/









































































































































































































































































Теги: химия
Год: 1960




Текст
и * л
Издательстве
иностранной
литературы
*
CHEMISTRY OF THE
RARE RADIOELEMENTS
POLONIUM-ACTINIUM
/С W. Bagnall, B. Sc., Ph. D.
A.E.R.E., Harwell
Sponsored by
The United Kingdom Atomic Energy Authority
Harwell
LONDON
BUTTERWORTHS SCIENTIFIC PUBLICATIONS
19 5 7
ХИМИЯ РЕДКИХ
РАДИОАКТИВНЫХ
ЭЛЕМЕНТОВ
ПОЛОНИЙ — АКТИНИЙ
Перевод с английского
В. М. Сахарова
с дополнениями
канд. хим. наук К. Г. Ш ее бе ль б лита
Под редакцией
канд. хим. наук К). В. Г а г ар и нс к о го
ИЗДАТЕЛЬСТВО ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, I960
АН НОТАЦИЯ
В книге описаны шесть редких радиоактивных эле-
ментов: полоний, астатин, франций, радон, радий
и актиний. Автор умело и полно обобщил опубли-
кованные данные, относящиеся к этим малоизвестным
до недавнего времени элементам, описал методы изу-
чения их химических свойств. Приведены сведения
о способах разделения этих элементов, технике работы
с ними, их свойствах, а также о свойствах их соеди-
нений. В русском издании книги помещены дополне-
ния, охватывающие основные данные, опубликованные
отечественными и зарубежными исследователями в по-
следние годы. Книга представляет интерес для хими-
ков-неоргаников и для лиц, занимающихся радио-
химическими исследованиями или работающих с радио-
активными веществами.
Редакция литературы по химии
ПРЕДИСЛОВИЕ
К РУССКОМУ ПЕРЕВОДУ
Открытия искусственной радиоактивности и деления
ядер, обусловившие возникновение новой отрасли науки и
техники — ядерной энергетики, стимулировали широкую по-
становку исследований химических и ядерных свойств
радиоактивных элементов. В этой области за последние два
десятилетия накоплен обширный материал, быстро расту-
щий с каждым годом.
Химическим и ядерным свойствам актиния и актинидов
было посвящено несколько монографий. Среди них «Акти-
ниды» Г. Т. Сиборга и Дж. Дж. Каца (перев. с англ., ИЛ,
1955), «Химия актинидных элементов» Дж. Дж. Каца
и Г. Т. Сиборга (перев. с англ., Атомиздат, 1960) «The
Transuranium Elements» Г. Т. Сиборга, 1958.
Книга К. У. Бэгнала является первой монографией,
охватывающей химические, физические и ядерные свойства
другой группы радиоактивных элементов — элементов от
полония до актиния.
Основную часть книги составляет химия полония (гл.
1—8)—автор книги является одним из крупнейших специа-
листов в этой области.
Первые сведения о химических свойствах полония со-
общает в своей диссертации М. Кюри-Склодовская, открыв-
шая этот элемент в урановой руде (1898 г.). Из ее данных
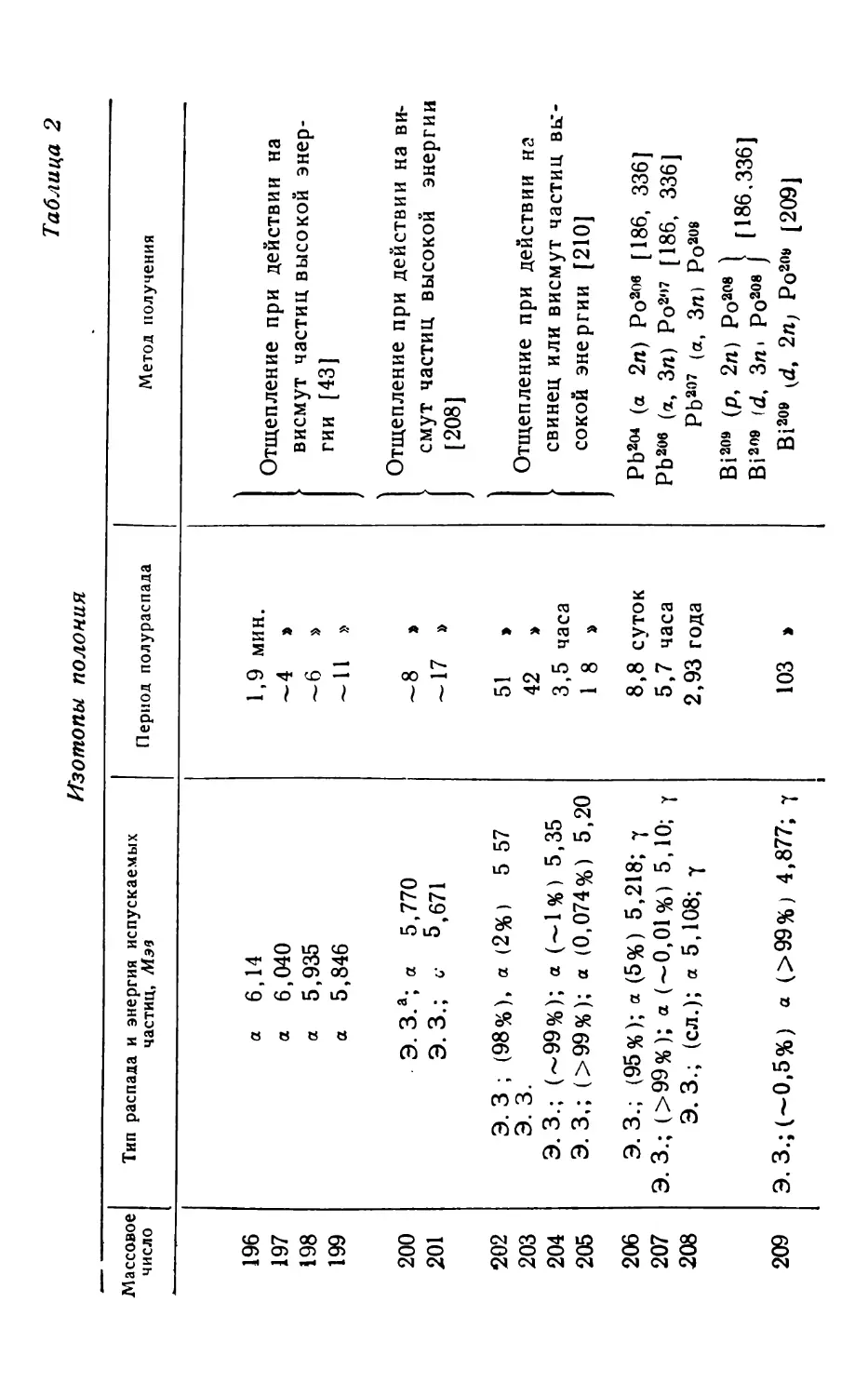
следовало, что по своим свойствам полоний похож на вис-
мут. Однако Марквальд показал, что полоний скорее яв-
ляется аналогом теллура, чем висмута. Свойства полония
еще до его открытия были предсказаны Д. И. Менделее-
вым, который называл этот элемент «двителлуром». Ре-
зультаты изучения химических свойств полония на микро-
количествах, добываемых из урановой руды, были изло-
жены в монографии «Le polonium» М. Гайсинского —
одного из учеников М. Кюри (1937 г.).
луч₽нП0С,о Д->Нпе вРемя был освоен метод искусственного по-
у ия Ро-1 облучением висмута нейтронами в ядерных
6
Предисловие к русскому переводу
реакторах, что позволило накопить его в сравнительно
больших количествах и более подробно исследовать его хи-
мические и физические свойства. Работы с миллиграммовы-
ми количествами полония особенно четко подтвердили
его сходство с теллуром.
В книге Бэгнала подведен итог всем работам как с ми-
кро-, так и с миллиграммовыми количествами полония.
Основное внимание автор уделяет исследованиям англий-
ских химиков, работающих вместе с ним в Харуэлле,
Менее полно излагаются им остальные работы, в том числе
и работы советских ученых. С работами советских радио-
химиков, посвященными изучению электрохимических и
коллоидных свойств полония, читатель может ознакомиться
в недавно вышедшей монографии И. Е. Старика «Основы
радиохимии» (1959 г.).
Во второй, сравнительно небольшой части книги Бэг-
нала (гл. 9) собран материал по астатину, францию и ра-
дону. Первые два из них (аналоги, соответственно, йода
и цезия) не имеют долгоживущих изотопов и наряду с наи-
более тяжелыми искусственно получаемыми трансурано-
выми элементами (менделевием и элементом 102) принадле-
жат к числу самых неустойчивых элементов периодической
системы.
Получить даже миллиграммовые количества астатина
и франция невозможно, и тем не менее химические свойства
их изучены довольно хорошо. Работы советских ученых по
францию отражены в обзоре А. К. Лаврухиной («Успехи
химии», т. 27, вып. 10, 1958).
Материал третьей части книги (гл. 10—12), посвящен-
ной радию и актинию, представляет несколько меньший
интерес, так как уже ранее были опубликованы моногра-
фии, содержащие обзор данных по этим элементам.
С момента издания книги Бэгнала в Англии и до выпуска
ее перевода на русский язык прошло около трех лет. В
столь быстро развивающейся отрасли знания за это время
появилось много новых сведений, и без них книга Бэгнала
в ее первоначальном объеме была бы в данный момент
в какой-то мере неполноценной. Чтобы хоть частично
исправить это положение, в перевод книги Бэгнала вклю-
чены дополнения, при составлении которых были исполь-
зованы работы, вышедшие с 1957 г., главным образом ра-
Предисловие к русскому переводу 7
боты самого Бэгнала с сотрудниками и Гайсинского, а
также советских исследователей школы В. Г. Хлопина —
И. Е. Старика, Д. М. Зива и др. Эти дополнения в об-
щем тексте выделены петитом. Соответствующим образом
дополнены имеющиеся в книге списки литературы.
В таблицы изотопов (табл. 2, 17-19, 24, 28) и в при-
ложения внесены исправления и дополнения, касающиеся
типов распада, энергии испускаемых частиц и периодов
полураспада изотопов рассматриваемых элементов, заим-
ствованные в основном из «Table of Isotopes» D. Stro-
minger, J. M. Hollander, G. T. Seaborg, Rev. Mod. Phys.,
30, № 2, part. 2, 1958, и из справочника «Схемы распада
радиоактивных ядер» Б. С. Джелепова и Л. К. Пекера
1958.
Ю. Гагаринский,
К. Швебельблит
ПРЕДИСЛОВИЕ
К АНГЛИЙСКОМУ ИЗДАНИЮ
Развитие исследований в области атомной энергии способ-
ствовало общему повышению интереса к неорганической
химии в послевоенный период. Ранее не известные химикам
элементы неожиданно приобрели большое техническое
значение, причем дальнейший прогресс в этом направле-
нии тормозится из-за недостатка сведений об этих элемен-
тах. Такое положение постепенно исправляется, но пред-
стоит еще многое сделать. В настоящей книге д-р Бэгнал,
который сам внес весьма существенный вклад в изучение
химии полония, делает попытку систематического изложе-
ния и анализа данных, полученных до настоящего времени
об элементах от полония до актиния (порядковые номера
в периодической системе элементов от 84 до 89).
Эта работа должна заинтересовать не только химиков-
неоргаников и радиохимиков, но также тех, кто имеет от-
ношение к новым отраслям техники, где часто приходится
судить о возможности применения или, наоборот, о спо?
собах удаления элементов, о которых до сих пор мало что
было известно, кроме их химических символов.
Р. Спенс
Харуэлл,
1957 г.
ПРЕДИСЛОВИЕ АВТОРА
О шести элементах, описанных в настоящей книге, лишь
весьма кратко упоминается в обычных учебниках неорга-
нической химии; эти элементы являются самыми редкими
и наиболее радиоактивными элементами периодической
системы. Оборудование, необходимое для работы с весо-
мыми количествами этих элементов, чрезвычайно дорого
и в большинстве случаев недоступно химическим факульте-
там университетов. Вследствие этого фундаментальные ра-
боты по исследованию данных элементов проводились пра-
вительственными учреждениями, такими, как Управление
по атомной энергии Великобритании и Комиссия по атом-
ной энергии Соединенных Штатов Америки.
До второй мировой войны только радий можно было
получить в чистом виде в весомых количествах; об актинии
и полонии в то время имелось мало достоверных данных.
Теперь же эти два элемента можно получить в чистом виде
нейтронным облучением висмута и радия соответственно
и, вероятно, есть все основания утверждать, что наши зна-
ния химии полония приближаются по объему к знаниям
химии теллура — ближайшего гомолога полония по пе-
риодической системе, несмотря на то, что преобладающая
часть химических исследований полония была проведена
на менее чем миллиграммовых количествах этого элемента.
Настоящая книга была написана с целью объединения
всех опубликованных данных об элементах от полония до
актиния и в какой-то мере описания методов, применяв-
шихся при изучении химии этих элементов.
В данных об этих элементах все еще имеются суще-
ственные пробелы, причем некоторые из них нельзя устра-
нить при использовании лишь применяемых в настоящее
мет°Д°в- Можно надеяться, что эта книга ока-
я полезной для будущих исследователей в области
10
Предисловие автора
неорганической химии, которые попытаются пополнить
сведения об этих элементах.
Автор глубоко благодарен д-ру Р. Спенсу, руководи-
телю Химического отдела Управления по атомной энергии,
за прочтение данной работы и за многие полезные замеча-
ния по существу излагаемых вопросов Автор хотел бы
также поблагодарить Л. Хоара из Фотографического от-
деления за оригинальные снимки, использованные для
изготовления репродукций, и У. Флетчера из Отдела об-
щей физики, сделавшего большую часть стеклянных при-
боров, применявшихся при работах с полонием.
К. У. Бэгнал
Харуэлл,
1957 г.
Часть I
ПОЛОНИЙ
(Z=84)
1
ОТКРЫТИЕ, ВЫДЕЛЕНИЕ И ПРИМЕНЕНИЕ
ПОЛОНИЯ
Открытие полония и распространение его в природе. Методы выде-
ления из старых радиевых солей и из смесей радия-D, радия-Е и
радия-F. Промышленное применение и использование в научно-ис-
следовательских работах.
ОТКРЫТИЕ ПОЛОНИЯ
При изучении радиоактивности различных минералов,
содержащих уран и торий, М. Кюри обнаружила, что на-
блюдаемая радиоактивность значительно выше той, которую
можно было ожидать, если исходить из известного содер-
жания урана и тория в этих минералах [79]. Для объясне-
ния этого было высказано предположение, что в указанных
минералах присутствует какой-то неизвестный до сих пор
элемент, более радиоактивный, чем уран и торий; хими-
ческой обработкой большого количества остатков урановой
руды после извлечения из нее урана был получен продукт
в несколько сот раз более радиоактивный, чем уран. Этот
продукт концентрируется в висмутовой фракции радио-
активных минералов, изучавшихся супругами Кюри, и при
действии сероводорода его сульфид осаждается из кислого
раствора вместе с сульфидом висмута. Было установлено,
что сульфид этого сильно радиоактивного элемента более
летуч, чем сульфид висмута, от которого его можно отде-
лить возгонкой в вакууме [83]. В результате всей этой ра-
боты были выделены крайне малые количества данного
вещества, и следить за процессами, применявшимися при
концентрировании продукта, можно было лишь по его ра-
диоактивности. Предполагалось, что новый элемент по хи-
мическим свойствам напоминает висмут; его назвали по-
76Н80М Я1 ЧяСТЬ родины МаРии Кюри — Польши [72, 75,
л 8 ’ 86]. Это был первый новый элемент, откры-
тый благодаря его радиоактивности.
14
Открытие, выделение и применение полония
Несмотря на то, что уже предварительная работа, ко-
торая привела к открытию полония, показала, что этот
элемент можно отделить от висмута, радиоактивные пре-
параты полония все еще считали «радиоактивным висму-
том» [124, 125], т. е. висмутом, который приобрел радио-
активные свойства в результате контакта с другими радио-
активными элементами. На первый взгляд такое представ-
ление подкреплялось следующим наблюдением: металли-
ческий висмут, погруженный в солянокислый раствор соли
радия, приобретал способность интенсивно излучать
а-частицы (см., например, [230]); вскоре, однако, было уста-
новлено, что это явление обусловлено самопроизвольным
выделением полония на висмуте.
В это же время Марквальд [231, 232] показал, что ра-
диоактивное вещество, концентрируемое вместе с висмутом,
можно отделить от него электрохимическим (на катоде)
или самопроизвольным выделением, а также осаждением
хлористым оловом. Полученный осадок содержал некото-
рое количество теллура, присутствующего в качестве
примеси в урановой руде; принимая во внимание, что изу-
чавшийся радиоактивный продукт по химическим свой-
ствам напоминал теллур, Марквальд предложил назвать
отделяемый продукт «радиотеллуром», чтобы не спутать
его с открытым Кюри полонием. Марквальд обнаружил
также, что радиотеллур можно отделить от теллура осажде-
нием последнего солянокислым гидразином [234].
Основное возражение против идентификации полония
и радиотеллура как одного элемента основывалось, по-ви-
димому, на очень большом различии в периодах полурас-
пада этих двух препаратов (см., например, [235]), каждый
из которых был загрязнен значительным количеством ра-
дия-D. Несмотря на это, Дебьерн [93] утверждал, что радио-
теллур Марквальда и радиоактивный висмут Гизеля со-
держат полоний, который и является их радиоактивной
составной частью.
Одновременно Резерфорд [299, 301 ] при изучении при-
роды остаточной радиоактивности, обусловленной распа-
дом радона, установил присутствие в радиоактивном осадке
долгоживущего ^-излучателя и короткоживущего а-излу-
чателя. Эти радиоактивные элементы вначале были на-
званы соответственно радием-D и радием-Е. Резерфорд
Открытие полония 15
считал что радий-Е является радиоактивной составляющей
полония и радиотеллура; Марквальд не был с этим согласен.
В результате более поздней работы 1300, 3021 в продуктах
распада радона был обнаружен второй р-излучатель, и
а-излучающий радий-Е был переименован в радий-F; таким
образом был завершен хорошо известный ряд продуктов
распада радия:
0 3 а
адРЬ«°(ЯаО)—-► цаВ1»10 (RaE)-► MPo2’°(RaF)-> s,Pb*>' (RaO).
22 г. 5 суток 138,4 суток
Первоначально в качестве а-излучающего радия-Е,
по-видимому, был идентифицирован продукт, загрязненный
другими членами данного радиоактивного ряда, поскольку
период полураспада его составлял примерно один год. При-
нимая во внимание, что радий-F не дает радиоактивных
продуктов распада, и исходя из предположения, что а-ча-
стица представляет собой ядро атома гелия, Резерфорд
[300] подсчитал, что продукт распада радия-F должен об-
ладать атомным весом, равным приблизительно 205, а по
близости этого значения к 206 заключил, что этим продук-
том мог быть свинец.
Идентичность полония и радиотеллура была оконча-
тельно подтверждена новыми измерениями периода полу-
распада, произведенными М. Кюри [82]; она установила,
что период полураспада этого продукта равен 140 суткам.
С этим значением хорошо согласовались результаты неза-
измеРений, проведенных Мейером и Швейдлером
[243], и больше уже не оставалось сомнений в том, что по-
лоний действительно представляет собой новый элемент.
Химические свойства полония, насколько они были из-
вестны к тому времени, в какой-то мере напоминали свой-
ства теллура, а поэтому можно было считать, что полоний
является неизвестным до тех пор двителлуром, существова-
ние которого было предсказано Менделеевым [241]. Однако
кончательно это стали считать решенным тогда, когда
Фаянсом и Содди был сформулирован закон сме-
я’ на основании которого оказалось возможным вы-
числить порядковый номер полония.
16
Открытие, выделение и применение полония
РАСПРОСТРАНЕНИЕ ПОЛОНИЯ В ПРИРОДЕ
В 1 пг урановой смоляной руды содержится менее 0,1 мг
полония [85, 235], и выделение этого элемента из такой
массы исходного материала в период исследовательских
работ, приведших к его открытию, было чрезвычайно труд-
ным делом, особенно если учесть, что в то время возможно-
сти для проведения экспериментальной работы были крайне
ограниченны.
Полоний можно также извлечь из старых радиевых со-
лей, содержащих по достижении радиоактивного равнове-
сия примерно 0,2 мг полония на 1 г радия, однако связан-
ная с таким извлечением опасность подвергнуться облуче-
нию и высокая стоимость работ не позволяют использовать
радий в качестве источника для получения полония.
До 1947 г. количества полония, обычно применявшиеся
для химических исследований, составляли от 10“10до 10"6г;
исключением является один образец, который, как было
установлено, содержал около 100 иг полония и несколько
миллиграммов примесей; в 1910 г. этот образец был приго-
товлен Кюри и Дебьерном [84] для спектроскопических
исследований.
Указанные микроколичества полония обычно получали
из старых радоновых ампул, в которых после практически
полного распада радона оставался радиоактивный осадок,
состоящий из радия-D-E-F. В более ранней литературе
по полонию много места уделялось описанию методов от-
деления полония от радия-D и радия-Е, а также от загряз-
нений, таких, как ртуть. Последняя часто присутствовала
в радиоактивном осадке; по-видимому, она попадала в ра-
доновые ампулы в виде паров из ртутных насосов, исполь-
зуемых для наполнения ампул.
। В настоящее время, когда в ядерных реакторах полу-
чают интенсивные потоки нейтронов, миллиграммовые ко-
личества полония можно получить облучением висмута
88Bi2W(«. т) 83Bi210 84po210
5 суток
Поперечное сечение захвата тепловых нейтронов висму-
том-209 равно приблизительно 0,020 барн. При облучении
в ядерном реакторе образуются большие количества изо-
Выделение полония
17
топа Bi210, и благодаря этому удалось показать, что неболь-
шая часть ядер этого изотопа (10-5%) также распадается
с испусканием я-частиц.
Некоторые методы, применяемые для выделения поло-
ния из облученного висмута, описаны в последующих
разделах; многие из них более детально изложены в докладе,
недавно опубликованном в США [249].
ВЫДЕЛЕНИЕ ПОЛОНИЯ
Классический метод выделения полония из урансодержа-
щих минералов в настоящее время уже не применяется,
однако многие наблюдения, сделанные в процессе первона-
чальной работы, которая привела к открытию этого эле-
мента, интересны в химическом отношении, и о них гово-
рится в соответствующих разделах книги. Методы, при
помощи которых производится выделение микроколичеств
полония, детально рассмотрены в обзоре Арбитрио [5]
и в справочнике Гмелина [126].
Выделение из старых радиевых солей
Полоний благодаря его самопроизвольному осаждению
на меди (см. далее, раздел 3) можно легко выделить из рас-
творов солей радия, однако получающийся при этом про-
дукт содержит значительное количество радия-Е.
Обычные методы получения полония из старых радиевых
солей основаны на выделении радия-D и выдерживании его
до тех пор, пока в нем не образуется полоний в таких коли-
чествах, что его извлечение становится целесообразным.
Согласно одной методике [290], радий-D выделяли из
азотнокислого раствора на платиновом аноде. Растворе-
нием в азотной кислоте и повторным анодным выделением
продукт очищали от следов присутствующего в нем радия.
Совершенно ясно, что этот метод не применим к радиево-
бариевым смесям, но в этом случае свинец, висмут и поло-
нии можно осадить сероводородом на сульфиде меди
в качестве носителя (см., например, [321]). Осаждение серо-
водородом требует известной осторожности, так как в ре-
ультате^ окисления некоторой части сульфида в сульфат
I ОД действ» облучения) может произойти частичное
2
К- Бэгна.
Открытие, выделение и применение полония
осаждение радия. Этот метод подробно описан в работе
Рона [290].
Радий-D также можно отделить от радия кристаллиза-
цией бромидов из концентрированных растворов бромисто-
водородной кислоты. Радий-D остается в растворе, по-
скольку бромид свинца не образует смешанных кристал-
лов с бромидом радия. Выделение из концентрированных
солянокислых растворов менее эффективно из-за внедре-
ния хлорида свинца в кристаллическую решетку хлорида
радия (происходит образование аномальных смешанных
кристаллов [146, стр. 102]).
Выделение из смесей ради я-D-E-F
Методы, применяемые для отделения полония от радио-
свинца, можно подразделить на четыре группы: первая
объединяет методы отделения свинца от полония; вторая —
осаждение полония из растворов на соответствующих но-
сителях или возгонку полония из твердого источника;
третья (наиболее широко применяемые методы) — исполь-
зование самопроизвольного или электрохимического выде-
ления на подходящей металлической подложке, от которой
полоний легко можно отделить химическим или физическим
методом; в четвертую группу входят разработанные в по-
следнее время методы экстрагирования растворителями,
ионного обмена и хроматографии на бумаге. Методы, вклю-
ченные в последнюю группу, применяли только для вы-
деления микроколичеств полония.
1. Отделение свинца (ради я-D
и нерадиоактивных изотопов)
Большинство препаратов радия-D-E-F содержат значи-
тельные количества неактивного свинца, большая часть
которого может быть удалена растворением исходного пре-
парата в разбавленной кислоте и последующим осажде-
нием концентрированной соляной [42, 93] или концентри-
рованной азотной кислотой [73, 74].
При отсутствии достаточного количества неактивного
свинца в качестве носителя можно использовать хлорид
бария. При этом радий-D можно снова извлечь и исполь-
Осаждение полония
19
зовать в дальнейшем в качестве источника полония путем
повторного растворения осажденных хлоридов и последую-
щего выделения свинца в виде гидроокиси [42]. Как со-
общалось, выход полония при использовании этого метода
достигает порядка 90%; указанный метод часто приме-
няют на предварительных стадиях при отделении полония
от больших количеств свинца.
Свинец можно также отделить диализом в слабокислом
или щелочном растворе, так как только он обладает свой-
ством проникать через полупроницаемую перегородку
[255, 258, 267, 268 стр. 1049]. Недостаток метода заклю-
чается в том, что пергамент, употребляемый в качестве
полупроницаемой мембраны, адсорбирует значительную
часть полония [153, 318]. Если диализ ведут в растворе
едкого натра, потери в результате адсорбции могут дости-
гать 70% полония; из аммиачного раствора адсорбируется
только 10% полония [153]. По наблюдениям Старика и
Комлева [319] большие количества полония адсорбируются
мембраной из кислых растворов и меньшие из щелочных;
минимальная адсорбция наблюдается в нейтральных рас-
творах.
2. Методы осаждения и возгонки
Методы соосаждения
Полоний можно отделить от висмута в очень слабом азотно-
кислом растворе путем осаждения с теллуратом свинца.
Для этого раствор, содержащий полоний, свинец и висмут,
нагревают с теллуровой кислотой; при выдерживании об-
разуется теллурат свинца, с которым осаждается большая
часть полония. Свинец удаляют, нагревая осадок с серной
кислотой и отфильтровывая нерастворимый сульфат
свинца. Фильтрат выпаривают до сухого остатка, который
растворяют в концентрированной соляной кислоте, и полу-
ченный раствор кипятят, чтобы восстановить теллур до
четырехвалентного состояния. Теллур затем можно осадить
в элементарном состоянии восстановлением сернистым га-
3[204 чего остается довольно чистый раствор полония
ший ’ ^аК соо^ают, этот метод позволяет получить об-
выход, составляющий примерно 97%. Возможно, что
2*
20
Открытие, выделение и применение полония
осаждение зависит от образования нерастворимого теллу-
рата полония.
Данный метод представляет значительный интерес бла-
годаря тому, что полоний в отличие от теллура не восста-
навливается сернистым газом до элементарного состояния
и осадок мелкораздробленного теллура не адсорбирует
микроколичеств полония. Марквальд [234 ] установил также,
что полоний можно отделить от теллура восстанов-
лением последнего гидразином и что отделение от теллура
методом восстановления осуществляется очень хорошо.
Однако в присутствии селена полоний осаждается почти
количественно из растворов в соляной [235] или плавиковой
кислоте [335] при действии таких восстановителей, как сер-
нистый газ или гидразин; это объясняли тем, что микро-
количества полония адсорбируются мелкораздробленным
селеном, образующимся в процессе восстановления. Осажде-
ние проходит почти столь же эффективно с миллиграммо-
выми количествами полония; это, по-видимому, обуслов-
лено образованием селенида полония, так как если бы
данное явление объяснялось адсорбцией, то аналогичные
эффекты наблюдались бы при осаждении теллура. Полоний
можно извлечь из осадка селенида полония, обрабатывая его
концентрированной соляной кислотой, после чего остается
почти не обладающий активностью селен; такое же положе-
ние наблюдалось в случае сульфида полония (см. стр. 124).
Полоний можно отделить от облученного висмута со-
осаждением его с элементарным теллуром, образующимся
в процессе восстановления хлористым оловом [247, 296].
Осадок растворяют в концентрированной азотной кислоте
и после добавления концентрированной соляной кислоты
осаждают теллур сернистым газом [247]. Затем полоний
можно очистить электрохимическим выделением на золоте
или используя самопроизвольное выделение его на серебре
(см. стр. 25—26).
Другой метод соосаждения основан на адсорбции микро-
количеств полония коллоидной платиной в очень разбав-
ленных кислых растворах [204]. Под действием ультрафио-
летовых лучей платина коагулирует, и полоний выделяют
из полученного осадка возгонкой в вакууме. С той же
целью предлагали использовать коллоидную кремневую
кислоту [100], однако осаждение полония из растворов
Возгонка полония
21
на коагулирующем серебре или гидроокиси железа, как
утверждают, оказывается более эффективным [33, 34].
У Полоний можно также выделить из слабокислых раство-
ров нитрата радиосвинца (pH 4) фильтрованием через слой
двуокиси титана; от двуокиси титана полоний отделяют
промыванием 3—6 н. соляной кислотой [249 стр 131].
Механизм данного процесса не изучен.
Смесь полония с радием-Е можно получить осаждением
с пирогаллатом сурьмы или висмута из 0,05 н. раствора азот-
ной кислоты [133, 149, 154, 155] или р-аминонафталидом
тиогликолевой кислоты (тионалидом) из нейтральных рас-
творов [324]. В литературе есть указания, что осадок
тионалида растворим в ацетоне. Осадок пирогаллата рас-
творяют в кислоте и затем электрохимическим методом
выделяют из раствора полоний.
В аммиачном растворе микроколичества полония почти
количественно захватываются гидроокисью висмута [83];
распределение полония между осадком и раствором изу-
чали детально [108, 114, 115], причем было установлено,
что в данном случае имеет место адсорбция. В качестве
носителей полония в щелочной среде использовали гидро-
окись железа Fe(OH)3 [115, 211], гидроокись дидима*
[42], гидроокись лантана (см., например, [10]) и гидро-
окись алюминия [211]. Из перечисленных носителей при
концентрировании разбавленных растворов полония наибо-
лее широко применяется, по-видимому, гидроокись лантана.
Имеются также сообщения о том, что микроколичества
полония соосаждаются с двуокисью марганца из раствора
в азотной кислоте [226].
Методы возгонки
М. Кюри [83] установила, что полоний можно выделить
из смеси сульфидов висмута и полония возгонкой в вакууме
при 700°. В настоящее время известно, что полученный та-
ким образом возгон представляет собой металлический
полоний [11]. Данный метод применим для быстрой очистки
осадков с большим содержанием полония и значительным
количеством свинца, образовавшегося в результате распада;
мали‘ 3^МэлемТн°^ИдМидим^—ИПрим.е°ДИЛЯ BPeM” ПРИН"
22
Открытие, выделение и применение полония
сульфиды свинца и полония осаждают сероводородом из
разбавленного солянокислого раствора и отфиль-
тровывают через стеклянный пористый фильтр. Осадок
нагревают в вакууме до 500° и отгоняют полоний, а суль-
фид свинца остается на пористом фильтре [47].
Возгонку микроколичеств полония на воздухе изучали
довольно подробно; полоний начинает возгоняться при
700° [294], и полностью переходит в парообразное состоя-
ние при 900° [294, 298], тогда как висмут (Ra-E) в анало-
гичных условиях вряд ли возгоняется при температуре
ниже 1100° [298]; однако в атмосфере водорода большая
часть висмута возгоняется при 900°. По всей вероятности,
продуктом сублимации на воздухе является двуокись по-
лония, возгоняющаяся при 885° [10].
Эти работы показали, что полоний более летуч, чем сви-
нец и висмут [184]; недавно была изучена возможность при-
менения метода фракционной возгонки для выделения мил-
лиграммовых количеств полония из облученного металли-
ческого висмута. Были получены значения коэффициентов
обогащения порядка 150—1000 с выходом полония 99,5%
при использовании дистилляторов, в которых расплав
висмута с полонием перемешивался барботированием ге-
лия. Методы простой дистилляции также вполне пригодны.
Одной из трудностей, с которыми связано применение дан-
ного метода, является выбор подходящего материала для
изготовления дистиллятора, поскольку многие металлы
корродируют под действием расплавленного висмута [117].
Облучение тонкого порошка окиси висмута можно ис-
пользовать в качестве более простого метода разделения.
Облученный материал, по-видимому, содержит металли-
ческий полоний и двуокись полония, которая в вакууме
при 500° разлагается на кислород и металл. Поэтому при
нагревании облученной окиси висмута до 500° в вакууме
достигается некоторое разделение, и, когда весь полоний от-
гонится, с ним вместе возгоняется примерно 1% висмута
[249, стр. 171]. Скорость дистилляции может быть до-
вольно небольшая, поскольку скорость всего процесса
определяется скоростью диффузии полония к поверхности
частиц окиси висмута. В то же время коэффициент обога-
щения в данном случае имеет большую величину, чем при
фракционной возгонке облученного металла.
Возгонка полония
23
Методы возгонки применяли главным образом для по-
лучения сравнительно сильных (порядка милликюри) источ-
ников полония из многочисленных более мелких источни-
ков получаемых электрохимическим выделением. Воз-
гонкой в токе водорода при 900—1000° [77] был получен
полоний в количестве нескольких милликюри на неболь-
ших кусках фольги или в виде точечных источников на
палладиевой проволоке [291, 294, 304, 322]. Золотую
фольгу считали неподходящей для использования в ка-
честве металлической подложки, на которой выделялись
бы электрохимическим путем небольшие количества поло-
ния; непригодность золота для этой цели обусловлена тем,
что оно при указанных температурах плавится и обволаки-
вает полоний [294, 304]; эту трудность можно преодолеть,
если возгонку проводить в вакууме при более низких тем-
пературах [11—13]. В качестве газа-носителя при воз
гонке микроколичеств полония можно использовать также
азот [77, 291, 292].
В тех случаях, когда перегонку микроколичеств полония
проводят в кварцевой трубке в токе газа, а в холодную
часть трубки помещают палладиевую или платиновую
фольгу, почти весь полоний конденсируется на фольге,
хотя ее поверхность крайне мала по сравнению с поверх-
ностью трубки [268]. Полоний, по-видимому, предпочти-
тельнее конденсируется на платине и палладии; на медной
или никелевой фольге он почти не осаждается, даже если
ее поместить рядом. Этот факт объясняли тем, что полоний
реагирует с водородом, содержащимся в первых двух метал-
лах, образуя гидрид полония, однако это соединение весьма
нестабильно и приведенное выше объяснение, видимо, сле-
дует считать маловероятным. Возможно, что в указанных
условиях происходит образование полонидов платины и
палладия, однако все-таки нельзя привести убедительных
объяснений тому, что полоний не конденсируется на меди
или никеле и не соединяется с ними в тех же условиях.
При изучении процесса испарения микроколичеств по-
*гпсИЯ9д?1ЛИ полУчены в какой-то мере ошибочные данные
I □ 28], что частично явилось результатом неполного
удаления газа из металла, служившего подложкой. Полоний
292Г]°НЯеТСЯ опРеделенно более легко с золота [11, 291,
и никеля [287 ], чем с платины. Вполне возможно,
24
Открытие, выделение и применение полония
что наблюдаемые для микроколичеств полония отклонения
от нормального хода перегонки являются результатом диф-
фузии полония в материал подложки [28]. Из работы Боне-
Мори [28] следует, что отгонка большего количества поло-
ния была сопряжена с большими трудностями, однако,
поданным автора [10]» отгонка миллиграммовых количеств
протекает совершенно нормально.
3. Методы электрохимического
и самопроизвольного выделения
Электрохимическое выделение
В большинстве опубликованных работ при попытках
отделить полоний от радиосвинца использовали источники,
содержащие большие количества свинца как активного,
так и неактивного, который в первую очередь надлежало
удалить одним из методов, описанных в разделе 1.
Эти источники обычно содержали большие количества дру-
гих примесей, которые могли мешать электрохимическому
выделению, и поэтому данный метод нельзя рекомендовать
для проведения первичного разделения [290].
Микроколичества полония можно выделять из уксусно-
кислого раствора на платиновом катоде при плотности тока
4 ра1см2\ висмут осаждается при более высоких плотно-
стях тока (~10 рьа/слс2) [244]. Полоний можно также вы-
делить из раствора в трихлоруксусной кислоте [42].
Сообщения о катодном выделении микроколичеств по-
лония из азотнокислых растворов носят противоречивый
характер. Выделение, как сообщается, происходит при та-
ких условиях, при которых свинец осаждается на аноде
[268], при этом на золотом катоде выделение полония
происходит лучше [262]. Рекомендовалось также исполь-
зование в качестве катода вращающегося диска [275].
В литературе имеются сообщения о применении плотностей
тока порядка 30 ^а/см2 [219, 269], и, хотя при этом полоний
и отделяется от свинца, продукт содержит большое коли-
чество висмута. Во всяком случае, И. Кюри [73] установи-
ла, что катодное выделение полония из азотнокислого
раствора в общем не дает удовлетворительных результа-
тов, поскольку при этом часть полония выделяется на ка-
тоде, смесь радия-D и полония выделяется на аноде и пере-
менные количества полония остаются в растворе, откуда
Электрохимическое выделение полония
25
нельзя было извлечь электрохимическим путем; коли-
чественные выходы достигались только в тех случаях,
когда полоний осаждался на аноде. Другие исследователи
(см например, [294, 304]) сообщают о достаточно эффек-
тивных выходах при катодном электровыделении, получа-
емых в случае применения плотностей тока порядка 10—
20 ^а/см2 (см., например, [266, 283, 343]); В присутствии
примесей, таких, как пыль или кремневая кислота, по-
видимому, целесообразнее выделять микроколичества по-
лония на аноде, хотя при этом будет также выделяться
и свинец, если в растворе содержится азотная кислота в вы-
сокой концентрации. В процессе анодного выделения по-
лоний очищается от случайных примесей ртути, золота и
платины [183, 269]. Электрохимическое выделение поло-
ния из раствора в 1 н. плавиковой кислоте не так давно
было рекомендовано для приготовления источников с вы-
соким содержанием полония (30 кюри/см2) [249, стр. 181 ];
согласно опубликованным сообщениям, при этом достига-
лось хорошее отделение от висмута, однако условия опыта
в статьях не указаны.
Методы электрохимического выделения часто исполь-
зуют для окончательной очистки миллиграммовых количеств
полония после предварительного отделения от свинца,
облученного висмута и т. д. [10, 11, 38, 159, 275]. Недавно
сообщалось [10] об успешном выделении полония на пла-
тине из растворов, содержащих — 50 мкюри/мл Ро21э в 1 н.
азотной кислоте, при плотности тока 1 и напряже-
нии 2,2 в. В период выделения раствор надлежит переме-
шивать; лучшим способом перемешивания является бар-
ботирование инертного газа через раствор. Таким мето-
дом за1 час можно выделить 80% полония и получить одно-
родный серый или серовато-черный осадок с поверхностной
активностью порядка 500 мкюри/см2. Следует отметить,
что на катоде значительно легче выделить миллиграммовые
количества полония, чем микроколичества, и первый про-
цесс подвержен значительно меньше случайным изменениям.
Выделение полония протекает более гладко на золоте,
ем на платине [199, 342], и это объясняли поверхностным
кисленисм платины в условиях интенсивной бомбарди-
сорли а’частИ1*ами [199] или образованием какого-то
нения полония о платиной [157]. Так как миллиграм-
26
Открытие, выделение и применение полония
мовые количества полония возгоняются в вакууме с золота
лучше, чем с платины, поэтому при обычном выделении по-
лония в количествах порядка кюри часто пользуются золо-
той проволокой или золотой фольгой [11]. Не так давно
сообщалось [140] об образовании полонида платины PtPo2,
и возможно, что отмечавшиеся выше явления, наблюда-
емые при процессах выделения и возгонки, обусловлены
именно образованием этого соединения. Имеются сообще-
ния о том, что электрохимическое выделение микроколи-
честв полония на молибдене является весьма эффективным
[178], однако этот металл, по-видимому, не применяли в
сколько-нибудь значительных масштабах.
В работе [349] приведены данные о совместном выделении по-
лония с Ag, Au, Hg и Pt из 1,5 н. HNO3 на золотых и платиновых
электродах. Слои в 3 кюри/см2 получали при потенциале 0,0 в (отно-
сительно нормального каломельного электрода). За 6 час. выделя-
лось ~ 85% полония, но для 99%-ного выхода требовалось уже
12 час.
Самопроизвольное выделение
Большинство обычно применяемых способов разделения
включает самопроизвольное выделение полония на менее
благородных металлах. Условия выделения и отделения
полония от наиболее широко используемых металлов опи-
саны ниже.
Серебро. Марквальд [235] нашел, что полоний очень легко
выделяется на серебре из разбавленного азотнокислого или
солянокислого раствора; в процессе выделения на серебре
появляется черная пленка, которая может состоять как из
окиси, так и из перекиси серебра. Окисная пленка, согласно
имеющимся сообщениям, предотвращает дальнейшее вы-
деление [73 ] и на ней образуются включения полония [111],
которые отделяются с большим трудом. Выделение проте-
кает значительно лучше, и серебряная фольга бывает зна-
чительно чище, если раствор предварительно прокипятить
с сернистым газом [344] или если выделение вести в при-
сутствии гидразина [158].
Микроколичества полония выделяются на серебре из
растворов в разбавленной азотной кислоте при взбалтыва-
нии за 1—2 часа [204], и образующийся при этом осадок
не содержит свинпа (RaD). Источники со сравнительно
Самопроизвольное выделение полония 27
сокой поверхностной активностью (30 мкюри/см2) были по-
ВЬ'чены в результате выделения полония из раствора в 0,1 н.
азотной кислоте в присутствии небольших количеств
соляной кислоты [144]. Может оказаться полезным пред-
варительное нагревание фольги с металлическим никелем,
а также травление фольги 3 н. азотной кислотой, чтобы
свести до минимума количество находящегося на поверх-
ности сульфида серебра. Кюри и Жолио [77] рекомендо-
вали проводить выделение из раствора 0,5 н. HNOS — 0,1 н.
НС1; они считали также, что для приготовления чистого
полония необходимо применять аппарат из кварцевого
стекла и специально очищенные реагенты; однако автор
настоящего исследования установил, что при работе с мил-
лиграммовыми количествами полония применение аппарата
из кварцевого стекла не дает больших преимуществ.
Кинетику выделения микроколичеств полония на се-
ребре из разбавленной азотной кислоты изучали довольно
тщательно [23, 111, 332, 334]; скорость выделения (dxldt)
из 0,7%-ного раствора азотной кислоты можно выразить
следующим соотношением:
dxjdt=k{a—x) (s/v),
где а — начальное количество присутствующего полония,
$ — поверхность фольги, v — объем раствора и k = D/3,
где D — коэффициент диффузии ионов полония ио —
толщина диффузионного слоя на фольге [334 ]. Значение k
различно для каждого металла, используемого в качестве
подложки, однако оно, вероятно, не зависит от электрохи-
мического потенциала данного металла, хотя Жолио [199]
показывает, что вероятность выделения является функцией
этого потенциала. Процесс выделения, по-видимому, до-
вольно сложен.
В последнее время было показано, что при выделении
макроколичеств полония на серебре из раствора в разбав-
ленной азотной кислоте два атома серебра замещают один
ион полония [337 ]. Хотя и можно ожидать, что в растворе
тьп данных Условиях опыта присутствует полоний в че-
татеХВаЛеНТН0М состоянии» весьма возможно, что в резуль-
рода Рад,И0ЛИЗ? растворителя образуется перекись водо-
полоний при этом восстанавливается до двухвалент-
28
Открытие, выделение и применение полония
ного состояния. Если предположить, что полоний присут-
ствует в растворе в виде ионов Ро+ \ то электродный потен-
циал Е” Ро/Ро+ + по расчетам должен быть порядка
+ 0,7 в. При данном процессе, однако, может происходить
разрядка ионов РоО+ + . До настоящего времени еще с уве-
ренностью нельзя утверждать, какой именно ион прини-
мает участие в данном процессе.
Полоний также выделяется на серебре из растворов
в уксусной кислоте, причем выделение облегчается в при-
сутствии небольших количеств соляной кислоты [73, 74].
Выделение полония из солянокислых растворов эффек-
тивнее и протекает более гладко, чем из растворов в азот-
ной, серной и уксусной кислотах, хотя выделение на серебре
из азотнокислого раствора, как сообщают, сопровождается
меньшим поверхностным окислением серебряной фольги [73];
причина этого явления неизвестна. Большую часть поло-
ния можно извлечь из раствора в соляной кислоте, периоди-
чески очищая фольгу, чтобы ее поверхность была свежей;
общий выход в данном случае обычно достигает порядка
90% (см., например, [209]).
Выделение микроколичеств полония из солянокислых
растворов, очевидно, не зависит от концентрации кислоты,
если кислотность превышает 0,1 н. [108]. Однако скорости
выделения миллиграммовых количеств полония все же
в известной мере изменяются с изменением концентрации
кислоты (см. рис. 1). Если в растворе содержится золото,
ртуть, платина или теллур, то выделение замедляется,
и указанные примеси необходимо предварительно удалить
восстановлением гидразином в 20%-ном растворе соляной
или уксусной кислоты [158]. Согласно имеющимся сведе-
ниям, отделение от теллура лучше всего происходит из
4,1—4,3 н. солянокислых растворов [65]. Небольшие
количества ионов трехвалентного железа также замедляют
выделение, но это можно предотвратить восстановлением
железа аскорбиновой кислотой [330] или сернистым газом
[344], а также если связать его в комплексное соединение
с ионами фтора [161 ].
Выделение протекает предельно гладко при повышен-
ных температурах [108, 112] и из 0,5 н. солянокислого
раствора уже при 70—80° выделяется около 99% полония
Самопроизвольное выделение полония 29
1112]. В осадке обнаруживаются лишь следы радия-D
и радия-Е; большие количества висмута не оказывают
влияния на ход процесса [112, 113] Облегчение выделения
при повышенных температурах может быть обусловлено
Рис. 1. Скорость выделения полония на се-
ребряной фольге (размером 2 см2) из соля-
нокислого раствора.
(По данным [46].)
удалением из раствора озона, образующегося в результате
омбардировки а-частицами молекул воды или растворен-
ного кислорода, что предотвращает образование окиси или
перекиси на поверхности серебра. Миллиграммовые коли-
ества полония легко выделяются на серебряной вате,
орошке или фольге из разбавленных солянокислых рас-
чая^°В Ю «лучшие результаты получаются в тех слу-
сутст К0ГДа ВыДеление проводят из горячих растворов в при-
и ииаИИ восстановителей (гидразина или сернистого газа)
цианистоводородной кислоты, которая растворяет хло-
30
Открытие, выделение и применение полония
рид серебра, образующийся при электрохимическом за-
мещении [46].
Метод выделения на серебре можно применять при от-
делении полония от облученного висмута; вполне успеш-
ным оказалось применение покрытой серебром стеклянной
ваты, порошка и серебряной фольги [249, стр. 173].
Микроколичества полония выделяются также на се-
ребре из 0,1 н. раствора едкого кали или едкого натра;
осадок легко удаляется с серебра при обработке горячей
концентрированной щелочью. Полоний не выделяется на
серебре из аммиачных растворов [108]. Микроколичества
полония медленно выделяются на серебре из растворов
ацетилацетоната в хлороформе, однако это явление, воз-
можно, обусловлено распадом ацетилацетоната [313].
Полоний можно извлечь из серебряной фольги рас-
творением ее в азотной кислоте и осаждением серебра
в виде хлорида серебра действием соляной кислоты.
В этих условиях незначительные количества полония
адсорбируются осадком хлорида серебра. Извлечение мил-
лиграммовых количеств полония удобнее проводить, отде*
ляя его возгонкой в вакууме со свежеприготовленной фоль-
ги [10]. Крайне трудно осуществить возгонку поло-
ния, выделенного на серебре, если осадок в течение неко-
торого времени находился в контакте с воздухом или
с раствором, из которого он был извлечен. Возможно, что
это объясняется включением полония в поверхностную
окисную пленку серебра или образованием соединения
(например, полонида или полонита серебра). Аналогичное
явление, как сообщают, наблюдается в случае выделения
микроколичеств полония на золоте [2911.
Более позднее исследование процесса возгонки полония с се-
ребряной подложки показало [3501, что на поверхности фольги по-
лоний связывается пленкой AgCl. Тем не менее при нагревании та-
кой фольги в вакууме при 450° в течение 5 мин. в присутствии H2S
или Н2 можно извлечь 50% полония.
Если полоний, осажденный на серебряной фольге, нагревать
при 500° в атмосфере окиси углерода, то часть полония возгоняется
в виде металла и около 10% уносится током СО, вероятно, в виде
сравнительно устойчивого карбонильного соединения.
Определение следов полония в биологических жидко-
стях обычно предполагает в качестве последней стадии
самопроизвольное выделение полония на серебряных
Самопроизвольное выделение полония 31
дисках, которые используют в дальнейшем для подсчета
а-частиц; такое выделение обычно проводят при высоких
температурах из разбавленных солянокислых растворов
[122, 123, 330].
Медь. Некоторые ранее разработанные методы разделения
включали выделение полония на меди [84, 268, стр. 1050].
Этот процесс вполне успешно применяли при выделении
полония из старых радиевых растворов [298], однако при
этом получали продукт, загрязненный радием-Е и, воз-
м)жно, содержащий радий [17]; данный метод применяли
также при извлечении полония из солянокислых раство-
ров радиосвинца [17, 42, 70]. Выделение замедляется
ртутью, которую поэтому следует предварительно удалять
восстановлением (см. выше процесс выделения на серебре).
В осадках всегда содержится радий-Е.
Полоний можно выделить из солянокислых растворов
старых радиевых солей пропусканием этих растворов че-
рез небольшую колонку (поперечное сечение 3—4 мм2),
наполненную медным порошком с размером частиц 150—
200 меш [213]. Медный порошок предварительно промы-
вают разбавленным (1:5) раствором соляной кислоты до
тех пор, пока порошок не станет блестящим; затем его про-
мывают водой, этиловым спиртом и, наконец, эфиром. На
медном порошке также выделяется приблизительно 75%
висмута (RaE), однако часть его можно удалить повторным
промыванием разбавленным раствором соляной кислоты.
Этот метод первоначально был разработан с целью приго-
товления чистого радия для a-эталонов, но он оказался
приемлемым и для извлечения полония в отсутствие ви-
смута. Миллиграммовые количества металлического по-
лония, полученные возгонкой осадка с медной подложки,
часто оказываются загрязненными хлористой медью, не-
См?ТРя на тщательную промывку осадка перед возгонкой
UOJ. Если в сублимате содержатся соли меди, то их можно
выделить растворением сублимата в соляной кислоте и
последующим осаждением полония в виде гидроокиси
в присутствии избытка аммиака; медь остается в растворе
в виде аммиаката.
П*^на. Полоний самопроизвольно выделяется на
тине в восстановительной среде. Эрбахер [105—107]
32 Открытие, выделение и применение полония
наблюдал эффективное выделение полония и радия-Е на
насыщенной водородом платине из растворов в разбавлен-
ной соляной кислоте. В этих условиях платина служит
в качестве водородного электрода, окислительный потен-
циал которого составляет приблизительно —0,06 в. При-
сутствие окислителей, таких, как бром или азотная кислота,
отрицательно сказывается на выделении. При этом выде-
ляется весьма незначительное количество радия-D, кото-
рый может быть полностью отделен при повторном раство-
рении осадка и вторичном выделении в присутствии свинца,
служащего носителем. Выделение на платине, при котором
ионы металла не переходят в раствор, не страдает поэтому
одним из основных недостатков методов разделения, вклю-
чающих электрохимическое замещение; отработанный рас-
твор, следовательно, не нуждается в допол нител ьной очистке,
после того как из него извлечен полоний. Эрбахер считает,
что полученные таким методом осадки содержат 98%
полония, а остальное, вероятно, составляет висмут. Со-
общалось, что таким методом удалось получить источники,
содержащие 12 мкюри полония на 20 мм2 фольги, однако
Канн [203] установил, что данным методом трудно приго-
товить источники с такой поверхностной активностью.
Тем не менее этим способом получали высокоактивные
источники, содержащие 100—200 мкюри, причем выходы
были сравнимы с теми выходами, о которых сообщал Эр-
бахер [274, 276].
Полоний также выделяется на платине из раствора в 0,1 н.
соляной кислоте в присутствии хинона и гидрохинона [348];
теллуровая кислота при этом мешает. Выделение полония
на платине из разбавленного кислого раствора в отсутствие
восстановителей происходит довольно неравномерно [28,
109], что, по-видимому, объясняется наличием на платине
водорода, освобождающегося при действии а-частиц на
раствор [109].
Палладий. Полоний и радий-Е выделяются на насыщен-
ном водородом палладии из раствора в 0,1 н. соляной ки-
слоте [203]. По имеющимся сообщениям, выделение по-
лония на палладии происходит легче, чем на платине,
при гех же условиях.
Самопроизвольное выделение полония
33
Золото. Полоний выделяется на золоте из раствора
в 1 н. соляной кислоте в присутствии тиомочевины (0,9Л4);
этот метод, по-видимому, позволяет добиться успешного
отделения полония от радия-Е. В растворе указанного со-
става золото имеет окислительный потенциал, равный
примерно + 0,20 в, определяемый стабильностью комплекс-
ного соединения золота с тиомочевиной. Приведенное зна-
чение лежит между значениями потенциалов висмута и
полония при данных концентрациях, вследствие чего на
золоте выделяется только полоний [106, 107]. В отсутствие
комплексообразующего агента выделяется лишь весьма
незначительное количество полония [283, 290], и это вы-
деление, вероятно, является результатом адсорбции; обра-
зующийся осадок легко стирается с золота [70]. Количе-
ство полония, выделяющегося из разбавленного кислого
раствора на сплавах золота с медью, по имеющимся со-
общениям, пропорционально содержанию золота в сплаве;
в процессе выделения происходит окисление меди на по-
верхности сплава [332].
Никель. Микроколичества полония легко выделяются на
никеле из разбавленных солянокислых растворов; макси-
мальное выделение наблюдается для растворов с концен-
трацией соляной кислоты в пределах 0,1—1 н. [28]. В
этих условиях столь же хорошо выделяются и миллиграм-
мовые количества полония [10]. Присутствие солей никеля
не влияет на выделение микроколичеств полония, и при
указанных концентрациях кислоты количество выделен-
ного металла линейно растет со временем [28]. Эрбахер
[105, 106] наблюдал почти полное выделение полония и
радия-Е на никеле из горячих растворов в 0,1 н. НО, Со-
гласно его сообщениям, осадок не содержал радия-D, однако
Кук [70] отмечает, что некоторое количество радия-D все
же при этом выделялось. Выделение из растворов в других
кислотах (азотной, серной и уксусной) протекает менее
Удовлетворительно. Выделение из азотнокислого раствора
может сопровождаться образованием окисла на поверхно-
сти металла [28], тем не менее относительно большие ко-
личества полония можно выделить из растворов в 0,1—0,2 н.
[199]. Полоний можно отделить от никеля возгонкой
в вакууме [10, 77, 78, 222, 287, 351 ].
3 к с
Бэгнал
34
Открытие, выделение и применение полония
Висмут. Микроколичества полония медленно выделяются
на висмуте из солянокислых [231 ] или сернокислых раство-
ров [299, 3011; при этом висмут принимает бронзовую
□краску [185].
Недавно сообщалось [38] о выделении миллиграммовых
количеств полония из больших количеств облученного
висмута; метод предусматривает применение промежуточ-
ных стадий концентрирования, включающих процесс вы-
деления полония на висмуте. Облученный висмут раство-
ряют в смеси соляной и азотной кислот, и после удаления
избытка ионов нитрата полоний выделяется на порошке
висмута, взятом в количестве нескольких граммов. Процесс
повторяют несколько раз, причем всякий раз берут все
меньшие количества висмута, пока концентрация висмута
станет достаточно низкой для проведения окончательной
очистки. Выделенный продукт, как и прежде, растворяют
в кислоте, и свободный от носителя полоний получают в виде
металла, проводя осаждение хлоридом олова. Окончатель-
ную очистку проводят растворением металлического поло-
ния в азотной кислоте с последующим электрохимическим
выделением на платине. Уравнения, приводимые в рас-
сматриваемой статье, как сообщается, основаны на резуль-
татах работ, проведенных с микроколичествами полония
[126], и к ним в свете более поздних работ следует отно-
ситься с известной осторожностью.
Другие металлы. Микроколичества полония самопроиз-
вольно выделяются из разбавленных кислых растворов на
железе [84, 334], но не выделяются на хроме [334]. Неко-
торые выводы, основанные на более ранних наблюдениях
за выделением а-излучателей на иридии, родии и на некото-
рых других металлах, могут относиться и к полонию [ 185 ].
4. Новейшие методы
Экстрагирование растворителями
Растворы трибутил фосфата (ТБФ) в органических ра-
створителях можно использовать для извлечения полония
из растворов, содержащих висмут. Чистый ТБФ нельзя
применять для экстрагирования из водной среды, поскольку
его плотность очень близка к плотности воды; по этой при-
Экстрагирование полония
35
чине ТБФ следует разбавить несмешивающимся с водой
растворителем, обладающим более низкой плотностью.
Опубликованы данные, согласно которым коэффициент
разделения, равный ПО в пользу органической фазы, до-
стигнут при экстрагировании полония из раствора в 6 н.
Р и с. 2. Зависимость коэффициента распре-
деления полония в количествах порядка не-
скольких милликюри от концентрации соля-
ной кислоты при экстрагировании 10об.%-ным
раствором ТБФ в декалине при 22°.
(По данным [15].)
соляной кислоте 20%-ным раствором ТБФ в бутиловом
эфире [210]. Некоторое количество висмута, перешедшее
| органическую фазу, удаляется при обратной промывке
с н’ соляной кислотой; при экстрагировании висмута ки-
кра^°И Указанн°й концентрации коэффициент разделения
каютНеббЛаГ0»Риятен для в°Дн°й Ф^ы. Полоний извле-
рован° »атн°й промывкой органической фазы концентри-
н°и азотной кислотой. Трибутил фосфат извлекает
3*
36
Открытие, выделение и применение полония
полоний также из растворов, содержащих серную кислоту
и сульфат закисного железа [247].
Экстрагирование из солянокислого раствора в значи-
тельной мере зависит от концентрации кислоты в водной
фазе (рис. 2) и достигает максимума для растворов, в ко-
торых концентрация НС1 лежит в пределах 7—9 н. [15].
Учитывая, что из разбавленных кислых растворов полоний
экстрагируется в весьма незначительной степени, извле-
чение его из органической фазы можно проводить, промывая
ее 0,5—1 н. соляной кислотой.
Декалин, по-видимому, является наилучшим раствори-
телем для разбавления ТБФ, поскольку дибутиловый эфир
довольно легко окисляется при интенсивном а-облучении.
Опыты по изучению экстрагирования полония в количе-
ствах порядка милликюри не дали воспроизводимых ре-
зультатов вследствие окисления, вызываемого действием
облучения. Экстрагируемый комплекс, вероятно, пред-
ставляет собой РоС14-2ТБФ, и константа равновесия для
реакции
РоС14 + 2ТБФ РоС14 • 2ТБФ
при комнатной температуре равна 0,04 [15].
Растворы дитизона в хлороформе применяли для экстра-
гирования микроколичеств полония из кислых растворов
радиосвинца [32, 194, 195], при этом около 95% полония
экстрагировалось из азотнокислых и солянокислых раство-
ров, имеющих pH в пределах 0,2—5 [32]. При экстрагиро-
вании «весомых» количеств полония из растворов невысокой
кислотности (pH > 2) получаются крайне невоспроизво-
димые результаты, что, по-видимому, обусловлено гидро-
лизом соединения полония, присутствующего в растворе.
Микроколичества полония можно, очевидно, извлечь из
органической фазы промыванием 4 н. соляной кислотой.
Для извлечения микроколичеств полония из аммиачных
растворов цианистого калия или из растворов цитрата ам-
мония можно применять также дитизон [195]; этот реак-
тив применяли для отделения радиохимически чистого
радия-Е от радиосвинца [21 ]. Кривая, показывающая
зависимость количества экстрагируемого полония (порядка
милликюри) от концентрации соляной кислоты (рис. 3),
несколько отличается от кривой, полученной для микро-
Экстрагирование полония
37
количеств полония; результаты не являются вполне воспро-
изводимыми из-за быстрого радиационного разложения
дитизона [15].
Купферрон в растворе амилацетата, по имеющимся
сведениям, экстрагирует микроколичества полония из 3 н.
растворов минеральных кислот в присутствии сернистой
кислоты [226]. Полоний можно отделить от висмута
Рис. 3. Зависимость степени извлечения полония
в количествах порядка нескольких милликюри от
концентрации соляной кислоты при экстрагировании
растворами дитизона в хлороформе.
(По данным [15].)
экстрагированием 0,25 М раствором теноилтрифторацетона
(ТТА) в бензоле из водных растворов, имеющих pH в пре-
делах 0—2. Почти полный переход в органическую фазу
происходит при pH 2. Висмут экстрагируется в пределах
pH 1—3, а свинец — в пределах pH 3—5 [145]. Микроко-
личества полония экстрагируются также из водных раство-
ров Диизопропилкетоном [68] и окисью мезитила [236].
Почти полное экстрагирование миллиграммовых количеств
полония из солянокислого раствора достигается при об-
работке раствора ацетил ацетоном и метил изобутил кетоном
«гексоном), что, вероятно, обусловлено образованием ста-
бильного соединения с экстрагентом [12]. Полоний из рас-
твора в этих кетонах извлекается с трудом.
ни п. работе [3521 изучено экстрагирование микроколичеств поло-
стей 0 И3 азотн°киелых растворов (1,75—8 н. HNO3) в присут-
Други ВОсстановителей, окислителей, комплексообразователей и
38
Открытие, выделение и применение полония
Полоний-210 в четырехвалентной форме не экстрагируется
эфиром, не содержащим восстановителей.
При применении эфира, содержащего перекись водорода, а
также при добавлении к раствору перекиси водорода, или несколь-
ких миллиграммов гидразина или гидроксиламина, или после про-
пускания в раствор SO2 часть полония переходит в органическую
фазу, вероятно, вследствие восстановления четырехвалептного по-
лония в двухвалентный; при пропускании SO2 в концентрированные
растворы HNO3 экстрагирование не происходит, по-видимому,
вследствие окисления восстановителя азотной кислотой.
В присутствии сильных окислителей эфир не экстрагирует по-
лония. Наличие комплексообразующих веществ ухудшает экстраги-
рование. При добавлении щавелевой кислоты полоний эфиром не
экстрагируется. Диизопропиловый эфир и изоамиловый спирт в при-
сутствии восстановителей экстрагируют полоний, но в меньшей
степени. н-Амиловый спирт и метилизобутилкетон экстрагируют по-
лоний без добавления восстановителя, но имеются данные, что
при проведении опытов в указанных растворах присутствовали вос-
становители. Безкислородные растворители (CCU, СНС1з) ни при
каких условиях не экстрагируют полоний из азотнокислых рас-
творов.
Ионный обмен
Полоний можно отделить от висмута (радия-Е) погло-
щением на ионообменной смоле дауэкс-50 (50—100 меш)
из растворов в 0,1—0,3 н. соляной кислоте с последующим
элюированием висмута 2 н. азотной кислотой и полония 2 н.
соляной кислотой [282]. Разделение можно также произ-
вести на ионообменной смоле IR-1 (40—60 меш), из кото-
рой висмут можно элюировать 3 н. азотной кислотой [39].
Следы полония можно отделить от других элементов
подгруппы селена на смоле дауэкс IX-4 (менее 180 меш)
поглощением из раствора в 12 н. соляной кислоте с после-
дующим элюированием селена 6 н. соляной кислотой, тел-
лура — 2 н. соляной кислотой и полония — 1 н. азотной
кислотой. Полоний можно также элюировать 2 н. хлорной
кислотой, однако если полоний простоит в колонке 50 час.,
то лишь 10% его элюируется хлорной кислотой, тогда как
3 н. азотная кислота продолжает действовать в этом случае
вполне эффективно [327].
При выделении миллиграммовых количеств полония на
ионообменных смолах могут возникнуть затруднения, свя-
занные с воздействием a-излучения. Газы, выделяющиеся
в результате радиолиза растворителя, образуют воздушные
пробки; это затруднение можно частично преодолеть, если
Химическое выделение полония
39
держать колонку в горизонтальном положении или под-
водить раствор и растворитель в колонку снизу.
Хроматография на бумаге
Опубликовано сообщение [120] о выделении микроко-
личеств радия-D, радия-Е и радия-F из смеси бутилового
спирта, пиридина, соляной и уксусной кислот. Выделение
из смеси бутилового спирта концентрированной соляной
и концентрированной серной кислот протекает удовлетво-
рительно лишь в присутствии свинца и висмута в каче-
стве носителей.
В работе [353] описан метод полного хроматографического раз-
деления на бумаге четверной смеси радиоизотопов Se75. Те127,129,
Bi210 и Ро210, а в работах [354—356] — различные варианты разде-
ления смеси Ra-D, Е, F.
Методы химического выделения
полония
Выше уже были описаны методы отделения полония от
других элементов подгруппы селена, основанные на ионном
обмене; существуют еще два метода, основанные на хими-
ческих приемах, разработанных для анализа селената по-
лония [48].
Первый из них основан на окислении серы, селена и
теллура до шестивалентного состояния. Смесь после оки-
сления обрабатывают концентрированной щелочью и от-
фильтровывают осаждающуюся «гидроокись» полония. Вод-
ная фаза содержит всю серу и весь селен, а также большую
часть теллура; некоторое количество теллура, попавшее
в осадок, можно удалить растворением осадка в разбавлен-
ной соляной кислоте и осаждением гидразином. Второй
метод основан на восстановлении серы и селена до газо-
образных гидридов смесью фосфорноватистой кислоты и
концентрированных соляной и йодистоводородной кислот
при нагревании с обратным холодильником; в этих усло-
виях полоний и теллур осаждаются в элементарном состоя-
нии, причем теллур можно выделить из осадка описанным
выше методом.
Осаждение сульфида полония из раствора в разбавленной
соляной кислоте обусловливает возможность отделения его
от большого, числа элементов. Осадок сложного состава
40
Открытие, выделение и применение полония
отмывают сернистым аммонием для удаления мышьяка,
сурьмы, олова и теллура, а затем нагревают в вакууме
до 500°. В этих условиях возгоняются только полоний и
висмут; полоний можно отделить от висмута, воспользо-
вавшись самопроизвольным выделением полония на се-
ребряной фольге.
ПРИМЕНЕНИЕ ПОЛОНИЯ
Включение полония в состав стандартных электродных
сплавов, применяемых для изготовления свечей двигателей
внутреннего сгорания, облегчает, по имеющимся сведе-
ниям, зажигание свечей в холодном состоянии [94]. Это,
возможно, обусловлено понижением напряжения искро-
образования под действием излучаемых полонием а-ча-
стиц, однако небольшие количества полония, используе-
мые с этой целью, оказывают, по-видимому, весьма незна-
чительное влияние в направлении улучшения качества
свечей [101]. Кроме того, полоний, находящийся на по-
верхности электродов свечей (где только и можно ожидать
от него полезного действия, если учесть небольшой пробег
а-частиц), будет почти наверное испаряться при температу-
рах работы свечей, а выделение полония в течение длитель-
ного времени с отработанными газами в местах скопления
людей не может быть допущено. Электродные сплавы приго-
товляют добавлением никелевой фольги с осадком полония к
расплаву, используемому при изготовлении электродов [95].
Недавно сообщалось о применении полония в промыш-
ленности для снятия статических зарядов. Однако было
обнаружено, что при этом происходит радиоактивное за-
грязнение в результате недостаточной изоляции полония
[30, 326]. При более строгом соблюдении надлежащих
условий изготовления и применения источников масштабы
такого рода использования полония могут возрасти. По-
лоний также использовали для метеорологических наблю-
дений за электрическим потенциалом воздуха [197].
Сравнительно большие радиоактивные источники
(160 мкюри) можно применять для определения микроко-
личеств некоторых элементов путем а-активационного ана-
лиза [252, 357 ]. Так, например, не превышающие 1 мг
количества фтора и алюминия можно определить по най-
Применение полония
41
денному методом счета количеству Na22 и Рзв образующихся
по реакции
F19 (а, п) Na22 и А127 (а, п) Р30.
Этот метод менее чувствителен по сравнению с методом акти-
вации нейтронами, и только несколько элементов могут
быть определены таким образом.
Основное применение полоний находит в области ядер-
ной физики, в частности для получения не обладающих
у-излучением источников нейтронов. С этой целью полоний
сплавляют с одним из элементов, который имеет изотопы
с высоким поперечным сечением а, и-реакции; для этого
чаще всего используют бериллий. Сплав полония с бе-
риллием можно получить, используя самопроизвольное
выделение полония на бериллиевых дисках, однако изго-
товленные таким образом источники дают, как правило,
невысокий выход нейтронов; его можно увеличить спека-
нием продукта. Лучшие результаты получаются также,
по-видимому, при нанесении полония на бериллий мето-
дом возгонки [249].
В работе [394] приведен метод изготовления полониевых ис-
точников на основе неорганических эмалей.
Небольшие количества полония использовали при ис-
следовательских работах в области радиационной химии
[3, 4, 71, 223 и многие другие]. Было высказано предполо-
жение, что следы полония катализируют разложение пе-
рекиси водорода [29], хотя эта реакция могла быть обу-
словлена наличием примесей в используемых растворах [4].
В работе [358] описан прибор для исследования радиационно-
химических процессов в жидкостях под действием а-излучения.
Источником излучения служит Ро-210, нанесенный возгонкой на
танталовую подложку. Источник заключен в латунный корпус
со слюдяным окошком (~ 1 мг/см2) и непрерывно продувается то-
ком гелия под давлением 0,35 ат, так как поглощение а-частиц в ге-
лии в шесть раз меньше, чем в воздухе. Исследуемую жидкость по-
мещают в стеклянную ячейку, также имеющую слюдяное окошко
Для впуска а-частиц.
Полоний находит также широкое применение в работах
по изучению физиологического влияния a-излучения на
живые организмы [3971. Обзор ранних работ в этой области
Дается в справочнике Гмелина [126],
2
химия микроколичеств полония
В РАСТВОРАХ
Реакции с кислотами, нейтральными солями и восстановителями.
Диффузия в водных растворах. Свойства щелочных растворов. Гид-
ролиз. Образование радиоколлоидов.
Существует обширная литература, посвященная работам
по изучению химического поведения полония в растворах,
проведенным на микроколичествах полония, однако ввиду
отсутствия аналитических данных выводы, сделанные на
основании полученных результатов, часто носят спекуля-
тивный характер. В более ранний период исследователи
в этой области, по-видимому, редко обращали внимание
на химическое сходство полония с теллуром, ближайшим
гомологом полония, и сосредоточивали основное внимание
на более слабо выраженном его сходстве с висмутом, со-
седним с полонием элементом в периодической системе.
Основные методы, применяемые в работах по изучению
химических свойств полония в микроконцентрациях, вклю-
чают соосаждение, центрифугирование и наблюдение за
изменением критического потенциала выделения полония
при добавлении различных реагентов. В последнем случае
заметные изменения потенциала могут быть обусловлены
восстановлением полония до более низкой валентности или
осаждением его в виде нерастворимого соединения, и по-
этому данный метод не требует дальнейших объяснений.
Центрифугирование в отсутствие носителя позволяет по-
лучить некоторое представление об образовании нераство-
римых соединений полония (см. свойства радиоактивных
коллоидов, стр. 49—53), в частности об образовании про-
дуктов гидролиза солей полония при низких концентра-
циях кислоты.
Метод совместного осаждения заключается в добавле-
нии осадителя к раствору, содержащему микроколичества
полония и элемент, соединение которого соосаждается и
выполняет роль носителя. В качестве носителя выбирают
элемент, химические свойства которого аналогичны свой?
Реакции с кислотами и нейтральными солями
43
ствам полония (следует отметить, что до сих пор недо-
статочно ясно, почему теллур не применяют с этой
пелью). Увлечение полония осадком, которое обнаружи-
вают, определяя a-активность осадка и раствора, можно
объяснить образованием полонием соединения, изоморф-
ного соединению, образуемому носителем (изоморфное
включение). В этом случае оказывается применимым закон
распределения Бертло—Нернста, и отношение концентра-
ции микроколичеств полония в твердой фазе к его концен-
трации в растворе должно быть постоянным при изменении
количества носителя. Данный вопрос детально рассмотрен
Валем и Боннером [341, стр. 104].
Подробные обзоры исследований, проведенных на микро-
количествах полония, даны в справочнике Гмелина [126],
а также в работах Гайсинского [161, 162] и автора настоя-
щей книги [9, 48]. Ряд таблиц (от 6А до 6F), напечатанных
в работе Валя и Боннера [341 ], чрезвычайно удобны для
быстрого нахождения справочных данных.
РЕАКЦИИ С КИСЛОТАМИ И НЕЙТРАЛЬНЫМИ СОЛЯМИ
Работы, проведенные на микроколичествах полония по
изучению его хлоридов, сульфатов и нитратов, описаны
в последующих главах. В следующих разделах данной
главы дается краткий обзор других работ, относящихся
к отдельным изучавшимся реагентам.
Одна из наиболее интересных особенностей полония
заключается в том, что этот элемент образует комплексные
ионы почти в любой кислой среде. Предполагают, что эти
ионы образуются из катионов полония и кислотных ани-
онов, но могут также содержать и молекулы воды [168].
Щавелевая кислота. Гийо и Гайсинский [132, 133], а также
Жолио [198, 199] наблюдали заметное изменение в значе-
нии критического потенциала выделения микроколичеств
полония в присутствии щавелевой кислоты и причину этого
явления приписали восстановлению полония до более низ-
кого валентного состояния. Однако более вероятно, что
эти результаты обусловлены образованием комплексных
ионов [167], а опыты по изучению электролиза [152] по-
казали, что почти весь полоний в присутствии щавелевой
44
Химия микроколичеств полония в растворах
кислоты перемещается к аноду. Жолио [198] наблюдал,
что анодное выделение полония подавляется щавелевой
кислотой, однако механизм этого процесса остается не-
ясным. Следует отметить, что микроколичества полония
растворимы в нейтральном растворе, содержащем окса-
лат-ионы, и при этом не наблюдается признаков гидролиза
[137], что также свидетельствует об образовании оксалат-
ных комплексов. Сервинь, чтобы объяснить результаты
своих работ по изучению диффузии, постулировал суще-
ствование в растворе однозаряженного комплексного иона
[Ро|,,(С2О4)2, (Н2О)2]“, однако более вероятно, что в усло-
виях этих опытов полоний находился в четырехвалентном
состоянии.
Работы по изучению совместной кристаллизации с окса-
латами лантана, иттрия и скандия показали, по-видимому,
существование трехвалентного полония [310, 311], но этот
вывод вряд ли следует считать правильным в свете более
поздних работ с миллиграммовыми количествами полония,
в результате которых было установлено наличие у него
лишь двухвалентного и четырехвалентного состояний.
Фосфорная кислота. Работы по электрохимическому выде-
лению микроколичеств полония показывают, что в фосфор-
нокислых растворах образуются комплексные ионы этого
элемента [154, 1381.
Железисто- и железосинеродистый калий. Каждая из
этих двух солей подавляет электрохимическое выделение
микроколичеств полония из растворов, в которых присут-
ствуют указанные соли; такое влияние, возможно, является
результатом образования нерастворимых соединений по-
лония [138].
ДЕЙСТВИЕ ВОССТАНОВИТЕЛЕЙ
Хлористое олово. Под действием хлористого олова полоний
немедленно осаждается из растворов [232, 134], по-види-
мому, в металлическом состоянии [133, 134, 38].
Сернистый газ. Из раствора в соляной [235] или плавико-
вой [285] кислоте микроколичества полония осаждаются
вместе с селеном и теллуром при действии сернистого газа
Действие восстановителей
45
или сульфита; однако при восстановлении в концентриро-
ванном солянокислом растворе полоний с теллуром не
осаждается [204]. Гайсинский [160] считает, что в про-
цессе восстановления осаждение происходит в результате
адсорбции полония на тончайшем осадке селена (см. гл. I).
Из радиографических данных следует, что сернистый
газ осаждает полоний из сернокислых растворов, но не
осаждает из растворов в соляной кислоте [24]. Природа
реакции, протекающей в первом случае, неизвестна: осаж-
дения не наблюдалось при работе с миллиграммовыми ко-
личествами полония (см. гл. 6). Сернистый газ тормозит
электрохимическое выделение микроколичеств полония из
сернокислых растворов [198, 199]; это, вероятно, происхо-
дит из-за катодного восстановления сернистой кислоты до
дитионовой, которая затем восстанавливает полоний до
металла [133].
Гидразин, В концентрированных растворах соляной ки-
слоты полоний не восстанавливается гидразином до металла
даже при температуре 100° [235]. Критический потенциал
выделения полония заметно изменяется в присутствии ги-
дразина, и это изменение объясняли восстановлением
полония до трехвалентного состояния [167]; недавно про-
веденные работы показывают, однако, что полоний восста-
навливается в данном случае до двухвалентного состоя-
ния [11, 12]. В уксуснокислых холодных растворах гидра-
зин восстанавливает полоний до более низкого валентного
состояния [167], тогда как в растворах серной кислоты вос-
становление происходит при температуре кипения [132,
167]. Имеются сообщения о том, что из холодных щелоч-
ных растворов гидразин осаждает полоний в виде металла
[ 133 ], однако осадок может состоять из гидроокиси двух-
валентного полония.
Восстановление гидразином в солянокислом растворе
средней концентрации применяли для отделения микроко-
личесгв полония от теллура [234], а также от золота,
Ртути, платины и теллура [158]. Осаждение микроколи-
честв полония с селеном из раствора в плавиковой кислоте
При восстановлении гидразином [335], возможно, обуслов-
лено адсорбцией полония на выпадающем осадке селена
1160] или образованием селенида полония.
46
Химия микроколичеств полония в растворах
Другие восстановители. Имеются сообщения, что металли-
ческий полоний осаждается из кислых холодных растворов
его солей при действии трехвалентного титана [133] и
фосфорноватистой кислоты [132, 133, 167]. Дитионит
натрия немедленно осаждает микроколичества полония из
кислых растворов, причем полоний, по-видимому, осаж-
дается в виде металла [132, 133, 167]; присутствие дитио-
нита замедляет электрохимическое выделение полония
[133, 167]. Согласно имеющимся сообщениям, восстанов-
ление дитионитом в щелочном растворе, проводимое в атмо-
сфере водорода, приводит к образованию полонида [65,
305].
Электрохимическое выделение полония в присутствии
перекиси водорода может привести к окислению полония
до неустойчивого более высокого валентного состояния,
которое затем переходит в валентное состояние, более
низкое по сравнению с исходным (ср. с хромом, церием,
марганцем) [167]. Реакция с перекисью водорода, по-ви-
димому, протекает легче в уксусной кислоте, чем в серной
кислоте.
Согласно имеющимся данным [132, 167], гидроксиламин
восстанавливает микроколичества полония до более низ-
кого валентного состояния в уксуснокислых или серно-
кислых растворах при температуре кипения. Муравьиная
кислота и формальдегид не восстанавливают полония в
кислых растворах, но в щелочных растворах формальдегид
восстанавливает микроколичества полония до металла [132,
133, 167]. Сульфат двухвалентного железа не восстанавли-
вает четырехвалентного полония в растворах [178].
Незначительные количества нитрита натрия не реаги-
руют с полонием в разбавленном (0,1 н.) растворе азотной
кислоты, однако более концентрированные растворы ни-
трита (0,2—4 н. растворы нитрита натрия), по имеющимся
данным, осаждают большую часть присутствующего в рас-
творе полония [132, 167]. Природа осадка остается не-
известной. Нитриты применяли при разложении тетра-
йодида полония для анализа [13] без осаждения полония.
Работы по электрохимическому выделению полония
в растворе винной кислоты показали, что восстановление
полония до более низких валентных состояний сопрово-
ждается образованием комплексных ионов [148, 152, 154],
Осаждение из щелочных растворов
47
а радиографическое исследование указывает на то, что
в данном случае образуется нерастворимое соедине-
ние [24].
Влияние различных восстановителей на значение элек-
тродного потенциала полония можно в значительной мере
приписать образованию комплексных ионов [167]. Это
можно выяснить лишь после того, как будут выполнены
более обстоятельные работы по изучению такого рода си-
стем, причем желательно с миллиграммовыми количествами
полония.
ДИФФУЗИЯ В ВОДНЫХ РАСТВОРАХ
Некоторые из опубликованных данных, характеризующих
диффузию микроколичеств полония в растворах, приве-
дены в табл. 1.
Таблица 1
Диффузия микроколичеств полония
в различных растворах
Среда D, см2/сутки Литература
НС1 0,001н. 0,31 [260]
НС1 0,1н. 0,78 [314, 315]
HNO3 0,1н. 0,80 [314, 315]
H2SO4 0,1н. 0,47 [314, 315]
(СООН 2 0,1н. 1,06 [314, 315]
NH4OH 0,5н. 0,19а [175]
а Приведенное в таблице значение, вероятно, за-
вышено вследствие адсорбции части полония на стен-
ках сосуда, в котором он находился.
ОСАЖДЕНИЕ ИЗ ЩЕЛОЧНЫХ РАСТВОРОВ
Осаждение микроколичеств полония с нерастворимыми
гидроокисями довольно тщательно изучали Кюри и Дебьерн
\*1. Если в растворе присутствует висмут и теллур, то
Добавление щелочи приводит к тому, что первой осаждается
иДроокись висмута, затем полоний и, наконец, непосред-
48
Химия микроколичеств полония в растворах
ственно перед достижением нейтральной точки — теллу-
ристая кислота. В известных пределах можно произвести
отделение микроколичеств полония от гидроокиси висмута
обработкой осадка 2 н. раствором щелочи.
Гидроокись трехвалентного железа является лучшим
носителем для полония, чем гидроокись висмута. Работы
по изучению осаждения микроколичесгв полония показали,
что в случае осаждения его с этими гидроокисями имеет место
адсорбция [108, 114, 115]. Осаждение микроколичеств
полония на гидроокиси висмута в присутствии аммиака
происходит почти количественно [83].
Имеются сведения, что микроколичества полония не-
растворимы в водном растворе аммиака [235] и не выде-
ляются на серебре из суспензий в водном растворе аммиака
[108], в то время как они легко выделяются из раство-
ров в едком натре. Растворимость микроколичеств поло-
ния в щелочной среде достигает, по-видимому, минималь-
ного значения в случае 0,1 н. аммиачного раствора или
0,007 н. раствора едкого натра [63].
ГИДРОЛИЗ СОЛЕЙ ПОЛОНИЯ в ВОДНЫХ РАСТВОРАХ
Полоний можно отделить от висмута фракционным гидро-
лизом растворов нитратов полония и висмута, причем оса-
док, выпадающий вначале, содержит основную часть по-
лония [83]. Этот осадок зачастую имеет желтую или крас-
ную окраску и нерастворим в разбавленных или концен-
трированных кислотах.
Опыты по центрифугированию показали, что полоний
не гидролизуется в 1 н. растворе соляной кислоты. Гидро-
лиз происходит в значительной степени в 0,01 н. растворе
соляной кислоты [59, 135] и осаждение микроколичеств
полония из нейтрального раствора, по-видимому, должно
заметно усиливаться при добавлении нитратов серебра или
свинца [62]. Состав этих осадков неизвестен. Жолио [199]
наблюдал осаждение микроколичеств полония из раствора
в разбавленной азотной кислоте (менее чем 0,1 н.); макси-
мальное осаждение достигается при концентрации азот-
ной кислоты в пределах 2—4,7-10“5 н. [209]. Доля поло-
ния, осаждающегося при центрифугировании из разбавлен-
ного азотнокислого раствора, возрастает с продолжитель-
Образование радиоколлоидов 49
ностью выдерживания раствора и, по-видимому, умень-
шается при увеличении концентрации полония в раство-
рах 0,05 н. кислоты, тогда как обратная зависимость имеет
место для растворов полония в более разбавленной кис-
лоте [60]; причина этого явления неясна. Однако если
стенки сосуда, в котором находится раствор, покрыть
парафином, то очень малые количества полония можно
выделить центрифугированием из раствора в 6-10“4 н.
азотной кислоте [295], а это свидетельствует о влиянии на
осаждение полония следов двуокиси кремния, попадающей
в раствор из стенок сосуда. Опубликованы сообщения об
изучении радиографическим методом адсорбции полония
из раствора на парафине [237].
Микроколичества полония очень легко адсорбируются
на стекле из растворов в разбавленных кислотах (см., напри-
мер, [73]), и поэтому крайне трудно удалить адсорбиро-
ванный полоний промыванием кислотой [224]. В соответ-
ствии с данными Старика [319 ], адсорбция наиболее заметна
в случае растворов в 0,1 н. кислоте и минимальна вблизи
точки нейтрализации; возможно, что это является резуль-
татом образования растворимого полонита.
Легкость, с которой микроколичества полония можно
выделить центрифугированием из раствора в разбавленной
кислоте, может означать, что гидролизованные соли поло-
ния присутствуют в виде тончайшей суспензии [135, 156].
Если это действительно так, то растворимость участвую-
щих в данном процессе соединений полония должна быть
чрезвычайно малой и загрязнения (пыль, двуокись крем-
ния и т. д.), попадающие в раствор, вероятно, действуют
как носители и центры адсорбции [257, 260, 266, стр. 55,
318, 209, 159].
ОБРАЗОВАНИЕ РАДИО КОЛЛОИДОВ
При изучении методов приготовления источников полония
из растворов радиосвинца Панет [255] обнаружил, что
микроколичества полония в нейтральном растворе не диф-
фундируют через пергаментную мембрану. В последующих
работах [256, 257, 259, 260] было установлено, что в ней-
тральных, слабокислых или щелочных растворах поло-
ний, видимо, находится в коллоидальном состоянии, при-
К. Бэгнал
50
Химия мйкрокбличеств полония 6 растворах
чем коллоидные частицы имеют лишь молекулярные раз’
меры. Вскоре после этого Годлевский [127] показал, что
частицы полония (Ra-A, Ро218) в этих условиях могут ад-
сорбировать заряженные ионы и увеличивать свой заряд,
что характерно для истинных коллоидов. Однако, по-
скольку не было достоверно известно, что в данном случае
имели дело с истинными коллоидами, термин «радиокол-
лоид» стали применять по отношению к элементам, подоб-
ным полонию и висмуту, которые в указанных усло-
виях при микроконцентрациях проявляют свойства колло-
идов.
На основании химических данных Панет высказал пред-
положение, что образование полониевых коллоидов может
быть обусловлено присутствием крайне нерастворимых
продуктов гидролиза, образующихся из присутствующих
в растворе солей полония. Весьма знаменательно, что наи-
более заметную способность к образованию радиоколлои-
дов обнаруживают элементы, которые при гидролизе дают
нерастворимые гидроокиси или основные соли. Слабая
сторона такого толкования заключается в том, что произве-
дение растворимости соединения полония, образующегося
в результате гидролиза, вообще нельзя превзойти в рабо-
тах с микроколичествами полония, при которых отмеча-
лось образование радиоколлоидов, так как количество
присутствующего полония весьма незначительно. Для пре-
одоления этого затруднения воспользовались предположе-
нием [260], что полоний существует в растворе в ионной
форме и что ионы полония адсорбируются другими кол-
лоидами, следы которых присутствуют в данном растворе.
Эти посторонние коллоиды предположительно состоят из
кремневой кислоты, образующейся в результате химиче-
ского или радиационного воздействия на стекло сосуда,
а также из частиц пыли или даже примесей присутствую-
щих в первоначальном растворе, которые не были удалены
при химической обработке раствора.
Имеются многочисленные подтверждения того, что при-
меси оказывают заметное влияние на образование радио-
коллоидов; было установлено, что тщательная очистка
перед опытом применявшихся реагентов, особенно путем
центрифугирования, заметно снижает количество полония,
осаждающегося при последующем центрифугировании; пол-
Образование радиоколлоидов
51
ностью предотвратить образование радиоколлоидов никогда
не удавалось. Образование полониевых коллоидов также,
по-видимому, заметно задерживается при покрытии пара-
фином внутренних стенок сосудов, в которых находится
раствор [295]; это наблюдение, как видно, подтверждает
теорию о роли посторонних коллоидов. В отсутствие пре-
дохраняющего покрытия были вновь получены такие же
результаты, как и в более ранних работах (см., например,
[62]).
Литература, посвященная коллоидным свойствам ми-
кроколичеств полония, весьма обширна (см.,, например,
[52, 146, 2591); подтверждение способности полония обра-
зовывать коллоиды получено методами седиментации и
центрифугирования [215, 621, диффузии [260, 174, 176],
диализа [153, 256, 257, 259, 260, 268, стр. 1049] и радио-
графии [24, 36, 51—56, 64].
Метод радиографии применяли также при исследова-
ниях радиоколлоидных свойств микроколичеств висмута
(ThC) [1471.
Радиографические наблюдения производили следующим
образом: каплю раствора, содержащего полоний, наносили
на тонкий листок слюды, помещенный на фотографическую
пластинку (листок слюды должен быть достаточно тонким,
чтобы через него могли проходить а-частицы, образую-
щиеся при радиоактивном распаде). Если фотографическая
пластинка не оказывалась равномерно почерневшей под
действием a-излучения, то предполагали, что полоний
присутствует в растворе в виде радиоколлоида. Были
получены экспериментальные данные, свидетельствующие
о том, что описанный метод не является вполне надежным,
поскольку незначительные количества полония, присут-
ствующие в растворе, могут адсорбироваться на слюде или
на других материалах, применяемых для экранирования
фотографической пластинки [172, 328]. Имеются также
сообщения, согласно которым в процессе электрохимиче-
ского выделения образуются агрегаты атомов полония [219 ];
в связи с этим высказывалось предположение, что пер-
вые атомы полония служат зародышами для дальней-
шего выделения. Радиографическим методом было также
подтверждено существование агрегатов в осадках возогнан-
ного полония [6, 171 ].
52
Химия микроколичеств полония в растворах
По имеющимся сообщениям, нейтральные, слабощелоч-
ные или слабокислые растворы микроколичеств полония
обладают в известной мере способностью расслаиваться
при стоянии, причем более тяжелые частицы суспензии
оседают [215], однако это наблюдение требует проверки
[318].
Таким образом, имеются две теории, объясняющие об-
разование радиоколлоидов — теория коллоидов, соглас-
но которой это явление обусловлено образованием
истинных коллоидов, и теория адсорбции, которая данное
явление рассматривает как результат адсорбции ионов
полония на других коллоидах, присутствующих в растворе.
Трудно, конечно, совместить представление об образова-
нии истинных коллоидных суспензий в столь сильно раз-
бавленных растворах с тем фактом, что произведение ра-
створимости рассматриваемых соединений полония почти
наверное не достигается в этих растворах.
Обстоятельный обзор по данному вопросу читатель най-
дет в специальной литературе [126, 159, 181, 246 и 316].
В работах советских радиохимиков [359—370] приведены экс-
периментальные данные, подтверждающие самостоятельное сущест-
вование полония в коллоидном состоянии в очень разбавленных
растворах (10“9—10~13 М). Показано, что образование коллоидов
радиоактивных элементов не зависит от специфических свойств,
связанных с радиоактивной природой этих элементов [361].
Эти выводы основаны на результатах опытов по электрохими-
ческому выделению полония на меди при различных концентра-
циях полония и pH раствора, на изучении адсорбции и десорбции
на стекле различного состава, на применении методов центрифуги-
рования, ультрафильтрования и радиографии.
В кислых, слабокислых, нейтральных, слабощелочных и ще-
лочных средах полоний существует в различных состояниях. При
pH 1—4 полоний находится в ионном негидролизованном состоянии;
при pH 6—7 начинается гидролиз и образование положительно
заряженных коллоидных частиц; при pH 8—13 образуются нераст-
воримые соединения и устанавливается равновесие с ионами поло-
ния в растворе.
Существование собственно радиоколлоидов полония подтверж-
дается склонностью его солей к гидролизу и очень малой величиной
произведения растворимости его гидроокиси.
Приведены данные, указывающие на существование в коллоид-
ных растворах полония двух групп частиц (порядка нескольких
десятков микронов и около 1 р.) и высказывается предполо-
жение, что грубодисперсные частицы представляют собой радиоак-
тивный элемент, адсорбированный на загрязнении, а мелкодисперс-
Образование радиоколлоидов 53
ные частицы образованы самим радиоактивным элементом. Этим са-
мым как бы сглаживаются противоречия двух точек зрения [359].
В работе [363] приведены данные в пользу высказанного ранее
предположения [328], что результаты радиографического изучения
полониевых растворов, нанесенных непосредственно на фотоэмуль-
сию или поверхность желатины, стекла, слюды, позволяют судить
не о природе ионов полония, существующих в растворе, а о харак-
тере адсорбции его ионов на этих поверхностях. Все эти вопросы
очень обстоятельно изложены в монографии И. Е. Старика «Основы
радиохимии».
ТЕХНИКА РАБОТЫ С ПОЛОНИЕМ
Вредность для здоровья. Практика лабораторной работы с полонием
в количествах порядка нескольких кюри. Оборудование и работа
в защитных камерах с перчатками. Агрегатная отдача. Действие
излучения и химические аспекты работы в условиях высокой радио-
активности.
ВРЕДНОСТЬ ДЛЯ ЗДОРОВЬЯ
Возможность получения сравнительно больших (милли-
граммовых) количеств полония в результате нейтронного
облучения висмута облегчила работу в области химии по-
лония: однако из-за высокой удельной активности Ро210 —
изотопа полония, применяемого во всех последних работах
по изучению данного элемента, возникает ряд трудностей,
преодоление которых требует применения специальных
методов и оборудования.
С источниками небольшой активности порядка несколь-
ких милликюри в виде растворов можно работать при со-
блюдении необходимых мер предосторюжности под вытяж-
ными колпаками или в вытяжном шкафу с надлежащей
тягой. С сухими источниками, даже если активность их
составляет всего несколько милликюри, не рекомендуется
работать иначе как в закрытых камерах. Учитывая, что
максимально допустимое количество Ро210, которое
можно ввести в организм человека, составляет всего
лишь 0,02 ркюри (4,5-10“12 г), во всех случаях, когда это
возможно, надо пользоваться защитной камерой с пер-
чатками. Удельная активность только что названного изо-
топа полония составляет 4,5 кюри/мг, что соответствует
1013 распад/мин • мг\ отсюда следует, что работа с миллиграм-
мовыми количествами полония может привести к зна-
чительным загрязнениям и быть опасной для здоровья.
Всю работу с веществами, обладающими такой активно-
стью, необходимо выполнять в защитных камерах, описа-
ние которых дано в последующих разделах данной главы.
Сухие источники полония высокой активности следует
содержать в герметичной таре и работать с ними в защит-
Лабораторная работа с полонием
55
ных камерах, поскольку многие соединения полония замет-
но летучи при комнатной температуре (например, гало-
гениды полония). Сам полоний, а также его соединения
при стоянии на воздухе или в кислороде легко образуют
двуокись полония. Реакция, по-видимому, идет с участием
озона, образующегося в результате бомбардировки кисло-
рода а-частицами. Конечным продуктом окисления метал-
лического полония на воздухе является белое твердое
вещество, которое может быть основным нитратом, полу^
чающимся в результате взаимодействия двуокиси полония—
начального продукта окисления — с окислами азота, обра-
зующимися при бомбардировке воздуха а-частицами [10].
ПРАКТИКА ЛАБОРАТОРНОЙ РАБОТЫ
С ПОЛОНИЕМ В КОЛИЧЕСТВАХ ПОРЯДКА
НЕСКОЛЬКИХ КЮРИ
Безопасная и эффективная работа с высокорадиоактивными
веществами в радиохимической лаборатории в большой сте-
пени зависит от лиц, работающих в ней, поскольку одно
неосторожное действие может вывести из строя на дли-
тельный период всю лабораторию, не говоря уже о том,
что будет поставлено под угрозу здоровье всего персонала.
Технику безопасности следует строго соблюдать в работе
точно так же, как и все другие правила, имея в виду, что
радиоактивные материалы обладают большей токсичностью,
чем обычные химикалии (если брать их в тех же весовых
количествах), и что несчастный случай, вызванный одним
из работающих, неизбежно вызовет поражение всего пер-
сонала лаборатории.
Подсчеты показывают, что опасностью внешнего облуче-
ния сильно радиоактивными источниками полония, не превы-
шающими 125 кюри (эквивалентных ~1 мкюри у-излучения
с энергией 0,8 Мэе), можно пренебречь; количества поло-
ния, превышающие необходимые для химических экспери-
ментов, не следует держать в лаборатории, ибо загрязне-
ние радиоактивностью, которое может произойти в резуль-
тате пожара или иного несчастного случая, весьма опасно.
Основная опасность при работе с большими количествами
полония заключается в возможности поражения кожи и
внутреннего облучения. В связи с этим при работе с поло-
56
Техника работы, с полонием
нием следует иметь в виду возможность случайных загряз-
нений лаборатории; кроме того, должен быть налажен
непрерывный контроль за зараженностью воздуха и поверх-
ности предметов. При проведении такого контроля за-
грязненности особое внимание следует обращать на про-
верку труднодоступных мест лаборатории.
Особенно тщательно нужно удалять в лаборатории пыль,
поскольку она может быть причиной попадания в организм
недопустимо больших количеств радиоактивных загрязне-
ний. По этой причине поступающий в лабораторию воздух
пропускают через систему фильтров и вводят в помещение
через вентиляционную систему, обеспечивая таким обра-
зом возможно более полную очистку воздуха от пыли.
Некоторое количество пыли, все же проникающее в лабо-
раторию, убирают мокрой тряпкой. Рекомендуется также
поддерживать высокую скорость обмена воздуха в лабора-
тории (30—40 раз в час).
Курить и принимать пищу в лаборатории запрещается.
В такой лаборатории все следует считать загрязненным,
пока не будет доказано обратное; что касается лаборатор-
ного оборудования, и в частности пипеток, то его ни в коем
случае не следует брать в рот. Без тщательной проверки
на загрязненность оборудование нельзя выносить из ла-
боратории для работы с обычными веществами; если даже
проверка покажет отсутствие загрязненности, то перенос
оборудования все же нежелателен, ибо наличие необнару-
женного дефекта в контрольно-измерительных приборах
может привести к тому, что сильно загрязненные части при-
боров окажутся вынесенными в чистую зону.
Весь персонал лаборатории должен иметь специальную
одежду для работы с радиоактивными веществами, и в слу-
чае попадания на нее радиоактивных веществ она должна
быть выстирана или уничтожена в зависимости от степени
загрязнения.
В лаборатории должно быть помещение, в котором рас-
полагаются душевые и контрольно-измерительная аппа-
ратура для определения загрязненности; от лаборатории
это помещение должно быть отгорожено невысоким барье-
ром. Обувь, в которой находился работающий в лабора-
тории, должна сниматься до выхода в чистую зону. Весь
персонал в обязательном порядке должен перед выходом из
Лабораторная работа с полонием
57
лаборатории проходить проверку на-загрязненность радио-
активными веществами рук и одежды. Сотрудникам не раз-
решается работать с радиоактивными материалами, если
на незащищенной одеждой участках кожи имеются порезы.
Пол лаборатории полония в Харуэлле сделан из бетона
и покрыт линолеумом; на него нанесен прочный слой пла-
стика (в случае необходимости его можно снять),
подобного тому, который применяется для покрытия обо-
рудования подводных телевизионных установок. В случае
попадания на пол радиоактивного материала на загрязнен-
ное место можно нанести еще один слой пластика; образо-
вавшийся таким образом «сандвич» после высыхания сле-
дует вырезать, а пол затем покрыть новым слоем пластика.
Данный прием пригоден в случае работы с короткоживу-
щими высокорадиоактивными ^-излучателями. Существует
и другой прием: на линолеум сначала наносят слой твер-
дого воска, а затем слой воска, растворимого в том или ином
растворителе. В случае загрязнения пола радиоактивными
веществами поверхностный слой воска вместе с загрязне-
ниями смывают подходящим растворителем, а затем пол
снова покрывают воском. Очистка подобного рода поверх-
ностей от загрязнений не всегда бывает успешной на прак-
тике, так как в процессе очистки часть радиоактивных ве-
ществ проникает в слой твердого воска, а затем и в линолеум.
Чем бы ни был покрыт пол, весьма существенно, чтобы его
поверхность не имела трещин и царапин, в которых может
накапливаться разлитый или просыпанный радиоактивный
материал или радиоактивная пыль.
Удаление загрязнений с других поверхностей в лабо-
ратории обычно производят протиранием их влажной
тряпкой. Для изготовления лабораторного оборудования
по возможности не следует применять дерево или другие
пористые материалы, так как разлитые радиоактивные
препараты будут впитываться в такие поверхности. Во-
прос о загрязнениях растворами полония активностью
порядка 6—9 кюри рассмотрен в работе [91 ].
Все химические операции с препаратами полония
большой активности следует проводить в защитных ка-
Мерах с перчатками, известных ранее под названием сухих
камер, поскольку первоначально они были сконструиро-
ваны для работы с ядовитыми или гигроскопическими
68
Техника работы с полонием
материалами в сухой атмосфере. Такие камеры в соответ-
ствии со своим назначением представляют собой большие
ящики с окнами из органического стекла, в которые
вделаны длинные резиновые перчатки. Пользуясь этими
Рис. 4. Защитная камера с перчатками для работы
с полонием [49].
перчатками, с полониевыми источниками работают непо-
средственно руками.
Ввиду того что полоний быстро проникает через рези-
новые перчатки, вделанные в защитные камеры, в целях
дополнительной предосторожности следует надевать их на
хирургические перчатки и обе пары перчаток чаще про-
верять на загрязненность. Перчатки, по-видимому, про-
пускают радиоактивность не в результате диффузии, а
вследствие проникновения радиоактивных веществ через
поры или микроскопические отверстия в резине, открываю-
щиеся только тогда, когда резина находится в растянутом
состоянии [10]. Неопреновые перчатки также применяют
для этой цели, и они оказываются значительно более стой-
Работа в защитной камере с перчатками и ее оборудование 59
кими к проникновению полония, хотя менее прочными,
чем перчатки из природного каучука. Работа с небольшими
предметами, особенно с рентгеновскими капиллярами,
в двух парах резиновых перчаток требует достаточного
практического навыка и терпения.
Типичная защитная камера с перчатками, используемая
в лаборатории полония в Харуэлле, показана на рис. 4.
Внутренняя часть камеры — рабочая — изготовлена из
листов органического стекла, смонтированных на сталь-
ной раме; рама покрыта пластиком, позволяющим удалять
загрязнения описанным выше методом «сандвича». Во всех
защитных камерах при помощи воздушных насосов поддер-
живается небольшое разрежение, что обусловливает попа-
дание воздуха только из помещения в камеру, но не наобо-
рот. Откачиваемый из защитных камер воздух фильтруется
перед выпуском в вентиляционную систему.
Внутренняя защитная камера помещается во внешней
камере подобной же конструкции и в ней также поддержи-
вается несколько пониженное давление. Таким образом,
радиоактивные загрязнения, проникающие из внутренней
камеры, должны попасть в вентиляционную систему с по-
током воздуха, циркулирующего между стенками внутрен-
ней и внешней камер, или же на дно внешней камеры, что
предотвращает возможность попадания загрязнений в по-
мещение лаборатории.
Стены лаборатории покрывают специальным лаком,
слой которого в случае необходимости можно снять, и, по-
добно полу, они не должны иметь трещин, в которых могла
бы собираться пыль. В остальной части лаборатории можно
производить обычную химическую работу; в лаборатории
должно иметься оборудование для работы с р и т-актив-
ными веществами и для их хранения. Более подробное
описание устройства лаборатории и мер по технике безопас-
ности можно найти в работах [16, 89, 121, 143, 227 и 317].
РАБОТА В ЗАЩИТНОЙ КАМЕРЕ С ПЕРЧАТКАМИ
И ЕЕ ОБОРУДОВАНИЕ
В целях уменьшения опасности загрязнения лаборатории
каждая защитная камера или система камер снабжает-
ся форкамерой, служащей тамбуром между областью с
60
Техника работы с полонием
высокой радиоактивностью, — самой защитной камерой —
и окружающим пространством. Все оборудование, необхо-
димое для работы в камере, вносят в нее через фор-
камеру. При работе с сильно радиоактивными препаратами
полония безопаснее иметь «буферную систему», представ-
ляющую собой двухступенчатую форкамеру; таким путем
между защитной камерой и окружающим пространством
создается область с небольшой радиоактивностью. Из за-
щитной камеры материалы не выносят непосредственно
в лабораторию, а помещают в мешок из пластика, герме-
тично прикрепленный к горловине люка внутренней ка-
меры. Этот мешок затем заклеивают при помощи аппарата
для высокочастотной сварки и заклеенную часть, в которой
находится материал, предназначенный для удаления, отре-
зают вдоль склеенного шва. Разбитые стеклянные детали
помещают в специальные банки с тем, чтобы предотвратить
возможность пореза мешка, перчаток или рук осколками
стекла.
Защитные камеры по существу являются миниатюрными
лабораториями, в которых имеется все необходимое обору-
дование и реагенты, требующиеся для проведения разно-
образных экспериментов. В каждой камере имеется центри-
фуга, электроплитка для выпаривания жидких пре-
паратов для счета а-частиц, штепсельные гнезда, подводка
сжатого воздуха, кислорода, газа и т д. Различные специ-
альные приспособления находятся в отдельных камерах,
соединенных с рабочей камерой перемычками из органиче-
ского стекла, сообщение по которым осуществляется по-
средством тележек. Защитная камера, показанная на рис. 4,
является камерой специального назначения; она имеет со-
ответствующую вакуумную систему для работы с полонием
в количестве порядка нескольких кюри. Вакуумный насос
находится в небольшой защитной камере, помещенной под
вакуумной системой; доступ к нему осуществляется в слу-
чае необходимости через отверстия для перчаток нижней
камеры. В случае полной неисправности насоса нижнюю
камеру можно отделить для очистки и ремонта насоса,
причем эта операция не связана с опасностью загрязнения
помещения.
В защитных камерах имеются ввинченные в дно стойки
для пробирок для предотвращения возможности случай-
Работа в защитной камере с перчатками и ее оборудование 61
ного проливания радиоактивных растворов, а также стойки
для поршневых пипеток и принадлежностей для отбора
проб. Дно защитных камер покрывают фильтровальной
бумагой с тем, чтобы предотвратить растекание слу-
чайно пролитых радиоактивных растворов; исключение
представляют камеры, в которых производят стеклодувные
работы, где дно застилают асбестовыми листами; воспла-
меняющиеся растворители никогда не применяют в такого
рода защитных камерах. Давление, необходимое для стекло-
дувных работ, создается сжатым воздухом. В защитных
камерах, в которых возможно неожиданное повышение
давления (например, в результате расплескивания жидкого
воздуха или взрыва газа вследствие утечки его из газо-
подводящей системы), устанавливают предохранительный
клапан, расположенный на крышке и непосредственно
соединенный с вытяжной вентиляцией. При отсутствии
такого приспособления увеличение давления в защитной
камере может привести к разрыву перчаток или в исключи-
тельных случаях к разрушению стенок камеры.
Большинство работ, выполненных в лаборатории автора,
было связано с получением соединений полония в твердом
состоянии весом порядка 0,1—3 мг. На рис. 5 показаны
две детали аппаратуры, применявшейся при получении
галогенидов полония; на этом рисунке А — дистилляцион-
ная трубка, используемая при приготовлении образцов со-
единений полония для съемки рентгенограмм методом по-
рошков. Приблизительно 1 кюри (0,22 мг) металлического
полония, электрохимически выделенного на золотой про-
волоке (диаметром 0,25 мм), помещали в открытый конец
трубки а, внутренним диаметром 4 мм, и запаивали ее.
Трубку затем эвакуировали, причем металлический поло-
ний возгонялся в среднюю часть капилляра б, внутрен-
ний диаметр которого равен ~ 0,4 мм. Конец трубки а,
в котором находилась освобожденная от полония золотая
проволока, после этого отпаивали и в эвакуированную
трубку впускали необходимый для реакции газ. В неко-
торых случаях удобнее отламывать кончик капилляра
после заполнения реагирующим газом и затем пропускать
Дополнительное количество газа через капилляр; в этом
случае рекомендуется улавливать выходящий газ с тем,
чтобы предотвратить возможность потери части полония.
62
Техника работы с полонием
Рис. 5. Стеклянные детали, применяемые при приготовле-
нии образцов полониевых соединений для съемки рентгено-
грамм методом порошков.
1 — фланец; 2 — трубка из стекла пирекс с наружным диаметром 6—8 мм;
S — рентгеновский капилляр с наружным диаметром 0,5 лмс; /— филь-
трующий слой (порошок стекла пирекс); 5— осколки тонких капилляров.
Когда реакция заканчивается, капилляр запаивают,
очищают от загрязнений промыванием в соляной кислоте
и извлекают из камеры для съемки рентгенограммы. Ми-
крофильтр Б, изготовленный из капилляра для съемки
рентгенограмм, укрепляют в небольшой центрифужной
трубке при помощи резинового кольца [11]; микрофильт-
ром в виде чашечки с капилляром можно также пользо-
ваться таким образом, что он своими флянцами опирается
Агрегатная отдача 63
на закраину центрифужной трубки. Реакция осаждения
при этом происходит в чашечке над капилляром, и осадок
собирается на фильтре при центрифугировании. Капил-
лярный фильтр данного типа применяли при приготовле-
нии образцов для съемки рентгенограмм методом порошков
и для определения растворимости на небольших количест-
вах исследуемых соединений. Описание прочей применя-
емой аппаратуры см. в работах [16, 89, 238]. Описание
оборудования, необходимого для работы с препаратами
полония высокой активности, приведено в недавно вы-
шедшем докладе [249].
АГРЕГАТНАЯ ОТДАЧА
Многие исследователи [143, 144, 218, 220, 221, 294, 297,
304] сообщали о появлении значительного загрязнения
вблизи открытых источников полония. Согласно Лоусону
[218, 219], распространение загрязнения является резуль-
татом агрегатной отдачи, вследствие которой полоний как
бы становится летучим при комнатной температуре. Это
предположение, по-видимому, подтверждается наблюде-
ниями [309], свидетельствующими о том, что распростра-
няющееся количество радиоактивных веществ не зависит
от температуры в интервале от 100 до 350°. Лоусон считает,
что самые большие группы атомов должны первыми от-
деляться в процессе агрегатной отдачи, поскольку коли-
чество полония, теряемое источником в вакууме или при
атмосферном давлении за небольшие отрезки времени, по-
видимому, пропорционально протекшему времени. Пред-
полагали, что распространение загрязнения можно умень-
шить, если источник полония покрыть тонкой фольгой, и
в единственном известном случае, когда был проделан та-
кой опыт [228], радиоактивность на фольге не была обна-
ружена.
Распространение радиоактивности наиболее заметно
в вакууме, и, хотя в основном его объясняют агрегатной
отдачей, некоторые авторы [294, 304] не исключают воз-
можности, что данное явление может быть следствием лету-
чести полония или одного из его соединений. В ряде более
Ранних статей по этому вопросу отмечалось, что потери
полония из влажных источников были значительно меньше:
64
Техника работы с полбнием
это могло свидетельствовать об образовании при гидролизе
нелетучего соединения полония. Галогениды полония,
например, легко гидролизуются во влажном воздухе [11,
12, 200].
Наблюдения над количествами полония порядка не-
скольких кюри позволяют сделать вывод, что распро-
странение загрязнения из открытых источников полония
происходит главным образом вследствие того, что у исполь-
зуемых соединений полония высокое давление пара. В ла-
боратории автора были случаи, когда рассыпали зна-
чительные количества соединений полония (порядка не-
скольких кюри), например в результате повреждения
капилляров с образцами для съемки. Когда этими соедине-
ниями были летучие галогениды, загрязнение распростра-
нялось настолько сильно, что требовалась полная замена
защитного покрытия на полу и стенках вытяжных шкафов,
причем эту работу выполняли в противогазах. В этих слу-
чаях контрольно-измерительные приборы показывали при-
сутствие значительных количеств переносимых воздухом
радиоактивных веществ. Если рассыпанные или разлитые
вещества были относительно нелетучими (например, дву-
окись полония), то применяли те же меры предосторож-
ности, но при этом в воздухе не отмечалось радиоактив-
ности, и распространение активности с тех мест, где были
просыпаны радиоактивные вещества, происходило только
за счет переноса вещества на обуви персонала. Пятно
на лицевой стороне вытяжного шкафа, содержащее при-
мерно 100 мкюри Ро, не обнаружило признаков распростра-
нения активности даже по прошествии нескольких суток.
Если бы распространение активности являлось резуль-
татом одного лишь процесса агрегатной отдачи, то оно не
должно было бы зависеть от природы применяемых соеди-
нений.
ПОТЕРИ ПРИ УПАРИВАНИИ
Обычно количество полония определяют выпариванием не-
больших аликвотных частей раствора до получения сухого
остатка с последующим измерением числа а-частиц, испу-
скаемых этим источником в единицу времени. Ряд авторов
сообщают, что такой способ приводит к потере части радио-
активного вещества, обусловливающей неточность в опре-
Действие излучения при высоких уровнях активности
65
делении полония. Потери наблюдались из солянокислого
раствора, нагретого до температуры, превышающей 150®
[330], из сернокислого раствора при температуре, превы-
шающей 200° [122]; в одной недавно опубликованной
статье есть указания о том, что потери полония составляют
приблизительно 10% при нагревании сернокислого раствора
до 300° или фосфорнокислого раствора до температуры,
превышающей 500° [331 ]. Если для определения исполь-
зуют высокорадиоактивные растворы полония в соляной
кислоте, то аликвотные части пробы упаривают досуха
с небольшим количеством концентрированной азотной ки-
слоты; в этом случае не наблюдается заметных потерь
при температурах до 200°. Можно с уверенностью утвер-
ждать, что наблюдавшиеся потери никоим образом не объ-
ясняются испарением, особенно если учесть, что сульфаты
полония, как было установлено, относительно нелетучи
при температурах, не превышающих 400°. Возможно, что
по крайней мере часть потерь обусловлена разбрызгива-
нием раствора при его нагревании.
В работе [371] отмечена значительная летучесть некоторых ор-
ганических соединений полония (см. также стр. 136 этой книги).
ДЕЙСТВИЕ ИЗЛУЧЕНИЯ ПРИ ВЫСОКИХ УРОВНЯХ
АКТИВНОСТИ
Энергия испускаемых полонием (Ро210) а-частиц составляет
5,3 Мэв, и поэтому совершенно очевидно, что они способны
инициировать химические реакции, энергии которых зна-
чительно ниже. Радиационные эффекты a-излучения сильно
радиоактивных препаратов полония могут маскировать
изучаемые химические реакции. Даже приготовление срав-
нительно слабых источников в 45 ркюри(= 1СГ8 г)1мм2
связано с трудностями, обусловленными действием излу-
чения [73, 74]. Работа с растворами полониевых соедине-
ний осложняется тем, что под действием излучения происхо-
дит разложение растворителя, которое в случае высоко-
радиоактивных растворов (порядка нескольких кюри на
1 мл растворителя) приводит к непрерывному выделению
газа [84,10,366]. Следует указать, что 0,001Л4 рас-
твор полония содержит немного менее 1 кюри Ро21е
на 1 мл. Работа с осадками нерастворимых соединений
& К. Бвгыад
66
Техника работы с полонием
полония сопровождается разрушением частиц твердого ве-
щества, происходящим, вероятно, в результате радиолиза
растворителя, захваченного осадком, и это увеличивает
трудности отделения таких осадков от растворителя.
Действие излучения
на стекло
Стеклянная или кварцевая аппаратура, в которой нахо-
дится полоний в количествах порядка нескольких кюри
(вне зависимости от то-
Р и с. 6. Растрескивание стекла,
вызванное a-излучением высокой
интенсивности [49].
го, в каком состоя-
нии—твердом или жид-
ком), поддействием 7-из-
лучения разъедается че-
рез несколько суток и
на ее поверхности во-
круг радиоактивного об-
разца появляются не-
большие трещины. По-
добного рода явления
были хорошо известны
и ранее исследователям
радия (см., например,
[92, 225]). При работе
с сильно радиоактив-
ными растворами (^ 4
кюри/мл) эрозия проте-
кает очень быстро, и
тонкий осадок кремне-
кислоты появляется на
дне стеклянного сосуда
уже по прошествии все-
го лишь нескольких су-
ток. Такие концентриро-
ванные растворы не ре-
комендуется хранить в
одном стеклянном со-
суде более двух недель, так как его дно постепенно раз-
рушается. Это обстоятельство—одна из весьма важных при-
чин, почему сильно радиоактивные растворы хранят в двой-
ных контейнерах. На рис. 6 видны тонкие трещины на
Действие излучения при высоких уровнях активности 67
трех трубках из стекла пирекс, применявшихся при опре-
делениях растворимости; в этих трубках в течение несколь-
ких недель содержалось максимум 100 мкюри сульфата по-
лония. Стекло, находящееся в контакте с твердыми препа-
ратами полония в количестве порядка нескольких кюри,
становится весьма хрупким, и приблизительно через неделю
тонкостенные рентгеновские капилляры трескаются вблизи
места соприкосновения с радиоактивным образцом.
В ряде случаев рентгеновские капилляры разрывались
под действием нарастающего в них давления газа. Если
считать, что капилляр имеет длину 10 мм и внутренний
диаметр 0,2 мм, то объем такого капилляра составляет
0,3 лш3. Гелий, выделяемый в течение суток источником
в 1 кюри (0,2222 мг) полония, при нормальных температуре
и давлении занимает объем 0,12 лш3 и оказывает давление
на стенки капилляра, равное 0,4 ат. Если работу ведут
с галогенидом полония, например с тетрахлоридом поло-
ния, то при превращении четырехвалентного полония
в двухвалентный свинец выделяющийся свободный галоид
будет оказывать такое же давление, и общее давление в по-
добных капиллярах за неделю может возрасти приблизи-
тельно до 6 ат. При таком расчете не учитывается влияние
разогревания, происходящего в результате торможения
в образце выделяющихся а-частиц, что также приводит к
увеличению давления. Опасность, связанную с быстрым
нарастанием давления, можно уменьшить, если пользо-
ваться капиллярами большего объема и принять дополни-
тельные меры предосторожности, покрыв капилляр тонким
слоем пластика, пропускающего рентгеновские лучи [48].
В настоящее время невозможно получить рентгеновский
снимок порошка гидратированных соединений полония пу-
тем применения обычных методов, поскольку этому пре-
пятствует очень высокое давление, развивающееся вслед-
ствие образования газа в результате радиолиза.
Влияние a-и злучения на коррозию
некоторых металлов
Влияние a-активности на скорость коррозии платины и цирко-
ния показано на образцах в виде фольги толщиной 25—100 ц [372 |.
Образцы подвергали воздействию a-излучения Ро210 в растворах
бромистоводородной кислоты (41-и 47%-ной )при комнатной темпе-
б*
68
Техника работы с полонием
ратуре, а также при 80, 100 и 127°. Концентрация Ро210 в
растворе менялась от 0,3 до 1 кюри/мл. Платина оказалась более
устойчивой по отношению к коррозии под действием излучения,
чем цирконий. Так, наличие a-активности порядка 1,0 кюри/мл не
приводит к заметному увеличению скорости коррозии платины при
комнатной температуре. Однако при наличии в растворе Ро 210 в ко-
личествах 0,25—0,3 кюри/мл скорость коррозии циркония при ком-
натной температуре возрастает примерно в 100 раз. При повышении
температуры до 80° в растворах с активностью 0,3 кюри/мл скорость
коррозии платины возрастает незначительно. Увеличение скорости
коррозии в присутствии Ро210 авторы объясняют влиянием про-
дуктов радиолиза раствора бром и стоводородной кислоты.
Обугливание органических веществ
Старицкий [320] наблюдал быстрое обесцвечивание соеди-
нений полония, содержащих органические радикалы; ана-
логичный эффект был отмечен также в лаборатории автора
при работе с органическими материалами («перспекс»,
линолеум, вата и т. д.), на которые попадали сильно радио-
активные препараты. На рис. 7 приведена фотография
стаканчика для взвешивания, в котором находилась вата,
сильно обуглившаяся после разрушения рентгеновского
капилляра, содержавшего ~ 500 мкюри металлического
полония. Этот снимок был сделан через несколько месяцев
после разрушения капилляра, однако обугливание наблю-
далось уже через несколько суток. Попытка извлечь
полоний из ваты после аналогичного случая, происшед-
шего ранее, не увенчалась успехом. Следует отметить,
что полоний не распространялся далее с первоначально
образовавшихся пятен, как можно было бы ожидать, если
исходить из того, что его распространение обусловлено
агрегатной отдачей; металл и продукты его окисления от-
носительно нелетучи, а поэтому и надо было ожидать не-
большого распространения активности в данном случае.
Нагревание, обусловленное
излучением
Температура любого образца полония всегда будет выше
температуры окружающей среды вследствие нагревания,
обусловленного торможением в самом образце испускаемых
полонием а-частиц. Поэтому свежеприготовленные пре-
Действие излучения при высоких уровнях активности
69
параты соединений полония, существующих в нескольких
модификациях, всегда получаются в высокотемпературной
Рис. 7. Вата, обуглившаяся под действием про-
сыпанного порошка двуокиси полония.
форме (например, р-полоний в случае металла [19, 20],
тетрагональная форма в случае двуокиси [10, 238]).*
Первоначальные измерения теплоты, выделяемой пре-
* В работе (10] рентгенограммы двуокиси полония снимали
сразу же после ее получения (нагреванием металла в атмосфере кис-
лорода), при этом наряду с высокотемпературной тетрагональной
формой обнаруживалась низкотемпературная гранецентрированная
кубическая форма PoOj.— Прим. ред.
70
Техника работы с полонием
паратами полония, дали значение, равное 24 кал/час-кюри
[96]; вычисленное значение этой величины составляет
27,4 кал/час-кюри, или 140 впг/г Ро210. Калориметрические
определения теплоты, выделяемой в единицу времени
большими источниками полония, часто используют в ка-
честве удобного метода определения содержания полония;
для этой цели недавно был рекомендован простой диффе-
ренциальный калориметр [347].
Свечение
Точно определить цвет того или иного соединения полония
довольно сложно из-за голубого свечения, вызываемого
возбуждением присутствующего газа о.-частицами; под
Рис. 8. Образец металлического полония, запаянный при раз-
режении.
действием излучения возникает также флуоресценция квар-
цевых или стеклянных сосудов [10, 249]. Наиболее трудные
условия возникают, когда приходится иметь дело с окра-
шенными парами галоидных соединений; в этих случаях,
например, бывает крайне трудно решить, является ли пар
Действие излучения при высоких уровнях активности
71
пурпурно-коричневым или просто коричневым, освещенным
голубым свечением.
Спектр света, испускаемого небольшими (^4 мкюри)
источниками полония на платиновой фольге, находящимися
в контакте с воздухом, состоит из линий кислорода и дру-
гих присутствующих газов [2541. Разбавленные растворы
полония слабо люминесцируют [221; суспензии нераство-
римых соединений полония в количествах порядка не-
скольких кюри имеют характерный вид. Помещенная на су-
перобложке книги фотография и рис. 8 представляют со-
бой снимки образца металлического полония, запаянного
в трубку из стекла пирекс при пониженном давлении 10 |л,
снятые в свете, испускаемом самим полонием *, и в отра-
женном свете; в первом случае на снимке вполне отчетливо
видно все, что окружает образец. Температура поверх-
ности стекла, соприкасающегося со слоем полония, была
равна 75°. Свежеприготовленный образец металлического
полония, показанный на рис. 8, имел вид блестящего се-
ребристого зеркала, плотно прилегающего к стеклу. Од-
нако по истечении некоторого времени от стекла начинают
отделяться чешуйки и их легко наблюдать в виде темных
пятен на металле. Данное явление можно объяснить воз-
никновением местных скоплений газа, по-видимому гелия.
* Как ясно из сказанного в начале данного раздела, этот свет
испускается материалом ампулы, в которой находится полоний,
флуоресцирующим под действием а-частиц.— Прим. ред.
4
ЭЛЕМЕНТАРНЫЙ ПОЛОНИЙ
Изотопы. Получение и свойства металлического полония. Валентные
состояния. Полониды. Нитрид полония. Карбонил полония. Соеди-
нение полония с гелием. Гидрид полония,
ИЗОТОПЫ
Насчитывается двадцать три изотопа полония с массовыми
числами от 196 до 218, причем все эти изотопы радиоактивны.
Ядерные свойства и методы получения каждого изотопа
приведены в табл. 2 (стр. 74). 1
Хевеши с сотрудниками [178, 179, 2801 не смогли найти
каких-либо признаков существования нерадиоактивного
изотопа полония, однако Хулубей и его сотрудники [187,
188, 303] считают, что в результате рентгенографических
исследований им удалось обнаружить присутствие неак-
тивного или долгоживущего изотопа полония в петците и
алтаите (последний из месторождения Стария в Трансиль-
вании). Оба этих минерала содержат теллур. Сообщения
Де [97, 98] о наличии долгоживущего изотопа полония
(ThE) в траванкорском монаците, по-видимому, имеют сом-
нительную достоверность. Массовое число неактивного изо-
топа вряд ли может быть меньше 198 или больше 218, по-
скольку существование стабильного ядра за пределами
этой области маловероятно, и любой долгоживущий или не-
активный изотоп должен, следовательно, быть ядерным изо-
мером одного из известных изотопов. Во всяком случае,
Мейтнер [240], основываясь на законе Фаянса, предпола-
гает, что такой изотоп может иметь массу около 218.
Изотоп полония с массовым числом 210 является един-
ственным известным изотопом, доступным в достаточном
количестве для макроскопического изучения химии даН’
ного элемента, и почти все сведения, собранные в этой книге,
получены при работе именно с этим изотопом. Изотоп с мас-
совым числом 209 был бы более удобным для химических
исследований, если бы его можно было получать в мил-
лиграммовых количествах, так как удельная активность его
Получение и свойства металлического полония 73
равняется только 8 кюри/г, тогда как удельная активность
изотопа Ро210 составляет 4500 кюри/г, и, следовательно,
радиационное воздействие Ро 09 почти в 600 раз меньше,
чем Ро210, если сравнивать одинаковые массы этих изотопов.
Период полураспада Ро210
Точное значение периода полураспада Ро210 особенно важно
знать потому, что массу полония, используемую в любом
опыте, нельзя определить непосредственно взвешиванием
из-за интенсивного местного разогревания, возникающего
в результате поглощения испускаемых самим полонием
а-частиц. Массу полония поэтому рассчитывают по его ак-
тивности, которую определяют либо непосредственно сче-
том а-частиц, ли^о косвенно — калориметрическими из-
мерениями*, но такого рода расчет требует точного знания
константы распада полония. Трудности взвешивания
полония, казалось бы, можно преодолеть, производя взве-
шивание в вакууме, но о такого рода попытках сообщалось
всего лишь раз [249], и этот метод, по-видимому, связан
с другими неудобствами, вызываемыми летучестью и неста-
бильностью многих соединений полония, в частности его
галоидопроизводных.
В течение многих лет считали, что период полураспада
Ро210 составляет приблизительно 138 суток (микрокалори-
метрическим методом были найдены значения 138,7 и 139,6
суток [307 ]). Не так давно в результате весьма тщательных
измерений для периода полураспада Ро210 было установлено
значение 138,40 суток [18, 87, 102, 374]. Удельная ак-
тивность, рассчитанная по этому значению, соответст-
вует 222,2 ^г/кюри.
ПОЛУЧЕНИЕ И СВОЙСТВА МЕТАЛЛИЧЕСКОГО ПОЛОНИЯ
Проводились многочисленные исследования процесса воз-
гонки микроколичеств полония, выделенного на различ-
ных металлах, однако, ввиду того что в этих опытах исполь-
* Влияние побочных физико-химических процессов на кало-
риметрические измерения радиоактивных веществ рассмотрено в ра*
ботах [375, 376].— Прим, ред.
Изотопы полония
Таблица 2
Массовое число Тип распада и энергия испускаемых частиц, Мэз Период полураспада Метод получения
196 197 198 199 200 201 202 203 204 205 206 207 208 209 а 6,14 а 6,040 а 5,935 а 5,846 Э. З.а; а 5,770 Э. 3.; с 5,671 Э, 3 ; (98%), а (2%) 5 57 Э. 3. Э. 3.; (~99%); а (~1%) 5,35 Э. 3,; (>99%); a (0,074%) 5,20 Э. 3.; (95%); a (5%) 5,218; 7 Э. 3.; (>99%); а ( — 0,01 %) 5,10; 7 Э. 3.; (сл.); а 5,108; 7 Э. 3,;(~0,5%) a (>99%) 4,877; 7 1,9 МИН. — 4 » — 6 » ~11 » ~8 » ~17 » 51 » 42 » 3,5 часа 1 8 » 8,8 суток 5,7 часа 2,93 года 103 » Отщепление при действии на висмут частиц высокой энер- гии [43] Отщепление при действии на ви- смут частиц высокой энергии [208] Отщепление при действии на свинец или висмут частиц вы- сокой энергии [210] РЬ2<* (а 2п) Ро2™ [186, 336] РЬ206 (а, Зп) Ро207 [186, 336] РЬ207 (а, Зп) Ро208 BiMe (р, 2п) Ро™ | Bi20» Id, Зп. Ро20’ J 1 Bi2”» 2п, Ро20» [209]
210 а 5,305; тО.г-Ю-3’'’) 0,8е 138,40 суток Естественно-радиоактивный изотоп RaF Bi?09 (n, 7) Bi21aP-->Po210
211м а 7,14 (90,5%); 7 25 сек. Bi“> (d, n) Po«° 1 [336] Pb*1» (a, 2/1) Po210 J
211 а 8,70 (7,0% , 7,85 (2,5%) 7,442 (99% >, 7 0,52 » Естественно-радиоактивный изотоп AcC'
-212 а 6,895 (0,50%) 6,569 .0,53%) 8,780 3,04-Ю-7 » Бомбардировка свинца a-частицами Естественно-радиоактивный изотоп ThC'
213 а 8,35 4,2-10“ * 6 » Дочерний Rn^ 17(248] или Bi213[i93]
214 а 7 683 1,58-Ю-4 » Естественно-радиоактивный изотоп RaG' в в АсА
215 и (>99% 7,36; (5-10—4 % ) 1.8310-3 >
216 а (>99%) 6,775; !)“ (0,014%) 0,158 » . . . ThA
217 а 6,54 <10 »
218 а (>99%) 5,996; ^0,02%) 3,05 мин. Естественно-радиоактивныый изотоп RaA
а Э. 3.— электронный захват. — Прим. ред.
6 у-Излучение Роаю с энергией 0,8 Мэв, как было установлено, обусловливается переходом с первого возбужденного состоя-
ния РЬ306 в основное состояние [8]; в последнее время удалось обнаружить [99J ожидаемую группу а-частиц более низкой
анергии (~4,5 /Мэе).
76
Элементарный полоний
зовали весьма малые количества полония, нельзя было с
уверенностью утверждать, являлся ли получаемый продукт
металлическим полонием или его окислом. Бимер и Мак-
суэлл [19, 20] впервые получили и идентифицировали чи-
стый металлический полоний в весомых количествах, поль-
зуясь методом, основанным на возгонке в вакууме полония,
электрохимически выделенного на платине. Полоний можно
также получить возгонкой в вакууме осадков этого металла,
образующихся в результате самопроизвольного выделения
на серебре и никеле [10] или путем электрохимического
выделения на золоте [11 ], однако металлический полоний,
полученный методом возгонки из отложений на меди, ча-
сто оказывался загрязненным хлористой медью [10]; спо-
собы приготовления таких осадков описаны в гл. 1.
Если выделять полоний на меди из азотнокислых растворов, то
после возгонки образуется продукт очень высокой чистоты [377,
340].
Более простой способ получения металлического поло-
ния заключается в осаждении сульфида полония из разбав-
ленного солянокислого раствора с последующим нагрева-
нием осадка до 500° в вакууме; сульфид в этих условиях
разлагается и чистый металлический полоний возгоняется,
а в осадке остается сульфид свинца (изотоп РЬ20в, образую-
щийся при распаде полония) [И, 47].
По данным Бэгнала и сотрудников [3781, металлический поло-
ний можно получить:
а) действием аммиачной воды (более 5н.) на гидроокись полония;
индукционный период реакции уменьшается с увеличением концен-
трации аммиака;
б) действием жидкого аммиака на гидроокись полония и неко-
торые другие соединения;
в) восстановлением гидроокиси полония гидразином, гидроксил-
амином или дитионитом натрия в щелочной среде*;
г) восстановлением тетрахлорида полония хлоридами олова
или титана в разбавленном растворе соляной кислоты;
д) восстановлением дитионитом натрия в солянокислом рас-
творе. Металл как в кислой, так и в щелочной среде выделяется в
двух аллотропических формах.
Было показано, что замещенные аммониевые соли также вое'
станавливают полоний, причем индукционный период реакции уве-
личивается в такой последовательности:
EtNH2 < Et2NH < Et<N+ < EtiNOH,
* Способы а, б и в пригодны лишь для Ро11®.
Получение и свойства металлического полония
77
Последние два реагента не способны восстановить заметное ко-
личество гидроокиси полония даже за 40 мин
В настоящее время авторы затрудняются объяснить механизм
этой реакции, но предполагают, что восстановление происходит
вследствие радиолиза раствора а-частицами.
По-видимому, этот вопрос можно будет уточнить, когда будут
получены достаточные количества Ро208 или Ро209.
Металлический полоний
Плотные слои металлического полония (~ 0,5 мг/см2), по-
лучаемые методом возгонки в вакууме, имеют вид сереб-
ристого металла (см. рис. 8); более тонкие слои почти про-
зрачны и имеют вид дымчатой пленки [10]. Согласно
Бербиджу [38], тонкие слои металлического полония
имеют желтый цвет, причем их окраска изменяется
от оранжевой до черной по мере увеличения толщины слоя.
Обработка более плотных металлических осадков, имею-
щих вид зеркала, небольшим количеством 8 н. азотной кис-
лоты приводит к тому, что часть полония отделяется от
стекла в виде тонкой фольги. По-видимому, полоний яв-
ляется довольно мягким металлом [249, стр. 18].
Существуют по меньшей мере две аллотропические формы
металлического полония: низкотемпературная модифика-
ция— а-полоний—с простой кубической решеткой и высоко-
температурная модификация—р-полоний—с простой ромбо-
эдрической решеткой; эти формы были идентифицированы
методом порошковой рентгенографии [19, 20]. Фазовое
превращение, по первым сообщениям, происходит примерно
при 75° [20]. Однако более позднее исследование [139, 396]
показало, что превращение происходит приблизительно при
36° и что обе фазы могут сосуществовать в интервале 18—
54°. Превращение а -> р, по имеющимся сведениям, со-
провождается увеличением объема приблизительно на
3% [20], но это маловероятно, поскольку p-форма имеет
большую плотность. При комнатной температуре свежепри-
готовленные образцы металла остаются в p-форме в течение
нескольких суток благодаря выделению тепла в самом об-
разце при торможении испускаемых полонием а-частиц.
По мере распада полония наблюдается медленное превра-
щение его в a-форму вследствие уменьшения выделения
тепла со временем. Рентгенографические данные свидетель-
78
Элементарный полоний
ствуют о том, что свинец (продукт распада Ро210) образует
твердый раствор с а-полонием с предельным содержанием
свинца по крайней мере до 50 ат. % [20] (см. также полонид
свинца). Кристаллографические данные для двух аллотро-
пических форм полония приведены в табл. 3.
Таблица 3
Кристаллографические даннные для а- и ^-полония
а-Полоний, простая куби-
ческая решетка
З-Полоний, простая ромбо-
эдрическая решетка3
Параметры решетки
Вычисленная плот-
ность, г/сж8
Пространственная
группа
Атомный объем [20]
а=3,345А [20],
3.359А [141]
9,32 [20], 9,196
при 36° [141]
О»
22,5
а=3,359А [20],
3.366А [141]
а = 98° 13' [20],
98° 5' [141]
9,51 [20], 9,398 при
39° [141]
22,1
Может быть также описана как гексагональная с периодами л—5.084А,
с -= 4,943 А [141].
Особенности рентгеноструктурного анализа соединений поло-
ния^ вызываемые действием его излучения, рассмотрены в работе
Атомный диаметр полония, экстраполированный к ну-
левому содержанию свинца, составляет 3,288 А [201 или
3,38 А [140]; более ранние расчеты атомного диаметра да-
вали значения 3,4 А [288] или 3,9 А ([1101; эти расчеты
основывались на предположении, что координационное
число равно 12). Вычисленный атомный объем полония со-
ставляет 22,7 [289 ].
Более ранняя попытка определить кристаллическую
структуру металла электронографическим методом при ис-
пользовании очень малых образцов, возогнанных на колло-
Получение и свойства металлического полония 79
диевую подложку [287, 289], позволила установить, что
кристаллическая структура полония может быть моноклин-
ной с элементарной ячейкой, содержащей 12 атомов (про-
странственная группа Cjj [52]. Кристаллическую решетку
полония можно представить также как сильно искаженную
решетку ртути (псевдогексагональную [289]). В данной
модификации решетка полония напоминает решетку тел-
лура [288]. Гайсинский [161] считает, что эти структуры
могут относиться к окиси полония, однако Роллиер [288]
выдвигает возражение против подобного утверждения и
отмечает, что использованные в указанных опытах образцы
были приготовлены возгонкой в вакууме или в атмосфере
водорода [289]. Вычисленные плотности этих двух форм
полония равны 9,24 г/см? (моноклинная) и 9,39 г! см? (псев-
догексагонал ьна я).
Октаэдрический ковалентный радиус полония, вычис-
ленный на основании данных по диффракции рентгеновских
лучей для комплексных галогенидов этого элемента, ра-
вен 1,52 А. Это значение приблизительно на 4% больше зна-
чения 1,46 А, установленного путем экстраполяции для
тетраэдрического радиуса. Такое небольшое различие на-
ходится в согласии с известным фактом постепенного умень-
шения различия между октаэдрическими и тетраэдриче-
скими радиусами при переходе от селена к теллуру [13].
Физические свойства
металлического полония
Некоторые физические свойства этого элемента установ-
лены Максуэллом [239] и другими исследователями; по-
лученные данные приведены в табл. 4. Эти данные пока-
зывают, что по физическим свойствам полоний в большей
мере напоминает стоящие рядом с ним в периодической си-
стеме элементы таллий, свинец и висмут, чем его низший
гомолог теллур [239]. Однако по химическим свойствам
он не похож на соседние элементы и в большинстве случаев
обнаруживает поразительное сходство со свойствами тел-
лура (см. гл. 8).
Давление пара металлического полония было измерено
80
Элементарный полоний
Таблица 4
Физические свойства металлического полония
Плотность (р-Ро\ г/см3 9,4 [239]
Точка плавления, °C 254а [239]
» кипения, °C 962 137]
Скрытая теплота парообразования, ккал/моль Удельное электросопротивление, 24,597 ±0,031 [37]
[1 ом-см при 0°
а-Ро 42± 10 [239]
₽-Ро Температурный коэффициент электросо- 44± 10 [239]
противления
а-Ро . 0,0046 [239]
₽-Ро Коэффициент линейного расширения 0,0070 [239]
(—196—30°)6 2,35-10"5 1142]
а По теоретической оценке, сделанной в более ранних работах, точка плавле-
ния этого элемента должна быть значительно выше (см., например, [339]).
& Ряд других значений приведен в недавно опубликованном американском
докладе [249].
в интервале 438—745°; зависимость давления пара полония
от температуры выражается уравнением [37]
Igp = -5377,8 ±6,7 + 7 2345 + 0,0068.
Радиографическим методом было найдено, что
давление пара над микрообразцом полония, выделен-
ного на серебре, при комнатной температуре составляет
5-10“15 мм рт.ст. [7]. Однако из приведенного выше уравне-
ния следует, что давление пара полония при 20° должно иметь
значение порядка 10“пмм рт.ст и возможно, что металл,
применявшийся в измерениях [7], был окислен. Имеется со-
общение [250] об изучении эффекта Холла в металлическом
полонии.
Получение и свойства металлического полония
81
Рентгеновский спектр полония
Некоторые L-линии полония были отмечены в ранних ра-
ботах, проведенных на микроколичествах полония [189,
323]; недавно был опубликован обзор этих работ [325].
Спектры Ки L были получены в опытах с медной мишенью,
покрытой 2—3 мг (10—15 кюри) металлического полония;
обнаруженные в этом случае линии перечислены в табл. 5
[273].
Таблица 5
Рентгеновский спектр полония
Серии Идентифика- ция линий Длины волн, Х-единицы
эксперименталь- ные значения расчетные значения по данным более ранних наблюдений
к а2 160,5 160,89
al 155,6 156,12
pl 137,9 138,28
Р2 133,0 134,04
L 1 1280,2 — —
а2 1123,7 1123,10 1123,29
al 1112,2 1111,59 1111,52
₽6 965,3 — —
₽2 927,0 927,31 927,44
919,4 919,79 919,89
03 905,8 — —
71 784,5 785,52 785,86
72 769,9 — —
73 762 — —
Оптический спектр полония
Все линии полония, отмеченные в работе Чапека [88], а
также Карлик и Петерсона [205], и одна из линий, наблю-
давшихся Кюри и Дебьерном, были подтверждены недавно
и одновременно проводившимися исследованиями Чарльса
и сотрудников [90], а также Верного, Зайделя и Шве-
бельблита [340]; результаты этих исследований находятся
в очень хорошем согласии. Для возбуждения спектра был
использован высокочастотный безэлектродный разряд при
б К Ьвгмал
82
Элементарный полоний
200—800°, а также искровой разряд [90]. Более 100 линий,
лежащих в интервале 1920—9375 А, наблюдались обеими
группами исследователей, и 35 из этих линий вместе с до-
полнительными 13 новыми линиями обнаружены в спектре
искрового разряда между графитовыми электродами, на
торцы которых был нанесен полоний. Было найдено, что
потенциал ионизации для нейтрального атома полония ра-
вен 8,43 эв [90]; это значение хорошо согласуется с опубли-
кованными ранее вычисленными значениями 8,4 эв [119],
8,3 эв [118], 8,46 эв [277] и 9,44 эв [338]. Потенциал иони-
зации однократно ионизованного атома полония, как по-
казывает вычисление, равен 18,2 эв [119]. Остальные спек-
троскопические данные, включая колебательные энерге-
тические уровни молекулы Ро^10, приведены в недавно опуб-
ликованном докладе [249]*.
Растворимость металлического
полония в кислотах
Микроколичества полония. Разбавленная
(1 н.) соляная кислота лишь частично растворяет микроко-
личества полония, осажденные на кварце [222], и, чтобы
добиться полного растворения, необходимо применять смесь
соляной и плавиковой кислот. Разбавленная серная кис-
лота растворяет полоний, содержащийся в осадках, на-
капливающихся в старых радоновых трубках [299, 301 ],
где можно ожидать наличия полония. Уксусная, хромовая,
щавелевая и винная кислоты применялись для растворения
полония, электрохимически выделенного на золоте; действие
таких относительно слабых кислот свидетельствует о том,
что в данном случае полоний присутствует в виде окиси,
а не в виде металла. Имеющиеся данные о растворении по-
лония с золотых электродов при действии азотной кислоты
носят противоречивый характер. Как правило, для этих
целей достаточно применять разбавленную кислоту, однако
некоторые авторы [215, 318] утверждают, что следует поль-
зоваться кипящей концентрированной кислотой. Полное
удаление полония с платины при обработке ее кислотой
• См. также [380J.— Прим, ред.
Получение и свойства металлического полония
83
связано с чрезвычайно большими трудностями, и, вероятно,
в этом случае лучше применять возгонку в вакууме при
высоких температурах.
Миллиграммовые количества. В милли-
граммовых количествах полоний медленно растворяется
в 2 н. соляной кислоте, образуя раствор, содержащий
двухвалентный полоний, который под действием интен-
сивного а-излучения быстро окисляется и переходит
в четырехвалентное состояние. Скорость растворения воз-
растает при добавлении хлорной воды, что, вероятно, свя-
зано с выделением хлора под действием a-излучения. Ме-
таллический полоний быстро растворяется в концентриро-
ванной азотной кислоте с выделением окислов азота, однако
разбавленная кислота действует на него медленно [10].
Возможно, что нитрат полония образует защитную пленку
на поверхности металла, поскольку он лишь умеренно ра-
створим в разбавленной кислоте [253].
С у л ь ф о т р е х о к и с ь и с е л е н о т р е х о к и с ь
полония
Металлический полоний реагирует с дымящей серной кис-
лотой и с концентрированной селеновой кислотой или с со-
ответствующими этим кислотам ангидридами (трехокисями
серы и селена); при этом образуются чрезвычайно нестой-
кие темно-красные твердые вещества, состав которых, по-
видимсму, соответствует формулам PoSO3 и PoSeO3, по ана-
логии с другими элементами подгруппы селена. Оба этих
соединения быстро разлагаются с образованием черного
твердого вещества, которое, по-видимому, является моно-
окисью полония. Указанное соединение полония с серой
растворимо в концентрированной серной кислоте; образую-
щийся при этом раствор окрашен в красный цвет. Соответ-
ствующее соединение полония с селеном не растворяется
в селеновой кислоте [48].
Диффузия полония в металлы
Определение периода полураспада полония прямым счетом
а-частиц, испускаемых образцами полония, нанесенными на
различные подложки, привело к несовпадающим результа-
6*
84
Элементарный полоний
там, причем наблюдалась некоторая неоднородность а-из-
лучения, испускаемого такими источниками (см., напри-
мер, [206]). Эти наблюдения могут свидетельствовать о су-
ществовании диффузии полония в металл, на котором он
находится [218, 228, 229, 346]; было высказано предполо-
жение, что в основе механизма диффузии лежит агрегатная
отдача [219]; чтобы происходила диффузия, возможно,
необходимо присутствие следов кислот [245]. Вертенштейн
и Добровольская [343] установили, что коэффициент диффу-
зии полония в золото и платину при 470° равен приблизи-
тельно 10-9 смЧсутки и что при комнатной температуре
скорость диффузии полония в эти металлы и в серебро
пренебрежимо мала. Рона и Шмидт [293] для верхнего
предела коэффициента диффузии полония в алюминий, же-
лезо, медь, никель, серебро, золото и свинец при комнат-
ной температуре приводят значение, равное 10~14 смЧсутки;
этим исследователям не удалось обнаружить какой бы то
ни было неоднородности a-излучения, испускаемого поло-
ниевыми источниками на палладии даже по прошествии не-
скольких месяцев [294 ]. Радиографические опыты, в которых
полоний отлагался на золотой и серебряной фольге из 10“ 13Л4
раствора, свидетельствуют о возможном проникновении
полония в металл подложки [67]. По-видимому, полоний
в какой-то мере диффундирует в насыщенную водородом
платину и в насыщенный водородом палладий; это явление
объясняют образованием гидрида полония [219]. При от-
качивании источников, содержащих микроколичества по-
лония, выделенного на этих металлах, полоний улетучи-
вался с платины вместе с водородом, тогда как на палла-
дии полоний удерживался; причиной такого явления, ви-
димо, является образование сплава полония с палла-
дием.
Изучение самодиффузии свинца [129] с применением
меченых атомов радия-D не дало никаких указаний о том,
что происходит какая бы то ни было диффузия полония даже
после выдерживания в течение 400 суток при 280°. Более
поздние измерения скорости диффузии полония в свинцо-
вую фольгу для коэффициента диффузии при 310® дали зна-
чение, равное 1,3-10“5 смЧсутки [1801. Опубликованные
данные о диффузии полония приведены в табл. 6.
Валентные состояния полония
85
Таблица 6
Диффузия полония в некоторых металлах
Металл Температура, °C Коэффициент диффузии Литература
Никель Комнатная 10“13 см2/сутки (281]
Свинец 150 ю-9 > Там же
> 200 10“7 » » »
> 310 1,3-10-5 » [180)
Алюминий 20 ЗЮ-13 см2/се к [249, стр. 27]
» 500 5-Ю-11 » Там же
Висмут 150 5-Ю-11 » »
» 200 5-Ю-10 » > »
Диффузию полония изучали также в ряде других ме-
таллов [128]; опубликованы данные о поверхностной диф-
фузии полония на меди [171 ], золоте [309] и платине [196].
ВАЛЕНТНЫЕ СОСТОЯНИЯ ПОЛОНИЯ
Количества полония, применявшиеся в более ранних иссле-
дованиях химических свойств этого элемента, были настолько
малы, что непосредственно свойства самого полония и его
соединений нельзя было определить. Выводы относительно
возможных валентных состояний полония были сделаны,
например, на основании аналогии в химических свойствах,
установленной в результате опытов по соосаждению. К дан-
ным, полученным в этих опытах, следует подходить осто-
рожно; такого рода методы исследования, как замечает
Хан [146, стр. 84], даже тогда, когда происходит образо-
вание смешанных кристаллов, в лучшем случае позволяют
судить лишь о координационной валентности данного эле-
мента, а порой и для этого не дают достаточных оснований.
Данные о критическом потенциале выделения полония
в присутствии различных восстановителей свидетель-
ствуют о том, что полоний существует в растворе по край-
ней мере в двух валентных состояниях, однако при работе
86
Элементарный полоний
с микроколичествами полония численные значения этих
валентностей нельзя было определить.
В основном состоянии нейтральные атомы полония
имеют, как установлено, электронную конфигурацию,
5s25pG5d106s 6p4(T2) [341, стр. 198]*, подобную конфигу-
рации селена или теллура. На основании этого у полония
можно ожидать нижеследующие устойчивые степени окис-
ления: 2 — ,2 + ,4 + и 6 +. По-видимому, имеется мало
теоретических оснований для утверждения существования
трехвалентного полония, хотя о нем неоднократно упоми-
налось. Ниже приведены данные о валентных состояниях
полония, полученные как в опытах с миллиграммовыми ко-
личествами полония, так и в опытах с микроколичествами
этого элемента.
Шестивалентный полоний
Анодное выделение микроколичеств полония из кислого рас-
твора, по имеющимся сообщениям, приводит к образова-
нию перекиси [182, 183], предположительно имеющей со-
став РоО3. Насколько известно, экспериментальных данных
о строении этого соединения, а также о наличии (или отсут-
ствии) у него перекисной связи не получено. Так или иначе,
выделенные на аноде микроколичества полония могут быть
смыты с электрода перекисью водорода [165], что опреде-
ленно свидетельствует о существовании высшего окисла
полония. До настоящего времени еще нет сообщений о по-
лучении этого окисла в миллиграммовых количествах. Дру-
гие данные, подтверждающие существование полония в ше-
стивалентном состоянии, основаны на результатах опытов
по соосаждению, однако следует отметить еще раз, что вы-
воды, сделанные на основании таких опытов, несомненно,
носят условный характер. Процесс соосаждения микроколи-
честв полония с гексахлорплатинатом аммония из раствора в
разбавленной соляной кислоте, по-видимому, замедляется
в присутствии свободного хлора, который не оказывает яв-
ного влияния на осаждение из концентрированной кислоты.
* Об электронной конфигурации атомов полония см. [3811.—?
Прим, ред*
Валентные состояния полония
87
Отсюда делают вывод, что хлор в разбавленной кислоте
окисляет четырехвалентный полоний до более высокого
валентного состояния (предположительно шестивалентного)
[137]. Однако недавнее сообщение [190], в котором приве-
дены данные спектроскопических исследований миллиграм-
мовых количеств полония в разбавленных солянокислых
растворах, показывает, что, несмотря на образование сво-
бодного хлора под действием а-частиц, признаки окисле-
ния полония до более высокого валентного состояния
отсутствуют. Кроме того, миллиграммовые количества не-
растворимых гексахлорполонитов обычно получают в ре-
зультате осаждения из разбавленного (2 н.) солянокис-
лого раствора [11, 320].
Данные об осаждении полония с теллуратами (Те VI)
довольно противоречивы. Гийо [137, стр. 122] установил,
что полоний не осаждается с шестивалентным теллуром
после окисления хромовой кислотой смеси теллура с микро-
количествами полония. Самарцева [306] отмечает, что по-
лоний кристаллизуется с теллуратами свинца и калия
в условиях, благоприятствующих проявлению изомор-
физма; Карл [204] использовал соосаждение с теллуратом
свинца в качестве метода отделения полония от смесей
радия-D, Е, F. Не исключено, что полученные при этом
осадки содержат теллурат полония.
Радиус иона Ро6+,по расчетным данным, равен 0,67 А [2].
В работе Зива Д. М. и сотрудников [385] существование шести-
валентного иона Ро устанавливают из факта диспропорционирова-
ния, обнаруженного по кривым разложения.
Мацура и Гайсинский [390] показали, что распределение че-
тырехвалентного полония при концентрациях порядка 1О“10 М. ме-
жду его азотнокислыми или солянокислыми растворами и метил-
изобутилкетовом мало зависит от кислотности в интервале рН1—6.
Примерно 80% его при обычной температуре находится в органиче-
ской фазе. В сернокислом растворе процент извлечения при изме-
нении кислотности проходит через максимум и минимум. Однако,
какова бы ни была природа кислоты, присутствие сильного окис-
лителя (Се4+, Сг6+) значительно сдвигает равновесие в сторону вод-
ной фазы. Разрушение окислителя перекисью водорода восстанав-
ливает первоначальное распределение в случае Се4+, а с бихррматом
приводит к промежуточным значениям коэффициента распределе-
ния. Несомненно, при этом происходит неполное восстановление.
В действительности распределение зависит от отношения Сг3+/Сг6^
88
Элементарный полоний
или Се3+/Се4+ в растворе. Эти эксперименты более определенно
указывают на существование шестивалентного полония. Для потен-
циала пары Ро4 ь/Ро6+ Гайсинским было вычислено значение, близ-
кое к 1,5 в. Гайсинский правильно замечает, что в данном случае к
обычной неустойчивости присоединяется еще один фактор — дей-
ствие собственного а-излучення. Так, трехокись плутония не из-
вестна, двуокись кюрия была получена только на 19-годичном изо-
топе. Поэтому не удивительно, что не удается получить в миллиграм-
мовых количествах полоний в шестивалентном состоянии. Однако
если двуокись полония сплавлять со смесью хлората калия и ед-
кого кали, то образуется бесцветный плав, который при охлаждении
становится бледно-синим. Это изменение цвета обратимо. Полоний
после такой обработки более растворим в воде и образует бесцвет-
ный раствор; возможно, что этот раствор содержит полонат калия
(КаРоО4) [350, стр. 11].
Четырехвалентный полоний
Четырехвалентное состояние для данного элемента было
установлено на основании анализа двуокиси полония [238],
соответствующих галогенидов и их производных [10—13,
38, 320], а также сульфата и селената полония [48]. Ве-
роятность существования этого валентного состояния у по-
лония была высказана Гийо [137] на основании опытов
с микроколичествами этого элемента, в которых полоний со-
осаждался из солянокислого раствора с аммониевыми солями
типа (ЫН4)гМС1в (М = Pb, Sn, Те, Pt) при условиях, ха-
рактерных для изоморфизма; однако соосаждение микроко-
личеств полония с соответствующей солью трехвалентного
иридия также наблюдалось при аналогичных условиях, так
что из этих опытов существование четырехвалентного со-
стояния полония в солянокислых растворах со всей несом-
ненностью не может быть установлено. Ацетилацетонат по-
лония, по имеющимся сообщениям, кристаллизуется с со-
ответствующим производным четырехвалентного тория [311,
313]; электрохимические наблюдения при изучении микро-
количеств полония также позволяют сделать вывод, что
в растворе полоний находится в четырехвалентном состоя-
нии [157].
Вычисленное значение радиуса иона Ро4+ лежит в ин-
тервале 1,02—1,15 А [288]. Наблюдаемые значения радиуса
этого иона в двуокиси полония составляют 1,04 А [10] и
1,02 А [238].
Валентные состояния полония
89
Трехвалентный полоний
Работы по изучению совместной кристаллизации диэтилди-
тиокарбамата полония с соответствующими солями висмута^
трехвалентного кобальта, меди и никеля показали, что сме-
шанные кристаллы с никелевыми и медными солями не об-
разуются; полоний однородно распределяется в соли ко-
бальта и неоднородно — в соли висмута [136]. Нельзя счи-
тать, что это подтверждает существование трехвалентного
полония в растворе до начала эксперимента, поскольку вос-
становление могло произойти при добавлении органиче-
ского реагента. Существование трехвалентного полония было
подтверждено данными о совместной кристаллизации его
с оксалатами лантана, скандия, иттрия и стронция [310,
313], а также работами по соосаждению полония с солью
трехвалентного иридия—гексахлориридитом аммония [137 ].
Ацетилацетонат полония, по-видимому, кристаллизуется
с соответствующим соединением скандия [311, 313], и по-
этому вполне вероятно, что при взаимодействии «гидро-
окиси» полония и ацетилацетоната образуются ацетилацето-
наты как четырех-, так и трехвалентного полония. Работа
с миллиграммовыми количествами ацетилацетоната полония
показала, однако, что в этом случае образуется соединение
четырехвалентного полония, которое по своей структуре
может быть похоже на соответствующее соединение теллура
[12]; если эта точка зрения правильна, то структура этого
соединения не имеет никакого отношения к структуре со-
ответствующих соединений тория и скандия.
Работа с миллиграммовыми количествами полония не
подтвердила существования соединений трехвалентного по-
лония в твердом состоянии, хотя возможно, что трихлорид
полония может образоваться в качестве промежуточного
соединения при окислении двухвалентного полония до
четырехвалентного под действием собственного излуче-
ния [11].
Вычисленный радиус иона Ро3+ равен 1,04 А [288].
Двухвалентный полоний
Существование двухвалентного полония было установлено
после того, как были получены и подвергнуты анализу
галогениды этого элемента в двухвалентном состоянии
90
Элементарный полоний
[11, 12, 38, 200]. В растворе эти галоидные соединения бы-
стро окисляются, причем полоний переходит в четырехва-
лентное состояние [11, 12] в результате сильного окисляю-
щего эффекта облучения а-частицами, чем собственно и
обусловливается трудность работы по изучению свойств
двухвалентного полония.
Присутствие положительно заряженных ионов двухва-
лентного полония в солянокислом растворе, содержащем
микроколичества полония, было установлено в работах по
изучению диффузии и подвижности ионов [175—177];
весьма возможно, что такими ионами являются гидролизо-
ванные или частично ионизированные производные тетра-
хлорида полония (например, РоО' +, РоС1* +).
В соответствии с данными Эрбахера и Кединга [111]
полоний по своему положению в периодической системе и
электрохимическим свойствам в нормальных условиях дол-
жен находиться в двухвалентном состоянии. Легкость раз-
ложения галогенидов четырехвалентного полония позво-
ляет полагать, что двухвалентное состояние полония яв-
ляется стабильным при условии отсутствия ионизирую-
щего излучения. Гийо, однако, не считает, что полоний
можно восстановить до двухвалентного состояния; поскольку
в микроколичествах полоний не кристаллизуется с хлори-
дом свинца и поскольку на совместное осаждение микроко-
личеств полония с комплексным хлоридом трехвалентного
иридия не влияет щавелевая кислота, которая в этих ус-
ловиях должна действовать как восстановитель [137,
стр. 120). Полоний также образует двухвалентный ион
Ро““ в полонидах.
ПОЛОНИДЫ
В тех случаях, когда микроколичества полония сплавляют
с серебром или медью при 1100°, лишь небольшая доля
полония возгоняется из расплава [333], хотя в то же время
микроколичества полония обычно возгоняются с этих ме-
таллов при 900—1000°; этот факт может свидетельствовать
об образовании соединения. Микроколичества полония са-
мопроизвольно выделяются на ртути из растворов в соля-
ной или уксусной кислотах, а также из растворов в целом
ряде кетонов; возможно, что растворение сопровождается
Образованием амальгамы [57, .58, 61 ] или подоцида.
Таблица 7
Кристаллографические данные для некоторых полонидов
Соеди- нение Структура Параметры решетки Вычислен- ная плот- ность, г/см* Темпера- тура раз- ложения, °C
РЬРо Гранецентриро- ванная куби- ческая (типа NaCl) а = 6,590А (345] а = 6,599А [140] 9,6 [345] 600 [ 140] 1
HgPo То же а = 6,250А [345] 11,1[345]
СаРо В В а = 6,51А [249. стр. 91]
ZnPo Гранецентриро- ванная куби- ческая (типа ZriS) а = 6,28А [140] 7,39[249, стр. 91] Возго- няется при 400 [140]
Na2Po Гранецентриро- ванная куби- ческая (типа CaF2) а = 7,473А [140] 4,08(249, стр. 91]
PtPo2 Гексагональная а = 4,104А 12,47(249, 450—600
[типа Cd(OH)2] С = 1,366“ [140] стр. 91] [140]
NiPo AgPo? Гексагональная (типа NiAs) Ромбическая (или моно- клинная?) а = 3,973А С = 1,425“ [140] А = 1,327® 6 = 5,565А С = 1,404® [140] 11,53[249, стр. 91] 560[140]
BePo Г ранецентриро- ванная куби- ческая а = 5,827А [249, Стр. 91] 7,35[249, стр. 91]
А = у; с •= -у •-прим. лед.
92 Элементарный полоний
Некоторые полониды сравнительно недавно были по-
лучены в миллиграммовых количествах и идентифициро-
ваны рентгеноструктурным методом. Полониды свинца и
ртути черного цвета; они образуются из элементов при
325—350° [345]. Полониды цинка, платины и никеля об-
разуются при осаждении на этих металлах полония, воз-
гоняемого в вакууме; в тех же условиях тантал, золото,
бериллий, железо и алюминий не реагируют с полонием, а
висмут, по-видимому, образует сплав; по имеющимся дан-
ным, полоний и висмут смешиваются в любых отношениях
[140]. Имеются сведения, что полонид натрия образуется,
если оставить полоний в трубке из стекла пирекс [140];
характер протекающей в данном случае реакции неясен.
Полонид цинка возгоняется в вакууме при 400* [140];
поскольку давление пара составляющих его металлов
почти одинаково, возможно, что при нагревании происхо-
дит разложение полонида, а при охлаждении составляющие
его элементы соединяются вновь. Химические свойства
этих соединений не изучены. Кристаллографические дан-
ные приведены в табл. 7.
Изучение совместной кристаллизации с теллуридом на-
трия проводили восстановлением смеси микроколичеств по-
лония и теллура под действием дитионита натрия в щелоч-
ном растворе в атмосфере водорода; полученные результаты
свидетельствуют об образовании полонида натрия [65, 305].
НИТРИД полония
Металлический полоний возгоняется в атмосфере азота, не
претерпевая при этом никаких изменений [292], это поз-
воляет предполагать, что нитрид не образуется из элемен-
тов. При нагревании гексабромполонита аммония до 200*
получается соединение черного цвета, способное разлагаться
со взрывом. По-видимому, в этих условиях [12] образуется
взрывчатый нитрид, подобный нитриду теллура Te3N4.
КАРБОНИЛ ПОЛОНИЯ
Опыты, при которых производилась возгонка микрбколи-
честв полония в присутствии окиси углерода [78, 222],
свидетельствуют о том, что в этих условиях происходит
Гидрид полония
93
образование летучего карбонила полония неизвестного со-
става.
Имеется указание [350, стр. 3], что сравнительно устойчивый
карбонил полония образуется при взаимодействии металла, выде-
ленного на серебряной фольге, с окисью углерода при 500°; серебро,
по-видимому, является катализатором этой реакции.
СОЕДИНЕНИЕ ПОЛОНИЯ С ГЕЛИЕМ
Имеются сообщения о том, что летучее соединение полония
с гелием образуется непосредственно из элементов под дей-
ствием электрического разряда при пониженном давлении
(3—8 мм рт. ст.). Данной реакции благоприятствует высокая
температура [242].
ГИДРИД ПОЛОНИЯ
Панет показал, что можно получить летучий гидрид (с не-
большим выходом) действием 0,2 н. соляной кислоты на
магниевую фольгу, на которую электрохимическим выде-
лением или возгонкой нанесены следы полония [261, 262,
269]. Газообразный гидрид выделяется из раствора при
пропускании тока азота, водорода или кислорода. Имеются
также сообщения, согласно которым гидрид образуется
с выходом примерно 0,2% при добавлении порошка маг-
ния к кислому раствору, содержащему микроколичества
полония [270], и с еще меньшими выходами при действии
кислоты на цинк, на который нанесены микроколичества
полония. Гидрид можно также получить в незначительном
количестве при электролизе, осуществляемом при напря-
жении 220 в в 0,2 н. серной кислоте с применением катода
с нанесенным на него полонием [263, 264, 269]. Гидрид не
образуется из элементов при нагревании [38] или при дей-
ствии водорода на полоний, нанесенный на никель, при ком-
натной температуре [199], хотя Лоусон [217] при изуче-
нии ионизации, вызванной в водороде действием а-частиц,
испускаемых полонием, получил некоторые данные, сви-
детельствующие об образовании гидрида. Продукт описан-
ной выше реакции конденсируется при —84° [261, 262]
и, по-видимому, является весьма нестойким, поскольку
лишь 0,6% сконденсировавшегося вещества сохраняется
без разложения к моменту, когда сосуд, в котором происхо-
94
Элементарный полоний
дит конденсация, нагревается до комнатной температуры.
Некоторые физические свойства гидрида полония, найден-
ные расчетным путем, приведены в табл. 8.
Таблица 8
Физические свойства гидрида полония
Точка плавле- ния, °К Точка кипения, °К Теплота паро- образования, ккал • hPb (константа Трутона) Первый иони- зационный по- тенциал, эв
237 [272] 308,5 [272] 310 [266, стр. 106; 271] 6,19 [272] 20,06 [272] 8,6 [279]
Сухой гидрид полония, по имеющимся данным, более
стабилен, чем в смеси с водяными парами, и поскольку он
разлагается при действии многих осушителей (например,
хлористого кальция, пятиокиси фосфора [1, 269]), процесс
осушки гидрида может быть связан с некоторыми трудно-
стями. Разложение сухого гидрида может быть неполным
даже при 350—400° [1 ].
Лоусон [216, 217] высказал мнение, что гидрид должен
самопроизвольно разлагаться при контакте с озоном, обра-
зующимся при действии а-частиц на присутствующий кисло-
род, однако Панет [261, 262] установил, что кислород мо-
жет служить в качестве газообразного носителя для данного
соединения. Гидрид поглощается 0,1 н. едким натром или
раствором азотнокислого серебра, но не поглощается во-
дой, не содержащей воздуха; гидрид разлагается также под
действием смазки, применяемой для смазывания кранов.
Гидрид полония менее устойчив, чем гидрид висмута [271 ],
и, по-видимому, похож на гидрид теллура [269].
5
ЭЛЕКТРОХИМИЯ ПОЛОНИЯ
Выделение на катоде. Выделение на аноде. Электрод Ро/Ро““.
ВЫДЕЛЕНИЕ НА КАТОДЕ
Первоначально электрохимические работы с микроколи-
чествами полония были затруднены отсутствием данных об
основных химических свойствах этого элемента. Работа
затруднялась также адсорбцией, которая может полностью
замаскировать электродные процессы в чрезвычайно
разбавленных растворах (Ю-10 А4). Изучение электрохи-
мических процессов при столь низких концентрациях пред-
ставляет значительный интерес ввиду того, что при этом
могут быть получены сведения относительно нижнего пре-
дела концентраций, для которых применимы электрохими-
ческие законы. Так, электрохимические исследования мик-
роколичеств полония, проведенные Гайсинским и другими
в Институте радия в Париже, позволили расширить область
применения законов электрохимии до крайне низких кон-
центраций.
Несмотря на чрезвычайно малые количества полония,
имевшиеся для проведения этих работ, были получены до-
вольно надежные данные относительно электродного по-
тенциала Е” Ро/Ро4-, который оказался равным +0,75 в
[163]. В крайне разбавленных растворах нормальный элек-
тродный потенциал можно найти лишь путем экстраполя-
ции значения критического потенциала выделения методом,
впервые примененным Хевеши и Панетом [182] и впослед-
ствии усовершенствованным Жолио [199]. Критический
потенциал выделения был определен по зависимости ско-
рости выделения от катодного потенциала; типичная кривая
выделения показана на рис. 9. В аппарате, сконструиро-
ванном Жолио [199], электроды из металлической фольги,
достаточно тонкой, чтобы обеспечить прохождение а-частиц,
испускаемых выделенным на ней полонием, образуют про-
тивоположные стенки ячейки. Ионизационные камеры были
96
Электрохимия полония
расположены за электродами, что позволяло непрерывно
следить за выделением полония.
Данный метод применяли для определения электродных
потенциалов свинца и висмута, причем в качестве радиоак-
тивных индикаторов использовали соответственно торий-В
и торий-С [157]; по всей вероятности, нет оснований сомне-
ваться в точности результатов, полученных этим методом.
Рис. 9. Кривая электрохимического выделе-
ния микроколичеств полония.
а — выделение на катоде из 0,1 н. раствора HNOa<
(По данным [199].)
При работе с растворами микроколичеств полония (концен-
трация ~ 10“9 М) в разбавленной азотной кислоте для нор-
мального электродного потенциала было получено значение,
равное приблизительно +0,6 в (относительно нормального
каломельного электрода) [182, 183, 308]. Тем не менее счи-
талось, что полоний должен стоять в электрохимическом
ряду между висмутом и теллуром и что указанное значение
нормального электродного потенциала не согласуется с тем
фактом, что гидразин не осаждает полония в виде металла
из раствора; недавно проведенные исследования не подтвер-
ждают, однако, этого предположения.
Критический потенциал выделения полония (при концен-
трации — 10-9 М) из 0,1 н. растворов серной, азотной и
Выделение на катоде ft?
уксусной кислот, а также из раствора 1 н. фосфорной кис-
лоты составляет +0,37 в (относительно нормального ка-
ломельного электрода); совершенно ясно, что в каждой из
этих кислот полоний находится в виде одних и тех же ка-
тионов [198]. В присутствии восстановителей значение
этого потенциала падает до +0,10 в [167].
Данные, полученные в некоторых работах с микроко-
личествами полония, позволили предположить, что в
этом случае видоизмененное уравнение Нернста [£ =
= Ео + (RT/nF)\n а] может и не соблюдаться [170, 199,
342]; последующие более точные измерения показали
[164] некоторые отклонения от уравнения Нернста, но при
этом следует подчеркнуть, что при весьма низких концен-
трациях полония (10“8—10"13 2И), с которыми производи-
лась работа в этих опытах, электрод лишь частично был
покрытосадком полония и активность твердой фазы нельзя
было принимать равной единице [191]. Был выдвинут ряд
теорий выделения полония из растворов, содержащих этот
элемент в микроконцентрациях (см., например, [44, 192]);
было проведено значительное количество работ по электро-
химическому выделению полония из растворов, в которых
концентрация этого элемента составляла 1О-10—10-14 М
[66, 67, 69, 164, 166 ], однако до настоящего времени не разра-
ботано еще исчерпывающей теории, которая позволила бы
количественно предсказать электрохимическое поведение
того или иного элемента в таких крайне разбавленных
растворах.
Электродный потенциал полония нельзя точно устано-
вить на основании опытов по изучению самопроизвольного
выделения полония на менее благородных металлах, ча-
стично вследствие того, что остается невыясненной природа
участвующих в данном процессе ионов полония, частично
из-за неуверенности в значениях электродных потенциа-
лов металлов, используемых в качестве подложки, по-
скольку концентрация ионов этих металлов в растворе при
тех условиях, в которых производились опыты [173, 199],
была неопределенной.
Обзор работ по электрохимическому изучению раство-
ров, содержащих микроколичества полония, сделан Гай-
синским [162, 168, 1921.
7
К. Бэгнал
9Й
Электрохимия полонил
Первая попытка непосредственно измерить обратимый
электродный потенциал при использовании около 10 рьг
полония не увенчалась успехом [157] ввиду нестабильно-
сти электрода, состоявшего из выделенного на золоте по-
лония. Неудача была приписана окислению полония (даже
в отсутствие свободного кислорода) продуктами, образую-
щимися в результате действия а-частиц на растворитель.
Применение больших количеств полония усугубляетэти труд-
ности, поскольку интенсивное действие а-частиц вызывает
химические реакции, которые могут полностью замаскиро-
вать электродный процесс. В одной из последних работ [14 ]
в качестве полониевого электрода применяли золотую про-
волоку диаметром 0,25 мм, покрытую металлическим по-
лонием методом электрохимического выделения; количество
полония составляло 0,2 мг (1 кюри). Поток а-частиц через
поверхность электрода в этих опытах был порядка
10п частиц 1см2-сек и, следовательно, выделяемая энергия
составляла 31,6 мет, что обусловливало разложение при-
близительно 1016 молекул растворителя в 1 сек. Влияние
этого излучения на потенциал неизвестно, однако не уди-
вительно, что попытка непосредственно измерить электрод-
ный потенциал Е” Ро/Ро+ + оказалась неудачной, по-
скольку вероятность нахождения вблизи электрода каких
бы то ни было ионов полония в восстановленном состоянии
была исключительно мала. Тем не менее были определены
следующие электродные потенциалы:
Е“Ро/Ро4+ (в разбавленном азотнокислом растворе)
£;‘Po/PoCle“, £HPt/PoCl;-/PoCle” и Е”Ро РоСГ’.
Значения потенциалов для солянокислых растворов
(6-10~6—10"4Л1 Ро) приписывали комплексным ионам
РоС1““ (Ро IV) и РоС1”“ (Ро II), но эти значения в равной
мере могли бы относиться и к комплексным ионам РоС1“
и РоС1“ соответственно. Пока еще нельзя доказать, какие
из этих комплексных ионов полония в действительности
присутствуют в солянокислом растворе.
выделение на катоде
99
Электрохимическое выделение полония из раствора в 4Л1
соляной кислоте не так давно изучалось видоизмененным
методом Жолио [278]; при этом на кривой, выражающей
зависимость между скоростью выделения и потенциалом,
наблюдалось четыре изгиба. Было высказано предположе-
ние, что эти изгибы отражают переходы из одного окисли-
тельного состояния в другое, однако они не достаточно
четко выражены, и химическое поведение полония в соля-
нокислом растворе не подтверждает этого предположения.
Д. М. Зив [382] предложил новый метод определения потенциа-
лов выделения радиоактивных элементов из разбавленных раство-
ров, основанный на использовании внутреннего электролиза с об-
ратимыми окислительно-восстановительными системами в анодном
пространстве. В дальнейшем этот метод был развит Б. П. Николь-
ским, Д. М. Зивом и Г. С. Синицыной [383—389].
Этим методом была проведена серия электрохимических иссле-
дований с очень разбавленными растворами полония (10-7—10“14 Л4)
[383—3891. Исследования дали следующие результаты:
1. Уравнение Нернста применимо к солянокислым растворам
полония до концентрации 5«10~14 М при использовании золотых
электродов. Выделение полония на золоте протекает обратимо
[384, 388].
2. Уравнение Нернста применимо и к азотнокислым растворам
полония, только величина потенциала сильно зависит от концентра-
ции кислоты*. Увеличение катодного потенциала при концентрации
1,5 н. объясняется появлением состояния окисления полония Ров+;
в 3,0 н. HNOy наблюдается снижение потенциала, вероятно, за счет
образования комплексных ионов [388].
3. Увеличение концентрации кислоты в солянокислых и уксус-
нокислых растворах выше 0,1 н. приводит к снижению потенциа-
ла выделения, и это объясняется образованием комплексных ио-
нов [388].
4. Величина потенциала Ро/Ро4+ на золоте в разбавленных ра-
створах соляной кислоты при изменении ее концентрации от 10-1 до
10“3 н. остается приблизительно постоянной [388].
5. Потенциал Ро/Ро4+ на золоте в 0,01 н. НС1 для концентра-
ции полония 1О“10 М равен + 0,617 ± 0,002 в [386].
6. Нормальный потенциал четырехвалентного полония Ро/Ро4+
в 0,1 н. НС1 на золоте (при изменении концентрации полония от
1,46-10“7 до 5-Ю”14 М) равен 4-0,765 ± 0,006 в. Постоянство по-
тенциала в значительной области изменения концентрации поло-
ния, по-видимому, указывает на то, что потенциал выделения не
* В разбавленных растворах HNOa (10 3 и.) преобладают ионы
четырехвалентного полония,
100
Электрохимия полония
зависит от количества полония, находящегося на электроде. Это
авторы истолковывают так, что полоний образует на электроде само-
стоятельную твердую фазу в виде отдельных агрегатов — «микро-
электродов» [384].
7. Нормальный потенциал Ро/Ро2+ на золоте в 0,1 н. НС1 в
атмосфере SO2 (при изменении концентрации полония от 1,46-10“7
до 5-Ю”14 М) равен +0,68 ± 0,0 в [384].
8. В растворах серной кислоты (10”4 н.) при концентрации по-
лония 10-12 М наблюдается процесс диспропорционирования ионов
четырехвалентного полония с образованием ионов двух- и шести-
валентного полония. Константа диспропорционирования прини-
мается равной 10-8. По-видимому, этот эффект имеет место и в раст-
воре других кислот [385].
9. Потенциал выделения полония на платине в 0,1 н. HCI при
концентрации полония Ю"10 М равен 1,327 ± 0,003 в. Выделение
на платине, как и на золоте, происходит обратимо, но на платине
наблюдается значительное «недонапряжение» и явление старения
[387].
ВЫДЕЛЕНИЕ НА АНОДЕ
Потенциал перекиси полония был определен для растворов,
содержащих микроколичества полония. Полученные зна-
чения при использовании каломельного электрода сравне-
ния равны +0,89 в [183] и +0,82 в [308]. Для электрода
РоО3/РоО“" было вычислено значение Е" = +0,55 в
[162].
ЭЛЕКТРОД Ро/Ро“-
Потенциал электрода Е“ Ро/Ро”“ по расчету должен быть
равен —1,0 в [212 ]; это значение установлено путем экстра-
поляции потенциалов других элементов подгруппы селена.
Данные, полученные для анодного потенциала выделения,
свидетельствуют о том, что выделение происходит не в ре-
зультате разрядки ионов Ро-- [150].
Известные в настоящее время электрохимические данные
для полония приведены в табл. 9.
Из экспериментально полученных значений электрод-
ного потенциала Е“ Ро/Ро (IV) следует, что полоний в элек-
трохимическом ряду занимает место между теллуром и
серебром; это соответствует поведению полония в растворе
в присутствии восстановителей [133].
Электрод PolPcr
101
Таблица 9
Электродные потенциалы полонияа {Ь)
РоН4/Ро Po/Po(IV) Ро/Ро(И) Ро/Ро(1П) o' а. o' а. Po(II)/Po(IV) Po(IV)/Po(Vl)
< -1.1 +0,8 + 0,6 — — +1.1 + 1.1 [341,
стр. 199)
— +0,75 * 6 — +0,56 6 + 1,42 6 — -(163)
— +0,76 +0,38в — — +0,72 в -[14]
— + 0,55 в — — — — -[14)
— + 0,775 6 — +0,56 6 + 1.42® — -1157]
а Ряд других данных приведен в недавно опубликованном докладе (249, стр. 90).
6 Данные получены экстраполяцией значений, относящихся к растворам микро-
количеств полония, на 1тИ раствор.
в Измерения производили в солянокислом растворе, значения потенциалов от-
носятся к комплексным ионам.
ГАЛОГЕНИДЫ И ГАЛОГЕНАТЫ ПОЛОНИЯ
Фториды. Хлориды. Работа с микроколичествами. Тетра- и дихло-
риды полония, гексахлорполониты. Бромиды. Тетра- и дибромиды
полония, гексабромполониты. Тетрайодид полония и гексайодо-
полонит цезия. Смешанные галогениды. Галогенаты.
По химическим свойствам галогениды полония обнаружи-
вают большое сходство с галоидными соединениями теллура,
низшего гомолога полония по подгруппе селена; при этом
галогениды четырехвалентного полония обладают меньшей
устойчивостью, а галогениды двухвалентного полония зна-
чительно большей устойчивостью, чем соответствующие соли
теллура. Как полагают Эрбахер и Кединг [111], в от-
сутствие ионизирующего излучения двухвалентное состоя-
ние полония было бы более стабильным. Все галоидные соеди-
нения полония, подобно соответствующим соединениям тел-
лура, легко гидролизуются; комплексные соли четырех-
валентного полония типа М2РоХб (X = Cl, Вг, J) анало-
гичны соответствующим солям теллура, которым они изо-
морфны, причем цезиевые соли в каждом случае наименее
растворимы. Имеется мало данных' о существовании гало-
генидов трехвалентного полония, аналогичных по свойст-
вам, соответствующим производным висмута — элемента,
соседнего с полонием в периодической системе. Точки плав-
ления и кипения галогенидов полония, полученных в ве-
сомых количествах, указаны в табл 10
Таблица 10
Точки плавления и кипения некоторых галогенидов полония3
РоС14 РоС12 РоВг4 | РоВг2
Точка плавле- ния, °C . . Точка кипе- ния, °C . . 294 [200] 300 [И] 390 [200] 355 [200] Возг. 190 [200] 330 [12] 324 [201] 360^200 мм рт.ст.)[201] 270—280 (12] ПО (З Ю-2 мм рт. ст.) [12]
• Тоцци плавления определяли в запаянных трубках,
Фториды
103
ФТОРИДЫ
Попытки получить летучий фторид полония как в микро-
[103], так и в миллиграммовых количествах [38, 249] ока-
зались безуспешными. По аналогии с фторидами теллура
должны существовать фториды двухвалентного, четырех-
валентного и шестивалентного полония, причем последний
фторид должен быть летучим. Миллиграммовые количества
полония легко растворяются в плавиковой кислоте и, воз-
можно, в этом растворе образуются комплексные ионы фтор-
полонита [249, стр. 48]. Считали, что по аналогии с вис-
мутом будет происходить осаждение трифторида полония
при обработке микроколичеств фторидов полония серни-
стым газом, однако осаждения в этом случае не происходило
[160]. Возможно поэтому, что существует растворимый ди-
фторид полония. Плавиковую кислоту применяли при рас-
творении без видимых потерь микроколичеств полония, об-
разовавшегося в старых радоновых ампулах [23]; продук-
том реакции мог быть нелетучий тетрафторид полония.
Имеются лишь весьма скудные данные, подтверждающие
существование указанных фторидов, и поэтому необходима
дальнейшая работа в этой области.
Вопрос о существовании фторида шестивалентного полония не-
давно обсуждался Гайсинским и Мацура [390]. По-видимому, они
правы в том, что в миллиграммовых количествах вряд ли удастся осу-
ществить синтез PoFe. Если PuFe диссоциирует (под действием
собственного излучения) на PuF4 ф F2. то для PoFe подобная дис-
социация еще более вероятна, так как удельная активность полония
в 6-104 раз выше, чем у плутония. Слабая устойчивость шестива-
лентного состояния полония (по терминологии Менделеева <дви-
теллур») предсказывалась еще Менделеевым [391].
По вопросу о существовании других фторидов полония Бэгнал
с сотрудниками [350, стр. 4] отмечают, что белое твердое вещество,
образующееся при действии разбавленной плавиковой кислоты на
гидроокись полония или на его тетрахлорид, представляет, по-ви-
димому, тетрафторид полония. При взаимодействии с сернистым га-
зом это вещество приобретает голубовато-ссрый цвет, вероятно, в
связи с восстановлением до двухвалентного состояния; при стоянии
голубовато-серое вещество вновь приобретает белую окраску.
Указанные исследователи отмечают, что растворимость четы-
рехвалентного полония в плавиковой кислоте возрастает с повыше-
нием концентрации HF; это, по-видимому, связано с образованием
комплексных ионов.
Точное определение микрограммовых количеств фтора затруд-
нительно, и поэтому была сделана попытка оценить содержание
фтора во фторидах полония по выходу нейтронов в реакции F19
104
Галогениды и галогенаты полония
(а, л) Na22, однако полученные результаты оказались неубеди-
тельными. Таким образом, состав фторидов полония определен
неточно.
ХЛОРИДЫ
Работа с микроколичествами полония
На протяжении многих лет было известно, что полоний в со-
лянокислом растворе образует комплексные ионы. Изуче-
ние микроколичеств полония показало, что электрохимиче-
ское выделение этого элемента из солянокислого раствора
в основном происходит на аноде даже в 0,2 н. кислоте и что
доля полония, выделенного на аноде, возрастает с повыше-
нием концентрации кислоты [265]. Подтверждение сущест-
вования комплексных ионов (РоС!^- или PoCll”) также
было получено в работах по изучению соосаждения микро-
количеств полония [137]. Данные, полученные при экстра-
гировании микроколичеств полония растворителями, также
можно объяснить, допустив образование комплексных
ионов [68].
В более разбавленных солянокислых растворах при
электролизе полоний перемещается к катоду [41]. Резуль-
таты изучения диффузии и подвижности ионов, проводив-
шегося на микроколичествах полония в 0,01 и 0,1 н. раст-
ворах соляной кислоты, свидетельствуют о присутствии в
этих растворах двухвалентных катионов [174—177, 314,
315]. Хевеши [174, 175, 177] высказывал предположение,
что ими могут быть ионы Ро+ +, образующиеся в результате
диссоциации дихлорида полония, однако возможно также,
что эти ионы образуются в результате частичной ионизации
или гидролиза тетрахлорида полония (РоО+ + или РоС12++).
Высказывалось также предположение, что в солянокислых
растворах, концентрация которых лежит в пределах 1—3 н.,
может присутствовать растворимый основной хлорид [137]
и что осаждение происходит в более разбавленной кислоте,
возможно, в результате образования гидроокиси. Работа
с макроколичествами показывает, что комплексные ионы
образуются даже в 1 н. солянокислом растворе [11, 14, 190,
320].
Опыты по соосаждению микроколичеств полония с хло-
Хлориды
105
ристым серебром показали, что очень мало полония осаж-
дается с носителем при высоких концентрациях кислоты
или в присутствии больших концентраций растворимых
хлоридов [108]. Наблюдаемое соосаждение, по всей вероят-
ности, обусловлено адсорбцией на поверхности частиц
осадка. Микроколичества полония не осаждаются с хло-
ридом свинца из раствора в 10 н. соляной кислоте [137],
но в процессе кристаллизации хлорида свинца упаривани-
ем раствора в 0,1 н. соляной кислоте в осадок переходит
приблизительно 20% находящегося в растворе полония.
Возможно, что это происходит в результате механического
включения галогенида полония в кристаллы хлористого
свинца. Микроколичества полония не осаждаются с хлорис-
той ртутью в качестве носителя [34].
Тетрахлорид полония
Тетрахлорид полония — твердое вещество ярко-желтого
цвета, плавящееся в атмосфере хлора приблизительно при
300° [200, 11]. В расплавленном состоянии соль имеет
бледно-желтый цвет, который сохраняется вплоть до 350°,
после чего расплав становится алым, возможно, в результате
разложения до дихлорида; расплав кипит при 390° с выде-
лением пурпурно-коричневых паров, которые при темпе-
ратуре выше 500° приобретают голубовато-зеленую окраску
[11 ]. Изменение окраски паров может происходить вследст-
вие разложения с образованием низших галогенидов или
в результате изменения степени ассоциации.
Изучение летучести микроколичеств тетрахлорида по-
лония показало, что при нагревании сухих образцов выше
150° происходит потеря полония [330]. Это должно сказы-
ваться при получении образцов по методике, включающей
процесс выпаривания аликвотных проб растворов, содер-
жащих тетрахлорид полония, и следовательно, в этих слу-
чаях необходимо принимать меры предосторожности, чтобы
исключить возможность перегрева (см. гл. 3).
Тетрахлорид полония можно получить выпариванием
раствора металлического полония в соляной кислоте [11,
38, 200], нагреванием двуокиси полония в парах четырех-
хлористого углерода при 200° [38, 200], нагреванием ме-
таллического полония в сухом хлоре при 200° [11, 38, 2001,
106
Галогениды и галогенаты полония
а также нагреванием двуокиси полония в сухом газообраз-
ном хлористом водороде, в парах хлористого тионила или
с пятихлористым фосфором [11].
Тетрахлорид полония гигроскопичен так же, как и тет-
рахлорид теллура, и во влажном воздухе, не содержащем
галогенов, быстро гидролизуется с образованием белого
твердого вещества неопределенного состава [200]. Это ве-
щество, возможно, представляет собой смесь основного хло-
рида и гидроокиси или гидратированной окиси [И]. Ана-
логичный продукт образуется при гидролизе тетрахлорида
полония в кипящей воде.
Утверждают, что при выпаривании досуха раствора мик-
роколичеств полония в разбавленной соляной кислоте по-
лучается оксихлорид полония [30]. Растворимость полу-
ченного таким путем соединения изучали в работе [31 ].
В этих препаратах, вероятно, содержалась некоторая доля
тетрахлорида полония.
Тетрахлорид полония растворим в соляной кислоте,
в воде (в которой он медленно гидролизуется) и в хлористом
тиониле. Он обладает умеренной растворимостью в этило-
вом спирте, ацетоне и некоторых других кетонах. Раство-
римость в кетонах может быть обусловлена образованием
соединений с ними [12] (см. гл. 7).
Твердый тетрахлорид полония разлагается в разбавлен-
ной (0,1 н.) азотной кислоте с образованием белого нерас-
творимого твердого вещества, не содержащего хлора. Это
соединение, возможно, является основным нитратом, однако
состав этого соединения еще на установлен. Тетрахлорид
полония превращается в двуокись при нагревании на воз-
духе или в кислороде при 300° или же при длительном пре-
бывании в сухом кислороде [11 ]; пребывание тетрахлорида
полония в сухом воздухе приводит к образованию белого
вещества, получающегося в результате реакции с окислами
азота, которые образуются под действием а-частиц; это со-
единение также может быть основным нитратом.
Растворы тетрахлорида полония имеют ярко-желтую
окраску, и эта окраска в солянокислом растворе заметна
даже тогда, когда концентрация полония составляет всего
лишь 5-10“5 М. В солянокислом растворе образуются
комплексные ионы полония; добавление к раствору тетра-
хлорида полония в 2 н. соляной кислоте спиртового раствора
Хлориды
107
хлористого цезия вызывает немедленное осаждение зелено-
вато-желтого гексахлорполонита цезия [320, 11] (см.
также гл. 5).
Комплексное соединение
тетрахлорида полония
с трибутилфосфатом (ТБФ)
Работы по изучению растворимости тетрахлорида полония
в трибутилфосфате (ТБФ) показывают, что в системе, со-
стоящей из этих двух веществ, образуется комплекс
РоС14-2ТБФ [15].
Взаимодействие с аммиаком
При действии сухого газообразного аммиака на твердый
тетрахлорид полония последний приобретает оранжевую
окраску, появление которой может быть обусловлено либо
образованием аммиаката тетрахлорида полония, либо ча-
стичным восстановлением до дихлорида полония, окрашен-
ного в красный цвет. Тетрахлорид полония почти полно-
стью восстанавливается до металла в атмосфере сухого га-
зообразного аммиака при комнатной температуре, однако
в образующемся при этом продукте рентгеноструктурным
методом были обнаружены следы неизвестного соединения.
Это соединение может быть аммиакатом дихлорида полония.
Желтый гексахлорполонит аммония образуется при на-
гревании тетрахлорида полония в атмосфере газообразного
аммиака при 100° Если учесть, что аммонийная комплекс-
ная соль легко образуется в аналогичный условиях из хло-
ристого аммония и тетрахлорида полония [11], то вполне
вероятно, что следы хлора, адсорбированные на стенках
реакционной трубки, окисляют аммиак с последующим
образованием хлористого аммония, который затем соеди-
няется с тетрахлоридом полония.
Из кислых растворов тетрахлорида полония аммиачная
вода осаждает светло-желтую гидроокись или гидратиро-
ванную окись полония (см. гл. 7).
Дихлорид полония
Дихлорид полония — твердое вещество темного рубиново-
красного цвета; оно возгоняется в атмосфере азота при
190° с незначительным разложением. Дихлорид полония
108
Галогениды и галогенаты полония
получают термическим разложением тетрахлорида поло-
ния в вакууме при 200° [11, 38, 2001, а также восстановле-
нием тетрахлорида полония водородом при 200° [38, 2001,
сернистым газом без нагревания или сероводородом и
окисью углерода при 150° [111. Дихлорид полония восста-
навливается до металла при продолжительном нагревании
в атмосфере водорода [38, 200] или сероводорода [11].
В разбавленной соляной кислоте он дает ярко-розовый
раствор; в таком растворе под действием интенсивного a-из-
лучения полоний быстро (в течение 8 мин.) окисляется
до четырехвалентного состояния. Эффективное окисление
происходит также при действии перекиси водорода или
хлорной воды. Дихлорид полония растворяется в 0,1 н.
растворе азотной кислоты с образованием темно-красного
раствора, который быстро разлагается с выделением белого
хлопьевидного осадка неизвестного состава [11 ].
Дихлорид и тетрахлорид полония реагируют с газооб-
разным аммиаком при 200°, образуя коричневые вещества,
которые, как было установлено по их рентгенограммам,
снятым методом порошков, имеют идентичный состав; та-
ким соединением, вероятно, может быть аммиакат
PoC12-2NH3.
Растворы дихлорида полония можно приготовить вос-
становлением тетрахлорида полония в кислом растворе
действием сернистого газа или гидразина без нагревания,
а также действием мышьяковистого ангидрида при нагре-
вании.
Изучено взаимодействие тетрахлорида полония с жидким сер-
нистым ангидридом |350, стр. 47]. При этом отмечена незначитель-
ная растворимость тетрахлорида без химической реакции с ра-
створителем.
Дихлорид полония не образуется при обработке раство-
ров тетрахлорида полония гидроксиламином или щавелевой
кислотой даже при температуре кипения, и наблюдаемое
Жолио поведение полония в присутствии щавелевой кис-
лоты [199, стр. 147], возможно, связано с образованием
щавелевокислого комплекса, а не с восстановлением че-
тырехвалентного полония.
Самопроизвольное окисление двухвалентного полония
В солянокислом растворе использовали для определения
Хлориды
iOS
окислительно-восстановительного потенциала (см. гл. 5);
изменение потенциала со временем при водородном элек-
троде сравнения показано на рис. 10. Изгиб кривой, воз-
можно, соответствует промежуточному образованию три-
хлорида полония, однако других данных, подтверждающих
Рис. 10. Окисление полония под дей-
ствием собственного излучения; 5*10-4 М
раствор полония в 1 н. соляной кислоте.
(По данным [14].)
образование этого соединения, нет; кроме того, положение
изгиба, по-видимому, зависит от кислотности раствора.
В умеренно кислых растворах дихлорида полония в соля-
ной кислоте присутствуют комплексные ионы (например,
РоС1“ или РоО^-) 114].
Двухвалентное состояние восстановленного полония в
растворе можно было бы подтвердить, вычислив время, не-
обходимое для образования двух эквивалентов окислителя
под действием испускаемого полонием a-излучения. Если
110
Галогениды и галогенаты полония
предположить, что окисление происходит под действием
перекиси водорода по реакции
Ро++ + 2Н+ + Н2О2 -+ Ро4+ + 2Н2О,
и принять выход перекиси водорода G равным единице (об-
разуется одна молекула Н2О2 на 100 эв ионизирующего из-
лучения, поглощенного раствором), тогда время, необходи-
мое для окончания реакции, должно быть равно
6,023-1023-1,06-10“6-100
—----------:---------= 320 сек.,
5,4-106-3,7-Юю
поскольку 1 кюри Ро210 = 1,06-10~6 моль = 3,7-Ю10 а-ча-
стиц/сек с энергией 5,3 Мэв. Позднейшие определения вы-
хода перекиси водорода в растворах хлоридов, подвергав-
шихся облучению а-частицами, дают значение G, равное
0,6 [223], тогда вычисляемое время будет равным 530 сек.,
а это значение уже более близко к наблюдаемому. Окисле-
ние, вероятно,— сложный процесс.
Гексахлорполониты
Работы с микроколичествами полония показывают, что
в водных растворах существуют ионы гексахлорполонита
[137 ]; это подтверждается недавним получением гексахлор-
полонитов цезия, рубидия, калия, аммония и тетраметил-
аммония [320]. Эти соли имеют желтую окраску, и все
они изоморфны соответствующим солям теллура и другим
аналогичным комплексным солям четырехвалентных ме-
таллов. Препараты для кристаллографического исследова-
ния получали осаждением (соль цезия [320, 111), упарива-
нием солянокислых растворов тетрахлорида полония с со-
ответствующим галогенидом одновалентного металла [320]
или нагреванием твердого тетрахлорида полония с этим га-
логенидом (соль аммония [11]).
В результате разложения под действием излучения кри-
сталлы тетраметиламмония становятся оптически неодно-
родными; через 5—10 мин. такое изменение сопровождается
потускнением кристаллов. Рентгенограммы порошков со-
лей цезия [320, 11 ] и аммония [11] показали, что эти со-
Хлориды
1И
единения имеют гранецентрированную кубическую решетку.
Кристаллографические данные, полученные при изучении
хлоридов полония, приведены в табл. 11.
Таблица 11
Кристаллическая структура хлоридов полония
РоС14 РоС1а о о Р. Я О б л Е Z о о Z Е О
Структура Моноклин- ная или триклинная [200] Ромбиче- ческаяа [200,11] Г ранецентрированная кубическая
Вычислен- ная плот- ность, г/см3 6,50 [11] 3,82[320] 2,76 [11]
Периоды ре- шетки, kX а=3,66; 6=4,34 с=4,49 [11] а=4,33; 6=8,94 с=7,29 [200] а=10,59 [320] а=10,33 [И]
Показатель преломле- ния (5893 А) — — 1,86(8) [320] -— 1,618 [320]
а На элементарную ячейку дихлорида полония, очевидно, приходится рдна
молекула PoCL;вероятно, поэтому данная ячейка представляет собой псевдоячейку,
и структура рассматриваемого соединения может принадлежать к пространственной
группе более низкой симметрии, относящейся к моноклинной или триклинной систе-
ме с углом или углами, близкими к 90°.
Опубликованы результаты спектроскопических иссле-
дований растворов тетрахлорида полония в соляной кис-
112
Галогениды. и галогенаты полонил
лоте [190], однако объяснение этих результатов связано
с известными трудностями ввиду ненадежности сведений
о природе ионов, присутствующих в растворе. Мольный
коэффициент поглощения для комплекса хлорида поло-
ния, образующегося в 12,2 н. соляной кислоте + равен (1,058,
±0,028) 10-4 (для длины волны 418 мр).
Константа равновесия реакции
Ро4+ + 6СГ = РоС1~"
была вычислена по электрохимическим данным; она имеет
порядок 1014 [14].
Н итрозилхлорид
При взаимодействии тетрахлорида полония с жидким хлористым
нитрозилом после выпаривания образуется желтый твердый оста-
ток. Вещество легко разлагается в разбавленном водном растворе
(0,5 н.) едкого кали. Анализ желтого остатка показал, что на один
атом полония приходится 4 атома азота. Это позволяет приписать
соединению одну из следующих формул: (NO)2PoCle-2NOCl или
(NO)2PoCle-N2O4, однако для окончательного решения вопроса не-
обходимы дальнейшие исследования [350, стр. 4].
БРОМИДЫ
Тетрабромид полония
Тетрабромид полония — ярко-красное твердое вещество,
плавящееся в атмосфере паров брома приблизительно при
330° [12, 201] с образованием темной жидкости, кипящей
при 360° под давлением 200 мм рт. ст. [201 ]. Тетрабромид
полония получают нагреванием металлического полония
в течение 1 часа в парах брома под давлением 200 мм рт. ст.
и температуре 250° [201, 38], а также нагреванием металла
в токе сухих паров брома, разбавленных азотом, в течение
5 мин. при 200—250°, растворением металлического поло-
ния или двуокиси полония в бромистоводородной кислоте
и выпариванием раствора до получения сухого остатка или
же нагреванием двуокиси в газообразном сухом бромистом
водороде [12]. Металлический полоний без нагревания ре-
агирует с бромом (жидким или парообразным) менее энер-
гично.
Бромиды
113
Тетрабромид полония растворяется в разбавленной бро-
мистоводородной кислоте с образованием оранжево-крас-
ного (10“3 М РоВг4) или карминово-красного (0,025 М
РоВг4) раствора. Эти растворы, очевидно, содержат ком-
плексные ионы гексабромполонита (РоВг~~), так как
добавление бромида цезия в разбавленный раствор броми-
стоводородной кислоты вызывает немедленное осаждение
темно-красной соли цезия. Растворы в разбавленной бро-
мистоводородной кислоте при охлаждении до —30® выде-
ляют темно-коричневый осадок; возможно, что этот осадок
является комплексной гексабромполониевой кислотой.
Это соединение, которое, по-видимому, гидратировано по-
добно соответствующему соединению теллура, неустойчиво
при комнатной температуре и разлагается с образованием
раствора тетрабромида полония в бромистоводородной кис-
лоте [12].
Тетрабромид полония растворим в этиловом спирте,
ацетоне и ряде других кетонов; умеренно растворим в жид-
ком броме. Он не растворим в бензоле, хлороформе и четы-
реххлористом углероде. Тетрабромид полония гигроско-
пичен и при гидролизе образует белое твердое вещество
неопределенного состава, предположительно являющееся
основным бромидом, который может содержать некоторое
количество гидроокиси или гидратированной окиси.
Тетрабромид полония реагирует с аммиаком при ком-
натной температуре, образуя желтый аммиакат, который
при стоянии разлагается с образованием смеси дибромида
полония и металлического полония. Возможно образование
летучего бесцветного аммиаката [12]. При 100° основным
продуктом взаимодействия с аммиаком является гексабром-
полонит аммония (ср. с тетрахлоридом полония).
Дибромид полония
Дибромид полония — пурпурно-коричневое твердое веще-
ство, возгоняющееся с незначительным разложением при
110° и давлении 3-10”2 мм рт. ст.: при плавлении в атмо-
сфере азота при 270—280°, по-видимому, происходит дис-
пропорционирование этого соединения [12]. Дибромид по-
лония получают термическим разложением тетрабромида
$ К. Вэгнал
114
Галогениды и галогенаты полония
полония при 200° или восстановлением тетрабромида поло-
ния газообразным сероводородом без нагревания. Серни-
стый газ восстанавливает твердый тетрабромид полония
при нагревании. Растворы дибромида полония можно при-
готовить восстановлением растворов тетрабромида полония
сернистым газом или гидразином без нагревания. В отличие
от хлорида полония в данном случае нет оснований полагать,
что в процессе окисления до четырехвалентного состоя-
ния под действием собственного излучения происходит
промежуточное образование трибромида полония [12].
Дибромид полония растворяется в бромистоводородной
кислоте и в ряде кегонов, образуя растворы пурпурного
цвета, в которых полоний быстро окисляется до четырехва-
лентного состояния. При нагревании твердого дибромида
полония в газообразном аммиаке сразу же происходит
восстановление до металла; какие-либо указания о сущест-
вовании аммиакатов дибромида полония не получены.
Гексабромполониты
Гексабромполонит цезия красного цвета был получен осаж-
дением бромидом цезия из растворов тетрабромида поло-
ния в разбавленной соляной кислоте.
Красная аммонийная соль образуется при нагревании
до 100° тетрабромида полония в газообразном аммиаке;
если нагревание производить в закрытом сосуде, то эта соль
чернеет и разлагается со взрывом, возможно в результате
образования взрывчатого нитрида полония [12 ].
Кристаллическая структура бромидов
Тетрабромид полония, по-видимому, имеет гранецентриро-
ванную кубическую решетку с периодом 5,60 kX\ элемен-
тарная ячейка этого соединения содержит только одну
молекулу РоВг4, и возможно, что эта ячейка является псев-
доячейкой. Кристаллическая структура дибромида не уста-
новлена. Кристаллы гексабромполонитов имеют гране-
центрированную кубическую структуру; они изоморфны
гексахлорполонитам и гексабромтеллуритам. Периоды ре-
шетки этих солей соответственно равны 10,82 kX [(NH4)2
РоВгв 1 и 10,99 kX (Cs2PoBr6). Вычисленные значения плот-
ности соответственно равны 3,78 и 4,75 г/см* [121.
Тетрайодид полония 115
ГЕТРАЙОДИД ПОЛОНИЯ
Тетрайодид полония — летучее твердое вещество черного
цвета, возгоняющееся в атмосфере азота при 200° с частич-
ным разложением до металла. Тетрайодид полония можно
получить из элементов при нагревании до 40® под давлением
1 мм рт. ст., а также в результате взаимодействия гидро-
окиси или двуокиси полония с 0,1 н. раствором йодистово-
дородной кислоты или при добавлении 0,1 н. раствора йоди-
стоводородной кислоты к раствору тетрахлорида полония
в разбавленной соляной кислоте. Это соединение можно
получить в виде черного сублимата при нагревании двуокиси
полония в сухом газообразном йодистом водороде прибли-
зительно при 200°; без нагревания образуется черное моле-
кулярное соединение (PoO2xHJ). Металлический полоний
не реагирует с йодом, растворенным в четыреххлористом
углероде, однако с йодом, растворенным в бензоле, поло-
ний образует соединение, состав которого не установлен.
Тетрайодид полония слабо растворим в ацетоне и этило-
вом спирте (1 г PoJ4 на 1 л), но не растворим в соляной
(2 н.) и азотной (1 и 2 н.) кислотах, уксусной кислоте, хло-
роформе, бензоле, четыреххлористом углероде, этиловом
и бутиловом эфирах. В воде тетрс йодид полония медленно
гидролизуется. Состав белых продуктов гидролиза не уста-
новлен, но, вероятно, они похожи на продукты гидролиза
тетрахлорида и тетрабромида полония; при стоянии они
становятся коричневыми, возможно в результате адсорб-
ции йода, выделяющегося под действием облучения. Тет-
райодид полония разлагается при действии горячей кон-
центрированной азотной кислоты, гипохлорита натрия,
хлора и подкисленного раствора нитрита натрия; процесс
разложения идет медленнее при действии концентрирован-
ного раствора едкого кали. Результаты изучения раствори-
мости тетрайодида полония в растворе йодистоводородной
кислоты свидетельствуют о присутствии в растворе ионов
гексайодполонита (PoJ^““). Значение константы равнове-
сия реакции
PoJ4 +2J’^PoJe"
равно (5,9 ± 0,2) 10‘3 при 22°. Зависимость растворимости
тетрайодида полония при постоянной концентрации йодисто-
водородной кислоты от температуры показана на рис. 11.
8*
116
Галогениды и галогенаты полония
Возможно, что при низких концентрациях кислоты
([HJ ] <0,02 н.) образуются ионы пентайодполонита по
реакции
PoJ4+J" = PoJ;.
Константа равновесия этой реакции при 22° равна прибли-
зительно 6,7-10-5. Растворы гексайодполониевой кислоты
Рис. 11. Зависимость растворимости
тетрайодида полония от температуры
при постоянной концентрации (0,3 н.)
соляной кислоты.
(По данным [13].)
в соляной кислоте имеют зеленую окраску при 0* и стано-
вятся красно-коричневыми при 20°.
Тетрайодид полония восстанавливается до металла при
нагревании в газообразном сероводороде и не реагирует
с аммиаком. Суспензии в 0,1 н. йодистоводородной кислоте
не восстанавливаются гидразином и сернистым газом даже
при кипячении; реакция не идет и в том случае, когда к рас-
твору дихлорида полония в разбавленной соляной кислоте
добавляют водный раствор йодистоводородной кислоты или
йодистого калия. Таким образом, данных об образовании
дийодида полония не получено [13].
Смешанные галогенсодержащие соединения
117
Гексайодполонит цезия
Это соединение получают в виде черного осадка при добав-
лении раствора йодида цезия в йодистоводородной кислоте
к раствору тетрайодида полония в 2 н. йодистоводородной
кислоте [9, 13]. Гексайодполонит цезия имеет гранецен-
трированную кубическую решетку (а = 11,77 kX)\ он изо-
структурен аналогичным соединениям теллура. При нагре-
вании в вакууме это соединение разлагается на йодид це-
зия и тетрайодид полония; оно легко гидролизуется водой
[131. Есть сообщения об изучении совместной кристалли-
зации микроколичеств полония с гексайодтеллуритом це-
зия [202].
СМЕШАННЫЕ ГАЛОГЕНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ
Дихлорид и дибромид полония при взаимодействии с йодом
в растворе четыреххлористого углерода могут образовать
неустойчивые смешанные галогениды [131. Дихлорид по-
лония реагирует с парами брома при комнатной температуре;
в результате реакции образуется оранжево-розовый про-
дукт, являющийся, по-видимому, дихлордибромидом по-
лония. Дихлорид и дибромид полония не реагируют с па-
рами йода при нагревании [12].
Галогенаты полония
О галогенатах полония известно пока очень мало.
Перхлорат [3921
При испарении хлорнокислого раствора, содержащего ионы четы-
рехвалентного полония, выделяется белый хлопьевидный осадок,
нерастворимый в разбавленной хлорной кислоте. Возможно, что он
является перхлоратом полония.
Бромат [350. стр. 4]
Гетрахлорид полония при взаимодействии с 15%-ной бромноватой
кислотой образует белое твердое вещество, которое при нагревании
образует двуокись полония; возможно, что это вещество является
продуктом гидролиза тетрахлорида в бромноватой кислоте, а не бро-
матом полония. Растворимость его в 15%-ной бромноватой кислоте
отвечает 12.5 мг PoOi на I л бромноватой кислоты.
118
Галогениды и галогенаты полония
Двуокись и гидроокись полония не реагирует с 10%-ной бром-
новатой кислотой, однако часть продукта растворяется в ней (2,5 мг
РоОа на 1 л кислоты).
Если 3%-ный раствор бромноватой кислоты в 4 н. HNOa добав-
лять к раствору нитрата полония в 4 н. HNOa, то никаких измене-
ний не наблюдается.
Металлический полоний не реагирует с 15% ной бромноватой
кислотой при 70°, возможно, в связи с образованием на поверхности
металла тонкой защитной пленки двуокиси полония.
Йодат 1350, стр. 5J
При взаимодействии насыщенного раствора йодноватой кислоты
в 4 н. HNOa и раствора нитрата полония в 4 н. HNOs с последующим
упариванием смеси на водяной бане образуются белые кристаллы
йодата полония (аналогия с йодатом тория). При нагревании йодата
полония до 350—400° соединение разлагается с выделением йода и
пятиокиси йода.
Свежеприготовленный белый йодат полония по истечении 2 час.
становится кирпично-красным и выделяет свободный йод в резуль-
тате разложения под действием собственного a-излучения полония.
После 12 час. йодат полония чернеет и на его дебаеграмме обнару-
живается a-фаза полония.
Йодат полония нельзя получить осаждением из 8 н. (и выше)
раствора азотной кислоты, так как в этих условиях, возможно, про-
исходит подавление диссоциации йодноватой кислоты или образо-
вание устойчивого нитратного комплекса.
В приведенной ниже таблице представлены приблизительные
данные по растворимости йодата полония в 2 н. HNO8 и зависимости
ее от избытка йодноватой кислоты.
HJO3, н.
0,045
0,083
0,115
0,143
0,167
Растворимость,
мкюри! мл
43
17
16
12,5
7,3
Йодат полония растворим в 2 н. соляной кислоте.
При взаимодействии гидроокиси полония с разбавленной (0,2 н.)
йодноватой кислотой не наблюдалось образования белых кристал-
лов йодата полония.
Надйодная кислота в растворе азотной кислоты не дает осадка
с нитратом полония в азотной кислоте (отличие от тория).
ОКИСЛЫ И ДРУГИЕ СОЕДИНЕНИЯ ПОЛОНИЯ
Окислы и гидроокиси. Сульфид. Сульфаты. Селенат. Нитрат. Соли
органических кислот и металлорганические соединения.
ОКИСЛЫ ПОЛОНИЯ
Трехокись полония
Из’положения полония в периодической системе элементов
можно предположить о существовании нестойкой трехокиси
полония, имеющей преимущественно кислотные свойства
[241 1. О приготовлении макроколичеств этой окиси не со-
общалось, но некоторые данные, подтверждающие сущест-
вование трехокиси, получены при изучении электрохими-
ческого выделения микроколичеств полония Хевеши и
Панет [182, 1831 считают, что продукт, получаемый при вы-
делении на аноде микроколичеств полония, представляет
собой высший окисел и что поведение полония в этом отно-
шении аналогично поведению свинца и марганца. Выделе-
нию на аноде мешает перекись водорода [198, 165] и винная
кислота [1481; причиной такого действия этих реагентов
может быть восстановление высшего окисла на этом элек-
троде или возможное образование комплексных ионов в
случае винной кислоты. Показательно также, что анодное
выделение становится более эффективным в присутствии
таких окислителей, как трехокись хрома [198, 199, 152].
Двуокись полония
Двуокись полония образуется непосредственно из элемен-
тов при 250э. Состав этого окисла был установлен измере-
нием падения давления кислорода, находящегося в контакте
с металлическим полонием [238, 251 1. Двуокись существует
в двух кристаллических модификациях [238, 101: тетраго-
нальная (красная) и гранецентрированная кубическая (жел-
тая). Последняя, по-видимому, представляет собой низко-
температурную форму; превращение тетрагональной формы
в гранецентрированную кубическую зависит от темперд-
120
Окислы и другие соединения полония
туры и обратимо. Гранецентрированная кубическая моди-
фикация, по-видимому, является окислом типа UO2 с пере-
менным содержанием кислорода; период решетки ее изме-
няется в интервале 5,626—5,687 kX [10]. Периоды решет-
ки тетрагональной модификации равны: а = 5,44 kX и
с = 8,34 kX.
Радиус иона Ро4^, вычисленный по рентгенографиче-
ским данным, равен 1,02 А [238] или 1,04 А [10]; вычислен-
ное значение плотности гранецентрированной кубической
модификации составляет 8,96 г!см? [10] или 9,18 г! см?
[238]. Отношение ионных радиусов Ро4+/О2-в этом соеди-
нении равно 0,73, что соответствует нижнему пределу устой-
чивости для кубической координации, и это может объяс-
нить существование двух кристаллических форм [238].
Свежеприготовленные образцы двуокиси всегда состоят из
высокотемпературной (тетрагональной) формы, а гранецен-
трированная кубическая форма образуется после выдер-
живания образца в течение нескольких суток [238, 101 *
или после сильного охлаждения [10]. Двуокись полония
темнеет при нагревании, приобретая шоколадный цвет при
температуре возгонки (885°), она разлагается на элементы
при нагревании до 500° в вакууме [10] и медленно восста-
навливается до металла в водороде при 200° [251 ]. Хими-
ческие свойства двуокиси полония аналогичны химическим
свойствам двуокиси теллура; при взаимодействии этих со-
единений с кислотами образуются соли четырехвалентного
полония (см. выше).
Двуокись полония при температуре 250° реагирует с газообраз-
ным аммиаком и сероводородом с образованием темной массы нео-
пределенного состава; рентгенограмма продукта, снятая методом
порошков, показывает присутствие в нем непревращенной двуокиси,
и поэтому черная окраска продукта, возможно, обусловлена при-
сутствием тонкораздробленного металлического полония на поверх-
ности твердой двуокиси [350, стр. 3].
При нагревании двуокиси до 250° в атмосфере сернистого газа
образуется твердое белое вещество со следами красноватой примеси,
которой, возможно, является PoSO3 или Po(SO4)2.
Большая часть полония остается при этом в четырехвалентном
состоянии и белое твердое вещество является, по-видимому, суль-
* См. примечание на стр. 69, Прим, ред^
Окислы полония
121
фитом, так как сульфат полония при 250° окрашен в пурпурный или
желтый цвет.
Двуокись полония не реагирует с жидким сернистым ангид
ридом.
Недавно были опубликованы некоторые данные по рас-
творимости окисленного полония в ряде реагентов [249,
стр. 53]. Эти данные приведены в табл. 12. Химический со-
став соединений, образующиеся в этих условиях, неизве-
стен.
Таблица 12
Растворимость полония в различных реагентах
Реагент Концентрация, М Растворимость полония, МКЮри/МЛ
Азотнокислый натрий 1,5 1,4
Углекислый > 0,5 1,4
> > 0,05 1,6
» аммоний 0,75 24,0
> . » 0,25 0,4
Ортофосфорная кислота 0,5 30,0
Однозамещенный фосфорнокислый ка-
лий 1,0 4,0
Двузамещенный фосфорнокислый ам-
моний 1,0 1,7
Двузамещенный фосфорнокислый нат-
рий 1,0 4,8
Уксуснокислый натрий • . 1,0 0
Щавелевая кислота 0,5 42
Кислый щавелевокислый калий .... 0,4 62
Лимонная кислота 1,0 46
Кислый виннокислый калий 1,0 41
Теплота образования двуокиси полония по расчетным
Данным равна — 30 ккал/моль [50].
Моноокись полония
Это соединение может образовываться в виде черного твер-
дого вещества при самопроизвольном разложен и сульфо-
трехокиси и селенотрехокиси полония [48 L
122
Окисли и другие соединения полония
ГИДРООКИСИ
О гидроокисях полония не было опубликовано ана-
литических данных, и возможно, что соединения, о которых
сообщалось в литературе как о гидроокисях, в действитель-
ности представляют собой гидратированные окислы.
Четырехвалентный полоний
По аналогии с теллуром гидроокись полония должна обла-
дать слабыми кислотными свойствами [241 ], и такая оценка,
по-видимому, подтверждается опытными данными, полу-
ченными при работе с растворами, содержащими микроко-
личества полония (см., например, [153, 2651). Весьма веро-
ятно, что гидроокись полония (IV) имеет формулу
РоО(ОН)2 [137] и при растворении этого соединения в ще-
лочи могут образоваться ионы полонита РоО* + [136, 137,
151 1; как показывают радиографические исследования, мик-
роколичества полония действительно растворяются в рас-
творе едкого кали [24 1, и, как найдено в других работах,
эта растворимость повышается с увеличением концентрации
щелочи [134, 135, 150, 153]. Работы по изучению анодного
растворения микроколичеств полония с золотых электро-
дов в растворе едкого натра также позволили получить не-
которое подтверждение амфотерного характера данного
соединения [266, стр. 51 ], а изучение методом центрифу-
гирования, по-видимому, также свидетельствует о его сход-
стве с теллуровой кислотой [134]. Микроколичества поло-
ния осаждаются на серебре из раствора в 0,1 н. едком на-
тре, природа осадка точно неизвестна, однако осадок можно
снова растворить, если обработать его кипящим концентри-
рованным раствором едкого натра [108].
Подобно этому, осадки микроколичеств полония на зо
лотых электродах, по имеющимся сведениям, растворяются
в кипящей воде [215]. Вполне вероятно, что растворы в
обоих случаях содержат гидроокись полония.
В опытах с миллиграммовыми количествами при добав-
лении аммиачной воды или раствора щелочи к кислым рас-
творам свежеприготовленного тетрахлорида или тетрабро-
мида полония был получен бледно-желтый хлопьевидный
осадок. Осадки из старых растворов, содержащих относи-
Гидроокиси
123
тельно большие количества свинца, образовавшегося в про-
цессе распада, имеют окраску, изменяющуюся от красно-
вато-желтой до светло-коричневой. Изучение растворимо-
сти этого соединения в растворе едкого кали показало его
слабокислотный характер (данные о растворимости гидро-
окиси полония в водных щелочных растворах приведены
в табл. 13). Константа равновесия реакции
РоО (ОН)2 + 2КОН=К2РоО3 + 2Н2О
Кс= 1/ [КОН F при 22° равна (8,2 ± 0,4)-10"5.
Таблица 13
Растворимость гидроокиси полония в водных
щелочных растворах при 22°
Щелочи Концентрация, молъ/л Растворимость, мкюри/мл Литература
кон 0 0,060 [45]
0,260 4,7 Там же
0,433 14 » »
0,605 26 » >
0,865 55 » >
1,30 135 » >
1,73 240 » »
NH4OH 18 0,20 » >
18 1,0 [249, стр. 53]
Подобно теллуровой кислоте, гидроокись полония значи-
тельно менее растворима в аммиаке, чем в водных щелочных
растворах [45].
Растворимость гидроокиси полония в воде и в водных раство-
рах аммиака определялась также и другими исследователями (360,
365, 366]. При этом было установлено, что гидроокись полония яв-
ляется груднорастворимым соединением. Однако остается не ясным,
всегда ли авторы указанных работ имели дело именное гидроокисью,
так как недавно Бэгналом было показано [378], что при осаждений
гидроокиси аммиаком происходит выделение металлического поло-
ния и этот процесс идет тем быстрее, чем выше концентрация аммиака
и, по-видимому, концентрация полония. Вероятно, однозначный
ответ о величине растворимости гидроокиси полония *можно будет
получить лишь при использовании его более долгоживущего изо-
топа.
124
Окислы и другие соединения полония
Двухвалентный полоний
Добавление раствора едкого кали к свежеприготовленному
раствору дихлорида полония в разбавленной соляной кис-
лоте вызывает выпадение темно-коричневого осадка (раство-
римость по Ро210 составляет 1,4 который быстро окис-
ляется до четырехвалентного состояния [11 ]. Состав этого
осадка неизвестен, но возможно, он представляет собой гид-
роокись двухвалентного полония. Анализ этого соединения
затруднен окислением полония до четырехвалентного со-
стояния, вызываемым, по-видимому, продуктами радио-
лиза растворителя, остающегося в осадке.
СУЛЬФИД ПОЛОНИЯ
Еще в процессе наиболее ранних работ по обнаружению и
выделению полония было установлено, что полоний можно
осадить действием сероводорода из кислого раствора вместе
с другими нерастворимыми сульфидами (свинца, меди, вис-
мута и т. д.); в случае микроконцентраций полнота осажде-
ния достигает приблизительно 97% [134]. Осажденный
сульфид полония нерастворим в сернистом аммонии и, по-
видимому, обладает большей летучестью, чем сульфид вис-
мута, от которого его можно отделить возгонкой в вакууме
при 700° [83]. В одной из последующих работ было пока-
зано, что сульфиды висмута и полония в этих условиях раз-
лагаются до металла и разделение, о котором сообщалось
в печати, можно поэтому представить как фракционную
возгонку этих двух металлов. Микроколичества сульфида
полония нерастворимы в растворе щелочных сульфидов,
но растворяются в концентрированной [34] или в 4 н. со-
ляной кислоте [24]; они менее растворимы в кислоте по
сравнению с сульфидом висмута [83]. Из больших коли-
честв сульфида висмута [178] или сульфида теллура [135]
микроколичества полония можно выделить выщелачива-
нием осадка 1 н. соляной кислотой.
Поскольку в период проведения упоминавшихся выше
работ полоний был доступен лишь в незначительных коли-
чествах, анализировать осажденный сульфид полония было
невозможно. Гийо [1371 полагает, что действие сероводо-
рода на кислый раствор, содержащий микроколичества че-
Сульфаты полония
125
тырехвалентного полония, должно приводить к образова-
нию Po2S3; кроме того, считали также, что это соединение
должно образовываться при медленном разложении ди-
этилдитиокарбамата полония, растворенного в хлороформе.
Тем не менее достаточно веских доказательств для призна-
ния того, что в этом соединении полоний является трехва-
лентным, по-видимому, не имеется.
Более поздние работы с миллиграммовыми количествами
полония [И] показали, что при действии сероводорода на
10"3 М раствор четыреххлористого полония в разбавленной
соляной кислоте выпадает осадок черного цвета. Образую-
щийся продукт представляет собой моносульфид полония
PoS, нерастворимый в сернистом аммонии, этиловом спирте,
толуоле и разбавленной соляной кислоте [47 ]. При стоянии,
однако, он медленно растворяется в разбавленной соляной
кислоте, возможно благодаря действию хлора, выделяюще-
гося из кислоты под воздействием а-частиц. Он легко раз-
лагается при действии брома, царской водки и раствора
гипохлорита натрия. Моносульфид полония сравнительно
хорошо растворим в концентрированной соляной кислоте;
произведение растворимости его равно приблизительно
5,5-10-29.
Моносульфид полония в вакууме при 275° разлагается
с выделением металлического полония; это свойство может
быть использовано для приготовления чистого полония из
старых осадков, а также для аналитического выделения
полония (см. гл. I). Моносульфид образуется при дейстзии
сероводорода на кислые растворы дихлорида полония 147 ]
или при обработке гидроокиси полония сернистым аммо-
нием, однако это соединение нельзя получить из элементов
[38]; дисульфид полония неизвестен.
СУЛЬФАТЫ ПОЛОНИЯ
Работы по изучению диффузии микроколичеств полония
в разбавленной серной кислоте позволяют сделать вывод
о наличии в растворе двух- и трехвалентных ионов [314,
315]; знак заряда этих ионов неизвестен. Измерения потен-
циала выделения микроколичеств полония из нейтрального
или подкисленного раствора сульфата калия [138,- 152]
и данные экстрагирования растворителями [68] указывают
126
Окисли и другие соединения полония
на присутствие комплексных ионов; однако в концентриро-
ванной серной кислоте сульфатный комплекс, по-види-
мому, неустойчив [154]. Микроколичества полония можно
осадить с сульфатом свинца [83] и выделить обработкой
осадка дымящей серной кислотой. Соосаждение, возможно,
обусловлено адсорбцией.
Работы с большими количествами полония показали,
что можно получить сульфаты четырехвалентного полония
двух видов: 2PoO2-SO8 и Po(SO4)2 [48].
Дисульфат полония
Гидратированный дисульфат полония образуется в виде бе-
лого твердого вещества при обработке твердого тетрахло-
рида полония или гидроокиси полония серной кислотой
Рис. 12. Растворимость сульфатов полония
в серной кислоте и селената полония в селе-
новой кислоте.
(По данным [48].)
(более 0,5 н.); дисульфат умеренно растворим в разбавлен-
ной серной кислоте, но растворимость его возрастает с уве-
Сульфаты полония
127
личением концентрации кислоты, что, вероятно, можно
объяснить образованием комплексных ионов (рис. 12).
Выделенное из раствора соединение при стоянии дегидра-
тируется, при нагревании дегидратация происходит быстро
и при 200° соединение становится розовым, а выше 380° —
темно-пурпурным. Дисульфат можно также дегидратировать
промыванием безводным эфиром. Темно-пурпурная безвод-
ная соль относительно устойчива к нагреванию и разла-
гается до двуокиси при температуре, превышающей 550е.
Дисульфат полония нерастворим в ацетоне и этиловом
спирте; при действии спирта происходит гидролиз. Он очень
хорошо растворяется в разбавленной соляной кислоте [48].
Основной сульфат полония
Это соединение, имеющее состав 2PoO2SO3, образуется
в виде белого твердого вещества (желтого выше 250°) при
взаимодействии тех же веществ, при которых образуется
дисульфат, но при меньшей концентрации серной кислоты
(0,02—0,25 н.) и может также образоваться в результате
гидролиза дисульфата. Основной сульфат устойчив до тем-
пературы, превышающей 400°, и распадается до двуокиси
приблизительно при 550е. Он лучше растворим в разбав-
ленной серной кислоте, чем дисульфат (см. рис. 12), и легко
растворяется в разбавленной соляной кислоте. Изучение
растворимости основного сульфата показало, что это соеди-
нение метастабильно в контакте с разбавленной (0,1—0,5 н.)
серной кислотой [481.
Сульфат двухвалентного полония
Суспензии дисульфата полония в 1—2 н. серной кислоте
растворяются при кипячении с гидроксиламином, образуя
при этом розовый раствор; такое изменение цвета харак-
терно для двухвалентного полония. Этот результат согла-
суется с наблюдениями над поведением микроколичеств
полония [132, 167]. Дисульфат вновь выпадает в осадок
при охлаждении в течение нескольких минут; это происхо-
дит даже в присутствии избытка гидроксиламина [48].
128
Окислы и другие соединения полония
СЕЛЕНАТ ПОЛОНИЯ
Основной селенат полония 2PoO2SeO3 образуется в вид<
белого твердого вещества (темно-желтого при температуре
выше 250°) при обработке тетрахлорида полония или «гид
роокиси» селеновой кислотой (0,015—5,0 н.). Это соедине
ние устойчиво до температуры, превышающей 400°, и очеш
хорошо растворимо в разбавленной соляной кислоте. Рас
творимость его в разбавленной селеновой кислоте уве
личивается по мере увеличения концентрации кислоть
(см. рис. 12).
Хромат полония (350, стр. 6]
Водный 1 М раствор трехокиси хрома не реагирует с металли-
ческим полонием. Однако при действии 1 М раствора СгОз на гидро
окись или тетрахлорид полония образуется оранжево-желтое твер
дое вещество, представляющее собой, по-видимому, хромат,
Ро (СгО4)2. Хромат полония нерастворим в избытке реактива и раз-
лагается при промывании водой или сухим ацетоном. Образующийся
темно-коричневый продукт разложения слегка растворим в воде
(4 мкюри/мл)\ по данным анализа, его состав близок к 2РоО2.-СгОз
(по аналогии с сульфатом или селенатом полония). Как хромат поло
иия, так и продукт его разложения легко растворяются в разбавлен-
ной соляной кислоте (2 и.) с образованием тетрахлорида. При нагре-
вании в вакууме до 500° оба продукта становятся полностью нерас-
творимыми в кислотах и щелочах и полоний из них извлечь нельзя
Причины этого явления не ясны. Попытки определить их раствори
мость в водном растворе хромата калия оказались неудачными вслед
ствие восстановления хрома под действием a-излучения. Если же
хромат полония находится в контакте с избытком раствора трех-
окиси хрома (1Л4.) в течение ночи, то весь хромат полония раство-
ряется, образуя темно-коричневый раствор. Добавлением к этому
раствору аммиачной воды осаждают почти весь полоний в виде
красно-коричневого вещества. Этот осадок медленно растворяется
в 2 н. соляной кислоте и полностью — в водном 2 н. растворе едкого
кали. Образующийся в этом опыте зеленый раствор содержит ам-
миакат трехвалентного хрома, аквокомплекс хрома и шестивалент-
ный хром.
Так как растворимость гидроокиси двух- и четырехвалентного
полония весьма мала, то валентность полония в этом растворе, по-
видимому, равна не четырем и не двум, а шести и он находится или
в виде полоната калия, или, что более вероятно, по аналогии с ше-
стивалентным теллуром, в виде комплексной соли хромополониевой
кислоты. Дополнительным доказательством присутствия в растворе
шестивалентного полония является тот факт, что при добавлении
перекиси водорода и кипячении полоний из этого раствора осаж-
дается в виде четырехвалентной гидроокиси при одновременном осаж-
дении трехвалентного хрома; это происходит также при длительном
Нитрат полония
129
хранении раствора; по-видимому, под действием a-излучения в ре-
зультате радиолиза в растворе образуется перекись водорода. Та-
кое поведение полония указывает на его высокое валентное состоя-
ние.
НИТРАТ ПОЛОНИЯ
Данные, полученные при изучении диффузии микроколи-
честв полония в 0,1 н. растворе азотной кислоты, свидетель-
ствуют о существовании в этом растворе двухвалентных
ионов [314, 315]. При высоких концентрациях азотной кис-
лоты могут присутствовать комплексные ионы Ро (NO3)““
[152, 154], а в разбавленном растворе можно ожидать при-
сутствия катионов Ро4+, РоО + [154], хотя, вероятно,
доля катионов четырехвалентного полония очень мала.
Результаты, полученные при изучении потенциалов выде-
ления в нейтральных 3,5 н. растворах нитрата натрия
[138], а также при экстрагировании растворителями [68],
могут быть объяснены в предположении существования
комплексных нитратных ионов полония.
Не так давно была определена растворимость нитрата
полония в азотной кислоте в широких пределах концентра-
ций и в большом интервале температур. Применявшийся
метод сводился к определению полония в кислом растворе,
находящемся в контакте с выделенным на платиновой
фольге полонием. Состав образующегося нитрата был не-
известен [253], но в растворе полоний, вероятно, нахо-
дился в четырехвалентном состоянии. Наблюдаемые значе-
ния растворимостей крайне низки и имеют тот же порядок,
что и растворимость сульфата бария, а это свидетельствует
о присутствии основного нитрата [такого, например, как
РоО (NO3)2]. Значения растворимостей, о которых сообща-
лось в печати, частично подтверждаются результатами не-
опубликованной работы, выполненной в лаборатории авто-
ра. Увеличение растворимости с повышением концентрации
азотной кислоты свидетельствует об образовании комплекс-
ных ионов; было высказано предположение о присутствии
в растворе ионов Ро (NO^r [253].
При действии окислов азота (образующихся в резуль-
тате бомбардировки воздуха а-частицами) на двуокись и
некоторые другие соединения полония получается твердое
вещество белого цвета, которое, вероятно, представляет
9 К R
К- Bai нал
130
Окислы и другие соединения полония
собой основной нитрат. Подобный же продукт образуется
при действии разбавленной азотной кислоты на галогениды
полония [10—12].
Бэгнал с сотрудниками [393] описали три формы нитрата по-
лония с отношением NO^": Ро равным 1,5 : 1(1); 0,5 : 1(11) и
4 : 1 (III).
Соединение 1 в виде белого кристаллического продукта было по-
лучено при длительном взаимодействии (12 час.) гидроокиси или тет-
рахлорида полония с 0,5 н. HNOa. Высушивание кристаллов произ-
водили при небольшом разрежении. Продукт, образующийся при
длительном стоянии (72 часа) металлического полония в атмосфере
NO2 4* О2, также имел состав с отношением NO^" : Ро = 1,5 : 1.
Соединение II получали при упаривании раствора гидроокиси
Полония в 2 н. азотной кислоте до небольшого объема с окончатель-
ной подсушкой в вакууме (аналогия с нитратом теллура). Кристаллы
этого нитрата имели желтовато-белый цвет.
Соединение I легко разлагается водой, разбавленным раствором
гидроокиси калия и соляной кислотой с образованием гидроокиси
и хлорида полония соответственно. При нагревании до 100° продукт
I превращается в продукт II, который при дальнейшем нагревании
до 1306 разлагается с образованием двуокиси полония. Собственное
a-излучение препарата мешало точному определению температур
разложения.
Нитраты I и II при длительном стоянии в атмосфере сухого азота
или в вакууме разлагаются с образованием металла. Соединения
эти, возможно, представляют димерные или полимерные продукты,
в которых атомы полония соединены кислородным мостиком.
При взаимодействии двуокиси или тетрахлорида полония с
жидкой N2O4 образуется белый продукт состава Ро(МОз)4-N2O4.
Этот нитрат почти не растворим в жидкой N2O4 и очень легко гидро-
лизуется. Нитрат III медленно разлагается на воздухе при комнат-
ной температуре с образованием основной соли I. Превращение про-
исходит полностью за 1,5—2 часа в вакууме (15,0.).
Двуокись полония после нагревания выше 250° плохо реаги-
рует как с газообразной, так и с жидкой N2O4, а металлический по-
лоний совсем не реагирует с ними, а также с раствором жидкой N2O4
в этилацетате.
При ионном обмене полония на катионитах из азотнокислых
растворов коэффициент распределения (КД определяемый как от-
ношение концентрации полония на 1 г смолы к концентрации поло-
ния на 1 мл раствора, сильно уменьшается при увеличении концен
трации кислоты.
Фосфат полония [350, стр. 8]
Фосфат полония получают в виде белого желатинообразного твер-
дого вещества при действии: а) водного раствора двузамещенного
фосфата аммония (1 А4) на хлорид полония или б) фосфорной кис-
лоты (2 н.) на гидроокись полония. Растворимость фосфата полония
Нитрат полония
131
в фосфорной кислоте (0,5 Л4) составляет 30 мкюри/мл, что. эквива-
лентно 3«10”5 М в расчете на полоний.
Фосфат полония не изменяется при промывании водой или вод-
ным раствором аммиака, но реагирует с 0,1 н. раствором едкого кали,
образуя гидроокись полония (IV). Фосфат полония легко раство-
ряется в соляной кислоте (2 н.) и не осаждается при добавлении
фосфорной кислоты к раствору нитрата полония в азотной кислоте
(2 н.)
По аналитическим данным состав фосфата полония близок к
2РоО2-Н8РО4.
Ванадат полония [350, стр. 9]
Ванадат полония в виде коричневато-желтого вещества образуется
при действии водного раствора ванадата аммония на гидроокись
полония. Вследствие отсутствия подходящего метода анализа со-
став соединения не выяснен. Ванадат полония легко гидролизуется.
Карбонат полония [350, стр. 9]
При действии на гидроокись полония водой, насыщенной СО2, об-
разуется неустойчивое белое твердое вещество, которое, возможно,
является карбонатом полония.
Цианид полония
При обработке твердой гидроокиси полония или тетрахло-
рида полония водным раствором цианистоводородной кис-
лоты образуется белое кристаллическое вещество, предпо-
Таблица 14
Растворимость цианида полония в водных
растворах цианистого калия при 22°
Концентрация цианистого калия, моль/л Раствори- мость, мкюри/мл Литература
0,02 0,07 [45]
0,03 0,16 Там же
0,05 0,39 » »
0,07 0,44 » »
0,10 0,52 » »
0,15 1,00 » »
0,20 1,05 » »
0,30 1,70 » »
0,50 2,25 » »
0,70 3,1 » »
1,0 4,1 » »
1.0 3,8 [249,стр.53]
1,5 5,4 [45]
9*
132
Окисли и другие соединения полония
ложительно цианид полония. Состав этого соединения не
удалось определить ввиду быстрого разложения его под
действием излучения [45]. Данные, полученные при изу-
чении растворимости цианида полония, могут свидетель-
ствовать об образовании комплексных ионов (см. табл. 14).
Другие соли полония [350, стр. 9]
Арсенат, борат, феррицианид, ферроцианид, перекись водорода,
молибдат, перманганат, добавленные в раствор нитрата полония
в 2 н. азотной кислоте, осадка не образуют. Это позволяет считать,
что образующиеся при действии этих реагентов соли полония ра-
створимы в 2 н. HNO3.
СОЛИ ОРГАНИЧЕСКИХ КИСЛОТ
И МЕТАЛЛОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Ацетат полония
Это соединение образуется в виде белых кристаллов при
обработке твердой гидроокиси полония или тетрахлорида
полония водным раствором уксусной кислоты. Раствори-
мость его заметно повышается с увеличением концентрации
уксусной кислоты, что свидетельствует об образовании ком-
плексных ионов [45]. Состав образующегося ацетата не-
известен. Данные, характеризующие растворимость этого
соединения, приведены в табл. 15.
Таблица 15
Растворимость ацетата полония в водном
растворе уксусной кислоты при 22е
Концентрация кислоты, моль/л Раствори- мость, мкюри/мл Литература
0,1 0,88 [45]
0,2 2,3 Там же
0,3 5,0 » »
0,4 11,8 » »
0,5 22,5 » »
0,6 44 » »
0,75 50 » »
0,9 90 » »
1,0 110 » »
1,0 128 [249, стр.53J
1,25 156 [45]
1,5 270 Там же
2,0 375 » »
Соли органических кислот и металларганические соединения 133
Ацетилацетонат полония
Согласно опубликованным данным, при действии ацетил-
ацетона на микроколичества гидроокиси полония образуется
смесь ацетилацетонатов трехвалентного и четырехвалент-
ного полония [311, 313]. Валентность полония в этих со-
единениях была установлена при изучении совместной кри-
сталлизации их с ацетилацетонатами тория, алюминия и
скандия.
Имеются сведения, согласно которым данное соединение
(или смесь соединений) возгоняется с частичным разложе-
нием при 230° и давлении 10 мм рт. ст. Оно плохо раство-
ряется в воде, но значительно лучше в водных растворах
ацетил ацетона и очень хорошо растворимо в горячем хлоро-
форме, бензоле, этиловом спирте, ацетоне и ацетилацетоне
[313]. В одной из последних работ [12] с миллиграммовыми
количествами полония получены результаты, позволяющие
сделать вывод, что продукт реакции ацетилацетона с тетра-
хлоридом полония имеет циклическое строение, обуслов-
ленное конденсацией по месту конечных метильных групп
дикетона. Было получено два ряда соединений: одно с со-
держанием двух атомов галоида (хлорид — желтого цвета
и бромид — оранжевого) и второе, не содержащее га-
лоида (пурпурно-фиолетового цвета), образующееся из сое-
динений первого ряда при их нагревании или взбалтывании
с раствором едкого кали, а также в результате реакции
между дигалогенидами полония и ацетилацетоном. Анало-
гичные соединения были получены и с монокетонами [12].
Эти соединения, по-видимому, похожи на соответствующие
производные теллура.
Более подробные сведения об ацетилацетонатах полония изло-
жены в работе [350, стр. 9—11].
Этилендиаминтетраацетат (ЭДТА) полония
Об образовании этилендиаминтетраацетата судили по поведению
полония в разбавленном растворе HNCh, содержащем этиленди-
аминтетр а ацетат натрия, при ионном обмене на смоле амберлит
1R-120 в водородной форме. Количество реагента изменялось в пре-
делах 10-7—10~5 моля. Значение Кд уменьшалось с увеличением
количества реагента [731 ].
Этилендиаминтетраацетат полония можно получить также из
Тетрахлорида. В растворе это соединение бесцветно, в то время как
134 Окислы и другие соединения полония
тетрахлорид полония в 2 и. НС1 имеет желтую окраску; он может
быть оттитрован 0,0015 н. раствором ЭДТА до обесцвечивания в ко-
нечной точке 1350, стр. 12 £ Количество реагента, уходящего на тит-
рование, соответствует двум молекулам ЭДТА на один атом поло-
ния. Однако изучение растворимости тетрахлорида полония
в 0,001 М ЭДТА указывает на образование соединения с соотноше-
нием ЭДТА к Ро, равным 1 : 1.
Этилендиаминтетраацетат полония получается также растворе-
нием гидроокиси полония в водном 0,1 М растворе ЭДТА или ней-
трализацией 2 н. соляной кислотой растворов тетрахлорида полония
и аммонийной соли ЭДТА (0,1 М). Соединение растворимо в щелочи
и более устойчиво в щелочной среде, чем в кислой. Под действием
a-излучения оно быстро разрушается.
Камфорат полония
Спиртовый раствор камфарной кислоты реагирует, как со-
общают, со щелочными суспензиями полония, и приблизи-
тельно 40% образующегося продукта реакции можно экс-
трагировать бензолом [312]. Получающийся продукт рас-
творим также в хлороформе.
Диметил- и дибензилполоний
По имеющимся данным, микроколичества дибензилполония
образуются с выходом приблизительно 16% при действии
хлорида диметилфенилбензиламмония на смесь полонида
и теллурида натрия в воде, насыщенной водородом. Про-
дукт реакции экстрагируется эфиром [65, 305]. Диметилпо-
лоний образуется также при действии диметил сульфата на
смеси полонида и теллурида натрия; при этом около 10%
полония обнаруживается в диметил теллуровой фракции
[305]. Диметилполоний, как предполагают, должен обра-
зовываться при распаде радиоактивного тетраметилсвинца
РЬ^(СН3)4 [233J.
Диэтилдитиокарбамат полония
Микроколичества полония осаждаются из слабощелочного
раствора при действии диэтилдитиокарбамата натрия с вы-
ходом около 87%. Изучение совместной кристаллизации
с диэтилдитиокарбаматами меди, никеля, висмута и кобальта
(III) показало, что полоний равномерно распределяется
в осадке соединений висмута и кобальта, но концентри-
Соли органических кислот и металлорганические соединения 135
руется на поверхности осадков соединений меди и никеля.
Диэтилдитиокарбамат полония растворим в хлороформе
[130, 134, 135].
По данным [371J, полоний можно экстрагировать хлороформом,
четыреххлористым углеродом или амиловым спиртом из раствора
уксусной кислоты, содержащего диэтилдитиокарбамат калия и аце-
тат аммония. Состав образующегося при этом соединения, установ-
ленный двумя методами (экстракции и ионного обмена), соответст-
вует формуле, в которой на один ион полония приходится один
анион диэтилдитиокарбамата. Диэтилдитиокарбамат полония, как
и ряд других органических соединений этого элемента, возгоняется
при температуре выше 110°.
Дитизонат полония
Результаты изучения экстрагирования растворителями ми-
кроколичеств полония из разбавленного азотнокислого ра-
створа [194] и весомых количеств полония из соляноки-
слого раствора [15] свидетельствуют об образовании дитизо-
ната полония, содержащего две молекулы дитизона и, по-ви-
димому, имеющего состав PoODz2 (Dz—дитизон), поскольку
можно ожидать, что в условиях опыта полоний находится
в четырехвалентном состоянии. Дитизонат полония воз-
гоняется приблизительно при 120° и давлении 1 ат [214].
В работе 1371] показано, что полоний экстрагируется дифенил-
тиокарбазоном (раствор дитизона в CCU) из следующих водных рас-
творов: слабощелочного раствора, содержащего цианид калия и
цитрат аммония; слабощелочного раствора, содержащего цианид
калия и виннокислый натрий-калий; слабокислого раствора, со-
держащего цианид калия и ацетат натрия; разбавленной азотной
кислоты (~ 2,4 об.%).
Оксихинолят полония
Исследовалось [371] экстрагирование полония из азотнокис-
лого водного раствора хлороформом и раствором оксихино-
лина в хлороформе при изменении pH от 0 до 8,7. Показано, что
максимальное извлечение достигается при pH 3—4 с образованием
оксихинолята полония. Соединение содержит на один ион полония
один анион оксихинолина. Оксихинолят полония возгоняется при
температуре выше 140°.
Тионалид полония [371 ]
Полоний экстрагируется раствором тионалида в хлороформе из
уксуснокислого раствора, извлечение достигает 100% при pH 4.
Из зависимости степени извлечения от количества реагента при pH 4
установлено, что соль содержит на один ион полония два органиче-
ских аниона.
136
Окислы и другие соединения полония
Летучесть органических соединений
полония
В работе [371] собраны данные, характеризующие летучесть раз-
личных органических соединений полония. При проведении опытов
на водный раствор полония действовали соответствующим органи-
ческим реагентом, причем образующееся соединение экстрагиро-
валось в органическую фазу. Полученный раствор наносили на сталь-
ные диски, которые затем нагревали до определенной температуры.
О количестве полония, оставшегося на дисках после нагревания,
судили по его активности. Результаты этих опытов представлены
в приведенной ниже таблице.
Результаты опытов по нагреванию различных органических
соединений полония, характеризующие их летучесть
№ п/п Экспериментальные условия экстрагирования полония Активность, остав- шаяся после 15-минут- ного нагревания, % Низшая тем- пература воз- гонки, °C
реагент | растворитель 100° 150° | 200°
1 о-Оксихинолин Хлороформ 97 82 63 140
2 Диэтилдитиокарбамат натрия > 99 79 43 110
3 Теноилтрифторацетон Бензол 98 70 70
4 Тиомочевина Хлороформ 104 69 41 120
5 делен-Т и ом о ч е в и н а » 99 90 87 125
6 Ти семикарбазид > 101 81 29 120
7 1-Фенилтиосемн кар- базид Дифенил карбазон > 103 78 48 130
8 » 91 33 15 95
9 Дифеннлкарбазид » 77 19 10 90
10 Дифенилтиокарбазон Четырех- хлористый . 104 22 7 115
11 Ди-Р-нафтилтиокарба- зон углерод » 99 91 44 150
12 KJ, НС1 Изопропи- ловый эфир 99 105 100 —
13 KJ, HQ Метил изо- бутил кетон 116 114 102 —
8
ХИМИЧЕСКИЕ СВОЙСТВА ПОЛОНИЯ И ЕГО МЕСТО
В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ
Хорошо известно, что в пределах каждой группы периоди-
ческой системы с увеличением атомного веса элементов
обнаруживается заметное усиление их основных свойств.
Полоний является самым тяжелым элементом правой под-
группы шестой группы периодической системы. Интересно
провести сравнение полония с другими элементами этой
подгруппы — серой, селеном и теллуром, а также с сосед-
ними элементами периодической системы — свинцом и вис-
мутом, поскольку в литературе часто проводится аналогия
между полонием и этими элементами.
Элементарный полоний обладает более ярко выражен-
ными металлическими свойствами, чем теллур, и его физи-
ческие свойства ближе к свойствам таллия и висмута, чем
к свойствам теллура. Однако химические свойства элемен-
тарного полония весьма близки к химическим свойствам
теллура, причем наиболее поразительное сходство про-
является в образовании неустойчивых сульфотрехокиси
или селенотрехокиси полония из металла и серной или се-
леновой кислот или их ангидридов.
По свойствам галогениды полония также весьма близки
соответствующим соединениям теллура: они являются столь
же летучими, легко гидролизующимися ковалентными
соединениями, образующими устойчивые комплексы с
ионами галогенов в растворе. Комплексные галогениды
четырехвалентного полония и цезия, подобно соответ-
ствующим соединениям теллура, наименее растворимы
среди подобного рода соединений щелочных металлов;
комплексные соли полония изоморфны аналогичным солям
теллура. Тетрахлорид и тетрабромид полония, по-види-
мому, менее устойчивы, чем соответствующие соединения
теллура; они легко распадаются с образованием галоидных
Соединений двухвалентного полония, которые, наоборот,
138
Химические свойства полония
значительно более устойчивы, чем галогениды двухвалент-
ного теллура. Физические свойства галогенидов и дву-
окисей элементов правой подгруппы шестой группы пе-
риодической системы приведены в табл. 16.
Из элементов правой подгруппы шестой группы перио-
дической системы наиболее сильными кислотными свой-
ствами обладает сера, и, как и следовало ожидать, кислот-
ные свойства элементов данной подгруппы становятся все
более слабыми при продвижении к теллуру, который уже
обнаруживает свойства слабого основания, проявляющи-
еся в том, что могут быть получены его виннокислая соль
и некоторые основные соли (например, нитрат и сульфат).
Полоний и в этом отношении проявляет заметное сходство
с теллуром, поскольку он образует основные сульфаты и
селенаты (2PoO2SO3 и 2PoO2SeO3), однако наличие
у полония более сильных основных свойств сказывается
в том, что он образует устойчивый дисульфат Po(SO4)2,
тогда как основная соль полония метастабильна. Теллур
не образует устойчивого дисульфата. Свойства галоидных
соединений полония не позволяют утверждать, что поло-
ний обладает более сильными основными свойствами по
сравнению с теллуром, однако двуокись и «гидроокись»
полония обладают в известной мере менее сильными ки-
слотными свойствами, нежели двуокись теллура и теллу-
ровая кислота; до настоящего времени еще не получено
достаточных доказательств, подтверждающих существо-
вание трехокиси полония, обладающей преимущественно
кислотными свойствами. Более сильные основные свойства
двуокиси и гидроокиси полония проявляются в легкости,
с которой они вступают в реакцию с такими сравнительно
слабыми кислотами, как уксусная и щавелевая; эти соеди-
нения проявляют способность к комплексообразованию
с кислотными анионами в растворе [45]. Гайсинский [152],
имея для этого известные основания, отмечает, что химия
полония является химией комплексных соединений.
Опубликован целый ряд работ по химии микроколи-
честв полония, авторы которых считали, что полоний можно
восстановить до трехвалентного состояния в растворе и
что этим доказывается аналогия между химическими свой-
ствами полония и висмута. Основания для такого рода вы-
водов не очень убедительны, а работы, проведенные с мил-
Таблица 16
Физические свойства галогенидов и двуокисей серы и элементов
подгруппы селена
Свойство Элементы
s 1 Se Те Ро
Точка* плавления, °C . . 119 217,4 449,8 254
» кипения, °C . . . 444,6 684,8 1390 962
ДЯисп.- ккал 29,1 29,5 24,0 24,6
Атомный радиус, А . . 1,04 1,17 1,37 1,64
Ионный радиус (М--), А Двуокиси 1,90 2,02 2,22 2,30 [345]
точка плавления, °C —75,5 340 ? ?
» кипения, °C -10 315а 450 885а
Дйхлориды >3556
точка плавления, °C —78 — 208
» кипения, °C 59в — 308 190а
Дибромиды 270—2806
точка плавления,°C — 227в? 288?
» кипения, °C — — 339 110 при 3-10“2 мм рт.ст.
Тетрахлориды
точка плавления, °C —31 305 214 294—300е
» кипения, °C в 196а 414 390
Тетрабромиды
точка плавления, °C — — 380 324е
» кипения, °C — — 421в 360 при 200 мм рт.ст.
Тетрайодиды
точка плавления,°C — — 259б в
» кипения, °C — — ~ 100я -200
а Возгоняется.
В запаянной трубке.
в С разложением.
140
Химические свойства полония
лиграммовыми количествами полония, о результатах ко-
торых недавно сообщалось в печати, не дают достаточных
подтверждений предполагаемого сходства полония с ви-
смутом.
Целый ряд элементов правых подгрупп периодической
системы ведут себя в известном отношении так, будто у
них отсутствуют два валентных электрона: о таких элек-
тронах говорят как об «инертной паре» электронов.
В результате химические свойства этих элементов имеют
некоторое сходство с химическими свойствами элементов,
находящихся на две клетки левее в периодической таблице
(например, JC13, SbCl3; JF6, SbFJ. Можно было ожидать,
что наличие инертной пары будет более заметно у полония,
чем у теллура, и тем самым будет обусловлено большее
сходство химических свойств полония с химическими свой-
ствами свинца (но не висмута), однако по химическим свой-
ствам двухвалентный полоний мало напоминает свинец.
При современном уровне знаний в области химии по-
лония есть все основания утверждать, что поведение по-
лония и его соединений соответствует его положению в пе-
риодической системе элементов.
ЛИТЕРАТУРА
1. Adler Е., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 147, 197
(1938).
2. Ahrens L. H., Geochim. cosmochim. Acta, 2, 155 (1952).
3. Anta M. C., Haissinsky M., J. Chim. phys., 51, 33 (1954).
4. Anta M. C., Lefort M., J. Chim., phys., 51, 29 (1954).
5. Arbi trio F., Riv. fis. mat. sci. nat., 12, 178 (1938).
6. Archinard M.-I., J. Chim. phys., 30, 56 (1933).
7. A u s 1 a n d e r J. S., Georgescu I. I., Proc. int. Conf., Geneva,
1955, 7, 389, U. N., New York, 1956.
8. A1 b u r g e r D. E., Friedlander G., Phys. Rev., 81, 523 (1951).
9. Bagnall K. W., Proc. int. Conf., Geneva, 1955, 7, 386, U. N.,
New York, 1956.
10. Bagnall K. W., D’Eye R. W. M., J. Chem. Soc., 4295 (1954).
11. В a g n a 11 K. W., D ’ E у e R. W. M., F г e e m a n J. H., J. chem.
Soc., 2320 (1955).
12. В a g n a 11 K. W., D ’ E у e R. W. M., Freeman J. H., J. chem.
Soc., 3959 (1955).
13. В a g n a 11 K. W., D * E у e R. W. M., Freeman J. H., J. chem.
Soc., 3385 (1956).
14. В a g n a 11 K. W., Freeman J. H., J. chem. Soc., 2770 (1956).
15. В a g n a 11 K. W., Robertson D. S., J. chem. Soc., 509 (1957)
16. Bagnall K. W., Spragg W. T., Atomics, 0, 71, 125 (1955).
17. Bates L. F., Rogers J. F., Proc. Roy. Soc., A105, 360 (1924).
18. В e a m e r W. H., Easton W. E., J. chem. Phys., 17, 1298
(1949).
19. В e a m e r W. H., Maxwell C. R., J. chem. Phys., 14,569 (1946).
20. Beamer W. H., Maxwell C. R., J. chem. Phys., 17, 1293
(1949).
21. Begem a nn E. F., H о u t e r m a n s F. G., Z. Naturforsch., A7,
143 (1952).
22. В e 1 c h e r E. H., Proc. Roy. Soc., A216, 90 (1953).
23. В j e r g e T., Z. Physik., 89, 277 (1934).
24. В 1 a u M., Ron a E., Sitzungsber. Akad. Wiss., Wien, Abt. Ila,
139, 275 (1930).
25. Bonet-Maury P., C. R. Acad. Sci., Paris, 184, 1376 (1927).
26. Bonet-Maury P., C. R. Acad. Sci., Paris, 185, 204 (1927).
27. Bo n£t -Maury P., C. R. Acad. Sci., Paris, 187, 115 (1928).
142
Литература
28. В о п ё t - Ma и г у Р:, Ann. Phys., Paris [10], 11, 253 (1929).
29. Bonet-Maury P., Lefort M., C. R., Acad. Sci., Paris, 226,
173 (1948).
30. В о u i s s i ё r e s G., Bull. Soc. chim. Fr., 536 (1952).
31. Boussieres G., Ch astel R., Vigneron L., C. R. Acad.
Sci., Paris, 224, 43 (1947).
32. Bouissieres G., Ferradini C., Anal. Chim. Acta, 4, 610,
(1950).
33. Brennan J. H., C. R. Acad. Sci., Paris, 179, 161 (1924).
34. Brennan J. H., Ann. Chim. [10], 3, 390 (1925).
35. Briggs G. H., Rev. mod. Phys., 26, 1 (1954).
36. В rod a E., Epstein F., Monatsch., 81, 355 (1950).
37. Brooks L. S., J. Amer. chem. Soc., 77, 3211 (1955).
38. Burbage J. J., Rec. chem. Prog., 14, 157 (1953).
39. Broido A., Terese J. D., Tompkins P. C., Amer. Rep.
MDDC —598 (CH —3631 —C), 1946.
40. Bryan F. A., Silverman L. B., Amer. Rep. AECU-343
(UCLA-18), 1949.
41. Benjamin H., Dissert., Berlin, 1925, pp. 55, 63 (according to
[29]).
42. В rod a E., Wright P. K., Brit. Rep. BR 641, 1946.
43. Bennett W. E., Phys. Rev., 94, 997 (1954).
44. В у r n e J. T., Rogers L.B., G r i e s s J. G., J. electrochem.
Soc., 98, 452 (1951).
45. В a g n a 11 K. W., Freeman J. H., J. chem. Soc., 2161 (1957).
46. В a g n a 11 K. W., D’ Eу e R. W. M., Freeman J. H., Ro-
bertson D. S., Stewart M. A. (unpublished data).
47. Bagnall K. W., Robertson D. S., J. chem. Soc., 1044 (1957).
48. В a g n a 11 K. W., Freeman J. H., J. chem. Soc., 4579 (1956).
49. В a g n a 11 K. W., Guart. Rev., 11, 30 (1957).
50. Brewer L., Chem. Rev., 52, 1, (1953).
51. Chamie С., C. R. Acad. Sci., Paris, 184, 1243 (1927).
52. Chamie С., C. R. Acad. Sci., Paris, 185, 770 (1927).
53. Chamie С., C. R. Acad. Sci., Paris, 185, 1277 (1927).
54. C h a m i ё С., C. R. Acad. Sci., Paris, 186, 1838 (1928).
55. Chamie C., J. Phys. Radium [6],10, 44 (1929).
56. Chamie С., C. R. Acad. Sci. Paris, 208, 1300 (1939).
57. C h a m i ё С., C. R. Acad. Sci. Paris, 224, 1282 (1947).
58. Ch ami ё C., F i 1 9 а к о v a H., J. Chim. phys., 46, 174 (1949).
59. C h a m i ё C., Guillot M., C. R. Acad. Sci., Paris, 190, 1187
(1930).
Литература
143
60. Chamie С., Haissinsky М., С. R. Acad. Sci., Paris, 198,
1229 (1934).
61. Chamie C., Haissinsky M., J. Chim. phys., 44, 197 (1947).
62. Chamie C., Korvezee A., C. R. Acad. Sci., Paris, 192, 1227
(1931).
63. C h a m i ё C., Korvezee A., C. R. Acad. Sci., Paris, 194, 1488
(1932).
64. Chamie C., Marques В. E., C. R. Acad. Sci., Paris, 209, 877
(1939).
65. Хл о п и н В. Г., С а м а р ц е в а А. Г., ДАН СССР, 4, 433 (1934).
66. Coche A., J. Chim phys., 48, 135 (1951).
67. Coche A., Faraggi Н., Avignon Р., Haissinsky М.,
J. Phys. Radium, 10, 312 (1949).
68. Cairo A. E., Proc. int. Conf., Geneva, 1955, 7, 331, U. N.,
New York, 1956.
69. Coche A., Haissinsky M., C. R. Acad. Sci., Raris, 222, 1284
(1946).
70. Cook D. L., J. chem. Educ., 11, 313 (1934).
71. Cotti n M., J. Chim. phys., 51, 404, (1954).
72. Curie M., Recherches sur les Substances Radioactives, These.
Faculte des Sciences de Paris, Gauthier-Villars, Paris, 1904
73. Curie I., J. Chim. phys., 22, 471 (1925).
74. Curie I., Ann. Phys., Paris [10], 3, 299 (1925).
75. Curie M., Rev. Gen. des Sciences, 10, 41 (L899).
76. Curie M., Rev. Sci., Paris, (4), 14,65 (1900).
77. Curie I., Jo Hot F., J. Chim. phys., 28, 201 (1931).
78. Curie I., Lecoin M., C. R. Acad. Sci., Paris, 192, 1453 (1931)
79. Curie M., C. R. Acad. Sci., Paris, 126, 1101 (1898).
80. Curie M., Phys. Z., 4, 231 (1903).
81. Curie M., Ann. Chim. Phys., [7], 30, 99 (1903).
82. Curie M., C. R. Acad. Sci., Paris, 142, 273 (1906).
83. Curie P., Curie M., C. R. Acad. Sci., Paris, 127, 175 (1898)
84. Curie M., Debier ne A., C. R. Acad. Sci., Paris, 150, 386
(1910).
85. Curie M., Debi erne A., Le Radium, 7, 38 (1910).
86. Curie M., Swiatlo, 1,54 (1898).
87. C u r t i s M. L., Phys. Rev., 92, 1489 (1953).
88. Cz а рек A., Sutzungsber. Akad. Wiss., Wien, Abt. Ila, 139, 591
(1930).
89. C u n n i n g h a m В. B., Nucleonics, 5, № 5, 62 (1949).
144
Литература
90. С h а г 1 е s G. W., Н u n t D. J., Р i s h G., T i n n a D. L., J. Opt.
Soc. Amer., 45, 869 (1955).
91. Callihan D., Ross D., Amer. Rep. ORNL-1381, 1952.
92. Curtiss L. F., Nature bond., 120, 406 (1927).
93. Debierne A., C. R. Acad. Sci., Paris, 139, 281 (1904).
94. Dillon J. H., J. Appl. Phys., 11,291,562 (1940).
95. Dillon J. H., Street J. N.,J. Appl. Phys., 13, 189 (1942).
96. D о r a b i a 1 s к a A., C. R. Acad, Sci., Paris, 189, 988 (1929).
97. De R., Separate, Bani Press, Dacca, 1937.
98. De R., Separate, University of Dacca, 1947.
99. De В e n e d e 11 i S., Minton G. H., Phys. Rev., 85, 944 (1952).
100. Ebler E., Fellner M., Ber., 44, 2332 (1911).
101. Evans R. D., J. Appl. Phys., 11, 561 (1940).
102. Eichelberger J. F., Jordan К. C., Orr S. R., Parks
J. R., Phys. Rev., 96, 719 (1954).
103. E mel eus H., Maddock A. G., Miles G. L., S h a r p e A.,
J. chem. Soc., 1991 (1948).
104. E n s к о g D., Z. Phys., 52, 203 (1929).
105. Erbach er O., Z. phys. Chem., A156, 142 (1931).
106. Erbacher O., Naturwiss., 20, 390 (1932).
107. Erbacher O., Z. Elektrochem., 38, 532 (1932).
108. Escher-Desrivieres J., Ann. Chim., [10], 5, 251 (1926).
109. Erbacher O., Z. phys. Chem., A163, 196 (1933).
110. Erbacher O., Z. phys. Chem., A163, 215 (1933).
111. Erbacher О., К a d i n g H., Z. phys. Chem., A165, 421 (1933).
112. Erbacher O., Philipp K., Z. Phys. 51, 309 (1928).
113. Erbacher O., Philipp K., Z. phys. Chem., A150, 214 (1930).
114. Escher M., C. R. Acad. Sci., Paris, 177, 172 (1923).
115. Escher-Desrivieres J., C. R. Acad. Sci., Paris, 178, 1713
(1924).
116. Escher-Desrivieres J., C. R. Acad. Sci., Paris, 179, 158
(1924).
117. E n d e b г о с к R. W., Engle P. M., Amer. Rep. AECD-4146,
1953.
118. F i n ke 1 n b e r g W., Z. Naturf., A2, 16 (1947).
119. Finke In berg W., Phys. Rev., 77,303 (1950).
120. Frierson W. J., Jones J. W., Analyt. Chem., 23, 1447 (1951).
121. Fields P. R., Youngquist С. H., Proc. int. Conf., Geneva,
1955, 7, 44 U.N., New York, 1956.
122. Finkel M. P., Norris W. P., Kisieleski W. E.,
H i г s c h G. M., Amer. J. Roentgenol., 70, 477 (1953).
Литература
145
123. Frisch М., Feldman I., Amer. Rep. UR-426, 1956.
124. G i e s e 1 F., Ber., 33, 1665 (1900).
125. G i e s e 1 F., Ber., 36, 2368 (1903).
126. Gm el in, Handbuch der anorganischen Chemie, System-num-
mer 12, Verlag Chemie, G.m.b.H., Berlin, 1941.
127. God lewski T., Kolloid Z., 14,229 (1914).
128. Gray J. F., Hinds J. F., Phys. Rev., 47, 813 (1935).
129. Groh J., H eves у G., Ann. Phys. Lpz., (4), 65, 216 (1921).
130. Guillot M„ C. R. Acad. Sci., Paris, 190, 127 (1930).
131. Guillot M., C. R. Acad. Sci., Paris, 190, 590 (1930).
132. Guillot M., Haissinsky M., C. R. Acad. Sci., Paris, 198,
1911 (1934).
133. Guillot M., Haissinsky M., Bull. Soc. chim. Fr., [5], 2,
239 (1935).
134. Guillot M., C. R. Acad. Sci., Paris, 190, 1553 (1930).
135. G u i 11 о t M., J. Chim. phys., 28, 14 (1931).
136. Guillot M., J. Chim. phys., 28, 92 (1931).
137. Guillot M., J. Chim. phys., 28, 107 (1931).
138. Guillot M., Haissinsky M., C. R. Acad. Sci., Paris, 198,
1758 (1934).
139. Goode J. M., Amer. Rep. MLM 615, 1951.
140. Goode J. M., Amer. Rep. MLM 677, 1952.
141. Goode J. M., Amer. Rep. MLM 808, 1953.
142. Goode J. M., Amer. Rep. MLM-484-1, page 55, 1950.
143. H a f s t a d L. R., Phys. Rev., 44, 201 (1933).
144. H a f s t a d L. R., J. Franklin Inst., 221, 191 (1936).
145. H a g e m a n n F., J. Amer. chem. Soc., 72, 768 (1950).
146. Hahn O., Applied Radiochemistry, Cornell Univ. Press, Ithaca:
N. Y., 1936.
147. Hahn O., Werner O., Naturwiss., 17,961 (1929).
148. Haissinsky M., C. R. Acad. Sci., Paris, 192, 1448 (1931)
149. Haissinsky M., C. R. Acad. Sci., Paris, 192, 1645 (1931).
150. Haissinsky M., C. R. Acad. Sci., Paris, 194, 275 (1932).
151. Haissinsky M., C. R. Acad. Sci., Paris, 194, 1917 (1932).
152. Haissinsky M., C. R. Acad. Sci., Paris, 195, 131 (1932).
153. Haissinsky M., J. Chim. phys., 29, 453 (1932).
154 Haissinsky M., J. Chim. phys., 30, 27 (1933).
155. Haissinsky M., J. Chim. phys., 31, 43 (1934).
156. Haissinsky M., J. Chim. phys., 31, 469 (1934).
157. Haissinsky M., J. Chim. phys., 32, 116 (1935)-
158. Haissinsky M., J. Chim. phys., 33, 97 (1936).
Ю К Бэгнал
146
Литература
159. Haissinsky М., Trans. Amer, electrochem. Soc., 70, 343 (1936).
160. Haissinsky M., J. Chim. phys., 34, 94 (1937).
161. Haissinsky M., Le Polonium, Act. Sci., 517, Hermann et Cie.,
Paris, 1937.
162. Haissinsky M., Electrochimie des Substances radio-actives,
Act. Sci., 1009, Hermann et Cie., Paris, 1946.
163. Haissinsky M., Comite intern, thermodymna. et cinet. electro-
chim., Compt. rend, reunion, 1951, 214 (1952).
164. Haissinsky M., Coche A., J. chem. Soc., S397 (1949).
165. Haissinsky M., С о 11 i n M., C. R. Acad. Sci., Paris, 224, 467
(1947).
166. Haissinsky M., Faraggi H., Coche A., Avignon P.,
Phys. Rev., 75, 1963 (1949).
167. Haissinsky M., Guillot M., J. Phys. Radium, [7], 5, 419
(1934).
168. Haissinsky M., Mazumdar A. S. G., Bull. cent, electro-
chem. Inst., Karaikudi, 1, № 4, 5 (1954).
169. Hayward R. W., Hoppes D. D., M a n n W. B., J. Res. nat.
Bur. Stand., 54, 47 (1955).
170. Heal H. G., Canad. Rep., MC-33 (NRC-1567), 1943.
171. Henderson G. H., Proc. Camb. phil. Soc., 25, 344 (1928—
1929).
172. Herszfinkiel H., Jedrzejowski H., C. R. Acad. Sci.,
Paris, 188, 1167 (1929).
173. Hevesy G., Phil. Mag., [6], 23, 628 (1912).
174. H e v e s у G., Phil. Mag., [6], 27, 586 (1914).
175. Hevesy G., Phys. Z., 14, 49 (1913).
176. Hevesy G., Phys. Z., 14, 1202 (1913).
177. Hevesy G., Phyl. Mag., [6], 25, 390 (1913).
178. Hevesy G., Guenther A., Z. anorg. Chem., 194, 162 (1930).
179. Hevesy G., Guenther A., Nature bond., 125, 744 (1930).
180. Hevesy G., Obrutsheva A., Nature Lond., 115, 674 (1925).
181. Haissinsky M., Les Radiocolloides, Act. Sci., 185, Hermann
et Cie., Paris, 1934.
182. Hevesy G., P a n e t h F., Sitzungsber. Akad. Wiss., Wien, Abt.
Ha, 123, 1619 (1914).
183. Hevesy G., Paneth F., Monatsch., 36, 45 (1915).
184. Hoffer M., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 144, 393
(1935).
185. Hofmann K. A., Gon d er L., Wolfl V., Ann, Phys, Lpz.,
[4], 15, 615 (1904).
Литература
14?
186. Howland J. J., Т е m р 1 е t о n D. Н., Р е г 1 m a n n I., Phys.
Rev., 71,552 (1947).
187. Hulubei H., Cauchois Y., C. R. Acad. Sci., Paris, 210, 761
(1940).
188. Hulubei H., Cauchois Y., C. R. Acad. Sci., Paris, 224, 1265
(1947).
189. Hulubei H., Cauchois Y., С о t e 11 e S., C. R. Acad. Sci.,
Paris, 207, 1204 (1938).
190. Hunt D. J., Amer. Rep., MLM 979, 1954.
191. Herz f eld K- F., Phys. Z., 14,29(1913).
192. Haissinsky M., Experientia, 8, 125 (1952).
193. Hagemann F., Katzin L. I., Studier M. H., S e a-
borg G. T., Ghiorso A., Phys. Rev., 79, 435 (1950).
194. Ishimori T., Bull. chem. Soc. Japan, 27, 520 (1954).
195. Ishimori T., Sakaguchi H., J. chem. Soc. Japan, 71, 327
(1950).
196. Jedrzejowski H., C. R. Acad. Sci., Paris, 194, 1340 (1932).
197* Jeffries Z., Min. & Metall., N. Y., 22, 82 (1941).
198. J о 1 i о t F., C. R. Acad. Sci., Paris, 189, 986 (1929).
199. Joliot F., J. Chim. phys., 27, 119 (1930).
200. Joy E. F., Amer. Rep. M-4123, 1947 (declassified 1955).
201. Joy E. F., Chem. Engng. News, 32, 3848 (1954).
202. Jankovic О. M., Bull. Inst. Nuclear Sci. «Boris Kidrich», 6.
№6, 143 (1956).
203. К a n n e W. R., Phys. Rev., 52, 380 (1937).
204. Karl A., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 140, 199
(1931).
205. Karlik B., Petterson H., Sitzungsber. Akad. Wiss., Wien,
Abt. Ila, 143, 379 (1934).
206. Karlik B., Ron a E., Sitzungsber. Akad. Wiss., Wien, Abt. Ila,
143, 217 (1934).
207. Kelly E. L., Segre E., Phys. Rev., 75, 999 (1949).
208. Karracker D. G., Ghiorso A., Templeton D. H.,
Phys. Rev., 83, 390 (1951).
209. К о r v e z e e A., J. Chim. phys., 30, 130 (1933).
Karracker D. G., T e m p 1 e t о n D. H., Phys. Rev, 81,510
(1951).
Ku lie C., Coll. Trav. chim. Tchecoslovaquie, 4, 247 (1932),
2,2. Kasarnovsky J., Z. anorg. Chem., 128, 33 (1923).
213- Kirby H. W., Amer. Rep. MLM 675, 1952.
10*
148
Литература
214. Kimura К., Mabuchi Н., Bull. chem. Soc., Japan, 28, 535
(1955).
215. Lachs H., Wertenstein M., Phys. Z., 23, 318 (1922).
216. Lawson R. W., Monatsch., 36, 845 (1915).
217. Lawson R. W., Sitzungsber. Akad. Wiss., Wien, Abt. Ha, 124,
509 (1915).
218. Lawson R. W., Sitzugsber. Akad. Wiss., Wien, Abt. Ila, 124,
637 (1915).
219. Lawson R. W., Sitzungsber. Akad. Wiss., Wien. Abt. Ila, 128,
795 (1919).
220. Lawson R. W., Nature, Lond., 102, 464 (1919).
221. La ws on R. W., Nature, Lond., 114, 121 (1924).
222. Lecoin M., J. Chim. phys., 28, 411 (1931).
223. Lefort M., J. Chim. phys., 51, 351 (1954).
224. Leng H., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 136, 19
(1927).
225. Lind S. C., Science, 68, 643 (1928).
226. Maddock A. G., Miles G. L., J. chem. Soc., S252 (1949).
227. Manov G. G., В i z z e 11 О. M., Symposium on Radioactivity,
A.S.T.M. Spec. Tech. PubL, 159, 9, 1953.
228. Maracineanu S., C. R. Acad. Sci., Paris, 176, 1879 (1923).
229. Maracineanu S., C. R. Acad. Sci., Paris, 177, 1215 (1923).
230. Marckwald W., Phys. Z., 4, 51 (1903).
231. Marekwa Id W., Ber., 35, 2285 (1902).
232. Marckwald W., Ber., 35, 4239 (1902).
233. Mortensen R. A., Leighton P. A., J. Amer. chem. Soc.,
56, 2397 (1934).
234. M а г с к w a 1 d W., Ber., 36, 2662 (1903).
235. Marckwald W., Ber., 38, 591 (1905).
236. M а г e ch a 1 - С о r n i 1 J., Picciotto E., Bull. Soc. chim.
Belg., 62, 372 (1953).
237. Marques В. E., Rev. Quim. pura e aplicada, 33, 123 (1953).
238. Martin A. W., J. phys. Chem., 58, 911 (1954).
239. Maxwell C. R., J. chem. Phys., 17, 1288 (1949).
240. Meitner L., Naturwiss., 14,719 (1926).
241. Mendeleev D., J. chem. Soc., 55, T634 (1889).
242. Mendez R., An. Asoc. quim. argent., 27, 130 (1939).
243. Meyer S., Schweidler E., Sitzungsber. Akad. Wiss., Wien,
Abt. Ila, 115, 63 (1906).
244. Meyer S., Schweidler E., Sitzungsber. Akad. Wiss., Wien,
Abt. Ila, 115, 697 (1906).
Литература
149
245. Mo n t е 1 E., J. Phys. Radium, [6], 10, 78 (1929).
246. Morrow P.E., Della Rosa R. J., C a s a r e 11 L. J., Mil-
ler C. G., Amer. Rep. UR-363, 1954.
247. Meinke W. W., Amer. Rep. AECD-2738, Sec. 84-1, 1949.
248. Meinke W. W., G h i о r s о A., S e a b о r g G. T., Phys. Rev.,
81,782 (1951).
249. Moyer H. V., Ed., Polonium Amer. Rep. TID-5221, 1956.
250. Manning E. R., Wehmeyer D. B., Amer. Rep. MLM 509,
page 17 (according to [249]).
251. Moulton G., Farr J., Vi er D., Amer. Rep. LA-1523, 1953.
252. О d e b 1 a d Ё., Acta radiol. Stock., 42, 391 (1954).
253. Orban E., Amer. Rep. MLM 973, 1954.
254. Ortner G., Salim S., Nature, Lond., 169, 1060 (1952).
255. Paneth F., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 121, 2193
(1912).
256. Paneth F., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 122, 1079
(1913).
257. Paneth F., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 122, 1637
(1913).
258. Paneth F., Monatsch., 34, 401 (1913).
259. Paneth F., Kolloid. Z., 13,1 (1913).
260. Paneth F., Kolloid Z., 13, 297 (1913).
261. Paneth F., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 127, 1729
(1918).
262. Paneth F., Ben, 51, 1704 (1918).
263. Paneth F., Z. Elektrochem., 26, 452 (1920).
264. Paneth F., Chem. Z., 44, 341 (1920).
265. Paneth F., Benjamin H., Z. Elektrochem., 31, 572 (1925).
266. Paneth F., Radioelements as Indicators, McGraw Hill, New
York, 1928.
267. Paneth F., Hevesy G., Monatsch., 34, 1605 (1913).
268. Paneth F., Hevesy G., Sitzungsber. Akad. Wiss., Wien, Abt.
Ila, 122, 1049 (1913).
269. Paneth F., Johannsen A., Ber., 55, 2622 (1922).
270. Paneth F., Johannsen A., Matthies M., Ber., 55, 769
(1922).
271. Paneth F., R a b i n о w i t s c h E., Ber., 58, 1138 (1925).
272. Pearson T. G., Robinson P. L., J. chem. Soc., 736 (1934).
273. Peed W. F., Burkhart L. E., S t a n i f о r t h R. A., F a u -
ble L. G., Amer. Rep. ORNL-1317, 1952.
274. P г e s t w о о d R. J., Amer. Rep. AECD-2839, 1944.
150
Литература
275. Pegram G. В., DunningJ. R., Phys. Rev., 47, 325 (1935).
276. P r e s t w о о d R. J., Amer. Rep. AECD-3380, 1944.
277. P i с с а г d i G., Atti Accad. Lincei, [6], 6, 428 (1927).
278. Powe г W. H., Amer. Rep. MLM 909, 1953.
279. Price W. С., T e e g a n J. P., W a 1 s h A. D., Proc. Roy. Soc.,
A201, 600 (1950).
280. P г о s z t J., Vendl M., Mitt. berg. Hut. Hochschul. Berg-Fort-
wesen, 314 (1930).
281. Poole J. H. J., White J. N. T., Sci. Proc. Royal Dublin Soc,.
27, 1 (1955).
282. Radhakrishna P., J. Chim. phys., 51, 354 (1954).
283. Raudnitz H., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 136,
447 (1927).
284. R e i c h i n s t e i n D., Z. phys. Chem., 97, 288 (1921).
285. Reymond F., Tcheng da Tchang, C. R. Acad. Sci. Paris,
192, 1723 (1931).
286. R i о u M., J. Phys. Radium, 13, 244 (1952).
287. Rolli er M. A., Gazzetta, 66, 797 (1936).
288. Rolli er M. A., Atti X° Congr. internal, chim. Roma, 2, 770
(1938).
289. Rollier M. A., Hendricks S. B., Maxwell C. R., J.
chem. Phys., 4, 648 (1936).
290. Ron a E., Sitzungsber. Akad. Wiss., Wien, Abt. Ha, 137, 227
(1928).
291. Ron a E., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 141,533
(1932).
292. Ron a E., Hoffer M., Sitzungsber. Akad. Wiss., Wien, Abt.
Ila, 144, 397 (1935). English translation, M.O.S., TPA 3/TIB
Trans № P (2222(c))
293. Ron a E., Schmidt E. A. W., Sitzungsber. Akad. Wiss.,
Wien, Abt. Ila, 136, 65 (1927).
294. Ron a E., Schmidt E. A. W., Sitzungsber. Akad. Wiss.
Wien, Abt. Ila, 137, 103 (1928).
295. Rosenblum C., Kaiser E. W., J. phys. Chem., 39, 797
(1935).
296. Rollier M. A., Gazzetta, 84, 658 (1954).
297. Rothensteiner J. P., Sitzungsber. Akad. Wiss., Wien, Abt.
Ila, 125, 1237 (1916).
298. Russell A. S., Chadwick J., Phil. Mag., [6], 27, 112 (1914).
299. Rutherford E., Phil. Mag., [6], 8, 636 (1904).
300. Rutherford E., Phil. Mag., [6], 10, 290 (1905).
Литература
151
301. Rutherford Е., Phil. Trans., A204, 169 (1905).
302. Rutherford E., Nature, Lond., 71, 341 (1905).
303. R i p a n R., P a 1 a d i R., H u 1 u b e i H., Proc. int. Conf., Ge-
neva, 1955, 7, 392, U. N., New York, 1956.
304. Ron a E., Schmidt E. A. W., Z. Phys., 48, 784 (1928).
305. С а м a p ц e в а А. Г., Труды гос. радиевого инет. им. В. Г. Хло-
пина АН СССР, 4, 253 (1938); [С. А., 33, 4512 (1939)].
306. С а м а р ц е в а А. Г., ДАН СССР, 33, 498 (1941).
307. Sanielevici A. S., J. Chim. phys., 33, 759 (1936).
308. Schneidt S., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 138,
755 (1929).
309. Schwarz K., Z. Phys. Chem., A168, 241 (1934).
310. S e r v i g n e M., C. R. Acad. Sci., Paris, 195, 41 (1932).
311. Servigne M., C. R, Acad. Sci., Paris, 196, 264 (1933).
312. Servigne M., C. R. Acad. Sci., Paris, 198, 731 (1934).
313. S e r v i g n e M., J. Chim. phys., 31, 47 (1934).
314. S e г v i g n e M., J. Chim. phys., 31, 147 (1934).
315. Servigne M., J. Chim. phys., 31, 211 (1934).
316. Schweitzer G. K., Jackson M., J. chem. Educ., 29, 513
(1952).
317. Spence R., Proc. int. Conf., Geneva, 1955, 7, 39, U. N., New
York, 1956.
318. S t a г i к I. E., Z. phys. Chem., A157, 269 (1931).
319. Старик И. E. Комлев Л. В., Труды гос. радиевого инет,
им. В. Г. Хлопина АН СССР, 2, 91 (1933); [С. А., 28,6045
(1934)].
320. S t а г i t z к у Е., Amer. Rep., LA 1286, 1951.
321. Strutt R. J., Nature, Lond., 70, 627 (1904).
322. S z a 1 a у A., Z. Phys. 112, 29 (1939).
323. Siegbahn M., Friman E., Phys. Z., 17, 61 (1916).
324. Spiess F. N., Amer. Rep. UCRL 1494 (1951).
325. Sachs F. L., Amer.-Rep. Y-B4-49, 1951.
326. Silverman L. B., Bryan F. A., Amer. Rep. UCLA 84,
1951.
327. Sasaki Y., Bull. chem. Soc. Japan, 28, 89 (1955).
328. Старик И. E., Дейзенрот-Мысовская M. Ю., ДАН
СССР, 1,543 (1934).
329. Spiess F. N., Phys. Rev., 94, 1292 (1954).
330. Smith F. A., Della Rosa R. J., C a s a г e 11 L. J., Amer.
Rep., UR-305, 1955.
331. Scott R. G., Stannard J/N.. Amer. Rep.. UR-235, 1953.
152
Литература
332. Tammann G., Reinacker W., Z. anorg. Chem., 156, 275
(1926).
333. Tammann G., von L 6 w i s, Z. anorg. Chem., 205, 145
(1932).
334. Tammann G., W i 1 s о n C., Z. anorg. Chem., 173, 137 (1928).
335. Tcheng da Tchang, J. Chinese chem. Soc., 3, 381 (1935).
336. T e m p 1 e t о n D. H., Howland J. J., Perlman I., Phys.
Rev., 72, 758 (1947).
337. T r e i m a n L. H., В 1 w m a n M. G., Amer. Rep., LA-1550,
1953.
338. V a г s h n i Y. P., Z. Phys., 135, 512 (1953).
339. V i n a s s a P., Atti Acad. Lincei, [6], 8, 121 (1928).
340. Верный E. А., Зайдель A. H., Швебельблит К. Г.,
ДАН СССР, 104, 710 (1955).
341. Wahl А. С., Bonner N. A., Radioactivity Applied to Che-
mistry, New York, Wiley, 1951.
342. Werten stein L., C. R. Soc. sci. Varsovie, 10, 771 (1917).
343. Wertenstein L., Dobrolska H., J. Phys. Radium, [6], 4,
324 (1923).
344. Whitaker M. D., Bjorksted W., Mitchell A. C. G.,
Phys. Rev., 46,629 (1934).
345. W i 11 e m a n n W. G., G i о r g i A. L., V i e r D. T., Amer. Rep.
LA 1890, 1955.
346. W a d e у W. G., Phys. Rev., 74, 1846 (1948).
347. W h i t e A. G., J. sci. Instrum., 33 ,230 (1956).
348. 3 и в Д. M., ДАН СССР, 25, 743 (1939).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
349. Schulte J. W., Monsanto Chemical Со., Dayton, Ohio. Research
on the Electrodeposition of Polonium for period — November I,
1943—May 1, 1946. Decl. with deletions, Dec. 6, 1957, 85 p.
(M-3-D 537 Del. 2) Final Report № 14 (Del. 2) NSA V12,
№ 16, 10474 (1958); цит. no [395].
350. Bagnall K. W., Freeman J. H., Rо ber t sо n D. S., Ro-
bin s о n P., Stewart M. A. A., The Polonium Chemistry
Project (August 1953 — April 1958), unclassified, A. E. R. E. C/R
2566, 4—5.
351. Ездим Б., Гласник хемиског ДРУШТВА, 18, 325—328 (1953).
352. D а п о n J., Z a m i t h A. L., Nature, 147, № 4512, 746—747
(1956).
Литература
153
353. Crouthamel С. Е., Taianta, 1, 39—40 (1958).
354. Lima F. W., J. Chem. Educat., 31, 153—156 (1954).
355. Dickey E. E., J. Chem. Educat., 30, 525 (1953).
356. Ishimori T., Bull. Chem. Soc., Japan, 28, 432—435 (1955).
357. Odeblad E., Analyt. Chim. acta, 15, 114—117 (1956).
358. Hart E., T e n a г d у J., Rev. Sci. Instrum., 29, 962—965 (1958).
359. Ратнер А. П., Розовская H. Г., Гохман В., Труды
гос. радиевого инет. им. В. Г. Хлопина АН СССР, том VIII,
158—162 (1958).
360. Старик И. Е., Алексеенко Н. И., Розовская Н. Г.,
Изв. АН СССР, Отд. хим. наук, № 7, 755—763 (1956).
361. Старик И. Е., Р о з о в с к а я Н. Г., ДАН СССР, 107, 850
(1956).
362. Старик И. Е., Розовская Н. Г., Журн. неорг. химии, I,
стр. 598 (1956).
363. Старик И. Е., А л е к с е е н к о Н. И., Журн. неорг. химии, 1,
стр. 1676—1679 (1956).
364. Старик И. Е., Сб. Изотопы и излучения в химии, изд. АН
СССР, 282—293 (1958). Москва.
365. 3 и в Д. М., 3 и в В. С., Синицына Т. С., Труды гос. радие-
вого инет. им. В. Г. Хлопина АН СССР, том VIII, 158—162
(1958).
366. Зив Д. М., Эфрос И. А., Радиохимия, 1, 290 (1959).
367. Старик И. Е., Ампелогова Н. И., Гинзбург и др.,
Радиохимия, 1, 370 (1959).
368. Старик И. Е., А м п е л о г о в а Н. И., Радиохимия, 1, 414—
418, 419—424, 425—429 (1959).
369. Старик И. Е., Химическая наука и промышленность, т. IV,
448—455 (1959).
370. Старик И. Е., Основы радиохимии, изд. АН СССР (1959).
371. Kimura К., Ishimori Т., Вторая международная конфер.
по мирному использованию атомной энергии (1958), докл.
№ 1322.
372. Зив Д. М., Эфрос И. А., Атомная энергия, 4, 293—294
(1958).
373. Atterling Н., For sling W., Arkiv fis., 15, 81—88 (1959).
374. Ginnings D. С., В a 11 A. F., V i e г D. T., J. Res. Nat. Bur.
Stand., 50, 75—79 (1953).
375. Dora bj al ska A., Nucleonika, III, 369—391 (1958).
376. Шиманская H. С., Труды гос. радиевого инет. им. В. Г. Хло-
пина АН СССР, т. IX, 131 (1959); т. VII, вып. 1, 198—215 (1956).
154
Литература
377. Ершова 3. В., Владимирова М. В., Атомная энергия,
V, 546—549 (1958).
378. Bagnall К. W., Robinson Р. S., Stewart М. А. А.,
J. Chem. Soc., 3426—3428 (1958).
379. D’Eye R. W. М., J. Inorg. Nucl. Chem., 4, 367—369 (1957).
380. Marozowski E., J. Opt. Soc. America, 46, 663 (1956).
381. G a s p a г R., Molnar K., Acta Physica, Acad. Sci. Hung, 455
(1957).
382. 3 и в Д. M., Диссертация, РИАН СССР, 1948.
383. 3 и в Д. М., Труды гос. радиевого инет. им. В. Г. Хлопина, АН
СССР, т. VIII, 127—137 (1958).
384. Никольский Б. П., Синицына Г. С., Зив Д. М.,
Труды гос. радиевого инет. им. В. Г. Хлопина АН СССР, т. VIII,
141—151 (1958).
385. Зив Д. М., Колесникова Н. С., Синицына Г. С.,
Труды гос. радиевого инет. им. В. Г. Хлопина АН СССР, т. VIII,
163—165 (1958).
386. Зив Д. М., Синицына Г. С., Труды гос. радиевого инет,
им. В. Г. Хлопина АН СССР, т. VIII, 127—137 (1958).
387. Зив Д. М., Синицына Г. С., Труды гос. радиевого инет,
им. В. Г. Хлопина, АН СССР, т. VIII, 138—140 (1958).
388. Никольский Б. П., Зив Д. М., Шестаков Б. И., Си-
ницына Г. С., Труды гос. радиевого инет. им. В. Г. Хлопина,
АН СССР, т. VII, 153—157 (1958).
389. Зив Д. М. Синицына Г. С., Сб. Изотопы и излучение
в химии, стр. 309—317, 1958. Изд. АН СССР, Москва.
390. Matsuura N., Haissinsky М., J. Chim. Phys., 55, 475
(1958).
391. М е н д е л е е в Д. И., Периодический закон, Москва, АН СССР,
226 (1958).
392. Wehrman R., Report MLM-18, November 1947 (klassifiziert),
цит. no [395].
393. Bagnall K. W., Robertson D. S., Stewart M. A. A.,
J. Chem. Soc., 3633—3636 (1958).
394. Попова M. С., Швебельблит К. Г. и др., Proc. Intern.
Conf. UNESCO, Paris, 1957, ed. Extermann, Pergamon Press, v. 1
(1958).
395. Weigel F., Angew. Chemie. 71, 289—299 (1959).
396. Goode J M., J. Chem. Phys., 26, 1269—1271 (1957).
397. Мороз В. В., Смирнова Н. Р., Медицинская радиология,
4, № 9, 66—74. 1959.
Часть II
АСТАТИН
(Z=85)
ФРАНЦИЙ
(Z=87)
РАДОН
(Z = 86)
9
АСТАТИН, ФРАНЦИЙ, РАДОН
Астатин: изотопы, методы выделения, количественное определение,
химические свойства. Франций: изотопы, методы выделения, хи-
мические свойства. Радон: изотопы, применение, методы выделения,
обращение с радоном, количественное определение, физические
свойства, соединения
АСТАТИН И ФРАНЦИЙ
Представление о химических свойствах астатина и франция
было основано на результатах радиохимического изучения
поведения этих двух элементов при соосаждении их соот-
ветственно с йодом и цезием. Исследования проводили
только с растворами незначительной концентрации (10-11—
10"15.Л4); поскольку долгоживущие изотопы астатина и
франция не известны, то проводить химические исследова-
ния на весомых количествах этих элементов при помощи
методов, применяемых, например, в области химии поло-
ния, оказывается невозможным.
Астатин и франций наименее устойчивы из всех эле-
ментов периодической системы; из известных изотопов
этих элементов наиболее долгоживущие имеют периоды
полураспада, равные 8,3 часа (At 10) и 21 мин.* (Fr223,
АсК). Вести химическое исследование миллиграммовых
количеств этих изотопов совершенно невозможно даже
в том случае, если бы они и были получены в таких коли-
чествах, поскольку удельная активность At210 составляет
приблизительно 2000 кюри/мг, а удельная активность
Fr223 — 45 000 кюри/мг\ столь высокая активность обу-
словливала бы исключительно большие тепловые и радиа-
ционные эффекты.
Как показывают недавно проведенные расчеты, астатин
и франций являются крайне редкими элементами, даже по
сравнению с такими малораспространенными элементами,
• Хайд [178] не исключает возможности существования более
долгоживущих изомерных форм известных изотопов астатина и
франция.— Прим. ред.
158 Астатин
как полоний и актиний, так, в поверхностном слое земной
коры толщиной 1,6 км содержится 69 мг астатина (At218)
и 24,5 г франция (Fr223), тогда как полоний содержится
в этом слое в количестве 4000 m (Ро 10), а актиний (Ас2-7) —
в количестве 11 300 ш [5].
В природе изотопы астатина и франция встречаются в урановых
и ториевых рудах, где они образуются в результате разветвленного
распада Th-32, U236, U239 [178]. Главная ветвь распада проходит
через элементы 85 и 87 лишь в нептуниевом радиоактивном ряду
(4п 4- 1), в котором нет ни одного достаточно долгоживущего члена,
который не распался бы полностью за время существования Земли
(см. приложения). Члены этого ряда At21/ и Fr221 все же находятся
в весьма небольших количествах в урановых и ториевых рудах, как
продукты распада Np237 и U233, постоянно образующихся в резуль-
тате естественных процессов захвата нейтронов ураном-238 и то-
рием-232 [178, 179].
АСТАТИН
Первые сообщения о том, что элемент с порядковым номе-
ром 85 обнаружен в природе (его называли алабамином [21,
гельвецием [102], англо-гельвецием [92], см. также [64,
651), впоследствии не подтвердились [77, 791. Критиче-
ские обзоры по этому вопросу опубликованы Карлик [761
и Хулубеем [631. Не удалось обнаружить астатин в при-
роде Хевеши [551 и Бух-Андерсену [161, однако, как было
показано впоследствии, используемые ими методы и не могли
привести к выделению астатина, а потому полученные
указанными исследователями данные не позволяют сделать
какого-либо определенного вывода [20]. Сравнительно
недавно Карлик и Бернерт [78, 80—831 показали, что
природные изотопы At215, At216 и At218, отличающиеся
очень короткой продолжительностью жизни, образуются
при разветвленном распаде соответственно АсА (Ро216),
ThA (Ро216) и РаА (Ро218).
Астатин-219, образующийся в побочной ветви при распаде АсК,
является единственным изотопом астатина, выделенным химическим
путем из природного материала и самым долгоживущим из природ-
ных изотопов астатина [178].
Корсон, Мак-Кензи и Сегре впервые идентифицировали
элемент 85 как продукт, образующийся при бомбардировке
Изотопы астатина
159
висмута а-частицами высокой энергии по следующей ре-
акции:
Bi209 (a, 2n)At211.
Ученые, открывшие этот элемент, впоследствии назвали
его астатином (неустойчивым) [211. Выход астатина при
бомбардировке толстой висмутовой мишени а-частицами,
обладающими энергией 21—28 Мэв, составляет приблизи-
тельно 1,2- W распадов/мин-ра-час. Астатин обладает очень
высокой летучестью, и поэтому мишень (из висмута или из
окиси висмута) в процессе облучения целесообразно охлаж-
дать. Некоторое количество At21* образуется также [84 ]
в результате реакции
Bi209 (a, 3n)At210.
Эта реакция протекает в том случае, если энергия а-чавтиц
превышает 28 Мэв\ образующийся при этом изотоп в резуль-
тате электронного захвата превращается в Ро210.
Астатин является самым тяжелым из галоидов, и по хи-
мическим свойствам он должен походить на йод; последний,
по-видимому, должен быть наилучшим носителем для ми-
кроколичеств астатина, получаемого в результате бомбар-
дировок ускоренными частицами на циклотроне. Однако
сходство между этими двумя элементами неполное; воз-
можно, конечно, что некоторые различия, установленные
в свойствах йода и астатина, объясняются тем, что эти два
элемента сравнивали в различных состояниях окисления.
Физиологическое действие астатина подобно физиологи-
ческому действию йода [51 ].
В ряду F -► Cl — Вг -> J -► At электроположительный харак-
тер элементов возрастает и в соответствии с этим At, помимо метал-
лоидных свойств, обладает так же и некоторыми свойствами металлов
(осаждается из кислых растворов сероводородом, выделяется в эле-
ментарной форме на катоде). Эта двойственная природа At делает
его одним из наиболее интересных элементов периодической системы
ИЗОТОПЫ АСТАТИНА
Известно 19 изотопов астатина — они перечислены в
табл. 17. Проще всего получить At211, поэтому именно этот
изотоп использовали при проведении всех химических ис-
следований, описанных в настоящей книге.
160
Астатин
Изотопы астатина а
Таблица 17
Массо- вое чи- сло Период полураспада Тип распада и энергия испускаемых частиц. Мэе
<202 43 сек. Э. 3.; а 6,50
<203 1,7 мин. Э. 3.; а 6,35
203 7 » Э. 3., а 6,10
204 -25 » Э. 3.
205 25 > Э. 3.; а 5,90
206 2,9 часа Э. 3.
207 1,8 » Э. 3. (-90%); а(—10%) 5,75
208 6,3 > Э. 3.
208 м 1,6 > Э. 3. о 99%); а(~ 0,5%) 5,65
209 5,5 > Э. 3. (-95%); а(~5%) 5,65п
210 8,3 > Э. 3. (>99%); а(0,17) 5,519 (32%); 7
5,437(31%)
5,355 <37%)
211 7,20 » Э. 3. (59,1%); а(40,9%) 5,862; 7
212 0,22 сек. а
213 (Короткий) а 9,2
214 ~2-10”6 сек- a 8,78
215 -Ю-4 » а -8,00
216 - ЗЮ-4 » а 7,79
217 0,018 > а 7,05
218 1,5—2,0 » а(>99%) 6,63; р" (0.1%)
219 0,9 мин. а(-97%) 6,27; р“ (-3%)
а Изотопы астатина с массовыми числами от < 202 до 212 образуются
при бомбардировке висмута а-частицами высоких энергий. Бомбардировкой
золота ионами С13 можно получить At803 и At306, нонами С18 — At8®3 (101)
ВЫДЕЛЕНИЕ АСТАТИНА
Выделить астатин из металлического висмута, облучен-
ного а-частицами, а также из полония можно возгонкой
в вакууме при температуре плавления висмута [20, 731;
сублимат при этом собирают в охлаждаемой ловушке.
Повторной возгонкой можно- получить чистый продукт,
Количественное определение At211
161
свободный от примесей элемента носителя. Применяли и
другую методику, по которой облученный висмут или со-
ответственно облученную окись висмута растворяли в хлор-
ной кислоте, и из раствора висмут осаждали в виде фос-
фата; некоторые количества полония, образующегося в ре-
зультате распада At210, также осаждаются вместе с висму-
том. Чтобы на этой стадии удержать астатин в растворе
и уменьшить его потери за счет адсорбции на стенках со-
суда, рекомендуется добавлять йод [7].
Астатин можно выделить также методом экстрагирова-
ния растворителями; из концентрированных солянокислых
растворов астатин экстрагируется в присутствии солей
двухвалентного железа диизопропиловым эфиром. Микро-
количества полония, висмута и свинца, которые могут
перейти при этом в органическую фазу, можно количе-
ственно удалить, если добавить немного 20%-ного рас-
твора ТБФ в изобутиловом эфире и образовавшуюся объе-
диненную органическую фазу промыть смесью 2 н. азот-
ной и 4 н. соляной кислот [12]. Для отделения астатина
от висмута можно также использовать метод фракционного
гидролиза.
Нейман [180] разработал метод выделения астатина из облу-
ченных висмутовых мишеней *. Для этого висмут растворяют в азот-
ной кислоте, раствор упаривают, добавляют концентрированную
НС1 до 8 н., охлаждают и экстрагируют изопропиловым эфиром.
Выход составляет более 90%; в экстракте содержится менее 0,01%
висмута.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ At211
Количество изотопа At211 можно определить счетом испу-
скаемых а-частиц или измерением потока его рентгенов-
ского излучения, причем метод счета а-частиц, по-видимому,
более точен. В результате захвата электронов приблизи-
тельно 60% распадающихся атомов At211 превращается
в Ро211 (АсС'), который, распадаясь, в свою очередь испу-
скает а-частицы (период полураспада 0,005 сек.). Таким
образом, при распаде одного атома данного изотопа аста-
тина происходит испускание одной а-частицы.
Подготовка образцов, используемых для измерения
* При облучении висмутовых мишеней а-частицами с энергией
43 Мэв образуются At210 и At211.— Прим. ред.
И К. Бэгнал
162
Астатин
а-активности, требует некоторых мер предосторожности,
причем астатин лучше наносить на металлическую под-
ложку, а не на стекло, так как со стеклянной поверхности
астатин испаряется быстрее. Потери астатина с золотых
и платиновых подложек * значительно меньше, нежели со
стеклянной; менее летучими являются, по-видимому, об-
разцы, приготовленные растворением астатина в концен-
трированной азотной кислоте с последующим выпарива-
нием при температуре 80°.
ХИМИЧЕСКИЕ СВОЙСТВА АСТАТИНА
Нейтральные атомы астатина в их основном состоянии
имеют электронную конфигурацию 5s2 5/?6 5rf10 6s2 5р6 ^2P3/J.
Этому элементу, вероятно, свойственно несколько со-
стояний окисления. Данные по соосаждению микроколи-
честв астатина свидетельствуют о том, что астатин обра-
зует ионы астатида (At-) и астатата (AtOg") и имеет еще
одно окислительное состояние, которое пока не идентифи-
цировано.
Элементарный астатин
Астатин несколько менее летуч, чем йод, и в отличие от
йода не отгоняется из разбавленных азотнокислых раство-
ров, но из концентрированных сернокислых растворов
удается отогнать приблизительно 50% астатина. Из раство-
ров в холодной концентрированной HNO3, где, как пред-
полагают, астатин находится в элементарном состоянии,
он экстрагируется бензолом и четыреххлористым углеродом.
Своей хорошей растворимостью в органических растворителях
элементарный астатин похож на йод. Помимо бензола и четыреххло-
ристого углерода он растворяется в углеводородах, эфирах, трибу-
тилфосфате и других кислородсодержащих растворителях. Спо-
собность астатина экстрагироваться органическими растворителями
используют для получения количественных сведений по химии ас-
татина [178|.
Из щелочных растворов, подобно йоду, астатин не эк-
страгируется органическими растворителями.
* Пары астатина сильно адсорбируются поверхностями таких
металлов, как Ag, Au и Pt, и слабо Ni и Си [178].—Прим. ред9
Химические свойства астатина
163
Астатиды
Панет высказал предположение [113], что астатин
можно было бы легко идентифицировать по его способности
к образованию летучего гидрида. Однако об образовании
астатином летучего гидрида имеются лишь сведения,
опубликованные исследователями, открывшими этот эле-
мент [20], и требуется дальнейшее изучение этого вопроса.
В 1 М серной кислоте мышьяковистый ангидрид
и цинк, вероятно, восстанавливают астатин до астатид-
иона, что подтверждается дальнейшим соосаждением аста-
тина с йодидом серебра. Следует отметить, однако, что
соосаждение астатина на йодиде серебра после восста-
новления сернистым газом в 0,01 н. азотной кислоте
можно объяснить выделением элементарного астатина на
металлическом серебре, образующемся при восстановле-
нии йодида серебра. В тех случаях, когда восстановление
проводили в более концентрированной азотной кислоте,
данные получались весьма изменчивыми [7]. Астатин
осаждается также вместе с йодом на солях палладия в
присутствии сульфита (астатид палладия). Однако и это
осаждение может быть результатом выделения аста-
тина на металлическом палладии, образующемся при вос-
становлении сульфитом [7]. Станнит натрия точно так же
восстанавливает астатин до иона астатида.
Электрохимическим методом, подобным тому, который
применяли при исследованиях микроколичеств полония,
было установлено значение потенциала выделения аста-
тина на аноде в 0,066 н. азотной кислоте [73], равное
приблизительно + 1,45 в. Вычисленный на основании этого
значения электродный потенциал Е* Af/At равен при-
мерно + 0,7 в [50]. Однако выделение на аноде не является,
по-видимому, обратимым процессом, и поэтому указанное
выше значение потенциала сомнительно. Астатин в рас-
творе мигрирует в направлении обоих электродов; природа
участвующих в этом процессе ионов не установлена.
Межгалоидные соединения
Если в водной фазе присутствуют хлориды, бромиды или йодиды,
То способность астатина экстрагироваться растворителем типа СС14
Уменьшается, в то время как для кислородсодержащих растворите-
11*
164
Астатин
лей, например диизопропилового эфира, этого не наблюдается.
Из 8 н. раствора НС1, например, эфиром извлекается 90% астатина.
Нейман полагает, что по аналогии с JClrT и JCIJ' астатин экстраги-
руется в виде AtCl<^~ и AtCl^-.
При пропускании тока хлора в солянокислый раствор астатина
образуется комплекс, экстрагируемый изопропиловым эфиром. Мак-
симальная степень экстрагирования наблюдается при концентрации
НС! 8—-9 н., фториды не уменьшают эффективности экстрагирования.
Состав комплекса, вероятно, отвечает формуле AtCl^~, т. е. ас-
татин находится в состоянии окисления (1 +).
Астататы
Окисление микроколичеств астатина горячим раствором
персульфата калия или хлорноватистой кислотой приво-
дит, вероятно, к образованию ионов астатата АЮ“, ибо
окисленный таким образом астатин соосаждается с йода-
том серебра.
К такому же результату приводит окисление йодной кислотой,
ионами церия и висмутатом натрия [178]. Эппельман окислял аста-
тин перечисленными выше окислителями, а затем поочередно осаж-
дал йодатом калия и йодатом бария; осадки образовывались лишь
с йодатом бария [178].
Менее сильные окислители, например ванадат-ионы, бром,
хлор или бихромат калия, окисляют астатин до валентного состоя-
ния, в котором он не экстрагируется четыреххлористым углеродом
и не осаждается с нерастворимыми йодатами.
Нитрат трехвалентного железа вызывает частичное окис-
ление астатина в 0,01 н. азотной кислоте; астатин окис-
ляется также раствором йодата калия. Окисление астатина
бромом протекает быстрее, и можно предположить, что
при этом образуются ионы АЮ“, хотя точно конечное
состояние не известно. Соли двухвалентного железа легко
восстанавливают астатин, присутствующий в растворе в раз-
личных состояниях окисления.
Астатин, как и йод, можно количественно извлечь из изопропи-
лового эфира раствором NaOH, при этом, по-видимому, образуется
ион AtO“, устойчивый в области малых концентраций щелочи.
Растворы HAtO реагируют с фенолом и бензолом с образованием ор-
ганических соединений астатина, аналогичных соединениям HJO и
НВгО [178].
Ионы АЮ“ в растворе 0,1 А4 K2S2O8 0,5 М H2SO4 при 80°
окисляются до высших степеней окисления.
Химические свойства астатина
165
В приведенной ниже таблице представлены наиболее вероятные
состояния окисления астатина; там же указаны восстановители и
окислители, под действием которых он может переходить из одного
состояния в другое.
Состояния окисления астатина
Восстановители
Состояния окисления
Окислители
At1-
(в виде иона At-)
Н—атомарный (Zn + H)
SO2
SO^~ (в щелочном растворе)
H3AsO3 (медленно)
О2
At0
(элементарный At)
Fe+ +
SO2
Fe3+ (медленно)
Вг2 (быстро)
конц. HNO3
At+?
Fe2+ +
so2
нею
K2S2O8 (при нагрева-
нии)
At5+
(в виде иона AtO^")
Данные по соосаждению и другие
сведения
При действии сероводорода астатин осаждается из соляно-
кислого раствора вместе с нерастворимыми сульфидами
(например, с сульфидами висмута и сурьмы); некоторую
часть астатина можно извлечь из сульфидного осадка обра-
166
Франций
боткой его сернистым аммонием. Химическая форма аста-
тина в этих условиях не установлена.
Астатин количественно осаждается (по всей вероятности,
в элементарной форме) из солянокислых или сернокислых
растворов при действии сернистого газа, цинка или хло-
ристого олова. Осаждение сернистым газом можно принять
в основу метода отделения астатина от полония с приме-
нением теллура в качестве носителя.
Из раствора в 0,25 н. азотной кислоте астатин выделяется
на меди, с которой он трудно удаляется при нагревании.
Это можно объяснить образованием астатида меди. Неко-
торое количество астатина выделяется также на металли-
ческом висмуте в присутствии нитрата висмута [7, 73].
По данным [178], из раствора в разбавленной азотной кислоте
астатин, как и полоний, выделяется на меди, золоте и висмуте.
Нерастворимыми гидроокисями астатин извлекается из раство-
ров лишь частично. После окисления персульфатом калия астатин
извлекается на 97—99% из азотнокислого раствора с гидроокисями
железа или лантана (осадитель — едкий натр) [181].
Вычисленный на основе теоретических соображений
радиус иона At7-1" равен 0,62 А [1 ]. Расчет потенциала ио-
низации астатина дает следующие значения: 10,4 эв [165],
9,65 эв [124] и 9,5 эв [39]; вычисленный потенциал иони-
зации иона At+ равен 18,2 эв [39].
Работы по радиохимии астатина освещены в целом ряде
обзоров [7, 36, 68, 73, 159 и 167, стр. 200).
ФРАНЦИЙ
Сообщения об открытии стабильного изотопа элемента 87
в самарските [11, 115], в лепидолите и полюците (когда
этот элемент был назван виргинием [3] и молдавием [60—
62]), а также в солях цезия (когда его назвали алкали-
нием [94—96]), как было установлено впоследствии, ока-
зались необоснованными [56, 57].
Магнито-оптический метод, применявшийся в работе
по изучению Виргиния, по-видимому, весьма ненадежен;
в другом случае [115], при использовании спектрального
метода, ненадежность полученных данных была обуслов-
лена наличием трещины в использованном кристалле
Франций
167
Ас К
ДО/ 22 мин.
Асггт 21,6 года
ДсХ(«агм)
Rd Ас х
(Th227)
18,2 суток
Рис. 13. Схема разветвленного
распада Ас227
кальцита [56]. Элемент, образующийся в природе в резуль-
тате разветвленного а-распада Ас227 (рис. 13), был открыт
в 1939 г. Маргаритой Перей [116, 117] и впоследствии на-
зван францием в честь родины ученой, открывшей этот эле-
мент. Наличие а-ветви при распаде Ас227 было впоследствии
подтверждено радиографическим методом [48]. В 1914 г.
Мейер, Гесс и Панет [100] наблюдали небольшую ^-актив-
ность у тщательно очищенного актиния, наличие которой
было приписано протакти-
нию после открытия этого
элемента в 1918 г.
Возможно, что неболь-
шие количества изотопа
франция образуются и
при а-распаде мезотория-2
(Ас228); этот процесс впер-
вые наблюдал Крэнстон
в 1913 г. [22]. Следует
отметить, что Хан [70] и
Хевеши [54] этого не под-
твердили, причем Хевеши
показал, что франций не
образуется при p-распаде радона, хотя впоследствии Ге-
бен [46, 47 ] подтвердил наблюдение, сделанное Кран-
стоном.
Образующийся при а-распаде Ас227 p-активный изотоп
AcK(Fr223) с периодом полураспада 22 мин.—наиболее
хорошо изученный изотоп франция. Этот изотоп обычно
применяли при радиохимических исследованиях соосажде-
ния франция, которые в основном были выполнены Перей.
Работы с этим изотопом ограничиваются во времени из-за
его небольшой продолжительности жизни, а поэтому все
эксперименты, требующие длительной кристаллизации, не
могли быть проведены. Франций представляет собой самый
тяжелый из щелочных металлов, и поэтому его химические
свойства должны быть подобны химическим свойствам це-
зия — ближайшего более легкого гомолога франция. В
связи с тем что большинство солей щелочных металлов
хорошо растворимы в воде, работы по соосаждению фран-
ция не могут проводиться в широких масштабах. В то же
время хорошая растворимость большинства солей щелоч-
168
Франций
ных металлов может быть весьма выгодно использована при
радиохимических исследованиях благодаря тому, что это
их свойство позволяет относительно легко получить радио-
активно-чистый франций последовательным осаждением
примесей на соответствующих носителях.
ИЗОТОПЫ
Известно восемь изотопов франция; их свойства указаны
в табл. 18.
Таблица 18
Изотопы франция
Массовое число Период полураспада Тип распада и энергия испускаемых частиц, Мэв
212 19,3 мин. Э. 3. (56%); а (44%) 6,411 (37%);
6,387 (39%);
6.342 (24%)
217 (Короткий) а 8,3
218 5-10“3 сек. а 7,85
219 0,02 » а 7,30
220 27,5 » а 6,69
221 4,8 мин. а 6,30 (~75%); у
6,05 (-25%)
222 14,8 » а (0,01—0,1%) 6,00; (>99%) 2,04
223 22 » а (6-10“3%) 5,34; 1,15; f
ВЫДЕЛЕНИЕ ФРАНЦИЯ
Разделение франция и актиния можно осуществить осажде-
нием актиния аммиаком, сернистым аммонием, карбонатом
натрия или плавиковой кислотой с лантаном в качестве
носителя; франций при этом остается в растворе. Радий
(АсХ, Ra223) удаляют, осаждая его с бариевым носителем
при добавлении карбоната натрия, а таллий (АсС" ,Т1207)
соосаждается при добавлении тартрата или сульфида ка-
лия, АсХ и АсС" можно также удалить осаждением хро-
матом бария из щелочного раствора. Весь франций при
этом остается в растворе. Франций можно отделить от акти-
ния, пользуясь методом хроматографии на бумаге. Обра-
Выделение франция
169
зец оставляют на полоске фильтровальной бумаги, которую
в течение 15 мин. выдерживают в атмосфере насыщенного
водяного пара при 60°; один конец бумажной полоски по-
гружают в 10%-ный раствор карбоната аммония. Франций
и АсС" движутся по полоске бумаги, и последний затем
отделяется химическим путем или распадается (период
полураспада АсС" равен 4,76 мин.); после удаления АсС"
остается сравнительно чистый Fr223, который затем можно
отделить с фильтровальной бумаги водой [120, 1291.
В лаборатории ядерных проблем Объединенного института ядер-
ных исследований при облучении 1 г урана протонным пучком 600
Мэе в течение 15 мин. по реакции
92 и 238 (р, 6р, 21n) 87 Fr212
было получено 5-10“13г Fr212 с активностью 2,5-107 распад]мин.
Метод извлечения франция заключался в следующем: облучен-
ный продукт растворяли в азотной кислоте в присутствии незначи-
тельного количества перекиси водорода и хлорида цезия в качестве
носителя. В результате ряда операций по отделению урана и других
тяжелых металлов, получали раствор перхлората цезия, содержав-
ший франций. Описанный метод выделения франция из облученной
мишени занимал не более 30—50 мин. [183].
Другой метод применяли при выделении франция из
облученных ториевых мишеней. Мишень растворяли в,кон-
центрированной соляной кислоте, содержащей небольшое
количество фторсиликата аммония, после чего франций
осаждали либо кремневольфраматом цезия, либо свобод-
ной кремневольфрамовсй кислотой. В тех случаях, когда
применяли соль цезия, осадок упаривали с 70%-ной хлор-
ной кислотой и осадившиеся кремневую и вольфрамо-
вую кислоты удаляли центрифугированием. После выпа-
ривания и охлаждения к раствору добавляли этиловый
спирт и осаждали цезий и франций в виде перхлоратов.
Метод обработки осадка, полученного действием крем-
невольфрамовой кислоты, более прост. Осадок обрабатывают
концентрированной соляной кислотой и франций отделяют
на смоле дауэкс-50 (250—500 меш). Франций поглощается
смолой при концентрациях соляной кислоты, не превышаю-
щих 0,5 н., а элюируется концентрированной соляной
кислотой. Данный метод можно применять также и для
выделения АсК [67 ].
170
Франций
Применяя для осаждения фосфорновольфрамовую или кремне-
вольфрамовую кислоту в концентрированной НС1, достигают прак-
тически 100%-ного выделения франция-212, полученного облуче-
нием тория по реакции
лоТЬ232 (р, 4р17п) етЕг212(183].
Реакция с гетерополи кислотами с последующим отделением
франция-212 на смоле позволяет получить изотоп в чистом виде (при
этом отделяется и рубидий). Описанный метод требует 25—30 мин.
Для удаления примесей в технологии выделения франция можно
использовать также экстрагирование диэтиловым эфиром в НС1 и
HNOa.
Франций не соосаждается с гидроокисями и сульфидами раз-
личных элементов, а также с ВаСОз, BaSO4, МпО2 и AgCl.
Испарением Fr в сухой смеси хлоридов в течение 15 мин. при
900—1000° можно получить препарат франция высокой чисто-
ты [183].
В работе [184] обсуждается вопрос об отделении франция от
щелочных элементов двумя методами — соосаждением и хромато-
графией.
Для разделения франция и цезия методом соосаждения наиболее
перспективным считается применение ферроцианидов и гетербполи-
кислот, особенно тригетерополикислот.
Полагают, что метод вытеснительной хроматографии также мо-
жет оказаться полезным, так как известно, что способность катио-
нов щелочных металлов сорбироваться смолой возрастает в ряду:
К < Rb < Cs. Поэтому можно считать, что среди щелочных ме-
таллов франций будет обладать наибольшим сродством к смоле
[184].
ХИМИЧЕСКИЕ СВОЙСТВА ФРАНЦИЯ
Наиболее вероятной электронной конфигурацией франция
является конфигурация 5s25p65rf10 6s2 бр6 7s (2S1/J , откуда
следует, что франций должен иметь только одно состояние
окисления (Fr+). Вычисленное значение радиуса иона
Fr+ равно 1,80 А [1]; для потенциала ионизации франция
найдены величины 3,83 эв [165], 4 эв [39] и 4,24 эв [124],
а для потенциала ионизации иона Fr+ 21,5 эв [39].
Так как франций является высшим гомологом цезия и
рубидия, соли указанных элементов используют в исследо-
ваниях по совместной кристаллизации. Франций кристал-
лизуется с перхлоратом, пикратом, йодатом и тартратом
цезия, гексахлорплатинатом, пентахлорвисмутатом и пен-
тахлорантимонатами цезия или рубидия, а также с ко-
Радон
171
бальтинитритом натрия и цезия. Даже в присутствии зна-
чительно более растворимой соли рубидия франций пол-
ностью соосаждается с гексахлорстаннатом цезия. Это сви-
детельствует о том, что франций по химическим свойствам
более подобен цезию, нежели рубидию, что и следовало
ожидать [116—119].
По химии франция опубликован ряд работ (см. [36,
125, 128, 159 и 167, стр. 207]).
РАДОН
Резерфорд [147] показал, что выделяемая торием эманация
(торон) в обычных условиях является газом; несколько
позже Дорн [31 ] пришел к аналогичному заключению
относительно эманации, выделяемой радийсодержащими
солями бария. До последнего времени, однако, химическим
путем не была доказана идентичность этих двух изотопов
[1501. М. Кюри [231 обнаружила, что воздух, в контакте
с которым находятся соединения радия, приобретает за-
метную радиоактивность; позже было отмечено, что воздух
лаборатории, в которой велись работы по изучению радия,
обладал довольно большой электропроводностью*. Выска-
зывалось предположение о том, что «радиоактивная энер-
гия» передается излучением или проводимостью какой-то
особой, неизвестной формы [251; подобные предположения,
по-видимому, были возможны постольку, поскольку пред-
ставление о газовой природе эманации к тому времени
еще не нашло признания. В печати опубликован целый ряд
работ о природе эманаций (см. [26, 1531).
Опыты по изучению радона показали, что этот газ со-
стоит из незаряженных частиц [99] и по химическим свой-
ствам является инертным газом [134, 135, 152, 75]; спектр
радона (см. стр. 179) подобен спектру инертных газов.
Учитывая химическую инертность эманации тория, Резер-
форд и Содди пришли к выводу, что она относится к группе
* Максимально допустимое содержание радона в воздухе со-
ставляет 10”10 кюри!л\ в воздухе лаборатории, где вела работы
М. Кюри, содержалось такое количество радона, которое превы-
шало допустимые нормы, существующие в настоящее время.
172
Радон
инертных газов. После того как были разработаны пред-
ставления об изотопах, все другие виды эманаций стали
считать изотопами элемента, который должен занять в пе-
риодической системе свободное место наиболее тяжелого
члена нулевой группы.
Резерфорд назвал этот элемент эманацией; это название
во многих отношениях предпочтительнее названия «радон»,
принятого благодаря тому, что оно показывает генетиче-
ское родство этого элемента с радием [71 ]. Эманации, об-
разующиеся из радия, актиния и тория были названы Рам-
заем соответственно «эксрадий», «эксактиний» и «эксторий»
1133, стр. 470]; позднее Рамзай назвал эманацию радия
«нитоном» вследствие ее способности люминесцировать
в конденсированном состоянии [136].
Радон и радий в небольших количествах содержатся
во всех природных водах; единственным источником отно-
сительно больших количеств радона является сам радий.
По сравнению с астатином и францием радон является
более распространенным элементом; содержание Rn222 в
верхних слоях земной коры, толщиной 1,6 км, по прибли-
зительной оценке равно 115 tn [5].
В работе [185] изучена эмалирующая способность минералов.
Показано, что для различных видов минералов она лежит в преде-
лах 0,10—98,5%.
ИЗОТОПЫ
Известно шестнадцать изотопов радона (см. табл. 19), из
которых наибольшее применение получил Rn222 с периодом
полураспада 3,825 суток. Этот изотоп обладает удельной
активностью, равной при нормальных условиях прибли-
зительно 1700 кюри/мл. Возможности проведения экспери-
ментальных работ по изучению этого элемента чрезвычайно
ограничены, поскольку доступные количества радона со-
ставляют приблизительно 1 мм3.
ПРИМЕНЕНИЕ РАДОНА
Радон применяют главным образом в медицине, в основном
для лечения рака [72]. Ценность радия и радона при те-
рапевтическом и промышленном использовании обусловлена
наличием у их дочерних изотопов проникающего ;-излу-
Применение радона
173
Изотопы радона
Таблица 19
Массовое число Период полураспада Тип распада и энергия испускаемых частиц, Мэв
204 3 МИН. а 6,28
206 6,2 » Э. 3. (35%); а (65%) 6,25
207 11 » Э. 3. (96%); а (4%) 6,14
208 23 » Э. 3. (-80%); а (-20%) 6,141
209 30 » Э. 3. (83%); а (17%) 6,037
210 2,7 часа Э. 3. (-4%); а (-96%) 6,037
211 16 час. Э. 3. (74%); а (26%) 5,779 (64,5%)
5,847 133,5%)
5,613 (2%)
212 23 мин. а 6,264
215 ~10-6 сек. а 8,6
216 -10“4 » а 8,01
217 ~10"3 » а 7,74
218 0,019 » а 7,13
219 3,92 » а 6,813 (82%); 7
(Актинон) 6,547 (13%)
6,419 (5%)
220 51,5 » а 6,278 (99,7%); 7
(Торон) 5,747 (0,3%)
221 25 мин. а (~20%) 6,0; 3~(~80%)
222 3,825 суток а 5,482; 7
(Радон)
чения — в первую очередь у RaC и в меньшей степени
у RaB и RaC" (см. приложение, радиоактивные ряды). Для
использования в медицинских целях радон запаивают в ам-
пулы в виде капилляров, называемые «зернами»; радон при-
меняют также в качестве составной части мази, состоящей
из раствора радона в вазелине [72, стр. 24]. В неболь-
ших количествах радон применяют в промышленности для
обнаружения трещин в металле методом у-радиографии [38 ].
Этот метод позволяет обнаружить внутренние дефекты
изделия, а также неполноценные швы или трещины, вы-
являемые по неравномерной плотности изображения, по-
лучаемого на фотопластинке или пленке. Отчетливость изо-
174
Радон
бражений, получаемых на радиографических снимках,
повышается, если между фотографической пластинкой
и снимаемым объектом поместить тонкий экран из листо-
вого свинца. Это явление обусловлено комптоновскими
электронами и фотоэлектронами, которые образуются в
свинце под действием у-излучения.
Радон используют также при определении времени про-
хождения газа в доменных печах [166] и для обнаружения
утечки из зарытых в землю трубопроводов [41 ]. Одно время
применяли также радон-бериллиевые источники ней-
тронов [34, 35].
Методы, основанные на использовании радона, могут
найти широкое применение при проведении физико-хими-
ческих исследований; исследование процессов дегидратации,
реакций в твердом состоянии и других процессов, при ко-
торых происходят изменения поверхности и кристалли-
ческой структуры, может быть проведено путем введения
радия в состав исследуемых твердых веществ и определе-
ния количества радона, выделяющегося в условиях
опыта [49].
ОТДЕЛЕНИЕ РАДОНА
Коловрат [87—89] изучал выделение радона из солей ра-
дия при высоких температурах. Он установил, что при тем-
пературах ниже точки плавления изучавшихся солей вы-
деляется только часть радона. Более эффективным является
способ выделения радона из водных растворов. Обычно
применяют следующий метод: радий в виде раствора в раз-
бавленной кислоте помещают в небольшую экранирован-
ную колбу, присоединенную к вакуумной линии, и перио-
дически, через небольшие промежутки времени, откачи-
вают образующийся радон. На рис. 14 показана вычи-
сленная кривая роста количества радона, образующегося
из источника радия активностью 1 кюри, в зависимости от
времени. Не следует допускать накопления радона про-
должительное время, поскольку в этом случае в резуль-
тате радиолиза растворителя в системе образуются сравни-
тельно большие количества газа, под давлением которого
пробка крана может сместиться, что приведет к утечке ра-
дона из прибора и значительному загрязнению воздуха
дочерними продуктами распада радона (активный осадок).
Отделение радона
175
Если раствор радия оставляют стоять длительное время
в установке, то желательно, чтобы она имела спускное уст-
ройство для предотвращения возможности случайного попа-
дания радона в воздух рабочего помещения. Выходящий че-
рез это устройство газ должен поступать в ловушку с углем
Рис. 14. Нарастание количества радона
в радиевом (Ra22*) источнике в 1 кюри.
или отводиться в вытяжную вентиляцию. Если радон не со-
бирают для использования, то накапливающийся газообраз-
ный продукт следует удалять из раствора радия откачива-
нием через определенные промежутки времени, зависящие
от количества присутствующего в растворе радия и от объ-
ема свободного пространства над раствором.
В газовой смеси, полученной из раствора радия, имеются
такие примеси, как водород, кислород, водяные пары,
гелий, двуокись углерода и пары органических соединений,
образующихся при действии a-излучения на органические
вещества, например на вакуумную смазку (о разложении
вакуумной смазки под действием a-излучения имеется ряд
сообщений, см., например, [169]). В газовой смеси содер-
жатся также пары кислоты, в которой был растворен ра-
дий, и водород, всегда присутствующий в избытке благо-
даря радиолитическому образованию перекиси водорода
в растворе. Следует отметить, что при а-распаде радия и его
176
Радон
дочерних продуктов гелий образуется в таком количестве,
которое превышает количество образующегося радона.
Отделения кислорода и части водорода от образующейся
смеси газов можно достигнуть действием разрядов или про-
пусканием газовой смеси над соответствующим катализа-
тором, причем полностью отделить эти газы можно при
пропускании газовой смеси над нагретой медью или окисью
меди. При пропускании газов над нагретым бихроматом
свинца органические соединения окисляются: воду удаляют
фосфорным ангидридом, двуокись углерода и пары кислот
поглощают едким кали, после чего из полученной газовой
смеси радон вымораживают жидким кислородом; остаю-
щиеся водород и гелий откачивают. На последней стадии
радон собирают в капилляры, охлаждаемые жидким кисло-
родом. Опубликованы многочисленные данные относи-
тельно аппаратуры, применявшейся при подобных иссле-
дованиях [10, 33, 37, 93, 97, 103, 112, 126, 141, 158 и 164].
В литературе имеется описание более простой установки
для непрерывного извлечения радона [28]. В этой установке
смесь газов проходит над едким кали и фосфорным ангидри-
дом для удаления двуокиси углерода и воды, после чего
радон конденсируется в змеевике, охлаждаемом жидким кис-
лородом, в то время как другие газы удаляются из системы;
в этой системе водород и кислород не рекомбинируют. Суще-
ствует совершенно другой метод, основанный на адсорбции
радона углем, охлаждаемым жидким воздухом, после пред-
варительного удаления двуокиси углерода и воды. Радон
после этого можно десорбировать нагреванием угля до тем-
пературы приблизительно 350° [66].
В работе [1861 описан новый метод очистки радона от примесей,
основанный на инертности радона.
МЕРЫ ПРЕДОСТОРОЖНОСТИ ПРИ РАБОТЕ С РАДОНОМ
Радон сразу после отделения является чистым а-излучате-
лем, однако в нем очень быстро происходит накопление
трех его дочерних -^-активных продуктов (RaB, RaC и
RaC"). Возрастание у-активности радонового источника
в зависимости от времени показано на рис. 15. Если прихо-
дится работать с большими количествами радона, то в це-
тях безопасности экспериментатора необходимо экраниро-
Физические свойства радона
177
вать вакуумную систему, используемую для откачки ра-
дона (см. гл. 10). Учитывая, что максимально допустимое
содержание радона в воздухе составляет всего лишь
Рис. 15. Нарастание ^-активности в свежевыделенном
радоне
(По данным [72, табл. 3, стр. 210].)
Ю-10 кюри/л-, необходимо принимать все меры, предотвра-
щающие случайные поломки аппаратуры. По вопросу о
заболеваниях, вызываемых вдыханием радона, см. работы
[130, 131].
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ РАДОНА
Количество радона можно определить прямым счетом его
а-частиц в газовой фазе в ионизационной камере с воздуш-
ным наполнением или измерением -[-излучения короткожи-
вущих дочерних продуктов распада радона. Количества
радона порядка милликюри обычно определяют сравнением
[-излучения образца, содержащего радон, с -[-излучением
радиевых эталонов. В литературе имеется описание не-
скольких счетных устройств, используемых для такого
рода измерений [12, 32, 37, 69].
ФИЗИЧЕСКИЕ СВОЙСТВА РАДОНА
Радон представляет собой бесцветный одноатомный газ,
подчиняющийся закону Войля [132]. Вычисленные для
радона значения постоянных Ван-дер-Ваальса а и b равны
12 К. Бэгнал
178
Радон
соответственно 4,93-Ю* 6 и 62,1 (для одного моля и при
условии, что давление выражается в метрах [144]). Ука-
занные значения постоянных имеют тот же порядок, что
и значения соответствующих постоянных для других инерт-
ных газов. Жидкий радон является бесцветной фосфоресци-
рующей жидкостью, плотность которой равна приблизи-
тельно 5 г/см3 [149]; в твердом состоянии радон испускает
ярко-голубое свечение (ср. с полонием) [171]. Физические
свойства радона указаны в табл. 20.
Таблица 20
Физические и ядерные свойства радона
Точка кипения, °C.................... —65 [149]
» плавления, °C................... —71 [171] —113 [114]
Критическая температура, 7крит .... 104,5 [171]
Критическое давление, ат............. 62,4 [171]
Мольная теплота плавления, кал....... 650 [52]
» » испарения, »........... 3960®, 43876 [144]
4600а [170]
Диаметр молекулы, А . . ......... 3,64 [144], 3,70 [154]
Вязкость (при 0°), пуаз ......... 2,13-10—4 [ 156]
» (при Ткрит ), пуаз............. 2,954-10"4 [156]
Поперечное сечение поглощения тепловых
нейтронов (Rn222), барн.............. 0,72 [9]
а Значения, вычисленные из температурной зависимости давления пара.
6 Значения, вычисленные по правилу Трутона.
При попытках измерить показатель преломления радона
на очень небольших образцах (образец в 200 мкюри занимает
объем, равный лишь долям кубического миллиметра) были
получены ненадежные результаты вследствие присутствия
примесей в радоне [127]. Для температурной зависимости
давления пара радона было предложено два уравнения:
lgp=—0,5 lg Т- +11,54
(для интервала 91 —121° К [170])
и
1g р = 0,3021 lg Т— ?бу7- +1,158
(для интервала 85—111° К [90]).
Физические свойства радона
179
Данные, первоначально полученные Резерфордом [149],
удовлетворяют уравнению:
lgРмм = — + 10,16 (для интервала 146 —208° К),
которое для точки кипения радона дает значение —64,7®.
Экспериментальные трудности не позволяют точно устано-
вить, какое из приведенных уравнений является лучшим.
Влияние различных факторов на получаемые результаты
рассмотрено в работе Вертенштейна [1701.
Плотность и атомный вес радона (Rn222)
Первыми исследователями радона, пользовавшимися мето-
дами газовой диффузии, получены крайне противоречивые
значения для плотности и атомного веса этого элемента.
Найденные значения атомного веса колеблются в пределах
40—234 [19, 98, 121, 150]. Столь большие расхождения
можно объяснить главным образом присутствием в образце
неизвестных примесей. Дебьерн в своих более поздних ра-
ботах [29, 30], выполненных с применением метода диффу-
зии, нашел, что атомный вес радона равен 220—221. Это
значение согласуется со значением, полученным Уайтлоу-
Греем и Рамзаем [172, 136] путем определения плотности
при помощи микровесов.
Спектр радона
Изучению спектра радона были посвящены многочисленные
работы [18, 27, 123, 133 стр. 346, 142, 143, 145, 151, 168,
175], причем наиболее полные данные по этому вопросу
получены Нисвандером и др. [НО, 1111. Спектр радона
напоминает спектры других инертных газов. Ионизацион-
ный потенциал радона, как показывает расчет, равен при-
близительно 10,6 эв [43, 59, 138, 177]. Другие полученные
значения ионизационного потенциала приведены в рабо-
тах [14, 104, 124, 139, 140, 146, 160, 161 и 163]. Вычислен-
ное значение ионизационного потенциала иона Rnb равно
19,9 эв [39].
12*
180
Радон
Адсорбция
Радон хорошо адсорбируется на угле (см., например,
[148, 174]) и на силикагеле (см., например, [122]). На этом
свойстве радона может быть основан метод отделения ра-
дона от газообразных примесей. При нагревании до 350°
радон десорбируется из угля [66]. Вычисленное значение
теплоты адсорбции равно 7510 кал/моль для активирован-
ного древесного угля [45] и 6800 кал/моль [45] или
9000 кал/моль [17] для силикагеля.
Растворимость в жидкостях
В органических жидкостях радон растворяется лучше, чем
в воде. Соответствующие данные, полученные Рамстедтом
[137], приведены в табл. 21.
Таблица 21
Растворимость радона в различных жидкостях
Растворитель Отношение концентраций радона в жидкой и в газовой фазе
18° 0° -18°
Вода 0,285 0,52 —
Ацетон 6,30 7,99 10,8
Анилин 3,80 4,43 —
Бензол 12,82 (16,34 приЗ°) —
Сероуглерод .... 23,14 33,4 50,3
Хлороформ 15,08 20,5 28,5
Эфир 15,08 20,9 29,1
Этилацетат 7,35 9,41 13,6
Этиловый спирт . . 6,17 8,28 11,4
Глицерин 0,21 — —
Гексан 16,56 23,4 35,2
Толуол 13,24 18,4 27,0
Ксилол 12,75 — —
Экспериментальные результаты Рамстедта качественно
согласуются с соответствующими вычисленными значе-
Физические свойства радона
181
ниями [157]. Растворимость радона в спиртах и жирных
кислотах возрастает с увеличением числа атомов углерода
в молекуле растворителя [17], а растворимость радона
в смеси этилового спирта с водой возрастает по мере увели-
чения содержания спирта [85]. Имеются также некоторые
данные, характеризующие температурную зависимость ра-
створимости радона в воде для широкого интервала темпе-
ратур [861. Присутствие радона во льду и в заморожен-
ном бензоле можно объяснить, видимо, явлением окклю-
зии, а не растворимостью [173]. Растворимость радона
в воде, как и следовало ожидать, не влияет на элек-
тропроводность воды [42, 44]. Ряд других данных, харак-
теризующих растворимость радона в жидкостях, см. в ра-
ботах [4, 15, 91, 162].
Диффузия радона в жидкостях
Диффузия радона в различных жидкостях изучалась рядом
исследователей, и полученные ими результаты сведены в
табл. 22. Обзор экспериментальных работ по диффузии
радона опубликован Рамстедтом [155]. После того как
были получены данные, приведенные в табл. 22, методы
исследования диффузии в жидкостях были значительно
усовершенствованы. Поэтому желательно провести иссле-
дования диффузии радона с применением этих более совре-
менных методов.
Таблица 22
Диффузия радона в жидкостях
Температура, °C D, см2;су тки Литература
вода толуол
Комнатная 0,066 0,168 [176]
14 0,820“, 0.8536 — [155]
16 0,199®, 0,401г — [74]
18 0,985д — [154]
а 10%-ный раствор желатины в воде.
6 2,5%-ный раствор желатины в воде.
в 0,1 М раствор КО в воде.
г 0,5 М раствор КО в воде.
д раствор, содержащий небольшое количество мочевины или глицерина,
182
Радон
СОЕДИНЕНИЯ РАДОНА
Нейтральные атомы радона в их основном состоянии имеют
электронную конфигурацию 5s25p65d106s26p6 [8]. Дан-
ная конфигурация устойчива, и поэтому в химическом от-
ношении радон должен быть инертным.
На основании изучения распределения радона между
твердой и газообразной фазами гексагидратов сернистого
газа и сероводорода можно сделать вывод о существовании
гидрата Rn-6H2O [105], давление диссоциации которого
приблизительно равно 1 ат при 0°. Точно так же имеются
сообщения о существовании подобного рода соединений
с фенолом Rn-2CeH6OH [106] или Rn-3CeH5OH [109]
и с n-хлорфенолом Rn-3CeH4OHCl [108] (см. также [107]).
Считают, что подобные соединения образуются и другими
инертными газами. Обзор по химии радона опубликован
Валем и Боннером [167, стр. 206].
ЛИТЕРАТУРА
1. Ahrens L. Н., Geochim. cosmochim. Acta, 2, 155 (1952).
2. A 11 i s о n F., В i s h о p E., S о m m e r A., J. Amer, chem Soc.,
54, 616 (1932).
3. A 1 1 i s о n F., Murphy E. J., Phys. Rev., 35, 285 (1930).
4. A n t г о p о f f A., von Z. Elektrochem., 25, 269 (1919).
5. A s i m о v I., J. chem. Educ., 30, 616 (1953).
6. Aston F., Proc. Roy. Soc., A103, 462 (1923).
7. Aten A. H. W., Doorgeest T., Hollstein V., Moe-
ken H. P., Analyst, 77, 774 (1952).
8. В a ch er R., Goudsmit S., Atomic Energy States, McGraw
Hill, New York, 1932.
9. В a erg A. P., Phys. Rev., 90, 1121 (1953).
10. В a n n о n J., J. Cancer Res. Comm. Univ. Sydney, 3, 86 (1931).
11. Barnes L. L., Gibbs R. C., Phys. Rev., 40, 318 (1932).
12. Barton G. W., Ghiorso A., Perlman I., Phys. Rev., 82,
13 (1951).
13. Bates G. L., V о 1 c h о к H. L., К u 1 p J. L., Rev. sci. Instrum.,
25, 153 (1954).
14. Biswas S. C., Phil. Mag. [7], 5, 1094 (1928).
15. Boyle R. W., Phil. Mag. [6], 22, 840 (1911).
16. В u c h - A n d e r s e n E., K. danske vidensk. Selsk., 16, 5 (1938).
17. Burtt В. P., К u r b a t о v J. D., J. Amer. chem. Soc., 70, 2278
(1948).
18. Cameron, A. T., Ramsay W., Proc. Roy. Soc., A81, 210
(1908).
19. C h a u m о n t L., Le Radium, 6, 106 (1909).
20. Corson D. R., MacKenzie K. R., Segre E., Phys. Rev.,
58, 672 (1940).
21. С о r s о n D. R., MacKenzie K. R., Segre E., Nature, Lond.,
159, 24 (1947).
22. Cranston J. A., Phil. Mag., (6), 25, 712 (1913).
23. Curie M., Recherches sur les Substances Radioactives, These,
Faculte des Sciences de Paris Gauthier — Villars, Paris, 1904.
?4- Curie P., D e b i e r n e A. C. R. Acad. Sci., Paris, 132, 7G8 (1901).
184
Литература
25. Curie Р., Debierne А., С. R. Acad. Sci., Paris, 133, 276
(1901).
26. Curie P., Danne J., C. R. Acad. Sci., Paris, 136, 1314 (1903).
27. Cameron A. T., Ramsay W., J. chem. Soc., 93, 992 (1908).
28. Dawson J. A. T., J. sci. Instrum., 23, 138, (1946).
29. Debierne A., C. R. Acad. Sci., Paris, 150, 1740 (1910).
30. Debierne A., Ann. Phys., Paris, [9], 3, 62 (1915).
31. Dorn F. Е.» Abh. Naturf. Ges. Halle, 23, 1 (1901) [according
to Partington J. R., Nature, bond., 179, 912 (1957)].
32. Drigo A., Marco L., de Atti Accad. Sci. Ferrara, 27, 135
(1949—50).
33. Duane W., Phys. Rev., 5, 311 (1915).
34. Dunning J. R., Pegram G. B., Phys. Rev., 43, 497 (1933).
35. Dunning J. R., Pegram G. B., Phys. Rev., 44, 317 (1933).
36. E m e 1 e u s H. J., Sci., Progr., 38, 609 (1950).
37. Evans R. D., Rev. sci. Instrum., 4, 216 (1933).
38. Ferguson С. B., Metal Ind., 78, 439 (1951).
39. Finkelnburg W., Phys. Rev., 77, 303 (1950).
40. Frota-Pessda E., An Acad. bras. Cienc., 25, 337 (1953).
41. Gemant A., Hines E., Alexanderson E. L., J. appl.
Phys., 22, 460 (1951).
42. G 1 e d i t s c h E., G 1 e d i t s c h L., J. Chim. phys., 25, 290 (1928).
43 G1 о с к 1 e r G., Phyl. Mag., [6], 50, 997 (1925).
44. G r a s s i U., Atti Accad. Lincei, [5], 16, 179 (1907).
45. Giibeli-Litscher O., Stammbach K., Helv. chim. Acta,
34, 1257 (1951).
46. Gueben G., Ann. Soc. Sci. Bruxelles, 52, 66 (1932).
47. Gueben G., Ann. Soc. Sci. Bruxelles, 53, 115 (1933).
48. Guillot M., Perey M. C. R. Acad. Sci. Paris, 225, 330 (1947).
49. Hahn O., Pros. int. Symp. Reactivity of Solids, Gothenburg
Pt. 1, p. 21, 1952.
50. H a i s s i n s к у M., Comite intern thermodynam. et cinet. electro-
chim., Compt. rend, reunion, 1951, 218 (1952).
51. Hamilton J. G., So ley M. H., Proc. Nat. Acad. Sci., 26,483
(1940).
52. Harbutt J., Phys. Z., 22, 52 (1921).
53. Hen riot E., Le Radium, 5, 41 (1908).
54. Hevesy G., K. danske vidensk. Selsk., 7, № 11, 1 (1926).
55. Hevesy G., Hobbie R., Z. anorg. Chem., 208, 107 (1932).
56. Hirsh F., Phys. Rev., 51, 584 (1937).
57. Hirsh F., Phys. Rev., 63, 93 (1943).
Литература
185
58. Hofbauer G., Beibl. Ann. Physik, 39, 263 (1915).
59. Hol wee к F., We r ten stein L., Nature, Lond., 126, 433
(1930).
60. H u 1 u be i H., C. R. Acad. Sci., Paris, 202, 1927 (1936).
61. Hulubei H., C. R. Acad. Sci., Paris, 205, 854 (1937).
62. Hulubei H., C. R. Acad. Sci., Paris, 209, 675 (1939).
63. H u 1 u b e i H., J. Chim. phys., 44, 225 (1947).
64. H u 1 u b e i H., С a u c h о i s Y., C. R. Acad. Sci., Paris, 209, 39
(1939).
65. Hulubei H., С a u c h о i s Y., C. R. Acad. Sci., Paris, 210, 696
(1940).
66. Hursh J. B., Nucleonics, 12, № 1,62 (1954).
67. Hyde E. K., J. Amer. chem. Soc., 74, 4181 (1952).
68. Hyde E. K.,J. phys. Chem., 58, 21 (1954).
69. Hudgens J. E., Benging R. O., Cali J. P., Mey-
er R. S., Nelson L. C., Nucleonics, 9, № 2, 14 (1951).
70. Hahn O., Naturwiss, 14, 158 (1926).
71. Int. Committee on Chemical Elements, 1923, J. Amer. chem. Soc.
45,867 (1923).
72. J e n n i n g s W. A., Russ S., Radon, its Technique and Use,
John Murray, London, 1948.
73. Johnston G. L., Leininger F. F., Segre E., J. chem
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
Phys., 17, 1 (1949).
Jahn M., Dissert., Halle, 1914 (according to [155]).
К a di ng H., Riehl N., Naturwiss, 21, 479 (1933).
К a r 1 i к В., Monatsh., 77, 348 (1947).
К a r 1 i к В., В e г n e г t T., Sutzungsber. Akad. Wiss., Wien, Abt
Ila, 151, 255 (1942).
Karlik B., Bernert T., Sitzungsber. Akad. Wiss. Wien, Abt.
Ila, 152, 103 (1943).
Karlik B., Bernert T., Naturwiss., 30, 685 (1942).
Karlik B., Bernert T., Naturwiss., 31, 298, 413 (1943).
Karlik B., Bernert T., Naturwiss., 32, 44 (1944).
Karlik B., Bernert T., Naturwiss., 33, 23 (1946).
Karlik В., В e r n e r t T., Z. Phys., 123, 51 (1944).
Kelly E. L., Segre E., Phys. Rev., 72, 746 (1947).
Kof ler M., Phys. Z., 9,6 (1908).
Kofler M., Monatsh., 34, 389 (1913).
К о 1 о w r a t L., Le Radium, 4, 317 (1907).
К о I о w r a t L., Le Radium, 6, 321 (1909).
Ko 1 о w г a t L., Le Radium, 7, 266 (1910).
186
Литература
90. Kovari к A. F., Phil. Mag., (7), 4, 1262 (1927).
91. Lawrence J. H., Loomis W. F., Tobias C. A., Tur-
pin F. H., J. Physiol., 105, 197 (1946).
92. L e i g h - S m i t h A., Minder W., Nature, Lond., 150, 767
(1942).
93. Livingstone R., Rev. sci., Instrum., 4, 15 (1933).
94. Loring F. H., Chem. News, 131, 371 (1925).
95. Loring F. H., Chem. News, 140, 178, 202 (1930).
96. Loring F. H., Druce J. G. F., Chem. News, 131, 289, 305
(1925).
97. M a c d о n a 1 d P. A., Margolese M. S., Rev. sci. Instrum.,
12, 320 (1941).
98. M а к о w e r W., Phil. Mag., [6], 9,,56 (1905).
99. McClelland J. A., Phil. Mag., [6], 7, 355 (1904).
100. Meyer S., Hess V. F., Paneth F., Sitzugsber. Akad. Wiss.,
Wien, Abt. Ila, 123, 1459 (1914).
101. Miller J. F., Hamilton J. G., P u г n a m T. M., Hay-
mond H. R., Rossi G. B., Phys. Rev., 80, 486 (1950).
102. Minder W., Helv. phys. Acta, 13, 144 (1940).
103. Mund W., Bull. soc. chim. Belg., 32, 256 (1924).
104. Nielsen H. H., Naturwiss., 18, 620 (1930).
105. Nikitin B. A., Z. anorg. Chem., 227, 81 (1936).
106. Никитин Б. А., ДАН СССР, 29, 571 (1940).
107. Никитин Б. А., Иоффе Е. М., ДАН СССР, 60, 595 (1948).
108. Никитин Б. А., Иоффе Е. М., ДАН СССР, 85, 809 (1952).
109. Никитин Б. А., Ковальская М. П., Изв. Акад. Наук
СССР, Отд. хим. наук, 24 (1952).
ПО. N у s w a n d е г R. R., Lind S. С., Moore R. В., Phys. Rev.,
15, 239 (1920).
111. N у s w a n d е г R. R., Tind S. C., Moore R. B., Astro-
phys. J., 54, 285 (1921).
112. Odd ie T. H., Brit. J. Radiol., 10,348 (1937).
113. Paneth F., Nature, Lond., 149,567 (1942).
111. Paneth F., Rabi n о wit sc h E., Ber., 58, 1138 (1925).
115. Pa pish J., Wainer E., J. Amer. chem. Soc., 53, 3818 (1931).
116. Perey M., C. R. Acad. Sci., Paris, 208, 97 (1939).
117. Perey M., J. Phys. Radium, 10,435 (1939).
118. Perey M., Int. Congr. pure appl. Chem., 11, 159 (1947).
119. Perey M., Bull. Soc. chim. Fr., 779 (1951).
120. Perey M., A d 1 о I f J. P., C. R. Acad. Sci., Paris, 236, 1163
(1953).
Литература
187
121. Perkins Р. В., Amer. J. Sci., 25, 461 (1908).
122. Peters К., Weil К., Z. phys. Chem., A148, 1 (1930).
123. Petterson H., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 143,
303 (1934).
124. Piccardi G., Atti Accad. Lincei [6], 6, 428 (1927).
125. Perey M., J. Chim. phys., 43, 155, 262 (1946).
126. Palacios J., Baptista A. M., Riv. fac. cienc. Univ. Lisboa,
3, 49 (1954).
127. Porter A. W., Cuthbertson C., Nature, Lond., 82, 7
(1909—1910).
128. Perey M., Scientia, 88, 267 (1953).
129. Perey M., A d 1 о f f. J. P., C. R. Acad. Sci., Paris, 239, 1389
(1954).
130. Bajewski B., Schraub A., Schraub E., Naturwiss., 30,
733 (1942).
131. Rajewski B., Schraub A., Kahlau G., Naturwiss., 31,
170 (1943).
132. Ramsay W., C. R. Acad. Sci., Paris, 138, 1388 (1904).
133. Ramsay W., Collie J. N., Proc. Roy. Soc., A73, 346, 470
(1904).
134. Ramsay W., Soddy F., Chem. News, 88, 100 (1903).
135. Ramsay W., Soddy F., Proc. Roy. Soc., A72, 204 (1903).
136. Ramsay W., Whytlaw Gray R., C. R. Acad. Sci., Paris,
151, 126 (1910).
137. R a m s t e d t E., Le Radium, 8, 253 (1911).
138. Rasmussen E., Naturwiss., 18, 84 (1930).
139. Rasmussen E., Z. Phys., 62, 494 (1930).
140. Rasmussen E., Z. Phys., 80, 726 (1933).
141. Rose J. E., Swain R. W., J. Nat. Cancer Inst., 10, 605 (1949).
142. R о у d s T., Proc. Roy. Soc., A82, 22 (1909).
143. R о у d s T., Phil. Mag. [6], 17, 202 (1909).
144. R u d о r f G., Z. Elektorchem., 15, 748 (1909).
145. Runge G., Precht J., Ann. Phys. Lpz., 12, 407 (1903).
146. Russell H. N., Phys. Rev., 46, 989 (1934).
147. Rutherford E., Phil. Mag., [5], 49, 1, 161 (1900).
148. Rutherford E., Mem. Manchester phil. Soc., 53, № 2, 1
(1908—1909).
149. Rutherford E., Phil. Mag., [6], 17, 723 (1909).
150. Rutherford E., Brooks H. T., Chem. News, 85, 196 (1902).
151. Rutherford E., Royds T., Phil. Mag., [6], 16, 313 (1908).
152. Rutherford E., Soddy F., Phil. Mag., [6], 4, 580 (1902).
188
Литература
153. Rutherford Е., Soddy F., Phil. Mag., [6], 5, 561 (1903).
154. R о n a E., Z. phys. Chem., 92, 213 (1917).
155. Ramstedt E., Medd. Nobelinst., 5, № 5, 1 (1919).
156. R a n к i n e A. O., Phil. Mag., [6], 21, 45 (1911).
157. Schulze A., Z. phys. Chem., 95, 257 (1920).
158. Shiflett С. H., S t e i d 1 i t z M. E., Davis W., Rev. sci.
Instrum., 21, 842 (1950).
159. Sprousen J. W., van. Chem. Weekblad, 47, 55 (1951).
160. S t r u w e F„ Z. Phys., 36, 410 (1926).
161. S t r u w e F., Z. Phys., 37, 859 (1926).
162. Szeparowicz M., Sitzungsber. Akad. Wiss., Wien, Abt. Ila,
129, 437 (1920).
163. Turner L. A., Phil. Mag. [6], 48, 1010 (1924).
164. Underwood J. E., Schlundt H., Trans. Amer, electro-
chem. Soc., 34, 203 (1918).
165. Varshni T. P., Z. Phys., 135,512 (1953).
166. Voice E. W., Steel, 129, № 11, 102 (1951).
167. Wahl А. С., В о n n e r N. A., Radioactivity Applied to Chemistry
Wiley, New York, 1951.
168. Watson H. E., Proc. Roy. Soc., A83, 50 (1910).
169. Weinzierl P., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 161,
251 (1952).
170. Wertenstein L., Proc. Roy. Soc., A150, 395 (1935).
171. Whytlaw-Gray R., Ramsay W., J. chem. Soc., 95, 1073
(1909).
172. Whytlaw-Gray R., Ramsay W., Proc. Roy. Soc., A84, 536
(1911).
173. Witt F., Sitzungsber. Akad. Wiss., Wien, Abt. Ila, 139, 195
(1930).
174. Wolf P. M., Riehl N., Naturwiss., 17,566 (1929).
175. Wolf S., Z. Phys., 48, 790 (1928).
176. W a 11 s t a b e F., Phys. Z., 4, 721 (1903).
177. Y a g о d a H., Phys. Rev., 40, 316 (1932).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
178. X у d e E., J. Chem. Educ., 36, 15—21 (1959).
179. Krakkay T., Fortschr. chem. Forsch., 3, 738—821 (1958).
180. Neumanh H., J. Inorg. Nucl. Chem., 4, 349—353 (1957).
181. Кузнецова M. Я., Кузнецов В. H. и др., Материалы со-
вещания по применению радиохимических методов изучения
Литература
189
ядерных реакций в спектроскопии нейтронодефицитных ядер. 1,
Дубна, 25 (1958), цит. по [182].
182. Г е р л и т Ю. Б., П а в л о ц к а я Ф. И., Родин С. С., Химиче-
ская наука и промышленность, т. IV, 465—471 (1959).
183. Лаврухина А. К., Родин С. С., Труды комиссии по ана-
лит. химии, т. IX, (XII), 274—283 (1958).
184. Л а в р у х и н а А. К., Успехи химии, т. XXVII, 1209—1219 (1958).
185. Старик И. Е., Меликова О. С., Труды гос. радиевого
инет. им. В. Г. Хлопина АН СССР, т. V, в. 2, 184—202 (1957).
186. Szucs S., Ann. Soc. Sci. Bruxelles, Ser. I, 72, 13—139 (1958).
Часть III
РАДИЙ
(Z = 88)
АКТИНИЙ
(Z = 89)
10
ТЕХНИКА РАБОТЫ С РАДИЕМ И АКТИНИЕМ
Вредность для здоровья. Защита от излучения. Экранированные за-
щитные камеры с перчатками. Смотровые устройства защитных ка-
мер. Химическое действие 7-излучения. Техника работы в защитных
камерах.
ВРЕДНОСТЬ ДЛЯ ЗДОРОВЬЯ
Радиоактивные вещества в условиях высокого уровня ак-
тивности могут оказывать вредное воздействие на челове-
ческий организм в результате: 1) местного загрязнения
кожи, вызванного, например, проливанием или рассыпанием
радиоактивного материала; 2) внутреннего облучения вслед-
ствие попадания в организм радиоактивных изотопов и
3) внешнего чрезмерного облучения р- и 7-излучением.
Как правило, наименее серьезным является попадание
радиоактивного вещества на кожу при условии, что оно
сразу же удалено и не попало в организм в процессе дыха-
ния или через порезы на коже. Повторное загрязнение
кожи радиоактивным веществом или задержка с удале-
нием его может привести к длительному поражению
кожи.
Вдыхание радиоактивного вещества в виде пыли или
аэрозолей (в случае, когда радиоактивный элемент изби-
рательно накапливается в том или ином органе) приводит
к продолжительному местному внутреннему облучению;
это, в частности, происходит при попадании в организм ра-
дия, который избирательно концентрируется в костях. Ма-
ксимальная допустимая доза при вдыхании радия состав-
ляет всего лишь 0,1 ркюри (0,1 р.г Ra22G), а для актиния эта
доза составляет 0,01 ркюри (1,3-Ю”10 г Ас227).
Некоторые вопросы, касающиеся работы с а-активными
веществами, такими, как Ро210, в количествах порядка не-
скольких кюри, уже обсуждались в гл. 3. Опасность, свя-
занная как с загрязнением кожи, так и с попаданием радио-
активного вещества внутрь организма при работе с радием
и актинием, еще более усугубляется тем, что в цепи распада 13
13 К. Бэгнал
194 Техника работы с радием и актинием
этих элементов образуются газообразные радиоактивные
вещества (изотопы радона). Защитные камеры, применяемые
при работе с ощутимыми количествами радия, должны
быть поэтому по возможности газонепроницаемыми. Ис-
точники радия следует контролировать более строго, чем
в случае полония, ибо утечка радона из препарата приведет
к общему загрязнению защитной камеры и в конце концов
может обесценить действие экрана, применяемого для за-
щиты от у-излучения, так как ^-излучатели в виде активных
осадков, образующихся при радиоактивном распаде радона,
могут появиться на той стороне защитного экрана, где на-
ходится оператор. Это обстоятельство имеет меньшее зна-
чение в случае актиния, поскольку выделяемая им эмана-
ция имеет период полураспада, равный всего лишь 3,92 сек.,
а способность растворов актиния выделять эманацию очень
низка. (О распространении загрязнения, вызванного 40 мг
радия, и о способах удаления загрязнения, применявшихся
в данном случае, см. работу [116].)
Внешнее облучение р- и ^-излучением может привести
к серьезным последствиям, например к радиационным ожо-
гам, которые обычно появляются через несколько суток
после облучения и которые медленно заживают; такое об-
лучение может также вызывать ослабление деятельности
костного мозга (белокровие) (см. работу Кэгла [4]). Слу-
чаи, имевшие место с рабочими, занятыми производством
светящихся красок (например, для циферблатов), хорошо
известны.
Максимально допустимая недельная доза общего облу-
чения организма составляет 300 мр\ часто также указы-
вается ежедневная доза, равная 0,1 бэр (биологический эк-
вивалент рентгена) для 8-часового рабочего дня. Знание не-
дельной дозы, по-видимому, представляет больший интерес,
поскольку она допускает некоторое превышение дозы об-
лучения в какой-либо из рабочих дней. Приведенные выше
данные относятся к облучению всего тела человека, и до-
пускают, например, для рук большую дозу облучения, рав-
ную 1,5 фэр (физический эквивалент рентгена) в неделю,
при условии, что доза общего облучения всего тела остается
ниже допустимых пределов.
На практике крайне трудно сопоставить единицу дозы
(рентген) с единицей радиоактивности (кюри). Мощность
Вредность для здоровья
195
дозы Р, выраженную в рентгенах за 1 час, на расстоянии
0,3 м от незащищенного точечного источника 7-излучения,
приблизительно можно вычислить из эмпирического соот-
ношения
Р = аЕА,
в котором значение константы а принимают равным 6—7
(более высокое значение, вероятно, предпочтительнее, по-
скольку безопаснее более надежно защитить работающего),
Е — энергия 7-фотона (Мэв) [для значений энергии, лежа-
щих в интервале 0,2—1,5 Мэв] и А— 7-активность источника
(кюри). Точные расчеты мощности дозы 7-излучения могут,
однако, оказаться крайне сложными, поскольку при таких
расчетах необходимо учитывать энергии различных лучей,
испускаемых данным источником, и делать поправки на
поглощение излучения средой, через которую оно прохо-
дит (например, жидкость, в которой растворено соединение,
испускающее 7-излучение, воздух между сосудом и опера-
тором и т. д.). При этих расчетах следует также принимать
во внимание необходимость защиты работающего от рассеян-
ного излучения, появляющегося в результате изменения на-
правления первичных 7-лучей при взаимодействии с мате-
риалом сосуда, в котором находится радиоактивное веще-
ство, с окружающим воздухом и т. д.
Как правило, мощность дозы бокового и обратного рас-
сеянного излучения составляет лишь небольшую часть мощ-
ности дозы первичного излучения; учет такого облучения
очень сложен. Кроме того, в таких расчетах необходимо
принимать во внимание размеры источников, если их нельзя
рассматривать как точечные, что допустимо лишь в исклю-
чительных случаях.
Чтобы перекрыть возможные неточности при проведении
всех этих расчетов, желательно вычисленную мощность дозы
увеличить на один порядок, принимая во внимание опас-
ность занижения вычисленной дозы по сравнению с истин-
ной. Пожалуй, единственным надежным способом, позво-
ляющим установить наличие опасности облучения, является
прямое измерение интенсивности излучения данного источ-
ника одним из имеющихся приборов для измерения 7-излу-
чения; один из таких удобных и портативных приборов с пи-
танием от батарей описан Уитингом и др. [211; этот прибор
13*
196
Техника работы с радием и актинием
по существу представляет собой портативную ионизацион-
ную камеру.
Степень облучения, которому подвергся человек, обычно
определяют по почернению кусочка фотопленки, которую
в кассете носят при себе лица, работающие в условиях воз-
можного облучения. Экранированием этой пленки материа-
лами, имеющими соответствующий порядковый номер, можно
установить, какова общая доза облучения, обусловленная
различными видами излучения (мягкие рентгеновские лучи,
7-лучи, р-частицы). Для определения степени облучения
можно пользоваться карманным дозиметром той или иной
конструкции. Таким дозиметром обычно является неболь-
шой электроскоп, который периодически перезаряжают.
Совершенно очевидно, что такого рода измерительные при-
боры должны постоянно носить лица, работающие в ра-
диохимической лаборатории.
ЗАЩИТА ОТ ИЗЛУЧЕНИЯ
Учитывая, что радий и актиний в весомых количествах об-
ладают высокой 7-радиоактивностью, непосредственно ра-
ботать с ними и наблюдать за ними нельзя. При работе
с радием, правда, непрерывной вентиляцией можно удалять
радон, продукты распада которого обладают жестким ]-из-
лучением (см. гл. 9). Однако в таких условиях можно про-
изводить лишь некоторые химические операции.
Степень внешнего облучения, воздействующего на ра-
ботающего, можно снизить увеличением расстояния между
работающим и источниками излучения; это достигается
применением шпаговых манипуляторов (при расчетах сте-
пени облучения можно использовать закон обратной про-
порциональности квадрату расстояния, поскольку размеры
источника малы по сравнению с расстоянием от источника
до оператора) или уменьшением времени, в течение кото-
рого работающий подвергается облучению. Можно, напри-
мер, работать в течение 2 час. в день при дозе облучения,
в четыре раза превышающей дозу, допустимую при работе
на протяжении восьми часов. Если уровень радиации на-
столько высок, что ни один из этих приемов не достигает
цели, тогда между источником излучения и работающим
устанавливают защитный экран.
Защита от излучения
197
В зависимости от необходимости проводить те или иные
операции обычно применяют один из следующих двух спо-
собов экранирования. При «местном» экранировании каж-
дый элемент оборудования в защитной камере экранируют
отдельно. При работе с источниками у-излучения высокой
активности следует отдать предпочтение «барьерному» эк-
ранированию, заключающемуся в возведении вокруг за-
щитной камеры высокой стенки из материала подходящей
плотности. Важно при этом экранировать фильтры вытяж-
ной системы защитной камеры, поскольку адсорбированный
радон может повысить опасность облучения. Барьерное
экранирование неудобно, так как оно массивно и дорого,
а в то же время оно должно быть постоянной принадлеж-
ностью лаборатории, ибо, к сожалению, не существует
иного выхода в тех случаях, когда приходится работать с ис-
точниками у-излучения большой активности.
Для материалов не очень большой плотности толщина
экрана, способного поглотить заданную часть падающего
излучения, приблизительно обратно пропорциональна плот-
ности используемого материала. Обычно эффективность
защиты от излучения характеризуют толщиной экрана,
необходимой для уменьшения интенсивности падающих
7-л у чей в десять или в два раза (толщина слоя десятикрат-
ного или половинного ослабления). Некоторые значения
толщины слоя десятикратного или половинного ослабления
для экранов, изготовляемых из различных материалов, при-
ведены в табл. 23. Эти значения относятся к жесткому ?-из-
лучению, испускаемому выдержанным изотопом Ra226
(1,8 Мэв); толщина слоя десятикратного ослабления должна
Таблица 23
Защитные свойства различных материалов
Свинец Железо Алюми- ний Бетон Вода
Плотность, г!см3 Толщина слоя десятикратного и.з 7,86 2,7 2,35 1
ослабления, см Толщина слоя половинного 4,70 7,37 20,3 22,9 47,0
ослабления, см 1,42 2,21 6,1 6,86 14,2
125
Энергия %-излучения t Мэв
Р И с. 16. Толщина слоя десятикратного
ослабления для узкого пучка 7-лучей.
(По данным [18, раздел 6].)
Рис. 17. Толщина слоя десяти-
кратного ослабления для широ-
кого пучка у-лучей.
(По данным [18, раздел 6, рис. 7 — 10].)
Экранированные защитные камеры с перчатками
199
быть в 3,3 раза больше толщины слоя половинного ослаб-
ления.
Некоторые данные о защитных свойствах различных ма-
териалов от у-излучения различной энергии приведены на
рис. 16 и 17. Другие данные по этому вопросу можно найти
в работах [8, 13, 18 и 22].
ЭКРАНИРОВАННЫЕ защитные камеры с перчатками
Прежде чем конструировать защитную камеру и оборудо-
вание, необходимое для проведения работы, следует опре-
делить, какая защита потребуется для обеспечения безо-
пасности работающих, ибо толщина экранов может вызвать
известные изменения предусмотренных методов ведения ра-
боты. Для проведения наиболее часто применяемых опера-
ций лучше всего использовать защитную камеру такого
типа, который описан в гл. 3, снабженную, помимо лю-
ков для перчаток, шпатовыми манипуляторами, огородив
ее защитным экраном с тем, чтобы необходимые операции
можно было производить с любой стороны. Такой способ
имеет, однако, тот недостаток, что он не позволяет изме-
нять методику работы без снятия экрана и изготовления
новой защитной камеры.
В экранированной защитной камере несколько улуч-
шенного типа [17] имеется хорошо экранированный внут-
ренний шкаф, в который можно помещать высокоактивные
источники у-излучения в тех случаях, когда возникает не-
обходимость изменить оборудование защитной камеры.
В стенках камеры с двух противоположных сторон вделаны
специальные люки для перчаток, а также шпаговые мани-
пуляторы. По бокам защитной камеры имеются экраниро-
ванные приспособления для введения материалов в камеру,
а к люку в дне камеры присоединен экранированный ме-
шок для сбрасывания отходов. В тех случаях, когда ока-
зывается необходимым заменить оборудование в защитной
камере такого типа, препарат, являющийся источником
f-излучения, при помощи шпаговых манипуляторов по-
мещают в свинцовый шкаф, и защитную камеру после этого
используют обычным образом, как при работе с полонием,
если контрольные приборы показывают, что уровень радиа-
200 Техника работы с радием и актинием
ции соответствует условиям безопасности. По окончании
замены оборудования снова устанавливают экраны, и ра-
бота продолжается с применением дистанционного управ-
ления. Необходимо отметить, что радон быстро диффунди-
рует через защитные перчатки из естественного каучука,
однако неопреновые перчатки не пропускают радона, а по-
этому везде, где только возможно, следует применять нео-
прен (см. также гл. 3).
Существует возможность дальнейшего усовершенство-
вания конструкции защитных камер для выполнения ра-
бот такого типа.
СМОТРОВЫЕ УСТРОЙСТВА ЗАЩИТНЫХ КАМЕР
Прямое наблюдение за работой с у-активными материалами
можно осуществлять через толстые окна из свинцового
стекла (плотностью 4,3—6,3), толщина которого должна
в два-три раза превосходить толщину применяемого свин-
цового экрана. Такие окна чрезвычайно дороги, и поэтому
вместо них часто применяют трубы с вмонтированными
стеклами по концам, заполняемые прозрачным раствором,
обладающим высокой плотностью. Чаще всего для этой
цели применяют насыщенный раствор бромистого цинка
(плотность 2,52); под действием облучения в таком растворе
выделяется бром, который окрашивает раствор. Этого можно
избежать, если при приготовлении раствора добавить со-
лянокислый гидроксиламин [7 ]. Весьма важно, чтобы раст-
вор бромистого цинка не содержал сульфида, в противном
случае раствор будет мутнеть от серы, выделяющейся при
действии излучения на сульфид, причем эта сера не восста-
навливается гидроксиламином. Существенно также, чтобы
стекло, закрывающее внутренний конец трубы, служащей
окном, не окрашивалось под действием излучения. Для этой
цели подходит имеющееся в продаже стекло, содержащее
1—2% церия. В печати опубликованы также некоторые
данные по коррозии материалов, из которых изготовляются
подобные окна, происходящей в случае контакта их с бро-
мистым цинком [6].
Неудобство применения окон с жидкостным заполнен
#ием заключается в том, что прц прорыве стенок трубы»
Смотровые устройства защитных камер
201
в которой находится жидкость, или в случае поломки кон-
цевых стекол сразу же теряется защитное действие такого
окна; растрескивание же смотровых стекол приводит к
порче окна и ухудшению возможности ведения наблюдения,
однако при этом окно сохраняет защитное действие, по-
скольку в массивном стекле прямолинейное растрескивание,
пропускающее излучение, маловероятно.
Окно для наблюдения можно также сделать из стеклян-
ных пластин толщиной 2,5 мм, погруженных в минеральное
масло, имеющее коэффициент преломления, близкий к ко-
эффициенту преломления применяемого стекла. Такой спо-
соб, по-видимому, представляет наиболее удачное и эконо-
мичное решение вопроса об обеспечении дистанционного
наблюдения.
Конструкция окна для наблюдения имеет огромное зна-
чение; наиболее часто делают окна квадратного сечения со
стороной около 15 см и толщиной приблизительно 20 см,
а также цилиндрические — диаметром 30—45 см\ такие
окна не подходят для работы в больших камерах, для ко-
торых более удобными являются удлиненные окна. При
небольшом размере высокий коэффициент преломления
стекла или раствора крайне затрудняет манипулирование,
особенно если приходится производить работу на некото-
ром расстоянии от окна.
Для наблюдения на расстоянии можно в некоторых слу-
чаях пользоваться перископами и зеркалами, однако не
подлежит сомнению, что дистанционное управление аппа-
ратурой при таком способе наблюдения является очень
трудной операцией.
Несмотря на то, что разработке стекла для смотровых
окон, устойчивого к действию излучения, уделялось боль-
шое внимание, до настоящего времени еще мало сделано
в области производства специальной стеклянной посуды
для радиоактивных материалов. Действие радия в количе-
стве порядка нескольких кюри быстро вызывает такое окра-
шивание обычной лабораторной посуды, что содержимое
того или иного сосуда уже перестает быть видимым. Такое
окрашивание, однако, можно устранить, подвергнув стекло
действию ультрафиолетового облучения.
(По вопросам дистанционного наблюдения см. рабо-
ту 111.)
202
Техника работы с радием и актинием
ХИМИЧЕСКОЕ ДЕЙСТВИЕ 7-ИЗЛУЧЕНИЯ
Химическое действие у-излучения высокой интенсивности
похоже на химическое действие a-излучения высокой ин-
тенсивности (см. гл. 3).
Особая трудность, возникающая при работе с -[-актив-
ными материалами, заключается в том, что для них нельзя
получить хорошую рентгенограмму из-за почернения пленки
под действием излучения. Для актиния это затруднение
было преодолено использованием очень малых (около
10 мг) образцов свежеприготовленного материала, в кото-
ром отсутствуют дочерние продукты; даже в этом случае
рентгенограмму следует снимать не позже, чем через 24 часа
после очистки.
ТЕХНИКА РАБОТЫ
Учитывая трудности, связанные с необходимостью дистан-
ционного манипулирования, каждый опыт следует плани-
ровать так, чтобы он выполнялся возможно более просто.
Для каждого опыта можно было бы конструировать спе-
циальную систему дистанционного управления, но это
обошлось бы слишком дорого и потребовало бы затраты
большего времени, так как даже в обычных химических ис-
следованиях требуется выполнение целого ряда совершенно
различных химических операций, при которых может ока-
заться необходимой очень сложная аппаратура.
Существуют копирующие манипуляторы общего назна-
чения, в которых исполнительный механизм воспроизводит
на расстоянии движение руки оператора, но они довольно
дороги. Требования, предъявляемые к этому типу манипу-
лятора, описаны Гертцом [3, 9]. Дешевле обходится при-
менение стандартного щипцевидного манипулятора, снаб-
женного съемным набором головок, размещаемых на полке
в защитной камере. Манипулятор этого типа описан Ритчи
и Спраггом [14]. Ствол манипулятора проходит через свин-
цовый шарнир во внешней защите и герметически присое-
диняется к рукаву, который в свою очередь соединен со
стандартным входным отверстием с лицевой стороны защит-
ной камеры [17].
Внесение материалов или оборудования в защитную ка-
меру этого типа является довольно трудной операцией;
Техника работы
203
применение для этой цели обычных форкамер при работе
с радием или актинием слишком опасно, так как через нее
может легко произойти утечка изотопов радона из камеры.
Лучшим способом, по-видимому, является использование
для этой цели запечатанных мешков, подобных тем, которые
описаны в гл. 3 при рассмотрении вопроса об удалении из
камер a-активных отходов. Новые детали, необходимые для
работы в защитной камере, помещают в мешок из пластика,
который затем присоединяют к входному люку защитной
камеры. Входной люк открывают изнутри камеры, и детали
переносят внутрь камеры при помощи манипулятора. Ис-
пользованный мешок затем разрезают высокочастотным сва-
рочным аппаратом и отъединяют от камеры, а запаянный
его отросток оставляют присоединенным к камере до тех
пор, пока снова не потребуется вводить в нее какие-либо
детали.
В этом случае новый мешок с необходимыми для работы
принадлежностями прикрепляют поверх оставленного от-
ростка таким образом, чтобы этот отросток можно было
втянуть в защитную камеру и вскрыть [17]. Размеры пе-
реносимых таким образом в защитную камеру предметов
ограничиваются размерами входного люка, который поэ-
тому должен быть таким, чтобы через него проходили са-
мые большие детали установки, предназначенной для про-
ведения тех или иных экспериментов в данной камере. Это
условие кажется совершенно очевидным, однако оно часто
не соблюдается.
Библиография по вопросам защиты от облучения, а
также по вопросам конструирования и работы радиохими-
ческих лабораторий имеется в работе Уиттлези [20]. Для
получения более детальных данных по вопросу о методах
обращения с сильно радиоактивными веществами следует
обратиться к другим работам [2, 5, 10—12, 15 и 19].
ЛИТЕРАТУРА
1. Argonne National Lab. Amer. Rep. ANL-4903 (1952).
2. В a g n a 11 K. W., Spragg W. T., Atomics, 6, 71, 125 (1955).
3. Burnett J. R., Goertz R. C., Thompson W. M., Proc,
int. Conf., Geneva, 1955 U. N. New York, 14, 116 (1956).
4. Cagle C. D., Amer. Rep. ORNL-CF-51-4-173 (1951).
5. С о о к G. В., Duncan J. F., Modern Radiochemical Practice,
Clarendon Press, Oxford, 1952.
6. Draley J. E., Drugas P. G., Amer. Rep. ANL-4837 (1949).
7. Ferguson K. R., Nucleonics, 10, № 11,46 (1952).
8. F r a z i e r P. M., В u c h a n о n C. R., Morgan G. W., Amer.
Rep. AECU-2967 (1954).
9. Goertz R. C., Nucleonics, 10, № 11,36 (1952).
10. Henriques R. C., Schreiber A. P., Nucleonics 2, № 3, 1
(1948).
11. Kellex Corporation., Amer. Rep. KLX-08 (1951).
12. Oak Ridge National Lab., Health Physics Div. Amer. Rep. AECU-
817 (1950).
13. Parker H. M., Nucleonics, 3, № 4,44 (1948).
14. Ritchie A. B., Spragg W. T., J. sci. Instrum., 28, 214 (1951).
15. Schweitzer G. K., Whitney I. B., Radioactive Tracer Tech-
niques, Van Nostrand, New York, 1949.
16. S к о w R. К., V a n d i v e r t V. V., Holden F. R., Nucleonics,
11, № 8, 45, (1953).
17. Spragg W. T., Private communication.
18. Sherwood R. J., Dunster H. J., Ed. Brit. Rep. A.E.R.E.
HP/L 23 (1956) H.M.S.O. 91-3-2-74.
19. Tompkins P. C., Amer. Rep. MDDC-1527 (1947).
20. Whittlesey E., Givens E., Amer. Rep. AECU-1020 (1950).
21. Whiting F. B., Thomas D. G. A., Marsh J. B., J. sci.
Instrum., 27, 157 (1950).
22. Whitehouse W. J., Putman J. L., Radioactive Isotopes,
Clarendon Press, Oxford, 1953.
11
РАДИЙ
Открытие. Применение. Отделение от других элементов. Определе-
ние Ra22e. Изотопы. Спектр. Металлический радий. Атомный вес.
Соединения.
ИСТОРИЯ ОТКРЫТИЯ
Вскоре после открытия полония в висмутовой фракции ос-
татков, образующихся после переработки урановой руды,
Пьер и Мария Кюри заметили, что в бариевой фракции этих
остатков концентрируется новое радиоактивное вещество
[27, 33, 35]. После дальнейшего концентрирования Демарсе
удалось изучить спектр этого вещества, в ультрафиолетовой
области которого была обнаружена новая линия [40—42].
Радиоактивность полученного препарата была приписана
новому элементу, по химическим свойствам близкому к ба-
рию; в связи с тем, что новый элемент испускал излучение
(радиацию), супруги Кюри назвали его радием. Сообщение
об открытии радия было встречено некоторыми кругами
с явным недоверием; высказывалось мнение [ПО], что это
не новый элемент, а радиоактивная форма бария; что же
касается полония, то его рассматривали как радиоактивный
висмут (гл. 1). Вскоре после этого супруги Кюри при по-
средничестве Венской академии наук получили большое ко-
личество урановой смоляной руды из яхимовских урановых
месторождений (Чехословакия), которые тогда были собст-
венностью австрийского правительства. Работая в самых
примитивных и тяжелых условиях, супруги Кюри все
же в конце концов методом дробной кристаллизации по-
лучили образец хлористого радия, очищенного от хлори-
стого бария. Выделение радия в таких условиях было од-
ним из самых замечательных научных подвигов того вре-
мени.
Одновременно с успешным концентрированием радия
М. Кюри производила наблюдения его радиоактивности и
определение атомного веса, а Демарсе проводил спектроско-
пические исследования. Спектр очищенного материала
206
Радий
имел заметное сходство со спектрами щелочноземельных
металлов, а поскольку данный элемент обнаруживал хи-
мические свойства, близкие свойствам бария, то в периоди-
ческой таблице элементов его поместили на свободное место
во второй группе как самый тяжелый щелочноземельный
металл. Результаты определения атомного веса, проведен-
ные на очищенных образцах, вскоре рассеяли всякие сом-
нения относительно правильности предположений о том,
что радий представляет собой новый элемент. Развитие ра-
диевой промышленности во Франции, Германии, Австрии
и Англии относится к 1902—1903 гг. (исторический обзор
развития промышленности радия дан в справочнике Гме-
лина [72, стр. 27 и далее]).
Ra226 является членом радиоактивного ряда урана—
радия (4м + 2) (см. приложение) и встречается во всех ми-
нералах, содержащих уран; урановая смоляная руда со-
держит приблизительно 400 мг радия на 1 /и, и урановые
руды представляют собой главный источник получения этого
элемента. Радий содержится во многих природных водах,
и отмечен смертельный случай от воды, содержащей радий
[97, стр. 11]. В водах океанов содержится примерно
20 000 т радия, а в верхнем слое земной коры толщиной
1,6 км содержится, как показывает расчет, примерно
1,8 • 107 tn этого элемента [2].
ПРИМЕНЕНИЕ РАДИЯ
Радий применяли главным образом в медицине при терапии
7-лучами (например, при лечении рака); у-лучи испускаются
дочерними продуктами радона, и поэтому в клинических
условиях радон часто применяют отдельно в тонких ампу-
лах или «зернах». у-Излучение использовали в промышлен-
ности для определения дефектов литья (см., например,
[184]), а также при определении толщины слоя катализа-
тора в аппаратах для крекинга нефти [169]. Благодаря
свойству радия (и других радиоактивных элементов) испу-
скать лучи, ионизирующие воздух, небольшие количества
солей радия, по имеющимся данным, находят применение
в качестве веществ, способствующих повышению эффектив-
ности громоотводов; при этом соли радия наносят на концы
Отделение радия от других элементов
207
громоотводных стержней [161 ]. Это же свойство радия ис-
пользовали в промышленности для снятия статических за-
рядов [175]. Применение радия в этих целях уменьшается,
поскольку в настоящее время более дешевые препараты
можно получить облучением в реакторах.
Небольшие количества радия находят применение при
производстве светящихся красок, при этом радий исполь-
зуют вместе с такими сцинтилляторами, как сульфид
цинка. Смеси солей радия с бериллием применяют в ка-
честве источников нейтронов (см., например, [145]).
В работе [186] описан способ изготовления устойчивых радие-
вых а-, р- и ^-источников на основе неорганических эмалей.
В геологии адсорбцию бромида радия глинами использо-
вали в качестве метода их отличия от неадсорбирующих
минералов [146]; были проведены работы по изучению
диффузии радия в минералах [147].
Наблюдение за у-активностью воздуха применяют при
разведке урана и в известной мере при разведке нефти.
Такой способ определения нефтеносных полей основан на
обнаружении у-излучения, испускаемого сульфатом ра-
дия, выпавшим в осадок из подземных вод вблизи поверх-
ности; области, прилегающие к нефтеносным слоям, обычно
показывают более высокий уровень у-активности, нежели
собственно нефтеносные слои. Заметно более низкий уро-
вень у-активности показывают также озера, реки и болота
[114].
ОТДЕЛЕНИЕ РАДИЯ ОТ ДРУГИХ ЭЛЕМЕНТОВ
Сульфатные остатки от переработки урановой смоляной
руды превращают в карбонаты кипячением с концентри-
рованным раствором карбоната натрия, и полученный про-
дукт растворяют в разбавленной соляной кислоте. Полоний
и висмут осаждают пропусканием через раствор сероводо-
рода, после чего, добавляя аммиачную воду, осаждают
актиний и редкоземельные элементы. Затем раствор обра-
батывают серной кислотой с целью осаждения радия и ба-
рия в виде сульфатов, которые переводят в хлориды ранее
описанным способом. Из 1 tn остатков от переработки ура-
новой смоляной руды получают приблизительно 10—20 кг
208
Радий
сульфатов, содержащих немногим менее 0,5 г радия [27,
132].
Отделение радия от бария связано с известными труд-
ностями, поскольку оба элемента обладают весьма близ-
кими химическими свойствами. Первоначальные методы
их разделения основывались, как правило, на проведении
дробной кристаллизации или дробного осаждения, при ко-
торых использовали разницу в растворимости солей этих
двух элементов, особенно их хлоридов [12, 24, 35, 50,
124], бромидов [119, 171, 177], хроматов [79, 121, 131,
155, 156], йодатов [70, 135] и сульфатов [7, 45, 62, 121];
опубликовано очень много работ по изучению соосаждения
указанных солей радия и бария (см., например, [13, 18,
20, 21, 118, 123, 134]).
Работы по изучению подвижностей катионов бария и
радия показали, что некоторого отделения радия от ба-
рия можно достигнуть, применяя метод, основанный на
различии в подвижности ионов бария и радия в раство-
рах агар-агара; установлено, что катионы радия обладают
заметно большей подвижностью, чем катионы бария [100,
106]. Однако данный метод не нашел какого-либо при-
менения.
Один из наиболее интересных методов, применяемых
для обогащения соединений бария, содержащих радий,
основан на термической устойчивости карбоната радия;
некоторого обогащения радием можно достигнуть, по-види-
мому, при нагревании смеси карбонатов до 400—800° в ва-
кууме. Образующуюся при этом окись бария можно впо-
следствии выщелочить водой [180].
Методы дробной возгонки еще не применяли при отде-
лении радия от бария, взятых в виде относительно летучих
галогенидов, несмотря на то, что такие методы практически
осуществимы, поскольку бромид радия менее летуч, чем
бромид бария [165]. В печати, однако, имеются сообщения
[87 ] о выделении хлорида радия из урановых руд методом
возгонки. Есть указания о некотором обогащении радием,
достигаемом при электролизе смеси хлоридов на ртутном
катоде [31, 185] или даже восстановлением водных раст-
воров смеси галогенидов амальгамой натрия [117].
Отделение радия на пермутите и цеолите [115, 164] и
на глиноземе [116] было известно уже сравнительно давно,
Отделение радия от других элементов
209
а недавно для окончательного отделения радия от бария
после предварительного обогащения дробной кристалли-
зацией или дробным осаждением был применен метод ион-
ного обмена [138, 173, 176]. Обычная процедура сводится
Рис. 18. Влияние концентрации со-
ляной кислоты на коэффициент распре-
деления ионов щелочноземельных эле-
ментов между раствором и ионообмен-
ной смолой дауэкс-50.
(По данным [47].)
к пропусканию раствора, содержащего барий и радий, че-
рез колонку в таких условиях, при которых поглощается
предпочтительно радий; первый элюат, таким образом, ока-
зывается обогащенным барием. Фракцию, более богатую
радием, извлекают из колонки обработкой минеральной
кислотой, после чего элюат разбавляют и процесс повто-
ряют на новой колонке.
Разделение радия, бария и стронция на смоле дауэкс-50
(70—100 меш) путем элюирования раствором цитрата ам-
14
К. Багнал
210
Радий
монйя основано на различии констант диссоциации комп-
лексов, образуемых этими тремя элементами с анионами
лимонной кислоты [172]; данный метод оказался лучшим
способом разделения, известным в настоящее время. Ту же
смолу применяли для отделения радия от тория в ра-
створе 0,1 н. соляной кислоты; торий, как указывали, элюи-
руется 7%-ным раствором щавелевой кислоты при 80°, а ра-
дий — Зн. раствором соляной кислоты при комнатной темпе-
ратуре [153]. Влияние концентрации соляной кислоты на
коэффициент распределения радия между смолой и рас-
твором показано на рис. 18. Обратный порядок элюирова-
ния при высоких концентрациях кислоты обусловлен, по-
видимому, частичной дегидратацией ионов в растворе [47 ].
Недавно была описана [142] полузаводская установка
для отделения радия от бария методом ионного обмена
в больших масштабах; вполне возможно, что этот метод
найдет более широкое применение в будущем.
Метод отделения Ra от Ва при помощи ионного обмена описан
также в работе [193]. Анализируемый раствор (0,2 н. по НС1 или
HNO3) вводят в колонку высотой 250 мм и диаметром 13 мм, запол-
ненную катионитом дауэкс-50 в аммонийной форме с размером зе-
рен 50—100 меш. Затем производят элюирование, пропуская 1—6 н.
НС1, 3,5 н. NH4. Cl или 0,32 н. раствор цитрата аммония с pH 5,6 со
скоростью 3 мл/см2*мин.
Однако при работе с очень большими количествами ра-
дия ионообменная смола может в какой-то мере разла-
гаться под действием излучения, а при использовании суль-
фосмол можно ожидать также образования свободной сер-
ной кислоты; в этом случае радий будет осаждаться в ко-
лонке и его элюирование сильно затруднится. Кроме того,
в результате радиолиза может выделяться газ, создающий
пробки в колонке и затрудняющий, таким образом, прове-
дение непрерывного процесса (см. гл. 1).
В печати имеются сообщения [17] о результатах изу-
чения распределения радиевых солей между кристалличе-
ским нитратом кальция и его расплавом при высоких тем-
пературах. В расплавленных смесях сульфата и хлорида
бария при 700—1040° радий концентрируется в хлориде
бария [61 ]. Результаты, полученные при изучении рас-
пределения радия между расплавом и твердой фазой изо-
морфных солей, были истолкованы таким образом, будтс
Отделение радия от других элементов
211
они свидетельствуют о том, что скорость всего процесса
определяется скоростью диффузии в кристаллах [15], од-
нако коэффициент диффузии микроколичеств радия в нит-
рате бария при 300° имеет порядок лишь 10-12 см2/сутки,
[16].
Поведение радия при кристаллизации нитратных, хлоридных
и фторидных расплавов изучалось в работах [187—191].
Адсорбция микроколичеств радия на силикагеле, суль-
фате бария [54], коллоидной двуокиси кремния и двуокиси
марганца [52] изучалась в качестве возможного метода
разделения, однако эти способы разделения в заводском
масштабе не применялись.
В работе [192] приведены данные по адсорбции радия серно-
кислым свинцом. Автор приходит к выводу, что основным процес-
сом является обмен с ионами на поверхности твердой фазы. Но на-
ряду с адсорбцией наблюдается также распределение радия в крис-
таллах сернокислого свинца в процессе перекристаллизации
адсорбента.
Подробное описание применяемых методов выделения
радия дано в работе Арбитрио [1] и в справочнике Гме-
лина [72].
Отделение радия от его дочерних
производных
Методом электрохроматографии радий можно отделить от
его дочерних производных в растворе молочной кислоты;
скорости миграции присутствующих в данном случае ионов
образуют такую последовательность [157]:
радий > свинец > висмут > полоний.
Хроматография на бумаге с применением растворов, со-
держащих 80% ацетона, 10% 1 н. соляной кислоты и 10%
1 н. азотной кислоты, по имеющимся сведениям, также дает
эффективное разделение [168]. Возможны и чисто химиче-
ские способы разделения; радий можно осадить с некото-
рым количеством свинца действием концентрированной
соляной кислоты, при этом висмут и полоний остаются
в растворе. Осадок, содержащий радий, растворяют и от-
деляют RaD осаждением его в виде гидроокиси с добавле-
14*
212
Радий
нием свинца в качестве носителя; лучше осаждение вести
сероводородом в восстановительной среде (чтобы предот-
вратить возможность образования сульфата в результате
окисления под действием радиоактивного излучения).
В работе [194] описан метод отделения Ra22e от долгоживущих
активных осадков RaD, RaE и Ро, основанный на способности по-
следних в отличие от Ra22fl образовывать прочные комплексы с ком-
плексоном-I II. Из анализируемых растворов в присутствии комплек-
сона-111 сульфат аммония осаждает только Ra22e в форме RaSO4.
Анализируемую соль Ra растворяют в 100 мл воды, прибавляют
3 капли 20%-ной НС1, 10 мл ацетатного буферного раствора (130 мл
концентрированной СН3СООН, 200 мл NH4OH, 200 мл бидистиллята)
с pH 4,5—5 и 10 мл 1%-ного раствора комплексона; смесь кипятят
для количественного перевода RaD, RaE и Ро в комплексы с ком-
плексоном-111, прибавляют 3 мл 10%-ного раствора (NH4)2SO4
и выдерживают 6 час. Образующийся осадок RaSO4 отфильтровы-
вают (фильтр с синей лентой) и промывают 1%-ным раствором
(NH4)2SO4.
Установлено, что осадок RaSO4 растворим при кипячении в
3—5%-ном аммиачном растворе этилендиаминтетрауксусной кислоты.
После подкисления такого раствора уксусной кислотой по метило-
вому красному, а также кипячения и продолжительного выдержи-
вания при комнатной температуре образуется крупнокристалли-
ческий осадок RaSO4. Определением Ra-226 по 7-, RaD и RaE по
0- и Ро по a-активности установлено, что потери Ra при указанных
операциях составляют 10“б г на 100 г воды, а потери RaD, RaE и
Ро —3%.
ОПРЕДЕЛЕНИЕ РАДИЯ (Ra22«)
При работе с весомыми количествами радия, как это было
при определении атомного веса в опытах М. Кюри, Гониг-
шмида и других, радий определяют в виде хлорида или
бромида; внесение поправок на влияние повышенной тем-
пературы соли радия, являющейся результатом поглоще-
ния части излучения самим образцом, не вызывает затруд-
нений [5]. Малые количества радия обычно определяют
счетом а-частиц; этот метод осложняется тем обстоятель-
ством, что первым членом в цепи распада радия является
газообразный радон. В то же время образование радона
использовали для косвенного определения радия так на-
зываемым эманационным методом [4, 29, 49, 65, 67, 95,
111], который дает хорошую точность [66]. Количество
радона Q„ образующееся к моменту времени t9 можно вы-
разить уравнением
Q,= Q. (1- е-“),
Определение радия (Ra™)
213
где — количество радона, образующееся к моменту
достижения радиоактивного равновесия. можно при-
равнять количеству радия, присутствующего в источнике,
если его выразить в единицах активности. Данный спо-
соб определения радия предполагает выделение радона
из раствора, содержащего радий, кипячением и определе-
ние количества радона при использовании соответствую-
щего счетчика. Установка, необходимая для определения
радия таким методом, довольно сложна, и работа на ней
требует большого внимания. Применение этого метода мо-
жет также привести к ошибке в том случае, если из рас-
твора случайно выпадут нерастворимые соли (например,
сульфат или карбонат), так как радон не выделяется ко-
личественно из этих осадков (см., например, [10]). Однако
содержащие радий бариевые соли жирных кислот (напри-
мер, пальмитиновой и стеариновой) выделяют радон почти
количественно [75] и их можно применять при такого рода
определениях.
Количество радия можно определить непосредственно
сравнением у-излучения, испускаемого образцом, с излу-
чением эталонного радиевого источника [82, 112, 162];
в этом случае для получения точных результатов нужно
отфильтровывать p-излучение и тщательно контролировать
геометрию эксперимента, в частности следить за одинако-
вым расположением исследуемого образца и эталона от-
носительно счетной трубки. Данный метод эффективен в тех
случаях, когда исследуемый образец и эталон содержат
почти равные количества радия, а поэтому в лаборатории
обычно имеют набор эталонов различной активности (на-
пример, в соотношении 1 : 10, 1 : 100 и т. д.). Сравнение
с радиевым эталоном можно также производить по почер-
нению фотопленки, вызванному действием двух источни-
ков [1221, однако этот способ дает менее точные резуль-
таты.
Не так давно Кёрби [101] описал своеобразный метод
прямого счета а-частиц, испускаемых радием; этот метод
предполагает удаление радия-F (Ро210) путем выделения
его на меди. Для этого раствор радия в соляной кислоте
пропускают через небольшую колонку, заполненную мед-
ным порошком. а-Счет аликвотных проб полученного рас-
214
Радий
твора производят приблизительно через 5 час. после при-
готовления пробы и повторяют через 24 часа; по скорости
нарастания активности определяют поправочный коэффи-
циент на радон и его дочерние производные. Другой спо-
соб предусматривает прямой a-счет раствора после удале-
ния радона нагреванием и аэрацией; однако данный спо-
соб можно применять только в отсутствие радия-D, радия-Е
Рис. 19. Типичный спектр а-частиц для Ra22e.
и радия-F. Существует также метод, по которому радий
осаждают с сульфатом бария после удаления радона и оп-
ределения p-излучения, испускаемого радием-В и радием-С;
при пользовании этим методом необходимо принимать в рас-
чет поглощение излучения самим осадком [174].
Развитие за последние несколько лет электронной тех-
ники привело к созданию приборов, которые в ряде слу-
чаев исключают процессы химического разделения. Для
непосредственного определения радия в присутствии его
дочерних производных в настоящее время можно применять
одноканальные и многоканальные импульсные анализаторы.
Эти приборы по существу являются высокоскоростными
электронными анализирующими машинами, используемыми
в сочетании с ионизационными камерами для определения
Металлический радий
215
числа испускаемых частиц и их энергий, поскольку энер-
гия испускаемой частицы является характерной величиной
для каждого изотопа. Типичные кривые, полученные при
помощи импульсных анализаторов для радиевого образца,
приведены на рис. 19. Описание обычно применяющихся
типов импульсных анализаторов дано в работе [93].
Применявшиеся ранее методы количественного опреде-
ления радия описаны Фрезениусом и Яндером [63]; эти же
методы описаны в справочнике Гмелина [72].
ИЗОТОПЫ РАДИЯ
Известно тринадцать изотопов радия (табл. 24), причем
наиболее широко применяется изотоп Ra226; некоторое при-
менение в качестве радиоактивных индикаторов находят
также и изотопы Ra223, Ra224 и Ra228. Нерадиоактивные
изотопы радия неизвестны; поиски неактивного изотопа
в витерите (минерал, представляющий собой углекислый
барий) оказались безуспешными [76].
СПЕКТР РАДИЯ
Первые сведения о спектре радия, полученные в результате
наблюдений Демарсе [40—42], были подтверждены более
поздними исследованиями [23, 36, 37, 91, 148, 149, 151,
152, 178, 179, 182, 183]. Спектр радия подобен спектрам
остальных щелочноземельных элементов; вычисленный иони-
зационный потенциал радия равен 5,35 эв [133] или 5,252 эв
[154]. Галогениды радия окрашивают пламя газовой го-
релки в карминово-красный цвет [69, 74, 150].
МЕТАЛЛИЧЕСКИЙ РАДИЙ
Коен [22] высказал предположение, что при электролизе
растворов, содержащих в метиловом спирте радий-барие-
вые соли, на амальгамированном катоде образуется ра-
диево-бариевая амальгама.
Металлический радий впервые получили Кюри и Де-
бьерн [30] электролизом раствора хлорида радия с при-
менением ртутного катода и платино-иридиевого анода.
Получавшуюся амальгаму помещали в железную лодочку
Изотопы радия
Таблица 24
Массовое число Период полураспада Тип распада и энергия излучаемых частиц, Мэв
213 2,7 МИН. а 6,90
219 -10~3 сек. а 8,0
220 0,03 » а 7,43
221 30 » а 6,71
222 38 » а 6,55 (95%); 7
223 (АсХ)‘ 11,68 суток 6,23 (4,4%) а 5,712 (50%); 7
224 (ThX) 3,64 > 5,602 (24%) 5,742 (10,5%) 5,534 (10,3%) 5,429 (2,4%) 5,867 (1%) 5,497 (0,9%) а 5,681 (95,1%); •]
225 14,8 » 5,445 (4,9%) Г 0.32; 7
226а 1622 года6 а 4,777 (94,3%); 7
227 41,2 мин. 4,589 (5,7%) Г 1,31; 7
228 (MsThj) 6,7 года 0,012; 7
229 230 (Короткий) 1 час сч 1 1 ах ах
8 Поперечное сечение захвата тепловых нейтронов для реакции
Ra223 (л, j) Ra224 составляет 125 барн [78], а для реакции Ra226 (л, у)
R а227 -23 барн [9].
6 Период полураспада Ra22® был измерен также калориметрическим
методом на источнике высокой чистоты. Получено значение, равное
1677 ± 9 лет [195].— Прим. ред.
Металлический радий
217
и нагревали в очищенном водороде до 270—700° для уда-
ления ртути; при температуре 700° радий начинал улету-
чиваться и разъедал стенки кварцевой трубки, в которой
находилась лодочка с амальгамой.
Свежеприготовленный металлический радий обладает
металлическим блеском, однако под действием воздуха
металл чернеет, возможно в результате образования нит-
рида (Ra8N2?). Металлический радий разлагает воду с вы-
делением водорода; вычисленное значение теплоты этой
реакции составляет приблизительно 90 ккал/г-атом [38].
Образующаяся при этой реакции гидроокись радия пол-
ностью растворима в воде, однако после ее растворения
остается темный осадок, растворимый в разбавленной со-
ляной кислоте; природа этого осадка окончательно не
установлена.
По одним данным, металл плавится приблизительно при
700° [30], по другим [94], — при 960° и отличается боль-
шей летучестью, чем барий; вероятно, поэтому возгонкой
в вакууме металлический радий можно отделить от до-
черних продуктов и от бария. По имеющимся сообщениям,
металлический радий образуется при разложении его азида
в вакууме при 180—250° [48], однако получающийся при
этом продукт сильно загрязнен [80]. Амальгама радия
может образовываться в какой-то мере при взаимодействии
амальгамы бария [22] или амальгамы натрия [117] с вод-
ными растворами солей радия.
Желтовато-белый сплав радия и серебра образуется,
очевидно, при восстановлении углеродом при высоких
температурах смеси хлорида серебра, сульфата радия и
карбоната кальция [46]; никаких данных о составе этого
сплава не опубликовано.
Потенциал выделения радия из нормальных растворов
его солей, как показывает расчет, должен быть равен
— 1,718 в по отношению к нормальному каломельному
электроду [85]. Опубликованы сообщения [77] о изуче-
нии электролиза растворенных в ацетоне солей бария,
содержащих радий, с целью получения источников радия
в виде тонкого слоя, однако состав получаемых при этом
отложений неизвестен. Атомный парахор радия равен при-
близительно 140 [109].
Имеются весьма скудные данные о физических свойст-
218
Радий
вах металлического радия и эти данные основаны лишь на
нескольких экспериментах, выполненных приблизительно
пятьдесят лет назад и в последующие годы не повторяв-
шихся. Маловероятно, чтобы металлический радий обла-
дал свойствами, весьма близкими к свойствам бария; во
всяком случае, целесообразно провести дальнейшее изу-
чение радия в элементарном состоянии.
АТОМНЫЙ ВЕС РАДИЯ (Ra22®)
Почти все аналитические данные, относящиеся к соедине-
ниям радия, получены в процессе определения атомного
веса; большинство этих работ было проведено вскоре после
открытия данного элемента. Указанные работы сводились
к анализу весомых образцов хлорида или бромида радия.
В 1902 г. М. Кюри [26] получила значение атомного веса
радия, равное 225, а позже [28], работая с более чистым
образцом, получила значение 226,18 (Ag = 107,8). Торп
[170] нашел, что атомный вес радия равен 226,7. Это зна-
чение еще выше значения, полученного Уайтло-Греем и
Рамзаем, и равного 226,26 ([181 ] пересчитано Гонигшми-
дом [89]). При этом никаких поправок на влияние теплоты,
выделяемой радием, не делалось. Гонигшмид для атомного
веса радия определил значение, равное 225,95 [88, 89];
позже он нашел значение, равное 226,05, которое и при-
нято в настоящее время [90].
СОЕДИНЕНИЯ РАДИЯ
Нейтральный атом радия в основном состоянии, имеет
электронную конфигурацию 5s2 5р6 5d10 6s2 бр6 7s2 (Sq);
эта конфигурация аналогична электронным конфигура-
циям других щелочноземельных металлов. Ожидаемое окис-
лительное состояние 2+ было экспериментально под-
тверждено анализом галогенидов, а также данными, по-
лученными в работах по изучению диффузии и подвижно-
сти ионов [64, 83, 84]; никаких признаков существования
других окислительных состояний радия не имеется. Ра-
диус иона Ra+ +, по расчетным данным, составляет 2,45 А
[144]; однако рентгенографическое изучение кристаллов
Соединения радия
219
фторида радия приводит к значению 1,52 А в предположе-
нии, что координационное число равно 6 [1601.
Почти все соли радия окрашены в белый цвет, но под
действием собственного интенсивного излучения все они
[68], так же как и стеклянные сосуды, в которых они на-
ходятся [34], при стоянии изменяют окраску. Все соеди-
нения радия испускают на воздухе голубое свечение; спектр
этого свечения состоит из спектральных полос азота [81,
86, 87] (ср. с полонием, гл. 3).
В печати опубликованы результаты изучения адсорб-
ции микроколичеств радия из раствора на стеклянной по-
верхности [130, 163]; адсорбция в какой-то мере наблю-
дается даже в тех случаях, когда концентрация кислоты
составляет 0,17 н. [167]. Возможно, что такого рода ад-
сорбция является результатом присутствия в растворе
загрязнений, например пыли [163]. При стоянии раствора
в нем также может происходить в какой-то мере седимен-
тация радия [107] (см. также гл. 2).
Химические свойства радия очень похожи на свойства
бария, и поэтому последний, как правило, применяют
в качестве носителя для микроколичеств радия. Столь же
хорошим носителем для радия, вероятно, был бы и строн-
ций, применение которого имело бы то преимущество, что
радий легче отделяется от стронция, чем от бария, напри-
мер при дробном осаждении сульфатов [71 ].
Хотя уже на протяжении многих лет радий имеется в
количествах, измеряемых граммами, тем не менее проведено
очень мало кристаллографических исследований соедине-
ний радия и, как это ни странно, аналитические данные
о составе каких бы то ни было соединений радия, кроме
хлорида, бромида и фторбериллата, отсутствуют. Такая
ограниченность точных сведений по химии радия, вероятно,
связана с существованием предположения, что в химиче-
ском отношении радий аналогичен барию. Однако имеется
еще широкое поле деятельности для дальнейших иссле-
дований.
Галогениды радия
Хлорид. Безводный хлорид радия можно получать
нагреванием сульфата радия в смеси паров соляной кис-
лоты и четыреххлористого углерода при температуре крас-
220
Радий
кого каления. Плотность безводной соли составляет
4,9 г/см3 [181 ]. Дигидрат хлорида радия получают раство-
рением карбоната радия в соляной кислоте, после чего
соли дают выкристаллизоваться; кристаллизационная вода
удаляется при 100°. Обычно эти соли бывают бесцветными,
однако в присутствии бария они при стоянии становятся
желтыми или розовыми [26].
Хлорид радия плавится приблизительно при 900° без
разложения [88, 89, 92, 103—105], причем между 830 и
920°, возможно, происходит фазовое превращение. Хлорид
радия хуже растворяется в воде, чем хлорид бария [24];
в 100 г воды при 20° растворяется 24,5 г RaCl2 [59];
каких бы то ни было признаков образования комп-
лексных ионов в растворе соляной кислоты не отмечено
[56]. Теплота образования хлорида радия по расчетным
данным составляет —230 ккал/моль [99]; более ранние
расчетные данные приведены в работе [39]. При стоянии
во влажной атмосфере из этой соли, по-видимому, выде-
ляется хлор, поскольку имеются сообщения о том, что они
обладают запахом, подобным запаху гипохлорита [25].
Магнитная восприимчивость хлорида радия составляет
+ 1,05-10-6, и, следовательно, можно заключить, что ра-
дий обладает слабыми парамагнитными свойствами; в то
же время барий диамагнитен и загрязнение радиевой соли
барием приведет к искажению результатов при определе-
нии магнитной восприимчивости [32]. В литературе имеется
описание полосатого спектра хлорида радия; по расчетным
данным энергия диссоциации для основного состояния со-
ставляет приблизительно 2,9 эв [108].
Бромид. Безводный бромид радия можно получать
нагреванием хлорида радия в газообразном бромистом во-
дороде при температуре красного каления. Плотность без-
водной соли равна 5,78 г/см3 [181]. Могут существовать
два кристаллогидрата бромида радия: RaBr2-2H2O и
RaBr2-6H2O [139, 140]; в процессе разделения радия и
бария дробной кристаллизацией бромидов может образо-
ваться двойная соль RaBr2-2BaBr2-6H2O [158]. Дигид-
рат кристаллизуется в моноклинной системе с соотноше-
нием осей а.: b : с = 1,4485 : 1 : 1,1749 и 0 = 65О24'; эта
соль изоморфна дигидрату бромида бария [143].
Соединения радия
221
Свежеприготовленный бромид радия образует бесцвет-
ные кристаллы, однако при стоянии соль темнеет, выде-
ляет бром и в конце концов превращается в карбонат, ве-
роятно, с промежуточным образованием гидроокиси [55,
69, 137, 141].
Бромид радия плавится при 728° и, по-видимому, раз-
лагается при более высокой температуре с образованием
стекловидного вещества, которое, по имеющимся сообще-
ниям, не растворяется в соляной кислоте [98]; бромид
радия возгоняется приблизительно при 900° и, следова-
тельно, менее летуч, чем бромид бария, от которого он мо-
жет быть отделен дробной возгонкой [165].
Кольрауш и Хеннинг [102] производили измерения
электропроводности растворов бромида радия; полученные
ими результаты приведены в табл. 25.
Таблица 25
Эквивалентная электропроводность растворов
бромида радия
Концентрация раствора, г-эк в Ra/4a 4б а18 Температурный коэффициент (18°)
0,0000872 124,1 —
0,0003043 122,2 —
0,001356 118,3 0,0225
. 0,008395 111,0 —
0,01713 106,8 —
0,04901 99,5 0,0221
а Атомный вес радия принят равным 225.
6 А 1 -58; Лм -125-126
Ra+ +
Фторид. Эта соль была получена растворением карбо-
ната радия в плавиковой кислоте с последующим выпари-
ванием образующегося раствора досуха. Кристаллическая
структура фторида определена рентгенографическим ме-
тодом (методом порошка); соединение имеет решетку типа
флюорита с периодом а = 6,368 kX\ вычисленная плотность
равна 6,75 г/см3 [160].
222
Радий
Другие соли радия
Азид радия представляет собой белое кристаллическое
вещество, получаемое растворением карбоната радия в вод-
ном растворе азотистоводородной кислоты с последующим
выпариванием образующегося раствора. Можно предпо-
ложить, что образующееся соединение имеет состав Ra(N3)2
и подобно азиду бария при нагревании разлагается с вы-
делением металла [48]. Возможно, что это соединение раз-
лагается также под действием излучения, испускаемого
радием [80].
Карбонат радия получают осаждением из нейтраль-
ных растворов солей радия при действии на них карбоната
аммония. Карбонат радия лучше растворяется в концен-
трированных растворах карбоната аммония, чем анало-
гичная соль бария [127].
Хромат радия в отличие от хромата бария не превра-
щается в карбонат при нагревании с разбавленным рас-
твором карбоната натрия [127].
Фторбериллат радия RaBeF4 образуется в виде
белого осадка при добавлении раствора фторбериллата
калия к горячему раствору соли радия в 0,2 н. соляной
кислоте. Он, по-видимому, менее растворим, чем соответст-
вующая соль бария [8]. Это одна из немногих солей радия,
анализ которой производили с целью определения состава.
Йодат радия выпадает в осадок при добавлении йодата
калия к водным растворам хлорида радия; произведены
определения растворимости йодата радия в воде при раз-
личной температуре [136]; полученные результаты при-
ведены в табл. 26.
Изучена растворимость йодата радия в концентриро-
ванных растворах нитрата калия, нитрата кальция и йодата
калия; вычислены коэффициенты активности при различ-
ных значениях ионной силы [136]. Выполнен ряд работ
по изучению кристаллических форм этого соединения оп-
Соединения радия
223
тическими методами; при этом использовали микроколи-
чества данного соединения [431.
Таблица 26
Растворимость йодата радия в воде
Температура, °C Растворимость
мг/л молъ/л
0 176 0,000305
25 437 0,000760
78 1244 0,00216
100 1705 0,00296
Нитрат. Растворимость этой соли при 20® составляет
13,9 г на 100 г воды [59].
Окись. Вычислено, что теплота образования окиси ра-
дия равна 130 ккал/моль [II].
Платиноцианид радия, по имеющимся сведе-
ниям, представляет собой соль зеленого цвета, которая
в растворе быстро становится дихроичной.
Селена т, теллурат и другие соедине-
ния.
В связи с применением их в качестве лечебных средств были
запатентованы способы получения селената, селенита, тел-
лурата и теллурита радия и бария, а также селената ра-
дия и натрия методом осаждения [120].
Сульфат радия менее растворим, чем сульфат бария;
наиболее надежное значение растворимости этого соедине-
ния в воде равно 2,1 -10-4 г на 100 г воды при 20° (произ-
ведение растворимости 4,25-10-11) [129]. Ранние опыты
[14, 60, 113, 128] давали значительно более низкие зна-
чения растворимости в воде; это объясняется тем, что боль-
шая часть присутствующего радия адсорбировалась филь-
трующей средой, использовавшейся при отделении твер-
224
Радий
дого сульфата радия от раствора. Данное соединение обычно
приготовляют осаждением. Изучалась также адсорбция
радия на сульфате бария в 1 н. растворе соляной кислоты;
данное явление имеет важное значение, поскольку реагенты,
применяющиеся при экстрагировании радия в заводском
масштабе, перед использованием обычно очищают от суль-
фата обработкой хлоридом бария, и возможно, что неболь-
шие количества осажденного сульфата бария в дальнейшем
технологическом процессе извлечения радия могут последо-
вать за ним. Полученные результаты свидетельствуют о том,
что в процессе адсорбции соблюдаются правила Крокера
и Фрейндлиха [73].
Сульфид радия можно получить восстановлением
сульфата гидридом кальция [51], карбидом кальция или
смесью гидрида и карбида кальция [53]. Чтобы вызвать
реакцию, применяют термитный запал. Образующийся
продукт растворяется в соляной или в уксусной кислоте.
Комплексные соли. Были получены нераство-
римые комплексные соединения радия с алкилендиамин-
тетрауксусными кислотами (С2—Св) [3] и комплексы с ли-
монной, яблочной и винной кислотами [159]. Константа
диссоциации комплексного иона радия с лимонной кисло-
той равна 9,97-10 “3 при 25°; это значение было определено
методом ионного обмена [166].
В работе [196] приведены данные по исследованию комплексо-
образования Ra и Ва в растворах трилона В в интервале pH 2—8
методом ионного обмена с применением изотопов В а140 и Ra2?e в ка-
честве меченых атомов. Установлено, что Ra в растворах трилона В
при pH 5,5 — 6,9 образует анионные комплексы состава (RaA)2”.
Образование трилонатов Ва и Ra в заметных количествах начинается
при pH ~ 4, а при pH > 8 свободные ионы Ва и Ra в заметных ко-
личествах в растворе не обнаруживаются.
Из экспериментальных данных рассчитаны значения констант
нестойкости трилонатов Ва и Ra. Для трилоната бария получено
хорошее совпадение значения констант нестойкости (рК = 7,69) с
данными Шварценбаха (рК = 7,76, потенциометрический метод), а
также Шемид и Райлери (рК = 7,90, полярографический метод).
Установлена несколько большая прочность комплексного соеди-
нения Ва (рКВа = 7,69) по сравнению с соответствующим ком-
плексным соединением Ra (pKRa — 7,12).
Соединения радия
•225
При исследовании процесса комплексообразования Ra и Ва в
растворах нитрилтриуксусной кислоты в интервале pH 4—8 мето-
дом ионного обмена с применением в микроконцентрациях изотопов
Ва140 и Raaae установлено [197], что в растворе нитрилтриуксусной
кислоты при pH 6—8 образуются анионные комплексы состава
fRaX]“H [Ва(Х)|~, где X — анион нитрилтриуксусной кислоты.
Образование комплексных ионов начинается в заметных коли-
чествах (> 5%) при концентрациях ионов X3" ~ 4« 10“8 моль/л,
а при концентрации 2« 10“5моль/л, концентрация свободных ионов
Ва““ в растворе < 5% общей концентрации этого элемента.
Соответствующие пределы концентраций лиганда для радия со-
ставляют 10“7—5-10“5 Af. Из экспериментальных данных рассчи-
таны значения констант нестойкости радиевого и бариевого комплек-
сов (p/CRa=5,75; рКВа = 6,17).
Различные предсказанные значения теплот образова-
ния и растворимости см. в работах [38, 39 и 99]. Некоторые
данные по вопросу соосаждения микроколичеств радия
содержатся в обзоре Боннера и Кана [61; весьма полный
обзор работ, выполненных до 1928 г., опубликован в спра-
вочнике Гмелина [72].
15 к. Б'Я'мад
ЛИТЕРАТУРА
1. Arbi tri о F., Riv. fis. mat. sci. nat., 12, 178 (1938).
2. A s i m о v L, J. chem. Educ., 30, 616 (1953).
3. Auer ges A. G., белы. пат. 448736 (1943).
4. Baumgarten С. E., Barker H. H., Industr. engn. Chem.,
15, 597 (1923).
5. Baxter G. P., H о n i g s c h m i d O., Le Beau P., J. Amer.
. chem. Soc., 60, 737 (1938).
^.Bonner N. A., Kahn M., Nucleonics, 8, № 2, 46 (1951).
7, Bredt О. P. С., пат. США H54230—1154231 (1915).
^JBxetscher E., Cook G* B., Martin G. R., Wilkin
u son D. H., Pros. Roy. Soc., A196, 436 (1949).
9. Butler J. P., Adam J. S., Phys. Rev., 91, 1219 (1953).
10. Becker A., Z. Phys., 83, 701 (1933).
11. Brewer L., Chem. Rev., 52, 1 (1953).
12. Ch lo pin V. G., Bull. Soc. chim. Fr., 33, 1547 (1923).
13. C h 1 о p i n V. G., Z. anorg. Chem., 143, 97 (1925).
14. C h 1 о p i n V. G„ Naturwiss., 17, 959 (1929).
15. X лопин В. Г., К л окман В. Р., Изв. АН СССР, Отд. хим.
наук, 254 (1949).
16. Хлоп ин В. Г., К л окман В. Р., Мурин А. В., Нефе-
дов В. Д., Изв. АН СССР, Отд. хим. наук, 127 (1950).
17. Хлоп ин В. Г., К л окман В. Р., Пакельная Е. Г., Изв.
АН СССР, Отд. хим. наук, 250 (1953) (английский перевод
см. Brit. Rep. TIB/T4550, 1956).
18. Хлоп ин В. Г., Меркулова М. С., ЖФХ, 13, 1282 (1939).
19. Хл опин В. Г., Меркулова М. С., ДАН СССР, 71, 689
(1950).
20. Chlopin V. G., Nikitin В. A., Z. phys. Chem., А145, 137
(1929).
21. Chlopin V. G., Р о 1 е s i t s k i i A., Z. anorg. Chem., 172, 310
(1928).
22. Coehn A., Ber., 37, 811 (1904).
23. Crookes W., Proc. Roy. Soc., 72, 295, 413 (1903).
Литература
227
24. Curie M., C. R. Acad. Sci., Paris, 129, 760 (1899).
25. Curie M., C. R. Acad. Sci., Paris, 131, 382 (1900).
26. Curie M., C. R. Acad. Sci., Paris, 135, 161 (1902).
27. Curie M., Recherches sur les Substances Radioactives Th6se,
Faculte des Sciences de Paris Gauthier — Villars, Paris (1904).
28. Curie M., C. R. Acad. Sci., Paris, 145, 422 (1907).
29. Curie M., Le Radium, 7, 65 (1910).
30. Curie M., Debi erne A., C. R. Acad. Sci., Paris, 151, 523
(1910).
31. Curie P., J. Chim. phys., 1, 409, (1903).
32. C u r i e P., Cheenveau C., Bull. Soc. Phys., 1 (1903).
33. Curie P., Curie M., C. R. Acad. Sci., Paris, 127, 175 (1898).
34. C u r i e P., Curie M., C. R. Acad. Sci., Paris, 129, 823 (1899).
35. Curie P., Curie M., В emo nt G., C. R. Acad. Sci., Paris,
127, 1215 (1898).
36. De Broglie M., C. R. Acad. Sci., Paris, 168, 854 (1919).
37. De Broglie M., C. R. Acad. Sci., Paris, 169, 134 (1919).
38. De Fore rand R., Ann. Chim. Phys., [8], 24, 256 (1911).
39. De Fore rand R., C. R. Acad. Sci., Paris, 152, 66 (1911).
40. Demar?ay E., C. R. Acad. Sci., Paris, 127, 1218 (1898).
41. Dem ar? а у E., C. R. Acad. Sci., Paris, 129, 716 (1899).
42. Dem ar? а у E., C. R. Acad. Sci., Paris, 131, 258 (1900).
43. Deniges G., C. R. Acad. Sci., Paris, 171,633 (1920).
44. D i a m о n d R. M., Street K., S e a b о r g G. T., J. Amer,
chem. Soc., 76, 1461 (1954).
45. Doer n er H. A., Hoskins W. M., J. Amer. chem. Soc., 47,
662 (1925).
46. De Mare F., Jacobs C., Bull. Acad. roy. Sci. Belg. Classe Sci.,
53 (1912).
47. D i a m о n d R. M., J. Amer. chem. Soc., 77, 2978 (1955).
48. Ebler E., Ber., 43, 2613 (1910).
49. Ebler E., Z. angew. Chem., 26, 658 (1913).
50. Ebler E., Bender W., Ber., 46, 1571 (1913).
51. E b 1 e r E., Bender W., Z. anorg. Chem., 83, 149 (1913).
52. Ebler E., В e n d e r W., Z. anorg. Chem., 84, 77 (1913).
53. E b 1 e r E., Bender W., Z. anorg. Chem., 88, 255 (1914).
54. Ebler E., Van Rhyn A. J., Ber., 54, 2896 (1922).
55. Eger ton A. C. G., Nature, Lond., 76, 174 (1907).
56. Eliseev A. G., Ann. Inst. Anal. Phys. Chem., 3, 443 (1926).
57. E n g s t г о m S. R. AM Metallges A. G., герм. пат. 658254
(1938).
15*
228
Литература
58. Е р h г a i m F., Inorganic Chemistry (P. Thorne and E. Roberts,
Ed.), Oliver and Boyd, Edinburgh, 1943.
59. ErbacherO., Ber., 63, 141 (1930).
60. Erbach er O., Nikitin B. A., Z. Phys. Chem., A158, 216
(1932).
61. Ершова 3., Никольский В., Розенблюм А., Редкие
металлы, 7, № 1, 37 (1938).
62. Foyn Е., Gleditsch Е., Hanneborg S., Arch. Math. Na-
turvidenskab., 41, № 12 (1938).
63. Fresenius R.Jander G., Handbuch der analytischen Che-
mie II. 3, vol. Ha, page 411, J. Springer, Berlin, 1940.
64. F r e u n d 1 i c h H, von Elissafoff G., Phys. Z., 14, 1052
(1913).
65. Fulton R. G., J. Ass. Offic. Agr. Chem., 13, 497 (1930).
66. Fineman P., Weissbourd В. B., Anderson H. H.,
Sedlet J, Ames D. P., Kohma n T. P., Nat. Nucl. Energy
Series IV-14B, p. 1206, McGraw Hill, New York, 1949.
67. G e r m a n n F. E. E., Science, 59, 340 (1924).
68. Giesel F., Ann. Phys, chim., 69, 91 (1899).
69. Giesel F., Ber., 35, 3608 (1902).
70. Goldschmidt B., J. Chim. phys., 35, 407 (1938).
71. G о 1 d s c h m i d t B., Ann. Chim., 13, 88 (1940).
72. Gmelin, Handbuch der anorganischen Chemie, System №31,
8th Edn. 1928, Verlag Chemie, G.m.b.H., Berlin.
73. G e r m a ri n F. E. E., J. Amer. chem. Soc., 43, 1615 (1921).
74. Giesel F., Phys. Z., 3, 578 (1902).
75. Gregory J. N., M о о г b a t h S., Trans. Faraday Soc., 47, 1064
(1951).
76. Hahn O., Donat K, Z. Phys. Chem., A139, 143 (1928).
77. Haissinsky M., J. Chim. phys., 34, 321 (1937).
78. Harbottle G., J. inorg. nuclear Chem., 1, 253 (1955).
79. H e n d e r s о n L. M., К г a c e k F. C., J. Amer. chem. Soc., 49,
739 (1927).
80. H e r c h f i n k e 1 H., Le Radium, 8, 299 (1911).
81. Huggins W., Huggins Lady W., Proc. Roy. Soc., A72,
196 (1903).
82. Hess V. F., Phys. Z., 14, 1135 (1913).
83. Hevesy G., Phys. Z., 14,49 (1913).
84. Hevesy G., Phil. Mag., [6], 25, 390 (1913).
85 Heyrovsky J., Berezicky S., Coll. Trav. chim. Tchecorl.,
L 19 (1929).
Литература
229
86. Himstedt F., М е у е г G., Phys. Z., 6, 688 (1905).
87. Himstedt F., Meyer G., Phys. Z., 7, 762 (1906).
88. H о n i g s c h m i d O., Monatsch., 33, 253 (1912).
89. H о n i g s c h m i d O., Monatsch., 34, 283 (1913).
90. H о n i g s c h m i d O., Sachtleben R., Z. anorg. Chem., 221,
65 (1934).
91. Hulubei H„ C. R. Acad. Sci., Paris., 203, 399, 542, 665 (1936).
92. H о n i g s c h m i d O., Sitzungsber. Akad. Wiss., Wien, Abt. Ila,
120, 1661 (1911).
93. H i g i n b о t h a m W. A., Nucleonics, 14, № 4, 61 (1956).
94. International Critical Tables, Vol. I, p. 104, Nat. Res. Council
Mc-Graw-Hill, New York, 1928.
95. J a n i t z к у A., Kolloid Z., 89, 316 (1939).
96. J e n к i n s W. A., S e a b о r g G. T., Phys. Rev., 85, 758 (1952).
97. Jennings W. A., Russ S., Radon, Its Technique and Use,
John Murray, London, 1948.
98. Joly C. J., Nature Lond., 70, 31 (1904).
99. Ka пу стински й А. Ф., Изв. АН СССР, Отд. хим. наук,
568 (1948).
100. Kendall J., Science, 67, 163 (1928).
101. Kirby H. W., Analyt. Chem., 25, 1238 (1953).
102. Kohlrausch F., Henning F., Ann. Phys. Lpz., [4], 20, 96,
(1906).
103. К о 1 о w r a t L., Le Radium, 4, 317 (1907).
104. К о 1 о w r a t L., Le Radium, 6, 321 (1909).
105. К о 1 о w r a t L., Le Radium, 7, 266 (1910).
106. Kendall J., J e 11 e E. R., W e s t W., J. Amer. chem. Soc., 48,
3114 (1926). ‘
107. Lachs H., Wertenstein M., Phys. Z., 23, 318 (1922).
108. Lagerqvist A., Arkiv Fysik., 6, 141 (1952).
109. Lakhani J. V., Daroga R. P., J. Indian chem. Soc., 15, 604
(1938).
110. Lengyel B., von, Ber., 33, 1237 (1900).
111. Lind S. C., J. Industr. engng Chem., 7, 1024 (1915).
112. Lind S. C., J. Industr. engng. Chem., 12, 469 (1920).
113. Lind S. C., Underwood J. E., Whittemore C. F., J.
Amer. chem. Soc., 40, 465 (1918).
114. L u n d b о r g H., Oil Gas J., 50, № 49, 165 (1952).
115. L о n g c h a m b о n L., франи. пат. 679086 (1928).
116. Lindner R., Naturwiss., 33, 119 (1946).
117. Marckwald W., Ber., 37, 88 (1904).
230
Литература
118. Marques В. Е, J. Chim phys., 33, 97 (1936).
119. Marques В., E., J. Chim phys., 33, 306 (1936)
120. Merck E., Eicholz W., герм. пат. DR-P256666 (1913);
австр. пат. 63166 (1913).
121. Меркулова М. С., Труды гос. радиевого инет. им. В. Г. Хло-
пина АН СССР, 3, 141 (1937); С А., 31, 4590 (1937).
122. Meyer Н., Brit. J. Radiol., 15,85 (1942).
123. М u m b г a u е г R., Z. phys. Chem., А156, 113 (1931).
124. Marques В. E., C. R. Acad. Sci., Paris., 196, 1309 (1933).
125. Neidrach L. W., Mitchell A. M., Rodden C. J., Nat.
Nucl. Energy Series VIII-I, p. 368, McGraw Hill, New York, 1950.
126. N i e r m a n J L , J. phys. Chem , 24, 192 (1920).
127. Никитин Б. А., Труды гос. радиевого инет. им. В. Г Хло-
пина АН СССР, 3, 228 (1937); С. А., 31, 4590 (1937).
128. Nikitin В. A., Erba с he г О., Z. phys. Chem., А158, 231
(1932).
129. Nikitin В. А., Т о I m а с h е v Р., Z. phys. Chem., А167, 260
(1933).
130. Никитин Б. А., Вдовенко В. М., Труды гос. радиевого
инет. им. В. Г. Хлопина АН СССР, 3, 256 (1937); С. А., 31, 4589
(1937).
131. Nikitin В. А., С. R. Acad. Sci. U.R.S.S., 1, 19 (1934).
132. Ра week Н. Z., Elektrochem, 14,619 (1908).
133. Р i с с а г d i G., Atti accad. Lincei, [6], 6, 428 (1927).
134. P i e t e n p о 1 W. В., J. chem. Phys., 10, 211 (1942).
135. P о 1 e s i t s k i i A., Karataeva A., Acta Physicochim.
U.R.S.S., 8, 251 (1938).
136. Polesitskii A., Tolmachev P., C. R. Acad. Sci. U.R.S.S.,
3 (12), 319 (1936).
137. Porter A. W., Nature, Lond., 76, 151 (1907).
138. Power W. H., Kirby H. W., Me Luggage W. C., Nel-
son G. D., Payne J. H., Amer. Rep. MLM 833 (1953).
139. Precht J., Ber., 8, 436 (1906).
140. Precht J., Phys. Z., 7, 836 (1906).
141. Ramsay W., Monatsch., 29, 1013 (1908),
142. Reid A. F., Industr. engng. Chem., 40, 76 (1948).
143. R i n n e F., Centr. Min., 134 (1903).
144. Rinne F., Z. phys. Chem., 100, 411 (1922).
145 R о 11 i e r M. A., Gazzetta, 75, 97 (1945).
146. Rosenqvist I. T., Norsk, geol. Tdissk., 28, 21 (1949).
Литература
231
147. Rosenqvist I. T., Univ. Bergen Arbok, Naturvit. Rekke № 4
(1952).
148. Runge C., Ann. Phys. Lpz., [4], 2, 742 (1900).
149. Runge C., Phil. Mag., [6]. 6, 698 (1903).
150. Runge C., Precht J., Ann. Phys. Lpz., [4], 10, 655 (1903).
151. Runge C.. Precht J., Ann. Phys. Lpz., [4], 14, 418 (1904).
152. Runge C., Precht J., Phil. Mag., [6], 5, 476 (1903).
153. Radhakrishna P., J. Chim. phys., 51, 354 (1954).
154. Russell H. N., Phys. Rev., 46, 989 (1934).
155. Salutsky M. L., Stites J. G., Industr. engng. Chem., 47,
2162 (1955).
156 Salutsky M. L., Stites J. G., Martin A. W., Analyt.
Chem., 25, 1677, 1938 (1953).
157. Sato T. R., Norris W. P., Strain H. H., Analyt. Chem.,
26, 267 (1954).
158. Scholl С. E., J. Amer. chem. Soc., 42, 889 (1920).
159. Schubert J., J. Amer. chem. Soc., 76, 3442 (1954).
160. Schulze G. E. R., Z. phys. Chem., B32, 430 (1936).
161. S i e v e к i n g H., Naturwiss., 2, 973 (1914).
162. Sokolov V. A., Z. Phys., 54, 385 (1929).
163. Старик И. E., Гуревич A. M., Труды гос. радиевого инет,
им. В. Г. Хлопина АН СССР, 3, 241 (1937); С. А. 31, 4588 (1937).
164. Stern Н., герм. пат. 280694 (1914).
165. Stock A., Heynemann Н„ Вег., 42, 4088 (1909).
166. Schubert J., Russel Е. R., Myers L. S., Amer Rep.
AECU-542 (1949).
167. Старик И. E.,Гуревич A. M., ДАН СССР, № 12.331 (1931).
168. Sa vic P., Cvjeticanin D. N., Bull. Inst. NucL Sci. «Boris
Kidrich», 4, 21 (1954).
169. Thornton D. P., Petroleum Processing, 5, 941 (1950).
170. T h о г p e T. E., Proc. Roy. Soc , A80, 298 (1908).
171. Толмачев П. И., Труды гос. радиевого инет. им. В. Г. Хло-
пина АН СССР. 3, 239 (1937) ; С. А., 31, 4590 (1937).
172. Tompkins Е. R., J. Amer. chem. Soc., 70, 3520 (1948).
173. Tompkins E. R., пат. США 2554649 (1951).
174. T о m p k i n s P. C., N о г r i s W. P., Wish L., F i n k 1 e R. D.,
Amer. Rep. MDDC-699 (1946).
175. Tyler P. M., Inf Circular № 6312, page 5, U. S. Bureau of
Mines (1930).
176. Vermeulen T., Heister N. K., Industr. engng. Chem., 44(
636 (1952).
232
Литература
177. Walter Z. Т., S с h 1 u n d t H., J. Amer. chem. Soc., 50, 3266
(1928).
178. Watts W. M.. Phil. Mag., [6], 6, 64 (1903).
179. Watts W. M„ Phil. Mag., [6], 18,411 (1909).
180. Weil K., Peters К., пат. США. 1927726 (1933).
181. Whytlaw-Gray R., Ramsay W., Proc. Roy. Soc., A86,
270 (1912).
182. Wilde H., Mem. Manchester Phil. Soc., 52, 1 (1907).
183. Wilde H., Phil. Mag., [6], 16, 824 (1908).
184. Woods R. C., Iron Age, 138, № 3, 49 (1936).
185. W e d e к i n d E., Chem. Ztg., 28, 269 (1904).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
186. Зив Д. М., Синицына Г. С. и др., Атомная энергия, 4,
469—470, 1958; Kernenegrie, 2, 295—296 (1959).
187. Клокма н В. Р., Г а р м а ш е в Ю. М., Радиохимия, 1, 26—31
(1959).
188. Клокман В. Р., Радиохимия, 1, 32—35 (1959).
189. Клокман В. Р., Мельникова А. А., Радиохимия, 1, 514,
1959.
190. Клокман В. Р., Мельникова А. А., Радиохимия, 1, 241
(1959).
191. Клокман В. Р., Мельникова А. А., Урюпина Н. И.,
Изв. АН СССР, Отд. хим. наук, 953—957 (1954).
192. Гребенщикова В. И., Труды гос. радиевого инет. им.
В. Г. Хлопина АН СССР, т. V, 134-147 (1957).
193. Power W. Н., Kirby W. W., McC luggage W. С., Апа-
lyt. Chem., 31, 1077—1079 (1959).
194. Vebersik V., Но го v а К., Z. Analyt. Chem., 162, 401—407
(1958).
195. Горшков Г. В., Гритченко 3. Г., Шиманская Н. С.,
Ж- эксп. и теорет. физ., 34, 756—757 (1958).
196. Н и к о л ь с к и й Б. П., Трофимов А. М., Высоко ост-
ро века я Н. Б., Радиохимия, 1, 147—154 (1959).
197. Никольский Б. П., Трофимов А. М., Высокоост-
ровская Н. Б., Радиохимия. 1, 155—161 (1959).
12
АКТИНИЙ
Открытие и распространенность в природе. Отделение от лантана, це-
рия, тория и радия. Определение Ас227‘-Спектр. Изотопы. Металли-
ческий актиний. Соединения актиния.
ОТКРЫТИЕ АКТИНИЯ И РАСПРОСТРАНЕННОСТЬ ЕГО
В ПРИРОДЕ
Первые работы супругов Кюри, приведшие к открытию
полония и радия, вызвали интерес к другим радиоактив-
ным веществам, содержащимся в природных урановых ру-
дах. Вскоре после сообщения об открытии радия Дебьерн
[16, 17] опубликовал данные о том, что редкоземельная
фракция отходов от переработки урановой смоляной руды
обладает слабой радиоактивностью. Наличие этой радио-
активности было объяснено присутствием нового элемента,
названного Дебьерном актинием; полагали, что новый
элемент по своим химическим свойствам аналогичен то-
рию, поскольку было установлено, что эти два элемента
трудно разделить. Вскоре после этого Гизель [27—30],
работавший независимо, из остатка урановой смоляной
руды выделил радиоактивное вещество, которое он вначале
принял за актиний [30]; поскольку этот препарат выде-
лял радиоактивную эманацию (явление, которое не было
отмечено Дебьерном), Гизель впоследствии назвал получен-
ный им продукт «эманием» [31, 32]. В статьях Гизеля содер-
жится более подробное описание этого сильнорадиоактив-
ного элемента, чем в статье Дебьерна, причем никаких на-
блюдений относительно его химического сходства с торием
отмечено не было. В процессе дальнейшего изучения пре-
паратов эмания и актиния было установлено, что оба этих
препарата выделяют крайне короткоживущую эманацию,
заметно отличающуюся от эманации радия [181; идентич-
ность этих двух препаратов была доказана точным измере-
нием периода полураспада выделяемых ими эманаций
[10, 19 и др. ].
Большинство свойств, приписываемых Дебьерном ак-
234
Актиний
тинию, было, по-видимому, обусловлено присутствием
иония (Th230) в полученном им препарате [5—7]. Загряз-
нение препаратов актиния ионием послужило, по-види-
мому, причиной того, что актиний считали материнским
элементом радия (Ra22e) [4].
Дальнейшая работа по изучению актиния показала,
что его эманация (актинон) является дочерним производным
актиния-Х (Ra223), и позволила построить схему превра-
щения Ас227 (см. приложение 1). Более ранние работы по
изучению актиния весьма полно изложены в справочнике
Гмелина [35], а более поздний обзор [38] охватывает боль-
шинство работ, выполненных до 1954 г.
Актиний значительно менее распространен в природе,
чем радий и, подобно полонию, лишь небольшие количества
его доступны для химического исследования, что делает
практически невозможным прямое определение его хими-
ческих свойств. До настоящего времени весомые количе-
ства актиния не были получены в чистом виде из природных
источников, что обусловлено частично весьма низкой рас-
пространенностью актиния, частично сходством его хими-
ческих свойств со свойствами редкоземельных элементов,
с которыми он находится в урановой смоляной руде. Ра-
бота по изучению химических свойств актиния сводилась
поэтому к опытам по соосаждению, проведение которых
связано с исключительными трудностями ввиду того, что
Ас227 обладает очень мягким ^-излучением, которое почти
нельзя обнаружить. Испускаемые актинием р-частицы не
могли быть обнаружены до 1935 г. [51 ], и в течение многих
лет полагали, что радиоактивный распад этого изотопа.
(Ас227) не сопровождается излучением (см., например,
[39]). Вследствие этого за актинием в опытах с микроко-
личествами этого элемента можно было следить лишь по
возрастающей активности дочерних продуктов или путем
измерения выделяющегося актинона.
Вопрос о происхождении актиния в урановой смоляной
;>уде обсуждался в течение многих лет и был окончательно
решен в 1918 г. после открытия долгоживущего (период
полураспада 3200 лет) материнского вещества — Ра231 Ха-
ном и Мейтнер [43, 44] и одновременно Содди и Крен-
стоном [87, 88]. Актиний (Ас2?7) является одним из чле-
нов цепи распада долгоживущего изотопа урана U235, со-
Отделение актиния от других элементов
235
держание которого в природном уране составляет лишь
1 ч. на 140 ч. В равновесном состоянии 1 tn урановой смоля-
ной руды содержит только 0,15 мг актиния, тогда как со-
держание радия достигает приблизительно 400 мг. Другой
природный изотоп актиния — мезоторий-2 (MsTh2, Ас228) —
является членом радиоактивного ряда тория, причем 1 tn
тория содержит 5-Ю”8 г данного изотопа. Чистый протак-
тиний (Ра231) был бы более ценным источником актиния,
нежели остаток от переработки урановой смоляной руды,
однако в настоящее время не имеется достаточных количеств
этого элемента.
Недавно в результате нейтронного облучения Ra22e были
получены миллиграммовые количества чистого изотопа
Ас227 [37, 77, стр. 1393] по схеме
88Ra^e(«. i)-»88RaM’X89Ac«’,
причем поперечное сечение захвата нейтронов радием-226
имеет порядок 23 барн. Значительная часть работ по хи-
мии актиния в твердом состоянии в настоящее время вы-
полнена с применением чистого актиния, полученного вы-
шеуказанным способом, однако новых данных относительно
химических свойств этого элемента в растворенном состоя-
нии получено мало. До настоящего времени не опублико-
вано никаких результатов, например, о растворимости
труднорастворимых солей актиния (например, фторида) и
остается широкое поле деятельности по дальнейшему изу-
чению данного элемента.
ОТДЕЛЕНИЕ АКТИНИЯ ОТ ДРУГИХ ЭЛЕМЕНТОВ
Отделение от лантана. Актиний по химическим свой-
ствам очень близок лантану, и поэтому последний является
превосходным носителем для микроколичеств актиния.
Однако благодаря химическому сходству этих двух эле-
ментов получение актиния, свободного от носителя, путем
выделения из осадков лантана, содержащих актиний, чрез-
вычайно сложно и его не удалось осуществить при исполь-
зовании материалов, извлеченных из урановых руд, кото-
рые содержат сравнительно большие количества редкозе-
мельных элементов. Классические методы выделения ак-
236
Актиний
тиния из остатков урановых руд описаны достаточно под-
робно в справочнике Гмелина [35, стр. 11 и след. ], а также
Вельсбахом [93] и Пламом [781; в настоящее время эти
способы находят незначительное применение, поскольку
актиний можно более просто получить нейтронным облу-
чением радия.
Делались попытки отделения актиния от редкоземель-
ных элементов путем дробной кристаллизации двойных
нитратов актиния с нитратами магния или марганца —
при этом актиний концентрировался в средней фракции
между неодимом и самарием [15, 74 стр. 1551, а также
путем дробной кристаллизации их двойных солей с нитра-
том аммония из растворов в азотной кислоте — при этом
актиний концентрировался в наименее растворимой фрак-
ции [13, 14, стр. 138, 15, 961.
Другие методы включают обогащение лантана, содер-
жащего актиний, дробным осаждением смешанных оксала-
тов из разбавленного азотнокислого раствора; при этом
актиний накапливается в маточном растворе [74, стр. 155].
Дробное осаждение гидроокисей свободным от карбоната
аммиаком также приводит к некоторому обогащению, по-
скольку гидроокись актиния более растворима, чем гидро-
окись лантана [1, 58, 74, стр. 155, 75, 96].
Адсорбцию актиния на осадках сульфата бария изу-
чал Имре [551, который установил, что 99,5% присутст-
вующего актиния (MsTh2) адсорбируется на осадке из
растворов в 0,01 н. соляной кислоте; скорость адсорбции
уменьшается при добавлении солей бария или соляной
кислоты и увеличивается при добавлении ионов сульфата.
Лантан адсорбируется в меньшей степени, чем актиний
[1, 2], и поэтому такой метод можно исйользовать для обо-
гащения препарата актинием.
Ни один из описанных выше методов не приводит к пол-
ному отделению актиния от лантана, хотя при этом до-
стигается известное обогащение препарата актинием. Ме-
тоды дробной кристаллизации или дробного осаждения
оказываются, однако, пригодными для выделения актиния
вместе с лантаном из смеси с другими редкоземельными
элементами.
Методы ионного обмена и электрофореза на бумаге при-
меняли в последнее время при выделении как миллиграм-
Отделение актиния от других элементов
237
мовых количеств, так и микроколичеств химически чистого
актиния. Этот элемент можно отделить от лантана погло-
щением на смоле амберлит IR-100 (в аммиачной форме)
с последующим элюированием раствором цитрата аммония
при pH 5,5. Сначала элюируется лантан, затем актиний
[94, 95]. Для отделения актиния от лантана применяли
также катионообменную смолу амберлит IR-1 (20—65 меш);
в этом случае лантан элюировали 0,25 М раствором цит-
рата аммония при pH 3,09, а актиний — при pH 3,76 [70,
стр. 1385]. Обзор ионообменных методов, применяющихся
для отделения актиния от редкоземельных элементов, дан
в работе Хайда [52]. Опубликованы сообщения о том, что
достигнуто хорошее отделение актиния от лантана при при-
менении непрерывного электрофореза на бумаге с исполь-
зованием растворов в 1%-ой лимонной кислоте при pH
7—8 [59, 67]; хроматография на бумаге растворов в вод-
ном бутиловом спирте, ацетилацетоне и уксусной кислоте
[59] также приводит к хорошему разделению.
В работе [100] показано, что очистка La(NO3)3 от сопутствую-
щих микроколичеств Ас и его продуктов распада, многие из которых
активны, достигается при экстрагировании микропримесей 0,25 М
раствором теноилтрифторацетона в бензоле или толуоле. Экстраги-
рование проводят в две стадии: при pH 1 для выделения Th и при
pH 5 для выделения Ас и его дочерних продуктов.
Радий удаляют из водной фазы соосаждением с BaSO4.
Данным способом удается уменьшить a-активность La (NO3)3
от > 50 до ~ 5 распад I чао мг.
В работе 11011 изучено поведение актиния и лантанидов на
анионите дауэкс-1 в растворах LiNO.i с целью отделения микроко-
личеств актиния от лантана. Нитраты Ас-228 и лантана растворяли
в концентрированных растворах нитрата лития, вводили в колонку
и элюировали более разбавленным раствором нитрата лития. При
этом 3 мл 8,5 М раствора нитрата лития, содержащего следы ак-
тиния-228 и 30 мг LiNOa, медленно вливали в колонку (30 см X
х 0,25 см*)\ смолу в нитратной форме с размером зерен 50—100
меш предварительно промывали 8,5 М раствором нитрата лития;
элюирование проводили 4,4 М раствором нитрата лития со скоро-
стью 0,4 см/мин; при этом достигалось удовлетворительное отделение
актиния от лантана. Коэффициент распределения актиния и лан-
тана между смолой и раствором увеличивался с ростом концентра-
ции раствора нитрата лития. ,
По способности сорбироваться на анионите актиний распола-
гается между самарием и неодимом в ряду
Yb < Tb < Eu < Sm < Pm< Nd < Рг < Се < La,
что, по мнению автора, является неожиданным.
238
Актиний
В результате сопоставления ряда данных высказано мнение, что
порядок, в котором располагаются актиниды по их способности
сорбироваться из нитратных растворов, противоположен порядку,
найденному для лантанидов.
Отделение от церия. Актиний, не содержащий носителя,
можно получить при использовании трехвалентного це-
рия вместо лантана в качестве носителя для актиния [77,
стр. 1391 ]. Отделение от церия основано на его окислении
до четырехвалентного состояния и осаждении подходящей
нерастворимой соли церия (IV). В одном из методов смесь
нитратов церия (III) и актиния экстрагируют спиртом,
спиртовый раствор выпаривают досуха и остаток про-
каливают. Затем продукт растворяют в смеси 1 н. азотной
кислоты и 28%-ной перекиси водорода, причем избыток
перекиси разлагается при выпаривании досуха. Получен-
ный таким образом продукт растворяют в 1 н. азотной
кислоте, добавляют с некоторым избытком окись серебра
и раствор разбавляют 1 н. азотной кислотой. Серебро
и церий после этого осаждают йодноватой кислотой, при-
чем применение избытка йодноватой кислоты не допу-
скается. Затем фильтрат выпаривают досуха и прокали-
вают, после чего актиний растворяют в соответствующей
кислоте. Трехвалентный церий можно также удалить окис-
лением до четырехвалентного состояния и осаждением в
нейтральном растворе перманганатом калия [13, 34] или
персульфатом аммония [96].
Отделение от тория. До известной степени торий можно
отделить от актиния осаждением из разбавленного кис-
лого раствора тиосульфатом натрия при 60—80° [5, 7, 8,
33, 68], однако осаждение часто бывает неполным, и по-
видимому, этим методом трудно достигнуть полного раз-
деления. В тех случаях, когда раствор, из которого дол-
жен быть осажден торий, содержит кислоту в чрезмерной
концентрации или слишком нагрет, некоторое количество
актиния может захватываться осаждающейся серой [82].
Лучшее разделение достигается осаждением гидратиро-
ванной перекиси тория из весьма разбавленного соляно-
кислого раствора при добавлении избытка перекиси
водорода [51, 69, 91]; осадок выдерживают в течение
нескольких минут при температуре ~ 80е, прежде чем начать
Отделение актиния от других элементов
239
фильтрование. Торий также можно отделить от актиния
осаждением его пиридином из спиртового раствора их нит-
ратов [47, 70, стр. 1388] или дробным осаждением гидро-
окисей (см., например, [40]). Более просто разделение
достигается нагреванием смеси хлоридов, поскольку тетра-
хлорид тория значительно более летуч, чем трихлорид
актиния [54].
Для отделения тория от актиния можно также исполь-
зовать метод ионного обмена; торий и микроколичества
актиния поглощаются на смоле дауэкс-50 (50—100 меш)
из раствора в 0,1 н. соляной кислоте, причем торий элюи-
руют 7%-ной щавелевой кислотой при 80°, а актиний —
5%-ной лимонной кислотой (pH 3) при 81—82° [80].
В работе 1102] для отделения актиния от RaAc (Th) использо-
ван перекисный метод, проверенный предварительно на отделении
Th от La: осадок перекисного соединения тория адсорбирует не
более 1,5% MsTh2.
Изучено отделение оксалатным методом Ас от Th с использо-
ванием изотопа актиния MsTh2; степень выделения MsTh2 равна
97—98%. Метод использован для выделения актиния из урановой
смоляной руды. Из раствора последней проводили осаждение
La2(C2O4)3, увлекавшего Ас и RaAc. После прокаливания осадок
растворяли в HNO3. В раствор добавляли Th(NO8)4 и перекисью
водорода отделяли Th(RaAc) от La и Ас. Из фильтрата щавелевой
кислотой осаждали La и Ас. Степень выделения актиния, свободного
от RaAc и АсХ, составляла 92%.
Отделение от радия. Актиний можно отделить от радия
экстрагированием его из смеси твердых нитратов этиловым
[47] или изопропиловым спиртом [70, стр. 1388], поскольку
нитрат радия нерастворим в этих спиртах. Если количества
присутствующего нитрата радия очень невелики, то реко-
мендуется добавлять небольшое количество нитрата бария,
действующего в качестве удерживающего агента перед экс-
трагированием актиния. Радий можно также успешно от-
делить от актиния осаждением концентрированной соляной
кислотой [69], при этом актиний остается в растворе. Осаж-
дение радия в виде бромида или нитрата действием соответ-
ствующей концентрированной кислоты также может быть
эффективным. Радий достаточно полно осаждается также
в виде хромата из кипящего раствора, содержащего в ка-
честве буфера ацетат натрия; актиний не осаждается в этих
условиях 151 ]. Актиний удавалось отделить от радия дей-
1М0 Актиний
ствием сернистого аммония [84], однако осаждение в дан-
ном случае не является количественным. Полученный оса-
док состоит, по-видимому, из гидроокиси актиния. Не яв-
ляются количественными также методы, основанные на
осаждении актиния гидроокисью лантана; кроме того, при
применении этих методов на осаждающейся гидроокиси
может адсорбироваться некоторое количество радия [51 ].
Выделение актиния из облученного радия включает от-
деление его от радия и от долгоживущих дочерних продук-
тов как радия, так и актиния (изотопы тория, полония, свин-
ца и висмута). Метод, разработанный для первоначального
разделения, включает экстрагирование 0,25 М раствором
теноилтрифторацетона (ТТА) в бензоле при регулируемом
значении pH. Актиний вместе с микроколичествами радия
экстрагируется при pH 6 и извлекается при обратном
промывании 6 н. соляной кислотой. Получающийся при
этом раствор содержит актиний, радиоактиний (Th227), ра-
дий-D (РЬ210) и микроколичества других элементов. Ра-
створ выпаривают досуха, растворяют остаток в 0,1 н. со-
ляной кислоте и экстрагируют свежим раствором ТТА
с целью удаления тория; после этого pH раствора дово-
дят до 6, экстрагируют актиний свежей порцией ТТА и
извлекают его из полученного раствора обратной промыв-
кой, как и ранее. Полученный в результате этой операции
раствор содержит актиний, свинец и немного радия, а также
некоторые другие элементы. После двукратного повторе-
ния цикла очистки свинец, полоний и висмут удаляют осаж-
дением в виде сульфидов на неактивном свинцовом носи-
теле, а актиний окончательно отделяют, осаждая его в виде
гидроокиси [37]. После начального экстрагирования тория
раствором ТТА в бензоле из 0,1 н. раствора соляной кислоты
очистку актиния можно проводить поглощением на смоле
дауэкс-50; при этом радий элюируют 4 н. азотной кислотой,
а актиний — 6 н. азотной кислотой; элюат содержит неко-
торые количества смолы или продуктов ее радиолиза;
актиний извлекают выпариванием раствора досуха с после-
дующим растворением осадка в разбавленной азотной кис-
лоте и осаждением актиния в виде оксалата действием на-
сыщенного раствора щавелевой кислоты. Оксалат затем пре-
вращают в окись актиния растворением в 8 н. азотной ки-
слоте, выпариванием досуха и прокаливанием при 1000° [21 ].
Определение актиния (Ас227)
241
В работе (102] для оценки степени разделения актиния и ак-
тиния-X (Ra) исследована адсорбция ThX (Ra) осадком La2(C2O4)3;
установлено, что при двукратном переосаждении La2(C2O4)3 коли-
чество адсорбированного ThX снижается до 0,2—0,3%.
Проведен ряд опытов по отделению актиния от актиния-Х.
К раствору La (NO3)3 прибавляли раствор актиния (азотнокислый
раствор урановой руды) и проводили осаждение La2(C2O4)3. Осадок
прокаливали, растворяли в HNO3 и проводили повторное осаждение
оксалата La и Ас из 0,3 н. HNO3 Эта методика позволяет выделять
~ 97% Ас, свободного от АсХ.
Для отделения актиния от радия методом ионного об-
мена применяли катионообменную смолу амберлит 1R-1,
причем радий элюировали 3 н. соляной кислотой, а акти-
ний — 0,25 М раствором цитрата аммония (при pH — 3).
Поглощение и элюирование повторяли трижды, чтобы очи-
стить актиний от примесей радия, причем актиний оконча-
тельно элюировали 3 н. соляной кислотой; свинец и висмут
удаляли затем осаждением сероводородом, как было опи-
сано ранее [77 стр. 1393].
В работе 11031 описан метод отделения Ас227 от его дочерних
продуктов ионообменной хроматографией и метод абсолютного оп-
ределения Ас2-’7 измерением скорости нарастания a-активности пер-
воначально чистого препарата.
ОПРЕДЕЛЕНИЕ АКТИНИЯ (Ас227)
P-Излучение Ас227 обладает слишком малой энергией, чтобы
его можно было измерить непосредственно при помощи
стандартных счетчиков, и поэтому определение актиния-227
основано на измерении излучения, испускаемого продук-
тами его распада; данное обстоятельство требует, чтобы пре-
парат актиния не содержал иония и радия, ибо дочерние
продукты распада последних будут оказывать влияние на
результаты измерения. Радиохимически чистый образец
актиния непосредственно после его приготовления должен
казаться почти неактивным и, чтобы достигнуть радиоак-
тивного равновесия между актинием и его дочерними про-
дуктами, нужен промежуток в 3—4 мес. Наиболее широко
применяемый метод количественного определения актиния
после его отделения от дочерних продуктов заключается
в сравнении скорости нарастания дочерней активности с
активностью, рассчитанной по периодам полураспада
’/<16 К. Бэгнал
242
Актиний
актиния и его дочерних продуктов; такой способ требует
приблизительно 10 суток. В связи с этим часто оказыва-
ется более удобным следить за поведением актиния, по-
мечая его мезоторием-2 (Ас228), который испускает легко
наблюдаемые ^-частицы; однако этот метод нельзя применять
для количественного определения Ас227.
Более быстрые методы количественного определения ак-
тиния в каком-либо источнике основаны на выделении и
счете одного из нижеприводимых его короткоживущих до-
черних продуктов.
Франций (АсК). Франций легко отделяется от актиния
осаждением последнего в виде карбоната, гидроокиси или
фторида (см. гл. 9). После выделения франция строят
кривую его распада, которую экстраполируют к нулевому
времени (время выделения); количество франция в нуле-
вой момент времени пропорционально исходному коли-
честву актиния [57, 73, 74].
Актинон (Rn21<J). Актинон легко определить, пропуская
воздух через раствор актиния в ионизационную камеру.
Актинон (период полураспада 3,92 сек.) практически пол-
ностью распадается через 1 мин., т. е. за время, в течение
которого активность торона уменьшается приблизительно
наполовину, тогда как активность радона остается по су-
ществу неизменной. Данный метод изучал довольно под-
робно Баранов [3 ], однако полученные им результаты трудно
поддаются объяснению, и метод не нашел широкого при-
менения.
Радиоактиний (Th227). Количественное определение ак-
тиния можно проводить выделением радиоактиния (см. от-
деление от тория) и последующего наблюдения за измене-
нием его активности во времени; активность выделенного
продукта приблизительно через 20 суток нарастает до ма-
ксимума и затем уменьшается с периодом полураспада 18,6
суток [91 ].
Актиний-В (РЬ211). Этот изотоп можно отделить осажде-
нием сероводородом на свинцовом носителе из растворов
актиния в разбавленной кислоте; Ас227 не адсорбируется
Спектр актиния
243-
осадком. Период полураспада АсВ 36 мин., и количества
присутствующего АсВ в нулевой момент времени устанав-
ливается так же, как указано для франция. Данный ме-
тод применим только при отсутствии радия и тория [76].
Актиний в равновесии с его дочерними продуктами
можно определить калориметрическим методом [57, 85,
86], а также сравнением его ^-активности с -(-активностью-
радиевого эталона [57]; эти методы применимы только а
случае сравнительно больших источников.
Обзор методов количественного определения актиния
см. [26, стр. 192] и [35, стр. 53], [57].
СПЕКТР АКТИНИЯ
Ранние исследования спектра актиния [64—66] позволили
выявить около семи линий в интервале 4088—4812 А, ко-
торые могли быть приписаны актинию, и по этим данным
путем интерполяции были установлены значения термов
для оболочек от К до Р [83]. Существование этих линий
было подтверждено данными последних наблюдений спектра
испускания, опубликованными Меггерсом и др. [71 ]; в.
этой работе около 1 мг Ас227 было помещено в полый алю-
миниевый катод; спектр возбуждался ионами аргона. До-
полнительные спектрограммы получали при высоковольт-
ном искровом разряде между медными электродами с нане-
сенными на их торцы небольшими количествами (4—70
актиния. Наиболее интенсивные линии спектра актиния
приведены в табл. 27. Сравнение со спектрами редкоземель-
ных элементов показывает, что Ac (II) более похож на Y (II),
чем на Sc (II) или La(II). Наличие полос, также отмеченных
в спектре, вероятно, обусловлено молекулами АсО.
Таблица 27
Спектр актиния, А
Ас1 4179,98 4183,12 6359,87 6691,26
Acll 3863,11 4088,43 4168,40 5910,87
AcIII 3392,77 3487,60
В основном состоянии актиний имеет электронную кон-
фигурацию 6d 7s2 (2D3/2); спин Ас227 равен 8/2.
'Л 16*
2Я
Дктцний
ИЗОТОПЫ АКТИНИЯ
Известно десять изотопов актиния; их ядерные свойства
указаны в табл. 28.
Таблица 28
Изотопы актиния
Массовое число Период полураспада Тип распада и энергия испускаемых частиц, Мэв
221 222 5,5 сек. а 7,6 а 6,96
223 2,2 мин. Э. 3. (1%); а (99%) 6,64
224 225 2,9 часа 1Q.0 суток Э. 3. (—90%); а ( — 10%) 6,17; 7 а 5,818 (54%); 7 5,782 (28%) 5,721 (9,5%) 5,627 (3,8%) 5,713 (2,6%)
2?6 29 час. 3. 3. (~29^); Г(~ЭД%1 ».17; Т
?27а 228(MsTh2) 229 230 21.6 года6 6,13 часа 66 мин. < 1 > (-99%) 0,046; 1 (1,2%) 4,942; 7 Г 1,11 (53%); 7 0,45 (13%) 2,18 (10%) 1.85 (9,6%) 0,64 (7,6%) 1,7 (6,7%) р- р~, 2,2
Поперечное
Поперечное сеченне поглощения тепловых нейтронов для ДсА
равно 500 ± 35 барн (79].
В ранних определениях для перирда полураспада Ас227 были получены
значения от 7 до 22 лет.'
-Наиболее широкое применение находят изотопы Ас227
(период полураспада 21,6 года) и Ас228 (период полураспада
6,13 часз); последний изотоп, открытый в 1QQ8 Г. Ханом
[411, часто применяют в качестве индикатора ддр Ас’27,
Соединения актиния
245
поскольку высокая энергия его p-излучения позволяет легко
определять этот изотоп. Другие изотопы, получаемые об-
лучением, не имеют особой ценности для химических иссле-
дований.
МЕТАЛЛИЧЕСКИЙ АКТИНИЙ
Актиний в виде металла был получен в миллиграммовых
количествах восстановлением трифторида актиния метал-
лическим литием при 1200° [89], а также восстановлением
трихлорида актиния парами калия при 350* [21 ]. Ак-
тиний— серебристо-белый металл, напоминающий лантан;
он испускает слабое голубоватое свечение, заметное в тем-
ноте (ср. с полонием, гл. 3). Актиний легко окисляется
во влажном воздухе с образованием белой окисной пленки,
предотвращающей дальнейшее окисление. Металлический
актиний плавится при 1050 + 50* [89]; точка кипения его,
вероятно, лежит в области 3300* [24]. Давление паров ме-
талла составляет приблизительно 0,007 мм рт. ст. при 1600*
[24].
Металл может существовать в двух кристаллических
формах, подобно лантану, однако имеются сведения о
структуре лишь одной из них — кубической гранецентри-
рованной с периодом решетки а = 5,311 А [21].
Вследствие того что актиний является сильно электро-
положительным элементом, он не может выделяться на
катоде в виде металла из водных растворов. При электро-
лизе раствора нитратов актиния и лантана в смеси
этилового спирта и ацетона с применением серебряного
катода образуются катодные осадки неизвестного состава.
Невесомые осадки актиния получаются при электролизе
аналогичных растворов, содержащих нитрат натрия вме-
сто нитрата лантана [11]. Эти наблюдения не имеют от-
ношения к электродному потенциалу актиния.
СОЕДИНЕНИЯ АКТИНИЯ
Большую часть наблюдений, о которых сообщалось в пе-
чати, проводили с применением образцов, содержащих
микроколичества актиния и включавших большие коли-
17 К. Бэгнал
246
Актиний
чества редкоземельных элементов; имеющиеся данные о
свойствах соединений актиния получены в результате тех
же опытов по соосаждению, какие были поставлены при
исследовании микроколичеств полония (гл. 2). Эти иссле-
дования показывают, что актиний обладает свойствами,
аналогичными свойствам лантана; это соответствует пред-
ставлению об актинии как о высшем гомологе лантана.
Хевеши [48, 49] на основании работ по изучению диф-
фузии микроколичеств актиния в разбавленной соляной
кислоте пришел к выводу, что актиний в этих растворах
трехвалентен; проведенные в последнее время кристалло-
графические работы [98] показывают, что радиус иона
Ас3+ равен 1,1 А (радиус иона La3+ равен 1,04 А).
Гидроокись, карбонат, фторид, фторсиликат, оксалат и
фосфат актиния — нерастворимы; за исключением суль-
фида, все соединения актиния белого цвета, а их растворы
бесцветны. Большинство соединений актиния, которые были
достоверно идентифицированы, изоморфны аналогичным
соединениям лантана. Актиний обладает более выражен-
ными основными свойствами, чем лантан; соединения ак-
тиния, как правило, гидролизуются с большим трудом,
чем соответствующие соли лантана.
Окись актиния (Ас2О3)
Окись актиния образуется при прокаливании его оксалата
в атмосфере кислорода при 1000—1100°; кристаллическая
структура окиси актиния — гексагональная (типа La2O3)
с периодами решетки а = 4,07 А и с = 6,29 А; вычислен-
ная плотность равна 9,19 e/cjw3 [25]. По расчетным данным,
теплота образования Ас2О3 из элементов равна примерно
148 ккал/моль [9].
Гидроокись актиния [Ас(ОН)3]
По сообщениям ряда авторов, гидроокись актиния обладает
большей растворимостью, чем гидроокись лантана, и не
полностью осаждается аммиаком [36, 40, 45, 60—63]; от-
сюда следует, что для количественного осаждения следов
актиния может потребоваться много лантана в качестве
Соединения актиния
247
носителя. Для этой цели может служить также гидроокись
трехвалентного церия; носителем актиния не может быть
перекись урана [70, стр. 1371]. Высокая концентрация
аммонийных солей снижает эффективность осаждения гид-
роокиси актиния (см., например, [51]); осаждение про-
ходит более полно при нагревании раствора до кипения
перед добавлением аммиака и выдерживании полученной
суспензии в течение 2 час. на водяной бане. Оставшиеся
микроколичества актиния можно осадить на гидроокиси
трехвалентного железа [46] или на гидроокиси алюминия
[69, 78]; последнюю можно удалить растворением в раст-
воре едкого натра [34]. Гидроокись циркония также можно
использовать в качестве носителя для актиния [41 ]. Мик-
роколичества актиния, как и редкоземельные элементы
(церий, лантан, празеодим, неодим), не осаждаются из
щелочного раствора при обработке бромом или хлором
[68, 34, 96]. В миллиграммовых количествах белая жела-
тинообразная гидроокись актиния осаждается из растворов
солей актиния при добавлении растворов едкого натра или
аммиачной воды [25].
Гидрид актиния (АсН2)
Гидрид актиния, имеющий, по-видимому, состав АсН2,
может образоваться в процессе получения металлического
актиния при восстановлении трихлорида актиния парами
калия; образование этого соединения объясняется взаимо-
действием с хлористым аммонием, следы которого присутст-
вуют в виде загрязнения. Гидрид актиния имеет гранецент-
рированную кубическую решетку с периодом а = 5, 670 А
[21].
Сульфид актиния (Ac2S3)
Микроколичества актиния не осаждаются с сульфидами
ртути, свинца и висмута при действии сероводорода в кис-
лом растворе [69], а при обработке солей актиния серни-
стым аммонием, вероятно, осаждается гидроокись актиния
[84]. При нагревании миллиграммовых количеств окса-
лата актиния или окиси актиния до 1400° в смеси паров
17*
248
Актиний
сероводорода и сероуглерода образуется окрашенный в
темный цвет Ac2S3. Кристаллическая структура сульфида
актиния — кубическая (типа Ce2S3) с периодом решетки
а = 8,97 А и вычисленной плотностью, равной 6,75 г/см3
[25].
Галогениды актиния
Трифторид (AcF3)
Микроколичества актиния полностью осаждаются с три-
фторидом лантана при действии плавиковой кислоты [56];
кремнефтористоводородная кислота и 5М нитрат аммония
мешают осаждению [70, стр. 1371 ]. Актиний можно оса-
дить с тетрафторидом тория в тех случаях, когда приме-
нение лантана в качестве носителя нежелательно [52].
Трифторид получают в миллиграммовых количествах либо
осаждением плавиковой кислотой, либо действием паров
плавиковой кислоты при 700° на гидроокись актиния [25].
Кристаллическая структура AcF3 — гексагональная (типа
тизонита ЬаРз) с периодами решетки а = 4,27 А, с=7,53А.
Вычисленная плотность равна 7,88 г/си3 [97].
Оксифторид (AcOF).
Это соединение образуется при гидролизе трифторида
актиния в смеси аммиака и паров воды при 900—1000®.
В противоположность оксифториду лантана AcOF не об-
разуется при нагревании трифторида на воздухе при 800®.
Кристаллическая структура AcOF — кубическая (типа флю-
орита) с периодом решетки а = 5,931 А; вычисленная
плотность равна 8,28 г/см3 [25, 99].
Трихлорид (АсС13)
Препараты, содержащие микроколичества этого со-
единения, были получены при нагревании смесей окислов
актиния и редкоземельных элементов до температуры крас-
ного каления в смеси хлора и двухлористой серы [54].
Безводный трихлорид был получен в миллиграммовых ко-
личествах нагреванием гидроокиси актиния с хлористым
Соединения актиния
249
аммонием до 250* в вакууме [21 ] или нагреванием при
более высоких температурах оксалата или гидроокиси ак-
тиния в парах четыреххлористого углерода. Трихлорид
актиния возгоняется в вакууме при 960* [25]. Кристалли-
ческая структура АсС13 — гексагональная (типа UC1S)
с периодами решетки а = 7,62 А, с = 4,55 А; вычисленная
плотность равна 4,81 г!<м3 [25].
Спектр поглощения трихлорида в растворе 1 н. соля-
ной кислоты имеет пик при 50 мр [711; результаты иссле-
дований в области 230 — 1000 мр и кривая поглощения
приведены в обзоре Хагемана [38, стр. 30]. Опыты по
ионному обмену, проведенные в растворах с высокой кон-
центрацией соляной кислоты, свидетельствуют о том, что
трихлорид образует комплексные ионы [20]. Трихлорид
актиния нерастворим в эфире [53].
Оксихлорид (AcOCl)
Это соединение получается точно так же, как и окси-
фторид; оно также образуется при нагревании на воздухе
до 1000° гидроокиси актиния, осажденной аммиаком из
раствора в 0,1 н. соляной кислоте. Нельзя с уверенно-
стью сказать, осаждается ли оксихлорид в результате пер-
вичного процесса или же образуется при взаимодействии
гидроокиси с адсорбированным хлористым аммонием. Кри-
сталлическая структура AcOCl — тетрагональная (типа
PbFCl) с периодами решетки а = 4,24 А, с — 7,07 А. Вы-
численная плотность — 7,23 г/см.3 [25].
Трибромид (АсВг3)
Данное соединение образуется при нагревании окиси
актиния или оксалата с бромидом алюминия при 750*.
Оно возгоняется в вакууме при 800°. Кристаллическая
структура АсВг3 — гексагональная (типа UC13) с перио-
дами решетки а = 8,06 А, с = 4,68 А. Вычисленная плот-
ность— 5,85 г/см3 [25].
Оксибромид (АсОВг)
Это соединение образуется при нагревании трибромида
в смеси паров воды и аммиака при 500°. Кристаллическая
структура его — тетрагональная (типа PbFCl) с периодами
250
Актиний
решетки а = 4,27 А, с = 7,40 А. Вычисленная плотность —
7,89 г/см3 [25].
Трийодид (AcJ3).
Предполагают, что при нагревании окиси актиния или
оксалата актиния с йодидом алюминия или йодидом аммо-
ния при 500—700° образуется трийодид актиния. Он воз-
гоняется в вакууме при 700—800° [25].
Оксийодид (AcOJ).
Соединение, которое быть может является оксийодидом
актиния, образуется точно так же, как и оксифторид при
700° [25].
Другие соединения актиния
Азид
Микроколичества актиния не соосаждаются с торием
при добавлении азида калия [23].
Карбонат
Микроколичества актиния осаждаются количественно
с карбонатом лантана (см., например, [35]).
Хромат
Микроколичества актиния не соосаждаются с радием
из уксуснокислого раствора при действии хроматов и по-
этому возможно, что хромат актиния растворим в уксус-
ной кислоте; однако хромат бария может служить носите-
лем для актиния в слабокислом растворе [51 ].
Фторсиликат
Микроколичества актиния осаждаются количественно
с лантаном при действии кремнефтористоводородной кис-
лоты [72, 92, 93 ].
Йодат
Степень соосаждения актиния с йодатом циркония
сильно зависит от концентрации ионов йодата; в присутст-
вии нейтральных солей количество соосаждающегося ак-
тиния понижается [70, стр. 1371 ].
Соединения актиния
251
Нитрат
Это соединение хорошо растворимо в этиловом или изо-
пропиловом спирте [11, 47, 70, стр. 1388].
Оксалат
Актиний по сравнению с лантаном менее полно осаж-
дается щавелевой кислотой; в присутствии солей трехва-
лентного железа для осаждения нужно брать щавелевую
кислоту в значительном избытке ввиду образования комп-
лексного оксалата железа. Осаждение более эффективно,
если его проводить из менее кислых растворов [42, стр. 89],
[96]; процесс осаждения протекает значительно лучше
в случае применения концентрированного раствора окса-
лата аммония [25, 70, стр. 1371]. Хан [42, стр. 90],
используя данные о совместной кристаллизации, подсчи-
тал, что в 100 г 0,1 н. раствора соляной кислоты при 25°
растворяется примерно 0,05 г оксалата актиния. Согласно
более поздним данным, растворимость оксалата актиния
в растворе 0,1 н. HNO3 — 0,5 н. Н2С2О4 составляет
0,024 мг!мл. Оксалат актиния получают также, используя
гидролиз диметилоксалата при 60—70° в качестве источ-
ника щавелевой кислоты [90].
Фосфат (АсРО4)
Степень соосаждения микроколичеств актиния с фосфа-
том висмута понижается в присутствии ионов нитрата
[70, стр. 1371 ]; в 1 н. растворе азотной кислоты фосфат
циркония не может служить носителем для актиния [70,
стр. 1371]. Полугидрат фосфата актиния AcPO4-V2 Н2О
осаждается из раствора трихлорида актиния в 0,1 н. со-
ляной кислоте при добавлении однозамещенного фосфата
натрия. Фосфат актиния имеет гексагональную структуру
(типа LaPO4-1/2 Н2О) с периодами решетки а = 7,21 А и
с = 6,64 А; вычисленная плотность его равна 5,48 e/ui3.
Безводная соль не изоморфна соответствующей соли лан-
тана [25].
Сульфат
Микроколичества актиния соосаждаются с сульфатами
редкоземельных элементов цериевой группы [96] и с суль-
252 Актиний
фатом свинца из горячего разбавленного раствора серной
кислоты [70, стр. 1371 ], [81 ]. Работы с миллиграммовыми
количествами актиния показали, что из растворов актиния
в 0,1 н. серной кислоте при добавлении насыщенного вод-
ного раствора сульфата калия выпадает белая кристалли-
ческая двойная соль актиния и калия [25].
ЛИТЕРАТУРА
1. Bachelet М., Ann. Chim., 12,348 (1939).
2. Bachelet M., J. Chim. phys., 42, 98 (1945).
3. Baranov V. I., Akad. Vernad. Pyatid. Nauk. Deyatel. 1. 499
(1936). (English transl. Amer. Rep. NP-2236).
4. Bolt wood B.B., Amer. J. Sci., 22, 537 (1906).
5. Boltwood В. B., Amer. J. Sci., 24, 370 (1907).
6. В о 11 w о о d В. В.» Amer. J. Sci., 25, 289, 365 (1908).
7. В о 11 w о о d В. B., Nature, bond., 76, 544 (1907).
8. Bolt wo о d В. B., Le Radium, 8, 104 (1912).
9. Brewer L., Chem. Rev., 52, 1 (1953).
10. Brooks H., Phil. Mag., 8, 373 (1904).
11. Cote lie S., Haissinsky М.» C. R. Acad. Sci., Paris, 206,
1644 (1938).
12. Curie L., Bouissieres G., Cahiers de Physique, № 26, I
(1944).
13. Curie Marie, J. Chem. phys., 27, 1 (1930).
14. Curie Marie, Traite de Radioactivite Hermann, Paris, 1935.
15. C u r i e, Maurice, Takvorian S., C. R. Acad. Sci., Paris,
198, 1687 (1934).
16. Debierne A., C. R. Acad. Sci., Paris, 129, 593 (1899).
17. Debierne A., C. R. Acad. Sci., Paris, 130, 906 (1900).
18. Debierne A., C. R. Acad. Sci., Paris, 136, 446 (1903).
19. Debierne A., C. R. Acad. Sci., Paris, 138, 539 (1904).
20. Diamond R. M., Street K., Seaborg G. T., J. Amer.
chem. So<\, 76, 1461 (1954).
21. Farr J. D., Giorgi A. L., Bowman M. G., Money R. K-,
Amer. Rep., LA-1545 (1953).
22. Finkelnburg W., Phys. Rev., 77, 303 (1950).
23. Fleck A., J. chem. Soc., 103, 388 (1913).
24. Foster K. W., Amer. Rep., MLM-901 (1953).
25. F r i e d S., H a g e m a n n F., Z a c h a r i a s e n W. H., J. Amer,
chem. Soc., 72, 771 (1950).
26. F r e s e n i u s R., J a n d e r G., Handbuch der analytischen Che-
(nie, Tl. 2. vol. 3, Springer, Berlin, 1944
254
Литература
27. G i е s е 1 F., Вег., 33, 3569 (1900).
28. G i e s e 1 F., Ber., 34, 3776 (1901).
29. G i e s e 1 F., Ber., 35, 3608 (1902).
30. G i e s e 1 F., Ber., 36, 342 (1903).
31. Giese 1 F., Ber., 37, 1696 (1904).
32. G i e s e 1 F., Ber., 37, 3963 (1904).
33. Giese 1 F., B«r., 40, 3011 (1907).
34. G 1 e d i t s c h E., Chamie С., C. R. Acad. Sci., Paris, 182, 380
(1926).
35. G m e 1 i n, Handbuch der anorganischen Chemie, System — num-
mer 40, Verlag Chemie, G.m.b.H., Berlin, 1942 (English translation
Amer. Rep. AEC-tr-1734, 1954).
36. God lewski T., Phil. Mag., 10,35 (1905).
37. H a g e m a n n F., J. Amer. chem. Soc., 72, 768 (1950).
38. Hagemann F., in (G. T. Seaborg and J. J. Katz, Eds) Nat.
Nuclear Energy Series IV—14A, page 14, McGraw Hill, New York,
1954.
39. Hahn O., Ber., 39, 1606 (1906).
40. Hahn O., Phil. Mag., 13, 165 (1907).
41. Hahn O., Phys. Z., 9, 246 (1908).
42. Hahn O., Applied Radiochemistry, Cornell Univ. Press, Ithaca,
New York, 1936.
43. Hahn O., Meitner L., Naturwiss., 6, 324 (1918).
44. Hahn O., Meitner L., Phys. Z., 19,208 (1918).
45. Hahn O., Meitner L., Phys. Z., 20, 529 (1919).
46. Hahn O., Rothenbach M., Phys. Z., 14, 410 (1913).
47. Haissinsky M., C. R. Acad. Sci., Paris, 196, 1788 (1933).
48. Hevesy G., Phys. Z., 14, 1204 (1913).
49. Hevesy G., Phil. Mag., 27, 591 (1914).
50. Hollander J. M., Leininger R. F., Phys. Rev., 80, 915
(1950).
51. H u 11 D. E., Libby W. F., Latimer W. M., J. Amer. chem.
Soc., 57, 593, 1649 (1935).
52. Hyde E. K., in (G. T. Seaborg and J. J. Katz, Ed.) Nat. Nuclear
Energy, Series IV—14A, page 545, McGraw Hill, New York, 1954.
53. Imre L., Z. anorg. Chem., 166, 1 (1927).
54. Imre L., Z. anorg. Chem., 175, 142 (1928).
55. Imre L., Z. phys. Chem., A153, 127, 262 (1931).
56 Keetman B., Dissert. Berlin, 1909 (according to [35]).
57. Lecoin M., Perey M., Pompei A., J. Chim. phys., 46, 158
(1949),
Литература
255
58. L ecoin M., Регеу М., Riou М., Teillac J., J. Phys.
Radium, 11, 227 (1950).
59. Lederer M., C. R. Acad. Sci., Paris, 236, 200, 1557 (1953).
60. Levin M., Phys. Z., 7,513 (1906).
61. Levin M., Phil. Mag., 12, 177 (1906).
62. Levin M., Phys. Z., 8, 129 (1907).
63. Levin M., Ber., 5, 410, (1907).
64. Lub W. A., Konink. ned. Akad. Wetensk., 40, 584 (1937).
65. Lub W. A., J. Phys. Radium, 8, 366 (1937).
66. Lub W. A., C. R. Acad. Sci., Paris, 204, 1417 (1937).
67. Lederer M., Anal. Chim. Acta, II, 145 (1954).
68. Marckwald W., Ber., 38, 2264 (1905).
69. McCoy H. N, Viol С. H., Phil. Mag., 25, 333 (1913).
70. McLane С. K., Peterson S., in (G. T. Seaborg J. J. Katz
and W. M. Manning, Eds) Nat. Nuclear Energy, Series IV—14B,
pages 1371, 1385 and 1388, McGraw Hill, New York, 1949.
71. Meggers W. F., Fred M., T о m к i n s F. S., J. Opt. Soc.
Amer., 41,867 (1951).
72. Meyer S., Sitzungsber. Akad. Wiss., Wien, Abt. Ha, 129, 483
(1920).
73. Регеу M., C. R. Acad. Sci., Paris, 214, 797 (1942).
74. Регеу M., J. Chim. phys., 43, 155, 269 (1946).
75. Регеу M., J. Chim. phys., 46, 485 (1949).
76. Регеу M., Het tier A., C. R. Acad. Sci., Paris, 242, 2552
(1956).
77. P e t e r s о n S., in (G. T. S e a b о r g, J. J. Katz and W. M.
Manning, Eds) Nat. Nuclear Energy, Series IV—14B, pages 1391
and 1393, McGraw Hill, New York, 1949.
78. Plum H. M., J. Amer. chem. Soc., 37, 1797 (1915).
79. Pomerance H., Amer. Rep., ORNL-1415 (1952).
80. R a d h а к r i s h n a P., J. Chem. phys., 51, 354 (1954).
81. Rogers N. E., Watrous R. M., Amer. Rep., MLM-967
(1954).
82. R о t h e n b a c h M., Dissert., Berlin, 1912 (according to [35]).
83. Ruark A. E., Maxfield F. A., Phys. Rev., 47, 107 (1935).
84. Rutherford E., Phil. Mag., 14, 733 (1907).
85. Sanielevici A., J. Chim. phys., 33, 759 (1936).
86. S a n i e 1 e v i c i A., C. R. Acad. Sci., Paris, 202, 1055 (1936).
87. Sod d у F., Cranston J., Nature, Lond., 100, 498 (1918).
88. Sod d у F., Cranston J„ Proc. Roy. Soc., A94, 384 (1918).
256
Литература
89. S t i t е s J. G., S a 1 u t s к у M. L., S t о n e B. D., J. Amer. chem.
Soc., 77, 237 (1955).
90. Sal ut sky M. L., Kirby H. W., Analyt. Chem., 28, 1780
(1956).
91. Teleschow E., Dissert., Berlin, 1912 (according to [35]).
92. Ulrich C., Z. angew. Chem., 36, 54 (1923).
93. Wei sb a ch I. C. A. von, Z. anorg. Chem., 69, 353 (1911).
94. Young J. T., J. Chim. phys., 47, 805 (1950).
95. Young J. T., Haissinsky M., Bull. Soc. chim. Fr., 546
(1949).
96. Yovanovitch D. K., J. Chim. phys., 23, 15 (1926).
97. Z a c h a г i a s e n W. H., Amer. Rep., MDDC-1572 (1947).
98. Z a c h a г i a s e n W. H., Phys. Rev., 73, 1104 (1948).
99. Z a ch a r i a s e n W. H., Acta cryst. Camb., 4, 231 (1951).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
100. Mariott J., E.R.A.E. Res. Estab. NMED/R, 2720, p. 5 (1958).
101. D a n о n J., J. Inorg. Nucl. Chem., 7, 422—424 (1958).
102. Старик И. E., С т a p и к А. С. и др., Труды гос. радиевого
инет. им. В. Г. Хлопина АН СССР, т. VIII, 170—176 (1958).
103. Cabell М. J., Canad. J. Chem., 1094—1103 (1959).
ПРИЛОЖЕНИЯ
Радиоактивный ряд тория (4/г)
Радиоактивный ряд нептуния (4/г+1)
Радиоактивный ряд урана —радия (4«+2)
Радиоактивный ряд урана — актиния (4/г-(-3)
СОДЕРЖАНИЕ
Предисловие к русскому переводу.............. 5
Предисловие к английскому изданию............ 8
Предисловие автора........................... 9
Часть I. ПОЛОНИЙ ....................................11
I. Открытие, выделение и применение полония .... 13
2. Химия микроколичеств полония в растворах .... 42
3. Техника работы с полонием...................54
4. Элементарный полоний .......................72
5. Электрохимия полония .......................95
6. Галогениды и галогенаты полония............102
7. Окислы и другие соединения полония.........119
8. Химические свойства полония и его место в периоди-
ческой системе элементов .....................137
Литература .................................141
Часть II. АСТАТИН, ФРАНЦИЙ, РАДОН ..................155
9. Астатин, франций, радон....................157
Литература .................................183
Часть III. РАДИЙ, АКТИНИЙ ..........................191
10. Техника работы с радием и актинием........193
Литература..................................204
11. Радий ....................................205
Литература .................................226
12. Актиний ..................................233
Литература .................................253
Приложения..........................................257
К. Б э г н а л
ХИМИЯ РЕДКИХ
РАДИОАКТИВНЫХ ЭЛЕМЕНТОВ
Полоний — актиний
Редактор Г. М. Мануйлова
Художник Н. А. Липин
Художественный редактор
Е. И. Поамаръкова
Технический редактор А. Г. Резоухова
Корректор Р. Я. Новик
Сдано в производство 14/IV 1960 г.
Подписано к печати 31/VIII 1960 г.
Бумага ?4х108|/за-4,3 бум. л.
14,1 печ. л., в т. ч. вкл. 1
Уч.-изд. л. 13,1. Изд. №3/5247
Цена 11 р. 15 к. С 1/1-1961 г. цена 1 р. 12 к.
Зак. 835
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
1-й Рижский пер., 2
Типография № 4 УПП Ленсовнархоза.
Ленинград, Социалистическая, 14.
ОПЕЧАТКИ
Страница | Строка Напечатано Следует читать
74 Массовое число 202 6 57 5,57
74 » » 205 1 8 1.8
75 » » 210 Bi210P ро210 в,™Лро210
75 > » 214 7 683 7,683
133 3 снизу [731] [371]
160 7 > с12-аРэ С12 —At29*
К. Бэгяал
Радиоактивный ряд урана-актиния (4/7 + 3)
Массовое число 233 231 229 227 225 223 221 219 217 215 di3 211 209 207
Элемент Порядко- вый номер и235 (AcU) 7,1-10влет \a Pa231 3,43-10 лет P Th231 (UY) 25,65 часа X^ Th227 (RdAc) 18,2 суток p Ac227 21,6 года X a 1.2 %X • n 223 Ra v (AcX) 11,68 суток P Fr223 (AcK) 22 мин. 6-10 % D 219 Rn (Актинон) 3,92 сек. Г|з% At219 0,9 мин. x®^ x^ 97% At215 JO"4 сек. p|5-io"4% Po213 (AcA) 1,83-IO3 сек. P Bi215 8 мин . i i i i i 1 1 1 1 a a Po2" (AcC') 0,52 сек. pA 0,32% Bi2" (AcC) 2,16 мин. ♦ 3 Pb211 <AcB) 36,1 мин . X^ X^ Pb207 (AcD) If- у j 207 (AcC") 4,79 мин.
и Ра Th Ас Ra Fr Rn At Ро Bi Pb T1 92 91 90 89 88 87 86 85 84 83 82 81
Бэгнал
I
Радиоактивный ряд урана-радия (4л4-2)
Массовое ч исло 238 236 234 232 230 228 226 224 222 220 218 216 214 212 210 208 206
Элемент Порядко- вый номер и238 и234
и 92 (Ul) (ип) ।
4,51-Ю9 лет 2,48• 105 лет Is
т? t А v
' UX2 ; UZ ’
Ра 91
Х^^ 6,66часа{ 1,175мин
Ра234 Х^
1?
Th 90 Th234 Th230
(UXj) (Io)
24,1 суток 80 000 лет
Ас 89 Х^
Ra 88 Ra226
1620 лет
Fr 87
Rn 86 Rn222 Rn218 1
3,825 суток 0,019 сек Р |о,| к 1 *
At 85 Х^ At218 2,0 сек. Х^ 14
р|о,о2% Ро" (Ка( Ро210
Ро 84 Ро218 х. а (RaF)
(RaA) х^. 1.64-Ю" сек 138,4 суток
3,05 мин. р
Bi 83 * 21 Bi ! Bi210 X^a^
(RaC ) (RaE)
19,7 5 ин. 5,013 суток
Pb 82 И nu2 14 р Pb210 (RaD) 19,4 года a <> \5-10 % .Pb206
Pb (RaB 26,8 м ) ин. Х^а^ 0,04% (RaG) P -r • 206
T1 81 Tl210 тг
(RaC") 4,23 мин.
—4 1,32 мин
Радиоактивный ряд нептуния (4л + 1)
Массовое число 241 239 237 235 233 231 229 227 225 223 221 219 217 215 । 213 211 209
Элемент Порядко- вый номер • ! '
Ат Ри 95 94 * 241 Am 458 лет Ри24' 13 лет
Np 93 Np237 2,2*10влет
и Ра 92 91 340“% ^и237 6,75суток и233 1,62Ч05лет 13 233 Ра233 27,4суток
Th 90 Th229 7340 лет
Ас Ra Fr 89 88 87 Ас225 10,0 суток ^Ra225 14,8 суток Fr221 4,8 мин •
Rn 86
At 85 At217 0,018 сек.
Ро Bi 84 83 Ро2'3 сек. Ц|98% Bl213 47 мин. Bi209 In
Pb Ti 82 81 \о 2% \ Pb209 3,3 часа ;> 2,2 мич.
Радиоактивный ряд тория (4л)
Массовое число 232 230 228 226 224 222 220 218 216 214 212 210 208
Эле- мент Поряд- ковый номер
Th Ас Ra Fr 90 89 88 87 Th232 1,39-Ю,0лет Th228 (RdTh) 1,90 года f₽ a 228 Ac (MsTh2) 6,13 часа (MsThj) 6,7 года Ra224 (ThX) 3,б4суток
Rn At Ро Bi 86 85 84 83 n 220 Rn (To рои) 51,5 сек.. At216 3*10”4сек. 0,014% r» 2,6 Po (ThA) 0,158 сек. D 212 Po (Th C’) 3,04 - 10"7cck ftf 64% Bl21* (ThC) 60,5 мин
Pb Tl 82 81 31'>12 Pb-12 (ThB) 1U,64 часа 36% Pb208 b Tl208 - (ThC") 3,1 м и н .
ХИМИЯ РЕДКИХ
РАДИОАКТИВНЫХ
ЭЛЕМЕНТОВ
ПОЛОНИЙ-АКТИНИЙ
К Б Э Г Н А Л
ХИМИЯ РЕДКИХ
РАДИОАКТИВНЫХ
ЭЛЕМЕНТОВ
ПОЛОНИЙ'АКТИН ИЙ