











































































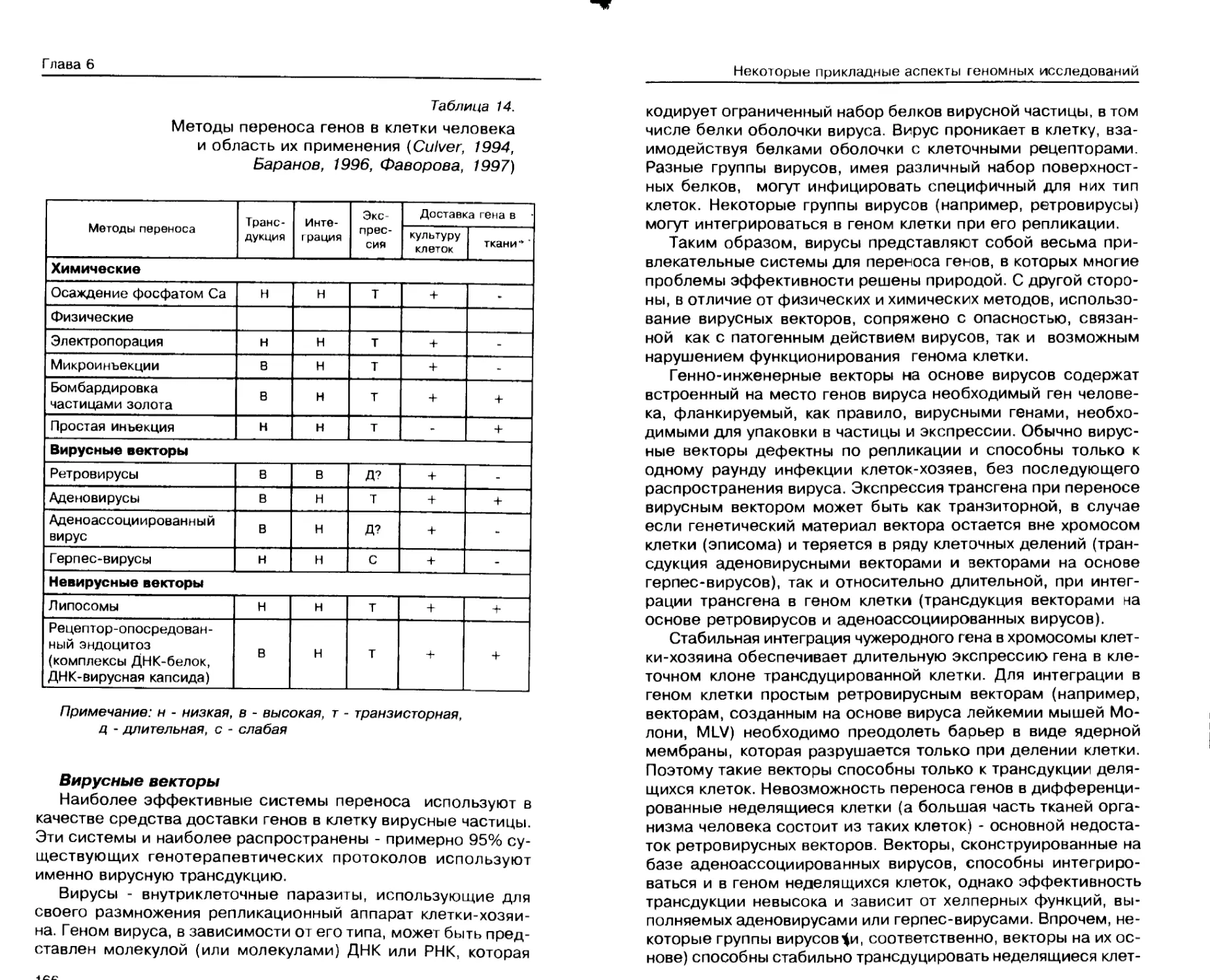


























Автор: Пузырёв В.П. Степанов В.А.
Теги: общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез общая патология анатомия человека патофизиология
ISBN: 5-02-031442-0
Год: 1997




УДК 575
ББК 52.5
t
П 88 Патологическая анатомия генома человека / Пузырев В. П.,
Степанов В, А — Новосибирск: Наука. Сиб. предприятие РАН,
1997.- 224 с.
ISBN 5-02-031442-0.
Монография посвящена одному из новейших и перспек-
тивных направлений медицинской генетики — патологической
анатомии генома человека. Рассматриваются современные тех-
нологии изучения генов человека (физическое и генетическое
картирование, секвенирование генома), вопросы генетической
классификации болезней человека, проблемы картирования
монотонных и мультифакториальных болезней. Большое внима-
ние уделено феноменам нетрадиционного наследования (мито-
хондриальные болезни геномный импринтинг, антиципация).
Впервые представлена версия хромосомных карт болезней чело-
века на русском языке.
Подробно описываются прикладные медицинские аспекты
геномных исследований — ДНК-диагностика монотонных бо-
лезней, молекулярная диагностика тонких хромосомных нару-
шений, гемотерапия. Обсуждаются теоретические основы
прикладного использования ДНК-технологий.
Монография представляет собой исчерпывающее обобще-
ние современных данных (список литературы включает около
400 работ ведущих российских и зарубежных ученых) по иссле-
дованию геномных основ патологии человека.
Книга рассчитана на специалистов в области генетики, сту-
дентов-медиков и биологов, практических врачей и всех, инте-
ресующихся вопросами медицинской генетики.
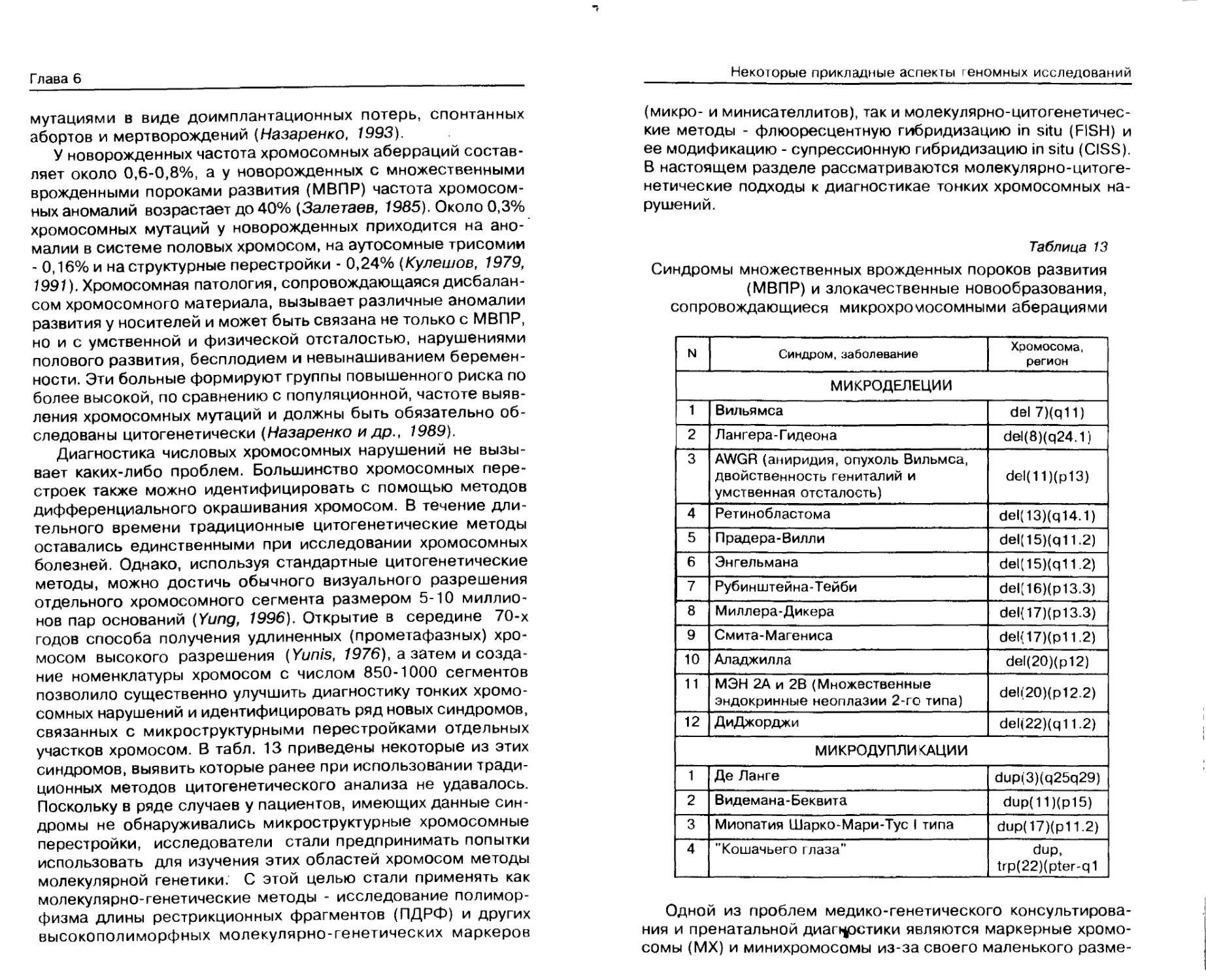
Табл. 19. Ил. 32. Библиогр.: 391 наэв.
ISBN 5-02-031442-0
© В. П. Пузырев,
В. А. Степанов,
1997
© STT, 1997
Оглавление
Предисловие...................................... 4
Глава 1. Наследственность и патология
человека: генетические аспекты
классификации болезней человека................... 6
Глава 2. Геном человека и методы его
анатомирования................................... 13
ДНК, гены, хромосомы .................... 13
Анатомирование генома: картирование
и секвенирование......................... 17
Размер генома человека и структура гена. 31
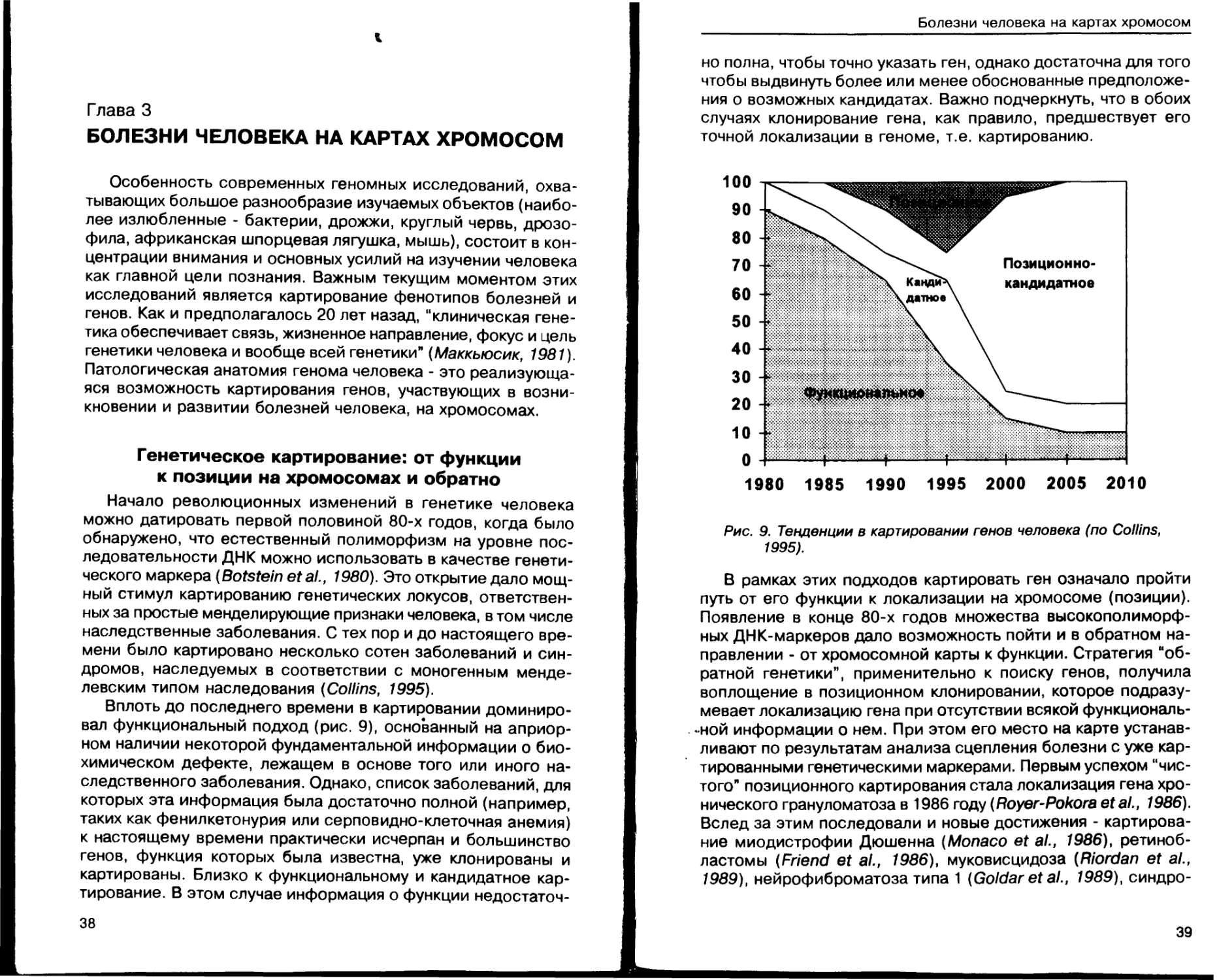
Глава 3. Болезни человека на картах хромосом... 38
Генетическое картирование: от функции
к позиции на хромосомах и обратно........ 38
Карты хромосом человека.................. 43
Глава 4. Генетическое картирование
мультифакториальных заболеваний.................. 70
Принципы генетического картирования
сложнонаследуемых признаков.............. 74
Кандидатные гены широко
распространенных заболеваний............. 83
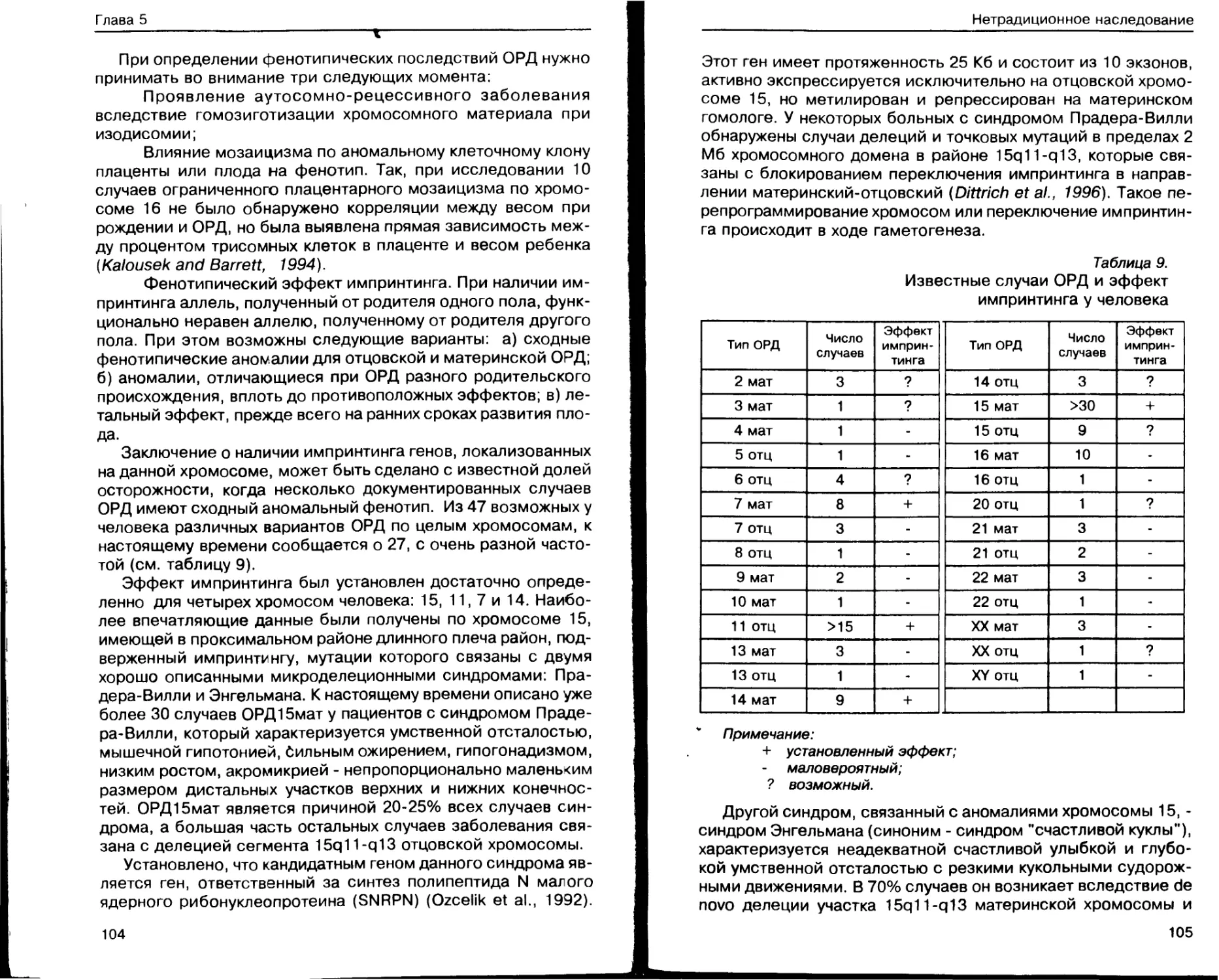
Глава 5. Нетрадиционное наследование............ 99
Геномный импринтинг и болезни
импринтинга ............................ 100
Митохондриальные болезни................ 109
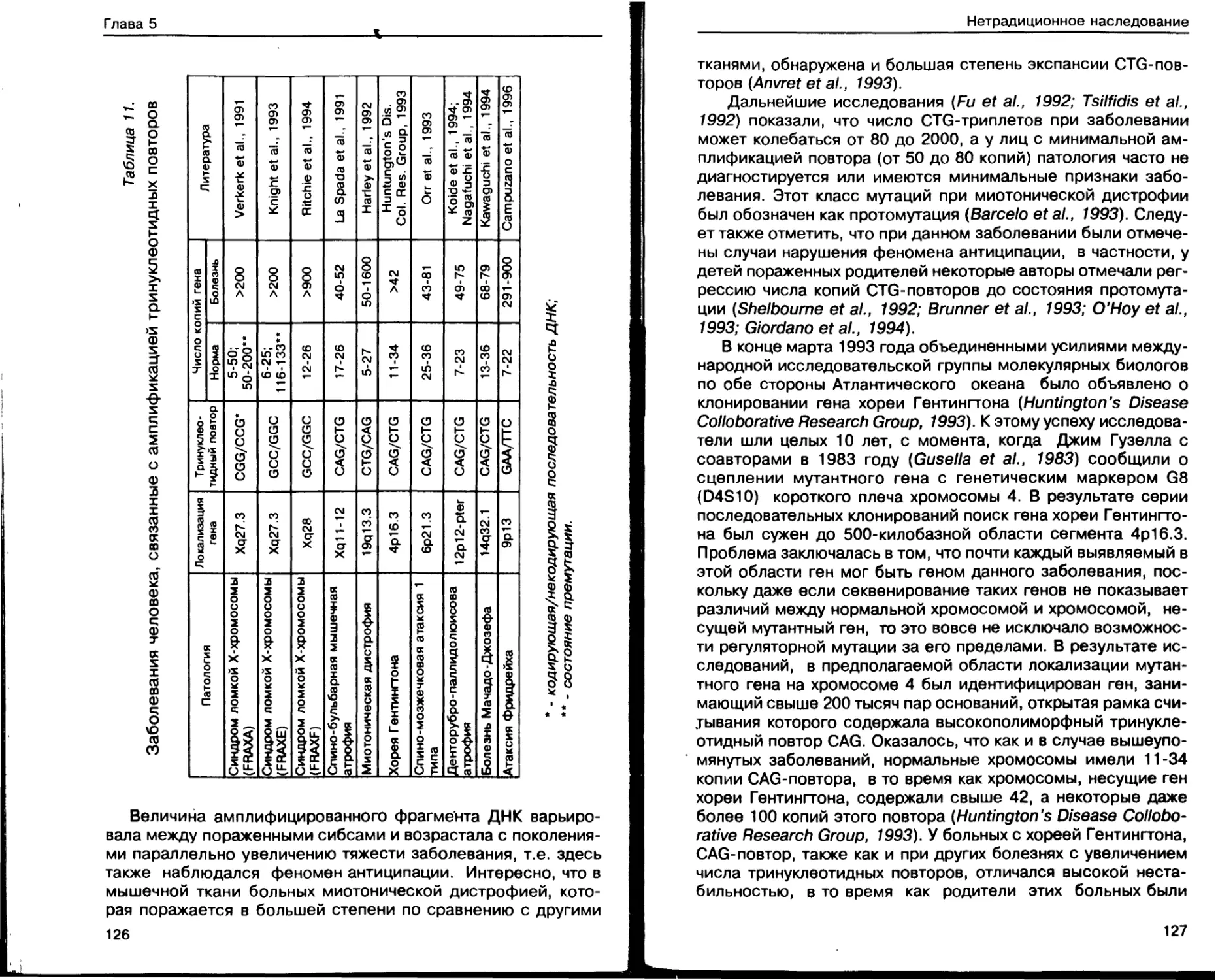
Болезни экспансии тринуклеотидных
повторов и антиципация.................. 119
Глава 6. Некоторые прикладные аспекты геномных
исследований.................................. 132
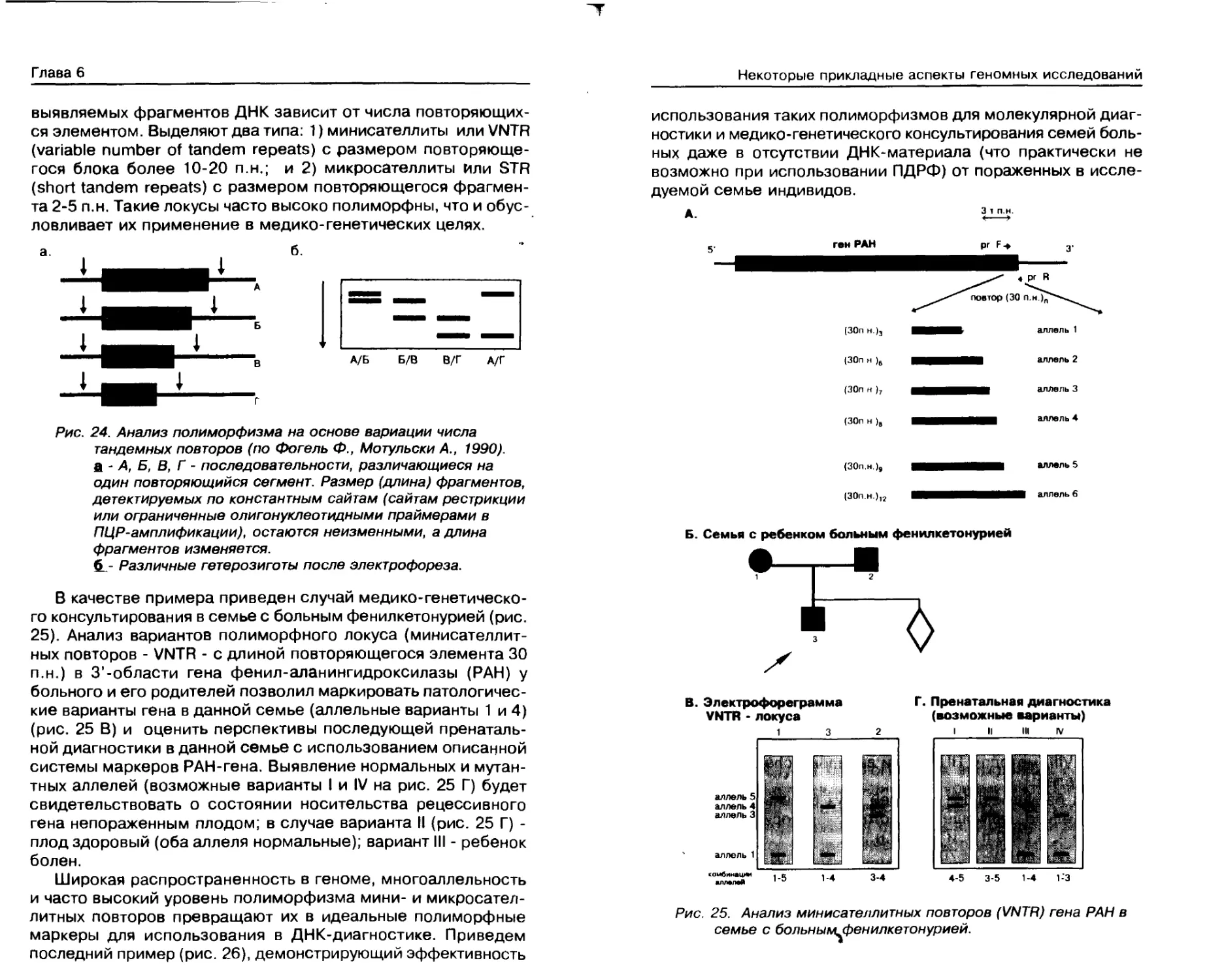
ДНК-диагностика монотонных болезней..... 132
Молекулярно-цитогенетическая диагностика
тонких хромосомных нарушений............ 151
Генотерапия ............................ 162
Глава 7. Патологическая анатомия генома
человека и общая патология
(вместо заключения)........................... 185
Приложение. Словарь терминов................... 196
Список литературы.............................. 204
3
УДК 575
ББК 52.5
t
П 88 Патологическая анатомия генома человека / Пузырев В. П.,
Степанов В, А — Новосибирск: Наука. Сиб. предприятие РАН,
1997.- 224 с.
ISBN 5-02-031442-0.
Монография посвящена одному из новейших и перспек-
тивных направлений медицинской генетики — патологической
анатомии генома человека. Рассматриваются современные тех-
нологии изучения генов человека (физическое и генетическое
картирование, секвенирование генома), вопросы генетической
классификации болезней человека, проблемы картирования
монотонных и мультифакториальных болезней. Большое внима-
ние уделено феноменам нетрадиционного наследования (мито-
хондриальные болезни геномный импринтинг, антиципация).
Впервые представлена версия хромосомных карт болезней чело-
века на русском языке.
Подробно описываются прикладные медицинские аспекты
геномных исследований — ДНК-диагностика монотонных бо-
лезней, молекулярная диагностика тонких хромосомных нару-
шений, генотерапия. Обсуждаются теоретические основы
прикладного использования ДНК-технологий.
Монография представляет собой исчерпывающее обобще-
ние современных данных (список литературы включает около
400 работ ведущих российских и зарубежных ученых) по иссле-
дованию геномных основ патологии человека.
Книга рассчитана на специалистов в области генетики, сту-
дентов-медиков и биологов, практических врачей и всех, инте-
ресующихся вопросами медицинской генетики.
Табл. 19. Ил. 32. Библиогр.: 391 наэв.
ISBN 5-02-031442-0
© В. П. Пузырев,
В. А. Степанов,
1997
© STT, 1997
Оглавление
Предисловие...................................... 4
Глава 1. Наследственность и патология
человека: генетические аспекты
классификации болезней человека................... 6
Глава 2. Геном человека и методы его
анатомирования................................... 13
ДНК, гены, хромосомы .................... 13
Анатомирование генома: картирование
и секвенирование......................... 17
Размер генома человека и структура гена. 31
Глава 3. Болезни человека на картах хромосом... 38
Генетическое картирование: от функции
к позиции на хромосомах и обратно........ 38
Карты хромосом человека.................. 43
Глава 4. Генетическое картирование
мультифакториальных заболеваний.................. 70
Принципы генетического картирования
сложнонаследуемых признаков.............. 74
Кандидатные гены широко
распространенных заболеваний............. 83
Глава 5. Нетрадиционное наследование............ 99
Геномный импринтинг и болезни
импринтинга ............................ 100
Митохондриальные болезни................ 109
Болезни экспансии тринуклеотидных
повторов и антиципация.................. 119
Глава 6. Некоторые прикладные аспекты геномных
исследований.................................. 132
ДНК-диагностика монотонных болезней..... 132
Молекулярно-цитогенетическая диагностика
тонких хромосомных нарушений............ 151
Генотерапия ............................ 162
Глава 7. Патологическая анатомия генома
человека и общая патология
(вместо заключения)........................... 185
Приложение. Словарь терминов................... 196
Список литературы.............................. 204
3
ПРЕДИСЛОВИЕ
Современная общая патология, основанная на концепции
организма человека как единого целого, непрерывно обога-
щается новыми фактами из области активно развивающихся
направлений медико-биологической науки. Генетика челове-
ка и медицинская генетика - одни из них, которые в последние
годы в значительной степени сконцентрировали свои усилия
на изучении молекулярного уровня организации человека. Осу-
ществление проекта “Геном человека”, над которым работают
ученые многих стран, в том числе отечественные, позволит
уже в начале приближающегося XXI века решить проблему
картирования генов человека (в том числе, имеющих отноше-
ние к патологии) и секвенирования всего генома (3 млрд, пар
нуклеотидов) человека.
Этот аспект геномных исследований для клиницистов обоз-
начен В.А.Маккьюсиком термином “патологическая анатомия
генома человека”. Впервые анатомическая метафора в обсуж-
дении генетических карт человека была им использована в
одной из своих лекций в 1979 году (McKusick, 1980). Действи-
тельно, определение расположения генов болезней человека
на хромосомах осуществляется приемом, который давно из-
вестен медицине, и название его происходит от греческого
глагола “анатемно” (рассекать, расчленять). В этом значении
слова структура ДНК и строение тела человека изучаются сход-
но, только орудием анатомирования в этом случае служат
ферменты (рестриктазы) и весь арсенал технологий современ-
ной молекулярной генетики, а раньше - скальпель. Кстати, ге-
нетиками позаимствован и другой медицинский термин (би-
опсия) для обозначения процедуры молекулярно-генетичес-
кой диагностики болезней - "диагностическая биопсия генома
человека”. Словом, современный врач должен быть знаком с
новыми возможностями генетической науки. В настоящей книге
изложены основные принципы и методы геномных исследова-
ний.
Патологическая анатомия генома человека, названная
Н.П.Бочковым (1983) “новой главой в генетике человека”, с
одной стороны, может рассматриваться в ряду существующих
разделов анатомии человека (нормальной, патологической,
топографической). Значимость их для медицинского образо-
вания очевидна. Вспомним слова великого русского естество-
испытателя М.В.Ломоносова: “Как можно рассуждать о теле
человека, не зная ни сложения костей и суставов для его ук-
репления, ни союза, ни положения мышц для чувствования, ни
расположения внутренностей для приготовления питательных
соков, ни протяжения жил для обращения крови, ни прочих
органов сего чудного строения". Этот путь пройден медицин-
ской наукой, а благодаря этому мировая практика врачевания
достигла поразительных успехов. С другой стороны, новые
возможности на этом пути открывают успехи геномных иссле-
дований, а необходимость их усвоить, понять и принять звучат
в Нобелевской лекции Пола Берга (Berg, 1981): “Как наша со-
временная медицинская практика опирается на утонченные
знания в области анатомии человека, физиологии и биохи-
мии, так в будущем изучение генетических болезней потребу-
ет детального понимания молекулярной патологии, физиоло-
гии и биохимии генома человека. Нам потребуются врачи на-
столько осведомленные в молекулярной анатомии и физиоло-
гии хромосом и генов, насколько кардиохирург знает работу
сердца и структуру сосудистого древа".
В настоящей книге впервые представлена русскоязычная
версия карт хромосом человека с указанием локализации ге-
нов, ответственных за возникновение и развитие болезней
человека, а также список картированных генов болезней че-
ловека. Это позволяет использовать книгу как справочное ру-
ководство. Есть еще одна задача у этой книги. Каждое новое
направление науки, касающееся проблемы природы болезней
человека, должно быть осмыслено с позиций включенности
его в современные концепции общей патологии. Обсудить эти
вопросы особенно важно с общебиологических позиций, пос-
кольку “перемещение “центра тяжести” медико-биологичес-
ких исследований на молекулярный уровень... означает оче-
редной период в истории развития общей патологии - пери-
од, который рано или поздно должен будет смениться или,
скорее всего, дополниться периодом синтетическим...” (Сар-
кисов и др., 1995). Эти вопросы обсуждаются в конце книги.
В заключение заметим, что до сих пор главным источником
развития теории общей патологии было обобщение фактов,
полученных на организменном, органном, тканевом, клеточ-
ном и внутриклеточном уровнях организации, но в меньшей
степени на геномном и популяционном. Сегодня активно ис-
следуется молекулярный уровень организации. Однако, в силу
новизны молекулярно-генетических материалов и данных ге-
нетической эпидемиологии, они еще-слабо ассимилированы
общей патологией человека. А именно результаты этих иссле-
дований (геномных и популяционных) составляют, главным
образом, “биологический аспект осмысления медицинских
проблем”.
5
I
Глава 1
НАСЛЕДСТВЕННОСТЬ И ПАТОЛОГИЯ
ЧЕЛОВЕКА: ГЕНЕТИЧЕСКИЕ АСПЕКТЫ
КЛАССИФИКАЦИИ БОЛЕЗНЕЙ ЧЕЛОВЕКА
Проблема “Наследственность и патология человека” столь
многообразна, что велика опасность при ее обсуждении увяз-
нуть в многочисленных подробностях, потеряв основную идею
книги - раскрыть современные молекулярно-генетические под-
ходы к анатомированию генома человека и локализации “па-
тологических генов” на хромосомах, обратить внимание на
недавно описанные неканонические механизмы формирова-
ния генетической основы некоторых форм патологии, осве-
тить медицинские прикладные аспекты геномных исследова-
ний. В этом контексте обсуждение вопросов классификаций
болезней человека (генетических ли аспектов классификации
всех болезней или классификации только наследственных -
не так важно, ибо это уже семантическая сторона вопроса)
как способа упорядочения накопленного исследовательского
материала по этиопатогенезу болезней, кажется, спасает си-
туацию. Систематизировать, распределить известные патоло-
гические фенотипы по классам - значит уяснить общие свой-
ства, фиксирующие закономерные связи между ними. В этом
и состоит одна из задач общей патологии.
И все же не избежать необходимости, хотя бы кратко и без
претензии на всеобъемлемость, упомянуть некоторые положе-
ния о роли наследственности в патологии человека, главным
образом, те из них, которые имеют прямое отношение к об-
суждаемым в книге вопросам.
1. Важно, что при относительной редкости наследственных
моногенных болезней (см. табл. 1) частота “патологических”
генов в популяции достаточно высока. Так, если частота фе-
нилкетонурии (ФКУ) составляет 1 на 10.000 новорожденных,
то частота лиц - носителей гена этой болезни (гетерозиготы)
в данной популяции - 1:50. Следовательно, с учетом всей со-
вокупности менделирующих болезней, потребность выявле-
ния гетерозиготного носительства мутантного гена в условиях
медико-генетического консультирования велика, а молекуляр-
но-генетические методы тестирования таких индивидов ста-
новятся ведущими среди различных подходов.
6
Наследственность и патология человека
Другой аспект этого положения заключается в том, что ге-
терозиготы по ФКУ, не имея клинических проявлений этой тя-
желой болезни, имеют ряд других отклонений в состоянии
здоровья: по IQ и другим психологическим тестам; отклоне-
ние от нормы в электроэнцефалограмме; у них высок риск
заболеть шизофренией определенного типа; для женщин по-
вышен риск спонтанных абортов и мертворождений (Vogel,
1984). Эти два аспекта лишь проиллюстрированы на одном
примере (ФКУ), но являются общими для многих наследствен-
ных болезней.
Таблица 1.
Частота генетически детерминированных
болезней (Е. Passarge, 1995)
Формы наследственной патологии Частота на 1000
1. Моногенные 4.5 - 15.0
аутосомно-доминантные 2.0 - 9.5
аутосомно-рецессивные 2.0 -3.5
Х-сцепленные 0.5 - 2.0
2. Хромосомные болезни 5.0 - 7.0
3. Мультифакториально обусловленные* 7.0 - 10.0
4. Врожденные пороки развития* 19.0 - 22.00
ВСЕГО 35-54
Примечание:
* вклад генетических факторов варьирует
2. Наследственная болезнь не является прерогативой только
детства, т.е. не всякая наследственная болезнь врожденная.
Некоторые из них (хорея Гентингтона, мышечная дистрофия,
поликистоз почек) проявляются в зрелом возрасте. В целом
довольно значительная доля наследственной патологии при-
сутствует среди взрослого населения (табл. 2). Об этом сле-
дует помнить клиницисту в процессе дифференциальной ди-
агностики и шире включать в логические дифференциально-
диагностические схемы наследственные болезни и синдромы
(Бочков, 1995).
7
Глава 1
Таблица 2.
Относительно частые менделевские болезни
взрослых (Гольдштейн и Браун, 1993)
Аутосомно-доминантные болезни
Семейная гиперхолестеринемия
Наследственная геморрагическая телангиэктазия
Синдром Марфана
Наследственный сфероцитоз
Поликистоз почек взрослых
Хорея Гентингтона
Острая перемежающаяся порфирия
Остеогенез несовершенный поздний
Болезнь Виллебранда
Миотоническая дистрофия
Идиопатический гипертрофический субаортальный
стеноз (ИГСС)
Нунан синдром
Нейрофиброматоз
Туберозный склероз
Аутосомно-рецессивные болезни
Глухота
Альбинизм
Болезнь Вильсона
Г емохроматоз
Серповидно-клеточная анемия
р-Талассемия
кистозный фиброз
Наследственная энфизема (дефицит а-антитрипсина)
Г омоцистинурия
Семейная средиземноморская лихорадка
Атаксия Фридрейха
Фенилкетонурия
Х-сцепленные болезни
Гемофилия А
Дефицит глюкозо-6-фосфатдегидрогензаы
Болезнь Фабри
Глазной альбинизм
Тестикулярная феминизация
Хронический гранулематоз
Гипофосфатемический рахит
Цветовая слепота
3. Не все наследственные болезни наследуются из преды-
дущих поколений. Так, индивиды с доминантным заболевани-
ями, приводящими к ранней смерти, влияющими на выживае-
мость или исключающие репродукцию, чаще являются мутан-
тами de novo. В случае болезней с поздним проявлением му-
тантного гена мутанты de novo редки или вообще отсутствуют.
Некоторые примеры по доле лиц с аутосомно-доминантными
заболеваниями, обусловленными мутациями de novo приве-
дены в табл. 3.
4. Наконец, схематично обозначим взаимосвязь положений
генетики и общей патологии, которые обосновывают утвер-
ждение о том, что именно они составляют важнейшую часть
8
Наследственность и патология человека
теоретической медицины (Бочков и др., 1984). Среди них: на-
следственный биохимический полиморфизм и индивидуаль-
ность в клинических проявлениях болезни (возраст начала
болезни, характер течения болезни, ее исход); адаптивная
норма, сбалансированное взаимодействие конкретных генов
и гомеостаз; факторы популяционной динамики (отбор, мута-
ционный процесс, миграция, изоляция, инбридинг, дрейф ге-
нов), геногеография и спектр патологии в различных этносах;
медико-генетическое консультирование, дородовая диагнос-
тика, скрининг наследственных дефектов и разные концепции
профилактической медицины (профилактика в соответствии с
уникальным генотипом индивида или направленность ее на
популяцию в целом).
Таблица 3.
Примерная доля больных, пораженных новыми
мутациями при некоторых аутосомно-доминантных
болезнях (Гольдштейн и Браун, 1993)
Болезнь Доля (%)
Ахондроплазия 80
Туберозный склероз 80
Нейрофиброматоз 40
Синдром Марфана 30
Миотоническая дистрофия 25
Хорея Гентингтона 4
Поликистоз почек взрослых 1
Семейная гиперхолестеринемия Очень мало
Сегодня известно более 20 тыс. нозологических форм па-
тологии человека (Смольянников, 1981), классификация кото-
. рых представляет большую проблему. Классификация - это
традиционно описательный метод упорядочения исследуемо-
го материала. Этому определению соответствуют многочис-
ленные международные классификации болезней как резуль-
тат более десятка пересмотров специальным комитетом Все-
мирной организации здравоохранения “перечня классов и групп
с номерами входящих в них трехзначных рубрик”, широко из-
вестных вариантов “Номенклатуры и классификации болезней
9
Глава 1
----------------------------------%---------------------
человека”. По сути дела подобного рода классификации, раз-
рабатываемые на протяжении всего 20 века, имеют ту же на-
правленность, что и первые известные классификации 19 века
В.Фара, М.Эспина, Дж.Бертильона на Западе и М.Я.Мудрова,
Н.Е.Дядьковского, Общества русских врачей в память Н.Н.Пи-
рогова в России. Они не стали той вехой на пути классифика-
ции болезней человека, которой была первая научная класси-
фикация болезней, предложенная Морганьи в 18 веке, по-
ложившего в основу ее патологоанатомический принцип пре-
имущественной локализации болезненных процессов, что при-
нципиально отличало ее от классификаций прошлого, исхо-
дивших из виталистических концепций.
Значимость подобного подхода к классификации болезней
несомненна, но необходимо иметь ввиду, что классификации
выполняют задачи медицинской статистики населения, а так-
же деятельности учреждений здравоохранения. В них практи-
чески не обозначено место для генетических болезней. Врож-
денные и наследственные болезни в соответствии с принятым
подходом, должны быть “втиснуты” либо в раздел “Врожден-
ные аномалии (пороки развития)”, либо в рубрикаторы разде-
лов, поименованных по названию органов и систем человека.
Это обстоятельство затрудняет оценку истинного “груза” на-
следственных болезней в популяциях человека, число генети-
ческих нозологических форм которых составляет сейчас не ме-
нее 4 тысяч (McKusick, 1996).
Pinsky (1974; 1975) указал на возможность и полезность
классификации наследственных синдромов, основанной на их
“фенотипической особенности”, как результата предположи-
тельной общности дисморфогенетических механизмов фор-
мирования синдромов. Этиология их может быть менделевс-
кой, хромосомной или совершенно неизвестной. Основанием
для объединения синдромов в кластеры по общности феноти-
пических черт служит допущение о том, что независимо от
этиологии патогенетические (т.е. дисморфогенетические) ме-
ханизмы очень сходны или идентичны (ранее в 1969 г. для сход-
ного случая J.M.Opitz и др. предложили термин “синдром фор-
мального генеза”). Основная научная ценность такого класси-
фикационного подхода к недифференцированным врожденным
синдромам заключается в стимулировании изучения дисмор-
фологических основ данной общности синдромов. В практи-
ческом плане этот подход позволяет более объективно вери-
фицировать “новые” или редкие синдромы (обосновать реаль-
ность или только видимость новизны). И все же этот подход
является описательным, а значит остается риск ошибок в сис-
тематике исследуемого материала до тех пор, пока не станет
ясна фундаментальная природа нарушения.
10
Наследственность и патология человека
Один из генетических аспектов классификации болезней
человека заключается в упорядочении их по критерию участия
наследственных и средовых факторов в этиологии, патогене-
зе и рекреации (восстановление утраченного ‘'качества” здо-
ровья). С этих позиций выделено четыре группы патологии,
между которыми нет резких границ (Бочков, 1978; Бочков и
цр., 1984): Г- собственно наследственные болезни (моноген-
ные и хромосомные); II - наследственные болезни, для прояв-
ления которых необходимо специфическое воздействие сре-
ды (экогенетические реакции); III - болезни с наследственным
предрасположением, объединяющие подавляющее большин-
ство распространенных болезней, особенно болезней зрело-
го и преклонного возраста; IV - сравнительно немногие фор-
мы патологии, в этиологии которых исключительную роль иг-
рают факторы среды (травмы, ожоги, обморожения, особо
опасные инфекции), но генетические факторы в этом случае
определяют особенности клинического течения болезни и ее
исход.
McKusick (1988) в основу своей классификации наслед-
ственных болезней положил два варианта мутаций - в поло-
вых и соматических клетках (табл. 4), тем самым утверждая
целесообразность выделения трех форм наследственной па-
тологии: собственно наследственные болезни, которые потен-
циально могут передаваться по наследству; генетические бо-
лезни соматических клеток, включающие большинство злока-
чественных опухолей, возможно, значительную часть аутои-
мунных нарушений, старение и врожденные аномалии разви-
тия; болезни, обусловленные комбинацией мутаций одновре-
менно в соматических и половых клетках.
Автор данной классификации подчеркивает наряду с ее
очевидной полезностью, и некоторую условность. Так, высо-
кая разрешающая способность современных цитогенетичес-
ких методов позволила установить хромосомную природу та-
ких заболеваний, как ретинобластома и некоторых других, счи-
тавшихся менделирующими (синдром Лангера-Гидеона, Пра-
дера-Вилли). Кроме того, гены большинства менделирующих
Заболеваний находятся под контролем генетического фона
пораженного индивида и средовых факторов, что позволяет,
по мнению автора, утверждать - все генетические нарушения
(болезни) в некотором смысле мультифакториальны.
11
Таблица 4.
Классификация наследственных болезней
_______________________________(по V.A.McKusick, 1988)
Болезни вследствие мутаций в половых клетках
(собственно наследственные болезни)
хромосомные (например, синдром Дауна)
мутации в отдельном гене (см, MIM1)
мультилокусные (полигенные, мультифакториальные)
Болезни вследствие мутаций в соматических клетках
(наследственные болезни соматических клеток: опухоли,
некоторые аутоиммунные болезни, старение, некоторые
врожденные пороки развития)
хромосомные
генные
мультифакториальные
Болезни, представляющие комбинацию мутаций
в половых и соматических клетках
(например, семейная ретинобластома)
Примечание:
*MIM - Mendelian Inheritance in Man - периодически издающий-
ся и обновляющийся каталог (V.A.McKusick, 1996)
Недавно генетиками предложена еще одна рабочая клас-
сификация болезней человека, включающая (Nora и др., 1994).
1. Болезни, вызванные мутацией отдельного гена (менде-
левские).
2. Синдромы, обусловленные хромосомными нарушениями.
3. Мультифакториальные заболевания как результат взаи-
модействия генетических и средовых факторов.
4. Болезни с нетрадиционным типом наследования.
5. Генетические болезни соматических клеток (новообра-
зования, старение, аутосомные болезни).
Особенностью данной классификации является выделение
болезней, обусловленных относительно недавно сформиро-
вавшимся научным интересом к таким феноменам как мито-
хондриальная наследственность, геномный импринтинг, одно-
родительская дисомия, экспансия тринуклеотидных повторов,
а также к связи их с патологией (Hall, 1990; 1996). “Нетрадици-
онная наследственность” - условное название новой интерес-
нейшей области генетики человека.
От описательных классификаций фенотипов наследствен-
ных болезней и синдромов к сущностным, в основе которых
лежат детальные знания дефекта гена (генов), - такова общая
тенденция современной медицины и клинической генетики в
систематике болезней и “наведении порядка” в концепциях
заболеваний. Эта тенденция поддерживается и развивается в
фарватере геномных исследований, анатомирования и струк-
турно-функционального изучения генома человека.
12
Глава 2
ГЕНОМ ЧЕЛОВЕКА И МЕТОДЫ
ЕГО АНАТОМИРОВАНИЯ
Геномом называют всю совокупность генетического мате-
риала организма, все множество его генов. Иными словами,
это полный набор инструкций, необходимых для его построе-
ния и функционирования. Физическим носителем генетичес-
кой информации является дезоксирибонуклеиновая кислота
(ДНК). Молекула ДНК вместе со связанными с ней белками
образует плотно упакованные структуры, называемые хромо-
сомами. Ядро каждой из клеток человека, исключая половые
клетки (гаметы) и зрелые эритроциты, содержит 23 пары хро-
мосом - по одной отцовской и материнской хромосоме в каж-
дой паре. Чтобы понять, каким образом все множество ин-
формации, необходимой для развития и жизнедеятельности
организма хранится и реализуется, напомним структурную
организацию ДНК, генов и хромосом, а затем рассмотрим те
методы, которые и представляют собой молекулярно-генети-
ческий “скальпель”, посредством которого ученые анатоми-
руют геном. Это важно сделать, на наш взгляд, еще и потому,
что постоянно накапливающаяся информация по структуре и
организации генома человека требует оперативной система-
тизации, что позволит врачам и студентам-медикам находит-
ся в курсе последних достижений молекулярной генетики и
поможет воспринять последующие разделы, в которых пойдет
речь о генетических основах болезней человека, молекуляр-
ной диагностике и генотерапии.
ДНК, гены, хромосомы
ДНК
Молекула ДНК человека и других высших организмов пос-
троена из двух спирально закрученных друг на друга цепочек,
каждая из которых состоит из множества мономеров - нукле-
отидов (рис. 1). Нуклеотид - это химическое вещество, состо-
ящее из одной молекулы сахара, остатка молекулы фосфор-
ной кислоты и молекулы азот-содержащего органического
соединения - азотистого основания. Соседние нуклеотиды
одной цепочки связаны друг с другом сахаро-фосфатными
связями между остатком фосфорной кислоты одного нуклео-
13
Глава 2
тида и молекулой сахара соседнего. Две цепочки удержива-
ются вместе благодаря слабым водородным связям между
азотистыми основаниями.
Рис. 1. Структура молекулы ДНК
Всего в состав ДНК входят четыре азотистых основания -
аденин (А), тимин (Т), цитозин (С) и гуанин (G). Порядок, в
котором чередуются азотистые основания в цепочке, называ-
ется последовательностью ДНК. Именно в последовательнос-
ти нуклеотидов ДНК и закодирована генетическая информа-
ция, которая наделяет организм присущими ему индивидуаль-
ными признаками.
Азотистые основания двух цепочек, взаимодействуя друг с
другом водородными связями, образуют пары оснований. При-
чем, каждое из четырех оснований способно образовать пару
не с любым из трех оставшихся, а только с одним из них. Это
свойство называется комплиментарностью. Аденин может об-
разовывать пару с тимином, иначе говоря, А комплиментарен
Т, а цитозин формирует пару с гуанином (С комплиментарен
G).
Цепь ДНК линейна, однако ее концы и направления от од-
ного конца к другому не равнозначны. Нуклеотид образовыва-
ет связь со следующим нуклеотидом в цепи от атома углерода
в составе дезоксирибозы с номером 3’ к атому 5’ следующего
нуклеотида. Следовательно, у первого нуклеотида в цепи 5'-
атом не формирует связи (т.е. свободным оказывается 5’-ко-
нец), а у последнего не участвует в формировании связи 3’-
атом (свободен З’-конец).
Размер генома принято выражать в парах оснований (п.о.).
Весь геном человека содержит примерно 3 миллиарда п.о. Если
бы всю эту информацию можно было напечатать в виде пос-
ледовательности ДНК, то она заняла бы 200 1000-страничных
14
Геном человека и методы его анатомирования
томов (рис. 2). Для сравнения, геном бактерии кишечной па-
лочки (Е. coli) уместился бы на 300 страницах, а геном дрож-
Рис. 2. Сравнительные размеры генома человека и некоторых
других организмов (по “DOE Primer on Molecular Genetics”,
с изменениями).
Каждая клетка человека, за некоторыми исключениями,
имеет полную копию генома. И каждый раз, когда клетка де-
лится, две дочерние клетки получают точно такую же генети-
ческую информацию, как и родительская клетка. В процессе
клеточного деления слабые связи между двумя цепочками
молекулы ДНК разрываются и цепочки разделяются. Затем на
каждой из этих цепочек как на матрице благодаря комплимен-
тарное™ оснований из свободных нуклеотидов строится вто-
рая (комплиментарная) цепочка. Таким образом, из одной ис-
ходной молекулы ДНК образуются две, совершенно одинако-
вые. Такой процесс матричного синтеза называется реплика-
цией ДНК и именно он позволяет сохранить постоянство гене-
тической информации от поколения к поколению.
Гены
Молекула ДНК может содержать множество генов. Ген -
это участок молекулы ДНК, который занимает на хромосоме
строго определенную позицию и последовательность ДНК
которого содержит информацию, необходимую для синтеза
белка. Геном человека включает, по приблизительным оценкам
50-100 тысяч генов, каждый из которых выполняет
специфическую функцию - кодирует определенный белок
15
Глава 2
(например, ферменты или структурные белки клетки) или
молекулу РНК.
Гены человека сильно варьируют по размерам - от несколь-
ких сотен до нескольких миллионов пар оснований. Известно,
что кодирующие белок последовательности занимают лишь
около 10% генома. Остальные 90% приходятся на долю неко-
дирующих участков. Большая часть генов состоит из перемежа-
ющихся кодирующих (экзоны) и некодирующих (интроны) час-
тей. Некодирующие районы между генами называют спейсе-
рами. В некодирующих участках располагаются последователь-
ности, регулирующие активность генов. Однако функция боль-
шей части некодирующей ДНК не ясна.
Основным "строительным материалом" живых организмов
является белок. Клетки человека способны синтезировать око-
ло 100 тысяч различных белков. Белок представляет из себя
сложную молекулу, состоящую из одной или более цепочек,
построенных из остатков аминокислот. Последовательность
аминокислот в белке закодирована в последовательности ос-
нований гена. Три подряд расположенных нуклеотида пред-
ставляют собой кодон, который и определяет, какая из амино-
кислот будет располагаться в данной позиции белка. Напри-
мер, последовательность оснований ATG является кодоном для
аминокислоты метионина, последовательность ТТТ кодирует
фенилаланин и т.д. Соответствие триплетов аминокислотам
называется генетическим кодом. Поскольку одна аминокис-
лота "зашифрована" тремя нуклеотидами гена, зная размеры
гена можно выяснить и размер белка (и наоборот). Например,
белок, кодируемый геном, экзоны которого занимают 3000 п.о.,
будет содержать 1000 аминокислотных остатков.
Реализация генетической информации - путь от гена к бел-
ку - проходит две стадии - транскрипцию и трансляцию. На
первой стадии информация с молекулы ДНК "переписывает-
ся" (транскрибируется) на молекулу рибонуклеиновой кисло-
ты (РНК). Молекула РНК по структуре очень близка к одноце-
почечной ДНК. РНК синтезируется на одноцепочечной ДНК-
матрице в соответствии с принципом комплиментарности. Та-
кая молекула РНК, содержащая копию гена, называется мат-
ричной (мРНК). Молекула мРНК затем переносится из ядра в
цитоплазму клетки, где на специализированных клеточных
органеллах - рибосомах - происходит процесс синтеза белка
(трансляция).
Хромосомы
Все 3 миллиарда пар оснований, составляющих геном че-
ловека, распределены по 23 хромосомам. Каждая хромосома
представляет собой одну гигантскую молекулу ДНК, связан-
16
Геном человека и методы его анатомирования
ную с множеством специализированных белковых молекул.
Гены на хромосоме расположены линейно - т.е. последова-
тельно друг за другом. Каждая клетка человека, за исключе-
нием половых клеток и зрелых эритроцитов, содержит по 2
полных набора хромосом (одного материнского и одного от-
цовского). В каждом наборе по 23 хромосомы - 22 аутосо-
мы и по одной половой хромосоме (X или Y). В хромосомном
наборе женщины - две Х-хромосомы, у мужчины - одна X- и
одна Y-хромосома. То есть, всего кариотип (набор хромосом)
человека состоит из 46 хромосом. Хромосомы неодинаковы
по размерам и содержат разное количество ДНК - от прибли-
зительно 50 до приблизительно 250 миллионов пар основа-
ний. В среднем, на одну хромосому приходится около 150
миллионов п.о. В кариотипе человека аутосомы нумеруются
по уменьшению размера - от 1 до 22 хромосомы.
Хромосомы можно увидеть в обычный световой микрос-
коп. При окрашивании некоторыми красителями хромосомы
предстают в виде чередующихся светлых и темных участков
(бэндов) - в зависимости от соотношения А-Т и G-С пар в дан-
ном сегменте. Такое окрашивание хромосом (оно называется
дифференциальным) позволяет не только отличать хромосо-
мы одну от другой, но и выделять на хромосомах отдельные
участки - сегменты и подсегменты.
На каждой хромосоме присутствует первичная перетяжка
(центромера), которая разделяет хромосому на два плеча -
короткое (р) и длинное (q). При делении клетки к центромере
прикрепляются нити веретена. Концевые участки хромосом -
теломеры - представляют собой специализированные струк-
туры, функция которых - обеспечивать целостность и стабиль-
ность хромосомы. Выявляемые при дифференциальном окра-
шивании сегменты хромосомы на каждом из плеч нумеруют
арабскими цифрами от центромеры к теломере. Внутри сег-
ментов выделяют отдельные подсегменты.
Анатомирование генома:
картирование и секвенирование
Одной из основных целей исследования генома человека
является построение его точной и подробной карты. Генети-
ческая карта - это схема, описывающая порядок расположе-
ния на хромосоме генов и других генетических маркеров, а
также расстояние между ними. Карты генома, как и географи-
ческие карты, можно строить в разном масштабе, т.е. с раз-
ным уровнем разрешения. Примером самой мелкомасштаб-
ной карты - карты с минимумом деталей - является картина
дифференциального окрашивания хромосом. Максимально
17
2 Заказ №127
Г лава 2
возможный уровень разрешения на генетической карте - один
нуклеотид. То есть, самой крупномасштабной картой генома
является полная последовательность нуклеотидов.
Карты генома принято подразделять на карты генетическо-
го сцепления (собственно генетические карты) и физические
карты. Различаются они методами построения. Карты генети-
ческого сцепления строятся на основе анализа данных по на-
следованию гена или маркера в ряду поколений, физические
карты основываются на прямом исследовании носителей ге-
нетической информации - хромосом, генов, молекул ДНК. Упо-
минавшееся выше сегментирование хромосом с помощью
дифференциальной окраски - пример физического картиро-
вания. Существуют и другие типы физических карт - рестрик-
ционная карта, карта контиг и т.д.
К настоящему времени для всех хромосом человека пос-
троены мелкомасштабные генетические карты с расстоянием
между соседними маркерами в 7-10 миллионов п.о. (мегабаз).
Для некоторых районов генома, представляющих особый ин-
терес, имеются и подробные физические карты. Всего сейчас
известна локализация около 5,5 тысяч генов.
Несколько лет назад была сформирована международная
генетическая программа ’Теном человека", которая объеди-
нила усилия ученых, работающих в области картирования ге-
нов человека. Конечной целью этого проекта является пос-
троение подробной карты генома. Для достижения этой цели
предстоит построить детальную генетическую карту (с уров-
нем разрешения в 2 Мб), затем создать физические карты
фрагментов генома с разрешением до 5 Кб, и наконец, пол-
ностью секвенировать всю ДНК генома человека. Создание
такой карты генома может совершить настоящую революцию
в познании биологии человека. Данные, полученные при кар-
тировании и полном секвенировании генома, позволят выявить
все гены человека, помогут определить их функции и разра-
ботать средства для коррекции наследственных нарушений.
Карты генетического сцепления
Генетические карты показывают относительное располо-
жение на хромосоме генов и других маркеров. Любой насле-
дуемый признак, будь то цвет глаз или различия в длине фраг-
ментов ДНК, потенциально может служить генетическим мар-
кером. Единственные требования к маркеру - межиндивиду-
альные различия по нему и легкость выявления этих различий.
Маркерами могут быть как экспрессируемые участки ДНК
(гены), так и некодирующие последовательности, наследова-
ние которых можно проследить.
18
Геном человека и методы его анатомирования
Для того, чтобы маркер можно было использовать при кар-
тировании, он должен быть полиморфным, т.е. должны сущест-
вовать альтернативные формы признака, которые можно было
бы легко различить и описать у членов семьи. Полиморфизм
ДНК - довольно распространенное явление. В среднем, пос-
ледовательность ДНК двух случайно выбранных людей будет
различаться по одному из каждых 300-500 нуклеотидов. Вари-
ации ДНК в экзонах могут приводить к изменениям структуры
белка и проявляться в виде внешних фенотипических призна-
ков, например, в различиях по группе крови или предрасполо-
женности к той или иной болезни. Поскольку экзоны занима-
ют лишь небольшую часть генома, большинство различий в
последовательности ДНК сосредоточено в интронах и не про-
является во внешних фенотипических признаках. В то же вре-
мя эти изменения легко определяемы на уровне ДНК и тоже
могут использоваться как маркеры. Примерами таких марке-
ров, где признаком служат сами характеристики последова-
тельности ДНК, являются полиморфизм длин рестрикционных
фрагментов (ПДРФ) и полиморфизм по числу тандемных пов-
торов.
ПДРФ является результатом полиморфизма ДНК в местах
(сайтах), которые можно разрезать рестрикционными фермен-
тами. Сайты узнавания рестрикционными нуклеазами - корот-
кие (как правило, 4-6 членные) строго определенные после-
довательности нуклеотидов, в которых происходит разреза-
ние ДНК рестриктазой. В результате образуется набор фраг-
ментов ДНК определенной длины. Любое изменение сайта
узнавания (вставка, делеция или замена нуклеотида) приво-
дит к тому, что он становится "невидимым” для фермента,
молекула ДНК не разрезается в этом месте и образуется дру-
гой набор фрагментов. Второй тип ДНК-маркеров - полимор-
физм по числу тандемных повторов - является результатом
межиндивидуальных различий по числу копий коротких повто-
ряющихся последовательностей ДНК, что проявляется в раз-
личиях по длине данного участка ДНК.
Карты генетического сцепления строят на основе совмест-
ного наследования генов или маркеров. Два маркера, распо-
ложенные на одной хромосоме вблизи друг от друга имеют
тенденцию передаваться от родителей ребенку совместно. Во
время нормальных процессов развития сперматозоидов и яй-
цеклеток, цепочка ДНК случайным образом разрывается и вос-
соединяется в различных местах той же хромосомы или ее
копии (т.е. гомологичной хромосомы). Этот процесс, называ-
емый мейотической рекомбинацией, может приводить к раз-
делению двух маркеров, первоначально расположенных на
одной хромосоме. Чем ближе расположены маркеры друг к
19
Г лава 2
другу - чем ’’теснее” они сцеплены, тем менее вероятно, что
процесс рекомбинации разделит их. Таким образом, по час-
тоте рекомбинации можно оценить расстояние между двумя
маркерами.
На рисунке 3 проиллюстрирован принцип построения кар-
ты генетического сцепления. Вертикальные линии на диаграм-
ме изображают пары 4 хромосомы у каждого из членов семьи.
У отца имеется два генетических локуса, ДНК-маркер М и ген
хореи Гентингтона (ХГ), каждый из них можно обнаружить и у
любого ребенка, который их унаследует. Один из детей унас-
ледовал только маркер М, но не хорею Гентингтона, ген кото-
рой располагался на той же отцовской хромосоме. Этот факт
свидетельствует, что генетический материал отца рекомбини-
ровал в процессе сперматогенеза. При наблюдении наследо-
вания маркера М и хореи Гентингтона во многих семьях мож-
но установить частоту, с которой происходит рекомбинация
между этими локусами и установить расстояние между ними.
Маркер
М иХГ
ОТЕЦ
М --
ХГ --
МАТЬ
Маркер
М иХГ
Только Маркер
маркер М* М и ХГ
*Рекомбинант. Частота рекомбинации зависит
от расстояния между маркером М и геном ХГ
Рис. 3. Анализ генетического сцепления (по “DOE Primer on
Molecular Genetics”, с изменениями).
На генетической карте расстояния между маркерами из-
меряются в сантиморганидах (сМ), названных так в честь
американского генетика Томаса Ханта Моргана. Когда часто-
та рекомбинации между двумя маркерами равна 1%, говорят,
что они находятся на расстоянии 1 сМ. Генетическое расстоя-
20
Геном человека и методы его анатомирования
ние в 1 сМ примерно равно физической дистанции в 1 милли-
он пар оснований (1 мегабаза (Мб)).
С помощью анализа сцепления на генетической карте можно
локализовать и наследственные заболевания, гены которых или
их молекулярные основы неизвестны. Генетическое картиро-
вание помогло найти точное хромосомное расположение ге-
нов многих важных болезней. Кроме уже упоминавшейся хо-
реи Гентинпгона, это - муковисцидоз, серповидно-клеточная
анемия, болезнь Тея-Сакса, поликистоз почек, синдром лом-
кой Х-хромосомы, миотоническая дистрофия и многие другие
заболевания (см. главу 3).
Одна из ближайших промежуточных целей проекта «Геном
человека» - построить генетическую карту человека с высо-
ким разрешением (2-5 сМ) (на современных картах большин-
ства хромосом маркеры расположены в среднем через каж-
дые 7-10 сМ). Разрешение генетических карт можно увели-
чить, используя технологию рекомбинантных ДНК, фрагмен-
тацию хромосом под действием радиации и гибридизацию
клеток (слияние клеток человека с клетками других видов) для
создания панелей клеток, содержащих отдельные части хро-
мосом человека. Оценивая, какие маркеры и как часто оста-
ются совместно после радиационной фрагментации ДНК, мож-
но установить порядок их расположения и расстояние между
маркерами. Поскольку для анализа требуется лишь одна ко-
пия хромосомы, этот метод может картировать и неполимор-
фные маркеры.
Физическое картирование
Локализация генов при анализе генетического сцепления
или межвидовой гибридизации клеток представляет собой не-
прямые методы картирования, поскольку основывается на кор-
реляции между признаком и хромосомой при передаче в ряду
поколений или клеточных клонов. Молекулярная генетика, в
особенности технология рекомбинантных ДНК, дает в руки
исследователей инструмент, позволяющий работать с носи-
телями генетической информации (хромосомами, молекула-
ми ДНК, отдельными генами) непосредственно. Генетические
карты, которые строятся на основе прямого исследования ге-
нетического материала, называют физическими.
Различные методы физического картирования позволяют
получить карты с различной степенью детализации. Общая
закономерность такова, что чем более детальна карта, тем
меньший участок генома можно картировать в ходе отдельно-
го эксперимента. Карта с самым низким уровнем разрешения
- хромосомная (ее иногда называют цитогенетической), кото-
21
Глава 2
рая отражает различия в окраске бэндов при наблюдении в
световой микроскоп и позволяет охватить весь геном. Карта
кДНК показывает расположение экспрессируемых участков
(экзонов) на хромосомной карте. Более подробная карта кос-
мидных контиг отражает локализацию наборов перекрываю-
щихся фрагментов ДНК, охватывающих небольшие участки
генома. Рестрикционные карты фиксируют порядок располо-
жения на молекуле ДНК сайтов рестрикции и расстояние меж-
ду ними. Самая подробная из физических карт - последова-
тельность нуклеотидов. Однако, одномоментно можно секве-
нировать лишь очень небольшие (по сравнению со всем гено-
мом) фрагменты ДНК.
Мелкомасштабные физические карты
К мелкомасштабным картам (физическим картам с низким
уровнем разрешения) относятся хромосомные карты и карты
кДНК.
При определении положения гена на хромосоме первона-
чально стоит задача приписать его к определенному хромо-
сомному сегменту - бэнду или группе бэндов, который можно
идентифицировать цитогенетически. Сейчас для этого исполь-
зуют метод гибридизации in situ. Клонированную копию нуж-
ного гена помечают радиоактивной (например, тритиевой или
фосфорной) или флуоресцентной меткой. Меченый таким об-
разом фрагмент ДНК называют зондом. Затем зонд инкубиру-
ют с метафазным препаратом хромосом и, после того как он
свяжется с комплиментарной цепочкой ДНК на интактной хро-
мосоме, устанавливают его точную локализацию. С помощью
гибридизации in situ было картировано множество генов чело-
века, в том числе гены альбумина, коллагена, альфа-глобина,
гормона роста.
До недавнего времени даже самые подробные из хромо-
сомных карт позволяли локализовать фрагмент ДНК с точ-
ностью до региона размером примерно 10 Мб (средний раз-
мер хромосомного бэнда). Усовершенствования метода флу-
оресцентной гибридизации in situ (FISH) позволили увеличить
разрешение хромосомных карт до 2-5 Мб. Модификации ме-
тодов гибридизации, использующие хромосомы на стадии
интерфазы, когда они менее компактны, дают возможность
локализовать зонд до участка размером в 100000 п.о. (0,1 Мб).
Карта кДНК отражает расположение на хромосоме кодиру-
ющих последовательностей. ДНК-копию (кДНК) синтезируют
в лаборатории, используя в качестве матрицы молекулы мРНК,
вносят метку и локализуют кДНК-зонды методом гибридиза-
ции in situ. Представляя экспрессируемые участки, кДНК поз-
22
Геном человека и методы его анатомирования
воляет выявить наиболее значимые с биологической и меди-
цинской точек зрения районы генома. Карта кДНК может по-
мочь в хромосомной локализации генов, функция которых еще
неизвестна. ДНК-копию можно получить и в том случае, если
белок синтезируется в очень небольших количествах и выде-
лить специфическую мРНК из общей смеси мРНК клетки не
удается. Зная последовательность аминокислот в белке, мож-
но «воссоздать» последовательность гена искусственно или
выделить его из клонированной геномной ДНК при гибриди-
зации со смесью олигонуклеотидов, соответствующих всем
возможным кодирующим последовательностям гена (вслед-
ствие вырожденности генетического кода их может быть не-
сколько).
Крупномасштабные физические карты
Существует два общих подхода к построению физической
карты участка генома с высоким уровнем разрешения: карти-
рование ’’сверху вниз” (макрорестрикционная карта) и ’’снизу
вверх” (карта контиг). В обоих случаях карта представляет со-
бой упорядоченный набор фрагментов ДНК. Фрагменты раз-
множают клонированием или с помощью полимеразной цеп-
ной реакции, разделяют гель-электрофорезом и выявляют при
окрашивании или гибридизации с меченым зондом. Для вос-
создания оригинальной последовательности фрагментов (той
последовательности, в которой они находятся в геноме) ис-
пользуют различные методы поиска перекрывающихся участ-
ков.
Методы физического картирования можно применять к изо-
лированным хромосомам человека. Отдельные изолированные
хромосомы получают путем сортировки хромосом в потоке
(метод проточной цитометрии) по их размеру. Существуют
также гибридные клеточные линии, содержащие отдельные
хромосомы человека. Для получения таких линий клетки чело-
века гибридизуют с опухолевыми клетками млекопитающих (как
правило, грызунов). В процессе деления гибридные клетки
теряют предпочтительно хромосомы человека. Когда в гиб-
ридной клетке останется лишь одна человеческая хромосома,
ее размножают и поддерживают как клеточную линию.
Необходимой ступенью при построении крупномасштабных
физических карт является получение большого количества
фрагментов ДНК картируемого участка. Существует два ос-
новных способа амплификации (’’размножения”) ДНК - клони-
рование и амплификация в полимеразной цепной реакции.
Клонирование (амплификация ДНК in vivo). Клонирова-
ние представляет собой размножение фрагментов в составе
23
Г лава 2
рекомбинантных молекул ДНК в клетках чужеродного хозяина.
Фрагменты ДНК человека (полученные чаще всего при рес-
трикции геномной ДНК) объединяют с кольцевой молекулой
(вектором), разрезанной той же рестриктазой. Полученную
рекомбинантную (т.е. содержащую ДНК разного происхожде-
ния) молекулу затем вводят в подходящие клетки, которые
можно размножать в практически неограниченных количест-
вах. Векторами служат обычно молекулы ДНК вирусного, бак-
териального или дрожжевого происхождения. Они могут нес-
ти вставки чужеродной ДНК от 12000 п.о. (бактериальные плаз-
миды и космиды) до 1 Мб (искусственные хромосомы дрож-
жей). В качестве клеток-хозяев используют обычно бактерии,
хотя применяются и клетки млекопитающих. В процессе деле-
ния клеток-хозяев (роста клона) ДНК человека в составе век-
тора реплицируется вместе с геномом клетки.
Набор клонированных фрагментов ДНК называют библио-
текой клонов. Геномная библиотека представляет собой на-
бор клонов, содержащих перекрывающиеся участки ДНК, ох-
ватывающий весь геном. Существуют и хромосомные библио-
теки - набор клонов, содержащий фрагменты ДНК определен-
ной хромосомы.
Полимеразная цепная реакция (амплификация ДНК in
vitro). Полимеразная цепная реакция (ПЦР) представляет со-
бой ферментативный синтез фрагментов ДНК. В пробирке с
помощью ДНК-полимеразы синтезируют цепочку ДНК, компли-
ментарную исходной молекуле-матрице. Реакционная смесь
содержит, кроме фермента и матрицы 4 дезоксинуклеотид-
трифосфата, и 2 короткие цепочки ДНК (праймеры), компли-
ментарные концам размножаемого участка. Реакционную смесь
вначале нагревают, чтобы разделить двухцепочечную матри-
цу, затем охлаждают, чтобы праймеры связались с компли-
ментарной им последовательностью, и полимераза построи-
ла вслед за праймерами всю цепочку. Циклическое повторе-
ние нагревания и охлаждения приводит к экспоненциальному
росту числа копий матрицы, поскольку каждая новая двухце-
почечная копия вновь разделяется и сама служит матрицей
для следующего цикла синтеза. За полтора часа можно про-
вести 30 таких циклов, что приведет к увеличению числа коп-
ий нужного фрагмента в миллионы раз.
Большая скорость амплификации, точность и простота (что
дает возможность полностью автоматизировать реакцию) вы-
годно отличают ее от технологии рекомбинантных ДНК. Одна-
ко ПЦР имеет и некоторые недостатки. Основные из них - не-
обходимость иметь предварительную информацию об ампли-
фицируемом участке (нужно знать последовательность прай-
24
Геном человека и методы его анатомирования
меров) и невозможность амплифицировать большие фрагменты
(больше нескольких тысяч п.о.).
Макрорестрикционная карта. При картировании "сверху
вниз" отдельную хромосому разрезают редкощепящей рес-
триктазой на относительно большие части, которые упорядо-
чивают и снова делят на более мелкие фрагменты, которые
затем тоже упорядочивают. В результате получают карту сай-
тов рестрикции крупнощепящих ферментов - порядок их рас-
положения и расстояние между ними (рис. 4). Этот метод поз-
воляет получить протяженные, полные (не содержащие про-
белов) карты достаточно больших регионов хромосомы. Од-
нако разрешение такой карты меньше, чем карты контиг и не
позволяет искать конкретные гены. В настоящее время этот
метод позволяет локализовать фрагменты ДНК до региона раз-
мером от 100000 п.о. до 1 Мб. Значительный прогресс в рес-
трикционном картировании был достигнут благодаря раз-
работке методов гель-электрофореза в пульсирующем поле
(PFGE). Тогда как обычным гель-электрофорезом можно раз-
делять фрагменты ДНК размером менее 40 Кб, электрофорез
в пульсирующем поле позволяет различать молекулы дости-
гающие 10 Мб, что делает возможным картирование протя-
женных регионов генома.
Карта контиг. При картировании ’’снизу вверх" хромосому
разделяют на множество небольших фрагментов, которые за-
тем клонируют и упорядочивают (рис. 4).
(а)
Хромосома
Сверху
вниз
Полная, но не детальная
Рис. 4. Методы физического картирования: картирование
“сверху вниз” (макрорестрикционная карта) и картирование
“снизу вверх” (карта контиг) (по Billings, 1991).
(6)
Карта контиг
Библиотека клонов
Набор упорядоченных фрагментов, покрывающий опреде-
ленный участок хромосомы называют контигой. Локализация
контиги на хромосоме может быть верифицирована с помощью
25
Глава 2
FISH. Карта контиг, таким образом, представляет собой биб-
лиотеку перекрывающихся клонов, покрывающих сегмент хро-
мосомы. Поскольку большие регионы не поддаются клониро-
ванию, карту контиг трудно расширить до крупных участков
хромосомы. Для заполнения пробелов можно использовать
технологию ДНК-зондов, хотя это требует значительных за-
трат времени.
В последнее время совершенствование методов картиро-
вания ’’снизу вверх” связано с разработкой средств для кло-
нирования больших - до 1 Мб - участков ДНК. Эти векторы
поддерживают в клетках дрожжей как искусственные хромо-
сомы (YAC - искусственная дрожжевая хромосома). До внед-
рения YAC самые крупные векторы (космиды) могли нести
вставки максимум в 20-40 Кб. Применение YAC позволяет зна-
чительно уменьшить число клонов, которые нужно упорядочи-
вать. Подробную карту YACa можно получить при субклониро-
вании - процессе, в котором фрагменты первоначальной боль-
шой вставки клонируются в более мелких векторах. Парал-
лельно идет и разработка вместительных бактериальных век-
торов.
Заполнение пробелов на карте и поиск генов
Описанные выше методы позволяют создавать подробные
карты лишь для относительно небольших участков хромосо-
мы, оставляя на карте многочисленные пробелы. Один из под-
ходов к заполнению пробелов - микрорассечение отдельных
хромосом. Из хромосомы вырезают небольшой район, кото-
рый затем разбивают на мелкие части и клонируют или ам-
плифицируют при помощи ПЦР. Полученные таким образом
фрагменты можно затем картировать обычными способами.
Еще одна стратегия заполнения пробелов - прогулка по
хромосоме. Прогулку начинают с участка уже известной пос-
ледовательности, по которой синтезируют праймер. Праймер
гибридизуют с клоном из неупорядоченной геномной библио-
теки и синтезируют в продолжение праймера короткую цепь,
комплиментарную вставке данного клона. Эту цепь затем сек-
венируют и используют в качестве праймера для следующего
шага по хромосоме. Таким образом, хромосома или ее учас-
ток систематически секвенируется. Недостаток этого метода
- слишком большое количество шагов, которые нужно сделать
чтобы пройти относительно протяженный район хромосомы.
Прогулка по хромосоме применяется и для точной локали-
зации генов (рис. 5). Когда методами анализа генетического
сцепления найден маркер, расположенный в пределах 1 сМ от
гена, это расстояние можно пройти с помощью прогулки по
26
Геном человека и методы его анатомирования
хромосоме. В начале с помощью зонда на сцепленный маркер
из геномной библиотеки выделяют клон, несущий фрагмент
хромосомы, в котором располагается маркер. Этот участок
разрезают рестриктазами и 3’-концевой рестрикционный фраг-
мент используют в качестве зонда для поиска следующего
клона, 5’-конец которого перекрывается с З’-концом вставки
первого. Этот процесс повторяют несколько раз, прогулива-
ясь по хромосоме до следующего маркера с 3’-конца от гена.
ФЛАНКИРУЮЩИЙ
СЦЕПЛЕННЫЙ
МАРКЕР
ФЛАНКИРУЮЩИЙ
СЦЕПЛЕННЫЙ
МАРКЕР
ГЕН БОЛЕЗНИ
находят фрагмент
в геномной
библиотеке
С помощью
юнда на 5'-
маркср
ФРАГМЕНТ X
ГЕНОМНОЙ ДНК L
С помощью юндов на 3’ коней
клонированных фрагментов
последовательно находят
перекрывающиеся фрагменты
I
Прогулку по хромосоме продолжают до тех пор,
пока нс найден фрагмент, содержащий 3'
фланкирующим маркер
Рис. 5. Клонирование гена с помощью “прогулки по хромосоме"
(по Micklos и Freyer, 1990, с изменениями).
Современная генетическая карта человека содержит при-
мерно 10000 маркеров, что означает, что маркеры находятся,
в среднем, на расстоянии 300000 п.о. друг от друга и что меж-
ду ними располагается около десятка генов. Для регионов ге-
нома, представляющих особый интерес, созданы, конечно,
более подробные карты, но тем не менее, большая часть ге-
нов человека еще не локализована.
27
Глава 2
На рисунке 6 схематически представлены типы геномных
карт. На самом низком уровне разрешения находится генети-
ческая карта, которая основывается на частоте рекомбинации
между сцепленными генами или полиморфными маркерами.
Хромосомное картирование, включая гибридизацию in situ,
позволяет соотнести генетические сайты с хромосомными
бэндами. Следующий уровень разрешения - крупные рестрик-
ционные фрагменты размером 1-2 Мб. Упорядоченные биб-
лиотеки космид или УАСов позволяют картировать вставки
размером 40-400 Кб. Последней ступенью физической карты
является последовательность оснований.
Ген или
полиморфизм
ГЕНЕТИЧЕСКАЯ |
КАРТА -П------------
Ген или
полиморфизм
ФРАГМЕНТЫ
рестрикции!""]! !Г~П1 If
УПОРЯДОЧЕННАЯ________________ _____ ____________________________________
БИБЛИОТЕКА
llllllllllllllllllUlllllllllllilllllllllllllllllllllllllllllllllllllllllllHIIIIII
ТЕЛЬНОСТЬ
ОСНОВАНИЙ
Рис. 6. Типы карт генома (по Billings, 1991).
Секвенирование
Секвенированием называют процесс выявления последо-
вательности нуклеотидов ДНК. Современные процедуры сек-
венирования включают, во-первых, субклонирование фрагмен-
тов ДНК из космид или библиотеки бактериофагов в специ-
альные векторы для секвенирования, которые несут неболь-
шие части клонированных фрагментов (рис. 7). Следующим
шагом является преобразование субклонированных фрагмен-
тов в набор молекул ДНК, различающихся по длине только на
28
Геном человека и методы его анатомирования
один нуклеотид. Высокоразрешающие методы электрофоре-
за позволяют разделить этот набор фрагментов и считать пос-
ледовательность оснований.
ХРОМОСОМА
ЧЕЛОВЕКА
В коеммде можно
клонирошгп, ф|м1гмснт
hj примерно 40000 п.о.
В YAC можно клониропать
фрагмент, содержащим
примерно 400000 п.о
ИСКУССТВЕННАЯ ДРОЖЖЕВАЯ
(ГСП Ц-'лабииа чежшека)
(iCiCACTGACTCTCTCTGC CTATTGGTCTATHTC ССAC СС TTAGGCTGCTGGTGGTCTrACXT
TGGACCCAGAGGTTCTTTGAGTCCTTTGGGGATCTGTCCACTCCTGATGCTGTTATGG
Рис. 7. Субклонирование фрагментов ДНК для секвенирования.
Путем последовательного клонирования фрагмента ДНК
из крупных векторов в более мелкие, получают фрагмент
относительно небольшого размера, последовательность
оснований в котором можно определить химическими
методами (по Micklos и Freyer, 1990).
29
Глава 2
I. Разделение фрагментов
в полиакпиламилном геле
для каждой реакции
2. Чтение последовательности
снизу вверх
Рис. 8. Секвенирование ДНК по
методу Сэнгера (по Micklos
и Freyer, 1990).
Каждая реакционная
пробирка (для Т, С, G,
А) при секвенировании
дидеокси-методом
(рис. 8) содержит:
- ДНК-матрицу, прай-
мер и ДНК-полимеразу
для инициации синтеза
новой цепочки в точке,
где праймер гибридизу-
ется с матрицей;
- 4 дезоксинуклеотид-
трифосфата (dATP,
dTTP, dCTP, dGTP), из
которых синтезируется
цепь ДНК;
- один радиоактивно
меченый дезоксинук-
леотидтрифосфат;
- один ди-дезокси-
нуклеотидтрифосфат,
при включении которо-
го в цепочку ее рост за-
вершается (в пробирке
А это di-dATP, в пробир-
ке С - di-dCTP и т.д.).
В каждой из пробирок
соотношение dNTP и di-
dNTP подобрано таким
образом, чтобы синте-
зировался набор фраг-
ментов ДНК с di-dNTP,
включенным в каждую
позицию данного осно-
вания на матричной
ДНК. Набор фрагментов
для каждого из нуклео-
тидов затем разделяют
в одном геле на четы-
рех параллельных до-
рожках. Фрагменты раз-
деляются по длине: чем
короче фрагмент (и,
следовательно, чем
ближе данное основа-
ние к началу секвениру-
30
Геном человека и методы его анатомирования
емого участка), тем быстрее он движется и тем ближе к концу
геля он оказывается. Последовательность читают снизу вверх.
Хотя усовершенствования методов Максама-Гилберта и
Сэнгера позволяют анализировать одновременно до 40 кло-
нов на одном секвенирующем геле, эти технологии секвенио-
вания (технологии первого поколения) не позволяют секвени-
ровать большие участки генома - затраты времени и средств
делают крупномасштабные проекты секвенирования с исполь-
зованием этих методов неприемлемыми. Даже самое лучшее
оборудование позволяет рабочей группе секвенировать толь-
ко 50 - 100 тысяч пар оснований в год при затратах 1 -2 долла-
ра за п.о. При таких темпах на секвенирование всего генома
человека (3 миллиарда п.о.) пришлось бы затратить 3000 лет
и 3 миллиарда долларов.
Поэтому, одной из основных проблем, которые нужно ре-
шить для построения подробной карты генома человека, яв-
ляется разработка автоматизированных, быстрых и экономич-
ных методов секвенирования. К технологиям секвенирования
второго поколения относятся высоковольтный капиллярный
электрофорез, позволяющий значительно улучшить разделе-
ние фрагментов, и ионизационная резонансная спектроско-
пия - метод, улучшающий выявление стабильных изотопных
меток. Эти технологии могут при понижении удельных затрат
ускорить секвенирование на порядок.
Однако, даже этого недостаточно для успешного секвени-
рования всего генома. Последовательность оснований боль-
шей его части будет, по-видимому, выявлена с помощью без-
гелевых технологий секвенирования третьего поколения, ко-
торые должны увеличить эффективность секвенирования на
несколько порядков. Эти развивающиеся в настоящее время
методы включают флуоресцентное определение индивидуаль-
ных меченых оснований при проточной цитометрии; прямое
чтение последовательности оснований с цепочки ДНК с по-
мощью сканирующей туннельной или атомной микроскопии;
масс-спектрометрию последовательностей ДНК; секвениро-
вание при гибридизации с панелями коротких олигонуклеоти-
'дов известной последовательности.
Размер генома человека и структура гена
Размеры генома
Размеры генома человека можно выразить в терминах ге-
нетической карты - в сантиморганидах и в терминах физичес-
кой карты - в парах оснований. Важной количественной харак-
теристикой является и общее число структурных генов.
31
Глава 2
Приблизительные оценки показывают, что гаплоидный ге-
ном человека содержит примерно 3.3 миллиарда п.о. Размер
генома в сантиморганидах, оцененный по цитогенетическим
данным (по числу хиазм), равен примерно 3000 сМ. Почти все
хромосомы человека превышают по размеру 100 сМ. Самая
маленькая из них - Y-хромосома - насчитывает 50 сМ, а самая
крупная - первая хромосома - 250 сМ. Генетические локусы,
расположенные на разных концах хромосомы, хотя и находят-
ся на одной нити ДНК (синтенны), могут вести себя как не-
сцепленные, демонстрируя независимое наследование при
генетическом анализе.
Размер генома человека можно оценить и в метрических
мерах длины. Расстояние между двумя соседними основания-
ми в цепочке ДНК составляет 3.4 ангстрема, или 3.4x10 8 см.
Умножив 3.3 миллиарда на эту цифру, получим длину всей ДНК
человека - около 110 см, то есть более чем метр!
По непрямым оценкам, человек имеет от 50 до 100 тысяч
структурных генов, т.е. генов, кодирующих аминокислотную
последовательность белков и нуклеотидную последователь-
ность различных видов РНК. По-видимому, наиболее вероят-
ная оценка общего числа генов находится в пределах 70 - 80
тысяч (Fields et al., 1994; Antonarakis, 1996). Некоторые пря-
мые наблюдения на подробно картированных участках генома
подтверждают эти оценки. Так, например, участок короткого
плеча хромосмы 11 размером 65 Кб содержит 5 структурных
генов бета-глобина-эпсилон, гамма-G, гамма-А, дельта и бета.
Если такая "плотность генов" характерна и для остальных учас-
тков генома, то предварительные оценки общего числа генов
человека верны.
Приведенные выше оценки позволяют сопоставить генети-
ческие и физические единицы размерности генома. Расстоя-
ние в 1 сМ на генетической карте эквивалентно физической
дистанции, в среднем, около 1 миллиона пар оснований (1
Мб). Оговорка "в среднем" здесь совсем не случайна, пос-
кольку рекомбинация на различных участках хромосомы про-
исходит с неодинаковой частотой. Во-первых, частота реком-
бинации ниже в прицентромерных районах хромосомы и по-
вышается при удалении от нее, во-вторых, существуют "горя-
чие точки" рекомбинации - небольшие районы хромосомы, для
которых характерна очень высокая частота разрывов. В гено-
ме человека найдено множество таких "горячих точек", напри-
мер, в районе главного комплекса гистосовместимости (хро-
мосома 6р), кластера генов бета-глобина (11р), в концевом
участке длинного плеча Х-хромосомы.
Современные методы дифференциального окрашивания
хромосом позволяют выявить на цитогенетической карте ге-
32
Геном человека и методы его анатомирования
нома около 600 бэндов. Это значит, что один бэнд имеет, в
среднем, размер, равный приблизительно 5 сМ, и содержит
от 100 до 200 генов.
К моменту написания этих строк на генетической карте че-
ловека известна точная локализация лишь 5-7% всех его ге-
нов. В геномной базе данных (Genome Data Base, GDB) - компь-
ютерном “хранилище”, которое объединяет всю информацию,
касающуюся исследований генома человека, на 26 июня 1997
года хранится информация о 6916 охарактеризованных генах,
5397 из них картированы. Кроме структурных генов GDB со-
держит информацию и о других картированных “ДНК-объек-
тах”. Всего в геномной базе данных их на сегодня 103296, в
том числе 18168 полиморфных ДНК-маркеров.
До выявления всех человеческих генов предстоит пройти
еще долгий путь. Важным подспорьем на этом пути могут слу-
жить так называемые метки экспрессируемых последователь-
ностей (expressed sequence tags, EST) - частично секвениро-
ванные участки кДНК, положение которых на карте известно.
Одним из наиболее впечатляющих достижений геномных ис-
следований является картирование более чем полмиллиона
EST (GDB сейчас содержит информацию о 553443 EST). При-
надлежность EST конкретным генам, как правило, неизвестна.
Однако, при наличии большой “библиотеки” EST картирова-
ние гена может свестись к поиску соответсвующих фрагмен-
тов в базе данных.
Митохондриальный геном
Утверждение, что весь генетический материал человека
находится в составе хромосом не совсем верно, поскольку
есть одно исключение - митохондриальный геном. Митохон-
дриальная ДНК (мтДНК) или, как ее еще иногда называют, хро-
мосома М, представляет собой небольшую кольцевую моле-
кулу длиной 16569 п.о. В отличие от ДНК ядерного генома она
не связана с белками, а существует в митохондриях в "чис-
том" виде. Еще одно важное отличие мтДНК от хромосомной -
очень высокая «плотность генов». В митохондриальных генах
отсутствуют интроны, а межгенные промежутки очень невели-
ки. В результате эта очень небольшая молекула содержит 13
генов, кодирующих белки (3 субъединицы цитохром с-оксида-
зы, 6 компонентов АТФ-азы и др.) и 22 гена транспортных РНК.
Каждый из ядерных генов присутствует в большинстве кле-
ток в двух копиях - по одной на каждой хромосоме, митохон-
дриальные же гены имеют гораздо большую копийность* - не-
сколько тысяч на клетку. Каждая митохондрия имеет от 2 до
10 копий мтДНК. Также как и для ядерной ДНК, для генома
митохондрий характерны межиндивидуальные различия. В то
33
3 Заказ №127
Глава 2
же время, мтДНК одного индивида, как правило, идентична.
На мтДНК в миниатюре достигнута та цель, которую пре-
следуют исследователи генома человека, - митохондриальная
ДНК полностью секвенирована и на ней выявлены все струк-
турные гены.
Структура гена
Функциональной единицей генома человека является от-
дельный ген. Средний по размеру ген человека имеет кодиру-
ющую часть общей длиной в несколько тысяч пар оснований.
Однако, общая длина гена значительно больше, поскольку кро-
ме экзонов (кодирующей части) в состав гена входят интроны
и участки, расположенные до (с 5’-конца) и после (с З'-конца)
кодирующей части.
Кодирующая часть большинства генов находится в пределах
1-3 тысяч п.о., что соответствует белковому продукту из 300-
1000 аминокислотных остатков. Существуют и очень малень-
кие белки (например, инсулин) и очень крупные (например,
аполипопротеин В состоит из более чем 4500 аминокислот, а
фактор свертывания VIII - из 3000). 5’-конец гена соответству-
ет N-концу полипептидной цепи, а З’-конец - карбоксильному.
У большинства генов кодирующая часть поделена на несколь-
ко экзонов, между которыми расположены некодирующие учас-
тки (интроны). Одним из немногих исключений из этого пра-
вила являются гены альфа-интерферона - в них интроны от-
сутствуют. Наиболее рациональной гипотезой, объясняющей
наличие интронов, является гипотеза Джилберта, который
предположил, что экзоны соответствуют доменам белка, и в
процессе эволюции гены белков с различной функцией соби-
рались из соответствующего "набора” экзонов. Вполне воз-
можно также, что функция интронов в том, чтобы обеспечить
место для кроссинговера без разрыва кодирующих фрагмен-
тов и, следовательно, без нарушения функции домена.
Интроны вырезаются из первичного транскрипта гена в про-
цессе формирования зрелой мРНК. Механизм этого процесса
(сплайсинга) до конца еще не ясен, однако известно, что на
3’- и 5'-концах интронов, т.е. в сайтах сплайсинга, имеются
короткие консервативные последовательности, которые, по-
видимому, и распознаются клеточными компонентами, осущес-
твляющими сплайсинг.
С учетом интронов общая длина генов возрастает подчас в
десятки раз. Например, ген альбумина имеет длину 25 Кб, из
которых 2.1 Кб заняты экзонами, а остальное приходится на
интроны. Есть и гены с очень небольшими интронами (напри-
мер, ген альфа-глобина, в котором длина интронов равна 300
п.о., а размер экзонов - 500 п.о.) и гигантские по протяжен-
34
Геном человека и методы его анатомирования
ности гены: ген фактора VIII имеет общую длину 186 Кб и со-
стоит из 26 экзонов длиной 9 Кб и 25 интронов общим разме-
ром 177 Кб, а ген дистрофина - самый большой из известных
генов человека - занимает 2500 Кб на хромосоме X, большая
часть которых, конечно, приходится на интроны.
Кодирующая часть гена начинается всегда с метиониново-
го кодона (из зрелого белка этот метионин удаляется). До на-
чала кодирующей части расположены участки регуляции тран-
скрипции. В районе 25-30 нуклеотидов "выше" (с 5'-конца)
сайта инициации транскрипции в генах большинства эукари-
от, в том числе и человека, расположен участок, обогащенный
А и Т, ТАТА-бокс. Еще одна регуляторная последовательность
- САТ-бокс - расположена в районе 70-80 нуклеотидов от сай-
та инициации транскрипции.
На З’-конце гена расположена последовательность ААТААА,
которая служит сигналом для присоединения полиА-хвоста к
мРНК, что по-видимому необходимо для транспорта мРНК че-
рез ядерную мембрану в цитоплазму клетки, где происходит
синтез белка.
Межгенные участки ДНК называются спейсерами и их фун-
кция, если она вообще имеется, неизвестна. Частично спей-
серы состоят из повторяющихся последовательностей различ-
ных типов. У человека, как и у других млекопитающих, самым
распространенным семейством таких последовательностей
являются Alu-повторы, названные так из-за того, что они со-
держат сайт рестрикции для нуклеазы Alul. В геноме человека
насчитывается несколько сотен тысяч копий Alu-повторов в
спейсерах и интронах.
Вариабельность генома человека
Последовательность ДНК в геноме человека не есть посто-
янная, раз и навсегда установленная величина. По современ-
ным оценкам, две случайно выбранные хромосомы отличают-
ся, в среднем, по 1 нуклеотиду из каждых 500. Иначе говоря,
последовательность ДНК двух неродственных людей отлича-
ется примерно на 0,2%. Именно эти, кажущиеся ничтожными,
различия и определяют всю гамму характеристик, по которым
один человек отличается от другого.
Вариации последовательности ДНК в геноме довольно ус-
ловно принято делить на мутации и полиморфизм. Под мута-
цией понимают изменение, которое возникает спонтанно или
редко и которое, как правило, сопровождается изменением
фенотипа. Полиморфизм, согласно классическому определе-
нию, - это наличие нескольких наследственных вариантов, на-
иболее редкий из которых встречается с частотой, превыша-
ющей частоту обратного мутирования. На практике, признак
35
Глава 2
или маркер считают полиморфным, если наиболее редкий ва-
риант встречается чаще, чем в 1% наблюдений.
Можно выделить несколько типов “нормальных” вариаций
последовательности ДНК:
- точечные мутации - замены отдельных нуклеотидов. Они
наиболее часты в интронах и фланкирующих районах генов, а
в кодирующей части встречаются реже. Если точечная мута-
ция приходится на сайт узнавания рестриктазой, то ее можно
выявить по различию в длине рестрикционных фрагментов
(ПДРФ). Эти вариации генома, как правило, диаллельны, т.е.
встречается 2 варианта.
- инсерции ^вставка) или делеции небольших фрагментов
ДНК.
- вариабельность по числу тандемных повторов (VNTR).
Полиморфизм относят к этому типу, если размер повторяю-
щегося элемента находится в пределах от 10 до 100 нуклеоти-
дов. Как правило, имеется множество аллелей, различающих-
ся по числу копий повтора.
- вариабельность по числу коротких тандемных повторов
(STR). Повторяющийся фрагмент состоит из 2, 3, 4, 5 или 6
нуклеотидов. Это наиболее высокополиморфный тип вариа-
ций.
- полиморфизм по наличию или отсутствию мобильных
генетических элементов (таких как Alu-повторы, ретротранспо-
зоны семейства LINES, псевдогены).
Мутации, имеющие фенотипические (в том числе и клини-
ческие) проявления, можно классифицировать в зависимости
от их эффекта на структуру белкового продукта гена и/или на
уровень экспрессии гена. По данным Маккьюсика (McKusick,
1992), мутации, изменяющие последовательность аминокис-
лот в белке, ответственны за 50-60% случаев монотонных бо-
лезней. Остальные 40-50% приходятся на долю мутаций, за-
трагивающих экспрессию гена. Изменение аминокислотного
состава белка проявляется на “качественном” уровне в выра-
женных клинических фенотипах, таких как метгемоглобинемия
или серповидно-клеточная анемия при мутациях гена бета-гло-
бина. Мутации, нарушающие нормальную экспрессию гена
приводят к изменению количества генного продукта и прояв-
ляются фенотипами, связанными с недостаточностью того или
иного белка.
Ген - это сложная структура, которая определяет не только
структуру белка, но и весь путь от ДНК через транскрипцию-
матричной РНК к трансляции белка. Мутации в различных струк-
турных частях гена могут проявляться на стадии транскрипции
или трансляции. Маккьюсик (McKusick, 1992) выделяет следу-
ющие типы мутаций:
36
Геном человека и методы его анатомирования
Мутации транскрипции:
- мутации в области промотора;
- мутации сплайсинга РНК, в том числе мутации в 5’-донор-
ном сайте интрона, мутации в З'-акцепторном сайте ин-
трона и мутации, приводящие к возникновению новых сай-
тов сплайсинга;
- мутации расщепления мРНК (мутации полиаденилирова-
ния);
- мутации кэп-сайта;
- делеции активирующих районов или энхансеров.
Мутации трансляции:
- мутации в инициирующем кодоне (ATG) или вблизи него;
- мутации сдвига рамки считывания (результат делеций или
инсерций);
- миссенс-мутации - замены одного аминокислотного ос-
татка в молекуле белка на другое. Миссенс-мутации тоже
могут приводить к нарушению экспрессии гена, поскольку
новая структура РНК или белка может оказаться нестабиль-
ной;
- нонсенс-мутации (“стоп”-мутации) - мутирование кодона
в терминирующий кодон, что приводит к образованию уко-
роченного белка;
- мутации в терминирующих кодонах (ТАА, TAG или TGA),
которые приводят к синтезу длинных (длиннее нормально-
го) белковых продуктов.
Существуют и другие типы изменений генетического мате-
риала, приводящие к фенотипическим изменениям. Например,
при транслокациях или других хромосомных перестройках
структурная часть гена и его премоторная области могут ока-
заться разделенными, что приведет к нарушению генной экс-
прессии (пример - различные типы лейкемии). Недавно отк-
рыт новый необычный тип мутаций - экспансия тринуклеотид-
ных повторов (значительное увеличение числа повторов у боль-
ных по сравнению нормой), ответственный за синдром фра-
гильной Х-хромосомы, миотоническую дистрофию и ряд дру-
гих болезней (см. главу 5).
Мутация будет передаваться по наследству только в том
случае, если она произошла в половых (герминальных) клет-
ках. Мутации же в соматических клетках (соматические мута-
ции) являются причиной спорадических (несемейных) случаев
различных форм рака. Некоторые из таких мутаций возникают
также и в половых клетках и передаются в поколениях как пред-
расположенность к раку (например, ретинобластома, синдром
Ли-Фромени).
В некоторых случаях мутации, летальные в половых клет-
ках, могут существовать в соматическом виде в части клеток
организма (мозаицизм), приводя к менее тяжелым аномалиям
(пример - наследственная остеодистрофия Олбрайта).
37
Г лава 3
БОЛЕЗНИ ЧЕЛОВЕКА НА КАРТАХ ХРОМОСОМ
Особенность современных геномных исследований, охва-
тывающих большое разнообразие изучаемых объектов (наибо-
лее излюбленные - бактерии, дрожжи, круглый червь, дрозо-
фила, африканская шпорцевая лягушка, мышь), состоит в кон-
центрации внимания и основных усилий на изучении человека
как главной цели познания. Важным текущим моментом этих
исследований является картирование фенотипов болезней и
генов. Как и предполагалось 20 лет назад, “клиническая гене-
тика обеспечивает связь, жизненное направление, фокус и цель
генетики человека и вообще всей генетики” (Маккьюсик, 1981}.
Патологическая анатомия генома человека - это реализующа-
яся возможность картирования генов, участвующих в возни-
кновении и развитии болезней человека, на хромосомах.
Генетическое картирование: от функции
к позиции на хромосомах и обратно
Начало революционных изменений в генетике человека
можно датировать первой половиной 80-х годов, когда было
обнаружено, что естественный полиморфизм на уровне пос-
ледовательности ДНК можно использовать в качестве генети-
ческого маркера (Botstein etaL, 1980}. Это открытие дало мощ-
ный стимул картированию генетических локусов, ответствен-
ных за простые менделирующие признаки человека, в том числе
наследственные заболевания. С тех пор и до настоящего вре-
мени было картировано несколько сотен заболеваний и син-
дромов, наследуемых в соответствии с монотонным менде-
левским типом наследования (Collins, 1995}.
Вплоть до последнего времени в картировании доминиро-
вал функциональный подход (рис. 9), основанный на априор-
ном наличии некоторой фундаментальной информации о био-
химическом дефекте, лежащем в основе того или иного на-
следственного заболевания. Однако, список заболеваний, для
которых эта информация была достаточно полной (например,
таких как фенилкетонурия или серповидно-клеточная анемия)
к настоящему времени практически исчерпан и большинство
генов, функция которых была известна, уже клонированы и
картированы. Близко к функциональному и кандидатное кар-
тирование. В этом случае информация о функции недостаточ-
38
Болезни человека на картах хромосом
но полна, чтобы точно указать ген, однако достаточна для того
чтобы выдвинуть более или менее обоснованные предположе-
ния о возможных кандидатах. Важно подчеркнуть, что в обоих
случаях клонирование гена, как правило, предшествует его
точной локализации в геноме, т.е. картированию.
1980 1985 1990 1995 2000 2005 2010
Рис. 9. Тенденции в картировании генов человека (по Collins,
1995).
В рамках этих подходов картировать ген означало пройти
путь от его функции к локализации на хромосоме (позиции).
Появление в конце 80-х годов множества высокополиморф-
ных ДНК-маркеров дало возможность пойти и в обратном на-
правлении - от хромосомной карты к функции. Стратегия “об-
ратной генетики”, применительно к поиску генов, получила
воплощение в позиционном клонировании, которое подразу-
мевает локализацию гена при отсутствии всякой функциональ-
ной информации о нем. При этом его место на карте устанав-
ливают по результатам анализа сцепления болезни с уже кар-
тированными генетическими маркерами. Первым успехом “чис-
того" позиционного картирования стала локализация гена хро-
нического гранулематоза в 1986 году (Royer-Pokora etal., 1986).
Вслед за этим последовали и новые достижения - картирова-
ние миодистрофии Дюшенна (Monaco et al., 1986), ретиноб-
ластомы (Friend et al., 1986), муковисцидоза (Riordan et al.,
1989), нейрофиброматоза типа 1 (Goldar et al., 1989), синдро-
39
Глава 3
--------------------------------1----------------------
ма фрагильной Х-хромосомы (Poustka etal., 1991} и т.д. Всего
же к настоящему времени с помощью позиционного картиро-
вания найдены гены 84 болезней человека (Bassett at al, 1997}.
Некоторые из них приведены в таблице 5.
Таблица 5.
Гены некоторых наследственных заболеваний,
найденные позиционным клонированием
Болезнь Хромосомные перестройки Тринуклеотид- ные повторы
1 2 3
Хронический грануломатоз
Мышечная дистрофия Дюшенна 4"-
Ретинобластома 4"
Муковисцидоз -
Опухоль Вильмса 4"-
Нейрофиброматоз, тип 1 4-
Хориодеремия 4"-
Синдром фрагильной Х-хр. 4" 4“
Семейный полипоз толстой кишки 4-
Синдром Каллмэна 4-
Аниридия
Миотоническая дистрофия -
Синдром Лоу 4"-
Синдром Норри 4"-
Болезнь Менкеса
Х-сцепл. агаммоглобулинемия 4"-
Недостаточность глицеролкиназы 4-
Адрено лейкодистрофия 4-
Рейрофиброматоз, тип 2 *
Болезнь Гентингтона — 4"-
Болезнь фон Гиппеля-Линдау —
Спиноцеребральная атаксия I - 4-
Лисэнцефалия 4"-
40
Болезни человека на картах хромосом
Таблица 5. Продолжение.
1 2 3
Болезнь Вильсона
Туберозный склероз 4“
Синдром Маклеода -J-
Поликистоз почек
Фрагильная Х-хр. Е 4"-
Ахондроплазия -
Синдром Вискотта-Олдрича -
Рак груди и яичиков, ранняя форма
Дистрофическая дисплазия -
Врожденная гипоплазия надпочечников 4"-
Синдром Орскога-Скотта 4" -
Мышечная дистрофия Эмери-Дрейфуса -
Болезнь Макадо-Джозефа - 4-
Спинальная мышечная атрофия -
Хондродисплазия пунктата
Глазной альбинизм
Главным ограничением “чистого” позиционного подхода
является низкая разрешающая способность генетической карты
человека - интервал между двумя соседними маркерами, в
котором локализован ген заболевания, может оказаться слиш-
ком велик и недоступен физическому картированию. И ген в
этом случае можно обнаружить только если есть какая-то “за-
цепка”. Для большинства заболеваний, которые были карти-
рованы позиционно до сегодняшнего дня, характерны круп-
ные структурные аномалии генома, что существенно облегча-
ет заключительную стадию поиска гена - физическое картиро-
вание.
Хронический грануломатоз, миодистрофия Дюшенна, ре-
тинобластома, семейный полипоз толстой кишки, врожденная
гипоплазия надпочечников, глазной альбинизм и некоторые
другие болезни характеризуются крупными хромосомными пе-
рестройками, легко обнаруживаемыми современными мето-
дами молекулярной цитогенетики. Для синдрома фрагильной
Х-хромосомы, миотонической дистрофии, болезни Гентингто-
41
Глава 3
-------------------------------1---------------------
на, спиноцеребральной атаксии, болезни Макадо-Джозефа и
некоторых других характерна экспансия тринуклеотидных пов-
торов.
Способом, который позволяет преодолеть ограничения по-
зиционного картирования, является объединение мощности
обратно-генетической стратегии с преимуществами кандидат-
ного подхода. Такой способ картирования, называемый пози-
ционно-кандидатным, постепенно приходит на смену чисто
позиционному и заключается в поиске на выявленном позици-
онно участке генома подходящих кандидатных генов. Одним
из первых успехов позиционно-кандидатного картирования
была локализация гена синдрома Марфана. Стандартными
генетическими методами (анализ сцепления в родословных)
синдром Марфана был картирован в районе 15q (Kainulainen
et al., 1990). Почти одновременно в этом же районе был лока-
лизован ген фибриллина - вполне подходящий по своим фун-
кциональным характеристикам на роль кандидатного для син-
дрома Марфана (Magenis et al., 1991). Дело оставалось за
малым - найти мутации фибриллина у больных с синдромом
Марфана, что и было довольно быстро сделано (Dietz et а!.,
1991). Таким же образом было показано, что за множествен-
ную эндокринную неоплазию типа II ответственен протоонко-
ген RET (Mulligan et al., 1993). А. чуть позже выяснилось, что
другие точечные мутации в этом же локусе приводят к совер-
шенно иному состоянию - болезни Хиршпрунга (Edery et al.,
1994). Список генов, картированных позиционно-кандидатным
способом будет, вероятно, расширяться весьма быстрыми
темпами. В этот список входят и гены таких сложных с генети-
ческой точки зрения признаков как гипертрофическая кардио-
миопатия, синдром удлиненного QT, неполипозный рак толс-
той кишки, некоторые формы болезни Альцгеймера, два типа
болезни Шарко-Мари-Тус, синдром Пфайффера, пигментный
ретинит и др.
Главной предпосылкой для выдвижения позиционно-кан-
дидатного картирования на первый план является создание
подробной генетической карты человека. Уже сейчас имеется
довольно плотная генетическая карта, охватывающая около
6000 локусов, расположенных в среднем через каждые 0,7 сМ.
Значительная часть этих локусов - высокополиморфные тан-
демные повторы, распределенные по всему геному более или
менее равномерно.
Не менее важным условием для успеха позиционного кар-
тирования является и создание подробной транскрипционной
карты. Сейчас несколько крупных исследовательских консор-
циумов в разных странах мира заняты созданием карт и баз
данных по EST (EST - expressed sequence tag, метка экспрес-
42
Болезни человека на картах хромосом
сируемых последовательностей). В самое ближайшее время
будет доступна карта, содержащая, по приблизительным оцен-
кам, от 50000 до 150000 EST, что даст “охотникам за генами”
неиссякаемый источник кандидатных генов.
Наличие ДНК-маркеров, покрывающих через примерно рав-
ные интервалы весь геном, позволяет тестировать на предмет
сцепления с болезнью каждый участок генома (т.е. проводить
своеобразное сканирование всего генома) и выявлять наибо-
лее вероятный участок или участки локализации гена в интер-
вале между двумя соседними маркерами. Альтернативой то-
тального сканирования, - в том случае, если есть какая-то пред-
варительная информация о кандидатном гене, источником ко-
торой могут быть знания патологических механизмов или эк-
спериментальная модель на животных, - является сканирова-
ние только кандидатного участка генома. После того как пред-
полагаемый ген локализован до интервала в несколько Мб,
можно проводить поиск кандидатных генов, используя тран-
скрипционную карту, цитогенетические методы, или анализи-
руя неравновесие по сцеплению в гомогенных изолированных
популяциях. Заключительной стадией является физическое
картирование и клонирование гена.
Карты хромосом человека
Для отечественных клиницистов, врачей разных специаль-
ностей, практически недоступна информация по этому вопро-
су. Нами (Пузырев, Степанов, 1997) подготовлена русскоязыч-
ная версия карт хромосом, на которых обозначены гены, от-
ветственные за развитие, главным образом, менделирующих
болезней, а также кандидатные гены некоторых мультифакто-
риальных заболеваний. В основу представленного ниже мате-
риала положены карты из “Mendelian Inheritance in Man” (MIM),
дополненные данными (текст для каждой хромосомы) по пос-
ледней CD-версии On-Line MIM от 6 июня 1996 года (McKu-
sick, 1996). Они включают информацию по 25 хромосомам: 22
- аутосомы, X- и Y-хромосомы, М (митохондриальная) - хро-
мосома. На картах хромосом обозначена локализация около
800 генов болезней человека. Эта исчерпывающая цифра на
сегодняшний день составляет всего лишь 1% от общего числа
фактически имеющихся генов у человека (Fields et al., 1994).
43
Глава 3
Условные обозначения на картах хромосом
I—। - аллельные состояния
[ ] - “неклинические” состояния
О - неоплазма со специфическими хромосомными
изменениями и/или имеющая отношение к
онкогену и/или потере гетерозиготности в
опухолевой ткани
ф - пороки с ограниченными хромосомными изме-
нениями
{ } - моногенная подверженность/устойчивость
курсив - несовместимость матери и плода
? - предполагаемая локализация на хромосоме
44
Болезни человека на картах хромосом
Р
21
32
31
В L
4
Хромосома 1
Рак груди (протоков)0
Недостаточность енолазы
. Нейробластома0
12 I
21
32
41
42
1pter-p36.13
1р36-р35
1р36-р34
1р32
1р21-р13
1р21-р13.3
1р11-q11
1р
1q21
1q21 -q31
? Гемолитическая анемия Rh-ноль
Эритробластоз плода_____________
Эллиптоцитоз-1
Эритрокератодермия вариабельная
. Гипофосфатазия, младенческая
Фукозидоз
Поздняя подкожная порфирия
Порфирия, гепатоэритропоэтическая
Недостаточность галактоэпимеразы
Ceroid lipofuscinos, нейронный-1, детский
Недостаточность С8. тип I и II
Лейкемия/лимфома, Т-клеточная
Недостаточность ацил-СоА-дегидрогеназы, средняя цепь
Болезнь кленового сиропа, тип 2
Недостаточность миоаденилатдеаминазы]
(Недостаточность АМФ-деаминазы эритроцитов
Гиперплазия надпочечников, тип II
Гипотироидизм, незобный_______________________
Миопатия из-за недостаточности ЦТФазы
Гипераммонемия из-за недостаточности ЦТФазы
ЯЖ—^^^111 ... 1
Болезнь Гоше, 2 или более типов
Гемолитическая анемия в результате недостаточности ПК
Эллиптоцитоз-2
Пиропойкилоцитоз
Сфероцитоз, рецессивный
Недостаточность антитромбина П1
Болезнь Шарко-Мари-Туз, тип 1b
Подверженность к Vivax malaria
Синдром Криглера-Наджара
Немалиновая миопатия
Недостаточность фактора V______
Lupus erythematosus, системный
Нейропения, иммунная___________
Катаракта опоясывающая, пулверулентная
Хронический грануломатоз в результате недоста-
точности NCF-2
Гликогеноз VII
Недостаточность фактора Х1ПВ
Недостаточность CR1
Недостаточность фактора Н
Синдром Ашера, тип 2
Синдром ван дер Вуда
Недостаточность фумаразы
Врожденная катаракта, тип Фолькмана
Нейропатия Шарко-Мари-Тус 2А
Синдром Пейтца-Егерса
Глухота, аутосомно-доминантная 2
Болезнь Штаргардта
Синдром Ваарденбурга, тип 2В
Кардиомиопатия, семейная дилатационная
с дефектом проводимости, 1
Феохромоцитома
Пикнодизостоз
Глаукома 1, открытоугольная
45
Глава 3
1q21-q31
1q31
1q31-q32.1
1q31-q42
1q41
1q42-q43
25
22
16
13
12
24
*
Гиперпаратироидизм (с опухолью челюсти)
Буллезный эпидермолиз 2А
Пигментный ретинит-12 (аутосомно-
рецессивный)
Болезнь Альцгеймера-4
Болезнь пульсирующих мышц
Аритмогенная дисплазия правого
желудочка-2
Хромосома 2
Недостаточность АКТ!
Аниридия -1
Г ипобеталипопротсинемия
9 Абсталипопротеинемия
Г ипербетал ипопротеинсмия
Аполипопротеин В-100, нарушения
Р
Иодинпсроксидазная недостаточность
щитовидной железы
Недостаточность карбомоилфосфатсинтстазы-1
14
21
22
24
31
32
12
36
37
Недостаточность белка С
Пигментная ксеродсрма, группа В
q
Синдром Элерса-Данлоса IV
Семейная аневризма
Катаоакта, Коппок-подобная
Лейкемия/лимфома, Т-клсточная
Недостаточность LACD
Синдром Элерса-Данлоса, тип X
Ксантоматоз, cerebiotentinous
^Рабдомиосаркома, альвеолярная0
—— Синдром Ваарденбурга, тип 1
2р23-р22 Глухота, аутосомно-рецессивная 9
2р21 Глаукома 3, первичная инфантильная
46
Болезни человека на картах хромосом
2р21 Голопрозэнцефалия-2, алобарная или
полулобарная
2р 11.2 Лиссэнцефалия, ген 2
2q Предрасположенность к микобактериальным
инфекциям
2q31 Инсулин-зависимый сахарный диабет-7
2q31 Синдактилия типа II (синполидактилия)
2q32 Синдром морщинистой кожи
2q37 Брахидактилия, тип Е
2q37 Синдром брахидактилия - умственная отсталость
Хромосома 3
Синдром фон Хиппеля-Линдау
Резистентность к тироидному гормону
Мелкоклеточная карцинома легких0
Псевдо-синдром Цслльвсгсра
GM 1 -ганглиозидоз
Синдром Моркио, тип В
21
14
12
11
13
21
24
26
25
29
Р
Пузырчатый дистрофический эпидермолиз
Карцинома клеток печени0
Недостаточность белка S
Гемолитическая анемия в результате
недостаточности глутатионпсроксидазы
Болезнь Rh-ноль
Оротикацидурия
ч
Пропионацидемия, рссВ-тип
Атрансфсрринемия
[ Гипоцсрулоплазминсмия, наследственная |
Пигментный ретинит-5
Постансстезическос апноэ
Непереносимость сахарозы
Блефарофимоз, обратный эпикант и птоз
Тромбофилия в результате избытка HRG
Недостаточность тиротропин-релизинг
гормона
47
Глава 3
Зр26-р22
3р25.3
Зр25
Зр25-р24.2
Зр25-р22
Зр21.1-р12
Зр21.1-р14.1
3р13-р12
3р11 ,1-q11.1
Зр
3q13
3q13-q22
3q2
3q21-q24
3q21-q25
3q26.3
3q28-q29
3
Анемия Фанкони, группа
комплементации В
Синдром Паллистера-Холла
Пигментная ксеродерма, группа
комплементации С
Синдром Марфана, тип II
Кардиомиопатия, семейная
дилатационная 2
Спиномозжечковая атаксия 7
Синдром Ларсена 1 (аутосомно-
доминантный)
Синдром Барде-Бидл я 3
Деменция, семейная неспецифическая
Глухота, аутосомно-рецессивная 6
Г ипопаратироидизм
Нейропатия Шарко-Мари-Тус 2В
Алкаптонурия
Хроническая доброкачественная
пузырчатка (болезнь Хейли-Хейли)
Синдром Ашера 3
Синдром Корнелиа де Ланге
Оптическая атрофия 1 (аутомосмно-
доминантная)
Подверженность вирусу герпеса
48
Болезни человека на картах хромосом
Хромосома 4
16
15
13
21
28
31
24
26
35
р
Болезнь Гентингтона
МПС1 (Синдромы Гурлера и Шейе)
Синдром Вольфа-Хиршхорна*
ФКУ в результате недостаточности
ди гидроптеридинреду ктазы
Юношеский периодонтит
Гиперцинкемия с дисальбуминемией
Гипертироксинемия с дисальбуминемией
Анальбуминемия______ ______
Наследственная персистенция альфа-фетопротеина
Недостаточность АФП, врожденная
. Пегость
Несовершенный дентиногенез-1
? Острая лимфоцитарная лейкемия °
„ Муколипидоз II
Муколипидоз ill
Синдром Ригера*
. Аспартил гл юкозаминурия
Недостаточность инактиватора СЗЬ
SCID в результате недостаточности ИЛ2
3 32
Дисфибриногенемия, гамма-тип
Гипофибриногенемия, гамма-тип
Дисфибриногенемия, альфа-тип
Дисфибриногенемия, бета-тип
Склероатрофический и ксратичсский дерматоз
конечностей
Мезенхимальный дисгенез переднего сегмента
Псевдогипоальдостеронизм
Карцинома клеток печени °
Мышечная дистрофия, фасциально-лопаточно-
плечевая
Недостаточность фактора XI
Недостаточность фактора Флетчера
4р16.3 Глухота, аутосомно-доминантная 6
4р16 Краниосиностоз, тип Аделаида
4р16 Синдром Эллиса-ван Кревельда
4р16-р14 Хондродисплазия пятнистая ризомелическая
4р Синдром Вольфрама
4q12 Мышечная дистрофия области поясницы и
конечностей, тип 2Е
4q25-q27 Синдром удлиненного QT, тип 4
4q35 Плече-лопаточно-лицевая мышечная
дистрофия 1А
4 Заказ №127
49
Глава 3
Хромосома 5
р
15
13
13
14
21
23
11
Недостаточность С6
Недостаточность С7
Недостаточность С9
Карликовость Ларона
Синдром Марото-Лами
(МПС VI, несколько форм)
Болезнь Верднига-Гоффмана
Спинальная мышечная атрофия II
Спинальная мышечная атрофия III
Болезнь Зандхофа
31
Полипоз толстой кишки, семейный
Синдром Гарднера
Рак прямой и толстой кишки_____
{Дифтерия, подверженность к|
Резистентность к кортизолу
Рефракторная макроцитарная анемия
(синдром 5q)
Глухота в диапазоне низких звуков
Мышечная дистрофия поясничного отдела
Синдром Тричера-Коллинза
Диастрофическая дисплазия
Недостаточность фактора XII
Контрактурная арахнодактилия
ОМ2-ганглиозидоз, вариант АВ
5q11.2 Синдром Киппеля-Фейла
5q11.2-q13.3 Шизофрения-1
5q21-q22 Аденоматозный полипоз толстой кишки
5 Синдром Коккейна 1
50
Болезни человека на картах хромосом
Хромосома 6
Р
21
22
/Недостаточность фактора XIIА
/ Рото-лицсвая расщелина
Болезнь кленового сиропа, тип 3
Спиноцеребральная атаксия-1
Дефект перегородки предсердия (одна из форм)
Недостаточность С2
Недостаточность С4
Врожденная гиперплазия надпочечников в результате
недостаточности 21-гидроксилазы
Юношеская миоклональная эпилепсия
Гемохроматоз
Почечная глюкозурия
Метилмалонинацидурия, один из типов
Пигментный ретинит, периферии-зависимый
q
25
24
Аргининемия
Рак груди
Макулярная дистрофия, ?витсллиновый тип
Дисплазминогенная тромбофилия
Плазминогеновая болезнь Тошиги
Недостаточность плазминогена I и II
Предрасположенность к ИБС
6р23
6р21.3
6р21.3
6р21.3
6р21.1 -р12
6cen-q14
6q
6q13-q15
6q14-q15
6q14-q16.2
6q22.3-q23.1
6q23-q25
Шизофрения 3
Дизлексия, специфичекая 2
Болезнь Педжета (костей)
Пигментный ретинит-14 (аутосомно-
рецессивный)
Поликистоз почек и болезнь печени-1
(аутосомно-рецессивный)
Болезнь Штаргардта 3 (аутосомно-
доминантная)
Прогрессивная двусторонняя
хориоретинальная атрофия
Глазной альбинизм, аутосомно-
рецессивный
Болезнь запасания сиаловой кислоты
Макулярная дистрофия, ретинальная 1
(тип Северная Каролина)
Наследственная персистенция фетального
гемоглобина, гетероцеллюлярная
Миоклональная эпилепсия, тип Лафора
51
Глава 3
6q24-q27
6q25-q26
6q25-q27
6q26-q27
Инсулин-зависимый сахарный диабет-5
Дистрофия колбочек сетчатки-1
Инсулин-зависимый сахарный диабет-8
Рак яичников, серозный
Хромосома 7
Р
I!
2 21
1 13
Краниосиностоз
Краниосиндактилия Г[к-ига
Миопатия в результате недостаточности
фосфоглицератмутазы
Аргининсукцинацндурия
Хронический гранулематоз в результате
недостаточности NCF-1
ьСиндром Цслльвегера
Мукополисахаридоз VII
Деформация кисти/стопы, тип 1
Синдром Элерса-Данлоса VII А2
Несовершенный остеогенез
Ч
15
11
Геморрагический диатез в результате
недостаточности PLAN HI
Тромбофилия в результате избытка
активатора плазминогена_________
Cutis laxa, марфаноцдный тип
Муковисцидоз
Нсдост. липоамиддегидрогеназы
Гемолитическая анемия в результате
недостаточности
дифосфоглицсратмутэзы
Недостаточность трипсиногена
Цветовая слепота, тритановая
Наследственная персистенция
фетального НЪ, один из типов
Голопрозэнцефалия, тип 3
Недостаточность 3-гидрокси -
ацил - СоА-дегидроге назы
7р21-р15
7p15-q13
7р15
7р
7р
7q
7q11io2-q21
7q11.2-q21.3
7q3
7q31
7q31-q35
Макулярная дистрофия, доминантная
кистозная
Пигментный ретинит-9
Глухота, аутосомно-доминантная 5
Синдром Голденхара
Спинальная мышечная атрофия,
дистальная, с преобладанием поражения
верхних конечностей
Гиперрефлексия
Церебральные кавернозные пороки
Эктодактилия, эктодермальная дисплазия,
расщепление губы и неба
Кардиомиопатия, гипертрофическая 6
Глухота, аутосомно-рецессивная 4
Пигментный ретинит-10 (аутосомно-
доминантный)
52
Болезни человека на картах хромосом
7q31.3 Ожирение
7q32.1 Синдром Смита-Лемли-Опитца
7q35-q36 Синдром удлиненного QT-2
7q36 Недоразвитие крестца, аутосомно-
доминантный (триада Куррарино)
Хромосома 8
Р
q
21
22
12
23
2 22
12
11
11
23
Гиперлипопротеинемия 1
Гемолитическая анемия в результате
недостаточности глутжтионрсдуктааы
Недостаточность активатора плазминогена
?Гипогонадотропный гипогонадизм
в результате недостаточности ГНРГ
Синдром Вернера
Сфероцитоз II
Пигментный ретинит-1
Плеоморфная аденома слюнных
желез °_____________________________
Врожденная гиперплазия надпочечников
в результате недостаточности
11 -бета-гидроксилазы
Недостаточность С МОП_______________
Ацидоз-остеопетрозный синдром
почечных канальцев
(Недостаточность карбоангидразы]
- Множественный экостаз
Синдром Лангера-Гидиона*
Трихоринофалангеальный синдром-1
Лимфома Беркитта0
Пузырчатый эпидермолиз, тип Огна
Макулярная дистрофия,
нетипичная вителлиформа
?3об, юношеский многоузелковый
Гипотироидизм, наследственный
врожденный (1 или более форм)
8pter-p22 Эпилепсия, прогрессивная, с умственной отсталостью
8q Хондрокальциноз 1 (болезнь отложения пирофосфата кальция)
8q Эпилепсия, доброкачественная неонаталь- ная (семейные доброкачественные конвульсии новорожденных)
8q13-q21.1 Нейропатия Шарко-Мари-Тус-4А (аутосомно-рецессивная)
8q22-q23 Синдром Коэна-1
8qJ>4 Буллезный эпидермолиз простой-1 (Огна)
8q24 Эпилепсия, генерализованная,
идиопатическая
53
Глава 3
Хромосома 9
Р
q
—П
2
21
В
31
12
21
22
34
Карцинома яичников
Острая лимфобластоилная
лейкемия0
Недостаточность a, i |,фа- и iirep<l)epoi ia
Галактоземия
Атаксия Фридрейха
Непереносимость фруктозы
Острая лимфобластическая
лейкемия-2
Амилоидоз, финский тип
Цитруллинемия
Острая печеночная порфирия
{чувствительность к
отравлению свинцом}
Дисплазия ногтей в сочетании
с гипоплазией коленной
чашечки
Гемолитическая анемия
в результате аденилаткиназной
недостаточности
Хроническая миелоидная
лейкемия0
Недостаточность С 5
Пигментная ксеродерма 1
Туберозный склероз-1
Торзионная дистония
Острая лимфобластоидная
Т-клеточная лейкемия
Копронорфирия
Г ардеропорфирия
9p21-q21
9р13
9р11
9р
9q13
9q13-q21
9q22.3
9q31
9q32
9q34.2-q34.3
Артрогрипоз, врожденный множественный,
дистальный, тип 1
Гипоплазия хрящей и волос
Синдром Мелкерссона-Розенталя
Венозные нарушения, множественные
кожные и слизистые
Кардиомиопатия, семейная
дилатационная 1
Глухота, аутосомно-рецессивная 7
Анемия Фанкони, группа
комплементации С
Синдром невоидной карциномы
базальных клеток
Акрофациальный дизостоз, тип Нагера
Синдром Элерса-Данлоса, тип If
54
Болезни человека на картах хромосом
Хромосома 10
р
Множественная глиобластома0
Гемолитическая анемия в результате
гексокиназной недостаточности
Тироидная папиллярная карцинома
15
12
23
25
21
Медуллярная тироидная карцинома0
Множественная неоплазия 0
эндокринных желез, тип II_______
Множественная неоплазия
эндокринных желез, тип III °
Метахром этическая лейкодистрофия
в результате недостат. SAP-1
Болезнь Гоше, вариантная форма
q
Ретинол-связ. белок, недостаточность
Острая лимфоцитарная Т-клсточная
лейкемия0______________
Болезнь Вольмана
Болезнь накопления эфиров
холестерина_______________
Круговая атрофия сосудистой
оболочки глаза и сетчатки, В6
зависимая и независимая
Порфирия, врожденная
эритропоэтическая
Недостаточность липазы
поджелудочной железы
Гиперплазия надпочечников
10pter-p11.2
10р12.1
10q
10q
10q23.1-q23.3
10q23.3-q24.1
10q24-q25
Болезнь Рефсума, взрослая форма с
усиленной пипеколикацидемией
Мегалобластическая анемия 1
Эпилепсия, парциальная
Прогрессивная наружная офтальмоплегия
Синдром Хермански-Пудлака
Спиномозжечковая атаксия 8
Порок расщепления кисти/стопы
(эктродактилия) тип 3
55
Глава 3
-------1-----
Хромосома 11
Болезнь Нимана-Пика
Рабдомиосаркома
Адренокортикальная карцинома0
Синдром Видемана- Бе квита0
Семейная гиперинсулинемия
Сахарный диабет, редкая форма
ГПФГ, гетероклеточный
t Серповидно-клеточная анемия
Бета-талассемия
Бета-метгемоглобинемия
Бета-эритремия
Бета-анемия телец Хайнца
ГПФГ, делеционный тип
ГПФГ, неделетированный тип A, G
Опухоль Вильмса, тип 2
Опухоль Вильмса0 г=----------------
. -• Гилопротромбинем!
, Аниридия-2 _ "
Акягалаэемия |ДиспрСТромби11еми1
Лейкемия, острая Т-клсточная°
4 Напряженная миоглобинурия в результате
лакгатцегидрогеназной недостаточности
Ангиоэдема, наследственная
. Атопия о
Множественная эндокринная неоплазия, тип I
Болезнь Макардля
чЛеЙкемия/лимфома В-клеток °
< Альбинизм
- Атаксия-телеан гиэктазия
Недостаточность пируваткарбоксилазы
Дистрофический буллезный эпидермолиз, рецессии.
Острая перемежающаяся порфирия
Совместная недостаточность ароА1/СЗ
Гипертриглицеридемия (1 форма)
Г ипоальфалипопротсинсмия
Амилоидоз, форма Айова
11 р15.5
11 р15.1
11р15.1-р14
11р15
11р13-р11.2
11р12-р11
11р11-q11
11q
11q13
11q13
11q13
11q13
Синдром удлиненного QT-1; Синдром
Уорда-Романо
Синдром Ашера-1С (аутосомно-
рецессивный, тяжелый)
Персистентная гиперинсулиновая
гипоглицемия детская (незидиобластоз)
Перипапиллярная хориоретинальная
дегенерация (Atrophia areata)
Рак простаты, метастазы
Экзостоз (множественный) 2
Спиномозжечковая атаксия 5
Синдром Якобсена
Синдром Барде-Бидля 1
Инсулин-зависимый сахарный диабет 4
Соматотрофинома
Вителлиформная макулярная дистрофия
(болезнь Беста)
56
Болезни человека на картах хромосом
11q13 Витреоретинопатия, неоваскулярная,
воспалительная
11q13-q23 Экссудативная витреоретинопатия-1
(аутосомно-доминантная, Синдром
Крисвика-Схепенса)
11q13.5 Глухота, аутосомно-рецессивная 2
11 q 14-q21 Мозжечковая атаксия-1
11q22-qter Карцинома анального канала
11q23 Рак груди, связанный с
транслокацией 11 ;22
Хромосома 12
Совместная недостаточность Clr/Cls
Гемолитическая анемия в результате
. недостат. триоэофосфатизомеразы
Болезнь фон Виллебоандга
.Эмфизема из-за недостаточности А2М
Аденома толстой и прямой кишки0
Рак толстой и прямой кишки0______
Синдром Стиклера
Врожденная спондилоэпифиэеальная
дисплазия
?Дисплазия Книста
Ахондрогенез Лангсра-Сальдино
Ранний остеоартроз_______________
Липома0
Микоидная липосаркома0
Синдром Санфилиппо D '
Псевдо-витамин D зависимый рахит
|Гистидинемия)
Туберозный склероз-3
Недостаточность короткой цепи ацил-
СоА-дегидрогеназы
Недостаточность иммунного интерферона
Фенилкетонурия
|гилерфенилаланмнемия, мягкая форма)
Острая непереносимость алкоголя
?Фетальный алкогольный синдром
12q Синдром Ослера-Рендю-Вебера
12q11-q13 Ладонно-подошвенная кератодерма,
тип Ботина
12q13.2-q24.1 Синдром поражения наружных мышц глаз,
врожденный
12q21.3-q22 Синдром Холта-Орама
12q22 Опухоль мужских половых клеток
12q22-qter Синдром Нунана-1
12q23-q24.1 Болезнь Дарье (фолликулярный кератоз)
12q24.1-q24.32 Диабет взрослых с проявлением в
молодом возрасте, тип III
57
Глава 3
------------
Хромосома 13
Ретинобластома °
Остеосаркома0
-----Болезнь Вилсона
-----Мегаколон
-----Пропион и и ацидемия, тип рссА
Недостаточность фактора VII
Недостаточность фактор X
.’Пигментная ксеродерма (1 тип)
Рак труди (протоков)
13q12
13q12
13q12.3
13q14-q31
13q34
13q34
Глухота, аутосомно-рецессивная 1
Мышечная дистрофия, Дюшенн-подобная,
аутосомно-рецессивная
Рак груди, ранняя форма
Болезнь Леттерера-Сиве
Синдром гиперорнитинемия-
гипераммонемия- гомоцитруллинемия
Болезнь Штаргардта 2
(аутосомно-доминантная)
Хромосома 14
га
21
24
31
11
11
32
Р
Синдром Ушера-1
Лейкемия/лимфома Т-клеток0
Гипертрофическая кардиомиопатия, одна из форм
Недостаточность нуклеозидфосфорилазы
Болезнь Краббе
Эллиптоцитоз-3
Сфероцитоз I
Гипотироидизм, нсэобный
Эмфизема циррозная
Эмфизема
Геморрагический диатез в результате наличия
антитромбина Питтсбург__________________
Недостаточность альфа-1-антихимотри псина
Лсйкемия/лимфома Т-клеток0
[Недостаточность транскортина|
Порфирия variegata
[Эктопическая экспрессия кератинкиназы В]
Гипогаммаглобулинемия, различные типы
Болезнь накопления гликогена, тип VI
58
Болезни человека на картах хромосом
14q
14q
14q11.2
14q23-q24
14q24-qter
14q24.3
14q24.3-q31
14q24.3-q31
14q32
14q32
14
Глухота, аутосомно-рецессивная 5
Миопатия, дистальная 1
Врожденный ихтиоз 2, неэритроматозный
чешуйчатый ихтиоз
Аритмогенная дисплазия правого
желудочка-1
Катаракта, передняя полярная 1
Болезнь Альцгеймера-3
Инсулин-зависимый сахарный диабет 11
Болезнь Макадо-Джозефа
(спиномозжечковая атаксия 3)
Синдром Ашера-1А
Порфирия смешаная
Мукополисахаридоз, тип IIIC
Хромосома 15
Р
Ч
га
21
22
И
И
26
?Дислексия 1
Синдром Прадера-Вилли*
Синдром Ангел ьм ан а
Изовалерианацидемия
Сфероцитоз наследственный,
японский тип
Мышечная листооФия поясничного
отдела, одна из форм
Недостаточность липазы печени
?Гинекомастия, семейная
Синдром Марфана
Гемодиализ*зависимый амилоидоз
Острая промиелоцитарная
лейкемия_____________________
Болезнь Тея-Сакса
GMl-ганглиозидоз, юношеский,
взрослый
[ Псевдонедостаточность
гексоаминидазы А] ______________
Глутарил ацидурия
Тирозинемия, тип I
Пигментная ксеродерма, гр. F
15q11-q13
15q11.1
15q22.3-q23
15q26.1
15
Наследственная остеодистрофия
Олбрайта-2
Спастическая параплегия
Синдром Барде-Бидля 4
Синдром Блума
Синдром уплотненной кожи
59
Глава 3
Хромосома 16
в
11
11
12
12
23
Альфа-анемия телец Хайнца
Альфа-талассемия
Альфа-эритремия
Альфа-метгемоглобинемия
Синдром умственной отсталости,
связанный с НЬН
Синдром Рубинштсйна-Тойби
Поликистоз почек
[Недостаточность глиоксалазы А]
Болезнь Баттена
Недостаточность БПЭХ
Катаракта, тип Марнера
Болезнь Норума
Болезнь рыбьих глаз
. Гирозинемия II
4 ?Недостаточность альдолазы А
Уролитиаз, 2,8-дигидрооксиаденин
Гранулематоз
[Цистатионурия]
Миопатия Броди
16р13.3 Катаракта врожденная с микрофтальмией
16р13.3 Поликистоз почек, тяжелый инфантильный
с туберозным склерозом
16р13.3-р13.2 Синдром углеводород-дефицитного
16q
16q
16q12-q13
16q12.1
16q21
16q24.3
гликопротеина
Спиномозжечковая атаксия 4
Опухоль Вильмса-3
Цилиндроматоз (множественная
доброкачественная кистозная эпителиома)
Синдром Таунса-Брокса
Синдром Барде-Бидл я 2
Анемия Фанкони-1
60
Болезни человека на картах хромосом
Хромосома 17
Синдром Бернара-Сулье
Лисс си цефалический Синдром Миллера-Дикера
13
11
21
Рак толстой и прямой кишки
Синдром Ли-Формен и°
Болезнь Шарко-Мари-Туз. тип
Синдром Смита-Маген иса__________________
Мужской псевдогермафродитизм с гинекомастией
Поликистоз яичников
Синдром Уотсона
. Нейрофиброматоз Рсклинхауэена0
Недостаточность ацетнл-СоА-карбоксилазы
Рак груди, ранняя форма
Недостаточность миелопероксидазы
Недостаточность галактокиназы_________________
Синдром Элерса-Данлоса, тип VII А1
Несовершенный остеогенез (две или более формы)
[Акантоцитоз, одна из форм]
|Эллиптошгтоз, малазийско-меланезийская форма]
Тромбоастения Гланцмана, тип А
Тромбоастсния Гланцмана, тип В
Болезнь Помпе
Недостаточность кислой мальтазы у взрослых
Недостаточность гормона роста,
тип Иллига 1А, тип Коварски
Недостаточность плацентарного лактогена
и
24
25
Врожденная пярямиотония
Врожденная миотония, атипическая
17р13.3
17р13
17р13
17р13-р12
17p12-q12
17р11.2
17q11-q12
17q11-q24
17q21
17q21
17q24
17q25
17q25
Пигментный ретинит-13
Катаракта, передняя полярная 2
Наследственный амавроз Лебера-1
Дистрофия колбочек сетчатки 5
Глухота, аутосомно-рецессивная 3
Синдром Сьогрена-Ларссона
Катаракта, врожденная зонулярная с
шовным помутнением
Предрасположенность к злокачественной
гипертермии 2
Рак груди-1, ранняя форма
Паркинсонизм-деменция с
паллидопонтонегральной дегенерацией
Катаракта, врожденная, церулиновый тип
Пигментный ретинит-15
Синдром Рассела-Сильвера
61
Глава 3
Хромосома 18
Р
q
Недостаточность ингибитора пл тмина
Семейная амилоидная нейропатия
(несколько типов)
Старческий амилоидоз_________
Лейкемия/лимфома В-клеток°
---Рак толстой и прямой кишки
Метгемоглобинемия в результате
недостаточности цитохрома Ь5
18р
18q11-q12
18q11-q12
18q21-q22
18q21-q22
18q21.1-q21.3
18q21.1-q22
18q22-qter
18q22.1
Биполярное аффективное расстройство
Семейный синдром рака Линча II
Болезнь Нимана-Пика, тип С
Рекуррентный внутрипеченочный холестаз
новорожденных
Прогрессивный семейный
внутрипеченочный холестаз
(болезнь Байлера)
Дистрофия колбочек сетчатки 1
(аутосомно-доминантная)
Семейный остеолизис
Множественный склероз
Синдром Жиля де ля Туретта
62
Болезни человека на картах хромосом
Хромосома 19
Р
q
Й2
Сахарный диабет, инсулин-устойчивый с папил-
лярно-пигментной дистрофией кожи
Лепре каунизм
’Синдром Рабсона-Мецденаля________________
Недостаточность СЗ
Лейкемия, острая лимфобластондная
Синдром Мюллеровых протоков
Семейная гиперхолестеринемия
Маннозидоз______________ _________________
Гемолитическая анемия из-за недостаточности
глюкоэофосфатиэомераэы
Водянка плода ____________________________
Недостаточность пролидазы
{Чувствительность к полиомиелиту}
Гипсрлипопротеинемия, тип IB
Гиперлипопротеинемия, тип III
Болезнь кленового сиропа
?Пигментная кссродерма (один из типов)____
’Мужской псевдогермафродитизм из-за дефекта
лютеинизирующего гормона
’Гипергонадотропный гипогонадизм__________
Лейкемия/лимфома В-клеток
Злокачественная гипертермия
Миотоническая дистрофия
19р13.3-р13.2
19р13.1
19cen-q13.2
19q13
19q13
19q13.1 -q13.2
19q13.2-q13.3
19q13.4
Предрасположенность к атеросклерозу
(связанная с липопротеинами)
Церебральная аутосомно-доминантная
артериопатия с субкортикальными
инфарктами
Болезнь Альцгеймера-2 (поздняя форма)
Глухота, аутосомно-доминантная 4
Орофациальное расщепление-3
Дистрофия колбочек сетчатки 2
(аутосомно-доминантная)
Миотоническая дистрофия
Пигментный ретинит-11 (аутосомно-
доминантный)
63
Глава 3
20р11.2
-----1------
Хромосома 20
Несахарный диабет, нейрогипофизарный
Болезнь Крсйцфельда-Якоба
Болезнь Герстиан на-Штраусс лера
[Недостаточность инозинтрифосфатана)
Артсриогсцатическая дисплазия
Церебральная амилоидная ангиопатия
Синдром Алагиля
'’Недостаточность гормона роста вследствие
дефекта РФГР
Анемия Фанкони-1
MODY, одна из форм____________________
Острый комбинированный иммунодефицит
из-за недостаточности деаминазы
Гемолитическая анемия из-за избытка АДА
Галакгосиалидоз_______________
Пссвдогипопарэтироцдизм, тип 1а
Дисплазия Маккьюна-Олбрайта
Питуитарная опухоль
Синдром Алагиля (артериогепатическая
дисплазия)
20p11.2-q11.2 Врожденная наследственная
эндотелиальная дистрофия роговицы
20p11.2-q11.2 Задняя полиморфная дистрофия роговицы
20q13 Диабет взрослых с ранним возрастом
проявления, тип I
Хромосома 21
Амиотрофический латеральный склероз
Амилоидоз, цеоебгюартсриальный
Голландский тип
Болезнь Альцгеймера, одна из форм
Острая миелоидная лейкемия
Снижение адгезивной способности
лейкоцитов
Гемолитическая анемия вследствие
недостаточности фосфофруктокиназы
Гомоцистинурия. Вб-зависимый и
независимый тип
Эпилепсия, прогрессивная
миоклональная
21q11.2
21q22.3
21q22.3
Миелопролиферативный синдром,
транзиторный
Глухота, аутосомно-рецессивная 8
Синдром Дауна, критический регион
64
Болезни человека на картах хромосом
Хромосома 22
Синдром кошачьих глаз*
Синдром Ди Георге
Недостаточность N-АГА
Р
q
Глутатионурия
Хроническая миелоидная лейкемия0
Тромбофилия из-за недостаточности
кофактораПгепарина
14 •“
11
zz>
13
Нейроэпителиома
Саркома Юинга0
Дебриэокин, чувствительность к
(?Паркинсонизм, подверженность к)
Аденилсукциназы недостаточность
Слуховая неврома0
Менингома0
Глюкозы/галактозы дефект поглощения
. Недостаточность транскобаламина II
Метгемоглобинемия энзимопатическая
Метахроматическая лейкодистрофия
22q11 Предрасположенность к шизофрении-3
22q11 Конотрункальные аномалии сердца
22q11.2 Синдром Опитца G, тип II
5 3аказ№127
65
Г лава 3
Х-Хромосома
22
21
11
13
21
25
28
X-сцепленный ихтиоз
Недостаточность сульфпаз» стероидов
Синдром Калльманна
X сцепленная рецессивная хондродисплазия, точечная
Наследственная гипофосфатемия
Синдром Амкар ди
Х-с цеп ленная первичная гипомагниемия
Альбинизм глазной
Ретиношизис
Мышечная дистрофия Дюшенна
Мышечная дистрофия Беккера
Синдром недержания пигмента
Синдром Вискотта-Олдрича
Синдром Менкеса
Первичная гипоплазия надпочечников
Недостаточность глицеролкиназы
Хронический гранулематоз
Пигментный рстинит-3
Недостаточность орнитин -карбамоилтрансферазы
Болезнь Норри
Пигментный ретинит-2
Синдром мужского бесплодия
Аигидротическая эктодермальная дисплазия
Агаммаглобулинемия
Болезнь Кеннеди
Болезнь Пелицеуса-Мерцбахера
Синдром Ал порта
Болезнь Фабри
Синдром Лоу
Гипериммуноглобулинемия М
Лимфопролиферативный синдром
Дункана____________________
Болезнь Хантера
Гемофилия В
Синдром альбинизма с глухотой
Синдром Мартина-Белла
Гемофилия А
Фавизм
Гемолитическая анемия
Сидеробластическая анемия
Синдром Аарскога-Скотта
Гемолитическая анемия (нсдост ФГК)
Болезнь Шарко-Мари-Туз
Тапето-хороидальная дистрофия
Х-сцспл спастическая параплегия
Расщепление неба, Х-сцепленное
Подагра, ФРПС-зависимая
Синдром Леша-Нихана
Подагра, ГПРТ-зависимая
Маниакально-депрессивный психоз, X-сцепленный
Цветовая слепота (несколько форм)
Дискератоз врожденный
Адрснолейкодистрофия
Адреномиелонейропатия
q
Миодистрофия Эмери-Дрейфусса
Диабет несахарный, нефрогенный
Х-сцепленная миопатия миотубуляркая, тип 1
Xpter-p22.2
Хр22.31
Хр22.31
Хр22.3-р21.1
Хр22.2
Краниофронтонозальная дисплазия
Дермальная гипоплазия, фокальная
Микрофтальмия “кожи в полоску”
Катаракто-дентальный синдром
Нэнса-Хорана
Болезнь Шарко-Мари-Тус, Х-сцепленная-2,
рецессивная
Хр22.2 Гипомагнеземия, вторичная гипокальцемия
Хр22.2-р22.1 Синдром Коффина-Лоури
Хр22.2-р22.1 Корнеальный дермоид
Хр22.2-р22.1 Синдром Партингтона
Хр22.13-р22.11 Пигментный ретинит 15
Хр22.11-р21.2 Гонадный дисгенез, XY женский тип
Хр22 Агаммаглобулинемия, Х-сцепленная 2
(с недостаточностью гормона роста)
Хр22 Наследственная гипофосфатемия 2
66
Болезни человека на картаххромосом
Хр22
Хр22
Хр22-р21
Хр21.3-р21.2
Хр21.2
Xp21.1-q22
Хр21
Хр21
Xp21-q13
Xp11.4-q11.23
Хр11.3
Умственная отсталость, Х-сцепленная-1,
без дисморфий
Синдром Опитца G, тип I
Пигментное расстройство, ретикулярное
Пигментный ретинит-6 (Х-сцепленный
рецессивный)
Глухота 4, врожденная нейросенсорная
Синдром Вильсона-Тернера
Недостаточность гонадотропина
Синдром Синдера-Робинсона
(Х-сцепленной умственной отсталости)
Умственная отсталость, Х-сцепленная 9
Болезнь глаз Аландских островов (глазной
альбинизм, тип Форсиуса-Эрикссона)
Врожденная стационарная ночная слепота
Хр 11.23-р11.22 Синдром Вискотта-Олдрича
Хр11.22 Нефролитиаз 1 (Х-сцепленный)
Хр11.2 Карцинома почеки, папиллярная
Хр11 Умственная отсталость, Х-сцепленная
неспецифическая с афазией
Хр11 Умственная отсталость, Х-сцепленная 20
Xp11-q21
Xp11-q21.3
Хр
Хр
Хр
Хр11-q12
Xq13
Xq13-q21
Xq13-q22
Xq13.1
Синдром Прието
Синдром Сазерленда-Хаана
Катаракта, врожденная тотальная
Синдром Ретта
Спинальная мышечная атрофия,
Х-сцепленная летальная инфантильная
Умственная отсталость, Х-сцепленная-2,
без дисморфий
Анемия, сидеробластная
со спиноцеребеллярной атаксией
Синдром Виккера-Вольфа
Синдром Майлса-Карпентера
Торзионная дистония-паркинсонизм,
тип Филипино
Xq21
Xq21.3-q22
Xq21.3-q22
Xq22
Xq22
Xq22-q28
Xq23-q24
Xq24-q27
Xq24-q27.1
Синдром умственной отсталости Аллана-
Херндона-Дадли
Мегалокорнеа-1, Х-сцепленная
Пангипопитуитаризм, Х-сцепленный
Глухота 1, прогрессивная
Синдром Мора-Тарнебьерга
Несовершенный амелогенез-3
Умственная отсталость, Х-сцепленная 23
Синдром Базекса
Гипертрихоз, врожденный
генерализованный
67
Глава 3
Xq25
Xq25-q26
Xq25-q26.1
Xq25-q27
Xq26
Xq26
Xq26
Xq26-q27
Xq26-q27
Xq26-q27
Xp11.3-q11.2
Xq27-q28
Xq27.3
Xq27-qter
Xq28
Xq28
Xq28
Xq28
Xq28
Xq28
Xq28
Xq28
Xq28
Xq28
Лимфопролиферативный синдром
Гетеротаксия-1
Торакоабдоминальный синдром
Синдром Петтигрю
Синдром умственной отсталости
Густавссона
Синдром дизморфии Симпсона
Расщепление кисти/стопы
(эктодактилия) 2
Синдром Берьесона-Форсманна-Лемана
Г ипопаратироидизм
Преждевременная недостаточность
яичников
Артрогрипоз множественный врожденный
Х-сцепленный
Анофтальмоз-1 (с умственной
отсталостью, но без аномалий)
Фрагильная Х-хромосома, умственная
отсталость
Буллезный эпидермолиз, макулярный тип
Адренолейкодистрофия
Цветовая слепота, голубой
цвет-монохромная
Кардиомиопатия, дилатационная
фатальная инфантильная Х-сцепленная
Хондродисплазия пятнистая-2,
Х-сцепленная доминантная (синдром
Гаппла)
Дискератоз врожденный
Эндокардиальный фиброэластоз-2
(синдром Барта)
Гомосексуальность, мужская
Недержание пигмента-2 (семейное,
летального типа)
Мажорное аффективное расстройство-2
Синдром Вайсмайна
Y-хромосом а
ХУгонадный дисгснс!
68
Болезни человека на картах хромосом
Yp Азооспермия, фактор 2
Yq11 Азооспермия, фактор 1
Yq12 Рост, Y-сцепленный
Митохондриальный геном (М-хромосома)
ATPase в
ADPD - Болезнь Альцгеймера/болезнь Паркинсона
DEAF - Нейросенсорная потеря слуха
LHON - Наследственная нейроофтальмопатия Лебера
LDYT - LHON. и дистония
MELAS - Митохондриальная миопатия, энцефалопатия,
' молочнокислый ацидоз и приступы судорог
MERRF - Миоклональная эпилепсия в сочетании
с необычно красными мышечными волокнами
NARP - Нейропатия, атаксия и пигментный ретинит
РЕМ - Летальная прогрессирующая энцефаломиопатия
69
Глава 4
ГЕНЕТИЧЕСКОЕ КАРТИРОВАНИЕ
МУЛЬТИФАКТОРИАЛЬНЫХ ЗАБОЛЕВАНИЙ
Развитие технологий картирования, построение подробной
генетической карты человека, разработка математических и
компьютерных методов и моделей позволили начать система-
тическое картирование генов, ответственных за сложнонас-
ледуемые (мультифакториальные) заболевания и признаки.
Описанный выше прогресс в картировании генов, отвечающих
за менделирующие болезни человека, рано или поздно (по не
самым оптимистичным оценкам - в течение ближайшего деся-
тилетия) приведет к тому, что гены всех более или менее час-
тых моногенных заболеваний человека будут идентифициро-
ваны. Более того, как следствие последних достижений в то-
пографии генома появилась возможность продвинуться на
доселе слабо исследованную территорию - генетику сложно-
наследуемых состояний. Действительно, с точки зрения гене-
тического анализа, большая часть наиболее распространен-
ных заболеваний человека и признаков, имеющих медицинс-
кую значимость, крайне неудобны - они не следуют простому
менделевскому принципу моногенного наследования. Наибо-
лее частые болезни человека являются результатом действия
многих генетических факторов в сочетании с факторами сре-
ды и случайными причинами, т.е. имеют мультифакториаль-
ную природу. В категорию мультифакториальных признаков
попадают все основные причины заболеваемости и смертности
в современных популяциях человека: атеросклероз, гиперто-
ния, многие формы рака, психические заболевания, диабет,
бронхиальная астма, ревматоидный артрит, наследственная
составляющая подверженности к инфекционным заболевани-
ям и, вероятно, значительная компонента общего процесса
старения.
К фенотипам, которые не проявляют моногенного доминан-
тного или рецессивного типов наследования, применяют тер-
мин “комплексные” или “сложнонаследуемые” признаки (т.е.
термин “сложнонаследуемый признак” является более общим
по отношению к термину “мультифакториальный признак”). В
чем же заключается сложность этих состояний? В общем, ос-
ложнения возникают всегда, когда нарушается простое сбот-
ветствие между генотипом и фенотипом: либо один и тот же
70
Генетическое картирование МФЗ
генотип в результате средовых влияний или взаимодействия с
другими генами дает разные фенотипы, либо различные гено-
типы проявляются одинаковым фенотипом.
В числе основных проблем, затрудняющих генетическое
картирование сложнонаследуемых признаков и мультифакто-
риальных заболеваний (МФЗ) (этот список далеко не полный)
можно выделить следующие (Weissman, 1995; Ландер, 1990;
Lander и Schork, 1994):
1) Генетические взаимодействия (полигения). Клинический
фенотип может быть результатом действия нескольких (мно-
гих) генетических локусов. Вклад каждого индивидуального гена
в общую картину формирования признака может быть очень
небольшим. Полигенные признаки можно подразделить на
дискретные, характеризующиеся наличием или отсутствием
определенного состояния (например, развития сахарного ди-
абета или инфаркта миокарда), и количественные, которые
описываются непрерывным рядом изменчивости (например,
уровень липидов плазмы, давление крови или концентрация
глюкозы) и определяются совокупным аддитивным эффектом
многих индивидуальных генетических локусов (QTL - локус
количественного признака). В свою очередь, дискретные по-
лигенные признаки могут быть результатом “порогового эф-
фекта” лежащего в их основе количественного признака, либо
результатом одновременного и совокупного действия мута-
ций нескольких локусов. Примеры первого варианта - инсу-
лин-зависимый сахарный диабет (ИЗСД), гипертония; второ-
го - одна из форм пигментного ретинита, требующая наличия
мутаций в двух локусах, кодирующих белки сетчатки (Kajiwara
etal., 1994).
2) Генетическая гетерогенность. Одна и та же фенотипи-
ческая картина может иметь причину в мутациях различных
генетических локусов - например, мутация, нарушающая функ-
цию любого из генов, контролирующих определенную мета-
болическую цепочку, приведет к отсутствию или уменьшению
концентрации конечного продукта этой цепочки. Генетическая
гетерогенность - явление характерное практически для любо-
го наследственного заболевания человека, в том числе для
МФЗ. Последствия генетической гетерогенности с точки зре-
ния картирования заключаются в том, что болезнь может ко-
сегрегировать с определенным генетическим маркером в од-
ной родословной и совершенно с другим - в другой. От со-
бственно генетической гетерогенности (гетерогенности локу-
сов) следует отличать аллельную гетерогенность, когда раз-
личные мутации одного локуса затрагивают одну и ту же фун-
кцию, но в разной степени и с разными клиническими послед-
71
Глава 4
------------------------------с-------------------------
ствиями. Аллельная гетерогенность, в отличие от локусной, не
является затруднением для генетического картирования.
3) Неполная пенетрантность, вследствие чего не все носи-
тели мутантного генотипа проявляют фенотип, отличный от
нормы. Мутантный ген может проявиться клиническим фено-
типом лишь с определенной, отличной от единицы, вероят-
ностью. Эта вероятность может определяться как генетичес-
ким окружением (“фоном”), так и негенетическими причинами
- полом, возрастом, средовыми влияниями. Тем самым, с оп-
ределенной вероятностью, мутантный аллель может присут-
ствовать у здоровых людей, а нормальный аллель - у больных.
4) Наличие фенокопий. Носители нормального генотипа
могут проявлять мутантный фенотип по причинам негенети-
ческого характера.
5) Неменделевские механизмы передачи генетической ин-
формации. В последние годы стало очевидно, что наследова-
ние по Менделю - лишь один из вариантов механизма переда-
чи генетической информации. Сейчас описано множество бо-
лезней, в основе которых лежит митохондриальное наследо-
вание, экспансия тринуклеотидных повторов, явление геном-
ного импринтинга.
6) Высокая частота в популяции аллеля, связанного с бо-
лезнью. Нежелательные последствия высокой частоты аллеля
для картирования заключаются в том, что в родословной мо-
жет присутствовать несколько копий одного и того же аллеля,
но разного происхождения. Например, попытки картировать
генетический локус болезни Альцгеймера методом анализа
сцепления в родословных долгое время были неудачными.
Причина этого стала очевидной, когда выяснилось, что основ-
ным генетическим фактором болезни является аллель Е4 гена
аполипопротеина Е, который в европеоидных популяциях встре-
чается с довольно высокой частотой (15-20%) (Corder et al.,
1993).
7) Редкость заболевания (очень низкая частота аллеля, свя-
занного с болезнью). При отсутствии родословных с несколь-
кими больными индивидами болезнь просто невозможно изу-
чать методами генетического анализа.
Кроме этих общих проблем генетического картирования
сложно наследуемых признаков существуют и более специ-
фические, связанные с методами набора материала и карти-
рования. Скажем, для генетического анализа очень важна пра-
вильная постановка диагноза. Ошибки в клинической диагнос-
тике, особенно характерные для случаев, когда диагностика
основывается на критериях, далеких от физиологических ос-
нов болезни (например, при психических заболеваниях), мо-
гут привести как к ложно-позитивной, так и ложно-негативной
72
Генетическое картирование МФЗ
классификации индивидов, что одинаково отрицательно ска-
зывается на перспективе картирования. Даже в тех случаях,
когда найдены значимые корреляции между признаком и ге-
нетическим маркером, дальнейший анализ может быть затруд-
нен чисто физическими причинами. Например, “подозревае-
мый” регион генома может оказаться слишком большим, а
значит, недоступным современным методам физического кар-
тирования. Такая ситуация сложилась сейчас с несколькими
локусами ИЗСД - их место на генетической карте определено,
однако эффективное физическое картирование пока невоз-
можно (Todd, 1995).
Затруднения иного рода возникают, если кандидатный ген
оказывается сцепленным с другим кандидатным геном. Эта
ситуация характерна для генетического картирования многих
аутоиммунных заболеваний, основные генетические локусы
которых расположены в районе главного комплекса гистосов-
местимости. Здесь, на участке размером всего 3 Мб, распо-
ложены гены 10 семейств, функционирующих в иммунном
процессе (Wei et al., 1993), и разобраться в этом комплексе
сцепленных локусов крайне сложно.
Отправной точкой для исследований на уровне ДНК - для
геномных исследований - служит генетическая эпидемиоло-
гия. Картирования никогда не начинается на пустом месте, ему
предшествуют исследования заболевания на популяционном
и семейном уровне. Подчеркнем, что данные генетико-эпиде-
миологических исследований позволяют оценить такие важ-
ные характеристики как частота болезни в популяции, доля
семейных случаев, уровень наследуемости и пенетрантности.
Наиболее важным, сточки зрения картирования, эпидемиоло-
гическим параметром является показатель относительного
риска U), который показывает насколько риск для родствен-
ника больного выше риска в общей популяции. Для моноген-
ных заболеваний этот показатель очень высок, например, при
муковисцидозе показатель относительного риска для сибса
(xs) равен 500, при болезни Хиршпрунга - 2000. Для мульти-
факториальных заболеваний относительный риск значитель-
но ниже. Так для ИЗСД сибсовый риск - 15, для шизофрении -
около 9, для диабета типа 2 - 3,5. Естественно, чем выше от-
носительный риск, тем выше доля генетических факторов в
этиопатогенезе болезни и тем больше вероятность выявить
генетические локусы заболевания.
Первой задачей при отборе клинического материала для
эксперимента по картированию является максимальное уве-
личение относительного риска. Универсальных рецептов для
этого нет, однако, чем жестче определение болезни и чем
73
Глава 4
---------------------------с-----------------------
более однородна группа больных, тем более близким менде-
левскому и более гомогенным может оказаться признак. Од-
нородный клинический фенотип, ранний возраст проявления
болезни, семейное накопление заболевания - вот те крите-
рии, которым стоит уделить внимание.
Еще один путь улучшить перспективу генетического карти-
рования комплексного признака - сфокусироваться на редких
этнических группах или изолированных популяциях. Ведь со-
гласно теоретическим положениям и данным популяционной
генетики для таких групп характерна большая генетическая и
аллельная гомогенность, чем для больших смешанных попу-
ляций.
Принципы генетического картирования
сложнонаследуемых признаков
Современные методы картирования сложнонаследуемых
признаков и МФЗ включают 4 категории: анализ сцепления,
основанный на проверке конкретной модели наследования
болезни в родословных; метод идентичных по происхождению
(общих) аллелей, который заключается в оценке того, насколько
часто больные родственники наследуют идентичный участок
генома; исследования ассоциаций в популяциях и семьях и
генетический анализ скрещиваний модельных организмов.
Анализ сцепления
Анализ сцепления - метод генетического картирования,
основанный на прослеживании косегрегации генов при пере-
даче от родителей к потомкам в ряду поколений. Мы не пре-
следуем цель детального описания тонкостей анализа сцеп-
ления и не будем вдаваться в сопутствующие статистические
проблемы, а лишь остановимся на самых общих положениях.
За детальной информацией по методам анализа генетическо-
го сцепления читатель может обратиться к соответствующим
работам (Ott, 1991; Terwilliger и Ott, 1994),
Анализ сцепления представляет собой проверку наблюда-
емой картины сегрегации признаков и генетических маркеров
в родословной на соответствие определенной модели насле-
дования. При этом рассчитываются шансы (вероятности) за и
против (т.е. при рекомбинантной фракции о = 0.5) сцепления в
данной семье. Количественным показателем сцепления явля-
ется логарифм соотношения шансов (правдоподобия) за и
против сцепления - лод-балл (от английского logariphm of odds
ratio). Лод-балл можно вычислить для различных значений ре-
комбинантной фракции (о) - от 0 до 0.5. То значение о, при
74
Генетическое картирование МФЗ
котором лод-балл максимален и есть наиболее вероятная оцен-
ка дистанции между генетическим маркером и предполагае-
мым геном болезни.
Сцепление считается подтвержденным, если лод-балл пре-
вышает определенное пороговое значение. В генетике чело-
века для единичного наблюдения принята величина порога 3.0,
что соответствует шансам за сцепление 1000:1. На самом деле,
как отмечает Ландер (1990), эта величина не так велика как
может показаться. Ведь в общем случае для двух случайно
выбранных локусов в геноме человека вероятность оказаться
несцепленными (т.е. находиться на разных хромосомах или
плечах) соотносится с вероятностью быть сцепленными при-
мерно как 50:1. То есть реально пороговое значение 3.0 озна-
чает, что вероятность наличия сцепления всего лишь в 20 раз
выше вероятности его отсутствия или, иными словами, 1 из 20
результатов, подтверждающих сцепление, оказывается лож-
ным.
Сейчас анализ сцепления проводят используя, как прави-
ло, большое число маркеров (мультилокусный анализ сцепле-
ния) - вплоть до 300 и более, распределенных по всему гено-
му. При этом рассчитывается соотношение вероятностей для
каждого интервала между двумя соседними маркерами. Ин-
тервалы со значением лод-балла, превышающим пороговое,
и есть наиболее вероятные области локализации гена (под-
тверждающее картирование). В интервалах, для которых лод-
балл ниже исключающего порога, локализация гена исключа-
ется (исключающее картирование). Мультилокусный анализ
сцепления требует обширных вычислений, которые берут на
себя специально разработанные программы - LINKAGE {Lath-
rop, et aL, 1984), FASTMAP {Curtis и Gurling, 1993), EXCLUDE
{Edwards, 1987) и другие.
Анализ сцепления является в настоящее время основным
методом генетического картирования простых моногенных
признаков. Этим методом были картированы сотни таких при-
знаков, включая многие наследственные болезни. Анализ сцеп-
ления помог картировать и локусы некоторых сложнонаследу-
емых заболеваний. Простейшая ситуация, в которой могут быть
преодолены трудности анализа сложных признаков - анализ
сцепления в одной большой разветвленной родословной.
Именно так были картированы гены поликистоза почек {Kim-
berling etal., 1993), ранней формы болезни Альцгеймера (Schel-
lenberg et al., 1992) и псориаза {Tomfohrde, 1994).
В других случаях могут помочь те способы увеличения от-
носительного риска, о которых мы говорили выше. Так, ген
ранней формы рака груди на хромосоме 17 был картирован
75
Глава 4
методом анализа сцепления, при ограничении возраста про-
явления болезни 47 годами (Hall, 1990). При этом был достиг-
нут лод-балл 6. При включении в анализ родословных с более
поздним проявлением лод-балл опускался ниже порогового
значения.
Анализ сцепления может применяться и в случаях, когда
более чем один ген вовлечен в наследование заболевания,
при этом просто отслеживается наследование нескольких ре-
гионов генома. Так было показано взаимодействие двух локу-
сов в проявлении рассеянного склероза. В большой финской
родословной рассеянный склероз оказался сцепленным од-
новременно с локусом HLA на хромосоме бис геном основно-
го белка миелина на хромосоме 18 (Tienari et al., 1994).
В целом, примеров успешного анализа сцепления для слож-
ных признаков не так много, поскольку классическим образом
сцепление может быть подтверждено лишь в относительно
простых, близких к менделевским, случаях.
Метод идентичных по происхождению
аллелей (IBD)
Классический анализ сцепления является параметрическим
методом, т е. основывается на заранее задаваемых характе-
ристиках, таких как способ наследования и уровень пенетран-
тности. Неверно заданные параметры могут привести, соот-
ветственно, и к неверным выводам. Скажем, неверно задан-
ный тип наследования приводит обычно к переоценке реком-
бинантной фракции (Ott, 1991; Terwilliger and Ott, 1994). При
IBD-анализе информацию о сцеплении получают только на
основе наследования маркеров в парах больных родственни-
ков без априорных предположений о типе наследования и дру-
гих характеристиках. Такой непараметрический подход менее
строг и, вероятно, более продуктивен: больные родственники
должны показывать избыток общих аллелей даже при непол-
ной пенетрантности, наличии фенокопий, генетической гете-
рогенности и высокой частоте аллеля болезни.
Метод общих аллелей заключается в оценке того, насколь-
ко чаще по сравнению со случайной сегрегацией пара боль-
ных родственников наследует одну и ту же (идентичную по
происхождению, IBD - identical by descent) копию участка ге-
нома (рис. 10). Скажем, в парах дед-внук, дядя-племянник или
в случае полусибсов эти пары родственников могут иметь 0
или 1 общий по происхождению аллель по каждому локусу.
Вероятность иметь общий аллель (или IBD-статистика, aR) при
случайной менделевской сегрегации равна 1/2. Если же мар-
керный локус сцеплен с болезнью, пары больных родственни-
76
Генетическое картирование МФЗ
ков должны иметь общий аллель чаще, чем в половине случа-
ев. Значимость отличий при этом может быть оценена про-
стым хи-квадрат тестом. Для количественного признака сте-
пень фенотипического сходства в парах родственников долж-
на коррелировать с долей аллелей, идентичных по происхож-
дению. Анализ общих по происхождению аллелей возможен
для любых пар близких родственников, однако на практике
чаще всего используют пары больных сибсов. IBD-анализ в
сибсовых парах получил название ASP-метода (от affected sib-
Рис. 10. Метод идентичных по происхождению аллелей (IBD).
IBD - число аллелей общего происхождения; ар - IBD-
статистика при случайной менделевской сегрегации
(вероятность иметь один идентичный аллель для данной
пары родственников).
* Сибсы рассматриваются как две полусибсовые пары.
С помощью метода идентичных аллелей был картирован
.один из локусов диабета типа 1 на длинном плече хромосомы
11 (Davies etal., 1994), показано сцепление гипертензии с ге-
ном ангиотензина (Jeunemaitre et al., 1992, ревматоидного
артрита - с локусом HLA (Hasstedt etal., 1994), поздней формы
болезни Альцгеймера - с локусом на хромосоме 19 (Pericak-
Vance et al., 1991), а гомосексуальной ориентации - с регио-
ном Xq28 (Hamer etal., 1993). С появлением подробной гене-
тической карты стало возможным использовать IBD-метод для
сканирования всего генома. Такое сканирование было прове-
77
Глава 4
-----------------------------------------------------------
дено пока лишь считанное число раз (Berrettini, 1994). Однако,
это положение в ближайшем будущем явно изменится.
В теории метод IBD довольно прост. Однако, на практике
ситуация выглядит несколько сложнее. Главным ограничени-
ем метода общих аллелей до недавнего времени была невоз-
можность в некоторых случаях отличить аллели, идентичные
по происхождению, от аллелей, идентичных по состоянию (IBS
- identical by state). Другими словами, невозможно было опре-
делить, имеют ли два одинаковых аллеля общее происхожде-
ния, или мы имеем дело с двумя копиями аллеля разного про-
исхождения.
Для преодоления этой трудности предлагались различные
методы. Простейший заключается в том, чтобы ограничиться
только теми парами родственников, для которых IBD-статус
можно определить однозначно. Предлагалось также исполь-
зовать ожидаемое значение IBD-статистики, рассчитываемое
на основе данных по маркерам, IBD-статус которых опреде-
ляется однозначно. Другой подход основан на использовании
IBS-статистики вместо IBD (Weeks and Lange, 1988; Brown et
aL, 1994). Оба подхода на практике дают иногда хорошие ре-
зультаты, однако их существенный недостаток в том, что они
не используют всю имеющуюся информацию о наследовании
маркеров, а, следовательно, теряют в продуктивности и мощ-
ности (Bishop and Williamson, 1990). Недавно предложен алго-
ритм, позволяющий извлечь всю возможную информацию из
данного набора родословных (Kruglyak et al., 1995; Kruglyak
and Lander, 1995). При этом для каждой точки генома на осно-
ве данных по маркерам и рекомбинантной фракции рассчиты-
вается вероятность быть идентичной по происхождению в дан-
ной паре родственников. Этот алгоритм реализован в програм-
мном пакете MAPMARKER/SIBS (Kruglyak and Lander, 1995).
Хорошей иллюстрацией достоинств IBD-метода является
работа Hugot и др. (1996) - один из первых примеров приме-
нения мультилокусного ASP-анализа сложнонаследуемого при-
знака для сканирования всего генома. Им удалось картиро-
вать один из генов болезни Крона (регионального энтерита,
MIM 266600) на хромосоме 16 между маркерами D16S409 и
D16S419. Авторы использовали две независимые панели се-
мей (всего 78 сибсовых пар) и 270 высокополиморфных мар-
керов, распределенных по всему геному. Первоначальный па-
раметрический анализ сцепления не позволил картировать ген.
Максимальный лод-балл (2.04 для локуса D16S409) был зна-
чительно ниже, чем предсказанный на основе аутосомно-ре-
цессивной модели. ASP-анализ на этом же наборе родослов-
ных и маркеров позволил картировать предполагаемый ген
78
Генетическое картирование МФЗ
болезни Крона на хромосоме 16 на уровне значимости р<0.01
для обоих независимых панелей семей, несмотря на относи-
тельно небольшое увеличение относительного риска, связан-
ное с этим локусом.
Интервал, в котором картирован ген болезни Крона, имеет
размер около 4 Мб - он слишком велик для эффективного
физического картирования. Современные технологии клони-
рования позволяют идентифицировать ген, если район его
предполагаемой локализации ограничен 1 сМ, что приблизи-
тельно соответствует 1 Мб. Какова же “разрешающая способ-
ность” метода общих аллелей? Kruglyak and Lander (1995) оце-
нили мощность IBD-метода для высокоразрешающего пози-
ционного клонирования, сопоставив долю IBD-аллелей с уров-
нем относительного риска. Признак с очень высокой долей
общих аллелей (ZL) в сибсовых парах соответствует простому
менделевскому (например, при ZL~0.975, xs =40). Для карти-
рования признака с ZL=0.975 с уровнем разрешения в 1 сМ с
95% вероятностью достаточно 170 мейозов (43 сибсовых пары).
Картирование признаков с xs от 6 до 40 с таким же разреше-
нием потребует примерно вдвое большую выборку семей. При
дальнейшем уменьшении относительного риска и, следова-
тельно, доли общих аллелей, требования к размеру выборки
резко возрастают.
Ассоциации в популяциях и семьях
Исследования ассоциаций, в отличие от двух предыдущих
методов генетического картирования, основаны не на анали-
зе косегрегации генетического материала в семьях, а на по-
иске популяционных корреляций в частотах аллелей. В клас-
сическом случае они представляют собой сравнение больных
индивидов со здоровыми из той же популяции. Генетический
маркер (аллель данного гена) считается ассоциированным с
болезнью, если его частота среди больных значимо выше, чем
в контрольной выборке.
Исторически исследования ассоциаций сыграли решающую
(золь в выявлении генетической компоненты аутоиммунных и
сердечно-сосудистых заболеваний. Анализ популяционных
ассоциаций показал вовлеченность комплекса HLA в этиоло-
гию таких связанных с иммунными нарушениями состояний как
диабет типа 1, множественный склероз, ревматоидный арт-
рит, системная волчанка и позволил выявить роль наследствен-
ной вариабельности генов метаболизма липидов (аполипоро-
теина В, рецептора липопротеинов низкой плотности, класте-
ра генов аполипопротинов А1-СЗ-А4 и др.) в этиопатогенезе
атеросклероза и нарушений липидного обмена (Breslow, 1988.;
79
Глава 4
Степанов и др., 1992; 1995). Из недавних примеров ассоциа-
ций можно назвать исследования, показавшие ключевую роль
гена АРОЕ в болезни Альцгеймера (Corder, 1993), и участие
ангиотензин-конвертирующего фермента в этиопатогенезе
инфаркта миокарда (Cambien et aL, 1992). Применительно к
моногенным признакам ассоциации успешно использовались
для дальнейшего уточнения локализации гена, первоначально
обнаруженного методами анализа сцепления. Такой подход
применялся при картировании муковисцидоза, хореи Гентинг-
тона, болезни Вилсона и др. (Jorde et aL, 1994).
В основе ассоциации генетического маркера с болезнью
могут лежать три причины. Во-первых, наличие ассоциации
может свидетельствовать о том, что ассоциированный локус и
есть ген или один из генов болезни. В этом случае следует
ожидать, что положительная ассоциация будет иметь место
во всех популяциях. Во-вторых, причиной ассоциации может
быть неравновесие по сцеплению между маркерным локусом
и локусом болезни. Это возможно в том случае, когда ассоци-
ированный аллель находился на предковой хромосоме, а сам
локус расположен достаточно близко к локусу болезни, чтобы
эта корреляция не была нарушена рекомбинацией за время
существования популяции. И, наконец, ассоциация может быть
артефактом, возникшим вследствие подразделенности попу-
ляции.
Для целей генетического картирования важно отделить два
первых случая от третьего. Этого можно добиться, исследуя
ассоциации в гомогенных изолированных популяциях, исполь-
зуя внутрисемейный контроль или проводя тест на неравнове-
сие при передаче - от гетерозиготных родителей к больным
детям ассоциированный аллель должен передаваться чаще,
чем неассоциированный (Julier et al.,1991).
Наиболее перспективны для идентификации генов комплек-
сных состояний исследования ассоциаций на семейном мате-
риале (Thomson, 1995; Falk and Rubinstein, 1987). При этом
контрольную популяцию аллелей составляют аллели здоро-
вых родителей, которые не передаются больным детям. Этот
подход называется AFBAC-методом (от английского Affected
Family-Based Control) (рис. 11). В первой родословной роди-
тели имеют генотипы АВ и CD, а больной ребенок - генотип
АС. Следовательно, аллели В и D, не передавшиеся потомку,
составят контрольную популяцию. Для второй родословной в
контрольную группу войдет аллель D. Если имеется большая
выборка подобных родословных, можно сравнить частоты ал-
лелей А, В, С и D у больных и в контрольной группе и выявить
возможные ассоциации. Поскольку используются ядерные
80
Генетическое картирование МФЗ
семьи, родительские аллели, несцепленные с геном или гена-
ми болезни, будут всегда сегрегировать независимо от алле-
лей гена предрасположенности к болезни. Тем самым, устра-
няются эффекты, связанные с подразделенностью популяции,
и выявляются ассоциации только для маркеров, физически
сцепленных с локусом болезни.
Рис. 11. Поиск ассоциаций на семейном материале. AFBAC-
метод: контрольную популяцию составляют маркерные
аллели родителей, которые не передаются больным
детям.
Исследования ассоциаций на ядерных семьях позволили
подтвердить роль гена инсулина при сахарном диабете типа
1, для которого ранее были показаны ассоциации на популя-
ционном уровне (Thomson etal., 1989), и помогли выявить мар-
керы, сцепленные с полипозом толстой кишки (Hastbacka et
al.,1994). В принципе, AFBAC-метод вполне приемлем и для
полного геномного скрининга, однако примеры таких иссле-
дований нам пока не известны.
Еще одной потенциально мощной стратегией генетическо-
го картирования является поиск ассоциаций в изолированных
популяциях с небольшим числом основателей. Этот подход
называется картированием с помощью неравновесия по сцеп-
лению. Лежащая в его основе идея проста: чем более высокая
степень генетической изоляции характерна для такой популя-
ции, тем больше доля больных, унаследовавших ген болезни
от общего предка, и, соответственно, тем больше вероятность,
что причиной ассоциации является физическое сцепление
маркера с геном болезни. Ассоциацию в изолированной попу-
ляции можно интерпретировать и с точки зрения метода об-
щих аллелей. Все члены изолированной популяции являются,
в определенной степени, дальними родственниками. Сравни-
вая частоту аллелей у больных и в контрольной группе, мы,
тем самым, оцениваем долю общих аллелей у дальних ро-
81
6 Заказ № 127
Глава 4
дственников, используя не только мейозы в данном поколе-
нии, но и все мейозы в истории популяции. Сейчас в работах
по картированию наиболее активно используется в качестве
генетического изолята финская популяция. Стратегия карти-
рования по неравновесию недавно была успешно применена
для поиска гена диастрофической дисплазии (DTD) - моноге-
ного менделевского заболевания. Традиционные методы ана-
лиза сцепления позволили локализовать ген DTD в районе
размером 2 Мб, а затем анализ неравновесия по сцеплению в
финской популяции сузил этот регион до 40 Кб, что позволило
клонировать ген (Hastbacka etal., 1994). Подобная технология
применима и для сложнонаследуемых состояний, хотя доля
больных, имеющих общего предка, в этом случае должна быть
меньше. Сейчас на финской популяции ведется картирование
по неравновесию локусов подверженности к системной во-
лчанке и некоторым другим заболеваниям (Krahe et al., 1996).
Экспериментальные скрещивания
модельных объектов
Экспериментальные скрещивания животных являются од-
ним из важнейших методов генетического картирования. Воз-
можность получить большое число потомков от одного скре-
щивания и использование чистых линий полностью устраняют
проблему генетической гетерогенности. Экспериментальные
манипуляции и скрещивания позволяют легко получить кон-
генные линии, т.е. линии, отличающиеся только по одному
нужному участку генома в плоть до одного локуса. Крайне при-
влекательной для исследования мультифакториальных состо-
яний является и возможность легко унифицировать условия
окружающей среды. Все эти преимущества дают возможность
исследовать на экспериментальных объектах гораздо более
сложные генетические взаимодействия, чем это возможно на
родословных человека. Особенно ярко это иллюстрируется на
примерах разложения количественных признаков до генети-
ческих составляющих - у человека исследования такого рода
пока невозможны. Сейчас, когда созданы подробные карты
генетического сцепления мыши и крысы (Copeland etal., 1993;
Dietrich et al., 1994), стало возможным интервальное карти-
рование локусов количественных признаков. Впервые карти-
рование QTL на всем геноме было проведено при исследова-
нии эпилепсии у мышей (Rise et al., 1991) и гипертонии у крыс
(Jacob, 1991). В последнем случае эти работы послужили ос-
нованием для параллельных исследований у человека, кото-
рые выявили сцепление с гипертонией гена ангиотензин-кон-
вертирующего фермента.
82
Генетическое картирование МФЗ
Главная проблема, связанная с генетическим картирова-
нием на экспериментальных объектах, - возможность перено-
са этих результатов на человека. Существуют два пути - фун-
кциональный, т.е. исследование генетического контроля ана-
логичных метаболических путей, и позиционный - исследова-
ние участков генома человека, гомологичных соответствую-
щим мышиным.
В настоящее время существуют экспериментальные моде-
ли многих комплексных болезней человека - инсулин-зависи-
мого (de Gouyon, 1993;. Bahary, 1990) и независимого (Gau-
guier et al., 1996; Galli et al., 1996) диабета, рака (Dietrich et al.,
1993), гипертонии (Jacob, 1991), атеросклероза (Breslow, 1993),
эпилепсии (Rise etal., 1991), множественного склероза (Roder
и Hickey, 1996), астмы (de Sanctis et al., 1995), дефектов нерв-
ной трубки (Neumann, 1994) и таких поведенческих количес-
твенных признаков как предпочтение пищевых субстратов (ал-
коголя, морфина, кокаина) (Berretini, 1994; Plomin et al., 1991).
Кандидатные гены широко
распространенных заболеваний
Недавно была представлена исследовательская парадиг-
ма (рис. 12) генетической этиологии коронарного атероскле-
роза (Boerwinkle etal., 1996). Однако предложенная схема но-
сит более универсальный характер и с некоторыми нюансами
может быть основой для изучения этиологии любых МФЗ.
Представим кратко комментарии к основным разделам дан-
ной схемы.
Локализация генов. Анализ генетического сцепления и ас-
социаций с использованием сотен маркеров, охватывающих
весь геном, позволяет определить локализацию генов подвер-
женности МФЗ. Для такого анализа используются два спосо-
ба - подход "кандидатных генов” и "геномный поиск” (geno-
me-wide search). В данной главе приведены результаты ис-
следований, основанные главным образом на подходе “кан-
дидатных генов”. К сожалению, до сих пор использование пол-
ногеномного анализа сцепления для МФЗ - редкость.
Идентификация генов. Локализованные до определенного
региона гены могут быть идентифицированы, используя зна-
ние о метаболизме и патофизиологии МФЗ, позиционным кло-
нированием или комбинацией подхода кандидатных генов и
позиционного клонирования (позиционно-кандидатный под-
ход). Решение этой задачи определяется успехами в детали-
зации геномных карт. Новым мощным средством для иденти-
83
Глава 4
фикации и характеристики генов, вносящих вклад в подвер-
женность МФЗ, являются метки экспрессируемых последова-
тельностей (EST).
Локализация генов
* Идентификация генов
Возможные функциональные мутации
•
Экспериментальные системы
Предска- зание болезни Генная терапия Взаимодейст- вие «генотип - среда»
Рис. 12 Схема исследовательской парадигмы для частых
хронических заболеваний (Boerwinkle etal., 1996).
Функциональные мутации. После обнаружения кандидат-
ного гена необходим поиск вариаций кДНК в кандидатном ре-
гионе и выяснение их точной природы с помощью секвениро-
вания ДНК. На этом этапе возникает задача дифференциации
функциональных вариантов от нейтрального полиморфизма.
Трудоемкость решения этой задачи может быть очевидна из
примера - известно более 150 мутаций гена рецептора LDL
(Hobbs etal., 1992).
Экспериментальные системы. Кажется устоялось мнение о
том, что модельные животные помогают проверить и уточ-
нить эффекты отдельных генов сложных признаков и МФЗ не-
зависимо от других локусов и средовых факторов. Трансген-
ная технология дает возможность проверить эффекты изме-
Генетическое картирование МФЗ
нения числа генного продукта, манипулируя числом копий
генов. Развитие моделей МФЗ на животных позволяет тести-
ровать функциональные варианты кандидатных генов.
Результаты фундаментальных исследований в области по-
иска кандидатных генов широко распространенных болезней
человека для практикующего врача важны, если они позволя-
ют предсказать болезнь у пока здорового человека или даже у
будущего ребенка (пренатальная диагностика), эффективно
проводить этиологическое лечение, включая генотерапию,
предупреждать манифестацию болезни, зная интимные меха-
низмы взаимодействия в системе "генотип - среда". Наука и
медицинская практика еще в начале этого пути, но движение
началось и уже проверена правильность избранного направ-
ления исследований. В настоящей главе будут представлены
последние сведения о кандидатных генах МФЗ: атеросклеро-
за, артериальной гипертензии и бронхиальной астмы.
Среди генетиков расхожим стало утверждение о том, что
кардиология позднее всех других клинических дисциплин ас-
симилировала идеи "новой биологии". Но последнее пятиле-
тие и в этой области медицины ознаменовалось рождением
нового направления - генетической кардиологии, интегриру-
ющей концепции и технологии молекулярной генетики для
познания этиологии, патогенеза и механизмов клинического
полиморфизма сердечно-сосудистых заболеваний человека
(Schwartz, 1994). Начало положили исследования молекуляр-
ных основ семейных случаев ЭКГ-синдрома удлиненного ин-
тервала QT (LQTS) и кардиомиопатий. Наряду с менделирую-
щими формами патологии, к которым относятся упомянутые
болезни, активно осуществляется поиск генов подверженности
распространенным МФЗ сердечно-сосудистой системы - эс-
сенциальной гипертензии (ЭГ) и атеросклерозу. Для того, что-
бы ощутить невероятную исследовательскую активность в этом
направлении, можно сопоставить информацию, изложенную в
первой отечественной монографии по этой проблеме (Дзи-
зинский, Пузырев, 1977) с тем, что достигнуто спустя 20 лет
после ее опубликования и представлено ниже.
Эссенциальная гипертензия (ЭГ)
В отношении ЭГ до сих пор ведутся дискуссии о генетичес-
кой природе болезни. Одни предполагают, что ЭГ - простой
менделевский доминантный признак (Kutz and Spence, 1993;
Platt, 1963); другие обосновывают мультифакториальный ха-
рактер наследования ЭГ (Pickering, 1990). В связи с предпол-
ожениями о существовании большого числа генов подвержен-
ности к ЭГ широко начаты исследования по идентификации
Глава 4
некоторых из них. К середине 1996 года картировано 9 канди-
датных генов ЭГ (McKusick, 1996), они представлены в табл. 6.
Таблица 6.
Кандидатные гены эссенциальной гипертензии
№ пп Название гена, символа, признака Локализация на хромосоме MIM* Литература
1. SCNN1B (1 бета субединица эпите- лиального Na канала) 16р13-р12 600760 Shimkets et al., 1994; Voilley et al.,1995
2. SCNN1G (1 гамма субединица эпите- лиального Na канала) 16р13-р12 600761 Voilley et al., 1995
3. АРЫН(Ыа+/Н+ антипортер) 1p36.1-p.35 107310 Mattei et al., 1988
4. REN (ренин) 1q25-q32 179820 McGill et al., 1987
5. AGT (ангиотензин I) 1q42-q43 106150 Isa et al., 1990
6. АСЕ (ангиотензин I конвертирующий фермент) 17q22-q24 106180 Jeunemaitre et al., 1993, Mattei et al., 1989
7. PLA2 (панкреати- ческая фосфоли- паза А2) 12q23-q24.1 172410 Gispert et al.,1993
8. SAH (гипертензия, обусловленная геном, экспресси- рующимся в почке) 16p13.11 145505 Iwai et al., 1994, Samani et al.,1994
9. NOS 3 (эндоте- лиальная синтаза окиси азота) • 7q35-q36 163729 Marsden et al., 1993
* MIM - номер по каталогу “Mendelian Inheritance in Man”
(McKusick, 1996).
Кроме указанных в таблице, исследуются и другие участки
ДНК, с которыми можно связать регуляцию АД. Среди них -
гипотетический ген, который условно обозначили SLC (socfi-
um-litium countertransport, Na-Li противоток). Из известных как
минимум семи электрохимических каналов для катионов, пе-
ремещающихся через клеточную мембрану, для двух доказа-
86
Генетическое картирование МФЗ
но существование трех генов, детерминирующих этот процесс.
Для Na+ - гены SCNN1B и SCNN1G, для Na+/H* - ген APNH,
которые имеют отношение к ЭГ (см. табл. 6). Показано, что у
сыновей с нормальным уровнем артериального давления от
больных родителей соотношение Na-Li было значительно выше,
чем у здоровых сыновей от здоровых родителей (Woods, 1982),
Получены доказательства на семейном материале, что два
аллеля главного локуса регулируют соотношение Na-Li (Has-
stedt, 1988). Однако до сих пор картировать этот ген не уда-
лось.
Из уже известных кандидатных генов ЭГ в последнее время
особое внимание привлекает ген NOS (ген синтазы окиси азо-
та), который определяет уровень окиси азота в стенке крове-
носных сосудов, регулируемый кальмодулин-зависимым ме-
ханизмом. Идентифицировано три изоформы синтазы окиси
азота: индуцибельная, невральная и эндотелиальная. Ген эн-
дотелиальной синтазы окиси азота (NOS3), участвуя в детер-
минации ЭГ, определяет, как недавно показано, и подвержен-
ность развитию ишемической болезни сердца и курению (Wang
etai., 1996).
Benediktsson и др. (1993) отметили у человека тесную связь
гипертензии с комбинацией двух признаков - низкого веса при
рождении и большого размеры плаценты. Известно, что пла-
центарная 11-бета-гидроксистероидная дегидрогеназа (11-р-
OHSD), которая превращает глюкокортикоиды в инактивные
продукты, защищает развитие плода в норме. Наряду с дан-
ными для человека авторы показали в эксперименте на кры-
сах, что между активностью 11-p-OHSD и ростом новорожден-
ного имеется положительная, а с размером плаценты - отри-
цательная корреляционная связь. Этот интересный факт нуж-
дается в детальном исследовании.
Среди различных форм АГ со сходным фенотипом, для
которых характерно семейное накопление, солезависимость
и низкий уровень ренина (все это свойственно и ЭГ), выделя-
ют несколько, имеющих особенности клинических проявлений,
и для них картированы специфические гены. К ним относится
' синдром Лиддла (псевдоальдостеронизм с АГ), синдром лож-
ной минералокортикоидной избыточности (АМЕ - apparent mi-
neralocorticoid excess) и семейный гиперальдостеронизм, под-
дающийся лечению небольшими дозами глюкокортикоидов
(McKusick, 1996). Ген синдрома Лиддла локализован на 16р13-
р12 (Hansson, 1995), синдрома АМЕ - на хромосоме 1 (Tannin,
1991) и 16q22 (Krozowski et al., 1995), а ген гиперальдостеро-
низма, коррегируемого глюкокортикоидами, - на 8q-21e об-
ласти гена цитохрома р-450 (Mornet and White, 1989).
87
Глава 4
Атеросклероз
Сложность этиопатогенеза атеросклероза, гипотетичность
многих механизмов развития его основных клинических про-
явлений является существенным препятствием к выяснению
генетических основ развития этой “эпидемии века”. В любом
случае любая концепция атерогенеза должна объяснить на-
копление липидов в сосудистой стенке. Этим и объясняется
особенно пристальное изучение механизмов нарушений ли-
пидного обмена при атеросклерозе. По этому вопросу доста-
точно много сведений. Однако здесь приведем резюмирую-
щие данные.
Атерогенный фенотип липопротеинов (ALP) рассматрива-
ется как общий наследуемый признак, характеризующийся
преобладанием частиц липопротеинов низкой плотности не-
больших размеров (LDL, подкласс В), высоким уровнем ли-
попротеинов, содержащих повышенное количество триглице-
ридов, пониженным уровнем липопротеинов высокой плотности
и 3-кратным увеличением риска инфаркта миокарда. Показа-
но тесное сцепление между ALP и локусом рецептора LDL;
максимальный лод-балл был равен 4.07 в модели со 100%
пенетрантностью ALP (Nishina et al., 1992). Авторы высказы-
вают гипотезу, по которой ген подверженности к атероскле-
розу (ATHS) отождествляется с геном LDLR (рецептор липоп-
ротеинов низкой плотности). Во всяком случае ген ATHS рас-
полагается дистальнее D19S76 вблизи или в локусе LDLR
(19р13.2-р13.12). В табл. 7 этот ген и обозначен в качестве
кандидатного гена атеросклероза (Lindgren et al., 1985).
Представляет интерес недавняя работа Rotter и др. (1996),
в которой с использованием непараметрических количествен-
ных методов анализа сибсовых пар и пар родственников в
семьях с коронарным атеросклерозом подтверждено сцепле-
ние с локусом LDLR. Однако, не найдено сцепление с 6 други-
ми кандидатными генами: АРОВ, АРОА2, LpA, АРОЕ, липазы
липопротеинов и белка, связывающего липопротеины высо-
кой плотности. Значимое сцепление найдено с локусом СЕТР
(транспортного белка эфиров холестерина) на хромосоме 16
и с локусом SOD 1 (супероксиддисмутаза 1) на хромосоме 6.
Предполагаемое сцепление обнаружено с кластером АРОА1/
АРОСЗ/АРОА4 на хромосоме 11.
К кандидатным генам атеросклероза (см. табл. 7) относит-
ся и ген недостаточности гепаринового кофактора II (HCF2).
Антикоагуляционное действие гепарина определяется мно-
гими гепариновыми кофакторами плазмы крови. Один из них
- антитромбин III (ATIII). Гепариновый кофактор II (НС II) отли-
чается от AT III по антигенным характеристикам. Известны се-
88
Генетическое картирование МФЗ
мейные случаи низкого уровня НС II у лиц с тромбозом вен и
внутрисосудистым диссеминированным тромбозом. Описан 61 -
летний мужчина, которому 4 раза делали ангиопластику по
поводу коронарного атеросклероза с рестенозом коронарных
сосудов; гепаринотерапия была неэффективна - лишь приме-
нение специфического ингибитора тромбина определило
эффективность последней ангиопластики (Matsuo etal., 1992).
В настоящее время ген HCF2 полностью секвенирован и лока-
лизован Ha22q11 (Herzog et al., 1991).
Таблица 7.
Некоторые кандидатные гены атеросклероза
№ пп Название гена, признака MIM* Локализация на хромосоме Источники
1. FHC (LDLR); семей- ная гиперхолестери- немия (рецептор липопротеинов низкой плотности) 14389 19р13.2-р13 Lindgren et al., 1985
2. HCF 2; недостаточ- ность гепаринового кофактора II (HCF II) 14236 22q11 Herzog et al., 1991
3. АСЕ 1; ангиотензин I конвертирующий энзим 106180 17q22-q24 Jeunemaitre et al., 1993; Mattei et all, 1989
4. CBS; недостаточ- ность фермента цистатионин-бета- синтаза 236200 21q22.3 Munke et al., 1988
5., MTHFR; недостаточ- ность N(5,1 (^-мети- лен-тетрагидрофо- латредуктазы (MTHFR) 236250 1р36.3 Goyette et al., 1994
6. NOS 3; эндоте- лиальная синтаза окиси азота 163729 7q35-q36 Marsden et al., 1993
* MIM - номер по каталогу “Mendelian Inheritance in Man”
(McKusick, 1996).
Известно, что ангиотензин I конвертирующий фермент (АСЕ)
играет важную роль в регуляции артериального давления и
электролитного баланса, гидролизуя ангиотензин I в ангио-
тензин II и инактивируя брадикинин. Он также является силь-
89
Глава 4
ным вазопрессором и регулятором синтеза альдостерон-сти-
мулирующего белка. АСЕ1-ген картирован на хромосоме 17q22-
q24 и также является кандидатным геном атеросклероза (Jeu-
nemaitre et а!., 1993; Mattei, 1989). После клонирования гена
АСЕ было показано, что 50% индивидуальной изменчивости
концентрации АСЕ в плазме крови определяется инсерцион-
но-делеционным (I/D) полиморфизмом в интроне 16 гена. Orishi
и др. (1993) показали, что генотип DD связан с увеличением
риска рестеноза после ангиопластики по поводу стеноза ко-
ронарных сосудов, вызванного атеросклерозом. Частота де-
леции в 16 интроне гена АСЕ у 132 неродственных индивидов
с инсулин-независимым сахарным диабетом (NIDDM), имев-
ших инфаркт миокарда или значительный стеноз коронарных
сосудов, в сравнении со 184 индивидуумами с NIDDM без ко-
ронарной болезни существенно выше (Ruiz etal., 1994). Авто-
ры утверждают, что аллель D является сильным и независи-
мым фактором риска коронарной болезни сердца у лиц с
NIDDM. Причем раннее развитие коронарной болезни сердца
у них в этом случае не зависит от уровня липидов и артери-
ального давления. Кроме того, недавно было обнаружена ас-
социация между гипертрофией левого желудочка, установлен-
ной по ЭКГ-критериям, и DD-генотипом АСЕ (Schunkert et al.,
1994). Эта связь сильнее у мужчин, чем у женщин, и более
четко проявляется у лиц с нормальным артериальным давле-
нием. Следовательно, генотип Появляется генетическим мар-
кером, ассоциированным с повышенным риском развития ги-
пертрофии левого желудочка у мужчин в среднем возрасте.
Давно был известен факт о том, что при гомоцистинурии,
наследственном заболевании, обусловленным нарушением
метаболизма серасодержащей аминокислоты метионина
вследствие недостаточности цистатионинсинтазы печени,
среди клинических проявлений (остеопороз, арахнодактилия,
артрит, эктопия хрусталика, умственная отсталость), наблю-
дается тромбоз артерий и вен. В 90-е годы была показана
положительная связь между недостаточностью термолабиль-
ной М(5,10)-метилентетрагидрофалат-редуктазы (MTHFR) и
развитием коронарной болезни сердца. У лиц с коронарной
болезнью сердца по сравнению с контрольной группой выяв-
ляется недостаточность термолабильной MTHFR (Krang et al.,
1991). Средний уровень гомоцистеина у лиц с недостаточ-
ностью термолабильной MTHFR был существенно (почти в 2
раза) выше такового у здоровых лиц. Среди 16 облигатных
гетерозигот с недостаточностью термолабильной MTHFR об-
наружено 4 человека, у которых активность этого фермента в
лимфоцитах составила менее 25% от нормальной. Биохими-
ческое обследование этих лиц позволило обосновать пред-
90
Генетическое картирование МФЗ
положение авторов о том, что эти индивиды являются генети-
ческими компаундами по аллельным мутациям термолабиль-
ной MTHFR и термостабильной MTHFR. Goyette et al. (1994)
локализовали ген MTHFR на 1р36.3.
Из недавних работ отметим исследование Kozich и др.
(1995), в котором показано, что 30% лиц с преждевременной
коронарной болезнью, имеющие гипергомоцистинурию, явля-
ются гетерозиготами с низкой активностью фермента циста-
тионин-бета-синтазы (CBS). Связь гипергомоцистинурии с
развитием атеросклероза была показана во многих работах
(Mudd, 1985), Более того, был известен весьма любопытный
факт “устойчивости” сосудов к атеросклеротическому пора-
жению у больных с синдромом Дауна (трисомии по 21 хромо-
соме) - его связывали с высокой активностью у них CBS (Mur-
doch et aL, 1977). Krauss et al. (1986) показали, что средняя
активность CBS в культуре фибробластов от больных с син-
дромом Дауна составляет 166% по сравнению с контролем.
Именно эти наблюдения позволили сосредоточить внимание
на исследовании 21 хромосомы как места возможной локали-
зации для гена CBS. Методом гибридизации соматических
клеток ген CBS картирован на 21q22.3 (Мипке et al., 1988).
Ранее упоминалась роль гена NOS3 (эндотелиальная син-
таза окиси азота) в качестве кандидатного для ЭГ. Эти сведе-
ния противоречивы. Так, недавняя работа (Bonnardeaux et al.,
1995) не обнаружила ассоциации ЭГ с высокополиморфными
(СА)П повторами в интроне 13 и двумя биаллельными марке-
рами в интроне 18 NOS3 гена. В то же время появились сведе-
ния о роли этого гена в этиопатогенезе коронарного атерос-
клероза при исследовании 27-членных повторов в 4-ом ин-
троне 5 конца NOS3 f-ена (Wang et aL, 1996). Было идентифи-
цировано 2 аллеля: большого размера (его частота 0.830) и
меньшего (его частота 0,170). Более крупный аллель имел 5
тандерных 27-членных повторов. Меньший аллель, в котором
явно отсутствовал третий повтор, оцененный минорным раз-
личием в нуклеотидной последовательности, имел только 4
повтора. При обследовании 549 лиц с коронарной болезнью и
^53 здоровых, но курящих (или куривших в прошлом), был об-
наружен значительный избыток гомозигот по редкому аллелю
у больных с тяжелым стенозом артерий. Этот генотип также
ассоциирован с инфарктом миокарда. Авторы отмечают, что
поскольку эндотелиально-зависимая вазодилятация опосре-
дуется освобождением NO, определяемой активностью эндо-
телиальной синтазы окиси азота, то повышенный риск коро-
нарной болезни у курильщиков, гомозигот по редкому алле-
лю, связан с предрасположением к эндотелиальной дисфун-
кции.
91
Глава 4
Бронхиальная астма
Сложность этиопатогенеза бронхиальной астмы (БА), вклю-
чающего взаимодействие трех основных его составляющих
(иммунологическая и воспалительная компоненты, а также
нейрогенный контроль) делает наиболее вероятной гипотезу
о мультифакториальном наследовании этого заболевания. Это
означает, что число генов, формирующих наследственную ос-
нову болезни достаточно важно. Рассмотрим “похромосом-
но” те из них, которые первыми “вышли в кандидаты” на
основании предположения об участии продуктов этих генов в
формировании иммуно-воспалительной и нейрогенной ком-
понент патогенеза астмы и атопии (табл 8).
Хромосома 5, К этой хромосоме у исследователей БА осо-
бенно повышен интерес. В участке 5q31-5q33 сосредоточены
гены, которые по своим функциональным характеристикам
обоснованно могли бы иметь отношение к БА: генный кластер
интерлейкина-4 (IL4), ген Ь2 адренорецептора (ADRB2), а
также (ближе к дистальному району длинного плеча хромосо-
мы) ген лейкотриена С4 синтазы или рецептора кортикосте-
роидов (GCCR). Действительно, в ряде работ были показаны
ассоциации и генетическое сцепление указанных локусов и
астмы. Так, при исследовании нидерландских семей выявле-
но сцепление маркеров около гена IL4 и гена ADRB2 с уров-
нем общего иммуноглобулина Е (IgE), бронхиальной гиперре-
активностью (BHR) и БА (Meyers etaL, 1994). На этих же семь-
ях (303 детей и внуков от 84 пробандов с БА), отобранных из
однородной популяции Postma et al. (1995), обнаружили кор-
реляцию между уровнем общего IgE и чувствительностью
бронхов к гистамину. Сибсовым методом авторы установили
связь между BHR и генетическими маркерами области 5q31-
q33, предварительно показав сцепление этого локуса с регу-
ляцией уровня общего IgE. В сибсовых парах, конкордантных
по BHR, получена тесная корреляция BHR и уровня IgE в це-
лом по исследуемой группе лиц. Анализ сибсовых пар пока-
зал сцепление BHR лишь с отдельными генетическими мар-
керами 5q, включая D5S436. Авторы резюмировали, что ген,
регулирующий BHR, локализован вблизи главного локуса, ко-
торый определяет уровень IgE в сыворотке крови.
Еще более впечатляющие результаты были получены при
изучении вариантов (полиморфизмов) р-адренорецептора. Так
аллель Arg 16 ассоциировал с ночной астмой (Turki et al., 1995),
Glu 27 аллель c BHR; Glu 16 гомозиготы проявляют более низ-
кий бронходилятаторный эффект на салбутамол; самая сла-
бая бронходилатация - у Arg 16 гомозигот (Hall, 1997).
Генетическое картирование МФЗ
Таблице 8.
Кадидатные гены бронхиальной астмы и атопии
№ пп Название гена, признака Локализация на хромосоме MIM* Источники
1 BHR1; ген гипер- реактивности брон- хиального дерева 5q 600807 Postma et al., 1995
2 ADRB2R’; ген бета-2 адренергического рецептора 5q32-q34 109690 Kibilka et al., 1987; Yang-Feng et al., 1990
3 JL4; ген интерлей- кина-4 (син.: кластер генов интерлейкина 4 - IL36 -4, -5, -9, -13, GM-CSF) 5q31-5q33 147780 Meyers et al., 1994
4 TNFA; ген альфа фактора некроза опухоли 6р21.3-р21.1 191160 Jacob, 1992; Spies et al., 1986
5 HLA-DR; ген HLA комплекса 6p 142860 Holgate, 1997
6 IGEL; ген уровня общего иммуно- глобулина Е 11q12-q13 147050 Cookson et al., 1989, 1992
7 FCERIb; ген бета иммуноглобулиново- го рецептора тучных клеток 11q12-q12 147138 Sanford et al., 1993; Shirakawa et al., 1994
8 IGIF; ген гамма интерферона 12q15-q24.1 600953 Barnes et al., 1996
9 NOS1; ген невраль- ной синтазы окиси азота 12q24.2-q24.3 163731 Meyers et al., 1994
10 ESD; ген эстеразы D 13q14.2-q14.3 133280 Holgate, 1997
11 TCRA; ген альфа цепи антигенного рецептора Т-клеток 14q11-14q12 186880 Maffatt et al., 1994
* MIM - номер по каталогу “Mendelian Inheritance in Man”
(McKusick, 1996).
Таким образом, имеется множество фактов, свидетель-
ствующих о том, что в хромосоме 5 локализуются по крайней
мере три гена БА: BHR1, ADRB2R и IGEL (ген уровня общего
IgE; синоним: ген атопии).^ отношении последнего парамет-
ра (IgE) следует отметить, что уровень IgE является сложно-
Глава 4
наследуемым признаком, а, значит, на хромосоме 5 распо-
ложен лишь один ген из многих, которые определяют концен-
трацию IgE в сыворотке крови, а, значит, и атопию.
Хромосома 6, Внимание к анализу этой хромосомы в ка-
честве кандидатной для БА естественно, так как на ней рас-
положены гены HLA-комплекса. Действительно, 20 лет назад
была показана тесная ассоциация HLA W6 с врожденной ас-
тмой (Bottazzo and Lendrum, 1976); и эта работа определила*
исследования по проверке гипотезы о доминантном исследо-
вании БА и атопии. Теперь ясно, что гены антигенного распоз-
навания являются важными кандидатами, и имеются сообще-
ния по нескольким специфическим антигенам: Amb aV при
сенной лихорадке; DqB1‘0101 при БА, вызываемой аллергена-
ми клеща домашней пыли; TDI (DQBT0503) при профессио-
нальной астме; DQ2 при астме, провоцируемой аспирином и
некоторыми другими лекарствами (Holgate, 1997).
В настоящее время активно исследуется роль а-фактора
некроза опухоли (TNFA), являющегося провоспалительным
цитокином, в связи с его центральной ролью (и близкого ему
TNFB) в иммунном и воспалительном ответе. Показана не-
адекватная продукция TNFA при целом ряде иммуновоспали-
тельных состояний, но и в норме существует заметная ме-
жиндивидуальная вариабельнность. У здоровых лиц высо-
копродуцирующие TNFA-фенотипы тесно корреллируют с
геном HLA-DR3 и DR4, которые локализованы на хромосоме
6р21.3-р21.1 (Jacob, 1992; Spies et al., 1986). Более того, пока-
зано, что некоторые мутации гена TNFA приводят к измене-
нию уровня TNFA. В настоящее время известно несколько
мутаций G_>A в промоторно-энхансорном регионе гена TNFA
в нуклеотидах -308, -238, -376, -163 (D’Alfonso and Richiardi,
1994;. Hamann et al., 1994). Для одной из них - TNFA 308A
показано, что гомозиготы по этому аллелю имеют относитель-
ный риск развития малярии в 7 раз выше, чем в среднем по
популяции (McCuize et al., 1994). К БА может иметь отноше-
ние мутация LTaNcol гена TNFA, при которой обнаружена
ассоциация между изменением в а-цепи Т-клеточного ре-
цептора и ответом на антиген клеща домашней пыли DerP2
(Cambell, 1995).
Хромосома 11. Астму, сенную лихорадку (аллергический
насморк) и экзему объединяют часто общим термином - ато-
пия, и развитие ее связывают с IgE-зависимым ответом на
взаимодействие распространенных аллергенов окружающей
среды. Десятилетняя исследовательская работа по поиску
генов, ответственных за уровень IgE крови, завершилась обос-
нованием их локализации в регионе 11q12-q13 (Cookson et
al., 1989; 1992). Эта группа исследователей располагала вы-
Генетическое картирование МФЗ
боркой из 239 лиц, принадлежащих к 40 семьям, и тремя об-
ширными родословными. У 9% детей с атопией в ядерных семь-
ях был обязательно болен один из родителей. В анализиро-
ванных родословных - в каждой была вертикальная передача
болезни и у 66% потомков от браков, в котором один из роди-
телей был болен, была атопия. Методом генетического сцеп-
ления атопии с гипервариабельными минисателлитными про-
бами из указанного выше региона хромосомы 11 были полу-
чены высокие оценки связи (лод-балл 6.39).
Эта же группа исследователей из Оксфордского универси-
тета (Cookson et al., 1992) попытались дать "генетическое”
объяснение известному факту о том, что риск атопии выше
для детей, у которых больны матери, чем для тех, кто имел
больного отца. Они нашли различия в распределении отцов-
ского и материнского 11 q13 локуса среди пар сибсов в семь-
ях, пораженных атопией. В 62% пораженных атопией сибсо-
вых пар присутствовали материнские аллели локусов 11 q13 и
D11S97. Это распределение отличалось от теоретически
ожидаемого 50/50 (Р=0,001). Отцовское наследование алле-
лей (46% и 54%) по распределению не отличалось. Авторы
ставят вопрос о необходимости объяснения установленного
факта, по крайней мере, в рамках двух гипотез: геномного
импринтинга, либо материнской модификации иммунного от-
вета.
Накапливаются данные о многообразии и сложности пато-
логических путей дисрегуляции иммуновоспалительного про-
цесса при атопии и БА. Оказалось, что существенную роль в
формировании патологии играет высокоспецифичный Fc ре-
цептор IgE тучных клеток - FcoR1. Sandford и др. (1993) пока-
зали, что в активно исследуемом интервале 11q, содержащем
ген атопии, располагаются два гомологичных гена - CD20 и р-
субъединицы FcoR1. Однако вскоре Shirakawa и др. (1994)
отклонили роль полиморфизма гена CD20 в развитии атопии
у детей, а детальное изучение ими полиморфизма р субъеди-
ницы FcoR1 привело к новым интересным фактам. У случайно
отобранных индивидов с атопией в 13% случаев отмечалась
замена Leu 181, и примерно у 50% из тех, кто имел атопию,
эта замена имела однородительское происхождение. 17% лиц
из 60 ядерных семей имели вариант Leu 181, унаследованный
от матери. В недавней работе эти же авторы (Shirakawa et al.,
1994), обнаружившие связь Leu 181 и других вариантов FcoR1 b
с атопией в британской и австралийской популяциях, не на-
шли подобной связи для жителей Японии.
Хромосома 12. В работах по идентификации генов, опре-
деляющих подверженности и особенности клинических про-
явлений атопии, включая БА, расположенных на 12q15 -q24.1,
Глава 4
внимание привлекают несколько. Среди них: интерферон - g
(IFNG), стволовой клеточный фактор (SCF; известный и как
фактор роста тучных клеток - MGF), инсулиноподобный ро-
стовой фактор-1 (IGF1) и р-субединица нуклеарного фактора
Y (NFYB). Недавно представлены результаты исследования
методом сибсовых пар 29 полных африкано-карибских семей
(общее число обследованных - 507 человек) и 24 ядерныхсе-
мей из популяции амишей (общее число обследованных,-. 209
индивидов), в котором использовалось около 20 генетических
маркеров на указанный выше участок хромосомы 12 (Barnes
etal., 1996). Было подтверждено, что регион 12 q15-q24.1 со-
держит ген(ы), контролирующий(ие) развитие астмы, ассоци-
ированной с высоким уровнем общего IgE сыворотки крови.
Дополнительно к перечисленным генам этого региона была
выявлена связь с геном лейкотриена А4 гидролазы (LTA4H),
расположенного также в этом участке хромосомы.
Хромосома 12 представляет интерес для исследователей
БА и тем, что на ней располагается ген конститутивной фор-
мы синтазы окиси азота (cNOS), названный NOS1. Окись азо-
та (NO; N=O) в 1992 году была названа “молекулой года” (Сио-
itta and Koshiand, 1992). Теперь ясно (см. обзор Nakaki, 1994),
что давно известный эндотелий-производный релаксирующий
фактор (EDRF) по сути представляет собой NO, образующую-
ся из L-аргинина при участии NO-синтазы (NOS). NOS имеет
уникальную структуру, ферментные свойства которой опреде-
ляются несколькими кофакторами (донорами электронов) -
FMN, FAD, кальмодулином, гемом и тетрагидробиоптерином.
Выделяют две формы этого фермента - cNOS и iNOS (индуци-
бельная NO синтаза). Форма cNOS обнаружена в эндотели-
альных клетках, тромбоцитах и мегакариобластах, в клетках
бронхиального эпителия, мозжечка, парасимпатических нервов
и macula densa, в нейронах пениса, иннервирующих каверноз-
ное тело, и в нервных сплетениях артерий пениса; iNOS широ-
ко представлена в макрофагах, эндотелии сосудов и гладко-
мышечных клетках, кардиомиоцитах, хондроцитах, глиальных
и купфферовских клетках. NO активирует гуаниновую циклазу
и стимулирует продукцию цГМФ. Показано значение NOS в
регуляции тонуса сосудов и артериального давления, аггре-
гационных свойств крови, в патогенезе атеросклероза, дила-
тационных кардиомиопатий, нарушений мозгового кровооб-
ращения, дисфункции сосудов сетчатки глаза, аллергическо-
го энцефалита, юношеского пилоростеноза, нарушении пол-
овых функций и сексуального поведения, регуляции транспорта
электронов митохондрий и энергетического обеспечения фун-
кций многих органов и систем, включая иммунную (Chung et
aL, 1996;. Nakaki, 1994; Nussler and Billiar, 1993).
Генетическое картирование МФЗ
К настоящему времени известны гены различных форм NOS
и их локализация на хромосомах: NOS1 - 12q24.2-q24.31;
NOS2A, NOS2B, NOS2C - в интервале 17p13.1-17q25; NOS3 -
7q35-q36 (см. McKusick, 1996). К первому из них (NOS1) при-
влечено внимание исследователей атопии и БА (Holgate, 1997).
Хромосомы 13 и 14. Обсуждается значение еще двух ло-
кусов, отнесенных к кандидатным генам атопии и БА, - ген
эстеразы D (13q14.2-q14.3) и ген а-цепи антигенного рецеп-
тора Т-клеток (TCRA) на 14q11-14q12, исследованный Maffatt
и др. (1994).
В настоящее время представляет интерес поиск гена им-
муноглобулин Е (1дЕ)-зависимого гистамин-освобождающего
фактора. Сам фактор получил обозначение HRF (Histamine-re-
leasing factor), а гену HRF присвоен номер MIM 600763 (Maffatt
et al., 1994;. McKusick, 1996). Однако ген до сих пор не клони-
рован и не локализован. Известно только, что продуцируемый
лимфоцитами у детей с атопией и присутствующий в биологи-
ческих жидкостях пациентов с аллергией, HRF имеет белко-
вую природу, показана его близкая гомология к мышиному
белку - р21, и описан его человеческий гомолог р23 (Bohm et
al.,1991). По-видимому перечисленные выше гены подвер-
женности БА - лишь небольшая часть в структуре ее генети-
ческой компоненты. Вся эта информация получена с исполь-
зованием подхода кандидатного картирования. Дополнитель-
ную информацию по “генам астмы” дает подход позиционного
клонирования, когда локализация гена конкретной болезни
(признака) устанавливается в отсутствие знаний о функции ге-
нетического маркера, используемого с этой целью, но уже кар-
тированного ранее. Недавно такое исследование было прове-
дено сотрудниками четырех центров, объединивших 10 науч-
ных коллективов США (A genome-wide search for asthma sus-
ceptibility loci in ethnically populations. The Collaborative Study on
the CSGA., 1997). Это впервые в мире осуществленный полно-
геномный поиск генов подверженности астме у человека.
Авторы анализировали сибсовые пары больных астмой в
140 семьях из трех этнических групп, проживающих в США -
африканцев (п-43), европеоидов (п=79) и испанцев (п=18).
Всего было 380 пораженных индивидов: 117 африканцев, 215
европеоидов и 48 испанцев. Многоточечный анализ сцепле-
ния проводился с использованием 360 полиморфных корот-
ких тандемных повторов (STRs) со средним расстоянием меж-
ду соседними маркерами в 10 сМ. В этом исследовании была
подтверждена связь астмы с известными ранее генами у ев-
ропеоидов: 5q23-3l; 6р21.3-23; l2q14-24.2. У африканцев ни
с одним из известных кандидатных генов сцепления с астмой
не было обнаружено. В >о же время были выявлены новые
участки ДНК, которые показывали генетическое сцепление с
астмой: у африканцев - 5р15; 17p11.1-q11.2; у европеоидов -
11р15; 19q13; у испанцев - 2q33; 21q21. Эти результаты под-
твердили, что количество и относительная важность генов под-
верженности БА может варьировать в различных этнических
группах, и эти генетические различия вместе с особенностя-
ми средовых факторов могут лежать в основе межпопуляци-
онной вариабельности заболеваемости бронхиальной астмой.*
Таким образом, изложенное в этой главе позволяет кон-
статировать, что современная генетика достигла такого уров-
ня развития фундаментальных знаний, ДНК-технологий, мате-
матических и компьютерных моделей, который позволяет на-
чать разгадку одной из самых интригующих головоломок со-
временной науки - генетических основ сложнонаследуемых
заболеваний человека. Отнюдь не исчерпывающий список МФЗ
и признаков, генетическое картирование которых находится в
состоянии разработки, включает сердечно-сосудистые забо-
левания (атеросклероз, гипертония, нарушения липидного
обмена), болезни аутоиммунной природы (диабет, астма, рев-
матоидный артрит), многие формы рака, психические заболе-
вания (шизофрения, маниакально-депрессивный психоз, эпи-
лепсия), поведенческие признаки. При этом применяется весь
спектр описанных в этой главе стратегий. И хотя завершенных
примеров картирования пока не так много, в самом ближай-
шем будущем следует ожидать настоящего прорыва, револю-
ции в картировании генов, вовлеченных в наследование ком-
плексных заболеваний и признаков человека. Именно эта за-
дача является сейчас одним из главных приоритетов в миро-
вом генетическом сообществе. Значение ее для медицины и
биологии трудно переоценить. Ведь именно картирование и
клонирование гена является тем узким местом, тем “горлыш-
ком бутылки”, пройдя через которое, можно выйти на широ-
кое поле биомедицинских приложений, включая понимание
физиологических механизмов болезни, диагностику, терапию
и превентивную медицину.
Глава 5
НЕТРАДИЦИОННОЕ НАСЛЕДОВАНИЕ
Клиницисту, врачу-генетику хорошо знакомы ситуации, ког-
да тот или другой анализируемый случай наследственной
(врожденной) патологии не попадает под традиционное гене-
тическое объяснение - хромосомная аномалия, моногенная
мутация или мультифакториальная болезнь. По мнению Hall
(1996) лишь для трети всех больных с наследственной патоло-
гией можно дать обоснованное генетическое объяснение в
рамках канонических форм наследования. Она же замечает,
что только треть всех признаков гороха - объекта, при изуче-
нии которого Мендель сформулировал законы передачи на-
следственной информации, подчиняются этим законам.
Явления, противоречащие менделевскому признаку экви-
валентности реципрокных скрещиваний, согласно которому у
потомства ген действует одинаково независимо от того, от
кого из родителей он унаследован, известны давно. Это пре-
жде всего признаки, определяемые генами, расположенными
в половых хромосомах, и неядерными генами (у человека -
митохондриальный геном). Относительно недавно стали из-
вестны новые эффекты родительского происхождения: геном-
ный импринтинг, однородительская дисомия, аллельная эк-
спансия, соматическая рекомбинация и некоторые другие (Hall,
1996). Среди неканонических генетических эффектов привле-
кает внимание новый класс мутаций - динамических мутаций,
обусловленных экспансией числа тринуклеотидных повторов,
лежащей в основе развития некоторых нейродегенеративных
заболеваний. Все эти новые явления объединяются сейчас
единым названием - “нетрадиционное наследование”. Иллюс-
трацией степени интереса к этим формам наследования мо-
жет служить одно событие, свидетелем которого был один из
авторов, участвовавший в работе 8 Международного конгрес-
са по генетике человека (Вашингтон, 1991): эта тема привлек-
ла так много генетиков в лекционный зал симпозиума, что
местные пожарные угрожали закрыть конгресс, пока избыток
публики не был распределен по двум дополнительным залам.
Изучение нетрадиционных способов наследования пред-
ставляет не только академический интерес, но имеет прямое
отношение к генетическому консультированию. Оценки риска
повторного появления анализируемого заболевания с немен-
делевским наследованием должны делаться с осторожностью
Глава 5
- этот риск может быть выше. Затрудняющим консультирова-
ние моментом является и то, что практически отсутствуют све-
дения о популяционной частоте болезней, для которых нетра-
диционное наследование составляет основу их развития.
Геномный импринтинг и
болезни импринтинга*
В генетическом смысле термин “импринтинг” впервые был
применен в 1960 году Х.Кроуз из Колумбийского университе-
та в США для описания селективной элиминации отцовских
хромосом у насекомых (Crouse, 1960). Долгое время этого
явления не замечали и лишь к середине 80-х годов интерес к
нему вспыхнул вновь, когда в нескольких лабораториях, ко-
торые экспериментировали с эмбрионами мышей, были полу-
чены новые факты, подтверждающие существование геном-
ного импринтинга (ГИ) млекопитающих. В частности, этим яв-
лением была объяснена селективная инактивация отцовской
Х-хромосомы у млекопитающих, а именно: в клетках произ-
водных трофобласта и внезародышевой энтодермы, начиная
со стадии бластоцисты, обнаруживается избирательное вы-
ключение Х-хромосомы отцовского происхождения, причем
этот эффект является тканеспецифичным, т.е< он ограничен
только провизорными органами, а непосредственно в тканях
зародыша имеет место равновероятная инактивация отцовс-
кой или материнской Х-хромосомы (Lyon and Rastan, 1984).
Факт существования такой тканеспецифичной памяти роди-
тельского происхождения Х-хромосомы свидетельствует о су-
ществовании специального механизма, детерминирующего ГИ,
и ставит ряд вопросов о его сущности, распространенности,
эволюции и т.д.
Под геномным импринтингом понимают эпигенетический
процесс, дифференциально маркирующий материнские и от-
цовские хромосомы, который приводит к разному фенотипи-
ческому проявлению мутаций у потомства, унаследованных от
матери или отца. Обычно у организмов имеется биаллельная
экспрессия генов, и лишь в участках генома, подверженных
импринтингу, экспрессируется только один отцовский или ма-
теринский аллель, т.е. наблюдается моноаллельная экспрес-
сия импринтированных генов. Установлено, что в основе ГИ
лежат специфические структурно-молекулярные изменения
отдельных участков хромосом, происходящие во время фор-
мирования мужских и женских половых клеток, которые при-
водят к стойким функциональным различиям экспрессии го-
* Раздел написан в соавторстве с проф. С.А.Назаренко
Нетрадиционное наследование
мологичных генов у потомства. Точные механизмы, лежащие в
основе ГИ, неизвестны, однако основную роль в этом процес-
се отводят специфическому метилированию цитозиновых ос-
нований ДНК, которое выключает транскрипцию генов (Razin
и Cedar, 1994). Вследствие ГИ для нормального развития ор-
ганизма в его диплоидном хромосомном наборе должны обя-
зательно присутствовать два разных гаплоидных набора хро-
мосом отцовского и материнского происхождения. Отклоне-
ние от этой закономерности, прежде всего по хромосомам,
содержащим импринтированные участки генома, влечет за со-
бой нарушение нормального процесса развития.
Особый интерес приобретает проблема генетического им-
принтинга в связи с наследственной патологией человека.
Примеров заболеваний, в основе этиологии которых лежит
нарушение функционирования импринтированных участков
генома, накопилось уже настолько много, что можно говорить
об особом классе болезней генетического импринтинга (Hall,
1990; Пузырев, 1995; Назаренко и Никитина, 1997).
Связь феномена ГИ с этиологией ряда наследственных
болезней человека можно проследить на разных уровнях ор-
ганизации генетического материала. Так, на уровне генома
доказательство роли ГИ в патологии дает анализ фенотипи-
ческого проявления триплоидий. Триплоиды, имеющие соот-
ношение гаплоидных наборов хромосом отца и матери как 2:1
(диандрические триплоиды), обладают сильно развитой кис-
тозной плацентой (частичный пузырный занос) с задержкой
развития собственно эмбриональных структур, в то время как
при обратном соотношении гаплоидных наборов родителей
{дигенические триплоиды) имеет место сильное угнетение
роста плацентарных тканей и отсутствие ее кистозной дегене-
рации. В то же время при удвоении гаплоидного набора от-
цовских хромосом и полном отсутствии хромосом матери у
зародышей с псевдонормальным кариотипом отцовского про-
исхождения (полный пузырный занос) наблюдается более вы-
раженное кистозное перерождение ткани хориона по сравне-
нию с диандрическими триплоидами, часто приводящее к ее
злокачественной трансформации - хорионэпителиоме (Szul-
man, 1984). Этот анализ показывает неэквивалентность фун-
кционирования гаплоидных наборов отцовских и материнских
хромосом у зародыша с наличием существенного вклада ге-
нома отца в развитие трофобласта и большего вклада генома
матери в развитие собственно зародышевых структур.
Связь феномена ГИ с патологией человека на уровне от-
дельных хромосом набора можно проследить на таком инте-
ресном явлении как одноцрдительская дисомия (ОРД). Под ней
понимается ошибочное происхождение диплоидии по отдель-
Глава 5
ной хромосоме или ее части из одного родительского источ-
ника у зародышей или организмов с "нормальным” кариоти-
пом. Существуют 2 типа ОРД: а) изодисомия, возникающая
при нерасхождении хромосом во II делении мейоза, причем
обе хромосомы являются редупликационными копиями и име-
ют идентичную последовательность ДНК с полной гомозиго-
тизацией хромосомного материала; б) гетеродисомия, нерас-
хождение хромосом в I мейотическом делении, с неидентич-
ными гомологами. Возможные механизмы возникновения ОРД
по целым хромосомам набора - гаметная комплиментация,
"коррекция" три- или моносомии и соматическая рекомбина-
ция.
Комплиментация гамет - дополнение нуллисомной по оп-
ределенной хромосоме набора одной гаметы дисомной по
этой же хромосоме другой гаметы. Несмотря на низкую веро-
ятность такого события, оно все же не является редким. Дей-
ствительно, если рассмотреть частоту хромосомных аберра-
ций в сперматозоидах и яйцеклетках, которая была оценена
на уровне 10 и 30%, соответственно, (Martin et а!., 1983; Martin
et al., 1986}, то на популяционном уровне частота ОРД стано-
вится достаточно высокой, порядка 1 на 3000 зигот (Engel и
DeLozier-Blanchet, 1991}.
"Коррекция" трисомии - редукция трисомии до дисомии
путем потери сверхчисленной хромосомы, происходящей от
одного родителя, с сохранением двух хромосом другого ро-
дителя. Редукция трисомии может происходить как при не-
расхождении хроматид в метафазе, так и при отставании хро-
мосомы в анафазе во время первого деления зиготы или в
более позднее время, с сохранением первоначальной хромо-
сомной конституции зиготы в части клеток. Коррекция моно-
сомии - дупликация единственной хромосомы, полученной от
одного родителя, при отсутствии гомологичной хромосомы
другого родителя.
Соматическая рекомбинация - обмен между хроматидами
гомологичных хромосом в соматических клетках. Этот меха-
низм ведет к ОРД по отдельным хромосомным сегментам.
Скорее всего, именно он отвечает за формирование отцовс-
кой ОРД по участку 11р15.5 в некоторых случаях синдрома
Видемана-Беквита (Henry et al., 1991}.
Как же соотносится одно родительская дисомия с генети-
ческим импринтингом? Если хромосома содержит импринти-
рованные участки, то, вследствие ОРД, аллели, локализован-
ные в этих участках, могут быть экспрессированы или супрес-
сированы, в зависимости от родительского происхождения
ОРД, что приводит к патологии развития организма. Если же
хромосома не содержит импринтированных участков генома,
Нетрадиционное наследование
то у индивидов, имеющих ОРД по данной хромосоме набора,
аномалий фенотипа не возникает.
Концепция ОРД в приложении к человеку и проблемам ме-
дицинской генетики была предложена в 1980 году Э.Энгелем
(Engel, 1980). С момента ее обоснования эта аномалия хромо-
сом описывается у все большего числа пациентов. Имеется
несколько групп индивидов с повышенным риском возникно-
вения ОРД: 1) аномальные носители семейных сбалансиро-
ванных транслокаций или центрических слияний (робертсо-
новских транслокаций); 2) носители мозаичных трисомий; 3)
плоды с пренатально подтвержденным диагнозом ограничен-
ного плацентарного мозаицизма.
Всего у человека возможно существование 47 типов одно-
родительского наследования целых хромосом: 44 типа ОРД
по 22 аутосомам от каждого родителя - материнская (мат.) и
отцовская (отц.) и 3 типа ОРД по половым хромосомам. Сле-
дует также указать на возможность возникновения неопреде-
ленного числа дупликаций сегментов хромосом однородитель-
ского происхождения.
Впервые ОРД у человека была продемонстрирована в 1988
г., когда Спенс и др. (Spence et al., 1988) у девочки с муко-
висцидозом и задержкой роста с помощью молекулярно-ге-
нетических маркеров обнаружили униродительскую дисомию
по хромосоме 7 материнского происхождения (ОРД7мат) при
отсутствии хромосомы 7 отца. Вскоре Voss et al. (1989) описа-
ли еще одного подобного пациента. Третий аналогичный слу-
чай подтвердил связь между ОРД7мат (изодисомией) и отста-
ванием в росте. Эта находка стимулировала специальный по-
иск ОРД в выборке пациентов с синдромом Рассела-Сильве-
ра (Kotzot et al., 1995). Это синдром неизвестной этиологии,
включающий значительное пре- и постнатальное отставание в
росте как основную черту. Кроме того, у больных отмечается
также гемигипертрофия, латеральная асимметрия, непропор-
ционально большая голова с широким лбом и сужающейся ни-
жней частью лица, синдактилия пальцев на ногах и ряд других
аномалий фенотипа. ОРД хромосомы 7 была обнаружена у 4
из 30 пациентов, т.е. была подтверждена высокая частота
ОРД7мат у больных с данным синдромом.
К настоящему времени накоплен большой объем факти-
ческого материала по механизмам формирования ОРД, кон-
кретным хромосомам, вовлеченным в ОРД у человека, и фе-
нотипическим эффектам отцовской и материнской ОРД по дан-
ным хромосомам, эффектам импринтинга, обнаруживаемым
через ОРД, и стратегии определения ОРД в практике клини-
ческой генетики.
Г лава 5
При определении фенотипических последствий ОРД нужно
принимать во внимание три следующих момента:
Проявление аутосомно-рецессивного заболевания
вследствие гомозиготизации хромосомного материала при
изодисомии;
Влияние мозаицизма по аномальному клеточному клону
плаценты или плода на фенотип. Так, при исследовании 10
случаев ограниченного плацентарного мозаицизма по хромо-
соме 16 не было обнаружено корреляции между весом при
рождении и ОРД, но была выявлена прямая зависимость меж-
ду процентом трисомных клеток в плаценте и весом ребенка
(Kalousek and Barrett, 1994).
Фенотипический эффект импринтинга. При наличии им-
принтинга аллель, полученный от родителя одного пола, функ-
ционально неравен аллелю, полученному от родителя другого
пола. При этом возможны следующие варианты: а) сходные
фенотипические аномалии для отцовской и материнской ОРД;
б) аномалии, отличающиеся при ОРД разного родительского
происхождения, вплоть до противоположных эффектов; в) ле-
тальный эффект, прежде всего на ранних сроках развития пло-
да.
Заключение о наличии импринтинга генов, локализованных
на данной хромосоме, может быть сделано с известной долей
осторожности, когда несколько документированных случаев
ОРД имеют сходный аномальный фенотип. Из 47 возможных у
человека различных вариантов ОРД по целым хромосомам, к
настоящему времени сообщается о 27, с очень разной часто-
той (см. таблицу 9).
Эффект импринтинга был установлен достаточно опреде-
ленно для четырех хромосом человека: 15, 11,7 и 14. Наибо-
лее впечатляющие данные были получены по хромосоме 15,
имеющей в проксимальном районе длинного плеча район, под-
верженный импринтингу, мутации которого связаны с двумя
хорошо описанными микроделеционными синдромами: Пра-
дера-Вилли и Энгельмана. К настоящему времени описано уже
более 30 случаев ОРД15мат у пациентов с синдромом Праде-
ра-Вилли, который характеризуется умственной отсталостью,
мышечной гипотонией, Сильным ожирением, гипогонадизмом,
низким ростом, акромикрией - непропорционально маленьким
размером дистальных участков верхних и нижних конечнос-
тей. ОРД15мат является причиной 20-25% всех случаев син-
дрома, а большая часть остальных случаев заболевания свя-
зана с делецией сегмента 15q11 -q13 отцовской хромосомы.
Установлено, что кандидатным геном данного синдрома яв-
ляется ген, ответственный за синтез полипептида N малого
ядерного рибонуклеопротеина (SNRPN) (Ozcelik et aL, 1992).
104
Нетрадиционное наследование
Этот ген имеет протяженность 25 Кб и состоит из 10 экзонов,
активно экспрессируется исключительно на отцовской хромо-
соме 15, но метилирован и репрессирован на материнском
гомологе. У некоторых больных с синдромом Прадера-Вилли
обнаружены случаи делеций и точковых мутаций в пределах 2
Мб хромосомного домена в районе 15q11 -q13, которые свя-
заны с блокированием переключения импринтинга в направ-
лении материнский-отцовский (Dittrich et al., 1996). Такое пе-
репрограммирование хромосом или переключение импринтин-
га происходит в ходе гаметогенеза.
Таблица 9.
Известные случаи ОРД и эффект
импринтинга у человека
Тип ОРД Число случаев Эффект имприн- тинга Тип ОРД Число случаев Эффект имприн- тинга
2 мат 3 ? 14 ОТЦ 3 9 •
3 мат 1 9 • 15 мат >30
4 мат 1 - 15 отц 9 9
5 отц 1 * 16 мат 10 *
6 отц 4 9 • 16 отц 1 -
7 мат 8 20 отц 1 9 •
7 отц 3 - 21 мат 3
8 отц 1 21 отц 2 -
9 мат 2 - 22 мат 3 а
10 мат 1 - 22 отц 1 4
11 отц >15 XX мат 3 М
13 мат 3 XX отц 1 9 •
13 отц 1 4 XY отц 1 -
14 мат 9
Примечание:
+ установленный эффект;
маловероятный;
? возможный.
Другой синдром, связанный с аномалиями хромосомы 15, -
синдром Энгельмана (синоним - синдром ’’счастливой куклы”),
характеризуется неадекватной счастливой улыбкой и глубо-
кой умственной отсталостью с резкими кукольными судорож-
ными движениями. В 70% случаев он возникает вследствие de
novo делеции участка 15q11-q13 материнской хромосомы и
105
Глава 5
-----------------------------1------------------------
лишь в 2% случаев обусловлен ОРД15 отцовского происхож-
дения. Установлено также, что около 3% случаев синдрома
связаны с "мутациями импринтинга", а оставшиеся 25% обус-
ловлены мутациями в локусе-мишени импринтинга. К настоя-
щему времени этот локус уже установлен. Им является убик-
витин-белковый лигазный ген (UBE3A) Е6-АР (Kishino et al.,
1997). Таким образом, в проксимальном районе хромосомы
15 имеются близко сцепленные, но противоположно им-
принтированные локусы, отвечающие за возникновение двух
фенотипически разных синдромов - Прадера-Вилли и Зигель-
мана. Очевидно, что участки генома, подверженные имприн-
тингу, кластеризованы в определенных хромосомных доменах
и регулируются с помощью региональных дистанционных цис-
действующих элементов - центров импринтинга.
Интересно отметить, что с помощью флюоресцентной гиб-
ридизации in situ в интерфазных ядрах клеток обнаружены раз-
личия в аллель-специфической репликации ДНК материнско-
го и отцовского локусов участка 15q11 -q13 (White et aL, 1996).
Пациенты с ОРД 15 имели около 3-10% клеток с асинхронной
репликацией, в то время как у пациентов без делеции и у нор-
мальных индивидов наблюдалась более высокая частота кле-
ток с асинхронной репликацией ДНК (23-36 %).
По меньшей мере 15 случаев ОРД11 отцовского происхож-
дения описано в связи с синдромом Видемана-Беквита (СВБ),
характеризующимся гигантизмом, макроглосеней, пупочной
грыжей (омфалоцеле) и увеличенным риском развития эмбри-
ональных опухолей. ОРД по хромосоме 11 как этиологический
фактор возникновения СВБ отмечена приблизительно у чет-
верти всех обследованных больных с этим синдромом (Led-
better и Engel, 1995). Особый интерес в плане выяснения ме-
ханизмов регуляции работы импринтированных генов представ-
ляют случаи заболеваний, не сочетающихся с ОРД. Конкрет-
ный ген, ответственный за СВБ, не выяснен, однако ряд на-
блюдений свидетельствует о том, что патогенез этого заболе-
вания связан с нарушением регуляции импринтированных ге-
нов, локализованных в этом районе хромосомы 11. К настоя-
щему времени в данном регионе картирован ряд генов и, в
частности, инсулиноподобный ростовой фактор 2 (IGF2) и Н19
с импринтированной отцовской и материнской экспрессией,
соответственно. Показано, что у некоторых пациентов с СВБ,
но без цитогенетических аномалий, обнаруживается аномаль-
ная экспрессия материнского аллеля IGF2. У части из этих
больных такая биаллельная экспрессия гена IGF2 сопровож-
дается репрессией и ДНК-метилированием материнской ко-
пии соседнего гена Н19 (Brown et al., 1996). Авторы полагают,
что мутации импринтинга гена Н19, приводящие к инактива-
106
Нетрадиционное наследование
ции материнского аллеля этого гена, могут косвенно влиять
на активацию материнского аллеля IGF2. Однако кроме мате-
ринских хромосомных транслокаций, расположенных в двух
кластерах, центромернее от гена IGF2 - в 200-400 Кб и в не-
скольких миллионах пар оснований, других мутаций в этой
области не обнаружено. Эти данные свидетельствуют о том,
что несмотря на то, что точки транслокации находятся на зна-
чительном расстоянии от импринтированных генов, они могут
нарушать регуляцию работы этих генов. Описана, однако, семья
с хромосомной транслокацией в сайте 11р15.5, в которой би-
аллельная экспрессия гена IGF2 у больных с СВБ сочеталась с
нормальным импринтингом гена Р19 (Brown et aL, 1996). Эти
данные говорят о сложной регуляции работы генов, располо-
женных внутри доменов хромосом, подверженных импринтин-
гу.
Имеется 9 сообщений о ОРД14мат, которая приводит к
формированию четко определяемого синдрома. Основными и
постоянными признаками данного синдрома являются низкий
вес и рост при рождении, гипотония, задержка моторного и
физического развития, а также преждевременное половое
развитие (Tomkins etal., 1996). Кроме того, у некоторых паци-
ентов обнаруживается гидроцефалия, умеренные лицевые
аномалии, маленькие руки и ноги, гиперрастяжимость суста-
вов, хронические отиты и сколиоз. По ОРД14отц имеются толь-
ко два сообщения (Diamond et aL, 1993; Wang et aL, 1991). У
больных отмечались множественные врожденные пороки раз-
вития и умственная отсталость. Ограниченность данных пока
не позволяет сделать выводы о характере фенотипических
признаков, связанных с данной аномалией, и очертить синд-
ром ОРД14отц.
Итак, в связи с картированием генома человека перед ис-
следователями встает задача поиска и картирования участ-
ков хромосом, подверженных импринтингу. Первым шагом на
пути решения этой задачи является поиск конкретных хромо-
сом набора, содержащих импринтированные гены.
Следующей задачей после нахождения фенотипических
эффектов ОРД по той или иной хромосоме является поиск
конкретных генов, локализованных в этих хромосомах и име-
ющих моноаллельную экспрессию. Так, на хромосоме 7 чело-
века, ОРД материнского происхождения которой, как было
отмечено выше, сочетается с пре- и постнатальной задерж-
кой роста, недавно был обнаружен высоко консервативный ген
MEST, экспрессирующийся только с отцовского аллеля в ме-
зодерме и ее производных (Nishita et aL, 1996). Авторы этой
работы предполагают, что данный ген и является тем самым
импринтированным геном, который вовлечен в патогенез син-
107
Глава 5
дрома Рассела-Сильвера и который отвечает за аномалию
роста у больных при ОРД7мат.
Основание для утверждения о возможном существовании
материнского и отцовского импринтинга по отдельным генам
можно получить из анализа родословных (Hall, 1990). Имприн-
тированный аллель передается менделевским путем, но его
экспрессия определяется полом передающего его родителя.
Отцовский импринтинг означает, что фенотипической экспрес-
сии аллеля не происходит при передаче его от отца, а мате-
ринский импринтинг означает, что экспрессии гена не проис-
ходит при передаче его от матери. Связь феномена ГИ с на-
следственной патологией человека на уровне отдельных ге-
нов отчетливо прослеживается на примере ряда моногенных
заболеваний. Так, в случае унаследования доминантных генов
от отца при хорее Гентингтона и спиномозжечковой атаксии I
типа, заболевания возникают раньше и протекают более тя-
жело, по сравнению со случаями наследования мутантных
генов от матери (Reik, 1989) При нейрофиброматозе I и II
типов, а также при миотонической дистрофии, наоборот, за-
болевания имеют более раннее начало и тяжелое течение при
унаследовании мутантных генов от матери.
В последнее время открылись новые возможности молеку-
лярно-генетических исследований мультифакториальных за-
болеваний. В комплексе генов, причастных к развитию этих
заболеваний, могут быть и гены, подверженные ГИ, и которые
могут модифицировать клиническое проявление болезней. В
настоящее время не вызывает сомнения причастность явле-
ния ГИ к этиологии опухолевого роста. Так, установлено, что
некоторые эмбриональные опухоли и, в частности, опухоль
Вильмса возникает при потере гетерозиготности материнского
происхождения по хромосоме 11. При потере гетерозигот-
ности по хромосоме 13 также материнского происхождения
возникают остеосаркома и ретинобластома (Hall, 1990; Reik,
1989). Таким образом, инактивация отцовского гена вследст-
вие импринтинга и потеря гетерозиготности вследствие мута-
ции другого (в данных случаях материнского) аллеля является
механизмом полной потери активности указанных генов суп-
рессоров опухоли (функциональная нуллисомия) и причиной
развития этих злокачественных новообразований. В связи с
картированием генов, причастных к возникновению диабета,
следует указать на находки ОРД по хромосоме 6 отцовского
происхождения у детей с транзиторным неонатальным сахар-
ным диабетом (James etal., 1995).
Подводя итог рассмотрению болезней человека, этиоло-
гически связанных с ГИ, следует отметить, что наши знания об
этом явлении ограничены, и точные механизмы возникнове-
108
Нетрадиционное наследование
ния заболеваний ГИ еще не ясны. Не вызывает сомнения, что
исследование этой фундаментальной проблемы позволит глуб-
же понять закономерности функционирования генома челове-
ка, расширит наши представления о патологии, связанной с
этим явлением, и будет иметь важное клиническое значение.
Митохондриальные болезни
В 1988 году были получены первые и убедительные дока-
зательства связи мутаций митохондриальной ДНК (мтДНК) с
конкретными болезнями человека: митохондриальной миопа-
тией (Holt et al., 1988) и наследственной нейроофтальмопа-
тией Лебера (Wallace et aL, 1988). Интерес к этой проблеме
позволил обнаружить большое число мутаций мтДНК, лежа-
щих в основе возникновения и развития целого ряда нейроде-
генеративных заболеваний, некоторых форм МФЗ (диабет,
рак), а также показать их связь с процессом старения.
В зависимости от типа мутаций была предложена класси-
фикация наследственных митохондриальных болезней (табл.
10), объединенных в 4 основных группы (Wallace et aL, 1992).
Она носит рабочий характер, ибо слишком коротка история
(менее 10 лет) объединения болезней в эти группы патоло-
гии. Интерес к проблеме “подогревается” и тем, что уж очень
велик соблазн достичь максимального понимания функции
митохондриального генома при полном сегодня знании струк-
туры мтДНК. А ведь, как отмечал McKusick (1988) “... описание
функций генома митохондрий представляет собой в миниа-
тюре цель, которая стоит перед исследователями ядерного
генома”.
Митохондриальные болезни,
вызванные миссенс-мутациями
Наследственная нейропатия зрительных нервов Лебера
(LHON) - одно из наиболее изученных заболеваний данной
группы. У больных с данной формой патологии наблюдается
острая и подострая билатеральная потеря зрения. В большин-
стве случаев у лиц с нейропатией зрительных нервов Лебера
выявляются точковые мутации в мтДНК, приводящие к заме-
не консервативных аминокислот. Описано несколько таких
мутаций: замена оснований G и А в положении 11778 мито-
хондриальной ДНК, что приводит к замене аргинина на гисти-
дин в 340 кодоне гена субъединицы 4 (NDA) НАДН-дегидроге-
назы (Wallace et aL, 1988); мутация в 3460 положении мтДНК,
в результате которой происходит замена Ala _> Thr в 52 кодоне
гена субъединицы 1 (ND1) НАДН-дегидрогеназы (Ниоропеп et
109
Глава 5
aL, 1991); мутация в положении 13708 (ген ND5), вызываю-
щая замену аланина на треонин (Brown etaL, 7992); мутация
в положениях 15257 и 15182 гена цитохрома b и в положении
5244 гена Nb2 (Brown et aL, 1992).
Таблица 10.
Митохондриальные наследственные болезни
у человека (по Wallace, 1992)
1. Миссенс-мутантные (аминокислотные замены
в компонентах I, III, IV комплексов дыхательной цепи):
- нейроофтальмопатия Лебера;
- пигментный ретинит.
2. Мутации в генах тРНК:
- синдром MERRF;
- синдром MELAS.
3. Делеции или дупликации участков мтДНК;
- наружная офтальмопатия;
- синдром Кернса-Сайра;
- синдром Пирсона;
- изолированный двусторонний асимметричный птоз;
- двусторонний птоз, сочетающийся с офталмопарезом
и слабостью мышц нижних конечностей;
- дилатационная кардиопатия.
4. Мутации, снижающие число копий мтДНК:
- летальная инфантильная дыхательная
недостаточность;
- синдром молочнокислого ацидоза.
Это далеко не все мутации мтДНК, ведущие к LHON, сей-
час их обнаружено 19 (Wallace, 1995). Во всех случаях мута-
ции затрагивают компоненты комплексов I и III дыхательной
цепи, поэтому предполагают, что развитие болезни Лебера
обусловлено не нарушением какого-то конкретного белка, а
общим изменением обмена энергии в митохондрии (Brown et
aL, 1992). Подтверждением этому также служит тот факт, что
клинические признаки у больных с различными точковыми
мутациями существенно не различаются. Единственным отли-
чием является то, что зрение сохраняется приблизительно у
20% больных при наличии мутации в положении 3460, тогда
как в случае мутации в положении 11778 сохранение зрения
наблюдается только в 4% случаев. Такое сходство фенотипов
объясняется нарушением нормального функционирования
одного и того же митохондриального фермента - НАДН-де-
гидрогеназы (Johns et aL, 1992).
В семьях с нейропатией зрительных нервов Лебера описа-
но явление гетероплазмии: количество мтДНК с мутацией
варьирует у разных больных от 5 до 100% всей ДНК (Vilkki et
110
Нетрадиционное наследование
al., 1990), а количество мутантной ДНК в клетках организма
коррелирует с тяжестью клинических симптомов (Isashiki and
Nakagawa, 1991; Zhu et al., 1992). В процессе эволюции гете-
роплазмия мтДНК может эффективно поддерживаться на про-
тяжении нескольких поколений, при этом у части лиц может
наблюдаться фиксация одной из форм мтДНК в результате
случайной полной элиминации другой формы в ходе оогене-
за. Так, в ряде случаев при наличии гетероплазмии у матери
мутантная мтДНК не передается детям.
Holt et al. (1990) описали еще одно заболевание, связан-
ное с нарушениями мтДНК, - пигментный ретинит. У больных
наблюдалась задержка развития, умственная осталось, пиг-
ментный ретинит, сенсорная нейропатия, атаксия и нейроген-
ная мышечная слабость. Несмотря на отсутствие признаков
типичной митохондриальной миопатии, анализ структуры ми-
тохондриальной ДНК выявил наличие точковой мутации в поло-
жении 8993 в субъединице 6 АТФ-азы, что привело к замене
гидрофобной аминокислоты лейцина на гидрофильную ами-
нокислоту аргинин, и, в связи с этим, к нарушению активности
фермента. Выраженность клинических признаков у больных
коррелировала с количеством мутантной митохондриальной
ДНК в клетках организма.
Митохондриальные болезни,
вызванные мутациями в генах тРНК
К этой группе относят синдром MERRF и синдром MELAS.
Около 30 мутаций генов тРНК в мтДНК связаны с различными
дегенеративными заболеваниями. Они варьируют по тяжести
от летальных до легких клинических отклонений.
Синдром MERRF (myoclonic epilepsy and ragged-red fiber -
миоклонус эпилепсия в сочетании с необычно красными ра-
зорванными мышечными волокнами) характеризуется прогрес-
сирующей дегенерацией нервной и мышечной ткани, которая
проявляется судорогами, атаксией, миопатией и потерей слу-
ха, а также поражением печени и почек (Wallace, 1992; Темин
и др., 1996). Биохимический дефект связан с недостаточностью
дыхательных комплексов I и II митохондрий. Примерно в 94%
случаев заболевание вызывается точковой мутацией в гене
лизиновой тРНК, локализующейся в положении 8344 и приво-
дящей к трансзиции А G (Shofner et al., 1990; Yoneda et al.,
1990). Выявляемость данной мутации не у всех больных ука-
зывает на генетическую гетерогенность данного патологичес-
кого состояния. Так, у одного больного с синдромом MERRF
выявлена мутация мтДНК в 8356 положении гена tPHKys, кото-
рая присутствовала в состоянии гомоплазмии в мышечных
111
Глава 5
клетках и в состоянии гетероплазмии в клетках крови больно-
го (Silvesri et al., 1992). Такая мутация не выявлялась у лиц с
другими формами митохондриальных болезней и контрольных
образцах крови, что, по мнению авторов, позволяет предпол-
агать, что мутации в гене tPHKys являются специфичными для
синдрома MERRF.
Риск передачи рассматриваемого синдрома в ряду поко-
лений зависит от доли мутантной ДНК в лимфоцитах матери;
тяжесть заболевания и степень выраженности биохимических
нарушений также определяется соотношением нормальной и
мутантной мтДНК в клетках больных: при наличии 94-96 %
мутантной формы мтДНК наблюдается снижение активности
ферментов дыхательной цепи, а при 61-92% - процесс окис-
лительного фосфорилирования протекает нормально (Larsson
etal., 1991).
Точковая транзиция А G в положении 3243 (Kobayashi et
aL, 1991), замена С Т в положении 3271 (Goto et al., 1991),
нарушающие функцию тРНК лейцина, приводят к развитию
синдрома MELAS (myopathy, encefalopathy, lactic acidosis, stro-
kelike episodes - миопатия, энцефалопатия, лактат-ацидоз,
инсулиноподобные эпизоды) (Николаева и др., 1997). Мута-
ции в этом гене найдены приблизительно у 80% больных и не
обнаружены у здоровых лиц. Обе мутации приводят к нару-
шению дыхательной функции митохондрий. Предполагается
также, что с синдромом MELAS связана замена А G в поло-
жении 11084, вследствие чего происходит замена триптофа-
на на аланин в ND4 дыхательного комплекса I (Letritetal., 1992).
В эксперименте in vitro было показано, что в митохондриях,
содержащих мутантные молекулы мтДНК, характерные для
синдрома MELAS, обнаруживается повышенное содержание
РНК-19 - интермедиата процессинга РНК-транскрипта тяже-
лой нити митохондриальной ДНК, содержащей последователь-
ность 16S-pPHK, тРНК лейцина и мРНК полипептида ND-1.
Клетки с данным типом митохондрий показали снижение бе-
локсинтезирующей активности митохондрий и снижение фун-
кциональной активности дыхательной цепи. На основании этого
выдвинуто предположение, что аномальная РНК-19 включает-
ся в рибосомы митохондрий, которые при этом становятся
функционально неактивными (Shon et al., 1992).
С точечной мутацией в гене тРНК лейцина связывают еще
одно из заболеваний, характеризующееся сочетанием: кар-
диомиопатия и миопатия скелетных мышц с поздним началом
развития патологических симптомов. У больных наблюдается
гетероплазмия по мутантной митохондриальной ДНК, причем
степень гетероплазмии хорошо коррелирует с тяжестью забо-
левания (Zeviani etal., 1991).
112
Нетрадиционное наследование
Митохондриальные болезни, вызванные
делециями или дупликациями участков
митохондриального генома
Размер и местоположение делеций в мтДНК очень сильно
варьируют, большинство делеций расположены между точка-
ми начала репликации Н- и L-цепей мтДНК и ограничено пов-
торами размером от 5 до 13 п.н.; некоторые из них захваты-
вают область начала репликации легкой цепи ДНК, и у таких
больных миопатия выражена более сильно и начинается очень
рано (в возрасте до 1 года). Делеции с затрагиванием точки
начала репликации легкой цепи появляются, видимо, в резуль-
тате нарушения нормальной репликации митохондриальной
ДНК при нарушении взаимодействия между ядерным и мито-
хондриальными геномами (Miyabayashi etal., 1991). Основная
масса делеций возникает спонтанно, механизмами их возни-
кновения могут являться гомологичная рекомбинация, топои-
зомеразное отщепление и слип-репликация. Подтверждено,
что крупные делеции митохондриальной ДНК блокируют тран-
крипцию всех митохондриальных генов дыхательной цепи. За-
болевания, вызванные делециями митохондриальной ДНК,
имеют тенденцию прогрессировать с возрастом {Harding and
Hammans, 1992).
Приведем несколько примеров митохондриальных заболе-
ваний, вызванных структурными перестройками мтДНК.
Синдром Кернса-Сейра - мультисистемное заболевание,
клинически характеризующееся развитием хронической наруж-
ной офтальмоплегии, нарушением сердечного ритма, пигмен-
тной дегенерацией сетчатки {Николаева и др., 1997). У боль-
ных с данной патологией выявляются большие (до 7 Кб) деле-
ции, локализованные в различных участках митохондриальной
ДНК; независимо от размера делеций наблюдается снижение
активности ферментов комплексов I, III и IV митохондрий. Кор-
реляция между размером, локализацией делеции и тяжестью
заболевания отсутствует. Показано, что делетированная мтДНК
транскрибируется, но трансляция образующихся транскрип-
тов отсутствует. Нарушение процессов трансляции в митохон-
дриальной ДНК объясняется тем, что делеции захватывают гены
тРНК (Grossman, 1990; Nakase et aL, 1990).
Содержание делетированной митохондриальной ДНК мо-
жет варьировать в различных клетках организма. Причиной
этого, возможно, является случайная сегрегация мутантной
мтДНК при созревании яйцеклетки и при дальнейшем эмбри-
ональном развитии, т.е. с реализацией так называемого эф-
фекта "горлышка бутылки". При незначительном содержании
делетированной митохондриальной ДНК в тканях организма
113
8 3аказ№127
Глава 5
клинические проявления болезни не фиксируются (Poulton et
al., 1991). Существует предположение, что синдром Пирсона,
описанный ранее как самостоятельное состояние, для кото-
рого характерны поражение костного мозга и поджелудочной
железы, и синдром Кернса-Сейра могут являться различными
фенотипическими проявлениями одного и того же дефекта
митохондриальной ДНК в зависимости от распределения и
относительного содержания мутантной ДНК в тканях организ-
ма (Harding and Hammans, 1992). При синдроме Пирсона де-
леции митохондриальной ДНК обнаружены в лимфоцитах, в
клетках костного мозга и мышц.
Помимо делеций, в митохондриальной ДНК обнаруживаются
крупные перестройки, связанные с дупликацией митохондри-
ального генома, которые также приводят к развитию патоло-
гических состояний. Две формы мтДНК - нормальная, разме-
ром 16,5 Кб, и аномальная, размером приблизительно 24 Кб,
возникшая за счет дупликации около 8 Кб, выявлены у боль-
ных с митохондриальной миопатией и множественными пора-
жениями других органов и систем организма. Локализация
участков дупликации у больных не совпадает, но в обоих слу-
чаях найдено удвоение точки начала репликации мтДНК, генов
рРНК и тРНК (Poulton etal., 1989). Более сложная перестройка
обнаружена в семье с проксимальной тубулопатией, пов-
реждениями кожи, диареей, инсулинзависимым сахарным
диабетом, атаксией, глухотой, слепотой и психомоторной за-
держкой развития, где заболевание наследовалось по мате-
ринской линии. Молекулярный анализ показал, что у больных
имеется удвоение митохондриального генома, включающее
одну полноразмерную молекулу ДНК и одну делетированную
молекулу ДНК, соединенные в области генов АТФ-азы 6 и ци-
тохрома b (Rottig etal., 1991).
Митохондриальные болезни, вызванные
мутациями, снижающими число копий
митохондриальной ДНК
К данной группе относятся летальная инфантильная дыха-
тельная недостаточность и синдром молочнокислого ацидо-
за. Эта группа болезней наименее изучена. Их генетическая
основа может быть проиллюстрирована следующим приме-
ром.
Moraes et al. (1991) описана семья с митохондриальным на-
следственным заболеванием, характеризующимся резким
снижением содержания митохондриальной ДНК в различных
тканях организма; снижение содержания мтДНК (1-2% от нор-
мы) было обнаружено в мышцах у больного с миопатией, в
114
Нетрадиционное наследование
почках у больного нефропатией и в печени у больного, стра-
дающего печеночной недостаточностью. Во всех случаях на-
блюдалось ослабление синтеза белков, кодируемых мтДНК, и
снижение активности цитохром-с-оксидазы, хотя не удалось
выявить каких-либо делеций, перестроек и точечных замен в
митохондриальной ДНК.
Подводя итог краткой характеристики митохондриальных
болезней, следует отметить, что вряд ли следует преувеличи-
вать исключительную самостоятельность генетических дефек-
тов митохондриальной ДНК в возникновении и клинических
проявлениях митохондриальных болезней. В последнее время
появляются данные о тесном взаимодействии ядерного и ми-
тохондриального генома в развитии целого ряда патологичес-
ких состояний. Предполагается, что причиной ряда митохон-
дриальных миопатий могут быть ядерные мутации, приводя-
щие к возникновению различных дефектов в митохондриаль-
ной ДНК, нарушающие процессы транскрипции и трансляции.
Фактором, дестабилизирующим мтДНК, и тем самым приво-
дящим к образованию множественных делеций, скорее всего,
является мутантный вариант белка, регулирующего реплика-
цию митохондриальной ДНК. Однако, ядерные гены, ответ-
ственные за появление делеций в мтДНК, не идентифициро-
ваны (Zeviani, 1992). Фенотипически данные мутации проявля-
ются в виде хронической прогрессирующей наружной офталь-
моплегии (ХПНО), наследуемой по аутосомно-доминантному
типу, аутосомно-рецессивной ХПНО, возвратной миоглобину-
рии, приступов метаболического ацидоза, наследующихся по
аутосомно-рецессивному типу миопатий, нефропатией, гепа-
топатий и энцефаломиопатий (Zeviani, 1992).
Описаны несколько семей, в которых прогрессирующая
наружная офтальмоплегия наследовалась как аутосомно-до-
минантное заболевание. Вместе с тем, в тканях больных по-
мимо нормальной мтДНК присутствовала мтДНК с делециями
различного размера (Zeviani et а!., 1990). Множественные де-
леции митохондриальной ДНК обнаружены у больного с тяже-
лой мышечной слабостью, атаксией, глухотой, умственной от-
сталостью, наследующихся по аутосомно-доминантному типу.
В различных тканях больного делеции отличаются по размеру
и локализации, но во всех случаях делегированные участки
фланкированы короткими (4 п.н.) прямыми повторами (Cormi-
er etal., 1990).
Нарушение структуры и функции митохондриального гено-
ма выявлены также для митохондриальной дистрофии - ауто-
сомно-доминантного заболевания, ген которого картирован на
длинном плече 19 хромосомы. У больных с данной патологией
обнаружены цитохром-с-оксидаза-негативные миофиламенты
115
Глава 5
и “рваные” миофибриллы (данные признаки характерны для
митохондриальных миопатий) и делеции митохондриальной
ДНК различного размера (Sahashi et al., 1992).
Существует также предположение, что некоторые заболе-
вания могут быть обусловлены действием двух локусов, один
из которых является ядерным, а другой - митохондриальным.
Так, генетический анализ семьи с нейросенсорной глухотой
(тканеспецифичное заболевание, проявляющееся потерей слу-
ха и не имеющее каких-либо других неврологических симпто-
мов) показал одновременное наличие митохондриальной и
аутосомно-рецессивной мутаций (Prezant et aL, 7992). Двуло-
кусная модель наследования не исключается также для на-
следственной нейропатии зрительных нервов Лебера. Пре-
обладание этой патологии у мужчин и сниженная пенетран-
тность у женщин, свидетельствует о том, что развитие болез-
ни Лебера может контролироваться также Х-сцепленным ре-
цессивным геном (Nakamura et al., 1992).
С другой стороны, нарушения митохондриальной ДНК ока-
зывают влияние на экспрессию ядерных генов митохондри-
альных белков. Так, при синдроме Кернса-Сейра делеция в
митохондриальной ДНК приводит к снижению синтеза мРНК
субъединиц 6-8 АТФ-азы, и, в то же время, наблюдается сни-
жение синтеза мРНК р-субъединицы АТФ-азы, кодируемой
ядерным геном (Hoddi et al., 1992).
Исследования по участию митохондриального генома в
формировании наследственной предрасположенности к муль-
тифакториальным заболеваниям приносят неопределенные
результаты. Имеются лишь догадки, предположения и немно-
гочисленные работы, связанные с данной проблемой. Однако
было бы неправильно пренебрегать первыми наблюдениями
в этой области общей патологии. Они прежде всего касаются
процессов старения, сахарного диабета, хронических пора-
жений миокарда, опухолевых заболеваний.
Как уже отмечалось, экспрессия митохондриального гено-
ма составляет основу поддержания биоэнергетической функ-
ции митохондрий. В связи с этим, не случайно, что патогенез
основных клинических проявлений наследственных митохон-
дриальных болезней определяется нарушением этих функций
органелл клеток. За состоявшейся мутацией следует период
временной гетероплазмии мтДНК; произвольный характер
цитоплазматической сегрегации в течение дальнейшего раз-
вития повышает степень мозаицизма клеток. Неравномерность
клеточного распределения мутировавших молекул мтДНК при-
водит к неодинаковости биоэнергетических характеристик кле-
ток самых различных тканей организма.
116
Нетрадиционное наследование
МтДНК и старение. Выдвинута гипотеза о том, что спон-
танное возникновение и накопление мутаций в митохондриях
с последующим цитоплазматическим распределением мутан-
тных генов приводит к появлению клеток с различными биоэ-
нергетическими свойствами и является звеном механизмов
старения и развития дегенеративных процессов у человека
(Linnane et al., 1989).
В митохондриальной ДНК у человека, выделенной из раз-
личных тканей организма (сердце, мозг, печень, почки, селе-
зенка, скелетная мускулатура и др.) была обнаружена возраст-
зависимая делеция (Cortopassi and Arnheim, 1990; Cortopassi
et al., 1992; Ikebe et al., 1990; Linnane et al., 7990). При этом
уровень делетированных мтДНК во взрослой мышце сердца,
диафрагме и головном мозге был значительно выше (в 16-64
раза), чем в печени, почке, коже, селезенке и легких, а среди
клеточных параметров обнаружена высокая скоррелли-
рованность уровня делетированных молекул с низкой митоти-
ческой активностью, большим размером клеток и повышен-
ной скоростью метаболизма (Cortopassi et al., 1992). Делети-
рованный фрагмент митохондриальной ДНК приходится на
участок, заключенный между 8470 и 13549 парами оснований,
где расположены гены, кодирующие 6 и 8 субъединицы АТФ-
азы, субъединицу СОЗ цитохром оксидазного комплексов и
субъединицы ND3, 4L, 4 и 5 HADH коэнзим-Q-редуктазного
комплекса (Linnane et al., 1990).
Вывод о неизбежном в жизни человека накоплении сома-
тически приобретаемого кислородного повреждения мтДНК
вместе с мутациями в мтДНК, связанными с возрастом и
ведущими к биоэнергетической недостаточности и слабости
сердечной мышцы, был сделан также на основании анализа
мтДНК индивидов в возрасте 24-97 лет с клиническими сим-
птомами дисфункции сердца (Hayakawa et al., 1992). В данной
группе лиц наблюдалось увеличение с возрастом (до 7%) со-
держания мтДНК, имеющей делецию 7,4 Кб.
Аналогичная ситуация (рост частоты мутантных молекул
мтДНК) выявляется при изучении точковых мутаций. Так, у по-
жилых людей обнаруживается транзиция A_>G в положении
8344 мтДНК гена тРНК лизина, характерная для синдрома
MERRF; относительное количество таких мутантных молекул
составляет в данной группе 1 -2% от общего количества мтДНК
(Мunscher et al., 1992).
МтДНК и сахарный диабет. Семейный анализ при сахар-
ном диабете выявляет преобладание материнского типа на-
следования, что позволяет предположить связь этого заболе-
вания с мутациями митохондриальной ДНК. Данный вопрос в
настоящее время практически не изучен, а имеющиеся сооб-
117
Глава 5
ков за счет ассортативности браков индивидов с высокой под-
верженностью патологии (Вартанян, 1983).
Последние открытия в исследовании ряда тяжелых невро-
логических заболеваний с использованием молекулярно-ге-
нетических методов анализа позволили пролить новый свет
на механизм возникновения антиципации. Идентификация в
1991 году первой мутации нестабильных тринуклеотидных пов-
торов, вызывающей наследственное заболевание у человека
с четким проявлением феномена антиципации, означала су-
щественное переосмысление основ и принципов мутационно-
го процесса в менделевской генетике. Открытие нового клас-
са наследственных заболеваний с экспансией числа тринук-
леотидных повторов, в основе которых лежит “динамическая
мутация” - резкое увеличение числа копий повторов у индиви-
дов в последующих поколениях родословной, не только про-
яснило механизм одного из возможных путей нетрадиционно-
го наследования признаков, но и поставило новые проблемы
в понимании сущности этих заболеваний (Korneluk, Narang,
1997; Назаренко и др., 1995).
Открытие феномена экспансии числа тринуклеотидных пов-
торов было сделано при исследовании синдрома Мартина-
Белла или синдрома фрагильной (ломкой) Х-хромосомы. Этот
синдром характеризуется ломкой Х-хромосомой в сайте Xq27.3
(выявляемой в специальных условиях культивирования в сре-
де с дефицитом фолатов), умственной отсталостью (IO ко-
леблется в диапазоне от 35 до 50), аутизмом, удлиненным
лицом (в сочетании с его отеком, утолщением тканей и про-
гнатией), оттопыренными ушами и макроохидизмом. Однако
такая комбинация признаков встречается лишь у 60% мужчин
с фрагильной Х-хромосомой. 10% пораженных индивидов не
имеют лицевых аномалий, 10% имеют только умственную от-
сталость без остальных признаков и до 30% пораженных не
имеют макроорхидизма. Отмечается также выраженное нару-
шение соединительной ткани с гиперрастяжимостью суставов,
а до 80% взрослых больных имеют пролапс митрального кла-
пана (Hirst, 1992).
Синдром ломкой Х-хромосомы уже давно привлекал вни-
мание исследователей в связи с его необычным характером
наследования и довольно широкой распространенностью в
популяции (порядка 1:1000), сравнимой с частотой часто встре-
чающегося синдрома Дауна. Необычность наследования дан-
ного синдрома заключается в том, что не все, а лишь 80%
мужчин носителей мутантного гена, имеют клинические и ци-
тогенетические признаки заболевания. Остальные же 20% как
клинически, так и цитогенетически нормальны, хотя после пе-
редачи мутации всем своим дочерям, они могут иметь пора-
120
Нетрадиционное наследование
женных внуков. Эти мужчины получили название трансмитте-
ров, т.е. передатчиков неэкс пресс и рован но го мутантного гена,
который становится экспрессируемым в последующих поко-
лениях. Кроме того, выявляется 2 типа женщин-гетерозигот
по данному гену (Fryns, 1989). Первый тип - дочери нормаль-
ных мужчин трансмиттеров, практически никогда не имею-
щие симптомов данного синдрома и у которых не выявляется
ломкая Х-хромосома при цитогенетическом обследовании.
Второй тип - внучки нормальных мужчин-трансмиттеров и сес-
тры пораженных мужчин, обнаруживающие клинические при-
знаки заболевания до 35% случаев. Таким образом, мутация
гена при синдроме ломкой Х-хромосомы существует как бы в
двух формах, отличающихся по своей пенетрантности: фено-
типически не проявляющаяся премутация (первая форма), ко-
торая переходит в полную мутацию (вторая форма) при про-
хождении через женский мейоз. При анализе большого числа
родословных была обнаружена четкая закономерность - веро-
ятность развития умственной отсталости зависит от положе-
ния индивида в родословной. Такой необычный способ насле-
дования мутантного гена, отличный от классического Х-сцеп-
ленного локуса, получил название парадокса Шермана (Sher-
man et al., 1984, 1985). При этом хорошо просматривался фе-
номен антиципации - более тяжелого проявления заболева-
ния в последующих поколениях.
В 1991 году Verkerk с соавторами (Verkerk et al., 1991) и
практически одновременно с ними ряд других авторов из
разных исследовательских групп (Bell, 1991; Nakahori, 1991;
Rousseau et al., 1991; Yu et al., 1991; Oberle et al., 1991; Kremer
et al., 1991; Heitz et al., 1991), в результате клонирования и
секвенирования локуса фрагильной Х-хромосомы, который
обозначили как ген FMR-1, сообщили, что в основе клиничес-
ких проявлений и цитогенетической нестабильности в локусе
Xq27.3, наблюдаемой при этом синдроме, лежит многократ-
ное увеличение в первом экзоне гена FMR-1 простого тринук-
леотидного повтора CGG. Было показано, что у нормальных
индивидов число этих повторов в Х-хромосоме колеблется
от 5 до 50. При возрастании числа CGG-повторов от 50 до 200
отмечается состояние премутации, а увеличение этого числа
свыше 200 приводит к клинической манифестации заболева-
ния и цитогенетической экспрессии ломкой Х-хромосомы. Ус-
тановлено, что пораженные лица имеют как полную мутацию,
т.е. увеличенное сверх критического число тринуклеотидных
повторов, так и аномальное метилирование ДНК. Метилиро-
ваны не только CGG-повторы, но и прилежащий к ним CpG-
островок (Bell, 1991; Vincent, 1991), a FMR-1 ген транскрипци-
онно неактивен (Pieretti, 1991). В то же время, нормальные
121
Глава 5
индивиды обнаруживают неметилированный CpG-островок в
этом участке Х-хромосомы. Лица с меньшим числом повторов
или не имеют умственной отсталости или имеют небольшой
риск этой патологии, но имеют высокий риск иметь поражен-
ных детей и внуков. Вызывает интерес то обстоятельство, что
переход от состояния премутации к полной мутации возника-
ет только при передаче гена от матери (Rousseau et. al., 1991).
Показано также, что экспансия CGG-повторов существенно
зависит от пола потомка (Loesch et aL, 1995). Она заметно
увеличена при передаче мутации от матери к сыну, по сравне-
нию с передачей мутантного аллеля от матери к дочери, что
свидетельствует о возможном взаимодействии в зиготе нор-
мального гомолога Х-хромосомы с последовательностью гена
FMR-1. Более того, экспансия триплетов является постзиго-
тическим событием и происходит на очень ранних стадиях
эмбриогенеза (Davys et al., 1992).
В свете явного прогресса в молекулярном исследовании
синдрома ломкой Х-хромосомы становится ясной и причина
вышеупомянутого парадокса Шермана (Fu et al., 1991). Име-
ется четкая корреляция между величиной амплифи-цирован-
ных CGG-повторов с тяжестью клинических проявлений забо-
левания и экспрессией ломкого сайта в Х-хромосоме (Rous-
seau et al., 1991). В состоянии премутации область CGG-пов-
торов является крайне нестабильной, с тенденцией к возрас-
танию числа этих повторов в последующих поколениях. Эта
прогрессия приводит к появлению у лиц с премутацией по-
томков с полной мутацией, что и объясняет более тяжелое
протекание заболевания у этих лиц, т.е. феномен антиципа-
ции. Точная природа увеличения числа CGG-повторов и воз-
никновения полной мутации гена FMR-1 остается неясной. Вы-
сказывались гипотезы, что в основе такого увеличения числа
тринуклеотидных повторов лежит амплификация повторяющих-
ся последовательностей с помощью неравного кроссингове-
ра (Nussbaum et al., 1986), аномальной рекомбинации в дан-
ном локусе Х-хромосомы (Pembrey et al., 1985), локальной
блокировки реактивации ранее инактивированной Х-хромосо-
мы (Laird, 1987). Предполагается также, что потеря AGG-трип-
летов, которые в норме всегда имеются в составе CGG-тракта
гена FMR-1, может быть фактором, предрасполагающим к не-
правильному спариванию гомологов и проскальзыванию реп-
ликации в пределах области тринуклеотидных повторов (Eich-
ler etal., 1994).
Открытие факта связи числа тринуклеотидных повторов с
синдромом ломкой Х-хромосомы позволило разработать мо-
лекулярный метод идентификации этой мутации с помощью
прямого анализа ДНК. Особый интерес в этом отношении пред-
122
Нетрадиционное наследование
ставляют полученные ДНК-зонды двух групп исследовате-
лей - французских, под руководством J.L.Mandel (Rousseau et
aL, 1991; 1992), и английских, под руководством K.E.Davies (Bell
et aL, 1991; Nakahori et aL, 1991; Hirst et aL, 1993; Knight et aL,
1992). Удобными как для постнатальной, так и для пренаталь-
ной диагностики синдрома являются ДНК-зонды 0x1.9,0x0.55
и ОхЕ20 (Hirst et aL, 1993; Knight et aL, 1992; 1993) и StB-12.3
(Rousseau et aL, 1991; 1992). Эти зонды при использовании
различных комбинаций рестриктаз дают хорошие результаты,
позволяющие определять как амплификацию тринуклеотидных
повторов, так и оценивать аномальное метилирование иссле-
дуемого гена.
Интересное наблюдение было сделано в первых отечес-
твенных молекулярно-генетических исследованиях семей с
синдромом Мартина-Белла (Баранов с соавт., 1993; Ракише-
ва, 1993). Показано, что выявляемость мутации гена FMR-1
молекулярно-генетическими методами на порядок выше су-
ществующих методов цитогенетической диагностики синдро-
ма. Эти данные свидетельствуют о необходимости перехода
на новый уровень диагностики заболевания.
Появились данные о возможности применения метода по-
лимеразной цепной реакции (ПЦР) для оценки числа CGG-пов-
торов (Erster et aL, 1992). Авторы данной работы показали,
что трудности амплификации CGG-мотива при полной мута-
ции гена синдрома фрагильной Х-хромосомы можно успешно
преодолеть при замещении dGTP (дезоксигуанозинтрифосфа-
та) на 7-deaza-dGTP (7-деазадезоксигуанозинтрифосфат).
Однако по мнению некоторых авторов ПЦР не позволяет об-
наруживать полную мутацию в связи с тем, что соматический
мозаицизм числа этих повторов, часто наблюдаемый при дан-
ном заболевании, может привести к ложным результатам.
Дальнейшие исследования показали, что синдром ломкой
Х-хромосомы может быть обусловлен не только вариабель-
ностью числа CGG-повторов ДНК в гене FMR-1 и аномальным
метилированием прилежащего CpG-островка. Ряд работ сви-
детельствует о существовании генетической гетерогенности
этого синдрома. Так, Ворле с соавт. (Wohrle et aL, 1992) был
описан мужчина с клиническим фенотипом синдрома ломкой
Х-хромосомы, имеющий микроделецию проксимальной части
гена FMR-1, протяженностью менее 250 Кб. Эти данные под-
держивают гипотезу о том, что потеря функции гена FMR-1
приводит к клиническому фенотипу синдрома ломкой Х-хро-
мосомы, который может быть обусловлен и другими патоге-
нетическими механизмами, отличными от амплификации CGG-
повторов.
123
Глава 5
Молекулярная гетерогенность синдрома ломкой Х-хромо-
сомы была обнаружена также Накахори с соавт. (Nakahori et
al., 1991), которые обследовали несколько семей, в которых
экспрессия ломкого сайта Xq27.3 не всегда была связана с
типичным клиническим фенотипом синдрома, или где наблю-
дались клинические признаки заболевания, но экспрессия
фрагильного сайта не отмечалась. При обследовании этих
семей авторы обнаружили пациента с изменением ДНК в об-
ласти, прилежащей к CGG-повтору и трех пациентов, имею-
щих амплификацию этих повторов лишь в небольшой популя-
ции клеток.
В ряде работ при исследовании большой группы семей с
синдромом ломкой Х-хромосомы были обнаружены родослов-
ные, в которых экспрессия ломкой Х-хромосомы возникала
независимо от умственной отсталости (Voelckel et al., 1988;
1989). Исследование этих семей на предмет мутаций в гене
FMR-1 не показало каких-либо изменений этого гена (Naka-
hori et а!., 1991; Dennis et al., 1992). Анализ ломкого сайта с
помощью метода флюоресцентной гибридизации in situ (FISH-
метод) позволил обнаружить новый ломкий участок, который
был обозначен FRAXE, в отличие от ранее известного ломкого
сайта в гене FMR-1, обозначенного FRAXA (Sutherland et aL,
1992). Существование нового ломкого участка было позднее
подтверждено Флинном с соавторами (Flynn et aL, 1993), ко-
торые показали, что сайт FRAXE также является фолат-чув-
ствительным и располагается приблизительно на расстоянии
500 тыс. пар оснований дистальнее сайта FRAXA. Эти же авто-
ры идентифицировали (Hirst et aL, 1993), а позднее клониро-
вали (Ritchie et aL, 1994) третий фрагильный сайт - FRAXF,
располагающийся более теломерно - на 1-2 миллиона пар
оснований дистальнее FRAXE, в области Xq28.
Клонирование и секвенирование ломкого участка FRAXE
показало, что его экспрессия связана с амплификацией GCC-
повтора, связанного с CpG-островком (Knight et aL, 1993).
Нормальные индивиды имеют 6-25 копий этого повтора, а у
умственно отсталых FRAXE-позитивных пациентов выявляется
свыше 200 копий тринуклеотида с метилированием CpG-oc-
тровка (табл. 11). Таким образом, отмечается сходная ситуа-
ция с локусом FRAXA, которая демонстрирует другой пример
экспансии числа тринуклеотидных повторов, связанный с на-
следственным заболеванием у человека. Однако, в отличие от
синдрома ломкой Х-хромосомы, число GCC-триплетов может
увеличиваться или уменьшаться и является нестабильным в
равной степени при передаче по мужской или женской линии.
В локусе FRAXF у нормальных индивидов число CGG-повторов
варьирует от 12 до 26 (в среднем 14), а у цитогенетически
124
Нетрадиционное наследование
позитивного мужчины с задержкой развития было обнаруже-
но увеличение числа триплетов свыше 900 (Ritchie et al., 1994).
Факт существования трех клинически значимых фолат-чувстви-
тельных сайтов в дистальной части длинного плеча Х-хромо-
сомы является интригующим и ставит вопрос об эволюцион-
ном значении таких участков хромосом в организации генома,
их структуре, функции и связи с патогенезом умственной от-
сталости.
Вскоре после открытия факта амплификации CGG-повто-
ров при синдроме ломкой Х-хромосомы было сделано еще
одно открытие: при исследовании Х-сцепленной спинобуль-
барной мышечной атрофии (болезни Кеннеди) Ла Спада с со-
авторами (La Spada et al., 1991; 1992) обнаружили очень похо-
жую амплификацию тринуклеотидных повторов, на сей раз
CAG-триплета в Х-сцепленном гене рецептора андрогена (табл.
11). Клинически это заболевание с поздним проявлением ха-
рактеризуется прогрессирующим ослаблением и атрофией
мышц, вследствие дегенерации спинальных и бульбарных
мотонейронов, но у пораженных мужчин отмечаются также
гинекомастия и снижение фертильности, связанные с дефек-
том функции рецептора андрогена. Молекулярно-генетичес-
кий анализ показал, что у нормальных лиц число CAG-повто-
ров колебалось в пределах 17-26, в то время как у больных
оно составило 40-52.
В 1992 году появилось несколько сообщений о результатах
молекулярно-генетического анализа другого аутосомно-доми-
нантного нейромышечного заболевания - миотонической дис-
трофии. Эта патология клинически характеризуется ослабле-
нием мышц головы и шеи, катарактой, гипогонадизмом, фрон-
тальным облысением и изменениями электрокардиограммы.
Миотония и ослабление мышц приводят к нарушению глота-
ния и речи. У 25-50% больных наблюдаются абдоминальные
симптомы, обусловленные холелитиазом. Наблюдается также
моторная и сенсорная нейропатия. Молекулярный анализ му-
тантного гена, локализованного в сегменте 19q13.3, показал,
что и при этом заболевании также наблюдается увеличение
числа CTG-тринуклеотидных повторов в З’-конце гена (Harley
et al., 1992; Buxton et al., 1992; Mahadevan et al., 1992). В нор-
ме число данных повторов составило 5-27, причем большин-
ство здоровых лиц имело или 5 или от 11 до 15 CTG-повторов,
т.е. составили 2 модальных класса по числу этих повторов.
При заболевании число CTG-повторов значительно увеличи-
валось и колебалось в пределах 50-1600.
125
Глава 5
3-
I
Величина амплифицированного фрагмента ДНК варьиро-
вала между пораженными сибсами и возрастала с поколения-
ми параллельно увеличению тяжести заболевания, т.е. здесь
также наблюдался феномен антиципации. Интересно, что в
мышечной ткани больных миотонической дистрофией, кото-
рая поражается в большей степени по сравнению с другими
126
Нетрадиционное наследование
тканями, обнаружена и большая степень экспансии CTG-пов-
торов (Anvret et al., 1993).
Дальнейшие исследования (Fu et al., 1992; Tsilfidis et al.,
1992) показали, что число CTG-триплетов при заболевании
может колебаться от 80 до 2000, а у лиц с минимальной ам-
плификацией повтора (от 50 до 80 копий) патология часто не
диагностируется или имеются минимальные признаки забо-
левания. Этот класс мутаций при миотонической дистрофии
был обозначен как протомутация (Barcelo etal., 1993). Следу-
ет также отметить, что при данном заболевании были отмече-
ны случаи нарушения феномена антиципации, в частности, у
детей пораженных родителей некоторые авторы отмечали рег-
рессию числа копий CTG-повторов до состояния протомута-
ции (Shelbourne et al., 1992; Brunner et aL, 1993; O'Hoy et aL,
1993; Giordano et aL, 1994).
В конце марта 1993 года объединенными усилиями между-
народной исследовательской группы молекулярных биологов
по обе стороны Атлантического океана было объявлено о
клонировании гена хореи Гентингтона (Huntington's Disease
Colloborative Research Group, 1993). К этому успеху исследова-
тели шли целых 10 лет, с момента, когда Джим Гузелла с
соавторами в 1983 году (Gusetla et al., 1983) сообщили о
сцеплении мутантного гена с генетическим маркером G8
(D4S10) короткого плеча хромосомы 4. В результате серии
последовательных клонирований поиск гена хореи Гентингто-
на был сужен до 500-килобазной области сегмента 4р16.3.
Проблема заключалась в том, что почти каждый выявляемый в
этой области ген мог быть геном данного заболевания, пос-
кольку даже если секвенирование таких генов не показывает
различий между нормальной хромосомой и хромосомой, не-
сущей мутантный ген, то это вовсе не исключало возможнос-
ти регуляторной мутации за его пределами. В результате ис-
следований, в предполагаемой области локализации мутан-
тного гена на хромосоме 4 был идентифицирован ген, зани-
мающий свыше 200 тысяч пар оснований, открытая рамка счи-
тывания которого содержала высокополиморфный тринукле-
отидный повтор CAG. Оказалось, что как и в случае вышеупо-
мянутых заболеваний, нормальные хромосомы имели 11-34
копии CAG-повтора, в то время как хромосомы, несущие ген
хореи Гентингтона, содержали свыше 42, а некоторые даже
более 100 копий этого повтора (Huntington's Disease Collobo-
rative Research Group, 1993). У больных с хореей Гентингтона,
CAG-повтор, также как и при других болезнях с увеличением
числа тринуклеотидных повторов, отличался высокой неста-
бильностью, в то время как родители этих больных были
127
Глава 5
клинически здоровы и имели число повторов на пределе вер-
хних значений нормального диапазона.
Интересно, что в ходе исследования этих больных были
найдены 2 новых мутации в предполагаемом гене данного за-
болевания, что с большой вероятностью свидетельствовало
о том, что этот ген и является искомым геном хореи Гентин-
гтона. Успешное клонирование данного гена позволит усовер-
шенствовать пресимптоматическую и дородовую диагностику
заболевания, а также будет способствовать дальнейшему
поиску новых мутаций гена и анализу генетической гетероген-
ности данной патологии в разных странах мира.
Вскоре после открытия гена хореи Гентингтона Орром с
соавторами (Orretal., 1993; Davies, 1993) было выявлено еще
одно наследственное неврологическое заболевание у чело-
века с экспансией числа тринуклеотидных повторов - спино-
мозжечковая атаксия первого типа (SCA1). Это заболевание
характеризуется нейродегенераций мозжечка, ствола мозга и
спинного мозга. Коварство этой болезни, также как и хореи
Гентингтона, заключается в том, что она не выявляется при
рождении, а становится очевидной только к юношескому воз-
расту, причем смерть наступает обычно через 10-20 лет после
начала заболевания. Следует указать на значительную гене-
тическую гетерогенность заболевания. К настоящему време-
ни идентифицированы гены уже не менее 6 разных форм дан-
ной патологии, располагающиеся на разных хромосомах - SCA1
- 6р23; SCA2 - 12q23-q24.1; SCA4 - 16q; SCA5 - 11cen; SCA7 -
Зр21.1; SCA8 - 10q23.3-q24.1 (McKusick, 1996). Одна из опи-
санных форм заболевания, обозначенная ранее как SCA3, ока-
залась аллельным вариантом болезни Мачадо-Джозефа, на
которой мы остановимся ниже (Twist et al., 1995).
Opp с соавторами напрямую скринировали космиды с ДНК
из критической области хромосомы 6 на предмет содержания
тринуклеотидных повторов при спиномозжечковой атаксии
первого типа (SCA1). В результате была изолирована неболь-
шая последовательность генома, которая гибридизовалась с
зондом, содержащим CAG-повтор. Было показано, что у здо-
ровых лиц число копий этого повтора колебалось от 25 до 36,
а у пораженных индивидов оно составило 43-81. Также как и в
других случаях подобных заболеваний наблюдается четкая
корреляция между количеством CAG-повторов и возрастом
начала заболевания. Показано, что пол передающего мутант-
ный ген родителя играет большую роль в распределении раз-
мера аллелей с экспансией числа тринуклеотидных повторов:
аллели с более чем 54 повторами передавались исключитель-
но пораженными отцами (Jodice et al., 1994), а носители этих
аллелей имели меньшую вероятность выживания.
128
Нетрадиционное наследование
В начале 1994 года две группы исследователей (R.Koide et
al., 1994; Nagafuchi et al., 1994) независимо друг от друга иден-
тифицировали экспансию нестабильного CAG-повтора при
другом наследственном заболевании - дентаторубро-палли-
долюисовой атрофии (DRPLA). Это редкое аутосомно-доми-
нантное нейродегенеративное заболевание, характеризующее-
ся вариабельной комбинацией прогрессирующей атаксии, хо-
реи, атетоза, миоклонус-эпилепсии и деменции. Это заболе-
вание почти не встречается в западных странах, но относи-
тельно часто встречается в Японии, с частотой приблизитель-
но 1 случай на миллион (Miwa, 1994). Мутантный ген, локали-
зованный на коротком плече хромосомы 12, был открыт вслед-
ствие наблюдения, что данное заболевание имеет клиничес-
кие и генетические характеристики сходные с другими забо-
леваниями с экспансией числа тринуклеотидных повторов. В
частности, отмечалось определенное сходство рассматрива-
емой патологии с хореей Гентингтона по следующим при-
знакам: 1) CAG-повтор локализован ближе к 5’-концу кодиру-
ющей области; 2) имеется хорошая корреляция между воз-
растом дебюта заболевания и числом CAG-триплетов; 3) по-
томки пораженных отцов обнаруживают более более тяжелое
клиническое течение заболевания, характеризующееся миок-
лонус-эпилепсией; 4) экспансия числа тринуклеотидных пов-
торов происходит в мужском гаметогенезе.
Японскими исследователями в 1994 году описана еще одна
динамическая мутация при другом аутосомно-доминантном
заболевании, сопровождающемся спино-мозжечковой дегене-
рацией - болезни Мачадо-Джозефа (Kawaguchi et al., 1994).
Авторами было обнаружена экспансия CAG-повторов в гене,
обозначенном MJD1. У нормальных индивидов ген содержал
13-36, а у пораженных - 68-79 повторов. Вызывает интерес
тот факт, что, несмотря на одинаковую длину повторов, у по-
раженных братьев заболевание возникает в среднем на 10 лет
раньше, чем у сестер.
В 1996 году Campuzano и др. (1996) изолировали дефект-
ный ген Х25, вызывающий атаксию Фридрейха. Ген Х25 коди-
рует белок с неизвестной функцией, состоящий из 210 амино-
кислот, который назвали “фратаксин”. Авторы обнаружили
уменьшение уровня мРНК фратаксина у пациентов с атаксией
Фридрейха и увеличенный нестабильный тринуклеотидный
GAA-повтор в первом интроне гена у пациентов с этим забо-
леванием. Нормальные хромосомы содержали 7-22 повтора,
а больные с атаксией - от 200 до 900. Кроме того, авторы
описали три редких точковых мутации гена Х25. Таким обра-
зом, атаксия Фридрейха является еще одним заболеванием с
экспансией GAA-повтора. Эти повторы обладали мейотичес-
129
9 Заказ № 127
Глава 5
кой нестабильностью со средней вариабельностью в 150 еди-
ниц (F/7/а et al., 1996). Длина как большого, так и малого алле-
лей у каждого пациента обратно коррелировала с возрастом
начала заболевания, причем меньшие аллели обнаруживали
более тесную корреляцию с этим признаком. Авторы показа-
ли также, что средняя длина аллелей с GAA-повторами была
значительно выше у больных с атаксией Фридрейха, имеющих
такие осложнения как диабет или кардиомиопатию.
Учитывая тот факт, что ряд неврологических заболеваний
человека связаны с белками, имеющими аномально длинный
полиглютаминовй тракт, Karlin и Bruge (1996) провели скри-
нинг имеющихся баз данных бейковых последовательностей
человека, мыши, дрозофилы, ячменя и кишечной палочки с
целью идентификации всех белков, содержащих множествен-
ные длинные гомопептиды. Оказалось, что такие белки, вклю-
чающие не только глютаминовые остатки, составили около 12%
у дрозофилы и только около 1,7% у человека, мыши и ячменя.
Белки кишечной палочки вообще не имели исследуемых по-
липептидных участков. Наличие в белках множественных длин-
ных гомопептидов сильно коррелировало с наличием других
необычных последовательностей, включающих множественные
заряженные белковые кластеры, перемежающиеся с протя-
женными незаряженными сегментами, и избыточное число
длинных повторяющихся гомопептидов с двумя и более ами-
нокислотными остатками. Вызывает интерес то обстоятельст-
во, что подавляющее большинство идентифицированных бел-
ков дрозофилы играют большую роль в онтогенезе и особен-
но важны для развития центральной или периферической нерв-
ной системы. Почти половина выявленных белков человека и
мыши оказались гомологами гомеозисных генов. По-видимо-
му длинные гомопептидные участки в белках играют важную
структурную роль в тонкой трехмерной конформации белко-
вых молекул, влияющей, в свою очередь, на их множествен-
ную функциональную активность. Избыточная экспансия го-
мопептидов в белке может изменить организацию белковых
доменов, что может привести к аберрантной внутримолеку-
лярной конформации и изменению межбелкового взаимодей-
ствия.
Заметное преобладание механизма экспансии числа три-
нуклеотидных повторов в этиологии заболеваний, связанных
с патологией нервной системы, заставило исследователей
предпринять попытки поиска нестабильных участков ДНК и при
других болезнях, обнаруживающих феномен антиципации, в
том числе и при классических мультифакториальных заболе-
ваниях. К такой патологии можно отнести шизофрению, эндо-
генные психозы, биполярные аффективные расстройства, ана-
130
Нетрадиционное наследование
лизу которых посвящается все нарастающее число работ (Труб-
ников, Голимбет, 1996; Bassett, Нопег, 1994; Sasaki et al., 1996).
Sasaki и др. (1996) исследовали ряд генов, обнаруживающих
полиморфизм тринуклеотидных повторов (В1, ВЗЗ, В37 и N-
кадерин), у больных с шизофренией и биполярными аффек-
тивными расстройствами. Авторы не обнаружили существен-
ной роли исследованных генов при данной патологии, однако
они не исключают возможности участия других генов с неста-
бильными участками в этиологии этих заболеваний. Некото-
рые авторы, однако, считают, что экспансию числа тринуклео-
тидных повторов нельзя рассматривать в качестве единствен-
ного механизма, лежащего в основе молекулярно-генетичес-
кой природы антиципации (Трубников, Голимбет, 1996).
Таким образом, подводя итог рассмотрения наследствен-
ных заболеваний с экспансией числа тринуклеотидных повто-
ров, следует отметить, что в течение короткого времени был
открыт новый класс наследственных болезней, чиоло которых
уже достигло 10. Происхождение мутации увеличения повто-
ров при всех рассматриваемых заболеваниях основывается,
вероятно, на общем неизвестном пока механизме. Не ясно
также как влияет экспансия числа тринуклеотидных повторов
на тонкие биохимические процессы, приводящие, в конечном
итоге, к развитию данных заболеваний. Возможные события
гиперэкспансии триплетов ДНК могут включать потерю или
изменение скорости транскрипции или трансляции, нестабиль-
ность мРНК, формирование аберрантных шпилечных структур
ДНК. Увеличение числа ДНК-повторов причинно связано с ги-
перметилированием ДНК, которое, как известно, выключает
транскрипцию. Однако даже если и произошла успешная тран-
сляция белка с длинным гомопептидным участком, то на уров-
не белка также могут возникать трудности успешной реализа-
ции наследственной информации вследствие агрегации бел-
ковых молекул путем прикрепления белков с длинным гомо-
пептидным трактом к неродственным молекулам, неуместной
мультимеризации синтезированных молекул и т.д. (Carlin, Bur-
ge, 1996).
Учитывая значительный интерес исследователей к расшиф-
ровке молекулярно-генетических механизмов наследственных
заболеваний, следует полагать, что число заболеваний с эк-
спансией числа тринуклеотидных повторов еще не является
окончательным. Имеет смысл проверка на подобный меха-
низм нарушения работы генов и других наследственных за-
болеваний с феноменом антиципации, материнским или от-
цовским эффектом, варьирующей пенетрантностью.
131
Глава 6
НЕКОТОРЫЕ ПРИКЛАДНЫЕ АСПЕКТЫ
ГЕНОМНЫХ ИССЛЕДОВАНИЙ
Геномные исследования • картирование генов человека,
идентификация полиморфных генетических маркеров, секве-
нирование генома - ключ к познанию фундаментальных зако-
номерностей организации и функционировния генома, меха-
низмов реализации генетической информации в норме и при
патологии. Анатомирование генома человека открывает путь
и для широкого спектра биомедицинскимх приложений - от
генодиагностики и терапии до идентификации личности с по-
мощью анализа ДНК в криминалистике. В задачу настоящей
книги не входит освещение всех возможных областей практи-
ческого применения ДНК-технологий. В этой главе мы оста-
новимся лишь на некоторых прикладных аспектах анализа ге-
нома, наиболее значимых для практической медицины. Мето-
ды ДНК-диагностики моногенных наследственных болезней,
молекулярно-цитогенетической диагностики тонких хромосом-
ных нарушений, генотерапевтические подходы - вот те ДНК-
приложения, которые уже вошли или войдут в ближайшем
будущем в арсенал практического врача.
ДНК-диагностика моногенных болезней*
В данном разделе будут рассмотрены вопросы ДНК-диаг-
ностики только моногенных наследственных болезней. ДНК-
диагностика мультифакториальных заболеваний также актив-
но разрабатывается, особенно в последние годы, но обычно
она может быть эффективной лишь при тех патологических
состояниях, для которых удается вычленить “главный" ген,
мутации которого приводят к болезни; в этих случаях страте-
гия диагностики мультифакториальной патологии сходна с ДНК-
исследованиями при моногенных болезнях.
Все разнообразие применяемых в настоящее время в мо-
лекулярной генетике подходов для идентификации определен-
ных генов или определенных фрагментов ДНК и их вариаций
основывается на двух основных методологических разработ-
* Раздел написан в соавторстве с к.б.н. О.Н.Одиноковой
Некоторые прикладные аспекты геномных исследований
ках - технологиях блот-гибридизации и амплификации отдель-
ных участков ДНК.
Методология блот-гибридизации используется уже более
20 лет без принципиальных модификаций. Принцип блот-гиб-
ридизации ДНК по Саузерну показан на рис. 13.
Образец
крови
Рис. 13. Блог-гибридизация по Саузерну.
Анализ начинается с выделения ДНК из любой ядросодер-
жащей ткани организма (из любого органа, ткани, культивиру-
емых клеток). Обычно у обследуемого берется несколько мил-
Глава 6
лилитров крови, а при пренатальной диагностике - амниоти-
ческая жидкость или биоптат хориона. Выделенная клеточная
ДНК обрабатывается одной из рестрикционных эндонуклеаз,
расщепляющей ДНК в строго определенных сайтах. В резуль-
тате получается характерный для данного человека набор из
огромного множества фрагментов различной длины, которые
при электрофоретическом фракционировании в агарозном или
полиакриламидном геле располагаются в зависимости от их
молекулярного веса; чем меньше фрагмент, тем выше ско-
рость его миграции в геле. На следующей стадии происходит
сам Саузерн-блоттинг (названный по имени доктора из Эдин-
бурга Эдмунда Саузерна, предложившего метод, а английс-
кое blot - означает промокать); фрагмент ДНК переносится ос-
мотическим током жидкости из влажного геля на помещен-
ный на него нитроцеллюлозный или нейлоновый фильтр. На
фильтре получается реплика ДНК-рестрикта. При переносе в
щелочной среде ДНК денатурирует, то есть переводится в
одноцепочечное состояние, в котором она может гибридизо-
ваться с меченым ДНК-зондом на исследуемый ген или ДНК-
фрагмент. В зависимости от способа мечения (радиоактив-
ный или флуоресцентный) гибридные фрагменты, соответству-
ющие изучаемой ДНК-последовательности, выявляются авто-
радиографически или с помощью флуоресцентной метки.
Методология амплификации дает возможность оперировать
с существенно меньшими количествами ДНК. Полимеразная
цепная реакция (ПЦР) позволяет проводить селективную ам-
плификацию отдельных регионов ДНК посредством имитации
in vitro репликации ДНК. Для реакции необходимы олигонук-
леотидные праймеры, комплиментарные фланкирующим пос-
ледовательностям анализируемого участка ДНК.
ДНК-цепь, комплиментарная взятой для исследования мат-
рице (одноцепочечной исследуемой ДНК), синтезируется при
участии фермента Taq-ДНК-полимеразы, начиная со свобод-
ного-3’- конца праймера. Для проведения ПЦР (рис. 14) в ре-
акционной смеси необходимо наличие анализируемой ДНК,
всех четырех дезоксинуклеотидтрифосфатов (dNTP; dATP,
dTTP, dGTP, dCTP), ДНК-полимеразы, двух олигонуклеотид-
ных праймеров - прямого (F-forward) и обратного (R - reverse).
Одноцепочечная ДНК-матрица легко образуется при теп-
ловой денатурации двойной цепи ДНК. Олигонуклеотидные
праймеры можно либо синтезировать в лаборатории, либо
приобрести. Обычно используемые для ПЦР реакционные
буферы содержат ионы Мдг+, моновалентные катионы и неко-
торые добавки. Добавки могут помогать стабилизировать фер-
мент, влиять на кинетику процесса и/или температуру плавле-
ния ДНК (Тт). Большинство используемых ДНК-полимераз яв-
Некоторые прикладные аспекты геномных исследований
ляются термостабильными и могут выдерживать нагревание
до 95° - 97° С.
Как правило, ПЦР требует три температурных режима: 1)
при 90-95° С осуществляется температурная денатурация двой-
ной нити ДНК на две комплиментарные цепи; 2) при 50-65° С
происходит отжиг праймеров (их гибридизация) на специфич-
ных праймерам последовательностях матрицы; 3) обычно при
72° С протекает ферментативный синтез цепи ДНК, то есть
праймер достраивается, начиная с 3’-конца, присоединением
dNTP. Последовательные денатурация, отжиг и синтез с опре-
деленным фиксированным временем каждого этапа называ-
ется циклом. Повторение таких циклов ведет к множествен-
ной амплификации ДНК.
исходная ДНК
dNTP (dATP, сПТР, dCTP, dGTP),
Taq-ДНК-полимераза,
Праймеры:
Цикл амплификации:
1. Денатурация: 90°-95°С
2. Отжиг праймеров: 50°-65“С
3. Синтез ДНК: 72°С
п - количество циклов реакции
Рис. 14. Схема псЛвимеразной цепной реакции (ПЦР).
Глава 6
Специфичность ПЦР обеспечивается в первую очередь
праймерами, которые синтезируются химическими методами
и обычно имеют длину 20-30 нуклеотидов. Подбор эффектив-
ных праймеров для ПЦР - процесс эмпирический, но соблю-
дение определенных требований увеличивает вероятность
получения праймеров пригодных для использования.
1. Выбирают праймеры с содержанием GC>>50 % и слу-
чайным распределением оснований. Следует избегать прай-
меров с протяженными полипуриновыми или полипиримиди-
новыми последовательностями.
2. Избегают последовательностей с устойчивой вторичной
структурой.
3. Проверяют затравки на комплиментарность друг другу.
Праймеры? отжигающиеся друг с другом, не смогут участво-
вать в амплификации (Анализ генома, 1990).
Праймеры ориентированы таким образом, чтобы синтез
протекал только между ними (см. рис. 14), удваивая количес-
тво копий этого участка ДНК. Поскольку праймеры физически
включаются в концы продуктов амплификации, они детерми-
нируют сам продукт реакции - фрагмент ДНК, равный по дли-
не расстоянию между 5’-концами праймеров на исследуемом
участке ДНК. В результате циклического повторения стадий
ПЦР происходит экспоненциальное увеличение количества
специфического фрагмента по формуле: (2П- 2п), где п - коли-
чество прошедших циклов амплификации.
Для постановки ПЦР используют специальные приборы -
ДНК-амплификаторы или термоциклеры, позволяющие про-
граммировать и поддерживать необходимый температурный
цикл, обеспечивающий оптимальное прохождение каждой ста-
дии реакции.
ПЦР позволяет в течение нескольких часов выделить и раз-
множить определенный фрагмент ДНК в количестве, превы-
шающем исходное примерно в 109 раз. Такая высокая степень
направленного обогащения значительно упрощает работу с
минимальными количествами ДНК-образцов. Реакция высоко
специфична и очень чувствительна, позволяет исследовать
даже единичную копию гена в исходном материале.
Для идентификации мутаций разработаны подходы, приве-
денные в таблице 12. Говоря о подходах к ДНК-диагностике
наследственных болезней, прежде всего, различают прямую
и непрямую молекулярную диагностику генных болезней.
В случае прямой диагностики объектом исследования яв-
ляются мутации определенного гена, идентификация которых
- главная задача анализа. Данный вариант диагностики наибо-
лее точен, но для его осуществления необходима информа-
ция о локализации, природе и частоте наиболее частых (ма-
Некоторые прикладные аспекты геномных исследований
жорных) мутаций гена или данные о наличии в гене “горячих
точек” или районов преимущественного мутирования. К пре-
имуществам прямых методов, кроме практически стопроцент-
ной точности диагностики у обследуемого, относится отсут-
ствие необходимости обследования других родственников
пробанда.
Таблица 12.
Подходы к ДНК-диагностике наследственных
болезней
I. Прямая диагностика мутаций - детекция крупных перестроек (мутаций) генов методами блот-гибридизации с использованием ДНК-зондов; - выявление крупных и мелких делеций генов ПЦР-амплификацией отдельных фрагментов, в том числе мультиплекс- ной ПЦР; - детекция внутригенных мутаций, измененяющих сайты узнавания определенных рестриктаз (и вследствие этого - размер фрагментов, выявляемых блот-гибридизацией или ПЦР-амплификацией); - аллель-специфическая гибридизация (амплификация) с использованием олигонуклеотидов, комплементарных нормальной или мутантной последовательности ДНК; - детекция конформационного полиморфизма одноцепочечной ДНК (single strand conformation polymorphism); - метод детекции ошибок спаривания (cleavage mismatch detection); - гетеродуплексный анализ фрагментов гена; - секвенирование гена или его фрагмента.
II.Косвенная (непрямая) молекуляр- ная диаг- ностика - Анализ косегрегации генетических маркеров (микро- и минисателлиты, ПДРФ), сцепленных с патологическим геном, и болезни в семьях
Прямая диагностика эффективно применяется пока толь-
ко для сравнительно небольшого числа наиболее распростра-
Глава 6
ненных наследственных заболеваний, в том числе: муковис-
цидоз, недостаточность а,-антитрипсина, фенилкетонурия,
миодистрофия Дюшенна, гемофилии А и В, недостаточность
21-гидроксилазы, талассемии, нейрофиброматоз, синдром
ломкой Х-хромосомы и некоторые другие, молекулярная при-
рода которых уже хорошо изучена.
Ниже мы подробнее проиллюстрируем основные подходы
к ДНК-диагностике.
Гибридизационную детекцию крупных ДНК-перестроек (де-
леций или дупликаций) продемонстрируем на примере мышеч-
ной дистрофии Дюшенна - МДД (рис. 15). Кодирующая часть
гена дистрофина (кДНК) длиной 14 Кб, условно подразделен-
ная, начиная с 5’-конца гена, на субфрагменты размером 1
Кб, была получена в виде 9 отдельных клонов: 1-2, 3, 4-5Ь, 6а-
7, 8, 9-10, 11а-12Ь, 13 и 14 (Koenig М. etal., 1987) (рис. 15 А).
Каждый из клонов может быть использован в качестве зонда
для ДНК-исследований. В приводимом примере в Hind lll-рест-
рицированной ДНК больного блот-гибридизацией с зондом
кДНК 8 выявлено отсутствие отдельных фрагментов, соответ-
ствующих центральной области гена (рис. 15 Б, В).
Подобные крупные мутации генов стало возможно легче и
быстрее определять, с развитием описанной выше ПЦР-тех-
нологии, причем в случае мультиплексной (мультилокусной)
ПЦР в одной пробирке можно провести тестирование сразу
нескольких разных ДНК-локусов. На рис. 16 показаны некото-
рые осуществленные нами случаи детекции делеций крупных
регионов (экзонов) гена дистрофина у больных МДД методом
мультиплексной амплификации с набором специфических
праймеров, фланкирующих отдельные области гена. Наличие
делеций устанавливается по отсутствию соответствующих про-
дуктов амплификации при электрофоретическом анализе ам-
плификатов. В приведенном примере можно видеть у больных
№2 и №6 отсутствие фрагмента ДНК, соответствующего экзо-
ну 45; у больного №3 - нет экзона 44; в образце ДНК больного
№4 выявляется более протяженная делеция, захватывающая
экзоны с 8 по 19; в случае №5 обнаружен дефект экзона 48.
Определяя точно размер анализируемого фрагмента ДНК,
можно анализировать и небольшие мутации гена. Рассмотрим
на примере ПЦР-диагностики самой частой мутации при му-
ковисцидозе (D F-508) - делеции трех пар нуклеотидов, коди-
рующих одну аминокислоту (фенилаланин) в 508-положении
белкового продукта гена муковисцидоза (рис. 17). На рис. 18
приведены результаты обследования семьи, в которой один
из сыновей - больной муковисцидозом. Молекулярно-генети-
ческий анализ показал наличие у больного (№2 - отмечен
стрелкой) вместо нормального фрагмента ДНК длиной 91 пара
Некоторые прикладные аспекты геномных исследований
оснований укороченного фрагмента длиной 88 п.н. Носителя-
ми такого же укороченного фрагмента гена являются родите-
ли пробанда (№1 и 4). Важно и то, что носителем патологи-
ческого гена является и здоровый брат пробанда. Ему (его
будущей семье) рекомендовано в последующем (при вступ-
лении в брак) медико-генетическое консультирование для про-
гноза потомства. Поскольку носителем мутаций гена муковис-
цидоза в европейских популяциях является один из 20-25 че-
ловек, высока вероятность появления у него больного ребен-
ка.
А. кДНК дистрофина и фрагменты, используемые как ДНК-зонды:
кДНК з 6а-7 9-10 13
клоны 1-2 ™ 4-5Ь 8 ^ив11а-12Ь^^ 14
Н^ММ ! ШМН М
т.н.п. о 2.5 5 7.5 10 12.5 14
регионы частых делеций
Б. Hind Ill-сайты (*) в области кДНК8:
* * * * * * *
10.0 1.25 3.8 1.6 3.7 3.1 7 0 (т.п.н.)
В. Блот гибридизация по Саузерну с зондом кДНК8:
1.25 тпн > ямм*
1.6 ТПН : UMHRf и
3.1 тпн МММ* МММ*
3.7 тпн ДВ ШД
3.8 тпн > МиМи
7.0 тпн i 4MMMW МММ*
10.0 тпн «МММ
норма больной
ММД
Рис. 15. Определение крупной делеции в гене дистрофина
блот-гибридизацией Hind 111-рестрицированной
ДНК больного с мышечной дистрофией
Дюшенна (с меченым зондом кДНК8).
У
Глава 6
Рис. 16. Делеционный анализ 8 отдельных регионов гена дис-
трофина (4, 8, 12, 19, 44, 45, 48 экзонов) у больных мышеч-
ной дистрофией Дюшенна методом мультиплексной ПЦР
(электрофореграмма продуктов амплификации):
1 - норма,
2 - делеция экзона 45,
3 - делеция экзона 44,
4 - протяженная делеция экзонов 8-19,
5 - делеция экзона 48,
6 - делеция экзонов 45-48.
Прямая детекция мутаций возможна и для нуклеотидных
мутаций ДНК, изменяющих сайты узнавания соответствующих
рестриктаз. Пример приведен на рис. 19. При серповиднокле-
точной анемии, вследствие мутационной замены в 6 кодоне
гена р-гемоглобина аденина (А) на тимин (Т), нормальная нук-
леотидная последовательность, узнаваемая ферментом Mst II
(CCTNTGG), изменена на последовательность, уже не распоз-
наваемую данным ферментом (CCTNAGG). В норме (ра) рес-
трикцией и гибридизацией с ДНК-зондом в данном регионе
гена выявляется фрагмент длиной 1.15 Кб, тогда как при опи-
санной мутации Саузерн-блотом определяется фрагмент (ps)
размером 1.35 Кб. Таким образом, при блот-гибридизации
могут быть четко дифференцированы нормальные гомозигот-
ные индивиды (АА), гетерозиготы (AS) и гомозиготы по серпо-
видноклеточной мутации (SS); при этом каждый из трех гено-
типов может быть точно определен.
Следующий подход к ДНК-диагностике - аллель-специфи-
ческая амплификация (или в другом варианте - гибридизация)
Некоторые прикладные аспекты геномных исследований
нормальной и мутантной_ДНК-последовательности проиллюс-
трируем на исследовании одной из основных мутаций в гене
фенилаланингидроксилазы (R408W: однонуклеотидная мута-
ция приводит к замене в белковом продукте кодируемой ами-
нокислоты), приводящей к фенилкетонурии (Dvorakova D. et
al., 1993).
Р S £ G К I к н S G К 7 S F С S 4%"
A .V И____________________________
561 CCTTCAGAGGGTAAAATTAAGCACAGTGGAAGAATTTCATTCTGTTCTC
AGTITTCCTGG
IMPGTIKENIIFG V S Y 516
DEYR_______________________________________
621 ATTATGCCTGGCACCATTAAACAAAATATCATCTTCGTGTTTCCTATGA
TGAATATAGA
Y R S V I К A ( Q L E E D / S К 536
F A E К____________________________________
681TACAGAAGCGTCATCAAAGCATGCCAACTAGAAGAGGACATCTCCAAGT
TGCAGAGAAA _____________________________
Рис. 17. Нуклеотидная последовательность участка кДНК гена
CFTR (нуклеотиды с 1561 по 1740 - указаны под чертой;
над чертой - соответствующие им аминокислоты). Цифры
слева указывают номер нуклеотида кДНК, а цифры справа
- номер кодируемой аминокислоты (Riordan et al., 1989).
Для исследования используются два ДНК-праймера, флан-
кирующих исследуемый 12-ый экзон гена (праймеры А и В), а
также олигонуклеотидные праймеры, комплиментарные нор-
мальной (праймер S) и мутантной последовательности (прай-
мер М) в локусе 408 (рис. 20).
Рис. 18. Детекция делеции DF-508 гена CFTR (делеция трех пар
нуклеотидов) ПЦР-методом в семье с муковисцидозом.
У
Г лава 6
Mst II - сайты рестрикции (CCTNAGG)
Б.
(CCTNTGG)
1,35 тпн
ДА AS SS
Примеры Саузен-блота
В.
Рис. 19. Детекция точковой мутации, нарушающей сайт
рестрикции фермента Mst II, при серповидно-клеточной
анемии (по E.Passarge, 1995).
Амплификация с различными комбинациями четырех прай-
меров (А/В - амплификация всего экзона, S/В - нормального
варианта, А/М - амплификация мутантной последовательнос-
ти) позволяет в одном эксперименте определить нормальное
состояние обоих аллелей, гетерозиготное носительство мута-
ции R408W и гомозиготу по мутации R408W в гене
фенилаланингидроксилазы.
Успешное развитие методов молекулярно-генетического
анализа последовательностей ДНК на основе технологии ПЦР
и различных модификаций электрофоретического анализа ДНК-
фрагментов позволяет характеризовать разнообразные гене-
Некоторые прикладные аспекты геномных исследований
тические нарушения, связанные с наследственной патологией
человека.
А.
интрон 11 экзон 12 интрон 12
pr. М: З'-GTT ATG GAA COG GGA AG-5'
рг. S: 5 -САА ТАС СТС GGC ССт ТС-3’
Б.
Гомозигота по
мутации R408W
Г етерозиготное
носительство
мутации R408W
Норма - отсутствие
мутации R408W
Рис. 20. Аллель-специфическая амплификация ДНК при диагнос-
тике мутации R408W в 12-ом экзоне гена фенилаланингид-
роксилазы.
Очень большая часть ДНК-дефектов в генах связаны с то-
чечными мутациями: нуклеотидными заменами, делециями,
дупликациями. Например, точечные мутации лежат в основе
95% нарушений гена фактора свертываемости VIII (гемофилия
A) (Berg et al., 1992), подавляющее большинство известных к
настоящему времени более чем 60G мутаций (Estivill, 1996) в
гене CFTR (ген трансмембранного белка муковисцидоза) - это
также точечные (нуклеотидные) мутации. При том, что эти
мутации в каждом конкретном случае при многих заболевани-
ях могут затрагивать разнообразные регионы гена, понятно,
что столь мелкие изменения ДНК не так легко определить.
Поэтому выбираемый подход анализа зависит, в первую оче-
редь, от целей исследования (Rolf et al., 1992): а) определе-
ние известных нуклеотидных мутаций в определенном фраг-
менте гена (как ранее бы>о проиллюстрировано на рис. 17 и
Глава 6
рис. 18); б) проведение скрининга отдельных участков гена на
наличие точковых мутаций; в) необходимость точного опреде-
ления мутаций в анализируемом фрагменте; д) определение
всех присутствующих мутаций и т.д. Разрабатывается много
различных экспериментальных процедур подобных исследо-
ваний. Спектр их постоянно пополняется. Из очень перспек-
тивных и довольно результативных (эффективных) опишем
здесь основы поиска единичных нуклеотидных мутаций в ге-
нах или ДНК-фрагментах через изучение конформационного
полиморфизма одноцепочечных фрагментов ДНК и гетеродуп-
лексный анализ.
Метод анализа конформационного полиморфизма одноце-
почечной ДНК (single strand conformation polymorphism) - SSCP-
анализа - основывается на различной подвижности однони-
тиевых ДНК-структур в неденатурирующем полиакриламидном
геле, зависящей от их первичной нуклеотидной последователь-
ности. Анализируемый фрагмент (рис. 21) амплифицируется в
ходе ПЦР. Затем продукт реакции подвергается термической
денатурации (при +95°С) в присутствии формамида и быстро-
му охлаждению, препятствующему реассоциации нитей ДНК.
Далее проводится электрофорез в полиакриламидном геле и
подвижность исследуемого ПЦР-продукта сравнивается с под-
вижностью аналогичных нормальных вариантов денатуриро-
ванной ДНК (или с подвижностью ДНК-вариантов с известной
нуклеотидной последовательностью). Наличие внутри иссле-
дуемой цепи ДНК одной точечной мутации уже приводит и
изменению электрофоретических характеристик вследствие
изменения вторичной структуры фрагмента ДНК в геле. Фраг-
менты одной длины, но разные по последовательности, име-
ют разные скорости миграции в ходе электрофореза. Таким
образом, в SSCP-анализе, детекция мутантной последователь-
ности основана на различиях миграции в геле вследствие из-
менения конформации ДНК.
Гетеродуплексный анализ фрагментов ДНК также позволя-
ет выявлять мелкие (нуклеотидные) мутации в последователь-
ностях ДНК.
При данном подходе анализируемый фрагмент гена ампли-
фицируется в реакции ПЦР, а затем создаются условия для
образования гетеродуплексов между различными аллелями,
имеющими единичные расхождения в последовательности
нуклеотидов. Для этого амплификаты денатурируют при +95аС,
а затем медленно охлаждают.
Некоторые прикладные аспеюы 1еномных исследований
геномная ДНК
5’ _ 5'
3---------------------------------- з-
" ПЦР t J‘P-dCTP
аллель 1 аллель 1 >2 аллель 2
---А---- А--- С---
---Т---- Т--- G---
-С-
— G - —
I
Термоденатурации
в присутствии формамида
Рис. 21. Схема SSCP-анализа (Rolfs et al., 1992).
При этом происходит реассоциация молекул ДНК, в том
числе двойные цепи ДНК образуются не только между иден-
тичными однонитиевыми фрагментами, но и не полностью
комплиментарными ДНК-последовательностями. Так, при на-
личии в смеси ПЦР-продуктов разных аллелей гетеродуплек-
сы формируются между диким типом (нормальным вариантом)
и мутантной ДНК. Гетеродуплексные молекулы, вследствие
нарушения их вторичной конфигурации, имеют, обычно, мень-
шую подвижность в гидролизующем полиакриламидном геле
(ПААГ), чем соответствующие гомодуплексы, и выявляются
по изменению расположения ДНК-фрагментов в геле (Prior et
al., 1993). Основные этапы гетеродуплексного анализа показа-
ны на рисунке 22. Среди преимуществ гетеродуплексного ана-
лиза следует выделить его высокую чувствительность в оп-
ределении замен одного основания, возможность быстрого
обследования (скринирования) на наличие потенциальных му-
таций в различных областях исследуемых генов.
Конечно, в идеальном варианте все подходы к выявлению
аномального поведения при электрофорезе, связанные с из-
менением в нуклеотидных последовательностях (кроме опи-
санных используют еще анализ фрагментов в денатурирую-
Г лава 6
щих гелях и др.), должны завершаться секвенированием гена
или его фрагментов для точного описания мутаций.
геномная ДНК
5'—---------------------------------------------”----- 5’
3----—------------------------------------------------- 3’
ПЦР
аллель 1 аллель 2
- К -
инкубация
смешивание
денатурация
Гетеродуплексы
денатурирующий (7-8 М мочевина) ПААГ
Рис. 22. Схема гетеродуплексного анализа.
Описанные методы не исчерпывают всех подходов к пря-
мой диагностике наследственных болезней. В определенных
ситуациях, если известно, что заболевание связано с малым
количеством потенциальных мутаций, возможна интенсифи-
кация молекулярного анализа. Например, для скрининга на
ограниченный в ряде популяций круг мутаций при муковисци-
дозе применяют обратную дот-блот-гибридизацию: в реакции
гибридизации тестируются меченые амплифицированные
фрагменты ДНК с иммобилизованными олигонуклеотидами,
соответствующими нормальному и мутантному аллелю.
Непрямые (косвенные) методы анализа ДНК в иссле-
довании наследственных болезней более универсальны,
поскольку могут применяться и в тех случаях, когда не извес-
тна природа мутаций, когда ген болезни точно не идентифи-
цирован, а также они могут быть успешно использованы для
заболеваний, при которых мутации в генах слишком разнооб-
разны и не отмечается ярко выраженных мажорных (основ-
ных) патологических мутаций. ДНК-диагностика в этом случае
строится на семейном анализе различных ДНК-полиморфных
вариантов.
Исторически описанный первым и наиболее широко при-
менявшийся до недавнего времени тип полиморфизма ДНК -
рестрикционный полиморфизм - полиморфизм длин рестрик-
ционных фрагментов (ПДРФ). Самый обычный вариант ПДРФ
Некоторые прикладные аспекты геномных исследований
- наличие или отсутствие сайта рестрикции (вследствие раз-
личий нуклеотидных последовательностей ДНК). Полученные
после обработки рестриктазой фрагменты ДНК различных
людей могут различаться по длине и массе, а, следовательно,
и по электрофоретической подвижности.
Конечно, предпочтительнее для точного молекулярного ана-
лиза использовать маркеры, наиболее тесно сцепленные с
исследуемым геном, либо при наличии информации исполь-
зовать внутригенные ПДРФ. На рис. 23 продемонстрирован
данный подход диагностики на примере МДД. В показанной
семье проведено исследование одного из внутригенных поли-
морфных локусов - pERT87-8. Его амплификация и рестрик-
ция ферментом Taq I выявила у больного (3) два фрагмента
ДНК длиной 74 и 71 п.н. (наличие сайта рестрикции). Такой же
вариант выявлен и у матери-носительницы дефектного гена
(/). А у двух сибсов пробанда (4, 5) определен другой ДНК-
вариант (145 п.н.) с отсутствием сайта узнавания для рестрик-
тазы Taql и, следовательно, отсутствие сцепленного с данной
маркерной системой дефектного гена.
Таким образом, при непрямой диагностике используются
как внутригенные ПДРФ маркеры (как на рис. 23), так и вне-
генные (фланкирующие или тесно сцепленные с исследуемым
геном), хотя в последнем случае точность диагностики снижа-
ется из-за возможных рекомбинационных событий.
Рис. 23. Анализ ПДРФ гена дистрофина в семье с больным
МДД.
Наиболее интенсивно в настоящее время ведутся иссле-
дования полиморфных локусов генома известных как тандем-
ные повторы с изменяющимся числом копий (рис. 24). Длина
Глава 6
выявляемых фрагментов ДНК зависит от числа повторяющих-
ся элементом. Выделяют два типа: 1) минисателлиты или VNTR
(variable number of tandem repeats) с размером повторяюще-
гося блока более 10-20 п.н.; и 2) микросателлиты пли STR
(short tandem repeats) с размером повторяющегося фрагмен-
та 2-5 п.н. Такие локусы часто высоко полиморфны, что и обус-
ловливает их применение в медико-генетических целях.
Рис. 24. Анализ полиморфизма на основе вариации числа
тандемных повторов (по Фогель Ф., Мотульски А., 1990).
а - А, Б, В, Г - последовательности, различающиеся на
один повторяющийся сегмент. Размер (длина) фрагментов,
детектируемых по константным сайтам (сайтам рестрикции
или ограниченные олигонуклеотидными праймерами в
ПЦР-амплификации), остаются неизменными, а длина
фрагментов изменяется.
(L- Различные гетерозиготы после электрофореза.
В качестве примера приведен случай медико-генетическо-
го консультирования в семье с больным фенилкетонурией (рис.
25). Анализ вариантов полиморфного локуса (минисателлит-
ных повторов - VNTR - с длиной повторяющегося элемента 30
п.н.) в З’-области гена фенил-аланингидроксилазы (РАН) у
больного и его родителей позволил маркировать патологичес-
кие варианты гена в данной семье (аллельные варианты 1 и 4)
(рис. 25 В) и оценить перспективы последующей пренаталь-
ной диагностики в данной семье с использованием описанной
системы маркеров РАН-гена. Выявление нормальных и мутан-
тных аллелей (возможные варианты I и IV на рис. 25 Г) будет
свидетельствовать о состоянии носительства рецессивного
гена непораженным плодом; в случае варианта II (рис. 25 Г) -
плод здоровый (оба аллеля нормальные); вариант III - ребенок
болен.
Широкая распространенность в геноме, многоаллельность
и часто высокий уровень полиморфизма мини- и микросател-
литных повторов превращают их в идеальные полиморфные
маркеры для использования в ДНК-диагностике. Приведем
последний пример (рис. 26), демонстрирующий эффективность
Некоторые прикладные аспекты геномных исследований
использования таких полиморфизмов для молекулярной диаг-
ностики и медико-генетического консультирования семей боль-
ных даже в отсутствии ДНК-материала (что практически не
возможно при использовании ПДРФ) от пораженных в иссле-
дуемой семье индивидов.
(ЗОп н.)3 аллель 1
(ЗОп н )6 аллель 2
(ЗОп н )7 аллель 3
(ЗОп н )в аллель 4
(30п.н.)8
(30п.н.)12
аллель 5
аллель 6
Б. Семья с ребенком больным фенилкетонурией
В. Электрофореграмма
Г. Пренатальная диагностика
VNTR - локуса (возможные варианты)
Рис. 25. Анализ минисателлитных повторов (VNTR) гена РАН в
семье с больным^фенилкетонурией.
Глава 6
Косвенную молекулярную диагностику с детекцией ДНК-
маркера, расположенного близко к исследуемому патологи-
ческому гену, иллюстрирует случай обращения в НИИ меди-
цинской генетики СО РАМН за медико-генетическим консуль-
тированием и пренатальной диагностикой гемофилии А у пло-
да беременной женщины (рис. 16, пробанд 111-1). Было извес-.
тно о двух больных гемофилией А в ее семье: заболеванием
страдали два уже умерших брата (11-1, П-2) матери пробанда.
Рис. 26. Медико-генетическое консультирование в семье,
отягощенной гемофилией А, с исследованием
ДНК-маркера (DXS52 или St14), сцепленного
с геном фактора свертываемости крови VIII.
При отсутствии ДНК-материала от самих больных, иссле-
дованием у пробанда и ближайших родственников ДНК-поли-
морфного локуса St14 (DXS52), сцепленного с локусом гена
фактора VIII, методом ПЦР были выявлены следующие вари-
анты аллелей:
- у бабушки (1-1), имевшей двух больных сыновей, а,
следовательно, являющейся достоверной носительницей ге-
мофилии А, - 1390 п.н. и 1570 п.н.;
- у деда (I-2) - 1570 п.н.;
- у их дочери (или матери обследуемой, П-3) - 1390 п.н.
и 1570 п.н.;
- у отца обследуемой (П-4) -1930 п.н.
Сравнительный семейный молекулярно-генетический ана-
лиз указывает на наследование пробандом (111-1) от ее матери
Некоторые прикладные аспекты геномных исследований
(11-3) Х-хромосомы с аллельным вариантом 1570 п.н., который
идентичен аллелю, выявленному у её деда (I-2), не страдав-
шего гемофилией. Это позволяет, с вероятностью 97%, ис-
ключить наследование обследуемой (111-1) Х-хромосомы от ее
бабушки (1-1), облигатной носительницы мутантного гена ге-
мофилии А. Вероятность ошибки исключения статуса носи-
тельства у 111-1 может быть оценена как 3%, что определяется
расстоянием между двумя исследуемыми локусами (локус
DXS52 и локус гена фактора свертываемости крови VIII) и тео-
ретической вероятностью кроссинговера между двумя Х-хро-
мосомами у матери (П-3) обследуемой.
Таким образом, у беременной женщины-лробанда (111-1)
были исключены носительство патологического гена, с кото-
рым связано заболевание в данной семье, и потребность в
исследовании плода инвазивными методами пренатальной
диагностики.
В заключение подчеркнем, что имеющиеся методы и под-
ходы ДНК-анализа позволяют изучать практически любой ген
или фрагмент ДНК генома человека. В силу своей универсаль-
ности (возможность исследования любой ткани организма, на
любом этапе онтогенеза, вне зависимости от состояния эк-
спрессивности гена) ДНК-диагностика позволяет выявить па-
тологическое состояние в пресимптоматическом периоде, в
том числе пренатально, и провести возможные профилакти-
ческие мероприятия. В медико-генетическом консультирова-
нии молекулярная ДНК-диагностика может обеспечить возмож-
ность определения состояние носительства патологического
(мутантного) гена и оценить необходимость пренатальной ди-
агностики.
Молекулярно-цитогенетическая диагностика
тонких хромосомных нарушений*
Хромосомные болезни занимают существенное место в
структуре наследственной и врожденной патологии человека.
Эта большая группа патологических врожденных состояний
обусловлена численными или структурными аномалиями хро-
мосом, которые фенотипически проявляются разнообразны-
ми нарушениями развития организма. Предполагается, что
более 55% зигот человека несут хромосомные аберрации (Ку-
лешов, 1979). Под действием естественного отбора во внут-
риутробном периоде развития элиминируется до 99% зигот с
Раздел написан в соавторстве с проф. С.А. Назаренко
Г лава 6
мутациями в виде доимплантационных потерь, спонтанных
абортов и мертворождений (Назаренко, 1993).
У новорожденных частота хромосомных аберраций состав-
ляет около 0,6-0,8%, а у новорожденных с множественными
врожденными пороками развития (МВПР) частота хромосом-
ных аномалий возрастает до 40% (Залетаев, 1985). Около 0,3%
хромосомных мутаций у новорожденных приходится на ано-
малии в системе половых хромосом, на аутосомные трисомии
- 0,16% и на структурные перестройки - 0,24% (Кулешов, 1979,
1991). Хромосомная патология, сопровождающаяся дисбалан-
сом хромосомного материала, вызывает различные аномалии
развития у носителей и может быть связана не только с МВПР,
но и с умственной и физической отсталостью, нарушениями
полового развития, бесплодием и невынашиванием беремен-
ности. Эти больные формируют группы повышенного риска по
более высокой, по сравнению с популяционной, частоте выяв-
ления хромосомных мутаций и должны быть обязательно об-
следованы цитогенетически (Назаренко и др., 1989).
Диагностика числовых хромосомных нарушений не вызы-
вает каких-либо проблем. Большинство хромосомных пере-
строек также можно идентифицировать с помощью методов
дифференциального окрашивания хромосом. В течение дли-
тельного времени традиционные цитогенетические методы
оставались единственными при исследовании хромосомных
болезней. Однако, используя стандартные цитогенетические
методы, можно достичь обычного визуального разрешения
отдельного хромосомного сегмента размером 5-10 миллио-
нов пар оснований (Yung, 1996). Открытие в середине 70-х
годов способа получения удлиненных (прометафазных) хро-
мосом высокого разрешения (Yunis, /976), а затем и созда-
ние номенклатуры хромосом с числом 850-1000 сегментов
позволило существенно улучшить диагностику тонких хромо-
сомных нарушений и идентифицировать ряд новых синдромов,
связанных с микроструктурными перестройками отдельных
участков хромосом. В табл. 13 приведены некоторые из этих
синдромов, выявить которые ранее при использовании тради-
ционных методов цитогенетического анализа не удавалось.
Поскольку в ряде случаев у пациентов, имеющих данные син-
дромы не обнаруживались микроструктурные хромосомные
перестройки, исследователи стали предпринимать попытки
использовать для изучения этих областей хромосом методы
молекулярной генетики. С этой целью стали применять как
молекулярно-генетические методы - исследование полимор-
физма длины рестрикционных фрагментов (ПДРФ) и других
высокополиморфных молекулярно-генетических маркеров
Некоторые прикладные аспекты геномных исследований
(микро- и минисателлитов), таки молекулярно-цитогенетичес-
кие методы - флюоресцентную гибридизацию in situ (FISH) и
ее модификацию - супрессионную гибридизацию in situ (CISS).
В настоящем разделе рассматриваются молекулярно-цитоге-
нетические подходы к диагностикае тонких хромосомных на-
рушений.
Таблица 13
Синдромы множественных врожденных пороков развития
(МВПР) и злокачественные новообразования,
сопровождающиеся микрохромосомными аберациями
N Синдром, заболевание Хромосома, регион
МИКРОДЕЛЕЦИИ
1 Вильямса del 7)(q11)
2 Лангера-Гидеона del(8)(q24.1)
3 AWGR (аниридия, опухоль Вильмса, двойственность гениталий и умственная отсталость) del( 11 )(p13)
4 Ретинобластома del( 13)(q14.1)
5 Прадера-Вилли del(15)(q11.2)
6 Энгельмана del(15)(q11.2)
7 Рубинштейна-Тейби del(16)(p13.3)
8 Миллера-Дикера del(17)(p13.3)
9 Смита-Магениса del(17)(p11.2)
10 Аладжилла del(20)(p12)
11 МЭН 2А и 2В (Множественные эндокринные неоплазии 2-го типа) del(20)(p12.2)
12 ДиДжорджи del(22)(q11.2)
МИКРОДУПЛИ КАЦИИ
1 Де Ланге dup(3)(q25q29)
2 Видемана-Беквита dup(11)(pl5)
3 Миопатия Шарко-Мари-Тус 1 типа dup(17)(p11.2)
4 "Кошачьего глаза" dup, trp(22)(pter-q1
Одной из проблем медико-генетического консультирова-
ния и пренатальной диагностики являются маркерные хромо-
сомы (MX) и минихромосомы из-за своего маленького разме-
Глава 6
ра и отсутствия различимой дифференциальной окраски. Не-
сбалансированные транслокации, возникшие de novo, также
трудно поддаются диагностике, что затрудняет оценку прогноза
для их носителей. И, наконец, третьей группой структурных
хромосомных мутаций, которая требует применения современ-
ных молекулярно-цитогенетических методов с целью уточне-
ния диагноза, являются микроструктурные хромосомные ано-
малии, приводящие к микроделеционным и микродупликаци-
онным синдромам.
Использование классических цитогенетических методов в
исследовании MX и несбалансированных транслокаций в ряде
случаев позволяет сузить группу поиска. Однако ни один из
этих методов не оказывается достаточным для выявления про-
исхождения маркера и эффективного медико-генетического
консультирования пробанда. Флюоресцентная гибридизация
in situ позволяет разрешить большинство цитогенетических
диагностических проблем. Впервые метод гибридизации in situ
был предложен Gall and Pardue в 1969 году. В течение дли-
тельного времени использовался радиоактивный вариант ме-
тода, однако позднее был отработан и нерадиоактивный спо-
соб, собственно флюоресцентная гибридизация in situ. Изна-
чально она использовалась для быстрой локализации клони-
рованных фрагментов ДНК на хромосомах. Дальнейшее раз-
витие метод получил в работах Пинкеля с соавторами (Pinkel
et al., 1986) и постепенно стал широко применяться в практи-
ке клинической цитогенетики. В тех случаях, когда цитогене-
тическое исследование хромосомной патологии с помощью
методов дифференциального окрашивания хромосом оказы-
вается безуспешным, FISH-метод позволяет быстро провести
скрининг всех возможных хромосом человека, предположи-
тельно участвующих в дисбалансе.
В качестве ДНК-проб для гибридизации in situ могут быть
использованы любые клонированные последовательности ДНК,
выделенные фрагменты ДНК, участки или целые хромосомы.
Применение ДНК-зондов, специфичных для центромерных и
теломерных районов хромосом, а также хромосоме- и район-
специфичных ДНК-библиотек, позволяет провести идентифи-
кацию различных типов хромосомных аберраций. Кроме это-
го, успешное использование для гибридизации in situ уникаль-
ных последовательностей ДНК в качестве ДНК-проб (космид-
ные клоны, YAC-пробы, анонимные последовательности, или
отдельные гены) обеспечивает детальное исследование гене-
тической структуры хромосомных перестроек, например, MX,
а также анализ точек разрывов хромосом в различных типах
транслокаций, делеций, дупликаций, инверсий, инсерций, ди-
центрических и кольцевых хромосом.
Некоторые прикладные аспекты геномных исследований
Идентификация хромосомных аберраций методом FISH
обычно проводится с помощью центромероспецифичных ДНК-
проб, представленных высокоповторяющимися последователь-
ностями ДНК, локализованными в центромерном и перицен-
трическом районах хромосом человека: lt-, р- и классические
сателлитные ДНК-пробы. Центромероспецифические ДНК-
пробы наиболее широко используются для анализа числовых
хромосомных перестроек как при анализе метафазных, так и
интерфазных клеток, а также для установления процента ано-
мальных клеток у пациентов с мозаичным вариантом хромо-
сомной числовой аномалии.
Высокоповторяющиеся последовательности ДНК находят
применение также и в качестве ДНК-проб для определения
происхождения маркерных и минихромосом. В лаборатории
цитогенетики НИИ медицинской генетики ТНЦ СО РАМН уже
накоплен опыт такой диагностики (Островерхова, 1997),
Иногда с помощью центромероспецифичных ДНК-проб уда-
ется установить точки разрывов сложных транслокаций (Can-
nizzaro et al., 1985). Однако, использование центромероспе-
цифичных ДНК-проб имеет некоторые ограничения. Например,
структурно-аномальные хромосомы не могут быть определе-
ны с помощью этих проб. Кроме того, некоторые центромер-
ные пробы чувствительны к условиям гибридизации и при зна-
чительном изменении ее жесткости может наблюдаться пере-
крестная гибридизация или вообще отсутствие сигнала. И,
наконец, при участии нескольких хромосом в сложной пере-
стройке, положительный результат при использовании центро-
мероспецифических последовательностей ДНК становится
проблематичным. В таких случаях хромосомоспецифические
библиотеки ДНК могут дать более полезную информацию о
происхождении хромосомной перестройки.
Хромосомоспецифичные ДНК-библиотеки представлены
смесью уникальных последовательностей ДНК, покрывающих
всю длину хромосомы (Yung, 1996). Когда совокупность таких
специфических последовательностей ДНК используется со-
вместно в одной гибридизации, окрашивается целая хромо-
сома, поэтому такие ДНК-пробы получили название
хромосомоспецифичных ДНК-проб для раскрашивания хромо-
сом (painting-probes). Такая модификация FISH-метода, поз-
воляющая детектировать совокупность уникальных последо-
вательностей ДНК на целой хромосоме или ее участке полу-
чила название супрессионной гибридизации in situ (CISS). Хро-
мосомоспецифичные ДНК-библиотеки используются не толь-
ко для идентификации целых хромосом или их фрагментов
(маркерных хромосом) втиетафазных пластинках, но и для ана-
лиза транслокационных событий, затрагивающих разные хро-
Глава 6
мосомы. При исследовании MX хромосомоспецифичные ДНК-
библиотеки позволяют исключить участие дополнительных
хромосом в их происхождении.
Следует, однако, отметить, что и применение центроме-
роспецифичных ДНК-проб или хромосомоспецифичных ДНК-
библиотек не всегда дает возможность гонкого анализа хро-
мосомных перестроек. Некоторые случаи хромосомных ано-
малий требуют более детального исследования с помощью
уникальных генов, космидных и YAC-клонов, а также ДНК-проб,
специфичных для терминальных последовательностей хромо-
сом. Чем успешнее будет осуществлено картирование одно-
копийных генов и уникальных ДНК-последовательностей на
аберрантной хромосоме, тем тщательнее будет исследован
каждый маркер или транслокационное событие (Blennowetal.,
1995).
Таким образом, все классы ДНК-зондов могут использо-
ваться как для целей определения их локализации непосред-
ственно на метафазных хромосомах, так и для Целей клини-
ческой цитогенетики, обнаруживая в ряде случаев как опре-
деленные преимущества, так и некоторые ограничения. Цен-
тромероспецифичные ДНК-пробы наиболее важны при диаг-
ностике числовых хромосомных нарушений, а хрОмосомоспе-
цифичные ДНК-библиотеки - при анализе сложных структур-
ных перестроек, вовлекающих несколько хромосом, а также
при анализе дополнительных MX. Уникальные ДНК-пробы и
ДНК-зонды, специфичные для теломерных районов хромосом
позволяют изучить генетическую структуру хромосомной абер-
рации, детектировать терминальные и интерстициальные мик-
роделеции, определять точки разрывов хромосом в случаях
сложных транслокаций, инсерций и кольцевых хромосом. Толь-
ко использование всех типов ДНК-проб при исследовании хро-
мосомной аномалии позволит точно идентифицировать хро-
мосомную перестройку и определить ее генетический состав.
Использование меченных разными цветами различных ДНК-
проб в одной гибридизации значительно расширило возмож-
ности FISH-метода. Так, двухцветная илй трехцветная FISH с
центромероспецифичными ДНК-зондами позволяет получить
ценную информацию о наличии у больного анеуплоидии по
двум или трем разным хромосомам набора одномоментно не
только в отдельных метафазных пластинках, но и в интерфаз-
ных ядрах клеток. Особой ценностью этот анализ обладает
при исследовании мозаицизма, в случаях когда клетки иссле-
дуемой ткани трудно поддаются культивированию, например,
при анализе анеуплоидии в опухолевых клетках или в амнио-
цитах при проведении пренатальной экспресс-диагностики.
Двухцветная FISH может также с успехом применяться для
1 RK
Некоторые прикладные аспекты геномных исследований
анализа структуры перестроенных хромосом. В качестве при-
мера можно привести собственные данные по исследованию
характера хромосомной перестройки у больного с МВПР и
кариотипом 46,XY,18p+. Проведение двухцветной FISH с од-
новременным использованием ДНК-проб, специфичных для
центромерных районов хромосомы 18 и Y показало, что хро-
мосома 18 с увеличенным коротким плечом является в дей-
ствительности дицентрической, с транслокацией материала
хромосомы Y, поврежденной в сегменте р11, на короткое пле-
чо хромосомы 18. Применение в многоцветной FISH в качес-
тве ДНК-зондов уникальных последовательностей ДНК позво-
ляет локализовать расположение этих последовательностей
на хромосоме относительно друг друга, что находит примене-
ние для целей генетического картирования тех или иных кло-
нированных последовательностей ДНК.
В последнее время в цитогенетике человека разработана
система одновременной гибридизации разного числа хромо-
сомоспецифических библиотек ДНК, меченных в различных
соотношениях различными флюорохромами, например, TRITC,
дающим красное свечение, FITC - зелено-желтое или АМСА -
голубое. Этот подход дал возможность специфического рас-
крашивания разных пар гомологичных метафазных хромосом
до 12 различных цветов (Inazawa, 1993). Метод двухцветной и
многоцветной гибридизации с хромосомоспецифическими
библиотеками ДНК стал применяться для анализа сложных
хромосомных транслокаций и MX у больных (Popp etal., 1993;
Wang et al., 1995) или при изучении воздействия радиации на
хромосомы при исследовании проблем экологической гене-
тики.
Успешный анализ хромосомных перестроек можно осу-
ществлять и с помощью метода "обратной” гибридизации (re-
verse painting), когда с помощью проточной сортировки хро-
мосом или микроманипулятора могут быть изолированы абер-
рантные хромосомы с последующей амплификацией ДНК, ме-
чением и использованием ее в качестве ДНК-пробы для FISH-
анализа на нормальных метафазных хромосомах (Raimondi et
al., 1991; Blennowetal., 1992; Rubtsovetal., /996). В результа-
те может быть получена детальная информация о происхож-
дении маркера с определением не только самой хромосомы,
но и ее частей, вовлеченных в перестройку. Этот метод хотя и
стал использоваться для детекции MX, однако он пока еще не
получил широкого распространения в связи с трудоемкостью
и сложностью методов микродиссекции и сортировки хромо-
сом.
1
Глава 6
В настоящее время метод FISH используется при диагнос-
тике разных типов структурных хромосомных нарушений (Cheu-
ing et al., 1990; Tharapel et aL, 1991; Cannizzaro et aL, 1985).
Однако идентификация тонких хромосомных нарушений мето-
дами молекулярной цитогенетики требует большого количес-
тва центромеро- и теломероспецифичных ДНК-проб и
хромосомоспецифичных библиотек ДНК. Один из подходов к
молекулярно-цитогенетической характеристике хромосомных
аномалий, получивший распространение в последнее время,
основан на одновременной гибридизации препаратов хромо-
сом с несколькими ДНК-пробами - мультиплексная гибриди-
зация in situ (M-FISH). Такая тактика позволяет более быстро
проводить диагностику хромосомной патологии и использо-
вать значительно меньшее число препаратов хромосом, но
требует большого количества ДНК-проб. Однако с точки зре-
ния экономичности, способы ДНК-диагностики, не требующие
использования большого количества ДНК-проб, имеют важ-
ное значение для анализа хромосомной патологии. Из опи-
санных в литературе методических приемов представляется
интересным подход к идентификации маркерных хромосом,
основанный на относительно уникальной природе организа-
ции «-сателлитной ДНК в разных хромосомах (Plattner er aL,
1993; Vorsanova et aL, 1994). В зависимости от числа мономе-
ров, образующих повторяющуюся единицу, альфоидная ДНК
делится на несколько субсемейств, локализованных на раз-
личных группах хромосом человека. При гибридизации в ус-
ловиях "низкой жесткости" определенные ДНК-пробы связы-
ваются с группой хромосом одного а-сателлитного субсемей-
ства. Проведение последующей гибридизации нуклеиновых
кислот в условиях "высокой жесткости" с хромосомо-специ-
фичными ДНК-пробами одной группы обеспечивает точную
идентификацию аномальной хромосомы. Таким образом, су-
ществование коллекции альфоидных ДНК-проб на все хромо-
сомы человека и использование гибридизации in situ в раз-
личных условиях жесткости позволяет проводить быстрый ана-
лиз хромосомных аберраций.
Следут отметить, что при хромосомных перестройках, не
несущих центромерного района, вышеуказанный подход не
позволяет идентифицировать аберрантную хромосому. Нами
был предложен новый подход к идентификации дополнитель-
ных маркерных хромосом и несбалансированных хромосом-
ных транслокаций, основанный на использовании компьютер-
ных диагностических систем с методологией FISH-анализа,
существенно сокращающий число ДНК-проб, необходимых для
постановки точного диагноза и применимый для идентифика-
Некоторые прикладные аспекты г еномных исследований
ции перестроек, не включающих центромерные районы хро-
мосом (Назаренко и др., 1997].
Другой подход, не требующий большого количества ДНК-
проб при молекулярно-цитогенетической диагностике хромо-
сомных мутаций, предполагает использование ДНК-библио-
тек, созданных на основе аномальных хромосом. Однако, этот
метод проводится с помощью специального оборудования для
сортинга или микродиссекции аберрантных хромосом с пос-
ледующим созданием маркер- или районспецифичных библи-
отек ДНК. Микродиссекция - прямой метод вырезания хромо-
сомного участка или сегмента из любой хромосомы. Эта тех-
ника обеспечивает вырезание фрагментов хромосом, содер-
жащих, по меньшей мере, 2-4 Mb ДНК (Cannizzaro, 1996). Пер-
вые попытки диссекции хромосомных участков начали про-
водиться в конце 80-х и выполнялись на неокрашенных хромо-
сомах дрозофилы и мыши. Микродиссекция в исследованиях
генома человека первоначально проводилась как средство
изолирования новых ДНК-последовательностей, чтобы запол-
нить пробелы при составлении физических карт некоторых
хромосом. Исследования с применением микродиссекции для
целей клинической генетики, для диагностики наследствен-
ных и онкологических заболеваний пока немногочисленны.
Однако микродиссекция позволяет получить информативные
генетические маркеры для дифференциальной диагностики
этих болезней. ДНК-библиотеки, созданные на основе ано-
мальных хромосом, включая MX, гомогенно окрашенных рай-
онов хромосом, фрагильных сайтов, транслокаций и т.д., поз-
воляют установить природу происхождения этих событий на
молекулярном уровне.
В сочетании с другими цитогенетическими и молекулярны-
ми методами, микродиссекция хромосом обеспечивает эффек-
тивный и всесторонний анализ хромосомных перестроек, изо-
лирование новых ДНК-маркеров, связанных с генетическими
и онкологическими нарушениями, а также детальную характе-
ристику неисследованных районов генома человека. Однако
метод диссекции является дорогостоящим и малодоступным
и, в некоторых случаях, недостаточно чувствительным. Так, при
идентификации кольцевой MX, произошедшей от хромосомы
4, с использованием метода микродиссекции MX и последую-
щей амплификацией ее ДНК методом ПЦР, не удалось обна-
ружить небольшой район эухроматина короткого плеча хро-
мосомы 4, хотя значительный район длинного плеча выявлял-
ся без особых проблем (Fang et al., 1995). Очевидно, это свя-
зано с тем, что ДНК-проба, полученная с помощью микродис-
секции, содержала ампль|фицированные высокоповторяющи-
еся центромерные и перицентрические последовательности
Глава 6
ДНК данной хромосомы. Присутствие этих повторов может
ограничивать чувствительность метода.
Из относительно новых методов молекулярно-цитогенети-
ческого анализа хромосомного дисбаланса можно отметить
также метод сравнительной геномной гибридизации - compra-
tive genomic hybridization - CGH (Bryndorf et al., 1995). Он осно-
ван на гибридизации in situ двух различно меченных геномных
ДНК, одна из которых выделена из анализируемой ткани, а
другая - из кариотилически нормальной. Испытуемая и рефе-
ренсная ДНК метятся различными флюорохромами (например,
флюоресцеином - зеленого цвета и тексас-ред - красным),
смешиваются в соотношении 1:1с добавлением избытка не-
меченной повторяющейся ДНК (Cotl-ДНК). После гибридиза-
ции in situ с нормальными хромосомами при наличии хромо-
сомного дисбаланса в исследуемой ДНК на метафазных хро-
мосомах изменяется соотношение зеленого и красного флю-
оресцентного свечения. В связи с тем, что этот метод не тре-
бует проведения хромосомного анализа исследуемой ткани,
он обладает большой ценностью для исследования генети-
ческой структуры опухолей, из которых трудно получить мета-
фазные хромосомы для анализа.
Следует указать на один из важных аспектов молекулярно-
цитогенетической диагностики тонких хромосомных наруше-
ний, который связан с картированием на хромосомах мутант-
ных генов, вызывающих моногенные заболевания человека.
Особый интерес в плане картирований мутантных генов с по-
мощью цитогенетического подхода вызывают некоторые спе-
цифические микроделеции, которые приводят к хорошо очер-
ченным клиническим фенотипам. Еще более важными с этой
точки зрения являются отдельные случаи патологии, фенотип
которых представлен совокупностью признаков, характерных
не для одного, а для двух и более сйндромов. Эти случаи
получили название смежных генных синдромов (contiguous gene
syndromes), т.к. в случае если делецйя затрагивает область
хромосомы, в которой локализован более чем один мутант-
ный ген, то фенотип больного может включать более чем один
доминантный признак. Подобные наблюдения позволяют не-
посредственно на хромосомах картировать локусы, ответствен-
ные за развитие того или иного аномального фенотипа. В на-
стоящее время выделяют несколько цйтогенетических мето-
дов картирования генов (Schinzel, 1995):
1. Картирование через микроделеции. Если у пациента с
микроделецией какой-либо аутосомы клиническая картина
включает признаки необычные для аутосомных хромосомных
аберраций, то можно предполагать локализацию гена или ге-
нов, ответственных за эти признаки, в области, затрагивав-
Некоторые прикладные аспекты геномных исследований
мой микроделецией. Часто такие пациенты имеют смежные
генные синдромы.
2. Картирование через сбалансированные хромосомные
перестройки. Многие Х-сцепленные гены были впервые лока-
лизованы благодаря наблюдению больных девочек, кариотип
которых обнаруживал возникшую de novo Х-аутосомную тран-
слокацию. В аутосомах косегрегация доминантного наруше-
ния со сбалансированной транслокацией может указывать на
наличие двух кандидатных локусов, ответственных за данную
патологию. Особенно полезным для привязки какого-либо гена
к конкретному хромосомному сегменту является наличие двух
или более наблюдений отдельных случаев заболевания или
семейных форм патологии, сочетающихся с транслокаций,
имеющей общую точку поломки в определенном хромосом-
ном сегменте.
3. Картирование с помощью флюоресцентной гибридиза-
ции in situ. Этот подход может быть реализован в случае на-
личия клонированного гена, локализация которого неизвест-
на, и если космида или ДНК-зонд доступны для проведения
FISH. Кроме того, данный метод может быть полезен для кар-
тирования генов человека, имеющих аналоги и уже картиро-
ванных у мышей.
4. Картирование импринтированных генов. Потеря хромо-
сомного гомолога с активным геном, локализованном в под-
верженной импринтингу области генома, благодаря унироди-
тельской дисомии по определенной хромосомной паре, мо-
жет вызывать фенотипические аномалии. Обнаружение таких
случаев с помощью методов молекулярно-гентического ана-
лиза позволяет в дальнейшем использовать различные стра-
тегии для идентификации этих генов.
Завершая рассмотрение методологии современной моле-
кулярно-цитогенетической диагностики тонких хромосомных
нарушений, следует сказать о существенном прогрессе в этой
области, достигнутом за последние годы. Существование раз-
нообразных ДНК-зондов на повторяющиеся и уникальные
последовательности ДНК, специфичные к центромерным, те-
ломерным и строго определенным участкам хромосом, а так-
же создание хромосомоспецифичных и районспецифичных
библиотек ДНК, богатый арсенал методов молекулярно-цито-
генетического анализа открывает большие возможности в изу-
чении тонкой структуры хромосомных нарушений, исследова-
нии фенокариотипических корреляций при хромосомном дис-
балансе, картировании на хромосомах новых клонированных
последовательностей ДНК и мутантных генов.
1
Глава 6
лишь организм пациента (точнее, определенную ткань или тип
клеток). Все современные подходы к генотерапии основаны
именно на терапии соматических клеток.
Заместительная и коррегирующая гемотерапия. Причина
любого генетического дефекта, в общем виде, заключается в
том, что определенный ген или группа генов в клетках больно-
го не работают или работают неправильно. То есть, клетка не
экспрессирует всю информацию, которую должна экспреоси-
ровать в Йорме, либо экспрессирует ее в искаженном виде.
Принципиально существует два пути “заставить” клетку выда-
вать недостающую информацию. Во-первых, дефектный ген
можно физически заменить его нормальной копией, скоррек-
тировав тём самым мутантный генотип к нормальному. Такой
подход называется коррегирующей (исправляющей) терапией.
Однако реально заменить дефектный участок хромосомы нор-
мальным й большой доле клеток крайне сложно. Существую-
щие лабораторные процедуры не позволяют этого делать с
высокой степенью эффективности. Гораздо проще ввести в
клетку дополнительную нормальную копию (или копии) гена,
который функционально (но не физически) заменил бы мутан-
тный ген в геноме клетки. Такой подход называется замести-
тельной тёрапией, и именно он используется во всех сущест-
вующих сёйчас генотерапевтических протоколах.
Оба подхода в итоге приводят к изменению мутантного
фенотипа на нормальный, то есть к его коррекции. Таким об-
разом, и Заместительная и корректирующая в отношении ге-
нотипа терапия в отношении фенотипа всегда является кор-
регирующёй. В этом, более широком, контексте термин “кор-
регирующая генотерапия" и будет использоваться в дальней-
шем.
Механизмы коррекции генетических дефектов. В отно-
шении механизмов коррекции генетических дефектов страте-
гии генной терапии, как отмечает Свердлов (1997), делятся на
3 группы:
- коррёкция утраченной функции клетки путем доставки в
клетку здорового гена, который “компенсирует” неработаю-
щий ген клетки;
- подавление избыточной функции клетки. Эту стратегию
применяют, когда болезнь возникает вследствие того, что клет-
ка приобретает несвойственную ей в норме функцию (напри-
мер, при опухолях клетка приобретает функцию деления, при
вирусных инфекциях - функцию репликации чужеродного ге-
нома);
- усиление иммунного ответа. Модификацию можно осу-
ществлять введением новой генетической информации либо в
клетки, против которых нужно усилить иммунный ответ (на-
Некоторые прикладные аспекты геномных исследований
пример, модифицируя антигены опухолевых клеток), либо в
клетки иммунной системы, которые осуществляют иммунные
реакции.
Методы переноса генов в клетки человека
Существует два общих подхода к доставке генов в сомати-
ческие клетки человека - перенос генов in vivo и ex vivo. При
переносе генов ex vivo осуществляется трансформация кле-
ток человека в культуре (in vitro). За^ем трансформированные
клетки переносят обратно в организм. При подходе in vivo ген
доставляется прямо в организм человека и переносится в клет-
ки in situ.
Различные методы доставки генсв в культивируемые клет-
ки и ткани человека (они перечислены в таблице 14) отлича-
ются по спектру возможного применения и эффективности.
Важнейшие из параметров, определяющие эффективность
генотерапии: эффективность трансдукции (перенос трансге-
нов в клетку), вероятность интеграции трансгена в геном клет-
ки, продолжительность экспрессии трансгена в клетке-хозяи-
не. Большая часть современных методов переноса генов пока
обеспечивает относительно низкую степень интеграции чуже-
родного гена в геном клетки (не интегрированный генетичес-
кий материал с неизбежностью теряется в ряду клеточных де-
лений) и временную (транзиторную) экспрессию гена.
Наиболее просты, с технической точки зрения, физические
и химические методы переноса генов. Химический метод за-
ключается в том, что ДНК осаждают добавлением ионов Са24
или других катионов. Образующийся осадок поглощается куль-
тивируемыми клетками. При этом лишь небольшая доля мо-
лекул ДНК достигает места назначения - ядра клетки и еще
меньшая их доля случайно интегрируется в геном. Физичес-
кие методы трансфекции, такие как электропорация (перенос
ДНК через клеточную мембрану посредством электрического
поля), микроинъекции, бомбардировка клеток микроскопичес-
кими частицами золота со связанными молекулами ДНК, не-
рколько более эффективны, однако ввиду присущих им недо-
статков (например, при электропорации высокое напряжение
серьезно повреждает клетки) и низкой вероятности интегра-
ции, в генотерапии практически не применяются. Исключение
составляют лишь инъекции раствора ДНК обычным шприцем
с иглой. Такой способ доставки генов может быть эффектив-
ным при переносе их в определенные ткани (например, в мыш-
цы).
1
Г лава 6
Таблица 14.
Методы переноса генов в клетки человека
и область их применения {Culver, 1994,
Баранов, 1996, Фаворова, 1997)
Методы переноса Транс- дукция Инте- грация Экс- прес- сия Доставка гена в •
культуру клеток ткани у ’
Химические
Осаждение фосфатом Са Н Н Т + *
Физические
Электропорация н Н т + -
Микроинъекции в Н т + -
Бомбардировка частицами золота в Н т + +
Простая инъекция н Н т - +
Вирусные векторы
Ретровирусы в В Д? + -
Аденовирусы в Н т + +
Аденоассоциированный вирус в н д? + -
Герпес-вирусы н н с + -
Невирусные векторы
Липосомы н н т +
Рецептор-опосредован- ный эндоцитоз (комплексы ДНК-белок, ДНК-вирусная капсида) в н т + +
Примечание: н - низкая, в - высокая, т - транзисторная,
д - длительная, с - слабая
Вирусные векторы
Наиболее эффективные системы переноса используют в
качестве средства доставки генов в клетку вирусные частицы.
Эти системы и наиболее распространены - примерно 95% су-
ществующих генотерапевтических протоколов используют
именно вирусную трансдукцию.
Вирусы - внутриклеточные паразиты, использующие для
своего размножения репликационный аппарат клетки-хозяи-
на. Геном вируса, в зависимости от его типа, может быть пред-
ставлен молекулой (или молекулами) ДНК или РНК, которая
Некоторые прикладные аспекты геномных исследований
кодирует ограниченный набор белков вирусной частицы, в том
числе белки оболочки вируса. Вирус проникает в клетку, вза-
имодействуя белками оболочки с клеточными рецепторами.
Разные группы вирусов, имея различный набор поверхност-
ных белков, могут инфицировать специфичный для них тип
клеток. Некоторые группы вирусов (например, ретровирусы)
могут интегрироваться в геном клетки при его репликации.
Таким образом, вирусы представляют собой весьма при-
влекательные системы для переноса генов, в которых многие
проблемы эффективности решены природой. С другой сторо-
ны, в отличие от физических и химических методов, использо-
вание вирусных векторов, сопряжено с опасностью, связан-
ной как с патогенным действием вирусов, так и возможным
нарушением функционирования генома клетки.
Генно-инженерные векторы на основе вирусов содержат
встроенный на место генов вируса необходимый ген челове-
ка, фланкируемый, как правило, вирусными генами, необхо-
димыми для упаковки в частицы и экспрессии. Обычно вирус-
ные векторы дефектны по репликации и способны только к
одному раунду инфекции клеток-хозяев, без последующего
распространения вируса. Экспрессия трансгена при переносе
вирусным вектором может быть как транзиторной, в случае
если генетический материал вектора остается вне хромосом
клетки (эписома) и теряется в ряду клеточных делений (тран-
сдукция аденовирусными векторами и векторами на основе
герпес-вирусов), так и относительно длительной, при интег-
рации трансгена в геном клетки (трансдукция векторами на
основе ретровирусов и аденоассоциированных вирусов).
Стабильная интеграция чужеродного гена в хромосомы клет-
ки-хозяина обеспечивает длительную экспрессию гена в кле-
точном клоне трансдуцированной клетки. Для интеграции в
геном клетки простым ретровирусным векторам (например,
векторам, созданным на основе вируса лейкемии мышей Мо-
лони, MLV) необходимо преодолеть барьер в виде ядерной
мембраны, которая разрушается только при делении клетки.
Поэтому такие векторы способны только к трансдукции деля-
щихся клеток. Невозможность переноса генов в дифференци-
рованные неделящиеся клетки (а большая часть тканей орга-
низма человека состоит из таких клеток) - основной недоста-
ток ретровирусных векторов. Векторы, сконструированные на
базе аденоассоциированных вирусов, способны интегриро-
ваться и в геном неделящихся клеток, однако эффективность
трансдукции невысока и зависит от хелперных функций, вы-
полняемых аденовирусами или герпес-вирусами. Впрочем, не-
которые группы вирусов^и, соответственно, векторы на их ос-
нове) способны стабильно трансдуцировать неделящиеся клет-
Глава 6
ки. Например, лентивирусы - подкласс ретровирусов, к кото-
рому относится и вирус СПИД.
Эффективность экспрессии трансгена и способность век-
тора к персистенции в клетке зависит от работы промотора
трансгена и иммунного ответа организма хозяина. Иммунный
ответ может бьнь направлен как против самого продукта гран-
сгена, так и против вирусных белков, синтезирующихся в транс-
дуцированной клетке. Если генным инженерам удается пода-
вить экспрессию вирусных генов, то шансы на длительное су-
ществование трансдуцированной клетки значительно возрас-
тают.
Ретровирусные векторы. Ретровирусы были описаны пер-
воначально как онкогенные агенты. Однако большая часть рет-
ровирусов непатогенна. Злокачественная трансформация клет-
ки онкогенными ретровирусами обуславливается экспрессией
вирусных онкогенов или интеграцией генетического материа-
ла вируса вблизи онкогенов генома клетки с последующей их
активацией.
Широкий спектр клеток-хозяев и стабильная интеграция в
геном делают ретровирусы очень привлекательным средст-
вом переноса генов. Размер вставки, которую могут перено-
сить ретровирусы ограничен 9 Кб, что соответствует неболь-
шому эукариотическому гену или фрагменту кДНК.
Геном ретровирусов представлен двумя идентичными мо-
лекулами РНК. Чтобы использовать клеточный механизм реп-
ликации, вирус должен на определенной стадии жизненного
цикла “перевести” свой геном из РНК в ДНК. Процесс синтеза
молекулы ДНК на РНК-матрице называется обратной тран-
скрипцией и осуществляется ферментом, который кодируется
вирусным геномом. Именно поэтому ретровирусы и получили
свое название (от лат. retro - обратно, назад).
Векторы на основе ретровирусов сохраняют, как правило,
лишь небольшую часть вирусного генома, необходимую для
упаковки в частицы и транскрипции ретровируса и не экспрес-
сируют вирусных белков, вызывающих иммунный ответ орга-
низма-хозяина. Специфичность вирусного вектора по отноше-
нию к хозяйской клетке определяется его геном env, продукт
которого распознается рецептором на поверхности клетки.
Замена этого гена у экотропных (т.е. узнающих только опре-
деленный тип рецепторов) вирусов на аналогичный ген ам-
фитропных (распознающих широкий спектр рецепторов) мо-
жет значительно расширить специфичность ретровирусных
векторов.
В отличие от некоторых других вирусных векторов, дефек-
тные по репликации ретровирусные векторы поражают клетки
только один раз и не способны к распространению в организ-
Некеморые прикладные аспекты геномных исследований
ме. Эю свойство, а также их относительно низкий титр (из 1
мл среды, в которой растет культура пакующих клеток, можно
получить примерно 10ь вирусных частиц) делают их мало при-
годными для генотерапии in vivo.
Для трансдукции неделящихся дифференцированных кле-
ток, в частности, нейронов ЦНС, разрабатываются векторы на
основе вируса иммунодефицита человека (HIV) (Blomer et al.,
1996). Как и другие лентивирусы, HIV способен инфицировать
неделящиеся клетки благодаря способности проникать через
поры ядерной мембраны. HIV-производные векторы исполь-
зуют не его “родные" белки оболочки, а аналогичные белки
вируса везикулярного стоматита (VSV G), что увеличивает ста-
бильность вектора и позволяет получать более высокие титры
(до 108). Пока векторы на основе HIV тестируются на модель-
ных линиях мышей и в клинических протоколах генотерапии
не применяются.
Аденовирусные векторы. Геном аденовирусов представ-
лен двухцепочечной молекулой ДНК размером примерно 36
Кб. Многие аденовирусы патогенны и вызывают иммунный
ответ, осуществляемый цитотоксическими Т-лимфоцитами.
Аденовирусы способны к транзиентной трансдукции неделя-
щихся клеток. В клеточной культуре можно получить большое
количество вирусных частиц (титр до 1012). Существуют два
типа аденовирусных векторов, дефектных по репликации и
инфекционному потенциалу. У векторов первой группы отсут-
ствуют вирусные гены области Е1 (Е1А и Е1В), ответственные
за ранние стадии жизненного цикла вируса. У аденовирусных
конструкций другого типа отсутствует регион ЕЗ, гены которо-
го ингибируют лизис клетки-хозяина цитотоксическими Т-лим-
фоцитами. На место одного из этих регионов может быть встав-
лен трансген размером до 10 Кб.
У аденовирусных векторов первого поколения экспрессия
вирусных генов подавлена не полностью, в результате чего
наблюдается существенный иммунный ответ на их введение в
организм. У второго поколения векторов район ЕЗ удален не
полностью. Вектор сохраняет способность экспрессировать
белок ЕЗ, ответственный за супрессию иммунного ответа, что
приводит к менее резкому сопротивлению организма-хозяи-
на. Сейчас разрабатываются аденовирусные векторы треть-
его поколения, в которых трансген встроен на место региона
Е4, отвечающего за онкогенную трансформацию клеток-хозя-
ев. Однако, удаление Е4 привело к резкому снижению уровня
экспрессии трансгена.
Векторы на основе аденоассоциированных вирусов.
Аденоассоциированный виЬус (AAV), геном которого представ-
лен одноцепочечной ДНК, не патогенен для человека. Почти
Глава 6
всю кодирующую часть генома AAV, исключая минимум, необ-
ходимый для трансдукции, можно удалить и заменить на тран-
сген. AAV может интегрировать в геном неделящихся клеток,
но эффективность интеграции очень низка. Для транскрипции
и трансляции генома AAV необходимы вирусы-помощники (хел-
перы), например, аденовирус или герпес-вирус. Именно в этом
и таится основная опасность использования AAV в качестве
вектора для переноса генов: больные, получившие трансген с
AAV-вектором, могут быть инфицированы вирусом-хелпером
дикого типа. В целом, низкий титр, слабая эффективность тран-
сдукции и зависимость от хелперов делают спектр примене-
ния AAV-векторов довольно узким.
Векторы на основе герпес-вируса. Вирус простого гер-
песа (HSV) имеет довольно большой геном (150 Кб), содержа-
щий около 70 генов, значительная часть которого может быть
заменена трансгеном. HSV имеет широкий спектр клеток-хо-
зяев, можно получить высокий титр вируса в культуре, ем-
кость его генома достаточно высока (размер вставки может
достигать 30 Кб). Однако регуляция его генома сложна, и ген-
ным инженерам пока не удалось полностью подавить репли-
кацию вируса и иммунный ответ организма против клеток-хо-
зяев.
Невирусные векторы. В последнее время активно разра-
батываются системы доставки чужеродной ДНК в клетку с по-
мощью невирусных векторов. Главное преимущество таких
систем в том, что отпадают опасности, связанные с патоген-
ным действием вирусов и возможностью повреждения генома
клетки. Однако при этом приходится решать те проблемы, ко-
торые при использовании вирусов решены природой - как
эффективно доставить трансген в клетку, преодолев при этом
двойной рубеж из клеточной и ядерной мембраны; как избе-
жать повреждения, деградации и расщепления ДНК клеточны-
ми ферментами; как обеспечить длительную экспрессию ге-
нетического материала трансгена.
Генные инженеры пытаются решить эти проблемы, вводя
молекулы ДНК в комплексы с биологическими или химически-
ми структурами, обеспечивающими проникновение комплек-
са в ядро. Один из путей невирусной доставки - комплексы
ДНК с липидами в составе липосом. Такие системы, благода-
ря взаимодействию липидного слоя с клеточной мембраной,
легко проникают в клетку, но большая часть генетического ма-
териала теряется, не достигнув ядра. Еще один способ до-
ставки - проникновение ДНК в клетку путем рецептор-опосре-
дованного эндоцитоза, т.е. поглощение ДНК за счет взаимо-
действия ДНК-белковых комплексов с клеточными рецепто-
Некоторые прикладные аспекты геномных исследований
рами. Для этого ДНК вводят в комплекс, включающий полика-
тионы (например, полилизин); лиганд, который распознается
поверхностным рецептором клетки (например, антитело, спе-
цифичное к какому-либо поверхностному белку клетки); и аген-
та, облегчающего высвобождение ДНК из комплекса с эндо-
сомами в цитоплазме. В качестве эндоосмотического агента
могут применяться и вирусные капсиды.
В последнее время широко обсуждается и изучается воз-
можность создания генотерапевтических векторов на основе
искусственных хромосом млекопитающих (mammalian artificial
chromosomes), аналогичных искусственным хромосомам дрож-
жей (YAC), способных автономно существовать в клетке, реп-
лицироваться при клеточном делении и стабильно экспресси-
ровать свой генетический материал.
Генотерапия с помощью
антисенс-олигонуклеотидов
В случае, если кодирующая последовательность гена из-
вестна (а для многих наследственных заболеваний это так),
экспрессию гена можно подавить, внося в клетку молекулы,
комплиментарные мРНК, транскрибируемой с этого гена. Свя-
зываясь с мРНК, такие молекулы будут ингибировать трансля-
цию, т.е. синтез белкового продукта (рис. 27). Кодирующая
цепочка ДНК или РНК, которая содержит информацию об ами-
нокислотной последовательности белка, называется смысло-
вой (англ, sense strand), а комплиментарная ей - антисенс-
(антисмысловой) цепочкой. Поскольку кодирующие участки
генома, как правило, уникальны, можно синтезировать анти-
смысловые нуклеотиды, которые будут связывать только нуж-
ную мРНК. Такие антисенс-молекулы можно использовать для
блокировки экспрессии как "нежелательных” генов в геноме
человека, так и геномов вирусов или других инфекционных
агентов.
Существует два пути терапевтического использования ан-
тисенс-нуклеотидов: химически синтезированные олигонукле-
отиды или экспрессируемые нуклеотиды. Олигонуклеотиды -
это короткие цепочки ДНК длиной 15-20 оснований, которые
можно синтезировать на автоматических синтезаторах ДНК и
вводить больному простой инъекцией. Экспрессируемый нук-
леотид необходимо встроить в вектор и доставить в клетки
или ткани больного. Этот путь теоретически привлекателен,
однако встают те же вопросы и проблемы, касающиеся без-
опасности и эффективности, что и при доставке любого транс-
гена.
1
Глава 6
Рис. 27. Блокировка трансляции с помощью антисенс-олиюнук-
ь леотидов (Askari, McDonnell, 1996).
!Vb- ) ..
Ч-
Отметим некоторые принципиальные возможности приме-
нения антисенс-олигонуклеотидной терапии, над реализацией
которых сейчас работают в различных лабораториях (Askari et
al., 1996). Одной из основных “мишеней" для антисмысловой
генной терапии является сейчас синдром приобретенного
иммунодефицита (СПИД) (рис. 28). Разрабатываются олиго-
нуклеотиды, способные блокировать различные участки гено-
ма вируса HIV. Созданы, например, олигонуклеотиды, связы-
вающиеся с длинным терминальным повтором (LTR) провиру-
са - регуляторным районом, который обеспечивает интегра-
цию провирусной ДНК в геном клетки. Разрабатываются так-
же системы, способные блокировать специфичные мРНК ви-
русных белков, таких как tat и rev, которые регулируют экспрес-
сию вируса. Уже показана возможность блокировать антисенс-
олигонуклеотидами репликацию вируса in vitro.
Разрабатывается и антисенс-терапия гипертензии (рис. 29).
Для этого применяют антисмысловые олигонуклеотиды, бло-
кирующие мРНК ангиотензиногена - ключевого звена ренин-
ангиотензиновой системы. Ангиотензиноген, экспрессирую-
щийся в клетках печени и мозга, является предшественником
ангиотензина I и II, участвующих в регуляции давления крови.
Высокий уровень ангиотензина II коррелирует с повышенным
давлением крови.
. • Щ ,............ * . * ......j
, I- , И; '< V. .,' - )
;Л( J .. 4, ?('. л ’ * < МГП <( I
Некоторые прикладные аспекты геномных исследований
Рис. 28. Антисенс- терапия против вируса СПИД (Askari, McDon-
nell, 1996).
Фармакологическая терапия гипертензии осуществляется,
как правило, ингибиторами ангиотензин-конвертирующего
фермента (АСЕ), который превращает ангиотензиноген в ан-
гиотензин. Антисенс-терапия с помощью олигонуклеотидов
направлена на предыдущую стадию этой биохимической це-
почки. Пока эта модель находится на экспериментальной ста-
дии. Показано, что введение антисмысловых олигонуклеоти-
дов, блокирующих мРНК ангиотензиногена, снижает уровень
этого белка в крови, блокирует формирование ангиотензина II
и приводит к уменьшению кровяного давления. Другие сер-
дечно-сосудистые состояния также являются потенциальны-
ми объектами для антисенс-терапии. Например, рассматри-
вается возможность использования олигонуклеотидов, блоки-
рующих пролиферацию сосудистых клеток для предотвраще-
ния рестеноза артерий после ангиопластики.
Основные проблемы антисенс-олигонуклеотидной терапии
- быстрое расщепление олигонуклеотидов ДНКазой в орга-
низме и большое количество олигонуклеотидов, необходимое
для достижения терапевтического эффекта. Стабильность
олигонуклеотидов в плазме можно повысить их химическими
модификациями, например, модифицируя фосфодиэфирные
Глава 6
связи между двумя соседними нуклеотидами или вводя оли-
гонуклеотиды в комплексы с поликатионами и лигандами для
клеточных рецепторов.
Рис. 29. Ингибирование ангиотензиногена с помощью антисенс-
олигонуклеотидов (Askari, McDonnell, 1996).
Существуют и другие пути доставки олигонуклеотидов в
нужное место. Так, на стадии клинических испытаний нахо-
дится протокол интравитерального введения олигонуклеоти-
дов для терапии цитомегаловирусного ретинита.
Генотерапия моногенных заболеваний
Первой документированной успешной попыткой генотера-
певтической коррекции наследственного заболевания были уже
упомянутые выше эксперименты по терапии ТКИД с помощью
гена аденозиндезаминазы. Ген АДА кодирует фермент, кото-
рый катализирует превращение аденозина в инозин и амми-
ак. У гомозигот по дефектому гену АДА развивается ТКИД,
Некоторые прикладные аспекты геномных исследований
гетерозиготы имеют пониженный уровень фермента без яв-
ных клинических нарушений. Схема генотерапевтического про-
токола АДА-недостаточности была следующей (Blease et al.,
1995). Методом лейкофореза у больных выделяли мононукле-
арные клетки. Затем их культивировали в условиях, благопри-
ятствующих пролиферации Т-лимфоцитов. Культуру с наро-
щенными Т-лимфоцитами трансдуцировали ретровирусным
вектором, несущим нормальный ген АДА (т.е. чужеродный ген
вводили в клетки ex vivo). Через несколько дней проводили
реинфузию модифицированных Т-лимфоцитов в кровоток боль-
ного. Эту процедуру повторяли каждые несколько месяцев.
Иммунная функция больного за это время значительно улуч-
шилась, а исследование лимфоцитов показало, что примерно
четверть Т-лимфоцитов являются генно-инженерными. Необ-
ходимость повторных введений модифицированных клеток
объясняется тем, что Т-лимфоциты - зрелые дифференциро-
ванные клетки, время жизни которых в организме невелико.
Более продолжительный эффект можно получить пересажи-
вая трансдуцированные гемапоэтические стволовые клетки
костного мозга - предшественники клеток крови всех типов.
Такие эксперименты в случае АДА-недотаточности также были
успешно проведены.
В настоящее время методы генотерапевтической коррек-
ции существуют или разрабатываются для более чем двух де-
сятков наследственных заболеваний, перечисленных в табли-
це 15. Остановимся на некоторых из них подробнее
Генотерапия семейной гиперхолестеринемии
Семейная гиперхолестеринемия (СГХ) - аутосомно-рецес-
сивное заболевание, встречающееся в популяции с довольно
высокой частотой (частота гетерозигот 1:500) (Фогель, Мо-
тульски, 1990). Болезнь вызывается различными мутациями в
гене рецептора липопротеинов низкой плотности (LDLR). Де-
фектный рецептор клеток печени больных не способен связы-
вать липопротеиновые частицы, что приводит к накоплению
холестерина в крови, его отложению в сосудистой стенке, ран-
нему атеросклерозу и инфаркту миокарда.
При СГХ клетками-мишенями для генно-инженерной моди-
фикации являются клетки печени (гепатоциты). Их трансдук-
цию проводят ex vivo, используя рекомбинантный ретровирус,
несущий нормальный ген LDLR. Клинический протокол генной
терапии СГХ (Wilson et al., 1992, Grossman et al., 1994) пред-
ставлен на рис. 30. Больному проводили частичную гелатоэк-
томию - удаляли левый латеральный сегмент печени (печень
обладает замечательной способностью к регенерации - через
Глава 6
несколько недель ее размеры полностью восстанавливаются).
Удаленный сегмент (а его вес был около 250 г, что соответ-
ствует примерно 15% общего веса печени) обрабатывали кол-
лагеназой, чтобы освободить гепатоциты. Полученные таким
образом примерно 3,2 миллиарда живых клеток (98% клеток
оказались живыми) культивировали в течение двух суток. За-
тем в среду добавляли рекомбинантный ретровирус, несущий
ген LDLR, промотор гена бета-актина курицы и энхансер из
генома цитомегаловируса (функция двух последних участков -
обеспечивать экспрессию трансгенного LDLR).
Рис. 30. Стратегия ex vivo терапии семейной гиперхолестери-
немии (Grossmann et al., 1994).
Инкубацию с вектором проводили в течение 12-18 часов,
после чего примерно 20% клеток оказались трансдуцирован-
ными. Через 3 дня после гепатоэктомии проводили реинфу-
зию модифицированных гепатоцитов в печень больного через
воротную вену. Спустя 4 месяца после генотерапевтических
процедур больному провели биопсию печени и гибридиза-
цией in situ с РНК-зондом, специфичным к рекомбинантному
гену LDLR, и показали, что в среднем 1 из 1000-10000 гепато-
цитов экспрессирует нормальный рецептор. Через полтора
года после начала генотерапии уровень липопротеинов низкой
плотности в крови больного снизился на 17%, что свидетель-
ствовало об активности LDLR, а соотношение липопротеинов
Некоторые прикладные аспекты геномных исследований
низкой плотности к липопротеинам высокой плотности (кри-
терий атерогеНности) снизилось с 10-13 до 5-8 и оставалось
стабильным в течение всего периода лечения.
Таблица 15.
Наследственные болезни, методы генетической
коррекции которых применяются или разрабатываются
(Culver, 1994; Баранов, 1996)
Болезнь Дефектный ген
Тяжелый комбинированный иммунодефицит (ТКИД) аденозиндезаминаза
Иммунодефицит пуриннуклеозид-фосфорилаза
Болезнь Гоше (сфинголипидоз) В-глюкоцереброзидаза
Болезнь Хантера и дурон ат- сул ьфатаза
Синдром Гурлера L-идуронидаза
Семейная гиперхолестеринемия рецептор липопротеинов низкой плотности
Гемофилия В фактор IX
Гемофилия А фактор VIII
Талассемия бета-глобин
Серповидноклеточная анемия бета-глобин
Эмфизема легких альфа-1 -антитрипсин
Муковисцидоз CF-трансмембранный регулятор
Фенилкетонурия фенилаланингидроксилаза
Г ипераммонеМия орнитинтранскарбамилаза
Цитрулинемия аргиносукци н атс и нтетаз а
Миодистрофий Дюшенна дистрофин
Респираторный дистресс-синдром сурфактант белок В
Хронический грануломатоз NADPH-оксидаза
Болезнь Альцгеймера белок-предшественник В-амилоида
Болезнь Паркйнсона тирозин-гидроксилаза
Метахроматическая лейкодистрофия арилсульфатаза А
Синдром Леша-Найана гипоксантин-фосфорибозил- трансфераза
Глава 6
In vivo-генотерапия муковисцидоза
Предыдущие примеры касались генной терапии ex vivo.
Клетки печени и Т-лимфоциты легко поддаются культивирова-
нию, поэтому трансдукция клеток ex vivo - вполне приемле-
мый метод, если трансген необходимо ввести именно в эти
клетки. Однако, отнюдь не все клетки и ткани человека на-
столько “удобны”. Например, при муковисцидозе необходимо
проводить генетическую коррекцию клеток легкого, а доступ-
ных методов их культивирования не существует. Единствен-
ный путь - проводить терапию in vivo.
Муковисцидоз - достаточно распространенное моногенное
наследственное заболевание) новорожденных. Причиной за-
болевания являются мутации в гене трансмембранного регу-
лятора проводимости (CFTR), который участвует в транспор-
тировке ионов хлора через клеточную мембрану. Клинические
последствия дефекта регулятора - хронические инфекции,
воспаления и нарушения дыхательного аппарата. Нормальный
ген CFTR в клетки легочного эпителия пытаются доставить либо
с помощью дефектных по репликации аденовирусов, либо в
комплексе с липосомами аэрозольным впрыскиванием век-
тора в дыхательные пути. Такая модель генотерапии отрабо-
тана на мыши и находится сейчас в стадии клинических испы-
таний. На людях обнадеживающие результаты получены на
клетках эпителия полости носа.
Другие наследственные заболевания
На стадии клинических испытаний находятся протоколы
генотерапии гемофилии В - болезни, вызываемой дефектом
фактора свертывания IX. Ех vivo-подход основан на трансдук-
ции культивируемых фибробластов кожи. При in vivo терапии
проводят трансдукцию гепатоцитов аденовирусным вектором
прямо в организме больного. Результаты в экспериментах на
собаках показывают, что такая процедура позволяет поддер-
живать терапевтический уровень фактора в крови в течение
двух месяцев. При гемофилии А, более распространенном
заболевании крови, ситуация осложняется тем, что ген факто-
ра VIII, мутации которого и вызывают болезнь, слишком велик
по размерам и целиком в вирусные вектора не помещается.
Генным инженерам удалось найти решение - в ретровирусный
вектор встроили укороченный ген, удалив из него регион, ко-
дирующий домен В, что почти не сказалось на функциональ-
ных характеристиках белка. Эксперименты по коррекции ге-
мофилии В пока проводятся на модельных линиях мышей.
Аналогичный подход к конструкции векторов применяют и
в экспериментах по генотерапии мышечной дистрофии Дю-
Некоторые прикладные аспекты геномных исследований
шенна. Ген дистрофина (DMD) тоже очень велик (размер его
кДНК - 14 Кб) и не помещается ни в один из известных векто-
ров. Поэтому культивируемые миобласты трансдуцировали
аденовирусным вектором, несущим укороченный, но функци-
ональный вариант гена. От 5 до 50% мышечных клеток мышей,
дефектных по DMD, экспрессировали нормальный дистрофин
после трансдукции. Еще один подход к генотерапии миодис-
трофии Дюшенна - внутримышечная инъекция раствора “го-
лой’’ ДНК гена дистрофина также дает обнадеживающие ре-
зультаты.
Подходы к генотерапии онкологических заболеваний
Большая часть наследственных болезней с простым мен-
делевским типом наследования - относительно редкие собы-
тия. Основная же доля заболеваемости и смертности в разви-
тых странах приходится на долю мультифакториальных или
приобретенных состояний, таких как сердечно-сосудистые
нарушения, онкологические заболевания, диабет, астма, раз-
личные инфекционные болезни.
Разработка генных подходов к терапии широко распрос-
траненных болезней современного общества - важная задача
генотерапии. Первые работы по терапии опухолей с помощью
чужеродного генетического материала начались в конце 80-х
годов. С тех пор достигнуты определенные успехи, во всяком
случае, число протоколов, применяемых для генной терапии
рака, значительно превышает таковое для наследственных
болезней. Основной принцип стратегии генотерапии рака -
активация генетическими методами иммунного ответа орга-
низма против опухолевых клеток. Для этого можно модифици-
ровать как геном самих опухолевых клеток, повышая их имму-
ногенность, так и генетически активировать деятельность кле-
ток иммунной системы.
В настоящее время применяются и разрабатываются сле-
дующие подходы к генотерапии онкологических заболеваний
(Culver, Blease, 1994; Свердлов, 1995-97):
1. Повышение иммунореактивности опухолевых клеток.
Принципиально подход выглядит так. Из опухоли больного
получают клетки, которые затем культивируют и трансдуциру-
ют в культуре геном белка-иммуностимулятора. Затем клетки
облучают, чтобы сделать их неактивными и снова вводят в
организм больного. Трансгеном, стимулирующим иммунный
ответ могут служить любые чужеродные для этих клеток анти-
гены, либо цитокины (интерлейкины, интерфероны, фактор
некроза опухолей и др.). В экспериментах на мышах примене-
ние этого подхода дало^орошие результаты: у мышей, кото-
17Q
Г лава 6
рым вводили модифицированные опухолевые клетки, опухоль
деградировала, а во многих случаях мыши становились не-
восприимчивы к опухоли при повторных введениях немодифи-
цированных опухолевых клеток.
2. Активация клеток иммунной системы. Гены цитокинов и
других стимуляторов иммунного ответа можно вводить непос-
редственно в клетки иммунной системы. Значительная часть
протоколов генотерапии опухолей основана именно на этом
подходе.
3. Введение в опухолевые клетки генов-убийц (генов, кото-
рые синтезируют продукт, приводящий к гибели опухолевых
клеток). Для этих целей широко используется ген тимидинки-
назы вируса простого герпеса. Этот фермент способен фос-
форелировать антивирусное соединение ганцинкловир, пре-
вращая его в трифосфат. Фосфорелированный ганцикловир
включается в ДНК при ее синтезе клеткой (т.е. при делении
клетки) на место нуклеозидтрифосфата. Тем самым, синтез
ДНК терминируется, и клетка гибнет. То есть, экспрессия ти-
мидинкиназы HSV в присутствии ганцикловира приводит к ги-
бели делящихся клеток, что и требуется для борьбы с опухо-
левой тканью. В эксперименте на мышах этот подход дал за-
мечательные результаты: гибли не только модифицированные
тимидинкиназой HSV клетки, но и нетрансдуцированные клет-
ки опухоли, по видимому, из-за межклеточной диффузии фос-
форелированного ганцикловира. Сейчас стратегия самоубий-
ства раковых клеток проходит клинические испытания на лю-
дях.
4. Блокирование экспрессии онкогенов. Существует два
пути реализации этого подхода. Первый - введение антисмыс-
ловых нуклеотидов, связывающих мРНК онкогенов. Второй -
так называемая внутриклеточная иммунизация. Для этого в
клетку вводят ген, кодирующий белки-антитела, которые свя-
зывают нежелательный белок внутри клетки. Такие антитела
получили название “интрабоди”.
5. Введение в опухолевые клетки генов-супрессоров. Сей-
час для этого используют антионкоген р53, который повреж-
ден во многих типах опухолевых клеток. В клетку переносят
его нормальную копию.
6. Защита стволовых клеток от токсических эффектов хи-
миотерапии. Основным фармакологическим средством борь-
бы с опухолью является химиотерапия - интенсивное "накачи-
вание” организма лекарствами, следствием которого являют-
ся различные токсические эффекты. При интенсивном лекар-
ственном вмешательстве опухолевые клетки начинают экспрес-
сировать ген MDR-1 (множественная устойчивость к лекарст-
Некоторые прикладные аспекты геномных исследований
вам). Если ген MDR-1 вводить в гемапоэтические клетки, то
можно ослабить токсические эффекты химиотерапии. Этот при-
ем проверен на экспериментальных линиях мышей, на людях
он находится в стадии клинических испытаний.
Все вышеперечисленное показывает, что арсенал генно-
инженерных средств борьбы с раком довольно велик. Ближай-
шая задача состоит в том, чтобы научиться использовать его
эффективно и без вреда для больного.
Генотерапия инфекционных заболеваний:
ДНК-вакцины
Основная проблема, затрудняющая иммунизацию против
хронических вирусных инфекций (таких как гепатит С, HSV, HIV)
связана с низкой иммуногенностью стандартных вакцйн. Эту
проблему можно попытаться решить, проводя вакцинацию не
белковыми продуктами, а с помощью ДНК (McDonnell, Askari,
1996).
ДНК-вакцина - это молекула ДНК, содержащая какой-либо
из генов вируса, обладающий антигенными свойствами, на-
пример, ген одного из белков оболочки. Попадая внутрь клет-
ки, ДНК-вакцина начинает синтезировать вирусный белок, ко-
торый вызывает иммунный ответ организма-хозяина. Принци-
пиальное отличие вакцины на основе ДНК от стандартной (как
правило, белковой) заключается именно в ее способности
проникнуть в клетку. Дело в том, что чужеродный белок, син-
тезированный внутри клетки, “обрабатывается” молекулами
главного комплекса гистосовместимости (МНС, main histocom-
patibility complex) типа I, которые активируют клеточный им-
мунный ответ. Компоненты же стандартных вакцин проникают
в клетку путем эндоцитоза или фагоцитоза и связываются
молекулами главного комплекса гистосовместимости типа II,
которые запускают иммунный ответ, опосредованный антите-
лами. Хотя клеточная и гуморальная система иммунного отве-
та в организме тесно взаимосвязаны, решающим условием
успешного действия вакцины является ее способность стиму-
лировать цитотоксические Т-лимфоциты.
Вирусные инфекции преимущественно внутриклеточны.
Внутри инфицированной клетки вирус защищен от антител, и
чтобы обнаружить и уничтожить вирус, необходима работа
клеточной системы иммунного ответа. Антитела играют важ-
ную роль в нейтрализации вирусных частиц, высвобождаемых
из клетки в кровоток или межклеточное пространство после
того, как клетка убита самим вирусом или цитотоксическими
Т-лимфоцитами. Антитела направлены обычно против повер-
хностных белков вируса. Однако вирусы выработали тактику
181
Г лава 6
защиты от антител путем высокой частоты мутирования по-
верхностных белков. Высокая изменчивость антигенов вируса
(а у одного и того же больного, инфицированного, например,
вирусом гриппа, гепатита или иммунодефицита (HIV) может
обнаруживаться множество разных форм вируса) часто помо-
гает вирусу избежать гуморальной системы иммунного отве-
та.
ДНК-вакцины создаются на основе “голой” (nacked) ДНК,
то есть ДНК, свободной от белков, с которыми она обычно
связана в нативном состоянии (в хромосомах) или в искус-
ственных векторах (вирусных или ДНК-белковых комплексах).
Нужный ген просто встраивают в кольцевую молекулу ДНК -
плазмиду, которую и вводят в ткани больного путем инъекции.
ДНК-вакцины на основе плазмид позволяют избежать таких
проблем, типичных для переноса вирусными и невирусными
векторами, как иммунный ответ против вектора и вопросы
безопасности, связанные с патогенным действием вируса.
Первой была разработана ДНК-вакцина против вируса грип-
па. Чтобы справиться с высокой частотой мутирования виру-
са, генные инженеры встроили в плазмиду ген нуклеопротеи-
на (он кодирует белок внутреннего ядра вирусной частицы, а
не оболочки), который меньше подвержен мутированию и схо-
ден у многих штаммов вируса. В экспериментах на мышах при-
менение этой вакцины обеспечивало иммунитет против ле-
тальной дозы гетерологичного (другого) штамма вируса, чего
стандартные вакцины обеспечить не в состоянии. Через год
после вакцинации мыши все еще оставались полностью за-
щищенными от летальных доз гетерологичного вируса грип-
па. По оптимистическим прогнозам авторов эксперимента это
дает надежду на разработку вакцины для человека, которая
могла бы защитить от любых штаммов вируса пожизненно.
Еще одно потенциальное и очень важное преимущество
ДНК-вакцин в том, что легко можно создать рекомбинантную
плазмиду, которая содержала бы гены различных инфекцион-
ных агентов. Это могло бы значительно снизить число вакци-
наций и, соответственно, затраты на них, при широком внед-
рении в медицинскую практику.
ДНК-вакцина против вируса гриппа - пример борьбы с ос-
трой вирусной инфекцией. Вакцинация с помощью ДНК мо-
жет, в принципе, помочь и в борьбе с хроническими инфекци-
ями, такими как гепатит или СПИД. Хроническое инфекцион-
ное заболевание представляет собой длительную борьбу им-
мунной системы организма с инфекционным агентом, которая
отнюдь не всегда заканчивается победой первого. Быстрое
мутирование антигенов вируса приводит к тому, что гумораль-
Некоторые прикладные аспекты геномных исследований
ная система иммунного ответа не справляется с вирусом, а
цитотоксические лимфоциты не успевают уничтожить все ин-
фицированные клетки до того как высвободившиеся частицы
вируса инфицируют новые.
Вакцина, способная активировать клетки иммунной систе-
мы, может сместить баланс в этой борьбе в сторону организ-
ма хозяина. Такая стратегия разрабатывается сейчас для борь-
бы с вирусом HIV. В качестве ДНК-вакцины используется век-
тор, несущий ген белка оболочки вируса - гликопротеина 160.
Сейчас эта вакцина находится в стадии клинических экспери-
ментов. Первая их фаза показала, что после внутримышечной
инъекции вакцины у больных наблюдается экспрессия векто-
ра и обнаруживаются цитотоксические Т-лимфоциты против
HIV-инфицированных клеток.
Несмотря на довольно оптимистические результаты тестов
ДНК-вакцин, остаются и проблемы. Прежде всего, неизвест-
но, может ли плазмида встраиваться в геном клетки. Если да,
то следствием этого может быть инсерционный мутагенез. Хотя
никаких доказательств интеграции плазмид в геном клетки нет,
необходима тщательная проверки последствий ДНК-вакцина-
ции. Неясными также остаются долговременные эффекты воз-
действия на иммунную систему ДНК-вакцин.
Перспективы генной терапии
Последние несколько лет ознаменовались бурным ростом
исследований по генотерапии. Генотерапия - одно из самых
молодых и бурно развивающихся направлений медицинской
генетики. Ее “документированная” история берет начало в кон-
це 80-х годов. Но уже сейчас в мире опробовано и утверждено
более 150 генотерапевтических протоколов (Unterhuber, 1996;
Friedmann, 1996). Лидером в этой области являются США, где
принято 136 протоколов генной терапии, которые на стадии
клинических испытаний применены уже к 750 больным. Ре-
зультаты этих исследований порождают надежду на успех ге-
нотерапевтических мероприятий применительно к таким тя-
желым наследственным болезням, как ТКИД, муковисцидоз,
семейная гиперхолестеринемия, миодистрофия Дюшенна, а
также к различным онкологическим и инфекционным заболе-
ваниям.
Однако в большинстве экспериментов по генотерапии пока
достигнуты лишь относительные успехи. А именно, пока уда-
ется добиться более или менее успешного переноса чужерод-
ного генетического материала в нужное место и относительно
продолжительной экспрессии потенциально терапевтическо-
го гена. Это само по себе уже не мало, однако действительно
183
терапевтический эффект генно-инженерных манипуляций в
отношении клинических проявлений наследственных наруше-
ний остается под вопросом.
Представленные в этой главе исследования показывают
пока лишь принципиальную возможность генной терапии. Ген-
ная терапия, если пользоваться определением, приведенным
в начале раздела, прошла концептуальную фазу. Теперь дело
за внедрением этой концепции в жизнь. Правильнее всего, пр
мнению Т.Фридмана (Friedmann, 1996), с которым мы соли-
дарны, будет интерпретировать текущие результаты генной
терапии как принципиальную проверку того, что а) даже от-
носительно примитивные средства, которыми располагают ге-
нетики сегодня, позволяют осуществлять перенос чужеродных
генов в клетки и ткани человека in vitro и in vivo; б) чужеродные
гены при определенных условиях могут экспрессироваться in
vivo достаточно продолжительное время, хотя зачастую и на
низком уровне; в) экспрессируемые трансгены способны, хотя
бы частично и не всегда на терапевтическом уровне, скорре-
гировать клинический фенотип.
Генотерапия уже вышла из пеленок, но чтобы попасть в
арсенал рутинных средств практического врача, ей предстоит
пройти еще долгий путь. Необходимо развитие всего идейно-
го и технологического базиса молекулярной генетики, генети-
ки человека и медицинской генетики, чтобы не только познать
механизмы наследственных болезней, но и изобрести способ
вылечить их.
1 ЯЛ
Глава 7
ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ ГЕНОМА
ЧЕЛОВЕКА И ОБЩАЯ ПАТОЛОГИЯ
(ВМЕСТО ЗАКЛЮЧЕНИЯ)
Экспансия генетических идей и понятий на современную
медицину столь велика, что представляется своевременным
обсудить взаимоотношения патологической анатомии генома
человека и общей патологии.
До второй половины XIX века форма и строение тела чело-
века изучались невооруженным глазом на макроскопическом
уровне. С развитием техники микроскопического исследова-
ния, а также химии и методов избирательной окраски сложны-
ми синтетическими красителями отдельных элементов тка-
ней человека произошло разделение на макроскопическую и
микроскопическую анатомию, гистологию и цитологию. Даль-
нейшее развитие методических возможностей привело к пе-
реходу от изучения формы и строения человеческого тела на
макроуровне к еще более мелким структурам (ультраструк-
туры клеток, субъединиц ультраструктур, наконец, фрагмен-
тов хромосом и отдельных генов, последовательности нукле-
отидов ДНК). Д.С.Саркисов (1993) справедливо подчеркива-
ет, что идея периодизации истории развития анатомии в свя-
зи с прогрессом методических возможностей не нова и при-
надлежит Р.Вирхову, высказавшему ее в 1895 году в речи “Мор-
ганьи и анатомическая мысль”. Созвучно, что автор понятия
“патологическая анатомия генома человека” В.А.Маккьюсик
считает, что в основе этого научного направления лежит "нео-
везалиевский фундамент” - как анатомия Везалия оказала вли-
яние на физиологию Гарвея и патологию Морганьи, так и зна-
ния структуры хромосом и картирование генов становятся
основой нашего понимания и управления болезнями человека
и, в первую очередь, наследственными (McKusick, 1986). Дей-
ствительно, геномные исследования легко и естественно в
своем понятийном аппарате запечатлили анатомические и
медицинские термины - “анатомия генома”, “биопсия гено-
ма”, “топография генов”, “методы анатомирования генома” и
Другие.
В этом контексте рассуждения патологическая анатомия
генома человека представляет собой дальнейшее развитие
классической патологической анатомии. Новизна определя-
ется включением в арсенаЬ аналитических приемов и методов
Глава 7
“новой генетики", которая дополнила медико-генетические
исследования широким использованием ДНК-технологий: ге-
нетическое и физическое картирование генома; идентифика-
ция генов, детерминирующих нормальные и патологические
признаки человека, секвенирование последовательностей ДНК
и т.д. (см. главу 2). В качестве яркого примера поразительно
быстрого развития морфологических исследований хромосом
человека можно привести следующий. До 1956 года еще не
было известно точное число хромосом у человека. Tjio и Le-
van (1956) уточнили, что в соматических клетках человека не
48, а 46 хромосом. В 1959 году Лежен с коллегами установи-
ли хромосомную этиологию синдрома Дауна (Leujeune et al.,
1959). Этот год считается рождением клинической цитогене-
тики. Именно с этого открытия начинается один из самых яр-
ких этапов в исследовании синдромов хромосомной этиоло-
гии - Тернера, Клайнфельтера, Эдварса-Смита, Патау, трип-
ло-Х, дубльУ и другие. В настоящее время цитогенетика чело-
века опирается уже на новые молекулярно-генетические ме-
тоды исследования. Среди них - метод флюоресцентной гиб-
ридизации in situ (FISH), о возможностях которого мы писали
выше (см. главу 6). Здесь же (табл. 16) укажем на области
применения мультиплексной FISH в клинике (медицинская ге-
нетика и онкология) и научных исследованиях (Le Beau, 1996).
Методический прорыв в изучении молекулярных основ жиз-
ни и болезней подавал надежду на новые крупные открытия в
области медицины. Были все основания полагать, что "... при-
ближается момент, когда перед взором морфолога предста-
нут молекулы... Наступление этого момента будет означать
завершение длительного исторического процесса сближения
“классической морфологии” с биохимией, их фактическое сли-
яние и, в связи с этим, появление возможности видеть весь
диапазон структур организма, начиная от самых крупных ана-
томических и кончая мельчайшими, молекулярными” (Сарки-
сов, 1993). Следует констатировать, что этот момент насту-
пил!
В настоящее время активно осуществляется каталогизация
генов человека, в том числе “патологических”. Темп накопле-
ния такой информации впечатляющий (рис. 31). McKusick
(1992), обобщая историю “открытия” генов, отмечает, что в
1911 году был известен единственный картированный ген че-
ловека - ген цветовой слепоты, отнесенный Э.Вильсоном к X-
хромосоме на основании анализа родословных. В последую-
щие годы была доказана локализация Х-сцепленных генов,
определяющих возникновение и развитие ещё около 60 бо-
лезней (признаков): гемофилия, мышечная дистрофия Дю-
шенна и другие. Но только через 57 лет, в 1968 году был кар-
1 QC
Патоло! ическая анатомия генома человека
тирован первый аутосомный ген - ген группы крови Даффи.
По последним данным картировано более 5 тыс. генов (рис.
32). Следует, однако, заметить, что несмотря на поразитель-
ные успехи геномных исследований, к настоящему времени
изучено не более 10% от общей протяженности генома чело-
века, а доля уже картированных генов человека составляет
1% от их общего предполагаемого числа.
Таблица 16.
Применение мультиплексной FISH
(по М.М. Le Beau, 1996)
Клиническое применение
Медицинская генетика:
- Детекция численных и структурных хромосомных аномалий;
- Идентификация маркерных хромосом (перестроенных
хромосом неясного происхождения.
Диагностика рака:
- Мониторинг эффектов лечения и обнаружение минимальных
остаточных явлений заболевания;
- Обнаружение раннего рецидива;
- Идентификация происхождения клеток костного мозга
после пересадки костного мозга;
- Идентификация происхождения неопластических клеток;
- Установление кариотипа неделящихся или интерфазных
клеток;
- Детекция амплификации генов.
Применение в научных исследованиях
- Молекулярный анализ хромосомных аномалий в опухолевых
тканях человека;
- Мультиплексная локализация генов и последовательностей
ДНК на хромосомах;
- Создание физических карт (включая определение порядка
последовательностей и расстояния между ними);
- Характеристика соматических клеточных гибридов;
- Идентификация хромосом, содержащих гены с желаемыми
характеристиками в растительных гибридах, и выделение этих
генов;
- Филогенетические исследования для определения локализа-
ции и протяженности синтенных участков у различных видов;
- Детекция амплифицированных генов;
- Детекция вирусных последовательностей в клетках;
- Количественная и качественная оценка мРНК или белков
на клеточном уровне;
- Анализ процессинга и транспорта РНК;
- Исследование организации хромосомных доменов или муль-
тигенных семейств на интерфазных ядрах в зависимости от
типа ткани, стадии развития, стадии клеточного цикла или
патологического состояния (реконструкция в 3-хмерном
пространстве);
- Анализ хромосомных аббераций в гено-токсикологических
исследованиях.
--------------------------,--------------------------------
187
Г лава Z
Рис, 31. Общее число менделирующих признаков человека в
MIM (McKusick, 1992; OMIM, 1996).
Для формирующейся канвы теории общей патологии, на-
щупывающей общие закономерности формирования болезней
человека, по-видимому, должны представлять интерес ряд
генетических феноменов, которые отнесены к категории не-
канонических генетических эффектов (Hall, 1996; Голубовский
и Чураев, 1997). О некоторых из них мы уже вели речь: геном-
ный импринтинг, генетическая антиципация, митохондриальная
наследственность.
Еще совсем недавно геномный импринтинг расценивался
как “генетический курьез”, а антиципация - артефактом. Те-
перь это доказанные механизмы в развитии целых групп бо-
лезней у человека и животных. Hall (1990) упоминает личное
сообщение Spire, который сделал предположение, что в рабо-
чих журналах по скрещиванию мышей скрыта масса еще не
востребованной информации по импринтингу, и не меньше ее,
считает он, скрыто в историях болезни человека. Он предла-
гает пересмотреть родословные по тем заболеваниям, для ко-
торых характерна необычная картина наследования.
Следует указать еще на один, давно известный феномен,
генетическую гетерогенность. Явление генетической гетеро-
генности, обоснование которого западными исследователями
связывается с работой Morton (1956), по-видимому было из-
вестно в клинике раньше (работы Давиденкова, 1930-е годы),
а к общим феноменам проявления генов “гетерогенные гены”
отнесены были Тимофеевым-Ресовским (1934). Однако толь-
ко молекулярно-генетические исследования последнего вре-
мени накопили огромный материал по доказательству этого
явления (Гайцхоки, 1996; Romeo and McKusick, 1994). В качес-
Паюлогическая анатомия генома человека
тве примера приведем данные по хондродисплазии и эпилеп-
сии (табл. 17-18).
Рис. 32. Динамика картирования генов человека (McKusick,
1992; GDB, 1997).
Все эти перечисленные отдельные феномены столь изящ-
но доказаны и объясняют самые существенные моменты раз-
вития болезней, что порой кажутся незначительными другие
факторы, соучаствующие в формировании патологии. Так со-
блазнительно согласиться с героем одного из романов
Ф.М.Достоевского: "... наплевать на все общие принципы, в
наше время - не общие принципы, а одни только частные слу-
чаи”. Для аналитиков в науке эта ситуация нормальная; ре-
дукционизм в исследовании необходимая (или вынужденная)
сторона познания. Но в этом же романе другой герой думает
иначе: "... объединить частности и найти хоть какой-нибудь
толк во всей общей бестолочи”. Не в этом ли состоит одна из
задач общей патологии и теории медицины?
Стремление патологов охватить одновременно в условиях
комплексного изучения патологических процессов все уровни
организации жизни - нелегкая задача. Вся сложность ее ре-
шения очевидна из рис. 33. В настоящей книге нами обсужда-
лась информация, накопленная на левом фланге этой иерар-
хической схемы: гены - генетические системы. Но это, безус-
ловно, лишь часть информации о структурной организации ге-
нома человека, объединенная новым направлением - патоло-
гической анатомией генома человека. Параллельно с ним раз-
вивается функциональная анатомия генома человека (McKu-
sick, 1988). Это перспективное и важное направление геном-
ных исследований только в начале пути. Еще не устоялись даже
термины. Наряду с обозначением этого направления как “фун-
кциональная анатомия генома человека”, предложены другие
1 ЯП
Глава 7
- физиологическая или функциональная геномика. Обсужде-
нию этих вопросов была посвящена недавняя конференция
“Functional Genomics” (Вашингтон, 1997; см. “Up the function”,
1997). Filds (1997) за этим направлением видит будущее ге-
номных исследований.
Таблица 17.
Гены хондродисплазии человека
(Horton, 1995)
Клинический фенотип Локус Хромосома Функцйя генного продукта
Ахондрогенез тип II Г ипохондрогенез Врожденный SED Дисплазия Книста Поздний SED Дисплазия Стиклера COL2A1 12q13.11 белок внеклеточ- ного Матрикса
MED COL9A2 1р33-р32. белок внеклеточ- ного Матрикса
Метафизарная хондродисплазия Шмида COL10A 6q21-q22. белок внеклеточ- ного Матрикса гипертрофичес- кий хрящ
Стиклер-подобная дисплазия COL11A 6р21.3 белок внеклеточ- ного Матрикса
Псевдоахондро- плазия MED Фэрбенкс СОМР 19р13.1 белок внеклеточ- ного Матрикса
Танатоформная дисплазия Ахондроплазия Г ипохондроплазия FGFR3 4р16.3 тирозйнкиназа трансмембранный рецептор фактора роста фибро- бластов
Метафизарная хондродисплазия Янсена PTHrPR Зр21-р22 G-протеиновый трансмембранный рецептор для РТН и РТНгР
Дистрофическая дисплазия Ахондрогенезия тип IB DTDST 5q31-q34 трансмембранный переносчик сульфатов
Хондродисплазия пунктата ARSE Хр22.3 арилсульфатаза
Кампомелическая дисплазия SOX9 17q24.1-q фактор транскрипции
Патологическая анатомия генома человека
Таблица 18.
Гены эпилепсии человека (Allen and Walsh, 1996)
Болезнь Локализация Ген
Идиопатические генерализованные эпилепсии
Ювенильная миоклональная эпилепсия (EJM1) 6р ?
Семейные неопасные конвульсии новорожденных (BFNC1) 20q CHRNA4 (?)
Семейные неопасные конвульсии новорожденных (BFNC2) 8q ?
Идиопатическая генерализованная эпилепсия (IGE) 8q ?
Идиопатические парциальные эпилепсии
Парциальная эпилепсия со слуховыми симптомами (ЕРТ) 10q ?
Ночная эпилепсия фронтальной доли (ADNFLE) 20q CHRNA4
Моногенные нарушения с симптоматикой эпилепсии в качестве основного признака
Болезнь Унферрихта-Лундборга (ЕРМ1) 21q22.3 цистатин В
Миоклональная эпилепсия с разорванно-красными мышечными волокнами (MERRF) митохон. геном TPHK(Lys)
Синдром северной эпилепсии (EPMR) 8p ?
Цероидный липофусциноз, ювенильный тип (CLN3) 16p CLN3
Миоклональная эпилепсия Лафора (MELF) 6q23 ?
Околожелудочковая гетеротопия (PH) Xq28 ?
Правый фланг представленной схемы (популяции - попу-
ляционные системы; сообщества - экосистемы) также должен
быть, на наш взгляд, в поле зрения общей патологии. Здесь
свои проблемы и сложности. Источником идей для обсужде-
ния исследовательского материала этого уровня сложности
1Q1
Глава 7
БИОТИЧЕСКИЕ
СИСТЕМЫ Гены Клетки Органы Организмы Популяции Сообщества
Рис. 33. Спектр уровней
организации жизни
(Одум, 1986).
биосистемы, в которую включен,
естественно, человек (здоровый
и больной), могут быть совре-
менные идеи коэволюции, кос-
мопланетарной Основы феноме-
на человека, синергетики (см.
Тимофеев-Ресовский, 1980;
Пригожин и Стейгерс, 1986. Мо -
исеев, 1987; Казначеев и Спи-
рин, 1991; Родин, 1991). На пути
познания феномена человека от
генома до биосферы предстоит
еще решить многие проблемы,
которые по плечу только меж-
дисциплинарным и комплексным
исследованиям. Ибо сложен
объект познания - человек. Как
заметил Ключевский в 80-е годы
прошлого столетия: “Весь этот
человек - игра природы и судь-
бы: обе эти причудницы хотели
доказать на нем, как они изобре-
тательны в своих капризах".
Недавно был проведен опрос
2,5 тыс. ученых разных специаль-
ностей о наиболее перспектив-
ных науках ближайшего десяти-
летия, охватывающего горизонт
начала XXI века. Практически все
опрошенные были едины во мне-
нии, что генетика и биотехноло-
гия будут представлять первос-
тепенный интерес и приоритет
(табл. 19). Более того, сейчас и
на ближайшую перспективу об-
суждаются возможности “бизне-
са на геноме" (см.; Capitalizing on
genome, 1996).
Наиболее крупные фармако-
логические и биотехнологичес-
кие компании сосредотачивают
свою деятельность по идентифи-
кации генов, имеющих отноше-
ние к болезням человека, тратя
сотни миллионов долларов на
поддержку геномных исследова-
1 оо
Патологическая анатомия генома человека
ний и стратегии разработки новых лекарственных препаратов
на основе успехов генетики. Особенно крупные денежные вкла-
ды осуществлены в исследования на выделение генов таких
заболеваний, как диабет, астма, онкологические и психичес-
кие болезни. Так, компания Amgen купила за 20 млн. долларов
права у Рокфеллеровского университета на исследования гена
ожирения и возможных (вполне реальных) коммерческих про-
дуктов (препараты для диагностики и терапии).
В заключение приведем интересные временные сопостав-
ления по развитию генетики человека, которые неожиданно
поразили одного из известных генетиков Чарльза Эпштейна.
В 1993 году он написал статью "Семь памятных лет", в кото-
рой были перечислены достижения генетики человека за пе-
риод с 1987 по 1993 годы [Epstein, 1993). Они включали:
- Позиционное и функциональное клонирование генов бо-
лее 25 болезней.
- Открытие болезней экспансии числа тринуклеотидных
повторов и "реабилитация" феномена антиципации.
- Аутосомный импринтинг и болезни импринтинга у чело-
века.
- Первые попытки генотерапии.
- Модели трансгенеза и гомологичной рекомбинации на мы-
шах.
- Математические и физические подходы к анализу сцепле-
ния.
- Болезни мтДНК.
- Проект “Геном Человека”.
- Клонирование и секвенирование кДНК.
- Применение анализа ДНК в криминалистике.
- Гены опухолевых супрессоров.
- Микроделеционные синдромы.
- Особенности половых хромосом.
- Эволюционный консерватизм генома.
- Эволюция и расселение популяций человека.
- Новые методы пренатального скрининга и диагностики.
- Аналитические методы и приборы.
- Озабоченность юридическими и этическими вопросами.
Поразительны успехи в последующие три года - к 1997 году:
- Более 60 генов идентифицировано позиционным клони-
рованием, а общее число генов, клонированных любыми спо-
собами, значительно больше.
- Полногеномный поиск генов сложнонаследуемых болез-
ней человека.
- Появились новые классы генов: факторов транскрипции,
транспортеров ионных кдналов, рецепторов факторов роста
фибробластов, геликазы.
193
Глава 7
- Найдены болезни человека, связанные с генами НОХ, РАХ
и SOX.
- Картирование генома человека продвигается вперед, на-
чинается глобальное секвенирование:
- Число геномных маркеров становится огромным - оно
измеряется десятками тысяч.
- Практически любой мутантный ген человека может быть
смоделирован у мыши (“нокаутированные” мыши).
- Разработаны мощные цитогенетические технологии - 24-
цветовая окраска хромосом, сравнительная геномная гибри-
дизация.
- Расширение скрининга наиболее распространенных хро-
мосомных синдромов с одновременным использованием трех
различных ДНК-зондов (трипл-маркеры).
Одно только перечисление новых достижений в области
молекулярной генетики, генетики человека и медицинской ге-
нетики свидетельствует о том, что накапливающийся факто-
логический материал, наряду с новыми идеями методологи-
ческого и философского характера в объяснении феномена
человека, должны стать ориентиром в обсуждении проблем
общей патологии и теории медицины.
Патологическая анатомия генома человека
Таблица 19.
Наиболее перспективные науки следующего десятилетия по опросу
американских ученых разных специальностей (Science, 1992, N 1735)
195
Приложение
СЛОВАРЬ ТЕРМИНОВ
Авторадиография. Метод выявления радиоактивной метки на фраг-
ментах ДНК с помощью рентгеновского излучения. Используется для оп-
ределения длины и числа молекул ДНК, разделенных гель-электрофоре-
зом.
Аденин (А). Азотистое основание, один из четырех мономеров ДНК.
Образует комплиментарную пару с тимином.
Азотистое основание. Азот-содержащая молекула, имеющая хими-
ческие свойства основания.
Аллель. Альтернативная форма гена.
Аминокислота. Мономер, образующий молекулу белка. В состав бел-
ка у живых организмов могут входить 20 разных аминокислот. Последова-
тельность аминокислот в белке определяется последовательностью нук-
леотидов гена.
Амплификация. Увеличение числа копий определенного фрагмента
ДНК (in vivo или in vitro). См: клонирование, полимеразная цепная реак-
ция.
Аутосома. Хромосома, не участвующая в определении пола. Диплоид-
ный набор хромосом человека состоит из 46 хромосом: 22 пар аутосом и
1 пары половых хромосом (X и Y).
Бактериофаг. См: фаг.
Белок. Молекула, построенная из одной или нескольких аминокислот-
ных цепочек, каждая из которых структурирована определенным образом.
Структура аминокислотной цепи определяется последовательностью ами-
нокислот в ней, что в свою очередь детерминируется последователь-
ностью нуклеотидов в гене. Каждый из белков выполняет определенную
функцию и необходим для построения, функционирования и регуляции
клеток, тканей и органов. Примеры: гормоны, антитела, ферменты.
Библиотека генов. См: геномная библиотека.
Библиотека клонов. Неупорядоченная коллекция клонов (т.е., клони-
рованной ДНК определенного организма). Взаимное расположение фраг-
ментов генома, содержащихся в клонах можно установить методами фи-
зического картирования. См. также геномная библиотека.
Вектор. Молекула ДНК вирусного или плазмидного происхождения или
клетка высших организмов, в которую интегрируют другой (чужеродный)
фрагмент ДНК. Вектор вводят в клетку-хозяина, где, используя способ-
ность вектора к саморепликации, встроенный фрагмент ДНК репродуци-
руется вместе с ДНК вектора. Примеры векторов: плазмиды, космиды,
искусственные дрожжевые хромосомы (YAC). Вектор является рекомби-
нантной молекулой, т.е. содержит ДНК разного происхождения.
Вирус. Неклеточная форма существования живого. Репродуцируется
только вместе с клеткой-хозяином. Состоит из молекулы ДНК и оболочки,
построенной из молекул белка.
Гамета. Зрелая мужская или женская половая клетка (сперматозоид
или яйцеклетка) с гаплоидным набором хромосом (у человека - 23 хромо-
сомы)
Гаплоид. Клетка или организм, содержащий гаплоидный набор хро-
мосом (т.е., каждая хромосома представлена одной копией). Гаплоидный
набор хромосом человека состоит из 23 хромосом.
Ген. Единица организации генетической информации. Представляет
Словарь терминов
собой определенную последовательность нуклеотидов, расположенную в
определенном месте хромосомы и кодирующую определенный продукт
(белок или молекулу ДНК). См также экспрессия гена.
Генетическая инженерия. См. технология рекомбинантных ДНК.
Генетическая карта. См. карта сцепления.
Генвтический код. Последовательность нуклеотидов в молекуле мат-
ричной РНК (комплиментарной копии гена) организована в триплеты (ко-
доны), каждый из которых кодирует одну аминокислоту. Тем самым, пос-
ледовательность аминокислот в белке закодирована в последовательнос-
ти триплетов мРНК. Соответствие триплетов аминокислотам и называет-
ся генетическим кодом.
Генйый продукт. Результат экспрессии гена - РНК или белок. По ко-
личеству продукта гена можно судить, активен ген или нет. Ненормально
большое или ненормально малое количество продукта может быть связа-
но с аллелями, ответственными за заболевание.
Геном. Весь генетический материал организма.
Геномная библиотека. Коллекция клонов, содержащих перекрываю-
щиеся фрагменты ДНК, охватывающая весь геном организма. См. также
библиотека клонов.
Генотерапия. Введение нормальной копии гена в клетку для коррек-
ции генетического дефекта.
Гетерозиготность. Наличие различных аллелей в одном или более
локусах гомологичных хромосом.
Гибридизация in situ. Использование ДНК- или РНК-зондов для обна-
ружения комплиментарного участка ДНК в клонированных бактериальных
клетках или в культивируемых клетках эукариот.
Гибридизация. Процесс образования двухцепочечной молекулы из двух
комплиментарных цепей ДНК или цепи ДНК и цепи РНК.
Гомологичные хромосомы. Пара хромосом, содержащих одинаковую
последовательность генов, каждая из которых получена от одного роди-
теля.
ГоМология. Сходство в последовательности ДНК или белка между ор-
ганизмами того же вида или между видами.
Гуанин (G). Азотистое основание, один из четырех мономеров ДНК.
Образует комплиментарную пару с цитозином.
Двойная спираль. Форма, которую принимает двухцепочечная моле-
кула ДНК благодаря взаимодействию оснований.
Дезоксирибонуклеотид. См. нуклеотид.
Диплоид. Организм или клетка с диплоидным набором хромосом.
ДиИлоидный набор хромосом. Полный набор генетического матери-
ала, состоящий из парных (гомологичных) хромосом - по одной от каждо-
го родителя. Большинство клеток животных, исключая гаметы, имеют дип-
лоидный набор хромосом. Диплоидный набор человека состоит из 46 хро-
мосом. См. также: гаплоид.
ДНК (дезоксирибонуклеиновая кислота). Молекула, кодирующая
генетическую информацию. ДНК - двухцепочечная молекула, в которой 2
комплиментарные цепочки связаны водородными связями. Каждая цепочка
состоит из последовательности нуклеотидов, в состав ДНК входят 4 азо-
тистЫх основания: аденин, тимин, гуанин и цитозин. Аденин комплимен-
тарен тимину, а цитозин - гуанину. Благодаря свойству комплиментарнос-
ти, последовательность оснований на одной из цепочек можно выявить,
зная последовательность оснований на комплиментарной цепи.
ДНК, комплиментарная (кДНК). ДНК, которая синтезируется на мат-
рице мРНК. Одноцепочечная форма кДНК часто используется в качестве
зонда при физическом картировании.
197
Приложение
Домен. Участок белка, имеющий собственную функцию.
Зойд. Одноцепочечная молекула ДНК или РНК с определенной после-
довательностью оснований, несущая метку (радиоактивную, флюоресцен-
тную, иммунологическую). Гибридизация зонда с комплиментарной ему
последовательностью позволяет найти в геноме нужный участок ДНК.
Интерфаза. Период клеточного цикла, в течение которого происходит
реплйкация ДНК. Предшествует митозу.
Интрон. Участок ДНК, находящийся между кодирующими областями
(экзонами) гена. Интроны транскрибируются в РНК, но перед трансля-
цией РНК в белок вырезаются из молекулы РНК.
Искусственная дрожжевая хромосома (YAC). Вектор, применяемый
для клонирования очень больших (до 400 Кб) фрагментов ДНК. Сконстру-
ирован из структур, необходимых для репликации хромосомы дрожжей
(теломеры, центромеры и ориджина репликации). См. также вектор.
Кариотип. Набор хромосом организма и их характеристики (число,
размер, форма). Используется на начальных стадиях физического карти-
рования, чтобы связать крупные хромосомные перестройки с той или иной
болезнью.
Карта генетического сцепления. Карта, показывающая относитель-
ное расположение генетических локусов на хромосоме. Карта строится
на осйове частоты рекомбинации между локусами. Расстояния на карте
генетйческого сцепления измеряются в сантиморганидах.
Карта контиг. Карта, показывающая расположение друг относительно
друга набора небольших перекрывающихся клонов, покрывающих полный
сегмент хромосомы.
Картирование генов. Определение взаимного расположения генов на
молекуле ДНК (например, на хромосоме или в плазмиде) и расстояния
между ними.
Картирование. См. картирование генов, карта генетического сцепле-
ния, физическое картирование.
Кб. См. килобаза.
кДНК. См. ДНК, комплиментарная.
Килобаза (Кб). Единица длины ДНК, равная 1000 пар оснований.
КлОн. Группа клеток, полученная путем деления единственной перво-
начальной клетки.
Клонирование. Процесс получения генетически идентичной группы
клеток (клона) из одной первоначальной клетки. Клонирование ДНК - про-
цесс получения множественных копий отдельного гена или фрагмента ДНК.
Код. См. генетический код.
Кодон. Триплет нуклеотидов, кодирующий аминокислоту. См. также
генетйческий код.
Комплиментарная последовательность. Последовательность осно-
ваний ДНК, которая может образовать двухцепочечную молекулу с данной
последовательностью благодаря комплиментарное™ пар оснований. На-
пример, последовательности GTAC комплиментарна последовательность
CATG.
Комплиментарность. Свойство нуклеотидов образовывать пары с оп-
ределенным нуклеотидом (А-Т, G-C).
Контиги. Группы клонов, охватывающих перекрывающиеся участки ге-
нома.
Космида. Искусственно созданный вектор, содержащий ген cos фага
лямбда. Космиды могут упаковываться в частицы фага лямбда для раз-
множения в бактерии Е. coli. В космидах можно клонировать большие (до
45 Кб) по сравнению с плазмидами фрагменты ДНК.
Кроссинговер. Процесс во время мейоза, когда происходит разрыв
198
Словарь терминов
двух гомологичных (материнской и отцовской) хромосом, обмен участком
ДНК и воссоединение хромосом. В результате происходит обмен аллеля-
ми между хромосомами. См. также рекомбинация.
Локализация. Определение расположения (локуса) гена или маркера
на хромосоме.
Локус. Позиция гена илй маркера на хромосоме, а также собственно
генетический материал (ДНК) в этой позиции.
Макрорестрикционная карта. Карта, показывающая расположение на
хромосоме сайтов рестрикции и расстояние между ними.
Маркер. Физический участок хромосомы, который можно тем или ийым
образом идентифицировать (например, ген или рестрикционный сайт) и
чье наследование можно проследить. Маркерами могут быть как экспрес-
сируемые участки ДНК (гень(), так и последовательности, не несущие фун-
кции кодирования. См. также: ПДРФ.
Матричная РНК. См. мРНК.
Мб. См. мегабаза.
Мегабаза (Мб). Единица измерения длины ДНК, равная миллиону нук-
леотидов (пар оснований). Физическая дистанция в 1 Мб приблизительно
эквивалентна 1 См на карте генетического сцепления.
Мейоз. Процесс двух последовательных делений клетки, а результате
которого из одной исходной диплоидной клетки образуются четыре пол-
овые клетки (гаметы), имеющие гаплоидный набор хромосом.
Метафаза. Стадия клеточного деления (митоза или мейоза), при кото-
рой хромосомы располагаются в экваториальной плоскости клетки.
Митоз. Процесс делений клетки, в результате которого из одной дип-
лоидной клетки образуются две с точно таким же набором хромосом, т.е.
генетически идентичные друг другу и родительской клетке.
Моногенный (менделирующий) признак. Наследственный признак
(в том числе и болезнь), определяемый одним геном (например, мышеч-
ная дистрофия Дюшенна, Серповидно-клеточная анемия, ретинобласто-
ма). См. также полигенный признак.
мРНК. Матричная или информационная РНК - молекула РНК, транскри-
бируемая с молекулы ДНК и служащая матрицей для синтеза белка.
Мультиплексирований. Технология секвенирования, при которой
одновременно секвенируюТся несколько смешанных образцов, что позво-
ляет значительно ускорить этот процесс.
МультифакториальныЙ (мультигенный) признак. См. полигенный
признак.
Мутация. Любое наследуемое изменение последовательности ДНК. См.
также полиморфизм.
Нуклеиновая кислота. Полимерное вещество, состоящее из нуклео-
тидов и имеющее химические свойства кислоты. Имеется два типа нукле-
иновых кислот - дезоксирибонуклеиновая кислота (ДНК) и рибонуклеино-
вая кислота (РНК), различающиеся по химическому составу. См. также
нуклеотид, ДНК, РНК.
Нуклеотид. Мономер, йз которого строится молекула нуклеиновой кис-
лоты. Состоит из азотистого основания (аденин, гуанин, тимин и ци+озин
для ДНК, аденин, гуанин, урацил и цитозин для РНК), остатка фосфорной
кислоты и молекулы сахара (дезоксирибозы в составе ДНК и рибозы в
составе РНК).
Онкоген. Ген, одна ийи несколько форм которого связаны с раком.
Нормальной функцией онкогенов является контроль за ростом и делени-
ем клеток.
Ориджин репликации.. Участок ДНК, с которого начинается реплика-
ция. }
199
Приложение
п.о.. См. пара оснований.
Пара оснований (п.о.). Два азотистых основания (А-Т или С-G), свя-
занных друг с другом водородными связями. Две цепочки ДНК (двойная
спираль) удерживаются вместе благодаря связям оснований в компли-
ментарных парах.
ПДРФ (полиморфизм длин рестрикционных фрагментов). Межин-
дивидуальные различия в длине фрагментов ДНК, образующихся при рас-
щеплении определенной рестриктазой, являющиеся результатом вариан-
тного наличия или отсутствия рестрикционных сайтов. Такие полиморф-
ные рестрикционные сайты могут использоваться как маркеры при физи-
ческом или генетическом картировании. ПДРФ обычно является резуль-
татом мутации в сайте узнавания рестриктазой. См. также маркер.
Перекрывающиеся клоны. Клоны, содержащие фрагменты ДНК, име-
ющие общие участки. См. также геномная библиотека.
Перенос (блоттинг) по Саузерну. Метод переноса фрагментов ДНК,
разделенных гель-электрофорезом, из геля на мембранный фильтр. Ис-
пользуется для поиска генов или других последовательностей ДНК с по-
мощью радиоактивно меченных комплиментарных зондов.
Пиримидий. Химическое соединение, содержащее азот, имеющее
двухкольцевую структуру и свойства основания. К пиримидинам принад-
лежат цитозин, тимин и урацил.
Плазмида. Автономно реплицирующаяся ВнехромосоМная кольцевая
молекула ДНК (существующая, как правило, в бактериальной клетке). Ге-
нетический Matepnan плазмиды не является частью нормального генома
бактерии и не существенен для выживания клетки в нормальных (неселек-
тивных) условйях. Некоторые плазмиды способны к интеграции в геном
клетки-хозяина. Большое количество искусственно созданных плазмид
используется в качестве векторов для клонирования.
Полигенный признак. Наследуемый признак (в том числе и болезнь),
являющийся результатом совместного действия более чем одного гена.
Такие признаки! передаются по наследству, однако для их проявления тре-
буется присутствие определенных аллелей нескольких (мйогих) локусов.
Поэтому, картйна передачи по наследству полигенных признаков, как пра-
вило, более сложна чем таковая для моногенных признаков. Полигонный
признак, чье проявление зависит и от негенетических факторов (внешней
среды) называют мультифакториальным. См. также монотонный признак.
Полимераза, ДНК или РНК. Фермент, который катализирует процесс
синтеза молекулы нуклеиновой кислоты из отдельных нуклеотидов на од-
ноцепочечной молекуле-матрице.
Полимеразная цепная реакция. Метод амплификацйи фрагментов
ДНК с использованием термостабильной ДНК-полимеразЫ и пары олиго-
нуклеотидных Лраймеров. Один из праймеров, имеющих длину, как пра-
вило, около 20 нуклеотидов, комплиментарен (+)-цепочке ДНК на одном
из концов последовательности, другой комплиментарен (-)-цепочке на
другом конце. Вновь синтезированные в ходе ПЦР цепочки ДНК могут
сами служить матрицей для праймеров, что позволяет, циклически повто-
ряя отжиг (гибридизацию с матрицей) праймеров, элонгацию цепочки под
действием полимеразы и денатурацию (расхождение комплиментарных
цепочек), быстро получить множество копий (миллионы) исходной после-
довательности ПЦР можно применять и для поиска Определенной после-
довательности в образце ДНК.
Полиморфизм длин рестрикционных фрагментов. См. ПДРФ.
Полиморфизм. Существование более чем одного варйанта последо-
вательности ДНК (гена, маркера). Генетические вариации, встречающие-
ся в популяции с частотой больше 1% могут использоваться при генети-
Словарь терминов
ческом анализе.
Половые хромосомы. Хромосомы, определяющие пол индивида. В
геноме человека это X и Y-хромосомы. Женщины имеют две Х-хромосомы
в диплоидном наборе, мужчины - по одной X и Y-хромосоме.
Последовательность ДНК. Относительное расположение пар осно-
ваний во фрагменте ДНК, гене, хромосоме или во всем геноме.
Последовательность оснований. Порядок чередования нуклеотидов
(азотистых оснований) в молекуле ДНК.
Праймер. Короткая одноцепочечная молекула нуклеиновой кислоты
(олигонуклеотид), к которой полимераза «пристраивает» новые нуклеоти-
ды в процессе синтеза ДНК или РНК.
Прокариота. Клетка или организм, у которого отсутствует ограничен-
ное мембраной ядро. К прокариотам относятся вирусы, бактерии и сине-
зеленые водоросли. См. также эукариота.
Промотор. Участок гена, ДНК или РНК, связываясь с которым молеку-
ла ДНК- или РНК-полимеразы инициирует процесс транскрипции.
Проточная цитометрия. Метод анализа биологического материала,
основанный на использовании светопоглощающих или флуоресцентных
свойствах клеток или их фракций (например, хромосом).
Пурин. Химическое соединение, содержащее азот, имеющее одноколь-
цевую структуру и свойства основания. В состав ДНК и РНК входят пурины
аденин и гуанин.
ПЦР. См. полимеразная цепная реакция.
Регуляторные последовательности (участки) ДНК. Часть гена или
отдельный ген, функцией которого является контроль генной экспрессии.
Рекомбинантная молекула ДНК. Молекула ДНК, в которой посредст-
вом технологии рекомбинантной ДНК объединены фрагменты ДНК раз-
личного происхождения.
Рекомбинантный клон. Клон, содержащий рекомбинантную молекулу
ДНК. См. также технология рекомбинантной ДНК.
Рекомбинация. Процесс, в результате которого потомки получают ком-
бинации генов, отличные от комбинаций у обоих родителей У высших
организмов в основе рекомбинации лежит кроссинговер.
Репликация ДНК. Процесс удвоения количества молекул ДНК при ко-
тором для синтеза новой молекулы уже существующая молекула ДНК ис-
пользуется как матрица.
Рестрикционная эндонуклеаза (рестриктаза). Фермент, который
распознает специфическую последовательность (сайт) ДНК и разрезает
молекулу в этом месте. К настоящему времени известно более 400 рес-
триктаз, выделенных из различных бактерий, которые распознают более
100 рестрикционных сайтов. См. также рестрикционный сайт.
Рестрикционный сайт. Определенная последовательность ДНК, в ко-
торой молекула может быть разрезана рестрикционным ферментом. Не-
которые сайты встречаются в ДНК довольно часто (через несколько сотен
пар оснований). Рестриктазы, распознающие эти сайты называют мелко-
щепящими, другие, более редкие сайты (чапример, один на 100000 пар
оснований), распознаются крупнощепящими рестриктазами.
Рибонуклеиновая кислота. См. РНК.
Рибонуклеотид. См. нуклеотид.
Рибосома. Небольшой органоид клетки, место синтеза белка. Рибо-
сома состоит из молекул белка и специализированной рибосомной РНК.
Рибосомная РНК (рРНК). РНК, входящая в состав рибосом.
РНК (рибонуклеиновая кислота). Химическое вещество, содержаще-
еся в ядре и цитоплазме клетки. РНК играет важную роль в синтезе белка
и других клеточных процесса!. По структуре молекула РНК сходна с ДНК.
201
Приложение
В зависимости от функции, выделяют несколько классов РНК; матричная
РНК, транспортная РНК, рибосомальная РНК.
Сантиморганида (Морганида). Единица измерения генетического
расстояния на основе частоты рекомбинации. Названа в честь американ-
ского генетика Томаса Ханта Моргана. Одна сантиморганида соответствует
1% вероятности того, что два генетических локуса или маркера будут раз-
делены в процессе кроссинговера за одно поколение. В геноме человека
1 сантиморганида приблизительно соответствует 1 миллиону пар основа-
ний.
Секвенирование. Определение порядка расположения оснований (нук-
леотидов) в молекуле ДНК или РНК или порядка аминокислот в белке.
Семейство генов. Группа родственных генов, которые кодируют про-
дукты с близкой функцией.
сМ. См. сантиморганида.
Соматическая клетка. Любая клетка тела, кроме половых клеток (га-
мет) и их предшественников.
Сцепление генов. Близкое взаимное расположение генов или других
генетических маркеров (например, ПДРФ) на одной хромосоме, приводя-
щее к тому, что гены, как правило, наследуются совместно. Чем ближе
гены или маркеры друг к другу, тем ниже вероятность того, что они будут
разделены в процессе репарации или репликации ДНК (деление надвое у
прокариот и митоз и мейоз у эукариот), и, следовательно, тем выше веро-
ятность совместного наследования.
Тандемные повторы. Множественные копии одной исходной после-
довательности ДНК, расположенные подряд. Один из типов генетических
маркеров, применяемых в картировании генома.
Теломера. Концевой участок хромосомы. Это специализированная
структура, обеспечивающая стабильность линейных молекул ДНК и участ-
вующая в репликации.
Технология рекомбинантной ДНК. Спектр молекулярно-генетических
методов для манипуляций с фрагментами ДНК («сшивания» и разделения
фрагментов ДНК различного происхождения) в бесклеточной системе (вне
клетки или организма). При определенных условиях рекомбинантную мо-
лекулу ДНК можно ввести в клетку, где она будет автономно реплициро-
ваться или интегрируется в геном клетки-хозяина.
Тимин (Т). Азотистое основание, один из четырех мономеров ДНК.
Комплиментарен аденину.
Транскрипция. Синтез РНК-копии на молекуле ДНК (гене). Первая ста-
дия экспрессии гена. См. также трансляция.
Трансляция. Процесс синтеза белка на рибосоме, в ходе которого
аминокислоты включаются в молекулу белка в последовательности, зако-
дированной в молекуле мРНК.
Транспортная РНК (тРНК). Класс молекул РНК, имеющих специаль-
ные участки, содержащие триплет нуклеотидов, комплиментарный трип-
лету кодирующей последовательности мРНК. Молекулы тРНК участвуют в
Синтезе белка. Их функция - связывание с аминокислотой и перенос ее на
рибосому, где триплет тРНК распознается триплетом мРНК и данная ами-
нокислота встраивается в синтезируемый белок.
Трансформация. Процесс изменения генетического материала клет-
ки путем введения в нее чужеродной ДНК.
тРНК. См. транспортная РНК.
Урацил. Азотистое основание, входящее в состав РНК, но не входящее
в ДНК. Формирует комплиментарную пару с аденином.
Уровень разрешения. Степень детализации физической или генети-
ческой карты генома.
Словарь терминов
Фаг. Вирус, хозяином которого является бактериальная клетка.
Фермент. Белок, действующий как катализатор, ускоряя процесс но
не изменяя направление и природу реакции.
Физическое картирование. Процесс построения карты, показываю-
щей локализацию идентифицируемых участков ДНК (генов, рестрикцион-
ных сайтов), независимо от их наследования. Расстояния на физической
карте измеряются в парах оснований. Физическая карта с самой низкой
разрешающей способностью - картина хромосомных бэндоз, образующа-
яся при дифференциальном окрашивании хромосом. Максимально воз-
можный уровень разрешения физической карты - полная последователь-
ность нуклеотидов.
Хромосома. Компонента клетки, несущая гены (т.е. линейную после-
довательность нуклеотидов ДНК) и способная к саморепликации. Состоит
из молекулы ДНК и связанных с ней белков. Прокариоты имеют одну коль-
цевую хромосому. Геном эукариот состоит из нескольких (многих) хромо-
сом.
Центромера. Особый участок хромосомы, к которому присоединяют-
ся нити веретена при делении клетки.
Цмтозин. Азотистое основание, один из четырех мономеров ДНК. Об-
разует комплиментарную пару с гуанином.
Чужеродная (экзогенная) ДНК. ДНК иного, чем данный организм, про-
исхожде ния.
Экзон. Кодирующий белок участок гена. См также интрон.
Экзонуклеаза. Фермент, расщепляющий молекулу ДНК на ее конце.
Экспрессия гена. Процесс, при котором кодируемая геном информа-
ция преобразуется в существующие и действующие в клетке структуры. К
экспрессируемым генам относятся гены, транскрибируемые в молекулы
мРНК, которые затем транслируются в молекулу белка, а также гены, ко-
торые кодируют РНК, но не транслируются в белок (гены транспортной и
рибосомной ДНК).
Электрофорез. Метод разделения больших молекул (ДНК или бел-
ков). При электрофорезе электрический ток пропускают через среду, со-
держащую смесь молекул, которые нужно разделить. Каждая из молекул
при этом перемещается в среде с различной скоростью, зависящей от
электрического заряда и размера молекулы, что и приводит к разделению
молекул. В качестве сред чаще всего используют агарозные или акрила-
мидные гели.
Эндонуклеаза. Фермент, который расщепляет молекулу нуклеиновой
кислоты во внутренней части.
Эукариота. Клетка или организм с ограниченным мембраной ядром и
другими клеточными компонентами. К эукариотам относятся все организ-
мы, исключая вирусы, бактерии и сине-зеленые водоросли. См. также:
прокариота.
Ядро. Органоид эукариотической клетки, содержащий генетический
материал.
Е. coli. Бактерия кишечной палочки. Один из основных модельных объ-
ектов в генетике.
FISH (флуоресцентная гибридизация in situ). Метод физического
картирования, который использует флуоресцентные метки для выявления
гибридизации зонда с метафазной хромосомой либо с менее конденси-
рованным соматическим интерфазным хроматином.
In vitro. Вне живого организма.
In vivo. На живом организме.
YAC. См. искусственная дрожжевая хромосома.
1
203
СПИСОК ЛИТЕРАТУРЫ
1. Анализ генома. Методы. Под ред. К.Дейвиса. Пер. с англ. М.: Мир,
1990. - 247 с.
2. Баранов В.С. Самая современная терапия - генная. Природа; 1996; 8;
25-33.
3. Баранов В.С., Асеев М.В., Ракишева З.Б. и др. Молекулярная диагнос-
тика синдрома фрагильной Х-хромосомы (синдром Мартина-Белла) у
больных отечественных популяций. Генетика; 1993; 29; 6; 1026-1034.
4. Бочков Н.П. Генетика человека (наследственность и патология). М.;
Медицина; 1978. - 377 с.
5. Бочков Н.П. Генетические технологии в педиатрии. Педиатрия; 1995;
4; 21-26.
6. Бочков Н.П. Медицинские и социальные аспекты генетики человека.
Природа; 1983; 5; 26-32.
V 7. Бочков Н.П., Захаров А.Ф., Иванов В.И. Медицинская генетика; М.:
Медицина, 1984. - 368 с.
8. Вартанян М.Е. Руководство по психиатрии. Под ред. А.В.Снежневско-
го. - М., 1983; 1; 127-129.
9. Гайцхоки В.С. Проблемы молекулярной генетики наследственных
болезней. Вестник РАМН, 1996; 9; 8-14.
10. Гиндилис В.М. Генетика шизофренических психозов: Дйс. ... д-ра
биол. наук. - М., 1979.
11. Голубовский М.Д., Чураев Р.Н. Динамическая наследственность и
эпигены. Природа, 1997; 4; 16-25.
12. Гольдштейн Дж. Л., Браун М.С. Генетические аспекты болезни,- В
кн. Внутренние болезни. М.: Медицина.- пер. с англ., 1993, 133-159
13. Дзизинский А.А., Пузырев В.П. Наследственность и атеросклероз.
Новосибирск: Наука, Сиб. отд-е, 1997. - 178 с.
14. Евграфов О.В. ДНК-диагностика наследственных заболеваний. В кн.:
Итоги науки и техники. Серия: Генетика человека. М., 1991; 9; 53-126.
15. Залетаев Д.В. Хромосомная патология у детей с олигофренией и
множественными признаками дизморфогенеза. Автореф. дисс...
канд. биол. наук. М., 1985. 24 с.
16. Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова ЕД. Новый
механизм мутации у человека: экспансия тринуклеотиднЫх повторов. -
Генетика, 1995; 31; 11; 1478-1483.
17. Казначеев В.П., Спирин Е.А. Космопланетарный феномен человека. -
Новосибирск: Наука. Сиб.отд-ние, 1991. - 304 с.
18. Калинин В.Н., Шмидт В., Поллер В. и др. Новая точечная мутация в
митохондриальном гене ND1, обнаруженная у больного диабетом
типа II. - Генетика, 1995; 31; 8; 1180-1182.
19. Ключевский В. О. Неопубликованные произведения. М: Наука, 1983;
292-294
20. Кулешов Н.П. Частота возникновения и судьба хромосомных анома-
лий в популяции человека. Автореф. дисс... докт. мед. наук. М., 1979;
45 с.
21. Ландер Э. Картирование сложно наследуемых признаков человека. В
кн: Дейвис К (ред.). Анализ генома. Методы. М: Мир, 1990; 214-239
22. Маккьюсик В. Вклад клинической генетики в генетику человека. В
кн.: Вопросы общей генетики. Пер. с англ. М.: Наука, 1981; 133-138
Список литературы
23. Маниатис Г, Фрич Э., Сэмбрук Дж. Методы генетической инжене-
рий. Молекулярное клонирование. -М.: Мир, 1984.
24. Моисеев Н.Н. Алгоритм развития. - М.: Наука, 1987. - 304 с.
25. Назаренко С.А. Изменчивость хромосом и развитие человека. Томск.
1993. 200 с.
26. Назаренко С А., Назаренко Л.П., Пузырев В.П. Наследственные
болезни человека с экспансией числа тринуклвотидных повторов -
Бюлл. ТНЦ СО РАМН, вып. 6, Томск, 1995; 109-123.
27. Назаренко С.А., Никитина Т.В. Униродительская дисомия в исследо-
вании проблемы геномного и импринтинга у человека. В сб.: Генети-
ка человека и патология. - Томск, 1997; 4; с. 36-48.
28. Назаренко С.А., Островская М.Г., Васильева Е.О. и др. Цитогенети-
ческая диагностика наследственной патологии в медико-генетичес-
ком консультировании. Бюлл. ТНЦ АМН СССР. Вып.1. Томск, 1989; с.
84-91.
29. Николаева Е.Н., Темин П.А., Никонорова М.Ю. Синдром Кернса-
Сейра: генетические аспекты, клинические проявлений, возможности
лёчения. - Российский вестник перинат. и пед., 1997; 2; 24-29.
30. Островерхова Н.В. Молекулярно-цитогенетическое исследование
структурных хромосомных перестроек в практике медйко-генетичес-
кого консультирования. Автореф. дисс...канд. биол. наук. М., 1997; 24
с.
31. Пригожин И.Р., Стенгере И. Порядок из хаоса: Пер.с англ. - М.:
Прогресс, 1986. - 431 с.
32. Пузырев В.П. Геномный импринтинг и болезни человека. Бюллетень
Томского научного центра СО РАМН. - Томск, 1995; с. 72-87.
33. Пузырев В.П., Степанов В.А. Болезни человека на картах хромосом.
- В сб.: Генетика человека и патология, вып. 4- Томск: Изд-во
Томского ун-та, 1997; 10-35.
34. Ракишева З.Б. Диагностические основы для медико-генетической
профилактики синдрома Мартина-Белла [С. FRA(Xq27)]: Дис. ... канд.
Мед. наук. Москва: Медико-генетический научный центр РАМН, 1993
35. Родин С.Н. Идея коэволюции. Новосибирск: Наука, Сиб.отд-ие,
1991,- 271 с.
36. Саркисов Д.С. Очерки истории общей патологии. М.: Медицина,
1993; 512 с.
37. Саркисов Д.С., Пальцев М.А., Хитрое Н.К. Общая патология челове-
ка. М.: Медицина, 1995; 275 с.
38. Свердлов Е.Д. Очерки современной молекулярной генетики по курсу
лекций для студентов биологического факультета МГУ. Молекулярная
генетика, микробиология и вирусология. 1997; 2; 3-28.
39. Смольянников А.В. Нозология. БМЭ,- М.: Советская энциклопедия,
1981; 17; 159-160.
40. Степанов В.А., Лемза С.В., Пузырев В.П. Генетика нарушений
Метаболизма липопротеинов. Томск: ТГУ. 1992; 141 с.
41. Степанов В.А., Пузырев В.П., Лемза С.В. и др. Взаимосвязь струк-
турных вариантов гена аполипопротеина с ишемической болезнью
сердца и уровнем липидов плазмы крови. Генетика, 1995; 31; 3; 405-
409.
42. Темин П.А., Никонорова М.Ю., Сухорукое В С. Миоклонус эпилепсия
с разорванными красными мышечными волокнами: клинический
полиморфизм, вопросы диагностики и лечения. - Российский вестник
перинат. и пед., 1996; 4; 35-41.
43. Тимофеев-Ресовский Н.В.^Генетика, эволюция и теоретическая
биология. Природа, 1980; Т; 62-65.
205
Список литературы
44. Тимофеев-Ресовский Н.В. Связь между геном и внешним признаком
(феноменология проявления генов). - Избранные труды под ред.
О.Г.Газенко и В.И. Иванова, 1996, 59-84.
45. Трубников В.И., Голимбет В.Е. Современные представления об
антиципации при эндогенных психозах. Вестник РАМН. 1996. N4.
С.11-13.
46. Фаворова О.О. Лечение генами - фантастика или реальность?
Соросовский образовательный журнал. 1997. 2: 21-27.
47. Фогель Ф., Мотульски А. Генетика человека. Пер. с англ. М: Мир,
1990; в 3 т.
48. A genome-wide search for asthma susceptibility loci in ethnically popula-
tions. The Collaborative Study on the CSGA. Nat. Genet., 1997; 15; 389-
392.
49. Alcolado J.C., Brockington M., Sweeney M.G. et al. Search for large
mitochondrial deletion in Type 2 diabetes. Diabet. Med., 1993a; 10;
Suppl.; 1; 42.
50. Alcolado J.C., Majid A., Morgan R., Rus A, Search for the 8344 bp
mitochondrial DNA mutation in subjects with NIDDM. Diabet. Med., 1993b;
10; Suppl.; 1; 28.
51. Allen K.M., Walsh Ch. Shaking down new epilepsy genes. Nature
Medicine, 1996; 2; 516-518.
52. Antonarakis S.E. Mapping by sequence homology. Eur. J. Hum. Genet.
1996; 4; 247-249.
53. Anvret V., Ahlberg G., Grandell U. et al. Larger expansions of the CTG
repeat in muscle compared to lymphocytes from patients with myotonic
dystrophy. Hum. Mol. Gen., 1993; 2; 1397-1400.
54. Askari F, McDonnell W.M. Antisense-oligonucleotide therapy. New Eng.
J. Med. 1996. 334: 316-318.
55. Bahary N, Leibel RL, Joseph L, et al. Molecular mapping of the mouse db
mutation. Proc Natl Acad Sci USA, 1990; 87; 8642-8646.
56. Barcelo J.M., Mahadevan M.S., Tsilfidis C. et al. Intergenerational
stability of the myotonic dystrophy protomutation. Hum. Mol. Genet.,
1993; 2; 705-709.
57. Barnes K.C., Neely J.D., Duffy D.L., et al. Linkage of asthma and total
serum IgE concentration to markers on chromosome 12q; evidence from
Afro-Caribben and Caucasian populations. Genomics, 1996; 37; 41-50.
58. Bassett A.S., Honer l/V.G. Evidence for anticipation in schizophrenia. Am.
J. Hum. Genet., 1994; 54; 864-870.
59. Bassett D.E., Boguski M.S., Spenser F. et al. Genome cross-referencing
and XREFdb: Implifications for the identification and analysis of genes
mutated in human disease. Nat. Genet., 15; 1997; 339-344.
60. Bell M.V., Hirst M.C., Nakahori Y. et al. Physical mapping across the
fragile X: hypermethylation and the fragile X syndrome. Cell, 1991; 64;
861-866.
61. Benediktsson R., Lindsay R.S., Noble J. et al. Glucocorticoid exposure in
utero. new model for adult hypertension. Lancet, 1993; 341; 339-341
62. Berg L.P., Miller D.S., Grundi C.B. et al. Applicacion of PCR to the
Detection and Analisis of point Mutacions in the Human Factor VIII Gene.
Human Moleculer Genetics, 1992; 4; 5; 363-367.
63. Berg P. Dissections and reconstructions of genes and chromosomes
(Nobel lecture). Science, 1981, 213; 296-303.
64. Berretini W.H., Ferrato T.N., Alexander R.C. et al. Quantitative trait loci
mapping of three loci controlling morphine preference using inbred mouse
strains. Nature Genet., 1994; 7; 54-58.
Список литературы
65. Berrettini WH, Ferraro TN, Goldin LR et al. Chromosome 18 DNA markers
and manic-depressive illness evidence for a susceptibility gene. Proc
Natl Acad Sci USA., 1994; 91; 5918-5921.
66. Billings P.R. New technologies for physical mapping of the human
genome. FASEB Journal. 1991; 5; 29.
'67 Bishop DT, Williamson JA. The power of identity-by-descent methods for
linkage analysis. Am J Hum Genet., 1990; 46; 254-265.
68 Blease M. at al. T lymphocyte-directed gene therapy for ADA-SCID: Initial
trial results after 4 years. Science, 1995; 270; 475-480.
69. Blennow E., Nielsen K.B., Telenius H., Carter N.P., et al. Fifty probands
with extra structurally abnormal chromosomes characterized by fluores-
cence in situ hybridization. Am.J.Med.Genet., 1995; 55; 85-94.
70. Blennow E., Telenius H., Larsson C. et al. Complete characterization of a
large marker chromosome by reverse and forward chromosome painting.
Hum.Genet., 1992; 90; 371-374.
71. Blomer U., Naldini L., Verma I.M. et al. Application of gene therapy to the
CNS. Hum. Mol. Genet., 1996; 5; 1397-1404.
72. Boerwinkle E., Ellsworth D.L., Hallman M. et al. Genetic analysis of
atherosclerosis: a research paradigm for the common chronic disease.
Hum. Genet., 1996; 5; 1405-1410.
73. Bohm H., Gross B., Gaestel M. et a!. The S-prime-untranslated region of
p23mRNA from the Ehrlich ascites tumor is involved in translation control
of the growth related protein p23. Biomed. Biochim. Acta, 1991; 12;
1193-1203.
74. Bonnardeaux A., Nadaud S., Charru A. et al. Lack of evidence for linkage
of the endothelial cell nitric oxide synthase gene to essential hypertension.
Circulation, 1995; 91; 96-102.
75. Botstein D., White D.L., Skolnick M. et al. Construction of a genetic
linkage map in man using restriction fragment length polymorphisms. Am
J Hum Genet., 1980; 32; 314-331.
76. Bottazzo G.F., Lendrum R. Separate autoantibodies to human pancreatic
glucagon and somatostatin cells. Lancet II., 1976; 873-876.
77. Breslow J.L. Apolipoprotein genetic variation and human disease. Physiol
Rev 1988; 68; 85-132.
78. Breslow J.L. Transgenic mouse models of lipoprotein metabolism and
atherosclerosis. Proc Natl Acad Sci USA, 1993; 90;. 8314-8318.
79. Brown DL, Gorin MB, Weeks DE. Efficient strategies for genomic sear-
ching using the the affected-pedigree-member method of linkage analy-
sis. Am J Hum Genet. 1994; 54; 544-552.
80. Brown K.W., Villar A. J., Bickmore 1/V. et al. Imprinting mutation in the
Beckwith-Wiedemann syndrome leads to biallelic IGF2 expression through
an H19-independent pathway. Hum. Mol. Genet.,1996; 5;12; 2027-2032.
81. Brown M.D., Voljaveo A.S., Lott M.T., etal. Mitochondrial DNA complex I
and II mutations associated with Leber’s hereditary optic neuropathy
Genetics, 1992; 130, 1, 163-173.
82. Brunner H.G., Jansen G., Nillesen W. et al. Reverse mutation in myotonic
dystrophy. N. Engl. J. Med, 1993; .328;.476-479.
83. Bryndorf T., Kirchhoff M., Rose H., etal. Comparative genomic hybridiza-
tion in clinical cytogenetics. Am. J. Hum. Genet., 1995; 57; 1211-1220.
84. Buxton J., Shelbourne P., Daview J. et at. Detection of an unstable
fragment of DNA specific to individuals with myotonic dystrophy. Nature,
1992; 355; 547-548.
85. Cambell D.A. et. Eur. J. Resp. Dis., 1995; 8; 552. - Пит. no Holgate S T.
1
207
Список литературы
86. Cambien F, Poirier О, Lecerf L et al. Deletion polymorphism in the gene
for angiotensin-converting enzyme is a potent risk factor for myocardial
infarction. Nature; 359; 641-644.
87. Campuzano V., Montermini L., Molto M.D. et al Fridreich’s ataxia:
autosomal recessive disease caused by an intronic GAA triplet repeat
expansion. Science. 1996; 271,1423-1427.
88. Cannizzaro L.A. Chromosomal microdissection: short rewier. Cytogenet
Cell Genet., 1996; 74;157-160.
89. Cannizzaro LA., Aronson M.M., Emanuel B.S. In situ hybridization and
translocation breakpoint mapping. Cytogenet. Cell Genet., 1985; 39;* 173-
178.
90. Capitalizing on the genome (Editorial article). Nat. Genet., 1996; 13; 1-5.
91. Carter C.O. The inheritance of congenital pyloric stenosis. Br. Med. Bull,
1961; 17; 251-254.
92. Cattanach B.M., Kirk M. Differential activity of maternally and paternally
derived chromosome regions in mice. Nature, 1985;. 315; 496-498.
93. Cheung S.W., Sun L., Featherstone T. Molecular cytogenetics evidence
to characterize breakpoint regions in Robertsonian translocation. Cytoge-
net.Cell Genet., 1990; 54; 97-102.
94. Collins F.S. Positional cloning moves from perditional to traditional.
Nature Genet, 95; 9; 347-350.
95. Cookson W.O.C.M., Sharp P.A., Faux J.A.et al. Linkage between immu-
noglobulin E responses underlying asthma and rhinitis and chromosome
11q. Lancet I, 1989; 1292-1295.
96. Cookson W.O.C.M., Young R.P., Sandford A.J.et al. Maternal inheritance
of atopic IgE responsiveness on chromosome 11q. Lancet, 1992; 340;
381-384.
97. Copeland N.G. et al. A genetic linkage map of the mouse. Science, 1993.
V. 262. P. 57-66.
98. Corder E.H., Saunders AM., Strittmatter W.J et al. Gene dose of
apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late
onset families. Science. 1993; 261; 921-923.
99. Cormier V., Tardieu M., Saudubray J. et al. Autosomal dominant deletions
of the mitochondrial genome in case of progressive encefalomyopathy.
Amer. J. Hum. Genet., Suppl., 1990; 47; 3;. A212.
100. Corral-Debrinski M., Shoffner J.M., Lott M.T. et al. Association of
mitochondrial DNA damage with aging and coronary atherosclerotic ytart
disease. Mutat. Res. DNAging: Gen. Instab, and Aging, 1992; 257; 3-6,
169-180.
101. Cortopassi G.A., Arnheim N. Detection of a specific mitochondrial DNA
deletion in tissue of older humans. Nucleic Acids Res., 1990; 18; 6927-
6933.
102. Cortopassi G.A., Shibata D., Soong N.-W.et al. A pattern of accumulati-
on of a somatic deletion of mitochondrial DNA in aging human tissues.
Proc. Nat. Acad. Sci. USA, 1992; 89; 16; 7370-7374.
103. Crouse H.V. The controlling element in sex chromosome behaviour in
Sciara. Genetics, 1960; 45; 1429-1443.
104. Culver K. Gene Therapy, a handbook for physicians. New York: Mary
Ann Liebert, Inc. 1994.
105. Culver К.I/., Blease M. Gene therapy for cancer. TIG, 1994; 10; 174-
178.
106. Cuoitta E., Koshiand D.E. NO news is good news. Science, 1992; 258;
1862-1992
,107. Curtis D, Gurling H. A procedure for combining two-point lod-scores
into a summary multipoint map. Hum Hered, 1993; 43; 173-185.
208
Список литературы
108. D’Alfonso S.D., Richiardi P.M. A polymorphic variation in a putative
regulation box of the TNFA promoter region. Immunogenetics; 1994; 39;
150-154.
109. Davies JL, Kawaguchi Y., Bennett ST et al. A genome-wide scan for
human type 1 diabetes susceptibility genes. Nature, 1994; 371; 130-136
110. Davies K. Trilpet repeats on the rise. Nature; 1993;.364; 88.
111. Davys D., Biancalana V., Rousseau F. et al. Analysis of full fragile X
mutations in fetal tissues and monozygotic twins indicate that abnormal
methylation and somatic heterogeneity are established in early develop-
ment Am. J. Med. Genet., 1992; 43; 208-216.
112. de Gouyon B, Melanitou E, Richard MF et al. Genetic analysis of
diabetes and insulitis in an interspecific cross of the nonobese diabetic
mouse with Mus spretus. Proc Natl Acad Sci USA, 1993; 90; 1877-1881.
113. de Sanctis GT, Mercant M, Beier DR et al. Quantitative locus analysis of
airway hyperresponsiveness in A/J and C57BL/6J mice. Nature Genet,
1995; 11; 150-154.
114. Dennis N., Curts G., Macpherson L. et al. Two families with Xq27.3
fragility, no detectable insert in the FMR-1 gene, mild mental impairment
and absence of the Martin-Bell phenotype. Am. J. Med. Genet, 1992; 43;
232-236.
115. Derbrinski M., Lott M.T., Shoffner J.M.et al. Accumulation of Mitochon-
drial DNA Damage in Chronic Heart an Brain Disease. Amer. J. Hum.
Genet. Suppl., 1991; 49; 4; 132.
116. Diamond T.M., Mueller O.T., Sutcliffe M. et al. Uniparental disomy for
chromosome 14. Evidence of imprinting effect. Am. J. Hum. Genet, 1993;
53; A541.
117. Dietrich WF et al. A genetic map of the mouse with 4.006 simple
sequence length polymorphisms. Nature Genet., 1994; 7; 220-245.
118. Dietrich WF et al. Genetic identification of Mom-1, a major modifier
locus affecting Min-induced intestinal neoplasia in the mouse. Cell, 1993;
75; 631-639.
119. Dietz HC, Cutting GR, Pyeritz RE et al. Marfan syndrome caused by a
recurrent de novo missense mutation in the fibrillin gene. Nature, 1991;
352; 337-339.
120. Dittrich B., Suiting K., Korn B. et al. Imprint switching on human
chromosome 15 may involve alternative transcripts of the SNRPN gene.
Nat. Genet.,1996; 14; 2; 163-170.
121. Dizier MH, Babron MC, Clerget-Drapoux F. Interactive effect of two
candidate genes in a disease: extension of the marker-association-
segregation chi-square method. Am J Hum Genet., 1994; 55; 1045-1049.
122. Donis-Kelier H. et al. A genetic linkage map of the human genome. Cell,
1987; 51; 319-337.
123. Dvorakova D., Fajkusova L. Direct analysis of R408W mutation in
phenylalanine hydroxylase gene by allele-specific PCR amplification.
Human Molecular Genetics, 1993; 2; 3 323.
124. Edery P, Lyonnet S, Mulligan LM et al. Mutations of the RET proto-
oncogene in Hirschsprung’s disease. Nature, 1994; 367; 378-380.
125. Edwards J.H. Exclusion mapping. J Med Genet., 1987; 24; 539-543.
126. Eichler E.E., Holden J.J.A., Popovic B.W. et al. Length of uninterrupted
CGG repeats determines instability in the FMR1 gene. Nat. Genet, 1994;
8;.88-94.
127. Engel E. A new genetic concept: Uniparental disomy and its potential
effect, isodisomy. Am. J. Med. Genet, 1980; 6; 137-143.
128. Engel E., DeLozier-Blanchet (J.D. Umparenal disomy, isodisomy, and
imprinting: probable effects in man and strategies for their detection. Am.
Список литературы
J. Med. Genet, 1991; 40; 432-439.
129. Epstein Ch.J. Seven momentous years. Am. J. Human Genet., 1993; 53;
1163-1166.
130. Epstein Ch.J. Toward the 21-st Century. Am. J. Human Genet., 1997;
60; 1-9.
131. Erster S.H., Brown W.T., Goonewardena P. et al. Polymerase chain
reaction analysis of fragile X mutations. Hum. Genet., 1992; 90;.55-61.
132. Estivill X. Complexity in a monogenic disease. Nature Genetics, 1996;
12; 4; 348-350.
133. Falk CT, Rubinstein P. Haplotype relative risk: an easy reliable way to .
construct a proper control sample for risk calculations. Ann Hum Genet,
1987; 51; 227-233. )
134. Fang Y.-Y., Eyre H.J., Bohlander S.K.et al. Mechanisms of small ring
formation suggested by the molecular characterization of two small
accessory ring chromosomes derived from chromosome 4. Am. J. Hum.
Genet., 1995; 57; 1137-1142.
135. Fields C., Adams M.D., White O.et al. How many genes in the human
genome? Nature Genetics, 1994, 7; 3; 345-346.
136. Fields S. The future is function. Nat. Genet., 1997; 151; 325-327.
137. Filla A., De Michele G., Cavalcanti F. et al. The relationship between
trinucleotide (GAA) repeat length and clinical features in Fridreich ataxia.
Am. J. Hum. Genet., 1996; 59; 554-560.
138. Flynn G.A., Hirst M.C., Knight S.J.L. et al. Identification of the FRAXE
fragile site in two families ascertained for X linked mental retardation. J.
Med. Genet. 1993; 30; 97-103.
139. Friedmann T. Human gene therapy - an immature genie, but certainly
out of the bottle. Nature Med., 1996; 2; 144-147.
140. Friend S.H., Bernards R, Rogelj S et al. A human DNA segment with
properties of the gene that predisposes to retinoblastoma and osteosar-
coma. Nature, 1986; 323; 643-646.
141. Fryns J.-P. X-linked mental retardation and the fragile X syndrome: a
clinical approach. The fragile X syndrome. Ed. Davies K.E. Oxford: Oxford
University Press, 1989; 1-39.
142. Fu Y.H., Pizzuti A., Fenwick R.J. et al. An unstable triplet repeat in a
gene related to myotonic dystrophy. Science, 1992; 255; 1256-1258.
143. Fu Y.-H., Kuhl D.P.A., Pizzuti A. et al. Variation of the CGG repeat at the
fragile X site results in genetic instability: resolution of the Sherman
paradox. Cell, 1991; 67; 1047-1058.
144. Galli J, Li LS, Glaser A. et al. Genetic analysis of non-insulin dependent
diabetes mellitus in the GK rat. Nature Genet., 1996., 12; 31-37.
145. Gauguier D, Froguel P, Parent V et al. Chromosomal mapping of genetic
loci associated with non-insulin dependent diabetes in the GK rat. Nature
Genet., 1996; 12; 38-43.
146. Giordano M., De Angelis M.S., Mutani R. et al. Origin of a regressed
myotonic dystrophy allele. J. Med. Genet., 1994; 31;. 130-132.
147. Goldgar D.E., Green P, Parry D.M.et al. Multipoint linkage analysis in
neurofibromatosis type I: an international collaboration. Am J Hum Genet.
1989; 44; 6-12.
148. Goltsov A.A., Eisensmith R.C., Konecki D.S. et al. Associations between
mutations and a VNTR in the human phenylalanine hydroxylase gene. Am.
J. Hum. Genet., 1992; 51; 627-636.
149. Goltsov A.A., Eisensmith R.C., Naughton E.R. et al. A single polymorphic
STR system in the human phenylalanine hydroxylase gene permits rapid
prenatal diagnosis and carrier screening for phenylketonuria. Human
Molecular Genetics, 1993; 2; 5; 577-581.
210
Список литературы
150. Goodwin J.F. Congestive and hypertrophic cardiomypathies Lancet,
1970; v 1; 731-739.
151. Goto Y., Nonaka I., Horai S. A new mtDNA mutation associated with
mitochondrial myopathy, encefalopathy, lactic acidosis and strikelike
episodes (MELAS). Annu.Rept. Nat. nst. Genet., Jap., 1991; 42;.95-96.
152. Goyette P., Sumner J.S., Milos R., et al. Human Methylenetetrahydrofo-
late reductase: isolation of cDNA, mapping and mutation identification.
Nat.Genet., 1994; 7; 195-200.
153. Grossman L.l. Mitochondrial DNA in sickness and in helth. Amer. J.
Hum. Genet., 1990; 46; 3; 415-9.
154. Grossmann M., Raper S., Kozarsky K. et al. Succesfull ex vivo gene
therapy directed to liver in a patient with familial hypercholesterolaemia.
Nature Genet., 1994; 6; 335-341.
155. Gusella J., Wexler N.S., Conneally P.M et al. A polymorphyc DNA
marker genetically linked to Huntington's disease. Nature, 1983; 306;
234-238.
156. Hall J. The challenges and opportunities of times of change. Am.J.
Hum. Genet., 1996; 58; 649-656.
157. Hall J.G. Genomic imprinting: rewiew and relevance to human diseases.
Am. J. Hum. Genet, 1990; 46; 857-873.
158. Hall J.M., Lee M.K., Newman B. et al. Linkage of early-onset familial
breast cancer to chromosome 17q2l. Science, 1990; 250; 1684-1689.
159. Hamann A., Mantrozoros C., Vidal-Puig A. et al. Genetic variability in the
TNF-a promoter region associated with susceptibility to cerebral malaria.
Nature, 1994; 371; 508-511.
160. Hamer D.H., Hu S., Magnuson V.L. et at. A linkage between DNA
markers on the X chromosome and male sexual orientation. Science,
1993; 261; 321-327.
161. Hansson J.H., Nelson-Williams C., Suzuki H. et al. Hypertension caused
by a truncated epithelial sodium channel gamma subunit: genetic hetero-
genety of Liddle syndrome. Nat. Genet., 1995; 11; 76-82.
162. Harding A.E., Hammans S.R. Deletions of the mitochondrial genome. J.
Inherit. Metab. Disease, 1992; 15; 4; 480-486.
163. Harley H.G., Brook J.D., Rundle S.A. et al. Expansion of an unstable
DNA region and phenotypic variation in myotonic dystrophy. Nature, 1992;
355; 545-546. ..
164. Hasstedt S.J., Wu L.L., Ash K. et al. The inheretance intraerytrocytic
sodium level. Am.J.Med., 1988; 29; 193-203.
165. Hasstedt SJ, Clegg DO, Ingles L. et al. HLA-linked rheumatoid arthritis.
Am J Hum Genet., 1994; 55; 738-746.
166. Hastbacka J, de la Chapelle A, Mahtani MM. The diastrophic dysplasia
gene encodes a novel sulfate transporter : positional cloning by fine-
structure linkage disequilibrium mapping. Cell, 1994; 78; 1073-1087.
167. Hayakawa M., Hatton K., Sugiyama S. et al. Age-associated oxygen
damage and mutations in mitochondrial DNA in human hearth. Biochem.
Biophys. Res. Commun., 1992; 189; 2;. 979-988.
168. Heitz D., Rousseau F., Devys D. et al. Isolation of sequences that span
the fragile X and identification of a fragile X-related CpG island. Science,
1991; 251; 1236-1239.
169. Henry I., Bonaiti-Pellie C., Chenhensse V. et al. Uniparental paternal
disomy in a genetic, cancer predisposing syndrome. Nature, 1991;. 351;
665-667.
T
91 1
Список литературы
170. Herzog R., Lutz S., Blin N. et al. Complete nucleotide sequence of the
gene for human cofactor II and mapping to chromosomal band 22q11
Biochemistry, 1991; 30; 1350-1357
171. Hirst M.C., Barnicoat A., Flynn G. et al. The identification of a third
fragile site, FRAXF, in Xq27-q28 distal to both FRAXA and FRAXE. Hum.
Mol. Genet., 1993; 2; 197-200.
172. Hirst M.C., Knight S.J.L., Bell M.V. et al. The fragile X syndrome. Clin.
Sci., 1992; 83; 255-264.
173. Hirst M.C., Knight S.J.L., Christodoulou Z. et al. Origins of the fragile X
syndrome mutation. J. Med. Genet., 1993; 30; 647-650.
174. Hobbs H.H., Brown M.S., Goldstein J.L. Molecular genetics of the LDL
receptor gene in familial hypercholesterolemia. Hum. Mut., 1992; 1; 445-
466.
175. Hoddi A., Stepien G., Lestienne N. Feed back regulation of nuclesr gene
expression in mitochondrial DNA myopathies. Europ. Soc. Hum. Genet..:
24th Annu. Meet., May 27-31; 1992; Copenhagen; 1992; 85.
176. Holgate S.T. Asthma genetics: waiting to exhale. Nat. Genet., 1997; 15;
227-229.
177. Holt I.J., Harding A.E., Morgan-Hughes J.A. Deletions of muscle
mitochondrial DNA in patients with mitochondrial myopathies. Nature,
1988; 331;717-719
178. Holt I.J., Harding A.E., Petty R.K.H., Morgan-Hughes J.A. A new
mitochondrial disease associated with a mitochondrial DNA heteroplasmy.
Amer. J. Hum. Genet., 1990; .46; N 3; 428-433.
179. Horton W.A. Molecular Genetics of the human chondrodysplasias -
1995. Eur. J. Human Genet., 1995; 3; 357-373.
180. Housman D. Human DNA polymorphism. N. Eng. J. Med, 1995; 332;
318-320.
181. Hugot J.-P., Laurent-Puig P., Gower-Rousseau C et al. Mapping of a
susceptibility locus for Crohn's disease on chromosome 16. Nature, 1996;
379; 821-823.
182. Huntington’s Disease Colloborative Research Group. A novel gene
containing a trinucleotide repeat that is expanded and unstable on
Huntington’s Disease chromosomes, 1993; 72; 971-983.
183. Huoponen K, Vilkki J., Aula P. et al. A mutation in ND1 gene of mitoc-
hondrial DNA is associated with Leber hereditary optic neuroretinopathy.
Cytogenetic. And Cell Genet., 1991; 58; 3-4;.2121.
184. Ikebe S., Tanaka M., Ohno K. Increase of deleted mitochondrial DNA in
the striatum in Parkinson’s disease and senescence.Biochem. Biophys.
Res. Commun., 1990; 170; 3; 1044-1048.
185. Inazawa J., Azuma T, AriyamaT. et al. A simple G-banding technique
adaptable for fluorescent in situ hybridization (FISH) and physical ordering
of human renin (REN) and cathepsin E (CTSE) genes by multi-color FISH.
Acta Histochem.Cytochem., 1993; 26; 4; 319-324.
186. Isashiki Y., Nakagawa M. Clinical correlation of mitochondrial DNA
heteroplasmy and Leber’s hereditary optic neuropathy. Jap., J. Ophtal-
moL, 1991; 35; 3; 256-259.
187. Jacob C O. Tumor necrosis factor a in autoimmunity; pretty giel or old
witch? Immunol. Today, 1992; 19; 122-125.
188. Jacob HJ et al. Genetig mapping of gene causing hypertension in the
stroke-prone spontaneously hypertensive rat. Cell, 1991; 647; 213-224.
189. James R.S, Temple I.K, Dennis N.R. et al. A search for uniparental
disomy in carriers of supernumerary marker chromosomes. Eur. J. Hum.
Genet. - 1995; 3; 21-26.
1
Список литературы
190. Jeunemaitre X, Soubner F, Kotelevtsev YV et al. Molecular basis of
human hypertension: role of angiotensinogen. Cell, 1992; 71; 169-180.
191. Jeunemaitre X., Litton R.P., Hunt S.C. et al. Absence of linkage beetwe-
en the angiotensin converting enzyme locus and human essential hyper-
tension. Nature Genet., 1993; 1; 72-75.
192. Jodice C., Malaspina FL, Persichetti F. et al. Effect of trinucleotide
repeat length and parental sex on phenotypic variation in spinicerebellar
ataxia 1. Am. J. Hum. Genet., 1994; 54; 959-965.
193. Johns D.R., Smith K.N., Miller N.R. Leber's hereditary optic neuropathy
Clinical menifestations of the 3460 mutation. Arch. Ophthalmol, 1992;
110; 11; 1577-1581.
194. Jorde LB, Watkins WS, Carlson M et al. Linkage disequilibrium predicts
physical distance in the adenomatous polyposis coli. Am J Hum Genet.,
1994; 54; 884-898.
195. Julier C, Hyer RN, Davies J et al. Insulin-IGF2 region on chromosome
11p encodes a gene implicated in HLA-DR4-dependent diabetes suscepti-
bility. Nature, 1991; 354; 155-159.
196. Kainulainen K, Pulkkinen L, Savolainen A. et al. Location of the gene
defect causing Marfan syndrome on chromosome 15. N Engl J Med.
1990; 323; 935-939.
197. Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to
mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 1994;
264; 1604-1608.
198. Kalousek D.K, Barrett I. Genomic imprinting related to prenatal diagno-
sis. Prenatal Diagnosis, 1994;. 4; 1191-1201.
199. Kawaguchi Y., Okamoto T., Taniwaki M. et al. CAG expansions in a
novel gene for Machado-Joseph disease at chromosome I4q32.1. Nature
Genet., 1994; 8; 221-228.
200. Kimberling W. J., Kumar S., Gabow P.A. et al. Autosomal dominant
polycystic kidney disease: localization of the second gene to chromosome
4q13-q23. Genomics, 1993; 18; 467-472.
201. King S.C., Stapleton P.M., Walker A.P. et al. Two dinucleotide repeat
polymorphisms at the DMD locus. Journal Hum Mol Genet, 1994; 3; 523
202. Kishino T., Laland M., Wagstaff J. UBE3A/E6-AP mutations cause
Angelman syndrome. Nature Genet., 1997; 15; 70-73.
203. Knight S.J.L., Hirst M.C., Roche A. et at. Molecular studies of the fragile
X syndrome. Am. J. Med. Genet., 1992; 43; 217-223.
204. Knight S.J.L., Flannery A.V., Hirst M.C. et al. Trinucleotide repeat
amplification and hypermethylation of a CpG island in FRAXE mental
retardation. Cell, 1993; 74; 127-134.
205. Kobayashi Y., Tulinius M.N., Holme E. et al. Respiration-decell are
caused by a single point mutation in the mitochondrial tRNA-Leu (UUR)
gene in mitochondrial myopathy, encefalopathy, lactic acidosis and
strikelike episodes (MELAS). Amer. J. Hum. Genet., 1991; 49; 3; 590-599.
206. Koenig M., Hoffman E.P., Bertelson C.J. et al. Complete Cloning of the
Duchenne Muscular Dystrophy (DMD) cDNA and Preliminary Genomic
Organization of te DMD Gene in Normal and Affected Individuals. Cell,
1987; 50; 509-517.
207. Koide R., Ikeuchi T., Onodera O., et al. Unstable expansion of CAG
repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nature
Genet., 1994;.6; 9-13.
208. Kotzot D., Schmitt S., Bernasconi F. et al. Uniparental disomy 7 in
Silver-Rassel syndrome and primordial growth retardation. Hum. Mol.
Genet., 1995; 4; 583-587.
1
213
Список литературы
209. Kozich I/., Kraus E., Franchis P., et al. Hyperhomocysteinemia in
premature arterial disease: examination of cystethionine beta-synthasa
alleles at the molecular level. Hum. Molec. Genet., 1995; 4; 623-629.
210. Krahe P, Julkunen H, Ignatius J. et al. Genetic dissection of complex
disease in isolated populations: linkage disequilibrium mapping of two SLE
candidate susceptibility genes in the Finnish population (abstract). Am J
Hum Genet., 1996; 59; A384.
211. Krang S.S., Wang P.W.K., Bock H.-G.O. et al. Intermediate hypergo-
mocysteinemia resulting from compaund heterozygosity of methylenetet-
rahydrofolate reductase mutetion. Am. J. Hum. Genet., 1991; 48; 546-
551.
212. Kraus J.P., Williamson C.L., Figaira F.A., et al. Cloning and screening
with nanogram amounts of immunopurified mRNAs: cDNA cloning and
chromosomal mapping of cystationine beta synthase and the beta subunit
of propionyl-CoA ceroboxylase. Proc. Nat. Acad. Sci., 1986; 83; 2047-
2051.
213. Kremer E.J., Pritchard M., Lynch M. et al. Mapping of DNA instability at
the fragile X to a trinucleotide repeat sequence p(CCG)n. Science,
1991;.252; 1711-1714.
214. Kroon A.M., Van Den Bogert C. The mitochondrial genome as a target
for chemotherapy of cancer. Achievements and Perspectives of Mitochon-
drial research., II: Biogenesis, 1985; p. 21-33.
215. Krozowski Z., Baker E., Obeyesekere V. et al. Localization of the gene
for human 11-beta-hydroxysteroid dehydrogenase type 2 (HCD11B2) to
chromosome band 16q22. Cytogenet Cell Genet., 1995; 71; 124-125.
216. Kruglyak L, Lander ES. Complete multipoint sib-pair analysis of qualitati-
ve and quantitative traits. Am J Hum Genet., 1995; 57; 439-454.
217. Kruglyak L, Daly MJ, Lander ES. Rapid multipoint linkage analysis of
recessive traits in nuclear families, including homozygosity mapping. Am J
Hum Genet. 1995; 56; 519-527.
218. Kruglyak L., Lander ES. High-resolution genetic mapping of complex
traits. Am J Hum Genet., 1995; 56; 1212-1223.
219. Kutz T.W., Spence M.A. Genetics of essential hypertension.
Am.J.Med.,1993; 94; 77-84.
220. La Spada A.R., Poling D.B., Harding A.E. et al. Meiotic stability and
genotype-phenotype correlation of the trinucleotide repeat in X-linked
spinal and bulbar muscular atrophy. Nature Genet., 1992; 2; 301-304.
221. La Spada A.R., Wilson E.M., Lubahn D.B. et al. Androgen receptor gene
mutations in X-linked spinal and bulbar muscular atrophy. Nature, 1991;
352; 77-79.
222. Laird C.D. Proposed mechanism of inheritance and expression of the
human fragile X syndrome of mental retardation. Genetics, 1987; 117;
587-599.
223. Lander ES, SchOrk NJ. Genetic dissection of complex traits. Science,
1994; 265; 2037-2048.
224. Larsson N.-G., Tilinius M.N., Holme E., et al. Segregation and manifes-
tation of the mtDNA tRNA^ A®G8344 mutation of myoclonus epilepsy and
ragged-red fibers (MERRF) syndrome. Amer. J. Hum. Genet., 1992; 51; 6;
1201-1212.
f 225. Lathrop G.M., Lalouel J.M., Julier C. et al. Strategies for multilocus
linkage analysis in humans. Proc Natl Acad Sci USA, 1984; 81; 3443-
3446.
/ 226. Le Beau M.M. One FISH, blue FISH. Nat. Genet., 1996; 12; 341-344.
214
Список литературы
227. Ledbetter D.H, Engel E. Uniparental disomy in humans: development of
an imprinting map and its implications for prenatal diagnosis. Hum. Mol.
Genet., 1995; 4; 1757-1764.
228. Lejeune J., Turpin FL, Gautier M. Le mongolisme, premiere exemple
d’aberration autosomique humaine. Ann.genet., 1959, 1, 41-49.
229. Letrit P., Noer A.S., Jean-Fracois M.J.B. et al. A new diseaserated
mutat on for mitochondrial encefalopathy lactic acidosis and strikelike
episodes (MELAS) syndrome affects the ND 4 subunit of the respiratory
complex I. Amer. J. Hum. Genet., 1992; 51; 3; 457-68.
230. Lindgren V., Luskey K.L., Russel D.W. et al. Human genes involved in
cholesterol metabolism: chromosomal mapping of the loci for the low
density lipoprotein receptor and 3-hydroxy-3-methylglutaryl-coenzyme A
reductase with cDNA probes. Proc.Nat Acad. Sci., 1985; 82; 8567-8571.
231. Linnane A.W., Baumer A., Maxwell R.J. et al. Mitochondrial gene
mutation: the ageing process and degenerative diseases. Biochem. Int.,
1990; 22; 1067-1076.
232. Linnane A. IV., Marzuki S., Ozawa T. et al. Mitochondrial DNA mutations
as an important contributor to ageing and degenerative diseases. Lancet,
1989; 1; 642-645.
233. Loesch D., Huggins R., Petrovic V. et al. Expansion of the CGG repeat
in fragile X in the FMRI gene depends on the sex of the offspring. Am. J.
Hum. Genet., 1995; 57; 1408-1413.
234. Lyon M.F., Rastan S. Parental source of chromosome imprinting and its
relevance for X-chromosome inactivation. Differentiation, 1984; 26; 1; 63-
67.
235. Maffatt M.E., Hill M.R., Cornells F.et a!. Genetic Linkage of T-call
receptor a/5 complex to specific IgE responses. Lancet, 1994; 343; 1597-
1600.
236. Magenis R.E., Maslen C.L., Smith L et al. Localization of the fibrillin
(FBN) gene to chromosome 15, band q2l.1. Genomics. 1991; 11; 346-
351.
237. Mahadevan M., Tsilfidis C., Sabourin L. et al. Myotonic dystrophy
mutation: an unstable CTG repeat in the 3’ untraslated region of the gene.
Science, 1992; 255; 1253-1255.
238. Martin R.H., Balkan IV., Burns K. et al. The chromosome constitution of
1000 human spermatozoa. Hum. Genet., 1983; 63; 305-309.
239. Martin R.H., Mahadevan M.M., Taylor P. J. et al. Chromosomal analysis
of unfertilized human oocytes. J. Reprod. Fertil; 1986; 78; 673-678.
240. Matsuo T., Kario K., Yamada T. et a!. Hereditary heparin cofactor II
deficiency and coronary artery disease. Tromb. Res., 1992; 65; 495-505.
241. Mattei M.-G., Hubert C., Alhenc-Gelas F. et al. Angiotensin -I converting
enzyme gene is on chromosome 17 (Abstract) Cytogenet. Cell Genet.,
1989; 51; 1041.
242. McCuize IV., Hill A.V.S., Afisopp C.E M. et al. Variation in the TNF-a
promoter region associated with susceptibility to cerebral malaria. Nature,
1994; 371; 508-511.
243. McDonald S.M., Rafnar T., Langdom J. etal. Molecular identification of
an IgE-dependent histamine-releasing factor. Science, 1995; 269; 688-
690.
244. McDonnell W.M., Askari F.K. DNA vaccines. New Eng. J. Med, 1996;
334; 46-49.
245. McGill J.R., Chirgwin J.M. , Moore C.M. et al. Chromosome localization
of the human renin gene (REN) by in sity hybridization. Cytogenet. Cell
Genet., 1987, 45; 55-57.
1
215
Список литературы
246. McInnis M.G., McMahon F.J., Chase G.A. et al. Anticipation in bipolar
affective disorders.Am. J. Hum. Genet ,1993; 53; 385-390.
247. McKusick V.A. The anatomy of human genome. J. Hered., 1980; 71;
370-391.
248. McKusick V.A. Mendelian Inheritance in Man. Baltimor and London:
J.Hopkins Univer., 1992.
249. McKusick V. On-line Mendelian Inheritance in Man (OMIM) on CD-ROM.
J. Hoipkins University Press. 6 june 1996.
250. McKusick V.A. The Morbid Anotomy of the Human Genome: A Review of
gene Mapping in Clinical Medicine.- Howard Hughes Medical Institute,
1988; 227 p.
251. Meyers D.A., Postma D.S., Panhuysen C.l. et al. Evidence for a locus
regulating total serus IgE levels mapping to chromosome 5. Genomics,
1994; 23; 164-170.
252. Micklos D.A., Freyer G.A. DNA science. A first course in recombinant
DNA technology. Burlington: Carolina Biological Supply Company, 1990.
253. Miwa Sh. Triplet repeats strike again. Nature Genet., 1994; 6; 3-4.
254. Miyabayashi S., Hanamizu H., Endo H. A new type of mitochondrial DNA
deletion in patients with encephalomyopathy. Annu. Rept. Nan. Inst.
Genet., Jap., 1991; 42; 96-97.
255. Monaco A.P., Neve R.L., Colletti-Feener C et al. Isolation of candidate
cDNAs for portions of the Duchenne muscular dystrophy gene Nature.
1986; 323; 646-650.
256. Moraes C.T., Shanske S., Trirschler H.J. et al. MtDNA deletion witn
variable tissue expression: A novel genetic abnormality in mitochondrial
diseases. Amer. J. Hum. Genet., 1991; 48; 3; 492-501.
257. Mornet E., White P.C. Analysis of genes encoding steroid 11-beta-
hydroxylase. (Absrtact) Cytogenet. Cell. Genet., 1989; 51; 1047.
258. Morton N.E. The delection and estimation of linkage between the genes
for elliptocytosis and the Rh blood type. Am. J. Hum. Genet., 1956; 8; 80-
96.
259. Mudd S.H. Vascular disease and homocysteine metabolism. New Eng.
J. Med., 1985; 313; 751-753.
260. Mulligan L.M., Kwok J.B.J, Healey C.S. et al. Germ-line mutations of the
RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature.
1993; 363; 458-460.
261. Munke M., Kraus J.P., Ohura T. et al. The gene for CBS maps to the
subtelomeric region of human chromosome 21 q and proximal mouse
chromosome 17. Am. J. Hum. Genet., 1988; 42; 550-559.
262. Munscher C., Rieger T., Kadenbach B. Identification of the point
mutations characteristic for MERFE disease in mitochondrial DNA of
tissues from aged humans.Biol. Chem./Hoppe-Seycer, 1992; 373; 9; 806.
263. Murdoch J.C., Rodger J.C., Roo S.S. et al. Down’s syndrome: an
atheroma-free model. Brit. Med. J., 1977; 226-228.
264. Nagafuchi S., Yanagisava H., Sato K. et al. Dentatorubral and pallidoluy-
sian atrophy expansion of an unstable CAG trinucleotide on chromosome
12p. Nature Genet., 1994; 6; 14-18.
265. Nakahori Y., Knight S.J.L., Holland J. et.al. Molecular heterogeneity of
the fragile X syndrome. Nucl. Acids Res., 1991; 19; 4355-4359.
266. Nakahori Y., Knight S.J.L., Holland J. et al. Molecular heterogeneity of
the fragile X syndrome. Nucl. Acids Res., 1991; 19; 4355-4359.
267. Nakaki T. Physiological and clinical significance of nitric oxide (Review).
Keio J.Med., 1994; 43;1;15-26.
91R
Список литературы
268. Nakamura М., Fujiwara К, Yamamoto М. The two locus control of Leber
hereditary optic neuropathy and a high ptnttrance in Japanese pedigrees
Hum. Genet., 1992; 91; 4; 339-341.
269. Nakase H., Moraes C.T., Rizzuto R. Trascription and translation of
deleted mitochondrial genomes in Kearns-Sayre syndrome: implication for
pathogenesis. Amer. J. Hum. Genet., 1990; 46; 3; 418-427
270. Nazarenko S.A., Puzyrev V.P., Sukhanova N.N. Parental origin of
Turner’s syndrome in the Siberian populations in connection with analysis
of chromosomal imprinting. Genomic imprinting. Florence, 1994; 100 p.
271. Neumann PE, Frankel WN, Letts VA et al. Multifactorial inheritance of
neural tube defects: localisation of the major gene and recognition of
modifiers in ct mutant mice. Nature Genet., 1994, 6; 357-362.
272. Nishina P.M., Johnson J.P., Naggel J.K. et al. Linkage of atherogenic
lipoprotein phenotype to the low density lipoprotein receptor locus on the
short arm of chromosome 19. Proc.Nat.Acad. Sci. 1992; 89; 708-712.
273. Nishita Y., Yoshida I., Sado T. et al Genomic imprinting and chromoso-
mal localization of the human MEST gene. Genomics, 1996; 36; 539-542.
'/ 274. Nora J.J., Fraser F.C. Medical Genetics: Principles and Practice. Lea
and Febriger, A Waverly company, 1994, - 467 p,
275. Nussbaum R.L., Airhart S.D., Ledbetter D.H. Recombination and
amplification of pyrimidine rich sequences may be resonable for initiation
and progression of the Xq27 fragile site: an hypothesis. Am. J. Hum.
Genet., 1986; 23; 715-721.
276. Nussler A.K., Billiar T.R. Inflammation, immunoregulation and inducible
nitric oxide. Journal Leukocyte Biol., 1993; 54; 171-178.
277. O'Hoy K.L., Tsilfidis C., Mahadevan M.S. et al. Reduction in size of the
myotonic dystrophy trinucleotide repeat mutation during transmission.
Science. 1993; 259; 809-812.
278. Oberle I., Rousseau F, Heitz D. et al. Instability of a 550-base pair DNA
segment and abnormal methylation in fragile X syndrome. Science, 1991;
252; 51-56.
279. Oka Y., Katagiri H., Yazaki Y., et al, Mitochondrial gene mutation in islet-
cell-antibody-positive patients who wera initially non-insulin-dependet
diabetics. Lancet, 1993; 8870; 527-528.
280. Opitz J.M., Hermann J., Dieker H. The study of malformation syndromes
in man.- Birth Defects Orig. Art., 1969; 2; 1-10.
281. Orishi M., Fujii K., Minamino T. et.al. A potent genetic risk factor
restenosis (Letter) Nat. Genet., 1993; 5; 324-325.
282. Orr H.T., Ghung M., Banfi S. et al. Expansion of an unstable trinucleoti-
de CAG repeat in spinocerebellar ataxia type 1. Nature Genet., 1993; 4;
221-226.
283. Ott J. Analysis of human genetic linkage. Baltimor: John Hopkins
University Press. 1991.
284. Ozcelik T., Left S., Robinson W. et al. Small nuclear ribonucleoprotein
polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome
critical region. Nature Genet, 1992; 2; 265-269.
285. Pardue M.L., Gall J.G. Molecular hybridization of radioactive DNA to the
DNA of cytological preparations. Proc. Natl. Acad. Sci. USA, 1969; 64;
600-604.
286. Passarge E. Color Atlas of Genetics.- George Thieme Verlag Stuttgart,
New York., 1995. - 411p.
287. Pembrey M.E., Winter R.M., Davies K.E. A premutation that generates a
defect at crossing over explanes the inheritance of fragile X mental
retardation. Am. J. Hum. Genet., 1985; 21; 709-717.
1
217
Список литературы
288. Pericak-Vance МА, Bebout JL, Gaskell PC et al. Linkage studies in
familial Alzheimer disease : evidence for chromosome 19 linkage. Am J
Hum Genet., 1991, 48; 1034-1050.
289. Pickering T.G. Inheretance of hypqrtension and blood pressure reactivi-
ty. Hypertension, 1990; 16; 498-500.
290. Pieretti M., Zang R, Fu Y.H. et al. Absence of expression of the FMR-1
gene in fragile X syndrome. Cell, 1991; 66; 817-822.
/291 . Pinkel D., Gray J.W., Trask B. et al. Cytogenetic analysis by in situ
hybridization with fluorescently labeled nucleic acid probes. Cold Spring
Harbor Symp. Quant. BioL, 1986; LI; 151-157.
292. Pinsky L. A community of human Malformation syndromes involving the
mullerian ducts, distal extremities, urinary tract and ears. Teratology,
1974; 9; 65-80.
293. Pinsky L. The community of human malformation syndromes that shares
ectodermal dysplasia and deformities of the hands and feet. Teratology;
1975; 11; 227-242.
294. Platt R. Heredity in hypertension. Lancet, 1963; 899-904.
295. Plattner R., Heerema N.A., Yurov Y.B. et al. Efficent identification of
marker chromosomes in 27 patients by stepwise hybridization with alpha-
satellite DNA probes. Hum.Genet., 1993; 91; 131-140.
296. Plomin R, McClearn GE, Gora-Maslak G. Use of recombinant inbred
strains to detect quantitative trait loci associated with behavior. Behav
Genet., 1991; 21; 99-116.
297. Popp S., Jauch A., Schindler D. et al. A strategy for the characterization
of minute chromosome rearrangements using multiple color fluorescence
in situ hybridization with chromosome-specific DNA libraries and YAC
clones. Hum.Genet., 1993; 92; 527-532.
298. Postma D.S., Bleecker ER., Amelung P.J. et al. Genetic susceptibility to
asthma: bronchial hyperresponsiveness coinhetited with a major gene for
atopy. New Eng. J. Med., 1995; 333; 894-900.
299. Poulton J., Deadman M.E., Gardiner R.M. Duplications of mitochondrial
DNA in mitochondrial myopathy. Laricet, 1989; 1; 8632; 236-240.
300. Poulton J., Deadman M.E., Ramarcharan S. Germ-line delations of
mtDNA in mitochondrial myopathy. Amer. J. Hum. Genet., 1991; 48; 4;
649-653.
301. Poustka A, Dietrich A, Langenstein G et al. Physical map of human
Xq27-qter: localizing the region of the fragile X mutation. Proc Natl Acad
Sci USA. 1991; 88; 8302-8306.
302. Prezant T.R., Shohat M., Jaber L et al. Biochemical characterization of
pedigree with mitochondrially inherited deafness. Amer.Med. Gene, 1992;
44; 4, 465-472
303. Primrose S.B. Principles of Genorhe Analysis. Blackwell Science Ltd.,
1995; 148 p.
304. Prior T.W., Papp A.C., Snyder P.J et al. Identification of two point
mutacions and a one base deletion in exon 19 of the distrofin gene by
heterodu-plexes formacion. Human Molecular Genetics, 1993; 2; 3; 311-
313.
305. Raimondi E., Ferretti L., Young B.D. et al. The origin of a morphological-
ly unidentifiable human supernumerary minichromosome traces throgh
sorting, molecular cloning, and in situ hybridisation. J.Med.Genet., 1991;
28; 92-96.
306. Razin F., Cedar H. DNA methylation and genomic imprinting. Cell, 1994;
77; 473-476.
Список литературы
307. Reardon W., Ross R.J.M., Sweeney M.G. et al. Diabetes mellites
associated with a pthogenic point mutation in mitochondrial DNA. Lancet,
1992; 8832; 1376-1379.
308. Reik W. Genomic ihnprinting and genetic disorders in man. Trends
Genet., 1989; 5; 10; 331-336.
309. Richards B., Heilig R., Oberle I. et al. Rapid PCR analysis of the St14
(DXS52) VNTR. NAR, 1991; 19; 8; 1944.
310. Riordan J.R., Rominens J.M, Kereni B. et al. Identification of the cystic
fibrosis gene: cloning and characterization of complementary DNA.
Science, 1989; 245; 1066-1073.
311. Riordan J.R., Rommens J.M., Kerem B.S. et al. Identification of the
cystic fibrosis gene :: cloning and characterization of complementary DNA.
Science, 1989; 245; 1066-1073.
312. Rise ML, Frankel WN, Coffin JM, Seyfried TN. Genes for epilepsy
mapped in the mouse. Science, 1991; 254; 669-673.
313, Ritchie R.J., Knight S.L.J., Hirst M.C. et al. The cloning of FRAXF:
trinucleotide repeat expansion and methylation at a third fragile site in
distal Xqter. Hum. Mol. Genet., 1994; 3; 2115-2121.
314. Robbed T. Mitochondrial DNA may hold a key to human degenerative
diseases.J. NIH Res., 1992; 4; 6;. 62-66,
315. Roder J, Hickey WF. Mouse models, immunology, multiple sclerosis and
myelination. Nature Genet., 1996; 12; 6-8.
316. Rolf A., Schuller I., Finckh U.et al. PCR: Clinical diagnostics and
research. Berlin: Springer-Verlag Berlin Heidelberg, 1992; 112-167.
317. Romeo G., McKusick V.A. Phenotypic diversity, allelic series and
modifier genes. Nat. Genet., 1994; 7; 451-453.
318. Rotter J.I., Bu.X., Cantor R.M. et al. Multilocus genetic determinants of
LDL particle size in coronary artery disease families. Am. J. Hum. Genet.,
1996; 58; 585-594.
319. Rottig A., Bessis J.L., Romero N., et al. Maternally inherited duplication
of mitochondrial gehome in a syndrome of proximal tubulopathy, diabetes
mellitus and cerebelar ataxia. Amer. J. Hum. Genet., SuppL, 1991; 49; 4;
104.
320. Rousseau F., Heitz D., Biancalana V. et al. Direct diagnosis by DNA
analysis of the fragile X syndrome of mental retardati- on. New Eng. J.
Med.: 1991; 325; 1673-1681.
321. Rousseau F., Heitz D., Biancalana V. et al. On some technical aspects
of direct DNA diagnosis of the fragile X syndrome. Am. J. Med. Genet.,
1992, 43; 197-207
322. Royer-Pokora B, Kunkel LM, Monaco AP et al. Cloning the gene for an
inherited human disorder - chronic granulamatous disease - on the basis
of its chromosomal location. Nature, 1986; 322; 32-38.
323. Rubtsov N., Senger ., Kuzcera H. et al. Interstitial deletion of chromoso-
me 6q: precise definition of the breakpoints by microdissection, DNA
amplification, and reverse painting. Hum,Genet., 1996; 97; 705-709.
324. Ruiz J., Blanche H., Cohen N. et al. Insertion/deletion polymorphism of
the angiotensin-cohverting enzyme gene is strongly associated with
coronary heart disease in non-insulih-dependent diabetes meelitus. Proc.
Nat. Acad. Sci., 1994; 91; 3662-3665.
325. Sahashi K., Tanaka M., Tashiro M. et al. Increased mitochondrial DNA
deletion in the skeletal muscle of myotonic dystrophy. Gerontology.
(Schweiz.), 1992; 38; 1-2; 18-29.
9 326. Saiki R., Scharf S., Faloona FA. et al. Enzymatic amplification of b-
globin genomic sequences and restriction site analysis for diagnosis of
sickle cell anemia. Science,^985; 230; 1350-1354.
219
Список литературы
327. Saiki R.K., Gelfand D.H., Stoffel S. et al. Primer-directed enzymatic
amplification of DNA with a thermostable DNA polymerase. Science, 1988;
239; 487-491.
328. Sandford A.J., Shirakawa T., Moffatt M.F. et al. Localisation of atopy
and b subunit of the high-affinity IgE receptor (FcoRI) on chromosome
11q. Lancet, 1993; 341; 332-334.
329. Sasaki T., Billett E., Petronis A. et al. PSichosis and genes with trinucle-
otide repeat polymorphism. Hum. Genet., 1996; 97; 244-246.
330. Schellenberg G. D., bird, T. D., Wijsman E. M. et al. Genetic linkage
evidence for a familial Alzheimer’s disease locus on chromosome 14.
Science, 1992; 258; 668-671.
331. Schinzel A. Cytogenetic methods for gene mapping (preprint). European
school of medical genetics, Sestri-Levante, Italy, 1995.
332. Schunkert H., Hense H.-W., Holmer S.R. et al. Association beetween a
deletion polymorphism of the angiotensm-converting-enzyme gene left
ventricular hypertrophy New Eng. J. Med., 1994; 330; 1634-1638.
333. Schwartz K. On the pulse of genetic cardiology. Nat.Genet., 1994; 8;
110-111.
334. Shelbourne P., WinqYist R., Kunert E. et al. Unstable DNA may be
responsible for the incomplete penetrance of the myotonic dysrtophy
phenotype.Hum. Mol. Genet., 1992; 1; 467-473.
335. Sherman S.L., Jacobs P.A., Morton N.E. et al. Further segregation
analysis of fragile X syhdrome with special reference to transmitting
males. Hum. Genet., 1985; 69; 289-299.
336. Sherman S.L., Mortdn N.E., Jacobs P.A. et al. The marker (X) syndro-
me: a cytogenetic and genetic analysis. Ann. Hum. Genet., 1984; 48; 21-
37.
337. Shirakawa T., Li A., bubowitz M., Dekker J.W. etal. Association
between atopy and variants of the b subunit of the hiah-affinity IgE
receptor. Nat.Genet., 1994; 7; 125-130.
338. Shofner J.M., Lott M.T., Lezza A.M.S. et al. Myoclonic epilepsy and
regged-red fiber disease (MERRF) is associated with a mitochondrial DNA
tRNALys mutation. Cell, 1990; 61, 931-937.
339. Shon E.A., Koga Y., Davidson M., et al. The mitochondrial tRNAL*u(UUR)
mutation in MELAS: a mode for pathogehesis.Biochinn. Et Biophys. Acta.
Bioenerg., 1992; 1101; 2; 206-209.
340. Silvesri G., Moraes C.T., Shanske S. et al. A new mtDNA mutation in the
tRNALys gene associated myoclonic epilepsy and regged-red fobers
(MERRF). Amer. J. Hum. Genet., 1992; 51; 6; 1213-1217.
341. Southern E.M. Detection of specific sequences among DNA fragments
separated by gel electrophoresis. J. Mol. Biol.,1975; 98; 4; 503-517.
342. Spence J. E., Perciaccante R.G., Greig G.M. etal. Uniparental isodisomy
as a mechanism for human genetic disease. Am. J. Hum. Genet., 1988;
42; 217-226.
343. Spies T., Morton C.C., Nedospasov S.A. et al. Genes for the tumor
necrosis factor alpha and beta are linked to the human major histocompa-
tibility complex. Proc. Nat. Acad. Sci., 1986; 83; 8699-8702.
344. Sutherland G.R., Baker E. Characterisation of a new rare fragile site
easily confused with the fragile X. Hum. Mol. Genet., 1992; 1; 111-113.
345. Szulman A.E. Syndrbmes of hydatidifotm moles. Partial vs. Complete. J.
Reprod. Med., 1984; 29; 11; 788-791.
346. Tannin G.M., Agarwal A.K., Monder C.t et al. The human gene for
human 11-beta-hydroxysteroid dehydrogenase: structure, tissue distributi-
on and chromosomal localisation. J.Biol. Chem., 1991; 266; 16653-
16658.
Список литературы
347. Terwilliger JD, Ott J. Handbook of human genetic linkage. Baltimor:
John Hopkins University Press., 1994.
348. Tjio J.H., Levan A, The chromosome number of man. Hereditas, 1956,
42, 1, 6.
349. Tharapel A. T., Qumsiyeh M B., Martens PR. et al. Identification of the
origin of centromeres in whole-arm translocations using fluorescent in situ
hybridization with a-satellite DNA probes. Am.J.of Med.Genet., 1991; 40;
117-120.
350. Thomson G. Mapping disease genes: family-based association studies.
Am J Hum Genet., 1995; 57; 487-498.
351. Thomson G., Robinson WP, Kuhner MK et al. HLA and insulin gene
associations with IDDM. Genet Epidemiol., 1989; 6;. 155-160.
352. Tienari PJ, Terwilliger JD, Ott J et al. Two-locus linkage analysis in
multiple sclerosis (MS). Genomics, 1994; 19; 320-325.
353. Todd JA. Genetic analysis of type 1 diabetes using whole genome
approaches.Proc Natl Acad Sci USA, 1995; 92; 8560-8565.
354. Tomfohrde J, Silverman A, Barnes R et al. Gene for familial psoriasis
susceptibility mapped to the distal end of human chromosome 17q.
Science, 1994; 264; 1141-1145.
355. Tomkins D.J., Roux A.-F, Waye L. et al. Maternal uniparental isodisomy
of human chromosome 14 associated with a paternal t(13ql4q) and
precocious puberty. Eur. J. Hum. Genet., 1996; 4; 153-159.
356. Tsilfidis C., MacKenzie A.E., Mettler G. et al. Correlation between CTG
trinucleotide repeat length and frequency of severe congenital myotonic
dysrtophy. Nature Genet., 1992; 1; 191-195.
357. Turki J., Pak J., Green S.A. et al. Genetic polymorphisms of the bets-
2adrenergic receptor in nocturnal and nonnocturnal asthma: evidence that
gly 16 correlates with the nocturnal phenotype. J. Clin. Invest., 1995; 95;
1635-1641.
358. Twist E.C., Casanbon L.K., Ruttledge M.H. et al. Machado Joseph
disease maps to the region of chromosome 14 as the spinocerebellar
ataxia type 3 locus. J. Med. Genet., 1995; 32; 25-31.
359. Unterhuber R. Germany playing “catch-up" in gene therapy. Nature
Med., 1996; 2; 136.
360. Up the function (Editorial article). Nature Genetics., 1997, 16; 2; 111-
112.
361. US Department of Energy. DOE Human Genome Program. Primer on
Molecular Genetics. Washington, 1992; 42 p.
362. Verkerk A.J.M.H., Pieretti M., Sutcliffe J.S. et al. Identification of a gene
(FMR-1) containing a CGG repeat coincident with a breakpoint cluster
region exhibiting length variation in fragile X syndrome. Cell, 1991; 65;
905-914.
363. VHkkt J., Savontaus M.L., Nikoskelainen E.K. Segregation of mitochon-
drial genomes in a heteroplasmic lineage with Leber hereditary optic
neuroretinopathy. Amer. J. Hum. Genet., 1990; 47, 1; 95-100.
364 Vincent A., Heitz D., Petit C. et al. Abnormal pattern detected in fragile
X patients by pulsed field gel electroforesis. Nature, 1991; 329; 624-626.
365. Voelckel M.A., Mattei M.G., N'Guyen C. et al. Dissipation between
mental retardation and fragile site expression in a family with fragile X-
linked mental retardation. Hum. Genet., 1988; 80; 375-378.
366. Voelckel M.A., Philip N., Piquet C. et al. Study of a family with a fragile
site of the X chromosome at Xq27-28 without mental retardation. Hum.
Genet., 1989; 81; 353-357.
367 Vogel F. Relevant diviations heterozygotes of autosomal-recessive
diseases. - Clin. Genet., 1^84; 25; 381-415.
221
Список литературы
368. Vorsanova S.G., Yurov Y.B., Soloviev I.V. et al. Rapid identification of
marker chromosomes by in situ hybridization under different stringency
conditions. Analytical Cell Pathology, 1994; 7; 251-258.
369. Uoss R., Ben-Simon E., Avital A. ef al. Isodisomy of chromosome 7 in a
patient with cystic fibrosis: Could uniparental disomy be common in
humans? Am. J. Hum. Genet., 1989; 45; 373-380.
370. Wallace D.C., Singh G., Lott M.T. et al. Mitochjndrial DNA mutation
associated with Leber hereditary optic neuropathy. Science, 1988; 242;
1427-1430.
371. Wallace D.C. Deseases of the mitochondrial DNA. Annu. Rev. Biochem.’
Palo Alto (Calif ), 1992; 61; 1175-1212.
372. Wallace D C. Mitochondrial DNA variation in human evolution, degene-
rative disease and aging. Amer. J. Hum. Genet. 1995; 57; 201-223.
373, Wang J.-C.C., Passarge M.B., Yen P.H. et al. Uniparental heterodisomy
for chromosome 14 in a phenotipically abnormal familial balanced 13/14
robertsonian translocation carrier. Am. J. Hum. Genet., 1991; 48; 1069-
1074.
374. Wang M.-R., Perissel B., Malet P. Simultaneous in situ hybridization with
biotin-labeled centromeric and library DNA probes: a useful method for
identifying translocations. Analytical Cellular Path., 1995; 8; 53-56.
375. Wang X.L., Sim A.S., Badenhop R.F. et al. A smoking-dependent risk of
coronary artery disease associated with a polymorphism of the endothelial
nitic oxide synthase gene. Nat. Med., 1996; 2; 41-45.
376. Weeks DE., Lange K. The affected-pedigree-member method of linkage
analysis. Am J Hum Genet., 1988.; 42; 315-326.
377. We/ H, Fan WF, Xu H et al. Genes in one megabase of the HLA class I
region. Proc Natl Acad Sci USA, 1993; 90; 11870-11874.
378. Weissman SM. Genetic bases of common polygenic diseases. Proc Natl
Acad Sci USA, 1995; 92; 8543-8544.
379. White L.M., Rogan P.K., Nicholls R.D. et al. Allele-specific replication of
15q11-q13 loci: A diagnostic test for detection of uniparental disomy. Am.
J. Hum. Genet., 1996; 59; 423-430.
380. Wilson J.M. et al. Clinical protocol: ex vivo gene therapy of familial
hypercholesterolaemia. Hum. Gene Ther., 1992; 3; 179-222.
381. Wohrle B., Kotzot D., Hirst M.C. et al. A microdeletion of less than 250
kb, including the proximal part of the FMR-1 gene and the fragile-X site, in
a male with the clinical phenotype of fragile-X syndrome. Am. J. Hum.
Genet., 1992; 51; 299-306.
382. Woods J.W., Falk R.J., Pittman A.W. et.al. Increased red call sodium-
lithium countertransport in normotensive sons of hypertensive patients.
New Eng. J. Med., 1982; 306; 593-595.
383. Yang-Feng T., Xue F, Zhong W. et al. Chromosomal organization of
adrenergic receptor genes. Proc. Nat. Acad. Sci., 1990; 87; 1516-1520.
384. Yoneda M., Tanno Y., Horai S., et al A common mitochondrial DNA
mutation in the tRNALys of patients with myoclonic epilepsy associated with
regged-red fibers. Biochem. International, 1990; 21; 5; 789-796.
385. Yu S., Pritchard M., Kremer E. et al. Fragile X genotype characterized
by an unstable region ot DNA. Science, 1991; 252;111-113.
386. Yung J.-F New FISH probes - the end in sight. Nature Genet., 1996;
14; 10-12.
387, Yunis J.J. High resolution of human chromosomes. Science, 1976; 191;
1268-1270.
388. Zeviani M. Nucleus-driven mutations test human mitochondrial DNA. J.
Inherit. Metab. Disease, 1992; 15; 4; 456-471.
Список литературы
389. Zeviani М , Bresolin N., Gellera C. et al. Nucleus-driven multiple large-
scale deletions of the human mitochondrial dominant diseases. Amer. J.
Hum. Genet., SUppl., 1990; 47; 6; 904-914.
390. Zeviani M, Gellera C., Antozzi C. et al. Maternally-inherited myopathy
and cardiomiopathy: a disorder due to a new mitochondrial DNA point
mutation. Ital. J. Neurol. Sci, 1991; 12; 5; 7.
391. Zhu D., Econdmou E.R., Antonorakis S.E. et al. Mitochondrial DNA
mutation and heteroplasmy in type I Leber hereditary optic neuropathy.
Amer. J. Med. Genet., 1992; 42; 2; 173-179.
223
Cr
36
36
37
37
37
37
37'
37.'
37(
37'
371
37!
Научное издание
Пузырев Валерий Павлович
38< Степанов Вадим Анатольевич
38 Патологическая анатомия генома человека
зе;
38-
38^
386
386
387
386
Художественный редактор и художник Е В. Хоружая
Технический редактор Е В. Кондурова
ЛР № 020297 от 23.06.97. Сдано в набор 10.08.97. Подписано в
печать 02.09.97. Бумага офсетная. Формат 84x108 1/32. Гарнитура
прагматика. Офсетная печать. Усл. печ. л. 7,0. Уч.-изд. л. 7,0.
Тираж 1000 экз. Заказ № 127.
Сибирское издательско-полиграфическое и книготорговое
предприятие «Наука* РАН.
630077, Новосибирск, ул. Станиславского, 25.
Редакционная подготовка и изготовление оригинал-макета.
Scientific & Technical Translations.
Томск, тел./факс (3822) 244-688.
e-mail: editor@stt.tomsk.ru
i