но связь
не может образоваться между парой концов,
если ни один из них не несет 5'-фосфат. Поэто-
а) Роль ДНК-лигазы in vivo
Отсутствующая фосфодиэфирная связь
i 111111 и 11ТТТПТ;,
1
Отсутствующая связь, синтезированная ДНК-лигазой
б) Лигирование in vitro
pl||l|ll|||||g rlJUlUHUUg
git......wii^ummumg
Две связи, синтезированные ДНК-лигазой
в) Лигирование липких концов более эффективно
^11111111111 ГГГПГП^
in । и и । П||1Г1гш и
Переходное спаривание между липкими концами
Две связи, синтезированные ДНК-лигазой
Рис. 2.12. Лигирование молекул ДНК с по-
мощью ДНК-лигазы. а) В живых клетках ДНК-
лигаза синтезирует отсутствующие фосфодиэ-
фирные связи в одной из нитей двунитевых
молекул ДНК. б) Чтобы связать две молекулы
ДНК in vitro, ДНК-лигаза должна образовать две
фосфодиэфирные связи, по одной в каждой из
нитей, в) Лигирование in vitro более эффек-
тивно, когда молекулы имеют совместимые
липкие концы, потому что переходное спари-
вание между этими концами скрепляет обе
молекулы между собой и таким образом уве-
личивает возможности ДНК-лигазы скрепить
их и синтезировать новые фосфодиэфирные
связи. Роль ДНК-лигазы во время репликации
ДНК in vivo показана на рис.15.18
54
Часть 1. Изучение геномов
му разумно используя щелочную фосфатазу можно направлять действие ДНК-лигазы
предопределенным образом, так чтобы были получены только желательные продукты
лигирования. Полинуклеотидкиназа Т4, получаемая из клеток Е. coli, зараженных бак-
териофагом Т4, исполняет обратную щелочной фосфатазе реакцию, добавляя фосфаты
к 5’-концам. Подобно щелочной фосфатазе, этот фермент используется в ходе сложных
экспериментов с лигированием, но его главное применение состоит в мечении концов
молекул ДНК (см. техническое примечание 2.1).
2.2. Клонирование ДНК
Клонирование ДНК — логическое продолжение методов манипулирования моле-
кулами ДНК с помощью эндонуклеаз рестрикции и лигаз. Вообразим, что ген животного
был получен в виде единого фрагмента рестрикции после переваривания более крупной
молекулы ферментом рестрикции ZtamHI, который оставляет липкие концы 5'-GATC-3’
(рис. 2.15). Вообразим также, что плазмида — маленькая кольцевая ДНК, способная
реплицироваться внутри бактерии, — была очищена из экстракта Е. coli и обработана
ZtamHI, которая разрезает плазмиду в одной определенной позиции. Кольцевая плазмида
Тупоконечная Линкеры
молекула ДНК
Линкеры, прикрепленные
к концам молекулы ДНК
Опознавательная
последовательность Вал?Н1
| BamHI
у"!!! 111111ITTT1Q г"
Липкий конец, произведенный BamHI
Рис. 2.13. Линкеры используются для наращивания липких концов на молекуле с тупыми концами.
В этом примере все линкеры содержат последовательность узнавания для эндонуклеазы рестрикции
BomHI. ДНК-лигаза прикрепляет линкеры к концам молекулы ступыми концами входе реакции, которая
получается относительно эффективной, потому что линкеры присутствуют в высокой концентрации.
Затем добавляется фермент рестрикции, чтобы расщепить линкеры и произвести липкие концы. Об-
ратите внимание, что в процессе лигирования линкеры лигируют друг друга, так что к каждому концу
тупоконечной молекулы прикрепляется ряд линкеров (сцепка). Когда добавляется фермент рестрикции,
такие линкерные сцепки разрезаются на сегменты, причем однонитевая половина наиболее удаленного
от края линкера остается прикрепленной к молекуле ДНК. Адаптеры подобны линкерам, но имеют по
одному тупому концу и по одному липкому концу. Поэтому тупоконечная ДНК снабжается липкими
концами путем простого лигирования ее с адаптерами: нет никакой необходимости выполнять про-
цедуру рестрикции
Глава 2. Изучение ДНК 55
Рис. 2.14. Прикрепление гомополимерного конца.
В этом примере на обоих концах тупоконечной молеку-
лы ДНК синтезируется поли-G хвост. Хвосты, состоящие
из других нуклеотидов, синтезируются путем введения
в реакционную смесь соответствующих дНТФ
была таким образом преобразована в линей-
ную молекулу, опять же с липкими концами
5’-GATC-3’. Смешаем эти две молекулы ДНК
вместе и добавим ДНК-лигазу. Будут получе-
ны различные продукты рекомбинантного
лигирования, один из которых представляет
собой свернутую кольцом плазмиду с геном
животного, вставленным в позицию, перво-
начально занимаемую участком рестрикции
EamHI. Если теперь повторно ввести такую
Тупоконечная молекула ДНК
ylJJ LULL! 11 Ll’y.
Концевая дезоксинуклеоти-
X дилтрансфераза + дГТФ
Поли-G хвост
рекомбинантную плазмиду в Е. coli и если вставленный ген не утратил свою реплика-
тивную способность, то плазмида со вставленным в нее геном будет реплицироваться,
а ее копии — переходить в дочерние бактерии после деления клетки. Большое число
циклов репликации плазмиды и деления клеток даст в конечном итоге колонию реком-
бинантных бактерий Е coli, причем каждая бактерия будет содержать многочисленные
копии этого гена животного. Данный ряд операций, представленный на рис. 2.15, и со-
ставляет процесс, названный клонированием генов или ДНК.
Когда клонирование ДНК было впервые
изобретено в начале 1970-х гг., оно произвело
коренной переворот в молекулярной биологии,
сделав возможными эксперименты, которые
до этого невозможно было и вообразить. Это
связано с тем, что клонирование позволяет по-
лучить чистый образец отдельного гена, отде-
ленного ото всех остальных генов клетки. Рас-
смотрим основную процедуру клонирования,
представленную в несколько ином ракурсе (рис.
2.16). В этом примере фрагмент ДНК, который
будет клонирован, — один из компонентов
смеси многих различных фрагментов, причем
каждый из них несет неповторимый ген или
часть гена. Такой смесью вполне может быть
представлен полный геном. Каждый из этих
фрагментов вставляется в отдельную молекулу
плазмиды, с тем чтобы произвести семейство
рекомбинантных плазмид, одна из которых не-
сет интересующий нас ген. Обычно в какую-либо
отдельно взятую клетку хозяина переносится
только одна рекомбинантная молекула. Таким
образом, хотя конечный набор клонов и может
содержать много различных рекомбинантных
молекул, каждый отдельный клон будет содер-
Участки рестрикции BamHI
ДНК животного Плазмида Е. coli
I BamHI 1
Ген животного^
Лигирование^
>L Ген животного
(у встраивается
. / в плазмиду
Бактерия
Е. coli
Рекомбинантная
плазмида внутри
I Реплицирование
1 внутри бактерии
— Колония Е. coli
Множество копий
рекомбинантной
плазмиды
Рис. 2.15. Схема клонирования генов
56
Часть 1. Изучение геномов
жать многочисленные копии только одной
рекомбинантной молекулы. Затем ген отде-
ляется от всех других генов, содержавшихся
в исходной смеси. Очистка рекомбинантной
молекулы от бактериальной колонии или от
жидкой культуры, выращенной из колонии,
даст количество ДНК порядка микрограмм,
что достаточно для ее анализа путем секвени-
рования ДНК или одним из несметного числа
других методов, изобретенных для изучения
клонированных генов, со многими из которых
мы ознакомимся в дальнейших главах.
2.2.1. Клонирующие векторы и их
применения
В экспериментах, показанных на рис. 2.15
и 2.16, плазмида выступает клонирующим
вектором, предоставляя свою репликатив-
ную способность, которая позволяет клони-
рованному гену размножаться в клетке хо-
зяина. Плазмиды эффективно реплицируются
в бактериальных хозяевах, потому что каждая
плазмида обладает областью инициации, ко-
торая опознается ДНК-полимеразами и други-
ми белками, обычно реплицирующими хромосомы бактерии (раздел 15.2.1). Поэтому
репликативный аппарат клетки хозяина размножает плазмиду и любые новые гены,
которые были в нее вставлены. Геномы бактериофагов также могут использоваться
как клонирующие векторы, потому что они тоже обладают областями инициации,
которые позволяют им быть размноженными в бактериях — или за счет ферментов
хозяина, или за счет ДНК-полимераз и других белков, кодируемых генами самого фага.
Следующие два раздела описывают, как плазмидные и фаговые векторы используются
для клонирования ДНК в Е. coli.
Плазмиды обыкновенно не знакомы эукариотам, хотя Saccharomyces cerevisiae об-
ладает одной, что иногда используется в целях клонирования; поэтому большинство
векторов для использования в клетках эукариотов основано на вирусных геномах.
Напротив, имея хозяина-эукариота, данное требование репликации можно обойти,
выполняя эксперимент таким образом, что ДНК, которая будет клонирована, вставля-
ется в одну из хромосом хозяина. Эти подходы к клонированию в клетках эукариотов
описаны далее в этой главе.
Плазмиды^ Фрагменты ДНК
Конструирование молекул рекомбинантной ДНК
Каждая несет отличный от других фрагмент
Трансформация
Высев на чашку Петри
Каждая колония содержит многочисленные копии
только одной молекулы рекомбинантной ДНК
Рис. 2.16. Клонирование позволяет получить
чистый образец гена
Векторы, построенные на плазмидах Е. coli
Самый легкий способ понять, как используется клонирующий вектор, это начать
с самых простых плазмидных векторов Е coli, которые иллюстрируют все основные
принципы клонирования ДНК. Тогда мы сможем обратить наше внимание на особые
свойства фаговых векторов и векторов, используемых с хозяевами-эукариотами.
Один из самых популярных плазмидных векторов — pUC8, член ряда векторов,
которые впервые были представлены в начале 1980-х гг. Ряд pUC получают из более
Глава 2. Изучение ДНК 57
давнего клонирующего вектора pBR322, который в свое время был построен путем
лигирования друг с другом фрагментов рестрикции из трех встречающихся в природе
плазмид Е. coli: Rl, R6,5 и pMBl. pUC8 — маленькая плазмида, включающая только
2,7 тысяч пар нуклеотидов (тыс.п.н.). Наряду со своей областью инициации, она несет
следующие два гена (рис. 2.17):
• Ген устойчивости к ампициллину. Присутствие этого гена означает, что бакте-
рия, содержащая плазмиду pUC8, способна синтезировать фермент, названный
Р-лактамазой, который позволяет клетке противостоять ингибирующему рост
воздействию антибиотика. Это означает, что клетки, содержащие плазмиды pUC8,
можно отличить от тех, которые их не содержат, высевая бактерии на агаровую
среду, содержащую ампициллин. Нормальные клетки Е coli чувствительны к ампи-
циллину и не могут расти в присутствии антибиотика. Таким образом, устойчивость
к ампициллину представляет собой селективный маркёр для pUC8.
• Ген lacZ’, который кодирует часть фермента р-галактозидазы. р-Галактозидаза —
один из ряда ферментов, вовлеченных в поломку связей между лактозой, глюкозой
и галактозой. Он обычно кодируется геном lacZ, который находится на хромосоме
Е coli. Некоторые штаммы Е coli имеют видоизмененный ген lacZ, такой, в котором
отсутствует сегмент, упоминаемый как lacZ' и кодирующий а-пептидную часть
Р-галактозидазы. Эти мутанты могут синтезировать фермент, только если они
содержат плазмиду типа pUC8, которая несет отсутствующий сегмент lacZ' этого
гена.
Чтобы провести эксперимент клонирования с pUC8, в пробирке с очищенной ДНК
выполняют манипуляции, показанные на рис. 2.15, приводящие к построению рекомби-
нантной плазмиды. Чистая ДНК плазмиды pUC8 может быть весьма легко получена из
вытяжек бактериальных клеток (техническое примечание 2.3), и после манипуляций
плазмиды могут быть повторно введены в Е coli посредством трансформации —
процесса, в ходе которого «голая» ДНК принимается бактериальной клеткой. Именно
Рис. 2.17. pUC8. Карта показывает по-
зиции гена устойчивости к ампицилли-
ну, гена lacZ', область репликации (or/)
и группы участков рестрикции в пределах
гена lacZ1
Группа уникальных участков рестрикции
58
Часть 1. Изучение геномов
Техническое примечание 2.3: очистка ДНК
Методы для подготовки чистых образцов ДНК из живых клеток играют
центральную роль в исследованиях молекулярной биологии
Первый шаг в очистке ДНК состоит в разрушении клеток, из которых должна
быть получена ДНК. С материалами некоторых типов этот шаг выполняется до-
вольно легко: например, выращенные в культуре животные клетки разрушаются
путем простого добавления детергента типа додецил сульфата натрия (ДСН), кото-
рый разрушает клеточные мембраны, выпуская содержимое клеток наружу. Клетки
других типов имеют прочные стенки и, таким образом, требуют более радикальной
обработки. Растительные клетки обычно замораживают и затем размалывают пе-
стиком в ступе, что является единственным эффективным способом разрушения их
целлюлозных стенок. Бактерии типа Escherichia coli могут быть лизированы сочета-
нием ферментативной и химической обработки. В качестве фермента применяют
лизоцим, получаемый из яичного белка, который разрушает полимерные вещества
в стенке бактериальной клетки, а употребляемый химикалий представлен этилен-
диаминтетраацетатом (ЭДТА), который образует комплекс с ионами магния и еще
более нарушает целостность клеточной стенки. Таким образом, разрушение клеточ-
ной мембраны путем добавления детергента вызывает разрывание клеток.
После разрушения клеток для очистки ДНК из полученного экстракта могут
применить два различных подхода. Первый состоит в расщеплении или удалении
всех остальных компонентов клетки, кроме ДНК; этот метод работает лучше всего,
если клетки не содержат большие количества жиров или углеводов. Сначала экс-
тракт центрифугируется на низкой скорости, с тем чтобы удалить мусор, такой
как частицы клеточной стенки, которые образуют каплю осадка на дне пробирки
(рис. Т2.3). Надосадочный слой переносится в другую пробирку и смешивается
с фенолом, который заставляет белок выпадать в осадок на границе раздела между
органическим и водным слоями. Водный слой, который содержит растворенные
нуклеиновые кислоты, собирают и добавляют фермент рибонуклеазу, разбивающий
РНК на смесь нуклеотидов и коротких олигонуклеотидов. Полинуклеотиды ДНК,
которые остаются неповрежденными, могут теперь быть переведены в осадок до-
бавлением этанола, осаждения путем центрифугирования и повторного перевода
во взвесь в соответствующем объеме буфера.
Во втором подходе к очистке ДНК, вместо того чтобы расщеплять все осталь-
ное, кроме ДНК, сама ДНК избирательно удаляется из экстракта. Один из способов
Клеточная ДНК, РНК,
вытяжка белок
Центрифуга
Перенос надосадочного
слоя в новую пробирку
I
Клеточный
мусор
Водный слой
Фенол <ДНК РНК> Этанол
1 Перенос водного слоя ।
в новую пробирку, I
выдержка в присутствии 4
! Центрифуга _________рибонуклеазы________ «Центрифуга
Ib Поверхность раздела
у* (коагулированные белки) " j
Фенол Осажденная ДНК
Рис. Т2.3. Очистка ДНК от клеточного экстракта путем расщепления или удаления всех остальных
компонентов
Глава 2. Изучение ДНК 59
достичь этого — ионообменная хрома-
тография, которая разделяет молекулы
согласно тому как сильно они связаны
с электрически заряженными частицами
в хроматографической смоле. ДНК и РНК,
а также некоторые белки заряжены от-
рицательно и поэтому связываются с по-
ложительно заряженной смолой. Самый
простой способ выполнить ионообменную
хроматографию состоит в том, чтобы по-
местить смолу в колонку и добавить кле-
точный экстракт сверху (рис. Т2.4). По мере
того как экстракт проходит через колонку,
все отрицательно заряженные молекулы
связываются со смолой. Связи, образован-
ные за счет ионного взаимодействия, как
известно, могут быть разрушены добавле-
нием солевого раствора, при этом менее
прочно связанные молекулы отделяются
от смолы при относительно низких концен-
трациях соли. Это означает, что если через
Клеточная
вытяжка
Соль
Ионообменная
смола
Белок и РНК
Больше^соли
I На выброс
ДНК
I
колонку проходит раствор с постепенно Рис. Т2.4. Очистка ДНК с помощью ионооб-
увеличивающейся концентрацией соли, менной хроматографии
то молекулы различных типов элюируют
в последовательности белок, РНК и ДНК, отражающей их относительные силы свя-
зывания. Фактически, столь тщательное разделение обычно не требуется, так что
используются только два солевых раствора. Первый содержит NaCl в концентрации
1,0 моль/л при pH 7,0 и достаточен для элюирования белка и РНК, после которого
связанной остается только ДНК. После него добавляют второй раствор, содержащий
NaCl в концентрации 1,25 моль/л при pH 8,5, который элюирует ДНК, теперь уже
свободную от примесей РНК и белка.
Оба подхода, описанных выше, позволяют выделять в чистом виде всю ДНК,
содержащуюся в клетке. Если задача состоит в том, чтобы получить из бактериаль-
ных клеток только плазмидную ДНК (например, рекомбинантные клонирующие
векторы), необходимы специальные методы. Один популярный метод основан
на том факте, что, хотя как плазмиды, так и бактериальная хромосома состоят из
сверхскрученной ДНК, лизис бактериальной клетки неизбежно ведет к частичному
разрушению бактериальной хромосомы, что ведет к потере сверхспирализации ДНК.
Поэтому клеточный экстракт содержит сверхскрученную ДНК плазмиды и несверх-
скрученную хромосомную ДНК, и плазмиды могут быть очищены методом, который
различает молекулы ДНК в этих различных конформациях. Один из таких методов
предполагает добавление гидроокиси натрия, пока pH клеточного экстракта не до-
стигнет величины 12,0-12,5, что заставляет пары оснований в несверхскрученной
ДНК разрываться. Полученные отдельные нити запутываются в нерастворимую сеть,
которая может быть удалена центрифугированием, после которого сверхскрученные
плазмиды остаются в надосадочном слое.
60 Часть 1. Изучение геномов
такую систему изучал Эвери со своими коллегами в экспериментах, показавших, что
бактериальные гены состоят из ДНК (раздел 1.1.1). У многих бактерий, включая Е. coli,
трансформация есть не особенно эффективный процесс, но скорость трансформа-
ции может быть значительно увеличена, если перед добавлением ДНК создать взвесь
клеток-хозяев в хлориде кальция, а после кратковременно выдержать смесь при 42 °C.
Даже после таких процедур лишь очень маленькая доля клеток принимает плазмиду.
Именно по этой причине настолько важен маркёр устойчивости к ампициллину — он
позволяет отбирать небольшое количество трансформантов из большой популяции
нетрансформированных клеток.
Карта pUC8, показанная на рис. 2.17, указывает, что ген lacZ1 содержит группу
уникальных участков рестрикции. Лигирование новой ДНК с любым из этих участков
приводит к инсерционной инактивации гена и, следовательно, к потере активности
Р-галактозидазы. Это ключ к различению рекомбинантной плазмиды, — содержащей
вставленный кусок ДНК, — и нерекомбинантной плазмиды, которая не несет в себе
никакой новой ДНК. Идентификация рекомбинантов важна, потому что манипуляции,
иллюстрированные на рис. 2.15 и 2.16, дают множество разнообразных продуктов ли-
гирования, а в их числе и плазмиды, которые повторно свернулись кольцом без вставки
новой ДНК. Массовый анализ на предмет присутствия или отсутствия р-галактозидазы
по сути своей весьма прост. Вместо проверки того, расщеплена ли лактоза на глюкозу
и галактозу, присутствие молекул функциональной р-галактозидазы в клетках про-
веряется гистохимическим анализом с помощью вещества по имени Х-гель (5-бромо-
4-хлоро-3-индолил-р-В-галактопиранозид), который преобразуется этим ферментом
в продукт синего цвета. Если Х-гель (плюс индуктор фермента, например изопропилтио-
галактозид IPTG) добавляется к агару наряду с ампициллином, то нерекомбинантные
колонии, клетки которых синтезируют р-галактозидазу, будут окрашены в синий цвет,
тогда как рекомбинанты с разрушенным геном lacZ', которые не способны производить
Р-галактозидазу, будут белы (рис. 2.18). Такую систему называют Lac-отбором.
Агар + ампициллин + Х-гель
Рис. 2.18. Выбор рекомбинантов с помощью pUC8
Глава 2. Изучение ДНК 61
Клонирующие векторы на основе геномов бактериофагов, специфичных
к Е. coli
Клонирующие векторы на основе бактериофагов Е. coli были сконструированы
на заре революции рекомбинантной ДНК. Главной причиной поиска вектора нового
типа была неспособность плазмид типа pUC8 вмещать фрагменты ДНК размером более
10 тыс. п. н., при этом более крупные вставки подвергались перестройкам или интерфе-
ренции репликационной системой плазмиды, в результате чего молекулы рекомбинант-
ной ДНК выпадали из клеток хозяина. Первые попытки разработать векторы, способные
нести в себе более крупные фрагменты ДНК, сосредоточились на бактериофаге X.
Чтобы реплицироваться, бактериофаг должен проникнуть в бактериальную клетку
и перенастроить ферменты бактерии на экспрессию информации, содержащейся в генах
фага, с тем чтобы бактерия начала синтезировать новые фаги. Как только репликация
завершена, новые фаги покидают бактерию, обычно вызывая ее гибель, и идут дальше,
чтобы заразить новые клетки (рис. 2.19, а). Такие действия фагов ученые называют
литическим циклом инфицирования, потому что он завершается лизисом бактерии.
Помимо литического цикла, бактериофаг X (в отличие от бактериофагов многих других
типов) может проходить также и лизогенный цикл инфицирования, в ходе которого
геном бактериофага X встраивается в хромосому бактерии, где он может оставаться в со-
стоянии покоя на протяжении многих поколений, реплицируясь наряду с хромосомой
хозяина при каждом делении клетки (рис. 2.19, б).
Размер генома бактериофага X — 48,5 тыс.п.н., из которых приблизительно
15 тыс.п.н. или около того являются необязательными, или «факультативными», —
в том смысле, что в этой области содержатся гены, которые необходимы лишь для
встраивания ДНК фага в хромосому Е. coli (рис. 2.20, а). Поэтому такие сегменты могут
быть удалены без снижения способности фага заражать бактерии и управлять синтезом
новых частиц бактериофага X в ходе литического цикла. На основе бактериофага X был и
разработаны векторы двух типов (рис. 2.20, б):
• векторы вставки, в которых вся дополнительная ДНК или часть ее была удалена
и в некоторой позиции в пределах урезанного генома введен уникальный сайт
рестрикции;
• векторы замены, в которых факультативная ДНК содержится в пределах нахо-
дящегося между парой участков рестрикции вкладочного фрагмента, который
заменяется лигируемой в вектор ДНК, предназначенной для клонирования.
Геном бактериофага X линеен, но два естественных конца молекулы имеют одно-
нитевые выступы длиной 12 нуклеотидов, названные cos-сайтами, которые имеют
комплементарные последовательности и, таким образом, могут спариваться друг с дру-
гом. Поэтому клонирующий вектор X может быть получен в виде кольцевой молекулы,
которой можно манипулировать в пробирке точно так же, как и плазмидой, и повторно
вводить в Е. coli путем трансфекции (этот термин употребляют для обозначения за-
ражения бактерий голой ДНК фага). В качестве альтернативы может использоваться
более эффективная система поглощения, называемая упаковкой in vitro. Эта проце-
дура начинается с отбора линейной версии клонирующего вектора, затем первичная
рестрикция разрезает молекулу на два сегмента — левое и правое плечо, каждое с cos-
участком на одном конце. После этого, чтобы произвести сшивку, в которой различные
фрагменты будут связаны друг с другом в порядке левое плечо-новая ДНК-правое
плечо, выполняют лигирование с тщательно сбалансированным количеством каждого
плеча и ДНК вставки (см. рис. 2.21). Затем эти сцепки добавляются к смеси для упаков-
62
Часть 1. Изучение геномов
Голова
Бактериофаг X
Хвост
I Бактериофаг X
* прикрепляется
к бактерии Е. coli
а) Литический цикл
инфицирования
ДНК бактериофага X
управляет синтезом
новых фагов
б) Лизогенный цикл
инфицирования
Встраивание ДНК
бактериофага X
в хромосому бактерии
ДНК
Белковая оболочка
Лизис клетки
ДНК бактериофага X,
встроенная в хромосому
бактерии
Новые фаги X
выходят наружу
ДНК бактериофага X
впрыскивается в клетку
Возвращение к
литическому циклу
по прошествии
бактерией многих
клеточных делений
Новые фаги X
выходят наружу
Рис. 2.19. Литический и лизогенный циклы инфицирования бактериофагом X. а) В литическом цикле
вскоре после инфицирования производятся новые фаги, б) Во время лизогенного цикла геном фага
встраивается в хромосомную ДНК бактерии, где он может оставаться в состоянии покоя на протяжении
многих поколений
ки in vitro, которая содержит все белки, необходимые для синтеза частицы Х-фага. Эти
белки спонтанно формируют фаговые частицы и помещают внутрь этих частиц любой
фрагмент ДНК длины от 37 тыс.п.н. до 52 тыс.п.н., ограниченный с обеих сторон cos-
участками. Поэтому упаковочная смесь вырезает из конструкций комбинации типа левое
Глава 2. Изучение ДНК 63
а) Геном бактериофага X содержит «факультативную» ДНК
Функции
генов
Белковая Встраивание в
оболочка ДНК хозяина
Репликация ДНК
Лизис клетки
I
Геном
бактериофага X
Удаления в этой области не
затрагивают ход литического цикла
б) Векторы вставки и замены
Р
Х-вектор
вставки
I Новая ДНК, встроенная
* в участок рестрикции
Р Р
Р Р
Х-вектор * м
замены
: Новая ДНК заменяет
вкладочный фрагмент
Р Р
Р = участок рестрикции
Рис. 2.20. Клонирующие векторы, основанные на бактериофаге X. а) В геноме бактериофага X гены
распределены на функциональные группы. Например, область, отмеченная как «белковая оболочка»,
включает 21 ген, кодирующий белки, которые или являются компонентами капсида фага, или требуются
для сборки капсида, а область «лизис клетки» включает четыре гена, вовлеченных в лизис бактерии
в конце литической стадии цикла инфицирования. Области генома, которые могут быть удалены без
вреда для способности фага проходить литический цикл, обозначены зеленым цветом, б) Различия
между Х-вектором вставки и Х-вектором замены
плечо-новая ДНК-правое плечо длиной 37-52 тыс. п. н. и строит Х-фаги на их основе. За-
тем фаги смешивают с клетками Е. coli, и в ходе естественного процесса инфицирования
вектор с новой ДНК переносится в бактерии.
После инфицирования клетки высеваются в чашку с агаром. Цель — получить не
отдельные колонии, но произвести ровный слой бактерий по всей поверхности агара.
Бактерии, которые были инфицированы упакованным клонирующим вектором, умира-
ют в течение приблизительно 20 минут, потому что гены бактериофага X, содержащиеся
в плечах вектора, направляют репликацию ДНК и синтез новых фагов при помощи
литического цикла, при этом каждый из этих новых фагов содержит свою собственную
копию вектора и копию клонированной ДНК. В результате гибели и лизиса бактерии
эти фаги выходят в окружающую среду, где они заражают новые клетки и начинают
новый круг репликации фага и лизиса. Конечный результат — чистая зона, названная
бляшкой, которая заметна на газоне бактерий, растущем в чашке с агаром (рис. 2.22).
При использовании некоторых Х-векторов все бляшки состоят из рекомбинантных фа-
гов, потому что лигирование двух плеч без вставки новой ДНК дает молекулу, которая
является слишком короткой и не может быть упакована. При работе с другими векторами
возникает необходимость отличать рекомбинантные бляшки от нерекомбинантных.
64 Часть 1. Изучение геномов
cos-Участки
к, I „ Линейная версия
;..,1'||||чн||'|Ч. вектора вставки
| Рестрикт
। iTTTTTT тпттг*
Левое плечо Правое плечо
I Лигирование с
клонируемой ДНК
.ЛТППУЩТППТГ'
Вставленная ДНК
Rcos L
Rcos L
Может быть упакована
т
Инфекционный бактериофаг X
R cos L R cos L
Слишком коротка
для упаковки
Рис. 2.21. Клонирование с помощью век-
тора вставки на бактериофаге X. Линей-
ная форма вектора показана в верхней
части схемы. Обработка соответствую-
щей эндонуклеазой рестрикции произ-
водит левое и правое плечи, каждое из
которых имеет один тупой конец и один
конец с 12-нуклеотидным выступом cos-
участка. ДНК, которая будет клонирована,
является тупоконечной и, таким образом,
вставляется между этими двумя плечами
в ходе шага лигирования. Эти плечи так-
же лигируются друг с другом через свои
cos-участки, формируя сцепку. Некоторые
части сцепки включают в себя группу типа
левое плечо-новая ДНК-правое плечо и,
при условии, что эта комбинация имеет
длину 37-52 тыс. п. н., будут заключены
в капсид посредством in v/tro-упаковочной
смеси. Части сцепки, состоящие из левого
плеча, лигированного непосредственно
к правому плечу, и не содержащие новую
ДНК, являются слишком короткими и не
будут упакованы
Для этого используются различные методы, в том числе система р-галактозидазы,
описанная выше на примере плазмидного вектора pUC8 (см. рис. 2.18), которая при-
менима также к тем Х-векторам, что несут в себе фрагмент гена lacZ со вставленной
в него клонируемой ДНК.
Векторы для размножения более длинных фрагментов ДНК
Частица фага X может вмещать в себя ДНК длиной до 52 тыс.п.н., так что если
из его генома удалить 15 тыс.п.н., то в ней можно клонировать вплоть до 18 тыс.п.н.
новой ДНК. Этот предел выше тако-
вого для плазмидных векторов, но
все еще очень маленький по сравне-
Инфекция, наблюдаемая в виде бляшки -
чистой зоны на газоне бактерий
Рис. 2.22. Инфекция бактериофагом визуализируется
в виде бляшки на газоне бактерий
нию с размерами полных геномов.
Такое сравнение важно, потому что
библиотека клонов — коллекция
клонов, вставки которых охватыва-
ют полный геном, — часто является
отправной точкой для проекта, на-
целенного на определение последо-
вательности этого генома (глава 4).
Если Х-вектор используется с ДНК
человека, то необходимо более по-
лумиллиона клонов для того, чтобы
был 95%-й шанс присутствия в би-
блиотеке всех специфических частей
генома (таблица 2.4). Конечно, воз-
Глава 2. Изучение ДНК 65
Таблица 2.4. Размеры геномных библиотек человека, приготовленных в клонирующих векторах раз-
личных типов
Число клонов*
Тип вектора Размер вставки, тыс. п. н. Р = 95% Р = 99%
X-Вектор замены 18 532 500 820 000
Космидный, фосмидный 40 240 000 370 000
Р1 125 77 000 118 000
ВАС, РАС 300 32 000 50 000
YAC 600 16 000 24 500
МегаУАС 1400 6 850 10 500
*3начения рассчитаны по уравнению: N = ln(l - Р)/In(1 - а/b), где N—необходимое число клонов,
Р—вероятность того, что любой данный сегмент генома присутствует в библиотеке, а — средний
размер фрагментов ДНК, вставленных в вектор, и b — размер генома.
можно подготовить библиотеку, включающую в себя полмиллиона клонов, особенно
если используются автоматизированные методы, но столь объемная коллекция далека
от идеала. Было бы намного лучше сократить число клонов, используя вектор, способный
оперировать с фрагментами ДНК длины более, чем 18 тыс. п. н. Многие разработки тех-
нологии клонирования за последние 20 лет были направлены на обнаружение способов
достичь именно этой цели.
Одна из возможностей состоит в том, чтобы использовать космиду — плазмиду,
которая несет в себе cos-участок Х-вектора (рис. 2.23). Сцепки молекул космид, связан-
ные своими cos-участками, служат субстратами для системы упаковки in vitro, потому
что cos-участок — единственная последова-
тельность, необходимая молекуле ДНК, чтобы
быть опознанной как «геном бактериофага X»
белками, которые упаковывают ДНК в части-
цы фага X. Частицы, содержащие космидную
ДНК, являются столь же инфекционными,
как и реальные фаги X, но как только космида
попадает в клетку, она не может направлять
синтез новых фаговых частиц и вместо этого
реплицируется, как и всякая плазмида. Поэто-
му рекомбинантная ДНК получается из коло-
ний, а не из бляшек. Как и в случае Х-векторов
других типов, верхний предел длины клони-
руемой ДНК устанавливается свободным про-
странством, имеющимся в пределах частицы
фага X. Сама космида может иметь размер
8 тыс.п.н. или даже меньше, так что в части-
цу фага X могут быть вставлены до 44 тыс. п. н.
новой ДНК, прежде чем будет достигнут ее
предел упаковки. Благодаря этому размер
Рис. 2.23. Типичная космида. pJB8 имеет в раз-
мере 5,4 тыс. п. н. и несет в себе ген устойчиво-
сти к ампициллину (ampR), сегмент ДНК фага X,
содержащий cos-участок, и область репликации
Escherichia coli (ori)
66
Часть 1. Изучение геномов
Позиции
истоков
репликации
Рис. 2.24. Ключевые структурные компоненты хромосомы
эукариота. За более подробной информацией относительно
этих структур обращайтесь к разделам 7.1.2 (центромеры
и теломеры) и 15.2.1 (область репликации)
геномной библиотеки человека
уменьшается приблизительно до
одной четверти миллиона клонов,
что является усовершенствовани-
ем по сравнению с библиотекой на
Х-векторах, но это число клонов,
с которыми приходится работать,
по-прежнему огромно.
Первое главное крупное до-
стижение в попытках клонировать
фрагменты ДНК намного длиннее,
чем 50 тыс. п. н., пришло с изобрете-
нием дрожжевых искусственных
хромосом, или YAC. Эти векторы
размножаются в S. cerevisiae, а не
в Е. coli и основаны на хромосомах, а не на плазмидах или вирусах. Первые YAC были
сконструированы после того, как исследования естественных хромосом показали, что,
наряду с содержащимися в ней генами, каждая хромосома несет в себе три важных
компонента (рис. 2.24):
• центромеру, которая играет ключевую роль во время деления ядра;
• теломеры — специальные последовательности, которые отмечают концы молекул
хромосомной ДНК;
• одну или несколько областей инициации, в которых начинается синтез новой
ДНК при делении хромосомы.
В YAC последовательности ДНК, которые лежат в основе этих компонентов хромо-
сомы, сцеплены с одним и более селективными маркёрами и по крайней мере с одним
участком рестрикции, в который может быть вставлена новая ДНК (рис. 2.25). Все эти
компоненты могут содержаться в молекуле ДНК размером 10-15 тыс. п. н. Естественные
хромосомы дрожжей варьируют в размере от 230 тыс. п. н. до более чем 1700 тыс. п. н., так
что YAC имеют потенциал для клонирования фрагментов ДНК размером порядка млн п. н.
Когда этот потенциал был осознан, создали стандартные YAC, способные клонировать
фрагменты размером 600 тыс. п. н., и специальные типы, способные обращаться с ДНК
длины до 1400 тыс.п.н. Это наибольшая емкость клонирующего вектора какого-либо
типа, и в нескольких первых проектах расшифровки генома YAC широко применялись.
К сожалению, оказалось, что YAC некоторых типов вызывают проблемы со стабиль-
ностью вставки, заключающиеся в том, что последовательность клонированной ДНК
перестраивается в новые комбинации. По этой причине молекулярные биологи имеют
большой интерес к векторам других типов — таким, которые не могут клонировать
столь большие части ДНК, но зато меньше страдают от проблем неустойчивости. К таким
векторам относятся нижеследующие.
• Бактериальные искусственные хромосомы, или ВАС, основанные на встречаю-
щейся в естественной среде обитания F-плазмиде Е. coli. В отличие от плазмид,
применявшихся для конструирования первых клонирующих векторов, F-плазмида
является относительно большой и основанные на ней векторы имеют более вы-
сокую емкость для вмещения в себя вставленной ДНК. ВАС разработаны таким
образом, что рекомбинанты могут быть идентифицированы Lac-отбором (см.
рис. 2.18) и благодаря этому очень удобны в применении. Они способны клони-
Глава 2. Изучение ДНК 67
ОБОЗНАЧЕНИЯ
CE/V4 Центромера из дрожжевой хромосомы IV
TEL Теломера
ori Область начала репликации
ТЯР1-]
SUP4 Селективные маркёры
URA3-*
Рис. 2.25. Работа с YAC. а) Клонирующий вектор pYAC3.6) Чтобы клонировать с помощью pYAC3, кольцевой
вектор переваривается с помощью ВатН\ и SnaBI. Рестрикция BomHI удаляет вкладочный фрагмент, на-
ходящийся между двумя теломерами в кольцевой молекуле. SnaBI делает разрез в пределах гена SUP4
и тем самым обеспечивает участок, в который будет вставлена новая ДНК. В результате лигирования
двух плеч вектора с новой ДНК образуется структура, показанная в основании схемы. Эта структура несет
функциональные копии селективных маркёров TRP1 и URA3. Штамм хозяина содержит инактивирован-
ные копии этих генов, что означает, что ему в качестве питательных веществ необходимы триптофан
и урацил. После трансформации клетки высеваются на минимальную среду, не содержащую триптофан
и урацил. На этой среде могут выжить и произвести колонии только клетки, которые содержат вектор и,
следовательно, способны синтезировать триптофан и урацил. Обратите внимание, что если вектор несет
в себе два правых плеча или два левых плеча, то он не даст начало колониям, потому что трансформи-
рованные клетки будут по-прежнему нуждаться в одном из питательных веществ. Присутствие вставки
ДНК в клонированных векторных молекулах проверяют путем теста на инактивацию гена SUP4. Его вы-
полняют пробой с окрашиванием: на соответствующей среде колонии, содержащие рекомбинантные
векторы (то есть со вставкой), окрашиваются в белый цвет; нерекомбинанты (содержащие векторы, но
не содержащие вставки) — в красный
ровать фрагменты длиной 300 тыс.п.н. и более, а вставки очень устойчивы. ВАС
интенсивно использовались в проекте «Геном человека» (раздел 4.3), и в настоящее
время они остаются самыми популярными векторами для клонирования больших
частей ДНК.
• Векторы на бактериофаге Р1 очень похожи на Х-векторы, будучи основаны на
урезанной версии генома природного фага, при этом вместимость клонирующего
вектора определяется размером делеции и объемом свободного пространства
внутри фаговой частицы. Геном фага Р1 больше генома бактериофага X, и фаговая
частица крупнее, так что Pl-вектор может клонировать более длинные фрагменты
ДНК, чем Х-вектор, — вплоть до 125 тыс.п.н. при существующей технологии.
68
Часть 1. Изучение геномов
• Полученные на основе бактериофага Р1 искусственные хромосомы, или РАС,
объединяют особенности Pl-векторов и ВАС и имеют вместимость до 300 тыс.п.н.
• Фосмиды содержат области инициации F-плазмиды и cos-участок бактериофага X.
Они подобны космидам, но имеют более низкое копийное число в Е. coli, что пред-
полагает их меньшую подверженность проблемам неустойчивости.
Размеры геномных библиотек человека, приготовленных в векторах этих типов,
приведены в таблице 2.4.
Клонирование в других организмах
Клонирование — не просто средство получения ДНК для секвенирования и других
типов анализа. Оно служит также инструментом для изучения механизма экспрессии
генов и способа регуляции их экспрессии, для выполнения экспериментов генной ин-
женерии, нацеленных на изменение биологических характеристик организма хозяина,
и для синтезирования важных животных белков (типа фармацевтических препаратов)
в новой клетке-хозяине, из которойэти белки могут быть выделены в больших количе-
ствах, чем это возможно при обычной очистке тканей животного. Для осуществления
всех этих многообразных целей часто бывает необходимо, чтобы гены клонировались
в других организмах, а не в Е. coli.
Клонирующие векторы на основе плазмид или фагов были разработаны для ра-
боты с большинством хорошо изученных видов бактерий типа Bacillus, Streptomyces
и Pseudomonas, притом эти векторы используются в точности таким же образом, что
и их аналоги на основе Е. coli. Также имеются плазмидные векторы для дрожжей
и грибов. Некоторые из них несут область инициации из 2-мкм-кольца — плазмиды,
присутствующей во многих штаммах S. cerevisiae, но другие плазмидные векторы имеют
только область инициации из Е. coli. Пример: У1р5-вектор для S. cerevisiae, являющийся
всего-навсего плазмидой Е. coli, которая содержит копию гена дрожжей по имени URA3
(рис. 2.26, а). Присутствие области инициации Е. coli означает, что YIр5 — челночный век-
тор, который может использоваться или с Е. coli, или с 5. cerevisiae в качестве организма-
хозяина. Это очень полезная особенность, потому что клонирование в S. cerevisiae есть
относительно неэффективный процесс и производство большого количества клонов
трудноосуществимо. Если эксперимент требует, чтобы в смеси клонов был идентифи-
цирован желаемый рекомбинант (как иллюстрировано на рис. 2.16), то может оказаться
невозможным получить достаточно много рекомбинантов, с тем чтобы было в чем
искать необходимый. Дабы избегнуть этой проблемы, конструирование молекул ре-
комбинантной ДНК и выбор правильного рекомбинанта выполняют с Е. coli в качестве
хозяина. Как только правильные клоны идентифицированы, рекомбинантные молекулы
YIp5 очищают и перемещают в S. cerevisiae, обычно путем смешивания ДНК с протопла-
стами — клетками дрожжей, стенки которых были удалены обработкой ферментами.
Без области инициации вектор не способен независимо размножаться внутри клеток
дрожжей, но он может сохраниться, если встроится в одну из хромосом дрожжей, что
может произойти путем гомологичной рекомбинации (раздел 5.2.2) между геном
URA3, несомым вектором, и хромосомной копией этого гена (рис. 2.26, б). Фактически
название «Yip» означает: «дрожжевая встраиваемая плазмида». Будучи встроен, вектор
Yip, а вместе с ним и любая ДНК, которая была в него вставлена, реплицируются наряду
с хромосомами хозяина.
Встраивание в хромосомную ДНК является особенностью также многих клони-
рующих систем, используемых с животными и растениями, и формирует основу кон-
Глава 2. Изучение ДНК 69
б) Вставка У1р5 в хромосомную ДНК дрожжей
а) Ylp5
Хромосомная ш
ДНК дрожжей
( Y|P5 )
\иядзJ
Мутация^^Гомологичная
рекомбинация
Мутация * Нет мутаций
URA3 Встроенная имз
ДНК Ylp5
hi
Рис. 2.26. Клонирование с помощью Yip. a) Y1 р5, типичная дрожжевая встраиваемая плазмида. Плазмида
содержит ген устойчивости к ампициллину (атря), ген устойчивости к тетрациклину (tet*), ген дрожжей
URA3 и область репликации Escherichia coli (ori). Присутствие ori Е. coli означает, что рекомбинантные
молекулы YIр5 могут быть построены в Е. coli перед их перемещением в клетки дрожжей. Поэтому Ylp5
представляет собой челночный вектор — он может перемещаться между двумя биологическими видами.
6) Ylp5 не имеет области репликации, которая может функционировать в клетках дрожжей, но может
сохраняться, если она встраивается вхромосомную ДНК дрожжей путем гомологичной рекомбинации
между плазмидой и хромосомными копиями гена URA3. Хромосомный ген несет маленькую мутацию,
что означает его нефункциональность, и клетки хозяина характеризуются генотипом ura3~. Один из пары
генов URA3, который формируется после встраивания ДНК плазмиды, мутированный, а другой — нет.
Поэтому рекомбинантные клетки обладают генотипом ura3+ и могут быть отобраны путем высева на
минимальную среду, лишенную урацила
струирования нокаутных мышей, которые используются для определения функций
новообнаруженных в геноме человека генов (раздел 5.2.2). Векторы представяют собой
животные эквиваленты У1ров. Когда цель состоит в том, чтобы вылечить генетическую
болезнь или рак гемотерапией, применяют клонирование генов животных при помо-
щи аденовирусов и ретровирусов. Подобный диапазон векторов был разработан и для
того, чтобы клонировать гены в растениях. Плазмиды могут быть введены в зародыши
растений бомбардировкой покрытыми ДНК микроснарядами — процесс, названный
биолистикой. Встраивание плазмидной ДНК в хромосомы растений, сопровождаемое
ростом зародыша, приводит к получению растения, которое содержит клонированную
ДНК в большинстве или всех своих клетках. Некоторый успех был достигнут также
и с растительными векторами, основанными на геномах вирусов мозаики цветной капу-
сты (ВМЦК) и геминивирусов, но самые интересные типы растительных клонирующих
векторов — полученные из Ti-плазмиды: большой бактериальной плазмиды, обнару-
женной в почвенном микроорганизме Agrobacterium tumefaciens. Часть Ti-плазмиды —
область, названная Т-ДНК, — встраивается в хромосому растения, когда эта бактерия
заражает стебель растения и вызывает появление корончатых галлов. Т-ДНК несет ряд
генов, которые экспрессируются в клетках растения и вызывают различные физиоло-
гические изменения, характерные для данной болезни. Так, были разработаны векторы
типа pBIN19 (рис. 2.27), использующие эту естественную систему генной инженерии.
Рекомбинантный вектор вводится в клетки A tumefaciens, которым позволяют заразить
взвесь клеток или культуру каллюса растения, а из них могут быть выращены зрелые
трансформированные растения.
70
Часть 1. Изучение геномов
Участки рестрикции
Левая граница
Т-ДНК
PBIN19
10 тыс. п. н.
Правая граница
Т-ДНК
Рис. 2.27. Растительный клонирующий вектор
pBIN19. pBIN19 несет ген lacZ' (см. рис. 2.18), ген
устойчивости к канамицину (kanR), область репли-
кации Escherichia coli (ori) и две граничные последо-
вательности из области Т-ДНК Ti-плазмиды. Эти две
граничные последовательности рекомбинируют
с хромосомной ДНК растения, в результате чего
расположенный между ними сегмент ДНК встав-
ляется в ДНК растения. Ориентация граничных
последовательностей в pBIN19 означает, что гены
lacZ и kanR, равно как и любая новая ДНК, лиги-
рованная в участки рестрикции в пределах lacZ',
перемещаются в ДНК растения. Рекомбинантные
клетки растения отбирают, высевая на содержа-
щий канамицин агар, и затем восстанавливают до
целых растений. Обратите внимание, что pBIN19
представляет собой еще один пример челночно-
го вектора, при этом рекомбинантные молекулы
конструируются в Е. coli с помощью системы от-
бора lacZ\ после чего переносятся в Agrobacterium
tumefaciens и далее — в растение
2.3. Полимеразная цепная реакция (ПЦР)
Клонирование ДНК — мощный метод и его вклад в наши знания о генах и геномах
трудно переоценить. Клонирование, однако, имеет и существенный недостаток: оно
отнимает много времени и, кое в чем, уйму сил. На выполнение манипуляций, необхо-
димых для вставки фрагментов ДНК в клонирующий вектор, последующего введения
лигированных молекул в клетки хозяина и отбора рекомбинантов уйдет несколько
дней. Если план эксперимента предполагает создание большой библиотеки клонов,
а затем и просмотр этой библиотеки с целью идентификации клона, который содержит
интересующий нас ген (см. техническое примечание 2.4), то может потребоваться не-
сколько недель, а то и месяцев для завершения проекта.
ПЦР дополняет клонирование ДНК в том, что она позволяет достичь того же резуль-
тата — выделения заданного фрагмента ДНК в чистом виде — за намного более короткое
время, возможно, всего лишь за несколько часов. Тем не менее ПЦР дополняет, но отнюдь
не заменяет клонирование, потому что она имеет свои собственные ограничения, наи-
более важное из которых состоит в необходимости знать последовательность по крайней
мере части фрагмента, который должен быть очищен. Несмотря на это ограничение,
ПЦР получила ведущую роль во многих областях исследований молекулярной биологии.
Сперва мы изучим сам метод, а затем рассмотрим варианты его применения.
2.3.1. Проведение ПЦР
ПЦР заключается в многократно повторяющемся копировании выбранной об-
ласти молекулы ДНК (см. рис. 2.3). В отличие от клонирования, ПЦР осуществляется
в пробирке и не предполагает использования живых клеток: копирование выполня-
ется не клеточными ферментами, а очищенной термостабильной ДНК-полимеразой
Т. aquaticus (раздел 2.1.1). Причина, по которой необходим термостабильный фермент,
станет ясна, когда мы более подробно рассмотрим события, происходящие во время
протекания ПЦР.
Глава 2. Изучение ДНК 71
Техническое примечание 2.4: работа с библиотекой клонов
Коллекции клонов используются как источник генов и других сегментов
ДНК
В качестве средства получения отдельных генов, а также и других сегментов
ДНК для дальнейшего изучения путем секвенирования и других методов технологии
рекомбинантной ДНК начиная с 1970-х гг. создавались библиотеки клонов на основе
материала из различных организмов. Такие библиотеки могут быть приготовлены
или из геномной ДНК, или из кДНК с помощью плазмидного или бактериофагово-
го вектора. Обычно клоны хранят в виде бактериальных колоний или бляшек на
агаровых пластинках размером 23x23 см и плотностью 100000-150000 клонов на
пластинку. Так, например, полная библиотека человека может уместиться всего
лишь на 1-8-ми пластинках в зависимости от типа используемого клонирующего
вектора (см. табл. 2.4). Клон, содержащий ген или другую часть ДНК, которую не-
обходимо найти, можно идентифицировать тремя методами:
Гибридизационный анализ может быть выполнен с меченым олигонуклео-
тидом или с другой молекулой ДНК, которая (что точно известно) комплементарна
искомой последовательности. Чтобы это сделать, нейлоновая или нитроцеллюлоз-
ная мембрана помещается на поверхность агаровой чашки и затем осторожно сни-
мается так, чтобы «поднять» колонии или бляшки. Обработка щелочью и протеазой
расщепляет клеточный материал, оставляя нетронутой ДНК из каждого клона,
которая затем прочно связывается с поверхностью мембраны путем нагревания или
ультрафиолетового излучения. После этого к мембране прикладывается меченый
зонд (точно так же, как при гибридизации по Саузерну (рис. 2.11)), и позиция, в ко-
торой зонд прикрепился, определяется соответствующим методом обнаружения.
При таком раскладе позиция сигнала гибридизации на мембране соответствует
местоположению интересующего нас клона на агаровой пластинке.
ПЦР (раздел 2.3) может использоваться для проверки клонов на содержание
интересующей нас последовательности. Это не может быть сделано in situ, так что
отдельные клоны должны быть перемещены в лунки лотка микротитра. Поэтому
основанный на ПЦР подход к идентификации клонов относительно труден, ибо на
одном лотке могут быть размещены лишь несколько сотен клонов. Поэтому проводят
серии ПЦР с затравками, специфичными к искомой последовательности, с каждым
клоном в свою очередь, по возможности применяя комбинаторный подход с целью
уменьшить число реакций ПЦР, которые необходимо провести, чтобы идентифици-
ровать ту, которая дает положительный результат (см. рис. 4.14).
Иммунологические методы могут использоваться, если искомая последова-
тельность представляет собой ген, экспрессируемый в клетке, в которой была при-
готовлена библиотека клонов. Если экспрессия гена происходит, то будет произведен
белковый продукт, а он может быть детектирован путем просмотра библиотеки
с помощью меченого антитела, которое связывается только с этим белком. Как
и в гибридизационном анализе, клоны сначала перемещают на мембрану и затем
обрабатывают, с тем чтобы разрушить клетки и связать белок с поверхностью мем-
браны. Затем на мембрану воздействуют меченым антителом, которое проявляет
позицию клона, содержащего интересующий нас ген.
72
Часть 1. Изучение геномов
Область для размножения
*'|| ГПТП 'ППТТТТ*. Целевая ДНК
Денатурация при 94 °C
5'ТТТГГТТТГПТПТГ 3‘
yLLIllLlLL-LLUUl у
Охлаждение до 50-60 °C
5'i । П I III IITJJJII i 3‘
__ Затравки
yililwtHIIHIi 5‘
| Синтез ДНК при 72 °C
f. JllllllllllinL.1 3'
I * •>/
«Длинные» продукты
Рис. 2.28. Первая стадия ПЦР
Чтобы выполнить эксперимент ПЦР, целевая
ДНК смешивается с ДНК-пол имеразой Taq, парой оли-
гонуклеотидных затравок и фондом нуклеотидов.
Количество целевой ДНК может быть очень малень-
ким, потому что ПЦР чрезвычайно чувствительна и
начнет работать даже с одной-единственной стар-
товой молекулой. Затравки необходимы для того,
чтобы инициировать реакции синтеза ДНК, которые
будут выполняться полимеразой Taq (см. рис. 2.6).
Они должны прикрепиться к целевой ДНК с каждой
стороны сегмента, который подлежит копированию:
поэтому последовательности этих сайтов связывания
должны быть известны, с тем чтобы можно было
синтезировать затравки с соответствующими по-
следовательностями.
Реакция начинается с нагрева смеси до 94 °C.
При этой температуре водородные связи, которые
скрепляют два полинуклеотида двойной спирали,
разрываются, так что целевая ДНК становится дена-
турированной до однонитевых молекул (рис. 2.28).
Затем температуру снижают до 50-60 °C, что приво-
дит к некоторому воссоединению отдельных нитей
целевой ДНК, но также позволяет затравкам прикре-
питься к своим позициям отжига. После этого может быть начат синтез ДНК, так что
температуру поднимают до 72 °C — оптимального для полимеразы Taq значения. На этой
первой стадии ПЦР на каждой из нитей целевой ДНК синтезируется набор «длинных»
продуктов. Эти полинуклеотиды имеют идентичные 5’-концы, но случайные З’-концы,
причем последние представляют позиции, в которых синтез ДНК заканчивается случай-
ным образом. Когда цикл денатурация-отжиг-синтез повторяется, длинные продукты
выступают в качестве матриц для синтеза новой ДНК и дают начало «коротким» про-
дуктам, 5’- и З’-концы которых определяются позициями отжига затравок (рис. 2.29).
В последующих циклах число коротких продуктов возрастает по экспоненциальному
закону (удваиваясь во время каждого цикла), пока один из компонентов реакции не
исчерпывается. Это означает, что после 30-ти циклов в реакционной смеси будет более
чем 250 миллионов коротких продуктов, полученных из каждой стартовой молекулы.
В реальных единицах это соответствует нескольким микрограммам продукта ПЦР из
нескольких нанограмм или даже меньшего количества целевой ДНК.
Результаты ПЦР могут быть определены различными способами. Обычно про-
дукты анализируют посредством электрофореза в агарозном геле, который покажет
единственную полосу, если ПЦР работала, как и ожидалось, и размножила единственный
сегмент целевой ДНК (рис. 2.30). Последовательность продукта может быть определена
и другими методами, описанными в разделе 4.1.1.
2.3.2. Применение ПЦР
ПЦР — настолько прямая и незамысловатая процедура, что иногда трудно по-
нять, как она смогла стать настолько важной в современном исследовании. Сначала мы
рассмотрим ее ограничения. Для того чтобы синтезировать затравки, которые в ходе
отжига прикрепятся в надлежащих позициях, должны быть известны последовательно-
Глава 2. Изучение ДНК 73
сти граничных областей ДНК, которая будет
размножена. Это означает, что ПЦР не может
быть использована для очистки фрагментов
генов либо каких-либо иных частей генома,
которые никогда не изучались прежде. Вто-
рое ограничение — длина отрезка ДНК, ко-
торый может быть скопирован. Области до
5 тыс.п.н. могут быть размножены без осо-
бой трудности, а размножение более длинных
фрагментов—до 40 тыс. п. н. — является воз-
можным при использовании модификаций
стандартной технологии. Однако фрагменты
более 100 тыс. п. н., которые необходимы для
проектов секвенирования геномов и которые
могут быть получены путем клонирования
в ВАС или другом векторе высокой емкости,
являются недосягаемыми для ПЦР.
Каковы сильные места ПЦР? Первое
среди них — простота, с которой продукты,
представляющие отдельный сегмент генома,
могут быть получены из ряда различных об-
разцов ДНК. С одним важным примером этого
преимущества мы встретимся в следующей
главе, когда будем рассматривать способы ти-
пизации маркёров ДНК в проектах генетиче-
ского картирования (раздел 3.2.2). Подобным
образом ПЦР используется для просмотра об-
разцов ДНК человека на предмет мутаций,
связанных с генетическими болезнями типа
талассемии и кистозного фиброза. Она также
формирует основу генетического профилиро-
вания, в котором типизируются изменения
длины микросателлитов (см. рис. 7.24).
Вторая важная особенность ПЦР — ее
способность работать с крохотными количе-
ствами отправной ДНК. Это означает, что ПЦР
5*- t _ з*
adllllllllimiy Продукты первого цикла
LHIHLUW
Денатурация
| Синтез ДНК
в 1111ИIII111Г Продукты второго цикла
ашщггг
| Денатурация
TTWWW
| Синтез ДНК
Тптгггпттг
I
Продукты третьего цикла
«Короткий» продукт -
накапливается экспоненциально
Рис. 2.29. Синтез «коротких» продуктов в ходе
ПЦР. Из продуктов первого цикла, показанных
наверху схемы, следующий цикл денатурация-
отжиг-синтез дает четыре продукта, два из
которых идентичны продуктам первого цикла,
а два — полностью состоят из новой ДНК. В ходе
третьего цикла последние дают начало «корот-
ким» продуктам, которые в последующих циклах
накапливаются по экспоненте
iiiiiiiiiiiiaii
। it 11II1111jggp
можно применять для получения последо-
вательностей из ничтожных количеств ДНК, которые присутствуют в волосах, пятнах
крови и других судебно-медицинских образцах, а также из костей и других останков,
сохранившихся в археологических участках. В клинической диагностике ПЦР способна
обнаружить присутствие вирусной ДНК задолго до того, как вирус достигнет уровней,
необходимых для инициирования ответной реакции. Это особенно важно для ранней
идентификации вызываемых вирусами раковых образований, потому что это дает шанс
начать программы лечения прежде, чем рак станет устойчивым.
Все вышеупомянутое — только несколько из возможных вариантов применения
ПЦР. В настоящее время этот метод является главным компонентом инструментария
молекулярного биолога, и мы обнаружим еще много примеров его использования, по
мере того как будем проходить очередные главы этой книги.
74
Часть 1. Изучение геномов
Электрофорез в
агарозном геле
1 2 3
Рис. 2.30. Анализ результатов ПЦР при помощи электрофореза в агарозном геле. ПЦР была выполнена
в микроцентрифужной пробирке. Образец загружают на дорожку 2 агарозного геля. Дорожка 1 содер-
жит маркёры размера ДНК, а дорожка 3 содержит образец ПЦР, выполненной вашим коллегой. После
электрофореза гель окрашивается бромистым этидием (см. техническое примечание 2.2). Дорожка 2
содержит единственную полосу ожидаемого размера, что свидетельствует об успешном проведении
ПЦР. На дорожке 3 нет ни одной полосы — значит, эта ПЦР не удалась
Заключение
За последние 35 лет молекулярные биологи создали всесторонний инструмента-
рий методов, которые могут использоваться для изучения ДНК. Эти методы образуют
основу технологии рекомбинантной ДНК и привели к разработке процедур клониро-
вания ДНК и полимеразной цепной реакции (ПЦР). Главной особенностью технологии
рекомбинантной ДНК является использование очищенных ферментов, чтобы произ-
водить заданные изменения в молекулах ДНК в пробирке. Четыре основных типа фер-
ментов, употребляемых для этой цели, представлены: ДНК-полимеразами, нуклеазами,
лигазами и ферментами модификации концов. ДНК-полимеразы синтезируют новые
полинуклеотиды ДНК и используются в таких процедурах, как секвенирование ДНК,
ПЦР и мечение ДНК. Самые важные нуклеазы — эндонуклеазы рестрикции, которые
разрезают двунитевые молекулы ДНК в определенных последовательностях нуклеоти-
дов и, следовательно, нарезают молекулу на предсказуемый набор фрагментов, размеры
которых могут быть определены при помощи электрофореза в агарозном геле. Лигазы
соединяют молекулы вместе, а ферменты модификации концов выполняют массу раз-
нообразных реакций, в том числе несколько, применяемых для мечения молекул ДНК.
Клонирование ДНК является средством получения чистого образца отдельного гена
или другого сегмента молекулы ДНК.
Многие различные типы клонирующих векторов были разработаны для исполь-
зования с Е. coli в качестве организма-хозяина, самые простые из них основаны на
маленьких плазмидах, которые несут маркёры типа гена lacZ1. Этот ген позволяет
идентифицировать рекомбинантные колонии, потому что они выглядят белыми, а не
синими, когда в среде для выращивания присутствует Х-гель. Бактериофаг X также при-
менялся как основа создания ряда клонирующих векторов для Е. coli, включая гибриды
типа плазмида-фаг, названные космидами, которые используются, чтобы клонировать
фрагменты ДНК до 44 тыс. п. н. длины. Векторы других типов, такие как бактериальные
искусственные хромосомы, могут быть использованы для клонирования ещё более
длинных частей ДНК, вплоть до 300 тыс.п.н. в длину. Подобные векторы высокой ем-
кости используются при построении библиотек клонов — коллекций клонов, вставки
которых охватывают полный геном и которые используются в качестве материального
фонда для проектов секвенирования геномов. В качестве хозяев для осуществления
клонирования ДНК вместо Е. coli могут быть использованы также и другие организмы.
Глава 2. Изучение ДНК 75
Векторы нескольких типов были разработаны для Saccharomyces cerevisiae, и созданы
специализированные методы клонирования ДНК в животных и растениях. ПЦР пред-
ставляет собой дополнение к клонированию ДНК, позволяя быстро очищать заданные
сегменты ДНК, но по крайней мере часть последовательности ДНК этого фрагмента
должна быть известна. В ходе ПЦР термостабильная ДНК-полимераза делает повторные
копии целевой последовательности и копий, произведенных на более ранних циклах
реакции. Начиная лишь с единственной целевой молекулы, более 250-ти миллионов
копий могут быть получены в течение 30-ти циклов ПЦР.
Задания для закрепления материала
* Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
2.1 *. Какие из следующих ферментов ис-
пользуются для расщепления моле-
кулДНК?
а. ДНК-полимеразы.
Ь. Нуклеазы.
с. Лигазы.
d. Киназы.
2.2 . Почему направляемой матрицей
ДНК-полимеразе для запуска син-
теза ДНК необходима затравка?
а. Этим полимеразам для присоеди-
нения нового нуклеотида нужна
5'-фосфатная группа.
Ь. Этим полимеразам для присоеди-
нения нового нуклеотида нужна
З'-гидроксильная группа.
с. Затравка требуется для того, что-
бы ДНК-полимераза могла при-
крепиться к матрице ДНК.
d. Затравка гидролизуется, с тем
чтобы обеспечить энергию, не-
обходимую для синтеза ДНК.
2.3 *. Действие экзонуклеазы 3'—>5’ при-
суще ДНК-полимеразе для того,
чтобы:
а. Удалять 5'-конец нити полинукле-
отида, присоединенного к копи-
руемой матричной нити.
Ь. Удалять поврежденные нуклео-
тиды с матричной нити во время
синтеза ДНК.
с. Удалять нуклеотиды с концов мо-
лекул ДНК, чтобы гарантировать
порождение тупых концов.
d. Удалять неправильные нуклеоти-
ды из недавно синтезированной
нити ДНК.
2.4 . Версия полимеразы Клёнова из
ДНК-полимеразы I Е. coli пригодна
для проведения исследований, по-
скольку она не обладает действием
экзонуклеазы 5>3 Ее пригодность
обусловлена тем, что действие экзо-
нуклеазы 5’—>3':
а. Сильнее, чем собственно акив-
ность полимеразы.
Ь. Предотвратит встраивание ради-
оактивных или флюоресцентных
меток в ДНК.
с. Может препятствовать проведе-
нию некоторых исследователь-
скихпроцеду^укорачивая 5’-кон-
цы молекул ДНК.
d. Не дает полимеразе обнаружи-
вать ошибки при встраивании
новых нуклеотидов.
76 Часть 1. Изучение геномов
2.5 *. Температура 75 °C прерывает синтез
ДНК, осуществляемый ДНК-поли-
меразой IЕ coli. Это связано с тем, что:
а. ДНК-полимераза I Е coli денату-
рируется при этой температуре.
Ь. ДНК денатурируется при этой
температуре.
с. Затравки денатурируют при этой
температуре.
d. Температура слишком высока для
протекания ферментативных ре-
акций.
2.6 . Какая из следующих характеристик
в точности описывает обратные
транскриптазы?
а. Они присутствуют во всех виру-
сах и представляют собой РНК-
зависимые ДНК-полимеразы.
Ь. Они присутствуют во всех РНК-
содержащих вирусах и представ-
ляют собой ДНК-зависимые РНК-
полимеразы.
с. Они присутствуют в ретровиру-
сах и представляют собой РНК-
зависимые ДНК-полимеразы.
d. Они присутствуют во всех ви-
русах и представляют собой
независимые от матрицы ДНК-
полимеразы.
2.7 *. Ферменты рестрикции всех трех
типов связываются с молекулами
ДНК в определенных последова-
тельностях; однако ферменты II
типа предпочтительны для иссле-
довательских целей. С которой из
следующих причин это связано?
а. Ферменты II типа разрезают ДНК
в определенном участке.
Ь. Ферменты II типа всегда разреза-
ют ДНК таким образом, чтобы по-
лучить тупоконечные молекулы.
с. Ферменты II типа всегда разре-
зают ДНК таким образом, чтобы
получить молекулы с липкими
концами.
d. Ферменты II типа — единствен-
ные ферменты рестрикции, спо-
собные расщеплять двунитевую
ДНК.
2.8 . Какой метод используется для раз-
деления фрагментов ДНК различных
размеров после перевариваривания
их ферментами рестрикции?
а. Секвенирование ДНК.
Ь. Гель-электрофорез.
с. Клонирование генов.
d. ПЦР.
2.9 *. Связи какого типа синтезирует ДНК-
лигаза?
а. Водородные связи между основа-
ниями.
Ь. Фосфодиэфирные связи между
нуклеотидами.
с. Связи междуоснованиями и остат-
ками сахара дезоксирибозы.
d. Пептидные связи между амино-
кислотами.
2.10 . Какая из следующих полимераз не
нуждается в матрице?
а. ДНК-полимераза I.
Ь. Секвеназа.
с. Обратная транскриптаза.
d. Концевая дезоксинуклеотидил-
трансфераза.
2.11 *. При помощи которого из следующих
методов клетки Е coli принимают
в себя ДНК плазмиды в лаборатор-
ных экспериментах?
а. Конъюгация.
Ь. Электрофорез.
с. Трансдукция.
d. Трансформация.
2.12 . Что такое геномная библиотека?
а. Коллекция рекомбинантных мо-
лекул со вставками, которые со-
держат все гены организма.
Ь. Коллекция рекомбинантных
молекул со вставками, которые
содержат полный геном организ-
ма.
с. Коллекция рекомбинантных мо-
лекул, которые экспрессируют все
гены организма.
Глава 2. Изучение ДНК 77
d. Коллекция рекомбинантных мо-
лекул, которые были секвениро-
ваны.
2.13 *. Вектор какого из следующих типов
был бы наиболее подходящим для
введения ДНК в клетку человека?
а. Плазмида.
Ь. Бактериофаг.
с. Космида.
d. Аденовирус.
2.14 . Что из следующего НЕ используется
для введения молекул рекомбинант-
ной ДНК в растения?
а. Биолистика.
Ь. Космиды.
с. Ti-плазмида.
d. Вирусы.
2.15 *. ПЦР выгодна для клонирования
генов по всем нижеперечисленным
причинам, кроме:
а. ПЦР не требует, чтобы последова-
тельность гена была известна.
Ь. ПЦР — очень быстрый метод вы-
деления того или иного гена.
с. ПЦР по сравнению с клонирова-
нием генов требует очень малень-
ких количеств стартовой ДНК.
d. ПЦР в высокой степени пригодна
для картирования маркёров ДНК.
Вопросы открытого типа
2.1 *. Что означает термин «клонирова-
ние генов»?
2.2 . Как исследователь может иденти-
фицировать отдельный содержащий
интересующий его ген фрагмент, по-
лученный при переваривании фер-
ментом рестрикции геномной ДНК,
которая содержит тысячи различ-
ных фрагментов рестрикции?
2.3 *. Назовите полезный и быстрый ме-
тод повышения эффективности ли-
гирования тупоконечных молекул
ДНК.
2.4 . Почему плазмиды являются удобны-
ми клонирующими векторами?
2.5 *. Для чего плазмиды содержат гены
устойчивости к антибиотикам?
2.6 . Что такое Х-гель, добавляемый
к среде в экспериментах с клониро-
ванием?
2.7 *. Почему бактериофаг X пригоден
в качестве клонирующего вектора?
2.8 . Почему для создания библиотек кло-
нов выгодны векторы, которые мо-
гут нести большие вставки ДНК?
2.9 *. Какиетрисвойстванормальныххро-
мосом должны иметь дрожжевые ис-
кусственные хромосомы, чтобы они
могли поддерживаться в клетках?
2.10 . Почему начальные продукты ПЦР, —
произведенные за несколько первых
циклов реакции, — длинные и раз-
ных размеров, а все конечные про-
дукты ПЦР имеют более короткий
и одинаковый размер?
2.11 *. Каким образом затравки определя-
ют специфику ПЦР?
2.12 . Какого типа последовательности
ДНК не могут быть размножены по-
средством ПЦР?
78
Часть 1. Изучение геномов
Изыскательские задачи
2.1 *. Вскоре после того, как в начале
1970-х гг. были проведены первые
эксперименты по клонированию,
ряд ученых отстаивал мнение о том,
что должен быть введен временный
мораторий на исследования такого
рода. Чем обосновывались опасения
этих ученых и до какой степени эти
опасения были оправданны?
2.2 . Какими свойствами должен об-
ладать идеальный клонирующий
вектор? До какой степени этим тре-
бованиям удовлетворяет любой из
существующих ныне клонирующих
векторов?
2.3 *. Каким способом, кроме расшифров-
ки последовательности молекулы,
можно было бы определить пози-
ции участков рестрикции в молеку-
леДНК?
2.4 . Рассмотрите примеры использова-
ния клонирования генов в произ-
водстве животных белков в клетках
бактерий.
2.5 *. Специфичность затравок — опре-
деляющее свойство успешной ПЦР.
Если затравки в ходе отжига при-
крепятся к целевой ДНК в более чем
одной позиции, то, кроме искомого,
будут синтезированы дополнитель-
ные продукты. Исследуйте факторы,
которые определяют специфичность
затравки, и оцените влияние темпе-
ратуры отжига на исход ПЦР.
Тесты по рисункам
2.1 *. Какова роль затравки (показана синим цветом) в реакциях синтеза ДНК, катали-
зируемых ДНК-полимеразой?
Для синтеза ДНК необходима затравка
Затравка
3f-LLl1............ 5' 3,ТТТ....................... 5'
ДНК-полимераза
Новая ДНК
I l. l l.LLJ-l IJLLUJJL
Синтез ДНК не происходит
imiiiiiii
Синтез ДНК
Участки рестрикции BamHI __
ДНК животного Плазмида Е. coli
±ВатН\ 1
Ген животного ( )
и4 /'О'
Лигирование
JL Ген животного
встраивается
в плазмиду
2.2. Что если разрезаемая BamHI плазмида, исполь-
зуемая в этом эксперименте, с высокой частотой
обратно лигируется сама с собой и поэтому будет
выделено очень мало рекомбинантных плазмид?
Каким образом можно улучшить реакцию лигирова-
ния, с тем чтобы увеличить выход рекомбинантных
плазмид?
Глава 2. Изучение ДНК 79
2.3*. Этот клонирующий вектор обладает
бактериальной областью инициа-
ции, селективным маркёром (ген
устойчивости к антибиотику) и cos-
участками бактериофага. К какому
типу относится этот клонирующий
вектор и какого размера вставлен-
ную молекулу он может в себе не-
сти?
2.4. Какого типа реакция представлена
на этом рисунке? Обозначьте этапы
этого процесса и укажите темпера-
туры для каждой его процедуры.
Область для размножения
уШШППТТШТу Целевая ДНК
I
5 Ti l l ГI 14 ITI Г1 III'3
уЛ t-tl 1 1Л.1 I 1 1 1 » 1 1 L
Willi ЦНИИ
«Длинные» продукты
Дополнительная литература
Учебники и практические справочники по методам, применяемым для иссле-
дования ДНК
Brown,Т. А. (2006) Gene Cloning and DNA Analysis: An Introduction, 5th Ed. Blackwell
Scientific Publishers, Oxford.
Brown,T.A. (ed.) (2000) Essential Molecular Biology: A Practical Approach, Vol. 1 and 2,
2nd Ed. Oxford University Press, Oxford. Includes detailed protocols for DNA cloning and PCR.
Dale, J. W. (2004) Molecular Genetics of Bacteria, 4th Ed. Wiley, Chichester. Provides
a detailed description of plasmids and bacteriophages.
Ферменты для манипулирования ДНК
Brown, Т.А. (1998) Molecular Biology Labfax. Volume I: Recombinant DNA, 2nd Ed.
Academic Press, London. Contains details of all types of enzymes used to manipulate DNA and
RNA.
REBASE: http://rebase.neb.com/rebase/ A comprehensive list of all the known restriction
endonucleases and their recognition sequences.
Smith, H.O. and Wilcox, K.W. (1970) A restriction enzyme from Haemophilus influenzae.
J. Mol. Biol. 51: 379-391. One of the first full descriptions of a restriction endonuclease.
80
Часть 1. Изучение геномов
Клонирование ДНК
Frischauf,A.-M., Lehrach, Н., Poustka,A. and Murray, N. (1983) Lambda replacement
vectors carrying polylinker sequences./. Mol. Biol. 170: 827-842.
Hohn, B. and Murray, K. (1977) Packaging recombinant DNA molecules into bacteriophage
particles in vitro. Proc. Natl Acad. Sci. USA 74: 3259-3263.
Vieira, J. and Messing, J. (1982) The pUC plasmids, an M13mp7-derived system for
insertion mutagenesis and sequencing with synthetic universal primers. Gene 19:259-268.
Клонирующие векторы высокой емкости
Burke, D. T., Carle, G. F. and Olson, M. V. (1987) Cloning of large segments of exogenous
DNA into yeast by means of artificial chromosome vectors. Science 236: 806-812. YACs.
loannou, P. A., Amemiya, С. T., Games, J., Kroisel, P. M., Shizuya, H., Chen, C., Batzer, M. A.
and de Jong, P. J. (1994) Pl-derived vector for the propagation of large human DNA fragments.
Nat Genet 6: 84-89. PACs.
Kim, U.-J., Shizuya, H., de Jong, P. J., Birreii, B. and Simon, M. I. (1992) Stable propagation
of cosmid and human DNA inserts in an F factor based vector. Nucleic Acids Res. 20:1083-1085.
Fosmids.
Monaco, A. P. and Larin, Z. (1994) YACs, BACs, PACs and MACs - artificial chromosomes
as research tools. Trends Biotechnol. 12: 280-286. A good review of high-capacity cloning
vectors.
Shizuya, H., Birren,B., Kim,U.J., Mancino,V., Slepak,T., Tachiiri,Y. and Simon, M.
(1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in
Escherichia coli using an F-factor-based vector. Proc. Natl Acad. Sci. USA 89: 8794-8797. The
first description ofaBAC.
Sternberg, N. (1990) Bacteriophage PI cloning system for the isolation, amplification,
and recovery of DNA fragments as large as 100 kilobase pairs. Proc. Natl Acad. Sci. USA 87:
103-107. Bacteriophage Pl vectors.
Клонирование в организмах животных и растений
Bevan, М. (1984) Binary Agrobacterium vectors for plant transformation. Nucleic Acids
Res. 12:8711-8721.
Colosimo,A., Goncz,K. K., Holmes, A. R., Kunzelmann, K., Novelli,G., Malone, R.W.,
Bennett, M. J. and Gruenert, D. C. (2000) T ransfer and expression of foreign genes in mammalian
cells. Biotechniques 29: 314-321.
Hansen,G. and Wright,M.S. (1999) Recent advances in the transformation of plants.
Trends Plant Sci. 4: 226-231.
Kost, T. A. and Condreay, J. P. (2002) Recombinant bac-uloviruses as mammalian cell
gene-delivery vectors. Trends Biotechnol. 20:173-180.
ПЦР
Mullis, K.B. (1990) The unusual origins of the polymerase chain reaction. Sci. Am. 262
(4): 56-65.
Saiki,R.K., Gelfand, D. H„ Stoffel, S., Scharf, S. J., Higuchi,R., Horn,G.T., Mullis, K.B.
and Erlich, H. A. (1988) Primer-directed enzymatic amplification of DNA with a thermostable
DNA polymerase. Science 239: 487-491.
Глава 3
Картирование геномов
По прочтении главы 3 вы сможете:
• Объяснять, почему карта представляет собой важное вспомогательное средство
при секвенировании геномов.
• Проводить различие между понятиями «генетическая карта» и «физическая
карта».
• Описывать маркёры различных типов, применяемые для построения генетиче-
ских карт, и излагать, как маркёры каждого типа получают очки.
• Обрисовывать принципы наследования согласно открытиям Менделя и по-
казывать, как последующие генетические исследования привели к развитию
анализа сцепления между генными локусами.
• Объяснять, каким образом анализ сцепления используется для построения
генетических карт, описывая подробности того, как такой анализ проводится
в организмах различных типов, включая людей и бактерии.
• Озвучивать ограничения генетического картирования.
• Оценивать сильные и слабые стороны различных методов, применяемых для
построения физических карт геномов.
• Описывать принципы составления рестрикционных карт.
• Описывать, как для построения физических карт используется флюоресцентная
гибридизация in situ (FISH), включая ее модификации, применяемые для повы-
шения чувствительности этого метода.
• Объяснять, что лежит в основе карт меченых участков последовательности
(STS), и перечислять различные последовательности ДНК, которые могут быть
использованы в качестве STS.
• Описывать, каким образом в картировании STS используются радиационные
гибриды и библиотеки клонов.
Следующие две главы описывают методы и стратегии, применяемые для рас-
шифровки последовательностей геномов. Секвенирование ДНК, очевидно, главнейший
среди этих методов, но секвенирование имеет одно главное ограничение: даже с самой
искушенной технологией за один эксперимент редко удается определить последова-
тельность размером более 750 п.н. или около того. Это означает, что последователь-
ность длинной молекулы ДНК приходится выстраивать из множества более коротких
последовательностей. Это осуществляется путем разбиения молекулы на фрагменты,
определения последовательности каждого из них и, наконец, поиска перекрытий и по-
строения основной последовательности с помощью компьютера (рис. 3.1). Этот метод
82
Часть 1. Изучение геномов
ДНК
500 п. н.
Рис. 3.1. Метод дробовика для сборки последовательно-
сти. Молекула ДНК разбивается на маленькие фрагменты,
каждый из которых секвенируется. Основная последова-
тельность собирается путем отыскания перекрытий между
последовательностями отдельных фрагментов. На практи-
ке, чтобы установить, что две последовательности должны
быть связаны вместе, необходимо перекрытие нескольких
десятков пар оснований
дробовика — стандартный подход
к секвенированию небольших гено-
мов прокариотов (раздел 4.2.1), но
с более крупными геномами он осу-
ществляется гораздо труднее, пото-
му что необходимый анализ данных
несоразмерно усложняется по мере
увеличения числа фрагментов (для
п фрагментов число возможных пе-
рекрытий определяется выражени-
ем вида 2п2 - 2п). Вторая проблема
с применением метода дробовика
состоит в том, что он может вести
к ошибкам при анализе повторных
областей генома. Когда повторяю-
щаяся последовательность раз-
деляется на фрагменты, многие
из получившихся частей содержат
одинаковые или весьма подобные
мотивы последовательности. Мож-
но запросто ошибочно собрать эти
последовательности таким обра-
зом, что часть повторной области
окажется неучтенной, или даже со-
единить друг с другом не имеющие
ничего общего в территориальном
смысле части одной и той же молекулы или даже разных хромосом (рис. 3.2).
В силу трудностей в применении метода дробовика к большой молекуле, которая
содержит весомую долю повторяющейся ДНК, этот подход не может быть непосредствен-
но использован для секвенирования генома эукариота. Вместо этого сначала должна
быть создана карта генома. Карта генома служит руководством для экспериментов
секвенирования, показывая позиции генов и других отличительных особенностей. Как
только карта генома получена, входящая в проект стадия секвенирования может пойти
по одному из двух путей (рис. 3.3).
• Согласно методу дробовика для полных геномов (раздел 4.2.3), который в целом
основан на том же самом подходе, что и стандартный метод дробовика, использу-
ются отличительные особенности на карте генома в качестве ориентиров, чтобы
помочь сборке основной последовательности из огромных чисел коротких после-
довательностей, которые будут получены в процессе работы. Обращение к карте
гарантирует также и то, что области, содержащие повторную ДНК, будут собраны
правильно. Метод дробовика для полных геномов — быстрый способ получить
схему последовательности генома эукариота.
• Следуя методу сборки контигов из клонов (раздел 4.2.2), геном разбивается на
поддающиеся манипулированию сегменты, каждый по несколько сотен тыс. п. н. или
несколько млн п. н. в длину, которые являются достаточно короткими для точного
секвенирования методом дробовика. Как только последовательность того или
иного сегмента расшифровывается, его помещают в надлежащее местоположение
Глава 3. Картирование геномов 83
Рис. 3.2. Проблемы метода дробовика, а) Молекула ДНК содержит тандемный повторный элемент,
состоящий из многих копий последовательности GATTA. В ходе просмотра последовательности ДНК
между двумя такими фрагментами идентифицируется перекрытие, но это копии с концов тандемно-
го повторения. Если ошибка не распознана, то внутренняя область тандемного повторения не будет
включена в основную последовательность. 6) Во втором примере молекула ДНК содержит две копии
повторяющегося элемента. Когда последовательности просматриваются, два фрагмента оказываются
перекрытыми, но один фрагмент содержит левую часть одного повтора, а другой фрагмент — правую
часть второго повтора. В таком случае неудача в распознавании ошибки приведет к тому, что сегмент
ДНК, расположенный между этими двумя повторениями, будет выброшен из основной последователь-
ности. Если эти два повторения были бы на разных хромосомах, то последовательности этих хромосом
могли бы по ошибке быть связаны вместе
на карте. Такой поэтапный подход требует больше времени, чем секвенирование
методом дробовика для полных геномов, но дает более точную и свободную от
ошибок последовательность.
При применении обоих методов карта обеспечивает контур для выполнения стадии
секвенирования в ходе проекта. Если карта указывает позиции генов, то она может быть
использована также и для того, чтобы на начальной стадии направлять проект сборки
контигов из клонов на вызывающие особый интерес области генома, с тем чтобы по-
следовательности важных генов были получены как можно быстрее.
3.1. Генетические и физические карты
Согласно общепринятой классификации, методы картирования геномов подраз-
деляют на две категории.
• Генетическое картирование базируется на использовании генетических мето-
дов, чтобы строить карты, показывающие позиции генов и других особенностей
последовательности генома. В число таких генетических методов входят экспе-
рименты по скрещиванию или, в случае людей, экспертиза семейных анамнезов
(родословных). Генетическое картирование описано в разделе 3.2.
• Физическое картирование использует методы молекулярной биологии, чтобы
исследовать молекулы ДНК непосредственно, с тем чтобы строить карты, показы-
вающие позиции особенностей последовательности, в том числе генов. Физическое
картирование описано в разделе 3.3.
84 Часть 1. Изучение геномов
500 тыс. п. н.
Метод сборки контигов ।__________.
из клонов
Маркёры Метод дробовика
! 1 I для полных геномов
F СН
* Карта генома
\ Секвенирование полного
генома методом дробовика
{Секвенирование нанесенного
на карту сегмента методом дробовика
Собранная вседик,
последовательность
Позиция
лоследовательчосги
уже известна
Собранные воедино
последовательности
Маркёры, употребляемые
/ для закрепления собранных
/ последовательностей на карте
Рис. 3.3. Альтернативные подходы к секвенированию генома. Геном, состоящий из линейной молеку-
лы ДНК размером 2,5 млн п. н., был нанесен на карту, и установлены позиции восьми маркёров (А-Н).
Слева метод сборки контигов из клонов начинает с сегмента ДНК, позиция которого на карте генома
была идентифицирована, потому что он содержит маркёры А и В. Этот сегмент секвенируется методом
дробовика, и основная последовательность помещается в ее известную позицию на карте. Справа метод
дробовика для полных геномов производит беспорядочное секвенирование полного генома. В резуль-
тате мы получаем куски с непрерывной последовательностью, возможно сотни тыс.п.н. в длину. Если
эта непрерывная последовательность содержит маркёр, то она может быть помещена на карту генома.
Обратите внимание, что в любом из методов чем больше будет маркёров на карте генома, тем лучше.
Более подробное описание этих стратегий секвенирования приведено в разделе 4.2
3.2. Составление генетических карт
Подобно любой другой карте, генетическая карта должна показывать позиции
каких-либо отличительных признаков. На географической карте такими маркёрами
служат узнаваемые особенности ландшафта, такие как реки, дороги и здания. Какие
же маркёры мы можем узреть на генетическом пейзаже?
3.2.1. Первыми известными маркёрами служили гены
При создании первых генетических карт в начале XX века объектами служили
организмы типа плодовой мушки, а в качестве маркёров на протяжении нескольких
десятилетий использовали гены. Чтобы быть полезным для генетического анализа, ген
должен существовать по крайней мере в двух формах, или аллелях, и при этом каждый из
них определял бы отличительный фенотип — например, длинные или короткие стебли
у растений гороха, первоначально изучаемых Грегором Менделем. Вначале единствен-
ными генами, которые могли быть изучены, были такие гены, которые определяют
зрительно легко различимые фенотипы. Так, например, первые карты плодовой мушки
показывали позиции генов, определяющих цвета тела, цвета глаз, формы крыльев и тому
Глава 3. Картирование геномов 85
подобные признаки, при этом все эти фенотипы различимы при простом наблюдении
мух в микроскоп малой мощности или при их осмотре невооруженным глазом. Этот
подход был прекрасен в эпоху становления генетики, но вскоре ученые мужи поняли,
что в их распоряжении имеется только ограниченное число визуальных фенотипов, на-
следование которых могло быть изучено, и во многих случаях их анализ был усложнен,
потому что на один фенотип может влиять сразу несколько генов. Например, к 1922 г.
на карту плодовой мушки было нанесено более 50-ти генов, расположенных на четырех
хромосомах, но девять из этих генов определял и цвет глаз. В более поздних исследовани-
ях генетикам, изучавшим плодовых мушек, пришлось научиться различать такие цвета
мушьих глаз, как красный, светло-красный, пунцовый, гранатовый, телесный, киновар-
ный, рубиновый, сепии, алый, розовый, пурпурный, бордовый, багряный и коричневый.
Чтобы строить более сложные карты генов, необходимо было найти характеристики,
которые различались бы более четко и однозначно, чем визуальные.
Решение было найдено в том, чтобы использовать в качестве фенотипических
признаков организма его биохимические особенности. Это было особенно важно в от-
ношении организмов двух типов — микробов и человека. Микробы, такие как бактерии
и дрожжи, имеют очень мало визуальных характеристик, так что картирование генов
этих организмов должно полагаться на биохимические фенотипы, например приведен-
ные в таблице 3.1. Что касается людей, то они, конечно, дают возможность использо-
вать визуальные характеристики, но начиная с 1920-х гг. исследования генетической
изменчивости человека базировались главным образом на биохимических фенотипах,
которые могут быть отмечены путем определения групп крови. Эти фенотипы вклю-
чают не только стандартные группы крови типа ряда АВО, но также и варианты белков
сыворотки крови и иммунологических белков типа лейкоцитарных антигенов человека
(система HLA). Большое преимущество этих маркёров заключается в том, что многие из
значимых генов имеют множественные аллели. Например, ген по имени HLA-DRB1 име-
ет по крайней мере 290 аллелей, a HLA-B — более 400. Это обстоятельство существенно
для способа, которым выполняется картирование генов человека (раздел 3.2.4). Вместо
того чтобы ставить запланированные эксперименты по размножению, которое является
обязательной процедурой для экспериментальных организмов типа плодовых мушек
или мышей, данные о наследовании генов человека приходится кропотливо собирать,
изучая фенотипы, проявляемые членами семейств, браки которых были устроены по
личным мотивам, а не из прихоти пытливого генетика. Если все члены некоторого се-
мейства несут один и тот же аллель изучаемого гена, то никакая полезная информация
не может быть получена. Поэтому для целей картирования генов необходимо подыскать
семейства, в которых браки заключались случайным образом и между индивидуумами
с различными аллелями. Этот принцип особенно эффективен, если изучаемый ген имеет,
скажем, не 2, а 290 аллелей.
3.2.2. ДНК-маркёры для составления генетических карт
Гены — очень полезные маркёры, но они ни в коем случае не идеальны. Одна про-
блема, особенно с большими геномами, например с геномами позвоночных животных
и цветковых растений, состоит в том, что карта, основанная исключительно на генах, не
может быть очень подробной. Эта проблема сохранялась бы даже в том случае, если бы
на карту могли быть нанесены все гены, потому что в большинстве геномов эукариотов
они разбросаны и отстоят далеко друг от друга с обширными пропусками между ними
(см. рис. 7.12). Проблема еще более осложняется тем фактом, что только часть от общего
86 Часть 1. Изучение геномов
Таблица 3.1. Типичные биохимические маркёры, используемые для генетического анализа Saccharomyces
cerevisiae
Маркёр Фенотип Метод опознавания клеток, несущих данный маркёр
ADE2 Требует аденин Растет только в среде, содержащей аденин
CAN1 Устойчив к канаванину Растет в присутствии канаванина
CUP1 Устойчив к меди Растет в присутствии меди
CYH1 Устойчив к циклогексимиду Растет в присутствии циклогексимида
LEU2 Требует лейцин Растет только в среде, содержащей лейцин
SUC2 Способен сбраживать саха- розу Растет, если сахароза — единственный углевод в среде
URA3 Требует урацил Растет только в среде, содержащей урацил
числа генов существует в удобно различимых аллельных формах. Поэтому карты генов
не всеобъемлющие. Нам по-прежнему нужны маркёры других типов.
Нанесенные на карту особенности, которые не являются генами, называют ДНК-
маркёрами. Как и в случае генов-маркёров, всякий маломальски пригодный ДНК-маркёр
должен иметь по крайней мере два аллеля. Есть три типа особенностей последователь-
ности ДНК, которые удовлетворяют этому требованию: полиморфизмы длины фраг-
ментов рестрикции (RFLP), полиморфизмы длины простых последовательностей
(SSLP) и полиморфизмы отдельных нуклеотидов (SNP).
Полиморфизмы длины рестриктов
RFLP были первым типом ДНК-маркёров, который был изучен. Вспомним, что
ферменты рестрикции разрезают молекулы ДНК в определенных сайтах узнавания (раз-
дел 2.1.2). Такая специфичность последовательности означает, что обработка молекулы
ДНК ферментом рестрикции должна всегда производить один и тот же набор фрагментов.
С молекулами геномной ДНК так происходит не всегда, потому что некоторые участки
рестрикции являются полиморфными и существуют в виде двух аллелей, причем один
аллель показывает правильную для участка рестрикции последовательность и поэтому
разрезается при обработке ДНК ферментом, а второй аллель несет видоизменение по-
следовательности, так что участок рестрикции уже не опознается. В результате видо-
изменения последовательности, после обработки ферментом два смежных фрагмента
рестрикции остаются связанными вместе, что ведет к полиморфизму длины (рис. 3.4).
Это RFLP, и его позиция на карте генома может быть получена по данным анализа
наследования его аллелей — точно так же, как это делают при использовании генов
в качестве маркёров. По оценкам ученых, в геноме типичного млекопитающего при-
сутствует приблизительно 105 RFLP.
Чтобы оценить счет RFLP, необходимо определить размер всего лишь одного или
двух отдельных фрагментов рестрикции на фоне многих несоответствующих фрагмен-
Глава 3. Картирование геномов 87
тов. Это нетривиальная пробле-
ма: фермент типа EcoR\, с сайтом
узнавания из шести нуклеоти-
дов, должен произвести прибли-
зительно один разрез на каждые
46 = 4096 п.н. и, таким образом,
дать почти 800000 фрагмен-
тов (если используется с ДНК
человека). После разделения
электрофорезом в агарозном
геле эти 800 000 фрагментов
Полиморфный участок
рестрикции
ДНК
(аллель 1)
А
ДНК
(аллель 2)
Добавление эндонуклеазы рестрикции
J F ... j L
4 фрагмента 3 фрагмента
Рис. 3.4. Полиморфизм длины рестрикта (RFLP). Молекула ДНК
слева имеет полиморфный участок рестрикции (отмеченный
звездочкой), который отсутствует в молекуле справа. RFLP об-
наруживается после обработки ферментом рестрикции, потому
что одна из молекул разрезается на четыре фрагмента, тогда как
другая разрезается на три фрагмента
производят размытое пятно.
ДНК и RFLP нельзя различить на
общем фоне. Одним из способов
визуализации RFLP является
гибридизация по Саузерну с ис-
пользованием зонда, который
покрывает полиморфный уча-
сток рестрикции (рис. 3.5, а), но
в наши дни чаще используется
ПЦР. Затравки для ПЦР разраба-
тывают таким образом, чтобы
они отжигались к обеим сторонам полиморфного участка, и RFLP типизируется путем
обработки размноженного фрагмента ферментом рестрикции и последующего пробега
образца в агарозном геле (рис. 3.5, б).
Полиморфизмы длины простых последовательностей
SSLP — множества повторных последовательностей, которые показывают изме-
нения длины, при этом различные аллели содержат разное число повторных единиц
(рис. 3.6, а). В отличие от RFLP, SSLP могут быть многоаллельными, поскольку каждый
SSLP может иметь целый ряд различных вариантов длины. Существуют SSLP двух ти-
пов:
• минисателлиты, также известные как переменное число тандемных повторе-
ний (VNTR), в которых повторная единица может иметь длину до 25 п. н.;
• микросателлиты, или простые тандемные повторения (STR), чей повторяю-
щийся элемент короче — обычно 13 п.н. или меньше1^.
ДНК-маркёры, основанные на микросаттелитах, завоевали большую популярность,
чем минисателлиты, по двум причинам. Во-первых, минисателлиты не распространены
равномерно по всему геному, а, как правило, чаще встречаются в теломерных областях
на концах хромосом. В географических понятиях это равносильно попытке использо-
вать карту с береговыми маяками, чтобы проложить себе путь в центральной части
острова. Микросателлиты более удобно распределены по геному. Во-вторых, самый
быстрый способ типизировать полиморфизм длины — прибегнуть к ПЦР, но быстрая
и точная типизация посредством ПЦР возможна при длине последовательностей не
более 300 п. н. Большинство минисателлитных аллелей длиннее этой величины, потому
Обычно границей для длины элемента микросателлитов считают 7 п. н. — все, что длиннее, относят
к минисателлитам. — Прим. ред.
88
Часть 1. Изучение геномов
а) Гибридизация по Саузерну полиморфный участок
Зонд ДНК
Гибридизационные
полосы
Нейлоновая мембрана
б) ПЦР
Карта участков
рестрикции
Авторадиография
Полиморфный участок
1
Rj ^2 «3 |/
i + Карта участков
рестрикции
Затравки ПЦР
ПЦР сопровождается
обработкой
рестриктазами
Электрофорез в агарозном геле
———————
1 2 3
Рис. 3.5. Два метода оценки счета RFLP. a) RFLP могут быть оценены гибридизацией по Саузерну. ДНК
переваривается соответствующим ферментом рестрикции и разделяется в агарозном геле. Пятно фраг-
ментов рестрикции переносится на нейлоновую мембрану и гибридизируется с частью ДНК, которая
покрывает полиморфный участок рестрикции. Если этотучасток отсутствует, то детектируется единствен-
ный фрагмент рестрикции (дорожка 2); если этотучасток присутствует, то детектируются два фрагмента
(дорожка 3). 6) RFLP может быть типизирован также и при помощи ПЦР с применением затравок, которые
отжигаются к обеим сторонам полиморфного участка рестрикции. После ПЦР продукты обрабатывают
соответствующим ферментом рестрикции и затем анализируют посредством электрофореза в агарозном
геле. Если искомый участок отсутствует, то на агарозном геле наблюдается одна полоса (дорожка 2); если
участок присутствует, то можно видеть две полосы (дорожка 3)
что их повторные единицы относительно велики и тяготеют к скоплению в целые мно-
жества, так что для их типизации необходимы продукты ПЦР длины несколько тыс.п.н.
Микросателлиты, используемые в роли ДНК-маркёров, обычно состоят из 10-30 копий
повторения, которое не больше чем 6 п. н. в длину, и, таким образом, намного более при-
годны для анализа посредством ПЦР. В геноме человека имеется 5-105 микросателлитов
с повторными единицами длины 6 п.н. и менее.
При исследовании посредством ПЦР аллель, присутствующий в STR, выявляется по
точной длине продукта ПЦР (рис. 3.6, б). Изменения длины могут визуализироваться при
помощи электрофореза в агарозном геле, но стандартный гель-электрофорез является
трудоемкой процедурой, которую трудно автоматизировать, и поэтому не подходит
для высокопроизводительного анализа, востребованного сообществом современных
исследователей геномов. Вместо него STR обычно типизируют капиллярным электро-
форезом в полиакриламидном геле (см. техническое примечание 4.1). Большинство
систем капиллярного электрофореза использует флюоресцентное детектирование,
так что перед началом проведения ПЦР к одной или обеим затравкам прикрепляется
флюоресцентная метка (техническое примечание 2.1). После ПЦР продукт загружается
в капиллярную систему и движется мимо датчика флюоресценции. Компьютер, соеди-
ненный с датчиком, сопоставляет время прохождения продукта ПЦР с соответствую-
Глава 3. Картирование геномов 89
Рис. 3.6. STR и способ их типизации, а) Два
аллеля короткого тандемного повторения,
называемого также микросателлитом. В ал-
леле 1 мотив «GA» повторяется три раза,
а в аллеле 2 он повторяется пять раз. б) Ти-
пизация STR при помощи ПЦР. STR и часть
окружающей его последовательности раз-
множается, и размер продукта определяется
посредством электрофореза в агарозном геле
или капиллярного электрофореза. В агароз-
ном геле дорожка А содержит продукт ПЦР,
а дорожка В содержит ДНК-маркёры, кото-
рые показывают размеры полос, полученных
после ПЦР этих двух аллелей. Полоса на до-
рожке А имеет тот же размер, что и больший
из двух ДНК-маркёров, что свидетельствует
о том, что ДНК, которая была тестирована, со-
держала аллель 2. Результаты капиллярного
электрофореза отображаются в виде элек-
трофореграммы, где позиция пика указывает
размер продукта ПЦР. Электрофореграмма
автоматически калибруется по маркёрам
длины, так что может быть рассчитана точная
длина продукта ПЦР. Изображение любезно
предоставлено Сьюзен Со
а) Два варианта некоторого STR
б) Типизация STR посредством ПЦР
Капиллярный
электрофорез
Электрофорез
в агарозном геле
щими данными для набора маркёров длины и таким образом точно идентифицирует
продукт по его длине.
Полиморфизмы отдельных нуклеотидов
Это позиции генома, в которых некоторые индивидуумы имеют один нуклеотид
(например, G), а другие имеют отличающийся от него нуклеотид (скажем, С) (рис. 3.7).
В каждом геноме имеется огромное число SNP (в геноме человека более четырех мил-
лионов), некоторые из них также порождают RFLP, но многие не делают этого, потому
что последовательность, в которой они лежат, не опознается никаким ферментом
рестрикции.
Во всякой позиции генома может присутствовать любой из четырех известных
нуклеотидов, так что можно вообразить, будто каждый SNP имеет четыре аллеля. Теоре-
тически это возможно, но на практике большинство SNP существует в виде всего лишь
двух вариантов. Это связано с тем, что все SNP возникают, когда в геноме происходят
точечные мутации (раздел 16.1), преобразовывающие один нуклеотид в другой. Если
такая мутация имеет место в репродуктивных клетках индивидуума, то один или более
потомков этого индивидуума могут унаследовать эту мутацию и, по смене многих по-
90 Часть 1. Изучение геномов
Аллель 1 а
|. .AGTCAGAAATC.,|
.AGTCACAAATC.,|
Аллель 2
Рис. 3.7. Полиморфизм отдельного нуклеотида (SNP)
колений, данный SNP может в конечном счете стать закрепленным в этой популяции.
Но существует только два аллеля — первоначальная последовательность и ее мутиро-
вавшая версия. Для того чтобы появился третий аллель, в той же самой позиции генома
в организме другого индивидуума должна произойти новая мутация и этот индивидуум,
а так же и его потомки, должны воспроизводиться таким образом, чтобы новый аллель
стал закрепленным. Хотя такой сценарий и правдоподобен, он маловероятен: вследствие
этого подавляющее большинство SNP биаллельно. Это неудобство более чем перевеши-
вается огромным числом SNP, присутствующих в каждом геноме, — по крайней мере
один на каждые 10 тыс.п.н. ДНК в большинстве эукариотов. Поэтому1) SNP позволяют
строить очень подробные карты геномов.
Значение, которое SNP приобрели в исследовании генома, подтолкнуло развитие
быстрых методов их типизации. Некоторые из этих методов базируются на анализе ги-
бридизацией олигонуклеотидов. Олигонуклеотид — короткая однонитевая молекула
ДНК, обычно менее 50 нуклеотидов в длину, которая синтезируется в пробирке. Если
условия являются абсолютно правильными, то олигонуклеотид гибридизируется с дру-
гой молекулой ДНК только в том случае, если он способен образовать с этой второй мо-
лекулой полностью спаренную структуру. Если имеет место хотя бы одно-единственное
несоответствие — единственная позиция в пределах олигонуклеотида, которая не
образует пару оснований, — то гибридизация не происходит (рис. 3.8, а). Поэтому ги-
бридизация олигонуклеотида позволяет различать два аллеля того или иного SNP. На
основе гибридизации олигонуклеотидов были изобретены различные стратегии про-
смотра, в том числе следующие.
• Технология чипов ДНК (техническое примечание 3.1) использует стеклянную
или кремниевую пластину-подложку площадью 2,0 см2 или меньше, несущую на
себе множество различных олигонуклеотидов в виде высокоплотной матрицы.
Предназначенная для тестирования ДНК помечается флюоресцентным маркёром
и наносится пипеткой на поверхность чипа. Гибридизация детектируется путем
анализа чипа с помощью флюоресцентного микроскопа, при этом позиции, в ко-
торых испускается флюоресцентный сигнал, показывают, какие олигонуклеотиды
гибридизировались с тестируемой ДНК. Поэтому в одном эксперименте может
быть типизировано целое множество SNP. * У
Эта цифра сильно занижена. В геноме человека плотность SNP составляет 1 на 1000 или даже 1 на
800 п. н. При этом надо понимать, что человек — молодой вид. У более старых видов плотность SNP выше.
У оболочников эта величина достигает 1 на 10 п. н.! — Прим. ред.
Глава 3. Картирование геномов 91
а) Гибридизация олигонуклеотида весьма специфична
Полностью спаренный
основаниями гибрид
стабилен Олигонуклеотид
CTGGTCGTCAGTCTTTAGTT
। । । । । । । । । । । । । । । । । । । ।
i===GACCAGCAGTCAGAAATCAA =«=.
Целевая ДНК
SNP
Одно несовпадение -
гибрид нестабилен
Несовпадение - пара оснований
не может образоваться
CTGGTCGTCAGTC*TTTAGTT
i
iS=GACCAGCAGTCACAAATCAA^— «
б) Детектирование гибридизации с помощью гашения флюоресцентного красителя
SNP
Флюоресцентный
сигнал
Рис. 3.8. Типизация SNP посредством анализа гибридизацией олигонуклеотидов, а) При очень жест-
ких условиях гибридизации устойчивый гибрид получается только в том случае, если олигонуклеотид
способен сформировать с целевой ДНК спаренную всеми основаниями структуру. Если есть хотя бы
одно-единственное несоответствие, то гибрид не образуется. Чтобы достигнуть такого уровня жестко-
сти, температура выдержки должна быть чуть ниже температуры плавления Тт олигонуклеотида. При
температурах выше Тт даже полностью спаренный основаниями гибрид неустойчив. При температурах
ниже Тт более чем на 5 °C, могут быть устойчивы и гибриды с несоответствиями. Тт для олигонуклео-
тида, представленного на рисунке, будет около 58 °C. Тт в °C рассчитывается по формуле Тт = (4 х число
нуклеотидов G и С) + (2 х число нуклеотидов А и Т). Эта формула дает приближенное значение Тт для
олигонуклеотидов длины 15-30 нуклеотидов. 6) Один из способов типизации SNP — гибридизация
в растворе. Олигонуклеотидный зонд несет две концевые метки. Одна из них — флюоресцентный
краситель, а другая — гасящее его вещество. Два конца олигонуклеотида спариваются основаниями
друг с другом, так что флюоресцентный сигнал гасится. Когда зонд гибридизируется со своей целевой
ДНК, концы молекулы отделяются друг от друга, позволяя флюоресцентному красителю испускать свой
сигнал. Такие две метки называют «молекулярными маяками»
92
Часть 1. Изучение геномов
Техническое примечание 3.1: микроматрицы ДНК и чипы ДНК
Высокоплотные массивы молекул ДНК для параллельных гибридизационных
анализов
Микроматрицы ДНК и чипы ДНК дают возможность выполнять большое число
экспериментов по гибридизации параллельно. Свое предназначение они осущест-
вляют главным образом в массовой типизации полиморфизмов типа SNP (раз-
дел 3.2.2) и в сравнении популяций РНК различных клеток (раздел 6.1.2).
Хотя терминология не совсем точна, микроматрицы и чипы, строго говоря,
представляют собой матрицы двух разных типов. В обеих архитектурах большое
число зондов ДНК, каждый с отличной от других последовательностью, закрепля-
ется в определенных позициях на твердой поверхности. Зондами могут служить
синтетические олигонуклеотиды или иные короткие молекулы ДНК наподобие
кДНК или продуктов ПЦР. При создании микроматрицы их могут наносить на пред-
метное стекло микроскопа или на отрезок нейлоновой мембраны. Эта технология
позволяет достигнуть лишь относительно низкой плотности — как правило, 6400
зондов в виде массива 80x80 единиц на площадке 18x18 мм, что достаточно для
исследования популяций РНК, но менее пригодно для высокопроизводительных
анализов, необходимых для типизации SNP.
Чтобы подготовить действительно высокоплотные массивы, олигонуклеотиды
синтезируются in situ на поверхности пластинки из стекла или кремния, в результате
чего получается чип ДНК Стандартный метод синтеза олигонуклеотидов состоит
в добавлении нуклеотидов один за другим к наращиваемому концу олигонуклео-
тида, при этом его последовательность определяется тем порядком, в котором
нуклеотидные субстраты добавляются в реакционную смесь. Если бы этот метод
применялся для синтеза на чипе, то все олигонуклеотиды имели бы одну и ту же
последовательность. Вместо этого используются видоизмененные нуклеотидные
Активируемый светом синтез
Присоединение только
к активированным
олигонуклеотидам
Рис. Т3.1. Синтез олигонуклеотидов на поверхности чипа ДНК
Глава 3. Картирование геномов 93
субстраты — такие, которые могут быть активированы светом прежде, чем они
прикрепятся к концу наращиваемого олигонуклеотида. Нуклеотиды добавляют-
ся один за другим к поверхности чипа, при этом используется фотолитография
[Аналогичная той, что используется при изготовлении микропроцессоров. — Прим,
ред.], чтобы направлять импульсы света на отдельные позиции в массиве и таким
образом определять, какой из наращиваемых олигонуклеотидов будет продолжен
путем присоединения определенного нуклеотида, добавляемого на каждом этапе
процесса (рис. Т3.1). При данной технологии удается достичь плотности до 300 000
олигонуклеотидов на см2, так что при использовании ДНК-чипов для типизации
SNP в одном эксперименте могут быть типизированы 150 000 полиморфизмов
(при том условии, что в наличии имеются олигонуклеотиды для обоих аллелей
каждого SNP).
Чипы и микроматрицы просты в применении. Чип или матрица выдержива-
ется с меченой целевой ДНК, чтобы произошла гибридизация. Позиции, в которых
происходит гибридизация с целевой ДНК, определяются путем сканирования по-
верхности и записи точек, в которых детектируется сигнал, испускаемый меткой.
Можно использовать радиоактивные метки, при этом сигналы детектируются
электронно при помощи
фосфорографии, но она
обеспечивает лишь низкое
разрешение и не подходит
для высокоплотных чипов.
Более высокое разрешение
может быть достигнуто ме-
чением флюоресцентными
метками и детектированием
их при помощи их лазерно-
го сканирования или флюо-
ресцентной конфокальной
микроскопии (рис. Т3.2).
Рис. Т3.2. Визуализация гибридизации зонда с флюоресцентной меткой на микроматрице. Метка
была детектирована конфокальным лазерным сканированием и интенсивность сигнала преоб-
разована в спектр псевдоцветов, где красный показывает самую полную гибридизацию, далее
следуют оранжевый, желтый, зеленый, голубой, синий и фиолетовый (последний представляет
фоновый уровень гибридизации). Каждое пятно на микроматрице — отличный от других клон
кДНК, приготовленный из мРНК клеток крови человека, а зонд — кДНК из мРНК костного мозга
человека. Дополнительная информация об использовании чипов и микроматриц ДНК для ис-
следования популяций мРНК приводится в разделе 6.1.2. Изображение любезно предоставлено
Томом Страчаном и перепечатано с разрешения журнала «Nature»
94
Часть 1. Изучение геномов
• Методы гибридизации в растворе выполняют в лунках лотка микротитра, где
каждая лунка содержит отличный от других олигонуклеотид, используя систему
детектирования, способную различать негибридизированную однонитевую ДНК
и двунитевой продукт, который получается в результате спаривания олигонуклео-
тида с тестируемой ДНК. Было разработано несколько таких систем. В одной из них
используется пара меток, в которую входит флюоресцентный краситель и веще-
ство, которое гасит флюоресцентный сигнал, когда сближается с испускающим его
красителем. Краситель прикрепляется к одному концу олигонуклеотида, а гасящее
вещество — к другому его концу. Обычно не наблюдается никакой флюоресценции,
потому что олигонуклеотид сконструирован таким образом, что оба его конца спа-
риваются основаниями друг с другом, в результате чего гаситель флюоресценции
оказывается рядом с красителем (рис. 3.8, б). Если же между олигонуклеотидом
и тестируемой ДНК происходит гибридизация, то данное спаривание оснований
нарушается и гаситель отдаляется от красителя, позволяя ему вырабатывать
флюоресцентный сигнал.
В других методах типизации используется олигонуклеотид, чье несовпадение с SNP
происходит в его 5’- или З'-оконечностях. При соответствующих условиях олигонуклео-
тид этакого типа гибридизируется с несовпадающей матричной ДНК с коротким, ни с чем
не спаренным своими основаниями «хвостом» (рис. 3.9, а). Эту особенность используют
двумя различными способами.
• Анализ лигированием олигонуклеотидов (OLA) основан на применении двух
олигонуклеотидов, которые отжигаются смежно друг к другу, при этом З'-конец
одного из этих олигонуклеотидов точно попадает в SNP. Этот олигонуклеотид
образует полностью спаренную основаниями структуру, если в матричной ДНК
присутствует одна версия SNP, и когда это происходит, данный олигонуклеотид
может быть лигирован к своему партнеру (рис. 3.9, б). Если исследуемая ДНК со-
держит другой аллель этого же SNP, то З’-нуклеотид исследуемого олигонуклеотида
не отожжется к матрице и никакого лигирования не произойдет. Поэтому аллель
типизируется путем определения того, синтезирован ли продукт лигирования —
а) Гибридизация с олигонуклеотидом
с концевым несоответствием
Полностью спаренный
основаниями гибрид ~
Олигонуклеотид
5' 3'
TCGGTCGCTGGTCGTCAGTC
*• — AGCCAGCGACCAGCAGTCAGe“=-a*
Целевая ДНК
Гибриде не спаренным
основаниями хвостом Олигонуклеотид
5' г 3'
TCGGTCGCTGGTCGTCAGTС
iie=AGCCAGCGACCAGCAGTCAC*=»«
Целевая ДНК
б) Анализ лигированием в) Тест ARMS
олигонуклеотидов
Нет несовпадений
.. 1111111111111пшш..днк
SNP ''1'1'''' ’'''''
I Лигирование _.ТПТ.............. lt._.
^происходит .
Продукт ПЦР синтезируется
Несовпадение Несовпадение
Нет лигирования Продукт ПЦР отсутствует
Рис. 3.9. Методы типизации SNP. а) При соответствующих условиях, олигонуклеотид, не совпадающий
с SNP своим 5'- или З'-концом, гибридизируется с несовпавшей матричной ДНК, оставляя короткий не
спаренный основаниями «хвост», б) Типизация SNP посредством анализа лигированием олигонуклео-
тидов. в) Тест ARMS
Глава 3. Картирование геномов 95
обычно пробегом прореагировавшей смеси в системе капиллярного электрофореза,
как было описано выше на примере типизации STR.
• Система размножения термостабильных мутаций, или тест ARMS, основан на
том же самом принципе, что и OLA, но в этом методе контрольным олигонуклеоти-
дом выступает одна из пары затравок ПЦР. Если контрольная затравка отжигается
к SNP, то он может быть продолжен с помощью полимеразы Taq и ПЦР может иметь
место, но если она не отжигается, из-за того что присутствует альтернативная
версия SNP, то никаких продуктов ПЦР не производится (рис. 3.9, в).
3.2.3. Анализ сцепления — основа составления генетических карт
Теперь, когда мы собрали набор маркёров для построения генетической карты, мы
можем пройти дальше и взглянуть на сами методы картирования. Все эти методы осно-
ваны на сцеплении генетических признаков, которое, в свою очередь, было выведено
на почве плодотворных открытий в генетике, совершенных в середине девятнадцатого
столетия Грегором Менделем.
Законы наследственности и открытие сцепления генетических признаков
Генетическое картирование основано на законах наследственности в их первозданном
виде, как они и были впервые описаны Грегором Менделем в 1865 г. На основании резуль-
татов своих опытов по размножению растений гороха, Мендель заключил, что каждое из
растений гороха обладает двумя аллелями каждого гена, но проявляет только один фенотип.
Прийти к выводу о том, что если растение размножается путем самоопыления (то есть го-
мозиготно по тому или иному анализируемому признаку), то оно обладает двумя идентич-
ными аллелями и поэтому показывает соответствующий фенотип, несложно (рис. 3.10, а).
Однако Мендель показал, что если скрещиваются два самоопыляющихся растения с раз-
личными фенотипами, то все потомство (поколение FJ проявляет один и тот же фенотип.
Эти растения Fx должны быть гетерозиготными, а это означает, что они обладают двумя
различными аллелями, по одному для каждого фенотипа — один аллель, унаследованный
от матери, и один — от отца. Мендель постулировал, что в таком гетерозиготном состоянии
один аллель преодолевает все проявления другого аллеля: исходя из этого, он описал фено-
тип, экспрессированный в растениях Fr как доминирующий над вторым, рецессивным
(подавляемым)фенотипом (рис. 3.10, б).
Менделевская интерпретация гетерозиготного состояния абсолютно верна для пар
аллелей, которые он изучал, но теперь мы знаем, что в некоторых случаях, с которыми
он не сталкивался, это простое правило доминирования-рецессивности может оказаться
гораздо сложнее. К таким случаям относятся:
• неполное доминирование, когда гетерозиготная форма проявляет фенотип,
промежуточный между двумя гомозиготными формами. Примером служит цвет
окраски цветков на растениях, таких как гвоздика (но не горох): когда красные
гвоздики скрещивают с белыми, гетерозиготы Fx оказываются ни красными, ни
белыми, но розовыми (рис. 3.11, а);
• кодоминирование, при котором гетерозиготная форма показывает оба гомо-
зиготных фенотипа. Группы крови человека представляют несколько примеров
кодоминирования. Например, две гомозиготные формы ряда MN суть М и N, при
этом такие индивидуумы синтезируют соответственно гликопротеины крови
только М- или N-типа. Гетерозиготы, однако, синтезируют оба гликопротеина и,
следовательно, обозначаются MN (рис. 3.11, б).
96
Часть 1. Изучение геномов
а) Самоопыление размножающихся
в чистой линии растений гороха
Родители
Поколение F,
Рис. 3.10. Гомозиготность и гетерозиготность.
Мендель изучал на своих растениях гороха
семь пар контрастных признаков, одной из
которых служили фиолетовый и белый цвет
окраски цветков, как показано на этой схеме.
а) Самоопыляющиеся растения всегда дают
цветки с родительским цветом. Эти растения —
гомозиготы, и каждое из них обладает парой
идентичных аллелей, обозначенных здесь как
VV (фиолетовые цветки) и WW (белые цвет-
ки). б) Когда два самоопыляющихся растения
скрещиваются между собой, в поколении на-
блюдается только один из фенотипов. Мендель
пришел к выводу, что генотип растений был
VW, так что аллель V — доминантный, а аллель
W — рецессивный
Поколение F2
Фиолетовые цветки Белые цветки
W WW
б) Перекрестное опыление двух чистых типов
Родители
Фиолетовые цветки X
W
Белые цветки
WW
Поколение F,
Фиолетовые цветки
VW
Наряду с открытием доминирова-
ния и рецессивности, Мендель провел
дополнительные эксперименты, кото-
рые позволили ему установить два за-
кона генетики, носящих его имя. Пер-
вый закон Менделя гласит, что аллели
распределяются независимо. Другими
словами, если аллели родителя — А и
а, то член поколения Fx имеет равные
шансы наследования одного из аллелей:
А или а. Второй закон Менделя утверж-
дает, что пары аллелей распределяют-
ся независимо, так что наследование
аллелей гена А не зависит от наследова-
ния аллелей гена В. Благодаря этим за-
конам исходы генетических скрещива-
ний вполне предсказуемы (рис. 3.12).
Когда труды Менделя были вновь
открыты в 1900 г., его второй закон
вол новал ранних генетиков, потому что
вскоре было установлено, что гены рас-
положены на хромосомах, и было признано, что все организмы имеют намного больше
генов, чем хромосом. Хромосомы наследуются как нерушимые единицы, так что был
сделан вывод, что аллели некоторых пар генов будут наследоваться совместно, потому
что они находятся на одной и той же хромосоме (рис. 3.13). Таким образом, был сфор-
мулирован принцип сцепления генетических признаков, и быстро была показана его
верность, хотя результаты не совсем соответствовали ожиданиям. Полное сцепление,
которое предвиделось между многими парами генов, не осуществилось. Пары генов
или наследовались независимо, как ожидалось для генов в разных хромосомах, или они
показывали лишь неполное сцепление: иногда они наследовались вместе, а иногда —
порознь (рис. 3.14). Разрешение этого противоречия между теорией и наблюдениями
было определяющим шагом в развитии методов составления генетических карт.
Глава 3. Картирование геномов 97
а) Неполное доминирование
Белые цветки
WW
Родители
Розовые цветки
RW
Поколение F,
б) Кодоминирование
-О Л Л®
0©® С©
© © ©О
ММ * NN
I
о
®©®
0 о
Родители
Поколение
MN
Рис. 3.11. Два типа взаимодействия аллелей, с которыми не сталкивался Мендель: а) неполное доми-
нирование и б) кодоминирование
Неполное сцепление объясняется поведением хромосом во время мейоза
Этот шаг удалось совершить Томасу Ханту Моргану который силою своего во-
ображения перемахнул через пропасть между неполным сцеплением и поведением
хромосом во время деления ядра клетки. В конце девятнадцатого столетия цитологи
выделяли два типа деления ядра: митоз и мейоз. Митоз более всеобычен, будучи
процессом, посредством которого диплоидное ядро соматической клетки делится на
98
Часть 1. Изучение геномов
Родители
Генотипы поколения
МОНОГИБРИДНОЕ СКРЕЩИВАНИЕ
Высокие Tt х Tt Высокие
Фенотипы поколения F} Звысоких:! низкому
ДИГИБРИДНОЕ СКРЕЩИВАНИЕ
Родители Высокие, гладкие TtRr х TtRr Высокие, гладкие
Генотипы поколения TR Tr tR tr
TR TTRR TTRr TtRR TtRr
Tr TTRr TTrr TTRr Ttrr
tR TtRR TtRr ttRR ttRr
tr TtRr Ttrr ttRr ttrr
Фенотипы поколения F1 Эвысоких, гладких: Звысоким, морщинистым:
Знизким, гладким:!низкому, морщинистому
Рис. 3.12. Законы Менделя позволяют предсказать исход генетических скрещиваний. Показаны два
скрещивания с их предсказуемыми результатами. При моногибридном скрещивании прослеживается
наследование аллелей одного гена — в нашем случае это аллель Т (высокие растения гороха) и аллель t
(низкие растения гороха). Аллель Т является доминантным, at — рецессивным. Сетка показывает
генотипы и фенотипы поколения Fv предсказанные на основании первого закона Менделя, который
гласит, что аллели распределяются независимо. Когда Мендель провел такое скрещивание, он получил
787 высоких растений гороха и 277 низких растений и вывел соотношение 2,84:1. При дигибридном
скрещивании отслеживается наследование двух генов. Второй ген определяет форму горошин, при этом
аллель R (гладкие горошины) — доминантный аллель, а г (морщинистые горошины) — рецессивный.
Представленные генотипы и фенотипы предсказаны согласно первому и второму законам Менделя,
последний из которых гласит, что пары аллелей распределяются независимо
два дочерних ядра, оба из которых тоже диплоидные (рис. 3.15). Чтобы произвести
все клетки, необходимые организму человека в течение всей его жизни, понадобится
приблизительно 1017 митозов. Перед началом митоза каждая хромосома в ядре репли-
цируется, но получающиеся дочерние хромосомы не расходятся друг от друга тут же.
Поначалу они остаются прикрепленными своими центромерами. Дочери не отделяются
друг от друга до того момента митоза, когда хромосомы распределяются между двумя
Рис. 3.13. Гены на одной и той же хромосоме должны показать
сцепление. Гены А и В находятся на одной и той же хромосоме и,
следовательно, должны быть унаследованы вместе. Поэтому второй
закон Менделя не должен распространяться на наследование генов
А и В. Ген С находится на другой хромосоме, так что второй закон
будет справедлив как для наследования генов А и С, так и для на-
следования генов В и С. Мендель не открыл сцепления, потому что
все семь генов, которые он изучал, были расположены на разных
хромосомах растений гороха
Глава 3. Картирование геномов 99
Гены
Цвет цветков
Аллели
СКРЕЩИВАНИЕ РОДИТЕЛЕЙ
Форма пыльцевого зерна
Фиолетовый
Красный
Фиолетовый,
продолговатая
Красный,
круглая
Продолговатая Круглая
Все фиолетовые,
продолговатые
Заключение
Фиолетовые цветки доминируют над красными
Продолговатые пыльцевые зерна доминируют над круглыми
X
СКРЕЩИВАНИЕ в поколении F,
Если гены не сцеплены -
Скрещивание в поколении даст соотношение
9 фиолетовые, продолговатые : 3 фиолетовые, круглые :
3 красные, продолговатые : 1 красные, круглые —
Если гены сцеплены -
Скрещивание в поколении F, даст соотношение
3 фиолетовые, продолговатые : 1 красные, круглые
Фиолетовые, ж Фиолетовые,
продолговатые продолговатые
I
Фактические результаты
Заключение:
Гены показывают неполное сцепление
4831 - фиолетовые, продолговаты'
________________ 390 - фиолетовые, круглые
391 - красные, продолговатые
1338 - красные, круглые
Рис. 3.14. Неполное сцепление. Неполное сцепление было открыто в начале двадцатого столетия. Пред-
ставленное на рисунке скрещивание было проведено Бэтсоном, Саундерсом и Пюннеттом в 1905 г.
с душистым горошком. Скрещивание родителей дает типичный дигибридный результат (см. рис. 3.12),
где все растения показывают один и тот же фенотип, что указывает на то, что фиолетовый цвет окраски
цветков и продолговатая форма пыльцевых зерен определяются доминантными аллелями. Скрещивание
между членами поколения дает неожиданные результаты, поскольку потомство не показывает ни со-
отношение 9:3:3:1 (ожидаемое для генов на различных хромосомах), ни соотношение 3:1 (ожидаемое,
если гены полностью сцеплены). Для неполного сцепления всегда типично необычное соотношение
новыми ядрами. Очевидно, это важно, чтобы каждое из новых ядер получило полный
набор хромосом, и большинство интриг, затеваемых клеткой во время митоза, как ока-
зывается, направлено на достижение именно этой конечной цели.
Митоз иллюстрирует основные события, происходящие во время деления ядра, но
здесь нас интересуют именно отличительные особенности мейоза. Мейоз происходит
только в репродуктивных клетках, и в результате диплоидная клетка дает начало четы-
рем гаплоидным гаметам, каждая из которых может впоследствии слиться с гаметой
противоположного пола в ходе полового воспроизводства. Тот факт, что в результате
мейоза образуются четыре гаплоидные клетки, тогда как митоз дает начало двум ди-
плоидным клеткам, легко объяснить: мейоз обусловливает два деления ядра, одно за
другим, тогда как митоз — только одно деление ядра. Это важное отличие, но опреде-
ляющее различие между митозом и мейозом является более тонким. Вспомним, что
в диплоидной клетке находится по две отдельные копии каждой хромосомы (глава 1).
100 Часть 1. Изучение геномов
ИНТЕРФАЗА
Рис. 3.15. Митоз. В течение интерфазы (период между делениями ядра) хромосомы находятся в растяну-
той форме (раздел 7.1.1). В начале митоза хромосомы уплотняются и к поздней профазе представляют
собой оформленные структуры, видимые в световом микроскопе. Каждая хромосома уже подверглась
репликации ДНК, но две дочерние хромосомы еще скреплены между собой центромерой. Во время
метафазы ядерная мембрана расходится (у большинства эукариотов), и хромосомы выстраиваются в ряд
в центре клетки. После этого микротрубочки подтягивают дочерние хромосомы к обоим концам клетки.
В телофазе вокруг каждого набора дочерних хромосом заново образуются ядерные мембраны. Итак,
родительское ядро дало начало двум идентичным дочерним ядрам. Для простоты показана только одна
пара гомологичных хромосом; один член пары красный, другой — синий
Мы обращаемся к ним как к парам гомологичных хромосом. В процессе митоза гомо-
логичные хромосомы остаются отделенными друг от друга, при этом каждый член пары
реплицируется и передается в дочернее ядро независимо от своего гомолога. В мейозе,
однако, пары гомологичных хромосом никоим образом не являются независимыми. Во
время профазы I все хромосомы выстраиваются в ряд со своими гомологами и образуют
биваленты (рис. 3.16). Это происходит после того, как каждая хромосома реплициро-
валась, но прежде, чем реплицированные структуры разделятся, так что бивалент фак-
тически содержит четыре копии хромосомы, каждой из которых предстоит найти свой
путь в одну из четырех гамет, которые будут произведены в конце мейоза. В пределах
бивалента нити хромосомы (хроматиды) могут подвергнуться физическому разбиению
и обмену сегментами ДНК. Этот процесс называют кроссинговером, или рекомбинаци-
ей, он был открыт бельгийским цитологом Янсенсом в 1909 г. Это событие произошло
лишь за два года до того, как у Моргана зародилась мысль о неполном сцеплении.
Каким образом открытие кроссинговера помогло Моргану объяснить неполное сце-
пление? Чтобы понять это, нам надо подумать об эффекте, который кроссинговер может
иметь на наследование генов. Рассмотрим два гена, каждый из которых имеет два аллеля.
Назовем первый ген А и его аллели А и а, а второй ген — Вс аллелями ВиЬ. Вообразим,
что эти два гена расположены на хромосоме номер 2 Drosophila melanogaster — вида
плодовой мушки, изучаемой Морганом. Мы будем следовать мейозу диплоидного ядра,
в котором одна копия хромосомы 2 несет аллели А и В, а вторая — а и Ь. Эта ситуация
иллюстрирована на рис. 3.17. Рассмотрим два альтернативных сценария.
Глава 3. Картирование геномов 101
Рис. 3.16. Мейоз. Показаны события, вовлекающие одну пару гомологичных хромосом; один член
пары выделен красным цветом, другой — синим. В начале мейоза хромосомы уплотняются, и каждая
гомологичная пара выстраивается в ряд, с тем чтобы сформировать бивалент. В пределах бивалента
может произойти кроссинговер, заключающийся в разбиении плеч хромосом и в обмене ДНК. Затем
мейоз продолжается парой митотических делений ядер, первое из которых дает два ядра (а в каждом
из них — по две копии всех хромосом, все еще скрепленные своими центромерами), а второе — четы-
ре ядра (и в каждом из них —уже по одной копии всех хромосом). Поэтому такие конечные продукты
мейоза — гаметы — являются гаплоидными
• Между генами А и В кроссинговер не возникает. Если происходит именно это, то
две из получившихся гамет будут содержать копии хромосомы с аллелями А и В,
а другие две будут содержать аллели а и Ь. Другими словами, две гаметы имеют
генотип АВ, а две — генотип ab.
• Между генами А и В кроссинговер происходит. Это приводит к тому, что гомологич-
ные хромосомы обмениваются между собой сегментами ДНК, содержащими ген В.
В конечном итоге каждая гамета имеет отличный от других генотип: одна — АВ,
другая — аВ, следующая —АЬ и последняя — ab.
Теперь мы подумаем о том, что случилось бы, если бы мы наблюдали результаты
мейоза в сотне идентичных клеток. Если кроссинговеры никогда не происходят, то по-
лучающиеся гаметы будут иметь следующие генотипы:
200 АВ
200 ab
Это полное сцепление: гены А и В ведут себя во время мейоза как единоцельная
единица. Но если (что более вероятно), в некоторых ядрах происходят кроссинговеры
между генами А и В, то пары аллелей не будут наследоваться как единые единицы.
Скажем, кроссинговеры происходят в ходе 40-ка из 100 мейозов. Тогда получатся сле-
дующие гаметы:
102 Часть 1. Изучение геномов
160 АВ
160 ab
40 АЬ
40 аВ
Сцепление неполное: оно лишь частичное. Помимо гамет с двумя родительскими
генотипами (АВ, ab), мы видим гаметы с рекомбинантными генотипами (АЬ, аВ).
От неполного сцепления до составления генетических карт
Как только Морган понял, как неполное сцепление можно объяснить кроссинго-
вером в ходе мейоза, он смог изобрести способ наносить на карту относительные по-
зиции генов на хромосоме. Фактически самая важная работа была проделана не самим
Морганом, а студентом из его лаборатории — Артуром Стёртевантом...1)
Стёртевант сделал допущение о том, что кроссинговер является случайным событи-
ем, так что он может произойти в любой позиции на протяжении пары вытянутых одна
вдоль другой хроматид. Если это допущение верно, то два гена, которые расположены
близко друг к другу, будут разделяться кроссинговерами реже, чем два гена, которые
сильнее отдалены друг от друга. Более того, частота, с которой гены разъединяются
кроссинговерами, будет прямо пропорциональна тому, как далеко друг от друга они
находятся на своей хромосоме. Поэтому частота рекомбинации является мерой рас-
стояния между двумя генами. Если определить частоты рекомбинации для различных
ПРОФАЗА I
Генотипы Генотипы
2АВ : 2ab 1 АВ: 1аВ : 1 АЬ: 1аЬ
пар генов, то можно построить карту их относи-
тельных позиций на хромосоме (рис. 3.18).
Оказалось, что допущение Стёртеванта
о случайном характере кроссинговеров верно
не во всех отношениях. Сравнения между гене-
тическими картами и фактическими позициями
генов на молекулах ДНК, определенными при
помощи физического картирования и секвени-
рования ДНК, показали, что в некоторых обла-
стях хромосом, названных горячими точками
рекомбинации, события кроссинговера более
вероятны, чем в других. Это означает, что рас-
стояние на генетической карте не обязательно
указывает физическое расстояние между двумя
маркёрами (см. рис. 3.25). Также мы теперь по-
Рис. 3.17. Влияние кроссинговера на сцепленные гены.
Рисунок показывает пару гомологичных хромосом: одна
выделена красным цветом, другая — синим. Буквами
А и В обозначены сцепленные гены, характеризуемые
аллелями А, а, В и Ь. Слева представлен мейоз без крос-
синговера между генами А и В: первые две из получив-
шихся гамет имеют генотип АВ, а другие две — ab. Спра-
ва показан кроссинговер между генами А и В: четыре
получившиеся гаметы демонстрируют все возможные
генотипы — АВ, аВ, АЬ и ab
...который так увлекся своими экспериментами, что сам претерпел трансформацию — в лауреата
учрежденной его же тезкой премии Альфреда Генри Стёртеванта. — Прим, персе.
Глава 3. Картирование геномов 103
Рис. 3.18. Создание генетической карты на основании частот
рекомбинации. Пример взят из оригинальных экспериментов,
проводимых Артуром Стёртевантом с плодовыми мушками.
Все четыре гена находятся на Х-хромосоме плодовой мушки.
Частоты рекомбинации между генами показаны наряду с их
выведенными позициями на карте
нимаем, что одна хроматида в одно и то же время
может участвовать в более чем одном кроссинго-
вере, и что есть ограничения на то, сколь близко
друг к другу эти кроссинговеры могут происходить;
все это ведет к еще большему числу погрешностей
в процедуре картирования. Несмотря на эти досад-
ные ограничения, анализ сцепления генетических
признаков обычно позволяет сделать правильные
выводы о порядке взаимного расположения генов
и получить оценки расстояний, достаточно точные
для построения генетических карт, которые имеют
ценность как основа для проектов секвенирования
генома1). Поэтому мы чуть углубимся в эту область
Гены
m
Белые глаза
Частоты рекомбинации
Миниатюрные крылья
Ярко-красные глаза
Между m и v = 3,0%
Между m и у = 33,7%
Между v и w = 29,4 %
Между w и у = 1,3%
Выведенные позиции на карте
У w v m
0 1,3 30,7 33,7
и посмотрим, как анализ сцепления генетических признаков выполняется с организ-
мами различных типов.
3.2.4. Анализ сцепления генетических признаков у организмов различ-
ного типа
Чтобы познакомиться с тем, как анализ сцепления генетических признаков вы-
полняется на практике, мы должны рассмотреть три совершенно разные ситуации:
• анализ сцепления генетических признаков у видов наподобие плодовой мушки
и мыши, с которыми мы можем выполнять запланированные эксперименты по
скрещиванию;
• анализ сцепления генетических признаков у людей, с которыми мы не можем
проводить запланированные эксперименты, но вместо этого можем изучать их
семейные родословные;
• анализ сцепления генетических признаков у бактерий, которые не подвергаются
мейозу.
Анализ сцепления генетических признаков при возможности проведения за-
планированных экспериментов по скрещиванию
Анализ сцепления генетических признаков первого типа — современный аналог
метода, разработанного Морганом и его коллегами. Данный метод основан на анализе
потомства от экспериментальных скрещиваний, поставленных между родителями
с известными генотипами, и применим, по крайней мере в теории, ко всем эукариотам.
Этические соображения не допускают применения такого подхода к человеку, а про-
Единица расстояния между генетическими маркёрами, когда вероятность сегрегации (расщепления)
признаков равна 1 %, называется сантиморганидой и примерно (со всеми оговорками) равна 1 млн п. н. —
Прим. ред.
104 Часть 1. Изучение геномов
блемы практического характера, такие как продолжительность периода беременности
и времени, необходимого новорожденному, чтобы достигнуть зрелости (и, следователь-
но, участвовать в последующих скрещиваниях), ограничивают эффективность этого
метода при работе с некоторыми животными и растениями.
Если мы вернемся к рис. 3.17, то увидим, что ключ к картированию гена таится в спо-
собности определять генотипы гамет, получаемых в ходе мейоза. В немногих ситуациях это
возможно путем непосредственного исследования гамет. Например, гаметы, производимые
некоторыми микробными эукариотами, в том числе дрожжами Saccharomyces cerevisiae,
могут быть выращены в колонии гаплоидных клеток, генотипы которых можно опреде-
лить биохимическими тестами. Прямая генотипизация гамет возможна также и с высшими
эукариотами, если использовать ДНК-маркёры, поскольку ПЦР можно проводить с ДНК из
отдельных сперматозоидов. Это позволяет типизировать длину фрагментов рестрикции,
длину простых повторов и SNP. К сожалению, типизация спермы весьма трудоемка. Поэтому
для высших эукариотов обычный анализ сцепления генетических признаков выполняют
не прямым исследованием гамет, но определением генотипов диплоидного потомства,
которое следует из слияния двух гамет — по одной от каждого члена родительской пары.
Другими словами, выполняется генетическое скрещивание.
Сложности с генетическим скрещиванием состоят в том, что получающееся ди-
плоидное потомство является продуктом не одного мейоза, а двух (по одному в обоих
родителях), и в большинстве организмов события кроссинговера происходят с равной
вероятностью при производстве мужских и женских гамет. Так или иначе мы должны
быть способны выпутать из генотипов диплоидного потомства события кроссоверов,
которые произошли в каждом из этих двух мейозов. Это означает, что скрещивание
следует планировать и проводить с особым вниманием. Стандартная процедура —
использовать анализирующее скрещивание. Оно представлено на рис. 3.19, где мы
поставили анализирующее скрещивание с целью нанести на карту два гена, которые
мы встречали ранее: ген А (аллели А и а) и ген В (аллели В и Ь), расположенные на хро-
мосоме 2 плодовой мушки. Определяющий признак анализирующего скрещивания —
генотипы обоих родителей:
• Один родитель — двойная гетерозигота. Это означает, что у этого родителя име-
ются все четыре аллеля: его генотип — АВ/ab. Это обозначение указывает на то,
что одна пара гомологичных хромосом несет аллели А и В, а другая — а и Ь. Двой-
ные гетерозиготы могут быть получены путем скрещивания двух скрещиваемых
в чистой линии организмов, например АВ/АВ х ab/ab.
• Второй родитель — размножаемая в чистой линии двойная гомозигота. В этом
родителе обе гомологичные копии хромосомы 2 одинаковые: в примере, показан-
ном на рис. 3.19, оба имеют аллели а и Ь, и генотип такого родителя — ab/ab.
Двойная гетерозигота имеет тот же самый генотип, что и клетка, мейоз которой мы
прослеживали на рис. 3.17. Поэтому наша цель состоит в том, чтобы вывести генотипы
гамет, произведенных этим родителем и вычислить долю рекомбинантов. Обратите
внимание, что все гаметы, произведенные вторым родителем (двойной гомозиготой)
будут иметь генотип ab независимо от того, являются л и они родительскими или реком-
бинантными гаметами. Аллели а и b оба рецессивные, так что мейоз в этом родителе не
отражается в каких-либо заметных фенотипических признаках потомства. Это означает,
что, как показано на рис. 3.19, фенотипы диплоидного потомства могут быть однозначно
преобразованы в генотипы гамет от родителя-двойной гетерозиготы. Поэтому анализи-
рующее скрещивание позволяет нам проводить прямой осмотр одного мейоза в отдель-
Глава 3. Картирование геномов 105
Рис. 3.19. Анализирующее скрещивание между ал-
лелями, показывающими доминирование и рецес-
сивность. А и В — маркёры с аллелями А, а, Ви Ь.
Получающееся потомство просчитывается наблюде-
нием его фенотипов. Поскольку родитель-двойная
гомозигота (родитель 2) имеет оба рецессивных
аллеля — а и Ь, — он фактически не вносит никакого
вклада в фенотипы потомства. Поэтому фенотип
каждого индивидуума в поколении тот же, что
и генотип гаметы от родителя 1, который дал начало
этому индивидууму
Аллели А и В доминируют над аллелями а и Ь
1
АВ/аЬ
РОДИТЕЛИ
X
2
аЬ/аЬ
Анализирующее
скрещивание
АВ
АЬ
аВ
аЬ
ab
аЬ
аЬ
аЬ
Гаметы
ГЕНОТИПЫ
ПОКОЛЕНИЯ F,
АВаЬ
АЬ аЬ
аВ аЬ
аЬ аЬ
ФЕНОТИПЫ
АВ
АЬ
аВ
ab
Каждый из фенотипов тот же, что и генотип
гаметы родителя № 1
ности и, следовател ьно, вычислять для двух
изучаемых генов частоту их рекомбинации
и расстояние на карте между ними.
Если в одном скрещивании отслежи-
вается более двух маркёров, то эффектив-
ность этого типа анализа сцепления гене-
тических признаков еще более возрастает.
Он не только быстрее, чем другие методы,
воспроизводит частоты рекомбинации, но
также позволяет определять относитель-
ный порядок маркёров на хромосоме путем простого просмотра данных. Это обуслов-
лено тем, что необходимы два события рекомбинации, чтобы отцепить центральный
маркёр от двух внешних маркёров в ряде из трех маркёров, тогда как любой из двух
внешних маркёров может быть отцеплен от них всего лишь путем одной рекомбинации
(рис. 3.20). Двойная рекомбинация менее вероятна, чем одинарная, так что выщепле-
ние центрального маркёра будет происходить относительно нечасто. Набор типичных
данных от скрещивания с тремя контрольными признаками представлен в таблице 3.2.
Анализирующее скрещивание было поставлено между тройной гетерозиготой [АВС/
abc) и тройной гомозиготой [abc/abc). Самое частое потомство — с одним из двух роди-
тельских генотипов, являющееся результатом отсутствия событий рекомбинации в об-
ласти, содержащей маркёры А, В и С. Два других класса потомства относительно часты
(в представленном примере — 51 и 63 потомка). Оба из них, как можно предположить,
являются результатом единственной рекомбинации. Анализ их генотипов показывает,
что в потомстве из первого класса маркёр А отцепился от маркёров В и С, а у потомков
Таблица 3.2. Набор типичных данных анализирующего скрещивания
по трем определительным признакам
Генотипы потомства Число потомков Выведенные события рекомбинации
ABC/abc abc/abc 987 Ни одного (родительские генотипы)
aBC/abc Abc/abc 51 Одно, между А и В/С
AbC/abc aBc/abc 63 Одно, между В и А/С
ABc/abc abC/abc 2 Два: одно между С и А,
второе — между С и В
106 Часть 1. Изучение геномов
Аллели А и В кодоминантны по отношению
к аллелям а и b
РОДИТЕЛИ
1 х 2 Анализирующее
АВ/ab АЬ/АЬ скрещивание
Единичный кроссовер
АВ
АЬ
аВ
ab
Гаметы
ГЕНОТИПЫ
ПОКОЛЕНИЯ F.
АВАЬ
АЬАЬ
аВАЬ
аЬАЬ
ОПРЕДЕЛЕННЫЕ
АЛЛЕЛИ
А+В+Ь
А+Ь
А+а+В+Ь
А+а+Ь
Двойной кроссовер
det d Е f
Рис. 3.20. Эффекты кроссинговеров в ходе диги-
бридного скрещивания. Для того чтобы любой
из двух внешних маркёров мог отцепиться от
остальных, достаточно лишь одного события ре-
комбинации, но для выщепления центрального
маркёра от двух внешних маркёров потребуется
две рекомбинации
Генотипы гамет родителя № 1 устанавливают
по определенным аллелям. Если определяется
только аллель А, то гамета родителя № 1 имеет
генотип А. Если определяются аллели А + а,
то гамета родителя № 1 имеет генотип а, и т. п.
Рис. 3.21. Анализирующее скрещивание между
аллелями, показывающими кодоминирование.
Гены А и В являются маркёрами, пары аллелей
которых кодоминантны. В этом конкретном при-
мере родитель-двойная гомозигота имеет гено-
тип АЬ/АЬ. Аллели, присутствующие в каждом
индивидууме поколения F1? определяются непо-
средственно — например, при помощи ПЦР. Эти
комбинации аллелей позволяют вывести генотип
гаметы родителя 1, которая дала начало всем ин-
дивидуумам
второго класса маркёр В отцепился от маркёров А и С. Следовательно, маркёры А и В яв-
ляются внешними маркёрами. Этот вывод подтверждается числом потомков, у которых
маркёр С отцепился от маркёров А и В. Таких потомков только два, а это говорит о том,
что для получения данного генотипа необходима двойная рекомбинация. Поэтому
маркёр С должен быть расположен между маркёрами А и В.
Осталось рассмотреть лишь один дополнительный момент. Если, как в примерах
на рис. 3.19 и в таблице 3.2, в анализирующем скрещивании исследуются гены, ал-
лели которых показывают доминантность и рецессивность, то двойная или тройная
гомозигота-родитель должен нести аллели для рецессивных фенотипов. Если, с другой
стороны, используются кодоминантные маркёры, то двойная гомозигота-родитель
может иметь любую комбинацию гомозиготных аллелей (например АВ/АВ, АЬ/АЬ,
aB/aB или ab/ab). Рис. 3.21, на котором представлен пример анализирующего скрещи-
вания такого типа, раскрывает причину этого. Обратите внимание, что ДНК-маркёры,
Глава 3. Картирование геномов 107
типизированные посредством ПЦР, показывают, что в действительности имеет место
кодоминирование: поэтому на рис. 3.21 развертывается типичный сценарий, наблю-
даемый в тех случаях, когда анализ сцепления генетических признаков выполняется
с ДНК-маркёрами.
Составление генетической карты на основе анализа родословной человека
Что касается человека, то, конечно, невозможно заранее подобрать желаемые ге-
нотипы родителей и поставить скрещивания, специально предназначенные для целей
картирования. Вместо этого данные для вычисления частот рекомбинации приходится
добывать путем анализа генотипов членов последовательных поколений существующих
семей. Это означает, что в распоряжении ученого имеются только ограниченные данные,
и их интерпретация часто бывает затруднена, потому что заключаемый между людьми
брак редко дает удобное анализирующее скрещивание и часто генотипы одного или
нескольких членов семейства неизвестны, потому что данные индивидуумы мертвы
или не склонны к сотрудничеству.
Эти проблемы проиллюстрированы на рис. 3.22. В данном примере мы изучаем
генетическую болезнь, присущую семейству из двух родителей и шестерых детей. Гене-
тические болезни часто используются как маркёры генов человека, при этом состояние
болезни является одним аллелем, а здоровое состояние — вторым аллелем. Родословная
на рис. 3.22, а показывает нам, что больна мать и четверо ее детей. Из анамнезов членов
ее семейства мы знаем, что бабушка по матери также страдала от этой болезни, но и она
сама и ее муж — дедушка по матери — уже умерли. Мы можем включить их в родослов-
ную, обозначив косыми чертами, указывающими на то, что их уже нет в живых, но мы
не можем получить никакой дополнительной информации об их генотипах. Мы знаем,
что ген болезни расположен на той же хромосоме, что и микросателлит, который мы
назовем М, четыре аллеля которого — Мг М2, М3 и М4 — присутствуют у ныне живу-
щих членов семейства. Наша цель состоит в том, чтобы нанести на карту позицию гена
болезни относительно этого микросателлита.
Чтобы установить частоту рекомбинации между геном болезни и микросател-
литом М, мы должны определить, сколько детей из числа отпрысков являются реком-
бинантами. При взгляде на генотипы этих шестерых детей, мы видим, что номера 1, 3
и 4 несут аллель болезни и аллель Мх микросателлита. Номера 2 и 5 обладают аллелем
здоровья и аллелем М2 микросателлита. Поэтому мы можем выстроить две альтернатив-
ные гипотезы. Согласно первой, две копии соответствующих гомологичных хромосом
в матери имеют генотипы болъной-М1 и здоровый-М2; поэтому дети 1,2,3,4 и 5 имеют
родительские генотипы, а ребенок 6 — один-единственный рекомбинант (рис. 3.22, б).
Это предполагает, что ген болезни и микросателлит сцеплены относительно тесно и что
кроссинговеры между ними происходят нечасто. Согласно альтернативной гипотезе
хромосомы матери имеют генотипы здоровый-М2 и болъной-М2; это означает, что дети
1-5 суть рекомбинанты, а ребенок 6 имеет родительский генотип. Это подразумевает,
что ген и микросателлит расположены на хромосоме относительно далеко друг от
друга. Мы не можем определить, какая из этих альтернативных гипотез верна: данные
разочаровывающе неоднозначны.
Наиболее удовлетворительное разрешение задачи, поставленной перед нами ро-
дословной на рис. 3.22, состояло бы в том, чтобы узнать генотип бабушки. Вообразим,
что это семейство из мыльной оперы и что бабушка на самом деле жива. Ко всеобщему
удивлению она вновь появляется на экране, и как раз вовремя, чтобы спасти снижаю-
щиеся рейтинги телезрителей. Ее генотип в отношении микросателлита М оказывается
108 Часть 1. Изучение геномов
а) Родословная
[ Обозначения?
О Здоровая женщина
ф Больная женщина
□ Здоровый мужчина
м}м3 М2М3 м.м< м,м3 М2М< М2М<
Больной мужчина
Мертв(а)
б) Возможные интерпретации родословной МАТЕРИНСКИЕ ХРОМОСОМЫ
Гипотеза 1 Больной Ц Гипотеза 2 Здоровый /И,
Здоровый М2 Больной М2
Ребенок 1 Больной Родительский Рекомбинантный
Ребенок 2 Здоровый М2 Родительский Рекомбинантный
Ребенок 3 Больной Родительский Рекомбинантный
Ребенок 4 Больной Родительский Рекомбинантный
Ребенок 5 Здоровый М2 Родительский Рекомбинантный
Ребенок 6 Больной М2 Рекомбинантный Родительский
Частота рекомбинации 1/6=16,7% 5/6 = 83,3 %
в) Восстановление генотипа бабушки по матери
AW | ^Аллель болезни должен быть сцеплен с М,
1 ВЕРНА ГИПОТЕЗА 1
м}м2
Рис. 3.22. Пример анализа родословной человека, а) Родословная показывает наследование генетической
болезни в семье, в которой оба родителя и шестеро их детей живы, а информация о бабушке и дедушке
со стороны матери известна из семейных хроник. Аллель болезни (закрашенные символы) является до-
минирующим над аллелем здорового состояния (пустые символы). Цель нашего анализа состоит в том,
чтобы определить степень сцепленности между геном болезни и микросателлитом М путем типизации
аллелей этого микросателлита (М1? М2 ит. д.) в геномах ныне живущих членов семейства. 6) Родословная
может быть интерпретирована двумя разными способами: гипотеза 1 дает низкую частоту рекомбина-
ции и полагает, что ген болезни сильно сцеплен с микросателлитом М. Гипотеза 2 предполагает, что ген
болезни и микросателлит связаны намного слабее. На виде в проблема решена повторным появлением
бабушки по матери, генотип микросателлита которой согласуется только с гипотезой 1
МгМ5 (рис. 3.22, в). Это говорит нам, что хромосома, унаследованная матерью, имеет
генотип болезнъ-Мг Поэтому мы можем с уверенностью заключить, что гипотеза 1
верна и что только ребенок 6 является рекомбинантом.
К сожалению, живущие в реальной жизни генетики не властны воскрешать клю-
чевые фигуры, задействованные в сценариях поставленных ими экспериментов, хотя
они могут получать ДНК из старых патологоанатомических образцов тканей, таких как
Глава 3. Картирование геномов 109
готовые препараты и карты Гатри. Неполные родословные анализируют статистически,
используя меру называемую счет Л ОШ. Аббревиатура Л ОШ означает «логарифмическое
отношение шансов» (при этом шанс выпадает на то, что гены сцеплены) и используется
главным образом, чтобы определить, лежат л и два изучаемых маркёра на одной и той же
хромосоме, другими словами, сцеплены ли рассматриваемые гены или нет. Если анализ
с помощью ЛОШ устанавливает сцепление, то он может обеспечить также и меру наи-
более вероятной частоты рекомбинации. В идеале имеющиеся данные должны быть
собраны путем анализа не одной, а нескольких родословных, что увеличило бы досто-
верность результата. Такой анализ более однозначен для семейств с большим числом
детей, и, как мы видели на рис. 3.22, важно, чтобы был известен генотип членов по
крайней мере трех поколений. По этой причине были учреждены коллекции семейств,
такие как поддерживаемая Centre d'Etudes du Polymorphisme Humaine (СЕРН) — Центр
исследований полиморфизмов человека в Париже. Собрание СЕРН содержит культиви-
руемые клеточные линии семейств, в которых могли быть получены образцы от обеих
бабушек и обоих дедушек, а также по крайней мере от восьми детей второго поколения.
Эта коллекция доступна для картирования при помощи ДНК-маркёров любому иссле-
дователю, который соглашается предоставить полученные данные центральной базе
данных СЕРНЧ
Составление генетических карт бактерий
И последний тип генетического картирования, который нам осталось рассмо-
треть, — стратегия, применяемая при работе с бактериями. Главная трудность, с которой
генетики столкнулись при попытке развить методы генетического картирования для
бактерий, состоит в том, что эти организмы, как правило, являются гаплоидными и,
таким образом, не подвергаются мейозу. Поэтому должен был быть изобретен какой-то
иной способ вызвать кроссинговеры между гомологичными сегментами ДНК бактерий.
Решение было найдено в использовании трех естественных способов, к которым при-
бегают бактерии для передачи частей ДНК от одного партнера другому (рис. 3.23):
• В процессе конъюгации две бактерии входят в физический контакт, и одна бакте-
рия (донор) передает ДНК второй бактерии (реципиенту). Переданная ДНК может
быть копией части или, возможно, всей хромосомы клетки-донора, или это может
быть сегмент хромосомной ДНК — до 1 млн п. н. в дл ину — встроенный в плазмиду.
Последнее называют передачей эписомы.
• Трансдукция заключается в передаче маленького сегмента ДНК — до 50 тыс. п. н.
или около того — от донора рецепиенту через бактериофаг.
• В ходе трансформации клетка реципиента вбирает из окружающей среды фраг-
мент ДНК, редко длиннее 50 тыс. п. н., выпущенный из клетки-донора.
Обычно используются биохимические маркёры, при этом доминирующий (ди-
кого типа) фенотип проявляется в обладании некоторой биохимической характери-
стикой (например, способностью синтезировать триптофан), а рецессивный фенотип
проявляется в обладании комплементарной характеристикой (например, неспособ-
ностью синтезировать триптофан). Передача гена обычно устанавливается между
штаммом доноров, который обладает аллелем дикого типа, и штаммом реципиентов
с рецессивным аллелем, причем факт передачи штамму реципиентов устанавливается
Развитие этой области столь стремительно, что уже сейчас можно послать образец своей ткани (на-
пример, соскоб со щеки) и получить генотип по многим маркёрам, найти родственников, которые уже есть
в базе данных, узнать предрасположенность к некоторым заболеваниям. — Прим. ред.
110 Часть 1. Изучение геномов
ДОНОР
а) Конъюгация
РЕЦИПИЕНТ
Рис. 3.23. Три способа осуществления пе-
реноса ДНК между бактериями, а) Конъ-
югация может привести к передаче ДНК
хромосомы или плазмиды от бактерии-
донора реципиенту. Конъюгация предпо-
лагает физический контакт между этими
двумя бактериями, причем передача, как
думают, происходит через узкую трубочку,
называемую пилем. 6) Трансдукция — пе-
редача реципиенту маленького сегмента
ДНК клетки-донора через бактериофаг.
в) Трансформация подобна трансдукции,
но в данном случае передается «голая»
ДНК. События, иллюстрированные на схе-
мах бив, часто сопровождаются гибелью
клетки-донора. В случае б происходит ги-
бель клетки, когда бактериофаги выходят
из клетки-донора. В ситуации в выход ДНК
из клетки-донора обычно является след-
ствием гибели клетки, вызванной есте-
ственными причинами
путем наблюдения за приобретением биохимиче-
ской функции, определяемой изучаемым геном.
Эта методика проиллюстрирована на рис. 3.24, а,
где мы видим, как функционально активный ген
биосинтеза триптофана передается от бактерии
дикого типа (генотип, описанный как trp+] реци-
пиенту, у которого отсутствует функционально
активная копия этого гена (trp"). Реципиент назы-
вается ауксотрофом по триптофану (слово, упо-
требляемое для описания мутантной бактерии,
которая может выжить, только если обеспечена пи-
тательным веществом — в данном случае трипто-
фаном, — не требуемым для бактерии дикого типа;
см. раздел 16.1.2). После передачи необходимы два
кроссинговера, чтобы встроить переданный ген
в хромосому клетки-реципиента, что преобразует
реципиента из trp~ в trp+.
Особенности процедуры картирования зави-
сят от выбранного способа передачи гена. Во время
конъюгации ДНК передается от донора рецепиенту
таким же образом, каким нить протягивается через
трубку. Поэтому относительные позиции маркёров
на молекуле ДНК могут быть нанесены на карту пу-
тем определения промежутков времени, через ко-
торые маркёры появляются в клетке-реципиенте.
В примере, показанном на рис. 3.24, б, маркёры А,
В и С передаются соответственно в течение 8, 20
и 30 минут после начала конъюгации. На передачу
целой хромосомы Escherichia coli уходит прибли-
зительно 100 минут. Напротив, картирование при
помощи трансдукции и трансформации позволяет
картировать гены, которые расположены отно-
сительно близко друг к другу, потому что пере-
даваемый сегмент ДНК короткий (<50 тыс.п.н.),
так что вероятность того, что два гена будут пере-
даны совместно, зависит от того, как близко друг
к другу они находятся на хромосоме бактерии
(рис. 3.24, в).
3.3. Составление физических карт
Полученная исключительно генетическими
методами карта едва ли будет достаточно точна,
чтобы служить ориентиром для проекта секве-
нирования на стадии расшифровки генома. Это
обусловлено двумя причинами.
• Разрешение генетической карты зависит от
числа кроссинговеров, которые были набраны. Для
Глава 3. Картирование геномов 111
а) Перенос ДНК между бактерией-
донором и бактерией-реципиентом
б) Последовательная передача
маркёров в ходе конъюгации
ДОНОР РЕЦИПИЕНТ
ДОНОР РЕЦИПИЕНТ
А~ВС АВС'
в) Совместный перенос тесно
сцепленных маркёров во время
трансдукции или трансформации
ДОНОР РЕЦИПИЕНТ
Время
переноса,
мин
Частота, с которой происходит
преобразование Д В -+А'В\
зависит от того, насколько
близко друг к другу расположены
маркёры Д и В на хромосоме
Рис. 3.24. Основа картирования генов бактерий, а) Перенос функционально активного гена для био-
синтеза триптофана от бактерии дикого типа (генотип, описываемый как trp+) реципиенту, у которого
отсутствует функционально активная копия этого гена (trp-). б) Картирование при помощи конъюгации,
в) Картирование путем трансдукции и трансформации
микроорганизмов это не главная проблема, потому что они могут быть получены
в огромных количествах, что позволяет йзучить много кроссинговеров и получить
очень подробную генетическую карту, на которой маркёры будут расположены
всего лишь на несколько тыс. п. н. друг от друга. Например, когда проект секвени-
рования генома Escherichia coli был начат в 1990 г., последняя генетическая карта
для этого организма включала более чем 1 400 маркёров, то есть в среднем 1 мар-
кёр на 3,3 тыс.п.н. Она была достаточно подробна, чтобы направлять программу
секвенирования без необходимости проводить физическое картирование в боль-
ших объемах. Точно так же проект определения генома Saccharomyces cerevisiae
поддерживался мелкомасштабной генетической картой (приблизительно 1 150
генетических маркёров, в среднем 1 на 10 тыс.п.н.). Проблема с людьми и боль-
шинством других эукариотов состоит в том, что попросту невозможно получить
большое число потомков, так что может быть изучено сравнительно мало мейозов,
и разрешающая способность анализа сцепления генетических признаков неиз-
бежно будет ограничена. Это означает, что гены, которые отстоят друг от друга
на несколько десятков тыс. п. н., могут появиться на генетической карте в одной
и той же позиции.
• Генетические карты имеют ограниченную точность. Мы затрагивали этот момент
в разделе 3.2.3, когда оценивали предположение Стёртеванта о том, что кроссинго-
веры по всей длине хромосомы происходят случайным образом. Это предположение
верно лишь отчасти, потому что наличие очагов рекомбинации означает, что в не-
которых точках кроссинговеры будут происходить с большей вероятностью, чем
в других. Влияние данного явления на точность генетической карты было показано
112 Часть 1. Изучение геномов
Физическая карта Генетическая карта
his4
SUP53 _
1еи2
Центромера < н
pgkl -
pet18 - -
cry1 -
MAT -
ABP1
Рис. 3.25. Сравнение между генетиче-
ской и физической картами хромосомы III
Saccharomyces cerevisiae. Сравнение пока-
зывает несоответствия между генетической
и физической картами, где последняя опреде-
лена секвенированием ДНК. Обратите вни-
мание, что порядок верхних двух маркёров
(glkl и chai) на генетической карте неверен
и есть также различия в относительном рас-
положении других пар маркёров
thr4
SUP61
ABP1
в 1992 г., когда была опубликована полная по-
следовательность хромосомы III Saccharomyces
cerevisiae, что позволило провести первое прямое
сравнение между генетической картой и факти-
ческими позициями маркёров, выявленными
путем секвенирования ДНК (рис. 3.25). Были
обнаружены значительные несоответствия,
причем даже до такой степени, что на основа-
нии результатов генетического анализа одна
пара генов была расположена в неправильном
порядке. Имейте в виду, что S. cerevisiae — один
из двух эукариотов (второй — плодовая мушка),
чьи геномы были подвергнуты интенсивному
генетическому картированию. Если генетиче-
ская карта дрожжей является неточной, то на-
сколько же точными могут быть генетические
карты организмов, подвергавшихся менее под-
робному анализу?!
Эти два ограничения генетического кар-
тирования подразумевают, что в проектах опре-
деления геномов большинства эукариотов их
генетические карты следует проверять и до-
полнять альтернативными методами картиро-
вания, прежде чем начинать крупномасштабное
секвенирование ДНК. С целью устранения этой
проблемы было развито изобилие методов фи-
зического картирования, из числа которых мы
упомянем следующие, самые важные:
• рестрикционное картирование, которое
определяет местонахождение на молекуле
ДНК относительных позиций сайтов узнава-
ния для эндонуклеаз рестрикции;
• флюоресцентная гибридизация in situ (FISH), в которой местоположения мар-
кёра наносятся на карту путем гибридизации содержащего этот маркёр зонда
с интактными хромосомами;
• картирование меченых участков последовательности (STS), при котором на
карту путем исследования коллекций фрагментов геномной ДНК при помощи
ПЦР и (или) гибридизационного анализа наносятся позиции коротких последо-
вательностей.
3.3.1. Составление рестрикционных карт
Генетическое картирование, использующее в качестве ДНК-маркёров RFLP, позво-
ляет определить в пределах генома местонахождение позиций полиморфных участков
рестрикции (раздел 3.2.2), но лишь немногие участки рестрикции в геноме полиморфны,
так что многие участки не могут быть картированы этим методом (рис. 3.26). Можно
ли увеличить плотность маркёров на карте генома, используя альтернативный метод
определения местонахождения позиций некоторых из неполиморфных участков ре-
Глава 3. Картирование геномов 113
стрикции? Именно этого и достигает рестрикционное картирование, хотя на практике
данная методика имеет ограничения, в силу которых она применима только к сравни-
тельно небольшим молекулам ДНК. Сначала мы рассмотрим сам метод, а затем оценим
его уместность для картирования геномов.
Основная методология составления рестрикционных карт
Простейший способ построить рестрикционную карту состоит в сравнении разме-
ров фрагментов, полученных в результате переваривания молекулы ДНК двумя разными
ферментами рестрикции, которые опознают различные целевые последовательности.
Пример с использованием ферментов рестрикции EcoRl и BamHI показан на рис. 3.27.
Сначала молекула ДНК переваривается только одним из этих ферментов, и размеры по-
лученных фрагментов измеряются посредством электрофореза в агарозном геле. Затем
молекула переваривается вторым ферментом, и полученные фрагменты вновь измеря-
ются в агарозном геле. Полученные на данный момент результаты позволяют установить
число участков рестрикции для каждого фермента, но не позволяют определить их отно-
сительные позиции. Поэтому добывается дополнительная информация по результатам
совместного переваривания молекулы ДНК обоими ферментами. В представленном на
рис. 3.27 примере такая двойная рестрикция позволяет нанести на карту три участка.
Однако возникает проблема с большим фрагментом рестрикции EcoRI, потому что он
содержит два участка рестрикции BamHI и есть две альтернативные возможности рас-
положения на карте внешнего из них. Эта проблема решается возвращением к исходной
молекуле ДНК и дополнительной ее обработкой ферментом Bam HI в отдельности, но на
сей раз предотвращая протекание переваривания до конца, — например, путём выдер-
живания реакционной смеси в течение малого промежутка времени или посредством
выбора условно оптимальной температуры выдержки. Это называется частичной ре-
стрикцией и ведет к более сложному набору продуктов, причем на этот раз продукты
полной рестрикции дополнены частично рестриктированными фрагментами, которые
по-прежнему содержат один или несколько неразрезанных участков рестрикции BamHI.
В примере, показанном на рис. 3.27, размер одного из фрагментов частичной рестрик-
ции является диагностическим фактором, и
его определение позволяет распознать среди
возможных карт единственно верную.
Частичная рестрикция обычно дает
информацию, необходимую для завершения
карты, но если число участков рестрикции
велико, то анализ такого типа становится
громоздким потому лишь, что приходится
рассматривать очень много различных фраг-
ментов. Альтернативная стратегия более про-
ста, потому что она позволяет игнорировать
большинство фрагментов. Это достигается
прикреплением маркёра радиоактивного
или иного типа к каждому концу стартовой
молекулы ДНК до начала частичного пере-
варивания. В результате многие продукты
частичной рестрикции становятся «невиди-
мыми», потому что они не содержат концевой
фрагмент и, таким образом, не обнаружива
1“ к R Полиморфный
участок рестрикции
□
R Неполиморфный
- R участок рестрикции
R
R
R
Рис. 3.26. Не все участки рестрикции полиморфны
114 Часть 1. Изучение геномов
4,9 тыс. п. н.
EcoRI у BamHI I \ EcoRI + BamHI
1,5 0,2 W
3,4 0,7 1,0 1,2 2,0 1,0
1,2 2,0
ИНТЕРПРЕТАЦИЯ ДВОЙНОЙ РЕСТРИКЦИИ
Фрагменты
Выводы
0,2 тыс. п. н., 0,5 тыс. п. н.
1,0 тыс. п. н.
1,0 тыс. п. н., 2,0 тыс. п. н.
Они должны получаться из фрагмента BamHI 0,7 тыс. п. н.,
который поэтому имеет внутренний участок рестрикции EcoRI:
В Ев
oJc 2
Это должен быть фрагмент BamHI без внутреннего участка EcoRI. Мы можем
объяснить фрагмент EcoR11,5 тыс. п. н., если мы поместим фрагмент
1,0 тыс. п. н. следующим образом:
1,5
Они также должны быть фрагментами BamHI без внутренних участков EcoRI.
Они должны лежать в пределах фрагмента EcoRI 3,4 тыс. п. н. Есть две
возможности:
—У?—°— и™„—у? °
0,7 1|2 2,0 0,7 2’° 1,2
ПРЕДСКАЗАННЫЕ РЕЗУЛЬТАТЫ ЧАСТИЧНОЙ РЕСТРИКЦИИ BamHI
Если верна карта I, то продукты частичной рестрикции будут включать
фрагмент 1,2 + 0,7 - 1,9 тыс. п. н.
Если верна карта II, то продукты частичной рестрикции будут включать
фрагмент 2,0 + 0,7 = 2,7 тыс. п. н.
4,9 тыс. п. н.
I BamHI, условно
* оптимальные условия
1,0 1,7 3,7
0.7 2,7 3,9
— — -------------------------------— 4'9__________________
2,0 3,2
~ ----------------------- ВЫВОД:
— ВЕРНА КАРТА II
Рис. 3.27. Рестрикционное картирование. Цель состоит в том, чтобы нанести на карту участки EcoRI (Е)
и BamHI (В) в линейной молекуле ДНК размера 4,9 тыс. п. н. Результаты одинарной и двойной рестрикции
показаны наверху. Размеры фрагментов, полученных после двойной рестрикции, позволяют построить
две альтернативные карты, как объяснено в центральной области, нерешенная проблема — позиция
одного из трех участков BamHI. Обе карты проверяются частичной рестрикцией BamHI (нижняя часть
рисунка), что показывает, что карта II — правильная
Глава 3. Картирование геномов 115
ются, когда агарозный гель про-
сматривается на наличие меченых
продуктов (рис. 3.28). Размеры тех
продуктов частичной рестрикции,
которые являются видимыми,
позволяют определять позиции
ненанесенных на карту участков
относительно концов исходной
молекулы1).
si Молекула ДНК
с конечной меткой
Видимые фрагменты
частичной рестрикции
Масштаб рестрикционных
карт ограничен длиной ре-
стриктов
Если число участков разре-
за для используемых ферментов
относительно невелико, то по-
строение карты рестрикции не
вызывает особых затруднений.
Рис. 3.28. Упрощение анализа частичной рестрикции путем
прикрепления маркёров к концам молекулы ДНК перед пере-
вариванием. Показан один конец помеченной на конце моле-
кулы ДНК. После частичной рестрикции детектируют только
те продукты, которые включают в себя концевой фрагмент.
Это значительно упрощает анализ и позволяет определять
позиции участков рестрикции непосредственно по длинам
меченых продуктов
Однако по мере увеличения числа участков разреза возрастает также и число продуктов
одинарной, двойной и частичной рестрикции, размеры которых придется определять
и сравнивать для построения карты. Конечно, может быть пущен в ход компьютерный
анализ, но в конечном счете все равно возникают проблемы* 2). В какой-то момент бу-
дет достигнута стадия переваривания, когда реакционная смесь содержит так много
фрагментов рестрикции, что отдельные полосы сольются в агарозном геле и увеличат
шансы того, что один или несколько фрагментов будут неправильно измерены или
вообще пропущены. Если несколько фрагментов имеют подобные размеры, то, даже
если все они могут быть идентифицированы, может оказаться невозможно собрать из
них однозначную карту.
Поэтому рестрикционное картирование более приемлемо для маленьких, а не боль-
ших молекул, причем верхний предел этого метода зависит от частоты участков рестрик-
ции в картируемой молекуле. Если выбрать ферменты с шестинуклеотидными сайтами
узнавания, то на практике обычно удается построить однозначную рестрикционную
карту для молекулы ДНК не более 50 тыс. п. н. в длину. Порог в пятьдесят тыс. п. н. ниже
минимального размера хромосом бактерий или эукариотов, хотя он вполне покрывает
геномы вирусов и органелл, и рестрикционные карты полного генома действительно
сыграли немалую роль в ориентации проектов секвенирования таких малых молекул.
Рестрикционные карты будут столь же полезны, если геномную ДНК бактерии или
эукариота клонировать и клонированные фрагменты будут менее 50 тыс.п.н. в длину,
потому что в таком случае можно создать подробную рестрикционную карту как пред-
посылку к секвенированию клонированной области. Это важная сфера применения
рестрикционного картирования в проектах секвенирования больших геномов, но есть
На самом деле суть проблемы заключается в экспоненциальном росте числа вариантов. Даже карта
с 10 сайтами рестрикции для каждой из двух рестриктаз не может быть однозначно восстановлена на прак-
тике. — Прим.ред.
2> Описанные методы носят, скорее, исторический характер и сейчас не применяются (разве что при
поиске новых рестриктаз). Подобное картирование рестриктных фрагментов возможно только, когда число
сайтов расщепления невелико. Однако это означает, что и сама молекула ДНК невелика (несколько тыс. п. н.).
В современных условиях проще ее попросту секвенировать! — Прим. ред.
116 Часть 1. Изучение геномов
ли какая-нибудь возможность использования рестрикционного анализа для более
общего картирования полных геномов размером более 50 тыс.п.н.?
Компетентный специалист ответит «да», потому что ограничения рестрикционно-
го картирования могут быть немного ослаблены за счет подбора ферментов, которые,
как можно ожидать, имеют редкие участки разрезания в целевой молекуле ДНК. Такие
«редкощепящие» рестриктазы подпадают под две категории.
• несколько ферментов рестрикции разрезают в семи- или восьминуклеотидных сай-
тах узнавания. Примеры — Sap\ (5’-GCTCTTC-3') uSgfl (5'-GCGATCGC-3'). Ферменты
с семинуклеотидными сайтами узнавания, как ожидается, должны разрезать мо-
лекулу ДНК с содержанием CG 50 % в среднем единожды на каждые 47 = 16 384 п. н.
Ферменты с восьминуклеотидными сайтами узнавания должны разрезать один
раз на каждые 48 =65536 п. н. Эти числа сравнимы с 46 = 4096 п. н. для ферментов
с шестинуклеотидными сайтами узнавания, такими как Вап?Н1 и EcoRI. Рестрик-
тазы с семи- и восьминуклеотидными сайтами узнавания часто используются
в рестрикционном картировании больших молекул, но в целом данный подход не
является столь распространенным, сколь он мог бы быть, попросту потому, что
известно мало таких ферментов.
• Можно использовать ферменты с сайтами узнавания, содержащими такие мотивы,
которые встречались бы в целевой ДНК достаточно редко. Молекулы геномной
ДНК не имеют случайных последовательностей, а у некоторых молекул крайний
дефицит некоторых мотивов. Например, последовательность 5'-CG-3' редка в ге-
номах позвоночных животных, потому что клетки позвоночных обладают фер-
ментом, который добавляет метильную группу к 5-углероду нуклеотида С во всех
таких последовательностях1). Дезаминирование полученного 5-метилцитозина
дает тимин (рис. 3.29). Вследствие этого в ходе эволюции позвоночных многие
из последовательностей 5'-CG-3', которые были первоначально в этих геномах,
были преобразованы в 5'-TG-3'. Поэтому ферменты рестрикции, которые опозна-
ют участок, содержащий мотив 5'-CG-3', разрезают ДНК позвоночного животного
относительно нечасто. Примеры — Smai (5'-CCCGGG-3'), который разрезает ДНК
человека в среднем один раз на каждые 78 тыс.п.н., и FssHII (5'-GCGCGC-3'), кото-
рый разрезает единожды на каждые 390 тыс.п.н. Обратите внимание, что Not\ —
рестриктаза с восьминуклеотидным сайтом узнавания — тоже нацеливается на
последовательности 5'-CG-3' (сайт узнавания 5'-GCGGCCGC-3') и разрезает ДНК
человека очень редко — приблизительно один раз на каждые 10 млнп.н.
Таким образом, потенциал рестрикционного картирования увеличивается за счет
использования редкощепящих рестриктаз. Строить рестрикционные карты геномов
животных и растений пока что не удается, но уже вполне возможно использовать эту
методику с большими клонированными фрагментами и с маленькими молекулами ДНК
прокариотов и низших эукариотов наподобие дрожжей и грибов.
Если используются редкощепящие рестриктазы, то для изучения получающихся
фрагментов рестрикции может возникнуть необходимость применения специального
типа электрофореза в агарозном геле. Это связано с тем, что зависимость между длиной
молекулы ДНК и ее скоростью миграции в электрофоретическом геле не линейна, и с воз-
растанием длины молекул разрешающая способность гель-электрофореза уменьшается
Это утверждение неверно. Метилируется только часть цитозинов и это метилирование тканеспеци-
фично и играет регуляторную роль. В зародышевой линии клеток (а только они передаются по наследству)
уровень метилирования невысок. — Прим. ред.
Глава 3. Картирование геномов 117
(рис. 3.30, а). Это означает, что раз-
делять молекулы более чем, скажем,
50 тыс. п. н. в длину, невозможно, по-
тому что в стандартном агарозном
геле все наиболее длинные моле-
кулы перемещаются в виде единой
медленно мигрирующей полосы.
Для их разделения необходимо за-
менить линейное электрическое
поле, применяемое в обычном гель-
электрофорезе, более сложным
полем. Одним из примеров таких
методов служит электрофорез
в геле с ортогонально чередую-
щимся полем — OFAGE [пульс-
форез1) — Прим, ред.], при котором
электрическое поле чередуется
между двумя парами электродов,
каждая из которых помещена под
углом 45° к продольной линии геля
(рис. 3.30,6). Молекулы ДНК движут-
ся через гель, как и обычно, но каж-
дое изменение в поле вынуждает
молекулы перестраиваться. Более
короткие молекулы перестраива-
ются быстрее, чем более длинные,
и, таким образом, мигрируют через
гель быстрее. За счет такого приема
могут быть разрешены молекулы
намного большей длины, чем раз-
CG
Цитозин NH2
хсн
о N NH2
5-Метилцитозин
Тимин
Рис. 3.29. Последовательность 5'-CG-3' является редкой в ДНК
позвоночных из-за метилирования С, сопровождаемого деза-
минированием и получением!. Неметилированный цитозин
также может быть дезаминирован, но продукт — урацил —
обнаруживается системой восстановления ДНК клеток по-
звоночных (раздел 16.2.2) и преобразуется назад в цитозин.
Напротив, тимин не опознается эффективно системой репа-
рации, так что эти нуклеотиды сохраняются в геноме
деляемые обычным гель-электрофорезом. К подобного рода методам относят также CHEF
(электрофорез в геле с ограниченными контуром однородными электрическими
полями) и FIGE (электрофорез в геле с обращением поля).
Непосредственное наблюдение участков рестрикции в молекулах ДНК
Помимо всего вышеописанного, для нанесения на карту участков рестрикции
в молекулах ДНК можно использовать методы, не связанные с электрофорезом. Со-
гласно методу, названному оптическим картированием, позиции участков рестрик-
ции определяются путем непосредственного наблюдения разрезанных молекул ДНК
в микроскоп. Сначала ДНК необходимо закрепить на предметном стекле таким способом,
чтобы отдельные молекулы стали вытянутыми, а не слипшимися между собой в единую
массу. Существуют различные способы осуществления этой процедуры, в том числе
вытягивание гелем и расчесывание молекул.
Для того чтобы приготовить вытянутые гелем волокна ДНК, хромосомную ДНК
переводят во взвесь в расплавленной агарозе и помещают на предметное стекло микро-
Всякий форез в переменном электрическом поле, когда катод и анод меняют свою полярность через
определенные промежутки времени, называют пульс-форезом. Существует много схем проведения пульс-
фореза, различающихся взаимным расположением полюсов, частотой «пульсации» и т. д. — Прим, перев.
118 Часть 1. Изучение геномов
а) Стандартный электрофорез в агарозном геле
Плохое разделение молекул
ДНК длины > 50 тыс. п. н.
б) Электрофорез в геле с ортогонально чередующимся полем (OFAGE)
Рис. З.ЗО. Обычный и нестандартный электрофорез в агарозном геле, а) В стандартном электрофорезе
в агарозном геле электроды помещают с обоих концов геля, и молекулы ДНК мигрируют по прямой
линии к положительному электроду. Этот способ не позволяет разделять молекулы длиннее примерно
50 тыс. п. н. 6) В OFAGE электроды размещают по углам плиты геля, причем поле чередуется между
парами электродов А и В. OFAGE позволяет разделять молекулы длины до 2 млн п. н.
скопа. По мере того как гель охлаждается и затвердевает, молекулы ДНК вытягиваются
(рис. 3.31, а). Согласно методике расчесывания молекул волокна ДНК приготовляют пу-
тем погружения покрытого силиконом покровного стекла в раствор ДНК, выдерживания
его в течение 5-ти минут (за это время молекулы ДНК прикрепляются к покровному
стеклу своими концами) и последующего вынимания стекла из раствора с постоянной
скоростью 0,3 мм в сек (рис. 3.31, б). Сила, необходимая для протягивания молекул ДНК
через поверхностный мениск, заставляет каждую из них вытягиваться в линию. На от-
крытом воздухе поверхность покровного стекла сразу же высыхает, при этом молекулы
ДНК остаются на нем в виде массива параллельных волокон. После вытягивания или
расчесывания закрепленные молекулы ДНК обрабатывают ферментом рестрикции и за-
тем проявляют, добавляя флюоресцентный краситель, такой как DAPI (4,6-диамино-2-
фенилиндолдигидрохлорид), который окрашивает ДНКтаким образом, чтобы ее волокна
были видимы при наблюдении препарата во флюоресцентный микроскоп с большим
увеличением. По мере стягивания волокон ДНК в силу их природной упругости, участки
рестрикции в вытянутых молекулах постепенно превращаются в разрывы, что позволяет
регистрировать относительные позиции разрезов.
Поначалу оптическое картирование применялось к большим фрагментам ДНК,
клонированным в векторах на YAC и ВАС (раздел 2.2.1). Позже применимость этой тех-
нологии к картированию геномной ДНК была доказана в ходе исследований хромосомы
длиной 1 млн п. н. малярийного паразита Plasmodium falciparum, а также двух хромосом
и одиночной мегаплазмиды бактерии Deinococcus radiodurans (см. табл. 8.2).
Глава 3. Картирование геномов 119
а) Вытягивание гелем
б) Расчесывание молекул
Предметное стекло
микроскопа, покрытое
ферментом рестрикции
Хромосомная ДНК
У Расплавленная
агароза
Раствор ДНК
Покровное
стекло
Молекулы ДНК прикрепляются
к покровному стеклу одним концом
I
Агароза затвердевает,
ДНК вытягивается
участками рестрикции
Рис. 3.31. Вытягивание гелем и расчесывание молекул, а) Чтобы выполнить вытягивание гелем, расплав-
ленная агароза, содержащая молекулы хромосомной ДНК, наносится пипеткой на предметное стекло
микроскопа, покрытое ферментом рестрикции. По мере затвердевания геля молекулы ДНК постепенно
вытягиваются. Остается непонятным, почему это происходит, но думают, что этот процесс может быть
обусловлен движением жидкости на стеклянной поверхности во время гелеобразования. Добавление
хлорида магния активирует фермент рестрикции, который разрезает молекулы ДНК. По мере того как
молекулы постепенно сворачиваются в спирали, разрывы, представляющие собой участки разрезания,
ширятся и становятся видимыми. 6) При расчесывании молекул покровное стекло опускают в раствор
ДНК. Молекулы ДНК прикрепляются к покровному стеклу своими концами, и стекло извлекается из рас-
твора со скоростью 0,3 мм в сек, в результате чего получается «гребенка» параллельных молекул
Молекулы ДНК
расчесываются
3.3.2. Флюоресцентная гибридизация in situ
Метод оптического картирования, описанный выше, прокладывает путь к процеду-
ре физического картирования второго типа, которую мы сейчас и рассмотрим, — FISH.
Как и при оптическом картировании, FISH позволяет непосредственно визуализиро-
вать позицию маркёра на хромосоме или вытянутой молекуле ДНК. При оптическом
картировании маркёром служит участок рестрикции, и он наблюдается в виде разрыва
в вытянутом волокне ДНК. В методе FISH маркёром является последовательность ДНК,
которая отображается путем гибридизации с флюоресцентным зондом.
120 Часть 1. Изучение геномов
Делящиеся клетки
с метафазными
хромосомами
Предметное
------ стекло микроскопа
Формамид |
Хромосомы
денатурируются
Добавление I
зонда *
Сигнал от зонда
Рис. 3.32. Флюоресцентная гибридиза-
ция in situ. Образец делящихся клеток
высушивается на предметном стекле
микроскопа и обрабатывается формами-
дом таким образом, чтобы хромосомы
денатурировались, но не потеряли свою
характерную метафазную морфологию
(см. раздел 7.1.2). Позиция, в которой
зонд гибридизируется с хромосомной
ДНК, визуализируется за счет детекти-
рования флюоресцентного сигнала, ис-
пускаемого меченой ДНК
Гибридизация радиоактивными или флюорес-
центными зондами in situ
Гибридизация in situ — вариант гибридизаци-
онного анализа (раздел 2.1.2), в котором ненарушен-
ная хромосома исследуется путем ее зондирования
меченой молекулой ДНК. Позиция на хромосоме,
в которой происходит гибридизация, дает инфор-
мацию о местоположении используемой в качестве
зонда последовательности ДНК на карте (рис. 3.32).
Для того чтобы этот метод работал, ДНК в хромосо-
ме должна быть переведена в однонитевую форму
(«денатурирована») путем разрушения пар основа-
ний, которые сплачивают нити в двойную спираль.
Только после этого хромосомная ДНК будет способна
гибридизироваться с зондом. Стандартный метод
денатурирования хромосомной ДНК без наруше-
ния морфологии хромосомы состоит в высушивании
препарата на предметном стекле микроскопа и по-
следующей обработке формамидом.
В ранних версиях метода гибридизации in situ
применялся зонд, меченный радиоактивной меткой,
но эти процедуры были неудовлетворительны, по-
тому что с радиоактивной меткой трудно добиться
и чувствительности, и разрешения — двух опреде-
ляющих факторов для успешной гибридизации in
situ. Для получения высокой чувствительности не-
обходимо, чтобы радиоактивная метка имела высо-
кую энергию эмиссии (пример такой радиоактивной
метки — 32Р), но если радиоактивная метка имеет
высокую энергию эмиссии, то она рассеивает свой
сигнал и, таким образом, дает низкое разрешение.
Высокое разрешение возможно в том случае, если ис-
пользуется радиоактивная метка с низкой энергией
эмиссии типа 3Н, но такие радиоактивные метки имеют столь низкую чувствительность,
что необходимы длительные экспозиции, которые приводят к появлению сильного фона
и, как следствие, — трудностей в различении подлинного сигнала.
Эти проблемы были решены в конце 1980-х гг. разработкой нерадиоактивных
флюоресцентных меток ДНК. Эти метки сочетают в себе высокую чувствительность
с высоким разрешением и потому идеальны для гибридизации in situ. Были разработаны
флюоресцентные метки с эмиссией различных цветов, что позволило гибридизировать
целый ряд различных зондов с одной хромосомой и различать их индивидуальные ги-
бридизационные сигналы, благодаря чему появилась возможность наносить на карту
относительные позиции последовательностей зондов (рис. 3.33). Чтобы максимально
повысить чувствительность, необходимо метить зонды настолько интенсивно, на-
сколько это возможно, что в прошлом подразумевало применение в качестве зондов
весьма длинных молекул ДНК, как правило, клонированных фрагментов ДНК длины по
крайней мере 40 тыс. п. н. Теперь, когда разработаны методы, позволяющие достигнуть
Глава 3. Картирование геномов 121
Рис. 3.33. Применение FISH в физическом картировании. 18 Различных
космидных клонов были помечены различными флюоресцентными | И |
маркёрами и гибридизированы содной парой гомологичных хромосом. |*1
Хромосомы выходят из метафазного ядра и, следовательно, соедине- f I !
ны своими центромерами. Относительные позиции флюоресцентных £ Д
сигналов позволяют определять местоположения всех тех фрагментов . * ||
ДНК на карте, которые содержатся в космидах. Изображение любезно W и ТЛ ® |
предоставлено Октавианом Хенегариу " I
интенсивного мечения с более короткими молекулами, тре- I * i
бованиекдлине зондов имеет меньшее значение. Поскольку । *
мы заинтересованы построением физической карты, клони- | ЧI
рованный фрагмент ДНК может рассматриваться всего лишь Ж
как маркёр другого типа, хотя на практике использование * е Л
клонов в качестве маркёров открывает для анализа второе |
измерение, потому что клонированная ДНК — это материал, « ’ |
по которому определяется последовательность ДНК. Поэтому f * I
картирование позиций клонов обеспечивает прямую связь t сJi
между картой генома и последовательностью его ДНК. ™
Если зонд представлен длинным фрагментом ДНК, -ж!
то одна из возможных проблем, — по крайней мере в слу-
чае высших эукариотов, — состоит в том, что такой зонд
с большой вероятностью будет содержать примеры повторных последовательностей
ДНК (глава 9) и, таким образом, может гибридизироваться со многими позициями
хромосомы, а не только в определенной точке, к которой он абсолютно подходит.
Дабы уменьшить такую неспецифическую гибридизацию, перед использованием
зонд смешивают с непомеченной ДНК из изучаемого организма. В качестве такой
ДНК может попросту быть взята полная ядерная ДНК (то есть представляющая пол-
ный геном), но лучше, если будет использована фракция, обогащенная повторными
последовательностями. Идея состоит в том, что непомеченная ДНК прогибридизи-
руется с повторными последовательностями ДНК в зонде и тем самым блокирует
их, с тем чтобы в последующей гибридизации in situ участие принимали исключи-
тельно уникальные последовательности. Поэтому неспецифическая гибридизация
уменьшается или полностью устраняется (рис. 3.34).
FISH в действии
Метод FISH первоначально использовался с метафазными хромосомами (раз-
дел 7.1). Такие хромосомы, приготовленные из ядер, которые претерпевают деление,
сильно сжаты, и каждая хромосома в наборе принимает узнаваемые черты, характери-
зуемые позицией ее центромеры, и картину распределения дисков, которая проявляется
после окрашивания хромосомного препарата (см. рис. 7.5). В случае метафазных хромосом
флюоресцентный сигнал, полученный с помощью FISH, наносится на карту, при этом
позиция на ней определяется путем измерения его расстояния от конца короткого плеча
хромосомы (значение FLpter). Неудобство этого метода заключается в том, что сильная
уплотненность метафазных хромосом означает, что возможна только картография с низ-
кой разрешающей способностью, при этом два маркёра должны находиться по крайней
мере на расстоянии 1 млн п. н. друг от друга, чтобы быть различимыми как отдельные
гибридизационные сигналы. Такая степень разрешения недостаточна для построе-
122 Часть 1. Изучение геномов
Повторная ДНК
...................... и 11 I 1.....
Гибридизирующий зонд
Добавление ДНК,
обогащенной повторной ДНК
Рис. 3.34. Метод блокирования повторных
последовательностей ДНК в гибридизи-
рующем зонде. В этом примере молекула
зонда содержит две повторные последо-
вательности (показаны красным цветом).
Если эти последовательности не блокиро-
ваны, то зонд неспецифично гибридизиру-
ется с любыми копиями этих повторений
в целевой ДНК. Чтобы блокировать по-
вторные последовательности, зонд пред-
варительно гибридизируется с фракцией
ДНК, обогащенной повторной ДНК
Теперь повторные последовательности в зонде блокированы
1 i 2_1 1 1 1 ^3 [д I I J I Т1
I
Гибридизация зонда теперь протекает лишь с участием
уникальных последовательностей
ния полезных хромосомных карт,
и основное назначение FISH в ме-
тафазу заключалось в определении
хромосомы, на которой расположен
новый маркёр, и в обеспечении при-
ближенного представления отно-
сительно его позиции на карте, то
есть в создании черновика для более крупномасштабного картирования при помощи
других методов.
На протяжении ряда лет в число этих «других методов» не входила ни одна форма
FISH, но с 1995 г. был разработан ряд методов FISH более высокого разрешения. В этих
методах более высокое разрешение достигается за счет изменения характера изучаемого
хромосомного препарата. Если метафазные хромосомы слишком сжаты для крупномас-
штабного картирования, то нам следует использовать хромосомы в более вытянутом
виде. Есть два способа сделать это.
• Механически вытянутые хромосомы могут быть получены за счет изменения
метода приготовления препарата, применяемого для выделения хромосом из мета-
фазных ядер. За счет включенной в него операции центрифугирования создаются
сдвигающие усилия, которые могут привести к вытягиванию хромосом вплоть до
20-ти раз от их нормальной длины. Отдельные хромосомы являются по-прежнему
распознаваемыми, и сигналы FISH могут быть нанесены на карту тем же самым
способом, как и при работе с обычными метафазными хромосомами. Разрешение
значительно улучшено и позволяет различать маркёры, которые отстоят друг от
друга на 200-300 тыс.п.н.
• Неметафазные хромосомы могут быть пущены вдело, потому что только во время
метафазы хромосомы сильно сжаты: в других стадиях клеточного цикла хромосомы
пребывают в естественном для них развернутом состоянии. Были сделаны попытки
использовать профазные ядра (см. рис. 3.15), потому что в них хромосомы все еще
достаточно уплотнены для опознавания отдельных хромосом. Однако на практике
такие препараты не дают никакого преимущества над механически вытянутыми
хромосомами. Интерфазные хромосомы более полезны, потому что эта стадия
клеточного цикла (между делениями ядра) протекает как раз в тот момент, когда
хромосомы наиболее развернуты. Возможно получить разрешение до 25 тыс. п. н.,
но при этом теряется морфология хромосомы и в силу этого не остается никаких
Глава 3. Картирование геномов 123
с обоими маркёрами с обоими маркёрами
Рис. 3.35. Множество фрагментов, подходящее для STS-картирования. Фрагменты охватывают полную
длину хромосомы, при этом каждая точка на хромосоме присутствует в среднем в пяти фрагментах. Два
синих маркёра находятся близко друг к другу на хромосомной карте, и высока вероятность того, что они
будут обнаружены на одном и том же фрагменте. Два зеленых маркёра более отдалены друг от друга и,
таким образом, с меньшей вероятностью могут наблюдаться на одном и том же фрагменте
внешних контрольных точек, относительно которых можно было бы наносить на
карту позицию зонда. Поэтому этот метод используется после построения предва-
рительной карты и обычно служит средством определения порядка расположения
ряда маркёров в маленькой области хромосомы.
В интерфазных хромосомах молекулы клеточной ДНК пребывают в наиболее раз-
вернутом состоянии. Поэтому для того чтобы улучшить разрешение FISH до лучшего,
чем 25 тыс. п. н., значения, необходимо отказаться от интактных хромосом и вместо этого
использовать очищенную ДНК. Данный подход, названный волоконным FISH, основан
на применении ДНК, приготовленной методом вытягивания гелем или расчесывания
молекул (см. рис. 3.31), и позволяет различать маркёры, которые отстоят друг от друга
менее чем на 10 тыс. п. н.
3.3.3. Картирование с помощью меченых участков последовательности
Чтобы произвести подробную физическую карту большого генома, нам нужна,
в идеале, такая методика картирования, которая обеспечивала бы высокое разрешение
и вместе с тем была бы производительной и не требовательной с технической точки
зрения. Ни один из двух методов, которые мы рассмотрели до этого, — рестрикционное
картирование и FISH — не отвечает этим требованиям. Рестрикционное картирование
проводится быстро, легко и обеспечивает подробную информацию, но оно не может
быть применено к большим геномам. FISH, напротив, пригоден для работы с большими
геномами, и такие его видоизмененные версии, как волоконный FISH, могут дать данные
с высоким разрешением, но процедура FISH сложна, а сбор данных осуществляется мед-
ленно, причем за один эксперимент позиции на карте определяются для не более чем
трех или четырех маркёров. Если подробным физическим картам и суждено появиться
на свет, то для этого необходим более действенный метод.
124 Часть 1. Изучение геномов
мРНК Поли-А хвост
АААААА
Г- ТТТТТТ
Добавление затравки олиго-дТ
$ ТТТТТГм 11 Л i5 fFi'WPH'i i1 АААААА
3ЛТТТТТ у
: Синтез первой нити при
помощи обратной транскриптазы
5' ___________ ...... 3*
I Рибонуклеаза Н расщепляет
4 большую часть РНК
у П«.и нТЬ н мТТш н н > tttttt у
: Синтез второй нити при
помощи ДНК-полимеразы I
Рис. 3.36. Один из методов приготовления кДНК. Боль-
шинство молекул мРНК эукариотов несет на своем
З'-конце поли-А хвост (раздел 12.2.1). Такой ряд нуклео-
тидов (А) используется в качестве участка затравлива-
ния для первой стадии синтеза кДНК, выполняемой об-
ратной транскриптазой — ДНК-полимеразой, которая
копирует матрицу РНК (раздел 2.1.1). Затравкой служит
короткий синтетический олигонуклеотид ДНК (обычно
20 нуклеотидов в длину), состоящий полностью из ну-
клеотидов Т («затравка олиго-дТ»). По завершении син-
теза первой нити препарат обрабатывают рибонуклеа-
зой Н, которая специфично расщепляет РНК-компонент
гибрида РНК-ДНК. При выбранных условиях фермент
расщепляет не всю РНК, а оставляет короткие сегменты,
которые затравливают реакцию синтеза второй нити
ДНК, катализируемую ДНК-полимеразой I
* Шг1 ггттг i rtri ш,1111 5/
I Завершение синтеза
4 второй нити
5, у В настоящее время самый мощный метод
1 и 11ниш ш 1fun1ИШ ill физического картирования, благодаря которо-
му уже были получены самые подробные кар-
ты больших геномов, — это STS-картирование.
Меченый участок последовательности, или STS, представляет из себя всего-навсего
короткую последовательность ДНК, обычно от 100 до 500 п.н. в длину, которая легко
опознается и лишь единожды встречается в хромосоме или изучаемом геноме. Чтобы
нанести на карту набор STS, необходимо располагать множеством перекрывающихся
фрагментов ДНК из отдельной хромосомы или полного генома. В примере, показанном
на рис. 3.35, коллекция фрагментов была приготовлена из единственной хромосомы,
при этом каждая точка на хромосоме представлена в коллекции в среднем пять раз.
Данные, на основе которых будет построена карта, собирают путем определения того,
какие фрагменты содержат какие STS. Это может быть выполнено посредством гибри-
дизационного анализа, но обычно используют ПЦР, потому что она более быстрая и, как
оказалось, более пригодна для автоматизации. Шансы на то, что два STS присутствуют
в одном и том же фрагменте, будут, конечно, зависеть от того, насколько близко друг
к другу они расположены в геноме. Если расстояние между ними мало, то высока веро-
ятность того, что они всегда будут на одном и том же фрагменте; если же они отстоят
дальше друг от друга, то иногда они будут на одном и том же фрагменте, а иногда — нет
(рис. 3.35). Поэтому такие данные могут быть использованы для вычисления расстояния
между двумя маркёрами, причём способ расчета аналогичен тому, которым определя-
ются расстояния на карте в ходе анализа сцепления генетических признаков (раздел
3.2.3). Вспомним, что при анализе сцепления генетических признаков расстояние на
карте рассчитывается по частоте, с которой между двумя маркёрами происходят пере-
кресты. STS-картирование, по существу, заключается в том же, за исключением лишь
того, что расстояние на карте базируется на частоте, с которой между двумя маркёрами
встречаются разрывы.
При описании STS-картирования, представленном выше, мы упустили из виду не-
которые ключевые вопросы, как то: что именно представляет собой STS? Как получают
коллекцию фрагментов ДНК?
Глава 3. Картирование геномов 125
Любая уникальная последовательность ДНК может быть использована
в роли STS
Чтобы быть квалифицированной как STS, последовательность ДНК должна удовлет-
ворять двум критериям. Согласно первому ее поеледовательность должна быть известна,
с тем чтобы мог быть поставлен анализ (посредством ПЦР) с целью проверки различных
фрагментов ДНК на присутствие или отсутствие в них заданного STS. Согласно второму
STS должен иметь уникальное местоположение на изучаемой хромосоме (или в геноме
в целом, если набор фрагментов ДНК покрывает полный геном). Если последователь-
ность STS наблюдается сразу в нескольких позициях, то данные картирования будут
неоднозначны. Поэтому необходимо удостовериться в том, что в число предполагаемых
STS не входят последовательности, встречающиеся в повторной ДНК.
Эти критерии легко удовлетворить, и STS могут быть получены многими способами;
самые распространенные их источники представлены ярлыками экспрессируемых
последовательностей, SSLP и случайными геномными последовательностями.
• Ярлыки экспрессируемой последовательности (expression sequence tag, EST).
Это короткие последовательности, получаемые анализом клонов кДНК. Комплемен-
тарная ДНК приготовляется путем преобразования препарата мРНК в двунитевую
ДНК (рис. 3.36). Поскольку имеющаяся в клетке мРНК синтезируется из кодирую-
щих белок генов, молекулы кДНК и EST, полученные из них, представляют гены,
которые экспрессировались в клетке, из которой был приготовлен препарат мРНК.
EST рассматриваются как быстрое средство получения доступа к последователь-
ностям важных генов, и они ценны, даже если их последовательности неполные.
EST может быть использован также и в качестве STS, при условии, что он будет
получен из уникального гена, а не из члена семейства генов, в котором все гены
имеют одинаковые или весьма подобные последовательности.
• Полиморфизмы длины простой последовательности (SSLP). В разделе 3.2.2 мы
постигали принципы использования микросателлитов и других SSLP в генетическом
картировании. SSLP могут быть использованы также и в качестве STS при физическом
картировании. SSLP, которые являются полиморфными и уже были нанесены на карту
при помощи анализа сцепления генетических признаков, особенно ценны, поскольку
они обеспечивают прямую связь между генетической и физической картами.
• Случайные геномные последовательности. Их получают секвенированием слу-
чайных частей клонированной геномной ДНК или же просто закачивают последо-
вательности, которые были помещены в базы данных ранее.
Фрагменты ДНК для картирования с помощью STS
Второй насущный компонент для процедуры STS-картирования — коллекция фраг-
ментов ДНК, покрывающих изучаемый объект, будь то хромосома или полный геном.
Такую коллекцию иногда называют реактивом для картирования, и в настоящее время
есть два способа, которыми она может быть собрана: в виде библиотеки клонов и в виде
группы радиационных гибридов. Вначале мы рассмотрим радиационные гибриды.
Радиационный гибрид — клетка грызуна, которая содержит фрагменты хромосом
какого-либо иного организма. На первых порах данная технология была (в 1970-х гг.)
разработана для хромосом человека, когда было обнаружено, что подвергание клеток
человека дозам рентгеновских лучей 3000-8000 рад вызывает беспорядочное разбиение
хромосомы на фрагменты, при этом большие дозы рентгеновского облучения дают более
мелкие фрагменты (рис. 3.37, а). Такая обработка, конечно, смертельна для клетокчелове-
126 Часть 1. Изучение геномов
а) Облучение хромосом
Высокая доза
Низкая доза
Разбитые на фрагменты хромосомы
б) Слияние клеток с целью получения лучевого гибрида
Облученное ядро
клетки человека
Ядро клетки хомяка
Гибридное ядро
Рис. 3.37. Радиационные гибриды, а) Результат облучения
клеток человека: хромосомы разрываются на фрагменты, при
этом более высокие дозы рентгеновского облучения дают
более мелкие фрагменты. 6) Радиационный гибрид получают
путем слияния облученной клетки человека с необработан-
ной клеткой хомяка. Из соображений наглядности показаны
ка, но фрагменты хромосомы мо-
гут быть размножены, если такие
облученные клетки впоследствии
слить с необлученными клетка-
ми хомяка или другого грызуна.
Слияние стимулируется или хими-
чески (полиэтиленгликолем), или
биологически — воздействием ви-
руса Сендай (рис. 3.37, б). Не все
клетки хомяка принимают фраг-
менты хромосомы, так что необ-
ходимо средство идентификации
гибридов. Обычный процесс отбо-
ра заключается в использовании
линии клеток хомяка, которые
неспособны синтезировать или
тимидинкиназу (ТК), или гипок-
сантинфосфорибозилтрансфера-
зу (HPRT), ибо дефицит в любом
из этих двух ферментов является
смертельным для клеток, когда их
выращивают в среде, содержащей
смесь гипоксантина, аминоптери-
на и тимидина (среда HAT).
Итак, после слияния клет-
ки помещают в среду НАТ. Те из
них, что растут, суть гибридные
клетки хомяка, которые приоб-
рели фрагменты ДНК человека
с генами ферментов ТК и HPRT
человека; последние синтезиру-
ются в гибридах и позволяют этим
клеткам расти в селективной сре-
де. В результате такой обработки
выживают гибридные клетки,
которые несут в себе случайную
одборку фрагментов ДНК чело-
только ядра клеток века, встроенных в хромосомы
хомяка. Обычно фрагменты име-
ют размер 5-10 млн п. н., при этом
каждая клетка содержит фрагменты, эквивалентные 15-35 % генома человека. Такую
коллекцию клеток называют группой радиационных гибридов и могут применять
в качестве реактива для STS-картирования, но при условии, что анализ посредством
ПЦР, применяемый для опознавания STS, не будет размножать эквивалентную область
ДНК из генома хомяка.
Может быть построена группа радиационных гибридов второго типа, содержа-
щих ДНК только из одной хромосомы человека; с этой целью облучению подвергают
клеточную линию не человека, а гибридов грызунов второго типа. Цитогенетики раз-
Глава 3. Картирование геномов 127
работали ряд клеточных линий грызунов, в кото-
рых единственная хромосома человека устойчиво
размножается в ядре клетки грызуна. Если кле-
точная линия такого типа (например, клеточная
линия мыши) будет облучена и слита с клетками
хомяка, то гибридные клетки хомяка, получен-
ные после отбора, будут содержать фрагменты
хромосомы или человека, или мыши, или смесь
обоих. Клетки, содержащие ДНК человека, могут
быть идентифицированы путем их зондирования
специфическим для человека повтором, таким как
короткий рассеянный ядерный элемент (SINE) по
имени Л/п (раздел 9.2.1), который имеет число ко-
пий чуть более 1 миллиона (см. табл. 9.3) и, таким
образом, встречается в геноме человека в среднем
единожды на каждые 3 тыс.п.н. Только клетки,
содержащие ДНК человека, будут гибридизиро-
ваться с зондами А1и, что позволяет неинтересные
гибриды мыши не принимать во внимание и на-
правлять STS-картирование на клетки, содержа-
щие фрагменты хромосомы человека.
Картирование генома человека при помощи
радиационных гибридов первоначально выпол-
нялось со специфичными к хромосоме группами
(панелями), а не с полногеномными панелями,
потому что бытовало мнение, что для того, чтобы
нанести на карту единственную хромосому, тре-
буется меньше гибридов, чем для картирования
полного генома. Оказывается, что карта с высо-
ким разрешением для единственной хромосомы
человека требует группы из 100-200 гибридов,
что является наибольшим числом, которое может
быть удобно обработано в программе визуали-
зации результатов ПЦР. Но группы для целого
генома и для единственной хромосомы строятся
по-разному: первая предполагает облучение толь-
ко ДНК человека, а последняя требует облучения
клетки мыши, содержащей большую долю ДНК
мыши и относительно мало ДНК человека. Это
означает, что в группе для единственной хромо-
сомы содержание ДНК человека на гибрид будет
намного ниже, чем в группе для целого генома.
Становится ясно, что подробное картирование
полного генома человека возможно выполнить
с менее чем 100 радиационными гибридами на
весь геном, так что картирование целого генома
не сложнее, чем картирование единственной хро-
мосомы. Как только это было осознано, радиаци-
Смесь хромосом - -
Образец,
содержащий
хромосомы
только одного
типа
Все остальные
хромосомы
Рис. 3.38. Разделение хромосом поточ-
ной цитометрией. Смесь флюоресцентно
окрашенных хромосом проходит сквозь
маленькое отверстие, так что каждая вы-
ходящая из него капля содержит только
одну хромосому. Датчик флюоресценции
идентифицирует сигнал от капель, содер-
жащих нужную хромосому, и сообщает
электрический заряд этим каплям. Когда
капли достигают электрических пластин,
заряженные капли отклоняются в отдель-
ную мензурку. Все остальные капли падают
мимо отклоняющих пластин прямо вниз
и собираются в мензурке для отходов
128 Часть 1. Изучение геномов
АВ CD EF G Н
Применение библиотеки
I клонов в качестве реактива
*для картирования
Секвенирование взаимно
X перекрывающихся клонов
АВ С DE F G Н I
Карта STS
!—TAGGCATCCATGCCCA—।
Последовательность ДНК
Рис. 3.39. Ценность библиотек клонов в проектах расшифровки геномов. Маленькая библиотека клонов,
показанная в этом примере, содержит достаточную информацию для построения карты STS и может
быть использована также как источник ДНК, которую предстоит секвенировать
онные гибриды для полного генома стали основным компонентом стадии картирования
в проекте «Геном человека» (раздел 4.3). Библиотеки полных геномов использовались
также и для STS-картирования геномов других млекопитающих, а также полосатого
данио и курицы.
Библиотека клонов также может быть использована в качестве реактива
картирования для анализа STS
Необходимой предпосылкой стадии секвенирования проекта расшифровки генома
является разрушение генома или выделенных хромосом на фрагменты и клонирование
каждого из них в векторе высокой емкости, который способен принимать большие фраг-
менты ДНК (раздел 2.2.1). В результате этого получают библиотеку клонов — коллекцию
фрагментов ДНК, которые, в случае проекта расшифровки генома, имеют средний размер
нескольких сот тыс.п.н. Наряду с поддержкой работ по секвенированию, библиотеки
клонов могут быть использованы также и в STS-анализе как реактив картирования.
Как и в случае групп радиационных гибридов, библиотека клонов может быть при-
готовлена из геномной ДНК и, таким образом, представлять полный геном, или может
быть создана библиотека, специфичная к хромосоме, если исходная ДНК получена только
из хромосомы одного типа. Последнее возможно, потому что отдельные хромосомы
могут быть разделены посредством поточной цитометрии. Дабы выполнить такое
разделение, делящиеся клетки (с уплотненными хромосомами) аккуратно разрушают,
с тем чтобы была получена смесь неповрежденных хромосом. Затем хромосомы окра-
шивают флюоресцентным красителем. Количество красителя, связываемого хромо-
сомой, зависит от ее размера, так что более крупные хромосомы связывают большие
количества красителя и флюоресцируют более ярко, чем таковые меньшего размера.
Далее хромосомный препарат растворяют и пропускают через маленькое отверстие,
производя поток капелек, каждая из которых содержит по одной хромосоме. Капельки
проходят через датчик, который измеряет количество флюоресценции и таким образом
Глава 3. Картирование геномов 129
распознает, какие капельки содержат определенную искомую хромосому. Электрический
заряд сообщается этим каплям и никаким другим (рис. 3.38), что позволяет отклонять
и отделять капельки, содержащие желательную хромосому, от всех остальных. Что если
две различные хромосомы имеют подобные размеры, как это имеет место с хромосомами
21 и 22 человека? Обычно они могут быть разделены, если вместо неспецифического
красителя использовать красители, имеющие предпочтение к областям, обогащенным
АТ или GC. Примеры таких красителей — соответственно Hoechst 33258 и хромомицин А3.
В двух хромосомах одного размера содержание GC редко бывает одинаковым и, таким
образом, их можно различать по количествам АТ- или GC-специфичных красителей,
которые они связывают.
По сравнению с группами радиационных гибридов, библиотеки клонов имеют одно
важное преимущество для картирования STS. Общеизвестен тот факт, что отдельные
клоны могут последовательно представлять очередные отрезки ДНК, которая фактиче-
ски секвенируется. Данные, следующие из анализа STS, на основании которых строится
физическая карта, могут быть с неменьшим успехом использованы для определения того,
какие клоны содержат перекрывающиеся фрагменты ДНК, что позволяет построить
сборку контигов из клонов (рис. 3.39; прочие методы сборки контигов из клонов опи-
саны в разделе 4.2.2). Такая сборка из перекрывающихся клонов может служить базовым
материалом для репарации длинной, непрерывной последовательности ДНК, и позже,
опираясь на данные анализа STS, можно закрепить эту последовательность в точной
позиции на физической карте. Если в число STS были включены также и SSLP, которые
были нанесены на карту при помощи анализа сцепления генетических признаков, то
последовательность ДНК, физическая карта и генетическая карта — все эти сведения
могут быть объединены воедино.
Заключение
Карты геномов служат опорной схемой для проектов секвенирования, потому что
они указывают позиции генов и других опознаваемых особенностей генома и, следова-
тельно, позволяют проверять точность собранной последовател ьности ДНК. Генетиче-
ские карты строят по результатам экспериментов по скрещиванию и анализа родослов-
ной, а физические карты — посредством прямого наблюдения молекул ДНК. В первых
генетических картах маркёрами выступали гены, аллели которых можно было легко
отличать, потому что они вызывали легко опознаваемые фенотипы, такие как разный
цвет глаз, или же гены, аллели которых можно было отличать биохимическими тестами.
Сегодня ДНК-маркёры также интенсивно используются, к ним относятся: полиморфизмы
длины фрагмента рестрикции (RFLP), полиморфизмы длины простой последователь-
ности (SSLP) и полиморфизмы отдельных нуклеотидов (SNP). Все они могут быть быстро
и легко типизированы посредством ПЦР. Относительные позиции генов и ДНК-маркёров
на хромосомах определяются при помощи анализа сцепления генетических признаков.
Этот метод базируется на оригинальных генетических открытиях, сделанных Менделем,
и был впервые разработан (для использования с плодовыми мушками) в самом начале
двадцатого столетия. Анализ сцепления генетических признаков позволяет определять
частоту рекомбинации между парой маркёров и обеспечивает данные, необходимые для
установления относительных позиций маркёров на генетической карте. Для многих
организмов анализ сцепления генетических признаков проводится путем прослежи-
вания наследования маркёров в запланированных экспериментах по скрещиванию, но
это невозможно с людьми. Вместо этого генетическое картирование генома человека
130 Часть 1. Изучение геномов
опирается на данные наблюдений за наследованием маркёров в многолюдных семей-
ствах, то есть на сведения, почерпнутые из анализа родословной. Генетические карты
имеют относительно низкое разрешение и, как правило, являются неточными, а по-
тому, если такую карту предстоит использовать в проекте секвенирования генома, ее
необходимо уточнить физическим картированием. В маленькой молекуле ДНК позиции
участков рестрикции могут быть определены путем рестрикционного картирования,
но с хромосомами эукариотов этот способ имеет лишь ограниченную ценность. Больше
пользы приносит флюоресцентная гибридизация in situ (FISH), в которой препарат из
интактных хромосом, возможно, таких, которые были вытянуты посредством меха-
нического вытягивания, зондируется маркёром, меченным флюоресцентной меткой.
Позиция, в которой происходит гибридизация, определяется путем наблюдения пре-
парата в конфокальный микроскоп. Наиболее подробные физические карты получают
при помощи картирования содержания меченых участков последовательности (STS),
при котором используется реактив для картирования — коллекция перекрывающихся
фрагментов ДНК, которые покрывают полную хромосому или геном. Позиция маркёра
на карте определяется путем определения того, какие именно фрагмены из коллекции
содержат копии маркёра. Материалом для картирования может быть библиотека клонов
или группа радиационных гибридов.
Глава 3. Картирование геномов 131
Задания для закрепления материала
* Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
3.1*. Главная проблема с вычислитель-
ной сборкой последовательностей
ДНК сложных геномов эукариотов
заключается в присутствии:
а. Многочисленных хромосом.
Ь. Митохондриальной ДНК.
с. Интронов в пределах генома.
d. Повторов.
3.2. В первых генетических картах мар-
кёрами служили гены, потому что:
а. Местоположения генов на хро-
мосомах можно было наблюдать
при помощи окрашивания ДНК
красителем.
Ь. Определяемые генами фенотипы
можно визуально опознавать и из-
учать их схемы наследования.
с. Отдельные гены, определяющие
легко опознаваемые фенотипиче-
ские признаки, могли быть легко
клонированы.
d. Полиморфизмы отдельных ну-
клеотидов использовались для
опознавания точечных мутаций,
которые вызывали явно замет-
ные фенотипические различия.
3.3 *. Какая из следующих причин НЕ
обусловила общепринятое приме-
нение биохимических фенотипов
для создания генетических карт
человека?
а. Люди не имеют никаких визуаль-
ных характеристик, пригодных
для генетического картирова-
ния.
Ь. Есть биохимические фенотипы,
которые легко визуализируются
посредством определения групп
крови.
с. Некоторые легко характеризуе-
мые биохимические фенотипы
определяются генами с очень
большим числом аллелей.
d. Неэтично проводить запланиро-
ванные эксперименты по скрещи-
ванию над людьми.
3.4 . Геномы эукариотов картируют с ис-
пользованием ДНК-маркёров в до-
полнение к генам, потому что:
а. ДНК-маркёры не требуют на-
личия двух и более аллелей для
картирования.
Ь. Генетические карты могут не по-
крывать большие области гено-
ма.
с. Большинство генов обладает мно-
жественными аллелями, которые
могут быть легко картированы.
d. ДНК-маркёры менее изменчивы,
чем генетические маркёры.
3.5 *. Как правило, в качестве ДНК-маркё-
ров чаще используются микросател-
литы, а не минисателлиты, потому
что:
а. Минисателлиты присутствуют
в слишком многих местоположе-
ниях в пределах генома.
Ь. Ферменты рестрикции могут
быть использованы для типиза-
ции микросателлитов, но никак
не минисателлитов.
с. В геномах эукариотов находится
очень немного микросателлитов,
так что их легко опознавать и ана-
лизировать.
d. Микросателлиты присутствуют
во всех областях генома эукарио-
тов и легко размножаются с по-
мощью ПЦР.
3.6 . Какие из следующих генетических
маркёров наиболее многочисленны
в геноме человека?
132 Часть 1. Изучение геномов
a. RFLP.
b. Минисателлиты.
с. Микросателлиты.
d. Полиморфизмы отдельных ну-
клеотидов.
3.7 *. Принцип анализа сцепления генети-
ческих признаков — это:
а. Тот факт, что разные аллели одно-
го и того же гена всегда располо-
жены в одной и той же позиции
хромосом.
Ь. Открытие того факта, что за неко-
торые признаки (такие как цвет
глаз у мух) отвечают целые груп-
пы генов.
с. Наблюдение, что некоторые гены,
если они расположены на одной
и той же хромосоме, наследуются
совместно.
d. Наблюдение, что темно окраши-
вающиеся области хромосом не
содержат никаких генов.
3.8 . Различие между митозом и мейозом
заключается в том, что митоз харак-
теризуется следующим:
а. Производством двух диплоидных
клеток, которые являются гене-
тически идентичными родитель-
ской клетке.
Ь. Обменом ДНК (кроссинговером)
между гомологичными хромосо-
мами.
с. Производством двух диплоидных
клеток, которые являются гене-
тически отличными от родитель-
ской клетки.
d. Производством четырех гапло-
идных клеток, которые являются
генетически отличными от роди-
тельской клетки.
3.9 *. Какое из следующих утверждений
правильно описывает понятие ча-
стоты рекомбинации между двумя
генами?
а. Чем ближе друг к другу два гена
расположены на хромосоме, тем
выше будет частота рекомбина-
ции между ними.
Ь. Чем дальше друг от друга два гена
расположены на хромосоме, тем
выше будет частота рекомбина-
ции между ними.
с. Если два гена расположены на
одной и той же хромосоме, то меж-
ду ними не могут произойти ни-
какие события рекомбинации.
d. Если два гена расположены на
разных хромосомах, то частота
рекомбинации между ними будет
высокой.
3.10 . При анализе родословной человека
с целью определения того, насколь-
ко тесно сцеплены два гена, наилуч-
шим будет:
а. Заключить, что наиболее распро-
страненные генотипы в потомстве
суть родительские генотипы.
Ь. Заключить, что самые распро-
страненные генотипы в потом-
стве — рекомбинанты.
с. Провести анализирующее скре-
щивание, с тем чтобы определить
сцепление генов.
d. Определить генотипы бабушки
и дедушки.
3.11 *. Какой из следующих факторов НЕ
ограничивает точность генетиче-
ских карт людей и других сложных
ядерных организмов?
а. От многих ядерных организмов
невозможно получить достаточно
потомства.
Ь. Очаги рекомбинации могут ме-
шать генетическому картирова-
нию.
с. В генетическом картировании ис-
пользуются только гены, а числа
имеющихся генов недостаточно
для картирования полных гено-
мов.
d. Гены или маркёры, которые в ге-
номе отстоят друг от друга на де-
сятки тысяч п. н., на генетической
карте могут появиться в одной
и той же позиции.
Глава 3. Картирование геномов 133
3.12 . Для флюоресцентной гибридизации
in situ первоначально использова-
ли метафазные хромосомы, но их
возможности оказались несколько
ограниченными в связи с тем, что:
а. Многие области хромосомы сжа-
ты и не могут гибридизироваться
с зондами.
Ь. Зонды предпочтительно скрещи-
ваются с повторными последова-
тельностями, присутствующими
на множественных хромосомах.
с. В сжатом состоянии хромосомы
неустойчивы, а когда они развер-
нуты, — сигнал рассеивается.
d. Поскольку хромосомы уплотне-
ны, картирование возможно лишь
с низкой разрешающей способно-
стью.
3.13 *. Интерфазные хромосомы полезны
для крупномасштабного картиро-
вания при помощи флюоресцент-
ной гибридизации in situ, потому
что они:
а. Пребывают в наименее сжатом
состоянии.
Ь. Легко отличаются друг от друга
своими структурами.
с. Имеют транскрипционно актив-
ные области хроматина, которые
необходимы для этой технологии.
d. Позволяют осуществлять физиче-
ское картирование геномов с раз-
решением вплоть до 1 тыс.п.н.
3.14 . Какими из следующих свойств об-
ладают меченые участки последо-
вательности (STS)?
а. Они присутствуют только еди-
ножды в пределах генома и об-
ладают участком RFLP.
Вопросы открытого типа
3.1*. Почему для секвенирования гено-
мов необходимы карты? Если бы
карта генома была недосягаема
для исследователя, то какие глав-
h. Они присутствуют только еди-
ножды в пределах генома, и их
последовательность известна.
с. Их последовательность известна,
и они должны содержать повтор-
ные последовательности ДНК.
d. Они должны содержать последо-
вательность гена, и не допускают
присутствия в них никаких по-
вторных последовательностей
ДНК.
3.15 *. Какая из следующих последователь-
ностей не может быть использована
в качестве меченого участка после-
довательности?
а. Ярлыки экспрессируемых после-
довательностей.
Ь. Случайные геномные последова-
тельности.
с. Полиморфизмы длины простых
последовательностей.
d. Полиморфизмы длины фрагмен-
тов рестрикции.
3.16 . Группы радиационных гибридов
обеспечивают хороший способ фи-
зического картирования, потому
что:
а. В любой заданной гибридной
клетке присутствует только
часть генома человека.
Ь. В клетках-хозяевах хомяка от-
сутствуют последовательности,
гомологичные генетическим мар-
кёрам человека.
с. Клетки-хозяева хомяка устойчи-
вы к облучению.
d. Известна полная физическая кар-
та генома хомяка.
ные затруднения в определении
последовательности генома он бы
испытывал?
134 Часть 1. Изучение геномов
3.2 . Четко объясните различия между
генетической и физической картами
генома.
3.3 *. Каким образом ПЦР сделала анализ
RFLP намного более быстрым и лег-
ко выполнимым? Что было необхо-
димо для нанесения RFLP на карту
до начала использования ПЦР с этой
целью?
3.4 . Каким образом ферменты рестрик-
ции используются для генетиче-
ского и физического картирования
генома?
3.5 *. Почему сцепление между генами
представляет собой столь важный
фактор для генетического карти-
рования? Обсудите, как генетиче-
ские маркёры могут быть сцепле-
ны, чтобы по этим данным можно
было строить карты отдельных хро-
мосом.
3.6 . Сформулируйте два генетических
закона Менделя. Какой компонент
генетического картирования не
охвачен законами Менделя?
3.7 *. Почему для анализирующих скре-
щиваний в экспериментах анализа
сцепления генетических признаков
используется двойная гомозигота?
Почему предпочтительно, чтобы ал-
лели гомозиготы были рецессивны
по проверяемым признакам?
3.8 . Рестрикционное картирование
молекул ДНК часто ограничивает-
ся молекулами размером не более
50 тыс. п. н. Почему для этой тех-
нологии именно данная величина
является пределом, и как такое огра-
ничение может быть сглажено, что-
бы можно было исследовать более
крупные молекулы ДНК?
3.9 *. Методы генетического картирова-
ния требуют наличия по крайней
мере двух аллелей для заданного
маркёра, тогда как методы физиче-
ского картирования не полагают-
ся на присутствие аллелей, чтобы
картировать геномы. Обсудите, как
технология флюоресцентной гибри-
дизации in situ может быть исполь-
зована для картирования позиций
в геноме, даже если во всякой задан-
ной позиции не наблюдается ника-
кой генетической изменчивости.
3.10 . Почему для картирования геномов
радиационные гибриды целого ге-
нома предпочтительнее гибридов
одинарной хромосомы?
3.11 *. * Как ученый может собрать библио-
теку клонов ДНК, имея в своем рас-
поряжении одну хромосому?
Изыскательские задачи
3.1 *. Каковыми в идеале должны быть
особенности ДНК-маркёра, исполь-
зуемого для построения генетиче-
ской карты? До какой степени RFLP,
SSLP или SNP могут рассматриваться
как «идеальные» ДНК-маркёры?
3.2 . Изучите и оцените возможности
и сферы применения технологии
чипов ДНК в биологических иссле-
дованиях.
3.3 *. Какие особенности были бы жела-
тел ьны для организма, который сле-
дует использовать для всесторонних
исследований наследственности?
3.4 . Какие проблемы могли бы возник-
нуть, если бы была сделана попытка
секвенировать геном до построения
генетической или физической кар-
ты?
3.5 *. Какая карта полезнее — генетиче-
ская или физическая?
Глава 3. Картирование геномов 135
Тесты по рисункам
3.1*. Каким образом эксперимент с кра- 3.2.
сителем-тушителем позволяет
определить, гибридизировался ли
олигонуклеотид к молекуле ДНК, со-
держащей полиморфизм отдельного
нуклеотида, или нет?
б) Детектирование гибридизации
с помощью гашения флюоресцентного красителя
Все нижеприведенные гены рас-
положены на одной хромосоме. Для
перечисленных ниже частот реком-
бинации постройте карту, показы-
вающую относительные положения
этих генов на хромосоме.
Гены
m
v Ярко-красные глаза
w Белые глаза
У Желтое тело
Частоты рекомбинации
Миниатюрные крылья
Флюоресцентный
сигнал
Между m и v
Между m и у
Между v и w
Между w и у
= 3,0 %
= 33,7 %
= 29,4 %
= 1,3%
3.3*. * Метод электрофореза, показанный
на этом рисунке, применяется для
разделения относительно больших
молекул ДНК (более 50 тыс. п. н.). На
чем основана данная технология?
3.4. Эта пара хромосом была гибридизирована с клони-
рованными молекулами, содержащими различные
флюоресцентные метки. К какому типу относится этот
метод и пример какого вида картирования он пред-
ставляет: генетического или физического?
136 Часть 1. Изучение геномов
Дополнительная литература
Книги по истории генетики
Orel,V. (1995) Gregor Mendel: The First Geneticist. Oxford University Press, Oxford.
Shine,!, and Wrobel,S. (1976) Thomas Hunt Morgan: Pioneer of Genetics. University
Press of Kentucky, Lexington, Kentucky.
Sturtevant, A. H. (1965) A History of Genetics. Harper and Row, New York. Describes the
early gene mapping work carried out by Morgan and his colleagues.
Генетические и ДНК-маркёры
Wang,D.G., Fan,J.-B., Siao,C.-J., et al. (1998) Large-scale identification, mapping, and
genotyping of single-nucleotide polymorphisms in the human genome. Science 280: 1077-
1082.
Yamamoto, F, Clausen, H., White,T., MarkenJ. and Hakamori,S. (1990) Molecular
genetic basis of the histo-blood group ABO system. Nature 345: 229-233.
Анализ сцепления генетических признаков
Morton, N. E. (1955) Sequential tests for the detection of linkage. Am. J. Hum. Genet. 7:
277-318. The use of lod scores in human pedigree analysis.
Strachan, T. and Read, A. P. (2004) Human Molecular Genetics, 3rd Ed. Garland, London.
Chapter 13 covers human genetic mapping.
Sturtevant, A. H. (1913) The linear arrangement of six sex-linked factors in Drosophila
as shown by mode of association./. Exp. Zool. 14: 39-45. Construction of the first linkage map
for the fruit fly.
Рестрикционное картирование
Hosoda, F., Arai, Y., Kitamura, E., etal. (1997) A complete Not\ restriction map covering
the entire long arm of human chromosome 11. Genes Cells 2: 345-357.
Ichikawa, H., Hosoda, R, Arai,Y., Shimizu, K., Ohira,M. and Ohki,M. (1993) Not\
restriction map of the entire long arm of human chromosome 21. Nat. Genet. 4: 361-366.
JingJ.P., Lai,Z.W., Aston,C., etal. (1999) Optical mapping of Plasmodium falciparum
chromosome 2. Genome Res. 9:175-181.
Lin, J., Qi, R., Aston, C., et al. (1999) Whole-genome shotgun optical mapping of Deinococcus
radiodurans. Science 285:1558-1562.
Michalet,X., Ekong, R., Fougerousse, F., et al. (1997) Dynamic molecular combing:
stretching the whole human genome for high-resolution studies. Science 277:1518-1523.
Zhou, S. G., Kvikstad, E., Kile, A., etal. (2003) Whole-genome shotgun optical mapping of
Rhodobactersphaeroides strain 2.4.1 and its use for whole-genome shotgun sequence assembly.
Genome Res. 13: 2142-2151.
FISH
Heiskanen,M., Peltonen,L. and Palotie,A. (1996) Visual mapping by high resolution
FISH. Trends Genet 12: 379-382.
Lichter, P. (1997) Multicolor FISHing: what's the catch? Trends Genet. 13: 475-479.
Romanov, M. N., Daniels, L. M., Dodgson, J. B. and Delany, M. E. (2005) Integration of
the cytogenetic and physical maps of chicken chromosome 17. Chromosome Res. 13: 215-222.
Describes an application of FISH carried out with ВАС probes.
Глава 3. Картирование геномов 137
Tsuchiya,D. and Taga,M. (2001) Application of fibre-FISH (fluorescence in situ
hybridization) to filamentous fungi: visualization of the rRNA gene cluster of the ascomycete
Cochliobolus heterostrophus. Microbiology 147:1183-1187.
Zelenin, A. V. (2004) Fluorescence in situ hybridization in studying the human genome.
Mol. Biol. 38:14-23.
Радиационные гибриды
Hudson, T.)., Church, D.M., Greenaway, S., et al. (2001) A radiation hybrid map of the
mouse genome. Nat. Genet. 29: 201-205.
Itoh, T., Watanabe, T., Ihara, N., Mariani, P., Beattie, C. W., Sugimoto, Y. and Takasuga, A.
(2005) A comprehensive radiation hybrid map of the bovine genome comprising 5593 loci.
Genomics 85: 413-424.
McCarthy,L. (1996) Whole genome radiation hybrid mapping. Trends Genet. 12: 491-
493.
Walter,M.A., Spillett,D.)., Thomas,P., Weissenbach,), and Goodfellow,P.N. (1994)
A method for constructing radiation hybrid maps of whole genomes. Nat. Genet. 7: 22-28.
Глава 4
Секвенирование геномов
По прочтении главы 4 вы сможете:
• Приводить подробные описания методов секвенирования ДНК с обрывом цепи
и в тепловом цикле.
• Вкратце описывать методы секвенирования путем химической деградации
и пиросеквенирования и обозначать возможности их применения.
• Показывать сильные и слабые стороны методов секвенирования геномов, как
то: дробовика, дробовика для полных геномов и сборки контигов из клонов.
• Описывать, каким образом маленький геном бактерии может быть секвениро-
ван методом дробовика (на примере проекта расшифровки генома Haemophilus
influenzae).
• Указывать различные способы построения сборки контигов из клонов.
• Объяснять сущность метода дробовика для секвенирования полных геномов,
ставя акцент на мерах, предпринимаемых с целью обеспечения точности по-
лучаемой последовательности.
• Давать сводку развития проектов расшифровки генома человека вплоть до пу-
бликации расшифрованных последовательностей хромосом в 2004-2005 гг.
• Обсуждать этические, юридические и социальные вопросы, поднятые в ходе
развития проектов расшифровки генома человека.
Конечная цель проекта секвенирования генома — полностью расшифрованная по-
следовательность ДНК изучаемого организма, в идеале объединенная с генетическими
и (или) физическими картами генома, с тем чтобы в пределах последовательности ДНК
могли быть найдены гены и другие интересные особенности. Эта глава описывает ме-
тоды и стратегии исследования, которые используются отцами проектов расшифровки
генома на стадии секвенирования, когда они ставят перед собой именно эту оконча-
тельную цель. Центральное место в этом контексте, несомненно, занимают методы
секвенирования ДНК, и мы начнем эту главу с подробнейшего изучения методологии
секвенирования. От этой методологии, однако, будет мало толку, если получаемые на
выходе отдельных экспериментов секвенирования короткие последовательности мы
не сможем объединить между собой в правильном порядке и таким образом восстано-
вить полные последовательности хромосом, входящих в состав генома. Поэтому вторая
часть этой главы описывает стратегии, применяемые с целью обеспечения правильной
сборки основной последовательности хромосомы.
Глава 4. Секвенирование геномов 139
4.1. Методология секвенирования ДНК
Существует несколько методов секвенирования ДНК, но несравненно более попу-
лярный среди них — метод секвенирования с обрывом цепи, впервые изобретенный
Фредом Сенгером и его группой в середине 1970-х гг. Секвенирование с обрывом цепи
получило превосходство по нескольким причинам, не в последнюю очередь благодаря
относительной простоте, с которой этот метод может быть автоматизирован. Как мы
увидим далее в этой главе, проект расшифровки генома вовлекает огромное число от-
дельных экспериментов секвенирования и на выполнение их всех вручную потребуются
многие годы. Поэтому если проект необходимо осуществить за разумный промежуток
времени, обязательно нужны автоматизированные методы секвенирования.
4.1.1. Секвенирование ДНК с обрывом цепи
Секвенирование ДНК с обрывом цепи основано на том принципе, что молекулы
однонитевой ДНК, которые отличаются друг от друга по длине только на один нуклеотид,
могут быть разделены между собой посредством электрофореза в полиакриламидном
геле (техническое примечание 4.1). Это означает, что возможно разрешать семейство
молекул, представляющее весь диапазон длин от 10 до 1500 нуклеотидов, в виде ряда
полос в плоском или капиллярном геле (рис. 4.1).
Сущность секвенирования с обрывом цепи
Исходным материалом для эксперимента секвенирования с обрывом цепи служит
препарат из идентичных однонитевых молекул ДНК. Первый шаг — отжечь короткий
олигонуклеотид к одной и той же позиции на каждой из этих молекул, причем этот
олигонуклеотид после этого выступает затравкой для синтеза новой нити ДНК, кото-
рая будет комплементарна матрице (рис. 4.2, а). Реакция синтеза нити, катализируемая
ферментом ДНК-полимеразой (см. ниже) и требующая в качестве субстратов четырех
дезоксирибонуклеотидтрифосфатов (дНТФ — дАТФ, дЦТФ, дГТФ и дТТФ), обычно про-
должается до той стадии, пока не будут полимеризованы несколько тысяч нуклеотидов.
В эксперименте секвенирования с обрывом цепи этого не происходит, потому что, наряду
Рис. 4.1. Электрофорез в полиакриламидном геле
позволяет разрешать молекулы однонитевой ДНК,
которые отличаются по длине только на один ну-
клеотид. На иллюстрации представлена картина
полос, полученная после разделения молекул
однонитевой ДНК при помощи электрофореза
в денатурирующем плоском полиакриламидном
геле. Молекулы были помечены радиоактивной
меткой, и полосы были визуализированы посред-
ством авторадиографии. В полиакриламидном
геле разбег между отдельными молекулами ДНК
по мере их миграции к положительному электроду
увеличивается. Поэтому полосы, наблюдаемые на
авторадиограмме, находятся все дальше друг от
друга в сторону основания лесенки. На практике,
если электрофорез продолжается достаточно долго,
удается разделять молекулы приблизительно до
1500 нуклеотидов в длину (будь это плоский гель
или капиллярная система)
50 нуклеотидов
10 нуклеотидов
140 Часть 1. Изучение геномов
Техническое примечание 4.1: электрофорез в полиакриламидном
геле
Разделение молекул ДНК, отличающихся по длине всего лишь на один
нуклеотид
Электрофорез в полиакриламидном геле используется для исследования семейств
молекул ДНК с оборванной цепью, полученных в ходе эксперимента секвенирования.
Электрофорез в агарозном геле (техническое примечание 2.2) не может быть использо-
ван для этой цели, потому что он не обладает разрешающей способностью, необходимой
для разделения однонитевых молекул ДНК, отличающихся по длине только на один
нуклеотид. Полиакриламидные гели имеют меньший размер пор, чем агарозные гели,
и позволяют точно разделять молекулы длины от 10 до 1 500 п.н. Наряду с секвени-
рованием ДНК, полиакриламидные гели используются также и для других целей, где
требуется разделение ДНК в крупном масштабе, — например, при изучении размно-
жения продуктов экспериментов ПЦР, направленных на локусы микросателлитов, где
продукты различных аллелей могут отличаться по размеру только двумя или тремя
парами оснований (см. рис. 3.6). Полиакриламидные гели могут быть приготовлены
в виде листов между двумя стеклянными пластинами, удерживаемыми на определен-
ном расстоянии распорками, или же в виде длинных, тонких колонок, подходящих для
капиллярного электрофореза (рис. Т4.1).
ПЛИТКА ГЕЛЯ
Передняя
пластина
КАПИЛЛЯРНЫЙ ГЕЛЬ
Гель, 0,1 мм
в диаметре
Капилляр, 50-80 см
в длину
Рис. Т4.1. Две конфигурации полиакриламидного геля для электрофореза
Полиакриламидный гель состоит из линейных цепей мономеров акриламида
(CH2=CH-CO-NH2), поперечно связанных единицами Ы,Н'-метиленбисакриламида
(CH2=CH-CO-NH-CH2-NH-CO-CH=CH2), последние обычно называют «бис». Раз-
мер пор геля определяется как общей концентрацией мономеров (акриламид +
бис), так и отношением акриламида к бису. В плитках геля толщины 1 мм, упо-
требляемых для секвенирования ДНК, обычно используется 6%-й гель с отно-
шением акриламида к бису 19:1, потому что это позволяет получать разрешение
однонитевых молекул ДНК в интервале длин от 100 до 750 нуклеотидов. Поэтому
на одном листе геля могут быть прочитаны приблизительно 650 нуклеотидов по-
Глава 4. Секвенирование геномов 141
следовательности. Концентрация геля может быть увеличена до 8 %, с тем чтобы
считывать последовательность ближе к затравке (разрешая молекулы 50-400
нуклеотидов в длину), или уменьшена до 4%, чтобы считывать более отдаленную
последовательность (500-1500 нуклеотидов от затравки). Полимеризация раствора
акриламид:бис инициируется персульфатом аммония и катализируется ТЕМЭДом
(Ы,М,М',М’-тетраметилэтилендиамином). Гели для секвенирования содержат также
мочевину, которая является денатурирующим агентом, который препятствует
формированию внутримолекулярных пар оснований в молекулах с оборванной
цепью. Это важно, потому что изменение конформации, следующее из спаривания
оснований, изменяет скорость миграции однонитевой молекулы, так что теряется
строгая зависимость между длиной молекулы и позицией ее полосы, являющаяся
определяющим фактором для считывания последовательности ДНК.
с четырьмя дезоксинуклеотидами, к реакции в небольших количествах добавляются
четыре дидезоксинуклеотидтрифосфата (ддНТФ — ддАТФ, ддЦТФ, ддГТФ и ддТТФ).
Каждый из этих дидезоксинуклеотидов бывает помечен отличной от других флюорес-
центной меткой.
а) Запуск синтеза нити б) Дидезоксинуклеотид
О-
ОСНОВАНИЕ
Затравка
5' з' Матричная ДНК °' °“
i 1.41 и», "0-р—о—р—о
з' т т т 5' R R
о о
8 й
н н
*
* Позиция, в которой группа -ОН того
или иного дНТФ заменена на -Н
е) Синтез нити обрывается
при включении в нее ддНТФ
1111111'11111ТТТ?ГГГ?х
т т т
ДДА
। ДДА
1 ДДА
Семейство «А»
Рис. 4.2. Секвенирование ДНК с обрывом цепи, а) Секвенирование с обрывом цепи состоит в синтезе
новых нитей ДНК, которые являются комплементарными однонитевой матрице, б) Синтез нити не
продолжается неопределенно долго, потому что реакционная смесь содержит маленькие количества
каждого из четырех дидезоксинуклеотидов, которые блокируют дальнейшую элонгацию, потому что
в них к З'-углероду присоединен водородный атом вместо гидроксильной группы, в) Включение в цепь
ддАТФ дает цепи, которые оборваны напротив нуклеотидов Т в матрице. Это производит семейство «А»
оборванных молекул. Включение других дидезоксинуклеотидов производит семейства «С», «G» и «Т»
142 Часть 1. Изучение геномов
ддА • ддЦ • ддНТФы - каждый с отличной от
ддТ ® дцГ • других флюоресцентной меткой
Реакции секвенирования,
* фракционирование продуктов
1- - - -пддте
< -...ДДА<
------гв
....ддГ • I L Система формирования
_____ддЦф [ изображения
““ДДЦ® Датчик
=» ддГ •
Флюоресцентные полосы
проходят мимо датчика
б)
Рис. 4.3. Чтение последовательности,
полученной в результате эксперимента
с обрывом цепи, а) Каждый дидезоксину-
клеотид помечен своим флюорофором.
В ходе электрофореза меченые молекулы
движутся мимо датчика флюоресценции,
который опознает, какой дидезоксину-
клеотид присутствует в каждой полосе.
Далее информация передается в систему
формирования изображения, б) Распе-
чатка результатов секвенирования ДНК.
Последовательность представлена рядом
пиков, по одному для каждой нуклеотид-
ной позиции. В этом примере зеленый пик
соответствует «А», синий — «С», коричне-
вый — «G» и красный — «Т»
Фермент полимераза не делает различий между дезокси- и дидезоксинуклеотида-
ми, и, будучи включен в цепь, дидезоксинуклеотид блокирует дальнейшую элонгацию
нити, потому что у него отсутствует З'-гидроксильная группа, необходимая для обра-
зования связи со следующим нуклеотидом (рис. 4.2, б). Поскольку в реакционной смеси
присутствуют также и нормальные дезоксинуклеотиды, и притом в больших количе-
ствах, чем дидезоксинуклеотиды, синтез нити не всегда заканчивается поблизости от
затравки: фактически несколько сотен нуклеотидов могут полимеризоваться прежде,
чем дидезоксинуклеотид будет наконец включен в цепь. В результате образуется набор
новых молекул различной длины, и каждая из них заканчивается дидезоксинуклеотидом,
Встройка ДНК
Рекомбинантный вектор
на фаге М13 (двунитевая ДНК)
Трансфекция в Е. coli
Белковая
оболочка
Сердцевина
из ДНК
Высвобождение
новых фагов
Рекомбинантный
фаг М13
Однонитевая ДНК
Рис. 4.4. Получение однонитевой ДНК путем клонирова-
ния в векторе на бактериофаге М13. Векторы на фаге М13
могут быть получены в двух формах: двунитевой реплика-
тивной молекулы и однонитевой версии, встречающейся
в частицах бактериофага. Репликативной формой можно
манипулировать таким же образом, что и плазмидным
клонирующим вектором (раздел 2.2.1), притом что новая
ДНК встраивается путем рестрикции, сопровождаемой
лигированием. Рекомбинантный вектор вводят в клетки
Escherichia coli посредством трансфекции. Как толь-
ко двунитевой вектор окажется внутри клетки Е. coli,
он реплицируется и направляет синтез однонитевых ко-
пий, которые упаковываются в фаговые частицы и выде-
ляются из клетки. Фаговые частицы могут быть собраны
из среды культивирования после ее центрифугирования
с целью осаждения бактерий. Белковые оболочки фагов
удаляются обработкой фенолом, и однонитевая версия
рекомбинантного вектора очищается для использования
в секвенировании ДНК
Глава 4. Секвенирование геномов 143
тип которого указывает на нуклеотид — А, С, G или Т, — который присутствует в той
же позиции в матричной ДНК (рис. 4.2, в).
Чтобы определить поеледовательность ДНК, все, что нужно сделать, — это опознать
дидезоксинуклеотид в конце каждой молекулы с оборванной цепью. Именно здесь в игру
входит полиакриламидный гель. Смесь ДНК загружают в лунку полиакриламидного
плоского геля или в трубку капиллярной гелевой системы и проводят электрофорез
с целью разделения молекул по их длине. После разделения молекулы продвигаются
мимо датчика флюоресценции, способного различать метки, прикрепленные к диде-
зоксинуклеотидам (рис. 4.3, а). Таким образом, датчик определяет, каким нуклеотидом
заканчивается каждая проходящая мимо него молекула: А, С, G или Т. Последователь-
ность может быть распечатана для изучения оператором (рис. 4.3, б) или введена не-
посредственно в запоминающее устройство для последующего анализа. Автоматиче-
ские секвенаторы с многочисленными работающими параллельно капиллярами могут
считывать до 96 различных последовательностей за двухчасовой период; это означает,
что при среднем числе 750 п.н. за отдельный эксперимент 864 тыс. п. н. информации
может быть получено на одну машину за один день. Для этого, конечно, требуется кру-
глосуточная техническая поддержка, в идеале с помощью робототехнических устройств,
применяемых для приготовления реакций секвенирования и загрузки продуктов ре-
акции в секвенаторы. Если такой промышленный подход может быть поставлен «на
конвейер» и поддерживаться в потоке, то данные, необходимые для секвенирования
полного генома, могут быть собраны в течение нескольких недель.
Для выполнения секвенирования с обрывом цепи необходима однонитевая
ДНК-матрица
Матрицей для эксперимента с обрывом цепи служит однонитевая версия молекулы
ДНК, предназначенной для секвенирования. Она может быть получена несколькими
способами:
• ДНК может быть клонирована в плазмидном векторе (раздел 2.2.1). Получающаяся
ДНК будет двунитевой и, таким образом, не может быть непосредственно использована
в секвенировании. Для этого она должна быть преобразована в однонитевую ДНК по-
средством денатурации щелочью или кипения. Этот метод получения матричной ДНК
для секвенирования стал общепринятым — в значительной степени благодаря тому,
что клонирование в плазмидном векторе является стандартной процедурой. Недоста-
ток его состоит в сложности приготовления плазмидной ДНК, не загрязненной малыми
количествами бактериальных ДНК и РНК, которые могут оказаться соответственно
ложными матрицами или затравками в эксперименте по секвенированию ДНК.
• ДНК может быть клонирована в векторе на бактериофаге Ml3. Векторы, основанные
на бактериофаге М13, специально разработаны для производства однонитевых
матриц для секвенирования ДНК. Бактериофаг М13 имеет геном из однонитевой
ДНК, который после инфицирования бактерии Escherichia coli преобразуется в дву-
нитевую репликативную форму. Репликативная форма копируется до тех пор, пока
в клетке не будут присутствовать более 100 молекул, и когда клетка делится, число
копий в новых клетках поддерживается за счет последующей репликации. В то же
время зараженные клетки непрерывно выделяют новые частицы фага М13 — при-
близительно 1000 за одно поколение, — причем эти фаги содержат однонитевую
версию генома (рис. 4.4). Клонирующие векторы, основанные на векторах М13,
представляют собой двунитевые молекулы ДНК, эквивалентные репликативной
144 Часть 1. Изучение геномов
форме генома бактериофага М13. Ими можно манипулировать в точности таким
же образом, что и плазмидным клонирующим вектором. Различие состоит в том,
что клетки, которые были подвергнуты трансфекции рекомбинантным вектором
М13, выделяют фаговые частицы, содержащие однонитевую ДНК, причем эта ДНК
включает в себя векторную молекулу плюс любую дополнительную ДНК, которая
была с ней лигирована. Таким образом, фаги поставляют матричную ДНК для
секвенирования с обрывом цепи. Единственный недостаток при клонировании
в векторе на фаге М13 заключается в том, что фрагменты ДНК длиннее прибли-
зительно 3 тыс.п.н. претерпевают удаления и перестройки, так что такая схема
может быть применима только с короткими частями ДНК.
• ДНК может быть клонирована в фагмиде. Это плазмидный клонирующий вектор,
который содержит в дополнение к своей плазмидной области инициации данную
область от фага М13 или любого другого бактериофага с геномом из одноните-
вой ДНК. Если клетка Е. coli содержит и фагмиду, и репликативную форму фага-
помощника, причем последний несет гены ферментов репликации фага и белков
капсида, то область инициации фага в фагмиде становится активированной, что
приводит к синтезу фаговых частиц, содержащих однонитевую версию этой фаг-
миды. Двунитевая плазмидная ДНК тем самым преобразуется в однонитевую
матричную ДНК для секвенирования ДНК. Эта система избегает неустойчивости
клонирования в векторе на фаге М13 и может быть использована с фрагментами
длиной до 10 тыс.п.н. и более.
ДНК-полимеразы для секвенирования с обрывом цепи
Любая направляемая матрицей ДНК-полимераза способна продолжать затравку,
отожженную к однонитевой молекуле ДНК, но не все полимеразы делают это таким об-
разом, который был бы пригоден для секвенирования ДНК. Фермент для секвенирования
должен удовлетворять в особенности трем следующим критериям.
• Высокая обрабатывающая способность (процессивность). Эта характеристика
относится к длине полинуклеотида, который синтезируется полимеразой прежде,
чем она заканчивает синтез в силу естественных причин. Подходящая для секве-
нирования полимераза должна иметь высокую процессивность, — так чтобы она
не отделялась от матрицы до включения в новую нить дидезоксинуклеотида.
• Незначительное или нулевое действие экзонуклеазы 5’->3'. Большинство ДНК-
полимераз обладают также действием экзонуклеаз, сие же означает, что, наряду
с синтезом полинуклеотидов ДНК, они могут также и расщеплять их (раздел 2.1.1;
см. рис. 2.7). В секвенировании ДНК это недостаток, потому что удаление нуклео-
тидов с 5'-концов новосинтезируемых нитей изменяет длины этих нитей, делая
невозможным достоверное определение последовательности.
• Пренебрежимо малое или нулевое действие экзонуклеазы 3'—>5’. Также желательно,
чтобы полимераза не удаляла дидезоксинуклеотид на конце законченной нити.
Если это случается, то такая нить может быть продолжена далее. В конечном ре-
зультате в реакционной смеси будет содержаться небольшое число коротких нитей,
и последовательность вблизи от затравки будет нечитабельна.
Эти требования строгие и полностью не выполняются никакой встречающейся
в природе ДНК-полимеразой. Вместо них обычно применяются искусственно видо-
измененные ферменты. Первым из таких ферментов была разработана полимераза
Клёнова, которая является версией ДНК-полимеразы I Escherichia coli, у которой было
Глава 4. Секвенирование геномов 145
устранено действие экзонуклеазы 5'—>3', присущее стандартному ферменту или пу-
тем расщепления соответствующей части белка, или средствами генной инженерии
(раздел 2.1.1). Полимераза Клёнова обладает относительно низкой процессивностью,
что ограничивает длину последовательности, которая может быть получена в одном
эксперименте, приблизительно до 250 п. н., и приводит к получению в реакции секвени-
рования неспецифических продуктов — нитей, которые были оборваны естественным
образом, а не из-за включения в них дидезоксинуклеотида. Поэтому фермент Клёнова
был заменен видоизмененной версией ДНК-полимеразы, кодируемой бактериофагом
Т7 (этот фермент известен под торговым названием «секвеназа»). Секвеназа имеет
высокую процессивность, не проявляет действие экзонуклеазы и обладает прочими
желательными свойствами, такими как высокая скорость протекания реакции.
Затравка определяет область ДНК-матрицы, которая будет секвенирована
Чтобы начать эксперимент по секвенированию с обрывом цепи, олигонуклеотидная
затравка отжигается на ДНК-матрицу. Затравка необходима, потому что направляемые
матрицей ДНК-полимеразы не могут начать синтез ДНК на молекуле, которая является
полностью однонитевой: на ней должна быть короткая двунитевая область, могущая
предоставить свой З'-конец, к которому фермент может добавлять новые нуклеотиды
(раздел 2.1.1).
Затравка играет ключевую роль также и в определении области матричной моле-
кулы, которая будет секвенирована. Для большинства экспериментов секвенирования
используется «универсальная» затравка, то есть такая, которая является комплемен-
тарной части векторной ДНК, непосредственно смежной с точкой, к которой лигиро-
вана новая ДНК (рис. 4.5, а). Поэтому одна и та же универсальная затравка может дать
последовательность любой части ДНК, которая была лигирована в вектор. Конечно же,
если такая встроенная ДНК будет длиннее 750 п.н. или около того, то только часть ее
последовательности будет получена, но обычно такой проблемы не возникает, потому
что проект в целом всего-навсего требует, чтобы было произведено большое число
а) Универсальная затравка
Затравка
Векторная ДНК Вставка ДНК Векторная ДНК
б) Внутренние затравки
м Универсальная затравка
Внутренняя затравка
Рис. 4.5. Затравки разного типа для секвенирования с обрывом цепи, о) Универсальная затравка отжи-
гается к векторной ДНК, прилегающей к позиции, в которой вставлена новая ДНК. Поэтому одиночная
универсальная затравка может использоваться для секвенирования любой вставки ДНК, но обеспечи-
вает только последовательность одного конца вставки, б) Один из способов получения более длинной
последовательности — выполнить серию экспериментов с обрывом цепи, каждый с новой, отличной от
других, внутренней затравкой, отжигаемой ко внутренним позициям вставки ДНК
146 Часть 1. Изучение геномов
коротких последовательностей и впоследствии собрано в непрерывную основную по-
следовательность. Неважно, будут ли эти короткие последовательности полными или
только частичными последовательностями фрагментов ДНК, используемых в качестве
матриц. Если в качестве матричного материала используется двунитевая ДНК плазми-
ды, то при необходимости можно прочитать последовательности (в большем объеме)
с противоположного конца вставки. В качестве альтернативы можно продолжить по-
следовательность в одном направлении, синтезировав неуниверсальную внутреннюю
затравку, предназначенную для отжига в позиции, находящейся внутри вставки ДНК
(рис. 4.5, б). Эксперимент с такой затравкой даст вторую короткую последовател ьность,
которая перекрывается с предыдущей.
Секвенирование в тепловом цикле предлагает альтернативу традиционной
методологии
Открытие термостабильных ДНК-полимераз, которые привели к развитию ПЦР
(разделы 2.1.1 и 2.3), дало также и новые методы секвенирования с обрывом цепи.
В частности, новшество, называемое секвенированием в тепловом цикле, имеет два
преимущества перед традиционным секвенированием с обрывом цепи. Во-первых, в нем
в качестве исходного материала употребляется двунитевая, а не однонитевая ДНК. Во-
вторых, требуется очень мало матричной ДНК, так что нет необходимости клонировать
ДНК перед ее секвенированием.
Секвенирование в тепловом цикле проводят подобным же образом, что и ПЦР, но
используется только одна затравка и реакционная смесь включает четыре дидезокси-
нуклеотида (рис. 4.6). Поскольку наличествует только одна затравка, копируется только
одна из нитей исходной молекулы и продукт накопляется по линейному закону, а не по
экспоненте, как это имеет место в настоящей ПЦР. Присутствие в реакционной смеси
дидезоксинуклеотидов вызывает обрыв цепи, как и в стандартной процедуре, и семей-
ство полученных нитей может быть проанализировано, а последовательность — считана
обычным способом.
4.1.2. Альтернативные методы секвенирования ДНК
Хотя по большей части секвенирование выполняют методом обрыва цепи, из-
вестны и другие методы, приберегаемые для специальных случаев. Мы изучим два из
таких альтернативных методов: метод химической деградации, который, подобно
секвенированию с обрывом цепи, был
изобретен в 1970-х гг., и пиросеквениро-
вание, которое является более молодым
изобретением.
ДНК-матрица
—ДДА
Нити с обрывом цепи - их число
эт при выполнении большего числа циклов
ДДА
ДДА
ддАТФ
ПЦР с одной-единственной
затравкой
Рис. 4.6. Секвенирование в тепловом цикле. ПЦР
выполняется только с одной затравкой и с че-
тырьмя дидезоксинуклеотидами, находящимися
в реакционной смеси. В результате получается
набор нитей с обрывом цепи — семейство «А»
в части реакции, показанной здесь. Эти нити, на-
ряду с продуктами реакций на С, G и Т, подвер-
гаются электрофорезу посредством стандартной
методологии (см. рис. 4.3)
Глава 4. Секвенирование геномов 147
Синтез нити
заблокирован
ДНК-матрица
Стебле-петельная
структура
Рис. 4.7. Внутринитевое спаривание оснований может затруднять
секвенирование с обрывом цепи. В этом примере ДНК-матрица мо-
жет образовывать стебле-петельную структуру, потому что ее по-
следовательность допускает образование ряда внутринитевых пар
оснований. Такая стебле-петельная структура блокирует продвижение
ДНК-полимеразы, что приводит к неспецифическому обрыву цепи
Секвенирование путем химической деградации
Одно из ограничений секвенирования с обрывом цепи
заключается в том, что с его помощью может оказаться
невозможным получить точную последовательность, если
матричная ДНК способна образовывать внутринитевые
пары оснований (рис. 4.7). Внутринитевые пары основа-
ний могут блокировать продвижение ДНК-полимеразы,
сокращая тем самым протяженность синтезируемой нити,
и могут изменить также и подвижность молекул с обо-
рванной цепью в ходе электрофореза, а это означает, что
порядок прохождения молекул мимо датчика более не будет определяться исключи-
тельно их длиной. Внутринитевые пары оснований не препятствуют секвенированию
путем химической деградации, так что этот метод может быть использован в качестве
альтернативы, когда возникают такие проблемы.
Метод химической деградации подобен секвенированию с обрывом цепи в том,
что последовательность определяется путем установления длины молекул, конечный
нуклеотид которых известен. Однако эти молекулы производятся абсолютно иным об-
разом — путем обработки химикалиями, которые специфично разрезают их в позиции
заданного нуклеотида. Это означает, что необходимо выполнять по крайней мере четыре
отдельные реакции секвенирования — по одной для каждого нуклеотида.
Исходный материал представлен двунитевой ДНК, которая сначала метится при-
креплением группы радиоактивного фосфора к 5’-концу каждой нити (рис. 4.8, а). После
этого добавляется ДМСО (диметилсульфоксид) и ДНК нагревается до 90 °C.
Это разрушает пары оснований, образованные между нитями и позволяет отделять
их друг от друга при помощи гель-электрофореза; это возможно благодаря тому, что одна
из нитей, вероятно, содержит больше пуриновых нуклеотидов, чем другая, и поэтому
немного тяжелее и, следовательно, в ходе электрофореза перемещается медленнее. Одна
нить очищается от геля и разделяется на четыре образца, каждый из которых обраба-
тывают одним из расщепляющих реактивов. Чтобы иллюстрировать данную процедуру,
мы проследим за ходом реакции «G» (рис. 4.8, б). Сначала молекул ы обрабатывают диме-
тилсульфатом, в результате чего метильная группа прикрепляется к пуриновому кольцу
нуклеотидов G. Добавляется только ограниченное количество диметил сульфата, чтобы
изменить в среднем только один остаток G на один полинуклеотид. На этой стадии нити
ДНК все еще целостны: расщепление не происходит до момента добавления второго
химиката — пиперидина. Пиперидин удаляет видоизмененное пуриновое кольцо и раз-
резает молекулу ДНК в фосфодиэфирной связи, находящейся непосредственно выше
новосозданного участка без основания. В результате мы получаем набор расщепленных
молекул ДНК, одни из которых помечены, а другие — нет. У всех помеченных молекул
один конец одинаковый, а другой конец определяется участками разрезания, при этом
последние указывают позиции нуклеотидов G в молекулах ДНК, которые были расще-
плены. Подобные же подходы используются и для получения дополнительных семейств
148 Часть 1. Изучение геномов
а) Мечение ДНК и разъединение нитей
Тяжелая
Легкая Т w
Меченый 5 -конец
| ДМСО, 90 °C
I Электрофорез
* в агарозном геле
Очистка одной
—' из нитей
б) Реакция «G»
Молекула для секвенирования
(множество копий)
| Диметилсульфат
Me
, ,7,, с
Me
*111111111 1г
Me
I Пиперидин
•n i । ii 11 i 111* *? rrt. mW
^^тгтттттттт?
* 11 ii 11111 m
в) Считывание последовательности
с авторадиограммы
G A+G С C+T
Рис. 4.8. Секвенирование путем химической деградации
расщепленных молекул, хотя обычно это не просто семейства «А», «Т» и «С», так как при
разработке способов химической обработки, позволяющей специфично разрезать цепи
полинуклеотидов в позициях А или Т, возникли проблемы. Поэтому четыре выполняе-
мых реакции обычно представлены набором «G», «А + G», «С» и «С + Т». Это усложняет
процедуру, но не влияет на точность определяемой последовательности.
Семейство молекул, произведенных в каждой реакции, загружается на дорожку
полиакриламидного плоского геля и после электрофореза позиции полос в геле ви-
зуализируются авторадиографией (см. техническое примечание 2.1). Полоса, которая
продвинулась наиболее далеко, представляет наименьший кусок ДНК. В примере, пред-
ставленном на рис. 4.8, в, эта полоса находится на дорожке «А + G». Поскольку на дорожке
«G» не имеется полосы, соответствующей такому же размеру, первый нуклеотид в по-
следовательности — «А». Следующая размерная позиция занята двумя полосами —
одна на дорожке «С», а другая на дорожке «С + Т»; поэтому второй нуклеотид — «С»
и последовательность на данный момент — «АС». Считывание последовательности
может быть продолжено подобным же образом вплоть до области геля, где отдельные
полосы не были разделены.
Пиросеквенирование применяется для быстрого определения очень корот-
ких последовательностей
Пиросеквенирование не требует электрофореза или любой другой процедуры
разделения фрагментов и, таким образом, работает быстрее и секвенирования с об-
рывом цепи, и секвенирования путем химической деградации. Оно позволяет получить
лишь несколько десятков п.н. за один эксперимент1), но становится важным методом
в ситуациях, где требуется определить большое число коротких последовательностей
как можно быстрее, например при типизации SNP (раздел 3.2.2).
При пиросеквенировании матрица копируется прямым способом без добавленных
дидезоксинуклеотидов. В то время как строится новая нить, порядок, в котором дезокси-
Современные приборы позволяют читать порядка 400 нуклеотидов. Кроме того, в настоящее время
активно используются другие приборы массового параллельного секвенирования, использующие иные
принципы. В стадии разработки и испытаний находится ряд технологий, позволяющих секвенировать ин-
дивидуальные молекулы. — Прим. ред.
Глава 4. Секвенирование геномов 149
нуклеотиды встраиваются в нее, устанавливается датчиком, так что последовательность
может «считываться» прямо в ходе реакции. Добавление дезоксинуклеотида к концу
растущей нити обнаружимо, потому что оно сопровождается высвобождением молекулы
пирофосфата, каковое событие может быть преобразовано ферментом сульфурилазой
во вспышку хемилюминесценции. Конечно, если бы все четыре дезоксинуклеотида
добавлялись одновременно, то вспышки света были бы заметны все время и никакой
полезной информации о последовательности не было бы получено. Поэтому каждый из
дезоксинуклеотидов добавляется по отдельности, один за другим, причем в реакционной
смеси присутствует также фермент нуклеотидаза, с тем чтобы быстро осуществлять
деградацию введенного дезоксинуклеотида, если он не был включен в полинуклеотид
прежде, чем будет добавлен следующий (рис. 4.9). Такая процедура позволяет отслежи-
вать порядок, в котором дезоксинуклеотиды встраиваются в наращиваемую нить. Судя
по описанию, этот метод представляется очень сложным, но для его осуществления
всего лишь необходимо последовательно добавлять в реакционную смесь реагенты,
то есть выполнять именно ту процедуру, которая легко может быть автоматизирована.
Обнаружение хемилюминесценции очень чувствительно, так что каждая реакция мо-
жет проводиться в очень малом объеме — возможно, только в одном пиколитре. Это
означает, что на пластинке площадью 6,4 см2 параллельно может быть выполнено до 1,6
миллиона реакций, что позволяет получить за четыре часа 25 миллионов нуклеотидов
последовательности, — такая скорость расшифровки последовательности приблизи-
тельно в 100 раз быстрее, чем позволяет достичь метод обрыва цепи.
Затравка
IIIIIIIIIIIIIIII
Матрица
дАТФ деградирует
дТТф —► деградирует
дГТФ f хемилюминесценция
+
дЦТФ —► деградирует
+
дАТФ I хемилюминесценция
+
iGA
IlliHillllllilfi
Рис. 4.9. Пиросеквенирование. Реакция синтеза нити выполняется при отсутствии дидезокси нуклеоти-
дов. Каждый дезоксинуклеотид добавляется индивидуально, совместно с ферментом нуклеотидазой,
который деградирует дезоксинуклеотид, если он не встроен в синтезируемую нить. Встраивание обна-
руживается вспышкой хемилюминесценции, вызванной пирофосфатом, высвобожденным из дезок-
синуклеотида. Порядок, в котором дезокси нуклеотиды добавляются к наращиваемой нити, поэтому
может быть отслежен
150 Часть 1. Изучение геномов
4.2. Сборка непрерывной последовательности ДНК
Следующий вопрос, к которому необходимо обратиться, — как основная последо-
вател ьность хромосомы, возможно, несколько десятков млн п. н. в длину, может быть со-
брана из множества коротких последовательностей, полученных в ходе секвенирования
с обрывом цепи. Мы обращались к этой проблеме в начале главы 3 и установили, что
относительно короткие геномы прокариотов могут быть собраны методом дробовика,
который состоит в разрушении молекулы ДНК на фрагменты, определении последо-
вательности каждого из них и поиске (с использованием компьютера) перекрытий, по
которым строится основная последовательность (см. рис. 3.1 и раздел 4.2.1). Этот подход
к настоящему времени был успешно применен к более чем 200 геномам прокариотов, но
если его применить к большим геномам эукариотов, он может привести к ошибкам, —
главным образом из-за того, что присутствие повторных последовательностей в геномах
эукариотов усложняет поиск перекрытий последовательности и может в результате дать
неправильно собранные сегменты генома (см. рис. 3.2). Чтобы избежать таких ошибок,
применяют подход, упоминаемый как метод дробовика для полных геномов, который
опирается на карту как на вспомогательное средство сборки основной последователь-
ности (см. рис. 3.3 и раздел 4.2.3). Секвенирование методом дробовика для полных
геномов было продуктивно использовано с несколькими геномами эукариотов, в том
числе с геномами плодовой мушки и человека, но, согласно оценкам многих специали-
стов, наибольшая степень точности может быть достигнута методом сборки контигов
из клонов. В этом подходе до начала секвенирования геном разбивается на сегменты
с известными позициями на карте этого генома (см. рис. 3.3 и раздел 4.2.2). Для начала
мы посмотрим, как метод дробовика применялся к геномам прокариотов.
4.2.1. Сборка последовательности методом дробовика
Прямой подход к сборке последовательности состоит в том, чтобы создать полную
последовательность непосредственно из коротких фрагментов, полученных в результате
индивидуальных экспериментов секвенирования, — путем простого поиска возможных
наложений этих фрагментов (см. рис. 3.1). Это называют методом дробовика. Метод не
требует никакого предварительного знания генома, и сборка может быть выполнена
при отсутствии генетической или физической карты.
Перспективность метода дробовика была доказана успешной расшифров-
кой последовательности Haemophilus influenzae
В первой половине 1990-х гг. шли серьезные дебаты о том, будет ли метод дробовика
работать на практике, при этом многие молекулярные биологи придерживались того
мнения, что объем обработки данных, необходимый для сравнения всех минипоследо-
вательностей и распознавания перекрытий, даже в случае наименьших геномов был бы
за пределами возможностей существующих компьютерных систем. Этим сомнениям был
положен конец в 1995 г., когда была опубликована последовательность генома бактерии
Haemophilus influenzae размером 1 830 тыс. п. н.
Геном Н. influenzae был секвенирован всецело методом дробовика, причем без обра-
щения к какой-либо информации с генетической или физической карты. Стратегия, при-
менявшаяся для получения этой последовательности, показана на рис. 4.10. Первый шаг
состоял в раздроблении геномной ДНК на фрагменты путем разрушения ультразвуком —
этот метод основан на использовании высокочастотных звуковых волн для произведения
случайных разрезов в молекулах ДНК. Затем фрагменты были подвергнуты электрофорезу,
Глава 4. Секвенирование геномов 151
Рис. 4.10. Схема применения метода дробовика
при определении последовательности ДНК гено-
ма Haemophilus influenzae. ДНКН. influenzae была
разрушена ультразвуком, и фрагменты размером
от 1,6 до 2,0 тыс. п. н. были очищены от агарозного
геля и лигированы в плазмидный вектор, чтобы
произвести библиотеку клонов. Были получены
конечные последовательности клонов, взятых
из этой библиотеки, и с помощью компьютера
распознаны перекрытия между этими последо-
вательностями. В результате было получено 140
контигов, которые были собраны в полную после-
довательность генома, как показано на рис. 4.11
и попавшие в размерный диапазон 1,6-2,0
тыс. п. н. были очищены от агарозного геля
и лигированы в плазмидный вектор. Из по-
лученной библиотеки были взяты наугад
19 687 клонов и было выполнено 28 643
экспериментов секвенирования; число
экспериментов секвенирования превыша-
Q Haemophilus influenzae
| Извлечение ДНК
^^|Ргк5рушение ультразвуком
.ZrZJTLTZ- Фрагменты ДНК
“ZLZ" -ZTZ7 различных размеров
1 Электрофорез в агарозном геле
ДОРОЖКА 1: Разрушенная ультразвуком
ДНК Н. influenzae
ДОРОЖКА 2: ДНК-маркёры
1 2
’I Очистка ДНК от геля
—3— — Фрагменты ДНК - 1,6-2,0 тыс. п. н.
| Приготовление библиотеки клонов
ет число плазмид, потому что у некоторых
вставок были секвенированы оба конца. Из
этих экспериментов секвенирования 16 %
были сочтены неудачными, потому что они
дали менее 400 п.н. последовательности.
Оставшиеся 24304 последовательности
в общей сложности дали 11631485 п.н.,
что соответствует шести длинам генома
Н. influenzae; такая степень избыточности
была сочтена необходимой для гарантии
полного покрытия. Сборка последователь-
ности потребовала 30 часов на компьютере
I Получение конечных
* последовательностей встроек ДНК
7
Конечные последовательности
। Построение последовательности
♦ контига
с объемом оперативной памяти (ОЗУ) 512 Мб и дала 140 длинных непрерывных последо-
вательностей, при этом каждая из таких последовательностей1^ представляла отдельную
неперекрывающуюся часть генома.
Следующим шагом было соединение пар контигов, для чего надо было прочитать
последовательности между контигами (рис. 4.11). Сначала была проверена библиоте-
ка, с тем чтобы увидеть, имеются ли в ней какие-либо клоны, в которых две конечные
последовательности были бы расположены в разных контигах. Если такой клон будет
идентифицирован, то дополнительное секвенирование его вставки закроет «кодовый
пропуск» между этими двумя контигами (рис. 4.11, а). Фактически было обнаружено 99
клонов этой категории, так что 99 из всех пропусков могли быть закрыты без особых
затруднений.
В конечном итоге осталось 42 зияющих пропуска, состоявших, по всей вероятно-
сти, из последовательностей ДНК, которые были неустойчивы в клонирующем векторе
^Такая последовательность называется контигом. — Прим. ред.
152 Часть 1. Изучение геномов
а) Заполнение «кодового пропуска»
1
2
м Клонированный фрагмент
< ДНК (заполняет пропуск)
контиги последовательности
Завершение расшифровки
I последовательности при
помощи внутренних затравок
б) Заполнение «физического пропуска»
1 2 3 И Т. Д. —
олигонуклеотиды
ВЫВОД: контиги 1 и 4 являются смежными
Рис. 4.11. Сборка последовательности полного генома Haemophilus influenzae путем заполнения про-
пусков между отдельными контигами последовательности, а) «Кодовые пропуски» таковы, что могут
быть заполнены дальнейшим секвенированием клонов, уже имеющихся в библиотеке. В данном при-
мере концы последовательностей контигов 1 и 2 лежат в пределах одного и того же плазмидного клона,
так что дальнейшее секвенирование этой вставки ДНК со внутренними затравками (см. рис. 4.5, б)
даст последовательность для заполнения этого пропуска, б) «Физические пропуски» — это отрезки
последовательности, которые не присутствуют в библиотеке клонов, вероятно, потому, что эти области
неустойчивы в используемом клонирующем векторе. Показаны две стратегии заполнения таких про-
пусков. Слева вторая библиотека клонов, приготовленная с помощью вектора на бактериофаге X вместо
плазмидного вектора, зондируется олигонуклеотидами, соответствующими концам контигов. Оба оли-
гонуклеотида — 1 и 7 — гибридизируются к одному и тому же клону, вставка которого должна поэтому
содержать ДНК, заполняющую пропуск между контигами 1 и 4. Справа эксперименты ПЦР выполнены
с парами олигонуклеотидов. Только номера 1 и 7 дают продукт ПЦР, подтверждая, что концы контигов,
представленные этими двумя олигонуклеотидами, находятся близко друг к другу в геноме. Для запол-
нения пропуска между контигами 1 и 4 могут быть секвенированы продукт ПЦР или вставка из клона
на бактериофаге X
Глава 4. Секвенирование геномов 153
и поэтому отсутствовали в библиотеке. Для того чтобы закрыть такие «физические
пропуски», была приготовлена вторая библиотека клонов, причем на этот раз с примене-
нием вектора иного типа. Вместо того чтобы использовать другую плазмиду, в которой
неклонированные последовательности, скорее всего, по-прежнему будут неустойчивы,
вторая библиотека была приготовлена в векторе на бактериофаге X (раздел 2.2.1). Эта
новая библиотека была прозондирована 84-мя олигонуклеотидами, по одному, причем
эти 84 олигонуклеотида имели последовательности, идентичные последовательностям
на концах несцепленных контигов (рис. 4.11, б). Разумное объяснение таково: если два
олигонуклеотида гибридизировались с одним и тем же Х-клоном, то концы контигов,
из которых они были получены, должны лежать в пределах этого клона, и поэтому
секвенирование ДНК Х-клона заполнит пропуск. Таким способом были заполнены 23
из 42 физических пропусков.
Вторая стратегия закрытия пропусков состояла в том, чтобы использовать пары
олигонуклеотидов из 84-олигонуклеотидного набора, описанного выше, в качестве
затравок для экспериментов ПЦР геномной ДНК Н. influenzae. Некоторые олигону-
клеотидные пары были отобраны наугад, и те, которые заполняют пропуск, были
идентифицированы попросту на основании того, дают ли они продукт ПЦР или нет (см.
рис. 4.11, б). Секвенирование этих продуктов ПЦР закрыло соответствующие пропуски.
Другие пары затравок были выбраны на более рациональном основании. Например,
олигонуклеотиды были использованы как зонды для гибридизации по Саузерну (см.
рис. 2.11) с ДНК Н. influenzae, разрезанной разнообразными эндонуклеазами рестрик-
ции, и были идентифицированы пары, которые гибридизировались после обработки
подобными наборами фрагментов рестрикции. Два члена пары олигонуклеотидов,
опознанных таким способом, должны содержаться в пределах одних и тех же фрагмен-
тов рестрикции и, таким образом, с большой вероятностью лежат близко друг к другу
в геноме. Это означает, что пара контигов, из которых получены олигонуклеотиды,
смежна, и пропуск между этими контигами может быть заполнен по результатам ПЦР
геномной ДНК, в которой в качестве затравок, обеспечивающих ДНК-матрицу для за-
крытия пропуска, используются эти два олигонуклеотида.
Демонстрация того факта, что маленький геном может быть секвенирован относи-
тельно быстро методом дробовика, привела к внезапному изобилию расшифрованных
геномов микробов. Эти проекты продемонстрировали, что секвенирование методом
дробовика может быть поставлено по типу поточной линии, где каждый член команды
имел бы свою индивидуальную задачу, состоящую, скажем, в приготовлении ДНК, в вы-
полнении реакций секвенирования или в проведении анализа данных. Эта стратегия
позволила секвенировать геном Micoplasmagenitalium из 580 тыс.п.н. с привлечением
пяти сотрудников всего лишь за восемь недель, и теперь все признают, что несколько
месяцев были бы вполне достаточным сроком, чтобы произвести полную последова-
тельность любого генома размером не более 5 млнп.н., даже если ничего не известно
о геноме до начала проекта. Итак, сильные стороны метода дробовика — его скорость
и способность работать в отсутствие генетической или физической карты.
4.2.2. Сборка последовательностей методом сборки контигов из клонов
Метод сборки контигов из клонов рассматривается как обычный метод получе-
ния последовательности генома эукариотов, и он также применялся с теми геномами
микробов, которые были до того картированы генетическими и (или) физическими
средствами. Согласно методу сборки контигов из клонов геном разбивается на фраг-
154 Часть 1. Изучение геномов
менты до 1,5 млн п. н. в длину обычно посредством неполной рестрикции (раздел 3.3.1),
и эти фрагменты клонируются в векторе высокой емкости типа ВАС (раздел 2.2.1).
Сборка контигов из клонов выстраивается путем опознавания клонов, содержащих пере-
крывающиеся фрагменты, которые после того по отдельности секвенируют методом
дробовика. В идеале клонированные фрагменты помещают на генетическую и (или)
физическую карту генома, с тем чтобы данные последовательности контигов могли
быть проверены и интерпретированы путем отыскания особенностей (например, STS,
SSLP, гены), о которых известно, что они присутствуют в определенной области.
Сборки контигов из клонов могут быть построены путем обхода хромосо-
мы, но этот метод весьма трудоемок
Простейший способ построить ряд перекрывающихся клонированных фрагментов
ДНК состоит в том, чтобы начать с одного клона из библиотеки, засим опознать второй
клон, вставка которого перекрывается со вставкой в первом клоне, затем опознать
третий клон, вставка которого перекрывается с таковой во втором клоне, и так далее.
Это основа метода обхода хромосомы, который был первым методом, изобретенным
для сборки контигов из клонов.
Первоначально обход хромосомы использовался, чтобы продвигаться по молекулам
ДНК на относительно короткие расстояния, используя библиотеки клонов, приготов-
ленные с помощью векторов на бактериофаге X или космидных векторов. Наиболее
прямой подход состоит в том, чтобы использовать ДНК вставки из исходного клона
в качестве гибридизирующего зонда, с тем чтобы перебрать все остальные клоны
в библиотеке. Клоны, вставки которых перекрываются с зондом, дают положительные
сигналы скрещивания, и их вставки могут быть использованы в качестве новых зондов,
чтобы продолжить обход (рис. 4.12).
Главная проблема, которая возникает при осуществлении этого подхода, состоит
в том, что если зонд содержит повторную последовательность, то он будет гибриди-
зироваться не только с перекрывающимися клонами, но также и с неперекрывающи-
мися клонами, вставки которых содержат копии этого повторения. Степень такой не-
специфической гибридизации может быть уменьшена путем блокирования повторных
последовательностей предварительной гибридизацией с непомеченной геномной ДНК
(см. рис. 3.34), но это полностью не решает проблему, особенно если обход выполняется
с длинными вставками из вектора высокой емкости типа ВАС. По этой причине неповреж-
денные вставки редко используются для обходов хромосом с ДНК человека и подобными
ДНК, которые имеют высокую частотность повторных последовательностей. Вместо
этого в качестве зонда используется фрагмент с конца вставки, ибо меньше шансов
встретить повторение в коротком концевом фрагменте, чем во всей вставке в целом.
Если требуется полная уверенность, то концевой фрагмент может быть секвенирован
перед его использованием, с тем чтобы убедиться в том, что никакой повторной ДНК
в нем не присутствует.
Если концевой фрагмент был секвенирован, то обход может быть ускорен за счет
применения для опознавания клонов с перекрывающимися вставками ПЦР, а не гибри-
дизации. Затравки конструируют из последовательности конечного фрагмента и исполь-
зуют в предпринятых экспериментах ПЦР со всеми другими клонами из библиотеки.
Клон, который дает продукт ПЦР правильного размера, должен содержать перекры-
вающуюся вставку (рис. 4.13). Чтобы ускорить процесс еще больше, вместо того чтобы
выполнять ПЦР с каждым отдельным клоном, группы клонов смешивают вместе таким
образом, чтобы однозначная идентификация перекрывающихся вставок могла быть
Глава 4. Секвенирование геномов 155
осуществлена наверняка. Данный метод иллюстрирован на рис. 4.14; в предложенном
примере библиотека из 960 клонов была приготовлена в десяти лотках микротитра,
где каждый лоток содержит 96 лунок в виде массива 8x12, по одному клону на лунку.
Эксперименты ПЦР выполняют следующим образом.
• Образцы каждого клона в ряду А первого лотка микротитра смешиваются вместе
и выполняется одна ПЦР. Это повторяют для каждого ряда каждого лотка — всего
80 экспериментов ПЦР.
• Образцы каждого клона в столбце 1 первого лотка микротитра смешиваются вместе
и выполняется одна ПЦР. Это повторяют для каждого столбца каждого лотка —
всего 120 экспериментов ПЦР.
• Клоны из лунки А1 каждого из десяти лотков микротитра смешиваются вместе
и выполняется одна ПЦР. Это повторяют для каждой лунки — всего 96 экспери-
ментов ПЦР.
Шаг 1
Шаг 2
1 J 3 4 5 в 7 I 9 10 11 12
Зонд: вставка из клона А1
Положительные сигналы:
клоны А1, Е7 и F6
Зонд: вставка из клона F6
Положительные сигналы:
клоны А1, В12 и F6
А1
Е7 F6
Рис. 4.12. Обход хромосомы. Библиотека включает 96 клонов, каждый из которых содержит свою,
отличную от других, вставку. Чтобы начать обход, вставка из одного из клонов используется как ги-
бридизирующий зонд относительно всех других клонов в библиотеке. В показанном примере зондом
является клон А1; он гибридизируется сам с собой и с клонами Е7 и F6. Поэтому вставки из последних
двух клонов должны перекрываться со вставкой из клона А1. Чтобы продолжить обход, зондирование
повторяется, но на сей раз со вставкой из клона F6. Гибридизация клонов Al, F6 и В12 показывает, что
вставка из клона В12 перекрывается со вставкой из клона F6
Вставка № 1 Олигонуклеотиды
ВЫВОД: вставки 1 и 15 перекрываются
Рис. 4.13. Обход хромосомы с помощью ПЦР. Два олигонуклеотида отжигаются в пределах конечной об-
ласти вставки номер 1. Они используются в экспериментах ПЦР со всеми другими клонами из библиотеки.
Только клон 15 дает продукт ПЦР, показывая, что вставки в клонах 1 и 15 перекрываются. Обход можно
продолжить секвенированием фрагмента с другого конца клона 15, конструированием второй пары оли-
гонуклеотидов и использованием их в новой серии экспериментов ПЦР со всеми прочими клонами
156 Часть 1. Изучение геномов
Столбец , 2 3 4 5 6 7 8 9 10 11 12
Смешивание, Смешивание с лунками А1 из
Смешивание,
ПЦР
Повтор по всем рядам на всех 10-ти
лотках = 80 экспериментов ПЦР
Повтор по всем столбцам на всех 10-ти
лотках = 120 экспериментов ПЦР
Повтор по всем лункам = 96
экспериментов ПЦР
ОБЩЕЕ ЧИСЛО ЭКСПЕРИМЕНТОВ
ПЦР = 296
Рис. 4.14. Комбинаторный отбор клонов на лотках микротитра. В этом примере библиотека из 960 клонов
должна быть просмотрена с помощью ПЦР. Вместо того чтобы выполнять 960 отдельных экспериментов
ПЦР, клоны группируют, как показано, и выполняется только 296 экспериментов ПЦР. В большинстве
случаев результаты позволяют однозначно опознавать «положительные» клоны. Фактически, если
положительных клонов немного, то иногда их можно опознать посредством всего лишь только экспе-
риментов ПЦР «по ряду» и «по столбцу». Например, если положительные эксперименты ПЦР получены
с рядом А лотка 2, рядом D лотка 6, столбцом 7 лотка 2 и столбцом 9 лотка 6, то можно заключить, что
есть два положительных клона: один в лунке А7 лотка 2 и второй в лунке D9 лотка 6. Эксперименты
ПЦР «по лункам» необходимы в том случае, если в одном и том же лотке обнаруживается два и более
положительных клона
Как объяснено в подписи к рис. 4.14, эти 296 экспериментов ПЦР обеспечивают
достаточно информации, чтобы определить, которые из 960 клонов дают продукты,
а которые — не дают. Неоднозначность возникает, только если число клонов, дающих
положительный результат, велико.
Более быстрые методы сборки контигов из клонов
Даже когда шаг отбора выполняется при помощи комбинаторного подхода с при-
влечением ПЦР, показанного на рис. 4.14, обход хромосомы оказывается медленным
процессом и редко позволяет собрать контиги более чем из 15-20 клонов. Эта процедура
была чрезвычайно ценна в позиционном клонировании, где цель состоит в том, чтобы
пройти от нанесенного на карту участка до интересующего гена, о котором заранее из-
вестно, что он расположен на расстоянии не более нескольких млн п. н. Он оказался менее
ценен для сборки контигов из клонов по полным геномам, особенно в случае сложных
геномов высших эукариотов. Так какие же альтернативные методы существуют?
Главная альтернатива состоит в использовании метода дактилоскопии клонов.
Снятие отпечатков пальцев у клонов дает информацию о физической структуре кло-
нированного фрагмента ДНК; эта физическая информация, или «отпечаток пальцев»,
Глава 4. Секвенирование геномов 157
сравнивается с эквивалентными данными от других клонов, что позволяет опознавать
клоны с подобиями — возможно, указывающими на перекрытия. Используется один из
следующих методов (или их сочетание) (рис. 4.15).
• Картины рестрикции могут быть получены путем переваривания клонов множе-
ством разнообразных ферментов рестрикции и разделения продуктов в агарозном
геле. Если два клона содержат перекрывающиеся вставки, то отпечатки пальцев их
рестриктов будут иметь общие полосы, поскольку оба будут содержать фрагменты,
полученные из области перекрытия.
• Отпечатки пальцев повторной ДНК могут быть приготовлены в ходе анализа на-
бора фрагментов рестрикции при помощи гибридизации по Саузерну (раздел 2.1.2)
с зондами, специфичными к повторным последовательностям одного или несколь-
ких типов. Как и в случае отпечатков пальцев рестриктов, перекрытия опознаются
путем отыскания двух клонов, имеющих несколько общих полос гибридизации.
• ПЦР с повторной ДНК, или ПЦР с рассеянными повторными элементами
(РПЭ-ПЦР), предполагает использование затравок, которые отжигаются в пределах
повторных последовательностей и таким образом размножают одну копию области
ДНК, находящейся между двумя соседними повторениями. Поскольку повторные
последовательности распределены по геному неравномерно, размеры продуктов,
полученных после ПЦР с повторной ДНК, могут быть использованы как отпечаток
пальцев при сравнениях с другими клонами, проводимыми с целью опознавания
потенциальных перекрытий. В случае ДНК человека часто используются повто-
рения, называемые элементами Alu (раздел 9.2.1), потому что они встречаются
в среднем один раз на каждые 3 тыс. п. н. Д/и-ПЦР вставки человека в ВАС размером
а) Отпечаток пальцев рестриктов б) Отпечаток пальцев повторных ДНК
R R RRR R RR
2 Клонированные
фрагменты ДНК
| Рестрикция
1 2
.^у/общие фрагменты
рестрикции
R RRRR R RR
/ * 1 . Рассеянное по
2 । геному повторение
I
1 2
______Рестрикция, гибридизация по
Саузерну с повторением-зондом
в) ПЦР с повторной ДНК г) Картирование содержания STSob
STS
* — Последовательность Alu
| ПЦР с затравками Alu
1 2
. Общие продукты
----------' ПЦР
| STS-специфическая ПЦР
Рис. 4.15. Четыре метода дактилоскопии клонов
158 Часть 1. Изучение геномов
150 тыс. п. н., как можно ожидать, даст приблизительно 50 продуктов ПЦР различ-
ного размера, из которых можно получить подробный отпечаток пальцев.
• Картирование содержания STS особенно полезно, потому что оно может дать
сборку контигов из клонов, которая закреплена на физической карте местопо-
ложений STS. Эксперименты ПЦР, направленные на отдельные STS (раздел 3.3.3),
выполняются с каждым членом библиотеки клонов. При допущении о том, что STS
есть единственная копия в геноме, можно считать, что все клоны, которые дают
продукты ПЦР, должны содержать перекрывающиеся вставки.
Как и в случае обхода хромосомы, эффективное применение этих методов снятия
отпечатков пальцев требует комбинаторного отбора расположенных на координатной
сетке клонов, в идеале с компьютеризированной методологией анализа полученных
данных.
4.2.3. Секвенирование методом дробовика для полных геномов
Метод дробовика для полных геномов был впервые предложен Крейгом Бентером
и коллегами как относительно быстрое средство получения данных о непрерывной
последовательности крупных геномов, таких как геном человека и других эукариотов.
Опыт с секвенированием обычным методом дробовика (раздел 4.2.1) показал, что если
полная длина фрагментов последовательности составляет от 6,5 до 8 длин изучаемого
генома, то полученные контиги последовательности охватят более 99,8 % генома, с не-
сколькими пропусками, которые могут быть закрыты такими методами, как развитые
в ходе проекта расшифровки генома Haemophilus influenzae (см. рис. 4.11). Это под-
разумевает, что 70-ти миллионов отдельных последовательностей (каждая по 500 п.н.
или около того в длину), соответствующих общему количеству 35 000 млн п. н., было бы
достаточно, если бы случайный подход был предпринят с геномом млекопитающего
размером от 3 000 до 3 500 млнп.н. Семьдесят миллионов последовательностей — не
есть нечто невозможное: фактически с 60-ю автоматическими секвенаторами, каждый
из которых определяет 96 последовательностей за двухчасовой период, работающими
все дни напролет, такая задача может быть выполнена за три года.
Могут ли 70 миллионов последовательностей быть собраны правильно? Если
с таким большим количеством фрагментов используется обычный метод дробовика
и не делается никакого обращения к карте генома, то ответ — конечно нет. Огромное
количество компьютерного времени, необходимого для опознавания перекрытий между
этими последовательностями, и ошибки или в лучшем случае неопределенности, вызван-
ные значительным содержанием повторной ДНК в большинстве геномов эукариотов
(см. рис. 3.2), могут сделать такую задачу невыполнимой. Однако при условии ссылки
на карту вполне можно правильно собирать минипоследовательности.
Основные характеристики секвенирования методом дробовика для полных
геномов
Наиболее времязатратная часть проекта секвенирования методом дробовика —
стадия, на которой отдельные контиги последовательности объединяются путем за-
крытия кодовых пропусков и физических пропусков между ними (см. рис. 4.11). Чтобы
минимизировать число пропусков, которые необходимо закрывать, метод дробовика
для полных геномов использует по крайней мере две библиотеки клонов, приготовлен-
ные на основе векторов различного типа. Необходимо использовать как минимум две
библиотеки, потому что при любом клонирующем векторе ожидается, что некоторые
Глава 4. Секвенирование геномов 159
фрагменты не будут клонированы из-за проблем несовместимости, которые препятству-
ют размножению векторов, содержащих такие фрагменты. Векторы различных типов
страдают от разных проблем, так что фрагменты, которые не могут быть клонированы
в одном векторе, нередко могут быть клонированы, если используется второй вектор.
Поэтому восстановление последовательности из фрагментов, клонированных в двух
различных векторах, должно улучшить полноту покрытия генома.
Что можно сказать о проблемах, вызываемых при сборке последовательности
повторными элементами? Мы освещали этот вопрос в главе 3 как главный аргумент
против применения метода дробовика для секвенирования геномов эукариотов ввиду
возможности потери частей повторной области из-за перескоков между повторными
единицами и возможности неправильного соединения между двумя отдельными частя-
ми одной и той же хромосомы или различных хромосом (см. рис. 3.2). Было предложено
несколько возможных решений этой проблемы, но самая успешная стратегия состоит
в том, чтобы гарантировать, что одна из библиотек клонов содержит фрагменты, кото-
рые длиннее, чем самые длинные повторные последовательности в изучаемом геноме.
Например, одна из плазмидных библиотек, использовавшаяся, когда этот метод был
применен к геному Drosophila, содержала вставки со средним размером 10 тыс.п.н.,
потому что большинство повторных последовательностей Drosophila имеет размер
8 тыс.п.н. или меньше. Перескоки по последовательности генома, от одной повторной
последовательности до другой, исключаются за счет гарантии того, что две конечные
последовательности каждой вставки размером 10 тыс.п.н. находятся в своих соответ-
ствующих позициях в основной последовательности (рис. 4.16).
Первичный результат сборки последовательности — ряд каркасов (рис. 4.17, а),
где каждый каркас состоит из набора контигов последовательности, разделенных
кодовыми пропусками, которые лежат между считываниями спаренных концов —
минипоследовательностями с двух концов отдельного клонированного фрагмента — и,
таким образом, являются промежутками, которые могут быть закрыты дальнейшим
секвенированием этого фрагмента (рис. 4.17, б). Сами каркасы разделены физически-
Рис. 4.16. Избежание ошибок при исполь-
зовании метода дробовика для полных ге-
номов. На рис. 3.2, б мы видели, как легко
можно «перескочить» между повторами при
сборке основной последовательности стан-
дартным методом дробовика. Результатом
такой ошибки будет потеря всей последова-
тельности между этими двумя повторения-
ми, которые были ошибочно соединены вме-
сте. Метод дробовика для полных геномов
позволяет избежать ошибок такого типа за
счет гарантии того, что обе конечные после-
довательности клонированного фрагмента
ДНК (размера 10 тыс.п.н. или около того)
появляются на основной последовательности
в своих ожидаемых позициях. Если одна из
конечных последовательностей отсутствует,
то, значит, при сборке основной последова-
тельности была допущена ошибка
а) Правильная сборка последовательности
Рассеянные по
10 тыс. п. н. геному повторения
Последовательность
ДНК
Обе конечные последовательности
вставки размером 10 тыс. п. н. могут
быть размещены на основной
последовательности
б) Неправильная сборка последовательности
Последовательность между
повторениями была пропущена
-------------—-----------------Последовательность
'------------------------------ДНК
Лишь одна конечная последовательность
встречается в основной последовательности
160 Часть 1. Изучение геномов
а) Каркасы
КАРКАС 1
КАРКАС 2
Контиг последовательности Физический пропуск Кодовые пропуски
15 тыс. п. н.
Рис. 4.17. Первичный результат сборки последовательности при помощи метода дробовика для полных
геномов, а) Каркасы — промежуточные звенья в сборке последовательности методом дробовика для
полных геномов. Показано два каркаса. Каждый включает ряд контигов последовательности, разделен-
ных кодовыми пропусками, при этом сами каркасы отделены друг от друга физическими пропусками.
6) Кодовые пропуски лежат между считываниями спаренных концов — парой минипоследовательностей
с двух концов отдельного клонированного фрагмента — и поэтому могут быть закрыты дальнейшим
секвенированием клонированной ДНК
ми пропусками, которые гораздо труднее закрыть, потому что они представляют по-
следовательности, отсутствующие в библиотеках клонов. Для того чтобы определить
позицию каждого такого каркаса на карте генома, используется анализ содержания
в нем маркёров. Например, если известны местоположения STS на карте генома, то
каркас может быть помещен на нее, если будет установлено, какие STS он содержит.
Если каркас содержит STS из двух несмежных частей генома, то, значит, в ходе сборки
последовательности произошла ошибка. Точность сборки последовательности может
быть далее проверена за счет получения концевых последовательностей из фрагментов
длиной 100 тыс. п. н. или более, которые были клонированы в векторе большой емкости.
Если пара таких концевых последовательностей, закрепленных в их ожидаемых пози-
циях друг относительно друга, не попадает в пределы отдельного каркаса, то опять же
это означает, что произошла ошибка в сборке.
Осуществимость метода дробовика для полных геномов была продемонстриро-
вана его применением к геномам плодовой мушки и человека. Но все еще остаются не-
решенными вопросы о достоверности последовательностей генома, полученных этим
методом. Сравнения между двумя версиями генома человека (раздел 4.3) показали, что
последовательность, полученная методом дробовика для полных геномов, содержит
значительное число недостающих сегментов, в общем 160 млн п. н., причем эти сегменты
были утеряны из последовательности из-за проблем, вызванных повторной ДНК. В ре-
Глава 4. Секвенирование геномов 161
зультате этих ошибок 36 генов были полностью потеряны и еще 67 — потеряны частично.
Возникли также предположения о том, что последовательность, полученная методом
дробовика для полных геномов, может не иметь желательной степени точности, и даже
в тех областях, которые были собраны правильно. Отчасти эта проблема связана с тем,
что беспорядочный характер получения последовательности означает, что некоторые
части генома покрыты несколькими полученными минипоследовательностями, тогда
как другие части представлены только единожды или дважды (рис. 4.18). Общепринято,
что каждая часть генома должна быть секвенирована по крайней мере четыре раза, что-
бы гарантировать приемлемый уровень точности, и что такое покрытие должно быть
увеличено в 8-10 раз, прежде чем последовательность можно рассматривать как полную.
Последовательность, полученная методом дробовика для полных геномов, вероятно,
превышает это требование во многих областях, но в других областях может не достигать
этого уровня. Если в таких областях находятся гены, то недостаток точности может
причинить серьезные проблемы при попытках определить местонахождение генов
и понять их функции (см. главу 5). Во всей серьезности этих проблем помог убедиться
анализ черновой последовательности Drosophila, полученной методом дробовика для
полных геномов, на основании которого было предположено, что из 13 600 генов целых
6 500 могут содержать существенные ошибки кода последовательности.
ii^=s==^^^^====ss==========ssi Последовательность генома
а нм ям ав ш аав ав мв на ав на ав
“ Минипоследовательности
1 тыс. п. н.
Рис. 4.18. Беспорядочный характер получения последовательности методом дробовика для полных
геномов означает, что одни части генома покрыты большим количеством минипоследовательностей,
чем другие
4.3. Проекты расшифровки генома человека
Дабы подвести итог нашему обзору картирования и секвенирования геномов, мы
посмотрим, как эти методы были применены к геному человека. Хотя каждый проект
расшифровки генома отличается ото всех остальных и имеет свои собственные труд-
ности и свои собственные решения этих затруднений, проекты, связанные с человеком,
иллюстрируют общие вопросы, к которым приходилось обращаться, чтобы секвениро-
вать большой геном эукариота, и во многих случаях иллюстрируют процедуры, кото-
рые в настоящее время расцениваются как передовые в этой области молекулярной
биологии.
4.3.1. Стадия картирования в проекте «Геном человека»
До начала 1980-х гг. полагали, что подробная карта генома человека является недо-
сягаемой целью. Хотя к тому времени уже были построены полные генетические карты
плодовых мушек и нескольких других организмов, проблемы, свойственные анализу
человеческих родословных (раздел 3.2.4), и сравнительно малое число полиморфных
генетических маркёров вызывало у большинства генетиков глубокие сомнения в том,
что генетическая карта человека может когда-либо быть получена. Первая надежда на
успешное генетическое картирование человека затеплилась с открытием RFLP, которые
162 Часть 1. Изучение геномов
были первыми высокополиморфными ДНК-маркёрами, распознанными в геномах жи-
вотных. В1987 г. была опубликована первая карта RFLP человека, включающая 393 RFLP
и 10 дополнительных полиморфных маркёров. Эта карта, построенная по результатам
анализа 21 семейства, имела среднюю плотность маркёров один на 10 млнп.н.
В конце 1980-х гг. был учрежден проект «Геном человека» в форме самофинансируе-
мого, но контролируемого государственными организациями совместного предприятия,
в котором участвовали генетики со всего мира. Одна из целей, которые проект поста-
вил сам по себе, была генетическая карта с плотностью один маркёр на 1 млн п. н., хотя
думалось, что реальным пределом могла быть плотность один маркёр на 2-5 млнп.н.
Фактически к 1994 г. международный консорциум достиг поставленной цели и даже
превзошел ее благодаря использованию RFLP и образцовых семейств из обширной
коллекции СЕРН (раздел 3.2.4). Карта 1994 г. содержала 5 800 маркёров, из которых бо-
лее чем 4000 были представлены RFLP, и имела плотность один маркёр на 0,7 млнп.н.
Последующая версия в еще большей степени уточнила карту 1994 г. за счет включения
в нее дополнительных 1250 RFLP.
Физическое картирование не отставало от генетического. В начале 1990-х гг. зна-
чительные усилия были направлены на создание карт контигов из клонов при по-
мощи отбора STS (раздел 3.3.3) наряду с другими методами дактилоскопии клонов
(раздел 4.2.2). Главным достижением этой стадии проекта физического картирования
была публикация карты сборки контигов из клонов полного генома, состоящей из
33 000 YAC, содержащих фрагменты со средним размером 0,9 млнп.н. Однако, когда
стало понято, что клоны YAC могут содержать две или больше части несмежной ДНК,
зародились сомнения о ценности карт контигов YAC (рис. 4.19). Использование таких
химерных клонов при построении карт контигов может привести к тому, что сегменты
ДНК, которые далеко отделены друг от друга в геноме, по ошибке будут нанесены на
карту в соседних позициях. Исходя из этих проблем, был взят курс (главным образом
Уайтхедовским институтом и Центром исследований генома при Массачусетском тех-
нологическом институте) на картирование радиационных гибридов маркёров STS (раз-
дел 3.3.3), который привел в конечном итоге к публикации в 1995 г. карты STS человека,
содержащей 15086 маркёров, со средней плотностью один маркёр на 199 тыс.п.н. Эта
карта была позже дополнена дополнительными 20104 STS, большинство из которых
было представлено EST и, следовательно, показывало позиции кодирующих белок генов
на физической карте. Достигнутая плотность карты приблизилась к пределу — один
маркёр на 100 тыс.п.н., — поставленному в качестве цели физического картирования
Объединенные карты STS включали пози-
ции почти 7 000 полиморфных SSLP, которые
были нанесены на карту генома также и гене-
тическими средствами. В результате физиче-
ская и генетическая карты могли быть непо-
средственно сравнены, и карты сборки контигов
из клонов, которые включали данные STS, могли
быть позиционированы на обеих картах. Конеч-
ным результатом была полная, объединенная
карта, которая могла быть использована в каче-
стве опорной схемы для стадии секвенирования
ДНК в ходе проекта «Геном человека».
в начале проекта «Геном человека».
ДНК
Клон YAC
Рис. 4.19. Некоторые клоны YAC содержат
сегменты ДНК из различных частей генома
человека
Глава 4. Секвенирование геномов 163
4.3.2. Секвенирование генома человека
Первоначальный план состоял в том, что стадия секвенирования проекта «Геном
человека» будет основана на библиотеках YAC, потому что вектор этого типа может быть
использован с более длинными фрагментами ДНК, чем могут вместить клонирующие
системы любого другого типа. От этой стратегии пришлось отказаться, когда было
открыто, что некоторые клоны YAC содержат несмежные фрагменты ДНК. Поэтому
проект обратил свое внимание к ВАС (раздел 2.2.1). Была произведена библиотека из
300 000 клонов ВАС, и эти клоны были нанесены на карту генома, в результате чего по-
лучилась карта «подручной последовательности», которая могла быть использована
как первооснова для стадии проекта секвенирования, в ходе которой вставку из каждой
ВАС будут полностью секвенировать методом дробовика.
Приблизительно в то же время, когда проект «Геном человека» был готов к пере-
ходу на стадию получения данных о последовательности, метод дробовика для полных
геномов был впервые предложен в качестве альтернативы более трудоемкому методу
сборки контигов из клонов, который к тому времени был общепринятым. Опасения
организаторов проекта «Геном человека» в плане того, что полученная ими последова-
тельность генома человека фактически не будет первой в своем роде, подтолкнула их
перенести запланированные ими даты завершения рабочего проекта последователь-
ности на более ранний срок. Первая черновая последовательность полной хромосомы
человека (номер 22) была опубликована в декабре 1999 г., а несколько месяцев спустя
появилась черновая последовательность хромосомы 21. Наконец, 26 июня 2000 г., в при-
сутствии президента Соединенных Штатов Америки, Фрэнсис Коллинз и Крейг Бентер,
руководители этих двух тем проекта, совместно объявили о завершении рабочих про-
ектов последовательностей, которые появились в печати восемь месяцев спустя.
Важно понимать, что две последовательности генома, опубликованные в 2001 г.,
были черновыми рабочими проектами, а не полными окончательными последователь-
ностями. Например, версия, полученная методом сборки контигов из клонов, охватывала
только 90 % генома, отсутствующие 320 млнп.н. лежали преимущественно в струк-
турном гетерохроматине (раздел 10.1.2) — области хромосом, в которых ДНК очень
сильно упакована и которые, как думают, если и содержат какие-либо гены, то очень
мало. В пределах охваченных 90 % генома каждая часть последовательности была
секвенирована по крайней мере четыре раза, что обеспечило «приемлемый» уровень
точности, и лишь 25 % было секвенировано 8-10 раз, что может дать основания считать
работу «готовой». Более того, эта черновая последовательность имела приблизительно
150 000 пропусков, и было признано, что некоторые сегменты, по всей вероятности, были
упорядочены неправильно. Международный консорциум по секвенированию генома
человека, управлявший заключительной стадией проекта, поставил его целью получе-
ние готовой последовательности — то есть такой, у которой по крайней мере в 95 %
эухроматина — части генома, в которой расположено большинство генов, — частота
появления ошибок была бы менее одной на 104 нуклеотидов, и все пропуски, кроме
наиболее трудноизлечимых, были бы заполнены. Достижение этой цели потребовало
дальнейшего секвенирования 46 000 клонов на ВАС, РАС, YAC, фосмидах и космидах (раз-
дел 2.2.1). Первые готовые последовательности хромосом начали появляться в 2004 г.,
при этом завершение расшифровки полной последовательности генома ожидали годом
позже. Эта последовательность имела полную длину 2 850 млн п. н. и в ней недоставало
лишь 28 млн п. н. эухроматина (последние приходятся на 308 пропусков, которые до сих
пор не поддаются никаким попыткам закрытия).
164 Часть 1. Изучение геномов
4.3.3. Будущее проектов по изучению генома человека
Полное завершение расшифровки последовательности — не единственная цель
консорциумов, работающих с геномом человека. Грандиозная задача, в решение ко-
торой вовлечены многие группы по всему миру, с привлечением различных методов
и подходов, которые будут описаны в следующих двух главах, — постижение последо-
вательности генома. Важный среди этих подходов — использование сравнительной
геномики, в которой две полные последовательности генома сравниваются, с тем
чтобы идентифицировать общие особенности, которые, будучи консервативными, по
всей вероятности, очень важны (см. раздел 5.1.1). В отношении генома человека срав-
нительная геномика сверх того предлагает ценную возможность, состоящую в том, что
она позволяет обнаруживать гены животных, аналогичные генам болезней человека,
и тем самым прокладывает путь к изучению генетической основы этих болезней с ис-
пользованием генов животных в качестве моделей соответствующих генов человека.
Схемы геномов мыши и крысы были опубликованы в 2002 г., а черновая карта шимпанзе
была получена в 2005 г. Будут запущены дополнительные проекты расшифровки генома
человека, нацеленные на создание каталога изменчивости последовательности в раз-
личных популяциях; полученные результаты, возможно, позволят вывести древние
истоки этих популяций (раздел 19.3.2).
Такие проекты изучения разнообразия человека ведут нас к спорным моментам
секвенирования генома. Большинство ученых ожидает, что данные о последовательно-
сти из различных популяций подчеркнут единство человеческой расы, показывая, что
схемы генетической изменчивости не отражают принадлежности людей к тем ил и иным
географическим и геополитическим группам, сложившимся за несколько последних
столетий. Но, несмотря на это, результаты этих проектов, несомненно, вызовут дебаты
в ненаучных кругах. Кроме того, спорным является вопрос о том, кому (если это вообще
возможно) должны принадлежать последовательности ДНК человека. Многие относят
идею о праве собственности на последовательность ДНК исключительно к области
юридических формулировок, но на информации, содержащейся в геноме человека, могут
быть сделаны большие деньги, например, за счёт использования последовательности
генов в качестве основы для разработки новых лекарственных препаратов и терапий
против рака и других болезней. Фармацевтические компании, вовлеченные в секвени-
рование генома, естественно, хотят защитить свои инвестиции, как они действовали бы
в случае любого другого исследовательского проекта, и в настоящее время единственный
способ добиться этого — патентование открытых ими последовательностей ДНК. К сожа-
лению, в прошлом при проработке финансовых вопросов, относящихся к исследованиям
с биологическим материалом человека, были сделаны ошибки; и в итоге индивидуум,
от которого был получен материал, не всегда является стороной в разделении прибыли.
Эти вопросы до сих пор не решены.
Проблемы, касающиеся общественного использования последовательностей
генома человека, оказались еще более трудноразрешимыми. Главное опасение связа-
но с возможностью того, что как только последовательность генома человека будет
интерпретирована, индивидуумы, последовательности которых будут по какой-либо
причине сочтены «нестандартными», могут подвергнуться дискриминации. Прочие
опасения простираются от увеличенных страховых взносов для индивидуумов, по-
следовательности которых включают мутации, предрасполагающие их к той или
иной генетической болезни, до возможности того, что расисты могли бы попытаться
обозначить «хорошие» и «плохие» особенности последовательности с удручающе
Глава 4. Секвенирование геномов 165
предсказуемыми последствиями для индивидуумов, которым «посчастливится»
попасть в категорию «плохих».
Два проекта расшифровки генома человека, особенно в Соединенных Штатах, про-
должают поддерживать исследовательские и обсуждать этические, юридические и со-
циальные вопросы, поднятые секвенированием генома. В частности, большое внимание
уделяется обеспечению гарантии того, что последовательности генома, полученные
в ходе проектов, невозможно будет идентифицировать ни с каким отдельно взятым
индивидуумом. ДНК, которая клонируется и секвенируется, берется только от индиви-
дуумов, которые дали согласие на использование их материала таким способом и для
кого можно гарантировать анонимность. Когда начали проводить такую политику, потре-
бовался более или менее значительный пересмотр проекта научно-исследовательской
работы, потому что более старые библиотеки клонов должны были быть разрушены
и существующие физические карты проверены согласно новому материалу. Было при-
знано, однако, что эта дополнительная работа была необходима, чтобы сохранить
и приумножить доверие общества к этим проектам.
Заключение
Процедуры для быстрого секвенирования ДНК были впервые изобретены в 1970-х гг.
Версия, которая сегодня используется наиболее часто, — метод обрыва цепи, который
завоевал популярность благодаря возможности автоматизации, что позволяет выпол-
нять большое число отдельных экспериментов за короткий период времени. Это важно,
потому что один эксперимент дает не более 750 п.н. последовательности, так что для
того, чтобы получить последовательность полного генома, должны быть выполнены
тысячи, если не миллионы, отдельных экспериментов. Другие методы секвенирования
ДНК, такие как метод химической деградации и пиросеквенирование, имеют более
специализированные функции1^. При секвенировании генома главная трудность со-
стоит в сборке всех минипоследовательностей, полученных в ходе многочисленных
экспериментов секвенирования, в правильном порядке. С маленьким геномом бакте-
рии сборка последовательности возможна методом дробовика, который состоит всего
лишь в исследовании минипоследовательностей на предмет наличия перекрытий. Этот
подход, который не требует никакого предварительного знания генома, был впервые
использован в 1995 г. для генома Haemophilus influenzae размером 1830 тыс. п. н. и впо-
следствии стал стандартным методом для секвенирования бактериальных геномов.
Попытки применить этот подход к более крупным геномам эукариотов осложняются
присутствием повторных последовательностей ДНК, каковые могут привести к не-
правильной сборке сегментов генома. Метод сборки контигов из клонов избегает этих
проблем за счет опознавания ряда клонов (в векторе большой емкости типа ВАС), кото-
рые содержат перекрывающиеся фрагменты, уже позиционированные на физической
и (или) генетической карте изучаемого генома. Короткие сборки контигов из клонов
могут быть построены путем обхода хромосомы, но более длинные сборки, применяемые
в проектах секвенирования, обычно собираются различными методами дактилоскопии
клонов. Затем фрагменты, присутствующие в отдельных клонах, секвенируются методом
дробовика. Именно этот подход был принят официальным проектом «Геном человека»,
но когда этот проект приблизился к концу стадии секвенирования, Крейг Вентер с кол-
В настоящее время пиросеквенирование и другие платформы для массового параллельного секвени-
рования становятся стандартом, в то время как секвенирование по Сенгеру (методом обрыва цепи) становится
вспомогательным методом. — Прим. ред.
166 Часть 1. Изучение геномов
легами показали, что геном человека и другие большие геномы могут быть секвениро-
ваны намного быстрее методом дробовика для полных геномов, который основан на
том же самом подходе, что и стандартный метод дробовика, но включает несколько мер
безопасности, таких как твердая опора на физическую карту, чтобы гарантировать, что
последовательности, смежные с областями повторной ДНК, будут собраны правильно.
Сравнение двух черновых последовательностей человека позволило предположить, что
метод сборки контигов из клонов обеспечивает более точную последовательность, но
что метод дробовика для полных геномов, благодаря его быстроте, является лучшими
средством для получения начальной черновой карты генома. Проекты генома человека
теперь перешли к стадии, на которой были опубликованы законченные последователь-
ности хромосом, покрывающие по крайней мере 95 % эухроматина каждой хромосомы,
с коэффициентом ошибок не более одной на 104 нуклеотидов. Этические, юридические
и социальные вопросы, поднятые к моменту завершения работы над последователь-
ностью человека, включают вопросы относительно права собственности, патентного
права и возможности дискриминации по генетическим признакам.
Глава 4. Секвенирование геномов 167
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
4.1 *. Что случилось бы, если бы в реак-
ции секвенирования с обрывом цепи
была слишком высокая концентра-
ция дидезоксинуклеотидов?
а. Реакции дали бы очень длинные
молекулы и было бы мало данных
о последовательности вблизи от
затравки.
Ь. Реакции дали бы очень короткие
молекулы.
с. Реакции не продолжались бы,
поскольку при высоких концен-
трациях дидезоксинуклеотидов
ингибируется ДНК-полимераза.
d. Флюоресценция продуктов секве-
нирования была бы слишком вы-
сока и трудна для считывания.
4.2 . Как помечают различные нуклеоти-
ды (А, С, G или Т) в реакции секвени-
рования с обрывом цепи?
а. Затравки для реакций метят флю-
оресцентными красителями.
Ь. Различные дезоксинуклеотиды
метят разными флюоресцентны-
ми красителями.
с. Различныедидезоксинуклеотиды
метят разными флюоресцентны-
ми красителями.
d. Различные продукты секвениро-
вания окрашивают антителами,
которые обнаруживают различ-
ные дидезоксинуклеотиды.
4.3 *. Почему до секвенирования фрагмен-
та ДНК с обрывом цепи выгодно его
клонировать?
а. Процесс секвенирования с обры-
вом цепи требует однонитевые
молекулы ДНК в качестве матриц.
Ь. Процесс секвенирования с обры-
вом цепи требует двунитевые мо-
лекулы ДНК в качестве матрицы.
с. Процесс секвенирования с обры-
вом цепи требует вектор, чтобы
стабилизировать ДНК-матрицу.
d. Дидезоксинуклеотиды встраи-
ваются только в клонированные
фрагменты ДНК.
4.4 . Почему фермент Клёнова является
плохим выбором для реакций секве-
нирования с обрывом цепи?
а. Фермент обладает сильным дей-
ствием экзонуклеазы 5’->3' и из-
меняет длину продуктов.
Ь. Фермент имеет сильное действие
экзонуклеазы 3'—>5' и удаляет
З'-дидезоксинуклеотиды из про-
дуктов.
с. Фермент не включает дидезокси-
нуклеотиды в матричную цепь.
d. Фермент обладает низкой про-
цессивностью, что ограничивает
длину полученной последова-
тельности.
4.5 *. Какая из перечисленных особен-
ностей секвенирования с обрывом
цепи является проблемой для спе-
циалистов?
а. Считывания последовательности
менее 100 п.н.
Ь. Последовательностичастосодер-
жат ошибки.
с. Внутринитевое соединение осно-
ваний может блокировать продви-
жение ДНК-полимеразы и, наряду
с этим, затруднить перемещение
молекул в ходе электрофореза.
d. Невозможно секвенировать обе
нити молекулы ДНК.
4.6 . Каково назначение нуклеотидазы
в реакции пиросеквенирования?
а. Она преобразует пирофосфат
в люминесцентный продукт.
168 Часть 1. Изучение геномов
Ь. Она деградирует молекулу ДНК,
высвобождая нуклеотиды, кото-
рые детектируются посредством
хемилюминесценции.
с. Она стабилизирует короткие про-
дукты ДНК, полученные этим ме-
тодом.
d. Она деградирует невстроенные
нуклеотиды, находящиеся в реак-
ционной смеси.
4.7 *. Многие ученые сомневались в том,
что секвенирование методом дробо-
вика сработает даже с наименьшими
геномами, потому что:
а. Не будет перекрытий между раз-
личными минипоследователь-
ностями.
Ь. Компьютеры не справятся с об-
работкой огромного количества
данных, произведенных проек-
том секвенирования методом
дробовика.
с. Маленькие геномы прокариотов
содержат большие количества по-
вторной ДНК.
d. Не существовало никакого метода
для разбиения геномной ДНК на
случайные фрагменты.
4.8 . Почему метод сборки контигов из
клонов полезен для секвенирования
геномов эукариотов?
а. Геномы попросту являются слиш-
ком большими для секвенирова-
ния методом дробовика.
Ь. Повторные последовательности
геномов эукариотов сделали бы
сборку контигов, полученных
исключительно методом дробо-
вика, трудной и подверженной
ошибкам задачей.
с. Попросту при использовании ме-
тода дробовика получилось бы
слишком много рекомбинантных
плазмид, которые надо было бы
выделять.
d. Метод сборки контигов из клонов
облегчает исследователям задачу
опознавания генов.
4.9 *. Выберите наилучшее описание об-
хода хромосомы:
а. Выравнивание последовательно-
стей ДНК компьютером, с тем что-
бы произвести сборку контигов.
Ь. Последовательноевыстраивание
карты хромосомы.
с. Опознавание клонов, вставки
которых перекрываются, с тем
чтобы произвести библиотеку
клонов, которые покрывают за-
данный сегмент ДНК.
d. Секвенирование генома по одно-
му клону за раз, чтобы гаранти-
ровать полное отсутствие про-
пусков в последовательности
к концу проекта.
4.10 . На чем из нижеследующего основа-
но позиционное клонирование?
а. Обход хромосомы от маркёра до
близлежащего гена.
Ь. Сборка контигов из клонов для
восстановления полного генома.
с. Идентификация генов, присут-
ствующих в геномных последо-
вательностях.
d. Снятие отпечатков пальцев у хро-
мосомы или фрагмента ДНК, что-
бы построить карту для секвени-
рования.
4.11 *. Какие приемы и условия исследо-
ватели обычно используют, чтобы
гарантировать, что в черновой по-
следовательности генома будет при-
сутствовать только минимальное
число кодовых и физических про-
пусков?
а. Общее число секвенированных
нуклеотидов соответствует раз-
меру генома.
Ь. Полный геном клонируется обхо-
дом хромосом, чтобы гарантиро-
вать полное покрытие.
с. Нет необходимости минимизиро-
вать число пропусков, поскольку
они легко заполняются после на-
чальной стадии секвенирования.
Глава 4. Секвенирование геномов 169
d. По крайней мере две библиотеки
клонов приготовляют и секвени-
руют в различных векторах.
4.12 . Какова самая трудная и времяза-
тратная часть проекта секвениро-
вания методом дробовика?
а. Получение библиотек клонов.
Ь. Секвенирование библиотек кло-
нов.
с. Получение сборки контигов из
последовательностей ДНК.
d. Закрытие кодовых и физических
пропусков между секвенирован-
ными сборками контигов.
4.13 *. Какой из следующих методов по-
зволил получить наилучшую кар-
ту генома человека к середине
1990-х гг.?
а. Генетическое картирование
RFLP.
b.Генетическое картирование
SSLP.
с. Физическое картирование STS.
d. Физическое картирование FISH.
4.14 . Почему дрожжевые искусственные
хромосомы (YAC) не использовались
на стадии секвенирования в ходе
проекта «Геном человека»?
а. Было обнаружено, что некоторые
Вопросы открытого типа
4.1 *. Какова функция дидезоксинуклео-
тидов в реакции секвенирования
с обрывом цепи?
4.2 . Как различные продукты секвени-
рования с обрывом цепи детектиру-
ются в ходе электрофореза?
4.3 *. Возможно ли секвенировать про-
дукт ПЦР непосредственно (не кло-
нируя его)? Как этого можно было
бы достичь?
4.4 . Какие три свойства должны иметь
ДНК-полимеразы, используемые
для секвенирования ДНК?
YAC содержат клонированные
фрагменты ДНК из различных
частей генома.
Ь. Вставки YAC были слишком боль-
шими, чтобы позволить собрать
поддающуюся управлению би-
блиотеку клонов для секвениро-
вания.
с. Было открыто, чтов YAC дрожже-
вая геномная ДНК рекомбинирует
с человеческой ДНК.
d. Было установлено, что через
какое-то время YAC теряют боль-
шие сегменты вставленной ДНК.
4.15 *. Какое из следующих заявлений
о завершении последовательности
генома человека в 2000 г. является
ЛОЖНЫМ?
а. В то время было секвенировано
только 90 % генома.
Ь. Последовательности генома,
опубликованные в 2000 г., были
черновыми планами последова-
тельности.
с. Была полностью расшифрована
вся последовательность эухро-
матина.
d. Значительное количество струк-
турного гетерохроматина еще не
было секвенировано.
4.5* . Каковы выгоды автоматизирован-
ных версий процедуры секвениро-
вания с обрывом цепи?
4.6. Каковы возможности применения
и каковы ограничения пиросекве-
нирования?
4.7* . Почему секвенирование методом
дробовика требует, чтобы число сек-
венируемых нуклеотидов было в не-
сколько раз больше, чем необходимо
для полного покрытия генома?
4.8. Объясните сущность секвенирова-
ния генома методом сборки конти-
гов из клонов.
170 Часть 1. Изучение геномов
4.9* . Какие методы могут быть использо-
ваны для снятия отпечатков паль-
цев ДНК при работе с клонами из
большого фрагмента ДНК, который
должен быть секвенирован?
4.10 . Каким образом при сборке последо-
вательностей генома используются
каркасы?
Изыскательские задачи
4.1* . В конце 1970-х гг. методы обрыва
цепи и химической деградации для
секвенирования ДНК, как оказалось,
были одинаково эффективными.
Но сегодня фактически все секве-
нирование проводится методом об-
рыва цепи. Почему секвенирование
с обрывом цепи стало преобладаю-
щим?
4.2. Вы выделили бактерию нового вида,
геном которой представлен един-
ственной молекулой ДНК размером
около 2,6 млн п. н. Напишите подроб-
ный план проекта секвенирования
генома этой бактерии, из которого
были бы видны все этапы, ведущие
к получению его последовательно-
сти.
4.3* . Критически оцените подход сборки
контигов из клонов как средство
секвенирования больших геномов
эукариотов.
4.11 *. Ошибки какого типа сопряжены
с секвенированием сложных гено-
мов эукариотов методом дробови-
ка?
4.12 . Какие выгоды будут получены из
сравнения геномов мышей, крыс
и шимпанзе с геномом человека?
4.4. Обсудите, как проект «Геном чело-
века» развивался со времени, когда
исследователи впервые помыслили
о создании карты генома, и до за-
вершения расшифровки последова-
тельности генома. Каковы главные
достижения, которые позволили
исследователям создать подробную
карту генома и затем закончить рас-
шифровку последовательности?
4.5* . Фармацевтическая компания инве-
стировала много времени и денег,
чтобы выделить ген генетической
болезни. Компания изучает ген и его
белковый продукт и работает над
созданием лекарственных препара-
тов, чтобы лечить эту болезнь. Име-
ет ли эта компания право (по вашему
мнению) запатентовать этот ген?
Обоснуйте ваш ответ.
Глава 4. Секвенирование геномов 171
Тесты по рисункам
4.1 *. Каковы преимущества использования универсальных или внутренних затравок
в проектах секвенирования?
а) Универсальная затравка
Затравка
Векторная ДНК Вставка ДНК
—е
Векторная ДНК
б) Внутренние затравки
= Универсальная затравка
Внутренняя затравка
4.2 . Метод секвенирования в тепловом цикле обычно используется для секвениро-
вания ДНК. Каковы выгоды от применения этого процесса?
ДНК-матрица
ПЦР с одной-единственной
затравкой
ДДА
Нити с обрывом цепи - их число
возрастает при выполнении большего числа циклов
172 Часть 1. Изучение геномов
4.3 *. К какому типу относится данная реакция секвенирования? Каковы преимущества
секвенирования этим способом?
а) Мечение ДНК и разъединение нитей
Тяжелая
б) Реакция «G» в) Считывание последовательности
с авторадиограммы
Молекула для секвенирования
(множество копий)
Меченый 5’-конец
| ДМСО, 90 °C
| Диметилсульфат
Ме
Me
Me
I Электрофорез
* в агарозном геле
.=- Очистка одной
—' из нитей
| Пиперидин
4.4. Обсудите, каким образом различные методы, представленные на рисунке, могут
быть употреблены с целью получения отпечатков пальцев клонов.
а) Отпечаток пальцев рестриктов б) Отпечаток пальцев повторных ДНК
R RRRR R RR
1 2 Клонированные
фрагменты ДНК
| Рестрикция
1 2
Общие фрагменты
рестрикции
R R RRR R RR
Рассеянное по
2 । геному повторение
Рестрикция, гибридизация по
Саузерну с повторением-зондом
в) ПЦР с повторной ДНК
г) Картографирование содержания STSob
STS
д * '» ** Последовательность Alu *
| ПЦР с затравками Alu
1 2
-=i------/Эбщие продукты
—---------' ПЦР
| STS-специфическая ПЦР
1 2
“ — / Общие STSbi
Глава 4. Секвенирование геномов 173
Дополнительная литература
Методология секвенирования ДНК
Brown,Т.А. (ed.) (2000) Essential Molecular Biology: A Practical Approach, Vol. 1 and 2,
2nd Ed. Oxford University Press, Oxford. Includes detailed protocols for DNA sequencing.
Maxam, A. M. and Gilbert, W. (1977) A new method for sequencing DNA. Proc. Natl Acad.
Sci. USA 74: 560-564. The chemical degradation method.
Prober, J. M., Trainor, G.L., Dam,R.J., Hobbs, F.W., Robertson, C.W., Zagursky,R.J.,
Cocuzza, A. J., Jensen, M. A. and Baumeister, K. (1987) A system for rapid DNA sequencing
with fluorescent chain-terminating dideoxynucleotides. Science 238: 336-341. The chain
termination method as used today.
Rogers, Y.-H. and Venter, J. C. (2005) Massively parallel sequencing. Nature 437:326-327.
Describes how over one million sequencing reactions can be carried out in parallel.
Ronaghi,M., Ehleen, M. and Nyrn,P. (1998) A sequencing method based on real-time
pyrophosphate. Science 281: 363-365. Pyrosequencing.
Sanger, F., Nicklen, S. and Coulson, A. R. (1977) DNA sequencing with chain terminating
inhibitors. Proc. Natl Acad. Sci. USA 74: 5463-5467. The first description of chain termination
sequencing.
Sears, L.E., Moran, L.S., Kisinger, C., Creasey,T., Perry-O'Keefe, H., Roskey,M.,
Sutherland, E. and Slatko,B.E. (1992) CircumVent thermal cycle sequencing and alternative
manual and automated DNA sequencing protocols using the highly thermostable Vent (exo")
DNA polymerase. Biotechniques 13: 626-633.
Примеры употребления метода дробовика для сборки последовательностей
Fleischmann, R. D., Adams, M.D., White, О., et al. (1995) Whole-genome random
sequencing and assembly of Haemophilus influenzae Rd. Science 269: 496-512.
Fraser, C.M., Gocayne, J.D., White, O., et al. (1995) The minimal gene complement of
Mycoplasmagenitalium. Science 270: 397-403.
Подход сборки контигов из клонов
IHGSC (International Human Genome Sequencing Consortium) (2001) Initial
sequencing and analysis of the human genome. Nature 409: 860-921.
Метод дробовика для полных геномов
Adams, М. A., Celniker, S. Е., Holt, R. A., etal. (2000) The genome sequence of Drosophila
melanogaster. Science 287: 2185-2195.
She, X., Jiang, Z., Clark, R. A., et al. (2004) Shotgun sequence assembly and recent segmental
duplications within the human genome. Nature 431: 927-930. Determines the accuracy of the
whole-genome shotgun approach in assembly of sequences containing repetitive DNA.
Venter,). C., Adams, M. D., Sutton, G. G., Kerlavage, A. R., Smith, H. O. and Hunkapiller, M.
(1998) Shotgun sequencing of the human genome. Science 280:1540-1542.
Venter, J. C., Adams, M. D., Myers, E. W., et al. (2001) The sequence of the human genome.
Science 291:1304-1351.
Weber,). L. and Myers, E. W. (1997) Human whole-genome shotgun sequencing. Genome
Res. 7: 401-409.
174 Часть 1. Изучение геномов
Основные вехи в развитии проектов расшифровки генома человека
Donis-Keller, Н., Green, Р., Helms, С., et al. (1987) A genetic map of the human genome.
Cell 51: 319-337. The first genetic map with a marker density of one per 10 Mb.
Cohen, D., Chumakov, I. and Weissenbach, J. (1993) A first-generation map of the human
genome. Nature 366: 698-701. The first YAC contig map.
Murray, J. C., Buetow, K.H., Weber, J. L., etal. (1994) A comprehensive human linkage
map with centimorgan density. Science 265; 2049-2054. The genetic map with a density of one
marker per 0.7 Mb.
Hudson, T. J., Stein, L.D., Gerety,S.S., et al. (1995) An STS-based map of the human
genome. Science 270:1945-1954. The physical map with a density of one marker per 199 kb.
Dib,C., Faure, S., Fizames,C., et al. (1996) A comprehensive genetic map of the human
genome based on 5 264 microsatellites. Nature 380:152-154. The most comprehensive genetic
map.
Schuler, G. D., Boguski, M. S., Stewart, E. A., etal. (1996) A gene map of the human genome.
Science 274: 540-546. Refinement of the Hudson map, marker density close to one per 100 kb.
Deloukas,P., Schuler,G.D., Gyapay,G., et al. (1998) A physical map of 30000 genes.
Science 282: 744-746. The integrated map used as the framework for DNA sequencing.
IHGSC (International Human Genome Sequencing Consortium) (2001) Initial
sequencing and analysis of the human genome. Nature 409: 860-921. The draft sequence
obtained by the «official» Human Genome Project
Venter, J. C., Adams, M.D., Myers, E.W., et al. (2001) The sequence of the human
genome. Science 291: 1304-1351. The draft sequence obtained by the whole-genome shotgun
approach.
IHGSC (International Human Genome Sequencing Consortium) (2004) Finishing the
euchromatic sequence of the human genome. Nature 431: 931-945. Review of the finishing
process and its outcomes.
Ross,M.T., Grafham,D. V., Coffey, A. J., et al. (2005) The DNA sequence of the human
X chromosome. Nature 434: 325-337. Description of the finished sequence of a human
chromosome.
Этические, юридические и социальные вопросы, связанные с последователь-
ностями генома человека
Davies,К. (2001) Cracking the Genome: Inside the Race to Unlock Human DNA. Free
Press, New York. (Published in the UK as The Sequence: Inside the Race for the Human Genome.
Weidenfeld and Nicholson, London.) A history of the human genome projects.
Garver, K.L. and Garver, B. (1994) The Human Genome Project and eugenic concerns.
Am. J. Hum. Genet 54:148-158.
Wilkie,T. (1993) Perilous Knowledge: The Human Genome Project and its Implications.
Faber and Faber, New York. A view of the social impact of the Human Genome Project
Глава 5
Определение структуры
последовательности генома
и функций, ее составляющих
По прочтении главы 5 вы сможете:
• Описывать сильные и слабые стороны вычислительных и экспериментальных
методов, применяемых для анализа последовательностей геномов.
• Описывать идеи, лежащие в основе поиска открытых рамок считывания (ОРС,
ORF), и объяснять, почему этот подход не всегда успешен при определении ме-
стоположения генов в геномах эукариотов.
• Объяснять, как гены, кодирующие функциональную РНК, расположены в по-
следовательности генома.
• Давать определение термину «гомология» и объяснять, как гомология и срав-
нительная геномика используются для определения местонахождения генов
в последовательности генома.
• Характеризовать различные экспериментальные методы, применяемые для
опознавания частей последовательности генома, которые кодируют молекулы
РНК.
• Оценивать сильные стороны и ограничения анализа гомологии как средства
приписывания функций к генам.
• Описывать методы, применяемые для инактивации или сверхэкспрессии от-
дельных генов дрожжей и млекопитающих, и объяснять, каким образом эти
методы могут обеспечить распознавание функции гена.
• Приводить схематичные описания методов, которые могут быть использованы
для получения более подробной информации о деятельности белка, кодируемого
неизвестным геном.
• Давать сводку применяемых методов и основных достижений в области анно-
тирования последовательности генома Saccharomyces cerevisiae.
Последовательность генома не есть самоцель. Главная задача, которую все еще
предстоит решить, состоит в понимании того, что именно содержит геном и как геном
экспрессируется. Попытки понять, что геном содержит, осуществляются с применением
комбинации компьютерного анализа и экспериментальных методов и ставят основны-
ми целями определение местоположения генов и установление их функций. Эта глава
посвящена методам, применяемым для решения этих вопросов. Вторая задача — по-
нимание механизма экспрессии генома — есть в некоторой степени не что иное, как
одна из формулировок цели молекулярной биологии, которую она ставила перед собой
176 Часть 1. Изучение геномов
последние 30 лет. Отличие состоит в том, что в прошлом внимание было направлено на
пути экспрессии отдельных генов, притом что группы генов рассматривались только
в том случае, если экспрессия одного гена явно была связана с таковой другого. Теперь
этот вопрос стал более общим и относится к экспрессии генома в целокупности. Методы,
применяемые для решения данной задачи в таком ракурсе, будут освещены в главе 6.
5.1. Определение местоположения генов в последовательно-
сти генома
Как только последовательность ДНК будет установлена, будь это последователь-
ность отдельного клонированного фрагмента или полной хромосомы, то могут быть
применены различные методы для определения местоположения генов, которые в ней
присутствуют. Эти методы могут быть подразделены на такие, которые подразумева-
ют простой просмотр последовательности с помощью зрения или, чаще, компьютера,
с тем чтобы отыскать специфические особенности последовательности, соотносимые
с генами, и на такие методы, которые определяют местонахождение генов путем экс-
периментального анализа последовательности ДНК. Компьютерные методы составляют
часть методологии, названной биоинформатикой, и именно с них мы и мы начнем
наш экскурс.
5.1.1. Картирование генов с помощью анализа последовательности
Просмотр последовательности может быть использован для определения местона-
хождения генов, потому что гены представляют собой не случайный ряд нуклеотидов,
но напротив, имеют отличительные особенности. В настоящее время мы до конца не
понимаем природу всех этих специфических особенностей, и просмотр последователь-
ности поэтому не является безусловно надежным способом определения местонахож-
дения генов, но все же это мощный инструмент и, как правило, первый метод, который
применяют к анализу новой последовательности генома.
Кодирующие области генов суть открытые рамки считывания
Ген
|ATG GGC AGT GCA-]------------|ТТТ TAg|
Открытая рамка считывания
Гены, кодирующие белки, включают открытые рамки считывания (ОРС, ORF), со-
стоящие из ряда кодонов, которые определяют аминокислотную последовательность
белка, кодируемую этим геном (рис. 5.1). ОРС начинается со старт-кодона — обычно
(но не всегда) ATG — и заканчивается
стоп-кодоном: TAA, TAG или TGA (раз-
дел 1.3.2). Поэтому один из способов
поиска генов — отыскание в последо-
вательности ДНК ОРС, которые начи-
наются кодоном ATG и заканчиваются
стоп-кодоном. Такой анализ усложняе-
стя тем фактом, что каждая последо-
вательность ДНК имеет шесть рамок
считывания — три в одном направле-
нии и три в обратном направлении на
комплементарной нити (рис. 5.2), но
компьютеры вполне способны к про-
Рис. 5.1. Кодирующий белок ген — открытая рамка
считывания триплетных кодонов. Показаны первые
четыре и последние два кодона этого гена. Первые
четыре кодона определяют метионин/инициация-гли-
цин-серин-аланин, а последние два определяют
фенилаланин-стоп
Глава 5. Определение структуры последовательности генома 177
GAC—4
TGA—►
ATG—4
Г-ATG ACGAGAGAGCAGCCATTTTAG-3'
З'—TACTGCTCTCTCGTCGGTAAAATC— 5'
4—АТС
>-ААТ
4—AAA
Рис. 5.2. Молекула двунитевой ДНК имеет шесть рамок
считывания. Обе нити считываются в направлении 5'->3'.
Каждая нить имеет три рамки считывания, — зависящие
от того, который из нуклеотидов выбран в качестве стар-
товой позиции
смотру всех шести рамок считывания
с целью отыскания ОРС. Насколько
это эффективно как средство уста-
новления местоположения гена?
Ключ к успеху просмотра на
ОРС — частотность, с которой стоп-
кодоны встречаются в последова-
тельности ДНК. Если ДНК имеет
случайную последовательность и со-
держание GC в ней составляет 50 %,
то каждый из трех стоп-кодонов —
TAA, TAG и TGA — будет встречаться
в среднем единожды на каждые 43=
= 64 п.н. Если содержание GC боль-
ше чем 50 %, то стоп-кодоны, будучи
обогащенными АТ, будут встречаться
реже, но тем не менее будет ожидать-
ся один стоп-кодон на каждые 100-200 п. н. Это означает, что случайная ДНК не должна
показывать много ОРС длиннее 50 кодонов, особенно если присутствие стартового
триплета ATG используется как один из факторов определения ОРС. С другой стороны,
большинство генов длиннее 50 кодонов: средняя длина — 317 кодонов у Escherichia coli,
483 кодона у Saccharomyces cerevisiae и приблизительно 450 кодонов у человека. Поэтому
просмотр на ОРС, в простейшем виде, принимает цифру, скажем, 100 кодонов как самую
короткую длину предполагаемого гена и регистрирует положительные результаты для
всех ОРС длиннее этого значения.
Насколько хорошо эта стратегия работает на практике? В случае геномов бактерий
простой просмотр на ОРС является эффективным способом определения местонахожде-
ния большинства генов в последовательности ДНК. Это иллюстрировано на рис. 5.3, на
котором показан сегмент генома Е. coli, где все ОРС длиннее 50-ти кодонов подчеркнуты.
Истинные гены в последовательности не могут быть ошибочно пропущены, потому что
они намного длиннее 50-ти кодонов. В случае бактерий анализ еще более упрощен тем
фактом, что гены очень близко расположены друг к другу, и, следовательно, в геноме
имеется относительно мало межгенной ДНК (только 11 % у Е. coli', см. раздел 8.2.1). Если
мы делаем допущение о том, что истинные гены не перекрываются (а это справедливо
для большинства генов бактерий), то только в межгенных областях есть возможность
принятия короткой ложной ОРС за истинный ген. Так что если доля межгенной состав-
ляющей генома мала, то и вероятность ошибочной интерпретации результатов простого
просмотра на ОРС1) будет невысока.
В случае ДНК высших эукариотов простые просмотры на ОРС оказываются
менее эффективными
Хотя в случае геномов бактерий просмотры на ОРС работают хорошо, для опреде-
ления местонахождения генов в последовательностях ДНК высших эукариотов они
менее эффективны. Отчасти это связано с тем, что в геноме эукариотов есть существен-
^Здесь есть один источник ошибок — кодон ATG кодирует метионин и может встретиться внутри
гена, равно как по случайным причинам может встретиться и вне гена. Поэтому точное картирование старта
трансляции не очевидно. С другой стороны, существуют и очень короткие гены (до 7 кодонов), эти гены не-
возможно обнаружить описанным способом. — Прим. ред.
178 Часть 1. Изучение геномов
2 ’ М I i М I I i i I I I I M 1 E 3 I 8 5 3 s 3 5 1 ? 3 3 3 I 3 3 H JI 1 1 1 1 II 1 1 II II i H II 111111 11 1 I 11II 1 II II 1 1 1 1 HI II 1 I M H И H i i N н I И 1 I | 1 I II ! 1 I II I 1 I I Hi! Il I : 111 I 11 l. 11 ll i i,l. 111 H i 11 1 a i I I i I i I e § ! I M II MM! ! I I M as 3 3 3 3 3 3 I 3 5 I 5 I I i t | | I | 1 M | I Il lill 1 1 ill i 1 1,1 1 И hll 1 1 1 Illi 3 3 1 , 1 ё I | 3 I MH H I I M 1 ? 1 i Mil 1 I i ! И 1 ! 1 ? M ! I M I I ! | I si 1 M M M 1 1 3 M 1 MM II I 1 II M I M M M II I I i 3 1 i I I i lill MM! 1 MM M 1 1 i M 1 I II M H M H M Hi MM । | м ! 1 I II 1 1 1 1 M ! - i i 1 1 i МММ; M MM ! з 3 з з I 1 ? I I 3 ! ? I 33 3 3 3 3 3 3 s 8 3 3 ё s M MM i M M III I i I M h M ! I M I 4 I I м г H H M I I I и П H И II 1 1 1 M ! 1 II 1 ! 1 1 i II i i II I
Рис. 5.3. Просмотр на ОРС — эффективный способ определения местонахождения генов в геноме бакте-
рии. На схеме показан отрезок оперона лактозы Escherichia coli длиной 4 522 п. н., где отмечены все ОРС
длиннее 50 кодонов. Последовательность содержит два истинных гена — lacZ и lacY, — обозначенных
красными линиями. Эти истинные гены не могут быть опознаны ошибочно, потому что они намного
длиннее, чем ложные ОРС, показанные желтым цветом. См. рис. 8.8, а, на котором представлена под-
робная структура оперона лактозы
но больше места между истинными генами (например, приблизительно 62 % генома
человека приходится на межгенную ДНК), что увеличивает вероятность отыскания
ложных ОРС. Но главная проблема с геномом человека и с геномами высших эукариотов
в общем состоит в том, что их гены часто разделены интронами (раздел 1.2.3) и, таким
образом, встречаются в последовательности ДНК не в виде непрерывных ОРС. Многие
экзоны короче 100 кодонов, некоторые состоят из менее чем 50-ти кодонов, и продол-
жение рамки считывания в интрон обычно ведет к стоп-кодону, который, как известно,
завершает ОРС (рис. 5.4). Другими словами, гены высшего эукариота присутствуют
в последовательности генома не в виде длинных ОРС, и простой просмотр на ОРС не
позволяет определить их местонахождение.
Решение проблемы, связанной с интронами, — главнейшая задача для биоин-
форматиков, пишущих новые программы для отыскания местоположения ОРС. Были
приняты три модификации основной процедуры просмотра на ОРС.
• Отклонение частоты использования кодонов принимается во внимание. «От-
клонение частоты кодонов» относится к тому факту, что не все кодоны использу-
ются с одинаковой частотой в генах определенного организма. Например, согласно
генетическому коду лейцин определяется шестью кодонами (ТТА, TTG, СТТ, СТС,
СТА и CTG; см. рис. 1.20), но в генах человека лейцин наиболее часто кодируется
триплетом CTG и только изредка описывается кодонами ТТА или СТА. Подобным
же образом из четырех кодонов валина гены человека используют триплет GTG
в четыре раза чаще, чем триплет GTA. Биологическая причина отклонения частоты
Глава 5. Определение структуры последовательности генома 179
Экзон Интрон Экзон
ATGGGCAATGCAAGGTACGGTGAGCAGGTAAGTGATTAATGCATTTCTCGCAGTGGCTAGACGATGCATAG
М G N A R Y G Е Q ....................... WL D D А ♦
MGNARYGEQ V S D *
Рис. 5.4. Просмотры на ОРСусложнены интронами. Показана последовательность нуклеотидов короткого
гена, содержащего единственный интрон. Правильная аминокислотная последовательностьбелка, транс-
лированного с этого гена, представлена непосредственно под последовательностью нуклеотидов: в этой
последовательности интрон не показан, потому что он был удален из транскрипта до трансляции мРНК
в белок. В нижней линии последовательность была транслирована без учета присутствия в ней интрона.
В результате этой ошибки последовательность аминокислот оказалась законченной в пределах интро-
на. Последовательности аминокислот были записаны с помощью однобуквенного кода (см. табл. 1.2).
Звездочки указывают позиции стоп-кодонов. Интроны будут подробно рассмотрены в разделе 12.2.2
использования кодонов не ясна, но все организмы имеют такое отклонение, которое
различно у разных видов. Истинные экзоны ожидаемо показывают отклонение
кодонов, тогда как случайный ряд триплетов — нет. Поэтому в программное обе-
спечение поиска ОРС заложены характеристики отклонения частоты кодонов,
свойственные изучаемым организмам.
• Интрон-экзонные границы могут отыскиваться, поскольку они имеют отличи-
тельные особенности последовательности, хотя, к сожалению, различительная
сила этих последовательностей не является столь большой, чтобы сделать поиск
их местоположения тривиальной задачей. Последовательность вышерасположен-
ной интрон-экзонной границы [в начале интрона, донорный сайт. — Прим, ред.]
обычно описывается как
5'-AG4<GTAAGT-3',
где стрелка указывает точную точку границы. Однако только пара «GT», стоящая непо-
средственно за стрелкой, является неизменной: где-либо в другом месте последователь-
ности другие нуклеотиды, вместо показанных в данном случае, встречаются довольно
часто. Другими словами, такая последовательность есть консенсус. Это подразумевает,
что последовательность в каждой своей позиции показывает нуклеотид, встречающийся
наиболее часто в соответствующей позиции во всех известных вышерасположенных
интрон-экзонных границах, но что в любой конкретной граничной последовательности
одна или более из таких позиций может иметь отличный от стоящих в других последова-
тельностях нуклеотид (рис. 5.5). Нижерасположенная интрон-экзонная граница [конец
интрона, акцепторный сайт. — Прим, ред.] определена еще менее четко:
5'-PyPyPyPyPyPyNCAGJ—3',
где «Ру»1) означает один из пиримидиновых нуклеотидов (Т или С), a «N» — любой ну-
клеотид. Простой поиск таких консенсусных последовательностей не позволит опреде-
лить местонахождение больше чем нескольких интрон-экзонных границ, потому что
большая их часть имеет не такие последовательности, как выбранная в качестве при-
мера. Создание соответствующего программного обеспечения, которое принимало бы
Сейчас вместо Ру принято писать Y. — Прим. ред.
180 Часть 1. Изучение геномов
5'ia^S AG^GTAAGT^Sai 3'
Согласованная
последовательность
S'iSSS AG^GTTAGTSSai 3' "
S'iJSSBAG GTAACT3' Фактические
последовательности
5' i«=S TTGTAAGT 3'
5Ч1^ИАС^GTACGTSBai г.
Рис. 5.5. Связь между консенсус-
ной последовательностью выше-
расположенной интрон-экзонной
границы и фактическими ее по-
следовательностями, встречаю-
щимися в истинных генах. Отличия
от консенсусной последователь-
ности показаны красным цветом.
В вышерасположенных интрон-
экзонных границах (в донорных
сайтах) только нуклеотидная пара
«GT», стоящая непосредственно
после участка сплайсинга (пока-
занного стрелкой), является инва-
риантной
во внимание известные переменные, оказалось трудной задачей, и в настоящее время
определение местонахождения интрон-экзонных границ путем просмотра последова-
тельности является, по сути, не чем иным, как методом проб и ошибок1).
• Вышерасположенные регуляторные последовательности могут использо-
ваться, чтобы определить местонахождение областей, в которых начинаются
гены. Это связано с тем, что такие регуляторные последовательности наподобие
интрон-экзонных границ имеют отличительные особенности последовательности,
которыми они обладают, чтобы выполнять свою роль опознавательных сигналов
для ДНК-связывающих белков, вовлеченных в экспрессию генов (глава 11). К сожа-
лению, как и в случае интрон-экзонных границ, регуляторные последовательности
являются изменчивыми, причем в большей степени у эукариотов, чем у прокарио-
тов, и у эукариотов не все гены имеют один и тот же набор регуляторных после-
довательностей. Поэтому их использование для определения местонахождения
генов проблематично.
Эти три расширения способа простого просмотра на ОРС, несмотря на их ограниче-
ния, являются, в общем, применимыми к геномам всех высших эукариотов. Для работы
с отдельными организмами возможны также дополнительные стратегии, основанные
на специфических особенностях их геномов. Например, геномы позвоночных содержат
перед многими генами островки CpG; они представлены последовательностями при-
бл изител ьно 1 тыс. п. н., в которых содержание GC больше, чем среднее на геном в целом.
Приблизительно 40-50 % генов человека связаны с вышераспооженным CpG-островком.
Благодаря своей отличительности эти последовательности легко находятся, и когда
в ДНК позвоночного установлено местоположение того или иного CpG-островка, можно
обоснованно предположить, что в данной области непосредственно ниже него начина-
ется некоторый ген.
Определение местоположения генов, кодирующих функциональные РНК
Итак, просмотр на ОРС подходит для кодирующих белок генов; но что можно сказать
о генах, кодирующих функциональные РНК, такие как рРНК и тРНК (раздел 1.2.2)? Эти
гены не включают открытые рамки считывания, и, следовательно, их местоположение
не будет установлено методами, описанными выше. Однако молекулы функциональной
РНК, вместо этого, имеют свои собственные отличительные особенности, которые можно
Современные программы поиска генов в эукариотах дают точность примерно 85-90 %. — Прим. ред.
Глава 5. Определение структуры последовательности генома 181
учитывать для облегчения их отыскания в последовательности генома. Наиболее важной
из таких особенностей является способность сворачиваться во вторичную структуру
наподобие «клеверного листа», принимаемую молекулами тРНК (рис. 5.6, а). Такие
вторичные структуры скрепляются воедино спариванием оснований не между двумя
отдельными молекулами, как в двойной спирали ДНК, а между различными частями
одной и той же молекулы — то есть теми силами, которые мы называем внутримолеку-
лярным спариванием оснований. Для того чтобы образовались внутримолекулярные
пары оснований, последовательности нуклеотидов в обеих частях молекулы должны
быть комплементарны, и чтобы построить сложную структуру типа клеверного листа,
компоненты таких пар комплементарных последовательностей должны быть вы-
строены в характерном порядке в пределах последовательности РНК (рис. 5.6, б). Эти
особенности обеспечивают море информации, которая может быть использована для
определения местонахождения генов тРНК в последовательности генома, и программы,
разработанные для этой конкретной цели, обычно оказываются очень удачными.
а) Структура тРНК в виде клеверного листа
б) Последовательность одного из генов tRNAleu Escherichia coli
5'gccgaagtggcgaaatcggtagtcgcagttgattcaaaatcaaccgtagaaatacgtgccggttcgagtccggccttcggcacca 3'
Рис. 5.6. Отличительные особенности молекул тРНК помогают устанавливать местоположение генов,
кодирующих эти функциональные РНК. а) Все тРНК сворачиваются в структуру клеверного листа, которая
поддерживает свою целостность за счет внутримолекулярного спаривания оснований в четырех выде-
ленных цветом областях, б) Последовательность ДНК гена одной из тРНК Escherichia coli, специфичной
к аминокислоте лейцину. Выделенные цветом сегменты соответствуют областям внутримолекулярного
спаривания оснований, показанного на виде а. Специфические сегменты последовательности, способ-
ные формировать характерную вторичную структуру, выполняют роль особенностей, которые можно
отыскивать при помощи компьютерных программ, разработанных для определения местонахождения
генов тРНК. Дополнительная информация о структуре тРНК приведена в разделе 13.1.1
A G
U U
C—G
G — C
A—U
U “А
G--C
G--C
C~G
IIHAUA GGCHII
Рис. 5.7. Типичная структура РНК
типа стебель-петля
182 Часть 1. Изучение геномов
Так же, как и молекулы тРНК, молекулы рРНК и часть маленьких функциональных
РНК (раздел 1.2.2) принимают вторичные структуры, которые имеют достаточную слож-
ность, чтобы позволить их генам быть идентифицированными без особых затруднений.
Местонахождение генов других функциональных РНК определить труднее, потому что
РНК принимают структуры, которые предполагают спаривание относительно малого
числа оснований, или же спаривание оснований происходит не по регулярной схеме.
Для определения местоположения генов, кодирующих эти виды РНК, используются
три подхода.
• Хотя некоторые функциональные РНК не принимают сложных вторичных структур,
большая часть из них содержит одну или более структур типа стебель-петля (или
шпилек), которые образуются в результате внутримо-
лекулярного спаривания оснований простейшего типа
(рис. 5.7). Поэтому программы, которые просматривают
последовательности ДНК на такие структуры, опознают
области, в которых могут присутствовать гены функ-
циональной РНК. В эти программы заложены термо-
динамические правила, которые позволяют оценивать
стабильность структур типа стебель-петля, принимая
во внимание такие особенности, как размер петли, число
пар оснований в стебле и доля пар оснований G-С (они
являются более устойчивыми, чем пары А-Т, поскольку
скрепляются воедино тремя, а не двумя водородными
связями; см. рис. 1.8). Предполагаемую структуру типа
стебель-петля с оцениваемой стабильностью выше вы-
бранного предела рассматривают возможным индика-
тором присутствия гена функциональной РНК.
• Как и в случае кодирующих белок генов, может быть проведен поиск регуляторных
последовательностей, связанных с генами функциональных РНК. Эти регуляторные
последовательности отличаются от свойственных кодирующим белок генам и мо-
гут присутствовать в пределах гена функциональной РНК, а также выше него.
• В компактных геномах внимание направлено на области, которые остаются после
всестороннего поиска кодирующих белок генов. Зачастую эти «пустоты» не совсем
пусты, и тщательный просмотр выявит присутствие одного или нескольких генов
функциональной РНК.
Поиск гомологии и сравнительная геномика открывают дополнительное
измерение для просмотра последовательностей
Большинство различных программ, предназначенных для определения местопо-
ложения гена путем просмотра на ОРС, могут опознать до 95 % кодирующих областей
в геноме эукариота, но даже лучшие из них имеют тенденцию делать частые ошибки
в производимом ими позиционировании экзон-интронных границ, и опознавание лож-
ных ОРС в качестве истинных генов — все еще главная проблема1). Эти ограничения
могут быть до некоторой степени компенсированы за счет поиска гомологии с целью
проверки того, является ли ряд триплетов истинным экзоном или случайной последо-
1 }Другая важная проблема связана с тем, что программы поиска генов зачастую сливают два соседних
гена в один, объявляя межгенные промежутки интронами, и наоборот, объявляют интроны межгенными
промежутками, тем самым разбивая ген на части. — Прим. ред.
Глава 5. Определение структуры последовательности генома 183
вательностью. В ходе такого анализа производится поиск в базах данных ДНК с целью
определить, идентична ли исследуемая последовательность (или хотя бы подобна)
каким-либо генам, которые уже были секвенированы ранее. Очевидно, что если иссле-
дуемая последовательностьявляется частью гена, который уже был секвенирован кем-то
другим, то будет найдено идентичное соответствие, но это не цель поиска гомологии.
Вместо этого, намерение состоит в том, чтобы определить, подобна ли абсолютно новая
последовательность каким-либо известным генам, потому что если это так, то есть шанс,
что исследуемая и совпавшая с ней последовательности являются гомологичными, а это
означает, что они представляют гены, которые являются эволюционно связанными.
Главное применение поиска гомологии состоит в приписывании функций к недавно
открытым генам, и поэтому мы возвратимся к этому, когда будем рассматривать эту
составляющую анализа генома чуть позже в этой главе (раздел 5.2.1). Данный метод
сфокусирован также на определении местоположения гена, потому что он позволяет
проверять предварительные последовательности экзонов, местоположение коих было
установлено просмотром на ОРС, на наличие функций. Если предварительная после-
довательность экзона дает одно или более положительных совпадений после поиска
гомологии, то весьма вероятно, что это истинный экзон, но если она не дает ни одного
совпадения, тогда ее подлинность должна остаться под сомнением, пока она не будет
оценена тем или иным экспериментальным методом определения местоположения
генов.
Более точная версия поиска гомологии возможна, когда в наличии имеются после-
довател ьности геномов двух или более родственных видов. В геномах родственных видов
имеются общие для них области сходства (унаследованные ими от их общего предка),
на которые наложились видоспецифические различия, возникавшие с тех пор, как эти
виды начали эволюционировать независимо (рис. 5.8). В результате естественного от-
бора (раздел 19.3.2) такие области сходства последовательностей между родственными
геномами выражены наиболее ярко в пределах генов и наименее ярко в межгенных
областях. Поэтому при сравнении родственных геномов гомологичные гены легко
а) Взаимная организация генов
А В С D Е
Общий предок
АВ D Е
В С D Е
Эволюционно связанные виды
б) Последовательности ДНК
--GACAGTTAGCAATCGGAT--
Общий предок
--GATAGTTATCAATCCGAT-- --GACAGCTATCAATCCGAA--
Эволюционно связанные виды
Рис. 5.8. Эволюционно связанные виды имеют подобные геномы, а) Иллюстрация того, как организация
генов может измениться, когда два вида расходятся от их общего предка. Общий предок имеет пять
генов, обозначенных буквами от А до Е. У одного из производных видов ген С больше не присутствует,
а у другого вида ген А стал усеченным, б) Эволюционно связанные виды показывают подобия после-
довательности ДНК. На схеме представлен короткий отрезок последовательности гена из предкового
организма наряду с гомологичными этому сегменту гена последовательностями из производных видов.
Более полное описание принципов эволюции геномов приводится в главе 18
184 Часть 1. Изучение геномов
опознаются, потому что они имеют сильное подобие последовательностей, и у любой
ОРС, которая не имеет четкого гомолога во втором геноме, может быть снижен счет
как у почти наверняка являющейся случайной последовательностью, а не подлинным
геном. Анализ такого типа, — называемый сравнительной геномикой, — оказывается
очень ценным для определения местонахождения генов в геномеSaccharomyces cerevisiae
(раздел 5.3), поскольку полные или частичные последовательности теперь имеются
не только для этих дрожжей, но также и для 16 других членов группы полусумчатых
грибов (Hemiascomycetes), в том числе Saccharomyces paradoxus, Saccharomyces mikatae
и Saccharomyces bayanus — видов, состоящих в наиболее близком родстве с S. cerevisiae.
Сравнения между этими геномами подтвердили подлинность ряда ОРС S. cerevisiae и,
кроме того, позвол ил и удалить из каталога S. cerevisiae почти 500 предполагаемых ОРС на
том основании, что они не имеют никаких эквивалентов в родственных геномах. Анализ
становится еще более мощным за счет синтении — консервативности порядка взаим-
ного расположения генов, — показываемой геномами этих родственных дрожжей. Хотя
каждый геном подвергся своим собственным видоспецифическим перестройкам, есть
все же обширные области, в которых порядок генов в геноме S cerevisiae тот же самый,
что и в одном или нескольких родственных геномах. Это позволяет легко опознавать
гомологичные гены, но, что еще более важно, дает возможность отбрасывать ложные
ОРС, в особенности короткие, с большой достоверностью, потому что ожидаемое местопо-
ложение подобной ОРС в родственном геноме может быть детально проанализировано,
с тем чтобы убедиться в том, что в нем не имеется никакого ее аналога (рис. 5.9).
Аннотируемый геном
Короткая ОРС
Родственные геномы
Рис. 5.9. Применение сравнений с сингенными геномами с целью проверки подлинности короткой
ОРС. В данном примере ОРС присутствует в трех из четырех родственных геномов и, следовательно,
с большой вероятностью является подлинным геном
Автоматическое аннотирование последовательностей генома
Одно большое преимущество компьютерных подходов к определению местополо-
жения генов — возможность объединения набора аналитических программ в единую,
объединенную систему. Таким образом, различные подходы к определению место-
положения генов могут выполняться параллельно, а результаты — автоматически
сравниваться, с тем чтобы изучаемый геном был быстро и всесторонне аннотирован.
Аннотация генома с помощью таких систем начинается с анализа последовательности
при помощи программ просмотра на наличие ОРС, экзон-интронных границ и выше-
расположенных регуляторных последовательностей, далее ОРС проверяют на предмет
гомологии к генам в базах данных и ищут повторные последовательности и характер-
ные особенности генов, кодирующих функциональные РНК. Базы данных последо-
вательностей кДНК (см. ниже) также могут быть просмотрены на наличие любых из
вышеперечисленных совпадающих сегментов последовательности генома, ибо любые
такие совпадения показывают область, которая транскрибируется в мРНК. Затем ин-
Глава 5. Определение структуры последовательности генома 185
формация объединяется на экране компьютера, который показывает местоположения
различных особенностей последовательности, определенные разными программами
(рис. 5.10). К полученной информации открыт свободный доступ по сети Интернет,
благодаря чему ею могут пользоваться другие исследователи, строящие планы своих
собственных, более подробных вычислительных или экспериментальных исследований
определенных областей генома.
Promoters
GooPoftlrit*
EST hits
Repeats
Genscan
Genefinder
GRAIL
Genie
GanBank CDS
ORFs
ORFs
GenBcmk CDS
Genie
GRAIL
Gone finchw
Genscan
Repeats
EST hits
GeoPoptrJts
Promoters
i i iibibi
in i hi
Рис. 5.10. Экран типичной системы аннотации генома. Показанный пример — составленная обозре-
вателем Genotator аннотация сегмента 15 тыс.п.н. генома человека, содержащего ген фактора ткани.
В ряду сверху вниз приведены результаты анализа следующих видов: местоположение возможных
промоторов (вышерасположенных регуляторных элементов); последовательности, соответствующие
белкам в базе данных GenPept; последовательности, соответствующие известным EST (см. раздел 3.3.3);
местоположение известных повторов человека; предсказания экзонов посредством программ Genscan,
Genefinder, GRAIL и Genie; последовательности, соответствующие известным генам в базе данных
GenBank; ОРС в каждой из трех рамок считывания. Под черной чертой анализ всех видов повторен
для обратного комплемента этой последовательности ДНК. Аннотация показывает позиции пяти воз-
можных экзонов, обозначенные черными стрелками в верхней части рисунка. Иллюстрация любезно
предоставлена Номи Харрисом
5.1.2. Экспериментальные методы определения местоположения генов
Большинство экспериментальных методов определения местоположения генов
основаны не на прямом просмотре молекул ДНК, но вместо этого полагаются на обна-
ружение молекул РНК, которые транскрибируются с генов. Все гены транскрибируются
в РНК, и если ген прерывист, то первичный транскрипт впоследствии обрабатывается,
чтобы удалить интроны и соединить экзоны (разделы 1.2.3 и 12.2.2). Поэтому для опре-
деления местонахождения экзонов и полных генов могут быть использованы методы,
которые позволяют наносить на карту позиции транскрибируемых последовательностей
во фрагменте ДНК. Единственная проблема, которую необходимо учитывать, состоит
в том, что транскрипт обычно длиннее кодирующей части гена, потому что последняя
начинается на несколько десятков нуклеотидов выше старт-кодона и продолжается на
несколько десятков или сотен нуклеотидов ниже стоп-кодона. Поэтому анализ транс-
186 Часть 1. Изучение геномов
крипта не дает точного определения начала и конца кодирующей области гена, но он
достоверно сообщает, что в данной области присутствует некоторый ген, и позволяет
определить местонахождение экзон-интронных границ. Часто этой информации до-
статочно, для того чтобы очертить границы кодирующей области.
Реакции гибридизации позволяют определить, содержит ли данный фраг-
мент транскрибируемые последовательности
Самые простые процедуры для изучения транскрибируемых последовательно-
стей основаны на гибридизационном анализе. Молекулы РНК могут быть разделены
специализированными формами электрофореза в агарозном геле, перемещены на
нитроцеллюлозную или нейлоновую мембрану и исследованы с помощью процедуры,
названной нозерн-гибридизацией (см. техническое примечание 5.1). Она отличается
от гибридизации по Саузерну (раздел 2.1.2) только специфическими условиями, при ко-
торых выполняется перемещение, и тем фактом, что она не была изобретена доктором
Нозерном и, таким образом, не имеет права быть именованой с заглавной буквы «Н».
Если нозерн-отпечаток клеточной РНК зондируется меченым фрагментом генома, то
молекулы РНК, транскрибируемые с генов в пределах этого фрагмента, будут обнару-
жены датчиком (рис. 5.11). Поэтому теоретически нозерн-гибридизация есть средство
определения числа генов, присутствующих во фрагменте ДНК, и размера каждой коди-
рующей области. У этого подхода есть два слабых места.
• Некоторые отдельные гены дают начало двум и более транскриптам разной длины,
потому что некоторые из их экзонов являются факультативными и могут сохра-
ниться или не сохраниться в зрелой РНК (раздел 12.2.2). Если дело обстоит именно
так, то фрагмент, который содержит только один ген, может обнаружить две или
больше гибридизационные полосы в нозерн-отпечатке. Подобная проблема может
возникнуть, если ген состоит членом многогенного семейства (раздел 7.2.3).
• При работе с многими видами не практикуется делать препарат мРНК из полного ор-
ганизма, так что экстракт получают из отдельного органа или ткани. Следовательно,
Клетки
| Извлечение РНК
I Денатурация и электрофорез
в агарозном геле
Полосы рРНК
Перенос промоканием,
I нозерн-гибридизация,
авторадиография
Зонд ДНК гибридизируется
к единственному транскрипту РНК
Рис. 5.11. Нозерн-гибридизация. Экстракт РНК
подвергается электрофорезу в агарозном геле при
денатурирующих условиях (см. техническое при-
мечание 5.1). После окрашивания бромистым эти-
дием наблюдаются две полосы. Они представлены
двумя наиболее крупными молекулами рРНК (раз-
дел 1.2.2), которыми изобилует большинство клеток.
Меньшие молекулы рРНК, которые также обильны,
не заметны, потому что они настолько коротки, что
выбегают из основания пластины геля, и в боль-
шинстве клеток ни одна из молекул мРНК не встре-
чается в достаточно больших количествах, чтобы
формировать полосу, видимую после окрашивания
бромистым этидием. Гель переносится промокани-
ем на нейлоновую мембрану и, в данном примере,
зондируется радиоактивно меченным фрагментом
ДНК. На авторадиограмме видна единственная по-
лоса, а это показывает, что фрагмент ДНК, исполь-
зуемый в качестве зонда, содержит часть или всю
транскрибируемую последовательность
Глава 5. Определение структуры последовательности генома 187
любые гены, не экспрессируемые в этом органе или ткани, не будут представлены
в популяции РНК и, таким образом, не будут обнаружены при зондировании РНК
изучаемым фрагментом ДНК. Даже если используется целый организм, не все
гены дадут сигналы гибридизации, потому что многие экспрессируются только
на определенной стадии развития, а иные экспрессируются слабо, а это означает,
что их РНК-продукты присутствуют в слишком малых количествах, чтобы быть
обнаруженными гибридизационным анализом.
Гибридизационный анализ второго типа избегает проблем со слабо экспрессируе-
мыми и тканеспецифическими генами за счет поиска не молекул РНК, а родственных
последовательностей в ДНК других организмов. Подобно поиску гомологии этот подход
основан на том факте, что гомологичные гены в эволюционно связанных организмах
имеют подобные последовательности, тогда как межгенная ДНК обычно весьма отлична.
Если фрагмент ДНК от одного вида используется для зондирования перенесенных по
Саузерну молекул ДНК от эволюционно связанных видов и получен один или несколько
сигналов гибридизации, то весьма вероятно, что этот зонд содержит один или несколько
генов (рис. 5.12). Это называют зоогибридизацией.
Секвенирование кДНК позволяет наносить на карту гены в пределах фраг-
ментов ДНК
Нозерн-гибридизация и зоогибридизация позволяют определять присутствие
или отсутствие генов во фрагменте ДНК, но не дают никакой позиционной информа-
ции о местоположении этих генов в последовательности ДНК. Самый простой путь
Кролик
Человек Шимпанзе Корова
Рис. 5.12. Зоогибридизация. Цель состоит в том,
чтобы определить, гибридизируется ли фрагмент
ДНК человека с ДНК эволюционно связанных видов.
Поэтому образцы ДНК человека, шимпанзе, коровы
и кролика приготовляются, обрабатываются рестрик-
тазами и подвергаются электрофорезу в агарозном
геле. Затем выполняется гибридизация по Саузерну
с зондом в лице фрагмента ДНК человека. Положи-
тельный гибридизационный сигнал наблюдается
с ДНК каждого животного, а это предполагает, что
фрагмент ДНК человека содержит экспрессируемый
ген. Обратите внимание, что гибридизирующиеся
фрагменты рестрикции от ДНК коровы и кролика
меньше, чем гибридизирующиеся фрагменты в об-
разцах шимпанзе и человека. Это показывает, что
рестрикционная карта вокруг транскрибируемой по-
следовательности отлична у коров и кроликов, но не
умаляет обоснованность заключения о том, что у всех
четырех видов имеется некий гомологичный ген
1 Гибридизация по Саузерну
с зондом из ДНК человека
Зонд гибридизируется со
всеми образцами ДНК
188 Часть 1. Изучение геномов
Техническое примечание 5.1: методы изучения РНК
Многие методы, изобретенные для изучения молекул ДНК, могут быть
приспособлены для исследования РНК
Электрофорез в агарозном геле выполняется с РНК после ее денатурации,
с тем чтобы скорость миграции каждой молекулы зависела исключительно от ее
длины и на нее не влияли внутримолекулярные пары оснований, которые могут об-
разовываться во многих РНК (например, рис. 13.2). Денатурирующий агент (обычно
формальдегид или глиоксаль) добавляется к образцу до его загрузки в гель.
Нозерн-гибридизация относится к процедуре, посредством которой гель с РНК
переносится промоканием на нейлоновую мембрану и гибридизируется с меченым
зондом (см. рис. 5.11). Это эквивалентно гибридизации по Саузерну (см. рис. 2.11)
и выполняется подобным же образом.
Меченые молекулы РНК обычно приготовляются копированием матрицы
ДНК в РНК в присутствии меченого рибонуклеотида. Используются ферменты
РНК-полимеразы бактериофагов SP6, ТЗ или Т7, потому что они могут произвести
до 30 мкг меченой РНК из 1 мкг ДНК за 30 минут. РНК может также быть помечена
на концах обработкой очищенной поли-А-полимеразой (раздел 12.2.1).
ПЦР молекул РНК требует модификации первого шага обычной реакции. Поли-
мераза Taq не может копировать молекулу РНК, так что первый шаг катализируется
обратной транскриптазой, которая производит ДНК-копию матрицы РНК. Затем
эта ДНК-копия размножается Taq-полимеразой. Этот метод называют ревертазной
ПЦР, или РТ-ПЦР. Открытие термостабильных ферментов, способных производить
ДНК-копии как РНК-матриц, так и ДНК-матриц (например, ДНК-полимераза Tth
из бактерии Thermus thermophilus) увеличивает возможность выполнения РТ-ПЦР
в виде единой реакции и с участием только одного фермента.
Методы секвенирования РНК существуют, но трудны в исполнении и при-
менимы только к маленьким молекулам. Эти методы подобны секвенированию
ДНК путём химической деградации (раздел 4.1.2), но для получения расщеплен-
ных молекул используют специфичные к последовательности эндонуклеазы, а не
химикалии. На практике последовательность молекулы РНК обычно получают
путем преобразования ее в кДНК (см. рис. 3.36) и последующего секвенирования
ее методом обрыва цепи (раздел 4.1.1).
Специальные методы были разработаны для картирования позиций молекул
РНК на последовательностях ДНК, например, с тем чтобы определить точки начала
и конца транскрипции и определить местонахождение позиций интронов в после-
довательности ДНК. Эти методы описаны в разделе 5.1.2.
Единственный главный недостаток в инструментарии для изучения РНК —
отсутствие ферментов со степенью специфичности последовательности, выказы-
ваемой эндонуклеазами рестрикции, которые так важны при манипуляциях с ДНК.
Кроме этого, единственное препятствие при работе с РНК — легкость, с которой
деградация молекул РНК осуществляется рибонуклеазами, высвобождающимися
при разрушении клеток (что имеет место в ходе извлечения РНК) и, помимо того,
присутствующими на руках сотрудников лаборатории, стенках стеклянной посуды
и в растворах. Это означает, что для сохранения молекул РНК нетронутыми должны
быть приняты строгие лабораторные процедуры (например, очистка стеклянной
посуды химикалиями, разрушающими рибонуклеазы).
Глава 5. Определение структуры последовательности генома 189
к получению такой информации — секвенирование соответствующих молекул кДНК.
кДНК представляет собой копию мРНК (см. рис. 3.36) и, таким образом, соответствует
кодирующей области гена с какими-либо предшествующими и последующими [назы-
ваемыми 5'-UTR и З'-UTR, или 5'- и З'-нетранслируемыми областями — НТО. — Прим,
ред.] последовательностями, которые также транскрибируются. Поэтому сравнение
последовательности кДНК с последовательностью геномной ДНК очерчивает позицию
соответствующего гена и показывает экзон-интронные границы.
Для того чтобы получить отдельную кДНК, сначала должна быть приготовлена
библиотека кДНК изо всех мРНК в изучаемой ткани. Как только библиотека будет готова,
успех секвенирования кДНК как средства определения местоположения гена зависит от
двух факторов. Первый относится к частоте встречаемости желательных молекул кДНК
в библиотеке. Как и в случае нозерн-гибридизации, проблема проистекает из различия
уровней экспрессии разных генов. Если изучаемый фрагмент ДНК содержит один или
несколько слабо экспрессируемых генов, то соответствующие молекулы кДНК будут
редки в данной библиотеке, и может возникнуть необходимость перебора множества
клонов, прежде чем будет опознан желаемый. Чтобы обойти эту проблему, были изо-
бретены различные методы захвата кДНК, или отбора кДНК, в которых изучаемый
фрагмент ДНК неоднократно гибридизируется с совокупностью молекул кДНК, с тем
чтобы обогатить эту смесь желательными клонами. Поскольку пул кДНК содержит так
много различных последовательностей, обычно невозможно отбраковать все несоот-
ветствующие клоны такими повторными гибридизациями, но возможно значительно
увеличить частотность тех клонов, которые специфично гибридизируются с фрагментом
ДНК. Это уменьшает размер библиотеки, которую впоследствии необходимо будет про-
сматривать при строгих условиях с целью опознавания желательных клонов.
Второй фактор, который определяет успех или неудачу, — полнота отдельных мо-
лекул кДНК. Обычно молекулы кДНК получают путем копирования молекул РНК в одно-
нитевую ДНК с помощью обратной транскриптазы и последующего преобразования
однонитевой ДНК в двунитевую ДНК посредством ДНК-полимеразы (см. рис. 3.36). Всегда
есть шанс, что та или иная реакция синтеза нити не будет протекать до его завершения,
что даст усеченную кДНК. Присутствие внутримолекулярных пар оснований в РНК может
привести также к неполному копированию. Усеченные молекулы кДНК могут страдать
отсутствием части информации, необходимой для определения местонахождения точек
начала и конца гена и всех его экзон-интронных границ.
Известные методы точного картирования концов транскриптов
Проблемы с неполными молекулами кДНК означают, что для точного определе-
ния местонахождения начальных и конечных точек транскриптов генов необходимы
более устойчивые методы. Одна из возможностей — ПЦР специального типа, в которой
в качестве исходного материала используется не ДНК, а РНК. Первый шаг в ПЦР этого
типа — преобразование РНК в кДНК с помощью обратной транскриптазы, после чего
кДНК размножается при помощи полимеразы Taq тем же самым способом, как и в нор-
мальной ПЦР. Эти методы идут под общим названием ревертазная ПЦР (РТ-ПЦР), но
конкретная версия, которая интересует нас в настоящее время, — быстрое размноже-
ние концов кДНК (RACE). В простейшей форме этого метода одна из затравок является
специфичной ко внутренней области, расположенной вблизи от начала изучаемого
гена. Эта затравка прикрепляется к мРНК, соответствующей этому гену, и направляет
первую катализируемую обратной транскриптазой стадию процесса, в течение которой
производится кДНК, соответствующая началу мРНК (рис. 5.13). Поскольку копируется
190 Часть 1. Изучение геномов
-----3' РНК
J Отжиг затравки ДНК
• Синтез кДНК обратной
* транскриптазой
3'
1 Денатурация
mnnnnnttttt
it
7 Добавление ряда А к 3'-концу
4 посредством концевой трансферазы
AAAAAN NNBII.
1 Отжиг закрепляемой затравки
AAAiANMNNMIl
Рис. 5.13. RACE — быстрое размножение концов
кДНК. Изучаемая РНК преобразуется в частичную
кДНК путем продолжения затравки ДНК, которая
отжигается ко внутренней позиции, не слишком
далеко от 5'-конца молекулы. Далее З'-конец кДНК
продлевается обработкой концевой дезоксинукле-
отидилтрансферазой (раздел 2.1.4) в присутствии
дАТФ, в результате чего к кДНК присоединяется ряд
нуклеотидов А. Этот ряд А действует как участок
отжига для закрепляемой затравки. Продолжение
якорной затравки ведеткдвунитевой молекуле ДНК,
которая после ее синтеза может быть размножена
стандартной ПЦР. Это 5'-RACE, названный так по-
тому, что он обеспечивает размножение 5'-концов
исходной РНК. Подобный метод — З'-RACE — может
быть использован, если желательно получить по-
следовательности с З'-концами
। Синтез второй нити посредством
1 полимеразы Taq
I
Продолжение соответственно
стандартной ПЦР
только маленький сегмент мРНК, ожидае-
мый результат таков: синтез кДНК не будет
оборван преждевременно, так что один ко-
нец кДНК будет в точности соответство-
вать началу мРНК. Как только кДНК будет
произведена, к ее З’-концу присоединяется
короткий поли-А хвост. Вторая затравка
отжигается к этой поли-А последовательности, и в ходе первого круга нормальной
ПЦР преобразует однонитевую кДНК в двунитевую молекулу, которая впоследствии
размножается в ходе ПЦР. Последовательность этой размноженной молекулы покажет
точную позицию начала транскрипта.
К другим методам точного картирования транскриптов относится гетеродуплекс-
ный анализ. Если изучаемая область ДНК клонируется в виде фрагмента рестрикции
в векторе М13 (раздел 4.1.1), то она может быть получена в виде однонитевой ДНК. Буду-
чи смешана с соответствующим препаратом РНК, транскрибируемая последовательность
в клонируемой ДНК гибридизируется с соответствующей мРНК и образует двунитевой
гетеродуплекс. В примере, показанном на рис. 5.14, начало этой мРНК лежит в пределах
клонированного фрагмента рестрикции, так что какая-то часть клонированного фраг-
мента участвует в гетеродуплексе, а остальная — нет. Однонитевые области могут быть
переварены в ходе обработки специфичной к однонитевой ДНК нуклеазой, такой как S1.
Размер гетеродуплекса определяется расщеплением РНКового компонента щелочью
и подверганием полученной однонитевой ДНК электрофорезу в агарозном геле. Затем
такое измерение размера используется, чтобы поместить начало транскрипта относи-
тельно участка рестрикции, расположенного в конце клонированного фрагмента.
Местоположение границ между интронами и экзонами также может быть
определено с достаточной точностью
Гетеродуплексный анализ может быть использован также и для определения
местонахождения экзон-интронных границ. Данный метод почти не отличается от
представленного на рис. 5.14, за исключением лишь того, что клонированный фрагмент
Глава 5. Определение структуры последовательности генома 191
Рис. 5.14. Sl-картирование. Этот метод картирования
транскриптов использует нуклеазу S1, фермент, который
расщепляет полинуклеотиды однонитевой ДНК или РНК,
в том числе однонитевые области в преимущественно
двунитевых молекулах, но не имеет никакого действия
на двунитевую ДНК или на гибриды ДНК-РНК. В пока-
занном примере фрагмент рестрикции, который покры-
вает начало единицы транскрипции, лигируется в вектор
М13, и полученная однонитевая ДНК гибридизируется
с препаратом РНК. После обработки нуклеазой S1 по-
лученный гетеродуплекс имеет один конец, отмеченный
началом транскрипта, и другой — нижерасположенным
участком рестрикции (R2). Размер непереваренного
фрагмента ДНК измеряется путем гель-электрофореза,
с тем чтобы определить позицию начала единицы транс-
крипции относительно нижерасположенного участка
рестрикции
Ген
R1 R2
Фрагмент рестрикции
размером 400 п. н.
{Клонирование фрагмента
рестрикции в векторе М13
| Отжиг РНК
(Расщепление всех однонитевых
* областей посредством нуклеазы S1
R2
рестрикции покрывает картируемую экзон-
интронную границу, а не начало транскрипта.
Второй метод отыскания экзонов в по-
следовательности генома называют уловлени-
ем экзонов. Он требует вектора специального
типа, который содержит миниген, состоящий
из двух экзонов, окружающих последователь-
ность интрона, причем первому экзону предше-
ствуют сигналы последовательности, необхо-
| Расщепление РНК щелочью
R2
I Измерение ДНК посредством
электрофореза - скажем, 200 п. н.
* I *
R1 t________R2
/ 200 п. н.
Точка начала
транскрипции
димыедля инициации транскрипции в клетке
эукариота (рис. 5.15). Чтобы использовать такой вектор, часть ДНК, предназначенная
для изучения, вставляется в участок рестрикции, расположенный в пределах интронной
области вектора. Затем вектор вводится в подходящую клеточную линию эукариотов,
где он транскрибируется, а произведенная из него РНК — сплайсируется. В результате
любой экзон, содержащийся в геномном фрагменте, прикрепляется между выше- и ни-
жерасположенными экзонами из минигена. Теперь применяется РТ-ПЦР с затравками,
отжигаемыми в пределах двух экзонов минигена, с тем чтобы размножить секвени-
руемый фрагмент ДНК. Поскольку последовательность минигена уже известна, могут
быть определены позиции нуклеотидов, в которых вставленный экзон начинается
и заканчивается, и которые тем самым точно очерчивают границы этого экзона.
5.2. Определение функций отдельных генов
Как только в последовательности генома устанавливают местоположение ново-
го гена, сразу же возникает вопрос о его функции. Это становится важной предметной
областью геномики, потому что завершенные проекты секвенирования показали, что
мы, скорее, знаем меньше, чем мы думали о содержании отдельных геномов. Например,
Escherichia coli и Saccharomyces cerevisiae интенсивно изучались обычным генетическим
анализом до появления проектов секвенирования, и одно время генетики были вполне
уверены в том, что большинство генов этих организмов уже было опознано. Последо-
192 Часть 1. Изучение геномов
Улавливающий
экзон вектор R
| Встраивание новой ДНК
R R
I Введение в эукариота-хозяина,
* транскрипция
Рис. 5.15. Уловление экзонов. Улавливающий экзон
вектор состоит из двух экзонных последователь-
ностей, которым предшествуют промоторные по-
следовательности — сигналы, необходимые для
экспрессии гена в эукариоте-хозяине (раздел 11.2.2).
Новая ДНК, содержащая ненанесенный на карту
экзон, лигируется в вектор и рекомбинантную мо-
лекулу, введенную в клетку хозяина. Затем получен-
ный в результате этого транскрипт РНК исследуется
при помощи РТ-ПЦР, стем чтобы опознать границы
ненанесенного на карту экзона
Транскрипт РНК
| Сплайсинг
Затравки для РТ-ПЦР
Последовательности промоторов
4* Экзон
R
afas Участок рестрикции
вательности геномов показали, что в нашем
знании фактически есть большие провалы.
Из 4288 белок-кодирующих генов в геноме
Е coli было ранее идентифицировано только
1853 (43 % от общего числа). Для S. cerevisiae
эта цифра составила лишь 30 %.
Как и в случае определения местополо-
жения генов, попытки определить функции
неизвестных генов осуществляются путем
компьютерного анализа и эксперименталь-
ных исследований.
5.2.1. Компьютерный анализ функций гена
Мы уже видели, что компьютерный анализ играет важную роль в определении ме-
стоположения генов в последовательностях ДНК, и что одно из самых мощных средств,
имеющихся для этой цели, есть поиск гомологии, при котором местонахождение генов
определяется путем сравнения изучаемой последовательности ДНК со всеми осталь-
ными последовательностями ДНК в базах данных1). Основа поиска гомологии в том, что
эволюционно связанные гены имеют подобные последовательности, и, следовательно,
новый ген может быть открыт на основании его подобия некоторому его эквивален-
ту — уже секвенированному гену какого-либо иного организма. Теперь мы более при-
стально взглянем на анализ гомологии и рассмотрим, как он может быть использован
для приписывания функции к новому гену.
Гомология отражает эволюционные отношения
Гомологичные гены имеют общего эволюционного предка и выявляются на осно-
вании подобия их последовательностей. Эти подобия представляют собой данные, на
почве которых вырастают молекулярные филогенетические деревья; мы увидим их
в главе 19. Гомологичные гены подпадают под две категории (рис. 5.16).
• Ортологичные гены — такие гомологи, которые присутствуют в разных организ-
мах и общий предок этих генов предшествует расхождению видов. Ортологичные
гены обычно имеют одинаковые или очень подобные функции. Например, орто-
логами являются гены миоглобина людей и шимпанзе.
На самом деле сравнивают аминокислотные последовательности, которые могут быть получены
в результате трансляции. — Прим. ред.
Глава 5. Определение структуры последовательности генома 193
Предковый ген
Дублирование гена
ОРТОЛОГИ
Рис. 5.16. Ортологичные и паралогичные гены
ПАРАЛОГИ
• Паралогичные гены присутствуют в одном и том же организме и зачастую принад-
лежат общему многогенному семейству (раздел 7.2.3), причем общий предок этих
генов может предшествовать, а может и не предшествовать появлению вида, в ко-
тором такие гены теперь обнаруживаются. Например, гены миоглобина и р-глобина
людей суть паралоги: они порождены дупликацией некоторого предкового гена
приблизительно 550 миллионов лет назад (раздел 18.2.1).
Нуклеотидные последовательности пары гомологичных генов обычно не иден-
тичны, потому что эти два гена подвергаются различным случайным изменениям, обу-
словленным мутациями, но являются подобными, потому что эти случайные изменения
происходили на разных путях эволюции из одной общей исходной последовательности
общего предкового гена. Поиск гомологии основан как раз на таких подобиях после-
довательностей. В основе данного анализа лежит тот факт, что если недавно секвени-
рованный ген оказывается подобным какому-либо ранее секвенированному гену, то
может быть предсказана их эволюционная связь и функция нового гена, вероятно, будет
идентична или по крайней мере подобна функции уже известного гена.
Важно не путать слова гомология и подобие. Неправильно описывать пару эволю-
ционно связанных генов как «на 80 % гомологичные», если их нуклеотидные после-
довательности имеют 80%-ю идентичность (рис. 5.17). Гены рассматриваемой пары
являются или эволюционно связанными, или нет; не может быть никаких промежу-
точных ситуаций, и поэтому бессмысленно приписывать паре гомологов степень го-
мологичности в процентах.
Последовательность 1 ggtgagggtatcatcccatctgactacacctcatcgggagacggagcagt
Последовательность 2 ggtcaggatatgattccatcacactacaccttatcccgagtcggagcagt
Идентичность *** *** *** ** ***** ********* *** *** *********
Рис. 5.17. Две последовательности ДНК с 80%-й идентичностью последовательностей
Анализ гомологии может предоставить информацию о функции целого гена
или же о функциях расположенных в его пределах сегментов
Поиск гомологии можно проводить с последовательностью ДНК, но обычно перед
выполнением поиска исследуемая последовательность гена преобразуется в последо-
вательность аминокислот. Одна из причин этого состоит в том, что в белках есть 20
различных аминокислот, а в ДНК — только четыре нуклеотида, так что гены, которые
не связаны между собой никаким родством, обычно показывают более разительные
отличия друг от друга, когда сравниваются их аминокислотные последовательности
194 Часть 1. Изучение геномов
GAPGMWLRLAAGSFEHAG
Последовательность 1 ggtgcacccggtatgtgactgcgattagcagcgggatcatttcagcatgcaggg
★ ★ ★★★★★ ★★★★ ★★★★ ★★ ★★★ **** ★★★ ★★ ★★ ★
Последовательность 2 gatacaccccgtatttgacagcaatttgcagggggatgattgcaccatggagcg
DTPRIWEEFAGGWLHHGA
Рис. 5.18. Часто отсутствие гомологии между двумя последовательностями проявляется более оче-
видно, когда сравнения проводятся на уровне аминокислотной последовательности. Показаны две
последовательности нуклеотидов, где нуклеотиды, которые являются идентичными в этих двух после-
довательностях, выделены зеленым цветом, а не идентичные — красным. Эти две последовательности
нуклеотидов идентичны на 76 %, как показано звездочками. Это могло бы быть взято как свидетельство
в пользу того, что последовательности являются гомологичными. Однако, когда последовательности
транслированы в аминокислоты, степень идентичности снижается до 28 %. Идентичные аминокислоты
выделены желтым цветом, а не идентичные — коричневым. Сравнение между аминокислотными по-
следовательностями показывает, что гены не гомологичны и что подобие на уровне нуклеотидов было
случайным1). Последовательности аминокислот были написаны с использованием однобуквенного
кода (см. табл. 1.2)
(рис. 5.18). Поэтому менее вероятно, что поиск гомологии даст ложные результаты, если
оценивается последовательность аминокислот.
Программа поиска гомологии начинает анализ с построения выравниваний между
последовательностью запроса и последовательностями из баз данных. Для каждого вы-
равнивания рассчитывается счет [часто называемый весом выравнивания. — Прим, ред.],
по которому исследователь может оценить вероятность того, что последовательность
запроса и сверяемые с ней суть гомологи. Есть два способа начисления счета.
• Простейшие программы считают число позиций, в которых одна и та же аминокис-
лота присутствует в обеих последовательностях. Это число, будучи преобразовано
в проценты, дает степень идентичности двух последовательностей.
• Более искушенные программы при назначении счета каждой позиции в выравни-
вании учитывают химическое сходство между неидентичными аминокислотами
и начисляют более высокий счет для идентичных или аналогичных аминокислот
(например, лейцина и изолейцина или аспарагиновой кислоты и аспарагина)
и более низкий счет для аминокислот с меньшей степенью аналогии (например,
цистеина и тирозина или фенилаланина и серина). Данный метод анализа опреде-
ляет степень подобия между парой последовательностей.
Чтобы достичь возможно большего счета, алгоритм вводит пропуски в различных
позициях в одной или обеих последовательностях (до пределов, установленных исследо-
вателем). Считается, что в ходе эволюции генов происходят похожие процессы, — когда
целые блоки нуклеотидов, кодирующие отдельные или смежные аминокислоты, могут
быть вставлены в ген или удалены из него.
Практические стороны поиска гомологии нисколько не устрашают. Существует
несколько программ для анализа такого типа, и наиболее популярная среди них —
BLAST (основное средство поиска локальных выравниваний). Для проведения анализа
необходимо всего лишь соединиться с веб-узлом одной из баз данных ДНК и ввести по-
следовательность в окно инструмента оперативного поиска. Стандартная программа
BLAST эффективна при опознавании гомологичных генов, обладающих более 30-40%-м
Такие ситуации крайне редки. Обычно если нет сходства аминокислот, то и нуклеотидные последо-
вательности не похожи. Сравнивать аминокислоты правильнее именно потому, что при отсутствии сходства
на уровне нуклеотидов можно обнаружить его на уровне белков. — Прим. ред.
Глава 5. Определение структуры последовательности генома 195
подобием последовательностей, но менее эффективна при распознавании эволюцион-
ных отношений, если подобие ниже этой величины. Видоизмененная версия по имени
PSI-BLAST (позиционный итерационный BLAST) опознает более отдаленно связанные
последовательности, объединяя гомологичные последовательности из поиска стандарт-
ной программы BLAST в профиль, особенности которого используются для опознавания
дополнительных гомологичных последовательностей, которые не были обнаружены
в первоначальном поиске.
Поиск гомологии с помощью BLAST и подобных программ приобрел огромное
значение в исследованиях геномики, но необходимо признать и его ограничения. Нарас-
тающая проблема — присутствие в базах данных генов, заявленные функции которых
не соответствуют действительности. Если один из таких генов будет опознан в качестве
гомолога последовательности запроса, то такая неправильная функция будет приписана
и этой новой последовательности,усугубляя означенную проблему. Есть также несколько
случаев, когда гомологичные гены имеют весьма различные биологические функции,
примером чего служат кристаллины хрусталика глаза, некоторые из которых гомоло-
гичны метаболическим ферментам. Поэтому гомология между последовательностью
запроса и некоторым кристалл ином не подразумевает, что последовательность запроса
есть кристаллин, и, точно так же, очевидная гомология между последовательностью за-
проса и метаболическим ферментом может вовсе не означать, что последовательность
запроса — метаболический фермент.
Известны также примеры генов, которые обладают подобными последователь-
ностями, но не имеют никакой очевидной эволюционной связи. Иногда это объясня-
ется тем, что, хотя гены являются несвязанными, их белки имеют подобные функции
и общая часть их последовательностей кодирует некоторый домен в каждом из белков,
который является главным в определении этой общей функции. Хотя сами гены не име-
ют никакого общего предка, домены имеют, но их общий предок жил в очень древнее
время, и гомологичные домены впоследствии эволюционировали не только за счет
единичных изменений нуклеотидов, но также и посредством более сложных перестроек,
которые привели к созданию новых генов, в пределах которых эти домены расположе-
ны (раздел 18.2.1). Будучи распознана, гомология этого типа может быть чрезвычайно
информативна. Типичный пример представлен доменом tudor — мотивом приблизи-
тельно из 120 аминокислот, который был сначала опознан в последовательности гена
Drosophila melanogaster, называемого tudor. Кодируемый геном tudor белок, функция
которого неизвестна, состоит из десяти копий домена tudor, расположенных одна за
другой (рис. 5.19). Поиск гомологии с употреблением домена tudor в качестве пробы
показал, что несколько известных белков содержат этот домен. Последовательности
этих белков не очень подобны друг другу и нет никакого признака, что они являются
истинными гомологами, но все они обладают доменом tudor. В числе этих белков со-
стоят: белок, вовлеченный в транспорт РНК в ходе оогенеза Drosophila, белок человека
с определенной ролью в метаболизме РНК и другие белки, действия которых, кажется,
так или иначе связаны с РНК. Поэтому анализ гомологии позволяет предположить, что
последовательность tudor играет некоторую роль во взаимодействии между белком и его
РНКовым субстратом. Информация от компьютерного анализа неполна сама по себе, но
она указывает путь к экспериментам, которые необходимо провести, чтобы получить
более определенные данные относительно функции домена tudor1).
На самом деле существует еще множество дополнительных компьютерных методов определения
и уточнения функций генов. — Прим. ред.
196 Часть 1. Изучение геномов
Ген tudor Drosophila
Ген homeless Drosophila
Ген АКАР149 человека
Рис. 5.19. Домен tudor. В верхней части рисунка показана
структура белка tudor Drosophila, который содержит
десять копий домена tudor. Этот домен обнаружен
также и во втором белке Drosophila (homeless) и в опор-
ном белке А-киназы человека (АКАР149), который играет
роль в метаболизме РНК. В остальном, за исключением
присутствия доменов tudor, эти белки имеют несхожие
структуры. Деятельность всех этих белков так или иначе
связана с РНК
Применение поиска гомологии для приписывания функций генам, связанным
с болезнями человека
Чтобы проиллюстрировать важность и значение поиска гомологии в геномике,
мы исследуем вопрос о том, какую помощь этот метод оказал исследователям генети-
ческих болезней человека. Одна из главных причин секвенирования генома человека
состояла в том, чтобы получить доступ к генам, вовлеченным в болезни человека. Есть
определенные надежды, что последовательность гена болезни обеспечит понимание
биохимической основы этой болезни и, следовательно,укажет путь предупреждения или
лечения этой болезни. Поиск гомологии играет присущую ему важную роль в изучении
генов болезней, потому что открытие гомолога гена болезни человека в каком-либо
ином организме часто дает ключ к пониманию биохимической функции гена челове-
ка. Если такой гомолог уже был охарактеризован, то информация, необходимая для
понимания биохимической роли гена человека, может уже быть под рукой; если он
еще не был охарактеризован, то намеченное исследование может быть направлено на
выявленный гомолог.
Для того чтобы гомолог был полезным в изучении болезни человека, вовсе не
обязательно, чтобы он присутствовал в близкородственном виде. Многообещающей
в этом отношении является Drosophila, поскольку фенотипические эффекты многих генов
Drosophila хорошо известны, так что уже имеются данные для того, чтобы определить
механизм действия генов, которые имеют гомологов в геноме Drosophila. Но наибольший
успех был достигнут с дрожжами. В геноме S. cerevisiae имеются гомологи нескольких
генов болезней человека (таблица 5.1). Эти гены болезней включают гены, вовлеченные
в рак, кистозный фиброз, а также неврологические синдромы, и в некоторых случаях
дрожжевой гомолог имеет известную функцию, которая обеспечивает ясный признак
биохимического действия гена человека. В некоторых случаях даже было возможно про-
демонстрировать физиологическое подобие между действием гена у людей и дрожжей.
Например, ген дрожжей SGS1 является гомологом гена человека, вовлеченного в болезни,
называемые синдромами Блума и Вернера, которые характеризуются нарушениями ро-
ста. Дрожжи с мутантным геном SGS1 живут более короткие промежутки времени, чем
нормальные дрожжи, и показывают признаки ускоренного начала старения, например
бесплодие. Ген дрожжей, как было установлено, кодирует одну из пары родственных
ДНК-геликаз, которые необходимы для транскрипции генов рРНК и для репликации
ДНК. Таким образом, связь между SGS1 и генами синдромов Блума и Вернера, установ-
ленная на основании поиска гомологии, указала на возможную биохимическую основу
этих болезней человека.
Глава 5. Определение структуры последовательности генома 197
Таблица 5.1. Примеры генов болезней человека, которые имеют гомологов в геноме Saccharomyces
cerevisiae
Ген болезни человека Гомолог дрожжей Функция гена дрожжей
Боковой амиотрофический склероз S0D1 Защита от пероксида (02")
Атаксическая телеангиэктазия TEL1 Кодирует протеинкиназу
Рак ободочной кишки MSH2, MLH1 Репарация ДНК
Кистозный фиброз YCF1 Устойчивость к металлам
Миотоническая дистрофия YPK1 Кодирует протеинкиназу
Нейрофиброматоз 1-го типа IRA2 Кодирует регуляторный белок
Синдром Блума, синдром Вернера SGS1 ДНК-геликаза
Болезнь Вильсона ССС2 Перенос меди?
5.2.2. Экспериментальное определение функций гена
Ясно, что анализ гомологии не есть панацея, способная опознать функции всех
новых генов. Поэтому, чтобы дополнить и расширить результаты изучения гомологич-
ности, необходимы экспериментальные методы. Это оказывается одним из крупней-
ших вызовов исследовательским задачам геномики, и многие молекулярные биологи
согласны в том, что методологии и стратегии, используемые в настоящее время, не
в полной мере адекватны для того, чтобы приписывать функции к огромному числу
неизвестных генов, открываемых в ходе проектов секвенирования. Проблема состоит
в том, что цель — провести путь от гена к его функции — является обратной направ-
лению, обычно принимаемому генетическим анализом, в котором отправной точкой
является фенотип и цель состоит в том, чтобы опознать лежащий в его основе ген или
гены. Данная же проблема, к решению которой мы в настоящее время подходим, ведет
нас в обратном направлении: мы начинаем с нового гена и, надо надеяться, движемся1)
к опознаванию связанного с ним фенотипа.
Функциональный анализ посредством инактивации гена
В обычном генетическом анализе генетическая основа фенотипа обычно изучается
посредством поиска мутантных организмов, в которых данный фенотип стал видоизме-
ненным. Такие мутанты могут быть получены экспериментально — например, путем
обработки популяции организмов (скажем, культуры бактерий) ультрафиолетовым из-
лучением или мутагенным химикатом (см. раздел 16.1.1), — или же могут быть обнару-
жены в естественной популяции. Затем ген или гены, которые были видоизменены в му-
тантном организме, изучаются при помощи генетических скрещиваний (раздел 3.2.4),
посредством которых можно определить местонахождение позиции некоторого гена
в геноме, а также определить, является ли этот ген таким же, как тот, который уже был
охарактеризован ранее. Затем данный ген может быть изучен подробнее методами
молекулярной биологии, например клонированием и секвенированием.
Общий принцип описанного нами стандартного метода анализа состоит в том,
что гены, ответственные за фенотип, могут быть опознаны путем определения того,
какие гены инактивированы в организмах, которые показывают мутантную версию
Иногда это направление называют обратной генетикой. — Прим. ред.
198 Часть 1. Изучение геномов
этого фенотипа. Если отправной точкой служит ген, а не фенотип, то эквивалентная
стратегия состояла бы в том, чтобы мутировать ген и опознавать фенотипическое из-
менение, которое из него проистекает. Это основа большинства методов, употребляемых
для приписывания функций к неизвестным генам.
Отдельные гены могут быть инактивированы гомологичной рекомбинацией
Наиболее простой способ инактивировать определенный ген состоит в том, чтобы
разрушить его с помощью не связанного с ним сегмента ДНК (рис. 5.20). Этого можно
достичь гомологичной рекомбинацией между хромосомной копией этого гена и какой-
либо иной частью ДНК, которая обладает некоторой идентичностью последовательности
с целевым геном. Гомологичная рекомбинация и рекомбинация других типов — сложные
события, которые мы рассмотрим подробнее в главе 17. На данный момент достаточно
знать, что если две молекулы ДНК имеют подобные последовательности, то рекомби-
нация может привести к взаимному обмену молекул своими сегментами.
Как инактивацию генов выполняют на практике? Мы рассмотрим два примера,
и первый — с S. cerevisiae. Со времени завершения расшифровки последовательности
генома в 1996 г. работающие с дрожжами молекулярные биологи осуществили скоор-
динированный международный проект по определению функций как можно больше-
го числа неизвестных генов (раздел 5.3). Один из применяемых методов показан на
рис. 5.21. Центральный компонент—«кассетаудаления», которая несет ген устойчивости
к антибиотику. Этот ген не есть нормальный компонент генома дрожжей, но если его
вставить в хромосому дрожжей, он будет работать и даст начало трансформированной
клетке дрожжей, устойчивой к антибиотику генетицину. Перед использованием кассеты
удаления к обоим ее концам в виде хвостов прикрепляются новые сегменты ДНК. По-
следовател ьности этих сегментов идентичны частям гена дрожжей, которому предстоит
инактивация. После того как видоизмененная кассета введена в клетку дрожжей, между
хвостами ДНК и хромосомной копией гена дрожжей происходит гомологичная рекомби-
нация. Поэтому клетки, которые подверглись замене, отбираются путем высева культуры
на агаровую среду, содержащую генетицин. Полученные колонии не обладают действием
Хромосомная ДНК
целевого гена, и их фенотипы могут
быть исследованы, с тем чтобы по-
лучить некоторое представление
о функции этого гена.
Второй пример инактивации
гена представляет аналогичный
процесс, но с мышами, а не с дрож-
жами. Мышь часто используется
в качестве модельного организма
человека, потому что геном мыши
подобен геному человека и они со-
Хромосомная ДНК
Ген разрушен
Рис. 5.20. Инактивация гена гомологичной рекомбинацией.
Хромосомная копия целевого гена рекомбинируете разру-
шенной версией гена, несомого клонирующим вектором.
В результате целевой ген становится инактивированным
держат много одинаковых генов.
Поэтому функциональный анализ
неизвестных генов человека вы-
полняется в значительной степени
путем инактивации эквивалентных
генов у мыши, поскольку подобные
эксперименты с людьми были бы
с этической точки зрения немые-
Глава 5. Определение структуры последовательности генома 199
лимы. Этап гомологичной реком-
бинации в ходе общей процедуры
идентичен описанному для дрож-
жей и вновь дает клетку в которой
целевой ген был инактивирован.
Проблема в данном случае состоит
в том, что мы хотим получить не
одну-единственную мутированную
клетку а целую мутантную мышь,
поскольку полную оценку эффек-
та инактивации гена на фенотип
мы можем провести только на ор-
ганизме в целом. Чтобы достичь
этого, необходимо использовать
клетки мыши специального типа:
зародышевые стволовые клетки,
или ЗС-клетки. В отличие от боль-
шинства клеток мыши, ЗС-клетки
являются полипотенциальными,
что означает, что они не приписа-
ны к какому-либо одному пути раз-
вития и поэтому могут породить
дифференцированные клетки
всех типов. Итак, сконструирован-
ная ЗС-клетка вводится в эмбрион
мыши, который продолжает раз-
виваться и в конечном счете дает
начало химере — мыши, клетки
которой представляют собой смесь
из мутантных клеток, полученных
из сконструированных ЗС-клеток,
и немутантных клеток, получен-
ных из всех остальных клеток эм-
бриона. Это все еще не совсем то,
чего мы хотим, так что химерным
мышам позволяют спариваться
друг с другом. Часть потомства
получается вследствие слияния
двух мутантных гамет, и поэтому
она будет нехимерной, поскольку
каждая из их клеток будет нести
инактивированный ген. Это нока-
утные мыши, и в случае успеха их
фенотипы обеспечат желаемую ин-
формацию о функции изучаемого
гена. Такой прием хорошо работает
для инактиваций многих генов, но
Кассета удаления
ИНЦМ
R1
кап' Векторная ДНК
R2
i Вставка дрожжевой ДНК
в участки рестрикции
Введение в клетки дрожжей
Хромосомная копия целевого гена
R1 R1 R2 R2
Гомологичная рекомбинация
разрушает целевой ген за счет
введения в него последовательности
гена кап'
I
Экспрессия гена кап
ОБОЗНАЧЕНИЯ
кап' Ген устойчивости к генетицину
«ф. Промоторные последовательности дрожжей
R1, R2 Участки рестрикции
Рис. 5.21. Использование кассеты удаления с дрожжами.
Кассета удаления состоит из гена устойчивости к антибиотику,
которому предшествуют промоторные последовательности,
необходимые для его экспрессии в дрожжах, и к которому
с обеих сторон примыкают участки рестрикции. Начальный
и конечный сегменты целевого гена вставлены в эти участки
рестрикции, и вектор введен в клетки дрожжей. Рекомби-
нация между сегментами гена в векторе и хромосомной
копией целевого гена приводит к разрушению последнего.
Клетки, в которых такое разрушение произошло, являются
опознаваемыми, потому что они теперь экспрессируют ген
устойчивости к антибиотику и, таким образом, будут расти
на агаровой среде, содержащей генетицин. Обозначение
гена «капг» — сокращение названия «kanamycin resistance»
(устойчивость к канамицину), при этом канамицин — общее
название группы антибиотиков, в которую входит и генети-
цин
некоторые оказываются смертель-
200 Часть 1. Изучение геномов
ними и посему не могут быть изучены в гомозиготной нокаутной мыши. Вместо этого,
получают гетерозиготную мышь как продукт слияния между одной нормальной и одной
мутантной гаметой в надежде, что эффект инактивации гена на фенотип будет заметным
даже при том, что мышь будет иметь одну правильную копию изучаемого гена.
Инактивация генов без гомологичной рекомбинации
Гомологичная рекомбинация — не единственный способ разрушить ген с целью
изучения его функции. Одна из альтернатив — использование мечения транспозонами,
в котором инактивация достигается вставкой в изучаемый ген мобильного генетиче-
ского элемента, или транспозона. Большинство геномов содержит мобильные генетиче-
ские элементы (раздел 9.2), и хотя большая часть их является бездействующей, обычно
есть немногие, которые сохраняют свою способность перемещаться в новые позиции
в геноме. При нормальных обстоятельствах транспозиция есть относительно редкое
явление, но иногда возможно использовать методы технологии рекомбинантной ДНК,
чтобы произвести видоизмененные транспозоны, которые изменяют свою позицию
в ответ на внешний раздражитель. Один из способов достижения этой цели, основанный
на транспозоне дрожжей 7у1, показан на рис. 5.22. Мечение транспозонами сыграло
важную роль также и в анализе генома плодовой мушки с использованием эндогенного
транспозона Drosophila, названного P-элементом. Слабое место мечения транспозонами
заключается в том, что трудно метить отдельные гены, потому что транспозиция есть
более или менее случайное событие, и невозможно предсказать, куда попадет транс-
позон после своего скачка. Если цель состоит в том, чтобы инактивировать какой-либо
конкретный ген, то необходимо вызвать существенное число транспозиций и затем
просмотреть все полученные организмы, с тем чтобы найти один с надлежащей встав-
кой. Поэтому мечение транспозонами более применимо к глобальным исследованиям
функции генома, в которых гены инактивируются наугад и опознаются группы генов
с подобными функциями [либо гены, связанные в один путь. — Прим, ред.] в ходе про-
смотра потомства на предмет интересующих исследователя изменений фенотипа.
Чувствительный
к галактозе промотор
1 Перенос клеток на среду,
содержащую галактозу
Транскрибированная
с элемента Ту] иРНК
Транспозиция
Рис. 5.22. Искусственное вызывание
транспозиции. Чтобы поместить в геном
дрожжей чувствительную к галактозе
премоторную последовательность выше
элемента Ту1, были использованы ме-
тоды технологии рекомбинантной ДНК.
Когда галактоза отсутствует, элемент Ту1
не транскрибируется и, таким образом,
остается в состоянии покоя. Когда клетки
переносят на среду культивирования, со-
держащую галактозу, промотор активи-
руется и элемент Ту1 транскрибируется,
запуская процесс транспозиции. За до-
полнительной информацией об актива-
ции промоторов у эукариотов обращай-
тесь к разделу 11.3, а за подробными
сведениями о процессе транспозиции
элемента Ту1 — к разделу 17.3.2
Глава 5. Определение структуры последовательности генома 201
Совершенно иной подход к инактивации гена основан на РНК-интерференции,
или РНК-и, — одном из ряда естественных процессов, посредством которых короткие
молекулы РНК влияют на экспрессию генов в живых клетках (раздел 12.2.6). При употре-
блении в исследованиях геномики РНК-и служит средством инактивирования целевого
гена без разрушения самого гена, за счет уничтожения его мРНК. Этого достигают, вводя
в клетку короткие молекулы двунитевой РНК, последовательности которых совпадают
с таковой мРНК-мишени. Молекулы двунитевой РНК разбиваются на более короткие
молекулы, которые вызывают деградацию целевой мРНК (рис. 5.23). Эффективная
работа данного процесса первоначально была показана на черве Caenorhabditis elegans,
геном которого был полностью секвенирован и который рассматривается как важный
модельный организм для высших эукариотов (раздел 14.3.3). Фактически все 19 000
предсказанных генов в геноме С. elegans были по одному инактивированы при помощи
РНК-интерференции. Ключевым шагом в любом эксперименте РНК-и является введение
в исследуемый организм молекулы двунитевой РНК, которая даст начало молекулам
однонитевой интерферирующей РНК. С С. elegans этого можно достигнуть, скармливая
РНК червям. С. elegans питается бактериями, в том числе и Escherichia coli, и часто вы-
ращивается на газоне бактерий в чашке с агаровой средой. Если бактерии содержат
клонированный ген, который направляет экспрессию двунитевой РНК с той же после-
довательностью, что и у гена С. elegans, то после приема пищи начинает работать путь
РНК-и. В качестве альтернативы двунитевая РНК может быть непосредственно введена
в червя посредством микроинъекции, но этот способ отнимает больше времени.
РНК-интерференция, как известно, происходит естественным образом у ряда эу-
кариотов, но ожидалось, что применение ее к клеткам млекопитающих будет трудным,
потому что эти организмы проявляют сопутствующую реакцию на двунитевую РНК,
в результате которой синтез белка, как правило, ингибируется, что приводит к смерти
клетки. Эту проблему обошли за счет использования молекул двунитевой РНК длины
лишь 21-22 п.н. — достаточно длинных, чтобы быть специфичными к отдельному
Двунитевая РНК
i Нуклеаза «Дайсер»
разрезает двунитевую РНК
W W Tff ПТ ПТ W МолекУлы короткой
111 111 ill ill ill ill интерферирующей РНК (киРНК)
Рис. 5.23. РНК-интерференция.
Молекула двунитевой РНК
расщепляется рибонуклеазой
«Дайсер» на молекулы «корот-
кой интерферирующей РНК»
(si PH К) длиной 21-25 п. н. Одна
нить каждой si PH К спаривается
с целевой мРНК, которая после
того деградируется нуклеа-
зой RDE-1
I Молекулы однонитевой
киРНК прикрепляются к иРНК
„ттг.ттт.ттт.ттт.ттг.ттт
3' иРНК
Деградация нуклеазой RDE-1
202 Часть 1. Изучение геномов
целевому гену и активировать процесс РНК-и, но слишком коротких, чтобы вызвать
реакцию, заключающуюся в ингибировании синтеза белка. Этот подход, основанный
на использовании ретровирусных векторов для доставки клонированных генов, экс-
прессирующих молекулы РНК, в культивируемые клетки, позволил по отдельности
инактивировать более 8000 из 35000 генов человека. На данный момент основной
задачей, каковую необходимо решить для осуществления РНК-и у млекопитающих, —
переход от систем на основе отдельных клеток, которые позволяют оценивать лишь
некоторые фенотипы, к нокаутным мышам, которые дают возможность наблюдать
полный диапазон фенотипов. Это требует развития клонирующих систем для долго-
срочного, устойчивого синтеза молекул интерферирующей РНК, которые должны при-
сутствовать в клетке на значительно высоком уровне, чтобы поддерживать целевой ген
в инактивированном состоянии.
Сверхэкспрессия гена также может быть использована для оценки его функ-
ции
До сей поры мы фокусировали наше внимание на методах, обеспечивающих инак-
тивацию изучаемого гена («потерю функции»). Известен альтернативный подход, со-
стоящий в том, чтобы сконструировать некий организм, в котором иследуемый ген был
бы намного более активен, чем в нормальном («усиление функции»), и определить,
какие изменения, если таковые имеются, это вызывает в фенотипе. Результаты таких
экспериментов нужно интерпретировать с особым вниманием из-за необходимости
проводить различие между изменением фенотипа, которое происходит из-за специфи-
ческой функции сверхэкспрессируемого гена, и менее специфическим изменением фе-
нотипа, которое отражает ненормальность ситуации, где единственный продукт гена
синтезируется в чрезмерных количествах и, возможно, даже в тех тканях, в которых этот
ген обычно бездействует. Несмотря на такую особенность, сверхэкспрессия позволила
получить некоторую важную информацию о функции генов.
Чтобы сверхэкспрессировать ген, необходимо использовать клонирующий вектор
специального типа. Он должен обеспечивать синтез как можно большего количества
исследуемого белка с клонируемого гена. Поэтому такой вектор многокопийный, то
есть он размножается в организме хозяина до степени 40-200 копий на клетку, так что
в последнем присутствует множество копий изучаемого гена. Вектор должен также
содержать очень активный промотор (раздел 11.2.2), с тем чтобы каждая копия иссле-
дуемого гена была преобразована клеткой в большие количества мРНК, — опять же
гарантируя, что белок будет производиться в возможно больших количествах. В примере,
показанном на рис. 5.24, клонирующий вектор содержит высокоактивный промотор,
который экспрессируется только в печени, так что каждая трансгенная мышь сверх-
экспрессирует исследуемый ген в своей печени. Этот подход использовался с генами,
последовательности которых предполагали, что они кодируют белки, которые вы-
деляются в кровоток. После синтеза в печени трансгенной мыши исследуемый белок
выделяется и рассматривается фенотип трансгенной мыши в поиске ключей к функции
клонированного гена. Было сделано интересное открытие, когда стало понятно, что
одна трансгенная мышь имела кости, которые были значительно плотнее, чем таковые
у нормальных мышей. Это было важно по двум причинам: во-первых, это позволило
идентифицировать соответствующий ген как вовлеченный в синтез костной ткани;
во-вторых, открытие белка, который увеличивает ее плотность, имеет значение для
разработки методов лечения остеопороза человека — болезни ломкости костей.
Глава 5. Определение структуры последовательности генома 203
Высокоактивный специфичный
к печени промотор
1кДНК
Многокопийный
клонирующий вектор
J Клонирование в организме мыши
Клетки печени
Белок, выделяемый
в кровоток
—► Наблюдение
фенотипа
Рис. 5.24. Функциональный анализ путем сверхэкспрессии гена. Цель состоит в том, чтобы определить,
оказывает ли сверхэкспрессия изучаемого гена какое-либо влияние на фенотип трансгенной мыши.
Поэтому кДНК гена вставляется в клонирующий вектор, несущий очень активную премоторную после-
довательность, которая направляет экспрессию клонируемого гена в клетках печени мыши. Использу-
ется кДНК, а не геномная копия гена, поскольку первая не содержит интроны и, таким образом, короче
второй, а посему ею легче манипулировать в пробирке
Влияние инактивации или сверхэкспрессии гена на фенотип может быть
трудно различимо
Определяющий момент эксперимента с инактивацией или сверхэкспрессией гена—
необходимость идентифицировать изменение фенотипа, характер которого и дает ключ
к функции манипулируемого гена. На деле это может оказаться намного труднее, чем
оно звучит. У любого организма диапазон фенотипов, который предстоит исследовать,
огромен. Даже у одноклеточного организма типа дрожжей этот список весьма продол-
жителен (таблица 5.2, а), а у многоклеточных эукариотов он намного длиннее (табли-
ца 5.2, б). У высших же организмов некоторые фенотипы (например, поведенческие)
трудно, если вообще возможно, оценить всесторонне.
Более того, эффект инактивации гена может быть очень тонким и в силу этого не
быть распознан при исследовании фенотипа. Хороший пример проблем, воникающих
в данной области, представлен самым длинным геном на хромосоме III дрожжей, кото-
рый при своих 2167 кодонах и типичном для дрожжей отклонении частоты использо-
вания кодонов попросту должен был быть функциональным геном, а не ложной ОРС.
Инактивация этого гена не имела никакого очевидного эффекта — мутантные клетки
дрожжей, казалось, имели фенотип, идентичный таковому нормальных дрожжей. Не-
которое время думали, что этот ген, возможно, незначащий и его белковый продукт
или вовлечен в отправление некоторой абсолютно несущественной функции, или
имеет функцию, дублируемую каким-либо вторым геном. В конечном же счете было
показано, что мутанты умирают, когда они выращиваются при низком pH в присутствии
глюкозы и уксусной кислоты, что нормальные дрожжи могут переносить, и был сделан
вывод о том, что этот ген кодирует белок, который выкачивает ацетат из клетки. Это,
несомненно, существенная функция, поскольку этот ген играет жизненно важную роль
защиты дрожжей от вызываемого уксусной кислотой повреждения, но эту его важность
было трудно выследить в опытах с фенотипом.
204 Часть 1. Изучение геномов
Таблица 5.2. Типичные фенотипы, оцениваемые при анализе генов Saccharomyces cerevisiae или
Caenorhabditis elegans
Фенотип
а) Все гены Saccharomyces cerevisiae
Синтез ДНК и клеточный цикл
Синтез и обработка РНК
Синтез белка
Ответы на раздражения
Синтез клеточной стенки и морфогенез
Транспорт биохимических веществ внутри клетки
Энергетический и углеводный обмен веществ
Жировой обмен веществ
Восстановление и рекомбинация ДНК
Развитие
Мейоз
Структура хромосом
Строение клетки
Выделение и транспорт белков
б) Гены, участвующие в раннем эмбриогенезе Caenorhabditis elegans
Стерильность/сниженная фертильность родителей
Осмотическая целостность
Полярное вытягивание тела
Прохождение через мейоз
Вход в интерфазу
Корковая динамика
Внешний вид проядра/ядра
Прикрепление центросомы
Миграция проядра
Сборка веретена деления
Вытягивание/целостность веретена деления
Расхождение сестринских хроматид
Внешний вид ядра
Расхождение хромосом
Цитокинез
Асимметрия деления
Темп деления клетки
Общий темп развития
Серьезные плейотропные дефекты
Целостность прикрепленных к мембране органелл
Размер яйца
Аберрантные цитоплазматические структуры
Сложное сочетание дефектов
Глава 5. Определение структуры последовательности генома 205
Таблица 5.3. Примеры генов-репортёров
Ген Продукт гена Испытание
lacZ Р-галактозидаза Гистохимическое испытание
uidA Р-глюкуронидаза Гистохимическое испытание
lux Люцифераза Биолюминесценция
GFP Зеленый флюоресцентный белок Флюоресценция
Даже когда выполняется самый тщательный анализ, инактивации многих генов,
как оказывается, не дают никакого заметного фенотипического изменения. Почти
5 000 из 6 000 генов дрожжей могут быть по отдельности инактивированы, не вызывая
смерть клетки, и инактивация многих из этих 5 000 генов не имеет никакого обнару-
жимого эффекта на метаболические свойства клетки при нормальных условиях роста.
Что касается С. elegans, то в ходе проектов крупномасштабной инактивации генов до
сих пор было приписано фенотипов к менее чем 10 % из 19000 предсказанных генов.
Ввиду такого «успеха» напрашивается вывод о том, что в обоих организмах большинство
генов играет специфические роли и опознавание такой роли для каждого гена грозит
оказаться длинным и трудным процессом.
5.2.3. Более подробные исследования активности белка, кодируемого
неизвестным геном
Инактивация и сверхэкспрессия генов являются основными методами, используе-
мыми исследователями генома, чтобы определить функцию нового гена, но это не един-
ственные процедуры, которые могут дать информацию о деятельности интересующего
их гена. Другие методы могут дополнить и уточнить результаты инактивации и сверх-
экспрессии. Они могут использоваться, чтобы обеспечить дополнительную информацию,
которая поможет опознаванию функции гена или могла бы формировать основу более
полного анализа активности белка, ген которого уже был охарактеризован.
Направленный мутагенез может быть применен для подробного исследова-
ния функции гена
Инактивация и сверхэкспрессия позволяют определить общую функцию гена, но
они не могут дать подробную информацию об активности белка, кодируемого этим
геном. Например, можно предположить, что часть гена кодирует последовательность
аминокислот, которая либо направляет его белковый продукт к определенному ком-
партменту в клетке, либо ответственна за способность белка отвечать на химический
или физический сигнал. Чтобы проверить эти гипотезы, необходимо удалить или видо-
изменить соответствующую часть последовательности гена, но оставить неизменной
большую часть, так чтобы белок по-прежнему синтезировался и сохранял основную
суть своей функции. Чтобы произвести эти тонкие изменения, могут быть использова-
ны различные процедуры сайт-специфического мутагенеза или мутагенеза in vitro
(техническое примечание 5.2). Это важные методы, возможности применения которых
лежат не только в области изучения деятельности гена, но также и в сфере белковой
инженерии, где цель состоит в создании новых белков со свойствами, которые лучше
отвечают промышленным или клиническим целям.
206 Часть 1. Изучение геномов
Целевой ген Хромосомная ДНК
X Векторная ДНК
II
Промотор Ген устойчивости к антибиотику
| Замена гена на первом шаге
_____ Хромосомная ДНК
X Векторная ДНК
in
Мутантный ген
| Замена гена на втором шаге
Хромосомная ДНК
Рис. 5.25. Двушаговая процедура замены гена
После мутагенеза последователь-
ность гена должна быть введена в клет-
ку хозяина так, чтобы в результате гомо-
логичной рекомбинации существующая
копия гена могла быть заменена его ви-
доизмененной версией. Это представляет
проблему потому что мы должны каким-
либо образом узнавать, в каких клетках
гомологичная рекомбинация произошла,
а в каких — нет. Даже у дрожжей это бу-
дет только долей от общего числа клеток,
а у мышей эта доля будет очень мала. Обыч-
но эту проблему решают, помещая рядом
с мутантным геном некий маркёрный ген
(например, кодирующий устойчивость
к антибиотику) и отыскивая клетки, ко-
торые принимают фенотип, переданный
этим маркёром. В большинстве случаев
клетки, которые вставляют маркёрный ген
в свой геном, также вставляют вблизи него
и прикрепленный мутантный ген и, таким
образом, являются именно теми клетками,
которые нам и нужны. Проблема состоит в том, что при выполнении эксперимента сайт-
специфического мутагенеза мы должны убедиться в том, что всякое изменение работы
изучаемого гена есть результат специфичной мутации, которая была введена в ген, а не
косвенный результат изменения его окружающей среды в геноме за счет вставки мар-
кёрного гена рядом с ним. Ответ — использовать более сложную, двушаговую замену
гена (рис. 5.25). В этой процедуре целевой ген сначала заменяется маркёрным геном
самим по себе. Клетки, в которых эта рекомбинация произошла, опознаются отбором
по фенотипу маркёрного гена. Затем эти клетки используются во второй стадии проце-
дуры замены гена, когда маркёрный ген заменяется мутантным геном, причем успех на
данном этапе проверяют поиском клеток, которые потеряли фенотип маркёрного гена.
Такие клетки содержат мутантный ген, и их фенотипы могут быть исследованы, чтобы
определить эффект направленной мутации на деятельность белкового продукта.
Гены-репортеры и иммуноцитохимия могут быть использованы для опреде-
ления места и времени экспрессии генов
Нередко ключи к функции гена могут быть получены путем определения того,
где и когда этот ген бывает активен. Если экспрессия гена ограничена определенным
органом, или тканью многоклеточного организма, или же отдельным набором клеток
в пределах органа или ткани, то на основании этой позиционной информации можно
вывести общую роль продукта этого гена. То же самое справедливо и для информации,
касающейся стадии развития, в которую данный ген экспрессируется. Анализ такого
типа оказался особенно полезным в понимании действий генов, вовлеченных в самые
ранние стадии развития Drosophila (раздел 14.3.4), и все чаще используется, чтобы
разгадать таинство генетики развития млекопитающих. Он применим также и к тем
одноклеточным организмам (наподобие дрожжей), которые имеют отличительные
стадии развития в своем жизненном цикле.
Глава 5. Определение структуры последовательности генома 207
Определение картины экспрессии гена в пределах организма возможно с помощью
гена-репортера. Это ген, экспрессия которого может быть отслеживаема удобным спо-
собом, в идеале визуальным наблюдением (таблица 5.3), когда клетки, экспрессирующие
ген-репортер, становятся синими, флюоресцирующими или испускающими некоторый
другой видимый сигнал. Для того чтобы ген-репортер дал надежный признак того, где
и когда опытный ген экспрессируется, репортер должен подчиняться тем же регулятор-
ным сигналам, что и исследуемый ген. Этого достигают, заменяя последовательность
(ОРС) исследуемого гена на последовательность (ОРС) гена-репортера (рис. 5.26). Боль-
шинство регуляторных сигналов, которые управляют экспрессией гена, содержится
в области ДНК выше этой ОРС, так что ген-репортер должен теперь показывать ту
же картину экспрессии, что и опытный ген. Поэтому картина экспрессии может быть
определена путем исследования организма на наличие сигнала репортера.
Промотор
I Гомологичная рекомбинация заменяет
целевой ген геном-репортёром
Ген-репортёр
Рис. 5.26. Ген-репортер. Последовательность (ОРС) гена-репортера заменяет последовательность (ОРС)
изучаемого гена. В результате ген-репортер помещается под контроль регуляторных последовательно-
стей, которые обычно управляют картиной экспрессии опытного гена. Для получения дополнительной
информации об этих регуляторных последовательностях см. разделы 11.2 и 11.3. Обратите внимание,
что стратегия применения генов-репортеров предполагает, что важные регуляторные последователь-
ности действительно лежат выше данного гена. В генах эукариотов это не всегда так
Наряду с информацией о том, в каких клетках ген экспрессируется, часто полезно
определить местонахождение позиции в пределах клетки, в которой белок, кодируемый
этим геном, обнаруживается. Например, ключевые данные относительно функции гена
могут быть получены путем отслеживания расположения белкового продукта — в ми-
тохондриях, в ядре или на поверхности клетки. Здесь гены-репортеры не могут помочь,
потому что последовательность ДНК перед данным геном — последовательность, к ко-
торой ген-репортер прикреплен, — не участвует в направлении белкового продукта к его
верному внутриклеточному местоположению. Вместо него именно аминокислотная по-
следовател ьность самого белка является в данном случае определяюще важной. Поэтому
единственный способ определить, где белок расположен, состоит в том, чтобы отыскать
его непосредственно. Это может быть сделано иммуноцитохимией, которая использует
антитело, специфичное к интересующему белку и, таким образом, связывающееся только
с этим белком и ни с каким другим. Антитело метится так, чтобы его позиция в клетке
и, следовательно, позиция целевого белка могла быть визуализирована (рис. 5.27). Для
208 Часть 1. Изучение геномов
Рис. 5.27. Иммуноцитохимия. Клетку обрабатывают анти-
телом, которое мечено красным флюоресцентным краси-
телем. Наблюдение клетки показывает, что флюоресцент-
ный сигнал связан с внутренней мембраной митохондрий.
Рабочая гипотеза поэтому такова: целевой белок вовлечен
в перенос электронов и окислительное фосфорилирование,
поскольку это главные биохимические функции внутренней
митохондриальной мембраны
J Зонд с меченым антителом
Метка обнаруживается
внутри митохондрий
исследований с низкой разрешающей способно-
стью используются мечение флюоресцентными
метками и конфокальная микроскопия, а дающая
высокое разрешение иммуноцитохимия может
быть выполнена при помощи электронной микро-
скопии с использованием плотной для электронов
метки типа коллоидного золота1).
5.3. Наглядный пример: аннотиро-
вание последовательности генома
Sacharomyces cerevisiae
В этой главе мы рассмотрели диапазон методов (как компьютерных, так и экс-
периментальных) определения местонахождения генов в последовательности генома
и определения их функций. Каждый из этих методов подходит далеко не ко всем, а лишь
к определенным организмам; выбор зачастую определяется техническими соображе-
ниями, например, РНК-интерференция интенсивно использовалась с С. elegans отчасти
ввиду легкости, с которой молекулы двунитевой РНК могут быть введены в червей по-
средством обычного кормления последних (раздел 5.2.2). Чтобы проиллюстрировать,
как эти различные методы сочетаются, мы заключим эту главу обзором достижений,
свершенных в аннотировании генома дрожжей S. cerevisiae. В масштабе эукариотов ге-
ном дрожжей относительно несложен, ибо он содержит относительно мало межгенной
ДНК и очень немного интронов. Это упрощает опознавание ОРС, но не дает никакого
преимущества, когда дело касается определения функций отдельных генов.
5.3.1. Аннотирование последовательности генома дрожжей
Проект секвенирования S. cerevisiae был завершен в 1996 г. Первичный анализ, для
которого минимальный размерный предел для потенциального гена был установлен
равным 100 кодонам, позволил опознать 6274 ОРС, приблизительно 30 % из которых,
как было известно, были истинными генами, потому что они были ранее опознаны
путем обычного генетического анализа еще до того, как этот проект секвенирова-
ния был запущен. Остальные 70 % изучались посредством анализа гомологии, когда
Сейчас большую популярность приобрели флюоресцентные белки. Наиболее популярен GFP — зеле-
ный флюоресцентный белок из медузы. Их гены через короткий линкер присоединяют к исследуемому гену
так, чтобы при трансляции получился слитый белок. Локализацию такого белка легко наблюдать в микро-
скоп. — Прим. ред.
Глава 5. Определение структуры последовательности генома 209
Техническое примечание 5.2: сайт-специфический мутагенез
Методы получения точного видоизменения в последовательности гена
с целью изменения структуры и, возможно, функций белка
Изменения в структуре белка могут быть спроектированы и осуществлены
методами сайт-специфического мутагенеза, которые дают в результате заданные
видоизменения, произведенные в нуклеотидной последовательности гена, коди-
рующего интересующий нас белок. Эти методы позволяют исследовать функции
различных частей белка и, кроме того, нашли широкое применение в разработке
новых ферментов для биотехнологических целей.
Обычный мутагенез — случайный процесс, который вызывает изменения
в произвольных (не заданных исследователем) позициях в молекуле ДНК. Чтобы
найти интересующую нас мутацию, необходим просмотр большого числа мутантных
организмов. Даже в случае микробов, которые могут быть проверены в огромных
количествах, лучшее, на что можно надеяться, — диапазон мутаций в нужном гене,
одна из которых могла бы затронуть часть изучаемого белка. Сайт-специфический
мутагенез предлагает средство получения намного более специфичных мутаций.
Наиболее важные из этих методов следующие.
• Специфичный к олигонуклеотидам мутагенез, в ходе которого олигонуклео-
тид, содержащий желаемую мутацию, отжигается к однонитевой версии соот-
ветствующего гена, при этом последний получают клонированием в векторе
М13 (раздел 4.1.1). Этот олигонуклеотид затравливает реакцию синтеза нити,
которой позволяют продолжаться полностью вокруг кольцевой матричной
молекулы (рис. Т5.1, а). После введения в Escherichia coli репликация ДНК
производит многочисленные копии этой молекулы рекомбинантной ДНК,
б) Опознавание мутантных фагов
Несоответствие
Двунитевая ДНК
Инфицированная Е. coli
Частицы фага высевают
на чашку, с тем чтобы
получить бляшки
Перенос промоканием,
зондирование
Сигнал гибридизации -
бляшка мутантных фагов
Рис. Т5.1. Мутагенез, специфичный к олигонуклеотидам
210 Часть 1. Изучение геномов
при этом половина из них является копиями исходной нити ДНК и полови-
на — копиями нити, которая содержит мутированную последовательность.
Все эти двунитевые молекулы направляют синтез частиц фага, так что при-
бл изител ьно половина фагов, высвобождаемых из зараженных бактерий, несет
однонитевую версию мутированной молекулы. Фаги высевают на твердый
агар и таким образом получают бляшки, после чего мутантные фаги опозна-
ются их гибридизационным зондированием с исходным олигонуклеотидом
(рис. Т5.1, б). Затем мутантный ген может быть помещен назад в его перво-
начального хозяина посредством гомологичной рекомбинации, как описано
в разделе 5.2.3, или перемещен в вектор Е. coli, предназначенный для синтеза
белка из клонированной ДНК, и, таким образом, может быть получен образец
мутантного белка.
• Синтез искусственных генов предполагает конструирование гена в пробирке
с помещением мутаций во все желаемые позиции. Ген конструируется синте-
зом ряда частично перекрывающихся олигонуклеотидов (каждый до 150 ну-
клеотидов в длину). Далее ген собирается путем заполнения пропусков между
перекрытиями при помощи ДНК-полимеразы и лигируется в клонирующий
вектор до введения в организм-хозяин или в Е coli.
• ПЦР также может использоваться, чтобы создавать мутации в клонированных
генах, хотя, подобно специфичному к олигонуклеотиду мутагенезу, за один
эксперимент может быть введена только одна мутация.
В представленном на рис. Т5.2 методе проводятся две реакции ПЦР — каждая
с одной нормальной затравкой (которая образует полностью спаренный основа-
ниями гибрид с ДНК-матрицей)
s* Т11П11Н П11111Г3‘. и °Дн°й мутагенной затрав-
кой (которая содержит един-
| ственное несоответствие пары
оснований, соответствующее
111111111111 и 111 л „
— мутации). Поэтому эта мута-
Ш.'.мл.чн»1,!мутацию ция изначально присутствует
в продуктах обеих ПЦР, каждый
соответствует одной половине
стартовой молекулы ДНК. Эти
два продукта ПЦР затем сме-
шивают вместе, и выполняет-
ся заключительный цикл ПЦР,
с тем чтобы построить полно-
размерную мутантную молеку-
луДНК.
,,111111II III птттт^
। Денатурация,
I отжиг затравок
г,,.Тн-ТГ,1,г,г ЗзтрйВКИ
I ПЦР | ПЦР
I Отжиг мутированных I
нитей
Заключительный
I цикл ПЦР
Мутация
Рис. Т5.2. Один метод сайт-специфи-
ческого мутагенеза посредством ПЦР
Глава 5. Определение структуры последовательности генома 211
последовательность генома уже была полностью расшифрована, и дали следующие
результаты (рис. 5.28).
• Почти к 30 % генов в геноме могли быть приписаны функции после поиска го-
мологии в базах данных последовательностей. Приблизительно половина имела
менее яркие подобия, при этом у многих из их числа подобия были ограничены
отдельными доменами. Для всех этих генов анализ гомологии мог быть сочтен
успешным, но все же имел различные степени точности. Для некоторых генов опо-
знавание гомолога позволило в полной мере определить функции гена дрожжей:
к таким примерам относится распознавание генов дрожжей, кодирующих субъе-
диницы ДНК-полимеразы. Для других генов приписывание функций могло быть
осуществлено только в масштабе широкой категории типа «ген протеинкиназы»:
другими словами, могли быть выведены биохимические свойства продукта гена,
но отнюдь не конкретная роль этого белка в клетке. Некоторые результаты рас-
познавания были первоначально озадачивающими, и лучшим примером этого
служит открытие дрожжевого гомолога гена бактерии, участвующего в усвоении
азота. Дрожжи не усваивают азот, так что это не могло быть функцией гена дрожжей.
В данном случае открытие дрожжевого гомолога перефокусировало внимание на
ранее охарактеризованный ген бактерии и привело к последующему осознанию
того, что, наряду с участием в усвоении азота, основная роль продукта этого гена
бактерии заключается в синтезе металлосодержащих белков, которые играют
многоплановые роли во всех организмах, а не только в усваивающих азот.
• Оказалось, что около 10 % всех генов дрожжей имеют гомологов в базах данных,
но функции этих гомологов были неизвестны. Поэтому анализ гомологии был
неспособен помочь приписыванию функций к этим генам дрожжей. Такие гены
дрожжей и их гомологи были названы сиротскими семействами.
• Остальные гены дрожжей, приблизительно 30 % от общего количества, не имели
никаких гомологов в базах данных. Доля из них (приблизительно 7 % от общего
количества) была представлена сомнительными ОРС, которые могли не быть истин-
ными генами, будучи довольно короткими или имеющими необычное отклонение
частоты использования кодонов. В числе оставшихся были последовательности,
напоминавшие гены, но представленные в единичном варианте. Их назвали оди-
ночными сиротами.
После первичной аннотации последовательности генома дрожжей необходимо
рассмотреть два вопроса: во-первых, сколько из одиночных сирот является истинными
генами и, во-вторых, существуют ли какие-либо истинные гены меньше чем 100 кодо-
нов в длину и, следовательно, не опознанные в ходе этого предварительного анализа?
Последний вопрос важен: хотя в геноме дрожжей есть только 6274 ОРС длиной 100
кодонов и более, существует более 100000 ОРС длиной 15 кодонов и более, причем
большинство из них показывают модель использования кодонов, неотличимую от свой-
ственной истинным генам дрожжей. Поэтому потенциал для поиска новых, коротких
генов был огромен.
При уточнении набора генов дрожжей были использованы три подхода, все осно-
ваны на методах, с коими мы уже встречались в этой главе.
• Сравнительная геномика, которая использует набор последовательностей гено-
ма родственных видов дрожжей, была использована, чтобы оценить истинность
многих коротких ОРС.
212 Часть 1. Изучение геномов
Сомнительные ОРС
Неопознанные члены
сиротских семейств
Рис. 5.28. Итоговая сводка результатов первоначальной аннотации генома дрожжей
• Данные транскрипции собирались секвенированием молекул кДНК, включая би-
блиотеки ярлыков экспрессируемых последовательностей (EST), — которые пред-
ставлены короткими и обычно неполными кДНК, получаемыми из концов транс-
крибируемых последовательностей (раздел 3.3.3), — последовательным анализом
экспрессии генов (SAGE; раздел 6.1.1), а также исследованиями на микроматрицах
и микрочипах (раздел 6.1.2).
• Мечение транспозонами, применяемое также для инактивации генов в ходе
функционального анализа, было использовано для опознавания ОРС, которые
являются истинными генами. В данной работе использовался транспозон, содер-
жащий копию гена lacZбез его старт-кодона (рис. 5.29). В нормальных клетках ген
lacZ поэтому является неактивным, так что при испытании в Х-геле (раздел 2.2.1)
колонии выглядят белыми. Ген lacZстановится активированным, если транспози-
ция перемещает его в позицию внутри истинного гена дрожжей, и вставка приво-
дит к слиянию кодонов гена дрожжей, причем в одной рамке с геном lacZ. Теперь
колонии выглядят синими. Сомнительные ОРС могут поэтому быть оценены по
цвету колоний после того, как ОРС стала меченой транспозоном.
Ген lacZ (без кодона запуска)
Транспозон
Транспозиция
в ген дрожжей
Рамочное
слияние
Ген lacZ активен
Рис. 5.29. Применение мечения транспозо-
нами для опознавания генов дрожжей. Если
транспозон перемещается в функциональ-
ный ген дрожжей, так что между началом
гена дрожжей и геном lacZ, присутствующим
в транспозоне, образуется открытая рамка
считывания, то ген lacZ получает возмож-
ность быть экспрессированным. Полученный
белок р-галактозид содержит в себе сегмент
белка, кодируемого геном дрожжей, смеж-
но с его N-концом, но часто это не влияет
на деятельность фермента. Следовательно,
транспозиция в функциональный ген дрож-
жей обнаружима проверкой Х-гелем
Глава 5. Определение структуры последовательности генома 213
Эти эксперименты продолжаются, но их результат пока что сводится к сокращению
каталога генов дрожжей приблизительно до 6120 ОРС, что примерно на 150 единиц
меньше первоначальной оценки. Такое сокращение произошло в результате отбрасыва-
ния многих из первоначальных одиночных сирот, которые больше не рассматриваются
как возможные гены. При этом в список добавилось несколько новых генов, которые
короче 100 кодонов, но все же оказались истинными генами.
5.3.2. Приписывание функций генам дрожжей
S. cerevisiae имеет две особенности, которые помогают попыткам приписать функ-
ции к неизвестным генам в его геноме. Первая из них — высокая природная склонность
к гомологичной рекомбинации, благодаря которой относительно легко использовать
этот подход для инактивации отдельных генов (см. рис. 5.20). Вторая — присутствие
в геноме транспозонов семейства Ту, которые позволяют использовать мечение транс-
позонами как альтернативное средство разрушения генов. Оба подхода были интенсивно
использованы исследователями дрожжей. Теперь основная задача заключается не в раз-
работке методов инактивации отдельных генов дрожжей, но в изобретении способов
просмотра огромных количеств появляющихся мутантов на предмет наличия новых
фенотипических особенностей, которые могли бы указать функцию инактивированного
гена. Выполнение многих параллельных экспериментов, каждый со своим мутантом,
занимает много времени, особенно когда есть такое большое количество фенотипов для
оценки. Поэтому необходимы стратегии массового отборочного анализа.
Наиболее успешным из таких методов просмотра является методика штрих-
кодированных кассет удаления. Это модификация основной системы кассеты удале-
ния, показанной на рис. 5.21; отличие состоит в том, что кассета, помимо всего прочего,
включает в себя также две 20-нуклеотидные последовательности «штрихового кода»,
уникальные для каждого удаления, которые служат ярлыками для каждого конкрет-
ного мутанта (рис. 5.30). К каждому штриховому коду примыкает с обеих сторон одна
и та же пара последовательностей, так что он может быть размножен единой ПЦР. Это
означает, что группы мутантных линий дрожжей (каждая с иным инактивированным
геном) могут быть смешаны вместе и их фенотипы — просматриваться в едином экс-
перименте. Например, чтобы опознать гены, необходимые для роста в обогащенной
глюкозой среде, можно смешать вместе коллекцию мутантов и культивировать при
Сегмент гена Сегмент гена
дрожжей капг дрожжей
Молекулярные штрих-коды
S Затравочные участки ПЦР
Рис. 5.30. Кассета удаления, используемая в методике штрихового кода. Сравните с кассетой, показан-
ной на рис. 5.2.1. Два молекулярных штриховых кода представлены последовательностями длины 20
нуклеотидов, иных для каждой кассеты, которые могут быть размножены при помощи ПЦР. В ходе гомо-
логичной рекомбинации штриховые коды вставляются в геном дрожжей наряду с геном устойчивости
к канамицину. Поэтому штриховые коды представляют собой специфичные ярлыки для обозначения
удаления каждого отдельного гена
214 Часть 1. Изучение геномов
этих условиях. После инкубации из культуры выделяется ДНК и выполняется штрих-
кодовая ПЦР. Результат — смесь продуктов ПЦР, где каждый из них представляет собой
отдельный штриховой код, а относительное содержание каждого штрихового кода по-
казывает содержание каждого мутанта после роста в обогащенной глюкозой среде. Те
штриховые коды, которые отсутствуют или присутствуют только в малом количестве,
показывают мутанты, инактивированные гены которых были необходимы для роста
при этих условиях.
Как и распознавание генов дрожжей, их функциональная характеризация продол-
жается, и пройдет несколько лет, прежде чем этот проект войдет в завершающую фазу.
Но мало-помалу работа продвигается вперед. Приблизительно 55 % всех генов дрожжей
теперь имеют хорошо охарактеризованную функцию, которая была приписана на осно-
ве одного или нескольких экспериментальных методов. Это приблизительно на 1500
генов больше, чем имело место, когда геном был впервые секвенирован. Другие 2 000
генов — 33 % от общего числа — имеют функции, которые были приписаны на основе
анализа гомологии. Сюда относится лишь 500 ОРС, которые, как думают, являются ис-
тинными генами, но не имеют никакой приписанной функции, и еще 300 сомнительных
ОРС, которые могут не быть истинными генами1).
Заключение
Для опознавания генов в геноме и определения функций этих генов было изобрете-
но целое разнообразие методов. Из числа этих методов одни являются компьютерными,
а другие вовлекают экспериментирование. После определения последовательности
генома ставится задача определить местонахождение позиций всех ее генов. Для коди-
рующих белок генов эту задачу можно попытаться решить путем поиска открытых рамок
считывания (ОРС), хотя у эукариотов этот путь усложняется присутствием интронов,
граничные последовательности которых являются изменчивыми и не могут быть опо-
знаны точно. Также может быть определено местоположение генов функциональных
РНК посредством поиска их характерных особенностей и прежде всего — отвечающих
за способность молекул РНК сворачиваться во вторичные структуры за счет форми-
рования спаренных основаниями стебле-петельных структур. Местоположение генов
может быть определено также анализом гомологии, который использует присутствие
эквивалентного гена во втором геноме как свидетельство в пользу того, что предпола-
гаемый ген в испытываемом геноме является истинным. Анализ гомологии становится
более мощным, если в наличии имеется полностью расшифрованная последовательность
родственного генома. Экспериментальные методы определения местоположения генов
основаны на обнаружении молекул РНК, транскрибируемых с генома. К таким методам
относится секвенирование кДНК и картирование транскриптов посредством ревертаз-
ной ПЦР (РТ-ПЦР) или путем гетеродуплексного анализа. Функции гена могут быть при-
писаны экспериментально — по результатам анализа гомологии, потому что гомологи
эволюционно связаны и часто, хотя и не всегда, имеют подобные функции. Большинство
экспериментальных методов определения функций генов предполагает изучение эф-
фекта, который инактивация гена оказывает на фенотип организма. Инактивация может
быть достигнута различными способами: гомологичной рекомбинацией с дефектной
копией гена, вставкой транспозона в ген или РНК-интерференцией (последний из этих
подходов был особенно успешен с Caenorhabditis elegans). Сверхэкспрессия гена также
Впрочем, они могут быть истинными генами, просто исследователи пока не смогли найти такие
условия, когда они экспрессируются. — Прим. ред.
Глава 5. Определение структуры последовательности генома 215
может быть использована для оценки функции, но и в случае инактивации, и в случае
сверхэкспрессии может быть трудно различить изменение фенотипа, и точная функция
гена может остаться неуловимой. Более подробные исследования функции гена могут
быть выполнены сайт-специфическим мутагенезом, и местоположение белка в клетке
может быть установлено по экспрессии гена-репортера или при помощи иммуноцито-
химии. Когда последовательность генома Saccharomyces cerevisiae была окончательно
расшифрована в 1996 г., 6274 ОРС, которые могли быть генами, были опознаны, но эта
цифра была к настоящему времени пересмотрена до 6120 ОРС на основе данных экспе-
риментального анализа и сравнений с геномами других дрожжей. Первоначально только
30 % этих генов имели хорошо охарактеризованные функции, но это число постепенно
было увеличено за счет анализа гомологии и применения высокопроизводительных
функциональных отборочных испытаний, таких как методика штрих-кодированных
кассет удаления.
216 Часть 1. Изучение геномов
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
5.1 *. Что такое открытая рамка считыва-
ния (ОРС)?
а. Все нуклеотиды гена, которые
транскрибируются в мРНК.
Ь. Нуклеотиды гена, которые об-
разуют кодоны, определяющие
аминокислоты.
с. Нуклеотиды молекулы мРНК до
удаления из нее интронов.
d. Аминокислотная последователь-
ность полипептида.
5.2 . Что понимают под отклонением ча-
стоты использования кодонов?
а. Одни из кодонов, задающих не-
которую аминокислоту, использу-
ются у всех видов чаще других.
Ь. Некоторые аминокислоты редко
используются в белках некоторых
организмов.
с. Одни из кодонов, задающих не-
которую аминокислоту, исполь-
зуются чаще других, и такое от-
клонение различно у разных
видов.
d. У иных видов некоторые кодоны
кодируют редкие аминокислоты
типа селеноцистеина.
5.3 *. Консенсусная последовательность
экзон-интронной границы или про-
мотора гена — это:
а. Точная последовательность ну-
клеотидов, необходимая для
функционирования этой после-
довательности.
Ь. Последовательность нуклеоти-
дов, наблюдаемая в этих участках
наиболее часто.
с. Наикратчайшая последователь-
ность нуклеотидов, необходимая
для функционирования этой по-
следовательности.
d. Последовательность нуклеоти-
дов, окружающая сайты сплай-
синга интронов или сайты ини-
циации транскрипции.
5.4 . Почему просмотр на ОРС не приго-
ден при поиске генов, кодирующих
молекулы функциональной РНК?
а. Кодоны для молекул функцио-
нальной РНК изменяются от вида
к виду.
Ь. Гены молекул функциональной
РНК содержат интроны, которые
делают просмотр на ОРС невоз-
можным.
с. Кодоны в генах функциональной
РНК имеют лишь по два нуклео-
тида в длину.
d. Гены функциональной РНК не со-
держат кодонов.
5.5 *. Какова цель выполнения поиска го-
мологии последовательности ДНК?
а. Определить, присутствуют ли
в базах данных ДНК какие-либо
гены с подобными последова-
тельностями.
Ь. Определить, находится ли уже
данная последовательность
в базе данных.
с. Искать согласованные экзон-
интронные границы.
d. Определить отклонение частоты
использования кодонов в опреде-
ленном гене.
5.6. Какая из следующих формулировок
является правильным определени-
ем синтении?
а. Процент идентичности нуклео-
тидных последовательностей
двух геномов.
Ь. Процент идентичности амино-
кислотных последовательностей,
кодируемых двумя геномами.
Глава 5. Определение структуры последовательности генома 217
с. Консервативность порядка следо-
вания генов в двух геномах.
d. Консервативность функций генов
в двух геномах.
5.7* . Размножение мРНК посредством
ПЦР называют:
а. ПЦР в режиме реального време-
ни.
Ь. Ревертазная ПЦР.
с. Транскрипционная ПЦР.
d. Трансляционная ПЦР.
5.8. По определению гомологичные
гены — это гены, которые:
а. Имеют общую функцию.
Ь. Имеют общего эволюционного
предка.
с. Экспрессируются в подобных
условиях.
d. Имеют по крайней мере 50%-ю
идентичность последовательно-
стей нуклеотидов.
5.9* . Аминокислотная последовательность
a-цепи гемоглобина более подобна
аминокислотной последовательности
P-цепи гемоглобина, чем последова-
тельности аминокислот миоглобина.
Все эти гены имеют общего эволюци-
онного предка. Какое из следующих
утверждений верно описывает отно-
шения между генами, кодирующими
эти полипептиды?
а. Ген, кодирующий a-цепь, имеет
более высокую степень подобия
с геном p-цепи, чем с геном мио-
глобина.
Ь. Гены, кодирующие эти три поли-
пептида, — все гомологи.
с. Ген миоглобина не является гомо-
логом двух остальных генов.
d. Все эти гены суть гомологи, если
они присутствуют в организме
одного вида.
5.10. Почему инактивация — полезный
метод определения функции гена?
а. Инактивация генов дает инфор-
мацию об экспрессии исследуе-
мого гена.
Ь. Инактивация генов дает инфор-
мацию о местоположении продук-
та исследуемого гена в клетке.
с. Инактивация генов дает возмож-
ность опознать изменения фено-
типа, связанные с потерей функ-
ционального гена.
d. Инактивация генов дает инфор-
мацию о структуре продукта ис-
следуемого гена.
5.11 *. Зародышевые стволовые клетки
мыши используются в эксперимен-
тах с инактивацией генов, потому
что они:
а. Могут быть клонированы, чтобы
дать начало устойчивой клеточ-
ной линии.
Ь. Являются химерными и произво-
дят клетки, гетерозиготные по ис-
следуемому гену.
с. Являются единственными клет-
ками мыши, которые можно гене-
тически конструировать, с целью
инактивации генов.
d. Являются полифункциональны-
ми и способны дать начало диф-
ференцированным клеткам всех
типов.
5.12 . Которым из следующих методов ра-
ботает РНК-интерференция?
а. Использование антисмысловых
молекул РНК, с тем чтобы блокиро-
вать трансляцию молекул мРНК.
Ь. Использование ингибиторов РНК-
полимеразы, с тем чтобы блоки-
ровать транскрипцию определен-
ных генов.
с. Использование коротких молекул
двунитевой РНК, которые вы-
зывают деградацию молекулы
мРНК.
d. Использование видоизмененных
молекул тРНК, с тем чтобы бло-
кировать трансляцию молекул
мРНК.
5.13 *. Какой из нижеперечисленных спо-
собов является наилучшим для опо-
218 Часть 1. Изучение геномов
знавания местоположения белка
в клетке?
а. Поместить рядом с промотором
гена, кодирующего этот белок,
ген-репортер и опознать место-
положение соответствующего
белка-репортера в клетке.
Ь. Использовать меченое антитело,
с тем чтобы опознать местополо-
жение белка в клетке.
с. Отделить клеточные компартмен-
ты центрифугированием и зонди-
ровать различные компартменты
антителом.
Вопросы открытого типа
5.1 *. Почему опознавать ОРС в геномах
прокариотов при помощи компью-
терного анализа относительно лег-
ко?
5.2 . Каковы две главные проблемы, ко-
торые возникают при попытке ис-
пользовать компьютерный анализ
для опознавания ОРС в геномах выс-
ших эукариотов?
5.3 *. Каковы три основные модификации,
которые могут быть применены,
с тем чтобы улучшить определение
местоположения ОРС компьютер-
ным анализом?
5.4 . Какие структурные особенности мо-
лекул функциональной РНК (таких
как тРНК и рРНК) можно отыскивать
в последовательности генома, что-
бы опознать гены, кодирующие эти
молекулы РНК?
5.5 *. Каковы два ограничения, которые
возникают, когда для определе-
ния числа генов, присутствующих
во фрагменте ДНК, используется
нозерн-гибридизационный ана-
лиз?
d. Пометить белок флюоресцентны-
ми аминокислотами и опознать
его местоположение в клетке
флюоресцентной микроскопией.
5.14. Сиротские семейства — семейства
генов, которые:
а. Не имеют гомологов в других ви-
дах.
Ь. Не имеют никакой известной
функции.
с. Не проявляют никакого измене-
ния фенотипа после инактивации
гена.
d. Еще не изучались.
5.6 . Опишите применение метода бы-
строго размножения концов кДНК
(RACE) для картирования участка
начала транскрипции гена.
5.7 *. В чем состоит различие между орто-
логичными и паралогичными гена-
ми?
5.8 . Почему иногда делаются ошибки
при приписывании функции к гену
на основании результатов поиска
BLAST?
5.9 *. Какую информацию о болезнях че-
ловека можно извлечь из результа-
тов изучения гомологичных генов?
5.10 . Каково правдоподобное объяснение
того, что после спаривания между
гетерозиготными родителями не
получаются нокаутные мыши?
5.11 *. Какие подходы могут быть исполь-
зованы при анализе последователь-
ности генома, чтобы опознать ис-
тинные гены длиной не более 100
кодонов?
Глава 5. Определение структуры последовательности генома 219
Изыскательские задачи
5.1 *. До какой, вы думаете, степени в бу-
дущем будет возможно использовать
биоинформатику, чтобы получать
полное описание местоположений
и функций кодирующих белок генов
в последовательности генома всяко-
го эукариота?
5.2 . Исследования инактивации генов
предполагали, что по крайней мере
некоторые гены в геноме избыточ-
ны, то есть имеют одну и ту же функ-
цию и, таким образом, могут быть
инактивированы без затрагивания
фенотипа организма. Какие эволю-
ционные вопросы возникают в свя-
зи с генетической избыточностью?
Каковы возможные ответы на эти
вопросы?
5.3 *. Выполните поиск BLAST со следую-
щей последовательностью амино-
кислот: IRLFKGHPETLEKFDKFKHL.
Что за белок содержит эту амино-
кислотную последовательность? Яв-
ляются ли гомологичные последова-
тельности, опознанные в ходе этого
поиска, большей частью ортолога-
ми или паралогами? (Поиск BLAST
может быть выполнен на веб-узле
www.ncbi.nlm.nih.gov/BLAST/).
5.4 . Пока что сверхэкспрессия гена обе-
спечила хотя и неполную, но важную
информацию о функциях неизвест-
ных генов. Оцените общий потен-
циал этого подхода, применяемого
в функциональном анализе.
Тесты по рисункам
5.1 *. Как компьютерная программа может определить различие между стоп-кодоном
в интроне и фактическим стоп-кодоном в конце экзона?
Экзон
Интрон
Экзон
ATGGGCAATGCAAGGTACGGTGAGCAGGTAAGTGATTAATGCATTTCTCGCAGTGGCTAGACGATGCATAG
М G N A R У G Е Q ........................ WL D D А *
MGNARYGEQVSD*
220 Часть 1. Изучение геномов
5.2. Какова цель выполнения гибриди-
зации по Саузерну с геномной ДНК
различных организмов?
5.3*. Какова цель помещения гена, ко-
дирующего GFP (зеленый флюо-
ресцентный белок), под промотор
интересующего нас гена?
Промотор
1 Гомологичная рекомбинация заменяет
целевой ген геном-репортёром
Ген-репортёр
1 Гибридизация по Саузерну
с зондом из ДНК человека
Зонд гибридизируется со
всеми образцами ДНК
5.4. Объясните, каким образом методика
штрих-кодированных кассет удале-
ния применяется для опознавания
фенотипических свойств делецион-
ных мутантов дрожжей.
J Затравочные участки ПЦР
Глава 5. Определение структуры последовательности генома 221
Дополнительная литература
Определение местоположения генов посредством компьютерного анализа
Fickett, J. W. (1996) Finding genes by computer: the state of the art. Trends Genet1). 12:
316-320.
Kellis, M., Patterson, N., Birren, B. and Lander, E. S. (2003) Sequencing and comparison of
yeast species to identify genes and regulatory elements. Nature 423'.241-254. Using comparative
genomics to annotate the yeast genome sequence.
Ohler,U. and Niemann, H. (2001) Identification and analysis of eukaryotic promoters:
recent computational approaches. Trends Genet 17: 56-60.
Pavesi,G., Mauril,G., Stefanil,M. and Pesole,G. (2004) RNA Profile: an algorithm for
finding conserved secondary structure motifs in unaligned RNA sequences. Nucleic Acids Res.
32: 3258-3269. Locating functional RNA genes.
Экспериментальные методы определения местоположения генов
Church,D.M., Stotler,С.J., Rutter,J.L, Murrell,J.R., TrofatterJ.A. and Buckler,A.).
(1994) Isolation of genes from complex sources of mammalian genomic DNA using exon
amplification. Nat Genet 6: 98-105. Exon trapping.
Frohman,M.A., Dush,M.K. and Martin, G.R. (1988) Rapid production of full-length
cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer.
Proc. Natl Acad. Sci. USA 85: 8998-9002. RT-PCR.
Lovett, M. (1994) Fishing for complements: finding genes by direct selection. Trends
Genet 10: 352-357. cDNA capture.
Приписывание функции на основании анализа гомологии
Altschul, S. F., Gish,W., Miller, W., Myers, E.W. and Lipman, D.). (1990) Basic local
alignment search tool./. Mol. Biol. 215: 403-410. The BLAST program.
Bassett, D. E., Boguski, M. S. and Hieter, P. (1996) Yeast genes and human disease. Nature
379: 589-590. Studying human disease genes by comparisons with the yeast genome.
Henikoff,S. and HenikoffJ.G. (1992) Amino acid substitution matrices from protein
blocks. Proc. NatlAcad.Sci. USA 89:10915-10919. Describes the chemical relationships between
amino acids, from which sequence similarity scores are calculated.
Исследования с применением РНК-интерференции
Fraser, A. G., Kamath, R. S., Zipperlen, P., Martinez-Campos, M., Sohrmann,M. and
AhringerJ. (2000) Functional genomic analysis of C. elegans chromosome I by systematic
RNA interference. Nature 408: 325-330.
Kittier, R., Putz, G., Pelletier, L., etal. (2004) An endoribonuclease-prepared siRNA screen
in human cells identifies genes essential for cell division. Nature 432:1036-1040.
Novina,C.D. and Sharp, P. A. (2004) The RNAi revolution. Nature 430:161-164.
Sonnichsen, B., Koski, L. B., Walsh, A., et al. (2005) Full-genome RNAi profiling of early
embryogenesis in Caenorhabditis elegans. Nature 434: 462-469.
По-видимому, ключевыми работами здесь является Burge С., Karlin S. (1997). Prediction of complete gene
structures in human genomic DNA. J Mol Biol 268:78-94 и Besemer J., Borodovsky M. (2005)GeneMark: web software
for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. Jul l;33(Web Server issue):W451-4. —
Прим. ped.
222 Часть 1. Изучение геномов
Прочие методы инактивации генов
Evans, М. J., Carlton, М. В. L and Russ, А. Р. (1997) Gene trapping and functional genomics.
Trends Genet 13: 370-374. The use ofES cells.
Ross-Macdonald, P., Coelho, P. S. R., Roemer, T., et al. (1999) Large-scale analysis of the
yeast genome by transposon tagging and gene disruption. Nature 402: 413-418.
Wach, A., Brachat, A., Pohlmann, R. and Philippsen, P. (1994) New heterologous modules
for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793-1808.
Gene inactivation by homologous recombination.
Аннотирование последовательности генома дрожжей
Dujon, В. (1996) The yeast genome project: what did we learn? Trends Genet 12:263-270.
A summary of the initial annotation.
Giaever, G., Chu, A. M., Connelly, C., et al. (2002) Functional profiling of the Saccharomyces
cerevisiae genome. Nature 418: 387-391. The barcode deletion strategy.
Snyder, M. and Gerstein, M. (2003) Defining genes in the genomics era. Science 300:
258-260. Summarizes the methods used to annotate the yeast genome, and the progress made
up to 2003.
Глава 6
Постижение механизмов
функционирования генома
По прочтении главы 6 вы сможете:
• Описывать возможные пути изучения транскриптома средствами анализа по-
следовательностей молекул кДНК.
• Оценивать сильные и слабые места технологии микроматриц и чипов в изуче-
нии транскриптомов и объяснять, как сравнивать профили экспресии генов,
выявляемыми в результате этих исследований.
• Приводить примеры вкладов, которые исследования транскриптома внесли
в наше понимание биологии дрожжей и рака человека.
• Сравнивать характер информации, получаемой в ходе изучения транскриптомов
и протеомов.
• Описывать методику выполнения анализа белкового профиля.
• Сравнивать и сопоставлять методы, применяемые для опознавания пар и групп
белков, которые взаимодействуют друг с другом в живых клетках, в частности,
проводя различие между методами, с помощью которых опознают физические
взаимодействия, и методами, при помощи которых опознают функциональные
взаимодействия.
• Приводить примеры карт белок-белкового взаимодействия и обсуждать их
важные особенности.
• Объяснять основу и важность анализа биохимических профилей.
• Озвучивать принципы и цели системной биологии.
Из предыдущей главы мы узнали, как использовать разнообразие вычислитель-
ных и экспериментальных методов для приписывания функций к генам, найденным
в последовательности генома, и что применение этих методов к геному Saccharomyces
cerevisiae почти удвоило число генов дрожжей, для которых были определены функции.
Такого типа аннотация генома — грандиозное предприятие, но даже если каждый ген
в геноме может быть опознан и наделен функцией, все еще остается одна проблема. Она
заключается в необходимости понять, как геном в целом работает в пределах клетки,
определяя и координируя различные биохимические процессы, которые в ней протека-
ют. Эти глобальные исследования деятельности генома должны обратиться не только
к самому геному, но также и к транскриптому и протеому, которые синтезируются и под-
держиваются геномом. Они должны обратиться также к пути, которым транскриптом
и протеом устанавливают и координируют конечную цель экспрессии генома — сеть
связанных биохимических путей и процессов, которые составляют биохимию живой
224 Часть 1. Изучение геномов
клетки. В этой главе мы рассмотрим методы, применяемые в ходе таких глобальных
исследований функционирования генома.
6.1. Изучение транскриптома
Транскриптом включает в себя молекулы мРНК, которые присутствуют в клетке
в определенное время. Транскриптомы могут иметь очень сложные составы, пред-
ставленные сотнями или тысячами различных мРНК, где каждая из них составляет
особенную долю в общей совокупности (раздел 1.2.4). Поэтому чтобы охарактеризовать
транскриптом, необходимо опознать виды мРНК, которые он содержит и, в идеале,
определить их относительное содержание.
6.1.1. Изучение транскриптома посредством анализа последовательно-
сти
Самый прямой способ охарактеризовать транскриптом состоит в том, чтобы преоб-
разовать составляющие его мРНК в кДНК (см. рис. 3.36) и затем секвенировать каждый
клон в полученной библиотеке кДНК. Сравнения между последовательностями кДНК
и последовательностью генома покажут тождества генов, мРНК которых присутствуют
в транскриптоме. Этот подход выполним, но он трудоемок, причем необходимо опреде-
лить множество последовательностей различных кДНК прежде, чем (почти полная)
картина состава транскриптома начнет вырисовываться. Если сравниваются два и бо-
лее транскриптомов, то время, необходимое для завершения проекта, увеличивается.
Можно ли применить какие-либо упрощения, чтобы получить важнейшую информацию
о последовательности сколько-нибудь быстрее?
Последовательный анализ экспрессии генов (SAGE) предлагает решение этого
вопроса. Вместо того чтобы изучать полные кДНК, SAGE дает короткие последователь-
ности (до 12 п.н. в длину), каждая из которых представляет мРНК, присутствующую
в транскриптоме. Основа данного метода в том, что эти последовательности длины
12 п. н., несмотря на их короткость, являются достаточными, чтобы опознать ген, кото-
рый кодирует мРНК. Обоснованием такого подхода является тот факт, что всякая после-
довательность длиной 12 п.н. должна появляться в геноме единожды на каждые 412 =
= 16 777216 п. н. Средний размер мРНК эукариота составляет приблизительно 1500 п. н.,
так что 412 п. н. эквивалентно совокупной длине более чем 11 000 транскриптов. Это число
превышает число транскриптов, ожидаемых во всех, кроме наиболее сложных, транс-
криптомах, так что ярлыки последовательности длины 12 п. н. должны быть способны
опознавать гены, кодирующие молекулы мРНК всех производимых клеткой видов.
Процедура, используемая для получения ярлыков длины 12 п. н., представлена на
рис. 6.1. Сначала мРНК закрепляется в хроматографической колонке отжигом поли-А
хвостов, имеющихся на 3'-концах этих молекул, к олиго-дТ нитям, которые были при-
соединены к целлюлозным бусинам. мРНК преобразуется в двунитевую кДНК и затем
обрабатывается ферментом рестрикции, который распознает целевой участок длины
4 п. н. и таким образом производит частые разрезы в каждой кДНК. Конечный фрагмент
рестрикции каждой кДНК остается прикрепленным к целлюлозным бусинам, позволяя
элюировать и отбросить все остальные фрагменты. Короткий олигонуклеотид теперь
присоединяют к свободному концу каждой кДНК, этот олигонуклеотид содержит сайт
узнавания для BsmFI. Этот фермент рестрикции необычен тем, что вместо того чтобы раз-
резать в пределах своего сайта узнавания, он разрезает в позиции на 10-14 нуклеотидов
Глава 6. Постижение механизмов функционирования геномов 225
Рис. 6.1. SAGE. В этом примере первый фермент рестрик-
ции, который используется, — А1и\, он опознает целевой
участок 5'-AGCT-3' длиной 4 п. н. Олигонуклеотид, который
лигируется к кДНК, содержит сайт узнавания для BsmFI,
который производит разрез в 10-14 нуклеотидах ниже
и таким образом отщепляет фрагмент кДНК. Фрагменты
различных кДНК лигируются, с тем чтобы произвести сцеп-
ку, которая секвенируется. Образуемая при использовании
этого метода сцепка частично состоит из последователь-
ностей олигонуклеотидов, полученных перевариванием
BsmFI. Чтобы избежать этого, то есть получить сцепку, со-
стоящую полностью из фрагментов кДНК, олигонуклеотид
может быть сконструирован так, чтобы конец, который
лигируется к кДНК, содержал сайт узнавания для третьего
фермента рестрикции. Обработка этим ферментом отще-
пляет олигонуклеотид от фрагмента кДНК
ниже. Поэтому обработка HsmFI удаляет с конца
каждой кДНК фрагмент со средней длиной 12 п. н.
Фрагменты собирают, лигируют по принципу «го-
лова к хвосту», чтобы произвести сцепку, и секве-
нируют. Отдельные последовательности-ярлыки
опознаются в пределах сцепки и сравниваются
Целлюлозная
бусина
Олиго-дТ нить тттт -J
| Добавление РНК
nui/ 5 тттт
иРНК АААА^Ч^
кДнк’;
ПТТТТ
I Преобразование
* в двунитевую кДНК
тттт
АААА^^
Аперевариваривание Alul
ШПШПаааа
Сбпягыяячмр |Лигирование олигонуклеотида,
Ь?пчрччпгг> ^содержащего опознавательную
конечного последовательность для BsmFI
фрагмента £тттт
ЩШШиаааа
фрагмента
I Переваривание
* посредством BsmFI
нпшп шПИ
Собрание, лигирование
с другими фрагментами,
секвенирование
IIIIIIIIIIIIIIIH
с последовательностями генов в геноме.
6.1.2. Изучение транскриптома с помощью анализа на микроматрицах
или чипах
Чипы и микроматрицы ДНК (см. техническое примечание 3.1) также могут быть
использованы для изучения транскриптомов. Вспомним, что разница между ними
в том, что чип несет массив закрепленных олигонуклеотидов, синтезируемых in situ на
поверхности стеклянной или кремниевой подложки, а микроматрицы содержат моле-
кулы ДНК — обычно продукты ПЦР или молекулы кДНК, — которые были нанесены на
поверхность стеклянной пластинки или нейлоновой мембраны. Как микроматрицы, так
и чипы используются одним и тем же способом (рис. 6.2). Совокупность молекул мРНК,
которые составляют транскриптом, преобразуется в смесь кДНК, меченых (обычно
флюоресцентной меткой) и нанесенных на микроматрицу или чип, и детектируются
позиции, в которых происходит гибридизация. По сравнению с SAGE, этот подход имеет
то преимущество, что быстрая оценка различий между двумя и более транскриптомами
может быть сделана за счет гибризизации различных препаратов кДНК с идентичными
массивами и сравнения гибридизационных картин. Дальнейшее усовершенствование
может быть достигнуто за счет зондирования массива кДНК, приготовленного из доли
мРНК, которая связывается с рибосомами в изучаемых клетках, а не из общей массы
мРНК. Связанные молекулы мРНК соответствуют части транскриптома, которая активно
направляет синтез белка и дает несколько отличную картину деятельности генома.
Сначала мы рассмотрим технические вопросы, связанные с исследованиями на
микроматрицах и чипах, а затем обсудим некоторые варианты применения такого рода
анализа.
226 Часть 1. Изучение геномов
Микроматрица
1 Гибридизация
с меченой кДНК
Рис. 6.2. Анализ на микроматрицах. Препарат кДНК метится
флюоресцентной меткой и гибридизируется на микроматрицу.
Метка обнаруживается конфокальным лазерным сканером,
и интенсивность сигнала преобразуется в спектр псевдоцве-
тов, где красный означает наибольшую гибридизацию, за ко-
торым следуют оранжевый, желтый, зеленый, голубой, синий
и фиолетовый (последний представляет фоновый уровень
гибридизации). Для получения дополнительной информации
о приготовлении препарата и использовании микроматрицы
см. техническое примечание 3.1. Изображение любезно пре-
доставлено Томом Страчаном и перепечатано с разрешения
журнала Nature
Применение микроматрицы или чипа для изучения одного или нескольких
транскриптомов
При анализе транскриптома ставятся две ключевые задачи: опознать гены, мРНК
которых присутствуют в транскриптоме, и определить относительные количества этих
различных мРНК. Первая задача требует, чтобы каждый значимый ген был представлен
в массиве по крайней мере одним зондом. С микроматрицей это достигается за счет ис-
пользования продуктов ПЦР или молекул кДНК, которые получены из интересующих
нас генов, а с чипом ДНК — за счет синтеза в каждой позиции смеси олигонуклеотидов
(всего возможно до 20 штук различных единиц), последовательности которых соот-
ветствуют различным позициям в значимом гене (рис. 6.3).
Второе требование — способность определять относительные количества от-
дельных мРНК в транскриптоме — выполняется, потому что каждая позиция в микро-
матрице или чипе содержит до 109 копий молекул зонда. Это выше, чем ожидаемое
число копий любой мРНК в маленьком количестве транскриптома, которое помещено
в массив, а это означает, что никакая позиция никогда не станет насыщенной (то есть не
дойдет до той степени гибридизации, когда каждая молекула зонда спарится с целевой
молекулой). Поэтому степень гибризизации является переменной величиной: интенсив-
ность сигнала в каждой позиции зависит от количества каждой отдельно взятой мРНК
в транскриптоме (рис. 6.4).
Из прочтенного выше, процедуры анализа на микроматрицах и чипах могут пока-
заться незатейливыми. Однако на практике возникает ряд осложнений. Первое состоит
в том, что со всеми, кроме самых простейших, транскриптомами гибридизационный
анализ будет иметь незначительную специфичность, в силу которой появляется изби-
рательность в отношении каждой мРНК, присутствующей в транскриптоме. Это связано
с тем, что две различные мРНК могут иметь подобные последовательности и в таком
случае могут перекрестно гибридизироваться со специфичными к ним зондами в масси-
Глава 6. Постижение механизмов функционирования геномов 227
Рис. 6.3. Микроматрицы и чипы ДНК. Каждая позиция микроматрицы содержит кДНК или продукт ПЦР
из интересующего гена, тогда как каждая позиция в чипе ДНК содержит смесь олигонуклеотидов, по-
следовательности которых соответствуют различным сегментам исследуемого гена
ве. Это часто случается, когда в одной и той же ткани активны два или несколько пара-
логичных гена (раздел 5.2.1). Транскриптом тогда содержит группу родственных мРНК,
каждая из которых способна гибридизироваться до некоторой степени с различными
членами семейства генов. Различение относительных количеств каждой мРНК или даже
установление того, какие именно мРНК присутствуют в транскриптоме, может в таком
случае быть затруднено. Подобная проблема возникает, когда из одного и того же гена
получены две и более различных мРНК. Такая ситуация возникает сравнительно часто
у позвоночных из-за альтернативного сплайсинга — процесса, в ходе которого экзоны
из пре-мРНК собираются в различных сочетаниях и синтезируется ряд родственных, но
различных молекул мРНК (рис. 6.5). Поэтому массив должен конструироваться очень
тщательно, ибо необходимо обнаружить и в точности определить количественно все
эти варианты.
Дальнейшие осложнения возникают, если цель состоит в сравнении двух и более
транскриптомов (что, как мы увидим ниже, является частым сценарием). Для того
чтобы сравнения были обоснованными, различия между интенсивностями гибриди-
Более
интенсивный
Цель насыщена: повышение степени
fTTirrnn гибридизации при добавлении большего
_________количества зонда не наблюдается
Цель не насыщена: сигнал гибридизации
пропорционален количеству зонда
Количество зонда
Рис. 6.4. Зависимость между интенсивностью гибридизации и количеством зонда
228 Часть 1. Изучение геномов
Рис. 6.5. Альтернативный сплайсинг. Альтернативный сплайсинг дает различные сочетания соединенных
друг с другом экзонов, что дает в конечном итоге различные белки, синтезируемые с одних и тех же
пре-мРНК
зации, определенными для одного и того же гена на двух разных микроматрицах или
чипах, должны отражать истинные различия в количестве мРНК и не должны быть
обусловлены экспериментальными факторами, такими как количество целевой ДНК
в массиве, эффективность, с которой зонд был помечен, или эффективность процесса
гибридизации. Даже в пределах одной лаборатории эти факторы редко можно контро-
лировать с абсолютной точностью, и точная воспроизводимость между различными
лабораториями в большей или меньшей степени невозможна. Это означает, что анализ
данных должен включать процедуры нормализации, которые позволяли бы точно срав-
нивать результаты экспериментов, выполненных на разных массивах. Для этого массивы
должны включать контрольные элементы отрицательного (с тем чтобы в каждом экс-
перименте мог быть определен фон) и положительного контролей (которые должны
всегда давать идентичные сигналы гибридизации). Для транскриптомов позвоночных
в качестве положительного контроля часто используется ген актина, поскольку его уро-
вень экспрессии, как правило, является довольно постоянным в определенной ткани,
независимо от стадии развития или болезненного состояния. Более удовлетворительная
альтернатива — так спроектировать эксперимент, чтобы два транскриптома могли быть
непосредственно сравнены в ходе единого анализа, выполняемого на одном массиве.
Это достигается мечением препаратов кДНК различными флюоресцентными зондами
и последующим сканированием массива при соответствующих длинах волн, чтобы
в каждой позиции определить относительные интенсивности двух флюоресцентных
сигналов и, следовательно, определить различия между содержанием мРНК двух транс-
криптомов (рис. 6.6).
Глава 6. Постижение механизмов функционирования геномов 229
Рис. 6.6. Сравнение двух транскрип-
томов в едином эксперименте. Изо-
бражение любезно предоставлено
Томом Страчаном и перепечатано
с разрешения журнала Nature
Исходя из предположе-
ния о том, что между двумя
и более транскриптомами
могут быть сделаны точные
сравнения, можно наблю-
дать и сопоставлять весьма
сложные картины экспрес-
сии генов. Гены, которые
показывают подобные про-
фили экспрессии, вероятно,
будут обладать и связанными
функциями, и для опознава-
Микроматрица,
гибридизированная /йШИ®
двумя препаратами кДНК
Сканирование при
/разной длине волны\
Транскриптом 1 Транскриптом 2
ния таких групп необходим точный метод. Стандартный метод, который называют
иерархической группировкой, производит сравнение уровней экспрессии для каждой
пары генов во всех проанализированныхтранскриптомах и приписывает веса, которые
показывают степень подобия между этими уровнями экспрессии. Эти данные могут быть
выражены в виде древовидной диаграммы, в которой гены с подобными профилями
экспрессии группируются вместе (рис. 6.7). Древовидная диаграмма дает наглядное
представление о функциональных связях между генами.
г гЪ гЪ
ч % Ъ V b <о Л
И Ген А
К Ген В
~Ген
: Ген D
ГенЕ
Рис. 6.7. Сравнение профилей экспрессии пяти генов в семи транскриптомах. Семь транскриптомов
были приготовлены из клеток в различные периоды после добавления богатого энергией питательного
вещества в среду для выращивания. После анализа данных посредством иерархической группировки,
выстраивается древовидная диаграмма, показывающая степень подобия между профилями экспрессии
этих пяти генов
Исследования транскриптома дрожжей
Имея чуть более 6000 генов, дрожжи Saccharomyces cerevisiae идеально подходят
для изучения транскриптома, и многие из ранних проектов были выполнены именно
с этим организмом. Одним из первых открытий было то, что благодаря тому, что моле-
кулы мРНК все время деградируют и повторно синтезируются, состав транскриптома
дрожжей подвергается очень небольшим изменениям, даже если биохимические осо-
бенности окружающей среды остаются постоянными. Когда дрожжи выращиваются
в богатой глюкозой среде, которая позволяет клеткам делиться с их максимальной
скоростью, транскриптом почти абсолютно устойчив, и только 19 видов мРНК пока-
230 Часть 1. Изучение геномов
зывают более чем двукратное изменение в относительном содержании за период двух
часов. Значительные изменения в транскриптоме наблюдаются только в том случае,
если глюкоза в среде для выращивания исчерпывается и клетки вынуждены переклю-
читься с аэробного дыхания на анаэробное. В ходе такого переключения уровни более
чем 700 видов мРНК возрастают в два и более раз, а других 1000 мРНК — снижаются
до менее чем половины их первоначального количества. Изменяющаяся окружающая
среда, очевидно, приводит к перестройке транскриптома для того, чтобы он мог удо-
влетворять новые биохимические потребности клетки.
Транскриптом дрожжей подвергается перестройке также в течение дифференци-
ровки клетки. Это было установлено при изучении спорообразования, которое вызывает-
ся голоданием и другими стрессовыми условиями окружающей среды. Путь споруляции
может быть разделен на четыре стадии — раннюю, среднюю, средне-позднюю и позд-
нюю — на основе морфологических и биохимических событий, которые происходят в его
ходе (рис. 6.8). Предварительные исследования показали (что не было неожиданностью),
что каждая стадия характеризуется экспрессией своего специфического набора генов.
Исследования транскриптома внесли в наше понимание процесса споруляции вклад,
состоящий из нескольких важных фактов. Наиболее значительный из них тот, что из-
менения, которые происходят в составе транскриптома, указывают, что ранняя стадия
споруляции может быть подразделена на три различные фазы, названные ранняя (I),
ранняя (II) и ранне-средняя. Уровни более чем 250 мРНК значительно увеличиваются
во время ранней споруляции, а других 158 мРНК определенно увеличиваются во время
средней стадии. Относительное содержание следующих 61 мРНКувеличивается во время
Диплоидная клетка
дрожжей
I
Прорастание, слияние спор
РАННЯЯ:
Репликация и рекомбинация ДНК
СРЕДНЯЯ:
Завершение мейоза I
СРЕДНЕ-ПОЗДНЯЯ:
Мейоз II
ПОЗДНЯЯ:
Синтез стенки споры,
прочие биохимические изменения
Рис. 6.8. Путь споруляции Saccharomyces cerevisiae. Средние три рисунка показывают деления ядра,
которые происходят в ходе спорообразования. См. рис. 3.16 для подробного представления событий,
происходящих во время мейоза I и мейоза II
Глава 6. Постижение механизмов функционирования геномов 231
средне-поздней стадии и еще 5-ти — во время поздней стадии. Есть также 600 мРНК,
относительное содержание которых уменьшается в течение споруляции. Возможно
они кодируют белки, которые необходимы в период вегетативного роста, но их синтез
должен быть выключен, когда формируются споры.
Эта работа над спорообразованием дрожжей важна по двум причинам. Во-первых,
благодаря описанию изменений в экспрессии генома, происходящих во время споро-
образования, исследования транскриптома открывают путь к изучению взаимодействий
между геномом и воздействиями факторов окружающей среды, которые инициируют
механизм спорообразования. Исследования такого рода, проводимые над относитель-
но простым организмом типа дрожжей, служат ценной моделью для изучения более
сложных процессов развития, которые протекают у высших эукариотов, включая че-
ловека. Во-вторых, некоторые из тех видов мРНК, уровень которых высок во время
спорообразования, представляют собой транскрипты генов, функции которых были
ранее неизвестны. Поэтому исследования транскриптома помогают аннотировать по-
следовательность генома, облегчая опознавание генов, роли которых в геноме не были
определены другими методами.
Транскриптом человека
Будучи обусловлен в пять раз большим числом генов, транскриптом человека
значительно более сложен, чем транскриптом дрожжей, и исследования его состава все
еще переживают период младенчества. Однако были получены некоторые интересные
результаты. Например, транскриптомы клеток различных типов были картированы
на последовательность генома человека, что дало общую картину экспрессии генов
по всем хромосомам. Это привело к тому важному открытию, что есть транскрипты,
которые картируются на области генома, о которых известно, что там никаких генов
нет. Например, были приготовлены ДНК-чипы, в которых целевые позиции отдельных
олигонуклеотидов наблюдаются в среднем один раз на каждые 35 нуклеотидов на
протяжении хромосом 21 и 22. Такие выстилающие массивы включают более одного
миллиона зондов, но только 26 000 из них лежат в пределах экзонов, присутствующих
в этих хромосомах. Однако более чем 350000 из этих зондов обнаружили мРНК1) в по
крайней мере одном из 11 изучавшихся транскриптомов человека, взятых из различных
клеточных линий (рис. 6.9). По всему геному было опознано приблизительно 10500
транскрибируемых последовательностей, причем из областей генома, про которые
ранее не думали, что они содержат какие-либо гены. Эта работа иллюстрирует важную
роль, которую анализ транскриптомов может играть в аннотации генома.
Анализ транскриптомов оказывает существенное влияние также и на исследования
болезней человека, в особенности рака. Перестройка структуры транскриптома в результате
рака впервые была открыта в 1997 г., когда было показано, что 289 мРНК присутствуют
в разительно отличающихся количествах в транскриптомах нормальных эпителиальных
клеток ободочной кишки по сравнению со злокачественными клетками ободочной кишки,
и что приблизительно половина этих мРНК показывает измененное относительное содер-
жание также и в раковых клетках поджелудочной железы. Эти наблюдения очень важны,
так как понимание различий между транскриптомами нормальных и злокачественных
клеток указывает на различия в их биохимиях и, следовательно, на новые пути к лечению
различных видов рака. Исследования транскриптома имеют применения также в диагно-
стике рака. Первое крупное достижение в этом отношении датируется 1999 годом, когда
Здесь лучше использовать термин «транскрипты», поскольку вовсе не обязательно эти РНК соот-
ветствуют белок-кодирующим генам. — Прим. ред.
232 Часть 1. Изучение геномов
15 20 25 30 35 40 45
Позиция, млн п. н.
Рис. 6.9. Анализ транскриптома хромосом 21 и 22 человека. Показана часть каждой хромосомы с её
G-картиной распределения дисков (раздел 7.1.2) и с проставленными позициями на карте (21р11.2
и т.д.). Для каждой хромосомы верхний график показывает, где расположены известные экзоны; эта
информация выражена как плотность экзонов для каждого «окна» ДНК размером 5,7 млн п. н. Нижний
график показывает позиции, в которых мРНК были обнаружены в 11 исследуемых транскриптомах; опять
же данные выражены как плотность покрытия в окне размером 5,7 млн п. н. Перепечатано с разрешения
Kapranov et al., Science, 296, 916-919. © 2002 AAAS
было показано, что транскриптом клеток острого лимфобластного лейкоза отличается от
такового клеток острой миелоидной лейкемии. Были изучены двадцать семь лимфобласт-
ных и одиннадцать миелоидных раковых образований, и, хотя все транскриптомы оказались
немного отличными, различия между двумя типами были достаточны для осуществления
однозначных идентификаций. Значение этой работы заключается в улучшенных темпах
ремиссии, которые являются достижимыми, если рак будет точно идентифицирован на
ранней стадии, прежде чем станут заметны явные морфологические признаки. Это не
относится к этим двум типам лейкемии, потому что их можно отличить негенетическими
средствами, но это важно для других видов рака, таких как неходжкинские лимфомы. Самую
распространенную разновидность этой болезни называют диффузной большой В-клеточной
лимфомой, и многие годы считалось, что все опухоли этого типа были одинаковыми. Иссле-
дования транскриптома изменили это представление и показали, что В-клеточная лимфома
может быть подразделена на два различных подтипа. Различия между транскриптомами
этих двух подтипов позволяют каждому быть связанным с различного класса В-клетками,
что стимулирует и направляет поиск специфических методов лечения, которые можно будет
подбирать для каждой лимфомы.
Глава 6. Постижение механизмов функционирования геномов 233
6.2. Изучение протеома
Исследования протеома важны из-за центральной роли, которую протеом играет
в деле наведения моста между геномом и биохимическими возможностями клетки
(раздел 1.3.2). Поэтому характеризация протеомов различных клеток является ключом
к пониманию того, как геном работает и каким образом дисфункциональная деятель-
ность генома может привести к болезням. Исследования транскриптома могут лишь
частично ответить на эти вопросы. Изучение транскриптома дает точную картину того,
какие гены активны в конкретной клетке, но дает менее точный список белков, которые
в ней присутствуют. Это связано с тем, что к факторам, которые влияют на содержание
белка, относится не только количество свободной мРНК, но также и скорость, с которой
присутствующие мРНК транслируются в белок, и скорость, с которой белки деградируют.
Кроме того, белок, который является первичным продуктом трансляции, может быть
не активным, так как некоторые белки должны подвергнуться физической и (или) хи-
мической модификации, перед тем как стать функционально активными (раздел 13.3).
Поэтому определяюще важным для понимания биохимии клетки или ткани является
определение количества активной формы белка.
Методологию, применяемую для изучения протеомов, называют протеомикой.
Строго говоря, протеомика — совокупность разнообразных методов, которые связаны
между собой только на основании их способности обеспечивать информацию о протео-
ме, при этом такая информация включает не только тождества белков-составляющих,
которые присутствуют в протеоме, но также и факторы типа функций отдельных белков
и их локализации в пределах клетки. Специальный метод, применяемый для изучения
состава протеома, называют определением профилей белков, или экспрессионной
протеомикой.
6.2.1. Определение профилей белков — методология для опознавания
белков в протеоме
Определение профилей белков базируется на двух методах — электрофорезе
белков и масс-спектрометрии, — имеющих длинные родословные, но редко применяв-
шихся вместе в эру до появления геномики. Сегодня они были объединены под эгидой
одной из главных развивающихся областей на ниве современных изысканий.
Выделение отдельных белков из числа составляющих протеом
Чтобы охарактеризовать тот или иной протеом, в первую очередь необходимо
приготовить чистые образцы составляющих его белков. Это далеко не тривиальное
мероприятие ввиду сложности среднестатистического протеома: вспомним, что клетка
млекопитающих может содержать 10000-20000 различных белков (раздел 1.3.2).
Электрофорез в полиакриламидном геле (см. техническое примечание 4.1) пред-
ставляет собой стандартный метод разделения смеси белков. В зависимости от состава
геля и условий, при которых выполняется электрофорез, различные химические и фи-
зические свойства белков могут быть положены в основу для их разделения. В наиболее
распространенном методе используется детергент, называемый додецилсульфатом
натрия, который денатурирует белки и сообщает им отрицательный заряд, который
приблизительно эквивалентен длине развернутого полипептида. При таких условиях
белки разделяются согласно их молекулярным массам, при этом наименьшие белки
мигрируют к положительному электроду быстрее всех. Как альтернатива, белки могут
быть разделены посредством изоэлектрофокусировки в геле, который содержит хи-
234 Часть 1. Изучение геномов
микаты, устанавливающие градиент pH при приложении электрического заряда. В геле
такого типа белок мигрирует к своей изоэлектрической точке — позиции в градиенте,
в которой его результирующий заряд равен нулю. При определении профилей белков
эти методы объединяются в двумерном гель-электрофорезе. В первом измерении
белки разделяются посредством изоэлектрофокусировки. Затем гель погружают в до-
децилсульфат натрия, поворачивают на 90°, и второй электрофорез, разделяющий
белки по их размерам, выполняют под прямым углом к первому (рис. 6.10). Этот подход
позволяет разделить несколько тысяч белков в одном геле.
Установка образца белка
1 _ _ _ —
= lllllllllll IIIII
Первый = Поворот Второй '• *
электрофорез = электрофорез
Рис. 6.10. Двумерный гель-электрофорез
После электрофореза окрашивание геля показывает сложную картину пятен, каж-
дое из которых содержит свой характерный белок (рис. 6.11). При сравнении двух гелей1)
различия в картинах пятен и в интенсивности соответствующих пятен показывают
различия в видах и относительных количествах отдельных белков в двух изучаемых
протеомах. Интересные пятна могут поэтому быть намечены на вторую стадию опреде-
ления профилей, во время которой определяется конкретный вид белков, как будет
описано ниже. Однако до перехода к рассмотрению этой стадии мы должны признать,
что двумерный гель-электрофорез имеет ограничения, которые могут оказать значи-
тельное влияние на общую пригодность определения профилей белков как средства
изучения протеома. Наиболее значительная проблема состоит в том, что в геле будут
видимы не все белки протеома — в частности, белки, которые не растворимы в водном
буфере (такие как многие из белков, присутствующих в клеточных мембранах), будут не
видны. Для изучения этих компонентов протеома должны быть использованы специ-
альные буферы и составы геля, что означает, что должно быть выполнено несколько
параллельных экспериментов, если мы хотим изучать протеом в его целостности. Есть
также проблемы с воспроизводимостью двумерного гель-электрофореза и трудности
в изобретении процедур контроля, которые позволяли бы нормализовывать данные
от таких гелей при сравнении двух протеомов. По этим причинам ученые ищут альтер-
нативные методы разделения, причем внимание в настоящее время сосредоточено на
высокоэффективной жидкостной хроматографии (ВЖХ) и изоэлектрофокусировке со
свободным потоком.
Опознавание белков протеома
В результате двумерного гель-электрофореза мы получаем сложную картину
пятен, каждое из которых представляет отдельный белок. Как мы опознаем, какой
белок присутствует в том или ином пятне? Это было трудной задачей, но достижения
в масс-спектрометрии обеспечили быструю и точную процедуру опознавания, про-
Часто в одном пятне находится смесь нескольких белков. — Прим. ред.
Глава 6. Постижение механизмов функционирования геномов 235
диктованную требованиями исследователей геномов. Масс-спектрометрия была изна-
чально разработана как средство распознавания состава по отношениям массы к заряду
ионизированных форм, которые производятся, когда молекулы состава подвергаются
воздействию поля высокой энергии. Стандартный метод не мог применяться к белкам,
потому что они являются слишком крупными для эффективной ионизации. Новая про-
цедура, названная времяпролетной масс-спектрометрией с лазерной десорбционной
ионизацией матрицы (MALDI-TOF), позволяет обойти эту проблему, по крайней мере
с пептидами длиной до 50 аминокислот. Конечно, большинство белков намного длиннее
50 аминокислот, и поэтому их необходимо разбивать на фрагменты перед исследова-
нием при помощи MALDI-TOF. Стандартный подход состоит в очистке белка от пятна
и переваривании его специфичной к последовательности протеазой, такой как трипсин,
который расщепляет белки непосредственно после остатков аргинина или лизина. Для
большинства белков этот подход дает ряд пептидов длиной 5-75 аминокислот.
После ионизации отношение массы к заряду всякого пептида определяется по
его «времени пролета» в недрах масс-спектрометра, когда он проходит от источника
ионизации к датчику (рис. 6.12). Отношение массы к заряду позволяет установить
молекулярную массу, что, в свою очередь, позволяет вывести аминокислотный состав
пептида. Если анализируется ряд пептидов из одного-единственного белкового пятна
в двумерном геле, то полученная информация о составе может быть соотнесена с по-
следовательностью генома, с тем чтобы опознать ген, который кодирует этот белок.
Также можно использовать аминокислотные составы пептидов, полученных из одного-
единственного белка, для того, чтобы проверить, насколько правильно определены
24 см
Рис. 6.11. Результат двумерного гель-электрофореза. Белки мышиной печени были разделены изоэлек-
трофокусировкой в диапазоне pH 5-6 в первом измерении и согласно молекулярной массе во втором
измерении. Белковые пятна были проявлены посредством окрашивания их раствором серебра. Пере-
печатано с разрешения Gorg et al., Electrophoresis, 21,1037-1053. © 2000 Wiley-VCH Verlag
236 Часть 1. Изучение геномов
а) Масс-спектрометрия MALDI-TOF
Траектории
б) Спектр MALDI-TOF
Отношение массы к заряду
Рис. 6.12. Применение MALDI-TOF при определении профилей белков. После двумерного гель-
электрофореза интересующий нас белок извлекается из геля и переваривается протеазой, например,
трипсина. Переваривание разбивает белок на ряд пептидов, которые могут быть проанализированы
посредством MALDI-TOF. а) В масс-спектрометре пептиды ионизируются импульсом энергии от лазера,
затем ускоряются по мере продвижения по туннелю к отражателю и далее попадают на датчик. Время
пролета каждого пептида зависит от свойственного ему отношения массы к заряду. Данные визуали-
зируются в виде спектра. 6) Компьютер содержит базу данных предсказанных молекулярных масс всех
полученных посредством трипсина фрагментов каждого белка, кодируемого геномом изучаемого ор-
ганизма. Компьютер сравнивает массы обнаруженных датчиком пептидов с базой данных и опознает
наиболее вероятный исходный белок
последовательности гена, и, в частности, с тем чтобы убедиться в том, что местополо-
жение экзон-интронных границ было установлено правильно. Это средство не только
помогает отмечать точную позицию гена в геноме (раздел 5.1.1), оно позволяет также
опознавать пути альтернативного сплайсинга в случаях, где из одного и того же гена
получаются два и более белка.
Если сравниваются два протеома, то определяющее требование состоит в том,
чтобы белки, которые присутствуют в различных количествах, могли быть опознаны.
Глава 6. Постижение механизмов функционирования геномов 237
Если различия сравнительно велики, то они будут заметны в ходе простого осмотра
окрашенных гелей после двумерного электрофореза. Однако важные изменения в био-
химических свойствах протеома могут быть вызваны относительно незначительными
изменениями в количествах отдельных белков, поэтому существенно необходимы
методы обнаружения изменений малого масштаба. Одна возможность состоит в том,
чтобы метить составляющие двух протеомов различными флюоресцентными метками
и затем проводить совместный электрофорез в одном двумерном геле. Эта стратегия
сродни той, что используется для сравнения пары транскриптомов (см. рис. 6.6). Ви-
зуализация двумерного геля в волнах разной длины позволяет более точно оценивать
интенсивности эквивалентных пятен, чем это возможно, когда получают два отдельных
геля. Более точная альтернатива — метить каждый протеом изотопным ярлыком по
сродству (ICAT). Такие маркёры могут быть получены в двух формах: одна содержит
атомы обычного водорода, а другая—дейтерий, тяжелый изотоп водорода. Нормальную
и тяжелую формы можно отличить масс-спектрометрией, что позволяет определять
относительные количества белка в двух протеомах, которые были смешаны вместе,
в ходе стадии MALDI-TOF процедуры определения профиля (рис. 6.13)4
6.2.2. Опознавание межбелковых взаимодействий
Важные данные, имеющие отношение к деятельности генома, могут быть полу-
чены также путем опознавания пар и групп взаимодействующих друг с другом белков.
На подробном уровне эта информация часто бывает ценна, когда предпринимаются
попытки приписать функцию к недавно открытому гену или белку (раздел 5.2), пото-
му что взаимодействие со вторым, хорошо охарактеризованным белком, может часто
указывать на роль неизвестного белка. Например, взаимодействие с белком, который
расположен на поверхности клетки, может указывать на то, что неизвестный белок
вовлечен в передачу сигналов между клетками (раздел 14.1). На глобальном уровне
построение карт взаимодействий белков рассматривается как важный шаг в установ-
лении связей протеома с биохимией клетки.
Рис. 6.13. Анализ двух протеомов по-
средством ICAT. В спектре MALDI-TOF
пики, соответствующие пептидам,
содержащим атомы обычного водо-
рода, показаны синим цветом, а со-
ответствующие пептидам, содержа-
щим дейтерий, показаны красным.
Исследуемый белок приблизитель-
но в 1,5 раза более обилен в том
протеоме, который был помечен
дейтерием
Отношение массы к заряду
Проблема состоит в том, что значительная часть белков имеет пост-трансляционные модификации,
такие как метилирование, фосфорилирование, гликозилирование и многое другое. Причем один и тот же
белок может быть модифицирован по-разному. Все это может сильно изменить спектр. — Прим. ред.
238 Часть 1. Изучение геномов
Опознавание пар взаимодействующих белков путем фагового дисплея и ис-
следований с двумя гибридами
Существует несколько методов изучения межбелковых взаимодействий, и два
наиболее полезных из них — фаговый дисплей и дрожжевая двугибридная система.
В фаговом дисплее используется клонирующий вектор специального типа—основанный
либо на бактериофаге К либо на одном из нитевидных бактериофагов типа М13. Вектор
разработан таким образом, чтобы новый ген, который клонируется в нем, экспрессиро-
вался таким способом, что его белковый продукт был соединен с одним из белков обо-
лочки фага (рис. 6.14, а). Поэтому белок фага переносит чужеродный белок в оболочку
а) Получение выставляющего фага
Участок Ген белковой
рестрикции оболочки фага
ii Векторная ДНК
{Встройка ДНК, кодирующей
белок для выставления
ЭкспрессияХ
-ТТА ATCtGGA GCC-
Составная рамка считывания
Составной
белок
Выставляющий фаг
Белок, выставленный
в оболочке фага
б) Использование библиотеки фагового дисплея
Рис. 6.14. Фаговый дисплей, а) Клонирующий вектор, используемый для фагового дисплея, — геном
бактериофага с уникальным участком рестрикции, расположенным в пределах гена, кодирующего
белок оболочки фага. Этот метод первоначально выполняли с кодируемым геном III белком оболочки
нитевидного фага, названного fl, но позже он был распространен и на другие фаги, в том числе X. Чтобы
создать выставляющий фаг, последовательность ДНК, кодирующая опытный белок, лигируется в участок
рестрикции так, чтобы была произведена составная рамка считывания, такая, в которой ряд кодонов
продолжается неразрывно от опытного гена в ген белка оболочки. После трансформации Escherichia
coli эта рекомбинантная молекула направляет синтез гибридного белка, состоящего из опытного белка,
слиянного с белком оболочки. Поэтому фаговые частицы, производимые такими трансформированными
бактериями, выставляют опытный белок на своих оболочках, б) Использование библиотеки фагового
дисплея. Опытный белок фиксируется в пределах лунки лотка микротитра и добавляется библиотека
фагового дисплея. После промывания в лунке остаются те фаги, которые выставляют белок, взаимодей-
ствующий с опытным белком
Глава 6. Постижение механизмов функционирования геномов 239
фага, где он «выставляется напоказ» в форме, которая позволяет ему взаимодействовать
с другими белками, с которыми этот фаг соприкасается. Есть несколько путей, которыми
фаговый дисплей может быть использован для изучения межбелковых взаимодействий.
В одном методе выставляется исследуемый белок и отыскиваются его взаимодействия
с рядом очищенных белков или белковых фрагментов с известной функцией. Этот под-
ход ограничен, потому что требуется время, чтобы выполнить каждый опыт, так что он
удобоисполним, только если была получена некоторая предварительная информация
о вероятных взаимодействиях. Более действенный метод состоит в том, чтобы приго-
товить библиотеку фагового дисплея — коллекцию клонов, показывающих диапазон
белков, — и определить, какие члены библиотеки взаимодействуют с исследуемым
белком (рис. 6.14, б).
Дрожжевая дигибридная система обнаруживает взаимодействия белков более
сложным способом. В разделе 11.3.2 мы увидим, что есть белки, называемые актива-
торами, ответственные за управление экспрессией генов у эукариотов. Чтобы выпол-
нять эту функцию, активатор должен связываться с последовательностью ДНК выше
гена и стимулировать фермент РНК-полимеразу, который копирует этот ген в РНК.
Эти две способности — связывание с ДНК и активация полимеразы — определяются
различными частями активатора, и некоторые активаторы могут работать даже по-
сле разделения на две доли: одна доля содержит ДНК-связывающий домен, а другая
содержит активационный домен. В клетке эти две доли взаимодействуют и образуют
дееспособный активатор.
Дигибридная система использует штамм Saccharomyces cerevisiae, у представи-
телей которого отсутствует активатор гена-репортера. Поэтому этот ген всегда вы-
ключен. Искусственный ген, который кодирует ДНК-связывающий домен активатора,
лигируется к гену, кодирующему белок, взаимодействия которого мы желаем изучить.
Этот белок может быть взят у любого организма, не только у дрожжей: в приведенном
на рис. 6.15 примере это белок человека. После введения в дрожжи эта структура
определяет синтез составного белка, составленного из ДНК-связывающего домена
активатора, присоединенного к белку человека. Штамм рекомбинантных дрожжей
все еще неспособен экспрессировать ген-репортер, потому что видоизмененный ак-
тиватор лишь связывается с ДНК; он не может влиять на РНК-полимеразу. Активация
происходит только после того, как штамм дрожжей будет совместно трансформиро-
ван второй структурой, которая включает в себя кодирующую последовательность
активационного домена, слиянную с фрагментом ДНК, который определяет белок,
способный взаимодействовать с проверяемым нами белком человека (рис. 6.15, б).
Как и в случае фагового дисплея, если есть какая-либо предварительная информа-
ция о возможных взаимодействиях, то отдельные фрагменты ДНК могут быть про-
верены один за другим в дигибридной системе. Обычно, однако, ген, кодирующий
активационный домен, лигируется со смесью фрагментов ДНК, с тем чтобы получить
множество различных структур. После трансформации клетки высевают и опознают
те из них, которые экспрессируют ген-репортер. Это клетки, которые вобрали в себя
копию гена, кодирующего активационный домен, слитую с фрагментом ДНК, который
кодирует белок, способный взаимодействовать с проверяемым белком.
Опознавание компонентов многобелковых комплексов
Фаговый дисплей и дрожжевая дигибридная система являются эффективными
методами опознавания пар взаимодействующих друг с другом белков, но опознавание
таких связей выявляет только основной уровень межбелковых взаимодействий. Многие
240 Часть 1. Изучение геномов
а) Дигибридная система
ГИБРИД 1 ГИБРИД 2
ДНК-связываю- Активационный
щий домен домен
б) Перебор с целью отыскания межбелковых
взаимодействий с помощью дигибридной системы
Взаимодействие
между белками
человека
Экспрессия
гена
РНК-полимераза
Взаимодействие между
белками человека
не наблюдается
Экспрессия гена
не происходит
РНК-полимераза
активируется
ОБОЗНАЧЕНИЯ:
ж Ген дрожжей
ж Ген человека
Домены белков
дрожжей
Домены белков
Рис. 6.15. Дрожжевая двугибридная система, а) Слева показан ген, кодирующий белок человека, лиги-
рованный с геном, кодирующим ДНК-связывающий домен активатора дрожжей. После трансформации
дрожжей эта конструкция определяет составной белок: частью белок человека, а частью — активатор
транскрипции дрожжей. Справа показаны различные фрагменты ДНК человека, которые были лигирова-
ны с геном, кодирующим активационный домен активатора: эти структуры определяют разнообразные
составные белки. 6) Эти два набора составных структур смешиваются и совместно трансформируются
в дрожжах. Колония, в которой экспрессируется ген-репортер, содержит составные белки, в которых
фрагменты белков человека взаимодействуют, за счет чего связывающиеся с ДНК и активационные
домены приходят в тесный контакт и стимулируют РНК-полимеразу. Более подробные сведения об
активаторах можно почерпнуть в разделе 11.3.2
функции клетки выполняются многобелковыми комплексами, такими как медиатор,
который играет центральную роль в регулировании транскрипции гена (раздел 11.3.2),
или сплайсосома, которая отвечает за удаление интронов из пре-мРНК (раздел 12.2.2).
Подобного рода комплексы обычно включают набор ядерных белков (которые при-
сутствуют в них всегда), наряду с разнообразием вспомогательных белков (которые
связываются с комплексами при специфических обстоятельствах). Опознавание ядерных
и вспомогательных белков есть определяюще важный шаг к пониманию того, как именно
эти комплексы выполняют возложенные на них функции. Эти белки можно было бы
опознавать пара за парой посредством длинного ряда экспериментов с дигибридной
системой, но, очевидно, необходимо проторить более прямой путь к определению со-
става многобелковых комплексов.
В принципе, для опознавания членов многобелкового комплекса может быть ис-
пользована библиотека фагового дисплея, так как согласно этой процедуре все белки,
которые взаимодействуют с опытным белком, опознаются в едином эксперименте (см.
рис. 6.14). Проблема состоит в том, что большие белки выставляются неэффективно, по-
скольку они прерывают цикл репликации фага, и чтобы обойти эту проблему, как прави-
Глава 6. Постижение механизмов функционирования геномов 241
Рис. 6.16. Фаговый дисплей может быть не в состоянии
обнаружить всех членов многобелкового комплекса.
Комплекс состоит из центрального белка, который
взаимодействует с пятью белками меньшего разме-
ра. В нижней части рисунка показано, как пептид из
центрального белка используется в эксперименте фа-
гового дисплея. Этот пептид обнаруживает два из взаи-
модействующих белков, но другие три белка остаются
пропущенными, потому что их участки связывания
лежат в другой части центрального белка
ло, необходимо выставить короткий пептид,
представляющий часть клеточного белка, а не
целый белок. Поэтому выставляемый пептид
Белки не
определяются
Многобелковый
комплекс
Выставленный пептид
не взаимодействует со
всеми членами комплекса
может быть неспособен взаимодействовать со всеми членами комплекса, в пределах
которого расположен нативный белок, потому что в данном пептиде отсутствуют неко-
торые сайты связывания белков, имеющиеся в интактной форме (рис. 6.16). Метод, кото-
рый позволяет избегнуть этой проблемы, потому что он работает с нативными белками,
называется аффинной хроматографией. В аффиной хроматографии опытный белок
прикрепляется к хроматографической смоле и помещается в колонку (см. техническое
примечание 2.3). Клеточный экстракт проходит через колонку в малосолевом буфере,
который позволяет формироваться водородным связям, которые удерживают белки
вместе в едином комплексе (рис. 6.17, а). Поэтому белки, которые взаимодействуют со
связанным исследуемым белком, остаются в колонке, тогда как все остальные вымыва-
ются. Взаимодействующие белки затем элюируют многосолевым буфером. Неудобство
этой процедуры заключается в необходимости очищать исследуемый белок, что является
времязатратным и трудным в использовании как основа для масштабной программы
перебора. В более сложном методе, названном тандемная аффинная очистка (ТАР),
который был разработан как средство изучения белковых комплексов у S. cerevisiae, ген,
кодирующий исследуемый белок, видоизменяют таким образом, чтобы белок, будучи
синтезирован, имел на С-конце продолжение, которое связывается со вторым белком,
называемым кальмодулином. Клеточный экстракт приготовляется в мягких условиях,
с тем чтобы многобелковые комплексы не разрушались, и экстракт затем проходит
через колонку аффинной хроматографии, наполненную смолой, содержащей закре-
пленные в ней молекулы кальмодулина. В результате закрепляются как исследуемый
белок, так и другие, с которыми он связан (рис. 6.17, б). В обоих методах особенности
очищенных белков определяются масс-спектрометрией. При применении в массовом
переборном анализе 1739 генов дрожжей ТАР опознал 232 многобелковых комплекса,
позволив тем самым познать функции новых 344 генов, многие из которых не были до
того охарактеризованы экспериментальными средствами.
Неудобство работы с методами аффинной хроматографии состоит в том, что от-
дельный член многобелкового комплекса используется как «приманка» для выделения
других белков из этого комплекса. На практике оказывается, что если член комплек-
са не взаимодействует непосредственно с приманкой, то он не может быть выделен
(рис. 6.18). Поэтому эти методы дают возможность опознать группы белков, которые
присутствуют в комплексе, но не всегда позволяют опознать весь комплекс таких белков.
Поэтому развитие способов очистки интактных комплексов является важной задачей
сегодняшней научно-исследовательской деятельности. В совместном иммунном осаж-
242 Часть 1. Изучение геномов
а) Стандартная аффинная хроматография
Клеточная
вытяжка
Слабосолевой
буфер
Смола
с прикрепленным
опытным белком ®
Сильно-
солевой
буфер
I
б) Тандемная аффинная хроматография
Клеточная
Смесь опытного
На выброс и взаимодействующих
с ним белков
Смесь опытного
На выброс и взаимодействующих
с ним белков
Рис. 6.17. Методы аффиной хроматографии, применяемые для очистки многобелковых комплексов.
а) В стандартной аффиной хроматографии опытный белок прикрепляется к смоле. Клеточный экстракт
подается в слабосолевой буфер, так что различные члены многобелкового комплекса связываются
с опытным белком. Белки затем элюируют в сильносолевом буфере. 6) В ТАР (очистке по тандемному
сродству) клеточный экстракт подается в буфер, содержащий 2 ммоль/л СаС12; такие условия способ-
ствуют прикреплению видоизмененного опытного белка и белков, с которыми он взаимодействует,
к молекулам кальмодулина, прикрепленным к хроматографической смоле. Белки затем элюируют
буфером, который вообще не содержит СаС12
дении клеточный экстракт приготовляется в мягких условиях, с тем чтобы комплексы
остались интактными. Затем добавляется антитело, специфичное к опытному белку,
в результате чего происходит осаждение этого белка и всех других членов комплекса,
в котором он находится. Более сложным является метод многомерного опознавания
белков (MudPIT), который сочетает в себе различные хроматографические методы
Многобелковый
комплекс
(например, противофазная жидкостная хрома-
тография в паре или с катионообменной, или
с исключающей по размеру хроматографией),
с тем чтобы выделять интактные комплексы.
После этого компоненты комплекса могут быть
опознаны масс-спектрометрией. Этот метод
Приманка плюс
прикрепленные белки
Неопознанные белки
Рис. 6.18. Недостаток аффиной хроматографии. Если
белок-приманка (отмеченный буквой «В») не взаимо-
действует непосредственно с одним или несколькими
белками комплекса, то эти белки не могут быть вы-
делены
Глава 6. Постижение механизмов функционирования геномов 243
сначала использовался для изучения большой субъединицы рибосомы дрожжей и позво-
лил опознать 11 белков, о связи которых с этим комплексом ранее не было известно.
Опознавание функционально взаимодействующих друг с другом белков
Белки не обязательно должны образовывать физические ассоциации друг с другом,
чтобы иметь возможность функционально взаимодействовать между собой. Например,
в бактериях наподобие Escherichia coli ферменты лактопермеаза и р-галактозидаза имеют
функциональное взаимодействие, в котором они обе вовлечены в переработку лактозы
как источника углерода. Но нет никакого физического взаимодействия между этими
двумя белками: пермеаза расположена в клеточной мембране и переносит лактозу
в клетку, а р-галактозидаза, которая расщепляет лактозу на глюкозу и галактозу, нахо-
дится в цитоплазме клетки (см. рис. 8.8, а). Многие ферменты, которые работают вместе
в одном и том же биохимическом пути, никогда не образуют физических взаимодействий
друг с другом, и если бы исследования должны были базироваться исключительно
на обнаружении физических ассоциаций между белками, то многие функциональные
взаимодействия были бы упущены из вида.
Для опознавания белков, которые имеют функциональные взаимодействия, мо-
жет быть использовано несколько методов. Большинство из них не предполагает не-
посредственного изучения самих белков и, следовательно, строго говоря, не идет под
общим заголовком «протеомика». Однако удобно рассмотреть их здесь же, потому что
информация, которую они дают, часто включается, наряду с результатами исследований
в рамках собственно протеомики, в карты взаимодействия белков. В числе этих методов
мы видим следующие.
• Сравнительная геномика может быть использована различными способами, с тем
чтобы опознавать группы белков, состоящих в функциональных отношениях. Один
подход базируется на том наблюдении, что пары белков, которые являются отдель-
ными молекулами в одних организмах, слияются в единую полипептидную цепочку
в других. Пример представляет ген дрожжей HIS2, который кодирует фермент, вовле-
ченный в биосинтез гистидина. У Е coli гену HIS2 гомологичны два гена. Один из них,
который сам называется his2, имеет подобие последовательности с 5’-областью гена
дрожжей, а второй, hislO, подобен З'-области (рис. 6.19). Вывод: белки, кодируемые
генами his2 и hisW, взаимодействуют в протеоме Е coli, за счет чего выполняют часть
работы по биосинтезу гистидина. Анализ баз данных поеледовательностей показывает
множество примеров подобного типа, где два белка одного организма стали слитыми
в единый белок в другом организме. Подобный подход базируется на анализе оперонов
бактерий. Оперон состоит из двух и более генов, которые транскрибируются вместе
и обычно имеют функциональные отношения (раздел 8.2). Например, гены, кодирую-
щие лактопермеазу и р-галактозидазу, в организме Е coli находятся в одном и том же
опероне, наряду с геном, кодирующим третий белок, вовлеченный в использование
лактозы. Поэтому для того, чтобы вывести функциональные взаимодействия между
белками, кодируемыми гомологичными генами в геноме эукариота, могут использо-
ваться особенности генов в оперонах бактерий.
Рис. 6.19. Применение анализа гомологии для 1ПЛЛ
Область гена HIS2 дрожжей
выявления межоелковых взаимодействии. г
5'-Область гена HIS2 дрожжей гомологична Область his2 Е. coli
области his2 Escherichia coli, а З'-область го-
мологична области hislO Е. coli Область his10 Е. coli
244 Часть 1. Изучение геномов
• Исследования транскриптома позволяют опознать функциональные взаимодей-
ствия между белками, поскольку молекулы мРНК, соответствующие функциональ-
но связанным белкам, часто показывают подобные профили экспрессии в разных
условиях.
• Исследования с инактивацией генов также могут быть информативны. Если измене-
ние в фенотипе наблюдается только тогда, когда два и более гена инактивируются
вместе, то можно сделать вывод о том, что эти гены функционируют совместно
в деле создания фенотипа1).
Карты взаимодействий белков
Карты межбелковых взаимодействий, также называемые сетями взаимодействий,
показывают все взаимодействия, которые происходят между компонентами протеома.
Первые из таких карт были построены в 2001 г. для относительно простых протеомов
почти полностью на основании экспериментов с дигибридными системами. В их числе
были: карта бактерии Helicobacter pylori, охватывающая более чем 1200 взаимодействий,
вовлекающих почти половину белков протеома, и карта 2 240 взаимодействий между
1870 белками из протеома S. cerevisiae (рис. 6.20, а). Позже применение дополнительных
методов привело к более подробным версиям карты S. cerevisiae, включая такие, в кото-
рых иллюстрированы взаимодействия между многобелковыми группами (а не только
в их пределах) (рис. 6.20, б). Также впервые были построены карты для более сложных
видов наподобие Caenorhabditis elegans.
Какие интересные особенности удалось открыть благодаря всем этим картам
межбелковых взаимодействий? Наиболее занятное открытие — то, что каждая такая
сеть построена вокруг небольшого числа белков, которые имеют много взаимодействий
и которые формируют ядра (хабы) в сети, наряду с намного большим числом белков
с немногочисленными отдельными связями (рис. 6.21, а)* 2). Такая архитектура, как дума-
ют, минимизирует влияние на протеом разрушающих действий мутаций, которые могли
бы инактивировать отдельные белки. Только в том случае, если мутация затрагивает
один из белков в сильно связанном узле, сеть будет повреждена в целом. Эта гипотеза
согласуется с открытием, совершенным по результатам исследований с инактивацией
генов (раздел 5.2.2), того факта, что значительное число белков дрожжей явно избыточ-
но, то есть если активность белка нарушена, протеом в целом продолжает нормально
функционировать без заметного воздействия на фенотип клетки. Анализ профилей
экспрессии белков, образующих ядра, и их непосредственных партнеров позволяет раз-
делить эти ядра на две группы. Первая группа — те белки ядер, которые взаимодействуют
со всеми своими партнерами одновременно. Их называют «ядрами-компанейщиками»,
и их удаление оказывает небольшое воздействие на общую структуру сети (рис. 6.21, б).
Напротив, удаление второй группы — «ядер-свиданщиков», которые взаимодействуют
с различными партнерами в разное время, — приводит к разделению сети на ряд ма-
леньких подсетей (рис. 6.21, в). Исходя из этого, можно прийти к заключению о том, что
ядра-компанейщики работают в русле отдельных биологических процессов и не вносят
особо значительный вклад в общую организацию протеома, а ядра-свиданщики, напро-
тив, являются ключевыми игроками, которые обусловливают организацию протеома,
связывая биологические процессы друг с другом.
Это спорное утверждение. Действительно, если инактивировать только один из белков комплекса,
то перестанет работать весь комплекс. — Прим. ред.
2> Интересно, что сеть ссылок веб-страниц в Интернете имеет аналогичную архитектуру. — Прим. ред.
Глава 6. Постижение механизмов функционирования геномов 245
а)
б)
Рис. 6.20. Версии карты межбелковых взаимодействий Saccharomyces cerevisiae. а) Первоначальная карта,
впервые опубликованная в 2001 г. Каждая точка представляет белок, а соединительные линии указы-
вают взаимодействия между белками пары. Красные точки — незаменимые белки: инактивирующая
мутация в гене, кодирующем один из таких белков, смертельна. Мутации в генах белков, обозначенных
зелеными точками, не смертельны, а мутации в генах белков, показанных оранжевым цветом, приводят
к замедлению роста. Влияния мутаций в генах белков, показанных желтыми точками, не были извест-
ны во время построения карты, б) Более полная карта, опубликованная в 2002 г. В этой карте каждый
овал — белковый комплекс, а связи показаны между комплексами, у которых есть по крайней мере один
общий белок. Комплексы закодированы цветом согласно их функции следующим образом: красный —
клеточный цикл; темно-зеленый — передача сигналов; темно-синий — транскрипция, поддержание ДНК
и (или) структуры хроматина; розовый — перенос белков и (или) РНК; оранжевый — метаболизм РНК;
светло-зеленый — синтез и (или) оборот белков; коричневый — полярность и (или) структура клетки;
фиолетовый — метаболизм промежуточных продуктов обмена веществ и (или) энергетический обмен
веществ; светло-синий — биогенез мембран и (или) мембранный транспорт. Изображение а любезно
предоставлено Хавоуном Цзеуном. Переиздано с разрешения Jeong et al., (2001), Nature, 411, 41-42.
Изображение б любезно предоставлено Энн Клодом Гэвином. Переиздано с разрешения Gavin et al.,
(2002), Nature, 415,141-147
6.3. За пределами протеома
Протеом традиционно рассматривается как конечный продукт экспрессии гено-
ма, но такой взгляд оставляет в тени истинную роль протеома как связующего звена,
которое соединяет геном с биохимией клетки (рис. 6.22). Исследование природы этой
связи оказывается одним из самых захватывающих и плодотворных направлений со-
временной биологии.
6.3.1. Метаболом
Очень многие крупные достижения в биологии свершались не в ходе каких-нибудь
претенциозных исследовательских проектов вселенского масштаба, а в умах биологов,
пришедших к новому осмыслению стоящей перед ними научной проблемы. Пример
тому — введение концепции метаболома. Определение понятия «метаболом» звучит
как: «полная совокупность метаболитов, присутствующих в клетке или ткани при
специфическом наборе условий». Иными словами, метаболом — биохимическая матрица,
и его изучение, которое называют метаболомикой, или определением биохимиче-
ского профиля, дает точное описание биохимии, лежащей в основе различных физио-
246 Часть 1. Изучение геномов
а) Полная сеть
б) Удалены ядра-компанейщики
Рис. 6.21. Ядра на карте взаимодействий белков Saccharomyces cerevisiae. Эта карта была опублико-
вана в 2004 г. Ядра отчетливо видны на полной карте (а). После удаления ядер-компанейщиков
сеть остается почти неповрежденной (б), но после того, как были удалены ядра-свиданщики,
сеть разбилась на разрозненные подсети (в). Изображение любезно предоставлено Николасом
Бертином. Перепечатано с разрешения Han et al., (2004) Nature, 430,88-93
логических состояний, в том числе болезненных состояний, в которых могут пребывать
клетки или ткани. Преобразуя биохимию клетки в посписочный набор метаболитов,
метаболомика обеспечивает набор данных, который может быть напрямую увязан
с эквивалентной, расписанной попунктно информацией,
которая рождается протеомикой и прочими областями
| Транскрипция исследования экспрессии генома.
Метаболом может быть охарактеризован химически-
ми методами типа инфракрасной спектроскопии, масс-
| Трансляция спектрометрии и спектроскопии ядерного магнитного
ГЕНОМ
ТРАНСКРИПТОМ
ПРОТЕОМ
I Деятельность
I протеома
Рис. 6.22. Протеом является частью
конечной связи, которая связывает
геном с биохимией клетки
резонанса, которые, в отдельности и в сочетании, могут
опознать и определить количественно различные малые
молекулы, выступающие в роли метаболитов клетки. Когда
эти данные объединяются со знанием о скоростях реакций,
протекающих на различных этапах в подробно охаракте-
ризованных биохимических путях, таких как гликолиз
и цикл трикарбоновой кислоты (ТКК), возможно моде-
лировать метаболический поток — скорость потока
Глава 6. Постижение механизмов функционирования геномов 247
метаболитов через сеть путей, которые составляют клеточную биохимию. Тогда изме-
нения в метаболоме могут быть определены в понятиях возмущений в потоке метабо-
литов через одну или несколько частей сети, обеспечивая очень сложное, комплексное
описание биохимической основы изменений физиологического состояния. Это ведет
к возможности появления и осуществления метаболической инженерии, согласно
которой в геном посредством методов мутирования или технологии рекомбинантной
ДНК вносятся изменения, с тем чтобы влиять на биохимию клетки предопределенным
образом, например, чтобы увеличить синтез антибиотика микроорганизмом.
К настоящему времени метаболомика больше всего продвинулась в изучении ор-
ганизмов с относительно простой биохимией, таких как бактерии и дрожжи. Сегодня
над метаболомом человека проводится серьезная научно-исследовательская работа,
имеющая своей целью описание метаболических профилей здоровых тканей, тканей
в болезненном состоянии и тканей пациентов, проходящих курс медикаментозного ле-
чения. Есть надежда, что, когда эти исследования достигнут зрелости, метаболическую
информацию будет возможно использовать для разработки лекарственных препаратов,
которые обращают вспять или хотя бы смягчают отклонения в заданном потоке, про-
исходящие в болезненном состоянии. Определение биохимических профилей может
также указать какие-либо нежелател ьные побочные эффекты лечения лекарственными
препаратами, позволяя вносить изменения в химическую структуру препарата или в его
способ применения, с тем чтобы эти побочные эффекты были сведены на нет.
6.3.2. Постижение биологических систем
Акцент, ставимый теперь в картах взаимодействий белков и в метаболомике,
ведет нас к заключительной составляющей функции генома, которую нам осталось
рассмотреть. Это необходимость описать и постигнуть экспрессию генома не в поня-
тиях молекул — РНК, белков и метаболитов, — синтезом которых управляет геном, но
в понятиях биологических систем, которые следуют из согласованной деятельности
этих молекул. Это сущность рывка, сделанного в последние годы от генов к геномам.
Одним из основополагающих принципов догеномной эры молекулярной биологии
служила гипотеза «один ген — один фермент», впервые выдвинутая Джорджем Бидлом
и Эдвардом Тейтумом в 1940-е гг. Фразой «один ген — один фермент» Бидл и Тейтум
подчеркивали, что отдельный ген кодирует один-единственный белок, который, если
он фермент, обслуживает единственную же биохимическую реакцию. Например, ген
trpC Escherichia coli кодирует фермент индол-3-глицеринфосфатсинтетазу, который
преобразует 1-(о-карбоксифениламино)-1’-дезоксирибулозо-5’-фосфат в индол-3-
глицеринфосфат. Однако этот фермент не работает в отдельности: его деятельность
формирует часть биохимического пути, который завершается синтезом триптофана,
другие ферменты в этом пути задаются генами trpA, B.DwE, которые вместе с trpC фор-
мируют оперон триптофана Е. coli (см. рис. 8.8, б). Поэтому путь биосинтеза триптофана
представляет собой простую биологическую систему, а оперон триптофана — набор
генов, который определяет этот путь. Но простая транскрипция и трансляция генов
оперона не приведет к синтезу триптофана. Для успешной работы системы требуется,
чтобы биосинтезирующие ферменты присутствовали в клетке в должном месте, в не-
обходимых относительных количествах и в соответствующее время. Поэтому такая
система зависит от таких факторов, как скорость синтеза белков, кодируемых генами,
правильное сворачивание этих белков в функционально активные ферменты, скорость
деградации молекул фермента, их размещение в клетке и наличие в необходимых
количествах метаболитов, выполняющих роли субстратов и кофакторов для синтеза
248 Часть 1. Изучение геномов
триптофана. Эта простая биологическая система при таком взгляде начинает приоб-
ретать довольно значительную сложность. И это при том, что на примере этой системы
мы рассматриваем только 5 из 4405 генов, имеющихся в геноме Е. coli.
J\q настоящего времени в ряде областей системной биологии был сделан прогресс,
и в том числе в постижении биологической системы, отвечающей за синтез жгутика
Е. coli. Исследования догеномной эры показали, что синтез жгутика требует 51-го гена,
организованного в 12 оперонов, которые активируются тремя группами (рис. 6.23).
Группа, которая активируется в первую очередь, включает единственный оперон, со-
держащий два гена, кодирующих белок, который действует как главный регулятор,
включающий экспрессию второй группы из семи оперонов, гены которых вместе опреде-
ляют компоненты базальной структуры жгутика. Один из этих генов кодирует второй
регуляторный белок, включающий оставшиеся четыре оперона, которые направляют
синтез нити жгутика и биохимической системы, позволяющей бактерии отвечать на
химические раздражения вращением своего жгутика, с тем чтобы плыть к раздражителю.
Аккуратное использование генов-репортеров, прикрепленных к отдельным оперонам,
показало точный порядок, в котором опероны в каждой группе активируются. Это
позволило коэффициентам активации — мерам относительных скоростей экспрес-
сии — быть приписанными к каждому оперону. Полученная информация достаточна
для моделирования системы на компьютере, что позволяет досконально определить
роли этих двух регуляторных белков. Компьютерное моделирование дает возможность
предсказывать влияния тонких изменений в системе (например, изменения свойств
одного из регуляторов). Затем эти предсказания могут быть проверены последую-
щими экспериментами с биологической системой. Хочется верить, что именно такого
рода научно-исследовательская деятельность однажды позволит работать с клетками
человека, с тем чтобы понять точную основу отклонений и, надеемся, разрабатывать
способы возвращения больной ткани к ее нормальному состоянию. Пропорциональное
увеличение масштаба до таких все более сложных биологических систем, с великой це-
лью однажды понять, как работает клетка бактерии или эукариота, еще на протяжении
многих десятилетий будет испытывать находчивость и изобретательность биологов.
Однако уже был сделан первый шаг, состоящий в переносе акцента с функционирования
flhDC
£ Главный регуляторный белок
| Включает
fliGHklK ME fliLMNOPQR fliAZY flhBAE ffgAMN flgCDEFGHIJ
j Включает
flgKL fl™
Vr" Базальная
Ц структура
Рис. 6.23. Система, отвечающая за биосинтез жгутика Escherichia coli
Глава 6. Постижение механизмов функционирования геномов 249
отдельных генов на функционирование целых геномов и в утверждении методов изуче-
ния транскриптомов, протеомов и метаболомов, которые, вместе взятые, представляют
компоненты этих биологических систем.
Заключение
Главное испытание постгеномики — попытка понять, как геном задает и согласует
различные биохимические процессы, которые имеют место в живой клетке. Центральное
место в этой работе занимают исследования транскриптома и протеома, которые синте-
зируются и поддерживаются геномом1). Хотя транскриптомы можно изучать путем секве-
нирования кДНК, включая использование методов типа SAGE, которые позволяют читать
минипоследовательности многих кДНК в одном эксперименте, самые важные достижения
получаются за счет использования технологии микроматриц и чипов* 2). Гибридизация
дифференцированно меченных кДНК, приготовленных из двух и более транскриптомов
на микроматрице или чипе, дает информацию о картинах экспрессии генов, которые могут
быть проанализированы методом иерархической группировки, чтобы выявить функцио-
нал ьные отношения между этими генами. Изучение транскриптомов помогает нам понять
генетическую основу путей развития организма и возникновения болезней человека, в том
числе различных видов рака. Исследования протеома не менее важны, потому что анализ
транскриптома лишь показывает, какие гены экспрессируются в конкретной клетке, и не
дает точной картины белков, которые в ней присутствуют. Определение профилей белков
проводят при помощи двумерного гель-электрофореза, сопровождаемого MALDI-TOF вы-
деленных фрагментов пептидов, с тем чтобы охарактеризовать белки в протеоме. Чтобы
понять, как протеом работает внутри клетки, полезно знать, какие белки взаимодействуют
друг с другом. Наиболее часто используемыми методами опознавания пар белков, которые
формируют физические ассоциации, являются фаговый дисплей и дрожжевая дигибридная
система, а чтобы выделять интактные многобелковые комплексы, могут использоваться,
например, методы совместного иммунного осаждения. Функциональные взаимодействия,
которые не всегда требуют, чтобы белки пары образовывали физический контакт, могут
быть выведены сравнительной геномикой, анализом профилей экспрессии генов и посред-
ством исследований с инактивацией генов. Полученная информация позволяет составлять
карты взаимодействий белков, показывающие все взаимодействия, происходящие в едином
протеоме. В таких картах обычно наблюдаются характерные структуры вокруг относитель-
но небольшого числа белков, имеющих много взаимодействий и образующих ядра в сети,
причем одни ядра представляют отдельные биологические процессы, а другие связывают
такие биологические процессы друг с другом. Протеом поддерживает метаболом — полную
коллекцию метаболитов, находящихся в клетке или ткани, — и мы полны ожиданий, что
исследования метаболома выявят точную биохимическую основу болезненных состояний
и нежелательных побочных эффектов лечения медикаментозными препаратами. Работа
с транскриптомами, протеомами и метаболомами ведет биологов к системной биологии,
в которой делаются попытки осмыслить экспрессию генома не в понятиях молекул, синтез
которых направляет геном, а в понятиях биологических систем, являющихся результатом
согласованной деятельности этих молекул.
В последнее время все больше внимания уделяется эпигеному — системе модификации хроматина
(включая метилирование ДНК и модификации гитонов), которая оказывает существенное влияние на функ-
ционирование генома. — Прим. ред.
2) В самые последние годы все более важные результаты получаются с применением технологий па-
раллельного секвенирования. — Прим. ред.
250 Часть 1. Изучение геномов
Задания для закрепления материала
* Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
6.1 *. В чем состоит преимущество зон-
дирования транскриптома молеку-
лами мРНК, прикрепленными к ри-
босомам?
а. Молекулы мРНК эукариотов труд-
но выделять, если они не в соста-
ве комплексов с рибосомой.
Ь. Молекулы мРНК, выделенные из
рибосом, представляют те из них,
которые активно транслируются
в белки.
с. Молекулы мРНК, выделенные из
рибосом, более устойчивы, чем
другие молекулы мРНК.
d. Молекулы мРНК, которые не
транслируются рибосомами, все
еще содержат в себе последова-
тельности интронов.
6.2 . Каким образом можно использовать
микроматрицы для измерения уров-
ней экспрессии отдельных генов?
а. Каждая позиция на микроматри-
це содержит больше копий после-
довательности зонда, чем ожида-
емое число идентичных молекул
мРНК в транскриптоме.
Ь. Каждая последовательность зон-
да на микроматрице присутству-
ет в многочисленных позициях на
матричной нити.
с. После гибридизации молекулы
кДНК элюируются и определя-
ются количественно из каждой
позиции в микроматрице.
d. Молекулы кДНК секвенируются
после гибридизации и количе-
ственного определения флюо-
ресценции сигналов секвениро-
вания.
6.3 *. Почему в качестве индикатора для
изучения транскриптомов позво-
ночных используется актин?
а. Он используется как отрицатель-
ный индикатор, поскольку ген не
экспрессируется в организмах по-
звоночных.
Ь. Он используется как отрицатель-
ный индикатор, поскольку мРНК
актина быстро деградирует.
с. Он используется как положитель-
ный индикатор, поскольку экс-
прессия актина довольно посто-
янна в клетках различного типа.
d. Он используется как положитель-
ный индикатор, поскольку этот ген
наиболее интенсивно экспрессиру-
ется в клетках всех типов.
6.4 . Каким образом два разных транс-
криптома можно изучать с помощью
одной-единственной микроматри-
цы?
а. Сначала гибридизируется и из-
учается один транскриптом,
а затем его последовательности
удаляются и на той же самой
микроматрице изучается второй
транскриптом.
Ь. Только один из транскриптомов
метится, и он конкурирует со вто-
рым, непомеченным транскрип-
том при связывании с последова-
тельностями зондов.
с. Транскриптомы гибридизируют-
ся друг с другом до анализа ми-
кроматрицы, с тем чтобы удалить
присутствующие кДНК из клеток
обоих типов.
d. Оба транскриптома метятся раз-
личными флюоресцентными зон-
дами и гибридизируются одно-
временно.
6.5 *. По какому принципу гены группи-
руются при иерархической группи-
ровке?
Глава 6. Постижение механизмов функционирования геномов 251
а. По картинам экспрессии.
Ь. По гомологии.
с. По сходству последовательно-
стей.
d. По подобию белковых доменов.
6.6 . Какие изменения, как показали ис-
следования Saccharomyces cerevisiae,
происходят в транскриптоме, когда
клетки выращивают при устойчи-
вых, богатых энергией условиях?
а. Есть значительные изменения
в уровнях мРНК из-за меняю-
щихся скоростей деградации
и синтеза.
Ь. Уровни большинства мРНК оста-
ются постоянными, но некоторые
сильно колеблются на протяже-
нии клеточного цикла.
с. Почти у всех мРНК уровни оста-
ются постоянными, и только у не-
многих они изменяются значи-
тельно.
d. Уровни всех мРНК остаются по-
стоянными при этих условиях.
6.7 *. Как могут исследования транскрип-
тома помочь в диагностике видов
рака человека?
а. Все виды рака показывают уве-
личенную экспрессию специфи-
ческого набора генов.
Ь. Рак каждого вида обладает своим
собственным уникальным транс-
криптомом.
с. Гены, которые вызывают опухо-
ли, не экспрессируются в здоро-
вых клетках.
d. Исследования транскриптома
могут указать скорость деления
клетки.
6.8 . По какому из следующих критериев
электрофорез в полиакриламидном
геле в присутствии додецилсульфа-
та натрия отделяет белки?
а. По отношению заряда к массе.
Ь. По конформации.
с. По изоэлектрической точке.
d. По размеру.
6.9 *. Изоэлектрическая точка белка опре-
деляется в науке как:
а. pH, при котором суммарный за-
ряд белка равен нулю.
Ь. pH, при котором белок теряет
свою активность.
с. pH, при котором белок имеет мак-
симальную активность.
d. pH, при котором все аминокисло-
ты белка ионизируются.
6.10 . Для опознавания каких из следу-
ющих сущностей предназначена
дрожжевая дигибридная систе-
ма?
а. Всех компонентов многобелково-
го комплекса.
Ь. Белков человека, которые не-
обходимы для связывания РНК-
полимеразы.
с. Двух белков, которые непосред-
ственно взаимодействуют друг
с другом.
d. Двух белков, которые вовлечены
в один и тот же метаболический
путь.
6.11 *. Вид хроматографии, в ходе которой
белок связывается со смолой и по-
мещается в колонку, с тем чтобы
определить, какие белки с ним свя-
зываются, называется:
а. Хроматографией с гель-фильтра-
цией.
Ь. Ионообменной хроматографией,
с. Аффиной хроматографией.
d. Изоэлектрической хроматогра-
фией.
6.12 . Что представляют собой ядра в сети
взаимодействий белков?
а. Это белки, которые регулируют
работу клетки.
Ь. Это белки, которые формируют
каркас клетки.
с. Это белки, которые взаимодей-
ствуют со многими другими бел-
ками клетки.
d. Это белки, которые направляют
экспрессию генов в клетке.
252 Часть 1. Изучение геномов
6.13 *. Что понимают под метаболомом
клетки?
а. Все белки и нуклеиновые кисло-
ты клетки.
Ь. Все метаболиты клетки при опре-
деленном наборе условий.
Вопросы открытого типа
6.1 *. Почему исследователи стремятся
продолжить изучение геномов, даже
если ко всем их генам уже были при-
писаны функции?
6.2 . Объясните, почему последователь-
ность кДНК длиной всего 12 п. н. мо-
жет быть использована для опозна-
вания гена, который ее кодирует.
6.3 *. Обсудите проблемы, вызываемые
паралогичными генами в исследо-
ваниях на микроматрицах. Какие
условия эксперимента позволяют
преодолеть эти проблемы?
6.4 . Какие трудности в исследовании
транскриптома ткани могут возник-
нуть из-за альтернативного сплай-
синга? Какие подходы могут быть
использованы для опознавания
различных продуктов сплайсинга,
получаемых от одного гена?
6.5 *. Какую информацию о функциях ге-
нов могут предоставить исследова-
ния транскриптома?
6.6 . Каким образом выстилающие масси-
вы используются, чтобы просматри-
вать хромосомы на предмет наличия
экспрессируемых последовательно-
стей?
Изыскательские задачи
6.1 *. Часто исследователей интересуют
сравнения картин экспрессии гено-
ма в организме или ткани на различ-
ных стадиях развития или их изме-
нение в ответ на различные условия
окружающей среды. Какие подходы
с. Все потенциальные метаболиты,
которые могут быть произведены
клеткой.
d. Все макромолекулы клетки.
6.7 *. Почему транскриптом не дает пол-
ного и точного представления о про-
теоме клетки?
6.8 . Как могут небольшие различия
в уровнях белка быть определены
количественно при помощи двумер-
ного гель-электрофореза?
6.9 *. Как с помощью экспериментов фа-
гового дисплея проверяются взаи-
модействия между белками?
6.10 . В чем разница между белками, ко-
торые функционируют как «ядра-
компанейщики», и белками, которые
являются «ядрами-свиданщиками»
в сети межбелковых взаимодей-
ствий?
6.11 *. Какой вклад исследования метабо-
лома могут внести в лечение болез-
ней человека?
6.12 . На чем сосредоточена системная
биология и что явствует из ее срав-
нения с исследованиями регуляции
генов, проводимыми в рамках моле-
кулярной биологии еще до того, как
были секвенированы геномы?
являются наиболее действенными
для сравнительного исследования
такого рода?
6.2. После выполнения двумерного гель-
электрофореза протеомов двух ор-
ганизмов одного вида, выращенных
Глава 6. Постижение механизмов функционирования геномов 253
при разных условиях, вы опознаете
некий белок, который присутствует
в одном протеоме, но отсутствует
во втором. Какие эксперименты вы
должны выполнить, с тем чтобы
опознать ген, который кодирует
этот белок?
6.3* . При каких обстоятельствах белки
пары могут иметь функциональные
отношения, но никакого физическо-
го взаимодействия? Есть ли возмож-
Тесты по рисункам
6.1*. Опишите экспериментальный под-
ход к визуализации транскриптома,
примененный на этом рисунке.
ные сценарии, где обратное могло
бы быть верным — пара белков,
показывающих физическое взаи-
модействие, но никаких функцио-
нальных отношений?
6.4. Обсудите роль ядер на карте меж-
белковых взаимодействий.
6.5*. Объясните, почему системная био-
логия является предметом такого
большого внимания в настоящее
время.
Микроматрица
I Гибридизация
А с меченой кДНК
6.2. На основании какого признака в представленной на этом рисунке дендрограмме
группированы гены?
Г Л гЪ гЪ
ч Ъ ь Ъ <о Л
I Ген А
И Ген В
Ген
i ГенО
7 ||генЕ
254 Часть 1. Изучение геномов
6.3 *. Объясните принцип разделения молекул белка в двумерном гель-электрофо-
резе.
6.4 . Каким образом дрожжевая дигибридная система позволяет проверить взаимо-
действия между двумя разными белками? Объясните причину и роль активации
РНК-полимеразы в данном эксперименте.
Перебор с целью отыскания межбелковых
взаимодействий с помощью дигибридной системы
Взаимодействие
между белками
человека
Экспрессия
гена
РНК-полимераза
Взаимодействие между
\ белками человека
| не наблюдается
Экспрессия гена
не происходит
РНК-полимераза
активируется
Дополнительная литература
Исследования транскриптомов — методология
Leung, Y.F. and Cavalieri,D. (2003) Fundamentals of кДНК microarray data analysis.
Trends Genet 19:649-659.
Velculescu, V.E., Vogelstein, B. and Kinzler,K.W. (2000) Analysing uncharted
transcriptomes with SAGE. Trends Genet 16: 423-425.
Исследования транскриптомов — примеры
Alizadeh, A. A., Eisen, M. B., Davis, R. E., etal. (2000) Distinct types of diffuse large B-cell
lymphoma identified by gene expression profiling. Nature 403: 503-511.
Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P. O. and Herskowitz, I.
(1988) The transcriptional program of sporulation in budding yeast. Science 282: 699-705.
DeRisi,J.L., Iyer,V.R. and Brown,P.O. (1997) Exploring the metabolic and genetic
control of gene expression on a genomic scale. Science 278: 680-686. One of the first studies
of the yeast transcrip tome.
Глава 6. Постижение механизмов функционирования геномов 255
Golub, T.R., Slonim, D.K., Tamayo, Р., etal. (1999) Molecular classification of cancer:
class discovery and class prediction by gene expression monitoring. Science 286: 531-537.
Zhang, L., Zhou,W., Velculescu, V. E., Kern,S.E., Hruban, R. H., Hamilton, S.R.,
Vogelstein, B. and Kinzler,K. W. (1997) Gene expression in normal and cancer cells. Science
276:1268-1272.
Определение профилей белков
Fields,S. (2001) Proteomics in genomeland. Science 291: 1221-1224. Explains the
importance of proteomics in understanding the human genome sequence.
Mann, M., Hendrickson, R. C. and Pandey, A. (2001) Analysis of proteins and proteomes
by mass spectrometry. Annu. Rev. Biochem. 70: 437-473.
Phizicky, E., Bastiaens, P. I. H., Zhu, H., Snyder, M. and Fields, S. (2003) Protein analysis
on a proteomics scale. Nature 422: 208-215. Reviews all aspects of proteomics.
Yates, J.R. (2000) Mass spectrometry: from genomics to proteomics. Trends Genet 16:
5-8.
Zhu,H., Bilgin,M. and Snyder,M. (2003) Proteomics. Annu. Rev. Biochem. 72: 783-
812.
Изучение межбелковых взаимодействий
Clackson,T. and Wells, J. A. (1994) In vitro selection from protein and peptide libraries.
Trends Biotechnol. 12:173-184. Phage display.
Enright, A. J., Iliopoulos, L, Kyrpides, N. C. and Ouzounis, C. A. (1999) Protein interaction
maps for complete genomes based on gene fusion events. Nature 402:86-90. Using comparative
genomics to identify functional interactions.
Fields, S. and Sternglanz, R. (1994) The two-hybrid system: an assay for protein-protein
interactions. Trends Genet 10: 286-292.
Карты взаимодействий белков
Gavin, A.-C., Bdsche,M., Krause, R., et al. (2002) Functional organization of the yeast
proteome by systematic analysis of protein complexes. Nature 415: 141-147. A recent yeast
protein interaction map.
Han, J.-D. J., Bertin, N., Hao, T., et al. (2004) Evidence for dynamically organized modularity
in the yeast protein-protein interaction network. Nature 430: 88-93. Defines party and date
hubs.
Jeong, H., Mason, S. P., Barabasi, A.-L. and Oltvai, Z. N. (2 001) Lethality and centrality in
protein networks. Nature 411: 41-42. The first version of the yeast protein interaction map.
Lee, L, Date, S. V., Adai, A. T. and Marcotte, E. M. (2004) A probabilistic functional network
of yeast genes. Science 306:1555-1558.
Legrain,P., Wojcik,), and Gauthier, J.-M. (2001) Protein-protein interaction maps: alead
towards cellular functions. Trends Genet 17: 346-352.
Метаболомика и системная биология
Covert, M.W., Schilling, С. Н., Famili,!., Edwards, J.S., Goryanin, I. L, Seikov,E. and
Palsson,B.O. (2001) Metabolic modelling of microbial strains in silico. Trends Biochem. Sci.
26:179-186. Explains the concept of metabolic flux.
Kalir,S. and Alon,U. (2004) Using a quantitative blueprint to reprogram the dynamics
of the flageOa gene network. Cell 117: 713-720.
Kirschner, M.W. (2005) The meaning of systems biology. Cell 121: 503-504.
Часть 2
Анатомия генома
Часть 2 — Анатомия генома — содер-
жит сведения об организации генома, которая
была открыта, главным образом, в течение по-
следних десяти лет благодаря использованию
методов, описанных в части 1. В главе 7 мы
исследуем ядерные геномы эукариотов, с ак-
центом на геноме человека, который, мало того
что является нашим собственным геномом,
есть самый сложный геном из секвенирован-
ных до сей поры. Глава 8 посвящена геномам
прокариотов и органелл эукариотов, причем
последние рассматриваются здесь же ввиду их
прокариотического происхождения. В главе 9
мы устремляем наш взор на геномы вирусов
и на мобильные элементы генома, сгруппи-
рованные вместе, потому что некоторые из
таких мобильных генетических элементов
имеют эволюционное отношение к геномам
вирусов.
Глава 7
Ядерные геномы
эукариотов
Глава 8
Геномы прокариотов
и органелл эукариотов
Глава 9
Геномы вирусов
и мобильные
элементы генома
Глава 7
Ядерные геномы эукариотов
По прочтении главы 7 вы сможете:
• Описывать ДНК-белковые взаимодействия, которые дают начало нуклеосомам,
хроматосомам и 30-нм волокнам хроматина.
• Называть функции центромер и теломер и описывать специфические взаимо-
действия ДНК с белком, которые происходят в этих структурах.
• Объяснять, каким образом картины исчерченности хромосом и модель изохор
показывают, что в хромосомах эукариотов гены распределены неравномерно.
• Сравнивать организацию генов в ядерных геномах разных эукариотов и рас-
суждать о зависимости между организацией генов и размером генома.
• Описывать в общем виде полный состав генома человека.
• Описывать различные способы категоризации функций генов эукариотов и вы-
делять важные особенности, выявляемые в ходе сравнений каталогов генов
различных эукариотов.
• Объяснять, сопровождая примерами, что подразумевают под термином «муль-
тигенное семейство».
• Проводить различие между обычными и процессированными псевдогенами,
а также пережитками эволюции других типов.
• Различать тандемные повторы ДНК и рассеянные повторы ДНК и описывать важ-
ные особенности сателлитной, минисателлитной и микросателлитной ДНК.
В следующих трех главах мы совершим обзор анатомии геномов разного типа,
которые встречаются на нашей планете. Я написал именно три главы, потому что су-
ществует три типа геномов, которые нам необходимо рассмотреть.
• Ядерные геномы эукариотов (эта глава), среди которых геном человека пред-
ставляет для нас наибольший интерес.
• Геномы прокариотов и органелл эукариотов (глава 8), которые мы будем рас-
сматривать совместно, потому что органеллы эукариотов произошли от древних
прокариотов.
• Геномы вирусов и мобильные элементы генома (глава 9) сгруппированы вме-
сте, потому что некоторые мобильные элементы имеют отношение к геномам
вирусов.
260 Часть 2. Анатомия генома
7.1. Ядерные геномы содержатся в хромосомах
Ядерный геном разбит на набор линейных молекул ДНК, каждая из которых со-
держится в хромосоме. Не известны никакие исключения этой модели: все доселе
изученные эукариоты имеют по крайней мере две хромосомы, а молекулы ДНК всегда
линейны. Единственная изменчивость на этом уровне структуры генома эукариотов
заключается в числе хромосом, которое, как оказывается, не связано с биологическими
особенностями организма. Например, дрожжи имеют 16 хромосом, что в четыре раза
больше, чем у плодовой мушки. Не связано число хромосом и с размером генома: неко-
торые саламандры имеют геномы, в 30 раз превышающие геном человека, но разбитые
на меньшее вполовину число хромосом. Эти сравнения интересны, но в настоящее время
не сообщают нам ничего полезного о самих геномах; они, скорее, отражают многооб-
разие эволюционных событий, которые формировали архитектуру генома в разных
организмах1^.
Очищенный хроматин
Белковый комплекс
Обработка нуклеазой -
неограничивающие условия
Обработка нуклеазой -
ограничивающие условия
Деградация белка, анализ
ДНК посредством
ель-электрофореза
400 п. н
ДОРОЖКА 1: ДНК-маркеры
ДОРОЖКА 2: Полосы фрагментов
длины 200 п. н., 400 п. н. и т. д.
ДОРОЖКА 3: Единственная полоса
фрагментов длины 146 п. н.
Рис. 7.1. Эксперимент с защитой от нуклеазы по анализу хроматина из ядер клеток человека. Хроматин
осторожно очищается от ядер и обрабатывается ферментом нуклеазой. Слева показана обработка
нуклеазой при ограничивающих условиях, так что ДНК разрезается в среднем только единожды в каж-
дой из линкерных областей между связующими белками. После удаления белка фрагменты ДНК были
проанализированы электрофорезом в агарозном геле и оказались 200 п. н. в длину или кратными этой
величине. Представленная справа обработка нуклеазой протекает до конца, так что вся ДНК в лин-
керных областях переваривается. Все остающиеся фрагменты ДНК имеют длину 146 п.н. Результаты
свидетельствуют о том, что в этой форме хроматина белковые комплексы разнесены по ДНК с равными
интервалами — один на каждые 200 п. н., при этом отрезки ДНК по 146 п. н. прочно привязаны к каждому
белковому комплексу
Более того, существуют виды, разные особи которых (в пределах вида) имеют разное число хромо-
сом. — Прим. ред.
Глава 7. Ядерные геномы эукариотов 261
7.1.1. Упаковка ДНК в хромосомах
Хромосомы физически намного короче, чем молекулы ДНК, которые они вмещают:
средняя хромосома человека содержит в себе ДНК длиной чуть менее 5 см. Поэтому, чтобы
уместить молекулу ДНК в ее хромосому, необходима высокоорганизованная упаковочная
система. Прежде чем дерзновенно помыслить о том, как же геномы функционируют, мы
должны разобраться в этой упаковочной системе, потому что характер упаковки оказывает
влияние на процессы, отвечающие за экспрессию отдельных генов (глава 10).
Важные крупные достижения в понимании упаковки ДНК были сделаны в начале
1970-х гг., во многом благодаря сочетанию методов биохимического анализа и электрон-
ной микроскопии. К тому времени уже было известно, что ядерная ДНК связана с ДНК-
связывающими белками, названными гистонами, но точный характер этой связи еще
не был обрисован. В 1973-1974 гг. несколько исследовательских групп выполняли над
хроматином (ДНК-гистонными комплексами) эксперименты с защитой от нуклеазы.
Комплексы были осторожно извлечены из ядер методами, разработанными с целью
сохранить как можно больше структуры хроматина. В эксперименте с защитой от ну-
клеазы комплекс обрабатывают ферментом, который разрезает ДНК в позициях, «не
защищенных» прикреплением к белку. Размеры полученных фрагментов ДНК показы-
вают расположение белковых комплексов на исходной молекуле ДНК (рис. 7.1). После
ограниченной обработки очищенного хроматина нуклеазой большая часть фрагментов
ДНК имеет длину приблизительно 200 п. н. или кратную этой величине, что предполагает
равномерное расположение белков-гистонов по ДНК.
В 1974 г. эти биохимические результаты были дополнены электронными микро-
фотоснимками очищенного хроматина, которые позволили зрительно наблюдать регу-
лярное размещение белка в виде бусин на нити ДНК (выведенное поданным эксперимен-
тов с защитой от нуклеазы) (рис. 7.2, а). Последующий биохимический анализ показал,
что каждая такая бусина, или нуклеосома, содержит восемь молекул белков-гистонов
четырех типов Н2А, Н2В, НЗ и Н4 (по два белка каждого типа). Исследования структу-
ры показали, что эти восемь белков формируют
цилиндрообразный стержневой октамер, вокруг
которого с внешней стороны дважды обернута
ДНК (рис. 7.2, б). С каждой нуклеосомной частицей
связан отрезок ДНК длиной от 140 до 150 п.н.
а)
Рис. 7.2. Нуклеосомы, а) Электронный микрофотоснимок
нити очищенного хроматина, показывающий структуру
типа «бусины на нити». 6) Модель структуры типа «бусины
на нити», в которой каждая бусина представлена цилин-
дрической нуклеосомой, вокруг которой с внешней сторо-
ны дважды обернута ДНК. Каждая нуклеосома состоит из
восьми белков: центральный тетрамер из двух субъединиц
из гистона НЗ и двух субъединиц из гистона Н4, плюс пара
димеров Н2А-Н2В, один выше и один ниже центрального
тетрамера (см. рис. 10.13). в) Точная позиция линкерного
гистона относительно нуклеосомы не известна, но, как
показано здесь, линкерный гистон, скорее всего, служит
своего рода зажимом, препятствующим отделению ДНК
от внешней стороны нуклеосомы. Изображение а любезно
предоставлено доктором Барбарой Хамкало
б) ДНК
Нуклеосомы
Линкерный гистон
262 Часть 2. Анатомия генома
(в зависимости от вида организма), и все нуклеосомы отделены друг от друга отрезком
линкерной ДНК длиной 50-70 п. н., что дает длину повторений 190-220 п. н., ранее по-
казанную экспериментами с защитой от нуклеазы.
Помимо белков стержневого октамера, есть группа дополнительных гистонов, все
члены которой тесно связаны друг с другом и вместе названы линкерными гистонами.
У позвоночных они включают гистоны Н1а-е, Н1°, Hit и Н5. К каждой нуклеосоме при-
соединен единственный линкерный гистон и вместе они образуют хроматосому, но
точное расположение этого линкерного гистона не известно. Структурные исследования
поддерживают традиционную модель, в которой линкерный гистон действует как за-
жим, препятствуя отделению намотанной ДНК от нуклеосомы (рис. 7.2, в). Однако другие
результаты предполагают, что по крайней мере в некоторых организмах линкерный
гистон не расположен на самой поверхности комплекса нуклеосома-ДНК, как следовало
а) Модель типа б) Модель типа закрученной
соленоида винтом ленты
Рис. 7.3. Две модели 30-нм хроматинового волок-
на. Модель типа соленоида (а) была в предпочте-
нии в течение ряда лет, но недавно полученные
экспериментальные данные поддерживают мо-
дель типа закрученной винтом ленты (6). Пере-
печатано с разрешения Dorigo et al., Science, 306,
1571-1573. Copyright 2004 AAAS
бы ожидать, если бы он действительно был
зажимом, но вместо этого вставлен между
стержневым октамером и ДНК.
Структура типа «бусины на нити», пока-
занная на рис. 7.2, а, как думают, представляет
распакованную форму хроматина, которая
наблюдается в живых ядрах лишь изредка.
Очень мягкие методы разрушения клеток,
разработанные в середине 1970-х гг., позво-
л ил и обнаружить более уплотненную версию
комплекса, названную 30-нм волокном (его
ширина приблизительно равна 30 нм). Точ-
ный способ, которым нуклеосомы объединя-
ются и образуют 30-нм волокно, не известен,
но было предложено несколько моделей, две
из которых показаны на рис. 7.3. Отдельные
нуклеосомы в пределах 30-нм волокна могут
удерживаться вместе за счет взаимодействий
между линкерными гистонами, или же в скре-
плениях могут участвовать стержневые ги-
стоны, белковые «хвосты» которых выдаются
из нуклеосомы (см. рис. 10.13). Последняя
гипотеза привлекательна, потому что хими-
ческая модификация этих хвостов приводит
к раскрытию 30-нм волокна, что позволяет
генам, содержащимся в нем, быть активиро-
ванными (раздел 10.2.1).
7.1.2. Особенности хромосом в период метафазы
30-нм волокно есть, вероятно, главный тип хроматина в ядре во время интер-
фазы — периода между делениями ядра. Когда ядро делится, ДНК принимает более
компактную форму упаковки, дающую высокоуплотненные метафазные хромосомы,
которые можно наблюдать в световой микроскоп и которые предстают перед нами в том
характерном внешнем облике, каковой мы обычно ассоциируем со словом «хромосома»
(рис. 7.4). Метафазные хромосомы формируются на следующей за протеканием репли-
кации ДНК стадии клеточного цикла, и, таким образом, каждая содержит две копии
Глава 7. Ядерные геномы эукариотов 263
Теломера
Рис. 7.4. Типичный вид метафазной хромосомы. Метафазные хромосомы образуются по завершении
репликации ДНК, так что каждая в действительности представлена двумя хромосомами, связанными
вместе в центромере. Плечи называют хроматидами. Теломера — оконечность хроматиды
своей молекулы хромосомной ДНК. Эти две копии скрепляются вместе в центромере,
которая имеет определенную позицию в пределах каждой хромосомы. Плечи хромосомы,
которые называют хроматидами, и концевые структуры, называемые теломерами,
в разных хромосомах имеют различную длину. Поэтому конкретные хромосомы могут
быть опознаны по длинам их хроматид и по местоположению центромеры относитель-
но теломер. Следующие отличительные особенности выявляются при окрашивании
хромосом. Существует ряд различных методов окрашивания (таблица 7.1), каждый из
которых дает картину исчерченности, характерную для каждой отдельной хромосомы.
Это означает, что набор хромосом, имеющийся в некотором организме, может быть пред-
ставлен в виде кариотипа, в котором будет отображена полосчатая структура каждой
из них. Кариотип человека показан на рис. 7.5.
Таблица 7.1. Методы окрашивания, применяемые для получения картин исчерченности хромосом
Метод Процедура
G-исчерчивание Умеренный протеолиз, сопровождае-
мый окрашиванием красителем Гимза
R-исчерчивание
Q-исчерчивание
С-исчерчивание
Тепловая денатурация, сопровождае-
мая окрашиванием красителем Гимза
Окрашивание хинакрином
Денатурация гидроокисью бария и по-
следующее окрашивание красителем
Гимза
Картина исчерченности
Темные полосы обогащены АТ
Светлые полосы обогащены GC
Темные полосы обогащены GC
Светлые полосы обогащены АТ
Темные полосы обогащены АТ
Светлые полосы обогащены GC
Темные полосы содержат струк-
турный гетерохроматин (см
раздел 10.1.2)
Кариотип человека типичен для подавляющего большинства эукариотов, но не-
которые организмы проявляют необычные кариотипические черты, не наблюдаемые
у человека. К таковым относятся следующие из нижеперечисленных.
• Минихромосомы относительно коротки в длине, но богаты генами. Геном кури-
цы, например, разбит на 39 хромосом: шесть макрохромосом, содержащих 66 %
ДНК, но только 25 % генов, и 33 минихромосомы, содержащих оставшуюся одну
264 Часть 2. Анатомия генома
Рис. 7.5. Кариотип человека. Хромосомы изображены в виде картины G-исчерченности, полученной по-
сле окрашивания красителем Гимза. Под каждой из приведенных структур указан номер (или буквенное
обозначение) хромосомы, а слева — номера полос. «рДНК» представляет собой область, содержащую
группу повторных единиц генов рибосомной РНК (раздел 1.2.2). «Структурный гетерохроматин» — сильно
компактный хроматин, который содержит мало или вообще никаких генов (раздел 10.1.2)
Глава 7. Ядерные геномы эукариотов 265
треть генома и 75 % генов. Поэтому плотность генов в минихромосомах примерно
в шесть раз больше, чем в макрохромосомах.
• В-хромосомы — дополнительные хромосомы, имеющиеся у некоторых индиви-
дуумов в популяции, но не у всех. Они обычны для растений и встречаются также
у грибов, насекомых и животных. По всей вероятности, В-хромосомы являются
фрагментарными версиями нормальных хромосом и появляются в результате
ненормальных событий в ходе деления ядера. Некоторые часто содержат гены,
кодирующие рРНК, но не ясно, являются ли эти гены активными. Присутствие
В-хромосом может затронуть биологические характеристики организма, особенно
у растений, где они имеют отношение к уменьшению жизнеспособности. Пред-
полагается, что В-хромосомы постепенно исчезают из клеточных линий в силу
изъянов в схеме их наследования.
• Голоцентрические хромосомы имеют не одинарную центромеру, а, вместо этого,
многочисленные структуры, рассеянные по всей их длине. Например, обладателем
голоцентрических хромосом является червь нематоды Caenorhabditis elegans.
Взаимодействия белка с ДНК в центромерах и теломерах
ДНК, содержащаяся в пределах центромер и теломер, и белки, прикрепленные
к этой ДНК, имеют специфические особенности, связанные со специальными функциями
этих структур.
Последовательность нуклеотидов ДНК центромеры у высших эукариотов лучше
всего понята у растения Arabidopsis thaliana, благоприятные для генетического анализа
свойства которого позволил и определить местоположение позиций центромер на после-
довател ьности ДНК более или менее точно. Также специальное усилие было направлено
на секвенирование этих центромерных областей, которые иногда не попадают в прочи-
танные последовательности генома из-за проблем с обеспечением точного считывания
сильно повторных структур, которые характерны для таких областей. Центромеры
Arabidopsis простираются на 0,9-1,2 млнп.н. ДНК, и каждая состоит большей частью
из повторных последовательностей длиной 180 п.н. У людей эквивалентные последо-
вательности имеют 171 п.н. в длину и называются альфоидной ДНК с 1500-30000
копиями на центромеру. До того как последовательности Arabidopsis были получены,
считалось, что эти повторные последовательности были самым главным и основным
компонентом ДНК центромеры. Однако центромеры Arabidopsis содержат также и много-
кратные копии рассеянных повторов, наряду с несколькими генами, причем последние
характеризуются плотностью 7-9 генов на 100 тыс.п.н. по сравнению с 25 генами на
100 тыс. п. н. для нецентромерных областей хромосом Arabidopsis. Открытие того факта,
что ДНК центромеры содержит гены, стало большой неожиданностью, ибо в ту пору
бытовало мнение, что эти области являются генетически бездействующими.
Основная схема строения ДНК центромеры у Arabidopsis и человека демонстрирует
общие черты строения ДНК центромеры, присущие фактически всем эукариотам, но ин-
тересная вариация встречается у дрожжей Saccharomyces cerevisiae, центромера которых
определяется единственной последовательностью, приблизительно 125 п.н. в длину.
Эта последовательность состоит из двух коротких элементов, названных CDEI и CDEIII,
которые с двух сторон примыкают к более длинному элементу, называемому CDEII
(рис. 7.6). Последовательность CDEII является изменчивой, хотя всегда весьма обогащена
нуклеотидами А и Т, тогда как последовательности CDEI и CDEIII сильно консервативны,
что означает, что их нуклеотидные последовательности являются весьма подобными
266 Часть 2. Анатомия генома
CDEI
CDEII CDEIII
во всех 16 хромосомах дрожжей. Мутации в CDEII редко затра-
гивают функцию центромеры, но мутация в CDEI или CDEIII
Рис. 7.6. Центромера
Saccharomyces cerevisiae.
CDEI 9 п.н. в длину, CDEII —
80-90 п.н. и CDEIII-11 п.н.
Дополнительные последо-
обычно препятствует формированию центромеры. Короткий
неповторный характер ДНК центромеры дрожжей позволил
продвинуться в понимании того, как ДНК должна взаимодей-
ствовать с белками, чтобы сформировать функционально
активную центромеру. Ключевую роль здесь играет специ-
вательности, примыкающие
с обеих сторон к показанной
здесь области, рассматрива-
ются как часть ДНК центро-
меры, полная длина которой
составляет около 125 п. н.
альный хромосомный белок по имени Cse4 (подобный по
структуре гистону НЗ), каковой, совместно со вторым белком
по имени Mif2, образует стержень, вокруг которого обернута
последовательность CDEII (рис. 7.7). ДНК, как показано, удер-
живается на своем месте двумя следующими белками: Cbfl,
который опознает последовательность CDEI и прикрепляется
к ней, и Cbf3 (фактически тетрамер о четырех белках), кото-
Белок СЬТЗ прикреплен
к области CDEIII
Белок Cbf 1 прикреплен
к области CDEI
рый прикрепляется к последова-
тельности CDEIII. Белки Cbfl и Cbf3
связываются также и с комплексом,
содержащим по крайней мере при-
мерно 20 или около того дополни-
тельных белков. Они формируют
кинетохор — структуру, которая
действует как точка прикрепления
для микротрубочек, утягивающих
разделенные хромосомы в дочер-
ние ядра (рис. 7.8). До какой степени
эта модель центромеры дрожжей
приемлема для других эукариотов,
еще не ясно. Центромеры высших
эукариотов весьма различны, по-
скольку они содержат нуклеосомы,
подобные нуклеосомам в других
областях этой же хромосомы. Но
некоторые из них содержат белок
CENP-A вместо гистона НЗ. Нуклео-
сомы, содержащие белок CENP-A,
Рис. 7.7. Взаимодействия ДНК с белком в центромере дрож- более компактны и структурно
жей. Рисунок является исключительно схематичным, по- жестки, чем те, что содержат гистон
скольку точное расположение белков и компонентов ДНК НЗ. В связи с этим было высказано
неизвестно
предположение, что расположение
нуклеосом с CENP-A и с НЗ по ДНК
является таким, что нуклеосомы с CENP-A расположены на поверхности центромеры, где
они формируют внешнюю оболочку. На этом кожухе и строится кинетохор (рис. 7.9).
Вторая важная часть хромосомы — ее конечная область, или теломера. Теломе-
ры важны, потому что они отмечают концы хромосом и поэтому позволяют клетке
отличить реальный конец от неестественного конца, вызванного разрывом хромосо-
мы, — существенно важное требование, потому что клетка должна восстанавливать
последние, но никак не первые. ДНК теломер состоит из сотен копий повторного мотива
(у людей 5’-TTAGGG-3’) с коротким продолжением на З’-конце двунитевой молекулы
Глава 7. Ядерные геномы эукариотов 267
Рис. 7.8. Роль кинетохоров в процессе де-
ления ядер. В течение периода анафазы
деления ядер отдельные хромосомы рас-
тягиваются в разные стороны за счет со-
кращения микротрубочек, прикрепленных
к кинетохорам
ДНК (рис. 7.10). С повторными после-
довательностями в теломерах свя-
зываются два специальных белка
человека. Их называют TRF1 (помо-
гает регулировать длину теломеры)
и TRF2 (поддерживает однонитевое
продолжение). Если TRF2 инактиви-
рован, то это продолжение теряет-
ся и два полинуклеотида сливаются
вместе в ковалентное соединение.
Другие теломерные белки, как ду-
мают, формируют соединение между
теломерой и периферией ядра — об-
ластью, с которой связаны концы
хромосомы. Следующие белки опо-
средствуют ферментативную актив-
ность, которая поддерживает длину
каждой теломеры в ходе репликации
ДНК. Мы возвратимся к этой актив-
ности в разделе 15.2.4: она является
критически важной для выживания
хромосомы и может быть ключом
к пониманию механизмов старения
и смерти клетки.
Рис. 7.9. Центромеры млекопитающих со-
держат CENP-A- и НЗ-нуклеосомы. Одна воз-
можность состоит в том, что НЗ-нуклеосомы
расположены главным образом в цен-
тральном стержне центромеры, при этом
их CENP-A-версии формируют внешнюю
оболочку, на которой зиждется структура
кинетохора
1 Стадия анафазы процесса
деления ядра
7.2. Генетические характеристики ядерных геномов эукариотов
В главе 5 мы исследовали спектр методов биоинформатики и экспериментальных
методик, которые могут использоваться для определения местонахождения генов
в последовательности генома и для определения их функций. Теперь мы обратим наше
внимание ко всему тому, что эти методы поведали нам о генетических особенностях
ядерных геномов эукариотов.
268 Часть 2. Анатомия генома
5' 3
is.AGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAG
ТСССААТСССААТСССААТСССААТСС
Рис. 7.10. Теломеры. Последовательность на конце теломеры человека. В каждой теломере длина
З'-продолжений различна. См. раздел 15.2.4 при желании получить больше сведений о ДНК теломер
7.2.1. Где именно в ядерном геноме эукариотов находятся гены?
В предыдущем разделе мы узнали, что центромеры Arabidopsis содержат гены, но
с меньшей плотностью, чем свойственно остальной части хромосом. Это говорит нам
о том факте, что гены расположены неравномерно по длине хромосомы. В большинстве
организмов гены, оказывается, распределены более или менее случайно, со значитель-
ными колебаниями плотности генов в различных позициях хромосомы. Средняя плот-
ность генов Arabidopsis — 25 генов на 100 тыс.п.н., но даже вне центромер и теломер
плотность изменяется от 1 до 38 генов на 100 тыс.п.н., как показано на рис. 7.11 на
примере наиболее крупной из пяти хромосом этого растения. То же самое верно и для
хромосом человека, где плотность варьирует от 0 до 64 генов на 100 тыс. п. н.
Неравномерное распределение генов по длине хромосом человека подозревалось
в течение нескольких лет, прежде чем их последовательность была полностью секве-
нирована. В распоряжении ученых было два канала данных, один из которых связан
с картинами исчерченности, которые получаются при окрашивании хромосом. Краси-
тели, применяемые в этих процедурах (см. табл. 7.1), связываются с молекулами ДНК, но
в большинстве случаев с предпочтением к некоторым парам оснований. Краситель Гимза,
например, имеет большее сродство к областям ДНК, которые обогащены нуклеотида-
ми А и Т. Поэтому полагают, что темные G-полосы на кариотипе человека (см. рис. 7.5)
соответствуют обогащенным АТ областям генома. Состав оснований генома в целом
59,7 % А + Т, так что в темных G-полосах содержание АТ должно значительно превышать
60 %. Исходя из этого, цитогенетики предсказывали, что в темных G-полосах должно
быть меньше генов, потому что гены, как правило, имеют содержание АТ 45-50 %. Это
100 тыс. п. н.
I__।_।__।__।__।__।__।__।__।___I_।__।_।__।__।__।__।__।__।__I__।_।__।__।__।__।__।__।
man umiiiiiIiiiiiiihimi । г и и ни и inn
Спектр псевдоцветов: ——-
Высокая плотность Низкая плотность
Рис. 7.11. Плотность генов на протяжении наибольшей из пяти хромосом Arabidopsis thaliana. Пред-
ставлена хромосома 1, которая имеет длину 29,1 млн п. н., где секвенированные части показаны светло-
зеленым, а центромера и теломеры — темно-зеленым цветом. Генетическая карта ниже хромосомы
представляет плотность генов псевдоцветом: от темно-синего (низкая плотность) до красного (высокая
плотность). Плотность изменяется от 1 до 38 генов на 100 тыс. п. н. Перепечатано с разрешения AGI (The
Arabidopsis Genome Initiative), Nature, 408, 797-815. © 2000 Macmillan Magazines Limited
Глава 7. Ядерные геномы эукариотов 269
предсказание было подтверждено, когда последовательность генома была сравнена
с кариотипом человека.
Второй канал данных, указывающих на неравномерное распределение генов, имел
отношение к изохорной модели организации генома. Согласно этой модели геномы по-
звоночных и растений (а возможно, и остальных эукариотов) суть мозаики сегментов
ДНК (по крайней мере по 300 тыс.п.н. в длину), причем каждый такой сегмент имеет
однородный состав оснований, который отличается от такового смежных сегментов.
Подтверждение изохорной модели пришло из экспериментов, в которых геномная ДНК
разбивается на фрагменты длиной приблизительно 100 тыс. п. н., обрабатывается краси-
телями, которые специфично связываются с обогащенными АТ или GC областями, и эти
части разделяются при помощи центрифугирования в градиенте плотности (техническое
примечание 7.1). Когда этот эксперимент проводят с ДНК человека, можно наблюдать
пять фракций, каждая из которых представляет различный тип изохора с отличитель-
ным составом оснований: два обогащенных АТ изохора, названные L1 и L2, и три обо-
гащенных GC класса с названием Hl, Н2 и НЗ. Последний из них, НЗ, является наименее
изобильным в геноме человека, составляя лишь 3 % от общего количества, но содержит
более чем 25 % генов. Это ясный признак того, что гены не распределены равномерно по
всему геному человека. Фактически просмотр последовательности генома показывает,
что теория изохоров чрезмерно упрощает в действительности намного более сложную
модель изменения состава оснований по протяжению каждой хромосомы человека. Но
даже если она окажется неправильной моделью, теория изохор будет полезным непра-
вильным представлением, поскольку она во многом помогла молекулярным биологам
эры до расшифровки последовательностей понять структуру генома.
7.2.2. Каким образом организованы гены в ядерном геноме эукариотов?
Изменения плотности генов, которые просматриваются по длине хромосомы вся-
кого эукариота, наводят исследователя на мысль о том, как трудно ему будет опознать
области, в которых организация генов может рассматриваться как «типичная» для
генома в целом. Несмотря на эту трудность, ясно, что общая схема организации генов
у разных эукариотов сильно отличается, и мы должны понять эти различия, потому что
они отражают важные отличия между генетическими особенностями и эволюционными
историями этих геномов. Дабы проиллюстрировать таковые различия, мы в этом раз-
деле подробно рассмотрим маленькую часть генома человека и сравним этот сегмент
со столь же малыми частями геномов других организмов. При чтении и осмыслении
этого материала следует сосредоточиться на отличительных особенностях, которые
мы обнаружим, но помнить, что изменчивость в пределах отдельной хромосомы под-
разумевает, что невозможно сделать твердые и быстрые выводы о схемах организации
генов, которые обнаруживаются или не обнаруживаются в интересующем нас геноме.
Гены составляют лишь малую часть генома человека
Так как же организованы гены в ядерном геноме человека? Чтобы ответить на
этот вопрос, мы исследуем сегмент хромосомы 12 длиной 50 тыс.п.н. (рис. 7.12). Этот
сегмент обладает следующими генетическими особенностями.
• Четыре гена, как то:
• РКР2, который кодирует плакофилин 2 — белок, вовлеченный в синтез десмо-
сом (структур, играющих роль участков соединения, скрепления между собой
смежных клеток млекопитающих);
270 Часть 2. Анатомия генома
Техническое примечание 7.1: методы ультрацентрифугирования
Методология разделения компонентов клетки и крупных молекул
Создание высокоскоростных центрифуг в 1920-х гг. привело к появлению ме-
тодов разделения органелл и других фракций в массе разрушенных клеток. Первым
из подобных методов применялось дифференциальное центрифугирование,
в котором последовательно собирались капли осадка всё более легких компонен-
тов клетки посредством центрифугирования клеточных экстрактов при разных
скоростях. Неповрежденные ядра, например, являются относительно крупными
и могут быть собраны из клеточного экстракта центрифугированием при 1000 g1)
в течение 10 минут; митохондрии, будучи легче, требуют центрифугирования при
20 000 g в течение 20 минут. Посредством точного подбора параметров центрифу-
гирования различные компоненты клетки могут быть получены в довольно чистой
форме.
Центрифугирование в градиенте плотности впервые использовалось
в 1951 г. В этой процедуре клеточная фракция не центрифугируется в нормальном
водном растворе. Вместо этого, раствор сахарозы расслаивается в пробирке таким
образом, что создается градиент плотности, причем более концентрированный и,
следовательно, более плотный раствор располагается ближе к основанию пробирки.
Клеточная фракция помещается сверху градиента, и пробирка центрифугируется
с очень высокой скоростью: по крайней мере 500 000 g в течение нескольких часов.
При таких условиях скорость перемещения компонента клетки через градиент
зависит от его коэффициента седиментации, который, в свою очередь, зависит от
его молекулярной массы и формы. Например, рибосомы эукариотов имеют коэффи-
циент седиментации 80 S (S означает единицу Сведберга; Сведберг был шведским
ученым, первопроходцем в области применения ультрацентрифугирования для
нужд биологии), тогда как рибосомы бактерий, будучи меньше, имеют коэффициент
седиментации 70 S.
При центрифугировании в градиенте плотности второго типа используется
раствор хлорида цезия 8 моль/л, который является значительно более плотным,
чем раствор сахарозы, применяемый для измерения значений S. Исходный раствор
однороден, градиент устанавливается во время центрифугирования. Компоненты
клетки мигрируют вниз по центрифужной пробирке, но молекулы типа ДНК и белков
не достигают ее дна; вместо этого, каждая из них останавливается в положении, где
плотность матрицы (взвешивающего раствора) равняется ее собственной плавучей
плотности (см. рис. 7.23). Этот метод нашел множество применений в молекуляр-
ной биологии, будучи способным разделять фрагменты ДНК с различным составом
оснований и молекулы ДНК с разными конформациями (например: сверхспиральная,
кольцевая и линейная ДНК). С его помощью можно даже различать также нормаль-
ную ДНК и ДНК, меченную тяжелым изотопом азота (раздел 15.1.1).
g — гравитационное ускорение, или ускорение свободного падения, равное на широте Москвы
на уровне моря 981,56 см/с2. — Прим, перев.
Глава 7. Ядерные геномы эукариотов 271
• SYB1, определяющий ассоциированный с пузырьками мембранный белок, роль
которого состоит в обеспечении слияния пузырьков с их правильными целевыми
мембранами в клетке;
• ген, функция которого еще не была опознана, названный FLJ10143;
• CD27, кодирующий члена надсемейства рецепторов фактора некроза опухоли —
группы белков, которые регулируют пути передачи сигналов, задействованные
в апоптозе (запрограммированной смерти клетки) и в дифференцировке клет-
ки.
• Обратите внимание, что каждый из этих четырех генов прерывен, число интронов
находится в пределах от двух (в SYB1) до восьми (в РКР2).
• 88 рассеянных повторов. Это последовательности, которые вновь и вновь встре-
чаются во многих местах генома. Есть четыре главных типа рассеянных повторов,
названных LINE (длинные рассеянные ядерные элементы), SINE (короткие рассе-
янные ядерные элементы), элементы LTR (длинный концевой повтор) и транс-
позоны ДНК. В этом коротком сегменте генома наблюдаются примеры каждого
типа. Большинство рассеянных повторов расположено в межгенных областях, но
несколько лежит в пределах интронов.
• Семь микросателлитов, которые, как описано в разделе 3.2.2, представляют собой
последовательности, где короткий мотив повторяется в тандеме. Один из микро-
сателлитов, наблюдаемых здесь, имеет мотив СА, повторяющийся 12 раз^ давая
последовательность
5’-САСАСАСАСАСАСАСАСАСАСАСА-3'
3’-GTGTGTGTGTGTGTGTGTGTGTGT-5'.
РКР2
SYB1 ’ ‘ ‘ '
FLJ10143 25 тыс^п^н.
-. . . ср27 —
50 тыс. п. н.
ОБОЗНАЧЕНИЯ:
Гри _ Прочие рассеянные
ЭкзонI Иитппи .- «.к .г- Элемент Транспозон по всему геному
ИНТР°Н LINE SINE ДКП ДНК повторения Микроспутник
Рис. 7.12. Отдельно взятый сегмент генома человека. Эта карта показывает местоположение генов,
сегментов генов, рассеянных по всему геному повторений, и микросателлитов в сегменте хромосомы
12 человека длиной 50 тыс. п. н.
272 Часть 2. Анатомия генома
• Еще шесть микросателлитов, как то: повторения соответственно СААА, CCTG,
CTGGGG, CAAAA, TG и TTTG. Четыре из этих семи микросателлитов расположены
внутри интронов.
• Наконец, приблизительно 30 % нашего сегмента генома человека длиной 50 тыс. п. н.
состоит из отрезков негенной, неповторной, единственной копии ДНК, не харак-
теризующейся никакой известной функцией или значением.
Самая поразительная особенность этого сегмента генома человека длиной
50 тыс.п.н. — относительно малое количество места, занимаемого генами. Суммарная
длина экзонов — частей этих четырех генов, которые содержат биологическую инфор-
мацию1^, — равна 4745 п.н., что эквиваленте 9,5 % от 50-ти тыс.п.н. этого сегмента.
Фактически данный сегмент еще довольно богат генами: все экзоны в геноме человека
составляют л ишь 48 млн п. н., то есть всего 1,5 % от общего количества. В противополож-
ность, 44 % генома занято рассеянными повторами (рис. 7.13). [Для более детального
знакомства с анатомией генома человека рекомендую обратиться к веб-ресурсу UCSC
genome browser (http://genome.ucsc.edu/). — Прим. ред.].
Геном человека
3200 млн п. н.
Гены и относящиеся к генам
последовательности 1200 млн п. н.
Межгенная ДНК
2000 млн п. н.
Экзоны I Относящиеся к генам I Рассеянные по всему
i нпи п и последовательности I геному повторения
| 1152 млн п.н. | 1400 млн пн.
Псевдогены
’рагменты
генов
Интроны,
НТО
Микроспутники
90 млн п, н,
ДРЯЭ
640 млн п. н.
Элементы ДКП
250 млн п. н.
КРЯЭ
420 млн п. н.
Транспозоны ДНК
90 млн п. н.
Рис. 7.13. Состав генома человека. Аббревиатурное сокращение «НТО» обозначает нетранслируемые
области
Геном дрожжей очень компактен
Насколько же велики различия в организации генов среди эукариотов? Имеют
место, несомненно, очень значительные различия в размере генома: наименьшие ге-
номы эукариотов менее 10 млн п. н. в длину, а наибольшие — более 100 000 млн п. н. Как
можно видеть из рис. 7.14 и таблицы 7.2, этот диапазон размеров согласуется, до неко-
торой степени, со сложностью организма: простейшие эукариоты типа грибов имеют
наименьшие геномы, а высшие эукариоты типа позвоночных и цветковых растений
имеют наибольшие. Может казаться, что это имеет смысл, поскольку разумно было
Это слишком сильное утверждение. Биологическая информация заложена не только в белок-
кодирующих областях. — Прим. ред.
Глава 7. Ядерные геномы эукариотов 273
Грибы > ~<
Водоросли >
Протозойные ।----------------------------1
Насекомые 1-----------------1
Моллюски •-------1
Рыбы 1—1
Земноводные •
Пресмыкающиеся —
Птицы
Млекопитающие
Цветковые растения <---------------------1
1 ю юо юоо юооо юоооо юооооо юоооооо 100000000
Размер генома, млн п. н.
Рис. 7.14. Диапазон размеров геномов в различных группах эукариотов
бы ожидать, что сложность организма связана с числом генов в его геноме — высшим
эукариотам нужны более крупные геномы, чтобы вместить дополнительные гены. Од-
нако соответствие отнюдь не столь однозначное: если бы оно было таким, то ядерный
геном дрожжей Saccharomyces cerevisiae, который, имея 12 млн п. н., составляет 0,004 от
ядерного генома человека, как следовало бы ожидать, содержал бы 0,004-35 000 генов,
то есть всего 140 генов. Фактически же геном S. cerevisiae содержит около 6000 генов.
Многие годы отсутствие точной корреляции между сложностью организма и раз-
мером его генома рассматривалось как своего рода загадка—так называемый парадокс
показателя С. Фактически ответ весьма прост: в геномах менее сложных организмов
сэкономлено место, потому что гены упакованы более плотно1). Геном S. cerevisiae ил-
люстрирует этот пункт, как можно видеть из двух верхних частей рис. 7.15, где сегмент
генома человека размером 50 тыс.п.н., который мы только что рассматривали, срав-
нивается с сегментом генома дрожжей размером 50 тыс. п. н. Сегмент генома дрожжей,
который взят из хромосомы III (первая хромосома эукариотов, которая была секвени-
рована), имеет следующие отличительные особенности.
• Он содержит больше генов, чем сегмент человека. Эта область хромосомы III дрож-
жей содержит 26 генов, которые, как думают, кодируют белки, и еще два гена,
кодирующих транспортные РНК.
• Относительно немногие из генов дрожжей содержат интроны. В данном сегменте
хромосомы III ни один из генов неявляется их носителем. В полном геноме дрожжей
есть только 239 интронов по сравнению с более чем 300000 в геноме человека.
• В нем меньше рассеянных повторов. Эта часть хромосомы III содержит единствен-
ный элемент LTR, названный 7у2, и четыре усеченных элемента LTR, названных
дельта-последовательностями. Эти пять рассеянных повторов составляют 13,5 %
от сегмента длиной 50 тыс.п.н., но эта цифра не типична для генома дрожжей
в целом. Когда рассматривают все 16 хромосом дрожжей, общая длина последова-
тельностей, занимаемых рассеянными повторами, составляет лишь только 3,4 %
от общей длины генома.
Прямой корреляции ожидать трудно, но важно понимать, что сложность генома определяется не
только количеством белок-кодирующих генов, но также (и в большей степени) разного рода регуляторными
элементами, которые лежат вне генов. — Прим. ред.
274 Часть 2. Анатомия генома
Таблица 7.2. Размеры геномов эукариотов
Биологический вид Размер генома, млн п. н.
Грибы Saccharomyces cerevisiae 12,1
Aspergillus nidulans 25,4
Протисгы Tetrahymena pyriformis 190
Беспозвоночные Caenorhabditis elegans 97
Drosophila melanogaster 180
Bombyxmori (тутовый шелкопряд) 490
Strongylocentrotus purpuratus (морской еж) 845
Locusta migratoria (саранча) 5000
Позвоночные Takifiigu rubripes (бурый скалозуб) 400
Homo sapiens 3200
Mus musculus (мышь) 3300
Растения Arabidopsis thaliana (резушка Таля) 125
Oryza sativa (рис) 466
Zea mays (кукуруза) 2 500
Pisum sativum (горох) 4800
Triticum aestivum (пшеница) 16000
Fritillaria assyriaca (рябчик) 120000
Картина, которая вырисовывается, говорит о том, что генетическая организация
генома дрожжей намного более экономична, чем таковая генома человека. Во-первых,
сами гены более компактны (имеют меньше интронов), во-вторых, промежутки между
генами сравнительно короче и, в-третьих, намного меньше места занято рассеянными
повторами и прочими некодирующими последовательностями.
Организация генов у других эукариотов
Гипотеза о том, что более сложные эукариоты имеют менее компактные геномы, под-
тверждается и при рассмотрении геномов организмов других видов. Третья часть рис. 7.15
показывает сегмент генома плодовой мушки длиной 50 тыс.п.н. Если мы согласны с тем,
что плодовая мушка более сложна, чем клетка дрожжей, но менее сложна, чем человек, то
мы ожидали бы организацию генома плодовой мушки промежуточной между таковой
у дрожжей и у людей. Именно это мы видим на рис. 7.15, в: данный сегмент генома плодовой
мушки размером 50 тыс.п.н. имеет 11 генов — больше, чем в сегменте человека, но мень-
Глава 7. Ядерные геномы эукариотов 275
а) Человек
ОБОЗНАЧЕНИЯ:
экзон интрон LINE
Прочие рассеянные
Элемент Транспозон по всему геному
SINE LTR ДНК повторения Микросателлит Ген тРНК
---- - <? 1g t
Рис. 7.15. Сравнение геномов людей, дрожжей, плодовых мушек и кукурузы, а) Сегмент хромосомы
12 человека длиной 50 тыс. п. н., представленный ранее. Он сравнивается с сегментами по 50 тыс. п. н.
геномов б) Saccharomyces cerevisiae, в) Drosophila mehnogaster и г) кукурузы
ше, чем в последовательности дрожжей. Все эти гены прерывны, но семь имеют только по
одному интрону. Подобная картина наблюдается и при сравнении последовательностей
полных геномов этих трех организмов (таблица 7.3). Плотность генов в геноме плодовой
мушки является промежуточной между плотностью генов у дрожжей и у людей, и средний
ген плодовой мушки имеет намного больше интронов, чем средний ген дрожжей, но по-
прежнему в три раза меньше, чем средний ген человека.
Сравнение между геномами дрожжей, плодовой мушки и человека показывает ана-
логичную картину и тогда, когда мы рассматриваем рассеянные повторы (см. табл. 7.3).
Они составляют 3,4 % генома дрожжей, приблизительно 12 % генома плодовой мушки
и 44 % генома человека. Начинает становиться ясным, что рассеянные повторы играют
любопытную роль в предписывании компактности или разреженности генома. Это
поразительно иллюстрировано геномом кукурузы, который при своих 2500 млнп.н.
является относительно маленьким для цветкового растения. И хотя к настоящему
времени секвенировано только несколько областей генома кукурузы1), были полу-
чены некоторые примечательные результаты, показавшие, что геном этого растения
находится во власти повторных элементов. На рис. 7.15, г показан сегмент этого генома
размером 50 тыс. п. н., содержащий ген из одного семейства генов, кодирующих фермен-
ты алкогольдегидрогеназы. Это единственный ген в этом сегменте длиной 50 тыс. п. н.,
хотя есть и второй, неизвестной функции, отстоящий приблизительно на 100 тыс.п.н.
от правого конца последовательности, показанной здесь. Вместо генов преобладающая
Геном кукурузы секвенирован полностью. — Прим. ред.
276 Часть 2. Анатомия генома
Таблица 7.3. Компактность геномов дрожжей, плодовой мушки и человека
Показатель Дрожжи Плодовая мушка Человек
Плотность генов (среднее число на млн п. н.) 496 76 11
Число интронов в гене (среднее) 0,04 3 9
Количество генома, занятого рассеянными по всему геному повторениями (в %) 3,4 12 44
особенность этого сегмента генома — рассеянные повторы, которые были описаны
как образующие море, в котором расположены островки генов. Рассеянные повторы
относятся к типу элементов LTR, которые охватывают фактически всю некодирующую
часть сегмента и сами по себе, согласно оценке, составляют приблизительно 50 % генома
кукурузы. Становится ясно, что в геномах некоторых видов одно или несколько семейств
рассеянных повторов подверглись массовому распространению. Этим фактом можно
объяснить наиболее озадачивающий момент парадокса показателя С, который состоит
не в общем увеличении размера генома, которое наблюдается во все более и более слож-
ных организмах, но в том факте, что подобные друг другу организмы могут сильно от-
личаться по размеру генома. Хороший пример показывает Amoeba dubia, которая, будучи
протистой, как можно было бы ожидать, должна, подобно другим протистам, таким как
Tetrahymena pyriformis (см. табл. 7.2), иметь геном размером 100-500 тыс. п. н. Фактически
же геном Amoeba составляет более чем 200 000 млн п. н. [почти в 60 раз длиннее генома
человека! — Прим. ред.]. Точно так же мы могли бы предположить, что геномы сверчков
будут подобны в размере таковым других насекомых, но эти жучки имеют геномы при-
близительно 2000 млнп.н., что в 11 раз больше, чем геном плодовой мушки.
7.2.3. Сколько генов находится в ядерном геноме эукариотов и каковы
их функции?
Наиболее подробные аннотации полностью секвенированных последователь-
ностей хромосом человека предполагают, что геном человека содержит 30000-40000
геновЧ неопределенность возникает из-за трудности, освещенной в разделе 5.1.1, в рас-
познавании того, какие последовательности являются генами, а какие — нет. Это число
намного меньше, чем первоначально ожидалось, поскольку «наиболее правдоподобная
оценка» в 80 000-100 000 генов все еще была популярна вплоть до нескольких месяцев
до того, как в 2000 г. были получены черновые последовательности. Эти ранние оценки
были высоки, потому что они основывались на предположении о том, что в большин-
стве случаев один ген определяет единственную мРНК и единственный белок. Согласно
этой модели число генов в геноме человека должно быть тождественно числу белков
в клетках человека, что и привело к оценкам в 80000-100000 генов. Открытие того
факта, что число генов намного ниже этих цифр, указывает на то, что альтернативный
сплайсинг — процесс, посредством которого экзоны из пре-мРНК собираются в раз-
личных сочетаниях, так что одним геном может быть закодировано более одного белка
(см. рис. 6.5) — более распространенное явление, чем полагали вначале. Числа генов
в ядерных геномах разнообразных эукариотов приведены в таблице 7.4, но вы должны
О Современные данные указывают на число около 25 тыс. генов. — Прим. ред.
Глава 7. Ядерные геномы эукариотов 277
иметь в виду, что из-за альтернативного сплайсинга вопрос «сколько там генов?» не
имеет никакого действительного биологического смысла, поскольку число генов не
указывает на число белков, которые могут быть синтезированы, и, следовательно, не
является мерой биологической сложности генома.
Хотя альтернативный сплайсинг позволяет одному гену определять несколько
белков, по крайней мере некоторые части аминокислотных последовательностей этих
белков будут общими, и, таким образом, эти белки будут обычно наделены подобны-
ми или связанными функциями. Поэтому категоризация генов согласно их функциям
может дать значимую информацию о диапазоне биохимических действий, задаваемых
геномом, даже если для многих генов варианты сплайсинга либо не были опознаны,
либо к ним не были приписаны отдельные функции. Даже каталоги генов не решают
эту проблему полностью (ввиду трудностей в опознавании функций) в отношении
сравнительно простых организмов типа Saccharomyces cerevisiae. Весьма вероятно, что
некоторые категории генов недостаточно представлены в существующих каталогах,
потому что эти гены имеют функции, которые особенно трудно опознать. Имея в виду
вышеизложенное, мы сначала оглядим каталог генов человека.
Каталог генов человека
Из 30 000-40 000 генов человека функции более чем половины известны или могут
быть выведены с разумной степенью достоверности. Подавляющее большинство коди-
рует белки; менее 2 500 определяют функциональные РНК различных типов. Почти одна
четверть кодирующих белки генов участвует в экспрессии, репликации и поддержании
генома (рис. 7.16), и другие 21 % определяют компоненты путей передачи сигналов,
которые регулируют экспрессию генома и прочие действия клетки в ответ на сигналы,
полученные клеткой извне (раздел 14.1.2). Все эти гены могут рассматриваться как
носители некой функции, которая так или иначе вовлечена в деятельность генома.
Следующие 17,5 % известных генов кодируют ферменты, ответственные за общие био-
Экспрессия, репликация
и поддержание генома
Рис. 7.16. Категории каталога генов человека. Круговая диаграмма показывает классификацию опо-
знанных кодирующих белок генов человека. В ней опущено приблизительно 13000 генов, функции
которых еще не известны. Сегмент, характеризуемый как «разные другие действия», включает, наряду
с другими, белки, вовлеченные в биохимические процессы переноса веществ и сворачивание белка,
иммунологические белки и структурные белки
278 Часть 2. Анатомия генома
Таблица 7.4. Размер геномов и число генов в геномах различных эукариотов
Вид Размер генома, млн п.н. Приблизительное число генов
Saccharomyces cerevisiae (почкующиеся дрожжи) 12,1 6100
Schizosaccharomycespombe (делящиеся дрожжи) 12,5 4900
Caenorhabditis elegans (червь нематоды) 97 19000
Arabidopsis thaliana (растение) 125 25500
Drosophila melanogaster (плодовая мушка) 180 13600
Oryza sativa (рис) 466 40000
Gallusgallus (курица) 1200 20000-23000
Homo sapiens (человек) 3200 30000-40000
химические функции клетки, а оставшиеся вовлечены в такие действия, как транспорт
соединений внутрь и наружу клеток, сворачивание белков в их правильные трехмер-
ные структуры, иммунный ответ и синтез структурных белков, например, образующих
клеточный скелет или формирующих мышцы. Возможно, что как только каталог генов
человека станет более полным, относительные доли генов в трех главных категориях
на рис. 7.16 уменьшатся. Это связано с тем, что эти главные категории представляют
наиболее изученные области клеточной биологии, а это означает, что многие из соот-
ветствующих генов могут быть опознаны, потому что их белковые продукты известны.
Гены, продукты которых еще не были опознаны, скорее всего, вовлечены в менее хорошо
изученные области клеточной деятельности.
Один вопрос, на который каталог генов не может дать нам ответ и не сможет отве-
тить даже тогда, когда будет полным, это вопрос о том, что делает человека человеком.
Минималистскому подходу к молекулярной биологии, в свете которого ожидается, что
изучение отдельных генов или групп генов приведет в конечном счете к полному био-
молекулярному описанию того, как человек построен и функционирует, был нанесен
серьезный удар расшифровкой последовательности генома человека. В ней не было
открыто никаких удивительных особенностей, которые отличали бы людей от челове-
кообразных обезьян. Даже при том, что геном шимпанзе был полностью секвенирован,
все еще невозможно просто из сравнений наших геномов определить, что делает нас
людьми (раздел 18.4). Если судить по числу генов, то мы только в три раза сложнее,
чем плодовая мушка, и только вдвое сложнее микроскопического червя Caenorhabditis
elegans. Более доскональные исследования принципов функционирования генома че-
ловека могут вскрыть ключевые особенности, которые лежат в основе некоторых
специфических черт из числа неотъемлемых признаков людей, но геномика никогда
не объяснит природу человека.
Каталоги генов показывают отличительные особенности различных орга-
низмов
Существуют различные способы категоризировать гены в геноме эукариотов. Одна
из возможностей состоит в том, чтобы классифицировать гены согласно их функции, как
показано на рис. 7.16 на примере генома человека. Эта система имеет то преимущество,
Глава 7. Ядерные геномы эукариотов 279
что довольно широкие функциональные категории, используемые на рис. 7.16, могут
быть далее подразделены, с тем чтобы произвести иерархию все более и более специфи-
ческих описаний для все меньших и меньших наборов генов. Слабина этого подхода
кроется в том, что ко многим генам эукариотов еще не были приписаны функции, так
что классификация такого типа оставляет неучтенной некоторую долю полного набора
генов. Более мощный метод состоит в том, чтобы основывать классификацию не на
функциях генов, а на структурах белков, ими определяемых. Молекула белка построена
из ряда доменов, каждый из которых выполняет специфическую биохимическую функ-
цию. Примеры — цинковый палец, который является одним из нескольких доменов,
позволяющих белку связываться с молекулой ДНК (раздел 11.1.1), и «домен смерти»,
который присутствует во многих белках, вовлеченных в апоптоз. Каждому домену при-
суща характерная последовательность аминокислот — возможно, последовательности
во всех экземплярах этого домена не в точности одинаковы, но достаточно подобны
для того, чтобы присутствие специфического домена было распознаваемым на осно-
вании исследования аминокислотной последовательности белка. Последовательность
аминокислот белка определяется последовательностью нуклеотидов кодирующего ее
гена, так что домены, присутствующие в белке, могут быть определены по последо-
вательности нуклеотидов гена, который кодирует этот белок. Поэтому гены в геноме
могут быть категоризированы согласно белковым доменам, которые они определяют.
Этот метод имеет то преимущество, что он может быть применен к генам, функции
которых не известны, и, следовательно, может охватить большую долю набора генов,
составляющих геном.
Схемы классификации, что используют домены для выведения функции гена,
предполагают, что все эукариоты обладают одним и тем же базовым набором генов, но
более сложные виды имеют большее число генов в каждой категории. Например, люди
имеют самое большое число генов во всех категориях, кроме одной из представленных
на рис. 7.17, исключение пало на рубрику «метаболизм», где Arabidopsis обходит всех
вследствие своей фотосинтетической способности, которая требует большого набора
генов, отсутствующих в других четырех геномах, включенных в это сравнение. Эта
функциональная классификация являет нам и другие интересные особенности — в осо-
бенности, что С. elegans имеет относительно высокое число генов, вовлеченных в пере-
дачу сигналов между клетками; это удивительно, ведь данный организм имеет лишь
959 клеток. У человека, при его 1013 клеток, число генов для передачи межклеточных
сигналов больше лишь на 250 штук. Вообще, такого рода анализ подчеркивает подо-
бия между геномами, но не показывает генетическую основу чрезвычайно различных
типов биологической информации, содержащейся в геномах, скажем, плодовых мушек
и людей. Однако доменный подход в данном отношении выглядит многообещающим,
поскольку он показывает, что геном человека кодирует ряд белковых доменов, кото-
рые отсутствуют в геномах других организмов; эти домены представляют несколько
типов функций, как то: клеточная адгезия, электрическая связь между клетками и рост
нервных клеток (таблица 7.5). Эти функции интересны, потому что именно их мы рас-
сматриваем как придающие отличительные особенности позвоночным по сравнению
с другими типами эукариотов.
Возможно ли опознать набор генов, которые присутствуют у позвоночных, но
отсутствуют у остальных эукариотов? В настоящее время такой анализ может быть
проведен лишь приближенно, потому что в нашем распоряжении имеется только не-
сколько последовательностей геномов. На данный момент ясно, что приблизительно от
одной пятой до одной четвертой генов в геноме человека свойственны исключительно
280 Часть 2. Анатомия генома
5000
4500
4000
ОБОЗНАЧЕНИЯ:
S. cerevisiae
g 3500
£ 3000
§ 2500
* 2000
A. thaliana
С. elegans
Плодовая мушка
Человек
Рис. 7.17. Сравнение каталогов генов Saccharomyces cerevisiae, Arabidopsis thaliana, Caenorhabditis
elegans, плодовой мушки и человека. Гены категоризированы согласно их функциям, выведенным из
белковых доменов, определяемых каждым геном
Таблица 7.5. Примеры белковых доменов, задаваемых различными геномами
Число генов в геноме, содержащих домен
Домен Функция Человек Плодовая мушка Caeno- rhabditis Arabi- dopsis Дрожжи
Цинковый палец типа С2Н2 Связывание ДНК 564 234 68 21 34
Цинковый палец типа GATA Связывание ДНК 11 5 8 26 9
Гомеобокс Регулирование генов в ходе развития 160 100 82 66 6
Смерти Запрограм- мированная смерть клетки 16 5 7 0 0
Коннексин Электрическая связь между клетками 14 0 0 0 0
Эфрин Рост нервных клеток 7 2 4 0 0
Примечание: за дополнительной информацией о цинковых пальцах и домене гомеобокс обращайтесь
к разделу 11.1.
Глава 7. Ядерные геномы эукариотов 281
Только
приматы (1 %)
Только позвоночные
Позвоночные
и другие животные
Эукариоты
и прокариоты
Животные и другие эукариоты
Рис. 7.18. Сравнение каталогов генов человека с генетическими каталогами организмов из других групп.
Круговая диаграмма категоризирует каталог генов человека согласно распределению отдельных генов
в других организмах. Диаграмма показывает, например, что 22 % каталога генов человека состоит из
генов, специфичных для позвоночных, и что другие 24 % включают гены, специфичные для позвоночных
и других животных
позвоночным, а другая четверть встречается только у позвоночных и других животных
(рис. 7.18).
Семейства генов
Начиная с первых дней секвенирования ДНК было известно, что мультигенные
семейства — группы генов с идентичной или подобной последовательностью — яв-
ляются общими особенностями многих геномов. Например, все эукариоты, которые
были изучены (равно как и все бактерии, кроме самых простых), имеют множественные
копии генов, кодирующих рибосомную РНК. Это иллюстрировано геномом человека,
который содержит приблизительно 2000 генов, задающих 5S рРНК (так названную,
потому что ее коэффициент седиментации равен 5S; см. техническое примечание 7.1),
при этом все они расположены в одной группе на хромосоме 1. Есть также около 280
копий повторной единицы, содержащей гены 28S рРНК, 5,8S и 18S, сгруппированные
в пять групп из 50-70 повторений, по одной на каждой из хромосом 13,14,15,21 и 22 (см.
рис. 7.5). Рибосомные РНК — компоненты синтезирующих белок частиц, называемых
рибосомами, и предполагается, что кодирующие их гены присутствуют во множестве
копий, потому что есть большой спрос на синтез рРНК в ходе деления клетки, когда
должно быть собрано несколько десятков тысяч новых рибосом.
Гены рРНК представляют примеры «простых», или «классических», мультигенных
семейств, в которых все члены имеют идентичные или почти идентичные последова-
тельности. Эти семейства, как полагают, возникли путем дублирования (дупликации)
гена, а последовател ьности отдельных членов сохранились идентичными за счет эволю-
ционного процесса, который пока еще не был полностью описан (раздел 18.2.1). Другие
мультигенные семейства, более распространенные у высших эукариотов, чем у низших
эукариотов, называют «сложными», потому что отдельные члены, хотя и подобны в по-
следовательности, являются достаточно различными в отношении продуктов гена,
чтобы иметь отл ичител ьные свойства. Один из лучших примеров мул ьтигенного семей-
282 Часть 2. Анатомия генома
ства такого типа — гены глобина млекопитающих. Глобины — белки крови, которые
объединяются и образуют гемоглобин, при этом каждая молекула гемоглобина состоит
из двух а- и двух р-глобинов. У людей а-глобины кодируются маленьким мультиген-
ным семейством на хромосоме 16, а р-глобины — вторым семейством на хромосоме 11
(рис. 7.19). Эти гены были среди первых секвенированных генов еще в конце 1970-х гг.
Данные секвенирования показали, что гены в каждом семействе подобны друг другу,
но никоим образом не идентичны. Фактически последовательности нуклеотидов двух
наиболее разнящихся генов в группе p-типа, кодирующих р- и Е-глобины, имеют только
79,1 % идентичности. Хотя это достаточно высокий уровень сходства белков, чтобы быть
отнесенным к р-глобинам, этого различия достаточно, чтобы они имели отличительные
биохимические свойства. Подобные различия наблюдаются и в а-группе.
Почему члены семейств генов глобина столь отличны друг от друга? Ответ был
найден в ходе изучения картин экспрессии отдельных генов. Было открыто, что гены
экспрессируются на различных стадиях развития человека, например: в группе р-типа
ген е экспрессируется в раннем эмбрионе, гены Gy и Ау (чьи белковые продукты отли-
чаются только одной аминокислотой) — в плоде, а гены 5 и Р — во взрослом организме
(рис. 7.19). Различные биохимические свойства синтезируемых белков глобина, как
думают, отражают небольшие изменения в физиологической роли, которую гемоглобин
играет в ходе развития человека.
В одних мультигенных семействах отдельные члены группируются так же, как гены
глобина, но в других гены рассеяны по геному. Пример рассеянного семейства — пять
генов человека, кодирующих альдолазу (фермент, вовлеченный в выработку энергии),
которые расположены на хромосомах 3,9,10,16 и 17. Отметим, что, несмотря на рассе-
янный характер, члены этого мультигенного семейства обладают сходством последова-
тел ьностей, которое указывает на их общее эволюционное происхождение. Если сделать
сравнение последовательностей, то иногда возможно увидеть эволюционную связь не
только в пределах одного семейства генов, но также и между различными семействами.
Все гены семейства а- и р-глобинов, например, обладают некоторым сходством после-
довательностей и, как думают, эволюционировали от единственного предкового гена
t 5 тыс, п. н. |
Ъ VX’Va2Vai a2 a1 0
--- -----------—мм т-si Гены a-глобина
е с, А % 5 ₽ . „ ,
д — ш ~ Гены р-глобина
ОБОЗНАЧЕНИЯ:
Ген зародыша ВВ Ген взрослого
ВВ Ген плода ^В Псевдоген
Рис. 7.19. Группы генов а- и р-глобинов человека. Группа a-глобина расположена на хромосоме 16,
а p-группа — на хромосоме 11. Обе группы содержат гены, которые экспрессируются на различных
стадиях развития, и каждая включает по крайней мере один псевдоген. Обратите внимание, что экс-
прессия гена a-типа х2 начинается в эмбрионе и продолжается в течение эмбрионального периода;
не существует специфичного для эмбриона a-глобина. Псевдоген 0 экспрессируется, но его белковый
продукт является неактивным. Ни один из других псевдогенов не экспрессируется. За дополнительной
информацией о регулировании генов р-глобина в ходе развития следует обращаться к разделу 10.1.2
Глава 7. Ядерные геномы эукариотов 283
глобина. Поэтому мы обращаемся к этим двум мультигенным семействам как к членам
общего надсемейства генов глобинов, и на основании подобий между отдельными
генами мы можем картировать события дупликаций, которые привели к появлению
множества генов, которое мы видим сегодня (раздел 18.2.1).
Псевдогены и другие эволюционные реликты
Группы генов глобина человека содержат пять генов, которые более уже не активны.
Это псевдогены — функционально неактивные копии генов. Псевдогены суть своего
рода эволюционные реликты, признак того, что геномы непрерывно подвергаются из-
менению. Есть два главных типа псевдогенов.
• Обычный псевдоген — ген, который был инактивирован, потому что его нуклео-
тидная последовательность изменилась вследствие мутации (глава 16). Многие
мутации оказывают лишь незначительные воздействия на деятельность гена, но
некоторые являются более существенными, и весьма возможно, что в результате
изменения отдельного нуклеотида ген станет полностью функционально неактив-
ным. Как только псевдоген становится неактивным, он начинает деградировать
за счет накопления все большего числа мутаций и в конечном счете больше не
будет распознаваем как останок гена. Псевдогены глобина — примеры обычных
псевдогенов.
• Процессированный псевдоген возникает не за счет эволюционного распада, а в ре-
зультате ненормального хода экспрессии гена. Такой обработанный псевдоген полу-
чается из копии мРНК того или иного гена посредством синтеза копии кДНК, которая
впоследствии повторно вставляется в геном (рис. 7.20)4 Поскольку обработанный
псевдоген есть копия молекулы мРНК, он не содержит ни одного интрона из тех,
которые присутствовали в его родительском гене. В нем также отсутствуют последо-
вательности нуклеотидов, лежащие непосредственно выше родительского гена, где
находится область, в которой расположены сигналы, используемые для включения
экспрессии родительского гена. Отсутствие
таких сигналов означает, что обработан-
ный псевдоген является неактивным.
Наряду с псевдогенами, геномы содер-
жат и другие эволюционные реликты в форме
укороченных генов (в которых отсутствует
больший или меньший отрезок с одного кон-
ца полного гена) и фрагментов гена, то есть
коротких обособленных областей внутренней
части гена (рис. 7.21).
Функционально
активный ген
Транскрипция^
Обратная транскрипция
Рис. 7.20. Происхождение процессированного псев-
догена. Процессированный псевдоген, как думают,
возникает путем встраивания в геном копии мРНК,
транскрибированной с функционально активного гена.
Эта мРНК обратно транскрибируется в кДНК-копию,
которая может встроиться в ту же хромосому, где на-
ходится ее функционально активный родитель, а воз-
можно, и в какую-либо другую хромосому
ДНК
J Повторное встраивание
Псевдоген
Функционально
активный ген
Это событие называется ретропозицией. — Прим. ред.
284 Часть 2. Анатомия генома
Функционально
активный ген
Укороченный ген Фрагмент гена
Рис. 7.21. Укороченный ген и фрагмент гена
7.2.4. Содержание повторов ДНК в ядерных геномах эукариотов
Последовательность генома человека показала, что приблизительно 62 % гено-
ма человека занимают межгенные области — части генома, которые лежат между
генами и не обладают никакой известной функцией. Такие последовательности было
принято называть избыточной ДНК, но этот термин выходит из употребления. Это от-
части связано с рядом неожиданных открытий, совершенных при исследовании генома
в течение последних нескольких лет. Эти открытия привели к тому, что молекулярные
биологи стали менее уверенно утверждать о незначимости какой-либо части генома
просто потому, что мы в настоящее время не знаем, какова могла бы быть ее функция.
Как мы увидели, у большинства организмов большая часть межгенной ДНК состоит
из повторных последовательностей того или иного типа. Повторная ДНК может быть
подразделена на две категории (рис. 7.22): разбросанные по всему геному, или рас-
сеянные повторы, отдельные повторные единицы которых распределены по геному
явно случайным образом, и тандемные повторы ДНК, повторные единицы которой
помещены рядом друг с другом в виде массива.
Тандемные повторения ДНК встречаются в центромерах и других местах
в хромосомах эукариотов
Тандемно повторная ДНК также называется сателлитной ДНК, потому что фраг-
менты ДНК, содержащие тандемно повторные последовательности, образуют «сател-
литовые» полосы, когда геномная ДНК разделяется на фракции центрифугированием
в градиенте плотности (см. техническое примечание 7.1). Например, когда ДНК человека
разбита на фрагменты по 50-100 тыс. п. н. в длину, то она образует главную полосу (пла-
вучей плотности 1,701 гем-3) и три сателлитные полосы (1,687, 1,693 и 1,697 гем-3).
Главная полоса содержит фрагменты ДНК, состоящие главным образом из однокопийных
последовательностей с содержанием GC, близким к 40,3 % — среднему значению для
повторы ДНК
Рис. 7.22. Два типа повторной ДНК: рассеянные повторы и тандемные повторы ДНК
Глава 7. Ядерные геномы эукариотов 285
Рис. 7.23. Сателлитная ДНК из генома челове-
ка. ДНК человека имеет среднее содержание
GC 40,3 % и среднюю плавучую плотность
1,701 гем-3. Фрагменты, состоящие главным 1’60
образом из однокопийной ДНК, имеют со-
держание GC, близкое к приведенному здесь
среднему числу, и содержатся в главной поло-
се в градиенте плотности. Сателлитные полосы ’*5
в положениях 1,687,1,693 и 1,697 гем-3 состоят
из фрагментов, содержащих повторную ДНК.
Содержание GC в этих фрагментах зависит от их
мотивов повторов и отличается от среднего чис- ’
ла для генома, означая, что эти фрагменты имеют
отличающиеся плавучие плотности от таковой
однокопийной ДНК и мигрируют к различным
позициям в градиенте плотности
генома человека. Сателлитные полосы
содержат фрагменты повторной ДНК и, гем
следовательно, имеют содержание GC
и плавучие плотности, нетипичные для
генома в целом (рис. 7.23). Эта повторная ДНК состоит из длинного ряда тандемных по-
вторений, возможно, сотни тыс. п. н. в длину. Один геном может содержать сателлитную
ДНК нескольких разных типов, каждый с иной повторной единицей, причем эти единицы
могут иметь любую длину в размахе от менее 5 до более 200 п.н. Три сателлитные по-
лосы в ДНК человека включают повторы по крайней мере четырех разных типов.
Мы уже встречались с сателлитной ДНК человека одного типа — альфоидными
повторениями ДНК, находящимися в центромерных областях хромосом (раздел 7.1.2).
Хотя некоторая сателлитная ДНК рассеяна по геному, большая часть расположена в цен-
тромерах, где она, быть может, играет структурную роль (возможно, работает в качестве
сайтов связывания для одного или более специальных центромерных белков).
Минисателлиты и микросателлиты
Хотя тандемно повторные ДНК еще двух других типов и не наблюдаются в сател-
литных полосах градиентов плотности, они также классифицируются как «сателлитная»
ДНК. Это минисателлиты и микросателлиты. Минисателлиты формируют группы
до 20 тыс.п.н. в длину с повторными единицами от 25 п.н. в длину и выше; микроса-
теллитные группы короче, обычно менее 150 п.н., а длина повторных единиц обычно
13 п.н. или меньше.
Минисателлитная ДНК — повторная ДНК второго типа, с которой мы уже знако-
мы ввиду ее привязки к структурным особенностям хромосом. ДНК теломер, которая
у людей включает сотни копий мотива 5’-TTAGGG-3’ (см. рис. 7.10), являет нам пример
минисателлита. Мы имеем определенные знания о том, как теломерная ДНК формиру-
ется, и мы знаем, что она выполняет важную функцию в репликации ДНК (раздел 15.2.4).
В дополнение к теломерным минисателлитам некоторые геномы эукариотов содержат
различные другие группы минисателлитной ДНК, причем многие, хотя и не все, находятся
около концов хромосом. Функции этих других минисателлитных последовательностей
еще не были распознаны.
286 Часть 2. Анатомия генома
Микросателлиты также представляют примеры тандемно повторной ДНК. В микро-
сателлите повторная единица коротка — до 13 п. н. в длину. Самый распространенный тип
присущего человеку микросателлита — динуклеотидные повторения приблизительно со
140000 копиями в геноме в целом, при этом примерно половина из них суть повторения
мотива «СА». Мононуклеотидные повторения (например, ААААА) следующие по распро-
страненности (всего приблизительно 120000 копий). Как и с рассеянными повторами,
неясно, имеют ли микросателлиты какую-либо функцию. Известно, что они возникают
вследствие ошибки в процессе, ответственном за репликацию генома входе деления клетки
(раздел 16.1.1), и попросту могут быть неизбежными продуктами репликации генома.
Хотя их функция, если таковая вообще имеется, и неизвестна, микросателлиты
оказались очень полезными для генетиков. Многие микросателлиты являются изменчи-
выми (полиморфными). Это означает, что у различных представителей биологического
вида число повторных единиц в массиве разное. Это связано с тем, что в ходе репликации
ДНК иногда происходит «промах» при копировании микросателлита, что ведет ко встав-
Рис. 7.24. Использование анализа микросателлитов
в определении генетических профилей. В этом примере
микросателлиты, расположенные на коротком плече
хромосомы 6, были размножены посредством ПЦР.
Продукты ПЦР помечены синей или зеленой флюорес-
центной меткой и пробегают в полиакриламидном геле,
где каждая дорожка показывает генетический профиль
отдельного индивидуума. Никакие два индивидуума не
имеют одинакового генетического профиля, потому что
каждый человек имеет уникальный набор вариантов
длин микросателлитов, после ПЦР эти варианты дают
полосы различных размеров. Красные полосы — маркё-
ры размера ДНК. Изображение любезно предоставлено
Applied Biosystems, Warrington, Великобритания
ке или, менее часто, к удалению одной
или нескольких повторных единиц
(см. рис. 16.5). Никакие два человека,
из ныне живущих, не имеют в точно-
сти одинаковой комбинации вариантов
длины всех микросателлитов: если ис-
следовать достаточное число микро-
сателлитов, то для каждого человека
может быть построен уникальный ге-
нетический профиль. Единственные
исключения — генетически идентич-
ные близнецы. Определение генетиче-
ских профилей широко известно как
инструмент в судебно-медицинской
экспертизе (рис. 7.24), но опознавание
преступников—довольно тривиальное
применение изменчивости микросател-
литов. Более совершенная методология
использует тот факт, что генетический
профиль человека унаследован частич-
но от матери и частично от отца. Это
означает, что микросателлиты могут
использоваться, чтобы установить от-
ношения родства и эвлюционной близо-
сти популяций не только для людей, но
также и для животных и растений.
7.2.5. Рассеянные повторы
Последовательности тандемно повторной ДНК, как думают, возникли вследствие
распространения отрезка последовательности предка—либо из-за промаха репликации,
описанного выше на примере микросателлитов, либо в силу процессов рекомбинации
ДНК (глава 17). По всей вероятности, оба этих события скорее дают в результате ряд
связанных повторений, а не сонм отдельных повторных единиц, рассеянных по геному.
Глава 7. Ядерные геномы эукариотов 287
Поэтому рассеянные повторы, должно быть, возникли другим механизмом, таким, в ре-
зультате работы которого копия повторной единицы появляется в геноме в позиции,
отдаленной от местоположения исходной последовательности. Самый частый путь,
которым это происходит, — транспозиция, и большинство рассеянных повторов об-
ладают самостоятельной транспозиционной активностью. Транспозиция свойственна
и некоторым вирусным геномам, которые способны встраиваться в геном зараженной
клетки и затем перемещаться с места на место в пределах этого генома. Некоторые рас-
сеянные повторы явно происходят от транспонируемых вирусов, и ввиду этой связи мы
отложим обсуждение рассеянных повторов этого и других типов до главы 9, к которой мы
подступим лишь после того, как подробно рассмотрим особенности геномов вирусов.
Заключение
Ядерный геном эукариотов разделен на набор линейных молекул ДНК, каждая из
которых содержится в хромосоме. В пределах хромосомы ДНК упакована за счет связи
с белками-гистонами и образует нуклеосомы, которые взаимодействуют друг с другом
и собираются в 30-нм волокно и структуры хроматина более высокого порядка. Самая
компактная организация характерна для метафазных хромосом, которые можно на-
блюдать посредством оптической микроскопии делящихся клеток и которые являют
нам характерные картины исчерченности после окрашивания. Центромеры, заметные
в метафазных хромосомах, содержат специальные белки, образующие кинетохор — точку
прикрепления для микротрубочек, которые оттягивают разделенные хромосомы в до-
черние ядра. У S’, cerevisiae ДНК центромеры, которая действует как сайт связывания для
этих белков, имеет длину приблизительно 125 п. н., но у большинства других эукариотов
эта область ДНК намного длиннее и состоит из повторной ДНК. Теломеры — струк-
туры, которые поддерживают концы хромосомы, — также содержат повторную ДНК
и специальные связывающиеся белки. Гены не равномерно распространены по длине
хромосом, плотность в хромосомах человека изменяется в пределах от 0 до 64 генов на
100 тыс. п. н. Кодирующие части генов охватывают лишь малую часть генома человека
(менее 1,5 %), при этом 44 % генома состоит из последовательностей повторной ДНК
различных типов. Напротив, геном S. cerevisiae намного более компактен: в нем только
3,4 % занято повторными последовательностями. Вообще, более крупные геномы менее
компактны, что объясняет, почему организмы с подобным числом генов могут иметь
геномы очень разных размеров. Люди имеют 30 000-40 000 генов, примерно вдвое боль-
ше, чем червь нематоды Caenorhabditis elegans, и приблизительно столько же, что и рис.
Сравнения каталогов генов, перечисляющих функции генов в геноме, показывают, что
все эукариоты обладают одним и тем же базовым набором генов, но при этом более
сложные виды имеют большее число генов в каждой функциональной категории. Многие
гены организованы в мультигенные семейства, члены которых имеют подобные или
идентичные последовательности, а в некоторых семействах (например, генов глобинов
позвоночных) разные члены экспрессируются на разных стадиях развития. Ядерные
геномы эукариотов содержат также эволюционные реликты, например, функционально
неактивных псевдогенов и фрагментов генов. Содержимое генома в лице повторной
ДНК может быть подразделено на рассеянную ДНК, большая часть которой обладает
транспозиционной активностью, и тандемно повторную ДНК, к которой относится
сателлитная ДНК, находящаяся в центромерах, минисателлиты типа ДНК теломер
и микросателлиты, которые используются учеными в области судебно-медицинской
экспертизы при определении генетических профилей.
288 Часть 2. Анатомия генома
Задания для закрепления материала
♦Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
7.1 *. Белки, которые связываются с ДНК
в нуклеосоме и образуют стержне-
вой октамер, называют:
а. Гистидинами.
Ь. Гистонами.
с. Хроматином.
d. Хроматосомой.
7.2 . Каким образом ДНК, как думают,
упакована в течение интерфазы?
а. В виде отдельных нуклеосом, ко-
торые можно видеть как «бусины
на нити».
Ь. В виде 30-нм волокон.
с. В высокоуплотненном состоянии,
видимом в световой микроскоп.
d. ДНК не упакована и не связана
с нуклеосомами в течение ин-
терфазы.
7.3 *. Что такое центромера хромосомы?
а. Это конец хромосомы.
Ь. Это несвернутая область хромосо-
мы, которая содержит активные
гены.
с. Это зауженная область хромосо-
мы, в которой две копии скрепле-
ны вместе.
d. Это свернутые, транскрипционно
неактивные области хромосом.
7.4 . Какие из следующих хромосом от-
носят к голоцентрическим?
а. Хромосомы со множественными
центромерами.
Ь. Дополнительные хромосомы,
присущие некоторым индиви-
дуумам в популяции.
с. Короткие хромосомы с много-
численными генами, например,
наблюдаемые у курицы.
d. Кольцевые хромосомы, встре-
чающиеся у некоторых низших
эукариотов.
7.5 *. Почему центромеры часто не вклю-
чаются в черновую последователь-
ность генома?
а. Чрезвычайно трудно клониро-
вать эту ДНК, потому что она
очень скручена.
Ь. Исследователинезаинтересованы
секвенированием областей ДНК,
в которых отсутствуют гены.
с. Последовательности центромер
одинаковы во всех организмах.
d. Трудно получить точную после-
довательность для этих длинных
областей повторной ДНК.
7.6 . Что ученые наблюдали в характере
распределения генов в геномах эу-
кариотов?
а. В геномах эукариотов гены рас-
пределены равномерно.
Ь. В геномах эукариотов гены рас-
пределены в определенных ме-
стоположениях.
с. В геномах эукариотов всегда най-
дется по крайней мере 10 генов
на 100 тыс.п.н. последователь-
ности.
d. Гены в геномах оказались бес-
порядочно распределены, и их
плотность сильно изменяется.
7.7 *. Размер генома дрожжей в сравне-
нии с геномом человека составляет
0,004, в то время как количество ге-
нов в геноме дрожжей составляет
0,2 от количества генов человека.
Верное объяснение этого факта:
а. По сравнению с генами человека
гены дрожжей содержат гораздо
меньше кодонов.
Ь. Хромосомы дрожжей содержат
намного меньше центромер и те-
ломер.
Глава 7. Ядерные геномы эукариотов 289
с. Геном дрожжей содержит на-
много меньше межгенной ДНК
и меньше интронов.
d. Геном дрожжей содержит много
перекрывающихся генов.
7.8. В чем состоит парадокс показателя С?
а. Отсутствие корреляции между
сложностью организма и разме-
ром его генома.
Ь. Отсутствие корреляции между
сложностью организма и числом
его хромосом.
с. Отсутствие корреляции между
сложностью организма и числом
его генов.
d. Отсутствие корреляции между
числом генов и числом хромосом
в организмах.
7.9* . Какая из следующих структур явля-
ется примером белкового домена?
а. р-лист.
Ь. Цинковый палец.
с. Экзон.
d. Белок глобина.
7.10. Что именно методы классификации,
основанные на функциях генов, го-
ворят исследователям об организ-
мах различных эукариотов?
а. Организмы всех эукариотов име-
ют одинаковые числа генов в каж-
дой функциональной категории;
сложные организмы имеют боль-
шее число неизвестных генов.
Ь. Сложные организмы имеют
большие числа генов в каждой
функциональной категории.
с. Более простые организмы, в срав-
нении со сложными, содержат
много меньше типов генов.
d. Все организмы эукариотов име-
ют приблизительно одинаковое
число генов.
7.11* . Что классификация генов, основан-
ная на белковых доменах, рассказы-
вает о геноме человека?
а. Геном человека не кодирует ника-
ких белковых доменов, которые
присущи исключительно людям.
Ь. Геном человека кодирует неболь-
шое число белковых доменов, ко-
торые являются уникальными
для позвоночных.
с. Геном человека кодирует много
белковых доменов, которые явля-
ются уникальными для людей.
d. Белковые домены, кодируемые
геномом человека, уникальны
для людей и отсутствуют у дру-
гих организмов.
7.12. Какие из следующих характеристик
не относятся к мультигенным семей-
ствам рибосомной РНК в геноме че-
ловека?
а. Семейства генов для каждой ри-
босомной субъединицы присут-
ствуют по всему геному на каждой
хромосоме.
Ь. Различные члены семейств генов
все имеют идентичные или поч-
ти идентичные последователь-
ности.
с. Эти семейства генов, как думают,
возникли вследствие дупликации
гена.
d. В них, как думают, имеются боль-
шие числа генов ввиду большого
спроса на новые рибосомы в ходе
деления клетки.
7.13* . Что такое псевдоген?
а. Ген, который экспрессируется
только на некоторых стадиях
развития.
Ь. Функционально неактивный ген.
с. Ген, который содержит мутацию, но
все еще функционально активен.
d. ПоследовательностьДНК,которая
медленно эволюционирует на пути
становления активным геном.
7.14. Какая область хромосомы эукарио-
тов характеризуется самой высокой
плотностью генов?
а. Центромера.
Ь. Уплотненный гетерохроматин.
с. Эухроматин.
d. Теломера.
290 Часть 2. Анатомия генома
Вопросы открытого типа
7.1 *. Что обработка хроматина эукарио-
тов нуклеазами говорит об упаковке
ДНК эукариотов?
7.2 . Что известно о форме 30-нм волокна
хроматина? Что известно об упаков-
ке нуклеосом в этом волокне?
7.3 *. Чем минихромосомы отличаются от
макрохромосом?
7.4 . Что исследователи обнаружили, ког-
да они секвенировали центромеры
Arabidopsis? Что неожиданного было
в этом открытии?
7.5 *. Почему важно, чтобы хромосомы
имели теломеры на своих концах?
7.6 . Какие два наблюдения были сдела-
ны еще до окончательной расшиф-
ровки последовательности генома
человека, которые привели иссле-
дователей к заключению, что в ге-
номе человека гены распределены
неравномерно?
7.7 *. Какие различия в распределении ге-
нов и в содержании повторной ДНК
заметны при сравнении хромосом
дрожжей и человека?
7.8 . Геном человека содержит приблизи-
тельно на 50 000 меньше генов, чем
было предсказано многими исследо-
вателями. Почему эти первоначаль-
ные оценки были столь высоки?
7.9 *. Назовите различные методы, приме-
няемые для составления каталогов
генов: каковы преимущества и не-
достатки этих методов?
7.10 . Какова функция различных генов
в семействах генов глобинов чело-
века?
7.11 *. В чем заключается разница между
обычным псевдогеном и процесси-
рованным псевдогеном?
7.12 . Какие типы повторной ДНК присут-
ствуют в геноме человека?
Изыскательские задачи
7.1 *. Какое воздействие упаковка ДНК,
вероятно, оказывает на экспрессию
отдельных генов?
7.2 . Выступите в защиту изохорной мо-
дели или раскритикуйте ее.
7.3 *. Обсудите возможные функции меж-
генной составляющей генома чело-
века.
7.4 . До какой степени возможно описать
«типичные» особенности генома эу-
кариотов?
Глава 7. Ядерные геномы эукариотов 291
Тесты по рисункам
7.1*. Что представлено на этой картине?
Чем отличаются разные хромосомы?
7.3*. Какого типа псевдоген показан на
этой схеме? Почему недавно встро-
енная копия гена функционально
неактивна?
7.2. Это схематичное изображение цен-
тромеры дрожжей и связанных
с нею белков. Какова функция этих
последовательностей и белков?
Функционально
активный ген
ТранскрипцияJ
/ Обратная транскрипция
ДНК
I Повторное встраивание
7.4. На этом рисунке показан результат
изучения генетического профиля,
который можно было бы использо-
ваться при опознании преступника
или установлении отцовства. Какие
типы последовательностей иссле-
дуются при определении профилей
ДНК и почему они наиболее пригод-
ны для этой цели?
Псевдоген Функционально
активный ген
292 Часть 2. Анатомия генома
Дополнительная литература
Основные первые публикации последовательностей геномов, включая пер-
вые каталоги генов
Adams, М. A., Celniker, S. Е., Holt, R. A., et al. (2000) The genome sequence of Drosophila
melanogaster. Science 287: 2185-2195.
AGI (The Arabidopsis Genome Initiative) (2000) Analysis of the genome sequence of
the flowering plant Arabidopsis thaliana. Nature 408: 796-815.
CESC (The C. elegans Sequencing Consortium) (1998) Genome sequence ofthe nematode
C. elegans: a platform for investigating biology. Science 282: 2012-2018.
ICGSC (International Chicken Genome Sequencing Consortium) (2004) Sequence
and comparative analysis of the chicken genome provide unique perspectives on vertebrate
evolution. Nature 432: 695-716.
IHGSC (International Human Genome Sequencing Consortium) (2001) Initial
sequencing and analysis of the human genome. Nature 409: 860-921.
Venter, J. C., Adams, M. D., Myers, E. W., et al. (2 001) The sequence of the human genome.
Science 291:1304-1351.
Структура хромосомы
Cleveland, D.W., Mao,Y. and Sullivan, K. F. (2003) Centromeres andkinetochores: from
epigenetics to mitotic checkpoint signalling. Cell 112: 407-423. Describes the DNA-protein
interactions in the yeast and mammalian centromeres.
Copenhaver, G. P., Nickel, K., Kuromori,T., etal. (1999) Genetic definition and sequence
analysis of Arabidopsis centromeres. Science 286: 2468-2474.
Dorigo,B., Schalch,T., Kulangara,A., Duda,S., Schroeder, R. R. and Richmond, T.).
(2004) Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science
306:1571-1573. New models ofthe 30 nm fiber.
Ramakrishnan, V. (1997) Histone Hl and chromatin higher-order structure. Crit Rev. Eukaryot
Gene Expr. 7: 215-230. Detailed descriptions of models for the 30 nm chromatin fiber.
Schueler,M.G., Higgins,A.W., Rudd,M.K., Gustashaw,K. and Willard,H.W. (2001)
Genomic and genetic definition of a functional human centromere. Science 294: 109-115.
Details ofthe sequence features of human centromeres.
Travers, A. (1999) The location ofthe linker histone on the nucleosome. Trends Biochem.
Sci. 24: 4-7.
van Steensel,B., Smogorzewska, A. and de Lange,T. (1998) TRF2 protects human
telomeres from end-to-end fusions. Cell 92: 401-413.
Генетические признаки
Balakirev, E.S. and Ayala, F.). (2003) Pseudogenes: are they «junk» or functional DNA?
Annu. Rev. Biochem. 37:123-151.
Csink, A. K. and Henikoff, S. (1998) Something from nothing: the evolution and utility of
satellite repeats. Trends Genet 14: 200-204.
Fritsch, E. F., Lawn, R. M. and Maniatis, T. (1980) Molecular cloning and characterization
of the human а-like globin gene cluster. Cell 19: 959-972.
Gardiner, K. (1996) Base composition and gene distribution: critical patterns in mammalian
genome organization. Trends Genet 12: 519-524. The isochore model.
Petrov, D. A. (2001) Evolution of genome size: new approaches to an old problem. Trends
Genet 17: 23-28. Reviews the C-value paradox and the genetic processes that might result in
differences in genome size.
Глава 8
Геномы прокариотов и органелл
эукариотов
По прочтении главы 8 вы сможете:
• Описывать принцип упаковки бактериальной ДНК в нуклеоид и представлять
экспериментальные данные в пользу доменной модели нуклеоида Escherichia
coli.
• Приводить примеры геномов прокариотов, которые являются линейными и (или)
многокомпонентными, и объяснять, почему присутствие плазмид в некоторых
клетках прокариотов усложняет определение того, что составляет «геном».
• Упоминать важные особенности организации генов в геномах прокариотов.
• Давать формулировку (сопровождая примерами) термину «оперон».
• Обсуждать взаимосвязь между числом генов и размером генома прокариотов
и рассуждать о содержании минимального генома прокариотов и об идентич-
ности генов, отвечающих за отличительные признаки.
• Обсуждать гипотезу об эндосимбионтах, объясняющую происхождение геномов
органелл.
• Описывать физические характеристики и содержание генов в геномах митохон-
дрий и хлоропластов.
Прокариоты — организмы, клетки которых не имеют развитых внутренних ком-
партментов. Есть две очень разные группы прокариотов, различие между которыми
проводится на основании характерных генетических и биохимических особенностей:
• бактерии, к которым относят львиную долю прокариотов, коих мы повсеместно
встречаем, — таких как грамм-отрицательные (например, Escherichia coli) и грамм-
положительные (например, Bacillussubtilis), цианобактерии (например, Anabaena)
и многие другие;
• археи, которые менее хорошо изучены и были обнаружены главным образом
в предельных окружающих средах, таких как горячие источники, соленые озера
и дно анаэробных озер.
В этой главе мы исследуем геномы прокариотов, а также митохондрий и хлоро-
пластов эукариотов, которые, поскольку они произошли от бактерий, имеют геномы,
демонстрирующие многие особенности, присущие прокариотам. Из-за относительно
294 Часть 2. Анатомия генома
малых размеров геномов прокариотов за последние несколько лет было опубликовано
несколько сотен полных последовательностей геномов различных бактерий и архей.
В результате мы начинаем понимать многое об анатомии геномов прокариотов, и в не-
которых отношениях мы знаем больше об этих организмах, чем об эукариотах. Карти-
на, которая вырисовывается в нашем сознании, показывает огромную изменчивость
среди прокариотов в целом и в некоторых случаях даже между близкородственными
видами.
8.1. Физические характеристики геномов прокариотов
Геномы прокариотов сильно отличаются от геномов эукариотов, в особенности
в отношении физической организации генома в клетке. Хотя для описания структур
типа белок-ДНК, имеющихся в клетках прокариотов, и употребляется слово «хромосо-
ма», это неправильное употребление данного термина, поскольку сия структура имеет
мало общего с хромосомой эукариотов.
8.1.1. Хромосомы прокариотов
Традиционное представление состояло в том, что у типичного прокариота геном
содержится в единственной кольцевой молекуле ДНК, ограниченной в пределах ну-
клеоида — светло окрашенной (в остальном — без обнаруживаемых особенностей)
области клетки прокариотов (рис. 8.1). Это определенно верно для Е. coli и многих дру-
гих обычно изучаемых бактерий. Однако, как мы увидим, наши все более совершенные
знания о геномах прокариотов наводят на мысль о том, чтобы подвергнуть сомнению
некоторые из предубеждений, утвердившихся в течение предгеномной эры микробио-
логии. Эти предубеждения относятся и к физической структуре генома прокариотов,
и к свойственной ему организации генов.
1 мкм
Рис. 8.1. Нуклеоид Escherichia col1. На этой микрофотографии, полученной с помощью просвечивающего
электронного микроскопа, показан поперечный разрез делящейся клетки Е. coli. Нуклеоид — светло
окрашенная область в центре клетки. Изображение любезно предоставлено Конрадом Уолдрингом
Традиционное представление о хромосоме прокариотов
Как и в хромосомах эукариотов, геном прокариотов должен втиснуться в относи-
тельно микроскопическое пространство (кольцевая хромосома Е coli имеет окружность
1,6 мм, тогда как клетка Е coli по своим габаритам всего лишь 1,0x2,0 мкм), и, как и у
эукариотов, это достигается с помощью ДНК-связывающих белков, которые упаковы-
вают геном в организованную структуру.
Глава 8. Геномы прокариотов и органелл эукариотов 295
Большая часть из того, что мы знаем об организации ДНК в нуклеоиде, была полу-
чена в ходе исследований Е. coli. Первая распознанная особенность была та, что кольце-
вой геном Е. coli сверхспирализован. Сверхспирализация происходит, когда в двойную
спираль ДНК добавляются дополнительные витки (правосторонняя сверхспираль) или
если витки удаляются из нее (левосторонняя сверхспираль). В линейной молекуле на-
пряжение кручения, вводимое перенамоткой или недомоткой, немедленно снимается
поворотом концов молекулы ДНК, но кольцевая молекула, не имеющая никаких концов,
не может уменьшить напряжение таким образом. Вместо этого, кольцевая молекулаотве-
чает закручиванием вокруг самой себя и образует более компактную структуру (рис. 8.2).
Поэтому сверхспирализация представляет идеальный способ упаковки кольцевой мо-
лекулы в малом пространстве. Данные о том, что в основе упаковки кольцевого генома
Е. coliлежит сверхспирализация, были впервые получены в 1970-х гг. при изучении вы-
деленных в чистом виде нуклеоидов, и впоследствии подтверждены как особенность
ДНК в живых клетках в 1981 г. У Е. coli сверхспирализация, как думают, осуществляется
и управляется двумя ферментами — ДНК-гиразой и ДНК-топоизомеразой I, — на ко-
торые мы взглянем более пристально в разделе 15.1.2 в свете постижения ролей этих
ферментов в репликации ДНК.
Исследования выделенных в чистом виде нуклеоидов показывают, что молекула
ДНК Е. coli утрачивает способность к неограниченному закручиванию (сверхспирали-
зации), как только в ней происходит разрыв. Наиболее правдоподобное объяснение —
бактериальная ДНК прикреплена к белкам, которые ограничивают ее способность
к релаксации, так что раскручивание в участке разрыва приводит к потере сверхспираль-
ности лишь маленького сегмента молекулы (рис. 8.3). Веские доказательства в пользу
этой доменной модели были получены в ходе экспериментов, которые основаны на
способности триметил псоралена различать между сверхзакрученной и релаксированной
ДНК. При фотоактивировании импульсом света длины волны 360 нм триметилпсорален
связывается с двунитевой ДНК со скоростью, прямо пропорциональной степени на-
пряжения кручения, испытываемого молекулой. Поэтому степень сверхскрученности
может быть установлена путем измерения количества триметилпсоралена, которое
связывается с молекулой за единицу времени. При облучении клеток Е coli &ля полу-
чения однонитевых надрывов в их молекулах ДНК, количество связывающегося три-
метилпсоралена пропорционально дозе облучения (рис. 8.4). Это ответ, предсказанный
доменной моделью, согласно которой общая сверхспиральность молекулы постепенно
уменьшается, по мере того как все большие дозы облучения вызывают разрывы во все
возрастающем числе доменов. Напротив, если бы нуклеоид Е. coli не был организован
ч
$ Кольцевая,
Й двунитевая ДНК
? Удаление 8
И нескольких %
витков %
двойной спирали %
8 Молекула
g образует
£ левостороннюю '§5^3
г сверхспираль g*
Рис. 8.2. Сверхспирализация. На схеме показано, как частичное раскручивание кольцевой молекулы
двунитевой ДНК дает левостороннюю сверхспираль
296 Часть 2. Анатомия генома
Белковое
ядро
Петли сверхзакрученной ДНК
Петля с разрывом -
отсутствие
сверхзакрученности
Рис. 8.3. Модель структуры нуклеоида Escherichia coli. От 40 до БОсверхзакрученных петель ДНК расходят-
ся радиальными лучами из центрального белкового ядра. Одна из петель показана в кольцевой форме,
указывая на то, что разрыв произошел в этом сегменте ДНК и привел к потере сверхзакрученности
ОООООоооос
в виде доменов, то единственный разрыв в молекуле ДНК привел бы к полной потере
сверхзакрученности: облучение в таком случае оказывало бы воздействие на связывание
триметилпсоралена по принципу «все или ничего».
В данной модели ДНК Е. coli прикреплена к белковому ядру, от которого к перифе-
рии клетки радиально исходят 40-50 сверхзакрученных петель. Каждая петля содержит
приблизительно 100 тыс.п.н. сверхзакрученной ДНК (количество ДНК, которая стано-
вится раскрученной после единственного разрыва). Белковый компонент нуклеоида
включает ДНК-гиразу и ДНК-топоизомеразу I — два фермента, которые главным об-
разом ответственны за поддержание сверхзакрученного состояния, — а также набор
Рис. 8.4. График, показывающий зависимость
между дозой облучения и интенсивностью
связывания триметилпсоралена
из по крайней мере четырех белков, которые,
как верят, играют более специализированную
роль в упаковке бактериальной ДНК. Наиболее
обильный из этих упаковочных белков — HU,
структурно он очень отличается от гистонов
эукариотов, но действует подобным образом,
образуя тетрамер, вокруг которого наматыва-
ется приблизительно 60 п. н. ДНК. В одной клет-
ке Е coli есть приблизительно 60 000 молекул
белка HU; этого достаточно для того, чтобы по-
крыть приблизительно одну пятую молекулы
ДНК, но неизвестно, равномерно ли эти тетра-
меры распределены по ДНК или ограничены
областью ядра нуклеоида.
Приведенные выше соображения относят-
ся к хромосоме Е coli, которую мы полагаем ти-
пичной для всякой бактериальной хромосомы.
Но мы должны проявлять осторожность при
сравнении бактериальной хромосомы и хромо-
сомы второй группы прокариотов, архей. Одна
из причин, по которой археи рассматриваются
Глава 8. Геномы прокариотов и органелл эукариотов 297
как отдельная группа организмов, отличающихся от бактерий, состоит в том, что археи
не обладают упаковочными белками типа HU, но вместо этого имеют белки, которые
намного больше подобны гистонам. Они формируют тетрамер, который связывается
приблизительно с 80 п. н. ДНК, и образуют структуру, подобную нуклеосоме эукариотов
(см. рис. 7.2). В настоящее время мы располагаем весьма скудной информацией о ну-
клеоиде архей, однако можем предположить, что эти гистоноподобные белки играют
центральную роль в упаковке ДНК.
Некоторые бактерии имеют линейные или многораздельные геномы
Геном Е. coli, как было описано выше, представлен единственной кольцевой мо-
лекулой ДНК. Такое устройство характерно также и для подавляющего большинства
хромосом доселе изученных бактерий и архей, однако со временем обнаруживается все
возрастающее число линейных версий. Первая из них, присущая Borrelia burgdorferi —
организму, вызывающему болезнь Лайма, — была описана в 1989 г., а в последующие
годы подобные открытия были сделаны для Streptomyces coelicolor и Agrobacterium
tumefaciens. Линейные молекулы имеют свободные концы, которые следует отличать от
разрывов ДНК, так что этим хромосомам необходимы специальные концевые структуры,
эквивалентные теломерам хромосом эукариотов (раздел 7.1.2). У Borrelia и Agrobacterium
настоящие концы хромосомы различимы, потому что образуется ковалентная связь
между 5'- и З’-концами полинуклеотидов в двойной спирали ДНК, а у Streptomyces концы
оказались отмеченными специальными связывающими белками.
Вторая и более широко распространенная особенность структуры генома прока-
риотов — присутствие у некоторых из них многораздельных геномов — геномов, кото-
рые разделены на две и более молекулы ДНК. С такими многораздельными геномами
часто возникает проблема в различении подлинной части генома и плазмиды. Плаз-
мида — это маленькая часть ДНК, часто, но не всегда кольцевая, которая сосуществует
с главной хромосомой в бактериальной клетке (рис. 8.5). Плазмиды некоторых типов
способны встраиваться в основной геном, но другие, как думают, являются постоянно
независимыми. Плазмиды несут гены, которые обычно отсутствуют в главной хромо-
соме, но во многих случаях эти гены незначительны
для бактерии, ибо кодируют такие характеристики,
как устойчивость к антибиотикам, в каковой бакте-
рия не нуждается, если условия окружающей среды
приемлемы (таблица 8.1). Наряду с этой очевидной
незначительностью, многие плазмиды обладают
способностью передаваться от одной клетки другой,
и одни и те же плазмиды иногда встречаются в бакте-
риях, которые принадлежат к различным видам. Столь
разнообразные особенности плазмид предполагают,
что они независимые сущности и что в большинстве
случаев содержимое плазмид в клетке прокариотов
не должно включаться в содержание ее генома.
Так, у такой бактерии, как Е. coli К12, которая имеет хромосому длиной 4,6 млн п. н.
и может вбирать в себя различные сочетания плазмид, ни одна из коих не превышает
нескольких тыс.п.н. в размере1) и не несет никаких значимых функций, приемлемо
Однако, есть плазмида E.coli, называемая F-фактором. Она имеет размер около 100 тыс. п. н. Эта
плазмида определяет способность клетки, ее несущей, к конъюгации и передаче собственного генома другой
клетке — аналог полового процесса. — Прим. ред.
Нуклеоид
Плазмиды
Рис. 8.5. Плазмиды являют собой ма-
ленькие кольцевые молекулы ДНК,
встречающиеся в клетках некоторых
прокариотов
298 Часть 2. Анатомия генома
Таблица 8.1. Особенности типичных плазмид
Черта плазмиды Функции генов Примеры
Устойчивость Устойчивость к антибиотику Rbk Escherichia coli и других бактерий
Фертильность Конъюгация и передача ДНК между бактериями F Е. coli
Убийца Синтез токсинов, которые убивают другие бактерии Col Е. coli ввиду выработки колицина
Вызывающие распад Ферменты для метаболизма необычных молекул TOL Pseudomonas putida (метаболизм толуола)
Вирулентность Патогенность Ti Agrobacterium tumefaciens, придаю- щий способность вызывать поражение корончатым цецидием на двудольных растениях
определить главную хромосому как «геном». С другими прокариотами это не столь
просто (таблица 8.2).
Vibrio cholerae — болезнетворная бактерия, которая вызывает холеру, — имеет две
кольцевые молекулы ДНК: одна из них 2,96 млн п. н., а другая —1,07 млн п. н., при этом 71 %
от 3 885 генов этого организма находится в большей из них. Казалось бы очевидным, что
эти две молекулы ДНК, вместе взятые, и составляют геном Vibrio, но более пристальный
взгляд показывает, что большинство генов, отвечающих за центральные отправления
клетки, например экспрессии генома и выработки энергии, а также львиная доля генов,
которые обусловливают патогенность, расположены на большей из двух молекул. Меньшая
молекула содержит много существенных генов, но имеет также и некоторые особенности,
которые считают характерными для плазмид, особенно присутствие интегрона — набора
генов и прочих последовательностей ДНК, которые позволяют плазмидам захватывать гены
бактериофагов и других плазмид. Поэтому оказывается возможным, что меньший геном
является «мегаплазмидой», которая была приобретена для Vibrio ее предком в некоторый
период эволюционного прошлого этой бактерии. Deinococcus radiodurans R1, чей геном
представляет особый интерес, потому что он содержит много генов, которые помогают
этой бактерии сопротивляться вредному воздействию радиации, построен по той же схеме,
где существенные гены распределены по двум кольцевым хромосомам и двум плазмидам.
Однако геномы Vibrio и Deinococcus относительно несложные по сравнению с таковым как
Borrelia burgdorferi В31, чья линейная хромосома размером 911 тыс.п.н., несущая 853 гена,
сопровождается 17 или 18 линейными и кольцевыми плазмидами, которые вместе вносят
в геном остальные 533 тыс.п. н. и как минимум 430 генов. Хотя функции большей части этих
генов неизвестны, те, что были распознаны, включают несколько таких, которые обычно
не считались бы несущественными, например, гены мембранных белков и биосинтеза
пуринов. Вывод: по крайней мере некоторые из плазмид Borrelia являются необходимыми
компонентами генома, что наводит на мысль о возможности того, что некоторые прокариоты
имеют чрезвычайно многораздельные геномы, включающие ряд отдельных молекул ДНК,
а это более похоже на то, что мы видим в ядре эукариотов, чем на «типичное» устройство
генома прокариота. Данная интерпретация генома Borrelia все еще спорна и усложняется тем
фактом, что родственная бактерия Treponema pallidum, чей геном представлен единственной
кольцевой молекулой ДНК размером 1138 тыс.п.н., содержащей 1041 ген, не содержит ни
одного гена, присутствующего в плазмидах Borrelia.
Глава 8. Геномы прокариотов и органелл эукариотов 299
Таблица 8.2. Примеры организации геномов прокариотов
Организация генома
Вид Молекулы ДНК Размер, млн п.н. Число генов
Escherichia coli К12 Одна кольцевая молекула 4,639 4405
Vibrio cholerae El Tor N16961 Две кольцевые молекулы
Главная хромосома 2,961 2 770
Мегаплазмида 1,073 1115
Deinococcus radiodurans R1 Четыре кольцевые молекулы
Хромосома 1 2,649 2633
Хромосома 2 0,412 369
Мегаплазмида 0,177 145
Плазмида 0,046 40
Borrelia burgdorferi B31 Семь или восемь кольцевых
молекул, одиннадцать линейных
молекул
Линейная хромосома 0,911 853
Кольцевая плазмида ср9 0,009 12
Кольцевая плазмида ср26 0,026 29
Кольцевая плазмида ср32* 0,032 Неизвестно
Линейная плазмида 1р17 0,017 25
Линейная плазмида 1р25 0,024 32
Линейная плазмида 1р28-1 0,027 32
Линейная плазмида 1р28-2 0,030 34
Линейная плазмида 1р28-3 0,029 41
Линейная плазмида 1р28-4 0,027 43
Линейная плазмида 1р36 0,037 54
Линейная плазмида 1р38 0,039 52
Линейная плазмида 1р54 0,054 76
Линейная плазмида 1р56 0,056 Неизвестно
Примечание: существует пять или шесть подобных версий плазмиды ср32 в одной бактерии.
8.2. Генетические характеристики геномов прокариотов
Определение местоположения генов путем просмотра последовательности намного
легче для прокариотов по сравнению с эукариотами (раздел 5.1.1), и для большинства
геномов прокариотов, которые были секвенированы, мы имеем достаточно точные
оценки числа генов и довольно исчерпывающие списки функций генов. Результаты
этих исследований были неожиданны и вынудили микробиологов пересмотреть зна-
чение термина «вид» применительно к прокариотам. Мы разберем эти эволюционные
300 Часть 2. Анатомия генома
вопросы в разделе 8.2.3. Сначала же нам необходимо осветить принцип обустройства
генов в геноме прокариотов.
8.2.1. Каким образом организованы гены в геноме прокариотов?
Мы уже имеем представление о том, что геномам бактерий свойственны компакт-
ные организации генов с очень небольшим местом между генами, поскольку это было
важной частью нашего обсуждения сильных и слабых сторон просмотра на ОРС как
средства распознавания генов в последовательности генома (см. рис. 5.3). Чтобы еще раз
подчеркнуть этот пункт, на рис. 8.6 представлена полная круговая карта генов генома
Е. coli К12. В геноме Е. coli наличествует некодирующая ДНК, но она составляет всего
лишь 11 % от общего количества и распределена по геному маленькими сегментами,
которые не видны, когда карта нарисована в таком масштабе. В этом отношении Е coli
типична для всех прокариотов, геномы которых были секвенированы к настоящему
времени, — геномы прокариотов имеют очень немного такого неиспользуемого пустого
пространства. Существуют теории, что такая компактная организация выгодна про-
кариотам, ибо, скажем, позволяет геному реплицироваться относительно быстро, но
эти представления никогда не были подтверждены надежными экспериментальными
данными.
Организация генов в геноме Е. coli
Рассмотрим геном Е. coli подробнее. Типичный сегмент размером 50 тыс.п.н. по-
казан на рис. 8.7. При сравнении этого сегмента с типичной частью генома человека
(см. рис. 7.12) сразу становится очевидно, что в сегменте Е. coli содержится больше
генов и намного меньше места между ними — 43 гена занимают 85,9 % этого сегмента.
Некоторые гены не имеют фактически никакого места между ними: thrA и thrB, напри-
мер, отделены одним-единственным нуклеотидом, a thrC начинается с нуклеотида,
следующего непосредственно после последнего нуклеотида гена thrB1). Эти три гена
являют нам пример оперона — группы генов, входящих в единый биохимический путь
(в данном случае это синтез аминокислоты треонина) и экспрессируемых совместно
друг с другом. Вообще, гены прокариотов короче своих присущих эукариотам аналогов:
средняя длина бактериального гена равна примерно двум третям гена эукариотов, даже
после того как интроны были удалены из последнего. Гены бактерий, кажется, немного
длиннее генов архей.
Две другие особенности геномов прокариотов можно увидеть на рис. 8.7. Во-первых,
в генах, находящихся в этом сегменте генома Е. coli, нет ни одного интрона В геноме
Е. coli вообще не имеется прерывных генов, и, как правило, полагают, что прерывные
гены фактически отсутствуют у прокариотов* 2), немногие исключения из этого правила
встречаются главным образом среди архей. Вторая особенность — редкость повторов.
Большинство геномов прокариотов не имеет ничего эквивалентного семействам много-
копийных рассеянных повторов, встречающихся в геномах эукариотов. Однако, вместо
этого, они обладают некоторыми последовательностями, которые могут повторяться
в любом месте в геноме, примеры представлены вставными последовательностями
[IS-элементами. — Прим, ред.] IS1 и IS186, которые можно заметить в сегменте длиной
Для бактерий характерны перекрывающиеся стоп- и старт-кодоны смежных генов, например ATGA
или TGATG. Такое перекрывание, по-видимому, обеспечивает реинициацию трансляции без сборки рибосо-
мы. — Прим. ред.
2) В геномах прокариот нет и механизма для вырезания интронов, за исключением самосплайсирую-
щихся РНК, — но это совсем другая история. — Прим. ред.
Глава 8. Геномы прокариотов и органелл эукариотов 301
Рис. 8.6. Геном Escherichia coli К12. Карта показана с областью
репликации (раздел 15.2.1), помещенной сверху. Гены на внеш-
ней стороне круга транскрибируются в направлении почасовой
стрелке, а гены на внутренней стороне — в направлении против
часовой стрелки. Изображение любезно предоставлено докто-
ром Ф. Р. Блеттнером
50 тыс.п.н., показанном на рис. 8.7. Они суть приме-
ры транспонируемых элементов — последователь-
ностей, которые имеют способность перемещаться
по геному и, в случае IS-элементов, передаваться от
одного организма другому, даже иногда между двумя
разными видами (раздел 9.2.2). Позиции элементов
IS1 и IS 186, показанных на рис. 8.7, относятся только
к конкретному штамму Е coli, из которого эта последо-
вательность была получена: если проанализировать
другой штамм, то последовательности IS вполне могли бы быть в иных позициях или
же могли бы совершенно отсутствовать в геноме. Геномы большинства других прока-
риотов имеют очень немного повторов (практически ни одного в геноме Campylobacter
jejuni NCTC11168 размером 1,64 млн п.н.), но есть исключения, и особенно характерное
среди них — бактерия менингита Neisseria meningitidis72491, которая имеет свыше 3700
копий повторов 15-ти различных типов (вместе взятые, они составляют почти 11 %
генома размером 2,18 млнп.н.).
thrB
л I thrO IS186 IS1
▼£ dnaK | carB
I—i—т—i—i—।—i—i—i—i—[—i—i—i—i—|—i—г
Точка старта репликации
fixA
50 тыс. п. h.
ОБОЗНАЧЕНИЯ:
Ген
Рассеянное
по всему геному
повторение
Рис. 8.7. Сегмент генома Escherichia coli размером 50 тыс. п. н.
Характерные особенности геномов прокариотов — опероны
Одна из характерных особенностей геномов прокариотов, илл юстрируемых Е coli, —
наличие оперонов. До того как последовательности геномов стали известны, мы думали,
что очень хорошо понимаем опероны; теперь мы не столь уверены в этом.
Оперон — группа генов, которые расположены в геноме смежно друг с другом
(возможно, между концом одного гена и началом следующего втиснут только один или
два нуклеотида). Все гены в опероне экспрессируются как единая единица. Организация
такого типа свойственна геномам прокариотов. Типичный пример, представляемый
геномом Е. coli, — оперон лактозы; этот оперон был открыт первым, он содержит три
гена, участвующих в преобразовании дисахарида лактозы в ее моносахаридные едини-
302 Часть 2. Анатомия генома
а) Оперон лактозы
1 тыс. п. н.
Лактопермеаза переносит
лактозу в клетку
P-галактозидаза и трансацетилаза
расщепляют лактозу на галактозу и глюкозу
Н ОН Н ОН
Лактоза
б) Оперон триптофана
1 тыс. п. н.
Хоризмовая кислота —> —> —> —► Триптофан
Рис. 8.8. Два оперона Escherichia coli. а) Оперон лактозы. Входящие в него три гена называют lacZ, lacY
и lacA, первые два отделены промежутком длиной 52 п.н., а вторые два — 64 п.н. Все три гена экс-
прессируются вместе: lacY кодирует лактопермеазу, которая переносит лактозу в клетку, a lacZ и 1асА
кодируют ферменты, которые расщепляют лактозу на составляющие ее сахара — галактозу и глюкозу, б)
Оперон триптофана, который содержит пять генов, кодирующих ферменты, вовлеченные в многоэтап-
ный биохимический путь превращения хоризмовой кислоты в аминокислоту триптофан. Гены вопероне
триптофана расположены ближе друг к другу, чем гены в опероне лактозы: trpE и trpD перекрываются на
1 п. н., так же как и trpB и trpA; trpD и trpCотделены промежутком длиной 4 п. н., a trpC и trpB — 12 п. н.
Более досконально вопросы регулирования этих оперонов рассмотрены в разделах 11.3.1 и 14.1.1
цы — глюкозу и галактозу (рис. 8.8, а). Данные моносахариды являются субстратами
для производящего энергию гликолитического пути, так что функция генов в опероне
лактозы заключается в преобразовании лактозы в форму, которая может быть исполь-
зована Е. coli как источник энергии. Лактоза не есть обычный компонент естественной
окружающей среды Е. coli, так что большую часть времени оперон не экспрессируется
и ферменты переработки лактозы не синтезируются бактерией. Когда лактоза по-
является в меню Е. coli, она включает оперон; все три гена экспрессируются вместе,
в результате чего осуществляется согласованный синтез перерабатывающих лактозу
Глава 8. Геномы прокариотов и органелл эукариотов 303
gate recA pilU emk pgsA recJ
Рис. 8.9. Типичный оперон в геноме Aquifex aeolicus. Входящие в него гены кодируют следующие белки:
gate — субъединица С глутамил-тРНК-аминотрансферазы, которая играет роль в синтезе белка; гесА —
белок рекомбинации RecA; pilU — двигательный белок; стк — цитидилаткиназа, требуемая для синтеза
нуклеотидов цитидина; pgsA — фосфотидилглицерофосфатсинтетаза — фермент, вовлеченный в био-
синтез жиров, и red — специфичная к однонитевым областям эндонуклеаза Red, которая является еще
одним белком, способствующим рекомбинации
ферментов. Это классический пример регуляции генов у бактерий и подробно мы его
разберем в разделе 11.3.1.
Всего в геноме Е. coli К12 имеется почти 600 оперонов, каждый содержит два и бо-
лее генов, и подобное их число имеется у Bacillus subtilis. В большинстве случаев гены
в опероне функционально связаны, кодируя набор белков, которые вовлечены в один
биохимический путь наподобие использования сахара в качестве источника энергии
или синтеза некоторой аминокислоты. Пример последнего — оперон триптофана Е. coli
(рис. 8.8, б). Специалистов в области генетики микробов привлекает простота этой
системы, посредством которой бактерия способна управлять своими разнообразными
биохимическими действиями путем регулирования экспрессии групп сцепленных генов,
связанных между собой в оперонах. Это может быть верной интерпретацией функции
оперонов у Е. coli, Bacillus subtilis и многих других прокариотов, но по крайней мере
у некоторых видов эта схема не такая прямая. Как архея Methanococcus jannaschii, так
и бактерия Aquifex aeolicus имеют опероны, но у них гены в отдельном опероне редко
имеют какую-либо биохимическую связь. Например, один из оперонов в геноме A aeolicus
содержит шесть сцепленных генов, где эти гены кодируют соответственно два белка,
вовлеченных в рекомбинацию ДНК, фермент, используемый в синтезе белка, белок,
требуемый для двигательной активности, фермент, вовлеченный в синтез нуклеотидов,
и фермент для синтеза жиров (рис. 8.9). Это типично для структуры оперона в геномах
A aeolicus и М. jannaschii. Другими словами, мысль о том, что экспрессия оперона при-
водит к согласованному синтезу ферментов, необходимых для одного биохимического
пути, у этих видов не подтверждается.
Итак, проекты расшифровки геномов лишь спутали все наши представления об
оперонах. Конечно слишком рано отказываться от теории, что во многих бактериях
опероны играют центральную роль в биохимическом регулировании, но нам необхо-
димо объяснить неожиданные особенности оперонов у A aeolicus и М. jannaschii. Было
показано, что и A aeolicus, и М. jannaschii являются автотрофами, что означает, что, в от-
личие от многих прокариотов, они способны синтезировать органические вещества из
углекислого газа, но как такое подобие между этими видами может помочь объяснить
обнаруживаемые у них структуры оперонов, не ясно1).
8.2.2. Сколько генов имеется в геноме прокариотов и каковы их функ-
ции?
Хотя размеры наиболее крупных геномов прокариотов и наименьших геномов эука-
риотов и перекрываются, в общем геномы прокариотов намного скромнее (таблица 8.3).
Есть еще одна особенность — некоторые виды бактерий имеют собственный генетический код, не-
сколько отличный от универсального. — Прим. ред.
304 Часть 2. Анатомия генома
Например, геном Е. coli К12 состоит только из 4639 тыс.п.н., что составляет две пятые
размера генома дрожжей, и имеет только 4405 генов. Большинство геномов прокарио-
тов меньше чем 5 млн п. н. в размере, но общий размах среди секвенированных геномов
составляет от 491 тыс.п.н. у Nanoarchaeum equitans до 9,1 млнп.н. у Bradyrhizobium
japonicum, а несколько несеквенированнных геномов значительно крупнее этого: Bacillus
megaterium, например, имеет огромный геном размером 30 млнп.н.
Большинство этих геномов организовано по подобной схеме, что и у Е. coli; это
означает, что размер генома пропорционален числу генов при средней плотности при-
близительно 950 генов на 1 млн п. н. ДНК. Поэтому число генов изменяется в широком
диапазоне и отражает характер экологической ниши, в пределах которой прокариот
данного вида обитает. Наибольшие геномы, как правило, принадлежат свободно жи-
вущим видам, обнаруживаемым в почве — среде, которая обычно рассматривается
как обеспечивающая широчайший диапазон физических и биологических условий, на
которые геномы этих видов должны так или иначе реагировать. На другом конце шкалы
находятся многие из наименьших геномов, принадлежащих видам, которые являются
облигатными паразитами, такими как Mycoplasmagenitalium, которая имеет всего лишь
470 генов в геноме размером 0,58 млнп.н. Ограниченная способность кодирования
у этих маленьких геномов означает, что эти виды неспособны синтезировать многие
питательные вещества и, следовательно, должны получать их от своих хозяев. Это
проиллюстрировано в таблице 8.4, в которой сравнивается каталог генов М. genitalium
с таковым Е. coli. Мы видим, что, например, Е. coli имеет 131 ген для биосинтеза амино-
кислот, тогда как М. genitalium имеет только один, что Е. coli имеет 103 гена для синтеза
кофакторов по сравнению с пятью у М. genitalium и так далее.
Эти сравнения навели на размышления о наименьшем числе генов, которое не-
обходимо для описания строения и биологических функций свободно живущей клетки.
Теоретические соображения первоначально привели к предположению, что 256 генов
являются необходимым минимумом, но эксперименты, в которых последовательно мути-
ровалось все большее число генов Mycoplasma, показывают, что их необходимо 265-350.
Был подобный интерес в поиске генов «отличительности» — таких, которые отличают
один вид от другого. Из 470 генов в геноме М. genitalium 350 присутствуют также и у
дальнеродственной бактерии Bacillus subtilis, а это предполагает, что биохимические
и структурные особенности, которые отличают Mycoplasma от Bacillus, закодированы
приблизительно в 120 генах, которые являются уникальными для первого из сравни-
ваемых организмов. К сожалению, природа этих предполагаемых генов отл ичительности
не дает никаких очевидных ключей к тому, что именно придает бактерии Mycoplasma
ее неповторимые черты.
8.2.3. Геномы прокариотов и понятие биологического вида
Проекты расшифровки геномов привели в беспорядок наше понимание того, что
составляет «вид» в царстве доядерных. Это всегда было проблемой в микробиологии,
потому что стандартные биологические определения вида было трудно применять
к микроорганизмам. Первые таксономисты, такие как Линней, описывали вид в мор-
фологических понятиях, согласно чему все члены одного вида имеют одинаковые или
весьма подобные особенности строения. Эта форма классификации была в моде до
начала двадцатого столетия и была впервые применена к микроорганизмам в 1880-е
гг. Робертом Кохом и другими учеными, которые использовали окрашивание и биохи-
мические опыты, чтобы устанавливать различия между видами бактерий. Однако было
Глава 8. Геномы прокариотов и органелл эукариотов 305
Таблица 8.3. Размеры геномов и число генов у разных прокариотов
Вид Размер генома, млнп. н. Приблизительное число генов
Бактерии
Mycoplasma genitalium 0,58 500
Streptococcus pneumoniae 2,16 2 300
Vibrio cholerae El Tor N16961 4,03 4000
Mycobacterium tuberculosis H37Rv 4,41 4000
Escherichia coli KI2 4,64 4400
Yersinia pestis CO92 4,65 4100
Pseudomonas aeruginosa PA01 6,26 5 700
Археи
Methanococcus jannaschii 1,66 1750
Archaeoglobusfulgidus 2,18 2 500
Примечание: для видов бактерий обозначение штамма (например, «К12») приводится в том случае, если
оно определено группой, которая секвенировала этот геном. У многих видов бактерий различные штаммы
имеют геномы разного размера и разные наборы генов (раздел 8.2.3).
признано, что классификация такого типа неточна, потому что многие из выделенных
видов состояли из разнообразия типов с весьма различными характеристиками. Пример
представляет Е. coli, которая, подобно многим видам бактерий, включает штаммы с ха-
рактерными болезнетворными свойствами, в пределах от безвредных до смертельных.
В двадцатом столетии биологи переопредел ил и термин «вид» в эволюционных понятиях,
и мы теперь рассматриваем вид как группу организмов, которые могут скрещиваться
друг с другом. Во всяком случае, в этой новой трактовке понятие вида более проблема-
тично соотносится лишь с микроорганизмами, потому что существует несколько разных
способов, которыми прокариоты могут обмениваться генами между собой, причем даже
такие, которые, согласно их биохимическим и физиологическим свойствам, относятся
к разным видам (см. рис. 3.23). Таким образом, барьер потоку генов, который является
центральным понятием в концепции вида, для прокариотов не существует.
Секвенирование геномов подчеркнуло трудности в применении понятия вида
к прокариотам. Стало ясно, что различные штаммы отдельного вида могут иметь очень
отличающиеся последовательности геномов, и даже могут иметь индивидуальные на-
боры специфичных для штамма генов. Это впервые было показано сравнением между
двумя штаммами Helicobacter pylori, который вызывает язвы желудка и другие болезни
пищеварительного тракта человека. Были выделены два штамма соответственно, в Ве-
ликобритании и Соединенных Штатах с геномами 1,67 млн п. н. и 1,64 млн п. н. Больший
геном содержит 1552 гена, а меньший — 1495 генов, при этом 1406 из этих генов
присутствуют в обоих штаммах. Другими словами, около 6-9 % содержания генов каж-
дого генома уникально для этого штамма. Намного более разительное отличие между
штаммами было обнаружено, когда последовательность генома обычного лаборатор-
ного штамма Е coli (К12) была сравнена с последовательностью одного из наиболее
болезнетворных штаммов О157:Н7. Длины этих двух геномов значительно отличают-
306 Часть 2. Анатомия генома
Таблица 8.4. Частичные каталоги генов Escherichia coli К12 и Mycoplasma genitalium
Категория Число генов в геноме
E.coli К12 М. genitalium
Всего кодирующих белок генов 4288 470
Биосинтез аминокислот 131 1
Биосинтез кофакторов 103 5
Биосинтез нуклеотидов 58 19
Белки клеточной оболочки 237 17
Энергетический обмен веществ 243 31
Промежуточный обмен веществ 188 6
Жировой обмен веществ 48 6
Репликация, рекомбинация и восстановление ДНК 115 32
Сворачивание белков 9 7
Регуляторные белки 178 7
Транскрипция 55 12
Трансляция 182 101
Захват молекул из окружающей среды 427 34
Примечание: приведенные числа относятся только к тем генам, к которым при секвенировании геномов
могли быть приписаны функции. Приписывание функций к некоторым из остальных генов, осуществленное
впоследствии, не изменило общей картины.
ся — 4,64 млн п. н. для К12 и 5,53 млн п. н. для О157:Н7, — при этом дополнительная ДНК
в болезнетворном штамме разбросана по геному почти в 200 отдельных локусах. Эти
«О-островки» содержат 1387 генов, отсутствующих у Е coli К12, причем многие из этих
генов кодируют токсины и другие белки, которые явно вовлечены в болезнетворные
свойства О157:Н7. Но это не просто тот случай, что бактерия из штамма О157:Н7 со-
держит дополнительные гены, которые делают ее болезнетворной. Штамму К12 также
присущи 234 сегмента своей собственной уникальной ДНК, и хотя эти «К-островки»
в среднем меньшего размера, чем О-островки, они тем не менее содержат 528 генов,
которые отсутствуют у бактерий штамма О157:Н7. Итак, ситуация такова, что и Е coli
О157:Н7, и Е. coli К12 имеют набор специфичных для штамма генов, которые составляют,
соответственно 26 % и 12 % каталога их генов. Это значительно большая изменчивость,
чем может допустить понятие вида применительно к высшим организмам, и ее трудно
примирить также и с любым определением вида из числа придуманных к настоящему
времени для микроорганизмов.
Подобного рода трудности проявляются еще сильнее, когда исследуются геномы
других бактерий и архей. Ввиду легкости, с которой гены могут перетекать между раз-
личными видами прокариотов, ожидалось, что одни и те же гены будут время от вре-
мени обнаруживаться в разных видах, но степень горизонтального переноса генов,
выявленная секвенированием, захватила всех врасплох. Большинство геномов содержит
по несколько сотен тыс.п.н. ДНК, приобретенной непосредственно от другого вида,
и в некоторых случаях эта цифра выше: 12,8 % генома Е. coli К12, что соответствует
Глава 8. Геномы прокариотов и органелл эукариотов 307
Escherichia coli К12
Mycobacterium tuberculosis
Bacillus subtilis
Synechocystis PCC6803
Deinococcus radiodurans
Archaeoglobus fulgidus
Aeropyrum pemix
Thermotoga maritime
Pyrococcus horikoshii
Methanobacterium thermoautrophicum
Haemophilus influenzae
Helicobacter pylori 26695
Aquifex aeolicus
Methanococcus jannaschii
Treponema pallidum
Borrelia burgdorferi
Rickettsia prowazekii
Mycoplasma pneumoniae
Mycoplasma genitalium
Рис. 8.10. Вклад горизонтального переноса генов в содержание геномов прокариотов. ДНК, которая яв-
ляется уникальной для данного вида, на диаграмме показана синим, а ДНК, которая была приобретена
посредством горизонтального переноса генов, — красным. Число в конце каждой полосы показывает
(в процентах) долю генома, полученную путем горизонтального переноса генов. Обратите внимание,
что межгенные области не были включены в анализ
0,59 млнп.н., было получено таким способом (рис. 8.10). Второй неожиданностью был
тот факт, что перенос генов происходил даже между очень различными видами, такими
как бактерии и археи. Например, термофильная бактерия Thermotoga maritima имеет
1877 генов, 451 из которых, как оказалось, получен от архей. Перемещение в другом на-
правлении, от бактерий к археям, не менее распространенное явление. Картина, которая
вырисовывается, показывает, что прокариоты, живущие в подобных экологических
нишах, обмениваются генами друг с другом, чтобы увеличить свою собственную при-
способленность к выживанию в своей специфической окружающей среде. Многие из
генов Thermotoga, которые были получены от архей, вероятно, помогли этой бактерии
приобрести характерную для неё способность переносить высокие температуры.
Секвенирование геномов показало также и то, что многие виды бактерий остаются
неопознанными. Микробиологи подозревали это много лет, понимая, что искусствен-
ные условия выращивания культур, применяемые для выделения бактерий из их есте-
ственных сред обитания, не подходят для всех видов, и что многие не растут при этих
условиях и, следовательно, остаются необнаруженными. Специалисты в новой научной
области — метагеномике — обращаются к этой проблеме, получая последовательности
ДНК от всех геномов в специфической среде обитания, например из морской воды или из
кислой почвы. В одном таком исследовании из 1500 литров воды поверхностного слоя
Саргассова моря было получено более 1 млнп.н. последовательности бактериальной
ДНК. Эта последовательность включала сегменты геномов более чем 1800 видов, из
которых 148 были совершенно новыми. Метагеномика открывает перед нами захваты-
вающую возможность, что в будущем мы сможем иметь последовательности геномов
организмов, которых никто никогда не видел.
8.3. Геномы органелл эукариотов
Теперь мы возвратимся к миру эукариотов, чтобы исследовать геномы, нахо-
дящиеся в их митохондриях и хлоропластах. Возможность того, что некоторые гены
308 Часть 2. Анатомия генома
могут быть расположены вне ядра — внехромосомные гены, как их первоначально
называли, — была впервые высказана в 1950-х гг. как средство объяснения необычных
схем наследования некоторых генов у гриба Neurospora crassa, дрожжей Saccharomyces
cerevisiae и фотосинтетической морской водоросли Chlamydomonas reinhardtii. Электрон-
ная микроскопия и биохимические исследования примерно в одно и то же время навели
ученых на мысль о том, что молекулы ДНК могут присутствовать в митохондриях и хло-
ропластах. В конечном счете, в начале 1960-х гг., эти различные каналы данных были
сведены воедино и существование геномов митохондрий и хлоропластов, независимых
и отличающихся от ядерного генома эукариотов, было признано.
8.3.1. Происхождение геномов органелл
Открытие геномов органелл породило множество предположений об их проис-
хождении. Сегодня большинство биологов признает, что теория эндосимбионтов верна
по крайней мере в общем виде — даже при том, что она считалась весьма оригинальной,
когда впервые была предложена в 1960-х гг. Теория эндосимбионтов основана на том
наблюдении, что процессы экспрессии генов, происходящие в органеллах, подобны во
многих отношениях эквивалентным процессам у бактерий. Кроме того, при сравнении
последовательностей нуклеотидов оказалось, что гены органелл более подобны эквива-
лентным генам бактерий, чем ядерным генам эукариотов. Поэтому теория эндосимбион-
тов считает, что митохондрии и хлоропласты — потомки свободно живущих бактерий,
которые образовали симбиотический союз с предтечей ядерной клетки, сущей целую
вечность назад во времена самых первых шагов эволюции.
Поддержка теории эндосимбионтов пришла от открытия организмов, которые
явно показывают стадии эндосимбиоза, менее развитые, чем наблюдаемые у митохон-
дрий и хлоропластов. Например, раннюю стадию в эндосимбиозе показывает протиста
Cyanophora paradoxa, чьи фотосинтетические структуры, названные цианеллами, от-
личаются от хлоропластов и вместо органелл напоминают проглоченные цианобакте-
рии. Точно так же Rickettsia, которая живет внутри ядерных клеток, может относиться
к современным штаммам бактерий, в свое время давшим начало митохондриям. Было
также предположено, что гидрогеносомы трихомонад (одноклеточные микробы, многие
из которых суть паразиты) представляют усовершенствованный тип митохондриаль-
ного эндосимбиоза, так как некоторые гидрогеносомы обладают геномом, тогда как
большинство — нет.
Если митохондрии и хлоропласты были когда-то свободно живущими бактерия-
ми, то с тех пор как установился эндосимбиоз, должно было быть перемещение генов
из органеллы в ядро. Мы не знаем, было ли массовое перемещение множества генов
из одного генома в другой на самом деле, и не понимаем, как оно могло произойти —
сразу или постепенно. Но мы знаем наверняка, что перемещение ДНК из органеллы
в ядро и между органеллами все еще происходит. Это было открыто в начале 1980-х гг.,
когда были получены первые частичные последовательности геномов хлоропластов.
Обнаружилось, что в некоторых растениях геном хлоропластов содержит сегменты
ДНК, несущие полные гены, которые являются копиями частей митохондриального
генома. Исходя из этого наблюдения, был сделан вывод о том, что эта так называемая
смешанная ДНК была перемещена из одной органеллы в другую. Теперь мы знаем, что
это не единственный тип перемещения, которое может произойти. Митохондриальный
геном Arabidopsis содержит различные сегменты ядерной ДНК, а также 16 фрагментов
хлоропластного генома, включающих шесть генов тРНК, которые сохранили свою ак-
Глава 8. Геномы прокариотов и органелл эукариотов 309
тивность после перемещения в митохондрию. Ядерный геном этого растения включает
несколько коротких сегментов хлоропластного и митохондриального геномов, а также
часть митохондриальной ДНК (длиной 270 тыс. п. н.), расположенную в пределах области
центромеры хромосомы 2. Было отмечено также перемещение митохондриальной ДНК
в ядерные геномы позвоночных.
8.3.2. Физические характеристики геномов органелл
Почти все эукариоты имеют митохондриальные геномы, а все фотосинтезирующие
эукариоты имеют хлоропластные геномы. Первоначально считалось, что фактически все
геномы органелл представлены кольцевыми молекулами ДНК. Электронная микроско-
пия показала в некоторых органеллах и кольцевую, и линейную ДНК, но было сделано
допущение о том, что линейные молекулы были попросту фрагментами кольцевых
геномов, которые разрушались в ходе подготовки препарата для электронной микро-
скопии. Мы все еще полагаем, что большинство митохондриальных и хлоропластных
геномов являются кольцевыми, но теперь мы признаем, что среди разных организмов
имеет место сильная изменчивость. У многих эукариотов кольцевые геномы сосуще-
ствуют в органеллах с линейными версиями и, в случае хлоропластов, с меньшими
кольцами, которые содержат подкомпоненты полного генома. Последняя схема наиболее
выражена у морских водорослей, названных динофлагеллатами, чьи хлоропластные ге-
номы раздроблены на множество маленьких колец, каждое из которых содержит лишь
один-единственный ген. Также мы теперь понимаем, что митохондриальные геномы
некоторых микробных эукариотов (например, Paramecium, Chlamydomonas и нескольких
видов дрожжей) всегда линейны.
Числа копий для геномов органелл объяснены не до конца. Каждая митохондрия
человека содержит приблизительно 10 идентичных молекул, а это означает, что на
клетку приходится приблизительно 8000 копий генома митохондрии, но у S’, cerevisiae
общее число, вероятно, меньше (приблизительно 6500) даже при том, что у нее на
митохондрию может приходиться более чем 100 геномов. Фотосинтезирующие микро-
организмы типа Chlamydomonas имеют приблизительно 1000 хлоропластных геномов
на клетку, что составляет примерно одну пятую числа геномов, находящихся в клетке
высшего растения. Одна загадка, которая возникла в 1950-е гг. и удовлетворительное
решение которой так и не было найдено, состоит в том, что, когда гены органеллы
изучаются путем генетических скрещиваний, результаты предполагают, что на клетку
приходится только одна копия митохондриального генома или генома хлоропластов.
Это явно не так, а посему эта загадка показывает, что наше понимание передачи геномов
органелл от родителя к потомству далеко не совершенно.
Размеры митохондриальных геномов являются самыми разными (таблица 8.5)
и не связаны со сложностью организма. Большинство многоклеточных животных
имеет маленькие митохондриальные геномы с компактной генетической органи-
зацией, где гены расположены близко друг к другу с небольшими промежутками
между ними. Митохондриальный геном человека (рис. 8.11) размером 16 569 п.н.
является типичным для организации такого типа. Большинство низших эукариотов
наподобие S. cerevisiae (рис. 8.12), а также цветковые растения имеют более крупные
и менее компактные митохондриальные геномы, где ряд генов содержит интроны.
Геномы хлоропластов имеют менее разнокалиберные размеры (таблица 8.5) и боль-
шей частью имеют структуру, подобную таковой у хлоропластного генома риса,
представленной на рис. 8.13.
310 Часть 2. Анатомия генома
Таблица 8.5. Размеры митохондриальных и хлоропластных геномов
Вид Тип организма Размер генома, тыс.п.н.
Митохондриальные геномы Plasmodium falciparum Протиста (малярийный паразит) 6
Chlamydomonas reinhardtii Зелёная водоросль 16
Mus musculus Позвоночное (мышь) 16
Homo sapiens Позвоночное (человек) 17
Metridium senile Беспозвоночное (морской анемон) 17
Drosophila melanogaster Беспозвоночное (плодовая мушка) 19
Chondrus crispus Бурая водоросль 26
Aspergillus nidulans Гриб аскомицет 33
Reclinomonas americana Протиста 69
Saccharomyces cerevisiae Дрожжи 75
Suillus grisellus Гриб базидиомицет 121
Brassica oleracea Цветковое растение (капуста) 160
Arabidopsis thaliana Цветковое растение (резушка Таля) 367
Zea mays Цветковое растение (кукуруза) 570
Cucumis melo Цветковое растение (дыня) 2500
Хлоропластные геномы Pisum sativum Цветковое растение (горох) 120
Marchantia polymorpha Печёночник 121
Oryza sativa Цветковое растение (рис) 136
Nicotiana tabacum Цветковое растение (табак) 156
Chlamydomonas reinhardtii Зелёная водоросль 195
ЛВ Ген дыхательного комплекса
ЯБ Ген рибосомной РНК
. Ген транспортной РНК
Рис. 8.11. Митохондриальный геном человека. Митохон-
дриальный геном человека является маленьким и ком-
пактным, с небольшим пустым пространством — настоль-
ко, что гены АТР6 и АТР8 перекрываются. Сокращения:
АТР6, АТР8 — гены субъединиц 6 и 8 АТФазы; COI, СОН,
COHI — гены субъединиц I, II и III цитохром-с-оксидазы;
Cytb — ген апоцитохрома b; ND1-ND6 — гены субъединиц
1-6 НАДВ-гидрогеназы
Глава 8. Геномы прокариотов и органелл эукариотов 311
ОБОЗНАЧЕНИЯ:
Ген дыхательного комплекса «=► Ген рибосомного белка Интрон
Ген рибосомной РНК • Ген транспортной РНК Ген прочей РНК
Рис. 8.12. Митохондриальный геномSaccharomycescerevisiae. Благодаря своим относительно небольшим
размерам многие митохондриальные геномы были полностью секвенированы. В митохондриальном
геноме дрожжей гены более далеко разнесены, чем в митохондриальном геноме человека, а некото-
рые гены имеют интроны. Организация такого типа типична для многих низших эукариотов и растений.
Митохондриальный геном дрожжей содержит пять дополнительных открытых рамок считывания (не
показанных на этой карте), причем до сих пор еще не было показано, что они кодируют функционально
активные продукты генов, и также имеется несколько генов, расположенных в пределах интронов пре-
рывных генов. Большинство последних кодируют белки-матуразы, вовлеченные в вырезание интронов
из транскриптов этих генов. Сокращения: АТР6, АТР8, АТР9 — гены субъединиц 6,8 и 9 АТФазы; COI, СОИ,
СОШ — гены субъединиц I, II и III цитохром-с-оксидазы; Cytb — ген апоцитохрома b; varl — ген белка,
связанного с рибосомой. Ген РНК 9 S определяет РНКовый компонент фермента рибонуклеазы Р (раз-
дел 12.1.3)
8.3.3. Генетическое содержание геномов органелл
Геномы органелл намного меньше своих ядерных аналогов, и поэтому мы ожи-
даем, что их генетическое содержание намного скромнее, что действительно так. Ми-
тохондриальные геномы показывают более заметную изменчивость, генетическое1^
содержание изменяется в пределах от пяти для малярийного паразита Р. falciparum до
92 для протесты Reclinomonas americana (таблица 8.6). Все митохондриальные геномы
содержат гены некодирующих рРНК и по крайней мере некоторые из белковых компо-
нентов дыхательной цепи (последняя является главной биохимической особенностью
митохондрии). Более богатые генами геномы кодируют вдобавок тРНК, рибосомные
белки и белки, вовлеченные в транскрипцию, трансляцию и транспорт прочих белков
в митохондрию из окружающей цитоплазмы (таблица 8.6). Большинство хлоропластных
Особо отметим, что огранеллы имеют собственные системы транскрипции, трансляции и репликации
(пусть в основном и закодированные в ядерном геноме). При этом они имеют собственный генетический код,
несколько отличный от универсального, а органеллы разных видов имеют различающиеся генетические
коды. — Прим. ред.
312 Часть 2. Анатомия генома
Таблица 8.6. Особенности митохондриальных геномов
Особенность Plasmodium falciparum Chlamidomonas reinhardtii Homo sapiens Saccharomices cerevisiae Arabidopsis thaliana Reclinomonas americana
Общее число генов 5 12 37 35 52 92
Типы генов
Кодирующие белок гены 3 7 13 8 27 62
Дыхательный комплекс 3 7 13 7 17 24
Рибосомные белки 0 0 0 1 7 27
Транспортные белки 0 0 0 0 3 6
РНК-полимераза 0 0 0 0 0 4
Фактор трансляции 0 0 0 0 0 1
Гены функциональной РНК 2 5 24 27 25 30
Гены рибосомной РНК 2 2 2 2 3 3
Гены транспортной РНК 0 3 22 24 22 26
Гены прочих РНК 0 0 0 1 0 1
Число интронов 0 1 0 8 23 1
Размер генома, тыс. п. н. 6 16 17 75 367 69
геномов, как видно, обладает одним и тем же набором из приблизительно 200 генов,
опять же кодирующих рРНК и тРНК, а также рибосомные белки и белки, вовлеченные
в фотосинтез (см. рис. 8.13).
Общая особенность геномов органелл прослеживается в таблице 8.6. Эти геномы
определяют некоторые из белков, находящихся в органелле, но не все из них. Остальные
белки кодируются ядерными генами, синтезируются в цитоплазме и переносятся в ор-
ганеллу. Если клетка имеет механизмы переноса белков в митохондрии и хлоропласты,
то почему бы не всем белкам органеллы быть закодированными ядерным геномом? Мы
еще не имеем убедительного ответа на этот вопрос, хотя было высказано предположение,
что по крайней мере некоторые из белков, кодируемых геномами органелл, являются
чрезвычайно гидрофобными и не могут переноситься через мембраны, что окружают
митохондрии и хлоропласты, и, таким образом, попросту не могут быть перемещены
в органеллу из цитоплазмы. Единственный путь, которым клетка может получить их
в органелле, состоит в том, чтобы производить их прямо там.
Заключение
К прокариотам относят организмы двух разных таксономических групп —
бактерии и археи. Бактериальный геном размещен в пределах нуклеоида — светло
окрашенной области, — в остальном же лишён характерных черт генома доядерной
Глава 8. Геномы прокариотов и органелл эукариотов 313
ОБОЗНАЧЕНИЯ:
Ген фотосинтеза Ген рибосомного белка
ЛВ Ген рибосомной РНК 4В Ген РНК-полимеразы
транспортной
РНК
Рис. 8.13. Хлоропластный геном риса. Показаны только гены с известными функциями. Ряд представ-
ленных генов содержит интроны, которые не обозначены на этой карте. К таким прерывным генам
относится несколько кодирующих тРНК генов, по каковой причине гены тРНК имеют различную длину
даже при том, что сами тРНК, которые они определяют, все имеют одинаковый размер
клетки. ДНК прикреплена к ядру из ДНК-связывающих белков, от которого 40-50
сверхспиральных петель ДНК расходятся лучами к периферии клетки. Мы знаем
намного меньше об аналогичных структурах архей, но они, вероятно, окажутся
устроенными совершенно иначе, поскольку археи обладают белками, подобными
гистонам эукариотов, а не белкам бактериального нуклеоида. Геном Е. coli — един-
ственная кольцевая молекула ДНК, но некоторые прокариоты имеют линейные
геномы, а некоторые — многораздельные геномы, состоящие из двух и более коль-
цевых и (или) линейных молекул. В более сложных случаях может быть трудно
различить, какие молекулы являются подлинными частями генома, а которые яв-
ляются несущественными плазмидами. Геномы прокариотов очень компактны,
314 Часть 2. Анатомия генома
с небольшим количеством повторной ДНК. Многие гены организованы в опероны,
члены которых экспрессируются совместно и могут иметь функциональную связь.
Число генов связано с размером генома. Наиболее крупные геномы принадлежат
свободно живущим видам, встречающимся в почве — окружающей среде, обеспечи-
вающей широкий диапазон физических и биологических условий, на которые эти
виды должны быть способны реагировать. Наименьшие геномы принадлежат видам,
которые являются облигатными паразитами типа Mycoplasma genitalium, имеющей
всего лишь 470 генов. От 250 до 350 генов, как думают, составляют минимальный
набор, необходимый для описания свободно живущей клетки. Исследования геномов
прокариотов усложнили понятие вида для этих организмов, поскольку геномы раз-
личных штаммов одних и тех же видов зачастую имеют очень различное генетическое
содержимое и между различными видами может происходить горизонтальный пере-
нос генов. Метагеномика — изучение всех геномов в определенной среде обитания
(типа морской воды) — показывает, что значительная доля видов, которые при-
сутствуют в специфической среде обитания, никогда еще не была опознана. Геномы
в митохондриях и хлоропластах ядерных клеток имеют свойственные прокариотам
особенности, потому что они произошли от свободно живущих бактерий, которые
образовали симбиотическое объединение с предтечей ядерной клетки. Большей
частью митохондриальные и хлоропластные геномы являются кольцевыми, возмож-
но многораздельными, с числом копий 1000-10 000 на клетку. Митохондриальные
геномы изменяются в размере от 6 до 2 500 тыс. п. н. и содержат 5-100 генов, в числе
которых гены митохондриальных рРНК, тРНК и белков типа компонентов дыха-
тельного комплекса. Геномы хлоропластов разнятся менее заметно, большинство
характеризуется размером 100-200 тыс. п. н. и имеет одинаковый для всех набор из
приблизительно 200 генов, основная доля которых кодирует функциональные РНК
и белки для фотосинтеза.
Глава 8. Геномы прокариотов и органелл эукариотов 315
Задания для закрепления материала
* Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
8.1 *. Как упакована ДНК в клетках бакте-
рий типа Е coli?
а. Она упакована в комплексы ну-
клеосом, содержащие белки-
гистоны.
Ь. Она упакована в нуклеоидные
структуры, содержащие белки-
гистоны.
с. Она упакована в комплексы ну-
клеосом, содержащие ДНК-гиразу
и ДНК-топоизомеразу.
d. Она сверхспирализована ДНК-
гиразой и ДНК-топоизомеразой.
8.2 . Что из себя представляет бактери-
альный нуклеоид?
а. Это прикрепленная к мембране
органелла, которая содержит ге-
номную ДНК.
Ь. Это светло окрашенная область
бактериальной клетки, которая
содержит геномную ДНК.
с. Это белковый комплекс бактери-
альной клетки, который связыва-
ет геномную ДНК.
d. Это прикрепленный к мембране
комплекс, который содержит ри-
босомы бактерии.
8.3 *. Что такое плазмида?
а. Маленькая, обычно кольцевая мо-
лекула ДНК, которая независима
от основной хромосомы.
Ь. Маленькая, обычно кольцевая
молекула ДНК, которая содержит
незаменимые гены.
с. Маленькая, обычно кольцевая мо-
лекула ДНК, которая стабилизи-
рует бактериальную хромосому.
d. Доядерный вирус, который может
заразить бактериальные клетки.
8.4 . Что такое интегрон?
а. Плазмида, которая может встроить-
ся в бактериальную хромосому.
Ь. Плазмида, которая может перей-
ти к другим бактериям.
с. Набор генов и последователь-
ностей ДНК, которые позволяют
плазмиде захватывать гены бак-
териофагов и других плазмид.
d. Клонирующий вектор, который
содержит последовательности
бактериофага и плазмиды.
8.5 *. Что такое бактериальный оперон?
а. Группа генов, которые имеют за-
висимые биохимические функ-
ции.
Ь. Группа генов, которые являются
эволюционно родственными.
с. Группа генов, которые вовлече-
ны в единый биохимический путь
и экспрессируются совместно.
d. Группа генов, которые экспресси-
руются с различных промоторов,
но регулируются одними и теми
же белками-репрессорами.
8.6 . Какого типа повторные последова-
тельности присутствуют в бактери-
альных геномах?
а. И микросателлиты, и минисател-
литы.
Ь. Мобильные генетические эле-
менты.
с. LINE (длинные рассеянные ядер-
ные элементы).
d. Геномы прокариотов не обладают
никакими повторными последо-
вательностями.
8.7 *. Какая из следующих особенностей
не свойственна типичному бакте-
риальному оперону?
а. Гены транслируются в единый
полипептид.
316 Часть 2. Анатомия генома
Ь. Гены в опероне транскрибиру-
ются в единственную молекулу
мРНК.
с. Гены часто кодируют белки, ко-
торые вовлечены в один биохи-
мический путь.
d. Гены находятся под контролем
единственного промотора.
8.8 . Как доядерные организмы сначала
классифицировались на виды?
а. Окрашивание и биохимические
пробы.
Ь. Генетические опыты.
с. Наблюдение в микроскоп.
d. Исследования последовательно-
стей ДНК.
8.9 *. Какого из следующих типов обмен
ДНК НЕ относится к горизонталь-
ному переносу генов?
а. Перемещение генов от бактерий
к археям.
Ь. Перемещение генов от архей
к бактериям.
с. Слияние бактерий двух видов
и произведение диплоидного
потомства.
d. Перемещение гена от одного вида
к другому.
8.10 . Какие из следующих данных не мо-
гут быть сочтены подтверждением
теории эндосимбионтов?
а. Митохондрии и хлоропласты
имеют внешние структуры, по-
добные стенкам бактериальных
клеток.
Ь. Гены в этих органеллах подобны
генам бактерий.
с. Процессы экспрессии генов в этих
органеллах подобны процессам,
протекающим в клетках бакте-
рий.
d. Рибосомы органелл напоминают
рибосомы бактерий.
8.11 *. Наименьший бактериальный ге-
ном — несколько сотен тысяч п.н.
в длину, тогда как митохондри-
альный геном человека — менее
17 000 п. н. Каким из следующих фак-
торов обусловлен меньший размер
митохондриального генома?
а. Митохондриальный геном чело-
века потерял свои кодирующие
белок гены.
Ь. Митохондриальный геном чело-
века потерял свои гены функцио-
нальной РНК.
с. Митохондриальный геном чело-
века функционально не активен
и представляет собой эволюци-
онный реликт.
d. Гены из митохондриального ге-
нома человека переместились
в ядро.
8.12 . Сколько идентичных копий своей
молекулы ДНК содержит типичная
митохондрия человека?
а. Одну.
Ь. Десять.
с. Сто.
d. Восемь тысяч.
8.13 *. Гены какого из следующих типов не
были обнаружены ни в каком мито-
хондриальном геноме?
а. Гены тРНК.
Ь. Гены дыхательной цепи.
с. Гены гликолиза.
d. Гены рРНК.
8.14 . Каково правдоподобное объяснение
присутствия некоторых генов в ми-
тохондрии, тогда как все гены могли
бы переместиться в ядро?
а. Некоторые белки являются слиш-
ком большими, чтобы их можно
было перенести в митохондрии.
Ь. Некоторые белки состоят из мно-
жественных субъединиц.
с. Некоторые белки успели бы де-
градировать еще до их переноса
в митохондрии.
d. Некоторые белки слишком гидро-
фобии, чтобы быть перенесенны-
ми в митохондрии.
Глава 8. Геномы прокариотов и органелл эукариотов 317
Вопросы открытого типа
8.1 *. Каковы различия между хромосомой
эукариотов и хромосомой Е. coin
8.2 . Какие экспериментальные данные
предполагают, что хромосома Е. coli
организована в сверхспирализован-
ные домены и прикреплена к бел-
кам, которые ограничивают ее воз-
можность развернуться?
8.3 *. Каковы подобия между белками HU
Е coli и белками-гистонами эукарио-
тов?
8.4 . Геном Е. coli — единственная коль-
цевая молекула ДНК. Каких еще ти-
пов структуры генома встречаются
среди прокариотов?
8.5 *. Как гены и другие особенности по-
следовательности организованы
в типичном геноме прокариота? Ка-
кие различия наблюдаются в плот-
ности генов, в числе интронов и в
содержании повторной ДНК при
сравнении геномов прокариотов
и млекопитающих?
8.6 . Чем опероны Methanococcusjannaschii
и Aquifex aeolicus отличаются от та-
ковых Е. coin
8.7 *. Облигатная внутриклеточная па-
разитическая бактерия Mycoplasma
genitalium имеет всего лишь 470
генов. Почему этот организм до-
вольствуется столь малым числом
генов?
8.8 . Чем обусловлена зависимость меж-
ду размером генома и числом генов
у прокариотов?
8.9 *. Почему понятие вида, применяемое
к эукариотам, — то есть группа орга-
низмов, которые могут скрещивать-
ся друг с другом, — не применимо
к прокариотам?
8.10 . Каким образом можно секвениро-
вать геном организма, который ни-
когда не был выделен?
Изыскательские задачи
8.1 *. Следует ли нам отказаться от тради-
ционного представления о геноме
прокариотов как о единственной
молекуле кольцевой ДНК? Если это
так, то какое новое определение
«генома прокариотов» должно быть
принято?
8.2 . Поразмыслите о том, какая инфор-
мация может быть закодирована
в 250-350 генах, которые входят
в минимальный набор генома вся-
кой свободно живущей клетки.
8.3 *. Может ли концепция вида бактерий
пережить секвенирование их гено-
мов?
8.4 . Для чего вообще нужны геномы ор-
ганелл?
318 Часть 2. Анатомия генома
Тесты по рисункам
8.1 *. Здесь представлена модель нуклеоида Е coli. Каким образом геном Е coliупакован
в нуклеоиде?
8.2 . На рисунке показан сегмент генома Е coli размером 50 тыс. п. н. Каковы главные
особенности этой последовател ьности и что показывает ее сравнение с типичным
сегментом генома человека размером 50 тыс.п.н.?
8.3*. Исходя из того, что все эти гены уча- 8.4.
ствуют в биосинтезе триптофана,
выскажите предположение о том,
как будет согласована их экспрес-
сия у Е. coli?
На этом рисунке представлен ми-
тохондриальный геном человека.
Каковы главные особенности этого
генома? Где находятся остальные
гены, кодирующие белки митохон-
1 тыс. п. н.
trpE trpD trpC trpB trpA
Хоризмовая кислота —► —► —► —► Триптофан
Глава 8. Геномы прокариотов и органелл эукариотов 319
Дополнительная литература
Нуклеоиды прокариотов
Drlica, К. and Riley, М. (1990) The Bacterial Chromosome. American Society for
Microbiology, Washington, DC. A source of information on all aspects of bacterial DNA.
Sinden, R. R. and Pettijohn, D. E. (1981) Chromosomes in living Escherichia coli cells are
segregated into domains of supercoiling. Proc. Natl Acad. Sci. USA 78: 224-228.
White, M. F. and Bell, S. D. (2002) Holding it all together: chromatin in the Archaea. Trends
Genet. 18: 621-626.
Многораздельные геномы
Bentley,S.D. and Parkhill,). (2004) Comparative genome structure of prokaryotes.
Anno. Rev. Genet. 38: 771-791. Review of many ofthe aspects of prokaryotic genomes discussed
in this chapter.
Fraser, С. M., Casjens, S., Huang, W. M., et al. (1997) Genomic sequence of a Lyme disease
spirochaete, Borrelia burgdorferi. Nature 390: 580-586.
Heidelberg, J.F., Eisen, J. A., Nelson, W.C., etal. (2000) DNA sequence of both chromo-
somes of the cholera pathogen Vibrio cholerae. Nature 406: 477-483.
White, O., Eisen, J. A., Heidelberg, J. F., etal. (1999) Genome sequence ofthe radioresistant
bacterium Deinococcus rodiodurans Rl. Science 286:1571-1577.
Примеры организации генов
Blattner, F. R., Plunkett, G., Bloch, C. A., et al. (1997) The complete genome sequence of
Escherichia coli K-12. Science 277:1453-1462.
Bult,C.)., White, O., Olsen, G.)., et al. (1996) Complete genome sequence of the
methanogenic archaeon Methanococcus jannaschii. Science 273:1058-1073.
Deckert,G., Warren, P.V., Gaasterland,T., et al. (1998) The complete genome of the
hyperthermophile bacterium Aquifex aeolicus. Nature 392: 353-358.
Parkhill,)., Achtman,M., James,K.D., et al. (2000) Complete genome sequence of
a serogroup A strain of Neisseria meningitidis Z2491. Nature 404: 502-506.
Parkhill,)., Wren, B. W., Mungall, K., etal. (2000) The genome sequence ofthe food-borne
pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665-668.
Минимальное содержание геномов
Koonin,E.V. (2000) How many genes can make a cell: the minimal-gene-set concept.
Annu. Rev. Genomics Hum. Genet. 1: 99-116. Describes the experimental and theoretical work
that is being done on the minimal genome.
Проблемы, связанные с концепцией вида
Alm,R.A., Ling, L.-S. L., Moir,D.T., etal. (1999) Genomic-sequence comparison of two
unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176-180.
Boucher, Y., Douady,C.)., Papke, R.T., Walsh, D. A., Boudreau, M. E., Nesbo,C.L.,
Case,R. J. and Doolittle, W.F. (2003) Lateral gene transfer and the origins of prokaryotic
groups. Annu. Rev. Genet 37: 283-328.
Nelson, K.E., Clayton, R. A., Gill,S.R., et al. (1999) Evidence for lateral gene transfer
between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:
323-329.
Ochman,H., Lawrence, J. G. and Groisman,E.A. (2000) Lateral gene transfer and the
nature of bacterial innovation. Nature 405: 299-304.
320 Часть 2. Анатомия генома
Рета, N.T., Plunkett, G.,Burland,V.,eta/. (2001) Genomesequenceofenterohaemorrhagic
Escherichia coli O157:H7. Nature 409: 529-532.
Метагеномика
Riesenfeld, C. S., Schloss, P.D. and HandelsmanJ. (2004) Metagenomics: genomic
analysis of microbial communities. Annu. Rev. Genet 38: 525-552.
Venter, J. C., Remington, K., Heidelberg, J. E, et al. (2004 Environmental genome shotgun
sequencing of the Sargasso Sea. Science 304: 66-74.
Геномы органелл
Lang, В. V., Gray,M. W. and Burger, G. (1999) Mitochondrial genome evolution and the
origin of eukaryotes. Annu. Rev. Genet 33: 351-397.
Margulis, L. (1970) Origin of Eukaryotic Cells. Yale University Press, New Haven,
Connecticut. The first description ofthe endosymbiont theory for the origin of mitochondria and
chloroplasts.
Palmer, J.D. (1985) Comparative organization of chloroplast genomes. Annu. Rev. Genet
32:437-459.
Глава 9
Геномы вирусов и мобильные
элементы генома
По прочтении главы 9 вы сможете:
• Описывать диапазон структур капсидов, наблюдаемых у бактериофагов и ви-
русов других типов.
• Давать краткий обзор разнообразных вариантов организации геномов бакте-
риофагов.
• Проводить различие между литическим и лизогенным путями инфицирования
и приводить подробности ключевых шагов каждого из них.
• Описывать основные схемы репликации, используемые вирусами эукариотов
и, в частности, вирусными ретроэлементами.
• Обсуждать особенности сателлитных РНК, вирусоидов, вироидов и прионов.
• Демонстрировать знание отличий между консервативной и репликативной
транспозицией.
• Приводить подробное описание структур LTR-ретроэлементов и различных
типов связей между ними.
• Обсуждать, с примерами, главные особенности LINE и SINE.
• Описывать диапазон транспозонов ДНК, встречающихся у прокариотов.
• Объяснять, почему ДНК-транспозоны эукариотов сыграли важную роль в раз-
витии наших представлений о транспозиции.
• Приводить примеры транспозонов ДНК у растений и Drosophila melanogaster.
Вирусы — наинизшая и наипростейшая форма жизни, геномы которой мы и со-
бираемся исследовать в этой главе. Фактически вирусы настолько просты в биоло-
гическом смысле, что мы должны спросить себя, можно ли о них всерьез думать как
о живых организмах. Сомнения возникают отчасти потому, что вирусы построены по
образу, отличному ото всех остальных форм жизни (вирусы не есть клетки), и отчасти
из-за характера жизненного цикла вируса. Вирусы суть облигатные паразиты самой
крайней формы: они воспроизводятся только внутри клетки хозяина и для того, что-
бы реплицировать и экспрессировать свои геномы, должны использовать по крайней
мере часть генетических механизмов хозяина для своих собственных целей. Некоторые
вирусы обладают генами, кодирующими свои собственные ферменты ДНК-полимеразу
и РНК-полимеразу, но многие зависят от ферментов хозяина, связанных с репликацией
322 Часть 2. Анатомия генома
и транскрипцией генома. Все вирусы используют рибосомы и трансляционный аппарат
хозяина для синтеза полипептидов, из которых образуются белковые оболочки их по-
томства. Это означает, что гены вирусов должны соответствовать генетической системе
хозяина. Поэтому вирусы весьма специфичны к определенным организмам и отдельные
типы не могут заражать широкий спектр видов.
В этой главе мы рассмотрим также мобильные генетические элементы, которые
составляют существенную часть повторного компонента геномов эукариотов и прока-
риотов. Мы связываем эти элементы с вирусными геномами, потому что, как стало ясно
в последние годы, по крайней мере некоторые из этих повторных последовательностей
произошли от вирусов и являются, в сущности, вирусными геномами, утратившими
способность покидать клетки своего хозяина.
9.1. Геномы бактериофагов и вирусов эукариотов
Есть множество различных типов вирусов, однако наибольшее внимание генетиков
привлекли те из них, что заражают бактерии. Их называют бактериофагами и изучают
весьма досконально с 1930-х гг., когда первые молекулярные биологи, особенно Макс
Дельбрюк, выбрали фаги в качестве удобных опытных организмов, на которых можно
изучать гены. Мы проследуем путем, выбранным Дельбрюком, и используем бактерио-
фаги как отправную точку нашей экскурсии по миру вирусных геномов.
9.1.1. Геномы бактериофагов
Бактериофаги построены из двух основных компонентов: белка и нуклеиновой
кислоты. Белок формирует оболочку, или капсид, внутри которого содержится геном из
нуклеиновой кислоты. Существует три основных типа структуры капсида (рис. 9.1).
Двадцатигранный Нитевидный Головка с отростком
Рис. 9.1. Структуры капсида трех типов, обычно демонстри-
руемые бактериофагами
• Двадцатигранный, в котором
отдельные полипептидные
субъединицы (протомеры)
образуют трехмерную геоме-
трическую структуру, окружа-
ющую нуклеиновую кислоту.
Примеры — фаг MS2, кото-
рый заражает Escherichia coli,
и фаг РМ2, который заражает
Pseudomonas aeruginosa.
• Нитевидный, или винтовой,
в котором протомеры устроены
в виде винтовой спирали и соз-
дают имеющую форму стержня
структуру. Пример — фаг М13,
заражающий Е coli.
• Головка с отростком — сочетание двадцатигранной головки, содержащей ну-
клеиновую кислоту, и прикрепленного к ней нитевидного отростка и, возможно,
дополнительных структур, которые облегчают введение нуклеиновой кислоты
в клетку хозяина. Это общая структура, присущая, например, специфичным к Е coli
фагам Т4 и X, а также фагу SP01, тяготеющему к Bacillus subtilis.
Глава 9. Геномы вирусов и мобильные элементы генома 323
Геномам бактериофагов свойственно разнообразие структур и вариантов
организации
При обращении к геномам фагов должен употребляться термин «нуклеиновая
кислота», потому что в некоторых случаях их молекулы состоят из РНК. Вирусы — одна
из форм «жизни», которая противоречит заключениям Эвери и его коллег, а также Хер-
ши и Чейза, согласно которым генетический материал представлен ДНК (раздел 1.1.1).
Фаги и другие вирусы нарушают также и другое правило: их геномы, будь они из ДНК
или РНК, могут быть и однонитевыми, и двунитевыми. В сообществе фагов встречается
целый диапазон различных структур генома (сведены в таблицу 9.1). У фагов большин-
ства типов есть единственная молекула ДНК или РНК, которая включает полный геном.
Однако это не всегда так и несколько РНК-фагов имеют сегментированные геномы, то
есть их гены содержатся во множестве различных молекул РНК. Размеры геномов фагов
характеризуются чрезвычайно широким диапазоном — от приблизительно 1,6 тыс. п. н.
для наименьших фагов до более чем 150 тыс.п.н. для наибольших типа Т2, Т4 и Тб.
Геномы бактериофагов, будучи относительно маленькими, были одними из первых,
всесторонне изученных быстрыми и эффективными методами секвенирования ДНК,
которые были разработаны в конце 1970-х гг. Число генов изменяется от всего лишь
трех в случае MS2 до более 200 у более сложных фагов типа головки с отростком (см.
табл. 9.1). Меньшие геномы фагов, конечно же, содержат относительно мало генов, но
они могут быть организованы по очень сложной модели. Фаг фХ174, например, уму-
Таблица 9.1. Особенности некоторых типичных бактериофагов и их геномов
Фаг Хозяин Структура капсида Структура генома Размер гено- ма, тыс. п. н. Число генов
X Escherichia coli Головка с отростком Двунитевая линейная ДНК 49,5 48
фХ174 Е. coli Двадцатигранный Однонитевая кольцевая ДНК 5,4 11
f6 Pseudomonas phaseolicola Двадцатигранный Двунитевая сегментированная линейная РНК 2,9,4,0, 6,4 13
М13 E. coli Нитевидный Однонитевая кольцевая ДНК 6,4 10
MS2 E. coli Двадцатигранная Однонитевая линейная РНК 3,6 3
РМ2 Pseudomonas aeruginosa Двадцатигранная Двунитевая линейная ДНК 10,0 *21
SP01 Bacillus subtilis Головка с отростком Двунитевая линейная ДНК 150 100+
Т2,Т4, Тб E. coli Головка с отростком Двунитевая линейная ДНК 166 150+
Т7 E. coli Головка с отростком Двунитевая линейная ДНК 39,9 55+
Примечание: приведенная структура генома относится к содержимому капсида фага; некоторые геномы
существуют в иных формах внутри клетки хозяина
324 Часть 2. Анатомия генома
дряется упаковать в свой геном «дополнительную» биологическую информацию, так
как несколько из его генов перекрываются (рис. 9.2). Такие перекрывающиеся гены
имеют общие последовательности нуклеотидов (ген В, например, содержится полностью
в пределах гена А), но кодируют различные продукты генов, поскольку транскрипты
транслируются с различных начальных позиций и в различных рамках считывания. Пере-
крывающиеся гены не редки у вирусов. Более крупные геномы фагов содержат больше
генов, что отражает более сложные структуры капсидов этих фагов и зависимость от
большего числа кодируемых фагом ферментов в ходе цикла инфицирования. Геном
Т4, например, включает около 50 генов, вовлеченных исключительно в строительство
капсида фага (рис. 9.3). Несмотря на их сложную организацию, даже такие большие фаги
опять же требуют по крайней мере некоторых кодируемых хозяином белков и РНК,
чтобы осуществлять свои циклы инфицирования.
Рис. 9.2. Геном фХ174 содержит перекрывающиеся гены. Геном состоит из однонитевой ДНК. В увели-
ченной области показаны начало и конец перекрытия между генами EuD
Стратегии репликации в геномах бактериофагов
Бактериофаги по стилю их жизненного цикла классифицируются на две группы:
литические и лизогенные. Фундаментальное различие между этими группами заклю-
чается в том, что литический фаг убивает свою бактерию-хозяина очень скоро после
первичного инфицирования, тогда как лизогенный фаг может оставаться в состоянии
покоя внутри своего хозяина в течение длительного периода времени, даже на протя-
жении множества поколений клетки-хозяина. Типичные примеры этих двух вариантов
жизненного цикла олицетворены двумя фагами Е. coli: литический (или вирулентный)
Т4 и лизогенный (или умеренный) X.
Глава 9. Геномы вирусов и мобильные элементы генома 325
Рис. 9.3. Геном фага Т4. Геном состоит из двунитевой ДНК и по-
казан в кольцевой форме, существующей в клетке хозяина.
Показаны только гены, кодирующие компоненты капсида фага; У
около 100 прочих генов, вовлеченных вдругие события жизнен- X X
ного цикла фага, опущены /
Т4
гж, 1 - - I 166 тыс. п.н.
Т-группа фагов Е. coll (Т1-Т7) первой попала 1 I
в руки молекулярных генетиков и стала предметом V g
интенсивного изучения. Их литический цикл инфици- ’Ь jjr
рования был впервые исследован в 1939 г. Эмери Эл- /
лисом и Максом Дельбрюком, которые добавили фаги
Т4 к культуре Е. coll, выждали 3 минуты, чтобы фаги
прикрепились к бактериям, и затем измеряли число
зараженных клеток в течение 60 минут. Их результаты
(рис. 9.4, а) показали, что нет никакого изменения числа зараженных клеток в течение
первых 22 минут инфицирования (этот латентный период времени необходим фагам
для воспроизводства внутри своих хозяев). После 22 минут число зараженных клеток
начало увеличиваться, показывая, что лизис первоначальных хозяев произошел и что
новые фаги, которые были произведены, теперь заражают другие клетки в культуре.
Молекулярные события, происходящие на различных стадиях этой одноступенчатой
кривой роста, показаны на рис. 9.4, б. Начальное событие — прикрепление фаговой
частицы к рецепторному белку на внешней стороне бактерии. Фаги разных типов име-
ют различные рецепторы: например, у Т4 рецептор представлен белком, названным
ОтрС («Отр»1) означает «белок внешней мембраны»), который относится к классу
поринов — белков, образующих каналы во внешней мембране клетки и облегчающих
ей поглощение питательных веществ. После прикрепления фаг вводит свой ДНК-геном
в клетку через имеющийся у него нитевидный отросток. Сразу после введения ДНК
фага синтез ДНК, РНК и белков хозяина останавливается и начинается транскрипция
генома фага. В течение 5 минут молекула бактериальной ДНК деполимеризуется и по-
лученные нуклеотиды используются в репликации генома Т4. Через 12 минут начинают
появляться новые белки капсида фага и собираются первые готовые фаговые частицы.
Наконец, в конце латентного периода клетка разрывается и новые фаговые частицы
высвобождаются. За один типичный цикл инфицирования производится от 200 до 300
фагов Т4 на клетку, и все они могут отправиться заражать другие бактерии.
В большинстве своем фаги могут следовать литическим циклом инфицирования,
но некоторые, например X, могут пойти также и лизогенным циклом. В разделе 2.2.1,
когда мы смотрели на использование фагов X в качестве клонирующих векторов, мы
обнаружили, что в ходе лизогенного цикла геном фага встраивается в ДНК хозяина. Это
происходит немедленно после входа ДНК фага в клетку и порождает дремлющую фор-
му бактериофага, называемую профагом (рис. 9.5, а). Встраивание происходит путём
сайт-специфической рекомбинации (раздел 17.2) между идентичными участками
последовательности размера 15 п.н., имеющимися в геномах X и Е. coli. Обратите вни-
мание, что это означает, что геном X встраивается в молекулу ДНК Е coli всегда в одной
и той же позиции. Встроенный профаг может сохраняться в молекуле ДНК хозяина на
протяжении многих поколений клетки, реплицируясь наряду с бактериальным геномом
и передаваясь с ним дочерним клеткам. Однако если профаг индуцируется любым из
Outer membrane protein. — Прим, перев.
326 Часть 2. Анатомия генома
б) Литический цикл инфицирования
ДНК
Белковая
оболочка
Бактериофаг Т4
Бактериофаг Т4
прикрепляется
к бактерии Е. coli
--------------►
| Транскрипция ДНК
▼ фага началась
а) Одноступенчатая кривая роста
О 0 10 15 20 25 30 35
Время, мин
Рис. 9.4. Литический цикл инфицирования бактериофага Т4.
а) Одноступенчатая кривая роста, построенная по данным
эксперимента, проведенного Эллисом и Дельбрюком, б) Мо-
лекулярные события, происходящие в ходе литического
цикла инфицирования
мин
ДНК-кРНК
| Репликация
▼ ДНК фага
ДНК-кДНК РНК
I Синтез белков
▼ капсида
мин
РНК -► белок
мин
I Клетка-хозяин разрывается,
новые фаговые частицы
высвобождаются
нескольких химических или физических раздражителей, происходит его переключение
на литический способ инфицирования. Каждый из них, кажется, связан с повреждением
ДНК и, возможно, поэтому сигнализирует о неизбежной смерти хозяина по естествен-
ным причинам. В ответ на эти раздражители второе событие рекомбинации вырезает
геном фага из ДНК хозяина, начинается репликация ДНК фага и синтезируются белки
оболочки фага (рис. 9.5, б). В конечном счете клетка разрывается и высвобождаются
новые фаги X. Лизогения привносит дополнительный уровень сложности в жизненный
цикл фага и дает фагу возможность избрать специфическую стратегию инфицирования,
наиболее подходящую в свете преобладающих условий.
Глава 9. Геномы вирусов и мобильные элементы генома 327
Рис. 9.5. Лизогенный цикл инфицирова-
ния на примере бактериофага X. После
индукции цикл инфицирования подобен
литическому. В разделе 14.3.1 приведено
описание того, как экспрессия генома X ре-
гулируется в ходе лизогении
а) Встраивание в ДНК хозяина
j Бактериофаг X
Е. coli
9.1.2. Геномы вирусов эука-
риотов
Капсиды вирусов эукариотов
являются или двадцатигранными
или нитевидными: структура типа
головки с отростком присуща ис-
ключительно бактериофагам. Одна
из отличных особенностей вирусов
эукариотов, в особенности таких,
у которых хозяева-животные, состо-
ит в том, что капсид их может быть
окружен липидной мембраной, об-
разующей дополнительный компо-
нент структуры вируса (рис. 9.6). Эта
мембрана заимствуется у хозяина,
когда новая вирусная частица остав-
ляет клетку, и может впоследствии
. Сайт-специфическая
Участок встраивания \ рекомбинация
в ДНК Е. coli я
Профаг X
/ Многократные
г деления клетки
6)
Вырезание и синтез
новых фагов
Вырезанная
У Индукция
/ профага
/Экспрессия генов, репликация ДНК
и синтез капсида фага
—• Высвобождение
Г* новых фагов X
быть видоизменена вставкой специфических белков вируса.
Структуры геномов вирусов эукариотов и присущие им стратегии реплика-
ции
Геномы вирусов эукариотов показывают большое разнообразие структур (табли-
ца 9.2). Они могут быть из ДНК или из РНК, одно- или двунитевыми (или частично двуни-
тевыми с однонитевыми областями), линейными или кольцевыми, сегментированными
или несегментированными. По причинам, которые никому не известны, подавляющее
большинство вирусов растений имеет геномы из РНК. Размеры геномов охватыва-
ют приблизительно тот же диапазон, что
и наблюдаемый у фагов, хотя наибольшие
геномы вирусов (например, геном вируса
коровьей оспы имеет размер 240 тыс.п.н.)
несколько больше, нежели наибольшие ге-
номы фагов.
Хотя большинство вирусов эукарио-
тов следует только литическому циклу
инфицирования, немногие принимают на
себя управление генетическим механиз-
Шипы
Мембрана
РНК
Белок капсида
Рис. 9.6. Структура ретровируса эукариотов. Капсид
окружен липидной мембраной, к которой прикре-
плены дополнительные вирусные белки
мом клетки-хозяина — до степени, кото-
рую может позволить себе только бакте-
риофаг. Многие вирусы сосуществуют с их
328 Часть 2. Анатомия генома
Таблица 9.2. Особенности некоторых типичных вирусов эукариотов и их геномов
Вирус Хозяин Структура генома Размер генома, тыс. п.н. Число генов
Аденовирус Млекопитающие Двунитевая линейная ДНК 36,0 30
Гепатита В Млекопитающие Частично двунитевая кольцевая ДНК 3,2 4
Вирус гриппа Млекопитающие Однонитевая сегментированная линейная РНК 22,0 12
Парвовирус Млекопитающие Однонитевая линейная ДНК 1,6 5
Полиовирус Млекопитающие Однонитевая линейная РНК 7,6 8
Реовирус Млекопитающие Двунитевая сегментированная линейная РНК 22,5 22
Ретровирусы Млекопитающие, птицы Однонитевая линейная РНК 6,0-9,0 3
SV40 Обезьяны Двунитевая кольцевая ДНК 5,0 5
Вирус табачной мозаики Растения Однонитевая линейная РНК 6,4 6
Вирус коровьей оспы Млекопитающие Двунитевая кольцевая ДНК 240 240
Примечание: приведенная структура генома относится к содержимому капсида; некоторые геномы
существуют в различных формах внутри клетки хозяина.
клетками-хозяевами в течение долгих периодов (возможно, годами), при этом функции
клетки-хозяина прекращаются только к концу цикла инфицирования, когда вирусное
потомство, которое сохранялось в клетке, высвобождается. У других вирусов циклы
инфицирования подобны поведению фага М13 в Е. coli (раздел 4.1.1), и они непрерывно
синтезируют новые вирусные частицы, которые выселяются из клетки. Такие долго-
срочные заражения могут происходить даже если вирусный геном не встраивается
в ДНК хозяина, но это не подразумевает, что не существует вирусов эукариотов, эквива-
лентных лизогенным бактериофагам. Ряд ДНК- и РНК-вирусов способны встраиваться
в геномы своих хозяев, иногда оказывая сильное воздействие на клетку-хозяина. При-
меры встраивающихся вирусов эукариотов — вирусные ретроэлементы. В их пути
репликации входит новый шаг, за счет которого РНК-версия генома преобразуется
в ДНК. Известно два вида вирусных ретроэлементов.* ретровирусы, капсиды которых
содержат РНК-версию генома, и параретровирусы, чей заключенный в капсид геном
состоит из ДНК. Способность вирусных ретроэлементов преобразовывать РНК в ДНК
была подтверждена независимо в 1970 г. Говардом Темином и Дэвидом Балтимором.
Работая с клетками, зараженными ретровирусами, и Темин и Балтимор выделили
фермент, ныне называемый обратной транскриптазой, который способен произво-
дить ДНК-копию матрицы РНК (и приносит огромную пользу в экспериментальном
исследовании геномов — раздел 2.1.1). Типичный геном ретровируса представляет со-
бой однонитевую молекулу РНК длиной 6 000-9 000 нуклеотидов. После входа в клетку
геном копируется в двунитевую ДНК с помощью нескольких молекул обратной транс-
Глава 9. Геномы вирусов и мобильные элементы генома 329
Рис. 9.7. Встраивание генома ретровируса в хромосому хозяина
криптазы, которые вирус несет в своем капсиде. После этого двунитевая версия генома
встраивается в ДНК хозяина (рис. 9.7). В отличие от генома фага К геном ретровируса
нисколько не обладает подобием последовательностей с предписанным ему участком
вставки в ДНК хозяина. Встраивание вирусного генома в ДНК хозяина — необходимая
предпосылка для экспрессии генов ретровируса. Имеется три таких гена, названныхgqg,
pol и env (рис. 9.8). Каждый из них кодирует полипротеин, который после трансляции
расщепляется на два и более функционально активных продукта генов. Эти продукты
включают белки оболочки вируса (с гена env) и обратной транскриптазы (с гена роГ).
Белковые продукты объединяются с полноразмерными РНК-транскриптами генома
ретровируса и производят новые вирусные частицы.
В1983-84 гг. было показано, что возбудителями СПИДа (синдрома приобретенного
иммунодефицита) являются ретровирусы. Первый вирус СПИДа был выделен незави-
симо двумя группами, возглавляемыми Люком Монтанье и Робером Галло. Этот вирус
называют вирусом иммунодефицита человека, или ВИЧ 1, и он отвечает за самую распро-
страненную и болезнетворную форму СПИДа. Родственный ему вирус ВИЧ 2, открытый
Монтанье в 1985 г., распространен менее широко и вызывает более умеренную форму
этой болезни. Вирусы ВИЧ поражают находящиеся в кровотоке лимфоциты некоторых
типов и тем самым снижают иммунную реакцию хозяина. Эти лимфоциты несут на своих
поверхностях многочисленные копии белка по имени CD4, который является рецептором
для вируса. Частица ВИЧ связывается с белком CD4 и затем входит в лимфоцит после
слияния между его липидной оболочкой и мембраной клетки.
Геном — «краеугольный камень» жизни
Вирусы находятся на рубеже между живым и неживым мирами. На самом краю
этой границы — или, возможно, за ее пределами — располагается целое разнообразие
молекул нуклеиновых кислот, которые и можно и нельзя было бы классифицировать
как геномы. Примеры — сателлитные РНК и вирусоиды. Это молекулы РНК длиной
Рис. 9.8. Геном ретровируса. Каждое LTR — длин-
ное концевое повторение протяженностью
250-1400 п.н., которое играет важную роль в ре-
пликации генома (см. раздел 17.3.2)
LTR gag pol
env LTR
330 Часть 2. Анатомия генома
а) Автокаталитическое расщепление РНК
вироидов и вирусоидов
Геномы, сцепленные голова к хвосту
i Автокаталитическое
расщепление
Отдельные линейные геномы
б) Структура расщепления
'c-G/ Расщепление
Формирование кольца
ООО
Рис. 9.9. Автокаталитическое расщепление сцепленных геномов в ходе репликации вироидов и вирусои-
дов. а) Путь репликации, б) Структура типа «головки молотка», которая формируется на каждом участке
расщепления и которая обладает ферментативной активностью. N — любой нуклеотид
около 320-400 нуклеотидов, которые не кодируют белки своего собственного капсида,
а вместо этого кочуют из клетки в клетку внутри капсидов вирусов-помощников. Раз-
личие между этими двумя группами в том, что сателлитный вирус пользуется капси-
дом вируса-помощника за счет генома последнего, тогда как молекула РНК вирусоида
заключается в его капсид самостоятельно. Их обычно рассматривают как паразитов
своих вирусов-помощников, хотя известно по крайней мере несколько случаев, где
вирус-помощник не может реплицироваться без сателлитной РНК или вирусоида, что
позволяет предположить, что по крайней мере некоторые из имеющих место взаимо-
отношений являются симбиотическими. И сателлитные РНК и вирусоиды встречаются
преимущественно у растений, равно как и самая крайняя группа так называемых ви-
роидов. Это молекулы РНК длиной 240-375 нуклеотидов, которые не содержат никаких
генов и никогда не бывают заключены в капсид, а распространяются от клетки к клетке
в виде голой РНК. К ним относятся некоторые экономически ощутимые патогены, та-
кие как вироид наружной кожуры цитрусовых, который задерживает рост у деревьев
цитрусовых. Молекулы вироидов и вирусоидов являются кольцевыми и однонитевыми
и реплицируются ферментами, кодируемыми геномами хозяина или вируса-помощника.
В результате процесса репликации получается ряд РНК, соединенных голова к хвосту,
и, в случае некоторых вироидов и вирусоидов, они расщепляются автокаталитической
реакцией, в которой ферментом служит сама молекула РНК (рис. 9.9). Мы изучим такие
РНК-ферменты более подробно в разделе 12.2.4.
Возможно, молекулы нуклеиновых кислот, которые реплицируются внутри клеток
растений, и можно рассматривать как геномы, даже если они не содержат никаких ге-
нов. Того же нельзя сказать о прионах, поскольку эти заразные, вызывающие болезнь
частицы не содержат никакой нуклеиновой кислоты. Прионы являются причиной
бешенства у овец и коз, а их передача крупному рогатому скоту привела к новой болез-
ни, названной БГЭ (бычьей губчатой энцефалопатией). Вопрос о том, вызывает ли их
дальнейшая передача людям вариантную форму болезни Крейцфельда-Якоба (БКЯ),
спорен, но многие биологи отвечают на него утвердительно. Сначала думали, что прионы
относятся к вирусам, но теперь ясно, что они состоят исключительно из белка. Нормаль-
ная версия белка приона, называемая РгРс, кодируется ядерным геном млекопитающих
и синтезируется в мозгу, хотя его функция неизвестна. РгРс легко переваривается про-
Глава 9. Геномы вирусов и мобильные элементы генома 331
Рис. 9.10. Способ действия приона. У нормальной, здоровой овцы
в мозге находятся белки РгРс. Инфицирование молекулами PrPsc
приводит к преобразованию новосинтезированных белков РгРс
в PrPsc, что ввергает овцу в болезненное состояние
теазами, тогда как заразная версия, PrPsc, имеет более
насыщенную 0-листами структуру, которая является
устойчивой к протеазам и формирует фибриллярные
агрегаты, наблюдаемые в зараженных тканях. Попав
в клетку, молекулы PrPsc способны преобразовывать
новосинтезированные белки РгРс в инфекционную
форму (механизм такого превращения еще не понят),
что заканчивается болезненным состоянием. Пере-
дача одного или нескольких таких белков PrPsc ново-
му животному приводит к накоплению новых белков
PrPsc в мозгу этого животного и к передаче болезни
(рис. 9.10). Заразные белки с подобными свойствами
известны у низших эукариотов, примеры представ-
лены прионами Ure3 и Psi+ Saccharomyces cerevisiae.
Ясно, однако, что прионы являются продуктами генов,
а не генетическим материалом, и, несмотря на свои
инфекционные свойства, которые привели к первоначальной путанице относительно
их статуса, они не имеют отношения к вирусам ил и к субвирусным частицам наподобие
вироидов и вирусоидов.
9.2. Мобильные элементы генома
В главах 7 и 8 мы узнали, что геномы эукариотов и — в меньшей степени — тако-
вые прокариотов содержат разбросанные по всему геному, или рассеянные, повторы,
некоторые с числом копий несколько тысяч на геном, при этом отдельные повторные
единицы распределены явно случайным образом (раздел 7.2.4). Для многих рассеянных
повторов закон распределения по всему геному задается транспозицией — процессом,
посредством которого сегмент ДНК может перемещаться по геному из одной позиции
в другую. Эти мобильные сегменты называют транспонируемыми элементами, или
транспозонами. Некоторые типы перемещаются посредством консервативного про-
цесса, который вовлекает вырезание последовательности из ее первоначальной по-
зиции и последующую ее повторную вставку в каком-либо другом месте. В результате
консервативной транспозиции транспозон попросту меняет свою позицию в геноме
без увеличения присущего ему числа копий (рис. 9.11). Репликативная транспозиция,
Консервативная
Репликативная
X
Рис. 9.11. Консервативная и репликативная транспозиция
332 Часть 2. Анатомия генома
Рис. 9.12. Ретротранспозиция. Сравните с рис. 7.20
и обратите внимание, что события по существу те
же самые, что и те, в результате которых получается
обработанный псевдоген
Ретротранспозон
Транскрипция
напротив, приводит к увеличению числа
копий, потому что в ходе этого процесса
исходный элемент остается на месте, а его
копия вставляется в новой позиции. Поэто-
му такой репликативный процесс может
привести к быстрому увеличению числа
копий транспозона в разбросанных по все-
му геному позициях.
Транспозиция обоих типов вовлекает
рекомбинацию, и поэтому мы рассмотрим
особенности этих процессов при изучении
рекомбинации и связанных с нею типов
-ЯШШКШВ- Однонитевая РНК
Обратная транскрипция
Двунитевая ДНК
Повторная встройка
Ретротранспозон
Копия
ретротранспозона
перестройки генома в главе 17. Что нас интересует сейчас, так это разнообразие струк-
тур, показываемое транспонируемыми элементами, имеющимися в геномах эукариотов
и прокариотов, и связь, которая существует между этими элементами и вирусными
геномами.
9.2.1. Транспозиция через РНК-посредник
Репликативные транспозоны могут быть далее подразделены нате, которые пере-
мещаются через РНК-посредник, и нате, которые в нем не нуждаются. Процесс, который
вовлекает РНК-посредник и называется ретротранспозицией [или ретропозицией. —
Прим, ред.], начинается с синтеза РНК-копии транспозона путем нормального процесса
транскрипции (рис. 9.12). Далее полученный транскрипт копируется в двунитевую
ДНК, которая изначально существует в виде независимой молекулы вне генома. Нако-
нец, ДНК-копия нашего транспозона встраивается в геном, возможно, обратно в ту же
самую хромосому, занимаемую исходной единицей, а возможно, и в какую-либо иную
хромосому. В конечном итоге мы получаем две копии исходного транспозона в различ-
ных точках генома.
Если мы сравним механизм ретротранспозиции с механизмом репликации вирус-
ного ретроэлемента, представленным на рис. 9.7, то увидим, что эти два процесса очень
подобны, одно существенное отличие состоит в том, что молекула РНК, которая ини-
циирует процесс, транскрибируется в ходе ретротранспозиции с эндогенной геномной
последовательности, а в ходе репликации вирусного ретроэлемента — с экзогенного
вирусного генома. Столь близкое подобие предупреждает нас о связи, существующей
между элементами этих двух типов.
РНК-транспозоны с длинными концевыми повторениями связаны с вирусны-
ми ретроэлементами
РНК-транспозоны, или ретроэлементы, являются особенностями геномов эука-
риотов и были не так давно открыты у прокариотов. Они могут быть классифицированы
на два основных типа: те, которые обладают длинными концевыми повторениями
(LTR), и те, которые ими не обладают. Длинные концевые повторения, которые играют
Глава 9. Геномы вирусов и мобильные элементы генома 333
Рис. 9.13. Гомологичная рекомбинация между LTR на обоих
концах Ту-элемента может дать дельта-последовательность
Ту-элемент
I Рекомбинация
центральную роль в процессе, посредством которого между LTR
РНК-копия LTR-элемента обратно транскрибируется
в двунитевую ДНК (раздел 17.3.2), имеются также и у * ^3
вирусных ретроэлементов (см. рис. 9.8). Теперь ясно, Одиночный дельта-элемент
что эти вирусы являются одним членом надсемейства
элементов, в которое входят также и эндогенные LTR-
транспозоны. Первым из открытых эндогенных элементов была последовательность
дрожжей Ту, которая имеет 6,3 тыс.п.н. в длину и число копий 25-35 в большинстве
геномов Saccharomyces cerevisiae, — вспомним, что один такой элемент присутствовал
в сегменте генома дрожжей длиной 50 тыс.п.н., который мы исследовали в разде-
ле 7.2.2 (см. рис. 7.15, б). Геномы дрожжей содержат также 100 или около того допол-
нительных копий LTR 7у-элементов длиной 330 п.н.; эти самостоятельные «дельта»-
последовательности, вероятно, возникают за счет гомологичной рекомбинации между
двумя LTR 7у-элемента, за счет которой может быть вырезана большая часть элемента
и оставлено единственный LTR (рис. 9.13). Это событие вырезания, вероятно, не связано
с транспозицией 7у-элемента, которая происходит посредством опосредствованного
РНК процесса, представленного на рис. 9.12.
В геномах дрожжей есть 7у-элементы нескольких типов. Наиболее представлен-
ный из них, Tyl, подобен ретроэлементу copia плодовой мушки. Поэтому теперь эти
элементы в совокупности называют семейством Tyl/copia. Если мы сравним структуру
ретроэлемента Tyl/copia со структурой вирусного ретроэлемента, то увидим явные се-
мейственные отношения (рис. 9.14, а и б). Каждый элемент Tyl/copia содержит два гена
(названные у дрожжей Ту А и ТуВ), которые являются подобными генам дад и pol вирусно-
го ретроэлемента. В частности, ТуВ кодирует полипротеин, который включает обратную
транскриптазу, играющую центральную
роль в транспозиции элемента Tyl/copia.
Обратите внимание, однако, что в элементе
Tyl/copia отсутствует эквивалент вирус-
ному гену env — тому, что кодирует белки
оболочки вируса. Это означает, что ретроэ-
лементы Tyl/copia не могут образовывать
заразные вирусные частицы и поэтому не
могут покинуть клетку своего хозяина. Они,
однако, образуют вирусоподобные частицы
(ВПЧ), состоящие из РНК- и ДНК-копий ре-
троэлементов, прикрепленных к стержне-
вым белкам, полученным из полипротеина
с гена ТуА. В отличие от этих элементов,
члены второго семейства ретроэлемен-
тов LTR, названные ТуЗ/gypsy (опять же
по аналогии с версиями, встречающимися
у дрожжей и плодовой мушки), имеют экви-
валент гена env (рис. 9.14, в), и по крайней
мере некоторые из них могут образовывать
а) Вирусный ретроэлемент
LTR gag pol
env LTR
~7 тыс. п. н.
6) Ретроэлемент Ty1/copia
LTR gag pol LTR
в) Ретроэлемент Ty3/gypsy
LTR gag pol
~7 тыс. п. h.
“env” LTR
~7 тыс. п. h.
Рис. 9.14. Структуры геномов ретроэлементов с LTR
334 Часть 2. Анатомия генома
Таблица 9.3. Транспонируемые элементы в геноме человека
Класс Семейство Приблизительное число копий Доля генома, %
SINE Alu 1200000 10,7
MIR 450000 2,5
MIR3 85000 0,4
LINE LINE-1 600000 17,3
LINE-2 370000 3,3
LINE-3 44000 0,3
Ретроэлементы с LTR ERV 240000 4,7
MaLR 285000 3,8
ДНК-транспозоны MER-1 213000 1,4
MER-2 68000 1,0
Прочие 60000 0,4
заразные вирусы. Хотя они классифицированы как эндогенные транспозоны, эти ин-
фекционные версии должны рассматриваться как вирусные ретроэлементы.
Ретроэлементы с LTR составляют существенные части многих геномов эукариотов
и особо изобильны в крупнейших геномах растений, в особенности в геномах злаков,
например кукурузы (см. рис. 7.15, г). Они составляют важный компонент также геномов
беспозвоночных и некоторых позвоночных, но в геномах людей и других млекопитающих
все LTR-элементы, как оказалось, являются распавшимися вирусными ретроэлементами,
а не истинными транспозонами. Эти последовательности называют эндогенными ре-
тровирусами (ЭРВ), и, имея число копий приблизительно 240 000, они составляют 4,7 %
генома человека (таблица 9.3). У человека ЭРВ имеют 6-11 тыс.п.н. в длину и содержат
копии генов gag, pol и env. Хотя большинство содержит мутации или делеции, которые
инактивируют один или несколько этих генов, несколько членов группы эндогенных
ретровирусов человека HERV-K имеют функционально активные последовательности.
При сравнении позиций элементов группы HERV-K в геномах различных индивидуумов,
было выявлено, что по крайней мере некоторые из них являются активными транс-
позонами. Большинство таких элементов человека, однако, являются неактивными
последовательностями, которые не способны дополнительно пролиферировать.
РНК-транспозоны с отсутствием LTR
РНК-транспозоны не всех типов имеют LTR-элементы. У млекопитающих самые
важные типы не содержащих LTR ретроэлементов, или ретропозонов, представлены
LINE (длинными рассеянными ядерными элементами) и SINE (короткими рассе-
янными ядерными элементами). SINE имеют наивысшее число копий среди рассеян-
ных повторных ДНК любого типа в геноме человека — более чем 1,7 миллиона копий,
охватывающих 14 % генома в целом (таблица 9.3). LINE менее распространены, имеют
лишь чуть более 1 миллиона копий, но, будучи длиннее, они составляют более весомую
долю генома — более 20 %. Относительное содержание LINE и SINE в геноме человека
подчеркивается их частотой встречаемости в сегменте длины 50 тыс.п.н., котрый мы
рассматривали в разделе 7.2.2 (см. рис. 7.12).
Глава 9. Геномы вирусов и мобильные элементы генома 335
Известно три семейства LINE в геноме че-
ловека, из которых одна группа, LINE-1, является
и наиболее частой, и единственным типом, ко-
торый способен транспонироваться, семейства
LINE-2 и LINE-3 состоят из неактивных релик-
тов. Полноразмерный элемент LINE-1 имеет
длину 6,1 тыс.п.н. и содержит два гена, один
из которых кодирует полипротеин, подобный
продукту вирусного гена pol (рис. 9.15, а). В нем
нет ни одного LTR, но З'-конец LINE отмечен
рядом пар оснований А-Т, обычно упоминаемым
как последовательность поли-А (хотя, конечно,
это последовательность поли-Т на другой нити
ДНК). Не все копии LINE-1 полноразмерны, по-
тому что обратная транскриптаза, кодируемая
LINE, не всегда производит полную ДНК-копию
a) LINE
поли-А
~6 тыс. п. н.
б) SINE
поли-А
~0,3 тыс. п. н.
Рис. 9.15. Не содержащие LTR ретроэлемен-
ты. Как LINE, так и SINE имеют поли-А после-
довательности на своих З'-концах
исходного РНК-транскрипта, а это означает, что часть З’-конца LINE может быть поте-
ряна. Такое событие усечения столь распространено, что только 1 % элементов LINE-1
в геноме человека представлен полномерными версиями, при этом средний размер
всех копий равен всего лишь 900 п.н. Хотя транспозиция LINE-1 случается редко, она
наблюдалась в культивируемых клетках и, как оказалось, ответственна за гемофилию
у некоторых пациентов из-за перемещения последовательности LINE-1 в ген фактора
VIII, прерывающего этот ген и, следовательно, препятствующего синтезу этого факто-
ра — важного белка свертывания крови.
SINE намного короче, чем LINE, будучи всего лишь 100-400 п. н., и не содержат никаких
генов, сие же означает, что SINE не производят свои собственные ферменты обратные транс-
криптазы (рис. 9.15, б). Вместо этого они «заимствуют» обратные транскриптазы, которые
были синтезированы LINE. Наиболее распространенный SINE в геномах приматов — Alu,
который имеет число копий приблизительно 1,2 миллионау людей (таблица 9.3). Элемент
Alu состоит из двух половин, каждая половина состоит из подобных последовательностей
длины 120 п.н., с 31-32 п.н. вставкой в правой половине (рис. 9.16). Геном мыши имеет
родственный элемент, названный В1, длиной 130 п.н. и эквивалентный одной половине
последовательности Alu. Некоторые элементы Alu активно копируются в РНК, что обеспе-
чивает возможность для быстрого размножения элемента.
Alu получается из гена для РНК 7SL — некодирующей РНК, участвующей в передви-
жении белков по клетке. Первый элемент Alu, возможно, возник в результате случайной
обратной транскрипции молекулы РНК
7SL и встраивания этой ДНК-копии в ге-
ном человека. Другие SINE получаются
из генов тРНК, которые, подобно гену
для РНК 7SL, транскрибируются РНК-
полимеразой III в клетках эукариотов
(раздел 11.2.1), из чего можно предполо-
жить, что некоторая особенность транс-
криптов, синтезируемых этой полиме-
разой, делает эти молекулы склонными
к случайному преобразованию в ретро-
позоны.
Левая половина Правая половина
1 АААаААЛД
Вставка
Рис. 9.16. Структура элемента Alu. Данный элемент
состоит из двух половин, каждая по 120 п. н., с 31-
32 п. н. вставкой в правой половине и поли-А хвостом
на З'-конце. Эти две половины (исключая вставку) об-
ладают приблизительно 85 %-й идентичностью после-
довательностей
336 Часть 2. Анатомия генома
9.2.2. ДНК-транспозоны
Не все транспозоны требуют РНК-посредника. Многие способны транспонироваться
более прямым способом «ДНК в ДНК». У эукариотов такие ДНК-транспозоны менее рас-
пространены, чем ретротранспозоны, но они занимают особое место в генетике, потому
что семейство ДНК-транспозонов растений — элементов Ac/Ds кукурузы — стали пер-
выми известными транспонируемыми элементами, открытыми Барбарой Макклинток
в 1950-е гг. Ее выводы о том, что некоторые гены мобильны и могут перемещаться по
хромосоме из одной позиции в другую, были основаны на результатах изящных гене-
тических экспериментов, но молекулярная основа транспозиции не была объяснена
до конца 1970-х гг.
ДНК-транспозоны распространены среди геномов прокариотов
ДНК-транспозоны являются важным компонентом многих геномов прокариотов.
Последовательности вставок, IS1 и IS186, присутствующие в сегменте ДНК Е. со//длиной
50 тыс.п.н., который мы исследовали в разделе 8.2.1 (см. рис. 8.7), являют собой при-
меры ДНК-транспозонов, и один-единственный геном Е. coli может содержать целых 20
таких объектов различных типов. Большая часть последовательности IS занята одним
или двумя генами, которые определяют фермент транспозазу, который катализирует
ее транспозицию (рис. 9.17, а). На обоих концах каждого элемента IS находится пара
инвертированных повторов длины от 9 до 41 п.н., в зависимости от типа 1S, и вставка
этого элемента в целевую ДНК создает пару коротких (4-13 п.н.) прямых повторений
в геноме хозяина. Элементы IS могут транспонироваться как репликативно, так и кон-
сервативно.
Элементы IS являются компонентами также и ДНК-транспозона второго типа,
впервые охарактеризованного у Е. coli, и, как теперь известно, распространены у мно-
гих прокариотов. Такие составные транспозоны состоят из пары IS-элементов, с двух
сторон примыкающих к сегменту ДНК, обычно содержащему один или несколько генов,
чаще всего кодирующих устойчивость к антибиотику (рис. 9.17, б). Например, транспо-
зон Тп10 несет ген устойчивости к тетрациклину, а транспозоны Тп5 и Тп903 несут ген
устойчивости к канамицину. Некоторые составные транспозоны имеют идентичные
IS-элементы на обоих концах, а у других один элемент одного типа, а второй — второго.
В некоторых случаях IS-элементы ориентированы как прямые повторы, а иногда — как
обратные. Кажется, что эти варианты не затрагивают механизма транспозиции состав-
ного транспозона, который является консервативным по природе и катализируется
транспозазой, кодируемой одним или обоими IS-элементами.
У прокариотов известны также и другие классы ДНК-транспозонов. Два дополни-
тельных важных типа, присущих Е coli, следующие:
• транспозоны типа ТпЗ, которые имеют свой собственный ген транспозазы и, та-
ким образом, не нуждаются для транспонирования в окаймляющих IS-элементах
(рис. 9.17, в). Элементы ТпЗ транспонируются репликативно;
• транспонируемые фаги [фаги-транспозоны. — Прим, ред.], которые являются
бактериальными вирусами, транспонируются репликативно, что является частью
их нормального цикла инфицирования (рис. 9.17, г).
В геномах эукариотов ДНК-транспозоны встречаются реже
Геном человека содержит приблизительно 350 000 ДНК-транспозонов различного
типа (таблица 9.3) — все с инвертированными концевыми повторами и все содержат
Глава 9. Геномы вирусов и мобильные элементы генома 337
а) Последовательность вставки
ITR Транспозаза ITR
-0,3 тыс. п. н.
а) Транспозон типа ТпЗ
ITR Резольваза ITR
Транспозаза Ген устойчивости
к антибиотику
а) Транспонируемый фаг
ITR Гены лизиса ITR
Гены встраивания гены белковой
и репликации оболочки
Рис. 9.17. ДНК-транспозоны прокариотов. Показаны четыре типа. Последовательности вставки, транс-
позоны ТпЗ-типа и транспонируемые фаги ограничены с двух сторон последовательностями коротких
(< 50 п. н.) инвертированных концевых повторов. Ген резольвазы транспозона ТпЗ-типа кодирует белок,
вовлеченный в процесс транспозиции
ген фермента транспозазы, который катализирует событие транспозиции. Однако
львиная доля этих элементов является неактивной — или потому, что ген транспозазы
функционально неактивен, или потому, что последовательности на концах транспозона,
которые необходимы для активной транспозиции, отсутствуют или мутировали.
Активные ДНК-транспозоны чаще встречаются у растений и включают транспозон
Ac/Ds, который был первым из открытых Б. Макклинток, и элемент Spm, оба из которых
обнаружены в кукурузе. Интересная особенность этих транспозонов растений заключа-
ется в том, что они работают вместе в группах семейств. Например, элемент Ас кодирует
активную транспозазу, которая распознает и элементы Ас, и последовательности Ds.
Последние представляют собой версии Ас, несущие внутренние делеции, в результате
которых удаляется часть гена транспозазы, в силу чего элемент Ds не может произво-
дить свою собственную транспозазу и способен перемещаться только за счет действия
транспозазы, синтезируемой полномерным элементом Ас (рис. 9.18). Подобным же об-
разом и полномерным элементам Spm сопутствуют версии, которые содержат делеции
и транспонируются посредством использования ферментов-транспозаз, кодируемых
неповрежденными элементами. Действие элементов Ас четко просматривается в тече-
338 Часть 2. Анатомия генома
Элемент Ds
IR IR
Удаленный ген
транспозазы
Транспозиция
Рис. 9.18. Семейство Ac/Ds транспозонов куку-
рузы. Полномерный элемент Ас имеет длину
4,2 тыс. п. н. и содержит функционально актив-
ный ген транспозазы. Транспозаза распознает
инвертированные повторы длиной по 11 п.н.
на обоих концах последовательности Ас и ката-
лизирует ее транспозицию. Элемент Ds содер-
жит внутреннюю делецию и, таким образом,
не синтезирует свою собственную транспозазу.
Но он все равно содержит последовательности
инвертированных повторов, которые распозна-
ются транспозазой, производимой элементом
Ас. Следовательно, элемент Ds также способен
транспонироваться. Существует приблизительно
десять различных типов элементов Ds в геноме
кукурузы, делеции в которых имеют размеры
в диапазоне от 194 п. н. до нескольких тыс. п. н.
ние нормального жизненного цикла растения кукурузы, при этом транспозиция в со-
матических клетках приводит к изменениям экспрессии генов, которые проявляются,
например, в пестрой пигментации зерен кукурузы (рис. 9.19).
Прозрение Барбары Макклинток о том, что геном кукурузы содержит транспонируе-
мые элементы, пришло от ее исследований генетической подоплеки различных цветных
узоров, образуемых зернами. Подобным образом был открыт P-элемент (ДНК-транспозон
у Drosophila melanogastef) в ходе изучения необычного генетического события, которое,
как оказалось, является результатом транспозиции. Это явление назвали гибридным
дисгенезом, оно происходит, когда самки из лабораторных линий D. melanogaster скре-
щиваются с самцами из диких популяций. Потомство, следующее из таких скрещиваний,
бесплодно и имеет хромосомные отклонения наряду с другими разнообразными гене-
тическими дисфункциями. Это объясняется тем, что геномы диких плодовых мушек
содержат неактивные версии Р-элементов—типичных ДНК-транспозонов, включающих
Рис. 9.19. Пестрая пигментация зерен кукурузы, вызванная транспозицией в соматических клетках.
Многоцветные формы Zea mays общеизвестны как «индейская кукуруза». Изображение любезно
предоставлено Леной Струве
Глава 9. Геномы вирусов и мобильные элементы генома 339
Рис. 9.20. Гибридный дисгенез. Скрещивания
между мужскими лабораторными мухами
и женскими дикими мухами дают нормаль-
ное потомство, но когда мужской партнер
является дикой мухой, потомство бесплодно.
Одним из возможных объяснений гибридного
дисгенеза может быть то, что цитоплазма мух
с P-элементами (на этой схеме — Р+) содержит
ген-репрессор, который предотвращает транс-
позицию P-элемента. Оплодотворенное яйцо,
следующее из скрещивания между самкой Р+
мухи и самцом Р мухи, будет содержать этот
ген-репрессор и, следовательно, даст нор-
мальное потомство. Однако ген-репрессор не
будет содержаться в сперме самца Р+ мухи, так
что в оплодотворенном яйце от скрещивания
между самцом Р+ и самкой Р‘ мухи не будет
гена-репрессора, что позволит произойти транс-
позиции P-элемента и получить потомство, по-
казывающее гибридный дисгенез
ген транспозазы, зажатый между инвер-
тированными концевыми повторами, —
тогда как в лабораторных линиях эти
элементы отсутствуют. После скрещива-
Самец Самка
лабораторной мухи дикой мухи
Р“ Р+
Нормальное потомство
Отсутствие
прерываний гена
Самка
лабораторной мухи Самец дикой мухи
Р‘ Р+
ния элементы, унаследованные от диких
мух, становятся активными в оплодотво-
ренных яйцах, транспонируясь в различные новые позиции и порождая прерывания
генов, которые характеризуют гибридный дисгенез (рис. 9.20). Почему происходит такая
активация, точно не известно, но более интересный вопрос — почему геномы диких по-
пуляций D. melanogaster содержат P-элементы, тогда как в лабораторных линиях таковые
отсутствуют. Большинство лабораторных линий ведут свое начало от мух, собранных
Томасом Хантом Морганом около 90 лет назад и использовавшихся Морганом и его кол-
легами в первых экспериментах по картированию генов (раздел 3.2.3). Из этого явствует,
что дикие популяции в то время, по-видимому, не имели P-элементов, которые так или
иначе распространились в диких геномах за последние 90 лет. Неспособность диких
и лабораторных мух производить жизнеспособное потомство означает, что эти две по-
пуляции не соответствуют одному из главных критериев, используемых для опознавания
биологического вида, — способности всех индивидуумов спариваться продуктивно. Это
допускает занимательную возможность, что видообразование могло, по крайней мере
у некоторых организмов, быть движимо быстрым дифференциальным распростране-
нием транспонируемых элементов в геномах членов различных популяций.
Заключение
Первые исследования вирусов были сосредоточены главным образом на бакте-
риофагах — вирусах, что заражают бактерии. Бактериофаги построены из белка и ну-
клеиновой кислоты — белок образует капсид, в котором заключен геном фага. Известно
три основных типа структуры капсида и множество типов организации генома; разные
340 Часть 2. Анатомия генома
фаги имеют геномы из одно- или двунитевой ДНК или РНК, у некоторых полный геном
содержится в одной-единственной молекуле, а у некоторых геном сегментирован. Бак-
териофаги следуют двумя различными циклами инфицирования. Все фаги способны
заражать бактерии через литический цикл, который приводит к незамедлительному
синтезу новых бактериофагов, обычно сопровождаемому смертью клетки-хозяина.
Некоторые могут следовать также и лизогенным циклом, в ходе которого копия ге-
нома фага встраивается в ДНК хозяина, где может оставаться в дремлющей форме на
протяжении многих поколений клеток. Вирусы эукариотов столь же многообразны
в отношении организации генома, но показывают только два варианта структуры
капсида. Большинство вирусов эукариотов следует литическим циклом инфицирова-
ния, но он не всегда заканчивается незамедлительной гибелью клетки хозяина. Ряд
ДНК- и РНК-вирусов могут встраивать свои геномы в хромосомы эукариотов способом,
подобным лизогенному бактериофагу. Вирусные ретроэлементы, в числе которых со-
стоит ВИЧ, возбудитель СПИДа, являются примерами встраивающихся РНК-вирусов.
Сателлитные РНК и вирусоиды суть инфекционные молекулы РНК различного типа,
которые не содержат ни одного гена и зависят от других вирусов в плане их передачи.
Вироиды — маленькие инфекционные молекулы РНК, которые никогда не бывают
заключены в капсид, а прионы — инфекционные белки. Некоторые мобильные гене-
тические элементы, которые представлены последовательностями ДНК, способными
транспонироваться в пределах генома, но при этом не могут покинуть клетку, сродни
РНК-вирусам. Эти элементы транспонируются через РНК-посредник по пути, подобному
процессу инфицирования, свойственному вирусным ретроэлементам. Ретроэлементы
Tyl/copia и ТуЗ/gypsy, а также эндогенные ретровирусы млекопитающих являются мо-
бильными элементами, состоящими в наиболее тесном родстве с РНК-вирусами. Геномы
млекопитающих содержат также и РНК-транспозоны других типов, названные LINE
и SINE, большая часть которых утеряла свою способность к транспонированию.
ДНК-транспозоны не используют РНК-посредник на своем пути транспозиции. Та-
кие транспозоны распространены у бактерий, в которых они являются ответственными
за распространение генов, кодирующих устойчивость к антибиотикам. ДНК-транспозоны
менее распространены среди эукариотов, но включают некоторые важные примеры,
такие как транспозон Ac/Ds кукурузы — первый в своем роде транспозон, который был
изучен досконально, — и P-элемент Drosophila melanogaster, который является причиной
гибридного дисгенеза, происходящего, когда самок лабораторных плодовых мушек
скрещивают с самцами диких мух.
Глава 9. Геномы вирусов и мобильные элементы генома 341
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
9.1 *. В какого типа структуре капси-
да бактериофага полипептидные
субъединицы устроены в виде
специфической структуры, кото-
рая окружает ядро с нуклеиновой
кислотой, и нитевидного отростка,
который облегчает проникновение
в клетку?
а. Двадцатигранная.
Ь. Нитевидная.
с. Головка с отростком.
d. Сегментированная.
9.2 . В какого типа структуре капсида
бактериофага полипептидные субъ-
единицы расположены в виде спи-
рали, в результате чего образуется
стержневидная структура?
а. Двадцатигранная.
Ь. Нитевидная.
с. Головка с отростком.
d. Сегментированная.
9.3 *. В какого типа жизненном цикле
бактериофага клетка хозяина уби-
вается вскоре после первичного за-
ражения?
а. Литическом.
Ь. Лизогенном.
с. Умеренном.
d. Профага.
9.4 . Профаг определяется как:
а. Новая фаговая частица, которая
собирается в клетке хозяина в те-
чение периода инфицирования.
Ь. Молекула РНК, которая не коди-
рует белки своего собственного
капсида.
с. Фаг с геномом из РНК, который
преобразуется в ДНК с помощью
фермента обратной транскрип-
тазы.
d. Бактериофаг в состоянии покоя,
который встроен в геном клетки
хозяина.
9.5 *. Каким образом вирусы эукариотов
приобретают липидные мембра-
ны?
а. Необходимые липиды синтези-
руются белками, кодируемыми
генами вируса.
Ь. Капсид вируса приобретает мем-
брану, когда он покидает клетку
хозяина.
с. Капсид вируса приобретает мем-
брану при сборке в клетке хозяи-
на.
d. Капсид вируса приобретает мем-
брану в момент его связывания
с клеткой хозяина.
9.6 . В вирусах которого типа присутству-
ет фермент обратная транскрипта-
за?
а. Прионы.
Ь. Профаги.
с. Ретровирусы.
d. Вирусоиды.
9.7 *. Какие из следующих сущностей
являются молекулами РНК, кото-
рые не кодируют белки своего соб-
ственного капсида и переходят из
клетки в клетку с помощью вирусов-
помощников?
а. Прионы.
Ь. Профаги.
с. Ретровирусы.
d. Вирусоиды.
9.8 . Каким образом вироиды способны
реплицироваться и переходить из
клетки в клетку, притом что они не
содержат ни одного гена и никогда
не окружают себя капсидом?
342 Часть 2. Анатомия генома
а. Они реплицируются и перемеща-
ются из клетки в клетку с помо-
щью вируса-помощника.
Ь. Они реплицируются ферментами
клетки хозяина и передвигаются
от клетки к клетке с помощью
вируса-помощника.
с. Они реплицируются фермента-
ми клетки хозяина или вируса-
помощника и переходят из клетки
в клетку в виде голой РНК.
d. Они реплицируются с помощью
вируса-помощника и перескаки-
вают из одной клетки в другую
в виде голой ДНК.
9.9 *. Прионами называют инфекцион-
ные, болезнетворные частицы, ко-
торые:
а. Содержат только РНК.
Ь. Содержат только ДНК.
с. Содержат только белки (никаких
нуклеиновых кислот).
d. Содержат только жиры (никаких
нуклеиновых кислот).
9.10 . Какой из следующих сценариев со-
бытий характерен для консерватив-
ной транспозиции?
а. Вырезание транспозона из одного
местоположения и его последую-
щая вставка в иное местоположе-
ние.
Ь. Репликация транспозона, такая,
что исходная последователь-
ность остается на месте, а новая
последовательность вставляется
в каком-либо другом местополо-
жении.
с. Передвижение транспозона из
одной клетки в другую.
d. Репликация повторных после-
довательностей ДНК из-за про-
скальзывания ДНК-полимеразы.
9.11 *. Какой из следующих ферментов
определяется геном, находящимся
в РНК-транспозонах?
а. ДНК-полимераза.
Ь. РНК-полимераза.
с. Обратная транскриптаза.
d. Теломераза.
9.12 . Какие из следующих объектов явля-
ются РНК-транспозонами, в которых
отсутствуют длинные концевые по-
вторения (LTR) и которые неспособ-
ны синтезировать свои собственные
обратные транскриптазы?
а. Ретроэлементы.
Ь. Эндогенные ретровирусы (ЭРВ).
с. Длинные рассеянные ядерные
элементы (LINE).
d. Короткие рассеянные ядерные
элементы (SINE).
9.13 *. Что, как думают, является источни-
ком РНК-транспозона А1и?
а. Он, как думают, произошел от ре-
тровируса.
Ь. Он, как думают, произошел от ко-
дирующего белок гена.
с. Он, как думают, возник из кле-
точной некодирующей молекулы
РНК.
d. Он, как думают, появился из ДНК-
вируса.
9.14 . Какой фермент определяется геном,
находящимся в ДНК-транспозонах?
а. ДНК-полимераза.
Ь. РНК-полимераза.
с. Обратная транскриптаза.
d. Транспозаза.
9.15*. Назовите исследователя, который
впервые опознал транспозоны,
и организм, который он (или она)
изучал.
а. Дэвид Балтимор и ретровирусы.
Ь. Барбара Макклинток и кукуруза.
с. Томас Хант Морган и плодовая
мушка.
d. Крейг Вентер и человек.
Глава 9. Геномы вирусов и мобильные элементы генома 343
Вопросы открытого типа
9.1 *. Чем вирусы отличаются от клеток?
Уместно ли рассматривать вирусы
как живые организмы?
9.2 . Чем геномы вирусов отличаются от
геномов клеток?
9.3 *. Что из себя представляют перекры-
вающиеся гены, обнаруженные в не-
которых вирусных геномах?
9.4 . Какое время требуется литическо-
му бактериофагу, чтобы лизировать
клетку хозяина после первичного за-
ражения? Как выглядит временной
график литического цикла инфици-
рования для фага Т4?
9.5 *. Обсудите различия между капсида-
ми бактериофагов и вирусов эука-
риотов.
9.6 . Обсудите жизненный цикл ретро-
вирусов.
9.7 *. Что такое транспозон?
9.8 . Каковы характеристики ретроэле-
ментов с LTR, имеющихся в геноме
человека?
9.9 *. Обсудите свойства и типы ретро-
позонов, присутствующих в геноме
человека.
9.10 . Каковы общие свойства составных
транспозонов?
9.11 *. Каковы важные особенности ДНК-
транспозонов, облюбовавших рас-
тения?
9.12 . Опишите генетическую основу ги-
бридного дисгенеза у плодовых му-
шек.
Изыскательские задачи
9.1 *. До какой степени вирусы можно счи-
тать формой жизни?
9.2 . Бактериофаги с маленькими гено-
мами (например, фХ1574) способны
реплицироваться очень успешно
в своих хозяевах. Зачем же тогда
другие бактериофаги, такие как Т4,
имеют большие и усложненные ге-
номы?
9.3 *. Генетические элементы, которые
воспроизводятся внутри генома хо-
зяина или наряду с ним, но не дают
никакой выгоды хозяину, иногда
называют «эгоистичной» ДНК. Об-
судите это понятие и, в частности,
насколько оно имеет отношение
к транспозонам.
9.4 . Некоторые бактериофаги (например,
Т4) видоизменяют РНК-полимеразу
хозяина после заражения, так что
эта полимераза больше не опознает
гены Е. coli, но транскрибирует вме-
сто них гены бактериофага. Каким
образом такая модификация может
быть выполнена?
9.5 *. Почему ретроэлементы с LTR имеют
длинные концевые повторения?
344 Часть 2. Анатомия генома
Тесты по рисункам
9.1 *. Опознайте структуры капсида
бактериофага трех типов.
Белок Нуклеиновая
9.2. Какого типа жизненный цикл
бактериофага воспроизведен на
данном рисунке?
а) Встраивание в ДНК хозяина
J Бактериофаг Л
Е. coli
. Сайт-специфическая
Участок встраивания \ рекомбинация
в ДНК Е. coli я
Профаг Л
9.3*. Какого типа вирусное заражение показано на рисунке?
РНК вируса ДНК вируса
Ретровирус
Впрыск
Обратная
транскрипция
РНК
Обратная----
транскриптаза
Цитоплазма
^страивани
Ядро
Глава 9. Геномы вирусов и мобильные элементы генома 345
9.4 . Геном какого типа вируса представлен на данном рисунке? В чем заключаются
функции последовательностей LTR?
LTR gag pol
env LTR
9.5 *. Назовите имя исследователя, который впервые описал элементы Ас и Ds. В чем
состоит различие между этими элементами?
Элемент Ас
IR IR
Ген транспозазы
Элемент Ds
IR IR
Удаленный ген
транспозазы
Транспозиция
Транспозиция
Дополнительная литература
Классические труды по генетике бактериофагов
Delbriick,M. (1940) The growth of bacteriophage and lysis of the host./. Gen. Physiol.
23: 643-660.
Doermann,A.H. (1952) The intracellular growth of bacteriophage./. Gen. Physiol. 35:
645-656.
Ellis, E.L. and Delbriick,M. (1939) The growth of bacteriophage./. Gen. Physiol. 22:
365-383.
Lwoff, A. (1953) Lysogeny. Bacteriol. Rev. 17: 269-337.
Последовательности геномов бактериофагов
Dunn, J. J. and Studier,F. W. (1983) Complete nucleotide sequence of bacteriophage T7
DNA and the locations of T7 genetic elements./. Mol. Biol. 166:477-535.
Sanger, F., Air,G. M., Barrell, B. G., Brown, N.L., Coulson, A. R., Fiddes,C.A.,
Hutchison, C. A., Slocombe, P. M. and Smith, M. (1977) Nucleotide sequence of bacteriophage
ФХ174 DNA. Nature 265:687-695.
Sanger, F., Coulson, A. R., Hong,G.F., Hill,D.F. and Petersen, G.B. (1982) Nucleotide
sequence of bacteriophage X DNA./. Mol. Biol. 162: 729-773.
346 Часть 2. Анатомия генома
Вирусы эукариотов
Baltimore, D. (1970) RNA-dependent DNA polymerase in virions of RNA tumour viruses.
Nature 226:1209-1211.
Dimmock, N. J., Easton, A. J. and Leppard, K. N. (2001) An Introduction to Modern Virology,
5th Edn. Blackwell Scientific Publishers, Oxford. The best general text on viruses.
Temin, H. M. and Mizutani, S. (1970) RNA-dependent DNA polymerase in virions of Rous
sarcoma virus. Nature 226:1211-1213.
Varmus,H. and Brown, P. (1989) Retroviruses. In: Mobile DNA (eds D. E.Berg and
M. Howe). American Society for Microbiology, Washington, DC, pp. 3-108.
Прионы
Prusiner, S. В. (1996) Molecular biology and pathogenesis of prion diseases. Trends
Biochem. Sci. 21: 482-487.
РНК-транспозоны
Kumar,A. and BennetzenJ.L. (1999) Plant retrotransposons. Annu. Rev. Genet. 33:
479-532. Detailed review of this subject
Ostertag,E. M. and Kazazian,H.H. (2005) LINEs in mind. Nature 435: 890-891. Brief
review of recent research into LINEs.
Patience, C., Wilkinson, D. A. and Weiss, R. A. (1997) Our retroviral heritage. Trends
Genet 13:116-120. ERVs.
Peterson-Burch,B. D., Wright, D.A., Laten,H.M. and Voytas,D.F. (2000) Retroviruses
in plants? Trends Genet 16:151-152.
Song, S. U., Gerasimova, T., Kurkulos, M., Boeke, J. D. and Corces, V. G. (1994) An env-like
protein encoded by a Drosophila retroelement: evidence that gypsy is an infectious retrovirus.
Genes Dev. 8: 2046-2057.
Volff,J.-N., Bouneau, L., Ozouf-Costaz, C. and Fischer, C. (2003) Diversity of
retrotransposable elements in compact pufferfish genomes. Trends Genet 19: 674-678.
ДНК-транспозоны
Comfort, N. C. (2001) The Tangled Field: Barbara McClintock's Search for the Patterns of
Genetic Control. Harvard University Press, Cambridge, MA. A biography of the geneticist who
discovered transposable elements; for a highly condensed version, see Trends Genet. 17: 475-
478.
Engels, W.R. (1983) The P family of transposable elements in Drosophila. Annu. Rev.
Genet 17:315-344.
Gierl, A., Saedler, H. and Peterson, P. A. (1989) Maize transposable elements. Annu. Rev.
Genet 23: 71-85.
Kleckner,N. (1981) Transposable elements in prokaryotes. Annu. Rev. Genet. 15: 341-
404.
Часть 3
Принципы
функционирования
геномов
Часть 3 — принципы функционирова-
ния геномов — исследует события, которые
приводят к передаче биологической инфор-
мации от генома к транскриптому и далее
к протеому. Мы начнем с генома как такового
и со способа, которым структура хроматина
влияет на общую картину экспрессии генома
(глава 10). После этого мы исследуем струк-
туры комплексов инициации транскрипции
у эукариотов и прокариотов и разыщем пути,
согласно которым осуществляется сборка этих
структур (глава 11), прежде чем проследуем
к событиям, ответственным за синтез и обра-
ботку компонентов транскриптома (глава 12)
и протеома (глава 13). Наконец, в главе 14 мы
рассмотрим различные стратегии, которые
используются клетками, чтобы регулировать
экспрессию своих геномов, ибо эти стратегии
позволяют претворять деятельность генома во
внеклеточные сигналы и дают многоклеточ-
ным организмам возможность продвигаться
по сложным путям развития.
Глава 10
Доступ к геному
Глава 11
Сборка комплекса
инициации транскрипции
Глава 12
Синтез
и созревание РНК
Глава 13
Синтез и процессинг
протеома
Глава 14
Регулирование
активности генома
Глава 10
Доступ к геному
По прочтении главы 10 вы сможете:
• Объяснить, каким образом структура хроматина влияет на экспрессию гено-
ма.
• Описывать внутреннее строение ядра эукариотов.
• Видеть отличие между терминами «структурный гетерохроматин», «факульта-
тивный гетерохроматин» и «эухроматин».
• Обсуждать ключевые особенности функциональных доменов, инсуляторов
и управляющих локусом областей, а также описывать экспериментальные дан-
ные, подтверждающие наши теперешние знания об этих структурах.
• Указывать особенности протекания процессов ацетилирования и дезацитили-
рования гистонов и объяснять, как эти модификации влияют на экспрессию
генома.
• Описывать химическую модификацию других типов, которая может быть при-
менена к гистоновым белкам, и связывать эту информацию с концепцией «ги-
стонного кода».
• Излагать, почему расположение нуклеосомы важно в экспрессии генома, и кон-
статировать особенности белковых комплексов, вовлеченных в перестройку
нуклеосомы.
• Объяснять, как происходит метилирование ДНК, и давать оценку значению
метилирования в подавлении генома.
• Сообщать подробности причастности метилирования ДНК к геномному имприн-
тингу и инактивации Х-хромосомы.
Для того чтобы клетка могла использовать биологическую информацию, содержа-
щуюся в ее геноме, необходимо согласованно экспрессировать группы генов, где каждый
ген представлял бы одну единицу информации. Такая согласованная экспрессия генов
задает состав транскриптома, который, в свою очередь, определяет характер протеома
и предопределяет действия, которые клетка способна выполнять. В части 3 «Геномов»
мы исследуем события, которые обусловливают передачу биологической информации
от генома к протеому. Наши знания об этих событиях мы первоначально получали в ходе
изучения отдельных генов, часто в виде «голой» ДНК в опытах, выполняемых в пробирке.
Эти эксперименты обеспечили интерпретацию экспрессии генов, в материю которой
в последние годы был вплетен орамент более тонких и замысловатых исследований,
в большей степени учитывающих тот факт, что в действительности экспрессируется
350 Часть 3. Принципы функционирования геномов
именно геном, а не отдельные гены, и что такая экспрессия происходит (и возможна)
в живых клетках, а не в пробирке.
Наше расследование экспрессии генома мы начнем здесь, в главе 10, с исследования
существенного и важного воздействия, которое окружающая среда ядра оказывает на
употребление биологической информации, содержащейся в геномах эукариотов, при-
том что доступность этой информации зависит от способа упаковки ДНК в хроматин
и является чувствительной к процессам, которые могут подавить или инактивировать
часть или всю хромосому. Далее, в главе 11, мы описываем события, вовлеченные в ини-
циацию транскрипции, и еще более подчеркиваем определяющую роль, которую ДНК-
связывающие белки играют на ранних стадиях экспрессии генома. В главе 12 мы будем
иметь дело с синтезом транскриптов и их последующим созреванием в функциональные
РНК, а в главе 13 мы охватим события, которые ведут к синтезу протеома. По прочтении
глав 10-13 вы обнаружите, что управление составом транскриптома и протеома может
осуществляться на различных стадиях в ходе целой череды событий, которые составляют
экспрессию генома. Эти регуляторные пути сойдутся в главе 14, в которой мы исследуем
вопрос о том, как активность генома изменяется в ответ на внеклеточные сигналы и в
ходе дифференцировки и развития.
10.1. Внутри ядра
Когда мы оглядываем последовательность генома, расписанную в виде символов
А, С, G и Т или нарисованную на карте (как на рис. 7.12, например), у нас, как правило,
возникает представление, что все части генома легко доступны для связывающихся
с ДНК белков, которые отвечают за его экспрессию. В действительности ситуация со-
вершенно иная. ДНК в ядре клетки эукариотов или в нуклеоиде прокариота прикреплена
к разнообразным белкам, которые непосредственно не вовлечены в экспрессию генома
и которые должны быть смещены, для того чтобы РНК-полимераза и другие экспрес-
сионные белки получили доступ к генам. Мы знаем очень немного об этих событиях
у прокариотов, что является отражением нашего в общем-то бедного знания о физи-
ческой организации генома прокариотов (раздел 8.1.1), но мы начинаем понимать, как
упаковка ДНК в хроматин (раздел 7.1.1) влияет на экспрессию генома у эукариотов. Это
захватывающая область молекулярной биологии, недавние изыскания в которой пока-
зали, что гистоны и другие упаковочные белки представляют собой не просто инертные
структуры, вокруг которых обвита ДНК, но, напротив, активные участники процессов,
которые определяют, какие части генома будут экспрессироваться в отдельно взятой
клетке. Многие открытия в этой области проистекли из новых прозрений относительно
субструктуры ядра. Именно с этой темы мы и начинаем эту главу.
10.1.1. Внутреннее строение ядра эукариотов
Внутреннее строение ядра вначале исследовал и посредством световой и электронной
микроскопии. Кажущееся отсутствие структуры, которое обнаружилось в ходе этих исследо-
ваний, привело к представлению о том, что внутренняя часть ядра является относительно
однородной и представляет собой типичный «черный ящик», говоря простым языком.
В последние годы данная интерпретация была низвергнута, и теперь мы признаем, что
ядро имеет сложную внутреннюю структуру, которая связана со множеством разнообраз-
ных биохимических действий, которые оно должно выполнять. В самом деле, внутренняя
часть ядра столь же сложна, что и цитоплазма клетки, единственная разница состоит в том,
что, в отличие от цитоплазмы, функциональные полости в пределах ядра не заключены
Глава 10. Доступ к геному 351
в отдельности в мембраны и поэтому не заметны, когда клетку рассматривают, применяя
обычные методы световой или электронной микроскопии.
Ядро обладает высокоупорядоченной внутренней структурой
Эта пересмотренная картина структуры ядра появилась в результате двух новых
видов микроскопического анализа. Первый метод отличается тем, что обычная электрон-
ная микроскопия сочетается с наблюдением клеток млекопитающих, которые были
приготовлены специальным способом. После растворения мембран путем выдерживания
в умеренном, неионогенном детергенте типа одного из составов Твина, последующей
обработки дезоксирибонуклеазой с целью деградации ядерной ДНК и экстрагирования
в солевом растворе, с тем чтобы удалить химически основные гистоновые белки, суб-
структура ядра выявилась в виде сложной сети волоконец из белка и РНК, названной
ядерным матриксом (рис. 10.1, а). Этот матрикс пронизывает все ядро и включает
области, определяемые как хромосомный каркас, который изменяет структуру хро-
мосом в ходе деления клетки и обусловливает уплотнение последних в их метафазные
формы (см. рис. 7.4).
Второй новейший метод микроскопии предполагает использование флюорес-
центного мечения и разработан специально для обнаружения областей внутри ядра,
в которых протекают конкретные биохимические действия. Ядрышко (рис. 10.1, б),
которое является центром синтеза и созревания молекул рРНК, распознано уже давно,
поскольку это единственная структура в пределах ядра, которую можно увидеть по-
средством обычной электронной микроскопии. Флюоресцентное мечение, направленное
на белки, вовлеченные в сплайсинг РНК (раздел 12.2.2), показало, что эта активность
также локализована в определенных областях (рис. 10.1, в), хотя они более широко рас-
средоточены и хуже определяются, чем ядрышки. Другие структуры, такие как тельца
Кахаля (заметные на рис. 10.1, б), которые, вероятно, вовлечены в синтез малых ядерных
РНК (раздел 12.2.2), также заметны после флюоресцентного мечения.
Сложность ядерного матрикса, как видно на рис. 10.1, а, можно принять за признак
того, что ядро имеет статическую внутреннюю среду с ограниченным передвижением
молекул от одного участка к другому. Еще один новый метод микроскопии, названный
восстановлением флюоресценции после фотообесцвечивания (ВФПФ; техническое
примечание 10.1), который позволяет визуализировать передвижение белков внутри
ядра, показывает, что это не так. Перемещение ядерных белков не происходит так
быстро, как ожидалось бы, если их движение было бы абсолютно беспрепятственно,
что вполне ожидается ввиду больших количеств ДНК и РНК в ядре, но все же белок
может пересечь полный диаметр ядра в течение нескольких минут. Поэтому белки,
вовлеченные в экспрессию генома, имеют свободу, необходимую для передвижения из
участка одного рода деятельности к другому, в соответствии с тем, что предписывают
изменяющиеся требования клетки. В частности, линкерные гистоны (раздел 7.1.1)
непрерывно отделяются и снова прикрепляются к своим сайтам связывания в геноме.
Это открытие важно, ибо оно подчеркивает, что ДНК-белковые комплексы, из которых
состоит хроматин, являются динамическими, — наблюдение, которое имеет прямое
отношение к экспрессии генома, в чем мы убедимся далее в этой главе.
Каждая хромосома имеет свою собственную территорию в пределах ядра
Первоначально думали, что хромосомы распределены беспорядочно в пределах
ядра эукариотов. Теперь мы знаем, что это представление неверно и что каждая хро-
мосома занимает свое собственное место, или территорию. Это можно увидеть при
352 Часть 3. Принципы функционирования геномов
а)
Цитоплазматические волокна
Граница цитоплазмы
ядра
Сеть волокон
внутри ядра
Ядрышко
б)
в)
Рис. 10.1. Внутреннее строение ядра эукариотов, а) Микрофотография просвечивающей электронной
микроскопии, показывающая ядерный матрикс культивируемой клетки HeLa человека. Клетки были об-
работаны неионогенным детергентом с целью удаления мембран, переварены дезоксирибонуклеазой,
с тем чтобы деградировать большую часть ДНК, и подвергнуты экстрагированию в сульфате аммония,
чтобы удалить гистоны и другие связанные с хроматином белки, б) и в) Изображения живых ядер, со-
держащих флюоресцентно меченные белки (см. техническое примечание 10.1). На снимке б ядрышко
показано синим, а тельца Кахаля — желтым цветом. Фиолетовые области на снимке в показывают позиции
белков, вовлеченных в сплайсинг РНК. Изображение а перепечатано с разрешения Penman et al., Symp.
Quant. Biol., 46,1013. Copyright 1982 Cold Spring Harbor Laboratory Press. Изображения бив пере-
печатаны с разрешения Misteli (2001), Science, 291, 843-847
помощи раскрашивания хромосом, которое представляет собой версию флюорес-
центной гибридизации in situ (FISH; раздел 3.3.2), в которой гибридизационный зонд
представлен смесью молекул ДНК, каждая из которых специфична к отдельной области
одной из хромосом. При применении к интерфазным ядрам раскрашивание хромосом
показывает территории, занимаемые отдельными хромосомами (рис. 10.2). Эти терри-
тории занимают большую часть пространства внутри ядра, но отделены друг от друга
бесхроматиновыми областями, в пределах которых расположены ферменты и другие
белки, вовлеченные в экспрессию генома.
Территории хромосом, как оказалось, довольно статические в пределах отдель-
ного ядра. Это заключение было сделано по данным экспериментов, в которых белки
CENP-B — компоненты центромер (раздел 7.1.2) — метятся зеленым флюоресцентным
Глава 10. Доступ к геному 353
Техническое примечание 10.1: восстановление флюоресценции по-
сле фотообесцвечивания (ВФПФ)
Зрительное отображение подвижности белка в живых ядрах
ВФПФ, возможно, представляет собой наиболее информативный из различных
новаторских методов микроскопии, которые приоткрыли нам дверь к пониманию
субструктуры ядра. Он впервые позволил зрительно отображать передвижение
белков внутри живых ядер, и получаемые данные об этом позволяют проверять
биофизические модели динамизма белков.
Отправная точка эксперимента ВФПФ — ядро, в котором каждая копия интере-
сующего нас белка несет флюоресцентную метку. Мечение белковых молекул in vitro
и последующее повторное введение их в ядро невозможно, так что организм хозяина
должен быть генетически спроектирован так, чтобы флюоресцентная метка была
неотъемлемой частью белка, который синтезируется in vivo. Это достигается лиги-
рованием последовательности, кодирующей зеленый флюоресцентный белок,
к гену изучаемого белка. Затем используются стандартные методы клонирования,
чтобы вставить видоизмененный ген в геном хозяина, что дает рекомбинантную
клетку, которая синтезирует флюоресцентную версию белка. В результате наблюде-
ние клетки при помощи флюоресцентного микроскопа показывает распределение
меченого белка в пределах ядра.
Чтобы изучить подвижность белка, маленькая область ядра фотообесцвечи-
вается с помощью узко сфокусированного светового импульса от лазера с высокой
энергией излучения. Лазерный импульс инактивирует флюоресцентный сигнал
в облучаемой зоне, оставляя область, которая выглядит обесцвеченной в изобра-
жении микроскопа. Эта обесцвеченная область постепенно восстанавливает свой
флюоресцентный сигнал, причем не за счет обращения эффекта обесцвечивания,
а в силу перемещения в обесцвеченную область флюоресцентных белков из не-
подвергнутой облучению области ядра. Поэтому скорое восстановление флюорес-
центного сигнала в обесцвеченной области указывает на то, что меченые белки
довольно подвижны, тогда как медленное его восстановление указывает, что белки
являются относительно статическими. Таким образом, кинетика восстановления
сигнала может быть использована для проверки теоретических моделей подвиж-
ности белков, оцениваемого из таких биофизических параметров, как константа
связывания и скорость потока.
белком (см. техническое примечание 10.1), и местоположения этих белков и, следова-
тельно, центромер, наблюдаются в течение некоторого периода времени. В целом, от-
дельные центромеры остаются неподвижными на всем протяжении клеточного цикла,
хотя проявляют эпизодические порывы к медленному движению. Хотя они и являются
довольно статическими в течение жизненного цикла клетки, результаты большинства
исследований позволяют предположить, что относительное расположение территорий
не сохраняется после деления клетки, поскольку в ядрах дочерних клеток наблюдаются
иные картины. Могут, однако, быть определенные ограничения на местоположения тер-
риторий, поскольку уже несколько лет известно, что транслокации хромосом, в резуль-
тате которых сегмент одной хромосомы становится прикрепленным к другой хромосоме,
354 Часть 3. Принципы функционирования геномов
HSA18
HSA19
HSA19
HSA18
Рис. 10.2. Территории хромосом. Хромосомы
человека 18 (HSA 18) и 19 (HSA 19) были раскра-
шены соответственно в зеленый и красный цвета.
Каждая занимает свою собственную обособлен-
ную территорию в пределах ядра. Изображение
любезно предоставлено Уэнди Бикмор
более часты между одними парами, чем
между другими. Например, транслока-
ция между хромосомами человека 9 и 22,
приводящая к неправильному продукту,
названному филадельфийской хро-
мосомой, является обычной причиной
хронического миелоидного лейкоза (рис.
10.3). Повторное возникновение одной
и той же транслокации предполагает, что
территории хромосом взаимодействующей пары часто бывают расположены в ядре
близко друг к другу. Есть также данные, что по крайней мере в некоторых организмах
некоторые хромосомы предпочтительно занимают территории вблизи к периферии
ядра. В этой области происходит относительно слабая экспрессия генома, и часто здесь
находятся именно те хромосомы, которые содержат немного активных генов; примеры
представлены макрохромосомами генома курицы (раздел 7.1.2).
Расположение активных генов в пределах отдельных территорий хромосом — еще
одна тема дебатов. Одно время думали, что активные гены расположены на поверхности
территории, смежной с бесхроматиновой областью и, следовательно, в пределах легкой
досягаемости ферментов и белков, вовлеченных в транскрипцию генов (рис. 10.4). Теперь
это представление подвергается сомнению, отчасти на основе данных экспериментов,
которые показали, что РНК-транскрипты распределены внутри территорий, также как
и на их поверхностях. Более тонкое микроскопическое исследование показало, что сквозь
Нормальные
хромосомы
Продукты
транслокации
22
9
Рис. 10.3. Продукты транслокации между хромосома-
ми 9 и 22 человека. Нормальные хромосомы 9 и 22
человека показаны слева, а продукты транслокации —
справа. Филадельфийская хромосома — меньшая из
двух продуктов транслокации. Хромосомы 9 и 22 обыч-
но разрываются в обозначенных позициях. Часто раз-
рывы правильно устраняются, но иногда неправиль-
ное их устранение порождает гибридные продукты.
Думают, что относительно высокая частота, с которой
возникает филадельфийская хромосома, указывает
на то, что хромосомы 9 и 22 занимают взаимно при-
легающие территории в ядре клетки человека. Точка
разрыва хромосомы 9 лежит в пределах гена ABL, про-
дукт которого вовлечен в передачу сигналов между
клетками (раздел 14.1.2). В результате транслокации
новая кодирующая последовательность прикрепляет-
ся к началу этого гена и синтезируется неправильный
белок, который вызывает трансформацию клетки
и дает начало хроническому миелоидному лейкозу
Глава 10. Доступ к геному 355
территории хромосом пробегают каналы, связывая различные части бесхроматиновых
областей и обеспечивая средство, при помощи которого транскрипционные машины
могут проникать во внутренние части этих территорий.
10.1.2. Хроматиновые домены
Из раздела 7.1.1 мы узнали, что хроматин — комплекс геномной ДНК и хромосомных
белков, находящихся в ядре эукариотов. Структура хроматина является иерархической,
поднимаясь от двух низших уровней упаковки ДНК — нуклеосомы и 30-нм хромати-
нового волокна (см. рис. 7.2 и 7.3) — до метафазных хромосом, которые представляют
самую компактную форму хроматина у эукариотов и появляются только во время де-
ления ядра. После деления хромосомы становятся менее компактными и не могут быть
различимы как отдельные структуры, если не использовать специальные методы, на-
пример, раскрашивание хромосом. Когда неделящиеся ядра рассматриваются световой
микроскопией, все, что можно увидеть, — смесь светло и темно окрашенных областей
внутри ядра. Темные области называются гетерохроматином и содержат ДНК, которая
находится все еще в относительно компактной организации, хотя и менее компактной,
чем в метафазной структуре. Различают гетерохроматин двух типов:
• структурный (постоянный) гетерохроматин — постоянная составляющая всех
клеток, представленная ДНК, которая не содержит никаких генов и, таким обра-
зом, всегда может пребывать в компактной организации. Эта фракция включает
центромерную и теломерную ДНК, а так же определенные области некоторых
других хромосом. Например, большая часть Y-хромосомы человека состоит из
структурного гетерохроматина (см. рис. 7.5);
• факультативный гетерохроматин — не является постоянным компонентом кле-
ток, но обнаруживается в некоторых из них в определенное время. Факультативный
гетерохроматин, как думают, содержит гены, которые являются неактивными
в некоторых клетках или в некоторые периоды клеточного цикла. Когда эти гены
неактивны, соответствующие им области ДНК уплотняются в гетерохроматин.
Предполагается, что организация гетерохроматина настолько компактна, что
белки, вовлеченные в экспрессию генома, попросту
не могут получить доступ к ДНК такого типа. Напро-
тив, части хромосомной ДНК, в которых расположены
активные гены, менее компактны и допускают доступ
белков, осуществляющих экспрессию. Эти области
называют эухроматином. Точная организация ДНК
в пределах эухроматина неизвестна^, но с помощью
электронного микроскопа в пределах областей эух-
роматина можно видеть петли ДНК, каждая длиной
от 40 тыс. п. н. до 100 тыс. п. н. и преобладающе в фор-
Рис. 10.4. Территории хромосом. Вид слева показывает первона-
чальную модель, в которой каждая территория образует единый
Ядерная пора
массив, из чего следует, что активные гены расположены на
периферии территории. Вид справа показывает исправленную
модель, в которой территории пронизаны каналами
В последнее время появились новые методы определения структуры хроматина в ядре, основанные на
методах параллельного секвенирвания. См., напр., (Lieberman-Aiden Е., et al., Comprehensive mapping of longrange
interactions reveals folding principles of the human genome. Science. 2009 Oct 9; 326 (5950): 289-293). — Прим. ped.
356 Часть 3. Принципы функционирования геномов
Ядро
30-нм хроматиновое волокно
Петли
ДНК
Ядерный матрикс
ДНК
Белок
Рис. 10.5. Схема организации ДНК в ядре. Ядер-
ный матрикс — волокнистая структура на основе
белков, точный состав которой и расположение
в ядре до сих пор не были описаны. Эухроматин,
преимущественно в форме 30-нм хроматиновых
волокон (см. рис. 7.3), как думают, прикреплён
к матриксу обогащенными АТ последовательно-
стями, названными связанными с матриксом об-
ластями, или областями прикрепления к каркасу
(соответственно MAR, или SAR)
ме 30-нм хроматинового волокна. Петли
прикреплены к ядерному матриксу через
обогащенные АТ сегменты ДНК, назван-
ные связанными с матриксом областя-
ми (MAR), или областями прикрепления
к каркасу (SAR) (рис. 10.5).
Петли ДНК между точками прикре-
пления к ядерному матриксу называют
Обогащённые АТ области структурными доменами. Любопытный
= MAR, или SAR
вопрос — точная зависимость между оны-
ми и функциональными доменами, кото-
рые можно различить, когда рассматрива-
ется область ДНК вокруг экспрессируемого
гена или набора генов. Функциональный
домен выделяется обработкой области
очищенного хроматина дезоксирибону-
клеазой I (ДНКазой I), которая, будучи
ДНК-связывающим белком, не может по-
лучить доступ к более уплотненным об-
ластям ДНК (рис. 10.6). Области, чувствительные к ДНКазе I, простираются с обеих
сторон гена или набора генов, которые экспрессируются, показывая, что в этой области
хроматин имеет более открытую организацию, хотя не ясно, представлена ли эта орга-
низация 30-нм волокном или структурой типа «бусины на нити» (рис. 7.2, а). Интуиция
подсказывает, что должно быть какое-то соответствие между структурными и функцио-
нальными доменами, и это представление подтверждается местоположением некоторых
MAR, которое отмечает пределы структурного домена, на границе функционального
домена. Но такое соответствие кажется неполным, потому что некоторые структурные
домены содержат гены, которые не экспрессируются в одно и то же время, а границы
некоторых структурных доменов лежат в пределах генов.
Ген
Нечувствительная —
к ДНКазе! 1П Чувствительная
1 и тыс, п. н. к ДНКазе । область
Нечувствительная
к ДНКазе I
Рис. 10.6. Функциональный домен в чувствительной к ДНКазе I области
Глава 10. Доступ к геному 357
Функциональные домены отграничиваются инсуляторами
Границы функциональных доменов отмечены последовательностями длиной
1-2 тыс.п.н., называемыми инсуляторами. Последовательности инсуляторов впервые
были открыты у Drosophila и к настоящему времени опознаны у целого ряда эукариотов.
Лучше всего изучена пара последовательностей, названных scs и scs’ (scs означает «спе-
циализированная структура хроматина1^»), которые расположены по обе стороны от двух
генов hsp70 в геноме плодовой мушки (рис. 10.7).
Инсуляторы выказывают два специфических hsp70 hsp70
свойства, проистекающих из их роли разделителей S»———
функциональных доменов. Первое — их способность scs * * scs'
преодолевать позиционный эффект, который воз- 1 п* н-
никает в ходе эксперимента по клонированию ге-
нов с хозяином из царства эукариотов. Позиционный
эффект проявляется в изменении экспрессии гена
после его вставки в хромосому эукариотов. Он, как
думают, следует из случайного характера события
вставки, в силу чего ген может быть доставлен в об-
ласть плотно упакованного хроматина, где он будет
неактивным, или в область открытого хроматина, где
он будет экспрессироваться (рис. 10.8, а). Способность
последовательностей scs и scs' преодолевать позици-
онный эффект была продемонстрирована следующим
экспериментом. Их поместили по обе стороны гена
Рис. 10.7. Последовательности инсуля-
торов в геноме плодовой мушки. На
схеме представлена область генома
Drosophila, содержащая два гена hsp70.
Последовательности инсуляторов scs
и scs' расположены по обе стороны от
пары генов. Стрелки под этими двумя
генами показывают, что они лежат на
разных нитях двойной спирали и, таким
образом, транскрибируются в противо-
положных направлениях
плодовой мушки, отвечающего за
цвет глаз. Будучи заключенным
а) Позиционный эффект
между инсуляторами, этот ген
всегда сильно экспрессировал-
ся, когда был вставлен в геном
Рис. 10.8. Позиционный эффект.
а) Копируемый ген, который встав-
лен в область плотно упакованного
хроматина, будет неактивным, но, бу-
дучи вставлен в раскрытый хроматин,
будет экспрессироваться. 6) Результа-
Новый ген
вставлен сюда
Экспрессия слабая
Или же сюда
I
I
или отсутствует
Высокий уровень
экспрессии
ты экспериментов по клонированию
без последовательностей инсулято-
ров (красный) и с ними (синий). Когда
инсуляторы отсутствуют, уровень экс-
прессии клонированного гена явля-
ется изменчивым, в зависимости от
того, вставлен ли он в упакованный
или в раскрытый хроматин. Будучи
обрамлён инсуляторами, клониро-
ванный ген показывает единообразно
высокий уровень экспрессии, потому
что инсуляторы образуют функцио-
нальный домен на участке вставки
6) Инсуляторы преодолевают позиционный эффект
Клонируемые гены
Без инсуляторов С инсуляторами
Specialized chromatin structure. — Прим, перев
358 Часть 3. Принципы функционирования геномов
Drosophila, в отличие от измененной экспрессии, которая наблюдалась, когда этот ген
был клонирован без инсуляторов (рис. 10.8, б). Вывод из этого и подобных экспери-
ментов был тот, что инсуляторы могут вызывать модификации в упаковке хроматина
и, следовательно, образовывать функциональный домен при вставке в новый участок
последовательности генома.
Инсуляторы также поддерживают независимость каждого функционального до-
мена, предотвращая «перекрестное взаимодействие» между смежными доменами. Если
последовательностьзсз или scs’ вырезана из ее нормального местоположения и повторно
вставлена между геном и вышерасположенными регуляторными модулями, которым
подвластна экспрессия этого гена, то ген более не реагирует на сигналы назначенных ему
регуляторных модулей: он становится «изолированным» от их воздействий (рис. 10.9, а).
Это наблюдение предполагает, что в своих нормальных позициях инсуляторы предо-
храняют гены в пределах домена от влияния регуляторных модулей, расположенных
в смежной области (рис. 10.9, б).
Как инсуляторы выполняют свои роли, еще не известно, но предполагается, что
функциональный компонент представлен не самой последовательностью инсулятора,
но ДНК-связывающими белками, такими как Su(Hw) у Drosophila, которые специфично
прикрепляются к инсуляторам. Кроме связывания с инсуляторами, эти белки образуют
объединения с ядерным матриксом, а это, возможно, указывает на то, что функцио-
а) Инсуляторы блокируют регуляторные сигналы,
управляющие экспрессией гена
нальные домены, которые они определяют, также являются структурными доменами
в хроматине. Это привлекательная гипотеза, которая может быть увязана со способно-
стью инсуляторов образовывать раскрытые области хроматина и предотвращать пере-
крестное взаимодействие между
функциональными доменами, но
она подразумевает, что инсулято-
ры содержат последовательности
MAR, что не было доказано. Поэто-
му эквивалентность между функ-
циональными и структурными до-
менами остается иллюзорной.
Регуляторные сигналы влияют
на экспрессию гена
Регуляторные модули
Ген
Сигналы блокированы
Инсулятор
б) Инсуляторы предотвращают перекрестное
взаимодействие между функциональными доменами
Рис. 10.9. Инсуляторы поддерживают не-
зависимость функционального домена.
а) Будучи помещена между геном и его
вышерасположенными регуляторными
модулями, последовательность инсу-
лятора препятствует достижению регу-
ляторными сигналами гена, б) В своих
нормальных позициях инсуляторы
предотвращают перекрестное взаимо-
действие между функциональными до-
менами, так что регуляторные модули
одного гена не влияют на экспрессию
какого-либо гена в каком-либо ином до-
мене
Глава 10. Доступ к геному 359
Некоторые функциональные домены содержат управляющие локусом об-
ласти
Формирование и поддержание открытого функционального домена, по крайней
мере для некоторых доменов, является прерогативой последовательности ДНК, назы-
ваемой управляющей локусом областью, или LCR. Подобно инсуляторам, LCR может
преодолевать позиционный эффект при связывании с новым геном, который вставлен
в хромосому эукариотов. В отличие от инсуляторов, LCR также стимулирует экспрессию
генов, содержащихся в пределах ее функционального домена.
LCR были впервые открыты в ходе изучения генов р-глобина человека (раздел 7.2.3)
и, как теперь думают, вовлечены в экспрессию многих генов, которые являются ак-
тивными только в некоторых тканях или в течение некоторых стадий развития. LCR
глобина содержится в отрезке ДНК длиной около 12 тыс. п. н., размещенной выше генов
в функциональном домене р-глобина длиной 60 тыс. п. н. (рис. 10.10). LCR были первона-
чально опознаны в ходе исследований индивидуумов с талассемией — болезнью крови,
которая обусловлена дефектами в белках а- или р-глобина. Многие случаи талассемии
порождаются мутациями в кодирующих областях генов глобина, но некоторые, как было
показано, расположены на карте в пределах области протяженностью 12 тыс.п.н., рас-
положенной выше группы генов р-глобина; эту область теперь называют LCR. Способ-
ность мутаций в LCR вызывать талассемию есть явный признак того, что разрушение
LCR приводит к потере экспрессии генов глобина.
Более доскональное изучение LCR р-глобина показало, что она содержит пять от-
дельных сверхчувствительных к ДНКазе I участков—коротких областей Д Н К, которые
расщепляются ДНКазой I более легко, чем другие части функционального домена. Эти
участки, как думают, совпадают с позициями, в которых нуклеосомы были видоизмене-
ны или отсутствуют и которые поэтому являются доступными для ДНК-связывающих
белков, прикрепляющихся к ДНК. Именно эти белки, а не последовательность LCR в ДНК,
управляют структурой хроматина в пределах функционального домена1). Каким в точ-
ности образом и в ответ на какие биохимические сигналы — неизвестно.
Сверхчувствительные
к ДНКазе! участки £ G. А 6 Ц г енов
¥ » » --------»--------------•-------•- Лглобина
Управляющая локусом
последовательность
10 тыс. п. н.
Рис. 10.10. Сверхчувствительные к ДНКазе I участки указывают положение управляющей локусом области,
соответствующей группе генов р-глобина человека. Ряд сверхчувствительных участков расположен на
20 тыс. п. н. ДНК выше начала группы генов р-глобина. Эти участки отмечают положение управляющей
локусом области. Дополнительные сверхчувствительные участки наблюдаются непосредственно выше
каждого гена — в позиции, где РНК-полимераза прикрепляется кДНК. Эти сверхчувствительные участки
являются специфическими для различных стадий развития и заметны только в течение фазы развития,
когда смежный ген активен. Область размером 60 тыс.п.н., показанная здесь, представляет полный
функциональный домен р-глобина. На рис. 7.19 показана более подробная картина регулирования
экспрессии группы генов р-глобина в ходе развития
На самом деле последовательность важна, ведь ДНК-связывающие белки связываются не любой ДНК,
а вполне определенными участками. Может быть здесь играют роль не отдельные сайты на ДНК, а некоторые
общие свойства последовательности? — Прим. ред.
360 Часть 3. Принципы функционирования геномов
Сверхчувствительные к ДНКазе I участки встречаются также сразу же выше каждого
из генов в функциональном домене 0-глобина (см. рис. 10.10) в позициях, где на ДНК
собирается комплекс инициации транскрипции (раздел 11.2.2). Эти позиции сборки ил-
люстрируют интересную особенность сверхчувствительных к ДНКазе I участков: они не
инвариантные компоненты функционального домена. Вспомним, что гены 0-глобинов
различного типа экспрессируются на разных стадиях цикла развития человека: с являет-
ся активным в раннем эмбрионе, Gy и Ау — в плоде, а 5 и 0 — во взрослом организме (см.
рис. 7.19). Только в тот период, когда ген активен, соответствующая ему позиция сборки
комплекса инициации транскрипции отмечена сверхчувствительным участком. Вначале
думали, что это было следствием дифференциальной экспрессии этих генов; другими
словами, что в отсутствие активности гена нуклеосомы имеют возможность покрыть
участок сборки и, возможно, быть отодвинутым к одной из сторон вбок, когда настает
время экспрессировать ген. Теперь думают, что присутствие или отсутствие нуклеосом
есть причина экспрессии гена, и ген выключается, если нуклеосомы покрывают участок
сборки, или включается, если доступ к участку открыт.
10.2. Модификации хроматина и экспрессия генома
Предыдущие разделы указали нам два пути, которыми структура хроматина может
влиять на экспрессию генома (рис. 10.11).
• Степень упаковки хроматина, показываемая сегментом хромосомы, определяет,
могут ли гены в пределах этого сегмента экспрессироваться.
• Если ген доступен, то на его транскрипцию влияет конкретная природа и точное
расположение нуклеосом в области, где комплекс инициации транскрипции будет
собран.
За последние годы были сделаны значительные успехи в понимании обоих типов
модификации хроматина. Мы начнем с процессов, которые влияют на упаковку хрома-
тина.
СТРУКТУРА А:
Нуклеосомы разделены
равными интервалами
СТРУКТУРА В:
Нуклеосомы
передвинулись
Рис. 10.11. Два возможных пути влия-
ния структуры хроматина на экспрессию
гена. Область неупакованного хрома-
тина, в которой гены являются доступ-
ными, находится между двумя более
компактными сегментами. В пределах
неупакованной области расположение
нуклеосом влияет на экспрессию генов.
Изображенные слева нуклеосомы Име-
ют регулярный интервал, который от-
ражен в типичной структуре «бусины
на нити». Справа расположение нуклео-
сом изменилось и открылся короткий
отрезок ДНК длиной приблизительно
300 п.н.
Глава 10. Доступ к геному 361
10.2.1. Химическая модификация гистонов
Нуклеосомы являются очевидными главными определителями деятельности
генома эукариотов, не только посредством их расположения на нити ДНК, но также
и потому, что точно заданная химическая структура гистоновых белков, содержащихся
в нуклеосомах, есть главный фактор, определяющий степень упаковки, присущей соот-
ветствующему сегменту хроматина.
Ацетилирование гистонов влияет на многие отправления ядра, в том числе
и на экспрессию генома
Гистоновые белки могут подвергаться модификациям различных типов, и наиболее
хорошо изученный из них — ацетилирование гистонов — прикрепление ацетильных
групп к лизиновым аминокислотам в N-концевых областях каждой из стержневых моле-
кул (рис. 10.12). Такие N-концы формируют хвосты, которые высовываются из октамера
ядра нуклеосомы (рис. 10.13), и их ацетилирование снижает сродство гистонов к ДНК
и, возможно, также ослабляет взаимодействие между отдельными нуклеосомами, деста-
билизируя тем самым 30-нм хроматиновое волокно. Гистоны в гетерохроматине в целом
неацетилированы, тогда как их собратья в функциональных доменах — ацетилированы,
что есть явный признак того, что модификация этого типа связана с упаковкой ДНК.
Значимость ацетилирования гистонов в экспрессии генома была подчеркнута
в 1996 г., когда после нескольких лет попыток были обнаружены первые примеры
гистонацетилтрансфераз (HAT) — ферментов, которые прикрепляют ацетильные
группы к гистонам. Стало понятно, что некоторые белки, которые, что уже было по-
казано, оказывают важные влияния на экспрессию генома, имеют активность HAT.
Например, одной из первых открытых НАТ был белок Tetrahymena, названный р55;
было показано, что он является гомологом белка дрожжей GCN5, который, как было
известно, активизировал сборку комплекса инициации транскрипции (раздел 11.3.2).
Подобным же образом, белок млекопитающих, названный рЗОО/СВР, которому была
приписана четко определенная роль в активации целого ряда генов, оказался НАТ. Эти
Ац Ац
Н2А SGRGKQGGKARAKAKTRSSR
10 20
Н2В
НЗ
Ац Ац Ац Ац
I III
PEPSKSAPAPKKGSKKAITKA
10 20
Ац Ац Ац Ац
I I I
ARTKQTARKSTGGKAPRKQLATKARKSAP
10 20
Ац Ац Ац Ац
Н4 SGRGKGGKGLGKGGAKRHRK
10 20
Рис. 10.12. Позиции, в которых ацетильные группы присоединены в пределах N-концевых областей четы-
рех образующих ядро гистонов. Каждая последовательность начинается с N-концевой аминокислоты
362 Часть 3. Принципы функционирования геномов
Рис. 10.13. Два вида стержневого октамера нуклеосомы. Слева расположен вид сверху на имеющий
форму цилиндра октамер; справа помещен его же вид сбоку. Две нити двойной спирали ДНК, обернутые
вокруг октамера, показаны коричневым и зеленым цветами. В октамер входят центральный тетрамер
из двух субъединиц из гистонов НЗ (синий) и двух субъединиц из гистонов Н4 (ярко-зеленый), плюс пара
димеров Н2А (желтый)-Н2В (красный) — один выше, а другой ниже центрального тетрамера. Обратите
внимание на N-концевые хвосты гистоновых белков, высовывающиеся из стержневого октамера. Пере-
печатано с разрешения Luger et al., (1997), Nature, 389, 251-260
наблюдения плюс демонстрация того факта, что клетки различного типа показывают
различные схемы ацетилирования гистонов, подчеркивают выдающуюся роль, которую
ацетилирование гистонов играет в регулировании экспрессии генома.
Отдельные HAT могут ацетилировать гистоны в пробирке, но большинство имеет
незначительную активность на интактных нуклеосомах, указывая на то, что в ядре HAT
почти наверняка не работают независимо, но вместо этого образуют многобелковые
комплексы, такие как комплексы SAGA и ADA дрожжей и комплекс TFTC людей. Эти
комплексы типичны для больших многобелковых структур, которые катализируют
и регулируют различные шаги в экспрессии генома, со многими примерами чего мы
встретимся по мере нашего продвижения по нескольким следующим главам. SAGA,
например, включает по крайней мере 15 белков с общей молекулярной массой 1,8 мил-
лионов. Комплекс 18x28 нм в размере, что означает, что он крупнее ядерного октамера
нуклеосомы, который вместе со связанной им ДНК имеет размер 11x13 нм, и сопоста-
вим в одном измерении с 30-нм хроматиновым волокном. Также как и GCN5 — белок,
обладающий активностью HAT, — комплекс SAGA содержит набор белков, имеющих
отношение к связывающему ТАТА-блок белку (ТВР), которые инициируют процесс,
в ходе которого ген транскрибируется (раздел 11.2.3), а также пять из связанных с ТВР
факторов (TAF), которые помогают ТВР выполнять его роль. Сложность комплека SAGA
и других комплексов HAT и присутствие в этих комплексах белков с разными ролями
в инициации экспрессии гена указывают на то, что отдельные события, в результате
которых ген становится активным, тесно связаны, причем ацетилирование гистонов
является неотъемлемой частью, но лишь одной из частей общего процесса.
Есть по крайней мере пять разных семейств белков НАТ. Родственные GCN5 аце-
тилтрансферазы, или GNAT, которые являются компонентами SAGA, ADA и TFTC, явно
имеют отношение к активации транскрипции гена, но также вовлечены и в репарацию
некоторых типов поврежденной ДНК, в частности, двунитевых разрывов и поврежде-
ний, следующих из ультрафиолетового облучения (раздел 16.2.4). Второе семейство
Глава 10. Доступ к геному 363
белков HAT, названное MYST по первым буквам четырех белков этого семейства, также
вовлечено в активацию транскрипции и в репарацию ДНК, участвует в управлении кле-
точным циклом, хотя это может просто быть другим аспектом функции репарации ДНК,
поскольку клеточный цикл приостанавливается, если геном сильно поврежден (раздел
15.3.2). Оказывается, разные комплексы ацетилируют различные гистоны, а некоторые
могут также ацетилировать другие белки, вовлеченные в экспрессию генома, такие как
общие факторы транскрипции TFIIE и TFIIF, с которыми мы встретимся в разделе 11.2.3.
НАТы поэтому предстают пред нами как многофункциональные белки, которые могут
иметь разнообразные функции в экспрессии, репликации и в поддержании генома.
Деацетилирование гистонов подавляет активные области генома
Активация генов должна быть обратимой, иначе гены, которые станут включен-
ными, останутся постоянно активными. Следовательно, это не удивительно, что есть
набор ферментов, которые могут удалять ацетильные группы от гистоновых хвостов,
отменяя активизирующие транскрипцию эффекты НАТов, описанные выше. Это роль
гистондезацетилаз (HDACee). Связь между деятельностью HDAC и подавлением генов
была установлена в 1996 г., когда было показано, что HDAC1 млекопитающих, первый
из таких открытых ферментов, связана с белком дрожжей по имени Rpd3, который,
как известно, является геном-репрессором транскрипции. Таким образом, связь между
деацетилированием гистонов и репрессией транскрипции была установлена тем же
самым способом, что и связь между ацетилированием и активацией, показывая, что два
белка, которые, как первоначально думали, имеют различные действия, фактически
связаны. Это хорошие примеры важной роли анализа гомологии в изучении функций
генов и белков (раздел 5.2.1).
HDACbi, подобно НАТам, содержатся в многобелковых комплексах. Один из них —
комплекс Sin3 млекопитающих, который включает по меньшей мере семь белков, в том
числе HDAC1 и HDAC2, наряду с другими, которые не обладают действием дезацетилазы,
но обеспечивают вспомогательные функции, существенные для протекания соответ-
ствующего процесса. Примеры вспомогательных белков представлены RbAp46 и RbAp48,
которые являются членами комплекса Sin3 и, как считается, вносят вклад в его способ-
ность связываться с гистонами. Белки RbAp46 и RbAp48 были впервые опознаны благо-
даря их ассоциации с белком ретинобластомы, который управляет разрастанием клеток,
ингибируя экспрессию различных генов, пока их действия не требуются, и который,
будучи мутирован, ведет к раку. Эта связь между комплексом Sin3 и белком, причастным
к раку, обеспечивает мощный довод в пользу большого значения деацетилирования
гистонов в подавлении генов. К другим деацетилирующим комплексам относятся NuRD
у млекопитающих, который объединяет в себе HDAC1 и HDAC2 с различным набором
вспомогательных белков, и Sir2 у дрожжей, который отличается от других HDACee тем,
что ему требуется энергия. Отличительные особенности комплекса Sir2 показывают,
что HDACbi более разнообразны, чем первоначально думали; возможно, это указывает
на то, что нам еще предстоит открыть новые роли деацетилирования гистонов.
Исследования комплексов HDAC начинают раскрывать связи между различными
механизмами активации и подавления генома. И Sin3 и NuRD содержат белки, которые
связываются с метилированной ДНК (раздел 10.3.1), a NuRD содержит белки, которые
являются очень подобными компонентам комплекса перестройки нуклеосомы Swi/Snf
(раздел 10.2.2). NuRD фактически действует как типичная перестраивающая нуклео-
сому машина in vitro. Дальнейшее исследование почти наверняка обнаружит новые
связи между тем, на что мы в настоящее время смотрим как на системы модификации
364 Часть 3. Принципы функционирования геномов
хроматина различных типов, но которые в действительности могут попросту быть раз-
личными вариантами единого грандиозного механизма.
Ацетилирование — не единственный тип модификации гистонов
Ацетилирование/деацетилирование лизина — лучше всего изученная форма
модификации гистонов, но это ни в коем случае не единственный тип. Известна также
ковалентная модификация следующих трех видов.
• Метилирование остатков лизина и аргинина в N-концевых областях гистонов НЗ
и Н4. Метилирование, как думали вначале, является необратимым и, следовательно,
отвечает за постоянные изменения в структуре хроматина. Такому представлению
бросило вызов открытие ферментов, которые деметилируют остатки лизина
и аргинина, но все еще признается, что эффекты метилирования относительно
долгосрочны.
• Фосфорилирование остатков серина в N-концевых областях гистонов Н2А, Н2В,
НЗ и Н4.
• Убиквитинирование остатков лизина в С-концах гистонов Н2А и Н2В. Эта модифи-
кация вовлекает прикрепление маленького распространенного («вездесущего»)
белка, называемого убиквитином, или родственного ему белка СУМО, хотя такое
название, пожалуй, звучит несколько тяжеловесно.
Как и в случае ацетилирования, модификации этих других типов влияют на струк-
туру хроматина и оказывают значительное воздействие на клеточную деятельность.
Например, фосфорилирование гистона НЗ и линкерного гистона связали с формирова-
нием метафазных хромосом, а убиквитинирование гистона Н2В — часть общей роли,
которую убиквитин играет в управлении клеточным циклом. Эффекты метилирования
пары аминокислот лизина в четвертой и девятой позициях, считая от N-конца гистона
НЗ, особенно интересны. Метилирование лизина-9 образует сайт связывания для белка
НР1, что вызывает упаковку хроматина и подавляет экспрессию гена, но это событие
блокируется присутствием двух или трех метильных групп, прикрепленных к лизину-4.
Поэтому метилирование лизина-4 благоволит к открытой структуре хроматина и свя-
зано с активными генами. В пределах функционального домена р-глобина и, вероятно,
во всех прочих местах метилирование лизина-4 также предотвращает связывание
NuRD-дезацетилазы с гистоном НЗ, в силу чего этот гистон остается ацетилированным.
Поэтому метилирование лизина-4 может работать рука об руку с ацетилированием
гистона, с тем чтобы активизировать области хроматина.
В целом, 29 участков в N- и С-концевых областях четырех ядерных гистонов, как
известно, претерпевают ковалентную модификацию (рис. 10.14). Наша растущая осве-
Ме Me МеАцФ Ац Me Ац Ац МеМеФ
II \/ I I II 1111
НЗ ARTKQTARKSTGGKAPRKQLATKARKSAP
10 20
Ф Me Ац Ац Ац Ац Me
I I I I I I ;
Н4 SGRGKGGKGLGKGGAKRHRK
10 20
Рис. 10.14. Модификации N-концевых областей гистонов НЗ и Н4 млекопитающих. Показаны все из-
вестные модификации, происходящие в этих областях. Сокращения: Ац — ацетилирование; Me — ме-
тилирование; Ф — фосфорилирование
Глава 10. Доступ к геному 365
домленность о множестве разного вида модификаций гистонов, которые происходят, и о
пути, в котором различные модификации работают вместе, привела к предположению
о том, что существует некий гистонный код, посредством которого схема химических
модификаций определяет, какие области генома экспрессируются в конкретное время,
и прописывает другие моменты биологии генома, такие как восстановление поврежден-
ныхучастков и согласование репликации генома с клеточным циклом. Это представление
все еще не доказано, но ясно, что схема определенных модификаций гистонов в преде-
лах генома тесно связана с деятельностью гена. Например, исследования хромосом 21
и 22 человека показали, что области в пределах этих хромосом, где лизин-4 гистона НЗ
триметилирован, а лизин-9 и лизин-14 — ацетилированы, соответствуют точкам на-
чала транскрипции активных генов, и что диметилированный лизин-4 также иногда
встречается в этих областях (рис. 10.15). Как и во всех случаях модификации хроматина,
ключевой вопрос здесь состоит в том, что есть причина, а что — следствие: являются ли
эти схемы модификации гистонов причиной, по которой эти специфические гены актив-
ны, или простым побочным продуктом процессов, отвечающих за их активацию?
Хромосома 21
Диметилированный лизин-4 ।
Триметилированный лизин-4 |
Ацетилированные лизины-9,-14 I
IFNGR2
I
I I
II
C21orf4
C21orf55
Диметилированный лизин-4
Триметилированный лизин-4
Ацетилированные лизины-9, -14
Хромосома 22
I
И I
__________ __II
HIRA ^ЬОСивЭТТ^
MRPL40
UFD1L
I
II
Н+Ч----1---HI I ►
CDC45L
Рис. 10.15. Схема модификаций гистонов связана с активностью гена. Показаны сегменты хромосом
человека 21 и 22, каждый сегмент 100 тыс. п. н. в длину. Области, которые обогащены диметил и рован-
ным лизином-4, триметилированным лизином-4, а также ацетилированным лизином-9 и лизином-14
в фибробластах легкого, показаны относительно местоположений известных генов. Стрелки показывают
направления, в которых гены транскрибируются
10.2.2. Влияние перестройки нуклеосом на экспрессию генома
Модификация хроматина второго типа, которая может влиять на экспрессию
генома, — перестройка нуклеосом. Этот термин относится к модификации или пере-
становке нуклеосом в пределах короткой области генома, с тем чтобы связывающие
ДНК белки могли получить доступ к своим сайтам связывания. Как оказалось, это не
является существенным требованием для транскрипции всех генов, и по крайней мере
в некоторых случаях белок, который включает экспрессию гена, может осуществить
свое действие, либо связываясь с поверхностями нуклеосом, либо так или иначе взаи-
модействуя с линкерной ДНК, не затрагивая позиции нуклеосом. В других примерах
перестановка нуклеосом, как было ясно показано, есть необходимая предпосылка для
активации гена. Например, транскрипция гена hsp70 Drosophila melanogaster, который
366 Часть 3. Принципы функционирования геномов
Гэн hsp70
I Активация белком
I GAGA
(UI
ПЛ
Рис. 10.16. Активация гена hsp70 сопряжена с созданием сверх-
чувствительной к ДНКазе I области. На схеме показаны ну-
клеосомы непосредственно выше начала гена и они же после
перестановки при активации этого гена
кодирует белок, вовлеченный в сворачивание дру-
гих белков (раздел 13.3.1), активизируется белком
GAGA в ответ на тепловой шок. Активация сопряжена
с созданием сверхчувствительной к ДНКазе I области
(раздел 10.1.2) выше гена hsp70 — ясный признак
того, что нуклеосомы в этой области были передвинуты и был открыт сегмент голой
ДНК (рис. 10.16).
В отличие от ацетилирования и других химических модификаций, описанных
в предыдущем разделе, перестройка нуклеосом не предполагает ковалентных изменений
молекул гистонов. Вместо этого, перестройка вызывается энергозависимым процессом,
который ослабляет контакт между нуклеосомой и ДНК, с которой она связана. Могут
произойти изменения трех разных типов (рис. 10.17).
• Перестройка, в строгом смысле, предполагает изменение в структуре нуклеосомы,
но никакого изменения в ее позиции. Характер изменения структуры неизвестен,
но когда она вызвана in vitro, то происходит удвоение размера нуклеосомы и воз-
никает повышенная чувствительность прикрепленной ДНК к ДНКазе.
• Сдвиг, или c/s-смещение, физически передвигает нуклеосому по ДНК.
• Перемещение, или trans-смещение, приводит к перемещению нуклеосомы на дру-
гую молекулу ДНК или на несмежную часть той же самой молекулы.
Как и в случае гистонацетилтрансфераз, белки, ответственные за перестройку
нуклеосом, работают совместно в составе больших комплексов. Один из них — Swi/Snf,
который состоит по крайней мере из 11 белков и встречается у многих эукариотов. Не-
много в настоящее время известно о пути, которым Swi/Snf или любой другой комплекс
перестроки нуклеосом выполняет свою роль, заключающуюся в расширении доступа
к геному. Ясно, что ни один из компонентов комплекса Swi/Snf не имеет способности
связывания с ДНК, так что этот комплекс должен быть доставляем к своему целевому
участку дополнительными белками. Между Swi/Snf и НАТами были обнаружены взаи-
модействия; это наводит на мысль о том, что перестройка нуклеосом может происходить
сопряженно с ацетилированием гистонов. Это привлекательная гипотеза, потому что
она связывает два действия, которые в настоящее время рассматриваются как опреде-
ляющие для активации генома. Но с этой гипотезой есть проблемы, потому что комплекс
Перестройка
I
Сдвиг
I
Перемещение
Swi/Snf, как оказалось, не оказы-
вает глобального эффекта по все-
му геному, а напротив, влияет на
экспрессию генов только в огра-
ниченном числе позиций: в слу-
чае дрожжей, возможно, не более
Рис. 10.17. Перестройка, сдвиг и пере-
мещение нуклеосом
Глава 10. Доступ к геному 367
чем в 6 % всех генов, имеющихся в геноме. Это наблюдение предполагает, что более
важные взаимодействия, осуществляемые комплексом Swi/Snf, могут быть реализованы
не с НАТами, которые работают по всему геному, но с другими белками, нацеленными на
ограниченный набор генов. Наиболее вероятные представители этих других белков —
активаторы транскрипции (раздел 11.3.2), каждый из которых является специфичным
к небольшому числу генов и некоторые из которых образуют ассоциации с комплексом
Swi/Snf in vitro.
10.3. Модификация ДНК и экспрессия генома
Важные изменения в активности генома могут быть достигнуты также привне-
сением химических изменений в саму ДНК. Эти изменения связаны с полупостоянным
подавлением областей генома, возможно даже целых хромосом, и часто видоизмененное
состояние наследуется потомством, появляющимся в результате деления клетки. Такие
модификации совершаются путём метилирования ДНК.
10.3.1. Подавление генома путем метилирования ДНК
У эукариотов цитозиновые основания в молекулах хромосомной ДНК иногда из-
меняются в 5-метилцитозин добавлением метильных групп ферментами, названны-
ми ДНК-метилтрансферазами (см. рис. 3.29). У низших эукариотов метилирование
цитозина — относительно редкое явление, но у позвоночных метилировано до 10 %
от общего числа цитозиновых оснований в геноме, а у растений эта цифра может до-
стигать 30 %. Схема метилирования не случайна, а ограничена цитозином в некото-
рых копиях последовательности 5'-CG-3'
и, у растений, — 5’-CNG-3'. Различают
два типа действий по метилированию
(рис. 10.18). Первый — поддерживаю-
щее метилирование, которое, следуя
репликации генома, отвечает за до-
бавление метильных групп к недавно
синтезированной нити ДНК в позици-
ях напротив метилированных участков
на родительской нити (раздел 16.2.3).
Поэтому поддерживающее действие
гарантирует, что две дочерние молеку-
лы ДНК сохранят схему метилирования
родительской молекулы, а это означа-
ет, что такая схема может быть унасле-
дована после деления клетки. Второе
действие — метилирование de novo,
которое добавляет метильные группы
в абсолютно новых позициях и таким
образом изменяет схему метилирова-
ния в ограниченной области генома.
I Репликация ДНК
Me
Me
Me
Me
Me
Me
I Поддерживающее
метилирование
3-Me
= -Me
Me
Me
I Метилирование
ф de novo
3-Me
--Me
Рис. 10.18. Поддерживающее метилирование
и метилирование de novo
Me
Me
368 Часть 3. Принципы функционирования геномов
ДНК-метилтрансферазы и подавление активности генома
ДНК-метилтрансферазы были интенсивно изучены и являются подобными во всех
организмах: от бактерий (которые метилируют свою ДНК, чтобы защитить ее от дегра-
дации их же собственными эндонуклеазами рестрикции, что позволяет им направлять
эти ферменты на вторгшуюся в их естество ДНК бактериофага) и до млекопитающих,
в том числе и человека. Хотя в течение ряда лет была проделана большая работа по
изучению этих ферментов, очевидное присутствие только одной ДНК-метилтрансферазы
в клетках млекопитающих было своего рода загадкой. Этот фермент, теперь называемый
ДНК-метилтрансферазой 1 (Dnmtl), отвечает за поддерживающее метилирование, а не
за метилирование de novo, поскольку нокаутные мыши с инактивированными генами
белка Dnmtl по-прежнему способны к выполнению метилирования de novo, как показала
их способность добавлять метильные группы к ДНК-геному заражающего их ретрови-
руса. В конце 1990-х гг., когда большинство генов в геномах человека и мыши уже было
картировано в виде ESTob (раздел 3.3.3), в подходящих базах данных были проведены
поиски гомологов белка Dnmtl и обнаружены два гена — Dnmt3a и Dnmt3b, — которые,
как теперь известно, кодируют de novo-метилтрансферазы. Мыши с нокаутом по этим
генам неспособны завершать свои полные пути развития, причем мыши с инактиви-
рованным геном Dnmt3b умирают всего лишь спустя несколько дней после рождения,
а те, у которых недостает гена Dnmt3a, живут лишь на 1-2 недели дольше. Анализ схем
метилирования в ДНК эмбрионов в течение периода перед смертью показывает, что
у нокаутных мышей степень метилирования ДНК составляет только половину наблюдае-
мой у нормальных мышей (рис. 10.19), указывая на то, что происходит поддерживающее
метилирование, являющееся результатом действия белка Dnmtl, а метилирование de
novo отсутствует, и, следовательно, у этих мышей общий уровень метилирования ДНК
с течением времени не возрастает.
Как поддерживающее, так и de novo-метилирование приводят к подавлению ак-
тивности гена. Это было показано в ходе экспериментов, в которых метилированные
или неметилированные гены вводились в клетки путем клонирования и их уровни
экспрессии измерялись: экспрессия не происходит, если последовательность ДНК ме-
тилирована. Связь с экспрессией генов прослеживается также и при исследовании схем
метилирования в хромосомных ДНК; они показывают, что активные гены расположены
в неметилированных областях. Например, у людей 40-50 % всех генов расположены
вблизи CpG-островков (раздел 5.1.1), причем состояние метилирования CpG-островка
отражает картину экспрессии прилегающего к нему гена. Гены домашнего хозяйства —
те, которые экспрессируются во всех тканях, — имеют неметилированные CpG-островки,
тогда как CpG-островки, связанные с тканеспецифическими генами, неметилированы
Возраст зародыша, дни
Рис. 10.19. Экспериментальные данные, согласно кото-
рым ДНК-метилтрансферазы За и ЗЬ являются de novo-
метилазами. График показывает общий уровень ме-
тилирования ДНК у эмбрионов нормальных (дикий
тип) мышей и у нокаутных мышей с инактивирован-
ным геном Dnmt3. У нормальных эмбрионов уровень
метилирования ДНКувеличивается из-за метилиро-
вания de novo, выполняемого ДНК-метилтрансферазами
За и ЗЬ, но в нокаутных эмбрионах уровень метилирова-
ния ДНК остается на своем первоначальном значении
Глава 10. Доступ к геному 369
только в тех тканях, в которых этот ген экспрессируется. Обратите внимание, что, по-
скольку схема метилирования поддерживается после деления клетки, информация,
определяющая, какие гены должны быть экспрессируемы, наследуется дочерними
клетками; это гарантирует, что в дифференцированной ткани соответствующая кар-
тина экспрессии генов сохраняется даже при том, что клетки в этой ткани заменяются
и (или) даже множатся новыми клетками1).
Важность метилирования ДНК подчеркивается исследованиями болезней чело-
века. Синдром, называемый ИНЦАЛ (иммунодефицита, нестабильности центромер
и аномального (строения) лица), который, как предполагает название, имеет широкий
диапазон фенотипических эффектов, связан с недометилированием (гипометилирова-
нием) различных областей генома и вызван мутацией в гене, кодирующем белок Dnmt3b.
Противоположная ситуация — переметилирование (гиперметилирование) — наблюда-
ется в пределах CpG-островков генов, которые показывают видоизмененные картины
экспрессии при раке некоторых типов, хотя в этих случаях ненормальное метилирование
может с тем же успехом быть следствием, а не причиной болезненного состояния.
Как именно метилирование влияет на экспрессию генома, было загадкой многие
годы. Теперь известно, что в числе компонентов гистондезацетилазных комплексов
(и Sin3 и NuRD) состоят связывающиеся с метилированными CpG-островками бел-
ки (МеСР). Это открытие привело к построению модели, в которой метилированные
CpG-островки являются целевыми участками для прикрепления комплексов HDAC,
которые видоизменяют окружающий хроматин, с тем чтобы подавлять примыкающие
к ним гены (рис. 10.20).
Метилирование вовлечено в геномный импринтинг и инактивацию
Х-хромосомы
Дальнейшие данные, подтверждающие связь между метилированием ДНК и по-
давлением активности генома, связаны с двумя любопытными явлениями, названными
геномным импринтингом и инактивацией Х-хромосомы.
Рис. 10.20. Модель связи между
метилированием ДНК и экс-
прессией генома. Метилиро-
вание CpG-островка выше не-
которого гена обеспечивает
опознавательные сигналы для
метил-Срб-связывающих белков
(МеСР) — компонентов гистонде-
зацетилазного комплекса (HDAC).
HDAC видоизменяет хроматин
в области CpG-островка и, сле-
довательно, инактивирует ген.
Имейте в виду, что относительные
позиции и размеры CpG-островка
и гена нарисованы без учета мас-
штаба
CpG-островок
Ген
Me Me
CpG-островок
* метилирован
ГДАЦ прикрепляется
посредством MeCPs
Упакованный хроматин
Недавние работы показали, что в редких случаях метилирование ДНК может приводить к активации
генов. — Прим. ред.
370 Часть 3. Принципы функционирования геномов
Геномный импринтинг — относительно редкая, но важная особенность геномов
млекопитающих, в которых только один из пары генов, находящихся на гомологичных
хромосомах в диплоидном ядре, экспрессируется, при этом второй подавляется мети-
лированием. Он встречается также у некоторых насекомых (хотя точно не у Drosophila
melanogastef) и некоторых растений. Всегда один и тот же член пары генов бывает
импринтирован и, следовательно, не активен: для некоторых генов это версия, уна-
следованная от матери, а для других генов это — отцовская версия. Немногим более 60
генов у человека и мыши, как было показано, показывают импринтинг, причем в это
число вошли и кодирующие белок гены, и гены функциональной РНК. Импринтирован-
ные гены распределены по всему геному, но тяготеют к образованию групп. Например,
у людей есть сегмент хромосомы 15 длиной 2,2 млн п. н., в пределах которого находится
по крайней мере десять импринтированных генов, и меньшая область хромосомы 11
длиной 1 млнп.н., которая содержит восемь импринтированных генов.
Пример импринтированного гена у человека представлен геном Igf2, который
кодирует фактор роста — белок, вовлеченный в передачу сигналов между клетками
(раздел 14.1). Только отцовский ген активен (рис. 10.21), потому что на хромосоме,
унаследованной от матери, различные сегменты ДНК в области 1д/2 метилированы,
что предотвращает экспрессию этой копии гена. Второй импринтированный ген, Н19,
расположен на расстоянии около 90 тыс. п. н. от 1д/2, но у него импринтинг осуществлен
наоборот: материнская версия гена Н19 активна, а отцовская версия подавлена. Им-
принтингом руководят элементы управления импринтами — последовательности
ДНК, находящиеся в пределах нескольких тыс. п. н. групп импринтированных генов. Эти
центры опосредствуют метилирование импринтированных областей, но механизм, по-
средством которого они делают это, еще не был описан подробно. Есть также неясность
относительно функции импринтинга. Одна возможность состоит в том, что он играет
роль в развитии, потому что искусственно созданные партеногенетические мыши, кото-
рые имеют две копии материнского генома, не способны развиться должным образом.
Также были предложены более тонкие и хитроумные объяснения, основанные на эво-
люционных конфликтах между самками и самцами в пределах биологических видов.
Инактивация Х-хромосомы менее загадочна. Это специальная форма импринтин-
га, которая ведет к почти полной инактивации одной из Х-хромосом в женской клетке
млекопитающих. Она происходит потому, что самки имеют две Х-хромосомы, тогда как
самцы имеют только одну. Если бы обе из Х-хромосом самки были активны, то белки,
закодированные генами на Х-хромосоме, могли
бы синтезироваться в двойной норме у самок по
сравнению с самцами. Дабы избегнуть такого не-
желател ьного положения дел, одна из Х-хромосом
самки подавляется и наблюдается в ядре в виде
уплотненной структуры, названной тельцем Бар-
ра, которое полностью состоит из гетерохрома-
тина. Большинство генов на инактивированной
Отцовская хромосома 11
Igf2 Ме Н19 Ме
Материнская хромосома 11
Me Me
ОБОЗНАЧЕНИЯ:
Активный ген
«jBSGSU* Импринтированный ген
Рис. 10.21. Пара импринтированных генов на хромосоме
11 человека. Ген Igf2 импринтирован на хромосоме, уна-
следованной от матери, а Н19 импринтирован на отече-
ской хромосоме. Рисунок выполнен без учета масштаба:
эти гены расположены на расстоянии около 90 тыс. п. н.
друг от друга
Глава 10. Доступ к геному 371
Х-хромосоме становится подавленным, но по причинам, которые остаются неизвестны-
ми, около 20 % уклоняются от этого процесса и остаются функционально активными.
Подавление происходит на ранней стадии развития эмбриона и управляется цен-
тром инактивации Х-хромосомы (Xie) — обособленной областью, находящейся на каждой
Х-хромосоме. В клетке, проходящей инактивацию Х-хромосомы, центр инактивации на
одной из Х-хромосом запускает формирование гетерохроматина, процесс распростра-
няется из центра его образования до тех пор, пока вся хромосома не будет затронута,
за исключением нескольких коротких сегментов, содержащих те гены, которые оста-
ются активными. По продолжительности данный процесс занимает несколько дней.
Точный механизм не понят, но он вовлекает, хотя и не зависит от него полностью,
расположенный в центре инактивации ген по имени Xist, который транскрибируется
в некодирующую РНК размером 25 тыс. п. н., копии которой покрывают хромосому
по мере того, как формируется гетерохроматин. В то же время происходят различные
модификации гистонов. Лизин-9 гистона НЗ становится метилированным (вспомним,
что эта модификация сопряжена с инактивацией генома — раздел 10.2.1), гистон Н4
становится деацетилированным (поскольку обычно встречается в гетерохроматине),
и молекулы гистона Н2А заменяются специальным гистоном макроН2А1. Некоторые по-
следовательности ДНК становятся гиперметилированными ДНК-метилтрансферазой За,
хотя это определенно происходит после того, как установится неактивное состояние.
Инактивация Х-хромосомы наследственна и свойственна всем клеткам, ведущим свое
начало от исходной, в которой инактивация имела место1).
У нормальной диплоидной самки однаХ-хромосома инактивирована, а другая остает-
ся активной. Примечательно, что у диплоидных самок с необычными составами половых
хромосом этот процесс по-прежнему оставляет только одну Х-хромосому активной. Напри-
мер, у тех редких индивидуумов, которые обладают только единственной Х-хромосомой,
никакая инактивация не происходит, а у индивидуумов с кариотипом XXX инактивиру-
ются две из трех Х-хромосом (рис. 10.22, а). Это означает, что должен существовать некий
а) Инактивация в необычных кариотипах
б) Инактивация предполагает подсчёт хромосом
АА
хххх
АААА
ХХХХ
АААА
Рис. 10.22. Инактивация Х-хромосомы. а) Если в геноме присутствует единственная Х-хромосома,
то никакой инактивации не происходит; если есть три Х-хромосомы, то инактивируются две из них.
б) В клетке, которая имеет диплоидный комплемент аутосом (АА), но четыре Х-хромосомы, инактиви-
руются три Х-хромосомы. И обратно: если клетка тетраплоидная (АААА), то инактивируются только две
Х-хромосомы
Инактивация Х-хромосомы происходит в разных клетках независимо. Поэтому в разных клетках
взрослой особи могут быть инактивированы разные Х-хромосомы, иными словами, взрослая самка гене-
тически неоднородна! Например, пятнистые рыже-белые кошки — продукт неоднородной инактивации
Х-хромосомы, — в части клеток работает «белый» ген из одной хромосомы, а в другой части — «рыжий» ген
из другой хромосомы. — Прим. ред.
372 Часть 3. Принципы функционирования геномов
механизм, с помощью которого Х-хромосомы в ядре подсчитываются и должное их число
инактивируется. Фактически этот механизм не просто считает Х-хромосомы; он также
считает аутосомы и сравнивает эти два числа. Это явствует из того, что если клетка имеет
четыре Х-хромосомы, но в другом отношении является диплоидной, то инактивируются
три Х-хромосомы, но если она тетраплоидна (то есть имеет четыре Х-хромосомы и также
четыре копии каждой аутосомы), то инактивируются две Х-хромосомы (рис. 10.22, б). Как
клетка подсчитывает свои хромосомы, озадачивало цитогенетиков многие годы и продол-
жает озадачивать нас, но самое недавнее исследование предполагает, что этим процессом
управляют два гена в пределах центра инактивации Х-хромосомы, названные Tsix и Xite,
ибо удаление или сверхэкспрессия одного или обоих из этих генов приводит к тому, что
инактивируется неправильное число хромосом.
Заключение
Среда ядра оказывает значительное и важное воздействие на экспрессию генома.
Ядро эукариотов имеет высокоупорядоченную внутреннюю структуру, которая вклю-
чает сложную сеть волоконец из белка и РНК, названную ядерным матриксом. Каждая
хромосома имеет свою собственную территорию в пределах ядра, и эти территории
отделены друг от друга бесхроматиновыми областями, в пределах которых находятся
ферменты и другие белки, вовлеченные в экспрессию генома. Наиболее компактная
формахроматина — гетерохроматин, в котором гены являются недоступными и не мо-
гут быть экспрессированы. Структурный гетерохроматин — постоянная составляющая
всех клеток и представляет собой ДНК, которая не содержит никаких генов, тогда как
факультативный гетерохроматин появляется в клетках лишь на определенное время и,
как думают, содержит гены, которые являются неактивными в некоторых тканях или
на некоторых стадиях клеточного цикла. Хроматин более открытого типа, названный
эухроматином, организован в петли, прикрепленные к ядерному матриксу; эти петли,
возможно, соответствуют функциональным доменам, в которые организуются гены,
находящиеся в геноме. Каждый функциональный домен ограничен парой инсуляторов,
а некоторые содержат управляющие локусом области, которые вовлечены в регули-
рование экспрессии генов, содержащихся в этих доменах. У эукариотов нуклеосомы
предстают перед нами как главные определители деятельности генома — не только
посредством их расположения на нити ДНК, но также и потому, что точно заданная
химическая структура гистоновых белков, содержащихся в нуклеосомах, есть главный
фактор, определяющий степень упаковки, присущую тому или иному сегменту хромати-
на. Ацетилирование аминокислот лизинов в N-концевых областях каждого из ядерных
гистонов связано с активацией некоторой области генома, а деацетилирование ведет
к подавлению генома. Гистоны могут быть видоизменены также метилированием,
фосфорилированием и убиквитинированием, причем каждое из этих событий оказы-
вает определенное, специфическое воздействие на активность примыкающих к ним
генов. Возможно, даже существует гистонный код, посредством которого комбинации
этих различных модификаций интерпретируются геномом. Перестановка нуклеосом
необходима для экспрессии некоторых, но не всех генов. Области генома могут быть по-
давлены также метилированием ДНК, при этом соответствующие ферменты, возможно,
работают в союзе с гистондезацетилазами. Метилирование ответственно за геномный
импринтинг, в результате которого один из пары генов на гомологичных хромосомах
становится подавленным, а инактивация Х-хромосомы ведет к почти полной инакти-
вации одной из Х-хромосом в ядре клетки женской особи.
Глава 10. Доступ к геному 373
Задания для закрепления материала
* Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
10.1 *. Что такое ядерный матрикс?
а. Комплекс гистоновых белков
и ДНК, который образует струк-
турную сеть по всему ядру.
Ь. Однородная смесь ДНК, РНК
и белков, которые формируют
состав ядра.
с. Микротрубочки, которые обе-
спечивают структурную основу
ядра.
d. Сложная сеть волоконец из бел-
ка и РНК, которые образуют суб-
структуру ядра.
10.2 . Какова функция ядрышка?
а. Это участок для экспрессии генов,
кодирующих белки.
Ь. Это хромосомный каркас, кото-
рый изменяет свою структуру,
с тем чтобы уплотнить хромосо-
мы в ходе деления клетки.
с. Это участок для синтеза и созре-
вания молекул рРНК.
d. Это участок для обработки моле-
кул мРНК.
10.3 *. Какой из следующих методов явля-
ется полезным в определении пере-
движения белков внутри ядра?
а. Электронная микроскопия.
Ь. Восстановление флюоресцен-
ции после фотообесцвечивания
(FRAP).
с. Флюоресцентная гибридизация
in situ (FISH).
d. Конфокальная оптическая микро-
скопия.
10.4 . Определение гетерохроматина зву-
чит как:
а. Хроматин, который состоит из ге-
терогенных последовательностей
нуклеотидов.
Ь. Хроматин, который содержит ге-
терогенные белки.
с. Хроматин, который относительно
уплотнен и содержит неактивные
гены.
d. Хроматин, который относитель-
но раскрыт и содержит активные
гены.
10.5 *. В хроматине какого типа содержатся
гены, которые активно экспресси-
руются?
а. Эухроматин.
Ь. Факультативный гетерохроматин,
с. Структурный гетерохроматин.
d. Все вышеперечисленные.
10.6 . Которая из нижеследующих сущно-
стей является областью ДНК эука-
риотов, содержащей один или не-
сколько активных генов, очертания
которой можно установить путем
обработки ДНКазой I?
а. Эухроматин.
Ь. Гетерохроматин.
с. Функциональный домен.
d. Структурный домен.
10.7 *. Которая из следующих структур
способна предотвратить экспрес-
сию гена, будучи вставлена между
геном и его регуляторными после-
довательностями?
а. Функциональный домен.
Ь. Структурный домен.
с. Последовательность инсулятора.
d. Управляющая локусом область.
10.8 . Какова роль управляющих локусом
областей (LCR) в регулировании экс-
прессии генов?
а. Связывающиеся с ДНК белки при-
крепляются к LCR и видоизменя-
ют структуру хроматина.
374 Часть 3. Принципы функционирования геномов
Ь. Факторы транскрипции связыва-
ются с LCR и способствуют экс-
прессии гена.
с. Связывающиеся с ДНК белки при-
крепляются к LCR и стимулируют
метилирование ДНК.
d. LCR образуют места прикрепле-
ния к ядерному матриксу.
10.9 *. Какого типа аминокислота ацети-
лируется в N-концевых областях
гистоновых белков?
а. Аргинин.
Ь. Лизин.
с. Серин.
d. Тирозин.
10.10 . Какой из следующих типов модифи-
кации не относится к модификации
гистонов?
а. Ацетилирование.
Ь. АДФ-рибозилирование.
с. Метилирование.
d. Фосфорилирование.
10.11 *. В результате какого из следую-
щих событий перестройки нуклео-
сома переходит на другую молекулу
ДНК?
а. Ацетилирование.
Ь. Перестройка.
с. Сдвиг.
d. Перемещение.
10.12 . Вследствие которого из следующих
типов модификации ДНК подавляет-
ся область генома таким способом,
что подавление может быть пере-
дано потомству?
а. Ацетилирование.
Ь. Метилирование.
с. Фосфорилирование.
d. Убиквитинирование.
10.13 *. Какое из нижеследующих опреде-
лений метилирования ДНК de novo
верно?
а. Добавление метильных групп
к ДНК в новых позициях, с тем
чтобы изменить схему метили-
рования генома.
Ь. Добавление метильных групп
к недавно синтезированным ни-
тям ДНК, чтобы гарантировать,
что дочерние нити будут обладать
теми же схемами метилирования,
что и родительские нити.
с. Добавление метильных групп
к промоторам генов, с тем чтобы
активировать экспрессию генов.
d. Добавление метильных групп
к изоляторным областям, чтобы
подавить экспрессию гена.
10.14 . Каково состояние метилирования
CpG-островков генов домашнего
хозяйства?
а. Они имеют переметилированные
CpG-островки.
Ь. Они метилированы в некоторых
тканях, но не во всех.
с. Они имеют неметилированные
CpG-островки.
d. Эти гены не имеют CpG-остров-
ков.
10.15 *. Геномный импринтинг имеет ме-
сто, когда:
а. Схемы метилирования ДНК в ге-
номе передаются потомству.
Ь. Гены неправильно подавляются
из-за метилирования ДНК, что
приводит к видоизмененным
фенотипам.
с. Только один из пары генов экс-
прессируется, второй метилиро-
ван и подавлен.
d. Метилирование ДНК снимается,
что позволяет генам, которые
должны быть подавлены, экс-
прессироваться.
10.16 . Если диплоидный индивидуум
имеет три Х-хромосомы, то сколько
Х-хромосом будет инактивирова-
но?
а. Одна.
Ь. Две.
с. Три.
d. Это число изменяется и может
быть равно одному или двум.
Глава 10. Доступ к геному 375
Вопросы открытого типа
10.1 *. Какие типы микроскопического ана-
лиза привели к подвижкам в нашем
понимании структурной организа-
ции ядра?
10.2 . Что показало раскрашивание хро-
мосом о местоположении хромосом
в пределах ядра?
10.3 *. Транслокации происходят более
часто между некоторыми парами
хромосом. Что это говорит нам о рас-
пределении хромосом по ядру?
10.4 . В чем разница между структурным
гетерохроматином и факультатив-
ным гетерохроматином?
10.5 *. Каково объяснение позиционно-
го эффекта, который иногда про-
исходит, когда ген клонируется
в хозяине-эукариоте?
10.6 . Что такое изоляторные последова-
тельности и какими уникальными
свойствами они обладают?
10.7 *. Обсудите подобия и различия между
изоляторными последовательностя-
ми и управляющими локусом обла-
стями (LCR).
10.8 . Что мы можем заключить из откры-
тия того факта, что гистонацетил-
трансферазы (НАТы) имеют низкую
активность по отношению к непо-
врежденным нуклеосомам?
10.9 *. Какова роль гистондезацетилаз
(HDACee) в регулировании экспрес-
сии генома?
10.10 . Что собой представляет «гистонный
код»?
10.11 *. Почему для изучения изменений
в структуре хроматина используется
ДНКаза I? Что восприимчивость ДНК
к расщеплению ДНКазой I говорит
нам об экспрессии генов?
10.12 . Какие изменения происходят в ну-
клеосомах в ходе инактивации
Х-хромосомы?
Изыскательские задачи
10.1* . До какой степени можно предпо-
лагать, что картина архитектуры
ядра, вырисованная современной
электронной микроскопией, точ-
но описывает истинную структуру
ядра, а не является артефактом1) ме-
тодов, применяемых для подготовки
клеток к наблюдению?
10.2. Во многих областях биологии труд-
но отличить причину от следствия.
Оцените справедливость этого
утверждения в отношении пере-
стройки нуклеосом и экспрессии
генома — действительно ли пере-
стройка нуклеосом причиняет из-
Артефакт — свойство, привнесенное проце-
дурой исследования. — Прим, перев.
менения в экспрессии генома, или
же она следует из этих изменений
экспрессии?
10.3* . Исследуйте и оцените гипотезу о ги-
стонном коде.
10.4. Поддерживающее метилирование
гарантирует, что схема метилиро-
вания ДНК, наблюдаемая у двух до-
черних молекул ДНК, будет той же,
что и схема, присущая родительской
молекуле. Другими словами, схема
метилирования и информация об
экспрессии генов, которую она пере-
дает, наследуются. Другие особен-
ности структуры хроматина могли
бы также наследоваться подобным
образом. В какой мере эти явления
376 Часть 3. Принципы функционирования геномов
опровергают менделевское пред-
ставление о том, что наследование
определяется генами?
10.5* . Какого рода могут быть средства, ко-
торыми число Х-хромосом и аутосом
в ядре подсчитывается, с тем чтобы
было инактивировано должное чис-
ло Х-хромосом?
Тесты по рисункам
10.1*. На рисунке показаны два
участка, в которых ген
может быть вставлен
в геном. Какой уровень
экспрессии гена ожидает-
ся после вставки в область
плотноупакованного хро-
матина и в область рас-
крытого хроматина?
Новый ген
вставлен сюда
Экспрессия слабая
или отсутствует
Или же сюда
I
I
10.2. Обсудите, как изоляторные после-
довательности увеличивают уро-
вень экспрессии гена, если они при-
цеплены к клонированному гену,
который вставлен в геном.
Высокий уровень
экспрессии
Без изоляторов С изоляторами
10.3*. Как влияет метилиро-
вание CpG-островка на
экспрессию соседнего
гена?
CpG-островок
Ген
Me Ме
CpG-островок
метилирован
ГДАЦ прикрепляется
посредством MeCPs
Упакованный хроматин
4Н
Глава 10. Доступ к геному 377
10.4. Обсудите различия в картинах экспрессии
генов, унаследованных от каждого из ро-
дителей.
Отцовская хромосома 11
Igf2 Ме Н19 Ме
Материнская хромосома 11
Me Ме
Дополнительная литература
Внутренняя структура ядра
Gerlich,D., Beaudouin, J., Kalbfuss,B., Daigle, N., Eils,R. and EllenbergJ. (2003)
Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 112:
751-764.
Misteli,T. (2001) Protein dynamics: implications for nuclear architecture and gene
expression. Science 291: 843-847.
Williams, R. R. E. (2003) Transcription and the territory: the ins and outs of gene
positioning. Trends Genet 19: 298-302. Chromosome territories.
Хроматиновые домены
Bell, A. C., West, A. G. and Felsenfeld,G. (2001) Insulators and boundaries: versatile
regulatory elements in the eukaryotic genome. Science 291:447-450.
Gerasimova, T. I., Byrd,K. and Corces,V.G. (2000) A chromatin insulator determines
the nuclear localization of DNA. Mol. Cell 6:1025-1035.
Li, Q., Harju, S. and Peterson, K. R. (1999) Locus control regions: coming of age at a decade
plus. Trends Genet 15: 403-408.
Ковалентная модификация гистонов
AhringerJ. (2000) NuRD and SIN3: histone deacetylase complexes in development.
Trends Genet 16: 351-356.
Bannister, A.), and Kouzarides,T. (2005) Reversing histone methylation. Nature 436:
1103-1106.
Bernstein, В. E., Kamal, M., lindblad-Toh, K., etal. (2005) Genomic maps and comparative
analysis of histone modifications in human and mouse. Cell 120:169-181. Correlates the positions
of histone modifications in chromosomes 21 and 22 with gene activity.
Carrozza,M. J., Utley, R.T., Workman, J. L. and Cote, J. (2003) The diverse functions of
histone acetyltransferase complexes. Trends Genet 19: 321-329.
Imai,S., Armstrong, С. M., Kaeberlein, M. and Guarente,L. (2000) Transcriptional
silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:
795-800.
Jenuwein,T. and Allis,C.D. (2001) Translating the histone code. Science 293: 1074-
1080.
Khorasanizedeh,S. (2004) The nucleosome: from genomic organization to genomic
regulation. Cell 116: 259-272. Review of histone modification, nucleosome remodeling, and
DNA methylation.
378 Часть 3. Принципы функционирования геномов
Lachner, М., O'Carroll, D., Rea, S., Mechtler, К. and Jenuwein, Т. (2001) Methylation of
histone НЗ lysine 9 creates a binding site for HP1 proteins. Nature 410:116-120.
Sims, R.J., Nishioka, K. and Reinberg,D. (2003) Histone lysine methylation: a signature
for chromatin function. Trends Genet 19: 629-639.
Strahl, B. D. and Allis, D. (2000) The language of covalent histone modifications. Nature
403: 41-45.
Taunton,)., Hassig,C.A. and Schreiber,S.L. (1996) A mammalian histone deacetylase
related to the yeast transcriptional regulator Rpd3p. Science 272: 408-411.
Timmers, H.T. and Tora,L. (2005) SAGA unveiled. Trends Biochem. Sci. 30: 7-10.
Verdin, E., Dequiedt,F. and Kasler,H.G. (2003) Class II histone deacetylases: versatile
regulators. Trends Genet 19: 286-293.
Перестройка нуклеосом
AalfsJ.D. and Kingston, R. E. (2000) What does 'chromatin remodelling' mean? Trends
Biochem. Sci. 25: 548-555. Stimulating discussion of histone modification and nucleosome
remodeling.
Sudarsanam,P. and Winston,F. (2000) The Swi/Snf family: nucleosome-remodeling
complexes and transcriptional control. Trends Genet. 16: 345-351.
Открытие ДНК-метилтрансфераз млекопитающих
Bird, A. (1999) DNA methylation de novo. Science 286: 2287-2288.
Okano,M., Bell,D.W., Haber, D. A. and Li,E. (1999) DNA methyltransferases Dnmt3a
and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:
247-257.
Xu,G.-L., Bestor,T.H., Bourc'his, D., et at (1999) Chromosome instability and
immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature
402:187-191.
Импринтинг
Feil,R. and Khosia,S. (1999) Genomic imprinting in mammals: an interplay between
chromatin and DNA methylation? Trends Genet 15: 431-434.
Jeppesen, P. and Turner, B.M. (1993) The inactive X chromosome in female mammals
is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression.
Cell 74: 281-289.
Инактивация Х-хромосомы
Ballabio,A. and Willard, H.F. (1992) Mammalian X-chromosome inactivation and the
XIST gene. Curr. Opin. Genet Devel. 2: 439-448.
Brown, C. J. and Greally, J. M. (2003) A stain upon the silence: genes escapingX inactivation.
Trends Genet 19:432-438.
Costanzi, C. and Pehrson, J. R. (1998) Histone macroH2Al is concentrated in the inactive
X chromosome of female mammals. Nature 393: 599-601.
Heard, E., Clerc, P. and Avner, P. (1997) X-chromosome inactivation in mammals. Annu.
Rev. Genet 31: 571-610.
Lee, J.T. (2005) Regulation ofX-chromosome counting by TsixandXite sequences. Science
309: 768-771.
Глава 11
Сборка комплекса инициации
транскрипции
По прочтении главы 11 вы сможете:
• Описывать главные структурные мотивы, которые позволяют белкам прикре-
пляться к молекулам ДНК специфично к их последовательности.
• Вкратце описывать различные методы, применяемые для определения ме-
стонахождения позиции, в которой ДНК-связывающий белок прикрепляется
к молекуле ДНК.
• Рассуждать об особенностях двойной спирали, которые имеют важное значение
во взаимодействиях между ДНК и связывающимися с ней белками, и подроб-
но описывать химические события, которые лежат в основе этих взаимодей-
ствий.
• Определять главные особенности различных РНК-полимераз прокариотов
и эукариотов, а также описывать структуры опознаваемых ими промоторных
последовательностей.
• Приводить доскональное описание процесса сборки комплекса инициации транс-
крипции у Escherichia coli и обсуждать различные способы управления этим
процессом, в частности, проводя различие между механизмами структурного
и регуляторного управления.
• Приводить исчерпывающее описание сборки комплекса инициации транскрип-
ции РНК-полимеразой II у эукариотов и в общих чертах обрисовывать аналогич-
ные процессы для РНК-полимераз I и III.
• Описывать, с примерами, модульную структуру промоторов у эукариотов.
• Объяснять, каким образом активаторы и репрессоры влияют на сборку ком-
плексов инициации транскрипции у эукариотов.
• Обсуждать наше понимание структур посреднических комплексов и их роль
в инициации транскрипции у эукариотов.
Первичным продуктом экспрессии генома является транскриптом — совокупность
молекул РНК, транскрибированных с тех кодирующих белок генов, биологическая ин-
формация которых востребована клеткой в данное время (см. рис. 1.2). Транскриптом
поддерживается за счет процесса, названного транскрипцией, в ходе которого отдельные
гены копируются с ДНК в молекулы РНК. Одно время транскрипцию рассматривали
как простой процесс «создания РНК из ДНК», и, в сущности, именно это и происходит,
но теперь мы осознаем, что процесс, который ведет от генома к транскриптому, намно-
го сложнее, чем предлагает это банальное утверждение. Эту часть экспрессии генома
теперь разделяют на две ключевые стадии (рис. 11.1).
Ген
ШАГ 1 - запуск
транскрипции
Комплекс
запуска
транскрипции
ШАГ 2 - Синтез
и обработка РНК
РНК
Рис. 11.1. Две стадии процесса, который ведет
от генома к транскриптому
380 Часть 3. Принципы функционирования геномов
• Инициация транскрипции, которая завер-
шается сборкой (перед геном) белкового
комплекса, включающего фермент РНК-
полимеразу и различные дополнительные
белки. Этот комплекс впоследствии копи-
рует ген в РНК-транскрипт. На этом этапе
происходят события, которые определят,
будет ли этот ген фактически транскриби-
роваться.
• Синтез и обработка (процессинг) РНК, которые
начинаются, когда РНК-полимераза оставляет
область инициации и начинает производить
РНК-копию гена, и заканчиваются после за-
вершения (терминации) событий обработки
и модификации, которые преобразуют исхо-
дный транскрипт в зрелую молекулу РНК, спо-
собную выполнять свою функцию в клетке.
В этой главе мы рассуждаем об инициации транскрипции, а в главе 12 поговорим
о синтезе и процессинге РНК. Но прежде чем перейти к этим темам, нам нужно вооружить-
ся неоходимыми материалами по данному делу. Центральные игроки в транскрипции
представлены ДНК-связываюшими белками, которые прикрепляются к геному, с тем
чтобы исполнять свои биохимические функции (см. табл. 11.1). Гистоны — примеры
ДНК-связывающих белков, и мы столкнемся со многими другими позже в этой главе,
когда будем созерцать сборку инициаторных комплексов у прокариотов и эукариотов.
Есть также ДНК-связывающие белки, которые вовлечены в репликацию, репарацию
и рекомбинацию ДНК, а также большая группа родственных им белков, которые свя-
зываются с РНК, а не с ДНК (см. табл. 11.1). Многие ДНК-связывающие белки опознают
определенные последовательности нуклеотидов и связываются преимущественно
с этими целевыми участками, тогда как другие связываются неспецифически в раз-
личных позициях последовательности генома.
Способ действия ДНК-связывающих белков является определяющим для инициа-
ции транскрипции, и без знания того, как они функционируют, нам нечего и надеяться
постичь принципы использования информации, сокрытой в геноме. А посему мы по-
жертвуем толикой нашего времени на изучение всего того, что человечеству известно
о ДНК-связывающих белках и механизмах их взаимодействия с геномом.
11.1. ДНК-связывающие белки и их сайты связывания
Наш главный интерес сосредоточен на тех белках, которые способны нацеливать-
ся на специфичные последовательности нуклеотидов и, следовательно, связываются
с ограниченным числом позиций на молекуле ДНК, причем такой тип взаимодействия
наиболее важен в экспрессии генома. Чтобы специфично связаться подобным образом,
белок должен вступить в контакт с двойной спиралью таким способом, чтобы после-
довательность нуклеотидов могла быть опознана, для чего обычно требуется проник-
новение части белка в большую и (или) малую бороздку спирали (см. рис. 1.8, а и 1.9),
с тем чтобы достигнуть прямого считывания последовательности (раздел 11.1.3). Это
обычно сопровождается более общими взаимодействиями с поверхностью молекулы
Глава 11. Сборка комплекса инициации транскрипции 381
ДНК, которые могут попросту стабилизировать комплекс белок-ДНК или могут получить
доступ к косвенной информации о последовательности нуклеотидов, которую можно
вывести из конформации спирали.
11.1.1. Особые характеристики ДНК-связывающих белков
Структуры многих белков, в том числе более 100, которые связываются с ДНК
или РНК, были определены такими методами, как рентгеноструктурный анализ
и спектроскопия ядерного магнитного резонанса (ЯМР-спектроскопия) (техниче-
ское примечание 11.1). При сравнении структур специфичных к последовательности
ДНК-связывающих белков сразу становится очевидно, что семейство таковых в целом
может быть разделено на ограниченное число различных групп на основании струк-
туры сегмента белка, который взаимодействует с молекулой ДНК (таблица 11.2). Каж-
дый из таких ДНК-связывающих мотивов присутствует в ряде белков, зачастую из
значительно разнящихся организмов, и по крайней мере некоторые из них, вероятно,
эволюционировали более одного раза. Два из них мы рассмотрим во всех подробно-
стях — мотив спираль-изгиб-спираль (НТН) и цинковый палец — и затем бегло
оглядим остальные.
Мотив спираль-изгиб-спираль присутствует в белках прокариотов и эука-
риотов
Мотив НТН стал первой опознанной ДНК-связывающей структурой. Как предпо-
лагает название, мотив состоит из двух а-спиралей, разделенных изгибом (рис. 11.2).
Последний — не произвольная конформация, а особая структура, которая называется
P-изгибом и состоит из четырех аминокислот, вторая из которых обычно представ-
лена глицином. Этот изгиб, вкупе с первой а-спиралью, помещает вторую а-спираль
на поверхности белка в такую ориентацию, которая позволяет ей войти в большую
бороздку молекулы ДНК. Эта вторая а-спираль, таким образом, является распознаю-
щей спиралью и образует имеющие огромное значение контакты, которые позволяют
считывать последовательность ДНК. Структура НТН обычно состоит из 20 или около
того аминокислот в длину и, таким образом, является лишь малой частью всего белка
в целом. Некоторые остальные части этого белка образуют прикрепления с поверхно-
стью молекулы ДНК, главным образом для того, чтобы обеспечить правильное рас-
положение распознающей спирали в главной бороздке.
Многие ДНК-связывающие белки прокариотов и эукариотов используют мотив
НТН. У бактерий мотивы НТН присутствуют в некоторых из наиболее изученных ре-
гуляторных белков, которые включают и выключают
экспрессию отдельных генов. Пример — лактозный
репрессор, который регулирует экспрессию оперо-
на лактозы (раздел 11.3.1). В число различных НТН-
содержащих белков эукариотов входят многие, свойства
которых (связываться с ДНК) важны в возрастном регу-
Рис. 11.2. Мотив спираль-изгиб-спираль. На рисунке показана
ориентация мотива спираль-изгиб-спираль (фиолетовым) гена-
репрессора бактериофага 434 Escherichia coli в большой бороздке
двойной спирали ДНК. Буквы «N» и «С» обозначают соответствен-
но N- и С-конец мотива
382 Часть 3. Принципы функционирования геномов
Таблица 11.1. Функции связывающихся с ДНК и РНК белков11
Функции Примеры
Связывающиеся с ДНК белки Экспрессия генома Инициация транскрипции Связывающийся с блоком ТАТА белок эукариотов (раздел 11.2.3) о-субъединица бактериальной РНК-полимеразы (раздел 11.2.3)
Синтез РНК РНК-полимеразы (раздел 11.2.1)
Регулирование транскрипции Активаторы и репрессоры эукариотов (раздел 11.3.2) Бактериальные репрессоры (раздел 11.3.1)
Упаковка ДНК Гистоны эукариотов (раздел (7.1.1) Белки нуклеоида бактерий (раздел 8.1.1)
Рекомбинация ДНК Восстановление ДНК Репликация ДНК RecA (раздел 17.1.2) ДНК-гликозилазы, нуклеазы (раздел 16.2.2) Белки опознавания старта репликации (раздел 15.2.1) ДНК-полимеразы и лигазы (разделы 2.1.1,2.1.3 и 15.2.2) Белки, связывающие однонитевую ДНК (раздел 15.2.2) ДНК-топоизомеразы (раздел 15.1.2)
Прочие Связывающиеся с РНК белки Экспрессия генома Сплайсинг интронов Полиаденилирование мРНК Редактирование мРНК Процессинг рРНК и тРНК Трансляция Эндонуклеазы рестрикции прокариотов (раздел 2.1.2) Белки мяРНП (раздел 12.2.2) CPSF, CstF (раздел 12.2.1) Аденозиндезаминазы (раздел 12.2.5) Рибонуклеазы (разделы 12.1.3 и 12.2.4) Аминоацил-тРНК-синтетазы (раздел 13.1.1) Факторы трансляции (разделы 13.2.2,13.2.3 и 13.2.4)
Деградация РНК Структура рибосомы Рибонуклеазы (разделы 12.1.4 и 12.2.6) Рибосомные белки (раздел 13.2.1)
лировании экспрессии генома, например, такие как гомеодомен-содержащие белки, чьи
роли мы исследуем в разделе 14.3.4. Гомеодомен — расширенный мотив НТН, входящий
в состав всех этих белков. Он состоит из 60 аминокислот, которые образуют четыре
а-спирали: номера 2 и 3 разделены p-изгибом, номер 3 действует как распознающая
спираль, а номер 1 образует контакты в пределах малой бороздки (рис. 11.3). К осталь-
ным вариантам мотива НТН, встречающимся у эукариотов, относятся:
Здесь приведены только основные классы полинуклеотид-связывающих белков и комплексов. Существует
еще великое множество типов белков, взаимодействующих с нуклеиновыми кислотами. — Прим. ред.
Глава 11. Сборка комплекса инициации транскрипции 383
Рис. 11.3. Мотив гомеодомен. Первые три спирали типичного гомео-
домена показаны так, что спираль 3 ориентирована параллельно
большой бороздке, а спираль 1 образует контакты в малой бороздке.
Спирали 1-3 бегут в направлении N—>С мотива
• домен POU, который обычно находится в белках, со-
держащих также гомеодомен, причем эти два мотива,
вероятно, работают совместно, связываясь с разными
областями двойной спирали. Название «POU» обра-
зовано из первых букв названий белков, в которых
этот мотив был впервые обнаружен;
• крыловатый мотив спираль-изгиб-спираль
представляет собой еще один расширенный вари-
ант основной структуры НТН и содержит третью
а-спираль на одной стороне мотива НТН и p-лист на другой его стороне.
Многие белки, как принадлежащие прокариотам, так и присущие эукариотам, об-
ладают мотивом НТН, но особенности взаимодействия распознающей спирали с большой
бороздкой несколько отличаются в разных случаях. Длина распознающей спирали из-
меняется (она, как правило, длиннее в белках эукариотов), ориентация спирали в боль-
шой бороздке не всегда одинакова, и позиции тех аминокислот в последовательности
распознающей спирали, которые образуют контакты с нуклеотидами, также бывают
разными.
Цинковые пальцы распространены среди белков эукариотов
ДНК-связывающий мотив второго типа, который мы детально рассмотрим, —
цинковый палец, который редок в белках прокариотов, но очень часто встречаетя
у эукариотов. Оказывается, что в черве Caenorhabditis elegans из общего числа 19000
белков имеется более 500 различных белков с цинковыми пальцами и, по оценкам, 1 %
всех генов млекопитающих кодируют белки с цинковыми пальцами.
Известно по крайней мере шесть разных вариантов цинкового пальца. Первым
подробно изученным был палец Cis2His2, который включает ряд из 12 или около того
аминокислот, в том числе два цистеина и два гистидина, образующих сегмент р-листа,
за которым следует а-спираль. Эти две структуры, которые
формируют «палец», выдающийся из поверхности белка,
удерживают между собой связанный атом цинка, располо-
женный симметрично к двум цистеинам и двум гистидинам
(рис. 11.4). а-спираль составляет часть мотива, которая
и образует важные контакты в большой бороздке; ее рас-
положение в бороздке определяется p-листом, который
взаимодействует с сахаро-фосфатной основой цепью ДНК
Рис. 11.4. Цинковый палец Cis2His2. Этот специфический цинковый
палец из белка SWI5 дрожжей. Атом цинка удерживается между двумя
цистеинами, находящимися в p-листе мотива, и двумя гистидинами
из а-спирали. Оранжевые линии показывают боковые группы этих
аминокислот. Буквы «N» и «С» показывают соответственно N- и С-конец
мотива
384 Часть 3. Принципы функционирования геномов
Техническое примечание 11.1: рентгеноструктурный ана-
лиз и спектроскопия ядерного магнитного резонанса
Методы изучения структур белков и комплексов белков с нуклеиновыми
кислотами
После очистки ДНК- или РНК-связывающего белка можно предпринимать
попытки определить его структуру — взятого в отдельности или прикрепленного
к своему участку связывания. Это позволяет определить точную структуру систе-
мы нуклеиновая кислота-связывающаяся часть белка и позволяет прояснить род
и характер контактов с молекулой ДНК или РНК. Оба метода — рентгеноструктур-
ный анализ и спектроскопия ядерного магнитного резонанса (ЯМР) — занимают
центральное место в данной области исследования.
Рентгеноструктурный анализ, который является давно утвердившимся мето-
дом и ведет свою родословную с конца девятнадцатого столетия, основан на диф-
ракции рентгеновских лучей. Рентгеновские лучи имеют очень короткую длину
волны — от 0,01 нм до 10 нм — что в 4 000 раз короче видимого света и сопоставимо
с межатомными расстояниями в химических структурах. Когда пучок рентгеновских
лучей направлен на кристалл, некоторые из рентгеновских лучей проходят прямо
через него, а другие дифрагируют и выходят из кристалла под разными углами, отли-
чающимися от угла их вхождения в кристалл (рис. Т11.1, а). Если кристалл состоит из
многочисленных копий одной и той же молекулы, расположенных в виде регулярной
решетки, то разные рентгеновские лучи дифрагируют подобным образом, в силу
чего возникают перекрывающиеся круги дифрагированных волн, которые интер-
ферируют друг с другом. Чувствительная к рентгеновскому излучению фотографи-
ческая пленка (или электронный датчик), помещенная поперек лучей, показывает
картину пятен (рис. Т11.1, б), то есть дифракционную рентгенограмму, по которой
может быть восстановлена структура молекулы в кристалле. Это возможно потому,
что относительное взаимное положение пятен показывает расположение молекул
в кристалле, а их относительная интенсивность дает информацию о структуре мо-
лекулы. Чем сложнее молекула, тем больше число пятен и больше число сравнений,
которые должны быть сделаны между ними. Поэтому для всех, кроме самых простых,
молекул требуется помощь вычислительных средств. В случае успеха мы получаем
карту электронной плотности (рис. Т11.1, в и г), которая, в случае белка, дает карту
свернутого полипептида, по которой может быть определено расположение таких
особенностей структуры, кака-спирали и p-листы. Если карта достаточно подробна,
то могут быть опознаны боковые группы отдельных аминокислот в полипептиде
и установлена их ориентация друг относительно друга, что позволяет сделать вы-
воды о водородных связях и прочих химических взаимодействиях, имеющих место
в структуре исследуемого белка. В случае удачи эти выводы позволят построить
подробную трехмерную модель белка.
Подобно рентгеноструктурному анализу, ЯМР — давно устоявшийся метод,
появление которого датируется началом двадцатого столетия, а первое описание —
1936 годом. Принцип данного метода основан на том, что вращение заряженного
химического ядра наводит магнитный момент. Будучи помещено в приложенное
электромагнитное поле, вертящееся вокруг своей оси ядро ориентируется одним
Глава 11. Сборка комплекса инициации транскрипции 385
а) Получение дифракционной картины
а) Интерпретация карты электронной
плотности - карта электронной плотности
с разрешением 2 А позволяет различить
R-группу тирозина
Рис. Т11.1. Рентгеноструктурный анализ, а) Рентгеновская дифрактограмма получается за счет про-
пускания рентгеновских лучей через кристалл изучаемой молекулы, б) Дифрактограмма кристаллов
рибонуклеазы, в) Часть карты электронной плотности, построенная по этой дифракционной карти-
не. г) Если карта электронной плотности имеет достаточное разрешение, то возможно распознать
боковые группы отдельных аминокислот, как показано здесь на примере тирозина
из двух способов, названных аир (рис. Т11.2), а-ориентация (которая выровнена
по магнитному полю) обладает немного более низкой энергией. В спектроскопии
ЯМР величина этого разделения по энергии определяется путем измерения частоты
электромагнитного излучения, необходимого для вызова перехода из состояния
а в Р; это значение описывается как резонансная частота изучаемого ядра. Важно
отметить, что хотя ядро каждого типа (например, 1Н, 13С, 15N) имеет свою собствен-
ную специфическую резонансную частоту, измеренная частота нередко немного
отличается от стандартного значения (типично менее чем 10 частей на миллион),
потому что электроны вблизи вращающегося ядра экранируют его до некоторой
степени от приложенного магнитного поля. Такой химический сдвиг (разность
между наблюдаемой энергией резонанса и стандартным значением для изучаемого
ядра) позволяет сделать выводы о химической среде в окрестности ядра и, следо-
вательно, дает информацию о структуре. Конкретные типы анализа (названные
COSY и TOCSY) позволяют опознавать атомы, соединенные химическими связями
с вертящимся ядром; другие виды анализа (например, NOESY) позволяют опознавать
386 Часть 3. Принципы функционирования геномов
атомы, которые близко расположены к вертящемуся ядру в пространстве, но не
связаны с ним непосредственно. Далеко не все химические ядра подходят для ЯМР.
Большинство проектов по ЯМР-спектроскопии белков основаны на исследовании
ядер 1Н и имеют целью определить химическое окружение и ковалентные связи
I
w I 0-спин Повышение
Электромагнитное f ▼ энергии
излучение
НеобХОДИМОЙ
частоты
। а-спин
Рис. Т11.2. Основа ЯМР. Вращающееся ядро может принимать любую из двух ориентаций в приложен-
ном электромагнитном поле. Измеряя частоту электромагнитного излучения, необходимую для вы-
зова перехода а->0, можно определить разнесение по энергии между состояниями а- и 0-спинов
всех атомов водорода, а на основании этой информации — восстановить полную
структуру белка. Эти исследования часто сопровождаются исследованиями белков
с замещением, в которых по крайней мере часть атомов углерода и (или) азота была
замещена редкими изотопами 13С и 15N, так как они тоже дают хорошие результаты
ЯМР-спектроскопии.
При благоприятном стечении обстоятельств ЯМР дает такой же уровень раз-
решения, что и рентгеноструктурный анализ, и, таким образом, предоставляет очень
подробную информацию о структуре белка. Главное преимущество ЯМР состоит
в том, что он работает с молекулами в растворе и, таким образом, избегает проблем,
которые иногда возникают при попытке получить кристаллы белка для рентге-
ноструктурного анализа. Исследования растворов также предлагают большую
гибкость, если цель состоит в том, чтобы исследовать изменения структуры белка,
происходящие во время его сворачивания или в ответ на добавление субстрата. Не-
достаток ЯМР — то, что он подходит только для относительно небольших белков.
Есть несколько причин для этого, одна из которых заключается в необходимости
определения резонансной частоты для всех или, возможно, большего числа ядер
2Н (или других изучаемых ядер). Это связано с тем, что различные ядра имеют
различные химические сдвиги, так что их частоты не должны накладываться. Чем
крупнее белок, тем больше число ядер и больше вероятность того, что частоты будут
накладываться и информация о структуре будет потеряна. Хотя это и ограничивает
применимость ЯМР, этот метод тем не менее очень ценный. Известно множество
интересных белков, которые достаточно малы и пригодны для исследования ме-
тодами ЯМР-спектроскопии, и важная информация может также быть получена
структурным анализом пептидов, которые, хотя и не являются полными белками,
всё же могут служить моделями различных функций активности белка, таких как,
скажем, связывание с нуклеиновой кислотой.
Глава 11. Сборка комплекса инициации транскрипции 387
Таблица 11.2. Связывающиеся с ДНК мотивы
Мотив Примеры белков с таким мотивом
Специфичные к последовательности связы- вающиеся с ДНК мотивы
Семейство спираль-изгиб-спираль [часто его на- зывают сокращенно НТН-домен. — Прим, ред.]
Стандартный спираль-изгиб-спираль Репрессор лактозы у Escherichia coli, репрессор триптофана
Гомеодомен Белок Antennapedia у Drosophila
Парный гомеодомен Фактор транскрипции Рах у позвоночных
Домен POU Регуляторные белки Pit-1, Oct-1 и Oct-2 у по- звоночных
Крыловатый спираль-изгиб-спираль Регуляторный белок GABP высших эукариотов
Домен группы (белков) высокой подвиж- ности (HMG) Белок определения пола SRY у млекопитаю- щих
Семейство цинковых пальцев
Палец Cis2His2 Фактор транскрипции TFIIIA у эукариотов
Многоцистеиновый цинковый палец Семейство стероидных рецепторов у высших эукариотов
Цинковая двуядерная трупа Фактор транскрипции GAL4 у дрожжей
Основный домен Фактор транскрипции GCN4 у дрожжей
Лента-спираль-спираль Репрессоры Met), Arc и Mnt у бактерий
Домен ТВР Связывающийся с блоком ТАТА белок эука- риотов
Димер р-цилиндров Белок папилломовируса Е2
Домен гомологичный Rel (RHB) Фактор транскрипции NF-кВ у млекопитающих
Неспецифичные к последовательности, свя- зывающиеся с ДНК мотивы
Гистонная свертка Гистоны эукариотов
Мотив HU/IHF* Белки HU и IHF бактерий
Полимеразная расселина ДНК- и РНК-полимеразы
♦Примечание: мотив HU/IHF представляет собой неспецифичный к последовательности ДНК-
связывающий мотив в бактериальных белках HU (нуклеоидные упаковочные белки; раздел 8.1.1), но на-
правляет специфичное к последовательности связывание белка HIF (хозяйского фактора встраивания)
(раздел 17.2.1).
и с атомом цинка, который, в свою очередь, удерживает p-лист и а-спираль в над-
лежащих положениях друг относительно друга. Другие варианты цинкового пальца
отличаются структурой пальца, в некоторых отсутствует компонент в лице р-листа,
и они состоят просто из одной или нескольких а-спиралей; точный способ удержания
атома цинка на своем месте также разнится. Например, в многоцистеиновых цинковых
пальцах отсутствуют гистидины и цинковый атом расположен симметрично между
четырьмя цистеинами.
388 Часть 3. Принципы функционирования геномов
Рис. 11.5. Цинковый палец рецептора стероидов. Бо-
ковые группы аминокислот, вовлеченных во взаимо-
действия с атомами цинка, показаны оранжевыми ли-
ниями. Буквы «N» и «С» показывают соответственно
N- и С-конец мотива
Интересная особенность цинкового
пальца — то, что многочисленные копии это-
го пальца иногда встречаются на одном бел-
ке. Несколько белков имеют по два, три или
по четыре пальца, но есть примеры с гораздо
большим числом — 37 в одном из белков жабы.
В большинстве случаев отдельные цинковые
пальцы, как думают, образуют независимые
контакты с молекулой ДНК, но в некоторых слу-
чаях отношения между различными пальцами более сложные. В одной специфической
группе белков — семействе ядерных, или стероидных, рецепторов — две а-спирали,
содержащие шесть цистеинов, совместно удерживают два цинковых атома в одном
ДНК-связывающем домене, более крупном, чем стандартный цинковый палец (рис. 11.5).
Одна из а-спиралей этого мотива, по-видимому, входит в большую бороздку, тогда как
вторая контактирует с прочими белками.
Другие мотивы, связывающиеся с нуклеиновыми кислотами
Были обнаружены другие разнообразные связывающиеся с ДНК мотивы, которые
представлены в различных белках.
• Основный домен — в нем распознающая ДНК структура представлена а-спирал ью,
которая содержит большое число основных аминокислот (например, аргинин,
серин и треонин). Своеобразие этого мотива в том, что а-спираль формируется,
только когда белок взаимодействует с ДНК: в несвязанном состоянии спираль
имеет разорганизованную структуру. Основные домены встречаются в ряде белков
эукариотов, принимающих участие в транскрипции ДНК в РНК.
• Мотив лента-спираль-спираль является одним из немногих мотивов, который
достигает специфичного к последовательности связывания с ДНК, без употре-
бления а-спирали в качестве распознающей структуры. Вместо нее в контакт
с большой бороздкой вступает лента (то есть две нити 0-листа) (рис. 11.6). Моти-
вы лента-спираль-спираль встречаются в некоторых регулирующих гены белках
у бактерий.
• Домен ТВР — к настоящему времени он был открыт только в связывающем блок
ТАТА белке (раздел 11.2.3), от которого он и заимствовал свое название. Как и в
случае мотива лента-спираль-спираль, роль распознающей структуры выполняет
0-лист, но в данном случае главные контакты образуются с малой, а не с большой
бороздкой молекулы ДНК.
Связывающиеся с РНК белки также содержат специфические мотивы, которые
обеспечивают их прикрепление к молекуле РНК. Наиболее важные из них следующие:
• рибонуклеопротеиновый (РНП) домен включает четыре 0-нити и две а-спирали
в порядке 0-а-0-0-а-0. Две центральные 0-нити образуют определяющие контак-
Глава 11. Сборка комплекса инициации транскрипции 389
Рис. 11.6. Мотив лента-спираль-спираль. На рисунке пред-
ставлен мотив лента-сп и рал ь-сп и рал ь гена-репрессора Metl
Escherichia coli, он является димером из двух идентичных бел-
ковых молекул, одна из которых показана серым, а другая —
фиолетовым. р-Нити в левой части структуры вступают в контакт
с большой бороздкой двойной спирали. Буквы «N» и «С» по-
казывают соответственно N- и С-конец мотива
ты с молекулой РНК. РНП домен наиболее рас-
пространенный из РНК-связывающих мотивов
и был обнаружен в более чем 250 белках;
• домен связывания с двунитевой РНК (dsRBD)
подобен РНП домену, но имеет структуру ос—р—р—
Р~а. Функция связывания с РНК заложена между
P-нитью и а-спиралью на конце структуры. Как
подразумевает его название, этот мотив встре-
чается в белках, которые связываются с двуни-
тевой РНК;
• домен к-гомологии имеет структуру р-а-а-р~р-а с функцией связывания между
парой а-спиралей. Он относительно редок, но имеется по крайней мере в одном из
РНК-связывающих ядерных белков.
Помимо этого, ДНК-связывающий гомеодомен может обладать также и связываю-
щим РНК действием в некоторых белках. Один рибосомный белок использует структуру,
подобную гомеодомену, чтобы прикрепляться к рРНК, а некоторые содержащие гомеодо-
мен белки, такие как Bicoid у Drosophila melanogaster (раздел 14.3.4), могут связываться
как с ДНК, так и с РНК.
Связывающиеся с ДНК белки
11.1.2. Определение позиций сайтов связывания с ДНК в геноме
Нередко первое, что удается открыть о ДНК-связывающем белке, — не род белка
сам по себе, но особенности последовательности ДНК, которые этот белок опознает. Это
связано с тем, что генетические эксперименты и исследования молекулярной биологии,
которые мы рассмотрим чуть позже в этой главе, показали, что многие из белков, во-
влеченных в экспрессию генома, связываются с короткими последовательностями ДНК,
лежащими непосредственно выше генов, на которые они воздействуют (рис. 11.7). Это
означает, что последовательность недавно откры-
того гена, при допущении о том, что она включает
и кодирующую ДНК и вышерасположенные области,
обеспечивает непосредственный доступ к сайтам
связывания по крайней мере некоторых из белков,
ответственных за экспрессию этого гена. Исходя из
этого, был разработан ряд методов для определе-
ния местонахождения сайтов связывания с белком
в пределах фрагментов ДНК до нескольких тыс.п.н.
в длину, причем эти методы великолепно работают,
даже если подходящие ДНК-связывающие белки не
были опознаны.
Ген
200 п. н.
Рис. 11.7. Сайты связывания ДНК-
связывающих белков расположены
непосредственно выше гена
390 Часть 3. Принципы функционирования геномов
Торможение в геле позволяет опознавать фрагменты ДНК, которые связы-
ваются с белками
Первый из таких методов основан на существенном различии между электрофоре-
тическими свойствами фрагмента «голой» ДНК и ДНК, связанной с белком. Вспомним,
что фрагменты ДНК разделяются электрофорезом в агарозном геле, потому что меньшие
фрагменты мигрируют через пористую структуру геля быстрее, чем большие фрагменты
(см. техническое примечание 2.2). Если фрагмент ДНК несет связанный с ним белок, то
его подвижность в геле будет затруднена: поэтому комплекс белок-ДНК образует полосу
в положении более близком к отправной точке (рис. 11.8). Это называют торможением
в геле. На практике эксперимент проводят с набором фрагментов рестрикции, кото-
рые покрывают область, предположительно содержащую сайт связывания с белком.
Гидролизат смешивают с экстрактом ядерных белков (при допущении о том, что объ-
ектом исследования служит эукариот) и заторможенные фрагменты опознаются путем
сравнения картины полос, полученной после электрофореза с таковой для фрагментов
рестрикции, которые не были смешаны с белками. Ядерный экстракт используется, по-
тому что на этой стадии проекта ДНК-связывающий белок обычно еще не очищен. Если,
однако, белок все же имеется в наличии, то эксперимент может быть проведен столь же
легко с чистым белком, как и со смешанным экстрактом.
Методы анализа с защитой позволяют определять местоположение сай-
тов связывания с большей точностью
Торможение в геле дает общую картину местоположения связывающегося с бел-
ком участка в последовательности ДНК, но не указывает такой участок с большой
точностью. Часто заторможенный фрагмент имеет несколько сот п.н. в длину, по
сравнению с ожидаемой длиной участка связывания в несколько десятков п. н. самое
большее, и никак не показывает, где именно в заторможенном фрагменте находится
участок связывания. Кроме того, если заторможенный фрагмент длинный, то он
может содержать отдельные участки связывания для нескольких белков, или же,
если он достаточно мал, то есть возможность, что сайт связывания включает также
нуклеотиды смежных фрагментов, которые сами по себе не образуют устойчивый
комплекс с белком и, таким образом, не дадут торможение в геле. Поэтому методы
анализа с торможением в геле служат отправной точкой, но для получения более
точной информации необходимы другие методы.
Методы анализа с защитой от модификации могут выйти на передний план
в сценах, где методы торможения в геле остаются за кулисами. Эти методы основаны
Фрагменты Фрагменты рестрикции
рестрикции + ядерный белок
на том факте, что если молекула ДНК несет на
себе связанный белок, то часть ее нуклеотидной
Рис. 11.8. Анализ с торможением в геле. Экстракт ядер был
смешан с гидролизатом рестрикции ДНК, и связывающий-
ся с ДНК белок в экстракте прикрепился к одному из фраг-
ментов рестрикции. Комплекс ДНК-белок имеет большую
молекулярную массу, чем «голая» ДНК и, таким образом,
продвигается более медленно в ходе гель-электрофореза.
В результате полоса от этого фрагмента отстает и может
быть опознана путем сравнения с картиной полос, об-
разованной фрагментами рестрикции, которые не были
смешаны с экстрактом ядер
Глава 11. Сборка комплекса инициации транскрипции 391
последовательности будет защищена от модификации. Известно два способа осущест-
вления модификации.
• Путем обработки нуклеазой, которая расщепляет все фосфодиэфирные связи,
кроме защищенных связанным белком.
• Посредством подвергания действию метилирующего агента, такого как диметил-
сульфат, который добавляет метильные группы к гуаниновым нуклеотидам. Все
гуанины, защищенные связанным белком, не будут метилированы.
Практические стороны этих двух методов представлены на рис. 11.9 и 11.10. Оба
опираются на экспериментальный подход, названный футпринтингом. В нуклеазном
футпринтинге исследуемый фрагмент ДНК метится на одном конце, объединяется
в комплекс со связывающим белком (в виде ядерного экстракта или чистого белка)
и обрабатывается дезоксирибонуклеазой I (ДНКазой I). Обычно ДНКаза I расщепляет
все фосфодиэфирные связи, оставляя только сегмент ДНК, защищенный связавшимся
с ним белком. Это не очень удобно, потому что может оказаться трудным секвенировать
столь малый ее фрагмент. Быстрее использовать более тонкий подход, представленный
на рис. 11.9. Обработка нуклеазой выполняется при ограничивающих условиях (низкая
Помеченные на одном конце
фрагменты рестрикции
Концевая метка »
i Ограниченное । Ограниченное
переваривание ДНКазой I переваривание ДНКазой I
/Переваривание белка,
гель-электрофорез,
авторадиография
Гель-электрофорез,
авторадиография
Рис. 11.9. Футпринтинге ДНКазой I. Фрагменты рестрикции, используемые в начале процедуры, должны
быть помечены только на одном конце. Этого обычно достигают, обрабатывая набор более длинных
фрагментов рестрикции ферментом, который прикрепляет метки к обоим концам, а затем разрезая эти
помеченные молекулы вторым ферментом рестрикции и очищая один из наборов концевых фрагментов.
Обработка ДНКазой I выполняется в присутствии соли марганца, которая побуждает фермент делать слу-
чайные двунитевые разрезы в целевых молекулах и оставлять после себя тупоконечные фрагменты
392 Часть 3. Принципы функционирования геномов
Помеченные на одном конце
фрагменты рестрикции
Концевая метка *— *—
обработка ДМС
Пиперидин
i Переваривание белка,
пиперидин
Денатурирующий
гель-электрофорез,
авторадиография
Денатурирующий
гель-электрофорез,
авторадиография
Рис. 11.10. Анализ с защитой от модификации диметилсульфатом (ДМС). Метод подобен футпринтингу
с ДНКазой I. Вместо переваривания ДНКазой I фрагменты обрабатываются ограниченными количествами
ДМС, так что в каждом фрагменте метилируется одно гуаниновое основание. Гуанины, которые защищены
связанным белком, не могут быть видоизменены. После удаления белка ДНК обрабатывают пипериди-
ном, который разрезает ее в позициях видоизмененных нуклеотидов. Для простоты диаграмма показыва-
ет двунитевые молекулы, расщепляемые на этой стадии. Фактически они только претерпевают надрезы,
поскольку пиперидин разрезает только нить, которая видоизменена, а не делает двунитевой разрез по
всему сечению молекулы. Поэтому образцы исследуются денатурирующим гель-электрофорезом, с тем
чтобы две нити отделились друг от друга. Полученная в конечном итоге авторадиограмма показывает
размеры нитей, которые имеют один помеченный конец и второй конец, созданный пиперидиновым
надрезом. Картина полос для контрольных нитей ДНК — не выдержанных в я дерном экстракте — по-
казывает позиции гуанинов (G) во фрагменте рестрикции, и футпринт просматривается в картине полос
для опытного образца и показывает, какие основания G были защищены
температура и (или) весьма малое количество фермента), так чтобы в среднем каждая
копия фрагмента ДНК претерпела одно-единственное «попадание», то есть чтобы она
была расщеплена только в одной позиции по ее длине. Хотя каждый фрагмент раз-
резается лишь однократно, во всей совокупности фрагментов будут расщеплены все
связи за изъятием защищенных связанным белком. После этого белок удаляется, смесь
Помеченные на одном конце
фрагменты рестрикции
Концевая метка
। Ограниченная обработка
диметилсульфатом
г г-------Видоизмененный G
I Добавление экстракта ядер
▼ Связывающийся
• 1 с ДНК белок
Глава 11. Сборка комплекса инициации транскрипции 393
подвергается электрофорезу, и меченые фрагменты отображаются. Каждый из этих
фрагментов несет метку на одном конце и участок расщепления на другом. В резуль-
тате мы получаем лесенку полос, соответствующих фрагментам, которые отличаются
по длине на один нуклеотид, а лесенка прерывается чистой областью, на которой не
наблюдается ни одной меченой полосы. Эта чистая область, или «след», соответствует
позициям защищенных фосфодиэфирных связей и, следовательно, связанного белка
в исходной ДНК.
Модификационное препятствование позволяет опознавать нуклеотиды,
критически важные для связывания с белком
Защиту от модификации не следует путать с модификационным препятство-
ванием — другим методом, который открывает дополнительное измерение в изуче-
нии связывания с белком. Моди-
фикационное препятствование
основывается на том, что если
нуклеотид, определяюще важный
для связывания белка, будет видо-
изменен (например, добавлением
метильной группы), то связывание
может быть предотвращено. Один
метод из этой группы представ-
лен на рис. 11.11. Фрагмент ДНК,
помеченный на одном конце, об-
рабатывается модифицирующим
реактивом, в данном случае ди-
метилсульфатом, при ограничи-
вающих условиях так, чтобы был
метилирован только один гуанин
на фрагмент. Теперь добавляются
исследуемый белок или ядерный
экстракт и фрагменты разделяются
гель-электрофорезом. Наблюдают-
ся две полосы: одна соответствует
комплексу белок-ДНК, а вторая
содержит ДНК без связанного бел-
ка. Последняя содержит молеку-
лы, связывание которых с белком
было предотвращено, потому что
метилирующая обработка моди-
фицировала один или более гуа-
нинов, которые являются опреде-
ляюще важными для связывания.
Для того чтобы определить, какие
именно гуанины модифицирова-
лись, фрагмент очищают от геля
и обрабатывают пиперидином —
соединением, которое расщепля-
ет ДНК в позициях нуклеотидов
днк- - Связывание не происходит
маркеры (участок блокирован)
Гель-электрофорез
Незаторможенная полоса
(без связанного белка)
| Пиперидин
ДНК-
маркеры
\| Гель-электрофорез,
▼ авторадиография
Полоса меченого фрагмента
соответствует 200 п. н.,
поэтому...
200 п. н.
Рис. 11.11. Анализ с защитой от модификации диметилсуль-
фатом (ДМС). См. подпись к рис. 11.9 для описания про-
цедуры, используемой для получения фрагментов ДНК, по-
меченных только на одном конце
394 Часть 3. Принципы функционирования геномов
метилгуанинов. В результате такой обработки каждый фрагмент разрезается на два
сегмента, один из которых несет метку. Длины меченых сегментов, определенные во
втором круге электрофореза, говорят нам, какие нуклеотиды в первоначальном фраг-
менте были метилированы, и, следовательно, позволяет опознать позиции гуанинов
в последовательности ДНК, которые участвуют в реакции связывания. Подобные ме-
тоды могут быть использованы для опознавания нуклеотидов А, С и Т, участвующих
в связывании1^.
11.1.3. Взаимодействие между ДНК и связывающимися с ней белками
В последние годы наше понимание роли, играемой молекулой ДНК во взаимодей-
ствии со связывающимся с ней белком, начало изменяться. Всегда признавалось, что
белки, которые опознают специфическую последовательность в качестве своего сайта
связывания, могут определить местонахождение этого сайта путем наведения контактов
с химическими группами, прикрепленными к основаниям, которые выходят на вну-
треннюю поверхность большой и малой бороздок, вьющихся вокруг двойной спирали
(см. рис. 1.8). Теперь признано, что последовательность нуклеотидов также влияет на
точную конформацию каждой области спирали и что такие особенности конформации
представляют собой второй, менее прямой путь, которым последовательность ДНК
может влиять на связывание с белком.
Прямое считывание последовательности нуклеотидов
Исходя из структуры двойной спирали, описанной Уотсоном и Криком (раздел 1.1.2),
было ясно, что хотя основания нуклеотидов находятся внутри молекулы ДНК, они не
полностью погребены в ее недрах, и некоторые из химических групп, прикрепленных
к пуриновым и пиримидиновым основаниям, доступны с внешней стороны спирали.
Поэтому должно быть возможно прямое считывание последовательности нуклеоти-
дов — без разрыва пар оснований и раскрытия молекулы.
Для того чтобы образовывать химические связи с группами, прикрепленными
к основаниям нуклеотидов, связывающийся белок должен образовывать контакты
в пределах одной или обеих бороздок на поверхности спирали. В случае В-формы ДНК,
род и ориентация открытых частей оснований в пределах большой бороздки таковы,
что большая часть последовательностей может считываться однозначно, тогда как
в пределах малой бороздки возможно определить, представлена ли каждая очередная
пара оснований парой А-Т или G-С, но трудно распознать, какой нуклеотид пары на-
ходится в которой из нитей спирали (рис. 11.12). Поэтому прямое считывание В-формы
преимущественно вовлекает контакты в большой бороздке. Что до прочих типов ДНК,
то имеется намного меньше информации о контактах, образуемых со связывающимися
Здесь не описан еще один популярный метод - SELEX. Идея метода заключается в следующем. Синте-
зируется множество коротких случайных молекул ДНК (размером около 20-30 п.н.]. Эти случайные молекулы
фланкируются фиксированными сегментами (по 15-20 п.н.]. Исследуемый белок иммобилизируется. Затем
в предельно мягких условиях эта смесь большого количества разнообразных молекул прокачивается через
белок. Часть наших фрагментов, которая имеет чуть большее сродство к исследуемому белку, свяжется с ним.
Мы смываем связавшиеся молекулы ДНК и делаем ПЦР, причем так, чтобы реакция делала ошибки. Тем са-
мым мы размножаем и мутируем молекулы, которые имеют хоть какое сродство к нашему белку. Процедуру
повторяем — прокачиваем-смываем-размножаем с ошибками. На каждом цикле происходит обогащение
нашей смеси молекулами ДНК, которые имеют сродство к белку. После 5-7 циклов секвенируем то, что оста-
лось, и получаем набор последовательностей, связывающихся с белком. Еще один, популярный в последнее
время метод — иммуноперципитация хроматина СЫР, при котором манипуляции производятся в условиях,
наиболее приближенных к условиям в клетке. После манипуляций используют либо микрочипы (ChlP-Chip)
либо массовое секвенирование с помощью приборов нового поколения (ChlP-Seq). — Прим. ред.
Глава 11. Сборка комплекса инициации транскрипции 395
Рис. 11.12. Опознавание пары оснований А-Т в двойной спи-
рали В-формы. Пара оснований А-Т показана схематично
(рис. 1.8, б), где стрелки указывают химические особенности,
которые могут быть опознаны при доступе к этой паре осно-
ваний через большую бороздку (вверху) и малую бороздку
(внизу). В большой бороздке химические особенности асим-
метричны, и ориентация пары А-Т может быть распознана
связывающимся с ДНК белком. Некоторое время полагали,
что это невозможно в малой бороздке, потому что только две
особенности, показанные серым цветом, как думали, имели
место, а они являются симметричными. Используя эти две
особенности, связывающийся белок мог распознать пару осно-
ваний А-Т, но не мог знать, какой из ее нуклеотидов находится
в которой из нитей спирали. Недавно были обнаружены асим-
метричные особенности, показанные зеленым; это позволяет
предположить, что ориентация пары может фактически быть
определимой через малую бороздку. Сокращения: а — акцеп-
тор водородной связи; д — донор водородной связи; вдВ —
взаимодействие ван дер Ваальса
белками, но картина, вероятно, будет совершенно иной. В A-форме, например, большая
бороздка глубокая и узкая и менее удобна для проникновения в нее какой-либо части
белковой молекулы (см. табл. 1.1). Поэтому более мелкая малая бороздка с большей
вероятностью будет исполнять ведущую партию в постановке прямого считывания.
В Z-ДНК большая бороздка фактически отсутствует и до некоторой степени возможно
осуществить прямое считывание, оставаясь на поверхности спирали.
Последовательность нуклеотидов оказывает ряд косвенных воздействий
на структуру спирали
Первоначально думали, что молекулы клеточной ДНК имеют довольно однородные
структуры, представленные главным образом В-формой двойной спирали. Некоторые
короткие сегменты могли находиться в A-форме, и могли быть некоторые отрезки
Z-ДНК, в особенности около концов молекулы, но большая часть длины двойной спи-
рали находится в виде неизменной В-ДНК. Теперь мы признаем, что ДНК намного более
полиморфна и что в пределах одной молекулы ДНК возможно сосуществование раз-
личных конфигураций (А-, В- и Z-ДНК), а также промежуточных форм, при этом разные
части молекулы будут иметь различную структуру. Такие конформационные варианты
зависят от последовательности, будучи в значительной степени результатом стекинг-
взаимодействия оснований, которые происходят между смежными парами оснований.
Помимо ответственности за устойчивость спирали, наряду со спариванием оснований,
стекинг оснований влияет также на угол поворота, происходящего вокруг ковалентных
связей в пределах отдельных нуклеотидов, и, следовательно, определяет конформацию
спирали в каждой конкретной позиции. На свободу вращения одной пары оснований
влияет, посредством стекинг-взаимодействий оснований, род соседних пар оснований.
Это означает, что последовательность нуклеотидов косвенно оказывает влияние на
общую конформацию спирали и, возможно, обеспечивает тем самым структурную ин-
формацию, которую связывающийся белок может использовать, чтобы облегчить себе
определение местонахождения соответствующего сайта связывания на молекуле ДНК.
В настоящее время это лишь теоретическая возможность, так как не было установлено
396 Часть 3. Принципы функционирования геномов
ни одного белка, который бы специфично опознавал спираль не В-формы, но многие
исследователи находят весьма вероятным, что конформация спирали играет некоторую
роль во взаимодействии между ДНК и белком.
Изменение конформации второго типа — изгибание ДНК. Оно относится не к есте-
ственной гибкости ДНК, которая позволяет ей образовывать кольца и сверхспирали,
а к соседним позициям, в которых последовательность нуклеотидов заставляет ДНК
изгибаться. Подобно другим конформационным вариантам, изгибание ДНК зависит
от последовательности. В частности, молекула ДНК, в которой один полинуклеотид
содержит две и более группы повторных аденинов, — каждая группа включает 3-5
аденинов, а отдельные группы отделены друг от друга 10 или 11 нуклеотидами, — изо-
гнется вблизи З'-конца обогащенной аденином области. Как и с конформацией спирали,
еще не известно, до какой степени изгибание ДНК влияет на связывание с белком, хотя
вызванное белком изгибание в гибких участках имеет явно демонстрируемую функцию
в регулировании некоторых генов (раздел 11.3.2).
Точки соприкосновения между ДНК и белками
Контакты, образуемые между ДНК и связывающимися с нею белками, являются
нековалентными. В пределах большой бороздки между основаниями нуклеотидов
и боковыми группами аминокислот в распознающей структуре белка образуются водо-
родные связи, тогда как в малой бороздке первостепенную роль играют гидрофобные
взаимодействия. На поверхности спирали ДНК основными взаимодействиями являются
электростатические — между отрицательными зарядами на фосфатной составляющей
каждого нуклеотида и положительными зарядами на боковых группах аминокислот, та-
ких как лизин и аргинин, — хотя отчасти происходит и образование водородных связей.
В некоторых случаях водородные связи на поверхности спирали ил и в большой бороздке
образуются напрямую между ДНК и белком; в других они опосредствуются молекулами
воды. Можно сделать ряд обобщений: на этом уровне взаимодействия между белком
и ДНК каждый пример имеет свои собственные уникальные качества и особенности
образования связей необходимо устанавливать посредством структурного анализа, а не
выводить на основании сравнения с другими белками.
Большинство белков, которые опознают специфические последовательности ну-
клеотидов, способны также неспецифично связываться с другими частями молекулы
ДНК. Фактически было предположено, что количество ДНК в клетке является настолько
большим, а число молекул каждого ДНК-связывающего белка столь мало, что белки
проводят если и не все свое время, то по крайней мере львиную долю его в состоянии не-
специфичного связывания с ДНК. Различие между формами неспецифичного и специфич-
ного связывания состоит в том, что последнее более благоприятно в термодинамическом
отношении. В результате белок способен связываться со своим специфичным участком
даже при том, что имеются буквально миллионы других участков, с которыми он мог бы
связаться неспецифично. Чтобы достичь такого термодинамически благоприятного по-
ложения, процесс специфичного связывания должен вовлекать наибольшее возможное
число контактов ДНК-белок, что объясняет отчасти, по какой причине распознающие
структуры многих ДНК-связывающих мотивов эволюционировали таким образом,
чтобы плотно входить в большую бороздку спирали, где возможность образования
контактов ДНК-белок является наибольшей. Это объясняет также, почему некоторые
взаимодействия ДНК-белок вызывают конформационные изменения у одного или
второго партнера, еще более увеличивая комплементарность взаимодействующих по-
верхностей и способствуя образованию дополнительных связей.
Глава 11. Сборка комплекса инициации транскрипции 397
Необходимость увеличить число контактов, с тем чтобы обеспечить специфич-
ность, также является одной из причин, по которой многие ДНК-связывающие белки
представляют собой димеры, состоящие из двух белков, присоединенных друг к другу.
Дело обстоит так для большинства белков с мотивом НТН и для многих с цинковыми
пальцами. Димеризация происходит таким образом, что ДНК-связывающие мотивы этих
двух белков оба способны получить доступ к спирали, возможно с некоторой степенью
кооперативности между ними, так что фактическое число контактов на деле оказывается
больше, чем удвоенное число, достижимое мономером. Так же как их ДНК-связывающие
мотивы, многие белки содержат дополнительные характерные домены, участвующие
в межбелковых контактах, которые обусловливают образование димера. Один из таких
доменов — лейциновая застежка-молния, который представляет собой а-спираль,
скрученную сильнее, чем обычно, и выставляющую ряд лейцинов на одной из своих
сторон. Они могут образовывать контакты с лейцинами застежки-молнии на втором
белке, формируя димер (рис. 11.13). Второй димеризующий домен, который, пожалуй,
неудачно назван мотивом спираль-петля-спираль (не путать с ДНК-связывающим
мотивом спираль-изгиб-спираль!).
Интригующий вопрос: может ли специфичность связывания с ДНК быть понята
достаточно исчерпывающе, чтобы последовательность целевого участка белка можно
было предсказывать на основании данных анализа структуры распознающей спирали
ДНК-связывающего мотива? До сих пор эта цель почти совершенно ускользала от нас,
но оказалось возможным вывести некоторые правила взаимодействия, вовлекающего
цинковые пальцы некоторых типов. В этих белках четыре аминокислоты — три в рас-
познающей спирали и одна непосредственно смежная с ней — образуют критически
важные контакты с основаниями нуклеотидов целевого участка. Некоторые из этих
соединений вовлекают одну аминокислоту с одним основанием, другие вовлекают две
аминокислоты с одним основанием. Путем сравнения последовательности аминокислот
в распознающих спиралях различных цинковых пальцев с последовательностями ну-
клеотидов на сайтах связывания было возможно определить набор правил, управляющих
таким взаимодействием. Они позволяют предсказывать специфичность нового белка
с цинковыми пальцами к последователь-
ности нуклеотидов (правда, возможна
некоторая неопределенность), если со-
став аминокислот распознающей спи-
рали этого белка известен.
Рис. 11.13. Лейциновая застежка-молния. Это
лейциновая застежка-молния типа bZIP. Красные
и оранжевые структуры — части разных бел-
ков. Каждый набор сфер представляет боковые
группы лейциновой аминокислоты. Лейцины из
обеих спиралей связываются друг с другом че-
рез гидрофобные взаимодействия, с тем чтобы
скрепить эти два белка в единый димер. В этом
примере димеризирующиеся спирали вытяну-
ты, чтобы формировать пару связывающихся
с ДНК мотивов основного домена, как показано,
образующих контакты в большой бороздке
398 Часть 3. Принципы функционирования геномов
11.2. Взаимодействия белков с ДНК в ходе инициации транс-
крипции
Теперь, когда мы установили, что взаимодействия между белком и ДНК являются
ключом к пониманию инициации транскрипции, мы можем продвинуться дальше и на-
чать обзор событий, происходящих в ходе сборки инициаторного комплекса. Мы сделаем
это в два этапа. Сначала мы изучим взаимодействия типа белок-ДНК, которые вовлечены
в инициацию транскрипции. Затем, в разделе 11.3, мы исследуем, как сборка инициа-
торного комплекса и его способность инициировать транскрипцию могут управляться
различными дополнительными белками, которые реагируют на сигналы, поступающие
изнутри или извне клетки, и гарантируют, что в соответствующие моменты времени
будут транскрибироваться надлежащие гены.
11.2.1. РНК-полимеразы
В разделе 1.2.1 мы узнали, что ферменты, ответственные за транскрипцию ДНК
в РНК, называют ДНК-зависимыми РНК-полимеразами. Транскрипция ядерных ге-
нов эукариотов требует три различные РНК-полимеразы: РНК-полимеразу I, РНК-
полимеразу II и РНК-полимеразу III. Каждая из них представляет собой многосубъеди-
ничный белок (8-12 субъединиц) с молекулярной массой свыше 500 кДа. В структурном
отношении эти полимеразы весьма подобны друг другу—три наибольшие субъединицы
близкородственны, а некоторые из меньших были обнаружены в более чем одном фер-
менте; однако что касаемо функций, то они весьма различны. Каждая обслуживает свой
набор генов, без какой-либо взаимозаменяемости (таблица 11.3). Наибольшее внимание
исследователей было направлено на РНК-полимеразу II, поскольку именно этот фер-
мент транскрибирует гены, кодирующие белки. Она работает также с набором генов,
определяющих мяРНК, которые вовлечены в обработку РНК, и с генами для миРНК.
РНК-полимераза III транскрибирует остальные гены малых РНК, в том числе гены тРНК.
РНК-полимераза I транскрибирует многокопийные повторные единицы, содержащие
гены рРНК 28S, 5,8S и 18S. Функции всех этих РНК были подытожены в разделе 1.2.2
и подробно описываются в главах 12 и 13.
Археи обладают единственной РНК-полимеразой, которая по своей структуре
весьма подобна ферментам эукариотов. Но это не типично для прокариотов в целом,
потому что бактериальная РНК-полимераза весьма многолика, хотя и состоит всего лишь
из пяти субъединиц, при этом ее состав описывается как а2РР'о (две а-субъединицы,
по одной р- и родственной ей Р’- и одна о-субъединица). Субъединицы а, р и Р' эквива-
лентны трем наибольшим субъединицам РНК-полимераз эукариотов, но о-субъединица
имеет свои собственные особые свойства, как в отношении ее структуры, так и, как мы
Таблица 11.3. Функции трех ядерных РНК-полимераз эукариотов
Полимераза Транскрибируемые гены
РНК-полимераза I Гены рибосомных РНК (рРНК) 28S, 5,8S и 18S
РНК-полимераза II Кодирующие белок гены, большая часть генов малой ядерной РНК (мяРНК), гены микроРНК (миРНК)
РНК-полимераза III Гены транспортных РНК (тРНК), рРНК 5 S, мяРНК U6, малой ядрышковой РНК (мякРНК)
Глава 11. Сборка комплекса инициации транскрипции 399
увидим в следующем разделе, в плане ее функции. РНК-полимераза, очень похожая на
бактериальный фермент, обнаружена также в хлоропластах, что отражает бактериальное
происхождение этих органелл (раздел 8.3.1). Интересно, однако, что митохондриальная
РНК-полимераза, которая состоит из одной-единственной субъединицы с молекулярной
массой 140 кДа, имеет более близкое родство с РНК-полимеразами некоторых бакте-
риофагов, чем со стандартной бактериальной версией1^.
11.2.2. Последовательности, распознаваемые при инициации транс-
крипции
Очень важно, чтобы комплексы инициации транскрипции возводились на моле-
кулах ДНК в правильных позициях. Эти позиции отмечены целевыми последователь-
ностями, которые опознаются или самой РНК-полимеразой, или ДНК-связывающим
белком, который, как только прикрепится к ДНК, образует площадку, на которую и са-
дится РНК-полимераза (см. рис. 11.14).
а) Непосредственное прикрепление Опосредствованное
РНК-полимеразы прикрепление РНК-полимеразы
РНК-полимераза
РНК-полимераза
Площадка, образуемая
связывающимся с ДНК белком
Рис. 11.14. Два способа, которыми РНК-полимеразы связываются со своими промоторами, а) Прямое
опознавание промотора РНК-полимеразой, что имеет место у бактерий, б) Опознавание промотора
связывающимся с ДНК белком, что формирует платформу, на которую садится РНК-полимераза. Этот
косвенный механизм работает в случае РНК-полимераз эукариотов и архей
Бактериальные РНК-полимеразы связываются с последовательностями
промоторов
У бактерий целевую последовательность для прикрепления РНК-полимеразы
называют промотором. Этот термин впервые был употреблен генетиками в 1964 г.
для описания функции локуса, лежащего непосредственно выше трех генов оперона
лактозы (рис. 11.15). Когда этот локус был инактивирован мутацией, гены в опероне
не экспрессировались; поэтому казалось, что этот локус способствовал* 2^ экспрессии
этих генов. Теперь мы знаем, что это связано с тем, что данный локус является сайтом
связывания для РНК-полимеразы, которая транскрибирует данный оперон.
Последовательности, из которых состоит промотор Escherichia coli, вначале были
определены путем сравнения областей, расположенных выше более чем 100 генов.
Предполагалось, что последовательности промоторов будут весьма подобны для всех
Есть еще целый ряд полимераз и субъединиц полимераз из бактериофагов. — Прим. ред.
2) В английском языке слова promote (способствовать) и promotor (способствующий агент) являются
однокоренными и выступают звеньями единой логической цепочки. Это как если бы наши законодатели
биологической терминологии нарекли саму последовательность споспешником, а ее действие наименовали
споспешествованием! — Прим, перев.
400 Часть 3. Принципы функционирования геномов
Рис. 11.15. Промотор оперона лактозы Escherichia coli. Промотор расположен непосредственно выше
lacZ, первого гена в опероне. Последовательность ДНК показывает позиции блоков -35 и -10 — двух
различных компонентов последовательности промотора. Сравните эти последовательности с консен-
сусными последовательностями, описанными в тексте. Более полные сведения об опероне лактозы
можно разглядеть на рис. 8.8, а
генов и, следовательно, должны быть распознаваемыми при сравнении областей, ле-
жащих выше различных генов. Эти исследования показали, что промотор Е. coli состоит
из двух сегментов, оба из шести нуклеотидов, описываемых следующим образом (см.
рис. 11.15):
Блок-35 5’-TTGACA-3'
Блок-10 5'-ТАТААТ-3'
Это не что иное, как консенсусные последовательности и, таким образом, они
описывают «усредненную» промоторную последовательность Е. coli: фактические по-
следовательности выше всякого конкретного гена могут немного отличаться (табли-
ца 11.4). Названия блоков указывают на их позиции относительно точки начала транс-
крипции. Нуклеотид в этой точке отмечен «+1» и находится где-нибудь между 20 и 600
нуклеотидами выше начала кодирующей области гена. Интервал между этими двумя
блоками важен, потому что он размещает эти два мотива на одной и той же стороне
двойной спирали, что облегчает их взаимодействие с ДНК-связывающим компонентом
РНК-полимеразы (раздел 11.2.3).
Промоторы эукариотов более сложные
В отношении эукариотов термин «промотор» употребляется для описания всех
последовательностей, играющих важную роль в инииации транскрипции гена. Для
некоторых генов такие последовательности могут быть многочисленными и обладать
разнообразными функциями, включая не только центральный промотор, иногда
называемый основным, или базальным промотором, представляющий собой уча-
сток, на котором собирается инициаторный комплекс, но также и один или несколько
вышележащих промоторных элементов, которые, как подразумевает их название,
лежат выше базального промотора. Сборка инициаторного комплекса на базальном
промоторе, как правило, может происходить и в отсутствие вышележащих элементов, но
лишь неэффективным способом. Это указывает на то, что белки, которые связываются
с вышележащими элементами, включают по крайней мере некоторые, которые явля-
ются активаторами транскрипции и которые поэтому «способствуют» экспрессии гена.
Поэтому вложение этих последовательностей в тело «промотора» вполне оправдано.
Каждая из трех РНК-полимераз эукариотов опознает промоторные последователь-
ности своего типа; в самом деле, именно различие между промоторами определяет, какие
Глава 11. Сборка комплекса инициации транскрипции 401
Таблица 11.4. Последовательности промоторов Escherichia coli
Промотор Последовательность
Блок -35 Блок -10
Консенсус 5'-TTGACA-3' 5'-ТАТААТ-3'
Оперон лактозы 5'-ТТТАСА-3' 5’-TATGTT-3’
Оперон триптофана 5’-ТТСАСА-3' 5’-ТТААСТ-3’
гены транскрибируются которыми полимеразами. Ниже приведены подробности для
промоторов позвоночных (рис. 11.16).
• Промоторы для РНК-полимеразы I состоят из базального промотора, покрываю-
щего точку начала транскрипции, на отрезке между нуклеотидами -45 и +20,
и вышележащего элемента управления (ВЭУ), расположенного примерно на
100 п.н. еще выше.
• Промоторы для РНК-полимеразы II изменчивы и могут простираться на несколь-
ко тыс. п. н. выше участка начала транскрипции. Базальный промотор состоит из
двух главных сегментов: блока -25, или ТАТА-блока (консенсус 5'-TATAWAAR-3',
где W — А или Т, a R — А или G), и инициаторной последовательности (кон-
сенсус у млекопитающих S'-YCANTYY-B’, где У — С или Т, a N — любой нуклео-
тид), расположенных вокруг нуклеотида +1. Некоторые гены, транскрибируемые
РНК-полимеразой II, имеют только один из этих двух компонентов базального
промотора, а некоторые, как это ни удивительно, не имеют ни одного. Последних
величают «нулевыми» генами. Они тем не менее транскрибируются, хотя позиция
начала транскрипции у них более изменчива, чем у гена с ТАТА-блоком и (или)
с инициаторной последовательностью. Некоторые гены имеют дополнительные
последовательности, которые иногда рассматриваются как часть базального про-
мотора. Примеры:
• Нижележащий промоторный элемент (НПЭ; расположенный в позициях с +28 по
+32), который обладает изменчивой последовательностью, но был определен бла-
годаря его способности связывать TFIID — белковый комплекс, который играет
центральную роль в предынициаторном комплексе (раздел 11.2.3).
Рис. 11.16. Структуры промоторов эукариотов. Расшифровка сокращений приведена в тексте
402 Часть 3. Принципы функционирования геномов
• Обогащенный GC мотив длиной 7 п. н., примыкающий сверху к ТАТА-блоку, который
опознается комплексом TFIIB — еще одним компонентом предынициаторного
комплекса.
• Проксимальный элемент последовательности, который расположен меж-
ду позициями -45 и -60 выше тех генов мяРНК, которые транскрибируются
РНК-полимеразой II.
Наряду с компонентами базального промотора, гены, транскрибируемые РНК-
полимеразой II, имеют различные вышележащие промоторные элементы, функции
которых описаны в разделе 11.3.2.
• Промоторы для РНК-полимеразы III являются изменчивыми и подпадают по край-
ней мере под три категории. Две из этих категорий необычны в том, что важные
последовательности расположены в пределах генов, транскрипции которых они
способствуют. Обычно эти последовательности охватывают 50-100 п. н. и включают
два консервативных блока, разделенных изменчивой областью. Третья категория
промоторов для РНК-полимеразы III подобна таковым для РНК-полимеразы II;
они имеют ТАТА-блок и веер дополнительных промоторных элементов (иногда
включающих упомянутый выше проксимальный элемент), расположенных выше
целевого гена. Интересно, что такое расположение наблюдается у гена U6, который
является единственным геном мяРНК, транскрибируемым РНК-полимеразой 111;
все остальные гены мяРНК транскрибируются РНК-полимеразой II.
11.2.3. Сборка комплекса инициации транскрипции
В общем смысле инициация транскрипции работает по одной и той же схеме
с РНК-полимеразами всех четырех типов, которые мы рассматривали (рис. 11.17). Бак-
териальная полимераза и три фермента эукариотов одинаково начинают свое дело
с прикрепления (непосредственно или через вспомогательные белки) к последователь-
ности своего базального промотора. Далее, этот закрытый промоторный комплекс
преобразуется в открытый промоторный комплекс путем разрыва ограниченного
числа пар оснований вокруг сайта инициации транскрипции. Наконец, РНК-полимераза
отодвигается от промотора. Этот последний шаг более сложен, чем может показаться,
потому что некоторые попытки полимеразы достичь очистки промотора неудачны
и ведут к усеченным транскриптам, которые деградируют вскоре после своего синтеза.
Поэтому настоящее завершение стадии инициации транскрипции наступает при уста-
новлении устойчивого комплекса транскрипции, который активно транскрибирует ген,
к которому он присоединен.
Хотя схема, показанная на рис. 11.17, верна в общем виде для всех четырех РНК-
полимераз, отдельные моменты отличны для каждой из них. Мы начнем с более простых
событий, происходящих у Е coli и других бактерий, а уж затем перейдем к усложненным
вариантам инициации транскрипции у эукариотов.
Инициация транскрипции у Е. coli
У Е coli образуется прямой контакт между РНК-полимеразой и промотором. Спец-
ифичность полимеразы к последовательности определяется ее п-субъединицей: «цен-
тральный фермент», у которого отсутствует этот компонент, может осуществлять л ишь
неплотные и неспецифичные прикрепления кДНК.
Мутационный анализ промоторов Е. coli показал, что изменения в последователь-
ности блока -35 нарушают способность РНК-полимеразы связываться, тогда как изме-
Глава 11. Сборка комплекса инициации транскрипции 403
Рис. 11.17. Обобщенная схема событий,
происходящих в ходе инициации транс-
крипции. Центральный промотор показан
зеленым, а участок инициации транскрип-
ции обозначен красной точкой. После при-
крепления РНК-полимеразы закрытый ком-
плекс преобразуется в открытый комплекс
за счет разрыва пар оснований в пределах
короткой области двойной спирали ДНК.
Синтез РНК начинается, но успешная ини-
циация не может быть достигнута, пока по-
лимераза не отодвинется от премоторной
области
Центральный промотор Позиция +1
ДНК I /
. ........... 1’ТНППГГГТТт
I РНК-полимераза прикрепляется
РНК-полимераза ▼ к центральному промотору
I I ПИШИ I IIIIIIIIIII
I Преобразование закрытого
▼ премоторного комплекса в открытый
нения в блоке -10 подрывают преоб-
разование закрытого промоторного
I Запуск
ф синтеза РНК
комплекса в его открытую форму. На
основании этих результатов была
построена модель инициации транс-
крипции у Е. coli, представленная на
рис. 11.18, в которой опознавание
промотора происходит за счет взаи-
модействия между о-субъединицей
и блоком -35, после чего образуется
закрытый промоторный комплекс,
в котором РНК-полимераза охва-
тывает приблизительно 80 п.н. от
вышележащего блока -35 до нижележащего
блока -10. Закрытый промоторный комплекс
преобразуется в открытую форму за счет со-
вместного действия Р'- и о-субъединиц, кото-
рые разрывают пары оснований в пределах
блока -10. Модель согласуется с тем фактом,
что блоки -10 различных промоторов состоят
главным образом (или даже полностью) из пар
блок-35 блок-10 Позиция+1
дик \ I /
ПНИЩ
РНК-полимераза
опознает блок -35
и..... К
Рис. 11.18. Инициация транскрипции у Escherichia coli.
РНК-полимераза Е. coli опознает блок -35 в качестве
своей последовательности для связывания. После
прикрепления к ДНК переход из закрытого в откры-
тый комплекс запускается разрывом пар оснований
в обогащенном АТ блоке -10. Обратите внимание, что,
хотя полимераза показана в виде цельной структуры,
именно а-субъединица обладает специфичным к после-
довательности действием связывания с ДНК и поэтому
опознает последовательность блока -35. Последующие
события, ведущие к очистке промотора, изображены
на рис. 11.17
ГГТТТТТТТТП1ЩТ1 (III 1?! ГТТГГТ1 ГГ Г11
I Раскрытие спирали
ф происходит в блоке -10
404 Часть 3. Принципы функционирования геномов
оснований А-Т, которые слабее, чем пары G-С, будучи связаны всего двумя водородными
связями в противоположность трем (см. рис. 1.8, б).
Раскрытие спирали вовлекает контакты между полимеразой и нематричной ни-
тью (то есть той, что не копируется в РНК), и снова центральную роль здесь играет
о-субъединица. Однако п-субъединица далеко не всеопределяющая, потому что она
обычно (но не всегда) диссоциирует вскоре после завершения инициации, преобразуя
голофермент (а2РР’о) в центральный фермент (а2РР'), который выполняет стадию
элонгации транскрипции (раздел 12.1.1). Первоначально центральный фермент по-
крывает приблизительно 60 п.н. ДНК, но вскоре после начала элонгации полимераза
подвергается второму конформационному изменению, сокращая свою зону охвата до
всего лишь 30-40 п. н.
Инициация транскрипции для РНК-полимеразы //
Что показывает сравнение простого для понимания ряда событий, происходящих
у Е. coli, с аналогичными процессами у эукариотов? Изучение РНК-полимеразы II пока-
жет нам, что инициация транскрипции у эукариотов вовлекает больше белков и имеет
прочие сложности.
Первое отличие между инициацией транскрипции у Е coli и у эукариотов заключает-
ся в том, что РНК-полимеразы эукариотов не опознают непосредственно последователь-
ности своих базальных промоторов. Для генов, транскрибируемых РНК-полимеразой II,
первоначальный контакт осуществляется общим фактором транскрипции (GTF) TFIID,
который является комплексом, состоящим из связывающегося с ТАТА-блоком белка
(ТВР) и по крайней мере 12 связанных с ТВР факторов, или TAF. ТВР — специфичный
к последовательности белок, который связывается с ДНК через его необычный ТВР-
домен (раздел 11.1.1), который вступает в контакт с малой бороздкой в области ТАТА-
блока. Рентгеноструктурный анализ ТВР показывает, что он имеет седловидную форму,
которая частично облегает двойную спираль, образуя площадку, на которой может быть
собрана остальная часть инициаторного комплекса (рис. 11.19).
Рис. 11.19. В результате прикрепления ТВР к блоку
ТАТА формируется площадка, на которой может быть
собран комплекс инициации. Димер из белков ТВР
показан коричневым цветом, а ДНК — серебряным.
Изображение любезно предоставлено Суном Танем
из университета штата Пенсильвания
TAF содействуют прикреплению
ТВР к ТАТА-блоку и в единении с други-
ми белками, названными зависимыми
от TAF и инициаторной последова-
тельности кофакторами (TIC), воз-
можно, также участвуют в опознавании
инициаторной последовательности,
в особенности в тех промоторах, в кото-
рых отсутствует ТАТА-блок. Белки TAF
любопытны и, кажется, играют мно-
жество разнообразных ролей во время
инициации транскрипции, а также в ходе
других событий, которые предполагают
сборку многобелковых комплексов на
геноме. Пять из TAF дрожжей присут-
ствуют также и в SAGA — одном из ги-
стонацетилтрансферазных комплексов,
которые мы встречали в разделе 10.2.1,
a TAF1 Drosophila melanogaster обладает
действием киназы, которое позволяет
Глава 11. Сборка комплекса инициации транскрипции 405
ему фосфорилировать серин-33 гистона Н2В, активируя тем самым экспрессию смежных
генов (раздел 10.2.1). Белки TAF были причастны также и к управлению клеточным
циклом у различных эукариотов и регулированию возрастных изменений, которые
приводят к образованию гамет у животных. Ключ к пониманию того, как белки TAF
выполняют свои многообразные роли, был выкован в ходе структурного анализа,
который показал, что по крайней мере три из них содержат гистонную свертку — не-
специфичный к последовательности ДНК-связывающий мотив, состоящий из длинной
а-спирали, продолжаемой с обеих сторон двумя более короткими а-спиралями, что
является характерной особенностью гистонов (см. табл. 11.2). В комплексе, образуемом
между TAFn42 и TAFn62, две гистонные свертки ориентированы почти так же, как и в ги-
стоновом димере НЗ/Н4, расположенном в центральном тетрамере стержневой частицы
нуклеосомы (рис. 11.20). Было предположено, что эти TAF могут обладать способностью
образовывать ДНК-связывающую структуру, напоминающую нуклеосому, и что такая
псевдонуклеосома служит площадкой для сборки инициаторного комплекса. Эта догадка
привлекательна, но необходимы дальнейшие исследования, чтобы выяснить, распро-
страняются ли подобия между TAF и гистонами на элементы связывания инициаторного
комплекса и его целевой ДНК. В TAF отсутствуют некоторые аминокислоты, которые
считаются существенно необходимыми для стабилизации контактов между настоящи-
ми нуклеосомами и ДНК, и обнаруженные подобия могут попросту отражать аналогию
в межбелковых взаимодействиях в пределах инициаторного комплекса, и нуклеосомы
и вовсе не имеют прямого отношения к собственно связыванию ДНК.
После прикрепления ТВР к центральному промотору образуется предынициатор-
ный комплекс (PIC) путем привлечения дополнительных GTFob, кратко охарактери-
зованные роли которых сведены в таблицу 11.5. Присоединение ТВР вызывает форми-
рование в ДНК изгиба приблизительно 80°, что расширяет малую бороздку в области
ТАТА-блока (рис. 11.21). Тогда TFIIB прикрепляется к комплексу, это присоединение во-
влекает контакты с ТАТА-блоком через расширенную малую бороздку и через большую
бороздку, при этом распознающий мотив TFIIB располагается непосредственно выше
ТАТА-блока (раздел 11.2.2). В результате этих прикреплений обеспечивается правиль-
ное расположение (относительно сайта инициации транскрипции) РНК-полимеразы II,
которая привносится в комплекс фактором TFIIF. Предынициаторный комплекс за-
вершается добавлением в него факторов TFIIE и TFIIH, последний из которых обладает
действием геликазы и, как думают, разрушает пары оснований в ДНК, а следовательно,
переводит промотор в открытую
форму. TFIIH — интересный белок,
поскольку он играет роль также
и в репарации ДНК, некоторые
типы которой сопряжены с транс-
крипцией, и мы встретимся с ним
снова при изучении механизмов
репарации ДНК в разделе 16.2.2.
Рис. 11.20. Димеры, образованные меж-
ду ТАГцДг/ТАРцбг и гистонами НЗ/Н4. Ри-
сунки показывают ориентации гистонных
сверток пары белков в обоих комплек-
сах
TAF„42
406 Часть 3. Принципы функционирования геномов
Рис. 11.21. Прикрепление ТВР вызывает изгиб молекулы
ДНК. ТВР показан фиолетовым цветом, а ДНК — зеле-
ным. Изгиб в ДНК открывает малую бороздку, облегчая
прикреплениеTFIIB. Фотография любезно предоставле-
на Стефаном К. Берли
Активация инициаторного комплекса
требует присоединения фосфатных групп
к С-концевому домену (CTD) — наибольшей
субъединицы РНК-полимеразы II. У млекопи-
тающих этот домен состоит из 52 повторов
семиаминокислотной последовательности
Tyr-Ser-Pro-Thr-Ser-Pro-Ser. Два из трех се-
ринов в каждой повторной единице могут быть
модифицированы присоединением фосфатной
группы, что вызывает существенное изменение
в ионных свойствах полимеразы. Будучи фос-
форилирована, полимераза способна оставить
инициаторный комплекс и начать синтез РНК.
Фосфорилирование может осуществляться фактором TFIIH, который обладает соответ-
ствующим действием протеинкиназы, или же это может быть функцией посредника
(раздел 11.3.2), который передает сигналы от активаторных белков, регулирующих
экспрессию отдельных генов. По отбытии полимеразы по крайней мере некоторые из
GTFob отделяются от базального промотора, но TFIID, TFIIA и TFIIH остаются на нем, что
позволяет осуществлять повторную инициацию без необходимости восстановления
полной сборки с самого начала. В связи с этим повторная инициация представляет со-
бой более быстрый процесс, чем первичная инициация, а это означает, что, как только
ген включен, транскрипты могут инициироваться с его промотора с относительной
простотой до того времени, покуда новая группа сигналов не выключит этот ген.
Таблица 11.5. Функции общих факторов транскрипции (GFT) человека
GFT Функция
TFIID (компонент ТВР) TFIID (TAF) TFIIA Опознавание блока TATA и, возможно, инициаторной последовательности; образует площадку для посадки TFIIB Опознавание центрального промотора; регулировка связывания ТВР Стабилизирует прикрепление ТВР и TAF
TFIIB Посредник в привлечении РНК-полимеразы II; влияет на выбор точки начала транскрипции
TFIIF Привлечение РНК-полимеразы II; взаимодействие с нематричной нитью
TFIIE Посредник в привлечении TFIIH; опосредствует различные действия TFIIH
TFIIH Действие геликазы, обусловливающее переход промоторного комплек- са из закрытой в открытую форму; возможно, влияет на разрешающий отступ промотора посредством фосфорилирования С-концевого домена наибольшей субъединицы РНК-полимеразы II
Глава 11. Сборка комплекса инициации транскрипции 407
Инициатор транскрипции для РНК-полимераз I и ///
Инициация транскрипции на промоторах РНК-полимераз I и III вовлекает со-
бытия, подобные происходящим в случае РНК-полимеразы II, но некоторые детали
отличаются. Одно из самых поразительных подобий — то, что ТВР, вначале определен-
ный как ключевой специфичный к последовательности ДНК-связывающий компонент
предынициаторного комплекса для РНК-полимеразы II, вовлечен также и в инициацию
транскрипции для двух других РНК-полимераз эукариотов.
Инициаторный комплекс для РНК-полимеразы I включает в себя, в дополнение
к самой полимеразе, четыре белковых комплекса. Один из них, UBF, являет собой димер
из идентичных белков, который взаимодействует и с базальным промотором, и с выше-
лежащим элементом управления (см. рис. 11.16). UBF — другой белок, который, подобно
некоторым TAF РНК-полимеразы II, напоминает собою гистон и может образовывать
в области промотора подобную нуклеосоме структуру. Второй белковый комплекс, на-
званный SL1 у людей и TIFIB у мышей, содержит ТВР и вместе с UBF направляет РНК-
полимеразу I и последние два комплекса, TIFIA и TIFIC, к промотору Первоначально
думали, что инициаторный комплекс возводится поэтапно, но недавно полученные
результаты предполагают, что РНК-полимераза I связывает четыре белковых комплекса
перед опознаванием промотора, и такая полноценная сборка прикрепляется кДНК за
один прием.
Промоторы РНК-полимеразы III изменчивы в структуре (см. рис. 11.16), и это от-
ражено разнородностью процессов их опознавания. Инициация на относящихся к раз-
личным категориям промоторах для РНК-полимеразы III требует различных наборов
GTFob, но в процессах инициации всех типов участвует TFII1B, одной из субъединиц
которого является ТВР. В случае промоторов того типа, что и в гене мяРНК U6, которые
содержат последовательность ТАТА, ТВР, вероятно, напрямую связывается с ДНК. В тех
промоторах для РНК-полимеразы III, которые лежат внутри генов и которые не имеют
последовательности ТАТА, закрепление ТВР осуществляется через пару факторов сборки,
названных TFIIIA и TFIIIC. Название «фактор сборки» указывает на то, что эти два белка
необходимы только для прикрепления ТВР к промотору и не нужны для последующего
приземления РНК-полимеразы III.
11.3. Регулирование инициации транскрипции
По мере продвижения по следующим нескольким главам мы познакомимся с рядом
стратегий, к которым организмы прибегают для регулирования экспрессии отдель-
ных генов. Мы обнаружим, что фактически каждый шаг на пути от генома к протеому
подвержен (в большей или меньшей степени) управлению. Кажется, что изо всех этих
регуляторных систем именно инициация транскрипции есть этап, на котором осущест-
вляются определяющие управляющие воздействия на экспрессию отдельных генов
(то есть такие управляющие воздействия, которые оказывают наибольшее влияние на
биохимические свойства клетки). Это вполне объяснимо. Вполне разумно, что инициа-
ция транскрипции, будучи первым шагом в экспрессии генома, должна быть этапом,
на котором происходит «первичное» регулирование (это уровень регулирования, ко-
торый определяет, какие гены будут экспрессироваться). Последующие звенья в этой
цепи, можно ожидать, должны реагировать на «вторичное» регулирование, функция
которого заключается не во включении или выключении генов, а в модулировании
экспрессии за счет внесения небольших изменений в скорость, с которой белковый про-
408 Часть 3. Принципы функционирования геномов
дукт синтезируется, или, возможно, за счет изменения некоторым образом характера
этого продукта (рис. 11.22).
Из главы 10 мы узнали о том, как структура хроматина может влиять на экспрессию
гена посредством управления доступностью промоторных последовательностей для
РНК-полимеразы и связанных с ней белков. Это всего лишь один способ, которым мож-
но регулировать инициацию транскрипции. Чтобы получить более широкую картину,
мы установим некоторые общие принципы на примере бактерий, а затем исследуем
события, происходящие у эукариотов.
__аиК-Ж-аивв_ДНК
I ПурОВЕНЬЙ транскрипции
------ РНК
ВТОРИЧНЫЙ
УРОВЕНЬ
Последующие стадии
экспрессии генов
ш ш
Ткань Ткань
1 2
-1-1
-ли
Ткань
1
4-Л
1 1 Белок
-л-л
Ткань
2
Рис. 11.22. Первичный и вторичный уровни регулирования генома. Согласно этой схеме «первичное»
регулирование экспрессии генома происходит на уровне инициации транскрипции — этот этап опреде-
ляет, какие гены экспрессируются в данной клетке в данное время и устанавливает относительные
уровни экспрессии тех генов, которые включены. «Вторичное» регулирование вовлекает все этапы на
пути экспрессии генома после инициации транскрипции и служит, чтобы корректировать количество
синтезируемого белка или некоторым образом изменять характер белка, например, путем его хими-
ческой модификации
11.3.1. Стратегии управления инициацией транскрипции у бактерий
У бактерий, таких как Е. coli, мы выделяем два разных способа управления ини-
циацией транскрипции:
• структурное управление, которое зависит от структуры промотора;
• регуляторное управление, которое зависит от влияния регуляторных белков.
Структура промотора определяет основной уровень инициации транскрип-
ции
Консенсусная последовательность для промотора Е. coli (раздел 11.2.2) сильно
изменчива, с диапазоном различных мотивов, допустимых и в блоке -35, и в блоке -10
(см. табл. 11.4). Такая изменчивость, наряду с менее четкими особенностями последова-
тельности около участка сайта инициации транскрипции и в пределах первых 50-ти или
около того нуклеотидов транскрипта, отрицательно влияет на эффективность промо-
тора. Эффективность определяется числом продуктивных инициаций, осуществляемых
промотором в секунду; продуктивной инициацией считается такая, в результате которой
Глава 11. Сборка комплекса инициации транскрипции 409
РНК-полимераза очищает промотор и начинает синтез полновесного транскрипта. Точ-
ный механизм, которым последовательность промотора воздействует на инициацию,
не известен, но из нашего обзора событий, вовлеченных в инициацию транскрипции
(раздел 11.2.3), мы можем интуитивно ожидать, что специфичная последовательность
блока -35 влияет на опознавание ДНК о-субъединицей и, следовательно, на скорость
посадки РНК-полимеразы, что переход от закрытого к открытому промоторному ком-
плексу, возможно, зависит от последовательности блока -10, и что частота неудачных
инициаций (таких, которые прерываются прежде, чем РНК-полимеразы успеют про-
двинуться достаточно далеко по единице транскрипции) может быть обусловлена
последовательностью, расположенной около (причем непосредственно ниже) нуклео-
тида +1. Все это лишь размышления, но это обоснованная «рабочая гипотеза». Ясно то,
что различные промоторы разнятся в своей эффективности в 1 000 крат, и наиболее
эффективные промоторы (названные сильными промоторами) направляют в 1 000
раз больше продуктивных инициаций, чем наиболее слабые промоторы. Мы называем
это различиями в основной скорости инициации транскрипции.
Обратите внимание, что основная скорость инициации транскрипции для гена
предопределена последовательностью его промотора и, таким образом, при нормальных
обстоятельствах не может быть изменена. Она может быть изменена мутацией, которая
видоизменяет определяющий нуклеотид в промоторе, и, несомненно, это случается вре-
мя от времени, но это не такое событие, которым бактерия может управлять. Бактерия
может, однако, определять, каким из промоторных последовательностей будет отдавать-
ся предпочтение, изменяя о-субъединицу своей РНК-полимеразы. п-Субъединица —
часть полимеразы, которая обладает способностью специфичного к последовательности
связывания с ДНК (раздел 11.2.3), так что замена одного варианта этой субъединицы
на иной вариант с немного отличным ДНК-связывающим мотивом и, следовательно,
с видоизмененной специфичностью к последовательности приводит к тому, что будет
опознаваться другой набор промоторов. У Е. coli стандартная п-субъединица, которая
опознает консенсусную последовательность промотора и, следовательно, направляет
транскрипцию большинства генов, называется и70 (ее молекулярная масса приблизи-
тельно равна 70 кДа). Е coli имеет также вторую п-субъединицу, и32, которая произво-
дится, когда бактерия подвергается тепловому шоку. Во время теплового шока Е. coli,
аналогично другим организмам, включает набор генов, кодирующих специальные белки,
которые помогают бактерии выдержать стресс (рис. 11.23). Эти гены имеют особые про-
моторные последовательности, специфично опознаваемые п32-субъединицей. Поэтому
бактерия способна включать целый диапазон специальных генов, производя одно про-
стое видоизменение в структуре своей РНК-полимеразы. Такая система распространена
у бактерий: например, Klebsiella pneumoniae использует ее для управления экспрессией
генов, участвующих в усвоении азота, на сей раз с помощью п54-субъединицы, а вид
Bacillus использует целый диапазон различных о-субъединиц, чтобы включать и вы-
ключать группы генов во время переключения с нормального роста на образование
спор (раздел 14.3.2).
Регулирующее управление инициацией транскрипции у бактерий
Структура промотора определяет основную скорость инициации транскрипции бак-
териального гена, но, за исключением опознавания альтернативными п-субъединицами,
не обеспечивает никаких общих средств, которыми экспрессия гена могла бы реагиро-
вать на изменения в окружающей среде или на биохимические потребности клетки.
Для этого необходим регуляторный контроль других типов.
410 Часть 3. Принципы функционирования геномов
а) Ген теплового шока Е. coli
Промотор —।
теплового шока Ген теплового шока
Л Л 4- - 32
б) Опознавание субъединицеи сг
РНК-полимераза а70 не способна связываться
-44 1 -36 -10 I
...CTGCCACCC I . CCATNT...
32
РНК-полимераза а связывается
с промотором теплового шока
Рис. 11.23. Опознавание гена теплового шока Escherichia coli субъединицей а32, а) Последовательность
промотора теплового шока отличается от таковой нормального промотора Е. coli (сравните с табли-
цей 11.4). б) Промотор теплового шока не распознается нормальной РНК-полимеразой Е. соИ, содержащей
субъединицу а70, но опознается РНК-полимеразой о32, которая является активной во время теплового
шока. Сокращение: N — любой нуклеотид. Более подробные сведения о применении бактериями ново-
синтезированных a-факторов можно найти в разделе 14.3.2
Основа нашего понимания регуляторного управления инициацией транскрипции
у бактерий была заложена в начале 1960-х Франсуа Жакобом, Жаком Моно и другими
генетиками, которые изучали оперон лактозы и другие модельные системы. Мы уже
видели, что эта работа привела к открытию промотора оперона лактозы (раздел 11.2.2).
В ее итоге также был обнаружен оператор — область, смежная с промотором и регули-
рующая инициацию транскрипции оперона (рис. 11.24, а). В первоначальную модель
было заложено представление о том, что ДНК-связывающий белок — лактозный ре-
прессор — прикрепляется к оператору и мешает РНК-полимеразе связываться с про-
мотором, попросту препятствуя ее доступу к надлежащему сегменту ДНК (рис. 11.24,6).
Свяжется репрессор или нет, зависит от присутствия в клетке аллолактозы (изомера
лактозы), причем последняя является субстратом для биохимического пути, выпол-
няемого ферментами, кодируемыми тремя генами в этом опероне. Аллолактоза слу-
жит индуктором оперона лактозы. Когда аллолактоза присутствует, она связывается
с лактозным репрессором, вызывая небольшое структурное изменение, которое пре-
пятствует мотивам НТН репрессора опознать оператор в качестве сайта связывания
с ДНК. Комплекс аллолактоза-репрессор поэтому не может связаться с оператором,
что позволяет РНК-полимеразе получить доступ к промотору. Когда ресурсы лактозы
исчерпываются и совсем не остается аллолактозы, чтобы связаться с репрессором,
репрессор снова прикрепляется к оператору и предотвращает транскрипцию. Поэтому
оперон экспрессируется только тогда, когда ферменты, кодируемые этим опероном,
необходимы бактерии.
Первоначальная схема регулирования оперона лактозы почти полностью была
подтверждена секвенированием регуляторной области ДНК и структурным анализом
репрессора, прикрепленного к своему оператору. Одним осложнением было открытие
того, что репрессор имеет три потенциальных сайта связывания, сосредоточенных
в позициях нуклеотидов -82, +11 и +412. Оператором, определенным генетическим
анализом, служит последовательность, расположенная в позиции +11 (см. рис. 11.24, а),
Глава 11. Сборка комплекса инициации транскрипции 411
а) Оператор лактозы
lacZ lacY lacA
-40
-30
-20
-10
лактозы
+10
+20
iAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCAC
блок-35 блок-10 Оператор
б) Исходная модель регуляции лактозы
В ОТСУТСТВИЕ ЛАКТОЗЫ
В ПРИСУТСТВИИ ЛАКТОЗЫ
Лактозный репрессор
РНК-полимераза прикрепляется
не может сесть к оператору
на промотор ф
Комплекс репрессор-аллолактоза
РНК-полимераза не может прикрепиться к оператору
может связаться
с промотором
Лактозный
репрессор
Лактозный
репрессор I
Аллолактоза
Рис. 11.24. Регулирование оперона лактозы Escherichia coli. а) последовательность оператора лежит
непосредственно ниже промотора оперона лактозы. Обратите внимание, что эта последовательность
обладает симметрией инверсии: при считывании в направлении 5*—>3' последовательность получается
одной и той же в обеих нитях. Это позволяет двум субъединицам тетрамерного репрессорного белка
вступать в контакт с одной последовательностью оператора, б) В исходной модели регулирования лак-
тозы репрессор лактозы рассматривался как простое блокирующее устройство, которое связывается
с оператором и предотвращает получение РНК-полимеразой доступа к промотору. Поэтому все три
гена в опероне выключаются. Эта ситуация имеет место при отсутствии лактозы, хотя транскрипция
не полностью блокирована, потому что репрессор иногда отделяется, позволяя произвести несколько
транскриптов. Из-за этого основного уровня транскрипции бактерия всегда обладает несколькими ко-
пиями (возможно, не более пяти) каждого из трех ферментов, кодируемых опероном (см. рис. 8.8, а).
Это означает, что, когда бактерия встречает источник лактозы, она способна транспортировать несколько
молекул в клетку и расщепить их на глюкозу и галактозу. Промежуточный продукт этой реакции — изомер
лактозы аллолактоза — вызывает экспрессию оперона лактозы, связываясь с репрессором и вызывая
изменение в конформации последнего, так что он более не способен прикрепляться к оператору. Это
позволяет РНК-полимеразе связаться с промотором и транскрибировать все три гена оперона. При
полной активации в клетке находится приблизительно по 5000 молекул каждого белкового продукта.
Когда запас лактозы израсходован и аллолактоза больше не присутствует, репрессор снова прикрепляется
к оператору и оперон выключается. Транскрипты оперона, которые имеют время полураспада менее 3
минут, распадаются и ферменты более не производятся. Имейте в виду, что показанные здесь формы
структур репрессора и полимеразы являются чисто схематическими
и это лишь один из трех участков, овладение которыми репрессором, как можно ожи-
дать, предотвратит доступ РНК-полимеразы к промотору. Но другие два участка также
играют некоторую роль в репрессии, поскольку их удаление, по отдельности или вместе,
значительно снижает способность репрессора выключать экспрессию гена. Репрессор
представляет собой тетрамер из четырех идентичных субъединиц, которые работа-
ют попарно, чтобы прикрепиться к отдельному оператору, так что репрессор имеет
способность связываться с двумя из трех операторных участков одновременно. Одна
возможность — что связывание одной пары субъединиц с участком +11 усиливается
412 Часть 3. Принципы функционирования геномов
или стабилизируется прикреплением другой пары субъединиц к участку-82 или +412.
Также возможно, что репрессор может связываться с парой операторных последователь-
ностей таким образом, что он не блокирует присоединение полимеразы к промотору,
но предотвращает последующий шаг в инициации, такой как образование открытого
промоторного комплекса.
Основополагающий принцип, на который опирается регуляторное управление
инициацией транскрипции, как демонстрирует нам оперон лактозы, состоит в том,
что прикрепление ДНК-связывающего белка к своему специфичному сайту узнавания
может влиять на события, вовлеченные в сборку комплекса инициации транскрипции
и (или) в инициацию продуктивного синтеза РНК, осуществляемого РНК-полимеразой.
У других генов бактерий встречается ряд разновидностей этой схемы.
• Некоторые репрессоры реагируют не на индуктор, а на сорепрессор. Пример пред-
ставляет оперон триптофана Е. coli, который кодирует набор генов, вовлеченных
в синтез триптофана (см. рис. 8.8, б). В отличие от оперона лактозы, регуляторной
молекулой для оперона триптофана служит не субстрат для соответствующего
биохимического пути, но его продукт — сам триптофан (рис. 11.25). Только в том
случае, когда триптофан присоединен к триптофановому репрессору, последний
может связаться с оператором. Поэтому оперон триптофана выключается в при-
сутствии триптофана и включается, когда триптофан необходим бактерии.
• Некоторые ДНК-связывающие белки являются активаторами, а не репрессорами
инициации транскрипции. Лучше всего изученный пример у Е. coli — белок-
активатор катаболитных оперонов, который связывается с участками, располо-
женными выше некоторых оперонов, в том числе оперона лактозы, и увеличивает
эффективность инициации транскрипции, вероятно, образуя прямой контакт
с РНК-полимеразой. Биологическая роль белка-активатора катаболитных оперонов
описана в разделе 14.1.1.
• Один и тот же репрессор или активатор может управлять двумя и более промото-
рами. Например, у Е. coli триптофановый репрессор управляет опероном триптофа-
на — геном агоН (который определяет фермент для раннего этапа биохимического
РНК-полимераза
может связаться
с промотором
ТРИПТОФАН ОТСУТСТВУЕТ
Триптофановый репрессор
не может прикрепиться I
к оператору »
РНК-полимераза
не может связаться
с промотором
ТРИПТОФАН НАЛИЧЕСТВУЕТ
Комплекс репрессор-триптофан
прикрепляется к оператору
trpE
Триптофановый
репрессор
Триптофановый Триптофан
репрессор
Рис. 11.25. Регулирование оперона триптофана Escherichia coli. Регулирование происходит через систему
репрессор-оператор, подобную описанной на примере оперона лактозы, ностой разницей, что в да ином
случае оперон подавляется регулирующей молекулой — триптофаном, — который является продуктом
биохимического пути, описываемого генами в опероне (см. рис. 8.8, б). Когда триптофан присутствует и,
таким образом, его синтез не требуется, оперон выключен, потому что комплекс репрессор-триптофан
связывается с оператором. В отсутствие триптофана репрессор не может присоединиться к оператору,
и оперон экспрессируется
Глава 11. Сборка комплекса инициации транскрипции 413
пути синтеза триптофана) и геном trpR (который является собственным геном
триптофанового репрессора, а это означает, что белок репрессора синтезируется
только тогда, когда это необходимо) (рис. 11.26).
• Опознавательные последовательности для ДНК-связывающих белков могут рабо-
тать по отдельности или вместе и усиливать или подавлять транскрипцию генов,
с которыми они близко не связаны. Такие энхансеры (усилители) и сайленсеры
(глушители) не свойственны бактериям, но известно несколько примеров, в том
числе энхансер, который действует на гены теплового шока Е. coli, промоторы ко-
торых опознаются вариантом РНК-полимеразы с о32-субъединицей. Поскольку они
находятся далеко от генов, которыми управляют, то могут образовывать контакты
с РНК-полимеразой, только если ДНК образует петлю. Характерной особенностью
является то, что единственный энхансер или сайленсер может управлять экспрес-
сией более чем одного гена.
Все эти основополагающие принципы регулирования генов относятся не только
к бактериям, но и, как мы увидим в следующем разделе, также к эукариотам.
Оперон триптофана
Управляет синтезом ферментов
для заключительных шагов
синтеза триптофана
агоН
Управляет синтезом фермента,
необходимого на первых шагах
синтеза ароматических
аминокислот
Рис. 11.26. Многочисленные цели триптофанового репрессора
11.3.2. Регуляция инициации транскрипции у эукариотов
Главный урок, который мы усвоили из нашего обзора управления транскрипцией
у бактерий, состоит в том, что на инициацию транскрипции могут влиять ДНК-свя-
зывающие белки, которые опознают специфичные последовательности, расположенные
около сайта связывания РНК-полимеразы. Такая же схема лежит в основе управления
транскрипцией у эукариотов, с двумя отличиями, первое из которых касается основной
скорости инициации транскрипции. Бактериальная РНК-полимераза обладает сильным
сродством к своему промотору, и основная скорость инициации транскрипции отно-
сительно высока для всех, кроме самых слабых промоторов. Для большинства генов
эукариотов верно обратное. Предынициаторные комплексы для РНК-полимераз II и III
не собираются эффективно, и основная скорость инициации транскрипции поэтому
очень низка, вне зависимости от того, насколько «силен» промотор. Чтобы достигнуть
эффективной инициации, образование комплекса должно быть активировано дополни-
тельными белками. Это означает, что по сравнению с бактериями эукариоты используют
414 Часть 3. Принципы функционирования геномов
иные стратегии управления инициацией транскрипции, где активаторы играют намного
более видную роль, чем белки-репрессоры.
Второе отличие состоит в том, что, как и во всех остальных моментах экспрессии
генома, процессы, которые регулируют инициацию транскрипции у эукариотов, значи-
тельно сложнее, чем аналогичные процессы у бактерий.
Промоторы эукариотов содержат регуляторные модули
Такая более значительная сложность становится очевидной при взгляде на про-
моторы эукариотов. На инициацию транскрипции типичного кодирующего белок гена
влияет целый сонм различных биохимических сигналов, которые действуют совместно,
чтобы гарантировать, что ген экспрессируется в точности на том уровне, который в наи-
более полной мере соответствует существующим условиям во внутренней и внешней
среде клетки, в которой этот ген расположен. Промотор РНК-полимеразы II можно
рассматривать как ряд модулей, каждый из которых включает короткую последова-
тельность нуклеотидов и действует как сайт связывания для белка, который влияет на
сборку комплекса инициации транскрипции. Много различных генов транскрибируются
РНК-полимеразой II (у человека — более 30 000), но имеется лишь ограниченное число
промоторных модулей для этой полимеразы. Поэтому картина экспрессии для всякого
гена определяется не каким-либо отдельным модулем, а сочетанием модулей в составе
его промотора и, возможно, их относительными позициями. Количество производимых
инициаций транскрипции зависит от того, какие модули заняты связывающимися
с ними белками в данное время.
Модули для промотора РНК-полимеразы II могут быть категоризированы различ-
ными способами. Одна из схем следующая:
• Модули базального промотора (раздел 11.2.2), наиболее важные из которых
представлены ТАТА-блоком и инициаторной последовательностью.
• Основные промоторные элементы — модули, которые присутствуют во многих
промоторах для РНК-полимеразы II и устанавливают основной уровень инициации
транскрипции, не отвечая ни на какие тканеспецифические сигналы или сигналы,
связанные с развитием. К ним относятся: блок СААТ (консенсус 5'-GGCCAATCT-3’),
опознаваемый активаторами NF-1 и NF-Y; блок GC (консенсус 5'-GGGCGG-3'), опо-
знаваемый активатором Spl, и октамерный модуль (консенсус 5'-ATGCAAAT-3’J,
опознаваемый активатором Oct-1.
• Модули ответных реакций располагаются выше различных генов и позволя-
ют инициации транскрипции реагировать на основные сигналы, поступающие
извне клетки. Примеры: модуль реакции на циклический АМФ CRE (консенсус
5'-WCGTCA-3', где W—А или Т), опознаваемый активатором CREB; модуль теплового
шока (консенсус 5'-CTNGAATNTTCTAGA-3', где N — любой нуклеотид), опознавае-
мый Hsp70 и другими активаторами, и модуль сывороточной реакции (консенсус
5'-CCWWWWWWGG-3'), опознаваемый фактором сывороточной реакции.
• Клеткоспецифические модули расположены в промоторах генов, которые экс-
прессируются только в ткани определенного типа. Примеры включают: эритроид-
ный модуль (консенсус 5’-WGATAR-3', где R — А или G), который является сайтом
связывания для активатора GATA-1; модуль гипофизарных клеток (консенсус
5'-АТАТТСАТ-3'), опознаваемый активатором Pit-1; миобластный модуль (консенсус
5'-CAACTGAC-3'), опознаваемый активатором MyoD, и модуль лимфоидных клеток,
или участок кВ (консенсус 5'-GGGACTTTCC-3'), опознаваемый активатором NF-kB.
Глава 11. Сборка комплекса инициации транскрипции 415
Следует помнить, что в лимфоидных клетках октамерный модуль опознается
тканеспецифическим активатором Oct-2.
• Модули для регуляторов развития опосредствуют экспрессию генов, которые яв-
ляются активными на определенных этапах развития. Два примера таких модулей
у Drosophila представлены модулем Bicoid (консенсус 5’-ТССТААТССС-3') и модулем
Antennapedia (консенсус 5'-ТААТААТААТААТАА-3’) (раздел 14.3.4).
Модульная структура типичного промотора РНК-полимеразы II представлена
на рис. 11.27. Так же, как модули в области непосредственно выше гена, те же самые
и другие модули могут содержаться еще и в пределах энхансеров, которые имеют дли-
ну 200-300 п.н. и могут быть расположены на некотором расстоянии выше или ниже
своего целевого гена. Сайленсеры подобны энхансерам, но, как предполагает их назва-
ние, их модули оказывают отрицательное, а не усиливающее влияние на инициацию
транскрипции.
GC А5 OCT Е2 АЗ CRE А2 Е1 А1 G1 ТАТА Ген
50 п. н.
ОБОЗНАЧЕНИЯ:
GC, А5 — Названия модулей
Основные промоторные элементы
Специфические для клетки модули
Рис. 11.27. Модульная структура промотора гена инсулина человека
Дополнительный уровень сложности встречается у некоторых генов, которые
имеют альтернативные промоторы, которые дают начало различным версиям транс-
крипта, определяемого геном. Пример представлен геном дистрофина человека, кото-
рый был объектом интенсивного изучения, потому что дефекты в этом гене приводят
к генетической болезни, известной под названием мышечной дистрофии Дюшенна.
Ген дистрофина — один из наибольших известных в геноме человека, протяженностью
более 2,4 млн п. н., содержит 78 интронов. Он имеет по крайней мере семь альтернатив-
ных промоторов (рис. 11.28). Три из них лежат выше гена и дают начало полномерным
транскриптам, которые отличаются только типом первого экзона; эти три промотора
активны, соответственно, в ткани коры головного мозга, мышцах и мозжечке. Другие
четыре промотора расположены в пределах гена и дают укороченные транскрипты,
которые опять же синтезируются специфично к той или иной ткани. Каждый промотор
имеет свою собственную модульную структуру, хотя каждый подвержен влиянию одних
и тех же последовательностей энхансера и сайленсера. Альтернативные промоторы ис-
пользуются также для порождения родственных версий некоторых белков на различных
этапах развития и для того, чтобы позволить одной клетке синтезировать подобные
белки со слегка разными биохимическими свойствами. Последний момент указывает
на то, что, хотя их обычно называют «альтернативными» промоторами, более правиль-
ным будет называть их «множественными» промоторами, поскольку в одно и то же
время могут быть активными сразу несколько из них. Действительно, это может быть
нормальной ситуацией для многих генов. Например, обзор всего генома показал, что
416 Часть 3. Принципы функционирования геномов
Альтернативные промоторы
К М Mo С
t 1 - ’______________1—
О 500 1000
ЦНС
Г .
1500
Ш О
С_____иС
2000
2500 тыс. п. н.
*-ПТИГjW&iWiiii l iiM
Ген дистрофина человека
Рис. 11.28. Альтернативные промоторы. Показаны позиции семи альтернативных промоторов гена
дистрофина человека. Сокращения указывают ткань, в которой соответствующий промотор является
активным: К — ткань коры головного мозга; М — мышечная; Мо — мозжечок; С — ткань сетчатки глаза
(и также мозговая и сердечная ткань); ЦНС — центральная нервная система (и также почки); Ш — [ива-
новские клетки; О — общие (большинство тканей, кроме мышечной)
в клетках фибробластов человека активными являются 10 500 промоторов, но что эти
промоторы способствуют экспрессии менее чем 8000 генов, а это указывает на то, что
значительное число генов в этих клетках экспрессируется с подачи сразу двух и более
промоторов.
Активаторы и коактиваторы инициации транскрипции у эукариотов
Белок, который стимулирует инициацию транскрипции, называют активатором,
если это специфичный к последовательности ДНК-связывающий белок, или коактива-
тором, если он связывается с ДНК неспецифично или работает через межбелковые взаи-
модействия. Некоторые активаторы опознают вышележащие промоторные элементы
и влияют на инициацию транскрипции только в промоторе, к которому эти элементы
прикреплены, а другие активаторы нацеливаются на участки в пределах энхансеров
и влияют на транскрипцию сразу нескольких генов (рис. 11.29). Как и у бактерий,
энхансеры эукариотов могут находиться на некотором расстоянии от своих генов, их
специфичность к цели гарантируется присутствием инсуляторов по обе стороны каждо-
го функционального домена, предотвращающих влияние энхансеров в пределах этого
домена на экспрессию генов в смежных доменах (раздел 10.1.2). Будь он связан с вы-
шележащим промоторным элементом или с более отдаленным энхансером, активатор
стабилизирует предынициаторный комплекс.
Коактиваторы намного более разнообразны. В их число входят комплексы аце-
тилирования гистонов типа SAGA (раздел 10.2.1) и комплексы перестройки нуклеосом
типа Swi/Snf (раздел 10.2.2). Другие белки, классифицируемые как коактиваторы, влия-
ют на экспрессию генов, привнося изгибы и другие деформации в ДНК, — возможно,
в качестве прелюдии к модификации хроматина, а возможно, с целью свести вместе
белки, прикрепленные к несмежным участкам, дабы позволить связанным факторам
Активатор
Усилитель
Рис. 11.29. Активаторы инициации транскрипции у эукариотов. Голубой активатор присоединен к регу-
лирующему модулю, расположенному выше гена, и влияет на инициацию транскрипции только одного
этого гена. Зеленый активатор присоединен к участку в пределах усилителя (энхансера) и влияет на
транскрипцию всех трех генов
Глава 11. Сборка комплекса инициации транскрипции 417
работать совместно в структуре, которую назвали энхансосомой. Пример коактиватора,
работающего таким образом, — SRY, который является главным белком, ответственным
за определение пола у млекопитающих.
Активаторы рассматривались как важные сайты инициации транскрипции для
РНК-полимераз II и III, но их роль в промоторах РНК-полимеразы I была менее четко
определена. РНК-полимераза I является необычной в том, что она транскрибирует
только один-единственный набор генов: множественные копии единицы транскрипции,
содержащей последовательности рРНК 28 S, 5,8 S и 18S (раздел 7.2.3). В большинстве
клеток эти гены экспрессируются непрерывно, но на протяжении клеточного цикла
скорость транскрипции изменяется и подвержена в некоторой степени тканеспецифи-
ческому регулированию. Регулирующий механизм не был описан подробно, но недав-
ние исследования предположили роль факторов терминации для РНК-полимеразы I.
Этот фактор, названный TTF-1 у мышей и Reblp у Saccharomyces cerevisiae, был сначала
опознан как активатор транскрипции для РНК-полимеразы II. Очевидно, что фактор
терминации может также активировать транскрипцию для РНК-полимеразы I, ибо сайт
связывания для него был обнаружен непосредственно выше промотора для единицы
транскрипции рРНК.
Посредник образует контакт между активатором и предынициаторным
комплексом для РНК-полимеразы II
Отличительная особенность активаторов «традиционного» типа — тех, которые
связываются с вышележащими промоторными элементами или с энхансерами, есть
контакт, образуемый с предынициаторным комплексом. Часть активатора, участвующую
в этом контакте, называют активационным доменом. Структурный анализ показал,
что, хотя активационные домены изменчивы, большинство из них подпадает под одну
из трех категорий:
• кислые домены, которые являются относительно богатыми кислыми аминокисло-
тами (аспарагиновая кислота и глутаминовая кислота). Это самая распространенная
категория активационных доменов;
• обогащенные глутамином домены часто обнаруживаются в активаторах, ДНК-
связывающие мотивы которых относятся к типу гомеодоменов или POU (раз-
дел 11.1.1);
• обогащенные пролином области менее распространены.
Детали взаимодействия между активаторами и предынициаторным комплексом
для РНК-полимеразы II были неясны в течение нескольких лет, при этом в ходе работ
с различными организмами были получены явно противоречивые данные. Ряд иссле-
дований межбелкового взаимодействия позволял предположить, что прямые контакты
могли быть образованы между различными активаторами и различными частями ком-
плекса, при этом ТВР, всеразличные TAFbi, TFIIB, TFIIH и РНК-полимераза II участвуют как
партнеры в разных взаимодействиях. Решение этой неразрешимой загадки было пред-
ложено, когда крупный белковый комплекс, названный посредником (медиатором),
был обнаружен у дрожжей. Посредник дрожжей состоит из 21 субъединицы, которые
формируют структуру с отчетливо выраженными головным, срединным и хвостовым
доменами. Хвост образует физический контакт с активаторным белком, прикреплен-
ным к своему сайту узнавания ДНК, а срединный и головной отделы взаимодействуют
с предынициаторным комплексом (рис. 11.30). Поэтому активатор связывается с пред-
ынициаторным комплексом не напрямую, а косвенно, при этом активационный сигнал
418 Часть 3. Принципы функционирования геномов
Рис. 11.30. Взаимодействие между посредником дрожжей
и предынициаторным комплексом РНК-полимеразы II. Комплекс
предзапуска показан белым цветом, а посредник — оранжевым.
Перепечатано из Molecular Cell, Vol. 10, Davis et al., 'Structure of
the Yeast RNA Polymerase II Holoenzyme', 409-415, 2002, с раз-
решения издательства Elsevier
передается через медиатор. Первоначально думали,
что медиатор активирует инициацию транскрипции
путем прямого фосфорилирования С-концевого до-
мена РНК-полимеразы II, стимулируя таким образом
очистку промотора (раздел 11.2.3), но теперь известно,
что эта деятельность киназы осуществляется Kin28 —
субъединицей фактора TFIIH. Есть данные, что медиа-
тор способен стимулировать деятельность Kin28, но
это не единственное его взаимодействие с предынициаторным комплексом, и точный
путь, которым он регулирует инициацию транскрипции, еще не ясен. Посредник при-
сутствует, когда ТВР прикрепляется к ТАТА-блоку и может формировать часть площадки,
на которой собирается остальная часть предынициаторного комплекса. Усложняющий
фактор — то, что медиатор передает сигналы не только от активаторов, но также и от
репрессоров, а это означает, что его влияние на предынициаторный комплекс должно
быть и положительным, и отрицательным.
У высших эукариотов посредники крупнее, чем аналогичный комплекс у дрожжей;
так, например, вариант медиатора у человека состоит из 30 и более белков. Одна особен-
ность медиатора млекопитающих — то, что его белковый состав является изменчивым,
что увеличивает возможность наличия нескольких вариантов, каждый из которых реа-
гирует на свой, хотя, возможно, и перекрывающийся с остальными, набор активаторов.
Бытующее сегодня мнение тяготеет к представлению о том, что медиатор является обя-
зательным компонентом предынициаторного комплекса для РНК-полимеразы II и что
стимулирующие воздействия всех активаторов проходят через медиатор. Не следует,
однако, исключать возможность, что некоторые активаторы обходят медиатор и ока-
зывают прямое воздействие на ту или иную часть предынициаторного комплекса.
Репрессоры инициации транскрипции у эукариотов
Исследования механизма регулирования инициации транскрипции у эукариотов
в основном сосредоточились на активации, отчасти потому, что низкий уровень основ-
ной инициации, наблюдаемый на промоторах для РНК-полимераз II и III, предполагает,
что репрессия инициации, которая является столь важной у бактерий (раздел 11.3.1),
вряд ли играет главную роль в управлении транскрипцией у эукариотов. Это пред-
ставление, возможно, неправильное, потому что обнаруживается все большее число
ДНК-связывающих белков, которые подавляют инициацию транскрипции, причем
эти белки закрепляются на вышележащих промоторных элементах или на более от-
даленных участках в сайленсерах. Некоторые влияют на экспрессию генома обычным
способом — через деацетилирование гистонов (раздел 10.2.1) или метилирование ДНК
(раздел 10.3.1), — но другие оказывают более специфические воздействия в отдельных
промоторах. Например, репрессоры дрожжей, называемые Motl и NC2, ингибируют сбор-
ку предынициаторного комплекса, связываясь непосредственно с ТВР и пресекая его
активность. Motl вынуждает ТВР отделяться от ДНК, a NC2 предотвращает дальнейшую
Глава 11. Сборка комплекса инициации транскрипции 419
сборку комплекса на связанном ТВР. Оба эти репрессора имеют широкий спектр дей-
ствия и инактивируют значительный набор генов, как поступает репрессор Ssn6-Tupl,
который является одним из главных сайленсеров генов у дрожжей Schizosaccharomyces
pombe и который имеет гомологов у многих других эукариотов.
Другим признаком большого значения репрессии для транскрипции у эукариотов
является тот установленный нами факт, что некоторые белки могут в зависимости от об-
стоятельств проявлять и активирующие, и подавляющие эффекты. Репрессор NC2, например,
подавляет инициацию транскрипции на промоторах с ТАТА-блоком, но оказывает активи-
рующее воздействие на промоторы без последовательности ТАТА. Pit-1, который является
первым из этих трех белков, по названиям которых был назван домен POU (раздел 11.1.1),
активирует одни гены и подавляет другие — в зависимости от последовательности сайта
связывания с ДНК. Присутствие в этом участке двух дополнительных нуклеотидов вызы-
вает изменение конформации Pit-1, позволяя ему взаимодействовать со вторым белком по
имени N-CoR и подавлять транскрипцию целевого гена (рис. 11.31).
Рис. 11.31. Конформация доменов POU активатора Pit-1, прикрепленного к своим целевым участкам
выше генов пролактина (слева) и гормона роста (срава). Pit-1 представляет собой димер, и каждый
его мономер состоит из двух доменов POU. Два домена одного мономера показаны красным цветом,
а два домена второго мономера показаны синим. Цилиндры — ot-спирали, где спираль аЗ в каждом
домене является опознавательной. Обратите внимание на разницу между конформациями доменов
при прикреплении к двум участкам связывания. Более открытая структура, принимаемая на участке
гормона роста, позволяет димеру Pit-1 взаимодействовать с N-CoR и другими белками, чтобы подавлять
транскрипцию гена гормона роста. Pit-1 поэтому активирует ген пролактина, но подавляет ген гормона
роста. Перепечатано с разрешения Scully et al., Science, 290,1127-1131. © 2000 AAAS
Управление действиями активаторов и репрессоров
Для того чтобы гарантировать, что клеткой будет экспрессироваться надлежащий
набор генов, необходимо управлять работой отдельных активаторов и репрессоров.
Мы возвратимся к этой теме в главе 14, когда она образует основной маршрут нашего
изыскания путей, которыми активность генома регулируется в ответ на внеклеточные
сигналы, а также в ходе дифференцировки и развития.
420 Часть 3. Принципы функционирования геномов
Есть несколько способов, которыми можно регулировать активатор или репрессор.
Одна возможность состоит в том, чтобы управлять его синтезом, но она не допускает
быстрых изменений в экспрессии генома, потому что необходимо время, чтобы нако-
пить активатор или репрессор в клетке или же уничтожить его, когда он становится
не нужен. Поэтому управление такого типа затрагивает активаторы и репрессоры,
ответственные за поддержание устойчивых картин экспрессии генома, например, тех,
которые лежат в основе дифференцировки клеток и некоторых особенностей развития.
Альтернативный способ управления активатором или репрессором — химическая моди-
фикация, например фосфорилирование, или вызывание изменения в его конформации.
Такие изменения происходят намного быстрее, чем синтез de novo, и позволяют клетке
реагировать на внеклеточные сигнальные соединения, которые вызывают преходящие
изменения в экспрессии генома. Детали этих различных регуляторных механизмов мы
разберем в главе 14.
Заключение
Основными игроками в транскрипции и остальных процессах активности генома
являются ДНК-связывающие белки,^которые прикрепляются к геному, чтобы испол-
нять свои биохимические функции. Многие из этих белков способны прикрепляться
к специфическим последовательностям ДНК посредством ДНК-связывающих мотивов,
таких как структура спираль-изгиб-спираль или цинковый палец. Их позиции связыва-
ния на молекулах ДНК могут быть опознаны при помощи анализа с торможением в геле
и очерчены более точно с помощью анализа с защитой от модификации и анализа с мо-
дификационным препятствованием. Некоторые белки опознают свои сайты связывания
прямым считыванием последовательности ДНК, которое является возможным за счет
образования контактов в пределах большой бороздки двойной спирали, так как здесь род
нуклеотидов может быть определен по положениям химических групп, присоединенных
к пуриновым и пиримидиновым основаниям. Прямое считывание может подвергаться
влиянию различных косвенных воздействий, которые последовательность нуклеотидов
оказывает на конформацию спирали, включая формирование изгибов в обогащенных
аденином последовательностях. Многие ДНК-связывающие белки действуют как диме-
ры, контактируя со спиралью в двух позициях одновременно. Специальные структуры
на поверхности белка, такие как лейциновые застежки-молнии, содействуют димери-
зации. Бактерии имеют единственную РНК-полимеразу, которая транскрибирует все
гены в геноме, но эукариоты имеют три разные ядерные РНК-полимеразы и ещё один
фермент, который работает в митохондриях. Промоторные последовательности от-
мечают позиции, на которых комплекс инициации транскрипции должен быть собран.
У бактерий промотор состоит из двух сегментов последовательности, но промоторы
эукариотов намного сложнее, обладают модульной структурой, в которую входят по-
следовательности, которые опознаются инициаторным комплексом и другими, кото-
рые действуют как сайты связывания для регуляторных белков. Бактериальная РНК-
полимераза непосредственно прикрепляется к своему промотору, но инициаторные
комплексы для каждого типа РНК-полимераз эукариотов содержат вспомогательные
белки и должны возводиться в определенном порядке. Успешная инициация дает поли-
меразный комплекс, который способен очистить промотор (достичь его разрешающего
отступа) и начать процесс транскрипции. Инициация транскрипции — определяющий
шаг, на котором регулируется экспрессия генома. Некоторые бактерии способны ви-
доизменять структуру своей РНК-полимеразы, с тем чтобы фермент опознавал иной
Глава 11. Сборка комплекса инициации транскрипции 421
набор промоторов и, следовательно, транскрибировал другой набор генов. Экспрессия
отдельных генов у бактерий может управляться также репрессорными и активаторны-
ми белками. То же самое верно и для эукариотов, хотя белки-активаторы оказываются
здесь более важными, чем репрессоры. Регуляторные белки прикрепляются к сайтам,
смежным с промоторами, которыми они управляют, или на некотором расстоянии от
них. У эукариотов активация комплекса инициации транскрипции происходит через
медиатор — многосубъединичный белок, который образует физический мостик между
активатором и РНК-полимеразой.
422 Часть 3. Принципы функционирования геномов
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
11.1 *. Каким образом белки способны свя-
зываться с ДНК в специфических по-
следовательностях?
а. Взаимодействуя с сахаро-фосфат-
ной основной цепью.
Ь. Раскрывая двойную спираль и об-
разуя связи с основаниями.
с. Взаимодействуя с основаниями
через гистоновые белки.
d. Взаимодействуя с основаниями
в большой и малой бороздках
двойной спирали.
11.2 . Который из следующих ДНК-свя-
зывающих доменов образует свой
главный контакт с основаниями
нуклеотидов через малую бороздку
двойной спирали?
а. Спираль-изгиб-спираль.
Ь. Цинковый палец.
с. Основный домен.
d. Домен ТВР.
11.3 *. Какой из следующих методов, осно-
ванный на перемещении фрагмен-
тов ДНК в геле в присутствии или
в отсутствие белков, применяется
для опознавания белков, которые
связываются с ДНК?
а. Спектроскопия ядерного магнит-
ного резонанса.
Ь. Торможение в геле.
с. Защита от нуклеазы.
d. Футпринтинг ДНК.
11.4 . Которая из следующих методик при-
меняется в анализе с модификацион-
ным препятствованием для опозна-
вания нуклеотидов, определяюще
важных для связывания белка?
а. Комплекс ДНК-белок обрабаты-
вают нуклеазами с целью дегра-
дации незащищенных фосфодиэ-
фирных связей.
Ь. Комплекс ДНК-белок обрабаты-
вают метилирующими агентами,
чтобы отграничить сайт связы-
вания.
с. ДНК обрабатывают метилирую-
щими агентами до прикрепления
белка.
d. Белок обрабатывают метилиру-
ющими агентами до связывания
с ДНК.
11.5 *. Какая из следующих РНК-полимераз
отвечает за транскрипцию кодирую-
щих белок генов у эукариотов?
а. РНК-полимераза!.
Ь. РНК-полимераза II.
с. РНК-полимераза III.
d. РНК-полимераза IV.
11.6. Участок посадки для РНК-полиме-
разы у бактерий называют:
а. Инициатором.
Ь. Оператором.
с. Промотором.
d. Старт-кодоном.
11.7*. Какой из субъединиц обусловлена
специфичность бактериальных РНК-
полимераз к своим промоторам?
а. а.
Ь. ₽.
с. у.
d. о.
11.8. Первый белковый комплекс, кото-
рый связывается с базальным про-
мотором для кодирующего белок
гена у эукариотов, есть:
а. РНК-полимераза II.
Ь. Общий фактор транскрипции
TFIIB.
с. Общий фактор транскрипции
TFIID.
d. Общий фактор транскрипции
TFIIE.
Глава 11. Сборка комплекса инициации транскрипции 423
11.9 *. Какого типа модификация должна
быть выполнена с РНК-полимера-
зой II, с тем чтобы активировать
предынициаторный комплекс?
а. Ацетилирование.
Ь. Метилирование.
с. Фосфорилирование.
d. Убиквитинирование.
11.10 . Почему повторная инициация
транскрипции в промоторе для РНК-
полимеразы II — более быстрый
процесс, чем событие первичной
инициации?
а. Все общие факторы транскрип-
ции остаются в промоторе и об-
легчают повторную инициацию.
Ь. Некоторые из общих факторов
транскрипции остаются в про-
моторе и облегчают повторную
инициацию.
с. Все общие факторы транскрип-
ции отделяются от промотора,
но промотор все еще выставлен,
и это позволяет осуществить бы-
струю повторную сборку инициа-
торного комплекса.
d. Ни одно из вышеупомянутого.
11.11 *. Как называют последователь-
ность ДНК, которая расположена
около промотора оперона лактозы
и которая регулирует экспрессию
оперона у Е. coli?
а. Активатор.
Ь. Индуктор.
с. Оператор.
d. Репрессор.
11.12 . В качестве которого из следующих
агентов действует аллолактоза в ре-
гулировании экспрессии оперона
лактозы?
Вопросы открытого типа
11.1* . Как мотив гомеодомена связывается
со специфичными последователь-
ностями ДНК?
а. Активатор.
Ь. Индуктор.
с. Оператор.
d. Репрессор.
11.13 *. Какие из следующих модулей по-
следовательности не являются эле-
ментом основного промотора?
а. БлокСААТ.
Ь. Блок GC.
с. Октамерный модуль.
d. ТАТА-блок.
11.14 . Какого из следующих типов мо-
дуль последовательности позволя-
ет транскрипции реагировать на
общие сигналы из внешней среды
клетки?
а. Клеткоспецифические модули.
Ь. Определяемые возрастом регуля-
торные модули.
с. Репрессорные модули.
d. Реагирующие модули.
11.15 *. Какая из следующих последова-
тельностей ДНК может увеличить
скорость инициации транскрипции
и может быть расположена в сотнях
п. н. выше или ниже от генов, ею ре-
гулируемых?
а. Активаторы.
Ь. Энхансеры.
с. Сайленсеры.
d. Терминаторы.
11.16 . Который из следующих доменов не
является доменом активационного
типа?
а. Кислые домены.
Ь. Богатые глутамином домены.
с. Домены лейциновой застежки-
молнии.
d. Богатые пролином домены.
11.2. Каковы общие свойства мотива
Cis2His2 цинкового пальца и как этот
мотив связывается с ДНК?
424 Часть 3. Принципы функционирования геномов
11.3* . Какие два варианта анализа с защи-
той от модификации имеются в ар-
сенале исследователя?
11.4. Обсудите различные пути, которы-
ми белки могут входить в контакт
с основаниями в А-, В- и Z-ДНК.
11.5* . Опишите типы связей и взаимодей-
ствий, которые происходят между
белками и молекулами ДНК.
11.6. Что случилось бы, если компоненты
-10 и -35 промотора Е. coli были бы
сдвинуты или раздвинуты друг от-
носительно друга? Обоснуйте свой
ответ.
11.7. * Объясните, в чем состоит различие
между базальными и вышележащи-
ми промоторными элементами эу-
кариотов.
11.8. Какие факторы управляют основной
скоростью инициации транскрип-
ции в промоторе бактерий?
11.9. * Как аллолактоза взаимодействует
с репрессорным белком, чтобы ре-
гулировать транскрипцию оперона
лактозы?
11.10. Назовите два фундаментальных
различия между управлением транс-
крипцией у бактерий и эквивалент-
ными событиями у эукариотов.
11.11. * Почему некоторые гены имеют
альтернативные, или множествен-
ные, промоторы?
11.12. Каковы различия между белками-
активаторами и белками-коактива-
торами?
Изыскательские задачи
11.1. * Опираясь на свои знания техно-
логии чипов ДНК и микроматриц
(техническое примечание 3.1), изо-
бретите метод опознавания сайтов
связывания для ДНК-связывающего
белка по всей хромосоме, в отличие
от методов их опознавания лишь
в пределах области выше того или
иного гена.
11.2. Постройте гипотезу, чтобы объяс-
нить, почему эукариоты имеют три
РНК-полимеразы. Каким образом
можно проверить вашу гипотезу?
11.3. * До какой степени Е. coli служит хо-
рошей моделью для регуляции ини-
циации транскрипции у эукариотов?
Подтвердите ваше мнение, приводя
характерные примеры того, как экс-
траполяции от Е. coli были полезны
и (или) бесполезны в развитии на-
шего понимания равнозначных со-
бытий у эукариотов.
11.4. Модель управления транскрипцией
оперона лактозы у Е coli была впер-
вые предложена Франсуа Жакобом
и Жаком Моно в 1961 г. (см. раздел
«Дополнительная литература»).
Объясните, до какой степени их
работа, которая почти полностью
основывалась на генетическом ана-
лизе, обеспечила точное описание
молекулярных событий, которые,
как теперь известно, действитель-
но происходят.
11.5. * Оценитеточность и полезность кон-
цепции модульной структуры про-
моторов для РНК-полимеразы II.
Глава 11. Сборка комплекса инициации транскрипции 425
Тесты по рисункам
11.1*. На этом рисунке представлено
взаимодействие между репрессо-
ром бактериофага 434 Е. coli и по-
следовательностью ДНК, с которой
он связывается. Назовите тип ДНК-
связывающего мотива, имеющегося
в этом белке, и опишите, как этот
домен взаимодействует с ДНК.
11.3*. Каково значение контактных участ-
ков, показанных на рисунке, для
пары оснований А-Т?
11.2. Обсудите эксперимент, показанный
на этом рисунке. Как этот экспери-
мент поставлен и какова его цель?
Фрагменты Фрагменты рестрикции
рестрикции + ядерный белок
11.4. На рисунке показаны два механизма,
которыми РНК-полимераза может
прикрепиться к своему промотору.
Определите, какой механизм ис-
пользуется бактериальной РНК-
полимеразой, а который — РНК-
полимеразой II у эукариотов.
а) Непосредственное прикрепление
РНК-полимеразы
РНК-полимераза
б) Опосредствованное
прикрепление РНК-полимеразы
РНК-полимераза
Площадка, образуемая
связывающимся с ДНК белком
426 Часть 3. Принципы функционирования геномов
11.5* . Опишите процесс присоединения бактериальной РНК-полимеразы к промотору,
представленный на данном рисунке.
блок -35 блок -10 Позиция +1
ДНК \ I /
niUJlll
з
liiiiiiiiiiiirtimiuaiiimiiiiiiii
।и и 1111111 imii min 11n11ТТПТП
IkUlUU'
Дополнительная литература
ДНК- и PH К-связы воющие мотивы
Fierro-Monti, I. and Mathews, М. В. (2000) Proteins binding to duplexed RNA: one motif,
multiple functions. Trends Biochem. Sci. 25: 241-246.
Gangloff,Y.G., Romier,C., Thuault,S., Werten,S. and Davidson,!. (2001) The histone
fold is a key structural motif of transcription factor TFIID. Trends Biochem. Sci. 26: 250-257.
Harrison, S.C. and Aggarwal,A.K. (1990) DNA recognition by proteins with the helix-
turn-helix motif. Annu. Rev. Biochem. 59:933-969.
Herr, W., Sturm, R. A., Clerc, R. G., et al. (1988) The POU domain: a large conserved region
in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes
Dev. 2:1513-1516.
Mackay, J. P. and Crossley, M. (1998) Zinc fingers are sticking together. Trends Biochem.
Sci. 23:1-4.
Методы изучения ДНК-связывающих белков
Galas, D. and Schmitz, A. (1978) DNase footprinting: a simple method for the detection
of protein-DNA binding specificity. Nucleic Acids Res. 5: 3157-3170.
Garner, M.M. and Revzin, A. (1981) A gel electrophoretic method for quantifying the
binding of proteins to specific DNA regions: application to components of the Escherichia coli
lactose operon regulatory system. Nucleic Acids Res. 9: 3047-3060. Gel retardation.
Глава 11. Сборка комплекса инициации транскрипции 427
Взаимодействия между ДНК и ДНК-связывающими белками
Kielkopf,C.L., White, S., Szewczyk, J. W., Turner, J. M., Baird, E.E., Dervan, P.B. and
Rees, D. C. (1998) A structural basis for recognition of AT and TA base pairs in the minor groove
of B-DNA. Science 282:111-115.
Stormo, G. D. and Fields, D. S. (1998) Specificity, free energy and information content in
protein-DNA interactions. Trends Biochem. Sci. 23:109-113.
РНК-полимеразы и их промоторы
Geiduschek, Е. Р. and Kassavetis,G.A. (2001) The RNA polymerase 111 transcription
apparatus./. Mol. Biol. 310:1-26.
Kim,Т.Н., Barrera,L.О., Zheng,M., Qu,C., Singer,M.A., Richmond,T.A., Wu,Y.,
Green, R.D. and Ren,B. (2005) A high-resolution map of active promoters in the human
genome. Nature 436: 876-880. Reveals the extent to which alternative promoters are used in
the human genome.
Russell, ).andZomerdijkJ.C.B.M. (2005) RNA-polymerase-l-directedrDNA transcription,
life and works. Trends Biochem. Sci. 30: 87-96.
Seither,P., Iben,S. and Grummt,!. (1998) Mammalian RNA polymerase I exists as
a holoenzyme with associated basal transcription factors./. Mol. Biol. 275: 43-53.
Smale, S.T. and KadonagaJ.T. (2003) The RNA polymerase 11 core promoter. Annu.
Rev. Biochem. 72: 449-479.
Young, B. A., Gruber, T. M. and Gross, C. A. (2004) Minimal machinery of RNA polymerase
holoenzyme sufficient for promoter melting. Science 303:1382-1384. Probes the fine structure
of the bacterial RNA polymerase.
Сборка комплекса инициации транскрипции
Dieci,G. and Sentenac,A. (2003) Detours and shortcuts to transcription reinitiation.
Trends Biochem. Sci. 28: 202-209.
Green,M.R. (2000) TBP-associated factors (TAFlls): multiple, selective transcriptional
mediators in common complexes. Trends Biochem. Sci. 25: 59-63.
KadonagaJ.T. (2004) Regulation of RNA polymerase 11 transcription by sequence-specific
DNA binding factors. Cell 116: 247-257.
Kim, T.-K., Ebright, R. H. and Reinberg, D. (2000) Mechanism of АТФ-dependent promoter
melting by transcription factor 11H. Science 288:1418-1421.
Verrijzer,C.P. (2001) Transcription factor 11D — not so basal after all. Science 293:
2010-2011.
Xie,X., Kokubo,T., Cohen, S.L., Mirza, U. A., Hoffmann, A., Chait, B.T., Roeder, R.G.,
Nakatani, Y. and Burley, S. K. (1996) Structural similarity between TAFs and the heterotetrameric
core of the histone octamer. Nature 380: 316-322.
Управление инициацией транскрипции у бактерий
facob, F. and Monod, J. (1961) Genetic regulatory mechanisms in the synthesis of proteins.
/. Mol. Biol. 3: 318-389. The original proposal of the operon theory for control of bacterial gene
expression.
Oehler, S., Eismann, E. R., Kramer, H. and Miiller-Hill, B. (1990) The three operators of
the lac operon cooperate in repression. EMBOJ. 9: 973-979.
Schleif,R. (2000) Regulation of the L-arabinose operon of Escherichia coli. Trends Genet
16: 559-565. Gives details of one example of bacterial gene regulation.
428 Часть 3. Принципы функционирования геномов
Управление инициацией транскрипции у эукариотов
Hanna-Rose, W. and Hansen, U. (1996) Active repression mechanisms of eukaryotic
transcription repressors. Trends Genet 12: 229-234.
Kim, Y.-J. and Lis, J.T. (2005) Interactions between subunits of Drosophila Mediator and
activator proteins. Trends Biochem. Sci. 30: 245-249.
Latchman, D. S. (2001) Transcription factors: bound to activate or repress. Trends Biochem.
Sci. 26: 211-213. Short review of proteins that combine activation with repression.
Scully, К, M., Jacobson, E. M., Jepsen, K., et al. (2000) Allosteric effects of Pit-1 DNA sites
on long-term repression in cell type specification. Science 290:1127-1131. Describes how Pit-1
acts as an activator of some genes and a repressor of others.
Wolffe, A. P. (1994) Architectural transcription factors. Science 264:1100-1101. Proteins
such as SRY which introduce bends into DNA.
Глава 12
Синтез и созревание РНК
По прочтении главы 12 вы сможете:
• Приводить подробности стадий элонгации и терминации транскрипции
у Escherichia coli и объяснять, как они регулируются антитерминацией, атте-
нюацией (ослаблением) и белками расщепления транскриптов.
• Описывать события разрезания и химической модификации, производимые
в ходе обработки функциональной РНК у бактерий.
• Подытоживать наши сегодняшние знания о деградации РНК у бактерий.
• Описывать подробности элонгации и терминации транскриптов эукариотов,
в том числе процессы, отвечающие за кэпирование и полиаденилирование
молекул мРНК эукариотов.
• Проводить различие между путями сплайсинга интронов различных типов и,
в частности, давать подробное описание сплайсинга интронов GU-AG, в том
числе примеры альтернативного сплайсинга и транссплайсинга.
• Описывать синтез и процессинг функциональной РНК у эукариотов.
• Давать определение термину «рибозим» и приводить примеры рибозимов.
• Объяснять, как молекулы рРНК эукариотов химически модифицируются в спец-
ифических позициях нуклеотидов.
• Приводить примеры редактирования РНК у млекопитающих и обрисовывать
более сложные типы редактирования РНК, которые происходяту разных других
эукариотов.
• Описывать механизмы деградации РНК у эукариотов с акцентом на роли siPHK
и миРНК в подавлении РНК.
• Вкратце описывать события, вовлеченные в перенос молекул РНК эукариотов
из ядра в цитоплазму.
Инициация транскрипции, кульминацией которой является уход РНК-полимеразы
с промотора и начало синтеза молекулы РНК, является всего лишь первым шагом на
пути экспрессии генома. В этой и в следующей главах мы будем продвигаться по этому
пути вперед и исследовать, как транскрипция и трансляция в конце него приведут нас
к синтезу протеома. Мы начнем со взгляда на синтез и процессинг РНК, в том числе
молекул мРНК, которые составляют транскриптом и определяют белковое содержание
клетки, и функциональных РНК, которые играют важные роли в экспрессии генома и дру-
гих процессах биологии клетки (раздел 1.2.2). Лежащие в их основе события подобны
у прокариотов и эукариотов, но есть значительные различия в некоторых деталях, и,
каки с другими моментами экспрессии генома, в процессы, происходящие у эукариотов,
привнесено больше сложностей. Поэтому мы начнем наш путь с рассмотрения синтеза
и созревания, или обработки (процессинга), РНК у бактерий.
430 Часть 3. Принципы функционирования геномов
12.1. Синтез и созревание РНК бактерий
Поскольку существует только одна бактериальная РНК-полимераза (раздел 11.2.1),
общий механизм синтеза РНК один и тот же для всех бактериальных генов. Однако при
рассмотрении механизмов регуляции синтеза транскриптов с отдельных генов все же
обнаруживаются некоторые отличия.
12.1.1. Синтез транскриптов в организмах бактерий
Химическая основа направляемого матрицей синтеза РНК представлена на
рис. 12.1. Рибонуклеотиды добавляются один за другим к наращиваемому З’-концу
РНК-транскрипта, род каждого нуклеотида при этом определяется правилами спари-
вания оснований: А спаривается с Т или U; G спаривается с С. Во время присоединения
Рис. 12.1. Химическая основа синтеза РНК. Сравните эту реакцию с полимеризацией ДНК, представлен-
ной на рис. 1.6
Глава 12. Синтез и созревание РНК 431
каждого очередного нуклеотида 0- и у-фосфаты удаляются из поступающего нуклео-
тида и гидроксильная группа удаляется с З’-атома углерода нуклеотида, находящегося
на конце цепи.
Удлинение транскриптов бактериальной РНК-полимеразой
В течение стадии элонгации транскрипции бактериальная РНК-полимераза нахо-
дится в свойственной ей форме центрального фермента, состоящего из четырех белков:
двух относительно небольших (приблизительно 35 кДа) а-субъединиц и по одной из
родственных субъединиц Р и Р' (обе приблизительно 150 кДа); с-субъединица, которая
играет в инициации транскрипции ключевую роль, обычно оставляет комплекс к на-
чалу этой стадии (раздел 11.2.3). РНК-полимераза покрывает приблизительно 30 п.н.
матричной ДНК, в том числе транскрипционный пузырек длиной 12-14 п. н., в пределах
которого наращиваемый транскрипт привязан к матричной нити ДНК приблизительно
восемью парами оснований ДНК-РНК (рис. 12.2).
РНК-полимераза должна крепко удерживать и ДНК-матрицу, и производимую ею
РНК, чтобы препятствовать комплексу транскрипции развалиться прежде, чем будет
достигнут конец гена. Однако это удержание не должно быть столь крепким, чтобы пре-
пятствовать полимеразе передвигаться по ДНК. С целью понять, как выполняются эти
явно противоречивые требования, взаимодействия между полимеразой, ДНК-матрицей
и РНК-транскриптом были исследованы рентгеноструктурным анализом (см. техниче-
ское примечание 11.1) в сочетании с экспериментами с перекрестным сшиванием, в ко-
торых ковалентные связи образуются между ДНК или РНК и полимеразой, при чем эти
связи позволяют опознавать аминокислоты, которые располагаются наиболее близко
кДНК и РНК. Эксперименты с перекрестным сшиванием используют разнообразие фото-
реагентов, которые могут прикрепляться к синтетическим молекулам ДНК и РНК. После
сборки комплекса метки активируются импульсом света, в результате чего они образуют
перекрестные сшивки между нуклеиновой кислотой и любыми аминокислотами в теле
полимеразы, которые расположены близко к позиции метки. Некоторые метки образуют
перекрестные сшивки с любой аминокислотой, а другие более избирательны и обра-
зуют сшивки только, например, с лизинами. После образования перекрестных сшивок
РНК-полимераза разбирается на субъединицы и каждая субъединица обрабатывается
бромцианом, который разрезает полипептиды специфично в остатках метионина, давая
характерный набор фрагментов. Тогда фрагменты, перекрестно сшитые с нуклеиновой
кислотой, опознаются. Эти фрагменты должны содержать аминокислоты, которые были
смежными с меткой в неповрежденном комплексе элонгации транскрипции. Если опо-
знать как можно больше перекрестных сшивок, то можно построить подробную карту
контактов (рис. 12.3). Эту информацию можно затем объединить с данными рентгено-
структурного анализа, чтобы построить модель комплекса элонгации транскрипции,
показывающую точное расположение различных частей двойной спирали ДНК и РНК-
транскрипта в пределах полимеразы (рис. 12.4). Эти эксперименты показали, что двойная
спираль находится между субъединицами 0 и 0' в пределах впадины на внутренней по-
Рис. 12.2. Схематичное представление комплекса элонгации
транскрипции у Escherichia coli. РНК-полимераза покрывает
приблизительно 30 п.н. ДНК, включая транскрипционный
пузырек длиной 12-14 п. н., при этом РНК прикреплена к ма-
тричной нити ДНК восемью или около того парами оснований
РНК-ДНК. Стрелка показывает направление, в котором РНК-
полимераза перемещается по ДНК
РНК-полимераза
432 Часть 3. Принципы функционирования геномов
200 400 600 800 1000 1200
200 400 600 800 1000 1200 1400
Рис. 12.3. Взаимодействия внутри бактериальной РНК-полимеразы. р- И Р'-субъединицы РНК-полимеразы
показаны оранжевым цветом; числа показывают позиции аминокислот, которые, согласно учению об
образовании поперечных (межмолекулярных) связей, лежат близко к одному и более полинуклеотидам
ДНК/РНК в пределах комплекса. Взаимодействия, проявляющиеся поперечными связями, показаны
тонкими розовыми линиями
верхности Р'-субъединицы. Активный участок для синтеза РНК также находится между
этими двумя субъединицами, при этом нематричная нить ДНК далеко выпетливается
из активного участка и удерживается внутри р-субъединицы. РНК-транскрипт выходит
из комплекса через канал, сформированный частично р- и частично Р'-субъединицей
(в правой нижней части структур, представленных на рис. 12.4).
Полимераза не синтезирует свой подопечный транскрипт с постоянной скоростью.
Вместо этого, синтез прерывист, с периодами быстрой элонгации, перемежающимися
краткими паузами, в течение которых активный участок полимеразы подвергается
небольшой структурной перестройке. Пауза редко длится дольше чем 6 миллисекунд
и может сопровождаться обратным перемещением полимеразы по матрице (возврат).
Паузы наступают случайно, а не обусловлены какой-либо специфической особенностью
матричной ДНК. Наличие пауз играет важную роль в обрыве транскрипта, как описано
ниже, но является ли это их единственной функцией, в настоящее время не известно.
Терминация бактериального транскрипта
Современное мышление рассматривает транскрипцию как прерывный процесс,
в ходе которого полимераза делает регулярные паузы и осуществляет «выбор» между
Глава 12. Синтез и созревание РНК 433
продолжением элонгации (и до-
бавлением следующих рибону-
клеотидов к транскрипту) или
терминацией (и отделением от ма-
трицы). Какой выбор будет пред-
почтен, зависит от того, которая
из альтернатив более благопри-
ятна в термодинамическом отно-
шении. Эта модель подчеркивает,
что, для того чтобы терминация
произошла, полимераза должна
достигнуть такой позиции на ма-
трице, в которой отсоединение
будет более благоприятно, чем
продолжение синтеза РНК.
Бактерии, кажется, использу-
ют две различные стратегии для
терминации транскрипции. Около
половины позиций у Escherichia coli,
в которых завершается транскрип-
Рис. 12.4. Модель комплекса элонгации транскрипции у бакте-
рий. 0- И Р'-субъединицы РНК-полимеразы изображены, соот-
ветственно синим/зеленым и розовым, двойная спираль окра-
шена красным (матричная нить) и желтым (нематричная нить),
а РНК-транскрипт — золотым. Два вида показывают, что двой-
ная спираль находится между р- и Р'-субъединицами в преде-
лах впадины на внутренней поверхности Р'-субъединицы.
Стрелки показывают направление, в котором ДНК перемеща-
ется сквозь полимеразу. Перепечатано с разрешения Korzheva
et al., Science, 289, 619-625. © 2000 AAAS
ция, соответствуют последователь-
ностям ДНК, где матричная нить
содержит инвертированный повтор, сопровождаемый вереницей дезоксиаденозиновых
нуклеотидов (рис. 12.5). Такие внутренние терминаторы, как думают, используют два
процесса, чтобы способствовать отсоединению полимеразы и дестабилизировать при-
крепление растущего транскрипта к матрице. Сначала, когда инвертированный повтор
транскрибируется, последовательность РНК сворачивается в устойчивую шпильку; такое
спаривание оснований РНК-РНК более благоприятно, чем спаривание ДНК-РНК, которое
обычно происходит в транскрипционном пузырьке. Вследствие этого уменьшается число
Матричная нить
3'
л НК 3' in । т9999^79999 ттттт9999*Т9999 тaaaaaaaaaajтттт
Дn g. 11111 CCGGTAGGCG -^—^CGCCTACCGG ДтТТТТТТТТТ 1LLLL 3'
Транскрипция
Рис. 12.5. Терминация транскрипции во внутреннем терминаторе. Присутствие обратного палиндрома
в последовательности ДНК приводит к образованию шпильки в транскрипте
434 Часть 3. Принципы функционирования геномов
контактов, образованных между матрицей и транскриптом, ослабляется взаимодействие
в целом и становится более благоприятным разобщение. Во вторую очередь, взаимодействие
еще более ослабляется, когда транскрибируется отрезок нуклеотидов А в матрице, потому
что получающиеся пары оснований A-U имеют только по две водородные связи каждая,
в отличие от трех в каждой паре G-С. В конечном счете терминация становится более пред-
почтительной, нежели дальнейшая элонгация. При известных свойствах гибридов ДНК-РНК
этой модели легко дать разумное и логически стройное обоснование, но альтернативная
гипотеза оформилась из результатов экспериментов с перекрестным сшиванием, которые
показали, что шпилька РНК вступает в контакт со структурой откидной створки на внеш-
ней поверхности 0-субъединицы РНК-полимеразы, смежной с местом выхода канала, через
который РНК выходит из комплекса (рис. 12.6). Хотя структура откидной створки весьма
отдалена (приблизительно на 6,5 нм) от активного участка полимеразы, между ними обра-
зуется прямое соединение посредством сегмента 0-листа в пределах 0-субъединицы. Поэто-
му движение откидной створки может влиять на расположение аминокислот в пределах
активного центра, возможно, оно ведет к разрыву пар оснований ДНК-РНК и к терминации
РНК выходит
из канала
Расселина активного участка
Спираль
на конце
створки
НТФ
Мд2+
Нижележащая
ДНК
Домен
створки
Соединитель
Рис. 12.6. Структура типа откидной створки на поверхности РНК-полимеразы может опосредствовать
терминацию транскрипции. Шпилька, которая формируется в РНК-транскрипте, по достижении об-
ласти терминации образует контакт со структурой типа откидной створки на внешней поверхности
0-субъединицы РНК-полимеразы, смежной с местом выхода из канала, через который РНК выходит
из комплекса. Часть полипептида 0-субъединицы, которая формирует откидную створку, окрашена
в темно-синий цвет, остальная часть 0-субъединицы — в светло-синий, 0'-субъединица — в розовый,
а а-субъединица — в белый. Имейте в виду, что, хотя структура типа откидной створки расположена
на поверхности полимеразы, область 0-полипептида, которая образует откидную створку, соединена
непосредственно с активным участком, обозначенным ионом магния сиреневого цвета и субстратом
в виде нуклеозид-5'-трифосфата — зеленого. Взаимодействие между шпилькой РНК и откидной створкой
может поэтому затрагивать расположение аминокислот в пределах активного участка, возможно, по-
буждая пары оснований ДНК-РНК разрываться, что приводит к терминации транскрипции. Перепечатано
с разрешения Toulokhonov et al., Science, 292, 730-733. © 2001 AAAs
Глава 12. Синтез и созревание РНК 435
транскрипции. Дополнительные данные в подтверждение этой модели были получены,
когда было показано, что белок, называемый NusA, который усиливает терминацию во
внутренних терминаторах, взаимодействует со шпилькой и структурой откидной створки
и может стабилизировать контакт между матрицей и транскриптом.
Сигнал терминации транскрипции
второго типа у бактерий является Rho-
зависимым. Такие сигналы обычно со-
храняют структуру шпильки внутренних
терминаторов, хотя шпилька менее устой-
чива и нет никакого отрезка нуклеотидов
А в матрице. Для терминации необходимо
действие белка, названного Rho, который
прикрепляется к транскрипту и перемеща-
ется по РНК в сторону полимеразы. Если по-
лимераза продолжает синтезировать РНК,
то она держится впереди преследующего ее
Rho, но на сигнале терминации полимераза
стопорится (см. рис. 12.7). Точная причина,
по которой это происходит, еще не была объ-
яснена — предположительно, за это неко-
торым образом отвечает шпилька, которая
формируется в РНК, — но результат ясен:
Rho способен догнать. Rho представляет со-
бой геликазу, а это означает, что он актив-
но разрывает пары оснований — в данном
случае между матрицей и транскриптом, что
приводит к терминации транскрипции.
Rho
РНК , г
>ЛГПТ!ТГТ^
iitiiiiiiiii -------лпггпттт
I Комплекс продления
застревает на шпильке
Rho разрывает пары
оснований РНК-ДНК
Рис. 12.7. Rho-зависимая терминация транскрип-
ции. Rho — это геликаза, которая следует за РНК-
полимеразой по транскрипту. Когда полимераза
застревает на шпильке, Rho догоняет ее и раз-
рывает пары оснований РНК-ДНК, высвобождая
транскрипт. Имейте в виду, что рисунок является
схематическим и не отражает относительные раз-
меры Rho и РНК-полимеразы
12.1.2. Управление выбором между элонгацией и терминацией
Можно ли повлиять как-либо на выбор, который делает взявшая паузу полимераза
между продолжением элонгации транскрипта и терминацией транскрипции путем от-
деления от матрицы? Ответ — да, причем это первейшее средство, с помощью которого
регулируется синтез (но никак не инициация) бактериальных транскриптов.
Антитерминация приводит к игнорированию сигналов терминации
. Первый из таких регулирующих процессов называют антитерминацией. Он имеет
место, когда РНК-полимераза игнорирует сигнал терминации и продолжает удлинять
свой транскрипт, пока не будет достигнут второй сигнал (рис. 12.8). Это обеспечивает
механизм, посредством коего один или несколько генов в конце оперона могут быть
выключены или включены в зависимости от того, опознает полимераза или не опознает
сигнал терминации, расположенный выше этих генов. Антитерминация управляется
антитерминаторным белком, который прикрепляется к ДНК около начала оперона
и затем переходит к РНК-пол имеразе, когда она проходит мимо на пути к первому сигналу
терминации. Присутствие антитерминаторного белка вынуждает фермент игнорировать
сигнал терминации — или противостоя дестабилизирующим свойствам внутреннего
терминатора, или предотвращая застопоривание на Rho-зависимом терминаторе.
Хотя механика антитерминации неясна, воздействие, которое она может оказать
на экспрессию гена, было описано досконально, в особенности события, развивающие-
436 Часть 3. Принципы функционирования геномов
Антитерминирующий белок
прикрепляется к РНК-
полимеразе и предотвращает
завершение в сигнале номер 1
Рис. 12.8. Антитерминация. Антитерминирующий белок прикрепляется к ДНК и переходит к РНК-
полимеразе, когда она проходит через него, впоследствии позволяя полимеразе продолжить транс-
крипцию через сигнал терминации номер 1, так что второй из пары генов в этом опероне транскриби-
руется
ся в ходе цикла инфицирования бактериофагом X. Незамедлительно после вторжения
в клетку Е. coli транскрипция генома бактериофага X инициируется присоединением
бактериальной РНК-полимеразы к двум промоторам — PL и PR — и синтезом двух
«немедленно-ранних» мРНК, они обрываются в позициях tL1 и tR1 (рис. 12.9, а). мРНК,
транскрибированная с PR по tR1, кодирует белок по имени Сто — один из главных регу-
ляторных белков, участвующих в цикле инфицирования бактериофага X. Вторая мРНК
определяет белок N, который является антитерминатором. Белок N прикрепляется
к геному фага X в участках nutL и nutR и переходит к РНК-полимеразе, когда она про-
ходит мимо. Теперь РНК-полимераза игнорирует терминаторы tL1 и tR1 и продолжает
транскрипцию вниз от этих точек. Получающиеся мРНК кодируют «задержанно-ранние»
белки (рис. 12.9, б). Поэтому антитерминация, управляемая белком N, гарантирует, что
немедленно-ранние и задержанно-ранние белки синтезируются в надлежащие моменты
времени в ходе цикла инфицирования фагом X. Один из задержанно-ранних белков, Q,
является вторым антитерминатором, который управляет переключением к поздней
стадии цикла инфицирования.
fL1 nutL PL PR nutR fR1
«Немедленно-ранние»
транскрипты
б) Гены N его CHOP Q
f|_l nutL P\_ PR nutR Jri Jr2 Jr3
«Задержанно-ранние»
транскрипты
Рис. 12.9. Антитерминация в ходе цикла инфицирования бактериофага X
Глава 12. Синтез и созревание РНК 437
Ослабление приводит к преждевременной терминации
Бактериальные мРНК не подвергаются никаким серьезным формам процессинга:
первичный транскрипт, который синтезируется РНК-полимеразой, сам по себе является
зрелой мРНК, и его трансляция обычно начинается еще до терминации транскрип-
ции (рис. 12.10). Такое сопряжение транскрипции и трансляции важно в том, что оно
позволяет применять управление специального типа, названное ослаблением, или
аттенюацией, к регулированию синтеза бактериальных мРНК.
Аттенюация работает в основном с оперонами, которые кодируют ферменты,
участвующие в биосинтезе аминокислот, но известно также несколько других приме-
ров. Оперон триптофана Е. coli (раздел 11.3.1)
иллюстрирует работу этого механизма. В этом
опероне в области между началом транскрип-
та и началом trpE могут образовываться две
шпильки. Меньшая из этих шпилек действует
как сигнал терминации, но большая шпилька,
которая ближе к началу транскрипта, более
устойчива. Большая шпилька перекрывается со
шпилькой терминации, так что только одна из
этих двух шпилек может образовываться в тот
или иной отдельный момент времени. Какая из
шпилек образуется, зависит от относительного
расположения РНК-полимеразы и рибосомы,
которая прикрепляется к 5’-концу транскрипта,
как только он будет синтезирован, с тем чтобы
транслировать гены в белок (рис. 12.11). Если
рибосома стопорится и таким образом отстает
Рис. 12.10. У бактерий транскрипция и транс-
ляция часто совмещаются
от полимеразы, то образуется большая шпилька и транскрипция продолжается. Однако
если рибосома идет в ногу с РНК-полимеразой, то она разрушает большую шпильку за
счет прикрепления к РНК, которая формирует часть стебля этой шпильки. Когда это
происходит, может образоваться терминаторная шпилька, а транскрипция — остано-
виться. Остановка рибосомы может произойти, потому что выше сигнала терминации
расположена короткая открытая рамка считывания, кодирующая 14-аминокислотный
пептид, который включает два триптофана. Если количество свободного триптофана
ограничено, то рибосома стопорится, поскольку она пытается синтезировать этот пептид,
в то время как полимераза продолжает изготовлять свой транскрипт. Поскольку этот
транскрипт содержит копии генов, кодирующих биосинтез триптофана, его элонгация
удовлетворяет спрос клетки в этой аминокислоте. Когда количество триптофана в клетке
достигает удовлетворительного уровня, система аттенюации предотвращает дальней-
шую транскрипцию оперона триптофана, потому что теперь рибосома не застревает на
производстве короткого пептида, но вместо этого идет наравне с полимеразой, позволяя
образоваться сигналу терминации.
Оперон триптофана Е coliуправляется не только аттенюацией, но также и репрессо-
ром (раздел 11.3.1). Как именно аттенюация и репрессия совмещают свое действие, чтобы
регулировать экспрессию оперона, в точности не известно, но думается, что репрессия
обеспечивает основной переключатель типа «включено-выключено», а аттенюация
модулирует точный уровень экспрессии гена, осуществляемой в случае включения. Дру-
гие опероны Е coli, такие, как например, отвечающие за биосинтез гистидина, лейцина
438 Часть 3. Принципы функционирования геномов
trpE trpD trpC trpB trpA
ДНК..
Короткая открытая Начало Короткая открытая Начало
рамка считывания trpE рамка считывания trpE
Сигнал завершения
Ограниченные количества
триптофана
Достаточное количество
триптофана
РИБОСОМА ЗАСТРЕВАЕТ
Образуется большая петля шпильки,
транскрипция продолжается
РИБОСОМА НЕ ЗАСТРЕВАЕТ
Формируется сигнал завершения,
транскрипция заканчивается
Рис. 12.11. Ослабление в триптофановом опероне
и треонина, управляются исключительно аттенюацией. Интересно, что у некоторых
бактерий, в том числе у Bacillus subtilis, оперон триптофана — один из тех, который
не имеет репрессорной системы и, таким образом, регулируется только аттенюацией.
У таких бактерий аттенюация опосредствуется не скоростью, с которой рибосома про-
катывается по мРНК, а РНК-связываюгцим белком, названным РНК-связывающим
белком аттенюации trp (TRAP), который, в присутствии триптофана, прикрепляется
к мРНК в области, соответствующей короткой ОРС в транскрипте Е. coli (рис. 12.12).
Присоединение TRAP ведет к формированию сигнала терминации и приостановке
транскрипции1^.
Недавно обнаружены другие системы аттенюации. Так, рибопереключатели образуют структуру на
мРНК, которая непосредственно взаимодействует с лигандом (например, с флавинами) и стабилизируется,
позволяя образоваться антитерминаторной шпильке. В отсутствии лиганда структура не стабильна, анти-
терминаторная шпилька не может образоваться и возникает активный терминатор транскрипции. Еще одна
система аттенюации, Т-боксы. Они непосредственно взаимодействуют с тРНК. — Прим. ред.
Глава 12. Синтез и созревание РНК 439
а)
Ограниченные количества
триптофана
Достаточное количество
триптофана
TRAP НЕ СВЯЗЫВАЕТСЯ
Образуется большая петля шпильки,
транскрипция продолжается
TRAP СВЯЗЫВАЕТСЯ
Формируется сигнал завершения,
транскрипция останавливается
Рис. 12.12. Регулирование оперона триптофана
у Bacillus subtilis. а) Центры регуляции на белке,
названном TRAP, который прикрепляется к ли-
дирующей области транскрипта, когда уровень
триптофана достаточен. Будучи прикреплен,
TRAP предотвращает формирование большой
шпильки и, таким образом, допускает формиро-
вание сигнала терминации. Сравните с рис. 12.12.
б) Структура комплекса TRAP-PHK. TRAP включает
11 идентичных субъединиц, каждая из которых
состоит преимущественно из p-листов, которые
объединяются вместе и образуют кольцевую струк-
туру диаметром 8 нм. 11 субъединиц TRAP пока-
заны разными цветами, каждая с прикрепленной
молекулой триптофана, обозначенной красными,
желтыми и синими сферическими структурами. Мо-
лекула РНК показана в виде структуры из шариков
и палочек, обернутой вокруг комплекса TRAP. Часть
б перепечатана с разрешения Antson et al., (1999),
Nature, 401, 235-242
440 Часть 3. Принципы функционирования геномов
Белки расщепления транскрипта могут предотвратить застопоривание
возвратившейся полимеразы
Возврат происходит, когда взявший паузу фермент полимераза отходит назад на
короткое расстояние по матричной нити ДНК (раздел 12.1.1). Это смещает новосинтези-
рованную молекулу РНК, 3'-конец которой отделяется от матрицы. Чтобы предотвратить
застревание полимеразы в этой точке, отделившаяся часть молекулы РНК должна быть
I Расщепление РНК с целью
предотвращения застревания
Рис. 12.13. Предотвращение застревания возвратив-
шейся РНК-полимеразы. Возврат отделяет короткую
часть недавно синтезированной РНК. Расщепле-
ние этого отделившегося сегмента необходимо для
срезана (рис. 12.13). Полимераза обладает
необходимой активностью расщепления
РНК, но обычно это действие не является
благоприятным, и, следовательно, застопо-
ривание более вероятно. Застопоривание
предотвращается парой факторов расщепле-
ния транскриптов, названных GreA и GreB,
которые, несмотря на их название, фактиче-
ски не разрезают РНК, но вместо этого про-
воцируют полимеразу, чтобы она сделала
это сама. Эти два белка, вероятно, работают
путем изменения положения одного из двух
ионов магния, которые находятся в актив-
ном участке полимеразы.
Роль GreA и GreB в предотвращении
задержания полимеразы стала ясна лишь
недавно, и никакой связи до сих пор не
было установлено между этими факторами
и каким-либо специфическим регуляторным
процессом. Известны, однако, интригующие
моменты их принципа действия, которые
предполагают, что они могут служить ме-
предотвращения застревания полимеразы
Рис. 12.14. Взаимодействие между GreB и бак-
териальной РНК-полимеразой
диаторами для регуляторных сигналов того
или иного рода. Главные части белков GreA
и GreB представлены двумя а-спиралями, отде-
ленными друг от друга коротким изгибом, со-
держащим одну аспарагиновую кислоту и одну
глутаминовую кислоту. Эти две спирали форми-
руют иглообразную структуру, которая проника-
ет в РНК-полимеразу через вторичный канал,
ведущий с поверхности фермента к активному
участку внутри белкового комплекса (рис. 12.14).
Две кислые аминокислоты на кончике иглы, как
думают, взаимодействуют с парой ионов магния
в активном участке, способствуя расщеплению
отделившегося сегмента РНК. Структура GreA
и GreB поразительно подобна таковой третьего
белка, названного DksA, который опосредствует
реакцию на стресс у Е coli и других бактерий. Эта
ответная реакция активизируется, когда в жизни
бактерии наступают неблагоприятные условия
Глава 12. Синтез и созревание РНК 441
роста, такие как низкое содержание незаменимых аминокислот в ее рационе. Дабы сохра-
нить запасы, бактерия снижает скорость транскрипции, в частности, синтез рРНК и тРНК,
прибл изител ьно до 5 % от нормального уровня. В активизации реакции на стресс участвуют
ppGpp и pppGpp (рис. 12.15) — два необычных нуклеотида, называемых также алармо-
нами, — которые синтезируются белком RelA в ответ на голодание по аминокислотам.
Алармоны работают в содружестве с DksA, чтобы приглушить транскрипцию. Как именно
это происходит, пока что можно лишь предполагать, но подобие между структурой DksA
и таковой GreA и GreB наводит на мысль о том, что DksA, подобно факторам расщепления
транскриптов, вводит свою иглообразную структуру во вторичный канал полимеразы и
посредством пары кислых аминокислот на ее конце (в данном случае—две аспарагиновые
кислоты) влияет на активность полимеразы, взаимодействуя с определяющими ситуацию
ионами магния в активном участке. Современная модель приписывает алармонам прямую
роль в этом процессе, предусматривая, что ppGpp входит в канал на конце иглы DksA и уча-
ствует в контакте с ионами магния.
Шаг транскрипции, который ингибируется во время реакции на стресс, точно не
известен — некоторые данные указывают на события, происходящие в ходе процесса
б) pppGpp
a) ppGpp
О Н
О=Р-О-
I
о
I
О=Р-О-
I
О’
О’ О’
О-
"О- Р- о- Р- о- р- о - сн2 EI
:о.
0 V?
о н
I
О=Р-О-
I
о
о=р-о-
।
О’
о
о
Рис. 12.15. Структуры алармонов ppGpp и pppGpp. Сравните со стандартными структурами нуклеотидов,
показанными на рис. 1.4
инициации, а другие результаты предполагают, что мишенью воздействия является
полимераза во время паузы. Важный момент, однако, заключается в том, что роль DksA
в этом регуляторном процессе указывает на то, что GreA и GreB, которые действуют по
схеме, почти неотличимой от принципа действия DksA, могут участвовать также и в
других, пока еще неоткрытых, механизмах регулирования синтеза РНК у бактерий.
12.1.3. Созревание бактериальных РНК
Из раздела 1.2.3 мы узнали, что большинство молекул РНК синтезируется в виде
предшественников, которые должны быть обработаны прежде, чем они смогут вы-
полнять свои функции в клетке. Бактериальная мРНК — одно исключение из этого
правила: первичные транскрипты большинства бактериальных кодирующих белок
генов являются функционально активными мРНК, которые незамедлительно могут
быть транслированы (см. рис. 12.10). Бактериальные тРНК и рРНК, с другой стороны,
сначала транскрибируются в виде пре-РНК, которые становятся функционально актив-
ными только после ряда событий разрезания и химической модификации.
442 Часть 3. Принципы функционирования геномов
тРНК
Пре-рРНК
Последовательность Последовательность Последовательность
рРНК 16S рРНК 23S рРНК 5S
Рис. 12.16. Обработка пре-рРНК у Escherichia coli. Обратите внимание, что тРНК расположена между
последовательностями 16 S и 23 S в этой пре-рРНК Е. coli. Эта тРНК обрабатывается, как показано на
рис. 12.17
События разрезания высвобождают зрелые рРНК и тРНК из молекул-
предшественников
Бактерии синтезируют три различные рРНК, называемые 5S рРНК, 16S рРНК и 23S
рРНК (названия сообщают размеры молекул, определенные при помощи осаждающего
анализа; см. техническое примечание 7.1). Три гена для этих рРНК сцеплены в единую
единицу транскрипции (которая обычно присутствует в виде многочисленных копий:
у Е. coli — семь), и, таким образом, пре-рРНК содержит копии всех трех рРНК. Поэтому,
чтобы выпустить зрелые рРНК, необходимы события разрезания. Эти разрезы произво-
дятся рибонуклеазами III, Р и F в позициях, предписанных двунитевыми областями, об-
разованными спариванием оснований между различными частями пре-рРНК (рис. 12.16).
Края разрезов впоследствии обрезаются за счет экзонуклеазного действия рибонуклеаз
М16, М23 и М5 и в конечном счете получаются зрелые рРНК.
Гены молекул тРНК распределены по геному бактерий, некоторые сами по себе,
а некоторые в виде тандемных множеств в мульти-тРНКовых единицах транскрипции.
У некоторых бактерий гены тРНК находятся также и в качестве резидентов в единицах
транскрипции рРНК, как это имеет место у Е. coli, которая имеет один или два гена тРНК
между генами 16S рРНК и 23S рРНК в каждой из ее семи единиц транскрипции рРНК
(см. рис. 12.16). Все пре-тРНК обрабатываются подобными, хотя и не идентичными
способами. Пример, показанный на рис. 12.17, относится к предшественнику молеку-
лы тРНКТуг Е. coli. Последовательность тРНК в молекуле предшественника принимает
присущую ей структуру спаренных оснований в форме клеверного листа (см. рис. 13.2),
и образуются две дополнительные структуры шпильки, по одной на каждой из сторон
тРНК. Процессинг начинается с разреза рибонуклеазой Е или F, образующего новый
З’-конец непосредственно выше одной из шпилек. Рибонуклеаза D, которая является
экзонуклеазой, обрезает семь нуклеотидов с этого нового З'-конца и затем берет паузу,
в то время как рибонуклеаза Р делает разрез в начале клеверного листа, образуя 5’-конец
зрелой тРНК. Затем рибонуклеаза D удаляет еще два нуклеотида, создавая З'-конец
Глава 12. Синтез и созревание РНК 443
Рис. 12.17. Обработка пре-тРНК Escherichia coli. В качестве примера показан синтез тРНКТуг. Разрез, отде-
ляющий предмет нашего интереса от некоторых других пре-тРНК и произведенный РНКазой Z, показан
красным. Сокращение: РНКаза — рибонуклеаза
зрелой молекулы. Все зрелые тРНК должны оканчиваться тринуклеотидом 5'-ССА-3’.
В случае тРНКТуг конечный тринуклеотид ССА присутствует в пре-РНК и не удаляется
рибонуклеазой D, но в некоторых других пре-тРНК эта последовательность отсутствует
или удаляется обрабатывающими рибонуклеазами. Это имеет место у большинства тех
пре-тРНК, З'-концы которых создаются эндонуклеазой, названной рибонуклеазой Z,
которая делает разрез, примыкающий к первой паре оснований в нашем трилистнике
тРНК (см. рис. 12.17), и, следовательно, удаляет область, которая содержала бы оконеч-
ный тринуклеотид ССА. Когда такой триплет ССА отсутствует, он должен быть добавлен
одной или несколькими не зависимыми от матрицы РНК-полимеразами, например
тРНК-нуклеотидилтрансферазы.
Из различных нуклеаз, упомянутых выше, рибонуклеаза Р особенно интересна,
потому что в число различных субъединиц этого фермента входит такая, которая
образована из РНК, а не из белка. Субъединицы из РНК присутствуют в нескольких
ферментах, участвующих в процессинге РНК, включая рибонуклеазу MRP, которая
работает с рибонуклеазой Р в процессинге пре-рРНК эукариотов (раздел 12.2.4). Такие
гибридные белково-РНКовые ферменты могут быть реликтами, оставшимися от мира
РНК — раннего периода эволюции, в котором все биологические реакции были заме-
шаны на РНК (раздел 18.1.1).
Модификации нуклеотидов расширяют спектр химических свойств тРНК
и рРНК
Процессинг последнего типа, который происходит с пре-РНК, — химическая мо-
дификация нуклеотидов транскрипта. Она происходит и с пре-рРНК, и с пре-тРНК.
Широкий спектр химических изменений был обнаружен у разных пре-РНК: всего из-
вестно более 50-ти модификаций (рис. 12.18). Большинство из них выполняется прямо
444 Часть 3. Принципы функционирования геномов
МЕТИЛИРОВАНИЕ
ДЕЗАМИНИРОВАНИЕ
СН3 о
J <?
'''NH
< И 1
" ЬГ 4NH2
Сахар
ЗАМЕЩЕНИЕ
СЕРОЙ
S
II
С.
HN СН
I II
/СН
о hr
I
Сахар
Сахар
7-Метил гуанозин
Инозин
4-Тиоуридин
ИЗОМЕРИЗАЦИЯ
ОСНОВАНИЯ
НАСЫЩЕНИЕ
ДВОЙНОЙ СВЯЗИ
О
II
С.
ньг сн2
.С\ /СН2
о Nx
।
ЗАМЕЩЕНИЕ
НУКЛЕОТИДА
Псевдоуридин
Сахар
Ди гидроуридин
NH
сн2 °
I л
// С NH
Сахар
NH2
Квеозин
Рис. 12.18. Примеры химических модификаций, происходящих с нуклеотидами в рРНК и тРНК. Метили-
рование есть добавление одной или более групп -СН3 к основанию или сахару. Дезаминирование —
удаление аминогруппы (-NHJ от основания: инозин — дезаминированная форма аденозина. Замещение
серой вовлекает замену кислорода серой. Изомеризация основания происходит, когда положения
атомов в циклическом компоненте основания изменяются: изомеризация уридина дает псевдоуридин.
Насыщение двойной связи преобразует двойную связь в одинарную: например, при преобразовании
уридина в дигидроуридин. Замещение нуклеотида — замена существующего нуклеотида новым (типа
квеозина)
на существующем нуклеотиде в транскрипте, но два модифицированных нуклеотида,
квеозин и виозин, ставятся на место путем вырезания целого нуклеотида и замены его
на видоизмененный вариант.
Многие из таких необычных нуклеотидов были впервые обнаружены в тРНК, в ко-
торых приблизительно один из десяти нуклеотидов претерпевает видоизменение. Эти
модификации, как думают, опосредствуют опознавание отдельных тРНК ферментами,
которые присоединяют аминокислоты к этим молекулам (раздел 13.1.1) и увеличивают
спектр взаимодействий, которые могут произойти между тРНК и кодонами в ходе транс-
ляции, позволяя одной тРНК опознавать более одного кодона (раздел 13.1.2). Мы знаем
относительно немного о том, как выполняются модификации тРНК, помимо того факта,
Глава 12. Синтез и созревание РНК 445
что есть ряд ферментов, которые катализируют эти изменения. Предполагается, что
для опознавания тех нуклеотидов, которые надлежит модифицировать, эти ферменты
используют характерные особенности структуры спаренных оснований тРНК.
Рибосомные РНК модифицируются двумя способами: добавлением метильных
групп, главным образом, к группе 2’-0Н на сахарах нуклеотидов и превращением уридина
в псевдоуридин (см. рис. 12.18). Одна и та же модификация происходит в одной и той
же позиции на всех копиях молекулы рРНК, и эти видоизмененные позиции до некото-
рой степени одни и те же у разных видов. Некоторые подобия в схемах модификации
наблюдаются даже при сравнении бактерий и эукариотов, хотя бактериальные рРНК
видоизменяются менее значительно, чем таковые эукариотов. Функции модификаций
не были определены, хотя большая часть происходит в тех частях рРНК, которые пред-
положительно являются наиболее важными для активности этих молекул в рибосомах
(раздел 13.2.1). Модифицированные нуклеотиды могут, например, участвовать в ка-
тализируемых рРНК реакциях, таких как синтез пептидных связей. У эукариотов для
модификации молекул рРНК существует сложный механизм (раздел 12.2.5), но такой
механизм отсутствует у бактерий, рРНК которых модифицируются ферментами, кото-
рые напрямую опознают последовательность и (или) характерные структуры областей
РНК, содержащие нуклеотиды, которые необходимо модифицировать. Часто в одной
и той же области модифицируются сразу два или больше нуклеотидов. Модификация
рРНК у бактерий поэтому подобна системам модификации молекул тРНК и у бактерий,
и у эукариотов.
12.1.4. Деградация бактериальных РНК
До сих пор эта глава была сосредоточена на синтезе молекул РНК. Их деградация
столь же важна, особенно в отношении молекул мРНК, присутствие или отсутствие кото-
рых в клетке определяет, какие белки будут синтезированы. Деградация определенных
молекул мРНК может быть мощным средством регулирования экспрессии генома.
Скорость деградации мРНК может быть оценена определением ее периода по-
лураспада в клетке. Оценки показывают, что есть значительные отличия как между
различными организмами, так и в пределах одних и тех же организмов. Бактериальные
мРНК, как правило, оборачиваются очень быстро, их периоды полураспада редко пре-
вышают несколько минут, что является отражением быстрых изменений в картинах
синтеза белков, могущих происходить в активно растущей бактерии со временем смены
поколений 20 минут или около того. Молекулы мРНК эукариотов живут дольше и имеют
период полураспада в среднем 10-20 минут у дрожжей и несколько часов — у млеко-
питающих1).
Бактериальные мРНК деградируют в направлении 3'^5'
В ходе исследований мутантных бактерий, мРНК которых имеют увеличенные
периоды полураспада, был опознан ряд рибонуклеаз и других ферментов деградации
РНК, которые, как думают, участвуют в деградации мРНК. К таковым относятся:
• РНКаза Е и РНКаза III, которые являются эндонуклеазами, делающими внутренние
разрезы в молекулах РНК;
• РНКаза II, которая является экзонуклеазой, удаляющей нуклеотиды в направлении
3'—>5';
Отметим, что характерное время жизни мРНК примерно такое же, как и время синтеза — небольшие
минуты для бактерий, несколько часов для млекопитающих. — Прим. ред.
446 Часть 3. Принципы функционирования геномов
Терминирующая шпилька
3'
РНКаза II, ПНФаза
РНКаза Е и (или)
РНКаза II, ПНФаза
Рис. 12.19. Деградация бактериальной РНК.
Терминирующая шпилька блокирует дей-
ствия экзонуклеазы у РНКазы II и у ПНФазы
и, таким образом, должна быть удалена
действием эндонуклеазы (у РНКазы Е и
(или) у РНКазы III) прежде, чем деградация
сможет продолжиться
• полинуклеотидфосфорилаза (ПНФаза), которая также удаляет нуклеотиды по-
следовательно с З’-конца мРНК, но, в отличие от истинных нуклеаз, требует не-
органический фосфат в качестве сосубстрата.
Ни один фермент, способный деградиро-
вать РНК в направлении 5'->3', не был до сих
пор выделен из бактерий. Это отсутствие ведет
к предположению о том, что главный процесс
деградации бактериальных мРНК является уда-
лением нуклеотидов с З'-конца. Это невозможно
в нормальных условиях, потому что большинство
молекул мРНК имеет структуру шпильки около
З’-конца — та же самая шпилька, которая вы-
зывает терминацию транскрипции (см. рис. 12.5
и 12.7). Эта структура блокирует продвижение
РНКазы II и ПНФазы, препятствуя их доступу
к кодирующей части транскрипта (рис. 12.19).
Поэтому модель деградации мРНК начинается
с удаления З'-конечной области, в том числе
шпильки, одной из эндонуклеаз, в результате чего
выставляется новый конец, с которого РНКаза II
и ПНФаза могут забраться в кодирующую область
и нарушить функциональную активность мРНК.
Полиаденилирование также может сыграть свою
роль. Хотя изначально поли-А хвосты рассматри-
вались как особенность мРНК эукариотов (раз-
дел 12.2.1), начиная с 1975 г. стало известно, что
многие бактериальные транскрипты имеют их
на некоторой стадии своего существования, но что эти хвосты быстро деградируют.
В настоящее время не ясно, предшествует ли полиаденилирование деградации мРНК
или оно происходит на разных промежуточных стадиях уже после того, как деградация
началась.
В клетке РНКаза Е и ПНФаза расположены в многобелковом комплексе, названном
деградосомой. К остальным компонентам деградосомы относится РНК-геликаза, ко-
торая, как думают, помогает деградации, раскручивая двойные спирали структур РНК.
Фрагменты рРНК иногда совыделяются с деградосомой, что предполагает, что этот
комплекс может участвовать в деградациии и рРНК и мРНК. Но точная роль деградосо-
мы все еще не ясна, и немногочисленный ряд исследователей скептически относится
к факту ее существования, указывая на то, что белки, неявно участвующие в деградации
мРНК, такие как фермент гликолиза енолаза, оказались компонентами деградосомы,
что, возможно, указывает на то, что этот комплекс является артефактом, производимым
в ходе извлечения белков из клеток бактерий. Более значительный пробел в нашем
знании касается пути, которым деградация специфично нацеливается на отдельные
молекулы мРНК. О том, что происходит именно специфическая деградация, мы знаем
потому, что деградация мРНК оказалась причастной к регулированию нескольких на-
боров бактериальных генов, таких как оперон pap Е. coli, который кодирует белки, уча-
ствующие в синтезе ворсинок на поверхности клетки. К сожалению, процесс, которым
такое управление осуществляется, остается необъяснимой загадкой.
Глава 12. Синтез и созревание РНК 447
12.2. Синтез и созревание РНК эукариотов
На самом фундаментальном уровне транскрипция у бактерий и эукариотов подобна.
Химия полимеризации РНК идентична в организмах всех типов, и все три ядерные РНК-
полимеразы эукариотов структурно связаны с РНК-полимеразой Е. coli, три их наиболь-
шие субъединицы эквивалентны субъединицам а, 0 и 0’ фермента бактерии. Контакты
между РНК-полимеразой II эукариотов, матричной ДНК и РНК-транскриптом, как показа-
ли рентгеноструктурный анализ и эксперименты с перекрестным сшиванием, подобны
взаимодействиям, описанным на примере транскрипции у бактерий (раздел 12.1.1),
а основной принцип, согласно которому транскрипция является последовательным
соревнованием между элонгацией и терминацией, также остается верным.
12.2.1. Синтез мРНК эукариотов РНК-полимеразой II
Несмотря на лежащие в основе подобия, в целом процессы синтеза мРНК у бакте-
рий и эукариотов весьма различны. Самое разительное отличие — степень, до которой
молекулы мРНК эукариотов обрабатываются в ходе транскрипции. У бактерий транс-
крипты кодирующих белок генов не обрабатываются вообще: первичные транскрипты
представляют собой зрелые молекулы мРНК. Напротив, все мРНК эукариотов имеют
кэп, насаженный на 5'-конец, большинство также полиаденилируется добавлением
ряда аденозинов к З'-концу, многие содержат интроны и, таким образом, подвергаются
сплайсингу, а некоторым назначается процедура редактирования РНК. Если к копирова-
нию была приписана функция, то цель полиаденилирования почти полностью остается
под покровом тайны. Что до сплайсинга и редактирования, то мы можем понять, почему
эти события происходят — первое удаляет интроны, которые блокируют трансляцию
мРНК, а второе изменяет кодирующие свойства мРНК, — но мы не можем постичь того,
для чего эти механизмы эволюционировали. Во-первых, почему гены имеют интроны?
Зачем нужно редактировать мРНК, вместо того чтобы сразу кодировать желаемые по-
следовательности с ДНК?
Молекулы мРНК эукариотов обрабатываются одновременно со своим синтезом.
Кэп добавляется, как только транскрипция была запущена, сплайсинг и редактирование
начинаются, когда транскрипт еще продолжает синтезироваться, а полиаденилирова-
ние — встроенная часть механизма терминации для РНК-полимеразы II. Знакомство
со всеми этими событиями вкупе привело бы в замешательство, ибо слишком много
разных вещей пришлось бы воспринимать сразу. Поэтому мы отложим изучение ре-
дактирования РНК до конца главы, а это означает, что его можно будет рассмотреть
в связке с подобными формами химической модификации, которые происходят в ходе
процессинга рРНК и тРНК, и мы посвятим отдельный раздел сплайсингу после изучения
кэпирования, элонгации и полиаденилирования.
Кэпирование произведенных РНК-полимеразой II транскриптов Происходит
сразу после инициации
Хотя фосфорилирование С-концевого домена наибольшей субъединицы РНК-
полимеразы II — заключительный шаг в инициации транскрипции кодирующих мРНК
генову эукариотов (раздел 11.2.3), оно не сопровождается незамедлительным началом
элонгации. Своего рода белое пятно существует в нашем понимании событий, отли-
чающих очистку промотора, которая относится к переходу от предынициаторного
комплекса к комплексу, начавшему синтезировать РНК, и уход от промотора, в ходе
которого полимераза отодвигается от промоторной области и предается созданию транс-
448 Часть 3. Принципы функционирования геномов
крипта (рис. 12.20). Противоположные воздействия отрицательных и положительных
факторов элонгации влияют на способность полимеразы начать продуктивный синтез
РНК, и если отрицательные факторы преобладают, то транскрипция останавливается
прежде, чем полимераза переместится более чем на 30 нуклеотидов от точки инициации.
Поэтому уход от промотора может быть важной контрольной точкой, но каким образом
регулирование применяется на этой стадии, еще неизвестно.
Успешный уход от промотора может быть связан с кэпированием, так как это событие
процессинга завершается до того, как транскрипт достигнет 30 нуклеотидов в длину. Первым
шагом кэпирования является добавление дополнительного гуанозина к самому 5'-концу
РНК. Вместо того чтобы происходить как нормальная полимеризация РНК, кэпирование
предполагает реакцию между 5’-трифосфатом оконечного нуклеотида и трифосфатом
нуклеотида ГТФ. у-фосфат оконечного нуклеотида (наиболее удаленный фосфат) удаляет-
ся, как и 0- и у-фосфаты ГТФ, в результате чего образуется связь 5-5’ (рис. 12.21). Реакция
выполняется ферментом 1уанилилтрансферазой. Второй шаг реакции кэпирования пре-
вращает новый оконечный гуанозин в 7-метилгуанозин присоединением метильной группы
к атому азота номер 7 пуринового кольца, эта модификация катализируется 1уанинметил-
трансферазой. Два копирующих фермента образуют точки прикрепления с С-концевым
доменом, и вполне возможно, что они являются собственными компонентами комплекса
РНК-полимеразы II во время очистки промотора.
Структуру 7-метилгуанозина называют кэпом 0-го типа, и она является наиболее
распространенной формой у дрожжей. У высших эукариотов встречаются дополнитель-
ные модификации (см. рис. 12.21).
• Второе метилирование заменяет водород группы 2'-0Н того, что является теперь
вторым нуклеотидом в транскрипте. В результате этого образуется кэп 1-го типа.
• Если этот второй нуклеотид представлен аденозином, то аминогруппа, присоеди-
ненная к атому углерода номер 6 пуринового кольца, также может быть метили-
рована.
• Метилирование других групп 2'-0Н может иметь место в позиции третьего ну-
клеотида, что дает кэп 2-го типа.
Все РНК, синтезируемые РНК-полимеразой II,
так или иначе кэпируются. Это означает, что, как
и молекулы мРНК, мяРНК, которые транскри-
бируются этим ферментом, также блокируются
кэпом (см. табл. 11.3). Кэп может быть важен для
экспорта мРНК и мяРНК из ядра (раздел 12.2.7), но
его наиболее очевидная роль состоит в трансля-
ции молекул мРНК, что будет рассмотрено в раз-
деле 13.2.2.
Рис. 12.20. Очистка промотора и уход от промотора. Очист-
ка промотора — переход от предынициаторного комплекса
к комплексу, который начал синтезировать РНК. Уход от
промотора происходит, когда полимераза отодвигается
от области промотора и предается созданию транскрипта.
Имейте в виду, что рисунок является схематическим и не
предназначен для отображения формы или состава субъе-
диниц комплекса РНК-полимеразы II, который синтезирует
этот транскрипт
Предзапускающий комплекс
1 ДНК
Промоторная область
| Очистка промотора
РНК., -----
РНК-полимераза II
| Уход от промотора
РНК —»
РНК-полимераза II
теперь предана
синтезу транскрипта
Глава 12. Синтез и созревание РНК 449
Gppp+pppN -ч
Гуанилилтрансфераза
GpppN -х
Гуанинметилтрансфераза
О = Р—0“
О
О = Р—О"
о
иРНК
Рис. 12.21. Кэпирование мРНК эукариотов. В верхней части рисунка схематично показана реакция кэ-
пирования. Молекула ГТФ (обозначена Gppp) реагирует с 5'-концом мРНК и дает трифосфатную связь.
На втором шаге процесса оконечный G метилируется в атоме азота номер 7. В нижней части рисунка
показана химическая структура кэпа 0-го типа, где звездочками отмечены позиции, в которых может
произойти дополнительное метилирование и получиться структура кэпа 1-го типа или 2-го типа
Удлинение мРНК эукариотов
Как было упомянуто выше, фундаментальные основы элонгации транскрипта у бак-
терий и эукариотов одни и те же. Одно главное отличие касается длины синтезируемого
транскрипта, который должен быть синтезирован. Самые длинные бактериальные гены
имеют только несколько тыс.п.н. в длину и могут быть транскрибированы в течение
450 Часть 3. Принципы функционирования геномов
нескольких минут бактериальной РНК-полимеразой, которая обладает скоростью по-
лимеризации несколько сот нуклеотидов в минуту. В отличие от нее, РНК-полимеразе II
могут понадобиться часы, чтобы транскрибировать один-единственный ген, даже при
том, что она может работать со скоростью до 2 000 нуклеотидов в минуту. Это связано
с тем, что присутствие многочисленных интронов во многих генах эукариотов (раз-
дел 12.2.2) означает, что должны быть скопированы отрезки ДНК значительной длины.
Например, пре-мРНК гена дистрофина человека имеет 2400 тыс.п.н. в длину и на её
синтез затрачивается около 20 часов.
Необычайная длина генов эукариотов налагает требования на устойчивость
комплекса транскрипции. РНК-полимераза II сама по себе не способна удовлетворить
этим требованиям: когда очищенный фермент изучается in vitro, его скорость поли-
меризации составляет менее 300 нуклеотидов в минуту, потому что фермент часто
берет паузу на матрице, а иногда совсем останавливается. В ядре паузы и остановы
сокращены благодаря действию ряда факторов элонгации — белков, которые
соединяются с полимеразой после того, как она очистила промотор и оставила по-
зади факторы транскрипции, участвующие в инициации. В клетках млекопитающих
в настоящее время известно тринадцать факторов элонгации, что говорит о разно-
образии их функций (таблица 12.1). Их важность показывают последствия мутаций,
которые нарушают активность одного или другого фактора. Инактивация CSB, на-
пример, приводит к развитию синдрома Кокейна — болезни, характеризующейся
пороками развития, такими как задержка умственного развития, а разрушение ELL
вызывает острую миелоидную лейкемию.
Второе отличие между элонгацией транскрипта у бактерий и эукариотов — что
РНК-полимераза II, равно как и другие ядерные полимеразы эукариотов, должна преодо-
леть нуклеосомы, которые присоединены к транскрибируемой матричной ДНК. На
первый взгляд, трудно вообразить, как полимераза может удлинять свой транскрипт
через область ДНК, обернутую вокруг нуклеосомы (см. рис. 7.2). Решение этой пробле-
мы, вероятно, обеспечивают факторы элонгации, которые способны модифицировать
структуру хроматина некоторым образом. У млекопитающих фактор элонгации FACT,
как было показано, взаимодействует с гистонами Н2А и Н2В, возможно, влияя на рас-
положение нуклеосом, и для других факторов были продемонстрированы менее ясные
взаимодействия. Дрожжи обладают фактором, названным элонгатором, которому была
приписана роль модификации хроматина, потому что он содержит субъединицу, обла-
дающую действием гистонацетилтрансферазы, но пока что гомолог этого комплекса
не был обнаружен у млекопитающих. Интригующий вопрос — является ли первая по-
лимераза, которая транскрибирует некоторый ген, «первопроходцем» с экипажем в лице
специального фактора элонгации, который открывает структуру хроматина, а после-
Таблица 12.1. Примеры факторов элонгации для РНК-полимеразы II млекопитающих
Фактор элонгации Функции
TFIIF, CSB, ELL, Elongin Эти факторы подавляют паузы РНК-полимеразы, что может прои-
зойти, когда фермент транскрибирует область, где внутринитевые
пары оснований (например, петля шпильки) могут образоваться
TFIIS Предотвращает задержание (полную приостановку элонгации)
FACT Предположительно модифицирует хроматин, с тем чтобы содей-
ствовать элонгации
Глава 12. Синтез и созревание РНК 451
дующие круги транскрипции выполняются стандартными комплексами полимеразы,
которые пользуются изменениями, произведенными пионером.
Терминация синтеза большинства мРНК сопряжена с полиаденилированием
Большинство молекул мРНК эукариотов имеет на своих З’-концах ряд до 250 адено-
зинов. Эти поли-А тракты не задаются ДНК и добавляются к транскрипту независимой от
матрицы РНК-полимеразой, названной поли-А-полимеразой. Эта полимераза воздействует
не на самый З'-конец транскрипта, но на внутренний участок, который расщепляется, с тем
чтобы создать новый З'-конец, к которому и добавляется поли-А хвост.
Основные особенности полиаденилирования были поняты некоторое время назад.
У млекопитающих полиаденилирование направляется сигнальной последовател ьностью
в мРНК, почти неизменно 5'-AAUAAA-3'. Эта последовательность расположена между
10 и 30 нуклеотидами выше участка полиаденилирования и часто непосредственно
лежит за динуклеотидом 5'-СА-3' и сопровождается 10-20 нуклеотидами спустя обо-
гащенной GU области. И сигнальная последовательность поли-А и обогащенная GU
область служат сайтами связывания для многосубъединичных белковых комплексов,
которые, соответственно, представляют собой фактор специфичности расщепления
и полиаденилирования (CPSF) и фактор стимуляции расщепления (CstF). Поли-А-
полимераза и по крайней мере два других белковых фактора должны соединиться со
связанными CPSF и CstF для того, чтобы произошло полиаденилирование (рис. 12.22).
Эти дополнительные факторы включают связывающийся с полиаденилатом белок
(PADP), который помогает полимеразе добавлять аденозины, возможно, влияет на длину
синтезируемого поли-А хвоста и, кажется, играет роль в поддержании хвоста после син-
теза. У дрожжей сигнальные последовательности в транскрипте немного отличаются,
но белковые комплексы подобны таковым у млекопитающих, и полиаденилирование,
как думают, осуществляется более или менее схожим механизмом.
Когда-то полиаденилирование рассматривали как «посттранскрипционное» со-
бытие, но теперь общепризнано, что процесс является собственной частью механизма
Пре-мРНК
5' 3'
AAUAAA ~ " СА Я^ВК GU ВВ^В
i Сборка комплекса
полиаденилирования
Связывающийся с полиаденилатом белок
Полиаденилированная
мРНК
Рис. 12.22. Полиаденилирование мРНК эукариотов. Имейте в виду, что рисунок является схематическим
и не имеет целью показать ни относительные размеры и формы различных белковых комплексов, ни
их точное расположение, хотя CPSF и CstF, как думают, связываются, соответственно, с сигнальной по-
следовательностью 5'-AAUAAA-3' и обогащенными GU последовательностями, как здесь и показано.
Учтите, что «GU» обозначает обогащенную GU последовательность, а не динуклеотид 5'-GU-3'
452 Часть 3. Принципы функционирования геномов
РНК-полимераза II
1
РНК -— —►
ДНК------------------------------------------------------------------
Последовательность сигнала
Т полиаденилирования
CPSF I
I CPSF прикрепляется
к сигнальной последовательности
I
Терминация становится
предпочтительнее
элонгации
Рис. 12.23. Связь между полиаденилированием и терминацией транскрипции РНК-полимеразой II. CPSF
показан прикрепленным к комплексу элонгации РНК-полимеразы II, который синтезирует РНК. CPSF свя-
зывается с последовательностью сигнала полиаденилирования, как только она транскрибирована. Это
изменяет взаимодействие между CPSF и С-концевым доменом РНК-полимеразы II, так что терминация
транскрипции теперь предпочтительна по сравнению с продолжающейся элонгацией. Учтите, что это
схематическое представление и оно игнорирует возможность, что CstF может быть компонентом также
комплекса элонгации. На этой схеме показан также CPSF, покидающий комплекс с тем, чтобы связаться
с сигналом полиаденилирования, тогда как в действительности он может поддерживать свой контакт
с РНК-полимеразой II во время процесса полиаденилирования
терминации транскрипции для РНК-полимеразы II. Известно, что CPSF взаимодействует
с TFIID и привлекается в комплекс полимеразы во время стадии инициации транс-
крипции. Прокатываясь по матрице на РНК-полимеразе II, CPSF способен связаться
с сигнальной последовательностью поли-А, как только она будет транскрибирована,
и запустить реакцию полиаденилирования (рис. 12.23). И CPSF и CstF образуют кон-
такты с С-концевым доменом полимеразы. Было высказано предположение о том, что
характер этих контактов изменяется, когда обнаруживается сигнальная последователь-
ность поли-А, и что это изменение видоизменяет свойства комплекса элонгации, так
что терминация становится более предпочтительной, чем продолжение синтеза РНК.
В результате транскрипция останавливается вскоре после транскрибирования сигналь-
ной последовательности поли-А.
Даже при том, что полиаденилирование может быть признано неотъемлемой
частью процесса терминации, это не объясняет, почему необходимо добавлять поли-А
хвост к транскрипту. Роль поли-А хвоста пытались установить в течение нескольких лет,
но никаких убедительных данных не было найдено в пользу ни одного из различных
предположений, которые были высказаны на этот счет. Эти предположения включают
влияние на стабильность мРНК, что кажется маловероятным, так как некоторые устой-
чивые транскрипты имеют очень короткие поли-А хвосты, а также роль в инициации
трансляции. Последнее предположение подтверждается исследованием, показавшим,
что активность поли-А-полимеразы подавляется на протяжении тех периодов клеточ-
ного цикла, когда происходит сравнительно скудный синтез белка.
Глава 12. Синтез и созревание РНК 453
Рис. 12.24. Обработка З'-концов мРНК
гистонов, а) Структура З'-области гисто-
новой мРНК, показывающая шпильки
и консенсусную последовательность из
9 нуклеотидов, б) Консенсусная после-
довательность образует пары оснований
с короткой областью 117-лляРНК. Прикре-
пление стабилизируется связывающимся
с петлей шпильки белком (не показан на
этом рисунке), а разрез делается неопо-
знанным белком в позиции четыре или
пять нуклеотидов ниже петли шпильки
Роль, которая в конечном
счете приписана полиаденили-
рованию, должна быть прописа-
на с учетом того факта, что не все
мРНК эукариотов имеют поли-А
хвост. Неполиаденилированные
б) В расщеплении участвует U7-mbPHK
мРНК составляют маленькую группу, но они включают несколько важных членов,
наиболее значительные из них те мРНК, что задают гистоновые белки. З’-концы таких
неполиаденилированных мРНК, подобно полиаденилированным вариантам, создаются
путем расщепления первичного транскрипта в специфической позиции, но сигналы
расщепления и белки, участвующие в этом процессе, совершенно иные. В пределах
транскрипта находятся два сигнала расщепления — первый из них шпилька, которая
образуется ниже кодирующей области (рис. 12.24, а). Шпилька всегда включает 6 п.н.
в стебле и 4 нуклеотида в петле, при этом такая специфическая конфигурация важна для
успешного расщепления, хотя сама последовательность нуклеотидов в такой структуре
может изменяться. Второй сигнал представлен 9-нуклеотидной последовательностью
(консенсус 5’-CAAGAAAGA-3'), помещенной около 12 нуклеотидов ниже шпильки. Эта
последовательность спаривается основаниями с частью 117-мяРНК (рис. 12.24, б), одной
из семейства малых ядерных РНК, члены которого вовлечены в различные операции
процессинга РНК, включая вырезание интронов, как мы увидим в следующем разделе.
Спаривание стабилизируется связывающимся со шпилькой белком, и разрез произво-
дится четырьмя или пятью нуклеотидами ниже шпильки. Как этот разрез в точности
осуществляется, неизвестно: ни одного белка с необходимой активностью еще не было
опознано.
Регулирование синтеза мРНК у эукариотов
Когда мы рассматривали синтез бактериальных мРНК, мы встретились с рядом
процессов, посредством которых может осуществляться управление экспрессией генома
за счет регулирования на стадии элонгации-терминации процесса транскрипции (раз-
дел 12.1.2). Эквивалентные регулирующие механизмы у эукариотов оказались неуло-
вимыми для нашего взора и, с нашей современной точки зрения, процессы управления
транскрипцией работают почти исключительно на стадии инициации, как описано
в разделе 11.3.2, а не во время стадий элонгации и терминации синтеза мРНК.
Однако, следует упомянуть три потенциальных механизма управления.^Первый,
фактор элонгации для РНК-полимеразы II, названный TFIIS, хотя структурно и отлича-
454 Часть 3. Принципы функционирования геномов
ется от бактериальных белков Gre, которые мы встретили в разделе 12.1.2, действует
подобным образом, добираясь до активного участка РНК-полимеразы II за счет введения
игловидной структуры через канал, который бежит к нему с поверхности ферментного
комплекса. Работая подобным образом, TFIIS, а также GreA и GreB, способен повторно
запустить застопоренную РНК-полимеразу, стимулируя расщепление отделившегося
З’-сегмента РНК-транскрипта. Мы решили (в разделе 12.1.2), что, хотя никакой специфи-
ческий регулирующий процесс не был еще приписан к белкам Gre, такая роль возможна,
а путем экстраполяции это заключение распространяется также и на TFIIS.
Второй потенциальный механизм регулирования элонгации и (или) термина-
ции синтеза мРНК эукариотов сосредоточивается на состоянии фосфорилирования
С-концевого домена РНК-полимеразы II. Вспомним, что в ходе процесса инициации транс-
крипции фосфорилирование серинов в пределах повторного компонента С-концевого
домена активирует полимеразу и стимулирует очистку промотора (раздел 11.2.3). Это
подразумевает, что фосфорилированный С-концевой домен может быть существенно
важен для эффективного синтеза РНК. Поэтому наблюдение, что состояние фосфорили-
рования С-концевого домена не остается постоянным в течение процесса транскрипции,
предполагает, что изменения в этом состоянии могут играть некоторую роль в регу-
лировании транскрипции. Было опознано несколько киназ, которые фосфорилируют
С-концевой домен, и известно по крайней мере три фосфатазы, которые могут выполнять
обратную реакцию, удаляя фосфаты из структуры. Следовательно, существует некото-
рый механизм изменения состояния фосфорилирования С-концевого домена во время
элонгации и терминации, но пока что никакая связь с регулированием транскрипции
не была раскрыта.
Наконец, имеется все больше данных о том, что управление полиаденилировани-
ем мРНК может быть важным регулирующим механизмом у эукариотов. Многие гены
эукариотов имеют более одной сигнальной последовательности полиаденилирова-
ния, что означает, что терминация может произойти в различных позициях и в итоге
дать молекулы мРНК с идентичными кодирующими свойствами, но с отличающимися
З'-концами. По-видимому, в разных тканях используются различные сигналы полиаде-
нилирования, а это наводит на мысль о том, что альтернативное полиаденилирова-
ние может служить важным механизмом установления специфичных к тканям картин
экспрессии генома.
12.2.2. Удаление интронов из ядерной пре-мРНК
О существовании интронов не подозревали до 1977 г., когда секвенирование ДНК
было впервые применено к генам эукариотов и стало понятно, что многие из них содер-
жат «вклинивающиеся последовательности», которые отделяют различные сегменты
кодирующей ДНК друг от друга (рис. 12.25). Теперь мы различаем семь различных типов
интронов у эукариотов и дополнительные формы у архей (таблица 12.2). Два из этих
100 п. н.
ОБОЗНАЧЕНИЯ:
Экзоны
Интроны
типов — интроны GU-AG и AU-AC — встречаются
в кодирующих белок генах эукариотов и рассматри-
ваются в этом разделе; другие типы будут охвачены
далее в этой главе.
Рис. 12.25. Интроны. Показана структура гена р-глобина чело-
века. Этот ген 1423 п. н. в длину и содержит два интрона (один
131 п.н., а второй 851 п.н.), которые вместе составляют 69%
длины гена
Глава 12. Синтез и созревание РНК 455
Таблица 12.2. Типы интронов
Тип интрона Где встречаются
Интроны GU-AG Интроны AU-AC Группа I Группа II Группа III Твинтроны Интроны пре-тРНК Интроны архей Ядерная пре-мРНК эукариотов Ядерная пре-мРНК эукариотов Ядерная пре-рРНК эукариотов, РНК органелл, некоторые РНК бактерий РНК органелл, некоторые РНК прокариотов РНК органелл РНК органелл Ядерные пре-тРНК эукариотов Различные РНК
Можно установить несколько правил распределения интронов в кодирующих бе-
лок генах в дополнение к известному факту что интроны реже встречаются у низших
эукариотов: 6000 генов в геноме дрожжей содержат в своей совокупности всего лишь
239 интронов, тогда как многие отдельные гены млекопитающих содержат по 50 и бо-
лее интронов. Когда один и тот же ген сравнивается у родственных видов, мы обычно
обнаруживаем, что некоторые из интронов находятся в идентичных позициях, но что
каждый вид имеет один или несколько уникальных интронов. Это подразумевает, что
некоторые интроны остаются на своем месте в течение миллионов лет, сохраняя свои
позиции, в то время как виды множат свое многообразие, тогда как другие появляются
или исчезают в течение того же периода. Из этих наблюдений проистекают важные
следствия для теорий, касающихся эволюции геномов (раздел 18.3.2). Однако немалое
значение имеет тот факт, что пре-мРНК эукариотов может содержать много интронов,
возможно, более 100, занимающих значительную длину транскрипта (таблица 12.3),
и что эти интроны должны быть вырезаны, а экзоны соединены вместе в правильном
порядке, прежде чем транскрипт сможет функционировать как зрелая мРНК.
Консервативные мотивы последовательности указывают ключевые участ-
ки в интронах GU-AG
В подавляющем большинстве интронов пре-мРНК первые два нуклеотида после-
довательности интрона — 5’-GU-3', а последние два — 5'-AG-3’. Поэтому их называют
Таблица 12.3. Интроны в генах человека
Ген Длина, тыс. п.н. Число интронов Доля гена, занимае- мая интронами, %
Инсулин 1,4 2 69
Р-глобин 1,6 2 61
Альбумин сыворотки 18 13 79
Коллаген VII типа 31 117 72
Фактор VIII 186 25 95
Дистрофин 2400 78 98
456 Часть 3. Принципы функционирования геномов
интронами «GU-AG» и все члены этого класса вырезаются одинаковым способом. Эти
консервативные мотивы были опознаны вскоре после того, как были открыты интро-
ны, и незамедлительно было предпоположено, что они должны быть важны в процессе
сплайсинга. Как только последовательности интронов начали накапливаться в базах
данных, было обнаружено, что мотивы GU-AG попросту являются частями более длин-
ных консенсусных последовательностей, которые охватывают 5’- и З'-сайты сплайсинга.
Эти консенсусные последовательности изменяются у различных типов эукариотов;
у позвоночных они могут быть описаны последовательностями:
5’-сайт сплайсинга: 5 '-AG^GUAAGU-S'
З’-сайт сплайсинга: 5'-YYYYYYNCAG4-3'
В данных обозначениях «У» — один из двух пиримидиновых нуклеотидов (U или С),
«N» — любой нуклеотид, а стрелка указывает экзон-интронную границу. 5'-сайт сплай-
синга известен также как донорный участок, а З'-сайт сплайсинга — как акцепторный
участок.
У некоторых, но не у всех эукариотов присутствуют другие консервативные после-
довательности. Интроны у высших эукариотов обычно имеют полипиримидиновый
тракт — обогащенную пиримидинами область, расположенную сразу выше З'-конца
последовательности интрона (рис. 12.26). Эта полоса реже наблюдается в интронах
дрожжей, но дрожжи имеют инвариантную последовательность 5’-UACUAAC-3', располо-
женную между 18 и 140 нуклеотидами выше З'-сайта сплайсинга, которая отсутствует
у высших эукариотов. Полипиримидиновая полоса и последовательность 5’-UACUAAC-3'
не эквивалентны в функциональном отношении, о чем будет сказано в последующих
двух разделах.
Пре-мРНК
Рис. 12.26. Консервативные последовательности в интронах позвоночных. Более длинные консенсусные
последовательности вокруг участков сплайсинга приведены в тексте. Сокращение: Y — пиримидиновый
нуклеотид (U или С)
Сущность процесса сплайсинга интронов GU-AG
Консервативные мотивы последовательностей указывают важные области ин-
тронов GU-AG — области, от которых мы будем ожидать или роли сайта узнавания для
РНК-связывающихся белков, участвующих в сплайсинге, или некоторой другой веду-
щей партии в этом процессе. Первым попыткам осмыслить сплайсинг препятствовали
технические проблемы (в частности, трудности в разработке бесклеточной системы
сплайсинга, с которой процесс мог быть исследован подробно), но в течение 1990-х гг.
был бум информации. Эти работы показали, что путь сплайсинга может быть разделен
на два этапа (рис. 12.27).
• расщепление 5'-сайта сплайсинга происходит при помощи реакции трансэфиро-
образования, активизируемой гидроксильной группой, присоединенной к 2’-атому
углерода нуклеотида аденозина, расположенного в пределах последовательности
Глава 12. Синтез и созревание РНК 457
интрона. У дрожжей этот аденозин стоит последним в консервативной последо-
вательности 5'-UACUAAC-3'. Результат воздействия гидроксила — расщепление
фосфодиэфирной связи в 5'-сайте сплайсинга, сопровождаемое образованием новой
фосфодиэфирной связи 5'-2', соединяющей первый нуклеотид интрона (G мотива
5-GU-3') с внутренним аденозином. Это означает, что интрон был теперь завернут
петлей сам на себя, с образованием структуры лассо.
• Расщепление З'-сайта сплайсинга и соединение экзонов следует из второй реак-
ции трансэфирообразования, активизируемой группой З'-ОН, прикрепленной
к концу вышележащего экзона. Эта группа воздействует на фосфодиэфирную
связь в З'-сайте сплайсинга, расщепляет его и таким образом высвобождает ин-
трон в виде структуры лассо, которая впоследствии преобразуется обратно в ли-
нейную РНК и деградирует. В то же самое время З’-конец вышележащего экзона
присоединяется к новообразованному 5’-концу нижележащего экзона, завершая
процесс сплайсинга.
Пре-мРНК
Воздействие гидроксила
S'________________________________________S'
ли _
—► Потеря разветвленности,
^вАИ“А<3 деградация
Рис. 12.27. Общая схема сплайсинга. Расщепление 5'-сайта сплайсинга промотируется гидроксилом
(ОН), присоединенным к 2'-углероду нуклеотида аденозина в пределах последовательности интрона.
В результате образуется структура лассо, и далее З'-ОН группа вышерасположенного экзона вызывает
расщепление З'-участка сращения. Это позволяет двум экзонам быть лигированными, при этом высво-
божденный интрон теряет разветвленную структуру и деградирует
В химическом смысле вырезание интрона — несложная задача для клетки. Это
всего лишь двойная реакция трансэфирообразования — не более сложная, чем многие
другие биохимические реакции, за которые берутся отдельные ферменты. Однако для
ее решения в ходе эволюции был развит сложный механизм. Сложность заключается
в топологических задачах.
Первая из них — значительное расстояние, которое может простираться между
сайтами сплайсинга, возможно, несколько десятков тыс. п. н. (что соответствует 100 нм
и более, если мРНК находится в форме линейной цепи). Поэтому необходимо некоторое
средство сближения сайтов сплайсинга.
Вторая топологическая задача касается выбора правильного сайта сплайсинга.
Все сайты сплайсинга подобны, так что если пре-мРНК содержит два и более интрона,
то есть возможность, что будут соединены не те, которые надо, сайты сплайсинга, а это
приведет к пропуску экзона — потере экзона в зрелой мРНК (рис. 12.28, а). Одинаково
неудачным будет выбор скрытого сайта сплайсинга — сайта в пределах интрона или
экзона, который имеет подобие последовательности с консенсусными мотивами ис-
458 Часть 3. Принципы функционирования геномов
а) Пропуск экзона
1 3
б) Выбор скрытого участка сращения
Часть
1 экзона 2
Рис. 12.28. Две ненормальные формы сплайсинга, а) При пропуске экзона ненормальный сплайсинг
приводит к потере экзона в мРНК 6) При выборе скрытого участка сплайсинга часть экзона может быть
потеряна в мРНК, как показано здесь, или если скрытый участок лежит в пределах интрона, то сегмент
этого интрона сохранится в мРНК
тинных сайтов сплайсинга (рис. 12.28, б). Скрытые сайты присутствуют в большинстве
пре-мРНК и должны игнорироваться аппаратом сплайсинга.
Молекулы мяРНК и связанные с ними белки — центральные компоненты
аппарата сплайсинга
Центральные компоненты аппарата сплайсинга для интронов GU-AG представлены
мяРНК, названными Ul, U2, U4, U5 и U6. Это короткие молекулы (от 106 нуклеотидов [U6]
до 185 нуклеотидов [U2] у позвоночных), которые соединяются с белками и образуют ма-
лые ядерные рибонуклеопротеиды (мяРНП) (рис. 12.29). мяРНП совместно с другими
вспомогательными белками прикрепляются к транскрипту и образуют ряд комплексов,
последний из которых — сплайсосома (структура, в пределах которой происходят факти-
ческие реакции сплайсинга). Процесс работает следующим образом (рис. 12.30).
• Комплекс СС (commitment complex), или фиксационный комплекс, инициирует
действие сплайсинга. Этот комплекс включает Ul-мяРНП, который прикрепляется
к 5'-сайту сплайсинга—частично спариванием оснований РНК-РНК, а частично бел-
ковыми факторами SF1, U2AF35 и U2AF65, которые образуют контакты белок-РНК
Рис. 12.29. Структура Ul-мяРНП. Ком-
плекс Ul-мяРНП млекопитающих вклю-
чает 165-нуклеотидную Ul-мяРНК плюс
десять белков. Три из этих бел ков (U1-70К,
Ul-А и Ul-С) являются специфическими
для этого мяРНП, остальные семь —
белки Sm, которые обнаруживаются во
всех мяРНП, участвующих в сплайсинге.
Ul-мяРНК формирует соединенную па-
рами оснований структуру представлен-
ного здесь облика. Белки U1-70K и U1-A
прикреплены к двум из главных петель
стебля этой спаренной основаниями
структуры, а Ul-С прикрепляется посред-
ством межбелкового взаимодействия.
Белки Sm прикрепляются к участку Sm
Глава 12. Синтез и созревание РНК 459
соответственно с участком ветвления, полипиримидиновой полосой и З’-сайтом
сплайсинга.
• Предсплайсосомный комплекс включает фиксационный комплекс плюс U2-
мяРНП, последний прикрепляется к участку ветвления. На этой стадии ассоциация
между Ul-мяРНП и Н2-мяРНП приносит 5'-сайт сплайсинга в непосредственную
близость с точкой ветвления.
• Сплайсосома образуется, когда и4/иб-мяРНП (единый мяРНП, содержащий две
мяРНК) и US-мяРНП присоединяются к предсплайсосомному комплексу. Это при-
водит к дополнительным взаимодействиям, которые близко сводят З'-сайт сплай-
синга с 5’-сайтом и точкой ветвления. Все три ключевые позиции в интроне нахо-
дятся теперь вблизи друг относительно друга, и две реакции трансэстерификации
происходят как связанная в одну реакция (возможно, катализируемая Нб-мяРНП),
завершая процесс сплайсинга.
Ряд событий, показанных на рис. 12.30, не дает никаких ключей к тому, как вы-
бираются правильные сайты сплайсинга, так чтобы экзоны не потерялись во время
сплайсинга, а скрытые сайты были проигнорированы. Эта сторона сплайсинга все еще
плохо понята, но стало ясным, что набор факторов сплайсинга, названных белками SR,
является важным в выборе сайтов сплайсинга. Белки SR, названные именно так, потому
что их С-концевые домены содержат область, обогащенную серином (буква S в назва-
нии) и аргинином (R), были впервые привязаны к сплайсингу, когда было обнаружено,
что они являются компонентами сплайсосомы. Они, кажется, имеют несколько функ-
ций, в том числе установление связи между связанным Ul-мяРНП и связанным U2AF
Комплекс вербовки
f— 112-мяРНП
Предсплайсосомный комплекс
I Притяжение между U1-мяРНП
I и 112-мяРНП
Рис. 12.30. Роли мяРНП и связанных белков в ходе сплайсинга. Есть несколько оставшихся без ответа во-
просов о череде событий, происходящих в ходе сплайсинга, и маловероятно, что показанная здесь схема
абсолютно точна. Важный пункт —что ассоциации между мяРНП, как думают, приносяттри ответственные
детали интрона — два сайта сплайсинга и точку ветвления — в непосредственную близость
460 Часть 3. Принципы функционирования геномов
белками в фиксационном комплексе. Это, возможно, ключ к их роли в выборе сайтов
сплайсинга, при этом образование фиксационного комплекса является определяющей
стадией процесса сплайсинга, поскольку именно это событие опознает сайты, предна-
значенные для соединения.
Белки SR взаимодействуют также с экзонными энхансерами сплайсинга, кото-
рые представляют собой обогащенные пурином последовательности, расположенные
в экзонных областях транскрипта. Мы все еще на ранней стадии в нашем понимании
энхансеров сплайсинга и их коллег, экзонных сайленсеров сплайсинга, но их важное
значение в управлении сплайсингом явствует из открытия того факта, что несколько
болезней человека, в том числе один из типов мышечной дистрофии, вызваны мутациями
в последовательностях энхансеров сплайсинга. Местоположение энхансеров сплайсинга
и сайленсеров сплайсинга указывает на то, что сборка сплайсосомы определяется не
просто контактами в пределах интрона, но также и взаимодействиями со смежными
экзонами. Фактически, возможно, что отдельный фиксационный комплекс не собира-
ется в пределах интрона, как это показано на рис. 12.30, но первоначально соединяет
экзон (рис. 12.31). Эта модель привлекательна не только потому, что она моделирует
средство, которым контакт между энхансерами или сайленсерами сплайсинга и бел-
ком SR может влиять на сплайсинг, но также и потому, что она принимает во внимание
большую несоразмерность между длинами экзонов и интронов в генах позвоночных.
В геноме человека, например, экзоны имеют среднюю длину 145 нуклеотидов по срав-
нению с 3 365 нуклеотидами для интронов. Поэтому первичная сборка фиксационного
комплекса на экзоне может быть менее трудной задачей, чем сборка на намного более
длинном интроне.
Есть один заключительный момент, касающийся белков SR, к которому мы долж-
ны обратиться. Это возможность, что поднабор этих белков SR, названных CASP (SR-
подобные белки, связанные с С-концевым доменом) или SCAF (SR-подобные фак-
торы, связанные с С-концевым доменом), образуют физическое соединение между
сплайсосомой и С-концевым доменом комплекса транскрипции для РНК-полимеразы II
и, следовательно, обеспечивают связь между элонгацией транскрипта и процессингом.
Как и в случае полиаденилирования некоторых белков (раздел 12.2.1), весьма вероятно,
что эти факторы сплайсинга едут на полимеразе, покуда она синтезирует транскрипт,
и высаживаются на своих надлежащих позициях в интронных сайтах сплайсинга, как
Экзонный усилитель
сплайсинга
Экзонный усилитель
сплайсинга
Рис. 12.31. Альтернативная модель сборки фиксационного комплекса. В этой модели каждый отдельный
фиксационный комплекс (один показан оранжевым цветом, а другой — красным) выстраивается вдоль
экзона, принося комплекс в тесную связь с присущим данному экзону энхансером или сайленсером
сплайсинга и прикрепленными к нему белками SR. Как только комплексы возведены на смежных экзонах,
сплайсинг протекает по пути, иллюстрированному на рис. 12.30, с тем лишь отличием, что созданная
сплайсосома состоит из компонентов смежных фиксационных комплексов, вместо того чтобы быть по-
лученной из отдельного фиксационного комплекса, показанного на рис. 12.30
Глава 12. Синтез и созревание РНК 461
только они транскрибируются. Исследования с помощью электронной микроскопии
показали, что транскрипция и сплайсинг происходят совместно, а открытие факторов
сплайсинга, которые обладают сродством к РНК-полимеразе II, подводит биохимическую
основу под это наблюдение.
Альтернативный сплайсинг распространен среди многих эукариотов
Когда интроны были впервые открыты, бытовало представление о том, что каж-
дый ген всегда дает начало одной и той же мРНК: другими словами, что существует
единственный путь сплайсинга для каждого первичного транскрипта (рис. 12.32, а).
В 1980-е гг. обнаружилось, что это предположение было неверным, и было показано,
что первичные транскрипты некоторых генов могут следовать двумя и более путями
альтернативного сплайсинга, позволяющего одному транскрипту быть обработанным
и созреть в родственные, но различные мРНК и, следовательно, направлять синтез целого
ряда белков (рис. 12.32, б). У некоторых организмов альтернативный сплайсинг встреча-
ется крайне редко (у Saccharomyces cerevisiae известно только три примера), но у высших
эукариотов он намного более распространен. Это впервые стало очевидным, когда была
изучена черновая последовательность генома Drosophila melanogasteru стало ясно, что
плодовые мушки имеют меньше генов, чем микроскопический червь Caenorhabditis
elegans (см. табл. 7.4), несмотря на очевидно большую физическую сложность Drosophila,
которая должна быть отражена в более разнообразном протеоме. Наиболее вероятное
объяснение отсутствия соответствия между числом генов в геноме Drosophila и числом
белков в ее протеоме — то, что за счет альтернативного сплайсинга значительное число
генов дает начало многочисленным белкам. Приблизительно в то же самое время, когда
были сделаны эти наблюдения, были получены первые последовательности хромосом
человека, и было установлено, что вместо того чтобы иметь 80000-100000 генов, как
а) Единственная схема сплайсинга
Рис. 12.32. Когда было открыто явление альтернативного сплайсинга, было показано, что предположение
о том, что каждая пре-мРНК следует единственной схеме сплайсинга, является неверным
462 Часть 3. Принципы функционирования геномов
а) Определяемый полом альтернативный
сплайсинг пре-мРНК sxl
б) Белок SXL вызывает выбор скрытого участка
сплайсинга в пре-мРНК tra
Белок TRA не синтезируется
Белок TRA
синтезируется
в) Белок TRA вызывает альтернативный сплайсинг
в пред-иРНК dsx
3 4 5
Пре-мРНК dsx
Специфичный для женской
особи белок DSX
Рис. 12.33. Регулирование сплайсинга в ходе экспрессии генов, вовлеченных в определение пола
у Drosophila, а) Каскад начинается с определяемого полом альтернативного сплайсинга пре-мРНК sxl.
У мужских особей все экзоны присутствуют в мРНК, но это означает, что производится усеченный белок,
потому что экзон 3 содержит стоп-кодон. У женских особей экзон 3 пропускается, что ведет к полно-
мерному, функционально активному белку SXL. б) В организме самок SXL блокирует З'-сайт сплайсинга
в первом интроне пре-мРНК tra. U2AF65 неспособен определить местонахождение этого сайта и вместо
этого направляет сплайсинг на скрытый сайт в экзоне 2. В результате этого получается мРНК, кодирующая
функционально активный белок TRA. У самцов нет белка SXL, так что сайт сплайсинга 3' не блокируется
и производится дисфункциональная мРНК. в) У мужских особей экзон 4 из пре-мРНК dsx пропускается.
Получающаяся в результате мРНК кодирует специфичный для особи мужского пола белок DSX. У женских
особей белок TRA стабилизирует прикрепление белков SR к экзонному энхансеру сплайсинга, располо-
женному в экзоне 4, так что этот экзон не пропускается, и в результате получается мРНК, которая кодирует
специфичный для особи женского пола белок DSX. Эти два варианта DSX суть первичные определители
мужской и женской физиологии. Женская мРНК dsx оканчивается экзоном 4, потому что интрон между
экзонами 4 и 5 не имеет сайта сплайсинга 5', а это означает, что экзон 5 не может быть лигирован к концу
экзона 4. Вместо этого, у женских особей опознан сайт полиаденилирования в конце экзона 4. Следует
учитывать, что рисунок является схематическим и что интроны изображены не в масштабе
Глава 12. Синтез и созревание РНК 463
предполагалось исходя из размера человеческого протеома, люди имеют только 35 000
генов или около того. Теперь считается, что по крайней мере 35 % генов в геноме человека
подвергаются альтернативному сплайсингу1^ принцип «один ген — один белок» — био-
логическая догма, господствующая с 1940-х гг., — полностью опровергнут.
Теперь альтернативный сплайсинг рассматривается как определяющее новшество
на пути экспрессии генома. Нтобы иллюстрировать его важность, будет достаточно двух
примеров. Первый из них касается определения пола — фундаментального вопроса
биологии всякого организма, — который у Drosophila определяется каскадом альтер-
нативного сплайсинга. Первый ген в этом каскаде — sxl, транскрипт которого содержит
необязательный экзон, который, будучи сращен с предыдущим, дает неактивную версию
белка SXL. У самок путь сплайсинга такой, что этот экзон пропускается и, таким образом,
производится функционально активный белок SXL (рис. 12.33). Белок SXL стимулиру-
ет выбор скрытого сайта сплайсинга во втором транскрипте, tra, направляя U2AF65
в сторону от ее нормального З'-сайта сплайсинга ко второму участку по направлению
вниз. Полученный специфичный для женских особей белок TRA снова вовлекается
в альтернативный сплайсинг, на сей раз взаимодействуя с белками SR, в результате чего
образуется многофакторный комплекс, который прикрепляется к энхансеру сплайсинга
в пределах экзона третьей пре-мРНК, dsx, стимулируя выбор вторичного, специфичного
для женских особей сайта сплайсинга в этом транскрипте. Мужские и женские версии
белка DSX суть первостепенные определители пола у Drosophila.
Второй пример альтернативного сплайсинга иллюстрирует многообразие молекул
мРНК, синтезируемых с некоторых первичных транскриптов. У человека ген slo кодирует
мембранный белок, который регулирует вход и выход ионов натрия в клетки и из клеток.
Этот ген имеет 35 экзонов, восемь из которых участвуют в событиях альтернативного
сплайсинга (рис. 12.34). Пути альтернативного сплайсинга включают различные сочета-
ния восьми необязательных экзонов и ведут к более чем 500 различным мРНК, каждая
из которых задает мембранный белок с несколько отличающимися функциональными
свойствами. Гены slo человека активны во внутреннем ухе и определяют особенности
слухового восприятия волосковых клеток на основной мембране улитки внутреннего
уха. Различные волосковые клетки реагируют на разные звуковые частоты в диапазоне
от 20 Гц до 20000 Гц, причем их индивидуальные способности определяются отчасти
в а «им а — на* иа-кьт..
Рис. 12.34. Ген slo человека. Этот ген включает 35 экзонов, показанных в виде квадратиков, восемь из
которых (зеленые) являются необязательными (факультативными) и встречаются в разных мРНК slo
в различных сочетаниях. Существует 8! = 40320 возможных схем сплайсинга2) и, следовательно, 40320
возможных мРНК, но только около 500 из них, как думают, синтезируется в улитке человеческого уха * 2 *
Эти оценки разнятся. Как правило, они основываются на анализе вариантов EST. Есть множество
минорных форм—таких форм РНК, молекул которых мало. В зависимости от того, какую представительность
мы будем считать значимой, мы будем получать разные оценки. Если принимать все наблюденные формы,
то получим, что почти 99 % генов альтернативно сплайсируются. Надо иметь в виду, что все молекулярные
машины, в том числе и РНК-полимераза и сплайсосома, имеют заметный уровень ошибок. Так что некоторое
количество наблюденных изоформ — аберрантные. — Прим. ред.
2> На самом деле это не перестановки! Всего возможно 28 = 256 вариантов, что тоже не мало, но меньше
чем 500! — Прим. ред.
464 Часть 3. Принципы функционирования геномов
свойствами их белков Slo. Поэтому альтернативный сплайсинг генов slo в волосковых
клетках улитки внутреннего уха определяет слуховой диапазон людей.
В настоящее время мы не знаем, как альтернативный сплайсинг регулируется, и не
можем описать процесс, который определяет, которым из нескольких путей сплайсинга
предстоит проследовать тому или иному транскрипту. Игроками здесь, как думают,
являются белки SR в соединении с энхансерами и сайленсерами сплайсинга, но способ,
которым они управляют выбором сайтов сплайсинга, не известен.
Транссплайсинг связывает экзоны из разных единиц транскрипции
В примерах сплайсинга, которые мы успели рассмотреть, оба экзона, которые соеди-
няются вместе, расположены в пределах одного и того же транскрипта. У нескольких
организмов сплайсинг имеет место также между экзонами, которые содержатся в разных
молекулах РНК. Это называют транссплайсингом, и он происходит в хлоропластах не-
которых растений, в некоторых генах С. elegans и у трипаносом — простейших паразитов
позвоночных животных, которые вызывают сонную болезнь у людей.
Все примеры транссплайсинга, которые были изучены к настоящему времени,
подобны в том, что они дают начало одному и тому же короткому лидерному сегменту,
который становится присоединенным к 5'-концу каждого члена набора молекул мРНК
(рис. 12.35). Транскрипт, который отдает этот лидерный сегмент, называют сплайс-
лидерной РНК (слРНК). У С. elegans такая слРНК имеет около 100 нуклеотидов в длину
и содержит 22-нуклеотидную последовательность, которая присоединяется к 5'-концам
целевых мРНК. Реакция сплайсинга протекает в манере, очень подобной стандартной
схеме, представленной на рис. 12.27, хотя, поскольку партнерами сплайсинга выступают
различные молекулы, вместо лассо образуется разветвленная структура. Единствен-
ное осложнение состоит в том, что слРНК способна свернуться в структуру спаренных
оснований, подобную мяРНК, и, согласно некоторым моделям транссплайсинга, слРНК
заменяет Ul-мяРНП в процессе сплайсинга.
Целевые РНК
Продукты транссплайсинга
Рис. 12.35. Транссплайсинг. Ведущий экзон единственной слРНК прикрепляется посредством сплайсинга
к множеству различных целевых РНК
Транссплайсингу С. elegans имеет другой интересный момент. Некоторые из мРНК,
участвующих в транссплайсинге, содержат два гена, которые транскрибируются со-
вместно, голова к хвосту, с одного промотора. Если одна из таких мРНК с двумя генами
у С. elegans не подвергается транссплайсингу, то только вышележащий ген может быть
транслирован, потому что, как мы увидим в главе 13, рибосома, которая транслирует
мРНК эукариотов, прикрепляется к самому 5’-концу молекулы и обычно отделяется от
транскрипта, когда она достигает стоп-кодона. Следовательно, в несращиваемой мРНК
нижележащий ген недоступен для аппарата трансляции. Поэтому в отношении этих
мРНК транссплайсинг представляет собой процесс, который активирует трансляцию
Глава 12. Синтез и созревание РНК 465
Рис. 12.36. Регулирование генов посредством транссплай-
синга. На верхнем рисунке лидерный экзон транссплай-
сируется с геном А. В результате ген Б не экспрессируется,
потому что рибосома не может пересечь промежуток
между концом гена А и началом гена Б. На нижнем рисун-
ке транссплайсинг происходите геном Б, который теперь
экспрессируется
нижележащего гена за счет создания нового
5'-конца, к которому рибосома может прикре-
питься (рис. 12.36).
Интроны AU-AC подобны интронам
GU-AG, но требуют иного аппарата
сплайсинга
слРНК
А Б
Двугенная иРНК
Ген Б
не экспрессируется
Одним из более удивительных событий
последних лет было открытие нескольких ин- |
тронов в молекулах пре-мРНК эукариотов, ко- Ген Б
торые не подпадают под категорию GU-AG, ибо экспрессируется
имеют иные консенсусные последовательности
в своих сайтах сплайсинга. Это интроны AU-AC,
которые к настоящему времени были обнаружены приблизительно в 20 генах у таких
разных организмов, как человек, растения и Drosophila.
Так же как и мотивы последовательности в их сайтах сплайсинга, интроны AU-AC
имеют консервативную (хотя и не инвариантную) последовательность участка вет-
вления с консенсусом S'-UCCUUAAC-S'; последний аденозин в этом мотиве является
тем, который участвует в первой реакции транеэфирообразования. Это направляет
нас к замечательной особенности интронов AU-AC: их путь сплайсинга весьма подобен
таковому для интронов GU-AG, но вовлекает другой набор факторов сплайсинга. Только
US-мяРНП участвует в механизмах сплайсинга интронов обоих типов. Для интронов
AU-AC роли Ul-мяРНП и и2-мяРНП исполняются и11/и12-мяРНП — ранее открытым
комплексом, которому никогда не приписывали никакой функции, и, чтобы дополнить
картину, впоследствии были выделены совершенно новые U4atac/U6atac-MnPHn.
Пути сплайсинга для интронов «старшего» и «младшего» типов не идентичны, но
многие взаимодействия между транскриптом и молекулами мяРНП и других белков
сплайсинга удивительно подобны. Это означает, что интроны AU-AC, вместо того чтобы
просто быть диковинкой, оказываются полезными для проверки моделей взаимодей-
ствий, происходящих в ходе сплайсинга интронов GU-AG. Довод—то, что предсказанное
взаимодействие между двумя компонентами сплайсосомы GU-AG может быть проверено
путем наблюдения того, возможно ли такое же взаимодействие между эквивалентными
компонентами AU-AC. Такая процедура уже давала значимую информацию, помог-
шую определить структуру спаренных оснований, образуемую между U2- и иб-мяРНК
в сплайсосоме GU-AG.
12.2.3. Синтез функциональных РНК у эукариотов
В общем, мы знаем меньше об элонгации транскрипта и терминации транскрипции
с РНК-полимеразами I и III, чем ведаем об эквивалентных процессах с РНК-полимеразой II.
466 Часть 3. Принципы функционирования геномов
Reb1p/TTF-I
ДНК
РНК-полимераза I
РНК
Рис. 12.37. Возможная схема nthvbyfwbb
транскрипции, осуществляемой РНК-
полимеразой I
Взаимодействие полимеразы с матрицей и транскриптом во время элонгации оказы-
вается подобным у всех трех ферментов, что является отражением структурной связи
трех наибольших субъединиц в этих РНК-полимеразах. Первое отличие — скорость
транскрипции: например, РНК-полимераза I, будучи намного медленнее, чем РНК-
полимераза II, справляется со скоростью полимеризации лишь 20 нуклеотидов в минуту
по сравнению со скоростью до 2 000 нуклеотидов в минуту, свойственной синтезу мРНК.
Второе отличие состоит в том, что ни транскрипты РНК-полимеразы I, ни транскрипты
РНК-полимеразы III не кэпируются. Были выделены различные белки, которые могут
действовать как факторы элонгации для РНК-полимеразы 1 или III, в том числе SGS1
и SRS2 у дрожжей, которые кодируют две родственные ДНК-геликазы. Мутации в генах
SGS1 и SRS2 подавляют транскрипцию РНК-полимеразой I, а также репликацию ДНК.
SGS1 интересен тем, что он является гомологом пары белков человека, дефекты по кото-
рым приводят к нарушениям роста, таким как синдромы Блума и Вернера (раздел 5.2.1),
но точное участие SGS1 и SRS2, а также других предполагаемых факторов элонгации
в транскрипции с РНК-полимеразами I и III, не известно.
Главные отличия между этими тремя РНК-
полимеразами выступают на первый план при
сравнении процессов терминации. Система по-
лиаденилирования для терминации транскрип-
ции с РНК-полимеразой II (раздел 12.2.1) является
уникальной для этого фермента, и для других двух
РНК-полимераз эквивалент не был описан. В тер-
минации транскрипции с РНК-полимеразой I уча-
ствует ДНК-связывающий белок, названный Reblp
у Saccharomyces cerevisiae и TTF-I у мышей, прикре-
пляющийся кДНК в сайте узнавания, расположен-
ном в интервале 12-20 п.н. ниже точки, в которой
завершается транскрипция (рис. 12.37). Как имен-
но связанный белок вызывает терминацию, в точ-
ности не известно, но была предложена модель,
в которой полимераза становится застопоренной
из-за блокирующего действия Reblp/TTF-I. Второй
белок, PTRF (фактор высвобождения полимеразы
I и транскрипта), как думают, вызывает отсоединение полимеразы и транскрипта от
матрицы ДНК. Еще меньше известно о терминации осуществляемой РНК-полимеразой
III транскрипции: в этом деле замешана вереница аденозинов в матрице, но в данном
случае процесс не нуждается в шпильке и, таким образом, не является аналогичным
терминации у бактерий.
12.2.4. Сплайсинг пре-рРНК и пре-тРНК у эукариотов
У эукариотов есть четыре рРНК. Одна из них, 5S рРНК, транскрибируется РНК-
полимеразой III и не подвергается процессингу. Остальные три (5,8S, 18S и 28S рРНК)
транскрибируются РНК-полимеразой I в виде единого транскрипта, в результате чего
получается пре-рРНК, которая, как и бактериальные пре-рРНК, обрабатывается путем
нарезки и подрезки концов. Требуется несколько нуклеаз, в том числе многофунк-
циональная рибонуклеаза MRP, которая, наряду с процессингом 5,8S рРНК, участвует
в репликации митохондриальной ДНК и управлении клеточным циклом. Гены молекул
Глава 12. Синтез и созревание РНК 467
тРНК существуют обособленно и в виде многогенных единиц транскрипции, а обраба-
тываются в манере, весьма подобной наблюдаемой у бактерий (см. рис. 12.17). Главное
отличие между процессингом функциональной РНК у бактерий и эукариотов состоит
в том, что первичные транскрипты некоторых рРНК и тРНК эукариотов содержат интро-
ны. Интроны ни одного из этих типов не подобны интронам GU-AG и AU-AC пре-мРНК,
и поэтому мы должны уделить некоторое время их изучению.
Интроны в пре-рРНК эукариотов автокаталитические
В пре-рРНК эукариотов интроны встречаются крайне редко, но несколько их видов
известно у микробных эукариотов, таких как Tetrahymena. Эти интроны принадлежат
семейству I группы (см. табл. 12.2) и также встречаются в геномах митохондрий и хло-
ропластов, в которых они обнаруживаются как в пре-мРНК, так и в пре-рРНК. Несколь-
ко отдельных примеров известно у бактерий, например, в гене тРНК цианобактерии
Anabaena и в гене тимидилатсинтетазы бактериофага Т4 Е coli.
Путь сплайсинга для интронов I группы подобен таковому для пре-мРНК интронов
в том, что происходит две реакции трансэстерификации. Первая вызывается не ну-
клеотидом в пределах интрона, но свободным нуклеозидом или нуклеотидом — каким
угодно гуанозином или гуанозинмоно-, ди- или трифосфатом (рис. 12.38). Группа З'-ОН
этого кофактора поражает фосфодиэфирную связь в 5'-сайте сплайсинга и расщепляет
ее с переносом G на 5'-конец интрона. Вторая реакция трансэфирообразования вовлекает
группу З'-ОН на конце экзона, которая поражает фосфодиэфирную связь в З'-сайте сплай-
синга, производя расщепление, соединение двух экзонов и высвобождение интрона.
Высвобожденный интрон линейный, а не лассообразный (что свойственно интронам
Таблица 12.4. Примеры рибозимов
Рибозим Описание
Самовырезающиеся интроны Некоторые интроны I, II и III групп самовырезаются за счет автокаталити- ческого процесса. Имеется также все больше данных, что путь сплайсинга интронов GU-AG включает по крайней мере некоторые шаги, которые катализируются мяРНК
Рибонуклеаза Р Фермент, который создает 5'-концы бактериальных тРНК (см. раз- дел 12.1.3), состоит из РНКовой субъединицы и белковой субъединицы, при этом каталитическое действие сосредоточено в РНК
Рибосомная РНК Действие пептидилтрансферазы, необходимое для образования пеп- тидной связи в ходе синтеза белка (раздел 13.2.3), связано с рРНК 23 S большой субъединицы рибосомы
тРНКРЬе Подвергается самокатализируемому расщеплению в присутствии ионов двухвалентного свинца
Геномы вирусов Репликация геномов РНК некоторых вирусов вовлекает самокатализи- руемое расщепление цепей новосинтезированных геномов, сцепленных голова к хвосту. Примеры представлены вироидами и вирусоидами рас- тений (раздел 9.1.2) и дельта-вирусом гепатита животных. Эти вирусы образуют разнообразную группу с действием саморасщепл ения, опреде- ляемым множеством всеразличных спаренных основаниями структур, в том числе хорошо изученной структуры, напоминающей головку мо- лотка (см. рис. 9.9)
468 Часть 3. Принципы функционирования геномов
Воздействие гидроксила
5'
Пре-рРНК -ЛВНШН
Сплайсированная рРНК
I
Замыкание в кольцо,
деградация
Рис. 12.38. Схема сплайсинга интрона рРНК Tetrahymena
пре-мРНК), но в ходе процесса своей деградации может подвергаться дополнительным
реакциям трансэфирообразования, дающим кольцевые продукты.
Примечательная особенность пути сплайсинга интронов I группы заключается
в том, что он протекает в отсутствие белков и, следовательно, является автокаталити-
ческим — сама РНК обладает ферментативным действием. Это было первым примером
фермента из РНК, или рибозима, открытого еще в начале 1980-х гг. Первоначально
это произвело сенсацию, но сейчас стало общеизвестно, что есть несколько примеров
рибозимов, хотя эти примеры и немногочисленны (таблица 12.4). Действие самовыре-
зания, присущее интронам I группы, сосредоточено в структуре спаренных оснований,
принимаемой РНК. Эта структура была сначала описана в двух измерениях на основа-
нии сравнения последовательностей различных интронов I группы и восстановления
общего расположения спаренных оснований, которое может быть принято всеми ва-
риантами. В итоге была построена модель, включающая девять главных структурных
областей (рис. 12.39). Позже была определена трехмерная структура при помощи рент-
геноструктурного анализа. Рибозим состоит из каталитического ядра, образованного
двумя доменами, каждый из которых включает две структурные области, причем сайты
сплайсинга сближены за счет взаимодействий между двумя другими частями вторичной
структуры. Хотя такая структура РНК достаточна для сплайсинга, вполне возможно, что
в случае некоторых интронов стабильность рибозима усилена некаталитическими бел-
ковыми факторами, которые связываются с ним. О таком поведении интронов I группы
в генах органелл подозревали уже давно, ибо многие из них содержат открытую рамку
считывания, кодирующую белок, названный матуразой, который, оказывается, играет
определенную роль в сплайсинге.
Удаление интронов из пре-тРНК эукариотов
Интроны транспортной РНК относительно распространены у низших эукариотов,
но менее часты у позвоночных — интроны присутствуют только в 6% всех генов тРНК
человека. Интроны в молекулах пре-тРНК эукариотов имеют длину 14-60 нуклеотидов
и обычно находятся в одной и той же позиции транскрипта (в пределах антикодоновой
петли, на один нуклеотид непосредственно ниже антикодона). Последовательность
Глава 12. Синтез и созревание РНК 469
aacuuugaga-u-g-g-ccuu°
? I I I I I I I I III с
ArGGGACUCU GGA*
w G nG A
\-UA
C-G
C-G
A-U
G-CGA
G-C
C-G
U G
A C
cg-ca
G-C
A-U
C-G
C-G
A-U
A-U
A-U
C-G
U-G
A-U
U-A
U-G
U-A
U-A
U-A
C-G
C-G-A-A-A-A-U—A—G-CAAGACCGUCAAAUU-A-----^C^^y^U^Gy^^^A^
>' gcuggcugu duga UAGG dagu£gucdDC
GUAGAAGGG aaucagaca a
11111
AuUCUUCUCA U a a G a U AUAGUC-GGACC-G--C-G—-G—A-UUUGGAGUACUCGuaag
U-A U-G A-U r q
C-G A-U C-G °
U | G-C U-A I
C-G C-G A-U
а-u
U-A
U G
и и
G U
и-А
A-U
U-A
G-U
C-G
G A
Сайты сплайсинга
Рис. 12.39. Структура спаренных оснований (вторичная структура) интрона рРНК Tetrahymena. Последователь-
ность интрона показана заглавными буквами, а экзона — строчными. За счет дополнительных взаимодействий
интрон свертывается в трехмерную структуру, посредством чего эти два сайта сплайсинга сводятся близко
друг к другу
интрона является изменчивой, но содержит короткую область, комплементарную анти-
кодону и, возможно, одному или двум смежным с ним нуклеотидам. Спаривание осно-
ваний между комплементарными последовательностями образует короткий стебель
между двумя петлями в несплайсированной пре-тРНК (рис. 12.40).
В отличие от сплайсинга интронов всех остальных типов, встречающихся у эука-
риотов, сплайсинг интронов пре-тРНК не предполагает протекания реакций трансэфи-
рообразования. Вместо этого, два сайта сплайсинга разрезаются эндонуклеазой. Этот
фермент содержит четыре неидентичные субъединицы, одна из которых использует
структуру спаренного основаниями интрона в качестве направляющей, с тем чтобы
опознать правильные позиции, в которых РНК должна быть разрезана. Затем двумя
другими субъединицами фермента производятся выше- и нижерасположенные раз-
резы. После расщепления на З'-конце вышележащего экзона остается структура ци-
470 Часть 3. Принципы функционирования геномов
Рис. 12.40. Сплайсинг пре-тРНКТир у Saccharomyces cerevisiae
клического фосфата, а на 5'-конце нижележащего экзона — гидроксильная группа (рис.
12.40). Циклический фосфат превращается в З'-ОН-конец посредством фосфодиэстеразы,
а 5'-0Н-конец превращается в 5'-Р-конец действием киназы. Эти два конца удерживаются
в тесной близости за счет естественной структуры спаренных оснований, принадлежа-
щей последовательности тРНК, и лигируются РНК-лигазой. Действия фосфодиэстеразы,
киназы и лигазы обусловливаются одним-единственным белком.
Интроны других типов
Известно восемь различных типов интронов (см. табл. 12.2). Четыре из них уже
были описаны в этой главе: интроны ядерной пре-мРНК классов GU-AG и AU-AC, самовы-
резающиеся интроны I группы и интроны генов пре-тРНК эукариотов. Дабы завершить
картину, мы обрисуем особенности остальных четырех категорий.
Глава 12. Синтез и созревание РНК 471
• Интроны II группы находятся в геномах органелл грибов и растений, в пре-мРНК
и в пре-рРНК, а несколько известно у прокариотов. Интроны II группы принимают
характерную вторичную структуру и способны самовырезаться в пробирке, однако
они отличаются от интронов I группы. Хотя вторичная структура и отлична от других,
механизм сплайсинга более сходен с таковым у интронов пре-мРНК, при этом пер-
вичное трансэфирообразование стимулируется гидроксильной группой внутреннего
аденозинового нуклеотида и интрон преобразуется в структуру лассо. Эти подобия
навели на мысль, что интроны II группы и интроны пре-мРНК могли иметь общее
эволюционное начало (см. раздел 18.3.2). Некоторые интроны II группы являются
мобильными генетическими элементами, которые транспонируются замечательным
процессом, названным ретрорепатриацией, в ходе которой вырезанный интрон, кото-
рый, несомненно, представляет собой молекулу однонитевой РНК, встраивается непо-
средственно в геном органеллы до того, как будет скопирован в свой ДНК-вариант.
• Интроны III группы также обнаруживают себя в геномах органелл, и они самовы-
резаются с помощью механизма, очень подобного таковому у интронов II группы, но
интроны III группы меньше в размере и имеют свою собственную отличительную
вторичную структуру. Подобие с интронами II группы снова позволяет предполо-
жить некую эволюционную связь.
• Твинтроны — смешанные структуры, состоящие из двух и более интронов II
и (или) III группы. Простейшие твинтроны состоят из одного интрона, вложенного
в другой, но более сложные содержат множественные вложенные интроны. Отдель-
ные интроны, которые составляют твинтрон, обычно вырезаются в определенной
последовательности.
• Интроны архей присутствуют в генах тРНК и рРНК. Они расщепляются рибону-
клеазой, подобной той, что участвует в сплайсинге пре-тРНК эукариотов.
12.2.5. Химическая модификация РНК эукариотов
Молекулы тРНК и рРНК эукариотов подвергаются химической модификации тех
же типов, что и молекулы бактерий (раздел 12.1.3). В случае молекул тРНК ферменты,
которые осуществляют химические модификации, направляются к соответствующим
нуклеотидам вторичной структуры тРНК подобно тому, как эндонуклеазы, которые
вырезают интроны тРНК, используют вторичную структуру в качестве направляющей.
Что касается рРНК эукариотов, то ситуация несколько отлична.
Роль направляющих для химической модификации молекул рРНК эукариотов
выполняют малые ядрышковые РНК
Очень трудно чисто интуитивно представить, как может быть обеспечена специфич-
ность модификации рРНК. Например, пре-рРНК человека подвергается 106 метилирова-
ниям и 95 псевдоуридинилированиям, при этом каждое видоизменение осуществляется
в заданной позиции, без очевидного сходства последовательности, которые могли бы
быть приняты за целевые мотивы для модифицирующих ферментов. Неудивительно,
что в самом начале прогресс понимания модификации рРНК был медленным. Скачок
стал возможен лишь тогда, когда было показано, что у эукариотов в процессе модифи-
кации замешаны короткие РНК, названные мякРНК. Эти молекулы имеют длину 70-100
нуклеотидов и расположены в ядрышке — области внутри ядра, в которой происходит
процессинг рРНК. Изначальное открытие состояло в том, что путем спаривания осно-
ваниями с соответствующей областью молекулы мякРНК точно указывают позиции,
472 Часть 3. Принципы функционирования геномов
в которых пре-рРНК должна быть метилирована. В спаривании оснований участвует
только несколько нуклеотидов, а не полная длина мякРНК, но эти нуклеотиды всегда
расположены непосредственно выше консервативной последовательности, названной
блоком D (рис. 12.41, а). Пара оснований, содержащая нуклеотид, которому предстоит
модификация, отстоит на пять позиций от блока D. Гипотетически блок D является
сайтом узнавания для метилирующего фермента, который поэтому направляется к со-
ответствующему нуклеотиду. После этих первых открытий в отношении метилирова-
ния было показано, что иное семейство мякРНК исполняет ту же направляющую роль
в превращении уридинов в псевдоуридины. Эти мякРНК не имеют блоков D, но, подобно
своим сотоварищам, содержат консервативные мотивы, которые могут опознаваться
модифицирующим ферментом, и каждая способна образовывать специфичное взаимо-
действие типа спаривания основаниями со своим целевым участком, отмечая нуклеотид,
предназначенный для модификации.
Вследствие этого существует своя мякРНК для каждой видоизмененной позиции
в пре-рРНК, за исключением, быть может, нескольких участков, которые расположены
достаточно близко друг к другу, для того чтобы могли общаться сообща с одной мякРНК.
Это означает, что в каждой клетке должно быть несколько сотен мякРНК. Одно время
это казалось неправдоподобным, потому что удалось обнаружить очень мало генов
мякРНК, но теперь ясно, что с этих стандартных генов транскрибируется только доля
всех мякРНК, большая же их часть задается последовательностями в пределах интронов
других генов и высвобождается за счет разрезания интрона на куски после сплайсинга
(рис. 12.41, б).
а) Метилирование посредством
1124-мякРНК дрожжей
Видоизмененный нуклеотид
б) Синтез 1116-мякРНК человека
Ген рибосомного белка L1
AGAUCUUGGUGGUA ***
I I I I I I I I I I I I I I
UCUAGAACCACUAU^^
С ^^5'
U мякРНК
Блок D
/
3'
Интрон 3
ДНК ..
Последовательность
1116-мякРНК
Рис. 12.41. Метилирование рРНК посредством мякРНК.
а) Этот пример показывает метилирование С в позиции
1436 в 25S рРНК Saccharomyces cerevisiae (эквивалент
28S рРНК позвоночных), управляемое 1124-мякРНК. Блок
D мякРНК выделен цветом. Модификация всегда про-
исходит в паре оснований на пять позиций от блока
D. Обратите внимание, что во взаимодействии между
рРНК и мякРНК участвует необычная пара оснований
G-U, которая допустима между полинуклеотидами РНК.
б) Многие мякРНК синтезируются из РНК интрона (как
показано здесь на примере U16-snoPHК человека). U16-
snoPHK определяется последовательностью в интроне
3 гена рибосомного белка L1
Процессинг
интрона
1116-мякРНК
Глава 12. Синтез и созревание РНК 473
Таблица 12.5. Примеры редактирования РНК у млекопитающих
Ткань Целевая РНК Изменение Комментарии
Кишечника мРНК аполипопротеина В с->и Превращает кодон глутамина в стоп-кодон
Мышечная мРНК а-галактозидазы U-»A Превращает кодон фенилала- нина в кодон тирозина
Семенники, опухоли мРНК опухоли Вильмса 1 и->с Превращает кодон лейцина в кодон пролина
Опухоли мРНК нейрофиброматоза 1-го типа с->и Превращает кодон аргинина в стоп-кодон
В-лимфоциты мРНК иммуноглобулина Различные Вносит вклад в обеспечение разнообразия антител
Зараженные ВИЧ клетки Транскрипт ВИЧ-1 G-»A, C->U Вовлечено в регулирова- ние цикла инфицирования ВИЧ-1
Мозга мРНК рецептора глутамата А-»инозин Многочисленные позиции, что ведет к изменению раз- личных кодонов
Редактирование РНК
Поскольку рРНК и тРНК являются некодирующими, химические модификации их
нуклеотидов затрагивают только структурные особенности и, возможно, каталитиче-
ские действия этих молекул. В случае мРНК ситуация совершенно иная: химическая
модификация имеет потенциал изменять кодирующие свойства транскрипта, что за-
канчивается соответствующим изменением аминокислотной последовательности белка,
который задается этим транскриптом. Этот процесс называют редактированием РНК.
Интересный пример редактирования РНК наблюдается с мРНК человека, кодирующей
аполипопротеин В. Ген этого белка кодирует полипептид из 4 563 аминокислот, назван-
ный аполипопротеином В100, который синтезируется в клетках печени и выделяется
в кровоток, где он переносит липиды по всему организму. Родственный ему белок апо-
липопротеин В48 образуется в клетках кишечника. Этот белок имеет длину только 2153
аминокислоты и синтезируется с отредактированной версии мРНК полномерного белка
(рис. 12.42). В клетках кишечника эта мРНК модифицируется путем дезаминирования
цитозина, превращающего его в урацил.
В итоге кодон САА, определяющий глу-
тамин, превращается в кодон UAA, ко-
торый вызывает останов трансляции и,
в конечном счете, дает усеченный белок.
5'
_САА
Клетки печени -
полипептид из 4563
аминокислот
Редактирование
Рис. 12.42. Редактирование мРНК аполипо-
протеина В человека. В результате превраще-
ния С в U появляется стоп-кодон, что приводит
к синтезу укороченной формы аполипопротеи-
на В в клетках кишечника
1 г
5' 3'
7 UAA'. , »
Клетки кишечника -
полипептид из 2153
аминокислот
474 Часть 3. Принципы функционирования геномов
Дезаминирование производится РНК-связывающим ферментом, который (в соединении
с набором вспомогательных белковых факторов) связывается с последовательностью
непосредственно ниже позиции модификации в пределах мРНК.
Хотя редактирование РНК и встречается нечасто, оно происходит в ряде раз-
личных организмов и охватывает множество разнородных изменений нуклеотидов
(таблица 12.5). Некоторые события редактирования оказывают существенное воздей-
ствие на организм: у людей редактирование РНК отчасти ответственно за обеспечение
разнообразия антител (раздел 14.2.1) и было также замечено за управлением циклом
инфицирования ВИЧ-1. Редактирование РНК одного особенно интересного типа — де-
заминирование аденозина до инозина, которое выполняется ферментами, названными
воздействующими на РНК аденозиндезаминазами (ADAR). Некоторые из целевых
мРНК для этих ферментов избирательно редактируются в ограниченном числе позиций.
Эти позиции явно указываются двунитевыми сегментами пре-мРНК, образуемыми спа-
риванием оснований между участком модификации и последовательностями в смежных
интронах. Редактирование этого типа происходит, например, при процессинге молекул
мРНК рецепторов глутамата у млекопитающих. Есть данные, что редактирование РНК-
аденозиндезаминазами тесно связано с синтезом РНК, так как некоторые нуклеотиды
в интронах редактируются (указание на то, что редактирование происходит до выреза-
ния интронов), и если в С-концевой домен РНК-полимеразы II искусственно привносятся
изменения, то эффективность редактирования уменьшается.
От избирательного редактирования разительно отличается осуществляемая РНК-
аденозиндезаминазами модификация второго типа, при которой целевые молекулы
интенсивно дезаминируются и более 50% аденозинов в РНК превращается в ино-
зины. Такое гиперредактирование до сих пор наблюдалось главным образом, хотя
и не исключительно, в РНК вирусов и, как думают, происходит случайно; эти РНК при-
нимают вторичные структуры, которые по воле случая служат субстратами для РНК-
аденозиндезаминаз. Оно может, однако, иметь физиологическое значение в этиологии
болезней, вызываемых отредактированными вирусами. Эта возможность возрастает
в силу открытия, что вирусные РНК, связываемые с хроническими инфекциями кори
(в противоположность более распространенной преходящей форме этой болезни), под-
вергаются гиперредактированию.
Вышеупомянутые примеры редактирования РНК являют собой относительно
простые события, которые, за исключением гиперредактирования, ведут к изменениям
нуклеотидов в одной или в ограниченном числе позиций в отобранных мРНК. Известны
также более сложные типы редактирования РНК.
• Панредактирование заключается в повсеместной вставке нуклеотидов в усечен-
ные РНК с целью порождения функционально активных молекул. Оно особенно
часто в митохондриях трипаносом. Многие из РНК, транскрибируемых в митохон-
дриях трипаносом, определяются скрытыми генами — последовательностями
с отсутствием некоторых нуклеотидов, находящимися в зрелой РНК. Молеку-
лы пре-РНК, транскрибируемые с таких скрытых генов, обрабатываются путем
многочисленных вставок нуклеотидов U в позициях, определяемых короткими
направляющими РНК. Они представляют собой короткие РНК, которые могут
спариваться основаниями с пре-РНК и которые содержат нуклеотиды А в позициях,
куда должны быть вставлены нуклеотиды U (рис. 12.43).
• Менее масштабное инсерционное редактирование происходит в некоторых
вирусных РНК. Например, ген парамиксовируса Р дает начало по крайней мере
Глава 12. Синтез и созревание РНК 475
Рис. 12.43. Роль направляющей РНК в панредактиро-
вании
двум различным белкам за счет вставки
нуклеотидов G в определенных позициях
в мРНК. Эти встройки не определяются
направляющей РНК: вместо этого они
добавляются РНК-полимеразой по мере
синтеза мРНК.
• Полиаденилирующее редактирование
обнаруживает себя во многих митохон-
дриальных мРНК животных. Из числа
5' 3' Пред-РНК
^^GAGGCCAGC '
I I I
aCGGUCG^ Направляющая
3Z 5' РНК
Вставка U
5' 3'
GAGUGCCAGC
I I I I I I I
^-ACGGTTCG-----
3' 5'
молекул мРНК, транскрибируемых с митохондриального генома человека, пять
оканчиваются только U или UA, а не одним из стоп-кодонов (UAA и UAG в мито-
хондриальном генетическом коде человека). Полиаденилирование превращает
оконечный U или UA в UAAAA... и, таким образом, создает стоп-кодон. Это лишь одна
из нескольких особенностей, которые, кажется, эволюционировали, чтобы сделать
митохондриальные геномы позвоночных как можно более компактными.
12.2.6. Деградация РНК эукариотов
Молекулы мРНК эукариотов живут дольше своих бактериальных аналогов и ха-
рактеризуются периодом полураспада в среднем 10-20 минут для мРНК дрожжей
и несколько часов для мРНК млекопитающих. В пределах отдельных клеток разброс
почти столь же поразителен: некоторые мРНК дрожжей имеют период полураспада
всего лишь 1 минуту, тогда как для других эта цифра составляет более чем 35 минут.
Эти наблюдения порождают два вопроса: 1) какие процессы лежат в основе деградации
мРНК; 2) как эти процессы управляются?
Эукариоты обладают разнообразными механизмами деградации РНК
Наибольший прогресс в понимании деградации мРНК среди эукариотов был осу-
ществлен на примере дрожжей. Были определены по крайней мере четыре ее пути.
На одном из них подвизается многобелковый комплекс, названный экзосомой, кото-
рая разрушает транскрипты в направлении 3’->5' и содержит нуклеазы, родственные
ферментам бактериальной деградосомы. Экзосомы, вероятно, присутствуют также и в
клетках млекопитающих и определенно важны, но они мало изучены. Их роль, возмож-
но, заключается не в деградации мРНК perse, а в отслеживании полиаденилирования
и обеспечении своевременной проверки наличия у собирающихся покинуть ядро транс-
криптов надлежащих поли-А хвостов.
Несколько больше известно о двух других процессах деградации мРНК эукарио-
тов. Первый из них называется зависимым от дезаденилирования декэпированием
(рис. 12.44) и инициируется удалением поли-А хвоста, возможно, экзонуклеазным расще-
плением или, возможно, потерей связывающегося с полиаденилатом белка, который ста-
билизирует хвост (раздел 12.2.1).Удалениеполи-Ахвостасопровождается расщеплением
5'-кэпадекэпирующим ферментом Вср1р.Декэпированиепрепятствуеттранслированию
мРНК (раздел 13.2.2) и таким образом оканчивает ее функциональную жизнь. После этого
мРНК подвергается быстрому перевариванию экзонуклеазой со стороны 5’-конца. Будет
476 Часть 3. Принципы функционирования геномов
Удаление
поли-А хвоста
7mG---- ААААААААААД
Декапирование ।
t
l
Переваривание экзонуклеазой
Рис. 12.44. Способ деградации мРНК путем зависимого
от дезаденилирования декэпирования
ли отдельная мРНК деградировать, вероятно,
определяется способностью Dcplp получить
доступ к структуре кэпа, что, в свою очередь,
зависит от связывания между кэпом и кэп-
связывающими белками, необходимыми для
инициации трансляции (раздел 13.2.2). Также
на деградацию влияют, по крайней мере в слу-
чае некоторых мРНК дрожжей, последователь-
ности, названные элементами неустойчиво-
сти и расположенные в пределах транскриптов.
Важное значение этих последовательностей
было продемонстрировано экспериментами,
в которых такой элемент искусственно удалял-
ся, что приводило к усиленной трансляции и ослабленной деградации мРНК.
Вторая хорошо изученная система деградации мРНК эукариотов называется опо-
средствованным бессмысленным кодоном распадом РНК (NMD), или надзором за
мРНК. Первое из этих названий дает ключ к его функции, потому что на жаргоне моле-
кулярных биологов «бессмысленная» последовательность означает стоп-кодон. NMD
обеспечивает специфичную деградацию молекул мРНК, в которых стоп-кодон стоит
в неправильной позиции — или потому, что ген претерпел мутацию, или как результат
неправильного сплайсинга. Неправильный кодон, как думают, будет обнаружен механиз-
мом надзора, который включает комплекс белков, которые просматривают мРНК и так
или иначе способны видеть разницу между правильным стоп-кодоном, расположенным
в конце кодирующей области транскрипта, и таковым, находящимся в неположенном
месте (рис. 12.45, а). С этой моделью возникает ряд концептуальных трудностей, потому
что трудно представить, как комплекс наблюдения может проводить различие между
а) Надзор за мРНК позволяет определить
местонахождение неправильных стоп-кодонов
Неправильный Правильный
стоп-кодон стоп-кодон
51 I 3-
° - UGA... UAA — °
Опознанный системой надзора
в качестве неправильного
б) Влияние экзон-интронной границы
Правильный стоп-кодон
5' 3'
1—| * Экзон-интронная граница
Опознанный в качестве неправильного
системой надзора
правильными и неправильными стоп-
кодонами. Современные гипотезы
основываются на демонстрации того
факта, что правильный стоп-кодон
опознается в качестве ненормально-
го, если транскрипт сконструирован
таким образом, что экзон-интронная
граница помещена ниже этого стоп-
кодона (рис. 12.45, б). Ферменты над-
зора могут поэтому использовать
экзон-интронные границы в качестве
ориентационных позиций, чтобы от-
личать правильный стоп-кодон, ко-
торый обычно располагается ниже
последнего интрона. Также были
предложены альтернативные схемы,
Рис. 12.45. Система надзора за мРНК
Глава 12. Синтез и созревание РНК 477
в которых важное значение придается не позиции стоп-кодона, атомному характеру со-
бытий, происходящих при терминации трансляции на преждевременном стоп-кодоне,
в отличие от аналогичного процесса на стоп-кодоне, расположенном в правильной
позиции. Каким бы ни был этот механизм, опознавание неправильного стоп-кодона
вызывает расщепление кэпа и деградацию экзонуклеазой в направлении 5’—>3' (без
предшествующего удаления поли-А хвоста) белками, отличающимися от участвующих
в зависимом от дезаденилирования декэпировании. Хотя система NMD предназначена
прежде всего для деградации молекул мРНК, видоизмененных мутацией или непра-
вильно сплайсированных, есть данные, что этот путь отвечает также за деградацию
нормальных мРНК, но, вероятно, не таким способом, который ведет к управлению экс-
прессией того или иного отдельно взятого гена.
Подавление РНК поначалу считали средством разрушения чужеродной вирус-
ной РНК
Вышеописанные системы используются клетками эукариотов для деградации
эндогенных мРНК. Несколько лет назад стало известно, что эукариоты обладают так-
же иными механизмами деградации РНК, которые призваны защищать их клетки от
нападения чужеродных РНК, таких как геномы вирусов. Изначально названный пода-
влением РНК, этот процесс теперь знаком нам под его альтернативным названием —
РНК-интерференция, — поскольку лежащий в его основе механизм использовался
исследователями геномов как средство избирательного инактивирования генов с целью
изучения их функции (раздел 5.2.2).
Мишень для подавления РНК должна быть двунитевая, что исключает клеточные
мРНК, но охватывает вирусные геномы, многие из которых являются или двунитевыми
РНК в их природном состоянии, или реплицируются через двунитевого РНК-посредника
(раздел 9.1.2). Двунитевая РНК опознается связывающимися белками, которые образуют
посадочный участок для рибонуклеазы по имени Dicer, которая нарезает молекулу на
короткие интерферирующие РНК (siPHK) длиной 21-28 нуклеотидов (рис. 12.46).
Это инактивирует вирусный геном, но что, если вирусные гены уже были транскриби-
рованы? Если бы это произошло, то вредные действия вируса уже были бы запущены
и подавление РНК оказалось бы бесполезным в своей попытке защитить клетку от
повреждения. Одно из самых заме-
чательных открытий последних лет
показало вторую стадию процесса
интерференции, которая направлена
определенно на вирусные мРНК. Мо-
лекулы siPHK, произведенные рас-
щеплением вирусного генома, раз-
деляются на отдельные нити, одна
нить каждой siPHK впоследствии
спаривается основаниями с любы-
ми вирусными мРНК, которые при-
сутствуют в клетке. Образованные
двунитевые области становятся
мишенью для сборки наведенного
..............III? ХиТрНК
: Нуклеаза Dicer
разрезает двунитевую РНК
Ш Ш Ш Ш Ш Ш рнрк™нк)Терфе₽ирующие
I Однонитевые киРНК
▼ прикрепляются к мРНК
г ТТТТТГ.ТТТ.ТТТ.ТТТ.ТТТ мрнк
I
Деградация посредством RISC
Рис. 12.46. Путь РНК-интерференции
478 Часть 3. Принципы функционирования геномов
РНК комплекса подавления (RISC), который включает в себя РНК-связывающий белок
семейства Argonaut и нуклеазу (ею может быть или не быть сам Argonaut), которая рас-
щепляет и, следовательно, подавляет мРНК.
Работа, завершившаяся первичным описанием молекулярного процесса, лежащего
в основе РНК-интерференции, была выполнена в конце 1990-х гг. с С. elegans. С тех пор
было показано, что РНК-интерференция происходит у всех эукариотов (с несколькими
исключениями, включающими S. cerevisiae) и она была связана с различными события-
ми, которые вовлекают деградацию РНК, но которые ранее считались никак с нею не
связанными. Например, перемещение мобильных генетических элементов некоторых
типов вовлекает двунитевой РНК-посредник, который может быть деградирован про-
цессом, который, как теперь известно, представляет собой РНК-интерференцию. Это
один из способов, которым эукариоты предотвращают массовое распространение транс-
позонов по своим геномам.Генные инженеры были озадачены также способностью не-
которых организмов, особенно растений, подавлять новые гены, которые встраивались
в их геномы методами клонирования. Теперь мы знаем, что подавление такого типа
может произойти, если трансген будет случайно вставлен выше промотора, который
направляет синтез антисмысловой копии РНК всего гена или его части, эта РНК затем
спаривается основаниями со смысловой мРНК, произведенной с собственного промотора
трансгена, в результате чего образуется двунитевая РНК, которая запускает путь РНК-
интерференции (рис. 12.47). Другие явления в разнообразных организмах, по-разному
называемые подавлением, сосдерживанием и посттранскрипционным подавлением
гена, как известно, являют собой различные облики РНК-интерференции.
Промотор трансгена
Эндогенный промотор -
Встроенный ген обратная ориентация
мРНК
Антисмысловая мРНК
umiiiiiimii
Двунитевая РНК
I
Запускает РНК-интерференцию
Рис. 12.47. РНК-интерференция объясняет, почему трансгены являются иногда неактивными. Для ясно-
сти мРНК и антисмысловая РНК показаны транскрибируемыми с разных копий вставленного трансгена.
Они могут также быть получены с одного трансгена, который транскрибируется и с его собственного
промотора, и с эндогенного промотора
МикроРНК регулируют экспрессию генома, вызывая деградацию специфиче-
ских мРНК-мишеней
Во многих организмах были обнаружены белки Dicer более одного типа. Drosophila
melanogaster, например, имеет два родственных фермента Dicer, a Arabidopsis thaliana —
четыре. Множественность белков Dicer со слегка отличающимися свойствами преду-
преждает нас о возможности того, что могут иметь место дополнительные процессы
Глава 12. Синтез и созревание РНК 479
деградации РНК, связанные с фор-
мой РНК-интерференции, описанной
выше, но, возможно, отличные от нее.
Оказывается, что Dicer второго типа
у Drosophila работает не с молекулами
двунитевой РНК, произведенными пу-
тем, иллюстрированным на рис. 12.46,
но с микроРНК (миРНК), которые ко-
дируются геномом плодовой мушки
и синтезируются РНК-полимеразой II.
МикроРНК изначально синтезируют-
ся как молекулы-предшественники,
названные структурированными
РНК; это название указывает на то,
что эти РНК могут образовывать
внутринитевые пары оснований, да-
ющие одну или несколько внутрен-
них шпилечных структур (рис. 12.48).
Внутри ядра такие складчатые РНК
разрезаются ферментом Drosha на
отдельные двунитевые спиральные
фрагменты (шпильки), которые пере-
носятся в цитоплазму. Тогда двуните-
вой РНК-компонент стебля стимули-
рует путь РНК-интерференции, при
этом второй из двух ферментов Dicer
Drosophila расщепляет молекулу на
молекулы миРНК длиной около 21
нуклеотида. Каждая миРНК компле-
ментарна части клеточной мРНК и,
миРНК заглушают
эндогенные иРНК
следовательно, спаривается основа- Рис. 12.48. Схема интерференции микроРНК
ниями с этой мишенью, стимулируя
сборку микрорибонуклеопротеино-
вого (миРНП) комплекса, который функционально идентичен RISC и содержит много тех
же белков. Это ведет к расщеплению мРНК. Часто участок отжига миРНК присутствует
в З'-нетранслируемой области целевой мРНК, иногда в виде множественных копий
(рис. 12.49). Поэтому расщепление посредством миРНП не нарушает кодирующую
область мРНК, но приводит к отделению поли-А хвоста. Это может помешать про-
цессу инициации трансляции, в котором принимает активное участие поли-А хвост
(раздел 13.2.2), или отметить мРНК в качестве мишени для деградации посредством
зависимого от дезаденилирования декэпирования. Каким бы в точности ни был сам
механизм, расщепление посредством миРНП ведет к подавлению мРНК.
Первая охарактеризованная система подавления миРНК вовлекает гены С. elegans,
названные Ип-4 и let-7, оба из которых кодируют структурированные РНК, которые
производят молекулы миРНК после расщепления ферментом Dicer. Мутация в одном
из этих двух генов вызывает нарушения в пути развития червя, указывая на то, что
деградация РНК такого типа не есть просто средство избавления от нежелательных
или потенциально опасных молекул мРНК, но, напротив, играет фундаментальную роль
480 Часть 3. Принципы функционирования геномов
5'
3'
АААА
Многочисленные миРНК прикрепляются
к З'-нетранслируемой области
Рис. 12.49. Целевые сайты для микроРН К
часто находятся в З'-нетранслируемой
области целевой мРНК
Ш1Л АААА
Расщепление удаляет поли-А хвост
Нет запуска
трансляции?
Деградация путем зависимого
от дезаденилирования декапирования?
в регулировании экспрессии гено-
ма. Дальнейшее подтверждение
этой концепции было обеспечено
другими исследованиями миРНК
С. elegans, которые показали, что
эти молекулы вовлечены в такие
разнообразные биологические со-
бытия, как смерть клетки, опре-
деление типов нервных клеток
и управление жировыми запаса-
ми. Анализ генома показывает,
что большинство животных имеет способность синтезировать по крайней мере 100-200
различных миРНК, а возможно, и намного больше. Хотя пока еще очень немногие из ми-
шеней для этих миРНК были определены, система миРНК выступает в очевидной роли
всестороннего и чрезвычайно важного механизма регулирования генома. В прошлом
внимание ученых было сфокусировано главным образом на пути, которым экспрессия
генома регулируется белками, и открытие того факта, что молекулы РНК могут быть
столь же важны в этом отношении, привело к значительному сдвигу в нашем восприятии
того, как осуществляется управление составом протеома клетки.
12.2.7. Перенос РНК в пределах клетки эукариотов
В типичной клетке млекопитающих около 14% всей РНК находится в ядре. Около
80% этой ядерной доли представлены РНК, которая обрабатывается перед отбытием
в цитоплазму. Остальные 20 % — мяРНК и мякРНК, играющие активную роль в событиях
процессинга, по крайней мере некоторые из этих молекул уже побывали в цитоплазме,
где они были покрыты белковыми молекулами, прежде чем были перенесены обратно
в ядро. Другими словами, молекулы РНК эукариотов непрерывно перемещаются из ядра
в цитоплазму и, возможно, снова назад в ядро.
Единственный путь для РНК, которым она могла бы выйти или войти в ядро, —
через один из многочисленных ядерных поровых комплексов, которые покрывают
ядерную мембрану (рис. 12.50). Первоначально рассматриваемые как нечто чуть более
сложное, чем отверстие в мембране, теперь поровые комплексы расцениваются как
сложные структуры, которые играют активную роль в движении молекул в ядро и из
него. Маленькие молекулы могут двигаться беспрепятственно через поровый комплекс,
но РНК и большинство белков слишком велики, чтобы просочиться через него без по-
сторонней помощи и, таким образом, должны переноситься сквозь него посредством
некоторого энергозависимого процесса. Как во многих биохимических системах, энер-
гия вырабатывается путем гидролиза одной из высокоэнергетических межфосфатных
связей в рибонуклеотидтрифосфате, в этом случае путем преобразования ГТФ в ГДФ
(в других процессах имеет место превращение АТФ -> АДФ). Выработка энергии осущест-
вляется белком, называемым Ran, а перенос требует рецепторных белков, именуемых
Глава 12. Синтез и созревание РНК 481
Рис. 12.50. РНК эукариотов должны транспортиро-
ваться через ядерные поровые комплексы. У эу-
кариотов рРНК, тРНК и мРНК переносятся из ядра
в цитоплазму, где эти молекулы выполняют свои
необходимые клетке функции. По крайней мере не-
которые из мяРНК и мякРНКтакже транспортируются
в цитоплазму, где они покрываются белками перед
возвращением в ядро, чтобы выполнять свои роли
в обработке РНК. Ядерная пора — не просто отвер-
стие в ядерной мембране. Она содержит белковый
комплекс, включающий вложенное в пору кольцо, от
которого отходят структуры и в ядро, и в цитоплазму.
Что не показано на этой схеме, так это комплекс
центрального канала — белок 12 кДа, который, как
думают, располагается в канале, соединяющем цито-
плазму с ядром. Считается, что в типичной животной
клетке на поверхности ядра имеется приблизитель-
ноЗООО пор
кариоферинами, или экспортинами и им-
портиками в зависимости от направления
их переносной деятельности. Известно по
крайней мере 20 различных кариоферинов
человека, каждый отвечает за перенос мо-
лекул своего класса — мРНК, рРНК и так да-
лее. Примеры — экспортин-t, который был
определен как кариоферин для экспорта тРНК у дрожжей и млекопитающих. Транспорт-
ные РНК непосредственно опознаются экспортином-t, но РНК других типов, вероятно,
экспортируются специфичными к белкам кариоферинами, которые опознают белки,
связанные с РНК, а не сами РНК. Это, кажется, имеет место также и для импорта мяРНК
из цитоплазмы в ядро, в котором используется импортин 0 — компонент одного из
путей белкового переноса.
Экспорт мРНК инициируется терминацией пути сплайсинга, возможно, через
действие белка, названного Yralp у дрожжей и Aly у животных. Оказавшиеся вне ядра
мРНК ввергают себя механизмам, которые гарантированно доставляют их к предна-
значенным для них местам в клетке. Неизвестно, до какой степени местоположение
белка в пределах клетки обусловлено трансляцией мРНК в определенной позиции
или же передвижением белка после того, как он был синтезирован, но ясно, что по
крайней мере некоторые мРНК транслируются в предписанных им местах. Например,
мРНК, кодирующие белки, которым надлежит быть перемещенными в митохондрию,
транслируются рибосомами, расположенными на поверхности этой органеллы. Пред-
полагается, что белковые «адресные ярлыки» прикрепляются к молекулам мРНК, с тем
чтобы направлять их к надлежащим местам после того, как они переносятся из ядра,
но об этом процессе известно очень мало.
482 Часть 3. Принципы функционирования геномов
Заключение
Исследования структуры начинают открывать точный характер контактов, об-
разуемых между РНК-полимеразой, матричной ДНК и РНК-транскриптом во время
стадии элонгации транскрипции. РНК-полимераза не синтезирует свой транскрипт
с постоянной скоростью. Вместо этого, синтез прерывен, и периоды быстрой элонгации
перемежаются краткими паузами, в течение которых активный участок полимеразы
подвергается небольшой структурной перестройке. Обрыв бактериальных транскрип-
тов может произойти одним из двух методов; один из них требует вспомогательный
белок, называемый Rho. Бактерии обладают различными механизмами регулирования
терминации: или прочитывание сквозь сигналы терминации, так что нижележащие
последовательности транскрибируются, что является определяющим для экспрессии
генома фага X, или останавливая транскрипцию прежде, чем ген или оперон будет транс-
крибирован, если продукты этого гена не востребованы. Рибосомные и транспортные
РНК у бактерий изначально синтезируются в виде молекул-предшественников, которые
обрабатываются событиями нарезки и обрезки, чтобы выпустить функциональные
РНК. Эти РНК также химически модифицируются в различных позициях нуклеотидов.
В управляемую деградацию бактериальных РНК вовлечено целое разнообразие фер-
ментов. У эукариотов мРНК, произведенные РНК-полимеразой II, копируются путем
добавления 7-метилгуанозина к 5’-концу и полиаденилируются на З'-конце присоедине-
нием ряда адениновых нуклеотидов. Многие пре-мРНК эукариотов содержат интроны,
которые вырезаются из транскриптов в ходе сложного пути, включающего маленькие
ядерные рибонуклеопротеиды, действующие в структуре, названной сплайсосомой.
Пути альтернативного сплайсинга позволяют синтезировать с одного-единственного
гена более одного белка и важны в различных физиологических процессах, в том числе
в определении полгу Drosophila melanogaster. Молекулы пре-рРНК и пре-тРНК эукариотов
могут содержать также интроны. Таковые в пре-рРНК самовырезаются и, следовательно,
представляют примеры рибозимов. У эукариотов рРНК химически модифицируется
в ходе процесса, в котором маленькие ядрышковые РНК выступают направляющими,
с тем чтобы указать позиции, в которых модификации должны быть осуществлены.
Химическая модификация мРНК менее распространена, но приводит к изменению за-
кодированной инструкции, что происходит в процессе синтеза варианта для печени
и варианта для кишечника аполипопротеина В у млекопитающих. Эукариоты имеют
разнообразные механизмы деградации РНК, в том числе процесс, известный под назва-
нием подавления РНК или РНК-интерференции, в котором маленькие интерферирующие
РНК и микроРНК способствуют разрушению и, следовательно, подавляют вторгшиеся
вирусные РНК, а также мРНК, транскрибированные с генов клетки, продукты которых
более не нужны.
Глава 12. Синтез и созревание РНК 483
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
12.1 *.Сколько приблизительно п.н. об-
разуют связь между ДНК-матрицей
и РНК-транскриптом в ходе транс-
крипции у прокариотов?
а. 8.
Ь. 12-14.
с. 30.
d. Вся молекула РНК остаётся спа-
ренной основаниями с матрицей
до тех пор, пока транскрипция не
будет завершена.
12.2 . Какой фактор, как думают, являет-
ся наиболее важным в определении
того, продолжает или заканчивает
транскрипцию бактериальная РНК-
полимераза?
а. Концентрация нуклеотида.
Ь. Структура полимеразы.
с. Метилирование последователь-
ностей терминатора.
d. Термодинамические события.
12.3 *. Какова роль белка Rho в термина-
ции транскрипции?
а. Это геликаза, которая активно
разрывает пары оснований между
матрицей и транскриптом.
Ь. Это ДНК-связывающий белок, ко-
торый блокирует движение РНК-
полимеразы по матрице.
с. Это субъединица РНК-полиме-
разы, которая связывается со
шпильками РНК и стопорит
транскрипцию.
d. Это нуклеаза, которая деградиру-
ет З’-концы РНК-транскриптов.
12.4 . Антитерминация вовлечена в регу-
лирование которых из следующих
объектов?
а. Оперонов, кодирующих фермен-
ты, участвующие в биосинтезе
аминокислот, с регулированием,
зависящим от концентрации ами-
нокислот.
Ь. Оперонов, кодирующих фермен-
ты, участвующие в деградации
метаболитов, регулирование зави-
сит от присутствия метаболита.
с. Генов, расположенных в области
выше оперона.
d. Генов, расположенных в области
ниже оперона.
12.5 *. Каково главное транскрипционное
изменение, которое происходит во
время реакции на стресс у Е coli?
а. Скорость транскрипции увеличи-
вается для большинства генов.
Ь. Скорость транскрипции увеличи-
вается только для оперонов био-
синтеза аминокислот.
с. Скорость транскрипции уменьша-
ется для большинства генов.
d. Скорость транскрипции уменьша-
ется только для оперонов биосин-
теза аминокислот.
12.6 . Какая из следующих, как думают,
причин не обусловливает модифи-
кацию нуклеотидов тРНК?
а. Достижение более сильного спа-
ривания оснований между рибо-
нуклеотидами.
Ь. Облегчение опознавания различ-
ных молекул тРНК аминоацил-
тРНК-синтетазами.
с. Расширение диапазона взаимо-
действий, которые могут прои-
зойти между молекулами тРНК
и кодонами.
d. Возможность опознания несколь-
ких кодонов одной молекулой
тРНК.
484 Часть 3. Принципы функционирования геномов
12.7 *. Уход от промотора у эукариотов свя-
зан с:
а. Переходом РНК-полимеразы из
предынициаторного комплек-
са в комплекс, синтезирующий
РНК.
Ь. Отодвиганием РНК-полимеразы
от промоторной области и ее ста-
новлением на путь создания РНК-
транскриптов.
с. Высвобождением РНК-полиме-
разы из предынициаторного ком-
плекса так, чтобы никакой транс-
крипт не был синтезирован.
d. Терминацией транскрипции,
вызванной отделением РНК-
полимеразы от матричной ДНК.
12.8 . Каким образом образуется структу-
ра лассо в ходе вырезания интрона
GU-AG?
а. После расщепления 5’-участка
сплайсинга образуется новая
фосфодиэфирная связь между
5’-нуклеотидом и 2’-углеродом
нуклеотида в З'-участке сплай-
синга.
Ь. После расщепления 5'-участка
сплайсинга образуется новая
фосфодиэфирная связь между
5'-нуклеотидом и 2'-углеродом
внутреннего аденозина.
с. После расщепления З'-участка
сплайсинга образуется новая
фосфодиэфирная связь между
5'-нуклеотидом и 2'-углеродом
нуклеотида в 5'-участке сплай-
синга.
d. После расщепления З'-участка
сплайсинга образуется новая
фосфодиэфирная связь между
5'-нуклеотидом и 2'-углеродом
внутреннего аденозина.
12.9 *. Какие сайты сплайсинга называют
скрытыми?
а. Это сайты сплайсинга, которые
используются в одних клетках
и не используются в других.
Ь. Это сайты сплайсинга, которые
всегда используются.
с. Это сайты, которые принимают
участие в альтернативном сплай-
синге и приводят к удалению экзо-
нов из некоторых молекул мРНК.
d. Это последовательности в преде-
лах экзонов или интронов, которые
напоминают согласованные сигна-
лы сплайсинга, но не являются ис-
тинными сайтами сплайсинга.
12.10 . Какое утверждение правильно опи-
сывает транссплайсинг?
а. Порядок экзонов в РНК-транс-
крипте перестраивается, с тем
чтобы дать новую последователь-
ность мРНК.
Ь. Экзоны удаляются из одних РНК-
транскриптов, но не удаляются из
Других.
с. Последовательностиинтроновне
удаляются из РНК-транскриптов
и транслируются в белки.
d. Экзоны из различных РНК-
транскриптов соединяются друг
с другом.
12.11 *. Интроны I группы примечательны
потому, что:
а. Они вырезаются внешними моле-
кулами РНК без участия белков.
Ь. Они вырезаются молекулами
белка при отсутствии внешних
молекул РНК.
с. Они являются автокаталитиче-
скими.
d. Они присутствуют только в ге-
номах митохондрий и хлоропла-
стов.
12.12 . Химическая модификация молекул
рРНК эукариотов имеет место в:
а. Цитоплазме.
Ь. Эндоплазматической сети.
с. Ядерной оболочке.
d. Ядрышке.
12.13 *. Который из следующих процессов
представляет пример редактирова-
ния РНК?
Глава 12. Синтез и созревание РНК 485
а. Удаление интронов из РНК-
транскрипта.
Ь. Деградация молекулы РНК ну-
клеазами.
с. Видоизменение последователь-
ности нуклеотидов молекулы
РНК.
d. Кэпирование 5’-конца РНК-
транскрипта.
12.14 . Опосредствованный бессмыслен-
ным кодоном распад РНК (NMD)
является системой деградации мо-
лекул мРНК эукариотов с какими
особенностями?
a. NMD деградирует молекулы мРНК
со стоп-кодонами в неправиль-
ных позициях.
b. NMD деградирует молекулы
мРНК, которые кодируют неак-
тивные белки.
с. NMD деградирует молекулы
мРНК, у которых отсутствует
старт-кодон.
d. NMD деградирует молекулы
мРНК, у которых отсутствует
стоп-кодон.
12.15 *. Какой из следующих процессов
характерен для РНК-интерферен-
ции?
Вопросы открытого типа
12.1 *. Опишите процесс Rho-зависимой
терминации транскрипции у Е. coli.
12.2 . Как антитерминаторные белки
предположительно предотвраща-
ют отсоединение РНК-полимеразы
в сигналах терминации?
12.3 *. Почему в организмах эукариотов
аттенюация отсутствует?
12.4 . Опишите факторы, которые опреде-
ляют, которая из двух шпилек обра-
зуется во время транскрипции об-
ласти, лежащей выше trpEв опероне
триптофана. Каким образом эти
а. Молекулы антисмысловой РНК
блокируют трансляцию молекул
мРНК.
Ь. Двунитевые молекулы РНК свя-
зываются белками, которые бло-
кируют их трансляцию.
с. Двунитевые молекулы РНК рас-
щепляются нуклеазой в молеку-
лы коротких интерферирующих
РНК.
d. Молекулы коротких интерфери-
рующих РНК связываются с ри-
босомой, чтобы предотвратить
трансляцию вирусных мРНК.
12.16. Каким способом молекулы РНК
переносятся из ядра?
а. Пассивная диффузия через мем-
брану.
Ь. Через мембранные поры посред-
ством не зависимого от энергии
процесса.
с. Через мембранные поры при по-
мощи энергозависимого процес-
са.
d. Через канал в мембране, кото-
рый ведет к эндоплазматической
сети.
шпильки регулируют экспрессию
оперона триптофана?
12.5*. Как зрелые молекулы тРНК созре-
вают в результате процессинга пре-
тРНК у Е. coli? Какие ферменты уча-
ствуют в этом процессе?
12.6. Почему деградация РНК играет важ-
ную роль в регулировании экспрес-
сии генома?
12.7*. Как РНК-экзонуклеазы деградируют
бактериальную мРНК с ее З'-конца,
если структура шпильки, которая
вызвала терминацию транскрипции,
486 Часть 3. Принципы функционирования геномов
присутствует и блокирует действие
этих ферментов?
12.8. Каковы обычные модификации, про-
изводимые с транскриптами коди-
рующих белок генов у эукариотов?
12.9*. Каким образом структура кэпа 0-го
типа прикрепляется к мРНК эука-
риотов?
12.10. Обсудите, как несколько сот различ-
ных молекул мРНК могут быть про-
изведены из одного-единственного
гена эукариотов, такого как ген slo
человека.
12.11* . Какова роль молекул маленькой
ядрышковой РНК (мякРНК) в мо-
дификации молекул пре-рРНК эу-
кариотов?
12.12. За счет чего микроРНК могут регу-
лировать экспрессию генома эука-
риотов, связываясь с нетрансл ируе-
мым З’-концом молекулы мРНК?
Изыскательские задачи
12.1 *. «Современное мышление рассма-
тривает транскрипцию как прерыв-
ный процесс, в котором полимераза
регулярно берет паузу и делает «вы-
бор» между продолжением элонга-
ции (и добавлением большего числа
рибонуклеотидов к транскрипту)
или терминацией (и отделением
от матрицы). Какой выбор будет
сделан, зависит от того, которая из
альтернатив более благоприятна
в термодинамическом отношении»
(страница 433). Оцените этот взгляд
на транскрипцию.
12.2 . До какой степени изучение интронов
AU-AC позволило понять особенно-
сти вырезания интронов GU-AG?
12.3 *. Прерывные гены распространены
среди высших организмов, но прак-
тически отсутствуют у бактерий.
Порассуждайте о возможных при-
чинах этого.
12.4 . Обсудите вопросы, поднятые откры-
тием редактирования РНК.
12.5 *. Существование рибозимов рассма-
тривается как свидетельство того,
что РНК появилась в ходе эволюции
до белков, и поэтому одно время, на
самых ранних этапах эволюции, все
ферменты состояли из РНК. Прини-
мая эту гипотезу за верную, объяс-
ните, почему некоторые рибозимы
сохранились до сегодняшнего дня.
Глава 12. Синтез и созревание РНК 487
Тесты по рисункам
12.1 *. Обсудите механизм терминации транскрипции на внутреннем терминаторе у бак-
терий.
Матричная нить
ДНК H I I Т9999*7999? тАААААЛаХАА-pi-p-- 5'
д 54 j1 i Lccggtaggcg ?-!..!, А ! cgcctaccgg IttttttttttIHII 3'
I Транскрипция
РНК 5'
Sft rUUUUUUUUUJSSJ 3'
c-
c-
G-
G-
u-
A-
G-
G’
c-
Пары оснований
РНК-полимераза
12.2 . Обсудите механизм полиаденилирования молекул мРНК у эукариотов.
5'
Пре-мРНК _
Полиаденилированная
мРНК
3'
—.... AAUAAA ... , СА_____GU______
. Сборка комплекса
1 полиаденилирования
Связывающийся с полиаденилатом белок
AAJAAA —СА --------- GU-----
Полиаденилирование
AAUAAA САААААААААААААА
488 Часть 3. Принципы функционирования геномов
12.3 *. Обсудите шаги удаления изображенного на рисунке интрона.
5'
Пре-мРНК ж
3'
[U
G
SAMSA. |
—> Потеря разветвлённости,
BA^“AG деградация
12.4. Обсудите путь дезаденилирования
для деградации мРНК эукариотов.
Начиная с какой точки молекулы
мРНК эукариотов более не транс-
лируются?
12.5*. Назовите этот путь и обсудите про-
цессы, представленные на рисунке.
3' 5' Двунитевая
б» ДПШИШШП3' вирусная РНК
Удаление
поли-А хвоста
ТТТ ПТ ТТТ ПТ ПТ ТТТ
7mG
.ммААААААмА
Декапирование
7mG
5' з' мрнк
Переваривание экзонуклеазой
Глава 12. Синтез и созревание РНК 489
Дополнительная литература
Синтез РНК бактериальной РНК-полимеразой
Klug, А. (2001) A marvellous machine for making messages. Science 292:1844-1846.
Korzheva,N., Mustaev,A., Kozlov, M., Malhotra, A., Nikiforov, V., Goldfarb, A. and
Darst,S. A. (2000) A structural model of transcription elongation. Science 289: 619-625.
Toulokhonov,!., Artsimovitch, I. and Landick,R. (2001) Allosteric control of RNA
polymerase by a site that contacts nascent RNA hairpins. Science 292: 730-733. A model for
termination of transcription.
Управление синтезом мРНК у бактерий
Henkin, Т.М. (1996) Control of transcription termination in prokaryotes. Annu. Rev.
Genet 30: 35-57. A detailed account of antitermination and attenuation.
Losick,R.L. and Sonenshein,A.L. (2001) Turning gene regulation on its head. Science
293: 2018-2019. Describes the attenuation systems at the tryptophan operons ofE. coli and B.
subtilis.
Nickels, B.E. and Hochschild,A. (2004) Regulation of RNA polymerase through the
secondary channel. Cell 118: 281-284. The mode of action of transcript cleavage proteins.
Синтез и процессинг мРНК эукариотов
Arndt, К. М. and Kane, С. М. (2003) Running with RNA polymerase: eukaryotic transcript
elongation. Trends Genet 19: 543-550. Includes details of the roles of elongation factors.
Conaway, J. W., Shilatifard, A., Dvir, A. and Conaway, R. C. (2000) Control of elongation
by RNA polymerase II. Trends Biochem. Sci. 25: 375-380.
Conaway,R.C., Kong,S.E. and Conaway, J.W. (2003) TFIIS and GreB: two like-minded
transcription elongation factors with sticky fingers. Cell 114:272-273. A eukaryotic transcript
cleavage protein.
Cougot,N., van Dijk,E., Babajko,S. and Seeraphin,B. (2004) 'Cap-tabolism'. Trends
Biochem. Sci. 29: 436-444. mRNA capping.
Manley, J.L. and Takagaki,Y. (1996) The end of the message — another link between
yeast and mammals. Science 274:1481-1482. Polyadenylation.
Proudfoot,N. (2000) Connecting transcription to messenger RNA processing. Trends
Biochem. Sci. 25: 290-293.
Shilatifard,A., Conaway,R.C. and Conaway,J.W. (2003) The RNA polymerase II
elongation complex. Annu. Rev. Biochem. 72: 693-716. Includes details of elongation factors.
Studitsky, V. M., Walter, W., Kireeva, M., Kashlev, M. and Felsenfeld, G. (2004) Chromatin
remodelingby RNA polymerases. Trends Biochem. Sci. 29:127-135. Possible ways by which RNA
polymerases deal with nucleosomes attached to the DNA being transcribed.
Сплайсинг пре-мРНК
Black, D.L. (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev.
Biochem. 72: 291-336.
Blencowe, B. J. (2000) Exonic splicing enhancers: mechanism of action, diversity and role
in human genetic diseases. Trends Biochem. Sci. 25:106-110.
CordenJ.L. and Patturajan, M. (1997) A CTD function linking transcription to splicing.
Trends Biochem. Sci. 22: 413-416.
Graveley,B.R. (2001) Alternative splicing: increasing diversity in the proteomic world.
Trends Genet 17:100-107.
490 Часть 3. Принципы функционирования геномов
StetefeldJ. and Ruegg,M.A. (2005) Structural and functional diversity generated by
alternative mRNA splicing. Trends Biochem. Sci. 30: 515-521.
Tam, W.-Y. and Steitz, J. A. (1997) Pre-mRNA splicing: the discovery of a new spliceosome
doubles the challenge. Trends Biochem. Sci. 22:132-137. AU-AC introns.
Valcarcel, J. and Green, M. R. (1996) The SR protein family: pleiotropic functions in pre-
mRNA splicing. Trends Biochem. Sci. 21: 296-301.
Интроны других типов
Burke, J. M., Belfort, M., Cech,T. R., Davies, R.W., Schweyen, R. J., Shub, D.A.,
SzostakJ.W. and Tabak,H.F. (1987) Structural conventions for Group I introns. Nucleic
Acids Res. 15: 7217-7221. The nomenclature for the two-dimensional representation of the
Group I intron structure.
Cech, T.R. (1990) Self-splicing of group I introns. Annu. Rev. Biochem. 59:543-568. Written
by one of the discoverers of autocatalytic RNA.
Copertino,D.W. and Hallick,R.B. (1993) Group II and Group III introns of twintrons:
potential relationships with nuclear pre-mRNA introns. Trends Biochem. Sci. 18: 467-471.
Lambowitx,A.M. and Zimmerly,S. (2004) Mobile Group II introns. Annu. Rev. Genet
38:1-35.
Lykke-Andersen,J., Aagaard,C., Semionenkov,M. and Garrett,R.A. (1997) Archaeal
introns: splicing, intercellular mobility and evolution. Trends Biochem. Sci. 22: 326-331.
Транскрипция посредством РНК-полимераз / и ///
Geiduschek,Е.Р. and Kassavetis,G.A. (2001) The RNA polymerase III transcription
apparatus./. Mol. Biol. 310:1-26.
Reeder, R.H. and Lang,W.H. (1997) Terminating transcription in eukaryotes: lessons
learned from RNA polymerase I. Trends Biochem. Sci. 22: 473-477.
Russell, J.andZomerdijk, J. C.B.M. (2005) RNA-polymerase-I-directed rDN A transcription,
life and works. Trends Biochem. Sci. 30: 87-96.
Процессинг функциональной РНК у бактерий и эукариотов
Tollervey,D. (1996) Small nucleolar RNAs guide ribosomal RNA methylation. Science
273:1056-1057.
Venema, J. and Tollervey, D. (1999) Ribosome synthesis in Saccharomycescerevisiae. Annu.
Rev. Genet. 33: 261-311. Extensive details on rRNA processing.
Редактирование РНК
Bass, В. L. (1997) RNA editing and hypermutation by adenosine deamination. Trends
Biochem. Sci. 22:157-162.
Bourara,K., Litvak, S. and Araya, A. (2000) Generation of G-to-A and C-to-U changes in
HIV-1 transcripts by RNA editing. Science 289:1564-1566.
GottJ.M. and Emeson,R.B. (2000) Functions and mechanisms of RNA editing. Annu.
Rev. Genet 34:499-531.
Stuart, K. D., Schnaufer, A., Ernst, N. L. and Panigrahi, A. K. (2005) Complex management:
RNA editing in trypanosomes. Trends Biochem. Sci. 30: 97-105.
Деградация РНК у бактерий и эукариотов
Carpousis, A. J., Vanzo, N. F. and Raynal, L. С. (1999) mRNA degradation: a tale of poly(A)
and multiprotein machines. Trends Genet. 15: 24-28.
CollerJ. and Parker,R. (2004) Eukaryotic mRNA decapping. Annu. Rev. Biochem. 73:
861-890.
Hilleren,P., McCarthy,!., Rosbach,M., Parker,R. and Jensen,Т.Н. (2001) Quality
control of mRNA З'-end processing is linked to the nuclear exosome. Nature 413: 538-542.
Singh, G. and Lykke-Andersen, J. (2003) New insights into the formation of active
nonsense-mediated decay complexes. Trends Biochem. Sci. 28: 464-466.
Подавление РНК
Mello, С. C. and Conte, D. (2004) Revealing the world of RNA interference. Nature 431-.
338-342.
Sontheimer,E.J. and Carthew,R.W. (2005) Silence from within: endogenous siRNAs
and miRNAs. Cell 122: 9-12.
Zamore,P.D. and Haley, B. (2005) Ribo-genome: the big world of small RNAs. Science
309:1519-1524.
Перенос РНК
Fahrenkrog, В., KoserJ. and Aebi,U. (2004) The nuclear pore complex: a jack of all
trades. Trends Biochem. Sci. 29:175-182.
Nigg,E.A. (1997) Nucleocytoplasmic transport: signals, mechanisms and regulation.
Nature 386: 779-787.
Weis,K. (1998) Importins and exportins: how to get in and out of the nucleus. Trends
Biochem. Sci. 23:185-189.
Глава 13
Синтез и процессинг протеома
По прочтении главы 13 вы сможете:
• Изображать общую структуру тРНК и объяснять, каким образом эта структура
позволяет тРНК играть как физическую, так и информационную роли в ходе
синтеза белка.
• Описывать процесс прикрепления аминокислоты к тРНК и обрисовывать собы-
тия, которые гарантируют образование сочетаний только между правильными
парами аминокислот с тРНК.
• Объяснять принцип взаимодействия кодонов с антикодонами и рассуждать
о влиянии неканонического спаривания на это взаимодействие.
• Вкратце описывать методы, которые применялись для изучения структуры рибо-
сомы, и подытоживать информацию, полученную в итоге этих исследований.
• Приводить подробное описание процесса трансляции у бактерий и эукариотов,
с акцентом на роли различных факторов трансляции.
• Описывать экспериментальные данные, которые позволили прийти к заклю-
чению о том, что пептидилтрансфераза является рибозимом.
• Объяснять, как именно регулируется трансляция, и сообщать основные сведе-
ния о таких необычных событиях, как сдвиг рамки считывания, которые могут
произойти во время стадии элонгации.
• Объяснять, по какой причине посттрансляционный процессинг белков является
важной составляющей пути экспрессии генома, и описывать главные особен-
ности сворачивания белка, обработки белка протеолитическим расщеплением
и химической модификацией, а также сплайсинга интеинов.
• Описывать основные процессы, ответственные за деградацию белка у бактерий
и эукариотов.
Конечный результат экспрессии генома есть протеом — коллекция функциональ-
но активных белков, синтезируемых живой клеткой. Род и относительное содержание
отдельных белков в протеоме отражает равновесие между синтезом новых белков
и деградацией существующих. Биохимические возможности протеома могут изменять-
ся также химической модификацией и другими операциями процессинга. Сочетание
синтеза, деградации и модификации/процессинга позволяет протеому удовлетворять
изменяющиеся требования клетки и отвечать на внешние раздражения.
В этой главе мы изучим синтез, процессинг и деградацию компонентов протеома.
Чтобы понять синтез белка, мы сначала изучим роль молекул тРНК в расшифровке
генетического кода и затем исследуем события, происходящие в рибосоме и обуслов-
ливающие полимеризацию аминокислот в полипептиды. Соотносимые с рибосомой
Глава 13. Синтез и процессинг протеома 493
события иногда рассматривают как заключительную стадию экспрессии генома, но
полипептид, представляющий собой первичный продукт синтеза, будет бездействовать,
пока не свернется, и возможно даже, ему потребуется пройти необходимые процедуры
подрезки и химической модификации, прежде чем он станет функционально активным.
Эти операции процессинга мы изучим в разделе 13.3. В конце главы мы исследуем, как
клетка уничтожает белки, в которых она более не нуждается.
13.1. Роль тРНК в синтезе белка
Транспортные РНК играют главную роль в трансляции. Они представляют собой
сопрягающие молекулы, существование которых было предсказано Фрэнсисом Криком
в 1956 г., которые образуют связь между мРНК и синтезируемым полипептидом. Эта связь
является одновременно физической, так как молекулы тРНК связываются и с мРНК и с
наращиваемым полипептидом, и информационной, ибо молекулы тРНК позволяют син-
тезировать полипептид с аминокислотной последовательностью, определяемой, через
генетический код, последовательностью нуклеотидов в молекуле мРНК (рис. 13.1). Чтобы
понять, каким образом молекулы тРНК играют такую двоякую роль, нам необходимо
рассмотреть аминоацилирование — процесс, посредством которого к каждой молекуле
тРНК присоединяется нужная аминокислота, — и опознавание кодон-антикодон при
взаимодействии между тРНК и мРНК.
13.1.1. Аминоацилирование: присоединение аминокислот к молекулам
тРНК
Бактерии содержат 30-45 различных тРНК, а эукариоты имеют до 50 тРНК. По-
скольку генетическим кодом определяется только 20 аминокислот, это означает, что
все организмы имеют по крайней мере несколько изоакцепторныхтРНК—различных
тРНК, которые специфичны к одной и той же аминокислоте. Согласно терминологии,
употребляемой для описания тРНК, следует указывать специфичность к аминокислоте
суффиксом в надстрочном индексе, используя цифры 1, 2 и так далее, с тем чтобы от-
личать разные изоакцепторы: например, две тРНК, специфичные к глицину, следовало
бы описывать как TPHKG,yl и тРНКс1у2.
Все тРНК имеют подобную структуру
Наименьшие тРНК имеют лишь 74 нукле-
отида в длину, а наибольшие — редко более 90
нуклеотидов. Ввиду их небольшого размера и в
силу возможности выделять отдельные тРНК
они были среди первых нуклеиновых кислот,
еще в 1965 г. секвенированных Робертом Холли
и коллегами. Их последовательности обнару-
Рис. 13.1. Сопрягающая роль тРНК в процессе трансля-
ции. На верхнем рисунке показана физическая рольтРНК,
опосредствующей прикрепление между полипептидом
и мРНК. На нижнем рисунке показана информационная
связь, где тРНК переносит аминокислоту, определяемую
кодоном, к которому она прикрепляется
494 Часть 3. Принципы функционирования геномов
жили одну неожиданную особенность: наряду со стандартными нуклеотидами РНК
(А, С, G и U), тРНК содержат ряд видоизмененных нуклеотидов — 5-10 в любой отдельно
взятой тРНК, в целом же известно более 50 различных модификаций (раздел 12.1.3).
Изучение первой же последовательности тРНК (тРНКА1ау Saccharomyces cerevisiae}
показало, что эта молекула могла принимать различные вторичные структуры спарен-
ных оснований. После того как было секвенировано большее число тРНК, стало ясно, что
для всех тРНК существует общая специфическая вторичная структура, напоминающая
клеверный лист (рис. 13.2) и обладающая следующими особенностями.
• Акцепторный стебель образован семью парами оснований между 5’- и З’-концами
молекулы. Аминокислота присоединяется к выступающему З'-концу тРНК, в дан-
ном случае аденозин к инвариантной конечной последовательности ССА (раз-
дел 12.1.3).
• D-стебель, именованный так по названию видоизмененного нуклеозида дигидроу-
ридина (см. рис. 12.18), который всегда присутствует в этой структуре.
• Антикодоновый стебель содержит триплет нуклеотидов, названный антикодоном,
который спаривается основаниями с мРНК в ходе трансляции.
• V-петля содержит 3-5 нуклеотидов в молекулах тРНК 1-го класса или 13-21 ну-
клеотид в молекулах тРНК 2-го класса.
• ТТС-стебель, названный так по последовательности тимидин-псевдоуридин-
цитидин, которая неизменно присутствует в этих молекулах.
Структура типа клеверного листа может быть образована фактически всеми тРНК,
главные исключения представлены теми молекулами тРНК, которые работают в мито-
хондриях позвоночных, кодируются митохондриальным геномом и у которых иногда
недостает некоторых частей основной структуры. Пример — митохондриальная тРНК5ег
человека, у которой отсутствует D-стебель. Так же как и консервативная вторичная
3'
О
G
Э
5' •
ОС Акцепторный
D-стебель <
©оо ©°
о <
О. .О
О.
Т^С-стебель
:::Г°
оои
Антикодоновый •—•
стебель •
V-петля
Антикодон
структура, нуклеотиды в некоторых по-
зициях являются абсолютно инвариант-
ными (всегда один и тот же нуклеотид
в соответствующей позиции) или полу-
инвариантными (всегда пурин или всег-
Рис. 13.2. Структура тРНК в форме клеверного
листа. тРНК нарисована в общепринятой струк-
туре типа клеверного листа, с помеченными
различными компонентами. Обозначены инва-
риантные нуклеотиды (А, С, G, Т, U, Ф, где Ф—
псевдоуридин) и полуинвариантные нуклеоти-
ды (сокращения: R — пурин; Y — пиримидин).
Факультативные нуклеотиды, присутствующие
не во всех тРНК, показаны в виде маленьких
кружков. Стандартная система нумерации поме-
щает позицию 1 на 5'-конец, а позицию 76 — на
З'-конец; она включает некоторые, но не все фа-
культативные нуклеотиды. Инвариантные и по-
луинвариантные нуклеотиды стоят в позициях
8,11,14,15,18,19, 21, 24, 32, 33, 37, 48, 53, 54,
55, 56, 57, 58, 60, 61, 74, 75 и 76. Нуклеотиды
антикодона — в позициях 34, 35 и 36
О
о
О
Глава 13. Синтез и процессинг протеома 495
Рис. 13.3. Трехмерная структура тРНК. Дополнительные
пары оснований, показанные черным цветом и обра-
зующиеся главным образом между D- и ТТС-петлями,
сворачивают трилистниковидную структуру в представ-
ленную конфигурацию L-формы. В зависимости от ее
последовательности V-петля может также образовывать
взаимодействия с D-стеблем, что показано черными
линиями. Цветовая гамма та же, что и на рис. 13.2
да пиримидин), а позиции модифицированных
нуклеотидов почти всегда одни и те же.
Многие из инвариантных позиций ну-
клеотидов важны в третичной структуре
тРНК. Рентгеноструктурный анализ показал,
что нуклеотиды в D- и ТТС-петлях образуют
ТфС-петля
пары оснований, которые сворачивают тРНК
в компактную L-образную структуру (рис. 13.3). Каждый стебель L-образной струк-
туры имеет длину около 7 нм и диаметр примерно 2 нм, при этом сайт связывания
с аминокислотой расположен на конце одного стебля, а антикодон на конце другого.
Дополнительное спаривание оснований приводит к тому, что стекинг-взаимодействие
оснований (см. раздел 1.1.2) простирается почти непрерывно от одного конца тРНК до
другого, обеспечивая устойчивость структуры.
Аминоацил-тРНК-синтетазы прикрепляют аминокислоты к молекулам
тРНК
Прикрепление аминокислот к тРНК — «загрузка» на жаргоне молекулярных
биологов — является функцией группы ферментов, названных аминоацил-тРНК-
синтетазами. Химическая реакция аминоацилирования протекает в два этапа. Сначала
в ходе реакции между аминокислотой и АТФ формируется промежуточный продукт
в лице активированной аминокислоты, а затем аминокислота переносится на З’-конец
Таблица 13.1. Особенности аминоацил-тРНК-синтетаз
Особенности Ферменты I класса Ферменты II класса
Структура активного участ- ка фермента Параллельный р-лист Антипараллельный Р-лист
Взаимодействие с тРНК Малая бороздка акцептор- ного плеча Большая бороздка акцеп- торного плеча
Ориентация связанной тРНК V-петля, выдающаяся из молекулы фермента V-петля, обращенная внутрь молекулы фермента
Присоединение аминокис- лот К группе 2'-0Н оконечного нуклеотида в тРНК К группе З'-ОН оконечного нуклеотида тРНК
Ферменты для Apr, Цис, Глн, Глу, Иле, Лей, Лиз*, Мет, Трп, Тир, Вал Ала, Асн, Асп, Гл и, Г ис, Лиз*, Фен, Про, Трп, Сер
Примечание: аминоацил-тРНК-синтетаза для лизина представлена ферментом I класса у некоторых
архей и бактерий и ферментом II класса в организмах всех остальных видов.
496 Часть 3. Принципы функционирования геномов
Рис. 13.4. Аллиноацилирование тРНК. Показан резуль-
тат аминоацилирования аминоацил-тРНК-синтетазой
класса II, аминокислота присоединяется через свою
группу -СООН к группе З'-ОН оконечного нуклеотида
тРНК. Аминоацил-тРНК-синтетаза класса I прикрепляет
аминокислоту к группе 2'-ОН
тРНК, при этом образуется связь между груп-
пой -СООН аминокислоты и группой -ОН, при-
соединенной или к 2'-, или к 3’-атомууглерода на
сахаре последнего нуклеотида, который всегда
представлен аденином (рис. 13.4).
За малым числом исключений, организмы
О — Р = О имеют 20 аминоацил-тРНК-синтетаз, по одной
для каждой аминокислоты. Это означает, что
группы изоакцепторных тРНК аминоацилиру-
ются одним-единственным ферментом. Хотя
тРНК
основная химическая реакция одинакова для
всех аминокислот, эти 20 аминоацил-тРНК-
синтетаз подразделяются на две различные группы, I класса и II класса, с несколькими
важными различиями между ними (таблица 13.1). В частности, ферменты I класса
прикрепляют аминокислоту к группе 2'-ОН оконечного нуклеотида тРНК, тогда как
ферменты II класса присоединяют аминокислоту к группе З’-ОН.
Аминоацилирование должно выполняться с непременной точностью: нужная амино-
кислота должна присоединяться к нужной тРНК, если следовать правилам генетического
кода в процессе синтеза белка. Оказывается, аминоацил-тРНК-синтетаза имеет высокое
сродство к своей тРНК как результат обширного взаимодействия между этими молеку-
лами, охватывающего около 25 нм2 площади поверхности и вовлекающего акцепторный
стебель и антикодоновую петлю тРНК, а также отдельные нуклеотиды в D- и ТТС-стеблях.
Взаимодействие между ферментом и аминокислотой в силу необходимости менее обшир-
но — аминокислоты намного меньше, чем молекулы тРНК, и представляют большие про-
блемы в отношении специфичности, потому что несколько пар аминокислот структурно
подобны. Поэтому все-таки происходят ошибки, с очень низкой частотой появления для
большинства аминокислот, но, возможно, с частотой одно аминоацилирование из 80 для
трудных пар, таких как изолейцин и валин. Большинство ошибок исправляется самой же
аминоацил-тРНК-синтетазой в процессе редактирования, который отличается от аминоа-
цилирования образованием иных контактов с тРНК.
В большинстве организмов аминоацилирование выполняется согласно только
что описанному механизму, но было зарегистрировано несколько необычных случаев.
К ним относится ряд примеров, когда аминоацил-тРНК-синтетаза прикрепляет к тРНК
не ту аминокислоту, и эта аминокислота впоследствии преобразуется в ту, которую
надо, в ходе второй, самостоятельной химической реакции. Она была впервые открыта
у бактерии Bacillus megaterium при синтезе связки глутамин-тРНКс1п (то есть глутамина,
прикрепленного к своей тРНК). Это аминоацилирование осуществляет фермент, отве-
чающий за синтез связки глутаминовая кислота-тРНКС1и и первоначально устраивающий
прикрепление глутаминовой кислоты к TPHKGln (рис. 13.5, а). Затем эта глутаминовая
кислота превращается в глутамин за счет трансамидирования, катализируемого вторым
Глава 13. Синтез и процессинг протеома 497
О. ( f Ck nh2 с
( 1 :н2 сн2 1
( 1 зн2 1 сн2 1
— о—с—( II О 1 D-NH, Н — о— с—с—NH, II 1 + о н
Глутаминовая кислота Глутамин
Рис. 13.5. Необычные типы аминоацилирова-
ния. а) У некоторых бактерий TPHKGln амино-
ацил и руется глутаминовой кислотой, которая
затем преобразуется в глутамин посредством
трансамидирования, б) Специальная тРНК, ис-
пользуемая в инициации трансляции у бакте-
рий, аминоацилируется метионином, который
затем преобразуется к N-формилметионину.
в) TPHKSeCys в различных организмах первона-
чально аминоацилируется серином
сн3
S
I
сн2
сн2
—о—с—с—NH3
II I +
о н
Метионин
N-формилметионин
Серин
Селеноцистеин
ферментом. Подобный механизм используется и разными другими бактериями (хотя не
Escherichia coli), а также археями. Некоторые археи также прибегают к трансамидиро-
ванию для синтеза связки аспарагин-тРНКА5П из пары аспарагиновая кислота-тРНКА5П.
В обоих этих случаях аминокислота, которая синтезируется процессом модификации,
является одной из 20, которые определяются генетическим кодом. Известны также
два примера, где модификация дает необычную аминокислоту. Первый пример — пре-
вращение метионина в N-формилметионин (рис. 13.5, б), производящее особую связку
аминоацил-тРНК, используемую в инициации трансляции у бактерий (раздел 13.2.2).
Второй пример встречается как у прокариотов, так и у эукариотов и завершается син-
тезом селеноцистеина, который определяется контекстнозависимым способом некото-
рыми кодонами 5’-UGA-3’ (раздел 1.3.2). Эти кодоны опознаются специальной TPHKSeCys,
но не существует такой аминоацил-тРНК-синтетазы, которая могла бы присовокупить
селеноцистеин к этой тРНК. Вместо этого, тРНК аминоацетилируется вместе с серином
серил-тРНК-синтетазой и затем модифицируется путем замены группы -ОН серина
Рис. 13.6. Взаимодействие между
кодоном и антикодоном. Номера
указывают позиции нуклеотидов
в тРНК (см. рис. 13.2)
498 Часть 3. Принципы функционирования геномов
на -SeH, в результате чего и образуется селеноцистеин (рис. 13.5, в). Второе открытое
контекстнозависимое переназначение кодона — эпизодическое использование три-
плета 5'-UAG-3’ для кодирования пирролизина у архей (раздел 1.3.2). Оно не предпо-
лагает модификации предварительно загруженной тРНК. Вместо этого, наличествует
специфическая аминоацил-тРНК-синтетаза, которая непосредственно присоединяет
ПИррОЛИЗИН KTPHKpLys.
13.1.2. Взаимодействия кодон-антикодон: прикрепление молекул тРНК
к мРНК
Аминоацилирование представляет первый уровень специфичности тРНК. Второй
уровень — специфичность взаимодействия между антикодоном тРНК и транслируемой
мРНК. Эта специфичность гарантирует соблюдение правил генетического кода при
синтезе белка (см. рис. 1.20).
В принципе, узнавание кодон-антикодон — непосредственный процесс, основан-
ный на спаривании оснований между антикодоном тРНК и кодоном в мРНК (рис. 13.6).
Специфичность аминоацилирования гарантирует, что тРНК переносит аминокислоту,
обозначаемую кодоном, с которым она спаривается, а ри-
босома управляет топологией взаимодействия таким
образом, что только один триплет нуклеотидов доступен
для спаривания. Поскольку спаренные основаниями по-
линуклеотиды всегда антипараллельны и потому, что
мРНК считывается в направлении 5’—>3’, первый нуклео-
тид кодона спаривается с нуклеотидом 36 в тРНК, вто-
рой — с нуклеотидом 35, а третий — с нуклеотидом 34.
На практике опознавание кодона осложняется воз-
можностью отклонения — неканонического спарива-
ния, или неоднозначного соответствия. Это еще один из
принципов экспрессии генов, о котором изначально пред-
полагал Крик и который впоследствии был подтверж-
ден экспериментально. Поскольку антикодон находится
в петле РНК, триплет нуклеотидов слегка изогнут (см. рис. 13.2 и 13.3) и, таким образом,
не может образовать полностью равномерное выравнивание с кодоном. В результате
между третьим нуклеотидом кодона и первым нуклеотидом (номер 34) антикодона
может образоваться нестандартная пара оснований. Возможно множество различных
вариантов спаривания, в особенности если нуклеотид в позиции 34 видоизменен. У бак-
терий две главные особенности неканонического спаривания следующие.
• Допустимо образование пар оснований G-U. Это означает, что антикодон с последо-
вательностью З’-Ф ♦G-5' может спариться основаниями и с 5'-Ф ♦С-З’ и с 5’-< ♦U-3’.
Подобным же образом антикодон 3'-wU-5' может спариться основаниями
и с 5'-< ♦А-З' и с 5'-< ♦С-З’. Благодаря этому, вместо того чтобы иметь свою тРНК
для каждого кодона, четыре члена того или иного семейства кодонов (например,
все триплеты вида 5'-GCN-3' кодируют аланин) могут быть закодированы всего
лишь двумя разными тРНК (рис. 13.7, а).
• Инозин, сокращенный до I, представляет собой видоизмененный пурин (см.
рис. 12.18), который может спариваться основаниями с А, С и U. Инозин может
встретиться только в тРНК, потому что мРНК не модифицируется таким образом1).
За исключением случаев, описанных в разделе 12.2.5. — Прим. ред.
Глава 13. Синтез и процессинг протеома 499
Триплет 3'-UAI-5' иногда используется как антикодон в молекуле тРНКПе, потому
что он спаривается с кодонами 5’-AUA-3', 5'-AUC-3’ и 5'-AUU-3' (рис. 13.7, б), кото-
рые образуют трехкодонное семейство для этой аминокислоты в стандартном
генетическом коде.
Отклонение уменьшает число различных тРНК, необходимых клетке, позволяя
одной тРНК считывать два или, возможно, три кодона. Следовательно, бактерии мо-
гут декодировать свои мРНК, располагая не более чем 30-ю тРНК. Эукариоты также
используют неоднозначность, но в ограниченной степени. Геном человека, который
в этом отношении является довольно типичным для высших эукариотов, имеет в своем
Кодоны аланина
5" -QCG- “3-
Кодоны аланина
б) Инозин может спариваться с А, С или U
Инозин
Урацил
Рис. 13.7. Два примера неканонического спаривания у бактерий, а) Шатание, вовлекающее пару основа-
ний G-U, позволяет семейству из четырех кодонов для аланина быть расшифровываемым только двумя
тРНК. Обратите внимание, что использование шаткой пары G-U также допускает точное декодирова-
ние семейства с четырьмя кодонами, которое определяет две аминокислоты. Например, антикодон
3'-AAG-5' может декодировать кодоны 5'-UUC-3' и 5'-UUU-3', кодирующие фенилаланин (см. рис. 1.20),
а антикодон 3'-AAU-5' может декодировать другие два члена этого семейства, 5'-UUA-3' и 5'-UUG-3',
которые кодируют лейцин, б) Инозин может спариваться с А, С или U, а это означает, что единственная
тРНК может декодировать все три кодона для изолейцина. Пунктирные линии обозначают водородные
связи. Сокращение: I — инозин
500 Часть 3. Принципы функционирования геномов
арсенале 48 тРНК. Из них 16 предположительно используют неоднозначность, чтобы
расшифровывать по два кодона каждая, а остальные 32 являются специфичными только
к одному триплету каждая (рис. 13.8). К отличительным особенностям по сравнению
с неоднозначным спариванием у бактерий можно отнести следующие.
• Отклонение G-U используется с восемью тРНК, но в каждом случае неканоническое
спаривание вовлекает антикодон с последовательностью 3'- ♦ ♦ G-5'. Альтернатив-
ное спаривание G-U, где последовательность антикодона имеет вид 3’-wU-5',
как оказалось, не используется эукариотами — возможно потому, что это могло
бы привести к считыванию тРНКПе с антикодоном 3’-UAU-5' кодона метионина
5'-AUG-3’ (рис. 13.9). Эукариоты поэтому имеют средство предупреждения появ-
ления отклонения такого типа.
• Восемь других тРНК человека имеют антикодоны, содержащие инозин (З’-* ♦1-5’),
но они декодируют только кодоны 5'-* ♦С-З' и 5'-* ♦U-3’. Спаривание оснований
иии Фен UCU
иис UCC Сер
UUA Лей UCA
UUG UCG
UAU Тир UGU Цис
UAC UGC
UAA UGA | стоп
UAG стоп UGG Трп
сии CUC CUA CUG Лей CCU ССС ССА CCG Про
CAU Гис CGU
САС CGC Apr
САА CAG Глн CGA CGG
AUU AUC Иле ACU АСС Тре
AUA АСА
AUG | Мет ACG
AAU Асн AGU
ААС AGC
ААА AAG Лиз AGA AGG Apr
GUU GCU
GUC Вал GCC Апа
GUA GCA
GUG GCG
GAU Acn GGU Гли
GAC GGC
GAA GAG Глу GGA GGG
ОБОЗНАЧЕНИЯ:
Шатание G-U
Шатание I
Рис. 13.8. Предполагаемое использование неканонического спаривания при декодировании генома
человека. Пары кодонов, которые предположительно могут декодироваться единственной тРНК, исполь-
зующей шатание G-U, выделены розовым цветом, а пары, которые предположительно декодируются
шатанием, вовлекающим инозин, выделены желтым цветом. Кодоны, которые не выделены, имеют
свои собственные индивидуальные тРНК. Предположения основываются большей частью на данных
изучения последовательностей антикодонов тРНК, которые были обнаружены в последовательности
генома человека. Представленные здесь результаты анализа подразумевают, что в клетках человека
имеется 45 тРНК: 16 для неоднозначных пар и 29 единичных экземпляров. Фактически есть 48 тРНК.
Это связано с тем, что три кодона, о которых думали, что они декодируются с использованием неодно-
значности (5'-AAU-3', 5'-AUC-3' и 5'-UAU-3'), также имеют свои собственные индивидуальные тРНК, хотя
содержание их мало
Глава 13. Синтез и процессинг протеома 501
Рис. 13.9. тРНК с антикодоном 3'-UAU-5' может читать 3’ тРНК 5'
кодон изолейцина 5'-AUA-3' и также кодон метионина \ /
^UAU^
5’ III 3’
— а и А — Кодон изолеицина
-----------------AUG-------------- Кодон метионина
5'_______________3'
между I и А слабое, что означает, что кодоны 5’-Ф ♦А-З' могут только лишь неэф-
фективно опознаваться антикодоном 3’- ♦ ♦ 1-5’. Чтобы избежать такой неэффек-
тивности, в каждом примере неканонического спаривания, вовлекающего инозин
в наборе тРНК человека, кодон 5'-мА-3' опознается отдельной тРНК. Следует,
однако, иметь в виду, что опознавание отдельной тРНК не устраняет возможности
декодирования кодона 5'- ♦ ♦ А-З’ молекулой тРНК, содержащей антикодон 3'- ♦ ♦ 1-5’,
хотя и неэффективного. Это не подрывает доверия к специфичности генетического
кода, потому что неоднозначность с инозином возможна только в тех семействах
кодонов, в которых все три триплета, которые могут быть декодированы антико-
доном З'-Ф ♦1-5', определяют одну и ту же аминокислоту (см. рис. 13.8).
Другие генетические системы используют более резкие формы неоднозначности.
Митохондрии человека, например, используют только 22 тРНК. Для некоторых из этих
тРНК нуклеотид в шаткой позиции антикодона фактически избыточен, потому что он
может спариться основаниями с любым нуклеотидом, позволяя опознавать все четы-
ре кодона семейства одной и той же тРНК. Это явление назвали сверхотклонением
(сверхнеоднозначностью).
13.2. Роль рибосомы в синтезе белка
Клетка Е. coli содержит приблизительно 20 000 рибосом, распределенных по всему
объему цитоплазмы. Средняя клетка человека содержит немногим больше (еще нико-
му не удавалось сосчитать их все), причем некоторые свободно плавают в цитоплазме,
а некоторые прикреплены к внешней поверхности эндоплазматической сети (сеть
ограниченных мембранами канальцев и пузырьков, которая пронизывает клетку).
Первоначально на рибосомы смотрели как на пассивных партнеров в синтезе белка —
просто как на структуры, на которых происходит трансляция. Со временем это пред-
ставление изменилось, и рибосомы, как теперь полагают, играют две активные роли
в синтезе белка.
• Рибосомы согласовывают синтез белка, помещая мРНК, аминоацил-тРНК и связан-
ные белковые факторы в предписанные им положения друг относительно друга.
• Компоненты рибосом, в том числе и рРНК, катализируют по крайней мере неко-
торые из химических реакций, происходящих во время трансляции.
Чтобы понять, как рибосомы играют эти роли, мы сначала рассмотрим структурные
особенности рибосом бактерий и эукариотов, а затем исследуем все части механизма
синтеза белка в организмах этих двух типов.
13.2.1. Структура рибосомы
Наше понимание структуры рибосомы постепенно развивалось за последние
50 лет, по мере того как все более и более действенные методы применялись к решению
задачи ее познания. Изначально названные «микросомами» рибосомы были впервые
502 Часть 3. Принципы функционирования геномов
обнаружены зрительно в первые десятилетия двадцатого столетия как крошечные ча-
стицы, лежащие почти за пределами разрешающей способности световой микроскопии.
В 1940-е и 1950-е гг. первые электронные микрофотографии показали, что бактериаль-
ные рибосомы имеют овальную форму размерами 29х21нм — несколько меньше, чем
рибосомы эукариотов, последние немного изменяются в размере в зависимости от вида,
но в среднем составляют приблизительно 32x22 нм. В середине 1950-х гг. открытие, что
рибосомы являются участками синтеза белка, подстегнуло старание ученых определить
структуры этих частиц более досконально.
Для измерения размеров рибосом и их компонентов применялось ультрацен-
трифугирование
Первые подвижки в понимании подробностей структуры рибосомы были достиг-
нуты не за счет их наблюдения в электронный микроскоп, а из анализа их компонентов
ультрацентрифугированием (техническое примечание 7.1). Цельные рибосомы имеют
коэффициенты седиментации 80S для эукариотов и 70S для бактерий, и каждая может
быть разделена на меньшие компоненты (рис. 13.10).
• Каждая рибосома включает две субъединицы. У эукариотов эти субъединицы име-
ют коэффициенты седиментации 60S и 40S; у бактерий они составляют 50S и 30S.
Следует иметь в виду, что коэффициенты седиментации не аддитивны, потому что
они зависят не только от массы, но и от формы: для неповрежденной рибосомы аб-
солютно приемлемо иметь меньшее значение S, чем сумма двух ее субъединиц.
• Большая субъединица содержит три молекулы рРНК у эукариотов (28S, 5,8S
и 5S рРНК), но только две у бактерий (23S и 5S рРНК). У бактерий эквивалент
5,8S рРНК эукариотов содержится в 23S рРНК.
ЭУКАРИОТЫ БАКТЕРИИ
80S
I
60S
28S рРНК (4718 нуклеотидов)
5.8S рРНК (160 нуклеотидов)
5S рРНК (120 нуклеотидов)
50 белков
70S
50S
23S рРНК (2904 нуклеотида)
5S рРНК (120 нуклеотидов)
34 белка
40S
18S рРНК (1874 нуклеотида)
33 белка
30S
16S рРНК (1541 нуклеотид)
21 белок
Рис. 13.10. Состав рибосом эукариотов и бактерий. Детали относятся к «типичной» рибосоме эукарио-
тов и рибосоме Escherichia coli. Различия между различными видами главным образом касаются числа
рибосомных белков
Глава 13. Синтез и процессинг протеома 503
• Малая субъединица содержит одну рРНК у организмов обоих типов: 18S рРНК
у эукариотов и 16S рРНК у бактерий.
• Обе субъединицы содержат целое разнообразие рибосомных белков, их численное
содержание указано в рис. 13.10. Рибосомные белки малой субъединицы называют
SI, S2 и так далее; таковые большой субъединицы — LI, L2 и так далее. В рибосоме
имеется только по одному белку из каждой группы, кроме белков L7 и L12, которые
присутствуют в форме димеров.
Исследование тонкой структуры рибосомы
Центральный
домен
5'- Домен
3'- Большой
домен
Рис. 13.11. Вторичная структура рРНК 16S Escherichia
coli. В данном представлении, стандартные пары
оснований (G-C, A-U) показаны черточками; не-
стандартные пары оснований (например, G-U)
показаны точками
3'- Малый
домен
Как только был установлен основной
состав рибосом эукариотов и бактерий,
внимание было сосредоточено на спосо-
бе, которым различные молекулы рРНК
и белки пригнаны друг к другу. Важная
информация была извлечена из анали-
за первых последовательностей рРНК,
сравнения между которыми позволили
опознать консервативные области, мо-
гущие спариваться основаниями с обра-
зованием сложных двумерных структур
(рис. 13.11). На основании этого было
высказано предположение о том, что
молекулы рРНК образуют некий каркас
в структуре рибосомы, к которому прикре-
пляются белки, — в этой интерпретации
недооценивается активная роль молекул
рРНК в синтезе белка, но тем не менее она
послужила основанием, пригодным для
возведения на нем здания последующих
исследований.
Большая часть этой исследователь-
ской работы сосредоточилась на бакте-
риальной рибосоме, которая меньше эу-
кариотической рибосомы и может быть
получена в больших количествах из кле-
точных экстрактов, выращиваемых до
высокой плотности в жидких культурах. Для изучения бактериальной рибосомы был
применен ряд технических подходов:
• эксперименты с защитой от нуклеазы (раздел 7.1.1) позволяют опознавать
контакты между молекулами рРНК и белками;
• эксперименты с перекрестным сшиванием имеют целью определение пар или
групп белков, что расположены близко друг к другу в рибосоме;
• электронная микроскопия постепенно стала более сложной и позволяет разре-
шать полную структуру рибосомы с большей точностью. Например, для опреде-
ления позиций этих белков на поверхности рибосомы были использованы такие
новшества, как иммуноэлектроная микроскопия, в которой перед исследованием
рибосомы метят антителами, специфичными к отдельным рибосомным белкам;
504 Часть 3. Принципы функционирования геномов
• саит-направленное зондирование гидроксильными радикалами основано на
способности ионов Fe(II) производить гидроксильные радикалы, которые расще-
пляют фосфодиэфирные связи РНК, расположенные в пределах 1 нм от участка,
вырабатывающего радикалы. Эта технология была использована для опреде-
ления точного расположения рибосомных белков в рибосоме Е. coli. Например,
Рис. 13.12. Позиции в пределах 16S рРНК
Escherichia coli, которые образуют контакты
с рибосомным белком S5. Распределение
контактных позиций (показанных красным
цветом) для одного этого рибосомного белка
подчеркивает степень, до которой спаренная
основаниями вторичная структура рРНК далее
свернута в пределах трехмерной структуры
рибосомы
чтобы определить позицию белка S5, разные
аминокислоты, входящие в состав этого белка,
были помечены ионами Fe(II) и гидроксильны-
ми радикалами, порожденными в воссоздан-
ных рибосомах. Затем позиции, в которых
16S рРНК были расщеплены, использовались,
чтобы выявить топологию рРНК в окрестности
белка S5 (рис. 13.12).
В последние годы эти методы все более
и более дополнялись рентгеноструктурным
анализом (техническое примечание 11.1), бла-
годаря которому и свершились самые захва-
тывающие проникновения в структуру рибо-
сомы. Анализ громадных количеств данных,
почерпнутых из рентгеновских дифрактограмм
кристаллов объекта такого размера, как рибо-
сома, — задача огромной трудности, особенно
на уровне, необходимом для получения доста-
точно подробной структуры, позволяющей
понять принцип действия рибосомы. Это ис-
пытание было выдержано, и были выведены
структуры рибосомных белков, связанных со
своими сегментами рРНК, для большой и малой
субъединиц, а также для целой бактериальной
рибосомы, связанной с молекулой мРНК и моле-
кулами тРНК. Наряду с раскрытием структуры
рибосомы (рис. 13.13), этот недавний всплеск
информации имел важное воздействие на наше
понимание процесса трансляции.
13.2.2. Инициация трансляции
Хотя строение рибосом у бактерий и эукариотов схоже, есть различия в способе,
которым происходит трансляция в организмах этих двух типов. Наиболее важное из этих
различий наблюдается во время первой стадии трансляции, когда рибосома собирается
на мРНК в позиции выше старт-кодона.
Для инициации трансляции у бактерий необходим внутренний сайт связы-
вания рибосомы
Главное различие между инициацией трансляции у бактерий и эукариотов состоит
в том, что у бактерий комплекс инициации трансляции возводится непосредственно
на старт-кодоне (точке, в которой начнется синтез белка), тогда как эукариоты для
определения места точки инициации используют более косвенный процесс, как мы
увидим в следующем разделе.
Глава 13. Синтез и процессинг протеома 505
Рис. 13.13. Бактериальная рибосома. На кар-
тине изображена рибосома бактерии Thermus
thermophilus. Малая субъединица находится
сверху, 16S рРНК окрашена светло-синим, а ри-
босомные белки малой субъединицы — темно-
синим. рРНК большой субъединицы окрашена
серым, а белки — фиолетовым. Золотая об-
ласть — A-сайт (раздел 13.2.3) — точка, через
которую аминоацилированные тРНК входят в ри-
босому в процессе синтеза белка. Этот участок
и большая часть области, в пределах которой
синтез белка фактически происходит, располо-
жены в расселине между двумя субъединицами
рибосомы. Перепечатано из Trends Biochem. Sci.,
26, Mathews and Pe'ery, The machine that decodes
the Genome. 585-587, 2001, с разрешения из-
дательства Elsevier
В те периоды времени, когда рибо-
сомы не принимают активного участия
в синтезе белка, они разобщаются на
свои субъединицы, которые остаются в цитоплазме в ожидании входа в очередной круг
трансляции. У бактерий процесс инициируется, когда малая субъединица в сотрудни-
честве с фактором инициации трансляции IF-3 (таблица 13.2) прикрепляется к сайту
связывания рибосомы (также названному последовательностью Шайна-Дальгарно).
Это короткий целевой участок (консенсусная последовательностьу Е. coli 5'-AGGAGGU-3’
(таблица 13.3)), расположенный приблизительно на 3-10 нуклеотидов выше старт-
кодона — точки, в которой начнется трансляция (рис. 13.14). Сайт связывания рибосомы
комплементарен области на З’-конце 16S рРНК, находящейся в малой субъединице, и,
как думают, спаривание оснований между ними обусловливает прикрепление малой
субъединицы к мРНК.
В результате прикрепления к сайту связывания рибосомы малая субъединица
рибосомы помещается над старт-кодоном (рис. 13.15). Этот кодон обычно представлен
триплетом 5'-AUG-3', который кодирует метионин, хотя иногда используются кодоны
5’-GUG-3' и 5'-UUG-3’. Все три кодона могут быть опознаны одной и той же инициаторной
тРНК, последние два в силу неканонического спаривания оснований. Этой инициаторной
тРНК служит именно та, которая была аминоацилирована метионином и затем моди-
фицирована путем превращения метионина в N-формилметионин (см. рис. 13.5, б). В ре-
зультате такой модификации формильная группа (-СОН) присоединяется к аминогруппе,
а это означает, что только карбоксильная группа инициаторного метионина свободна
для участия в образовании пептидной связи. Благодаря этому синтез полипептида
может протекать только в направлении N -> С.
Участок посадки рибосомы
Рис. 13.14. Участок посадки рибосомы для трансляции
у бактерий. У Escherichia coli сайт связывания рибосомы
имеет консенсусную последовательность 5'-AGGAGGU-3'
и расположен на 3-10 нуклеотидов выше стартового
Стартовый кодон
I 3’
-------AGGAGGU------AUG---------
3-10 нуклеотидов
кодона
506 Часть 3. Принципы функционирования геномов
Участок посадки рибосомы
5' \ Старт-кодон
AUG"—
Малая субъединица, IF-3
Малая субъединица
Запускающая тРНК-----◄
Запускающая тРНК, IF-2, ГТФ
—AUG
Рис. 13.15. Инициация трансляции у Escherichia coli. Учтите, что различные компоненты инициаторного
комплекса нарисованы не в масштабе. Сокращение: фМ — N-формилметионин
Инициаторная TPHKMet доставляется к малой субъединице рибосомы вторым фактором
инициации, IF-2, наряду с молекулой ГТФ (последняя служит источником энергии для
заключительного шага инициирования). Формильная группа остается присоединенной
до тех пор, пока трансляция не перейдет в стадию элонгации, но тогда она удаляется из
наращиваемого полипептида или самостийно, или вместе с остальной частью началь-
ного метионина. Следует иметь в виду, что инициаторная TPHKMet способна расшиф-
ровывать только старт-кодон; она не может войти в полноценную рибосому во время
стадии элонгации трансляции, в ходе которой внутренние кодоны 5'-AUG-3' опознаются
иной TPHKMet, переносящей невидоизмененный метионин. Различие между этими раз-
новидностями тРНК заключается главным образом в двух необычных особенностях
инициаторной молекулы. Во-первых, в отличие от всех остальных бактериальныхтРНК,
которые были секвенированы, инициаторная тРНК имеет отрезок из трех пар оснований
G-С в своем антикодоновом стебле, который способствует прикреплению этой тРНК
к малой субъединице. Во-вторых, 5'-нуклеотид, который в большинстве тРНКучаствует
в первой паре оснований акцепторного стебля (см. рис. 13.2), не спарен в инициаторной
тРНК. Эта необычная особенность служит сигналом для фермента, который превращает
присоединенный метионин в N-формилметионин и, возможно, также предотвращает
инициаторную тРНК от входа в рибосому во время стадии элонгации.
Глава 13. Синтез и процессинг протеома 507
Завершение стадии инициации происходит, когда фактор IF-1 связывается с ини-
циаторным комплексом. Точная роль IF-1 не ясна (см. табл. 13.2), но он может вызывать
конформационное изменение в инициаторном комплексе, которое обусловливает при-
соединение большой субъединицы рибосомы. Прикрепление большой субъединицы
требует энергии, которая вырабатывается посредством гидролиза связанного ГТФ
и завершается высвобождением факторов инициации.
Инициация трансляции у эукариотов опосредствована структурами кэпа
и поли-А хвоста
Только небольшое количество молекул мРНК эукариотов имеет внутренние сайты
связывания рибосомы. Вместо этого, в большинстве мРНК малая субъединица рибосомы
производит исходную посадку на 5'-конце молекулы и затем продвигается по последо-
вательности и просматривает ее (сканирует последовательность), пока не обнаружит
старт-кодон. Данный процесс требует множества факторов инициации, и есть все еще
некоторая неясность относительно функций всех их (таблица 13.2). Особенности данного
процесса следующие (рис. 13.16).
На первом этапе происходит сборка предынициаторного комплекса. Эта струк-
тура включает в себя субъединицу 40S рибосомы, «тройной комплекс», состоящий из
фактора инициации eIF-2, связанного с инициаторной TPHKMet и молекулой ГТФ, и три
дополнительных фактора инициации: eIF-1, eIF-lA и eIF-З. Как и у бактерий, инициа-
торная тРНК отличается от нормальной TPHKMet, которая опознает внутренние кодоны
5'-AUG-3', но, в отличие от бактерий, она аминоацилируется нормальным метионином,
а не его формулированным вариантом.
После сборки предынициаторный комплекс связывается с 5'-концом мРНК. Для
этого шага необходим кэп-связывающий комплекс (иногда называемый eIF-4F), кото-
рый включает в себя факторы инициации eIF-4A, eIF-4E и eIF-4G. Контакт с кэпом мог бы
быть образован посредством одного лишь eIF-4E (как показано на рис. 13.16) или может
вовлечь более массовое взаимодействие с комплексом посадки на кэп. Фактор eIF-4G
служит мостом между eIF-4E, связанным с кэпом, и eIF-З, прикрепленным к предыни-
циаторному комплексу. В результате всего этого предынициаторный комплекс садится
на 5'-область мРНК. Посадка предынициаторного комплекса на мРНК также подвержена
влиянию поли-А хвоста на далеком З'-конце мРНК. Это взаимодействие, как думают,
опосредствуется связывающимся с полиаденилатом белком (PADPom), который при-
креплен к поли-А хвосту (раздел 12.2.1). У дрожжей и растений было показано, что PADP
может образовывать ассоциацию с eIF-4G, причем эта ассоциация требует, чтобы мРНК
загнулась обратно на себя. В случае искусственно декэпированных мРНК взаимодействия
PADP достаточно, чтобы поместить предынициаторный комплекс на 5’-конец мРНК,
но в нормальных условиях структура кэпа и поли-А хвост, вероятно, работают вместе.
Поли-А хвост может играть важную регуляторную роль, поскольку длина хвоста, как
оказалось, взаимосвязана с интенсивностью инициации, которая происходит на той
или иной рассматриваемой мРНК.
После прикрепления к 5’-концу мРНК инициаторный комплекс, как его теперь
следует называть, должен просмотреть молекулу мРНК и найти старт-кодон. Лидерные
области мРНК эукариотов могут иметь длину несколько десятков, а то и сотен нуклеоти-
дов и часто содержат области, которые образуют шпильки и другие структуры спаренных
оснований. Они, вероятно, удаляются комбинацией факторов eIF-4A и eIF-4B. Фактор
eIF-4A и, возможно, также eIF-4B обладают действием геликазы и, таким образом, спо-
собны разорвать внутримолекулярные пары оснований в мРНК, освобождая путь для
508 Часть 3. Принципы функционирования геномов
Таблица 13.2. Функции факторов инициации у бактерий и эукариотов
Фактор Бактерии IF-1 Функция Не ясна; рентгеноструктурный анализ показывает, что прикрепление IF-1 блокирует участок А, так что его функция может заключаться в предотвращении преждевременного входа молекул тРНК в участок А. Альтернативно IF-1 может вызывать конформационные изменения, которые подготавливают малую субъединицу к связыванию с большой субъединицей
IF-2 Направляет инициаторную тРНКМа к ее правильной позиции в ини- циаторном комплексе
IF-3 Предотвращает преждевременное повторное соединение большой и ма- лой субъединиц рибосомы
Эукариоты
eIF-1 Компонент предынициаторного комплекса
eIF-lA Компонент предынициаторного комплекса
eIF-2 Связывается с инициаторной TPHKMet внутри тройного комплекса — компонента предынициаторного комплекса; фосфорилирование eIF-2 приводит к глобальной репрессии трансляции
eIF-2B Восстанавливает комплекс е1Р2-ГТФ
eIF-З Компонент предынициаторного комплекса; образует прямой контакт с eIF-4G и таким образом образует связь с комплексом установки кэпа
eIF-4A Компонент кэп-связывающего комплекса; геликаза, которая облегчает просмотр, разрывая внутримолекулярные пары оснований в мРНК
eIF-4B Облегчает просмотр, возможно, действуя как геликаза, которая разры- вает внутримолекулярные пары оснований в мРНК
eIF-4E Компонент кэп-связывающего комплекса, возможно компонент, кото- рый образует прямой контакт со структурой кэпа на 5’-конце мРНК
eIF-4F Кэп-связывающий комплекс, включающий в себя факторы eIF-4A, elF- 4Е и eIF-4G и образующий первичный контакт со структурой кэпа на 5’-конце мРНК
eIF-4G Компонент кэп-связывающего комплекса; формирует мостик между кэп-связывающим комплексом и фактором eIF-З в предынициаторном комплексе. По крайней мере в некоторых организмах фактор eIF-4G связывается также с поли-А хвостом через связывающийся с полиаде- нилатом белок
eIF-4H У млекопитающих облегчает сканирование способом, подобным eIF-4B
eIF-5 Облегчает высвобождение других факторов при завершении инициа- ции
eIF-6 Связан с большой субъединицей рибосомы; препятствует прикреплению больших субъединиц к малым субъединицам в цитоплазме
Глава 13. Синтез и процессинг протеома 509
Таблица 13.3. Примеры последовательностей участков посадки рибосом у Escherichia coli
Ген Кодирует Последователь- ность участка посадки рибосомы Число ну- клеотидов до старт- кодона
Консенсус Е. coli — 5'-AGGAGGU-3' 3-10
Оперон Ферменты переработки лактозы 5'-AGGA-3' 7
лактозы
galE Гексозо-1-фосфатуридилтрансфераза 5'-GGAG-3' 6
rplj Рибосомный белок L10 5'-AGGAG-3’ 8
инициаторного комплекса (рис. 13.16, б). Старт-кодон, который обычно представлен
триплетом 5'-AUG-3’ у эукариотов, является опознаваемым, потому что он заключен
в короткую консенсусную последовательность 5'-ACCAUGG-3', известную под названием
консенсусная последовательность Козак.
а) Прикрепление предзапускающего
комплекса к мРНК
----Запускающая тРНК
2 elF-2
Малая субъединица Предынициаторный
комплекс
3 . elF-3
б) Сканирование
последовательность
elF-4A Козак
Садящийся на кэп комплекс
Рис. 13.16. Инициация трансляции у эукариотов, а) Сборка предынициаторного комплекса и его прикре-
пление к мРНК. Для ясности картины несколько белков, точные роли которых не ясны, были опущены.
Общая конфигурация комплекса не известна. 6) Предынициаторный комплекс сканирует мРНК, пока
не достигает старт-кодона, который является распознаваемым, потому что он расположен в пределах
консенусной последовательности Козак. Просмотру помогают факторы elF-4A и elF-4B, которые, как ду-
мают, обладают действием геликазы. Вероятно, что elF-З остается прикрепленным к предынициаторному
комплексу на протяжении сеанса просмотра, как показано здесь. Не ясно, остаются ли факторы elF-4E
и elF-4G также прикрепленными на этом этапе. Обратите внимание, что сканирование — энергозави-
симый процесс, который требует гидролиза АТФ. Сокращение: М — метионин
510 Часть 3. Принципы функционирования геномов
Как только запускающий комплекс распознает старт-кодон, присоединяется боль-
шая субъединица рибосомы. Как и у бактерий, эта операция требует гидролиза ГТФ
и ведет к высвобождению факторов инициации. На этой стадии в игру входят два по-
следних фактора инициации: eIF-5, который содействует высобождению других фак-
торов, и eIF-б, который связан с несвязанной большой субъединицей и предотвращает
ее прикрепление к малой субъединице в цитоплазме.
Инициация трансляции у эукариотов без просмотра
Система просмотра для инициации трансляции применима не ко всем мРНК эука-
риотов. Это впервые поняли при изучении пикорнавирусов — группы вирусов с геномами
из РНК, в которую входит полиовирус и риновирус человека (первый вызывает полио-
миелит, последний причиняет насморк). Транскрипты с этих вирусов не блокируются
кэпом, но вместо этого имеют внутренний сайт посадки рибосомы (IRES), который
является подобным по функции сайту связывания рибосомы у бактерий, хотя последо-
вательности IRES и их позиции относительно старт-кодона более изменчивы, чем у их
аналогов, присущих бактериям. Присутствие IRES на своих транскриптах означает, что
пикорнавирусы могут блокировать синтез белка в клетке хозяина путем инактивации
комплекса посадки на кэп, не затрагивающей трансляцию их собственных транскриптов,
хотя это не является обычной составляющей стратегии инфицирования, применяемой
всеми пикорнавирусами.
Примечательно, что для опознавания IRES рибосомой хозяина не требуется никаких
вирусных белков. Другими словами, нормальная клетка эукариотов обладает белками
и (или) другими факторами, которые позволяют ей запустить трансляцию с помощью
IRES. Из-за изменчивости IRES их трудно опознать в ходе анализа последовательностей
ДНК, но становится ясно, что транскрипты нескольких ядерных генов обладают ими
и что они транслируются, по крайней мере при определенных условиях, посредством
своих IRES, а не путем просмотра. Примеры представлены молекулами мРНК для белка,
связывающегося с тяжелой цепью иммуноглобулина млекопитающих белка Antennapedia
у Drosophila (раздел 14.3.4). IRES также обнаружены в нескольких мРНК, белковые
продукты которых транслируются, когда клетка подвержена стрессу, например при
воздействии высокой температуры, облучения или в условиях низкого содержания
кислорода. При таких обстоятельствах зависимая от кэпа трансляция глобально пода-
вляется (описано в следующем разделе). Поэтому присутствие IRES в молекулах мРНК
«выживания» позволяет им подвергаться предпочтительной трансляции в те периоды
времени, когда их продукты необходимы.
Регулирование инициации трансляции
Инициация трансляции — важная контрольная точка в синтезе белка, в которой
может быть осуществлено регулирование двух различных типов. Первый из них —
глобальное регулирование, которое вовлекает общее видоизменение количества
происходящего синтеза белка, при котором в равной степени затрагиваются все
мРНК, транслируемые кэп-зависимым механизмом. Это обычно достигается фосфо-
рилированием фактора eIF-2, что приводит к подавлению инициации трансляции
за счет предотвращения связывания фактором eIF-2 молекулы ГТФ, что необходимо
ему, перед тем как он сможет транспортировать инициаторную тРНК к малой субъе-
динице рибосомы. Фосфорилирование eIF-2 происходит во время стрессов, таких
как тепловой шок, когда общий уровень синтеза белка снижается и на первый план
выходит опосредствуемая IRES трансляция.
Глава 13. Синтез и процессинг протеома 511
Специфичное к транскриптам регулирование вовлекает механизмы, которые
действуют на один транскрипт или маленькую группу транскриптов, кодирующих
функционально связанные белки. Наиболее часто приводимый пример специфичного
к транскриптам регулирования относится к оперонам генов рибосомных белков Е. coli
(рис. 13.17, а). Лидерная область мРНК, транскрибируемая с каждого оперона, содержит
последовательность, которая служит сайтом связывания для одного из белков, коди-
руемых этим опероном. Когда этот белок синтезируется, он может или прикрепиться
к своей позиции на рибосомной РНК, или связаться с лидерной областью мРНК. При-
соединение к рРНК более багоприятно и происходит, если в клетке имеются свободные
рРНК. Как только все свободные рРНК будут собраны в рибосомы, рибосомный белок
связывается со своей мРНК, блокируя инициацию трансляции и, следовательно, выклю-
чая дальнейший синтез рибосомных белков, закодированных в данной мРНК. Подобные
события с участием других мРНК гарантируют, что синтез каждого рибосомного белка
будет согласован с количеством свободной рРНК в клетке. Другие белки, обладающие
способностью связывания с РНК, такие как некоторые аминоацил-тРНК-синтетазы,
также используют специфичное к транскриптам регулирование в манере, подобной
рибосомным белкам.
Второй пример регулирования, специфичного к транскриптам, имеет место у мле-
копитающих и регулирует трансляцию ферритина — белка, участвующего в запасании
железа (рис. 13.17, б). При отсутствии железа синтез ферритина ингибируется белками,
которые связываются с последовательностями, названными реагирующими на железо
элементами и расположенными в лидерной области мРНК ферритина. Эти белки блоки-
руют рибосому, когда она пытается сканировать мРНК в поисках стартового кодона. Когда
железо присутствует, связывающиеся белки
отделяются и мРНК транслируется. Инте-
ресно, что мРНК функционально связан-
ного белка — рецептора трансферрина, во-
влеченного в поглощение железа, — также
имеет реагирующие на железо элементы, но
в данном случае отделение связывающихся
белков в присутствии железа приводит не
к трансляции мРНК, а к ее деградации. Это
вполне логично, потому что, когда железо
а) Авторегуляция синтеза рибосомных белков
Рис. 13.17. Определяемое транскриптами регули-
рование инициации трансляции, а) Регулирование
синтеза рибосомного белка у бактерий. Оперон L11
Escherichia coliтранскрибируется в мРНК, несущую
копии генов рибосомных белков L11 и L1. Когда по-
садочные участки для L1 на имеющихся в наличии
молекулах рРНК 23 S заполняются, L1 связывается
с 5'-нетранслируемой областью мРНК, блокируя
дальнейшую инициацию трансляции. 6) Регулиро-
вание синтеза белка ферритина у млекопитающих.
Когда железо отсутствует, белок реакции на железо
связывается с 5'-нетранслируемой областью мРНК
ферритина и предотвращает тем самым синтез
ферритина
Блокирует дальнейшую
трансляцию
б) Регуляция реагирующими
на железо элементами
Реагирующий на железо
белок ^Железо
5' 3'
Трансляция
512 Часть 3. Принципы функционирования геномов
Аминоацил-тРНК,
EF-IA-ГТФ
Рис. 13.18. Элонгация трансляции. На схеме пока-
заны события, происходящие в ходе одного цикла
элонгации у Escherichia coli. В тексте сообщены под-
робности о трансляции у эукариотов. Сокращения:
фМ — N-формилметионин; Т — треонин
Вход аминоацил-тРНК
в акцепторный сайт
Пептидная связь
Образование
пептидной связи
Дезацилированная
тРНК, EF-2, ГДФ
Транслокация
EF-2, ГТФ
присутствует в клетке, потребность в актив-
ности рецептора трансферрина снижена, так
как в данный период необходимость импор-
тировать железо извне мала.
Инициация трансляции некоторых
бактериальных мРНК может также регули-
роваться короткими РНК, которые прикре-
пляются к сайтам узнавания в молекулах
мРНК. Это не всегда приводит к предот-
вращению трансляции, так как некоторые
короткие РНК могут также активировать
трансляцию одной или нескольких своих це-
левых мРНК. Пример — РНК Е coli, называе-
мая OxyS, которая имеет длину 109 нуклео-
тидов и регулирует трансляцию около 40
мРНК. Синтез OxyS активируется перекисью
водорода и другими реакционно активными
соединениями кислорода, которые могут
причинить окислительное повреждение
клетке. Будучи синтезирована, OxyS включа-
ет трансляцию некоторых мРНК, продукты
которых помогают защитить бактерию от
окислительного повреждения, и выклю-
чает трансляцию других мРНК, продукты
которых были бы вредны при таких обстоятельствах. Хотя были описаны структуры,
образуемые, когда OxyS и другие короткие регуляторные РНК связываются со своими
целевыми мРНК, они не дают нам никаких существенных представлений о том, как
осуществляется регуляторный процесс, тем более что структуры, образуемые на по-
давляемых мРНК, и которые, возможно, действуют путем блокирования доступа малой
субъединицы рибосомы к мРНК, не отличаются заметно от структур, образуемых на
мРНК, трансляция которых активируется.
13.2.3. Стадия элонгации в ходе трансляции
Главные различия между трансляцией у бактерий и у эукариотов наблюдаются на
стадии инициации: события, происходящие после связывания большой субъединицы
рибосомы с инициаторным комплексом, подобны в организмах обоих типов. Поэтому мы
можем рассматривать их вместе — смотреть, что происходит у бактерий и соотносить
их с отличительными особенностями трансляции у эукариотов там, где это уместно.
Элонгация у бактерий и эукариотов
После присоединения большой субъединицы остается занять два участка, ожи-
дающие связывания с молекулами аминоацил-тРНК. Первый из них, пептидильный,
Глава 13. Синтез и процессинг протеома 513
или P-сайт, уже занят инициаторной тРНКМе\ нагруженной N-формилметионином или
метионином и спаренной основаниями со старт-кодоном. Второй участок, аминоациль-
ный, или A-сайт, покрывает второй кодон в открытой рамке считывания (рис. 13.18).
Структуры, выявленные рентгеноструктурным анализом, показывают, что эти участки
расположены в расселине между большой и малой субъединицами рибосомы, взаимо-
действия типа кодон-антикодон связывают саму тРНК с малой субъединицей, а ами-
ноацильный конец тРНК с большой субъединицей (рис. 13.19).
A-сайт заполняется соответствующей связкой аминоацил-тРНК, которая у Е. coli
подносится к должной позиции фактором элонгации EF-1A, гарантирующим, что только
те тРНК, которые несут предписанную аминокислоту, смогут войти в рибосому, неза-
груженные же тРНК отвергаются в этой точке. Фактор EF-1A — пример G-белка, а это
значит, что он связывает молекулу ГТФ, которую сам же может гидролизировать, с тем
чтобы высвободить энергию. У эукариотов эквивалентный фактор называется eEF-1
и является комплексом из четырех субъединиц: eEF-la, eEF-lb, eEF-ld и eEF-lg (табли-
ца 13.4). Первая из них существует по крайней мере в двух формах, eEF-lal и eEF-la2,
которые являются весьма подобными белками и, вероятно, имеют эквивалентные
функции в различных тканях. Специфичные контакты между тРНК, мРНК и рРНК малой
субъединицы в пределах A-сайта гарантируют, что только нужной тРНК будет оказан
прием. Эти контакты способны различать взаимодействие кодон-антикодон, в кото-
ром образовались все три пары оснований, и такое, в котором присутствуют одна или
несколько ошибочных пар (последнее сигнализирует о том, что пришла неправильная
тРНК). Это, вероятно, только одна часть ряда мер предосторожности, которые гаранти-
руют точность процесса трансляции.
Когда аминоацил-тРНК входит в A-участок, между этими двумя аминокислотами
образуется пептидная связь. В этом деле замешан фермент пептидилтрансфераза, ко-
торый высвобождает аминокислоту из хватки инициаторной тРНКМа и затем образует
пептидную связь между этой аминокислотой и той, что присоединена ко второй тРНК.
Реакция является энергозависимой и требует гидролиза ГТФ, прикрепленного к фак-
тору EF-1A (eEF-1 у эукариотов). Это инактивирует EF-1A, который выталкивается из
рибосомы и восстанавливается при помощи EF-1B. Присущий эукариотам эквивалент
EF-1B не был опознан, и вполне возможно, что одна из субъединиц фактора eEF-1 об-
ладает восстанавливающим действием.
Теперь дипептид, соответствующий первым двум кодонам в открытой рамке считы-
вания, прикреплен к тРНК в A-сайте. Следующий шаг — транслокация (см. рис. 13.18),
в ходе которой рибосома передвигается по мРНК на три нуклеотида, так что новый кодон
входит в A-сайт. При этом связка дипептид-тРНК перемещается в P-сайт, что, в свою оче-
редь, вытесняет дезацил ированную тРНК. У эукариотов дезацил ированная тРНК просто
выталкивается из рибосомы, но у бактерий дезацилированная тРНК отбывает с третьей
позиции — участка выхода, или Е-сайта. Этот участок первоначально рассматривали
как простую точку выхода из рибосомы, но сейчас известно, что она имеет важную роль
в обеспечении того, что в результате транслокации рибосома перемещается по мРНК
точно на три нуклеотида и, таким образом, гарантирует, что рибосома придерживается
правильной рамки считывания.
Для осуществления транслокации необходим гидролиз молекулы ГТФ, и она опо-
средствуется фактором EF-2 у бактерий и eEF-2 у эукариотов. Электронная микроскопия
рибосом на различных промежуточных стадиях транслокации показывает, что во вре-
мя транслокации рибосома принимает менее компактную структуру, при которой две
субъединицы немного поворачиваются в противоположных направлениях, открывая
514 Часть 3. Принципы функционирования геномов
Рис. 13.19. Важные участки в рибосоме. Структура слева — большая субъединица рибосомы Thermus
thermophilus-, таковая справа — малая субъединица. Представлены виды сверху на две поверхности,
которые входят в контакт друге другом, когда субъединицы сходятся друг с другом и получается цельная
рибосома. А- Р- и Е-сайты помечены соответствующими буквами и каждый заняттРНК, показанной крас-
ным или оранжевым. Основная часть каждой тРНК погружена в недра большой субъединицы и только
антикодоновые стебли и петли связаны с малой субъединицей. Те части рибосомы, которые образуют
важные сопрягающие контакты между двумя субъединицами, помечены как В1а и т. д. Перепечатано
с разрешения Yuspov et al., Science, 292, 883-896. © 2001AAAS
зазор между ними и позволяя рибосоме скользить по молекуле мРНК. В результате транс-
локации A-сайт становится свободным и готов принять новую связку аминоацил-тРНК.
Цикл элонгации теперь повторяется и продолжается до тех пор, пока не будет достигнут
конец открытой рамки считывания.
Пептидилтрансфераза — рибозим
Связанный с рибосомой белок, который обладает действием пептидилтрансфера-
зы, необходимой для синтеза пептидных связей в ходе трансляции, никогда не удава-
лось выделить. Причина такой неудачи теперь известна, по крайней мере касательно
бактерий: ферментативное действие определяется частью 23S рРНК и, следовательно,
представляет еще один пример действия рибозима (раздел 12.2, а).
Когда структуры спаренных оснований рРНК (см. рис. 13.11) были впервые опреде-
лены в начале 1980-х гг., возможность, что молекула РНК может обладать фермента-
тивным действием, была неслыханной, крупные открытия в области рибозимов были
сделаны лишь в период 1982-1986 гг. Поэтому рибосомные РНК были первоначально
наделены чисто структурными ролями в рибосоме, их спаренные основаниями кон-
формации рассматривались как каркасы, к которым прикреплялись поистине важные
компоненты рибосомы — белки. Проблемы с такой интерпретацией начали возникать
в конце 1980-х гг., когда ученые столкнулись с трудностями в опознании белка или белков,
отвечающих за главное каталитическое действие рибосомы — образование пептидных
связей. К настоящему времени существование рибозимов установлено, и молекулярные
биологи начали серьезно рассматривать возможность того, что ферментативную роль
в синтезе белка могут играть молекулы рРНК. Чтобы проверить эту гипотезу, было не-
обходимо определить точное местонахождение в рибосоме участка с действием пепти-
дилтрансферазы. Через несколько лет антибиотики и другие ингибиторы синтеза белка
Глава 13. Синтез и процессинг протеома 515
Таблица 13.4. Факторы элонгации трансляции у бактерий и эукариотов
Фактор Функция
Бактерии EF-1A Направляет следующую тРНК к надлежащей позиции в рибосоме
EF-1B Восстанавливает фактор EF-1A после того, как последний отдал энер-
EF-2 гию, содержавшуюся в присоединенной к нему молекуле ГТФ Опосредствует транслокацию
Эукариоты eEF-1 Комплекс из четырех субъединиц (eEF-la, eEF-lb, eEF-ld и eEF-lg);
eEF-2 направляет следующую тРНК к надлежащей позиции в рибосоме Опосредствует транслокацию
Примечание: следует иметь в виду, что бактериальные факторы элонгации были недавно переимено-
ваны. Обозначения старой системы EF-Tu, EF-Ts и EF-G соответствуют EF-1A, EF-1B и EF-2 в новой.
сыграли важную роль в исследованиях функции рибосомы. В 1995 г. был синтезирован
новый ингибитор, названный CCdA-фосфатпуромицином. Это соединение является ана-
логом промежуточной структуры, образуемой, когда две аминокислоты соединяются
за счет образования пептидной связи во время синтеза белка. CCdA-фосфатпуромицин
сильно связывается с бактериальной рибосомой, и в силу своей структуры, этот сайт
связывания должен быть расположен точно в позиции, где в действующей рибосоме
образуются пептидные связи. Чтобы показать точно, где в пределах большой субъеди-
ницы связывается CCdA-фосфатпуромицин, был использован рентгеноструктурный
анализ. Его пристанище обнаружилось в самой глубине тела субъединицы и оказалось
тесно связаннным с рРНК 23 S большой субъединицы, но на 1,84 нм от наиболее близле-
жащего белка L3 и на немного большем расстоянии от белков L2, L4 и L10 (рис. 13.20).
В атомном масштабе 1-2 нм — огромное расстояние, которое делает немыслимой воз-
можность, что синтез пептидной связи мог быть катализируем одним из этих четырех
белков. Поэтому такое расположение CCdA-фосфатпуромицина представляет убеди-
тельное доказательство того, что
пептидилтрансфераза является
рибозимом.
Теперь, когда это свидетель-
ство было получено, исследова-
тели идут дальше и стремятся
Рис. 13.20. Местоположение молекулы
CCdA-фосфатпуромицина в пределах
большой субъединицы бактериальной
рибосомы. Показаны части из рибосом-
ных белков L2, L3, L4 и L10. Расстояния
в нанометрах между молекулой CCdA-
фосфатпуромицина (показанной в виде
красной точки) и каждым из белков обо-
значены цифрами
516 Часть 3. Принципы функционирования геномов
определить, как именно основная цепь рРНК выполняет функцию рибозима в образо-
вании пептидной связи. Внимание было первоначально сосредоточено на нуклеотиде
аденине в позиции 2451 в 23S рРНК Е. coli, потому что этот аденин имеет необычные
свойства заряда по сравнению с другими нуклеотидами. Согласно выдвинутой гипотезе,
взаимодействие между этим аденином и близлежащим гуанином, стоящим в позиции
2447, является ключом к синтезу белка. Но эта модель была подвергнута сомнению дан-
ными мутационного анализа, которые показали, что, хотя при замене А2451 урацилом
скорость синтеза пептидной связи снижается на 99 %, как А2451, так и G2447 могут
быть заменены другими нуклеотидами без какого-либо заметного эффекта. Внимание
поэтому устремляется к другим частям 23S рРНК, которые расположены в окрестности
активного участка.
Сдвиг рамки считывания и другие необычные события в ходе элонгации
Прямая трансляция мРНК кодон за кодоном рассматривается как стандартный
способ синтеза белков. Но обнаруживается все большее число необычных событий
элонгации. Одно из них — сдвиг рамки считывания, который происходит, когда ри-
босома берет паузу в середине мРНК, сдвигается обратно на один нуклеотид или, что
менее часто, вперед на один нуклеотид и затем продолжает трансляцию. В результате
этого кодоны, которые считываются после паузы, не смежны с предыдущим набором
кодонов: они лежат в иной рамке считывания (рис. 13.21, а).
Самопроизвольные сдвиги рамки считывания происходят беспорядочно и вредо-
носны, потому что полипептид, синтезируемый после сдвига рамки считывания, имеет
неправильную последовательность аминокислот. Но не все сдвиги рамки считывания
самопроизвольны: некоторые мРНК используют запрограммированный сдвиг рамки
считывания, чтобы побудить рибосому изменить рамку в определенной точке в преде-
лах транскрипта. Запрограммированный сдвиг рамки считывания происходит в орга-
низмах всех типов, от бактерий до человека, а также в ходе экспрессии ряда вирусных
геномов. Пример наблюдается во время синтеза у Е. coli ДНК-полимеразы III — главного
фермента, участвующего в репликации ДНК (раздел 15.2.2). Две из субъединиц ДНК-
полимеразы 111 (у и т) кодируются единым геном dnaX. Субъединица т — полномерный
продукт трансляции мРНК dnaX, а субъединица у — его укороченный вариант. Синтез
субъединицы у вовлекает сдвиг рамки считывания в середине мРНК dnaX, при этом
рибосома встречает стоп-кодон сразу же после сдвига рамки считывания и, таким об-
разом, производит усеченный у-вариант продукта трансляции. Думают, что сдвиг рамки
считывания вызывают следующие три особенности мРНК dnaX:
• шпилька, которая расположена непосредственно после позиции сдвига рамки
считывания и которая стопорит рибосому;
• последовательность, подобная сайту связывания рибосомы и лежащая непосред-
ственно выше позиции сдвига рамки считывания, которая, как думают, спаривается
основаниями с рРНК 16 S (что делает истинный сайт связывания рибосомы), опять
же заставляя рибосому остановиться;
• кодон 5'-AAG-3' в позиции сдвига рамки считывания. Присутствие видоизмененного
нуклеотида в позиции неканонического спаривания TPHKLys, которая декодирует
5'-AAG-3', означает, что взаимодействие кодон-антикодон в этой позиции относи-
тельно слабое, и позволяет произойти сдвигу рамки считывания.
Подобное явление—трансляционное проскальзывание — позволяет одной ри-
босоме транслировать мРНК, которая содержит копии двух и более генов (рис. 13.21, б).
Глава 13. Синтез и процессинг протеома 517
а) Запрограммированный сдвиг рамки
считывания в мРНК dnaX
т-субъединица
TKAKKSEPAA
...ACCAAAGCAAAAAAGAGUGAACCGGCAGCC...
Т К А К К 1----11-1
стоп 7-субъединица
Сдвиг рамки считывания
6) Трансляционное проскальзывание в мРНК оперона лактозы
Конец lacZ Начало lacY
. . UGUCAAAAAUAAUAAUAACCGGGCAGGCCAUGUCUGCCCGUAUUUCGCGUAAAGGAAAUCCAUUAUGUACUAUUUA. . .
С Q К стоп М Y Y L
-------------------------------------------------►
Проскальзывание
е) Трансляционный обход в мРНК гена 60 бактериофага Т4
...GAUGGAUAGCCUUCGGGCUAUCUAUAGAAAUACCUCAUAAUUAAGAGAUUAUUGGAUUAGGC...
D G стоп
G L G
Обход
Рис. 13.21. Три необычных события элонгации трансляции, встречающихся у Escherichia coli. а) Запрограм-
мированный сдвиг рамки считывания во время трансляции мРНК dnaX. В ходе синтеза у-субъединицы
рибосома смещается назад на один нуклеотид немедленно после прохождения ряда из А. Рибосома
вставляет глутаминовую кислоту в полипептид и затем встречает стоп-кодон. 6) Проскальзывание между
генами lacZ и lacY мРНК оперона лактозы, в) Обход в течение трансляции мРНК гена 60 бактериофага
Т4 состоит в перескоке между двумя кодонами глицина. В табл. 1.2 приведены однобуквенные сокра-
щения аминокислот
Это означает, что, например, одна рибосома может синтезировать каждый из пяти белков,
кодируемых мРНК, транскрибированной с оперона триптофана Е. coli (см. рис. 8.8, б).
Когда рибосома достигает конца одной группы кодонов, она высвобождает белок,
только что ею произведенный, скользит к следующему старт-кодону и начинает син-
тезировать следующий белок. Более крайняя форма проскальзывания — трансляци-
онный обход, при котором большая часть транскрипта, возможно, несколько десятков
п.н., пропускается, и элонгация исходного белка продолжается после события обхода
(рис. 13.21, в). Обход начинается и заканчивается или в двух идентичных кодонах, или
в двух кодонах, которые могут быть транслированы одной и той же тРНК за счет неодно-
значного спаривания. Это предполагает, что скачок управляется тРНК, прикрепленной
к наращиваемому полипептиду, которая просматривает мРНК, по мере того как рибосома
продвигается вперед, и останавливает обход, когда новый кодон, с которым она может
518 Часть 3. Принципы функционирования геномов
спариться основаниями, будет достигнут. Так, у Е. coli в ходе трансляции мРНК для гена
60 бактериофага Т4, который кодирует субъединицу ДНК-топоизомеразы, происходит
трансляционный обход 44 нуклеотидов. Подобные события были замечены также у дру-
гих различных бактерий. В результате обхода с одной мРНК могут быть синтезированы
два различных белка — один белок от нормальной трансляции и один от обхода, — но
является ли это главной функцией данного механизма, еще не известно.
13.2.4. Терминация трансляции
Синтез белка завершается по достижении одного из трех стоп-кодонов (рис. 13.22).
В A-сайт теперь входит не тРНК, а фактор высвобождения белка (таблица 13.5). Бак-
терии располагают тремя такими факторами: RF-1, который опознает стоп-кодоны
5’-UAA-3’ и 5’-UAG-3’, RF-2, который опознает стоп-кодоны 5'-UAA-3’ и 5'-UGA-3’, и RF-3,
стимулирующий высвобождение RF-1 и RF-2 из рибосомы после терминации (в ходе
реакции, требующей энергии от гидролиза ГТФ). Эукариоты имеют в своем арсенале
только два фактора высвобождения: eRF-1, который опознает все три стоп-кодона,
и eRF-З, который, возможно, играет ту же роль, что и RF-3, хотя это не было доказано.
Структура eRF-1 была определена рентгеноструктурным анализом, который показал, что
структура этого белка во многом подобна структуре тРНК (рис. 13.23). Это привело к по-
строению модели, в которой фактор высвобождения повторяет структуру тРНК и, следо-
Стоп-кодон входит
в А-участок
RF1 + RF3 или RF2 + RF3
Законченный полипептид
вательно, способен войти в А-сайт,
когда будет достигнут стоп-кодон.
Эта модель привлекательна, но дру-
гие исследования предполагают, что
фактор высвобождения принимает
иную конформацию, когда связыва-
ется с рибосомой, — такую, которая
менее подобна форме тРНК.
Факторы высвобождения за-
вершают трансляцию, но они, ка-
жется, не отвечают за разъединение
рибосомных субъединиц (по край-
ней мере не у бактерий). Это пре-
рогатива дополнительного белка,
названного фактором повторного
использования рибосомы (RRF),
который, подобно eRF-1, имеет по-
добную тРНК структуру. RRF, веро-
ятно, входит в Р- или в A-сайт и «раз-
мыкает» рибосому (см. рис. 13.22).
Рис. 13.22. Терминация трансляции. Пока-
зано завершение у Escherichia coli. О раз-
личиях с эукариотами сообщается в тексте.
Аминокислота, помеченная «А», — аланин.
Сокращения: RF — фактор высвобождения;
RRF — фактор повторного использования
рибосомы
Глава 13. Синтез и процессинг протеома 519
Таблица 13.5. Факторы высвобождения и повторного использования рибосомы у бактерий и эукариотов
Фактор
Бактерии
Функция
RF-1 Опознает стоп-кодоны 5'-UAA-3’ и 5'-UAG-3’
RF-2 Опознает стоп-кодоны 5'-UAA-3’ и 5'-UGA-3’
RF-3 Стимулирует отделение RF-1 и RF-2 от рибосомы после терминации
RRF Фактор высвобождения рибосомы, ответственный за отсоединение субъединиц рибосомы после терминации трансляции
Эукариоты eRF-1 Опознает кодон терминации
eRF-3 Возможно, стимулирует отделение eRF-1 от рибосомы после терми- нации; возможно, вынуждает субъединицы рибосомы разъединяться после терминации трансляции
Разъединение требует энергии, которая высвобождается из ГТФ при участии EF-2
(одного из факторов элонгации), и требует также фактора инициации IF-3, который
препятствует повторному объединению субъединиц. Имеющийся у эукариотов эк-
вивалент фактора RRF не был обнаружен, и это может быть одной из функций eRF-3.
Разъединенные субъединицы рибосомы входят в цитоплазматический фонд, где они
остаются, пока не будут использованы снова в следующем круге трансляции.
Рис. 13.23. Структура фактора высвобождения eRF-1 эукариотов подобна структуре тРНК. На левой
картинке показан eRF-1, а на правой — тРНК. Часть eRF-1, которая напоминает тРНК, выделена белым
цветом. Фиолетовая доля eRF-1 взаимодействует со вторым фактором высвобождения эукариотов —
eRF-З. Перепечатано из Trends Biochem. Sci., 25, Kisselev and Buckinham, Transitional termination comes of
age, 561-566, 2000, с разрешения издательства Elsevier
13.2.5. Трансляция у археи
Описания вышеизложенных событий относятся к трансляции у бактерий и эука-
риотов. Мы не должны игнорировать вторую группу прокариотов, археи, и, таким об-
разом, прежде чем идти дальше, мы вкратце рассмотрим то, что известно о трансляции
в этих организмах.
520 Часть 3. Принципы функционирования геномов
Во многих отношениях трансляция у архей больше напоминает соответствующие
события у эукариотов, а не у бактерий. Одно очевидное исключение — то, что рибосома
архей при коэффициенте седиментации 70 S сопоставима в размере с бактериальной ри-
босомой и, подобно рибосомам бактерий, содержит 23S рРНК, 16S рРНК и 5S рРНК. Это
кажущееся подобие иллюзорно, потому что молекулы рРНК архей образуют спаренные
основаниями вторичные структуры, которые значительно отличаются от эквивалентных
структур у бактерий. Структуры рРНК архей также отличаются от свойственных эукариотам
вариантов, но рибосомные белки, которые прикрепляются к рРНК, являются гомологами
белков эукариотов.
Молекулы мРНК архей блокируются и полиаденилируются, а инициация трансляции,
как думают, вовлекает процесс просмотра, подобный описанному для мРНК эукариотов.
Молекулы тРНК архей проявляют несколько уникальных особенностей, в том числе отсут-
ствие тимидина в так называемом ТТС-стебле клеверного листа и присутствие в различных
позициях видоизмененных нуклеотидов, не наблюдаемых ни у бактерий, ни у эукариотов.
Метионин, переносимый инициаторной тРНК, не N-формилируется, а факторы инициации
и элонгации напоминают соответствующие молекулы эукариотов.
13.3. Посттрансляционный процессинг белков
Трансляция — не конец пути экспрессии генома. Полипептид, который появляет-
ся из рибосомы, является неактивным и, прежде чем он примет возложенную на него
функциональную роль в клетке, должен подвергнуться посттрансляционной обработке
(посттрансляционному процессингу) по крайней мере первого из следующих четырех
типов (рис. 13.24).
• Сворачивание белка. Полипептид является неактивным, пока не свернется в пра-
вильную третичную структуру.
• Протеолитическое расщепление. Некоторые белки в ходе созревания подвер-
гаются специфическому разрезанию, выполняемому ферментами, названными
протеазами. Такие события разрезания могут удалять сегменты с одного или
обоих концов полипептида и давать укороченную форму белка, или они могут
нарезать полипептид на несколько различных сегментов, все или некоторые из
которых активны.
• Химическая модификация. Отдельные аминокислоты в полипептиде могут быть
модифицированы путем присоединения новых химических групп.
• Вырезание интеинов. Интеины представляют собой вклинивающиеся после-
довательности в некоторых белках, подобные по характеру поведения интронам
в мРНК. Они должны быть удалены, а экстеины лигированы, для того чтобы белок
стал функционально активным.
Часто события процессинга этих различных типов происходят совместно — по-
липептид разрезается и модифицируется одновременно со сворачиванием. Если дело
обстоит именно так, то события разрезания, модификации и (или) вырезания могут
быть необходимы для того, чтобы полипептид принял свою правильную трехмерную
конформацию, потому что она зависит частично от относительного расположения раз-
личных химических групп вдоль молекулы. Альтернативно событие разрезания или
химическая модификация могут произойти после того, как белок был свернут, возможно,
как часть регулирующего механизма, который превращает свернутый, но неактивный
белок в активную форму.
Глава 13. Синтез и процессинг протеома 521
ПоЛИПеПТИД «ммнмм»
Протеолитическое
расщепление
Химическая
модификация
Вырезание интеина
Раздел
13.3.2
Раздел
13.3.3
I
Новая химическая группа
Раздел
13.3.4
Позиция удалённого
интеина
Рис. 13.24. Схематичное представление четырех типов событий посттрансляционной обработки. Не все
события происходят во всех организмах
13.3.1. Сворачивание белка
Вспомним, что в разделе 1.3.1 мы изучили четыре уровня структуры белка (первич-
ный, вторичный, третичный и четвертичный) и узнали, что вся информация, которая
нужна полипептиду для сворачивания в его правильную трехмерную структуру, содер-
жится в пределах его аминокислотной последовательности. Это один из центральных
принципов молекулярной биологии. Поэтому мы должны исследовать его эксперимен-
тальную основу и рассмотреть то, как информация, содержащаяся в последовательности
аминокислот, используется в течение процесса сворачивания нового транслированного
полипептида.
Не все белки самопроизвольно сворачиваются в пробирке
Концепция, согласно которой последовательность аминокислот содержит всю
информацию, необходимую полипептиду для сворачивания в его правильную третич-
ную структуру, основана на экспериментах, проведенных с рибонуклеазой в 1960-е гг.
Рибонуклеаза — маленький белок, всего лишь 124 аминокислоты в длину, содержащий
четыре дисульфидных мостика и обладающий третичной структурой, которая состоит
преимущественно из p-листа и дополнена очень небольшой а-спиралью. Исследования
его сворачивания были выполнены над рибонуклеазой, которая была очищена из под-
желудочной железы коровы и взвешена в виде суспензии в буфере. Добавление мочевины
(соединения, которое разрушает водородные связи) привело к уменьшению активности
этого фермента (измеряемой его способностью разезать РНК) и к увеличению вязкости
раствора (рис. 13.25), что указывает на то, что белок денатурировал, развернувшись
в неструктурированную полипептидую цепь. Важное наблюдение состояло в том, что,
когда мочевина была удалена посредством диализа, вязкость уменьшилась и активность
фермента восстановилась. Заключение — то, что белок самопроизвольно повторно сво-
рачивается, когда денатурирующий агент (в данном случае мочевина) удаляется. В этих
исходных экспериментах четыре дисульфидные связи оставались неповрежденными,
потому что они не разрушались мочевиной, но тот же самый результат был получен,
когда обработка мочевиной сочеталась с добавлением восстановителя, долженствую-
щего разорвать дисульфидные связи: активность по-прежнему восстанавливалась
при ренатурации. Это показывает, что дисульфидные связи не являются критически
522 Часть 3. Принципы функционирования геномов
Рис. 13.25. Денатурация и са-
мопроизвольная ренатурация
маленького белка. При увели-
чении концентрации мочевины
до 8 моль/л белок становится
денатурированным, развертыва-
ясь: его активность уменьшается
и вязкость раствора увеличива-
ется. Когда мочевина удаляется
диализом, этот маленький белок
повторно принимает свою свер-
нутую конформацию. Активность
белка увеличивается назад к пер-
воначальному уровню, а вязкость
раствора уменьшается
важными для способности белка повторно сворачиваться, они просто стабилизируют
третичную структуру, после того как ее примет белок.
Более доскональное исследование путей самопроизвольного сворачивания для
рибонуклеазы и других маленьких белков привело к следующему общему описанию
двухэтапного процесса:
• В течение нескольких миллисекунд после удаления денатурирующего агента в цепи
полипептида образуются мотивы вторичной структуры. Этот шаг сопровождается
складыванием белка в компактную, но не полностью свёрнутую нативную струк-
туру с огражденными от воды гидрофобными группами внутри молекулы.
• В течение следующих нескольких секунд или минут такие мотивы вторичной
структуры взаимодействуют друг с другом и третичная структура постепенно
принимает свою форму, часто через ряд промежуточных конформаций. Другими
словами, белок проходит путь сворачивания. Однако может быть несколько воз-
можных путей, которыми белок может проследовать и достигнуть своей правильно
свернутой структуры. Также пути могут иметь боковые ответвления, в которые
белок может отклониться и прийти к неправильной структуре. Если неправильная
структура достаточно неустойчива, то может произойти частичное или полное раз-
вертывание, предоставляя белку вторую возможность проследовать продуктивным
маршрутом к его правильной конформации (рис. 13.26).
В течение ряда лет большее или меньшее распространение получало допущение
о том, что все белки самопроизвольно свертываются в пробирке, но эксперименты по-
казали, что только небольшие белки с менее сложными структурами обладают этой
способностью. Два фактора, кажется, препятствуют большим белкам самопроизвольно
сворачиваться. Первый из них — их стремление образовывать нерастворимые агрегаты,
когда денатурирующий агент удаляется: полипептиды могут складываться в сцеплен-
ные сети, когда они пытаются защитить свои гидрофобные группы от воды в шаге
первом общего пути сворачивания. В эксперименте этого можно избежать, используя
сильное разбавление белка, но это не та возможность, которую клетка может исполь-
зовать, с тем чтобы препятствовать объединению её развернутых белков в агрегаты.
Вторым фактором, который препятствует сворачиванию, является стремление больших
белков устремляться в непродуктивные боковые ответвления своих путей сворачивания
Глава 13. Синтез и процессинг протеома 523
Рис. 13.26. Неправильно свернутый белок может быть
способен повторно свернуться в свою правильную кон-
формацию. Черная стрелка представляет правильный
путь свертывания, ведущий от развернутого белка слева
к активному белку справа. Красная стрелка ведет к не-
правильносвернутой конформации, но эта конформация
неустойчива и белок способен частично развернуться,
возвратиться к его правильному пути сворачивания и,
в конечном счете, прийти к своей активной конфор-
мации
и застревать в них, принимая промежуточную
форму, которая неправильно свернута, но явля-
ется слишком устойчивой, чтобы развернуться Неправильно свёрнутый
до сколь-нибудь значительной степени. Кроме
того, возникли вопросы об адекватности сво-
рачивания in vitro, которое применялось с рибонуклеазой для сворачивания белков
в клетке, естественным процессам в клетке, потому что клеточный белок может начать
сворачиваться прежде, чем он будет полностью синтезирован. Если происходит первич-
ное сворачивание, когда имеется только часть полипептида, то может быть достаточно
велика вероятность следования неправильными путями сворачивания. Эти различные
соображения направили исследования на сворачивание белка в живых клетках.
В клетках процессу сворачивания помогают молекулярные шапероны
Большая часть нашего понимания сворачивания белка в клетке основана на от-
крытии белков, которые помогают другим белкам сворачиваться. Их назвали молеку-
лярными шаперонами и наиболее досконально изучили у Е coli. Ясно, что и эукариоты,
и археи обладают эквивалентными белками, хотя некоторые особенности принципа их
работы у них отличаются.
Молекулярные шапероны могут быть разделены на две группы:
• шапероны Hsp70, которые включают белки по имени Hsp70 (кодируемый ге-
ном dnaK у Е. coli и иногда называемый белком DnaK), Hsp40 (кодируемый dnaj)
и GrpE;
• шаперонины, главный вариант которых — комплекс GroEL/GroES, имеющийся
у бактерий и эукариотов, и TRiC, встречающийся только у эукариотов.
Молекулярные шапероны не определяют третичную структуру белка, они просто
помогают белку найти эту правильную структуру. Шапероны двух типов делают это
различными способами. Представители семейства Hsp70 связываются с гидрофобны-
ми областями развернутых белков, в том числе и белков, которые все еще продолжают
транслироваться (рис. 13.27). Они удерживают белок в раскрытой конформации и со-
действуют сворачиванию, возможно, опосредствуя соединение между теми частями
полипептида, которые образуют взаимодействия в свернутом белке. Как именно это
достигается, в точности не понято, но оно вовлекает повторное закрепление и отделение
белка Hsp70, каждый цикл которого требует энергии, обеспечиваемой гидролизом АТФ.
Белок Hsp70, наряду со связыванием с целевым полипептидом, обладает действием
АТФазы и, следовательно, может высвободить эту энергию, но он может эффективно
функционировать только с помощью белков Hsp40 и GrpE. Hsp40 стимулирует действие
АТФазы белка Hsp70, a GrpE удаляет из комплекса молекулу АДФ (в который АТФ пре-
524 Часть 3. Принципы функционирования геномов
Рис. 13.27. Система шаперонов Hsp70. Шапероны
Hsp70 связываются с гидрофобными областями
в развернутых полипептидах, в том числе тех,
которые все еще транслируются, и удерживают
белок в открытой конформации, с тем чтобы
помочь его свертыванию
вращается при высвобождении энергии)
и позволяет циклу начаться снова. Поми-
мо сворачивания белка, шапероны Hsp70
участвуют также и в других процессах,
требующих ограждения гидрофобных
областей в белках, таких как перенос че-
рез мембраны, объединение белков в многосубъединичные комплексы и разобщение
белков, которые были повреждены тепловым стрессом.
Шаперонины действуют совершенно иным образом. GroEL и GroES образуют много-
субъединичную структуру, которая напоминает пустотелую пулю с центральным кана-
лом (рис. 13.28). Единичный развернутый белок входит в канал и выходит свернутым.
Механизм этого не известен, но был выдвинут постулат, согласно которому комплекс
GroEL/GroES служит клеткой, которая препятствует развернутому белку объединиться
с другими белками, и что внутренняя поверхность канала изменяется с гидрофобной на
гидрофильную таким образом, чтобы способствовать заглублению гидрофобных ами-
нокислот внутрь белковой молекулы. Это не единственная гипотеза: другие исследова-
тели придерживаются того мнения, что канал развертывает белки, которые свернулись
неправильно, и возвращает такие развернутые белки назад в цитоплазму, так что они
могут иметь вторую попытку принятия своей правильной третичной структуры.
Хотя у эукариотов имеется и семейство шаперонов Hsp70 и шаперонины
GroEL/GroES, кажется, что в этих организмах сворачивание белка зависит главным обра-
зом от действия белков Hsp70. Это, вероятно, верно также и для бактерий, хотя главную
Рис. 13.28. Шаперонин GroEL/GroES. Слева показан вид сверху, а справа — вид сбоку. 1А равен 0,1 нм.
Часть структуры GroES состоит из семи идентичных белковых субъединиц и показана золотым цветом.
Компоненты GroEL состоят из 14 идентичных белков, скомпонованных в виде двух колец (показаны
красным и зеленым), каждое из которых содержит семь субъединиц. Главный вход в центральный
канал — через основание структуры, показанной справа. Перепечатано с разрешения Xu et al., (1997),
Nature, 388, 741-750
Глава 13. Синтез и процессинг протеома 525
роль в сворачивании метаболических ферментов и белков, участвующих в транскрипции
и трансляции, играют шаперонины GroEL/GroES.
13.3.2. Обработка протеолитическим расщеплением
Протеолитическое расщепление имеет две функции в посттрансляционом про-
цессинге белков (рис. 13.29).
• Оно используется для удаления коротких отрезков с N- и (или) С-концевых об-
ластей полипептидов, после чего остается одна укороченная молекула, которая
сворачивается в активный белок.
• Оно используется, чтобы разрезать полипротеины на сегменты, все из которых
(или хотя бы некоторые) являются активными белками.
Эти события относительно обычны для эукариотов, но менее часты у бактерий.
Расщепление концов полипептидов
Обработка расщеплением обычна для секретируемых полипептидов, биохимиче-
ские действия которых могут быть вредны для клетки, производящей белок. Пример
представлен мелиттином — наиболее изобильным белком в пчелином яде, вызы-
вающим лизис клеток после впрыскивания яда из жала пчелы в уязвленного человека
или ужаленное животное. Мелиттин лизирует клетки и у пчел, и у животных и, таким
образом, должен изначально синтезироваться в виде неактивного предшественни-
ка. Этот предшественник, промелиттин, имеет 22 дополнительные аминокислоты на
своем N-конце. Предпоследовательность удаляется внеклеточной протеазой, которая
разрезает ее в 11 позициях, высвобождая активный белок яда. Протеаза не расщепляет
активную последовательность, потому что ее способ действия — расщеплять дипептиды
с последовательностью X-Y, где X — аланин, аспарагиновая кислота или глутаминовая
кислота, a Y — аланин или пролин; эти мотивы не встречаются в активной последова-
тельности (рис. 13.30).
Подобный тип процессинга происходит с инсулином — белком, производимым
в островках Лангерганса в поджелудочной железе позвоночных и отвечающим за управ-
ление уровнем сахара в крови. Инсулин синтезируется в виде пре-про-инсулина, который
имеет длину 105 аминокислот (рис. 13.31). В процедуре процессинга предусмотрено уда-
ление первых 24 аминокислот, в результате чего образуется проинсулин, за которым сле-
дуют два дополнительных разреза, вырезающих центральный сегмент и оставляющих
две активные части белка (А- и В-цепи), которые связываются друг с другом за счет об-
разования двух дисульфидных связей, в результате чего образуется зрелый инсулин. Пер-
вый сегмент, который должен быть
удален (24 аминокислоты с N-конца),
является сигнальным пептидом —
Полипептид
N С
Рис. 13.29. Обработка белка посредством
протеолитического расщепления. Слева бе-
лок обрабатывается удалением N-концевого
сегмента. У некоторых белков происходит
также и С-концевая обработка. Справа по-
липротеин обрабатывается, чтобы дать три
различных белка. Не все белки подвергаются
протеолитическому расщеплению
Обработка концов Обработка полипротеина
Отброшенный
оконечный сегмент
Активный белок 3 активных-белка
526 Часть 3. Принципы функционирования геномов
Участки разреза
пшшш
APEPEPAPEPEAEADAEADPEAGIGAVLKVLTTGLPALISWIKRKRQQG
Рис. 13.30. Обработка проллелиттина — пчелиного яда. Стрелки указывают участки разреза
Сигнальный
пептид А-цепь
t
Разрез
В-цепь С-цепь
______________ -С Пре-про-инсулин
Удаление сигнального
пептида
_____________________________— Проинсулин
I Образование
дисульфидной связи
Разрез
Разрез
I Удаление
В-цепи
Инсулин
s — S
Рис. 13.31. Обработка пре-про-инсулина
чрезвычайно гидрофобным отрезком аминокислот, которым белок-предшественник
прикрепляется к мембране до переноса через эту мембрану и выхода из клетки. Сиг-
нальные пептиды обычно присутствуют в белках, которые связываются с мембранами
и (или) проходят через мембраны (как у эукариотов, так и у прокариотов).
Протеолитический процессинг полипротеинов
В примерах, показанных на рис. 13.30 и 13.31, протеолитический процессинг дает
один зрелый белок. Это не всегда так. Некоторые белки изначально синтезируются в виде
полипротеинов — длинных полипептидов, которые содержат ряд зрелых белков, сце-
пленных вместе по принципу голова к хвосту. Расщепление полипротеина высвобождает
отдельные белки, которые могут иметь сильно отличающиеся друг от друга функции.
Полипротеины знакомы эукариотам. Вирусы нескольких типов, заражающие клетки
эукариотов, используют их как способ сокращения размеров своих геномов, ибо единствен-
ный ген полипротеина с одним промотором и одним терминатором занимает меньше места,
чем ряд отдельных генов. Полипротеины участвуют также в синтезе пептидных гормонов
Глава 13. Синтез и процессинг протеома 527
Рис. 13.32. Обработка полипротеина проопиомелано-
кортина. Сокращения: АКТГ — адренокортикотропный
гормон; КБСДГ — кортикотропиноподобный белок сред-
ней доли гипофиза; ЭНДО — эндорфин; Л ПТ — липотро-
пин; МЭ — мет-эн кефалин; МЛТ — меланотропин
Проопиомеланокортин
Ч3МЛТ АКТГ
(3-ЛПТ
N
С
^МЛТ аМЛТ КБСДГ ^-ППТ 3-ЭНДО
0МЛТ МЭ
у позвоночных. Например, полипротеин, назван-
ный проопиомеланокортином и производимый
в гипофизе, содержит по крайней мере десять
различных пептидных гормонов. Они высво-
бождаются протеолитическим расщеплением
полипротеина (рис. 13.32), но не все могут быть
произведены сразу из-за перекрытий между последовательностями отдельных пептидов.
Вместо этого, в различных клетках используются разные схемы расщепления.
13.3.3. Обработка химической модификацией
Геномы имеют способность кодировать 22 различные аминокислоты: 20 зада-
ваемых стандартным генетическим кодом, селеноцистеин и (по крайней мере у ар-
хей) пирролизин; последние две вставляются в полипептиды контекстнозависимым
переназначением кодонов — соответственно 5'-UGA-3’ и 5’-UAG-3' (раздел 1.3.2). Этот
ассортимент заметно расширяется посттрансляционной химической модификацией
белков, которая дает обширное множество различных типов аминокислот. Модификация
простейших типов происходит во всех организмах; более сложные ее разновидности,
в особенности гликозилирование, являются редкими у бактерий.
К наиболее простым типам химической Модификации относится присоединение
маленькой химической группы (например, ацетильной, метильной или фосфатной
группы; таблица 13.6) к боковой цепи аминокислоты или же к амино- или карбоксиль-
ным группам конечных аминокислот полипептида. Более 150 различных модифици-
рованных аминокислот было зарегистрировано в различных белках, при этом каждая
модификация выполняется весьма специфическим образом, одни и те же аминокислоты
модифицируются одинаковым способом во всех копиях белка. Это иллюстрировано на
рис. 13.33 на примере гистона НЗ. Данный пример напоминает нам, что химическая
модификация часто играет центральную роль в определении точного биохимического
действия целевого белка: мы видели в разделе 10.2.1, как ацетилирование, метилиро-
вание и фосфорилирование гистона НЗ и других гистонов сильно влияют на структуру
хроматина и, следовательно, на экспрессию генома. Химическая модификация имеет
несколько других регуляторных ролей — в частности, фосфорилирование используется
для активирования многих белков, участвующих в передаче сигналов (раздел 14.1.2).
К модификациям более сложного типа относят гликозилирование — присоеди-
нение больших углеводородных боковых цепей к полипептидам. Есть два главных типа
гликозилирования (рис. 13.34):
Me Me Ме Ацф Ац Ме Ац Ац Me Ме Ф
II \/ I I \ / I \ I /
НЗ ARTKQTARKSTGGKAPRKQLATKARKSAP
10 20
Рис. 13.33. Посттрансляционная модификация гистона НЗ млекопитающих. Показаны все известные
модификации, происходящие в этой области. Сокращения: Ац — ацетилирование; Ме — метилирова-
ние; Ф — фосфорилирование
528 Часть 3. Принципы функционирования геномов
Таблица 13.6. Примеры посттрансляционной химической модификации
Модификация Подлежащие модифика- ции аминокислоты Примеры белков
Добавление маленьких химических групп
Ацетилирование Лизин Гистоны
Метилирование Лизин Гистоны
Фосфорилирование Серин, треонин, тирозин Некоторые белки, вовлечен- ные в передачу сигналов
Гидроксилирование Пролин, лизин Коллаген
N-формилирование Добавление сахарных боковых цепей N-концевой глицин Мелиттин
0-связанное гликозилиро- вание Серин, треонин Многие мембранные бел- ки и секретируемые белки
N-связанное гликозилиро- вание Добавление липидных боковых цепей Аспарагин Многие мембранные бел- ки и секретируемые белки
Ацилирование Серин, треонин, цистеин Многие мембранные белки
N-миристоилирование Добавление биотина N-концевой глицин Некоторые протеинкина- зы, участвующие в переда- че сигналов
Биотинилирование Лизин Различные ферменты семейства карбоксилаз
• 0-гликозилирование — присоединение сахарной боковой цепи через гидроксиль-
ную группу аминокислоты треонина или серина;
• N-гликозилирование состоит в прикреплении сахарной боковой цепи через ами-
ногруппу на боковой цепи аспарагина.
Гликозилирование может выразиться в присоединении к белку крупных струк-
тур, включающих разветвленные сети из 10-20 сахарных единиц различных типов.
Эти боковые цепи помогают нацеливать белки на специфические участки в клетках
и определять стабильность белков, обращающихся в кровотоке. Другого типа круп-
номасштабная модификация заключается в прикреплении длинных липидов, часто
к аминокислотам цистеину или серину. Этот процесс называют ацилированием, и он
происходит со многими белками, которые становятся связанными с мембранами. Менее
распространенная модификация — биотинилирование — заключается в присоеди-
нении молекулы биотина к небольшому числу ферментов, которые катализируют кар-
Глава 13. Синтез и процессинг протеома 529
Рис. 13.34. Гликозилирование, а) О-гликозилирование. Показан-
ная структура встречается у ряда гликопротеинов. Она нарисова-
на здесь прикрепленной к аминокислоте серину, но она может
также быть связана с треонином, б) N-гликозилирование обычно
порождает более крупные сахарные структуры, чем получаемые
в результате О-гликозилирования. Рисунок показывает типичный
пример комплексного гликана, соединенного с аминокислотой аспа-
рагином. Сокращения: Фук — фукоза; Гал — галактоза; ГалМАц —
N-ацетилгалактозамин; ГлкМАц — N-ацетилглюкозамин; Ман — ман-
ноза; Сиа — сиаловая кислота
боксилирование органических кислот, таких как уксусная
и пропионовая.
а) О-гликозилирование
Сиа
Гал
ГалЫАц-Сиа
I
О
I
О сн2
II I
----С—С—N---------
I I
н н
13.3.4. Интеины
И последний тип посттрансляционного процессинга,
который мы должны рассмотреть, это вырезание интеи-
нов — белковый вариант более обширного вырезания
интронов, происходящего с молекулами пре-РНК. Интеи-
ны представляют собой внутренние сегменты белков,
которые удаляются вскоре после трансляции, при этом
два внешних сегмента, или экстеина, сцепляются вместе
(рис. 13.35). Первый интеин был открыт у S. cerevisiae
в 1990 г., и с тех пор было достоверно опознано только 100
других интеинов. Несмотря на их редкостность, интеины
широко распространены среди различных организмов.
Больше всего их известно у бактерий и архей, но есть
также примеры у низших эукариотов. В редких случаях
в одном белке содержится более одного интеина.
Большинство интеинов имеют длину около 150 ами-
нокислот, и, подобно интронам пре-мРНК (раздел 12.2.2),
последовательности в участках вырезания интеинов об-
ладают некоторым подобием в большинстве известных
примеров. В частности, первая аминокислота нижеле-
жащего экстеина представлена цистеином, серином или
треонином. Несколько других аминокислот в пределах
б) N-гликозилирование
Сиа Сиа Сиа
Гал Гал Гал
ГлкЫАц ГлкЫАц ГлкЫАц
Ман Ман
ГлкЫАц
ГлкЫАц-Фук
NH
с=о
о сн2
II I
---С—С—N------
Н Н
последовательности интеина также консервативны. Эти консервативные аминокислоты
участвуют в процессе вырезания, который самокатализируется самим интеином.
Недавно обнаружились две интересные особенности интеинов. Первая из них
открылась, когда структуры двух интеинов были
определены рентгеноструктурным анализом. Эти
структуры подобны в некотором отношении струк-
туре белка Drosophila по имени Hedgehog, который
участвует в развитии схемы сегментации эмбриона
мухи. Hedgehog — автообрабатывающийся белок,
который саморазрезается надвое. Структурное по-
добие с интеинами заключается в той части белка
Hedgehog, что катализирует его саморасщепление.
Возможно, одна и та же структура белка эволюциони-
Интеин
N 1 С
Сплайсинг
N С
Рис. 13.35. Вырезание интеина в ходе
сплайсинга
530 Часть 3. Принципы функционирования геномов
ровала дважды, или, возможно, интеины и Hedgehog имели общую связь на некотором
этапе эволюционного прошлого.
Вторая интересная особенность выражается в том, что в некоторых интеинах вы-
резаемый сегмент представлен специфичной к последовательности эндонуклеазой.
Интеин разрезает ДНК в последовательности, соответствующей его участку вставки
в гене, кодирующем свободный от интеинов вариант белка, из которого он образуется
(рис. 13.36). Если клетка содержит также ген, кодирующий содержащий интеин белок, то
последовател ьность ДНК для этого интеина способна запрыгнуть в участок разреза, пре-
образуя ген интеин-минус в его вариант интеин-плюс; этот процесс назвали репатриа-
цией интеина. Событие подобного типа происходит с некоторыми интронами I группы
(раздел 12.2.4), которые кодируют белки, направляющие репатриацию интронов.
Возможно, что перенос интеинов и интронов I группы может также происходить между
клетками или даже между видами. Думают, что посредством именно этого механизма
эгоистичная ДНК получает возможность распространяться (раздел 18.3).
Содержащий интеин ген
Запрыгивание
интеиновой ДНК
Разрез 4
интеином |
Сплайсинг
— «ъ
Интеин
Ген «интеин-минус»
Рис. 13.36. Репатриация интеина. Клетка гетерозиготна по содержащему интеин гену: обладает одним
аллелем с интеином и вторым аллелем без интеина. После сплайсинга белка интеин разрезает ген
интеин-минус в соответствующем месте, что позволяет копии ДНК-последовательности интеина впрыг-
нуть в этот ген, преобразуя его в вариант интеин-плюс
13.4. Деградация белка
Синтез белка и процессинг, которые мы успели рассмотреть в этой главе, дают но-
вые, функционально активные белки, занимающие свое место в протеоме клетки. Эти
белки или заменяют существующие, которые достигли конца своего срока службы, или
обеспечивают новые белковые функции в ответ на изменяющиеся требования клетки.
Концепция, согласно которой протеом клетки может изменяться со временем, предпо-
лагает не только синтез белка de novo, но также и удаление белков, функции которых
больше не требуются. Такое удаление должно быть чрезвычайно избирательным, с тем
чтобы только заданные белки подвергались деградации, и должно также быть быстрым,
Глава 13. Синтез и процессинг протеома 531
чтобы соответствовать резким изменениям, которые происходят при некоторых усло-
виях, например во время ключевых переходов в клеточном цикле.
Многие годы деградация белка была немодным предметом исследования и только
в 1990-е гг. произошел настоящий прогресс в понимании того, как события специфично-
го протеолиза связаны с такими процессами, как клеточный цикл и дифференцировка
клеток. Даже теперь наши знания сосредоточены главным образом на описаниях общих
путей распада белков и в меньшей степени — на регулировании путей и механизмов,
используемых для нацеливания специфичных белков. Кажется, существует множество
различных типов путей распада, взаимосвязанности которых еще не были прослежены.
Это особенно верно для бактерий, которые, кажется, имеют спектр протеаз, совместно
работающих в управляемой деградации белков. У эукариотов большая часть распада
осуществляется единственной системой, в которую входят убиквитин и протеасома.
Связь между убиквитином и деградацией белка была впервые установлена в 1975 г.,
когда было показано, что этот обильный белок с 76 аминокислотами участвует в энергоза-
висимых реакциях протеолиза в клетках кролика. В ходе дальнейших исследований была
опознана группа из трех ферментов, которые присоединяют молекулы убиквитина по
отдельности или цепями к аминокислотам лизина в белках, которые намечены на распад.
Известны также убиквитиноподобные белки, такие как СУМО, которые действуют таким
же образом, что и убиквитин. Будет ли белок убиквитинирован, зависит от присутствия или
отсутствия в нем аминокислотных мотивов, которые служат сигналами восприимчивости
к деградации. Эти сигналы еще не были полностью охарактеризованы, но у 5. cerevisiae, как
думают, имеется по крайней мере десять различных типов, в том числе:
• N-дегрон — элемент последовательности, расположенный на N-конце белка.
• последовательности PEST — внутренние последовательности, которые обогаще-
ны пролином (Р), глутаминовой кислотой (Е), серином (S) и треонином (Т).
Эти последовательности являются постоянными составляющими белков, их со-
держащих, и, таким образом, не могут быть прямыми «сигналами к деградации»: если
бы они были ими, то эти белки были бы разложены сразу после их синтеза. Вместо
этого, они должны определять восприимчивость к деградации и, следовательно, общую
устойчивость белка в клетке. Как это может быть связано с управляемым распадом
избираемых белков в определенные моменты времени (например, на протяжении
клеточного цикла), еще не ясно.
Второй компонент пути зависимой от убиквитина деградации представлен протеа-
сомой — структурой, в недрах которой убиквитинирован-
ные белки разлагаются. У эукариотов протеасома пред-
ставляет собой большую, многосубъединичную структуру
с коэффициентом седиментации 26S, состоящую из поло-
го цилиндра величиной 20S и двух «колпачков» величи-
ной 19S (рис. 13.37). Археи также обладают протеасомами
примерно того же размера, но они менее сложны, будучи
составленными из многочисленных копий только двух
белков: протеасомы эукариотов содержат белковые субъе-
диницы 14-ти различных типов. Вход в канал протеасомы
узок, и белок должен быть развернут прежде, чем он сможет
Колпачок
Цилиндр
Колпачок
Рис. 13.37. Протеасома эукариотов. Белковые компоненты двух
колпачков показаны оранжевым и красным, а формирующих ци-
линдр — синим
532 Часть 3. Принципы функционирования геномов
в него войти. Такое развертывание, вероятно, происходит посредством энергозависимого
процесса и может вовлекать структуры, подобные шаперонинам (раздел 13.3.1), но с раз-
вертывающим, а не со свертывающим действием. После развертывания белок может войти
в протеасому, внутри которой он расщепляется на короткие пептиды длиной 4-10 аминокис-
лот. Они выпускаются обратно в цитоплазму, где распадаются на отдельные аминокислоты,
которые могут быть повторно использованы в синтезе белка.
Заключение
Конечным результатом экспрессии генома является протеом — совокупность функ-
ционирующих белков, синтезируемых живой клеткой. Род и относительное содержание
отдельных белков в протеоме отражает равновесие между синтезом новых белков и де-
градацией существующих. Транспортные РНК играют главную роль в синтезе белка, служа
сопрягающими молекулами, которые образуют физическую и информационную связь
между транслируемой мРНК и синтезируемым полипептидом. Фактически все тРНК сво-
рачиваются в одну и ту же структуру спаренных оснований, двумерное представление
которой называют клеверным листом. Нужная аминокислота присоединяется к З'-концу
тРНК при помощи аминоацил-тРНК-синтетазы. Аминоацилирование — очень точный про-
цесс, потому что каждая аминоацил-тРНК-синтетаза имеет высокое сродство к своей тРНК
и аминокислоте и потому что есть также корректирующий механизм, который проверяет,
что надлежащая аминокислота была присоединена к предписанной тРНК. Взаимодействия
типа кодон-антикодон обеспечивают соблюдение правил генетического кода. Хотя код
имеет 61 триплет, которые определяют аминокислоты, большинство клеток имеет мень-
шее, чем это, число молекул тРНК разного типа, потому что одна тРНК может считывать
два или несколько кодонов за счет процесса, называемого неканононическим спариванием
оснований. Структура рибосомы исследуется все с большим и большим разрешением раз-
личными методами, в том числе рентгеноструктурным анализом. Инициация трансляции
у бактерий вовлекает малую субъединицу рибосомы, опознающую внутренний сайт связы-
вания на мРНК, лежащий в нескольких нуклеотидах выше старт-кодона. У эукариотов лишь
несколько молекул мРНК имеют внутренние сайты связывания, более распространенный
процесс вовлекает присоединение малой субъединицы рибосомы к копированному 5'-концу
мРНК, за которым следует просмотр молекулы мРНК в поиске старт-кодона. Известны
различные механизмы, посредством которых может регулироваться инициация трансля-
ции — глобально или локально (в отношении определенных транскриптов). Присоединение
большой субъединицы рибосомы к инициаторному комплексу позволяет начаться стадии
элонгации трансляции. Основная деятельность в течение элонгации — синтез пептидных
связей. Образование пептидной связи катализируется пептидилтрансферазой, причем это
ферментативное действие исходит от одной из молекул рРНК, которая выполняет роль
рибозима. Необычные события элонгации включают запрограммированные изменения
рамки считывания и трансляционый обход; врезультате последнего значительный сегмент
мРНК пропускается рибосомой. В терминации трансляции участвуют специальные белки,
которые входят в рибосому по достижении стоп-кодона. Синтезированный полипептид
должен быть свернут в его правильную третичную структуру и, возможно, обработан
протеолитическим расщеплением и (или) химической модификацией. Очень небольшое
число белков содержит вклинивающиеся последовательности, названные интеинами,
которые должны быть удалены в ходе сплайсинга белка. Деградация белка у эукариотов
состоит в убиквитинировании белков, намеченных для разложения, сопровождаемом их
деградацией в протеасоме.
Глава 13. Синтез и процессинг протеома 533
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
13.1 *. Какое из следующих определений
относится к молекуле изоакцептор-
ной тРНК?
а. Отдельная молекула тРНК, которая
может взаимодействовать с раз-
личными кодонами, задающими
одну и ту же аминокислоту.
Ь. Различные молекулы тРНК, ко-
торые являются специфичными
к одной и той же аминокислоте.
с. Различные молекулы тРНК, ко-
торые опознают один и тот же
кодон.
d. Молекула тРНК, которая может
быть аминоацилирована различ-
ными аминокислотами.
13.2 . Какое из следующих утверждений
о специфичности аминоацил-тРНК-
синтетаз является ВЕРНЫМ?
а. Каждая аминоацил-тРНК-синте-
таза катализирует присоедине-
ние одной аминокислоты к одной
молекуле тРНК.
Ь. Каждая аминоацил-тРНК-синте-
таза катализирует присоединение
одной аминокислоты к одной или
нескольким молекулам тРНК.
с. Каждая аминоацил-тРНК-синте-
таза катализирует присоединение
одной или нескольких аминокис-
лот к одной молекуле тРНК.
d. Каждая аминоацил-тРНК-синте-
таза катализирует присоедине-
ние одной или нескольких ами-
нокислот к одной или нескольким
молекулам тРНК.
13.3 *. Взаимодействия типа кодон-анти-
кодон происходят за счет:
а. Ковалентных связей.
Ь. Электростатических взаимодей-
ствий.
с. Водородных связей.
d. Гидрофобных взаимодействий.
13.4 . Неканоническое спаривание между
кодоном и антикодоном происходит
между:
а. Первым нуклеотидом кодона и пер-
вым нуклеотидом антикодона.
Ь. Первым нуклеотидом кодона и тре-
тьим нуклеотидом антикодона.
с. Третим нуклеотидом кодона и пер-
вым нуклеотидом антикодона.
d. Третим нуклеотидом кодона и тре-
тьим нуклеотидом антикодона.
13.5 *. Неканоническое спаривание проис-
ходит из-за всего нижеследующего,
КРОМЕ того, что:
а. Антикодон находится в петле мо-
лекулы тРНК и потому не вырав-
нивается равномерно с кодоном.
Ь. Нуклеотид инозин в молекуле
тРНК может спариваться с А,
С и U в молекуле мРНК.
с. Нуклеотид инозин в молекуле
мРНК может спариваться с А,
С и U в молекуле тРНК.
d. Гуанин может спариваться с ура-
цилом.
13.6 . Каков первый шаг в инициации
трансляции у бактерий?
а. Малая субъединица рибосомы
связывается с 5'-кэпом мРНК
и просматривает мРНК в поиске
старт-кодона.
Ь. Большая субъединица рибосомы
связывается с сайтом связывания
рибосомы на молекуле мРНК.
с. Рибосома связывается со старт-
кодоном на молекуле мРНК.
d. Малая субъединица рибосомы
связывается с сайтом связывания
рибосомы на молекуле мРНК.
534 Часть 3. Принципы функционирования геномов
13.7 *. Какой функции служит формильная
группа, присоединенная к инициа-
торному метионину у бактерий?
а. Она связывает инициаторную тРНК
с большой субъединицей рибосомы
при инициации трансляции.
Ь. Она связывает молекулу ГТФ, не-
обходимую для сборки инициа-
торного комплекса.
с. Она блокирует аминогруппу ме-
тионина и гарантирует тем са-
мым, что синтез белка будет про-
исходить в направлении N —> С.
d. Она блокирует боковую цепь ме-
тионина, в результате чего он не
реагирует с фактором инициации
IF-3.
13.8. Каков общий механизм инициации
трансляции у эукариотов?
а. Малая субъединица рибосомы
связывается с 5'-кэпом мРНК
и просматривает мРНК в поиске
старт-кодона.
Ь. Большая субъединица рибосо-
мы связывается с 5'-кэпом мРНК
и просматривает мРНК в поиске
старт-кодона.
с. Рибосома связывается со старт-
кодоном на молекуле мРНК.
d. Малая субъединица рибосомы
связывается с сайтом связывания
рибосомы на молекуле мРНК.
13.9* . Какова функция фактора инициации
eIF-6?
а. Он связывается с инициаторной
TPHKMet и ГТФ во время сборки
предынициаторного комплекса.
Ь. Он служит мостом между 5'-кон-
цевым кэпом мРНК и предыни-
циаторным комплексом.
с. Он высвобождает другие факторы
инициации в ходе сборки рибосо-
мы на стартовом кодоне.
d. Он препятствует соединению
большой субъединицы рибосо-
мы с малой субъединицей в ци-
топлазме.
13.10. Какова функция фактора элонгации
EF-1A?
а. Он катализирует образование
пептидных связей.
Ь. Он гарантирует, что надлежащая
связка аминоацил-тРНК входит
в рибосому.
с. Он препятствует выходу молекул
тРНК из рибосомы до образова-
ния пептидных связей.
d. Он гидролизирует ГТФ, чтобы по-
мочь перемещению рибосомы по
молекуле мРНК.
13.11* . Как происходит сдвиг рамки счи-
тывания во время трансляции?
а. Рибосома транслирует молекулу
мРНК, в которой находится лиш-
ний или отсутствует необходи-
мый нуклеотид.
Ь. Рибосома пропускает кодон во
время трансляции молекулы
мРНК.
с. Рибосома делает паузу в ходе
трансляции и сдвигается обрат-
но или вперед на один нуклео-
тид и затем продолжает транс-
ляцию.
d. Рибосома завершает трансляцию
в кодоне, который обычно опреде-
ляет некоторую аминокислоту.
13.12. Как терминируется синтез белка?
а. Фактор высвобождения опознает
стоп-кодон и входит в А-сайт.
Ь. тРНК для стоп-кодона входит
в А-сайт.
с. тРНК для стоп-кодона входит
в Р-сайт.
d. Рибосома стопорится в стоп-
кодоне и катализирует освобож-
дение белка от тРНК.
13.13* . Какая из следующих причин не
предопределяет потребность бел-
ков в помощи при их сворачивании
в ходе трансляции или после дена-
турации?
а. После денатурации белки могут
образовывать нерастворимые
Глава 13. Синтез и процессинг протеома 535
агрегаты, что вызвано неспособ-
ностью (в развернутом состоя-
нии) оградить свои гидрофобные
группы от воды.
Ь. После денатурации белки могут
принять неправильно свернутое,
но устойчивое состояние.
с. В процессе трансляции частично
транслированный белок пред-
ставляет собой случайную спи-
раль, неспособную свернуться
ни в какую специфическую кон-
формацию.
d. В ходе трансляции частично
транслированные белки могут на-
чать неправильно сворачиваться
прежде, чем полный белок будет
синтезирован.
13.14. Какая из следующих функций не
свойственна молекулярным шапе-
ронам при сворачивании белка?
а. Молекулярные шапероны помо-
гают белкам отыскивать их пра-
вильные структуры.
Ь. Молекулярные шапероны опре-
деляют третичную структуру
белка.
с. Молекулярные шапероны могут
стабилизировать частично свер-
нутые белки и препятствовать
образованию их агрегатов с дру-
гими белками.
Вопросы открытого типа
13.1 *. Каким образом молекулы тРНК слу-
жат и физической, и информацион-
ной связью между транслируемой
молекулой мРНК и синтезируемым
белком?
13.2 . Опишите вкратце двухэтапную реак-
цию присоединения аминокислоты
к молекуле тРНК.
13.3 *. Что произойдет, если аминоацил-
тРНК-синтетаза ошибочно присое-
динит неправильную аминокислоту
к молекуле тРНК (например, ами-
d. Молекулярные шапероны могут
оградить и защитить выставлен-
ные на поверхность гидрофобные
области белков.
13.15 *. Какая из следующих операций не
является примером посттрансля-
ционной химической модификации
белков?
а. Гликозилирование.
Ь. Метилирование.
с. Фосфорилирование.
d. Протеолиз.
13.16 . Что такое интеины?
а. Внешние или внутренние сегмен-
ты белков, которые удаляются
посредством протеолиза, после
чего образуется функционально
активный белок.
Ь. Внешние сегменты белков, кото-
рые добавляются к другим бел-
кам протеинлигазами.
с. Внутренние сегменты белков,
которые удаляются после транс-
ляции, после чего внешние сег-
менты связываются вместе.
d. Внешние сегменты белков, кото-
рые ковалентно присоединяют-
ся к липидам для встраивания
в мембрану.
ноацилирует тРНК валина изолей-
цином)?
13.4 . Каковы две активные роли рибосом
в процессе синтеза белка?
13.5 *. Перечислите молекулы, входящие
в состав предынициаторного ком-
плекса, который собирается во вре-
мя первого шага инициации транс-
ляции у эукариотов.
13.6 . Какую роль приписывают поли-А
хвосту, участвующему в инициации
трансляции у эукариотов?
536 Часть 3. Принципы функционирования геномов
13.7 *. Каким образом клетки эукариотов
могут быстро подавлять трансля-
цию в ответ на стресс, например,
теплового шока?
13.8 . Какие особенности мРНК dnaX, как
думают, вызывают запрограмми-
рованный сдвиг рамки считывания
в процессе трансляции этой мРНК?
13.9 *. Какие два шага происходят в ходе
повторного сворачивания малень-
кого белка?
13.10 . Обсудите различия между путями,
которыми функционируют шапе-
Изыскательские задачи
13.1* . Почему существует два класса
аминоацил-тРНК-синтетаз?
13.2. В митохондриях человека для син-
теза белка требуется только 22 раз-
личные тРНК. Какие поправки это
вносит в правила, описывающие вза-
имодействия типа кодон-антикодон
в этой системе?
13.3* . Каким образом мог появиться гене-
тический код?
13.4. Большинство организмов проявля-
ет уникальное отклонение частоты
использования кодонов в своих ге-
нах. Например, в генах Saccharomyces
cerevisiae из четырех кодонов для
роны Hsp70 и шаперонин GroEL/
CroES.
13.11 *. Каким образом интеин удаляется
из белка?
13.12 . Опишите два сигнала, которые опо-
знаются ферментами, присоединяю-
щими молекулы убиквитина к бел-
кам, намеченным для деградации.
Что происходит с белками, которые
были убиквитинированы таким спо-
собом?
пролина только два — CCU и ССА —
встречаются достаточно часто, ССС
и CCG используются реже. Было
предположено, что ген, который со-
держит относительно высокое чис-
ло непредпочтительных кодонов,
может экспрессироваться с относи-
тельно низкой скоростью. Разъяс-
ните соображения, легшие в основу
этой гипотезы, и обсудите ее много-
значащие следствия.
13.5* . До какой степени исследования
структуры рибосомы были полез-
ны для полного понимания процесса
синтеза белков?
Тесты по рисункам
13.1* . В какой позиции антикодона тРНК иногда располагается инозин? Если инозин
присутствует, то какова его функция?
мРНК
Глава 13. Синтез и процессинг протеома 537
13.2. Опишите инициацию трансляции
у Е. coli.
5’ I Старт-кодон
* AUG————
13.3* . На рисунке показаны расстояния
между молекулой CCdA-фосфат-
пуромицина (красная точка) и бли-
жайшими белками в большой субъ-
единице рибосомы Е. coli. Что этот
рисунок рассказывает об образова-
нии пептидных связей в рибосоме?
13.4. Обсудите, как именно активность белка зависит от событий, происходящих после
трансляции.
Полипептид
Свёртывание
Раздел
13.3.1
Протеолитическое расщепление Химическая модификация
Раздел Раздел
13.3.2 13.3.3
▼ 1 Г
- , ч
I
Новая химическая группа
Вырезание интеина
Раздел
13.3.4
Позиция удалённого
интеина
538 Часть 3. Принципы функционирования геномов
Дополнительная литература
Структура и функции тРНК
Clark,В.F.С. (2001) The crystallization and structural determination of tRNA. Trends
Biochem. Sci. 26: 511-514.
Hale,S.P., Auld, D.S., Schmidt, E. and Schimmel, P. (1997) Discrete determinants in
transfer RNA for editing and aminoacylation. Science 276:1250-1252. Ensuring the accuracy
of aminoacylation.
Ibba,M. and Soll,D. (2000) Aminoacyl-tRNA synthetases. Annu. Rev. Biochem. 69: 617-
650.
Percudani, R. (2001) Restricted wobble rules for eukaryotic genomes. Trends Genet 17:
133-135.
Исследование структуры рибосомы
Ban,N., Nissen, P., Hansen,)., Moore,P.B. and Steitz,T.A. (2000) The complete atomic
structure of the large ribosomal subunit at 2,4 A resolution. Science 289: 905-920.
Heilek, G. M. and Noller, H. F. (1996) Site-directed hydroxyl radical probing of the rRNA
neighborhood of ribosomal protein S5. Science 272:1659-1662.
Moore,P.B. and Steitz,T.A. (2003) The structural basis of large ribosome subunit
function. Annu. Rev. Biochem. 72: 813-850.
Wimberly, B.T., Brodersen, D. E., Clemons, W.M., Morgan-Warren, R.J., Carter, A. P.,
Vonrhein, C., Hartsch,T. and Ramakrishnan, V. (2000) Structure ofthe 30S ribosomal subunit.
Nature 407: 327-339.
Yusupov, M. M., Yusupova, G.Z., Baucom, A., Lieberman, K., Earnest, T.N., Cate, J. H.
and Noller, H.F. (2001) Crystal structure of the ribosome at 5.5 A resolution. Science 292:
883-896.
Механика синтеза белка
Andersen, G. R., Nissen, P. and Nyborg, J. (2003) Elongation factors in protein biosynthesis.
Trends Biochem. Sci. 28: 434-441.
Frank,), and Agarwal,R.K. (2000) A ratchet-like inter-subunit reorganization of the
ribosome during translocation. Nature 406: 318-322.
Ibba, M. and Sol, D. (1999) Quality control mechanisms during translation. Science 286:
1893-1897.
Kapp,L.D. and LorschJ.R. (2004) The molecular mechanics of eukaryotic translation.
Annu. Rev. Biochem. 73: 657-704.
McCarthy, J. E. G. (1998) Posttranscriptional control of gene expression in yeast. Microbiol.
Mol. Biol. Rev. 62:1492-1553. Detailed review of translation and its control in yeast
Nakamura, Y. and Ito, K. (2003) Making sense ofmimic in translation termination. Trends
Biochem. Sci. 28:99-105. The mode of action of release and ribosome recycling factors.
Rodnina, M.V. and Wintermeyer,W. (2001) Ribosome fidelity: tRNA discrimination,
proofreading and induced fit. Trends Biochem. Sci. 26:124-130.
Пептидилтрансфераза является рибозимом
Nissen,P., Hansen,)., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis
of ribosome activity in peptide bond synthesis. Science 289: 920-930.
Polacek,N., Gaynor, M., Yassin, A. and Mankin, A. S. (2001) Ribosomal peptidyl transferase
can withstand mutations at the putative catalytic nucleotide. Nature 411: 498-501.
Глава 13. Синтез и процессинг протеома 539
Steitz,T.A. and Moore, Р.В. (2003) RNA, the first macromolecular catalyst: the ribosome
is a ribozyme. Trends Biochem. Sci. 28: 411-418.
Необычные события в ходе трансляции
Farabaugh, Р. J. (1996) Programmed translational frameshifting. Annu. Rev. Genet 30:
507-528.
Herr,A.J., Atkins, J. F. and Gesteland,R.F. (2000) Coupling of open reading frames by
translational bypassing. Annu. Rev. Biochem. 69: 343-372.
Сворачивание белка
Anfinsen, С. В. (1973) Principles that govern the folding of protein chains. Science 181:
223-230. The first experiments on protein folding.
Daggett, V. and Fersht,A.R. (2003) Is there a unifying mechanism for protein folding?
Trends Biochem. Sci. 28:18-25.
Frydman,). (2001) Folding of newly translated proteins in vivo: the role of molecular
chaperones. Annu. Rev. Biochem. 70: 603-649.
Xu,Z., Horwich,A.L. and Sigler, P.B. (1997) The crystal structure of the asymmetric
GroEL-GroES-(ADP)7 chaperonin complex. Nature 388; 741-750.
Процессинг и модификация белка
Chapman-Smith, A. and Cronan, J. Е. (1999) The enzymatic biotinylation of proteins:
a post-translational modification of exceptional specificity. Trends. Biochem. Sci. 24: 359-
363.
Drickamer, K. and Taylor, M. E. (1998) Evolving views of protein glycosylation. Trends
Biochem. Sci. 23: 321-324.
Paulus, H. (2000) Protein splicing and related forms of protein autoprocessing. Annu.
Rev. Biochem. 69: 447-496.
Деградация белка
Varshavsky, A. (1997) The ubiquitin system. Trends Biochem. Sci. 22: 383-387.
Voges,D., Zwickl,P. and Baumeister,W. (1999) The 26 S pro-teasome: a molecular
machine designed for controlled proteolysis. Annu. Rev. Biochem. 68:1015-1068.
Глава 14
Регулирование активности генома
По прочтении главы 14 вы сможете:
• Проводить различие между дифференцировкой и развитием и вкратце сооб-
щать о том, за счет чего регуляция экспрессии генома лежит в основе этих двух
процессов.
• Описывать, с примерами, различные способы, которыми импортированные
сигнальные соединения, такие как лактоферрин и стероидные гормоны, могут
вызывать переходные изменения в активности генома.
• Подробно описывать катаболитную репрессию у бактерий.
• Обсуждать различные пути, по которым сигналы рецепторов клеточной поверх-
ности передаются в геном.
• Описывать, с примерами, различные способы, которыми могут быть вызваны
постоянные и полупостоянные изменения в активности генома, проводя четкое
различие между теми процессами, которые предполагают перестройку генома,
теми, которые обусловлены изменениями в структуре хроматина, и, наконец,
теми, в которые включены петли обратной связи.
• Обсуждать, каким образом исследования лизогенного цикла инфицирования
бактериофага^ и спорообразования у Bacillus subtilis дают нам основную инфор-
мацию, имеющую отношение к дифференцировке и развитию.
• Объяснять, благодаря чему Caenorhabditis elegans весьма успешно служит модель-
ным организмом, и описывать, как путь дифференцировки клетки определяется
в ходе развития яйцевода у С. elegans.
• Описывать генетические события, лежащие в основе эмбриогенеза у Drosophila
melanogaster.
• Обсуждать роли гомеозисных генов в организмах D. melanogaster, позвоночных
и растений.
Мы проследовали путем, которым экспрессия генома определяет содержание про-
теома, которое, в свою очередь, определяет биохимическую сигнатуру клетки. Ни в одном
организме такая биохимическая сигнатура не остается совершенно постоянной. Даже
простейшие одноклеточные организмы способны изменять свои протеомы, с тем чтобы
отвечать изменениям в окружающей среде, так чтобы их биохимические возможности
неизбежно соответствовали имеющимся запасам пищи и преобладающим физическим
и химическим условиям. Клетки в многоклеточных организмах столь же восприимчи-
вы к изменениям во внеклеточной окружающей среде, единственное отличие состоит
в том, что, наряду с питательными веществами, главными раздражителями у них служат
гормоны и факторы роста. Преходящие непостоянные изменения в активности генома
Глава 14. Регулирование активности генома 541
позволяют повторно перестраивать протеом, с тем чтобы удовлетворять требованиям,
которые внешний мир предъявляет клетке (рис. 14.1). Другие изменения в активности
генома носят постоянный или по крайней мере полупостоянный характер и приводят
к изменению биохимической сигнатуры клетки, которое не является легко обратимым.
Эти изменения ведут к дифференцировке клеток — принятию клетками специальных
физиологических ролей. Пути дифференцировки известны у многих одноклеточных
организмов, пример представляет образование споровых клеток такими бактериями,
как Bacillus, но мы чаще связываем дифференцировку с многоклеточными организма-
ми, у которых многообразие клеток специализированных типов (у человека — более
250-ти типов) организовано в ткани и органы. Сборка столь сложных многоклеточных
структур и организма в целом требует согласования действий геномов в различных
клетках. Такое согласование предполагает как непостоянные, так и постоянные из-
менения и должно продолжаться в течение длительного периода времени в процессе
развития организма.
Существует множество уровней (этапов) в путях экспрессии отдельных генов, на
которых может быть осуществлено регулирование (таблица 14.1). Примеры биологиче-
ских ролей различных регуляторных механизмов были предъявлены в соответствующих
Переходное
Постоянное
или полупостоянное
ОБОЗНАЧЕНИЯ:
—МаЖ— Активный ген
-^ОБСО— Неактивный ген
Рис. 14.1. Два способа регулирования активности генома. Гены слева подвержены переходному регу-
лированию и включаются и выключаются в ответ на изменения во внеклеточной среде. Гены справа
подвержены постоянному или полупостоя иному изменению в их картине экспрессии, в результате чего
продолжают непрерывно экспресироваться эти же три гена
542 Часть 3. Принципы функционирования геномов
Таблица 14.1. Примеры этапов в пути экспрессии генома, на которых может осуществляться регулирование
Шаг Пример регулирования
Транскрипция Достижимость генов Управляющие локусом области определяют структуру хроматина в об- ластях, которые содержат гены (раздел 10.1.2) Модификации гистонов влияют на структуру хроматина и определяют, какие гены будут доступными (раздел 10.2.1) Размещение нуклеосомы управляет доступом РНК-полимеразы и фак- торов транскрипции к области промотора (раздел 10.2.2) Метилирование ДНК глушит области генома (раздел 10.3.1)
Инициация транс- крипции Синтез РНК Продуктивная инициация подвержена влиянию активаторов, репрес- соров и других систем управления (раздел 11.3) Прокариоты используют антитерминацию и аттенюацию, чтобы управлять количеством и характером отдельных транскриптов (раз- дел 12.1.2)
Процессинг мРНК эукариотов
Кэпирование Некоторые животные используют кэпирование как средство регули- рования синтеза белка во время созревания яйцеклетки
Полиаденилирование Альтернативные участки полиаденилированияуправляют цветением у Arabidopsis Трансляция мРНК bicoid в яйцеклетках Drosophila активизируется после оплодотворения продолжением поли-А хвоста (раздел 14.3.4)
Сплайсинг Выбор альтернативных сайтов сплайсинга управляет определением пола у Drosophila (раздел 12.2.2)
Химическая модифи- кация Редактирование РНК, осуществляемое над мРНК аполипопротеина-В, дает специфические для печени и для кишечника варианты этого белка (раздел 12.2.5)
Деградация мРНК Молекулы микроРНК управляют смертью клетки, дифференцировкой нервных клетоки на различные типы и управлением жировыми запа- сами у Caenorhabditis elegans, а также многими различными процессами у других эукариотов (раздел 12.2.6) Железо управляет деградацией мРНК рецептора трансферрина (раз- дел 13.2.2)
Синтез и процессинг белка
Инициация трансляции* Фосфорилирование eIF-2 влечет за собой общее снижение уровня ини- циации трансляции у эукариотов (раздел 13.2.2) Рибосомные белки у бактерий управляют своим собственным синте- зом, модулируя прикрепление рибосомы к кодирующим их молекулам мРНК (раздел 13.2.2) У некоторых эукариотов железо управляет пробегом рибосомы по мРНК ферритина (раздел 13.2.2)
* В последнее время выяснилось, что значительную роль в подавлении инициации трансляции в эука-
риотах играют siPHK. — Прим. ред.
Глава 14. Регулирование активности генома 543
Таблица 14.1. Окончание
Шаг Пример регулирования
Маленькие РНК в бактериях регулируют ответную реакцию на окисли-
тельное воздействие, модулируя инициацию трансляции различных
мРНК (раздел 13.2.2)
Синтез белка Сдвиг рамки считывания позволяет двум субъединицам ДНК-
полимеразы III быть транслированными с гена dnaX Escherichia coli
(раздел 13.2.3)
События разрезания Пути альтернативного расщепления для полипротеинов дают ткане-
специфические белковые продукты (раздел 13.3.2)
Химическая Многие белки, участвующие в передаче сигналов, активируются через
модификация фосфорилирование (раздел 14.1.2)
местах в главах 10-13. Цель этой главы состоит не в том, чтобы повторно описывать эти
геноспецифические системы управления, но в том, чтобы объяснить, как регулируется
активность генома в целом. При этом мы должны иметь в виду, что биосфера настолько
многообразна и число генов в отдельных геномах столь велико, что разумно предпо-
ложить, что для регулирования экспрессии генома в ходе эволюции мог развиться
какой-либо механизм, и что, вероятно, так и произошло. Поэтому неудивительно, что мы
можем назвать примеры регулирования для каждой точки на пути экспрессии генома.
Но равно ли значение всех этих контрольных точек в регулировании активности генома
в целом? Согласно нашим современным представлениям это не так. Наше понимание
может быть несовершенным, основаным в его сегодняшнем виде на исследовании
лишь ограниченного числа генов в немногих организмах, но ясно, что определяющие
управляющие воздействия на экспрессию генома — решения о том, которые гены
включены и которые выключены, — принимаются на уровне инициации транскрипции.
Для большинства генов управление, которое осуществляется на более поздних шагах,
служит для модулирования экспрессии, но не служит первичным определителем того,
находится ли ген во включенном или в выключенном состоянии (см. рис. 11.22). Поэтому
большая часть, но не все из того, что мы обсудим в этой главе, относится к управлению
активностью генома, осуществляемому механизмами, которые определяют, какие гены
будут транскрибированы, а какие подавлены. Мы обратимся к двум вопросам: к способам,
которыми производятся непостоянные и постоянные изменения в активности генома,
и к способам, которыми эти изменения увязываются между собой в путях развития во
времени и в пространстве.
14.1. Непостоянные изменения в активности генома
Непостоянные изменения в активности генома происходят преимущесвенно в от-
вет на внешние раздражители. Для одноклеточных организмов наиболее важные
внешние раздражители касаются наличия питательных веществ: эти клетки живут в из-
менчивых окружающих средах, в которых род и относительное количество питательных
веществ изменяются стечением времени. Поэтому геномы одноклеточных организмов
содержат гены для поглощения и усвоения разнообразных питательных веществ, и за
изменениями в содержании питательных веществ неотступно следуют изменения
в активности генома, так что в любой момент времени экспрессируются только те
Прямая активация - Косвенная активация -
сигнальное соединение сигнал передается через
проникает в клетку рецептор клеточной
поверхности
Рецептор клеточной
поверхности
544 Часть 3. Принципы функционирования геномов
Рис. 14.2 Два способа, которыми внеклеточное
сигнальное соединение может влиять на со-
бытия, происходящие в клетке
гены, которые необходимы для усвоения
наличествующих питательных веществ.
Большинство клеток в многоклеточных
организмах живет в менее изменчивых
средах, но поддержание каждой такой
среды требует согласования между ак-
тивностями различных клеток. Поэтому
для этих клеток главными внешними
раздражителями выступают гормоны,
факторы роста и связанные с ними соединения, которые передают сигналы в пределах
организма и активизируют согласованные изменения в активности генома.
Чтобы оказывать воздействие на активность генома, питательное вещество, гор-
мон, фактор роста или другое внеклеточное соединение, которое представляет собой
внешний раздражитель, должны влиять на события внутри клетки. Есть два способа,
которыми это может быть осуществлено (рис. 14.2):
• напрямую, действуя как сигнальное соединение, которое переносится через кле-
точную мембрану внутрь клетки;
• косвенно, связываясь с рецептором клеточной поверхности, который передает
сигнал внутрь клетки.
Передача сигнала прямыми или косвенным средствами является одной из главных
областей исследования в цитобиологии, где внимание, в частности, сосредоточено на ее
связи с ненормальными биохимическими активностями, которые лежат в основе раз-
вития рака. Было открыто множество примеров передачи сигналов, некоторые имеют
общую важную роль в целом разнообразии организмов, а другие ограничены только
несколькими видами. В первой части этой главы мы рассмотрим именно такие пути
передачи сигналов.
14.1.1. Передача сигнала посредством импорта внеклеточного сигналь-
ного соединения
В прямом методе передачи сигналов внеклеточное соединение, которое представ-
ляет собой внешний раздажитель, проходит сквозь клеточную мембрану и проникает
в клетку. После импорта в клетку сигнальное соединение может влиять на активность
генома любым из трех способов (рис. 14.3).
• Если сигнальное соединение представлено белком, то оно может действовать тем
же способом, что и любой из разнообразных регуляторных белков, с которыми
мы ознакомились в главах 10-13 — например, путём активации или подавления
сборки инициаторного комплекса транскрипции (раздел 11.3), либо посредством
взаимодействия с энхансером или сайленсером сплайсинга (раздел 12.2.2).
• Сигнальное соединение может непосредственно влиять на активность существую-
щего регуляторного белка. Такое сигнальное соединение не обязательно само
должно быть белком: оно может, теоретически, быть соединением любого типа.
Глава 14. Регулирование активности генома 545
Сигнальное соединение
непосредственно влияет
на белковый фактор
Сигнальное
соединение -
активатор или
репрессор
транскрипции
> Геном
Сигнальное соединение
косвенно влияет
на белковый фактор
Рис. 14.3. Три способа, которыми внеклеточное сигнальное соединение может влиять на активность
генома после проникновения в клетку
• Сигнальное соединение может влиять на активность существующего регулятор-
ного белка через одного или нескольких посредников, а не взаимодействуя с ним
напрямую.
Примеры каждого из этих трех способов действия описаны ниже.
Лактоферрин — внеклеточный сигнальный белок, который служит актива-
тором транскрипции
Если внеклеточное сигнальное соединение, которое импортируется в клетку, пред-
ставлено белком с подходящими свойствами, то оно может непосредственно затронуть
активность своих целевых генов, действуя как активатор или репрессор на некоторой
стадии пути экспрессии генома. Прямой способ регулирования активности генома может
показаться привлекательным, но это не распространенный механизм. Причина этого не
ясна, но, вероятно, связана, по крайней мере отчасти, с трудностью проектирования бел-
ка, который сочетал бы в себе гидрофобные свойства, необходимые для эффективного
переноса через мембрану с гидрофильными свойствами, нужными для перемещения
через водную цитоплазму к участку действия белка в ядре или на рибосоме.
Один наглядный пример сигнального соединения, которое может действовать
таким способом, представлен лактоферрином — белком млекопитающих, обнаруживае-
мым главным образом в молоке и, в меньшей степени, в кровотоке. Лактоферрин — акти-
ватор транскрипции (раздел 11.3.2). Его специфическая функция ускользает от ученых,
но кажется, что он играет роль в механизмах защиты организма от нападок всеразличных
микробов. Как предполагает его название, лактоферрин способен связывать железо,
и думается, что по крайней мере отчасти его защитная роль обусловлена его способно-
стью снижать уровни свободного железа в молоке и таким образом лишать вторгшихся
микробов этого жизненно необходимого им кофактора. Поэтому может показаться
неправдоподобным, что лактоферрин играет какую-либо роль в экспрессии генома,
но в начале 1980-х гг. стало известно, что этот белок обладает массой способностей и,
помимо прочих вещей, может связываться с ДНК. Это свойство было увязано со второй
функцией лактоферрина — возбуждением клеток крови, вовлеченных в иммунную
546 Часть 3. Принципы функционирования геномов
реакцию, — когда в 1992 г. было показано, что этот белок вбирается отвечающими за
иммунитет клетками, входит в их ядра и прикрепляется к геному. Позже было показано,
что связывание с ДНК является специфичным к последовательности и обеспечивает
стимулирование транскрипции; этот результат подтвердил, что лактоферрин является
истинным активатором транскрипции.
Некоторые импортированные сигнальные соединения непосредственно
влияют на активность уже находящихся в клетке регуляторных белков
Хотя лишь немногие импортированные сигнальные соединения способны сами
действовать как активаторы или репрессоры экспрессии генома, многие обладают
способностью напрямую влиять на активность регуляторных белков, которые уже
имеются в клетке. Мы столкнулись с одним примером регулирования такого типа в раз-
деле 11.3.1, когда изучали лактозный оперон Escherichia coli. Этот оперон реагирует на
внеклеточные уровни лактозы, причем это соединение служит сигнальной молекулой,
которая входит в клетку и, после превращения в свой изомер аллолактозу, влияет на
свойства связывания с ДНК лактозного репрессора и, следовательно, определяет, будет
оперон лактозы транскрибироваться или нет (см. рис. 11.24). Подобным же образом
управляются и многие другие опероны бактерий, кодирующие гены, что отвечают за
усвоение сахара.
Прямое взаимодействие между сигнальным соединением и активатором или ре-
прессором транскрипции является общепринятым средством регулирования активности
генома также и у эукариотов. Хороший пример представлен системой управления, кото-
рая поддерживает внутриклеточное содержание ионов металлов на соответствующем
уровне. Клеткам нужны ионы металлов, таких как медь и цинк, как кофакторы в био-
химических реакциях, но эти металлы становятся ядовитыми, если они накапливаются
в клетке выше определенного уровня. Поэтому их усвоение должно тщательно управ-
ляться, с тем чтобы клетка содержала достаточно ионов металлов, когда в окружающей
среде отсутствуют соединения металлов, но не накапливала бы ионы металлов сверх
меры, когда их концентрации во внешней среде являются высокими. Используемые
стратегии иллюстрированы системой управления содержанием меди у Saccharomyces
cerevisiae. Эти дрожжи имеют два зависимых от меди активатора транскрипции: Maclp
и Acelp. Оба этих активатора связывают ионы меди, причем это связывание вызывает
изменение конформации, которое позволяет фактору стимулировать экспрессию своих
целевых генов (рис. 14.4). У Maclp эти целевые гены кодируют белки усвоения меди,
тогда как у Acelp эти гены кодируют такие белки, как супероксиддисмутаза, которые
вовлечены в детоксификацию меди. Управляемое металлами равновесие между дей-
ствиями Maclp и Acelp гарантирует, что содержание меди в клетке остается в пределах
приемлемых уровней.
Активаторы транскрипции являются целями также и для стероидных гормонов,
представляющих собой сигнальные соединения, которые согласуют диапазон физио-
логических действий в клетках высших эукариотов. К ним относятся половые гормоны
(эстрогены для полового развития женских особей, андрогены для мужского полового
развития), а также глюкокортикоидные и минералокортикоидные гормоны. Стероиды
гидрофобны и, таким образом, легко проникают через клеточную мембрану. Оказав-
шись в клетке, каждый гормон связывается со специфическим белком-рецептором
стероидов, который обычно расположен в цитоплазме. После связывания активирован-
ный рецептор мигрирует в ядро, где он прикрепляется к реагирующему на гормоны
элементу, лежащему выше целевого гена. После такого связывания рецептор начина-
Глава 14. Регулирование активности генома 547
Рис. 14.4. Регулируемая медью экспрессия генов
у Saccharomyces cerevisiae. Дрожжам необходимы
небольшие количества меди, потому что несколько
присущих им ферментов (например, цитохром-с-
оксидаза и тирозиназа) суть медьсодержащие ме-
таллобелки, но слишком много меди ядовито для
клетки. Когда уровни меди низки, белковый фактор
Maclp активируется связыванием меди и включа-
ет экспрессию генов для поглощения меди. Когда
уровни меди слишком высоки, активируется второй
фактор, Acelp, включая экспрессию иного набора
генов, которые кодируют белки, вовлеченные в де-
токсификацию меди
Концентрация меди во внешней среде
Пищевая
Токсичная
Си
Си
Си
Си
Активен Mad р
Си Си Си
Си Си Си
Си Си Си
Си Си Си
Си
Активен Асе 1р
ет работать как активатор транскрипции.
Реагирующие элементы для каждого рецеп-
тора расположены выше на 50-100 генов,
часто в пределах энхансеров, так что один
стероидный гормон может вызвать круп-
номасштабное изменение в биохимических
свойствах клетки. Все рецепторы стерои-
дов структурно подобны, причем не только
в отношении своих ДНК-связывающих до-
менов, но также и в других частях их бел-
ковых структур (рис. 14.5). Распознавание
этих подобий привело к опознаванию ряда
предполагаемых или сиротских рецепторов стероидов, гормональные партнеры которых
и функции в клетке еще не известны. Структурные подобия показали также и то, что
второй набор рецепторных белков — надсемейство ядерных рецепторов — принад-
лежит к тому же общему классу, что и рецепторы стероидов, хотя гормоны, с которыми
они работают, не относятся к стероидам. Как предполагает их название, эти рецепторы
расположены в ядре, а не в цитоплазме. К ним относятся рецепторы витамина D3, роли
которых включают управление развитием костей
и тироксином, который стимулирует превращение
головастика в лягушку.
CTR1
CUP1
CTR3
CRS5
FRE1
SOD1
Синтез белков
поглощения меди
Синтез белков
детоксификации меди
Рецептор эстрогена
N
—С
Рецептор прогестерона
N С
Рис. 14.5. Все белки-рецепторы стероидных гормонов имеют
схожие структуры. Для сравнения показаны три рецепторных
белка. Все они показаны в виде развернутых полипептидов,
выровненных между собой по двум консервативным функцио-
нальным доменам. ДНК-связывающий домен очень подобен во
всех рецепторах стероидов и показывает 50-90%-ю идентич-
ность аминокислотных последовательностей. Домен связыва-
ния гормона (раздел 11.3.2) консервативен в меньшей степени
и обладает лишь 20-60%-й идентичностью последовательно-
стей. Домен активации лежит между N-концом и связывающим
ДНК доменом, но эта область показывает небольшое подобие
последовательностей при сравнении разных рецепторов
Рецептор глюкокортикоида
N С
200 аминокислот
ОБОЗНАЧЕНИЯ:
Изменчивая область
ДНК-связывающий домен
— Домен связывания гормона
548 Часть 3. Принципы функционирования геномов
Чувствительный элемент Рис. 14.6. Последовательности типичных элемен- тов отклика рецепторов стероидов и ядерных ре- цепторов. Рецептор ретиноевой кислоты необы- чен, поскольку присущие ему последовательности длиной 6 п. н. не являются точными повторениями
AGAACA TCTTGT nnn TGTTCT nnn £CAAGA Рецептор глюкокортикоида
AGGTCA TCCAG7 nnn TGACCT nnn ^GTfiGA Рецептор эстрогена и отделены друг от друга более чем четырьмя нуклеотидами
AGGTCA TQCAGT nnnnn AGACCA nnnnn ICTGGT Рецептор ретиноевой кислоты Рецепторы стероидов и ядерные рецепторы — димеры, каждая субъеди-
AGGR1K TCCAGT TGACCT ACTGGA Рецептор тироксина ница которых обладает одним из специ- альных цинковых пальцев, которые яв- ляются характерными для этой группы
AGGTCA TCCAGT nnn AGGTCA nnn TCCAGT Рецептор витамина Д белков (см. рис. 11.5). Каждый из таких цинковых пальцев опознает и связывает последовательность длиной 6 п.н. в сво-
ем реагирующем на гормон элементе. Для большинства рецепторов стероидов пара
таких последовательностей длиной 6 п.н. устроена как прямой или инвертированный
повторы, между которыми находится разделитель протяженностью 0-4 п. н. (рис. 14.6).
Элементы отклика для ядерных рецепторов подобны, за исключением того, что сайты
узнавания почти всегда являются прямыми повторами. Разделитель служит только
лишь для того, чтобы гарантировать, что расстояние между сайтами узнавания будет
соответствовать ориентации цинковых пальцев в рецепторном белке. Это означает, что
различные рецепторные белки могут обладать одинаковой парой цинковых пальцев,
но опознавать различные реагирующие элементы, при этом специфичность поддержи-
вается ориентацией пальцев и промежутком между сайтами узнавания.
Некоторые импортированные сигнальные соединения влияют на актив-
ность генома косвенно
Связь между сигнальной молекулой и регуляторными белками, отвечающими за
экспрессию генома, не обязательно должна быть столь же прямой, сколь в примерах,
описанных в предыдущем разделе. Сигнальные молекулы могут также влиять на актив-
ность генома косвенным манером через одного или нескольких посредников. Пример
представлен системой катаболитной репрессии у бактерий. Это средство, посредством
которого внеклеточные и внутриклеточные уровни глюкозы определяют, будут ли
включены опероны для усвоения других сахаров, когда эти альтернативные сахара
присутствуют в среде.
Это явление было открыто в 1941 г. Жаком Моно, который показал, что если Е coli
или Bacillus subtilis обеспечены смесью сахаров, то сначала будет усвоен один сахар,
и бактерия перейдет на следующий по шкале ее пристрастий сахар только тогда, ког-
да предыдущий будет израсходован. Чтобы описать это явление, Моно использовал
французское слово диауксия. Одно из сочетаний сахаров, которое вызывает диаукси-
ческую реакцию — глюкоза плюс лактоза, при этом глюкоза используется до лактозы
(рис. 14.7, а). Когда особенности оперона лактозы были определены около 20 лет спу-
стя (раздел 11.3.1), стало ясно, что диауксия между глюкозой и лактозой должна быть
обусловлена неким механизмом, посредством которого присутствие глюкозы может
пересилить активирующее воздействие, которое лактоза обычно оказывает на свой
Глава 14. Регулирование активности генома 549
оперон. В присутствии лактозы плюс глюкозы оперон лактозы выключен, даже при
том, что часть лактозы из смеси превращается в аллолактозу, которая связывается
с лактозным репрессором, так что в нормальных условиях оперон был бы транскриби-
руем (рис. 14.7, б).
Объяснить диауксическую реакцию можно тем, что глюкоза действует как сиг-
нальное соединение, которое подавляет экспрессию оперона лактозы, а также оперонов
усвоения других сахаров посредством косвенного влияния на белок-активатор ката-
болитных оперонов (называемый также активатором CRP). Этот белок связывается
с сайтом узнавания на различных участках в геноме бактерии и активирует инициацию
транскрипции в нижележащих промоторах, вероятно, взаимодействуя с а-субъединицей
РНК-полимеразы. Что свойственно этой активации, так это создание острого, 90°-го изги-
ба в двойной спирали в области сайта связывания, когда белок-активатор катаболитных
оперонов прикрепляется. Продуктивная инициация транскрипции в этих промоторах
зависит от присутствия связанного белка-активатора катаболитных оперонов: если
белок отсутствует, то гены, управляемые промотором, не транскрибируются.
Глюкоза сама по себе не взаимодействует с белком-активатором катаболитных
оперонов. Вместо этого, глюкоза управляет уровнем видоизмененного нуклеотида
в клетке — циклического АМФ (цАМФ; рис. 14.7, в). Она осуществляет это, ингибируя
активность аденилатциклазы — фермента, который синтезирует цАМФ из АТФ. Инги-
бирование опосредствуется белком под названием IIAGlc — компонентом многобелкового
комплекса, который переносит сахара внутрь бактерии. Когда глюкоза доставляется
в клетку, посредник IIAGlc становится дефосфорилированным (то есть дополнительные
фосфатные группы, ранее добавленные к белку в ходе посттрансляционной модифика-
ции, удаляются). Дефосфорил ированный вариант IIAGlc ингибирует действие аденилат-
циклазы. Это означает, что если уровни глюкозы высоки, содержание цАМФ в клетке
будет низким. Белок-активатор катаболитных оперонов может связаться со своими
целевыми участками только в присутствии цАМФ, так что когда глюкоза присутствует,
белок остается отделенным и опероны, которыми он управляет, выключены. В особом
случае диауксии с глюкозой и лактозой косвенный эффект глюкозы на белок-активатор
катаболитных оперонов означает, что оперон лактозы остается инактивированным,
даже при том что лактозный репрессор не связан, и, таким образом, глюкоза в среде
расходуется в первую очередь. Когда вся глюкоза выйдет, уровень цАМФ повышается
и белок-активатор катаболитных оперонов связывается со своими целевыми участ-
ками, в том числе с участком выше оперона лактозы, и транскрипция генов лактозы
активируется.
Белок 11 AGlc имеет также вторую роль в диауксической реакции — роль, которая не
имеет отношения к геному, но о которой мы должны упомянуть ради полноты описания.
Дефосфорилированная форма 11 AGlc предотвращает поглощение лактозы и другого сахара
путем ингибирования ферментов пермеаз, которые переносят эти сахара в клетку, —
вспомним, что лактопермеаза кодируется геном lacY, вторым в опероне лактозы (см.
рис. 8.8, а). Присутствие глюкозы поэтому имеет двоякое воздействие: опероны для
усвоения другого сахара выключены и поглощение этих сахаров предотвращено.
14.1.2. Передача сигналов опосредствуется рецепторами клеточной по-
верхности
Многие внеклеточные сигнальные соединения неспособны войти в клетку, потому
что они слишком гидрофильны, чтобы проникнуть через липидную мембрану, и клетка
550
Часть 3. Принципы функционирования геномов
б)
В ПРИСУТСТВИИ ЛАКТОЗЫ
РНК-полимераза
может сесть
Комплекс репрессор-
аллолактоза не может
прикрепиться к оператору
Репрессор
лактозы
ЕСЛИ ГЛЮКОЗА ТАКЖЕ В НАЛИЧИИ...
Транскрипция не происходит
Рис. 14.7. Катаболитная репрессия, а) Типичная
кривая диауксического роста, наблюдаемая,
когда Escherichia coli выращивается в среде, со-
держащей смесь глюкозы и лактозы. В течение
нескольких первых часов бактерии делятся по
экспоненте, используя глюкозу как источник
углерода и энергии. Когда глюкоза израсходо-
вана, есть краткий период задержки, в то время
как гены lac включаются перед возвращением
бактерий к экспоненциальному росту, теперь
потребляя лактозу, б) Глюкоза пересиливает
репрессор лактозы. Если лактоза присутствует,
то репрессор отделяется от оператора и опе-
рон лактозы должен транскрибироваться, но
остается в состоянии покоя, если глюкоза также
присутствует. На рис. 11.24, б подробно показан
механизм, посредством которого репрессор
лактозы управляет экспрессией оперона лак-
тозы. в) Глюкоза оказывает свое влияние на
оперон лактозы и другие целевые гены через
IIAGlc, который управляет активностью аде-
нилатциклазы и, следовательно, регулирует
количество цАМФ в клетке. Белок-активатор
катаболитов (САР) может связаться со своим
сайтом на ДНК только в присутствии цАМФ.
Если глюкоза присутствует, то уровень цАМФ
низок, так что САР не связывается с ДНК и не
активирует РНК-полимеразу. Как только глюко-
за израсходована, уровень цАМФ повышается,
позволяя САРу связаться с ДНК и активировать
транскрипцию оперона лактозы и других своих
целевых генов
в)
Уровень глюкозы высокий
- уровень цАМФ низкий
О’ О’ О’
lacZ -г
____ . Транскрипция
невозможна
Аденилатциклаза
ингибируется глюкозой
Il a Glc
через ПА
САР отсоединяется
О=Р—ОН ОН
О цАМФ
Уровень глюкозы низкий
- уровень цАМФ высокий
на участок прикрепления
Транскрипция
Глава 14. Регулирование активности генома 551
клеточнои поверхности
наружная поверхность
Клеточная мембрана
внутренняя поверхность
Внутриклеточный белок
Внеклеточное сигнальное
Рис. 14.8. Роль рецептора клеточной поверхности в пере-
даче сигналов. Прикрепление молекул внеклеточного
сигнального соединения к внешней поверхности белка-
рецептора вызывает конформационное изменение, часто
предполагающее димеризацию, которая дает активацию
внутриклеточного белка, например, за счет фосфорили-
рования. События, происходящие после этой начальной
активации белка, разнообразны и описаны в тексте. «Р»
обозначает фосфатную группу РО32-
не располагает специальным транспортным ме-
ханизмом их приемки. Для того чтобы влиять на
активность генома, такие сигнальные молекулы
должны связаться с рецепторами клеточной по-
верхности, которые переносят их сигналы через
клеточную мембрану. Эти рецепторы — белки,
пронизывающие всю мембрану, с участком для
связывания сигнального соединения на внеш-
ней поверхности. Связывание с сигнальным
соединением вызывает конформационные из-
менения в рецепторе. Часто такое изменение
конформации выражается в димеризации, благо жидкостный характер клеточной мем-
браны допускает боковое смещение мембранных белков в ограниченном масштабе, что
позволяет субъединицам димеров объединяться и разъединяться в ответ на присутствие
или отсутствие внеклеточного сигнала (рис. 14.8). Изменение конформации вызывает
биохимическое событие внутри клетки. Например, внутриклеточные сегменты многих
рецепторных белков обладают действием киназы, так что когда субъединицы объеди-
няются в димер, они фосфорилируют друг друга. Это биохимическое событие, будь то
взаимное фосфорилирование или некоторая другая реакция, составляет первый шаг
во внутриклеточной стадии пути передачи сигнала.
Были открыты рецепторы клеточной поверхности нескольких типов (таблица 14.2),
и внутриклеточные события, которые они инициируют, разнообразны, со многими ва-
риациями на каждую тему (не все они определенно участвуют в регулировании актив-
ности генома). Три примера помогут нам оценить сложность данной системы.
Передача сигнала между рецептором и геномом за один шаг
В некоторых системах передачи сигналов возбуждение рецептора клеточной по-
верхности за счет прикрепления внеклеточного сигнального соединения дает прямую
активацию белка, влияющего на активность генома. Это наипростейшая система, с по-
мощью которой внеклеточный сигнал может быть преобразован в реакцию генома.
Прямая система используется многими цитокинами, такими как интерлейкины
и интерфероны, которые являются внеклеточными сигнальными полипептидами,
управляющими ростом и делением клетки. Связывание этих полипептидов со своими
рецепторами клеточной поверхности обусловливает активацию фактора транскрипции
особого типа, называемого STAT (передатчик сигналов и активатор транскрипции).
Активация осуществляется фосфорилированием одной тирозиновой аминокислоты
в позиции вблизи С-конца полипептида STAT. Если рецептор клеточной поверхности
является членом семейства тирозинкиназ (см. табл. 14.2), то он способен напрямую ак-
552 Часть 3. Принципы функционирования геномов
Таблица 14.2. Белки рецепторов клеточной поверхности, участвующие в передаче сигналов внутрь
клеток эукариотов
Тип рецептора Описание Сигналы
Сопряженные с G-белком Активируют внутриклеточные G-белки, которые связывают ГТФ и управляют био- химическими действиями за счет превра- щения этого ГТФ в ГДФ с высвобождением энергии Разнообразные: адреналин, пептиды (например, глюка- гон), белковые гормоны, за- пахи, свет
Тирозинкиназы Активируют внутриклеточные белки пу- тем фосфорилирования тирозина Гормоны (например, инсулин), различные факторы роста
Связанные с тиро- зинкиназой Подобны рецепторам тирозинкиназы, но активируют внутриклеточные белки косвенно (например, см. описание STATbi в тексте) Гормоны, факторы роста
Серин- треонинкиназы Активируют внутриклеточные белки посредством фосфорилирования серина и (или) треонина Гормоны, факторы роста
Ионные каналы Управляют внутриклеточными действия- ми, регулируя движение ионов и других ма- леньких молекул внутрь и наружу клеток Химические раздражители (например, глутамат), элек- трические заряды
тивировать STAT (рис. 14.9, а). Если же это сопутствующий тирозинкиназам рецептор,
то сам он не имеет способности фосфорилировать STAT или любой другой внутрикле-
точный белок, но действует через посредников, называемых киназами Януса (JAK).
Связывание сигнальной молекулы с сопутствующим тирозинкиназам рецептором вы-
зывает изменение конформации рецептора, часто посредством вызова димеризации.
Это вынуждает JAK, которая связана с рецептором, фосфорилировать саму себя, такая
автоактивация сопровождается фосфорилированием фактора STAT посредником JAK
(рис. 14.9, б).
К настоящему времени семь STATob были опознаны у млекопитающих. Три из
них — STAT 2,4 и 6 — являются специфичными только к одному или двум внеклеточ-
ным цитокинам, но другие обладают широким спектром специфичности и могут быть
активированы несколькими различными интерлейкинами и интерферонами. Разли-
чение обеспечивается рецепторами клеточной поверхности: тот или иной отдельно
взятый рецептор связывается с цитокином только одного типа, а большинство клеток
имеет только один или несколько типов рецепторов цитокинов. Поэтому различные
клетки по-разному реагируют на присутствие определенных цитокинов, даже при том
что внутренний процесс передачи сигналов вовлекает только ограниченное число
факторов STAT.
Консенсусная последовательность сайтов связывания ДНК у STATob была опреде-
лена как 5'-TTN5 6AA-3' в значительной степени благодаря исследованиям, в которых
очищенные STATbi проверялись на предмет гибридизации с олигонуклеотидами с из-
вестной последовательностью. Связывающийся с ДНК домен белка STAT состоит из трех
петель, выходящих из цилиндрообразной структуры p-листа. Это ДНК-связывающий
домен необычного типа и он не был опознан в точно такой же форме в белках никако-
го другого типа, хотя он имеет подобия с ДНК-связывающими доменами активаторов
Глава 14. Регулирование активности генома 553
а) Прямая активация STAT
Переходит
в ядро и активирует
целевые гены
6) Активация посредством JAK
целевые гены
Рис. 14.9. В передаче сигналов участвуют STAT, а) Если рецептор принадлежит семейству тирозинкиназ,
то он может активировать STAT непосредственно. 6} Если рецептор относится к типу сопутствующих
тирозинкиназам, то он действует через киназу Януса (JAK), которая автофосфорилируется, когда вне-
клеточный сигнал связывается, и затем активирует STAT. Обратите внимание, что активация JAK обычно
вовлекает димеризацию — внеклеточный сигнал побуждает две субъединицы соединиться, давая вер-
сию JAK, обладающую фосфорилирующим действием. Димеризация также важна для активации STAT:
фосфорилирование вынуждает два STAT, не обязательно одного и того же типа, образовать димер. Этот
димер способен действовать как активатор транскрипции. «Р» обозначает фосфатную группу РО32-
транскрипции NK-кВ и Rel. Эти подобия относятся только к третичным структурам ДНК-
связывающих доменов, потому что STAT, NK-кВ и Rel в целом имеют очень небольшое
сходство аминокислотных последовательностей. Многие целевые гены активируются
факторами STAT, но общая реакция генома опосредствуется другими белками, которые
взаимодействуют со STAT и влияют на то, какие гены будут включены при существую-
щем наборе условий. Сложность вполне ожидаема, потому что клеточные процессы,
в которых факторы STAT выступают посредниками, — рост и деление — сами по себе
сложны, и мы предвидим, что для внесения изменений в эти процессы потребуется
значительная перестройка протеома и, следовательно, крупномасштабные изменения
в активности генома.
Передача сигнала между рецептором и геномом за несколько шагов
Простота системы, посредством которой рецептор клеточной поверхности акти-
вирует STAT, напрямую или через JAK, связанную с рецептором, разительно отличается
от более распространенных форм передачи сигнала, в которых рецептор представляет
собой только первый из последовательности шагов, ведущих в конечном счете к вклю-
чению или выключению одного или нескольких активаторов или репрессоров транс-
крипции. Ряд таких каскадных путей был выявлен в различных организмах. Ниже
приведены примеры таких путей, имеющие большое значение у млекопитающих.
• Система МАР- (активируемый митогеном белок) киназы (рис. 14.10) реагирует на
многие внеклеточные сигналы, в том числе на митогены — соединения с действия-
ми, подобными проявляемым цитокинами, но которые специфично стимулируют
деление клетки. Связывание сигнального соединения приводит к димеризации
рецептора митогенов и взаимному фосфорилированию внутренних частей двух
его субъединиц (см. рис. 14.8). Фосфорилирование стимулирует присоединение
к рецептору (с внутренней стороны мембраны) различных цитоплазматических
белков, один из которых — Raf — есть протеинкиназа, которая активируется при
554 Часть 3. Принципы функционирования геномов
Рецептор митогенов
. Связывание
сигнала
становится Р
фосфорилированным . Вербовка
I Raf
Передача
сигнала
Рис. 14.10. Передача сигнала путем MAP-киназы. «МК»
обозначает MAP-киназу, а «Р» — фосфатную группу РО32-.
Elk-1, с-Мус и CstF (фактор сывороточной реакции) — при-
меры факторов транскрипции, активизируемых в конце
пути
связывании с мембраной. Raf инициирует каскад
реакций фосфорилирования. Он фосфорилирует
Мек, активируя этот белок так, что он, в свою
очередь, фосфорилирует MAP-киназу. Активи-
рованная MAP-киназа теперь перемещается
в ядро, где она включает, снова фосфорилиро-
ванием, ряд активаторов транскрипции. Также
MAP-киназа фосфорилирует другую протеинки-
назу, называемую Rsk, которая фосфорилирует
и активирует второй набор факторов. Дополни-
тельная гибкость обеспечивается возможностью
замены одного или нескольких белков на пути
MAP-киназы родственными белками с немного
отличающимися особенностями и, следователь-
но, включения другого комплекта активаторов.
Путь MAP-киназы используется клетками по-
звоночных; у других организмов известны ана-
логичные пути, использующие посредников, по-
добных обнаруженным у млекопитающих (см.
примеры в разделах 14.2.1 и 14.3.3).
• Система Ras построена на белках Ras, три из
которых известны в клетках млекопитающих
(Н-, К- и N-Ras), и подобных им белках типа Rac
и Rho. Эти белки участвуют в регулировании
роста и в дифференцировке клетки и, как многие
белки в этой категории, будучи дисфункцио-
нальными, могут вызвать рак. Белки семейства
Ras не ограничены млекопитающими, примеры
известны у других эукариотов, например у пло-
довой мушки. Белки Ras — посредники в путях
передачи сигналов, которые инициируются с ав-
тофосфорилирования рецептора тирозинкина-
зы в ответ на внеклеточный сигнал. Фосфорилированный вариант рецептора об-
разует межбелковые комплексы с GNRP (белками, высвобождающими нуклеотид
гуанин) и GAP (активирующими ГТФазу белками), которые, соответственно,
активируют и инактивируют Ras (рис. 14.11). Поэтому внеклеточные сигналы
могут включать или выключать опосредствуемую Ras передачу сигналов. Выбор
между этими двумя действиями зависит от характера сигнала и относительных
количеств активных молекул GNRP и GAP в клетке. Будучи активирован, Ras сти-
мулирует активность Raf, так что фактически Ras обеспечивает вторую точку входа
в путь MAP-киназы, хотя вряд ли это единственная функция Ras, и, вероятно, он
Глава 14. Регулирование активности генома 555
Рис. 14.11. Система передачи сигнала Ras. Сокраще-
ния: GAP — активирующий ГТФазу белок; GNRP —
белок, высвобождающий нуклеотид гуанин. «Р»
обозначает фосфатную группу РО32-
также активирует белки, участвующие
в передаче сигналов вторичными по-
средниками (что описано в следующем
разделе).
• Система SAP- (активируемый стрессом
белок) киназы включается относящи-
мися к стрессовым сигналами типа
ультрафиолетового излучения, а так-
Пути
вторичных
посредников
Путь МАР-киназы
же факторами роста, связанными с воспалением. Этот путь еще не был подробно
описан, но он подобен системе МАР-киназы, хотя и нацелен на другой набор акти-
ваторов транскрипции.
На каждом этапе в этих каскадных путях происходит физическое взаимодействие
между двумя белками, часто выражающееся в фосфорилировании «нижестоящего» члена
пары. Фосфорилирование активирует нижестоящий белок, позволяя ему образовать
связь со следующим белком в каскаде. Эти взаимодействия вовлекают специальные
домены межбелкового связывания, такие как домены, названные SH2 и SH3, которые
связываются с рецепторными доменами, расположенными на белках — их партнерах.
Рецепторные домены содержат один или несколько тирозинов, которые должны быть
фосфорилированы, для того чтобы произошла стыковка. Следовательно, вышестоящий
белок содержит рецепторный домен, состояние фосфорилирования которого определяет,
может ли белок связаться со своим нижестоящим партнером и, следовательно, передать
сигнал далее (рис. 14.12).
Передача сигнала через вторичные посредники
Некоторые каскады передачи сигнала не обеспечивают прямую передачу внешнего
сигнала к геному, но вместо этого используют косвенное средство влияния на транс-
крипцию. Такая косвенность обеспечивается вторичными посредниками, которые
представляют собой менее специфичные внутренние сигнальные соединения, что
передают сигнал от рецептора клеточной поверхности в нескольких направлениях,
так что не только транскрипция, а целое разнообразие клеточных действий реагирует
на один сигнал.
В разделе 14.1.1 мы увидели, как глюкоза модулирует белок-активатор катабо-
литных оперонов, влияя на уровни цАМФ у бактерий (см. рис. 14.7). Циклические ну-
клеотиды служат важными вторичными посредниками также и в клетках эукариотов.
Некоторые рецепторы клеточной поверхности обладают действием гуанилатциклазы и,
таким образом, превращают ГТФ в цГМФ, но большинство рецепторов в этом семействе
работает косвенно, влияя на активность цитоплазматических циклаз и дециклаз. Эти
циклазы и дециклазы определяют клеточные уровни цГМФ и цАМФ, которые, в свою
очередь, управляют активностью различных целевых ферментов. К последним относит-
ся протеинкиназа А, которя стимулируется цАМФом. Одна из функций протеинкиназы
А состоит в фосфорилировании и, следовательно, в запуске активатора транскрипции
556 Часть 3. Принципы функционирования геномов
С Вышестоящий белок,
фосфорилирован
р
К Прикрепляется
нижний белок
р р
Е Нижний белок становится
фосфорилированным
р
wqj Нижний белок теперь
ф способен активировать
нижеследующий белок
в каскаде
Рис. 14.12. Схема взаимодействия белков в каскаде
передачи сигналов. Вышестоящий белок фосфори-
лирован и, следовательно, способен связываться со
своим нижестоящим партнером. Связывание ведет
к фосфорилированию рецепторного домена в ни-
жестоящем белке и тем самым к распространению
сигнала. «Р» обозначает фосфатную группу РО32~
по имени CREB. Это один из нескольких бел-
ков, которые влияют на активность цело-
го разнообразия генов, взаимодействуя со
вторичным активатором рЗОО/СВР, который
способен модифицировать гистоновые бел-
ки и таким образом затрагивать структуру
хроматина и расстановку нуклеосом (раз-
делы 10.2.1 и 10.2.2).
Кроме косвенного активирования
цАМФом, активатор рЗОО/СВР реагирует на
другой вторичный посредник — кальций.
Концентрация ионов кальция значительно
ниже внутри, чем снаружи клетки, так что
белки, которые открывают кальциевые каналы в клеточной мембране, позволяют
ионам кальция входить внутрь. Это может быть вызвано внеклеточными сигналами,
активирующими рецепторы тирозинкиназы, которые, в свою очередь, активируют
фосфолипазы, расщепляющие фосфатидилинозитол-4,5-бисфосфат (липидный ком-
понент внутренней клеточной мембраны) на инозитол-1,4,5-трисфосфат (Инз(1,4,5)Р3)
и 1,2-диацилглицерин (ДАГ). Инз(1,4,5)Р3 открывает кальциевые каналы (рис. 14.13).
Инз(1,4,5)Р3 и ДАГ — сами по себе вторичные посредники, которые могут запустить
другие каскады передачи сигналов. Как инициируемые кальцием, так и инициируемые
липидами каскады нацелены на активаторы транскрипции, но только косвенно: пер-
вичные цели — другие белки. Кальций, например, связывается с белком (и активирует
его), названным кальмодулином, который регулирует ферменты разнообразных типов,
в том числе протеинкиназы, АТФазы, фосфатазы и нуклеотидциклазы.
Распутывание путей передачи сигнала
Как цитобиологи распутывают хитросплетения путей передачи сигналов? Чтобы
ответить на этот вопрос, мы исследуем часть недавней исследовательской работы,
направленной на путь передачи сигналов, активируемый трансформирующим фак-
тором роста-p (TGF-Р), членом семейства около 30-ти родственных полипептидов,
которые управляют такими процессами, как деление и дифференцировка клеток по-
звоночных. Рецепторы клеточной поверхности для TGF-P — серин-треонинкиназы
(см. табл. 14.2), которые активируют целое разнообразие целевых белков в клетке.
Часть процесса передачи сигнала, инициируемого связыванием TGF-p, вовлекает набор
белков, называемых семейством SMAD; это название образовано сокращением наиме-
нования «связанные со SMA/MAD», относящегося соответственно к белкам Drosophila
melanogaster и Caenorhabditis elegans, которые были первыми выделенными членами
этого семейства.
Глава 14. Регулирование активности генома 557
Рис. 14.13. Индукция кальциевой системы вторичных
посредников. Сокращения: ДАГ—1,2-диацилглицерин;
Инз(1,4,5)Р3 — инозитол-1,4,5-трифосфат;
ФтдИнз(4,5)Р2 — фосфатидинозитол-4,5-бисфосфат
Первоначально пять белков SMAD были
открыты в клетках позвоночных. Четыре из
них — Smadl, Smad2, Smad3 и Smad5 — на-
зывают регулируемыми рецепторами бел-
ками SMAD, потому что они связываются
непосредственно с рецептором клеточной
поверхности. Каждый из этих белков SMAD
является специфичным к своему рецептору
серина-треонина и, следовательно, реагирует
на разные члены семейства TGF-p сигнальных
соединений. Связывание внеклеточного сиг-
нала побуждает рецептор фосфорилировать
свой SMAD, который после этого связывается
со Smad4, перемещается в ядро и через взаимо-
действия с ДНК-связывающими белками ак-
тивирует набор целевых генов (рис. 14.14, а).
Таким образом, Smad4 есть сопосредник, ко-
торый участвует в пути передачи сигналов
Рецептор тирозинкиназы
ФтдИнз(4,5)Р2
каждого из других четырех белков SMAD. Система SMAD — пример второго пути пере-
дачи сигналов за один прием между рецептором и геномом, подобного пути STAT,
описанному выше.
Данная интерпретация пути SMAD была осложнена открытием двух дополнитель-
ных белков SMAD — номер 6 и 7, — которые не вписываются в эту схему. У этих белков
SMAD отсутствует мотив последовательности аминокислот серин-серин-Х-серин (где
X — валин или метионин), расположенный в С-концевой области Smadl, Smad2, Smad3
и Smad5 и который фосфорилируется рецептором. Поэтому ясно, что Smad6 и Smad7 не
реагируют непосредственно на прикрепление внеклеточных сигналов к рецепторному
белку. Являются ли они сопосредниками наподобие Smad4 или играют какую-либо иную
роль в пути передачи сигналов TGF-P?
Первый шаг в понимании функций Smad6 и Smad7 состоял в определении влияния
сверхэкспрессии этих белков на путь TGF-p передачи сигналов. Сверхэкспрессия дости-
галась за счет присоединения гена Smad6 или Smad7 к сильному промотору и после-
дующего применения методов клонирования, чтобы ввести этот ген в культивируемые
клетки. В итоге ядерные гены, обычно включаемые TGF-p, стали нечувствительными
к внеклеточному сигналу в клетках, сверхэкспрессирующих Smad6 или Smad7. Этот
результат дал первый признак того, что Smad6 и Smad7 оказывают ингибирующие
воздействия на путь TGF-p.
Чтобы объяснить, как ингибирующие белки SMAD (так теперь называются Smad6
и Smad7) подавляют путь TGF-p, были предложены две модели (рис. 14.14, б). Первая
модель опирается на то наблюдение, что в клеточных экстрактах белки Smad6 и Smad7
связаны с внутриклеточными частями рецепторов клеточной поверхности. Гипотеза
состоит в том, что Smad6 и Smad7 ингибируют передачу сигналов, препятствуя активи-
558 Часть 3. Принципы функционирования геномов
а) Путь передачи сигналов SMAD
Рис. 14.14. Постижение пути передачи сигналов SMAD. а)
Общая схема пути передачи сигналов. Посредник Smadl
показан в момент его активации рецептором TGF-p. Та-
кой же путь приемлем для посредников Smad2, Smad3
и Smad5. б) Две модели ингибирующих воздействий
Smad6 (показан на этой схеме) и Smad7
Переходит в ядро
и активирует
целевые гены
6) Модели ингибирующего воздействия Smad6 и Smad7
Smad6
Smadl
Сигнал заблокирован
Smad6
Цель для сигнала
отсутствует
рованным рецепторам фосфорилировать другие белки SMAD. Эта модель, вероятно, объ-
ясняет ингибирующие воздействия сверхэкспрессии Smad6 и Smad7, но в нормальных
клетках может не быть достаточно копий этих белков, чтобы блокировать все рецепторы
клеточной поверхности. Поэтому была предложена альтернативная модель, в которой
ингибирующие белки SMAD связываются с одним или несколькими другими белками
SMAD, удаляя их из пути и тем самым останавливая передачу сигнала. Есть достоверные
данные о том, что взаимодействие такого типа служит объяснением ингибирующего
воздействия, которое Smad6 оказывает на Smadl. Исследования на дрожжевой дву-
гибридной системе (раздел 6.2.2) показали, что эти два белка SMAD ваимодействуют,
и после связывания с белком Smad6 белок Smadl не способен влиять на активаторы
транскрипции, которые он обычно стимулирует, даже после его фосфорилирования
рецепторами клеточной поверхности.
Каким бы ни был механизм действия Smad6 и Smad7, открытие этих ингибирую-
щих белков SMAD показывает, что путь передачи сигналов TGF-p через путь SMAD более
гибок, чем изначально представлялось. Вместо того чтобы быть реакцией по типу «всё
или ничего», активность регулируемых рецепторами белков SMAD может видоизме-
няться ингибирующими воздействиями Smad6 и Smad7, а эти белки, предположительно,
реагируют на пока что неопознанные внутри- и (или) межклеточные сигналы, чтобы
модулировать эффекты связывания TGF-p надлежащим образом.
Глава 14. Регулирование активности генома 559
14.2. Постоянные и полупостоянные изменения в активности
генома
Непостоянные изменения в активности генома являются по определению своему
легко обратимыми: картина экспрессии генома возвращается в ее первоначальное со-
стояние, когда внешний раздражитель удаляется или заменяется противодействующим
раздражителем. В отичие от этого, постоянные и полупостоянные изменения активности
генома, которые лежат в основе дифференцировки клеток, должны сохраняться в те-
чение длительных периодов времени и в идеале должны поддерживаться, даже когда
раздражитель, который первоначально вызвал их, исчезнет. Поэтому мы предвидим, что
регуляторные механизмы, вызывающие эти долгосрочные изменения, будут представ-
лены системами, дополняющими модуляцию активаторов и репрессоров транскрипции.
Это ожидание верно. Мы рассмотрим три механизма:
• изменения, следующие из физической перестройки генома;
• изменения, обусловленные структурой хроматина;
• изменения, поддерживаемые петлями обратной связи.
14.2.1. Перестройки генома
Изменение физической структуры генома есть очевидный, хотя и радикальный
способ произвести постоянное изменение в экспрессии генома. Это не распространенный
регуляторный механизм, но известно несколько важных примеров.
Типы спаривания дрожжей определяются событиями генной конверсии
Тип спаривания — эквивалент полового размножения, присущий дрожжам и дру-
гим относящимся к эукариотам микроорганизмам. Поскольку эти организмы воспроиз-
водятся главным образом путем вегетативного деления клеток, есть возможность, что
популяция клеток, происходящая только из одной или нескольких прародительских
клеток, будет в значительной степени или полностью состоять из одного типа спари-
вания и, таким образом, не будет способна воспроизводиться половым путем. Дрожжи
Saccharomyces cerevisiae и некоторые другие виды избегают этой проблемы, потому что
клетки способны изменять пол при помощи процесса, названного переключением
типа спаривания.
Два типа спаривания у S’, cerevisiae назвали а и а. Каждый тип спаривания выде-
ляет короткий полипептидный феромон (12 аминокислот в длину для а и 13 для а),
который связывается с рецепторами на поверхностях клеток противоположного типа
спаривания. Прикрепление феромона инициирует путь MAP-киназы передачи сигналов
(см. рис. 14.10), который изменяет профиль экспрессии генома в клетке, что ведет к тон-
ким морфологическим и физиологическим изменениям, которые превращают клетку
в гамету, способную участвовать в половом воспроизводстве. Поэтому смешивание
в одной культуре клеток двух гаплоидных штаммов противоположного типа спаривания
стимулирует образование гамет, которые слияются и производят диплоидную зиготу.
В зиготе происходит мейоз, дающий начало тетраде из четырех гаплоидных сумкоспор
(аскоспор), содержащихся в структуре, называемой сумкой (аском). Сумка вскрывается,
выпуская сумкоспоры, которые затем делятся митозом и порождают новые гаплоидные
вегетативные клетки (рис. 14.15).
Тип спаривания определяется геном МАТ, расположенным на хромосоме III. Этот
ген имеет два аллеля, МАТа и МА Та, при этом гаплоидная клетка дрожжей обнаруживает
560 Часть 3. Принципы функционирования геномов
Гаплоидные вегетативные клетки
Четыре
гаплоидные
аскоспоры
в аске
Прорастание,
митоз
Прорастание,
митоз
Рис. 14.15. Жизненный цикл дрожжей Saccharomyces
cerevisiae
тип спаривания, соответствующий тому аллелю,
которым она обладает. В каком-либо другом месте
на хромосоме III находятся два дополнительных
МЛТ-подобных гена, названных HMRa и HMLa
(рис. 14.16). Они имеют те же самые последо-
вательности, что и соответственно гены МАТа
и МАТа, но ни один ген не экспрессируется, потому
что выше каждого из них расположен сайленсер,
который подавляет инициацию транскрипции.
Эти два гена называют «подавленными кассетами
типа спаривания». В их подавлении участвуют
белки Sir, некоторые из их числа обладают дей-
ствием гистондезацетилазы (раздел 10.2.1), что
говорит о том, что подавление сопряжено с из-
менениями структуры хроматина в области генов
HMRa и HMLa.
Переключение типа спаривания иниции-
руется эндонуклеазой НО, которая производит
двунитевой разрез в последовательности дли-
ной 24 п. н., расположенной в пределах гена МАТ.
Благодаря этому становится возможным событие
генной конверсии. Мы досконально рассмотрим
генную конверсию в разделе 17.1.1; все, что ка-
сается нас в настоящее время — то, что один из свободных З'-концов, произведенных
эндонуклеазой, может быть продолжен посредством синтеза ДНК с использованием
одной из двух подавленных кассет в качестве матрицы (см. рис. 14.16). Новосинтези-
рованная ДНК впоследствии заменяет ДНК, находящуюся в локусе МАТ. В качестве
матрицы обычно выбирается та подавленная кассета, которая отличается от аллеля,
HMLa
МАТа
HMRa
. Хромосома III
। Эндонуклеаза НО
HMLa
HMRa
Двунитевой разрез
Превращение гена
HMLa МАТа HMRa
Рис. 14.16. Переключение между типами спаривания у дрожжей. В данном примере клетка начинает
свое существование как тип спаривания а. Эндонуклеаза НО вырезает локус МАТа, вызывая генную
конверсию на локус HMLa. В результате этого тип спаривания переключается на тип а. Молекулярная
основа генной конверсии раскроется во всех подробностях, если открыть раздел 17.1.1
Глава 14. Регулирование активности генома 561
первоначально находящегося в локусе МАТ, так что замена новосинтезированной ни-
тью превращает ген МАТ из МАТа в МАТа или наоборот. В результате этого происходит
переключение типа спаривания.
Гены МАТ кодируют регуляторные белки (один в случае МАТа и два в случае МАТа),
которые взаимодействуют с активатором транскрипции МСМ1 и таким образом опреде-
ляют, какой набор генов будет включен этим фактором. Продукты генов МАТа и МАТа
оказывают различные воздействия на МСМ1 и, таким образом, определяют различные
аллелеспецифические картины экспрессии генома. Эти картины экспрессии поддержи-
ваются полупостоянным способом, до тех пор пока не произойдет следующая генная
конверсия МАТ.
Перестройки генома ответственны за разнообразие иммуноглобулинов
и рецепторов Т-клеток
У позвоночных есть два поразительных примера использования перестроек ДНК,
чтобы достичь постоянных изменений в активности генома. Эти два примера, которые
очень подобны один другому, отвечают за порождение разнообразия иммуноглобул инов
и рецепторов Т-клеток.
Иммуноглобулины и рецепторы Т-клеток — родственные белки, которые син-
тезируются соответственно В- и Т-лимфоцитами. Белки обоих типов прикрепляются
к внешним поверхностям своих клеток, а иммуноглобулины также выпускаются в кро-
воток. Эти белки помогают защитить тело от вторжения бактерий, вирусов и других не-
желательных веществ, посредством связывания с этими антигенами, как их называют.
За время своей жизни организм может подвергаться нашествию любого числа и самого
широкого спектра антигенов, а это означает, что иммунная система должна быть способ-
на синтезировать одинаково широкий спектр иммуноглобулинов и белков рецепторов
Т-клеток. Фактически организм человека способен производить приблизительно 108
различных иммуноглобулинов и белков рецепторов Т-клеток. Но в геноме человека
имеется только 3,5-104 генов, так что возникает вопрос: откуда берутся все эти белки?
Чтобы найти ответ, мы взглянем на структуру типичного белка иммуноглобулина.
Каждый иммуноглобулин — тетрамер из четырех полипептидов, связанных дисульфид-
ными связями (рис. 14.17). Они представле-
ны двумя длинными «тяжелыми» цепями
и двумя короткими «легкими» цепями. При
Рис. 14.17. Структура иммуноглобулина. Каждый им-
муноглобулиновый белок состоит из двух тяжелых
и двух легких цепей, соединенных между собой ди-
сульфидными связями. Каждая тяжелая цепь — 446
аминокислот в длину и состоит из изменчивой об-
ласти (показана красным), охватывающей амино-
кислоты 1-108, и следующей за ней константной
области. Каждая легкая цепь — 214 аминокислот,
снова с N-концевой изменчивой областью длины
108 аминокислот. Дополнительные дисульфидные
мостики формируются между различными частя-
ми отдельных цепей: эти и другие взаимодействия
свертывают белок в более сложную трехмерную
структуру
Изменчивая область Тяжелая цепь
ОШв Константая область Легкая цепь
— Дисульфидная связь
562 Часть 3. Принципы функционирования геномов
сравнении последовательностей различных тяжелых цепей становится ясно, что их из-
менчивость сосредоточена главным образом в N-концевых областях этих полипептидов,
а С-концевые части являются во многом подобными, или «постоянными», во всех тяжелых
цепях. То же самое верно для легких цепей, за исключением того, что можно выделить
два семейства, к и X, отличающиеся последовательностями их постоянных областей.
В геномах позвоночных нет полных генов, кодирующих тяжелый и легкий поли-
пептиды иммуноглобулина. Вместо этого, эти белки определяются сегментами генов.
Сегменты тяжелой цепи находятся на хромосоме 14 и включают 11 сегментов генов
постоянной области (Сн), которым предшествуют 123-129 изменчивых сегментов VH
генов, 27 сегментов генов DH и 9 сегментов генов JH, эти последние три типа кодируют
различные версии V (изменчивых), D (разнообразных) и J (соединительных) компо-
нентов изменчивой части тяжелой цепи (таблица 14.3; рис. 14.18). Весь локус тяжелой
цепи тянется через несколько млн п.н. Подобное расположение наблюдается у локусов
легкой цепи на хромосомах 2 (локус к) и 22 (локус X), единственное отличие состоит
в том, что легкие цепи не имеют D-сегментов (таблица 14.3).
Хромосома 14
100 тыс. п. н.
123-129 V / 9J
сегментов генов /сегментов
27 D сегментов генов цс
генов сегментов генов
Рис. 14.18. Организация локуса IGH (тяжелая цепь) человека на хромосоме 14
На ранней стадии развития В-лимфоцита локусы иммуноглобулина в его геноме
подвергаются перестройкам. В пределах локуса тяжелой цепи эти перестройки связы-
вают один из генных сегментов VH с одним из генных сегментов DH и затем связывают
эту V-D комбинацию с генным сегментом JH (рис. 14.19). Такие перестройки происходят
рекомбинацией необычного типа, катализируемой парой белков по имени RAG1 и RAG2,
при этом позиции, в которых должны произойти реакции разреза и воссоединения,
приводящие к соединению генных сегментов, отмечены рядом консенсусных после-
довательностей длиной 8 и 9 п.н. В конечном результате создается экзон, который
содержит полную открытую рамку считывания, определяющую сегменты VH, DH и JH
белка иммуноглобулина. Этот экзон соединяется с экзоном сегмента Сн посредством
Таблица 14.3. Сегменты генов иммуноглобулина в геноме человека
Число сегментов генов
Компонент Локус Хромосома V D J С
Тяжелая цепь IGH 14 123-129 27 9 и
Легкая к-цепь IGK 2 76 0 5 1
Легкая Х-цепь IGL 22 70-71 0 7-11 7-11
Примечание: некоторые числа изменчивы в силу различий между генотипами человека. Неизвестно,
все ли генные сегменты являются функционально активными — некоторые могут быть представлены
псевдогенами.
Глава 14. Регулирование активности генома 563
сплайсинга в ходе процесса транскрипции, за счет чего создается полная мРНК тяжелой
цепи, которая может быть транслирована в белок иммуноглобулина, который является
специфичным только к этому одному лимфоциту. Подобный ряд перестроек ДНК обе-
спечивает построение экзона V-J легкой цепи лимфоцита или в локусе к, или в локусе X,
и опять же в ходе сплайсинга к нему подцепляется экзон сегмента С легкой цепи при
синтезе мРНК.
Несмотря на свое название, постоянная область не идентична во всех белках имму-
ноглобулина. Небольшие ее разновидности, которые имеют место, дают иммуноглобу-
лины пяти разных классов — IgA, IgD, IgE, IgG и IgM, — при этом представители каждого
из этих классов наделены своей собственной специализированной ролью в иммунной
системе. Первоначально каждый В-лимфоцит синтезирует молекулу IgM, сегмент Сн
которой определяется последовательностью Ст, которая лежит на 5’-конце группы
сегментов Сн. Как показано на рис. 14.19, позже в своем развитии незрелая клетка может
также синтезировать некоторые белки IgD, используя вторую последовательность Сн
в группе (С8); экзон для этой последовательности присоединяется к сегменту V-D-J
посредством альтернативного сплайсинга. На более поздних этапах своего жизненного
цикла, по достижении зрелости, некоторые В-лимфоциты подвергаются переключению
класса второго типа, в результате которого полностью изменяется тип иммуноглобули-
на, синтезируемого лимфоцитом. Это второе переключение класса требует еще одного
события рекомбинации, в ходе которого удаляется последовательность Ср и С8 наряду
с частью хромосомы между этой областью и сегментом Сн, определяющим класс имму-
ноглобулина, который лимфоцит теперь будет синтезировать. Например, для переклю-
чения лимфоцита к синтезу IgG — наиболее распространенного типа иммуноглобулина,
Транскрипция
Экзон 1 Экзон2 ЭкзонЗ
пре-мРнк ;• 12
I
Иммуноглобулин М
Альтернативный сплайсинг
1 3
I
Иммуноглобулин D
Рис. 14.19. Синтез специфического белка иммуноглобулина. Перестройка ДНК в пределах локуса тяжелой
цепи связывает сегменты V, D и J, которые затем соединяются с сегментом С посредством сплайсинга
мРНК. В незрелых B-клетках экзон V-D-J всегда становится связанным с экзоном Ср (экзон 2) и производит
мРНК, определяющую иммуноглобулин класса М. На ранней стадии развития В-клетки некоторые белки
иммуноглобулина D также производятся в ходе альтернативного сплайсинга, который связывает экзон
V-D-J с экзоном С5. Иммуноглобулины обоих типов становятся прикрепленными к мембране клетки
564 Часть 3. Принципы функционирования геномов
производимого зрелыми лимфоцитами, — в результате удаления один из сегментов
Cg, которые определяют тяжелую цепь IgG, помещается в 5’-конец группы (рис. 14.20).
Поэтому переключение класса представляет второй пример перестройки генома, про-
исходящей в ходе развития В-лимфоцита. Механизм отличается от соединения V-D-J,
и событие рекомбинации не вовлекает белки RAG.
J-область Сц С6 С^З С^1 ч|? Са1 гр 0,2 04 Се Са2
Удаление путем рекомбинации
0^204 Се Са2
Рис. 14.20. Переключение между классами иммуноглобулина. В этом примере удаляется семь сегментов
Сн, в результате чего сегмент Су2 помещается смежно с J-областью. Поэтому эта В-клетка синтезирует
молекулу иммуноглобулина G, которая выделяется клеткой. Два отмеченных буквой \|/ сегмента суть
псевдогены
Разнообразие рецепторов Т-клеток основано на подобных перестройках, которые
связывают генные сегменты V, D, J и С в различных сочетаниях, с тем чтобы произвести
клеткоспецифические гены. Каждый рецептор включает пару р-молекул, которые яв-
ляются подобными тяжелой цепи иммуноглобулина, и две а-молекулы, которые напо-
минают легкие цепи иммуноглобулинам Как и в случае иммуноглобулинов, рецепторы
Т-клеток погружаются в клеточную мембрану и позволяют лимфоциту опознавать
внеклеточные антигены и реагировать на них.
14.2.2. Изменения в структуре хроматина
Некоторые из воздействий, которые структура хроматина может оказывать на
экспрессию генома, были описаны в разделе 10.2. Они варьируют от модуляции ини-
циации транскрипции в отдельном промоторе посредством расстановки нуклеосом до
подавления крупных сегментов ДНК, запертой в структуре хроматина более высокого
порядка. Последнее выступает важным средством осуществления долгосрочных из-
менений в активности генома и принимает участие в ряде регуляторных мер. Одно
из них касается локусов типа спаривания дрожжей, которые мы рассматривали ранее
в этом разделе, подавление кассет HMRa и HMLa, проистекающее главным образом от
погружения этих локусов в недоступный хроматин в ответ на влияние последователь-
ностей их вышележащих сайленсеров. Инактивация Х-хромосомы (раздел 10.3.1) также
вовлекает образование недоступного хроматина, в данном случае фактически по всей
длине одной из двух Х-хромосом в ядре женской клетки.
Еще один пример подавления хроматином заслуживает внимания. Это система,
с которой мы снова встретимся далее в этой главе, когда будем рассматривать процессы
развития плодовой мушки. Она относится к семейству генов Polycomb, включающему
в себя около 30-ти генов, кодирующих белки, которые связываются с последователь-
ностями ДНК, называются реагирующими элементами Polycomb и вызывают формиро-
вание гетерохроматина — спирализованной формы хроматина, которая препятствует
транскрипции генов, в нем содержащихся (рис. 14.21). Каждый реагирующий элемент
имеет приблизительно 10 тыс. п. н. в длину, но, кажется, не содержит специфичных сайтов
связывания Polycomb, что позволяет предположить, что дополнительные белки служат
Глава 14. Регулирование активности генома 565
Чувствительный к Polycomb
элемент
Белок Polycomb
Образование гетерохроматина
Вся область заглушена
Рис. 14.21. Polycomb поддерживает подавление в областях генома Drosophila, запуская формирование
гетерохроматина. Имейте в виду что прикрепление белка Polycomb к его чувствительному элементу
опосредствуется дополнительными белками, не показанными на этом рисунке
посредниками в прикреплении Polycomb. Кандидат на эту посредническую роль — белок
non-Polycomb, DSP1 (Dorsal switch proteinl1)), который связывается с последовательно-
стями 5’-GAAAA-3', присутствующими в центральной области реагирующего элемента
Polycomb. Мутации в этих последовательностях, которые предотвращают связывание
DSP1, также препятствуют привлечению белков Polycomb в экспериментальных си-
стемах. Каков бы ни был механизм, в результате присоединения Polycomb происходит
зарождение гетерохроматина вокруг белков Polycomb, после чего гетерохроматин рас-
пространяется по ДНК на десятки тыс.п.н. в обоих направлениях.
Области, которые становятся подавленными, содержат гомеозисные гены, кото-
рые, как мы увидим в разделе 14.3.4, определяют развитие отдельных частей тела мухи.
Поскольку только одна часть тела должна быть задана в конкретной позиции в геноме
плодовой мушки, важно, чтобы клетка экспрессировала только надлежащий гомеозис-
ный ген. Это обеспечивается действием белка Polycomb, навсегда глушащего гомеозис-
ные гены, которые должны быть выключены. Белки Polycomb, однако, не определяют,
какие гены будут подавлены: экспрессия этих генов уже подавлена до того, как белки
Polycomb связываются со своими реагирующими элементами. Роль Polycomb поэтому
заключается в поддержании, а не вызывании подавления генов. Здесь важен тот момент,
что гетерохроматин, наведенный белком Polycomb, является наследственным: после
деления две новые клетки сохраняют гетерохроматин, утвердившийся в родительской
клетке. Поэтому регулирование активности генома этого типа является постоянным не
только в одной клетке, но также и во всей последовательности клеточных поколений.
Белки trithorax действуют в подобной Polycomb манере, но оказывают противо-
положное действие: они поддерживают раскрытое состояние хроматина в областях
активных генов, мишенями служат те же гомеозисные гены, что и те, которые глу-
шатся в различных частях тела белками Polycomb. Способы действия белков trithorax
и Polycomb могут быть тесно связаны в эволюционном отношении, так как есть данные,
что белки trithorax связываются со своими мишенями через посреднический белок, на-
званный GAGA, который связывается с последовательностями в пределах реагирующих
на Polycomb элементов. Некоторые мутации отменяют активность и Polycomb, и trithorax,
а это указывает на то, что эти две системы могут иметь общие компоненты.
Белок спинного переключения 1. — Прим, перев.
566 Часть 3. Принципы функционирования геномов
14.2.3. Регулирование генома петлями обратной связи
Последний из рассматриваемых нами механизмов, вызывающих долгосрочные
изменения в активности генома, предполагает использование петли обратной связи.
В этой системе регуляторный белок активирует свою собственную транскрипцию, так
что, как только его ген включится, он будет экспрессироваться непрерывно (рис. 14.22).
Известен ряд примеров такого регулирования петлями обратной связи.
• Активатор транскрипции MyoD, который участвует в развитии мышц, является
одним из наиболее изученных примеров дифференцировки клеток позвоночных
животных. Клетка направляется на путь становления мышечных клеток, когда
она начинает экспрессировать ген myoD. Продукт этого гена — активатор транс-
крипции, нацеленный на ряд других генов, кодирующих такие специфичные для
мышц белки, как актин и миозин, и также косвенно отвечает за одну из основных
характеристик мышечных клеток — отсутствие нормального клеточного цикла:
эти клетки останавливаются в фазе G1 (раздел 15.3.1). Белок MyoD связывается
также и выше гена myoD, гарантируя, что его собственный ген будет непрерывно
экспрессироваться. Результат этой петли положительной обратной связи — то, что
клетка продолжает синтезировать специфичные для мышц белки и остается мы-
шечной клеткой. Дифференцированное состояние наследуется, потому что деление
клетки сопровождается передачей белка MyoD дочерним клеткам, гарантируя, что
они тоже будут мышечными клетками.
• Белок Deformed у Drosophila — один из нескольких белков, кодируемых гомеозис-
ными избирательными генами, он отвечает за определение рода сегментов тела
плодовой мушки (раздел 14.3.4). Белок Deformed (Dfd) отвечает за род сегментов
головы. Чтобы выполнять эту функцию, ген Dfd должен непрерывно экспрессиро-
ваться в предписанных клетках. Это достигается за счет системы обратной связи,
в которой белок Dfd связывается с энхансером, расположенным выше гена Dfd.
Саморегуляция обратной связи также управляет экспрессией по крайней мере
некоторых гомеозисных избирательных генов позвоночных животных.
14.3. Регулирование активности генома в ходе развития орга-
низма
Путь развития многоклеточного эукариота начинается с оплодотворенной
яйцеклетки и завершается взрослой формой организма. Между ними простирается
сложная череда генетических, клеточных и физиологических событий, которые
Активирует свой _________ Ген активатора
собственный ген транскрипции
Активирует
целевые гены
должны происходить в правильном по-
рядке, в уместных клетках и в надлежа-
щие моменты времени, если путь должен
привести к успешному финалу. У людей
такой путь развития завершается взрос-
лым человеком, содержащим 1013 клеток,
дифференцированных приблизительно
на 250 специализированных типов, при-
Рис. 14.22. Регулирование экспрессии гена за счет
обратной связи
Глава 14. Регулирование активности генома 567
чем деятельность каждой отдельной клетки согласуется с действиями всех остальных
клеток. Процессы развития такой сложности могли бы показаться трудноразреши-
мыми даже для мощных исследовательских средств современной молекулярной
биологии, но в последние годы в их понимании произошел значительный прогресс.
Исследовательская работа, которая обусловила этот прогресс, была построена на
трех руководящих принципах.
• Должна быть возможность описывать и постигать генетические и биохимические
события, которые лежат в основе дифференцировки клеток на отдельные типы. Это,
в свою очередь, означает, что понимание того, как устроены специализированные
ткани и даже сложные части тела, должно быть в пределах досягаемости.
• Процессы передачи сигналов, которые согласуют события в различных клетках,
должны быть подвластны изучению. Мы увидели в разделе 14.1, что уже положено
начало описанию этих систем на молекулярном уровне.
• Между связанными с развитием процессами в различных организмах должны
быть подобия и параллели, отражающие их общее эволюционное происхождение.
Это означает, что информация, имеющая отношение к развитию человека, может
быть получена в ходе исследований модельных организмов, выбранных исходя из
относительной простоты путей развития последних.
Биология развития охватывает области генетики, молекулярной биологии, цито-
биологии, физиологии, биохимии и системной биологии. Нас интересует только роль
генома в развитии, и мы не будем пытаться дать всесторонний краткий обзор иссле-
дования развития во всех его обликах. Вместо этого, мы сосредоточимся на четырех
модельных системах возрастающей сложности, с тем чтобы исследовать типы изменений
в активности генома, которые происходят в ходе развития.
14.3.1. Лизогенный цикл бактериофага X
Бактериофаг, который заражает Escherichia coli, мог бы показаться странной от-
правной точкой для изучения регулирования генома в процессе развития. Но именно
с него молекулярные биологи и начали длинную программу исследований, которая
сегодня открывает геномный фундамент, лежащий в основе развития людей и других
позвоночных. Поэтому мы будем придерживаться того же курса — от относительно
простого к относительно сложному.
Из раздела 9.1.1 мы узнали, что лизогенные бактериофаги типа фага X могут пойти
по двум альтернативным путям репликации после инфицирования клетки-хозяина.
Помимо литического пути, в ходе которого новые фаги собираются и выходят из клет-
ки вскоре после первичного инфицирования (после 45 минут для X), эти фаги могут
также проследовать лизогенным циклом, для которого характерна встройка ДНК фага
в хромосому хозяина. Встроенный профаг остается в состоянии покоя на протяжении
многих поколений бактерий, пока химический или физический раздражитель, сопря-
женный с повреждением ДНК, не вызовет вырезание генома фага X, быструю сборку
фагов и лизис клетки-хозяина (см. рис. 9.4 и 9.5).
Бактериофаг X должен сделать выбор между лизисом и лизогенией
Способность таких бактериофагов, как фаг X, следовать л изогенным циклом инфи-
цирования поднимает три вопроса: как фаг «решает», следовать ли ему литическим или
лизогенным циклом, как лизогения поддерживается и что побуждает профага прервать
лизогению? Об экспрессии генома во время инфицирования фагом X известно доста-
568 Часть 3. Принципы функционирования геномов
точно много — настолько много, что можно дать вполне исчерпывающие и глубокие
ответы на эти вопросы.
Первый шаг в литическом цикле инфицирования — экспрессия двух немедленно-
ранних генов X, которые назвали N и его. Они транскрибируются с двух промоторов,
соответственно PL и PR (рис. 14.23, а). Белок N — антитерминатор, который позволяет
РНК-полимеразе хозяина игнорировать сигналы терминации, с которыми она сталкива-
ется немедленно ниже кодирующих последовательностей N и его, и транскрибировать
задержанно-ранние гены (см. рис. 12.9). Эти гены включают сП и сШ, которые совместно
активируют третий промотор, PRM, и тем самым обусловливают транскрипцию гена cl.
Это важный во всех отношениях ген, поскольку он кодирует белок-репрессор фага X —
ключевой выключатель, который предотвращает развитие литического цикла и под-
держивает лизогению. Репрессор делает это посредством связывания с операторами 0L
и 0R, смежными соответственно с промоторами PL и PR (рис. 14.23, б). В результате почти
весь геном фага к подавляется, потому что промоторы PL и PR направляют транскрипцию
не только немедленно-ранних и задержанно-ранних генов, но также и поздних генов,
которые кодируют белки, необходимые для сборки новых фагов и лизиса клетки-хозяина.
Один из немногих генов, остающихся активными, — int, который транскрибируется со
своего собственного промотора. Белок интегразы, кодируемый этим геном, катализиру-
ет сайт-специфическую рекомбинацию, в результате которой ДНК фага X встраивается
в геном хозяина. Лизогения поддерживается в течение многократных делений клеток,
потому что ген d непрерывно экспрессируется, хотя и на низком уровне, так что коли-
чество находящегося в клетке репрессора cl всегда достаточно, чтобы поддерживать
промоторы PL и PR выключенными. Такая непрерывная экспрессия гена с! происходит,
потому что репрессор cl, будучи связан с оператором 0R, не только блокирует транс-
крипцию с промотора PR, но также и стимулирует транскрипцию со своего собственного
промотора PRM. Поэтому ключом к лизогении является двоякая роль репрессора cl.
Когда ген cl экспрессируется, белок-репрессор предотвращает вход в литический
цикл и гарантирует, что лизогения установится и будет поддерживаться. Но фаг X не
всегда входит в лизогенный цикл — в некоторых случаях инфицирование незамедли-
тельно переходит к лизису хозяина. Это связано с активностью второго немедленно-
раннего гена, его, который также кодирует репрессор, но в данном случае такой, который
предотвращает транскрипцию гена d (рис. 14.23, в). Поэтому решение между лизисом
и лизогенией определяется итогом соревнования между генами d и его. Если репрессор
а) Синтез транскриптов
немедленно-ранних генов
N его
fL1 nutL PL PR nutR fR1
Транскрипты «немедленно-ранних» генов
б) Роль репрессора cl
„ cl PrM
Ранние Ранние
гены гены
•X «— -X-
Стимулирует Блокирует экспрессию
экспрессию cl ранних генов
Рис. 14.23. Генетические основания выбора, делаемого бак-
териофагом X между лизисом и лизогенией
в) Роль репрессора С го
Репрессор Сго
Экспрессия с/
Экспрессия
не происходит
ранних генов
Глава 14. Регулирование активности генома 569
cl синтезируется быстрее, чем репрессор Сго, то экспрессия генома блокируется и права
получает лизогения. Однако если ген его выигрывает гонку то репрессор Сго блокирует
экспрессию гена cl прежде, чем репрессор cl успеет синтезироваться в количестве, до-
статочном для подавления генома. В результате фаг входит в литический цикл инфици-
рования. Решение кажется случайным, зависящим от случайных событий, которые ведут
к более быстрому накоплению репрессора cl или Сго в клетке, хотя условия внешней
среды могут оказывать определенное влияние. Рост на богатой среде, например, смещает
равновесие к литическому циклу, предположительно потому, что выгодно производить
новые фаги, когда хозяйские клетки процветают. Такое смещение вызвано активацией
протеаз, которые деградируют белок сП, уменьшая способность комбинации репрессоров
сП-dll включать транскрипцию репрессорного гена d.
Если бактериофаг входит в лизогенный цикл, то это состояние поддерживается до
тех пор, пока репрессор cl связан с операторами 0L и 0R. Профаг будет поэтому активизи-
рован, если уровень активного репрессора cl снизится ниже определенного порога. Это
может произойти случайно и привести к самопроизвольной активизации, либо может
произойти в ответ на физические или химические раздражители. Такие раздражители
активируют главный защитный механизм Е. coli — реакцию SOS. Часть реакции SOS —
экспрессия гена Е. coli recA, продукт которого инактивирует репрессор cl, расщепляя
его надвое. Это включает экспрессию ранних генов, позволяя фагу войти в литический
цикл. Также инактивация репрессора cl означает, что транскрипция гена с! более не
стимулируется, что устраняет возможность повторного установления лизогении через
синтез большего количества репрессора cl. Инактивация репрессора cl поэтому ведет
к активизации профага.
Что нам удалось установить благодаря этой модельной системе?
• Простой генетический переключатель может определить, по какому из двух путей
развития пойдет клетка.
• Генетические перекл ючатели могут задействовать сочетание активации и репрес-
сии различных промоторов.
• Возможно перепрограммировать путь развития и перейти на альтернативный
путь в ответ на соответствующие раздражители.
14.3.2. Спорообразование у Bacillus
Вторая система, которую мы исследуем, — образование спор у бактерии Bacillus
subtilis. Как и в случае л изогении фага X, это, строго говоря, не путь развития, а всего лишь
тип дифференцировки клеток, но даный процесс иллюстрирует два фундаментальных
вопроса, к которым придется обратиться при изучении истинного развития многокле-
точных организмов. Эти вопросы касаются того, как череда изменений в активности
генома управляется с течением времени, и того, как передача сигналов устанавливает
согласованность между событиями, происходящими в различных клетках. Преимущества
Bacillus как модельной системы состоят в том, что его легко выращивать в лаборатории
и он поддается иследованиям при помощи таких методов генетики и молекулярной
биологии, как анализ мутантов и секвенирование генов.
Спорообразование предполагает взаимно согласованные действия в клетках
двух разных типов
Bacillus — один из нескольких родов бактерий, которые производят эндоспоры
в ответ на неблагоприятные условия внешней среды. Эти споры являются очень стой-
570 Часть 3. Принципы функционирования геномов
Bacillus
subtilis
Клеточная стенка
Клеточная мембрана
Перегородка
ВЕГЕТАТИВНОЕ
ДЕЛЕНИЕ КЛЕТКИ
Рис. 14.24. Спорообразование у Bacillus
subtilis. В верхней части рисунка пока-
зан нормальный вегетативный способ
деления клетки, предполагающий
формирование перегородки через
центр бактерии и дающий в итоге две
идентичные дочерние клетки. В ниж-
ней части рисунка показано спороо-
бразование, при котором перегород-
ка формируется около одного конца
клетки, что дает материнскую клетку
и проспору разного размера. Посте-
пенно материнская клетка полностью
охватывает проспору. В конце процесса
зрелая, стойкая спора выходит в окру-
жающий мир
Проспора
СПОРООБРАЗОВАНИЕ
Окружённая спора
спора
кими к непригодным для жизни
физическим и химическим усло-
вям и могут выживать в течение
многих десятилетий или даже
столетий — возможности зара-
жения спорами сибирской язвы,
образуемыми В. anthracis, очень
серьезно опасаются археологи,
раскапывающие участки, содержащие останки людей и животных. Устойчивость обу-
словлена специализированной природой оболочки споры, которая является непрони-
цаемой для многих химикатов, равно как и биохимическими изменениями внутри споры,
которые задерживают распад ДНК и других полимеров и позволяют споре пережить
длительный период покоя.
В лаборатории спорообразование обычно вызывается нехваткой питательных
веществ. Голод побуждает бактерий оставить их нормальный вегетативный способ
деления клеток, который предполагает синтез септы (или перегородки) в центре клет-
ки. Вместо этого, клетки строят необычную перегородку, более тонкую, чем обычная,
в одном конце клетки (рис. 14.24). В результате образуются две клеточные полости,
меньшая из которых называется проспорой, а большая — материнской клеткой. По
мере развития процесса спорообразования проспора становится полностью охваченной
материнской клеткой. К этому времени эти две клетки направлены на различные, но
при этом согласованные пути дифференцировки: проспора претерпевает биохимиче-
ские изменения, которые позволяют ей упокоиться, а материнская клетка выстраивает
устойчивую оболочку вокруг споры и в конечном счете погибает.
Во время спорообразования активностью генома управляют специальные
G-субъединицы
Изменения в активности генома во время спорообразования управляются глав-
ным образом синтезом специальных о-субъединиц, которые изменяют специфичность
РНК-полимеразы Bacillus к промоторам. Вспомним, что о-субъединица является частью
РНК-полимеразы, которая опознает последовательность промотора бактерии, и что
Глава 14. Регулирование активности генома 571
Внеклеточные сигналы
ООО
замена одной о-субъединицы на другую, с иной специфичностью связывания с ДНК,
может привести к транскрипции другого набора генов (раздел 11.3.1). Мы видели, как
эта простая система управления используется Е. coli в ответ на тепловой стресс (см.
рис. 11.23). Она является ключом также и к изменениям в активности генома, которые
происходят во время спорообразования.
Стандартные о-субъединицы В. subtilis называют оА и пн. Эти субъединицы синте-
зируются в вегетативных клетках и позволяют РНК-полимеразе опознавать промоторы
всех генов, которые она должна транскрибировать, с тем чтобы поддерживать нормаль-
ный рост и деление клетки. В проспоре и материнской клетке эти субъединицы заменя-
ются соответственно на субъединицы qf и ое, которые опознают последовательности
других промоторов и, таким образом, вызыва-
ют крупномасштабные изменения в картинах
экспрессии генома. Главным переключателем
с вегетативного роста на образование спор слу-
жит белок, называемый SpoOA, который присут-
ствует в вегетативных клетках, но в неактивной
форме. Этот белок активируется фосфорилиро-
ванием через каскад протеинкиназ, реагирую-
щих на различные внеклеточные сигналы, ко-
торые сообщают о наличии такого исходящего
из внешней среды стресса, как отсутствие пи-
тательных веществ. Первичная реакция обеспе-
чивается тремя киназами, называемыми KinA,
KinB и KinC, которые фосфорилируют сами себя
и затем передают фосфат через SpoOF и SpoOB
к SpoOA (рис. 14.25). Активированный SpoOA
является фактором транскрипции, который мо-
дулирует экспрессию различных генов, транс-
крибируемых вегетативной РНК-полимеразой
и, следовательно, опознаваемых обычными субъединицами оА и он. Гены, которые
включаются, включают таковые для субъединиц oF и оЕ, что приводит к переключению
на дифференцировку на проспору и материнскую клетку (рис. 14.26).
Изначально в каждой из двух дифференцирующихся клеток присутствуют обе
субъединицы — и aF и суе. Это не совсем то, что нужно, потому что oF — это специфическая
для проспор субъединица, и, следовательно, она должна быть активна только в такой
клетке, а суе — специфическая для материнских клеток субъединица. Поэтому необхо-
4i
Рис. 14.25. Каскад фосфорилирования, кото-
рый ведет к активации SpoOA. Сокращения:
А — KinA; В — KinB; С — KinC; OF — SpoOF;
OB — SpoOB; OA — SpoOA; P обозначает фос-
фатную группу PO32-
димо некоторое средство активирования
или инактивирования соответствующей
Рис. 14.26. Роль SpoOA в спорообразовании
у Bacillus. SpoOA фосфорилируется в ответ на вне-
клеточные сигналы, проистекающие из неблаго-
приятных воздействий окружающей среды, как по-
казано на рис. 14.25. Это активатор транскрипции
с ролями, которые включают активацию генов для
оЕ- и сЛсубъединиц РНК-полимеразы. Сокращения:
Е — оЕ; F — aF; OA — SpoOA; Р обозначает фосфат-
ную группу РО32-
Внеклеточные
сигналы
Активация
Ген субъединицы оЕ
Ген субъединицы о
572 Часть 3. Принципы функционирования геномов
субъединицы в заданной клетке. Это, как думают, достигается следующим образом
(рис. 14.27).
• Субъединица oF активируется высвобождением из комплекса совместно со вто-
рым белком — SpoIIAB. Этим управляет третий белок, SpoIIAA, который, будучи
нефосфорилирован, может связаться также и с SpoIIAB и препятствовать связы-
ванию последнего с субъединицей oF. Если SpoIIAA нефосфорилирован, то субъе-
диница oF высвобождается и является активной; когда SpoIIAA фосфорилирован,
то субъединица qf остается связанной с SpoIIAB и, таким образом, неактивной.
В материнской клетке SpoIIAB фосфорилирует SpoIIAA и тем самым удерживает
субъединицу qf в ее связанном, неактивном состоянии. Но в проспоре попыткам
SpoIIAB фосфорилировать SpoIIAA противодействует еще один белок, SpoIIE, так
что субъединица oF высвобождается и становится активной. Способность SpoIIE
противодействовать SpoIIAB в проспоре, но не делать этого в материнской клетке
объясняется тем фактом, что молекулы SpoIIE прикрепляются к мембране на по-
верхности перегородки. Поскольку проспора намного меньше материнской клетки,
но площадь поверхности перегородки одинакова в обеих, концентрация белка
SpoIIE выше в проспоре, и это позволяет ему противодействовать SpoIIAB.
• Субъединица оЕ активируется протеолитическим расщеплением белка-
предшественника. Выполняющая это расщепление протеаза представлена белком
SpoIIGA, который пронизывает перегородку между материнской клеткой и проспо-
а) Активация </ в проспоре
МАТЕРИНСКАЯ
КЛЕТКА
ПРОСПОРА
SpoIIE блокирует
6) Активация <уЕ в материнской клетке
КЛЕТКА
Рис. 14.27. Активация специфических для проспоры и для материнской клетки а-субъединиц в ходе
спорообразования у Bacillus, а) В материнской клетке субъединица aF является неактивной, потому что
она связана с белком SpoIIAB, который фосфорилирует SpoIIAA и предотвращает последующее высво-
бождение субъединицы aF. Активация aF в проспоре происходит за счет ее высвобождения из ее же
комплекса с белком SpoIIAB, что косвенно влияет на концентрацию прикрепленного к мембране SpoIIE.
6) В материнской клетке субъединица аЕ активируется протеолитическим расщеплением SpoIIGA, которое
происходит в ответ на присутствие в проспоре зависимого от aF белка SpollR. Сокращения: АА — SpoIIAA;
АВ - SpoIIAB; Е - аЕ; F - aF; GA - SpoIIGA; R - SpollR
Глава 14. Регулирование активности генома 573
рой. Домен протеазы, который находится на перегородке со стороны материнской
клетки, активируется связыванием SpoIIR с рецепторным доменом на стороне
проспоры. Это типичная опосредствуемая рецептором система передачи сигна-
ла (раздел 14.1.2). Ген для SpoIIR — один из тех, промотор которых специфично
опознается субъединицей oF, так что активация протеазы и превращение пре-оЕ
в активную субъединицу оЕ происходит, когда направляемая oF транскрипция
выполняется в проспоре.
Активация субъединиц qf иоЕ — только на-
чало истории. В проспоре спустя приблизительно
1 час после активации субъединица oF отвечает на
неизвестный сигнал (возможно, от материнской
клетки), что выливается в небольшое изменение
в активности генома в споре. Оно включает транс-
крипцию гена другой о-субъединицы, oG, которая
опознает промоторы, расположенные выше генов,
продукты которых требуются на последующих
стадиях дифференцировки споры. Один из этих
белков—SpoIVB—активирует другую связанную
с перегородкой протеазу SpoIVF (рис. 14.28). Эта
протеаза затем активирует вторую о-субъединицу
материнских клеток, ок, которая кодируется
транскрибируемым субъединицей оЕ геном, но
пребывает в материнской клетке в неактивной
форме, до тех пор пока сигнал для её активации
не будет получен от проспоры. Субъединица ок
направляет транскрипцию генов, продукты которых необходимы в течение последующих
стадий пути дифференцировки материнской клетки.
Итак, подведем итог главным особенностям спорообразования у Bacillus:
Главный белок, SpoOA, отвечает на внешние раздражители через каскад событий
фосфорилирования, чтобы определить, должно ли произойти переключение на спороо-
бразование, а если должно, то когда именно.
Череда п-субъединиц в проспоре и материнской клетке вызывает зависящие от
времени изменения в активности генома в обеих этих клетках.
Межклеточная передача сигналов гарантирует, что события, происходящие в про-
споре и материнской клетке, будут согласованы1).
КЛЕТКА
Рис. 14.28. Активация субъединицы ок в ходе
спорообразования у Bacillus. Обратите внима-
ние, что схема очень подобна процедуре акти-
вирования субъединицы оЕ (см. рис. 14.27, б).
Сокращения: G — aG; К — ак; IVB — SpoIVB;
IVF - SpoIVF
14.3.3. Развитие яйцевода у Caenorhabditis elegans
В. subtilis — одноклеточный организм, и, хотя спорообразование предполагает
согласованную дифференцировку клеток двух типов, вряд ли его можно считать сопо-
ставимым с процессами развития, которые происходят в многоклеточных организмах.
Спорообразование дает наводки на общие пути, которыми активность генома может
регулироваться в ходе развития многоклеточного организма, но оно не показывает
специфические события, которые можно ожидать. Поэтому нам необходимо изучить
развитие на примере простого многоклеточного эукариота.
Л Следует отметить, что достаточно близкие бактерии не имеют способности к споруляции, а другие
(даже родственные) спорообразующие бактерии имеют заметно отличную систему регуляции этого про-
цесса. — Прим. ред.
574 Часть 3. Принципы функционирования геномов
С. elegans — модель развития многоклеточных эукариотов
Исследования над микроскопическим червем-нематодой С. elegans (рис. 14.29) вош-
ли в практику с подачи Сидни Бреннера, начавшего в 1960-х гг. свои изыскания с целью
использования этого организма в качестве простой модели развития многоклеточных
эукариотов. С. elegansлегко выращивается в лаборатории и имеет короткое время смены
поколений (хотя и измеряемое днями, но все же удобное для генетического анализа).
Этот червь прозрачен на всех стадиях своего жизненного цикла, так что наблюдение
внутреннего строения возможно и без лишения животного жизни. Это важный момент,
потому что это позволило исследователям отслеживать полный процесс развития
червя на клеточном уровне. Каждое деление клеток на пути от оплодотворенного
яйца до взрослого червя было отмечено на картах, и была установлена каждая точка,
в которой клетка начинает выполнять специализированную функцию. Кроме того, на
карту была нанесена полная система (сеть) связей из 302 клеток, которые образуют
нервную систему червя.
Рис. 14.29. Червь нематоды Caenorhabditis elegans. На микрофотографии показан взрослый червь-
гермафродит, приблизительно 1 мм в длину. Яйцевод — маленький выступ, расположенный на нижней
стороне животного, примерно посередине. Яйцеклетки заметны внутри тела червя в области по обе
стороны от яйцевода. Перепечатано с разрешения Kendrew, J. (Ed.), Encylopaedia of Molecular Biology,
© 1994 Blackwell Publishing
Геном C. elegans относительно мал и составляет л ишь 97 млн п. н. (см. табл. 7.2), его
полная последовательность известна. Анализ последовательности при помощи многих
методов, описанных в главе 5, позволил нам начать приписывать функции к неизвест-
ным генам и устанавливать связи между активностью генома и путями развития. Цель
данного проекта состоит в полном генетическом описании развития С. elegans, и можно
надеяться, что меты сией достичь нам удастся в не столь отдаленном грядущем.
Определение пути дифференцировки клетки в ходе развития яйцевода
С. elegans
Главная особенность, которая обусловливает применимость С. elegans в качестве
инструмента исследований, — тот факт, что его развитие является более или менее
инвариантным: схема деления и дифференцировки клеток фактически одинакова
у всех особей. Это, кажется, связано в значительной степени с передачей межклеточных
сигналов, которая побуждает каждую клетку следовать назначенным ей путем диффе-
ренцировки. Чтобы иллюстрировать это, рассмотрим развитие яйцевода С. elegans.
Большинство червей С. elegans являются гермафродитами, а это означает, что они
имеют и мужские и женские половые органы. Яйцевод является частью половой системы
самок и представляет собой трубку, через которую вводится сперма и осуществляется
кладка оплодотворенных яиц. Взрослый яйцевод состоит из 22 клеток, суть потомков
трех предковых клеток, изначально располагавшихся в ряд на нижней поверхности
развивающегося червя (рис. 14.30). Каждая из этих предковых клеток направляется на
путь дифференцировки, который ведет к производству клеток яйцевода. Центральная
клетка, названная Рб.р, встает на «первичный путь дифференцировки клеток яйцевода»
и делится с порождением восьми новых клеток. Другие две клетки — Р5.р и Р7.р — из-
Глава 14. Регулирование активности генома 575
Предковые
клетки
Деления
клеток
Клетки
яйцевода
7 клеток 8 клеток 7 клеток
Яйцевод
Рис. 14.30. Деления клеток, в результате которых порождаются клетки яйцевода Caenorhabditis elegans.
Три предковые клетки делятся по запрограммированной схеме и производят 22 потомственные клетки,
которые реорганизуют свои положения относительно друг друга, с тем чтобы построить яйцевод
бирают «вторичный путь дифференцировки клеток яйцевода» и делятся на семь клеток
каждая. Эти 22 клетки затем реорганизуют свои позиции и выстраиваются в яйцевод.
Важный момент развития яйцевода заключается в том, что оно должно произойти
в правильном положении относительно гонады — структуры, содержащей яйцеклетки.
Если яйцевод разовьется в неправильном месте, то гонада не будет получать сперму
и яйцеклетки никогда не будут оплодотворены. Позиционная информация, необхо-
димая предшествующим клеткам яйцевода, предоставляется клеткой внутри гонады,
названной опорной клеткой (рис. 14.31). Важное значение опорной клетки было про-
демонстрировано в ходе экспериментов, в которых она искусственно разрушалась в за-
родыше червя: при отсутствии опорной клетки яйцевод не развивается. Из этого можно
заключить, что опорная клетка выделяет некое внеклеточное сигнальное соединение,
Опорная
клетка
Рис. 14.31. Постулированная роль опорной клетки в предопределении пути дифференциации клетки
в ходе развития яйцевода у Caenorhabditis elegans. Думается, что выпуск сигнального соединения LIN-3
опорной клеткой наставляет клетку Рб.р (показана красным), наиболее близкую к опорной клетке, на
первостепенный путь дифференциации клетки яйцевода. Клетки Р5.р и Р7.р (показаны желтым) на-
ходятся дальше от опорной клетки и, таким образом, подвержены более низкой концентрации LIN-3,
в силу чего становятся второстепенными клетками яйцевода. Как описано в тексте, есть данные, что на-
правление второстепенных клеток на их пути дифференциации также зависит от сигналов, посылаемых
первостепенной клеткой яйцевода
576 Часть 3. Принципы функционирования геномов
которое побуждает клетки Р5.р, Рб.р и Р7.р дифференцироваться. Это сигнальное соеди-
нение представлено белком, называемым LIN-3 и кодируемым геном Пп-З.
Почему клетка Рб.р избирает первичный путь дифференциации клетки, тогда как
клетки Р5.р и Р7.р выбирают вторичные ее пути? На то есть две возможные причины.
Первая состоит в том, что соединение LIN-3 образует градиент концентрации и поэтому
оказывает различные воздействия на клетку Рб.р (расположенную наиболее близко
к нему) и на более отдаленные от него клетки Р5.р и Р7.р, как показано на рис. 14.31.
Данные в пользу этого представления получены в ходе исследований, показавших, что
выделенные клетки выбирают вторичный путь, когда подвергнуты низким уровням
сигнала LIN-3. Альтернативно, сигнал, который наставляет клетки Р5.р и Р7.р на их вто-
ричные пути, возможно, поступает не напрямую от опорной клетки, а через клетку Рб.р
в форме иного внеклеточного сигнального соединения, синтез которого клеткой Рб.р
включается ее активацией белком LIN-3. Эта гипотеза подтверждается ненормальными
признаками, проявляемыми некоторыми мутантами, у которых на развитие яйцевода
направляется более трех клеток. У таких мутантов есть более одной первичной клетки,
но каждая из них неизменно окружена двумя вторичными клетками, что позволяет
предположить, что в живом черве выбор клеткой вторичного пути дифференцировки
зависит от присутствия смежной с ней первичной клетки.
Есть и другие поучительные особенности развития яйцевода у С. elegans. Первая
из них заключается в том, что процесс передачи сигналов, который направляет клетку
Рб.р на её первичный путь дифференцировки, имеет много подобий с системой передачи
сигналов MAP-киназы у позвоночных животных (см. рис. 14.10). Рецептор клеточной
поверхности для сигнального соединения LIN-3 — протеинкиназа, названная LET-23, ко-
торая, будучи активирована связыванием с LIN-3, инициирует череду внутриклеточных
реакций, ведущую к активации белка, подобного MAP-киназе, который, в свою очередь,
включает целое разнообразие активаторов транскрипции. К сожалению, целевые гены
еще не были определены как в первичных, так и во вторичных предшествующих клетках
яйцевода, но система открыта для дальнейшего изучения.
Вторая примечательная особенность состоит в том, что, наряду с сигналом актива-
ции от опорной клетки в форме LIN-3, предшествующие клетки яйцевода подвержены
также дезактивирующим воздействиям второго сигнального соединения, выделяемого
подкожной клеткой многоядерной оболочки, которая покрывает большую часть тела
червя. Этот репрессивный сигнал преодолевается положительными сигналами, которые
побуждают клетки Р5.р, Рб.р и Р7.р дифференцироваться, но предотвращает нежела-
тельную дифференциацию трех смежных клеток, РЗ.р, Р4.р и Р8.р, каждая из которых
может стать направленной на развитие яйцевода, если репрессивный сигнал даст сбой
(например, в мутантном черве).
В заключение приведем обобщенные понятия, рожденные в ходе изучения раз-
вития яйцевода С. elegans.
• В многоклеточном организме большое значение имеет позиционная информация:
организму нужно развивать правильные структуры и располагать их в соответ-
ствующих местах.
• Направление на дифференцировку небольшого числа клеток-предшественников
может привести к построению многоклеточной структуры.
• Передача межклеточных сигналов может быть основана на градиенте концентра-
ции и вызывать различные реакции в клетках, находящихся в различных позициях
по отношению к сигнализирующей клетке.
Глава 14. Регулирование активности генома 577
• Клетка может подвергаться воздействию конкурирующих сигналов, где один
сигнал уговаривает ее сделать одну вещь, а второй сигнал убеждает сотворить
нечто противоположное.
14.3.4. Развитие Drosophila melanogaster
Последний организм, развитие которого мы будем изучать,—Drosophila melanogaster.
История участия плодовой мушки в экспериментах начинается с 1910 г., когда Томас Хант
Морган впервые употребил этот организм в качестве модельной системы для генети-
ческих исследований. Морган обратил внимание на такие преимущества Drosophila,
как небольшой размер, позволяющий изучать большое число мушек в одном экспери-
менте, минимальные требования к пище (эти мухи обожают бананы) и присутствие
в природных популяциях случайных вариантов с легко опознаваемыми генетическими
признаками, такими как необычный цвет глаз. Морган не знал о других преимуще-
ствах — маленьком геноме (180 млн п. н.; см. табл. 7.2) и том факте, что выделение генов
облегчается наличием в слюнных железах «гигантских» хромосом. Они состоят из много-
численнных копий одной и той же молекулы ДНК, уложенных бок о бок, и показывают
картины полосчатости, которые могут быть соотнесены с физической картой каждой
хромосомы, чтобы точно определить позиции желаемых генов. Но Морган несомненно
предвидел, что Drosophila может стать важным организмом для исследования процессов
развития, — эта тема была ему столь же интересна, сколь интересует нас сегодня.
Главный вклад, который Drosophila внесла в наше понимание развития, заключается
в том, что она помогла установить механизм, посредством которого недифференциро-
ванный эмбрион приобретает позиционную информацию, которая в конечном счете
воплощается в построении сложных частей тела в положенных местах и в развитии
взрослого организма. Хотя в некоторых отношениях Drosophila весьма необычна органи-
зацией своего эмбриона (как мы увидим в следующем разделе), генетические механизмы,
которые определяют план мушьего тела, подобны таковым в других организмах, в том
числе у человека. Поэтому знание, почерпнутое у Drosophila, направило исследование
в те области развития человека, которые в течение длительного времени считались
недоступными. Повесть об этом мы начинаем с событий, которые происходят в раз-
вивающемся эмбрионе Drosophila.
Материнские гены устанавливают градиенты белка в эмбрионе дрозофилы
Необычная особенность раннего эмбриона Drosophila — то, что он состоит не из
большого числа клеток, как у большинства организмов, но вместо этого представляет
собой единое соклетие, содержащее большое количество цитоплазмы и многочислен-
ные ядра (рис. 14.32). Эта структура сохраняется, пока 13 последовательных кругов
деления ядер не произведут около 1500 ядер: только тогда отдельные одноядерные
клетки начинают появляться на внешней оболочке соклетия и образуют структуру,
названную бластодермой. Однако позиционная информация начинает определяться
еще до перехода к стадии бластодермы.
Первоначальная позиционная информация, необходимая эмбриону, — это опреде-
ление того, какой из концов является передним (антериорным), а который задним
(постериорным), а также подобная информация, касающаяся верхней (спинной, или
дорсальной) и нижней (брюшной, или вентральной) сторон. Эта информация обеспе-
чивается градиентами концентрации белков, которые устанавливаются в соклетии.
Большинство этих белков не синтезируются с генов в эмбрионе, но транслируются с мо-
578 Часть 3. Принципы функционирования геномов
Цитоплазма Ядра
1,25 часа
128 ядер
I Ядра
Рис. 14.32. Ранние стадии развития эмбриона Drosophila. Для
начала скажем, что эмбрион представляет собой отдельное
соклетие, содержащее постепенно увеличивающее число
ядер. Эти ядра мигрируют к периферии эмбриона после при-
близительно 2 часов, и в течение следующих 30 минут начина-
ют строиться клетки. Эмбрион имеет размер приблизительно
500 мкм в длину и 170 мкм в диаметре
Соклеточный эмбрион
Клетки
Клеточная бластодерма
2 часа
1500 ядер
2,5 часа
5000 клеток
Спинная
t
Передняя -* Задняя
I
Брюшная
лекул мРНК, вводимых в эмбрион матерью. Чтобы
увидеть, как такие гены материнского эффекта
работают, мы рассмотрим синтез Bicoid — одного
из четырех белков, участвующих в определении
передне-задней оси.
Ген bicoid транскрибируется в материнских
клетках-кормилицах, которые находятся в контакте
с яйцеклетками, и мРНК вводится в передний конец
неоплодотворенного яйца. Эта позиция определяет-
ся ориентацией яйцеклетки в яичниковом фоллику-
ле. мРНК bicoid остается в передней области яйце-
клетки, прикрепленной своей З'-нетранслируемой
областью к цитоскелету клетки. Она не транслиру-
ется немедленно, вероятно потому, что ее поли-А
хвост слишком короткий. Такой вывод был сделан, потому что трансляции, которая
происходит после оплодотворения яйца, предшествует продолжение поли-А хвоста
объединенными усилиями белков Cortex, Grauzone и Staufen, при этом все они синте-
зируются с генов яйцеклетки. После этого белок Bicoid диффундирует по соклетию,
устанавливая тем самым градиент концентрации — от наиболее высокой в переднем
конце до наиболее низкой в заднем конце (рис. 14.33).
В установление передне-заднего градиента вовлечены также продукты трех дру-
гих генов материнского эффекта. Это белки Hunchback, Nanos и Caudal. Все вводятся
в виде мРНК в переднюю область неоплодотворенной яйцеклетки. мРНК гена nanos
переносится к задней части яйца и прикрепляется к клеточному скелету в ожидании
трансляции. Молекулы мРНК генов hunchback и caudal равномерно распределяются по
всей цитоплазме, но их белки впоследствии образуют градиенты при соучастии белков
Bicoid и Nanos:
• Bicoid активирует ген hunchback в ядрах эмбриона, поставляет мРНК гена hunchback
в переднюю область и подавляет трансляцию материнской мРНК caudal. В результа-
Передняя
Задняя
Bicoid
Hunchback
в Nanos
Caudal
Рис. 14.33. Закладывание передне-задней оси в эмбрионе
Drosophila. Передне-задняя ось закладывается градиентами
белков Bicoid, Nanos, Caudal и Hunchback, как описано в тек-
сте. На этом рисунке градиенты концентрации обозначены
цветными полосами под контуром эмбриона*
* В настоящее время есть весьма подробные карты пространственного распределения сотен типов
мРНК, полученной гибридизацией in situ, для разных этапов раннего развития эмбриона. Эти изображения
доступны на сайте http://www.fruitfly.org/ex/Imaging.htm.
Глава 14. Регулирование активности генома 579
те этого в передней области возрастает концентрация белка Hunchback и снижается
концентрация белка Caudal;
• Nanos подавляет трансляцию мРНК гена hunchback, внося еще больший вклад
в установление передне-заднего градиента белка Hunchback.
В конечном итоге устанавливается градиент белков Bicoid и Hunchback, с большей
концентрацией в переднем конце, а также белков Nanos и Caudal, с более высоким со-
держанием в заднем конце (см. рис. 14.33). Градиент дополняется белком Torso — еще
одним продуктом гена материнского эффекта, который накапливается в самых переднем
и заднем концах. Подобные события дают спинно-брюшной градиент, преимущественно
из белка по имени Dorsal.
Каскад экспрессии гена преобразует позиционную информацию в схему сег-
ментации
План тела взрослой мухи, равно как и личинки, построен из ряда сегментов, при
этом каждый наделен своей структурной ролью. Это наиболее четко прослеживается
в грудной клетке, которая имеет три сегмента, где каждый несет одну пару ног, и в жи-
воте, который состоит из восьми сегментов, но также верно и для головы, даже при том,
что в голове сегментированная структура менее очевидна (рис. 14.34). Цель развития
эмбриона, таким образом, состоит в превращении в молодую личинку с правильной
схемой сегментации.
Градиенты, установленные в эмбрионе продуктами генов материнского эффек-
та, — первая стадия в формировании схемы сегментации. Эти градиенты обеспечива-
ют внутреннюю часть эмбриона основным количеством позиционной информации,
каждая точка в соклетии теперь имеет свою собственную уникальную химическую
сигнатуру, определяемую относительными количествами продуктов различных генов
материнского эффекта. Эта позиционная информация уточняется за счет экспрессии
gap-генов. Три из белков передне-заднего градиента — Bicoid, Hunchback и Caudal —
являются активаторами транскрипции, которые нацеливают gap-гены на ядра, теперь
выстроенные с внутренней стороны поверхности эмбриона (см. рис. 14.32). Род gap-генов,
экспрессируемых в каждом конретном
ядре, зависит от относительных концен-
траций градиентных белков и, следова-
тельно, от позиции ядра относительно
передне-задней оси. Некоторые gap-гены
активируются напрямую белками Bicoid,
Hunchback и Caudal, примеры представ-
лены генами buttonhead, empty spiracles
и orthodenticle, которые активируются
белком Bicoid. Другие gap-гены вклю-
чаются косвенно, как это имеет место
с huckebein и tailless, которые реагируют
Рис. 14.34. Схема сегментации взрослой
Drosophila melanogaster. Учтите, что голова так-
же сегментирована, но схема ее сегментации
трудно распознаваема по морфологии взрослой
мухи
580 Часть 3. Принципы функционирования геномов
голова ; грудь ; живот
Huckebein
Tailless МВННВ
Orthodenticle :
Empty spiracles
Buttonhead
Giant
Kruppel
Knirps
Рис. 14.35. Роль продуктов gap-
генов в предоставлении позицион-
ной информации в ходе развития
эмбриона Drosophila melanogaster.
Градиент концентрации продукта
каждого gap-гена обозначен цвет-
ными полосами. Отмечены части
эмбриона, которые дают начало
областям головы, груди и живота
взрослой мухи
на активаторы транскрипции, включаемые белком Torso. Имеют место также и ре-
прессирующие воздействия (например, белок Bicoid подавляет экспрессию гена knirps),
а продукты gap-генов регулируют свою собственную экспрессию различными способами.
В результате столь сложного взаимодействия эмбрион получает более точную позици-
онную информацию, отныне заложенную в относительных концентрациях продуктов
gap-генов (рис. 14.35).
Следующий набор генов, которые будут активированы, а именно генов pair-rule,
устанавливает основную схему сегментации. Транскрипция этих генов реагирует на
относительные концентрации продуктов gap-генов и происходит в ядрах, которые те-
перь заключены в клетки. Продукты генов pair-rule поэтому не диффундируют через
соклетие, а остаются в пределах клеток, которые их экспрессируют. В результате этого
эмбрион можно теперь рассматривать как включающий ряд полос, где каждая полоса
состоит из набора клеток, экспрессирующих специфический ген pair-rule. В следующем
круге активации генов включаются гены полярности сегментов, придающие полосам
большую определенность, устанавливая размеры и точные местоположения того, что
в конечном счете будет сегментами личинки мухи. Итак, мы постепенно преобразова-
ли неточную позиционную информацию градиентов продуктов генов материнского
эффекта во вполне определенную схему сегментации.
Род сегмента определяется гомеозисными избирательными генами
Гены pair-rule и гены полярности сегментов устанавливают схему сегментации
эмбриона, но не определяют род отдельных сегментов. Это прерогатива гомеозис-
ных избирательных генов, которые были впервые открыты благодаря причудливым
эффектам, которые мутации в них оказывают на облик взрослой мухи. Мутация гена
antennapedia, например, преобразует сегмент головы, из которого обычно формируется
стяжка, в такой, из которого образуется нога, так что мутантная муха имеет пару ног, там
где должны быть ее антенны. Первые генетики были очарованы этими чудовищными
гомеозисными мутантами, и многие из них были собраны в течение нескольких первых
десятилетий двадцатого столетия.
Генетическое картирование гомеозисных мутаций показало, что избирательные
гены образуют две группы на хромосоме 3. Эти группы назвали комплексом Antennapedia
(ANT-C), который содержит гены, участвующие в определении сегментов головы и груди,
и комплексом Bithorax (ВХ-С), который содержит гены сегментов живота (рис. 14.36).
Некоторые дополнительные (не избирательные) гены развития типа bicoid также рас-
Глава 14. Регулирование активности генома 581
Рис. 14.36. Генные комплексы Antennapedia и Bithorax Drosophila
melanogaster. Оба комплекса расположены на хромосоме 3 пло-
довой мушки, ANT-C выше ВХ-С. Гены обычно рисуют в указанном
порядке, хотя это означает, что они транскрибируются справа на-
лево. Рисунок не отражает фактическую длину генов. Полные на-
звания генов следующие: lab — labial palps; pb — proboscipedia;
Dfd — Deformed; Scr — Sex combs reduced; Antp — Antennapedia;
Ubx — Ultrabithorax; abdA — abdominal A; Abd В — Abdominal B.
В комплексе ANT-C неизбирательные гены zerknullt и bicoid обна-
руживаются между pb и Dfd, a fushi tarazu лежит между Scr и Antp
Комплекс Antennapedia (ANT-C)
lab pb Dfd Scr Antp
Комплекс Bithorax (BX-C)
Ubx abdA AbdB
положены в комплексе ANT-C. Одна интересная особенность групп ANT-C и ВХ-С, кото-
рую все еще не удается объяснить, заключается в том, что порядок генов соответствует
порядку сегментов в мухе: первый ген в ANT-C представлен геном labial palps, который
управляет крайним передним сегментом мухи, а последний ген в ВХ-С представлен
геном Abdominal В, который определяет оконечный задний сегмент.
В каждом сегменте экспрессируется надлежащий гомеозисный ген, потому что
активация каждого из них реагирует на позиционную информацию, представленную
распределениями продуктов gap-генов и генов pair-rule. Продукты гомеозисных генов
сами по себе являются активаторами транскрипции, каждый содержит гомеодомен-
ный вариант ДНК-связывающей структуры спираль-изгиб-спираль (раздел 11.1.1).
Каждый продукт избирательного гена, возможно в соединении с соактиватором типа
Extradenticle, включает набор генов, необходимых для инициации развития заданного
сегмента. Поддержание дифференцированного состояния обеспечивается частично
репрессивным воздействием, которое каждый продукт избирательного гена оказывает
на экспрессию других избирательных генов, и частично работой Polycomb, который,
как мы убедились в разделе 14.2.2, строит неактивный хроматин над избирательными
генами в тех клетках, в которых они не экспрессируются.
Гомеозисные гены — неотъемлемые составляющие системы развития выс-
ших эукариотов
Гомеодомены различных гомеозисных генов Drosophila поразительно схожи. Это на-
блюдение навеяло исследователям 1980-х гг. мысль о поиске других гомеозисных генов,
используя гомеодомен в качестве зонда в экспериментах с гибридизацией. Поиск начали
с генома Drosophila и выделили несколько ранее неизвестных содержащих гомеодомен
генов. Они оказались не гомеозисными генами, но генами другого типа, кодирующими
активаторы транскрипции, участвующие в развитии. Примеры включают гены pair-rule
even-skipped и fushi tarazu, а также ген полярности сегментов engrailed.
Когда были прозондированы геномы других организмов и стало понятно, что гомео-
домены присутствуют в генах самых разнообразных животных, в том числе и человека, по
научным кругам прокатилась настоящая сенсация. Еще более неожиданным было открытие,
что некоторые гомеодоменные гены в этих других организмах являются гомеозисными
генами, организованными в группы, подобные ANT-C и ВХ-С, и что эти гены имеют экви-
валентные функции, как у вариантов, присущих Drosophila, и определяют общее строение
организма. Например, в результате мутации в гене НохС8 мыши рождается животное, которое
имеет дополнительную пару ребер из-за превращения поясничного позвонка (обычно рас-
положенного в нижней части спины) в грудной позвонок (на котором растут ребра). Другие
мутации Нох у животных ведут к деформациям конечностей, таким как отсутствие задней
лапы или наличие лишних пальцев на руках или ногах.
582 Часть 3. Принципы функционирования геномов
lab pb Dfd Scr Antp UbxabdAAbdB
HOM-C ..ДВ-Л—Я-М-ВЫВ-В-а...
НохА ..
НохВ
НохС
HoxD
Рис. 14.37. Сравнение между генным комплексом HOM-C Drosophila и четырьмя группами Нох позво-
ночных. Гены, которые кодируют белки с эволюционно связанными структурами и функциями, обо-
значены цветами. Более подробные сведения относительно эволюции групп Нох содержатся в разделе
18.2.1. Рисунок не отражает фактическую длину генов. Названия генов в комплексе НОМ-С следующие:
lab — labial palps; pb - proboscipedia; Dfd — Deformed; Scr — Sex combs reduced; Antp — Antennapedia;
Ubx — Ultrabithorax; abdA — abdominal A; Abd В — Abdominal В
Теперь мы смотрим на группы ANT-C и ВХ-С гомеозисных генов Drosophila как на
две части единого комплекса, обычно именуемого комплексом гомеозисных генов,
или НОМ-С. У позвоночных животных есть четыре группы гомеозисных генов, назван-
ные HoxA-HoxD. Когда эти четыре группы выравниваются друг с другом и с НОМ-С
(рис. 14.37), обнаруживаются подобия между генами в эквивалентных позициях, так
что эволюционная история групп гомеозисных генов может быть прослежена от на-
секомых до человека (см. раздел 18.2.1). Как и у Drosophila, порядок генов в группах
у позвоночных отражает порядок предписываемых этими генами структур в общем
строении взрослого организма. Это хорошо заметно у группы НохВ мышей, которая
управляет развитием нервной системы (рис. 14.38). Примечательное заключение — то,
что на этом фундаментальном уровне процессы развития у плодовых мушек и других
«простых» эукариотов являются подобными процессам, протекающим у людей и других
«сложных» организмов. Открытие того факта, что исследования плодовых мушек имеют
прямое отношение к развитию человека, простирает перед нами широкие перспективы
для будущих исследований.
П С р1 р2 рЗ р4 рб рб р7 р8 Спинной мозг
НохВ1
НохВ2 Bi
НохВЗ «
НохВ4
НохВ5-8 ВВИВ
НохВЭ
Рис. 14.38. Специализация нервной системы мыши определяется гомеозисными генами группы НохВ.
Нервная система показана схематично и позиции, указуемые отдельными генами группы НохВ (с HoxBl
по НохВЭ), обозначены зелеными полосками. Компоненты нервной системы следующие: П — передний
мозг; С — средний мозг; р1-р8 — ромбомеры 1-8; и, наконец, спинной мозг. Ромбомеры — доли за-
днего мозга, наблюдаемые в ходе развития
Глава 14. Регулирование активности генома 583
Кольцо 4 - плодолистики
— Кольцо 3 - тычинки
— Кольцо 2 - лепестки
Г -----Кольцо 1 - чашелистики
Рис. 14.39. Цветки выстраиваются из четырех
концентрических колец
Гомеозисные гены лежат и в основе развития растений
Действенность Drosophila в качестве модельной системы развития распространя-
ется даже за пределы категории позвоночных животных. Процессы развития у расте-
ний во многих отношениях сильно отличаются от таковых у плодовых мушек и других
животных, но на генетическом уровне есть некоторые подобия, достаточные для того,
чтобы знания, полученные о развитии Drosophila, имели ценность в интерпретации
результатов подобного исследования, выполненного над растениями. В частности, осо-
знание того, что ограниченное число гомеозисных генов управляет общим строением
организма Drosophila, привело к модели развития растений, которая постулирует, что
структура цветка определяется небольшим числом гомеозисных генов.
Все цветы устроены аналогичным образом, будучи построены из четырех концен-
трических колец, каждому из которых соответствует свой цветковый орган (рис. 14.39).
Внешнее кольцо, номер 1, содержит чашелисти-
ки, представляющие собой видоизмененные
листья, которые окутывают и защищают поч-
ку на ранних стадиях ее развития. Следующее
кольцо, номер 2, содержит отличительные ле-
пестки и внутри них — кольца номер 3 (тычин-
ки, мужские репродуктивные органы) и номер
4 (плодолистики, женские репродуктивные
органы).
Исследования развития растений в основ-
ном были выполнены надАпйггЫпит (львиный
зев) и Arabidopsis thaliana — резушкой Таля, ко-
торая была принята в качестве модельного вида отчасти потому, что имеет геном лишь
125 млнп.н. (см. табл. 7.2), один из наименьших известных геномов среди цветковых
растений. Хотя эти растения и не содержат гомеодоменных белков, они имеют гены,
которые, будучи мутированы, ведут к гомеозисным изменениям в строении цветка,
таким как замена чашелистиков плодолистиками. Анализ таких мутантов привел к «мо-
дели АВС», которая гласит, что есть гомеозисные гены трех типов — А, В и С, — которые
управляют развитием цветка следующим образом:
• кольцо 1 определяется генами A-типа: примеры у Arabidopsis — apetalal
и apetala2;
• кольцо 2 определяется генами А, действующими сообща с генами В, примеры по-
следних включают apetala3 и pistillata;
• кольцо 3 определяется генами В плюс ген С agamous;
• кольцо 4 определяется геном С, действующим самостоятельно.
В соответствии с ожиданиями, произросшими на почве работ с Drosophila, продук-
ты гомеозисных генов А, В и С являются активаторами транскрипции. Все, кроме белка
APETALA2, содержат один и тот же ДНК-связывающий домен, блок MADS, который обна-
ружен также и в других белках, участвующих в развитии растений, таких как SEPALLATA1,
2 и 3, которые работают вместе с белками А, В и С на поприще определения подробной
структуры цветка. Другие компоненты системы развития цветка включают по крайней
мере один главный ген (названный floricaula у Antirrhinum и leafy у Arabidopsis), который
управляет переключением с вегетативного на репродуктивный рост, инициируя раз-
витие цветка, и играет роль также в установлении картины экспрессии гомеозисных
584 Часть 3. Принципы функционирования геномов
генов. У Arabidopsis есть также ген, названный curly leaf, продукт которого действует
как Polycomb Drosophila (раздел 14.2.2), поддерживая дифференцированное состояние
каждой клетки путем подавления тех гомеозисных генов, которые являются неактив-
ными в данном кольце.
Заключение
Пути экспрессии отдельных генов состоят из множества шагов, на которых может
осуществляться регулирование, но ключевые механизмы управления действуют в мо-
мент инициации транскрипции. Временные изменения в картинах экспрессии генома
происходят преимущественно в ответ на внешние раздражители, которые влияют на
транскрипцию отдельных генов. Некоторые внеклеточные сигнальные соединения
импортируются в клетку и напрямую влияют на транскрипцию, пример представлен
лактоферрином у млекопитающих. Стероидные гормоны также поступают в клетку, но
влияют на экспрессию генома через рецепторные белки, которые служат активаторами
транскрипции. При катаболитной репрессии оперонов у бактерий глюкоза влияет на экс-
прессию целого разнообразия генов, участвующих в усвоении сахара, косвенно управляя
уровнем циклического АМФ в клетке, который, в свою очередь, влияет на активность
активатора транскрипции, названного белком-активатором катаболитных оперонов.
Другие пути передачи сигналов опосредствуются рецепторами клеточной поверхности,
многие из которых димеризуются в ответ на внеклеточный сигнал, открывая тем самым
путь передачи сигнала, который ведет к геному. Один из таких примеров — путь МАР-
киназы, но есть несколько других, включая такие, в которых замешаны активаторы
транскрипции, получившие название STAT. Некоторые пути передачи сигналов ис-
пользуют вторичные посредники (например, циклических нуклеотидов и ионов каль-
ция), которые влияют на ряд отправлений клетки, в том числе на экспрессию генома.
Постоянные и полупостоянные изменения в экспрессии генома могут быть вызваны
перестройками генома, что и происходит в процессе переключения типа спаривания
у дрожжей и порождения разнообразия иммуноглобулинов и рецепторов Т-клеток у по-
звоночных животных. Также могут быть произведены постоянные и полупостоянные
изменения такими белками, как Polycomb Drosophila melanogaster, который вызывает
образование гетерохроматина над областями хромосомы, которые нужно подавить.
Нашему пониманию генетической подоплеки развития помогло изучение модельных
систем в относительно простых организмах, таких как бактерии, Caenorhabditis elegans
и Drosophila melanogaster. Лизогенный цикл инфицирования бактериофага X указывает
пути, которыми простые генетические переключатели могут определять, по какому из
двух путей развития пойдет клетка или организм, а исследования спорообразования
у Bacillussubtilis иллюстрировали, как могут быть вызваны зависящие от времени изме-
нения в экспрессии генома и как межклеточная передача сигналов может регулировать
путь дифференцировки. Механизмы определения пути дифференцировки клетки были
открыты в ходе исследований развития яйцевода у С. elegans. Наиболее информативным
путем для генетики развития явился эмбриогенез плодовой мушки, изучение которой
показало, как сложный план тела может определяться управляемыми картинами экс-
прессии генома. В ходе этой работы был обнаружен факт существования гомеозисных
генов, которые управляют процессами развития не только у мух, но также у позвоночных
животных и растений.
Глава 14. Регулирование активности генома 585
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
14.1 *. Которая из следующих формулиро-
вок служит определением термина
«дифференцировка»?
а. Изменения в экспрессии генома,
которые не изменяют протеом
клетки.
Ь. Непостоянные изменения в ак-
тивности генома клетки в ответ
на внеклеточные факторы.
с. Согласованный ряд изменений,
которые происходят в ходе жиз-
ненного цикла клетки.
d. Принятие специализированной
физиологической роли клеткой.
14.2 . Какая из контрольных точек для
регулирования экспрессии генома
является самой важной?
а. Инициация транскрипции.
Ь. Процессинг транскриптов.
с. Инициация трансляции.
d. Деградация белков и молекул РНК.
14.3 *. Какое из следующих событий не слу-
жит механизмом, которым сигналь-
ная молекула, как известно, влияет
на экспрессию генома, будучи им-
портирована в клетку?
а. Некоторые сигнальные молеку-
лы метилируют последователь-
ности ДНК, с тем чтобы подавить
определенные гены.
Ь. Некоторые сигнальные молекулы
представлены белками, которые
выполняют функцию регулято-
ров экспрессии генома.
с. Некоторые сигнальные молекулы
напрямую влияют на активность
регуляторных белков в клетке.
d. Некоторые сигнальные молекулы
влияют на активность регулятор-
ных белков в клетке косвенно, че-
рез промежуточные молекулы.
14.4 . Как стероидные гормоны, такие как
эстроген, модулируют экспрессию
генома в восприимчивых клетках?
а. Связываясь с последовательно-
стями энхансеров.
Ь. Связываясь с рецепторами в ци-
топлазме, которые затем мигри-
руют в ядро, где они связываются
с ДНК, с тем чтобы регулировать
экспрессию генома.
с. Связываясь с рецепторами в ядре,
которые активируются и затем
связываются с ДНК, чтобы регу-
лировать экспрессию генома.
d. Связываясь с рецепторами в кле-
точной мембране (сигнал после
этого передается в ядро через
путь передачи сигналов).
14.5* . В путях передачи сигналов какого
типа задействованы белки STAT?
а. Эти пути имеют один шаг между
рецептором и геномом.
Ь. Эти пути имеют несколько шагов
между рецептором и геномом.
с. Эти пути используют вторичные
посредники, чтобы передать сиг-
нал к геному.
d. Эти пути активируют рецептор,
который затем перемещается
в ядро, чтобы регулировать экс-
прессию генома.
14.6. Каков самый распространенный тип
ковалентной модификации, которая
активирует белки в путях передачи
сигналов?
а. Ацетилирование.
Ь. Гликозилирование.
с. Метилирование.
d. Фосфорилирование.
14.7. * Что собой представляют молекулы
вторичных посредников?
586 Часть 3. Принципы функционирования геномов
а. Это гормоны, которые открывают
путь передачи сигналов.
Ь. Это рецепторы, которые связывают-
ся с гормонами и активируют путь.
с. Это внутренние молекулы, которые
передают сигнал внутри клетки.
d. Это активаторы транскрипции, ко-
торые действуют в конце пути.
14.8. Процесс переключения типа спари-
вания, который происходит у дрож-
жей, служит примером которого из
следующих событий?
а. Альтернативного сплайсинга.
Ь. Изменения в петле обратной связи.
с. Изменения в схемах метилирова-
ния ДНК.
d. Изменения из-за физической пе-
рестройки генома.
14.9* . Какие изменения происходят
в В-клетках, когда они переключа-
ются с синтеза иммуноглобулинов
IgM или IgD на синтез иммуногло-
булина IgG?
а. Это изменение совершается за
счет альтернативного сплайсин-
га РНК-транскриптов.
Ь. Это изменение происходит в про-
теоме, поскольку постоянные об-
ласти IgM/lgD удаляются из белка
IgG протеолитическим расщепле-
нием.
с. Это изменение происходит в ге-
номе, поскольку гены, кодирую-
щие постоянные области для IgM
и IgD, удаляются белками RAG1
и RAG2.
d. Это изменение происходит в ге-
номе, поскольку гены, кодирую-
щие постоянные области для IgM
и IgD, удаляются независимо от
белков RAG.
14.10. В чем состоит функция белков
Polycomb у Drosophila:
а. Спирализация хроматина, чтобы
вызвать подавление гена, которое
передается дочерним клеткам.
Ь. Спирализация хроматина, чтобы
поддерживать подавление гена,
передаваемое дочерним клеткам.
с. Спирализация хроматина, чтобы
вызвать подавление гена, которое
не передается дочерним клеткам.
d. Спирализация хроматина, чтобы
поддерживать подавление гена,
которое не передается дочерним
клеткам.
14.11* . Посредством какого механизма
петли обратной связи могут вызвать
долгосрочные изменения в экспрес-
сии генома?
а. Регуляторный белок активирует
свою собственную транскрипцию
и, таким образом, непрерывно
экспрессируется.
Ь. Регуляторныйбелокподавляетсвою
собственную транскрипцию, так что
он постоянно подавляется.
с Регуляторный белок активирует
другой белок, который стимулирует
экспрессию регуляторного белка.
d. Всех описанных выше.
14.12. Как активируется путь спорообра-
зования у Bacillus?
а. Нехватка питательных веществ
сигнализирует активацию гена,
кодирующего белок SpoOA.
b. Недостаток питательных веществ
сигнализирует активацию белка
SpoOA путем протеолитического
расщепления.
с. Недостаток питательных веществ
сигнализирует активацию белка
SpoOA посредством ацетилиро-
вания.
d. Недостаток питательных веществ
сигнализирует активацию белка
SpoOA фосфорилированием.
14.13* . Что мешает клеткам, смежным
с предшествующими клетками яй-
цевода у С. elegans, дифференциро-
ваться в клетки яйцевода?
а. Эти клетки находяся слишком да-
леко от опорной клетки, чтобы
получить сигнал LIN-3.
Глава 14. Регулирование активности генома 587
Ь. Эти клетки не обладают способ-
ностью связывать сигнал LIN-3
и реагировать на него.
с. Эти клетки получают сигнальное
соединение от подкожной клет-
ки, которое дезактивирует сигнал
LIN-3.
d. Эти клетки уплотнили свой хро-
матин в областях, чувствитель-
ных к сигналу LIN-3.
14.14. Какова структура соклетия эмбрио-
на Drosophila?
а. Сильно уплотненная масса не-
дифференцированных клеток.
Ь. Продолговатая структура, кото-
рая содержит градиент концен-
трации белков, причастных к раз-
витию.
с. Море цитоплазмы и масса ядер.
d. Смесь диплоидных и гаплоидных
клеток, производимых митотиче-
Вопросы открытого типа
14.1 *. Вкратце перечислите различия меж-
ду дифференцировкой и развитием
и опишите основу этих отличий.
14.2 . Почему у одноклеточных организмов
изменения в условиях наличия и со-
става питательных веществ, как пра-
вило, вызывают более значительные
изменения в активности генома, чем
у многоклеточных организмов?
14.3 *. Объясните зависимость между пере-
носом глюкозы и уровнями цАМФа
у Е. coli.
14.4 . Как фосфорилируются белки STAT,
если рецептором является не тиро-
зинкиназа?
14.5 *. В чем состоит роль МАР-киназы
в процессе регулирования экспрес-
сии генома?
14.6 . Чем занимаются белки Ras в путях
передачи сигналов?
14.7 *. Какова основа переключения клас-
сов, которое происходит с некоторы-
ми В-лимфоцитами?
скими и мейотическими деления-
ми клеток.
14.15 *. Какие гены Drosophila обусловли-
вают самоопределение сегментов
личинки плодовой мушки?
а. gap-Гены.
Ь. Гены pair-rule.
с. Гены полярности сегментов.
d. Гомеозисние гены.
14.16 . Если мутация с потерей функции про-
исходит в генах В-типа Arabidopsis, то
каков будет состав колец цветка (от
кольца 1 к кольцу 4)?
а. Чашелистики-лепестки-тычин-
ки-плодолистики.
Ь. Чашелистики-чашелистики-ты-
чинки-плодолистики.
с. Чашелистики-чашелистики-пло-
долистики-плодолистики.
d. Лепестки-лепестки-тычинки-
тычинки.
14.8. За счет чего мышечные клетки оста-
ются дифференцированными на мы-
шечные клетки?
14.9* . Во время спорообразования у Bacillus
субъединицы оЕ и oF присутствуют
и в материнской клетке, и в проспо-
ре. Каким образом субъединица qf
активируется в проспоре?
14.10. Как опорная клетка С. elegans по-
буждает предшествующие клетки
яйцевода дифференцироваться
в клетки яйцевода? Почему клетки-
предшественники яйцевода следуют
различными путями после получе-
ния сигнала от опорной клетки?
14.11* . Как градиент концентрации белка
Bicoid устанавливается в соклетии
эмбриона Drosophila?
14.12. Каким образом мутации в комплек-
се генов ANT-C Drosophila и генов Нох
мыши снабдили исследователей ин-
формацией о функциях этих генов?
588 Часть 3. Принципы функционирования геномов
Изыскательские задачи
14.1 *. Расскажите о том, насколько иссле-
дования передачи сигналов расши-
рили наше понимание ненормаль-
ных биохимических отправлений,
которые лежат в основе развития
рака.
14.2 . Рассмотрите вопрос о влиянии пере-
дачи сигналов через вторичные по-
средники на регулирование актив-
ности генома.
14.3 *. Являются ли Caenorhabditis elegans
и Drosophila melanogaster хороши-
Тесты по рисункам
ми модельными организмами для
изучения развития высших эука-
риотов?
14.4 . Есть л и какое-то преимущество в ис-
пользовании модельных организ-
мов для изучения развития высших
эукариотов?
14.5 *. Какими основными особенностями
должен обладать идеальный мо-
дельный организм для изучения
развития высших эукариотов?
14.1 *. График показывает, что глюкоза
потребляется Е. coli до лактозы,
когда клетки выращивают в сре-
де, содержащей оба сахара. Какой
термин употребляется для описа-
ния этого явления? Опишите ме-
ханизм, который лежит в основе
этого процесса.
14.2 . На рисунке изображено изменение локуса МАТ с генотипа МАТа на генотип
МАТа. Какой термин используется для описания такого рода изменений и каков
механизм, посредством которого такое изменение происходит?
HMLa МАТа HMRa
В—>. Хромосома III
Эндонуклеаза НО
HMLa
HMRa
Превращение гена
Двунитевой разрез
HMLa
МАТа
HMRa
Глава 14. Регулирование активности генома 589
14.3 *. Обсудите процессы, благодаря которым В-клетками производится огромное
многообразие иммуноглобулинов.
I
Иммуноглобулин D
4
1 2
I
Иммуноглобулин М
14.4 . На рисунке показано распределение белков в ходе развития зародыша Drosophila.
Гены какого типа кодируют эти белки? Какие белки регулируют экспрессию этих
генов?
голова ; грудь ; живот
Huckebein
Tailless НМН
Orthodenticle
Empty spiracles
Buttonhead
Giant
Kriippel
Knirps
590 Часть 3. Принципы функционирования геномов
Дополнительная литература
Импортируемые внеклеточные сигнальные соединения
He,J. and Furmanski, Р. (1995) Sequence specificity and transcriptional activation in
the binding of lactoferrin to DNA. Nature 373: 721-724.
Tsai, M.-J. and O’Malley, B. W. (1994) Molecular mechanisms of action of steroid/thyroid
receptor superfamily members. Annu. Rev. Biochem. 63: 451-486.
Winge, D. R., Jensen, L T. and Srinivasan, C. (1998) Metal ion regulation of gene expression
in yeast. Curr. Opin. Chem. Biol. 2: 216-221.
Передача сигналов рецепторами клеточной поверхности
Horvath, С. М. (2000) STAT proteins and transcriptional responses to extracellular signals.
Trends Biochem. Sci. 25: 496-502.
Karin, M. and Hunter,T. (1995) Transcriptional control by protein phosphorylation:
signal transmission from the cell surface to the nucleus. Curr. Biol. 5: 747-757.
Maruta, H. and Burgess, A. W. (1994) Regulation of the Ras signaling network. Bioessays
16: 489-496.
Robinson, M. J. and Cobb,M.H. (1997) Mitogen-activated kinase pathways. Curr. Opin.
Cell Biol. 9:180-186.
Schlessinger, J. (1993) How receptor tyrosine kinases activate Ras. Trends Biochem. Sci.
18: 273-275.
Spiegel, S., Foster, D. and Kolesnick, R. (1996) Signal transduction through lipid second
messengers. Curr. Opin. Cell Biol. 8:159-167.
Whitman, M. (1998) Feedback from inhibitory SMADs. Nature 389: 549-551.
Перестройки генома
Alt,F.W., Blackwell, T. К. and Yancopoulos,G.D. (1987) Development of the primary
antibody repertoire. Science 238:1079-1087. Generation of immunoglobulin diversity.
Nasmyth, K, and Shore, D. (1987) Transcriptional regulation in the yeast life cycle. Science
237:1162-1170. Yeast mating-type switching.
Polycomb
Chan,C.S., Rastelli, L. and Pirrotta, V. (1994) A Polycomb response element in the Ubx
gene that determines an epigenetically inherited state of repression. EMBO J. 13: 2553-2564.
Dejardin, J., Rappailles,A., Cuvier, O., Grimaud,C., Decoville, M., Locker, D. and
Cavalli,G. (2005) Recruitment of Drosophila Polycomb group proteins to chromatin by DSP1.
Nature 434: 533-538.
Петли обратной связи
Popperl, H., Bienz, M., Studer, M., Chan, S. К., Aparicio, S., Brenner, S., Mann, R. S. and
Krumlauf,R. (1995) Segmental expression of HoxB-1 is controlled by a highly conserved
autoregulatory loop dependent upon exd/pbx. Cell 81:1031-1042.
Regulski,M., Dessain, S., McGinnis, N. and McGinnis, W. (1991) High affinity binding
sites for the Deformed protein are required for the function of an autoregulatory enhancer of
the deformed gene. Genes Devel. 5: 278-286.
Глава 14. Регулирование активности генома 591
Спорообразование у В. subtilis
ErringtonJ. (1996) Determination of cell fate in Bacillus subtilis. Trends Genet. 12:
31-34.
Sonenshein,A.L. (2000) Control of sporulation initiation in Bacillus subtilis. Curr. Opin.
Microbiol. 3: 561-566.
Stragier,P. and Losick,R. (1996) Molecular genetics of sporulation in Bacillus subtilis.
Annu. Rev. Genet 30: 297-341.
Развитие яйцевода у C. elegans
Aroian, R. V., Koga, M., Mendel, J. E., Ohshima, Y. and Sternberg, P. W. (1990) The let-23
gene necessary for Caenorh abditis elegans vulval induction encodes a tyrosine kinase of the
EGF receptor subfamily. Nature 348: 693-699.
Katz, W. S., Hill, R. J., Clandinin, T. R. and Sternberg, P. W. (1995) Different levels of the
C. elegans growth factor LIN-3 promote distinct vulval precursor fates. Cell 82: 297-307.
Kornfeld, K. (1997) Vulval development in Caenorhabditis elegans. Trends Genet. 13:
55-61.
Labouesse, M. and Mango, S. E. (1999) Patterning the C. elegans embryo: moving beyond
the cell lineage. Trends Genet 307-313. Reviews the developmental pathways ofC. elegans.
Sharma-Kishore, R., White, J.G., Southgate, E. and Podbilewicz, B. (1999) Formation
of the vulva in Caenorhabditis elegans-. a paradigm for organogenesis. Development 126:
691-699.
Эмбриогенезис плодовой мушки и гомеозисные избирательные гены позво-
ночных
Ingham, Р. W. (1988) The molecular genetics of embryo pattern formation in Drosophila.
Nature 335: 25-34.
Krumlauf, R. (1994) How genes in vertebrate development. Cell 78:191-201.
Maconochie, M., Nonchev, S., Morrison, A. and Krumlauf, R. (1996) Paralogous Hox
genes: function and regulation. Annu. Rev. Genet. 30: 529-556. Describes homeotic selector
genes in vertebrates.
Manowald, A.P. and Hardy, P. A. (1985) Genetics of Drosophila embryogenesis. Annu.
Rev. Genet 19:149-177.
Развитие растений
Goodrich, J., Puangsomlee, P., Martin, M., Long, D., Meyerowitz, E. M. and Coupland, G.
(1997) A Polycombgroup gene regulates homeotic gene expression in Arabidopsis. Nature
386: 44-51.
Ma, H. (1998) To be, or not to be, a flower - control of floral meristem identity. Trends
Genet 14: 26-32.
Parcy, F., Nilsson, O., Busch, M. A., Lee, I. and Weigel, D. (1998) A genetic framework
for floral patterning. Nature 395: 561-566.
Часть 4
Механизмы
репликации
и эволюции геномов
Часть 4 — Механизмы репликации
и эволюции геномов — связывает реплика-
цию, мутацию и рекомбинацию с последова-
тельной эволюцией геномов во времени. Мы
начнем наши изыскания с досконального зна-
комства с молекулярными процессами, лежа-
щими в основе репликации генома (глава 15),
мутации и репарации ДНК (глава 16), а также
рекомбинации (глава 17), после чего освоим
способы, при помощи которых эти процессы
сформировали структуры геномов и напол-
нили их генетическим содержимым за вре-
мя эволюции (глава 18). Наконец, в главе 19
приводится описание того, как молекулярные
филогенетики используют эволюционную ин-
формацию, содержащуюся в геномах, для того
чтобы исследовать такие вопросы, как отноше-
ния родства между людьми и другими прима-
тами, происхождение СПИДа и миграционные
маршруты, пройденные людьми в процессе
нашего распространения по планете из места
появления человека на свет, находившегося
где-то в Африке.
Глава 15
Репликация генома
Глава 16
Мутации
и репарация ДНК
Глава 17
Рекомбинация
Глава 18
Механизм эволюции
геномов
Глава 19
Молекулярная
филогенетика
Глава 15
Репликация генома
По прочтении главы 15 вы сможете:
• Сообщать о том, что понимается под топологической проблемой, и объяснять,
каким образом ДНК-топоизомеразы разрешают эту проблему.
• Описывать решающий эксперимент, который доказал, что репликация ДНК
происходит полуконсервативным процессом.
• Вкратце описывать такие способы репликации генома, как вытеснение и катя-
щееся кольцо.
• Обсуждать механизмы инициации репликации у бактерий, дрожжей и млеко-
питающих.
• Описывать ключевые особенности ДНК-полимераз бактерий и эукариотов.
• Объяснять причину, по которой опережающая и запаздывающая нити молекулы
ДНК должны реплицироваться в ходе различных процессов.
• Приводить подробное описание событий, происходящих в репликационной
вилке у бактерий, и показывать, чем эти события отличаются от тех, что имеют
место у эукариотов.
• Сообщать все, что в настоящее время известно о терминации репликации у бак-
терий и эукариотов.
• Объяснять, каким образом теломераза поддерживает концы молекулы хромо-
сомной ДНК эукариотов, и рассуждать о возможных зависимостях между длиной
теломеры, старением клетки и раком.
• Описывать, за счет чего репликация генома согласуется с клеточным циклом.
Первостепенная функция генома состоит в определении биохимической сигнатуры
клетки, в которой он размещается. Мы увидели, что геном достигает этой цели путем
скоординированной экспрессии генов и групп генов, обусловливающей поддержание
протеома, отдельные белковые компоненты которого выполняют и регулируют био-
химические отправления клетки. Чтобы непреложно выполнять эту функцию, геном
должен реплицироваться каждый раз, когда клетка делится. Это означает, что полное
содержимое ДНК клетки должно копироваться в соответствующий период клеточного
цикла и получающиеся молекулы ДНК должны быть распределены по дочерним клеткам
так, чтобы каждая из них получила полную копию генома. Сей сложный процесс, кото-
рый пересекает границы между молекулярной биологией, биохимией и цитобиологией,
и будет описан в этой главе.
Репликация генома служила объектом изучения с тех самых пор, как Уотсон и Крик
впервые открыли структуру двойной спирали ДНК еще в 1953 г. В последующие годы
ее исследование протекало в трех сообщающихся, но разных руслах.
596 Часть 4. Механизмы репликации и эволюции геномов
Родительская двойная спираль
Две дочерние двойные спирали
Рис. 15.1. Репликация ДНК согласно схеме, пред-
сказанной Уотсоном и Криком. Полинуклеотиды
родительской двойной спирали показаны чер-
ным. Оба служат матрицами для синтеза новых
нитей ДНК, показанных красным. Последова-
тельности этих новых нитей определяются спа-
риванием оснований с матричными молекула-
ми. Топологическая проблема возникает, потому
что два полинуклеотида родительской спирали
не могут просто быть разделены: спираль долж-
на быть раскручена некоторым образом
• Топологическая проблема была глав-
ным вопросом в годы с 1953 по 1958.
Эта проблема проистекает из необхо-
димости раскрутить двойную спираль,
с тем чтобы произвести копии ее двух
полинуклеотидов (см. рис. 15.1). Эта про-
блема встала во главу угла в середине
1950-х гг., потому что она была главным
камнем преткновения к принятию двой-
ной спирали в качестве достоверной
структуры ДНК, но отошла на задний
план в 1958 г., когда Мэтью Мезельсон
и Франклин Шталь продемонстрирова-
ли, что, несмотря на воспринимаемые
трудности, репликация ДНК у Escherichia
coli происходит способом, предполагае-
мым структурой двойной спирали. Экс-
перимент Мезельсона-Шталя позво-
лил исследованию репликации генома
продвинуться вперед, даже при том что
сама по себе топологическая проблема не была решена до начала 1980-х гг., когда
впервые был понят принцип действия ДНК-топоизомераз (раздел 15.1.2).
• Процесс репликации интенсивно изучался с 1958 г. В период 1960-х гг. были иден-
тифицированы ферменты и белки, участвующие в репликации у Е. coli, и были
определены их функции. В последующие годы подобный прогресс был сделан
в понимании деталей репликации генома эукариотов. Эта работа все еще про-
должается, при этом сегодня исследования сосредоточены на таких вопросах, как
инициация репликации и точные принципы действия белков, которые являются
активными в репликационной вилке.
• Регулирование репликации генома и, в частности, в контексте клеточного цикла,
стало преобладающей областью исследования в последние годы. Эта работа пока-
зала, что инициация — ключевая контрольная точка в репликации генома, и начала
объяснять, каким путем репликация синхронизирована с клеточным циклом, с тем
чтобы к моменту деления клетки дочерние геномы имелись в наличии.
В ходе нашего с вами изучения репликации генома мы рассмотрим каждый из этих
трех вопросов в вышеупомянутом порядке.
Глава 15. Репликация генома 597
15.1. Топологическая проблема
В своей статье в журнале Nature, объявляющей об открытии структуры двойной
спирали ДНК, Уотсон и Крик сделали одно из самых известных утверждений в молеку-
лярной биологии:
«От нашего внимания не ускользнул тот факт, что из постулированного нами
специфического спаривания непосредственно проистекает и возможный механизм
копирования генетического материала».
Процесс спаривания, к которому они обращаются, — тот, в котором каждая нить
двойной спирали служит матрицей для синтеза второй комплементарной нити, и в ко-
нечном результате обе дочерние двойные спирали являются идентичными родительской
молекуле (рис. 15.1). Эта схема почти явно прослеживается в структуре двойной спирали,
но ее принятие оставляет нерешенными проблемы, как признали Уотсон и Крик во вто-
рой статье, опубликованной в Nature лишь месяц спустя после сообщения об открытой
ими структуре. Эта статья описывает постулированный процесс репликации более
подробно, но указывает трудности, которые вытекают из необходимости раскрутить
двойную спираль. Наиболее тривиальная из этих трудностей — возможность запуты-
вания дочерних молекул. Более важная — вращение, которым сопровождалось бы рас-
кручивание: при одном витке, приходящемся на каждые 10 п. н. двойной спирали, полная
репликация молекулы ДНК в хромосоме человека 1, которая имеет длину 250 млн п. н.,
потребовала бы 25 миллионов оборотов хромосомной ДНК. Трудно вообразить, как это
могло бы осуществиться в ограниченном объеме ядра, хотя, в принципе, раскручивание
молекулы линейной хромосомной ДНК не является физически невозможным. Напро-
тив, кольцевая двунитевая молекула (например, геном бактерии или бактериофага),
не имея свободных концов, не была бы способна вращаться необходимым образом
и, исходя из этого, очевидно, не могла бы реплицироваться по схеме Уотсона и Крика.
Отыскание ответа на эту дилемму было главной заботой молекулярных биологов на
протяжении 1950-х гг.
15.1.1. Экспериментальное доказательство предложенной Уотсоном
и Криком схемы репликации ДНК
Некоторые молекулярные биологи, особенно Макс Дельбрюк, считали топологи-
ческую проблему настолько серьезной, что первоначально они оказывали некоторое
сопротивление принятию двойной спирали в качестве достоверной структуры ДНК. Эта
трудность касается плектонемного характера двойной спирали и относится к тополо-
гической структуре, которая делает невозможным разделение двух нитей спирали без ее
раскручивания. Поэтому эта проблема была бы решена, если бы двойная спираль была
фактически паранемной, потому что это подразумевало бы, что две нити могли быть
отделены друг от друга простым разведением их в разные стороны без раскручивания
молекулы. Было высказано предположение о том, что двойная спираль может быть пре-
образована в паранемную структуру путем скручивания в сверхспираль в направлении,
противоположном повороту самой спирали, или что в пределах молекулы ДНК право-
сторонняя спираль, предложенная Уотсоном и Криком, могла бы быть «уравновешена»
отрезками равной длины левой спиральной структуры. Возможность, что двунитевая
ДНК была вообще не спиралью, а лентообразной структурой с параллельно идущими
нитями, также рассматривалась непродолжительное время, причем эта идея, как это ни
удивительно, возродилась в конце 1970-х гг. Каждое из этих предложенных решений
топологической проблемы одно за другим отклонялось по той или другой причине,
598 Часть 4. Механизмы репликации и эволюции геномов
а) Разбросанная
iiiiiiiliiiiin
[И1ИИИИ1
Рис. 15.2. Три возможные схемы ре-
пликации ДНК. Ради ясности молекулы
ДНК нарисованы в виде лесенок, а не
спиралей
Родительская
двойная спираль
ОБОЗНАЧЕНИЯ:
гггт Родительская ДНК
ffW Новая ДНК
б) Полуконсервативная
......IIIH'IHl
IlilllillIlill lill II
в) Консервативная
НИИ III II Hill IIII
ШПШШШШШ
большинство из них потому, что
они предполагали видозменения
в структуре двойной спирали —
видоизменения, которые не были
совместимы с результатами рент-
геноструктурного анализа и дру-
гими экспериментальными дан-
ными о структуре ДНК.
Первая действительная
подвижка в решении тополо-
гической проблемы произошла
в 1954 г., когда Дельбрюк пред-
ложил модель «разрыва и воссоединения», описывающую принцип разделения
нитей двойной спирали. Согласно этой модели, нити отделяются друг от друга не
раскручиванием спирали с сопровождающим его вращением молекулы, но путем
разрыва одной из нитей, прохождения второй нити через разрыв и воссоединения
первой нити. Эта схема фактически очень близка к верному решению топологиче-
ской проблемы, будучи одним из путей, которым работают ДНК-топоизомеразы
(см. рис. 15.4, а), но, к сожалению, Дельбрюк излишне усложнил проблему, пытаясь
объединить процесс разрыва и воссоединения с синтезом ДНК, который происходит
во время процесса собственно репликации. Это привело его к модели репликации
ДНК, согласно которой каждый полинуклеотид в дочерней молекуле состоит частично
из родительской ДНК и частично из новосинтезированной ДНК (рис. 15.2, а). Этот
разбросанный способ репликации контрастирует с полуконсервативной систе-
мой, предложенной Уотсоном и Криком (рис. 15.2, б). Третья возможность — что
репликация полностью консервативная, и тогда одна из дочерних двойных спиралей
полностью состоит из новосинтезированной ДНК, а другая содержит две родитель-
ские нити (рис. 15.2, в). Модели консервативной репликации трудно построить, но
можно представить, что репликация такого типа могла бы быть достигнута без
раскручивания родительской спирали.
Эксперимент Мезельсона-Шталя
Построенная Дельбрюком модель разрыва и воссоединения была важна, по-
тому что она послужила отправной точкой для экспериментов, предназначенных
для проверки каждого из трех способов репликации ДНК, проиллюстрированных на
рис. 15.2. Незадолго до того времени в молекулярной биологии начали применять
радиоактивные изотопы, так что были предприняты попытки использовать мечение
ДНК (см. техническое примечание 2.1), чтобы отличить новосинтезированную ДНК от
родительских полинуклеотидов. Каждый способ репликации предполагает различное
распределение новосинтезированной ДНК и, следовательно, радиоактивной метки
в двойных спиралях, получаемых после двух или более кругов репликации. Поэтому
анализ радиоактивного содержимого этих молекул позволяет решить, которая из
а) Схема проведения эксперимента
6) Объяснение результатов эксперимента
Культивирование
Е. coli в содержащей
15NH4CI среде
Перенос
в содержащую
14NH4CI среду
--------►
Консервативная
Родительские
двойные спирали
Разбросанная Полуконсервативная
| ОБОЗНАЧЕНИЯ:
— Родительская
flHK(1SN)
— Новая ДНК ( N)
00:30 МИН 00:40 МИН
Одно клеточное
деление
в градиенте
Два
клеточных
деления
плотности
— 14N-1SN-flHK
— 14N-14N-flHK
14N-15N-flHK
Ожидаемые
полосы
Дочерние
молекулы
Ожидаемые
полосы
Молекулы второго поколения
Рис. 15.3. Эксперимент Мезельсона-Шталя. а) Эксперимент, выполненный Мезельсоном и Шталем, предполагал выращивание культуры Escherichia coli
в среде, содержащей 15NH4CI (хлорид аммония, помеченный тяжелым изотопом азота). Затем клетки были перенесены в нормальную среду (содержащую
14NH4CI) и образцы были взяты после 20 минут (одно деление клеток) и 40 минут (два деления клеток). ДНК была извлечена из каждого образца и молекулы
проанализированы центрифугированием в градиенте плотности. После 20 минут вся ДНК содержала подобные количества 14N и 15N, но после 40 минут
наблюдались две полосы: одна соответствовала гибриду 14N-15N-DNA, а другая — молекулам ДНК, построенным при употреблении исключительно 14N.
б) Предсказанный исход эксперимента показан для каждого из трех возможных способов репликации ДНК. Картина полос, наблюдаемая через 20 минут,
позволяет отвергнуть консервативную репликацию, потому что эта схема предсказывает, что из одного цикла репликации выйдут двойные спирали двух
различных типов: одна содержит только 15N, а другая содержит только 14N. Единственная 14N-15N-flHK полоса, которая фактически наблюдалась через 20
минут, согласуется и с разбросанной, и с полуконсервативной репликацией, но две полосы, наблюдаемые по прошествии 40 минут, совместимы только
с полуконсервативной репликацией. В результате продолжения разбросанной репликации после двух циклов репликации должны получаться гибридные
молекулы 14N-15N, тогда как в число молекул второго поколения, произведенных к этому этапу полуконсервативной репликацией, входит два варианта,
которые полностью состоят из 14Ы-ДНК
лава 15. Репликация генома 599
600 Часть 4. Механизмы репликации и эволюции геномов
Рис. 15.4. Принцип действия ДНК-топоизомеразы I типа. Топо-
изомераза I типа делает надрез в одной нити молекулы ДНК,
пропускает неповрежденную нить сквозь надрез и ликвидирует
брешь
схем репликации работает в живых клетках. К со-
жалению, оказалось невозможным получить четкий
результат, в значительной степени из-за трудности
в измерении точного количества радиоактивности
в молекулах ДНК: такой анализ осложнен быстрым распадом изотопа 32Р, который
применялся в качестве метки.
Крупное достижение было в конечном счете сделано Мэтью Мезельсоном и Фран-
клином Шталем, которые в 1958 г. провели необходимый эксперимент не с радиоак-
тивной меткой, а с 15N — нерадиоактивным «тяжелым» изотопом азота. Теперь было
возможно анализировать реплицированные двойные спирали центрифугированием
в градиенте плотности (см. техническое примечание 7.1), потому что молекула ДНК, по-
меченная 15N, имеет более высокую плавучую плотность, чем непомеченная молекула.
Мезельсон и Шталь начали с культуры клеток Е. coli, которые были выращены с 15NH4C1
и чьи молекулы ДНК поэтому содержали тяжелый азот. Клетки были перенесены на нор-
мальную среду, и образцы взяты после периодов 20 минут и 40 минут, соответствующих
одному и двум делениям клетки. ДНК была извлечена из каждого образца, и молекулы
исследованы центрифугированием в градиенте плотности (рис. 15.3, а). После одного
круга репликации ДНК дочерние молекулы, синтезированные в присутствии нор-
мального азота, образовали отдельную полосу в градиенте плотности, указывая на
то, что все двойные спирали состоят из равных количеств новосинтезированной
и родительской ДНК. Этот результат незамедлительно позволил отбросить консер-
вативный способ репликации, поскольку он предсказывает наличие двух полос после
одного круга репликации (рис. 15.3, б), но не позволил провести различие между
разбросанной моделью Дельбрюка и полуконсервативным процессом, которому от-
дали свое предпочтение Уотсон и Крик. Различение стало, однако, возможным, когда
были исследованы молекулы ДНК, полученные после двух кругов репликации. Теперь
градиент плотности показал две полосы ДНК, первая из которых соответствовала
гибриду, состоящему из равных частей новосинтезированной и старой ДНК, а вторая
соответствовала молекулам, полностью состоящим из новой ДНК. Этот результат
находится в согласии с полуконсервативной схемой, но несовместим с разбросанной
моделью репликации, ибо последняя предсказывает, что после двух кругов репли-
кации все молекулы должны быть гибридными.
15.1.2. ДНК-топоизомеразы решают топологическую проблему на деле
Эксперимент Мезельсона-Шталя доказал, что репликация ДНК в живых клетках
следует полуконсервативной схеме, предложенной Уотсоном и Криком, и, следовательно,
показал, что клетка должна иметь решение топологической проблемы. Это решение
было найдено молекулярными биологами лишь около 25 лет спустя, когда были оха-
рактеризованы действия ферментов, названных ДНК-топоизомеразами.
ДНК-топоизомеразы — ферменты, которые выполняют реакции разрыва и вос-
соединения, подобные, но не идентичные предсказанным Дельбрюком. Различают
ДНК-топоизомеразы двух типов (таблица 15.1):
Глава 15. Репликация генома 601
• топоизомеразы I типа производят разрыв в одном полинуклеотиде и пропускают
второй полинуклеотид через зазор, который образуется (рис. 15.4). Затем два
конца разорванной нити вновь лигируются. Такой принцип действия приводит
к изменению числа зацеплений (числа раз, которое одна нить пересекает другую
в кольцевой молекуле) на единицу;
• топоизомеразы II типа разрывают обе нити двойной спирали, создавая «ворота»,
через которые проходит второй сегмент спирали. Это изменяет число зацеплений
на два.
Разрыв одной или обеих нитей ДНК может показаться слишком жестким реше-
нием топологической проблемы, ведущим к возможности того, что топоизомераза
иногда будет не в состоянии воссоединить нить и, следовательно, может по оплош-
ности разорвать хромосому на две части. Такая возможность уменьшается за счет
самого принципа действия этих ферментов. Один конец каждого разрезанного по-
линуклеотида становится ковалентно прикрепленным к аминокислоте тирозину
на активном участке фермента, гарантируя, что этот конец полинуклеотида прочно
удерживается на месте, в то время как свободные концы подвергаются манипуляциям.
Топоизомеразы I и II типов подразделяются согласно точной химической структуре
связи полинуклеотид-тирозин: в случае ферментов IA и ПА связь вовлекает фосфат-
ную группу, присоединенную к свободному 5’-концу разрезанного полинуклеотида,
а в случае ферментов IB и ПВ такая связь осуществляется через З'-фосфатную груп-
пу. Топоизомеразы А и В, вероятно, эволюционировали независимо друг от друга.
В клетках эукариотов присутствуют оба типа, но у прокариотов ферменты IB и ИВ
встречаются крайне редко.
Следует иметь в виду, что сами по себе ДНК-топоизомеразы не раскручивают двой-
ную спираль. Вместо этого, они решают топологическую проблему, противодействуя
перекручиванию, которое в противном случае происходило бы в молекуле за счет про-
движения репликационной вилки. В результате спираль может быть «расстегнута», при
этом две нити растягиваются в разные стороны без необходимости вращать молекулу
(рис. 15.5). Репликация — не единственная деятельность, которая осложнена тополо-
гией двойной спирали, и становится все более и более ясно, что ДНК-топоизомеразы
имеют столь же важные роли в ходе транскрипции, рекомбинации и других процессов,
которые могут привести к пере- или недокручиванию ДНК. У эукариотов топоизомеразы
образуют главную часть ядерного матрикса — подобной каркасу сети, что пронизывает
ядро (раздел 10.1.1), — и отвечают за поддержание структуры хроматина и расцепле-
ние молекул ДНК в ходе деления хромосомы. Большинство топоизомераз способно
лишь расслабить ДНК, но некоторые ферменты прокариотов, такие как бактериальная
Таблица 15.1. ДНК-топоизомеразы
Тип Субстрат Примеры
Тип I Однонитевая ДНК Топоизомеразы I и III Escherichia coli', топоизомераза III дрожжей и человека; обратная гираза архей; топоизоме- раза I эукариотов
Тип II Двунитевая ДНК Топоизомеразы II (ДНК-гираза) и IV Е. coli', топоизомераза II эукариотов
602 Часть 4. Механизмы репликации и эволюции геномов
ДНК-топоизомераза I Репликационная
делает однонитевой вилка продвигается
надрез перед вперёд
репликационной вилкой
Синтез ДНК продолжается
Рис. 15.5. Расстегивание двойной спирали подобно молнии. В ходе репликации двойная спираль «рас-
стегивается» в результате действия ДНК-топоизомераз. Репликационная вилка поэтому способна про-
двигаться по молекуле без необходимости вращать спираль
ДНК-гираза и присущая археям обратная гираза, могут выполнять обратную реакцию
и вводить сверхспирали в молекулы ДНК.
15.1.3. Вариации на тему полуконсервативности
Ни одного исключения из полуконсервативного способа репликации ДНК не из-
вестно, но есть несколько вариаций на эту основную тему. Копирование ДНК через
репликационную вилку, как показано на рис. 15.1, является преобладающей системой,
используемой молекулами хромосомной ДНК эукариотов и кольцевыми геномами прока-
риотов. Некоторые меньшие кольцевые молекулы, такие как митохондриальные геномы
животных (раздел 8.3.2), используют несколько отличающийся процесс, названный вы-
теснительной репликацией. В этих молекулахточка, в которой начинается репликация,
отмечена D-петлей — областью длиной приблизительно 500 п. н., где двойная спираль
прервана за счет присутствия молекулы РНК, спаренной основаниями с одной из нитей
ДНК (рис. 15.6). Такая молекула РНК служит отправной точкой для синтеза одного из
дочерних полинуклеотидов. Этот полинуклеотид синтезируется путем непрерывного
копирования одной нити спирали, при этом вторая нить вытесняется и впоследствии
копируется, после того как синтез первого дочернего генома будет завершен.
Преимущество вытеснительной репликации в том виде, в котором она осущест-
вляется митохондриальной ДНК животных, не ясно. Напротив, специальный тип про-
цесса вытеснения, названный репликацией катящегося кольца, представляет собой
эффективный механизм быстрого синтеза многочисленных копий кольцевого генома.
Репликация катящегося кольца, которая используется фагом X и разными другими бак-
териофагами, инициируется в надрезе, который производится в одном из родительских
полинуклеотидов. Освободившийся З’-конец продолжается, вытесняя 5’-конец полину-
клеотида. Непрерывный синтез ДНК «скатывается» с полной копии генома, и дальней-
Глава 15. Репликация генома 603
Репликация
вытесненной нити
Завершение синтеза
первой нити
Рис. 15.6. Вытеснительная репликация. D-петля содержит короткую молекулу РНК, которая затравливает
синтез ДНК (см. раздел 15.2.2). После завершения синтеза первой нити вторая РНК-затравка прикрепля-
ется к вытесненной нити и запускает репликацию этой молекулы. На этом рисунке новосинтезированная
ДНК показана красным
Рис. 15.7. Репликация катящегося кольца
ший синтез в конечном счете дает ряд геномов, сцепленных по принципу голова к хвосту
(рис. 15.7). Эти геномы однонитевые и линейные, но могут легко быть преобразованы
в двунитевые кольцевые молекулы посредством синтеза комплементарной нити, со-
провождаемого расщеплением в точках сцепления между геномами и формированием
колец из полученных сегментов.
604 Часть 4. Механизмы репликации и эволюции геномов
15.2. Процесс репликации
Как и в случае многих процессов в молекулярной биологии, мы традиционно рас-
сматриваем репликацию генома как состоящую из трех этапов — инициации, элонгации
и терминации.
• Инициация (раздел 15.2.1) состоит в опознавании позиций на молекуле ДНК, в ко-
торых начнется репликация.
• Элонгация (раздел 15.2.2) касается событий, происходящих в репликационной
вилке, где копируются родительские полинуклеотиды.
• Терминация (раздел 15.2.3), о которой есть пока лишь расплывчатые представле-
ния, происходит, когда родительская молекула полностью скопирована.
Кроме этих трех стадий репликации, еще один вопрос требует внимания. Он каса-
ется укорочения хромосом в процессе репликации, которое, не будь оно исправляемо,
привело бы к тому, что линейные двунитевые молекулы ДНК становились бы короче
при каждом своем копировании. Решение этой проблемы, которая связана со структурой
и с синтезом теломер на концах хромосом (раздел 7.1.2), описано в разделе 15.2.4.
15.2.1. Инициация репликации генома
Инициация репликации — не случайный процесс и всегда начинается в одной
и той же позиции (или позициях) на молекуле ДНК; эти точки называют областями
начала (ориджинами) репликации, или истоками (ориджинами) репликации. После
инициации из точки начала репликации могут выйти две репликационные вилки и про-
двигаться по молекуле ДНК в противоположных направлениях, поэтому в большинстве
геномов репликация двунаправленная (рис. 15.8). Кольцевой бактериальный геном
имеет единственное начало репликации, и, таким образом, каждой репликационной
вилкой копируется несколько миллионов п. н. ДНК. Эта ситуация отличается от таковой,
наблюдаемой на примере хромосом эукариотов, которые имеют множественные старты
и чьи репликационные вилки продвигаются на более короткие расстояния. Дрожжи
Saccharomyces cerevisiae, например, имеют около 332 областей старта репликации, что
соответствует 1-му старту на 36 тыс.п.н. ДНК, а люди имеют приблизительно 20000
областей инициации, или 1 область на каждые 150 тыс.п.н. ДНК.
Инициация репликации у Е. coli
Мы знаем гораздо больше об инициации репликации у бактерий, чем у эукариотов.
Область инициации у Е coli получила название oriC. Путем переноса сегментов ДНК из об-
а) Репликация кольцевой б) Репликация линейной хромосомы эукариотов
бактериальной хромосомы d
◄— Направление репликации —►
Рис. 15.8. Двунаправленная репликация ДНК: а) кольцевой бактериальной хромосомы и б) линейной
хромосомы эукариотов
Глава 15. Репликация генома 605
ласти опСв плазмиды, у которых отсутствуют свои собственные ориджины, была получена
оценка, согласно которой ориджин репликации Е coli охватывает приблизительно 245 п. н.
ДНК. Анализ последовательности этого сегмента показывает, что он содержит два коротких
повторных мотива: один из девяти нуклеотидов, а второй из 13 нуклеотидов (рис. 15.9, а).
Девятинуклеотидное повторение, пять копий которого рассеяны по всей области старта
репликации опС, является сайтом связывания для белка, названного DnaA. Исходя из нали-
чия пяти копий последовательности связывания, можно представить, что пять копий DnaA
прикрепляются к области инициации, но фактически связанные белки DnaA кооперируют
с несвязанными молекулами, пока около 30 копий не будет связано с областью начала
репликации. Присоединение происходит только когда ДНК скручена в отрицательную
сверхспираль, что закономерно для хромосомы Е coli (раздел 8.1.1).
В результате закрепления DnaA двойная спираль раскрывается («плавится»)
в пределах тандемного массива из трех АТ-обогащенных 13-нуклеотидных повторов,
расположенных на одном конце последовательности опС (рис. 15.9, б). Точный механизм
неизвестен, но белок DnaA, кажется, не обладает ферментативным действием, необходи-
мым для разрыва пар оснований, и поэтому предполагается, что спираль плавится за счет
напряжений кручения, создаваемых присоединением белков DnaA. Привлекательная
модель построена из представления о том, что белки DnaA формируют подобную ци-
линдру структуру, вокруг которой обвита спираль. Расплавлению спирали способствует
белок HU — наиболее обильный из белков упаковки ДНК Е coli (раздел 8.1.1).
Расплавление спирали запускает череду событий, которые строят рождающуюся
репликационную вил ку с обоих концов открытой области. Первый шаг — присоединение
а) Структура ог/С
20 п. н.
Расплавленная Цилиндр
область из белков DnaA
Рис. 15.9. Область начала репликации у Escherichia coli. а) Область начала репликации Е. coli называется
oriC и имеет приблизительно 245 п.н. в длину. Он содержит три копии 13-нуклеотидного повторного
мотива (консенсусной последовательности S'-GATCTNTTNTTTT-S', где «N» — любой нуклеотид) и пять
копий девятинуклеотидного повторения (консенсусной последовательности 5'-ТТ(А/Т)Т(А/С)СА(А/С)А-3',
где (А/Т) — А или Т, а (А/С) — А или С). 13-нуклеотидные последовательности формируют тандемный
массив прямых повторений на одном конце oriC. Девяти нуклеотидные последовательности распре-
делены по oriC так, что три единицы формируют ряд прямых повторений, а две единицы в обратной
конфигурации, как обозначено стрелками. Три из девяти нуклеотидных повторений — номера 1, 3 и 5,
если считать с левого конца oriC, как нарисовано здесь, — расцениваются как главные сайты связывания
белков DnaA; другие два повторения — как второстепенные участки. Общая структура области начала
репликации подобна у всех бактерий, и последовательности повторений варьируют незначительно, б)
Модель прикрепления белков DnaA к oriC, сопровождающегося плавлением спирали в пределах обо-
гащенных АТ 13-нуклеотидных последовательностей
606 Часть 4. Механизмы репликации и эволюции геномов
предзатравочного комплекса к каждой из этих двух позиций. Каждый предзатравоч-
ный комплекс первоначально включает 12 белков (шесть копий DnaB и шесть копий
DnaC), но белок DnaC имеет кратковременную роль и высвобождается из комплекса
вскоре после его формирования: его функция, вероятно, попросту заключается в облегче-
нии прикрепления DnaB. Последний представляет собой геликазу—фермент, который
может разрывать пары оснований (см. раздел 15.2.2). Геликаза DnaB начинает расширять
однонитевую область в пределах области инициации, позволяя ферментам, вовлечен-
ным в стадию элонгации репликации генома, прикрепиться. На этом стадия инициации
репликации у Е. coli завершается, поскольку репликационные вилки отныне начинают
продвигаться от области инициации и копирование ДНК набирает обороты.
Области инициации у дрожжей были точно определены
Метод, применявшийся для определения последовательности oriC у Е. coli (пред-
полагающий перемещение сегментов ДНК в нереплицирующуюся плазмиду), ока-
зался ценным также и для опознавания областей инициации репликации у дрожжей
Saccharomyces cerevisiae. Области инициации, опознанные этим методом, называют
автономно реплицирующимися последовательностями, или ARS. Типичная область
начала репликации у дрожжей короче, чем oriCу Е. coli, будучи обычно менее 200 п.н.
в длину, но, подобно области инициации репликации Е. coli, содержит обособленные
сегменты с различными функциональными ролями, причем эти «субдомены» имеют
подобные последовательности в различных областях инициации (рис. 15.10, а). Всего
было опознано четыре субдомена. Два из них — субдомены А и В1 — составляют сайт
узнавания области инициации, отрезок общей длиной около 40 п. н., который является
сайтом связывания для комплекса опознавания области инициации (ORC), набора из
шести белков, которые прикрепляются к области инициации (рис. 15.10, б). ORC были
описаны как присущие дрожжам варианты белков DnaAE coli, но данная интерпретация,
вероятно, не строго верна, потому что ORC, как оказалось, остаются присоединенными
к областям инициации дрожжей в течение всего клеточного цикла. Вместо того чтобы
быть подлинными инициаторными белками, более вероятно, что ORC участвуют в ре-
гулировании репликации генома, служа посредниками между областями инициации
репликации и регуляторными сигналами, которые согласовывают инициацию репли-
кации ДНК с клеточным циклом (раздел 15.3.1).
Поэтому нам следует искать последовательности с функциями строго эквива-
лентными таковой oriC Е. coli в каком-либо ином месте в областях инициации дрожжей.
Это ведет нас к двум другим консервативным последовательностям в типичной об-
ласти инициации дрожжей — субдоменам В2 и ВЗ (см. рис. 15.10, а). Наши современ-
а) Структура истока репликации у дрожжей
В1 А
20 п. н.
Опознавательная
последовательность истока
б) Плавление спирали
ABF1 Расплавленная ORC
область
Рис. 15.10. Структура области инициации у дрожжей, а)
Структура ARS1 — типичной автономно реплицирую-
щейся последовательности (ARS), которая действует
как область инициации репликации у Saccharomyces
cerevisiae. Показаны относительные позиции функ-
циональных последовательностей A, Bl, B2 и ВЗ. б)
Плавление спирали происходит в пределах субдоме-
на B2 и вызывается прикреплением связывающегося
с ARS белка 1 (ABF1) к субдомену ВЗ. Белки комплекса
опознавания области инициации (ORC) постоянно при-
креплены к субдоменам А и B1
Глава 15. Репликация генома 607
ные представления позволяют предположить, что эти два субдомена функционируют
в манере, подобной области инициации Е. coli. Субдомен В2, кажется, соответствует
13-нуклеотидному повторному массиву области инициации Е coli, будучи позицией,
в которой две нити спирали начинают разделяться. Это расплавление активируется на-
пряжением кручения, привносимым присоединением ДНК-связывающего белка, фактора
связывания ARS1 (ABF1), который прикрепляется к субдомену ВЗ (см. рис. 15.10, б). Как
и у Е coli, расплавление спирали в пределах области инициации дрожжей сопровожда-
ется прикреплением геликазы и других репликационных ферментов к ДНК, завершая
процесс инициации и позволяя репликационным вилкам начать их продвижение по
молекуле ДНК, как описано в разделе 15.2.2.
Распознать области инициации у высших эукариотов было труднее
Попытки идентифицировать области инициации репликации у людей и других
высших эукариотов до недавнего времени были менее успешны. Области инициации
(части хромосомной ДНК, в которых запускается репликация) могут быть определены
различными биохимическими методами, например, репликации позволяют запуститься
в присутствии меченых нуклеотидов, затем задерживают процесс, очищая новосинте-
зированную ДНК и определяя позиции этих зарождающихся нитей в геноме. Эти экс-
перименты позволили предположить, что в хромосомах млекопитающих существуют
определенные области, в которых начинается репликация, но некоторые исследователи
высказывали сомнения относительно того, что эти области содержат области инициации,
эквивалентные таковым у дрожжей. Согласно одной альтернативной гипотезе репли-
кация инициируется белковыми структурами, которые имеют специфические позиции
в ядре, а хромосомные области инициации попросту являются сегментами ДНК, рас-
положенными близко к этим белковым структурам в трехмерной организации ядра.
Сомнения относительно областей инициации репликации у млекопитающих воз-
росли в связи с неудачей переноса репликативной способности областей инициации
млекопитающих в не обладающие этой способностью плазмиды, хотя эти эксперименты
не считались заключительными, потому что было признано, что область инициации
млекопитающих может быть слишком длинной, чтобы быть клонированной в плазмиде,
или может функционировать только при активации удаленными участками в хромосом-
ной ДНК. Решающим достижением была демонстрация того факта, что имеющий длину
8 тыс. п. н. сегмент области инициации у человека, будучи перенесен в геном обезьяны,
по-прежнему направляет репликацию, несмотря на то, что был отдален от какой-либо
гипотетической белковой структуры в ядре клетки человека. Анализ этой перемещен-
ной области инициации показал, что в пределах этой области есть первичные участки,
в которых инициация происходит с высокой частотой, окруженные вторичными участ-
ками, охватывающими всю область протяженностью 8 тыс. п. н., в которых репликация
запускается с более низкой частотой. Присутствие обособленных функциональных
доменов в пределах области инициации также было продемонстрировано в ходе ис-
следования воздействия удалений частей этой области на эффективность инициации
репликации.
Демонстрация того факта, что геном человека действительно содержит области
инициации репликаци, эквивалентные таковым у дрожжей, поднимает вопрос о том,
обладают ли млекопитающие эквивалентом ORC дрожжей. Ответом, кажется, будет
«да», так как несколько генов, белковые продукты которых имеют последовательно-
сти, подобные таковым белков ORC дрожжей, были определены у высших эукариотов,
и было показано, что некоторые из этих белков способны заменить эквивалентный
608 Часть 4. Механизмы репликации и эволюции геномов
белок дрожжей в их ORC. Эти результаты указывают на то, что инициация репликации
у дрожжей является хорошей моделью событий, происходящих у млекопитающих, при-
чем это заключение имеет большое значение для исследований управления инициацией
репликации, как мы увидим в разделе 15.3.
15.2.2. Стадия элонгации репликации
После инициации репликации репликационные вилки продвигаются по ДНК и уча-
ствуют в главном процессе репликации генома — синтезе новых нитей ДНК, которые
являются комплементарными родительским полинуклеотидам. На химическом уровне
направляемый матрицей синтез ДНК (рис. 15.11) весьма подобен направляемому ма-
трицей синтезу РНК, который происходит в ходе транскрипции (сравните с рис. 12.1).
Однако это подобие не должно вводить нас в заблуждение и сподвигать на проведение
широкой аналогии между транскрипцией и репликацией. Механика этих двух процессов
весьма различна, ибо репликация осложняется двумя факторами, которые не имеют
никакого отношения к транскрипции.
• В ходе репликации ДНК должны быть скопированы обе нити двойной спирали.
Это важное осложнение, потому что, как было отмечено в разделе 1.1.2, ферменты
ДНК-полимеразы способны синтезировать ДНК только в направлении 5'->3'. Это
означает, что одна нить родительской двойной спирали, названная опережающей
(лидирующей) нитью, может быть копируема непрерывно, но репликация от-
стающей (запаздывающей) нити должна быть выполняема прерывным способом,
то есть в виде ряда коротких сегментов, которые должны быть лигированы один
к другому, с тем чтобы произвести неповрежденную дочернюю нить (рис. 15.12).
• Второе осложнение имеет место, потому что зависимые от матрицы ДНК-полимеразы
не могут запустить синтез ДНК на всецело однонитевой молекуле: на ней должна
быть короткая двунитевая область, несущая 3'-конец, к которому фермент может
присоединять новые нуклеотиды. Это означает, что необходимы затравки: одна,
чтобы запустить синтез комплементарной нити на опережающем полинуклеотиде,
и по одной для каждого сегмента прерывной ДНК, синтезированной на отстающей
нити (рис. 15.12).
Прежде чем мы рассмотрим эти два осложнения, мы сначала изучим сами ферменты
ДНК-полимеразы.
ДНК-полимеразы бактерий и эукариотов
Основная химическая реакция, катализируемая ДНК-полимеразой, — синтез по-
линуклеотида ДНК в направлении 5'—>3', как показано на рис. 15.11. Из раздела 2.1.1 мы
узнали, что некоторые ДНК-полимеразы сочетают эту функцию с действием по крайней
мере одной экзонуклеазы, а это означает, что, помимо синтеза полинуклеотидов эти
ферменты могут деградировать их (см. рис. 2.7).
• Действием экзонуклеазы 3'—>5' обладают многие зависимые от матрицы ДНК-
полимеразы бактерий и эукариотов (таблица 15.2). Это действие позволяет фер-
менту удалять нуклеотиды с З'-конца нити, которую он только что синтезировал.
Этот процесс рассматривается как корректирующее действие, функция которого
заключается в исправлении случайных ошибок спаривания оснований, которые
могут произойти в процессе синтеза нити (см. раздел 16.1.1).
• Действие экзонуклеазы 5'—>3' менее распространено, но свойственно некоторым
полимеразам, функция которых в процессе репликации требует, чтобы они были
Глава 15. Репликация генома 609
Рис. 15.11. Направляемый матрицей синтез ДНК. Сравните эту реакцию с направляемым матрицей
синтезом РНК, представленным на рис. 12.1
способны удалять по крайней мере часть полинуклеотида, который уже присоеди-
нен к матричной нити, копируемой этой полимеразой. Такое действие используется
в ходе процесса, посредством которого соединяются вместе прерывные фрагменты
ДНК, синтезированные на отстающей нити во время репликации бактериальной
ДНК (см. рис. 15.18).
Поиск ДНК-полимераз начался в середине 1950-х гг., как только стало понятно,
что синтез ДНК был ключом к репликации генов. В то время считалось, что бактерии,
по всей вероятности, имеют только одну ДНК-полимеразу, и когда этот фермент, те-
перь называемый ДНК-полимеразой I, был выделен Артуром Корнбергом в 1957 г.,
широкое распространение получило предположение о том, что это главный репли-
610 Часть 4. Механизмы репликации и эволюции геномов
Рис. 15.12. Осложнения в процессе реплика-
ции ДНК. Когда реплицируется двунитевая
ДНК, должны быть решены два затруднения.
Во-первых, только опережающая нить может
непрерывно копироваться путем синтеза ДНК
в направлении 5'->3'; репликация отстающей
нити должна выполняться с перерывами. Во-
вторых, инициация синтеза ДНК требует затрав-
ки. Это верно как для синтеза клеточной ДНК,
как показано здесь, так и для реакций синтеза
ДНК, которые выполняются в пробирке (раз-
дел 2.1.1)
ОПЕРЕЖАЮЩАЯ ЗАПАЗДЫВАЮЩАЯ цирующий фермент. Поэтому открытие
нить НИТЬ того факта, что инактивация генаpolA Е.
coli, кодирующего ДНК-полимеразу I, не
была смертельна (клетки по-прежнему были способны реплицировать свои геномы),
вызвало удивление, особенно когда подобный результат был получен с инактивацией
гена polB, кодирующего второй фермент, ДНК-полимеразу II, который, как теперь
известно, участвует главным образом в репарации поврежденной ДНК, а не в реплика-
ции генома (раздел 16.2). Только в 1972 г. главная реплицирующая полимераза Е. coli,
ДНК-полимераза III, была, наконец, выделена. Как мы увидим в следующем разделе,
в репликации генома участвуют обе ДНК-полимеразы — и I, и III.
ДНК-полимеразы I и II представляют собой отдельные полипептиды, но ДНК-
полимераза III, дабы подобать своей роли главного реплицирующего фермента, являет
собой многосубъединичный белок с молекулярной массой приблизительно 900 кДа (табли-
ца 15.2). Три главные субъединицы, которые формируют основной фермент, названы а, с и
0, при этом действие полимеразы определяется а-субъединицей, а действие экзонуклеазы
3’—>5' — е-субъединицей. Функция 0-субъединицы не ясна: она может иметь исключи-
тельно структурную роль, состоящую в сведении воедино других двух основных субъе-
диниц, а также в сборке различных дополнительных субъединиц. К последним относятся
т- и у-субъединицы (причем обе кодируются одним и тем же геном—у-субъединица синте-
зируется путем трансляционного сдвига рамки считывания (раздел 13.2.3)), 0-субъединица,
которая служит своего рода «подвижным зажимом» и удерживает полимеразный комплекс
в тесном контакте с матрицей, а также 6-, 6’-, х- и у-субъединицы.
Эукариоты имеют по крайней мере девять ДНК-полимераз, которые у млеко-
питающих различают с помощью греческих суффиксов (а, 0, у, 6 и т.д.) — неудачный
выбор номенклатуры, поскольку она вызывает путаницу с тождественно названными
субъединицами ДНК-полимеразы III Е. coli. Главный реплицирующий фермент — ДНК-
полимераза 6 (таблица 15.2), которая имеет две субъединицы (согласно некоторым
исследователям — три) и работает совместно со вспомогательным белком, названным
ядерным антигеном пролиферирующих клеток (PCNA). PCNA — функциональный
эквивалент 0-субъединицы ДНК-полимеразы III Е. coli, он удерживает фермент в плот-
ном контакте с матрицей. ДНК-полимераза а также имеет важную функцию в синтезе
ДНК, будучи ферментом, что затравливает репликацию у эукариотов (см. рис. 15.13, б).
ДНК-полимераза у, хотя и кодируется ядерным геном, отвечает за реплицирование
митохондриального генома.
Глава 15. Репликация генома 611
Таблица 15.2. ДНК-полимеразы, участвующие в репликации геномов бактерий и эукариотов
Действия экзонуклеазы
Фермент субъединиц 3'—>5' 5'—>3' Функция
ДНК-полимеразы бактерий
ДНК-полимераза I 1 Да Да Восстановление и репликация ДНК
ДНК-полимераза III По крайней мере 10 Да Нет Главный реплицирующий фермент
ДНК-полимеразы эукариотов
ДНК-полимераза а 4 Нет Нет Затравливание в ходе репликации
ДНК-полимераза у 2 Да Нет Репликация митохондриальной ДНК
ДНК-полимераза 6 2 или 3 Да Нет Главный репликативный фермент
ДНК-полимераза к 1 7 7 Необходима для присоединения ко- гезиновых белков, которые удержи- вают сестринские хроматиды вместе вплоть до стадии анафазы процесса разделения ядер (раздел 15.2.3)
Примечание: бактерии и эукариоты обладают и другими ДНК-полимеразами, участвующими главным
образом в репарации поврежденной ДНК. Эти ферменты включают ДНК-полимеразы II, IV и V Escherichia coli
и ДНК-полимеразы эукариотов р, е, ц, 0 и i. Процессы репарации ДНК описаны в разделе 16.2.
Прерывный синтез нити и проблема затравливания
Ограничение, согласно которому ДНК-полимеразы могут синтезировать полину-
клеотиды только в направлении 5'->3', означает, что отстающая нить родительской
молекулы должна быть реплицируема прерывным образом, как показано на рис. 15.12.
Вывод, подразумеваемый этой моделью, о том, что первичные продукты репликации
отстающей нити представляют собой короткие сегменты полинуклеотида, был под-
тверждён в 1969 г., когда фрагменты Окадзаки, как эти сегменты теперь называют,
были впервые выделены из Е. coli. У бактерий фрагменты Окадзаки имеют 1000-2 000
нуклеотидов в длину, но у эукариотов эквивалентные фрагменты оказались намного
короче, возможно, менее 200 нуклеотидов в длину. Это интересное наблюдение, которое
может указывать на то, что каждый круг прерывного синтеза реплицирует ДНК, свя-
занную с одной нуклеосомой (от 140 до 150 п. н. обернутой вокруг стержневой частицы
плюс 50-70 п.н. линкерной ДНК; раздел 7.1.1).
Вторая трудность, иллюстрированная на рис. 15.12, — необходимость затравки
(праймера) для инициации синтеза каждого нового полинуклеотида. Не известно на-
верняка, почему ДНК-полимеразы не могут начать синтез на полностью однонитевой
матрице, но это может быть связано с корректирующим действием этих ферментов,
которое является определяюще важным для точности репликации. Как будет описано
в разделе 16.1.1, нуклеотид, который был вставлен неправильно в самый З’-конец на-
ращиваемой нити ДНК и, следовательно, не спаренный основанием с матричным по-
линуклеотидом, может быть удален за счет свойственного ДНК-полимеразе действия
экзонуклеазы 3'->5'. Это означает, что действие экзонуклеазы 3'->5’должно быть более
эффективным, чем действие полимеразы 5’->3’, когда З’-нуклеотид не спарен с матрицей.
612 Часть 4. Механизмы репликации и эволюции геномов
а) Затравливание синтеза ДНК у бактерий б) Затравливание синтеза ДНК у эукариотов
3х L^.LLU^.J. 5'
ДНК-матрица
3^1X!..U.LL!lJ.r ...........'5'
ДНК-матрица
РНК- I
затравка Праймаза
I
Новая ДНК ДНК-полимераза III
РНК- I
затравка Новая ДНК праймаза,
fvI L -------------ДНК-полимераза а
I
ДНК-полимераза 6
^lllllllllJIIIIIIJIIIIIIIIII1..5-
Рис. 15.13. Затравливание синтеза ДНК у а) бактерий и б) эукариотов. У эукариотов праймаза формирует
комплекс с ДНК-полимеразой а, который показан в момент синтеза РНК-затравки, сопровождаемого
первыми несколькими нуклеотидами ДНК
Вследствие этого полимераза может эффективно продолжать полинуклеотид, только
в том случае, если З’-нуклеотид спарен, что, в свою очередь, может служить причиной,
по которой полностью однонитевая матрица, в коей по определению отсутствует спа-
ренный основанием З'-нуклеотид, не может быть использована ДНК-полимеразой.
Как бы то ни было, затравливание — настоятельная потребность в процессе ре-
пликации ДНК, однако же не представляет слишком большой проблемы. Хотя ДНК-
полимеразы не могут работать с полностью однонитевой матрицей, РНК-полимеразы
не испытывают никаких трудностей в этом отношении, так что затравки для репли-
кации ДНК делают из РНК. У бактерий затравки синтезируются праймазой — специ-
альной РНК-полимеразой, не связанной с транскрибирующим ферментом, при этом
все затравки имеют 4-15 нуклеотидов в длину и большинство начинается с последова-
тельности 5-AG-3'. Как только синтез затравки завершен, синтез нити продолжается
ДНК-полимеразой III (рис. 15.13, а). У эукариотов ситуация немного сложнее, потому
что праймаза плотно скреплена с ДНК-полимеразой а и сотрудничает с этим фермен-
том в синтезе нескольких первых нуклеотидов нового полинуклеотида. Эта праймаза
синтезирует РНК-затравку из 8-12 нуклеотидов и затем передает полномочия ДНК-
полимеразе а, которая продолжает РНК-затравку, добавляя около 20 нуклеотидов ДНК.
Такой отрезок ДНК часто имеет несколько примешанных рибонуклеотидов, но не ясно,
встраиваются ли они ДНК-полимеразой а или перемежающимся действием праймазы.
По завершении синтеза РНК-ДНК-затравки синтез ДНК продолжается главным репли-
кативным ферментом — ДНК-полимеразой 6 (рис. 15.13, б).
На опережающей нити затравливание должно произойти только единожды, в пределах
начала репликации, потому что будучи затравлена, копия опережающей нити синтезируется
непрерывно, пока репликация не будет завершена. На запаздывающей нити затравливание
является повторяющимся процессом, который должен происходить каждый раз, когда ини-
циируется синтез очередного фрагмента Окадзаки. У Е coli, которая производит фрагменты
Окадзаки 1000-2000 нуклеотидов в длину, при каждой репликации генома необходимо
приблизительно 4000 событий затравливания. У эукариотов фрагменты Окадзаки намного
короче и затравливание происходит с большой частотой.
Глава 15. Репликация генома 613
События в репликационной вилке у бактерий
Теперь, когда мы рассмотрели осложнения, обусловленные прерывным синтезом нити
и проблемой затравливания, мы можем пойти дальше к изучению комбинации событий,
происходящих в репликационной вилке во время стадии элонгации репликации генома.
В разделе 15.2.1 мы определили присоединение геликазы DnaB, сопровождаемое
продолжением расплавленной области инициации, как представляющее конец стадии
инициации репликации у Е coli. В значительной мере разделение между инициацией
и элонгацией искусственно, ибо оба этих процесса плавно, без какого-либо резкого
перехода, перетекают один в другой. После того как геликаза свяжется с областью
инициации, с тем чтобы образовать предзатравочный комплекс, вербуется праймаза,
создавая праймосому, которая запускает репликацию опережающей нити. Она делает
это, синтезируя РНК-затравку, в которой нуждается ДНК-полимераза III, чтобы начать
копировать матрицу.
DnaB — главная геликаза, участвующая в репликации генома у Е. coli, но она далеко
не единственная геликаза, которой обладает эта бактерия: по последним оценкам, она
имеет одиннадцать геликаз. Объем совокупности этих ферментов отражает тот факт,
что раскручивание ДНК требуется не только в ходе репликации, но также и в течение
разнообразных процессов, таких как транскрипция, рекомбинация и репарация ДНК.
Принцип действия типичной геликазы не был точно определен, но думается, что эти
ферменты связываются с однонитевой, а не с двунитевой ДНК и передвигаются по по-
линуклеотиду в направлении 5’->3’ или 3'->5', в зависимости от специфики геликазы.
Разрыв пар оснований геликазой требует энергии, которая производится путем ги-
дролиза АТФ. Согласно этой модели единственная геликаза DnaB может передвигаться
по отстающей нити (DnaB является геликазой 5’—>3'), расстегивая спираль и создавая
репликационную вилку, при этом напряжение кручения, производимое раскручивающим
действием, снимается действием ДНК-топоизомеразы (рис. 15.14). Эта модель, вероятно,
является хорошим приближением того, что происходит фактически, хотя она не объяс-
няет функцию двух других геликаз Е. coli, которые, как думают, замешаны в репликации
генома. Обе из них, PriA и Rep, представляют собой геликазы 3'->5' и, таким образом, они,
вероятно, могут дополнять действие DnaB, продвигаясь по опережающей нити, но могут
играть и менее значимые роли. Причастность Rep к репликации ДНК может фактически
Рис. 15.14. Роль геликазы DnaB в ходе репликации ДНК у Escherichia coli. DnaB представляет собой гели-
казу 5'->3' и, таким образом, мигрирует по отстающей нити, разрушая пары оснований по мере своего
продвижения. Она работает в паре с ДНК-топоизомеразой и раскручивает спираль. Чтобы избежать
путаницы, фермент праймаза, обычно связанный с геликазой DnaB, не показан на этом рисунке
614 Часть 4. Механизмы репликации и эволюции геномов
a) SSB прикрепляются
к неспаренным полинуклеотидам
6) Структура RPA - SSB эукариотов
Рис. 15.15. Роль белков однонитевого связывания (SSB) в ходе репликации ДНК. a) SSB прикрепляются
к неспаренным полинуклеотидам, произведенным действием геликазы, и предотвращают нити от спа-
ривания друг с другом или от деградации посредством специфичной к однонитевым цепям нуклеазы.
б) Структура SSB эукариотов, называемого RPA. Белок содержит структуру 0-листа, которая формирует
канал, в котором связывается ДНК (показанная темно-оранжевым цветом, вид с торца). Перепечатано
с разрешения Bochkarev et al. (1997), Nature, 385,176-181
быть ограничена участием в процессе репликации катящегося кольца, используемой
фагом X и некоторыми другими бактериофагами Е. coli (раздел 15.1.3).
Однонитевая ДНК является по природе своей «липкой», и два полинуклеотида, от-
деленные действием геликазы, немедленно восстановили бы пары оснований после того,
как этот фермент прошел через них, если бы им это было позволено. Одинарные нити
также очень восприимчивы к воздействию нуклеазы и с большой вероятностью будут
деградированы ею, если не окажутся защищены некоторым образом. Дабы избегнуть
этих нежелательных исходов, белки однонитевого связывания (SSB) прикрепляются
к полинуклеотидам и препятствуют их воссоединению или деградации (рис. 15.15, а).
SSB Е. coli состоит из четырех идентичных субъединиц и, вероятно, работает подобно
главному SSB эукариотов, названному репликационным белком A (RPA), — заключая
полинуклеотид в канал, образуемый рядом SSB, присоединенных один за другим к нити
(рис. 15.15, б). Когда репликационный комплекс становится готовым копировать оди-
нарные нити, должно произойти отделение SSB, которое вызывается вторым набором
белков, названных репликационными посредническими белками (RMP). Как и в слу-
чае геликаз, SSB играют разнообразные роли в различных процессах, предполагающих
раскручивание ДНК.
После того, как 1000-2 000 нуклеотидов опережающей нити будет скопировано,
может начаться первый цикл прерывного синтеза нити на отстающей нити. Праймаза,
которая все еще связана с геликазой DnaB в праймосоме, производит РНК-затравку,
которая после этого продолжается ДНК-полимеразой III (рис. 15.16). Это тот же ком-
плекс ДНК-полимеразы III, который синтезирует копию опережающей нити, этот ком-
Глава 15. Репликация генома 615
Рис. 15.16. Праймирование и синтез копии ДНК
на отстающей нити в процессе репликации ДНК
у Escherichia coli
плекс включает фактически две копии по-
лимеразы, скрепляемые друг с другом парой
т-субъединиц. Это не полные две копии поли-
меразы, потому что существует только един-
ственный у-комплекс, содержащий субъеди-
ницу у в соединении с субъединицами 6, 6',
X и у. Главная функция у-комплекса (иногда
называемого «установщиком зажима») со-
стоит во взаимодействии с р-субъединицей
(«подвижным зажимом») в обеих половинах
комплекса и, следовательно, в управлении
прикреплением и удалением этого фермента
с матрицы; данная функция востребована
прежде всего в ходе репликации отстающей
нити, когда этот фермент должен присоеди-
няться и отделяться неоднократно в начале
и в конце каждого фрагмента Окадзаки. Не-
которые модели комплекса ДНК-полимеразы
III помещают эти два фермента в противопо-
ложных ориентациях, чтобы отразить раз-
личные направления, в которых происходит
синтез ДНК, в сторону к репликационной вилке на
опережающей нити и от нее — на отстающей нити.
Более вероятно, однако, что ферменты в паре смо-
трят в одну сторону и отстающая нить образует пет-
лю, так что синтез ДНК может протекать параллель-
но, по мере того как комплекс полимераз движется
вперед синхронно с продвижением репликационной
вилки (рис. 15.17).
Комбинацию димера ДНК-полимеразы III
и праймосомы, перемещающуюся по родительской
ДНК и выполняющую большинство репликационных
ДНК-полимеразы III
функций, называют реплисомой. После его прохода
процесс репликации должен быть завершен соедине-
Рис. 15.17. Модель параллельного синтеза копий ДНК на опере-
жающей и отстающей нитях димером из ферментов ДНК-
полимераз III. Как думают, отстающая нить продевается петлей
через свою копию фермента ДНК-полимеразы III показанным
здесь способом, так что и опережающая и отстающая нити
могут копироваться, по мере того как димер продвигается
по реплицируемой молекуле. Два компонента димера ДНК-
полимеразы III не идентичны, потому что в нем есть только
одна копия у-комплекса
Копия отстающей нити
616 Часть 4. Механизмы репликации и эволюции геномов
ДНК-полимераза III
Новая ДНК I Следующий фрагмент
1 Окадзаки
3'
5'
5'
3'
IfllllHIII
ДНК-полимераза III останавливается
по достижении РНК-затравки
£||||||||||||||||||||1111111П111т£
о ----------------------------о
Синтез продолжает
I ДНК-полимераза I
ДНК-лигаза соединяет
I два фрагмента ДНК
! Ill», i Ilin
3'
5'
Рис. 15.18. Череда событий, происходящих при
соединении смежных фрагментов Окадзаки
в ходе репликации ДНК у Escherichia coli. ДНК-
полимераза III не обладает действием экзонукле-
азы 5'->3' и, таким образом, прекращает делать
ДНК, когда достигает РНК-затравки следующего
фрагмента Окадзаки. В этой точке синтез ДНК
продолжается ДНК-полимеразой I, которая, на-
против, обладает действием экзонуклеазы 5'->3'
и работает в единении с РНКазой Н, необходи-
мой для удаления РНК-затравки и замены ее
на ДНК. ДНК-полимераза I обычно также заме-
няет часть ДНК из фрагмента Окадзаки перед
отделением от матрицы. В итоге остается одна
недостающая фосфодиэфирная связь, которая
синтезируется ДНК-лигазой, что завершает этот
шаг процесса репликации
нием отдельных фрагментов Окадзаки.
Это не тривиальное событие, потому что
к одному члену каждой пары смежных
фрагментов Окадзаки все еще прикре-
плена его РНК-затравка в точке, где должно произойти лигирование (рис. 15.18). Табли-
ца 15.2 показывает нам, что такая затравка не может быть удалена ДНК-полимеразой
III, потому что этот фермент не обладает необходимым действием экзонуклеазы 5'->3'.
В этой точке ДНК-полимераза III высвобождает отстающую нить и ее место занимает
ДНК-полимераза I, которая действительно обладает действием экзонуклеазы 5’—>3' и,
таким образом, удаляет затравку, и обычно начало ДНК-компонента фрагмента Окадза-
ки, наряду же с этим, продолжает З'-конец смежного фрагмента в ту область матрицы,
которая высовывается наружу. Эти фрагменты Окадзаки теперь примыкают друг к другу
(соединены встык), при этом конечные области обоих полностью состоят из ДНК. Все, что
осталось, — это поставить на (должное) место отсутствующую фосфодиэфирную связь
(что является функцией ДНК-лигазы), соединяющую оба фрагмента и довершающую
репликацию этой области отстающей нити.
Репликационная вилка у эукариотов: вариации на тему бактерий
Стадия элонгации репликации генома подобна у бактерий и эукариотов, хотя детали
отличаются. Продвижение репликационной вилки у эукариотов поддерживается действием
геликазы, но какая именно из нескольких геликаз эукариотов, которые были определены,
главным образом отвечает за раскручивание ДНК в ходе репликации, не было установлено.
Разъединенным полинуклеотидам препятствуют снова скрепиться друг с другом белки
однонитевого связывания, главный из них у эукариотов представлен белком RPA.
Когда мы исследуем метод, используемый для затравливания синтеза ДНК, мы
встречаем уникальные особенности процесса репликации у эукариотов. Как было
описано выше, присущая эукариотам ДНК-полимераза а сотрудничает с ферментом
праймазой, и вместе они ставят на место РНК-ДНК-затравки в начале копии в лице
ведущей нити и в начале каждого фрагмента Окадзаки. Однако ДНК-полимераза а не
способна осуществлять синтез длинной ДНК, возможно потому, что она не способна
оказывать стабилизирующее действие скользящего зажима, эквивалентное таковому
Глава 15. Репликация генома 617
Р-субъединицы ДНК-полимеразы III Е. coli или вспомогательного белка PCNA, который
помогает ДНК-полимеразе 8 эукариотов. Это означает, что, хотя ДНК-полимераза а спо-
собна продолжать исходную РНК-затравку приблизительно на 20 нуклеотидов ДНК,
она должна быть после этого заменена главным репликационным ферментом — ДНК-
полимеразой б (см. рис. 15.13, б).
Ферменты ДНК-полимеразы б, которые копируют опережающую и отстающую
нити у эукариотов, не связываются в димерный комплекс, эквивалентный образуемому
ДНК-полимеразой III в ходе репликации у Е. coli. Вместо этого, две копии полимеразы
остаются раздельными. Функция, выполняемая у-комплексом полимеразы Е. coli, —
управление прикреплением и отсоединением фермента от отстающей нити — осу-
ществляется многосубъединичным вспомогательным белком, названным фактором
репликации С (RFC).
Как и у Е. coli, для завершения синтеза отстающей нити необходимо удаление
РНК-затравки с каждого фрагмента Окадзаки. Как оказалось, у эукариотов нет ни одной
ДНК-полимеразы, обладающей действием экзонуклеазы 5'->3', необходимым для этой
цели, и данный процесс поэтому сильно отличается от описанного на примере клеток
бактерий. Главным игроком здесь выступает «эндонуклеаза откидной створки» FEN1
(ранее называемая MF1), которая связывается с комплексом ДНК-полимеразы б на
З’-конце фрагмента Окадзаки, с тем чтобы деградировать затравку с 5’-конца смеж-
ного фрагмента. Понять точно, как это происходит, сложно из-за неспособности FEN1
запускать деградацию затравки. Она не способна удалять рибонуклеотид на самом 5'-
конце затравки, так как этот рибонуклеотид несет 5’-трифосфатную группу, которая
блокирует действие FEN1 (рис. 15.19). Одна возможность состоит в том, что большая
часть РНК-компонента затравки удаляется РНКазой Н, которая может деградировать
РНКовую часть спаренного основаниями гибрида РНК-ДНК, но не может расщеплять
фосфодиэфирную связь между последним рибонуклеотидом и первым дезоксирибону-
клеотидом. Однако этот рибонуклеотид будет нести 5'-монофосфат, а не 5'-трифосфат и,
таким образом, может быть удален FEN1 (рис. 15.20, а). Эта модель имеет то неудобство,
что она приписывает значительную роль РНКазе Н, тогда как экспериментальные дан-
ные свидетельствуют о том, что клетки, в которых отсутствует РНКаза Н, по-прежнему
способны осуществлять репликацию отстающей нити. Альтернативная возможность
состоит в том, что геликаза разрывает пары оснований, удерживая затравку в контакте
Рис. 15.19. «Эндонуклеаза откидной створки» FEN1 не может начать деградацию затравки, потому что
ее активность блокируется трифосфатной группой, находящейся на 5'-конце РНК-затравки
618 Часть 4. Механизмы репликации и эволюции геномов
а) Модель с РНКазой Н
Следующий фрагмент
Новая ДНК Окадзаки
т 4. । т 5-
I
РНКаза Н удаляет затравку вплоть
до последнего рибонуклеотида
______________
1111II11111 ITTTTftlTlI 111ГП П1 IT
FEN1 удаляет этот
последний рибонуклеид
и некоторое количество ДНК
6) Модель с откидной створкой
Следующий фрагмент
Новая ДНК Окадзаки
* ТншмиинГ
3_.__.__.
tilllllllllllIII
ДНК-полимераза 6
+ геликаза отстраняет
затравку
IIIIIIHIIIIIIIIITHTM
FEN1 производит отрез
в точке ветвления
I Недостающая
" фосфодиэфирная
UIIIIII
ДНК-лигаза
соединяет оба
| фрагмента ДНК
1
Т11111 JI 11111111111111Г1И111 и I!
связь
IIIIIIIIIIIIIIIIITTTTT1TI
ilium
ДНК-лигаза
соединяет оба
I фрагмента ДНК
к
11! 111111! 11111111111111Т.II17
Рис. 15.20. Две модели завершения репликации отстающей нити у эукариотов. Новая ДНК (красная нить)
синтезируется ДНК-полимеразой 6, но этот фермент не показан, дабы не загромождать рисунки
с матричной нитью, что позволяет затравке быть отстраненной ДНК-полимеразой 6, по
мере того как она продолжает смежный фрагмент Окадзаки в освобождаемую таким
образом область (рис. 15.20, б). Откидная створка, которая получается при этом, может
быть отрезана FEN1, которая благодаря действию эндонуклеазы может расщепить
фосфодиэфирную связь в точке ветвления, где отстраненная область прикрепляет-
ся к части фрагмента, которая все еще спарена. Эта схема увеличивает вероятность,
что и РНК-затравка и вся ДНК, первоначально синтезируемая ДНК-полимеразой а,
удаляются. Такая модель привлекательна, потому что ДНК-полимераза а не обладает
корректирующим действием 3’->5’ (см. табл. 15.2) и поэтому синтезирует ДНК в отно-
сительно подверженной ошибкам манере. Удаление этой области как части откидной
створки, расщепляемой FEN 1, сопровождаемое повторным синтезом ДНК-полимеразой
6 (которая, напротив, обладает корректирующим действием и, таким образом, произ-
водит очень точную копию матрицы), препятствует этим ошибкам стать постоянными
составляющими дочерней двойной спирали.
И последнее отличие между репликацией у бактерий и эукариотов — то, что у эука-
риотов нет никакого эквивалента реплисоме бактерий. Вместо этого, ферменты и белки,
вовлеченные в репликацию, формируют внутри ядра структуры порядочного размера,
при этом каждая такая структура содержит сотни или тысячи отдельных репликационных
комплексов. Эти структуры неподвижны из-за прикрепления к ядерному матриксу, так что
молекулы ДНК продвигаются через комплексы по мере их репликации. Такие структуры
упоминаются как репликационные фабрики и фактически могут быть составляющими
процесса репликации также и у бактерий, по крайней мере у некоторых.
Глава 15. Репликация генома 619
Репликация генома у археи
Мы имеем мало прямой информации о репликации ДНК у архей, большая часть того,
что мы знаем, была выведена в процессе поиска в геномах архей генов и прочих после-
довательностей, подобных компонентам репликационного аппарата бактерий и (или)
эукариотов. Начальные попытки определять местонахождение областей инициации
в геномах архей путем поиска мотивов последовательности, обнаруженных в областях
инициации у бактерий или эукариотов, были неудачны. Впоследствии потенциальные
области инициации для целого ряда видов архей были определены статистическим
анализом частот встречаемости четырех нуклеотидов в различных частях всех исследуе-
мых геномов архей, при этом логическим обоснованием послужило то, что эти частоты
могут значительно разниться лишь с обеих сторон от всякой области инициации, как
это имеет место у бактерий. Для одного вида, Pyrococcus abyssi, потенциальная область
инициации, определенная анализом частоты встречаемости нуклеотидов, лежит в об-
ласти генома, которая реплицируется в первую очередь и, таким образом, может быть
истинной областью инициации.
Последовательности большинства белков, вовлеченных в стадию элонгации ре-
пликации геномов архей, как предсказано по их генам, являются подобными эквива-
лентным версиям таковых, свойственных эукариотам. В частности, археи имеют белки,
которые являются гомологами белков RFC и PCNA эукариотов. ДНК-полимераза архей
представляет интерес, потому что субъединица, которая определяет действие синтеза
ДНК, подобна эквивалентной субъединице ДНК-полимеразы 5 эукариотов, тогда как
корректирующая функция присуща белку, который оказался гомологом е-субъединицы
ДНК-полимеразы III Escherichia coli.
15.2.3. Терминация репликации
Репликационные вилки продвигаются вдоль линейных геномов, или вокруг кольцевых,
обычно беспрепятственно, кроме тех случаев, когда встречается транскрибируемая область.
Синтез ДНК происходит с приблизительно в пять раз большей скоростью, чем синтез РНК,
так что репликационный комплекс может легко настигнуть РНК-полимеразу, но это, веро-
ятно, не случается: вместо этого, как думают, репликационная вилка берет паузу позади
РНК-полимеразы и продвигается дальше только тогда, когда транскрипт будет завершен.
В конечном счете репликационная вилка достигает конца молекулы или встречает
вторую репликационную вилку, движущуюся во встречном направлении. Что случается
после этого — один из наименее понятых моментов репликации генома.
Репликация генома Е. coli завершается в пределах определенной области
Геномы бактерий реплицируются в обоих направлениях, начиная от одной точки
(см. рис. 15.8), а это означает, что две репликационные вилки должны встретиться в не-
которой позиции, диаметрально противоположной области инициации на карте генома.
Однако если одна вилка запаздывает (возможно потому, что ей приходится реплициро-
вать протяженные области, в которых происходит транскрипция), то может оказаться
так, что другая вилка проскочит лежащую на полпути точку и продолжит репликацию
на «другой стороне» генома (рис. 15.21). На первый взгляд не ясно, почему это должно
быть нежелательно, ибо дочерние молекулы, как кажется, никак от этого не пострадают,
но возникновению такой ситуации препятствуют стоп-последовательности. Шесть
из них были обнаружены в геноме Е. coli (рис. 15.22, а), при этом каждая из них служит
сайтом узнавания для специфичного к последовательности ДНК-связывающего белка
по имени Tus.
620 Часть 4. Механизмы репликации и эволюции геномов
Исток репликации Рис. 15.21. Ситуация, которая не должна происходить в ходе
О репликации кольцевого генома Escherichia coli. Одна из репли-
кационных вилок прошла некоторое расстояние за лежащую на
полпути точку. Это не случается в ходе репликации ДНК у Е. coli
благодаря действию белков Tus (см. рис. 15.22, б)
Принцип действия Tus весьма необычен. Будучи
связан со стоп-последовательностью, белок Tus по-
зволяет репликационной вилке проходить, если вилка
движется в одном направлении, но блокирует продви-
жение, если вилка движется во встречном направле-
нии вокруг генома. Направленность устанавливается
ориентацией белка Tus на двойной спирали. Когда
---------------------- геликаза DnaB приближается с одного направления,
Tus блокирует прохождение этого фермента, который
отвечает за продвижение репликационной вилки, потому что геликаза наталкивается
на «стенку» из 0-нитей, через которую она не может проникнуть. Но при приближении
с другого направления геликаза DnaB способна разрушить структуру белка Tus, воз-
можно, за счет воздействия, которое раскручивание двойной спирали оказывает на Tus,
и, таким образом, миновать эту преграду (рис. 15.22, б).
Ориентация стоп-последовательностей и, следовательно, связанных с ними белков
Tus в геноме Е. coli — такая, что обе репликационные вилки улавливаются в пределах
относительно короткой области на противоположной относительно области инициации
стороне генома (см. рис. 15.22, а). Это гарантирует, что терминация всегда происходит
в (или около) одной и той же позиции. Что случается, когда две репликационные вилки
встречаются, точно не известно, но это событие сопровождается разборкой реплисом
(или самопроизвольно, или управляемым способом). В результате получаются две
связанные между собой дочерние молекулы, которые отделяются друг от друга топо-
изомеразой IV.
О терминации репликации у эукариотов известно не много
У эукариотов не было обнаружено никаких последовательностей, эквивалентных
стоп-строкам бактерий, не были у них опознаны и белки, подобные Tus. Весьма возмож-
но, репликационные вилки встречаются в случайных позициях, и терминация попро-
сту состоит в лигировании концов новых полинуклеотидов. Мы знаем наверняка, что
репликационные комплексы не разбираются, потому что эти репликационные фабрики
являются постоянными составляющими ядра.
Вместо того, чтобы сосредоточиваться на молекулярных событиях, происходящих,
когда встречаются репликационные вилки, внимание было направлено на трудный во-
прос о том, как дочерние молекулы ДНК, произведенные в ядре эукариотов, не становятся
безнадежно запутанными. Хотя ДНК-топоизомеразы имеют способность распутывать
молекулы ДНК, существует мнение, что запутывание поддерживается на минимальном
уровне, с тем чтобы можно было избежать множественных реакций разрыва и воссоеди-
нения, катализируемых топоизомеразами (см. рис. 15.4). Чтобы решить эту проблему,
были предложены различные модели. Одна из них предполагает, что хромосома эукарио-
тов упакована не случайным образом в своей территории внутри ядра (раздел 10.1.1), но
упорядочена вокруг репликационных фабрик, которые, кажется, присутствуют только
в ограниченном числе случаев. Предполагается, что каждая фабрика реплицирует от-
Глава 15. Репликация генома 621
а) Стоп-последовательности в геноме E.coli
Область, в которой репликационные
вилки становятся «запертыми»
б) Роль Tus
I Репликационная вилка может
I пройти белок Tus, прикрепленный
" в одной ориентации,..
но блокируется белком Tus,
I прикрепленным в другой
▼ ориентации
Рис. 15.22. Роль стоп-последовательностей в процессе репликации ДНК у Escherichia coli. а) Показаны
позиции шести стоп-последовательностей в геноме Е. coli, причем стрелки указывают направление,
в котором каждая стоп-последовательность может быть пройдена репликационной вилкой, б) Связанные
белки Tus позволяют репликационной вилке проходить, когда вилка приближается с одного направления,
и не пропускают ее, когда она приближается с другого направления. На рисунке показана репликацион-
ная вилка, проходящая левый Tus, потому что геликаза DnaB, которая продвигает вилку вперед, может
разрушить белок Tus, когда она приближается к нему с этого направления. Затем эта вилка блокируется
вторым белком Tus, потому что он обращен своей непроницаемой стенкой из p-нитей лицом к вилке
дельную область ДНК, поддерживая дочерние молекулы в определенном взаимном
расположении, которое позволяет избежать их запутывания. Изначально две дочерние
молекулы скрепляются вместе белками когезинами, которые прикрепляются незамед-
лительно после прохода репликационной вилки посредством процесса, который, кажется,
вовлекает ДНК-полимеразу к — загадочный фермент, очень важный для репликации,
но единственная его известная роль не требует
явного действия ДНК-полимеразы. Когезины
поддерживают выравнивание сестринских хро-
матид до стадии анафазы деления ядра, когда
они расщепляются режущими белками, позво-
ляя дочерним хромосомам отделиться друг от
друга (рис. 15.23).
Рис. 15.23. Когезины. Белки когезины прикрепляют-
ся немедленно после прохода репликационной вилки
и удерживают дочерние молекулы вместе до наступления
анафазы. В течение анафазы когезины расщепляются, по-
зволяя реплицированным хромосомам отделиться друг
от друга до их распределения по дочерним ядрам (см.
рис. 3.15)
Репликация ДНК
Анафаза
I
ППППППи
622 Часть 4. Механизмы репликации и эволюции геномов
15.2.4. Поддержание концов линейной молекулы ДНК
И последняя проблема, которую нам осталось рассмотреть, прежде чем мы оста-
вим процесс репликации, касается шагов, которые должны быть предприняты, чтобы
оградить концы линейной двунитевой молекулы от постепенного укорочения в ходе
последовательных циклов репликации хромосомной ДНК. Есть два пути, которыми
такое укорочение может произойти,
• Крайний З'-конец отстающей нити может быть не скопирован, потому что послед-
ний фрагмент Окадзаки не может быть затравлен, так как естественная позиция
для участка его затравливания должна бы располагаться за концом матрицы
(рис. 15.24, а). Отсутствие этого фрагмента Окадзаки означает, что копия в лице
отстающей нити короче, чем она должна быть. Если копия сохраняет эту длину, то,
когда она служит родительским полинуклеотидом в следующем круге репликации,
полученная дочерняя молекула будет короче, чем ее прародитель.
• Если затравка для последнего фрагмента Окадзаки помещена на самом З'-конце
отстающей нити, то укорочение по-прежнему будет происходить, хотя и в меньшей
степени, потому что эта оконечная РНК-затравка не может быть преобразована
в ДНК стандартными процессами удаления затравки (рис. 15.24, б). Это связано
с тем, что методы для удаления затравки (подобные представленным на рис. 15.18
на примере бактерий и на рис. 15.20 на примере эукариотов) требуют продолжения
З'-конца смежного фрагмента Окадзаки, который не может пребывать на самом
конце молекулы.
Как только эта проблема была осознана учеными мужами, их внимание обратилось
к теломерам — необычным последовательностям ДНК на концах хромосом эукариотов.
В разделе 7.1.2 мы отмечали, что теломерная ДНК состоит из минисателлитной после-
довательности определенного типа, состоящей из множественных копий короткого
повторного мотива 5'-TTAGGG-3' у большинства высших эукариотов; несколько сотен
копий этой последовательности встречается в тандемных повторениях на обоих кон-
цах каждой хромосомы. Решение проблемы укорочения концов заключается в способе,
которым эта теломерная ДНК синтезируется.
Теломерная ДНК синтезируется ферментом теломеразой
Большая часть теломерной ДНК копируется обычным способом в процессе реплика-
ции ДНК, но это не единственный путь, которым она может быть синтезирована. Чтобы
компенсировать ограничения процесса репликации, теломеры могут быть продолжены
независимым механизмом, катализируемым ферментом теломеразой. Этот фермент
необычен тем, что он состоит из белка и РНК. В ферменте человека РНКовый компонент
имеет 450 нуклеотидов в длину и содержит около своего 5'-конца последовательность
5'-CUAACCCUAAC-3', центральная область которой является обратным комплементом
присущей теломере человека повторной последовательности 5'-TTAGGG-3'. Это позво-
ляет теломеразе продолжать теломерную ДНК на З'-конце полинуклеотида с помощью
механизма копирования, показанного на рис. 15.25, в котором теломеразная РНК ис-
пользуется в качестве матрицы для каждого шага продолжения, при этом синтез ДНК
осуществляется белковым компонентом фермента, представляющим собой обратную
транскриптазу. Достоверность этой модели подтверждается сравнениями между те-
ломерными повторными последовательностями и теломеразными РНК других видов
(таблица 15.3): во всех организмах, которые были изучены, теломеразная РНК содержит
последовательность, которая позволяет ей производить копии повторного мотива, при-
Глава 15. Репликация генома 623
а) Последний фрагмент Окадзаки не может быть затравлен
Опережающая нить Конец хромосомы
5 ТТ111П11Н|1>..........I 5- D
о/ 111111111111Родительская молекула
g.TT*l 111II11 Г. ----3
5------
Отстающая нить
Репликация
копии
опережающей
нити
15’
...................II..............
тт 11111111111111111111111111^
Две дочерние молекулы
___ЭуГ-Х
Недостающий фрагмент Окадзаки
Одна молекула второго поколения
Молекула стала короче
б) Затравка для последнего фрагмента Окадзаки
находится на самом 3'-конце отстающей нити
Опережающая нить Конец хромосомы
Отстающая нить
Родительская молекула
3' 5'
II IIIIIIIIIIIII......
° ° Две дочерние молекулы
5* - - 11' 111 1- -- ' Затравка последнего
Репликация / ; : фрагмента Окадзаки
копии I : :
опережающей I >
нити \
II Т] 11111111И 11111111111 Одна молекула второго поколения
Молекула стала короче
Рис. 15.24. Две причины, по которым линейные молекулы ДНК могут стать короче после репликации
ДНК. В обоих примерах родительская молекула реплицируется нормальным способом. Полная копия
получается из ее опережающей нити, но в примере а копия из отстающей нити неполна, потому что
последний фрагмент Окадзаки не был сделан. Это случилось потому, что затравки для фрагментов
Окадзаки синтезируются на отстающей нити в позициях, отстоящих приблизительно на 200 п. н. друг от
друга. Если один фрагмент Окадзаки начинается в какой-либо позиции, лежащей менее чем на 200 п. н.
от 3-конца отстающей нити, то не будет места для следующего затравливающего участка, и оставшийся
сегмент отстающей нити не будет скопирован. Поэтому полученная в итоге дочерняя молекула имеет
выступающий З'-конец и, когда реплицируется, дает начало молекуле второго поколения, которая короче,
чем исходная родительская молекула. В примере б последний фрагмент Окадзаки может быть помещен
на самом З'-конце отстающей нити, но его РНК-затравка не может быть преобразована в ДНК, потому
что это потребовало бы продолжения следующего фрагмента Окадзаки, помещенного за пределами
конца отстающей нити. Не ясно, может ли оконечная РНК-затравка сохраняться на протяжении всего
клеточного цикла, и также неясно, можетли сохранившаяся РНК-затравка быть скопирована в ДНК входе
последующего круга репликации ДНК. Если затравка не сохраняется или же не копируется в ДНК, то одна
из молекул второго поколения будет короче, чем первоначальная родительская молекула
624 Часть 4. Механизмы репликации и эволюции геномов
Хромосомная ДНК Теломеразная РНК
TTAGGGTTAGGG3
.CAAUCCCAAUC
3'
5'
^Синтез ДНК
Новая ДНК
5'
^==TTAGGGTTAGGGTTAG
CAAUCCCAAUC
3'
5'
| Перемещение
з'
’’^TTAGGGTTAGGGTTAG
Рис. 15.25. Наращивание конца хромосомы человека
посредством теломеразы. Показан З'-конец молеку-
лы ДНК хромосомы человека. Последовательность
включает повторения мотива 5'-TTAGGG-3' теломеры
человека. Теломеразная РНК спаривается с концом
молекулы ДНК, которая продолжается на короткое
расстояние, длина же этого продолжения, возмож-
но, определяется присутствием в теломеразной РНК
структуры в виде петли стебля. Теломеразная РНК
далее перемещается на новую позицию спаривания
немного далее по полинуклеотиду ДНК, и молекула
продолжается еще на несколько нуклеотидов. Этот
процесс может повторяться до тех пор, пока конец
хромосомы не будет продолжен на достаточное число
нуклеотидов
CAAUCCCAAUC
3'
’’^TTAGGGTTAGGGTTAGGGTTAG
I I I I I I I I I I I
^CAAUCCCAAUC^
3' 5'
I Очередной синтез ДНК сутствующего в теломерах организма. С этим
▼ связана интересная особенность, что во всех
организмах нить, синтезируемая теломера-
зой, характеризуется преобладанием в ней
нуклеотидов G и упоминается поэтому как
«обогащенная G нить».
Теломераза может синтезировать только такую обогащенную G нить. Не ясно, как
другой полинуклеотид — обогащенная С нить — продолжается, но предполагается, что,
когда обогащенная G нить достаточно длинна, праймаза — комплекс ДНК-полимера-
зы а— прикрепляется своим концом и запускает синтез комплементарной ДНК обычным
способом (рис. 15.26). Для этого необходимо использование новой РНК-затравки, так что
обогащенная С нить по-прежнему будет короче, чем обогащенная G, но важный момент
здесь состоит в том, что общая длина хромосомной ДНК не была уменьшена.
Ясно, что действие теломеразы должно управляться очень точно, чтобы гаран-
тировать, что на каждом конце хромосомы производится продолжение соответствую-
щей длины. Одна часть этого регулирующего механизма обеспечена белками TRF1,
которые связываются с повторными последовательностями теломер (раздел 7.1.2).
TRF1 вызывает формирование свернутой структуры хроматина, которая препятству-
^ППТТПТТГГТ?.т
□--- о
I Теломераза продолжает
выступ на З'-конце
ет доступу фермента теломеразы к концу
хромосомы. По мере укорочения теломеры
число связанных белков TRF1 уменьшается
и структура хроматина раскрывается, по-
т г- Новая ДНК
5~ ~~............
I После синтеза ДНК достаточной
длины может быть затравлен
новый фрагмент Окадзаки
Фрагмент Окадзаки Затравка
ТТ Т1 111 1 1 1 I 'l l'
Рис. 15.26. Завершение процесса наращивания
нити на конце хромосомы. Предполагается, что
после того как теломераза продолжит З'-конец на
достаточное расстояние, как показано на рис. 15.25,
новый фрагмент Окадзаки затравливается и синте-
зируется, преобразуя З'-продолжение в полностью
двунитевой конец
Глава 15. Репликация генома 625
Таблица 15.3. Последовательности теломерных повторений и теломеразных РНК у различных организ-
мов
Вид Последовательность теломерных повторений Матричная последовательность теломеразной РНК
Человек Oxytricha Tetrahymena 5'-TTAGGG-3' 5'-TTTTGGGG-3' 5'-TTGGGG-3' 5'-CUAACCCUAAC-3' 5'-СААААССССААААСС-3' 5'-СААССССАА-3'
Примечание: Oxytricha и Tetrahymena — простейшие, которые особенно полезны для изучения теломер,
потому что на некоторых стадиях развития их хромосомы распадаются на маленькие фрагменты, и при этом
все они содержат теломеры: поэтому они имеют много теломер в пересчете на одну клетку.
зволяя теломеразе прикрепиться к концу хромосомы и продолжить теломеру. По мере
элонгации теломеры белки TRF1 снова прикрепляются к ней, побуждая хроматин воз-
вратиться к его свернутой структуре, с тем чтобы теломераза снова была изгнана с конца
хромосомы. В результате белки TRF1 опосредствуют петлю отрицательной обратной
связи, которая регулирует действие теломеразы на всяком отдельно взятом конце хро-
мосомы. В клетках млекопитающих закрытая структура хроматина может предполагать
формирование «t-петли», в которой свободный З'-конец теломеры загибается петлей
назад, вторгается в двойную спираль и образует пары оснований с комплементарной
ей последовательностью на обогащенной С нити (рис. 15.27). Эта реакция катализиру-
ется вторым связывающимся с теломерой белком человека (TRF2) и может обеспечить
дополнительную стабилизацию конца хромосомы, который не требует дальнейшего
продолжения.
Длина теломеры причастна к старению клетки и раку
Как это ни удивительно, теломераза активна не во всех клетках млекопитающих.
Этот фермент является функционально активным в эмбрионе на ранних стадиях разви-
тия, но после рождения активен только в репродуктивных клетках и стволовых клетках.
Последние суть клетки-предшественники, которые непрерывно делятся в течение всего
периода жизни организма и производят новые клетки для поддержания органов и тканей
в функционально активном состоянии. Наиболее изученные примеры — кроветворные
стволовые клетки костного мозга, которые производят новые клетки крови.
Клетки, в которых отсутствует действие теломеразы, подвергаются укорочению
хромосом каждый раз, когда они делятся. В конечном счете, после многих клеточных
делений, концы хромосомы могут стать настолько обрезанными, что жизненно не-
обходимые гены будут потеряны, но это вряд ли бывает главной причиной дефектов,
которые могут произойти в клетках, испытывающих
недостаток в активности теломеразы. Вместо этого,
определяющий фактор — необходимость поддержи-
вать белковый «кэп» на каждом конце хромосомы,
дабы защитить эти концы от воздействий ферментов
Рис. 15.27. «t-петля», t-петля образуется, когда свободный
З'-конец теломеры загибается петлей назад и вторгается в двой-
ную спираль
626 Часть 4. Механизмы репликации и эволюции геномов
репарации ДНК, которые соединяют вместе декапированные концы, появляющиеся
в результате случайного разрыва хромосомы (раздел 16.2.4). Белки, которые форми-
руют этот защитный кэп (например, TRF2 у людей), опознают повторения в теломерах
в качестве своих последовательностей связывания и, таким образом, не имеют точек
присоединения после того, как теломеры были удалены. Если эти белки отсутствуют, то
ферменты репарации могут создать неуместные связи между концами неповрежденных,
хотя и укороченных хромосом; именно это, вероятно, и является основной причиной
прерывания клеточного цикла, которое следует за укорочением теломеры.
Поэтому укорочение теломеры будет приводить к пресечению последовательно-
сти клеточных поколений. В течение нескольких лет биологи пытались связать этот
процесс со старением клетки — явлением, первоначально наблюдаемым в клеточных
культурах. Все нормальные клеточные культуры имеют ограниченный срок жизни:
после некоторого числа делений клетки входят в состояние старения, в котором они
по-прежнему остаются живыми, но уже не могут делиться (рис. 15.28). В некоторых
последовательностях клеточных поколений у млекопитающих, в особенности в куль-
турах фибробластов (клеток соединительной ткани), старение может быть отсрочено,
если спроектировать клетки так, чтобы они синтезировали активную теломеразу. Эти
эксперименты предполагают явную зависимость между укорочением теломеры и ста-
рением, но непреложность такой зависимости была подвергнута сомнению, и любая
экстраполяция со старения клетки на старение организма преисполнена трудностей.
Не все клеточные линии проявляют старение. Злокачественные клетки способны
делиться непрерывно в культуре, их бессмертие рассматривается как аналогичное
росту опухоли в неповрежденном организме. При раке нескольких типов такое отсут-
ствие старения связано с активацией теломеразы, иногда до такой степени, что длина
теломеры поддерживается в течение многократных клеточных делений, но часто та-
ким образом, что теломеры ста-
новятся длиннее нормальных,
потому что теломераза сверхак-
тивна. Не ясно, является акти-
вация теломеразы причиной или
следствием рака, хотя первое ка-
жется более вероятным, потому
что по крайней мере рак одного
типа, врожденный дискератоз,
по всей очевидности, происте-
кает из мутации в гене, опреде-
ляющем РНК-компонент тело-
меразы человека. Этот вопрос
является определяюще важным
для понимания этиологии рака,
но имеет меньшее отношение
к терапевтической проблеме,
Клетки состарились -
деление прекращается
Рис. 15.28. Культивируемые клетки ста-
реют после многократных клеточных
делений
Глава 15. Репликация генома 627
которая сосредоточивается на том, может ли теломераза быть мишенью для лекарств,
разработанных для борьбы с раком. Такая терапия может быть успешной, даже если
активация теломеразы — следствие рака, потому что инактивация лекарствами вызвала
бы старение раковых клеток и, следовательно, предотвратила бы их разрастание.
Теломеры у Drosophila
Когда аминокислотные последовательности белковых субъединиц ферментов
теломераз сравнивают с таковыми других обратных транскриптаз, наиболее близкие
подобия наблюдаются с обратными транскриптазами, кодируемыми не содержащими
LTR ретроэлементами, названными ретропозонами (раздел 9.2.1). Это явление кажется
особенно интересным, когда рассматривается в контексте необычной структуры тело-
мер Drosophila. Эти теломеры не состоят из коротких повторяющихся последователь-
ностей, наблюдаемых в большинстве других организмов, но вместо этого состоят из
тандемных массивов намного более длинных повторений — 6 или 10 тыс. п. н. в длину.
Эти повторения представляют собой полномерные копии двух ретропозонов Drosophila,
родственных LINE 1 человека и названных НеТ-А и TART. Не известно, как эти теломеры
поддерживаются, но вполне возможно, что этот процесс аналогичен таковому, выполняе-
мому теломеразой, при этом матричная РНК производится транскрипцией теломерных
ретропозонов, копируемых обратной транскриптазой, кодируемой последовательно-
стями TART [НеТ-А не имеет гена обратной транскриптазы).
Необычная структура теломеры Drosophila может попросту быть причудой Матуш-
ки Природы, но привлекательная возможность того, что теломеры других организмов
представляют собой деградированные ретропозоны, как предполагают подобия между
теломеразой и обратными транскриптазами ретропозонов, нельзя сбрасывать со сче-
тов.
15.3. Регулирование репликации генома у эукариотов
Репликация генома в клетках эукариотов регулируется на двух уровнях:
• репликация согласовывается с клеточным циклом так, чтобы в момент деления
клетки в наличии имелись две копии генома;
• сам по себе процесс репликации может быть задержан при некоторых обстоятель-
ствах, например, если ДНК повреждена и должна быть восстановлена прежде, чем
может быть завершено копирование.
Мы завершаем эту главу рассмотрением этих регуляторных механизмов.
15.3.1. Согласование репликации генома и деления клетки
Концепция клеточного цикла возникла на основании исследований световой
микроскопии, проводимых первыми цитобиологами. Их наблюдения показали, что
делящиеся клетки проходят через повторяющиеся циклы митоза — периода, когда
происходит деление ядра и клетки (см. рис. 3.15), — и интерфазы — менее значитель-
ного периода, в котором с помощью светового микроскопа можно пронаблюдать лишь
немногие динамические изменения. Было обнаружено, что хромосомы делятся во
время интерфазы, так что, когда ДНК была отождествлена с генетическим материалом,
интерфазе было придано важное значение — как периоду, в который должна иметь
место репликация генома. Это привело к новой интерпретации клеточного цикла как
процесса из четырех стадий (рис. 15.29):
628 Часть 4. Механизмы репликации и эволюции геномов
Рис. 15.29. Клеточный цикл. Длины отдельных
фаз изменяются у разных клеток. Сокращения:
G1 и G2 — промежуточные фазы; М — митоз;
S — фаза синтеза
• митоз, или М-фаза — период, в кото-
ром ядро и клетка делятся;
• промежуток 1, или Gl-фаза — интер-
вал, во время которого происходят
транскрипция, трансляция и другие
общие клеточные отправления;
• синтез, или S-фаза — когда геном ре-
плицируется;
• промежуток 2, или GZ-фаза— второй
интервальный период.
Ясно, что важно, чтобы S-фаза
и М-фаза были согласованы так, чтобы
геном полностью реплицировался, но был реплицирован только один раз, прежде чем
произойдет митоз. Периоды непосредственно перед вхождением в S-фазу и М-фазу рас-
сматриваются как ключевые контрольные точки клеточного цикла, и именно в одной
из этих двух точек цикл становится задержанным, если критически важные гены, во-
влеченные в управление клеточным циклом, мутированы или если клетка подвергается
травме типа обширного повреждения ДНК. Поэтому попытки понять, как репликация
генома согласуется с митозом, были направлены на эти две контрольные точки, особенно
на контрольную точку npe-S — период непосредственно перед репликацией.
Образование предрепликационного комплекса позволяет начать реплика-
цию генома
Исследования (главным образом над Saccharomycescerevisiae) привели к модели управ-
ления выбором времени S-фазы, которая постулирует, что репликация генома требует
построения предрепликационных комплексов (предРК) в областях инициации; эти
предРК превращаются в постРК после начала репликации. ПостРК не способен запустить
репликацию и, таким образом, не может случайно перекопировать часть генома, прежде
чем произошел митоз. ORC — комплекс из шести белков, которые собираются на доменах
А и В1 области инициации дрожжей (см. рис. 15.10, б), — был первым претендентом на роль
предРК, но, вероятно, он не является центральным компонентом, так как ORC присутствуют
в точках начала репликации на всех стадиях клеточного цикла. Вместо этого, ORC рассма-
тривают как «посадочную площадку», на которой строится предРК.
В качестве компонентов предРК рассматривались белки различных типов. Пер-
вый — Cdc6p, который был первоначально опознан в дрожжах (а впоследствии было
показано, что он имеет гомологов у высших эукариотов). Cdc6p дрожжей синтезируется
в конце фазы G2, когда клетка входит в митоз, и становится связанным с хроматином
в начале фазы G1 перед исчезновением в конце фазы G1, когда начинается репликация
(рис. 15.30). Причастность Cdc6p к предРК предполагается на основании экспериментов,
в которых его ген подавляется и это приводит к отсутствию предРК, а также других
экспериментов, в которых Cdc6p производится в чрезмерных количествах, что ведет
к многократным репликациям генома в отсутствие митоза. Есть также биохимические
Глава 15. Репликация генома 629
Рис. 15.30. График, показывающий
количество Cdc6p в ядре на разных
стадиях клеточного цикла
данные о прямом взаимодействии между Cdc6p
и ORC дрожжей.
Второй компонент предРК, как думают, пред-
ставлен группой белков, названных факторами
лицензирования репликации (RLF)- Как и в случае
Cdc6p, первые примеры этих белков были опозна-
ны у дрожжей (семейство белков МСМ), а гомологи
у высших эукариотов открыты позднее. RLF связы-
ваются с хроматином ближе к концу М-фазы и оста-
ются на месте до начала S-фазы, после которой они
постепенно удаляются с ДНК по мере ее репликации.
Их удаление может быть ключевым событием, кото-
рое превращает предРК в постРК и, таким образом,
предотвращает повторную инициацию репликации в той области инициации, которая
уже направляла круг репликации.
Регулирование сборки предРК
Определение компонентов предРК несколько приближает нас к пониманию того,
как запускается репликация генома, но все еще оставляет открытым вопрос о том, как
репликация согласуется с другими событиями клеточного цикла. Управление клеточным
циклом — сложный процесс, опосредствованный главным образом протеинкиназами,
которые фосфорилируют и активируют ферменты и другие белки, которые имеют
определенные функции в ходе клеточного цикла. Те же самые протеинкиназы присут-
ствуют в ядре в течение всего клеточного цикла, так что они сами должны подвергаться
управлению. Это управление осуществляется частично белками, названными цикли-
нами (относительное содержание которых изменяется на разных стадиях клеточного
цикла), частично другими протеинкиназами, которые активируют циклинозависимые
протеинкиназы, и частично ингибирующими белками. Даже прежде, чем мы начнем
рассматривать регуляторы сборки предРК, мы можем ожидать, что система управления
окажется довольно замысловатой.
Ряд циклинов был связан с активацией репликации генома и с предотвращением
повторной сборки предРК после завершения репликации. К ним относятся митотические
циклины, главная функция которых, согласно первым предположениям, заключалась
в активации митоза, но которая предполагает и подавление репликации генома. Когда
воздействия этих циклинов блокируются (например, за счет перепроизводства белков,
которые подавляют их активность), клетка не только неспособна войти в М-фазу, но
вместе с этим подвергается повторной репликации генома. Существуют также более
специфичные циклины S-фазы, такие как С1Ь5р и С1Ь6р у S. cerevisiae, инактивация кото-
рых задерживает или предотвращает репликацию генома, и другие циклины, которые
являются активными в течение С2-фазы и предотвращают сборку предРК в период
после репликации генома и перед делением клетки (рис. 15.31).
В дополнение к этим циклинозависимым системам управления репликация генома
регулируется также и циклинонезависимой протеинкиназой Cdc7p-Dbf4p, встречающейся
в столь разнообразных организмах, как дрожжи и млекопитающие. Белки, активируемые
этой киназой, не были опознаны, и разрозненные данные позволяют предположить, что
и RLF и ORC являются ее мишенями. Каким бы ни был механизм, активность протеинкиназы
Cdc7p-Dbf4p есть необходимая предпосылка для репликации, а циклинозависимые процессы
сами по себе не достаточны, чтобы подтолкнуть клетку к вступлению в S-фазу.
630 Часть 4. Механизмы репликации и эволюции геномов
Митотические
циклины
Циклины S-фазы
Рис. 15.31. Контрольные точки клеточного цикла для циклинов, вовлеченных в регулирование репли-
кации генома
15.3.2. Управление во время S-фазы
Управление переходом G1-S можно рассматривать как главный процесс управления,
затрагивающий репликацию генома, но он не единственный. Определенные события,
происходящие во время S-фазы, также подчинены регулированию.
Ранние и поздние области инициации
Инициация репликации не происходит в одно и то же время во всех областях ини-
циации, и при этом «включение области инициации» не является абсолютно случайным
процессом. Некоторые части генома реплицируются в начале S-фазы, а некоторые позже,
при этом схема репликации остается постоянной на протяжении всей последователь-
ности делений клеток. Согласно общей схеме активно транскрибируемые гены и цен-
тромеры реплицируются в начале S-фазы, а теломеры и нетранскрибируемые области
генома реплицируются позже. Поэтому рано включаемые области инициации являются
тканеспецифическими и отражают картину экспрессии генома, свойственную клетке.
Этапы
репликации
хромосом
I Извлечение ДНК, центрифугирование
▼ в градиенте плотности
14N-15N-flHK—►
15N-15N-flHK—► —
Градиенты
плотности
^ Гибридизация реплицируемой ДНК на микроматрицах
Микроматрицы
Рис. 15.32. Анализ включения областей инициации у 5. cerevisiae, проводимый на микроматрицах
Глава 15. Репликация генома 631
Эти особенности репликации генома первоначально были экстраполированы
с данных исследования лишь нескольких областей инициации, но были недавно до-
полнены результатами анализа, опирающегося на технологию микроматриц. Анализ на
микроматрицах основан на гибридизационном зондировании, так что для того, чтобы
использовать микроматрицу с целью исследования схемы репликации генома, необхо-
димо изобрести способ отделения нереплицированной ДНК от реплицированной ДНК,
так чтобы та или иная фракция могла быть использована в качестве гибридизационно-
го зонда. Если, например, образец реплицированной ДНК получен из клеток, которые
только что вступили в S-фазу, то такая ДНК может быть использована для зондирования
микроматрицы с целью опознать те гены, которые уже были реплицированы на этой
ранней стадии. Второй зонд, приготовленный из реплицированной ДНК от немного позд-
него этапа S-фазы, позволил бы опознать следующий набор реплицируемых генов и так
далее. Для Saccharomyces cerevisiae эти фракции получают, выращивая клетки в среде
с тяжелым азотом, затем их переносят в нормальную среду и позволяют клеткам войти
в S-фазу. После этого ДНК извлекается в соответствующие периоды времени на протя-
жении S-фазы, обрабатывается эндонуклеазой рестрикции и разделяется на фракции
путем центрифугирования в градиенте плотности (рис. 15.32). В итоге наблюдаются две
полосы: одна состоит из фрагментов 15М-151М-ДНК, полученных из нереплицированного
компонента генома, а другая состоит из фрагментов 14М-15М-ДНК, полученных из об-
ластей, которые подверглись репликации. Последняя фракция очищается, помечается
флюоресцентной меткой и привносится на микроматрицу.
Такой анализ прост, но невероятно информативен. На рис. 15.33, а представлена
карта динамики включения областей инициации по всей длине хромосомы VI дрожжей
и показана область, лежащая на полпути по короткому плечу, в которой начинается
репликация этой хромосомы. Как показали предыдущие эксперименты, центромера
(обозначена кружком на оси X на рис. 15.33, а) реплицируется на раннем этапе S-фазы,
а теломеры — в конце репликационного цикла. Обратите внимание, что обе теломеры
реплицируются приблизительно в одно и то же время. Это наблюдение остается верным
для всех хромосом дрожжей, но не все хромосомы завершают свои репликационные
циклы одновременно. Некоторые (такие как хромосы XI и XV) полностью реплициру-
а) Репликация хромосомы VI б) Репликация хромосом XII и XV
Координаты на хромосоме, тыс. п. н. Время репликации,
минут от начала S-фазы
Рис. 15.33. Динамика включения областей инициации у S. cerevisiae. а) Хронометраж включения областей
инициации по длине хромосомы VI. Стрелка обозначает точку, в которой репликация этой хромосомы
начинается, а кружок на оси X — центромеру. Пустая область — область хромосомы, которую невозможно
было исследовать, б) Сравнение между динамиками репликации хромосом XII и XV
632 Часть 4. Механизмы репликации и эволюции геномов
Cdc25 ПРИВОДЫ
I
Реакция клетки
Рис. 15.34. Каскад событий, которые запускают соответствую-
щую реакцию клетки на повреждение ДНК
ются в начале S-фазы, а другие (например, VIII и IX)
реплицируются до конца к намного более позднему
периоду времени. Эта разница во времени не свя-
зана с длиной хромосомы, но указывает реальные
вариации в кинетике репликации хромосомы. Рис.
15.33, б подчеркивает этот момент, показывая, что
спустя 15 минут после начала S-фазы многие точки
старта в хромосоме XV были активированы, тогда
как репликация хромосомы XII к тому времени толь-
ко началась. Анализ на микроматрицах позволяет
также определить скорость перемещения отдель-
ных репликационных вилок и опять же показывает изменчивость. Средняя скорость
равна 2,9 тыс. п. н. в минуту, но некоторые вилки движутся намного быстрее, вплоть до
11 тыс.п.н. в минуту у самых резвых.
Понимание того, чем именно определяется время включения точки старта, оказы-
вается весьма трудным. Оно зависит не просто от последовательности областей инициа-
ции, потому что перенос сегмента ДНК из его нормальной позиции на другой участок
в той же самой или иной хромосоме может привести к изменению в схеме включения
стартов, содержащихся в этом сегменте. Этот позиционный эффект может быть связан
с организацией хроматина и, следовательно, подвергаться влиянию таких структур,
как управляющие локусом области (раздел 10.1.2), которые управляют упаковкой ДНК.
В данном отношении может быть важна также и позиция точки старта в ядре, ибо об-
ласти инициации, которые становятся активными в подобные периоды S-фазы, как
оказалось, группируются вместе, по крайней мере у млекопитающих.
Контрольные точки во время S-фазы
Заключительный момент регулирования репликации генома, который мы рас-
смотрим, — функция контрольных точек, которые работают во время S-фазы. Они
были впервые опознаны, когда было показано, что одна из реакций клеток дрожжей на
повреждение ДНК — замедление и, возможно, полная остановка процесса репликации
генома. Это связано с активацией генов, продукты которых участвуют в репарации ДНК
(раздел 16.2).
Помимо подталкивания клетки ко вхождению в S-фазу, циклинозависимые киназы
замешаны в регулировании контрольных точек S-фазы. Эти киназы реагируют на сиг-
налы от белков, связанных с репликационной вилкой. Белки репликационной вилки,
к которым относится PCNA и вспомогательный белок RFC, были первоначально наделены
ролями в обнаружении повреждения, но эта роль, вероятно, не исполняется версиями
этих белков, участвующими в синтезе ДНК. Чувствительная к повреждениям версия RFC
имеет отличающийся субъединичный состав от стандартного RFC, и чувствительный
белок, первоначально опознанный как родственник PCNA, оказался совершенно другим
белком (комплекс 9-1-1), который имеет подобную PCNA структуру и, вероятно, взаи-
модействует с двойной спиралью подобным же образом. Сигналы от чувствительных
к повреждению белков передаются киназами типа ATM, ATR, Chkl и Chk2 к исполнитель-
ным белкам типа Cdc25, которые взаимодействуют с циклинозависимыми киназами,
Глава 15. Репликация генома 633
чтобы вызвать соответствующую реакцию клетки (рис. 15.34). Процесс репликации
может быТь задержан путем подавления включения областей инициации, которые
обычно активируются на более поздних этапах S-фазы, или посредством замедления
продвижения существующих репликационных вилок. Если повреждение не чрезмерно,
то активируются процессы репарации ДНК (раздел 16.2); альтернативно, клетка может
быть переведена на путь запрограммированной смерти клетки, названной апоптозом,
ибо смерть единственной соматической клетки в результате повреждения ДНК обычно
является менее опасной, чем дозволение этой клетке реплицировать свою мутиро-
ванную ДНК и, возможно, вызывать развитие опухоли или других злокачественных
новообразований. У млекопитающих центральный игрок в индуцировании задержания
клеточного цикла и апоптоза — белок, названный р53. Он классифицируется как белок,
подавляющий опухоли, потому что, когда этот белок дефектен, клетки с поврежденны-
ми геномами могут обойти контрольные точки S-фазы и, возможно, разрастись в рак.
Белок р53 представляет собой специфичный к последовательности ДНК-связывающий
белок, активирующий ряд генов, которые, как думают, непосредственно отвечают за
задержание и апоптоз, а также подавляет экспрессию других генов, которые должны
быть выключены, с тем чтобы облегчить протекание этих процессов.
Заключение
Для продолжения выполнения своей функции геном должен реплицироваться при
каждом делении клетки. Уотсон и Крик указали, когда они впервые объявили о своем
открытии структуры ДНК, что специфичное спаривание оснований, которое удерживает
две нити двойной спирали вместе, обеспечивает средство для точного копирования каж-
дого из полинуклеотидов. Они предугадали полуконсервативный способ репликации,
в котором каждая родительская нить служит матрицей для синтеза комплементарной
дочерней нити. Эксперимент Мезельсона-Шталя показал, что данная интерпретация
верна, но все еще оставались проблемы в понимании того, как две нити спирали рас-
ходились, особенно в кольцевых молекулах, которые обладают малой свободой враще-
ния. Открытие ДНК-топоизомераз, которые разделяют нити двойной спирали путем
повторного разрыва и воссоединения одного или обоих полинуклеотидов, разрешило
эту проблему. Не известно ни одного исключения из полуконсервативного способа
репликации, хотя существуют специализированные версии репликации, такие как вы-
теснительная репликация и репликация катящегося кольца.
Инициация репликации генома происходит в обособленных областях инициации,
которые были хорошо охарактеризованы у бактерий и дрожжей, но которые менее ясно
поняты у высших эукариотов. После инициации репликации пара репликационных
вилок продвигается в противоположных направлениях по ДНК.
ДНК-полимераза может только синтезировать ДНК в направлении 5'->3', а это
означает, что хотя одна нить, названная опережающей нитью, может реплицироваться
непрерывно, вторая, отстающая нить должна быть копируема короткими сегментами.
Последние были названы фрагментами Окадзаки.
Синтез ДНК должен быть затравлен РНК-полимеразой, при этом спираль должна
быть раскручена, а отдельные нити стабилизированы геликазами и белками одно-
нитевого связывания. Репликационный комплекс, названный реплисомой у бактерий,
состоит из фермента ДНК-полимеразы наряду со вспомогательными белками, такими
как подвижный зажим, которые гарантируют, что связь между полимеразой и ДНК будет
надежна, но полимераза по-прежнему будет способна продвигаться по ДНК.
634 Часть 4. Механизмы репликации и эволюции геномов
Терминация репликации происходит в определенных областях в пределах бак-
териальной хромосомы, но в хромосомах эукариотов такие области очерчены менее
четко. Хромосомам эукариотов требуются специальные процессы для поддержания
их концов, поскольку в результате репликации теломеры постепенно укорачиваются.
Они удлиняются ферментом теломеразой, который несет в себе РНК-субъединицу, вы-
ступающую матрицей для синтеза новых теломерных повторных единиц. Репликация
генома должна быть согласована с клеточным циклом. Это достигается комбинацией
регуляторных белков, многие из которых активны только в определенные периоды
клеточного цикла. Сборка предрепликационного комплекса в областях инициации —
определяющий шаг, который регулируется, с тем чтобы гарантировать, что геном ре-
плицируется только один раз за один клеточный цикл. Как только репликация началась,
контрольные точки, работающие во время фазы синтеза, реагируют на повреждение
ДНК, с тем чтобы задержать или совсем закончить репликацию генома.
Глава 15. Репликация генома 635
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
15.1 *. К которому из следующих пунктов
относится топологическая проблема
репликации ДНК?
а. Блокировка участков репликации
ДНК нуклеосомами.
Ь. Трудность синтезирования ДНК
на отстающей нити.
с. Раскручивание двойной спирали
и вращение ДНК.
d. Синхронизация репликации ДНК
с делением клетки.
15.2 . Какой термин описывает топологи-
ческую структуру которая предот-
вращает разобщение нитей двойной
спирали без ее раскручивания?
а. Гелинемная.
Ь. Паранемная.
с. Плектонемная.
d. Топонемная.
15.3 *. Кто из исследователей впервые пред-
ложил модель «разрыва и воссоеди-
нения» для решения топологической
проблемы репликации ДНК?
а. Дельбрюк.
Ь. Корнберг.
с. Мезельсон и Шталь.
d. Уотсон и Крик.
15.4 . Которая из следующих функций
присуща ДНК-гиразе Е. coli?
а. Противодействие перекручива-
нию генома, которое происходит
в ходе репликации ДНК.
Ь. Противодействие перекручива-
нию генома, которое происходит
в процессе транскрипции.
с. Введение сверхспирали в молеку-
лы ДНК.
d. Все вышеупомянутое.
15.5 *. Молекулы ДНК каких типов копи-
руются с использованием процесса
репликации катящегося кольца?
а. Бактериальные хромосомы.
Ь. Геномы некоторых бактериофа-
гов (например, фага X).
с. Митохондриальные геномы.
d. Хромосомы дрожжей.
15.6 . Участок, в котором инициируется
репликация ДНК, называют:
а. Энхансером.
Ь. Инициатором.
с. Областью инициации.
d. Промотором.
15.7 *. Какова роль затравки в синтезе ДНК?
а. Она обеспечивает 5’-фосфатную
группу для прикрепления сле-
дующего нуклеотида.
Ь. Она обеспечивает 5'-фосфатные
группы, которые могут быть ги-
дролизированы, с тем чтобы вы-
свободить энергию, необходимую
для синтеза ДНК.
с. Она обеспечивает З’-гидроксиль-
ную группу для присоединения
следующего нуклеотида.
d. Она обеспечивает источник нукле-
отидов для синтеза нити ДНК.
15.8 . Действие которой из следующих ну-
клеаз используется ДНК-полимера-
зами, чтобы обеспечить корректи-
рующее действие во время синтеза
ДНК?
а. Экзонуклеазы 3'->5’.
Ь. Экзонуклеазы 5'->3'.
с. Специфичной к однонитевым це-
пям эндонуклеазы.
d. Специфичной к двунитевым це-
пям эндонуклеазы.
15.9 *. Что собой представляют фрагменты
Окадзаки?
а. Короткие сегменты полинуклео-
тида, синтезируемые на опере-
жающей нити ДНК.
636 Часть 4. Механизмы репликации и эволюции геномов
Ь. Короткие сегменты полинуклео-
тида, синтезируемые на отстаю-
щей нити ДНК.
с. Затравки, синтезируемые на от-
стающей нити, которые необхо-
димы для синтеза ДНК.
d. Протеолитические фрагменты
ДНК-полимеразы.
15.10 . Какие белки предотвращают дегра-
дацию или воссоединение одноните-
вой ДНК в репликационной вилке?
а. Геликазы.
Ь. Праймазы.
с. Белки однонитевого связывания,
d. Топоизомеразы.
15.11 *. Какой из следующих ферментов
бактерий удаляет РНК-затравки,
находящиеся в начале каждого
фрагмента Окадзаки на отстающей
нити?
а. ДНК-полимераза I.
Ь. ДНК-полимераза III.
с. ДНК-лигаза.
d. РНКаза Н.
15.12 . Какой белок участвует в разделе-
нии двух связанных дочерних хро-
мосом по терминации репликации
ДНК у Е. соШ
a. DnaB.
b. ДНК-полимераза.
с. Топоизомераза IV.
d. Tus.
Вопросы открытого типа
15.1 *. До проведения эксперимента Ме-
зельсона-Шталя не было известно,
является ли репликация ДНК раз-
бросанной, полуконсервативной
или консервативной. Опишите раз-
личия в содержании ДНК дочерних
молекул, получающихся в резуль-
тате репликации этими разными
способами.
15.2 . Опишите механизм вытеснительной
репликации молекулы ДНК.
15.13 *. Которое из следующих утвержде-
ний о теломеразе ВЕРНО?
а. Теломераза — это РНК-зависимая
ДНК-полимераза.
Ь. Теломераза — это РНК-зависимая
РНК-полимераза.
с. Теломераза — этоДНК-зависимая
ДНК-полимераза.
d. Теломераза — этоДНК-зависимая
РНК-полимераза.
15.14 . Которые из следующих сущно-
стей напоминают собой теломеры
Drosophila?
а. Центромеры.
Ь. Микросателлиты.
с. Ретропозоны.
d. Транспозоны ДНК.
15.15 *. В течение какой фазы клеточного
цикла происходит репликация ДНК?
а. М.
b. G1.
с. S.
d. G2.
15.16 . Какой метод был применен для из-
учения выбора времени инициации
репликации в различных областях
генома?
а. Флюоресцентная гибридизация
in situ.
b. Масс-спектроскопия.
с. Анализ на микроматрицах.
d. ПЦР.
15.3 *. Опишите механизм репликации ка-
тящегося кольца.
15.4 . Где и как белок DnaA связывается
с областью инициации у Е. coli?
15.5 *. Как инициируются репликацион-
ные вилки в области инициации
у Е. coli?
15.6 . Каковы функции субъединиц а, р и
с ДНК-полимеразы III Е. coli?
15.7 *. Опишите вкратце три фермента,
которые вовлечены в синтез копии
Глава 15. Репликация генома 637
ДНК в лице опережающей нити у эу-
кариотов.
15.8 . Что известно о терминации репли-
кации генома у Е. coli? Какие белки
и последовательности замешаны
в этом процессе?
15.9 *. Почему в ходе последовательных
кругов репликации ДНК у эукарио-
тов концы линейных хромосом ста-
новятся короче?
Изыскательские задачи
15.1* . Обсудите вопрос о том, почему полу-
консервативному способу реплика-
ции ДНК было отдано предпочтение
еще до того, как был проведен экс-
перимент Мезельсона-Шталя.
15.2. Оцените состояние текущих иссле-
дований областей инициации у мле-
копитающих.
15.3* . Напишите пространный доклад
о «ДНК-геликазах».
15.4. Наши современные знания о репли-
кации генома у эукариотов смещены
в сторону событий, происходящих
в репликационной вилке. Следую-
щая задача состоит в том, чтобы пре-
15.10 . Как активность теломеразы регули-
руется в клетках эукариотов?
15.11 *. Как изменения в экспрессии белка
Cdc6p дрожжей влияют на регулиро-
вание репликации генома?
15.12 . Какие общие схемы наблюдались
относительно выбора времени ре-
пликации различных частей генома
эукариотов?
образовать это сфокусированное на
ДНК описание репликации в модель,
которая описывала бы, как реплика-
ция организована в пределах ядра,
и обращалась бы к таким вопросам,
как роль репликационных фабрик
и процессы, используемые для того,
чтобы избегнуть запутывания до-
черних молекул. Изобретите план
исследований, который позволил бы
обратиться к одному или несколь-
ким подобным вопросам.
15.5* . Постарайтесь изыскать связи меж-
ду теломерами, старением клетки
и раком.
Тесты по рисункам
15.1*. Какого типа репли-
кация ДНК показана
на этом рисунке? Об-
судите процесс, по-
средством которого
ДНК реплицируется
согласно изображен-
ной системе.
638 Часть 4. Механизмы репликации и эволюции геномов
15.2. Какие два осложнения возникают в ходе осуществляемой ДНК-полимеразой
репликации ДНК?
ОПЕРЕЖАЮЩАЯ
НИТЬ
ЗАПАЗДЫВАЮЩАЯ
НИТЬ
15.3*. Обсудите механизм, при помощи ко-
торого смежные фрагменты Окадза-
ки соединяются в ходе репликации
отстающей нити ДНК у Е. coli.
15.4. Охарактеризуйте механизм, благо-
даря которому теломераза способна
продолжать З’-концы хромосом.
ДНК-полимераза III
Новая ДНК I Следующий фрагмент
1 Окадзаки
5'. 1П1НППП ГТ1‘
Хромосомная ДНК Теломеразная РНК
’^TTAGGGTTAGGG 3
I I I I I I I
CAAUCCCAAUCi^
3' 5'
| Синтез ДНК
Новая ДНК
Л
S==TTAGGGTTAGGGTTAG
mnifiiii
11111111111111111ТТГГ7 з;
| Перемещение
£ 'питии 'I П111!ц з‘.
О --------------- ----
3.11111 i 1111111111111111i111II1ГГГГ
5a^=TTAGGGTTAGGGTTAG
I I I I I
CAAUCCCAAUCi^
3' 5'
| Очередной синтез ДНК
3
5a^=TTAGGGTTAGGGTTAGGGTTAG
I I I I I I I I I I I
CAAUCCCAAUC
3'
5'
Глава 15. Репликация генома 639
Дополнительная литература
История исследования репликации генома
Crick,F.H.C., Wang,J.С. and Bauer,W.R. (1979) Is DNA really a double helix? J. Mol.
Biol. 129: 449-461. Crick's response to suggestions that DNA has a side-by-side rather than
helical conformation.
Holmes,F.L. (1998) The DNA replication problem, 1953-1958. Trends Biochem. Sci. 23:
117-120.
Kornberg, A. (1989) For the Love of Enzymes: The Odyssey of a Biochemist. Harvard
University Press, Boston, Massachusetts. A fascinating autobiography by the discoverer of DNA
polymerase.
Meselson,M. and Stahl, F. (1958) The replication of DNA in Escherichia coli. Proc. Natl
Acad. Sci. USA 44: 671-682. The Meselson-Stahl experiment.
Okazaki, T. and Okazaki, R. (1969) Mechanisms of DNA chain growth. Proc. Natl Acad.
Sci. USA 64:1242-1248. The discovery of Okazaki fragments.
Rodley, G. A., Scobie, R. S., Bates, R. H. T. and Lewitt, R. M. (1976) A possible conformation
for double-stranded polynucleotides. Proc. Natl Acad. Sci. USA 73: 2959-2963. A side-by-side
model for DNA structure.
Watson, ).D. and Crick, F.H.C. (1953) Genetical implications of the structure of
deoxyribonucleic acid. Nature 171: 964-967. Describes possible processes for DNA replication,
shortly after discovery of the double helix.
ДНК-топоизомеразы
Berger, J. M., Gamblin, S. J., Harrison, S. C. and Wang, J. C. (1996) Structure and mechanism
of DNA topoisomerase II. Nature 379: 225-232 and 380:179.
Champoux, J. J. (2001) DNA topoisomerases: structure, function, and mechanism. Annu.
Rev. Biochem. 70: 369-413.
Stewart, L, Redinbo,M.R., Qiu,X., Hol,W.G.J, and Champoux,).). (1998) A model for
the mechanism of human topoisomerase I. Science 279:1534-1541.
Области инициации
Aladjem, M. I., Rodewald, L. W., Kolman, J. L. and Wahl, G. M. (1998) Genetic dissection
of a mammalian replicator in the human 0-globin locus. Science 281:1005-1009.
DiffleyJ.F.X. and Cocker,J.H. (1992) Protein-DNA interactions at a yeast replication
origin. Nature 357:169-172.
Gilbert, D.M. (2001) Making sense of eukaryotic replication origins. Science 294:96-100.
ДНК-полимеразы и события в репликационной вилке
Bochkarev,A., Pfuetzner,R.A., Edwards,А. М. and Frappier,L. (1997) Structure of
the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 385:
176-181.
Hiibscher,U., Nasheuer, H.-P. and SyvaojaJ.E. (2000) Eukaryotic DNA polymerases:
a growing family. Trends Biochem. Sci. 25:143-147.
Johnson, A. and O'Donnell, M. (2005) Cellular DNA replicases: components and dynamics
at the replication fork. Annu. Rev. Biochem. 74: 283-315. Details of replication in bacteria and
eukaryotes.
Lemon, K.P. and Grossman, A. D. (1998) Localization of bacterial DNA polymerase:
evidence for a factory model of replication. Science 282:1516-1519.
640 Часть 4. Механизмы репликации и эволюции геномов
Liu, Y., Као, Н. I. and Bambara, R. А. (2004) Flap endonuclease I: a central component of
DNA metabolism. Annu. Rev. Biochem. 73: 589-615.
Myllykallio, H., Lopez, P., Lopez-Garcia, P., Heilig,R., Saurin, W., Zivanovic,Y.,
Philippe, H. and Forterre,P. (2000) Bacterial mode of replication with eukaryotic-like
machinery in a hyperthermophilic archaeon. Science 288: 2212-2215.
Soultanas,P. and Wigley, D.B. (2001) Unwinding the 'Gordian knot' of helicase action.
Trends Biochem. Sci. 26: 47-54.
Trakselis,M.A. and Bell,S.D. (2004) The loader of the rings. Nature 429: 708-709. The
sliding clamp and clamp loader.
Теломеры
Blackburn, E.H. (2000) Telomere states and cell fates. Nature 408: 53-56.
Cech,T. R. (2004) Beginning to understand the end of the chromosome. Cell 116:273-279.
Reviews all aspects of telomerase.
McEachern, M.)., Krauskopf,A. and Blackburn, E. H. (2000) Telomeres and their
control. Annu. Rev. Genet 34:331-358. Describes the processes involved in regulation of telomere
length.
Pardue, M.-L. and DeBaryshe, P. G. (2003) Retrotransposons provide an evolutionarily
robust non-telomerase mechanism to maintain telomeres. Annu. Rev. Genet. 37: 485-511.
Smogorzewska,A. and de Lange,T. (2004) Regulation of telomerase by telomeric
proteins. Annu. Rev. Biochem. 73:177-208.
Управление репликацией генома
Kelly, T. J. and Brown, G.W. (2000) Regulation of chromosome replication. Annu. Rev.
Biochem. 69: 829-880.
Raghuraman,M. K., Winzeler, E.A., Collingwood, D., etal. (2001) Replication dynamics
of the yeast genome. Science 294:115-121. Microarray studies of origin firing.
Sancar,A., Iindsey-Boltz,L.A., Unsal-Ka^maz, K. and Linn,S. (2004) Molecular
mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem.
73: 39-85.
Stillman, B. (1996) Cell cycle control of DNA replication. Science 274:1659-1664.
Zhou,B.-B.S. and Elledge, S.). (2000) The DNA damage response: putting checkpoints
in perspective. Nature 408: 433-439.
Глава 16
Мутации и репарация ДНК
По прочтении главы 16 вы сможете:
• Давать определение термину «мутация», а также различным терминам, которые
употребляются для обозначения мутаций разных типов.
• Описывать, с характерными примерами, механизм, посредством которого само-
произвольные ошибки репликации вызывают мутации.
• Представлять примеры химических и физических мутагенов, а также обрисовы-
вать видоизменения, которые они производят в молекулах ДНК.
• Перечислять, с отличительными примерами, воздействия мутаций на кодирую-
щие и некодирующие области геномов.
• Описывать возможные воздействия мутаций на многоклеточные организмы.
• Перечислять и описывать различные эффекты мутаций на микроорганизмы.
• Обсуждать биологическое значение сверхмутации и запрограммированных
мутаций.
• Проводить различие между механизмами репарации ДНК разных типов.
• Приводить подробные описания молекулярных событий, происходящих в ходе
прямой репарации, репарации вырезанием оснований и нуклеотидов, а также
репарации устранением несоответствий.
• Описывать, как залечиваются однонитевые и двунитевые разрывы ДНК.
• Сообщать основные сведения о механизме обхода поврежденного места ДНК
в ходе репликации генома.
• Подытоживать известные данные о связи между востановлением ДНК и болез-
нями человека.
Геномы суть динамические сущности, которые изменяются во времени в результате
кумулятивных воздействий мелкомасштабных видоизменений последовательности, вы-
зываемых мутациями (раздел 16.1). Мутация — это изменение в последовательности
нуклеотидов на протяжении короткой области генома (рис. 16.1, а). Многие мутации яв-
ляются точечными мутациями (также называемыми простыми мутациями или однону-
клеотидными мутациями), которые заменяют один нуклеотид другим. Точечные мутации
подразделяют на две категории: транзиции, которые представляют собой замены типа
пурин на пурин или пиримидин на пиримидин (A->G, G->A, С->Т или Т->С), и трансверсии,
которые являют собою замены типа пурин на пиримидин или пиримидин на пурин (А->С,
А—>Т, G—>С, G—>Т, С—>А, С—>G, Т->А или T->G). Остальные мутации являются результатом
вставки или удаления (делеции) одного или нескольких нуклеотидов.
Мутации проистекают или из ошибок в репликации ДНК, или из разрушительных
воздействий мутагенов, таких как химические агенты и радиация, которые реагируют
с ДНК и изменяют структуру отдельных нуклеотидов. Все клетки обладают ферментами
642 Часть 4. Механизмы репликации и эволюции геномов
а) Мутация
.. ' । . Молекула ДНК
...GACAGTACGA...
...CTGTCATGCT...
I Мелкомасштабное изменение
▼ в последовательности нуклеотидов
...GACAGTACGA...
...CTGTAATGCT...
6) Восстановление ДНК
| Ошибка исправлена
...GACAGTACGA...|
...CTGTCATGC Т...
*. ; Немутированная
молекула ДНК
. Мутированная
молекула ДНК
Рис. 16.1. Мутация и восстановление ДНК. а) Мутация — мелкомасштабное изменение в нуклеотидной
последовательности молекулы ДНК. Здесь показана точечная мутация, но есть мутации нескольких дру-
гих типов, которые описаны в тексте, б) Восстановление ДНК исправляет мутации, которые возникают
в виде ошибок репликации и в результате мутагенной активности
репарации ДНК, которые пытаются минимизировать число происходящих мутаций (раз-
дел 16.2). Эти ферменты работают двумя способами. Некоторые из них предрепликационные
и просматривают ДНК с целью отыскания нуклеотидов с необычными структурами, причем
такие нуклеотиды заменяются до начала репликации; другие—пострепликационные и они
проверяют новосинтезированную ДНК на предмет наличия ошибок и исправляют любые
ошибки, которые находят (рис. 16.1, б). Поэтому возможно сформулировать определение
мутации как недостаточность репарации ДНК.
Мутации могут оказывать значительные воздействия на клетку, в которой они
происходят: мутация в основном гене может выразиться в синтезе дефектного белка,
который, в свою очередь, может привести к смерти клетки. Другие мутации оказывают
менее существенное воздействие на фенотип клетки, а многие вообще никакого. Как мы
увидим в главе 18, все события, которые не являются гибельными для клетки, потенци-
ально могут способствовать эволюции генома, но для того чтобы это произошло, они
должны передаваться по наследству при воспроизводстве организма. В одноклеточном
организме, например бактерий или дрожжей, все видоизменения генома, которые не
являются смертельными и не исправляются, наследуются дочерними клетками и ста-
новятся постоянными составляющими клеточной линии, которая берет свое начало от
исходной клетки, в которой произошло данное видоизменение. В многоклеточном же
организме только те события, которые происходят в зародышевых клетках, сопряжены
с эволюцией генома. Изменения в геномах соматических клеток ничего не значат в эво-
люционном смысле, но они приобретают биологическую значимость, если порождают
порочный фенотип, который губительно сказывается на здоровье организма.
16.1. Мутации
Разбирая мутации, мы коснемся следующих вопросов: как они возникают; воздей-
ствия, которые они оказывают на геном и на организм, в котором этот геном находится,
и возможно ли, чтобы клетка в определенных условиях увеличивала частоту появления
мутаций и вызывала запрограммированные мутации.
Глава 16. Мутации и репарация ДНК 643
16.1.1. Причины мутаций
Мутации возникают по двум причинам.
• Некоторые мутации являются самопроизвольными ошибками репликации, кото-
рые избегают корректирующего действия ДНК-полимераз, синтезирующих новые
полинуклеотиды в репликационной вилке (раздел 15.2.2). Такие мутации называют
несоответствиями или заменами, потому что они представляют собой позиции,
в которых нуклеотид, который вставлен в дочерний полинуклеотид, заменен и
не соответствует, по правилу спаривания оснований, нуклеотиду в соответствую-
щей позиции в матричной ДНК (рис. 16.2, а). Если такое несоответствие не будет
исправлено в дочерней двойной спирали, то одна из молекул второго поколения,
произведенных в ходе следующего круга репликации ДНК, будет содержать по-
стоянную, двунитевую версию этой мутации.
• Другие мутации возникают, потому что с родительской ДНК прореагировал мута-
ген, вызвав структурное изменение, которое затрагивает способность спаривания
оснований видоизмененного нуклеотида. Обычно такое видоизменение затраги-
вает только одну из нитей родительской двойной спирали, так что только одна
из дочерних молекул несет мутацию, но обе молекулы второго поколения, произ-
веденные во время следующего круга репликации, будут ее нести (рис. 16.2, б).
Ошибки репликации — источник точечных мутаций
Если рассматривать комплементарное спаривание оснований исключительно как
химическую реакцию, то это не особо точный процесс. Никто еще не изобрел способ
осуществить направляемый матрицей синтез ДНК без помощи ферментов, но если бы
данный процесс мог быть выполнен просто как химическая реакция в пробирке, то
полученный полинуклеотид, вероятно, имел бы точечные мутации в 5-10 позициях
из каждой сотни. Это представляет коэффициент ошибок 5-10 %, который был бы
абсолютно неприемлем в ходе репликации генома. Поэтому направляемые матрицей
ДНК-полимеразы, которые выполняют репликацию ДНК, должны повышать точность
процесса на несколько порядков величины. Такое улучшение достигается двумя спо-
собами:
• ДНК-полимераза использует процесс подбора нуклеотидов, который резко уве-
личивает точность направляемого матрицей синтеза ДНК (рис. 16.3, а). Такой
процесс подбора, вероятно, действует на трех различных стадиях в ходе реакции
полимеризации: отсев неправильных нуклеотидов проявляется, когда нуклеотид
впервые связывается с ДНК-полимеразой, когда он сдвигается к активному участку
фермента и когда присоединяется к З'-концу синтезируемого полинуклеотида.
• Точность синтеза ДНК еще более увеличивается, если ДНК-полимераза обладает
действием экзонуклеазы 3'^5' и, таким образом, способна удалить неправильный
нуклеотид, который благополучно минует процесс подбора нуклеотидов и ста-
новится присоединенным к З'-концу нового полинуклеотида (см. рис. 2.7, б). Это
называют коррекцией (раздел 15.2.2), но данное название употребляется как раз
некорректно, так как данный процесс — это не просто механизм проверки. Вместо
этого, каждый шаг в синтезе полинуклеотида должен рассматриваться как сорев-
нование между полимеразной и экзонуклеазной функциями фермента, причем
полимераза обычно побеждает, потому что она более активна, чем экзонуклеаза, по
крайней мере когда З'-конечный нуклеотид спарен с матрицей. Но если оконечный
нуклеотид не спарен, то действие полимеразы менее эффективно и обусловленная
644 Часть 4. Механизмы репликации и эволюции геномов
а) Ошибка в ходе репликации
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
Мутированная молекула
РОДИТЕЛЬСКАЯ
МОЛЕКУЛА
Ошибка
репликации
I
...GACCTAGAAA...
...CTGAATCTTT...
ДОЧЕРНИЕ
МОЛЕКУЛЫ
...GACCTAGAAA...
...CTGGATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
МОЛЕКУЛЫ
ВТОРОГО ПОКОЛЕНИЯ
б) Одно из возможных воздействий мутагена
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAATCTTT...
...GACTTAGAAA...
...CTGAXTCTTT...
t
Видоизменённый
нуклеотид
РОДИТЕЛЬСКАЯ
МОЛЕКУЛА
Мутированная молекула
...GACTCAGAAA...
Мутированная
молекула
...GACTCAGAAA...
...CTGAXTCTTT...
...CTGAGTCTTT...
ДОЧЕРНИЕ
МОЛЕКУЛЫ
Мутированная молекула
* ...GACTCAGAAA...
...CTGAXTCTTT...
МОЛЕКУЛЫ
ВТОРОГО ПОКОЛЕНИЯ
Рис. 16.2. Примеры мутаций, а) Ошибка в репликации ведет к несоответствию в одной из дочерних
двойных спиралей: в данном случае произошла замена Т -> С, потому что один из нуклеотидов А в ма-
тричной ДНК был неверно скопирован. Когда молекула с несоответствием сама копируется, она дает одну
двойную спираль с правильной последовательностью и другую с мутированной последовательностью.
б) Мутаген изменил структуру А в нижней нити родительской молекулы, давая нуклеотид X, который не
спаривается с Т в другой нити, так что в итоге создается несоответствие. Когда родительская молекула
реплицируется, X образует пару с С, давая мутированную дочернюю молекулу. Когда эта дочерняя мо-
лекула реплицируется, обе молекулы второго поколения наследуют мутацию
Глава 16. Мутации и репарация ДНК 645
Техническое примечание 16.1: обнаружение мутаций
Быстрые процедуры обнаружения мутаций в молекулах ДНК
Многие генетические болезни вызываются точечными мутациями, которые
приводят к видоизменению или инактивации продукта гена. Методы обнаружения
таких мутаций важны в двух контекстах. Во-первых, когда ген, ответственный за
некоторую генетическую болезнь, опознается впервые, обычно бывает необходи-
мо исследовать много версий этого гена от различных индивидуумов, с тем чтобы
опознать мутацию или мутации, ответственные за это болезненное состояние.
Во-вторых, когда вызывающая болезнь мутация охарактеризована, необходимы
высокопроизводительные методы обнаружения, для того чтобы клинические врачи
могли просматривать множество образцов ДНК, с тем чтобы опознавать индивидуу-
мов, которые несут в себе данную мутацию и подвержены риску развития данной
болезни или передачи ее своим детям.
Любая мутация может быть опознана путем секвенирования ДНК, но секвени-
рование — относительно медленная процедура и вряд ли подходит для перебора
большого числа образцов. Технология чипов ДНК (техническое примечание 3.1)
также может быть взята на вооружение, но она все еще не является широко приме-
няемым средством. По этим причинам был разработан ряд относящихся к «низким
технологиям» методов. Они могут быть подразделены на две категории: методы
поиска мутаций, которые не требуют никакой предварительной информации
о позиции мутации, и методы детектирования мутаций, которые просматривают
последовательности на предмет наличия определенной мутации.
Большинство методов поиска мутаций основаны на анализе гетеродуплекса,
образованного между одинарной нитью исследуемой ДНК и комплементарной
нитью контрольной ДНК, которая имеет немутированную последовательность
(рис. Т16.1). Если анализируемая ДНК содержит мутацию, то в гетеродуплексе бу-
дет одна позиция несоответствия, в которой пара оснований не образуется. Чтобы
определить, присутствует такое несоответствие в гетеродуплексе или нет, могут
быть использованы различные методы:
• электрофорез или жидкостная хроматография высокого разрешения
(ЖХВР) позволяют обнаружить несоответствие за счет обнаружения раз-
личия в подвижности гибрида с несоответствием по сравнению с полностью
спаренным основаниями в полиакриламидном геле колонки ЖХВР. Этот под-
ход позволяет установить, присутствует несоответствие или нет, но не дает
никакой информации о том, где именно в анализируемой ДНК расположена
данная мутация;
Позиция несоответствия Опытная ДНК
____________ 1 1
ш । 111 пи 11 |уПТ
Контрольная ДНК
Рис. Т16.1. Гибридизация между комплементарными нитями ДНК, одна из которых содержит мута-
цию, дает двунитевую молекулу с несоответствием
646 Часть 4. Механизмы репликации и эволюции геномов
• расщепление гетеродуплекса в позиции несоответствия, сопровождаемое
гель-электрофорезом, позволяет определить местоположение позиции несоот-
ветствия. Если гетеродуплекс остается неповрежденным, то в нем не имеется
ни одного несоответствия; если он расщепляется, то, значит, содержит несоот-
ветствие, при этом позиция мутации в анализируемой ДНК определяется раз-
мерами продуктов расщепления. Расщепление выполняется путем обработки
ферментами или химикатами, которые производят разрез в однонитевых об-
ластях преимущественно двунитевой ДНК, или же специфичной к однонитевой
ДНК рибонуклеазой, например, S1 (см. рис. 5.14), в том случае если гибрид был
образован между контрольной ДНК и РНК-версией анализируемой ДНК.
Большинство методов детектирования для обнаружения определенных мута-
ций основаны на способности гибридизации олигонуклеотидами различать между
собой целевые ДНК, последовательности которых отличаются хотя бы в одной ну-
клеотидной позиции (см. рис. 3.8, а). В гибридизации аллелеспецифическими оли-
гонуклеотидами (АСО) образцы ДНК просматриваются путем зондирования олиго-
нуклеотидом, который гибридизируется только с мутантной последовательностью
(рис. Т16.2). Это эффективная процедура, но она излишне тягомотна. Образцы ДНК
обычно получают при помощи ПЦР клинических изолятов, так что более быстрая
альтернатива — использовать диагностический олигонуклеотид в качестве одной
из затравок ПЦР, так чтобы присутствие или отсутствие мутации в анализируемой
ДНК отмечалось бы синтезом продукта ПЦР или отсутствием такового.
Дот-блот - образцы ДНК
нанесены на нейлоновую
мембрану
Нейлоновая
мембрана • - - * -
{Гибридизация
АСО
Авторадиограмма
ДНК с мутированной
последовательностью
Рис. Т16.2. Гибридизация аллелеспецифическими олигонуклеотидами (АСО)
этим пауза в полимеризации позволяет действию экзонуклеазы возобладать, так
что неправильный нуклеотид удаляется (см. рис. 16.3, б).
Escherichia coli способна синтезировать ДНК с частотой появления ошибок л ишь 1 на
107 добавлений нуклеотидов. Что интересно, эти ошибки распределяются не равномерно
между двумя дочерними молекулами; продукт репликации отстающей нити подвержен
приблизительно в 20 раз большему числу ошибок, чем репликат опережающей нити.
Такая асимметрия может указывать на то, что ДНК-полимераза I, которая участвует
только в репликации отстающей нити (раздел 15.2.2), обладает менее эффективными
способностями подбора оснований и корректирования по сравнению с ДНК-полимеразой
III — главным реплицирующим ферментом.
Глава 16. Мутации и репарация ДНК 647
а) Подбор нуклеотидов
б) «Корректирование»
у Тнt; ;нпн11нн л.и .
х. ту* »
т
ДНК-полимераза
I1IIIIIIH1I
Последний нуклеотид спарился
ПОБЕЖДАЕТ ПОЛИМЕРАЗА
IIIIII
। П1111111.
•...............1 -1.5'
Последний нуклеотид не спарился
ПОБЕЖДАЕТ ЭКЗОНУКЛЕАЗА
Рис. 16.3. Механизмы обеспечения точности репликации ДНК. а) ДНК-полимераза активно выбирает
правильный нуклеотид для вставки в каждую позицию, б) Те ошибки, которые возникли, могут быть
исправлены «корректированием», если полимераза обладает действием экзонуклеазы 3'->5'. Если по-
следний нуклеотид, который был вставлен, спарился с матрицей, то преобладает действие полимеразы,
но если последний нуклеотид не образовал пары, то предпочтение отдается действию экзонуклеазы
Не за все ошибки, которые происходят во время синтеза ДНК, можно возложить
ответственность на ферменты полимеразы: иногда ошибка возникает даже при том, что
фермент добавляет «правильный» нуклеотид, то есть тот, что спаривается с матрицей.
Это связано с тем, что каждое нуклеотидное основание может существовать в виде одного
из двух альтернативных таутомеров — структурных изомеров, которые находятся в ди-
намическом равновесии. Например, тимин существует в виде двух таутомеров — кето-
и енольной формах, причем отдельные молекулы эпизодически претерпевают переход от
одного таутомера к другому. Равновесие сильно смещено к кето-форме, но в матричной
ДНК время от времени появляется енольная версия тимина, и иногда точно в тот момент,
когда репликационная вилка проходит через данный участок последовательности. Это
приводит к «ошибке», потому что енолтимин спаривается с G, а не с А (рис. 16.4). Та же
самая проблема может возникать с аденином, редкий имино-таутомер этого основания
предпочтительно образует пару с С и с гуанином, а енолгуанин спаривается с тимином.
После репликации редкий таутомер неизбежно возвращается к своей более обычной
форме, что ведет к несоответствию в дочерней двойной спирали.
Как было указано выше, частота появления ошибок при синтезе ДНК у Е. coli со-
ставляет 1 на 107. Однако общая частота ошибок репликации генома Е coli равна лишь
от 1 на 1О10 до 1 на 1011, такое улучшение по сравнению с коэффициентом ошибок
полимеразы является результатом работы системы устранения несоответствий (раз-
дел 16.2.3), которая просматривает новореплицируемую ДНК на предмет наличия
позиций, в которых основания неспарены, и, следовательно, исправляет немногочис-
ленные ошибки, которые допускают ферменты репликации. Это означает, что в среднем
возникает только одна неисправленная ошибка репликации на каждые 2 000 событий
копирования генома Е. coli.
648 Часть 4. Механизмы репликации и эволюции геномов
Таутомерный
О переход ОН
--------► I
С.. 'СН-, /CHj
HN С hr С
I II I II
1 сн ^С\1 СН
О N О N
I I
Кетотимин Энолтимин
I
СПАРИВАЕТСЯ
С G, АНЕСА
Рис. 16.4. Влияние таутомеризма на спаривание
оснований. В каждом из этих трех примеров две
таутомерные формы одного и того же основания
имеют различные свойства спаривания. Цитозин
также имеет амино- и имино-таутомеры, но оба
они образуют пару с гуанином
Аминоаденин
Таутомерный
переход
Иминоаденин
СПАРИВАЕТСЯ
С С, АНЕСТ
Таутомерный
0 переход 0Н
II ----------► II
// С NH // С
нс'9 II I HCg II I
С \9^С /(X
N NH2 N NH.
Кетогуанин Энолгуанин
СПАРИВАЕТСЯ
С Т, А НЕ С С
Ошибки репликации могут вести
также к мутациям типа вставки
и удаления
Не все ошибки в репликации выража-
ются точечными мутациями. Ошибочная
репликация может также привести к встав-
ке небольшого числа дополнительных ну-
клеотидов в синтезируемый полинуклео-
тид или к пропуску некоторых нуклеотидов
матрицы. Вставка или удаление, которое
происходит в пределах кодирующей обла-
сти, может привести к мутации со сдвигом
рамки считывания, которая изменяет рам-
ку считывания, используемую для транс-
ляции белка, описываемого данным геном
(см. рис. 16.12). Существует определенное
стремление использовать «сдвиг рамки
считывания», чтобы описывать все вставки
и удаления, но это неточно, так как вставка
или удаление трех нуклеотидов (или крат-
ного трем их числа) попросту добавляет
или удаляет кодоны или части смежных
кодонов, не затрагивая саму рамку считы-
вания. Также, конечно, многие вставки/
удаления происходят вне открытых рамок
считывания, в пределах межгенных обла-
стей генома.
Мутации типа вставок и удалений мо-
гут затронуть все части генома, но особен-
но распространены, когда матричная ДНК
содержит короткие повторяющиеся последовательности, наподобие обнаруженных
в микросателлитах (раздел 3.2.2). Это связано с тем, что повторы могут вызвать про-
скальзывание репликации, при котором матричная нить и ее копия сдвигают свои
относительные позиции таким образом, что некоторая часть матрицы либо копируется
дважды, либо пропускается. В результате новый полинуклеотид имеет, соответственно,
большее или меньшее число повторных единиц (рис. 16.5). Это главная причина, по
которой микросателлитные последовательности являются настолько изменчивыми:
проскальзывание репликации эпизодически производит вариант новой длины, допол-
няя коллекцию аллелей, представленную к тому времени в популяции.
Глава 16. Мутации и репарация ДНК 649
...САСАСАСАСА...
...САСАСАСАСА...
...GTGTGTGTGT...
..САСАСАСАСА...
..GTGTGTGTGT...
РОДИТЕЛЬСКАЯ
МОЛЕКУЛА
...САСАСАСАСА...
...GTGTGTGTGT...
Добавочная
Результат проскальзывания
репликации
...GTGTGTGTGT...
ДОЧЕРНИЕ
МОЛЕКУЛЫ
повторная единица
I
...САСАСАСАСАСА...
...GTGTGTGTGTGT...
...САСАСАСАСА...
...GTGTGTGTGT...
МОЛЕКУЛЫ
ВТОРОГО ПОКОЛЕНИЯ
Рис. 16.5. Проскальзывание репликации. На рисунке изображена репликация микросателлитов из пяти
повторных единиц СА. Проскальзывание произошло во время репликации родительской молекулы
и выразилось во вставке дополнительной повторной единицы в новосинтезированный полинуклеотид
одной из дочерних молекул. Когда эта дочерняя молекула реплицируется, она дает молекулу второго
поколения, микросателлитная последовательность которой на одну единицу длиннее, чем таковая ис-
ходной родительской молекулы
Проскальзывание репликации, вероятно, также ответственно за болезни, связан-
ные с расширением тринуклеотидных повторений, которые были открыты у людей
в последние годы. Каждая из таких нейродегенеративных болезней вызвана удлинением
относительно короткого ряда тринуклеотидных повторов в два и более раз относительно
своей нормальной длины. Например, ген HD человека содержит последовательность
5'-CAG-3', тандемно повторяющуюся от 6 до 35 раз и кодирующую ряд глутаминов в бел-
ковом продукте. При хорее Гентингтона это повторение расширяется до числа копий
36-121, увеличивая длину полиглутаминового отрезка и давая дисфункциональный
белок. Несколько других болезней человека также вызваны расширениями кодонов по-
лиглутамина (таблица 16.1). Некоторые болезни, связанные с умственной отсталостью,
проистекают из тринуклеотидных расширений в 5’-концевой области гена, в результате
чего образуется хрупкий участок — позиция, в которой вероятен разрыв хромосомы.
Также известны расширения, вовлекающие интронные и З’-концевые области.
Как происходят расширения триплетов, точно не известно. Размер вставки намного
больше, чем происходящий при нормальном проскальзывании репликации (например,
при наблюдаемом с микросателлитными последовательностями), и как только рас-
ширение достигает некоторой длины, оно, кажется, начинает тяготеть к дальнейшему
расширению в последующих циклах репликации, так что в последующих поколениях
болезнь становится все более и более серьезной. Была высказана гипотеза о том, что
650 Часть 4. Механизмы репликации и эволюции геномов
расширение сопряжено с формированием шпилечных петель в ДНК. Она основывалась
на том наблюдении, что известно только ограниченное число тринуклеотидных по-
следовательностей, которые подвергаются расширению, и все эти последовательности
обогащены динуклеотидом GC и, таким образом, могут формировать устойчивые вто-
ричные структуры. Есть также данные, что по крайней мере одна область расширения
триплетов — для наследственной атаксии Фридрейха — может формировать структуру
в виде тройной спирали. Исследования подобных расширений триплетов у дрожжей
показали, что они случаются гораздо чаще, когда инактивирован ген RAD27. Это на-
блюдение представляет особый интерес, поскольку RAD27—дрожжевая версия гена
млекопитающих, кодирующего FEN1 — белок, вовлеченный в процессинг фрагментов
Окадзаки (раздел 15.2.2). Это может указывать на то, что расширение тринуклеотидного
повторения вызывается нарушением синтеза отстающей нити.
Мутации могут быть вызваны также химическими и физическими мутаге-
нами
Многие химикаты, которые встречаются в окружающей природной среде, обладают
мутагенными свойствами, и к ним в последние годы прибавились другие химические
мутагены, которые проистекают из промышленной деятельности человека. Физические
Таблица 16.1. Примеры расширения тринуклеотидных повторений у человека
Локус Повтор Нормальный Мутированный Сопряженная болезнь
Полиглута HD AR DRPLA SCA1 SCA3 аминовые расшг (CAG)6.35 (cag)9 36 (САС)6_35 (cag)639 (CAG)12_40 [рения (все в коди (CAG)36121 (cag)38 62 (cag)49 88 (cag)39 82 (CAG)55 84 рующих областях генов) Хорея Гентингтона Атрофия мышц спины и глазного яблока Дентаторубрально-паллидолиузиа- нская атрофия Спинномозговая и мозжечковая атак- сия 1-го типа Синдром Машадо-Жозефа
Расширения хрупких участков (оба в нетранслируемых ведущих областях генов)
FRM1 (cggj6_53 (CGG)60cBbluje 230 Синдром хрупкой Х-хромосомы
FRM2 (gcc)6.35 (GCC)61CBbluje 200 Синдром умственной отсталости из- за хрупкой ХЕ-хромосомы
Прочие расширения (позиции указаны ниже)
DMPK (ctgj5_37 (CTG)50 3000 Миотоническая дистрофия
Х25 (GAA)7.34 (GAA)34_CBbIuie 200 Наследственная атаксия Фридрейха
Примечание: расширения DMPK и Х25 находятся соответственно в З'-концевых и интронных областях
своих генов и предположительно затрагивают процессинг РНК. Известно также несколько болезнетворных
мутаций, которые связаны с расширениями более длинных последовательностей, например, прогрессирую-
щая миоклоническая эпилепсия вызывается расширением (CCCCGCCCCGCG)2_3 до (CCCCGCCCCGCG)CBbluje 12
в области промотора локуса ЕРМ1.
Глава 16. Мутации и репарация ДНК 651
агенты, такие как радиация, также являются мутагенными. Большинство организмов
подвергается воздействию в большей или меньшей степени этих разнообразных мута-
генов, в результате чего геномы этих организмов повреждаются.
Определение термина «мутаген» сформулировано как химический или физический
агент, который вызывает мутации. Это определение важно, потому что оно отличает
мутагены от агентов внешней среды других типов, которые причиняют ущерб клеткам
иными способами, чем вызов мутаций (таблица 16.2). Эти категории перекрываются (на-
пример, некоторые мутагены также являются канцерогенными веществами), но агент
каждого типа имеет отличный от других веществ биологический эффект. Определение
«мутагена» указывает на различие также между истинными мутагенами и другими
агентами, которые повреждают ДНК, не вызывая мутации (например, вызывая раз-
рывы в молекулах ДНК). Повреждения такого типа могут блокировать репликацию
и спровоцировать смерть клетки, но это не мутация в строгом смысле этого термина,
и поэтому вызывающие их агенты не относятся к мутагенам.
Мутагены вызывают мутации тремя различными способами.
• Некоторые выступают в роли аналогов оснований и ошибочно используются
в качестве субстрата при синтезе новой ДНК в репликационной вилке.
• Некоторые непосредственно реагируют с ДНК, вызывая структурные изменения,
которые ведут к ошибкам копирования матричной нити в ходе репликации ДНК.
Такие структурные изменения разнообразны, что мы сможем увидеть, когда рас-
смотрим отдельные мутагены.
• Некоторые мутагены действуют на ДНК косвенно. Сами они не повреждают струк-
туру ДНК, но вместо этого побуждают клетку синтезировать химикаты типа пе-
роксидов, которые обладают прямым мутагенным действием.
Разнообразие мутагенов настолько велико, что трудно изобрести какую-либо
всеобъемлющую классификацию. Мы поэтому ограничим наше изучение наиболее рас-
пространенными типами. Из когорты химических мутагенов упомянем следующие.
• Аналоги оснований — пуриновые и пиримидиновые основания, достаточно по-
добные стандартным основаниям ДНК для включения их в нуклеотиды, синтези-
руемые клеткой. Получающиеся необычные нуклеотиды могут впоследствии быть
использованы в качестве субстрата для синтеза ДНК в ходе репликации генома.
Например, 5-бромоурацил (5-bU; рис. 16.6, а) обладает теми же свойствами спа-
ривания оснований, что и тимин, и нуклеотиды, содержащие это основание, могут
быть добавлены к дочернему полинуклеотиду в позициях напротив расположенных
в матрице аденинов. Мутагенный эффект возникает, потому что равновесие между
двумя таутомерами 5-bU сдвинуто в большей степени к более редкой енольной
Таблица 16.2. Категории агентов внешней среды, которые причиняют вред живым клеткам
Агент Воздействие на живые клетки
Канцероген Вызывает рак — злокачественное перерождение клеток эукариотов
Кластоген Вызывает фрагментацию хромосом
Мутаген Вызывает мутации
Онкоген Вызывает образование опухоли
Тератоген Вызывает пороки развития
652 Часть 4. Механизмы репликации и эволюции геномов
а) 5-бромоурацил
О
JL ,Вг
HN С
I II
''С 1 сн
О' N
I
Рис. 16.6. 5-бромоурацил и его мутагенное действие
6) Спаривание с 5-бромоурацилом
Кето-форма Аденин
5-бромоурацила
в) Мутагенное действие 5-бромоурацила
Енольная форма
5-бромоурацила
Гуанин
Переходит обратно
в кето-таутомер
...GATACTAG...
...CTATGATC...
...GATACTAG...
...CTATGATC...
Таутомерный переход
I
...GABACTAG...
I
...GABACTAG...
...CTATGATC...
\ Вставка
5-бромоурацила
...GABACTAG...
...CTATGATC...
...GACACTAG...
...CTGTGATC...
Мутированная молекула
...GATACTAG...
...CTATGATC...
форме, чем это имеет место у тимина. Это означает, что в ходе следующего круга ре-
пликации есть относительно высокий шанс на то, что полимераза встретит енол5Ьи,
который (подобно енолтимину) спаривается с G, а не с А (рис. 16.6, б). Это приводит
к появлению точечной мутации (рис. 16.6, в). 2-аминопурин действует подобным
образом: это аналог аденина, амино-таутомер которого спаривается с тимином,
а имино-таутомер спаривается с цитозином, при этом имино-форма встречается
чаще, чем аминоаденин, и, следовательно, вызывает переходы Т в С в процессе
репликации ДНК.
• Дезаминирующие агенты также вызывают точечные мутации. В некоторой сте-
пени дезаминирование оснований (удаление аминогруппы) в молекулах геномной
ДНК происходит самопроизвольно, при этом скорость данного процесса увеличи-
вается такими химикатами, как азотистая кислота, которая дезаминирует аденин,
цитозин и гуанин (тимин не имеет аминогруппы и поэтому не может быть деза-
минирован), а также бисульфит натрия, который действует только на цитозин.
Глава 16. Мутации и репарация ДНК 653
Дезаминирование гуанина не является мутагенным, потому что получающееся
в результате этого основание ксантин блокирует репликацию, когда появляет-
ся в матричном полинуклеотиде. Дезаминирование аденина дает гипоксантин
(рис. 16.7), который спаривается с С, а не с Т, а дезаминирование цитозина дает
урацил, который спаривается с А, а не с G. Поэтому дезаминирование этих двух
оснований приводит к возникновению точечных мутаций при копировании ма-
тричной нити.
• Алкилирующие агенты — мутагены третьего типа, которые могут породить
точечные мутации. Химикаты типа этилметансульфоната (ЭМС) и диметил нитро-
замина добавляют алкильные группы к нуклеотидам в молекулах ДНК, как и ме-
тилирующие агенты типа метилгалогенидов, которые присутствуют в атмосфере,
и продукты метаболизма нитритов. Эффект алкилирования зависит от позиции,
в которой нуклеотид видоизменен, и от типа присоединенной алкильной группы.
Метилирования, например, часто производят модифицированные нуклеотиды
с видоизмененными свойствами спаривания оснований и, таким образом, ведут
к точечным мутациям. Другие алкилирования блокируют репликацию, образуя
поперечные сшивки между двумя нитями молекулы ДНК или присоединяя круп-
ные алкильные группы, которые препятствуют продвижению репликационного
комплекса.
• Интеркалирующие агенты обычно связывают с мутациями типа вставок. Самый
известный мутаген этого типа — бромистый этидий, который флюоресцирует,
будучи подвергнут ультрафиолетовому (УФ-) излучению, и благодаря этому ис-
пользуется для выявления позиций полос ДНК после электрофореза в агарозном
геле (см. техническое примечание 2.2). Бромистый этидий и другие интеркали-
рующие агенты представляют собой плоские молекулы, которые могут протиски-
ваться между парами оснований в двойной спирали, слегка раскручивая спираль
и, следовательно, увеличивая расстояние между смежными парами оснований
(рис. 16.8).
Самые важные типы физических мутагенов следующие.
• Ультрафиолетовое излучение длины волны 260 нм вызывает димеризацию смеж-
ных пиримидиновых оснований, особенно если оба они представлены тимином
(рис. 16.9, а), давая в результате циклобутильный димер. Другие комбинации
пиримидинов также образуют димеры, их порядок в сторону убывания частоты
встречаемости следующий: 5'-СТ-3', 5’-ТС-3' и 5’-СС-3’. Пуриновые димеры встречаются намного реже. Вызванная УФ-излучением димеризация обычно приводит к появлению мутации типа удаления при копировании ви- доизмененной нити. Другой тип вызванного УФ-излучением фотопродукта — поврежде- ние (6-4), при котором атомы углерода номер 4 и 6 из смежных пиримидинов становятся ковалентно связанными (рис. 16.9, б). nh2 // нс || | ^СН ’j1 Дезаминирование О II ^NH
Рис. 16.7. Гипоксантин — дезаминированная форма аденина. Гипоксантин Нуклеозид, который содержит гипоксантин, называют ино- зином (см. рис. 12.18) М Jh !
654 Часть 4. Механизмы репликации и эволюции геномов
а) Бромистый этидий
б) Мутагенное действие
Бромистый
этидий
Рис. 16.8. Мутагенное действие бромистого этидия, о) Бромистый этидий — плоская, подобная пластин-
ке молекула, которая способна втиснуться между парами оснований двойной спирали, б) Молекулы
бромистого этидия показаны вставленными в спираль: молекулы рассматриваются сбоку. Обратите
внимание, что вставка увеличивает расстояние между соседними парами оснований
• Ионизирующее излучение оказывает различные воздействия на ДНК в зависимости
оттипа излучения и его интенсивности. Могут возникнуть мутации точечные, вставки
и (или) удаления, равно как и более серьезные формы повреждения ДНК, которые
препятствуют последующей репликации генома. Некоторые типы ионизирующего
излучения действуют непосредственно на ДНК, другие действуют косвенно, активи-
зируя образование в клетке реакционно активных молекул типа пероксидов.
• Высокая температура стимулирует вызываемое водой расщепление p-N-гликозидной
связи, которая скрепляет основание с сахарной составляющей нуклеотида (рис. 16.10, о).
Это происходит более часто с пуринами, чем с пиримидинами, и приводит к образо-
ванию АП (апуринового/апиримидинового) участка, или участка без основания.
Сахарофосфат, который остается, неустойчив и быстро деградирует, оставляя про-
пуск, если молекула ДНК двунитевая (рис. 16.10, б). Эта реакция обычно не является
мутагенной, потому что клетки имеют эффективные системы устранения пропусков
(раздел 16.2.2). Это звучит обнадеживающей темой на мрачном фоне того, что в каж-
дой человеческой клетке возникает 10 000 АП участков в день. Однако при некоторых
обстоятельствах (например, когда у Е coli активизируется реакция SOS) пропуски
ведут к мутациям: когда, скажем, пропуски заполняются аденинами независимо от
рода нуклеотида в другой нити (раздел 16.2.5).
16.1.2. Воздействия мутаций
При рассмотрении воздействий мутаций мы должны проводить различие между
прямым воздействием, которое мутация оказывает на функционирование генома, и ее
косвенным воздействием на фенотип организма, в котором она возникает. Прямое воз-
действие относительно легко оценить, потому что мы можем использовать наши знания
о структуре и экспрессии гена, чтобы предсказать воздействие, которое мутация окажет
на функции генома. Косвенные воздействия гораздо сложнее, потому что они касаются
фенотипа мутированного организма, который, как было описано в разделе 5.2.2, зача-
стую бывает трудно увязать с активностью отдельных генов.
Глава 16. Мутации и репарация ДНК 655
а) Тиминовый димер
6) Продукт облучения (6—4)
Рис. 16.9. Продукты облучения, порожденные освещением ультрафиолетовым светом. Показан сегмент
полинуклеотида, содержащий два смежных основания тимина, о) Тиминовый димер содержит две по-
рожденные УФ-излучением ковалентные связи: одна соединяет атомы углерода в позиции 6, а другая
соединяет атомы углерода в позиции 5. 6) Повреждение (6-4) состоит в формировании ковалентной
связи между атомами углерода 4 и 6 смежных нуклеотидов
Воздействия мутаций на геномы
Многие мутации вызывают изменения в последовательности нуклеотидов, которые не
оказывают никакого воздействия на функционирование генома. В число таких молчащих
мутаций входят фактически все те, которые возникают в межгенной ДНК, в некодирующих
компонентах генов и в относящихся к генам последовательностях. Другими словами, около
98,5 % генома человека может мутировать без существенных последствий.
Мутации в кодирующих областях генов намного более значимы. Сначала мы рассмо-
трим точечные мутации, которые изменяют последовательность триплетного кодона.
Мутация такого типа вызовет одно из четырех последствий (рис. 16.11).
656 Часть 4. Механизмы репликации и эволюции геномов
а) Вызванный высокой температурой гидролиз 0-М-гликозидной связи
О=Р-О-СН.
ОСНОВАНИЕ
О’
0= Р-О-СН.
Гидролиз
ОСНОВАНИЕ
Отщепленное
основание
0= Р-О-СН
ОСНОВАНИЕ
ОСНОВАНИЕ
0‘
О’
б) Последствия гидролиза в двунитевой ДНК
Рис. 16.10. Мутагенное действие высокой температуры, а) Высокая температура вызывает гидролиз
P-N-гликозидной связи, в результате чего в полинуклеотиде образуется безосновательный участок.
б) Схематическое представление влияния вызванного высокой температурой гидролиза в двунитевой
молекуле ДНК. Потерявший свое основание участок неустойчив и деградирует, оставляя на своем месте
пропуск в одной нити
• Она может привести к синонимичному изменению, при котором новый кодон
определяет ту же аминокислоту, что и немутированный кодон. Поэтому синони-
мичное изменение относится к молчащей мутации, так как оно не оказывает ника-
кого воздействия на кодирующую функцию генома: мутированный ген кодирует
в точности тот же самый белок, что и немутированный ген.
• Она может вызвать несинонимичное изменение, при котором мутация видо-
изменяет кодон таким образом, что он начинает определять совершенно иную
аминокислоту. Поэтому белок, кодируемый мутированным геном, содержит одну
замененную аминокислоту. Часто это не оказывает существенного эффекта на
биологическую активность белка, потому что в большинстве своем белки могут
допустить замены по крайней мере нескольких аминокислот без заметного влияния
на их способность функционировать в клетке, но замены некоторых аминокислот
(например, находящихся в активном участке фермента) имеют более ощутимое воз-
действие. Несинонимичное изменение называют также мутацией с изменением
смысла [миссенс-мутацией. — Прим. ред.].
Глава 16. Мутации и репарация ДНК 657
Несинонимичная
АТА
Иле
I С потерей смысла Сквозного считывания
Синонимичная \ ТЛ Л _л
...ATGGGCAAATATAGCATTCCATAAAAATATATA...
।__11__11__11_II_it__к___II_।
Мет Гли Лиз Тир Сер Иле Про стоп
Рис. 16.11. Влияние точечных мутаций на кодирующую область гена. Показаны четыре различных эф-
фекта точечных мутаций. Мутация типа сквозного считывания приводит к продолжению гена за пределы
конца показанной здесь последовательности; кодон лейцина, порожденный мутацией, сопровождается
кодонами ААА = Лиз, ТАТ = Тир и АТА = Иле. См. генетический код на рис. 1.20
• Мутация может преобразовать кодон, который определяет некоторую аминокис-
лоту, в стоп-кодон. Это мутация с потерей смысла и она дает укороченный белок,
потому что трансляция мРНК останавливается в этом новом стоп-кодоне, вместо
того чтобы продолжаться до нижележащего правильного стоп-кодона. Эффект
такой мутации на активность белка зависит от того, какая доля полипептида будет
потеряна: обычно эффект разителен и белок нефункционален.
• Мутация может преобразовать стоп-кодон в кодон, определяющий ту или иную
аминокислоту, что приводит к сквозному считыванию сигнала остановки, так что
белок прирастает дополнительным рядом аминокислот на своем С-конце. Боль-
шинство белков может допустить короткие продолжения без какого-либо влияния
на свою функцию, но более длинные продолжения могут помешать сворачиванию
белка и через это нарушить его функцию.
Мутации типа удаления и вставки также оказывают различные воздействия на
кодирующие способности генов (рис. 16.12). Если число удаленных или вставленных
нуклеотидов равно или кратно трем, то удаляются или вставляются один или несколько
кодонов, при этом обусловленная этим потеря или приобретение аминокислот имеет
различные последствия, сказывающиеся на функции кодируемого белка. Удаления
или вставки такого типа часто не влекут за собой никаких последствий, но возымеют
действие, если, например, аминокислоты, вовлеченные в активный участок фермента,
будут потеряны или если вставка прерывает важную вторичную структуру в белке.
В ином случае, если число удаленных или вставленных нуклеотидов не равно трем и не
кратно этой цифре, происходит сдвиг рамки считывания, и все кодоны, лежащие ниже
мутации, выбираются из иной рамки считывания, отличающейся от используемой в не-
мутированном гене. Обычно это оказывает значительное воздействие на функцию белка,
потому что большая или меньшая часть мутированного полипептида имеет полностью
отличающуюся последовательность от таковой нормального полипептида.
Труднее сделать обобщения о воздействиях мутаций, которые возникают вне ко-
дирующих областей генома. Любой сайт связывания с белком восприимчив к точечным
658 Часть 4. Механизмы репликации и эволюции геномов
Трёхнуклеотидное удаление
... AT G G G С ТАТА G С АТ Т С С АТА А А А АТ АТ АТ А...
Мет Гли Тир Сер Иле Про стоп
...ATGGGCAAATATAGCATTCCATAAAAATATATA...
।__11__11__11_II__।__к___к___।
Мет Гли Лиз Тир Сер Иле Про стоп
Однонуклеотидное удаление
...ATGGGAAATATAGCATTCCATAAAAATATA...
Мет Гли Асн Иле Ала Фен Гис Лиз Асн Иле
Рис. 16.12. Мутации типа удалений. В верхней последовательности удалены три нуклеотида, состав-
ляющие отдельный кодон. Это укорачивает конечный белковый продукт на одну аминокислоту, но не
затрагивает остальную часть его последовательности. В нижней части рисунка удален единственный
нуклеотид. Это приводит к сдвигу рамки считывания, так что изменяются все кодоны ниже удаления,
в том числе и стоп-кодон, который теперь считывается. Обратите внимание, что если трехнуклеотидное
удаление удаляет части смежных кодонов, то результат оказывается более пагубным, чем показанный
здесь. Рассмотрите, например, удаление тринуклеотида GCA из последовательности ...ATCGGCAAATAT...,
кодирующей олигопептид Мет-Гли-Лиз-Тир. Новая последовательность ...ATGGAATAT... кодирует три-
пептид Мет-Глу-Тир. Две аминокислоты были заменены на одну чужеродную
мутациям и к мутациям типа вставок или удалений, которые изменяют род или отно-
сительное расположение нуклеотидов, вовлеченных во взаимодействие ДНК с белком.
Поэтому такие мутации потенциально способны инактивировать промоторы или регу-
ляторные последовательности с предсказуемыми последствиями для экспрессии гена
(рис. 16.13; разделы 11.2 и 11.3). Области инициации могут, по-видимому, утратить свою
функциональную активность под действием мутаций, которые изменяют, удаляют или
прерывают последовательности, опознаваемые соответствующими связывающимися
белками (раздел 15.2.1), но такие события достоверно не зарегистрированы. Также име-
ется немного информации о потенциальном воздействии на экспрессию генов мутаций,
которые затрагивают положение нуклеосом (раздел 10.2.2).
Одна область, которая была лучше всего исследована, рассматривает мутации,
которые происходят в интронах или на экзон-интронных границах. В этих областях от-
дельные точечные мутации будут важны, если они изменяют нуклеотиды, участвующие
во взаимодействиях типа РНК-белок и РНК-РНК, которые происходят в ходе вырезания
различных типов интронов (разделы 12.2.2 и 12.2.4). Например, мутация G или Т в ДНК-
копии 5'-сайта вырезания интрона GU-AG или же мутация А либо G в З’-сайте вырезания
прервет процесс сплайсинга, потому что правильная экзон-интронная граница больше
не будет опознаваться. Это может означать, что интрон не будетудален из пре-мРНК, но
более вероятно, что в качестве альтернативы будет использован скрытый сайт сплайсин-
га (см. рис. 12.28, б). Также возможно, что мутация в пределах интрона или экзона создаст
новый скрытый участок, который будет предпочтен вместо истинного сайта Сплайсинга,
который сам по себе не мутирован. События обоих типов имеют одинаковый результат:
Глава 16. Мутации и репарация ДНК 659
Регуляторная Основной
последовательность промотор
Удаление
регуляторной
последовательности
НЕУПРАВЛЯЕМАЯ
ТРАНСКРИПЦИЯ
Удаление
основного
промотора
ТРАНСКРИПЦИЯ
ОТСУТСТВУЕТ
Рис. 16.13. Два возможных эффекта мутаций типа удаления в области, расположенной выше гена
изменение местоположения активного сайта сплайсинга, что ведет к ошибочному сплай-
сингу. В результате этого в ходе созревания может удалиться часть белка, добавиться
новый отрезок аминокислот или произойти сдвиг рамки считывания. Несколько вари-
антов р-талассемии (болезни крови) вызваны мутациями, которые приводят к выбору
скрытых сайтов сплайсинга в ходе процессинга транскриптов р-глобина.
Воздействия мутаций на многоклеточные организмы
Теперь мы обратимся к косвенным воздействиям, которые мутации оказывают
на организмы, начиная от многоклеточных диплоидных эукариотов, например людей.
Первая тема, которую необходимо рассмотреть, — относительная значимость одной
и той же мутации в соматической и в зародышевой клетках. Поскольку соматические
клетки не передают копии своих геномов следующему поколению, мутация в соматиче-
ской клетке имеет значение только для того организма, в котором она происходит: она
не обладает никаким потенциальным эволюционным эффектом. Фактически львиная
доля мутаций соматических клеток не оказывает никакого существенного воздействия,
даже если они вызывают смерть клетки, потому что в той же ткани имеется много дру-
гих тождественных клеток и потеря одной гиблой клетки является несущественной.
Совершенно иная картина возникает, когда мутация заставляет соматическую клетку
неправильно функционировать, причем таким образом, который является вредным
для организма, например, вызывая образование опухоли или другую злокачественную
активность.
Мутации в зародышевых клетках более существенны, потому что они могут быть
переданы членам следующего поколения и в таком случае будут присутствовать во всех
клетках того или иного индивидуума, который унаследует данную мутацию. Большин-
ство мутаций, включая все молчащие и многие из приписанных к кодирующим областям,
опять же не будут изменять фенотип организма каким-либо существенным образом. Те,
которые действительно имеют эффект, могут быть подразделены на две категории.
• Потеря функции — нормальный результат мутации, которая снижает или упразд-
няет активность белка. В большинстве своем мутации с потерей функции рецес-
сивны, потому что в гетерозиготе вторая копия хромосомы несет немутированную
версию гена, кодирующую полностью функционально активный белок, присут-
ствие которого компенсирует воздействие мутации (рис. 16.14). Есть некоторые
660 Часть 4. Механизмы репликации и эволюции геномов
Мутация с потерей
функции
Никакого белкового
продукта
Пара
гомологичных
хромосом
• Белковый продукт
Рис. 16.14. Мутация с потерей функции обычно
является рецессивной, потому что на второй копии
хромосомы присутствует функционально активный
вариант гена
исключения, когда мутация с потерей функции является доминантной; один из
примеров представлен гаплонедостаточностью, при которой организм не спо-
собен переносить приблизительно 50 %-е снижение активности белка, каковым
страдают гетерозиготы. Этим объясняется несколько генетических болезней чело-
века, в том числе синдром Марфана, который развивается на почве мутации в гене,
кодирующем белок соединительной ткани, называемый фибриллином.
• Мутации с приобретением функции встречаются намного реже. Такого рода
мутация должна сообщать неправильную активность белку. Многие мутации
с приобретением функции привязаны к регуляторным последовательностям, а не
к кодирующим областям, и могут поэтому иметь целый ряд последствий. Напри-
мер, мутация может привести к экспрессии одного или нескольких генов в несо-
ответственных тканях, причем эти ткани приобретают функции, которые обычно
им не свойственны. В других случаях мутация может привести к сверхэкспрессии
одного или нескольких генов, вовлеченных в управление клеточным циклом,
и, таким образом, вызвать безудержное деление клетки и, следовательно, рак.
В силу своей природы мутации с приобретением функции, как правило, являются
доминантными.
Оценка воздействий мутаций на фенотипы многоклеточных организмов может
быть трудна. Не все мутации проявляют незамедлительное воздействие: для некото-
рых характерно задержанное проявление и они придают видоизмененный фенотип
организму на поздних стадиях жизни индивидуума. Другие мутации выказывают непе-
нетрантность у некоторых индивидуумов и никогда не проявляются даже при том, что
индивидуум обладает доминантной мутацией или гомозиготен по рецессивной мутации.
У людей эти факторы усложняют попытки нанести на карту вызывающие болезнь мута-
ции путем анализа родословной (раздел 3.2.4), потому что они вносят неопределенность
в отношении того, который из членов семейства несет мутантный аллель.
Воздействия мутаций на микроорганизмы
Мутации в микробах типа бактерий и дрожжей также могут быть описаны как
мутации с потерей функции или с приобретением функции, но в случае микроорганиз-
мов это ни вполне адекватная, ни самая удачная схема классификации. Вместо этого,
более подробное описание фенотипа обычно предпринимается на основе свойств роста
мутированных клеток на различных питательных средах. Это позволяет распределить
большую часть мутаций по следующим четырем категориям:
• ауксотрофы — клетки, которые будут расти только будучи обеспечены питатель-
ным веществом, не требующимся немутированному организму. Например, Е coli
обычно производит свой собственный триптофан — благодаря действию фермен-
тов, кодируемых пятью генами в опероне триптофана (рис. 8.8, б). Если один из
этих генов мутировал таким образом, что его белковый продукт инактивирован,
Глава 16. Мутации и репарация ДНК 661
то клетка более не способна производить триптофан и, как следствие, становится
ауксотрофом по триптофану. Она не может выжить на среде, не содержащей трип-
тофан, и может расти только тогда, когда эта аминокислота наличествует в виде пи-
тательного вещества (рис. 16.15). Немутированные бактерии, которые не требуют
дополнительных присадок к их питательной среде, называют прототрофами:
• условно летальные мутанты не способны выдерживать определенные условия
роста: при разрешающих условиях они кажутся абсолютно нормальными, но ког-
да переносятся на ограничивающие условия, проявляется мутантный фенотип.
Чувствительные к температуре мутанты—типичные примеры условно леталь-
ных мутантов. Чувствительные к температуре мутанты при низких температурах
ведут себя подобно клеткам дикого типа, но проявляют свой мутантный фенотип,
когда температура поднимается выше определенного порога, который является
уникальным для каждого мутанта. Обычно это связано с тем, что мутация снижает
устойчивость белка, так что, когда температура поднимается, белок развертывается
и, следовательно, становится неактивным;
• устойчивые к ингибитору мутанты способны сопротивляться токсическому дей-
ствию антибиотика или ингибитора другого типа. Есть различные молекулярные
объяснения мутантов такого типа. В некоторых случаях мутация изменяет струк-
туру белка, который служит мишенью ингибитора, так что последний больше не
может связываться с белком и нарушать его функцию. Это основа устойчивости
Е. coli к стрептомицину, каковая обусловлена изменением структуры рибосомного
белка S12. Другая возможность состоит в том, что мутация изменяет свойства белка,
ответственного за перенос ингибитора в клетку, этим путем часто приобретается
устойчивость к токсичным металлам;
а) Ауксотроф по триптофану б) Перепечатывание реплики
Ауксотроф
по триптофану
Минимальная среда + Минимальная
триптофан среда
Колонии
на минимальной среде
с триптофаном
Минимальная
среда
Колонии
на минимальной
среде
Рис. 16.15. Ауксотрофный мутант по триптофану, а) Показаны две культуры на чашках Петри. В обеих
находится минимальная среда, которая удовлетворяет лишь основные пищевые потребности роста
бактерий (азот, углерод и источники энергии, плюс некоторые соли). В среду слева добавлен триптофан,
а среда справа его не содержит. В чашке слева могут расти немутированные бактерии плюс ауксотрофы
по триптофану, причем ауксотрофы растут, потому что среда снабжает их триптофаном, который они
не могут производить самостоятельно. В чашке справа ауксотрофы по триптофану расти не могут, по-
тому что она не содержит триптофан, б) Чтобы идентифицировать ауксотрофов по триптофану, колонии
сначала выращивают в чашке с минимальной средой и триптофаном, а затем переносят их в чашку
с минимальной средой путем перепечатывания реплики. После инкубации в чашке с минимальной
средой появляются колонии в тех же самых относительных позициях, что и в чашке, содержащей трип-
тофан, за исключением ауксотрофов по триптофану, которые не растут. Поэтому такие колонии могут
быть идентифицированы и образцы ауксотрофных по триптофану бактерий восстановлены (получены)
из чашки с минимальной средой и триптофаном
662 Часть 4. Механизмы репликации и эволюции геномов
• регуляторные мутанты имеют дефекты в промоторах и прочих регуляторных
последовательностях. Эта категория включает конститутивных мутантов, не-
прерывно экспрессирующих гены, которые обычно включаются и выключаются
в различных условиях. Например, мутация в операторной последовательности
оперона лактозы (раздел 11.3.1) может препятствовать репрессору связываться и,
таким образом, приводит к тому, что оперон лактозы экспрессируется все время,
даже когда лактоза отсутствует и соответствующие гены должны быть выключены
(рис. 16.16).
В дополнение к этим четырем категориям есть многие другие мутации, одни из
которых легальны и, как следствие, вызывают гибель мутантной клетки, тогда как
иные не оказывают никакого воздействия. Последние менее распространены в среде
микроорганизмов, чем среди высших эукариотов, потому что большинство микробных
геномов относительно компактны и содержат лишь небольшое количество некодирую-
щей ДНК. Мутации могут также быть расплывчатыми, означая, что мутантный фено-
тип выражается в менее резкой форме. Например, расплывчатая версия ауксотрофа по
триптофану, иллюстрированного на рис. 16.15, росла бы замедленно на минимальной
среде, а не прекратила бы рост вообще.
Репрессор лактозы не может связаться
с мутированным оператором
Мутация
в операторе
ОПЕРОН ЛАКТОЗЫ
ПОСТОЯННО
ЭКСПРЕССИРУЕТСЯ
Рис. 16.16. Влияние конститутивной мутации в операторе лактозы. Последовательность оператора была
видоизменена мутацией, и репрессор лактозы больше не может связываться с ним. В результате опе-
рон лактозы транскрибируется все время, даже когда лактоза отсутствует в среде. Это не единственный
путь, которым может возникнуть конститутивный /ас-мутант. Например, мутация может произойти
в гене, кодирующем репрессор лактозы, и изменить третичную структуру белка-репрессора так, что его
связывающий ДНК мотив будет прерван и он больше не сможет опознавать последовательность опе-
ратора, даже если последняя не мутировала. На рис. 11.24 можно увидеть более подробные сведения
о репрессоре лактозы и его регулирующем воздействии на экспрессию оперона лактозы
16.1.3. Сверхмутация и возможность запрограммированных мутаций
Могут ли клетки использовать мутации положительным образом — или увели-
чивая частоту, с которой мутации возникают в их геномах, или направляя мутации на
определенные гены? Возможны события обоих типов, что, на первый взгляд, противо-
речит здравому смыслу и представлениям о том, что мутации происходят случайно.
Случайность мутаций — важная концепция в биологии, потому что это требование
дарвинистского взгляда на эволюцию, согласно которому изменения в характеристи-
ках организма происходят случайно и не подвержены влиянию окружающей среды,
в которой организм находится. В противоположность ему Ламаркова теория эволюции,
которую биологи отвергли более чем столетие назад, заявляет, что организмы приобре-
Глава 16. Мутации и репарация ДНК 663
тают изменения, которые позволяют им приспособиться к окружающей их среде. Итак,
дарвинистское представление говорит, что мутации возникают случайным образом,
тогда как Ламаркова эволюция гласит, что мутации возникают в ответ на воздействие
окружающей среды.
Эти два явления, которые, как кажется на первый взгляд, противоречат концепции,
согласно которой мутации возникают случайно, были названы сверхмутациями и за-
программированными мутациями.
Сверхмутация обусловлена неправильными процессами репарации ДНК
Сверхмутация происходит, когда клетка позволяет частоте, с которой мутации про-
исходят в ее геноме, увеличиться. Известно несколько примеров сверхмутаций, причем
один из них являет собой часть механизма, используемого некоторыми позвоночными
животными, в том числе людьми, для того чтобы производить разнообразное множе-
ство белков иммуноглобулинов. Мы уже касались этого явления в разделе 14.2.1, когда
мы исследовали перестройки генома, которые приводят к соединению сегментов V, D,
J и С генов тяжелой и легкой цепей иммуноглобулина (см. рис. 14.19). Дополнительное
разнообразие привносится сверхмутацией сегментов V гена после сборки зрелого гена
иммуноглобулина (рис. 16.17), при этом частота появления мутаций в этих сегментах на
6-7 порядков величины больше, чем фоновая частота мутаций, свойственная остальной
части генома.
Точный механизм сверхмутации сегмента V гена неизвестен, и на основании име-
ющихся экспериментальных данных было предложено несколько моделей. Сначала
думали, что повышенная частота мутаций следует из необычного поведения системы
устранения несоответствий, которая обычно исправляет ошибки репликации (см. раз-
дел 16.2.3). Во всех других позициях генома система устранения несоответствий ис-
правляет ошибки репликации путем отыскания несоответствий и замены нуклеотида
в дочерней нити, то есть в нити, которая только что была синтезирована и, следова-
тельно, содержит ошибку. Думалось, что в сегментах V гена восстанавливающая система
заменяет нуклеотид в родительской нити и, таким образом, закрепляет мутацию, вместо
того чтобы исправлять ее (рис. 16.18, а). Позже было показано, что для сверхмутации
сегмента Vтребуются два фермента — цитозиндезаминаза и урацил-ДНК-гликозилаза.
Это привело к предположению о том, что сверхмутация возникает за счет преобразования
некоторых цитозиновых оснований в урацил (действием дезаминазы), сопровождае-
мого вырезанием урацилов (действием гликозилазы), в результате чего возникают АП
участки (рис. 16.18, б). Это напоминает ранние шаги процесса репарации вырезанием
оснований (раздел 16.2.2), в котором урацил, появляющийся в результате действия
дезаминирующего мутагена, вырезается урацил-ДНК-гликозилазой. При репарации вы-
V DJ С
i Гипермутация
Мутации
в сегментах V
Рис. 16.17. Сверхмутация сегмента V гена неповрежденного гена иммуноглобулина. На рис. 14.19 раз-
ворачиваются события, ведущие к сборке гена иммуноглобулина
664 Часть 4. Механизмы репликации и эволюции геномов
а) Гипермутация вследствие неправильного
устранения несоответствий
6) Гипермутация по причине
превращения цитозина в урацил
...GACTAGGAC...
...CTGACCCTG...
Несоответствие
с———\
Нормальное восстановление Гипермутация
I I
...GACTAGGAC.......GACTGGGAC...
...CTGAfCCTG.......CTGACCCTG...
Несоответствие Несоответствие
исправлено закрепилось
.GATACAGCA...
..CTATGTCGT...
I Цитозин дезаминируется
▼ в урацил
.GATAUAGCA...
..CTATGTCGT...
I Урацил вырезается,
АП участок ▼ оставляя АП участок
...GAD^AGCA... ^Репликация
...CTATGTCGT...
...GATA-AGCA...
Репликация ...CTATTTCGT...
/ Произвольный нуклеотид,
GATAAAGCA* вставленный напротив
..’.CTATTTCGT."' АП участка
Мутация закрепилась
Рис. 16.18. Две альтернативные схемы сверхмутации сегментов V гена иммуноглобулина
резанием оснований АП участок был бы затем заполнен ДНК-полимеразой р, с тем чтобы
восстановить первоначальную последовательность, но при сверхмутации АП участки
не устраняются. Это означает, что во время следующего круга репликации в дочернюю
нить напротив каждого АП участка может быть помещен любой из четырех нуклеоти-
дов (в примере на рис. 16.18, б — Т). После этого в дальнейшем цикле репликации эта
мутация будет закреплена.
Запрограммированные мутации, как оказалось, поддерживают теорию
эволюции Ламарка
Очевидное увеличение частоты мутаций, являющееся результатом модификаций
нормального процесса репарации ДНК, не противоречит догме о случайном характере
мутаций. С другой стороны, относящиеся к 1988 г. сообщения о предполагаемой способ-
ности Е coli направлять мутации на гены, мутация которых давала преимущество при
условиях внешней среды, в которых бактерия существует, объяснить гораздо труднее.
Случайный характер мутаций у бактерий впервые был продемонстрирован Лурией
и Дельбрюком в 1943 г. Они вырастили серию культур Е coli в различных колбах и затем до-
бавили бактериофаги Т1 в каждую. Большинство бактерий было убито фагами, но несколько
устойчивых к фагу Т1 мутантов оказались способными выжить. Они были опознаны путем
высева образцов из каждой культуры вскоре после инфицирования фагом Т1 на агаровую
среду. Если мутации, ведущие к устойчивости к фагу Т1, произошли случайным образом
в культурах прежде, чем были добавлены бактериофаги, то каждая культура содержала бы
различное число устойчивых мутантов, при этом их число зависело бы от того, насколько
рано в течение периода роста появились бы первые мутантные клетки (рис. 16.19). Те,
что возникли рано, успели бы поделиться много раз и дать начало большому количеству
устойчивого потомства в культуре к концу периода роста, тогда как те, которые возникли
позже, породили бы малое число потомков. Некоторые культуры поэтому содержали бы
много устойчивых к фагу Т1 клеток, а другие содержали бы лишь несколько. И наоборот,
если устойчивые бактерии появились в результате запрограммированной мутации и только
Глава 16. Мутации и репарация ДНК 665
Время, ч
О
КУЛЬТУРА 1
Ранняя мутация
4
КУЛЬТУРА 2
Поздняя мутация
КУЛЬТУРА КУЛЬТУРА
3 4
Добавление
। 1 фага Т1
8
СЛУЧАЙНЫЕ МУТАЦИИ
Разное число колоний
в различных культурах
ЗАПРОГРАММИРОВАННЫЕ
МУТАЦИИ
Одно и то же число колоний
в каждой из культур
Рис. 16.19. Случайные и запрограммиро-
ванные мутации. Результаты, полученные
Лури и Дельбрюком, показаны слева.
В течение роста культур Е. coli мутации,
которые придаютустойчивость к бактерио-
фагу Т1, возни кают беспорядочно в разное
время, означая, что каждая культура имеет
различное число устойчивых клеток, когда
фаги добавляются. Поэтому наблюдаются
различные числа колоний, когда бактерии
высеваются на твердую среду. Справа по-
казан результат, который был бы получен,
если бы происходили запрограммирован-
ные мутации. Теперь устойчивые к фагу
Т1 бактерии приобретают свои мутации
в ответ на их предание фагам, и каждая
чашка поэтому имеет одинаковое число
колоний
тогда, когда был добавлен фаг Т1, то
все культуры имели бы одинаковое
число мутантов. Лурия и Дельбрюк
установили, что каждая из их культур
содержала различное число устойчи-
вых к фагу Т1 бактерий; таким обра-
зом, они пришли к заключению о том,
что мутации происходят случайно,
а не в ответ на действия фага Т1.
Мысль о том, что вывод Лурии и Дельбрюка может быть неунивесально истинным
для мутаций Е coli, впервые возникла в ходе исследований штамма Е. coli, который несет
мутацию с потерей смысла в своем гене lacZ. Присутствие стоп-кодона в гене lacZ озна-
чает, что эти клетки неспособны синтезировать функционально активные ферменты
Р-галактозидазы и, следовательно, не могут использовать лактозу в качестве источника
углерода и энергии, поэтому они являются ауксотрофами по лактозе. Это не обязательно
постоянная ситуация, потому что клетка может подвергнуться мутации, которая пре-
вратит стоп-кодон обратно в кодон, определяющий некоторую аминокислоту. Эти новые
мутанты были бы способны производить р-галактозидазу и потреблять имеющуюся
в наличии лактозу. Согласно результатам Лурии и Дельбрюка, такие мутации должны
происходить случайно и не должны зависеть от присутствия лактозы в среде. Однако
исследования показали, что, когда ауксотрофы по лактозе высеваются на минимальную
среду, содержащую лактозу как единственный сахар, — те обстоятельства, которые
требуют, чтобы бактерии мутировали в прототрофы по лактозе, чтобы выжить, —
число появляющихся прототрофов по лактозе значительно выше, чем следовало бы
ожидать, если бы мутации возникали случайно. Другими словами, некоторые клетки
претерпевают запрограммированную мутацию и приобретают определенное измене-
ние в последовательности ДНК, необходимое им для того, чтобы выдержать давление
естественного отбора.
Эти эксперименты предполагают, что бактерии могут программировать мутации
согласно воздействиям естественного отбора, которым они подвергаются. Другими
666 Часть 4. Механизмы репликации и эволюции геномов
словами, окружающая среда может непосредственно влиять на фенотип организма, как
это и предполагал Ламарк, вместо того чтобы действовать через случайные процессы,
постулированные Дарвином. При таких радикальных следствиях не удивительно, что
эти эксперименты вызвали продолжительные споры с многочисленными попытками
обнаружить недостатки в плане их постановки или дать альтернативное истолкование
результатов. Разновидности системы эксперимента над геном lacZ привели к заклю-
чению о том, что оригинальные результаты являются достоверными, и были описаны
подобные события в других бактериях. В настоящее время проверяются модели, осно-
ванные на умножении гена, а не на селективной мутации, и, кроме того, внимание было
направлено на возможные роли в порождении запрограммированных мутаций1) таких
событий рекомбинации, как транспозиция перемещающихся встроенных элементов
генома.
16.2. Репарация ДНК
Ввиду тысяч событий повреждения, которые геномы переносят каждый день, равно
как и ошибок, которые происходят при репликации генома, существенно необходимо,
чтобы клетки обладали эффективными системами репарации. Без таких систем репара-
ции геном был бы способен поддерживать свои жизненно важные для клетки функции
не долее нескольких часов, после чего основные гены стали бы инактивированными
вследствие повреждения ДНК. Подобным же образом клеточные линии накопляли бы
ошибки репликации с такой скоростью, что их геномы стали бы дисфункциональными
после нескольких клеточных делений.
Большинство клеток обладает системами репарации ДНК четырех различных
категорий (рис. 16.20):
• системы прямого восстановления (раздел 16.2.1), как подразумевает название,
воздействуют непосредственно на поврежденные нуклеотиды, преобразуя их об-
ратно к исконным структурам;
• восстановление вырезанием (раздел 16.2.2) предполагает вырезание сегмента по-
линуклеотида, содержащего поврежденный участок, сопровождаемое ресинтезом
правильной последовательности нуклеотидов при помощи ДНК-полимеразы;
• системы устранения несоответствий (раздел 16.2.3) исправляют ошибки репли-
кации — опять же путем вырезания отрезка однонитевой ДНК, содержащего про-
блемный нуклеотид, и последующего восстановления полученного пропуска;
• негомологическое соединение концов (раздел 16.2.4) используется, чтобы ис-
править двунитевые разрывы (заштопать прорехи).
В большинстве своем (если не все) организмы обладают также системами, которые
позволяют им реплицировать поврежденные области своего генома без предшествую-
щего восстановления. Мы исследуем эти системы в разделе 16.2.5, а в разделе 16.2.6
мы рассмотрим болезни человека, которые следуют из дефектов в процессах репара-
ции ДНК.
1 Существуют еще гены-мутаторы, которые активизируются при неблагоприятных условиях и увеличи-
вают количество ошибок при репликации. В неблагоприятных условиях также активизируются события транс-
позиции. Все это говорит о том, что по крайней мере бактерии в тяжелых условиях начинают модифицировать
свой геном. Если учесть, что в колониях живут многие миллиарды клеток, то можно понять, как среди этого
множества могут появиться более приспособленные клетки. Они за счет более интенсивного размножения
вытеснят остальных, и у исследователя возникнет иллюзия направленного мутагенеза. — Прим. ред.
Глава 16. Мутации и репарация ДНК 667
а) Прямое восстановление
ппп ищи it —► iimimri гит
Поврежденный
нуклеотид
6) Вырезающее восстановление
Поврежденный
нуклеотид
Вырезанный
сегмент
III 1111IIII ГИТ
Ресинтезированная
ДНК
в) Устранение несоответствия
Несовпадение
Вырезанный
сегмент
—► 111I I11 l l'ITI'I IT
Ресинтезированная
ДНК
а) Негомологическое соединение концов
Двунитевой разрыв
тттптгггттт,гтт
Рис. 16.20. Четыре категории систем восстановления ДНК
16.2.1. Системы прямого восстановления заполняют однонитевые раз-
рывы и исправляют модификации нуклеотидов некоторых типов
Повреждения ДНК большинства типов, которые вызываются химическими или
физическими мутагенами (раздел 16.1.1), могут быть восстановлены только выреза-
нием поврежденного нуклеотида, сопровождаемым ресинтезом нового отрезка ДНК,
как показано на рис. 16.20, б. Лишь относящиеся к нескольким типам повреждения
нуклеотидов могут быть устранены непосредственно.
• Однонитевые разрывы могут быть устранены ДНК-лигазой, если все, что слу-
чилось, — разорвалась фосфодиэфирная связь без ущерба для 5'-фосфатной и 3'-
гидроксильной групп нуклеотидов по обе стороны от разрыва (рис. 16.21). Это часто
имеет место при однонитевых разрывах, получающихся в результате воздействия
ионизирующего излучения.
• Некоторые формы повреждения алкилированием напрямую восстанавливаются
ферментами, которые переносят алкильную группу с нуклеотида на свои собствен-
ные полипептидные цепи. Ферменты, способные это делать, обнаружены во многих
различных организмах и включают фермент Ada Е coli, который вовлечен в адаптив-
ный процесс, который эта бактерия способна активизировать в ответ на поврежде-
ние ДНК. Ada удаляет алкильные группы, присоединенные к кислородным группам
в позициях 4 и 6 соответственно тимина и гуанина, и может также восстанавливать
фосфодиэфирные связи, которые стали метилированными. Другие ферменты снятия
ал кил ированности имеют более ограниченную специфику, пример представлен MGMT
(06-метилгуанин-ДНК-метилтрансферазой) человека, которая, как явствует из ее на-
звания, удаляет алкильные группы только из позиции 6 гуанина.
• Циклобутильные димеры устраняются светозависимой прямой системой, на-
званной фотореактивацией. У Е coli этот процесс вовлекает фермент, названный
Надрыв
1111111111111 i jl 1111! 1111111
I
.— ДНК-лигаза
1111'11J111 f H Г.П1 ПТТТТТТТТ
l
ПТТШЛШТПТГГП H1111'1 f
Надрыв устранен
Рис. 16.21. Устранение надрыва
ДНК-лигазой
668 Часть 4. Механизмы репликации и эволюции геномов
ДНК-фотолиазой (более правильно называть дезоксири-
бодипиримидинфотолиазой). Будучи активирован све-
том с длиной волны от 300 до 500 нм, данный фермент
связывается с циклобутильными димерами и превра-
щает их обратно в исходные мономерные нуклеотиды.
Фотореактивация — восстановление широко распростра-
ненного, но не универсального типа: она обнаружена
у многих, но не у всех бактерий и также у порядочного
числа эукариотов, в том числе у некоторых позвоночных,
но отсутствует у людей и других плацентарных млеко-
питающих. Подобного типа фотореактивация вовлекает
фотопродукт (6-4)-фотолиазу и обеспечивает устране-
ние повреждения (6-4). Ни Е. coli, ни человек не обладают
этим ферментом, однако он наличествует у разных других
организмов.
16.2.2. Восстановление вырезанием
Механизмы устранения повреждений прямого типа, описанные выше, важны, но
они составляют весьма незначительную долю механизмов репарации ДНК большин-
ства организмов. Этот момент иллюстрирован последовательностью генома человека,
которая, как оказалось, содержит лишь один-единственный ген, кодирующий белок,
участвующий в прямой репарации (ген MGMT), но которая имеет по крайней мере
40 генов, кодирующих компоненты путей восстановления вырезанием. Эти пути под-
падают под две категории:
• восстановление вырезанием оснований заключается в вырезании короткой
части полинуклеотида вокруг поврежденного участка. Таким образом создается
АП участок. Затем происходит ресинтез с помощью ДНК-полимеразы;
• восстановление вырезанием нуклеотидов подобно восстановлению вырезани-
ем оснований, но ему не предшествует удаление поврежденного основания и оно
может действовать на более значительно поврежденные области ДНК.
Мы исследуем каждый из этих путей по очереди.
Система вырезания оснований восстанавливает повреждения нуклеотидов
многих типов
Вырезание оснований является наименее сложной из различных систем репара-
ции, которые предполагают удаление одного или нескольких нуклеотидов, сопрово-
ждаемое ресинтезом ДНК с целью заполнить получившийся пропуск. Она используется
для репарации многих видоизмененных нуклеотидов, основания которых перенесли
относительно незначительное повреждение, обусловленное, например, действием
алкилирующих агентов или ионизирующего излучения (раздел 16.1.1). Этот процесс
инициируется ДНК-гликозилазой, которая расщепляет p-N-гликозидную связь между
поврежденным основанием и сахарной составляющей нуклеотида (рис. 16.22, а). Каждая
ДНК-гликозилаза имеет ограниченную специфичность (таблица 16.3), и специфичности
гликозилаз, которыми обладает клетка, определяют охват поврежденных нуклеоти-
дов, которые могут быть восстановлены путем вырезания оснований. Большинство
организмов способно справляться с дезаминированными основаниями типа урацила
(дезаминированный цитозин) и гипоксантина (дезаминированный аденин), продукта-
Глава 16. Мутации и репарация ДНК 669
ми окисления типа 5-гидроксицитозина и тимингликоля, а также с метилированными
основаниями типа 3-метиладенина, 7-метилгуанина и 2-метилцитозина. Другие ДНК-
гликозилазы удаляют нормальные основания, выступая частью системы устранения
несоответствий (раздел 16.2.3). Большинство ДНК-гликозилаз,участвующих в репарации
вырезанием оснований, как думают, распространяется вдоль малой бороздки двойной
спирали ДНК в поиске поврежденных нуклеотидов, но некоторые могут быть связаны
с репликационными ферментами.
ДНК-гликозилазаудаляет поврежденное основание, «перещелкивая» соответствую-
щую структуру в положение вне (снаружи) спирали и затем отделяя ее от полинуклеоти-
да. В результате этого образуется АП участок, или участок без оснований, который пре-
вращается в однонуклеотидный пропуск на втором шаге пути репарации (рис. 16.22, б).
Этот шаг может быть выполнен множеством различных путей. Стандартный метод
использует АП-эндонуклеазу типа экзонуклеазы III, или эндонуклеазы IV Е coli, или
АРЕ1 человека, которая разрезает фосфодиэфирную связь с 5'-стороны АП участка. Не-
которые АП-эндонуклеазы могут также удалить сахар из АП участка, причем это все,
а) Удаление поврежденного основания ДНК-гликозилазой
б) Общая схема способа
111111 HI iiriffirnirinun'
к ДНК-гликозилаза (см. вид а)
АП участок
1111111II1111 1?Н 111111111 и
\АП-эндонуклеаза, возможно
с фосфодиэстеразой
к .Однонуклеотидный пропуск
1Ш1тп1ттгттт 11111 ПН ГI
\ДНК-полимераза +
ДНК-лигаза
ни»птгп гттптп ттгп'пт
Рис. 16.22. Восстановление вырезанием оснований, а) Вырезание поврежденного нуклеотида ДНК-
гликозилазой. б) Схематическое представление способа восстановления вырезанием оснований. Аль-
тернативные варианты этого способа описаны в тексте
670 Часть 4. Механизмы репликации и эволюции геномов
Таблица 16.3. Примеры ДНК-гликозилаз человека
ДНК-гликозилаза Объект специфичности
MBD4 Урацил
MPG Этеноаденин, гипоксантин, 3-метиладенин
NTH1 Цитозингликоль, ди гидроурацил, формамидопиримидин, тимингликоль
OGG1 Формамидопиримидин, 8-оксогуанин
SMUG1 Урацил
TDG Этеноцитозин, урацил
UNG Урацил, 5-гидроксиурацил
что остается от поврежденного нуклеотида, но у других недостает такой способности
и поэтому они работают совместно с обособленной фосфодиэстеразой. Альтернативный
путь преобразования АП участка в пропуск основан на действии эндонуклеазы, свой-
ственном некоторым ДНК-гликозилазам, благодаря которому возможно сделать разрез
с З'-стороны АП участка, вероятно, в то же самое время, когда поврежденное основание
удаляется, за чем опять же следует удаление сахара фосфодиэстеразой.
Однонуклеотидный пропуск заполняется ДНК-полимеразой за счет спаривания
основания с неповрежденным основанием в другой нити молекулы ДНК, что позволяет
гарантировать, что будет вставлен правильный нуклеотид. У Е coli пропуск заполняется
ДНК-полимеразой I, а у млекопитающих — ДНК-полимеразой р (см. табл. 15.2). Дрожжи
кажутся необычными в том, что они используют для этой цели свой главный реплици-
рующий ДНК фермент — ДНК-полимеразу 8. После заполнения пропуска завершающая
процесс фосфодиэфирная связь устанавливается на место ДНК-лигазой.
Восстановление ДНК вырезанием нуклеотидов используется для исправле-
ния более обширных повреждений
Восстановление путем вырезания нуклеотидов имеет намного более широкую
специфику, чем система вырезания оснований, и способна исправлять более тяжелые
формы повреждений типа межнитевых поперечных сшивок и оснований, которые стали
видоизмененными вследствие присоединения крупных химических групп. Оно также
способно исправить циклобутильные димеры при помощи процесса темнового вос-
становления, обеспечивая те организмы, которые не имеют системы фотореактивации
(например, людей) средством устранения повреждений такого типа.
При восстановлении вырезанием нуклеотидов сегмент однонитевой ДНК, содержа-
щий поврежденный нуклеотид (или нуклеотиды), вырезается и заменяется новой ДНК.
Этот процесс поэтому подобен восстановлению вырезанием оснований, за исключением
того, что ему не предшествует избирательное удаление оснований, а вырезается более
длинный отрезок полинуклеотида. Наиболее полно изученный пример восстановле-
ния вырезанием нуклеотидов — процесс латания короткими заплатами у Е. coli. Он
так назван, потому что область полинуклеотида, которая вырезается и впоследствии
«латается», относительно коротка (обычно 12 нуклеотидов в длину).
Восстановление короткими заплатами инициируется многоферментным комплек-
сом, названным UvrABC-эндонуклеазой, иногда также именуемый «эксинуклеазой».
На первой стадии процесса тример, состоящий из двух белков UvrA и одной копии UvrB,
Глава 16. Мутации и репарация ДНК 671
прикрепляется кДНК на поврежденном участке. Как такой участок опознается, не извест-
но, но широкая специфика процесса указывает на то, что отдельные типы повреждений
не являются прямыми мишенями, и что сей комплекс, должно быть, отыскивает более
общий признак повреждения ДНК наподобие искажения двойной спирали. Возможно,
UvrA является частью комплекса, наиболее вовлеченной в определение местоположения
повреждения, потому что он отделяется, как только такой участок будет найден, и не
играет никакой дальнейшей роли в процессе репарации. Отсоединение UvrA позволяет
связаться белку UvrC (рис. 16.23) с образованием димера UvrBC, который разрезает по-
линуклеотид по обе стороны от поврежденного участка. Первый разрез производится
UvrB на пятой фосфодиэфирной связи ниже поврежденного нуклеотида, а второй раз-
рез выполняется UvrC на восьмой фосфодиэфирной связи выше него, в результате чего
вырезается 12 нуклеотидов, хотя существует некоторая изменчивость, особенно в от-
ношении позиции участка разрезания белком UvrB. После этого вырезанный сегмент
удаляется (обычно в виде неповрежденного олигонуклеотида) ДНК-геликазой II, которая
предположительно отделяет такой сегмент, разрывая пары оснований, удерживающие
его на второй нити. UvrC также отделяется на данном этапе, но UvrB остается на месте
и покрывает пропуск, произведенный вырезанием. Связанный UvrB, как думают, препят-
ствует однонитевой области, которая стала открытой, спариваться основаниями самой
с собой, но роль UvrB может заключаться и в том, чтобы предотвратить повреждение
этой нити, или, возможно, в том, чтобы направлять ДНК-полимеразу к участку, который
требует восстановления. Как и при восстановлении вырезанием оснований, пропуск
заполняется ДНК-полимеразой I, а последняя фосфодиэфирная связь синтезируется
ДНК-лигазой.
Е coliтакже имеет систему восстановле-
ния вырезанием нуклеотидов и наложением
длинных заплат, которая вовлекает белки
Uvr, но отличается в том, что вырезаемая
часть ДНК может быть до 2 тыс.п.н. в дли-
ну. Восстановление длинными заплатами
было менее хорошо изучено, и сам процесс
не понят в мелочах, но он, как предполага-
ется, работает с более обширными формами
повреждения, возможно, с областями, где
видоизмененными оказались не отдельные
нуклеотиды, а целые их группы. У эукарио-
тов процесс восстановления вырезанием
нуклеотидов также называют «наложением
длинных заплат», но у них он обеспечивает
Рис. 16.23. Восстановление вырезанием нуклеотида
и наложением короткой заплаты у Escherichia coli.
Поврежденный нуклеотид показан искажающим
спираль, потому что он, как думают, является одним
из опознавательных сигналов для тримера UvrAB,
который начинает процесс короткого латания. В тек-
сте подробно описываются события, происходящие
в ходе восстановления. Сокращения: А — UvrA; В —
UvrB; С - UvrC
Повреждённый нуклеотид,
вызывающий искажение спирали
IIIHlIHiiiiiiii^iiiiiiiillllirrr
I Отсоединяется UvrA
Прикрепляется UvrC
Вырезание сегмента (разрезы,
произведённые UvrB и UvrC),
удаление одной вырезанной
нити ДНК-геликазой II
UvrB покрывает пропуск
5' \ 3'
ШЩттгп гттт,, ,111111111 U-ini
I ДНК-полимераза I +
ДНК-лигаза
TimmmTmTFi т.п гнпттгтттгпт
672 Часть 4. Механизмы репликации и эволюции геномов
Повреждённый нуклеотид,
вызывающий искажение спирали
Точка вырезания |
Точка вырезания
тптптггтдггт
Расплавляемая . вырезание повреждённой
область | области
Рис. 16.24. Общая схема событий, из которых
складывается восстановление вырезанием
нуклеотидов у эукариотов. Эндонуклеазы,
удаляющие поврежденную область, делают
разрезы специфично в месте перехода между
однонитевыми и двунитевыми областями мо-
лекулы ДНК. Поэтому ДНК, как думают, плавит-
ся с обеих сторон поврежденного нуклеотида,
как показано на схеме, возможно, в результате
действия геликазы TFIIH
замену только 24-29 нуклеотидов ДНК.
Фактически у эукариотов не существу-
i ДНК-полимераза +
ДНК-лигаза
ет системы с «котороткими заплатами»,
и название «длинные заплаты» употре-
бляется, чтобы отличать этот процесс
от восстановления вырезанием основа-
ний. Система более сложна, чем у Е. coli,
и связанные с ней ферменты, кажется, не являются гомологами белков Uvr. У людей
в эту систему замешано по крайней мере 16 белков, а нижележащий разрез произво-
дится в той же самой позиции, что и у Е. coli, — по пятой фосфодиэфирной связи, — но
вышележащий разрез более отдален, в силу чего получается более длинная вырезка.
Оба разреза осуществляются эндонуклеазами, которые воздействуют на однонитевую
ДНК специфично в месте ее стыка с двунитевой областью, указывая на то, что, прежде
чем производятся разрезы, ДНК вокруг участка повреждения была расплавлена, пред-
положительно геликазой (рис. 16.24). Это действие обеспечивается, по крайней мере от-
части, TFIIH — одним из компонентов инициаторного комплекса РНК-полимеразы II (см.
табл. 11.5). Сначала предполагалось, что TFIIH попросту играет двоякую роль в клетке,
функционируя отдельно и в транскрипции и в репарации, но теперь думается, что есть
более прямая связь между этими двумя процессами. Это представление подтверждается
открытием сопряженного с транскрипцией восстановления, которое восстанавливает
некоторые формы повреждения в матричных нитях генов, которые активно транскри-
бируются. Первым открытым типом сопряженного с транскрипцией восстановления
была видоизмененная версия вырезания нуклеотидов, но теперь известно, что восста-
новление вырезанием оснований также сопряжено с транскрипцией. Эти открытия не
подразумевают, что нетранскрибируемые области генома не восстанавливаются. Про-
цессы восстановления вырезанием защищают весь геном от повреждения, но вполне
логично, что специальные механизмы должны существовать для того, чтобы направлять
эти процессы на гены, которые транскрибируются. Матричные нити этих генов содер-
жат биологическую информацию генома и поддержание их целостности должно быть
высшим приоритетом для систем репарации.
16.2.3. Устранение несоответствий: исправление ошибок репликации
Каждая из систем репарации, которые мы до сих пор рассмотрели, — прямого, выре-
занием оснований и вырезанием нуклеотидов — опознает и работает над повреждением
ДНК, вызванным мутагенами. Это означает, что они отыскивают неправильные химические
структуры типа видоизмененных нуклеотидов, циклобутильных димеров и межнитевых
Глава 16. Мутации и репарация ДНК 673
Рис. 16.25. Метилирование новосинтезированной ДНК
у Escherichia coli не происходит немедленно после ре-
пликации и тем самым обеспечивает удобный момент
белкам устранения несоответствий опознать дочерние
нити и исправить ошибки репликации
РОДИТЕЛЬСКАЯ МОЛЕКУЛА
Полностью метилирована
• Метильные группы
перекрестных сшивок. Они не могут, однако, ис-
правлять несоответствия, следующие из ошибок
репликации, потому что несоответствующий ну-
клеотид не является аномальным ни в каком от-
ношении, это попросту А, С, G или Т, который был
вставлен в ненадлежащую позицию. Поскольку эти
нуклеотиды в точности походят на любой другой
нуклеотид, система устранения несоответствий,
которая исправляет ошибки репликации, должна
обнаруживать не сам по себе несоответствующий
нуклеотид, а отсутствие спаривания оснований
между родительской и дочерней нитями в пози-
ции такого нуклеотида. Как только система репа-
рации нашла несоответствие, она вырезает часть
дочернего полинуклеотида и заполняет пропуск,
способом, подобным восстановлению вырезанием
оснований и нуклеотидов.
Схема, описанная выше, оставляет без отве-
та один важный вопрос. Восстановление должно
быть произведено в дочернем полинуклеотиде,
ДОЧЕРНИЕ МОЛЕКУЛЫ
Новая ДНК еще
не метилирована
ДОЧЕРНИЕ МОЛЕКУЛЫ
Полностью метилированы
потому что ошибка произошла именно в этой
новосинтезированной нити: родительский по-
линуклеотид имеет правильную последовательность. Как процесс репарации узнает,
которая нить является которой? У Е. coli ответ заключается в том, что дочерняя нить
на этой стадии недометилирована и может поэтому быть отличима от родительского
полинуклеотида, который имеет полный набор метильных групп. ДНК Е. coli метили-
руется из-за действий ДНК-аденинметилазы (Dam), которая превращает аденины
в 6-метиладенины в последовательности 5’-GATC-3', и ДНК-цитозинметилазы (Dem),
которая превращает цитозины в 5-метилцитозины в последовательностях 5’-CCAGG-3'
и 5'-CCTGG-3’. Эти метилирования не являются мутагенными, и видоизмененные нуклео-
тиды обладают такими же свойствами спаривания оснований, что и невидоизмененные
версии. Между репликацией ДНК и метилированием дочерней нити имеет место простой,
и именно в течение этого удобного момента система репарации просматривает ДНК
в поиске несоответствий и вносит необходимые исправления в недометилированную
дочернюю нить (рис. 16.25).
Е. coli имеет по крайней мере три системы репарации несоответствий, названные
«длинной заплатой», «короткой заплатой» и «очень короткой заплатой»: эти назва-
ния отражают относительные длины вырезаемых и ресинтезируемых сегментов ДНК.
Система с длинными заплатами заменяет до тысячи пар нуклеотидов и более ДНК
и требует белки MutH, MutL и MutS, а также ДНК-геликазу II, которую мы встречали
в процессе восстановления вырезанием нуклеотидов. MutS опознает несоответствие,
674 Часть 4. Механизмы репликации и эволюции геномов
Несоответствие в дочерней нити
Метильная . Прикрепление
группа | MutH и MutS
Н S
11111111ИТПIГГ7У111111111111111
I MutH разрезает ДНК
I Отсоединение
и удаление нити
Экзонуклеаза —.^^ ДНК-геликаза II
IIIIIIITIIL......7Т
I ДНК-полимераза I +
4 ДНК-лигаза
IF; 111! и 1111111 И ! и 1111 М ! [ гпт
------------J------------------------
Рис. 16.26. Устранение несоответствий длинными заплата-
ми у Escherichia coli. Сокращения: Н — MutH; S — MutS
a MutH различает две нити, связываясь с немети-
лированными последовательностями 5'-GATC-3'
(рис. 16.26). Роль MutL не ясна, но он, возможно,
согласовывает действия других двух белков, так
что MutH связывается с неметилированными по-
следовательностями 5'-GATC-3' только вблизи
участков несоответствия, опознанных MutS. По-
сле связывания MutH разрезает фосфодиэфир-
ную связь непосредственно выше G в неметили-
рованной последовательности, и ДНК-геликаза
II отделяет одинарную нить. Как оказалось, не
существует фермента, который разрезает эту
нить ниже несоответствия; вместо этого, отде-
ленная однонитевая область деградирует под
действием экзонуклеазы, которая следует за ге-
ликазой и простирается за пределы участка не-
соответствия. После этого пропуск заполняется
ДНК-полимеразой I и ДНК-лигазой. Подобные
события, как думают, происходят в процессе ре-
парации несоответствий с латанием короткими
заплатами и очень короткими заплатами, отличие
заключается в специфичности белков, которые
опознают несоответствие. Система с короткими
заплатами, которая осуществляет вырезание сег-
ментов длины менее 10 нуклеотидов, начинает работать, когда MutY опознают несоот-
ветствия А-G или А-С, а система репарации с очень короткими заплатами исправляет
несоответствия G-Т, которые опознаются Vsr-эндонуклеазой.
Гомологи белков MutS и MutL, находящихся в арсенале Е. coli, присутствуют в эука-
риотах, и процессы восстановления несоответствий, вероятно, работают у них подобным
образом. Единственное отличие — отсутствие гомолога белка MutH, что предполагает,
что метилирование может не быть критерием, используемым для различения между
родительским и дочерним полинуклеотидами. Было обнаружено, что метилирование
участвует в репарации несоответствий в клетках млекопитающих, но ДНК некоторых
эукариотов, в том числе плодовых мушек и дрожжей, не метилируется интенсивно.
Поэтому думается, что в этих организмах для отличения дочерней нити, должно быть,
используется какой-либо иной метод. Возможно, это осуществляется объединением
восстанавливающих ферментов и репликационного комплекса, так что репарация
сопрягается с синтезом ДНК, или же привлечением белков однонитевого связывания,
которые отмечают родительскую нить.
16.2.4. Устранение разрывов ДНК
Однонитевой разрыв в двунитевой молекуле ДНК, наподобие тех, что произво-
дятся окислительными повреждениями некоторых типов, не представляет для клетки
серьезной проблемы, поскольку двойная спираль в общем остается неповрежденной.
Глава 16. Мутации и репарация ДНК 675
Открытая одинарная нить покрывается белками PARP1,
что защищает эту неповрежденную нить от разрушения
и препятствует ей участвовать в нежелательных собы-
тиях рекомбинации. После этого разрыв заполняется
ферментами, участвующими в системах восстановления
вырезанием (рис. 16.27).
Двунитевой разрыв более серьезен, потому что он
превращает исходную двойную спираль в два отдель-
ных фрагмента, которые должны быть снова соеди-
нены воедино, чтобы разрыв был ликвидирован. Два
оторванных конца должны быть защищены от дальней-
шей деградации, которая может привести к появлению
л'лгтпгтгь.
1 Белки PARP1
/\
птттттъ., л, мтп ш mi пт
| Устранение
imimmimiifvmiiiiiiii
Рис. 16.27. Устранение однонитевого
разрыва
мутации типа удаления в восстановленной точке разрыва. Процессы репарации должны
гарантировать также и то, что будут соединены правильные концы: если в ядре находятся
две разорванные хромосомы, то должны быть сведены правильные пары концов, с тем
чтобы были восстановлены первоначальные структуры. Экспериментальные исследо-
вания клеток мыши показывают, что достижение такого результата трудно, и если две
хромосомы разорваны, то довольно часто происходит неправильное восстановление,
дающее начало гибридным структурам. Даже если только одна хромосома будет разо-
рвана, все еще сохраняется возможность, что естественный конец хромосомы может
быть перепутан с обрывом и произойдет неправильное восстановление. Ошибки такого
типа не редкость, несмотря на присутствие специальных связывающихся с теломерами
белков, которые отмечают естественные концы хромосом (раздел 7.1.2).
Двунитевые разрывы возникают в результате действия ионизирующего из-
лучения и некоторых химических мутагенов, и в процессе репликации ДНК также
могут произойти разрывы. В большинстве своем (если не все) организмы имеют две
разные системы устранения двунитевых разрывов. Первая связана с гомологичной
рекомбинацией, и мы поэтому отложим ее анализ до той поры, пока не рассмотрим
основной путь гомологичной рекомбинации в главе 17. Вторую систему называют
негомологическим соединением концов (NHEJ). Прогресс в понимании NHE) под-
гонялся исследованиями линий мутантных клеток человека, благодаря которым
удалось распознать различные наборы генов, участвующих в данном процессе. Эти
гены определяют многокомпонентный белковый комплекс, который направляет
ДНК-лигазу к разрыву (рис. 16.28, а). Этот комплекс включает две копии белка Ku:
каждая из копий прикрепляется к каждому концу разорванной ДНК. Белки Ku могут
присоединяться только к обрезанным концам, но не к внутренним областям моле-
кулы ДНК, потому что молекула ДНК должна вписаться в петлю, формируемую за
счет ассоциации между двумя субъединицами, которые составляют каждый белок Ku
(рис. 16.28, б). Отдельные белки Ku обладают сродством друг к другу, а это означает,
что два оборванных конца молекулы ДНК приводятся в непосредственную близость.
Ku связывается с ДНК в ассоциации с протеинкиназой ДНК-РК^, что активирует тре-
тий белок — XRCC4, — который взаимодействует с ДНК-лигазой IV млекопитающих,
направляя этот восстанавливающий белок к двунитевому разрыву.
Система NHEJ, как думали, ограничена эукариотами, но поиск в базах данных белков
раскрыл бактериальных гомологов белков Ku млекопитающих, а экспериментальные
исследования показали, что они действуют в сотрудничестве с бактериальными лига-
зами в упрощенной версии процесса устранения двунитевых разрывов.
676 Часть 4. Механизмы репликации и эволюции геномов
а) Процесс восстановления негомологическим
соединением концов
Двунитевой разрыв
•~|| Ill'll 1 ШИН"
I Прикрепление
1 белков Ku
б) Структура комплекса Ки-ДНК
I Процесс
восстановления
-ПТТТТГ П 1111111Г'1Г‘
Восстановленная ДНК
Рис. 16.28. Негомологическое соединение концов (NHEJ) у людей, а) Процесс восстановления. В систему
NHEJ входят также дополнительные белки, не показанные на рисунке. К ним относятся протеинкиназы
ATM и ATR (раздел 15.3.2), главная роль которых может заключаться в сигнализировании клетке о том
факте, что произошел двунитевой разрыв и клеточный цикл должен быть приостановлен, пока разрыв
не будет устранен. Если клетка войдет в митоз с разорванной хромосомой, то часть этой хромосомы
будет потеряна, потому что только один из фрагментов будет содержать центромеру, а центромеры
исключительно необходимы для распределения хромосом по дочерним ядрам во время анафазы (см.
рис. 3.15). б) Структура комплекса Ku-ДНК. Слева показан вид сверху на оторванный конец двойной
спирали ДНК, а справа — вид сбоку, где оторванный конец молекулы ДНК расположен слева. Ku — это
гетеродимер, состоящий из субъединиц Ки70 и Ки80, цифры обозначают молекулярную массу в кДа.
Ки70 окрашена красным, а Ки80 — желтым. Молекула ДНК показана серым. Перепечатано с разрешения
Walker et al. (2001) Nature, 412, 607-614
16.2.5. Обход повреждения ДНК во время репликации генома
Если та или иная область генома перенесла обширное повреждение, то весьма
вероятно, что процессы репарации захлебнутся от непосильной работы. Клетка тогда
сталкивается с горьким выбором между смертью или попыткой реплицировать повреж-
денную область даже при том, что эта репликация может быть подвержена ошибкам
и дать начало мутированным дочерним молекулам. Когда клетки Е. coli сталкиваются
с таким выбором, они неизменно выбирают вторую возможность, вызывая одну из не-
скольких чрезвычайных процедур для обхода участков наибольшего повреждения.
Реакция SOS — крайняя мера, позволяющая справиться с повреждением генома
Наиболее полно изученный из таких процессов обхода проявляется как часть ре-
акции SOS, что позволяет клетке Е. coli реплицировать свою ДНК даже при том, что ма-
тричные полинуклеотиды содержат АП участки и (или) циклобутильныедимеры и дру-
гие фотопродукты, возникающие вследствие воздействия химических мутагенов или
Глава 16. Мутации и репарация ДНК 677
УФ-излучения, которые обычно блокировали
бы или по крайней мере задержали бы репли-
кационный комплекс. Обход таких участков
требует построения мутасомы, включающей
в себя комплекс UmuD'2C (также называемый
ДНК-полимеразой V — тример, состоящий из
двух белков UmuD' и одной копии UmuC) и не-
сколько копий белка RecA. Последний — пре-
жде всего белок однонитевого связывания,
который играет много ролей в репарации
и рекомбинации ДНК. В этой системе обхода
белок RecA покрывает поврежденные нити
полинуклеотида, позволяя комплексу UmuD’2C
сместить ДНК-полимеразу III и выполнять под-
верженный ошибкам синтез ДНК, до тех пор
пока поврежденная область не будет пройдена
и ДНК-полимераза III не сможет снова при-
няться за свое дело (рис. 16.29).
Помимо своего действия как белка одно-
нитевого связывания, который облегчает му-
тасоме процесс обхода, RecA выполняет также
и вторую функцию — активатора общей ре-
ДНК-пол имераза III
Сильно поврежденная
матричная ДНК
Вызывание реакции SOS
ДНК-полимераза V
I Подверженный ошибкам
▼ синтез ДНК
Ошибки репликации
IIIIIIIIlLiriH'HiiTTiTiT^
Рис. 16.29. Реакция SOS у Escherichia coli
акции SOS. Этот белок активируется химическими сигналами (еще не опознанными),
которые указывают на присутствие обширного повреждения ДНК. В ходе реакции белок
RecA расщепляет ряд целевых белков, прямо или косвенно, включая UmuD, при этом
расщепление превращает этот белок в его активную форму UmuD' и инициирует про-
цесс восстановления мутасомой. Также белок RecA расщепляет репрессорный белок по
имени LexA, тем самым включая или усиливая экспрессию ряда генов, обычно подавляе-
мых белком LexA, к ним относится сам ген гесА (что ведет к 50-кратному увеличению
синтеза белка RecA) и несколько других генов, продукты которых числятся в системах
репарации ДНК. Белок RecA расщепляет также и репрессор cl бактериофага X, так что
если встроенный профаг X присутствует в геноме, он может вырезать его и бежать
с корабля (раздел 14.3.1).
Реакция SOS прежде всего рассматривается как последняя наилучшая возможность
для бактерии реплицировать свою ДНК и, следовательно, выжить при неблагоприят-
ных условиях. Однако цена выживания — повысившаяся частота появления мутаций,
потому что мутасома не устраняет повреждение, она просто позволяет поврежденной
области полинуклеотида быть реплицированной. Когда она доходит до поврежденной
позиции в матричной ДНК, полимераза подбирает нуклеотид более или менее наугад,
хотя и с некоторым предпочтением поместить А напротив АП участка: вследствие этого
ошибочность процесса репликации повышается. Было высказано предположение о том,
что такая возросшая частота мутаций является целью реакции SOS, так как мутация
представляет собой в некотором отношении выгодную реакцию на повреждение ДНК,
однако это представление остается спорным.
Некоторое время думали, что реакция SOS является единственным процессом
обхода повреждения у бактерий, но теперь мы знаем, что по крайней мере ещё две
полимеразы Е. coli действуют подобным образом, хотя при повреждениях иных типов.
Это ДНК-полимераза II, которая может обойти нуклеотиды, связанные с мутагенными
678 Часть 4. Механизмы репликации и эволюции геномов
химикатами типа N-2-ацетиламинофлюорена, и ДНК-полимераза IV (также называемая
DinB), которая может продолжать репликацию через область матричной ДНК, в которой
два родительских полинуклеотида потеряли выравнивание друг относительно друга.
Обходящие полимеразы были открыты также и в клетках эукариотов. К таковым от-
носятся ДНК-полимеразы 8 и ц, которые способны обойти циклобутильные димеры,
и ДНК-полимеразы i и которые работают совместно и могут в творческом союзе
осуществлять репликацию в областях с фотопродуктами и АП участками.
16.2.6. Дефекты репарации ДНК лежат в основе многих болезней чело-
века, в том числе и появления раковых образований
О важном значении репарации ДНК говорит число и серьезность наследственных
болезней человека, которые были связаны с дефектами в одном из процессов репара-
ции. Одним из наиболее полно охарактеризованных нарушений такого рода является
пигментная ксеродерма, которая проистекает из мутации в каком-либо одном из не-
скольких генов, кодирующих белки, участвующие в восстановлении вырезанием ну-
клеотидов. Вырезание нуклеотидов — единственный путь, которым клетки человека
могут устранять циклобутильные димеры и другие фотопродукты, так что не удиви-
тельно, что в число признаков пигментной ксеродермы входит сверхчувствительность
к УФ-излучению, при этом пациенты переносят больше мутаций, чем нормальные люди
при освещении солнечным светом, и часто страдают от рака кожи. Трихотиодистрофия
также обусловлена дефектами в системе восстановления вырезанием нуклеотидов, но
это более комплексное расстройство, которое, хотя и не сопровождается раком, обычно
включает проблемы и с кожей, и с нервной системой.
Несколько болезней было связано с дефектами в сопряженной с транскрипцией
составляющей системы восстановления вырезанием нуклеотидов. К ним относятся рак
молочной железы и яичника (ген BRCA1, который сообщает восприимчивость к раку этих
видов, кодирует белок, который участвует, по крайней мере косвенно, в сопряженном
с транскрипцией репарации) и синдром Кокейна — комплексное заболевание, про-
являющееся в нарушениях роста и неврологических расстройствах. Недостаточность
сопряженной с транскрипцией репарации была установлена также у людей, страдающих
синдромом восприимчивости к раку, названным ННРПОК (наследственный неполипоз-
ный рак прямой и ободочной кишки), хотя эта болезнь была первоначально определена
как дефект в системе устранения несоответствий. Атаксия-телеангиэктазия, к признакам
которой относится чувствительность к ионизирующему излучению, возникает вслед-
ствие дефектов в гене АТХ, который вовлечен в процесс обнаружения повреждений.
Другие болезни, которые связаны с нарушениями процессов репарации ДНК, — син-
дромы Блума и Вернера, вызываемые инактивацией ДНК-геликазы, которая, возможно,
играет роль в NHEJ, анемия Фанкони, которая придает чувствительность к химикатам,
вызывающим поперечные сшивки в ДНК (но биохимическая основа их действия еще не
известна), и атаксия спинного мозга и мозжечка, которая следует из дефектов в системе,
используемой клетками для устранения однонитевых надрывов.
Заключение
Всякая мутация представляет собой то или иное изменение в нуклеотидной по-
следовательности молекулы ДНК, будь это точечная мутация, которая затрагивает
только один-единственный нуклеотид, или же вставка либо удаление одного или не-
Глава 16. Мутации и репарация ДНК 679
скольких смежных нуклеотидов. Мутации могут следовать из ошибок, произошедших
в ходе репликации ДНК, даже при том, что ДНК-полимеразы имеют в своем арсенале
процедуры подбора нуклеотидов и корректирующие функции, которые поддерживают
высокую степень точности. Эти механизмы проверки, однако, удается обойти, если в ма-
трице присутствует необычная таутомерная версия нуклеотида. Ошибка репликации
второго типа, называемая проскальзыванием, может привести к появлению мутаций
типа удаления или вставки. Существуют также химические или физические агенты
многих типов, которые могут вызвать мутации. Некоторые соединения выступают
в роли аналогов оснований и вызывают мутации, будучи принятыми репликационным
аппаратом за подлинные нуклеотиды. Дезаминирующие и алкилирующие агенты на-
прямую поражают молекулы ДНК, а вставочные агенты, например бромистого этидия
интеркалируют (втискиваются) между парами оснований, порождая вставки и удаления
во время репликации такой спирали. УФ-излучение побуждает смежные нуклеотиды
связываться вместе в димеры, а ионизирующее излучение и высокая температура
вызывают различные типы повреждений. Точечная мутация в пределах одного гена
может не иметь никаких последствий на кодирующие свойства в силу вырожденности
генетического кода, но некоторые мутации способны изменить смысл кодона, так что
кодон будет определять иную аминокислоту или, возможно, превратится в стоп-кодон.
Вставки и удаления могут вызывать сдвиги рамки считывания, которые ведут к пре-
ждевременному завершению трансляции или к сквозному считыванию правильного
стоп-кодона. Любая из таких мутаций может привести к мутации с потерей функции,
а некоторые могут вызвать мутацию с приобретением функции и, возможно, привести
к раку. У бактерий мутация может дать начало ауксотрофу — клетке, которая требует
подкормки для роста, не нужной дикому типу, или же породить мутанта, который вы-
казывает устойчивость к антибиотику или какому-то другому ингибитору. Утверждение
о том, что клетки при некоторых обстоятельствах могут увеличивать обычную для них
частоту мутаций или программировать мутации в ответ на изменения условий окру-
жающей среды, вызывает жаркие споры. Все клетки обладают процессами репарации
ДНК, которые позволяют исправлять многие мутации. Системы прямого восстановления
не получили широкого распространения, но немногие, которые известны, правят не-
которые типы повреждения оснований, включая удаление вызванных УФ-излучением
нуклеотидных димеров. Процессы восстановления вырезанием заключаются в выреза-
нии содержащего поврежденный участок сегмента полинуклеотида, сопровождаемом
ресинтезом правильной последовательности нуклеотидов ДНК-полимеразой. Система
устранения несоответствий исправляет ошибки репликации, опять же вырезая отрезок
однонитевой ДНК, содержащий мутацию, и ликвидируя получающийся пропуск. Него-
мологическое соединение концов применяется для исправления двунитевых разрывов.
Известны также процессы, позволяющие совершить обход участков повреждения ДНК
в ходе репликации, многие из которых служат аварийными системами спасения генома,
который оказался сильно мутированным.
680 Часть 4. Механизмы репликации и эволюции геномов
Задания для закрепления материала
♦Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
16.1 *. Какое из следующих утверждений
является НЕВЕРНЫМ?
а. Мутация — изменение в нуклео-
тидной последовательности ко-
роткой области генома.
Ь. Все мутации вызываются агента-
ми окружающей среды.
с. Многие мутации могут быть
устранены.
d. Некоторые мутации обусловлены
ошибками репликации.
16.2 . Которая из следующих мутаций при-
водит к замене одного нуклеотида
другим?
а. Мутация удаления.
Ь. Мутация вставки.
с. Точечная мутация.
d. Транслокация.
16.3 *. Самопроизвольные мутации явля-
ются результатом действия которо-
го (которой) из следующих агентов
(причин)?
а. Химические мутагены.
Ь. Ошибки репликации ДНК.
с. Высокая температура.
d. Радиация.
16.4 . Каким образом корректирование
повышает точность репликации
генома?
а. ДНК-полимераза отбраковывает
неправильные нуклеотиды, когда
они только связываются с этим
ферментом.
Ь. За счет действия экзонуклеазы
5'->3' ДНК-полимераза удаляет
нуклеотид, который был ошибоч-
но добавлен к концу синтезируе-
мого полинуклеотида.
с. Если посаженный на З’-конец ну-
клеотид не спарился основанием
с матричной ДНК, то он удаляется
за счет экзонуклеазного действия
ДНК-полимеразы.
d. Благодаря всему вышеперечис-
ленному.
16.5 *. Какая из следующих причин обычно
обусловливает появление ошибок
в ходе репликации генома?
а. Образование пар оснований G-U
в репликационной вилке.
Ь. Репликация областей генома, ко-
торые в этот момент транскри-
бируются.
с. Таутомерный сдвиг в нуклеотиде
матричной ДНК.
d. Присутствие нуклеосом, при-
соединенных к реплицируемой
ДНК.
16.6 . Изменчивость в какого типа повто-
рах обычно возникает за счет про-
скальзывания репликации?
а. В микросателлитах.
Ь. В минисателлитах.
с. В ретропозонах.
d. В транспозонах ДНК.
16.7 *. Какого типа химические мутагены
встраиваются в геном ДНК-полиме-
разой в процессе репликации гено-
ма?
а. Алкилирующие агенты.
Ь. Аналоги оснований.
с. Дезаминирующие агенты.
d. Интеркалирующие агенты.
16.8 . Какого типа повреждение ДНК вы-
зывается ультрафиолетовым излу-
чением?
а. Циклобутильные димеры.
Ь. АП (апуриновые/апиримидино-
вые) участки.
с. Дезаминирование оснований.
d. Таутомеризация оснований.
Глава 16. Мутации и репарация ДНК 681
16.9 *. Какого типа мутация превращает
кодон, определяющий некоторую
аминокислоту, в стоп-кодон?
а. С потерей смысла.
Ь. Несинонимичная.
с. Сквозного считывания.
d. Синонимичная.
16.10 . Что за штука — ауксотрофный му-
тант?
а. Мутант, который может расти на
минимальной среде.
Ь. Мутант, которому для роста не-
обходим антибиотик.
с. Мутант, которому нужны питатель-
ные вещества, в которых не нужда-
ются организмы дикого типа.
d. Мутант, который может расти при
ограничивающих температурах.
16.11 *. Которые из следующих типов по-
вреждения ДНК НЕ МОГУТ быть
устранены Е coli с помощью системы
прямого восстановления?
а. Алкилированные основания.
Ь. АП участки.
с. Циклобутильные димеры.
d. Отсутствующие фосфодиэфир-
ные связи.
16.12 . Какая из следующих процедур от-
носится к процессу восстановления
вырезанием нуклеотидов?
а. Область двунитевой ДНК, содер-
жащая поврежденные нуклеоти-
ды, удаляется и заменяется новой
ДНК.
Ь. Единственный поврежденный
нуклеотид удаляется и заменя-
ется новым нуклеотидом.
с. Отдельное поврежденное осно-
вание удаляется и заменяется
новым основанием.
d. Область однонитевой ДНК, содер-
жащая поврежденные нуклеоти-
ды, удаляется и заменяется новой
ДНК.
16.13 *. Которая из нижеследующих осо-
бенностей характерна для системы
устранения несоответствий?
а. Опознаются видоизмененные ну-
клеотиды.
Ь. Удаляются циклобутильные ди-
меры.
с. Различение родительской и до-
черней нитей новореплициро-
ванной ДНК.
d. Распознавание правильной рамки
считывания.
16.14 . За счет чего родительская и дочер-
няя нити недавно реплицированной
ДНК различаются у Е coli?
а. Дочерние нити метилируются
незамедлительно после их син-
теза.
Ь. Дочерние нити метилируются не
сразу по окончании их синтеза.
с. Дочерние нити не сразу прикре-
пляются к гистоновым белкам.
d. Дочерние нити содержат рибо-
нуклеотиды от РНК-затравок,
употребленных для инициации
синтеза ДНК.
16.15 *. Какими средствами клетка Е coli
пытается реплицировать повреж-
денную ДНК во время реакции
SOS?
а. Области поврежденной ДНК уда-
ляются из генома.
Ь. В поврежденныхучастках нуклео-
тиды встраиваются наугад.
с. Синтез ДНК полностью останав-
ливается, пока повреждение не
будет устранено.
d. Матричная РНК преобразуется
в ДНК, которая вставляется в по-
врежденныеучастки посредством
рекомбинации.
682 Часть 4. Механизмы репликации и эволюции геномов
Вопросы открытого типа
16.1 *. Каковы механизмы возникновения
мутаций в геноме?
16.2 . Каковы различия между предрепли-
кационным и пострепликационным
процессами репарации ДНК?
16.3 *. Каким образом ДНК-полимераза
подбирает правильный нуклеотид
во время синтеза ДНК?
16.4 . За счет чего аналог основания
2-аминопурин производит мутации
в ДНК?
16.5 *. Как именно высокая температура
нарушает структуру ДНК? Насколь-
ко распространено вызванное вы-
сокой температурой повреждение
ДНК и каковы эффекты такого по-
вреждения?
16.6 . Как могут мутации в некодирующих
последовательностях ДНК пагубно
повлиять на экспрессию генома?
16.7 *. У людей семейная гиперхолестери-
немия происходит от мутации с по-
терей функции в гене, кодирующем
Изыскательские задачи
16.1 *. Объясните, по какой причине точеч-
ную мутацию типа замены пурина
на пурин или пиримидина на пири-
мидин называют транзицией (пере-
ходом), тогда как замену типа пурин
на пиримидин (или наоборот) назы-
вают трансверсией (перебегом).
16.2 . Каково ожидаемое отношение числа
транзиций к числу трансверсий при
большом количестве мутаций?
16.3* . Изучите современные знания о бо-
лезнях, связанных с расширением
тринуклеотидных повторений, в том
связывающийся с рецептором ЛНП
домен аполипопротеина В-100, и на-
следуется по доминантному призна-
ку. Почему это так?
16.8. Как условно летальные мутации
могут быть использованы для опо-
знавания или характеризации про-
дуктов основных генов в микроор-
ганизме?
16.9* . Какова основа сверхмутации сег-
ментов V генов иммуноглобулина
человека?
16.10. Из каких этапов состоит процесс
восстановления вырезанием осно-
ваний?
16.11* . Что собой представляет процесс,
посредством которого двунитевые
разрывы в ДНК устраняются систе-
мой негомологического соединения
концов (NHEJ)?
16.12. Какова роль белка LexA в реакции
SOS у Е. coli?
числе гипотезы, которые призваны
объяснить, почему расширение три-
плетов в этих генах приводит к раз-
витию болезни.
16.4. Дайте оценку имеющимся данным
о запрограммированных мутациях.
16.5*. Бактерия Deinococcus radiodurans
является весьма устойчивой к ра-
диации, а также к другим физи-
ческим и химическим мутагенам.
Обсудите, как эти особые свойства
D. radiodurans отражены в последо-
вательности ее генома.
Глава 16. Мутации и репарация ДНК 683
Тесты по рисункам
16.1 *. Какого типа мутация (взгляните на окрашенные красным нуклеотиды) изобра-
жена на этом рисунке? Какова причина возникновения таких мутаций?
...САСАСАСАСА...
...САСАСАСАСА...
...GTGTGTGTGT...
...САСАСАСАСА...
...GTGTGTGTGT...
...САСАСАСАСА...
...GTGTGTGTGT...
РОДИТЕЛЬСКАЯ
МОЛЕКУЛА
I
СА
...САСАСАСАСА...
САСАСАСАСАСА
GTGTGTGTGTGT
...GTGTGTGTGT...
ДОЧЕРНИЕ
МОЛЕКУЛЫ
...САСАСАСАСА...
...GTGTGTGTGT...
МОЛЕКУЛЫ
ВТОРОГО ПОКОЛЕНИЯ
16.2 . Обсудите потенциальное воздействие различных типов мутаций, изображенных
на рисунке.
Несинонимичная
АТА
Иле
I С потерей смысла Сквозного считывания
Синонимичная \ _А
.ATGGGCAAATATAGCATTCCATAAAAATATATA...
।___11_11__11_к__к__II__II_।
Мет Гли Лиз Тир Сер Иле Про стоп
684 Часть 4. Механизмы репликации и эволюции геномов
16.3 *. Каково название фермента, который удаляет поврежденное основание из мо-
лекулы ДНК? Какой способ употребляется для репарации ДНК после удаления
основания?
16.4 . Какая схема репликации ДНК изображена на этом рисунке? В каких ситуациях
она инициируется клеткой?
ДНК-полимераза III
Linuiiimi
Сильно повреждённая
матричная ДНК
ДНК-полимераза V
llll'llllllllll1
Белки RecA
I Подверженный ошибкам
▼ синтез ДНК
Ошибки репликации
1111 (Lu IIII ll h i .ТТ.Т^Г*
Глава 16. Мутации и репарация ДНК 685
Дополнительная литература
Причины мутаций
Kunkel,Т. A. and Bebenek,K. (2000) DNA replication fidelity. Annu. Rev. Biochem. 69:
497-529. Covers the processes that ensure that the minimum number of errors are made during
DNA replication.
Болезни, вызванные расширением тринуклеотидных повторений
Ashley, С. Т. and Warren, S. Т. (1995) Trinucleotide repeat expansion and human disease.
Annu. Rev. Genet 29: 703-728.
Perutz, M.F. (1999) Glutamine repeats and neurodegenerative diseases: molecular
aspects. Trends Biochem. Sci. 24: 58-63.
Sutherland, G. R., Baker, E. and Richards, R. I. (1998) Fragile sites still breaking. Trends
Genet 14: 501-506.
Сверхмутация и запрограммированные мутации
Andersson, D. I., Slechta,E.S. and RothJ.R. (1998) Evidence that gene amplification
underlies adaptive mutability of the bacterial lac operon. Science 282:1133-1135.
Cairns,)., Overbaugh, J. and Miller, S. (1988) The origin of mutants. Nature 335:142-145.
The original experiments suggesting that bacteria can program mutations.
Chicurel,M. (2001) Can organisms speed their own mutation? Science 292: 1824-
1827.
NolaJ.D. and Neuberger,M.S. (2002) Altering the pathway of immunoglobulin
hypermutation by inhibiting uracil-DNA glycosylase. Nature 419: 43-48.
Восстановление вырезанием
Lehmann, A. R. (1995) Nucleotide excision repair and the link with transcription. Trends
Biochem. Sci. 20: 402-405.
Seeberg,E., Eide,L. and Bj0ras,M. (1995) The base excision repair pathway. Trends
Biochem. Sci. 20: 391-397.
Устранение несовпадений
Kolodner, R. D. (1995) Mismatch repair: mechanisms and relationship to cancer
susceptibility. Trends Biochem. Sci. 20: 397-401.
Kunkel,T.A. and Erie,D.A. (2005) DNA mismatch repair. Annu. Rev. Biochem. 74:681-710.
Shannon, M. and Weigert,M. (1998) Fixing mismatches. Science 279:1159-1160.
Устранение разрывов ДНК
Critchlow,S.E. and Jackson, S.P. (1998) DNA end-joining: from yeast to man. Trends
Biochem. Sci. 23: 394-398.
Walker,J.R., Corpina,R.A. and Goldberg,). (2001) Structure of the Ku heterodimer
bound to DNA and its implications for double-strand break repair. Nature 412: 607-614.
Wilson,T.E., Topper, L.M. and Palmbos, P. L. (2003) Non homologous end-joining:
bacteria join the chromosome breakdance. Trends Biochem. Sci. 28: 62-66. Evidence for NHEJ
in bacteria.
Обход повреждений ДНК
Hanaoka,F. (2001) SOS polymerases. Nature 409: 33-34.
686 Часть 4. Механизмы репликации и эволюции геномов
fohnson, R. Е., Prakash, S. and Prakash, L. (1999) Efficient bypass of a thymine-thymine
dimer by yeast DNA polymerase, Pols. Science 283:1001-1004.
fohnson, R. E., Washington, M. T., Haracska, L, Prakash, S. and Prakash, L (2000) Eukaryotic
polymerases i and £ act sequentially to bypass DNA lesions. Nature 406:1015-1019.
Sutton, M.D., Smith, B.T., Godoy, V.G. and Walker, G.C. (2000) The SOS response:
recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev.
Genet 34: 479-497.
Восстановление и болезни
Gowen, L. C., Avrutskaya,A.V., Latour, A. M., Roller, B.H. and Leadon,S.A. (1998)
BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science 281:
1009-1012.
Hanawalt, P. C. (2000) The bases for Cockayne syndrome. Nature 405: 415-416.
Глава 17
Рекомбинация
По прочтении главы 17 вы сможете:
• Описывать различные модели гомологичной рекомбинации и указывать раз-
личия между ними.
• Подробно описывать путь RecBCD гомологичной рекомбинации у Escherichia
coli.
• Описывать отличительные особенности путей RecE и RecF у Е. coli, а также
перечислять названия и функции белков эукариотов, которые подозреваются
в причастности к гомологичной рекомбинации.
• Рассказывать о том, как гомологичная рекомбинация используется для устра-
нения разрывов ДНК.
• Зачитывать подробный сценарий инфицирования бактериофагом X с описани-
ем роли сайт-специфической рекомбинации и объяснять, почему такая сайт-
специфическая рекомбинация важна в конструировании генетически модифи-
цированных зерновых культур.
• Иллюстрировать модели Шапиро для консервативной и репликативной транс-
позиции ДНК-транспозона.
• Описывать путь транспозиции для ретроэлемента с LTR.
• Кратко обсуждать возможные пути, которыми клетки минимизируют вредные
последствия транспозиции.
Рекомбинация — термин, первоначально употреблявшийся генетиками для опи-
сания исхода кроссинговера между парами гомологичных хромосом в ходе мейоза.
В результате кроссинговера получаются дочерние хромосомы, которые имеют отли-
чающиеся сочетания аллелей от таковых в родительских хромосомах (раздел 3.2.3).
В 1960-е гг. были предложены модели для описания молекулярных событий, которые
лежат в основе кроссинговера, и стало понятно, что определяющей операцией процесса
молекулярной рекомбинации является разрыв и последующее воссоединение молекул
ДНК. В настоящее время биологи понимают под «рекомбинацией» разнообразные про-
цессы, которые включают в себя разрыв и воссоединение полинуклеотидов, как то:
• Гомологичная рекомбинация, также называемая общей рекомбинацией, ко-
торая происходит между сегментами молекул ДНК, обладающих обширной гомо-
логичностью последовательностей. Такие сегменты могут находиться на разных
хромосомах или могут быть двумя частями одной хромосомы (рис. 17.1, а). Гомо-
688 Часть 4. Механизмы репликации и эволюции геномов
а) Гомологичная рекомбинация
Между разными молекулами В пределах одной молекулы
+Гомологичные хромосомы + J
* *
—-------------------- -—)
б) Сайт-специфическая рекомбинация
Короткая область гомологии
________L________
+
о
в) Транспозиция
Транспозон
Репликативная
Консервативная
Рис. 17.1. События рекомбинации трех различных типов
логичная рекомбинация лежит в основе кроссинговера, происходящего во время
мейоза, и первоначально изучалась именно в данном контексте, но сейчас полагают,
что ее первостепенная роль в клетке заключается в репарации ДНК.
• Сайт-специфическая рекомбинация, которая происходит между молекулами
ДНК, имеющими только короткие области подобия последовательностей — воз-
можно, лишь несколько п.н. (рис. 17.1, б). Сайт-специфическая рекомбинация от-
ветственна за встраивание геномов фагов, например генома пресловутого фага X,
в хромосомы бактерий.
• Транспозиция, которая состоит в перемещении некоторого сегмента ДНК из одной
позиции генома в другую (рис. 17.1, в).
Прочие разнообразные события, которые мы изучили, в том числе переключение
типа спаривания у дрожжей (см. рис. 14.16) и созревание генов иммуноглобулинов (см.
рис. 14.19), также являются результатами рекомбинации.
Не будь рекомбинации, геномы были бы относительно статическими структура-
ми, претерпевающими весьма незначительные изменения. Постепенное накопление
мутаций в течение длительного периода времени вылилось бы в мелкомасштабные
видоизменения нуклеотидной последовательности генома, но более значительная
перестройка структуры, в которой и состоит роль рекомбинации, не происходила бы,
и эволюционный потенциал генома был бы жестоко ограничен.
Глава 17. Рекомбинация 689
17.1. Гомологичная рекомбинация
При изучении гомологичной рекомбинации молекулярные биологи столкнулись
с двумя серьезными проблемами, ни одна из которых не была до сих пор полностью
решена. Первая проблема заключается в необходимости описать ряд взаимодействий,
в том числе разрыв и воссоединение полинуклеотидов, которые происходят во время
рекомбинации. Модели гомологичной рекомбинации, которые были построены в ходе
этой работы, описаны в разделе 17.1.1. Вторая проблема связана с тем фактом, что
рекомбинация является клеточным процессом, который, подобно другим клеточным
процессам с участием ДНК (например, транскрипция и репликация), осуществляется
и регулируется ферментами и другими белками. Биохимические исследования обна-
ружили ряд связанных путей рекомбинации, которые в общих чертах описаны в разде-
ле 17.1.2. Дальнейшее изучение этих путей привело к осознанию того, что гомологичная
рекомбинация лежит в основе нескольких важных типов репарации ДНК, причем эта
репарационная функция, вероятно, является более важной для клетки (особенно для
бактериальных клеток), чем возможность, которую гомологичная рекомбинация предо-
ставляет кроссинговеру между хромосомами. Мы исследуем такие процессы репарации
в разделе 17.1.3.
17.1.1. Модели гомологичной рекомбинации
Многие крупные достижения в понимании гомологичной рекомбинации были сде-
ланы Робином Холл идеем, Мэтью Мезельсоном, а также их коллегами в 1960-х и 1970-х гг.
В ходе этой работы был построен ряд моделей, которые показали, как разрыв и воссоеди-
нение молекул ДНК могут обусловливать механическую сторону обмена сегментами
хромосом, который, как известно, происходит во время кроссинговера. Поэтому мы
и начнем изучение гомологичной рекомбинации с рассмотрения этих моделей.
Модели гомологичной рекомбинации Холлидея и Мезельсона-Реддинга
Эти модели описывают рекомбинацию между двумя гомологичными двунитевыми
молекулами с идентичными или почти идентичными последовательностями. Главная
особенность этих моделей — формирование гетеродуплекса, следующего из обмена
сегментами полинуклеотидов между двумя гомологичными молекулами (рис. 17.2).
Гетеродуплекс первоначально стабилизируется за счет спаривания оснований между
каждой переданной нитью и исходными полинуклеотидами принимающих молекул;
такое спаривание оснований является возможным ввиду подобия последовательностей
между этими двумя молекулами. За сим ДНК-лигаза запечатывает промежутки и об-
разуется окончательная структура Холлидея. Эта структура является динамической,
и перемещение точки ветвления обусловливает обмен более длинными сегментами
ДНК, если обе спирали закручиваются в одном и том же направлении.
Разъединение, или разрешение, структуры Холлидея обратно на отдельные дву-
нитевые молекулы происходит путем расщепления в точке ветвления. Это ключевой
момент всего процесса, потому что разрез может быть сделан в любой из двух ориента-
ций, что становится очевидным, когда рассматривается трехмерная конфигурация, или
%-форма, структуры Холлидея (см. рис. 17.2). Эти два разреза дают совершенно разный
результат. Если разрез через %-форму сделан слева направо, как показано на рис. 17.2,
то между этими двумя молекулами будет обменян короткий сегмент полинуклеотида,
соответствующий расстоянию, пройденному точкой ветвления структуры Холлидея.
Разрез же сверху вниз приводит к обоюдному обмену нитями — двунитевая ДНК пере-
690 Часть 4. Механизмы репликации и эволюции геномов
Две гомологичные
молекулы ДНК
Обмен полинуклеотидами
создает гетеродуплекс
ДНК-лигаза
Структура Холлидея
а b
। Перемещение точки
▼ ветвления
НИ 11 • I II I М11111
«111111111111111111
• ‘ . I м о > . ♦ ।
а Ь а В
Рис. 17.2. Построенная Холлидеем модель гомологичной рекомбинации
мещается между двумя молекулами, так что конец одной молекулы обменивается на
конец другой молекулы. Это перемещение ДНК, наблюдаемое при кроссинговере.
До сих пор мы игнорировали один вопрос, не освещенный в этой модели. Это спо-
соб взаимодействия обеих двунитевых молекул, которое происходит в начале процесса
и приводит к образованию гетеродуплекса. В первоначальной схеме Холлидея обе мо-
лекулы выстраиваются друг против друга и в эквивалентных позициях обеих спиралей
появляются однонитевые разрывы. В результате этого появляются свободные, однони-
тевые концы, которые могут служить объектами обмена, и в конечном итоге образуется
гетеродуплекс (рис. 17.3, а). Эта особенность модели критиковалась, потому что не уда-
валось предложить ни одного механизма, который мог бы гарантировать, что разрывы
появятся точно в тождественных позициях обеих молекул. Усовершенствованная модель
Мезельсона-Реддинга предлагает более удовлетворительную схему, согласно которой
однонитевой надрыв происходит только в одной из двойных спиралей, полученный
в результате этого свободный конец «вторгается» в ненарушенную двойную спираль
в гомологичной позиции и вытесняет одну из ее нитей, формируя D-петлю (рис. 17.3, б).
Последующее расщепление вытесненной нити в месте перехода между ее однонитевой
и спаренной основаниями областями производит гетеродуплекс.
Глава 17. Рекомбинация 691
а) Первоначальная модель
6) Вариант Мезельсона-Реддинга
иадрывы 1111111111 ГУН IГТТТГ
в эквивалентных____*
Надрыв
только в одной
молекуле
HHHiHimiiiHi
Обмен нитями
Вторжение нити
111рН11|111111111
!ц1МтЙнЯЯ1
D-петля
; Формирование
гетеродуплекса
1»Ц|ИП|»И1|1И1
1П>П>Н|Ц1Ш!Г
Рис. 17.3. Две схемы инициации гомологичной рекомбинации, а) Инициация согласно первоначальной
модели гомологичной рекомбинации, б) Вариант Мезельсона-Реддинга, который предлагает более
правдоподобный ряд событий формирования гетеродуплекса
Модель гомологичной рекомбинации, предполагающая разрыв двойной цепи
Хотя модель гомологичной рекомбинации Холлидея, будь она в своей первоначаль-
ной форме или в усовершенствованном Мезельсоном и Реддингом виде, и объяснила,
каким образом может происходить кроссинговер во время мейоза, она имеет некото-
рые несоответствия, которые обусловили развитие альтернативных схем. В частности,
думалось, что модель Холлидея не способна объяснить генную конверсию — явление,
сначала описанное у дрожжей и грибов, но, как теперь известно, происходящее и у многих
эукариотов. У дрожжей слияние пары гамет дает зиготу, которая дает начало плодовой
сумке, содержащей четыре гаплоидные споры, генотипы которых могут быть индивиду-
ально определены (см. рис. 14.15). Если гаметы имеют различные аллели в определенном
локусе, то при нормальных обстоятельствах две споры покажут один генотип, а другие
две покажут другой генотип, но иногда такая ожидаемая закономерность распределения
признаков 2:2 изменяется на неожиданное соотношение 3:1 (рис. 17.4). Это явление
называют генной конверсией, потому что такое отношение можно объяснить лишь
«конверсией» одного из аллелей из одного типа в другой, предположительно путем
рекомбинации во время мейоза, который происходит после слияния гамет.
Модель с двунитевым разрывом (ДНР) объясняет возможность протекания со-
бытия генной конверсией в ходе рекомбинации. Согласно этой модели гомологичная
рекомбинация начинается не с однонитевого разрыва, как в схеме Мезельсона-Реддинга,
а с двунитевого разреза, который разрывает одного из партнеров рекомбинации на две
части (рис. 17.5). После двунитевого разреза одна нить в каждой половине молекулы
укорачивается, так что каждый конец теперь имеет 3'-выступ. Один из таких высту-
пов вторгается в гомологичную молекулу ДНК способом, подобным предполагаемому
в схеме Мезельсона-Реддинга, и тем самым формирует структуру Холлидея, которая
может перемещаться по гетеродуплексу, если вторгшаяся нить продолжается ДНК-
полимеразой. В завершение формирования гетеродуплекса другая разорванная нить
(та, что не входит в структуру Холл идея) тоже удлиняется. Следует иметь в виду, что обе
реакции синтеза ДНК предполагают продолжение нитей того партнера, который пере-
692 Часть 4. Механизмы репликации и эволюции геномов
Рис. 17.4. Генная конверсия. Одна гамета содержит
аллель А, а другая — аллель а. Они слияются и произ-
водят зиготу, которая дает начало четырем гаплоидным
спорам, все они содержатся в одном аске. В норме две
из этих спор будут иметь аллель А, а две будут иметь
аллель а, но если происходит генная конверсия, то
отношение изменяется, возможно, на ЗА:1а, как по-
казано здесь
нес двунитевой разрез, используя в качестве
матриц тождественные области нерассечен-
ного партнера. Это основа генной конверсии,
потому что это означает, что сегменты пол ину-
клеотида, изъятые из разрезанного партнера,
были замещены копиями ДНК из нетронутого
партнера. После лигирования получившийся
гетеродуплекс имеет пару структур Холлидея,
которые могут быть распущены различны-
ми путями, одни из которых ведут к генной
конверсии, а другие приводят к стандартному
обоюдному обмену нитями. Пример пути, за-
вершающегося генной конверсией, показан
на рис. 17.5.
Хотя первоначально модель с двуните-
вым разрывом была предложена как механизм
для объяснения явления генной конверсии
у дрожжей, теперь модель с ДНР рассматрива-
ГАМЕТЫ
ЗИГОТА
Без генной конверсии С генной конверсией
2А:2а
ЗАЛа
ется как по крайней мере близкое приближение к механизму протекания гомологичной
рекомбинации во всех организмах. Принятие этой модели было обусловлено двумя
причинами. Во-первых, в 1989 г. было открыто, что во время мейоза хромосомы подвер-
гаются двунитевым разрывам в 100-1 000 раз чаще, чем это имеет место в вегетативных
клетках. Вывод о том, что образование двунитевых разрывов есть неотъемлемая часть
мейоза, явно свидетельствует в пользу модели с ДНР по отношению к схемам, согласно
которым рекомбинация начинается с одного или нескольких однонитевых надрывов.
Вторым фактором, приведшим к принятию модели с ДНР, послужило осознание того
факта, что гомологичная рекомбинация вовлечена в репарацию ДНК, а именно от-
ветственна за устранение двунитевых разрывов, которые возникают как нарушения
в процессе репликации. Модели Холлидея и Мезельсона-Реддинга не объясняют этот
момент гомологичной рекомбинации, тогда как устранение двунитевых разрывов есть
безусловный атрибут модели с ДНР. Мы возвратимся кроли гомологичной рекомбинации
в процессе репарации ДНК в разделе 17.1.3.
17.1.2. Биохимия гомологичной рекомбинации
Гомологичная рекомбинация происходит во всех организмах, но, как и многие
другие процессы молекулярной биологии, первые подвижки в понимании механизма
протекания данного процесса в клетке произошли благодаря опытам с Е. coli. Иссле-
дования с применением искусственных мутаций позволили опознать ряд генов Е. coli,
которые, будучи инактивированы, вызывают дефекты в гомологичной рекомбинации,
Глава 17. Рекомбинация 693
Рис. 17.5. Модель гомологичной рекомбинации с двуни-
тевым разрывом. Эта модель объясняет возможный ме-
ханизм генной конверсии
что указывает на то, что их белковые продукты
участвуют в данном процессе тем или иным об-
разом. Были описаны три различные системы
рекомбинации, как то: пути RecBCD, RecE и RecF,
причем путь RecBCD со всей очевидностью явля-
ется наиболее важным у бактерий.
Путь RecBCD у Escherichia coli
В пути RecBCD рекомбинация опосредству-
ется ферментом RecBCD, который, как подразу-
мевает его название, состоит из трех различных
белков. Два из них — RecB и RecD — суть гели-
казы. Для инициации гомологичной рекомбина-
ции одна копия фермента RecBCD прикрепляется
к свободным концам хромосомы в месте двуните-
вого разрыва. ДНК раскручивается с обоих концов
за счет действия белка RecB, который продвигает-
ся по одной нити в направлении 3'—>5', и действия
белка RecD, движущегося в направлении 5'->3’ по
другой нити. Белок RecB, помимо функции гели-
казы, обладает также активностью экзонуклеазы
3’->5’ и, таким образом, постепенно деградирует
нить, по которой он продвигается вперед, — нить
со свободным З’-концом (рис. 17.6).
Фермент RecBCD продвигается по молеку-
ле ДНК со скоростью приблизительно 1 тыс.п.н.
в секунду, до тех пор пока не достигнет первой
копии восьминуклеотидной консенсусной по-
следовательности 5'-GCTGGTGG-3’, названной
Двунитевой разрез
А\
• Экзонуклеаза подрезает
▼ обрезанные нити
iiiiiiiiiiiiiiiiiiin
Вторжение нити
iiMiiinpiimiM
Продолжение нитей
illl|IWIIlllllJlllli
Лигаза соединяет зазоры
|lllfllllllllll|llll
BE ЗЯв я.
lllHiiiilllIllilllll
- Гетеродуплекс
• с двумя переходами
Холлидея
I Расщепление
▼ переходов Холлидея
А
iiiiiiiiiiiiiiiiiiii
а
А
IIIIIIIIIIIIIIIIIIII
А
• Устранение
▼ несоответствия
ПТТТТГ....IIIIII
А
IIIIIIIIIIIIIIIIIIII^
А
%-участком, который встречается в среднем один раз на каждые 6 тыс. п. н. в ДНК Е. coli.
В ^-участке конформация фермента RecBCD изменяется таким образом, что геликаза
RecD отцепляется от него и продвижение комплекса RecBCD замедляется примерно до
половины его первоначальной скорости. В точности, что происходит вслед за этим, не
ясно, но, согласно самой последней модели, изменение конформации фермента также
снижает или полностью отменяет присущее белку RecB действие экзонуклеазы 3'->5’ —
этот белок теперь делает одинарный эндонуклеолитический разрез в другой нити
молекулы ДНК, в близлежащей к х-участку позиции (см. рис. 17.6). Каким бы ни был
точный механизм, в результате всего этого фермент RecBCD производит двунитевую
молекулу с З'-выступом — в точности, как это предполагает модель с ДНР (см. второй
вид на рис. 17.5).
Следующий шаг — устройство гетеродуплекса. Эта стадия опосредствуется белком
RecA, который формирует покрытую белком нить ДНК, способную вторгаться в непо-
врежденную двойную спираль и образовывать D-петлю (рис. 17.7). Промежуточным
694 Часть 4. Механизмы репликации и эволюции геномов
Рис. 17.6. Способ гомологичной рекомбинации с
участием комплекса RecBCD у Е. coli. Эти события
ответственны за первый шаг в модели гомоло-
гичной рекомбинации с двунитевым разрывом,
показанной на рис. 17.5
звеном в образовании D-петли служит
триплексная структура (см. третий вид
на рис. 17.5) — трехнитевая спираль ДНК,
в которой вторгшийся полинуклеотид ле-
жит в большой бороздке исходной спира-
ли и образует водородные связи с парами
оснований, к которым он примыкает.
Перемещение точки ветвления ката-
лизируется белками RuvA и RuvB, оба из ко-
торых прикрепляются к точке ветвления
гетеродуплекса, сформированного втор-
жением З'-выступа в молекулу-партнер.
Продвижение RecBCD по молекуле ДНК
Х-участок RecBCD
Деградация
в направлении 3'—►б'
Kill
I Переключение активности
в х-участке
Субъединичная
структура RecBCD
Рентгеноструктурный анализ показывает, что четыре копии белка RuvA присоединя-
ются непосредственно к месту ветвления, формируя ядро, к которому прикрепляются
два кольца белка RuvB (каждое из которых состоит из восьми белков) — по одному
с обеих сторон (рис. 17.8). Полученная структура может работать как «молекулярный
двигатель», вращающий спирали в требуемой манере, так чтобы точка ветвления пере-
двигалась. Перемещение точки ветвления, как кажется, не случайный процесс, ибо она
останавливается предпочтительно на последовательности 5’-(A/T)TT(G/C)-3’ (где обо-
значение (А/Т) и т.д. означает, что в указанной позиции может присутствовать любой
из двух нуклеотидов). Такая последовательность часто встречается в геноме Е coli, так
что предположительно перемещение не всегда останавливается в первом достигнутом
экземпляре этого мотива. Когда перемещение точки ветвления завершилось, комплекс
RuvAB отделяется и замещается двумя белками RuvC (см. рис. 17.8), которые выполняют
расщепление, в результате чего структура Холлидея разрешается. Разрезы производятся
между вторым Т и (С/С)-компонентами сайта узнавания.
Обратите внимание, что приведенное выше
Прикрепление RecA
..........
11 | 111 111
Вторжение нити
описание не наделяет никакой определенной ро-
лью белок RecC в гомологичной рекомбинации,
следующей по пути RecBCD. Белок RecC фактически
довольно загадочен, поскольку его аминокислотная
последовательность отличается от последователь-
ности любого другого белка, синтезируемого Е coli.
Недавние исследования с помощью рентгенострук-
турного анализа показали, что третичная струк-
тура белка RecC напоминает структуру геликаз из
семейства SF1. Аминокислоты, которые являются
мд* "ЧДЯ ^iiiiiiii
тттт
11Ш1
Рис. 17.7. Роль белка RecA в формировании D-петли в ходе
гомологичной рекомбинации у Е. coli
Глава 17. Рекомбинация 695
Прикрепление Перемещение
к переходу точки ветвления Разрешение
Рис. 17.8. Роль белков Ruv в гомологичной рекомбинации у Escherichia coli. Перемещение точки ветвле-
ния вызывается структурой, включающей четыре копии белка RuvA, связанные со структурой Холлидея
и кольцами белков RuvB с обеих сторон. После отделения комплекса RuvAB два белка RuvC связываются
со структурой, при этом ориентация их прикрепления определяет направление разрезов, которые раз-
решают структуру
определяющими для действия геликазы, отсутствуют в составе RecC, хотя эквивалентная
область белка RecC, несмотря на это, образует контакт с молекулой ДНК. Кажется мало-
вероятным, что белок RecC обладает каким-либо остаточным действием геликазы, и он
поэтому не помогает белкам RecB и RecD в раскручивании двойной спирали, но одна
возможная роль состоит в том, что RecC приобрел функцию просмотра и отвечает за
опознавание х-участка и, следовательно, за инициацию конформационного изменения
в комплексе RecBCD, которое ведет к формированию гетеродуплекса.
Прочие пути гомологичной рекомбинации у Е. coli
Мутанты Е. coli, у которых отсутствуют компоненты системы RecBCD, по-прежнему
способны осуществлять гомологичную рекомбинацию, хотя и со сниженной эффек-
тивностью. Это связано с тем, что бактерия обладает по крайней мере двумя другими
путями гомологичной рекомбинации, названными RecE и RecF. В нормальных клетках
Е. coli гомологичная рекомбинация в основном протекает путем RecBCD, но если этот
путь инактивирован мутацией, система RecE способна принять на себя эти функции,
а если система RecE тоже будет инактивирована, ее место займет система RecF.
Детали пути RecF начинают проясняться, а общий механизм его, как становится
ясным, подобен описанному для RecBCD. Действие геликазы системы RecF обеспечива-
ется белком RecQ, а 5'-конец нити удаляется белком RecJ, оставляя З’-выступ, который
покрывают белки RecA благодаря объединенным усилиям белков RecF, RecJ, RecO, RecQ
и RecR. Существует значительная взаимозаменяемость между компонентами путей
RecBCD и RecF, и думается, что в некоторых мутантах, у которых недостает тех или
иных компонентов стандартных процессов, работают гибридные системы. Есть, однако,
отличия, так как только путь RecBCD инициирует рекомбинацию на х-участках, рас-
сеянных по всему геному Е. coli, и только путь RecF способен запустить рекомбинацию
между парой плазмид. Кроме того, RecF является главной системой, ответственной за
рекомбинационное устранение однонитевых пропусков, возникающих при репликации
сильно поврежденной ДНК (раздел 17.1.3). Путь RecE изучен намного хуже, но, судя по
признакам, он во многом перекрывается с системой RecF, причем сами по себе белки
RecJ, RecO, RecQ и RecF, как оказалось, присущи обоим путям.
696 Часть 4. Механизмы репликации и эволюции геномов
Наряду с вариантами RecBCD, RecE и RecF, функции которых состоят в установке
структуры гетеродуплекса, Е coli имеет также альтернативные средства для выполнения
этапа (функции) перемещения точки ветвления. Мутанты, у которых не имеется белков
RuvA или RuvB, по-прежнему способны выполнять гомологичную рекомбинацию, по-
тому что функция комплекса RuvAB может быть обеспечена также геликазой по имени
RecG. Еще не ясно, являются ли RuvAB и RecG попросту взаимозаменяемыми или же они
специализированы для различных сценариев рекомбинации. Мутанты с отсутствием
RuvC также способны выполнять гомологичную рекомбинацию, а это предполагает, что
Е. coli обладает другими белками, которые способны разрешать структуры Холлидея,
но род этого белка (а возможно, и белков) неизвестен.
Пути гомологичной рекомбинации у эукариотов
Модель гомологичной рекомбинации с двунитевым разрывом, как думают, уместна
для всех организмов, не только для Е. coli, — вспомним, что она была первоначально
построена для объяснения генной конверсии у Saccharomyces cerevisiae (раздел 17.1.1).
Биохимические события, лежащие в основе этого процесса, как оказалось, подобны во
всех организмах, и был идентифицирован ряд белков дрожжей, выполняющих функции,
эквивалентные функциям белков, которые встречаются в системе RecBCD у Е coli. В част-
ности, два белка дрожжей по имени RAD51 и DMC1 являются гомологами белка RecAE.
coli. Хотя имеются подозрения на особую роль белков RAD51 и DMC1, они, как думают,
работают совместно во многих событиях гомологичной рекомбинации. Это заключение
опирается на тот факт, что мутанты с отсутствием одного или другого белка имеют по-
добные фенотипы и эти два белка находятся вместе в одних и тех же местоположениях
внутри ядер, в которых происходит мейоз. Что интересно, в геноме S. cerevisiae существует
около 100 горячих точек рекомбинации (раздел 3.2.3), что предполагает возможность
наличия последовательностей, эквивалентных %-участкам Е coli, хотя и с более низкой
частотой встречаемости — одна на 40 тыс. п. н. в геноме дрожжей по сравнению с одним
участком на 6 тыс.п.н. у Е coli. Белки, гомологичные RAD51 и DMC1, известны также
у некоторых других эукариотов, в том числе у людей.
Одним озадачивающим моментом гомологичной рекомбинации у эукариотов был
механизм, которым разрешаются структуры Холлидея. Это связано с тем, что многие
годы предпринимались попытки поиска белков, гомологичных RuvC Е coli, однако безу-
спешно. Фактически белок RuvC не универсален даже среди бактерий. Некоторые виды
явно используют нуклеазу совершенно иного типа для разрешения структур Холл идея.
У архей эквивалентную функцию, как думают, выполняет белок Hjc, который не имеет
никакого сходства последовательности с RuvC, но, как было показано биохимическими
испытаниями, связывается со структурами Холлидея. У эукариотов было обнаружено
несколько белков, которые имеют структурное подобие с Hjc, в том числе RAD51C
S. cerevisiae, ген которого является членом многогенного семейства RAD51, и Mus81
человека. Также были предложены модели разрешения структур Холлидея сочетанием
действий геликазы и топоизомеразы, эти модели основаны на предположении о том,
что, возможно, не всем эукариотам нужны прямые эквиваленты белка RuvC.
17.1.3 . Гомологичная рекомбинация и репарация ДНК
Внимание, уделявшееся генетиками кроссинговеру как главной особенности по-
лового воспроизводства, неизбежно сместило первые исследования гомологичной
рекомбинации в сторону событий, происходящих во время мейоза. Альтернативная
роль гомологичной рекомбинации стала очевидной, когда впервые были исследованы
Глава 17. Рекомбинация 697
мутанты Е. coli, дефектные по компонентам RecBCD и других путей рекомбинации,
и было показано, что они имеют нарушения репарации ДНК. Сегодня мы полагаем, что
основная функция гомологичной рекомбинации заключается в пострепликационной
репарации, ее роль в кроссинговере для большинства клеток имеет второстепенное
значение.
Пострепликационная репрация происходит, когда в результате нарушений процесса
репликации в дочерних молекулах ДНК возникают разрывы. Одно из таких нарушений
может произойти, когда репликационный аппарат пытается копировать сегмент генома,
который сильно поврежден, в частности, те области, которые заполонил и циклобутиль-
ные димеры. Когда циклобутильный димер встречается, матричная нить не может быть
скопирована, и ДНК-полимераза попросту перескакивает через него вперед к ближай-
шей неповрежденной области, где снова начинает процесс репликации. В результате на
одном из дочерних полинуклеотидов зияет прореха (рис. 17.9). Один способ, которым
этот пропуск может быть восстановлен, — событие рекомбинации, переносящее в него
эквивалентный сегмент ДНК из родительского полинуклеотида, находящийся во второй
дочерней двойной спирали. Пропуск, который теперь присутствует во второй двойной
спирали, заполняется ДНК-полимеразой, использующей неповрежденный дочерний
полинуклеотид этой спирали в качестве матрицы. У Е. coli в такого типа устранении
однонитевых пропусков используется система рекомбинации RecF.
Если поврежденный участок не удается обойти, то дочерний пол инуклеотид вместо
получения пропуска оборвется (рис. 17.10). Есть несколько путей, которыми этот пропуск
может быть устранен. Одна из возможностей состоит в том, что репликационная вилка
останавливается и оборачивается назад на короткое расстояние, так что образуется
дуплекс между дочерними полинуклеотидами. Неполный полинуклеотид затем про-
должается ДНК-полимеразой, используя неповрежденную дочернюю нить в качестве
матрицы. Репликационная вилка после этого снова продвигается вперед процессом,
аналогичным этапу перемещения точки ветвления при гомологичной рекомбинации.
В результате поврежденный участок удается обойти и репликация может быть про-
должена.
Более серьезное нарушение происходит, если один из реплицируемых родительских
полинуклеотидов содержит однонитевой разрыв. В таком случае процесс репликации
приводит к двунитевому разрыву в одной из дочерних двойных спиралей и репликаци-
онная вилка уходит в небытие (рис. 17.11). Такой разрыв может быть устранен формой
гомологичной рекомбинации между оборванным концом и второй, неповрежденной
молекулой. На схеме, показанной на рис. 17.11, дочерний полинуклеотид в месте двуни-
тевого разрыва продолжается посредством реакции обмена нитями, которая позволяет
ему использовать другую родительскую нить в качестве матрицы. За перемещением
точки ветвления следует распускание структуры Холлидея и, наконец, восстановление
репликационной вилки.
17.2. Сайт-специфическая рекомбинация
Протяженная область гомологии не является исключительной предпосылкой ре-
комбинации: данный процесс может быть начат также и между двумя молекулами ДНК,
которые имеют только очень короткие отрезки тождественной последовательности.
В таком случае рекомбинацию называют сайт-специфической и ее интенсивно изучали
из-за партии, которую она исполняет в ходе цикла инфицирования бактериофагом X.
698 Часть 4. Механизмы репликации и эволюции геномов
5'
3'
111111111
Ц111£Ц±Ц\
!IJ IU U НТ/^'1'1'" 5‘
I Репликационная
| вилка обращается
Повреждение
Однонитевой
пропуск
Перенос ДНК
гг-г1ТгТтп!’|1
i Ресинтез
заимствованной ДНК
Рис. 17.9. Восстановление однонитевого про-
пуска с помощью системы RecF у Е. coli
Повреждение обойдено
Рис. 17.10. Оборванный дочерний поли-
нуклеотид может быть восстановлен об-
ращением репликационной вилки
. ..MiuHmim
17.2.1. Встраивание ДНК бактериофага X в геном £ coli
После впрыскивания своей ДНК в клетку Е. coli бактериофаг X может пойти любым
из двух путей инфицирования (раздел 14.3.1). Один из них — литический путь — ведет
к быстрому синтезу белков оболочки бактериофага X, сопряженному с репликацией
генома фага К что вызывает смерть бактерии и высвобождение новых фагов в течение
приблизительно 45 минут после первоначального заражения. Напротив, если фаг идет
лизогенным путем, новые фаги не появляются незамедлительно. Бактерия делится как
обычно, возможно, в течение многих клеточных делений, с фагом в состоянии покоя,
называемым профагом. Рано или поздно, быть может в результате повреждения ДНК
или некоторого другого толчка, фаг снова становится активным.
Глава 17. Рекомбинация 699
Во время лизогенной фазы геном фага X встраи-
вается в хромосому Е coli. Поэтому он реплицируется
всякий раз, когда ДНК Е coli копируется и передается
дочерним клеткам, как если бы он был стандартной
частью генома бактерии. Встраивание происходит
путем сайт-специфической рекомбинации между ме-
стами прикрепления или участками att, attP в геноме
фага X и attB на хромосоме Е coli. Каждый из таких
сайтов связывания имеет в своем центре идентич-
ную срединную последовательность длиной 15 п. н.,
называемую О (рис. 17.12), ограниченную с обеих
сторон изменчивыми последовательностями, на-
званными В и В' в бактериальном геноме и Р и Р*
в ДНК фага. Последовательности В и В' являются
весьма короткими, только по 4 п. н. каждая, что озна-
чает, что участок attB покрывает лишь 23 п. н. ДНК,
но последовательности Р и Р’ намного длиннее —
полная последовательность attP охватывает более
250 п.н. Мутации в срединной последовательности
неизбежно ведут к инактивации участка att, так что
он более не может участвовать в рекомбинации, но
мутации в примыкающих последовательностях име-
u nmirmiv________________з'
t ттптгт ri 111111 гпт
2 J Я11111 ГТТ/~ J °
Пропуск
I Репликационная
▼ вилка разрушается
ПТГПТТГГТПТТПТП ПТТТШТ1ТТТ
НШШ1Ш
Вторжение нити
3^1ТГ~Н|1ШГ
I Перемещение
▼ точки ветвления
Роспуск структуры
▼ Холлидея
IIIIIIIIM111II
III I lllfll11
III IIII llllllllll
Репликационная вилка восстановлена
ют менее серьезные последствия и только уменьша-
ют эффективность рекомбинации. Если attB — сайт
связывания в геноме Е coli — инактивируется, то
вставка ДНК фага X может произойти на вторичных
участках, которые обладают некоторым подобием
Рис. 17.11. Один из механизмов восста-
новления разрушенной репликационной
вилки с помощью гомологичной реком-
бинации
последовательности с подлинным локусом attB. Если используется вторичный участок,
то частота лизогении значительно уменьшается, при этом встраивание, возможно,
происходит с частотой, которая составляет менее 0,01 % от частоты, наблюдаемой в не-
мутированных клетках Е. coli.
Поскольку это рекомбинация между двумя кольцевыми молекулами, ее резуль-
татом является образование одного более крупного кольца: другими словами, ДНК
фага X встраивается в бактериальный геном (рис. 17.13). Событие рекомбинации ка-
тализируется специальной топоизомеразой I типа (раздел 15.1.2), называемой инте-
гразой и принадлежащей многообразному семейству рекомбиназ, представленных
в клетках бактерий, архей и дрожжей. Есть по крайней мере четыре сайта связывания
для интегразы в пределах участка attP, равно как и по крайней мере три участка для
второго белка, хозяйского фактора встраивания, или HIF. Вместе эти белки покрывают
сайт связывания фага. После этого инте-
граза делает ступенчатый, двунитевой
разрез в эквивалентных позициях участ-
ков att геномов фага X и бактерии. За-
тем молекулы ДНК обмениваются между
собой двумя короткими однонитевыми
выступами, производя структуру Холли-
дея, которая перемещается на несколько
п. н. по гетеродуплексу, прежде чем быть
---------G СТТТТТТАТАС ТАА
• ------С G A A AAAATATg| АТТ »
Рис. 17.12. Срединная последовательность участков
att, присутствующих в бактериофаге X и в хромосоме
Е. coli. Красная линия показывает ступенчатый разрез,
производимый в каждом участке att в ходе встраива-
ния и вырезания генома фага
700 Часть 4. Механизмы репликации и эволюции геномов
ВОР' РОВ'
............. H1IIII.........................II...........
Рис. 17.13. Встраивание генома бактериофага X в хромосомную ДНК Е. coli. ДНК и фага X, и Е. coli содержит
копию участка att, состоящую из идентичной центральной последовательности, названной «О», и при-
мыкающих к ней с обеих сторон последовательностей Р и Р’ (для участка att фага) или В и В’ (для участка
att бактерии). Рекомбинация между областями О встраивает геном фага X в бактериальную ДНК
расщепленной. Это расщепление, при условии, что оно производится в должной ори-
ентации, разрешает структуру Холлидея таким образом, что ДНК фага X оказывается
встроенной в геном Е coli.
Встраивание создает гибридные версии сайтов связывания, которые теперь назы-
вают attR (имеет структуру ВОР’) и attL (со структурой РОВ’). Вторая сайт-специфическая
рекомбинация между двумя участками att (теперь уже оба содержатся в одной и той же
молекуле) обращает первоначальный процесс вспять и высвобождает ДНК фага X. Эта
рекомбинация также катализируется интегразой, но в совокупности с белком, назван-
ным «эксцизионазой», и кодируемым геном xis бактериофага X, а не с IHF. Функции Xis
и IHF, состоящие соответственно в вырезании и во встраивании, являются, вероятно,
совершенно различными, и эти два белка не должны рассматриваться как играющие
эквивалентные роли в этих двух процессах. Ключевой момент здесь заключается в том,
что комбинация интегразы и эксцизионазы является способной свести участки attR и attL
друг с другом, с тем чтобы запустить внутримолекулярную рекомбинацию, посредством
которой вырезается геном фага X. После вырезания геном фага X возвращается к лити-
ческому способу инфицирования и руководит синтезом новых фагов.
17.2.2. Сайт-специфическая рекомбинация — помощь в генной инжене-
рии
Процессы, ответственные за встраивание и вырезание генома фага X, довольно
типичны для стратегий, используемых фагами для установления лизогении, хотя у не-
которых фагов молекулярные события менее сложны, чем наблюдаемые у фага X. Встраи-
вание и вырезание генома бактериофага Р1, например, требует лишь единственного
фермента, рекомбиназы Сге, которая опознает целевые участки протяженностью 34 п. н.,
названные 1охВ и 1охР, которые являются идентичными друг другу и не имеют никаких
специфических примыкающих последовательностей, эквивалентных В, В’ и т.д.
Глава 17. Рекомбинация 701
Простота системы фага Р1 привела к ее использованию в проектах генной инже-
нерии, в которых необходима сайт-специфическая рекомбинация. Важное применение
возникло в технологии, используемой для производства генетически модифицирован-
ных зерновых культур. Одна из главных причин беспокойства, возникшая из дебатов
вокруг генетически модифицированных растений, — возможные вредные воздействия
маркерных генов, используемых в растительных клонирующих векторах. Большин-
ство растительных векторов несут в себе копию гена устойчивости к канамицину (см.
рис. 2.27), позволяющего опознавать модифицированные растения в ходе процесса кло-
нирования. Ген kanR является бактериальным по происхождению и кодирует фермент
неомицинфосфотрансферазу II. Этот ген и его ферментативный продукт присутствуют
во всех клетках конструируемого растения. Опасение по поводу того, что неомицин-
фосфотрансфераза может быть ядовита для людей, было приуменьшено успешными
испытаниями на модельных животных, но все еще остается озабоченность в том, что
ген kanR, содержащийся в генетически модифицированных пищевых продуктах, может
быть передан бактериям, обитающим в кишечнике человека, и сделать их устойчивыми
к канамицину и родственным ему антибиотикам,
и в том, что ген kanR может перейти к другим орга-
низмам в окружающей среде с возможным ущер-
бом экосистеме.
Страхи вокруг использования гена kanR и дру-
гих маркерных генов побудили биотехнологов
изобрести способы удаления этих генов из ДНК
растения после того, как событие трансформации
было подтверждено. Одна из стратегий исполь-
зует рекомбиназу Сге. Чтобы использовать эту
систему, растение трансформируется с помощью
двух клонирующих векторов: первый несет ген,
добавляемый к растению, наряду с его селектив-
ным маркерным геном kanR, окруженным целевы-
ми последовательностями lox, а второй несет ген
рекомбиназы Сге. Поэтому после трансформации
экспрессия гена Сге приводит к вырезанию гена
kanR из ДНК растения (рис. 17.14).
Ген Сге
| Вырезание гена кап*
Рис. 17.14. Использование рекомбиназы
Сге в генной инженерии растений. Экс-
прессия гена Сге вызывает вырезание гена
kanR из ДНК растения
17.3. Транспозиция
Транспозиция не является разновидностью ре-
комбинации, а представляет собой самостоятельный
процесс, который часто использует рекомбинацию
и конечный результат которого — перемещение сег-
мента ДНК из одной позиции генома в другую. Харак-
терная особенность транспозиции состоит в том, что
перемещаемый сегмент ограничен парой коротких
прямых повторений (рис. 17.15), которые формиру-
ются во время процесса транспозиции.
В разделе 9.2 мы рассматривали различные типы
мобильных генетических элементов, встречающихся
у эукариотов и прокариотов, и обнаружили, что они
Хромосомная ДНК
повторения
Рис. 17.15. Встроенные подвижные ге-
нетические элементы втиснуты между
коротких прямых повторов. Этот кон-
кретный транспозон ограничен тетра-
нуклеотидными повторениями вида
5'-CTGG-3'. Другие транспозоны имеют
иные прямые повторы
702 Часть 4. Механизмы репликации и эволюции геномов
могут быть широко классифицированы на три категории на основании механизма их
транспозиции:
• ДНК-транспозоны, которые транспонируются репликативно, исходный транспозон
остается на своем месте, а новая копия появляется в любом другом месте генома
(рис. 17.16);
• ДНК-транспозоны, которыетранспонируются консервативно, исходный транспозон
перемещается на новый участок в ходе процесса типа вырезания и вставки;
• ретроэлементы, которые транспонируются репликативно через РНК-посредник.
Теперь мы обсудим события рекомбинации, которые отвечают за каждый из этих
трех типов транспозиции.
(
Репликативная
1
Консервативная
Рис. 17.16. Репликативная и консервативная транспозиция. ДНК-транспозоны пользуются или репли-
кативным, или консервативным способом (некоторые могут использовать оба). Ретроэлементы транс-
понируются репликативно через РНК-посредник
17.3.1. Репликативная и консервативная транспозиция
ДНК-транспозонов
За эти годы был предложен ряд моделей репликативной и консервативной транс-
позиции ДНК-транспозонов, но большая часть из них — модификации схемы, первона-
чально очерченной Шапиро в 1979 г. Согласно этой модели репликативная транспозиция
бактериального элемента, такого как транспозон ТпЗ-типа или транспонируемый фаг
(раздел 9.2.2), инициируется одной или несколькими эндонуклеазами, которые делают
однонитевые разрезы по обе стороны транспозона и в целевом участке, в который будет
вставлена новая копия данного элемента (рис. 17.17). На целевом участке оба разреза
отделяются друг от друга несколькими парами оснований, так что расщепленная дву-
нитевая молекула имеет короткие 5'-выступы.
Лигирование этих 5’-выступов к свободным З’-концам с обеих сторон транспозона
производит гибридную молекулу, в которой две исходные ДНК — одна, содержащая
транспозон, и вторая, содержащая целевой участок, — связаны вместе мобильным
генетическим элементом, ограниченным парой структур, напоминающих реплика-
ционные вилки. Синтез ДНК в этих репликационных вилках копирует мобильный
элемент и превращает первоначальный гибрид в совставку (коинтеграт), в которой
две исходные ДНК все еще связаны. Гомологичная рекомбинация между этими двумя
копиями данного транспозона расцепляет коинтеграт, отделяя исходную молекулу
ДНК (с ее копией транспозона, все еще стоящей на своем месте) от целевой молекулы,
которая теперь содержит копию этого транспозона. Таким образом и происходит ре-
пликативная транспозиция.
Видоизменение только что описанного процесса изменяет способ транспозиции
с репликативного на консервативный (см. рис. 17.17). Вместо того чтобы выполнять
синтез ДНК, гибридная структура преобразуется назад в две отдельные молекулы ДНК,
просто за счет создания дополнительных однонитевых надрезов с обеих сторон транс-
Глава 17. Рекомбинация 703
Транспозон
Лигирование
РЕПЛИКАТИВНАЯ
ТРАНСПОЗИЦИЯ
КОНСЕРВАТИВНАЯ
ТРАНСПОЗИЦИЯ
Рис. 7.17. Модель процесса, дающего репликативную и консервативную транспозицию
позона. В результате этого транспозон вырезается из его первоначальной молекулы
и остается «вставленным» в целевую ДНК.
17.3.2. Транспозиция ретроэлементов
Для человека самые важные ретроэлементы — ретровирусы, в число которых
входят вирусы иммунодефицита человека, которые вызывают СПИД, и другие раз-
личные типы вирусов. Большая часть наших знаний о ретротранспозиции пропитана
спецификой ретровирусов, хотя существует мнение, что другие ретроэлементы, такие
как ретротранспозоны семейств Tyl/copia и ТуЗ/gypsy, перемещаются подобными ме-
ханизмами. Эти механизмы не вовлекают рекомбинацию, но, удобства ради, мы рас-
смотрим их прямо здесь.
Первый шаг в ретротранспозиции — синтез РНК-копии встроенного ретроэле-
мента (рис. 17.18). Длинный концевой повтор (LTR) на 5’-конце данного элемента
содержит последовательность ТАТА, которая служит промотором для транскрипции
704 Часть 4. Механизмы репликации и эволюции геномов
5-ДКП
- -- V W ' <
Встроенный
। । р ретроэлемент
^Транскрипция
РНК-копия
3'
I тРНК затравливает синтез первой нити
R U5 * из R
Деградация конца РНК
▼ из R
RU5
U5
Первое переключение матрицы
RU5
Продолжение
синтеза нити
РНКаза Н
деградирует
большую часть
нити РНК
U5’
/ U3RAlI5
/ U51
I Затравливание синтеза
f второй нити
Второе переключение
матрицы
Разрыв пар
оснований
в области U3-R
второй нити
Рис. 17.18. Репликация РНК и ДНК во время транспозиции ретроэлемента. На этой схеме показано, как
встроенный ретроэлемент копируется в свободную версию двунитевой ДНК. Первый шаг — синтез
РНК-копии, которая затем преобразуется в двунитевую ДНК чередой событий, в которую входят два
переключения матрицы, как описано в тексте
РНК-полимеразой II (раздел 11.2.2). Некоторые ретроэлементы имеют также и после-
довательности энхансеров (раздел 11.3), которые, как думают, регулируют количество
производимой транскрипции. Транскрипция продолжается через всю длину элемента
вплоть до последовательности полиаденилирования (раздел 12.2.1) в З'-LTR.
Глава 17. Рекомбинация 705
Полученный транскрипт теперь выступает матрицей для направляемого РНК син-
теза ДНК, катализируемого ферментом обратной транскриптазой, кодируемой частью
гена pol ретроэлемента (см. рис. 9.14). Поскольку это синтез ДНК, необходима затравка
(раздел 15.2.2) и, как и в процессе репликации генома, затравка состоит из РНК, а не
из ДНК. Во время репликации генома затравка синтезируется de novo ферментом по-
лимеразой (см. рис. 15.13), но ретроэлементы не кодируют РНК-полимеразы и по этой
причине не могут производить затравки таким путем. Вместо этого, они используют
в качестве затравки одну из молекул тРНК клетки; которую именно — зависит от само-
го ретроэлемента: элементы семейства Tyl/copia всегда используют тРНКМет, а другие
ретроэлементы употребляют иные молекулы тРНК.
тРНК-затравка отжигается кучастку в пределах 5'-LTR (см. рис. 17.18). На первый взгляд
это место кажется странным для участка затравливания, потому что это означает, что синтез
ДНК направляется прочь от центральной области ретроэлемента и, таким образом, выра-
жается лишь в короткой копии части 5’-LTR. Фактически к тому времени, когда копия ДНК
была продолжена до конца LTR, часть РНК-матрицы деградирует, и выступ ДНК, который
производится, переотжигается к З'-LTR ретроэлемента, который, будучи длинным концевым
повтором, имеет ту же самую последовательность, что и 5'-LTR, и, таким образом, может
спариться основаниями с ДНК-копией. Синтез ДНК теперь продолжается по матрице РНК,
в конечном счете вытесняя тРНК-затравку. Обратите внимание, что результат—ДНК-копия
полной матрицы, в том числе и участка затравливания: переключение матрицы является,
в сущности, стратегией, которую ретроэлемент использует для решения проблемы «уко-
рочения концов»—той же проблемы, с которой хромосомные ДНК справляются благодаря
синтезу теломер (раздел 15.2.4).
Завершение синтеза первой нити ДНК
дает гибрид ДНК-РНК. РНК частично дегра-
дируется ферментом РНКазой Н, кодируемой
другой частью гена pol. РНК, которая не де-
градировала (обычно она представляет собой
только единственный фрагмент, прикреплен-
ный к короткой полипуриновой последова-
тельности, смежной с З'-LTR), затравливает
синтез второй нити ДНК, снова обратной
транскриптазой, которая способна действо-
вать и как РНК-, и как ДНК-зависимая ДНК-
полимераза. Как и в первом цикле синтеза
Ретроэлемент 111111111111|*
Хромосомная ДНК --11111'11'111 ITTTTTT"
Действие интегразы
Удаление _____ь»
двух нуклеотидов ’----------
3 °
Ступенчатый разрез
5' / 3'
“ТПТГТм Т|ГЧШТГ'
3' 5'
>111111111
Встраивание
1 ретроэлемента
Рис. 17.19. Встраивание в геном хозяина версии ретроэ-
лемента в виде двунитевой ДНК. Встраивание ретроэле-
мента дает четырехнуклеотидные прямые повторения
с обеих сторон встроенной последовательности. В случае
ретровирусов этот этап процесса транспозиции требует
и интегразу, и протеинкиназу ДНК-РК^, и белки Ku, кото-
рые участвуют также и в негомологическом соединении
концов NHEJ (раздел 16.2.4)
Выступы ретроэлемента
теряются
-ТТТТТТ....иТГГП'1ТПП'>"11111!
I Заполнение
1 пропусков
HillIIBIIIIIIIIIIIIIIIIIIIIIIII
Выступающие последовательности
дублировались
706 Часть 4. Механизмы репликации и эволюции геномов
ДНК, синтез второй нити в начале дает ДНК-копию только LTR, но второе переключение
матрицы, к другому концу молекулы, позволяет продолжить ДНК-копию до полной
длины. Это создает матрицу для дальнейшего продолжения первой нити ДНК, так что
окончательная двунитевая ДНК представляет собой полную копию внутренней области
ретроэлемента и обоих LTR.
Все, что остается, это встроить новую копию ретроэлемента в геном. Первоначаль-
но думали, что встраивание происходит случайным образом, но сейчас очевидно, что,
хотя никакая специфическая последовательность не используется в качестве целевого
участка, есть предпочтительные сайты встраивания. Встраивание предполагает удале-
ние двух нуклеотидов с З'-концов двунитевого ретроэлемента ферментом интегразой
(кодируемой еще одной частью гена pol). Эта интеграза также производит ступенчатый
разрез в геномной ДНК так, что теперь и ретроэлемент, и участок встраивания имеют
5'-выступы (рис. 17.19). Последовательности этих выступов могут и не быть комплемен-
тарны друг другу, но они тем не менее, кажется, взаимодействуют некоторым образом,
так что ретроэлемент встраивается в геномную ДНК. Такое взаимодействие приводит
к потере выступов ретроэлемента и к заполнению пропусков, которые остаются, а это
означает, что участок встраивания становится дублированным в пару прямых повто-
рений: по одному на каждом конце встроенного ретроэлемента.
17.3.3. Каким образом клетки минимизируют вредное влияние транс-
позиции?
Транспозиция может оказывать вредные воздействия на геном. Эти воздействия
явно преодолевают порог нарушения функционирования отдельного гена, которое про-
исходит, когда мобильный генетический элемент занимает новую позицию, лежащую
в пределах кодирующей области этого гена. Некоторые элементы, особенно ретротран-
спозоны, содержат промоторные и энхансерные последовательности, которые могут
видоизменить картину экспрессии смежных генов, а кроме того, транспозиция часто
предполагает создание двунитевых разрывов, которые, как мы увидели в разделе 16.2.4,
могут оказывать серьезные вредные воздействия на целостность генома. Рассеянные
повторы, по крайней мере из ряда мобильных, составляют почти половину каждого ге-
нома млекопитающих и значительно более весомую долю геномов некоторых растений.
Можно поэтому ожидать, что эти и другие геномы развили механизмы для ограничения
передвижения своих мобильных генетических элементов.
Один путь, которым транспозиция и ДНК-транспозонов и ретроэлементов может
быть предотвращена, — метилирование их последовательностей ДНК, ибо метилиро-
вание является обычным средством подавления геномных областей (раздел 10.3.1).
Многие последовательности мобильных генетических элементов действительно сверх-
метилированы — 90 % метилированных цитозинов в геноме человека расположены
в рассеянных повторах, — но экспериментальные данные, связывающие такое метили-
рование с подавлением транспозиции, было трудно получить. Недавно было показано,
что мутанты растения Arabidopsis thaliana, которые имеют дефектные системы мети-
лирования, страдают от количества транспозиции больше нормы, но такая усиленная
транспозиция может не быть показана всеми типами транспозонов в геноме растения.
В настоящее время проводится дальнейшая работа по исследованию роли метилиро-
вания у Arabidopsis, и подобные исследования распространяются на организмы других
типов.
Глава 17. Рекомбинация 707
Заключение
Рекомбинация первоначально использовалась для описания хода и объяснения
исхода кроссинговера между парами гомологичных хромосом во время мейоза. Теперь
этот термин употребляется также и для описания молекулярных событий, которые лежат
в основе этого процесса. Гомологичная рекомбинация происходит между сегментами
молекул ДНК, имеющих протяженные области гомологии последовательностей. Перво-
начальные модели гомологичной рекомбинации предполагали, что процесс рекомби-
нации начинается с надрезов, которые производятся в одной или обеих двунитевых
молекулах, но теперь думается, что отправная точка — двунитевой разрез в одной из
этих молекул. Обмен нитями ведет к структуре гетеродуплекса, которая разрешается рас-
щеплением, могущим привести или к обмену сегментами ДНК, ил и к генной конверсии.
Escherichia coli обладает по крайней мере тремя молекулярными путями гомологичной
рекомбинации. Один из них, который был изучен наиболее подробно, — путь RecBCD —
предполагает раскручивание одного партнера рекомбинации с помощью пары геликаз,
которые прикрепляются к двунитевому разрыву и продвигаются по молекуле. В сайте
узнавания, названном х-участком, комплекс RecBCD запускает обмен нитями, где белок
RecA играет центральную роль в перемещении вторгающейся нити в неповрежденную
двойную спираль. Перемещение точки ветвления в пределах гетеродуплекса и разре-
шение структуры катализируется белками Ruv. Пути RecE и RecF работают подобным
образом, причем ряд белков является общим для этих трех рекомбинационных систем.
У эукариотов известны эквивалентные им белки. Гомологичная рекомбинация ответ-
ственна за пострепликативное устранение разрывов ДНК. Сайт-специфическая реком-
бинация не требует протяженных областей гомологии между молекулами-партнерами.
Рекомбинация этого типа отвечает за встраивание геномов бактериофагов, таких как
геном фага X, в хромосому бактерии-хозяина. Встраивание генома фага X в ДНК Е. coli
происходит путем рекомбинации между парой последовательностей длиной 15 п. н., со-
держащихся в пределах более длинных сайтов связывания. Для встраивания необходима
интеграза, кодируемая геномом фага X, и хозяйский фактор встраивания, принадлежа-
щий Е. coli. Вырезание требует наличия интегразы и белка эксцизионазы.
Транспозиция ДНК-транспозонов происходит путем рекомбинации. Этот процесс
может быть или репликативным, или консервативным, но оба происходят в виде че-
реды событий, впервые описанных Шапиро в 1979 г. Ретроэлементы транспонируются
через РНК-посредник, который транскрибируется с родительской копии транспозона.
После копирования в двунитевую ДНК ретроэлемент перевстраивается в хромосому
хозяина.
708 Часть 4. Механизмы репликации и эволюции геномов
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
17.1 *. Который из нижеследующих приме-
ров относится к сайт-специфической
рекомбинации?
а. Кроссинговер во время мейоза.
Ь. Генная конверсия.
с. Встраивание генома бактериофа-
га X в хромосому Е coli.
d. Встраивание транспозона в но-
вый участок генома.
17.2 . Какого типа обмен ДНК происходит
в процессе кроссинговера во время
мейоза?
а. Однонитевой обмен.
Ь. Обоюдный обмен нитями.
с. Интегративный обмен нитями.
d. Репликативный обмен нитями.
17.3 *. Каким образом модель рекомбина-
ции Мезельсона-Реддинга объясня-
ет, как две молекулы ДНК взаимо-
действуют в начале гомологичной
рекомбинации?
а. Однонитевые надрезы появля-
ются в эквивалентных позициях
в каждой молекуле.
Ь. Специализированная топоизо-
мераза производит одноните-
вые надрывы в обеих молекулах
ДНК.
с. Обе молекулы ДНК начинают ре-
комбинацию без появления раз-
рывов.
d. Однонитевой разрыв появляется
в одной молекуле, создавая сво-
бодный конец, который вторга-
ется в другую молекулу, чтобы
вытеснить одну из ее нитей.
17.4 . Генная конверсия наблюдается, ког-
да:
а. Во время мейоза один аллель за-
меняется другим аллелем.
Ь. Во время мейоза ожидаемое соот-
ношение аллелей 2:2 изменяется
на отношение 4:0.
с. Во время мейоза ожидаемое соот-
ношение аллелей 2:2 изменяется
на отношение 3:1.
d. Все вышеперечисленное.
17.5 *. Что происходит на %-участках во
время гомологичной рекомбина-
ции, опосредствуемой ферментом
RecBCD у Е. coli?
а. Это место, где белки RecBCD свя-
зываются с ДНК, с тем чтобы за-
пустить рекомбинацию.
Ь. Это участок, который заставляет
фермент произвести двунитевой
разрыв ДНК.
с. Это участок, где действием ге-
ликазы этого фермента начина-
ет осуществляться деградация
ДНК.
d. Это участок, где заканчивается
перемещение точки ветвления.
17.6 . Какие белки катализируют пере-
мещение точки ветвления в про-
цессе гомологичной рекомбинации
у Е. coli?
a. RecA.
b. RecBCD.
с. RuvA и RuvB.
d. Топоизомераза ПВ.
17.7 *. В чем, как думают, состоит первич-
ная функция гомологичной реком-
бинации?
а. Кроссинговер во время мейоза.
Ь. Генная конверсия.
с. Встраивание геномов лизогенных
фагов.
d. Пострепликационная репарация
ДНК.
Глава 17. Рекомбинация 709
17.8 . Примером какого типа рекомбина-
ции является система Сге, используе-
мая в генной инженерии растений?
а. Гомологичной рекомбинации.
Ь. Ретротранспозиции.
с. Сайт-специфической рекомбина-
ции.
d. Транспозиции.
17.9 *. Что случается, если в последова-
тельностях attB у Е. coli происходят
инактивирующие мутации?
а. ДНК фага Сформирует частичную
структуру Холлидея с геномом
Е. coli и затем деградирует.
Ь. Фаг X может следовать лишь ли-
тическим циклом.
с ДНК фага X может быть встроена
в геном Е coli на вторичныхучастках
с намного более низкой частотой.
d. ДНК фага X встраивается в уча-
сток attB, но не может быть вы-
резана.
17.10 . Вырезание профага X из генома
Е coli есть:
а. Событие внутримолекулярной
рекомбинации.
Ь. Событие межмолекулярной ре-
комбинации.
с. Событие высвобождения нуклеа-
зы.
d. Опосредствуемое транспозоном
событие.
17.11 *. Какую роль гомологичная реком-
бинация играет в репликационной
транспозиции?
Вопросы открытого типа
17.1* . Какова роль рекомбинации в эволю-
ции генома?
17.2. Как может разрешение структуры
Холлидея приводить к двум различ-
ным результатам?
17.3* . Опишите, как модель с двунитевым
разрывом объясняет механизм со-
бытия генной конверсией.
а. Репликационная транспозиция
может происходить только между
гомологичными последователь-
ностями.
Ь. Белки, вовлеченные в гомологич-
ную рекомбинацию, требуются
для инициации репликационной
транспозиции.
с. После того, как транспозон был
реплицирован, свободная копия
его последовательности встраи-
вается в геном на новом участке
посредством гомологичной ре-
комбинации.
d. Репликация последовательности
транспозона превращает гибрид-
ную молекулу ДНК в коинтеграт,
который отцепляется посред-
ством гомологичной рекомбина-
ции.
17.12. Как клетки минимизируют по-
тенциально вредные воздействия
транспозиции?
а. Иммуноглобулины связываются
с кодируемыми транспозоном
белками.
Ь. Последовательноститранспозона
свертываются в плотноупакован-
ный хроматин.
с. Последовательности транспозо-
нов метилируются.
d. Белки транспозонов помечают-
ся убиквитином для деградации
в протеасомах.
17.4 . Некоторые штаммы Е coli, которые
используются для размножения ре-
комбинантных плазмид, содержат
мутации гена гесА. Почему дефекты
гена гесА могут быть полезными для
исследователей, работающих с ре-
комбинантными плазмидами?
710 Часть 4. Механизмы репликации и эволюции геномов
17.5 *. Исходя из того, что Е coli есть гапло-
идный организм, скажите: когда она
может иметь две гомологичные мо-
лекулы ДНК, которые могут участво-
вать в событиях рекомбинации?
17.6 . Какой из белков Е coli, вовлеченных
в гомологичную рекомбинацию,
имеет гомологов у дрожжей? Какой
белок Е coli, вовлеченный в этот про-
цесс, кажется, не имеет гомологов
у дрожжей?
Изыскательские задачи
17.1 *. Напишите подробный отчет о раз-
нообразных ролях белка RecA в био-
логии.
17.2 . Обсудите значение гомологичной
рекомбинации в биологии.
17.3 *. Определение структуры комплекса
RecBCD рассматривалось как ключе-
вой шаг в понимании молекулярной
основы гомологичной рекомбина-
ции. Объясните, почему знание
структуры этого комплекса было
настолько важно.
17.4. Система рекомбинации Сге лежит
в основе одного из наиболее спор-
ных моментов генной инженерии
растений — пресловутой ограни-
чительной технологии. Это один
из процессов, посредством которых
компании, которые продают гене-
тически модифицированные зерно-
вые культуры, пытаются защитить
Тесты по рисункам
17.1* . Что представлено на этом рисун-
ке — пример гомологичной реком-
бинации, сайт-специфической ре-
комбинации или транспозиции?
17.7* . Каковы свойства участков attPuattB,
которые опосредствуют встраива-
ние ДНК фага X в геном Е. coli?
17.8. Как новая копия ретроэлемента
встраивается в геном?
17.9* . Какова роль молекул тРНК в репли-
кации ретроэлементов?
17.10 . Приведите примеры вредных воз-
действий, которые транспозоны
могут оказывать на геном.
свои финансовые инвестиции, при-
нуждая фермеров покупать новые
семена каждый год, вместо того что-
бы просто собирать семя от урожая
и сеять это семя второго поколения
в следующем году. Ограничительная
технология основана на использова-
нии гена так называемого инакти-
вирующего рибосомы белка (НРБ).
Белок НРБ блокирует синтез белка,
разрезая одну из молекул рибосом-
ной РНК на два сегмента, а это озна-
чает, что любая клетка, в которой
белок НРБ будет активным, вскоре
умрет. На основании этой информа-
ции восстановите принцип работы
ограничительной технологии.
17.5* . Дайте подробный ответ на вопрос:
Как клетки уменьшают вредное воз-
действие транспозиции?
Короткая область гомологии
1
+
о
Глава 17. Рекомбинация 711
17.2. Что за событие представлено на 17.3*. На этом рисунке показано действие
этом рисунке и какие этапы про- цесса показаны? Двунитевой разрез 11111 i|imi 1111ГТПТ а а 1 П1111ЧЧ|||ШПГ 1Ш|,||1|1М1..ТТГТТ З'^б' 5' 1 гтт^**т^тттттт^^* 1 ГГТТГГГГТТТГГГТТГГ* 1 1_Ш 111 1 111 UJL11 f I ИНГ НИГТПТГП 1И rmiTH 1ГГТПТГТ I A iw xLH.. T 1 Г a A A I nnniiiiiimiinr A A белка в пути RecBCD у E. coli. Какой белок присоединяется к одноните- вой ДНК и опосредствует образова- ние D-петли? И '!• ^JIIIILLIIII1.1.11111111"11 3 3 5 •••! Т Г Г Г П1 Вторжение нити .......---АшиТПьД/ 17.4. На рисунке представлена часть пути транспозиции ретроэлемента. Об- судите этапы этого процесса, при- веденные на данном рисунке. 5 чдчУ, Встроенный 1 1 ретроэлемент ^Транскрипция R U5 из R г-н ____—РНК-копия 5' 3' R U5 U3 R U5 ▼ U3 R r7j5 из R U5 j RU5 1
712 Часть 4. Механизмы репликации и эволюции геномов
Дополнительная литература
Модели гомологичной рекомбинации
Eggleston, А. К. and West,S. С. (1996) Exchanging partners in E. coli. Trends Genet 12:
20-26.
Heyer, W. D., Ehmsen, К. T. and Solinger, J. A. (2003) Holliday junctions in the eukaryotic
nucleus: resolution in sight? Trends Biochem. Sci. 28: 548-557.
Holliday, R. (1964) A mechanism for gene conversion in fungi. Genet Res. 5: 282-304.
Kowalczykowski,S.C. (2000) Initiation of genetic recombination and recombination-
dependent replication. Trends Biochem. Sci. 25:156-165.
Meselson, M. and Radding, C.M.(1975)A general model for genetic recombination. Proc.
Natl Acad. Sci. USA 72: 358-361.
Shinagawa,H. and Iwasaki,H. (1996) Processing the Holliday junction in homologous
recombination. Trends Biochem. Sci. 21:107-111.
Молекулы, участвующие в гомологичной рекомбинации
Amundsen, S. К. and Smith, G.R. (2003) Interchangeable parts of the Escherichia coli
recombination machinery. Cell 112: 741-744. Hybrid pathways involving parts ofthe RecBCD
and RecF systems.
Baumann, P. and West,S.C. (1998) Role of the human RAD51 protein in homologous
recombination and double-stranded-break repair. Trends Biochem. Sci. 23: 247-251.
Masson, J.-Y. and West,S.C. (2001) The Rad51 and Dmcl recombinases: a non-identical
twin relationship. Trends Biochem. Sci. 26:131-136.
Pyle, A. M. (2004) Big engine finds small breaks. Nature 432:157-158. The structure of
the RecBCD complex.
Rafferty, J. B., Sedelnikova, S. E., Hargreaves, D., Artymiuk, P. J., Baker, P. J., Sharpies, G. J.,
Mahdi, A. A., Lloyd, R. G. and Rice, D. W. (1996) Crystal structure of DNA recombinations protein
RuvA and a model for its binding to the Holliday junction. Science 274: 415-421.
Symington, L.S. and Holloman, W.K. (2004) Resolving resolvases. Science 303:184-185.
Proteins for resolution of Holliday structures in eukaryotes.
West,S.C. (1997) Processing of recombination intermediate by the RuvABC proteins.
Annu. Rev. Genet 31: 213-244.
Сайт-специфическая рекомбинация
Kwon,H.)., Tirumalai, R., Landy,A. and Ellenberger,T. (1997) Flexibility in DNA
recombination: structure of the lambda integrase catalytic core. Science 276:126-131.
Транспозиция
Bushman, F. D. (2003) Targeting survival: integration site selection by retroviruses and
LTR-retrotransposons. Cell 115:135-138.
Shapiro,). A. (1979) Molecular model forthe transposing and replication ofbacteriophage
Mu and other transposabK elements. Proc. Natl Acad. Sci. USA 76:1933-1937.
Глава 18
Механизм эволюции геномов
По прочтении главы 18 вы сможете:
• Объяснять, исходя из чего биологи полагают, что первые геномы состояли из
РНК.
• Описывать события, которые предположительно привели к утверждению бел-
ковых катализаторов и геномов из ДНК первыми живыми клетками.
• Проводить различие между разными путями, которыми геномы могут приоб-
ретать новые гены.
• Приводить примеры, употребляя которые можно иллюстрировать роль дупли-
кации гена в эволюции мультигенных семейств.
• Обсуждать данные о дупликациях целых геномов в эволюционных историях
Saccharomyces cerevisiae, Arabidopsis thaliana и человека.
• Описывать воздействие, которое дупликация сегментов оказала на относительно
недавнюю эволюцию генома человека.
• Объяснять, каким образом новые гены могут возникнуть путем дупликации
домена и перетасовывания доменов.
• Оценивать вероятное воздействие горизонтального переноса генов на эволюцию
геномов бактерий и эукариотов.
• Кратко объяснять, как мобильные генетические элементы могли повлиять на
эволюцию генома.
• Раскрывать сущность гипотез «о ранних интронах» и «о поздних интронах»,
а также давать им обоснованную оценку.
• Перечислять различия между геномами человека и шимпанзе, а также рас-
суждать о том, в силу чего настолько подобные геномы могут дать начало столь
различным биологическим признакам.
Мутация и рекомбинация дают геному возможность эволюционировать, но мы
узнаем очень мало об эволюционных историях геномов, если будем просто изучать
эти события в живых клетках. Вместо этого, мы должны объединить наши знания
о мутации и рекомбинации со сравнениями между геномами разных организмов, чтобы
восстановить пути, по которым шла эволюция генома. Ясно, что такой подход является
неточным и неопределенным, но, как мы увидим, он опирается на удивительно большое
число достоверных данных, и мы можем иметь все основания для уверенности в том,
что по крайней мере в общих чертах картина, которая вырисовывается перед нами, не
слишком далека от истины.
В этой главе мы обсудим эволюцию геномов от самых истоков биохимических си-
стем и до сегодняшнего дня. Мы рассмотрим представления ученых о мире РНК, сущем
714 Часть 4. Механизмы репликации и эволюции геномов
до появления первых молекул ДНК, и затем посмотрим, как геномы из ДНК постепенно
становились все более и более сложными. Наконец, в разделе 18.4 мы сравним геном
человека с геномом шимпанзе, с тем чтобы установить эволюционные изменения, ко-
торые произошли за последние пять миллионов лет и которые, должно быть, так или
иначе придали нам наш теперешний облик.
18.1. Геномы: первые десять миллиардов лет
Космологи полагают, что Все-
ленная начала свое существование
около 14 миллиардов лет назад
с гигантского «первичного огнен-
ного шара», названного Большим
взрывом. Математические модели
предполагают, что по прошествии
приблизительно 4 миллиардов
лет из облаков газа, испускаемого
Большим взрывом, начали выде-
ляться галактики, и что в преде-
лах нашей собственной Галактики
так называемая околосолнечная
туманность уплотнилась и при-
мерно 4,6 миллиардов лет назад
из нее образовались Солнце и окру-
жающие его планеты (рис. 18.1).
Молодая Земля была покрыта во-
дой, и именно в этом безбрежном
мировом океане появились первые
биохимические системы, а ко вре-
мени, когда вздыбилась земная
Рис. 18.1. Происхождение Вселенной, галактик, Солнечной
системы и клеточной жизни
твердь и начали появляться конти-
нентальные массивы (приблизи-
тельно 3,5 миллиардов лет назад),
утвердилась и клеточная форма
жизни. Но клеточная жизнь была сравнительно поздним этапом биохимической эво-
люции, которому предшествовали самореплицирующиеся полинуклеотиды — праро-
дители первых геномов. С таких доклеточных систем мы и начнем постигать эволюцию
генома.
18.1.1. Происхождение геномов
По мнению ученых, солевой состав первых океанов был подобен составу океа-
нов, которые мы видим сегодня, но атмосфера Земли и, следовательно, растворенные
в океанах газы, были совершенно иными. Содержание кислорода в атмосфере остава-
лось очень низким до тех пор, пока в ходе эволюции не возник фотосинтез, и наиболее
изобильными газами были, вероятно, метан и аммиак. Эксперименты, пытающиеся
воссоздать условия в древней атмосфере, показали, что электрические разряды в смеси
метана с аммиаком вызывают химический синтез ряда аминокислот (таких как аланин,
глицин, валин и несколько других), входящих в состав белков. Также образуются циани-
Глава 18. Механизм эволюции геномов 715
стый водород (синильная кислота) и формальдегид. Они участвуют в дополнительных
реакциях, ведущих к синтезу других аминокислот, а также пуринов, пиримидинов и,
в меньшем количестве, сахаров. Таким образом, в древней хемосфере могли накапли-
ваться по крайней мере некоторые из составных частей биомолекул.
Первоначальные биохимические системы были основаны на РНК
Полимеризация стандартных кирпичиков в биомолекулы, возможно, произошла
в океанах, или, быть может, ей могли способствовать неоднократная конденсация и ис-
парение капелек воды в облаках. Альтернативно, полимеризация могла иметь место на
твердых поверхностях (возможно, между мономерами, осевшими на частицах глины)
или в гидротермальных источниках. Точный механизм полимеризации не должен нас
занимать: важным представляется тот факт, что можно представить себе искл ючительно
геохимические процессы, которые могли привести к синтезу полимерных биомолекул,
подобных тем, которые мы находим в живых системах. Вот о следующих этапах эволюции
нам и необходимо поразмыслить. Мы должны перейти от хаотичной совокупности био-
молекул к упорядоченному ансамблю, который показывает по крайней мере некоторые
из биохимических свойств, которые мы связываем с жизнью. Эти шаги никогда еще не
были воспроизведены экспериментально, и наши представления поэтому основаны,
главным образом, на умозрительном построении, разбавленном в некоторой степени
данными компьютерного моделирования. Важно отметить, что пищей для таких пред-
положений служит необузданное число возможных сценариев — мировой океан, воз-
можно, содержал вплоть до 1О10 биомолекул на литр, и в распоряжении гипотетичного
наблюдателя имеется миллиард летдля протекания необходимых событий. Это означает,
что даже наиболее неправдоподобные сценарии не могут быть сброшены со счетов.
Продвижение в понимании происхождения жизни первоначально наталкивалось на
очевидно неумолимые требования, согласно которым полинуклеотиды и полипептиды
должны работать в одной упряжке, чтобы произвести на свет самовоспроизводящуюся
биохимическую систему. Это связано с тем, что для катализа биохимических реакций
необходимы белки, но они не могут осуществлять свою собственную саморепликацию.
Полинуклеотиды, в принципе, могут определять синтез белков и самореплицироваться,
но в то время думали, что они не смогут сделать ни то, ни другое без помощи белков.
Казалось, что биохимическая система должна была возникнуть из неупорядоченной
совокупности биомолекул уже полностью сформированной, потому что любая про-
межуточная стадия не могла бы поддерживать свое существование. Мощный прорыв
через эту преграду был совершен в середине 1980-х гг., когда было открыто, что РНК
может обладать каталитическим действием.
Те рибозимы, которые мы знаем сегодня, выполняют биохимические реакции
трех типов:
• саморасщепление, какое показывают самовырезающиеся интроны групп I, II и III,
а также геномы некоторых вирусов (таблица 12.4 и раздел 12.2.4);
• расщепление других РНК, что выполняет, например, РНКаза Р (таблица 12.4 и раз-
дел 12.1.3);
• синтез пептидных связей, выполняемый рРНКовым компонентом рибосомы (раз-
дел 13.2.3).
В пробирке, как было показано, молекулы синтетической РНК выполняют и дру-
гие биологически значимые реакции, такие как синтез рибонуклеотидов, синтез и ко-
пирование молекул РНК, а также перенос связанной с РНК аминокислоты на вторую
716 Часть 4. Механизмы репликации и эволюции геномов
аминокислоту с образованием дипептида способом, аналогичным роли тРНК в синтезе
белка. Открытие этих каталитических свойств разрешило дилемму полинуклеотид-
полипептид, показав, что первые биохимические системы вполне могли быть всецело
основаны на РНК.
В последние годы представления о мире РНК начали принимать определенные
очертания. Теперь мы представляем себе, что молекулы РНК в начале реплицирова-
лись медленным и совершенно случайным способом, попросту служа матрицами для
связывания комплементарных нуклеотидов, которые полимеризировались самопроиз-
вольно (рис. 18.2). Такой процесс репликации должен был быть очень неточен, так что
производилось целое многообразие последовательностей РНК, в конечном счете давая
одну или несколько с зачаточными свойствами рибозима, которые были способны на-
правлять свою собственную, более точную саморепликацию. Возможно, что некоторая
форма естественного отбора проявляла свое действие, так что наиболее эффективные
реплицирующиеся системы начали господствовать, что, как было показано, происходит
в модельных системах. Большая точность репликации позволила РНК увеличиваться
в длине без потери своей специфики последовательности, дала потенциал для более
замысловатых каталитических свойств и, возможно, достигла своей наивысшей точки
в таких сложных структурах, как современные интроны группы I (см. рис. 12.39) и ри-
босомные РНК (см. рис. 13.11).
Называть ранние РНК «геномами» было бы в какой-то мере нелепо, но термин
протогеном имеет привлекательные стороны как отражение качеств молекул, ко-
торые самореплицировались и были способны направлять простые биохимические
реакции. В число таких реакций мог
входить, скажем, энергетический
обмен, основанный, как и сегод-
ня, на высвобождении свободной
энергии путем гидролиза фосфат-
фосфатных связей в рибонуклеоти-
дах АТФ и ГТФ, и со временем эти ре-
акции, возможно, стали протекать
в полостях, окруженных липидны-
ми мембранами, то есть постепен-
но сформировались первые струк-
туры, подобные клеточным. Есть
трудности в понимании того, как
длинноцепные, неразветвленные
липиды могли образоваться в ходе
реакций, катализируемых химиче-
Рис. 18.2. Копирование молекул РНК
в раннем мире РНК. До появления
РНК-полимераз в ходе эволюции ри-
бонуклеотиды, которые объединялись
с РНК-матрицей, должны были полимери-
зоваться самопроизвольно. Этот процесс
был неточен и производилось множество
различных последовательностей РНК
Глава 18. Механизм эволюции геномов 717
ски или рибозимом, однако, наличествуя в достаточных количествах, они должны были
самопроизвольно собираться в мембраны, возможно, охватывая один или несколько
протогеномов и обеспечивая РНК замкнутой средой, в которой могли быть выполняемы
более управляемые биохимические реакции.
Первые геномы на основе ДНК
Каким образом мир РНК развивался в мир ДНК? Первым определяющим измене-
нием было, вероятно, развитие белковых ферментов, которые дополнили и в конечном
счете заменили большинство каталитических действий рибозимов. Есть несколько
оставшихся без ответа вопросов, касающихся этой стадии биохимической эволюции,
к которым относится и причина, по которой прежде всего произошел переход от РНК
к белку. Изначально предполагалось, что 20 стандартных аминокислот в полипептидах
придали белкам большую химическую изменчивость, чем четыре рибонуклеотида в РНК,
что позволило белковым ферментам катализировать более широкий спектр биохими-
ческих реакций, но это объяснение становилось все менее привлекательным по мере
того, как все большее число катализируемых рибозимами реакций демонстрировалось
в пробирке. Более позднее предположение — что опосредствуемый белком катализ
более эффективен в силу свойственной им гибкости свернутых полипептидов по срав-
нению с большей жесткостью спаренных основаниями молекул РНК. Альтернативно,
обособление РНК-протогеномов в окруженные мембранами пузырьки, возможно, под-
толкнуло эволюцию первых белков, потому что молекулы РНК гидрофильны и должны
быть снабжены гидрофобной оболочкой (например, путем прикрепления к молекулам
пептида) прежде чем они смогут пройти сквозь мембрану или встроиться в нее.
Переход к опосредствованному белком катализу потребовал радикального из-
менения функции РНКовых протогеномов. Вместо того чтобы быть напрямую ответ-
ственными за биохимические реакции, происходящие в первых подобных клеточным
структурах, протогеномы стали кодирующими молекулами, главная функция которых
заключалась в руководстве построением каталитических белков. Сами ли рибозимы
стали кодирующими молекулами или же кодирующие молекулы синтезировались
рибозимами, не известно, хотя самые убедительные теории о началах синтеза белка
и генетического кода предполагают, что последняя альтернатива более правдоподобна
(рис. 18.3). Каким бы ни был сам механизм такого перевоплощения, результатом явилась
парадоксальная ситуация, когда РНКовые протогеномы сложил и с себя полномочия фер-
ментов, с обязанностями которых они блестяще справлялись, и приняли на себя функцию
кодирования, для которой они менее подходили ввиду относительной неустойчивости
фосфодиэфирных связей в РНК (раздел 1.2.1). Передача функции кодирования к более
устойчивой ДНК кажется почти неизбежной, и ее нетрудно было осуществить — вос-
становлением рибонуклеотидов до дезоксирибонуклеотидов, которые могли за этим
полимеризоваться в копии РНКовых протогеномов посредством катализируемой об-
ратной транскриптазой реакции (рис. 18.4). Замена урацила его метилированным про-
изводным тимином, вероятно, придала еще большую стабильность полинуклеотиду
ДНК, а принятию двунитевой ДНК в качестве кодирующей молекулы почти наверняка
способствовала возможность репарации повреждений ДНК путем копирования нити,
комплементарной ущербной (разделы 16.2.2 и 16.2.3).
Согласно этому сценарию первые ДНКовые геномы содержали много отдельных
молекул, каждая из которых определяла один-единственный белок и поэтому была
эквивалентна отдельному гену. Соединение воедино этих генов в первые хромосомы,
что могло начаться до перехода к ДНК, повысило эффективность распределения генов
718 Часть 4. Механизмы репликации и эволюции геномов
а) Рибозим, который является также
и кодирующей молекулой
Аминокислоты
Рис. 18.3. Два сценария эволюции первой кодирующей РНК. Рибозим, возможно, а) эволюционировал
так, чтобы иметь двойную функцию — каталитическую и кодирующую, или б) рибозим, возможно,
синтезировал кодирующую молекулу. В обоих примерах аминокислоты показаны прикрепленными
к кодирующей молекуле через маленькие сопрягающие РНК — предполагаемые прародители сегод-
няшних тРНК
б) Рибозим, который синтезирует
кодирующие молекулы
во время деления клетки, поскольку гораздо проще организовать равномерное рас-
пределение нескольких больших хромосом, чем множества разрозненных генов. Как и в
большинстве этапов эволюции раннего генома, было предложено несколько различных
механизмов, которыми гены могли стать связанными в хромосомы.
Насколько жизнь уникальна?
Если экспериментальное моделирование и компьютерные модели верны, то весьма
правдоподобно, что начальные стадии биохимической эволюции многократно про-
Рибонуклеотиды Дезоксирибонуклеотиды
< Восстановление
---------------►
Обратная
транскрипция
Кодирующая РНК
Первая молекула
кодирующей ДНК
исходили параллельно в океанах
или в атмосфере молодой Земли.
Поэтому весьма возможно, что
«жизнь» в свое время возникла
не в одном, а сразу в нескольких
очагах, хотя все современные ор-
ганизмы, кажется, происходят от
единого первоистока. На такое
общее происхождение указывает
поразительное подобие между
основными механизмами моле-
кулярной биологии и биохимии
в клетках бактерий, архей и эука-
Рис. 18.4. Преобразование молекулы ко-
дирующей РНК в прародителя первого
генома из ДНК
Глава 18. Механизм эволюции геномов 719
NH
Рис. 18.5. Короткий отрезок пептидной нуклеиновой кислоты.
Пептидная нуклеиновая кислота имеет амидную основную
цепь вместо сахаро-фосфатной структуры, обнаруживаемой
у стандартной нуклеиновой кислоты
риотов. Взять только один пример, кажется, нет
никакой очевидной биологической или химической
причины, по которой каждый из известных три-
плетов нуклеотидов должен кодировать какую-то
определенную аминокислоту, но генетический код
хотя и не универсален, является фактически одним
и тем же во всех организмах, которые были изучены
к настоящему времени. Если бы все эти организмы
произошли из двух и более источников, то мы бы
наблюдалие соответственно два и более сильно от-
личающихся кода.
Если возможны многочисленные источники,
но современная жизнь ведет свое начало только из
одного такого истока, то на каком этапе одна биохи-
мическая система начала преобладать над другими?
На этот вопрос нельзя ответить точно, но наиболее
вероятный сценарий — то, что преобладающая система была первой, которая развила
средства синтеза белковых ферментов и поэтому, вероятно, была также и первой, при-
нявшей геном из ДНК. Больший каталитический потенциал и более точная репликация,
сообщенные белковыми ферментами и ДНКовыми геномами, наверное, придали этим
клеткам значительное преимущество по сравнению с теми, которые все еще довольство-
вались РНКовыми протогеномами. ДНК-РНК-белковые клетки размножались быстрее,
что дало им возможность вытеснить РНКовые клетки в борьбе за пищевые ресурсы.
Вскоре и сами РНКовые клетки стали таким пищевым ресурсом.
Возможны ли формы жизни, основанные на каких-либо иных информационных
молекулах, кроме ДНК и РНК? Вполне возможно, что в самый ранний период биохими-
ческой эволюции молекулам РНК предшествовала какая-либо иная информационная
молекула. В частности, пиранозильная версия РНК, в которой сахар принимает немного
иную структуру, могла бы быть лучшим выбором для раннего прото генома, чем нормаль-
ная РНК, потому что спаренные основаниями молекулы, которые она образует, более
устойчивы. То же самое верно и для пептидной нуклеиновой кислоты (ПНК) — аналога
полинуклеотида, в котором сахаро-фосфатная основная цепь заменена амидными свя-
зями (рис. 18.5). Молекулы ПНК были синтезированы в пробирке, и было показано, что
она образует пары оснований с нормальными полинуклеотидами. Однако нет никаких
признаков того, что в процессе эволюции в первичном бульоне вероятность возникно-
вения пиранозильной РНК или ПНК была больше, чем вероятность появления РНК.
18.2. Приобретение новых генов
Хотя древние отпечатки на окаменелостях и трудно интерпретировать, есть впол-
не убедительные данные, что ко времени 3,5 миллиардов лет назад биохимические
системы эволюционировали в клетки, подобные по облику современным бактериям.
720 Часть 4. Механизмы репликации и эволюции геномов
Рис. 18.6. Эволюция жизни
Мы не можем прочитать в летописи ока-
менелостей, какого рода геномы имели
эти первые настоящие клеткиЧ но из рас-
суждений, коими полнится предыдущий
раздел, мы можем вывести, что они были
построены из двунитевой ДНК и состоя-
ли из небольшого количества хромосом,
возможно, лишь из одной, каждая из ко-
торых содержала множество сцепленных
генов.
Если мы проследуем за летописью
окаменелостей вперед по времени, то
мы увидим первые данные о ядерных
клетках — структурах, напоминаю-
щих одноклеточные водоросли, — око-
ло 1,4 миллиарда лет назад (рис. 18.6),
а о многоклеточных водорослях — при-
близительно 0,9 миллиардов лет назад.
Многоклеточные животные появились
приблизительно 640 миллионов лет на-
зад, хотя есть загадочные норы и таинственные ходы в породе, предполагающие, что
животные жили еще до этого времени. Кембрийская революция, когда бесхребетная
жизнь произвела на свет многие новые формы одушевленного бытия, произошла 530
миллионов лет назад и завершилась исчезновением многих из этих новых форм в мас-
совом вымирании, случившемся 500 миллионов лет назад. С тех пор эволюция шла
спорым ходом и сопровождалась увеличивающимся разнообразием: первые наземные
насекомые, животные и растения утвердились на суше 350 миллионов лет назад, ди-
нозавры жили и ушли в небытие к концу мелового периода (65 миллионов лет назад),
а первые гоминиды появились всего лишь 4,5 миллиона лет назад.
Морфологическая эволюция шла рука об руку с эволюцией генома. Опасно при-
равнивать эволюцию с «прогрессом», но бесспорно, что, когда мы восходим по эволюци-
онному дереву, мы видим все более и более сложные геномы. Один из признаков такой
сложности — число генов, которое изменяется от менее чем 1000 у некоторых бактерий
до 30000-40000 у позвоночных животных, таких, как например, человек. В пределах
отдельных ветвей, например, в мире бактерий, изменения в числе генов, вероятно,
были постепенными, когда приобретение новых генов уравновешивалось, по крайней
мере отчасти, потерей имевшихся. Мы должны помнить также и то, что эволюционные
пути некоторых организмов проходили с уменьшением, а не с увеличением числа генов
и привели к минимальным геномам, наблюдаемым у Mycoplasma и других паразитиче-
ских видов (раздел 8.2.2). Постепенные изменения в пределах отдельных ветвей были,
однако, прерваны по крайней мере двумя переходными периодами, когда появились
новые организмы со значительно возросшим числом генов. Один из таких переходов
сопровождал новорождение первых эукариотов приблизительно 1,4 миллиарда лет
Мы не можем даже определить, были это ДНК-белковые или РНК-клетки. — Прим. ред.
Глава 18. Механизм эволюции геномов 721
назад — эти клетки, вероятно, содержали по крайней мере 10 000 генов (минимум у со-
временных эукариотов) по сравнению с типичным для прокариотов числом 5000 или
меньше того. Второй переход был связан с появлением на свет первых позвоночных
животных вскоре после конца кембрийского периода — они, подобно современным
позвоночным, вероятно, имели по крайней мере 30000 генов.
Существует два в корне различных пути, которыми новые гены могут быть при-
обретены геномом:
• дупликация некоторых или всех имеющихся в геноме генов;
• приобретение генов от других видов.
Как мы увидим в следующих двух разделах, события обоих типов имели большое
значение в эволюции генома.
18.2.1. Приобретение новых генов в ходе событий дупликации
Предположение об определяющей роли дупликации генов в эволюции генома
впервые зародилось в 1970 г. Первичным результатом дупликации гена будут два
идентичных гена. Давление отбора обусловит, что один из этих генов сохранит свою
исходную последовательность нуклеотидов, по крайней мере весьма подобную ей, так
что он будет способен продолжать кодировать белок с функцией, которая выполнялась
в клетке благодаря единственной копии этого гена, прежде чем его дупликация имела
место. Возможно, что те же самые ограничения естественного отбора подействуют и на
второй ген, особенно если увеличение в скорости синтеза продукта этого гена, ставшее
возможным благодаря его дупликации, дает преимущества организму (рис. 18.7). Однако
чаще всего вторая копия не дает никакой выгоды организму и, следовательно, не будет
подвергнута тем же самым воздействиям естественного отбора и, таким образом, будет
накапливать случайные мутации. Данные показывают, что большинство новых генов,
возникающих путем дупликации, приобретают пагубные мутации, которые инактиви-
руют их, так что они становятся псевдогенами; при этом анализ существующих псевдо-
генов наводит на мысль о том, что наиболее распространенными инактивирующими
мутациями являются сдвиги рамки считывания и мутации с потерей смысла, которые
происходят в пределах кодирующей области гена. Иногда, однако, мутации могут не
приводить к инактивации гена, а вместо этого придать ему новую функцию, которая
будет полезной для организма (см. рис. 18.7).
| Дубликация гена
Давление естественного
отбора на оба гена
Давление естественного отбора
лишь на один из генов
Гены остаются Одна из копий деградирует Одна из копий приобретает
одинаковыми новую функцию
Рис. 18.7. Три сценария получения исхода дупликации гена
722 Часть 4. Механизмы репликации и эволюции геномов
Сначала мы рассмотрим данные о дупликациях гена, произошедших в прошлом
и оставивших следы, которые содержатся в современных последовательностях гено-
мов, а затем взглянем на механизмы, под действием которых дупликация гена может
произойти.
Последовательности геномов предоставляют исчерпывающие данные
о прошлых событиях дупликации генов
Самый поверхностный взгляд на последовательность генома обнаруживает впол-
не убедительные данные, говорящие о том, что многие гены возникли в ходе событий
дупликации. Важное значение первого сценария, иллюстрированного на рис. 18.7, где
увеличенное количество продукта гена, следующее из дупликации этого гена, выгодно
и упрочивает данную дупликацию, подтверждается многими примерами многогенных
семейств, состоящих из генов с идентичными или почти идентичными последователь-
ностями. Главные примеры — гены рРНК, число копий которых изменяется от двух
у Mycoplasma genitalium до 500 и более у Xenopus laevis, при этом все копии имеют фак-
тически одну и ту же последовательность. Такие множественные копии идентичных
генов предположительно отражают потребность в быстром синтезе молекул рРНК на
определенных этапах клеточного цикла. Обратите внимание, что существование таких
многогенных семейств указывает не только на то, что дупликации гена произошли
в прошлом, но также и на то, что должен существовать некий молекулярный механизм,
который гарантировал бы, что члены одного семейства сохранят свое лицо за время
эволюции. Это называют согласованной эволюцией. Если одна копия семейства при-
обретает выгодную мутацию, то возможно, что эта мутация распространится по всему
семейству, пока все члены не станут ее обладателями. Наиболее вероятный путь, кото-
рым это может быть достигнуто, — генная конверсия, в результате которого, как было
описано в разделе 17.1.1, последовательность одной копии гена может быть заменена
всей последовательностью (или частью ее) его второй копии. Многократные события
генной конверсии могут поэтому поддерживать идентичность среди последовательно-
стей отдельных членов многогенного семейства, особенно если эти члены организованы
в тандемные массивы.
Согласно третьему сценарию на рис. 18.7 дуплицированный ген накапливает мута-
ции, которые придают ему новую, полезную функцию. И снова мультигенные семейства
обнаруживают многочисленные признаки того, что такие события часто происходили
в прошлом. Мы уже видели, что дупликация гена в семействах генов глобина привела
к эволюции новых глобиновых белков, которые используются организмом на разных
этапах его развития (см. рис. 7.19). Мы отметили, что все гены глобинов, и а-, и 0-типа,
имеют родственные последовательности нуклеотидов и, следовательно, составляют
часть надсемейства, в которое входят гены, определяющие разные другие белки, кото-
рые, подобно глобинам крови, имеют способность связывать молекулы кислорода. По
степени подобия, наличествующего в парах генов в надсемействе, возможно вывести
схему дупликаций гена, которая привела к появлению генов, что мы видим сегодня,
а применяя к этим данным молекулярные часы (раздел 19.2.2), мы можем оценить,
сколько миллионов лет назад каждая такая дупликация имела место. Эти исследования
сообщают нам, что дупликация приблизительно 800 миллионов лет назад дала пару
предковых генов, один из которых эволюционировал в современный ген мозгового
белка нейроглобина, а другой дал начало всем остальным членам этого надсемейства
(рис. 18.8). Приблизительно 250 миллионов лет спустя произошла вторая дупликация
на пути, ведущем к глобинам крови, и одним из продуктов этой дупликации явился ген,
Глава 18. Механизм эволюции геномов 723
Рис. 18.8. Эволюция надсемейства генов глобинов человека. Члены этого надсемейства находятся теперь
на разных хромосомах. Ген нейроглобина находится на хромосоме 14, ген цитоглобина находится на
хромосоме 17, а ген миоглобина находится на хромосоме 22. Группа а-глобина находится на хромосо-
ме 16, а группа 0-глобина находится на хромосоме 11. Сокращение: млн — миллионов лет назад
который через очередную дупликацию дал начало миоглобину, проявляющему свою
активность в мышцах, и цитоглобину, присутствующему во многих тканях, но все еще
таящему от ученых свою функцию. Линии родословной прото-а и прото-p разошлись
дупликацией, которая произошла 450 миллионов лет назад, а дупликация в пределах
семейств генов а- и р-глобина имела место в течение последних 200 миллионов лет.
В пределах этого периода времени возможно вывести не только схему дупликации генов,
но также и некоторые из более подробных изменений, которые произошли в отдельных
генах. Так, были выведены события, ведущие к различным группам генов р-глобина,
присутствующим в геномах различных млекопитающих (рис. 18.9).
Мы наблюдаем подобные модели эволюции и при сравнении последовательностей
других генов. Гены трипсина и химотрипсина, например, связаны общим предковым
геном, который дублировался приблизительно 1500 миллионов лет назад. Оба теперь
кодируют протеазы, участвующие в разложении белка в пищеварительном тракте по-
звоночных: трипсин разрезает другие белки в позициях аминокислот аргинина и лизина,
а химотрипсин рассекает их по фенилаланинам, триптофанам и тирозинам. Эволюция
генома, таким образом, произвела две дополняющие друг друга функции белка там, где
первоначально была лишь одна.
Другой поразительный пример эволюции генов путем дупликации показыва-
ют гомеозисные гены — основные гены, связанные с развитием и ответственные за
задание общего строения организма животных. Как было описано в разделе 14.3.4,
Drosophila имеет единственную группу гомеозисных генов, названную НОМ-С, которая
состоит из восьми генов, каждый из которых содержит последовательность гомео-
домена, кодирующую ДНК-связывающий мотив в белковом продукте (см. рис. 14.37).
Эти восемь генов, равно как и другие гены гомеодоменов у Drosophila, как полагают,
возникли чередой дупликации генов, которая началась с некоторого предкового гена,
724 Часть 4. Механизмы репликации и эволюции геномов
Предок-
первозверь
Прото-Е П рото-3
Сумчатые
Плацентарный ,
пред™
Потеря г)
Потеря
преобразование 6 в 3;
инактивация 6
Дублирование
5 т и инактивация £
" ~ vb , 3 vb , /Г,
Превращение ^ДублированиеСлияние Дублирование
-4
еУ трЗЬО 3hi vi3h2i$h3 01 02
Мышь
Преобразование
6вЗ;
инактивация rj;
дублирование
Потеря tj;
преобразование
6 в 3;
инактивация fc
е Ч vb
Кролик
е G.
6 3
Человек
3
Опоссум
Утроение
Коза
Рис. 18.9. Эволюция генов р-глобина млекопитающих. Перепечатано из Genomics, Vol. 13, Tagle et al.,
'The p-globin gene cluster..., 741-760,1992, с разрешения издательства Elsevier
который существовал приблизительно 1 000 миллионов лет назад. Функции современ-
ных генов, каждый из которых определяет тип отдельного сегмента плодовой мушки,
служат нам манящим проблеском, высвечивающим картину событий дупликации ге-
Группа Нох
Дупликация Муха
Амфиокс
| Дупликация
Позвоночные
Дупликация,
▼ потеря одной группы
Лучеперые
рыбы
нов и расхождения последовательностей, которые
могли в данном случае быть основными процессами,
ответственными за увеличение морфологической
сложности ряда организмов в эволюционном дереве
Drosophila. Если мы затем поднимаемся далее вверх
по эволюционному древу, то видим, что позвоночные
имеют четыре группы генов Нох (см. рис. 14.37), каж-
дая из которых — узнаваемая копия принадлежащей
Drosophila группы, с подобными последовательно-
стями генов, лежащих в эквивалентных позициях.
Из этого следует, что в ветви позвоночных было две
дупликации, причем не отдельных генов Нох, а целой
группы (рис. 18.10). Не все гены Нох позвоночных
были наделены функциями, но мы полагаем, что до-
полнительные версии, имеющиеся у позвоночных, так
или иначе связаны с возросшей сложностью общего
строения организма позвоночных. Два наблюдения
подтверждают это заключение. Амфиокс, беспозво-
ночное, которое показывает некоторые примитивные
Рис. 18.10. Эволюция группы генов Нох от мух до лучеперых рыб
Глава 18. Механизм эволюции геномов 725
особенности позвоночных, имеет две группы Нох, что мы могли бы ожидать для при-
митивного «протопозвоночного». Лучеперые рыбы, возможно, самая разнообразная
группа позвоночных с обширным диапазоном различных разновидностей основного
общего строения организма, — имеют семь групп Нох.
К дупликации генов могли привести самые разнообразные процессы
Есть несколько способов, посредством которых короткий сегмент молекулы ДНК,
возможно, содержащий один ген или же их малочисленную группу, мог быть дуплици-
рован. К ним относятся:
• неравный кроссинговер, представляющий собой событие рекомбинации, которое
запускается подобными последовательностями нуклеотидов, расположенными не
в идентичных местах в паре гомологичных хромосом. Как показано на рис. 18.11, а,
результатом неравного кроссинговера может быть дупликация некоторого сегмен-
та ДНК в одном из продуктов рекомбинации;
• неравный обмен сестринскими хроматидами, который происходит тем же самым
механизмом, что и неравный кроссинговер, но вовлекает пару хроматид отдельно
взятой хромосомы (рис. 18.11, б);
• размножение ДНК, которое иногда используется в данном контексте для опи-
сания дупликации сегментов ДНК у бактерий и других гаплоидных организмов,
где дупликации возникают посредством неравной рекомбинации между двумя
дочерними молекулами ДНК в репликационном пузырьке (рис. 18.11, в);
а) Неравный кроссинговер
б) Неравный обмен сестринскими хроматидами
Пара
Повторные гомологичных
последовательности хромосом
в) В процессе репликации ДНК
Репликационная вилка Репликационная вилка
Рис. 18.11. Модели дупликации гена: а) неравным кроссинговером между гомологичными хромосо-
мами, б) неравным обменом сестринских хроматид и в) в ходе репликации бактериального генома.
В каждом случае рекомбинация происходит между двумя различными копиями короткого повтора,
ведя к дупликации последовательности между повторениями. Неравный кроссинговер и неравный
обмен сестринскими хроматидами по существу одно и то же, за исключением того, что первый вовле-
кает хроматиды из пары гомологичных хромосом, а второй вовлекает хроматиды из одной хромосомы.
В случае в) рекомбинация происходит между двумя дочерними двойными спиралями, которые только
что были синтезированы репликацией ДНК
726 Часть 4. Механизмы репликации и эволюции геномов
• проскальзывание репликации (см. рис. 16.5), которое может привести к дупли-
кации коротких сегментов типа микросателлитных последовательностей. Этот
процесс, в принципе, может дуплицировать область, достаточно большую, чтобы
содержать полный ген, но на практике это маловероятно.
Каждый из четырех вышеупомянутых процессов обусловливает тандемные ду-
пликации — такие, в которых два дублируемых сегмента лежат в геноме смежно друг
с другом. Такая схема прослеживается во многих многогенных семействах, таких как
семейство генов а-глобина на хромосоме человека 16 и семейство р-глобина на хромосо-
ме 11, но это не единственная возможность. Члены семейства не всегда сорасположены,
например, в геноме человека есть три функциональных гена для кодирования метабо-
лического фермента альдолазы и при этом каждый на своей хромосоме. Эти копии, воз-
можно, когда-то находились в виде тандемного множества и стали рассеянными в ходе
крупномасштабных реорганизаций генома, но также возможно, что такое отдаленное
расположение является последствием процесса дупликации. Это имело бы место, если
бы дупликация произошла путем ретротранспозиции в манере, подобной той, что, как
думают, привела к формированию процессированных псевдогенов (см. рис. 7.20). Обра-
ботанный (процессированный) псевдоген возникает, когда мРНК-копия некоторого гена
превращается в кДНК и повторно встраивается в геном. Получающаяся структура явля-
ется псевдогеном, потому что в нем отсутствует промоторная последовательность (ведь
ее не было в мРНК!). Такой псевдоген мог, очевидно, быть встроен смежно с промотором
какого-либо находящегося там гена и, следовательно, стать активным, порабощая этого
промотора для своих собственных нужд. Дубликаты гена, которые появляются таким
способом, называют ретрогенами, и опять же много примеров известно в различных
геномах (в геноме человека специфическая для семенников версия гена пируватдеги-
дрогеназы являет собой ретроген). Отличительной особенностью ретрогена является
то, что в нем нет ни одного интрона из имевшихся в родительской копии этого гена,
поскольку они отсутствуют в мРНК. Недавно стало понятно, что полные копии генов,
включающие не только интроны, но и некоторые (или даже все) промоторные после-
довательности, могут быть произведены также и посредством обратной транскрипции.
Это может произойти, когда РНК, которая обратно транскрибируется, представлена не
мРНК, но антисмысловой копией этого гена, полученной путем транскрипции «невер-
ного» полинуклеотида (рис. 18.12). Мы начинаем понимать, что антисмысловые РНК не
являются чем-то необыкновенным и могут действительно играть определенную роль
Промотор—» Промотор
5' 3' 3' 5'
I Со сплайсингом 1 Без сплайсинга
Рис. 18.12. Представленная антисмысловой РНК копия гена сохранит интроны этого гена. Слева ген
транскрибируется начиная от его стандартного промотора и дает пре-мРНК, интрон которой удаляется
входе сплайсинга. Справа антисмысловая РНК синтезируется начиная от нижележащего промотора. Эта
РНК не сплайсируется, потому что последовательности на ее экзон-интронных границах не относятся
к стандартным, опознаваемым сплайсосомой (раздел 12.2.2)
Глава 18. Механизм эволюции геномов 727
в регулировании гена — подобную, быть может, способу действия молекул микроРНК
(раздел 12.2.6). Будучи превращена в кДНК, антисмысловая РНК может обеспечить
полномерный, функционально активный дубликат гена для встройки в геном, причем
в позиции, которая может быть удалена от позиции исходной копии этого гена.
Дупликация целого генома тоже возможна
Процессы, описанные выше, обусловливают протекание дупликаций относительно
коротких отрезков ДНК, возможно, несколько десятков тыс. п. н. в длину. Возможны ли
более масштабные дупликации? Кажется маловероятным, что дуплицирование полных
хромосом играло какую-либо значительную роль в эволюции генома, потому что мы
знаем, что дупликация отдельных хромосом человека, приводящая к появлению клетки,
которая содержит три копии одной хромосомы и по две копии всех остальных (состоя-
ние, названное трисомией), или является смертельным, или вызывает генетическую
болезнь, такую как синдром Дауна, и подобные вредные воздействия наблюдались
в искусственно созданных трисомических мутантах Drosophila. Вероятно, получающее-
ся увеличение в числе копий некоторых генов, при сохранении данной величины для
остальных неизменной, ведет к нарушению равновесия продуктов этих генов и к на-
рушению биохимии клетки.
Пагубные воздействия трисомии не означают, что дублирование полного набора
хромосом в ядре как таковое следует считать чем-то порочным. Дупликация генома
может произойти, если ошибка в процессе мейоза ведет к производству гамет, которые
являются диплоидными, а не гаплоидными (рис. 18.13). Если две диплоидные гаметы
слияются, то результатом будет клетка автополиплоидного, в данном случае тетрапло-
идного типа, ядро которой содержит по четыре копии каждой хромосомы. Автополи-
плоидия, как и полиплоидия других типов (раздел 18.2.2), нередкое явление, особенно
среди растений. Автопол иплоиды часто жизнеспособны, потому что каждая хромосома
Рис. 18.13. Основа порождения автополиплоидии. Слева в схематичной форме показаны нормальные
события, происходящие в процессе мейоза (ср. с рис. 3.16). Справа на этапе между профазой I и про-
фазой II произошло отклонение и пары гомологичных хромосом не разошлись по разным ядрам. По-
лучившиеся гаметы будут диплоидными, а не гаплоидными
728 Часть 4. Механизмы репликации и эволюции геномов
Триплоидная клетка
ПРОФАЗА I
Две гомологичные
хромосомы образуют
бивалент
Третья копия
Рис. 18.14. Автополиплоиды не могут успешно
скрещиваться со своими родителями. Слияние
диплоидной гаметы, произведенной нарушенным
мейозом, показанным на рис. 18.13, с гаплоидной
гаметой, произведенной нормальным мейозом,
ведет к триплоидному ядру, то есть такому, которое
имеет по три копии каждой гомологичной хромо-
сомы. Во время профазы I следующего мейоза две
из этих гомологичных хромосом сформируют бива-
лент, но третья не найдет себе партнера. Это имеет
разрушающее действие на расхождение хромосом
во время анафазы (см. рис. 3.16) и обычно не по-
зволяет мейозу достигать успешного завершения.
Это означает, что гаметы не производятся и трипло-
идный организм бесплоден. Обратите внимание,
что бивалент может образоваться между любыми
двумя из трех гомологичных хромосом, а не только
между парой, показанной на этом рисунке
не нашла гомолога
по-прежнему имеет гомологичного партнера и, таким образом, может образовывать
бивалент во время мейоза. Это позволяет автополиплоиду успешно воспроизводиться,
но, как правило, препятствует внутривидовому скрещиванию с исходным организмом,
от которого он был получен. Это связано с тем, что скрещивание между, например,
тетраплоидом и диплоидом дало бы триплоидного потомка, который сам был бы не-
способен воспроизводиться, потому что один полный набор его хромосом не нашел бы
себе гомологичных партнеров (рис. 18.14). Поэтому автополиплоидия служит меха-
низмом, с помощью которого может происходить видообразование, то есть появление
пары видов, обычно определяемых как организмы, которые не способны скрещивать-
ся. Порождение новых видов растений посредством автополиплоидии давно знакомо
биологам. Особенно увлекся этим явлением Гуго де Фриз, один из переоткрывателей
законов Менделя. В ходе своей работы с вечерним первоцветом Oenothera lamarckiana
де Фриз выделил тетраплоидную версию этого обычно диплоидного растения, которую
он назвал Oenotheragigas. Среди животных автополиплоидия встречается реже, особен-
но у таковых с двумя разными полами, возможно, из-за проблем, которые возникают,
если ядро содержит более одной пары половых хромосом. Тем не менее это явление не
является невозможным, и по крайней мере одно млекопитающее — аргентинская крыса
красная вискача — имеет тетраплоидный геном.
Анализ современных геномов дает сведения о свершившихся в прошлом со-
бытиях дупликации генома
Автополиплоидия не ведет непосредственно к увеличению числа генов, потому что
первичный ее продукт — организм, который попросту имеет дополнительные копии
всех его генов, а не какие-либо новые гены. Она, однако же, обеспечивает потенциал для
такого увеличения, потому что добавочные гены не являются определяюще важными
для функционирования клетки и, таким образом, могут подвергнуться мутационному
изменению, не нанося вред жизнеспособности организма. Держа все эти факты в голо-
ве, мы задаемся вопросом: известны ли какие-либо данные о том, что дуплицирование
полного генома имело важное значение в крупномасштабном приобретении новых
генов в ходе эволюционных историй современных геномов?
Глава 18. Механизм эволюции геномов 729
Исходя из того, что мы знаем о путях изменения геномов во времени, мы можем
ожидать, что данные о дуплицировании полного генома добыть весьма трудно. Многие
из дополнительных копий гена, появляющихся в результате дупликации генома, рас-
падаются до такой степени, что их уже невозможно различить в последовательности
ДНК. Те гены, которые сохраняются, потому что их дуплицированная функция полезна
для организма или потому что они развили новые функции, должны быть опознаваемы,
но, как правило, трудно распознать, возникли ли они путем дуплицирования полного
генома или же посредством дупликации намного меньших сегментов. Для того чтобы
получить явные признаки дупликации генома, было бы необходимо найти большие
дублированные наборы генов с одним и тем же порядком расположения генов в обоих
наборах. До какой степени такие дублированные наборы все еще различимы в геноме,
зависит от того, как часто прошлые события рекомбинации перемещали гены на новые
позиции.
С целью отыскания свидетельств прошлых дупликаций в геноме Saccharomyces
cerevisiae был выполнен анализ гомологичности (раздел 5.2.1), в ходе которого каждый
ген дрожжей сравнивался со всеми остальными генами дрожжей. Чтобы быть сочтенны-
ми потомками события дупликации, два гена должны были показать по крайней мере
25 %-ю идентичность при сравнении предсказанных аминокислотных последователь-
ностей их белковых продуктов. Около 800 пар генов было опознано таким способом,
376 из которых могли быть помещены в 55 наборов дубликатов, при этом каждый из
таких наборов содержал по крайней мере три гена, стоящих в одном и том же порядке
(рис. 18.15), с возможным вкраплением между ними других генов, при том эти наборы
вместе взятые охватывали половину генома. Конечно, эти наборы могли возникнуть
и в результате дупликации сегментов, а не полного генома, но если так оно и было, то
можно было бы ожидать, что некоторые из таких генов были бы дуплицированы не
один раз. Поэтому тот факт, что было обнаружено только по две копии каждого гена,
и ни разу по три или по четыре, подтвердил представление о том, что все эти копии
возникли путем дупликации целого генома. Эта возможность стала более очевидной,
когда были получены полные последовательности геномов других видов дрожжей.
Сравнения между геномами S. cerevisiae, Kluyveromyces lactis и Ashbya gossypii были осо-
бенно информативны. Эти три вида произошли от
общего предка, который жил более 100 миллионов
лет назад, еще до времени события дупликации
генома, выведенного из анализа гомологичности.
Если бы эта дупликация действительно произо-
шла в ветви, ведущей к S. cerevisiae, то следовало
бы ожидать, что этот вид имел бы дублированные
копии многих генов, представленных в геномах
К. lactis и A. gossypii лишь однократно. Кажется,
Рис. 18.15. Пример дублированного набора генов в геноме
S. cerevisiae. Каждая из трех пар генов, которые отмечены
на рисунке, обладает высокой гомологичностью последо-
вательностей. Этот и другие дублированные наборы дают
сведения о произошедшей чуть менее 100 миллионов лет
назад дупликации генома в последовательности поколений
S. cerevisiae
Хромосома VII
Хромосома XVI
730 Часть 4. Механизмы репликации и эволюции геномов
именно так оно и есть, ибо этот новый анализ предполагает, что около 10 % генов
в современном геноме S. cerevisiae происходит от дупликации полного генома, которая
произошла чуть менее 100 миллионов лет назад.
Аналогичная работа была проведена с другими геномами, и картина, которая
проясняется, показывает, что дупликация целого генома была относительно частым
событием в эволюции многих групп организмов. Например, сравнения между после-
довательностью генома Arabidopsis thaliana и сегментами геномов других растений
предполагают, что предок генома A. thaliana прошел четыре круга дупликаций генома
на отрезке времени между 100 и 200 миллионами лет назад. Геном человека и геномы
других млекопитающих также содержат столь много дубликатов генов, что по крайней
мере одно событие дупликации полного генома, как думают, произошло в этой ветви
где-то между 350 и 600 миллионами лет назад.
В геномах человека и других организмов могут быть обнаружены также со-
бытия дупликации меньшего масштаба
Хотя самая поздняя дупликация целого генома в эволюционной ветви человека
произошла значительное время назад, геном человека не был заморожен в после-
дующий период. Более того, верно обратное. Одной из неожиданностей, явленных по-
следовательностью генома человека, было осознание того факта, что в относительно
недавнем прошлом в ней произошли протяженные и частые дупликации коротких
сегментов генома. Это иллюстрировано на рис. 18.16, который изображает события
дупликации, произошедшие в течение последних 35 миллионов лет эволюции длин-
ного плеча хромосомы 22 человека. Как и при изучении дрожжей, описанном выше,
такие дупликации идентифицировались путем сравнения различных частей генома,
Центромера
Теломера
Рис. 18.16. Дупликация сегмента в длинном плече хромосомы 22 человека. На рисунке представлен от-
резок в 34 млн п. н. длинного плеча хромосомы 22 в виде ряда тонких горизонтальных линий, каждая из
которых представляет отрезок в 1 млн п. н. последовательности ДНК, бегущей от центромеры (наверху
рисунка) к теломере (внизу рисунка). Этот анализ базировался на черновой последовательности генома
человека, которая содержала одиннадцать пропусков в этой области, показанных черными полосками.
Розовые блоки, которые составляют 3,9 % от 34 млнп.н., являются последовательностями, которые
дублированы в пределах этого плеча хромосомы, а синие блоки (6,4 % от общего количества) — дупли-
цирование областей из других хромосом
Глава 18. Механизм эволюции геномов 731
в данном случае дубликатами считались области более чем 1 тыс. п. н. в длину которые
показывают 90 %-е или более высокое подобие последовательностей нуклеотидов. Этот
анализ игнорирует рассеянные повторы и, таким образом, определяет местоположение
областей, которые, по всей вероятности, являются продуктами относительно недав-
них событий дупликации. Почти 200 сегментов, составляющие более чем 10 % этой
области генома человека протяженностью 34 млн п. н., как оказалось, возникли в ходе
дупликаций, которые произошли в течение последних 35 миллионов лет. Более 100 из
этих сегментов представляют собой дубликаты последовательностей, находящихся
на других хромосомах, а остальные — дубликаты, обе копии которых расположены
в пределах этого плеча.
Схема дупликаций в длинном плече хромосомы 22 довольно типична для генома
человека в целом. Отдельные дупликации варьируют в размере от 1 до 400 тыс.п.н.
в длину, и существует отчетливое тяготение дупликаций к областям, смежным с цен-
тромерами, при относительно малом их числе в более отдаленных областях обоих плеч
хромосомы. При рассмотрении размера таких дупликаций сегментов мы должны иметь
в виду, что средний ген человека имеет длину 20-25 тыс. п. н. и что его гены редко бы-
вают рассеяны по всему геному. При исследовании последовательностей дупликаций
сегментов, выясняется, что немногие из этих событий кончаются дуплицированием
полных генов, а некоторые затрагивают части генов, а вследствие иных дупликаций вы-
шележащие экзоны одного гена помещаются рядом с нижележащими экзонами второго
гена. Некоторые из таких новых комбинаций транскрибируются, но не ясно, являются
ли такие транскрипты функционально активными. Эволюционный потенциал дупли-
каций сегментов поэтому не ясен. Несомненно, однако, то, что рекомбинация между
парой внутрихромосомных дупликаций может привести к удалению области, лежащей
между этими дупликациями, и что удаления, произведенные таким способом, могут
обусловить развитие генетической болезни. Пример — синдром Шарко-Мари-Тута,
который развивается спустя 5-15 лет после рождения и характеризуется вырождением
периферийной нервной системы, что ведет к мышечной атрофии и нарушению коорди-
нации движений. Этот синдром обусловлен рекомбинацией между парой дупликаций
сегмента длиной 24 тыс. п. н. на хромосоме 17 человека, в результате которой из генома
удаляется сегмент протяженностью 1,5 млнп.н. Потеря генов, содержащихся в этом
сегменте, и вызывает эту болезнь.
Эволюция генома заключает в себе также перегруппировку уже существую-
щих генов
Диапазон размеров дупликаций сегментов, наблюдаемых в геноме человека, на-
водит на мысль о возможности того, что события дупликаций, иллюстрированные на
рис. 18.11, могут, помимо появления новых копий генов, вызывать видоизменения
в пределах существующих генов. Это было бы альтернативным путем, которым новые
функции белков могли развиться в ходе эволюции. Он вполне возможен, потому что
большинство белков состоит из структурных доменов, каждый из которых включает
сегмент полипептидной цепи и, следовательно, кодируется в целом прерывным рядом
нуклеотидов (рис. 18.17). Перегруппировка кодирующих домены сегментов генов могла
привести к появлению новых функций белка.
• Дупликация доменов происходит, когда сегмент гена, кодирующий структурный
домен, дублируется путем неравного кроссинговера, проскальзыванием реплика-
ции или одним из других рассмотренных нами методов дупликации последователь-
ностей ДНК (рис. 18.18, а). В результате дупликации структурный домен может
732 Часть 4. Механизмы репликации и эволюции геномов
Полипептид
Рис. 18.17. Каждый структурный домен — отдельная единица в полипептидной цепи и кодируется непре-
рывным рядом нуклеотидов. В этом упрощенном примере каждая вторичная структура в полипептиде
рассматривается как отдельный структурный домен. В действительности большинство структурных
доменов содержат две и более единиц вторичной структуры
повторяться в белке, что само по себе может быть выгодно и, например, сделать
белок более устойчивым. Дуплицированный домен может также изменяться во
времени по мере того, как его кодирующая последовательность накопляет мута-
ции, что ведет к видоизмененной структуре, которая может придать белку новое
действие. Обратите внимание, что дупликация домена вызывает удлинение гена.
Удлинение гена, кажется, есть общее следствие эволюции генома, и гены высших
эукариотов длиннее в среднем, чем таковые более низших организмов.
• Перетасовка доменов происходит, когда сегменты, кодирующие структурные
домены, принадлежащие совершенно разным генам, соединяются вместе и фор-
мируют новую кодирующую последовательность, которая определяет гибридный
или мозаичный белок, то есть такой, который имеет новую комбинацию структур-
ных особенностей и может обеспечить клетку совершенно новой биохимической
функцией (рис. 18.18, б).
Данные модели дупликации и перетасовки доменов основаны на допущении
о том, что необходимые сегменты гена разнесены, так что они могут сами по себе быть
перестроены и перетасованы. Это требование привело к привлекательному предпо-
ложению о том, что экзоны могут кодировать структурные домены. Взять некоторые
белки, так и в самом деле кажется, что дублирование или перетасовка экзонов привели
к появлению наблюдаемых сегодня структур. Пример представлен геном коллагена а2
I типа у позвоночных, который кодирует одну из трех полипептидных цепей коллагена.
Последовательности всех трех полипептидов коллагена сильно насыщены повтора-
ми трипептида глицин-X-Y, где X — обычно пролин, a Y — обычно гидроксипролин
(рис. 18.19). У курицы ген а2 I типа раздроблен на 52 экзона, 42 из которых покрывают
часть гена, кодирующего повторения типа глицин-X-Y. В пределах этой области каждый
экзон кодирует набор полных трипептидных повторений. Число повторений на экзон
изменяется, но остается в пределах значений 5 (в 5 экзонах), 6 (23 экзона), 11 (5 экзонов),
12 (8 экзонов) или 18 (1 экзон). Ясно, что этот ген, возможно, эволюционировал путем
дупликации экзонов, приведшей к повторению структурных доменов.
Явление перетасовки доменов иллюстрируется активатором профибринолизина
ткани (ТРА) — белком, обнаруженным в крови позвоночных животных, который вовлечен
в реакцию свертывания крови. Ген ТРА имеет четыре экзона, каждый из которых кодирует
отдельный структурный домен (рис. 18.20). Вышележащий экзон кодирует модуль «палец»,
который позволяет белку ТРА связываться с фибрином — волокнистым белком, присут-
ствующим в сгустках крови, который активирует ТРА. Этот экзон, кажется, произошел из
второго связывающегося с фибрином белка, фибронектина, и отсутствует в гене родствен-
ного белка, урокиназы, который не активируется фибрином. Второй экзон ТРА определяет
домен фактора роста, который, очевидно, был получен из гена фактора роста эпидермиса
и который может позволить ТРА стимулировать разрастание клетки. Последние два экзона
Глава 18. Механизм эволюции геномов 733
а) Дублирование домена
_ Домен А 32 Домен В _ Домен С _
' Дублирование
1 генного сегмента В
21 Домен A _ Домен В 25 Домен В _ Домен С S
б) Перетасовывание доменов
21 Домен А изДомен В М Домен С S
««Домен X Домен У»
в Домен A S- Домен В К Домен Y S
Рис. 18.18. Создание новых генов: а) дупликацией домена и б) перетасовыванием доменов
кодируют структуры «крингл»Ч которые ТРА использует для связывания со сгустками
фибрина; эти экзоны крингл прибыли из гена профибринолизина.
Коллаген I типа и ТРА представляют изящные примеры эволюции гена, но, к со-
жалению, явные связи, которые они показывают между структурными доменами и эк-
зонами, исключительны и редко наблюдаются у других генов. Многие другие гены,
кажется, эволюционировали путем дупликации и перетасовки сегментов, но в них
структурные домены кодируются сегментами генов, которые не совпадают с отдель-
ными экзонами или даже с группами экзонов. Дупликация и перетасовка доменов все
еще происходят, но, возможно, в менее точной манере, и многие из перестраиваемых
генов не имеют никакой полезной функции. Несмотря на случайный характер, про-
цесс работает четко, что проявляется, наряду с другими примерами, в числе белков
с общими ДНК-связывающими мотивами (раздел 11.1.11). Некоторые из этих мотивов,
возможно, возникли в ходе эволюции de novo в более чем одном случае (и тому была не
одна возможность), но ясно, что во многих случаях последовательность нуклеотидов,
кодирующая мотив, была передана множеству различных генов.
Интересно, что Крис Крингл (Kriss Kringle) — американский псевдоним Санта Клауса, или нашего Деда
Мороза (искаженное немецкое Christkindlein — «Христос-младенец»). Остается только гадать: или открывший
эту структуру в душе ребенок и ему покровительствует святой Николай, или он упорно работал в Сочельник
и ее открытие свершилось в самый канун Рождества, или же, наконец, эту структуру ему попросту напомнил
собой Красный Нос?! — Прим, перев.
734 Часть 4. Механизмы репликации и эволюции геномов
Полипептид коллагена
-Гли-Про-Гип-Гли-Ала-Гип-Гли-Про-Глн-Гли-Фен-Глн-
Рис. 18.19. Полипептид коллагена а2 типа I имеет повтор, который можно описать как Гли-X-Y. Каждая
третья аминокислота представлена глицином, X — часто пролин, a Y — часто гидрокси прол ин (Гип).
Гидрокси прол ин синтезируется путем посттрансляционной модификации пролина (раздел 13.3.3).
Полипептид коллагена имеет винтовую конформацию, но такую, которая более продолговата, чем
стандартная а-спираль
Один из возможных механизмов переноса сегментов генов по всему геному — ас-
социация с мобильными генетическими элементами. Транспозиция элемента LINE-1
(раздел 9.2.1) иногда может привести к переносу короткого отрезка смежной ДНК вместе
с транспозоном; этот процесс назвали З'-трансдукцией, при этом перемещаемый сег-
мент примыкает к З'-концу мобильного элемента. Элементы LINE-1 иногда обнаружи-
ваются в интронах, так что З'-трансдукция могла, очевидно, переместить нижележащие
экзоны в новые участки генома. Передвижение экзонов и других сегментов генов может
быть осуществлено также ДНК-транспозонами по имени Mutator-подобные мобильные
генетические элементы (MULE), которые имеются у многих эукариотов, но особенно
многочисленны у растений. MULE часто содержат в пределах своей последовател ьности
ДНК сегменты генов, захваченные у генома хозяина (рис. 18.21). Транспозиция MULE
поэтому перемещает захваченные сегменты в новое местоположение. MULE могут вби-
рать в себя сегменты различных генов в ходе своих странствий по геному, собирая новые
гибридные гены во время своего пути. MULE поэтому обеспечивают привлекательный
способ продвижения эволюции генов, но всё ещё остаётся несколько оставшихся без
ответа вопросов об их вкладе. В частности, ещё не ясно, как часто сегменты генов спо-
собны высвобождаться из MULE.
Модуль «палец»
Фибронектин и
Профибринолизин
Активатор
профибринолизина
ткани
Рис. 18.20. Модульная структура гена активатора профибринолизина ткани
Домен фактора роста
Фактор роста эпидермиса
Глава 18. Механизм эволюции геномов 735
Подобный оболочке
споры белок
Гипотетический
белок
Предполагаемый белок Предполагаемый белок,
тетратрикопептидного связанный с болезнетворной
повторения активностью
।_________।
1 тыс. п. н.
ОБОЗНАЧЕНИЯ:
Экзон
ИНТРОН
Последовательность MULE
Рис. 18.21. Элементы MULE часто содержат сегменты генов, захваченных из хромосом хозяина. На ри-
сунке показаны пять смежных MULE из хромосомы 1 риса. Цветные сегменты — экзоны гена, а серые
сегменты между экзонами и примыкающие к ним — интроны. Тонкие серые полоски на концах струк-
тур — концевые инвертированные повторы транспозонов MULE. Личности сегментов генов указаны,
коль известны
18.2.2 . Приобретение новых генов от других видов
Второй возможный путь, которым геном может приобрести новые гены, состоит
в получении их от другого вида. Сравнения последовательностей геномов бактерий
и архей предполагают, что главным событием в эволюции геномов прокариотов был
горизонтальный перенос генов (раздел 8.2.3). Геномы большинства бактерий и архей
содержат по крайней мере несколько сотен тыс. п. н. ДНК, представляющих десятки генов,
которые, кажется, были приобретены от какого-либо другого прокариота.
Есть несколько механизмов, которыми гены могут быть перенесены между прока-
риотами, но трудно сказать наверняка, насколько важны эти различные процессы были
в формировании геномов этих организмов. Конъюгация (раздел 3.2.4), например, по-
зволяет плазмидам перемещаться между бактериями и часто кончается приобретением
новых функций генов получателями. В повседневной жизни бактерий перенос плазмид
важен, потому что это средство, которым гены устойчивости к антибиотикам типа
хлорамфениколя, канамицина и стрептомицина распространяются по микрофлорам,
преодолевая межвидовые барьеры, но его эволюционная значимость является сомни-
тельной. Известно, что гены, переданные посредством конъюгации, могут встраиваться
в геном бактерии-получателя, но обычно гены переносятся составными транспозонами
(см. рис. 9.17, б), а это означает, что встраивание обратимо и, таким образом, может
и не вызвать постоянное изменение в геноме. Второй процесс передачи ДНК между
прокариотами — трансформация (раздел 3.2.4) — более вероятно, имел определенное
влияние на эволюцию генома. Лишь несколько бактерий, особенно члены родов Bacillus,
Pseudomonas и Streptococcus, имеют эффективные механизмы забора ДНК из окружающей
среды, но эффективность поглощения ДНК, вероятно, не имеет особого значения, когда
мы имеем дело с эволюционным масштабом времени. Более важен факт, что поток генов,
обусловленный трансформацией, может возникнуть между любой парой прокариотов,
а не только между близкородственными (как это имеет место при конъюгации) и, таким
образом, может служить объяснением событий переноса, которые, кажется, произошли
между геномами бактерий и архей (раздел 8.2.3).
У растений новые гены могут быть приобретены посредством порождения по-
липлоидии. Мы уже видели, как образование автополиплоидии может закончиться
дупликацией генома у растений (см. рис. 18.13). Аллополиплоидия, которая следует из
скрещивания организмов двух разных видов, также распространена и, подобно автопо-
липлоидии, может дать жизнеспособный гибрид. Обычно два вида, которые образуют
736 Часть 4. Механизмы репликации и эволюции геномов
аллополиплоид, состоят в близком родстве и имеют много общих генов, но каждый из
родителей обладает малым числом новых генов или по крайней мере разными алле-
лями общих генов. Например, мягкая, или летняя, пшеница Triticum aestivum является
гексаплоидом, который возник путем аллополиплоидизации между культурной тучной
пшеницей Т. turgidum, которая является тетраплоидной, и диплоидной дикорастущей
травой эгилопсом оттопыренным Aegilops squarrosa. Ядро дикой травы содержало но-
вые аллели генов глютенина, обладающего высокой молекулярной массой, которые,
объединившись с аллелями генов глютенина, уже имевшимися в тучной пшенице,
дали превосходные свойства для хлебопечения, демонстрируемые гексаплоидными
пшеницами. Аллополиплоидизация может поэтому рассматриваться как сочетание
дупликации генома с межвидовым переносом генов.
Среди животных межвидовые барьеры преодолеваются гораздо труднее, и сложно
найти наглядное свидетельство горизонтального переноса генов какого-либо вида. Не-
сколько генов эукариотов имеют особенности, соотносимые с последовательностями
архей или бактерий, но вместо того чтобы быть результатом горизонтального пере-
носа генов, эти подобия, как думают, следуют из сохранения (за счет консервативно-
сти) в течение миллионов лет параллельной эволюции. Большинство предположений
о переносе генов между видами животных сосредоточено на ретровирусах и мобильных
генетических элементах. Передача ретровирусов между видами животных многократно
зарегистрирована, равно как их способность переносить гены животных между особями
одного и того же вида, что позволяет предположить, что они могли бы быть возможными
посредниками в горизонтальном переносе генов. То же самое может быть верно и для
мобильных генетических элементов, таких как P-элементы, которые, как известно,
распространяются от одного вида Drosophila к другому, и элемент mariner, который
также, как было показано, переходит между видами Drosophila и который, возможно,
переправился от других видов в людей.
18.3. Некодирующая ДНК и эволюция генома
До сих пор мы направляли наше внимание на эволюцию кодирующего компонента
генома. Поскольку кодирующая ДНК составляет только 1,5 % генома человека, постольку
наш взгляд на эволюцию генома будет весьма неполным, если мы не посвятим некото-
рое время рассмотрению некодирующей ДНК. Присутствие значительных количеств
некодирующей ДНК в геномах эукариотов — загадка для исследователей молекулярной
эволюции. Почему эта явно излишняя избыточная ДНК допускается природой? Одна воз-
можность состоит в том, что некодирующая ДНК имеет некоторую функцию, которая еще
не была определена, и как таковая должна поддерживаться, потому что без нее клетка
была бы нежизнеспособной. Возможные функции не столь трудно распознать, как мож-
но было бы представить. В нескольких местах в предыдущих главах была подчеркнута
большая значимость структуры хроматина, в том числе обусловливающая прикрепле-
ние хроматина к участкам внутри ядра. Возможно, некоторая часть некодирующего
компонента генома участвует в этих моментах организации генома. Альтернативно,
некодирующая ДНК может иметь широкий спектр функций управления, которая пока
что ускользала от пристального взгляда молекулярных биологов. И последняя возмож-
ность — что некодирующая ДНК не имеет никакой функции, но допускается геномом,
потому что нет никакого давления естественного отбора, направленного на избавление
от нее. Если данная точка зрения верна, то обладание некодирующей ДНК не является
ни преимуществом, ни недостатком, и, таким образом, некодирующая ДНК просто рас-
Глава 18. Механизм эволюции геномов 737
пространяется и переходит по наследству наряду с кодирующей ДНК. Согласно этой
гипотезе некодирующая ДНК попросту может быть «бросовой ДНК» или, быть может,
паразитной «эгоистичной ДНК».
Об эволюции большей части некодирующей доли генома мы мало что можем ска-
зать. Мы представляем себе, что дупликация и другие перестройки происходили путем
рекомбинации и проскальзывания репликации и что последовательности расходились
по мере накопления мутаций, освобожденных от ограничивающих сил естественного
отбора, действующих на функциональные области генома. Мы признаем, что некоторые
части некодирующей ДНК, например регуляторные области выше генов, имеют важные
функции, но что касается многих других частей некодирующей ДНК, то все, что мы мо-
жем сказать, — что они эволюционируют, по-видимому, случайным образом. Но такой
случайный характер не относится ко всем компонентам некодирующей ДНК. В частности,
мобильные генетические элементы и интроны имеют любопытные истории эволюции
и играют важную роль в эволюции генома.
18.3.1. Мобильные генетические элементы и эволюция генома
Мобильные генетические элементы оказывают ряд воздействий на общую эво-
люцию генома. Наиболее значительное из них — способность транспозонов запускать
события рекомбинации, которые ведут к перестройкам генома. Она не имеет никакого
отношения к способности этих элементов перемещаться, а просто связана с тем фактом,
что различные копии одного и того же элемента имеют подобные последовательности
и могут поэтому запустить рекомбинацию между двумя частями одной и той же хромо-
сомы или между разными хромосомами (рис. 18.22). Во многих случаях происходящая
перестройка будет вредна, потому что важные гены будут удалены, но были зарегистри-
рованы и некоторые случаи, где результат был выгоден. Рекомбинация между парой
элементов LINE-1 (раздел 9.2.1) приблизительно 35 миллионов лет назад, как думают,
явилась причиной дупликации гена р-глобина, благодаря которому появились члены
Gg и Ag этого семейства генов (см. рис. 18.9).
Перемещение транспозонов из одного участка на другой также может оказать
воздействие на эволюцию генома. В разделе 18.2.1 было упомянуто о возможном пере-
селении сегментов гена в процессе транспозиции элементов LINE и MULE. Транспозиция
была связана также и с видоизмененными картинами экспрессии генов. Например, эф-
фективность, с которой ДНК-связывающие белки, прикрепляющиеся к вышележащим
регуляторным последовательностям, могут активировать транскрипцию гена, может
быть снижена, если какой-нибудь транспозон переместится на новый участок непо-
средственно выше этого гена (рис. 18.23). Присутствие промоторов и (или) энхансеров
Транспозон 1 Транспозон 2
I Удаление сегмента
между транспозонами
Рис. 18.22. Рекомбинация между парами повторных последовательностей, таких как транспозоны, может
закончиться удалением сегментов генома
738 Часть 4. Механизмы репликации и эволюции геномов
Активация РНК-полимераза
Вышележащие регуляторные
последовательности
Транспонируемый элемент
Рис. 18.23. Встраивание транспозона в область выше гена может затронуть способность связывающихся
с ДНК белков активировать транскрипцию
в пределах транспозона также может повредить транскрипции, навязав смежному
гену совершенно новый режим регулирования. Интересный пример направляемой
транспозоном экспрессии гена происходит с геном Sip мыши, который кодирует белок,
вовлеченный в иммунную реакцию. Специфичность Sip к тканям придается энхансером,
расположенным в пределах смежного ретротранспозона. Известны также примеры, где
встраивание транспозона в ген приводит к видоизменению схемы сплайсинга.
18.3.2. Происхождение интронов
С тех пор как интроны были впервые открыты в 1970-х гг., их происхождение явля-
ется предметом непрекращающихся споров. Есть несколько разногласий, окружающих
типы групп I, II и III (см. табл. 12.2), поскольку, согласно общепринятой версии, все эти
самовырезающиеся интроны возникли в ходе эволюции мира РНК и дожили до на-
стоящего времени, не претерпев никаких серьезных изменений. Проблемы окружают
происхождение интронов GU-AG — тех, что встречаются в большом числе в ядерных
геномах эукариотов.
«Ранние интроны» и «поздние интроны»: две конкурирующие гипотезы
О происхождении интронов GU-AG было высказано множество предположений, но
основным предметом спора, как полагают, являются две противоположные гипотезы:
• Гипотеза «о ранних интронах» гласит, что интроны являются очень древними
и постепенно выпадают из геномов эукариотов.
• Гипотеза «о поздних интронах» утверждает, что интроны появились в ходе эволю-
ции относительно недавно и постепенно накапливаются в геномах эукариотов.
Существует несколько различных моделей для каждой из этих гипотез. Для гипо-
тезы «о ранних интронах» самая убедительная модель — та, что известна также под на-
званием «экзонная теория генов», которая стоит на том, что интроны сформировались
после того, как были построены первые геномы из ДНК, вскоре после конца мира РНК. Эти
геномы, должно быть, содержали много коротких генов, каждый из которых образовался
из отдельной молекулы кодирующей РНК и каждый из них определял очень маленький
полипептид, возможно, лишь один структурный домен. Такие полипептиды, вероятно,
должны были связываться вместе в более крупные многодоменные белки, с тем чтобы
произвести ферменты со специфическими и эффективными каталитическими меха-
Глава 18. Механизм эволюции геномов 739
Рис. 18.24. «Экзонная теория генов». Короткие гены пер-
вых геномов, вероятно, кодировали однодоменные по-
липептиды, которые должны были объединяться вместе,
чтобы формировать многосубъединичный белок с целью
произвести эффективный фермент. Позже синтез этого
фермента, возможно, осуществлялся более эффективно
за счет связывания коротких генов вместе в один пре-
рывный ген, кодирующий многодоменный односубъе-
диничный белок
низмами (рис. 18.24). Дабы облегчить синтез
многодоменного фермента, скорее всего, было
выгодно, чтобы отдельные полипептиды этого
фермента стали сцепленными в единый белок,
что мы и наблюдаем сегодня. Предполагается,
что это было достигнуто путем сплайсинга друг
с другом транскриптов подходящих минигенов,
и этому процессу способствовало перестраива-
ние генома таким образом, что группы миниге-
Коооткие гены
\\!//
О Многосубъединичный
белок
Единый прерывный ген
нов, определяющих различные части отдельных
многодоменных белков, помещались рядом друг с другом. Другими словами, минигены
стали экзонами, а последовательности ДНК между ними стали интронами.
Согласно экзонной теории генов и сообразно другим моделям гипотезы «о ран-
них интронах», все геномы изначально обладали интронами. Но мы знаем, что геномы
бактерий не имеют интронов GU-AG, так что если эта гипотеза верна, то мы должны
принять допущение о том, что по какой-то причине интроны выпали из предкового
бактериального генома на ранней стадии его эволюции. Это камень преткновения, по-
тому что трудно представить, как большое число интронов могло выпасть из генома
без риска нарушения функций многих генов. Если интрон будет удален из какого-либо
гена с некоторой погрешностью, то часть кодирующей области будет потеряна или
произойдет мутация типа сдвига рамки считывания, и оба этих события, как можно
ожидать, приведут к инактивации гена. Гипотеза «о поздних интронах» обходит эту
проблему, предполагая, что вначале никакие гены не имели интронов, но эти струк-
туры вторглись в ранний ядерный геном эукариотов и впоследствии разрослись до
наблюдаемых сегодня количеств. Подобия между путями вырезания интронов GU-AG
и интронов группы II (раздел 12.2.4) дают основу предположению о том, что вторгшиеся
элементы, которые дали начало интронам GU-AG, вполне могли быть последователь-
ностями интронов группы II, которые убежали из геномов органелл. Однако подобие
между интронами GU-AG и интронами группы II не доказывают правоту представления
«о поздних интронах», потому что одинаково возможно изобрести модель для гипотезы
«о ранних интронах», отличающуюся от экзонной теории генов, в которой последова-
тельности интронов группы II давали бы начало интронам GU-AG, но на очень ранней
стадии эволюции генома.
Современные данные не опровергают ни одну из этих гипотез
Одна из причин, по которой полемика о происхождении интронов GU-AG длится уже
более 25 лет, заключается в том, что данные в поддержку первой или второй гипотезы
очень трудно собрать и зачастую они оказываются неоднозначными. Одно из предска-
740 Часть 4. Механизмы репликации и эволюции геномов
заний гипотезы «о ранних интронах» — что должно быть близкое подобие между пози-
циями интронов в гомологичных генах неродственных организмов, потому что все эти
гены происходят от некоего предкового содержащего интроны гена (рис. 18.25). Первое
свидетельство в пользу гипотезы «о ранних интронах» было получено, когда было пока-
зано, что это предсказание сбылось для четырех интронов в гене животных и растений,
кодирующем триозофосфатизомеразу. Однако, когда было исследовано большее число
видов, позиции интронов в этом гене стало труднее интерпретировать: казалось, что
интроны были потеряны в одних ветвях эволюционного древа и приобретены в других.
Этот сценарий соответствует и гипотезе «о ранних интронах», и гипотезе «о поздних
интронах», поскольку обе гипотезы допускают потерю, приобретение или перемещение
интронов в ходе событий рекомбинации, происходящих во всех ветвях эволюционного
дерева. Когда исследуется много генов во многих организмах, вырисовывается общая
картина, которая показывает, что в ходе эволюции геномов животных число интронов
постепенно увеличивалось; эти факты выдвигаются как данные в подтверждение ги-
потезы «о поздних интронах», несмотря на тот факт, что митохондриальные геномы
животных не содержат интронов группы II, которые могли пополнять существующие
ядерные интроны повторными вторжениями. Поэтому число интронов, скорее всего,
увеличилось за счет событий рекомбинации.
Предковый ген
Современные гены - в геномах неродственных организмов
Рис. 18.25. Одно из предсказаний «гипотезы о ранних интронах» гласит, что позиции интронов в гомоло-
гичных генах должны быть подобны у неродственных организмов, потому что все эти гены произошли
от предкового содержащего интроны гена
Альтернативный подход состоял в том, чтобы попытаться соотнести экзоны со
структурными доменами белков, поскольку гипотеза «о ранних интронах» предсказы-
вает, что такая связь должна быть очевидной даже при учете вуалирующих воздействий
эволюции, проявляющихся с тех пор, как примитивные минигены были собраны в пер-
вые настоящие гены. И снова первые факты, которые были получены, свидетельствовали
в пользу гипотезы «о ранних интронах». Исследование белков глобина позвоночных
завершилось заключением о том, что каждый из них включает четыре структурных
домена, первый соответствует экзону 1 гена глобина, второй и третий — экзону 2,
а четвертый — экзону 3 (рис. 18.26). Предсказание о том, что должны существовать
гены глобина с другим интроном, который разделяет второй и третий домены, ока-
залось верным, когда было показано, что ген леггемоглобина сои имеет интрон точно
в ожидаемой позиции. К сожалению, когда было секвенировано больше генов глобинов
и открыто больше интронов (всего более десяти), оказалось, что позиции большинства
из них не соответствуют стыкам между доменами.
Глава 18. Механизм эволюции геномов 741
Рис. 18.26. Ген глобина позвоночного, показываю- Экзоны Ген глобина позвоночного
щий связь между тремя экзонами и четырьмя до- / \
менами белка глобина . • • *
I I
Домен 1 I Домен 4
Гены глобина поэтому находятся домены 2 и 3
в согласии с общим принципом, который
сформировался в ходе нашего обсуждения
перетасовки доменов (раздел 18.2.1): в большинстве случаев не имеется никаких четких
связей между экзонами генов и структурными доменами белков. Но верно ли само по
себе наше определение «структурного домена»? Структурный домен в макромолекуле
белка может не просто соответствовать группе вторичных структур, таких как а-спирали
и 0-листы. В более тонкой интерпретации под структурным доменом следовало бы по-
нимать сегмент полипептида, аминокислоты которого в третичной структуре белка
отстоят друг от друга на расстояние, меньшее определенной величины. Было высказано
предположение о том, что если принять данное определение структурного домена, то
обнаружится большее соответствие между ними и экзонами.
18.4. Геном человека: последние пять миллионов лет
Хотя история эволюции человека полна противоречий, согласно общепринятому
мнению нашим ближайшим родственником среди приматов является шимпанзе, а самый
недавний предок, общий с шимпанзе, жил 4,6-5,0 миллионов лет назад. С момента раз-
ветвления ветвь человека включала два рода —Australopithecus и Ното — и ряд других
видов, не все из которых были на прямой линии, ведущей к Homo sapiens (рис. 18.27).
В конечном итоге появились мы, обладающие всем тем, что, по крайней мере на наш
взгляд, является биологически отличительными чертами, далеко выделяющими нас из
ряда всех остальных животных. Так насколько же мы отличаемся от шимпанзе?
Australopithecus afarensis
«Люси»
Прочие
австралопитеки
Australopithecus
africanus
Homo habilis
I
Homo erectus
Homo sapiens
Рис. 18.27. Одна из возможных схем эволюции современных людей от относящихся к австралопитекам
предков. В этой области исследования есть много разногласий, и несколько различных гипотез было
предложено для эволюционных отношений между различными ископаемыми видами. Сокращение:
млн — миллионов лет назад
742 Часть 4. Механизмы репликации и эволюции геномов
Человек
I II НИ Н III IBIIIB IBBB II
Шимпанзе
I IB Bill М II I
Рис. 18.28. Хромосома человека 2 — результат слияния двух хромосом, которые являются отдельными
у шимпанзе
На современном этапе изучения нашего генома можно дать ответ: на 1,73 % (это
степень несходства нуклеотидных последовательностей между человеком и шимпан-
зе). Действительно, при сравнении геномов человека и шимпанзе намного легче найти
подобия, нежели различия. Степень идентичности наших нуклеотидных последова-
тельностей в пределах кодирующей ДНК составляет более 98,5 %, при этом 29 % генов
в геноме человека кодируют белки, последовательности аминокислот которых иден-
тичны последовательностям их аналогов у шимпанзе. Даже в некодирующих областях
геномов человека и шимпанзе идентичность нуклеотидов редко опускается ниже 97 %.
Порядок генов в этих двух геномах почти один и тот же, а хромосомы имеют очень по-
добный внешний облик. На этом уровне самое разительное различие проявляется в том,
что хромосома 2 человека представлена двумя отдельными хромосомами у шимпанзе
(рис. 18.28), так что шимпанзе, равно как и другие обезьяны, имеет 24 пары хромосом,
тогда как люди имеют только 23 пары. Последовательности альфоидной ДНК, присут-
ствующие в центромерах хромосом человека (раздел 7.1.2), совершенно отличны от
эквивалентных последовательностей в хромосомах шимпанзе и гориллы, а элементы
Alu (раздел 9.2.1) более распространены в геноме человека, но эти особенности, вероят-
но, говорят нам больше об эволюции повторной ДНК, чем о различиях между людьми
и шимпанзе.
Сравнения геномов человека и шимпанзе оказались также не в состоянии показать
и изменения в отдельных генах, которые могли бы так или иначе явиться ключом к от-
личительным качествам людей. Исследования, проведенные с целью выявить гены,
которые подвергались положительному отбору на эволюционном пути человека, опо-
знали несколько, связанных с разложением аминокислот, что находится в соответствии
с большей долей мяса, поедаемого людьми по сравнению с шимпанзе. Гены, обеспечи-
вающие защиту против человеческих болезней типа туберкулеза и малярии, также
прошли положительный отбор. Но никаких генов с явными ролями в развитии мозга или
нервной системы не было раскрыто анализом такого рода. Единственное существенное
различие в структуре генов — то, что у людей отсутствует сегмент гена длиной 92 п. н.,
кодирующего гидроксилазу N-гликолил-нейраминовой кислоты и, таким образом, люди
не могут синтезировать гидроксилированную форму N-гликолил-нейраминовой кис-
лоты, которая присутствует на поверхностях некоторых клеток шимпанзе. Это может
оказывать влияние на способность некоторых патогенов проникать в клетки человека,
и может влиять на межклеточные взаимодействия некоторых типов, но это разли-
чие нельзя считать особо значимым. Еще два гена, которые функционально активны
у шимпанзе, кажется, были инактивированы точечными мутациями в геноме человека:
один из них кодирует рецептор Т-лимфоцитов, а другой — белок кератина волос, но ни
одно из этих изменений, вероятно, не оказало никакого существенного воздействия
на эволюцию человека. В любом случае кажется невозможным, что характерные осо-
бенности людей могли возникнуть в результате вышибания генов. Вместо этого, нам
нужно найти гены, действие которых изменилось, а не было утеряно. В этом отношении
Глава 18. Механизм эволюции геномов 743
значительный интерес был направлен на ген фактора транскрипции FOXP2, так как
дефекты в этом белке приводят к развитию немощи человека, названной дизартрией
и характеризующейся расстройством артикуляции речи. Поэтому этот ген мог лежать
в основе способности человека общаться с помощью речи. И в самом деле, есть разли-
чия в двух аминокислотах между генами FOXP2 человека и шимпанзе, что позволяет
предположить, что этот ген подвергся относительно недавнему изменению, но прямая
связь между этими различиями в аминокислотном составе и речевыми способностями
человека почти неуловима1).
Теперь становится ясно, что многие, если не все, определяющие различия между
людьми и шимпанзе, вероятно, заложены не в самих геномах, а в картинах экспрессии
этих геномов. Внимание поэтому переносится с геномов на транскриптомы и протео-
мы, и эти исследования начинают показывать, что картина экспрессии генома в мозгу
претерпела значительные изменения в эволюционной ветви человека с момента рас-
хождения людей и шимпанзе. Конечно, именно это мы и могли ожидать, поскольку ясно,
что не что иное, как наш мозг, отличает нас от шимпанзе и ото всех остальных животных.
Ключевой вопрос, которому еще не нашли ответа, — можно ли извлечь какую-либо
полезную информацию из опознавания генов, которые имеют больший или меньший
уровень экспрессии в мозгу человека по сравнению с мозгом, скажем, шимпанзе.
Заключение
Считается, что первые полинуклеотиды, которые появились в ходе эволюции не-
сколько миллиардов лет назад, состояли из РНК, а не из ДНК. Эти молекулы РНК, веро-
ятно, сочетали в себе способность к самореплицированию с некоторой ферментативной
активностью, и возможно, что обособление их в простых липидных оболочках дало
начало прародителям первых клеток. Эксперименты показали, что каталитическими
РНК могут быть построены короткие пептиды, и, очевидно, такие пептиды переняли
некоторые из ферментативных функций у рибозимов. ДНК, вероятно, была рождена
эволюцией как более устойчивая версия РНКовых протогеномов. В ходе эволюции было
по крайней мере два периода, когда появлялись более сложные геномы, в каждом случае
с возросшим числом генов. Дупликация генов, как известно, является важным событием,
в результате которого геномы могли приобретать новые гены. Надсемейство генов глоби-
на возникло рядом дупликаций гена, схемы и хронологию которых можно восстановить
путем сравнения между последовательностями генов глобина, существующих сегодня.
События дупликации также играли важную роль в ходе развития гомеозисных избира-
тельных генов, которые определяют общее строение организма эукариотов. Дупликация
полных геномов также возможна, и, как думают, она и произошла в эволюционных путях,
приведших к Saccharomyces cerevisiae и к Arabidopsis thaliana, а также к позвоночным
животным. В поздней эволюции генома человека регулярно происходили дупликации
меньшего масштаба — отрезков в несколько десятков тыс. п. н. Некоторые из них дали
новые сочетания экзонов, и некоторые гены, несомненно, возникли путем дупликации
или перетасовки белковых доменов, кодируемых отдельными экзонами. Экзоны могут
также переноситься по геному за счет прикрепления к мобильным генетическим эле-
ментам. Горизонтальный перенос генов выражается в приобретении генов от других
видов. Это было регулярным событием в эволюции геномов прокариотов, но намного
реже происходило у эукариотов, за исключением, возможно, растений, которые могут
образовывать новые полиплоиды путем слияния гамет родственных видов.
Интересно, что аналог этого гена отвечает за сложность пения у певчих птиц. — Прим. ред.
744 Часть 4. Механизмы репликации и эволюции геномов
Происхождение интронов недосягаемо для нашего взора, а данные, которые мы
имеем, подтверждают и гипотезу «о ранних интронах», и гипотезу «о поздних интро-
нах». Первая предполагает, что первые геномы изначально имели интроны, которые
постепенно выпадают из них, а последняя полагает, что число интронов в них, наоборот,
постепенно увеличивается.
Пять миллионов лет назад эволюционные пути человека и шимпанзе разошлись.
Геномы людей и шимпанзе все еще показывают 98,3 % идентичности последователь-
ностей нуклеотидов, а многие гены кодируют идентичные белковые продукты. Поиск
специфических особенностей генома человека, которые делают нас людьми, оказался
трудным, и теперь думают, что наиболее важные различия между людьми и шимпанзе
могут быть заложены не в самих геномах, а в способе экспрессии этих геномов.
Глава 18. Механизм эволюции геномов 745
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
18.1 *. Первые биохимические системы на
Земле были, возможно, основаны на
биомолекулах которого типа?
а. Углеводы.
Ь. ДНК.
с. Белки.
d. РНК.
18.2 . Термин «протогеном» описывает:
а. Первые геномы из ДНК.
Ь. Первые клеточные геномы из РНК.
с. Первичные молекулы РНК, кото-
рые могли самореплицировать-
ся и направлять биохимические
реакции.
d. Первые полимерные молекулы
РНК.
18.3 *. Какое из следующих утверждений
о переходе от РНКовых к ДНКовым
геномам является ЛОЖНЫМ?
а. Фосфодиэфирные связи в ДНК
более устойчивы, чем таковые
в РНК.
Ь. РНК легко окислялась содержав-
шимся в атмосфере Земли кисло-
родом с образованием ДНК.
с. Замена урацила тимином придала
ДНК большую стабильность.
d. Двунитевая ДНК обусловила по-
явление механизма репарации
поврежденного генетического
материала.
18.4 . Что понимается под согласованной
эволюцией?
а. Процесс, посредством которого
два продукта генов эволюциони-
руют, чтобы взаимодействовать
друг с другом.
Ь. Процесс, посредством которого
гены дублируются, чтобы обеспе-
чить дополнительные продукты
генов.
с. Процесс, посредством которого
гены мутируют таким образом,
что они могут войти в новые се-
мейства генов.
d. Процесс, в ходе которого члены
одного семейства генов сохраня-
ют одинаковые или подобные по-
следовательности нуклеотидов.
18.5 *. Какой из следующих процессов, как
думают, лежит в основе согласован-
ной эволюции?
а. Генная конверсия.
Ь. Горизонтальный перенос генов,
с. Запрограммированная мутация,
d. Транспозиция.
18.6 . Каким способом можно установить
схему дупликации генов в пределах
мультигенного семейства?
а. Сравнением нуклеотидных после-
довательностей входящих в него
генов.
Ь. Сравнением физиологических
функций продуктов этих генов.
с. Сравнением структур продуктов
этих генов.
d. Сравнением местоположений
этих генов в геноме.
18.7 *. Который из следующих процессов,
приводящих к дупликации генов,
происходит, когда имеет место об-
мен ДНК между парой хроматид
в пределах одной хромосомы?
а. Размножение ДНК.
Ь. Проскальзывание репликации.
с. Неравный кроссинговер.
d. Неравный обмен сестринскими
хроматидами.
18.8 . Каков результат автополиплоидии?
а. Ядро, полученное слиянием гамет
организмов двух разных видов.
746 Часть 4. Механизмы репликации и эволюции геномов
Ь. Ядро, содержащее дополнитель-
ные копии одной из хромосом.
с. Ядро, полученное слиянием двух
диплоидных гамет организмов
одного вида.
d. Ядро, которое содержит допол-
нительные копии половых хро-
мосом.
18. 9*. Какое из следующих событий могло
привести к появлению в ходе эволю-
ции нового гена, который содержит
экзоны из двух или нескольких дру-
гих генов?
а. Дупликация доменов.
Ь. Перетасовка доменов.
с. Превращение генов.
d. Дупликация генов.
18.1 0. Какой процесс описывает транспо-
зицию сегмента ДНК наряду с при-
мыкающим к нему транспозоном?
а. З'-Трансдукция.
Ь. Генная конверсия.
с. Ретротранспозиция.
d. Трансформация.
18.11 *. Который из следующих процессов
есть наиболее вероятная причина
Вопросы открытого типа
18.1* . Чем атмосфера Земли сегодня отли-
чается от атмосферы, существовав-
шей при зарождении жизни?
18.2. Каким образом могли некоторые
аминокислоты, азотистые основа-
ния и сахара синтезироваться до
возникновения жизни?
18.3* . Если все современные живые орга-
низмы эволюционировали от одно-
го первоисточника, то является ли
возможным или вероятным, что
существовали и другие источники
биологических систем на древней
Земле? Обоснуйте свой ответ.
18.4. Приведите временную шкалу хода
эволюции живых организмов от об-
горизонтального переноса генов
среди прокариотов?
а. Конъюгация.
Ь. Трансдукция.
с. Трансформация.
d. Транспозиция.
18.12 . Путь вырезания для интронов
GU-AG наиболее подобен пути са-
мовырезания интронов которой из
групп?
а. Группа!.
Ь. Группа II.
с. Группа III.
d. Ни одна из вышеупомянутых.
18.13 *. Наиболее вероятно, что различия
между людьми и шимпанзе обуслов-
лены:
а. Наличием дополнительных генов
в геноме человека.
Ь. Удалением генов шимпанзе из ге-
нома человека.
с. Изменениями в аминокислотных
последовательностях белков, от-
вечающих за развитие речи.
d. Различиями между картинами
экспрессии этих двух геномов.
разования Земли до появления пер-
вых гоминидов.
18.5 *. Какие эволюционные периоды свя-
зывают с событиями увеличения
числа генов в геномах живых орга-
низмов?
18.6 . Обрисуйте контекст, в котором го-
меозисные избирательные гены
представляют хороший пример
эволюции генома путем дуплика-
ции генов.
18.7 *. Каковы характеристики ретрогенов
и как они появляются в геноме?
18.8 . Какие данные говорят о дупликации
целого генома в эволюционной вет-
ви Saccharomyces cerevisiae?
Глава 18. Механизм эволюции геномов 747
18.9 *. Каким образом дупликация сегмен-
тов может произвести новые гены?
18.10 . В каких организмах возможно по-
явление аллополиплоидии?
18.11 *. Что собой представляет «экзонная
теория генов»?
18.12 . Почему люди и шимпанзе имеют
различное число хромосом?
Изыскательские задачи
18.1 *. Приведите данные, на основании
которых было сделано заключение
о том, что геном Arabidopsis thaliana
прошел четыре цикла дупликации
генома на отрезке времени между
100 и 200 миллионами лет назад.
18.2 . Относятся ли примеры дуплика-
ции доменов и перетасовки доме-
нов, приведенные в разделе 18.2.1,
к частным случаям или же к общим
событиям эволюции генома?
18.3 *. Одна из первоначальных публи-
каций черновой последователь-
ности генома человека (IHGSC
[International Human Genome
Sequencing Consortium] [2001] Initial
sequencing and analysis of the human
genome. Nature 409:860-921) позво-
лила предположить, что от 113 до
223 генов человека могли быть при-
обретены у бактерий путем горизон-
тального переноса генов. Впослед-
ствии был сделан вывод о том, что
такая интерпретация не верна и эти
гены не являются бактериальными
по происхождению. Какие данные
говорили в пользу горизонтального
переноса этих генов и почему эти
данные впоследствии были опро-
вергнуты?
18.4 . Оцените гипотезы «о ранних интро-
нах» и «о поздних интронах».
18.5 *. До какой степени, как вы полагаете,
будет возможно определить генети-
ческую основу особых черт человека
на почве сравнений между после-
довательностями геномов людей
и других приматов?
Тесты по рисункам
18.1 *. Опираясь на пути, прочерченные на данном рисунке, порассуждайте о том, как
новые гены могут возникнуть в результате дупликации гена. Который из этих
путей привел бы нас к псевдогену?
Дупликация гена
Давление естественного
отбора на оба гена
Гены остаются
одинаковыми
Давление естественного отбора
лишь на один из генов
Одна из копий
деградирует
Одна из копий приобретает
новую функцию
748 Часть 4. Механизмы репликации и эволюции геномов
18.2 . Которая из представленных ниже моделей (а, б или в) отражает дупликацию гена,
возникшую в результате неравного обмена сестринскими хроматидами?
Глава 18. Механизм эволюции геномов 749
18.3 *. Обсудите, каким образом представленные на рисунках процессы приводят к по-
явлению новых генов в ходе эволюции.
а)
Домен А___Домен В Домен С
I Дупликация
генного сегмента В
ж Домен А _ Домен В в Домен В _ . Домен С
_Домен А —Домен В Домен С
— Домен X —Домен У_
SДомен А — Домен В — Домен У —
18.4. В пользу которой из двух гипотез (о ран-
них или о поздних интронах) говорят
представленные на этом рисунке эволю-
ционные события? Что вообще показыва-
ет данная схема?
Короткие гены
Многосубъединичный
белок
Многодоменный,
j односубъединичный
белок
] Интроны
Единый прерывный ген
750 Часть 4. Механизмы репликации и эволюции геномов
Дополнительная литература
Фундаментальные книги
Maynard Smith, J. and Szathmary, Е. (1995) The Major Transitions in Evolution. WH
Freeman, Oxford. Begins with the origin of life and ends with the evolution of human language.
Ohno,S. (1970) Evolution by Gene Duplication. George Allen and Unwin, London.
Мир РНК и происхождение геномов
Bartel, D.P. and Unrau, Р. J. (1999) Constructing an RNA world. Trends Genet 12: M9-
M13.
Freeland, S.)., Knight, R.D. and Landweber, L. F. (1999) Do proteins predate DNA?
Science 286: 690-692.
Lohse, P. A. and Szostak, J. W. (1996) Ribozyme-catalysed amino-acid transfer reactions.
Nature 381: 442-444.
Miller, S. L. (1953) A production of amino acids under possible primitive Earth conditions.
Science 117: 528-529.
Orgel,L.E. (2000) A simpler nucleic acid. Science 290:1306-1307. Pyranosyl RNA.
Robertson, M.P. and Ellington, A. D. (1998) How to make a nucleotide. Nature 395:
223-225.
Unrau,P.J. and Bartel,D.P. (1998) RNA-catalysed nucleotide synthesis. Nature 395:
260-263.
Дупликация генов
Amores,A., Force, A., Yan,Y.-L., et al. (1998) Zebrafish hox clusters and vertebrate
genome evolution. Science 282:1711-1714.
Wagner, A. (2001) Birth and death of duplicated genes in completely sequenced eukaryotes.
Trends Genet. 17: 237-239.
Дупликация геномов и сегментов генов
Eichler, Е.Е. (2001) Recent duplication, domain accretion and dynamic mutation of the
human genome. Trends Genet. 17: 661-669.
Goffeau, A. (2004) Seeingdouble. Nature 430:25-26. Comparisons between differentyeast
species indicate a genome duplication in the Saccharomyces cerevisiae lineage.
Vision, T. J., Brown, D. G. and Tanksley, S. D. (2000) The origins of genomic duplications
in Arabidopsis. Science 290: 2114-2117.
Wolfe, K.H. and Shields, D.C. (1997) Molecular evidence for an ancient duplication of
the entire yeast genome. Nature 387: 708-713.
Мобильные генетические элементы и эволюция генома
fiang, N., Bao, Z., Zhang, X., Eddy, S. R. and Wessler, S. R. (2004) Pack-MULE transposable
elements mediate gene evolution in plants. Nature 431: 569-573.
Kazazian, H. H. (2000) LI retrotransposons shape the mammalian genome. Science 289:
1152-1153.
Происхождение интронов
de Souza, S. J., Long, M., Schoenbach, L., Roy,S. W. and Gilbert, W. (1996) Intron positions
correlate with module boundaries in ancient proteins. Proc. Natl Acad. Sci. USA 93: 14632-
14636.
Глава 18. Механизм эволюции геномов 751
Gilbert, W. (1987) The exon theory of genes. Cold Spring Harb. Symp. Quant Biol. 52:
901-905.
Gilbert, W., Marchionni,M. and McKnight,G. (1986) On the antiquity of introns. Cell
46:151-153.
Palmer, J.D. and Logsdon, J.M. (1991) The recent origin of introns. Curr. Opin. Genet.
Dev. 1: 470-477.
Люди и шимпанзе
Balter, M. (2005) Are human brains still evolving? Brain genes show signs of selection.
Science 309:1662-1663.
Chou,H.H.,Takematsu,H., Diaz,S., et al. (1998) A mutation in human CMP-sialic acid
hydroxylase occurred after the Homo-Pan divergence. Proc. Natl Acad. Sci. USA 95: 11751-
11756.
Khaitovich, P., Hellmann, I., Enard, W., Nowick, K., Leinweber, M., Franz, H., Weiss, G.,
Lachmann, M. and Paabo,S. (2005) Parallel patterns of evolution in the genomes and
transcriptomes of humans and chimpanzees. Science 309:1850-1854.
Li, W. H. and Saunders, M. A. (2005) The chimpanzee and us. Nature 437:50-51. Describes
the key differences between the human and chimpanzee genomes.
Muchmore,E.A., Diaz,S. and Varki,A. (1998) A structural difference between the cell
surfaces of humans and the great apes. Am. J. Phys. Anthropol. 107:187-198.
Глава 19
Молекулярная филогенетика
По прочтении главы 19 вы сможете:
• Подробнейше рассказывать о том, как таксономия привела к филогении, и об-
суждать причины, по которым молекулярные маркёры очень важны в фило-
генетике.
• Описывать главные особенности филогенетического дерева и проводить раз-
личия между выведенными деревьями, истинными деревьями, генетическими
деревьями и видовыми деревьями.
• Объяснять, как данные, по которым может быть восстановлено дерево, извле-
каются из множественного выравнивания последовательностей ДНК.
• Выявлять основания, на которых покоятся методы объединения соседей и мак-
симальной бережливости, применяемые для восстановления дерева.
• Описывать приемы оценки точности дерева.
• Обсуждать, с примерами, области применения и ограничения молекулярных
часов.
• Разъяснять, почему эволюционные отношения некоторых последовател ьностей
ДНК не могут быть описаны обычным деревом, и освещать способ использования
сетей, позволяющий преодолеть эти проблемы.
• Приводить примеры использования филогенетических деревьев в исследова-
ниях эволюции человека, а также эволюции вирусов иммунодефицита человека
и обезьян.
• Очерчивать принципы изучения генов в популяциях.
• Описывать, как молекулярная филогенетика используется для изучения проис-
хождения современных людей и их переселений в Европу и Новый Свет.
Если геномы эволюционируют за счет постепенного накопления мутаций, то ко-
личество различий в последовательности нуклеотидов между парой геномов должно
указывать на то, как давно эти два генома разошлись от общего предка. Два генома,
которые разошлись в недавнем прошлом, будут, как ожидается, иметь меньше различий,
чем пара геномов, общий предок которых является более древним. Это означает, что
путем сравнения трех и более геномов должно быть возможно установить эволюци-
онные отношения между ними. На поиск таких отношений и нацелена молекулярная
филогенетика.
Глава 19. Молекулярная филогенетика 753
19.1. От классификации до молекулярной филогенетики
Молекулярная филогенетика предшествует сек-
венированию ДНК на несколько десятилетий. Она
возникла на почве традиционного метода класси-
фикации организмов по их подобиям и различиям,
который вначале широко практиковался Линнеем
в восемнадцатом веке. Линней был систематиком, а не
эволюционистом, его цель состояла в том, чтобы поме-
стить все известные организмы в логически стройную
классификацию, которая, как он верил, покажет нам
великий план, используемый Создателем, — Systema
Naturae. Однако он невольно заложил основу для бо-
лее поздних эволюционных схем, распределив орга-
низмы в иерархический ряд таксономических кате-
горий, начиная с царства и спускаясь ниже через тип,
класс, порядок, семейство и род к виду. Натуралисты
восемнадцатого и начала девятнадцатого веков упо-
добляли эту иерархию «дереву жизни» (рис. 19.1) —
эта аналогия была принята Дарвином в «Происхожде-
нии Видов» как средство описания взаимосвязанных
эволюционных историй живых организмов. Таким
образом, классификационная схема, изобретенная
Линнеем, получила новое толкование как филогения,
показывающая не только подобия между видами, но
также и их эволюционные отношения.
Современные виды ВРЕМЯ
Предок
Рис. 19.1. Дерево жизни. Предковые
виды находятся в основании «ствола»
дерева. С течением времени новые
виды эволюционируют от более ран-
них, так что дерево постоянно ветвит-
ся, пока мы не достигаем настоящего
времени, когда нас окружает множе-
ство видов, ведущих свое начало от
предка
19.1.1. Зарождение молекулярной филогенетики
Состоит ли цель в том, чтобы построить классификацию или восстановить фило-
гению, необходимые данные получают путем наблюдения изменчивых признаков в срав-
ниваемых организмах. Первоначально этими признаками служили морфологические
особенности, но молекулярные данные были введены на удивительно ранней стадии.
В1904 г. Нутталл использовал иммунологические испытания, чтобы вывести отношения
между самыми разными животными, при этом одна из его целей состояла в том, чтобы
поместить людей в их соответствующее эволюционное положение относительно других
приматов — проблема, к которой мы возвратимся в разделе 19.3.1. Работа Нутталла по-
казала, что молекулярные данные могут быть использованы в филогенетике, но данный
подход не получил широкого распространения до конца 1950-х гг. Эта задержка была
связана в значительной степени с техническими ограничениями, но отчасти также и с
тем, что классификация и филогенетика должны были претерпеть свои собственные
эволюционные изменения, прежде чем ценность молекулярных данных могла быть
оценена в полной мере.
Фенетика и кладистика требуют больших наборов данных
Эти изменения начались в 1957 г. с введения фенетики — нового филогенети-
ческого метода, который бросил вызов преобладающему представлению о том, что
классификации должны быть основаны на сравнениях между ограниченным числом
признаков, которые таксономисты считали важными по той или иной причине. Фене-
754
Часть 4. Механизмы репликации и эволюции геномов
Рис. 19.2. Аномалии в филогенетике, а) Обладание крыльями птиц и летучих мышей — пример конвер-
гентной (сходящейся) эволюции, б) Единственный палец на ногах лошадей — приобретенное состояние
признака
тики в противовес такой позиции утверждали, что классификации должны охватывать
столько изменчивых признаков, сколько возможно, причем все такие признаки подсчи-
тывались численно и анализировались строгими математическими методами.
За введением фенетики последовал десять лет спустя еще один новый филогенети-
ческий подход, названный кладистикой, который также подчеркивает необходимость
больших наборов данных, но отличается от фенетики в том, что не присваивает равный
вес всем признакам. Основанием такого новшества служит то, что для выведения порядка
ветвления в филогении необходимо отличать те признаки, которые ясно указывают на
эволюционные отношения, от других признаков, которые могут вводить в заблужде-
ние. Это, как может показаться, возвращает нас обратно к дофенетическому подходу,
но кладистика намного менее субъективна: вместо того чтобы делать допущения
о том, какие признаки являются «важными», кладистика требует, чтобы эволюцион-
ная значимость отдельных признаков была определена и задана. В частности, ошибки
в схеме ветвления филогении сводятся к минимуму за счет распознавания двух типов
аномальных данных:
• конвергентная (сходящаяся) эволюция, или аналогия, имеет место, когда одно
и то же состояние признака появляется в ходе эволюции в двух отдельных ветвях.
Например, и птицы и летучие мыши обладают крыльями, но летучие мыши ближе
связаны с бескрылыми млекопитающими, чем с птицами (рис. 19.2, а). Поэтому
состояние признака «обладание крыльями» вводит в заблуждение в контексте
филогении позвоночных;
• унаследованные состояния признака нужно отличать от приобретенных состоя-
ний признака. Приобретенное (или плезиоморфное1)) состояние признака — то,
которым обладал дальний общий предок группы организмов, пример представлен
наличием пяти пальцев на конечностях позвоночных животных. Приобретенное
(или апоморфное* 2)) состояние признака — то, которое появилось в ходе эволюции
из унаследованного состояния более позднего общего предка и, таким образом,
наблюдается только в подгруппе видов изучаемой группы. Среди позвоночных
обладание единственным пальцем на конечностях, что свойственно современ-
ным лошадям, является приобретенным состоянием признака (рис. 19.2, б). Если
Буквально означает «имеющее аналогичную форму». — Прим, перев.
2) Буквально означает «имеющее отдалившуюся, отклонившуюся, отошедшую форму». — Прим, перев.
Глава 19. Молекулярная филогенетика 755
бы мы не осознали этого, то мы могли бы прийти к заключению, что люди ближе
связаны с ящерицами, которые подобно нам имеют пять пальцев на ногах, чем
с лошадьми.
Большие наборы данных могут быть получены посредством изучения моле-
кулярных признаков
Фенетика и кладистика имели довольно сложные взаимоотношения за последние
40 лет. Большинство современных ученых в области эволюционной биологии благово-
лит кладистике, даже при том что строго кладистический подход порождает некоторые
явно парадоксальные результаты, примечательный пример — противоречащее здра-
вому смыслу заключение о том, что птицы не должны иметь свой собственный класс
(птицы), а должны быть помещены в класс пресмыкающихся. Однако обе методологии
в равной степени нуждаются в подпитке большими наборами математических данных
как основным материалом для восстановления филогений. Трудности в получении
наборов данных такого рода при использовании морфологических признаков были
одной из главных движущих сил, обусловивших постепенный переход к использованию
молекулярных данных, которые имеют три преимущества по сравнению с филогенети-
ческой информацией других типов.
• При использовании молекулярных данных один эксперимент может дать информа-
цию о многих различных признаках: в последовательности ДНК, например, каждая
позиция нуклеотида представляет собой признак с четырьмя состояниями при-
знака — А, С, G и Т. Поэтому большие наборы молекулярных данных могут быть
произведены относительно быстро.
• Состояния молекулярных признаков однозначны: нуклеотиды А, С, G и Т легко
опознаются и один нуклеотид не может быть перепутан с другим. Некоторые
морфологические признаки, например, основанные на форме той или иной струк-
туры, могут оказаться труднее различимыми из-за наложений между различными
состояниями признака.
• Молекулярные данные легко переводятся в числовую форму и, следовательно,
поддаются математическому и статистическому анализу.
Последовательности молекул белка и ДНК обеспечивают самые подробные и одно-
значные данные для молекулярной филогенетики, но методы секвенирования белка
стали общедоступными лишь к концу 1960-х гг., а технологии быстрого секвенирования
ДНК были разработаны лишь спустя 10 лет после этого. Поэтому первые исследования
зависели в значительной степени от косвенных оценок изменчивости ДНК или белка,
получаемых одним из трех нижеследующих методов.
• Иммунологические данные, такие, как например, полученные Нутталлом, предпола-
гают измерения количества перекрестной реакционной способности, наблюдаемой,
когда антитело, специфичное к некоторому белку одного организма, смешивается
с тем же белком другого организма. Вспомним, что в разделе 14.2.1 мы узнали,
что антитела — иммуноглобулиновые белки, которые помогают защищать тело
от вторжения бактерий, вирусов и других нежелательных веществ, связываясь
с этими «антигенами». Белки также выступают в роли антигенов, так что если
ввести, например, р-глобин человека в организм кролика, то кролик выработает
антитело, которое будет специфически связываться с этим белком. Антитело будет
также перекрестно реагировать с р-глобинами других позвоночных, потому что
структуры этих р-глобинов подобны структуре р-глобина человека. Степень пере-
756 Часть 4. Механизмы репликации и эволюции геномов
крестной реакционной способности зависит от того, насколько подобен опытный
0-глобин белку человека, и тем самым дает информацию о подобии, используемую
в филогенетическом анализе.
• Электрофорез белка применяется для сравнения электрофоретических свойств
белков различных организмов и, следовательно, степени их подобия. Данный метод
оказался весьма действенным для сравнения близкородственных видов, а также
разновидностей среди членов одного вида.
• Данные ДНК-ДНК-гибридизации получают путем взаимной гибридизации образ-
цов ДНК, изъятых из двух сравниваемых организмов. Образцы ДНК денатурируют
и смешивают вместе, в результате чего образуются гибридные молекулы. Ста-
бильность таких гибридных молекул зависит от степени подобия нуклеотидных
последовательностей этих двух молекул ДНК и измеряется путем определения
температуры плавления (см. рис. 3.8), при этом устойчивый гибрид имеет более
высокую температуру плавления, чем менее устойчивый. Температуры плавления,
полученные для ДНК из различных пар организмов, дают данные, используемые
в филогенетическом анализе.
К концу 1960-х гг. эти косвенные методы были подкреплены возрастающим числом
исследований белковых последовательностей, и в течение 1980-х гг. основанная на ДНК
филогенетика достигла больших высот. Сегодня последовательности белков все еще ис-
пользуются в некоторых контекстах, но ДНК к настоящему времени стала безусловным
лидером среди изучаемых биомолекул. Главным образом это связано с тем, что ДНК дает
больше филогенетической информации, чем белок: нуклеотидные последовательно-
сти пары гомологичных генов несут в себе более высокий объем информации, нежели
аминокислотные последовательности соответствующих белков, потому что мутации,
которые вызывают синонимичные изменения, видо-
изменяют последовательность ДНК, но не затрагивают
последовательность аминокислот (рис. 19.3). Также
в ходе анализа последовательности ДНК может быть
получена совершенно уникальная информация, потому
что может быть исследована изменчивость и в коди-
рующих, и в некодирующих областях генома. Просто-
та, с которой образцы ДНК для секвенирования могут
быть приготовлены с помощью ПЦР (раздел 2.3), служит
очередной определяющей причиной, подкрепляющей
господство анализа ДНК в современной молекулярной
филогенетике1^.
Наряду с последовательностями ДНК, молекуляр-
ные филогенетики используют также и ДНК-маркёры,
такие как RFLP, SSLP и SNP (раздел 3.2.2), в особенности
для внутривидовых исследований типа нацеленных на
понимание переселений популяций доисторического
человека (раздел 19.3.2). Позже в этой главе мы рассмо-
На самом деле, использование ДНК для филогенетического анализа работает на сравнительно не-
больших эволюционных расстояниях (например, на уровне млекопитающих). Для более удаленных сравне-
ний (например, при анализе всех позвоночных или насекомых) количество изменений в ДНК весьма велико
и разрешающая способность резко падает (за исключением генов, кодирующих рибосомную РНК). Обычно
на таких расстояниях используют сравнение именно аминокислотных последовательностей, выведенных
из ДНК. — Прим. ред.
-Гли - Ала - Иле - Лей -Асл - Apr
-G G AG С С ATATTAG ATAG А-
-^GA^CA^TTJTT^AT^GA-
-Гли - Ала - Иле -Фен - Асл - Apr
•
Рис. 19.3. ДНК дает больше фило-
генетической информации, чем бе-
лок. Две последовательности ДНК
отличаются в трех позициях, но со-
ответствующие им аминокислотные
последовательности отличаются
только водной позиции. Эти позиции
обозначены синими точками. Две из
нуклеотидных замен поэтому сино-
нимические, а одна несинонимиче-
ская (см. рис. 16.11)
Глава 19. Молекулярная филогенетика 757
трим различные примеры использования и последовательностей ДНК и ДНК-маркёров
в молекулярной филогенетике, но сначала мы должны более досконально ознакомиться
с методологией, используемой в этой области исследования генома.
19.2. Восстановление филогенетических деревьев на основе
ДНК
Цель большинства филогенетических исследований состоит в восстановлении
древовидной схемы, которая описывает эволюционные отношения между изучаемыми
организмами. Наперед изучения разработанной для этого методологии мы должны
ближе ознакомиться с типичным деревом, с тем чтобы освоиться с основной термино-
логией, употребляемой в филогенетическом анализе.
19.2.1. Основные характеристики построенных на основе ДНК филоге-
нетических деревьев
Типичное филогенетическое дерево представлено на рис. 19.4, а. Это дерево могло
быть восстановлено по сравнительным данным любого типа, но поскольку нас интере-
суют последовательности ДНК, мы положим, что данное дерево показывает отношения
между четырьмя гомологичными генами, названными A, B,CnD. Топология этого дерева
включает четыре внешних узла [листа. — Прим, ред.], каждый из которых представляет
один из четырех сравниваемых генов, и два внутренних узла, представляющих пред-
ковые гены. Длины ветвей показывают степень различия между генами, представлен-
ными узлами. Степень различия рассчитывается при сравнении последовательностей,
как будет описано в разделе 19.2.2.
а) Некорневое дерево
Ветви
В Внутренние
узлы
Рис. 19.4. Филогенетические деревья, а) Некорневое дерево с че-
тырьмя внешними узлами, б) Пять корневых деревьев, которые
могут быть получены из некорневого дерева, показанного в ча-
сти а. Положения корней обозначены цифрами на общей схеме
корневого дерева
758 Часть 4. Механизмы репликации и эволюции геномов
Рис. 19.5. Использование внешнего узла для укоренения филоге-
нетического дерева. Дерево генов человека, шимпанзе, гориллы
и орангутана имеет общий корень с геном бабуина, потому что
мы знаем из летописи окаменелостей, что бабуины ответвились
от линии приматов до времени жизни общего предка остальных
четырех видов. Более подробные сведения о филогенетическом
анализе человека и других приматов можно почерпнуть в раз-
деле 19.3.1
Дерево на рис. 19.4, а некорневое, а это значит,
что оно являет собой лишь иллюстрацию отношений
между генами Л, В, С и D и ничего не говорит нам о череде
эволюционных событий, которые привели к появлению
этих генов. В данном случае возможны пять различных
эволюционных путей, и каждый из них изображается своим корневым деревом, как
показано на рис. 19.4, б. Для проведения различий между ними в филогенетический
анализ вводят по крайней мере один внешний (корневой) вид (узел), в данном случае
представленный гомологичным геном, который, по нашим данным, менее близко связан
с генами Л, В, С и D, чем эти четыре гена связаны между собой. Чуждый узел позволяет
установить корень дерева и определить правильный эволюционный путь. Критерии,
используемые при выборе внешнего вида, во многом зависят от вида выполняемого
анализа. Примера ради мы скажем, что четыре гомологичных гена в нашем дереве при-
надлежат человеку, шимпанзе, горилле и орангутану. В таком случае в качестве внешнего
узла мы можем использовать гомологичный ген от другого примата, например бабуина,
который, как мы знаем по палеонтологическим данным, сошел с восстанавливаемой
нами эволюционной ветви, ведущей к человеку, шимпанзе, горилле и орангутану, до
времени жизни общего предка этих четырех видов (рис. 19.5).
Филогенетическое дерево, построенное из генов приматов, показывает относитель-
но простую схему отношений. В большинстве же случаев деревья более сложные, и нам
необходимо усвоить новые термины, чтобы освоиться в гуще разного рода отношений,
в которую мы можем попасть (рис. 19.6). Говорят, что две и более последовательности
являются монофилетическими, если они происходят от единой общей предковой
последовательности ДНК. Группу монофилетических последовательностей называют
кладойЧ если она охватывает все последовательности, включенные в анализ, которые
ведут свое начало от предковой последовательности в корне клады. Если в группу не
входят некоторые члены клады, то такая группа является парафилетической. Альтер-
нативно, если две или несколько последовательностей ДНК происходят от различных
предковых последовательностей, то такую группу зовут полифилетической.
Корневое дерево, которое мы получаем в результате филогенетического анализа,
мы рассматриваем как выведенное дерево. Этим мы подчеркиваем, что оно изображает
ряд эволюционных событий, логически выведенных по данным, которые были проана-
лизированы, и может отличаться от истинного дерева, изображающего фактический
В нашем языке это понятие крайне разнородно, то есть употребляется и в женском (клада), и в мужском
(клад) роде. Если формулировать его определение как группу организмов, предположительно включающую
в себя всех эволюционных потомков некоторого общего предка, то удобнее говорить клада, однако если под
такой группой понимать монофилетический таксон, то удобнее говорить клад. Собственно, этимон этого
слова, греческое слово кХабос; (отвод, побег), имеет мужской род. Так что это просто целая кладезь противо-
речий! — Прим, перев.
Глава 19. Молекулярная филогенетика 759
Кенгуру
130
Свиньи
Овцы, козы
Крупный рогатый скот
Лошади______I
Полифилетические
Собаки
Кошки
Парафилетические
Кролики
Крысы
Мыши
Обезьяны Нового Света
(например, мартышки)
Обезьяны Старого Света
(например, макаки)
Орангутан
Горилла
Монофилетические
Шимпанзе
Человек—
Клада
Рис. 19.6. Филогения млекопитающих, иллюстрирующая ключевые термины, употребляемые для опи-
сания различного типа отношений, наблюдаемых в филогенетических деревьях. Кенгуру используется
как внешний вид (узел). Цифры показывают оцененные времена расхождения для указанных линий
в миллионах лет. Люди и шимпанзе монофилетические, потому что они имеют общего предка, который
жил 4,6-5,0 миллионов лет назад. Люди, шимпанзе, гориллы и орангутаны образуют кладу, поскольку
они все происходят от одного общего предка, который жил 13 миллионов лет назад. Обозначенная
группа парафилетическая, поскольку она исключает овец и коз, которые имеют того же самого общего
предка, что и другие члены этой группы. Свиньи и лошади полифилетические, поскольку они имеют
разных общих предков. У свиней этот предок жил 65 миллионов лет назад и дал начало также овцам,
козлам и крупному рогатому скоту, а у лошадей предок жил более 75 миллионов лет назад и породил
также кошек и собак
ряд событий, которые имели место в действительности. Иногда мы можем быть вполне
уверены, что выведенное дерево является истинным деревом, но во многих случаях
анализ филогенетических данных подвержен погрешностям, которые вполне могут
привести к тому, что выведенное дерево будет отличаться в некоторой степени от ис-
тинного дерева. В разделе 19.2.2 мы рассмотрим разнообразные методы, используемые
для присваивания степени достоверности к схеме ветвления в выведенном дереве,
а ближе к концу этой главы мы исследуем некоторые противоречия, которые возникли
в силу неточного характера филогенетического анализа.
Генетические деревья отнюдь не то же самое, что видовые деревья
Дерево, показанное на рис. 19.5, иллюстрирует обычного типа проект молекуляр-
ной филогенетики, где цель состоит в том, чтобы с помощью генетического дерева,
восстановленного по результатам сравнений между последовательностями ортологич-
ных генов (происходящих от одной предковой последовательности), сделать выводы
об эволюционной истории видов, от которых эти гены были взяты. Данный подход
760 Часть 4. Механизмы репликации и эволюции геномов
а) Дерево генов
Ген А
Рис. 19.7. Разница между деревом генов и деревом видов
основан на допущении о том, что генетическое дерево, построенное на молекулярных
данных со всеми их преимуществами, будет более точным и менее неопределенным
представлением видового дерева, чем дерево, построенное по итогам морфологических
сравнений. Это допущение часто оказывается верным, но оно отнюдь не подразумевает
того, что дерево генов есть то же самое, что и дерево видов.
Для того чтобы это имело место, внутренние узлы в генетическом и видовом де-
ревьях должны были бы быть точно эквивалентными. Обычно они не эквивалентны,
потому что:
• внутренний узел в генетическом дереве представляет расхождение предкового
гена на два аллеля с различными последовательностями ДНК: это происходит
путем мутации (рис. 19.7, а);
• внутренний узел в видовом дереве представляет событие видообразования
(рис. 19.7, б): оно происходит посредством разделения популяции организмов
предкового вида на две группы, которые не способны скрещиваться между собой,
часто потому, что они становятся географически изолированными.
Важный момент состоит в том, что эти два события — мутация и видообразова-
ние — как ожидается, не происходят в одно и то же время. Например, событие мутации
может предшествовать видообразованию. Это означает, что сперва оба аллеля мути-
рованного гена присутствуют в неразделенной популяции предкового вида (рис. 19.8).
Когда происходит разделение популяции,
весьма вероятно, что оба аллеля неизменно
будут присутствовать в каждой из двух обо-
собившихся групп. После разделения новые
популяции эволюционируют независимо
друг от друга. Может случиться и так, что
результаты случайного генетическо-
го дрейфа (раздел 19.3.2) ведут к потере
одного аллеля в одной популяции и к по-
Рис. 19.8. Мутация могла предшествовать видоо-
бразованию и дать неправильное время события
видообразования, если используются молекуляр-
ные часы
Глава 19. Молекулярная филогенетика 761
тере другого в другой популяции. В результате такого события устанавливаются две
отдельные генетические линии, которые мы выводим из филогенетического анализа
последовательностей генов, имеющихся у современных видов, которые возникли в итоге
длительной (и непрерывной) эволюции этих двух популяций.
Как эти соображения затрагивают взаимную эквивалентность генетического и ви-
дового деревьев? Есть различные следствия, два из которых приведены ниже.
• Если для установления момента времени, в который произошло расхождение ге-
нов, применяются молекулярные часы (раздел 19.2.2), то нельзя считать, что это
время соответствует и событию видообразования. Если датируемый узел является
древним — скажем 50 и более миллионов лет назад, — то ошибка может быть пре-
небрежимо мала. Но если событие видообразования произошло относительно не-
давно (как например, при сравнении приматов), то дата расхождения генов может
значительно отличаться от даты события видообразования.
• Если за первым событием видообразования скоро следует второе событие видо-
образования в одной из двух образовавшихся популяций, то порядок ветвления
генетического дерева может отличаться от такового в дереве видов. Это может
произойти, если гены в современном виде происходят из аллелей, которые уже
появились перед первым из этих двух событий видообразования, как показано
на рис. 19.9.
19.2.2. Восстановление дерева
В этом разделе мы осветим принцип восстановления деревьев по последователь-
ностям ДНК, уделяя особое внимание четырем главным шагам этой процедуры:
• выравнивание последовательностей ДНК и получение сравнительных данных,
которые будут использоваться для восстановления дерева;
Рис. 19.9. Дерево генов может иметь отли-
чающийся порядок ветвления оттакового
у дерева видов. В этом примере ген пре-
терпел две мутации в предковом виде:
первая мутация дала начало «красному»
аллелю, а вторая породила «синий» ал-
лель. В результате случайного дрейфа
генов, наряду с двумя последующими
событиями видообразования, в виде
А появляется зеленая линия аллелей,
синяя линия аллелей в виде В и красная
линия аллелей в виде С. Молекулярная
филогенетика, основанная на последо-
вательностях генов, покажет, что развет-
вление зеленый-красный произошло
до разветвления красный-синий, давая
дерево генов, показанное справа. Однако
фактическое дерево видов отличается от
него и показано слева
762 Часть 4. Механизмы репликации и эволюции геномов
• преобразование сравнительных данных в восстановленное дерево;
• оценка точности восстановленного дерева;
• применение молекулярных часов для приписывания дат к точкам ветвления
в дереве.
Выравнивание последовательностей — неотъемлемый предварительный
этап восстановления дерева
Данные, используемые при восстановлении филогенетического дерева на основе
ДНК, получают путем сравнения последовательностей нуклеотидов. Эти сравнения
проводят посредством выравнивая последовательностей, с тем чтобы отличиям ну-
клеотидов мог быть назначен счет. Это определяющий этап всего предприятия, потому
что если выравнивание окажется неточным, то полученное дерево уж точно не будет
истинным деревом.
Первый вопрос, который необходимо решить, — являются ли сравниваемые по-
следовательности гомологичными. Если они гомологичны, то по определению должны
проистекать из некоторой общей предковой последовательности (раздел 5.2.1), так что
есть веское основание для филогенетического исследования. Если же они не гомологич-
ны, то у них нет и быть не может никакого общего предка. Филогенетический анализ
всегда найдет общего предка, потому что даже если данные абсолютно ошибочны, ме-
тоды, используемые для восстановления деревьев, всегда производят некоторое дерево
с тем или иным описанием отношений, но полученное при такой подоплеке дерево не
будет иметь никакой биологической значимости. В отношении некоторых последова-
тельностей ДНК — например генов 0-глобина разных позвоночных, — не представляет
особой сложности увериться в том, что сравниваемые последовательности являются
гомологичными, но это не всегда так, и одна из наиболее распространенных ошибок,
которая появляется в ходе филогенетического анализа, — оплошное включение в него
негомологичной последовательности.
Как только будет установлено, что две последовательности ДНК являются дей-
ствительно гомологичными, следующим шагом будет выравнивание этих последова-
тельностей так, чтобы можно было сравнить гомологичные нуклеотиды. С некоторыми
парами последовательностей это тривиальная задача (рис. 19.10, а), но она не настолько
легка, если эти последовательности относительно несхожи и (или) разошлись в силу
накопления вставок и удалений, наряду с точечными
мутациями. Когда сравниваются пары последователь-
ностей, пнсерции (вставки) и делеции (удаления) нельзя
а) Простое выравнивание
последовательностей
AGCAATGGCCAGACAATAATG
AGCTATGGACAGACATTAATG
б) Более сложное выравнивание
последовательностей
GACGACCATAGACCAGCATAG
GACTACCATAGA - CTGCAAAG
*** ******** * *** **
GACGACCATAGACCAGCATAG
GACTACCATAGACT - GCAAAG
*** ********* *** **
- Возможные позиции
для помещения индела
Рис. 19.10. Выравнивание последовательностей, а) Две последо-
вательности, которые не разошлись до сколь-нибудь большой
степени, могут быть легко выровнены на глаз. 6) Более сложное
выравнивание, в котором невозможно определить правильную
позицию для индела. Если во множественном выравнивании сде-
ланы ошибки в размещении индела, то дерево, восстановленное
филогенетическим анализом, врядли будет правильным. На этой
схеме красные звездочки указывают нуклеотиды, которые явля-
ются одинаковыми в обеих последовательностях
Глава 19. Молекулярная филогенетика 763
Рис. 19.11. Метод точечных матриц для выравни-
вания последовательностей. Правильное вырав-
нивание выделяется на общем фоне, потому что
оно образует диагональ непрерывных точек, раз-
рываемую в точечных мутациях и смещающуюся
на другую диагональ матрицы в местах инделов
AGACATTTAGАССAAGСATAGССАТ
отличить друг от друга, поэтому мы будем
использовать термин индел (от комбина-
ции терминов инсерция и делеция). По-
мещение инделов в надлежащие позиции
зачастую оказывается самой трудной ча-
стью процесса выравнивания последова-
тельностей (рис. 19.10, б).
Некоторые пары последовательно-
стей могут быть надежно выравнены «на глаз». Для более сложных пар выравнивание
может быть возможно методом точечных матриц (рис. 19.11). Две выравниваемые
последовательности расписывают по осям х и у графика и помещают точки в ячейки
клетчатой бумаги в позициях, соответствующих идентичным нуклеотидам в этих двух
последовательностях. Выравнивание показывается диагональным рядом точек, преры-
ваемым пустыми квадратами, в которых последовательности имеют различия нуклео-
тидов, и перебегающим из одного столбца в другой в местах, где находятся инделы.
Также были изобретены более строгие математические подходы к выравниванию
последовательностей. Первый из них — метод подобий — стремится максимизировать
число совпадающих нуклеотидов — таких, которые являются идентичными в соот-
ветствующих позициях двух выравниваемых последовательностей. Дополняющий его
подход — метод расстояний, в котором цель состоит в том, чтобы минимизировать
число несовпадений. Часто эти две процедуры определяют одно и то же выравнивание
как наилучшее. Обычно в сравнении участвует более двух последовательностей, а это
означает, что требуется их множественное выравнивание. Лишь в редких случаях
его удается эффективно построить, вооружившись ручкой и бумагой, так что, как и на
всех шагах филогенетического анализа, используется компьютерная программа. Для
множественных выравниваний Clustal часто оказывается наиболее популярным вы-
бором. Clustal и другие программные пакеты для филогенетического анализа описаны
в техническом примечании 19.1.
Преобразование данных выравнивания в филогенетическое дерево
Выровняв последовательности надлежащим образом, можно попытаться вырас-
тить на их почве филогенетическое дерево. До настоящего времени никто не изобрел
совершенный метод восстановления деревьев, и обычно используются несколько раз-
личных процедур. Были проведены сравнительные испытания с искусственными данны-
ми, для которых истинное дерево известно, но они не позволили выделить какой-либо
конкретный метод как наилучший среди всех остальных.
Главное отличие между различными методами построения деревьев — способ, ко-
торым множественное выравнивание последовательностей преобразуется в числовые
данные, которые можно анализировать математически, с тем чтобы восстановить дерево.
764 Часть 4. Механизмы репликации и эволюции геномов
Техническое примечание 19.1: филогенетический анализ
Пакеты программ для построения филогенетических деревьев
Очень мало наборов последовательностей ДНК достаточно просты, чтобы быть
преобразованными в филогенетические деревья вручную, абсолютно не прибегая
к помощи машин. Фактически все исследования в этой области проводят с исполь-
зованием компьютеров при помощи любого из многообразия пакетов программ,
специально разработанных для того или иного этапа восстановления дерева.
Один из наиболее удобных в использовании и самых популярных пакетов —
Clustal, который изначально был написан в 1988 г. и перенес несколько обновлений
в последующие годы. Clustal — в первую очередь программа для выполнения множе-
ственных выравниваний последовательностей белков или ДНК, которые она способ-
на делать очень эффективно при условии, что сравниваемые последовательности не
содержат обширных внутренних повторных мотивов. Clustal обычно используется
совместно с NJplot — простой программой для восстановления деревьев методом
объединения соседей. Одно их важных преимуществ Clustal и NJplot состоит в том,
что они не требуют огромных количеств памяти компьютера и, таким образом,
могут работать на малых компьютерах — ПК или Макинтошах.
Более комплексные пакеты программ позволяют исследователю выбирать сре-
ди разнообразия различных методов восстановления деревьев лучший и выполнять
филогенетический анализ более сложного типа. Наиболее широко используемые
из таких пакетов — PAUP и PHYL1P. Программы построения деревьев, входящие
в пакет PAUP, часто рассматривают как самые точные из имеющихся в настоящее
время, при этом они способны обращаться с относительно большими наборами
данных. Достоинство PHYL1P — наличие ряда программных средств, которые не-
легко получить из других источников. В число популярных пакетов входят также
PAML, MacClade и HENNIG86.
Самый простой подход состоит в преобразовании информации о последовательностях
в матрицу расстояний, которая представляет собой не что иное, как простую таблицу,
показывающую эволюционные расстояния между всеми парами последовательностей
из набора данных (рис. 19.12). Эволюционное расстояние рассчитывается по числу от-
личающихся нуклеотидов между парой последовательностей и используется, чтобы
установить длины ветвей, соединяющих эти две последовательности в восстановленном
Множественное выравнивание Матрица расстояний
1 2 3 4
1 AGGCCAAGCCATAGCTGTCC 1 0,20 0,05 0,15
2 AGGCAAAGACATACCTGACC 2 - 0,15 0,05
3 AGGCCAAGACATAGCTGTCC 3 - 0,10
4 AGGCAAAGACATACCTGTCC 4 —
Рис. 19.12. Простая матрица расстояний. Матрица показывает эволюционное расстояние между каждой
парой последовательностей в выравнивании
Глава 19. Молекулярная филогенетика 765
дереве. В примере, показанном на рис. 19.12, эволюционное расстояние выражено как
число отличающихся нуклеотидов на нуклеотидный участок для каждой пары последо-
вательностей. Например, последовательности 1 и 2 имеют длину 20 нуклеотидов и со-
держат четыре несовпадения, что соответствует эволюционному расстоянию 4/20 = 0,2.
Следует иметь в виду, что этот анализ основан на допущении об отсутствии многократ-
ных замен (также называемых многократными попаданиями). Многократная замена
происходит, когда отдельный участок подвергается двум и более заменам (например,
предковая последовательность ...AIGT... дает начало двум современным последова-
тельностям: ...AfiGT... и ...ACGT...). При этом имеется только одно различие нуклеотидов
между двумя современными последовательностями, но на самом деле произошли две
замены нуклеотидов. Если такое многократное попадание не будет распознано, то
эволюционное расстояние между двумя современными последовательностями будет
в большой мере недооценено. Чтобы избежать этой проблемы, матрицы расстояний
для филогенетического анализа обычно составляются с применением математических
методов, которые включают статистические средства для оценки количества имевших
место многократных замен.
Метод объединения соседей — популярная процедура построения деревьев,
основанная на методе матриц расстояний. Чтобы начать восстановление, делается из-
начальное допущение о том, что существует только один внутренний узел, от которого
ветви, ведущие ко всем последовательностям ДНК, расходятся в виде звездообразной
схемы (рис. 19.13). Это фактически невозможно в эволюционном контексте, но такая
схема служит лишь отправной точкой. Затем наугад выбирается пара последователь-
ностей, удаляется из звезды и присоединяется ко второму внутреннему узлу, соединен-
ному ветвью с центром звезды. Теперь, чтобы вычислить полную длину ветвей в этом
новом «дереве», используется матрица расстояний. Эти последовательности после этого
возвращаются в их первоначальные положения, и следующая их пара выбирается и при-
соединяется ко второму внутреннему узлу, и снова рассчитывается полная длина ветвей.
Эта операция повторяется до тех пор, пока все возможные пары не будут испытаны, что
позволяет найти сочетание, которое дает дерево с наикратчайшей полной длиной ветвей.
Последовательности этой пары будут соседями в конечном дереве; на промежуточном
этапе они объединяются в целостную единицу, в результате чего создается новая звезда,
имеющая на одну ветвь меньше, чем исходная. Засим весь процесс выбора пары и вы-
числения длины дерева повторяется, так что отыскивается вторая пара соседствующих
последовательностей, затем повторяется снова, так что устанавливается третья пара,
и так далее. В конечном итоге восстанавливается полное дерево.
а) Отправная точка для метода
объединения соседей
б) Удаление двух последовательностей
Рис. 19.13. Манипуляции, выполняемые при применении метода объединения соседей для восстанов-
ления дерева
766 Часть 4. Механизмы репликации и эволюции геномов
Преимущество метода объединения соседей состоит в том, что обработка данных
является относительно легко выполнимой, во многом благодаря тому, что информация
о множественном выравнивании была сведена к ее простейшей форме. Недостаток его
проявляется в том, что часть информации теряется, в особенности касающаяся рода
предковых и приобретенных нуклеотидов (эквивалент предковых и приобретенных
состояний признака, описанных в разделе 19.1.1) в каждой позиции множественного
выравнивания. Метод максимальной бережливости принимает во внимание эту ин-
формацию, используя ее для воссоздания ряда замен нуклеотидов, которые, наиболее
вероятно, привели к появлению картины изменчивости, прорисовывающейся во мно-
жественном выравнивании. Термин бережливость1) изначально был философским;
согласно принципу максимальной бережливости при выборе между конкурирующими
гипотезами, предпочтение следует отдавать той, что содержит наименьшее число не-
связанных допущений. В молекулярной филогенетике бережливым считают подход,
согласно которому, выбирая между различными топологиями дерева, предпочитают ту,
которая отражает кратчайший эволюционный путь, то есть путь, который проходит от
предковой последовательности в корне дерева ко всем современным последователь-
ностям, которые сравниваются, через наименьшее число видоизмененных нуклеоти-
дов. Поэтому деревья строятся наугад и вычисляется число изменений нуклеотидов,
которые они вовлекают, пока все возможные топологии не будут перебраны и та, что
требует наименьшего числа шагов, не будет определена. Она-то и выдается за наиболее
правдоподобное выведенное дерево.
Метод максимальной бережливости более строг в своем подходе по сравнению
с методом объединения соседей, но такое увеличение в строгости неизбежно увели-
чивает и объемы обрабатываемых данных. Это серьезная проблема, потому что число
возможных деревьев, которые должны быть тщательно просчитаны, резко возрастает
по мере добавления новых последовательностей к набору данных. При всего лишь
пяти последовательностях существует только 15 возможных некорневых деревьев,
но для десяти последовательностей существует уже 2027 025 некорневых деревьев,
а для 50 последовательностей это число превышает число атомов во Вселенной. Даже
с быстродействующим компьютером невозможно проверить каждое из этих деревьев
за разумное время, если вообще возможно, так что часто метод максимальной береж-
ливости не способен выполнить всесторонний анализ. То же самое верно и для многих
других, еще более замысловатых методов восстановления деревьев.
Оценка точности восстановленного дерева
Ограничения методов, используемых в филогенетическом восстановлении, неиз-
бежно ведут к вопросам о достоверности получаемых деревьев. Были изобретены ста-
тистические испытания точности восстановленного дерева, но они неминуемо сложны,
потому что дерево является геометрическим, а не числовым объектом, и точность одной
части топологии может быть выше или ниже точности остальных ее частей.
Обычный метод для установления границ доверительного интервала различным
точкам ветвления в дереве состоит в проведении анализа на случайных выборках
с возвращением. Для его проведения нам нужно второе множественное выравнива-
ние, которое отличается от реального выравнивания, но при этом эквивалентно ему.
Это новое выравнивание выстраивается, выбирая столбцы наугад из действительного
выравнивания, как показано на рис. 19.14. Поэтому это новое выравнивание включает
1}Так, бережливый в отношении своего времени и сил молекулярный биолог, вооружившись бритвой
Оккама, отсекает у взращиваемого им филогенетического дерева все излишние рогульки. — Прим, перев.
Глава 19. Молекулярная филогенетика 767
123456789 10111213141516
ATAGCCATAGCААССТ
ATACCCATGACAACGA настоящее
АТАСССATAGCААССА множественное
АТА G С С АТА G С А А С G А выравнивание
АТССССATAGCААССТ
2 7 4 9 11 4 16 5
TAGACGTC
TACGCCAC новое выравнивание
ТАСАССАС
TAGACGAC
ТАСАССТС
Рис. 19.14. Построение нового множественного вы-
равнивания, с тем чтобы уточнить филогенетическое
дерево. Новое выравнивание выстраивается из взятых
наугад столбцов настоящего выравнивания. Обратите
внимание, что один и тот же столбец может быть вы-
бран не раз
последовательности, которые отличаются от
исходных, но оно имеет подобную картину
изменчивости. Это означает, что, когда мы
используем такое новое выравнивание для
восстановления дерева, мы не просто вос-
производим первоначальный анализ, а стараемся получить то же самое дерево.
На практике создается 1 000 новых выравниваний, так что восстанавливается 1000
реплик дерева. Тогда каждому внутреннему узлу в первоначальном дереве может быть
дана бутстрап-оценка — эта величина отражает число раз, которое схема ветвления,
наблюдаемая в этом узле, была воспроизведена в репликах дерева. Если оценка само-
совершенствования больше чем 700/1000, то мы можем установить разумную степень
достоверности для топологии в этом конкретном внутреннем узле.
Молекулярные часы позволяют определить время расхождения предковых
последовательностей
Когда мы проводим филогенетический анализ, наша первостепенная цель состоит
в том, чтобы вывести схему эволюционных отношений между сравниваемыми последо-
вательностями ДНК. Эти отношения отражаются топологией восстановленного дерева.
Часто мы имеем также и вторичную цель: установить, когда именно предковые после-
довательности разошлись и дали современные последовательности. Эта информация
интересна в контексте эволюции генома, в чем мы убедились, когда познакомились
с эволюционной историей генов глобина человека (см. рис. 18.8). Такая информация
бывает даже еще более интересна в случаях, когда мы можем приравнять дерево генов
к видовому дереву, потому что тогда моменты времени, в которых предковые последо-
вательности разошлись, приближаются к датам событий видообразования.
Для того чтобы приписать даты к точкам ветвления в филогенетическом дереве,
мы должны использовать молекулярные часы. Гипотеза о молекулярных часах, впервые
предложенная в начале 1960-х гг., заявляет, что замены нуклеотидов (или замены ами-
нокислот, если сравниваются белковые последовательности) происходят с постоянной
частотой. Это означает, что степень различия между двумя последовательностями может
использоваться для оценки момента времени, когда они эволюционно разошлись от их
предковой последовательности. Однако, чтобы справиться с этой задачей, молекулярные
часы должны быть откалиброваны так, чтобы мы знали, сколько замен нуклеотидов
ожидается за миллион лет. Калибровка обычно выполняется со ссылкой на ископаемые
окаменелости. Например, окаменелости предполагают, что самый недавний общий
предок людей и орангутанов жил 13 миллионов лет назад. Поэтому для калибровки
молекулярных часов человека мы сравниваем поеледовательности ДНК человека и оран-
гутана, чтобы определить количество произошедших замен нуклеотидов, и затем делим
эту цифру на 13 и еще на 2 и получаем частоту замен на миллион лет (рис. 19.15).
Одно время думали, что могут быть универсальные молекулярные часы, которые
подходят для всех генов во всех организмах. Теперь мы осознаем, что молекулярные
768 Часть 4. Механизмы репликации и эволюции геномов
Человек
Орангутан
13 миллионов лет
Число замен = х
11 х
Число на родовую линию = 2
Число на родовую линию х
за миллион лет = 2x1
2x13
Рис. 19.15. Калибровка молекулярных часов человека. Число
замен определяется для пары гомологичных генов человека
и орангутана: назовем это число х. Число замен на родо-
вую линию будет поэтому равно х/2, а число на миллион
лет — х/(213)
часы разные в различных организмах и изменчи-
вы даже в пределах одного организма. Различия
между организмами могут быть результатом вре-
мен смены поколений, потому что вид с коротким
временем смены поколений, вероятно, накопит
ошибки репликации ДНК с более быстрой ско-
ростью, чем вид с более длительным временем
смены поколений. Возможно, этим и объясняется
тот факт, что грызуны имеют более быстрые мо-
лекулярные часы, чем приматы. В пределах отдельного генома изменчивость молеку-
лярных часов имеет следующие виды.
• Несинонимические замены происходят с меньшей частотой, чем синонимические.
Это связано с тем, что мутация, которая приводит к изменению в аминокислот-
ной последовательности белка, может оказаться вредной для организма, так что
накопление несинонимических мутаций в популяции сдерживается процессами
естественного отбора. Это означает, что при сравнении последовательностей генов
организмов двух видов обычно обнаруживается меньше несинонимических, чем
синонимических замен.
• Молекулярные часы для митохондриальных генов быстрее, чем для генов в ядер-
ном геноме. Это, вероятно, обусловлено тем, что митохондриям недостает многих
систем репарации ДНК, которые работают с ядерными генами.
• Молекулярные часы, кажется, ускорили свой бег за последние 1-2 миллиона лет.
Это, как думают, является артефактом, возможно связанным с постоянством в те-
чение этого периода таких мутаций, которые не сильно вредят организму и потому
выбраковываются естественным отбором относительно медленно. Очевидное
ускорение означает, что если молекулярные часы будут калиброваны по отно-
сительно древним событиям, отраженным в ископаемых окаменелостях, могут
возникнуть погрешности.
Несмотря на эти осложнения, молекулярные часы стали весьма ценным дополне-
нием при анализе деревьев, как мы увидим в разделе 19.3 при взгляде на некоторые
типичные проекты молекулярной филогенетики.
Стандартное восстановление дерева не годится для всех наборов данных
о последовательностях ДНК
Обычное построение дерева основано на допущении о том, что отношения между
членами группы последовательностей ДНК могут быть описаны простой схемой ветвле-
ния. Это допущение не всегда уместно. Например, если некоторая группа последователь-
ностей способна рекомбинировать, то могут быть произведены новые последователь-
ности, которые сочетают в себе особенности (и действительно являются её потомками)
пары предковых последовательностей, которые занимают отдаленные позиции в обыч-
Глава 19. Молекулярная филогенетика 769
а) Проблемы, связанные с рекомбинацией
б) Проблемы, связанные с существованием
предковых последовательностей
Рис. 19.16. Две проблемы, которые усложняют обычное построение деревьев
?
ном филогенетическом дереве (рис. 19.16, а). Если продукты рекомбинации будут
включены в дерево, то дихотомическая схема ветвления начинает ломаться и отноше-
ния становятся трудно различимыми. Такая ситуация представляет особую проблему
при попытках определить эволюционные отношения между членами мультигенного
семейства, поскольку эти последовательности базируются в одном геноме, возможно,
в виде тандемного массива, и поэтому вполне возможно, что в прошлом между ними
произошла рекомбинация. Обычное построение дерева является неподходящим также
и в том случае, если предковые последовательности, представленные внутренними
узлами, все еще существуют в природе (рис. 19.16, б). Такая проблема возникает редко,
когда сравниваемые гены взяты у разных видов, поскольку в таком случае внутренние
узлы представляют последовательности, которые присутствовали в предковых видах,
к настоящему времени уже вымерших, но она становится очень значимой, когда срав-
ниваемые последовательности берут из одной популяции с короткой эволюционной
историей. Эта проблема часто возникает при изучении недавней эволюции человека.
Возможное решение этих проблем состоит в использовании сети, а не дерева, что-
бы иллюстрировать эволюционные отношения между рассматриваемыми последова-
тельностями (рис. 19.17). Сеть позволяет определить предковые последовательности,
которые все еще существуют (как например, последовательность 1 на рис. 19.17), и на-
глядно иллюстрирует их отношения с последовательностями, от них происходящими.
Последовательности, которые исчезли (или которые существуют в популяции, но не
присутствуют в наборе данных), также могут быть определены. Ячеи в сети, такие, как
например, связывающая последовательности 1,3,7 и 8 на рис. 19.17, являются особенно
интересными, поскольку они отмечают последовательности, которые возникли или
путем рекомбинации, или посредством параллельных мутаций (гомоплазии) в раз-
личных эволюционных линиях.
Простые сети могут быть построены вручную, но по мере добавления большего
числа последовательностей их топологии резко усложняются (рис. 19.18), особенно если
учитываются все возможные отношения между последовательностями. Поэтому были
изобретены алгоритмы для уменьшения сложности сети без потери филогенетической
информации. К ним относятся методы обрезки ветвей, которые приписывают веса
к различным путям между парами последовательностей и удаляют те связи, которые
представляют истинные отношения с меньшей вероятностью.
77$ Часть 4. Механизмы репликации и эволюции геномов
а) 1 5 10 15 20 25 30 35 40
1 GCATTACTTTCGGTAGCGCGGAAAGGCGACGGGACTTATA
2 .............С...С........С.....А.........
3 ...........Т..............................
4 .Т.........Т..............................
5 ......................Т...................
6 ......Т......С.....................G......
7 ..........Т.............G.................
8 ........С...............G.................
Рис. 19.17. Филогенетический анализ с помощью построения сети, а) Множественное выравнивание, из
которого построена сеть, где точки указывают на тождества с последовательностью 1. б) Сеть построена
следующим образом: I) последовательность 1 используется как отправная точка; II) последовательность 2
отличается от последовательности 1 в четырех позициях и соединяется с последовательностью 1 линией
в сети; III) последовательность 3 отличается от последовательности 1 в одной позиции, а эта позиция
отличается ото всех позиций в последовательности 2. Последовательность 3 поэтому соединяется непо-
средственно с последовательностью 1; IV) последовательность 4 имеет общую с последовательностью 3
замену С->Т в позиции 11, но имеет дополнительную замену в позиции 2. Линия, ведущая к последо-
вательности 3, поэтому продолжается и последовательность 4 помещается на ее конец; V) последова-
тельность 5 имеет одну замену по сравнению с последовательностью 1, причём эта замена уникальна
и до сих пор не встречалась в построении. Последовательность 5 поэтому соединяется напрямую с по-
следовательностью 1; VI) подобным же образом последовательность 6 имеет три уникальные замены
и соединяется прямо с последовательностью 1; VII) последовательность 7 имеет два отличия от после-
довательности 1, одно из них — замена С—>Т в позиции 11, встречаемая также в последовательностях
3 и 4. Последовательность 7 поэтому соединяется непосредственно с последовательностью 3. В этой
части сети линия между последовательностями 1 и 3 представляет замену в позиции 11, таковая между
последовательностями 3 и 4 — замену в позиции 2, а та, что между последовательностями 3 и 7, — за-
мену в позиции 24; VIII) последовательность 8 делите последовательностью 7 замену A—>G в позиции 24
и имеет уникальную замену (не замеченную ни в какой из предыдущих последовательностей) в позиции 9.
Последовательность 8 должна поэтому формировать ветвь от линии между последовательностями 1 и 7.
Эта ветвь создает «пустой узел», показанный в виде маленькой точки, который представляет последо-
вательность, не существующую в наборе данных (возможно потому, что она является вымершей)
Глава 19. Молекулярная филогенетика 771
Рис. 19.18. Сложная сеть. Эта сеть была постро-
ена из последовательностей 5S рРНК морской
свеклы (Beta vulgaris ssp. maritime). Серые круж-
ки — последовательности от растений, растущих
вокруг Пул-Харбор, Великобритания, а пустые
кружки — растения на вершинах берегового уте-
са, в нескольких километрах на юг. Сеть была
построена по выравниванию 110 последова-
тельностей длиной 289 п. н. Сеть первоначаль-
но содержала 6808 связей, но их число было
уменьшено до 655, показанных здесь, путём
упрощения посредством описанного в тексте
метода отсечения ветвей. Сеть любезно предо-
ставлена Сарой Дайер
19.3. Возможности применения молекулярной филогенетики
Начиная с начала 1990-х гг. молекулярная филогенетика переживает бурный рост,
в значительной степени благодаря развитию более строгих методов построения дере-
вьев, наряду со всплеском количества информации о последовательностях ДНК, получае-
мой первоначально при помощи анализа с ПЦР, а позже — в ходе проектов расшифровки
геномов. Значение молекулярной филогенетики возросло также и в связи с успешным
приложением методов восстановления деревьев и других филогенетических приемов
к некоторым из наиболее озадачивающих проблем биологии. В этом заключительном
разделе мы и рассмотрим некоторые из таких успешных решений.
19.3.1. Примеры использования филогенетических деревьев
Сначала мы познакомимся с двумя проектами, которые иллюстрируют различные
пути применения в современной молекулярной биологии обычного восстановления
деревьев.
Филогенетика ДНК прояснила эволюционные отношения между человеком
и другими приматами
Дарвин был первым биологом, который размышлял об эволюционных отношениях
между людьми и остальными приматами. Его взгляд — согласно которому люди тесно
связаны с шимпанзе, гориллой и орангутаном, — вызвал бурные споры, когда был
впервые высказан, и потерял к себе интерес, даже среди эволюционистов, в последую-
щие десятилетия. Это неудивительно: биологи того времени были среди самых ярых
защитников антропоцентрического взгляда на наше место в животном мире.
На основании исследований окаменелостей палеонтологи еще до 1960 г. пришли
к заключению о том, что шимпанзе и гориллы — наши ближайшие родственники, но что
эта связь является далекой: расхождение, ведущее к людям с одной стороны и к шимпан-
зе и гориллам с другой, произошло около 15 миллионов лет назад. Первые подробные
молекулярные данные, полученные в ходе иммунологических исследований в 1960-е гг.
772 Часть 4. Механизмы репликации и эволюции геномов
подтвердили, что люди, шимпанзе и гориллы действительно формируют единую кладу,
но позволили предположить, что наша связь намного ближе: молекулярные часы указа-
ли, что это разветвление произошло всего лишь 5 миллионов лет назад. Это была одна
из первых попыток применить молекулярные часы к филогенетическим данным, и к ее
результату, что весьма естественно, отнеслись несколько недоверчиво. Фактически воз-
никла острая дискуссия между палеонтологами, которые верили в древнее расхождение,
о котором свидетельствовали ископаемые окаменелости, и молекулярными биологами,
которые были больше уверены в недавней дате, на которую указывали молекулярные
данные. В конечном счете этот спор «выиграли» молекулярные биологи, чья точка
зрения, согласно которой расхождение произошло около 5 миллионов лет назад, стала
общепринятой.
По мере получения все большего и большего количества молекулярных данных,
трудности в установлении точной картины эволюционных событий, которые привели
к появлению людей, шимпанзе и горилл, стали очевидными. Сравнения митохондри-
альных геномов этих трех видов — путем составления карт рестриктов (раздел 3.3.1)
и секвенирования ДНК — показали, что шимпанзе и горилла ближе связаны друг с дру-
гом, чем кто-либо из них — с человеком (рис. 19.19, а), тогда как данные гибридизации
ДНК-ДНК свидетельствовали о более близких отношениях между человеком и шимпанзе
(рис. 19.19, б). Причина таких противоречивых результатов — сильное подобие между
последовательностями ДНК этих трех видов: различие составляет менее 3 % даже для
наиболее разошедшихся областей геномов (раздел 18.4). В силу этого становится очень
сложно установить отношения однозначно.
Решение этой проблемы состояло в том, чтобы проводить сравнения между как
можно большим числом различных генов и нацелиться на те локусы, которые согласно
ожиданиям покажут наибольшую степень различия. К1997 г. было получено 14 разных
молекулярныхнаборовданных,включающих последовательности изменчивых локусов
типа псевдогенов и некодирующих последовательностей. Анализ этих наборов данных
подтвердил, что шимпанзе наиболее близкий родственник человека и наши эволюци-
онные пути разошлись 4,6-5,0 миллионов лет назад. Горилла — немного более отдален-
ный родич по второму родству, ее линия отклонилась от линии человка и шимпанзе на
0,3-2,8 миллионов лет раньше (рис. 19.19, в).
а) Данные митохондриальной ДНК б) Данные гибризизации ДНК
-------------Человек ------Человек
------Шимпанзе Шимпанзе
------Горилла ------------Горилла
в) Объединенные наборы молекулярных данных
4,6-5,0 млн
0,3-2,8 млн
Человек
Шимпанзе
---------------Горилла
Рис. 19.19. Различные интерпретации эволюционных отношений между людьми, шимпанзе и гориллами.
Сокращение: млн — миллионов лет назад
Глава 19. Молекулярная филогенетика 773
Происхождение СПИДа
Глобальная эпидемия синдрома приобретенного иммунодефицита (СПИДа) кос-
нулась жизни каждого из нас. СПИД вызывается вирусом иммунодефицита человека 1
(ВИЧ-1) — ретровирусом (раздел 9.1.2), который заражает клетки, отвечающие за им-
мунную реакцию. После того как в начале 1980-х гг. было показано, что СПИД обусловлен
ВИЧ-1, ученые начали выстраивать теории о происхождении этой болезни. Эти теории
отталкивались от открытия того факта, что подобные вирусы иммунодефицита имеют-
ся у таких приматов, как шимпанзе, серый мангабей, мандрил и у различных обезьян.
Эти вирусы иммунодефицита обезьян (ВИО) не являются болезнетворными для своих
обычных хозяев, но, как думали, если такой вирус был передан человеку, то в пределах
этого нового вида этот вирус, возможно, приобрел новые свойства, такие как способ-
ность вызывать болезнь и быстро распространяться по всей популяции.
Геномы ретровирусов накапливают мутации относительно быстро, потому что
обратная транскриптаза — фермент, который копирует РНКовый геном, содержащий-
ся в вирусной частице, в ДНК-версию, которая встраивается в геном хозяина (см. раз-
дел 9.1.2), — не обладает эффективным корректирующим действием (раздел 15.2.2) и,
следовательно, часто делает ошибки, когда осуществляет РНК-зависимый синтез ДНК.
Это означает, что в ретровирусах молекулярные часы бегут быстро, и геномы, которые
разошлись совсем недавно, показывают достаточное для проведения филогенетического
анализа несходство нуклеотидных последовательностей. Даже при том что эволюци-
онный период, который нас интересует, имеет протяженность менее 100 лет, геномы
ВИЧ и ВИО содержат достаточно данных для того, чтобы было возможно вывести их
эволюционные отношения при помощи филогенетического анализа.
Отправной точкой для такого филогенетического анализа является РНК, извле-
ченная из вирусных частиц. Поэтому для преобразования РНК в ее ДНК-копию и по-
следующего размножения ДНК используется РТ-ПЦР (см. техническое примечание 5.1),
что позволяет получить достаточное количество материала для секвенирования. По
данным сравнения между последовательностями вирусных ДНК было восстановлено
дерево, представленное на рис. 19.20. Это дерево имеет ряд интересных особенностей.
Во-первых, оно показывает, что различные образцы ВИЧ-1 имеют несколько разнящие-
ся последовательности, при этом сами образцы формируют плотную группу — почти
звездообразную структуру, — которая расходится от одного из концов корневого дерева.
Такая звездообразная топология подразумевает, что мировая эпидемия СПИДа нача-
лась с очень небольшого числа вирусов (возмож-
но, только одного), которые распространились
и разнообразились начиная с момента попадания
в популяцию человека. Наиболее близкий виру-
су ВИЧ-1 родственник среди приматов — ВИО
шимпанзе; это означает, что этот вирус переско-
чил через межвидовой барьер между шимпанзе
Рис. 19.20. Филогенетическое дерево, восстановленное
из последовательностей геномов вирусов ВИЧ и ВИО.
Эпидемия СПИДа происходит из-за вируса иммунной не-
достаточности типа ВИЧ-IM. Вирус ZR59 помещен около
корня звездообразной структуры, формируемой генома-
ми этого типа
774 Часть 4. Механизмы репликации и эволюции геномов
и человеком и запустил эпидемию СПИДа. Однако эта эпидемия началась не сразу: от-
носительно длинная, непрерывная ветвь соединяет центр звезды ВИЧ-1 с внутренним
узлом, ведущим к соответствующей последовательности ВИО, предполагая, что после
передачи людям ВИЧ-1 подвергся инкубационному периоду, когда он оставался в рам-
ках маленькой части общемировой популяции человека, предположительно в Африке,
прежде чем начать свое быстрое распространение в остальные части света. Вирусы ВИО
прочих приматов не столь близко связаны с ВИЧ-1, но один из них, ВИО серого мангабея,
группируется на дереве вместе со вторым вирусом иммунодефицита человека ВИЧ-2.
Кажется, что ВИЧ-2 был перенесен в популяцию человека независимо от ВИЧ-1 и от
иной обезьяны-хозяина. ВИЧ-2 также способен вызывать СПИД, но пока еще не стал
эпидемическим в мировом масштабе.
Интригующее дополнение к дереву ВИЧ/ВИО было сделано в 1998 г., когда была
секвенирована РНК экстракта ВИЧ-1 из образца крови, взятого в 1959 г. у африканского
мужчины. РНК была сильно фрагментирована и могла быть получена только короткая
последовательность ДНК, но этого было достаточно для помещения этой последовател ь-
ности в филогенетическом дереве (см. рис. 19.20). Эта последовательность, названная
ZR59, прикрепляется к дереву короткой ветвью, которая выходит примерно из центра
радиальной структуры ВИЧ-1. Такое положение указывает на то, что последователь-
ность ZR59 представляет собой одну из самых ранних версий ВИЧ-1 и показывает, что
глобальное распространение ВИЧ-1 было уже в полном разгаре к 1959 г. Более поздний
и более всесторонний анализ последовательностей ВИЧ-1 показал, что его распростра-
нение началось в период между 1915 и 1941 гг., причем наилучшая оценка пришлась
на 1931 г. Точное установление даты таким способом позволилоэпидемиологам начать
исследование исторических и социальных условий, которые, возможно, обусловили
начало эпидемии СПИДа.
19.3.2. Молекулярная филогенетика как инструмент изучения предысто-
рии человека
Теперь мы обратим наше внимание на применение молекулярной филогенетики во
внутривидовых исследованиях: изучение эволюционной истории членов одного вида.
Мы могли бы выбрать любой из нескольких различных организмов, чтобы иллюстри-
ровать подходы и области приложения внутривидовых исследований, но многие люди
смотрят на Ното sapiens как на самый интересный организм, так что мы исследуем во-
прос о том, как молекулярная филогенетика используется, чтобы вывести источники
происхождения современных людей и географические картины их недавних переселений
в Старом и Новом Свете.
Изучение генов в популяциях
В любом из приложений молекулярной филогенетики гены, выбираемые для ана-
лиза, должны показывать изменчивость в исследуемых организмах. Если нет никакой
изменчивости, то нет и никакой филогенетической информации. Это представляет про-
блему во внутривидовых исследованиях, потому что все сравниваемые организмы суть
члены одного биологического вида и в силу этого обладают значительным генетическим
подобием, даже если данный вид разделился на популяции, которые скрещиваются лишь
время от времени. Это означает, что последовательности ДНК, которые используются
в филогенетическом анализе, должны быть наиболее изменчивыми из доступных.
У человека есть три основные категории таких последовательностей.
Глава 19. Молекулярная филогенетика 775
• Многоаллельные гены, такие как члены семейства HLA (раздел 3.2.1), которые
существуют во многих различных формах последовательности.
• Микросателлиты, которые эволюционируют не за счет мутаций, а путем проскаль-
зывания репликации (раздел 16.1.1). Клетки, кажется, не имеют никакого восстано-
вительного механизма для устранения последствий проскальзывания репликации,
так что новые микросателлитные аллели производятся относительно часто.
• Митохондриальная ДНК, которая, как было упомянуто в разделе 19.2.2, накапливает
замены нуклеотидов относительно быстро, потому что митохондрии не обладают
многими из систем репарации, которые замедляют молекулярные часы в ядрах
клеток человека. Варианты митохондриальной ДНК, имеющиеся у одного вида,
называют гаплогруппами.
Важно обратить внимание на то, что отнюдь не потенциальная возможность из-
менения является определяющей при решении вопроса о пригодности этих локусов
в филогенетическом анализе, а тот факт, что различные аллели или гаплогруппы каждого
локуса сосуществуют в пределах всей популяции. Поэтому такие локусы полиморфны,
и информация об отношениях между разными особями может быть получена путем
сравнения комбинаций аллелей и (или) гаплогрупп, которыми эти особи обладают. Но-
вые аллели и гаплогруппы появляются в популяции из-за мутаций, которые происходят
в репродуктивных клетках отдельных организмов. Каждый аллель имеет свою собствен-
ную частотность аллеля, которая изменяется во времени в результате естественного
отбора и случайного генетического дрейфа. Естественный отбор происходит из-за
различий в приспособленности (способности организма выживать и воспроизводить-
ся), которые, согласно Дарвину, приводят к «сохранению благоприятных изменений
и отбраковке неблагоприятных изменений». Поэтому естественный отбор уменьшает
частотности аллелей, которые уменьшают приспособленность организма, и увеличивает
частотность аллелей, которые улучшают приспособленность. В действительности только
некоторые новые аллели, которые появляются в популяции, дают положительный вклад
в приспособленность, в то время как большинство из новых алллей не находятся под
отбором, а их частота меняется из-за случайного генетического дрейфа, обусловленного
произвольным характером рождения, смерти и воспроизводства (рис. 19.21).
Будь то связано с естественным отбором или сопряжено со случайным генетическим
дрейфом, один аллель начинает преобладать в популяции и в конечном счете достигает
частоты 100 %. В таком случае данный аллель становится закрепленным. Математиче-
ские модели предсказывают, что с течением времени в популяции закрепляются разные
аллели, что выливается в череду смен аллелей. Если некоторый вид распадается на
две популяции, которые не могут интенсивно скрещиваться, то частоты аллелей в этих
двух популяциях будут изменяться по-разному, так что по смене нескольких десятков
Рис. 19.21. Влияние генетического дрейфа на частоту ал-
лелей. График показывает результаты моделирования
частотности пары аллелей более чем 100 поколений для
популяций, содержащих 20 и 1000 индивидуумов. В обе-
их популяциях один аллель в конечном счете становится
закрепленным, а другой теряется, но время, необходимое
для того чтобы это произошло, сильно зависит от разме-
ра популяции. Переработано из M.Jobling et al., Human
Evolutionary Genetics
776 Часть 4. Механизмы репликации и эволюции геномов
поколений эти две популяции будут иметь отличительные генетические признаки.
В конечном счете в этих двух популяциях начинают происходить смены аллелей разного
рода, но даже прежде, чем это случается, мы можем проводить различие между ними
по показателям разности частоты аллелей. Эти различия могут использоваться для
установления времени, когда произошел раскол популяции, и определить, испытали
ли одна или обе популяции явление бутылочного горлышка (период, когда размер
популяции значительно сокращается).
Происхождение современного человека — из Африки или нет?
Представляется вполне достоверным, что прародитель человека находился в Аф-
рике, потому что именно здесь были найдены в раскопках все наиболее древние ока-
менелости доисторического человека. Палеонтологические данные показывают, что
гоминиды впервые вышли за пределы Африки более 1 миллиона лет назад, но они не
были современными людьми, они принадлежали к более раннему виду, названному
Homo erectus (рис. 19.22). Они были первыми гоминидами, которые территориально
рассредоточились и в конечном счете распространились во все части Старого Света.
События, которые последовали за расселением Homo erectus, противоречивы. Ис-
ходя из сравнений, опирающихся на ископаемые черепа и кости, палеонтологи заклю-
чил и, что популяции Homo erectus, которые расположились в различных частях Старого
Света, дали начало современным популяциям человека этих областей в итоге процесса
так называемой полицентрической эволюции (рис. 19.23, а). Возможно, в некоторой
мере происходило скрещивание между людьми из различных географических областей,
но по большому счету эти различные популяции оставались обособленными в течение
всей своей эволюционной истории.
Первые сомнения относительно гипотезы о полицентричном
происхождении человека возникли в связи с новыми истолко-
ваниями данных об ископаемых окаменелостях. Дискуссия обо-
стрилась после публикации в 1987 г. филогенетического дерева,
восстановленного по данным митохондриальных полиморфизмов
длин фрагментов рестрикции (RFLP), полученных от 147 чело-
век, представляющих популяции со всех частей мира. Это дерево
(рис. 19.24) подтверждало, что предки современных людей жили
в Африке, но предполагало, что они все еще оставались там при-
близительно 200000 лет назад. Этот вывод был сделан, приме-
няя митохондриальные молекулярные часы к дереву, которое
показало, что предковая митохондриальная ДНК — та, от которой
произошли все современные митохондриальные ДНК, — существо-
вала между 140 000 и 290 000 лет назад. Дерево показало, что этот
митохондриальный геном находился в Африке, так что человек,
который обладал им, — так называемая митохондриальная Ева
(она должна была быть женщиной, потому что митохондриальная
ДНК наследуется только по женской линии), — должно быть, был
африканцем.
Рис. 19.22. Скелет юноши Homo erectus. Этот экземпляр, также называемый
«Нариокотомским мальчиком», имеет возраст около 1,6 миллиона лет и был
найден около озера Туркана в Кении. Изображение любезно предоставлено
библиотекой Американского музея естествознания
Глава 19. Молекулярная филогенетика 777
а) Полицентрическая эволюция
Homo erectus
б) Моноцентрическая гипотеза
о происхождении из Африки
АФРИКА
I------1—
ЕВРОПА АФРИКА
1 I
Ното Ното
sapiens sapiens
---у - 1 млн
АЗИЯ
I
Ното
sapiens
I
Современные люди
Homo erectus
I
АФРИКА
I--------1--------1 млн
ЕВРОПА АФРИКА АЗИЯ
I I
, Homo JL
sapiens
(--------1.............0,1 млн
Современные люди
Рис. 19.23. Две конкурирующие гипотезы о происхождении современных людей
Открытие прародительницы митохондриального генома породило новый сценарий
происхождения современных людей. Вместо того чтобы эволюционировать параллель-
но по всему миру как предполагает полицентрическая гипотеза, Homo sapiens, как за-
являет гипотеза о происхождении из Африки, появился в Африке, и члены этого вида
впоследствии переместились в остальную часть Старого Света между 100000 и 50000
лет назад, вытесняя потомков Homo erectus,
с которыми они сталкивались на своем пути
(см. рис. 19.23,6).
Столь радикальная перемена во взгля-
дах неизбежно вызвала споры. Когда дан-
ные о RFLP были исследованы другими мо-
лекулярными филогенетиками, стало ясно,
что первичный компьютерный анализ был
некорректен и что по этим данным могло
быть восстановлено несколько совершенно
Рис. 19.24. Филогенетическое дерево, восстанов-
ленное поданным о митохондриальных RFLP, полу-
ченных у 147 современных людей. Согласно выводу,
предковая митохондриальная ДНК существовала
в Африке из-за разветвления в дереве между се-
мью митохондриальными геномами современных
африканцев, помещенных под предковой последо-
вательностью, а все остальные геномы над ней. По-
скольку эта нижняя ветвь является чисто африкан-
ской, было выведено заключение, что предок был
также африканский. Шкалы в основании указывают
расхождение последовательностей, из которого,
используя митохондриальные молекулярные часы,
возможно приписать даты к точкам ветвления в де-
реве. Часы предполагают, что предковая последо-
вательность существовала от 140000 до 290000
лет назад. Перепечатано с разрешения Cann et al.
(1997), Nature, 325, 31-36
Расхождение
поел едовател ьностей, %
Расхождение
последовател ьностей, %
778 Часть 4. Механизмы репликации и эволюции геномов
разных деревьев, причем некоторые из них не имели корня в Африке. Этой критике
противостояли наборы более подробных данных о последовательностях митохондри-
альной ДНК, большинство которых согласуется с относительно недавним африканским
происхождением человека и, таким образом, поддерживают гипотезу о его происхожде-
нии из Африки, а не гипотезу о полицентрической эволюции. Интересное дополнение
«митохондриальной Еве» было получено в итоге исследований Y-хромосомы, которые
предполагают, что «Y-хромосомный Адам» также жил в Африке около 200 000 лет назад.
Конечно, эти Ева и Адам не равнозначны библейским героям и ни в коем случае не были
единственными людьми, жившими в то время: они попросту были индивидуумами, ко-
торые несли в себе предковые митохондриальную ДНК и Y-хромосомы, давшие начало
всем митохондриальным ДНК и Y-хромосомам, существующим сегодня. Важный момент,
что эти предковые ДНК все еще находились в Африке много позже распространения
Homo erectus по Евразии.
Исследования митохондриальной ДНК и Y-хромосомы, кажется, дают веские данные
в поддержку теории об африканском происхождении человека. Но в результате анализа
ядерных генов, расположенных не на Y-хромосоме, возникли осложнения. Например,
последовательности р-глобина дают намного более раннюю дату (800000 лет назад)
жизни общего предка, а исследования гена Х-хромосомы PDHA1 помещают предковую
последовательность в период 1900000 лет назад. В настоящее время молекулярные
антропологи дискутируют о значимости этих результатов. Для окончательного от-
вета на этот вопрос требуется больше данных, и, надо надеяться, когда-нибудь все же
произойдет некоторого рода Великий Синтез.
Неандертальцы не являются предками современных европейцев
Неандертальцы — вымершие гоминиды, жившие в Европе между 300 000 и 30 000
лет назад (рис. 19.25). Они происходили от популяций Homo erectus, которые оставили
Африку около 1 миллиона лет назад и, согласно гипотезе о происхождении человека
из Африки, были вытеснены, когда современные люди достигли Европы приблизи-
тел ьно 50 000 лет назад. Поэтому одно из
предсказаний гипотезы об африканском
происхождении человека гласит, что нет
никакой генетической непрерывности
между неандертальцем и современны-
ми людьми, живущими сегодня в Европе.
Если вспомнить, что последний неандер-
талец вымер 30 000 лет назад, то имеется
ли какой-либо способ, которым мы мо-
жем проверить эту гипотезу?
Древняя ДНК позволила ответить
на этот вопрос. Уже несколько лет было
известно, что молекулы ДНК могут пере-
Рис. 19.25. Череп 40-50-летнего мужчины неан-
дертальца. Возраст экземпляра приблизительно
50000 лет и он был найден в окрестности Ла-
Шапель-о-Сенс, Франция. Изображение любезно
предоставлено Джоном Ридером из Библиотеки
фотографий «Сайенс»
Глава 19. Молекулярная филогенетика 779
жить смерть организма, в котором они содержатся, будучи восстановимыми столетия
и, возможно, тысячелетия спустя в виде коротких деградированных фрагментов, со-
хранившихся в костях и других биологических остатках. В таком образце никогда не
бывает очень много древней ДНК — возможно, не больше чем несколько сотен геномов
на грамм костной ткани, — но это не должно нас беспокоить, потому что мы всегда можем
использовать ПЦР, чтобы приумножить эти ничтожно малые крохи в более ощутимые
количества, из которых мы можем получить последовательности ДНК.
Изучение древней ДНК сопровождалось спорами все последние 15 лет. В начале
1990-х гг. было много сообщений о ДНК древнего человека, обнаруженной в костях и дру-
гих археологических экземплярах, но часто оказывалось, что то, что было размножено
при помощи ПЦР, совершенно не являлось древней ДНК, но представляло собою загряз-
няющую современную ДНК, оставленную на экземпляре археологом, который выкопал
его, или молекулярным биологом, который извлекал ДНК из образца. Всемирный успех
фильма «Парк Юрского периода» породил сообщения о ДНК в насекомых, сохранив-
шихся в янтаре и даже в костях динозавров, но все эти заявления теперь подвергнуты
сомнению. Многие биологи начали задаваться вопросом о том, существует ли древняя
ДНК вообще, но постепенно стало ясно, что, если работа проводится с исключительной
тщательностью, то иногда возможно извлечь подлинную древнюю ДНК из экземпляров,
имеющих возраст вплоть до 50 000 лет или около того. Этот возрастной порог как раз
подходит для исследования нескольких неандертальцевых костей.
Первому экземпляру неандертальца, отобранному для изучения, как полагали, было
от 30 000 до 100 000 лет. Извлечение ДНК было выполнено из фрагмента кости весом око-
ло 400 мг. Чтобы определить, присутствовали ли молекулы ДНК человека и, если так, то
сколько, использовался метод, известный под названием количественная ПЦР. Результаты
показали, что фрагмент кости содержал приблизительно 1300 копий митохондриального
генома неандертальца, и этого было достаточно для того, чтобы уверить исследователей
в целесообразности проведения анализа последовательности. Процедуры ПЦР были поэтому
направлены на то, что, согласно ожиданиям, являлось наиболее изменчивой частью мито-
хондриального генома неандертальца. Поскольку ожидалось, что ДНК будет раздроблена
на очень короткие куски, последовательность выстраивалась по частям с выполнением
девяти перекрывающихся ПЦР, при этом ни в одной из них не размножались отрезки ДНК
длиннее 170 п.н., но вместе они дали полную длину 377 п.н.
По результатам анализа было построено филогенетическое дерево, чтобы сравнить
последовательность, полученную из кости неандертальца, с последовательностями
шести гаплогрупп митохондриальной ДНК от современных людей (рис. 19.26). По-
следовательность неандертальца была помещена в собственную ветвь, соединенную
с корнем дерева, но не связана непосредственно ни с какой из последовательностей со-
временного человека. Затем, чтобы сравнить последовательность неандертальца с 994
последовательностями от современных людей, было
построено огромное множественное выравнивание.
Различия были поразительны. Последовательность
неандертальца отличалась от современных последо-
вательностей в среднем на 27,2 ± 2,2 позиции нуклео-
тидов, тогда как современные последовательности,
Рис. 19.26. Отношения между неандертальцами и современ-
ными людьми, восстановленные подревней последователь-
ности ДНК, полученной из кости неандертальца
Неандерталец
Современные люди
780 Часть 4. Механизмы репликации и эволюции геномов
которые прибыли со всех континентов, не только из Европы, отличались друг от друга
лишь на 8,0 ±3,1 позиции. Когда митохондриальная ДНК от второго скелета неандерталь-
ца была исследована, были получены схожие результаты. Степень различия между ДНК
неандертальца и современного европейца несовместима с концепцией, согласно которой
современные европейцы происходят от неандертальцев, и решительно поддерживает
гипотезу об африканском происхождении человека1). Сторонники полицентрической
эволюции, однако, не имеют полной уверенности в этом, и дебаты о происхождении
современного человека продолжаются.
Схемы более поздних миграций в Европу также спорны
По какому бы эволюционному пути ни шел человек, а только 40 000 лет назад со-
временные люди уже населяли большую часть Европы. Об этом свидетельствуют ока-
менелости и археологические раскопки. Следующим спорным моментом предыстории
человека является вопрос о том, были ли эти популяции вытеснены примерно 30000
лет спустя другими людьми, переселявшимися в Европу из Среднего Востока.
Данный вопрос сосредоточен на процессе распространения земледелия в Европу.
Переход от охоты и собирательства к сельскому хозяйству произошел на Среднем Вос-
токе приблизительно 9000-10000 лет назад, когда ранние неолитические крестьяне
начали выращивать такие зерновые культуры, как пшеница и ячмень. После становления
на Среднем Востоке сельское хозяйство распространилось в Азию, Европу и Северную
Африку. В ходе поисков свидетельств земледелия в местах археологических раскопок
(например, попыток найти остатки выращиваемых растений или орудий, используемых
в сельском хозяйстве) оказалось возможным проследить распространение сельского
хозяйства в Европу по двум маршрутам: один вдоль побережья Средиземного моря в Ита-
лию и Испанию и второй через долины Дуная и Рейна в Северную Европу (рис. 19.27).
Как же распространялось сельское хозяйство? Самое простое объяснение — что
«фермеры» переселялись из одной части Европы в другую, взяв с собой свои орудия,
животных и зерно, и вытесняли аборигенные доземледельческие общины, которые
населяли Европу в то время. Эта модель волны продвижения была первоначально одо-
Рис. 19.27. Распространение
земледелия из Среднего Вос-
тока в Европу. Темно-зеленая об-
ласть обозначает так называемый
«Полумесяц плодородия» — об-
ласть Среднего Востока (Месопо-
тамская низменность), где многие
из сегодняшних зерновых куль-
тур (пшеница, ячмень и так далее)
произрастали в диком виде и где
эти растения, как думают, впер-
вые начали культивировать
Самые последние данные, связанные с секвенированием неандертальца и человека из Денисовой
пещеры (на Алтае), показывают, что все совсем не просто — по-видимому, кроманьонцы (современный вид
человека) после выхода из Африки скрещивались со своими родственными видами. — Прим. ред.
брена генетиками благода-
ря результатам крупномас-
штабного анализа частоты
Глава 19. Молекулярная филогенетика 781
аллелей 95 ядерных генов в популяциях, разбросанных по всей Европе. Столь объемный
и сложный набор данных не может быть проанализирован никаким значимым способом
при помощи обычного построения дерева, но вместо этого должен быть исследован
более прогрессивными статистическими методами, основанными более на биологии
популяций, нежели на филогенетике. Одна из таких процедур — анализ главных компо-
нентов, который заключается в попытках распознать в наборе данных те картины рас-
пределения, которые соответствуют неравномерному географическому распределению
аллелей (возможно, неравномерное распределение является показателем переселения
популяции в прошлом). Наиболее поразительная картина в пределах европейского на-
бора данных, охватывающая приблизительно 28 % общей генетической изменчивости,
представляет собой градацию значений частот аллелей по Европе (рис. 19.28). Такая
картина говорит о том, что переселение людей происходило или со Среднего Востока
к северо-востоку Европы, или в обратном направлении. Поскольку первая возможность
совпадает с распространением сельского хозяйства, что показывают археологические
свидетельства, этот первый главный компонент рассматривали как серьезное подкре-
пление модели волны продвижения.
Анализ выглядел убедительным, но были высказаны два возражения. Первое
говорило о том, что данные не содержали никакого указания на то, когда выведенное
логическим путем переселение имело место, так что связь между первым главным ком-
понентом и распространением земледелия основывалась исключительно на картине
градации аллелей и не опиралась на какие-либо дополнительные факты, относящиеся
к периоду, когда эта градация установилась. Второе критическое замечание относилось
к результатам второго исследования европейских популяций человека, которое вклю-
чало временное измерение. Это исследование было посвящено изучению гаплогрупп
митохондриальной ДНК в 821 индивидууме из различных популяций со всей Европы.
Его результаты не смогли подтвердить градацию частоты аллелей, обнаруженных в на-
боре данных о ядерной ДНК, и вместо этого позволили предположить, что европейские
Рис. 19.28. Генетическая градация в современной Европе
782 Часть 4. Механизмы репликации и эволюции геномов
популяции оставались относительно статическими за последние 20 000 лет. Уточнение
этой работы привело к открытию того факта, что в современной европейской популя-
ции преобладают одиннадцать гаплогрупп митохондриальной ДНК. Эти одиннадцать
гаплогрупп являются поднабором приблизительно 100 или около того гаплогрупп,
наличествующих в целом по всему миру (рис. 19.29). Большинство из этих гаплогрупп
связано с определенными географическими областями, и подробный статистический
анализ их распределений и отношений может быть использован для выведения времен
их происхождения, которые для европейских гаплогрупп, как думают, указывают дату,
в которую каждая из них проникла в Европу (рис. 19.30). Самая древняя гаплогруппа,
названная U, впервые появилась в Европе приблизительно 50000 лет назад, что со-
впадает с периодом, в который, согласно археологическим свидетельствам, первые
современные люди переместились на этот континент, когда ледовые щиты отошли на
север в конце последнего глобального оледенения. Самые молодые гаплогруппы, J и Т1,
которые при своем 9000-летнем возрасте могут соответствовать зарождению земледе-
лия, имеют лишь только 8,3 % обладателей среди популяции современных европейцев,
а это наводит на мысль о том, что распространение сельского хозяйства в Европу не
было подобно огромной волне продвижения, на которую указывает анализ главных
компонентов. Вместо этого, как теперь думают, сельское хозяйство было принесено
в Европу меньшей группой «первопроходцев», которые смешивались с существующими
доземледельческими общинами, а не вытесняли их.
Рис. 19.29. Гаплогруппы митохондриальной ДНК человека. Сеть показывает отношения между главными
митохондриальными гаплогруппами в современной популяции человека. Цветная маркировка указывает
географическую область, в пределах которой отмеченная гаплогруппа наиболее распространена
Глава 19. Молекулярная филогенетика 783
U (5,7 %)
HV (5,4 %)
I (1,7%)
U4 (3,0 %)
Н (37,7 %)
Н-16304 (3,9%)
Т (2,2 %)
К (4,6 %)
Т2 (2,9 %)
J (6,1 %)
Т1 (2,2 %)
Зарождение земледелия
0 10000 ' 20000 ’ 30000 * 40000 * 50000
Лет до настоящего времени
Рис. 19.30. Одиннадцать главных европейских митохондриальных гаплотипов. Показано рассчитанное
время происхождения каждого гаплотипа: закрашенные и пустые части каждой полосы показывают
различные степени достоверности. Проценты относятся к размерам современных европейских популя-
ций с каждым гаплотипом. Все гаплотипы, кроме J и Т1, пришли в Европу до зарождения земледелия,
датируемого периодом 9000-10000 лет назад
Миграции доисторического человека в Новый Свет
Наконец, мы исследуем совершенно иной набор противоречий, окружающих гипо-
тезы о схемах переселений человека, приведших к первому вхождению людей в Новый
Свет. Нет никаких данных о распространении Ното erectus в Северную и Южную Америки,
так что предполагается, что люди были не вхожи в Новый Свет, пока современный Ното
sapiens не появился в ходе эволюции в Азии или не мигрировал в нее. Берингов пролив
между Азией и Северной Америкой довольно мелкий, и если уровень моря понизится ещё
на 50 метров, будет возможно перейти его с одного континента на другой. Полагается,
что этот Берингов перешеек и был маршрутом, избранным первыми людьми, которые
отважились открыть для себя Новый Свет.
Уровень моря был на 50 или более метров ниже его нынешнего уровня большую часть
последнего ледникового периода (прибл изител ьно между60 000 и 11000 лет назад), но поч-
ти все это время данный маршрут был непроходим из-за нагромождения льда не на самом
перешейке, а на пространстве, где теперь расположены Аляска и северо-западная Канада.
Также свободные от ледника части Северной Америки, скорее всего, были арктиче-
скими в течение большей части этого периода и могли обеспечить мигрантов лишь малым
количеством дичи для охоты и весьма скудными запасами древесины, которую они могли
пустить на костры. Но в течение краткого периода приблизительно 12000 лет назад Бе-
рингов перешеек был открыт, в то время когда климат теплел и ледники отступали, так
что существовал свободный ото льда коридор, ведущий от Чукотки через Берингов про-
лив и Аляску в центральную часть Северной Америки (рис. 19.31). Эти соображения вкупе
с отсутствием археологических свидетельств присутствия людей в Северной Америке до
момента времени 11500 лет назад привели к принятию момента времени «около 12000
лет назад» как даты первого вступления людей в Новый Свет. Последние данные, говоря-
щие о занятии этой территории людьми раньше, чем 11500 лет назад, побудили ученых
в некоторой степени пересмотреть прежнюю гипотезу, но все еще принято считать, что
массовое переселение популяции людей в Северную Америку, от которой происходят все
современные коренные жители Америки, произошло около 12000 лет назад.
784 Часть 4. Механизмы репликации и эволюции геномов
Рис. 19.31. Возможный маршрут миграции людей в Новый Свет. В течение короткого периода приблизи-
тельно 12000 лет назад мост через Берингов пролив был открыт в то же самое время, что и свободный
ото льда коридор в центральную Северную Америку. Темно-зеленые области на рисунке показывают
области земли, открытой в то время, но в настоящее время опустившейся ниже уровня моря
Какую информацию дает молекулярная филогенетика? Первые исследования в этой
области были выполнены в конце 1980-х гг. при использовании данных RFLP митохон-
дриальных геномов. Результаты этого анализа указали на то, что коренные американцы
происходят от азиатских предков, и выявили четыре различные митохондриальные га-
плогруппы, названные А, В, С и D, среди популяции в целом. По итогам лингвистических
изысканий к тому времени уже было известно, что американские языки могут быть
подразделены на три разные группы, а это предполагает, что современные коренные
американцы происходят от трех групп людей, каждой из которых присущ свой язык.
Сделанный на основании молекулярных данных вывод о том, что, возможно, фактически
существовали четыре предковые популяции, не слишком встревожил исследователей.
Первый значительный набор данных о последовательностях митохондриальной ДНК
был получен в 1991 г. и позволил применить строгий метод молекулярных часов. Он
показал, что переселения в Северную Америку произошли на временном отрезке между
15000 и 8 000 лет назад, что согласуется с археологическими свидетельствами, согласно
которым люди не жили на этом континенте ранее 11500 лет назад.
Эти первые филогенетические анализы подтвердили дополнительные данные
(или по крайней мере не слишком им противоречили), полученные в ходе археологи-
ческих и лингвистических исследований. Однако добавочные молекулярные данные,
получаемые с 1992 г., скорее затуманивали, чем проясняли данный вопрос. Например,
различные наборы данных обеспечили самые разные оценки числа переселений в Север-
ную Америку. Один особенно всесторонний анализ, основанный на митохондриальной
ДНК, сообщил цифру всего лишь одного переселения и позволил предположить, что оно
произошло между 25 000 и 20 000 лет назад — намного раньше, чем традиционная дата
периода переселения. В ходе первых исследований Y-хромосом «коренному американ-
цу Адаму» — носителю Y-хромосомы, которая является предковой для большинства,
Глава 19. Молекулярная филогенетика 785
если не всех, Y-хромосом современных коренных американцев, — была приписана дата
приблизительно 22 500 лет назад. Эти результаты все еще служат предметом горячих
споров. Некоторые молекулярные антропологи принимают заключение о том, что
люди закрепились в Северной Америке около 20 000 лет назад, то есть намного ранее,
чем срок, обозначенный археологическими и предыдущими генетическими данными.
Другие полагают, что вместе взятые генетические данные согласуются с двумя пере-
селениями: первое произошло в период от 20 000 до 15000 лет назад и вобрало в себя
все четыре гаплогруппы, а второе переселение последовало в более поздний период,
включив в себя те же самые гаплогруппы и закончившееся видоизменением графических
распределений этих гаплогрупп в Северной Америке.
Заключение
Молекулярная филогенетика использует молекулярную информацию и выводит
из нее эволюционные отношения между генами и между организмами. Молекулярная
информация, такая как иммунологические данные, белковые электрофореграммы и дан-
ные гибридизации ДНК-ДНК, использовалась в филогенетике долгие годы, но сегодня
большинство сравнений проводится между последовательностями ДНК. Сначала вы-
равнивают группы последовательностей и определяют полиморфизмы. Эта информация
переводится в числовые данные (например, в матрицу расстояний), которые могут быть
проанализированы математически, с тем чтобы восстановить дерево, показывающее
эволюционные отношения между рассматриваемыми последовательностями. Суще-
ствует несколько методов восстановления деревьев (такие как методы объединения
соседей и максимальной бережливости), которые опираются на различные подходы
к определению наиболее правдоподобного дерева для конкретного набора данных.
Некоторые из таких методов требуют большого объема вычислений и имеют основное
ограничение, состоящее в том, что для проведения строгого анализа выравнивания
необходим колоссальный объем оперативной памяти. Иногда возможно применить
молекулярные часы к дереву, чтобы приписать даты к точкам ветвления, но скорость
бега молекулярных часов своя у каждого организма и даже в каждой из частей одного
генома, и, следовательно, нужна особая внимательность, чтобы избежать ошибок.
Некоторые наборы филогенетических данных, такие как последовательности членов
мультигенного семейства, которые могут рекомбинировать друг с другом, не могут быть
представлены обычным деревом, но могут быть изображены в виде сети.
Молекулярная филогенетика разрешила давнишние вопросы об эволюционных от-
ношениях между приматами и дает ценную информацию об эволюции ВИЧ и, посредством
вывода, об истории эпидемии СПИДа. Молекулярная филогенетика может быть приложена
также и к исследованиям предыстории человека. В ходе сравнения последовательностей
митохондриальных геномов и таковых Y-хромосом было выведено, что все современные
люди происходят от популяции, которая жила в Африке приблизительно 200000 лет на-
зад и которая переселилась из Африки 100 000-50 000 лет назад, вытесняя потомков Ното
erectus, живших всюду по Старому Свету в то время. Исследования сохранившейся ДНК из
окаменелостей показали, что современные европейцы не происходят напрямую от неандер-
тальцев. Появление сельского хозяйства в Европе совпадает с относительно мелкомасштаб-
ным передвижением людей из Среднего Востока, а не с крупномасштабным переселением,
сопровождаемым вытеснением местных охотников-собирателей, как сначала думали. Люди
вступили в Новый Свет из Азии во время последнего ледникового периода — они прошли
по перешейку, ныне покрытому водами Берингова пролива.
786 Часть 4. Механизмы репликации и эволюции геномов
Задания для закрепления материала
*Ответы к нечетным вопросам могут быть найдены в приложении
Вопросы закрытого типа
19.1 *. Которая из следующих особенно-
стей не относится к молекулярной
филогенетике?
а. Использование молекулярных
данных для восстановления фи-
логенетического дерева.
Ь. Использование молекулярных
данных, чтобы понять генети-
ческую подоплеку изменчивых
фенотипов.
с. Использование молекулярных
данных для выведения эволю-
ционных отношений между ге-
номами.
d. Применение строгих математиче-
ских методов к анализу изменчи-
вых признаков.
19.2 . Какой из молекулярных методов
был использован при выведении
отношений между организмами
раньше всех остальных?
а. Секвенирование ДНК.
Ь. Электрофорез белка.
с. Иммунологические пробы.
d. Гибридизация ДНК-ДНК.
19.3 *. Какой из следующих примеров пред-
ставляет конвергентную эволюцию
(гомоплазию)?
а. Крылья птиц и летучих мышей.
Ь. Семейство генов гемоглобина.
с. Гены рибосомной РНК.
d. Число пальцев на конечностях
лошадей и людей.
19.4 . Первые молекулярные подходы
к филогенетике в 1950-1960-е гг.
использовали все нижеизложенное,
КРОМЕ:
а. Данных гибридизации ДНК-ДНК.
Ь. Иммунологических данных.
с. Электрофореза белка.
d. Секвенирования белка.
19.5 *. Длины ветвей филогенетического
дерева, построенного по данным
расшифровки последовательности
ДНК, показывают:
а. Отрезок времени с момента рас-
хождения организмов.
Ь. Число синонимических замен
между генами.
с. Степень различия между генами,
представленными узлами.
d. Ни одно из вышеупомянутого.
19.6 . Если две и более последовательно-
сти ДНК происходят от различных
предковых последовательностей, то
их относят к:
а. Монофилетическим.
Ь. Ортофилетическим.
с. Парафилетическим.
d. Полифилетическим.
19.7 *. Которые из следующих генов явля-
ются ортологичными?
а. Гены, не происходящие от общего
предка.
Ь. Гомологичные гены, которые
присутствуют в геномах разных
организмов.
с. Гомологичные гены, которые
присутствуют в одном геноме.
d. Негомологичные гены, которые
возникли в итоге сходящейся эво-
люции.
19.8 . Какой метод восстановления дере-
вьев определяет топологию с крат-
чайшим эволюционным путем?
а. Матриц расстояний.
Ь. Максимальной бережливости.
с. Объединения соседей.
d. Анализ главных компонентов.
19.9 *. Которое из следующих осложнений
не сопряжено с применением моле-
кулярных часов?
Глава 19. Молекулярная филогенетика 787
а. Несинонимические мутации про-
исходят с меньшей частотой, чем
синонимические мутации.
Ь. Молекулярныечасывгенахэука-
риотов бегут быстрее, чем тако-
вые в генах прокариотов.
с. Молекулярные часы в митохон-
дриальных генах идут быстрее,
чем таковые в ядерных генах.
d. Молекулярные часы, кажется,
убыстрили свой ход за последние
1-2 миллиона лет.
19.10 . Какое из следующих событий ослож-
няет обычное построение деревьев,
но не является проблемой при по-
строении сетей?
а. Присутствие предковых после-
довательностей в исследуемом
наборе данных.
Ь. Присутствие последовательно-
стей, которые возникли путем
рекомбинации.
с. И а и Ь.
d. Ни а, ни Ь.
19.11 *. Филогенетические исследования
последовательностей генома ВИЧ
показали все из нижеследующего,
КРОМЕ:
а. ВИЧ-1 наиболее близко связан
с ВИО шимпанзе.
Ь. ВИЧ-1 и ВИЧ-2 были переданы лю-
дям независимо друг от друга.
с. ВИЧ-1 и ВИЧ-2 были переданы
людям от приматов одного и того
же вида.
d. Глобальное распространение
ВИЧ-1 уже набрало обороты
к 1959 г.
19.12 . Какие из следующих объектов не
имеют ценности для филогенети-
ческого анализа популяций челове-
ка?
а. Гены рибосомной РНК.
Ь. Микросателлиты.
с. Митохондриальная ДНК.
d. Многоаллельные гены.
19.13 *. Какие из следующих сущностей
наследуются только по материнской
линии?
а. Гены глобина.
Ь. Митохондриальная ДНК.
с. Х-хромосомы.
d. Y-хромосомы.
19.14 . Который из следующих методов мо-
жет быть применен для распозна-
вания неравномерного географиче-
ского распределения аллелей?
а. Матриц расстояний.
Ь. Максимальной бережливости.
с. Объединения соседей.
d. Анализ главных компонентов.
Вопросы открытого типа
19.1 *. Чем фенетика отличается от тради-
ционных методов классификации,
используемых до 1957 г.?
19.2 . Чем кладистика отличается от фе-
нетики?
19.3 *. В чем заключено различие между
унаследованным и приобретенным
состоянием признака?
19.4 . Почему в филогенетических иссле-
дованиях последовательности ДНК
предпочитаются последовательно-
стям белка?
19.5 *. Чем отличаются внутренниеузлы ге-
нетического и видового деревьев?
19.6 . Каковы различия между методом
подобий и методом расстояний, при-
меняющимися для выравнивания
последовательностей?
19.7 *. Что за факторы влияют на частоту
аллелей в популяции?
19.8 . Каковы различия между гипоте-
зой о полицентрической эволюции
и гипотезой об африканском проис-
хождении современных людей?
788 Часть 4. Механизмы репликации и эволюции геномов
19.9 *. Как был выполнен первый филоге-
нетический анализ последователь-
ностей ДНК неандертальцев и какое
было сделано заключение об отно-
шениях между неандертальцами
и современными людьми?
19.10 . Обсудите схему анализа главных
компонентов, проведенного для
изучения доисторического пересе-
Изыскательские задачи
19.1* . Может ли вообще дерево генов
когда-либо быть эквивалентно де-
реву видов?
19.2. Насколько надежны молекулярные
часы?
19.3* . Филогенетические исследования
митохондриальной ДНК основаны
на допущении о том, что этот геном
наследуется по материнской линии
и что между материнским и отцов-
ским геномами не происходит реком-
бинации. Оцените обоснованность
такого допущения и опишите, какие
изменения претерпели бы гипотезы
ления земледельцев в Европу. Какие
выводы были получены в итоге этих
исследований?
19.11 *. Какую информацию предоста-
вила молекулярная филогенетика
о доисторическом переселении по-
пуляций человека в Северную Аме-
рику?
о происхождении и о переселениях
современных людей, если бы было
показано, что рекомбинация между
материнским и отцовским геномами
все-таки происходит.
19.4. Каков потенциал древней ДНК в ис-
следованиях эволюции человека?
19.5* . Рассмотрите случаи, в которых мо-
лекулярная филогенетика приме-
нялась для изучения гаплотипов
митохондриальной ДНК, имеющих-
ся в современных европейских по-
пуляциях.
Тесты по рисункам
19.1* . На каком из рисунков — а или б — представлен пример приобретенного состоя-
ния признака?
С крыльями
С крыльями
5 пальцев 1 палец 5 пальцев
Без крыльев
Глава 19. Молекулярная филогенетика 789
19.2. Какой из объектов, входящих в это филогенетическое дерево, является внешним
видом? На примере данного дерева поясните термины «монофилетичность»,
«полифилетичность» и «парафилетичность».
Овцы, козы
Крупный рогатый скот
Свиньи 1
Лошади_____|
Полифилетические
Собаки
Кошки_________________
Кенгуру
Парафилетические
Кролики
--------Крысы
1-------Мыши
Обезьяны Нового Света
(например, мартышки)
Обезьяны Старого Света
(например, макаки)
Орангутан
Горилла
Монофилетические
Шимпанзе”
Человек---
Клада
19.3* . Почему в схемах ветвления дерева видов и дерева генов возможны различия?
Дерево видов Дерево генов
19.4. К какому типу относится представленный на рисунке филогенетический анализ?
Какова цель анализа такого рода?
123456789 10111213141516
ATAGCСATAGCААССТ
ATACCCATGACAACGA настоящее
АТАCCCATAGCAACCA множественное
ATAG С С ATAG С А AC G А выравнивание
ATCCCCATAGCAACCT
2 7 4 9 11 4 16 5
TAGACGTC
TACGCCAC новое выравнивание
ТАСАССАС
TAGACGAC
ТАСАССТС
790 Часть 4. Механизмы репликации и эволюции геномов
Дополнительная литература
Основные учебники и обзорные статьи
A vise, J. С. (2004) Molecular Markers, Natural History and Evolution, 2nd Ed. Chapman and
Hall, New York. A detailed description ofthe use of molecular data in studies of evolution.
Futuyama, D.J. (1998) Evolutionary Biology, 3rd Ed. Sinauer, Sunderland,
Massachusetts.
Hall, B. G. (2004) Phylogenetic Trees Made Easy: A How-To Manual for Molecular Biologists,
2nd Ed. Sinauer, Sunderland, Massachusetts.
Nei,M. (1996) Phylogenetic analysis in molecular evolutionary genetics. Annu. Rev. Genet.
30: 371-403. Brief review of tree-building techniques.
Восстановление деревьев
Doolittle, W.F. (1999) Phylogenetic classification and the universal tree. Science 284:
2124-2128. Discusses the strengths and weaknesses of molecular phylogenetics as a means of
inferring species trees.
Felsenstein, J. (1989) PHYLIP — Phylogeny Inference Package (Version 3.20). Cladistics
5:164-166.
feanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G. and Gibson, T. J. (1998) Multiple
sequence alignment with Clustal X. Trends Biochem. Sci. 23: 403-405.
Saitou, N. and Nei, M. (1987) The neighbor-joining method: a new method for reconstructing
phylogenetic trees. Mol. Biol. Evol. 4: 406-425.
Swofford, D. L. (1993) PAUP: Phylogenetic Analysis Using Parsimony. Illinois Natural
History Survey, Champaign, Illinois.
Whelan, S., Lio, P. and Goldman, N. (2001) Molecular phylogenetics: state-of-the-art
methods for looking into the past. Trends Genet. 17: 262-272.
Yang,Z. (1997) PAML: a program package for phylogenetic analysis by maximum
likelihood. CABIOS 13: 555-556.
Молекулярные часы
Gu,X. and Li,W.-H. (1992) Higher rates of amino acid substitution in rodents than in
humans. Mol. Phylogenet. Evol. 1: 211-214.
Penny, D. (2005) Relativity for molecular clocks. Nature 436: 183-184. The apparent
increase in the rates of molecular clocks over the last few million years.
Strauss, E. (1999) Can mitochondrial clocks keep time? Science 283:1435-1438.
Эволюционные отношения среди приматов
Ruvolo,M. (1997) Molecular phylogeny of the hominoids: inferences from multiple
independent DNA sequence data sets. Mol. Biol. Evol. 14: 248-265.
Sarich,V.M. and Wilson, A. C. (1967) Immunological time scale for hominid evolution.
Science 158:1200-1203.
Происхождение ВИЧ
Korber, В., Muldoon, M., Theiler,)., Gao, F., Gupta, R., Lapedes,A., Hahn, В. H.,
Wolinsky, S. and Bhattacharya, T. (2000) Timing the ancestor of the HIV-1 pandemic strains.
Science 288:1789-1796.
Leitner, T., Escanilla, D., Franzen, C., Uhlen,M. and Albert, J. (1996) Accurate
reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc.
Natl Acad. Sci. USA 93:10864-10869.
Глава 19. Молекулярная филогенетика 791
Zhu,T., Korber,В. Т., Nahmias,A.J., Hooper,Е., Sharp,P.M. and Ho,D.D. (1998) An
African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature
391: 594-597.
Происхождение современного человека
Cann,R.L., Stoneking,M. and Wilson, A. C. (1987) Mitochondrial DNA and human
evolution. Nature 325: 31-36. The first discovery of mitochondrial Eve.
Harding,R.M., Fullerton,S.M., Griffiths, R. C., Bond,)., Cox,M.)., Schneider,).A.,
Moulin, D. S. and Clegg, J. B. (1997) Archaic African and Asian lineages in the genetic ancestry
of modern humans. Am.]. Hum. Genet 60: 772-789. Studies of nuclear genes.
Ingman, M., Kaessmann, H., Paabo, S. and Gyllensten, U. (2000) Mitochondrial genome
variation and the origin of modern humans. Nature 408: 708-713.
Krings, M., Stone, A., Schmitz, R. W., Krainitzki, H., Stoneking, M. and Paabo, S. (1997)
Neandertal DNA sequences and the origin of modern humans. Cell 90:19-30.
Миграции человечества
Cavalli-Sforza, L. L. (1998) The DNA revolution in population genetics. Trends Genet 14;
60-65. Principal component analysis of human nuclear genes.
Chikhi, L, Destro-Bisol, G., Bertorelle, G., Pascali, V. and Barbujana, G. (2002) Y genetic
data support the Neolithic demic diffusion model. Proc. Natl Acad. Sci. USA 99:11008-11013.
Migrations in Europe.
Forster,P., Harding,R., Torroni,A. and Bandelt,H.). (1996) Origin and evolution of
native American mtDNA variation: a reappraisal. Am.]. Hum. Genet 59: 935-945.
Richards, M. (2003) The Neolithic invasion of Europe. Annu. Rev. Anthropol. 32: 135-
162.
Semino, O., Passarino, G., Oefner, P.)., et al. (2000) The genetic legacy of paleolithic Homo
sapiens in extant Europeans: a Y chromosome perspective. Science 290:1155-1159.
Silva,W.A., Bonatto,S.L., Holanda,A.)., etal. (2002) Mitochondrial genome diversity
of Native Americans supports a single early entry of founder populations into America. Am. J.
Hum. Genet 71:187-192.
Приложение
Ответы
Глава 1: Геномы, транскриптомы и протеомы
Вопросы закрытого типа
1.1 — с; 1.3 — а; 1.5 — с; 1.7 — с; 1.9 — а; 1.11 — с; 1.13 — а; 1.15 — Ь.
Вопросы открытого типа
1.1. ДНК была впервые открыта в 1869 г. (Мишер) и признана за источник гене-
тической информации в 1940-е гг. (Эвери, Маклеод и Маккарти, а также Херши и Чейз).
Структура двойной спирали была определена в 1953 г. (Уотсон и Крик), а первая полная
последовательность генома клеточного организма была получена в 1995 г.
1.3. Ограничение, согласно которому А может спариваться только с Т, a G может
спариваться только с С, означает, что в ходе репликации ДНК могут быть получены пол-
ные копии родительской молекулы посредством простой схемы действий, состоящей
в использовании последовательностей уже существующих нитей для задания последо-
вательностей новых нитей. Поэтому спаривание оснований позволяет реплицировать
молекулы ДНК в их совершенные копии.
1.5. Бактериальные мРНК имеют периоды полураспада не более нескольких минут,
а у эукариотов большая часть молекул мРНК деградирует спустя несколько часов после
синтеза. Такой быстрый оборот означает, что состав транскриптома не постоянен и может
быть быстро реструктурирован за счет изменения скорости синтеза отдельных видов
мРНК. Поэтому состав транскриптома может быть быстро подобран в соответствии
с потребностями клетки.
1.7. Каждая клетка получает часть своего транскриптома, когда она впервые по-
является на свет путем деления родительской клетки, и поддерживает транскриптом
на протяжении всей своей жизни. Поэтому транскрипция отдельных кодирующих бел-
ки генов приводит не к синтезу транскриптома, а лишь поддерживает транскриптом,
заменяя те молекулы мРНК, которые деградировали, и привносит изменения в состав
транскриптома путем включения и выключения различных наборов генов.
1.9. Белки структурно и функционально многообразны, потому что аминокислоты,
из которых они сделаны, химически разнообразны. Различные последовательности
аминокислот поэтому дают различные сочетания химической реакционной способности,
причем такие сочетания обусловливают не только общую структуру синтезируемого
белка, но также и расположение на поверхности структуры активных групп, которые
определяют химические свойства белка.
Приложение 793
1.11. Местоположение этого кодона важно для определения того, функционирует
ли он как стоп-кодон или задает селеноцистеин. Сразу ниже кодона селеноцистеина
находится структура типа шпильки, которая позволяет этой аминокислоте быть встро-
енной в наращиваемую полипептидную цепь.
Изыскательские задачи
1.1. Обоснование правомерности этого утверждения может быть найдено в «Двой-
ной спирали» Джеймса Уотсона или в большинстве различных книг, написанных об
истории ДНК, например в «Восьмом дне сотворения» Хораса Фриленда Джадсона (см.
список дополнительной литературы). Именно вечером субботы 7 марта 1953 г. Уотсон
и Крик завершили свою модель структуры двойной спирали, сделанную из тонких
кусочков гальванизированных металлических и медных прутов в масштабе 50 см на
1 нм, при этом полный виток модельной спирали занимал почти 2 м. Можно, однако,
возразить, что фактическое открытие было сделано неделей раньше, когда Уотсон
и Крик осознали, что скрепленные водородными связями структуры, образуемые А с
Т и G с С, имеют одну и ту же внешнюю форму (см. рис. 1.8, б), что позволяет этим
парам оснований накладываться друг на друга и образовывать правильную спираль
неизменной ширины. Крик вспоминает, что эта мысль осенила их, когда они постигли
значимость выведенных Чаргаффом отношений оснований, но Уотсон утверждает, что
их определяющее значение было оценено только после того, как они построили свои
первые модели пар нуклеотидов.
1.3. Истолкование генетического кода было самым важным достижением био-
логии в 1960-е гг., и, хотя эта работа была выполнена почти полстолетия назад, она все
еще представляет блестящий пример научного метода: как планировать стратегию
исследования (и вести его к намеченному концу) и как видоизменять эту стратегию,
чтобы использовать новые методы, разработанные за время работы над проектом. Та-
кие книги, как, например, «Восьмой день сотворения» (см. перечень дополнительной
литературы), дают подробное описание того, как генетический код был взломан, но
наилучшими отправными точками для обсуждения данного предмета на учебном заня-
тии являются три обзорные статьи в «Сайентифик америкен», которые были написаны
в разгаре проекта и в его конце: 1) Crick,F.H.C. (1962) The genetic code. Sci. Am. 207(4):
66-74; 2) Nirenberg, M.W. (1963) The genetic code II. Sci. Am. 208(3): 80-94; 3) Crick, F.H.C.
(1966) The genetic code III. Sci. Am. 215(4): 55-62.
Тесты по рисункам
1.1. Часть а показывает, что процессинг трансформирующего начала протеазой или
рибонуклеазой не оказывает никакого воздействия, но что трансформирующее начало
инактивируется дезоксирибонуклеазой. Трансформирующее начало, которое содержит гене-
тическую информацию, необходимую для преобразования безвредных бактерий в заразную
форму, должно было поэтому состоять из ДНК. Часть б показывает, что, когда бактериофаги
помечены атомами 32Р и 35S, в процессе инфицирования в клетки проникает большая часть
меченного 32Р материала (ДНК), но только 20 % меченного 35S материала (белок фага). По-
скольку гены бактериофага должны проникнуть в бактерию, чтобы задавать синтез новых
бактериофагов, постольку эти гены должны состоять из ДНК.
1.3. Модель показывает, что сахаро-фосфатная основная цепь находится на внешней
стороне молекулы и что основания расположены на внутренней её стороне. Основания
свободно доступны в большой и малой бороздках, где они могут опознаваться ДНК-
связывающими белками.
794 Приложение
Глава 2: Изучение ДНК
Вопросы закрытого типа
2.1 — Ь; 2.3 — d; 2.5 — а; 2.7 — а; 2.9 — Ь; 2.11 — d; 2.13 — d; 2.15 — а.
Вопросы открытого типа
2.1. Ген клонируется, когда фрагмент ДНК, содержащий его, встраивается в век-
торную молекулу ДНК (например, плазмиды или бактериофага) и затем реплицируется
в клетке хозяина.
2.3. Можно присоединить линкерные, или сопрягающие, последовательности
к концам тупоконечных молекул, чтобы произвести липкие концы, которые облегчают
лигирование.
2.5. Это обусловлено простотой отбора бактериальных клеток, которые были
трансформированы плазмидой.
2.7. Геном бактериофага содержит гены для лизогенного инфицирования Е. coli,
которые не являются основными и могут быть заменены новой ДНК. Бактериофаг может
быть использован для вмещения молекул ДНК длиной до 18 тыс. п. н.
2.9. Они требуют присутствия центромеры, теломер и по крайней мере одной об-
ласти инициации.
2.11. Затравки гибридизируются к специфическим последовательностям в матрич-
ной ДНК и определяют области, которые необходимо размножить.
Изыскательские задачи
2.1. Мораторий явился результатом Азиломарской конференции в 1975 г. и был
предложен Паулем Бергом и др. в итоговом заявлении: Berg,P., Baltimore, D., Brenner, S.,
Roblin,R.O. and Singer, M.F. (1975) Summary statement of the Asilomar Conference on
recombinant DNA molecules. Proc. Natl Acad. Sci. USA 72: 1981-1984. Описанная менее
специальным языком поднаготная моратория может быть найдена в книге CherfasJ.
(1982) Man Made Life. Blackwell Scientific Publishers, Oxford. Возникает соблазн заключить,
что поскольку опасения этих ученых никогда не оправдывались, то эти опасения были
необоснованными, но при всестороннем обсуждении этой проблемы должны также
приниматься во внимание и посылы моратория (например, развитие стратегий для
предотвращения выживания генетически конструируемых бактерий в естественной
окружающей среде), которые во многом содействовали тому, что опасности, которые
побудили ученых объявить мораторий, обошли нас стороной.
2.3. Этот вопрос предвосхищает обсуждение составления карт рестриктов в разде-
ле 3.3.1. Попытка «изобрести» методику составления карт рестриктов в ходе обсуждения
в классе — превосходный способ убедиться в полном усвоении принципов рестрикции,
гель-электрофореза и тому подобных процедур.
2.5. Если допустить, что целевой последовательностью для ПЦР является един-
ственная копия в исследуемом геноме, то длина затравок становится важной проблемой.
Если затравки слишком коротки, то они могут гибридизироваться к нецелевым участкам
и дать нежелательные продукты размножения. Лучший способ иллюстрировать этот
момент — вообразить, что в эксперименте ПЦР с парой затравок длиной по восемь
нуклеотидов используется полная ДНК человека. Вероятным результатом будет раз-
множение множества различных фрагментов. Это связано с тем, что сайты связывания
для этих затравок, согласно ожиданию, встречаются в среднем единожды на каждые
48 = 65536 п.н., что дает приблизительно 49000 возможных участков в нуклеотидной
Приложение 795
последовательности длиной 3 200 000 тыс.п.н., составляющей геном человека. Это
означает, что было бы крайне маловероятно, что пара восьминуклеотидных затравок
даст одновариантный, специфичный продукт размножения в случае ДНК человека, так
как будет не одна, а несколько позиций, в которых участки отжига случайно окажутся
в достаточной (чтобы дать продукт размножения) близости друг к другу. Напротив,
ожидаемая частота появления 17-нуклеотидной последовательности — один раз на
каждые 417=17179 869184 п. н. Эта цифра более чем в пять раз превышает длину генома
человека, так что пара 17-нуклеотидных затравок должна поэтому дать одновариантный,
специфический продукт размножения. Идеальная температура отжига должна быть
достаточно низкой, чтобы позволить осуществиться гибридизации между затравкой
и матрицей, но достаточно высокой, чтобы препятствовать образованию внутренне
несовпадающих гибридов. Информация, необходимая для понимания того, как выбрать
соответствующую температуру для пары затравок, приводится в подписи к рис. 3.8.
Желающим ознакомиться с этим вопросом подробнее мы рекомендуем обратиться
к произведению Brown, Т. А. (2006) Gene Cloning and DNA Analysis: An Introduction, 5-th Ed.
Blackwell Scientific Publishers, Oxford.
Тесты по рисункам
2.1. Затравка инициирует синтез ДНК, предоставляя группу З'-ОН, необходимую
для присоединения нуклеотида. Затравка может быть использована также и для зада-
ния участка синтеза ДНК на матричных молекулах (как например, при секвенировании
ДНК и проведении ПЦР).
2.3. Это космида, которая способна нести в себе встроенные молекулы длиной
вплоть до 44 тыс. п. н.
Глава 3: Картирование геномов
Вопросы закрытого типа
3.1 — d; 3.3 — а; 3.5 — d; 3.7 — с; 3.9 — Ь; 3.11 — с; 3.13 — а; 3.15 — d.
Вопросы открытого типа
3.1. Карта генома обеспечивает руководство для экспериментов по секвенирова-
нию, показывая позиции генов и других отличительных особенностей. Если карта от-
сутствует, то весьма вероятно, что при сборке последовательности генома будут сделаны
ошибки, особенно в областях, которые содержат повторную ДНК.
3.3. Затравки для ПЦР разрабатывают так, чтобы они отжигались по обе стороны
от полиморфного участка, и RFLP типизируется путем процессинга размноженного
фрагмента ферментом рестрикции и последующего прогона образца в агарозном геле.
До изобретения ПЦР RFLP типизировали при помощи гибридизации по Саузерну, на
которую уходит относительно много времени.
3.5. Если пара генов показывает сцепленность, то они должны находиться на одной
хромосоме. Если кроссинговер происходит случайным образом, то частота рекомбинации
между парой сцепленных генов служит мерой расстояния между ними на хромосоме.
Частоты рекомбинации для различных пар генов могут быть использованы для по-
строения карты их относительных позиций на хромосоме.
3.7. Двойная гомозигота произведет гаметы, все из которых будут генетически
тождественны, и если они рецессивны, то такой родитель не будет вносить вклад в фе-
нотип потомства.
796 Приложение
3.9. FISH использует в качестве зонда меченный флюоресцентным красителем
фрагмент ДНК, который связывается с интактной хромосомой. Позиция связывания
может быть определена, а эта информация использована при создании физической
карты хромосомы.
3.11. Отдельные хромосомы могут быть разделены при помощи проточной цито-
метрии. Делящиеся клетки осторожно вскрываются так, чтобы получить смесь непо-
врежденных хромосом. После этого хромосомы метят флюоресцентным красителем.
Количество красителя, связываемого хромосомой, зависит от ее размера, так что более
крупные хромосомы связывают больше красителя и флюоресцируют ярче, чем более
мелкие хромосомы. Препарат хромосом растворяют и пропускают через мелкое от-
верстие, производя поток капелек, в каждой из которой содержится одна хромосома.
Капельки минуют датчик, который измеряет степень флюоресценции и, следовательно,
определяет, какие капельки содержат конкретную искомую хромосому. К этим каплям,
и к никаким другим, прикладывается электрический заряд, позволяющий капелькам, со-
держащим желаемую хромосому, отклоняться и тем самым отделяться от остальных.
Изыскательские задачи
3.1. В тексте говорится, что к идеальным особенностям относятся высокая частота
встречаемости в изучаемом геноме, простота типизации и присутствие множественных
аллелей. Это подразумевает, что SSLP должны быть «идеальными» маркёрами, но в дей-
ствительности более популярны SNP. Обсуждение этого очевидного парадокса требует
рассмотрения относительной важности каждого из этих трех критериев и в собенности
осознания того факта, что определяющей особенностью «идеального» маркёра является
высокая плотность.
3.3. Многим преподавателям вспомнится, как они бились над этим вопросом в пору
своего студенчества, но ответ не изменился: короткое время смены поколений, большое
количество потомства, легко оцениваемые фенотипы и тому подобное. Поучительно
рассмотреть, до какой степени геномика добавила новые критерии к этому списку:
является ли полная последовательность генома особенностью организма, полезной
для использования в исследованиях наследственности?
3.5. Это открытый вопрос, допускающий столько ответов, сколько может подсказать
воображение. Он предназначен, чтобы вызвать обсуждение ряда тем, которые охвачены
в последующих главах. Обсуждение можно начать, спрашивая, для какой цели необходима
карта. Карта, предназначенная проекту секвенирования, может отличаться от карты,
разработанной для клонирования отдельных генов. Если будет сделан вывод, что для
целей секвенирования более полезна физическая карта, а генетическая карта факти-
чески имеет небольшую или никакую ценность (что является разумным заключением,
которого и следует ожидать от прочитавших главу 4), то обсуждение может повернуть
к вопросу о том, насколько легко или, наоборот, трудно определить местонахождение
генов в последовательности генома и приписать функции к тем генам, без каких-либо
предварительных данных о том, где именно расположены эти гены. Эти вопросы рас-
сматриваются в главе 5.
Тесты по рисункам
3.1. Когда олигонуклеотид не гибридизируется с целевой последовательностью,
флюоресцентная метка и молекула гасителя располагаются рядом друг с другом и флюо-
ресценция гасится. Когда олигонуклеотид связывается с целевой последовательностью,
флюоресцентная метка оказывается далеко от молекулы гасителя. Можно так подобрать
Приложение 797
условия гибридизации, что олигонуклеотид будет связываться с целевой последова-
тельностью только в том случае, если комплементарными будут все нуклеотиды.
3.3. Это электрофорез в геле с ортогонально чередующимся полем, в котором
электрическое поле чередуется между парами электродов. Молекулы ДНК продвигаются
вниз через гель, но каждое изменение поля вынуждает молекулы переориентироваться.
Более короткие молекулы переориентируются быстрее, чем более длинные, и в силу
этого мигрируют через гель быстрее. Таким образом, при помощи пульс-фореза могут
быть разделены намного более длинные молекулы, чем разделяемые обычным гель-
электрофорезом.
Глава 4: Секвенирование геномов
Вопросы закрытого типа
4.1 — Ь; 4.3 — а; 4.5 — с; 4.7 — Ь; 4.9 — с; 4.11 — d; 4.13 — с; 4.15 — с.
Вопросы открытого типа
4.1. Дидезоксинуклеотидам недостает З'-гидроксильных групп, и когда дидезок-
синуклеотиды встраиваются в ДНК, синтез нити останавливается.
4.3. Да, это возможно. Продукт ПЦР очищается и выполняется секвенирование
в тепловом цикле, при этом в качестве затравки для реакции секвенирования исполь-
зуется одна из затравок ПЦР.
4.5. Автоматизированные секвенаторы, в которых многочисленные капилляры
работают параллельно, могут считывать до 96 различных последовательностей за
2-часовой период, а это означает, что при среднем числе 750 п.н. на отдельный экс-
перимент 864 тыс. п. н. информации может быть произведено одной машиной за день.
Это позволяет данные, необходимые для расшифровки последовательности полного
генома, получить за период, измеряемый неделями.
4.7. Избыточность требуется, поскольку клоны для проекта секвенирования произ-
водятся случайным образом и затем секвенируются; таким образом, чтобы гарантировать
полное покрытие генома, необходимо секвенировать большое число нуклеотидов.
4.9. Фингерпринтинг клонов может быть основан на рестрикционных картах, отпе-
чатках пальцев повторной ДНК, ПЦР с повторной ДНК и картировании содержания STS.
4.11. Секвенирование сложных геномов эукариотов методом дробовика может
привести к тому, что сегменты ДНК, возможно, включающие гены или части генов, не
войдут в черновую последовательность. Существует также более высокая вероятность
того, что ошибки последовательности не будут распознаны.
Изыскательские задачи
4.1. В начале раздела 4.1 содержится заявление о том, что «секвенирование с об-
рывом цепи получило преимущество по нескольким причинам, и не в последнюю
очередь благодаря относительной простоте, с которой этот метод может быть автома-
тизирован», — это основная часть ответа на данный вопрос. Рассмотрение же вопроса
о том, по какой причине метод химической деградации оказался трудно поддающимся
автоматизации — хороший способ повысить осведомленность в отношении проблем,
неразрывно связанных с развитием автоматизированной технологии. Второй изъян
метода химической деградации — токсичность используемых химикатов, ибо токсич-
ность является неотъемлемым свойством любого соединения, способного связываться
798 Приложение
с молекулами ДНК и видоизменять их. Хотя это не чисто теоретический вопрос, призван-
ный служить пищей для жарких дискуссий теоретиков-академистов, к данному предмету
обсуждения можно обращаться для того, чтобы ввести участников обсуждения в круг
вопросов о риске и безопасности лабораторной молекулярной биологии.
4.3. Раздел 4.2.2 охватывает метод сборки контигов из клонов, но полезнее в своей
критической оценке отправляться от сравнения между методом сборки контигов из
клонов и методом дробовика для полных геномов, в особенности потому, что их при-
меняли к расшифровке генома человека. Из этого сравнения становится очевидно, что
строгий метод сборки контигов из клонов является относительно времязатратным,
но что в настоящее время он является единственным способом гарантировать коэф-
фициент ошибок менее одной на 104 нуклеотида, а эта цифра установлена в качестве
приемлемого максимума для «готовой» последовательности.
4.5. Основная проблема — противоречие между правом компании защищать свои
инвестиции, которое принимается без вопросов в большинстве других видов ком-
мерческой деятельности, и менее четко прописанными правами индивидуумов, гены
которых были использованы компанией в ходе исследований, завершившихся разра-
боткой лекарственного препарата. На этот вопрос существуют самые разные взгляды,
и профессиональное и разумное обоснование выраженного мнения — наиболее важная
составляющая ответа на этот вопрос.
Тесты по рисункам
4.1. Для большинства экспериментов по секвенированию используется универ-
сальная затравка, которая комплементарна части векторной ДНК, непосредственно
примыкающей к точке, в которой лигируется новая ДНК. Поэтому одна и та же уни-
версальная затравка может дать последовательность любой части ДНК, которая была
лигирована в вектор. Возможно продолжить последовательность в одном направлении,
синтезировав внутреннюю затравку, предназначенную для отжига в какой-либо позиции
в пределах встройки ДНК. Эксперимент с такой затравкой обеспечит вторую короткую
последовательность, которая накладывается на предыдущую.
4.3. Это секвенирование путем химической деградации, и оно дает хорошие ре-
зультаты, когда есть проблемы со стандартными реакциями секвенирования с обрывом
цепи (из-за блокирования ДНК-полимеразы или измененной подвижности продуктов
секвенирования в ходе электрофореза).
Глава 5: Поиск смысла, заложенного в последовательности
генома
Вопросы закрытого типа
5.1 — Ь; 5.3 — Ь; 5.5 — а; 5.7 — Ь; 5.9 — Ь; 5.11 — d; 5.13 — b.
Вопросы открытого типа
5.1. Компьютеры могут легко просматривать все шесть рамок считывания последо-
вательности ДНК с целью отыскания ОРС. Кроме того, поскольку случайная последова-
тельность ДНК будет содержать один стоп-кодон по крайней мере на каждые 100-200 п.н.,
а число кодонов в большинстве генов заметно превышает это значение, можно напрямую
распознавать кодирующие последовательности в бактериальных геномах, не имеющих
интронов и прочих существенных некодирующих последовательностей.
Приложение 799
5.3. Компьютерные программы можно видоизменять и нацеливать на поиск от-
клонения частоты использования кодонов, экзон-интронных границ и вышележащих
регуляторных последовательностей генов.
5.5. Некоторые гены содержат факультативные экзоны и могут кодировать мо-
лекулы мРНК различных размеров. Также возможно, что не все гены, находящиеся во
фрагменте ДНК, экспрессируются в клетках, из которых выделена РНК.
5.7. Ортологичные гены — гомологичные гены, находящиеся в разных организмах,
а паралогичные гены — гомологичные гены, имеющиеся у одного организма.
5.9. Если биохимическое действие какого-либо продукта гена известно у другого
вида, то оно может дать ключи к функции этого гена в организме человека. Другие
организмы могут быть использованы также и в экспериментальных исследованиях,
нацеленных на изучение функции этого гена.
5.11. Для определения того, являются ли короткие ОРС истинными генами, можно
прибегнуть к сравнительной геномике, к опознаванию экспрессируемых последователь-
ностей и к мечению транспозонами.
Изыскательские задачи
5.1. Это сложный вопрос, но рассмотрение основных принципов позволяет до-
стигнуть известного прогресса. В разделе 5.1.1 разъясняется, что опознавание экзон-
интронных границ есть основное препятствие для определения местоположения ге-
нов эукариотов методом прямого просмотра последовательности. Вероятно, разумно
заключить, что консенсусные последовательности, в настоящее время приписанные
к этим участкам, настолько точны, насколько это возможно, поскольку они основаны на
сравнениях между многочисленными экзон-интронными границами во многих организ-
мах. Следовательно, можно утверждать, что сам по себе просмотр последовательности
никогда не будет средством надежного опознавания последовательности гена. Обсуж-
дение должно поэтому сосредоточиться на возможностях поиска гомологии — как для
установления местоположения генов, так и для приписывания к ним функций. Важными
предметами обсуждения, вероятно, являются степень, до которой поиск гомологии
станет более действенным и более точным по мере того, как в базы данных вносится
все большее число последовательностей, и возможности реализации потенциала срав-
нительной геномики, проявившегося в полной мере в проектах с Sacchammyces cerevisiae
и родственными им дрожжами, при работе с другими организмами. Чтобы обратиться ко
второму предмету, рассмотрите вопрос о том, насколько тесно связанными должны быть
два генома, для того чтобы была применима сравнительная геномика, и вероятно ли, что
пары или группы геномов с требуемой степенью родства среди, например, полученных от
млекопитающих будут иметься под рукой исследователя в обозримом будущем.
5.3. Это последовательность фрагмента белка миоглобина человека. Остальные
последовательности, опознанные в ходе поиска программой «BLAST», относятся к орто-
логам белка человека.
Тесты по рисункам
5.1. Компьютерная программа будет искать экзон-интронную границу и опознавать
последовательность интрона.
5.3. Львиная доля регуляторных сигналов, управляющих экспрессией гена, нахо-
дится в области ДНК, расположенной выше ОРС, так что ген GFP теперь покажет ту же
картину экспрессии, что и опытный ген. Картина экспрессии этого гена может поэтому
быть определена путем оптического исследования организма на предмет присутствия
и расположения в нем GFP.
800 Приложение
Глава 6: Постижение механизмов функционирования генома
Вопросы закрытого типа
6.1 — Ь; 6.3 — с; 6.5 — а; 6.7 — Ь; 6.9 — а; 6.11 — с; 6.13 — Ь.
Вопросы открытого типа
6.1. Для того чтобы понять, как именно геном, являя собой единоцельную систе-
му, работает в клетке, определяя и согласовывая различные биохимические действия,
которые в ней протекают. Такие глобальные исследования активности генома должны
обратиться не только к самому геному, но также и к транскриптому с протеомом.
6.3. Если две разные мРНК имеют подобные последовательности, то каждая из них
может перекрестно гибридизироваться со специфическим для второй молекулы зондом
на матрице. Это часто случается, когда в одной и той же ткани активны два или несколь-
ко паралогичных генов. В таком случае транскриптом содержит группу родственных
молекул мРНК, каждая из которых способна гибридизироваться до некоторой степени
с различными членами данного семейства генов. Различение относительных количеств
каждой мРНК или хотя бы достоверные данные о том, какие конкретно молекулы мРНК
присутствуют, может при таком раскладе оказаться трудной задачей. Чтобы решить
эту проблему, необходимо спроектировать чип ДНК, который несет олигонуклеотиды,
специфичные к определенным последовательностям, являющимся уникальными для
каждого члена семейства паралогов.
6.5. Гены, которые показывают подобные профили экспрессии, вероятно, будут
иметь и связанные функции. Они могут быть распознаны при помощи иерархической
группировки, что предполагает сравнение уровней экспрессии каждой пары генов
в каждом проанализированном транскриптоме и присвоение таким парам величины,
которая показывает уровень функциональной связи между этими генами. Эти данные
могут быть выражены в виде древовидной схемы, в которой гены со сходными про-
филями экспрессии группируются вместе. Такая древовидная схема дает наглядное
зрительное представление о функциональных связях между генами.
6.7. Анализ состава транскриптома дает точное представление о том, какие гены
активны в той или иной рассматриваемой клетке, но дает менее точное представление
о белках, в ней присутствующих. Это объясняется тем, что к факторам, которые влия-
ют на содержание белка, относится не только количество имеющейся мРНК, но также
и скорость, с которой молекулы мРНК транслируются в белок, и скорость, с которой
находящиеся в клетке белки деградируют.
6.9. Клонирующий вектор, используемый для фагового дисплея, разработан таким
образом, чтобы новый ген, который клонируется в нем, экспрессировался бы таким об-
разом, что его белковый продукт становился бы слитым с одним из белков оболочки
фага. Таким образом, белок фага переносит чужеродный белок в оболочку фага, где он
представляется в форме, позволяющей ему взаимодействовать с другими белками,
с которыми фаг встречается.
6.11. Есть надежда, что, когда метаболомика достигнет зрелости, появится воз-
можность использовать информацию для разработки таких лекарственных препаратов,
которые боролись бы с болезнью путем обращения вспять или смягчения специфиче-
ских нарушений метаболических потоков, возникших в болезненном состоянии. Также
составление метаболических профилей могло бы указать любые нежелательные по-
бочные эффекты лекарственной терапии, позволяя делать модификации в химической
Приложение 801
структуре препарата или в его схеме приема, с тем чтобы такие побочные эффекты
были сведены к минимуму.
Изыскательские задачи
6.1. В разделе 6.1.2 описывается, как микроматрицы используются для сравнения
транскриптомов в двух и более тканях или же в одной и той же ткани, но при разных
условиях, а освоение списка дополнительной литературы позволит ознакомиться со вся-
кими подробностями и специальными примерами. Обсуждение трудностей применения
метода секвенирования кДНК, такого как SAGE, к сравнениям между транскриптомами
гарантирует, что определяющие различия между методами секвенирования и микро-
матриц будут взвешены, и подчеркивает действенность технологии микроматриц.
6.3. Первая часть вопроса является относительно простой, и в тексте приводится
один пример с Е. coll (лактопермеаза и р-галактозидаза). Вторая часть вопроса — белки,
которые имеют физические, но не функциональные взаимодействия, — не так быстро
позволит найти ответ, но есть примеры молекулярных шаперонов (раздел 13.3.1), кото-
рые применяют физические воздействия к своим подопечным белкам с целью помочь
тем белкам свернуться, и белков протеасомы (раздел 13.4), которые подобным же об-
разом образуют физические взаимодействия с деградируемыми ими белками. Пони-
мание специфики взаимодействий этих типов (например, какие специфические белки
сворачиваются которыми индивидуальными шаперонами) является главной целью
исследования белка, и, следовательно, эти типы взаимодействий столь же интересны,
как и таковые на функциональной основе.
6.5. Это открытый вопрос, подходящий для обсуждения в классе или в небольшой
группе, возможно, руководствуясь опорной статьей типа Kirschner, M.W. (2005) The
meaning of systems biology. Cell 121: 503-504 в качестве отправной точки.
Тесты по рисункам
6.1. Препарат кДНК метится флюоресцентной меткой и гибридизируется на микро-
матрице. Метка обнаруживается сканированием лазером с конфокальными зеркалами
и её интенсивность преобразуется в спектр псевдоцветов.
6.3. В первом измерении белки разделяются изоэлектрическим фокусированием.
Гель затем вымачивают в додецилсульфате натрия, поворачивают на 90° и проводят
второй (под прямым углом к первому) электрофорез, разделяющий белки по их раз-
мерам.
Глава 7: Ядерные геномы эукариотов
Вопросы закрытого типа
7.1 — Ь; 7.3 — с; 7.5 — d; 7.7 — с; 7.9 — Ь; 7.11 — Ь; 7.13 — Ь.
Вопросы открытого типа
7.1. Полное переваривание хроматина человека нуклеазами показывает, что по-
следовательности ДНК длиной 146 п.н. защищены от переваривания. Частичное пере-
варивание нуклеазами дает фрагменты ДНК длиной 200 п. н. и кратных длин.
7.3. Минихромосомы короче, чем макрохромосомы, но имеют намного более вы-
сокую плотность генов.
802 Приложение
7.5. Теломеры отмечают концы хромосом и позволяют клетке отличить реальный
конец от конца, образовавшегося в результате разрыва хромосомы.
7.7. Типичная область хромосомы человека будет содержать малое число генов
(большинство которых будет содержать интроны), несколько повторных последователь-
ностей и большое количество неповторной, негенной ДНК. Хромосомы дрожжей имеют
более высокую плотность генов (при этом очень немногие гены содержат интроны),
малое число повторных последовательностей и намного меньше негенной ДНК.
7.9. Каталоги генов могут быть основаны на известных функциях генов, но такие
каталоги неполные, потому что в большинстве геномов многие гены имеют неизвестные
функции. Каталоги генов, которые основаны на типе белковых доменов, кодируемых
генами, являются более исчерпывающими, поскольку они включают много генов,
специфические функции которых неизвестны.
7.11. Обычный псевдоген стал инактивированным из-за мутации, тогда как про-
цессированный псевдоген возник путем повторной встройки кДНК-копии мРНК.
Изыскательские задачи
7.1. Этот вопрос отправляет читателя к большей части материала, охваченного
в главе 10 «доступ к геному». Начиная с основных принципов должно быть ясно, что
гены, которые присутствуют в областях плотноупакованного хроматина, вероятно,
являются недоступными белкам, отвечающим за активирование и транскрибирование
гена, и что точное расположение нуклеосом также может быть важно в определении
степени доступности генов. В конце раздела 7.1.1 указывается, что химическая мо-
дификация гистонов имеет большое значение в определении структуры хроматина,
и эта тема может быть с пользой анонсирована на данном этапе до ее досконального
изложения в главе 10.
7.3. Рис. 7.13 должен послужить отправной точкой, а первой целью должна быть
формулировка определения «межгенной ДНК» и глубокое понимание того факта, что
она исключает все последовательности, содержащиеся в пределах генов (кодирующих
областей и интронов), относимые к генам (например, псевдогенов, фрагментов генов)
или необходимые для активности гена (например, области, лежащие непосредственно
выше генов). Однако традиционно к межгенной ДНК относят некоторые функцио-
нальные последовательности типа областей инициации (раздел 15.2.1) и последова-
тельности, которые прикрепляют хромосомы к субструктуре ядра (раздел 10.1.2). Как
только этот факт будет осознан, возникает следующий вопрос: «Имеет ли какой-либо
существенный повторный компонент какую-либо функцию?» Из раздела 7.1.2 должно
быть ясно, что по крайней мере некоторая часть сателлитной и минисателлитной ДНК
является функционально активной, а последний параграф главы 7 указывает на то, что
большая часть рассеянной повторной ДНК обладает транспозиционным действием, но
до какой степени это можно рассматривать как «функцию»?!
Тесты по рисункам
7.1. На рисунке показана часть кариограммы человека. Хромосомы отличаются
по размерам, расположению центромеры и картинам исчерченности, проявившимся
после окрашивания.
7.3. На рисунке показан процессированный псевдоген, который функционально
неактивен, потому что он получен из мРНК и, следовательно, не имеет нуклеотидных по-
следовательностей, необходимых для включения и регулирования экспрессии генов.
Приложение 803
Глава 8: Геномы прокариотов и органелл эукариотов
Вопросы закрытого типа
8.1 — d; 8.3 — а; 8.5 — с; 8.7 — а; 8.9 — с; 8.11 — d; 8.13 — с.
Вопросы открытого типа
8.1. Хромосомы эукариотов линейны и имеют гистоновые белки, которые участву-
ют в упаковке ДНК. Хромосомы эукариотов имеют множественные области инициации
и содержат центромеры и теломеры. Хромосома Е. coli — кольцевая молекула, которая
содержит единственную область инициации, упакована посредством сверхспирал изации
и не имеет центромер и теломер.
8.3. Белки HU по структуре отличаются от гистонов, но, подобно гистонам, образуют
тетрамеры, на которые навивается ДНК.
8.5. Геномы прокариотов характеризуются очень плотным расположением генов,
содержат очень короткие области межгенной ДНК и не имеют интронов и последова-
тельностей повторной ДНК.
8.7. Поскольку она является паразитом, многие из пищевых потребностей этой
бактерии обеспечиваются ее хозяином, и поэтому геном ее не содержит многих генов,
кодирующих белки, входящие в те или иные пути биосинтеза.
8.9. Поскольку возможен обмен ДНК между разными видами посредством гори-
зонтального переноса генов.
Изыскательские задачи
8.1. В разделе 8.1.1 высказывается убедительное мнение в том, что традицион-
ное представление о геноме прокариотов как о единственной молекуле кольцевой
ДНК должно быть действительно оставлено. Обсуждение того, какое определение
должно быть принято, вероятно, не позволит прийти к устойчивому заключению
(поскольку никакое такое заключение все еще не было достигнуто учеными в об-
ласти генетики микробов), но данное упражнение поучительно в том, что оно тре-
бует, чтобы участники дискуссии были способны проводить четкое различие между
плазмидами и геномами.
8.3. Из раздела 8.2.3 можно заключить, что ответ на этот вопрос — «нет», и об-
суждение должно охватить главные моменты, к которым мы обращаемся в данном
разделе: общая трудность в приложении понятий вида, сформулированных для
эукариотов, к прокариотам; существенные различия между генетическим содер-
жанием штаммов, традиционно расцениваемых как члены одного вида, которые
обнаружило секвенирование геномов; осложнения, привносимые горизонтальным
переносом генов.
Тесты по рисункам
8.1. Хромосома Е coli прикреплена к белковому стержню, от которого простираются
петли сверхспирализованной ДНК. Если в одной из петель происходит разрыв ДНК, то
только эта петля теряет сверхспирализацию.
8.3. Гены на этом рисунке находятся в опероне и, следовательно, будут транскри-
бированы в единую молекулу мРНК.
804 Приложение
Глава 9: Геномы вирусов и мобильные генетические элементы
Вопросы закрытого типа
9.1 — с; 9.3 — а; 9.5 — Ь; 9.7 — d; 9.9 — с; 9.11 — с; 9.13 — с; 9.15 — Ь.
Вопросы открытого типа
9.1. Вирусы — облигатные паразиты, зависящие от клеток хозяина в плане вос-
производства. Вирусы не имеют многих компонентов, существенных для жизнеспособ-
ности клеточных организмов; все вирусы используют рибосомы своих хозяев, и только
некоторые вирусы имеют гены ДНК- или РНК-полимераз.
9.3. Это гены, которые имеют одинаковые последовательности нуклеотидов, но
кодируют разные белки. Последовательности нуклеотидов перекрывающихся генов
транслируются в разных рамках считывания.
9.5. Только бактериофаги имеют капсиды типа головки с хвостиком. Вирусы эука-
риотов, в особенности те, которые заражают животных, могут быть покрыты липидной
мембраной.
9.7. Транспозон — сегмент ДНК, который может перемещаться по геному из одной
позиции в другую.
9.9. Длинные рассеянные ядерные элементы (LINE) составляют более 20 % генома
человека, и полномерный элемент содержит два гена, один из которых кодирует об-
ратную транскриптазу. Короткие рассеянные ядерные элементы (SINE) имеют самое
высокое число копий относительно любой из последовательностей в геноме человека
и не содержат генов. Чтобы транспонироваться, они должны использовать обратные
транскриптазы, синтезируемые LINE.
9.11. Активные транспозоны ДНК более распространены в геномах растений, чем
в геноме человека. Некоторые транспозоны растений работают вкупе, что имеет место
в семействе Ac/Ds, открытом Барбарой Макклинток. Элемент Ас кодирует транспозазу,
которая опознает последовательности и Ас, и Ds.
Изыскательские задачи
9.1. Традиционный вопрос, для которого невозможно дать пространного руко-
водства. Ключ к плодотворному обсуждению — не увязать в рассмотрении вирусов, но
сформулировать значимое определение «жизни» и затем решить, охватывает ли это
определение неклеточные системы.
9.3. Эгоистичная ДНК рассматривается как ДНК, которая не дает никакой выгоды
геному, но допускается им, потому что не происходит никакого давления естественного
отбора, направленного на избавление от нее. Если это представление верно, то обладание
транспозонами есть ни преимущество, ни недостаток, и, значит, эти элементы попро-
сту размножаются наряду с функциональными частями генома. См. статью Orgel,L.E.
and Crick,F.H.C. (1980) Selfish DNA: the ultimate parasite. Nature 284: 604-607. Обратите
внимание, что довод против доброкачественной природы транспозонов обосновыва-
ется попытками, которые некоторые организмы предпринимают с целью умерить их
активность, в особенности путем метилирования и, следовательно, инактивирования
этих последовательностей (раздел 17.3.3).
9.5. Ответ на этот вопрос мы дали в разделе 17.3.2, когда рассматривали про-
цесс транспозиции ретроэлемента LTR и обнаружили, что репликация этого элемента
предполагает два переключения: каждое от одного LTR к другому; эти переключения
Приложение 805
гарантируют, что полная последовательность ретроэлемента будет скопирована (см.
рис. 17.18).
Тесты по рисункам
9.1. Идем слева направо: двадцатигранная, нитевидная, головка с хвостиком.
9.3. Ретровирусная инфекция.
9.5. Элементы Ас и Ds были впервые охарактеризованы Барбарой Макклинток.
Элементы Ас обладают геном транспозазы, который отсутствует в элементах Ds. Транс-
позаза, кодируемая элементами Ас, отвечает за транспозицию элементов и Ас и Ds.
Глава 10: Доступ к геному
Вопросы закрытого типа
10.1 — d; 10.3 — b; 10.5 — а; 10.7 — с; 10.9 — Ь; 10.11 — d; 10.13 — а; 10.15 — с.
Вопросы открытого типа
10.1. Электронная микроскопия клеток, приготовленных посредством обработки
ДНКазой (для деградации ДНК) и солевого экстракта (чтобы удалить гистоновые белки),
выявила ядерный матрикс — сложную сеть белковых и РНКовых волоконец. Флюорес-
центное мечение определенных белков показало, что такие процессы, как сплайсинг
РНК, локализованы в различных областях ядра.
10.3. Это предполагает, что эти пары хромосом занимают в ядре смежные терри-
тории.
10.5. Позиционный эффект относится к изменчивости экспрессии гена, которая
происходит, когда ген был клонирован в хозяине-эукариоте. Это связано со случайным
характером встраивания, которое может поместить ген в область раскрытого или,
наоборот, плотноупакованного хроматина.
10.7. Инсуляторы и LCR одинаково могут преодолеть позиционный эффект, когда
сцеплены с генами, встроенными в ядерные клетки. LCRтакже стимулируют экспрессию
генов, которые находятся в пределах своих функциональных доменов; инсуляторы не
способны делать это.
10.9. HDAC подавляют экспрессию генов, удаляя ацетильные группы с гистоновых
белков.
10.11. ДНКаза I не способна расщеплять ДНК, которая является недоступной (напри-
мер, если она содержится в плотноупакованном хроматине). Участки, которые являются вос-
приимчивыми к расщеплению ДНКазой I, обычно смежны с экспрессируемыми генами.
Изыскательские задачи
10.1. Рассмотрение процедур, применяемых для приготовления клеток к элек-
тронной микроскопии, обычно ведет к заключению о том, что структуры, имеющиеся
в живых ядрах, вероятно, теряются, и что артефакты, которых нет в живых ядрах, могут
в них появиться. Встречный довод — общее соответствие между картиной внутреннего
строения, полученной электронной микроскопией, и интерпретациями, проистекшими
из более современных и в меньшей степени разрушающих методов, основанных на
конфокальной микроскопии.
10.3. Отправными точками могут послужить статьи 1) Strahl, В. D. и Allis,С. D. (2000)
The language of covalent histone modifications. Nature 403:41-45; 2) Jenuwein, T. and Allis, C. D.
(2001) Translating the histone code. Science 293:1074-1080.
806 Приложение
10.5. Ответ на этот занимательный вопрос лучше всего искать, изучая соответ-
ствующую научно-исследовательскую литературу, например: LeeJ.T. (2005) Regulation
of X-chromosome counting by Tsix and Xite sequences. Science 309: 768-771.
Тесты по рисункам
10.1. Уровень экспрессии гена будет наиболее высоким, если этот ген встроен
в область раскрытого хроматина. В областях уплотненного хроматина экспрессия гена
будет слабой или не будет происходить вообще.
10.3. Метилированный CpG-островок взаимодействует с метил-СрС-связывающим
белком, который является частью комплекса гистондезацетилазы, что инактивирует
соответствующий ген.
Глава 11: Сборка комплекса инициации транскрипции
Вопросы закрытого типа
11.1 — d; 11.3 — b; 11.5 — Ь; 11.7 — d; 11.9 — с; 11.11 — с; 11.13 — d; 11.15 — b.
Вопросы открытого типа
11.1. Гомеодомен — протяженный мотив спираль-изгиб-спираль, состоящий из
60 аминокислот, которые формируют четыре а-спирали: под номерами 2 и 3 отделены
0-изгибом, номер 3 служит распознающей спиралью, а номер 1 образует контакты
в пределах малой бороздки.
11.3. В анализе с защитой от модификации одного типа ДНК обрабатывается
нуклеазой, которая расщепляет все фосфодиэфирные связи, кроме защищенных свя-
занным белком. В анализе второго типа ДНК обрабатывают метилирующим агентом.
Нуклеотиды, защищенные связанным белком, не будут метилированы.
11.5. В пределах большой бороздки водородные связи формируются между осно-
ваниями нуклеотидов и боковыми группами аминокислот в распознающей структуре
белка, тогда как в малой бороздке более важную роль играют гидрофобные взаимо-
действия. На поверхности спирали основные взаимодействия являются электроста-
тическими (между отрицательными зарядами на фосфатной составляющей каждого
нуклеотида и положительными зарядами на боковых группах аминокислот, таких как
лизин и аргинин), хотя образуется также и некоторое количество водородных связей.
11.7. Базальный промотор — участок, где собирается комплекс инициации
транскрипции. Вышележащие промоторные элементы — сайты связывания для ДНК-
связывающих белков, которые регулируют сборку инициаторного комплекса.
11.9. Лактозный репрессор связывается с операторной последовательностью
оперона лактозы, чтобы предотвратить транскрипцию. Когда лактоза имеется в на-
личии, ее изомер аллолактоза связывается с репрессором. Когда аллолактоза связана,
структура репрессора изменяется таким образом, что он больше не может связываться
с оператором.
11.11. Присутствие альтернативных или множественных промоторов позволяет
одному гену порождать два и более транскрипта. В результате этого синтезируются
подобные, но не идентичные белки — возможно, в различных тканях или на разных
этапах развития или, возможно, одновременно в одной и той же клетке.
Изыскательские задачи
11.1. Есть разные возможности, основанные на закреплении клонированных фраг-
ментов ДНК, представляющих полные последовательности хромосом, сопровождаемом
Приложение 807
применением очищенного связывающегося белка или ядерных экстрактов; при этом
связывание обнаруживается обработкой микроматрицы меченым антителом, специфич-
ным к связывающемуся белку.
11.3. Ответ на первую часть вопроса может быть найден в разделах 11.3.1 и 11.3.2,
но для его обоснования необходимо еще немного поразмыслить. В конце раздела 11.3.1
есть ряд жирных точек, отмечающих принципы регулирования гена бактериальной
природы, а из текста следует, что эти принципы применимы также и к эукариотам.
Это верно, и бесспорное признание этих принципов способствовало развитию нашего
понимания регулирования генов у эукариотов. Однако рассмотрите также и ту воз-
можность, что перенос этих принципов с прокариотов на эукариотов может оказаться
бесполезным в том отношении, что в результате этого можно недооценить значение
некоторых моментов регулирования инициации транскрипции у эукариотов, потому
что у бактерий нет аналогичных процессов.
11.5. Рассмотрим преимущества модульной концепции: ясная картина сценария
регуляции генов, которая вырисовывается; различие между модулями разных типов,
опять же, обеспечивает ясность; наконец, и тот факт, что эти модули точно существуют.
Рассмотрим и ее недостатки: акцент ставится на последовательностях ДНК, тогда как
именно связывающиеся белки являются фактическими регуляторами; возможность того,
что кооперативность между связывающимися белками окажется труднораспознаваемой;
и, последнее, акцент, помещенный на область непосредственно выше гена, тогда как
важные регуляторные сигналы могут быть расположены на отдаленных участках. Из
этих вопросов наиболее важным представляется, возможно, акцент, который модуль-
ная система помещает на ДНК. Мало того, что это вводит в заблуждение — активными
игроками в регулировании генов все-таки являются связывающиеся белки, — но также
и затеняет роль модификации хроматина в регулировании генов1).
Тесты по рисункам
11.1. Репрессор 434 содержит мотив спираль-изгиб-спираль. Вторая спираль мо-
тива вписывается в большую бороздку ДНК, и боковые цепи аминокислоты образуют
специфические контакты с основаниями.
11.3. Как в большой, так и в малой бороздке химические особенности асимметричны
и ориентация пары А-Т может быть определена с помощью связывающегося белка.
11.5. РНК-полимераза Е. coliопознает блок -35 в качестве своей посадочной после-
довател ьности. После прикрепления кДНК переход от закрытого к открытому комплексу
инициируется раскрытием пар оснований в обогащенном АТ блоке -10.
Глава 12: Синтез и созревание РНК
Вопросы закрытого типа
12.1 — а; 12.3 — а; 12.5 — с; 12.7 — Ь; 12.9 — d; 12.11 — с; 12.13 — с; 12.15 — с.
Вопросы открытого типа
12.1. Белок Rho прикрепляется к транскрипту и продвигается по РНК в сторону
полимеразы. Если полимераза продолжает синтезировать РНК, то она держится впереди
преследующего ее белка Rho, но на сигнале завершения полимераза стопорится и Rho
Вызывает недоумение подчеркивание исключительной роли белков. Ведь регуляторные белки не
будут взаимодействовать с ДНК, если там нет сайтов связывания! И ДНК и белки являются равноправными
партнерами. — Прим. ред.
808 Приложение
получает возможность догнать ее. Rho представляет собой геликазу, а это означает, что
он активно разрывает пары оснований, в данном случае между матрицей и транскрип-
том, что приводит к завершению транскрипции.
12.3. Аттенюация работает через сопряжение транскрипции и трансляции, причем
эти процессы не сопряжены в организмах эукариотов, так как РНК транскрибируется
в ядре, а трансляция происходит в цитоплазме.
12.5. Последовательность тРНК в пределах молекулы предшественника принимает
свою вторичную структуру типа клеверного листа и образуются две дополнительные
шпильки: по одной с обеих сторон тРНК. Процессинг начинается с разреза рибонуклеа-
зой Е или F, образующего новый З'-конец сразу выше одной из шпилек. Рибонуклеаза D,
которая по природе своей является экзонуклеазой, обрезает семь нуклеотидов с этого
нового З'-конца и затем берет паузу, пока рибонуклеаза Р делает разрез в начале клевер-
ного листа, образуя 5'-конец зрелой мРНК. После этого рибонуклеаза D удаляет еще два
нуклеотида, создавая З'-конец зрелой молекулы. Все зрелые тРНК должны оканчиваться
тринуклеотидом 5'-ССА-3'. В некоторых пре-тРНК эта последовательность отсутствует
или удаляется обрабатывающими рибонуклеазами. Это происходит с большинством
тех пре-тРНК, З'-концы которых создаются эндонуклеазой, названной рибонуклеазой Z,
которая делает разрез в позиции, смежной с первой парой оснований в клеверном листе
тРНК и, следовательно, удаляет область, которая содержала бы оконечный триплет ССА.
Когда такой триплет ССА отсутствует, он добавляется одной или несколькими не зависи-
мыми от матрицы РНК-полимеразами (например, тРНК-нуклеотидилтрансферазой).
12.7. Деградация бактериальной мРНК начинается с удаления З'-концевой области,
в том числе шпильки (эндонуклеазой, или РНКазой Е, или РНКазой III), открывая новый
конец, с которого экзонуклеазы РНКаза II и ПНФаза могут деградировать оставшуюся
часть молекулы.
12.9. Первый шаг кэпирования—добавление дополнительного гуанозина к самому
5'-концу РНК. у-фосфат конечного нуклеотида удаляется, равно как и 0- и у-фосфаты ГТФ,
в результате чего образуется связь 5'—5'. Эта реакция выполняется ферментом гуанилил-
трансферазой. Второй шаг реакции кэпирования превращает новый оконечный гуанозин
в 7-метилгуанозин за счет присоединения метильной группы к атому азота номер 7
пуринового кольца; эта модификация катализируется гуанинметилтрансферазой.
12.11. Молекулы мякРНК спариваются основаниями с пре-рРНК, чтобы опознать
остатки, которые надлежит метилировать или псевдоуридинилировать. Для остатков,
предназначенных к метилированию, спаривание оснований происходит выше бло-
ка D.
Изыскательские задачи
12.1. Это представление наиболее ярко описано в статье von Hippel, Р. Н. (1998) Ап
integrated model of the transcription complex in elongation, termination, and editing. Science
281: 660-665, которую следует использовать как отправную точку для оценки данной
интерпретации транскрипции.
12.3. Этот вопрос отражен в обсуждении происхождения интронов в разделе 18.3.2.
Гипотеза «о поздних интронах» постулирует, что интроны появились в ходе эволюции
относительно недавно и постепенно накапливаются в геномах эукариотов. Согласно этой
модели, интроны отсутствуют у бактерий, потому что основной план бактериального
генома был утвержден прежде, чем появились первые интроны. В противовес ей гипотеза
«о ранних интронах» заявляет, что интроны являются очень древними и постепенно
исчезают из геномов эукариотов. Поэтому отсутствие интронов в геномах бактерий
Приложение 809
является камнем преткновения для поборников гипотезы о «ранних интронах», но, как
описано в разделе 18.3.2, им ведомы способы обойти эту проблему.
12.5. Мир РНК, в котором рибозимы были биологическими катализаторами един-
ственного типа, описан в разделе 18.1.1. По какой причине некоторые рибозимы сохра-
нились, не известно, но обращаем внимание, что большая часть функций рибозимов,
приведенных в табл. 12.4, заключается в специфическом к последовательности рас-
щеплении молекул РНК. Вспомним, что в техническом примечании 5.1 сообщается, что
«единственная серьезная недостача в инструментарии РНК есть отсутствие ферментов
со степенью специфичности к последовательности, выказываемой эндонуклеазами
рестрикции, которые столь важны для манипуляций с ДНК».
Тесты по рисункам
12.1. Когда транскрибируется обращенный палиндром терминатора, формируется
шпилька РНК. Формирование шпильки предпочтительно по сравнению со спариванием
оснований ДНК-РНК и приводит к образованию меньшего количества контактов ДНК-
РНК. Когда транскрибируется обогащенная аденинами последовательность матрицы,
есть несколько пар оснований A-U, которые имеют только по две водородные связи
каждая. Эти два фактора ослабляют взаимодействие между матрицей и транскриптом
и вызывают завершение.
12.3. Расщепление 5’-сайта сплайсинга происходит за счет реакции трансэфиро-
образования, активизируемой гидроксильной группой, присоединенной к 2'-углероду
нуклеотида аденозина, расположенного в пределах последовательности интрона. Резуль-
тат гидроксильного воздействия — расщепление фосфодиэфирной связи в 5'-сайте сра-
щения, сопровождаемое образованием новой фосфодиэфирной связи 5-2', связывающей
первый нуклеотид интрона (G мотива 5'-GU-3') со внутренним аденозином. Это означает,
что интрон завернулся петлей сам на себя же и образовал лассо-подобную структуру.
Расщепление З'-сайта сплайсинга и соединение экзонов происходит в результате второй
реакции трансэфирообразования, она активируется группой З'-ОН, присоединенной
к концу вышележащего экзона. Эта группа воздействует на фосфодиэфирную связь
в З'-сайте сплайсинга, расщепляет ее и таким образом высвобождает интрон в виде
лассоподобной структуры, которая впоследствии преобразуется обратно в линейную
РНК и деградирует. В то же самое время З'-конец вышележащего экзона присоединяется
к новообразованному 5'-концу нижележащего экзона, завершая процесс сплайсинга.
12.5. На рисунке показан путь РНК-интерференции. Молекулы двунитевой РНК
деградируют до коротких интерферирующих РНК (siPHK) ферментом Dicer. Молекулы
siPHK прикрепляются к мРНК, которая затем расщепляется наведенным РНК комплек-
сом подавления (RISC).
Глава 13: Синтез и процессинг протеома
Вопросы закрытого типа
13.1 — Ь; 13.3 — с; 13.5 — с; 13.7 — с; 13.9 — d; 13.11 — с; 13.13 — с; 13.15 — d.
Вопросы открытого типа
13.1. Транспортные РНК образуют связь между мРНК и синтезируемым поли-
пептидом. Эта связь имеет как физическую природу (молекулы тРНК связываются
и с мРНК, и с наращиваемым полипептидом), так и информационную (молекулы тРНК
810 Приложение
обеспечивают соответствие аминокислотной последовательности синтезируемого
полипептида, определяемое генетическим кодом, последовательности нуклеотидов
матричной мРНК).
13.3. Большинство ошибок исправляется самой аминоацил-тРНК-синтетазой, при-
чем данный процесс редактирования отличается от аминоацилирования выстраиванием
иных контактов с тРНК.
13.5. Предынициаторный комплекс включает субъединицу 40 S рибосомы — «трех-
частный комплекс», состоящий из фактора инициации eIF-2, связанного с инициаторной
TpHKMet и молекулой ГТФ, — и три дополнительных фактора инициации: eIF-1, eIF-lA
и eIF-З.
13.7. Фосфорилирование фактора инициации eIF-2 вызывает подавление инициа-
ции трансляции, поскольку оно препятствует этому фактору связывать молекулу ГТФ,
что необходимо для доставки инициаторной тРНК в малую субъединицу рибосомы.
13.9. Мотивы вторичной структуры в полипептидной цепи формируются в течение
нескольких миллисекунд. Этот шаг сопровождается складыванием белка в компактную,
но не свернутую организацию, в которой его гидрофобные группы повернуты внутрь
и ограждены от воды. В течение следующих нескольких секунд или минут мотивы вто-
ричной структуры взаимодействуют друг с другом и постепенно оформляется третичная
структура, нередко через ряд промежуточных конформаций.
13.11. Интеины способны к самовырезанию, так что они способны самоудаляться
из белка.
Изыскательские задачи
13.1. Хорошей отправной точкой для того, чтобы приняться за решение столь слож-
ной задачи, послужит статья Ribas de Pouplana,L. and Schimmel, P. (2001) Aminoacyl-tRNA
synthetases: potential markers of genetic code development Trends Biochem. Sci. 26: 591-596.
13.3. С тех самых пор, как ДНК была утверждена в качестве генетического материала
ещё в 1950-е гг., эволюция генетического кода вызывала споры. Многие генетики поддер-
живаюттеорию «замороженного случая», согласно которой кодоны аминокислотам были
назначены случайно на самых ранних стадиях эволюции и впоследствии этот набранный
код стал «замороженным», потому что любые его изменения вызывали многочисленные
нарушения аминокислотных последовательностей белков, но полученные из различных
источников данные показывают, что генетический код, возможно, эволюционировал
в менее случайной манере. Во-первых, противоречивые экспериментальные результаты
говорят о том, что по крайней мере некоторые аминокислоты связываются непосред-
ственно с молекулами РНК, содержащими соответствующие кодоны, и это происходит
в отсутствие тРНК, которая опосредствует взаимодействие в современных клетках. Если
это верно, то отсюда вытекает, что существует некоторого рода химическая зависимость
между аминокислотой и ее кодоном (ами). Во-вторых, отклонения от стандартного кода
(см. табл. 1.3) указывают на то, что одни и те же перераспределения кодонов происходили
не раз. Если зависимость между кодоном и аминокислотой носит совершенно случайный
характер, как это предполагает теория «замороженного случая», то можно не ожидать,
что мы будем наблюдать повторение тех же самых схем перераспределения кодонов при
внесении в них изменений. См. также: Knight, R. D, Freeland, S. J. and Landweber, L. F. (1999)
Selection, history and chemistry: the three faces of the genetic code. Trends Biochem. Sci. 24:
241-247; Szathmary,E. (1999) The origin of the genetic code: amino acids as cofactors in an
RNA world. Trendsjenet 15:223-229; и Yarns, M., CaporasoJ.G. and Knight, R. (2005) Origins
of the genetic code: the escaped triplet theory. Annu. Rev. Biochem. 74:179-198.
Приложение 811
13.5. К данному вопросу относятся следующие моменты: рано пришедшее по-
нимание того факта, что рибосомы состоят из большой и малой субъединиц, было
определяюще важным в разработке первоначальных моделей механики синтеза белка;
опознавание Р-, А- и Е-сайтов было ключом к более глубокому пониманию трансляции;
а структурные исследования лежат в основе проводимой в настоящее время работы по
активности пептидилтрансферазы.
Тесты по рисункам
13.1. Остаток инозина может располагаться на нуклеотиде номер 34. Инозин в этой
позиции может спариться основаниями с А, С или U в мРНК, что позволяет одной моле-
куле тРНК опознавать три разных кодона для определения аминокислоты.
13.3. CCdA-фосфатпуромицин является аналогом переходного состояния, которое
возникает во время образования пептидных связей, и связывается рибосомой в активном
участке пептидилтрансферазы. Поскольку никакие белки рибосомы не близки про-
странственно молекуле CCdA-фосфатпуромицина, это указывает на то, что образование
пептидных связей не катализируется белками.
Глава 14: Регулирование активности генома
Вопросы закрытого типа
14.1 — d; 14.3 — а; 14.5 — а; 14.7 — с; 14.9 — d; 14.11 — d; 14.13 — с; 14.15 — d.
Вопросы открытого типа
14.1. Дифференцировка относится к принятию специализированной физиологи-
ческой роли клеткой. Она осуществляется за счет постоянных изменений в экспрессии
генома, которые меняют биохимический состав клетки. Развитие — череда согласован-
ных изменений, которые происходят в течение жизненного цикла клетки или организма.
Эти изменения могут быть временными или постоянными и должны длиться в течение
продолжительного периода времени.
14.3. Когда глюкоза переносится в Е coli, белок переноса сахара IIAGlc становится
дефосфорил ированным. Дефосфорил ированная форма 11 AGlc ингибирует фермент адени-
латциклазу, которая производит цАМФ. Таким образом, в присутствии глюкозы уровни
содержания цАМФа низки, когда же глюкоза отсутствует, уровни цАМФа высоки.
14.5. MAP-киназа активируется, когда она фосфорилируется белком Мек. Фосфо-
рилированная MAP-киназа перемещается в ядро, где она фосфорилирует активаторы
транскрипции, производя реакцию, которая стимулирует деление клетки.
14.7. Переключение класса приводит к полному изменению типа иммуноглобули-
на, синтезируемого лимфоцитом. Это требует события рекомбинации, которая удаляет
последовательности Ср и С5 наряду с частью хромосомы между этой областью и сегмен-
том Сн, определяющим класс иммуноглобулина, который будет теперь синтезироваться
лимфоцитом. Например, для того чтобы лимфоцит переключился на синтез IgG (самого
распространенного типа иммуноглобулина, синтезируемого зрелыми лимфоцитами),
удаление должно поместить один из сегментов Су, которые определяют тяжелую цепь
IgG, на 5'-конец группы. Переключение класса отличается от соединения V-D-J и событие
рекомбинации не вовлекает белки RAG.
14.9. Субъединица qf активируется путем высвобождения из комплекса со SpoIIAB.
Этим событием управляет комплекс SpoIIAA, который, будучи нефосфорилирован, может
812 Приложение
также присоединиться к комплексу SpoIIAB и препятствовать последнему связываться
с субъединицей aF. Если SpoIIAA нефосфорилирован, то субъединица qf высвобожда-
ется и становится активной; когда SpoIIAA фосфорилирован, субъединица cF остается
связанной с комплексом SpoIIAB и, следовательно, неактивной. В материнской клетке
SpoIIAB фосфорилирует SpoIIAA и, таким образом, оставляет субъединицу qf в ее свя-
занном неактивном состоянии. Но в проспоре попыткам SpoIIAB фосфорилировать
SpoIIAA противодействует еще один белок, SpoIIE, и, таким образом, субъединица cF
высвобождается и становится активной. Способность белка SpoIIE противодействовать
комплексу SpoIIAB в проспоре, но не в материнской клетке, обусловлена тем фактом,
что молекулы белка SpoIIE связываются с мембраной на поверхности перегородки. По-
скольку проспора намного меньше, чем материнская клетка, но площадь поверхности
перегородки одинакова в обеих, концентрация белка SpoIIE выше в проспоре, и это
позволяет ему противодействовать комплексу SpoIIAB.
14.11. Ген bicoid транскрибируется в материнских клетках-кормилицах, и мРНК
вводится в передний конец неоплодотворенных яиц. мРНК bicoid остаётся в переднем
конце яйцеклетки, присоединенной к клеточному скелету своим З'-нетранслируемым
концом. Трансляция мРНК происходит после оплодотворения яйца, и белок Bicoid рас-
ходится по всему соклетию, устанавливая градиент концентрации с переднего конца
(высокая) к заднему концу (низкая).
Изыскательские задачи
14.1. Данная тема для большого доклада требует допол нительного чтения, хорошей
отправной точкой может послужить книжка: Berg, J. М., Timoczko, J. L. and Stryer, L. (2006)
Biochemistry, 6th Ed. W. H. Freeman, New York.
14.3. Для того чтобы подойти к этому вопросу наиболее эффективно, необходимо
рассмотреть степень, до которой наше современное понимание развития у высших
эукариотов основывается на информации, которой не было бы в нашем распоряжении,
если бы мы никогда не проводили эксперименты с С. elegans или D. melanogaster. С одной
стороны, изучение С. elegans во многом способствовало нашему пониманию молекуляр-
ной основы РНК-интерференции (раздел 12.2.6), но это понимание, наверно, пришло бы
ненамного позже, если бы С. elegans вообще не существовало. С другой стороны, было
бы трудно раскрыть роль гомеозисных генов у высших эукариотов (раздел 14.3.4) без
предварительного изучения этих генов у D. melanogaster. Возможны и другие мнения
по данному вопросу.
14.5. В отличие от эквивалентного вопроса касательно исследований наследствен-
ности (глава 3, изыскательская задача 3.3), описание идеального модельного организма,
служащего для представления развития у высших эукариотов, не может быть легким
делом. Свои доводы можно было бы приводить в том русле, что такой моделью должен
быть наименее сложный эукариот, который показывает специфические признаки раз-
вития в ходе его изучения. Следовательно, для отражения различных сторон явления
развития идеальными будут разные модели.
Тесты по рисункам
14.1. Данное явление называют диауксией, и оно проистекает из катаболитной
репрессии. Глюкоза подавляет экспрессию оперона лактозы через косвенное влияние на
белок-активатор катаболитных оперонов. Этот белок связывается с сайтом узнавания
на различных участках в бактериальном геноме и активирует инициацию транскрипции
в нижележащих промоторах. Продуктивная инициация транскрипции в этих промоторах
Приложение 813
зависит от присутствия связанного белка-активатора катаболитных оперонов: если
белок отсутствует, то гены, управляемые этим промотором, не транскрибируются. Сама
глюкоза не взаимодействует с белком-активатором катаболитных оперонов. Вместо
этого, глюкоза управляет уровнем цАМФа в клетке. Она делает это, ингибируя действие
аденилатциклазы — фермента, который синтезирует цАМФ из АТФ. Ингибирование
опосредствуется единицей IIAGlc — компонентом многобелкового комплекса, который
переносит сахара в бактерию. Когда глюкоза переносится в клетку, IIAGlc становит-
ся дефосфорилированным. Дефосфорилированная версия IIAGlc ингибирует действие
аденилатциклазы. Это означает, что, если уровни глюкозы высоки, содержание цАМФа
в клетке низкое. Белок-активатор катаболитных оперонов может связываться со своими
целевыми участками только в присутствии цАМФа, так что когда глюкоза присутствует,
белок остается неприкрепленным и опероны, которыми он управляет, выключены.
14.3. Во время ранней стадии развития В-лимфоцита локусы иммуноглобулина
в его геноме подвергаются перестройкам. В пределах локуса тяжелой цепи эти пере-
стройки сцепляют один из сегментов VH гена с одним из сегментов DH гена и затем свя-
зывают такую комбинацию V-D с сегментом JH гена. В конечном итоге получается экзон,
который содержит полную открытую рамку считывания, определяющую сегменты V,
D и J белка иммуноглобулина. Этот экзон сцепляется с экзоном сегмента С (посредством
сплайсинга во время процесса транскрипции), в результате чего образуется полная
мРНК тяжелой цепи, которая может быть транслирована в белок иммуноглобулина,
который будет специфическим только для одного лимфоцита. Подобная череда пере-
строек ДНК выливается в построение экзона V-J легкой цепи лимфоцита, и опять же
путем сплайсинга к новоявленной комбинации присоединяется экзон сегмента С во
время синтеза мРНК.
Глава 15: Репликация генома
Вопросы закрытого типа
15.1 — с; 15.3 — а; 15.5 — Ь; 15.7 — с; 15.9 — Ь; 15.11 — а; 15.13 — а; 15.15 — с.
Вопросы открытого типа
15.1. Модель разбросанной репликации предсказывает, что каждая дочерняя моле-
кула будет состоять частично из родительской ДНК и частично из новосинтезируемой
ДНК. В полуконсервативной репликации каждая дочерняя молекула состоит из одной
родительской нити и одной новосинтезированной нити. Если репликация консерватив-
ная, то одна из дочерних двойных спиралей полностью состоит из новосинтезированной
ДНК, а другая включает две родительские нити.
15.3. Репликация катящегося кольца запускается в надрезе, который производится
в одном из родительских полинуклеотидов. Свободный З’-конец, который образуется,
продолжается, вытесняя 5'-конец полинуклеотида. Продолжающийся синтез ДНК «ска-
тывает» полную копию генома, и дальнейший синтез в конечном счете дает вереницу
геномов, сцепленных голова к хвосту. Эти однонитевые, линейные геномы превращаются
в двунитевые кольцевые молекулы за счет синтеза комплементарной нити, за которым
следует расщепление в точках сопряжения между геномами и формирование колец из
полученных сегментов.
15.5. На каждом конце открытой, расплавленной области ДНК в области инициации
собирается предынициаторный комплекс белков DnaB и DnaC. Белок DnaB — геликаза,
814 Приложение
которая продолжает однонитевую область в области инициации, тем самым позволяя
прикрепиться другим репликационным белкам.
15.7. Праймаза синтезирует затравку, состоящую из 8-12 рибонуклеотидов. Нить
затем продолжается ДНК-полимеразой а, которая добавляет следующие 20 или около
того нуклеотидов (среди которых может затесаться несколько рибонуклеотидов).
Остальная часть копии опережающей нити синтезируется ДНК-полимеразой 5.
15.9. Хромосома станет укороченной, если самый З’-конец отстающей нити не
копируется, потому что последний фрагмент Окадзаки не может быть затравлен, так
как естественная позиция для участка праймирования находится далее конца матрицы.
Отсутствие такого фрагмента Окадзаки означает, что копия в лице отстающей нити
короче, чем она должна быть. Если копия сохранит ту же длину, то, когда она будет
служить родительским полинуклеотидом в следующем круге репликации, полученная
хромосома будет короче, чем ее предшественник второго колена. Укорочение, хотя и до
меньшей степени, будет происходить также и в том случае, если затравка для последнего
фрагмента Окадзаки разместится на самом З'-конце отстающей нити, потому что эта
оконечная РНК-затравка не может быть превращена в ДНК с помощью стандартных
процессов удаления затравки.
15.11. Когда ген, кодирующий белок Cdc6p, подавляется, предрепликационные
комплексы отсутствуют. Когда ген сверхэкспрессируется, имеют место многократные
репликации генома без митоза.
Изыскательские задачи
15.1. Причина для такого принятия приводится в знаменитом заключении Уотсона
и Крика в их статье в журнале Nature, объявляющей об открытии структуры двойной
спирали ДНК: «от нашего внимания не ускользнуло, что специфическое спаривание,
кое мы постулировали, непосредственно предполагает и возможный механизм копи-
рования генетического материала» (см. начало раздела 15.1). Как только этот вопрос
будет уяснен, рассмотрите причины, по которым полуконсервативная репликация была
принята, фактически, не непосредственно (раздел 15.1.1).
15.3. Информацию, почерпнутую из «Геномов», можно дополнить большим коли-
чеством подробностей из научно-исследовательского обзора, как например: Patel, S.S.
and Picha, К.М. (2000) Structure and function of hexameric helicases. Annu. Rev. Biochem. 69:
651-697.
15.5. В качестве основных источников мы рекомендуем: 1) Shay, J. W. and Wright, W. E.
(2005) Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 26:
867-874; 2) Shay, J.W. (2005) Meeting report: the role of telomeres and telomerase in cancer.
Cancer Res. 65: 3513-3517.
Тесты no рисункам
15.1. На рисунке показана вытеснительная репликация. Двойная спираль раз-
рывается в D-петле за счет присутствия молекулы РНК, спаренной с одной из нитей
ДНК. Эта молекула РНК выступает как отправная точка для синтеза одного из дочерних
полинуклеотидов. Этот полинуклеотид синтезируется путем непрерывного копирова-
ния одной из нитей спирали, тем временем вторая нить вытесняется и впоследствии
копируется по завершении синтеза первого из дочерних геномов.
15.3. ДНК-полимераза III не обладает действием экзонуклеазы 5'->3' и, таким об-
разом, отделяется от отстающей нити по достижении следующего фрагмента Окадзаки.
Приложение 815
Ее место занимает ДНК-полимераза I, которая обладает действием экзонуклеазы 5’->3'
и ввиду этого удаляет затравку и, как заведено, начало ДНК-компонента фрагмента Окад-
заки, продолжая З'-конец смежного фрагмента в открытую область матрицы. Теперь эти
два фрагмента Окадзаки примыкают друг к другу, и отсутствующая фосфодиэфирная
связь постановляется на должное место ДНК-лигазой.
Глава 16: Мутации и репарация ДНК
Вопросы закрытого типа
16.1 — Ь; 16.3 — Ь; 16.5 — с; 16.7 — Ь; 16.9 — а; 16.11 — Ь; 16.13 — с; 16.15 — Ь.
Вопросы открытого типа
16.1. Мутации возникают по причине ошибок, которые получаются в процессе
репликации генома, и по вине мутагенов, которыми служат химические или физические
агенты, реагирующие с ДНК и изменяющие структуру отдельных нуклеотидов.
16.3. ДНК-полимераза может вычислять неверный нуклеотид в случаях, когда этот
нуклеотид только связался с ДНК-полимеразой, когда он переместился на активный уча-
сток фермента и когда он присоединен к З'-концу синтезируемого полинуклеотида.
16.5. Высокая температура вызывает гидролиз p-N-гликозидной связи, которая
скрепляет основание с сахарной составляющей нуклеотида. Это чаще происходит с пу-
ринами, чем с пиримидинами, и приводит к образованию АП (апуринового/апири-
мидинового) или не содержащего основания участка. Лишенный всяких оснований
сахарофосфат неустойчив и быстро деградирует, оставляя после себя пропуск, если
молекула ДНК двунитевая. В каждой клетке человека за день возникает около 10 000
АП участков, но они редко ведут к мутациям, потому что клетки имеют эффективные
системы устранения пропусков.
16.7. Данный признак является доминантным, потому что гетерозиготный инди-
видуум способен синтезировать лишь около 50 % активного аполипопротеина В-100
по сравнению с его содержанием в клетках здорового человека. Такое сокращение вы-
зывает болезнь. Это пример гаплонедостаточности.
16.9. Гипермутация, как думают, проистекает из превращения некоторых цитозино-
вых оснований в урацилы действием цитозиндезаминазы, сопровождаемого вырезанием
урацилов из полинуклеотида действием урацил-ДНК-гликозилазы, в результате чего
в таких позициях образуются АП участки. При восстановлении вырезанием оснований
каждый АП участок после этого был бы заполнен при помощи ДНК-полимеразы 0 с вос-
становлением первоначальной последовательности, но при гипермутации АП участки
не устраняются. Это означает, что на следующем круге репликации любой из четырех
стандартных нуклеотидов может быть помещен в дочернюю нить супротив каждого
АП участка. В следующем круге репликации такая мутация закрепится.
16.11. При NHEJ многокомпонентный белковый комплекс направляет ДНК-лигазу
к месту разрыва. Комплекс включает белок Ku, который связывается с концами ДНК
по обе стороны от разрыва. Отдельные белки Ku обладают сродством друг к другу, что
означает, что два оборванных конца молекулы ДНК сближаются. Белок Ku связывается
с ДНК совместно с ДНК-РК^-протеинкиназой, которая активирует третий белок, XRCC4,
который взаимодействует с ДНК-лигазой IV млекопитающих, направляя этот восстано-
вительный белок к двунитевому разрыву.
816 Приложение
Изыскательские задачи
16.1. Транзиция (замена вида пурин на пурин или пиримидин на пиримидин)
не изменяет ориентацию пар пурин-пиримидин в двойной спирали. Трансверсия (за-
мена вида пурин на пиримидин или пиримидин на пурин) изменяет ориентацию пар
пурин-пиримидин на обратную.
16.3. Помимо статей, указанных в соответствующей части раздела дополнитель-
ной литературы, в качестве важного обзора можно рекомендовать Cummings, С.J. and
Zoghbi,H.Y. (2000) Trinucleotide repeats: mechanisms and pathophysiology. Annu. Rev.
Genomics Hum. Genet. 1: 281-328.
16.5. Отправной точкой для проработки данного вопроса послужит статья White, О.,
Eisen,). A., Heidelberg,). F., et al. (1999) Genome sequence of the radioresistant bacterium
Deinococcus radiodurans Rl. Science 286:1571-1577.
Тесты по рисункам
16.1. На рисунке изображено встраивание двух нуклеотидов в микросателлитную
последовательность в молекуле ДНК. Это пример проскальзывания репликации.
16.3. Фермент ДНК-гликозилаза удаляет поврежденное основание из ДНК. В резуль-
тате этого образуется участок без основания, и сахаро-фосфатная группа на этом участке
вырезается АП-эндонуклеазой. После этого пропуск заполняется ДНК-полимеразой и в
заключение фосфодиэфирная связь устанавливается на место ДНК-лигазой. Это схема
восстановления вырезанием оснований.
Глава 17: Рекомбинация
Вопросы закрытого типа
17.1 — с; 17.3 — d; 17.5 — b; 17.7 — d; 17.9 — с; 17.11 — d.
Вопросы открытого типа
17.1. Рекомбинация делает возможным значительные изменения и серьезное
реструктурирование геномов. При отсутствии рекомбинации геномы подвергались бы
лишь небольшим изменениям и были бы довольно статичными структурами.
17.3. Согласно модели с двунитевым разрывом гомологичная рекомбинация иници-
ируется с образования двунитевого разрыва в одной из молекул. Одна из нитей в каждой
половине разорванной хромосомы укорачивается, оставляя З'-выступы. Один из таких
выступов вторгается в другую неповрежденную молекулу ДНК и создает структуру
Холлидея. Укороченные нити ДНК продолжаются ДНК-полимеразой, при этом синтез
ДНК в преобразуемой области осуществляется с использованием в качестве матрицы
молекулы ДНК, которая не подвергалась первоначальному двунитевому разрыву.
17.5. Бактерии могут приобретать новые гены путем трансформации, трансдукции
и конъюгации. Если ДНК, которая поступает в клетку, подобна по своей последователь-
ности какой-либо части генома Е. coli, то может произойти гомологичная рекомбинация,
в результате которой возможно встраивание чужеродной ДНК в хромосому Е. coli.
V7.7. Каждый из участков att содержит идентичную срединную последовательность
длины 15 п.н. Срединные последовательности зажаты между изменчивых последова-
тельностей: В и В' (каждая из которых имеет длину лишь 4 п.н.) в геноме бактерий и Р
и Р' в ДНК фагов. Как последовательность Р, так и ее компаньонка Р' имеют длину более
100 п.н. Мутации в срединной последовательности инактивируют участок att, так что
он также более не может участвовать в рекомбинации.
Приложение 817
17.9. Первый шаг репликации всякого ретроэлемента — синтез его РНКовой ко-
пии. Затем эта молекула РНК превращается в двунитевую ДНК. Первая стадия в таком
превращении — синтез (посредством обратной транскрипции) копии этой молекулы
РНК в лице однонитевой ДНК. Эта реакция синтеза нити затравливается тРНК, которая
отжигается к участку в пределах 5'-длинного концевого повторения РНКовой копии
ретроэлемента.
Изыскательские задачи
17.1. Цель этого вопроса состоит в том, чтобы углубить понимание того факта, что,
хотя название RecA указывает на роль этого белка в рекомбинации, более правильно
рассматривать его как белок однонитевого связывания, обладающий дополнительной
способностью активировать действие протеазы, которая играет множество разнообраз-
ных ролей в молекулярной биологии бактерий. К наиболее стоящим литературным
источникам мы относим: 1) Kowalczykowski, S. С. and Eggleston, А. К. (1994) Homologous
pairing and DNA strand-exchange proteins. Annu. Rev. Biochem. 63: 991-1043; 2) Michel, B.
(2005) After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol. 3: e255;
3) Lusetti, S. L. and Cox, M. M. (2002) The bacterial RecA protein and the recombinational DNA
repair of stalled replication forks. Annu. Rev. Biochem. 71: 71-100.
17.3. Необходимую информацию по данному вопросу можно найти в статье Pyle, А. М.
(2004) DNA repair: big engine finds small breaks. Nature 432:157-158.
17.5. В разделе 17.3.3 сообщается о том, что метилирование ДНК есть главнейший
из распознанных процессов минимизации активности транспозонов. Исчерпывающие
литературные источники по данному предмету, пожалуй, отыскать нелегко, но хорошими
отправными точками могут послужить: 1) Yoder,).A., Walsh,С. Р. and Bestor,T.H. (1997)
Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335-340;
2) Rabinowicz, P. D., Palmer, L.E., May,B.P., Hemann,M.T., Lowe,S.W., McCombie,W.R. and
Martienssen,R.A. (2003) Genes and transposons are differentially methylated in plants, but
not in mammals. Genome Res. 13: 2658-2664.
Тесты по рисункам
17.1. Сайт-специфическая рекомбинация.
17.3. RecA.
Глава 18: Механизмы эволюции геномов
Вопросы закрытого типа
18.1 — d; 18.3 — b; 18.5 — а; 18.7 — d; 18.9 — b; 18.11 — с; 18.13 — d.
Вопросы открытого типа
18.1. Ранняя атмосфера Земли содержала очень низкие уровни кислорода и имела
высокие количества аммиака и метана, значительно отличаясь от атмосферы совре-
менной Земли.
18.3. Весьма возможно, что на древней Земле эволюционировала не одна, а не-
сколько биологических систем, даже при том, что все ныне живущие организмы, кажется,
произошли из одного источника. Согласно наиболее правдоподобному сценарию го-
сподствующей системой стала та, что первая развила средства синтезировать белковые
ферменты и, вероятно, также первой приняла геном из ДНК. Больший каталитический
818 Приложение
потенциал и более точная репликация, сообщенная белковыми ферментами и ДНКовыми
геномами, должно быть, придали таким клеткам значительное преимущество над теми,
которые все еще содержали РНКовые протогеномы. Клетки с набором ДНК-РНК-белок
размножались быстрее, что позволило им вытеснить клетки с РНК в борьбе за пита-
тельные вещества.
18.5. Один из таких переходов сопровождал появление первых эукариотов около
1,4 миллиарда лет назад; эти клетки, вероятно, содержали по крайней мере 10 000 генов
(минимум для современных эукариотов) по сравнению с 5 000 и меньше, типичными
для прокариотов. Второй переход был сопряжен с появлением первых позвоночных
вскоре по окончании кембрийского периода; они, подобно современным позвоночным,
по всей вероятности имели по крайней мере 30000 генов.
18.7. Ретроген возникает в результате превращения молекулы мРНК в кДНК, сопрово-
ждаемого повторным встраиванием этой кДНК обратно в геном. Обычно встроенная кДНК
дает начало псевдогену, так как ей недостает собственного промотора (а также и интронов).
Но если кДНК будет встроена по соседству с промотором некоторого существующего гена,
то она может стать активной, подчинив этот промотор собственным нуждам.
18.9. Дупликация сегментов может поместить экзоны одного гена смежно с экзона-
ми другого гена. Если эти экзоны будут после этого транскрибированы, то полученная
мРНК может дать начало новому генному продукту.
18.11. «Экзонная теория генов» стоит на том, что интроны сформировались, когда
были построены первые ДНКовые геномы, вскоре после крушения мира РНК. Эти ге-
номы, видимо, содержали много коротких генов, каждый из которых определял очень
маленький полипептид (возможно, только единственный структурный домен). Чтобы
облегчить синтез многодоменного белка, как можно предположить, этакие короткие
гены были помещены группами один за другим. Короткие гены стали экзонами, а по-
следовательности между ними стали интронами.
Изыскательские задачи
18.1. Подходящая статья, как отмечено в разделе дополнительной литературы, —
Vision, T.J., Brown, D.G. and Tanksley, S.D. (2000) The origins of genomic duplications in
Arabidopsis. Science 290: 2114-2117.
18.3. Первые статьи, в которых содержалось опровержение этого предложения,
были: 1) Salzberg,S. L., White,О., Peterson,), and Eisen, J. А. (2001). Microbial genes in the human
genome: lateral transfer or gene loss? Science 292: 1903-1906; 2) Stanhope, M. J., Lupas,A.,
Italia, M. J., Koretke, К. K., Volker, C. and Brown, J. R. (2001) Phylogenetic analyses do not support
horizontal gene transfers from bacteria to vertebrates. Nature 411: 940-944. Прояснению
этого вопроса будет способствовать уяснение некоторых моментов молекулярной фило-
генетики, отраженных в главе 19.
18.5. Данный вопрос относится к сравнениям между последовательностями гено-
мов. Если интерпретировать буквально, то информация, сообщаемая в разделе 18.4 и в
статьях, упомянутых в разделе дополнительной литературы, побуждает прийти к за-
ключению о том, что ответ будет «нет» (по крайней мере для сравнений между геномами
человека и шимпанзе). Может ли ответ быть иным, когда (и если) будут секвенированы
геномы гориллы и орангутана? Или если полная последовательность ДНК неандертальца
могла бы быть получена (раздел 19.3.2)? Если вопрос интерпретировать менее строго
и будут проведены постгеномные исследования, то в разделе 18.4 явственнее станет
проглядывать мысль о том, что может быть возможно определить по крайней мере не-
которые из тех факторов, которые делают нас человеком.
Приложение 819
Тесты по рисункам
18.1. Когда давление естественного отбора оказывается только на одну копию
пары дуплицированных генов, вторая копия может накапливать мутации, которые
могут привести к появлению новых действий или функций. Псевдогены появляются
при действии схемы, согласно которой вредные мутации происходят во второй копии
гена, так что эта копия деградирует с течением времени.
18.3. На верхнем виде представлен механизм образования нового гена путем ду-
плицирования домена, когда домен В дуплицируется с исходного гена. На нижнем виде
изображено тасование доменов, где домены из двух различных генов объединяются
и тем самым образуют новый ген.
Глава 19: Молекулярная филогенетика
Вопросы закрытого типа
19.1 — Ь; 19.3 — а; 19.5 — с; 19.7 — Ь; 19.9 — Ь; 19.11 — с; 19.13 — Ь.
Вопросы открытого типа
19.1. Фенетика — метод филогенетического анализа, в котором учитывается как
можно большее число переменных; согласно преобладавшему до развития фенетики
представлению, филогении должны основываться на ограниченном числе характери-
стик, которые считались важными.
19.3. Предковые состояния признаков имелись у отдаленного общего предка груп-
пы организмов, тогда как приобретенные состояния признака эволюционировали из
предкового состояния более близкого по времени общего предка.
19.5. Внутренний узел в дереве генов представляет расхождение предкового гена
на два аллеля за счет мутации. Внутренний узел в дереве видов указывает событие
видообразования, которое имело место, когда предковая группа разделяется на две
группы, которые не могут скрещиваться. Такие события мутаций и видообразований
навряд ли происходили в одно и то же время.
19.7. Частотность аллелей определяется естественным отбором и случайным ге-
нетическим дрейфом. Естественный отбор изменяет частоту тех аллелей, от которых
зависит приспособленность особи, тогда как случайный генетический дрейф изменяет
частоту аллелей из-за случайного характера событий рождения, смерти и воспроиз-
водства.
19.9. ДНК была извлечена из 400 мг костной ткани неандертальца, и ПЦР были
нацелены на наиболее изменчивую, согласно ожиданиям, часть митохондриального
генома неандертальца. Ожидалось, что ДНК должна быть деградированной, так что по-
следовател ьность выстраивалась сечениями — выполнялись девять перекрывающихся
ПЦР, ни в одной из которых не размножалась ДНК длиннее 170 п. н., но вместе они да-
вали общую длину 377 п. н. Чтобы сравнить последовательность, полученную из кости
неандертальца, с последовательностями шести гаплогрупп митохондриальной ДНК
современных людей, было построено филогенетическое дерево. Последовательность
неандертальца была помещена в отдельную ветвь, не связанную напрямую ни с одной
из последовательностей современного человека. Чтобы сравнить последовательность
неандертальца с 994 последовательностями ныне живущих людей, было проведено
множественное выравнивание. Последовательность неандертальца отличалась от со-
временных последовательностей в среднем на 27,2±2,2 нуклеотидных позиций, тогда
820 Приложение
как современные последовательности отличались друг от друга только лишь на 8,0±3,1
позиций. Степень различия между ДНК неандертальца и современного европейца не-
совместима с концепцией, согласно которой современные европейцы происходят от
неандертальцев, и убедительно подтверждает гипотезу об африканском происхождении
человека.
19.11. Первые исследования митохондриальной ДНК показали, что коренные
американцы ведут свое начало от азиатских предков, и позволили опознать четыре
различные митохондриальные гаплогруппы в пределах данной популяции в целом.
Переселение в Северную Америку было датировано периодом от 15 000 до 8000 лет на-
зад. Более поздний, всесторонний анализ митохондриальной ДНК отнес это переселение
к периоду от 25000 до 20000 лет назад. Первые исследования Y-хромосом приписали
дату приблизительно 22 500 лет назад «коренному американцу Адаму», носителю
Y-хромосомы, которая является предком большинства, если не всех, Y-хромосом совре-
менных коренных американцев. Выводы из этих разнящихся, а порой и противоречивых
результатов все еще служат пищей для ожесточенных споров.
Изыскательские задачи
19.1. Хотя на рис. 19.9 приведен пример того, когда дерево генов не совпадает с де-
ревом видов, совпадение между деревьями этих двух видов явно возможно. Следующий
вопрос—«каким образом можно определить, дает л и дерево генов точное представление
о дереве видов?» Теперь ответ на него требует рассмотрения, например, информации,
сведенной на рис. 19.19, который показывает, что для того чтобы получить согласованное
представление, должны быть изучены различные гены или коллекции генов.
19.3. Возможные отправные точки для исследования этой проблемы: 1) Ladouka-
kis,E.D. и Zouros,E. (2001) Recombination in animal mitochondrial DNA: evidence from
published sequences. Mol. Biol. Evol. 18: 2127-2131; 2) Meunier, J. and Eyre-Walker, A. (2001)
The correlation between linkage disequilibrium and distance: implications for recombinations
in hominid mitochondria. Mol. Biol. Evol. 18: 2132-2135. Это одни из самых первых статей,
в которых была озвучена возможность митохондриальной рекомбинации, и обсуждение
должно сопровождаться дальнейшим поиском более поздних публикаций, которые
ссылаются на одну или на обе эти статьи.
19.5. Подробное введение в эту тему приводится в работе Richards, М. (2003) The
Neolithic invasion of Europe. Annu. Rev. Anthropol. 32:135-162. Дабы ответить на этот во-
прос полностью, должна быть изучена статья, которая обеспечивает информацию, вы-
несенную на рис. 19.30: Richards,М., Macaulay,V., Hickey,Е., etal. (2000) Tracing European
founder lineages in the Near Eastern mtDNA pool. Am. J. Hum. Genet 67:1251-1276. Это до-
вольно сложная статья, и если вам удалось успешно в ней разобраться, то, значит, ваше
изучение «Геномов 3» не было напрасным.
Тесты по рисункам
19.1. На рис. б показано, что единственный палец ноги лошадей — приобретенное
состояние признака.
19.3. Если за первым событием видообразования сразу же следует второе событие
видообразования, в одной из двух обособившихся популяций, то порядок ветвления
дерева генов может отличаться от такового в дереве видов. Это может произойти, если
гены, присущие современному виду, получены из аллелей, которые уже появлялись до
первого из этих двух событий видообразования.
Словарь терминов
1 млрдп.н. — 1000000 тыс.п.н. = 1000000000 п.н.
2-аминопурин — аналог основания, который способен вызывать мутации, заменяя
аденин в молекуле ДНК.
2-мкм кольцо — плазмида, имеющаяся у дрожжей Saccharomyces cerevisiae и используе-
мая в качестве основы для создания ряда клонирующих векторов.
З'-трансдукция — перенос сегмента геномной ДНК с одного места в другое, обуслов-
ленный перемещением элемента LINE.
3*-нетранслируемая область — нетранслируемая область мРНК, лежащая ниже стоп-
кодона.
З'-ОН-конец — конец полинуклеотида, который оканчивается гидроксильной группой,
присоединенной к 3'-атому углерода молекулы сахара.
30-нм хроматиновое волокно — относительно разуплотненная форма хроматина,
состоящая из возможно сильнее спирализованного массива нуклеосом в виде во-
локна приблизительно 30 нм в диаметре.
5'-Р-конец — конец полинуклеотида, который оканчивается моно-, ди- или трифосфа-
том, присоединенным к 5'-атому углерода сахара.
5*-нетранслируемая область — нетранслируемая область мРНК, расположенная выше
старт-кодона.
5-бромурацил — аналог основания, который может вызвать мутации, заменяя тимин
в молекуле ДНК.
а-спираль — одна из наиболее распространенных конформаций вторичной структуры,
принимаемая сегментами полипептидов.
P-N-гликозидная связь — связь между основанием и сахаром нуклеотида.
(3-изгиб — последовательность из четырех аминокислот (вторая обычно глицин), в ко-
торой полипептид изменяет свое направление.
Р-лист — одна из наиболее распространенных конформаций вторичной структуры,
свойственная сегментам полипептидов.
%-участок — повторная последовательность нуклеотидов в геноме Escherichia coli,
которая участвует в запуске гомологичной рекомбинации.
Х-форма — промежуточная структура, наблюдаемая во время рекомбинации между
молекулами ДНК.
у-комплекс — компонент ДНК-полимеразы III, включающий в себя у-субъединицу,
объединенную с 8-, 8'-, х- и у-субъединицами.
к-домен гомологии — тип связывающегося с РНК домена.
л-л-взаимодействия — гидрофобные взаимодействия, которые возникают между
смежными парами оснований в двунитевой молекуле ДНК.
А-ДНК — структурная конфигурация двойной спирали, встречающаяся (хотя и не рас-
пространенная) среди клеточных ДНК.
А1и — SINE-элемент, встречающийся в геномах людей и родственных им млекопитаю-
щих.
822 Словарь терминов
Л/и-ПЦР — метод отпечатков пальцев клонов, в котором используется ПЦР, с тем чтобы
обнаружить относительные позиции последовательностей А/w в клонированных
фрагментах ДНК.
ARS — см. Автономно реплицирующаяся последовательность.
ВАС — см. Бактериальная искусственная хромосома (ВАС).
BLAST — алгоритм, часто используемый при поиске сходства.
В-ДНК — наиболее распространенная структурная конформация двойной спирали ДНК
в живых клетках.
В-хромосома — хромосома, присущая лишь некоторым членам популяции.
С-конец — конец полипептида, который имеет свободную карбоксильную группу.
СААТ — см. Блок СААТ.
CASP (связанные с С-КД SR-подобные белки) — белки, которые, как думают, играют
регуляторные роли во время вырезания интронов GU-AG.
CREB — важный фактор транскрипции.
Cis-смещение — передвижение нуклеосомы в новую позицию на молекуле ДНК.
CpG-островок — обогащенная динуклеотидами GC область ДНК. В геноме человека
приблизительно 56 % генов имеют перед собой такую область.
D-петля — промежуточная структура, образуемая согласно модели Мезельсона-Реддинга
во время гомологичной рекомбинации. Также область протяженностью прибли-
зительно 500 п.н., в которой двойная спираль разрывается за счет присутствия
молекулы РНК, спаренной основаниями с одной из нитей ДНК и служащей областью
инициации вытеснительной репликации.
D-стебель — структурная составляющая молекулы тРНК.
Dicer — рибонуклеаза, которая играет центральную роль в РНК-интерференции.
Е-сайт — позиция в пределах бактериальной рибосомы, к которой тРНК перемещается
незамедлительно после деацетилирования.
Elongator (Продлеватель) — белок дрожжей, возможно, обладающий действием гистон-
ацетилтрансферазы и вовлеченный в стадию элонгации (продления) процесса
транскрипции.
F-плазмида — связанная с воспроизводством плазмида, которая направляет осущест-
вляемую при конъюгации передачу ДНК между бактериями.
FEN1 — «эндонуклеаза откидной створки», участвующая в репликации отстающей
нити у эукариотов.
FISH с волокнами — специальная форма ФГНМ с высоким разрешением маркёров.
Gap-гены — отвечающие за развитие гены, которые играют роль в установлении по-
зиционной информации в эмбрионе Drosophila.
GAP-белки — см. Активирующие ГТФазу белки (GAP).
GAP (активирующие ГТФазу белки) — набор белков, которые являются промежуточ-
ными звеньями в пути передачи сигнала Ras.
GC-блок — см. Блок GC.
GNRP — см. Белки, высвобождающие нуклеотид гуанин.
GNRP (Белки, высвобождающие нуклеотид гуанин) — набор белков, которые явля-
ются промежуточными звеньями в пути передачи сигнала Ras.
IRES — см. Внутренний участок вхождения рибосомы (IRES).
LINE (длинный рассеянный ядерный элемент) — тип рассеянного по всему геному
повторения, нередко обладающий действием транспозонов.
Словарь терминов 823
LINE-1 — один из типов LINE человека.
М-фаза — стадия клеточного цикла, когда происходит митоз или мейоз.
MADS — см. Блок MADS.
МАР-киназа — путь передачи сигналов.
MGMT (О6-метилгуанин-ДНК-метилтрансфераза) — фермент, вовлеченный в прямое
устранение алкилационных мутаций.
N-дегрон — N-концевая последовательность аминокислот, которая влияет на деграда-
цию белка, в котором она находится.
N-конец — конец полипептида, который имеет свободную аминогруппу.
N-гликозилирование — прикрепление сахарных единиц к аспарагину в полипепти-
де.
0-гликозилирование — присоединение сахарных единиц к серину или треонину в по-
липептиде.
Р-элемент — ДНК-транспозон Drosophila.
PSI-BLAST — видоизмененная и более мощная версия алгоритма BLAST.
RACE — см. Быстрое размножение концов кДНК (RACE).
Ras — белок, вовлеченный в передачу сигналов.
RecA — белок Escherichia coli, участвующий в гомологичной рекомбинации.
Rho — белок, участвующий в терминации транскрипции некоторых генов бактерий.
Rho-зависимый терминатор — позиция в ДНК бактерий, где завершение транскрипции
происходит с участием белка Rho.
SAP-(активируемый стрессом белок) киназа — активируемый стрессом путь пере-
дачи сигнала.
SINE (короткий рассеянный ядерный элемент) — тип рассеянных по всему геному
повторений, в частности, Д/п-последовательности, распространенные в геноме
человека.
SOS-ответ — ряд биохимических изменений, которые происходят у Escherichia coli в от-
вет на повреждение генома и другие раздражения.
STAT (преобразователь сигнала и активатор транскрипции) — белок, который
реагирует на связывание внеклеточного сигнального соединения с рецептором
клеточной поверхности, активируя фактор транскрипции.
SR — см. Белок SR.
SR-подобные связанные с С-КД факторы (SCAF) — белки, которые, как думают, играют
регуляторные роли в процессе вырезания интронов GU-AG.
SSB — см. Белок однонитевого связывания.
Т-ДНК — часть плазмиды Ti, которая переносится в ДНК растения.
ТАТА — см. Блок ТАТА.
Тт — температура плавления.
Tus — белок, который связывается с бактериальным терминатором репликации и опо-
средствует терминацию репликации генома.
Т\|/С-стебель — структурная составляющая молекулы тРНК.
U-PHK — обогащенная урацилом молекула ядерной РНК, к которым относятся мяРНК
и мякРНК.
UvrABC эндонуклеаза — см. Эндонуклеаза UvrABC.
V-петля — структурная составляющая молекулы тРНК.
824 Словарь терминов
Z-ДНК — конформация ДНК, в которой два полинуклеотида закручены в левосторон-
нюю спираль.
Автономно реплицирующаяся последовательность (ARS) — последовательность
ДНК, особенно у дрожжей, которая придает способность к репликации нерепли-
цирующейся плазмиде.
Автополиплоид — полиплоидное ядро, полученное из слияния двух гамет от организ-
мов одного вида, ни один из которых не является гаплоидным.
Авторадиография — обнаружение меченных радиоактивной меткой молекул путем
засвечивания чувствительной к рентгеновскому излучению фотографической
плёнки.
Адаптер — синтетический двунитевой олигонуклеотид, используемый для присоеди-
нения липких концов к тупоконечной молекуле.
Аденилатциклаза — фермент, который превращает АТФ в циклический АМФ.
Аденин — пуриновое основание, входящее в состав ДНК и РНК.
Азотистое основание — один из пуринов или пиримидинов, который образует часть
молекулярной структуры нуклеотида.
Акридиновый краситель — химическое соединение, которое вызывает мутацию со
сдвигом рамки считывания, интеркаллируя (втискиваясь) между смежными парами
оснований двойной спирали.
Активатор — связывающийся с ДНК белок, который стабилизирует формирование
комплекса инициации транскрипции для РНК-полимеразы II.
Активационный домен — часть активатора, которая вступает в контакт с иниции-
рующим комплексом.
Активирующие ГТФазу белки (GAP) — набор белков, которые являются промежуточ-
ными звеньями в пути передачи сигнала Ras.
Акцепторный стебель — структурная составляющая молекулы тРНК.
Акцепторный участок — сайт сплайсинга на З'-конце интрона.
Алармон — один из активаторов реакции на стресс, ppGpp и pppGpp.
Алкилирующий агент — мутаген, действие которого проявляется в добавлении ал-
кильных групп к нуклеотидным основаниям.
Аллель — одна из двух и более альтернативных форм гена.
Аллополиплоид — полиплоидное ядро, порождённое слиянием гамет от организмов
разных видов.
Альтернативное полиаденилирование — использование двух и более различных
участков для полиаденилирования мРНК.
Альтернативный сплайсинг — производство двух и более разных молекул мРНК
из одинакового вида пре-мРНК за счёт соединения вместе экзонов в различных
комбинациях.
Альтернативный промотор — один из двух или нескольких разных промоторов, дей-
ствующих на один и тот же ген.
Альфоидная ДНК — тандемно повторяющиеся последовательности нуклеотидов, рас-
положенные в центромерных областях хромосом человека.
Аминоацил-тРНК-синтетаза — фермент, который катализирует аминоацилирование
одной и более тРНК.
Аминоацилирование — присоединение аминокислоты к акцепторному стеблю тРНК.
Словарь терминов 825
Аминоацильный, или А-сайт — участок в рибосоме, занятый аминоацил-тРНК во
время трансляции.
Аминокислота — одна из мономерных единиц молекулы белка.
Аминоконец — конец полипептида, который имеет свободную аминогруппу.
Анализ биохимического профиля — изучение метаболомов.
Анализ гибридизацией олигонуклеотидов — метод анализа с использованием оли-
гонуклеотида в качестве гибридизационного зонда.
Анализ главных компонентов — процедура, основанная на распознавании закономер-
ностей в большом наборе данных изменчивых состояний признака.
Анализ лигированием олигонуклеотидов (OLA) — методика типизации однонуклео-
тидных полиморфизмов (SNP), которая зависит от лигирования двух олигонуклео-
тидов, которые отжигаются смежно друг с другом, причем один из них покрывает
позицию SNP.
Анализ на связывание триплетов — методика определения специфичности кодиро-
вания рассматриваемого триплета нуклеотидов.
Анализ родословной — использование схемы родословной для анализа наследования
генетического маркёра или ДНК-маркёра в человеческой семье.
Анализ с модификацией — диапазон методов, используемых для определения место-
положения связанных белков на молекулах ДНК.
Анализ с торможением в геле — метод определения участков связывания с белком
на молекулах ДНК за счет воздействия, оказываемого связанным белком на под-
вижность фрагментов ДНК в процессе гель-электрофореза.
Анализ сцепления генетических признаков — процедура, применяемая для карти-
рования генов при помощи генетических скрещиваний.
Анализирующее скрещивание — генетическое скрещивание между двойной гетеро-
зиготой и двойной гомозиготой.
Аналог основания — соединение, структурное подобие которого одному из оснований
в ДНК позволяет ему действовать как мутаген.
Аналогия — ситуация, которая происходит, когда одно и то же состояние признака не-
зависимо появляется у организмов, идущих разными путями эволюции.
Антиген — вещество, которое вызывает иммунную реакцию.
Антикодон — триплет нуклеотидов в молекуле тРНК (в позициях 34-36), который
спаривается основаниями с кодоном в молекуле мРНК.
Антикодоновый стебель — структурная составляющая молекулы тРНК.
Антитерминаторный белок — белок, который прикрепляется к бактериальной ДНК
и блокирует терминацию транскрипции.
Антитерминация — развитый бактериями механизм регулирования терминации
транскрипции.
АП (апуриновый/апиримидиновый) участок—позиция в молекуле ДНК, где основная
составляющая нуклеотида отсутствует.
АП-эндонуклеаза — фермент, участвующий в процессе репарации вырезанием осно-
ваний.
Апоморфное состояние признака — состояние признака, которое появилось в ходе
эволюции у ближнего предка подгруппы организмов в изучаемой группе.
Апоптоз — запрограммированная смерть клетки.
826 Словарь терминов
Аркан (лассо) — относится к имеющему форму аркана интрону РНК, который образу-
ется при вырезании интрона GU-AG.
Археи — одна из двух главных групп доядерных, главным образом встречающаяся
в экстремальных средах.
Аттенюация (ослабление) — процесс, используемый некоторыми бактериями для регу-
лирования экспрессии оперона биосинтеза аминокислот в соответствии с уровнем
содержания аминокислот в клетке.
Ауксотроф — мутантный микроорганизм, который может расти только при условии на-
личия питательного вещества, без которого могут обойтись особи дикого типа.
Аутосома — хромосома, которая не относится к половым хромосомам.
Афинная хроматография — метод хроматографии на колонках, в котором используется
лиганд, связывающийся с очищаемой молекулой.
Ацетилирование гистонов — модификация структуры хроматина путем присоедине-
ния ацетильных групп к основным (срединным) гистонам.
Ацилирование — присоединение липидной боковой цепи к полипептиду.
Бактериальная искусственная хромосома (ВАС) — высокоемкий клонирующий вектор
на основе F-плазмиды Escherichia coli.
Бактерии — одна из двух главных групп доядерных.
Бактериофаг — вирус, который заражает бактерию.
Безосновный участок — позиция в молекуле ДНК, в которой отсутствует основная
составляющая нуклеотида.
Белки, высвобождающие нуклеотид гуанин (GNRP) — набор белков, которые явля-
ются промежуточными звеньями в пути передачи сигнала Ras.
Белковая инженерия — совокупность различных методов внесения целенаправлен-
ных изменений в молекулы белков, часто с целью улучшения свойств ферментов,
используемых в промышленных процессах.
Белок домашнего хозяйства — белок, который непрерывно экспрессируется во всех
или по крайней мере в большинстве клеток многоклеточного организма.
Белок SR — белок, который играет роль в выборе участков сплайсинга в ходе вырезания
интронов GU-AG.
Белок однонитевого связывания (SSB) — один из белков, которые прикрепляются к од-
нонитевой ДНК в области репликационной вилки, предотвращая образование пар
оснований между двумя родительскими нитями до момента их копирования.
Белок — полимерное соединение, построенное из аминокислотных мономеров.
Белок-активатор катаболитных оперонов — регуляторный белок, который связыва-
ется с различными участками в бактериальном геноме и активирует инициацию
транскрипции в нижележащих промоторах.
Бережливость — подход, на основании которого из различных топологий филогене-
тического дерева выбирается одна, которая включает в себя кратчайший эволю-
ционный путь.
Бесклеточная синтезирующая белок система — клеточный экстракт, содержащий
все компоненты, необходимые для синтеза белка, и способный транслировать
добавляемые в него молекулы мРНК.
Бесхроматиновая область — пространство, отделяющее территории хромосом в преде-
лах ядра.
Библиотека клонов — коллекция клонов (возможно, представляющая полный геном),
из которой набираются отдельные интересующие исследователя клоны.
Словарь терминов 827
Библиотека фагового дисплея — собрание клонов, несущих различные фрагменты
ДНК, используемые в фаговом дисплее.
Бивалент — структура, образуемая, когда пара гомологичных хромосом выстраивается
друг против друга во время мейоза.
Биоинформатика — наука о применении компьютерных методов в исследованиях
геномов.
Биолистика — средство введения ДНК в клетки путем их бомбардировки высокоско-
ростными микроснарядами, покрытыми ДНК.
Биологическая информация — информация, содержащаяся в геноме организма и на-
правляющая развитие и поддержание этого организма.
Биотехнология — использование живых организмов (часто, но не всегда микробов)
в промышленных процессах.
Биотинилирование — прикрепление биотиновой метки к молекуле РНК или ДНК.
Блок СААТ — основной промоторный элемент.
Блок GC — тип основного промоторного элемента.
Блок MADS — связывающийся с ДНК домен, встречающийся в нескольких факторах
транскрипции, вовлеченных в развитие растений.
Блок ТАТА — компонент основного промотора РНК-полимеразы II.
Блок Прибнова — компонент бактериального промотора.
Блок-35 — компонент бактериального промотора.
Бляшка — зона очистки на газоне бактерий, вызванной лизисом клеток заражающими
их бактериофагами.
Горизонтальный перенос гена — передача гена от одного вида другому.
Болезнь, связанная с расширением тринуклеотидных повторений — болезнь, ко-
торая следует из расширения группы тринуклеотидных повторений в пределах
или вблизи значимого гена.
Большая бороздка — большая из двух бороздок, которые закручиваются спиралью на
поверхности ДНК В-формы.
Бромистый этидий — один из вклинивающихся агентов, который вызывает мута-
ции, вставляясь между смежными парами оснований в молекуле двунитевой
ДНК.
Бросовая ДНК — одна из интерпретаций содержащейся в геноме межгенной ДНК.
Бусины на нити — разуплотненная форма хроматина, состоящая из нуклеосомных
бусин на нити ДНК.
Бутстрап-анализ — метод вывода степени достоверности, которая может быть при-
писана к точке ветвления в филогенетическом дереве.
Быстрое размножение концов кДНК (RACE) — основанный на ПЦР метод картирова-
ния конца молекулы РНК.
Ван дер Ваальсовы силы — притягивающие или отталкивающие нековалентные
взаимодействия специфического типа.
Вегетативная клетка — нерепродуктивная клетка; клетка, которая делится митозом.
Вектор замещения — вектор на основе бактериофага X, разработанный таким образом,
что встройка новой ДНК осуществляется заменой части несущественной области
молекулы ДНК фага X.
Вектор на бактериофаге Р1 — высокоемкий клонирующий вектор на основе бакте-
риофага Р1.
828 Словарь терминов
Вектор со вставкой — вектор на бактериофаге X, построенный удалением сегмента
несущественной ДНК.
Ветвь — компонент филогенетического дерева.
Вироид — молекула РНК 240-375 нуклеотидов в длину, которая не содержит никаких
генов, никогда не становится покрытой оболочкой и распространяется из клетки
в клетку в виде голой ДНК.
Вирулентный бактериофаг — бактериофаг, который следует литическим путем ин-
фицирования.
Вирус — инфекционная частица, состоящая из белка и нуклеиновой кислоты, которой
для собственной репликации необходимо паразитировать в клетке хозяина.
Вирусный ретроэлемент — вирус, процесс репликации генома которого включает
событие обратной транскрипции.
Вирусоид — молекула РНК около 320-400 нуклеотидов в длину, которая не кодирует
белки своего капсида, а вместо этого перемещается из клетки в клетку в капсиде
вируса-помощника.
Вкладочный фрагмент — фрагмент ДНК, содержащийся в векторе на основе бактерио-
фага X, который заменяется ДНК, предназначенной для клонирования.
Внехромосомный ген — ген в митохондриальном геноме или в геноме хлоропласта.
Внешний узел — конец ветви в филогенетическом дереве, представляющий один из
изучаемых объектов — организм или последовательность ДНК.
Внешняя группа — организм или последовательность ДНК, который используется для
укоренения филогенетического дерева.
Внутренний терминатор — позиция в бактериальной ДНК, где терминация транс-
крипции происходит без привлечения белка Rho.
Внутренний узел — точка ветвления в кроне филогенетического дерева, представ-
ляющая организм или последовательность ДНК, которая является предковой по
отношению к изучаемым.
Внутренний участок вхождения рибосомы (IRES) — последовательность нуклеоти-
дов, которая позволяет рибосоме собираться на внутренней позиции некоторых
мРНК эукариотов.
Внутримолекулярное спаривание оснований — спаривание оснований, которое про-
исходит между двумя частями одного и того же полинуклеотида ДНК или РНК.
Водородная связь — слабое электростатическое притяжение между одним электроо-
трицательным атомом (например, кислорода или азота) и атомом водорода, при-
соединенным к другому электроотрицательному атому.
Возврат — отход РНК-полимеразы в обратном направлении на короткое расстояние
по матричной нити ДНК.
Волна продвижения — гипотеза, согласно которой распространение сельского хозяй-
ства в Европе сопровождалось массовым переселением популяций человека.
Времяпролетная масс-спектрометрия с лазерной десорбционной ионизацией ма-
трицы (MALDI-TOF) — разновидность масс-спектрометрии, применяемая в про-
теомике.
Вставные последовательности — короткий подвижный генетический элемент, встре-
чающийся у бактерий.
Встраивающая инактивация — методика клонирования, посредством которой встройка
новой части ДНК в вектор инактивирует ген, который заложен в этом векторе.
Словарь терминов 829
Встраивающее редактирование — менее масштабная форма панредактирования, ко-
торая имеет место во время обработки (созревания) некоторых вирусных РНК.
Вторичная структура — конформации, такие как а-спираль и 0-лист, принимаемые
полипептидом.
Вторичный канал — канал, который ведет с поверхности бактериальной РНК-
полимеразы к активному участку в недрах белкового комплекса.
Вторичный посредник — промежуточное звено в путях передачи сигналов некоторых
типов.
Выведенное дерево — дерево, полученное в результате филогенетического анализа.
Вырожденность — относится к тому факту, что генетический код имеет более одного
кодона для кодирования большинства аминокислот.
Высокоподвижные белки группы N (HMGN) — группа ядерных белков, которые влияют
на структуру хроматина.
Вытеснительная репликация — способ репликации, который предполагает непрерыв-
ное копирование одной нити спирали при одновременном вытеснении второй нити
и последующем ее копировании по завершении синтеза первой дочерней нити.
Вытягивание гелем — методика приготовления обработанных рестриктазами молекул
ДНК к оптическому картированию.
Выше — в направлении 5’-конца полинуклеотида.
Вышележащий промоторный элемент — компонент промотора эукариотов, который
лежит выше позиции сборки инициаторного комплекса.
Вышележащий элемент управления (ВЭУ) — компонент промоторов для РНК-
полимеразы I.
Гамета — репродуктивная клетка, обычно гаплоидная, которая слияется со второй
гаметой с образованием новой клетки в процессе полового воспроизводства.
Гаплогруппа — один из главных классов последовательностей митохондриальной ДНК,
представленных в популяции человека.
Гаплоид — ядро, которое имеет по одной копии всех хромосом.
Гаплонедостаточность — ситуация, когда инактивация некоторого гена на одной из
членов пары гомологичных хромосом приводит к изменению фенотипа в мутант-
ном организме.
Гаплотип — отдельная последовательность митохондриальной ДНК.
Геликаза — фермент, который разрывает пары оснований в молекуле двунитевой
ДНК.
Гель-электрофорез — электрофорез, выполняемый в геле, с тем чтобы молекулы
с одноименным электрическим зарядом могли быть разделены по размеру.
Ген материнского эффекта — ген Drosophila, который экспрессируется в родителе
и мРНК которого впоследствии вводится в яйцеклетку, после чего он влияет на
развитие эмбриона.
Ген — сегмент ДНК, содержащий биологическую информацию и, следовательно, коди-
рующий молекулу РНК и (или) полипептида.
Ген-репортер — ген, фенотип которого может быть проанализирован и который может
поэтому быть использован для определения функции регуляторной последова-
тельности ДНК.
Генетика — отрасль биологии, посвященная изучению генов.
830 Словарь терминов
Генетическая избыточность — ситуация, которая возникает, когда два гена в одном
и том же геноме выполняют одну и ту же функцию.
Генетическое картирование — использование генетических методов для построения
карты генома.
Генетический код — правила, которые определяют, который триплет нуклеотидов
кодирует которую аминокислоту в процессе синтеза белка.
Генетический маркёр — ген, который существует в виде двух и более легко различимых
аллелей и наследование которого можно поэтому проследить во время генетиче-
ского скрещивания, что позволяет определить позицию этого гена на карте.
Генетический профиль — картина полосчатости, проявляемая после электрофореза
продуктов серии ПЦР, направленных на ряд микросателлитных локусов.
Генетический футпринтинг — метод быстрого функционального анализа одновре-
менно большого числа генов.
Генная конверсия — процесс, в результате которого четыре гаплоидных продукта
мейоза показывают необычную закономерность распределения признаков.
Геном — полный генетический набор (комплемент) живого организма.
Геномный импринтинг (геномное запечатление) — инактивация, осуществляемая
посредством метилирования определенного гена на одном из членов пары гомо-
логичных хромосом.
Генотерапия — клиническая процедура, в которой для лечения болезни применяется
ген или иная последовательность ДНК.
Генотип — описание генетического состава организма.
Гены полярности сегментов — отвечающие за развитие гены, которые придают
большую точность схеме сегментации эмбриона Drosophila, установленной дей-
ствием генов правила пар.
Гены правила пар (pair-rule) — отвечающие за развитие гены, которые определяют
основную схему сегментации эмбриона Drosophila.
Гены в генах — относится к гену, интрон которого содержит еще один ген.
Гетерогенная ядерная РНК (гяРНК) — фракция ядерной РНК, которая включает не-
обработанные транскрипты, синтезируемые РНК-полимеразой II.
Гетеродуплекс — гибрид вида ДНК-ДНК или ДНК-РНК.
Гетеродуплексный анализ — составление транскрипционных карт путем анализа
гибридов ДНК-РНК с помощью специфичной к однонитевым цепям нуклеазы,
такой как S1.
Гетерозиготность — вероятность того, что человек, выбранный из популяции наугад,
будет гетерозиготен по заданному маркёру.
Гетерозиготный — диплоидное ядро, которое содержит два разных аллеля некоторого
рассматриваемого гена.
Гетерополимер — искусственная РНК, состоящая из смеси различных нуклеотидов1).
Гетерохроматин — хроматин, который относительно уплотнен и, как думают, содержит
ДНК, которая в данный момент не транскрибируется.
Гибридизационное зондирование — метод, в котором используется меченая молекула
нуклеиновой кислоты в качестве зонда для опознавания комплементарных или
гомологичных молекул, с которыми он спаривается основаниями.
В обшем случае гетерополимер — это полимер из звеньев разного типа, в частности, белки и ДНК
также являются гетерополимерами. — Прим. ред.
Словарь терминов 831
Гибридизация аллелеспецифическими олигонуклеотидами (АСО) — использование
олигонуклеотидного зонда, чтобы определить, которая из двух альтернативных
нуклеотидных последовательностей содержится в молекуле ДНК.
Гибридизация по Саузерну — методика, используемая для обнаружения специфиче-
ского фрагмента рестрикции на фоне многих других фрагментов рестрикции.
Гибридизация — прикрепление друг к другу (спариванием оснований) двух взаимно
комплементарных полинуклеотидов.
Гибридный дисгенез — ситуация, которая возникает, когда самки из лабораторных
линий Drosophila melanogaster скрещиваются с самцами из природных популяций,
и потомство, следующее из таких скрещиваний, является бесплодным и имеет
хромосомные отклонения и прочие генетические сбои.
Гибридизация нуклеиновых кислот — формирование двунитевого гибрида за счет
спаривания оснований между взаимно комплементарными полинуклеотидами.
Гидрофобные явления — химические взаимодействия, в результате которых гидро-
фобные группы погружаются вглубь белковой молекулы.
Гипотеза «о поздних интронах» — гипотеза о том, что интроны эволюционировали
относительно поздно и постепенно накапливаются в геномах эукариотов.
Гипотеза «о ранних интронах» — гипотеза о том, что интроны эволюционировали
относительно рано и постепенно выпадают из геномов эукариотов.
Гипотеза о отклонении (неканоническом спаривании) — модель процесса, которым
одна тРНК может кодировать более одного кодона.
Гипотеза об африканском происхождении человека — гипотеза, которая предпо-
лагает, что современные люди эволюционировали в Африке и переместились
в остальную часть Старого Света в период от 100 000 до 50 000 лет назад, вытесняя
потомков Homo erectus, с которыми они сталкивались.
Гистон — один из основных белков, входящих в состав нуклеосом.
Гистонацетилтрансфераза (НАТ) — фермент, который присоединяет ацетильные
группы к стержневым гистонам.
Гистондезацетилаза (HDAC) — фермент, который снимает ацетильные группы со
стержневых гистонов.
Гистонный код — гипотеза о том, что схема химической модификации гистоновых
белков влияет на различные отправления клетки.
Главный комплекс гистосовместимости (ГКГС) — мультигенное семейство млеко-
питающих, кодирующее белки клеточной поверхности и включающее несколько
многоаллельных генов.
Гликозилирование — присоединение сахарных единиц к полипептиду.
Глобальное регулирование — главная понижающая регуляция в процессе синтеза
белка, которая происходит в ответ на различные сигналы.
Головка молотка — структура РНК с действием рибозима, которая встречается у не-
которых вирусов.
Голофермент — версия РНК-полимеразы Escherichia coli (субъединичный состав а2РР'о),
которая способна опознавать промоторные последовательности.
Голоцентрическая хромосома — хромосома, которая не имеет единственной цен-
тромеры, но вместо этого имеет многочисленные кинетохоры, распределенные
по ее длине.
Гомеодомен — связывающийся с ДНК мотив, обнаруживаемый во многих белках, от-
ветственных за регуляцию экспрессии генов развития.
832 Словарь терминов
Гомеозисная мутация — мутация, которая приводит к преобразованию одной части
тела в другую.
Гомеозисный избирательный ген — ген, который устанавливает род части тела, такой
как сегмент эмбриона Drosophila.
Гомозигота — диплоидное ядро, которое содержит два идентичных аллеля рассма-
триваемого гена.
Гомологичная рекомбинация — рекомбинация между двумя гомологичными мо-
лекулами двунитевой ДНК, то есть такими, которые характеризуются сильным
подобием нуклеотидных последовательностей.
Гомологичные гены — гены, которые имеют общего эволюционного предка.
Гомологичные хромосомы—две и более идентичные хромосомы, находящиеся в одном
ядре.
Гомополимер — искусственная РНК, состоящая из повторяющегося нуклеотида одного
рода.
Гомополимерное наращивание — присоединение последовательности идентичных
нуклеотидов (например, ААААА) к концу молекулы нуклеиновой кислоты; обыч-
но относится к синтезу однонитевого гомополимерного продолжения на концах
молекулы двунитевой ДНК.
Горизонтальный перенос гена — передача гена от одного вида другому.
Горячая точка рекомбинации — область хромосомы, в которой перекресты происходят
с более высокой частотой, чем среднее число для хромосомы в целом.
Готовая последовательность — последовательность хромосомы, которая является
почти полной, определяется для хромосом человека как та, что охватывает не менее
95 % эухроматина при коэффициенте ошибок менее одной на 104 нуклеотидов.
Гуанилилтрансфераза — фермент, который присоединяет ГТФ к 5'-концу мРНК эука-
риотов в начале реакции кэпирования.
Гуанин — один из пуриновых нуклеотидов, входящих в состав ДНК и РНК.
Гуанинметилтрансфераза — фермент, который прикрепляет метильную группу
к 5’-концу мРНК эукариотов в ходе реакции кэпирования.
ДНК — дезоксирибонуклеиновая кислота, одна из двух форм нуклеиновой кислоты
в живых клетках; генетический материал для всех клеточных форм жизни и мно-
гих вирусов.
ДНК-аденинметилаза (ДАМ) — фермент, вовлеченный в метилирование ДНК Escherichia
coli.
ДНК-гираза — топоизомераза II типа у Escherichia coli.
ДНК-гликозилаза — фермент, который расщепляет p-N-гликозидную связь между
основанием и сахарным компонентом нуклеотида как составляющая процессов
восстановления вырезанием оснований и устранением несоответствий. Данное
название неправильное и должно звучать как ДНК-гликолиаза, но такое непра-
вильное употребление укоренилось в научной литературе.
ДНК-зависимая ДНК-полимераза — фермент, который делает ДНКовую копию ма-
тричной ДНК.
ДНК-зависимая РНК-полимераза — фермент, который делает РНКовую копию ма-
трицы ДНК.
ДНК-лигаза — фермент, который синтезирует фосфодиэфирные связи как часть про-
цессов репликации, репарации и рекомбинации ДНК.
Словарь терминов 833
ДНК-маркёр — последовательность ДНК, которая существует в виде двух и более легко
различимых вариантов и которая поэтому может использоваться, чтобы отмечать
позицию на генетической, физической или объединенной карте генома.
ДНК-метилтрансфераза — фермент, который прикрепляет метильные группы к мо-
лекуле ДНК.
ДНК-полимераза I — бактериальный фермент, который завершает синтез фрагментов
Окадзаки во время репликации генома.
ДНК-полимераза II — бактериальная ДНК-полимераза, вовлеченная в репарацию
ДНК.
ДНК-полимераза III — главный реплицирующий ДНК фермент бактерий.
ДНК-полимераза а — фермент, что затравливает репликацию ДНК у эукариотов.
ДНК-полимераза 5 — главный реплицирующий ДНК фермент эукариотов.
ДНК-полимераза у — фермент, ответственный за репликацию митохондриального
генома.
ДНК-полимераза — фермент, который синтезирует ДНК.
ДНК-топоизомераза — фермент, который вводит или удаляет витки из двойной спирали
путем разрыва и воссоединения одного или обоих полинуклеотидов.
ДНК-фотолиаза — бактериальный фермент, вовлеченный в фотореактивационную
репарацию.
ДНК-цитозинметилаза (Dem) — фермент, вовлеченный в метилирование ДНК Escherichia
coli.
Двойная гетерозигота — ядро, которое является гетерозиготным по двум генам.
Двойная гомозигота — ядро, которое является гомозиготным по двум генам.
Двойная рестрикция — переваривание ДНК сразу двумя эндонуклеазами рестрик-
ции.
Двойная спираль — спаренная основаниями двунитевая структура, которая является
естественной формой ДНК в клетке.
Двумерный гель-электрофорез — метод разделения белков, особенно часто исполь-
зуемый в исследованиях протеомов.
Двунитевой — включающий два полинуклеотида, прикрепленных друг к другу за счет
спаривания оснований.
Деградосома — многоферментный комплекс, отвечающий за деградацию бактери-
альных мРНК.
Дезаминирующий агент — мутаген, который действует, удаляя аминогруппы от осно-
ваний нуклеотидов.
Дезоксирибонуклеаза — фермент, который расщепляет фосфодиэфирные связи в мо-
лекуле ДНК.
Денатурация — разрыв химическим или физическим путем нековалентных взаимодей-
ствий (например, водородных связей), которые поддерживают вторичный и более
высокие уровни структуры белков и нуклеиновых кислот.
Дерево видов — филогенетическое дерево, которое показывает эволюционные от-
ношения в пределах группы видов.
Дерево генов — филогенетическое дерево, которое показывает эволюционные отно-
шения в пределах группы генов или иных последовательностей ДНК.
Детектирование мутаций — набор методов для определения того, содержит ли моле-
кула ДНК определенную мутацию.
834 Словарь терминов
Диауксия — явление, посредством которого бактерия, будучи обеспечена смесью саха-
ров, полностью расходует один сахар, а уже потом начинает усваивать следующий
сахар.
Дигибридное скрещивание — половое скрещивание, которое сопровождается на-
следованием двух пар аллелей.
Дидезоксинуклеотид — видоизмененный нуклеотид, которому недостает 3'-гидро-
ксильной группы, и, таким образом, он прерывает синтез нити, будучи включен
в полинуклеотид.
Дикий тип — ген, клетка или организм, который показывает типичный для вида фе-
нотип и (или) генотип и поэтому принимается за стандарт.
Димер — белок или другая структура, которая включает две субъединицы.
Динамическая аллелеспецифическая гибридизация (DASH) — методика гибриди-
зации в растворе, используемая для типизации SNP.
Диплоид — ядро, которое содержит по две копии каждой хромосомы.
Дисульфидный мостик — ковалентная связь, соединяющая цистеиновые аминокис-
лоты, находящиеся на разных полипептидах или же в различных позициях одного
и того же полипептида.
Дифракция рентгеновских лучей — дифракция рентгеновских лучей, которая про-
является во время их прохождения через кристалл.
Дифференциальное центрифугирование — методика разделения компонентов клет-
ки путем центрифугирования экстракта на разных скоростях.
Дифференцировка — принятие клеткой специализированных биохимической и (или)
физиологической ролей.
Домен POU — связывающийся с ДНК мотив, встречающийся во множестве разнообраз-
ных белков.
Домен TBD — разновидность связывающегося с ДНК домена.
Домен связывания с двунитевой РНК (ДСднРНК) — распространенный РНК-связы-
вающий домен.
Домен свёртки — сегмент полипептида, который сворачивается независимо от других
сегментов.
Домен — сегмент полипептида, который сворачивается независимо от других сегментов;
также сегмент гена, кодирующий такой домен.
Доминантный аллель — аллель, который экспрессируется в гетерозиготе.
Донорный участок — сайт сплайсинга на 5'-конце интрона.
Древняя ДНК — ДНК, сохранившаяся в древнем биологическом материале.
Древовидная схема — дерево, которое начерчено, чтобы показать отношения между,
например, членами группы транскриптомов.
Дрожжевая искусственная хромосома (YAC) — высокоемкий клонирующий вектор,
построенный из компонентов хромосомы дрожжей.
Дрожжевая дигибридная система — средство опознавания белков, которые способны
взаимодействовать друг с другом.
Дупликация (дублирование) домена — дублирование сегмента гена, кодирующего
структурный домен в белковом продукте.
Единица S — единица измерения коэффициента седиментации (скорости осажде-
ния).
Словарь терминов 835
Естественный отбор — сохранение благоприятных аллелей и отбраковка неблаго-
приятных аллелей.
Жидкостная хроматография высокого разрешения (ЖХВР) — метод хроматографии
на колонках, широко применяющийся в биохимии.
Зависимое от дезаденилирования декэпирование — процесс деградации молекул
мРНК эукариотов, который запускается удалением поли-А хвоста.
Зависимый otTAF и инициаторной последовательности кофактор (TIC)—тип белка,
вовлеченного в инициацию транскрипции для РНК-полимеразы II.
Закодированный изотопом ярлык по сродству (ICAT) — маркёр, содержащий атомы
нормального водорода и дейтерия и применяемый для маркировки отдельных
протеомов.
Закрытый промоторный комплекс — структура, образуемая на начальном шаге сбор-
ки инициаторного комплекса транскрипции. Закрытый промоторный комплекс
состоит из РНК-полимеразы и (или) дополнительных белков, присоединенных
к промотору до того, как ДНК раскрывается путем раскрытия пар оснований.
Запрограммированные мутации — мутации, частоту возникновения которых в опреде-
ленном гене при некоторых обстоятельствах организм может увеличивать.
Запрограммированный сдвиг рамки считывания — управляемый переход рибосомы
из одной рамки считывания в другую во внутренней позиции гена.
Зародышевая стволовая (ЗС) клетка — полипотенциальная клетка из эмбриона мыши
или иного организма.
Затравка — короткий олигонуклеотид, который присоединяется к молекуле однони-
тевой ДНК, с тем чтобы обеспечить исходную позицию для синтеза нити.
Защита от модификации — метод, применяемый для опознания нуклеотидов, уча-
ствующих во взаимодействиях со связывающимся с ДНК белком.
Захват кДНК, или отбор кДНК — повторное гибридизационное зондирование группы
кДНК с целью получения подгруппы, обогащенной определенными последователь-
ностями.
Зеленый флюоресцентный белок — белок, который используется для мечения других
белков и чей ген используется в качестве гена-репортера.
Зигота — клетка, порождаемая слиянием гамет в процессе мейоза.
Зоогибридизация — метод, целью которого является определение того, содержит ли
фрагмент ДНК ген, гибридизируя этот фрагмент с препаратами ДНК от родственных
видов. Основанием такого подхода служит тот факт, что гены у родственных видов
имеют подобные последовательности и, таким образом, дают положительные ги-
бридизационные сигналы, тогда как области между генами имеют менее подобные
последовательности и, таким образом, не гибридизируются.
Инициация (запуск) транскрипции — сборка (научастке выше гена) комплекса белков,
который впоследствии будет копировать этот ген в РНК.
Инициаторная (запускающая) последовательность (Иск) — компонент основного
промотора для РНК-полимеразы II.
Инициаторная (запускающая) тРНК — тРНК, аминоацилируемая метионином у эу-
кариотов или N-формилметионином у бактерий, которая опознает старт-кодон
в процессе синтеза белка.
Инициаторный (запускающий) комплекс — комплекс белков, который запускает
транскрипцию. Также комплекс, который запускает трансляцию.
836 Словарь терминов
Иерархическая группировка — метод анализа транскриптомов на основе сравнений
между уровнями экспрессии пар генов.
Изгибание ДНК — тип конформационного изменения, производимого над молекулой
ДНК связывающимся с ней белком.
Изоакцепторные тРНК — две или несколько тРНК, которые нагружаются одной и той
же аминокислотой.
ИнДел (InDei) — позиция в выравнивании между двумя последовательностями ДНК,
в которой имело место событие вставки (инсерции) или удаления (делеции).
Инсулятор — сегмент ДНК, который служит граничной точкой между двумя функцио-
нальными доменами.
Интеркалирующий (втискивающийся) агент — соединение, которое может проник-
нуть в пространство между смежными парами оснований молекулы двунитевой
ДНК, часто вызывая тем самым мутации.
Изотоп — один из двух или нескольких атомов, которые имеют один и тот же атомный
номер, но разные атомные массы.
Изохора — сегмент геномной ДНК, имеющий однородный состав оснований, который
отличается от такового смежных с ним сегментов.
Изоэлектрическая точка — положение в градиенте pH, в котором результирующий
заряд белка равен нулю.
Изоэлектрическое фокусирование—разделение белков в геле, содержащем химикаты,
которые устанавливают градиент pH в приложенном электрическом заряде.
Иммуноцитохимия — методика определения местоположения белка в ткани за счет
зондирования антителами.
Иммуноэлектронная микроскопия — метод электронной микроскопии, в котором
применяется мечение антител с целью установления позиций специфичных к ним
белков на поверхности таких структур, как рибосомы.
Импортин — белок, участвующий в переносе молекул из цитоплазмы в ядро.
Инактивация Х-хромосомы — осуществляемая метилированием инактивация боль-
шинства генов на одной копии Х-хромосомы в ядре клетки самки.
Инвертированный повтор — две идентичные последовательности нуклеотидов, по-
вторяющиеся во взаимно встречной ориентации в молекуле ДНК.
Ингибирующий домен — часть репрессора эукариотов, которая вступает в контакт
с инициаторным комплексом.
Индуктор — молекула, которая вызывает экспрессию гена или оперона, связываясь
с белком-репрессором и предотвращая тем самым посадку репрессора на опера-
тор.
Инкубационный период — период между впрыскиванием генома фага в бактериаль-
ную клетку и временем, когда происходит лизис клетки.
Инозин — видоизмененная версия аденозина, иногда встречающаяся в неоднозначной
позиции антикодона.
Интеграза — топоизомераза I типа, которая катализирует встраивание генома бакте-
риофага X в ДНК Escherichia coli.
Интегрон — набор генов и иных последовательностей ДНК, который позволяет плаз-
мидам захватывать гены у бактериофагов и других плазмид.
Интеин — внутренний сегмент полипептида, который удаляется в ходе процесса сплай-
синга, протекающего после трансляции.
Интерфаза — период между делениями клетки.
Словарь терминов 837
Интерфазная хромосома — хромосома, находящаяся в клетке в течение периода между
делениями клетки, которая принимает относительно разуплотненную структуру
хроматина.
Интрон AU-AC — интрон, встречающийся в ядерных генах эукариотов: первые два
нуклеотида в этом интроне — 5'-AU-3', а последние два — 5'-АС-3’.
Интрон GU-AG — интрон наиболее распространенного типа в ядерных генах эукариотов.
Первые два нуклеотида этого интрона — 5'-GU-3', а последние два — 5'-AG-3'.
Интрон I группы — интрон, встречающийся главным образом в генах органелл.
Интрон II группы — интрон, встречающийся в генах органелл.
Интрон III группы — интрон, встречающийся в генах органелл.
Интрон — некодирующая область в пределах прерывного гена.
Информационная проблема — проблема, которой занимались первые молекулярные
биологи, касаемая природы генетического кода.
Ионообменная хроматография — метод разделения мол екул согласно тому, как сильно
они связываются с электрически заряженными частицами, взвешенными в хрома-
тографическом матриксе.
Искусственного гена синтез — создание искусственного гена из группы перекрываю-
щихся олигонуклеотидов.
Истинное дерево — филогенетическое дерево, которое отображает фактический ряд
эволюционных событий, которые привели к появлению группы изучаемых орга-
низмов или последовательностей ДНК.
кДНК — олицетворенная двунитевой ДНК копия молекулы мРНК.
Капиллярныйэлектрофорез—электрофорез в полиакриламидном геле, выполняемый
в тонкой капиллярной трубке, что обеспечивает высокое разрешение.
Капсид — белковая оболочка, которая окружает ДНКовый или РНКовый геном виру-
са.
Карбоксильный конец — конец полипептида, на котором имеется свободная карбок-
сильная группа.
Кариограмма — полный набор хромосом клетки, где каждая хромосома представлена
в ее метафазной форме.
Кариоферин — белок, вовлеченный в перенос РНК из ядра в цитоплазму или из цито-
плазмы в ядро.
Каркас — ряд контигов (НПО) последовательности, отделенных пропусками в этой
последовательности.
Карта межбелковых взаимодействий — карта, показывающая взаимодействия между
всеми или некоторыми белками протеома.
Карта — схема, показывающая позиции генетических и (или) физических маркёров
в геноме.
Картирование STS — процедура составления физической карты, состоящая в опреде-
лении позиций меченых участков последовательности (STS) в геноме.
Катаболитная репрессия — развитое бактериями средство, которым внеклеточные
уровни глюкозы диктуют, будут ли гены для усвоения сахара включены или вы-
ключены.
Квадрат Пюннетта — метод анализа табличных данных для предсказания генотипов
потомства, следующего из генетического скрещивания.
838 Словарь терминов
Киназа Януса (JAK) — разновидность киназы, которая играет посредническую роль
в некоторых системах передачи сигналов, вовлекающих ПСАТы (STAT).
Кинетохор — часть центромеры, к которому прикрепляются нити веретена деления.
Кислый домен — разновидность активационного домена.
Клада — группа монофилетичных организмов или последовательностей ДНК, в кото-
рую входят все включенные в анализ, что происходят от определенного общего
предка.
Кладистика — филогенетический подход, который подчеркивает важность понимания
эволюционной значимости изучаемых признаков.
Клеверный лист — двумерное представление структуры молекулы тРНК.
Клеткоспецифический модуль — мотив последовательности, находящийся в промо-
торах генов эукариотов и экспрессируемый в ткани только одного типа.
Клеточный цикл — череда событий, происходящих в клетке между одним делением
и следующим.
Клон — группа клеток, которые содержат одинаковую молекулу рекомбинантной ДНК.
Клонирование ДНК — встраивание фрагмента ДНК в клонирующий вектор и после-
дующее размножение молекулы рекомбинантной ДНК в организме хозяина.
Клонирование генов — встраивание фрагмента ДНК, содержащего ген, в клонирующий
вектор и последующее размножение молекулы рекомбинантной ДНК в организме
хозяина.
Клонирующий вектор — молекула ДНК, которая способна реплицироваться в клетке
хозяина и поэтому может быть использована для клонирования других фрагмен-
тов ДНК.
Коактиватор — белок, который стимулирует инициацию транскрипции путем неспе-
цифического связывания с ДНК или посредством межбелковых взаимодействий.
Когезин — белок, который удерживает сестринские хроматиды вместе в течение пе-
риода между репликацией генома и разделением ядра.
Кодирующая РНК — молекула РНК, которая кодирует белок; мРНК.
Кодоминирование — отношения между парой аллелей, которые оба вносят вклад
в фенотип гетерозиготы.
Кодон пунктуации — кодон, который определяет или начало, или конец гена.
Кодон — триплет нуклеотидов, кодирующий отдельную аминокислоту.
Количественная ПЦР — метод ПЦР, который позволяет оценить число молекул ДНК
в образце.
Компетентный — относится к культуре бактерий, которые были обработаны, напри-
мер, инкубацией с хлоридом кальция, так что их способность вбирать молекулы
ДНК возросла.
Комплекс опознавания начала репликации (ORC) — набор белков, который связы-
вается с опознавательной последовательностью области инициации.
Комплекс привлечения (вербовочный комплекс) — первичная структура, образуемая
во время вырезания интрона GU-AG.
Комплекс посадки на кэп — комплекс, который наводит первоначальный контакт со
структурой кэпа в начале стадии пробега трансляции у эукариотов.
Комплементарная ДНК (кДНК) — копия молекулы мРНК, представленная двунитевой
ДНК.
Словарь терминов 839
Комплементарный — относится к двум нуклеотидам или последовательностям ну-
клеотидов, которые способны спариваться основаниями друг с другом.
Конвергентная эволюция — ситуация, которая происходит, когда одно и то же состояние
признака независимо эволюционирует в двух ветвях эволюционного дерева.
Консенсусная (согласованная) последовательность Козак — последовательность
нуклеотидов, окружающая старт-кодон в мРНК эукариотов.
Консенсусная (согласованная) последовательность — последовательность нуклеоти-
дов, которая представляет «усредненную» последовательность ряда родственных,
но неидентичных последовательностей.
Консервативная репликация — гипотетический способ репликации ДНК, в котором
одна из дочерних двойных спиралей состоит из двух родительских полинуклеоти-
дов, а вторая состоит из двух новосинтезированных полинуклеотидов.
Консервативная транспозиция — транспозиция, которая не приводит к копированию
подвижного генетического элемента.
Контекстнозависимое переназначение функций кодонов — относится к ситуации,
посредством которой последовательность ДНК, окружающая некоторый кодон,
изменяет значение этого кодона.
Последовательность контига — непрерывная последовательность ДНК, полученная
как промежуточный продукт в проекте секвенирования генома.
Контиг — непрерывный набор перекрывающихся последовательностей ДНК (непре-
рывно покрытая область).
Контрольная точка клеточного цикла — период перед вступлением в S- или М-фазу
клеточного цикла — ключевая точка, в которой осуществляется регуляция.
Концевая дезоксинуклеотидилтрансфераза — фермент, который добавляет один
или несколько нуклеотидов к З'-концу молекулы ДНК.
Конъюгационное картирование — метод нанесения на карту бактериальных генов,
основанный на определении времени, необходимого для передачи каждого из этих
генов в процессе конъюгации.
Конъюгация — передача ДНК между двумя бактериями, которые входят в физический
контакт друг с другом.
Корневое — относится к филогенетическому дереву, которое дает информацию о про-
шлых эволюционных событиях, приведших к появлению изучаемых организмов
или последовательностей ДНК.
Короткая интерферирующая РНК (siPHK) — промежуточное звено на пути
РНК-интерференции.
Короткое тандемное повторение (КТП) — разновидность полиморфизма длины
простой последовательности, к коей относят тандемные копии, как правило, ди-,
три- или тетрануклеотидных повторных единиц. Называемое также микросател-
литом.
Корректирование — действие экзонуклеазы 3'—>5', присущее некоторым ДНК-
полимеразам, которое позволяет ферменту заменить ошибочно вставленный
нуклеотид.
Космида — высокоемкий клонирующий вектор, состоящий из участка cos бактериофа-
га X, встроенного в плазмиду.
Коэффициент седиментации — величина, употребляемая для выражения скорости,
с которой молекула или структура осаждается при центрифугировании в плотном
растворе.
840 Словарь терминов
Кроссинговер — обмен ДНК между хромосомами во время мейоза.
Крыловатый спираль-изгиб-спираль — разновидность связывающегося с ДНК до-
мена.
Кэпирование — присоединение кэпа (блокировки) к 5'-концу мРНК эукариотов.
Кэп 0-го типа — основная структура кэпа, состоящая из 7-метилгуанозина, присоеди-
ненного к 5'-концу мРНК.
Кэп 1-го типа — структура кэпа, включающая в себя основной 5'-концевой кэп плюс
дополнительное метилирование рибозы второго по счету нуклеотида.
Кэп 2-го типа — структура кэпа, включающая в себя основной 5'-концевой кэп плюс
метилирование рибоз второго и третьего по счету нуклеотидов.
Кэп — химическая модификация на 5'-конце большинства молекул мРНК эукариотов.
Лактозный репрессор—регуляторный белок, который управляет транскрипцией оперона
лактозы в ответ на присутствие или отсутствие лактозы в окружающей среде.
Лейциновая застежка-молния — димеризационный домен, как правило, обнаружи-
ваемый в связывающихся с ДНК белках.
Летальная мутация — мутация, которая вызывает гибель клетки или организма.
Лигаза — фермент, который синтезирует фосфодиэфирные связи в ходе процессов
репликации, репарации и рекомбинации ДНК.
Лидерный сегмент — нетранслируемая область мРНК, расположенная выше старт-
кодона.
Лизис — разрушение бактериальной клетки лизоцимом, что происходит в конце цикла
инфицирования литическим бактериофагом.
Лизогенный цикл инфицирования — тип заражения бактериофагом, который пред-
полагает встраивание генома фага в молекулу ДНК хозяина.
Лизоцим — белок, употребляемый для дестабилизации клеточной стенки бактерии
перед очисткой ДНК.
Липкий конец — конец молекулы двунитевой ДНК, на котором имеется однонитевое
продолжение.
Литический цикл инфицирования — тип заражения бактериофагом, который предпо-
лагает лизис клетки хозяина сразу после первичного инфицирования, без встраи-
вания молекулы ДНК фага в геном хозяина.
Локус — местоположение генетического маркёра или ДНК-маркёра на хромосоме.
Макрохромосома — одна из крупнейших хромосом с дефицитом генов, обнаруживаемая
в ядрах клеток курицы и разных других видов животных.
Малая бороздка — меньшая из двух бороздок, которые закручиваются спиралью на
поверхности ДНК В-формы.
Малая цитоплазматическая РНК (мцРНК) — тип короткой молекулы РНК эукариотов
с различными ролями в клетке.
Малая ядерная РНК (мяРНК) — разновидность короткой молекулы РНК эукариотов,
вовлеченная в вырезание интронов GU-AG и AU-AC, равно как и в другие события
обработки РНК.
Малая ядрышковая РНК (мякРНК) — разновидность короткой молекулы РНК эука-
риотов, участвующая в химической модификации рРНК.
Малый ядерный рибонуклеопротеид (мяРНП) — структура, принимающая участие
в вырезании интронов GU-AG и AU-AC, а также в других событиях обработки РНК.
Состоит из одной или двух молекул мяРНК в комплексе с белками.
Словарь терминов 841
Маркёр — отличительная особенность на карте генома. Также ген, заложенный в клони-
рующий вектор и кодирующий отличительный белковый продукт и (или) фенотип,
который, таким образом, может быть использован для того, чтобы определить,
содержит ли клетка копию этого клонирующего вектора.
Масс-спектрометрия — аналитический метод, в котором ионы разделяются согласно
их отношениям заряда к массе.
Массовый иммунологический анализ — использование антитела в качестве зонда
с целью обнаружения полипептида, синтезируемого клонированным геном.
Материал для картирования — коллекция фрагментов ДНК, охватывающая хромосому
или полный геном и используемая в картировании МУПов.
Матрица расстояний — таблица, показывающая эволюционные расстояния между
всеми парами нуклеотидных последовательностей из набора данных.
Матрица — полинуклеотид, по которому синтезируется другой полимер с помощью
ДНК- или РНК-полимеразы или рибосомы.
Матричная нить — полинуклеотид, который служит матрицей для синтеза РНК в про-
цессе транскрипции гена.
Матричная (информационная) РНК (мРНК) — транскрипт кодирующего белок
гена.
Матураза — белок, кодируемый содержащимся в интроне геном и, как думают, вовле-
ченный в сплайсинг.
Межбелковое поперечное сшивание — методика, в которой сшиваются вместе смеж-
ные белки, с тем чтобы опознать те белки, которые располагаются близко друг
к другу в структуре типа рибосомы.
Межгенная область — область генома, не содержащая никаких генов.
Мейоз — ряд событий, включающий в себя два ядерных деления, посредством которых
диплоидные ядра преобразуются в гаплоидные гаметы.
Метаболический поток — скорость потока метаболитов через сеть путей, которые
в совокупности составляют биохимию клетки.
Метаболомика — учение о метаболомах.
Метаболическая инженерия — процесс, которым в геном вносятся изменения при по-
мощи мутации или технологии рекомбинантной ДНК, чтобы влиять на биохимию
клетки предопределенным образом.
Метагеномика — совокупность методик исследования смеси геномов, представленных
в определенной среде обитания.
Метафазная хромосома — хромосома на стадии метафазы деления клетки, когда
хроматин принимает свою наиболее уплотненную структуру, и можно наблюдать
такие характерные особенности, как картина исчерченности.
Метилирование de novo — добавление метильных групп в новые позиции на мо-
лекуле ДНК.
Метилирование ДНК — относится к химической модификации ДНК путем присоеди-
нения к ней метильных групп.
Метод сборки контигов из клонов — метод секвенирования геномов, в котором мо-
лекулы, предназначенные для секвенирования, разрываются на более короткие
сегменты (каждый в несколько сотен тыс. п. н. или единиц млн п. н. в длину), которые
секвенируются по отдельности.
842 Словарь терминов
Метод дробовика для полных геномов — методика секвенирования генома, которая
сочетает беспорядочное секвенирование методом дробовика с картой генома: по-
следняя используется для облегчения сборки главной последовательности.
Метод дробовика — методика секвенирования генома, согласно которой молекулы,
подлежащие секвенированию, беспорядочно разбиваются на фрагменты, которые
затем секвенируются по отдельности.
Метод максимальной бережливости — один из методов восстановления филогене-
тических деревьев.
Метод многомерного опознавания белков (МеМОБ) — метод, который объединяет
в себе различные хроматографические методы и позволяет выделять неповреж-
денные белковые комплексы.
Метод объединения соседей — один из методов восстановления филогенетических
деревьев.
Метод отпечатков пальцев клонов — любой из нескольких методов, которые срав-
нивают клонированные фрагменты ДНК, с тем чтобы определить те из них, что
перекрываются между собой.
Метод отпечатков пальцев повторной ДНК—метод отпечатков пальцев клонов, кото-
рый предполагает определение позиций рассеянных по всему геному повторений
в клонированных фрагментах ДНК.
Метод подобия — строгий математический метод выравнивания последовательностей
нуклеотидов.
Метод расстояний — строгий математический подход к выравниванию последователь-
ностей нуклеотидов.
Метод секвенирования с обрывом цепи — метод секвенирования ДНК, который со-
стоит в ферментативном синтезе полинуклеотидных цепей, которые обрываются
в позициях определенных нуклеотидов.
Методика штрих-кодированных кассет удаления — метод, который был разрабо-
тан для массового отборочного анализа мутаций типа удаления у Saccharomyces
cerevisiae.
Мечение концов — прикрепление радиоактивной или иной метки к одному из концов
молекулы РНК или ДНК.
Мечение радиоактивными изотопами — методика прикрепления радиоактивного
атома к молекуле.
Мечение транспозонами — метод выделения генов, который состоит в инактивации
гена путем перемещения в его кодирующую последовательность транспозона и по-
следующего выделения копии меченого гена из библиотеки клонов при помощи
специфичного к такому транспозону гибридизационного зонда.
Меченый участок последовательности (STS) — последовательность ДНК, которая
является уникальной для генома.
Миграция точки ветвления — шаг в холлидеевой модели гомологичной рекомбинации,
на котором происходит обмен полинуклеотидами между парой рекомбинирующих
молекул двунитевой ДНК.
МикроРНК — класс коротких РНК, вовлеченных в регулирование экспрессии генов
у эукариотов и следующих путем, подобным РНК-интерференции.
Микроматрица — имеющий малую плотность массив молекул ДНК, используемый для
параллельного гибридизационного анализа.
Словарь терминов 843
Микросателлит — разновидность полиморфизма длины простой последовательности,
включающая тандемные копии, как правило, ди-, три- или тетрануклеотидных по-
вторных единиц. Также называется простым тандемным повторением (STR).
Миллион пар нуклеотидов (млн п. н.) — 1000 тыс. п. н.; 1 000 000 п. н.
Миниген — название, данное паре экзонов, заключенных в клонирующем векторе,
используемом в процедуре уловления экзонов.
Минимальная среда — среда, которая обеспечивает только минимальные пищевые
потребности для роста микроорганизма.
Минисателлит — разновидность полиморфизма длины простой последовательности,
включающая тандемные копии повторений, которые имеют длину несколько де-
сятков нуклеотидов. Также названный переменным числом тандемных повторений
(VNTR).
Минихромосома — одна из меньших, богатых генами хромосом, обнаруживаемых
в ядрах клеток кур и разных других видов животных.
МирРНК — ранний период эволюции, когда в основе всех биологических реакций были
молекулы РНК.
Митоз — ряд событий, который оканчивается делением ядра.
Митохондриальный геном — геном, содержащийся в митохондриях клеток эукариотов.
Митохондрия — одна из вырабатывающих энергию органелл, присущая клеткам
эукариотов.
Многокопийность — явление нахождения в одной клетке множественных копий гена,
клонирующего вектора или иного генетического элемента.
Многократное попадание или многократная замена — ситуация, которая возни-
кает, когда отдельный нуклеотид в последовательности ДНК подвергается двум
мутационным изменениям, порождая два новых аллеля, оба из которых в той
нуклеотидной позиции отличаются друг от друга и от родителя.
Многопозиционное скрещивание — генетическое скрещивание, сопровождающееся
наследованием трех и более маркёров.
Множественное выравнивание — выравнивание трех и более нуклеотидных после-
довательностей.
Множественные аллели — различные альтернативные формы гена, который имеет
более двух аллелей.
Мобильный (подвижной) генетический элемент — генетический элемент, который
может перемещаться из одной позиции молекулы ДНК в другую.
Модельный организм — организм, который сравнительно легко изучать и который,
следовательно, может использоваться, чтобы получить информацию (которая яв-
ляется значимой для биологии) о втором организме, который труднее изучать.
Модификационное препятствование — метод, применяемый для опознания нуклео-
тидов, вовлеченных во взаимодействия со связывающимся с ДНК белком.
Модификация концов — химическое видоизменение конца молекулы ДНК или РНК.
Мозаичный (выстилающий) массив — коллекция олигонуклеотидныхзондов, каждый
из которых нацелен на свою позицию в хромосоме или части хромосомы.
Молекула рекомбинантной ДНК — молекула ДНК, созданная в пробирке путем лиги-
рования частей ДНК, которые обычно не соединяются друг с другом.
Молекулярная филогенетика — набор методов, которые позволяют эволюционным
отношениям между последовательностями ДНК быть выведенными путем срав-
нений между этими последовательностями.
844 Словарь терминов
Молекулярная эволюция — постепенные изменения, которые происходят в геномах
стечением времени в результате накопления мутаций и структурных перестроек,
следующих из рекомбинации и транспозиции.
Молекулярные науки о жизни — область исследования, включающая молекулярную
биологию, биохимию и клеточную биологию, а также некоторые отрасли генетики
и физиологии.
Молекулярныечасы — средство, основанное на выведенной частоте мутаций, которое
позволяет приписать моменты времени к точкам ветвления в дереве генов.
Молекулярный биолог — человек, который изучает молекулярные отрасли наук
о жизни.
Молекулярный шаперон — белок, который помогает другим белкам свертываться.
Молчащая мутация — изменение в последовательности ДНК, которое не оказывает
никакого воздействия на экспрессию или функционирование какого-либо гена
или продукта гена.
Моногибридное скрещивание — межполовое скрещивание, сопровождающееся на-
следованием одной пары аллелей.
Монофилетический — относится к двум или нескольким организмам или последо-
вательностям ДНК, которые происходят от одного предка в лице организма или
последовательности ДНК.
Мотив лента-спираль-спираль — разновидность связывающегося с ДНК домена.
Мотив спираль-поворот-спираль — распространенный структурный мотив для
специфического связывания белка с молекулой ДНК.
Мотив спираль-петля-спираль — димеризационный домен, обычно встречающийся
в связывающихся с ДНК белках.
Мультигенное семейство — группа генов (сгруппированных или разбросанных) с эво-
люционно связанными последовательностями нуклеотидов.
Мутаген — химический или физический агент, который способен вызывать мутацию
в молекуле ДНК.
Мутагенез in vitro — методы, применяемые для получения заданной мутации в предо-
пределенной позиции молекулы ДНК.
Мутагенез — обработка группы клеток или организмов мутагеном как средство про-
воцирования мутаций.
Мутант — клетка или организм, который обладает мутацией.
Мутасома — белковый комплекс, который строится во время реакции SOS у Escherichia
coli.
Мутация с задержанным проявлением — мутация, действие которой не проявляется
до относительно поздней стадии в жизни мутантного организма.
Мутация с изменением смысла — видоизменение в последовательности нуклеотидов,
которое превращает кодон для одной аминокислоты в кодон для другой амино-
кислоты.
Мутация с потерей смысла — видоизменение в последовательности нуклеотидов, ко-
торое изменяет триплет, кодирующий некоторую аминокислоту, на стоп-кодон.
Мутация с потерей функции — мутация, которая снижает или полностью сводит на
нет активность белка.
Мутация с приобретением функции — мутация, в результате которой организм при-
обретает новую функцию.
Словарь терминов 845
Мутация сквозного считывания — мутация, которая изменяет стоп-кодон на кодон,
определяющий какую-либо аминокислоту, и, следовательно, приводит к сквозному
считыванию стоп-кодона.
Мутация со вставкой (инсерция) — мутация, которая возникает за счет вставки одного
и более нуклеотидов в последовательность ДНК.
Мутация со сдвигом рамки считывания — мутация, следующая из вставки или уда-
ления группы нуклеотидов, число которых не кратно трем, и которая поэтому
изменяет рамку, в которой производится трансляция.
Мутация типа замены — название, широко употребляемое как синоним точечной
мутации.
Мутация типа удаления (делеция) — мутация, следующая из удаления одного или
нескольких нуклеотидов из последовательности ДНК.
Мутация — видоизменение в нуклеотидной последовательности молекулы ДНК.
Надзор за мРНК — процесс деградации РНК у эукариотов.
Наведенный РНК комплекс глушения (RISC) — белковый комплекс, который расще-
пляет и, следовательно, глушит мРНК в процессе РНК-интерференции.
Надсемейство генов — группа из двух и более эволюционно связанных мультигенных
семейств.
Надсемейство ядерных рецепторов — семейство рецепторных белков, которые свя-
зывают гормоны на промежуточном этапе процесса опосредствования активности
генома этими гормонами.
Направленная эволюция — набор экспериментальных методов, который используется
для получения новых генов, кодирующих усовершенствованные продукты.
Направляемая матрицей ДНК-полимераза — фермент, который синтезирует ДНК
в соответствии с последовательностью матрицы.
Направляемая матрицей РНК-полимераза — фермент, который синтезирует РНК
в соответствии с последовательностью матрицы.
Направляемый матрицей синтез ДНК — синтез молекулы ДНК на ДНК- или РНК-
матрице.
Направляемый матрицей синтез РНК — синтез молекулы РНК на ДНК- или РНК-
матрице.
Направляемый олигонуклеотидом мутагенез — метод мутагенеза in vitro, в котором
для внесения предопределенного видоизменения нуклеотидов в ген, предназна-
ченный для мутирования, используется синтетический олигонуклеотид.
Направляющая РНК — короткая РНК, определяющая позиции, в которые один или
несколько нуклеотидов будут вставлены в урезанную РНК в ходе панредактиро-
вания.
Наследственный фактор — термин, введенный Менделем для обозначения единицы
наследственного материала, ныне называемой геном.
Не зависимая от матрицы ДНК-полимераза — фермент, который синтезирует ДНК
без обращения к матрице.
Не зависимая от матрицы РНК-полимераза — фермент, который синтезирует РНК
без оглядки на матрицу.
Негомологическое соединение концов (NHEJ) — одно из названий процесса устра-
нения двунитевых разрывов.
846 Словарь терминов
Незаконная рекомбинация — рекомбинация между двумя молекулами двунитевой
ДНК, которые имеют незначительное подобие (или вообще никакого) нуклеотид-
ных последовательностей.
Некодирующая РНК — молекула РНК, которая не кодирует белок.
Некорневое — относится к филогенетическому дереву, которое лишь илл юстрирует
отношения между изучаемыми организмами или последовательностями ДНК
и не дает никакой информации об эволюционных событиях, которые произошли
в прошлом.
Непенетрантность — ситуация, посредством которой эффект мутации никогда не на-
блюдается в течение периода жизни мутантного организма.
Неполное доминирование — относится к паре аллелей, ни один из которых не прояв-
ляет доминантности, и фенотип гетерозиготы получается промежуточным между
фенотипами двух гомозигот.
Неполное переваривание — переваривание ДНК эндонуклеазой рестрикции при
ограничивающих условиях, так что разрезы производятся не во всех участках
рестрикции.
Неполное сцепление — сцепление, обычно показываемое парой генетических и (или)
физических маркёров на одной хромосоме, при этом маркёры не всегда наследуются
совместно из-за возможности рекомбинации между ними.
Неполярная — гидрофобная (боящаяся воды) химическая группа.
Неравный кроссинговер — событие рекомбинации, которое приводит к дублированию
сегмента ДНК.
Неравный обмен сестринскими хроматидами — событие рекомбинации, которое
приводит к дублированию сегмента ДНК.
Несинонимическая мутация — мутация, которая превращает кодон для одной ами-
нокислоты в кодон для другой аминокислоты.
Несоответствие — позиция в молекуле двунитевой ДНК, где спаривание оснований не
происходит, потому что расположенные в ней нуклеотиды не комплементарны друг
другу; в частности, позиция с неспаренными основаниями появляется вследствие
ошибки репликации.
Нижележащий — со стороны 3'-конца полинуклеотида.
Нозерн-блоттинг — перенос РНК из электрофоретического геля на мембрану до начала
нозерн-гибридизации.
Нозерн-гибридизация — методика, применяемая для обнаружения определенной
молекулы РНК на фоне множества других молекул РНК.
Нокаутная (вышибная) мышь — мышь, которая сконструирована таким образом,
чтобы нести инактивированный (вышибленный) ген.
Нуклеаза S1 — фермент, который осуществляет деградацию молекул однонитевой
ДНК или РНК, в том числе однонитевых областей, в преимущественно двунитевых
молекулах.
Нуклеаза — фермент, который осуществляет деградацию молекулы нуклеиновой
кислоты.
Нуклеиновая кислота — термин, сначала употребляемый для описания кислотного
химического соединения, выделенного из ядер клеток эукариотов. Теперь упо-
требляется специализированно для описания полимерной молекулы, состоящей
из нуклеотидных мономеров, такой как ДНК и РНК.
Словарь терминов 847
Нуклеозид — скрепленное воедино пуриновое или пиримидиновое основание и сахар-
пентоза.
Нуклеоид — содержащая ДНК область доядерной клетки.
Нуклеосома — комплекс из гистонов и ДНК, который является основной структурной
единицей в хроматине.
Нуклеотид — мономерная единица ДНК и РНК, состоящая из пуринового или пирими-
динового основания, соединенного с пятиуглеродным сахаром, к которому, в свою
очередь, прикреплен моно-, ди- или трифосфат.
Область инициации — участок на молекуле ДНК, в котором запускается репликация.
Область прикрепления к каркасу (SAR) — обогащенный динуклеотидами АТ сегмент
генома эукариотов, который служит местом прикрепления к ядерному матриксу.
Обнаружение мутаций — набор методов для обнаружения мутаций в молекулах
ДНК.
Обогащенный глутамином домен — разновидность активационного домена.
Обогащенный пролином домен — разновидность активационного домена.
Обоюдный обмен нитями — обмен ДНК между двумя двунитевыми молекулами,
происходящий в результате рекомбинации, и такой, что конец одной молекулы
обменивается с концом другой молекулы.
Обрабатывающая способность (процессивность) — относится к объему синтеза ДНК,
который выполняется ДНК-полимеразой до отсоединения от матрицы.
Обработка (процессинг) мРНК — события химической и физической модификации,
которые происходят после синтеза мРНК.
Обратная транскриптаза — полимераза, которая синтезирует ДНК на матрице РНК.
Обход хромосомы — метод, который может быть использован для построения сборок
контигов из клонов путем опознания перекрывающихся фрагментов клониро-
ванной ДНК.
Общая рекомбинация — рекомбинация между двумя гомологичными молекулами
двунитевой ДНК.
Общий фактор транскрипции (GTF) — белок или белковый комплекс, который яв-
ляется временным или постоянным компонентом запускающего комплекса, об-
разуемого во время транскрипции у эукариотов.
Обычный псевдоген — ген, который стал неактивным из-за накопления мутаций.
Ограничивающие условия — условия, при которых условно летальный мутант не
способен выжить.
Одиночная сирота — единственный ген, не имеющий никаких гомологов, функция
которого неизвестна.
Однокопийная ДНК — последовательность ДНК, которая не повторяется ни в каком
другом месте генома.
Однонитевая — молекула ДНК или РНК, которая состоит только из одного полину-
клеотида.
Однонитевой разрыв — позиция в молекуле двунитевой ДНК, где один из полинуклео-
тидов разорван в результате отсутствия фосфодиэфирной связи.
Однонуклеотидный полиморфизм (SNP) — точечная мутация, которую несут неко-
торые особи в популяции.
Октамерный модуль — основной промоторный элемент.
Олигонуклеотид — короткая синтетическая однонитевая молекула ДНК.
848 Словарь терминов
Оператор — последовательность нуклеотидов, к которой репрессорный белок при-
соединяется, чтобы предотвратить транскрипцию гена или оперона.
Оперативная таксономическая единица (ОТЕ) — один из организмов, сравниваемых
в ходе филогенетического анализа.
Опережающая нить — нить двойной спирали, которая копируется непрерывным
способом во время репликации генома.
Оперон лактозы — группа из трех генов, которые кодируют ферменты, вовлеченные
в усвоение лактозы бактерией Escherichia coli.
Оперон — набор смежных генов в бактериальном геноме, транскрибируемый с одного
промотора и подверженный одному и тому же режиму регулирования.
Опознавание кодон-антикодон — взаимодействие между кодоном на молекуле мРНК
и соответствующим антикодоном на тРНК.
Опознавательная последовательность области инициации репликации — компо-
нент области инициации репликации у эукариотов.
Опосредствованный бессмысленным кодоном распад РНК (NMD) — процесс дегра-
дации мРНК эукариотов, который запускается присутствием внутреннего стоп-
кодона.
Определение профилей белков — методология, применяемая для опознавания белков
в протеоме.
Оптическое картирование — метод прямого зрительного наблюдения (осмотра) об-
работанных рестриктазами молекул ДНК.
Опухолеродный ДНК-вирус — вирус с геномом из ДНК, способный вызывать рак после
инфицирования животной клетки.
Ортологичный — относится к гомологичным генам, расположенным в геномах разных
организмов.
Очистка промотора — завершение успешной инициации транскрипции, которое про-
исходит, когда РНК-полимераза смещается с последовательности промотора.
Основная скорость транскрипции — число продуктивных запусков транскрипции,
совершающихся за единицу времени в отдельно взятом промоторе.
Основной промотор — позиция в пределах промотора эукариотов, на которой собира-
ется инициаторный комплекс.
Основной промоторный элемент (базальный промотор) — мотив последователь-
ности, который присутствует во многих промоторах эукариотов и устанавливает
основной (базовый) уровень инициации транскрипции.
Основной фермент — версия РНК-полимеразы Escherichia coli (с субъединичным соста-
вом а2рр')/ которая выполняет синтез РНК, но неспособна эффективно определять
местонахождение промоторов.
Основный домен — разновидность связывающегося с ДНК домена.
Отжиг — присоединение олигонуклеотидной затравки к ДНК- или РНК-матрице.
Отклонение частоты использования кодонов — относится к тому факту, что не все
кодоны используются с одинаковой частотой в генах того или иного организма.
Открытая рамка считывания (ОРС) — ряд кодонов, начинающийся со старт-кодона
и оканчивающийся стоп-кодоном. Часть кодирующего белок гена, которая транс-
лируется в белок.
Открытый промоторный комплекс — структура, формируемая во время сборки
комплекса инициации транскрипции (после раскрытия ДНК за счет разрыва пар
Словарь терминов 849
оснований) и состоящая из РНК-полимеразы и (или) дополнительных белков,
прикрепленных к промотору.
Отношение оснований — отношение А к Т или G к С в молекуле двунитевой ДНК. Чар-
гафф показал, что отношение оснований всегда близко к 1,0.
Отстающая нить — нить двойной спирали, которая копируется прерывным способом
во время репликации генома.
Оценка самосовершенствования — статистическая величина, полученная в итоге
анализа с самосовершенствованием.
Очистка по тандемному сродству (ТАР) — метод выделения белковых комплексов,
в котором используется исследуемый белок с С-конечным продолжением, который
связывается с кальмодулином.
ПЦР с повторной ДНК — метод отпечатков пальцев клонов, в котором используется
ПЦР с целью обнаружения в клонированных фрагментах ДНК относительных по-
зиций рассеянных по всему геному повторений.
ПЦР с рассеянными повторными элементами (РПЭ-ПЦР) — методика отпечатков
пальцев клонов, которая использует ПЦР, чтобы обнаружить относительные пози-
ции рассеянных по всему геному повторений в клонированных фрагментах ДНК.
Палец Cis2His2 — разновидность цинкового пальца — связывающегося с ДНК домена.
Панредактирование — массовая вставка нуклеотидов в сокращенную РНК с получе-
нием в итоге функционально активной молекулы.
Пара оснований — скрепленная водородными связями структура, образованная двумя
взаимно комплементарными нуклеотидами. В сокращенной форме обозначается
«п.н.» и служит кратчайшей единицей длины двунитевой молекулы ДНК.
Парадокс показателя С — неравносильность между размером генома и числом генов,
которая наблюдается при сравнении между некоторыми эукариотами.
Паралогичный — относится к двум или нескольким гомологичным генам, располо-
женным в одном геноме.
Паранемный — относится к спирали, нити которой могут быть разведены без рас-
кручивания.
Параретровирус — заключенный в оболочку вирусный ретроэлемент с геномом из
ДНК.
Парафилетический — группа последовательностей или таксонов в филогенетическом
дереве, в которую не входят некоторые члены клады.
Пентоза — сахар, молекула которого состоит из пяти атомов углерода.
Пептидилтрансфераза — фермент, который синтезирует пептидные связи в ходе
трансляции.
Пептидильный, или Р-участок—участок в рибосоме, занимаемый тРНК, прикрепляю-
щейся к наращиваемому полипептиду в ходе трансляции.
Пептидная нуклеиновая кислота (ПНК) — аналог полинуклеотида, в котором сахаро-
фосфатная основная цепь заменена амидными связями.
Пептидная связь — химическая связь между смежными аминокислотами в полипеп-
тиде.
Первичная структура — последовательность аминокислот в полипептиде.
Первичный транскрипт — первичный продукт транскрипции гена или группы генов,
впоследствии обрабатываемый с получением зрелого транскрипта (или несколь-
ких транскриптов).
850 Словарь терминов
Передача сигнала — управление активностью клетки, в том числе экспрессией генома,
через рецептор клеточной поверхности, который отвечает на внешний сигнал.
Переключение класса — процесс, который приводит к полному изменению типа им-
муноглобулина, синтезируемого В-лимфоцитом.
Переключение типа спаривания — способность клеток дрожжей изменять тип спа-
ривания с а на а или наоборот путем генной конверсии.
Перекрывающиеся гены — два гена, кодирующие области которых перекрываются.
Переменное число тандемных повторений (VNTR) — полиморфизм длины простой
последовательности, включающий в себя тандемные копии повторений, которые
имеют длину несколько десятков нуклеотидов. Также называется минисателлитом.
Перенос эписом — передача между клетками целой бактериальной хромосомы или
некоторой ее части за счет встраивания в плазмиду.
Перепечатывание реплики — методика переноса колоний из одной чашки Петри
в другую так, что их относительные позиции на поверхности агаровой среды со-
храняются.
Перестройка нуклеосомы — изменение конформации нуклеосомы, сопряженное с из-
менением доступности ДНК, на которой сидит нуклеосома.
Перетасовывание ДНК — основанная на ПЦР процедура, которая влечет за собой на-
правленную эволюцию последовательности ДНК.
Перетасовывание доменов — перестройка сегментов одного или нескольких генов
(причем каждый такой сегмент кодирует структурный домен в продукте гена)
с получением принципиально нового гена.
Пиль — структура, участвующая в сближении пары бактерий во время конъюгации;
возможно, трубка, через которую передается ДНК.
Пиримидин — азотистое основание одного из двух типов, входящее в состав нуклео-
тидов.
Пиросеквенирование — новый метод секвенирования ДНК, в котором присоедине-
ние нуклеотида к концу наращиваемого полинуклеотида обнаруживается непо-
средственно путем преобразования высвобождаемого пирофосфата во вспышку
хемилюминесценции.
Плавление — денатурация молекулы двунитевой ДНК.
Плавучая плотность — плотность, свойственная молекуле или частице и определяемая
при взвешивании ее в водном растворе соли или сахара.
Плазмида Ti — крупная плазмида, встречающаяся в тех клетках Agrobacterium tumefaciens,
которые способны вызывать образование корончатого галла на некоторых видах
растений.
Плазмида — как правило, кольцевой отрезок ДНК, часто встречающийся у бактерий
и в клетках некоторых других типов.
Плезиоморфное состояние признака—состояние признака, которым обладал дальний
общий предок группы организмов.
Плектонемная — относится к спирали, нити которой могут быть разведены только
путем раскручивания.
Повторная ДНК — последовательность ДНК, которая повторяется два и более раз
в молекуле ДНК или геноме.
Повреждение (6-4) — димер между двумя смежными пиримидиновыми основаниями
в полинуклеотиде, образуемый при действии ультрафиолетового излучения.
Подавление РНК — процесс деградации РНК у эукариотов.
Словарь терминов 851
Поддерживающее метилирование — добавление метильных групп в позиции на ново-
синтезированных нитях ДНК, которые соответствуют позициям метилирования
на родительской нити.
Позиционное клонирование — процедура, которая использует информацию о позиции
гена на карте, чтобы получить клон этого гена.
Позиционный эффект — относится к различным уровням экспрессии гена, которые
получаются после вставки этого гена в различных позициях в геном эукариотов.
Поиск гомологии — методика, в которой для определения функции неизвестного гена
отыскиваются гены с последовательностями, сходными с последовательностью
исследуемого гена.
Показатель FLpter — единица, используемая в методе FISH для описания позиции
гибридизационного сигнала относительно конца короткого плеча хромосомы.
Поли-А-полимераза — фермент, который присоединяет поли-А хвост к З'-концу мРНК
эукариотов.
Поли-А хвост — ряд нуклеотидов А, приложенных к З'-концу мРНК эукариотов.
Полиаденилирование — присоединение вереницы нуклеотидов А к З'-концу мРНК
эукариотов.
Полиаденилирующее редактирование — форма редактирования, которое проис-
ходит со многими животными митохондриальными РНК, в результате которого
стоп-кодон образуется добавлением поли-А хвоста к мРНК, оканчивающейся ну-
клеотидами U или UA.
Полимер — соединение, состоящее из длинной цепи идентичных или подобных еди-
ниц.
Полимераза Клёнова — фермент ДНК-полимераза, получаемый химической модифи-
кацией ДНК-полимеразы I Escherichia coli и применяемый прежде всего в секвени-
ровании ДНК с обрывом цепи.
Полимераза Корнберга — фермент ДНК-полимераза I Escherichia coli.
Полимеразная цепная реакция (ПЦР) — процедура, которая обеспечивает экспонен-
циальное увеличение числа избранной области молекулы ДНК.
Полиморфизм длины простой последовательности (SSLP) — тандемно повторяю-
щаяся последовательность, число повторов в которой вариьируется.
Полиморфизм длины рестрикта (RFLP) — фрагмент рестрикции, длина которого
является изменчивой из-за присутствия полиморфного участка рестрикции на
одном или на обоих концах.
Полиморфный — относится к локусу, который представлен множеством различных
аллелей или гаплотипов в популяции в целом.
Полинуклеотид — молекула однонитевой ДНК или РНК.
Полинуклеотидкиназа Т4 — фермент, который добавляет фосфатные группы
к 5'-концам молекул ДНК.
Полинуклеотидкиназа — фермент, который присоединяет фосфатные группы к 5'-кон-
цам молекул ДНК.
Полипептид — полимер из аминокислот.
Полипиримидиновый тракт — обогащенная пиримидином область около З'-конца
интрона GU-AG.
Полипотенциальная — относится к клетке, которая не наставлена на какой-либо
определенный путь развития и, следовательно, может давать начало дифферен-
цированным клеткам всех возможных типов.
852 Словарь терминов
Полипротеин — продукт трансляции, состоящий из ряда сцепленных белков, которые
обрабатываются протеолитическим расщеплением с высвобождением зрелых
белков.
Полисома — молекула мРНК, которая транслируется более чем одной рибосомой в одно
и то же время.
Полифилетическая — группа последовательностей ДНК, которые происходят от двух
или нескольких разных предковых последовательностей.
Полицентрическая эволюция — гипотеза, согласно которой современные люди Старо-
го Света происходят от популяций Ното erectus, которые оставили Африку более
1 миллиона лет назад.
Полицистеиновый цинковый палец — разновидность связывающегося с ДНК до-
мена — цинкового пальца.
Половая клетка — репродуктивная клетка; клетка, которая делится посредством
мейоза.
Половая хромосома — хромосома, которая отвечает за определение пола.
Полуконсервативная репликация — способ репликации ДНК, в котором каждая до-
черняя двойная спираль состоит из одного родительского полинуклеотида и одного
новосинтезированного полинуклеотида.
Полученная на основе фага Р1 искусственная хромосома (РАС) — вектор высокой
емкости, который объединяет в себе особенности векторов на бактериофаге Р1
и бактериальных искусственных хромосом.
Полярная — гидрофильньная (любящая воду) химическая группа.
Последовательности PEST — последовательности аминокислот, которые влияют на
деградацию белков, в которых они находятся.
Последовательность Шайна-Дальгарно — участок связывания рибосомы, располо-
женный выше гена Escherichia coli.
Последовательный анализ экспрессии генов (SAGE) — метод изучения состава транс-
криптома.
Посредник — белковый комплекс, который образует контакт между различными акти-
ваторами и С-концевым доменом наибольшей субъединицы РНК-полимеразы II.
Пострепликационное восстановление — процесс репарации, который устраняет
разрывы в дочерних молекулах ДНК, которые возникают в результате нарушений
в процессе репликации.
Пострепликационный комплекс (постРК) — комплекс белков, образующийся из
предРК, который формируется в точке начала репликации у эукариотов во время
процесса репликации и гарантирует, что данный старт репликации используется
только один раз за один клеточный цикл.
Поток генов — перемещение гена от одного организма к другому.
Праймаза — фермент РНК-полимераза, который синтезирует РНК-затравки в ходе
репликации ДНК у бактерий.
Праймосома — белковый комплекс, участвующий в репликации генома.
Пре-РНК — первичный продукт транскрипции гена или группы генов, который впо-
следствии обрабатывается с получением зрелого транскрипта (или нескольких
транскриптов).
Пре-мРНК — первичный транскрипт кодирующего белок гена.
Пре-рРНК — первичный транскрипт гена или группы генов, кодирующих молекулы
рРНК.
Словарь терминов 853
Пре-тРНК — первичный транскрипт гена или группы генов, кодирующих молекулы
тРНК.
Предынициаторный (предзапускающий) комплекс—структура, включающая в себя
малую субъединицу рибосомы, инициаторную тРНК плюс вспомогательные факто-
ры, которые формируют первичный контакт с мРНК во время синтеза белка. Также
структура, которая формируется в центральном промоторе гена, транскрибиуемого
РНК-полимеразой II.
Предзатравочный комплекс — белковый комплекс, образуемый во время запуска
репликации у бактерий.
Предрепликационный комплекс (предРК) — белковый комплекс, который соби-
рается в области начала репликации у эукариотов и обеспечивает инициацию
репликации.
Предсплайсосомный комплекс — промежуточное звено в пути вырезания для ин-
трона GU-AG.
Прерывный ген — ген, который разбит на экзоны и интроны.
Прибнова блок — см. Блок Прибнова.
Приобретенное состояние признака — состояние признака, которое в ходе эволю-
ции появилось у близкого предка поднабора организмов, входящих в изучаемую
группу.
Прион — необычный инфекционный агент, который состоит полностью из белка.
Приспособленность — способность организма или аллеля к выживанию и воспроиз-
водству.
Прицепной (хвостовой) сегмент — нетранслируемая область мРНК, расположенная
ниже стоп-кодона.
Проект «Геном человека» — финансируемый на общественные средства проект, в ходе
которого была получена одна из черновых последовательностей генома человека
и продолжают изучаться функции генов человека.
Прокариот — организм, клеткам которого недостает обособленного ядра.
Промежуточный период — один из двух промежуточных периодов в течение клеточ-
ного цикла.
Промотор — расположенная выше гена последовательность нуклеотидов, с которой
РНК-полимераза связывается, с тем чтобы запустить транскрипцию.
Пропуск экзона — аномальный сплайсинг, в котором один или несколько экзонов не
включаются в зрелую РНК.
Проскальзывание — перескок рибосомы через короткую некодирующую последо-
вательность нуклеотидов между стоп-кодоном одного гена и старт-кодоном сле-
дующего гена.
Проскальзывание репликации — ошибка репликации, которая ведет к увеличению
или уменьшению числа повторных единиц в тандемном повторении типа микро-
сателлита.
Процессированный псевдоген — псевдоген, который получается в результате встраи-
вания в геном обратно транскрибированной копии мРНК.
Поиск ОРС — просмотр последовательности ДНК на предмет наличия открытых рамок
считывания с целью определения местонахождения генов.
Протеаза — фермент, который осуществляет деградацию белка.
Протеасома — многосубъединичная белковая структура, которая участвует в дегра-
дации других белков.
854 Словарь терминов
Протеом — набор функционально активных белков, синтезируемых живой клеткой.
Протеомика — совокупность разнообразных методов изучения протеомов.
Протогеном — геном из РНК, который существовал в мире РНК.
Протопласт — клетка, у которой клеточная стенка была полностью удалена.
Прототроф — организм, который не имеет никаких пищевых потребностей, помимо
таковых у дикого типа, и который способен расти на минимальной среде.
Проточная цитометрия — метод разделения хромосом.
Профаг — встроенная форма генома лизогенного бактериофага.
Прямое восстановление — система репарации ДНК, которая воздействует непосред-
ственно на поврежденный нуклеотид.
Прямое повторение — последовательность нуклеотидов, которая повторяется дважды
(или более часто) в молекуле ДНК.
Прямое считывание — распознавание последовательности ДНК связывающимся с ней
белком, который образует контакты на внешней стороне двойной спирали.
Псевдоген — инактивированная и, следовательно, функционально неактивная копия
гена.
Пурин — азотистое основание одного из двух типов, входящее в состав нуклеотидов.
Путь сворачивания — череда событий, в которую входят частично свернутые про-
межуточные продукты и которая завершается свернутым белком, приобретшим
свою правильную трехмерную структуру.
Путь сплайсинга — ряд событий, которые превращают сплошную молекулу пре-мРНК
в функционально активную мРНК.
РНК — рибонуклеиновая кислота, одна из двух форм нуклеиновой кислоты в живых
клетках; генетический материал для некоторых вирусов.
РНК-аденозиндезаминаза (ADAR) — фермент, который редактирует различные мРНК
эукариотов путем дезаминирования аденозина в инозин.
РНК-зависимая ДНК-полимераза — фермент, который делает ДНК-копию матрицы
РНК; обратная транскриптаза.
РНК-зависимая РНК-полимераза — фермент, который делает РНК-копию матрицы
РНК.
РНК-интерференция (РНК-и) — процесс деградации РНК у эукариотов.
РНК-полимераза I — РНК-полимераза эукариотов, которая транскрибирует гены ри-
босомной РНК.
РНК-полимераза II — РНК-полимераза эукариотов, которая транскрибирует гены,
кодирующие белки и мяРНК.
РНК-полимераза III — РНК-полимераза эукариотов, которая транскрибирует гены
тРНК и прочие короткие гены.
РНК-полимераза — фермент, который синтезирует РНК на матрице РНК или ДНК.
РНК-транскрипт — РНК-копия гена.
Радиоактивный маркёр — введенный в молекулу радиоактивный атом, радиоактивное
излучение которого впоследствии используется для обнаружения и отслеживания
этой молекулы на протяжении биохимической реакции.
Радиационный гибрид — коллекция клеточных линий грызунов (которые содержат
различные фрагменты другого генома, построенные методикой, основанной на
облучении), используемая в качестве материала для картирования (например,
в исследованиях генома человека).
Словарь терминов 855
Развитие — согласованная череда преходящих и постоянных изменений, которые про-
исходят в течение всего периода жизни клетки или организма.
Разрешающие условия — условия, в которых условно летальный мутант способен
выжить.
Разрушение ультразвуком — процедура, в которой для образования случайных раз-
рывов в молекулах ДНК применяется ультразвук.
Рамка считывания — ряд триплетных кодонов в последовательности ДНК.
Раскрашивание хромосом — вариант флюоресцентной гибридизации in situ, при
котором в качестве гибридизационного зонда используется смесь молекул ДНК,
каждая из которых специфична к своим областям отдельной хромосомы.
Расплывчатая мутация — мутация, которая приводит не к полной, а л ишь к частичной
потере характеристики.
Распознающая спираль — а-спираль в ДНК-связывающем белке, которая отвечает за
опознавание целевой последовательности нуклеотидов.
Разрешение — разделение пары прорекомбинировавших молекул двунитевой ДНК.
Разбросанная репликация — гипотетический способ репликации ДНК, в котором оба
полинуклеотида каждой дочерней двойной спирали состоят частично из родитель-
ской ДНК и частично — из новосинтезированной ДНК.
Рассеянное по всему геному повторение — последовательность, которая повторяется
во многих разбросанных по всему геному позициях.
Рассеянное повторение — последовательность, которая повторяется во многих раз-
бросанных по геному позициях.
Расхождение — разделение гомологичных хромосом (или членов пар аллелей) по
разным гаметам в процессе мейоза.
Расчесывание молекул — методика приготовления обработанных рестриктазами
молекул ДНК к оптическому картированию.
Реагирующий модуль—мотив последовательности, располагающийся выше различных
генов и позволяющий согласовывать инициацию транскрипции РНК-полимеразой II
с основными сигналами, идущими с внешней поверхности клетки.
Реагирующий на железо элемент — разновидность реагирующего модуля.
Реакция на стресс — биохимическая и генетическая ответная реакция, проявляемая
Escherichia coli, когда бактерия попадает в неблагоприятные условия роста, такие
как низкие уровни содержания незаменимых аминокислот.
Ревертазная ПЦР (РТ-ПЦР) — ПЦР, в которой первый шаг выполняется обратной
транскриптазой, так что в качестве исходного материала может быть использо-
вана РНК.
Регуляторное управление — управление экспрессией бактериального гена, которое
зависит от влияния регуляторных белков.
Регуляторный мутант — мутант, который имеет дефект в промоторе или иной регу-
ляторной последовательности.
Редактирование РНК — процесс, посредством которого нуклеотиды, не кодируемые
геном, вводятся в определенные позиции молекулы РНК после транскрипции.
Рекомбиназа — разнообразное семейство ферментов, которые катализируют события
сайт-специфической рекомбинации.
Рекомбинант—член потомства, который не обладает ни одной из комбинаций аллелей,
присущих родителям.
856 Словарь терминов
Рекомбинантный белок — белок, синтезируемый в рекомбинантной клетке в резуль-
тате экспрессии клонированного гена.
Рекомбинационное восстановление — процесс репарации ДНК, который устраняет
двунитевые разрывы.
Рекомбинация — крупномасштабная перестройка молекулы ДНК.
Ренатурация — возвращение денатурированной молекулы в ее естественное состояние.
Рентгеновская дифрактограмма — картина, полученная в результате дифракции
рентгеновских лучей в кристалле.
Рентгеноструктурный анализ — метод определения трехмерной структуры крупной
молекулы.
Репарация ДНК — биохимические процессы, которые исправляют мутации, возникаю-
щие в результате ошибок репликации и воздействий мутагенных агентов.
Репарация вырезанием нуклеотидов — процесс репарации, который исправляет по-
вреждения ДНК различных типов путем вырезания и повторного синтеза области
полинуклеотида.
Репарация вырезанием оснований — процесс репарации ДНК, который заключается
в вырезании и замене неправильного основания.
Репарация вырезанием — процесс репарации ДНК за счет исправления повреждения
ДНК различных типов путем вырезания и повторного синтеза ущербной области
полинуклеотида.
Репарация короткими заплатами — процесс восстановления вырезанием нуклеотидов
у Escherichia coli, который заключается в вырезании и повторном синтезе отрезков
ДНК длиной около 12 нуклеотидов.
Репарация наложением длинных заплат — процесс восстановления вырезанием ну-
клеотидов, имеющийся у Escherichia coli, который состоит в вырезании и повторном
синтезе отрезков ДНК длиной до 2 тыс. п. н.
Репарация флюоресценции после фотообесцвечивания (ВФПФ) — технология, при-
меняемая для изучения подвижности ядерных белков.
Репатриация интеинов — генная конверсия, кодирующая белок с недостачей интеина,
в ген, кодирующий белок с интеином, катализируемая вырезанным (сплайсиро-
ванным) компонентом интеина.
Репатриация интронов — генная конверсия без интрона в ген, который содержит
интрон, катализируемая белком, кодируемым этим интроном.
Репликационная вилка — область молекулы двунитевой ДНК, которая раскрывается,
чтобы позволить произойти репликации ДНК.
Репликационная транспозиция — транспозиция, которая приводит к копированию
подвижного генетического элемента.
Репликационная фабрика — крупная структура, прикрепленная к ядерному матриксу;
участок репликации генома.
Репликационная форма — двунитевая форма молекулы ДНК фага М13, встречающаяся
среди зараженных клеток Escherichia coli.
Репликационный белок A (RPA) — главный белок однонитевого связывания, уча-
ствующий в репликации ДНК эукариотов.
Репликационный посреднический белок (RMP) — белок, ответственный за отсоеди-
нение белков однонитевого связывания в ходе репликации генома.
Репликация ДНК — синтез новой копии генома.
Словарь терминов 857
Репликация катящегося кольца—процесс репликации, состоящий в непрерывном син-
тезе полинуклеотида, который «скатывается» с кольцевой молекулы матрицы.
Реплисома — белковый комплекс, участвующий в репликации генома.
Рестрикционное картирование — определение позиций участков рестрикции в мо-
лекуле ДНК путем анализа размеров фрагментов рестрикции.
Ретровирус — вирус с геномом из РНК, который встраивается в геном клетки своего
хозяина.
Ретровирусоподобный элемент (РВПЭ) — усеченный ретровирусный геном, встро-
енный в хромосому хозяина.
Ретроген — дубликат гена, который возникает за счет вставки псевдогена смежно
с промотором существующего гена.
Ретропозон — ретроэлемент, который не содержит LTR.
Ретрорепатриация — процесс, в ходе которого вырезанный интрон, включая одно-
нитевую РНК, вставляется непосредственно в геном органеллы, прежде чем быть
скопированным в двунитевую ДНК.
Ретротранспозиция — транспозиция через РНК-посредник.
Ретротранспозон — рассеянное по всему геному повторение с последовательностью,
подобной встроенному ретровирусному геному, и, возможно, обладающее ретро-
транспозиционной активностью.
Ретроэлемент — генетический элемент, который транспонируется через РНК-пос-
редник.
Рецептор стероидных гормонов — белок, который связывает стероидный гормон
после проникновения последнего в клетку (как промежуточный шаг в опосред-
ствовании активности генома).
Рецессивный — аллель, который не экспрессируется в гетерозиготе.
Рибоза — сахарная составляющая рибонуклеотида.
Рибозим — молекула РНК, которая обладает каталитическим действием.
Рибонуклеаза D — фермент, вовлеченный в обработку пре-тРНК у бактерий.
Рибонуклеаза MRP — фермент, вовлеченный в обработку пре-рРНК эукариотов.
Рибонуклеаза Р — фермент, вовлеченный в обработку пре-тРНК у бактерий.
Рибонуклеаза — фермент, который осуществляет деградацию РНК.
Рибонуклеопротеидный (РНП) домен — распространенный РНК-связывающий домен.
Рибосома — один из РНК-белковых комплексов, в котором происходит трансляция.
Рибосомная РНК (рРНК) — молекулы РНК, служащие компонентами рибосом.
Рибосомный белок — один из белковых компонентов рибосомы.
Родительский генотип — генотип, присущий одному или обоим родителям, участвую-
щим в генетическом скрещивании.
Родословная — схема, показывающая генетические отношения между членами семьи
человека.
С-концевой домен (С-КД) — компонент наибольшей субъединицы РНК-полимеразы II,
важный в активации полимеразы.
Сайленсер — регуляторная последовательность, которая уменьшает скорость транс-
крипции гена или генов, расположенных на некотором расстоянии выше или ниже
от нее.
Сайт-специфический мутагенез — методы, применяемые для получения заданной
мутации в предопределенной позиции молекулы ДНК.
858 Словарь терминов
Сайт-специфическое зондирование гидроксильными радикалами — методика
определения позиции белка в комплексе белок-РНК, таком как рибосома, при ис-
пользовании способности ионов Fe2+ порождать гидроксильные радикалы, которые
расщепляют близлежащие фосфодиэфирные связи в РНК.
Сайт-специфическая рекомбинация — рекомбинация между двумя молекулами дву-
нитевой ДНК, которые имеют лишь короткие области подобия нуклеотидных
последовательностей.
Самопроизвольная мутация — мутация, которая является результатом ошибки ре-
пликации.
Сборка контигов из клонов — коллекция клонов, фрагменты ДНК которых перекры-
ваются.
Сверхотклонение — крайняя форма отклонения (неоднозначного соответствия, или не-
канонического спаривания), которая имеет место в митохондриях позвоночных.
Сверхмутация — увеличение частоты мутаций генома.
Сверхспирализация — конформационное состояние, в котором двойная спираль явля-
ется перекрученной или недокрученной, так что происходит скручивание в сверх-
спираль.
Сверхчувствительный к ДНКазе I участок — короткая область ДНК эукариотов, кото-
рая относительно легко расщепляется дезоксирибонуклеазой I; может совпадать
со свободными от нуклеосом позициями.
Связанная с матриксом область (MAR) — обогащенный динуклеотидами АТ сегмент
генома эукариотов, который служит точкой прикрепления к ядерному матриксу.
Связанный с С-КД SR-подобный белок (CASP) — белок, который, как думают, играет
регуляторную роль во время вырезания интронов GU-AG.
Связанный с ТВР фактор (TAF) — один из нескольких компонентов общего факто-
ра транскрипции TFIID, играющий вспомогательные роли в опознавании блока
ТАТА.
Связующая (линкерная) ДНК — ДНК, которая связывает нуклеосомы: «нить» в модели
«бусины на нити», описывающей структуру хроматина.
Связующий гистон — гистон типа Н1, который расположен на внешней стороне стерж-
невого октамера нуклеосомы.
Связывающийся с trp-PHK ослабляющий белок (TRAP) — белок, участвующий в осла-
бляющей регуляции некоторых оперонов в бактериях типа Bacillus subtilis.
Связывающийся с ДНК белок — белок, который прикрепляется к молекуле ДНК.
Связывающийся с ДНК мотив — часть связывающегося с ДНК белка, которая вступает
в контакт с двойной спиралью.
Связывающийся с блоком ТАТА белок (ТВР) — компонент общего фактора транскрип-
ции TFIID, часть, которая опознает блок ТАТА промотора для РНК-полимеразы II.
Связывающийся с метилированным CpG-островком белок (МеСР) — белок, который
связывается с метилированными CpG-островками и может влиять на ацетилиро-
вание близлежащих гистонов.
Связывающийся с полиаденилатом белок (PADP) — белок, который помогает поли-
А-полимеразе во время полиаденилирования молекул мРНК эукариотов и играет
определенную роль в поддержании поли-А хвоста после синтеза.
Связывающийся с теломерой белок (ТВР) — белок, который связывается с теломерой
и регулирует ее длину.
Сворачивание белка — принятие свернутой структуры полипептидом.
Словарь терминов 859
Сдвиг рамки считывания — смещение рибосомы из одной рамки считывания в другую
во внутренней позиции гена.
Сегментированный геном — вирусный геном, который разбит на две и более моле-
кулы ДНК или РНК.
Седиментационный анализ — метод центрифугирования, применяемый для измере-
ния коэффициента седиментации молекулы или структуры.
Секвеназа — фермент, используемый при секвенировании ДНК с обрывом цепи.
Секвенирование ДНК — методика определения порядка нуклеотидов в молекуле
ДНК.
Секвенирование в тепловом цикле — метод секвенирования ДНК, в котором исполь-
зуется ПЦР для получения полинуклеотидов с оборванной цепью.
Секвенирование путем химической деградации — метод секвенирования ДНК, кото-
рый предполагает использование химикатов, которые разрезают молекулы ДНК
в позициях определенных нуклеотидов.
Селективная среда — среда, которая поддерживает рост только тех клеток, которые
несут в себе определенный генетический маркёр.
Селективный маркёр — ген, заложенный в вектор и придающий легко распознаваемые
характеристики клетке, содержащей этот вектор или молекулу рекомбинантной
ДНК, полученную из этого вектора.
Семейство SMAD — группа белков, участвующих в передаче сигнала.
Сеть белок-белковых взаимодействий— карта, показывающая взаимодействия между
всеми или некоторыми из белков в протеоме.
Сигнальный пептид — короткая последовательность на N-конце некоторых белков,
которая проводит белок через мембрану.
Сильный промотор — промотор, который обеспечивает относительно большое число
продуктивных запусков в единицу времени.
Синонимическая мутация — мутация, которая изменяет один кодон на второй кодон,
который определяет ту же самую аминокислоту.
Синтения — относится к паре геномов, в которых по крайней мере некоторые гены
расположены в подобных картографических позициях.
Сиротское семейство — группа гомологичных генов, функции которых неизвестны.
Система размножения теплостойких мутаций (проба СРТМ) — метод типизации
SNP, в котором ПЦР направляется парой затравок, одна из которых покрывает
позицию SNP.
Системная биология — подход в биологии, который пытается связать метаболические
пути и внутриклеточные процессы с экспрессией генома.
Сканирование — система, используемая во время инициации трансляции у эукариотов,
в которой предзапускающий комплекс садится на структуру кэпа на 5'-конце мРНК
и затем пробегает по молекуле, пока не достигнет старт-кодона.
Скрытый ген — один из нескольких генов в митохондриальном геноме трипаносомы,
которые определяют сокращенные РНК, кои должны подвергнуться панредакти-
рованию, чтобы стать функционально активными.
Скрытый сайт сплайсинга — участок, последовательность которого напоминает ис-
тинный сайт сплайсинга и который может быть использован вместо подлинного
участка во время аномального сплайсинга.
Слабый промотор — промотор, который осуществляет сравнительно немного про-
дуктивных запусков в единицу времени.
860 Словарь терминов
Слаженная эволюция — эволюционный процесс, в результате которого члены мульти-
генного семейства сохраняют одинаковые или подобные последовательности.
Случайный генетический дрейф — процесс, который ведет к постепенному изменению
частотности аллелей в популяции.
Смена аллелей — замена аллеля, который однажды был закреплен в популяции, вторым
аллелем, причем такие вторые аллели возникают по причине мутации и увеличи-
вают частоту, пока сами не закрепятся в популяции.
Смешанная ДНК — ДНК, которая была передана от одного генома органеллы другому.
Смола — хроматографический матрикс.
Снятие последовательности — метод быстрого получения последовательности, в кото-
ром некоторое число случайных последовательностей получают из клонированного
фрагмента; основан этот метод на том, что если фрагмент содержит какие-либо
гены, то есть хороший шанс на то, что по крайней мере некоторые из них будут
показаны этими случайными последовательностями.
Совместная трансдукция — перенос двух и более генов от одной бактерии к другой
через трансдуцирующий фаг.
Совместная трансформация — вобрание двух и более генов на отдельной молекуле
ДНК во время трансформации бактерии.
Совместное иммунное осаждение — выделение всех членов белкового комплекса
с помощью антитела, специфичного только к одному из этих белков.
Совстройка (коинтеграт) — промежуточный продукт на пути, ведущем к репликаци-
онной транспозиции.
Содержание GC — процентное содержание (доля) нуклеотидов G или С в геноме.
Соклетие — подобная клетке структура, включающая в себя море цитоплазмы и массу
ядер.
Соматическая клетка — нерепродуктивная клетка; клетка, которая делится мито-
зом.
Сопряженное с транскрипцией восстановление — процесс восстановления выре-
занием нуклеотидов, который заключается в восстановлении матричных нитей
генов.
Сорепрессор — молекула, которая подавляет экспрессию гена или оперона за счет
того, что связывается с белком-репрессором и тем самым позволяет репрессору
связаться с оператором.
Составной белок — белок, который состоит из слияния двух полипептидов или частей
полипептидов, обычно кодируемых отдельными генами.
Составной транспозон — ДНК-транспозон, включающий пару вставных последова-
тельностей, примыкающих с обеих сторон к сегменту ДНК, обычно содержащему
один или несколько генов.
Состояние признака — одна из по крайней мере двух альтернативных форм признака,
учитываемого в филогенетическом анализе.
Спаривание оснований — присоединение одного полинуклеотида к другому или
одной части полинуклеотида к другой части того же самого полинуклеотида за
счет спаривания оснований.
Спектроскопия ядерного магнитного резонанса (ЯМР) — метод определения трех-
мерной структуры крупных молекул.
Словарь терминов 861
Специфичная ктранскриптам регуляция — регуляторные механизмы, которые управ-
ляют синтезом белка, воздействуя на отдельный транскрипт или на малую группу
транскриптов, кодирующих соответствующие белки.
Сплайсосома — белково-РНКовый комплекс, участвующий в вырезании интронов
GU-AG или AU-AC.
Сателлитная ДНК — повторная ДНК, которая образует сопутствующую полосу в гра-
диенте плотности.
Сателлитная РНК — молекула РНК около 320-400 нуклеотидов в длину, которая не
кодирует свои собственные белки капсида, а вместо этого переселяется из клетки
в клетку в капсиде вируса-помощника.
Сравнительная геномика — стратегия исследования, использующая информацию,
полученную в итоге изучения одного генома, для построения выводов о позициях
на карте и функциях генов во втором геноме.
Сплайсинг (сращивание) — удаление интронов из первичного транскрипта прерыв-
ного гена.
Сплайсированная лидерная РНК (слРНК) — транскрипт, который отдает ведущий
сегмент нескольким РНК путем транссращивания.
Старение клетки — период в последовательности клеточных поколений, когда клетки
еще живут, но уже не способны делиться.
Старт-кодон — кодон, обычно, хотя и не исключительно, 5’-AUG-3', расположенный
в начале кодирующей области гена.
Стволовая клетка — клетка-предшественник, которая непрерывно делится на про-
тяжении всей жизни организма.
Стекинг оснований — гидрофобные взаимодействия, которые происходят между
смежными парами оснований в молекуле двунитевой ДНК.
Стержневой октамер — центральный компонент нуклеосомы, состоящий из двух
субъединиц, каждая из которых, в свою очередь, состоит из гистонов Н2А, Н2В, НЗ
и Н4, вокруг которого обвита ДНК.
Стероидный гормон — разновидность внеклеточного сигнального соединения.
Стоп-кодон — один из трех кодонов, которые отмечают позицию, в которой трансляция
мРНК должна быть завершена.
Стоп-последовательность — одна из нескольких последовательностей в бактериаль-
ном геноме, задействованная в завершении репликации генома.
Структура стебли и петли (шпилька) — структура, состоящая из спаренного основа-
ниями стебля и не спаренной основаниями петли, которая может образоваться
в однонитевом полинуклеотиде, который содержит инвертированный повтор.
Структурное управление — управление экспрессией генов у бактерий, которое опреде-
ляется последовательностью промотора.
Структурный гетерохроматин — хроматин, который постоянно находится в компакт-
ной организации.
Структурный домен — сегмент полипептида, который сворачивается независимо от
других сегментов. Также петля ДНК эукариотов, преимущественно в форме 30-нм
хроматинового волокна, прикрепленная к ядерному матриксу.
СУМО — белок, связанный с убиквитином.
Сумка — структура, в которой содержится четыре сумкоспоры, произведенные одинар-
ным мейозом в дрожжах Saccharomyces cerevisiae.
862 Словарь терминов
Сумкоспора — один из гаплоидных продуктов мейоза в сумчатом грибе, например,
дрожжей Saccharomyces cerevisiae.
Супрессорная мутация — мутация в одном гене, которая изменяет действие мутации
во втором гене на противоположное.
Сцепка — молекула ДНК, составленная из линейных геномов или иных единиц ДНК,
сцепленных по принципу «голова к хвосту».
Сцепление генов — физическая связь между двумя генами, которые находятся на
одной и той же хромосоме.
Считывания спаренных концов — минипоследовательности с двух концов одного
клонированного фрагмента.
Счет ЛОШ — логарифм отношения шансов — статистическая мера сцепленности на
основе анализа родословной (и не только. — Прим. ред.).
тРНК-нуклеотидилтрансфераза — фермент, отвечающий за посттранскрипционное
присоединение триплета 5'-ССА-3' к З'-концу молекулы тРНК.
Тандемно повторяющаяся ДНК — мотивы последовательности ДНК, которые повто-
ряются по принципу «голова к хвосту».
Тандемное повторение — прямые повторения, которые примыкают друг к другу.
Таутомерный сдвиг — самопроизвольный переход молекулы от одного структурного
изомера к другому.
Таутомеры — структурные изомеры, которые находятся в динамическом равнове-
сии.
Твинтрон — составная структура, состоящая из двух и более интронов II и (или) III
группы, вложенных друг в друга.
Теломера — конец хромосомы эукариотов.
Теломераза — фермент, который поддерживает концы хромосом эукариотов, синтезируя
теломерные повторные последовательности.
Тельце Барра — структура плотноупакованного хроматина, принимаемая инактиви-
рованной Х-хромосомой.
Темновое восстановление — один из процессов восстановления вырезанием нуклео-
тидов, который устраняет циклобутильные димеры.
Температура плавления (Тт) — температура, при которой две нити молекулы двуни-
тевой нуклеиновой кислоты или спаренного основаниями гибрида отделяются
друг от друга в результате полного разрыва водородных связей.
Теория эндосимбионтов — теория, согласно которой митохондрии и хлоропласты эу-
кариотов ядерных клеток происходят от симбиотических доядерных бактерий.
Термостабильный — способный выдерживать высокие температуры.
Территория хромосомы — область ядра, занятая одной хромосомой.
Технология рекомбинантной ДНК — совокупность методов конструирования, изуче-
ния и использования молекул рекомбинантной ДНК.
Тимин — одно из пиримидиновых оснований, входящих в состав ДНК.
Тип спаривания — эквивалент самца и самки для ядерных микроорганизмов.
Топология — схема ветвления филогенетического дерева.
Точечная матрица — метод выравнивания последовательностей нуклеотидов.
Точечная мутация — мутация, которая возникает в результате изменения одного
нуклеотида в молекуле ДНК.
Словарь терминов 863
Транзиция (переход) — точечная мутация, в результате которой пурин заменяется
другим пурином или пиримидин — другим пиримидином.
Трансверсия (перебег) — точечная мутация, в результате которой пуриновое основание
заменяется пиримидиновым или наоборот.
Трансгенная мышь — мышь, которая несет в себе клонированный ген.
Трансдукционная картирование — использование трансдукции для нанесения на
карту относительных позиций генов в бактериальном геноме.
Трансдукция — передача бактериальных генов из одной клетки в другую путем упа-
ковки в фаговую частицу.
Транскрипт — РНК-копия гена.
Транскриптом — полный состав молекул мРНК клетки.
Транскрипционная фабрика — крупная структура, скрепленная с ядерным матриксом;
участок синтеза РНК.
Транскрипционный пузырек — не спаренная основаниями область двойной спирали,
поддерживаемая РНК-полимеразой, в пределах которой происходит транскрипция.
Транскрипция — синтез РНК-копии гена.
Транслокация — присоединение сегмента одной хромосомы к другой хромосоме. Также
передвижение рибосомы по молекуле мРНК в процессе трансляции.
Трансляционный обход — форма проскальзывания, при котором большая часть мРНК
пропускается в ходе трансляции, при этом за событием обхода следует продолже-
ние первичного белка.
Трансляция — синтез полипептида, последовательность аминокислот которого опреде-
ляется нуклеотидной последовательностью мРНК в соответствии с правилами
генетического кода.
Транспозаза — фермент, который катализирует транспозицию подвижного генетиче-
ского элемента.
Транспозиция — перемещение генетического элемента с одного участка молекулы
ДНК на другой.
Транспозон ТпЗ-типа — разновидность ДНК-транспозона, который не ограждает себя
вставными последовательностями.
Транспозон ДНК — транспозон, механизм транспозиции которого не вовлекает РНК-
посредника.
Транспозон — подвижной генетический элемент, который может перемещаться из
одной позиции молекулы ДНК в другую.
Транспортная РНК (тРНК) — маленькая молекула РНК, которая служит сопрягающим
звеном в ходе трансляции и отвечает за расшифровку генетического кода.
Транспортно-матричная РНК (тмРНК) — бактериальная РНК, участвующая в дегра-
дации белка.
Транссмещение — перемещение нуклеосомы из одной молекулы ДНК в другую.
Транссплайсинг — сплайсинг между экзонами, которые содержатся в разных моле-
кулах РНК.
Трансфекция — введение очищенных молекул ДНК фага в клетку бактерии.
Трансформант — клетка, которая была трансформирована введением в нее голой
ДНК.
Трансформационное картирование — использование трансформации для нанесения
на карту относительных позиций генов в геноме бактерии.
864 Словарь терминов
Трансформация клетки — видоизменение морфологических и биохимических свойств,
которое происходит, когда животная клетка заражается опухолеродным виру-
сом.
Трансформация — приобретение клеткой новых генов за счет введения в нее голой
ДНК.
Трансформирующее начало — соединение (как теперь установлено, представляю-
щее собой ДНК), обусловливающее трансформацию невирулентной бактерии
Streptococcus pneumoniae в вирулентную форму.
Третичная структура — структура, получающаяся в результате сворачивания единиц
вторичной структуры полипептида.
Триплекс — структура ДНК, состоящая из трех полинуклеотидов.
Трисомия — наличие трех копий некоторой гомологичной хромосомы в ядре, дипло-
идном по всем остальным хромосомам.
Тупой конец — конец молекулы двунитевой ДНК, которым обе нити оканчиваются
в одной и той же нуклеотидной позиции без какого-либо однонитевого продол-
жения.
Тысяча пар нуклеотидов (тыс. п. н.) — 1 000 п. н.
Убиквитин — состоящий из 76 аминокислот белок, который, будучи присоединен
к какому-либо другому белку, служит ярлыком, направляющим тот белок на де-
градацию.
Уловление экзонов — основанный на клонировании метод опознавания позиций эк-
зонов в последовательности ДНК.
Умеренный бактериофаг — бактериофаг, который способен следовать лизогенным
путем инфицирования.
Унаследованное состояние признака — состояние признака, присущее дальнему
общему предку группы организмов.
Упаковка in vitro — синтез инфекционных фагов X из препарата белков фага X и сцепки
молекул ДНК фага X.
Управляющая локусом область (УЛО) — последовательность ДНК, которая поддержи-
вает функциональный домен в раскрытой активной конфигурации.
Урацил — одно из пиримидиновых оснований, входящих в состав РНК.
Усеченный ген — остаток гена, в котором отсутствует сегмент с одного конца исходного
полного гена.
Условно летальная мутация — мутация, в результате которой клетка или организм
способны выживать только при разрешающих условиях.
Устойчивый к ингибитору мутант — мутант, который способен противостоять ядо-
витым воздействиям антибиотика или ингибитора иного вида.
Устранение двунитевых разрывов — процесс репарации ДНК, который исправляет
двунитевые разрывы.
Устранение несоответствий — процесс репарации ДНК, который исправляет несоот-
ветствующие пары нуклеотидов, заменяя неправильный нуклеотид в дочернем
полинуклеотиде на правильный.
Уход от промотора — стадия транскрипции, во время которой полимераза отодвигается
от промоторной области и предается созданию транскрипта.
Участок cos — одно из липких, однонитевых продолжений, находящихся на концах
молекул ДНК некоторых штаммов фага X.
Словарь терминов 865
Участок САР — участок связывания с ДНК для белка-активатора катаболитных опе-
ронов.
Участок выхода (Е-сайт) — позиция в пределах бактериальной рибосомы, в которую
тРНК перемещается сразу после дезацилирования.
Участок посадки рибосомы — последовательность нуклеотидов, которая служит
участком прикрепления для малой субъединицы рибосомы в момент инициации
трансляции у бактерий.
Учредительная мутация — мутация, в результате которой устанавливается непре-
рывная экспрессия гена или набора генов, которые обычно находятся под регу-
ляцией.
фМет—N-формилметионин, переносимая тРНК видоизмененная аминокислота, которая
используется во время запуска трансляции у бактерий.
Фаг — вирус, который заражает бактерию.
Фаг-помощник — фаг, который вводится в клетку-хозяина совместно со связанным
клонирующим вектором с целью обеспечения ферментов и прочих белков, необ-
ходимых для репликации клонирующего вектора.
Фаг-траспозон — бактериофаг, у которого в цикл инфицирования входит событие
транспозиции.
Фагмида — клонирующий вектор, представляющий собой смесь ДНК плазмиды
и фага.
Фаговый дисплей (выставление на фаге) — методика опознавания белков, взаимо-
действующих друг с другом.
Фаза G1 — первый промежуточный период клеточного цикла.
Фаза G2 — второй промежуточный период клеточного цикла.
Фаза S — фаза клеточного цикла, в которую происходит синтез ДНК.
Фактор высвобождения — белок, который играет вспомогательную роль во время
терминации трансляции.
Фактор терминации — белок, который играет вспомогательную роль в терминации
транскрипции.
Фактор инициации — белок, который играет вспомогательную роль в процессе ини-
циации трансляции.
Фактор повторного использования рибосомы (RRF) — белок, отвечающий за раз-
борку рибосомы в конце синтеза белка у бактерий.
Фактор элонгации — белок, который играет вспомогательную роль на стадии элонга-
ции процессов транскрипции или трансляции.
Фактор репликации С (RFC) — многосубъединичный дополнительный белок, уча-
ствующий в репликации генома эукариотов.
Фактор специфичности расщепления и полиаденилирования (CPSF) — белок,
который играет вспомогательную роль в процессе полиаденилирования мРНК
эукариотов.
Фактор стимуляции расщепления (CstF) — белок, который играет вспомогательную
роль в процессе полиаденилирования мРНК эукариотов.
Факторы лицензирования репликации (RLF) — набор белков, которые регулируют
репликацию генома, в частности, дают добро лишь на один круг репликации генома
за один клеточный цикл.
Факультативный гетерохроматин — хроматин, который имеет компактную органи-
зацию в некоторых, но не во всех клетках и, как думают, содержит гены, которые
866 Словарь терминов
являются неактивными в некоторых клетках или в некоторые периоды клеточного
цикла.
Фенетика — классификационный подход, основанный на числовой типизации как
можно большего числа признаков.
Феномен «бутылочного горлышка» — временное сокращение размера популяции
вида.
Фенотип — заметные глазу или прибору характеристики, проявляемые клеткой или
организмом.
Фермент RecBCD — ферментативный комплекс, участвующий в гомологичной реком-
бинации у Escherichia coli.
Фермент Ада (Ada) — фермент Escherichia coli, который участвует в прямом устранении
алкилационных мутаций.
Фермент модификации концов — фермент, применяемый в технологии рекомбинантной
ДНК, который видоизменяет химическую структуру на конце молекулы ДНК.
Физическое картирование — применение методов молекулярной биологии для по-
строения карты генома.
Фиксация — относится к ситуации, которая происходит, когда отдельный аллель до-
стигает частотности 100 % в рассматриваемой популяции.
Филадельфийскаяхромосома — ненормальная хромосома, получающаяся в результате
транслокации между хромосомами 9 и 22 человека, обычная причина хронического
миелоидного лейкоза.
Филогения — классификационная схема, которая показывает эволюционные отноше-
ния между организмами.
Флюоресцентная гибридизация на месте (FISH) — технология для определения ме-
стонахождения маркёров на хромосомах путем зрительного обнаружения позиций
гибридизации флюоресцентных меток.
Фосмида — высокоемкий вектор, несущий в себе участок инициации репликации
F-плазмиды и участок cos бактериофага X.
Фосфодиэстераза — фермент из рода способных разрывать фосфодиэфирные связи.
Фосфодиэфирная связь — химическая связь между смежными нуклеотидами в по-
линуклеотиде.
Фосфоревидение — электронный метод определения позиций радиоактивных мар-
кёров в микроматрице или на подложке для гибридизации.
Фотолиаза — фермент Escherichia coli, участвующий в восстановлении фотореактива-
цией.
Фотолиаза фотопродукта (6-4) — фермент, участвующий в восстановлении фото-
реактивацией.
Фотолитография — методика, что использует импульсы света, чтобы строить олиго-
нуклеотид из активируемых светом нуклеотидных субстратов.
Фотоотбеливание — составляющая методики FRAP для изучения подвижности белка
в ядре.
Фотопродукт — видоизмененный нуклеотид, получающийся в результате обработки
ДНК ультрафиолетовым излучением.
Фотореактивация — процесс репарации ДНК, в котором циклобутильные димеры
и фотопродукты (6-4) исправляются активируемым светом ферментом.
Фрагмент Окадзаки — один из коротких затравляемых РНК сегментов ДНК, синтези-
руемых в ходе репликации отстающей нити двойной спирали.
Словарь терминов 867
Фрагмент гена — остаток гена, состоящий из короткой области, выделенной из общей
структуры гена.
Функциональная РНК — РНК, которая играет функциональную роль в клетке, то есть
РНК всех видов, кроме мРНК.
Функциональный анализ — область исследования генома, посвященная установлению
функций неизвестных генов.
Функциональный домен — область ДНК эукариотов, которая расположена вокруг
гена или группы генов и может быть определена при помощи обработки дезок-
сирибонуклеазой I.
Футпринтинг — совокупность методов, используемых для определения местоположе-
ния связанных белков на молекулах ДНК.
Хаб — белок, который имеет множество взаимодействий на карте межбелковых взаи-
модействий.
Химера — организм, состоящий из клеток двух или нескольких генетически разных
типов.
Химический сдвиг — изменение во вращении химического ядра, используемое как
основа ЯМР.
Хлоропласт — одна из фотосинтезирующих органелл ядерной клетки.
Хлоропластный геном — геном, заложенный в хлоропласты фотосинтезирующей
ядерной клетки.
Холлидея структура — промежуточная структура, образуемая в процессе рекомбинации
между двумя молекулами ДНК.
Хроматида — плечо хромосомы.
Хроматин — комплекс ДНК и гистоновых белков, обнаруживаемый в хромосомах.
Хроматосома — подкомпонент хроматина, состоящий из стержневого октамера ну-
клеосомы со связанной ДНК и связующими гистонами.
Хромосома — одна из ДНК-белковых структур, которая содержит часть ядерного генома
эукариота. Менее точно, молекула (одна или несколько) ДНК, в которой содержится
геном прокариота.
Хромосомная теория — теория, впервые выдвинутая Саттоном в 1903 г., согласно
которой гены расположены в хромосомах.
Хромосомный каркас — компонент ядерного матрикса, который изменяет свою струк-
туру во время деления клетки, что приводит к уплотнению хромосом в их мета-
фазные формы.
Хрупкий участок — позиция в хромосоме, которая подвержена разрывам, потому что
она содержит расширенную последовательность тринуклеотидных повторений.
Центральный промотор — позиция в пределах промотора эукариотов, в которой со-
бирается инициаторный комплекс.
Центрифугирование в градиенте плотности — методика, в которой клеточная фрак-
ция центрифугируется в плотном растворе в форме градиента, так что отдельные
компоненты разделяются.
Центромера — суженная (перетянутая) область хромосомы, за счет которой пара хро-
матид скрепляется вместе.
Цианелла — фотосинтетическая органелла, которая напоминает поглощенную циа-
нобактерию.
868 Словарь терминов
Циклин — регуляторный белок, относительное содержание которого изменяется в тече-
ние клеточного цикла и который регулирует биохимические события специфично
к клеточному циклу.
Циклический АМФ (цАМФ) — видоизмененная версия АМФ, в которой внутримолеку-
лярная фосфодиэфирная связь соединяет 5'- и З’-атомы углерода.
Циклобутильный димер — димер между двумя смежными пиримидиновыми осно-
ваниями в полинуклеотиде, образуемый при воздействии ультрафиолетового
излучения.
Цинковый палец — распространенный структурный мотив для прикрепления белка
к молекуле ДНК.
Цитозин — одно из пиримидиновых оснований, входящих в состав ДНК и РНК.
Цитохимия — использование специфичных к соединениям красителей в сочетании
с микроскопией, чтобы определить биохимическое содержание клеточных струк-
тур.
Частота рекомбинации — доля рекомбинантного потомства, полученного в результате
генетического скрещивания.
Частотность аллеля — частота появления аллеля в популяции.
Челночный вектор — вектор, который способен реплицироваться в клетках более чем
одного организма (например, Escherichia coli и дрожжей).
Четвертичная структура — структура, возникающая в результате объединения двух
и более полипептидов.
Чип ДНК — высокоплотная матрица молекул ДНК, используемая для параллельного
гибридизационного анализа.
Чувствительная к температуре мутация — условно летальная мутация, которая про-
является только выше определенной пороговой температуры.
Чувствительный к гормонам элемент — последовательность нуклеотидов, располо-
женная выше гена, которая опосредствует регуляторное воздействие стероидного
гормона.
Шаперон Hsp70 — представитель семейства белков, которые связываются с гидрофоб-
ными областями на других белках, с тем чтобы помочь их сворачиванию.
Шаперонин — многосубъединичный белок, образующий структуру, которая помогает
сворачиванию других белков.
Шпилька — структура, состоящая из спаренного основаниями стебля и не спаренной
основаниями петли, которая может формироваться в однонитевом полинуклеотиде,
содержащем инвертированный повтор.
Щелочная фосфатаза — фермент, который удаляет фосфатные группы с 5'-концов
молекул ДНК.
Эгоистичная ДНК — ДНК, которая, кажется, не имеет никакой функции и, очевидно,
ничего не дает клетке, в которой она находится.
Экзон — кодирующая область в пределах прерывного гена.
Экзон-интронная граница — последовательность нуклеотидов на стыке между экзо-
ном и интроном.
Экзонная теория генов — гипотеза «о ранних интронах», которая утверждает, что
интроны образовались тогда, когда были построены первые геномы из ДНК.
Экзонный сайленсер (глушитель) сплайсинга — последовательность нуклеотидов,
которая играет отрицательную регуляторную роль во время вырезания интронов
GU-AG.
Словарь терминов 869
Экзонный энхансер (усилитель ) сплайсинга — последовательность нуклеотидов,
которая играет положительную регуляторную роль во время вырезания интронов
GU-AG.
Экзонуклеаза — фермент, который удаляет нуклеотиды с концов молекулы нуклеи-
новой кислоты.
Экзосома — многобелковый комплекс, вовлеченный в деградацию мРНК у эукарио-
тов.
Эндонуклеаза UvrABC — многоферментный комплекс, участвующий в процессе вос-
становления наложением коротких заплат у Escherichia coli.
Энхансер (усилитель) — регуляторная последовательность, которая увеличивает
скорость транскрипции гена или генов, расположенных на некотором расстоянии
поодаль в том или ином направлении.
Эксперимент Мезельсона-Шталя — эксперимент, который показал, что репликация
ДНК в клетке происходит полуконсервативным способом.
Эксперименты с защитой от нуклеазы — метод, в котором используется переварива-
ние нуклеазами, предназначенный для определения позиций белков на молекулах
РНК или ДНК.
Экспортин — белок, вовлеченный в перенос молекул из ядра в цитоплазму.
Экспрессия гена — ряд событий, которым биологическая информация, заложенная
в гене, высвобождается и становится доступной клетке.
Экспрессия генома — ряд событий, в ходе которых биологическая информация, зало-
женная в геноме, высвобождается и становится доступной клетке.
Экспрессионная протеомика — методология, используемая для опознавания белков
в протеоме.
Экстейн — функциональный компонент прерывного белка.
Электростатические взаимодействия — ионные связи, которые образуются между
заряженными химическими группами.
Электрофорез белков — разделение белков в электрофоретическом геле.
Электрофорез в геле с ограниченными контуром однородными электрическими
полями (CHEF) — электрофоретический метод, применяемый для разделения
крупных молекул ДНК.
Электрофорез в агарозном геле — электрофорез, выполняемый в агарозном геле
и применяемый для разделения молекул ДНК длиной от 100 п. н. до 50 тыс. п. н.
Электрофорез в геле с обращением поля (FIGE) — электрофоретический метод, при-
меняемый для разделения крупных молекул ДНК.
Электрофорез в геле с ортогонально чередующимся полем (OFAGE) — электро-
форетическая система, в которой поле чередуется между парами электродов, рас-
положенных под углом 45° относительно основного направления, применяемая
для разделения крупных молекул ДНК.
Электрофорез в полиакриламидном геле — электрофорез, выполняемый в полиа-
криламидном геле и применяемый для разделения молекул ДНК длиной от 10 до
1500 п.н.
Электрофорез — метод разделения молекул на основе их результирующего электри-
ческого заряда.
Элемент LTR — разновидность рассеянного по всему геному повторения с характер-
ными длинными концевыми повторами (LTR).
870 Словарь терминов
Элемент неустойчивости — последовательность, которая находится в молекулах мРНК
дрожжей и влияет на их деградацию.
Элемент управления импринтами — последовательность ДНК, встречающаяся в преде-
лах нескольких тыс. п. н. возле групп генов, находящихся под импринтингом, которая
управляет метилированием импринтируемых областей.
Элюирование — вымывание молекулы из хроматографической колонки.
Эндогенный ретровирус (ЭРВ) — активный или неактивный ретровирусный геном,
встроенный в хромосому хозяина.
Эндонуклеаза рестрикции — фермент, который разрезает молекулы ДНК в ограни-
ченном числе специфических последовательностей нуклеотидов.
Эндонуклеаза — фермент, который разрывает фосфодиэфирные связи во внутренних
позициях молекулы нуклеиновой кислоты.
Энхансосома — структура, образованная за счет изгибания ДНК и состоящая из со-
брания белков, вовлеченных в активацию комплекса инициации транскрипции
для РНК-полимеразы II.
Эписома — плазмида, которая способна встраиваться в хромосому клетки-хозяина.
Этилметансульфонат (ЭМС) — мутаген, который действует добавлением алкильных
групп к основаниям нуклеотидов.
Эукариот — организм, клетки которого содержат заключенные в мембрану ядра.
Эухроматин — области хромосомы эукариотов, которые относительно разуплотнены
и, как думают, содержат активные гены.
Ядерный антиген разрастающихся клеток (PCNA) — дополнительный белок, уча-
ствующий в репликация генома у эукариотов.
Ядерный геном — набор молекул ДНК, находящихся в ядре клетки эукариота.
Ядерный матрикс — белковоподобная напоминающая каркас сеть, которая пронизы-
вает ядро.
Ядерный поровый комплекс — комплекс белков, расположенный в ядерной поре.
Ядро — заключенная в мембрану структура клеток эукариотов, в которой находятся
хромосомы.
Ядрышко — область ядра клетки эукариотов, в которой происходит транскрипция
рРНК.
Ярлык экспрессируемой последовательности (EST) — кДНК, которая секвенируется,
чтобы получить быстрый доступ к генам в геноме.
Благодарности
«Геномы» написаны со ссылками на ранее изданную литературу и опираются на
работы многих научно-исследовательских групп. Если рисунок был воспроизведен не-
посредственно из другого источника, автор упоминал об этом в подписи к нему. Кроме
этого, автор желает выразить признательность за различные рисунки, которые были
перерисованы или созданы с опорой на информацию, приводимую в нижеследующих
книгах, статьях и ресурсах Интернета.
Рисунок 3.25: Oliver,S.G., vanderAart,Q.J.M., Agostini-Carbone, М. L., et al. (1992) The
complete DNA sequence of yeast chromosome III. Nature 357: 38-46.
Рисунок 5.19: Ponting, C.P. (1997) Tudor domains in proteins that interact with RNA.
Trends Biochem. Sci. 22: 51-52.
Рисунок 5.28: Dujon,B. (1996) The yeast genome project: what did we learn? Trends
Genet 12:263-270.
Рисунок 6.7: Leung, Y. F. and Cavalieri, D. (2003) Fundamentals of cDNA microarray data
analysis. Trends Genet 19: 649-659.
Рисунок 6.23: Kalir,S. and Alon,U. (2004) Using a quantitative blueprint to reprogram
the dynamics of the flagella gene network. Cell 117: 713-720.
Рисунок 7.5: Strachan, T. and Read, A. P. (2004) Human Molecular Genetics, 3rd Ed. Garland,
London.
Рисунок 7.12: Запись «Генбанка» HUMHBB.
Рисунок 7.15,6: Oliver, S. G., van der Aart, Q. J. M., Agostini-Carbone, M. L., et al. (1992) The
complete DNA sequence of yeast chromosome III. Nature 357: 38-46.
Рисунок 7.15, в: Adams, M. A., Celniker, S. E., Holt, R. A., etal. (2000) The genome sequence
of Drosophila melanogaster. Science 287: 2185-2195.
Рисунок 7.15,a: SanMiguel, P., Tikhonov, A., Jin, Y.-K., etal. (1996) Nested retrotransposons
in the intergenic regions of the maize genome. Science 274: 765-768.
Рисунок 7.16: Venter, J. C., Adams, M.D., Myers, E.W., et al. (2001) The sequence of the
human genome. Science 291:1304-1351.
Рисунок 7.17: МКСГЧ (Международный консорциум по секвенированию генома че-
ловека) (2001) Initial sequencing and analysis of the human genome. Nature 409: 860-921.
Рисунок 7.18: МКСГЧ (Международный консорциум по секвенированию генома че-
ловека) (2001) Initial sequencing and analysis of the human genome. Nature 409: 860-921.
Рисунок 8.4: Sinden,R.R. and Pettijohn,D.E. (1981) Chromosomes in living Escherichia
coli cells are segregated into domains of supercoiling. Proc. Natl Acad. Sci. USA 78: 224-228.
Рисунок 8.7: Blattner, F. R., Plunkett,G., Bloch,C. A., et al. (1997) The complete genome
sequence of Escherichia coli K-12. Science 277:1453-1462.
872 Благодарности
Рисунок 8.10: Ochman, Н., Lawrence,). G. and Groisman, E. A. (2000) Lateral gene transfer
and the nature of bacterial innovation. Nature 405: 299-304.
Рисунок 10.4: Williams, R.R.E. (2003) Transcription and the territory: the ins and outs
of gene positioning. Trends Genet 19: 298-302.
Рисунок 10.15: Bernstein, В. E., Kamal, M., Lindblad-Toh,K., et al. (2005) Genomic maps
and comparative analysis of histone modifications in human and mouse. Cell 120:169-181.
Рисунок 11.2: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Техническое примечание 11.1, рисунок Т11.1, части б & в: Campbell, N. (1993) Biology,
2nd Ed. Benjamin Cummings, San Francisco, CA.
Техническое примечание 11.1, Рисунок Т11.1, часть г: Zubay,G. (1997) Biochemistry,
4th Ed. McGraw-Hill, New York, NY.
Рисунок 11.3: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Рисунок 11.4: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Рисунок 11.5: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Рисунок 11.6: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Рисунок 11.12: Kielkopf, C. L., White, S., Szewczyk, J. W., Turner, J. M., Baird, E. E., Dervan, P. B.
and Rees, D. C. (1998) A structural basis for recognition of AT and TA base pairs in the minor
groove of B-DNA. Science 282:111-115.
Рисунок 11.13: Travers, A. (1993) DNA-Protein Interactions. Chapman & Hall, London.
Рисунок 11.20: Xie,X., Kokubo,T., Cohen, S.L., et al. (1996) Structural similarity between
TAFs and the heterotetrameric core of the histone octamer. Nature 380: 316-322.
Рисунок 12.3: Korzheva, N., Mustaev, A., Kozlov, M., Malhotra, A., Nikiforov, V., Goldfarb, A.
and Darst,S. A. (2000) A structural model of transcription elongation. Science 289: 619-625.
Рисунок 12.14: Nickels, В. E. and Hochschild,A. (2004) Regulation of RNA polymerase
through the secondary channel. Cell 118: 281-284.
Рисунок 12.29: Stark, H., Dube,P., Liihrmann,R. and Kastner, B. (2001) Arrangement of
RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature
409: 539-542.
Рисунок 12.34: Graveley, В. R. (2001) Alternative splicing: increasing diversity in the
proteomic world. Trends Genet. 17:100-107.
Рисунок 12.39: Burke,J. M., Belfort,M., Cech,T. R., etal. (1987) Structural conventions for
Group I introns. Nucleic. Acids Res. 15: 7217-7221.
Рисунок 13.3: Freifelder,D. (1986) Molecular Biology, 2nd Ed. Jones and Bartlett, Sudbury, MA.
Рисунок 13.12: Heilek, G. M. and Noller, H. F. (1996) Site-directed hydroxyl radical probing
of the pPHK neighborhood of ribosomal protein S5. Science 272:1659-1662.
Рисунок 13.20: Nissen,P., Hansen,)., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The
structural basis of ribosome activity in peptide bond synthesis. Science 289: 920-930.
Благодарности 873
Рисунок 15.33: Raghuraman, М. К., Winzeler,E. A., Collingwood, D., etal. (2001) Replication
dynamics of the yeast genome. Science 294:115-121.
Рисунок 17.8: Rafferty, J. B., Sedelnikova, S. E., Hargreaves, D., etal. (1996) Crystal structure
of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science
274:415-421.
Рисунок 18.15: Wolfe,К. H. and Shields, D.C. (1997) Molecular evidence for an ancient
duplication of the entire yeast genome. Nature 387: 708-713.
Рисунок 18.16: Eichler, E.E. (2001) Recent duplicate domain accretion and dynamic
mutation of the human genome. Trends Genet. 17: 661-669.
Рисунок 18.21: Jiang, N., Bao, Z., Zhang, X., Eddy, S. R. and Wessler, S. R. (2004) Pack-MULE
transposable elements mediate gene evolution in plants. Nature 431: 569-573.
Рисунок 19.6: Strachan,T. and Read, A. P. (2004) Human Molecular Genetics, 3rd Ed.
Garland, London.
Рисунок 19.8: Li,W.-H. (1997) Molecular Evolution. Sinauer, Sunderland, MA.
Рисунок 19.9: Li,W.-H. (1997) Molecular Evolution. Sinauer, Sunderland, MA.
Рисунок 19.20: Wain-Hobson,S. (1998) 1959 and all that. Nature 391: 531-532.
Рисунок 19.30: Richards, M., Macauley,V., Hickey, E., et al. (2000) Tracing European
founder lineages in the Near Eastern mtDNA pool. Am. J. Hum. Genet 67:1251-1276.
Рисунок 19.31: Jobling, M. A., Hurles, M. E. and Tyler-Smith, C. (2004) Human Evolutionary
Genetics: Origins, Peoples and Disease. Garland, London.
Предметный указатель
а-спирали
- связывающиеся с ДНК белки, 381, 386-
388, 397
- структура, 21-22р
а-субъединица РНК-полимеразы бактерий,
431, 435р
P-N-гликозидная связь, 8,
Р-галактозидаза, 56р, 59р, 242, 665
Р-изгиб, 381, 386,
Р-лист, 21-22, 22р, 387, 388,496т,
Р-полипептид, 435р
Р-субъединица РНК-полимеразы бактерий,
431р, 432р, 433,435р
Р-талассемия, 658
Р-цилиндровый димер, 386т
Р’-субъединица РНК-полимеразы бактерий,
431р, 432р, 435
/-участки, 693, 694р, 696, 697
/-форма, 689, 690р
6-вирус гепатита, 468т
^-последовательности, 331, 331р
/-комплекс, 614,
Х-бактериофаг, см. бактериофаг X
л-л-взаимодействия, 12,
а-субъединицы РНК-полимеразы бактерий,
402,409р, 572р, 569-573, 573р, 575р
е-глобин, 281р, 281-282
1-(о-карбоксифениламино)-Г-
дезоксирибулозо-5'-фосфат, 247
2'-дезоксирибоза, 8
2-аминопурин, 651,
2-метилцитозин, 669
2-мкм кольцо, 68,
З’-ОН-конец, 9р,
З’-RACE, 190р
З’-НТО, 27,
З’-трансдукция, 733-734,
3-метиладенин, 669, 670т
4-тиоуридин, 443р
5’-Р-конец, 9р,
5’-RACE, 190р
5’-НТО, 510р,
5-бромо-4-хлоро-3-индолил-р-В-
галактопиранозид (Х-гель), 59р
5-бромоурацил, 651р,
5-гидроксиурацил, 670т
5-гидроксицитозин, 669
5-метилцитозин, 116р, 367
7-метилгуанин, 669
7-метилгуанозин, 443р, 448р, 449
8-оксогуанин, 670т
9-1-1-белок, 632р
12 п. н. ярлыки последовательности, 224р
30-нм хроматиновое волокно, 261, 262р
А-ДНК, 15,15т, 16р, 394
А-топоизомеразы, 600
A-сайт, 512р, 513р, 512-514, 514р, 518
ABF1 (фактор связывания ARS 1), 606р
ADAR (действующие на РНК аденозиндеами-
назы), 474,
Aegilops squarrosa, 735
Aeropyrum pernix, 307p
Agrobacterium tumefaciens, 70p, 296, 297т
А/п-ПЦР, 157,
Anabaena, 466
Antirhinum, 582-583
Aquifex aeolicus, 302p, 302-303, 307p
Arabidopsis thaliana
- геномы органелл, 308-309
- гомеозисные избирательные гены, 582,583
- каталоги генов, 278р, 279т
- митохондриальные геномы, 310т, 311
- размер генома, 272т, 275т
- распределение генов, 267р
- свидетельства эволюции, 729
- транспозиция, 707
- ферменты Dicer, 479
- центромеры, 265
- число генов, 275т
Archaeoglobusfiilgidus, 303т, 307р
Argonaut, 478
ARS (автономно реплицирующиеся системы),
605, 606р
Ashby a gossypii, 729
Aspergillus nidulans, 272т, 310т
Australopithecus, 740, 740р
В-ДНК, 13р, 15т, 16р, 394
В-лимфоциты, 474т, 560-563
В-топоизомеразы, 600
В-форма ДНК, 394, 396р
В-хромосомы, 265,
Bacillus, 409
- В. anthracis, 569
- В. megaterium, 497р
- В. subtilis
Предметный указатель 875
- - бактериофаги, 322,323т
- - горизонтальный перенос генов, 307р
- - оперон триптофана, 438,439р
- -опероны, 302
- - подавление катаболитных оперонов,
548
- - спорообразование, 571р, 572р,
569-573,573р, 575р
- - сравнение с геномом М. genitalium, 304
ВАС (бактериальные искусственные хромосо-
мы), 64т, 66,117,153-154,162
BamHI, 48,48р, 53р, 53-55, 55р, 67р, 112, ИЗр
BLAST (основное средство поиска локальных
выравниваний), 193
Bombyx mori, 272т
Borrelia burgdorferi, 296,297т, 298,307р
Brassica oleracea, 310т
ВХ-С (комплекс Bithorax), 580-581, 581р
С-КД (С-концевой домен), 35,405,447,448,
451,454, 550р, 555
С-реакция, 147
С-конец, 21, 22р
С-концевой домен (С-КД), 405,447,448,451,454
Caenorhabditis elegans, 243
Campylobacter jejuni NCTC11168,301
CAP (белок-активатор катаболитных оперо-
нов), 411, 550p, 548-551
CASPs (связанные c CDT SR-подобные белки),
460
CCdA-фосфатпуромицин, 515p
CENP-A, 266,266p
CHEF (электрофорез в геле с ограниченными
контуром однородными электрическими
полями), 116
Chlamydomonas reinhardtii, 308,309, 310т, 311
Chondrus crispus, 310т
COSY, 384тп
CpG-островки, 180, 368-369, 369р,
CPSF (фактор специфичности расщепления и
полиаденилирования), 450-451,451р
CRE (модуль реакции на циклический АМФ),
414
CREB, 414, 555,
CstF (фактор стимуляции расщепления), 450,
451,451р
Cucumis melo, 310т
Cyanophora paradoxa, 308
D-петли
- рекомбинация, 690,692р, 694, 694р
- репликация, 602, бОЗр
D-стебель, 494,494р, 496
ДАГ, 555,556р
Dam (ДНК-аденинметилаза), 673
Dem (ДНК-цитозинметилаза), 673
Deinococcus radiodurans, 297т, 298, 307р
Dicer, 478,478р, 479,480
Dnmtl (ДНК-метилтрансфераза 1), 368
Dnmt3a (ДНК-метилтрансфераза За), 368,
368р, 370
Dnmt3b (ДНК-метилтрансфераза ЗЬ), 368,
368р, 369
Drosophila melanogaster
- TAF1,404
- ДНК-транспозоны, 338, 338р
- альтернативный сплайсинг, 460-462,
462р
- белок Antennapedia, 386т, 510
- белок Bicoid, 388
- белок Deformed, 565
- белок Hedgehog, 529
- генетические маркёры, 85
- генный комплекс Antennapedia, 580-581,
581р
- генный комплекс Bithorax, 580-581,581р
- генный комплекс НОМ-С, 581, 582р
- гомологи генов болезней человека, 196
- домен tudor, 195,195р
- достижения в экспериментах, 576-577
- инсуляторные последовательности, 356,
356р
- каталоги генов, 278р, 279т
- метод дробовика для полных геномов,
159-161
- митохондриальные геномы, 310т
- модуль Antennapedia, 414
- модуль Bicoid, 414
- перестройка нуклеосомы, 365,365р
- развитие, 206, 577р, 579р, 580р, 576-583
- размер генома, 272т, 275т
- рекомбинация, 101,102р
- составление генетических карт, 101,102р
- схема сегментации, 579р, 579-580
- теломеры, 626-627
- уплотнение генов, 273р, 274, 274т
- ферменты Dicer, 479
- число генов, 275т
- эндогенные ретроэлементы, 331-333, ЗЗЗр
Drosophila
- группы гена Нох, 725р
- межвидовой горизонтальный перенос
генов, 735
- семейство генов Polycomb, 564, 564р
- эволюция гомеозисных избирательных
генов, 725
Drosha, 479
dsRBD (домен связывания двунитевого рибо-
нуклеопротеида), 388
DSP1 (белок Dorsal switch 1), 564
876 Предметный указатель
Е-сайт, 513р, 514
EcoRI, 112,113р
eEF-1, 513, 513т, 514
eEF-2, 513т, 514
EF-1A, 513, 513т, 514
EF-1B, 513т, 514
EF-2, 513т, 514
elF-1, 506т, 509
elF-lA, 506т, 509
elF-2, 506т, 509, 509р, 510
elF-2B, 506т
elF-З, 506т, 509р
elF-4A, 506т, 509р
elF-4B, 506т, 509р
elF-4E, 506т, 509р
elF-4F, 506т, 509
elF-4G, 506т, 509р
elF-4H, 506т
elF-5, 506т, 509
elF-б, 506т, 509
ELL, 449т, 450
elongator, 450,
eRF-1,518, 519р, 519т
eRF-3, 518, 519т
Escherichia coli, см. также бактериофаги
- а-субъединицы, 409,409р
-OxyS, 512
- ДНК-лигазы, 52-53
- ДНК-полимераза 1,44,45,144
-ДНК-транспозоны, 335-337
- ДНК-фотолиаза, 668
- РНК-полимераза, 402,404р
- белок-активатор катаболитных оперонов,
411
- горизонтальный перенос генов, 307, 307р
- восстановление вырезанием нуклеотидов,
670р, 670-672
- ген trpC, 247-248
- ген теплового шока, 409,409р, 412
- гомологичная рекомбинация, 694р,
693-696, 696р
- терминация трансляции, 518р
- инициация транскрипции, 402,404р
- инициация трансляции, 508р
- исследования функции генов, 190
- комплекс продолжения транскрипта, 431р
- коэффициент ошибок синтеза ДНК, 646,
647
- молекулярные шапероны, 523
- мотив лента-спираль-спираль, 388р
- мотив спираль-изгиб-спираль, 381р
- мутантные типы, 661
- обработка пре-рРНК, 442,442р
- инвертированный повтор, 433
- оперон рар, 445
- оперон лактозы, 410р, 546
- оперон синтеза жгутика, 248, 248р
- оперон триптофана, 411,411р, 437р,
437-438
- опероны, 301-302, 302р
- организация генов, ЗООр, 301р, 300-303
- плазмиды, 296, 297т
- плазмиды для клонирования ДНК, 55р,
53-70
- репрессия катаболитных оперонов, 550р,
548-551
- последовательности участков посадки
рибосомы, 506р, 508т
- последовательность тРНК, 180р
- сканирование ОРС, 176,177,177р
- продление трансляции, 517р, 517-518
- промотор, 398-400,400р, 400т, 402
- рРНК 16 С, 504р, 505
- размер генома, 303т
- реакция SOS, 568, 654, 676-677, 677р
- реакция на стресс, 441
- репликация
- - ДНК-полимеразы, 609-610,610т
- - геликазы, 612р, 612-614
- - завершение, 619р, 619-620, 620р
- - область инициации, 604-605
- - синтез ДНК, 612р, 612-615, 615р
- - эксперимент Мезельсона-Шталя, 599р,
598-600
- репрессор лактозы, 386т
- сдвиг рамки считывания в процессе элон-
гации, 515-517, 517р
- случайные и запрограммированные мута-
ции, 663-665, 665р
- консенсус, 508т
- состав рибосом, 504р
- составление генетических карт, 109,111
- специфические для штаммов геномы, 305
- транскриптоспецифическая регуляция
рибосомного белка, 510, 510р
- устранение несоответствий, 673-674
- устранение однонитевых пропусков, 696р,
697
- сайт-направленное зондирование гидрок-
сильными радикалами, 504
- сайт-направленный мутагенез, 209тп
- фагмиды, 143
- фаза продления трансляции в ходе синте-
за белка, 512р, 513
- фермент Ада, 668
- физические особенности геномов, 294р,
295р, 294-296
- функциональные взаимодействия белка,
242-243, 243р
- число генов, 303т
Предметный указатель 877
- число рибосом в клетке, 501
ESE (экзонные энхансеры сплайсинга), 460,
460р
ESS (экзонные сайленсеры сплайсинга), 460
Extradenticle, 581
FACT, 449т, 450
FEN1 (эндонуклеаза откидной створки), 616,
617р, 650
FISH (флюоресцентная гибридизация на ме-
сте), 112,119р, 120р, 119-121, 352
FRAP (восстановление флюоресценции после
фотообесцвечивания), 351,352тп
Fritillaria assy паса, 272т
G-белки, 513
G-реакция, 147,148р
Gallus gallus, 275т
gap-гены, 579-580, 580р,
GAP (активирующие ГТФазу белки), 553, 555р
GNATs (связанные с GCN5 ацетилтрансфера-
зы, 361
GNRP (белки, высвобождающие нуклеотид
гуанин), 553, 555р
GTF (основные факторы транскрипции), 405т,
404-406
Haemophilus influenzae, 48,151р, 152р, 149-153,
307р
НАТ (гипоксантиновая, аминоптериновая и
тимидиновая) среда, 124
НАТ (гистонацетилтрансферазы), 361-363,
367
HDAC (гистондезацетилазные комплексы),
363, 369р
Helicobacter pylori, 243,305, 307р
Hinfl, 48
H1S2, сравнительная геномика, 243
HLA-DRB1, множественные аллели, 85-87
HMG (группа белков высокой подвижности),
386т
НОМ-С (комплекс гомеозисных генов), 581,
582р, 725
Homo erectus, 740р, 776, 776р, 777
Homo habilis, 740р
HPRT (гипоксантинфосфорибозилтрансфера-
за), 124
ICAT (закодированный изотопом ярлык по
сродству), 237, 237р
IF-1,506т, 508, 508р
IF-2, 506т, 508, 508р
IF-3, 506, 506т, 508р, 518
IRES (внутренние участки входа рибосомы), 510
Kin28,417
Klebsiella pneumoniae, 409
Kluyveromyces lactis, 729
Lac-отбор, 59, 59p
LCR (управляющие локусом области),
358-359,
LINE (длинные рассеянные ядерные элемен-
ты), 269р, 269-271, 333т, 334, 334р, 733-734
- LINE-1, 333т, 334, 626, 733-734, 737
Locusta migratoria, 272т
М-фаза клеточного цикла, 627, 628
MALDI-TOF (времяпролетная масс-спектро-
метрия с лазерной десорбционной иониза-
цией матрицы), 235, 237р, 238
Marchantia polymorpha, 310т
mariner, 735
MARs (связанные с матриксом области), 355,
355р, 356
MGMT (О6-метилгуанин-ДНК-
метилтрансферазы), 668,
MeCPs (связывающиеся с метил-CpG белки),
369, 369р
Methanobacterium thermoautrophicum, 307р
Methanococcusjannaschii, 302-303,303т, 307р
Metridium senile, 310т
miPHK (микроРНК), 18,18р, 398т, 479-480,
480р
miPHn (микрорибонуклеопротеид), 479
MudPIT (многомерный метод опознавания
белков), 242,
MULEs (подобные Mutator мобильные генети-
ческие элементы), 734, 734р
Mus musculus
- ген sip, 737
- ген НохС8, 581
- группа НохВ, 582, 582р
- митохондриальные геномы, 310т
- размер генома, 272т
- функциональный анализ генов, 199, 201,
202р
Mycobacterium tuberculosis, 303т, 307р
Mycoplasma genitalium
- горизонтальный перенос генов, 307р
- секвенирование методом дробовика, 153
- соотношение — размер генома : число
генов, 303т, 304, 304т
- число копий гена рРНК, 722
Mycoplasma pneumoniae, 307р
MyoD, 414, 565
MYST, 361
N-ацетилгалактозамин, 528р
N-ацетилглюкозамин, 528р
878 Предметный указатель
N-белок, 436
N-гликолилнейраминовая кислота, 741
N-дегрон, 530
N-конец, 21, 22р,
N-миристилирование, 527т
N-связанное гликозилирование, 527т, 528,
528р
N-формилирование, 527т
N-формилметионин (фМет), 497р, 498, 508
Neisseria meningitidis, 301
Neurospora crassa, 308
NF-кВ, 386т, 414
NHEJ (негомологическое соединение концов),
666, 674-676,676р
Nicotiana tabacum, 310т
NMD (опосредствуемое бессмысленным кодо-
ном разложение РНК), 476р, 476-478
NOESY, 384тп
О-последовательность, 698, 700р
О-направленное гликозилирование, 527т, 528,
528р
06-метилгуанин-ДНК-метилтрансфераза
(MGMT), 668
Oenothera lamarckiana, 728
OFAGE (электрофорез в геле с ортогонально
чередующимся полем), 116р
OLA (анализ лигированием олигонуклеоти-
дов), 94р
ORC (комплекс опознавания точки старта
репликации), 605,606р, 608, 628, 630
oriC, 604, 605р
Oryza sativa, 275т, 310т, 312р, 360т
OxyS, 512
Oxytricha, 625т
Р-мобильный генетический элемент, 338,
338р
Р-участок, 512, 512р, 513, 518
Р-элементы, 199,
рЗОО/СВР, 361, 555
PCNA (ядерный антиген разрастающихся
клеток), 610, 615, 632
PADP (связывающийся с полиаденилатом
белок), 450,509
Pisum sativum, 94р, 96,96р, 97,97р, 272т, 310т
Plasmodium falciparum, 310т, 311
Pseudomonas aeruginosa, 303т, 322, 323т
Pseudomonas phaseolicola, 323т
PSI-BLAST (позиционный итеративный
BLAST), 193,
Pyrococcus abyssii, 619
Pyrococcus horikoshii, 307p
Q-белок, 436
R-группы, 23p
RACE (быстрое размножение концов кДНК),
189,190р,
Ran, 480
RecG, 696
Reclinomonas americana, 310т, 311
RF-1, 518p, 519т
RF-2, 518p, 519т
RF-3, 518p, 519т
RFLP (полимофизмы длин рестриктов), 87p,
87-88,88p
- филогенетика, 757, 776, 777p, 783
RHB домен гомологии rel, 386т
Rho-зависимый терминатор, 435,435p, 436,
Rickettsia prowazekii, 307p, 308
RISC (наведенный РНК комплекс глушения),
478,478р
RMP (репликационный посреднический
белок), 614,
S-фаза, 627, 630-632,
S-фазные циклины, 628, бЗОр
Sau3AI, 48р
SnaBl, 67р
Saccharomyces
- S. cerevisiae
- - Reblp, 416
- - YIp5,68,68p
- - альтернативный сплайсинг, 460
- - белковые комплексы, 242, 242р
- - генетические и физические карты,
111,112р
- - генетические маркёры, 85т
- - гомологи генов болезней человека,
196т
- - гомологичная рекомбинация, 198-199,
199р, 696-697
- - данные о дупликации геномов,
728-729
- - данные об автополиплоидии, 728-729,
729р
- - дигибридная система, 239р, 238-241
- - дрожжевые искусственные хромосо-
мы, 64т, 66, 67р, 117,162,162р
- - жизненный цикл, 559р
- - терминация транскрипта РНК-
полимеразой 1,466
- - изучение транскриптома, 229-230,
230р
- - изучение функций генов, 190
- - инактивация гена RAD27, 650
- - интеины, 528
- - карта межбелковых взаимодействий,
246р
Предметный указатель 879
- - каталоги генов, 278р, 279т
- - метилирование рРНК посредством
мярРНК, 472р
- - митохондриальные геномы, 308, 309,
310т, ЗИр, 311т
- - переключение типа спаривания,
559-560
- - плазмиды для клонирования ДНК, 55
- - поиск гомологии, 183
- - генная конверсия, 692,696
- - пример аннотирования генов, 212р,
211-214
- - прионы, 330
- - просмотр на ОРС, 176
- - размер генома, 272т, 275т
- - регулируемая медью экспрессия генов,
546р, 547
- - репликация ДНК, 605-606, 606р,
627-628, 630-631,631р
- - ретроэлементы Ту, 331р, 331-333,
ЗЗЗр
- - сети межбелковых взаимодействий,
243, 245р
- - сигналы восприимчивости к деграда-
ции, 530
- - составление генетических карт, 103
- - сплайсинг пре-тРНКТуг, 471р
- - тРНКА1а, 493
- - уплотнение генов, 273р, 272-274
- - фенотипы, 204т, 205
- - центромеры, 265р, 265-266
- - число генов, 275т
-S. pombe, 275т
- поиск гомологии, 183
SAGE (последовательный анализ экспрессии
генов), 224р, 224-225
SAR (области прикрепления к каркасу), 355,
355р,
SCAF (SR-подобные связанные с С-КД факто-
ры), 460
Schizosaccharomyces pombe, 418
scs, 356,356р
scs', 356, 356р
siPHK (короткая интерферирующая РНК), 18р,
478р
SINE (короткие рассеянные ядерные элемен-
ты), 124, 269р, 269-271, 333т, 334, 334р
Sir2,363
SNP (полиморфизмы отдельных нуклеоти-
дов), 87,90р, 90-91,94,94р
SR-подобные связанные с С-КД факторы
(SCAF), 460
SSB (белки однонитевого связывания), 614р,
615р, 677
SSLP (полиморфизмы длин простых последо-
вательностей), 88р, 87-90
- проект «Геном человека», 161,162
STAT (передатчик сигналов и активатор
транскрипции), 552р, 552-553
STS (меченый участок последовательности)
- библиотеки клонов, 125-127,127р
- картирование, 112,123р, 121-127,127р
- картирование содержания, 157р, 157-158
- проект «Геном человека», 162
Streptococcus pneumoniae, 5,7р, 303т
Streptomyces coelicolor, 296
Strongylocentrotus purpuratus, 272т
Suillus grisellus, 310т
Synechocystis, 307p
Т-лимфоциты, 560, 563
Tm (температура плавления), 91p
TAF (связанный с ТВР фактор), 404
TAF„42,404,405p
TAFH62,404,405p
TAF1,404
TAP (тандемная аффинная хроматография),
242, 242p
Takifiigu rubripes, 272т
ТВР (связывающийся с блоком ТАТА белок),
404,405р, 406р, 417-418
TGF-p (трансформирующий фактор роста Р),
556-557
ТЕМЭД, 139тп
Теропета pallidum, 307р
Tetrahymena, 272т, 361,466,467р, 468,625т
TFIIA, 405т, 406
TFIIB, 404,405т, 406р
TFIID, 404,405т, 406,451
TFIIE, 361,405т
TFIIF, 361,405т, 449т
TFIIH, 405,405т, 406, 672, 672р
ТИПА, 386т, 406
TFIIIB, 406
TFIIIC, 406
TFIIS, 449т, 453-454
Thermotoga maritima, 307,307р
Thermus aquaticus, 45
Thermus thermophilus, 505p, 513p
TIC (зависимые от TAF и инициаторной по-
следовательности кофакторы), 404
TOCSY, 384тп
ТРА (активатор профибринолизина ткани),
733, 733р
TRAP (связывающийся с РНК trp ослабляю-
щий белок), 438,439р
Treponema pallidum, 298
Triticum aestivum, 272т, 735
Triticum turgidum, 735
trpE, 437,437p, 439p
880 Предметный указатель
U-PHK, 18,18р, см. также малые ядерные РНК;
малые ядрышковые РНК
Ul-мяРНК, 457р, 457-459,459р
и11/и12-мяРНП, 465
и16-мярРНК, 472р
и2-мяРНП, 459,459р
024-мярРНК, 472р
и4/иб-мяРНП, 459,459р
U4atac/U6atac-MHPHH, 465
US-мяРНП, 459,459р, 465
иб-мяРНП, 459
и7-мяРНП, 453,453р
V-петля, 494,494р, 496т,
Vibrio cholerae, 297т, 298, 303т
VNTR (переменное число тандемных повторе-
ний), 88,90
Х-гель (5-бромо-4-хлоро-3-индолил-0-В-
галактопиранозид), 59, 59р
Х-хромосома, 777
- инактивация, 370р, 369-372, 564
Xenopus laevis, 722
Y-хромосома, 776, 785
YAC (дрожжевые искусственные хромосомы),
64т, 66,67р
- оптическое картирование, 117
- проект «Геном человека», 162,162р
Yersinia pestis, 303т
YIp5,68,68р
Z-ДНК, 15,15т, 16р, 394
Zea mays
- ДНК-транспозоны, 337р, 337-338
- митохондриальные геномы, 310т
- размер генома, 272т
- уплотнение генов, 273р, 274
аббревиатуры, аминокислоты, 25т
автокаталитические интроны, 466-467
автоматизированные секвенаторы ДНК, 141р,
141-143
автономно реплицирующиеся последователь-
ности (ARS), 605,606р
автополиплоиды, 727р, 727-728, 728р
авторадиография
- гибридизационные зонды, 52
- мечение радиоактивными метками, 41тп,
52
- определение
- секвенирование способом химической
деградации, 148р
- электрофорез в полиакриламидном геле,
140р
агаровые пластинки, 63р, 661р
агенты окружающей среды, 650,650р
«Адам с Y-хромосомой», 776
аденилатциклаза, 548, 550р,
аденин, 8р, 12р, 13
аденозин-5’-трифосфат (АТФ), 17, 716
аденозины, 450
«адресные ярлыки», 482
азотистые основания, 8
акриламидные мономеры, 139тп
активатор
-CRP, 550р, 548-551
-GATA-1,414
-NF-1,414
- NF-Y, 414
- Oct-1,414
- Oct-2,414
-Spl, 414
- профибринолизина ткани (ТРА), 733,
733р
- транскрипции МСМ1, 559-560
- транскрипции NK-kB, 552
- транскрипции Rel, 552
- транскрипции при иммунной реакции,
546
активаторы
- взаимодействия предынициаторного ком-
плекса РНК-полимеразы II, 416-417
- дрожжедигибридной системы, 239р,
239-241
- инициации транскрипции у бактерий,
411-412
- инициации транскрипции у эукариотов,
416р, 415-420
- клеточной регуляции, 29
- определение,
активаторы транскрипции
- ацетилирование гистонов, 361-363
- внеклеточные сигнальные соединения,
546-547
- лактоферрин, 546
- содержание ионов металлов в клетке,
546р, 546-547
- стероидные гормоны, 547
активационные домены, 239р, 241,416-417
активация встройки, 56
активирующие ГТФазу белки (GAP), 553,555р
активные гены, 354
актин, 565
акцепторный стебель, 494р, 496т, 493-821
акцепторный участок, 455,
аланин, 23р
алармон ppGpp, 441,442р
алармон pppGp, 441,442р
алармоны, 441,442р,
Предметный указатель 881
алкилирующие агенты, 653,
аллели
- генетические маркёры, 85-87
- кодоминирование, 96,96р, 105,106р
- однонуклеотидные полиморфизмы (SNP),
90,90р
- случайное расщепление, 96
- типы взаимодействий, 96р, 94-97
аллолактоза, 410,410р, 546
аллолактозо-репрессорный комплекс, 410,410р
аллополиплоидия, 735,
альтернативное полиаденилирование, 454
альтернативные промоторы, 415,415р
альтернативный сплайсинг, 226р, 228, 275,
461р, 460-463,482
альфоидная ДНК, 265, 741
амино-конец, 21, 22р
аминоаденин, 647, 647р
аминоацил-тРНК, 498, 512р, 512-513
аминоацил-тРНК-синтетаза, 496р, 496т,
496-498,512,532
аминоацил-тРНК-синтетазы I класса, 496,
496т
аминоацил-тРНК-синтетазы II класса, 496,
496т
аминоацилирование, 496р, 497р, 493-498,532
аминоацильный участок, 512-514
аминокислоты
- R-группы, 23р
- аббревиатуры, 25т
- биосинтез, 437
- аттенюация, 437
- разнообразие, 22-23
- ранняя эволюция, 716-717
- расщепляющие ферменты, 741
- сравнения генов по сходству, 193,195р
- структура, 21, 21 р, 22
анализ
- генома, эволюционные данные, 728-733,
739-743
- главных компонентов, 781
- лигированием олигонуклеотидов (OLA),
94р
- митохондриальной ДНК
- - миграции в Европу, 781, 783р
- - миграции в Новый Свет, 783-785
- - молекулярные часы, 768
- - неандерталец, 778
- - популяции человека, 774, 781, 782р
- - происхождение человека, 776, 777р
- - родство человека и человекообразных
обезьян, 770, 772, 772р
- на микроматрицах
- - сравнения транскриптомов, 225р,
228р,229р, 225-233
- - транскриптом человека, 232р, 230-233
- - эксперименты над кинетикой репли-
кации, 630-631,631р
- на чипах, 91, 93тп, 225р, 228р, 225-233
- последовательностей, 175-221, 224-225
- родословной, 106-108,108р,
- родословной человека, 106-108,108р
- с защитой, 390-393
- с защитой от модификации диметилсуль-
фатом (ДМС), 393р
- с модификацией, 390-391, 391р, 393р
- с модификационным препятствованием
диметилсульфатом (ДМС), 391-393, 394р
- бутстрап, 767, 767р
- с торможением в геле, 390,390р
- сцепления генетических признаков,
94-106
анализирующее скрещивание по трем точкам,
105т
анализирующие скрещивания, 103-105,105р,
105т, 106р
аналоги оснований, 650-651, 651р
анатомия, 256-344
анафаза, 99р, 101р
анаэробное дыхание, 229
анемия Фанкони, 679
аннотирование генов, 184р, 212р, 211-214,
232
аннотирование генома, 184р, 212р, 211-214,
232
антигены, 560,
антикодоновый стебель, 494,494р,
антикодоны, 494, см. также взаимодействия
типа кодон-антикодон
антисмысловая РНК, 479,479р, 726, 727р
антитерминаторный белок, 436,436р
антитерминация, 436р, 435-436,436р
АП (апуриновые/апиримидиновые участки),
см. также безосновные участки
- восстановление вырезанием оснований,
669р, 670
- реакция SOS, 677
- сверхмутация, 663,663р
- тепловые мутации, 653-654,655р
АП-эндонуклеаза, 669р, 670
аполипопротеин 100,472
аполипопротеин В, 472,474р
аполипопротеин В48,472
апоморфные состояния признака, 754
апоптоз, 269, 632
апуриновые/апиримидиновые (АП) участки,
см. также безосновные участки
- восстановление вырезанием оснований,
669р, 670
- реакция SOS, 677
882 Предметный указатель
- сверхмутация, 663,663р
- тепловые мутации, 653-654,655р
арабидопсис, см. Arabidopsis thaliana
аргинин, 23р, 27р, 363-364
археи, 293
аспарагин, 23р, 27р
аспарагин-тРНКА5П, 497
аспарагиновая кислота, 23р, 27р
аспарагиновая кислота-тРНКА5р, 497
атаксия Фридрейха, 649т, 650
атаксия спинного мозга и мозжечка, 649, 679
атаксия-телеангиэктазия, 679
аттенюация (ослабление), 437р, 438,439р
атрофия мышц спины и глазного яблока, 649
АТФ (аденозин-5’-трифосфат), 17, 716
ауксотрофы, 109, 661, 661р, 665
ауксотрофы по триптофану, 109,111р, 661,
661р, 662
аутосомы, 3
аффинная хроматография, 241, 241р, 242,
242р
ацетилирование, ЗбОр, 360-363, 527т, 528р
ацилирование, 527т, 528
аэробное дыхание, 229
бабуины, 757, 758р
бактериальная РНК
- деградация, 444-445,445р
- интроны, 455т
- обработка, 442р, 443р, 441-444
- химическая модификация, 443р, 444
бактериальная рРНК, 442р, 441-443,443р, 444
бактериальная тРНК, 441-443,443р, 444,468т
бактериальные РНК-полимеразы, 398-400,
431р, 431-432,432р, 435-441
бактериальные искусственные хромосомы
(ВАС), 64т, 66,117,153-154,162,
бактериальные плазмиды, см. плазмиды
бактерии
- ДНК-белковые взаимодействия, 295р,
295-296
- ДНК-гираза, 600т, 602
- анатомия генома, 293-307
- воздействия мутаций, 659-662
- репарация ДНК, 668, 670-674
- генетические особенности, 298-308
- дублирование гена в ходе репликации,
726, 726р
- завершение репликации, 619р, 619-620,
620р
- инициация транскрипции, 408-412
- инициация трансляции, 506р, 505-508
- линейные геномы, 296, 297т, 298
- многораздельные геномы, 296, 297т, 298
- мотив спираль-изгиб-спираль, 386
- опероны, 301р, 302р, 300-303
- отсутствие интронов GU-AG, 738
- просмотр на ОРС, 176,177,177р
- процессы обхода повреждений, 654,
676-677, 677р
- размер генома/число генов, 303, 303т
- сверхспирализованные домены хромосом,
295
- синтез ДНК, 609-610, 610т, 612р, 611-615,
616р
- составление генетических карт, 108-109,
109р, 111р
- физические особенности геномов,
294-298
- шатание, 498-500, 500р
- эндосимбиотическая теория органелл, 308
бактериофаг
- X, 322, 323, 325, 326, 327р
- - встраивание (интеграция) ДНК, 698р,
698-700, 700р
- - геном, 436,436р
- - клонирование, 63р
- - расщепление репрессора cl, 677
- - репликация ДНК, 603
- - цикл инфицирования, 61р, 62, 62р,
436,436р, 568р, 567-569
-фХ174, 323, 323т, 325р
-фХ174, 323, 323т, 325р
- f6, 323т
- MS2, 322, 323, 323т
- РМ2, 322, 323т
- SP01, 322, 323т
- М13,143,143р, 192р, 322, 323т
- РМЗ, 322, 323т
- Т2, 5, 7р, 323, 323т
- Т4
- - ДНК-лигаза, 52
- - геном, 322, 323, 323т, 325р
- - интроны, 466
- - полинуклеотидкиназа, 53,
- - трансляционный обход, 517р, 518
- Тб, 323, 323т
-Т7, 323т
бактериофаги, 322-326, 338,
безосновные участки, 147,653,655р, 670, см.
также АП (апуриновые/апиримидиновые)
участки
белки, см. также отдельные белки; ДНК-
белковые взаимодействия; протеомы,
связывающиеся с ДНК белки
- Hsp40, 523, 524
- HU, 295-296, 386т, 605
- Ku, 676,676р
- PARP1, 674,674р
- Ras, 553, 555р,
Предметный указатель 883
- Sir, 559
- SR, 459-460,460р,
- trithorax, 565
- Tus, 619р, 619-620, 620p,
- U2AF, 459,460,460p, 461,462p
- биохимия клетки, 27-30
- блокирование линейной хромосомы,
625-626
- деградация, 529-530
-динамизм, 352
- изучение взаимодействий, 243р, 238-245,
245р
- каталоги генов эукариотов, 278р, 278-279
- киназы, 454
- обработка, 543т
- однонитевого связывания (SSB), 614, 614р,
615,615р, 677,
- опознавание компонентов комплексов,
241, 241р, 242, 242р
- определение,
- определение профилей, 234р, 237р,
233-238,
- передне-заднего градиента, 577-579
- подвижность ядер, 351-352
- посттрансляционная обработка, 520р,
527т, 519-529
- путаница с ДНК как с генетическим мате-
риалом, 5
- регуляция активности генома, 543т
- рецепторов Т-клеток, 560,563
- сравнение с рибозимами, 716-717
- струкура, 21-23
- филогенетическое секвенирование, 756
белки, высвобождающие нуклеотид гуанин
(GNRP), 553, 555р
белки домашнего хозяйства, 25
белковая инженерия, 205
белковые домены
- взаимосвязь экзонов, 739-740, 740р
- перестройки генов, 731р, 732р, 731-733,
733р
- появление интронов в ходе эволюции,
738р, 739-740
белковые факторы SF1,459
белковый комплекс
--SL1,406
- - TIFIA, 406
- -TIFIB, 406
- - TIFIC, 406
белок
- Acelp, 546р, 547
- Aly, 480
- Antennapedia, 386т, 510
- Bicoid, 388, 577-579, 579р
- Buttonhead, 580р
- Caudal, 579, 579р
- CD4, 327
- Cdc6p, 628, 628р
- Cortex, 577
- Cro, 436
- Deformed (Dfd), 565
- Dfd (Deformed), 565
- DksA, 441
- DMC1, 696
- DnaC, 605
- DnaK, 523
- DnaA, 604-605, 605p
- DnaB, 605, 612,612p, 615p
- Dorsal, 579
- Dorsal switch 1 (DSP1), 564
- DSX, 462,462p
- Empty spiracles, 580p
- GCN5, 361
- Giant, 580p
- Grauzone, 577
- GrpE, 523
- H-Ras, 553
- Hedgehog, 529
- Hjc, 697
- HP1, 364
- Huckebein, 580p
- Hunchback, 579, 579p
-IIAGlc, 550p, 548-551
- K-Ras, 553
- KinA, 571
- KinB, 571
-KinC, 571
- Knirps, 580p
- Kriippel, 580p
- LexA, 677
- Maclp, 546p, 547
- Mek, 553, 553p
- MutH, 673, 674p
- MutL, 673
- MutS, 673, 674p
- MutY, 674
- N-CoR, 418,418p
- N-Ras, 553
- Nanos, 579, 579p
- NusA, 435
- Oct-1, 386т
- Oct-2, 386т
- Orthodenticle, 580p
-p53,632
- p55, 361
- Pit-1, 386т, 414,418,418p
- Polycomb, 564p, 564-565
- Rac, 553
-RAD51, 696, 697
- Raf, 553,553p, 555p
884 Предметный указатель
-RAG1,561
-RAG2,561
- RbAp46, 363
- RbAp48, 363
- Reblp, 416,466,466p
- RecA, 677, 694, 694p, 696,
- RecC, 693, 694, 694p
- RelA, 441
- Rho, 482, 553,
- Rpd3,363
- Rsk, 553, 553p
- RuvA, 694, 696p
- RuvB, 694, 696p
- RuvC, 694,696p
- SGS1,465-466
- SpoIIAA, 572, 573p
- SpoIIAB, 572, 573p
- SpoIIE, 572, 573p
- SpoIIGA, 572, 573p
- SpoIIR, 572, 573p
- SpoIVB, 572, 575p
- SpoIVF, 572, 575p
-SpoOA, 571-572, 572p
- SpoOB, 572, 572p
-SpoOF, 571, 572p
- SRS2,465-466
- SRY, 386т, 416
- Staufen, 577
- Su(Hw), 356
- SWI5, 387p
- SXL, 461,462p
- Tailless, 580p
- toad, 387
- Torso, 579
- TRA, 461,462p
-TRF1,625
- TRF2,625
-TTF-1,416,466,466p
- U2AF35,459
-U2AF65,459,461,462p
- UBF, 406
- XRCC4, 676, 676p
- Yra lp, 480
- ВПГЫ (высокоподвижной группы N),
- E2, 386т
- СУМО, 364, 530,
- вируса папилломы Е2, 386т
- высокоподвижной группы N (HMGN),
- ретинобластомы, 363
белок-активатор катаболитных оперонов
(САР), 411, 550р, 548-551
бережливость, 765
бесклеточная синтезирующая белок система,
26
бесхроматиновые области, 352, 354
библиотеки, 189, см. также библиотеки клонов
- генома человека, 64т
- кДНК (комплементарных ДНК), 189
- фаговый дисплей, 238р, 239,241
библиотеки геномов, 64т
библиотеки клонов, 71тп
- анализ STS, 125-127,127р
- комбинаторный отбор генов, 156,156р
- обход хромосомы, 153,154р
- секвенирование генома Haemophilus
influenzae методом дробовика, 151р, 152
- фагового дисплея, 238р, 239,241
биваленты, 99,101р, 727, 728р
Бидл, Джордж, 247
биоинформатика, 176
биолистика, 70
биологическая информация, 1,3
биомолекулы, истоки жизни, 715-716, 716р
биотехнология, 208тп
биотинилирование, 527т, 528
биохимические маркёры, 85т, 85-87,109
бластодерма, 577
блок
--25
- СААТ, 414
- D, 472,472р
- GC, 414
- MADS, 583
- ТАТА, 401,401р, 404,405р, 414
бляшки, 63р
горизонтальный перенос генов, 307р,
734-735
более крупные фрагменты ДНК, 64-70
- исследования сверхэкспрессии, 202
- метод дробовика для полных геномов, 158
- однонитевая ДНК для секвенирования,
143,143р
- фаговый дисплей, 238р, 238-239, 241р
болезни, см. СПИД/ВИЧ; рак; генетические
болезни; болезни человека
болезни человека, см. также рак
- СПИД/ВИЧ, 327,474,474т, 772-773, 773р
- дефекты восстановления ДНК, 677-679
- обнаружение мутаций, 642тп
- поиск гомологии, 195-196,196т
- расширения тринуклеотидных повторе-
ний, 649т, 647-650
болезни, связанные с расширением тринукле-
отидных повторений, 649т, 647-650
болезнь Гентингтона, 647, 649т
болезнь Мачадо-Жозефа, 649
большая бороздка,
- ДНК В-формы, 13р, 15,15т, 16р, 394, 396р
- связывающиеся с ДНК белки, 381, 386,
386р, 387
Предметный указатель 885
Бреннер, Сидни, 573
бромциан, 432
бромистый этидий, 49р, 653, 653р,
бросовая ДНК, 282-284, 737,
бурые водоросли, 310т
бурый скалозуб (Takifiigu rubripes), 272т
бутылочное горлышко, 774
буферы, 235, 241, 242
быстрое размножение концов кДНК (RACE),
189,190р
быстрые методы сборки контигов из клонов,
157р, 156-158
БЭП (близлежащий элемент последователь-
ности), 401р
валин, 23р, 27р, 179
вегетативный рост, 569, 571, 571р
вектор замещения, 62р
векторы, см. векторы конкретных видов
векторы на бактериофаге Р1, 66, 700-701
величина S (коэффициент седиментации),
502, 530
Бентер, Крейг, 158,162
ветви филогенетических деревьев, 757, 757р,
760, 761р
взаимодействия типа кодон-антикодон, 513
- синтез белка, 498р, 500р, 498-501,501р
- тРНК, 498р, 500р, 498-501, 501р
видообразование
- автополиплоидия, 728, 728р
- мобильные генетические элементы, 338
- филогенетические деревья, 760р, 761
ВИО (вирусы иммунодефицита обезьян),
772-773, 773р
виозин, 444
вироиды, 329-330, ЗЗОр,
вирулентные бактериофаги, 325
вирус Сендай, 124
вирус иммунодефицита человека 1 (ВИЧ-1),
327,474т, 772-773, 773р
вирус иммунодефицита человека 2 (ВИЧ-2),
327, 773р
вирусная РНК, 1,474,475,478-479
вирусные ретроэлементы, 327, 329р, 331-333,
340,
вирусоиды, 327-330, ЗЗОр
вирусы иммунодефицита обезьян (ВИО),
772-773, 773р
вирусы эукариотов, 326-330, 338-340
вирусы-помощники, 329
витамин D3, 547
ВИЧ-1 (вирус иммунодефицита человека 1),
327, 474т, 772-773, 773р
ВИЧ-2 (вирус иммунодефицита человека 2),
327, 773р
вкладочный фрагмент, 62, 62р,
влияние температуры на ДНК-полимеразу, 45
внеклеточная среда, 541
внеклеточные сигнальные соединения, 545р,
545-557
внехромосомные гены, 308,
внешние раздражители, 542,545
внешние узлы, 757, 757р,
внешние элементы, 757р, 758
внутренние затравки, 145,145р
внутренние стоп-последовательности, 433р,
433-436
внутренние узлы, 757, 757р, 760, 760р
внутренние участки входа рибосомы (IRES),
510
внутримолекулярная рекомбинация, 700
внутримолекулярное спаривание оснований,
180р, 181
внутринитевое спаривание оснований, 147,
147р
водородные связи, 12,13р
возбуждаемые липидами каскады, 555
возврат РНК полимеразы, 432,438-441,441р
воздействия удалений, 731, 737р, 762р
возрастание сложности во времени, 720
возрастание числа генов в ходе эволюции,
720-735
волоконный FISH, 121
воспаление, 555
восстановление флюоресценции после
фотообесцвечивания (FRAP), 351, 352тп
ВПЖХ (высокопроизводительная жидкостная
хроматография), 642тп
временные изменения активности генома,
541р, 542-557
время полураспада мРНК, 444-445,475
времяпролетная масс-спектрометрия с лазер-
ной десорбционной ионизацией матрицы
(MALDI-TOF), 235, 237р, 238
времяпролетный, 235, 237р
вставочные агенты, 653,653р
встраивание
- бактериофага, 700, 700р
- транспозиция ретроэлементов, 705, 705р
встраивающие векторы, 62р, 63
встраивающиеся вирусные элементы, 326,
327, 327р, 329р, 340
встройки, перекрытие, 154,154р
вторгающиеся последовательности, 454
вторичная регуляция, 408,408т
вторичные каналы, 441,441р,
вторичные посредники, 555-556, 556р,
вторичные структуры, 21-22, 22р, 180р, 181
вторичный путь дифференциации клеток
яйцевода, 573-575, 576р
886 Предметный указатель
второй закон генетики, 96р, 97
выбор между элонгацией и терминацией,
435-441
выведенные деревья, 758, 762-765
вызываемое уксусной кислотой повреждение,
205
вызывающая корь инфекция, 474
выискивание мутаций, 642тп,
выравнивание
- последовательностей ДНК, 761-762, 762р
- последовательностей белка, 193
выравнивание последовательностей, 193,
761-762, 762р
вырезание, 701, 701р
вырожденность, 26
высокопроизводительная жидкостная хрома-
тография (ВПЖХ), 642тп
выстилающие массивы, 232, 232р
вытеснительная репликация, 602-603, бОЗр
вытягивание гелем, 116-117,117р
вышележащие промоторные элементы, 400,
401,416
вышележащие регуляторные последователь-
ности, 179-180
вышележащие элементы управления (ВЭУ),
401,401р, 406
ВЭУ (вышележащий элемент управления),
401,401р, 406
галактоза, 528р
галактозидазы, 56,56р, 59, 59р, 242,474т, 665
гаметы, 99,101р
- автополиплоидизация, 727р, 727-728,
728р
- анализ сцепления, 102,102р, 103
- генная конверсия, 692,692р
- человек, 3
гаплогруппы, 774, 781, 782р, 783, 783р
гаплоидные клетки, 3
гаплотипы, 783р
гашение флюоресцентного красителя, 91р
гексозо-1-фосфатуридилтрансфераза, 508т
гелевые блоки, 139тп, 141,147
геликаза
- PriA, 614
- RecB, 693, 694р
- RecD, 693, 694р
- Rep, 614
геликазы, 435,435р, 445,
- гомологичная рекомбинация, 694р,
693-696
- репликационные вилки, 605,612р,
612-615,615р
гель-электрофорез, 49тп, 51, 74р, 390р, см.
также полиакриламидный гель
- определение профилей белков, 234р,
233-235,237
- определение счетов RFLP, 88р
- последовательность 5’-CG-3', 116р
- редкощепящие рестриктазы, 116
гемоглобин, см. надсемейство генов глобинов
гемофилия, 334
ген
- AR, 649т
- АТХ, 679
- BRCA1,679
- DMPK, 663т
- DRPLA, 649т
- HMLa, 559, 560р, 563
- HMRa, 559, 560р, 563
- МАТ, 559-560, 560р
-SGS1,196
- агоН, 412,412р
- bicoid, 577, 580
- buttonhead, 579
- cl, 568р, 568-569
- сП, 568
- сШ, 568
- его, 568, 568р
- dnaj, 523
- dnaK, 523
- galE, 508т
- slo, 462-463,463р
- slo человека, 462-463,463р
- sip, 737
- ABL, 354р
- Abdominal В, 581, 581р
- Dfd, 565
- Н19, 370, 370р
- Igf2, 369-370, 370р
- RAD27,650
- Tsix, 372
- URA3, 68,68р
- Х25, 649т
-Xist, 370
-Xite, 372
- agamous, 583
- apetalal, 583
- apetala2, 583
- apetala3, 583
- curly leaf, 583
- empty spiracles, 579
- engrailed, 581
- even-skipped, 581
- floricaula, 583
- fushi tarazu, 581, 58lp
- hsp70,356,356p, 365, 365p
- huckebein, 579
- hunchback, 579
- int, 568
Предметный указатель 887
- kanR, 70р, 199р
- knirps, 580
- labial palps, 581, 581р
- lacY, 177р, 517р
- lacZ, 177р, 206т, 400р
- - запрограммированные мутации, 665
- - изучение случаев аннотирования
генов дрожжей, 212, 212р
- - мутации, 665
- - проскальзывание трансляции, 517р
- lacZ", 56, 56р
- leafy, 583
- let-7,480
- lin-3,575
- lin-4,480
- myoD, 565
- orthodenticle, 579
- pistillata, 583
- recA, 568
- rplj, 508т
- tailless, 579
- trpC, 247
- trpR, 412,412p
- CD27, 269, 269p
- FLJ10143, 269, 269p
- KanR, 701, 701p
- N, 568, 568p
- PKP2, 269, 269p
- SYB1, 269, 269p
- U6 мяРНП, 398т, 401,406
- актина, 228
- гормона роста, 418р
- дистрофина, 415,415р, 449,455т
- инсулина, 415р, 455т
- коллагена VII типа, 455т
- леггемоглобина, 740
- леггемоглобина сои, 740
- материнского эффекта, 577-579
- миоглобина, 192, 722р, 723
- нейроглобина, 722, 723р
- парамиксовируса Р, 475
- пролактина, Pit-1,418р
- сывороточного альбумина, 455т
- тимидилатсинтазы, 466
- устойчивости к ампициллину, 56, 56р, 68р
- фактора VIII, 455т
- цитоглобина, 722р, 723
генетика, определение, 838
генетическая избыточность, 219
генетические болезни
- дефекты восстановления ДНК, 677-679
- исследования функций генов, 195-196,
196т
- мутации, 642тп, 649т, 647-650, 677-679
- поиск гомологии, 195-196,196т
- расширения тринуклеотидных повторе-
ний, 649т, 647-650
- страховые взносы, 165
генетические маркёры, 84р, 84-87,123р,
121-127,127р
генетический код, 27р, 26-29, 29т, 719
генетический футпринтинг, 391р, 390-393,
393р
генная инженерия, 700-701, 701р
генная конверсия, 559-560, 560р, 692,692р,
722
геном человека
- альтернативный сплайсинг, 461
- библиотеки, 64т
- генный импринтинг, 369-370, 370р
- данные о дублировании сегментов, 729р,
729-731
- данные об автополиплоидии, 729
- интроны, 455т
- области инициации репликации, 608-609
- кариограмма, 263р
- каталоги генов, 278р, 279т
- компоненты, Зр
- межгенные области, 282-285
- микросателлиты, 269р, 271,285,285р
- митохондриальный, 309,309р, 310т, 311
- мобильные генетические элементы, 333,
333т, 334, 337
- организация генов, 269р, 268-271
- размер генома, 272т, 275т
- распределение генов, 267
- состав, 269р, 269-271, 271р
- составление карт радиационных гибри-
дов, 124-125
- сравнение с геномом шимпанзе, 740-743
- структура теломеры, 625р, 626
- уплотнение генов, 273, 273р, 274, 274т
- управляющие локусом области, 358р,
358-359
- хромосомы, 232р
- число генов, 275т, 275-277
- шатание, 500, 501р
- эволюция, 740р, 741р, 740-743
геномный импринтинг, 369-370, 372
геномы
- определение, 1-30
- функции, 223-254
геномы бактериофагов Escherichia coli, 61-64
геномы вирусов, 321-330, 338-340
- РНК, 1
- бактериофаги, 322-326
- вирусы эукариотов, 326-329
- ретровирусы, 45, 327, 327р, 329р
- рибозимы, 468т
- связь с рассеянными повторениями, 285
888 Предметный указатель
- связь с транспозонами, 331-333, ЗЗЗр,
335р, 337
геномы прокариотов
- взаимодействия ДНК с белком, 295р,
295-296
- генетические особенности, 298-307
- концепции вида, 305-307,307р
- метод дробовика, 151р, 152р, 149-153
- нестандартные генетические коды, 29т
- опероны, 301р, 302р, 300-303
- размер генома/число генов, 303, 303т
- секвенирование методом дробовика, 82р,
82-84, 84р
- физические особенности, 294-298
геномы рыб, 272т, 725, 725р
генотерапия, 70,
генотип Disease-М{}_1,106,108р
генотип Healthy-M_2t 106,108р
генотип trp+, 109, lllp
гены
- FRM, 649т
- HD, 647, 649т
- SCA, 649т
- р-глобина
- - интроны, 454р, 455т
- - мультигенное семейство, 192,281р,
281-282
- - происхождение человека, 777
- - управляющие локусом области, 358р,
358-359
- - эволюция, 657р, 723р, 723-725, 737
- pol
- - Escherichia coli, 609
- - ретровирусы, 327, ЗЗЗр, 334р
- - транспозиция ретроэлементов, 703,
705
- «отличительности», 304-305
- polA/polB, 609
- pair rule, 580,
- геном человека, 3
- глушение, 363,564-565
- доступность, 543т
- защиты от болезни, 741
- инактивация, 198р, 198-201,201р, 205,243
- коллагена, 732р, 732-733
- манипуляции с ДНК, 38р, 39-53
- межклеточного сообщения, 278,278р
- местоположения в последовательности,
176-190
- отдельные функции, 190-211
- открытие роли ДНК, 4-7
- плотность, 274т
- полярности сегментов, 580,
- распределение, 267-268
- под импринтингом, 369-370, 370р
- сверхэкспрессия, 201-202, 202р
- теплового шока, 409,409р, 412
- триозофосфатизомеразы, 739, 739р
-трипсина, 725
- упаковка, 273р, 273-274, 274т, 298-303
- химотрипсина, 725
- частота рекомбинации, 102,103р
гены-репортеры
- дрожжевая дигибридная система, 239, 241
- изучение функций новых генов, 206т,
206-209, 211р
- синтез жгутика Escherichia coli, 248р
гены домашнего хозяйства, 368
гермафродиты, 573
гетерогенная ядерная РНК (гяРНК), 20
гетеродупл ексный анализ, 190,
гетерозиготность, 94р, 96р, 94-97,97р
гетерохроматин, 354-355, 372
- гистоны, 361
- инактивация Х-хромосомы, 370
- чувствительные элементы Polycomb,
564р, 564-565
гибридизационные зонды, 51
гибридизационный анализ олигонуклеоти-
дов, 91р, 93тп
гибридизационный анализ, библиотеки
клонов, 71тп
гибридизация
- анализ на чипах, 225-233
- зоогибридизация, 187,189р
- интенсивность, 226р, 228, 228р
- нозерн-гибридизация, 186,186р
- олигонуклеотиды, 91р
- по Саузерну, 189р
- радиационные гибриды, 124,124р
- филогенетика, 756
- флюоресцентная на месте, 119р, 120р,
119-121
гибридизация аллелеспецифическими олиго-
нуклеотидами (АСО), 642тп
гибридизация на месте, 119р, 120р, 119-121
гибридизация нуклеиновой кислоты, см. так-
же гибридизация
гибридизация по Саузерну, 41тп, 51р, 51-52,
88,88р, 189р
гибридные гены в эволюции, 732р, 733р,
732-734, 734р
гибридный дисгенез, 338, 338р
гибриды ДНК-РНК, 705
гидрогеносомы, 308
гидроксилирование, 527т
гидролиз, 653,655р
гидрофобные взаимодействия, 396, 523, 523р
гиподермальные клетки, 575
гипоксантин, 653, 653р, 669
Предметный указатель 889
гипоксантиновая, аминоптериновая и тими-
диновая (HAT) среда, 124
гипоксантинфосфорибозилтрансфераза
(HPRT), 124
гипотеза «о поздних интронах», 738-740
гипотеза «о ранних интронах», 739р, 738-740
гипотеза «один ген — один фермент», 247
гипотеза о полицентрической эволюции, 776,
777р
гипотеза о шатании, 500р, 498-501, 501р, 508
гипотеза об африканском происхождении,
776, 777р, 779
гиразы, 600т, 602
гистидин, 23р, 27р, 438
гистон Н2А, ЗбОр, 370,450
гистон Н2В, ЗбОр, 364,404,450
гистон НЗ, ЗбОр, 364, 364р, 370
- инициация транскрипции, 404,405р
- посттрансляционная модификация, 528,
528р
гистон Н4, ЗбОр, 364р, 370,404,405р
гистон тасгоН2А1, 370
гистонацетилтрансферазы (HAT), 361-363,367
гистондезацетилазные комплексы (HDAC),
363, 369, 369р
гистонный код, 364, 365р, 372
гистоны, 372
- ацетилирование, ЗбОр, 360-363,416
- деацетилирование, 363,417
- димер НЗ/Н4,404,405р
- нуклеосомы, 261,261р, 266,266р
- петли шпилек в мРНК, 453р
- посттрансляционные химические моди-
фикации, 527т, 528, 528р
- свертки, 386т, 404
- химическая модификация, 360-365
- хроматин, 260-261, 261р
гликозилирование, 527т, 528, 528р
гликопротеиды, 528р
глицин, 23р, 27р
глобальная регуляция, 510
глобальная функция генома, 223-254
глутамин, 23р, 27р
глутамин-тРНКс1п, 497,497р
глутаминовая кислота, 23р, 27р
глутаминовая кислота-тРНКс|и, 497,497р
глушение РНК, 478-479
глушение генома, 369р, 367-372
глюкоза, 550р, 548-551
глюкокортикоидные гормоны, 547
голоферменты, 402,417р
голоцентрические хромосомы, 265
гомеодомены, 386, 386р, 386т, 388
гомеозисные гены, 564
гомеозисные избирательные гены
- Drosophila melanogaster, 580-581, 581р
- позвоночные, 581-582, 582р
- растения, 582-583
- эволюция, 725
гомеозисные мутации, 580-581
гомозиготность, 94р, 96р, 94-97,97р
гомологичная рекомбинация, 687-688, 688р,
689-698, 707
- Escherichia coli, 694р, 693-696, 696р
- репарация ДНК, 674, 697-698
- инактивация генов, 198,198р
- клонирование ДНК, 68
- модели, 689-693
- транспозиция, 702, 703р
- эукариоты, 696-697
гомологичные гены, 761-762,
гомологичные последовательности, 761-762
гомологичные хромосомы, 99
гомология последовательностей, 183р,
181-184, 687-688
гомоплазия, 754, 769, 769р
горизонтальный перенос генов, 307, 307р,
734-735
гориллы, 757, 758р, 770-772, 772р
горох (Pisum sativum}, 94р, 96,96р, 97,97р,
272т, 310т
горячие точки рекомбинации, 102,111
градиенты концентрации, 579р, 575-580,
580р
группа белков высокой подвижности (HMG),
386т
группы генов, 281р, 281-282
группы генов Нох, 581-582, 582р, 725
ГТФ (гуанозин-5’-трифосфат), 17, 716
гуанилилтрансфераза, 448р
гуанин, 8р, 12р, 13
гуанинметилтрансфераза, 448р
гуанозин-5’-трифосфат (ГТФ), 17, 716
гяРНК (гетерогенная ядерная РНК), 20
дарвиновская теория эволюции, 662,665, 770,
774
двадцатигранные капсиды, 322, 326
двойная рестрикция, 112,113р, 115
двойные гетерозиготы, 103
двойные гомозиготы, 103
двумерный гель-электрофорез, 234,234р, 237,
237р
двунаправленная репликация ДНК, 604р
двунитевая ДНК, 143,147
двунитевая РНК, 201р
двунитевой разрыв (ДНР)
- модель рекомбинации, 692-693, 693р, 694,
694р
- устранение, 674-676, 676р
деградация
890 Предметный указатель
- РНК, 444-445,445р, 475-480, 543т
- белков, 529, 530
деградосома, 445
дезаминированные основания, 443р, 669
дезаминирующие агенты, 653,653р
деацетилирование гистонов, 363
дезоксирибонуклеаза, 5
дезоксирибонуклеаза I (ДНКаза I), 45т,
390-391, 391р
дезоксирибонуклеиновая кислота, см. ДНК
действие гуанилатциклазы, 555
действие нуклеазы 3'^5'
- коррекция, 42, 646, 646р
- манипулирование ДНК, 42,44,44р
- репликация, 42, 609, 611
- секвенирование с обрывом цепи, 144
действие экзонуклеазы 5'—>3'
- манипулирование ДНК, 44р, 45
- репликация, 609, 615
- секвенирование с обрывом цепи, 144
действующие на РНК аденозиндезаминазы
(ADAR), 474,
дейтерий, 238
декэпирование, 409р, 475-476
декэпирующий фермент Dcplp, 475,476
деление клетки, 627-630
деление ядра, см. меоз
- кинетохоры, 265р, 266
- митоз, 97-99,99р, 627
- расщепление когезина, 622,622р
- согласование репликации ДНК, 627-630
Дельбрюк, Макс, 597, 599, 600, 663, 665р
денатурация, 45,143, 522, 522р
денатурирующий гель-электрофорез, 393
дентаторубральная-паллидолуизианская
атрофия, 649
дерево жизни, 753, 753р
деревья, см. филогенетические деревья
деревья видов, 758-760, 760р, 761
деревья генов, 758-760, 760р, 761
деформации конечностей, 581
дециклазы, 555
диауксия, 550р, 548-551,
дигибридное скрещивание, 96р,
дигидроурацил, 670т
дигидроуридин, 443р
дидезоксинуклеотидтрифосфаты, 140
дидезоксинуклеотиды, 140,141р, 144-145,
145р, 148,
дикий тип, 109,
димеризация
- активация STAT, 552, 552р
- рецепторы митогенов, 553
- роль рецепторов клеточной поверхности
в передаче сигналов, 551, 551р
- связывающиеся с ДНК белки, 397, 397р
- ультрафиолетовое излучение, 653,654р
димеры, 396-397,404,405р, 420, 547,
диметилсульфат (ДМС), 147,148р
диметилсульфоксид (ДМСО), 393р
динамика репликации, 630-631, 631р
диплоидные гаметы, 727р, 728
диплоидные клетки, 715, 716
дисульфидные мостики, 22,
дифракция, 383тп
дифракция рентгеновских лучей, 383тп
дифференциальное центрифугирование,
268тп, см. также центрифугирование по
градиенту плотности
дифференциальный сплайсинг, см. альтерна-
тивный сплайсинг
дифференцировка, 542, 569
длинные концевые повторения (LTR), 269р,
269-271, 329р, 331р, 331-333, 702-705
длинные рассеянные ядерные элементы
(LINE), 269р, 269-271, 333т, 334, 334р,
733-734
«длинные» продукты полимеразной цепной
реакции, 71р, 72
ДМС (диметилсульфат), 147,148р
ДМСО (диметилсульфоксид), 147
ДНК, см. также А-ДНК; В-ДНК; Z-ДНК
- взаимодействие с белком, 393-397
- выравнивание последовательностей,
761-762, 762р
- вытягивание гелем, 116-117,117р
- гены из, 4-7, 7р
- дупликация гена, 726, 726р
- изгибание, 396,
- исследования, 37-74
- маркёры, 87
- матрицы, 431р, 431-433,438
- мечение, 41тп, 52
- модифицирование, 367-372
- открытие, 4
- очистка, 58тп, 59р
- первые геномы, 716-717, 717р
- перенос бактериальной, 109,109р
- плавление, 672, 672р
- позиции на последовательности, 176-190
- полимеразная цепная реакция, 71р, 71-72,
72р
- размножение, 71р, 71-72, 72р, 726, 726р
- разрыв, 666, 674р, 674-676,676р, 687,
692-693, 693р
- расчесывание молекул, 116-117,117р
- сборка непрерывной последовательности,
149-161
- сборка непрерывной последовательности,
151р
Предметный указатель 891
- секвенирование, 138-172
- структура, 5,8-16
- структура двойной спирали, 5,13р, 15т,
9-16,16р
- структурные изменения, 643,650
- технология чипов, 91,93тп, 225р, 225-233
- ферменты для манипулирования, 39-53
- формы клеточной жизни, 1
- химическое сравнение с РНК, 17р
- электрофорез в агарозном геле, 49тп
ДНК-РСС5, 676, 676р
ДНК-аденинметилаза (ДАМ), 673
ДНК-белковые взаимодействия, 265р,
265-266, 266р, 296
- инициация транскрипции, 397-406
- мутации посадочных участков, 658
- нуклеосомы, 261,261р
- центромеры, 265р, 265-266
ДНК-гираза, 295, 600т, 602,
ДНК-гликозилазы, 669р, 669-670, 670т
ДНК-зависимые ДНК-полимеразы, 42р
ДНК-зависимые РНК-полимеразы, 17
ДНК-лигазы, 39р, 41, 52, 52р
ДНК-маркёры, 757
ДНК-метилтрансфераза 1 (Dnmtl), 368
ДНК-метилтрансфераза 3b (Dnmt3b), 368,
368р, 369
ДНК-метилтрансфераза За (Dnmt3a), 368,
368р, 370
ДНК-метилтрансферазы, 367-369
ДНК-полимераза а, 610,610т, 615-616
ДНК-полимераза 0, 670
ДНК-полимераза 6,610,610т, 615-616,617р, 670
ДНК-полимераза у, 610,610т
ДНК-полимераза к, 610т, 622
ДНК-полимераза 1,44т, 609, 610т, 615, 670,
670р, 672
ДНК-полимераза II, 609-610, 677
ДНК-полимераза III, 515-517, 610, 610т,
614-615, 616р
ДНК-полимераза IV, 677
ДНК-полимераза V, 677, 677р
ДНК-полимеразы, 39-45
- бактерии, 609-610, 610т, 614-615
- репарация ДНК, 610, 610т, 669р, 670, 670р
- затравки, 609,609р
- корректирование, 646,646р
- направленность, 609,609р
- обход повреждения ДНК, 677
- полимеразная расселина, 386
- секвенирование с обрывом цепи, 140-141,
141р, 144
- эукариоты, 610,610т
ДНК-топоизомераза, 295, 518, 596, 597, бООр,
600т, 600-602,602р
ДНК-транспозоны, 334-338, 340
- определение,
- организация генома человека, 269р,
269-271
- прокариоты, 335р, 335-337
- транспозиция, 701, 702
- эукариоты, 334-335, 337р, 337-338,338р
ДНК-фотолиаза, 668
ДНКаза I (дезоксирибонуклеаза I), 45т,
390-391, 391р
ДНР (двунитевой разрыв)
- репарация, 674-676, 676р
- модель рекомбинации, 692-693, 693р, 694,
694р
додецилсульфат натрия (ДСН), 233, 234
домен к-гомологии, 388
домен ТВР (связывающегося с блоком ТАТА
белка), 386т, 388
домен tudor, 195,195р
домен гомологии rel, 386т
домен связывания двунитевых рибонуклео-
протеидов (ДСднР), 388
домен связывающегося с блоком ТАТА белка
(домен ТВР), 386т, 388
домен смерти, 278, 279т
доменная модель сверхспирализации ДНК
прокариотов, 295, 295р
доменный подход к каталогам генов, 278р,
278-279
домены POU, 386т, 387,418,418р
доминантные аллели, 659
доминантный фенотип, 94р, 96,103-105,
105р, 105т
донорный сайт, 455
допущение Стёртеванта, 111
древняя ДНК, 777-778
древовидные схемы, 229р
дрейф генов, 774р
дрожжевые искусственные хромосомы (YAC),
64т, 66, 67р
- оптическое картирование, 117
- проект «Геном человека», 162,162р
дрожжевая дигибридная система, 239р, 238-241
дрожжи, см. также Saccharomyces cerevisiae
- встраивающаяся плазмида, 68,68р
- вызванное уксусной кислотой поврежде-
ние, 205
- интроны, 456
- комплекс белков-посредников, 417,417р
- межвидовые сравнения геномов, 729
- переключение типа спаривания, 559-560,
560р
- размеры геномов и число генов, 275
- репрессоры, 417-418
- фактор elongator, 450
892 Предметный указатель
ДСН (додецилсульфат натрия), 233, 234
дупликация генома в ходе эволюции, 729р,
726-731
дупликация домена, 731-732, 732р, 733
дупликация полного генома, 726-729
дупликация сегмента, 729р, 729-731
дупликация гена
- аллополиплоидия, 735
-данные, 722, 725
- процессы, 725-726, 726р
- эволюция, 721р, 721-726
дыня (Cucumis meld], 310т
енол-5-бромоурацил, 651,651р
енолаза, 445
енолгуанин, 647, 647р
енолтимин, 646, 647р
естественный отбор, 774
Жакоб, Франсуа, 409
железо, лактоферрин, 546
жизненные циклы, 559р
зависимое от дезаденилирования декэпиро-
вание, 475р, 475-476
зависимые от TAF и инициаторной последо-
вательности кофакторы (TIC), 404
задержанные мутации, 650
задержка умственного развития из-за хрупко-
го участка ХЕ, 649
закодированный изотопом ярлык по сродству
(ICAT), 237, 237р
законы наследования/открытие сцепления,
94-97
зафиксированные аллели, 774
закрытый промоторный комплекс, 402,402р
замена гена в два этапа, 199,206р
замена генов, 774
замены нуклеотидов, см. точечные мутации
замещение серой, 443р
запрограммированные мутации, 662-666
запрограммированный сдвиг рамки считыва-
ния, 515-517, 517р
зародышевые стволовые (ЗС) клетки, 199
застопоривание полимераз, 438,441,441р, 454
заторможенные фрагменты, 390
затравки
- RACE, 189,190р
- ДНК-полимераза а, 610,610т
- вытеснительная репликация, 602, бОЗр
- репликация, 609, 609р, 611-612, 612р, 615,
615р
- секвенирование в тепловом цикле, 145р
- секвенирование с обрывом цепи, 144-145,
145р
- синтез ДНК, 42р
- тест ARMS, 94р
захват, 189
захват ДНК при трансформации, 734-735
защита от модификации, 390-391, 393р
зеленые водоросли, 310т, 311
зеленый флюоресцентный белок, 352тп
зиготы, 692, 692р
значение FLpter, 120
значение бутстрап, 767
зонды
- FISH, 119р, 120р, 119-121,121р
- гибридизационные зонды, 51
- обход хромосомы, 153,154р
- отношение интенсивностей гибридиза-
ции, 226, 226р
зоогибридизация, 187,189р
ЗС (зародышевые стволовые) клетки, 199
инделы, 762, 762р
инициация
- репликации ДНК, 604-608, 630-632
- транскрипции, см. инициация транскрип-
ции
- трансляции, 505-512
инициация транскрипции, 380р, 379-420
- Escherichia coli, 402,404р
- ДНК-белковые взаимодействия, 397-406
- РНК-полимеразы, 402р, 402-406
- активаторы у бактерий, 411-412
- активаторы у эукариотов, 416р, 415-420
- обобщенная схема, 402р
- опознавательные последовательности,
398-401
- промоторы у бактерий, 398-400,400р,
400т, 402,409р, 410р, 408-412
- промоторы у эукариотов, 400-401,
404-406,414-415,415р
- регуляция, 406-420
- регуляция активности генома, 543т
- репрессоры у бактерий, 410р, 411р,
409-412,412р
- репрессоры у эукариотов, 418р, 417-420
- связывающиеся с ДНК белки и участки
прикрепления, 381-397
- соактиваторы у эукариотов, 415-416
- стратегии управления у бактерий,
408-412
- стратегии управления у эукариотов,
412-420
- структурное управление у бактерий,
408-409
инициаторная последовательность, 401,401р,
404,414
инициаторная тРНК, 508р, 509р, 508-512, 519
Предметный указатель 893
инициаторный комплекс, 405,405р, 406, 509
идентичность в поиске гомологии, 193
иерархическая группировка, 229,
избирательное редактирование, 474
изменения числа соединений, 600
изоакцепторная тРНК, 493,496
изолейцин, 23р, 27р
инсуляторы, 355-356, 356р, 358р, 416
изомеризация оснований, 443р
изотопы, 598-599, 599р, 630-631, 631р
изотопы 15N
- эксперимент Мезельсона-Шталя, 598,599р
- эксперименты по хронометражу реплика-
ции, 630-631,631р
изоэлектрическая точка, 234
изоэлектрическая фокусировка, 234, 234р
изучение популяций, 773-774, 781
изучение транскриптома
- Saccharomyces cerevisiae, 229-230,230р
- анализ последовательностей, 224-225
- методология, 224-233
- профили экспрессии, 229, 229р
- рак, 232-233
- сравнения на микроматрицах, 225р, 228р,
229р, 225-233
- функциональные взаимодействия белков,
243
иминоаденин, 647,647р
иммуноглобулины
- белок связывания с тяжелой цепью, 510
- мРНК, 474т
- разнообразие, 561р, 561т, 560-563, 563р,
564
- сверхмутация, 662р, 662-663,663р
- структура, 560,561р, 561т, 563р
иммунодефицит, неустойчивость центромер и
аномалии лица (ИНЦАЛ), 368-369
иммунологические методы, 71тп, 756
иммуноцитохимия, 206-209, 211р
иммуноэлектронная микроскопия, 504
импортины, 480
инактивация Х-хромосомы, 370р, 369-372, 564
инвертированные концевые повторы, 335р,
335-337
инвертированный повтор, 433,433р
ингибиторные SMADs, 557
индикаторы, 59, 59р
индол-3-глицеринфосфат, 247
индол-3-глицеринфосфатсинтаза, 247
индуцирование профагов, 326, 327р
метаболическая инженерия, 247,
инозин, 443р, 474,498, 500р, 501, 501р
инсулин, 525, 525р
интегрон, 298
интеины, 520,520р, 528-529, 529р
интерактомные сети, 243-245, 245р
интерлейкины, 552
интерфаза, 99р, 101р, 120-121
интерфазные хромосомы, 121, 261
интерфероны, 552
интроны, 454р, 455т, 470, см. также интроны
AU-AC; интроны GU-AU
-ДНК высших эукариотов, 177,179р
- внутривидовые сравнения, 739, 739р
- вырезание, 456р, 456-457,457р
- ген р-глобина, 454р
- гены человека, 455т
- дупликация гена, 726, 727р
- истоки эволюции, 737-740
- митохондриальные геномы, 311р, 311т
- мутации, 658
- плазмиды, 298
- пре-тРНК эукариотов, 467,470
- распределение, 454,455т
- самовырезающиеся интроны I группы,
466-467,468т
- сравнения между эукариотами, 273р,
273-274, 274т
- типы, 454,455т, 470
- удаление из ядерной пре-мРНК, 454-465
интроны AU-AC, 455т, 465
интроны GU-AG, 455т, 455-460
интроны GU-AG, происхождение в ходе эво-
люции, 738-740
интроны I группы
- происхождение в ходе эволюции, 737-738
- путь вырезания, 466-467,467р
- ранняя эволюция, 716
- распределение, 455т
- репатриация интронов, 529
- самовырезающее действие, 467,468т
интроны II группы, 468т, 470
- происхождение в ходе эволюции, 737-738
- происхождение интронов GU-AG, 739
- распределение, 455т
интроны III группы, 468т, 470
- происхождение в ходе эволюции, 737-738
- распределение, 455т
информационная проблема, 26
ИНЦАЛ (иммунодефицит, неустойчивость
центромер и аномальное лицо), 368-369
ионизирующее излучение, 653
ионообменная хроматография, 58тп, 59р
ионы металлов, 546р, 546-547
искусственная индукция, 201р
искусственные хромосомы на основе бакте-
риофага Р1 (ИХБ-Р1), 64т, 66
исследования ДНК на молекулярном уровне,
37-74
исследования РНК, 187тп
894 Предметный указатель
исследования взаимной гибридизации ДНК,
756, 770-772, 772р
исследования с двумя гибридами, 239р,
238-241
истинные деревья, филогенетика, 758,
истоки жизни, 714-719
история
- генетические маркёры, 85
- метод дробовика, 151
- открытие генов и ДНК, 4-7
- предыстория человека, 773-785
- проект «Геном человека», 161
- филогенетика, 754-757
кДНК (комплементарная ДНК), 45
кальмодулин, 242, 242р, 556
канцерогены, 650,650т
капиллярный электрофорез, 90,139тп, 141
капсиды, 322, 322р, 323, 323т, 326
капсиды типа головки с отростком, 322,326
капуста [Brassica oleracea), 310т
карбоксильный конец, 21,22р
кариограммы, 263,263р
кариотип XXX, 370,370р
кариоферрины, 480
каркасы, 159,159р
карта, 38
карта электронной плотности, 383тп, 384
картина G-исчерченности, 232р
картирование STS, 123
картирование при помощи конъюгации, 111р
картирующие реагенты, 124-127
карты межбелковых взаимодействий, 245р,
246
карты рестриктов, 157,157р
каскадные пути, 553-555,555р
кассеты удаления, 198-199,199р, 214, 214р
каталитическое действие РНК, 467,468т,
514-515, 715-717, 717р
каталоги генов, 277р, 278р, 279т, 277-279,
304т
каталоги функций генов, 277р, 278р, 275-282
квеозин, 443р, 444
кетогуанин, 647,647р
кетотимин, 646, 647р
киназы Януса (JAKs), 552р
кинетика репликации хромосомы, 630-631,
631р
кинетохоры, 265р, 266р, 267
кислые домены, 416
кладистика, 753-754
клады, 758, 758р
кластогены, 650т
клетка HeLa, 351р
клетки крови, 546
клетки рака поджелудочной железы, 232
клетки хомяка, 124,124р
клетки-предшественники, 575, 576
клеткоспецифические модули, 414,415р
клеточные культуры, 124, 626, 627р
клеточный цикл
- интерфазные хромосомы, 99р, 101р,
120-121, 261
- контрольные точки, 627, 632
- метафазные хромосомы, 99р, 101р, 262р,
263р, 261-266
- нарушение, 626, 627, 627р, 632,632р, 676р
- промежуточные периоды, 627
- регулирование репликации ДНК, 627р,
628р, 627-630, бЗОр
- фаза синтеза, 627
клонирование ДНК, 38, 38р, 55р, 53-70,833р
клонирование генов, 38, 38р, 53-70
клонирующие векторы, 38р, 53-70
клонирующий вектор pBR322,56
клонирующий вектор рУАСЗ, 67р
ковалентная модификация, 364,364р
когезиновые белки, 622,622р
кодирующая РНК, 17
кодоминирование, 96,96р, 105,106р
кодоны, 26, 27р, 176р, 177р, 176-180
кодоны пунктуации, 26, 27р
количественная ПЦР, 778
коллаген, 527т
коллаген а2 I типа, 732р, 732-733
Коллинз, Френсис, 162
кольца, гомеозисные избирательные гены,
582-583
кольцевые хромосомы
- терминация репликации ДНК, 619р,
619-620, 620р
- инициация репликации ДНК, 597, 604р,
602-605, 605р
- органеллы, 309
- сайт-специфическая рекомбинация, 700,
700р
комбинаторный отборочный анализ, 156,156р
компактные геномы, 181
комплекс
- ADA, 361
- Antennapedia (ANT-C), 580-581,581р
- Bithorax (ВХ-С), 580-581,581р
- NuRD, 363, 369
-SAGA, 361,404,416
- Sin3, 363, 369
- Swi/Snf, 363, 367,416
- TFTC, 361
- UmuD'22C, 677
- гомеозисных генов (HOM-C), 581, 582p,
725
Предметный указатель 895
- инициации транскрипции, 379-420
- опознавания областей инициации (ORC),
605, 606р, 608,628,630
- посадки кэпа, 509, 509р, 510
- привлечения, 459р, 459-460,460р
- продления транскрипции, 431р, 432,432р
- репрессор-триптофан, 411,411р
- эндонуклеазы UvrABC, 670р, 672,
комплексные мультигенные семейства,
281-282
комплексные филогенетические сети,
769-770, 770р
комплементарные нуклеотиды, 39,42,
компьютеры
- биоинформатика, 176
- выравнивание последовательностей, 193,
762, 764тп
- поиск гомологии, 193
- последовательность Haemophilus
influenzae, 151
- построение сетей, 770р
- секвенирование методом дробовика для
полных геномов, 158
- функция гена, 192-196
концевые последовательности, 151р, 158р
конкретный пример аннотирования генома,
212р, 211-214
конкурирующие сигналы, 575-576
консенсус, 179
консенсусная последовательность Козак, 509р
консенсусные последовательности, 180р,
400т, 408,409
консервативная репликация, 598р, 597-599,
599р
консервативная транспозиция, 330, 331р, 701,
702р, 703
консервативные последовательности,
455-456,456р
контекстнозависимое переназначение кодо-
нов, 27, 29т
контиги, 151р, 152р, 151-153, см. также сбор-
ка контигов из клонов
контрольные точки, 627,632
конформации спирали, 394-396, см. также
а-спирали; двойная спираль; тройная
спираль
концевая дезоксинуклеотидилтрансфераза,
52, 53р
концепции вида, 305-307
конъюгация, 109,109р, 111р, 297т, 734
корневые деревья, 757, 757р, 758, 758р
короткая интерферирующая РНК (siPHK), 18,
18р, 478,478р
«короткие» продукты полимеразной цепной
реакции, 72р
короткие (простые) тандемные повторения,
88р, 90, см. также микросателлиты
короткие последовательности ДНК
- затравки для секвенирования с обрывом
цепи, 145р
- клонирование в векторе М13,143
- метод дробовика, 151р, 152р, 149-153
- пиросеквенирование, 148,149р
короткие прямые повторения, 701, 701р
короткие рассеянные ядерные элементы
(SINE), 124, 269р, 269-271, 333т, 334, 334р
коррекция, 42,44р, 609, 611, 646, 646р
косвенные воздействия мутаций, 654
косвенные мутагены, 650
космиды, 64т, 64-66, 66р,
коэффициент седиментации (величина S),
502,530
красная вискача, 728
кривая одноэтапного роста, 325, 326р
Крик, Френсис, 8-12,493,498, 596р, 596-597,
600
криптогены, 475
кристаллины, 195
кроссинговер, 687, 688, 690, 690р
крупные наборы данных, филогенетика, 753,
754
крыловатая эндонуклеаза (FEN1), 616,617р,
650
крыловатый мотив спираль-изгиб-спираль,
386т, 387
кукуруза, см. Zea mays
курица (Gallus gallus), 275т
кэп 0-го типа, 449,
кэп 1-го типа, 449,
кэп 2-го типа, 449,
кэпирование мРНК эукариотов, 20, 20р, 448р,
447-449, 508-509, 543т
кэпирование транскриптов, 448р, 447-449
кэпы, 20, 20р, 449, 508-509
лактопермеаза, 242
лактоферрин, 546
Ламаркова теория эволюции, 662,663,665-666
латание длинными заплатами, 672-673,
латание короткими заплатами, 670р, 670-673
Лаурия и Дельбрюк, 663, 665р
лейкемия, 232, 354, 354р
лейцин
- группа R, 23р
- кодоны, 27р
- оперон Escherichia coli, 438
- отклонение частоты использования кодо-
нов, 179
- последовательность тРНК Escherichia coli,
180р
896 Предметный указатель
лейциновая застежка-молния, 397р
лейциновая застежка-молния типа bZIP, 397
летальные мутации, 661
летучие мыши, 754
лечение лекарственными препаратами, 247
лигазы, 39р, 41, 52, 66р
лизин
- аминоацил-тРНК-синтетазы, 496т
- ацетилирование, 363
- группа R, 23р
- деацетилирование, 363
- кодоны, 27р
- метилирование, 363-364
- убиквитинирование, 364
лигирование (6-4), 653, 654р
лизис, 62р
лизогенный цикл инфицирования, 323-326,
327р
- бактериофаг X, 61р, 62,62р, 568р, 567-569
- сайт-специфическая рекомбинация, 698р,
698-700, 700р
лизоцим, 58тп,
лимфома В-лимфоцитов, 233
линейные хромосомы
- бактерии, 296, 297т
- органеллы, 309
- поддержание концов, 622-627
- раскручивание, 597
линкерная ДНК, 261,261р,
линкерные гистоны, 261, 261р, 352,
линкеры, лигирование ДНК, 52, 53р
Линней, Карл фон, 753
липидные мембраны, 326, 716-717
липкие концы, 48,48р, 52р, 53
литический цикл инфицирования, 323-326,
326р, 327, 567-568,
- бактериофаг X, 61р, 62,62р
локус IGH, 561т, 563р
локус IGK, 561т
локус IGL, 561т
лотки микротитра, 156,156р
лошади, 754
лучеперые рыбы, 725, 725р
львиный зев (Antirrhinum), 582-583
мРНК а-галактозидазы, 474т
мРНК dnaX, 515-517,517р
мРНК (матричная РНК), 17-18,18р,
- анализ последовательностей в ходе изуче-
ния транскриптома, 224-228
- блокирование, 448р, 447-449
- взаимодействие кодон-антикодон, 498р,
500р, 498-501, 501р
- деградация, 444-445,445р, 475-480, 543т
- завершение синтеза, 451р, 450-453
- изучение транскриптома дрожжей,
229-230
- надзор, 476р, 476-478
- обработка, 543т
- перенос, 480-482,482р
- продолжение транскрипта, 449-450
- рак, 232-233
- регуляция синтеза, 453-454
- редактирование, 474р, 474т, 472-475
- синтез РНК-полимеразой II, 447-454
- транссплайсинг, 463р, 463-465,465р
мРНК аполипопротеина В, 472-474,474р, 474т
мРНК бактерий
- деградация, 444-445,445р
- обработка, 441
- аттенюация, 437р, 437-438,439р
мРНК нейрофиброматоза 1-го типа, 474т
мРНК опухоли-1 Вильмса, 474т
мРНК рецептора глутамата, 474т
мРНК эукариотов
- деградация, 475-480
- терминация синтеза, 451р, 450-453
- кэпирование, 448р, 447-449
- обработка, 543т
- перенос в клетках, 480-482,482р
- продолжение транскрипта, 449-450
- регуляция синтеза, 453-454
- редактирование РНК, 474т, 472-475,485р
- синтез РНК-полимеразой II, 447-454
- транссплайсинг, 463р, 463-465,465р
Макклинток, Барбара, 335, 337-338
макрохромосомы, 265,
малая бороздка,
- ДНК В-формы, 13р, 15,15т, 16р, 394, 396р
- связывающиеся с ДНК белки, 381,386,
386р, 387
малая ядерная РНК (мяРНК)
- РНК-полимераза II, 398т
- блокирование, 449
- перенос в клетке, 480,482р
- содержание в клетке, 18,18р
- сращивание, 457,460
- эукариотов, 449,480,482р
малая ядрышковая РНК (мякРНК)
- ген, 398т
- перенос в ядерных клетках, 482р
- содержание в клетке, 18,18р
- химическая модификация рРНК, 472,472р
малые ядерные рибонуклеопротеиды
(мяРНП), 457р, 459р, 457-460,465
манноза, 528р
маркёры
- анализ с неполной рестрикцией, 115,115р
- картирование меченых участков последо-
вательности, 123р, 121-127,127р
Предметный указатель 897
- перенос бактериями, 111р
- составление генетических карт, 84р, 85т,
84-87
масс-спектрометрия, 237р, 233-238,
масштаб карт рестриктов, 115-116
материнские клетки в процессе спорообразо-
вания, 571р, 569-573, 573р, 575р
матрицы, 39, 39р,
матрицы расстояний, 764р, 762-765,
матричная РНК, см. мРНК
матураза, 467,
мегаплазмиды, 297т, 298
межбелковое поперечное сшивание, 504,
межвидовой перенос генов, 735
межвидовые сравнения, 739, 739р
межгенные области, 177,282-285,
Международный консорциум по секвенирова-
нию генома человека, 163
межклеточное взаимодействие, 278, 278р,
573-576
межмолекулярная рекомбинация, 698р,
698-700, 700р
межфазный переход G1-S, 628-630
мейоз, 99,101р,
- двунитевые разрывы, 693
- кроссинговер, 687, 688
- неполное сцепление, 101р, 97-102,102р
- спорообразование у дрожжей, 230, 230р
мелиттин, 524-525, 527т
Мендель, Грегор, 85,94р, 96р, 94-97,97р
местоположение гена, 183р, 186р, 189р,
181-190,190р
местоположения генов в последовательности,
176-190
метаболический поток, 247
метаболомика, 246-247
метагеномика, 307
метафазные хромосомы, 99р, 101р, 262р, 263р,
261-266
метилирование
- ДНК, 367р, 369р, 367-372
- восстановление, 669
- геномный импринтинг, 369-370
- гистоны, 363-364
- инактивация Х-хромосомы, 369-372
- модификации нуклеотидов, 443р, 444
- мутагенные агенты, 653
- нормальная репликация ДНК, 673, 673р
- ограничивающая транспозиция, 707
- посттрансляционная обработка белков,
527т, 528р
- рРНК при помощи мярРНК, 472,472р
метилирование de novo, 367р, 368,368р
метилирование ДНК, 367р, 369р, 367-372, 417
метилирование лизина-4, 364
метилирование цитозинов, 333
метионин
- боковая группа, 23р
- инициация трансляции в процессе синте-
за белка, 508
- кодоны, 27р
- остатки, 432
- превращение в N-формилметионин, 497р,
498
метод сборки контигов из клонов, 149
- анализ STS, 127,127р
- быстрый, 157р, 156-158
- обход хромосомы, 153,154р
- сборка последовательности, 153-158,
158р
- секвенирование, 82-84,84р
метод выравнивания последовательностей на
основе расстояний, 762
метод дробовика для полных геномов, 82,84р,
149,158р, 158-161
метод максимальной бережливости, 765
метод объединения соседей, 764тп, 765,765р
метод отпечатков пальцев клонов, 156-157,
157р
метод подобия, 192,193, 762
метод точечных матриц, 762р
методология
- исследования РНК, 187тп
- исследования транскриптомов, 224-233
- метод сборки контигов из клонов,
153-158
- метод химической деградации, 145-148,
148р
- определение белковых профилей, 234р,
237р, 233-238
- пиросеквенирование, 148,149р
- препарат однонитевой матрицы ДНК, 143,
143р
- сборка непрерывной последовательности
ДНК, 158р, 149-161
- секвенирование ДНК, 139-148
- секвенирование ДНК с обрывом цепи,
140р, 141р, 140-145,145р
- секвенирование в тепловом цикле, 145,
145р
- секвенирование методом дробовика для
полных геномов, 158-161
- электрофорез в полиакриламидном геле,
139тп
методы гибридизации в растворе, 91,91р
методы исследования, изучение ДНК, 37-74
механически вытянутые хромосомы, 120-121
мечение, см. мечение флюоресцентными мет-
ками; мечение радиоактивными метками
мечение концов, 41тп, 53,187тп
898 Предметный указатель
мечение радиоактивными метками
- бактериофаги, 5, 7р
- гибридизационные зонды, 41тп, 52
- гибридизация in situ, 119
- секвенирование методом химической
деградации, 147
- эксперименты над режимами репликации,
598
мечение транспозонами, 199, 201р, 212,212р,
мечение флюоресцентными метками, 351,
351р
- ДНК, 41тп
- двумерный электрофорез, 237
- дидезоксинуклеотидтрифосфаты, 140,
141,141р
- иммуноцитохимия, 209, 211р
- исследования транскриптомов на чипах,
225, 225р
- технология чипов ДНК, 91,91р, 93тп
меченый участок последовательности (STS)
- библиотеки клонов, 125-127,127р
- картирование, 112,123р, 121-127,127р
- картирование содержимого, 157р,
157-158
- проект «Геном человека», 162
миграции в Новый Свет, 781-785
миелоидные раки, 232
микроРНК (миРНК), 18,18р, 398т, 479-480,
480р
микроматрицы, 91,93тп
микрорибонуклеопротеид (миРНП), 479
микросателлиты, 271, 284-285,
- геном человека, 269р, 271, 271р, 285, 285р
- определение генетических профилей, 285,
285р
- проскальзывание репликации, 647,649р
- типизация простых тандемных повторе-
ний, 88,88р, 90
- филогенетика человека, 774
микросомы, см. рибосомы
микротрубочки, 265р, 266
минералокортикоидные гормоны, 547
минигены, 190
минимальные генетические требования, 304
минимальные геномы, 720-721
минипоследовательности, 159р, 159-161
минисателлитная ДНК, 284-285, см. также
переменное число тандемных повторений
минихромосомы, 265
миобластный модуль, 414
миотоническая дистрофия, 649
миоцин, 565
мир РНК, 443, 715-716, 716р
митогены, 553,553р
митоз, 97-99,99р, 627
митотические циклины, 628, бЗОр
«митохондриальная Ева», 776, 777р
митохондриальная РНК-полимераза, 398
митохондриальные геномы, 308, 314
- генетическое содержимое, 309р, 311р,
311т, 309-312
- клетки человека, Зр
- нестандартные генетические коды, 29т
- перенос ДНК, 308-309
- размер генома, 310т
- репликация ДНК, 351-352, 610
- физические особенности, 309,309р
митохондрии грибов, 310т
Мишер, Иоганн Фридрих, 4
млекопитающие
- автополиплоидия, 728
- области инициации, 606-608
- филогенетическое дерево, 758
многоаллельные гены, 774
многобелковые комплексы, 241, 241р, 361,
363,404,445
многогенные единицы транскрипции, 466
многодоменные белки, 738, 738р, 739-740
многоклеточные организмы
- модель развития, 573-576
- последствия мутаций, 658-659
многократная генная конверсия, 722
многократные замены, 764
многократные попадания, 764
многомерный метод опознавания белков
(MudPIT), 242
многораздельные геномы, 296, 298, 309
многосубъединичные белковые комплексы,
450
многоцистеиновый цинковый палец, 386т,
387
«множественные промоторы», 415
множественные аллели, 85-87
множественные выравнивания последова-
тельностей ДНК, 762
множественные копии, 202
мобильные, 335р, 337
мобильные генетические элементы, 322,
330-338, 340
- LTR-ретроэлементы РНК, 331-333
- ДНК-транспозоны, 334-338
- горизонтальный перенос генов, 735
- геном человека, 333, 333т, 334, 337
- не содержащие LTR ретроэлементы РНК,
334
- прокариоты, 301
- сравнение вирусов и эукариотов, 331-333,
ЗЗЗр
- транспозиция, 701-707
- эволюция генов, 733-734, 734р
Предметный указатель 899
- эволюция генома, 737, 737р
модель «разрыва и воссоединения», 597
модель АВС, 583
модель Мезельсона-Реддинга, 689,690,692р
модель волнообразного продвижения, 779-781
модель изохор, 267-268
модельные организмы, 199
модификационное препятствование, 391-393,
394р
модифицирование концов, 20,20р
модули для промоторов РНК-полимеразы II,
414-415
модуль
- Antennapedia, 414
- Bicoid, 414
- гипофизных клеток, 414
- лимфоидной клетки, 414
- реакции на цАМФ (CRE), 414
- сывороточной реакции, 414
- теплового шока, 414
молекулы рекомбинантной ДНК, 38
молекулярная филогенетика, 752-791
- области инициации, 753-757
- области применения, 770-785
- филогенетические деревья, 753-770
молекулярная эволюция, 713-749
молекулярные
- моторы, 694
- шапероны, 523-524,
- науки о жизни,
- признаки, 753-757
- - деревья генов и видов, 760р
- - надсемейство генов глобинов, 722р,
723-725
- - определение,
- - ретровирусы, 772
- - филогенетический анализ, 767-768,
768р
- - эволюция человека, 770
молоко, 546
молчащие кассеты типа спаривания, 559
молчащие мутации, 655,
моменты включения точек старта реплика-
ции, 630-631, 631р
Моно, Жак, 409, 548
моногибридное скрещивание, 96р,
монофилетические последовательности,
757-758, 758р
Морган, Томас Хант, 97-103, 576-577
морской ёж (Strongylocentrotus purpuratus),
272т
морской анемон (Metridium senile), 310т
морфологическая эволюция, 719-720
морфологические признаки, 753-756,
758-760
мотив HU/IHF, 386т
мотив НТН (спираль-изгиб-спираль), 381р,
386т, 386-387,396-397,410
мотив лента-спираль-спираль, 386т, 388р,
мотив спираль-петля-спираль, 397
мотив цинковый палец, 381, 386т, 387, 397
- Caenorhabditis elegans, 387
- каталоги генов, 278, 279т
- мультицистеин, 386т, 387
- рецептор стероидных гормонов, 387,387р,
547
- ядерные рецепторы, 547
мочевина, 139тп, 522,522р
мультигенные семейства, 281р, 281-282, 722,
769
мутагенез, 205-206, 208-209тп
мутагенез in vitro, 205, 208-209
мутагены
- двунитевой разрыв, 674
- замены нуклеотидов, 642, 643, 668
- определение, 650
- реакция SOS, 677
- физические, 653-654,668
- химические, 650-653,668
мутанты, 641-666
мутасомы, 677
мутации, 641-666
- Нох, 581
- дублированные гены, 721
- зародышевых клеток, 642,659
- инженерия обмена веществ, 247
- карты межбелковых взаимодействий, 245,
245р
- методы отборочных испытаний, 642тп
- микроорганизмы, 659-662
- многоклеточные организмы, 658-659
- обнаружение, 642тп
- ошибки репликации, 646-650
- последствия, 654-666
- причины, 642-654
- псевдогены, 282
- реакция SOS, 677
- с гаплонедостаточностью, 659
- с потерей смысла, 655
- с потерей функции, 659,659р
- с приобретением функции, 659
- скорость, 767-768, 768р, 772
- стоп-кодона, 657,657р, 665
- типа замен, см. точечные мутации
- типа сдвига рамки считывания, 647, 657р,
657-658
- типа сквозного считывания, 657,657р
- типа удалений, 642, 647, 653, 657р, 658
- типы, 641-642
- филогенетические деревья, 760,760р
900 Предметный указатель
мутация antennapedia, 580
мутация с потерей смысла, 657,657р, 665
мутация типа вставки, 642, 647-650, 657, 762,
762р
мышечная дистрофия, 460
мышечная дистрофия Дюшенна, 415
мышь, см. Mus musculus
мяРНК, см. малая ядерная РНК
мяРНП (малые ядерные рибонуклеопротеи-
ды), 457р, 459р, 457-460,465
мякРНК, см. малая ядрышковая РНК
мякРНК эукариотов, 482р
наведенный РНК комплекс глушения (RISC),
478,478р
надсемейства генов, 282, 723-725, 739-740
надсемейство генов глобинов, 192, 281р,
281-282
- интроны, 454р, 455т
- картины экспрессии, 281р, 281-282
- происхождение человека, 777
- управляющие локусом области, 358р,
358-359
- эволюция, 722р, 723р, 723-725, 739-740,
740р
надсемейство рецепторов фактора некроза
опухолей, 269
надсемейство ядерных рецепторов, 547
наживочные белки, 242, 243р
наличие питательных веществ, 109,542,661,
661р, 665
наложение оснований, 12, 394,494
направленность транскрипции, 608р, 608-
609,609р, 611
направленный мутагенез, 205-206, 208-209
направляемая транспозонами экспрессия
генов, 737
направляемые матрицей ДНК-полимеразы,
42р, 41-44,44р, 144
направляемый матрицей синтез ДНК, 42р,
44т, 608р, 608-609
направляемый матрицей синтез РНК, 12,17р,
431р, 608, 608р
направляющая РНК, 475,475р
наращивание гомополимеров, 52, 53р
наследственные болезни, см. генетические
болезни
наследственный неполипоидный рак прямой
и ободочной кишки (ННРПОК), 679
шапероны Hsp70, 523, 523р, 524
насыщение двойной связи, 443р
нацеленный на олигонуклеотиды мутагенез,
208-209тп
независимые от матрицы ДНК-полимеразы,
52-53
несодержащие LTR-ретроэлементы, 334,334р
неспецифичные к последовательности связы-
вающиеся с ДНК мотивы, 386т
неандертальцы, 778р, 777-779
негомологическое соединение концов (NHEJ),
666, 674-676, 676р
недокручивание ДНК, 602
недометилирование, 369
неизвестные гены, изучение функциональной
активности, 205-209
некодирующая ДНК, 655, 658, 735-740, 756
некодирующая РНК, 17,18,18р
некорневые дереья, филогенетика, 757, 757р
неметафазные хромосомы, 99р, 101р, 120-
121,261
неметилированные нити ДНК, 673, 673р
неомицинфосфотрансфераза II, 701
неопознанные прокариоты, метагеномика,
307
непенетрантность, 659,
неполиморфные участки рестрикции, 112р
неполная рестрикция, 112,113р, 115,115р
неполное доминирование, 96,96р
неполное сцепление, 97р, 97-102
неполные кДНК, 189
неполярные группы, 23р,
непрерывная репликация ДНК, 609,609р, 615,
616р, 617
неравный кроссинговер, 725, 726р
неравный обмен сестринскими хроматидами,
725, 726р
нервная система, 582,582р
несинонимические мутации, 655,657р, 768
несоответствия, 642, 643р
неспецифичная гибридизация, 120,121р
нестандартные генетические коды, 26-27,29т
неходжкинская лимфома, 232
нижележащий промоторный элемент (НПЭ),
401,401р
нитевидные капсиды, 322, 326
ННРПОК (наследственный неполипоидный
рак прямой и ободочной кишки), 679
новые гены
- изучение функций, 208тп, 205-209, 211р
- эволюция, 720-735
нозерн-блоттинг, 186,
нозерн-гибридизация, 186,186р, 187тп,
нокаутные мыши, 70,199, 201
нуклеаза, 39р, 41,45-52
нуклеаза S1,45т, 192р
нуклеазный футпринтинг, 391р, 390-393,
393р
нуклеиновая кислота, 4, см. также ДНК; РНК
нуклеозиды, 8-9
нуклеоиды, 294, 294р
Предметный указатель 901
нуклеосомы
- археи, 296
- модификация гистонов, 360-365
- перестановка, 372
- перестройка, 365р, 365-367,416
- стержневой октамер, 360, ЗбОр
- структура, 261, 261р, 266, 266р
нуклеотиды
-ДНК/РНК,1
- мечение радиоактивными метками, 41тп,
52
- определение, 8-9,9р
- структурные изменения, 643, 643р
- таутомерия, 646-647, 647р
- химическая модификация, 443р, 444
нуклеотиды дезоксиаденозины, 433
«нулевые» гены, 401
обезьяны, 757, 758р, 772-773, 773р
области инициации, 606
области инициации репликации, 55,66,66р
- археи, 617-619
- бактерии, 604р, 604-605,605р
- моменты включения, 631р, 630-632
- эукариоты, 606р, 605-608,628
области прикрепления к каркасу (SAR), 355,
355р
область Т-ДНК, 70, 70р
облигатные паразиты, 304
обмен полинуклеотидами, 690, 690р, 692р
обогащенные G нити, 625
обогащенные глутамином домены, 416
обогащенные пролином домены, 416
обозреватель Genotator, 184р
оболочки спор, 569
обособленные популяции, 774
обоюдный обмен нитями, 690, 690р
обрабатывающая способность, 144
обработанные псевдогены, 282, 282р, 726
обработка
- РНК, 20, 20р, 441-444
- белка, 519-529, 543т
обработка концов полипептидов, 524р,
524-525
образование гетеродуплекса, 689,690,690р,
692р, 694, 694р
образование дипептида, 147
обратная гираза, 600т, 602
обратная транскриптаза, 327
- особенности, 44т
- ошибки, 772
- ранняя эволюция ДНК, 717
- репликация ретровирусов, 45
- транспозиция ретроэлементов, 703, 705
обратная транскрипция
- дублирование гена, 726, 727р
- обработанные псевдогены, 282,282р
- теломер Drosophila, 626-627
- теломераза, 622-625, 625р
обратное прослеживание, 432,438-441,441р
обратные повторения, 335-337, 337р, 547,
734р
обход поврежденной ДНК, 676-677
обход хромосомы, 153,154р
общие предки, 183р, 181-184,184р
обычные псевдогены, 282
ограничения, 139тп
ограничивающие условия роста, 661
одиночные сироты, 212
одноклеточные организмы, 542, 659-662
однонитевые матрицы ДНК, 143,143р
однонитевые разрывы, 668,668р
одноучастковые мутации, 641-642,642р, 643
окисление, 669
окрашивание по Гимзе, 262, 262т, 263р
окрашивание флюоресцентными красителя-
ми, 125,125р
окрашивание хромосом, 262, 262т
октамерный модуль, 414
олигонуклеотиды
- анализ на чипах, 225-233
- обход хромосомы с помощью ПЦР, 153,
154р
- олигонуклеотиды BsmFl, 224, 224р
- синтез ДНК, 42
- смыкание пропусков при секвенировании
методом дробовика, 152р, 152-153
онкогены, 650т
оператор, 410р, 409-411,
опережающая нить, 609,609р, 614,615,615р,
646
оперон рар, 445
оперон лактозы, 301, 302р, 508т, 546
- диауксическая реакция, 550р, 548-551
- промотор, 398-400,400р, 400т
- проскальзывание трансляции, 517р
- регуляторное управление инициации
транскрипции, 410р, 409-411
- контитутивная (постоянная) экспрессия,
661, 662р
оперон триптофана, 302, 302р
- Escherichia coli, 400т, 411,411р, 412,412р
- аттенюация, 437р, 437-438,439р
опероны, 302, см. также оперон лактозы; опе-
рон триптофана
- активация, 248, 248р
- антитерминация, 436
- археи, 302
- бактерии, 301р, 302р, 300-303
- оперон рар, 445
902 Предметный указатель
- определение,
- прокариоты, 301р, 302р, 300-303
- синтез жгутика, 248, 248р
- функциональные взаимодействия белков,
243
опознавание белков протеома, 235-243
опознавание белковых взаимодействий,
238-245
опознавание дочерней нити, 673, 673р, 674
опознавание кодон-антикодон, 493,498-501,
опознавание пары оснований А-Т, 394, 396р
опознавательная последовательность обла-
стей инициации, 605
опознавательные последовательности для
инициации транскрипции, 398-401,412
опознающая спираль, 386, 387
опорная клетка, 575, 576р
опосредствуемое бессмысленным кодоном
разложение РНК (NMD), 476р, 476-478
определение биохимических профилей,
246-247
определение генетических профилей, 285,
285р
определение пола, 461-462,462р
определение счетов RFLP, 87-88, 88р
оптическое картирование, 117р, 116-119
опухоли, 474, 650, 659
орангутан, 757, 758р
органеллы
- генетическое содержимое, 309р, 311р,
311т, 309-312,312р
- интроны, 455т, 467,470
- перенос ДНК, 308, 309
- происхождение, 308
- физические особенности генома, 309,
309р, 310т
организация генома, 322-323, 323т, 325р
- прокариоты, 298-303
- эукариоты, 267-275
ОРС (открытые рамки считывания), 176р,
184р
- гены-репортеры, 208
- застопоривание рибосомы, 438
- скольжение, 177р, 176-184
ортологичные гены, 192,193р, 758-760
основные домены, 386т, 388
основная рекомбинация, см. гомологичная
рекомбинация
основная скорость транскрипции, 409,412
основное средство поиска локальных вырав-
ниваний (BLAST), 193
базальный уровень инициации транскрип-
ции, 408,409,410р
основные промоторные элементы, 414,415р
основные промоторы, 400
основные факторы транскрипции (GTF), 405т,
404-406
остатки серина, 364
островки Лангерганса, 525
островки генов, 305
острый лимфобластный лейкоз, 232
острый миелоидный лейкоз, 450
отбор, 189
селективные маркёры, 56р
отжиг, 42, 823
отклонение частоты использования кодонов,
179
открытые рамки считывания, см. ОРС
открытый промоторный комплекс, 402,402р,
411
отношение акриламид:бис, 139тп
отношение массы к заряду, 235,237р
отрицательно заряженные боковые группы,
23р
отстающая нить, репликация ДНК
- затравливание, 611-612
- непрерывный синтез, 609,609р, 611-612,
612р,615р, 614-616, 617р
- определение,
- ошибки синтеза, 646, 650
- проблема, 609
- укороченная копия, 622, 623р
очистка ДНК, 58тп, 59р
очистка промотора, 402,402р, 447р, 447-448,
454
ошибки
- каркасы сборки последовательности, 159
- метод дробовика для полных геномов,
158р, 158-161
- обратной транскриптазы, 772
- репликации ДНК, 642, 643р, 646-650,
696р, 697-698, 698р
плазмидный вектор pUC8, 56, 56р, 59, 59р
палец Цис2Гис2, 386т, 387, 387р
панредактирование, 475,475р
парадокс показателя С, 273-275
паразитические виды, 720-721
параллельные мутации, 769, 769р
параллельный гибридизационный анализ,
93тп
паралогичные гены, 192,193р
паранемная структура, 597
параретровирусы, 327
парафилетические группы, 758,758р
парный гомеодоменный мотив, 386т
пары оснований РНК-ДНК, 435р
патогенность, 297т, 305
пептидилтрансфераза, 513-515
пептидильный (Р) участок, 512,512р
Предметный указатель 903
пептидная нуклеиновая кислота (ПНК), 719,
719р
пептидные связи, 21,22р
- образование, 512р, 513-515, 532, 715, 716
первичная регуляция, 408,408т
первичная структура, 21,21р
первые гоминиды, 740
первые европейцы, 778р, 779р, 777-781, 781р
первый закон генетики, 96,96р
перебежные точечные мутации, 642,
переваривание рестриктазами, 51р, 51-52
переваривание, фрагменты рестрикции ДНК,
48,48р
передатчик сигналов и активатор транскрип-
ции (STAT), 552р, 552-553
передача, 109
передача сигналов
- внеклеточные сигнальные соединения,
545-557
- интерпретация пути, 556р, 556-557
- каскадные пути, 553-555,555р
- множество шагов на пути от рецептора к
геному, 553-555
- один шаг между рецептором и геномом,
552-553
- по обходному пути, 545,545р, 551р,
551-557
- прямая, 545р, 545-551
- рецепторы клеточной поверхности, 551р,
551-557
- через вторичных посредников, 555-556,
556р
- число генов пути, 277, 277р
передне-задняя ось, 577-579,579р
переключение класса, 561-563,564р
переключение типов спаривания, 559-560,
560р
перекрестная связь, 356,358р
перекрестное опыление, 94р
сверхспирализация ДНК, 602
перекрывающиеся гены, 323, 325р
перекрывающиеся последовательности, 82,
82р, 84р, 154,154р
переменное число тандемных повторений
(VNTR), 88,90
перемещение точки ветвления, 689,690р, 694,
696, 696р
перенос, 109р, 111р, 733-734
перенос в эписомах, 109,109р
перенос генов бактерий, 735
перенос, перестройка нуклеосом, 365р, 367
перепечатывание реплики, 661р,
перестройка нуклеосом, 365р, 365-367,416
перестройки, 38р, 559-563
перестройки генов в ходе эволюции, 731-734
перестройки геномов, 559-563, 737, 737р
перетасовка доменов, 732, 732р, 733, 733р
переходная точечная мутация, 641-642
перешеек через Берингов пролив, 782-783,
783р
период инкубации, 325, 326р
период покоя, 569
пероксиды, 650, 653
персульфат аммония, 139тп
петли обратной связи, 565,565р
печеночники, 310т
пигментная ксеродерма, 679
пикорнавирусы, 509-510
пиль, 109р
пиперидин, 147,148р, 393
пиримидины, 8,179,455
пиросеквенирование, 145,148,149р
пирролизин, 22, 25р, 27, 29т, 498, 527
плавление двойной спирали, 604-605,605р,
606,606р
плавучая плотность, 268тп
плазмида F, 66
плазмида Ti, 70, 70р
плазмиды, 296, 296р, 297т, 298
- YIp5, 68, 68р
- клонирование ДНК, 38р, 55р, 53-70
- перенос генов, 734
- препарат однонитевой матрицы ДНК, 143,
143р
- эксперименты со стартами репликации,
604,606
плакофилин, 1, 269
плезиоморфные состояния признака, 754
плектонемная спираль, 597,
плодовые мушки, см. Drosophila melanogaster
ПНК (пептидная нуклеиновая кислота), 719,
719р
ПНФаза (полинуклеотидфосфорилаза), 445
повреждение ДНК
- агенты окружающей среды, 650,650т
- механизмы восстановления, 666-679
- мутагены, 642, 643, 650-654, 668
- обращение репликационной вилки, 697р,
697-698
- обход в ходе репликации, 676-677
- последствия репликации, 632,632р
повреждение клетки, 650,650т
повторная ДНК
- ПЦР, 157,157р,
- гибридизация in situ, 120,12lp
- метод «отпечатков пальцев», 157,157р
- метод дробовика для полных геномов,
158,159
- обход хромосомы, 153
- проблемы метода дробовика, 82,82р
904 Предметный указатель
- прокариоты, 301
- проскальзывание репликации, 647, 649р
- теломеры, 625т, 622-626
- транспозиция, 701, 701р
- ядерные геномы эукариотов, 284р,
282-285
повторное сворачивание, 522,522р
подавление, 479
подавление катаболитных оперонов, 550р,
548-551, 583
подавление роста опухолей, 632,632р
подбор нуклеотидов ДНК-полимеразами, 646,
646р
поддерживающее метилирование, 367р,
367-368,
подобные Mutator мобильные генетические
элементы (MULEs), 734, 734р
позвоночные
- гомеозисные избирательные гены,
581-582, 582р
- группы генов Нох, 725, 725р
- консервативные последовательности
интронов, 456р
- последовательность ДНК 5-CG-3’ 116,116р
- транскриптомы, 228
- уникальные гены, 279
- эволюция гена глобина, 739-740, 740р
поздние старты репликации, 630-632
позиционная информация в развитии,
575-580
позиционный итеративный BLAST (PSI-
BLAST), 193
позиционный эффект, 356, 356р
позционное клонирование, 156
поиск гомологии
- данные о дупликации гена, 728-729,729р
- применение для аннотирования генов
дрожжей, 211, 212р
- изучение функций генов, 193р, 192-196
- межбелковые взаимодействия, 243р
- просмотр последовательности, 183р,
181-184,184р
поиск гомологии среди родственных видов,
183р, 183-184,184р
поиск мутаций, 642тп
поли-G хвосты, 53, 53р
поли-А последовательности, 334, 334р,
450-451
поли-А хвосты, 20, 20р, 224, 224р, 445,
450-453
- RACE, 189,190р
- зависимое от дезаденилирования декэпи-
рование, 475,475р
- инициация трансляции в ходе синтеза
белка, 508-509
поли-А-полимераза, 450
полиаденилирование
- деградация бактериальных мРНК, 445
- терминация синтеза мРНК, 447,451р,
450-453
- регуляция активности генома, 543т
полиаденилирующее редактирование, 475
полимераза Taq, 44т, 71-72, 72р, 187тп
полимераза Клёнова, 44,44т, 45,144
полимераза Корнберга, 44,45
полимеразная расщелина, 386т
полимеразная цепная реакция (ПЦР), 39,39р,
71р, 72р, 70-74
- библиотеки клонов, 71тп
- изучение РНК, 187тп
- области применения, 72-74
- обнаружение мутаций, 642тп
- обход хромосомы, 153,154р
- определение счетов RFLP, 88р, 88-90
- первая стадия, 71р, 71-72, 72р
- сайт-специфический мутагенез, 209тп
полимеразы, см. ДНК-полимеразы; РНК-
полимеразы
полиморфизмы длин простых последователь-
ностей (STR), 88р, 87-90
- картирование МУПов, 123
- проект «Геном человека», 161,162
полиморфизмы длин рестриктов (RFLP), 87р,
87-88,88р
полиморфизмы отдельных нуклеотидов
(SNP), 87,90р, 90-91,94,94р
полиморфные локусы, 774, 774р
полиморфные участки рестрикции, 87-88,
88р, 112р
полинуклеотидкиназа, 53
полинуклеотидфосфорилаза (ПНФаза), 445
полинуклеотиды, 39-45
полиовирус, 510
полипептиды, 21-22, 22р, 524р, 524-525
полипиримидиновая полоса, 456,456р
полиплоидия
- автополиплоидия, 727р, 727-728, 728р
- аллополиплоидия, 735
полипотенциальные клетки, 199,
полипротеин проопиомеланокортина, 527,527р
полипротеины, 327, 524, 524р, 525-527
полифилетические группы, 758,758р
полнота кДНК, 189
полнота молекул, 189
половые гормоны, 547
половые клетки, 3
половые хромосомы, 3, см. также
Х-хромосома; Y-хромосома
положительно заряженные боковые группы,
23р
Предметный указатель 905
полуконсервативная репликация, 598р, 599р,
597-600, 602-603,632-634
полупостоянные изменения активности гено-
ма, 541р, 557-565
полярные группы, 23р
последовательности PEST, 530
последовательности встроек, 301,301р, 335,
335р
последовательности контигов, 151р, 152,158,
159р
последовательности нуклеотидов
- косвенные воздействия на структуру
спирали, 394-396
- прямое считывание, 394
- филогенетика, 761-762, 762р, 764р
последовательность 5’-CG-3' 116,116р
последовательность Шайна-Дальгарно, 506
последовательный анализ экспрессии генов
(SAGE), 224р, 224-225
посредники, 406,416-417,417р, 420
постоянные изменения активности генома,
541р, 557-565
постоянные нуклеотидные позиции, 494
пострепликационное восстановление, 642,
696р, 697, 698р
пострепликационные комплексы (постРК),
630
посттранскрипционное глушение генов, 479
посттрансляционная обработка, 520р,
519-529
- вырезание интеинов, 520, 520р, 528-529,
529р
- протеолитическое расщепление, 520,
520р, 524р, 524-527
- сворачивание белков, 520р, 522р, 523р,
520-524
- химическая модификация белков, 520,
520р, 527т, 527-528
поток генов, 305
почкующиеся дрожжи, см. Saccharomices
pombe
права на последовательности ДНК человека,
163-165
праймаза, 611, 614, 615,615р
праймосомы, 612,615
превращение цитозин-урацил, 663,663р, 669
пре-РНК (РНК-предшественник), 20,20р, см.
также пре-мРНК; пре-тРНК; пре-рРНК
пре-мРНК, 20, 20р
- альтернативный сплайсинг, 226р, 228,
461р, 460-463
- вырезание интронов AU-AC, 465
- ген дистрофина человека, 449
- удаление интронов, 455т, 454-465
- эукариоты, 455т, 461р, 454-465
пре-мРНК dsx, 462,462р
пре-мРНК sxl, 461,462р
пре-мРНК tra, 461,462р
пре-мРНК эукариотов, 455т, 461р, 454-465
пре-рРНК,
- бактерий, 441-442,442р, 443,444
- эукариотов, 467р, 466-470
пре-рРНК бактерий, 441-442,442р, 443,444
пре-рРНК эукариотов, 467р, 466-470
пре-тРНК,
- бактерий, 442-443,443р, 444
- эукариотов, 455т, 466-470,471р
пре-тРНК бактерий, 442-443,443р, 444
пре-тРНК эукариотов, 455т, 466-470,471р
пре-тРНКТуг, 471р
предРК (предрепликационные комплексы),
627-631
предынициаторный комплекс, 404-405,
416-417,447,447р, 509, 509р
предзатравливающий комплекс, 605
предковые клетки, 573, 576р
предковые последовательности, 193р,
192-196, 768р, 769
предковые состояния признака, 754
предполагаемые рецепторы стероидных
гормонов, 547
препроинсулин, 525, 525р
предрепликационные комплексы (предРК),
627-631
предсплайсосомный комплекс, 459,459р
преждевременная терминация, 437р, 437-438,
439р
преждевременная терминация синтеза РНК,
437р, 437-438,439р
препарат, 123,123р
прерывная репликация ДНК, 609,609р,
611-612, 612р, 615р, 622,623р
прерывные гены, 301, 738-740
приматы
- молекулярные часы, 767-768, 768р, 770
- происхождение СПИДа, 772-773, 773р
- филогенетическая связь, 757, 758р,
770-772, 772р
приобретенные состояния признака, 754,
754р
приобретение генов в ходе эволюции,
719-735
прионы, 330, ЗЗОр
приспособленность, 774
проба ARMS (система размножения тепло-
устойчивых мутаций), 94р
проблема укорочения концов, 625-627,705
программное обеспечение, см. компьютеры
- HENNIG86, 764тп
- MacClade, 764тп
906 Предметный указатель
- NJPlot, 764тп
- PAML, 764тп
- PAUP, 764тп
- PHYLIP, 764тп
- Clustal, 762, 764тп
продуктивные запуски, 408,409
проекты расшифровки генома человека,
161-165
проекты расшифровки геномов, 161-165, 770
проинсулин, 525, 525р
происхождение коренных американцев, 783р,
782-785
происхождение современного человека,
773-779
прокариоты
-ДНК-транспозоны, 335р, 335-337
- горизонтальный перенос генов, 734-735
- мобильные генетические элементы, 330
- репликация, 597,602, 609-615, 617-620
- связывающиеся с ДНК белки, 381, 386-387
пролин, 23р, 27
промежуточные периоды клеточного цикла,
627,628
промелиттин, 524-525, 525р
промоторы
- РНК-полимеразы, 398р, 400т, 398-401,
401р
- мутации, 658,658р
- направляемая транспозонами экспрессия
генов, 737
- у бактерий, 398-400,400р, 400т, 402,409р,
410р, 408-412
- у бактериофагов, 322
- у эукариотов, 400-401,404-406,414-415,
415р, 420
пропуск экзона, 457,457р
проскальзывание, 517, 517р
проскальзывание репликации, 647, 649р, 726
проскальзывание трансляции, 517, 517р
просмотр последовательности, 176р, 177р,
183р, 176-184
проспоры, 571р, 569-573, 573р, 575р
простейшие
- Reclinomonas americana, 310т, 311
- Tetrahymena, 272т, 361,466,467р, 468,
625т
простые (короткие) тандемные повторения
(STR), 88, 88р, 90, см. также микросателли-
ты
простые мультигенные семейства, 281
простые мутации, см. точечные мутации
протеазы, 5, 520
протеасомы, 530, 530р
протеинкиназа Cdc7p-Dbf4p, 630
протеинкиназа LET-23, 575
протеинкиназы, 553, 555, 559,575,628-630,
676
протеолитическое расщепление, 520, 520р,
524р, 524-527
протеомика, 233-245
протеомы
- деградация белка, 529-530
- определение, 1-30
- посттрансляционная обработка, 519-529
- роль рибосом, 501-519
-роль тРНК, 493-501
- составляющие элементы, 4, 23-30
- эволюция человека, 743
протогеномы, 716-717
протопласты, 68
прототрофы, 661,661р
проточная цитометрия, 125,125р
профаги, 326, 327р
профаза, 99р, 101р, 102
профили экспрессии, 229,229р
процессы восстановления, см. восстановление
ДНК
процессы темнового восстановления, 670
прямое восстановление, 666р, 668р
прямое считывание, 381, 394,420
прямые воздействия мутаций, 654
прямые повторения, 701, 701р
псевдогены, 282, 282р, 721
псевдоуридин, 443р, 444
псевдоуридинилирование, 472
птицы, 265,275т, 754
пурины, 8
индукторы, 410
путь RecBCD, 693-694,694р, 696,696р, 707
путь RecE, 694-696
путь RecF, 694-696,696р
путь биосинтеза триптофана, 247-248
путь дифференциации зародышевых кеток
яйцевода, 573, 575, 576р
путь зависимой от убиквитина деградации,
530
путь сворачивания, 522, 522р, 523
путь сплайсинга, 456-457,467р
путь экспрессии, 543т
ПЦР, см. полимеразная цепная реакция
ПЦР с рассеянными повторными элементами
(РПЭ-ПЦР), 157р
пчелиный яд, 524-525,525р
пшеница (Triticum aestivurri), 272т, 735
пшеница тучная (Triticum turgidum), 735
рДНК, 263р
рРНК (рибосомная РНК)
- РНК-полимераза 1,398т
- бактерий, 442р, 443р, 441-444
Предметный указатель 907
- единица транскрипции, 416
- кариограммы человека, 263р
- определение,
- перенос веществ в ядерных клетках, 480,
482р
- ранняя эволюция, 716
- реакция на стресс через синтез, 441
- рибозимы, 468т
- семейства генов, 281,722
- события разрезания, 442р, 441-443
- содержание в клетке, 18,18р
- химическая модификация, 443р, 444,
471-475
- эукариотов, 471-475,480,482р
рРНК 16S, 441,442,442р, 502, 504р, 505
рРНК 18S, 398т, 466, 502,504р
рРНК 23S, 441,442,442р, 468т, 502,504р
PPHK25S, 472р
рРНК 28S, 398т, 466, 502, 504р
рРНК 5S, 398т, 441,442р, 466, 502, 504р
рРНК 5,8S, 398т, 466, 502,504р
рРНК эукариотов, 471-475,480,482р
радиационные гибриды, 124,124р,
радиационные гибриды грызунов, 124
радиомечение ДНК, 5, 7р
разбор случая аннотации генов дрожжей, 212
разбросанная репликация, 598р, 599р,
597-600,
развитие
- Drosophila melanogaster, 577р, 579р, 580р,
576-583
- лизогенный цикл бактериофага X, 568р,
567-569
- регулирование активности генома, 414,
542,565-583
- регуляторы, 414
- семейств генов, 281,282
- спорообразование у Bacillus subtilis, 57lp,
572р, 569-573, 573р, 575р
- яйцевода у Caenorhabditis elegans, 575р,
573-576, 576р
развитие мышечной ткани, 565
развитие эмбриона, 281, 577р, 577-579
развитие яйцевода, 575р, 573-576,576р
размер генома, 272т, 275т, 303т
- бактериофаги, 323, 323т
- вирусы эукариотов, 326, 329т
- прокариоты, 303, 303т
- эукариоты, 272, 272р, 272т, 274-275, 275т
размытые мутации, 662,
разнообразие антител, 474,474т
разнообразие белков, 22-23,23р
разрешающие условия роста, 661,
разрешение электрофореза в полиакриламид-
ном геле, 140,140р
разрушение ультразвуком, 151,
рак
- активность теломеразы, 626
- анализ транскриптома, 232-233
- белки Ras, 553
- комплекс Sin3, 363
- мутации соматических клеток, 659
- повреждение ДНК, 632
- сбои системы восстановления вырезани-
ем нуклеотидов, 679
рак молочной железы, 679
рак прямой и ободочной кишки, 679
рак яичников, 679
рамки считывания, 176р, 176-180,
ранние старты репликации, 630, 632
ранняя эволюция биомолекул, 715-719
раскрашивание хромосом, 352,
раскручивание ДНК для репликации, 596р,
596-597, 600-602
распространение земледелия в Европу, 779р,
779-781
разрешение
- определение,
- структура Холлидея, 689,690р, 694,696р,
697
рассеянные по всему геному повторения, 82,
82р, 269р, 269-271, 274, 274т,
рассеянные повторения, 284, 284р, 285,
705-707, см. также рассеянные по всему
геному повторения
расстегивание ДНК, 602, 602р
растения
- LTR-ретроэлементы, 333
- ДНК-транспозоны, 335,337-338
- автополиплоидия, 727-728
- аллополиплоидия, 735
- вирусоиды и вироиды, 329-330,468т
- ген леггемоглобина сои, 740
- генная инженерия, 701, 701р
- гомеозисные гены в развитии, 582-583
- данные эволюции, 729
- митохондрии, 308-309
- подобные Mutator мобильные генетиче-
ские элементы, 734, 734р
- хлоропласты, 310т, 308-312, 312р
растительный клонирующий вектор pBIN19,
70, 70р
расхождение, 96,
расчесывание молекул, 116-117,117р,
расщепление
- обнаружение мутаций, 642тп
- протеолитическая посттрансляционная
обработка, 520, 520р, 524р, 524-527
- сигналы полиаденилирования, 453,453р
- структуры Холлидея, 689, 690р
908 Предметный указатель
расщепляющие белки, 438,441
РБА (репликационный белок А), 614, 614р,
615,
реагирующий на гормоны элемент, 547
реакция А + G, 147
реакция С + Т, 147
реакция SOS, 568
реакция на стресс, 441
реакция синтеза нити, 140,140р, 141,189
ревертазная ПЦР (РТ-ПЦР), 187тп, 189
регулируемая медью экспрессия гена, 546р,
546-547
регулируемые рецепторами SMADs, 556
регуляторное управление, 408-412
регуляторные мутации, 658
регуляторные последовательности, 179-181,
658,658р, 659,661
регуляторный белок GABP, 386т
регуляция
- активности генома в ходе развития,
565-583
- инициации транскрипции, 406-420
- инициации трансляции, 510-512
- регуляторные модули, 414-415
- репликации ДНК, 605-606, 627-632
- теломераза, 625
- транспозиции, 702-703, 705-707
- функций белка, 29-30, 248, 248р
регуляция активности генома
- ДНК-метилтрансферазы, 368-369
- в ходе развития, 565-583
- кратковременные изменения, 541р,
542-557
- модифицирование гистонов, 360-365
- перестройки генома, 559-563
- петли обратной связи, 565, 565р
- постоянные/полупостоянные изменения,
541р, 557-565
- структурные изменения хроматина,
563-565
регуляция генома, 408,408т
редактирование РНК, 474р, 474т, 472-475,
475р
редактирование встройки, 475
редкощепящие рестриктазы, 116
рекомбиназа Сге, 701, 701р
рекомбиназы, 700, 701, 701р
рекомбинация, 687-712
- гомологичная, 198,198р, 687-698, 707
- проблемы филогенетического анализа,
768р, 768-769
- составление генетических карт, 99,101,
101р, 102,102р, 106р
- типы, 687-688,688р
- транспозиция, 688, 701
- транспозоны, 330, 331р
- удаления между дублированиями сегмен-
тов, 731
- удаления между мобильными генетиче-
скими элементами, 737, 737р
- сайт-специфическая, 688, 700р, 698-701
- эволюция интронов, 739
реликты эволюции, 282, 282р
ренатурация, 522, 522р,
рентгеновские дифрактограммы, 11-12,
383тп, 384
рентгеновское излучение, радиационные
гибриды, 124,124р
рентгеноструктурная кристаллография, 381,
383-384тп, 431,432
- интеины, 529
- структура рибосомы, 505
репарация
- ДНК, 669р, 670р, 666-679, 833-834,
- - ДНК-полимеразы, 610, 610т
- - восстановление двунитевых разрывов,
693,698
- - гомологичная рекомбинация, 693,
696р, 697-698, 698р
- - недостаточность, 642
- - последствия укорочения теломеры,
626
- - простые мутации, 642, 642р
- - реакция на повреждение, 632
- - сбои, вызывающие болезни, 677-679
- - сверхмутация, 662-663
- - устранение однонитевых пропусков,
696р, 697
- алкилирования, 668
- вырезанием, 666, 666р, 669р, 670р, 670т,
672р, 668-673
- вырезанием нуклеотидов, 670р, 672р,
670-673, 679,
- вырезанием оснований, 669р, 668-670,
670т
- путем рекомбинации, 697-698,
репатриация интеинов, 529, 529р,
репатриация интронов, 529,
репликационная транспозиция, 330-331,
331р, 701-702, 702р, 703
репликационные вилки, 602,602р, 605, 614р,
612-617
- обращение, 697р, 697-698
- разрушение, 698, 698р
репликационные фабрики, 617,
репликационные формы, 143,
репликационный белок А (РБА), 614, 614р,
615,
репликационный посреднический белок
(RMP), 614,
Предметный указатель 909
репликация, см. также репликация ДНК
- первые геномы из РНК, 716, 716р
- стратегии вирусов, 323-327, 329р
репликация ДНК, бОЗр, 595-640,
- альтернативные способы, 598р, 599р,
597-600
- бактерии, 609-610, 610т, 612р, 611-615
- дублирование гена, 726, 726р
- инициация, 604-608,630-632
- комплекс опознавания областей инициа-
ции, 605-606
- линейные концы, 622-627
- непрерывная, 609, 609р, 611-612, 614-615,
615р
- обход поврежденной ДНК, 676-677
- ошибки, 642,643р, 646-650, 696р, 697-
698, 698р
- продление, 604,608-627
- процесс, 604-627
- регулирование, 605-606
- таутомерия оснований, 646-647, 647р,
651, 651р
- теломераза, 622,625р, 626,627
- терминация, 619-622
- топологическая проблема, 596-603
- устранение несоответствий, 673-674
- эукариоты, 606р, 605-608,610т, 611,612р,
615-617
репликация катящегося кольца, 603, бОЗр,
614,
репликация ретровирусов, обратные транс-
криптазы, 45
реплисомы, 615,
репрессор
- Аге, 386т
- cl, 568р, 568-569
- Сго, 568,568р
- Met), 386т, 388р
- Mnt, 386т
-Motl, 417-418
-NC2,417-418
- Ssn6-Tupl, 418
- лактозы, 386,386т, 410р, 409-411,
- триптофана, 386т, 411р, 412р
репрессоры
- инициация транскрипции у бактерий,
410р, 411р, 409-412,412р
- инициация транскрипции у эукариотов,
418р, 417-420
- оперон триптофана Escherichia coli, 438
рестриктаза Alul, 224, 224р
рестриктаза BsmFl, 224, 224р
ретровирусный ген gag, 327, 333, ЗЗЗр, 334,
334р
ретровирусный ген env, 327, 333, ЗЗЗр
ретровирусы, 327, 327р, 329р
- горизонтальный перенос генов, 735
- отношение к транспозонам, 331-333
- происхождение СПИДа, 772-773, 773р
- транспозиция, 702
- частота мутаций, 772
- эндогенные, 333
ретроген пируватдегидрогеназы, 726
ретрогены, 726,
ретропозон TART, 626-627
ретропозон НеТ-А, 626-627
ретропозоны, 334, 626-627
ретрорепатриация, 470
ретротранспозиция, 331р, 331-334, 726
ретротранспозоны, 702, 737
ретроэлемент copia, 331-333, ЗЗЗр
ретроэлемент gypsy, 333, ЗЗЗр
ретроэлементы, 331р, 331-333, 704р, 701-705,
705р
ретроэлементы Tyl/copia, 331-333, ЗЗЗр
ретроэлементы Ty3/gypsy, 333, ЗЗЗр
ретроэлементы Ту, 331-333, ЗЗЗр
рецептор ретиноевой кислоты, 547р
рецептор серин-треонинкиназы, 551т, 556
рецептор трансферрина, 512
рецепторы ионных каналов, 551т
рецепторы клеточной поверхности, 551р,
551-557
рецепторы стероидных гормонов, 386т, 387,
387р, 547, 547р
рецепторы тирозинкиназы, 551т, 552, 552р
рецепторы, связанные с тирозинкиназой,
551т, 552,552р
рецессивные аллели, 659,659р
рецессивные фенотипы, 94р, 96,103-105,
105р, 105т
рибозимы, 467,468т, 514-515, 715-717, 717р
рибонуклеаза
- бактерий, 441-445
- дифракционная рентгенограмма, 383тп
- карта электронной плотности, 383тп
- определение,
- сворачивание, 520-523
- эукариоты, 466
рибонуклеаза D, 442-443,443р,
рибонуклеаза Е, 442,443р, 445,445р
рибонуклеаза F, 441,442р, 443
рибонуклеаза II, 445
рибонуклеаза III, 441,442р, 445,445р
рибонуклеаза М16,442р
рибонуклеаза М23,442р
рибонуклеаза М5,442р
рибонуклеаза MRP, 443,466,
рибонуклеаза Р, 441,442р, 443р, 468т
рибонуклеаза Z, 443р
910 Предметный указатель
рибонуклеиновая кислота, см. РНК
рибонуклеопротеиновый (РНП) домен, 388,
рибонуклеотиды, 17,17р, 716
рибосомная РНК, см. рРНК
рибосомные белки, 502,504р, 508т, 510, 510р
рибосомы
- коэффициенты осаждения, 518
- аттенюация, 437р, 437-438
- роль в синтезе белка, 501-519
- структура, 504р, 502-505, 505р, 518-519
- ультрацентрифугирование, 502
рибосомы бактерий, 504р, 502-505, 505р
риновирус, 510
рис (Oryza sativa), 272т, 275т, 310т, 312р
РНК
- LTR-транспозоны, 331-333
- РНК-в, 201, 201р
- бактерий, 430р, 443р, 430-445,445р
- взаимодействия с мембранами, 717
- геномы вирусов, 1
- глушение, 478-479,
- деградация, 444-445,445р, 475-480
- домен tudor, 195,195р
- матрицы для ДНК-полимераз, 39,39р, 41,42
- методы определения местоположения
генов, 186р, 187тп, 186-190,190р
- транспозоны без LTR, 334, 334р
- обработка, 380, 380р, 441-444
- первые биомолекулы, 443,715-716, 716р,
717, 717р
- первые геномы, 715-716, 716р, 717
- перенос веществ в ядерных клетках,
480-482,482р
- рибозимное расщепление, 825
- рибонуклеотиды, 17р
- саморепликация, 716, 716р
- содержание в клетке, 17-18,18р
- структура, 17,17р
- тРНК, 180р, 180-181,181р
- теломераза, 622-625,625р
- транспозиция ретроэлементов, 701-705
- транспозоны, 331-334, 334р
- химические модификации, 443р, 444,
471-475
- шпилечные структуры, 433-435,435р
- эукариотов, 471-482,482р
РНК пиранозила, 719
РНК эукариотов, 447-482,482р
РНК-зависимая ДНК-полимераза, 45,123р,
РНК-зависимая РНК-полимераза, 17,
РНК-затравки
- вытеснительная репликация, 602, бОЗр
- репликация, 611, 612р, 614, 615, 615р
- транспозиция ретроэлементов, 703-704,
704р
- удаление, 615-616,617р
- укороченная копия, 622, 623р
РНК-интерференция (РНК-и), 201, 201р, 478р,
478-479,479р
РНК-полимераза, 398
РНК-полимераза I,
- инициация транскрипции, 397-398, 398т,
401,401р, 406
- промоторы, 416
- синтез функциональной РНК, 465-466,
466р
РНК-полимераза II,
- восстановление вырезанием нуклеотидов,
672
- терминация транскрипции, 451р, 450-453
- инициация транскрипции, 397-398, 398т,
401р, 402-404
- инициаторный комплекс, 672
- кэпирование транскриптов, 448р, 447-449
- предынициаторный комплекс, 416-417,
417р
- продолжение мРНК, 449-450
- промоторы, 414-416
- регуляция синтеза, 453-454
- синтез мРНК, 447-454
РНК-полимераза III,
- инициация транскрипции, 397-398, 398т,
401,401р, 406
- промоторы, 416
- синтез функциональной РНК, 465-466
РНК-полимеразы, 17,17р,
- Bacillus, 569-571
- Escherichia coli, 402,404р
- инициация транскрипции, 398р, 398т,
400т, 397-401,401р, 402
- полимеразная расселина, 386т
- продолжение транскрипта, 431-432
- промоторы, 410-411
- скорость транскрипции, 465
РНК-полимеразы эукариотов, 397-398, 398т,
400,401
РНК-предшественник (пре-РНК), 20, 20р, см.
также пре-мРНК; пре-тРНК; пре-рРНК
РНКовые геномы
- бактериофагов, 322-323
- вирусоидов/вироидов, 327-330, ЗЗОр
- ретровирусов, 327, 327р, 329р
РНП (рибонуклеопротеидный) домен, 388,
род сегмента, 580-581
родительские генотипы, 102,103,105,105р,
родословная, 106-108,108р,
РПЭ-ПЦР (ПЦР с рассеянными повторными
элементами), 157,157р
РТ-ПЦР, 187тп, 189
Предметный указатель 911
сайленсеры, 412,415
сайт-специфическая рекомбинация, 688,688р,
698р, 700р, 698-701, 707
- встраивание бактериофага, 326,327р,
698р, 698-700, 700р
- генная инженерия, 700-701, 701р
сайт-специфический мутагенез, 205-206,
208-209тп
сайт-специфическое зондирование гидрок-
сильными радикалами, 504
самовырезающиеся интроны, 467,468т, 715
самокатализируемое расщепление, 330, ЗЗОр,
715
самооплодотворение, 94р
самопроизвольное сворачивание белка,
520-523
самопроизвольные мутации, 642,643р,
сателлитная ДНК, 284, 284р,
сателлитные РНК, 327-329,
сахаро-фосфатная основная цепь ДНК, 13р
сборка непрерывной последовательности
ДНК, 151р, 149-161
сборка последовательности, 82р, 82-84,84р
сворачивание белков, 520р, 522р, 523р,
520-524
сворачивающиеся белки, 520р, 522р, 523р,
520-524
сверхкачание, 501,
сверхметилирование, 369
сверхмутация, 662р, 662-663, 663р, 677,
сверхредактирование, 474
сверхспирализация, 59р, 294р, 294-295, 295р,
597
сверхчувствительные к ДНКазе I участки, 355,
355р, 358р, 358-359, 365, 365р
сверхэкспрессия, 202р, 204т, 201-205
связанные с CTD SR-подобные белки (CASPs),
460
связанные с G-белком рецепторы, 551т
связанные с GCN5 ацетилтрансферазы
(GNATs), 361
связанные с ТВР (связывающимся с блоком
ТАТА белком) факторы (TAF), 404
связанные с ТВР факторы (TAF), 404
связанные с матриксом области (MARs), 355,
355р, 356
связывание, см. связывающиеся с ДНК белки
связывание триметилпсоралена, 295, 295р
связывающиеся с ДНК белки
- взаимодействие ДНК со связывающимися
белками, 393-397
- геномы прокариотов, 295р, 294-296
- инициация транскрипции, 420
- места соединения ДНК с белками, 396-397
- определение,
- определение позиций связывания, 388р,
390р, 391р, 388-393, 393р, 394
- отличительные особенности, 381,386-388
- подавление инициации транскрипции,
418,439
- участки прикрепления, 381-397
- функции, 380т
связывающиеся с ДНК домены, 239, 239р, 241,
552
связывающиеся с ДНК мотивы, 381, 381р,
386т, 386-388, 388р, 420, 733,
связывающиеся с РНК белки, 380т, 388
связывающиеся с метил-CpG белки (MeCPs),
369, 369р,
связывающийся с trp РНК аттенюаторный
белок (TRAP), 438,439р
связывающийся с блоком ТАТА белок (ТВР),
404,405р, 406,406р, 417-418
связывающийся с полиаденилатом белок
(PADP), 450, 509
связь между биохимией клетки и протеомом,
27-30
сдвиг рамки считывания, 515-517, 517р
сегмент гена V, 662р, 662-663,663р
сегментированные геномы, 323
сегменты легкой цепи, 560-561, 561р, 561т
сегменты тяжелой цепи иммуноглобулина,
560-561, 561р, 561т, 563р
седиментационный анализ, 441
секвеназа, 44,44т, 45
секвенирование ДНК, 41тп, 52, 756-757,
761-762
секвенирование в тепловом цикле, 145р
секвенирование геномов, 138-172
секвенирование методом химической дегра-
дации, 145,147-148,148р
секвенирование с обрывом цепи, 143р,
139-145,
селекционные эксперименты, 103-106
селеноцистеин, 22
- контекстнозависимое переназначение
кодонов, 27, 29т, 498, 527
- модификация тРНК, 497р, 498
- структура, 25р
семейства генов, 281-282, 723-725, 739-740,
семейство а-глобинов, 281р, 281-282, 722р,
723-725
семейство белков SMAD, 556р, 556-557,
семейство генов Polycomb, 564, 564р
семейство генов альдолаз, 282, 726
семейство транспозонов Ту, 199, 201р, 212
Сенгер, Фред, 139
серил-тРНК-синтетаза, 398
серин, 23р, 27р, 454,497р, 498
сети
912 Предметный указатель
- межбелковые взаимодействия, 243-245,
245р
- филогенетический анализ, 769р, 769-770,
770р
сиаловая кислота, 528р
сигналы восприимчивости к деградации, 530
сигнальные пептиды, 525
сильные промоторы, 409
симметрия фрагментов эндонуклеазной
рестрикции, 47т
синдром Блума, 466,679
синдром Вернера, 466,679
синдром Кокейна, 450, 679
синдром Шарко-Мари-Тута, 731
синдром хрупкости Х-хромосомы, 649
синонимические мутации, 655,657р, 756
синтез, см. репликация ДНК; синтез ДНК;
синтез белка; синтез РНК
синтез ДНК
- репарация, 674, 677, 677р
- гомологичная рекомбинация, 692-693,693р
- ретровирусы, 327, 329р, 772
- транспозиция, 702, 703, 703р, 704-705
синтез РНК, 380, 380р
- микроРНК, 479
- направляемый матрицей, 12,17р, 431р,
608, 608р
- регуляция, 453-454, 543т
- у бактерий, 430р, 430-441
- у эукариотов, 451р, 447-454
синтез бактериальной РНК, 430р, 430-441
синтез белка
- аминоацилирование, 496р, 497р, 493-498
- ваимодействия типа кодон-антикодон,
498р,500р, 498-501, 501р
- терминация трансляции, 518
- инициация трансляции, 505-512
- регуляция активности генома, 543т
- роль рибосом, 501-519
-роль тРНК, 493-501
- трансляция у архей, 518-519
- управление активатора и репрессора, 418
- фаза элонгации в ходе трансляции, 512р,
512-518
синтез жгутика, 248р
синтез искусственного гена, 209тп,
синтез ферритина, 510р, 512
синтения, 184,184р,
сиротские рецепторы стероидных гормонов,
547
сиротские семейства, 211,
система МАР (активируемого митогеном бел-
ка) киназы, 553, 553р, 559,
система SAP (активируемого стрессом белка)
киназы, 555
система АСБ (активируемого стрессом белка)
киназы,555
система активируемого митогеном белка
(МАР) киназы, 553, 553р, 559
система вторичного посредника-кальция,
555-556, 556р
система глушения миРНК, 480
система глушения на микроРНК, 480
система надзора, 476р, 476-478
система размножения теплостойких мутаций
(проба ARMS), 94р
системная биология, 4, 247-249,
системы обхода повреждений, 676-677
сканирование, 508-509, 509р,
складчатая РНК, 479,480
скользящие нуклеосомы, 365р, 367
«скользящий зажим», 0-субъединица ДНК-
полимеразы III, 614
скорость транскрипции, 465
скорость полимеризации, 449
скрытые участки сплайсинга, 457,457р, 658
согласованная эволюция, 722
слуховой диапазон, альтернативный сплай-
синг, 463
слуховые волосковые клетки, 463
случайное расхождение хромосом, 96,96р
случайные последовательности генома, 123
случайный дрейф генов, 774, 774р
смена матриц, 704р, 705
смешанная ДНК, 804
смолы, 58тп, 59р
смыкание пропуска, 152р, 151-153,158,159
соактиваторы, 415-416
события дублирования в ходе эволюции,
721-732
события разрезания
- обработка РНК, 20,20р, 442р, 441-443,
443р
- протеолитическое расщепление, 520,
520р, 524р, 524-527
- регулирование активности генома, 543т
- эндонуклеазы рестрикции, 38р, 47р, 47т,
47-48,48р, 51-52
совместное иммунное осаждение, 242
совстройки, 702
содержание GC, 115,125,176,180, 284, 284р
содержание воды в волокнах ДНК, 11
соклетие, 577, 579
сокращение спутанной ДНК, 620-622, 622р
соматические клетки, 3,658-659
сонная болезнь, 463
сопрягающие молекулы, 52, 53р, 532
сопряженное с транскрипцией восстановле-
ние, 672, 679
сорепрессоры, 411
Предметный указатель 913
составление генетических карт, 84,103,109р,
111р, 108-112,112р, 187-189
составление карт генов, 84-109
составление карт геномов, 81-135
- бактерии, 108-109,109р
- проекты расшифровки генома человека,
161-165
составление карт рестриктов, 112,113р, 115р,
115-116
составление карт транскрипции, 189,190,
190р, 192
составление физических карт, 109-127
- FISH, 119р,120р, 119-121
- картирование STS, 123р, 121-127,127р
- определение, 84
- секвенирование, 109-127
- составление карт рестриктов, 112,113р,
115р, 115-116
составные белки, 238р, 239, 241
составные транспозоны, 335р, 335-337
состояния признаков, 754
спаривание оснований
- взаимодействие типа кодон-антикодон,
498, 501
- вторичная структура тРНК, 180р, 181,181р
- интроны I группы, 467,468р
- основная цепь ДНК, 12р, 13
спаривание оснований ДНК с РНК, 433,435р
спаривание оснований РНК-РНК, 433
спектроскопия ядерного магнитного резонан-
са (ЯМР), 381, 383тп, 384
специфические для материнских клеток
субъединицы, 572
специфичные для штаммов геномы, 305
специфичные к последовательностям связы-
вающиеся с ДНК мотивы, 386т
специфичные к последовательностям эндону-
клеазы, 529
специфичные к проспорам субъединицы, 572
СПИД/ВИЧ, 327,474,474т, 772-773,773р
сплайсинг
- альтернативный, 461р, 460-463
- интеин, 520, 520р, 528-529, 529р
- интроны AU-AC, 465
- интроны GU-AU, 456-457
- интроны II группы, 470
- малая ядерная РНК, 457-460
- мутации экзон-интронных границ, 658
- обработка РНК, 20, 20р
- определение,
- последствия встраивания транспозонов,
737
- пре-рРНК, 467р, 466-470
- пре-тРНК, 466-470,471р
- регуляция активности генома, 543т
- транссплайсинг, 463р, 463-465,465р
- шаги на пути, 456р, 456-457
сплайсированная лидерная РНК (слРНК),
463р, 465р
сплайсосома, 459р, 457-460,
спорообразование
- Bacillus subtilis, 571р, 572р, 569-573, 573р,
575р
- изучение транскриптома дрожжей,
229-230, 230р
споры anthrax, 569
способ удаления штрих-кода, 214, 214р,
способы репликации, 323-326
способы секвенирования методом дробовика,
82р, 82-84,84р
- полных геномов, 82,84р, 149,158р,
158-161
- секвенирование коротких геномов, 151р,
152р, 149-153
сравнение случайных и запрограммирован-
ных мутаций, 665р, 663-666
сравнительная геномика
- взаимодействия функционально актив-
ных белков, 243р
- изучение аннотации генов дрожжей, 212
- поиск гомологии, 183р, 183-184,184р
- проекты расшифровки генома человека,
163
старение, 625-627,627р
старение клетки, 625-627,627р
старт-кодоны, 26, 27р, 176,176р, 508р,
505-509
стволовые клетки, 625
стебель ТЧ'С, 494,494р, 496
стебельно-петельные структуры, 147р, 181р
стержневой октамер, 261
стероидные гормоны, 547
стоп-кодоны, 26,27р, 176,176р
стоп-последовательности, 619р, 619-620,
620р
страховые взносы, 165
стрессовые условия, 510
структура «бусины на нити», 261р
структура t-петли теломеры, 625, 626р
структура Холлидея, 689-690, 690р, 694, 696р,
697
структура лассо, 456,456р, 470
структура двойной спирали, 5,13р, 15т, 9-16,
16р
- вставочные агенты, 653,653р
- проблемы репликации, 596р, 596-597
структура дезоксирибонуклеотида, 8р, 8-9
структура клеверного листа, 180р, 181,493,
494р
структура откидной створки, 433-435,435р
914 Предметный указатель
структура специализированного хроматина,
356р
структура триплекса, 694р
структурное разнообразие геномов вирусов,
322-323, 323т, 326, 329т
структурное управление, 408-409
структурные белки, 29
структурные домены, 355, 356
- определения, 740
- эволюция, 731р, 732р, 731-733, 733р, 740
структурный гетерохроматин, 162, 263р, 355,
372
структуры
- ДНК, 15т, 8-16,16р
- РНК, 17
- белков, 21-23
- дезоксирибонуклеотидов, 8р, 8-9
- иммуноглобулина, 560, 561р, 561т, 563р
структуры типа головки молотка, ЗЗОр
структуры тройных спиралей, 650
субдомены, 605-606,606р
субъединица ст32,409,409р, 412
субъединица ст54,409
субъединица ст70,409,409р
субъединица 571-572
субъединица аЕ, 571-572, 572р, 573
субъединица oF, 571-572, 572р, 573
субъединица oG, 572, 575р
субъединица стн, 571-572
субъединица стк, 572, 575р
субъединицы рибосом, 502
сумка, 559, 559р
сумкоспоры, 559, 559р
схема сегментации, 579р, 579-580
конвергентная эволюция, 754р
сцепки, 53р, 63р
сцепление, 94-106
сцепление генетических признаков, 94-106
сцепление полинуклеотид-тирозин, 600
счет ЛОШ (логарифмического отношения
шансов), 108
считывания парных концов, 159,159р
тРНК, 18,18р, 180р, 180-181,181р
- аминоацилирование, 496р, 497р, 493-498
- взаимодействия типа кодон-антикодон,
498р,500р, 498-501, 501р
- гены, 398т
- затравки транспозиции ретроэлементов,
704, 704р
- местоположения генов, 180р, 180-181
- перенос веществ в клетках эукариотов,
480,482р
- реакция на стресс через синтез, 441
- роль синтеза белков, 493-501
- события разрезания, у бактерий, 441-443,
443р
- содержание в клетке, 18,18р
- структура, 493-494,494р
- химическая модификация нуклеотидов, у
бактерий, 443р, 444
- химическая модификация, у эукариотов,
471-475
тРНК эукариотов, 471-475,480,482р
тРНКА1а, 493
TPHKAsn, 497
TPHKGln, 497,497р
tPHKGIu, 497,497р
тРНКНе, 500
тРНКеу5, 517
TPHKMet, 506т, 508, 509, 514
тРНКРЬе, 468т
тРНКР^5,498
TPHKSeCys, 497р, 498
TPHKSer, 494
тРНКТуг, 443,443р
тРНК-нуклеотидилтрансфераза, 443,
табак Nicotiana tabacum, 310т
таксономия, 753
талассемия, 358
тандемно повторяющаяся ДНК, 82, 82р, 284р,
284-285
тандемные
- дупликации, 725-726, 726р
- повторения, 626-627,647, 649р
тандемная аффинная хроматография (ТАР),
242, 242р
таутомерный сдвиг, 646-647, 647р, 651, 651р
таутомеры, 646-647, 647р, 651, 651р
твинтроны, 455т, 470
Тейтум, Эдвард, 247
теломераза, 625р, 625т, 626р, 622-627
теломеры, 66р, 67, 262р
- Drosophila, 626-627
- Oxytricha, 625т
- Tetrahymena, 625т
- ДНК-белковые взаимодействия, 266
- длина сокращения, 625-627
- минисаттелитная ДНК, 284-285
- последовательность, 266,266р
- продолжение теломеразой, 622-625,625р,
626
- хронометраж репликации, 630,631, 631р
- человек, 625т, 622-626
телофаза, 99р, 101р, 102
тельца Кахаля, 351, 351р
тельце Барра, 370,
температура плавления (Тш), 91р
температуры реакций, 45
теория эндосимбионтов, 308,
Предметный указатель 915
тепловые мутации, 653-654,655р
теплоустойчивые ДНК-полимеразы, 45,70,145
тератогены, 650т
терминирующая шпилька, 437-438,445,445р
терминация, 433р, 435р, 433-441
- РНК-полимераза 1,465-466,466р
- РНК-полимераза III, 465-466
- репликации ДНК, 619-622
- транскриптов у бактерий, 433р, 435р,
433-441
-трансляции, 518
территории, 352р, 354р
территории хромосом, 352р, 352-354,354р
тетраплоидия, 727-728
тетраплоидные клетки, 370р, 370-372
технология рекомбинантной ДНК
- ДНК-полимеразы, 41-45
- нуклеазы, 45-48, 51-52
- плазмиды, 55р, 53-70
- подбор pUC8, 59, 59р
- полимеразная цепная реакция, 70-74
- развитие, 38-39
технология чипов, 225, 225р, 228р
тимидинкиназа, 124
тимин, 8,8р, 12р, 13,116р
тимингликоль, 669, 670т
тиминовые димеры, 654р
типы спаривания, 559-560, 560р,
тирозин, 23р, 27р
тканеспецифические картины экспрессии,
454
топоизомераза I типа, бООр, 600т, 602р
топоизомераза II типа, 600т
топоизомеразы, 596, 597, бООр, 600т, 600-602,
602р
топоизомеразы I типа, 700
топология
- репликация ДНК, 596р, 596-597, 600-602
- филогенетические деревья, 757, 757р
точечные мутации, 641-642, 642р
- полиморфизмы отдельных нуклеотидов,
90
- причины, 642, 643р, 651-653
- филогенетика, 764, 767-768, 768р
точки управления, 228, 235
точность
- восстановления филогенетических дере-
вьев, 767-768
- генетических карт, 112р, 111-112
- метода дробовика для полных геномов,
158р, 158-161
транс-замена, 367
трансамидирование, 497,497р
трансгенные мыши, 202
трансгены, 479,479р
трансдукция, 109,109р
транскриптом
- определение, 4р, 1-30
- связь с протеомом, 25-26
- составляющая РНК, 20-21
- эволюция человека, 743
транскриптом человека, 232р, 230-233
транскриптоспецифическая регуляция, 510р,
510-512
транскрипционный пузырек, 431,433
транскрипция
- терминация, 433р, 435р, 433-441,465-466,
466р
- задержка репликации, 619
- основные этапы, 380р
- подавление, 363, 546
- процесс, 4
- регуляция активности генома, 543т
- репликация ретроэлемента, 702-703, 704р
- сравнение с репликацией, 608р, 608-609
транслокации, 354, 354р, 512р, 514,
трансляции
- археи, 518-519
- терминация, 518
- инициация, 505-512, 543т
- элонгация, 512-518
- процесс, 4
- регуляция, 543т
- сопрягающая функция тРНК, 493р
трансляционный обход, 517р, 517-518,
трансляция, синтез белка, 518-519
транспозиция, 688,688р, 701-707
- искусственная индукция, 201р
- мобильные генетические элементы, 330,
335
- рассеянные повторения, 285
транспозоны, 331-338, 737, 737р, 738
транспозоны типа ТпЗ, 335р, 337,
транспонируемые фаги, 335р, 337,
транспорт РНК в клетках эукариотов, 480-
482,482р
транспортная РНК, см. тРНК
транспортные белки, 29
транссплайсинг, 463р, 463-465,465р
трансфекция, 62,
трансформация
- клонирование ДНК, 56, 56р
- онкогенная,
- определение,
- роль эволюции, 734-735
- у бактерий, 109,109р, 111р
трансформация клетки, 354р,
трансформирующие начала, 5, 7р,
трансформирующий фактор роста 0 (TGF-0),
556-557
916 Предметный указатель
трансэстерификация, 456-457,466-467
треонин, 23р, 27,438
третичные структуры, 22, 23р, 494,
трипаносомы, 463,475
триплетный генетический код, 26-29
триплоидия, 728, 728р
трипсин, 235
триптофан, 23р, 27р
трисомия, 726-727,
трихомонады, 308
трихотиодистрофия, 679
тупые концы, 48,48р, 52р, 53,63р
тутовый шелкопряд (Bombyx mori), 272т
тяжелые изотопы азота, 598-599, 599р,
630-631,631р
- эксперимент Мезельсона-Шталя, 598-599,
599р
- эксперименты по хронометрированию
репликации, 630-631, 631р
Уайтхедовский институт, 162
убиквитин, 530,
убиквитинирование, 364
убиквитиноподобные белки, 530
убывающее число генов, 720-721
удаление маркерного гена, 701
удлинение гена в ходе эволюции, 732
узлы в сети межбелковых взаимодействий,
245р, 246
уловление экзонов, 190,193р
ультрафиолетовое (УФ) излучение, 555,653,
654р, 679
ультрацентрифугирование, 268тп, 502
умеренный бактериофаг, 325
универсальность генетического кода, 26-27,
719
«универсальные» затравки, 144-145,145р
Уотсон, Джеймс Д., 8-12, 596р, 596-597, 600
упаковка in vitro, 62,63, 63р, 64
управляющие локусом области (LCR), 358-359
урацил, 17р
- восстановление ДНК, 669, 670т
- мутации, 653, 663, 663р
уридин, 444
уридин-5'-трифосфат (УТФ), 17
усвоение азота, 211,212р, 409
усеченные гены, 282р
усилители (энхансеры), 412,415,416, 737
условия на молодой Земле, 714-715
условия окружающей среды, 540-542
условно летальные мутации, 661
«установщик зажима», 614
устойчивость к антибиотикам
- генная инженерия, 701
- кассеты удаления, 198-199,199р
- клонирующие векторы, 56, 56р, 68р
- мутации, 661
- плазмиды, 56, 56р, 68р, 296, 297т, 734
- транспозоны, 335р, 335-337
устойчивость к канамицину, 701
устойчивые к ингибитору мутанты, 661,
устранение однонитевых разрывов, 668,668р
устранение несоответствий, 663,663р, 666,
673р, 673-674, 674р
устранение несоответствий очень короткими
заплатами, 673
устранение однонитевых пропусков, 696,
696р, 697
устранение однонитевых разрывов, 674, 674р
устранение пропусков, 654,655р, 674-676
закрепившиеся мутации, 661,662р
УТФ (уридин-5’-трифосфат), 17
УФ (ультрафиолетовое) излучение, 555, 653,
654р, 679
уход от промотора, 447р, 447-448
участки att, 698р, 698-700, 700р
участки cos, 62, 63р
участки прикрепления, 381-397
участки рестрикции, 56, 56р, 62, 62р
участок кВ, 414
участок nutL, 436
участок nutR, 436
участок САР, 411, 548, 550р
участок выхода, 514
участок посадки рибосомы, 506р, 506-508,
508р, 508т
фМет (N-формилметионин), 497р, 498, 508
фаги, см. бактериофаги
фаги-помощники, 143
фагмиды, 143
фаговый дисплей, 238р, 238-239, 241р
фаза G1 клеточного цикла, 627,628
фаза G2 клеточного цикла, 627, 628
фаза синтеза в клеточном цикле, 627
«фактор сборки», 406
фактор высвобождения, 518,518р
фактор высвобождения полимеразы I и транс-
крипта (PTRF), 466
фактор терминации для РНК-полимеразы I
(TTF-I), 416
фактор лицензирования репликации (RLF),
628,630,
фактор повторного использования рибосомы
(RRF), 518, 519т
фактор продления CSB, 449т, 450
фактор расщепления транскриптов GreA, 438,
441
фактор расщепления транскриптов GreB, 438,
441,441р
Предметный указатель 917
фактор репликации С (RFC), 615
фактор связывания с автономно реплицирую-
щимися последовательностями 1 (ABF1),
606,606р
фактор специфичности расщепления и полиа-
денилирования (CPSF), 450-451,451р
фактор стимуляции расщепления (CstF), 450,
451,451р
фактор сывороточной реакции, 414
фактор транскрипции
- - FOXP2, эволюция человека, 741
- - GAL4, 386т
- - GCN4, 386т
- - Рах, 386т
факторы терминации, 416,
факторы инициации, 506т, 508р, 506-509,
509р
факторы продления, 449т, 449-450, 513, 513т
факторы расщепления транскриптов, 438,441
факультативный гетерохроматин, 355, 372
PTRF (фактор высвобождения полимеразы I и
транскрипта), 466
фенетика, 753
фенилаланин, 23р, 27р
фенотип, 202р, 204т, 202-205,654
фенотипы, 204т
фермент, см. также отдельные ферменты
- EcoRI, 87
- smal, 116
- Ada, 668
- RecBCD, 693, 694p
- РНКаза H, 616, 704p, 705
- биологические системы, 247-248
- интеграза, 700, 705, 705р
- манипулирование ДНК, 39-53
- сравнение между белком и РНК, 716-717
- транспозаза, 335, 337-338
ферменты карбоксилазы, 527т
ферменты модифицирования концов, 41,
52-53,53р
ферменты пермеазы, 551
ферменты предрепликационного восстанов-
ления ДНК, 642
ферменты рестрикции
- SAGE, 224р, 224-225
- редкосекущие резаки, 115-116
ферменты типа редкощепящих рестриктаз,
115-116
ферменты усвоения лактозы, 508т, 665
феромоны, 559
фибробласты, 415
физические мутагены, 653-654,654р
филадельфийская хромосома, 354,354р,
филогенетика, 752-791
- аномалии, 754, 754р, 768р, 768-769
- использование молекулярных данных,
753-757
- морфологические признаки, 753-756,
758-760
- области применения, 770-785
- секвенирование ДНК, 756-757, 761-762,
762р, 764р
филогенетика человека
-доисторические переселения, 773-785
- молекулярные часы, 767-768, 768р, 770
- проблемы анализа, 769
- происхождение Homo sapiens, 774-777,
777р
- сравнения с приматами, 770-772, 772р
филогенетические деревья
- ВИЧи, 773р
- восстановление, 761-770
- деревья генов и видов, 758-760, 760, 761
- методы построения деревьев, 764р,
762-765
- особенности, 757р, 758р, 757-760
- проблемы, 768р, 768-769
- эволюция человека, 770-772, 772р, 777,
778
филогения, 753,
ФЛРы (факторы лицензирования реплика-
ции), 628,630,
флюоресцентная гибридизация на месте
(FISH), 112,119р, 120р, 119-121, 352
формамидопиримидин, 670т
фосмиды, 64т, 66-67
фосфатные группы, 8р, 8-9,9р, 11р
фосфодиэстераза, 669р, 670
фосфодиэфирные связи, 9,9р, 52, 52р
фосфоиндикация, 41тп, 52,91,91р, 93тп
фосфорилирование
- С-концевой домен, 447,454
- elF-2, 510
- активация белка SpoOA, 571-572, 572р
- гистоны, 364
- остатки серина, 364
- посттрансляционная обработка белков,
527т, 528, 528т
- пути сигнальных каскадов, 555, 555р
- серины, 454
- система МАР-киназы, 553, 553р
- управление активатора и репрессора, 420
фотолиаза, 668
фотолиаза фотопродукта (6-4), 668
фотолитография, 93тп
фотообесцвечивание, 352тп
фотопродукты, 653, 654р, 677, 679
фотореактивация, 668
ФПИР (фактор повторного использования
рибосомы), 518, 519т
918 Предметный указатель
фрагмент Клёнова, 44
фрагментированная ДНК
- картирование STS, 123р, 124,124р
- метод дробовика, 82р, 82-84
- разрушение ультразвуком, 151,152р
- составление генетических карт, 187-189
- составление карт рестриктов, 115
фрагменты Окадзаки, 611, 614-616р, 617р,
622, 623р
фрагменты генов, 282, 282р
фрагменты рестрикции
- липкие концы, 48,48р
- масштаб карт рестриктов, 115-116
- торможение в геле, 390, 390р
- футпринтинг ДНКазы I, 391, 391р
Франклин, Розалина, 11,12
ФРС (фактор репликации С), 615,632
фукоза, 528р
функции геномов, 223-254
функции отдельных генов, 190-211
функциональная РНК, 18,18р, 180р, 180-181,
465-466
функционально неактивные копии гена, 282,
282р
функциональные взаимодействия белков,
242-245
функциональные домены, 355р, 356р, 358р,
355-359, 372
функциональный анализ
- инактивация генов, 198р, 198-201
- неизвестных генов, 205-209
- отдельных генов, 190-211
- сверхэкспрессия генов, 201-202, 202р
футпринтинг, 391р, 390-393, 393р
хемилюминесценция, 148,149р
химеры, 199
химические модификации
- гистоны, 360-365
- нуклеотиды, 443р, 444
- обработка РНК, 20, 20р, 443р, 444,471-475
- посттрансляционная обработка белков,
520, 520р, 527т, 527-528
- регулирование активности генома, 543т
- управление активатора/репрессора,
418-420
химические мутагены, 651р, 651-653,653р,
668
химический сдвиг, 384тп
хлоропласты, 310т, 312р, 308-314,463
хозяйский фактор встраивания (HIF), 386т,
700
Холли, Роберт, 493
хроматиды, 99,101р, 262, 262р
хроматин
- анализ с защитой от нуклеазы, 260р,
260-261
- гистоны, 360-364
-домены, 354-359
- инактивация Х-хромосомы, 564
- метилирование, 363-364
- модифицирование, 359-367
- перестройка нуклеосом, 365р, 365-367,
416
- структура, 261, 262р, 564р, 563-565
- топоизомеразы, 602
- факторы продления, 449т, 450
- фосфорилирование, 364
- экспрессия генома, 359р, 359-367
хроматография
- высокопроизводительная жидкостная,
642тп
- ионообменная, 58тп, 59р
- аффинная, 241, 241р, 242, 242р
хроматосомы, 261
хромосома 2, 741, 741р
хромосома 21, 22, 232р
хромосомная теория, 4
хромосомный каркас, 351
хромосомы
-FISH, 120-121
- гомологичные, 99
- дупликации, 726-727
- картины исчерченности, 262т, 262-263,
263р
- метафазные, 99р, 101р, 262р, 263р,
261-266
- митотические, 97-99,99р
- неполное сцепление, 97р, 97-102
- определение,
- прокариоты, 294-298
- разделение проточной цитометрией, 125,
125р
- ранняя эволюция, 717
- слияние, 741, 741р
- транслокация, 354, 354р
- упаковка ДНК, 260-261
- фрагментированные для картирования
STS, 124р
- человека, Зр
- число, 260
- ядерных геномов эукариотов, 260-266
хронический миелоидный лейкоз, 354, 354р
хронометраж репликации, 630,631,631р
хронометраж репликации ДНК, 630-632
хрупкие участки, 647, 649т
HIF (хозяйский фактор встраивания), 386т,
700
цАМФ (циклический АМФ), 548,550р, 555
Предметный указатель 919
цГМФ (циклический ГМФ), 555
цветки, 582-583
Центр изучения полиморфизмов человека
(ЦИПЧ), 108,161
центр инактивации Х-хромосомы (Х/с), 370,
372
центральные промоторы, 400-402,402р, 404,
406,414,658,658р
центральный фермент, 149
центрифугирование, см. центрифугирование
в градиенте плотности
центрифугирование в градиенте плотности,
268тп,
- микроматрицы реплицируемой ДНК, 631
- рибосомы, 502
- сателлитная ДНК, 284, 284р
- ультрацентрифугирование, 268тп, 502
- эксперимент Мезельсона-Шталя, 599р,
598-600
центромеры, 262, 262р
цианеллы, 308
цианобактерия, 466
циклазы, 555
циклин С1Ь5р, 628
циклин С1Ь6р, 628
циклинозависимые киназы, 628-630, 632
циклины, 628-630
циклины G2, 630
циклический АМФ (цАМФ), 548, 550р, 555
циклический ГМФ (цГМФ), 555
циклобутильные димеры
- УФ излучение, 653
- реакция SOS, 677
- устранение, 668
- устранение однонитевых пропусков, 696р,
697
циклы инфицирования
- бактериофаги, 61р, 62, 62р, 436,436р,
568р, 567-569
- вирусы, 323-326, 326р, 327, 327р
цинковая двуядерная группа, 386т
цинковый палец, 387
ЦИПЧ (Центр изучения полиморфизмов чело-
века), 108,161
циссмещение, 367
цистеин, 23р, 27р
цитидин-5'-трифосфат (ЦТФ), 17
цитозин, 8,8р, 12р, 13
цитозин гл и коль, 670т
цитокины, 552
цитоплазматические дециклазы, 555
цитоплазматические циклазы, 555
цитохимия, 4
ЦТФ (цитидин-5'-трифосфат), 17
частота рекомбинации, 102,103р
частотность аллелей, 774, 774р, 779-781
Чейз, Марта, 5, 7р
челночные векторы, 68,68р
человекообразные обезьяны, 757,758р,
770-772, 772р
червь нематоды, см. Caenorhabditis elegans
четвертичная структура, 22
чипы ДНК, 91, 91р, 93тп, 225р, 225-233
число генов, 275т, 303т
- геном человека, 275т, 275-277
- прокариоты, 303т, 303-304, 304т
- сравнения среди эукариотов, 275т
- эволюция, 720-721
чувствительные к железу элементы, 510р, 512
чувствительные к температуре мутации, 661
чувствительные модули, 414
чувствительные элементы, 547,547р
чувствительные элементы ядерных рецепто-
ров, 547, 547р
шаперонины, 523, 524
шаперонины GroEL/GroES, 523, 524, 524р
шаперонины TriC, 523
шапероны, 523-524
шатание G-U, 498, 500р, 500-501, 501р
HDACbi (гистондезацетилазные комплексы),
369
шимпанзе, 741р, 740-743, 757, 758, 770-772,
772р
шпильки
- терминация транскриптов у бактерий,
433-435,435р
- мРНК гистонов, 453р
- оперон тириптофана, 437р, 437-438
- расширение тринуклеотидных повторе-
ний, 649-650
-тРНК, 181,181р
щелочная фосфатаза, 53
эволюция, 713-749
- гомология последовательностей, 183,
183р, 193р, 192-196
- запрограммированная мутация, 662-666
- молекулярная филогенетика, 752-791
- некодирующая ДНК, 735-740
- приобретение генов, 719-735
- происхождение жизни, 715-719
- роль рекомбинации, 689
- сходящаяся, 754, 754р
- человек, 740-743, 770-779
эволюция гибридных белков, 732, 732р
эволюция клеточной мембраны, 716-717
эволюция мозаичных белков, 732, 732р
эволюция мозга, 743
920 Предметный указатель
эволюция языка, 741
эгоистичная ДНК, 529, 737,
ЭДТА (этилендиаминтетраацетат), 58тп
экзон-интронные границы, 179,179р, 189,
190, 658
«экзонная теория генов», 738,738р, 739-740
экзонные сайленсеры сплайсинга (ESS), 460
экзонные энхансеры сплайсинга (ESE), 460р
экзонуклеазы, 39р, 45,609, см. также действие
экзонуклеазы 3'^5'; действие экзонуклеа-
зы 5'^3’
экзоны, 179р, 732-733, 739-740, 740р
экзосома, 475
эксперимент Мезельсона-Шталя, 596,599р,
598-600
эксперимент Херши-Чейз, 5, 7р
эксперимент Эвери, 5, 7р
эксперименты Чаргаффа по определению от-
ношения оснований, 12р
эксперименты с защитой от нуклеазы, 260р,
260-261, 504,
эксперименты с отношением оснований, 12р
эксперименты с поперечным сшиванием,
431р, 432,433
экспортины, 480
экспрессионная протеомика, 234р, 233-238
экспрессия гена, см. также экспресия генома
экспрессия генома
- SAGE, 224р, 224-225
- метилирование ДНК, 369р, 367-372
- модифицирование ДНК, 367-372
- модифицирование хроматина, 359р, 359-367
- перестройка нуклеосом, 365р, 365-367
- протеомика, 234р, 233-238
- профили, 229, 229р
- процесс, 3,4р
- путь, 543т
- эволюция человека, 743
экстеины, 520, 528
эксцизионаза Xis, 700
электронная микроскопия, 351, 504
электростатические взаимодействия, 22
электрофорез, см. также агарозный гель; по-
лиакриламидный гель
- обнаружение мутаций, 642тп
- определение белковых профилей, 234р,
233-238
- определение счета RFLP, 88р
- последовательность 5'-CG-3', 116р
- простые тандемные повторения, 90
- редкощепящие рестриктазы, 116
- филогенетика, 756
электрофорез белков, 756,
электрофорез в агарозном геле, 49тп, 51, 74р,
390
электрофорез в геле с обращением поля
(FIGE), 116
электрофорез в геле с ограниченными конту-
ром однородными электрическими полями
(CHEF), 116
электрофорез в геле с ортогонально чередую-
щимся полем (OFAGE), 116р
электрофорез в полиакриламидном геле,
139тп
- капиллярный электрофорез, 90,139тп,
141
- определение белковых профилей, 234р,
233-235
- разрешение, 140р
- секвенирование методом химической
деградации, 147
электроэндосмос (ЭЭО), 49тп
элемент Spm, 337
элементы Alu, 334, 334р, 741
элементы Ac/Dc, 335, 337р, 337-338
элементы Tyl/copia, 702, 704
элементы неустойчивости, 476
элементы управления импринтингом, 370
элонгация
-ДНК, 604, 608-619
- РНК, 431р, 431-432,432р, 435-441,449т,
449-450,465-466
- полипептидов, 512, 518
элонгация транскрипции, 431р, 431-432,
432р, 435-441
- РНК-полимеразы, 465-466
- мРНК эукариотов, 449-450
- синтез бактериальной РНК, 431р, 431-432,
432р, 435-441
элонгин, 449т
элюция, 58тп
ЭМС (этилметансульфонат), 653
эндогенные LTR-транспозоны, 331-333
эндогенные ретровирусы (ЭРВы), 333
эндонуклеаза НО, 559, 560р
эндонуклеаза Vsr, 674
эндонуклеазы, 39р, 45,45р
- вырезание интронов из пре-тРНК, 470
- интеины, 529
- эндонуклеаза НО, 559, 560р
эндонуклеазы рестрикции
- особенности, 45т
- полиморфизмы длин рестриктов, 87р,
87-88,88р
- разрезы в ДНК, 38р, 47р, 47-48,48р, 51-52
- типы, 47т
эндоспоры, 569
энергетический обмен веществ, 716
энхансосома, 416
ЭРВы (эндогенные ретровирусы), 333
Предметный указатель 921
эритроидный модуль, 414
этеноаденин, 670т
этеноцитозин, 670т
этика, 163-165
этилендиаминтетраацетат (ЭДТА), 58тп
этилметансульфонат (ЭМС), 653
эукариоты
- ДНК-полимеразы, 610,610т, 615-616
- ДНК-транспозоны, 334-335, 337р, 337-
338, 338р
- РНК органелл, 455т
- РНК-транспозоны, 331-334
- альтернативный сплайсинг, 461р, 460-463
- анатомия ядерного генома, 259-291
- горизонтальный перенос генов, 735
- восстановление вырезанием нуклеотидов,
672-673
- генетические особенности, 266-287
- геномы органелл, 308-314
- гомологичная рекомбинация, 696-697
- инициация трансляции, 508-510
- исследования видоизменения фенотипа и
сверхэкспрессии, 204т
- старты репликации, 606р, 605-609
- межвидовые сравнения, 271-275, 278-279
- перенос ДНК между ядерным геномом и
органеллами, 308-309
- подвижные генетические элементы,
330-340
- последствия мутаций, 658-659
- распределение интронов, 454,455т
- регулирование репликации, 627-632
- репликация, 606р, 605-611, 612р, 615-617,
617р, 620-632
- связывающиеся с ДНК белки, 381, 386-387
- секвенирование геномов, 158р, 153-161
- стратегии управления инициации транс-
крипции, 412-420
- строение ядра, 351р, 350-359
- структура рибосом, 502-504, 504р
- структура хромосом, 260-266
- топоизомеразы, 600т, 602
- шатание, 500-501
- эволюционные истоки интронов, 738-740
- эволюция, 308, 720р, 719-721
- эндосимбиотическая теория органелл,
308
эухроматин, 163,355,355р, 372,
ЭЭО (электроэндосмос), 49тп
ядерные поровые комплексы, 480,482р
ядерный антиген разрастающихся клеток
(PCNA), 610,615,632
ядерный геном, 3, Зр, 259-291
ядерный матрикс, 351,351р, 372,602
ядра-компанейщики, 245,246
ядра-свиданщики, 245
ядро, 351р, 350-359
ядрышко, 351,351р
ЯМР (ядерного магнитного резонанса)
спектроскопия, 381,383тп, 384
ярлыки экспрессируемых
последовательностей (EST), 123
Интересующие Вас книги нашего издательства можно заказать почтой или элек-
тронной почтой:
subscribe@rcd.ru
Внимание: дешевле и быстрее всего книги можно приобрести через наш интернет-
магазин:
http://shop.rcd.ru
Книги также можно приобрести:
1. Москва, ИМАШ, ул. Бардина, д. 4, корп. 3, к. 414,
тел. (499) 135-54-37, (495) 641-69-38
2. МГУ им. Ломоносова (ГЗ, 1 этаж)
3. Магазины:
Москва: «Дом научно-технической книги» (Ленинский пр., 40, тел.: 137-06-33)
«Московский дом книги» (ул. Новый Арбат, 8, тел.: 290-45-07)
Книжный магазин «ФИЗМАТКНИГА» (г. Долгопрудный,
Новый корпус МФТИ, 1 этаж, тел. 409-93-28)
С.-Пб.: «С.-Пб. дом книги» (Невский пр., 28)
Терри А. Браун
Геномы
Дизайнер В. А. Толстолуцкая
Технический редактор А. В. Широбоков
Корректор А. А. Чукарева
Подписано в печать 25.03.2011. Формат 70x100 1/16.
Печать офсетная. Усл. печ. л. 76,11. Уч. изд. л. 75,12.
Гарнитура Cambria. Бумага офсетная № 1. Заказ №0063.
АНО «Ижевский институт компьютерных исследований»
426034, г. Ижевск, ул. Университетская, 1.
http://shop.rcd.ru E-mail: mail@rcd.ru Тел./факс:+7(3412)500-295
Отпечатано в ГУП УР «Ижевский полиграфический комбинат»
426039, г. Ижевск, Воткинское шоссе, 180.