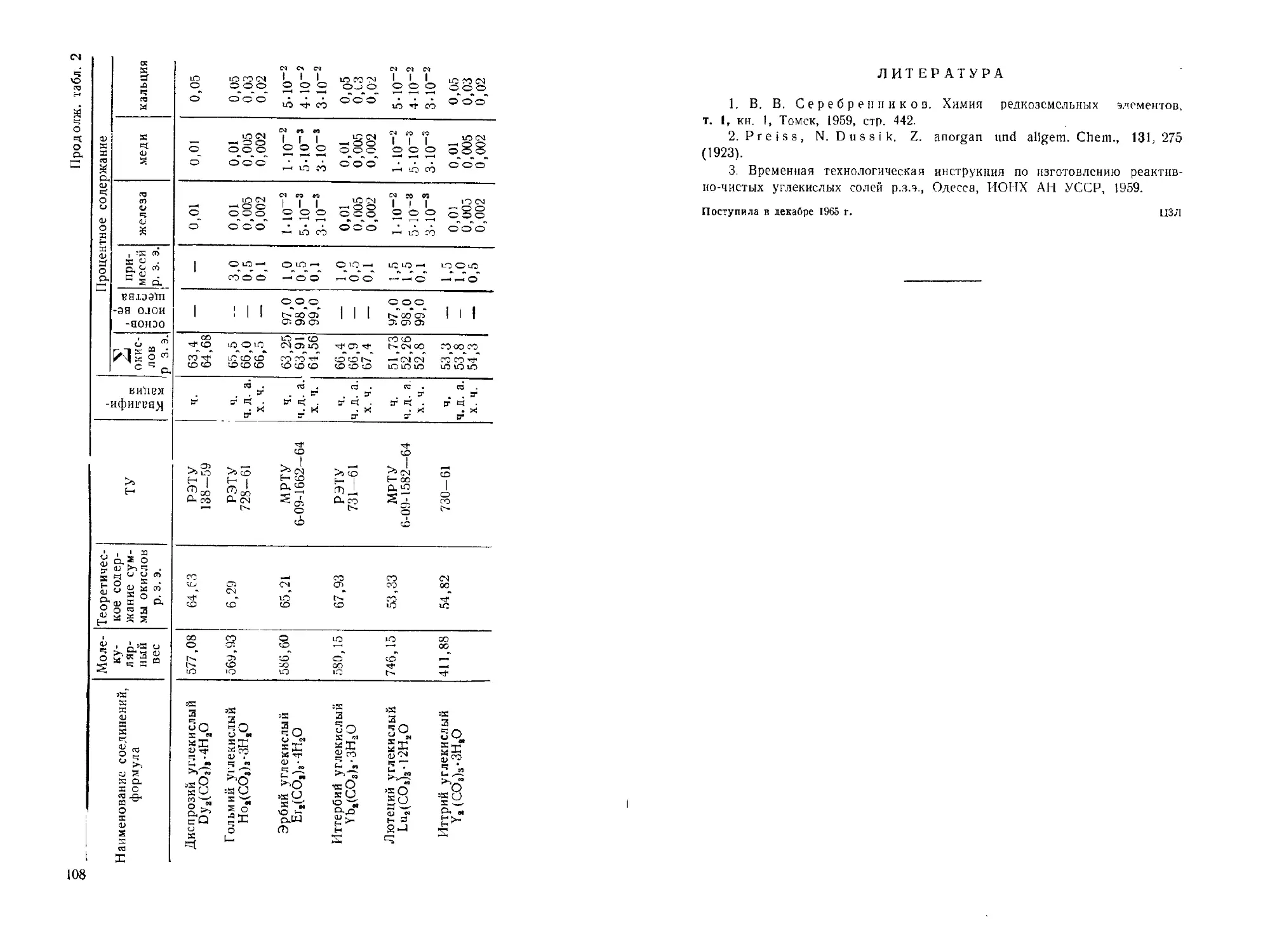
/
























































































Автор: Ластовский Р.П.
Теги: химия неорганическая химия химическая промышленность химические реакции
Год: 1967




Текст
ВСЕСОЮЗНЫЙ ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
научно-исследовательский институт
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ
химических веществ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 16
МОСКВА —1967
Настоящий выпуск содержит методики получения неор-
ганических реактивов и особо чистых веществ, преимущест-
венно соединений редких и цветных металлов. В большинст-
ве случаев предлагаемые методы являются новыми, более
совершенными и экономичными, чем описанные в литературе.
В тех случаях, когда использовался метод, заимствованный
из литературных источников, предлагаемая методика являет-
ся результатом его крупнолабораторной проверки с уточне-
нием технологических параметров и технико-экономических
показателей. Помещаемые методики обеспечивают полу-
чение продуктов реактивной квалификации (или особо чистых
препаратов) с выходом, как правило, не ниже 90%. Некото-
рые из предлагаемых методов могут быть использованы в ка-
честве типовых для разработки методик получения аналогич-
ных соединений (например, ионообменный синтез кислот, ва-
куум-термический синтез металлатных соединений, синтез
хлоридов переходных металлов, йодидов щелочных метал-
лов и др.).
Поскольку методики, включенные в сборник, предназна-
чены для воспроизведения в лабораторных масштабах и ус-
ловиях, описания синтезов, как правило, рассчитаны на один
или несколько циклов процесса и не включают всех данных,
необходимых для организации постоянных производственных
установок.
Сборник может представить интерес для широкого круга
химиков-производственников и исследователей, а также Для
специалистов, связанных с вопросами применения соединений
редких и цветных .металлов в народном хозяйстве.
СОДЕРЖАНИЕ
Ионообменное получение кислот из солей А. И. Вулих,
В. Л. Богатырев, В. А. Казьминская, Л. П. /Кероиенко .... 5
Метатитанаты щелочных металлов. С. А. Кутолин, А. И. Вулих 14
Метаниобаты щелочных металлов. С. А. Кутолин, А. И. Вулих,
А. Е. Шаммасова................................ . . 17
Метатанталаты щелочных металлов. С. А. Кутолин, А. И. Вулих,
А. Е, Шаммасова .................................... 21
Мапгаииты щелочных металлов. С. А. Кутолин, А. И. Вулих 24
Литий борогидрид. С. М Архипов,......................... 27
Литий тетрафторбориый. С. М. Архипов, П. Д. Комиссарова . . 31
Литий тетрафенилборат. Г. Е. Ревзин, А. И. Вулих, С. М. Архи-
пов, В. Д. Замедянская ............................. 33
Литий алюмогидрид. С. М, Архипов ............... 38
Карбид лития. М. Н. Короткевич, О. Н. Бреусов ...... • . . 43
Литий фенолят. Г. Е. Ревзин, В. Д. Замедянская.......... 45
Силикаты лития. А. А. Стаценко, М. Ф. Фокини............ 48
Литий цирконат. С А. Кутолин, А. И. Вулих............... 50
Литий нитрид. С. А, Кутолин, А. И. Вулих................ 52
Окись и безводная гидроокись лития. А. И. Вулих, М. И. Маковец-
кий, Л. Д Приходько................................. 54
Литий теллуристокислый. О. Н. Бреусов, Т. В. Ревзина...... Ь7
Литий феррит. С. А. Кутолин, А. И. Вулих................ 59
Рубидий борогидрид и цезий борогидрид. С. М. Архипов . . . . 61
Рубидий фенолят и цезий фенолят. Г. Е. Ревзин, В. Д. Замедян-
ская ............................................... 64
Рубидий и цезий мышьяковокислые однозамещенные особой чи-
стоты. Р. М. Шкловская, С. И. Романова.............. 67
Гидроокись цезия и гидроокись рубидия. П. Д. Комиссарова . . 70
Рубидий марганцовокислый и цезий марганцовокислый. /7. Д, Ко-
миссарова, С. А. Крестовникова .................... 72
Рубидий углекислый и цезий углекислый. А. И. Вулих, М. М. Се-
нявин, Б. И. Короткевич..............................74
Рубидий и цезий хлорновато-, бромновато- и йодноватокислые
особой чистоты. Р. М. Шкловская, А. И. Вулих ...... 77
Хроморубидиевые и хромоцезиевые квасцы. А. О. Лесовая, Н. И.
Кашина, В. А. Кузина........ ............ 85
Рубидий бромистый и цезнй бромистый, С. М. Архипов, А. И. Ву-
лих, Л. Г. Сидорова ................................ 88
Рубидий йодистый и цезий йодистый. С. М. Архипов, А И. Ву-
лих, Л. Г. Сидорова................................. 91
Цезий азотнокислый высокой чистоты. Р. М. Шкловская....... 94
Цезий фосфорноватистокислый. П. Д Комиссарова,.......... 97
Цезий железистосинеродистый. П. Ц. Комиссарова, С. А. Крес-
товникова .............. • • ....................... 99
Скандий оксигидрат. Ю. Б. Перковская.................. 101
3
Карбонаты редкоземельных элементов. Ю. Б. Перковская,
Н. П. Аношина, И. М Суханова..............................104
Сульфиды празеодима, неодима, самария. О. Н. Бреусов, М. Н. Ко-
роткевич..................._..............................110
Европий серпокислый закисный. Ю. Б. Перковская................112
Селенистокислые соли лантана, перш, празеодима и неодима.
ТО. Б. Перковская,........................................116
Селеновокислые соли редкоземельных элементов. ТО. Б. Перков-
ская, И. М. Суханова......................................120
Безводные хлориды редкоземельных элеменюв и скандия. Г. В. Рев-
зин ......................................................124
Галлий трехбромнетый. О. Н. Бреусов, В. Г. Лаврентьева .... 130
Индий хлорнокислый. И. М. Суханова. В. Д. Зимедянская .... 132
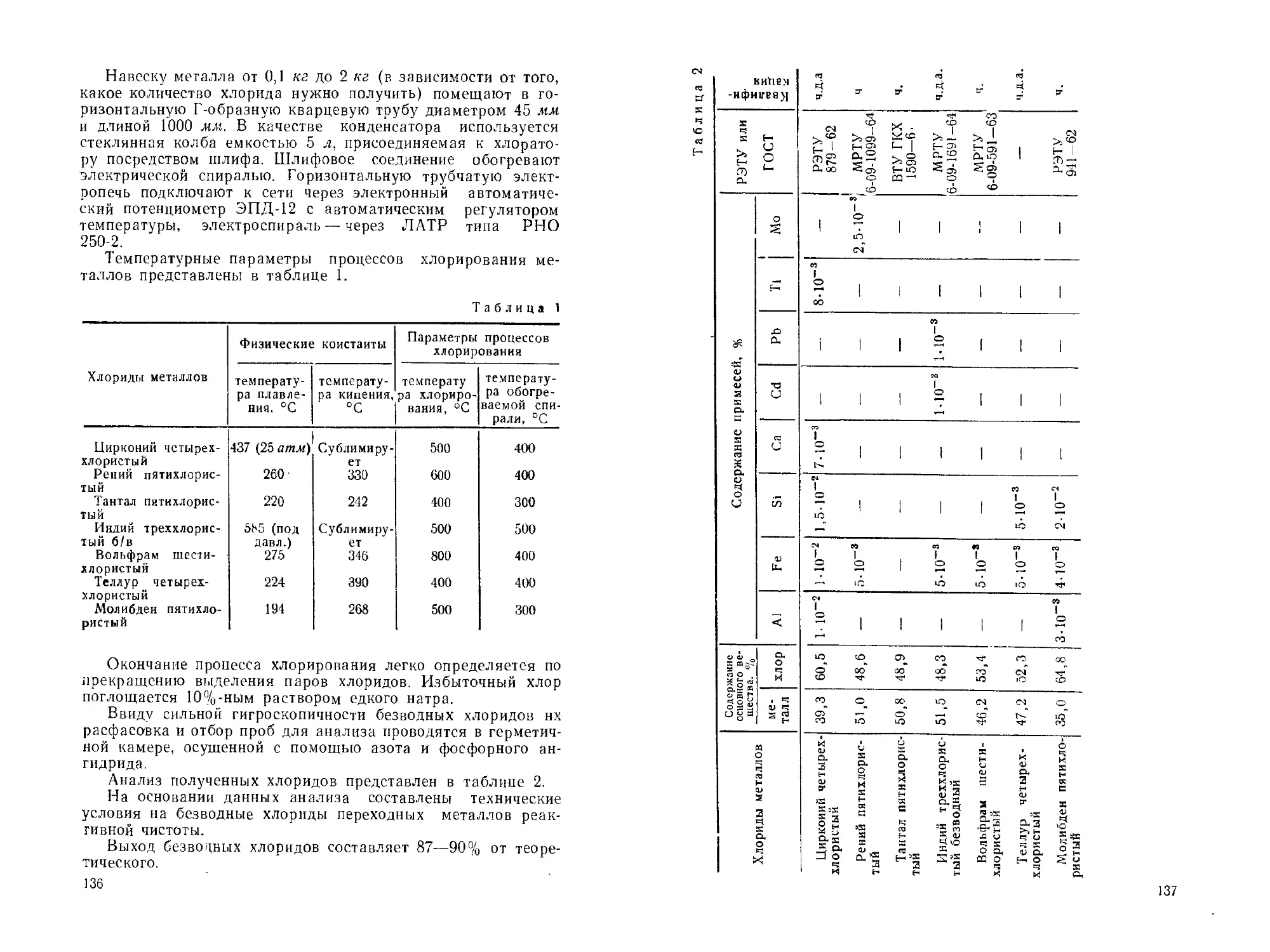
Безводные хлориды переходных металлов........................
Л. М. Петрова, О. Н. Бреусов..............................134
Германий дисульфид. О. Н. Бреусов, М. П. Короткевич..........139
Германий четыреххлорнстый. О. II. Бреусов, Л. М. Петрова ... 141
Германий четырехбромистый. О. Н. Бреусов, В. Г. Лаврентьева . . 143
Германий четырехйодистый. О. Н Биеусов, Л. М. Петрова ... 145
Свинец закись-окись, С. А. Кутолин, А. И. Вулих...............147
Ниобий треххлористый Л. М. Петрова, Б. Г. Коршунов...........149
Тантал четыреххлористый. Л. М. Петрова, Б. Г. Коршунов ... 151
Ниобий пятибромистый и тантал пятнбромистый. О. Н. Бреусов,
В. Г. Лаврентьева, А. И. Толкачев ........................154
Сурьма пятисернистая. И. А. Шигиенкова, Р. С. Бесяков, А. И. Ву-
лих ......................................................157
Калий-сурьма фтористая. О. Н. Бреусов, В. Г. Лаврентьева ... 160
Сурьма трехбромистая. Г. В. Ревзин, С. М. Архипов, В. Д Заме-
дянская ..................................................162
Сурьма трехйодистая. Г. Е. Ревзин, И. А. Шишенкова............165
Теллур двуокись. Г. Е. Ревзин.................................168
Теллуровая кислота. Г. Е. Ревзин.............................172
Молибден трехбромнетый. В. Г. Лаврентьева, О. Н. Бреусов ... 177
Рений семисернистый. О. Н. Бреусов, В. Г. Лаврентьева .... 180
Кобальт закись. А. И. Вулих, Д. А. Пахомов, С. А. Кутолин . . 182
Алфавитный перечень соединений, описанных в настоящем вы-
пуске ....................................................184
УДК 541.452:546.05:541.183.12:541.48
ИОНООБМЕННОЕ ПОЛУЧЕНИЕ КИСЛОТ ИЗ СОЛЕЙ
(Типовой метод)
А. И. ВУЛИХ, В. Л. БОГАТЫРЕВ, В. А. КАЗЬМИНСКАЯ,
Л. И. /КЕРДИЕНКО
НВг Бромистоводородная кислота (раствор) М.в. 80,92
HJ Йодистоводородная кислота (раствор) М.в. 127,91
НС1О3 Хлорноватая кислота (раствор) М.в. 84.45
НВгОд Бромноватая кислота (раствор) М.в. 128,'Ч
HJO3 Йодноватая кислота М.в. 175,91
HJO4 Йодная кислота (раствор) М.в. 191,90
HCNS Роданистоводородная кислота (раствор) М.в. 59.08
HReO4 Рениевая кислота (раствор) М.в. 251,20
H3AsO4 Мышьяковая кислота (раствор) М.в. 141,94
НаТеО4-2Н2О Теллуровая кислота М.в. 229,66
Синтез чистых кислот из солей химическими методами
представляет в каждом случае особый процесс, включающий
ряд более или менее сложных операций: осаждение, фильтра-
ция, выпаривание, дистилляция. Получаемая кислота даже
при использовании весьма чистых исходных реактивов обыч-
но требует дополнительной очистки От исходной соли, избыт-
ка реагента, побочных продуктов реакции, что осложняет
процесс и уменьшает выход чистой кислоты.
Поэтому в препаративной практике целесообразно исполь-
зовать универсальный и эффективный ионообменный метод
получения кислот [1, 2].
ОБЩАЯ СХЕМА СИНТЕЗА
Основной процесс:
RH+AX RA-ЬНХ
5
Регенерация ионита:
RA + HY — RH+AY
(R — радикал функциональной группы катионита, А —
катион исходной соли, X — анион исходной соли и получае-
мой кислоты, Y—анион регенерирующей кислоты).
Общее описание методики синтеза
Установка для ионообменного получения кислот представ-
лена на рис. 1.
Ионообменная колонка (У)
представляет собой трубу
(стеклянную, винипластовую,
металлическую гуммирован-
ную - в зависимости от необ-
ходимой производительности
и соответственно от размеров
колонки). Отношение длины
колонки к ее диаметру дол-
жно быть не менее 20 при
диаметре до 50 мм и не ме-
нее 10 при диаметре до
200 мм. В нижнем конце ко-
лонки закрепляется решетка
из кислотостойкого материа-
ла, на которую укладывается
редкая фильтровальная ткань,
например из стекловолокна,
или стеклянная цата. Оба кон-
ца колонки запираются флан-
цами, например из винипласта,
с патрубками для присоедине-
Рис. 1. Установка для ионообмеп- ния резиновых или пластмассо-
вого получения кислот вых трубок (см. примеча-
ние 1). Верхний конец
колонки соединяется с напорным бачком (2), а нижний — со
сливной Л-образной трубкой (3) с перегибом на уровне верх-
него края колонки (во избежание осушения смолы и попада-
ния в ее слой воздуха, создающего «пробки»). Свободный ко-
нец Л-образной трубки заканчивается краном (4), с помо-
щью которого можно регулировать скорость прохождения
раствора через слой ионита. Напорный сосуд (2) размеща-
ется на 1 —1,5 м выше верхнего края колонки.
В качестве ионита наиболее целесообразно использовать
стандартный катионит КУ-2 (МРТУ 6-05-903—65), предпоч-
тительно фракцию 0,5—1 мм. Перед загрузкой в колонку ио-
нит выдерживают в воде для набухания в течение 6—8 ча-
сов. Загрузку рассчитывают, исходя из насыпного веса на-
6
бухшего ионита 0,5 кг/л и коэффициента загрузки 0,7 (при
переходе из исходной Na-формы в Н-форму ионит увеличи-
вается в объеме на 15—20%). Предварительная подготовка
ионита заключается [3] в его промывке 5%-ным раствором
едкого натра (до исчезновения окраски фильтрата), водой,
10°/о-ной серной кислотой (до отсутствия качественной реак-
ции на железо с роданистым аммонием), дистиллированной
водой (до отсутствия реакции на хлор с азотнокислым сереб-
ром), этиловым спиртом (до исчезновения окраски фильтра-
та) и вновь дистиллированной водой (до отсутствия запаха
спирта от фильтрата). В результате этой процедуры катио-
нит полностью переводится в водородную форму и очищается
от примесей до уровня, позволяющего получать кислоты ре-
активной чистоты до квалификации «х. ч.» включительно.
Количество кислоты (в расчете на безводную), которое
теоретически возможно получить за 1 цикл работы ионооб-
менной установки, определяется формулой
Q = MEG* Г
где М — эквивалентный вес кислоты;
Е — емкость сухого катионита в Н-форме, г-экв/кг;
G—загрузка катионита, кг;
а — отношение веса сухого катионита в Н-форме к рас-
четному весу ионита.
Если нет необходимости точно определять емкость загру-
женной колонки, можно принять Е = 5 и а-=0,6, что соответ-
ствует 3 г-экв кислоты на 1 кг загруженного катионита или
1,5 г-экв кислоты на 1 дм3 объема колонки.
Получение кислоты из хорошо растворимой соли. Приго-
товляют 1—2 н. раствор соли, заливают в напорный бачок и
пропускают через колонку со скоростью 2—5 л/час на 1 кг
ионита, собирая порциями выходящий из колонки фильтрат.
Состав фильтрата в последовательных его порциях (выход-
ная кривая) представлен на рис. 2. Вначале из колонки и
Рис. 2. Типичная выходная кривая при ионообменном получении киелоты
(НХ) нз соли (АХ):
V — объем фильтрата; С — концентрация
7
Л-образной трубки вытесняется большая часть содержащей-
ся в них воды (без кислоты), затем в фильтрате появляется
кислота, концентрация которой быстро нарастает, достигая
(в г-экв) концентрации исходного раствора соли. В некото-
рый момент процесса в фильтрате появляется катион исход-
ной соли, так называемый «проскок», после чего концентра-
ция соли в фильтрате увеличивается, а кислоты — соответст-
венно уменьшается. При получении чистых кислот с появле-
нием «проскока» процесс прерывается и колонка переводится
на регенерацию. Поскольку до «проскока» используется не
вся обменная емкость ионита, практически получаемое коли-
чество кислоты меньше теоретического. При прочих равных
условиях выход чистой кислоты по отношению к полной ем-
кости ионита зависит от относительной сорбируемости катио-
на соли. Используя соли бария, можно получить для силь-
ных кислот выход >90%, соли кальция — 80—90%, соли ка-
лия— 70—80%, соли аммония — 60—70%, соли натрия —
50%. С другой стороны, уменьшение степени диссоциации
кислоты способствует вытеснению Н+ из ионита и повышает
выход чистой кислоты (в первом приближении неиспользо-
ванная емкость ионита пропорциональна степени ионизации
кислоты).
Выход чистой кислоты по отношению к исходной соли во
всех случаях тем ближе к 100%, чем более точно зафиксиро-
вано появление «проскока». Для этого при приближении к
моменту «проскока» (ориентировочно рассчитанному по объ-
ему фильтрата) отбирают сравнительно небольшие порции
фильтрата и качественно опробуют на присутствие катиона
соли.
При получении разовых относительно небольших партий
кислот проще использовать загрузку ионита, заведомо не-
сколько большую, чем необходимо для количественного пре-
вращения соли в кислоту. Это позволяет исключить контроль
за «проскоком». Процесс при этом сводится к пропусканию
раствора, содержащего рассчитанное количество соли, через
колонку и промывке колонки небольшим объемом воды для
извлечения оставшейся в ней кислоты. Фильтрат представ-
ляет собой раствор чистой кислоты, концентрация которого
несколько ниже, чем концентрация исходного раствора соли.
Получение кислоты из малорастворимой соли. В сосуд с
механической мешалкой загружают рассчитанные количест-
ва соли и катионита в Н-форме, подготовленного таким же
образом, как и катионит в колонке, и заливают воду. Коли-
чество ионита в г-экв обменной емкости обычно должно на
10—50% превышать количество соли, выраженное также в
г-экв, а объем воды рассчитывается на получение концентра-
ции 1—2 г-экв/л. Суспензию перемешивают до полного рас-
творения соли, затем раствор отделяют от ионита фильтра-
8
цией и промывают ионит водой для вытеснения оставшегося
в нем раствора. Объединенные фильтраты, содержащие не-
которое количество катиона металла, пропускают для отде-
ления последнего через колонку с катионитом в Н-форме и
получают чистую кислоту [4] (см. примечания 2, 3).
Регенерация ионита. Для регенерации используется сер-
ная или соляная кислота, квалификация которых определяет-
ся качеством синтезируемой кислоты. Регенерацию ведут
0,5—1,0 н. раствором серной или соляной кислоты, пропуская
ее со скоростью 5—10 л/час на 1 кг ионита. Пропускание ки-
слоты проводят до качественного отсутствия вытесняемого
катиона. Расход кислоты на регенерацию зависит главным
образом от природы десорбируемого катиона и составляет
(ка/ка катионита КУ-2): при вытеснении Na~0,8 кг,
К~1,2 кг, Са~2 кг (соляная кислота). Практический рас-
ход кислоты может быть уменьшен в 2 раза и более при
возвращении на регенерацию в следующем цикле последних
фракций кислоты, содержащих незначительное количество
катиона металла. В этом случае чистая кислота используется
лишь для заключительной стадии регенерации. Продолжи-
тельность регенерации от 1 до 4 часов. По окончании реге-
нерации катионит промывается дистиллированной водой до
отсутствия кислотности в фильтрате (по метилоранжу) или
до качественного отсутствия аниона SO42- или СП, после
чего может быть вновь использован для синтеза кислот.
Характеристика основного сырья
Калий бромистый, ч. или ч.д.а., ГОСТ 4160—48.
Калий йодистый, ч., ч.д.а. или х. ч., ГОСТ 4232—48.
Калий хлорноватокислый (бертолетова соль), техничес-
кий, высшего сорта, ГОСТ 2713—49.
Калий бромноватокислый, ч., ч.д.а или х.ч., ГОСТ 4457—
48.
Калий йодноватокислый, ч., ч.д.а. или х. ч., ГОСТ 4202 -
48.
Калий йоднокислый, ч. или ч.д.а., ВТУ МХП 3305—54.
Калий роданистый, ч., ч.д.а. или х.ч., ГОСТ 4139—48.
Калий рениевокислый, ч., ВТУ МГ УХП 201—58.
Кальций мышьяковокислый, ч., ВТУ ГКХ РУ 1864—62.
Условия получения отдельных кислот
Бромистоводородная кислота [6]. Для получения 1 кг
40%-ной бромистоводородной кислоты (ГОСТ 2062—43) рас-
творяют 620 г бромистого калия в 5 л воды. Полученный 1 н.
раствор бромистого калия пропускают через колонку, содер-
жащую ~1,5 кг катионита КУ-2 (здесь и далее в расчете на
9
сухой ионит в Н-форме), со скоростью 60 мл/мин. Затем Смо-
лу промывают 1 л дистиллированной воды, промывную воду
присоединяют к общему раствору и выпаривают до плотности
1,37 г/см3 (при 20°), получая 730 мл раствора 40%-ной кис-
лоты. Выход составляет 95%, считая на бромистый калий.
Кислоту хранят в склянках из темного стекла.
Иодистоводородная кислота [6]. Для получения 1. кг
45%-ной йодистоводородной кислоты (по ГОСТ 4200—48)
растворяют 610 г йодистого калия в 3,5 л воды. Полученный
1 н. раствор йодистого калия пропускают через колонку,
содержащую ~1,0 кг катионита КУ-2, со скоростью
60мл/мин. Затем смолу промывают 1 л дистиллированной
воды. Получают ~5 л раствора, содержащего 90 г/л йодисто-
водородной кислоты. Выход составляет 95%, считая на йоди-
стый калий. Концентрирование раствора кислоты проводят
путем упаривания в вакууме (см. примечание 4).
Хлорноватая кислота. Для получения 1 кг 25%-ной хлор-
новатой кислоты (по МРТУ 6-09-1473—64) растворяют 400 г
хлорноватокислого калия при температуре 50—60° в 6,5 л
воды. Полученный ~0,5 н. раствор хлорноватокислого калия
фильтруют и пропускают через колонку с 1 кг катионита со
скоростью 60 мл/мин. (Чтобы исключить возможность кри-
сталлизации соли ® колонке, перед пропусканием раствора
хлорноватой кислоты через колонку пропускают горячую во-
ду, пока температура наружной поверхности колонки не до-
стигнет 50—60°). Затем смолу промывают 1 л дистиллиро-
ванной воды, растворы объединяют, выпаривают в вакууме
до плотности 1,17 г/см3 (при 20°) и получают 850 мл раство-
ра 25%-ной хлорноватой кислоты, что соответствует выходу
90%, считая на хлорноватокислый калий. Хранят хлорнова-
тую кислоту в склянках из темного стекла.
Бромноватая кислота [7]. Для получения 1 кг 10%-ной
бромноватой кислоты 140 г бромноватокислого калия раство-
ряют при температуре 50—60° в 1,5 л воды и пропускают че-
рез колонку с 250 г катионита со скоростью 10 мл/мин. За-
тем смолу промывают 200 мл дистиллированной воды, рас-
творы объединяют и упаривают в вакууме до половины пер-
воначального объема. Выход бромноватой кислоты составля-
ет 90%> считая на бромноватокислый калий. Ввиду малой
устойчивости кислоту используют сразу же после ее получе-
ния.
Йодноватая кислота [7]. Для получения 1 кг 99,5%-ной
йодноватой кислоты (по ГОСТ 4213—48) 1,35 кг йодата ка-
лия растворяют при 60—70° в 10 л воды. Полученный 0,5 н.
раствор пропускают через колонку с 1,5 кг катионита со
скоростью 60 мл/мин. (Через колонку предварительно про-
пускают горячую воду до достижения температуры наружной
стенки 50—60°.) Затем смолу промывают ~ 1 л дистиллиро-
10
ванной воды и объединенный раствор упаривают в вакууме
(не выше 100°) до кристаллизации. Окончательно высушива-
ние образовавшейся массы, содержащей ~80% йодноватой
кислоты, производят в вакууме при 70- 80° или в эксикато-
ре с концентрированной серной кислотой.
Выход йодноватой кислоты составляет 90%, считая на
йодат калия.
Хранят кислоту в защищенном от света месте.
Иодная кислота [4, 8]. Для получения 1 кг 20%-ной йод-
ной кислоты готовят смесь из 280 г перйодата калия, 500 г
катионита КУ-2 в Н-форме и 2 л воды. Смесь нагревают до
40—50' и перемешивают в течение часа до полного раство-
рения соли. В случае, если осадок перйодата калия раство-
рился не полностью, приливают еще 500 мл воды и снова
перемешивают один час. После полного растворения осадка
катионит отделяют от раствора фильтрованием и промыва-
ют на фильтре 500 мл воды. Основной раствор и промывные
воды объединяют и пропускают через колонку с 200 г катио-
нита КУ-2 в Н-форме. Полученный раствор (~3 л) йодной
кислоты упаривают до плотности 1,2 г/см3.
Выход 20%-ного раствора йодной кислоты равен —830мл,
что составляет 85%, считая на йоднокислый калий.
Хранят кислоту в защищенном от света месте. Для полу-
чения кристаллической кислоты (HsJOg) раствор обрабаты-
вают по методике [8].
Роданистоводородная кислота. Для получения 1 кг 5%-
ной роданистоводородной кислоты (более концентрированные
водные растворы неустойчивы) растворяют 90 г роданистого
калия в 1 л воды, получая ~1 н. раствор вещества. Раствор
пропускают через колонку, содержащую 300 г катионита, со
скоростью 20 мл1мин. Затем катионит промывают 200 мл ди-
стиллированной воды, присоединяя промывную воду к ос-
новному фильтрату. Полученный раствор роданистоводород-
ной кислоты используется сразу же после его получения.
Выход составляет 90%, считая на роданистый калий.
Рениевая кислота. Для получения 1 кг 30%-ной рениевой
кислоты готовят смесь из 390 г перрената калия, 1 кг катио-
нита и 5 .г воды. Смесь нагревают до 40—50° и перемешива-
ют в течение часа. Полученную кислоту декантируют. Если
осадок рениевокислого калия полностью не растворился, то
добавляют еще 500 мл воды и вновь перемешивают 1 час.
После того, как осадок полностью растворится, катионит от-
фильтровывают и промывают на фильтре ~ 1,5 л воды. Ос-
новной фильтрат и промывную воду объединяют и пропус-
кают через колонку со 100 г катионита. Очищенную кислоту
(~7 л) упаривают до ‘Д первоначального объема и обраба-
тывают активированным углем в течение 30 минут. Затем
И
кислоту отфильтровывают и упаривают на водяной бане до
плотности ^1,25 г/см?.
Выход 30%-ной рениевой кислоты ранен 800 мл, что сос-
тавляет 90%, считая на ренисвокислый калий.
Мышьяковая кислота. Для получения 1 кг 25%-ной
мышьяковой кислоты готовят смесь из 480 г кальция мышья-
ковокислого, 1,5 кг катионита, 4 л воды и перемешивают в
течение часа. Полученную кислоту' декантируют. Если оса-
док арсената кальция растворился не полностью, добавляют
еще 1 л воды и вновь перемешивают 1 час. Затем катионит
отфильтровывают и промывают на воронке 1 л воды. Основ-
ной фильтрат и промывную воду объединяют и пропускают
через колонку со 100 г катионита. После этого кислоту
упаривают до плотности 1,19 г!см\
Выход 25%-ного раствора мышьяковой кислоты 840 мл,
что составляет 90%, считая на мышьяковокислый кальций
Ca3(AsO4)2 ЗН2О (см. примечание 5).
Теллуровая кислота. Для получения 1 кг 98,5%-ной тел-
луровой кислоты (по ТУ ГКХ РУ 1633—61) растворяют при
25—30° 1,65 кг теллуровокислого калия в 8 л воды, получая
1 н. раствор вещества. Раствор пропускают через колонку с
2 кг катионита со скоростью 60 мл!мин, затем промывают
ионит 2 л воды и фильтраты объединяют.
Выход 8—9%-иого раствора теллуровой кислоты равен
~ 10 л. Раствор выпаривают в вакууме (при 80—90°) до на-
чала кристаллизации, охлаждают до 10—20° и отфильтровы-
вают кристаллы Н2ТеО4 -2Н2О. Маточный раствор также упа-
ривают и повторно кристаллизуют. Кристаллическую теллу-
ровую кислоту высушивают в вакууме при 60—70°.
Общий выход кислоты равен 1 кг, что составляет 95%,
считая на теллуровокислый калий (К2ТеО4 5Н2О).
Примечания:
1. В стеклянных колонках диаметром до 30 мм иижний конец оття-
гивается до внутреннего диаметра 25 мм, а верхнее отверстие запира-
ется резиновой пробкой со стеклянной трубкой. Может быть использова-
на также стеклянная бюретка.
2. Если соль малорастворима при обычной температуре, но се раство-
римость существенно увеличивается при повышении температуры, целе-
сообразно непосредственно пропускать через колонку с ионитом горячий
раствор соли. При этом либо выбирают температуру и концентрацию
раствора 'так, чтобы при его охлаждении во время движения в колонке
кристаллизации соли ие происходило, либо теплоизолируют колонку. По-
вышение температуры является также эффективным приемом ускорения
конверсии малорастворимой соли в кислоту при перемешивании с иони-
том в водной среде.
3. Сочетание предварительного контактирования раствора соли с
ионитом (статический процесс) и последующей очистки полученного рав-
новесного раствора от катиона соли на колонке (динамически^ процесс)
может быть использовано также для повышения концентрации получае-
мой кислоты при использовании высокорастворимой соли [5].
12
4. Все операции проводят, обеспечивая защиту растворов йодистово-
дородной кислоты от света и воздуха (стеклянную колонку обертывают
темной бумагой и т. д.). Хранят кислоту в сосудах из темного стекла,
обернутых в черную бумагу, и в темном месте.
5. Так как соли мышьяка относятся к сильнодействующим ядам, при
получении мышьяковой кислоты следует соблюдать все меры предосто-
рожности, предусмотренные при работе с ядами.
ЛИТЕРАТУРА
1. А. И. В у л и х, В. А. Казьм инская, М. М. С е и я в и н , Авт,
свил. 145891; Бюлл. изобр., АГ» 7, 15 (1962).
2. М. М. С е и я в и н, А. И. Вулих, В, Л. Богатырев, Сб.
«Ионообменная технология», М„ «Наука», 1965, стр. 67.
3. К- М. С а л д а д з е, А. Б. Пашков, В. С. Титов. Ионооб-
менные высокомолекулярные соединения, М., Госхимиздат, 1960, стр. 40,
4. А. И. Вулих, В. А. Ка з ь м и н с к а я, Л. П. Жердиенко.
Промышленность химических реактивов. Информационный бюллетень,
вып. 2, М., ПРЕЛ, 1963, стр. 7.
5. А. И. Вулих, В. Л. Богатырев. Изв. Сибирского отд. АН
СССР, Сер. химич., выпуск 2,40 (1963); Промышленность химических ре-
активов, Информационный бюллетень, вып. 2, М., ИРЕА, 1963, стр. 14.
6. А. И. Вулих, В. Л. Богатырев. Изв. Сибирского отд. АН
СССР, № 8, 53 (1962).
7. В. Л. Богатырев, А. И. Вулих. Ж. прнкл. химии, 36, 220
(1963).
8. Руководство по препаративной неорганической химии. Под ред.
Г. Брауэра, М„ ИЛ, 1956, стр. 172.
Поступила в марте 1966 г.
ЦЗЛ
УДК 546.824'311.07
МЕТАТИТАНАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
С. А. КУТОЛИН, А. И. ВУЛИХ
LisTiO3 Литий титанат (мета) М.в. 109,77
Na2TiO3 Натрий титанат (мета) М.в. 141,88
KsTi()s Калий титанат (мета) М.в. 174,10
RbaT103 Рубидий титанат (мета) М.в. 266,84
CsjTiOg Цезий титанат (мета) М.в. 361,70
Титанаты щелочных металлов (титановокислые соли) на-
ходят применение в керамической и стекольной промышлен-
ности, электро- и радиотехнике [1—3].
Соединения этой группы представляют собой белые кри-
сталлические вещества с высокими показателями преломле-
ния [4]. В литературе описаны методы синтеза метатитанатов
щелочных металлов гидрохимическим путем из гидроокисей
металлов и метатитановой кислоты или путем сплавления
(при 1000° и выше) карбонатов или гидроокисей с двуокисью
титана [5—9].
Нами разработан метод синтеза титанатов щелочных ме-
таллов путем спекания при относительно низких температу-
рах, что оказывается возможным при применении вакуума
1Ю, 11].
СХЕМА СИНТЕЗА МЕТАТИТАНАТОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
2L1OH + Т1О3 + Li2TiO3 + Н2О
Na2CO3 Ч- TiO» - > Na3TiO3 + CO.
К2СОз + TiO2 - > K2T1O3 + CO;
Rb2COs + TIO, - » Rb2TiO8 + CO.
CSjCOg 4- TiO2 - Cs2TiO3 -|- CO;
14
Характеристика основного сырья
Литий едкий (моногидрат), ч. или ч.д.а., ЦМТУ 2062—48.
Натрий углекислый, безводный, ч.д.а., ГОСТ 83—41.
Калий углекислый, ч. или ч.д.а., ГОСТ 4221—57.
Рубидий углекислый, ч., МРТУ 6-09-227—63.
Цезии углекислый, ч. или ч.д.а., МРТУ 6-09-1721—64.
Титан двуокись, ч. или ч.д.а., ТУ МХП 3052—55.
Условия получения
Аппаратура для проведения процесса спекания в вакууме
описана в методике получения манганитов щелочных метал-
лов (стр. 24).
Литий метатитанат. Тщательно перемешивают 83 г. едко-
го лития (с содержанием 53% LiOH) с 73 г двуокиси тита-
на. Смесь загружают в корундовый или фарфоровый тигель,
который помещают в вакуумную печь и выдерживают в те-
чение 1 часа при 650° и атмосферном давлении, затем пони-
жают давление до 0,5—1 мм рт. ст. и выдерживают при той
же температуре в течение 2 часов. Полученный белый спек
измельчают.
Выход лития метатитаната равен 100 г, что составляет
98% от теоретического; соотношение Li2O:TiO2= 1 ±0,02.
По внешнему виду продукт представляет собой белый по-
рошок; его плотность 3,415 г/см3, показатель преломления
2,087; т. пл. 1320°; диэлектрическая постоянная 18,2; кристал-
лы кубической структуры с параметром ячейки а = 8,28 А.
Натрий метатитанат. Тщательно перемешивают 78,2 г
карбоната натрия с 58,9 г двуокиси титана. Шихту загружа-
ют в корундовый тигель и помещают в вакуумную печь. Тем-
пературу печи постепенно, в течение 1 часа, повышают до
850°, одновременно понижая давление до 1 мм. В этих усло-
виях выдерживают смесь в течение 3 часов. Спек измельча-
ют до получения белого порошка.
Выход метатитаната натрия равен 100 г, что соответству-
ет 98% от теоретического; соотношение Na2O:TiO2=
= 1,00 + 0,02.
Физические свойства продукта: плотность 3,20 г/см3; т. пл.
1025°; показатель преломления 1,804; диэлектрическая посто-
янная 12,2; кубические кристаллы (а = 7,60 А).
Калий метатитанат. Тщательно смешивают 82 г карбона-
та калия с 47,5 г двуокиси титана. Шихту загружают в ко-
рундовый тигель и помещают в вакуумную печь. Температу-
ру печи постепенно, >в течение 1 часа, повышают до 800°, од-
новременно понижая давление до 1 мм. В этих условиях вы-
держивают смесь в течение 2 часов. Спек измельчают до по-
лучения белого порошка.
Выход метатитаната калия равен 100 г, что составляет
98% от теоретического; соотношение К20:Т102=1,00±0,02.
Физические свойства продукта: плотность 3,58 г/с.и3; т. пл.
820°; показатель преломления 1,910; диэлектрическая посто-
янная 16,5; тетрагональные кристаллы с параметрами ячей-
ки: а=5,47 А, с = 16,6 А.
Рубидий метатитанат. Перемешивают 90 г карбоната ру-
бидия с 31,4 г двуокиси титана. Приготовленную смесь в ко-
рундовом или фарфоровом тигле помещают в вакуумную
печь. Температуру постепенно, в течение 1 часа, поднимают
до 600°, одновременно понижая давление до 1 мм. В этих ус-
ловиях дают выдержку в течение 2—2,5 часа; спек измельча-
ют до получения белого порошка.
Выход рубидия метатитаната равен 100 г, что составляет
97%, от теоретического; соотношение Rb2O:TiC>2=l,00 + 0,02.
Физические свойства продукта; плотность 3,01 г/сл13;т. пл.
755°; показатель преломления 1,818; диэлектрическая посто-
янная 13,5; тетрагональные кристаллы (а = 5,26 А, с=31,5А).
Цезий метатитанат. Перемешивают 93 г карбоната цезия
с 23 г двуокиси титана. Приготовленную смесь в корундовом
или фарфоровом тигле помещают в вакуумную печь. Темпе-
ратуру постепенно, в течение 1 часа, повышают до 650°, од-
новременно понижая давление до 1 мм. В этих условиях да-
ют выдержку в течение 2 часов, спек измельчают до получе-
ния белого порошка.
Выход цезня метатитаната равен 100 г, что составляет
97% от теоретического; соотношение CsaO:TiO2= 1 ±0,02.
Физические свойства продукта: плотность 3,39 г/см3, т. пл.
710°; показатель преломления 1,812; диэлектрическая посто-
янная 8,8; вещество рентгеноаморфно.
ЛИТЕРАТУРА
1. О. Lang. Z. anorgan. und allgem. Chem., 276, 77 (1954).
2. J, Schenk. Nucleonics, 10, 54 (1952).
3. P. Hup pert. Ceramic. Ind., 65, 70 (1955).
4. Gmelins Handbuch der anorg. Chemie, 8. Aufl., SN 41 (1951).
5. E. Kordes. Z, Kristallogr., 92, 139 (1935).
6. H. Remy. Lehrbucb der anorg. Chemie, В. II, 10. Aufl., Leipzig,
1959, S. 80.
7. F. Barblan, F. Brandenberger, P. Niggli. Helv. chim.
acta, 27, 88 (1944).
8. Франц, пат. 1108062 (1956).
9. Канад, пат. 363010 (1936).
10. С. А. Кчтолии, А. И. Вулих. Авт. свид. 157967; Бюлл изобр.,
№ 20 (1963).
II. С. А. Кутолин, А. И. Вулих. Ж. иеорган. химии, 10, 140
(1965).
Поступила в декабре 1965 г. ЦЗЛ
16
УДК 546.882.5'311.07
МЕТАНИОБАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
С. А. КУТОЛИН, А. И. ВУЛИХ, А. Е. ШАММАСОВА
LiNbO3 Литий ниобат (мета)
NaNbO3 Натрий ниобат (мета)
KNbO3 Калий ниобат (мета)
RbNbO3 Рубидий ниобат (мета)
CsNbOg Цезий ниобат (мета)
М.в. 147,84
М.в. 163,89
М.в. 180,00
М.в. 226,37
М.в. 273,80
Ниобаты щелочных металлов находят применение в ка-
честве сегнетоэлектриков [1—3].
В литературе описываются методы получения метаниоба-
тов путем сплавления или длительного спекания соответству-
ющих карбонатов с пятиокисью ниобия, иногда в присутствии
минерализаторов, в серебряных или платиновых тиглях при
высокой температуре. Существуют также гидрохимические
методы синтеза метаниобатов щелочных металлов с прока-
ливанием на заключительной стадии [4—9]. Эти методы дли-
тельны и неэкономичны для применения их в промышленно-
сти химических реактивов.
Нами разработана методика количественного синтеза ме-
таниобатов щелочных металлов путем спекания карбонатов
щелочных металлов с пятиокисью ниобия в вакууме без при-
менения минерализаторов при сравнительно низкой темпера-
туре [10, И].
СХЕМА СИНТЕЗА МАНГАНИТОВ ЛИТИЯ, НАТРИЯ, КАЛИЯ
РУБИДИЯ И ЦЕЗИЯ
LiaCO3 + Nb2O5 -> 2LiNbO3 Ц- СОа
Na2CO3 + NbaO6 -> 2NaNbO3 + СО2
KSCO3 +Nb2O6 -э- 2KNbO3 + СО2
Rb,CO3 + NbaO6 > 2 RbNbO3 Ц- CO2
Cs2CO3 + NbaO6 -» 2 CsNbO3 + COa
17
Характеристика основного сырья
Литий углекислый, ч., ЦМТУ 2006—48.
Натрий углекислый, ч.д.а., ГОСТ 83—41.
Калий углекислый, ч.д.а, ГОСТ 4221- 57.
Рубидий углекислый, ч., МРТУ 6-09-227—63.
Цезий углекислый, ч., МРТУ 6-09-1721—64.
Ниобий пятиокись, ч., РЭТУ 675—61.
Условия получения
Синтез всех соединений проводится в аппаратуре, опи-
санной в статье Метатанталаты щелочных металлов и поме-
щенной в настоящем сборнике.
Литий метаниобат. Тщательно смешивают 26,3 г карбо-
ната лития с 94,6 г пятиокиси ниобия. Приготовленную смесь
в фарфоровом сосуде помещают в вакуум-электрическую
печь и выдерживают при 750°/1 мм рт. ст. ® течение 2—2,5
часа. Полученный спек измельчают.
Выход лития метаниобата равен 100 г, что соответствует
97% от теоретического; плотность вещества 4,28 г/сж3; т. пл.
1140°: показатель преломления — 2,08. Соотношение
l,i20:Nb206=l±0,05.
По внешнему виду метаниобат лития представляет собой
белое кристаллическое вещество, нерастворимое в воде. Кри-
сталлы гексагональные с параметрами ячейки: а=5,15 А,
с= 13,82 А.
Натрий метаниобат. Тщательно смешивают 33,5 г углекис-
лого натрия с 84,4 г пятиокиси ниобия. Шихту в фарфоровом
тигле помещают в горизонтальную вакуум-электрическую
печь н выдерживают при температуре 800°/1 мм в течение
2,5—3 часов. Спек измельчают в фарфоровой ступке или
мельнице.
Выход натрия метаниобата равен 100 г, что составляет
98% от теоретического. Соотношение Na2O:Nb2O5= 1 ±0,05.
Метаниобат натрия представляет собой белый кристалли-
ческий порошок. В воде не. растворяется. Физические свойст-
ва; плотность 5,33 г/слт3; т. пл. 1300°; показатель преломле-
ния 2,21. Кристаллы орторомбические (а=Ь = 5,6 А;
с=16,0 А).
Калий метаниобат. Тщательно смешивают 39 г углекисло-
го калия с 77,7 а пятиокиси ниобия. Приготовленную шихту
в фарфоровой чашке загружают в вакуум-электрическую го-
ризонтальную печь и выдерживают при 700°/1 мм в течение
2—2,5 часа. Полученный спек измельчают.
Выход калия метаниобата равен 100 г, что составляет
97% от теоретического. Соотношение K2O:Nb2Os=l ±0,05.
По внешнему виду метаниобат калия представляет собой
желтое (иногда серое с голубым оттенком) кристаллическое
18
вещество, нерастворимое в воде. Его плотность равна
4,53 г/сж3; т. пл. 1160°; показатель преломления 2,17. Кри-
сталлы орторомбические (а=5,70 А, 6 = 5,74 А; с = 3,98 А).
Рубидий метаниобат. Тщательно перемешивают 52,61 г
рубидия углекислого с 61,55 г пятиокиси ниобия. Шихту в
фарфоровой чашке помещают в горизонтальную вакуум-элек-
трическую печь и выдерживают при температуре 750°/1 мм в
течение 2,5—3 часов. Образовавшийся спек измельчают,
Выход рубидия метаниобата равен 100 г, что составляет
98% от теоретического. Соотношение Rb2O:Nb2O5= 1 ±0,05.
По внешнему виду метаниобат рубидия представляет со-
бой белый кристаллический порошок, нерастворимый в воде;
его плотность 2,81 г/сж3; т. пл. 940°; показатель преломления
1,995; кристаллы кубические (д=7,47 А).
Цезий метаниобат. Тщательно смешивают 62,3 г цезия уг-
лекислого с 51,31 г ниобия пятиокиси. Смесь в фарфоровом
сосуде помещают в горизонтальную вакуум-электрическую
печь и выдерживают при температуре 650 —700°/1 мм в те-
чение 3—3,5 часа. Спек измельчают,
Выход цезия метаниобата равен 100 г, что составляет
97% от теоретического. Соотношение Cs2O:Nb2Os= 1 ±0,05.
По внешнему виду метаниобат цезия представляет собой
белый порошок. Его плотность равна 4,22 г/см3; т. пл. 1250°;
показатель преломления 2,01. В воде не растворяется; рент-
геноаморфен.
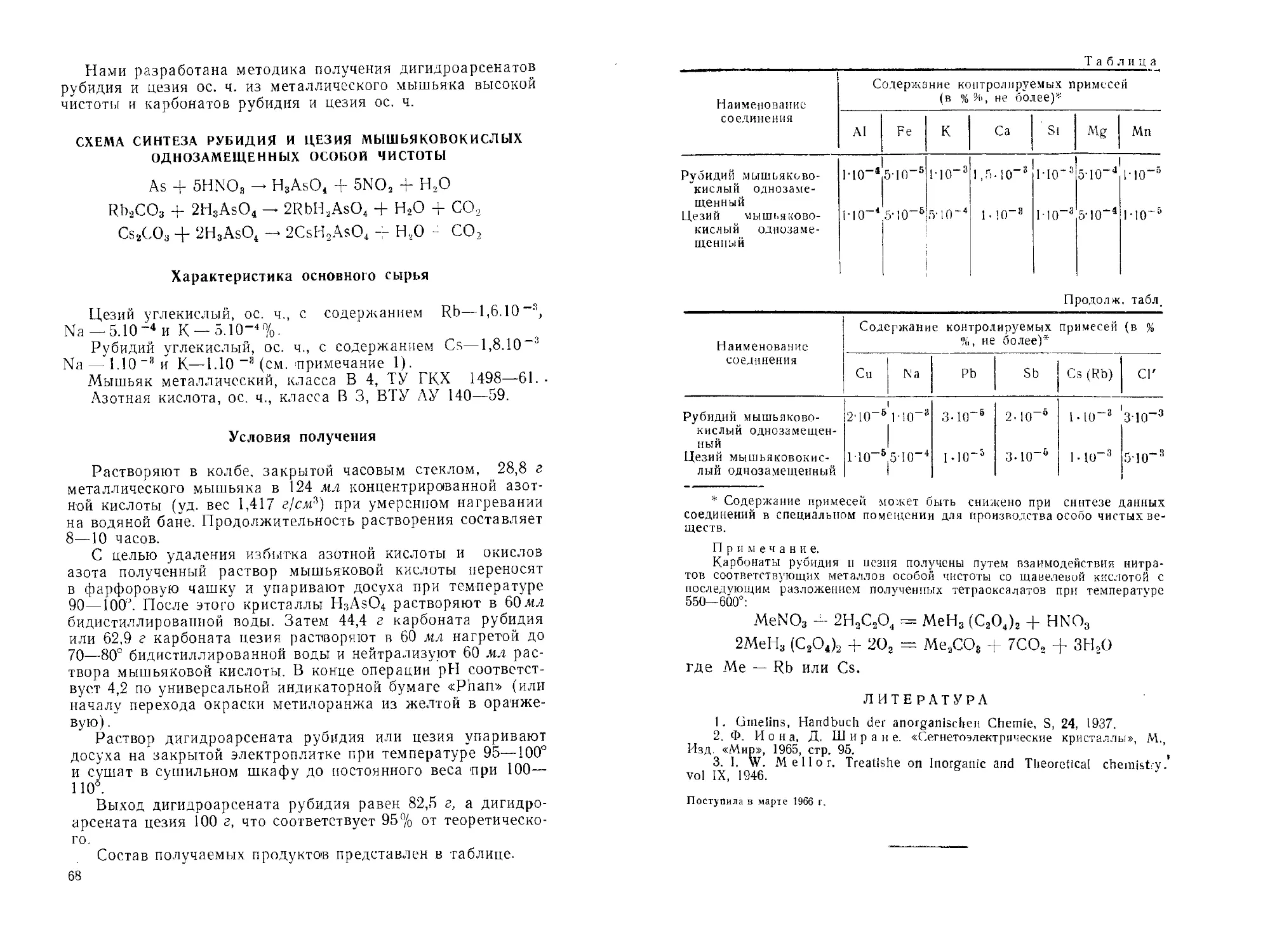
В нижеследующей таблице приведен химический состав
соединений, синтезированных по данной методике.
Состав метаниобатов щелочных металлов
Формула соединения Химический состав, %
Me2O ( + 1%) NbA (±1 %) Ta; Ti Fe; Мп; Zn Cu; Ni; Co
LlNbO3 10 90 <1 -IO-1 <5.IO*2 < 5.10“3
NaNbO3 19 81 каждой каждой каждой
KNbO, 26 74 примеси примеси примеси
RbNbO3 41 59
CsNbO3 51,5 48,5
ЛИТЕРАТУРА
I. Б. И К о г а и. Литий, области освоенного и возможного приме-
нения, М,, ВИНИТИ, I960, стр. 45.
2. V о 1 g е г. J. Research., 7, 6, 230 (1954).
3, И. Н. Беляев. Изв. АН СССР, Сер. физ,. 22, 12 (1958),
4, Руководство по препаративной неорганической химии, М., ИЛ.
1956, стр. 506.
19
5. J. Carriere, H. Gutter. Chem. Revs, 216, 568 (1943).
6. А. В. Л a и и цк и й, К). П. Симанов. Вести. Моск, ун-та, 2,69
(1954).
7. А. В. Лапицкий. Ж. общ. химии, 22, 38 (1952).
8. М. Л. Пчелкина, А. В. Лапицкий. Ж- общ. химии, 24, 1101
(1954).
9. А. В. Лапицкий, Ю. П. С и маков. Ж. физ. химии, 29, 1201
(1955).
10. С. А. Кутолин, А. И. Вулих, А. Е. Сергеева. Тезисы II
Всесоюзн. совещания по редким щелочным элементам, М.. изд. «Наука»,
1964, стр. 13.
11. С. А. Кутолин, А. И. Вулих, А. Е. Сергеева. Изв. АП
СССР, сер. Неорганнч. материалы, 1, 399 (1965).
Поступила в декабре 1965 г. ИЗЯ
УДК 546.883.5'311,07
МЕТАТАНТАЛАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
С. А. КУТОЛИН, А. И. ВУЛИХ, А. Е. ШАММАСОВА
LiTaO3 Литий танталат (мета) М.в. 235,89
NaTaO3 Натрий танталат (мета) М.в. 251,95
КТаО3 Калий тантала г (мета) М.в. 268,05
RbTaO3 Рубидий танталат (мета) М.в. 314,42
CsTaO3 Цезий танталат (мета) М.в. 361,85
Танталаты щелочных металлов могут найти применение в
качестве сегнетоэлектриков, как танталсодержащие микро-
удобрения и др. [1, 2].
В литературе описываются методы получения танталатов
путем нагревания соответствующих карбонатов с пятиоки-
сью тантала в платиновых сосудах при 800°. Этот процесс
весьма длителен; указывается, что он может быть ускорен
применением минерализаторов или нагреванием смеси в ва-
кууме, но конкретные условия не разработаны [1, 3—6].
Нами проверена и уточнена методика получения ряда ме-
татанталатов щелочных металлов путем спекания карбона-
тов щелочных металлов с пятиокисью тантала в вакууме.
СХЕМА СИНТЕЗА МЕТАТАНТАЛАТОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Ме2СО3 + Та2О5 -> 2МеТаО3 + СО2,
где Me = Li, Na, К, Rb, Cs.
Характеристика основного сырья
Литий углекислый, ч., ЦМТУ 2006—48.
Натрий углекислый, ч., ГОСТ 83—41.
Калий углекислый, ч„ ГОСТ 4221—57.
Рубидий углекислый, ч., МРТУ 6-09-227—63.
Цезий углекислый, ч., МРТУ 6-09-1721—64.
Тантал пятиокись, ч., ВТУ ГКХ 1589—61.
21
Условия получения
Для проведения синтезов используется горизонтальная
ретортная вакуум-электрическая печь с нихромовыми или си-
литовыми нагревателями. Реторта диаметром ~ 100 мм и
длиной ~600 мм изготавливается из нержавеющей стали.
Вакумм создается и поддерживается с помощью лаборатор-
ного вакуум-насоса, например ВН-461, и измеряется мано-
метром Мак-Леода. Температура поддерживается на задан-
ном уровне (с точностью ±20°) с помощью термопары и по-
тенциометра ЭПД-12.
Литий метатанталат. В фарфоровой ступке тщательно
смешивают 15,5 г карбоната лития с 95,1 г пятиокиси танта-
ла. Приготовленную смесь в фарфоровом тигле или чашке
загружают в вакуум-электрическую печь и выдерживают при
750“ и остаточном давлении около 1 мм рт. ст. в течение
2,5—-3 часов. Полученный спек измельчают.
Выход продукта равен 100 г, что составляет 97% от тео-
ретического; соотношение L 120:Та205=1,00±0,03. Метатан-
талат лития представляет собой белое кристаллическое веще-
ство, нерастворимое в воде; его плотность 7,1 г/см?, т. пл.
1325°.
Натрий метатанталат. Тщательно смешивают 21,2 г кар-
боната натрия с 88,5 г пятиокиси тантала. Приготовленную
шихту загружают в фарфоровый тигель и выдерживают в
вакууме при температуре 800° и остаточном давлении не бо-
лее 1 мм рт. ст. в течение 3—3,5 часа. Полученный спек из-
мельчают.
Выход продукта равен 100 г, что соответствует 98% от
теоретического; соотношение Na20:Ta205==l,00±0,03. Мета-
танталат натрия представляет собой белое кристаллическое
вещество, нерастворимое в воде; его плотность 7,08 efcM?,
т. пл. 1310°.
Калий метатанталат. Смешивают 26,2 г карбоната калия
с 83,7 г пятиокиси тантала. Смесь в фарфоровом тигле по-
мещают в вакуумную печь н выдерживают при 750° и оста-
точном давлении около 1 мм рт. ст. в течение 3—3,5 часа.
Спек измельчают.
Получают 100 г продукта, что составляет 98% от теорети-
ческого; соотношение К20:Та205=1,00±0,02.
Метатанталат калия представляет собой белое кристал-
лическое вещество; его плотность 5,43 г!смъ, т. пл. 1360°. В
воде труднорастворим, но медленно гидролизуется.
Рубидий метатанталат. Смешивают 38 г карбоната руби-
дия с 72 г пятиокиси тантала. Шихту загружают в фарфо-
ровый тигель, помещают в печь и выдерживают при темпе-
ратуре 750° в вакууме (остаточное давление не более 1 мм
рт. ст.) в течение 2,5—3 часов. Спек измельчают.
22
Получают 100 г продукта, что соответствует 98% от тео-
ретического выхода; соотношение Rb2O:Ta2O5= 1,00±0,02.
Рубидий метатанталат представляет собой мелкокристал-
лическое вещество белого цвета, нерастворимое в воде; его
плотность 4,32 а/см3, т. пл. 1320°.
Цезий метатанталат. Смешивают 48 г карбоната цезия и
62 г пятиокиси тантала. Смесь загружают в фарфоровый ти-
гель, помещают в вакуумную печь и выдерживают в тече-
ние 3 часов при 700° и остаточном давлении около 1 мм рт.
ст. Образовавшийся спек измельчают, получая 100 а про-
дукта, что составляет 98% от теоретического выхода; соот-
ношение Cs2O:Ta2O5= 1,00±0,02.
Метатанталат цезия — белое кристаллическое вещество;
его плотность 4,77 г/см3, т. пл. 1300°. В воде труднораство-
рим, но постепенно гидролизуется.
ЛИТЕРАТУРА
1. Я. Г. Горощенко. Химия ниобия п тантала, Киев, изд. «Нае-
кова думка», 1965, стр. 42, 179- 213.
2. D. Wai пег, С. Wentvorth. J. Amer. Ceram. Soc., 35, 8, 207
(1952).
3. L. Quill. Z. anorgan, und allgern, Chein., 208, 270 (1932).
4. M. А. Пчелкина, А. В. Лапицкий. Ж. общ. химии, 24, 110,5
(1954).
5. А. В. Лапицкий, 1О. В. Симанов. Ж. физ. химии, 29, 1201
(1955).
6. Руководство по препаративной неорганической химии, Под рец.
Г, Б pay ер а, М., ИЛ, 1950, стр. 608.
Поступила в декабре 1965 г.
цзл
УДК 546.714.311.07
МАНГАНИТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
С. А. КУТОЛИН, А. И. ВУЛИХ
ЫаМпО3 Литий манганит М.в. 116,81
Na2MnO3 Натрий манганит М.в. 148,91
К2МпОа Калий манганит М.в. 181,14
Манганиты щелочных металлов находят применение в ке-
рамической и анилино-красочной промышленности. Они мо-
гут также быть использованы как присадки в производстве
люминофоров, в качестве катализаторов [1, 2].
В литературе описан способ синтеза манганитов щелоч-
ных металлов путем сплавления карбонатов щелочного ме-
талла и марганца (11) при 1150—1300° в токе воздуха или
кислорода [3, 4], а для манганита калия — также сплавлени-
ем перманганата и хлорида калия при 1100’ [5]. Осуществле-
ние этих методов для получения чистых препаратов едва ли
возможно вследствие большой коррозионной активности ре-
акционной массы при высоких температурах.
Разработанный нами метод, основанный на взаимодейст-
вии карбонатов или гидроокисей щелочных металлов с дву-
окисью марганца в вакууме, позволяет получать манганиты
стехиометрического состава при температурах 550—770° [6].
СХЕМА СИНТЕЗА МЕТАНИОБАТОВ ЛИТИЯ, НАТРИЯ, КАЛИЯ,
2 L1OH + МпО2 -ч- Li3MnO3 + Н2О
Na2COs + MnO2 -> Na2MnO3+ СО2
К2СО3 МпО2 —> КаМпОа д- СО2
Характеристика основного сырья
Литий едкий, ч., ЦМТУ 2062—48.
Натрий углекислый, ч., ГОСТ 83—41.
Калий углекислый, ч., ГОСТ 4221—57.
Марганец перекись, ч., ГОСТ 4470—48.
24
Условия получения
Шихта, приготовленная тщательным смешением исход-
ных веществ, загружается в фарфоровый тигель, который по-
мешается в реторту вакуум-электрической печи. Вакуум соз-
дается лабораторным вакуум-насосом, например типа ВН-
461, и измеряется вакуум-метром А4ак-Леода. Температура
поддерживается на заданном уровне с помощью терморегу-
лятора (см. примечание 1).
Получение манганита лития. Шихту из 78 г едкого лития
(содержание LiOH 53%) и 104 г двуокиси марганца выдер-
живают в течение 3 часов при 550° и остаточном давлении
0,5—1 мм рт. ст. Измельчают образовавшийся спек и полу-
чают 100 г порошка (см. примечание 2) красно-коричневого
цвета, содержащего 99—99,5% лития манганита и не свыше
0,5—1% примесей исходных окислов; </25=3,007±0,005; т. пл.
1060°. Продукт представляет собой тетрагональные кристал-
лы с параметрами ячейки: о —4,82 А; с= 11,05 А. В воде ли-
тий манганит нерастворим, но постепенно гидролизуется.
Получение манганита натрия. Спекают шихту из 73 г кар-
боната натрия и 81 г двуокиси марганца в течение 3 часов
при 700’/1 льи. После измельчения спека получают 100 г про-
дукта в виде порошка черного цвета, содержащего 99% ман-
ганита натрия и не свыше 1% исходных окислов; </25 = 2,480 +
+ 0,005; т. пл. 1190°; тетрагональные кристаллы (а = 5,83 А;
с=11,70 А).
В воде манганит натрия растворяется с частичным гид-
ролизом.
Получение манганита калия. Смесь из 78 г карбоната ка-
лия и 66,5 г двуокиси марганца спекают при 700э/1 мм в те-
чение 3 часов. Измельчают спек и получают 100 г иссиня-
черного порошка, содержащего 98—99% манганита калия и
не свыше 1—2% исходных окислов; </25 = 3,172 ±0,005; т. пл.
1100°; тетрагональные кристаллы (а = 7,40 А; с= 11,70 А).
В воде манганит калия растворяется с частичным гидро-
лизом.
Примечания:
1. Для поглощения выделяющихся при реакции углекислоты и вла-
ги целесообразно в случае получения значительных количеств препара-
тов установить на линии между печью и вакуум-насосом ловушку с из-
мельченным едким кали.
2. Выход манганитов во всех случаях составляет 98—99% от теоре-
тического.
ЛИТЕРАТУРА
1. Б. И. Коган. Литий, области освоенного и возможного приме-
нения, М„ ВИНИТИ (1960).
2. Р. Huppert. Ceram. Ind., 65, 70 (1955).
25
3. H. Remy. Lehrbuch der anorganischen Chemie, В. II, 10. Anfl.,
Leipzig, 1959, 269.
4. Пат. США 2562705 (1951).
5. A. S. Cocosinchi. Z. anorg. Chem., 186, 176 (1930). там же,
189, .83 (1930).
6. С. А. Кутолин, А. И. Вулих, Н. А. Д р у з ь. Промышленность
химических реактивов, вып. 1 (7), М., ИРЕА, 1965, стр. 42.
Поступила в декабре 196э г.
УДК 546.34,271.07
ЛИТИЙ БОРОГИДРИД
С. /И. АРХИПОВ
LiBH4 М. в. 21,78
Литий борогидрид представляет собой белый кристалли-
ческий порошок, устойчивый в инертной атмосфере до 250—
275° [1]. Его стандартная теплота образования ДН°298 =
—46,37 ккал/моль, свободная энергия образования Af^s —
—30,75 ккал/моль [2].
Борогидрид лития растворяется в спиртах с разложением.
Наиболее подходящими для него растворителями являются
диэтиловый эфир, тетрагидрофуран, изопропиламин.
При 25° растворимость борогидрида лития составляет в
изопропиламине 3—4 г [3], в тетрагидрофуране 28 г [4], в ди-
этиловом эфире 3—4 г в 100 г растворителя [5].
Борогидрид лития чрезвычайно гигроскопичен и может
самовозгораться на влажном воздухе. При 0' он растворяет-
ся в воде со слабым разложением, при 20° разложение идет
быстрее, но выделяется лишь часть водорода, остальной во-
дород выделяется только при кислой реакции среды. Реакция
гидролиза катализируется ионом кобальта:
ЫВН4 + 2 Н2О -> LiBO, + 4Н4
Основное применение борогидрид лития находит в орга-
нической химии в качестве восстановителя [6]. Он восстанав-
ливает альдегиды и сложные эфиры до первичных спиртов,
кетоны до вторичных спиртов, но не. восстанавливает нитри-
лы, амиды, ароматические кислоты, Борогидрид лития ис-
пользуют для получения боразола, который применяется как
инициатор горения топлива [7], в электролитах для осажде-
ния циркония [8], как источник водорода при получении губ-
чатых материалов [9].
Все известные способы получения борогидрида лития от-
личаются друг от друга использованием различных гидриру-
ющих агентов, в качестве которых могут быть гидриды ще-
27
лочных металлов, бороводороды, комплексные гидриды или
водород. В соответствии с этим различают три типа реакций:
3 ЫГ 4- 2 B2HS -> 3 ивн4 + вг3
4UH4-Br3 ивн4+-3иг
ur + McBH4 -> ивн44-МеГ
где Me — щелочной металл, а Г — галогенид или органичес-
кий радикал.
Сведения о втором и третьем способах можно найти в ли-
тературе (10—13], но выделить чистый борогидрид лития эти-
ми способами затруднительно. Первый же способ позволяет
получать чистые препараты с хорошим выходом, поэтому он
нами описывается детально, при этом лучшие результаты
дает барботирование диборана через суспензию гидрида ли-
тия в эфире [14—15].
Диборан, в свою очередь, с хорошим выходом получается
путем взаимодействия гидрида лития с эфиратом трехфто-
ристого бора в присутствии катализатора — борогидрида ли-
тия.
Таким образом, целесообразно совмещать процессы полу-
чения диборана и борогидрида лития. В случае отсутствия бо-
рогидрида лития как катализатора процесс получения дйбо-
рана проводят таким образом, чтобы в растворе вначале на-
копился борогидрид, который далее служит катализатором
при получении диборана.
СХЕМА СИНТЕЗА БОРОГИДРИДА ЛИТИЯ
6L1H + 2BF3 6LiF + B2He
2 LiH фВ2Нв — 2LiBH4
Характеристика основного сырья
Гидрид лития, ЦМТУ 3106—52, измельчается в атмосфе-
ре азота до крупности 80—100 меш.
Диэтиловый эфир для наркоза, ГОСТ 6265—52, сушат
над хлористым кальцием и металлическим натрием.
Эфират трехфтористого бора с температурой кипения
125°, получают по методике [16].
Применяемая аппаратура
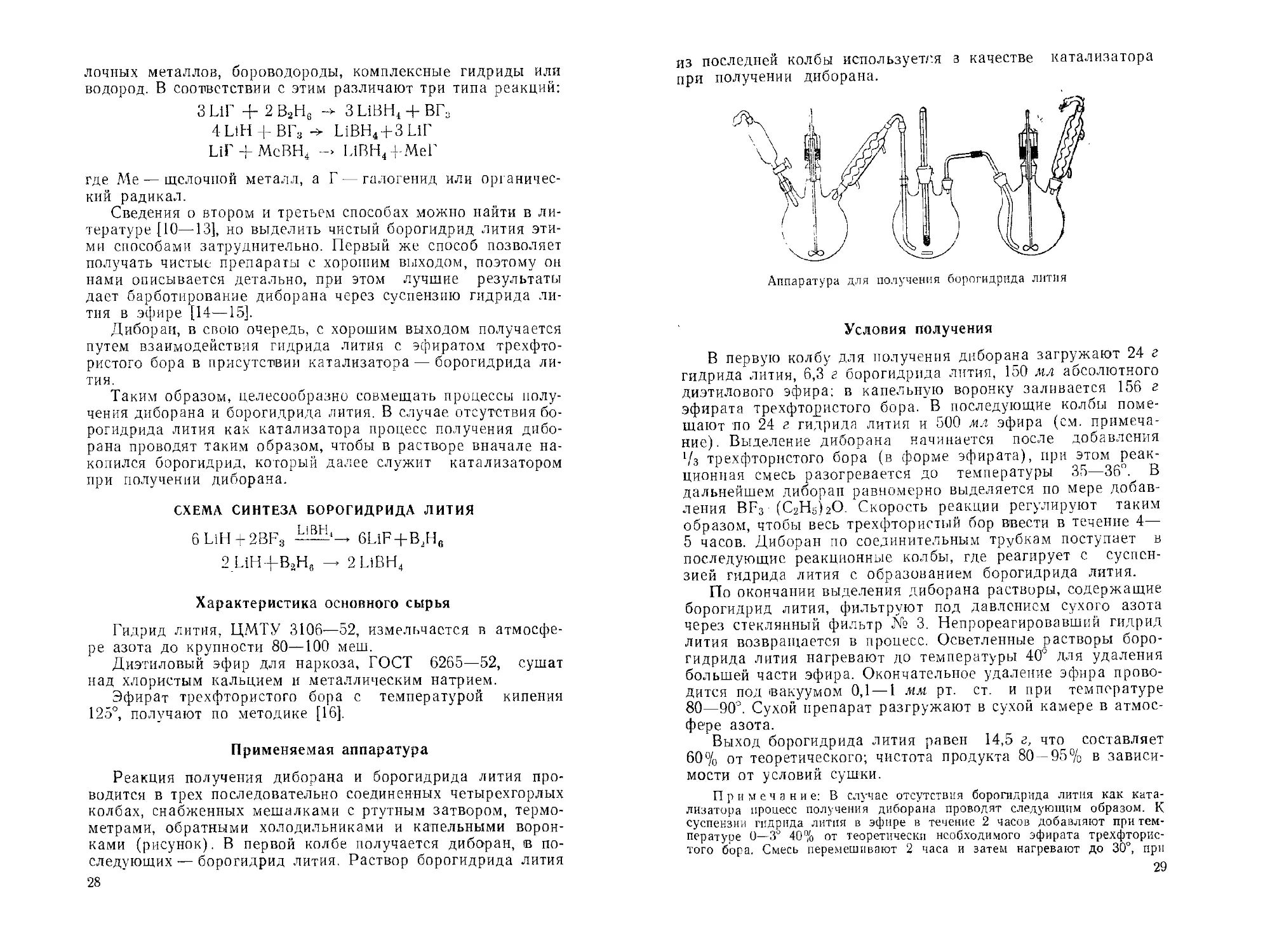
Реакция получения диборана и борогидрида лития про-
водится в трех последовательно соединенных четырехгорлых
колбах, снабженных мешалками с ртутным затвором, термо-
метрами, обратными холодильниками и капельными ворон-
ками (рисунок). В первой колбе получается диборан, в по-
следующих — борогидрид лития. Раствор борогидрида лития
28
из последней колбы используется з качестве катализатора
при получении диборана.
Аппаратура для получения борогидрида лития
Условия получения
В первую колбу для получения диборана загружают 24 г
гидрида лития, 6,3 г борогидрида лития, 150 мл абсолютного
диэтилового эфира; в капельную воронку заливается 156 г
эфирата трехфтористого бора. В последующие колбы поме-
шают по 24 г гидрида лития и 500 мл эфира (см. примеча-
ние). Выделение диборана начинается после добавления
*/з трехфтористого бора (в форме эфирата), при этом реак-
ционная смесь разогревается до температуры 35—36°. В
дальнейшем диборап равномерно выделяется по мере добав-
ления BF3 (CzHgjaO. Скорость реакции регулируют таким
образом, чтобы весь трехфтористый бор ввести в течение 4—
5 часов. Диборан по соединительным трубкам поступает в
последующие реакционные колбы, где реагирует с суспен-
зией гидрида лития с образованием борогидрида лития.
По окончании выделения диборана растворы, содержащие
борогидрид лития, фильтруют под давлением сухого азота
через стеклянный фильтр № 3. Непрореагировавший гидрид
лития возвращается в процесс. Осветленные растворы боро-
гидрида лития нагревают до температуры 40° для удаления
большей части эфира. Окончательное удаление эфира прово-
дится под вакуумом 0,1 — 1 мм рт. ст. и при температуре
80—90°. Сухой препарат разгружают в сухой камере в атмос-
фере азота.
Выход борогидрида лития равен 14,5 г, что составляет
60% от теоретического; чистота продукта 80 — 95% в зависи-
мости от условий сушки.
Примечание: В случае отсутствия борогидрида лития как ката-
лизатора процесс получения диборана проводят следующим образом. К
суспензии гидрида лития в эфире в течение 2 часов добавляют при тем-
пературе 0—3° 40% от теоретически необходимого эфирата трехфторис-
того бора. Смесь перемешивают 2 часа и затем нагревают до 30°, при
29
этом начинает бурно выделяться диборан. Далее процесс проводят, как
описано выше.
Борогидрид лития, полученный описанным методом, соответствует
требованиям МРТУ 6-09-2498—65 и содержит: лития—не менее 25,5%;
бора — не менее 40%; атомное отношение Li:B= 1,00±0,10.
ЛИТЕРАТУРА
1. Е. М. Ф е д н е в а, В. И. Алпатова, В. И. Михеева. Ж. не-
орган. химии, 9, 1519 (1964).
2. М. В. Smith, G. Е. Bass. J. Chem. Eng. Data, 8,342 (1963).
3. H. I. Schlesinger, H. C. Brown, H. R. Hoekstra, L. R.
Rapp. J. Amer. Chem. Soc., 75, 199 (1953).
4. J. R. Elliot, W. i.. Roth. Ci. F. R о c d e 1, E. M. Boldebuck.
J. Amer. Chem. Soc., 74, 5211 (1952).
5. T. L. Koi ski, H. B. Moore, L. B. Roth, K. J. Martin,
Q. W. Schaeffer. J. Amer. Chem. Soc. 80, 549 (1958).
6. H. Гейлорд. Восстановление комплексными гидридами метал-
лов, М, ИЛ, 1959.
7. В. И. Михеева, В. Ю. Маркина. Ж. иеорган .химии, 1, 2700
(1956).
8. W. Е. В е 1 i d, Y. М. Bish, A. Brenner, J. Elcctrochem.
Soc., 104, 21 (1957).
9. L. Tai a lay, J. О. T a 1 a 1 a y, Th. E Bush. Пат. США 2758980
(1956).
10. H. 1. Schlesinger, H. C. Brown, J. R. G i 11 b r e a th, J. Katz.
J. Amer. Client. Soc., 75, 195 (1953).
11. E. M. Феднева. Ж. иеорган. химии, 4, 286 (1959).
12. 11. 1. Schlesinger, H. C. Brown, E. К. H у d e„ J. Amer.
Chem. Soc., 75. 209 (1953).
13. J. К о 11 о п i t s c h, O. F u c h s. V. G a b e r. Nature, 173, 125(1954)
14. H. 1. Schlesinger. II. C. Brown. Пат. США 2545633(1951).
15. N. Noth, H. В e q e r. Chem. Ber. 93. 928 (1960).
16. Руководство по препаративной неорганической химии, Под ред.
Г. Брауэра. М., Г1Л, 1965, стр. 126.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.34'27'161.07
ЛИТИЙ ТЕТРАФТОРБОРНЫЙ
Борофторид лития
С. Л1. АРХИПОВ, П. Д. КОМИССАРОВА
LiBF4 М.в. 93,74
Борофторид лития представляет собой бесцветное крис-
таллическое вещество, хорошо растворимое в воде и низших
спиртах; в эфире его растворимость равна 1,3 г на 100 см3, в
тетрагидрофуране — 41,5% [1]. На воздухе расплывается и
дымит [2].
При нагревании борофторид лития разлагается, образуя
трехфтористый бор и фторид лития.
Теплота диссоциации вещества равна 3810 кал, давление
диссоциации при 324° достигает 760 мм [2].
Борофторид лития применяется в органическом синтезе в
качестве катализаторов и как компонент сварочных флюсов.
Препараты борофторида лития получают взаимодействи-
ем карбоната лития с эфиратом трехфтористого бора в эфи-
ре при 35° [1, 3]. В отсутствие растворителей борофторид ли-
тия получают пропусканием газообразного трехфтористого
бора над карбонатом лития при 300—400° [4]. В водных рас-
творах при взаимодействии фтороборной кислоты с карбона-
том лития можно получить продукт, содержащий в качестве
примеси 5—30% фтористого лития [4].
Ниже описан способ получения борофторида лития реак-
тивной чистоты, разработанный нами.
СХЕМА СИНТЕЗА БОРОФТОРИДА ЛИТИЯ
Li2CO3+8HF+2HsBO3 -> 2LiBF4%7H20 + C03
Характеристика основного сырья
Литий углекислый, ч., ТУ ЦМ 2006—48.
Борная кислота, ч., ГОСТ 9656—61.
31
Фтористоводородная кислота, ч., ТУ МХП 289—47.
Спирт этиловый, ГОСТ 5962—51.
Условия получения
В винипластовую чашу, содержащую 300 г 35%-ной пла-
виковой кислоты, добавляют небольшими порциями при по-
стоянном перемешивании и охлаждении 82 г борной кислоты.
К полученному раствору тетрафторборной кислоты прибавля-
ют при непрерывном перемешивании и охлаждении 49 г уг-
лекислого лития. Затем реакционную смесь упаривают под
инфракрасной лампой досуха и осадок переносят в двухгор-
лую колбу, снабженную механической мешалкой с масля-
ным затвором и обратным холодильником.
Осадок экстрагируют этиловым спиртом (1:2) в течение
часа при комнатной температуре. Нерастворимый осадок от-
фильтровывают, промывают на фильтре небольшим количе-
ством спирта.
Осветленный раствор вместе с промывными водами зали-
вают в перегонную колбу, снабженную холодильником Либи-
ха, и полностью отгоняют спирт.
Полученный сольват борофторида лития загружают в
фарфоровые чаши, закрытые стеклянными колпаками (во из-
бежание разбрызгивания), и помещают в вакуум-сушнльный
шкаф, где проводят десольватацию и сушку препарата при
температуре 80—90°/5—20 мм.
Выход борофторида лития равен 100 г, что составляет
80% от взятого в процесс углекислого лития.
Чистота полученного вещества соответствует 99% и по со-
держанию примесей отвечает требованиям РЭТУ 1215—64.
ЛИТЕРАТУРА
1. J. Shapiro, И. W е 1 s s. J. Amer. Chem. Soc., 75, 1753 (1953),
2. Г. Б у з, Д. Марти и. Химия трехфтористого бора и его произ-
водных, М., ИЛ, 1955, стр. 45.
3. J. R. Elliot, Е. М. Boldebuck. J. Amer. Cliem. Soc., 74, 5047
(1952).
4. II, I. Pure, Химия фтора и его неорганических соединений, М„
Госхимпздат, 1956, стр. 466.
Поступила в декабре 1965 г. ЦЗЛ
УДК 547.532'134'273.07
ЛИТИИ ТЕТРАФЕНИЛБОРАТ
Г. Е. РЕВЗИН, А. И. ВУЛИХ, С. М. АРХИПОВ, В. Д. ЗАМЕДЯНСКАЯ
Li[B(CeHs)4]-2НаО М. в. 362,20
Тетрафенилборат лития представляет собой мелкокрис-
таллический порошок белого цвета, иногда окрашен продук-
тами разложения в светло-бурый цвет; его плотность равна
1,128 г/сл13; препарат устойчив на воздухе до температуры
120°; при нагревании на воздухе до 520° количественно пере-
ходит в метаборат лития, L1BO2.
Тетрафенилборат лития очень хорошо растворим в воде,
спиртах, ацетоне, пиридине; плохо растворим в простых эфи-
рах, хлороформе, четыреххлористом углероде, нерастворим
в бензоле, толуоле и алифатических углеводородах. Водные
растворы его со временем гидролитически разлагаются по
реакции (1]:
L1[B(C.H6)J + 3H2O С«НвВ(ОН)3 + ЗС«Н6ф-ЫОН
Растворы тетрафенилбората лития сохраняются без изме-
нений в течение нескольких недель при pH > 8. В кислых
растворах гидролиз протекает с большой скоростью [1—3].
Водный раствор тетрафенилбората лития с растворами
солей аммония, калия, рубидия, цезия и таллия (I) мгновен-
но образует осадки нерастворимых в воде тетрафенилбора-
тов.
Тетрафенилборат лития может найти широкое применение
в аналитической химии, как и тетрафенилборат натрия, от
которого он выгодно отличается большей растворимостью
как в воде, так и в органических растворителях. Основные
области возможного применения тетрафенилбората лития—
весовое и объемное определение калия, аммония, рубидия,
цезия, таллия (I), а также органических соединений—ами-
нов, алкалоидов, некоторых обезболивающих и лекарствен-
ных веществ [1].
3 Зак. 1060 33
В литературе {2, 4—7] описаны следующие способы полу-
чения тетрафенилбората лития:
1) взаимодействие фенил-лития с трифенилбором в эфи-
ре [4]
С2Н5ОС2П5
LiCeH6 + B(CeH6)3------->Li[B(CaH5)4]
2) взаимодействие фенил-лития с эфиратом трехфторис-
того бора [5, 6]
4LiCeHe-j-BF3.(CsH5)20 Li[B(C6HJ4]+3UF+(C2Hs)2O;
3) обменная реакция соли лития с тетрафенилборатом
какого-либо металла [2|
LiAn+Me[B(CeH6)4] -> Li]B(CeH5)4] + MeAn;
4) ионообменный синтез в водной среде из тех же ве-
ществ [7].
Из описанных в литературе методов первые два непри-
годны для синтеза тетрафенилбората лития в сколько-нибудь
значительных количествах, так как исходным сырьем для
них является фенил-литий— металлоорганическое соединение.
Синтез и использование фенил-лития представляет сущест-
венные трудности, обусловленные чрезвычайно высокой его
реакционной способностью и бурным разложением в присут-
ствии следов влаги и кислорода.
Методы синтеза путем ионного обмена в водных раство-
рах требуют использования растворимых тетрафенилборатов,
получение которых связано с теми же трудностями, что и
прямой синтез тетрафенилбората лития. Кроме того, при кон-
центрировании водных растворов тетрафенилбората лития
происходит его частичное разложение.
Поэтому при разработке нами методики за основу был
принят ионообменный синтез тетрафенилбората лития из
тетрафенилбората калия в ацетоне.
Преимущества этого метода заключаются: а) в доступно-
сти исходного сырья — тетрафенилбората калия—и легкости
его получения в достаточно чистом состоянии; б) в значи-
тельной растворимости исходного сырья и продукта в ацето-
не; в) в большей величине константы ионного обмена ли-
тий—калий в ацетоне, что позволяет получать продукт высо-
кой чистоты с хорошим выходом; г) в простоте аппаратурно-
го оформления процесса и его контроля; д) в легкости вы-
деления продукта из раствора без заметного его разложения.
СХЕМА СИНТЕЗА ТЕТРАФЕНИЛБОРАТА ЛИТИЯ
СН3СОСН3 _
RLi + K[B(C6Hs)4[ -----> RK+Li[B(C6H5)4}
где R—радикал активного центра смолы.
34
Характеристика основного сырья
Литий гидрат окиси, ч., МРТУ 6-09-1471—64.
Ацетон, ч.д.а., ГОСТ 2603—51.
Смола КУ-2, техн., ВТУ МХП 661—55.
Калий тетрафенилборат (см. примечание 1).
Аппаратура
Прибор для ионообменного синтеза состоит из напорного
бачка (7), соединенного при помощи нормального шлифа (3)
с колонкой (4). Применение резиновых пробок и шлангов
недопустимо, так как ацетон разрушает резину.
В колонку загружают подготовленную смо-
лу в Li-форме так, чтобы вся смола была за-
лита ацетоном н между гранулами смолы от-
сутствовали пузырьки воздуха. Уровень ацето-
на должен быть выше уровня смолы на
25 — 30 мм.
Скорость вытекания раствора из колонки и
скорость подачи раствора в колонку регулиру-
ются хорошо притертыми стеклянными крана-
ми (2).
Под колонкой устанавливают приемник (5)
для сбора раствора, вытекающего из колонки.
Условия получения
Подготовка смолы. Готовят 1 и. раствор
гидрата окиси лития. Раствор заливают в на-
порный бачок. В колонку помещают катионит
КУ-2 в Н-форме и производят сорбцию лития
на смолу до полного насыщения смолы литием
и перевода ее в Li-форму; при этом фильтрат Рас. 1.
на выходе из колонки становится щелочным с
pH 9 — 10. Скорость пропускания раствора 1 л/час на 1 кг
воздушно-сухой смолы.
Затем смолу промывают дистиллированной водой до ней-
тральной реакции промывной воды по универсальной инди-
каторной бумаге. Промытую смолу сушат на листе фильтро-
вальной бумаги в течение 24 часов до воздушно-сухого сос-
тояния, переносят на воронку Бюхнера, промывают ацетоном
для удаления воды, отсасывают и сушат в вакуум-сушиль-
ном шкафу при температуре 40—50° и остаточном давлении
30—40 ля в течение 1 —1,5 часа.
Высушенную смолу заливают ацетоном н оставляют для
набухания в течение 2—3 часов.
35
Получение тетрафенилбората лития. Готовят 2,5%-ный
раствор тетрафенилбората калия в ацетоне растворением 50 г
ЩЩСбНаЫ в 1,95 кг (2,46 л) ацетона и порциями его зали-
вают в напорный бачок по мере вытекания из него раствора.
Загружают в колонку около 100 г смолы « Li-форме, как
описано выше, и проводят десорбцию лития со смолы со ско-
ростью 1 л/час на 1 кг смолы.
Десорбцию лития проводят до проскока иона калия в
фильтрат, что периодически, через каждые 25—50 мл филь-
трата, качественно контролируется следующим образом. От-
бирают пробу фильтрата объемом около 1 мл и разбавляют
ее 30—50 мл дистиллированной воды. Образование мути при
разбавлении фильтрата водой свидетельствует о появлении
в фильтрате нерастворимого в воде тетрафенилбората ка-
лия.
Раствор тетрафенилбората лития до проскока калия упа-
ривают в вакууме при остаточном давлении 10—20 мм и тем-
пературе 30—35° досуха.
Следующие порции фильтрата, содержащие смесь тетра-
фенилборатов лития и калия, направляют на десорбцию в
следующем цикле с новой загрузкой смолы КУ-2 в Li-фор-
ме.
Пробы, отбираемые для контроля, сливают в колбу, до-
бавляют избыток раствора хлористого калия до полного
осаждения тетрафенилборат-иона в виде K[B(C6Hs)J, кото-
рый отфильтровывают, высушивают в сушильном шкафу при
температуре 105—110° в течение 2—3 часов и возвращают в
голову процесса.
Выход тетрафенилбората лития равен 35,4 г, что состав-
ляет 70% от теоретического.
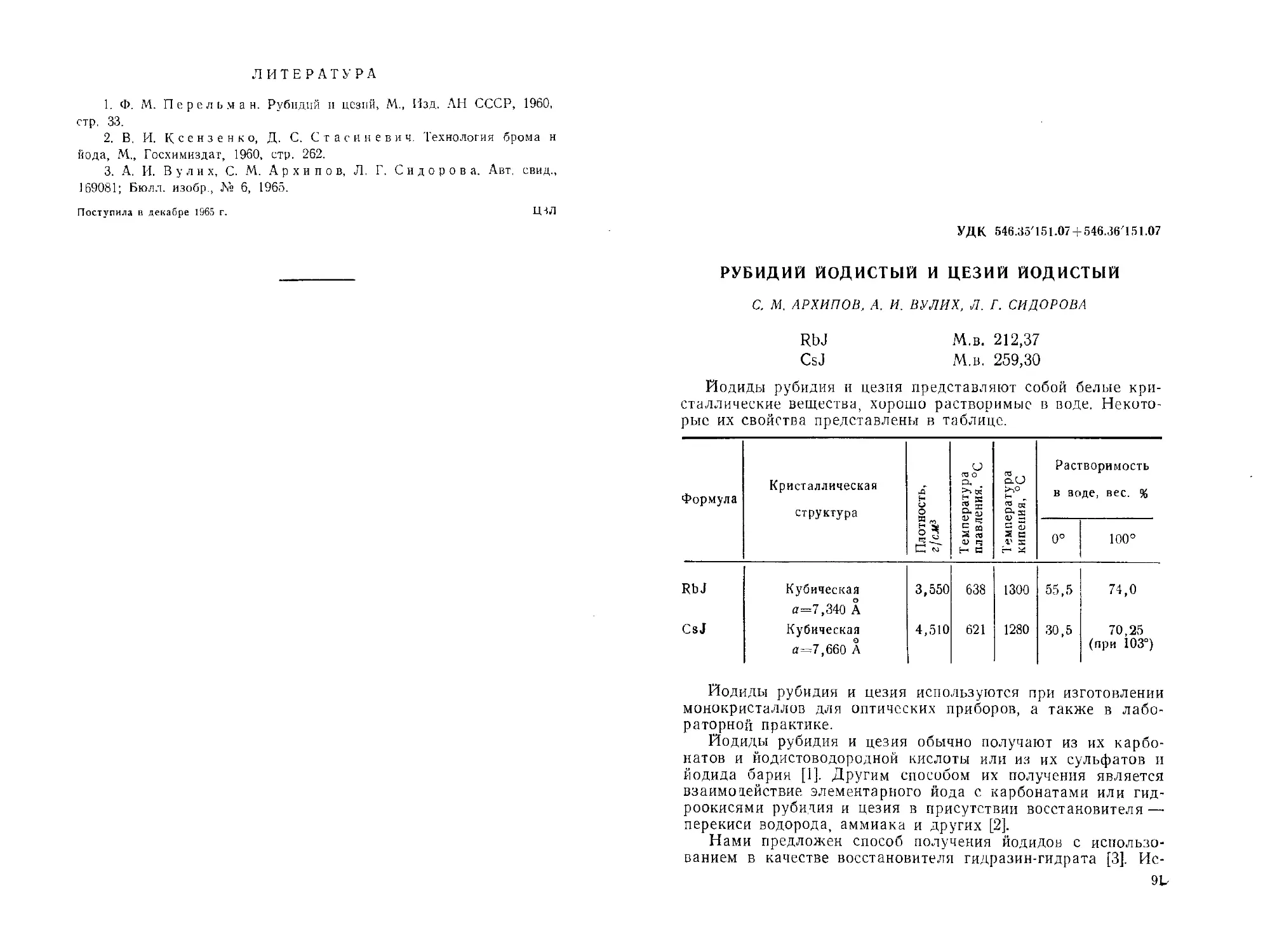
Чистота продукта достигает 83,94—88,08% Li[B(С6Н5)4],
что соответствует содержанию Li[B(CJ-L)J 2Н>О 93,2—
97,8% (см. примечание 2).
Примечания:
1. Тетрафенилборат калия готовят по методике [8] или получают из
хлористого калия и тетрафенилбората натрия, после чего сухой тетрафе-
нилборат калия растворяют в ацетоне, раствор осветляют активирован-
ным углем; отфильтровывают и осаждают тетрафенилборат калия добав-
лением пяти-шестикратного количества дистиллированной воды. Очи-
щенный таким образом препарат после высушивания имеет чистоту
98,5 99,8%.
2. Тетрафенилборат лития выпускается квалификации «чистый» и
«чистый для анализа» и должен содержать основного вещества в пере-
счете на безводный LifB (С6Н5)J не мепее 70% Для квалификаций «чи-
стый» п не менее 8О°/о Для квалификации «чистый для анализа».
ЛИТЕРАТУРА
1. Э. Ю. Янсон, А. Ф. И е в и н ь ш. Успехи химии, 28, 8, 980
(1959).
36
2. S. S. Cooper. Analyt. Chem., 29, 446 (1957).
3. S. S. Cooper. Chemist-Analyst, 43, 62 (1957).
4. G. Wittig. G. К e i c h e r, A. Rtickert. P. Raff. Liebigs Ann;
Chem, 563. 110 (1949).
5. G. Wittig, Angew. Chem., 62, 231 (1950).
6. G. Wittig, P. Raff. Liebigs Ann. Chem., 573. 195 (1951).
7. А. И. Вулих, В. Л. Казьминская, М. М. Сеиявин. Авт.
свид. 145891, Бюлл. изобр, № 7, 15 (1962).
8. А. Н. Несмеянов, В. А. Сазонова, Г, С. Либерман,
А. И. Емельянова. Изв. АН СССР, ОХИ, 1. 48 (1955),
Поступила в дек-лб] е 1965 г. НЗЛ
УДК 546.623'34'11.07
ЛИТИИ АЛЮМОГИДРИД
С. М АРХИПОВ
LiAlH4 М. в. 37,95
Литий алюмогидрид представляет собой белый мелкокри-
сталлический порошок, его плотность составляет 0,917 г/c.w3
|1]; теплота образования — 28,5 ккал/моль [2J. Препарат ус-
тойчив в вакууме до 150° [3], растворим в некоторых простых
и сложных эфирах [4]; при 25° эта растворимость в весовых
процентах такова: в диэтиловом эфире — 23; дибутиловом
эфире--2; тетрагидрофуране - 11,5; диоксане—0,1; димети-
ловом эфире диэтиленгликоля — 7.
В углеводородах, бензоле и толуоле алюмогидрид лития
нерастворим. Он энергично реагирует с соединениями, содер-
жащими активированный водород, — водой, спиртами.
Растворы в диэтиловом эфире метастабильны и могут са-
мопроизвольно разлагаться с выделением большого количе-
ства водорода. Разложение можно предотвратить введением
твердого гидрида лития или добавлением небольших коли-
честв диоксана и триметиламина.
Алюмогидрид лития находит применение в органической
химии как селективный восстановитель ряда функциональных
групп [5—7]. Кроме того, он применяется для получения гид-
ридов других элементов [8—11], в аналитической химии [12]
и для получения высокочистых веществ [13, 14].
Можно выделить три основных способа получения алюмо-
гидрида ЛИТИЯ;
1. Синтез из элементов:
растворитель
LI + А1 + 2Н2----------------> L1A1H*
100—120°. давление
2. Обменная реакция с алюмогидрндами других метал-
лов:
MeAlH* + LiCl — L1AIH4 + MeCl
где Me — Na, К.
38
3. Взаимодействие гидрида лития с галогенидами алюми-
ния:
4LiH + А1Г3 — L1A1H, -% 3LiT
где Г—Cl, Br, J.
Первые два способа пока не нашли 'применения; сведения
о них можно найти в литературе [4, 15, 16].
Наиболее полно изучена реакция гидрида лития с гало-
генидами алюминия и этот способ применяется при промыш-
ленном производстве алюмогидрида лития [17, 18]. В случае
использования хлористого алюминия обычно наблюдается ин-
дукционный период, что связано с нерастворимостью хлори-
стого лития в применяемых растворителях [19, 20]. Примене-
ние бромистого алюминия снимает затруднения при синтезе,
но выделить чистый алюмогидрид лития нельзя ввиду высо-
кой растворимости бромистого лития [21]. Можно применять
при синтезе гидрид алюминия [22] или этилат алюминия [23].
Влияние температуры на ход синтеза было подробно изу-
чено в работах [24, 25].
Алюмогидрид лития в зависимости от способа получения
может содержать примеси гидрида лития, хлористого лития,
гидрида алюминия, остатки растворителя. Для получения
чистых препаратов твердый продукт растворяют в эфире и
осаждают добавлением большого количества бензола [26].
Ниже приведены 3 методики получения алюмогидрида ли-
тия. Каждая из них имеет свои преимущества и недостатки.
По первому способу получаются чистые препараты, но для
успешного синтеза требуется большой избыток гидрида лития
и поддержание в процессе реакции низких температур. По
второму способу получаются препараты LiAlH<, содержащие
5—7% LiBr, однако способ удобен, так как не требуется со-
блюдения жесткого режима синтеза. По третьему способу
можно быстро и с высоким выходом получать растворы
ЫА1Н4, содержащие 25—30% бромистого лития. Если пре-
парат готовится для целей восстановления органических сое-
динений, то можно непосредственно использовать растворы
такого состава, так как бромистый литий не мешает в про-
цессах восстановления [21].
СХЕМА СИНТЕЗА АЛЮМОГИДРИДА ЛИТИЯ
4L1H + А1Г3 — L1A1H* + ЗЫГ
где Г = С1 или Вг.
Характеристика основного сырья
Гидрид лития, ЦМТУ 3106—52.
Хлористый алюминий, безводный, ВТУ МХП 3500—52.
Бромистый алюминий, безводный, ВТУ ЕУ 79—55.
39
Толуол, ГОСТ 5789—51.
Диэтиловый эфир, ГОСТ 4460—48.
Условия получения
Подготовка исходных веществ. Гидрид лития тщательно
измельчают в отсутствии влаги и воздуха до крупности
100 меш.
Диэтиловый эфир освобождают от перекисей встряхива-
нием с твердым едким кали и затем осушают настаиванием
над свежепрокаленным хлористым кальцием в течение 3 су-
ток. После этого фильтруют и окончательно освобождают от
влаги выстаиванием над металлическим натрием в течение
2—3 суток. Толуол осушается кипячением с гидридом лития
и перегонкой. Азот для фильтрования под давлением тща-
тельно высушивают.
Реакцию синтеза проводят в четырехгорлой колбе, снаб-
женной механической мешалкой с масляным затвором, об-
ратным холодильником, капельной воронкой и термометром.
Для охлаждения колбы служит ледяная баня.
Способ 1 [25]. В реакционную колбу загружают 12 г гид-
рида лития и 50 мл абсолютного эфира. Колбу охлаждают
до 0° тающим льдом. Медленно по каплям прибавляют эфир-
ный раствор хлористого алюминия, полученный из 36 г А1С13
и 125 мл абсолютного эфира, следя за тем, чтобы температу-
ра не поднималась выше 5°. Скорость приливания должна
быть 0,3 0,5 мл в минуту. После добавления 25 мл эфирата
смесь перемешивается 15—20 минут, после чего добавляют
вторую половину (50 мл) абсолютного эфира. Затем прибав-
ляют остальные 100 мл эфирата в течение 3 часов. В общей
сложности прикапывание эфирата (125 мл) продолжается
около 4,5 часа. После этого смесь перемешивают в течение
2 часов и фильтруют в цилиндрическую пробирку с отводом
на 0,5 л через воронку с пористым дном № 3 под давлением
сухого азота. Осадок на фильтре промывают 3—4 раза
абсолютным эфиром. Эфир отгоняют под вакуумом при на-
гревании до 40° и улавливают в ловушках, охлаждаемых
смесью ацетона и сухого льда. После отгона эфира продукт
окончательно высушивают под вакуумом при температуре
50—60е в течение 3 часов.
Выход алюмогидрида лития равен 8,5 г, что составляет
60% в пересчете на LiH; чистота вещества 90—95%.
Послереакционный остаток немедленно разлагается боль-
шим количеством воды (несколько литров).
Способ 2 [27]. Готовят эфирные растворы бромистого алю-
миния (5 г А1Вгз в 40 мл эфира) и хлористого алюминия
(50 г А1С1з в 150 мл эфира). Применяют гидрид лития дис-
персностью 60—100 меш.
40
В реакционную колбу помещают 18 г гидрида лития и
50 мл абсолютного эфира. Поддерживая температуру 18—
20°, добавляют эфирный раствор бромистого алюминия в те-
чение 30 минут. После кратковременного перемешивания к
реакционной суспензии добавляют в течение 4 часов эфир-
ный раствор хлористого алюминия. Суспензию фильтруют
в колбу с боковым отводом иод давлением сухого азота че-
рез стеклянный фильтр № 3. Эфир отгоняют на водяной бане
при температуре 40—45°, а затем под вакуумом при темпе-
ратуре 70° в течение 3—4 часов.
Выход алюмогидрида лития равен 14,2 г, что составляет
66% в пересчете на LiH; чистота вещества—90%.
Способ 3 [28]. В реакционную колбу загружают 16 г гид-
рида лития и 120 мл абсолютного эфира. В капельную во-
ронку заливают раствор бромистого алюминия, полученный
из 115 г А1Вг3 и 100 мл толуола.
При интенсивном перемешивании добавляют одну треть
раствора бромистого алюминия так, чтобы смесь равномерно
кипела. Затем добавляют 60 мл абсолютного эфира и при-
ливают оставшийся раствор бромистого алюминия. В конце
реакции температура реакционной массы достигает 56°. Об-
щее время синтеза 75 минут.
Раствор фильтруют под давлением азота и осадок промы-
вают 50 мл смеси эфир—толуол (1:4). Раствор алюмогидри-
да лития упаривают до объема 100 мл и охлаждают до 20°.
Выпавший осадок бромистого лития отделяют фильтровани-
ем и затем промывают 50 мл смеси эфир—толуол. Оконча-
тельно растворитель удаляют под вакуумом при температуре
90° в течение 4 часов.
Получается 25,2 г алюмогидрида лития в виде легкопере-
сыпающегося белого порошка, что составляет 97% от тео-
ретического выхода; чистота вещества — 63%•
Для очистки препарата от бромистого лития 20 г полу-
ченного алюмогидрида лития помещают в реакционную кол-
бу и растворяют в 200 мл смеси эфир—толуол (1:4) в тече-
ние 6—7 часов.
После фильтрации и отгонки растворителя получают 10,7 а
продукта, что соответствует 80,7% от теоретического выхода;
чистота вещества равна 95%.
Продукт, полученный описанными выше способами, удов-
летворяет требованиям РЭТУ 1067—63.
ЛИТЕРАТУРА
1. W. D. Davis, L. S. Mason, G. St eg eman. J. Amer, Chem.
Soc. 71, 2775 (1949).
2. W. B. S m i t h, G. E. В a s s. Chem. and Eng Data, 8 (3), 342
(1963).
3. В. И. Михеева, M. С. С e л и в о х и н а, О. Н. Крюкова.
Докл. АН СССР, 109, 541 (1956).
41
4. H. Clasen. Angew. Chemie, 10, 322 (1961).
5. H. Г. Гейлорд. Восстановление комплексными гидридами ме-
таллов, М., ИЛ, 1959.
6. В. М. Мигов и ч, М. Л. Михайлович. Алюмогидрид лития
и его применение в органической химии, М., ИЛ, 1958.
7. Е. В. Рогинская. Успехи химии, 21, 1 (1952).
8. В. И. Михеева. Успехи химии, 23, 831 (1954).
9. J. Aubry, G. М о n n i е г. Bull. Soc Chem. France, 10, 919(1953).
10. E. Wiberg, К. M 6 d r i t z e r. Z. Naturforsch., 11 b, 747 (1956).
11. J.Shapiro, H. G. Weiss, M. Schmidt, S. Skolnik,
G. B. L. Smith. J. Amer. Chem. Soc. 74, 901 (1952).
12. Д. И. Рябчиков, В. К- Беляева. Успехи химии, 24, 240
(1955).
13. Р. Roman, G. Mores. Rev. Chim. И. 461 (1960).
14. S. Sujshi, J. N. Keith. J. Amer. Chem Soc., 80, 4138 (1958).
15. E. C. Ashby, G. J. Brendel, N. E. Redman. Inorg. Chem.,
2 (3) 499 (1963),
16. А. И. Захаркин, В. В. Гавриленко. Изв. АН СССР, отд.
хим. наук, № 7, 1146 (1962).
17. Т. R. Р. gibb. Пат. США 2468260 (1949).
18. Т. R. Р. gibb. Kanad. пат. 474826 (1951).
19 R. Mirza. Nature, 170,669 (1952).
20. Е. Wiberg, R. Bauer, M Schmidt, R. L'son. Z. Naturforsch, 6в,
393 (1951).
21. E. Wiberg, M. Schmidt. Z. Naturforsch., 7b, 59 (1952).
22. J. E. Jonson, R. H. Blizzard, H. W. Carhart. J. Amer.
Chem. Soc., 70, 3664 (1948).
23. O. Schmit z-D u m о n t, V. Habernickel. Naturwissenschaften,
39, 20 (1952).
24. В. И. Михеева, M. С. С e л и в о х и н а, В. В. Леонова. Ж.
неорган. химии, 4, 2436 (1959).
25. В. И. Михеева, М. С. Сели во хин а, В. В. Леонова. Ж.
неорган. химии, 4, 2705 (1959).
26. W. D. Davis, L. S. Mason, G. S t с g е m a n. J. Amer. Chem.
Soc. 71, 277b (1919).
27. С. M. Архипов, В И. Михеева. Промышленность химиче-
ских реактивов, вып. 1 (7), М., ИРЕА, 1964, стр. 36.
28. С. М. Архипов. Изв. Сибирского отд. АН СССР, № 7, сер.
химии., 2, 138 (1964).
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.34'261.07
КАРБИД ЛИТИЯ
Ацетиленид лития
М. Н. КОРОТКЕВИЧ, О. Н. БРЕУСОВ
Li -С= С - L1
Li3C2 м.в. 37,90
Карбид лития представляет собой вещество желтовато-се-
рого цвета. Кристаллизуется в моноклинной сингонии с па-
раметрами ячейки: а = 7,807 А; Ь = 8,815 А; с= 10,865 А. Во-
дой разлагается с выделением ацетилена; плотность 1,65 [1].
В. литературе описано несколько способов получения кар-
бида лития:
1) непосредственным взаимодействием лития и углерода
или в атмосфере аргона [1], или в вакууме [2,5];
2) нагреванием лития в струе ацетилена или этилена
[3, 41,
3) восстановлением карбоната лития углем [4];
4) взаимодействием окисн и двуокиси углерода с литием
[3].
Указанные методы трудновоспроизводимы [1, 2] или слож-
ны в аппаратурном оформлении.
Нами разработан способ получения карбида лития, осно-
ванный на взаимодействии гидрида лития с сажей в вакууме
0,2 мм рт. ст. при температуре 650°.
СХЕМА СИНТЕЗА КАРБИДА ЛИТИЯ
2LiH + 2С = Li2C2 + Н.2 t
Характеристика основного сырья
Литий гидрид, ч., ЦМТУ 3106—52.
Сажа канальная, техн., ГОСТ 7885—56.
43
Условия получения
В сухой камере готовят шихту стехиометрического соста-
ва из 20 г гидрида лития и 30,25 г сажи. Сажу обезвожива-
ют предварительным прокаливанием в вакууме при 400° и ос-
таточном давлении 0,2 мм. Реторту с загруженной в нее
шихтой устанавливают в электрическую печь и присоединя-
ют к форвакуумному насосу. При достижении в системе ва-
куума 0,2 мм включают нагрев печи. Учитывая, что карбид
лития начинает диссоциировать на металлический литий и
углерод с 600°, температуру поднимают до 650° постепенно.
При 650° (допустимые отклонения температуры ±25°) шихту
выдерживают в течение шести часов, после чего охлаждают
в вакууме до комнатной температуры. При этом литий час-
тично уносится из зоны реакции.
Выгрузку образовавшегося карбида лития проводят в су-
хой камере. Карбид лития получают в виде желтовато-серо-
го спека, покрытого сверху налетом сажи. Куски карбида ли-
тия очищают от сажи и растирают в фарфоровой ступке.
Выход карбида лития равен 31 г, что составляет 65% от
теоретического.
Данная методика позволяет получать препарат, удовлет-
воряющий требованиям МРТУ 6-09-1910—64, что соответст-
вует:
1) содержанию основных компонентов (в %, не менее):
лития—36,0; углерода — 57,5;
2) атомному отношению литий: углерод = 1 ±0,1;
3) допустимому содержанию примесей (в %, не более);
натрия — 0,01; калия — 0,01; железа — 0,03.
ЛИТЕРАТУРА
1. D. R. Secris, L. О. W i s п у i. Acla crystallogr., 15, 10, 1042
(1962).
2. L. Vanino. Hundbucli der praparativen Chemie. B.l, Anorgan. teil,
Stuttgart, (1925).
3. Ю. И. Остроушко, П. И. Б у чихи н и др. Литий, его химия
и технология, М., Атомиздат, 1960, стр. 52.
4. Ю. Ньюлэнд, Р. Фог от. Химия ацетилена, М., ИЛ, 1947,
стр. 15.
5. Г. Реми. Курс неорганической химии, т. 1, М., ИЛ, 1963, стр .476.
Поступила в декабре 1965 г, ЦЛП
УДК 547.562.4'134.07
ЛИТИЙ ФЕНОЛЯТ
Г. Е. РЕВЗИН, В. Д. ЗАМЕДЯНСКАЯ
___/OLi-2H2O
C6H5OLi-2H2O М. в. 136,07
Фенолят лития представляет собой белые чешуйчатые
кристаллы с перламутровым блеском (1]. В сухом воздухе
продукт устойчив, но во влажном воздухе в присутствии уг-
лекислоты постепенно гидролизуется. При температуре вы-
ше 100° на воздухе начинает разлагаться, не плавясь; пол-
ное разложение наступает при температуре 150°.
Относительная плотность равна 1,28x0,01; растворимость
в воде составляет (в вес. %):
Температура, °C 0 10 20 25 30 35 40 50
Растворимость 10.92 11,31 11,92 12,57 13,10 13,64 14,18 15,47
Теплота растворения при 25° (ДН»™) равна 1,385 +
±0,005 ккал/моль.
Фенолят лития может применяться в лабораторной прак-
тике и органическом синтезе для синтеза простых и слож-
ных эфиров фенола [2—6].
СХЕМА СИНТЕЗА ФЕНОЛЯТА ЛИТИЯ
;он + LiOH + Н2О
/OLi-2H2O
45
Характеристика основного сырья
Литий гидрат окиси, ч,, ч.д.а., МРТУ 6-09-1471—64.
Фенол синтетический, ГОСТ 6417—52.
Условия получения
Растворяют при нагревании в 85 мл дистиллированной во-
ды 12 г (0,50 Л4) гидрата окиси лития (см. примечания 1,2);
при необходимости раствор фильтруют через стеклянный по-
ристый фильтр № 3 или № 4.
К нагретому до 70—80° раствору гидрата окиси лития
прибавляют при перемешивании раствор фенола (48 г в 2 мл
дистиллированной воды). При этом начинают выпадать в
осадок кристаллы фенолята лития. Затем реакционную мас-
су охлаждают до температуры 10—15° и на воронке Бюхне-
ра отфильтровывают выпавший осадок в виде снежно-белых
чешуйчатых кристаллов с перламутровым блеском. Кристал-
лы промывают на фильтре сначала 10 мл ацетона или спир-
та, а затем 10—15 мл бензола для удаления избытка фено-
ла. Хорошо отжатый на воронке Бюхнера продукт сушат на
воздухе между листами фильтровальной бумаги или в су-
шильном шкафу при температуре не выше 80°. Полученный
продукт по составу соответствует формуле СбН5ОБ1'2Н2О
(см. примечания 3,4).
Выход фенолята лития равен 58,5 г, что составляет 86%
от теоретического.
Указанный метод синтеза позволяет получать продукт,
удовлетворяющий требованиям РЭТУ 1140—63.
Примечания:
1. Лиши гидрат окиси (LiOH-НгО) реактивной чистоты содержит
LiQH от 52 до 56%, поэтому перед проведением синтеза необходимо оп-
ределить его содержание в исходном сырье и затем рассчитать потребное
количество кристаллогидрата, содержащее 12 г LiOH.
2. Для получения продуктов квалификаций «ч.» или «ч.д.а.» в каче-
стве исходного сырья используется гидрат окиси лития ч. нли ч.д.а. со-
ответственно.
3. Содержание основного вещества в феноляте лития определено
йодометрическим методом [8].
4. Содержание кристаллизационной воды в продукте определено ти-
трованием навески фенолята лития реактивом Фишера [8].
ЛИТЕРАТУРА
1. С. J, van Meurs, Z. phys. Chem., 91, 328, (1916).
2. Л. E. Чичибабин. Основные начала органической химии, т. II,
М., Госхимнздат, 1957, стр. 381.
3. Е. С. Хоти некий. Курс органической химии. Л., Госхимтехиз-
дат, 1933, стр. 356.
46
4. П. П. Ш о р ы г и н. Курс органической химии, М., Госхпмпздат,
1940, стр. 369.
5. П. Каррер. Курс органической химии, Л.. Госхимиздат, 1962,
стр. 540.
6. К. Д. Неницеску. Органическая химия, т. 1, Инлитиздат, 1962,
стр. 475.
7. К- Бауэр. Анализ органических соединений, М., Инлитиздат,
1963, стр. 97.
8. Д. Митчел. Д. Смит. Акваметрия, 54., Инлитиздат, 1952,
стр. 25.
Поступила в декабре 1965 г.
УДК 546.34'284.07
СИЛИКАТЫ ЛИТИЯ
A. A. СТАЦЕНКО, М. Ф. ФОКИНА
Li2SiO8 Литий метакремиевокислый М. в. 89,96
Li4SiO4 Литий ортокремневокислый М. в. 119,85
Силикаты лития могут быть использованы в керамичес-
кой промышленности, для производства специальных стекол,
электронных трубок, для изготовления бессвинцовой глазу-
ри с малой вязкостью и повышенным блеском |1].
Резко выраженная точка плавления (1201°) и хорошая
способность к кристаллизации метакремневокислого лития
позволяет пользоваться им в качестве калибровочного веще-
ства для термоэлементов [2].
По литературным данным метакремиевокислый и орто-
кремневокислый литий можно получить путем сплавления
стехиометрически рассчитанных количеств углекислого лития
и кварцевого песка в платиновом тигле [2].
Нами разработан метод получения силикатов лития пу-
тем непосредственного спекания гидроокиси лития и кремне-
вой кислоты при 800°. Данный метод позволяет понизить тем-
пературу процесса и использовать кварц в качестве материа-
ла для сосудов; кроме того, оба вещества получаются в виде
порошка, а не плава.
СХЕМА СИНТЕЗА СИЛИКАТОВ ЛИТИЯ
2L1OI1 + SiO2 -» Li2SiO3 + Н2О
4L1OH 4- SiO2 Li4SiO4 + 2НаО
Характеристика основного сырья
Литий гидрат окиси (моногидрат), ЦМ.ТУ 2062—48.
Кремневая кислота водная, ч., ГОСТ 4214—48.
48
Условия получения
В фарфоровой ступке или мельнице тщательно измельча-
ют и перемешивают до образования однородной массы 100 г
моногидрата едкого лития (около 53 г LiOH) и стехиометри-
чески рассчитанное количество кремневой кислоты: 67 г SiC>2
для получения метасиликата или 33,5 г SiOa для получения
ортосиликата.
Полученную смесь переносят в кварцевые кюветы или ча-
ши и помещают в муфельную печь; доводят температуру в
печи до 700—800°. По достижении указанной температуры
полупродукт подвергают прокалке в течение 5 часов.
После этого печь выключают и охлаждают. Кремневокис-
лый литий (мета или орто) получается в виде однородной
сыпучей массы кристаллов, легко поддающихся измельчению.
Выход метасиликата лития равен 96 г, выход ортосили-
ката лития равен 65 г, что составляет 97% от теоретическо-
го. Получаемый по данной методике метакремиевокислый ли-
тий соответствует РЭТУ 1128—63; ортокремневокислый ли-
тий— РТУ 1-7—61.
По литературным данным [3], для метасиликата лития:
плотность 2,52 г/сл’, т. пл. 120 Г ; для ортосиликата лития:
плотность 2,28 г/с.и3, т. пл. 1256°.
ЛИТЕРАТУРА
1. Сб. «Литий». Под ред. В. Е. Плющева, М., ИЛ, 1959, стр. 10.
2. Руководство по препаративной неорганической химии. Под ред.
Г. Брауэра, М., 1956, стр. 338.
3. Справочник химика, т. II, Л.-М., Госхимиздат, 1963, стр. 108.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.831.4'34.07
ЛИТИЙ ЦИРКОНАТ
С. А. КУТОЛИН, л. И. ВУЛИХ
Li2ZrO3 М. в. 153,09
Цирконат лития находит применение в керамической про-
мышленности и атомной технике [1—3].
В литературе описывается синтез цирконата лития сплав-
лением карбоната лития с двуокисью циркония, но этот про-
цесс требует сравнительно высоких температур, при которых
осложняется выбор материала для реакционных сосудов [4, 5].
Нами разработана методика синтеза стабильной формы
метацирконата лития спеканием едкого лития с двуокисью
циркония [6, 7].
СХЕМА СИНТЕЗА ЛИТИЯ ЦИРКОНАТА
2L1OH 4- ZrO2 — Li3ZrO3 %- Н2О
Характеристика основного сырья
Литий едкий, ч., ЦМТУ 2062—48.
Цирконий двуокись, я., МХТУ 2487—53.
Условия получения
Тщательно смешивают 61 г едкого лития (53% LiOH) с
85 а двуокиси Циркония и полученную шихту загружают в ко-
рундовый тигель, который помещают в муфельную печь. Спе-
кание производят в течение 4 часов при 950°. Спек измельча-
ют, получая белый порошок.
Выход лития цирконата равен 100 г, что составляет 97%
от теоретического. Продукт содержит около 98% лития цир-
коната; уд. в. 3,508 + 0,003; т. пл. 1530°. В воде постепенно
гидролизуется.
50
ЛИТЕРАТУРА
1. Сб. «Литий», Под ред. В. Е. Плющева, М., ИЛ, 1959, стр. 7.
2. Б. И. Коган. Литий, области освоенного и возможного примене-
ния, М., ВИНИТИ, 1960, стр. 23.
3. J. Schenk. Nucleonics, 10, 54 (1952).
4. А. А. Гризнк, В. Е. Плющев. Ж. неорган. химии, 6, 2249
(1961).
5. Н. A. Lehmann, Р. Erzberge г. Z. anorgan. und allgem Chem.,
301, 293 (1959).
6. С. А. Кутолин, H. А. Д р у з ь, А. И. Вулих. Ж. неорган. хи-
мии, 9, 2359 (1964).
7. С. А. Кутолин, А. И. Вулих, Н. А. Д р у з ь. Промышленность
химических реактивов, вып. 1 (7), М„ ИРЕА, 1965, стр. 42.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.34'171.1.07
ЛИТИЙ НИТРИД
С. А. КУТОЛИН, А. И. ВУЛИХ
Li3N ДА. в. 34,82
Нитрид лития может быть использован как катализатор
синтеза аммиака и как полупродукт при получении нитридов
металлов, плохо окклюдирующих азот [1]. Он может служить
также источником небольших количеств аммиака, выделяю-
щегося при действии на него воды.
В литературе описан ряд способов синтеза нитрида лития,
основанных на взаимодействии металлического лития с азо-
том: действие тока азота на расплавленный металл [2] или на
металл, тонко диспергированный в минеральном масле [3], а
также длительная выдержка металла под высоким давлением
азота при обычной температуре [4]. Во всех случаях указы-
вается необходимость тщательной очистки применяемого азо-
та от кислорода.
Найденные нами условия синтеза нитрида лития путем
взаимодействия металлического лития с азотом позволяют
быстро получать чистый препарат без расплавления или тон-
кого измельчения металла и без очистки азота.
СХЕМА СИНТЕЗА НИТРИДА ЛИТИЯ
6Li + N2 — 2L1SN
Характеристика основного сырья
Литий металлический, ГОСТ 8774—58.
Азот газообразный (из баллона), ТУ МХП 4280—54.
Условия получения
В стакан из нержавеющей стали емкостью 0,5—1 л поме-
щают 100 г металлического лития (около 200 см3), предвари-
тельно очищенного от поверхностной пленки и нарезанного
52
в форме пластинок толщиной 3—5 мм (см. примечание 1).
Стакан помещают в реторту, которую соединяют с баллоном
азота (см. примечание 2). В течение 1—2 часов температуру
постепенно повышают до 170—175° (не выше!) и давление
азота до 6—8 атмосфер. При этих условиях (давление азота
непрерывно поддерживают не ниже 5 атмосфер, температуру
не ниже 160° и не выше 175°) дают выдержку (в течение 4—6
часов (см. примечания 3, 4), затем реторту охлаждают и про-
дукт выгружают.
Выход нитрида лития равен 163—165 г, что составляет
98—99% от теоретического (см. примечание 5).
Нитрид лития получается в форме темно-красных или ко-
ричневых пластинок (сохраняющих форму исходного метал-
ла), которые затем измельчают в порошок; т. пл. 845°;
а?2о= 1,390. Отношение Li:N в получаемом нитриде 3,0±0,1.
По МРТУ 6-09-1105—64 нитрид лития должен содержать
не менее 35% азота и не более 0,05% примеси железа. Прак-
тически изложенный метод дает продукт, содержащий не ме-
нее 38% азота (по теории 40%) и не более 0,02% железа.
Содержание других примесей зависит от качества исходного
металла.
Примечания:
1. Предварительную механическую обработку и загрузку в стакан
металлического лития, а также измельчение и расфасовку нгприда сле-
дует проводить в инертной атмосфере (аргон) или в атмосфере азота.
Пластинки в сосуде помещают вертикально, с зазором (~ 1 мм) между
соседними пластинками.
2. Реторта до начала нагревания продувается током азота в течение
3—5 минут.
3. Реторта снабжается контактным термометром на 200—300°. тер-
морегулятором, манометром на 10—20 атмосфер и предохранительным
клапаном, отрегулированным на давление 8 атмосфер.
4. Температура плавления лития 180°, поэтому, особенно в первые
1—2 часа процесса, нельзя допускать перегрева реторты.
5. Хранить нитрид лития необходимо тщательно закупоренным, не
допуская контакта с влагой п с воздухом.
ЛИТЕРАТУРА
1. Е. Hoffman. Symp of, Nerver Metals., ASTM, 272, 195 (19.59).
2. F. Dafert, R. Miklauz. Monatsh., 31. 981 (1910); 32, 63 (1912)
3. Пат. США 2910347 (1959).
4. В. Neumann. C. Kroger, H. Habler. Z. апогеап. und allgein.
Chem, 204, 81 (1932).
Поступила в декабре i960 г.
ЦЗЛ
УДК 546.34-31.07+546.34-36.07
ОКИСЬ И БЕЗВОДНАЯ ГИДРООКИСЬ ЛИТИЯ
А. И. ВУЛИХ. М. И. МАКОВЕЦКИЙ, Л. Д. ПРИХОДЬКО
Ll,0 М. в. 29,87
LiOH М. в. 23,94
В литературе описаны методы синтеза окиси лития путем
окисления металла или разложения различных соединений
лития на воздухе [1—4]. Синтез чистого препарата по этим
методам требует большой продолжительности процесса и вы-
зывает аппаратурные трудности ввиду высокой агрессивно-
сти расплавленных литиевых продуктов при высоких темпе-
ратурах. Разложение карбоната лития в вакууме при 700°
требует нескольких суток {5], а прямое получение окиси ли-
тия из моногидрата гидроокиси лития (L1OHH2O) в ваку-
уме {6] также неудовлетворительно из-за агрессивности рас-
плава и улетучивания LiOH с водяным паром.
Свойства безводной гидроокиси и окиси лития (по лите-
ратурным данным i[7]): LiOH — плотность 1,43 г/см3, т. пл.
460—470°, т. кип. 925° (с разложением); LigO— плотность
2,01 г)см3, т. пл. >1500°, т. кип. 2600°.
Нами разработана методика получения окиси лития раз-
ложением гидроокиси лития в вакууме, особенность которой
заключается в промежуточном получении безводной гидро-
окиси [8].
СХЕМА СИНТЕЗА БЕЗВОДНОЙ ГИДРООКИСИ И ОКИСИ ЛИТИЯ
LiOH-H2O — LiOH + Н,О
2L1OH L12O + Н2О
Характеристика основного сырья
Литий едкий (моногидрат), ч., ЦМТУ 2062—48.
54
Условия получения
Получение безводной гидроокиси лития. В кварцевую кю-
вету или чашку помещают 314 г едкого лития (53% LiOH)
и выдерживают в вакуум-сушильном шкафу при 300 + 50°
и остаточном давлении 150—200 мм рт. ст. в течение 3 часов.
Обезвоженную гидроокись лития—белую пористую массу—
охлаждают в вакууме и измельчают в фарфоровой ступке
(см. примечание 1).
Выход безводной гидроокиси лития равен 163 г, что сос-
тавляет 98% от теоретического. Продукт содержит 97—98%
LiOH.
Получение окиси лития. В корундовый тигель емкостью
500 мл засыпают 163 г безводной гидроокиси лития и поме-
щают в вакуумную печь. Температуру повышают до 400° с
произвольной скоростью и одновременно понижают давление
до 5—10 мм. Далее, продолжая откачивание вакуум-насосом,
повышают температуру со средней скоростью 2—3 град[мин
до 900° (в течение 3—4 часов). При 900° выдерживают про-
дукт в течение 1 часа, понижая давление до 1—2 мм. Затем
повышают температуру до 950—1000° и выдерживают еще
в течение 30—60 минут. Полнота реакции подтверждается
сохранением вакуума при прекращении откачивания. Про-
дукт охлаждают в вакууме (или в токе азота) до комнатной
температуры и выгружают из тигля (см. примечание 2).
Окись лития представляет собой частично спекшуюся мас-
су, которую измельчают в фарфоровой ступке.
Выход продукта равен 100 г, что составляет 98%, от тео-
ретического, считая на безводную гидроокись, или 96% от ис-
ходного едкого лития.
Продукт согласно РЭТУ 797—61 (составленным на осно-
вании результатов, полученных по данному методу) пред-
ставляет собой белый порошок и содержит не менее 96%
LigO, не более 0,01% железа, 0,005% тяжелых металлов,
0,003% хлора, 0,1% веществ, нерастворимых в соляной кис-
лоте. Содержание LijO может достигнуть 98—99%, если ис-
ходный едкий литий свободен от карбоната. Содержание
алюминия (из корунда) не превышает 0,1% (см. примеча-
ние 3).
Примечания:
1. Измельчение как безводной гидроокиси, так и окиси лития сопро-
вождается образованием чрезвычайно едкой пыли, которая даже при не-
больших концентрациях в воздухе сильно раздражает глаза и органы
дыхания. Поэтому необходимо проводить измельчение и пересыпку этих
продуктов под тягой, в защитных очках и с респиратором. Лучше всего
воспользоваться боксом с защитной атмосферой, что благоприятно отра-
зится и на качестве препаратов.
2. Получение окнси лития из едкого лития моногидрата можно про-
водить и не выделяя обезвоживания моногидрата в обособленную опе-
рацию. В этом случае едкий литий моногидрат, помещенный в корундо-
55
вый тигель, выдерживают при условиях, указанных для стадии получе-
ния безводного LiOH, непосредственно в вакуумной печи, а затем повы-
шают температуру, как указано для стадии получения окиси лития. Од-
нако последующее измельчение окиси при этом труднее.
3. Хранить препараты следует в герметичной таре, предпочтительно
пластмассовой.
ЛИТЕРАТУРА
I. Ю. И. Остроушко и др. Литий, его химия и технология, М„
Атомиздат, 1960, стр. 44.
2. Р. Pierron. Bull. Soc. chim. France, 6, 235 (1939).
3. A. E. V a n A r k e 1. Canad. J. Chem., 31, 1009 (1953).
4. N. W. G r e g о r v, R. H. Mohr. J. Amer. Chem. Soc., 77, 2142
(1955).
5. E. Z I n 11, A. H а г d e r, B. D a u t h, Z. Elektrochem., 40, 258 (1934).
6. W. Klemm, H. J. Scharf. Z. anorgan, und allgem. Chem., 303.
263 (1960).
7. Справочник химика, т. 11. M., Госхимиздат, 1963, стр. 108, 110.
8. А. И. Вулих, М. И. Маковецкий, Л. Д. Приходько. Авт.
свид. 146730; Бюлл. изобр., № 9 (1962); Промышленность химических ре-
активов, Информационный бюллетень, вып. 2, М., ИРЕА, 1963, стр. 23.
Поступила в декабре 1965 г. цЗЛ
УДК 546.34'244.07
ЛИТИЙ ТЕЛЛУРИСТ^КИСЛЫЙ
О. И. БРЕУСОВ, Т. В. РЕВЗИНА
Li2TeO3 М. в. 189,47
Литий теллуристокислый 'представляет собой бесцветные
двуосные кристаллы с отрицательной индикатрисой; Ng> 1,78;
Np = 1,676 ±0,003; пикнометрическая плотность 3,83±0,02;
кристаллизуется в ромбической сингонии с параметрами
ячейки « = 8,79 кх; Ь = 10,52 кх; c = 7,\Q кх; г = 8; рентгенов-
ская плотность 3,836; вероятная пространственная группа
D^ — P 22,2; кристаллогидратов не образует и растворяется
в воде без разложения. При повышении температуры раство-
римость вещества уменьшается [1].
Растворимость лития теллуристокислого в воде
Температура, SC 30 40 50 60 70 80
Вес, % 12,65 10,44 8,96 7,72 6,66 5,86
В литературе [2] описан метод получения теллурита лития
сплавлением эквимолярных количеств двуокиси теллура и
карбоната лития. Однако в этих условиях возможно окисле-
ние Те4+ до Те5+ [3].
Описанный ниже способ основан на взаимодействии дву-
окиси теллура с гидратом окиси лития в водном растворе.
СХЕМА СИНТЕЗА ЛИТИЯ ТЕЛЛУРИСТОКИСЛОГО
Те + 4HNO3 = ТеО2 + 4NO3 + 2Н,0
ТеО2 + 2L1OH = LiaTeOs + Н2О
57
Характеристика исходного сырья
Теллур металлический, Т-1, ГОСТ 9514—60.
Литий гидрат окиси, ч.д.а., МРТУ 6—09—1471—64.
Азотная кислота, ч., ГОСТ 4461—48.
Условия получения
В фарфоровый стакан помещают 100 г порошкообразно-
го технического теллура и приливают небольшими порциями
(по 20 жл) 0,27 л азотной кислоты удельного веса 1,35—1,37.
Затем реакционную массу прогревают на песочной бане до
прекращения выделения окислов азота и фильтруют на во-
ронке Бюхнера через два слоя фильтровальной бумаги. От-
фильтрованный осадок двуокиси теллура промывают на
фильтре двумя литрами дистиллированной воды и переносят
в фарфоровый стакан емкостью 2,5 л. Туда же приливают
раствор 30 г едкого лития (в пересчете на 100%-ный) в 0,75л
воды. Растворение двуокиси теллура в едком литии проводят
при постоянном перемешивании в течение 1,5 часа при ком-
натной температуре. Затем раствор профильтровывают и упа-
ривают при периодическом перемешивании на песочной бане
при 120—130° до объема 60 мл. Выпавшую при упаривании
соль отделяют на воронке Бюхнера и промывают 30 мл аце-
тона. Промытую соль сушат в сушильном шкафу при тем-
пературе 90—100° в течение одного часа. Маточный раствор
упаривают досуха и выделенный теллурит лития подвергают
перекристаллизации из водного раствора. Полученный про-
дукт присоединяют к основной массе кристаллов. Из указан-
ной загрузки получают 108 г лития теллуристокислого.
Выход теллура в готовый продукт составляет 70—75%;
выход лития — 92%.
Полученный препарат соответствует квалификации «ч.» по
РЭТУ 1136—63.
ЛИТЕРАТУРА
1. О. Н. Бреусов, Т. В. Ревзина, Н. А. Д р у з ь, Ж- неорган.
химии, 10, 1990 (1965).
2. J. W. Mellor. A. Comprehensive Treatise on Inorganic and Theor-
etical chemistry, London, 1931, v. 11.
3. А. А. Кудрявцев. Химия и технология селена и теллура, М.,
Изд. «Высшая школа» 1961 г., стр. 89.
Поступила в декабре 1965 г. ЦВЛ
УДК 546.723'34-31.07
ЛИТИЙ ФЕРРИТ
С. А. КУТОЛИН, А. И. ВУЛИХ
LiFeO2 М. в. 94,786
Литий феррит аналогично ферритам других металлов мо-
жет найти применение в электро- и радиотехнической про-
мышленности, а также в металлургии. Имеется указание об
использовании твердого раствора ферритов лития и цинка в
радиотехнике [1].
В литературе описаны методы получения лития феррита
путем сплавления углекислого лития с окисью железа при
высоких температурах или нагревания концентрированного
раствора гидрата окиси лития с окисью железа под высоким
давлением [2].
Нами разработан метод получения феррита лития путем
спекания гидроокиси лития и окиси железа в вакууме.
СХЕМА СИНТЕЗА ЛИТИЯ ФЕРРИТА
2L1OH + FesO3 - 2LiFeO2 + Н2О
Характеристика основного сырья
Литий едкий, ч., ЦМТУ 2062—48.
Железо окись, ч.д.а., ГОСТ 4173—48.
Условия получения
В фарфоровой чашке тщательно перемешивают 49,1 г ед-
кого лития (53% LiOH) с 88,5 г окиси железа. Полученную
смесь помещают в муфельную вакуум-электрическую печь и
выдерживают в течение 3 часов при 650—700°/1 мм.
Выход продукта равен 100 г, что составляет 97% от тео-
ретического, с содержанием лития феррита 98—99%. По
59
внешнему виду вещество представляет собой красно-корич-
невый порошок, нерастворимый в воде, т. пл. 905°.
ЛИТЕРАТУРА
1 Б. И. Коган Литий, области освоенного и возможного приме-
нения, М„ ВИНИТИ, 1960, стр. 45.
2 . Руководство по препаративной неорганической химии, Под. ред.
Г. Брауэра, М., ИЛ, 1956, стр. 688.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.35'27'11.07 + 546.36'27'11.07
РУБИДИЙ БОРОГИДРИД И ЦЕЗИЙ БОРОГИДРИД
С. М. АРХИПОВ
RbBH4
CsBH4
М. в. 100,59
М. в. 148,03
Борогидриды рубидия и цезия представляют собой белые
кристаллические вещества, устойчивые в вакууме до 600°.
Некоторые их свойства представлены ниже:
Формула Плотность, г/ем3 Кристаллическая структура Теплота об- разования ккал! моль Показатель преломле- ния
RbBH4 1,71 [1] Кубическая гранецен- трированная а = 7,2 А -59 [2] 1,487 [1]
CsBH4 2,н m Кубическая гранецен- трированная а = 7,5 А -63 [2] 1,498 [I]
Борогидриды умеренно растворяются в метиловом спирте
и хорошо — в воде, причем борогидрид цезия растворим луч-
ше. В водных растворах оба соединения устойчивы и прак-
тически не разлагаются.
Борогидриды рубидия и цезия могут найти применение
как восстановители в неорганическом или органическом син-
тезе, а также в качестве высококалорийного твердого топли-
ва [3].
Единственный описанный в литературе [4] способ получе-
ния борогидридов рубидия и цезия заключается во взаим-
ном обмене борогидрида натрия с метилатами рубидия или
цезия в метиловом спирте. При этом менее растворимые бо-
рогидриды рубидия и цезия выпадают в осадок и отделяются
от раствора. Выход равен 85—95%.
61
Недостатком этого способа является сложность приготов-
ления метилатов, которые получают из металлов и абсолют-
ного метилового спирта.
Нами разработан способ получения борогидридов руби-
дия и цезия в водно-спиртовых растворах с использованием
более доступных соединений. Этот способ позволяет приме-
нять технический борогидрид натрия чистотой 85—90% без
предварительной его очистки.
СХЕМА СИНТЕЗА БОРОГИДРИДОВ РУБИДИЯ И ЦЕЗИЯ
NaBH4 + RbOH -* RbBH4 + NaOH
NaBHj % CsOH — CsBH4 + NaOH
Характеристика основного сырья
Борогидрид натрия, технический, с чистотой 85—90%.
Гидроокись рубидия, ч., РЭТУ 829—61.
Гидроокись цезия, ч., РЭТУ 658—60.
Метиловый спирт, ч., ГОСТ 2222—54.
Условия получения
Рубидий борогидрид. Растворяют 2,9 г борогидрида нат-
рия в 10 мл дистиллированной воды при температуре 0°.
Раствор фильтруют через стеклянный фильтр в колбу. Затем
растворяют 6 г гидроокиси рубидия в 16 мл метилового спир-
та и фильтруют. После охлаждения раствора гидроокиси ру-
бидия до 3—5° его прибавляют к осветленному раствору бо-
рогидрида натрия, при этом выпадает белый осадок боро-
гидрида рубидия, который отделяется от маточного раствора
фильтрацией. Осадок промывают на фильтре 100 мл метило-
вого спирта и сушат в вакууме при 30°. Промывной раствор
объединяется с маточным раствором и упаривается для вы-
деления большего количества борогидрида рубидия. Общий
выход продукта равен 4,5 г, что составляет 90% от теорети-
ческого.
Найдено, %: Rb—85,1; В—10,6; Н—4,1.
RbBH4. Вычислено, %: Rb-85,2; В-10,78; Н—4,02.
Борогидрид цезия получают по аналогичной методике с
выходом 80% от теоретического.
Найдено, %: Cs—89,5; В—7,3; Н-2,74.
CsBH4. Вычислено, %: Cs—89,95; В—7,32; Н—2,72.
62
ЛИТЕРАТУРА
1. М. О. В a n u s, R. W. Bragdon, A. A. Hinckley, d. Amer.
Chem. Soc., 76, 3848, (1954).
2. A. P. Altshu Her. J. Amer. Chem. Soc., 77, 5455 (1955).
3. Ф. M. Перельман. Рубидий и цезий, М., Изд. АН СССР, 1960,
стр. 9.
4. Англ. пат. 730263 (1955).
Поступила в декабре 1965 г.
ЦЗЛ
УДК 547.562.4'135.07+547.562.4' 136.07
РУБИДИИ ФЕНОЛЯТ И ЦЕЗИЙ ФЕНОЛЯТ
Г. Е. РЕВЗИН, В. Д. ЗАМЕДЯНСКАЯ
CeH8ORb-2H:O М.в. 214,61
C6H-OCs-2H3O М. в. 262,04
По фенолятам рубидия и цезия в литературе имеется не-
большое количество отрывочных сведений. Описаны способ
их получения [1] и некоторые свойства [2—4].
Феноляты рубидия и цезия могут представлять интерес
для синтеза оксикарбоновых кислот ароматического ряда по
методу Кольбе—Шмидта, простых и сложных эфиров фено-
лов [4—6], а также для синтеза некоторых соединений ру-
бидия и цезия.
Общий способ получения фенолятов щелочных металлов
заключается во взаимодействии фенола с гидроокисью ще-
лочного металла в водном, водно-спиртовом или спиртовом
растворе [1—9].
Нами уточнены условия получения фенолятов рубидия и
цезия и определены их плотности и температуры плавления.
СХЕМА СИНТЕЗА ФЕНОЛЯТОВ РУБИДИЯ И ЦЕЗИЯ
^ОН + МеОН-| Н2О ^ОМе-2НгО
Характеристика основного сырья
Рубидий гидрат окиси, ч., РЭТУ 829—61.
Цезий гидрат окиси, ч., РЭТУ 658—60.
Фенол синтетический, ч., ГОСТ 6417—52.
Этиловый спирт, ректиф., ГОСТ 5962—51.
Диэтиловый эфир, мед., Ф IX, ГОСТ 5963—51.
64
Условия получения
Смесь 25,6 г (0,25 М) гидрата окиси рубидия (или 37,5г
(0,25 Л4) гидрата окиси цезия), не содержащего карбонатов
(см. примечания 1,2), 15 мл этилового спирта и 23,5 г
(0,255 М) свежеперегнанного фенола помещают в стакан из
термостойкого стекла и подогревают при постоянном пере-
мешивании до полного растворения фенола и гидроокиси ру-
бидия (цезия) в спирте. После получения гомогенного рас-
твора его охлаждают до комнатной температуры и выделя-
ют фенолят рубидия (цезия), осаждая его добавлением
150 мл диэтилового эфира. Выделившийся в виде белого
кристаллического осадка с перламутровым блеском фенолят
рубидия (цезия) отфильтровывают и сушат в эксикаторе над
едким кали при комнатной температуре.
Выход фенолята рубидия цавен 52 г, что составляет 97%
от теоретического; выход фенолята цезия равен 64 г, что со-
ставляет 98% от теоретического.
Феноляты рубидия и цезия гигроскопичны и во влажном
воздухе расплываются; в сухом воздухе устойчивы. Фенолят
рубидия имеет т. пл. 61 ±0,5% dww—1,72±0,01; фенолят це-
зия—т. пл. 58±0,5°; ri2o20—1,93±0,01.
Указанный метод синтеза позволяет получать продукты, удовлетво-
ряющие следующим требованиям.
Фенолят рубидия:
содержание фенолята рубидия—не менее 98,0%
содержание примесей (в %, не более): железо—1.10-% сульфаты—
2,5.10 % хлориды—1 10*2.
Фенолят цезия:
содержание фенолята цезия -не менее 98%.
содержание примесей (в %, не более): железо—1.10 ~3,’ сульфаты—•
3.10 —3, хлориды—1.10-2.
Примечания:
I, Гидроокиси рубидия и цезия, не содержащие карбонатов, примесь
которых нежелательна при получении фенолятов, могут быть получены
методом электролиза раствора хлорида илн карбоната с ртутным като-
дом с последующим разложением амальгамы водой.
2. Гидроокиси рубидия и цезия по техническим условиям выпуска-
ются в виде моногидратов (RbOH.H2O и CsOH.H2O), содержащих колеб-
лющиеся количества МеОН, поэтому перед проведением синтеза необхо-
димо определить содержание RbOIl и CsOH в исходном сырье и затем
рассчитать потребные количества исходного сырья, содержащего 0,25 М
безводной гидроокиси в продукте.
ЛИТЕРАТУРА
1. F. Wessely, К. В е n е d i k t. Н. Bengcr, G. Friedrich’
F. Р г i 1 11 и g е г. Monatsh., 81, 1071 (1950).
2. H. В, Ворожцов. Основы синтеза промежуточных продуктов и
красителей, М., Госхимиздат, 1955, стр. 749.
3. Н. Я. Турова, А. В. Новоселов а. Усп. химии, 34, 385 (1965).
5 Зак 1060 65
4. A. H. Несмеянов, И. Ф. Луценко. Докл. АН СССР, 59, 707
(1948).
5. П. Каррер. Курс органической химии, М., Госхимиздат, 1962,
стр. 540.
6. А. Е. Ч и ч и б а б и н. Основные начала органической химии, т. II,
М„ Госхимиздат, 1957, стр. 384.
7. К. Д. Н е н и ц е с к . Органическая химия, т. 1, М„ ИЛ, 1962,
стр. 475.
8. Н. Erlenmeyer. Helv. Chim. Acta., 9, 648(1926).
9. A. N. М е 1 d г u m, М. М. Patel, J. Indian. Chetn. Soc. 5. 91
(1928).
Поступила в декабре 1965 г. П.ЗЛ
УД К 546.35' 195 = 482.07+546.36' 195 = 482.07
РУБИДИЙ И ЦЕЗИЙ МЫШЬЯКОВОКИСЛЫЙ
ОДНОЗАМЕЩЕННЫЕ ОСОБОЙ ЧИСТОТЫ
Р. М. ШКЛОВСКАЯ, С. И. РОМАНОВА
RbH2AsO4 М. в. 226,4
CsH2AsO4 М. в. 273,84
Рубидий и цезий мышьяковокислые однозамещенные (ди-
гидроарсенат рубидия и цезия представляют собой кристал-
лические вещества белого цвета, хорошо растворимые в воде
[1].
Растворимость RbH2AsO4 и CsH2AsO4 в воде составляет
И:
Соединение Температура, °C
0 25 | 50 | 75
RbH2AsO4 29,09 39,82 50,23 58,27
CsHjAsO, 66,62 71,63 76,23 79,92
Температура разложения RbH2AsO4 250° и CsH2AsO4 260°
[2]. При 150° CslI2AsO4 претерпевает полиморфное превраще-
ние [2].
Дигидроарсенаты щелочных металлов при пониженных
температурах обладают сегнетоэлектрическими свойствами и
в связи с этим представляют интерес для радиоэлектроники
[3]. Для этих целей требуются препараты высокой степени
чистоты.
Дигидроарсенат рубидия получают либо сплавлением
нитрата рубидия с трехокисью мышьяка, взятых в эквимо-
лекулярном соотношении, либо путем нейтрализации водно-
го раствора карбоната рубидия мышьяковой кислотой в при-
сутствии индикатора метилоранжа {1, 4]. Сведений по синте-
зу дигидроарсената цезия в литературе не имеется.
67
Нами разработана методика получения дигидроарсенатов
рубидия и цезия ос. ч. из металлического мышьяка высокой
чистоты и карбонатов рубидия и цезия ос. ч.
СХЕМА СИНТЕЗА РУБИДИЯ И ЦЕЗИЯ МЫШЬЯКОВОКИСЛЫХ
ОДНОЗАМЕЩЕННЫХ ОСОБОЙ ЧИСТОТЫ
As + 5HNOS — H3AsO4 + 5NO, + Н2О
Rb2CO3 4- 2H3AsO4 — 2RbH2AsO4 + H2O + CO2
Cs2CO3 + 2H3AsO4 — 2CsH2AsO4 - H,0 - CO,
Характеристика основного сырья
Цезий углекислый, ос. ч., с содержанием Rb—1,6.10—\
Na— 5.10~4и К — 5.10-*%.
Рубидий углекислый, ос. ч., с содержанием Cs—1,8.10“’3
Na — 1.10“8и К—1.10 “8 (см. примечание 1).
Мышьяк металлический, класса В 4, ТУ ГКХ 1498—61. •
Азотная кислота, ос. ч., класса В 3, ВТУ АУ 140—59.
Условия получения
Растворяют в колбе, закрытой часовым стеклом, 28,8 г
металлического мышьяка в 124 мл концентрированной азот-
ной кислоты (уд. вес 1,417 г/см3) при умеренном нагревании
на водяной бане. Продолжительность растворения составляет
8—10 часов.
С целью удаления избытка азотной кислоты и окислов
азота полученный раствор мышьяковой кислоты переносят
в фарфоровую чашку и упаривают досуха при температуре
90—1003 После этого кристаллы H3AsO4 растворяют в 60мл
бидистиллировашюй воды. Затем 44,4 г карбоната рубидия
или 62,9 г карбоната цезия растворяют в 60 мл нагретой до
70—80° бидистиллированной воды и нейтрализуют 60 мл рас-
твора мышьяковой кислоты. В конце операции pH соответст-
вует 4,2 по универсальной индикаторной бумаге «Phan» (или
началу перехода окраски метилоранжа из желтой в оранже-
вую) .
Раствор дигидроарсената рубидия или цезия упаривают
досуха на закрытой электроплитке при температуре 95—100°
и сушат в сушильном шкафу до постоянного веса при 100—
110°.
Выход дигидроарсената рубидия равен 82,5 г, а дигидро-
арсената цезия 100 г, что соответствует 95% от теоретическо-
го.
Состав получаемых продуктов представлен в таблице.
68
Таблица
Наименование соединения Содержание контролируемых примесей (в % %, не более)*
AI Ре К Са Si Mg Мп
Рубидий мышьяково- кислый однозаме- щенный НО-4 5I6- 5 I-I0-s 1,5-10-3 I-I0*3 5-Ю"4 1-10“ °
Цезий мышьяково- кислый однозаме- щенпый 1 10-4 5'10—6 5-1СГ4 1 10-8 I -10~3 5-Ю'1 1 • 10'°
Продолж. табл_
Наименование соединения Содержание контролируемых примесей (в % %, не более)*
Си | Na РЬ Sb Cs (Rb) Cl'
Рубидий мышьяково- кислый однозамещен- ный 2'10-6 I I0-8 3-Ю-6 2-10-6 1-10~3 ЗТ0-3
Цезий мышьяковокис- лый однозамещенный 1 10“6 5I0-4 1.10~° 3-Ю-6 1 -10“J 5 10~3
* Содержание примесей может быть
соединений в специальном помещении для
ществ.
снижено при синтезе данных
производства особо чистых ве-
Примечание.
Карбонаты рубидия н иезпя получены путем взаимодействия нитра-
тов соответствующих металлов особой чистоты со щавелевой кислотой с
последующим разложением полученных тетраоксалатов при температуре
550-600°:
MeNO3 - 2Н2С2О4 = МеН3 (С2О4)2 + HNO3
2МеН3 (С2О4)2 4- 2О2 = Ме2СО3 + ?СО2 + ЗН,0
где Me — Rb или Cs.
ЛИТЕРАТУРА
I . Gmelins, Handbuch der anorganischen Chemie, S, 24, 1937.
2. Ф. Иона, Д. Ширан e. «Сегнетоэлектрические кристаллы», M.,
Изд. «Мир», 1965, стр. 95.
3. I. W. Mellor. Treatishe on Inorganic and Theoretical chemisty.
vol IX, 1946.
Поступила в марте 1966 г.
УДК 546.36-36.07 + 546.35-36.07
гидроокись ЦЕЗИЯ И ГИДРООКИСЬ РУБИДИЯ
П. Д. КОМИССАРОВА
RbOHH2O М. в. 120,49
CsOH-H2O М. в. 167,92
Гидроокиси рубидия и цезия представляют собой белые
кристаллические вещества, плавящиеся при 145° и 180° со-
ответственно [1, 2].
На воздухе они жадно поглощают влагу и углекислоту,
расплываясь и переходя в углекислые рубидий и цезий.
Обезвоживание моногидратов возможно при длительном
нагревании в токе сухого 'водорода, не содержащего угле-
кислоты {2].
Гидроокиси рубидия и цезия применяются при получении
других соединений этих элементов, а также в виде растворов
в щелочных аккумуляторах [1].
В литературе описано много способов получения гидро-
окисей рубидия и цезия как из металлов, так и из различных
их солей [3]. Наиболее простым является метод, основанный
на взаимодействии углекислого рубидия (цезия) с гидро-
окисью бария. Нами уточнены условия синтеза по этому ме-
тоду.
СХЕМА СИНТЕЗА ГИДРООКИСЕЙ РУБИДИЯ И ЦЕЗИЯ
Rb2CO3 + Ва (ОН), = 2RbOH + ВаСО31
Cs2CO3 + Ва (ОН), = 2CsOH 4- ВаСО3 |
Характеристика основного сырья
Рубидий углекислый, ч., МРТУ 6-09-227—63.
Цезий углекислый, ч., МРТУ 6-09-1721—64.
Барий гидрат окиси, ч., ГОСТ 4107—48.
70
Условия получения
Растворяют 100 г углекислого рубидия или углекислого
цезия в 100 мл дистиллированной воды и к полученному рас-
твору добавляют при непрерывном перемешивании горячий
насыщенный раствор гидроокиси бария с небольшим избыт-
ком к стехиометрии. Отфильтрованные растворы гидроокисей
рубидия или цезия упаривают в серебряных или платиновых
чашах при непрерывном кипении с целью уменьшения кар-
бонизации. По мере упаривания небольшой избыток гидро-
окиси бария, введенный при каустификации, выпадает в оса-
док, так как в концентрированных растворах гидроокисей
рубидия или цезия гидрат окиси бария нерастворим.
Образовавшийся осадок гидроокиси бария отфильтровы-
вают и продолжают упаривание растворов гидроокисей руби-
дия и цезия до тех пор, пока не прекратится кипение.
Плав быстро охлаждают, помещая чашу в лед, и быстро
измельчают.
Выход гидроокиси рубидия равен 85 г и гидроокиси це-
зия— 82 г, что составляет 80% от теоретического.
Продукт хранят в винипластовых запарафинированных
банках.
Данная методика позволяет получать продукт, удовлетво-
ряющий требованиям технических условий МРТУ 6-09-224—
63 для гидроокиси рубидия и РЭТУ 1046—63 для гидроокиси
цезия.
ЛИТЕРАТУРА
1. Ф. М. Перельман. Рубидий и цезий, М„ Изд. АН СССР, 1960,
стр. 9, 26.
2. Руководство по препаративной неорганической химии, под ред.
Г. Брауера, М„ ИЛ, 1956, стр. 461.
3. Gmelins Handbuch der anorganischen Chernie 8. Aufl., SN 21
1937, стр. 106. SN 25, 1938, crp. 109.
Поступили в декабре 19бэг.
из Л
УДК 546.717'35.07 + 546,717'36.07
РУБИДИЙ МАРГАНЦОВОКИСЛЫЙ И ЦЕЗИЙ
МАРГАНЦОВОКИСЛЫЙ
Перманганаты рубидия и цезия
П. Д. КОМИССАРОВА, С. А. КРЕСТОВНИКОВА
RbMnO4 М. в. 204,40
CsMnO4 М, в. 251,84
Рубидий и цезий марганцовокислые относятся к малорас-
творимым соединениям. Растворимость перманганата руби-
дия при 0°—0,5 г, при 60°—4,7 г на 100 г воды; раствори-
мость перманганата цезия при 0°—0,1 г, при 60°— 1,3 г на
100 г воды. Синтез перманганатов рубидия и цезия основан
на осаждении их из растворов перманганатом калия и даль-
нейшей очистке путем перекристаллизации [1].
СХЕМА СИНТЕЗА КАРБОНАТОВ РУБИДИЯ И ЦЕЗИЯ
Rb2SO4 4- 2КМпО4 — 2RbMnO4 + K2SO4
Cs2SO4 + 2KMnO4 — 2CsMnO4 -f- K2SO4
Характеристика основного сырья
Рубидий сернокислый, техн., с содержанием основного ве-
щества 90%, калия до 5%, цезия 0,5%.
Цезий хлористый, ч., МРТУ 6-09-242-63.
Калий марганцовокислый, ч., ГОСТ 4527—48.
Условия получения
В 1 л горячей воды растворяют 100 г сернокислого руби-
дия и в 600 мл горячей воды растворяют 135 г марганцово-
кислого калия (избыток 10 г/л сверх теоретически рассчитан-
ного количества).
72
Приготовленные таким образом горячие растворы слива-
ют вместе и реакционную смесь медленно, без перемешива-
ния, охлаждают до комнатной температуры. Осадок пер-
манганата рубидия отфильтровывают и отмывают на фильт-
ре от маточного раствора небольшим количеством холодной
воды.
С целью очистки перманганата рубидия от калия и дру-
гих примесей осадок переносят в фарфоровый стакан емко-
стью 3—5 л, заливают горячей (80—90э) дистиллированной
водой, перемешивают до полного растворения и охлаждают
до 10—203.
Выкристаллизовавшийся осадок перманганата рубидия
отфильтровывают и сушат при 80°. Полученная соль пред-
ставляет собой игольчатые фиолетовые кристаллы.
Выход марганцовокислого рубидия, без переработки ма-
точных растворов и промывных вод, равен 107 г, что состав-
ляет 70% от теоретического.
Перманганат цезия получается по аналогичной методике,
с той лишь разницей, что перекристаллизация соли заменя-
ется двукратной промывкой горячей дистиллированной водой
при соотношении Т:Ж=1:2.
Данная методика позволяет получать перманганаты руби-
дия и цезия, соответствующие требованиям технических усло-
вий РЭТУ 909—62 (RbMnO4) и МРТУ 6-09-244—63 (CsMnO4).
ЛИТЕРАТУРА
1. А. М. Patterson. J. Amer. Chem. Soc., 28, 1735 (1906).
Поступила в декабре 1966 г.
цзл
УДК 546.35'264.07 + 546.36'264.07
РУБИДИЙ УГЛЕКИСЛЫЙ И ЦЕЗИЙ УГЛЕКИСЛЫЙ
А, И. ВУЛИХ, М. М. СЕН Я ВИ И, Б. И. КОРОТК.ЕВИЧ
Rb3CO3 М. в. 230.95
Cs2CO3 М. в. 325,82
Карбонаты рубидия и цезия являются исходными мате-
риалами для получения различных соединений этих метал-
лов, а также непосредственно применяются в аналитической
химии и органическом синтезе [1, 2].
Соли представляют собой бесцветные гигроскопичные ве-
щества, хорошо растворимые в воде. Растворимость карбо-
ната рубидия при 20° (вес. %): в воде 69, в этиловом спирте
0,7; т. пл. 8377 Растворимость карбоната цезия при 20° (вес.
%): в воде 72, в этиловом спирте 10; т. пл. >600° [1— 3].
В литературе описан ряд чисто химических методов полу-
чения карбонатов рубидия и цезия: термическим разложе-
нием оксалатов и других солей органических кислот, взаимо-
действием гидроокисей рубидия и цезия с углекислотой или
карбонатом аммония; по реакции между сульфатами руби-
дия и цезия с гидроокисью бария с последующей карбониза-
цией раствора [1, 2]. В частности, из хлоридов рубидия и це-
зия карбонаты этих металлов могут быть получены следую-
щими двумя способами: а) хлорид обрабатывают крепкой
азотной кислотой до удаления хлористого водорода и образо-
вавшийся нитрат прокаливают с 4-кратным избытком щаве-
левой кислоты [4]; б) хлорид обрабатывают концентрирован-
ной серной кислотой, полученный сульфат растворяют, до-
бавляют гидроокись бария, раствор отделяют от осадка суль-
фата бария, насыщают углекислотой, выпаривают досуха и
остаток прокаливают [2].
Описанный ниже ионообменный метод значительно более
эффективен, так как позволяет получить высокий выход кар-
боната рубидия при затрате дешевого реагента — карбоната
аммония [5].
74
СХЕМА СИНТЕЗА КАРБОНАТОВ РУБИДИЯ И ЦЕЗИЯ
MeCl + RNH4 -» RMe + NH4Cl
2RMe + (NH4)2CO3 — 2RNH4 -L- Me2COs
где Me = Rb или Cs, R — радикал функциональной группы
катионита.
Характеристика основного сырья
(см. примечание 1)
Рубидий хлористый, ч., ч.д.а. или х.ч., МРТУ 6-09-229—63.
Цезий хлористый, ч., ч.д.а. или х.ч., МРТУ 6-09-2607—65.
Аммоний углекислый, ч.д.а. или х.ч., ГОСТ 3770—47.
Условия получения
Подготовка катионита. Катионит КУ-2, предварительно
обработанный по методике, описанной в статье [6], переводят
в аммонийную форму путем контактирования с избытком
5%-ного раствора карбоната аммония. Об окончании реакции
судят по прекращению выделения углекислоты. Катионит от-
деляют от раствора фильтрованием и загружают в колонку
[6]. Для единовременного получения 100—150 г карбоната ру-
бидия или цезия подготавливают 300—400 г ионита (считая
на сухую смолу). Приготовленный ионит может использо-
ваться для многократного получения карбонатов без проме-
жуточной подготовки, так как в процессе десорбции происхо-
дит регенерация катионита с образованием исходной аммо-
нийной формы.
Получение рубидия или цезия углекислого. Растворяют
100 г хлористого рубидия или хлористого цезия в 1 л дистил-
лированной воды и полученный раствор пропускают через ко-
лонку с катионитом со скоростью 15—20 жл/лшн. Фильтрат,
представляющий собой чистый раствор хлористого аммо-
ния, выводится из процесса (см. примечание 2). По оконча-
нии пропускания раствора хлорида катионит промывают дис-
тиллированной водой до отсутствия иона хлора в фильтрате
(проба с азотнокислым серебром). Затем пропускают через
колонку 7—10%-ный раствор карбоната аммония со скоро-
стью 15—20 мл/мин до отсутствия иона рубидия или цезия в
фильтрате (проба с силикомолибденовой кислотой или тетра-
фенилборатом натрия). Фильтрат объемом 1—1,5 л содержит
наряду с карбонатом рубидия или цезия также избыток кар-
боната аммония. Фильтрат выпаривают досуха и прокалива-
ют в течение 1 часа при 400—500° в фарфоровой чашке.
Карбонат аммония при этом полностью улетучивается. По-
лучают около 100 г карбоната рубидия или цезия, что соот-
75
ветствует выходу 95—97% от теоретического. Квали-
фикация полученного препарата соответствует качеству ис-
ходной соли рубидия или цезия. Рубидий углекислый удов-
летворяет требованиям МРТУ 6-09-227 -63, цезий углекис-
лый МРТУ 6-09-1721—64.
Примечания:
1. В качестве исходного сырья может быть использован не только
хлорид, по и любая другая хороню растворимая соль рубидия или це-
зия.
2. При указанной выше загрузке катионита в колонке (300—400 г) на
стадии сорбции проскок рубидия или цезия в фильтрат не наблюдается.
Но во избежание случайных потерь редких металлов следует убедиться в
их отсутствии в фильтрате (проба с силикомолибденовой кислотой или
тетрафенилборатом натрия).
ЛИТЕРАТУРА
1. Краткая химическая энциклопедия, т. IV, Изд. «Советская энци-
клопедия», М„ 1965, стр. 716.
2. Ф. М. Перельма н. Рубидий и цезий, М., Изд. АН СССР, 1960,
стр. 8, 51.
3. Справочник химика, т. 11, Л.—М Госхимиздат, 1963, стр. 188,
248.
4. R. Shurmann. К. С 1 u s I и s. Z. anorg. Chem.. 152, 52(1926)
5. А. И. Вулих, М. М. С е н я в и н, Ю. Б, П е р к о в с к а я, Б. И.
К о р о т к е в и ч, Л. Г. Сидорова, Н. К. Галкина. Авт. свид. 128454;
Бюлл. изобр., № 10 (1960).
6. А. И. В у л и х, В. Л. Б о г а т ы р е в. В. Л. К а з ь м и н с к а я,
Л. П. Жердиепко. См. статью «Ионообменное получение кислот из
солей» в этом сборнике, стр. 5,
Поступила в впреле 1966 г. ц 1Л
УДК 546.35'125-482.07 + 546.36' 125-482.07
РУБИДИЙ И ЦЕЗИЙ ХЛОРНОВАТО-, БРОМНОВАТО-
И ЙОДНОВАТОКИСЛЫЕ ОСОБОЙ ЧИСТОТЫ
Р. М. ШКЛОВСКАЯ. А. И. ВУЛИХ
RbC103
Рубидий хлорноватокислый особой чистоты CsC103 М. в. 168,92
Цезий хлорноватокислый особой чистоты RbBrO3 М. в. 216,36
Рубидий бромноватокислый особой чистоты CsBrO3 м. в. 213,37
Цезий бромноватокислый особой чистоты RbJO3 м. в. 260,81
Рубидий йодноватокислый особой чистоты CsJO3 м. в. 260,37
Цезий йодноватокислый особой чистоты м. в. 307,80
Соединения типа АХО?„ где A--Rb или Cs, X—галоид,
представляют в настоящее время интерес по крайней мере в
двух отношениях. Во-первых, благодаря высокому темпера-
турному коэффициенту растворимости и сравнительно низ-
ким температурам термического разложения эти соединения
могут быть использованы для глубокой очистки рубидия и
цезия от примесей и последующего получения высоко чистых
галогенидов этих металлов — важнейших материалов для
специальной оптики и других областей новой техники. Во-
вторых, хлораты, броматы и йодаты рубидия и цезия могут
получить непосредственное применение благодаря собствен-
ным физическим свойствам, в частности пьезоэлектрическим.
В обоих случаях необходимы препараты высокой чистоты.
Наконец, очищенные соединения могут быть использованы
для получения других (кроме галогенидов) высоко чистых
солей рубидия и цезия.
В литературе методы получения особо чистых соединений
этой группы не описаны.
77
Указанные вещества представляют собой кристаллы бе-
лого цвета, устойчивые на воздухе. Некоторые их свойства
приведены в таблице 1 [1—3].
Физико-химические свойства
Таблица 1
хлоратов, броматов и йодатов рубидия и
цезия *
Наименование соединений
Свойства RbClO3 СзС1О3 RbBrO3 СзВгО3 RbJO3 CsJO,
Растворимость в воде (вес.%): при 15° при 25° при 100° 4,23 6,17 38,59 4,95 7,27 43,71 1,95 2,93 20,96 2,85 3,75 25,96 1,83 2,41 10,46 1,93 2,62 12,58
Температура плавления, °C Температура начала разло- жения, °C 340 534 392 548 | 428 458 534 531
* Броматы и йодаты рубидия и цезия плавятся разложением.
Предлагаемая методика включает приготовление водных
растворов солей из карбонатов рубидия и цезия и растворов
галоидокислородных кислот, полученных ионообменным ме-
тодом из соответствующих солей калия [4, 5]. Последующая
очистка проводится путем фракционной кристаллизации, ус-
ловия которой были подробно изучены для хлората рубидия
[6, 7] и дополнительно исследованы в приложении к осталь-
ным галоидокислородным солям рубидия и цезия.
Таблица 2
Коэффициент кристаллизационной очистки галоидокислородных солей
рубидия и цезия от щелочных примесей за одну кристаллизацию
Условия кристаллизации: температура исходного насыщенного раствора
90°. конечная температура охлаждения 15°. Исходное содержание
примесей 0,05—0,001%
Примесь Очищаемое соединение
RbC103 CsClO, RbBrO3 CsBiOj RbJO3 CsJO3
Na 4 —5 ~10 ~10 ~I0 ~5
К 2,9 5 3,1 1.5 3,1 1,6
Rb — 2,0 — 1,4 — 1,3
Cs 1,9 — 1,5 — 1,7 i
Кратность кристаллизационной очистки определяется ис-
ходным и конечным (заданным) содержанием примесей эле-
78
ментов-аналогов (К и Cs — в солях рубидия, К и Rb — в со-
лях цезия) и коэффициентом очистки, значения которого для
щелочных примесей приведены в таблице 2. Очистка от дру-
гих примесей проходит гораздо более эффективно и достига-
ется попутно.
В приведенных ниже методиках очистки отдельных солей
указано число операций кристаллизации, которое необходи-
мо провести для получения препарата с содержанием каж-
дой из щелочных примесей не более 0,001% при исходном
содержании, указанном в характеристике основного сырья.
Если исходное или заданное содержание примесей отличает-
ся от данных, кратность кристаллизации может быть опре-
делена по формуле:
п = Си - 1g Ск
IgAT
где С’и и Ск — исходное и конечное содержание примеси
(вес. %),
/ с
К — коэффициент очистки АГ = — для одной
. Ск
операции кристаллизации).
Характеристика основного сырья
Рубидий углекислый, х.ч. (содержание Na—0,008%, К—
0,05%, Cs —0,02%).
Цезий углекислый, ос. ч., МРТУ 6-09-241—63 (содержание
Na и К —по 0,003%, Rb— 0,005%).
Хлорноватая кислота, ~0%-ный раствор, получают [5] из
дважды перекристаллизованного калия хлорноватокислого
технического, высшего сорта, ГОСТ 2713—49, с содержанием
К не более 0,001 г/л.
Бромноватая кислота, ~5%-ный раствор, получают [5] из
калия бромноватокислого, ч.д.а. или х.ч., ГОСТ 4457—48, с
содержанием К не более 0,001 г/л.
йодноватая кислота, ~10%-ный раствор, получают [5] из
калия йодноватокислого, ч.д.а. или х.ч., ГОСТ 4202—48, с
содержанием К не более 0,001 г/л.
йодноватая кислота, х. ч., ГОСТ 4213—48.
Условия получения
Подробное описание процесса приводится только для ру-
бидия хлорноватокислого. При получении других солей сле-
дует обращаться к этому описанию для уточнения деталей
проведения операций.
79
Расход основных компонентов рассчитан на получение
100 г того или иного препарата с содержанием примесей,
указанным в таблице 3.
Рубидий хлорноватокислый. К 1 л 5 %-ной хлорноватой
кислоты прибавляют при перемешивании 110 г кристалличе-
ского рубидия углекислого. Затем постепенно приливают кис-
лоту до прекращения выделения углекислоты и установления
pH 5—5,5 (по универсальному индикатору). Общий расход
хлорноватой кислоты составляет около 1,5 л. Полученный
раствор упаривают до объема 320—340 мл и охлаждают прн
перемешивании до 15—20°, после чего продолжают переме-
шивание еще в течение 1—2 часов. Выпавшие кристаллы от-
фильтровывают от маточника на воронке под вакуумом и
промывают на фильтре 40—50 мл холодной дистиллирован-
ной воды. Получают около 150 г соли с влажностью ~10%.
Кристаллы вносят в 220—230 мл горячей (70—80°) биди-
стиллированной воды и нагревают при перемешивании до
полного растворения соли (до 95—100°), после чего раствор
охлаждают при перемешивании до температуры 15—20° (при
этом выпадает основная масса кристаллов), выдерживают в
течение 1—2 часов, отделяют кристаллы и промывают 30—
40 мл холодного бидистиллята. Перекристаллизацию прово-
дят трижды (на растворение для второй кристаллизации вво-
дят 200—210 мл, для третьей—180—190 мл бидистиллиро-
ванной воды).
Выход кристаллов соли после каждой операции составля-
ет около 90%. После третьей перекристаллизации кристаллы
высушивают в фарфоровой чашке при 100 110° в течение 2
часов.
Получают 100 г рубидия хлорноватокислого ос. ч., что со-
ответствует 65%, считая на исходный карбонат рубидия.
При упаривании маточного раствора от первоначальной
кристаллизации хлората рубидия может быть дополнительно
выделено 12—14 г соли квалификации «ч.д.а», а из маточно-
го раствора от 1-й кристаллизации 10—12 г хлората рубидия
х. ч. При получении значительных количеств рубидия хлор-
новатокислого ос. ч. целесообразно проведение процесса в не-
сколько циклов с использованием маточников в следующем
цикле (маточник от 1-й перекристаллизации присоединяют к
первоначальному раствору, от 2-й — для растворения соли на
1-ю перекристаллизацию и т. д.). Из процесса выводится
только первоначальный маточный раствор. При этом выход
особо чистой соли может быть доведен до 85% от теоретиче-
ского; кроме того, 8—10% рубидия будет получено в форме
препарата ч.д.а.
Цезий хлорноватокислый. Кристаллический карбонат це-
зия в количестве 100 г нейтрализуют при комнатной темпе-
ратуре хлорноватой кислотой (~1 л 5%-ного раствора
80
НСЮз) до pH 5—5,5. Раствор упаривают до объема 240—
260 мл и охлаждают при перемешивании до 15—20°. Выде-
лившиеся кристаллы вещества отфильтровывают и промыва-
ют 30—40 мл бидистиллированной воды. Получают ~ 130 г
хлората цезия с влажностью 10%.
Продукт дважды перекристаллизовывают, растворяя кри-
сталлы при нагревании в течение 10—15 минут до 100—105°
в 160—180 мл бидистиллированной воды и охлаждая раствор
при перемешивании до 15—20°.
Выход кристаллов на каждой операции составляет около
90%. Очищенную соль высушивают при 100—110° в течение
2 часов До постоянного веса и получают 100 г цезия хлорно-
ватокислого ос. ч., что составляет 75%, считая на исходный
карбонат цезия. При использовании маточных растворов вы-
ход особо чистого хлората цезия может быть доведен до
90%.
Рубидий бромноватокислый. Кристаллический карбонат
рубидия в количестве НО г при комнатной температуре ней-
трализуют при перемешивании бромноватой кислотой до pH
5—5,5 (~2,5 л 5%-ного раствора НВгО3). Полученный рас-
твор упаривают до объема 850—900 мл и охлаждают до 15—
20° при перемешивании. Образовавшиеся кристаллы отфиль-
тровывают и промывают 50—60 мл дистиллированной воды.
Получают около 200 г кристаллов бромноватокислого руби-
дия с влажностью ~10%.
Вещество подвергают 5-кратной перекристаллизации, рас-
творяя его при нагревании в течение 20—30 минут в биди-
стиллированной воде, затем охлаждая раствор до 15—20° и
отфильтровывая кристаллы от маточника. На 1-ю перекри-
сталлизацию берут ~750 мл воды, а на каждую следующую
~на 50 мл меньше, чем на предыдущую. После отделения
маточника соль промывают на фильтре 30—40 мл холодной
бидистиллированной воды.
Выход препарата на каждой операции перекристаллиза-
ции составляет 88—90%, а общий прямой выход вещества,
считая на исходный карбонат рубидия, составляет ~50%.
После высушивания очищенной соли при 100—110° получают
100 г рубидия бромноватокислого ос. ч. Из маточников мо-
жет быть дополнительно выделено до 50 г препарата ос. ч.
(общий выход до 75%) и около 20—30 г бромата рубидия
ч.д.а.
Цезий бромноватокислый. Кристаллический карбонат це-
зия в количестве 110 г нейтрализуют при комнатной темпе-
ратуре бромноватой кислотой (~1,8 л 5%-ного раствора
НВгО3) при перемешивании, доводя значение pH до 5—5,5.
Раствор упаривают до объема 600—620 мл н охлаждают при
перемешивании до 15—20°. Выпавшие кристаллы отфильтро-
вывают и промывают 40—50 мл холодной дистиллированной
6 Зак. 1060 81
воды. Получают около 180 г бромноватокислого цезия с вла-
жностью ~10%.
Продукт подвергают 3-кратной перекристаллизации, рас-
творяя вещество при нагревании (95—100°) в бидистиллиро-
ванной воде (500—520 мл воды на 1-ю перекристаллизацию.
450—470 мл на 2-ю, 400—420 мл на 3-ю) и охлаждая раствор
до 15—20°.
Выход препарата на каждой операции составляет 85—
87%. Очищенные кристаллы высушивают при 100—110° и по-
лучают 100 г цезия бромноватокислого ос. ч., что составляет
55%, считая на исходный карбонат цезия. Из маточных рас-
творов можно дополнительно получить до 40 г препарата ос.
ч. (общий выход до 75%) и 20—25 г квалификации «ч.д.а.».
Рубидий йодноватокислый. Разбавляют 1,5 л 10%-ной
йодноватой кислоты дистиллированной водой до 2,5—2,7 л
(или растворяют 165 г йодноватой кислоты в 2,4—2,5 л ди-
стиллированной воды) и к полученному раствору добавляют
при перемешивании НО а карбоната рубидия. Раствор нагре-
вают в течение 20—30 минут до полного растворения соли
(95—100°), а затем охлаждают до 15—20°.
Получают 220—230 г йодноватокислого рубидия с влаж-
ностью ~10%. Выделившиеся кристаллы подвергают 4-крат-
ной перекристаллизации. Для 1-й перекристаллизации веще-
ство растворяют при нагревании до 95—100° (продолжи-
тельность 20—30 минут) в 2 л бидистиллировапной воды, для
2-й — в 1,7 л, для 3-й — в 1,4 л, для 4-й — в 1,2 л. Охлажде-
ние проводят до 15—20°.
Выход препарата на каждой операции составляет 82—
84%. Очищенные кристаллы высушивают при 100—110° до
постоянного веса и получают 100 г рубидия йодноватокис-
лого ос. ч., что соответствует 40%, считая на исходный кар-
бонат рубидия. Из маточника дополнительно получают до
60 г соли ос. ч. (общий выход до 60%) и 60 -70 г препарата
ч.д.а.
Цезий йодноватокислый. Разбавляют 1,2 л 10%-ной йод-
новатой кислоты дистиллированной водой до 1,9—2 л (или
растворяют 130 г йодноватой кислоты в 1,8—1,9 л дистилли-
рованной воды) и к полученному раствору добавляют при
перемешивании 120 г карбоната цезия. Раствор нагревают до
полного растворения кристаллов и охлаждают до 15—20°.
Получают около 200 г йодноватокислого цезия с влажно-
стью ~10%.
Кристаллы очищают 3-кратной перекристаллизацией. Для
1-й операции соль растворяют в 1,3 л бидистиллированной
воды, для 2-й — в 1,1 л, для 3-й—в 0,9 л, нагревая раствор
в течение 20—30 минут до полного растворения кристаллов,
а затем охлаждая до 15—20°.
82
Выход препарата на каждой операции составляет 82—
84%. После высушивания при 100—110° до постоянного веса
получают 100 г цезия йодноватокислого ос. ч. (прямой выход
45%). Из маточника дополнительно получают до 50 г соли
ос. ч. (общий выход до 65%) и 40—50 г цезия йодноватокис-
лого ч.д.а.
Анализы получаемых продуктов приведены в таблице. 3.
Таблица 3
Допустимое содержание примесей в особо чистых
галоидокислородных солях рубидия и цезия
Наименование примеси Содержание, вес. % Примечание
Алюминий <1-10~4 Примесь Rb в солях
Железо Калий 1-5-10~5 5-10-10-4 цезия. Примесь Cs в солях рубидия
Кальций <5-10“4
Кремний <5-10“4
Магний < 1 • 10“4
.Марганец <1 ДО-5
Медь 1-5-10-5
Натрий <5-10’4
Никель <1-ю-5
Рубидий «1-10‘3
Свинец 1-5.10"6
Сульфаты 1-5-10-8
Сурьма <1-10-5
Титан < 1 • 10“ 6
Хром 1-5-10-6
Цезий «ЛЮ-3 ।
ЛИТЕРАТУРА
1. Gmelins, Handbuch dcr anorg. Chemie, 8 Aufl, SN 24, 1937, стр.
159, 180, 202; SN 25. 1938, стр. 168, 188, 213.
2. В. Л. Богатырев, А. И. Вулих. Ж. прикл. химии, 36, 220
(1963).
3. А. И. Вулих, В. Л. Богатырев, В. А. Казьминская,
Л. П. Жердиенко. См. статью «Ионообменное получение кислот ИЗ
солей» в этом сборнике, стр. 5.
4. А. О. Ле со в а я, В. А. Казьмин ска я, Л. П. Жердиенко,
83
А.. И. В у л их, Д. А. П а х о м о в. Промышленность химических реакти-
вов, вып. 1 (7), М., ИРЕА, 1965, стр. 25.
5. Р. М. Шкловская, А. И. Вулих, А. О. Л е с о в а я. Ж.
прикл. химии, 40, № 6 (1967).
Поступила в марте 1966 г. ЦЗЛ
УДК 546.76'35'226—325.05
546.76'36'226—325.05
ХРОМОРУБИДИЕВЫЕ И ХРОМОЦЕЗИЕВЫЕ КВАСЦЫ
А. О. ЛЕСОВАЯ, Н. И. КАШИНА. В. А. КУЗИНА
Rb2SO4-Cr2 (SO4)3-24H2O
М. в. 1091,53
Cs2SO4-Cr2 (SO4)3-24H2O
М. в. 1186,40
По хромовым квасцам рубидия и цезия в литературе име-
ется небольшое количество отрывочных сведений [1, 2]. Более
подробные данные имеются по получению и свойствам хромо-
вых квасцов натрия, калия и аммония [3—7], что было нами
использовано при разработке метода получения хроморуби-
диевых и хромоцезиевых квасцов.
По аналогии с другими квасцами [8] хромовые квасцы ру-
бидия и цезия могут представлять интерес в качестве сегне-
тоэлектрических материалов. Общий способ получения хро-
мовых квасцов щелочных металлов заключается в восстанов-
лении их бихроматов в серной кислоте [3]. В качестве восста-
новителей применяются органические соединения.
Хроморубидиевые квасцы представляют собой крупные
кристаллы темно-фиолетового цвета в отраженном свете и
красно-рубинового цвета в проходящем свете. Мелкокристал-
лическая соль сиреневого цвета.
Хроморубидиевые квасцы имеют кубическую решетку с
параметрами элементарной ячейки: а=12,24 А при N = 2;
</рент = 1,986 г/см3; ЙПИК|1 — 1,952 г/см3. Соль нерастворима в
спирте. Растворимость в воде при 25° — 2,7 вес. % (безвод-
ной соли). С повышением температуры растворимость увели-
чивается. Раствор имеет зеленый цвет. Кристаллизация соли
из раствора протекает медленно.
При продолжительном стоянии на воздухе квасцы теряют
часть воды и покрываются налетом сиреневого цвета. При
85
нагревании полное обезвоживание наступает при температу-
ре 360°. Безводная соль зеленого цвета.
Хромоцезиевые квасцы — крупные кристаллы темно-фио-
летового цвета в отраженном свете и красно-фиолетового цве-
та в проходящем свете. Мелкокристаллическая соль серого
цвета.
Хромоцезиевые квасцы кристаллизуются в кубической ре-
шетке с параметрами: а=12,40 A приП = 2; йСгнт= 2,074 а/елг3;
г/пикн=2,053 г/см3 [9].
Соль нерастворима в спирте. Растворимость (безводной
соли) в воде при 25°—0,7 вес. %; при 50° — 3,2 вес. %; в
водном растворе серной кислоты при 25°— 1,14 вес. % (25%-
ный раствор), 5,10 вес. % (5О°/о-иь1й раствор) и при 50° соот-
ветственно 6,0 вес. % и 22,6 вес. %.
Раствор имеет зеленый цвет. Кристаллизация соли из
раствора протекает медленно. При продолжительном стоя-
нии на воздухе квасцы теряют часть воды и покрываются
налетом светло-сиреневого цвета. При нагревании полное
обезвоживание наступает при температуре 400°. Безводная
соль зеленого цвета.
СХЕМА СИНТЕЗА ХРОМОВЫХ КВАСЦОВ РУБИДИЯ И ЦЕЗИЯ
2Ме2Сг2О7 ф- 8H2SO4 + 2СН2О —
—» 2Me2SO4-Cr2 (SO4)3 4- 10Н2О 2СО2 4~ О2
Характеристика основного сырья
Рубидий двухромовокислый, ч., МРТУ 6—09—221—63.
Цезий двухромовокислый, ч., ЦМ ТУ 4482—54.
Кислота серная, ч., ГОСТ 4204—48.
Формалин, технический, синтетический, ГОСТ 1625—54.
Условия получения
В фарфоровый стакан помещают 100 г бихромата руби-
дия или цезия, заливают дистиллированную воду (200 мл
для соли рубидия и 300 мл для соли цезия). В полученную
пульпу приливают серную кислоту (на 100 г Rb2Cr2O7—80 мд,
иа 100 г CssCrsO? — 70 мл кислоты уд. в. 1,83), после чего ос-
торожно, небольшими порциями приливают формалин (26л«л
и 28 мл соответственно).
Во избежание сильного разогрева реакционной смеси в
начале реакции формалин добавляют по каплям при непре-
рывном перемешивании. В конце реакции формалин добавля-
ют быстрее до полного растворения бихромата.
Температуру реакционной смеси поддерживают в преде-
лах 45—55°.
86
По окончании реакции раствор квасцов вместе с выпавши-
ми кристаллами охлаждают до 20° и выдерживают 15 суток
при комнатной температуре (20—22°). Затем кристаллы отде-
ляют декантацией, глубоко отжимают на воронке Бюхнера,
2—3 раза промывают на фильтре холодной дистиллирован-
ной водой с целью вытеснения маточника и снова отжимают.
Отжатые кристаллы высушивают на воздухе до рассыпча-
того состояния.
Выход хроморубидиевых квасцов равен 223 г, что состав-
ляет 80% от теоретического; (выход хромоцезиевых квасцов
равен 207 г, что составляет 85% от теоретического.
Изложенный метод синтеза позволяет получать препара-
ты, содержащие не менее 98% основного вещества
(Me2SO4 Cr2(SO4)3 • 24Н2О) и не более 0,5% примесей—ка-
тионов (включая щелочные металлы).
ЛИТЕРАТУРА
1. Ф. Эфраим. Неорганическая химия, М„ Госхнмтехпздат, 1932,
стр. 56.
2. Справочник по растворимости солевых систем, М., Госхимиздат,
т. III, 1961, стр. 1937, 1942, 2128, 2171, 2173.
3. М. Е. П о з п н. Технология минеральных солей, Л.—М., Госхимиз-
дат, 1961, стр. 417.
4. Ю. В. Карякин, И. И. Ангелов. Чистые химические реакти-
вы, М., Госхимиздат, 1955, стр. 232.
5. Д. П. 3 о си м о в и ч, А. И. Заяц, К. В. Клад ниц кая,
Л. К- Ч е б у к и н а. Ж- прикл. химии, 38, 979 (1965).
6. М. Г. Козловский, П. И. Забот ин. Изв. АН Каз. ССР, сер.
хим., 10, 40 (1956).
7. Д. П. Зое и мо вич, А. И. Заяц, Л. К. Рудая. Укр. хим. ж.,
28, 150 (1962).
8. Ф. Иона, Д. Ширане. Сегнетоэлектрические кристаллы, изд.
«Мир», М„ 1965, стр. 454.
9. Справочник химика, М., Госхимиздат, т. 1, 1963, стр. 432.
Поступила в апреле 1967 г.
ЦЗ Л
УДК 546.35'141.07 + 546.36'141.07
РУБИДИЙ БРОМИСТЫЙ И ЦЕЗИЙ БРОМИСТЫЙ
С. Л!. АРХИПОВ, А. И. ВУЛИХ, Л. Г. СИДОРОВА
RbBr
CsBr
М.в 165,38
М.в 212,81
Бромиды рубидия и цезия представляют собой белые кри-
сталлические вещества, хорошо растворимые в воде. Неко-
торые их свойства представлены ниже:
RbBr
CsBr
3,35
4,44
Кубическая,
а=6,868 А
Кубическая,
а=7,23 А
681 1350
627 1300
191
Бромиды рубидия и цезия используются для изготовле-
ния монокристаллов, применяемых в оптических приборах,
а также в лабораторной практике.
Бромиды рубидия и цезия обычно получают из их карбо-
натов и бромистоводородной кислоты или из их сульфатов и
бромида бария [1]. Другим способом их получения служит
взаимодействие элементарного брома с карбонатами или
гидроокисями рубидия и цезия в присутствии восстановите-
лей— перекиси водорода, аммиака iy др. [2].
88
Нами предложен способ получения бромидов с использо-
ванием в качестве восстановителя гидразин-гидрата [3]. Этот
метод прост, экономичен и исключает образование побочных
продуктов.
СХЕМА СИНТЕЗА БРОМИДОВ РУБИДИЯ И ЦЕЗИЯ
2 RbaCO3+2Br2-l-N2H4 -> 4Rb Br+N2+2CO2 + 2HSO
2 Cs2CO3R2Br24-N2H4 4CsBr+N2+2C0a+2H,O
Характеристика основного сырья
Рубидий углекислый, ч., МРТУ 6-09-227—63.
Цезий у1лекислый, ч., ч.д.а., МРТУ 6-09-1721—64.
Бром, ч.д.а., х.ч., ГОСТ 4109—48.
Гидразин-гидрат, ч., ч.д.а., ГОСТ 5832—51.
Условия получения
Рубидий бромистый. Растворяют 100 г углекислого руби-
дия в 400 мл воды и раствор отфильтровывают. В получен-
ный раствор добавля[от порциями по 20—30 г жидкий бром
(ч.д.а.). После введения каждой порции брома приливают
25%-ный водный раствор гидразин-гидрата до исчезновения
окрашивания. Время реакции с каждой порцией брома со-
ставляет 5—7 минут. Всего вводят 70—75 г жидкого брома
и около 12 г гидразин-гидрата (в пересчете на N2H4-H2O).
В конце процесса раствор должен быть нейтральным
(pH--6—7). Если среда щелочная, то добавляют небольшие
количества брома и гидрата-гидразина до установления нуж-
ного значения pH. Полученный раствор отфильтровывают и
упаривают досуха.
Выход бромистого рубидия равен 141—143 г, что состав-
ляет 98—99% от теоретического.
Цезий бромистый получают по аналогичной методике.
Для реакции берут 100 г углекислого цезия в 400 мл воды и
50 -55 г брома. Расход гидразин-гидрата 8,5 г.
Выход бромистого цезия равен 130—131 г, что составляет
99% от теоретического.
Квалификация получаемого бромистого цезия соответ-
ствует квалификации исходных реактивов: для квалифика-
ции «ч.» берут CS2CO3 ч., Вг2 ч.д.а., N'2H4 Н2О ч.; для ква-
лификации «ч.д.а.» — Cs2CO3 ч.д.а., Вг2 х.ч., N2Il4-H2O ч.;
для квалификации «х.ч.»—СйгСОз х.ч., Вг2 и N2H4 • Н2О ч.д.а.
(См. примечания в статье «Рубидий и цезий йодистые). По-
лученные бромиды рубидия и цезия удовлетворяют требова-
ниям МРТУ 6-09-223—6 и МРТУ 6-09-2288--65.
89
ЛИТЕРАТУРА
1. Ф. М. Перельман. Рубидий и цезий, М., Изд. АН СССР, 1960,
стр. 33.
2. В. И. К с е н з е н к о, Д. С. С т а с и и е в и ч. Технология брома н
йода, М., Госхимиздат, 1960, стр. 262.
3. А. И. Вулих, С. М. Архипов, Л. Г. Сидорова. Авт. свид.,
169081; Билл, изобр., № 6, 1965.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.35' 151.07 + 546.36 151.07
РУБИДИЙ ЙОДИСТЫЙ И ЦЕЗИЙ ЙОДИСТЫЙ
С. М. АРХИПОВ, А. И. ВУЛИХ, Л. Г. СИДОРОВА
RbJ М.в. 212,37
CsJ М.в. 259,30
Йодиды рубидия и цезия представляют собой белые кри-
сталлические вещества, хорошо растворимые в воде. Некото-
рые их свойства представлены в таблице.
Формула Кристаллическая структура Плотность, г/см3 Температура плавления. °C Температура кипения, °C Растворимость в воде, вес. %
0° 100°
RbJ Кубическая «=7,340 А 3,550 638 1300 55,5 74,0
CsJ Кубическая «=7,660 А 4,510 621 1280 30,5 70,25 (при 103°)
Йодиды рубидия и цезия используются при изготовлении
монокристаллов для оптических приборов, а также в лабо-
раторной практике.
Йодиды рубидия и цезия обычно получают из их карбо-
натов и йодистоводородной кислоты или из их сульфатов и
йодида бария [1]. Другим способом их получения является
взаимодействие, элементарного йода с карбонатами или гид-
роокисями рубидия и цезия в присутствии восстановителя—
перекиси водорода, аммиака и других [2].
Нами предложен способ получения йодидов с использо-
ванием в качестве восстановителя гидразин-гидрата [3]. Ис-
Эк
пользование этого метода исключает образование побочных
продуктов и уменьшает стоимость реагентов.
СХЕМА СИНТЕЗА ЙОДИДОВ РУБИДИЯ И ЦЕЗИЯ
2МеаСО3+2 J2 + N2H4 4MeJ + N2+2CO2+2 Н2О,
где Me=Rb или Cs.
Характеристика основного сырья
Рубидий углекислый, ч., ч.д.а., МРТУ 6-09-227—63.
Цезий углекислый, ч., ч.д.а., МРТУ 6-09-1721—64.
Иод металлический, ч., ч.д.а., ГОСТ 4159—48.
Гидразип-гидрат, ч., ч.д.а., ГОСТ 5832—51.
Условия получения
Рубидий йодистый. Растворяют 100 г углекислого руби-
дия в 400 мл дистиллированной воды (см. примечание 1) и
к полученному раствору добавляют 110 г йода. Суспензию
нагревают до 60—70° и постепенно, при перемешивании, вво-
дят 25%-ный водный раствор гидразин-гидрата до полного
растворения йода и обесцвечивания раствора (см. примеча-
ния 2, 3). Всего вводят около 12 г гидразин-гидрата (в пере-
счете на N'2H4-H2O). Конечный раствор должен иметь рИ 6;
если среда щелочная, то вводят дополнительно небольшое
количество йода и гидразин-гидрата. Полученный раствор
отфильтровывают и упаривают досуха. Остаток прокаливают
при 450° в течение 1 часа.
Выход йодистого рубидия квалификации «ч.» или «ч.д.а.»
(в зависимости от исходных реактивов) равен 180 г, что со-
ответствует 98% от теоретического.
Цезий йодистый получают аналогично. Для реакции берут
100 г углекислого цезия в 400 мл воды и 79 а йода. Расход
гидразин-гидрата равен 9,2 а. Получают 157 г йодистого це-
зия квалификации «ч.» или «ч.д.а.» в зависимости от исход-
ных реактивов, что соответствует 98% от теоретического
выхода.
Йодиды рубидия и цезия, получаемые по данной методи-
ке, удовлетворяют требованиям МРТУ 6-09-2742—65 и
ЦМТУ 4481—54.
Примечания:
1. Для предотвращения улетучивания гидразина, происходящего в
некоторой степени из щелочного раствора при повышенных температурах,
предварительно (до введения йода) насыщают раствор карбоната руби-
дия или цезия углекислотой. При этом образуется бикарбонат рубидия
или цезия и pH раствора понижается с 12 до 9.
2. Для предотвращения в растворе окисления гидразина кислородом
воздуха создают инертную атмосферу над раствором, проводя процесс
92
при небольшом избыточном давлении выделяющихся при реакции газов
(до 0,5 атмосферы).
При этих условиях потери гидразина практически исключаются.
3. Взаимодействие йода с гндразин-гидратом происходит бурно и
сопровождается выделением значительного количества газообразных про-
дуктов и вспениванием раствора, поэтому добавлять восстановитель
следует очень осторожно, небольшими порциями. Целесообразно также
предварительно разбавить раствор гидразин-гндрата водой (1:3).
ЛИТЕРАТУР А
1. Ф. М. Перельман. Рубидий и цезий, М„ Изд. АН СССР, 1960,
стр. 33.
2. В. И. Ксензенко, Д. С. С т а с и и е в и ч. Технология брома и
йода, М., Госхимиздат, 1960, стр. 277,
3. А. И. В у л н х, С. М. Архипов, Л. Г. Сидорова. Авт.
свид. 169081; Бюлл. изобр., № 6, 1965.
Поступила в дсгабрс 1965 г. ЦЗЛ
УДК 546.36'175 482.05
ЦЕЗИЙ АЗОТНОКИСЛЫЙ высокой ЧИСТОТЫ
Р. М. ШКЛОВСКАЯ
CsNO3 М.в. 194,91
Нитрат цезия высокой чистоты может использоваться не-
посредственно или для получения других высоко чистых со-
лей цезия, являющихся объектом исследования в новых на-
правлениях современной техники.
Получение нитрата цезия высокой чистоты сводится к
очистке соли от примесей тяжелых металлов и железа и
отделению цезия от примесей щелочных металлов (натрия,
калия, рубидия).
В предлагаемой методике для очистки нитрата цезия от
примесей тяжелых металлов и железа используется метод,
основанный на комплексообразовании этих примесей с ди-
этилдитиокарбаматом натрия и последующей сорбции полу-
ченных комплексов активированным углем.
Для отделения цезия от примесей щелочных металлов
используется метод фракционной кристаллизации нитрата
[1,2].
Характеристика основного сырья
Цезий азотнокислый, ч.д.а., ЦМТУ 4471---54.
Диэтилдитиокарбамат натрия, ч., ГОСТ 2706—51.
Активированный уголь марки А, щелочной, ГОСТ 4453—48.
Аммиак водный, 25%-ный, ч.д.а., ГОСТ 3760—47.
Азотная кислота, х.ч., ГОСТ 4451—48.
Соляная кислота, х.ч., ГОСТ 3118—48.
Условия получения
Растворяют при комнатной температуре 100 г азотнокис-
лого цезия в 400 мл дистиллированной воды.
Полученный раствор подщелачивают ~0,5 мл 25%-ного
раствора аммиака до значения рН~8, затем к нему прили-
94
вают 22 мл 3%-ного водного раствора диэтилдитиокарбамата
натрия и добавляют в два приема через 15 минут по 7 г акти-
вированного угля (см, примечание).
После 30-минутпого отстаивания осадок отфильтровывают
на воронке Бюхнера через бумажный фильтр, промытый
кипящей дистиллированной водой, и промывают ~5 мл
бидистиллированной воды.
Промывные воды присоединяют к основному раствору.
Фильтрат проверяют на полноту отделения тяжелых метал-
лов и избытка диэтилдитиокарбамат-иона. Для этого в две
пробы фильтрата (каждая по 5 .ил) добавляют по 2,5 мл
изоамилового спирта и в одном случае—1 мл 3%-ного ра-
створа диэтилдитиокарбамата натрия, а в другом — 1 мл
раствора сернокислой меди с концентрацией 1 г/л и взбал-
тывают. Обе вытяжки изоамилового спирта должны быть
бесцветны. Если вытяжки окрашены, проводят дополнитель-
ную очистку раствора азотнокислого цезия добавлением
3%-ного раствора диэтилдитиокарбамата натрия и угля.
Очищенный раствор нейтрализуют -—0,3 мл концентри-
рованной азотной кислоты до значения pH 5, упаривают до
появления плотной пленки кристаллов (уд. в. раствора
1,6 г!смл при 105’) и при перемешивании охлаждают до ком-
натной температуры.
Соль отфильтровывают на предварительно промытом ки-
пящей бидистиллированной водой плотном бумажном фильт-
ре, промывают мл бидистиллированной воды и подвер-
гают 2—3-кратной перекристаллизации. Для этого соль ра-
створяют в кипящей бидистиллированной воде при соотно-
шении 2 : 1 и раствор охлаждают до комнатной температуры.
Кристаллы отфильтровывают, промывают ~6 мл бидистил-
лированной воды от маточника и подвергают повторной пе-
рекристаллизации. Число кристаллизаций зависит от содер-
жания примесей щелочных металлов в исходной соли.
Очищенный продукт сушат в фарфоровой чашке при тем-
пературе НО—130° в течение 2 часов.
Выход цезия при трехкратной перекристаллизации исход-
ного нитрата цезия составляет 67 г или 67% от взятого в
процесс.
Маточные растворы, полученные при кристаллизации ни-
трата цезия и содержащие ~25% цезия от исходного коли-
чества, упаривают досуха. Остаток представляет собой про-
дукт реактивной чистоты.
Примечание. Активированный уголь предварительно очищают от
марганца, железа и других примесей. Для этого 90 а угля заливают
500 мл воды и 12 мл соляной кислоты. Смесь нагревают в течение 2—
3 часов, после чего уголь отфильтровывают и отмывают дистиллирован-
ной водой от хлор-нона.
Описанная методика позволяет получать продукт, соответствующий
техническим требованиям РЭ ТУ 930—62 (содержание щелочных при-
95
месей и щелочноземельных примесей — в пределах 2 10“3 каждой при-
меси, железа и тяжелых металлов — в пределах 1 • 10-5—1 . 10-4 каждой
примеси).
ЛИТЕРАТУРА
1. Ф. М. Перельман. Рубидий и цезий, М., АН СССР, I960,
стр. 46.
2. Сб. «Цезий», под редакцией В. Е. Плющева, М., ИЛ, 1963,
стр. 56.
Поступила в декабре Н'бт г. Ц'^Л
УДК 546.36'183.07
ЦЕЗИЙ ФОСФОРНОВАТИСТОКИСЛЫЙ
Гипофосфит цезия
П. Д. КОМИССАРОВА
CsH2PO2 М.в. 197,89
Гипофосфит цезия так же, как и другие соли фосфорно-
ватистой кислоты, является сильным восстановителем [1] и
может найти применение в технологии и аналитической хи-
мии цезия.
Гипофосфиты щелочных и щелочноземельных металлов
могут быть получены окислением фосфора в присутствии гид-
роокисей щелочных и щелочноземельных металлов [2]. Гипо-
фосфиты щелочных металлов могут быть получены обменным
путем из гипофосфитов кальция или бария [3].
Нами определены условия синтеза гипофосфита цезия на
основе последнего метода.
СХЕМА СИНТЕЗА ГИПОФОСФИТА ЦЕЗИЯ
CsoCOg -f- С а (Н2РОг) 2—2 СзНзРОг + СаСОд
Характеристика основного сырья
Цезий углекислый, ч., МРТУ 6-09-1721—64.
Кальций фосфорноватистокислый, ч., ВТУ МХП 2867—51.
Условия получения
Растворяют на холоду 100 г фосфорноватистокислого
кальция в 100 мл дистиллированной воды и к полученному
раствору добавляют концентрированный раствор углекисло-
го цезия (190 г в 100 г воды) до полного осаждения каль-
ция.
Реакционную смесь фильтруют и раствор подвергают
упариванию в две стадии; сначала на кипящей водяной бане
7 Зак. 1060 97
до сиропообразного состояния и затем досуха в вакуум-су-
шильном шкафу при температуре 60—80°.
При упаривании в вакууме.чашу с раствором необходи-
мо прикрывать стеклянным колпаком из-за сильного раз-
брызгивания.
Выход цезия фосфорноватистокислого равен 208 г, что
составляет 90% от теоретического.
Описанная методика обеспечивает получение продукта,
удовлетворяющего требованиям МРТУ 6-09-799—63, содер-
?кание СзНгРОа—97—98%.
ЛИТЕРАТУРА
1. Б. В. Некрасов. Курс общей химии, М., Госхимиздат, 1961,
стр. 383.
2. Франц, пат. 1164005 (1958).
3. Пат. США 2876174 (1959).
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.722'36'267.07
ЦЕЗИЙ ЖЕЛЕЗИСТОСИНЕРОДИСТЫЙ
Ферроцианид цезия
П. Д. КОМИССАРОВА, С. А. КРЕСТОВИИКОВА
Cs4Fe(CN)6
М.в. 743,57
Цезий железистосинеродистый представляет собой жел-
тое кристаллическое, вещество. Является труднорастворимым
соединением, особенно при образовании двойных солей с
другими ферроцианидами, что определяет его значение для
аналитической химии и технологии цезия [1].
Ферроцианид цезия получают из карбоната цезия и же-
лезистосинеродистой кислоты [2]. Однако предпочтительнее
использовать эфират железистосинеродистой кислоты, так
как он обладает сравнительно низкой растворимостью в воде
и легко может быть очищен перекристаллизацией из спирто-
вого раствора [3]. По разработанной нами методике ферро-
цианид цезия получается непосредственно из эфирата желе-
зистосинеродистой кислоты, без выделения последней в чи-
стом виде.
СХЕМА СИНТЕЗА ФЕРРОЦИАНИДА ЦЕЗИЯ
K4Fe(CN)6 + 4НС1+ 2(СгН6)гО = H4Fe(CN)e 2(СаНЕ)2О -ф 4КС1
2Cs2COj + H4Fe(CN)s-2(C2H8)2O = Cs4Fe(CN)e + 2(CSHB),O ф
+ 2H,0 -ф2СОа
Характеристика основного сырья
Цезий углекислый, ч., МРТУ 6-09-1721—64.
Калий железистосинеродистый, ч., ГОСТ 4207—48.
Диэтиловый эфир, медицинский, ГОСТ 6265—52.
Соляная кислота, ч., ГОСТ 3118—46.
99
Условия получения
Получение эфирата железистосинеродистой кислоты. В
фарфоровом стакане емкостью 2 л растворяют 100 г желези-
стосинеродистого калия в 800 мл дистиллированной воды.
К полученному раствору добавляют при непрерывном пере-
мешивании 240 мл соляной кислоты уд. веса 1,19. Выпавший
осадок хлорида калия переводят в раствор прибавлением не-
большого количества воды. К охлажденному до комнатной
температуры раствору добавляют 100 мл диэтилового эфира
и выдерживают реакционную смесь в течение 1 часа. Вы-
кристаллизовавшийся эфират железистосинеродистой кисло-
ты отфильтровывают и промывают на воронке 50—100 мл
эфира.
С целью очистки от хлористого калия эфират подвергают
перекристаллизации из спиртового раствора. Для этого оса-
док растворяют в 100 ли этилового спирта (см. примечание 1)
и отфильтровывают от нерастворимого в спирте хлористого
калия.
К отфильтрованному раствору добавляют 100 мл эфира
и выпавший осадок эфирата отфильтровывают.
Получение железистосинеродистого цезия. Растворяют
50 г углекислого цезия в 25 мл воды и к полученному ра-
створу добавляют небольшими порциями, при постоянном
перемешивании, свежеосажденнын эфират железистосинеро-
дистой кислоты до достижения pH 4 (см. примечание 2).
Для выделения ферроцианида цезия к раствору добав-
ляют 50 мл этилового спирта и реакционную смесь выдержи-
вают в течение 30 минут.
Выделившийся препарат цезия отфильтровывают и отмы-
вают от маточного раствора небольшим количеством спирта.
Выход ферроцианида цезия равен 51 г, что составляет
90% от теоретического. Состав продукта отвечает РЭТУ
1046—63.
Примечания:
1. Для очистки эфирата железистосинеродистой кислоты можно ис-
пользовать также пропиловый спирт.
2. Можно также использовать железистосинеродистую кислоту, по-
лученную разложением эфирата в вакууме при 40—60°.
ЛИТЕРАТУРА
1. Ф. М. Перельман. Рубидий и цезий, М., Изд. АН СССР, I960,
стр. 55.
2. И.В.Т а н а и а е в, Г. В. С е й ф е р. Ж- неорган. химии, 1, 64 (1956).
3. Руководство по препаративной неор:анической химии, под ред.
Г. Браус р а, М., ИЛ, 1956, стр. 691.
Поступила в декабре 1965 г. ЦЗЛ
100
УДК 546.633-31-36.07
СКАНДИЙ ОКСИГИДРАТ
Ю. Б. ПЕРКОВСКАЯ
ScO(OH) М.в. 77,96
Оксигидрат скандия представляет собой белый порошок,
нерастворимый в воде, легко растворимый в кислотах.
Оксигидрат скандия получают обезвоживанием гидрооки-
си скандия при 180° по реакции
Sc(OH)3=ScO(OH)tH2O. [1]
В литературе указано, что при термическом разложении
скандия-аммония углекислого в качестве промежуточного
продукта образуется оксигидрат скандия, существующий в
интервале температур 280—405" [2]. При дальнейшем нагре-
вании ScO(OH) разлагается с образованием окиси скандия.
Нами разработана методика синтеза оксигидрата скан-
дия, основанная на использовании указанных данных.
СХЕМА СИНТЕЗА ОКСИГИДРАТА СКАНДИЯ
Sc2O8+6HCl -> 2 ScCl3+3 Н2О
ScCl3-f-2(NH4)2CO3 ScNH4(CO,)2+3NH4C1
ScNH4(CO3)2 -> ScO(OH)+NH3+2CO4
Характеристика основного сырья
Скандий окись, ОС-99, ЦМТУ 4854—57.
Соляная кислота, х ч., ГОСТ 3118—-46.
Аммоний углекислый, х.ч., ГОСТ 3770—47.
Активированный уголь щелочной марки А, ГОСТ 4453—48.
Диэтилдитиокарбамат натрия, ч., ГОСТ 8864—58.
101
Условия получения
В фарфоровую чашу на песчаной бане заливают 9,8 л
разбавленной 1 : 1 соляной кислоты, нагревают до 80—90° и
при перемешивании вносят 0,983 кг (для квалификации
«ч.д.а.») или 0,962 кг (для квалификации «ч.») окиси скан-
дия. Нагревание и перемешивание ведут до полного
растворения окиси скандия. Для поддержания постоян-
ного объема в реакционную смесь периодически под-
ливают горячую дистиллированную воду. Полученный
раствор хлористого скандия разбавляют до уд. веса
1,06 а/с.м3 и осаждают 3%-ным раствором диэтилдитио-
карбамата натрия железо и тяжелые металлы. Раствор
диэтилдитиокарбамата натрия (330 мл) приливают тонень-
кой струйкой при интенсивном перемешивании основного ра-
створа. Затем в раствор добавляют 50 г активированного
угля, перемешивают 15 минут, добавляют елце 50 г активиро-
ванного угля и после 15-минутного перемешивания фильтруют.
Прозрачный раствор хлорида скандия с температурой нс
выше 25° нейтрализуют 25%-ным раствором углекислого ам-
мония до pH 7—7,5; при этом образуется объемистый осадок
основного карбоната скандия, который растворяют в избыт-
ке углекислого аммония. Раствор аммония углекислого сле-
дует добавлять небольшими порциями при тщательном пере-
мешивании до полного растворения первоначально выпавше-
го осадка (допустима легкая опалесценция). Далее раствор,
не фильтруй, нагревают до 60° и выдерживают при этой тем-
пературе 2 часа. Выпавшую двойную соль скандия-аммония
углекислого отфильтровывают через фильтр бумага-бязь и
промывают дистиллированной водой до отсутствия хлор-иона
в промывных водах. Отмытую и хорошо отжатую двойную
соль загружают в фарфоровые тигли или кварцевые кюветы
и прокаливают при 310—340° в течение 9 часов. В процессе
прокаливания соль необходимо дважды перемешивать. По-
лученный оксигидрат скандия быстро охлаждают и затари-
вают в банки с притертыми пробками или завинчивающими-
ся крышками.
Маточный раствор после осаждения двойной соли под-
кисляют соляной кислотой до pH = 2,0— 2,5 (с целью разруше-
ния избытка углекислого аммония) и аммиаком осаждают
скандий, не полностью выделившийся в виде двойной соли.
Полученную гидроокись скандия возвращают в голову про-
цесса.
Выход окиси скандия составляет для препарата квали-
фикации «ч.д.а.» 90%, для квалификации «ч.» 92,0%.
Описанный способ позволяет получить оксигидрат скан-
дия с содержанием основного вещества ScO(OH) 97—99%.
Содержание аммония — не более 1 • 10-2%, железа и тяже-
102
лых металлов — не более 5- 1О-зо/о. Содержание других при-
месей пропорционально их содержанию в исходной окиси
скандия.
ЛИТЕРАТУРА
1. М. W. Schafer, R. Roy. Z. anorgan. und allgem. Chem. 276,
275 (1954).
2 В. И. Спицын, Л. H. Комиссарова, В. M. Шацкий,
Г. Я. Пушкина. Ж- иеорган. химии, 5, 2223 (1960).
Поступила в марте 196G г. Ц?Л
УДК 546.651/.659'264.07
КАРБОНАТЫ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
Ю. Б. ПЕРКОВСКАЯ, Н. П. АНОШИНА, И. М. СУХАНОВА
Карбонаты редкоземельных элементов (р.з.э.) представ-
ляют собой мелкокристаллические или аморфные вещества,
практически нерастворимые в воде, но легко разлагающиеся
минеральными кислотами и рядом органических кислот. При
нагревании в соприкосновении с водой они гидролизуются и
переходят в основные карбонаты, образование которых уси-
ливается с понижением основности в ряду от лантана к лю-
тецию [1].
Существует несколько способов получения карбонатов
р.з.э.:
1) осаждение из раствора соли р.з.э. двууглекислым нат-
рием с последующим насыщением углекислотой [2];
2) насыщением углекислотой суспензий гидроокисей [1];
3) гидролизом трихлорацетатов р.з.э. [1];
4) осаждением из азотнокислых растворов р.з.э. раство-
ром углекислого аммония с последующим насыщением угле-
кислотой [3].
Нами проверен последний способ, предложенный ИОНХ
АН УССР, и с некоторыми изменениями (осаждение из со-
лянокислых растворов) внедрен в производство реактивных
солей р.з.э.
СХЕМА СИНТЕЗА КАРБОНАТОВ Р.З.Э.
R2O3+6HC1 -* 2RCl8+3HaO
2 RCl3+3 (NH4)aCO3 -> R2(COS)S + 6NH4C1
Характеристика основного сырья
Лантан окись, РЭТУ 96—59.
Церий гидроокись или церий углекислый, техн., соответст-
вующий марке Цо-1, РЭТУ 144—59.
104
Празеодим окись, РЭТУ 1075— 63.
Неодим окись, РЭТУ 1076—63.
Самарий окись, РЭТУ 1077—63.
Европий окись, РЭТУ 1078—63.
Гадолиний окись, РЭТУ 1079—63.
Тербий окись, РЭТУ 1080—63.
Диспрозий окись, РЭТУ 1081—63.
Гольмий окись, РЭТУ 1082—63.
Эрбий окись, РЭТУ 1083—63.
Иттербий окись, РЭТУ 1085—63.
Лютеций окись, РЭТУ 1086—63.
Иттрий окись, РЭТУ 1072—63.
Соляная кислота, х.ч., ГОСТ 3118—46.
Аммоний углекислый, ч.д.а., х.ч., ГОСТ 3770—47.
Углекислота пищевая, ГОСТ 8050—46.
Условия получения
Окись редкоземельного элемента помещают в химический
стакан емкостью 500 мл, смачивают дистиллированной водой
при соотношении Т:Ж=1:2 и растворяют в разбавленной
1 : 1 соляной кислоте. Сначала растворение ведут без нагре-
вания. Кислоту приливают небольшими порциями при пере-
мешивании (расход кислоты указан в таблице расходных
норм). После растворения основной массы окиси р.з.э. ра-
створ с осадком нагревают до 70—80° и добавляют остав-
шееся количество кислоты. По достижении полного растворе-
ния и установления pH 4 раствор прогревают до 90 100°,
охлаждают до комнатной температуры и фильтруют. При
этом с нерастворимым остатком отделяется железо в виде
гидроокиси, содержание которого в исходной окиси допус-
кается 1 • 10~2%.
Для карбоната церия вводится дополнительная очистка
от тяжелых металлов. В разбавленном растворе хлорида це-
рия (уд. вес 1,08) перед осаждением карбоната сероводоро-
дом осаждают тяжелые металлы до достижения полноты
осаждения. Осадок тяжелых металлов отфильтровывают,
промывают 2 раза дистиллированной водой и в прозрачном
фильтрате осаждают карбонат церия, как описано ниже.
Готовят 25°/о-ный раствор углекислого аммония (уд. вес
1,084 при 15°), растворяя в 3 частях воды 1 часть углекисло-
го аммония. Полученный раствор отфильтровывают от меха-
нических примесей. Раствор хлорида р.з.э. разбавляют до
уд. веса 1,02 (для иттриевых р.з.э.) или 1,08 (для цериевых
р.з.э.) и при энергичном перемешивании карбонат р.з.э.
осаждают раствором карбоната аммония. При этом выпадает
аморфный осадок. В полученную пульпу в течение 2 часов
со скоростью 12 л1,час пропускают углекислоту. Затем осадок
105
оставляют на сутки под раствором, при этом аморфный оса-
док переходит в кристаллический. (Для цериевых р.з.э. про-
пускание углекислоты не требуется, достаточно 24-часового
отстоя под раствором.) Через сутки раствор декантируют, а
осадок промывают дистиллированной водой сначала декан-
тацией 3—4 раза, затем на воронке Бюхнера от хлористого
аммония (качественная реакция на хлор-ион с азотнокислым
серебром в присутствии азотной кислоты). Отмытую, хорошо
отжатую соль сушат в сушильном шкафу при температуре
60—70° до достижения теоретического содержания суммы
окислов р.з.э.
Выход металла в соль составляет 93,0%.
В таблице 1 даны расходные нормы сырья на выпуск
100 г каждого продукта, а в таблице 2 отражено качество
получаемых продуктов (технические условия, указанные в
таблице, составлены на основании результатов, полученных
по настоящей методике).
Примечание. Дли получения препаратов х.ч., ч.д.а., и ч. исполь-
зуются окислы соответствующей марки— 1, 2 или 3.
Таблица 1
Расходные нормы сырья
Наименование соли Окись р.з.э Соляная кислота Аммоний углекислый Угле- кисло- та
теорети- ческий фактиче- ский теорсти- 1 ческий фактиче- ский теорети- ческий фактиче- ский фактиче- ' ский
Лантан углекислый 57,6 62,0 106,0 114,0 64,3 105,0 __
Церий углекислый 62,5* 70,0* 109,0 122,0 66,1 110,0 —
Празеодим углекислый 60,5 65,0 107,0 114,0 64,6 107,7
Неодим углекислый 62,2 66,9 111,0 119,0 67,3 112,0 —
Самарий углекислый 63,1 67,9 108,0 117,0 61 9 103,0 —
Европий углекислый 65,4 68,3 110,0 122,0 68,0 92,0 150
Гадолиний углекислый 66,0 71,0 108,0 120,0 65,5 90,0 150
Тербий углекислый 66,3 71,3 107,0 119,0 66,0 89,0 150
Диспрозий углекислый 64,6 69,5 102,0 113,0 63,3 86,0 150
Гольмий углекислый 67,0 72,0 105,0 116,0 6\0 88,0 150
Эрбий углекислый 65,21 70,1 101,0 112,0 62,1 84,0 150
Иттербий углекислый 68,0 73,0 102,0 114,0 64,0 85,0 150
Лютеций углекислый 53,3 57,3 80,0 89,0 48,5 66,0 150
Иттрий углекислый 55,0 59,0 144,0 160,0 89,0 120,0 150
* Церий углекислый или гидроокись церия в пересчете на СеО2.
106
кальция 0,08 0,05 1 1 0,05 0,03 сч сч 1 1 о © еч 54 1 1 © 2 © сч СО © со 0,05 0,03 0,02 сч О © см 03 1 1 о © CQ
<u s s я a меди 800*0 10*0 1 1 0,01 0,005 сч М 1 1 О о — © см М 1 1 О © «ч 1 © о К м 1 1 1 С © © — © СО 0,01 0,005 0,002 см ео м 1 I Ф © >-м >-н LQ СО
иное соде желе- за 0,005 0,003 0,003 0,002 0,005 0,003 1 s-oi-e 8_0Г1 сч «1 О О «“• © аз 0,01 0,005 0,002 1^ cq «е 1 1 ©> о Ю СО
о C при- месей р. 3. 3. 0,5 0,1 o' ю о” © с 1,о 0,5 । 1,0 0,5 О 1,0 0,5 0,1 © чэ — —* ©* о" СМ 1,0 0,1
Е01ЭЭТП -ЭЯ OJOH -аоиэо 0'66 0'86 98,5 99,0 1 1 98,0 99,0 । 97,0 98,0 99,0 © © © г* оо сп Ф © СП 1 1 1 ©^ © 98,0 99,0
2 окис- лов р. 3. э. 1 © ю © оо © © © 2 3 <© СП сч © — 00 CN © © © © © (о © 2 © © © Tf © © © ч.д. а. 64,96 х. ч. 165,63 ।
виПвх ифИ|ГВЯД ч. ч.д. а. S' ч. д. а. ч. ч.д. а. га sr ч ег га Л" S3 ег sr н ч. ч.д. а. X. ч. Ч. < ч. д. а.। X. Ч. 1 V
ТУ РЭТУ 798—61 МРТУ 6-09-250—63 РЭТУ 667-60 МРТУ 6-09-1583-64 МРТУ 6-09-1587-64 МРТУ 6-09-1578-64 РЭТУ 732 61 МРТУ I © 1 О1 ю 7 S । to
|теоретичес- кое содер- жание сум- мы окис- 1 лов р, 3. э. 57,56 1 62,54 59,74 62,24 63,08 65,41 66,04 66,29
I Моле- ку- ляр- иый и <L> а 565,93 550,34 СО © О> ю | 540,56 1 552,78 537,99 548,57 551,92
Наименование соединения, формула Лантан углекислый Las(CO3)3.6H,O Церий углекислый Се,(СО,)3-5НгО Празеодим углекислый Рг,(СО,),-6НгО Неодим углекислый Nd,(C0,),)-4H,0 Самарий углекислый Sm2(CO,),-4H,O Европий углекислый Eu,(COg).-3HsO Гадолиний углекислый Gd.CCOjJ.-SH.O Тербий углекислый ГЬ,(С0,),-ЗН,0
107
о 00 Продолж. табл. 2
Моле- Теоретичес- Процентное содержание
Наименование соединений, КУ- кое содер- К 2 (У ж при- месей
формула ляр- ный жание сум- мы окислов ТУ = w 2 х окис- са ш га железа меди кальция
вес р. 3, э. га £ а лов Р 3. э. 5 О и ока р. 3. э.
Диспрозий углекислый 577,08 64,63 РЭТУ Ч. 63,4 0,01 0,01 0,05
Dy2(CO3),-4H2O 138-59 64,68
Гольмий углекислый 569,93 6,29 РЭТУ ч. 65,5 __ 3,0 0,01 0,01 0,05
Но,(СО3)3-ЗН,О 728-61 ч. д. а. 66,0 — 0,5 0,005 0,005 0,03
х. ч. 66,5 — 0,1 0,002 0,002 0,02
Эрбий углекислый 586,60 65,21 МРТУ ч. 63,25 97,0 1,0 1.10"2 1 • 10~2 5-10~2
Ег2(СО,),-4Н2О 6-09-1662-64 ч.д. а. 63,91 98,0 0,5 5-Ю-3 5-Ю-3 4-Ю-2
X. ч. 64,56 99,0 0,1 З-Ю-3 З-Ю-3 З-Ю-2
Иттербий углекислый 580,15 67,93 РЭТУ ч. 66,4 1,0 0,01 0 01 0,05 0,13
Yb,(CO,)s-3H2O 731—61 ч. д. а. 66,9 — 0,5 о; 005 0,005
X. ч. 67,4 — 0,1 0,002 0,002 0,02
Лютеций углекислый 746,15 53,33 МРТУ ч. 51,73 97,0 1,5 1 • 10“ 2 1-10-2 5-Ю-2
Lus(CO3)3-Г2Н2О 6-09-1582-64 ч.д. а. X. ч. 52,26 52,8 98,0 99,0 1,5 0,1 5 10 ~3 5-10-3 4-10-2
3-10 3 3-10 3 3-10 2
Иттрий углекислый У,(СО3)3-ЗН,О 411,88 54,82 730-61 ч. ч.д. а. X. ч. 53,3 53,8 54,3 — 1,5 1,0 0,5 0,01 0,005 0,002 0,01 0,005 0,002 0,05 0,03 0,02
УДК 546.656.3'221.07+546.657.3'221.07 к 546.659.3'221.07
СУЛЬФИДЫ ПРАЗЕОДИМА, НЕОДИМА, САМАРИЯ
(Полуторные)
О. Н. БРЕУСОВ, М. Н. КОРОТКЕВИЧ
Pr3S3 Празеодим сернистый
Nd3S3 Неодим сернистый
Sm2S8 Самарий сернистый
М. в. 378,00.
М. в. 384,67
М. в. 396,89
Полуторные сульфиды редких земель представляют со-
бой весьма тугоплавкие соединения, чем и определяется ин-
терес, который они вызывают в последнее время. Одним из
основных методов синтеза этих соединений является взаи-
модействие соответствующих окислов с сероводородом при
высоких температурах [1—3]. Хотя в литературе указаны об-
щие условия синтеза, при разработке методик получения
отдельных соединений оказалось необходимым установить
конкретные параметры, различные для разных препаратов.
Нами определены оптимальные условия синтеза сульфи-
дов неодима, празеодима и самария из окислов этих эле-
ментов.
СХЕМА СИНТЕЗА ПОЛУТОРНЫХ СУЛЬФИДОВ Р.З.Э.
Э2О3 + 3HaS — 32S3 4- ЗН2О,
где Э=Рг, Nd, Sm.
Характеристика основного сырья
Празеодим окись, Пр-2. РЭТУ 103—59.
Неодим окись, Н-1, РЭТУ 95—59.
Самарий окись, См-3, РЭТУ 109—59.
НО
Условия получения
Сульфид празеодима. В кварцевую трубу диаметром 35—
40 мм, находящуюся в электрической печи с. силитовыми
нагревателями, помещают 30 г окиси празеодима.
Сероводород, полученный действием соляной кислоты на
сульфид железа, очищают от паров хлористого водорода и
влаги последовательным пропусканием его через дистилли-
рованную воду, безводный хлористый кальций и фосфорный
ангидрид, после чего сероводород поступает в трубу с окисью
празеодима. Температуру в печи доводят до 1300° и при этой
температуре пропускают сероводород в течение 5 часов. За-
тем печь охлаждают (в токе сероводорода) до комнатной
температуры и выгружают продукт сульфидирования.
Из указанной загрузки получают 32 г сульфида празео-
дима— порошка темно-бурого цвета, что соответствует 95%
от теоретического выхода. Состав препарата отвечает
МРТУ 6-09-794—63 (соотношение S : Рг=1,5±0,1).
Сульфид неодима. Синтез проводят по аналогии с мето-
дикой получения сульфида празеодима, но загрузка окиси
празеодима может быть увеличена до 50 г. Оптимальные
условия сульфидирования: температура 1150°, время реакции
3 часа.
Из указанной загрузки получают 54 г сульфида неоди-
ма— порошка зеленого цвета, что соответствует 95% от
теоретического выхода. Состав препарата соответствует
РЭТУ 1019—62 (соотношение S : Nd—1,5±0,1).
Сульфид самария. Окись самария сульфидируют по спо-
вобу, описанному для окиси празеодима. Загрузка окиси са-
мария составляет 50 а; оптимальными условиями являются:
температура 1300°, время сульфидирования 4 часа.
Из указанной загрузки получают 53 г сульфида сама-
рия— порошка темно-коричневого цвета, что соответствует
95% от теоретического выхода.
Полученный препарат соответствует РЭТУ 1045—63 (со-
отношение S : Sm=l,5±0,l).
ЛИТЕРАТУРА
1. Г. В. Самсонов, С. В. Радзиковская. Успехи химии, 30,
G0 (1961).
2. Н. Remy. Lehrbuch der anorganischen Chemie, В. II, Leipzig,
1959, стр. 594.
3. В. В. Серебренников. Химия редкоземельных элементов,
т. 1, Томск, ТГУ 1959, стр. 269.
Поступила в декабре [065 г. ЦЗЛ
УДК 546.661.2'226.07
ЕВРОПИЙ СЕРНОКИСЛЫЙ ЗАКИСНЫЙ
Ю. Б. ПЕРКОВСКАЯ
EuSO4 М.в. 248.02
Европий сернокислый закисный представляет собой кри-
сталлическое вещество белого цвета с уд. весом 4,98 при
25° [1]. Растворимость в воде при той же температуре состав-
ляет 6,24 г>л [2]; по данным других авторов, — 0,0185 г/л при
20° [3]. С увеличением температуры растворимость EuSO4
увеличивается [4]. Сухая соль устойчива на воздухе, но в со-
прикосновении с водой постепенно окисляется кислородом
воздуха [5].
Сульфат европия (24) получают катодным восстановле-
нием сульфата трехвалентного европия, восстановлением
амальгамой щелочных металлов или стронция, а также вос-
становлением хлорида европия (3+) в редукторе Джонса
амальгамированным цинком с взаимодействием вытекающе-
го раствора ЕнСЬ с серной кислотой [1, 2, 5, 6]. Описан спо-
соб получения европия сернокислого закисного путем элект-
ролиза ацетата европия и цитрата калия на ртутном катоде
с последующим разложением полученной амальгамы евро-
пня горячей разбавленной серной кислотой [3].
СХЕМА СИНТЕЗА ЕВРОПИЯ СЕРНОКИСЛОГО ЗАКИСНОГО
Еи2О3 -|- 6НС1 — 2EuCl3 + ЗН2О
2EuCl3+ZnHg —> 2EuCl2-r-ZnCl2 + Hg
EuC12 + H2SO4 -- EuSO4 + 2HC1
I
Характеристика основного сырья
Европий окись, РЭТУ 1078—63.
Соляная кислота, х,ч., ГОСТ 3118—46.
Цинк металлический, ГОСТ 989—41.
Ртуть окись, ч., ч.д.а,, ГОСТ 5230—50.
112
Серная кислота, х.ч., ГОСТ 4204—48.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Углекислота пищевая, ГОСТ 8050—46.
Условия получения
(Выпуск 25 г EuSOJ
В химический стакан помещают 22 г окиси европия, сма-
чивают 20 мл дистиллированной воды и растворяют в 61 мл
разбавленной 1 : 1 соляной кислоты. Растворение заканчи-
вают при нагревании на электроплитке до 80—90°. Кислоту
приливают небольшими порциями при перемешивании до
полного растворения окиси и установления pH 3—4. Полу-
ченный раствор отфильтровывают от механических примесей
и разбавляют до 1250 мл (0,1 М раствор ЕиС13).
Цинковый порошок в количестве 300 г полминуты обра-
батывают 300 мл разбавленной 1 : 3 соляной кислоты, до-
бавляют раствор хлорной ртути (3,3 г окиси ртути раство-
ряют в 3,5 мл соляной кислоты уд. в. 1,19) и перемешивают
до прекращения выделения пузырьков водорода и еще 3 ми-
нуты после прекращения выделения пузырьков водорода.
Полученный амальгамированный цинк промывают дистилли-
рованной водой до отсутствия хлор-иона в промывных водах
и загружают в редуктор Джонса. Конец трубки редуктора
доходит до дна приемного стакана.
Затем собирают установку, изображенную на этом ри-
сунке.
1 — редуктор Джонса, заполненный амальгамированным цинком;
2— химический стакан емкостью 2 л\
3 — ловушка, заполненная стеклянной ватой,
? — трубка с углем и медной стружкой;
5 — трубчатая печь, Дм а тс =800°;
6— промывалка с концентрированной серной кислотой;
7 — промывалка с дистиллированной водой;
8—аппарат Киппа или баллон с углекислотой
113
Перед работой колонку промывают 100 мл 0,1 н. соля-
ной кислоты. В колонке оставляют такое количество кисло-
ты, чтобы она только покрывала цинк. Затем выходное от-
верстие редуктора опускают в стакан емкостью 3 л, в кото-
рый залито 310 мл 8 н. серной кислоты. Стакан покрывают
кружком фильтровальной бумаги и удаляют воздух углекис-
лотой, осушенной серной кислотой и обескислороженной при
700° над древесным углем и медной стружкой.
Пропускают (1250 мл) 0,1 М раствора хлорида европия
со скоростью 4 мл/мин через редуктор Джонса в течение
5 часов. Вытекающий раствор хлорида двухвалентного евро-
пия взаимодействует с 8 н. серной кислотой, при этом обра-
зуется белый осадок «-модификации сульфата европия. Про-
цесс ведется при непрерывном токе углекислоты. Последнюю
порцию раствора хлорида европия из редуктора вымывают
150 мл 0,1 н. раствора соляной кислоты. Раствор с осадком
«-модификации сульфата европия нагревают в атмосфере
углекислоты до 80°, при этом a-модификация переходит в
более устойчивую плотную кристаллическую (3-модификацию,
осаждающуюся в виде компактной массы. Раствор с осад-
ком охлаждают до комнатной температуры в токе углекисло-
ты и фильтруют через тигель Шотта № 4. Осадок промывают
150 мл 0,05 н. соляной кислоты, а затем 50 мл этилового
спирта. Соль сушат при 75° до постоянного веса. Фильтрова-
ние и сушку проводят на воздухе. Прямой выход металла в
соль составляет 70% (25 г сульфата европия).
Маточный раствор и промывные воды объединяют, ней-
трализуют 25°/о-ным раствором аммиака до pH 2 и осаждают
горячим насыщенным раствором щавелевой кислоты оксалат
европия, который затем фильтруют, промывают дистиллиро-
ванной водой и прокаливают при 800° до окиси. Полученная
окись может быть использована для получения следующей
партии европия сернокислого закисного или любой другой
соли европия.
Общий выход металла с учетом переработки маточного
раствора и промывных вод составляет 85%.
Описанный способ позволяет получить продукт, соответ-
ствующий МРТУ 6-09-1590—64, с содержанием европия сер-
нокислого закисного до 98%. Содержание Еи3+ колеблется в
пределах 0,5—1,0%. Содержание контролируемых примесей
других металлов (железо, тяжелые металлы, цинк, кальций)
составляет для препарата квалификации «ч.» — 0,12%, для
препарата квалификации «ч.д.а.» — 0,06%. Содержание окис-
лов редкоземельных элементов (неодима, самария, гадоли-
ния) пропорционально их содержанию в исходной окиси
европия.
114
1ИТЕРАТУРА
1. В. В, Серебренников. Химия редкоземельных элементов,
т. 1, Томск, ТГУ, 1959, стр. 281.
2. Неорганические синтезы, т. 2. М.. ИЛ, 1951. стр. 70.
3. Ю. С. Скляренко Н. С. Строганова. Ж. неорган. химии,
10, 647 (1965).
4. Ю. А. Козьмин, Л П. Шульгин, В. Д. Пономарев. Ж-
неорган. химии, 9, 2532 (1964).
5. L. Gntema. J Amer. Chem. Soc. 52, 2782 (1930).
6. H. McCoy. J. Amer. Chem. Soc. 61, 9, 2455 (1939).
Поступила в декабре 1965 г.
Ц?Л
УДК 546.65l/.657'234.07
СЕЛЕНИСТОКИСЛЫЕ СОЛИ ЛАНТАНА, ЦЕРИЯ,
ПРАЗЕОДИМА И НЕОДИМА
Ю. Б. ПЕРК.ОВСКАЯ
Laa(SeO3)3 Селенистокислая соль лантана М. в. 658,69
Ce2(SeO3)3'4H2O Селенистокислая соль церия М. в. 733,17
Pr2(SeO3)3 Селенистокнслая соль празеодима М. в. 622,68
Nd2(SeO3)3-4HaO Селенистокислая соль неодима М. в. 741,41
Селенистокислые соли редкоземельных элементов (селе-
ниты р.з.э.) представляют собой аморфные порошки, плохо
растворимые в воде, разлагающиеся при нагревании с азот-
ной кислотой или соляной кислотой в присутствии перекиси
водорода.
Селениты р.з.э. получают путем осаждения их из суль-
фатных растворов раствором селенита натрия или действием
селенистой кислоты и спирта на концентрированные сульфат-
ные растворы, а также смешением стехиометрических коли-
честв растворов хлоридов р.з.э. с раствором селенистой ки-
слоты [1, 2]. Полученный селенит отделяют от маточного ра-
створа и промывают водой. Селенит церия получается при
обработке основного селенита церия селенистой кислотой [1].
Во всех случаях образуются аморфные объемистые осадки
с переменным содержанием кристаллизационной воды.
По литературным данным, лантан селенистокислый обра-
зует кристаллогидраты с 9 и 12 молекулами воды, церий
селенистокислый — с 12 молекулами воды [1]. Последний при
сушке над серной кислотой теряет девять молекул воды [1].
Селениты неодима, гадолиния и самария образуют 4—5-вод-
ные кристаллогидраты, полностью обезвоживающиеся при
230—290° [3].
Нами предлагается методика получения селенитов р.з.э.
путем осаждения их из слабокнслых хлоридных растворов
раствором селенита натрия. Получить селениты лантана и
116
празеодима с определенным содержанием кристаллизацион-
ной воды не представилось возможным.
Изучение термической устойчивости полученных кристал-
логидратов с переменным содержанием кристаллизационной
воды показало, что лантан и празеодим селенистокислые на-
чинают терять кристаллизационную воду при температуре
ниже 100° и полностью обезвоживаются соответственно при
360° и 240°, не образуя промежуточных устойчивых кристал-
логидратов. Безводный селенит лантана разлагается выше
720°, селенит празеодима выше 400’. Кристаллогидраты селе-
нитов церия и неодима начинают терять кристаллизационную
воду соответственно при температуре 60° и 80°, образуя че-
тырехводные кристаллогидраты, устойчивые в интервале тем-
ператур 200—330° для церия и 230—570° для неодима. Даль-
нейшее обезвоживание селенитов церия и неодима происхо-
дит с одновременным их разложением.
Окраска соединений зависит от входящего в его состав
редкоземельного иона: селенит лантана—белый, селенит
церия — белый с кремовым оттенком, селенит празеодима—
светло-зеленый, селенит неодима — розово-сиреневый.
СХЕМА СИНТЕЗА СЕЛЕНИТОВ Р.З.Э.
RaO34-6HCl -» 2RC1s + 3H2O;
Се2(СО3)3 + 6НС1 2СеС13 Д- ЗН3О + ЗСО8;
2NaOH -f- HaSeO3 -> Na2SeO3 + 2II3O
2RC13 4 3Na2SeO3 — Rs(SeO3)3 + 6NaCI,
где R—La, Ce, Pr, Nd.
Характеристика основного сырья
Лантан окись, Л-3, РЭТУ 96—59.
Церий углекислый, технический, соответствующий двуоки-
си церия марки Цо-1, РЭТУ 144—59.
Празеодим окись, По-3, РЭТУ 1075—63.
Неодим окись, Н-3, РЭТУ 1076—63.
Соляная кислота, х.ч., ГОСТ 3881—46.
Селенистая кислота, ч.д.а., ТУ ГКХ ОРУ 123—59.
Едкий натр, х.ч, ГОСТ 4328—48.
Спирт этиловый, гидролизный, СТУ 57-227—64.
Условия получения
Окись р.з.э. или технический карбонат церия загружают в
фарфоровую чашу, смачивают дистиллированной водой в
соотношении 1 : 1 и растворяют в концентрированной соля-
117
ной кислоте. После растворения основной массы окиси для
ускорения процесса раствор с осадком нагревают до 80—90°.
Процесс считают законченным по достижении полного раство-
рения и установлении pH 4. В растворе церия хлористого
проводят осаждение тяжелых металлов сероводородом до
полноты осаждения. Солянокислый раствор р.з.э. (La, Се, Рг,
N'd) отфильтровывают от механических примесей или тяже-
лых металлов и разбавляют до уд. веса 1,08.
Для приготовления 20%-ного раствора селенистокислого
натрия (уд. вес 1,22) готовят 40%-ный раствор едкого натра
уд. веса 1,4 (0,66 кг едкого натра в 990 мл дистиллированной
воды) и 22%-ный раствор селенистой кислоты уд. веса 1,18
(1 кг селенистой кислоты в 3,55 л дистиллированной воды)
и осторожно нейтрализуют кислоту щелочью до pH 7. По-
лученный раствор селенистокислого натрия отфильтровывают
от механических примесей и используют для осаждения се-
ленитов р.з.э.
К солянокислому раствору р.з.э. постепенно при переме-
шивании приливают раствор селенита натрия. Осадок остав-
ляют под раствором на сутки, затем фильтруют на воронке
Бюхнера и отмывают от хлористого натрия и избытка селе-
нистокислого натрия дистиллированной водой до отсутствия
хлор-иона в промывных водах (качественная реакция филь-
трата с азотнокислым серебром в присутствии азотной кисло-
ты). Отмытый осадок промывают спиртом (1:1) и сушат в
сушильном шкафу при 80—90° и остаточном давлении
10 мм рт. ст. (для церия) и 250° (для лантана, празеодима,
неодима) до постоянного веса. В маточном растворе после
осаждения селенитов р.з.э. и в первой промывной воде осаж-
дают 20%-ным раствором хлористого бария селенит бария,
который отфильтровывают и промывают от хлористого нат-
рия дистиллированной водой. Полученный селенит бария с
целью регенерации селенистой кислоты смешивают со смо-
лой КУ-2 в Н-форме в соотношении 1 : 3 и заливают 5-крат-
ным по отношению к смоле количеством дистиллированной
воды. Полученную пульпу перемешивают в течение 1 часа
(до полного растворения селенита бария). Смолу отфильтро-
вывают от раствора и промывают дистиллированной водой
до нейтральной реакции (pH 5 по универсальной индикатор-
ной бумаге). Основной раствор и промывные воды с целью
отделения следов иона бария пропускают через колонку со
смолой КУ-2 в Н-форме со скоростью 1—3 л/час, затем ко-
лонку промывают дистиллированной водой до нейтральной
реакции (pH 5—6). Полученный фильтрат представляет со-
бой разбавленный раствор селенистой кислоты, который мо-
х<ет быть использован для приготовления 22%-ного раствора
селенистой кислоты.
118
Из 100 г исходной окиси редкоземельного элемента (лан-
тана, празеодима, неодима) получают 180—185 г селенитов
лантана или празеодима и 200 г селенита неодима; из 100 г
карбоната церия получают 115—120 г селенита церия. Во
всех случаях выход составляет 92—95% от теоретического.
Химический состав селенитов редкоземельных элементов,
получаемых по данной методике: содержание основного ве-
щества— не менее 97%, содержание примесей (не более):
железа — 0,01%, кальция — 0,05%.
Примечание. Селенистая кислота и ее соединения являются
сильными ядами, подобно соединениям мышьяка. Поэтому работа с соеди-
нениями селена требует соблюдения особых мер предосторожности. Все
работы необходимо проводить в вытяжном шкафу при исправной венти-
ляции. Работать обязательно в перчатках. После окончания работы тща-
тельно мыть рукн.
ЛИТЕРАТУРА
1. В. В. Серебренников. Химия редкоземельных элементов, 1,
Томск. ТГУ, 1969. стр. 329.
2. А. И. Александрович. В. В. Серебренников. Тр. Том-
ского ун-та. Сер. хим., 154. 105 (1962).
3. Е. G 1 е s b г е с h t. М. Р е г г 1 е г, W. W с n d 1 a n d t. Anais Acad Bra-
sil. Cienc., 34, 1,37 (1962).
Поступила в декабре 19з5 r.
Ц:-Л
УДК 546.651/.659'236.07
СЕЛЕНОВОКИСЛЫЕ СОЛИ РЕДКОЗЕМЕЛЬНЫХ
ЭЛЕМЕНТОВ
л
Селенаты р.з.э.
Ю. Б. ПЕРКОВСКАЯ, И. М. СУХАНОВА
Селенаты р.з.э. по своим свойствам аналогичны соответ-
ствующим сульфатам, но обладают несколько большей ра-
створимостью. Это устойчивые па воздухе соединения, кри-
сталлизующиеся из водных растворов с различным содержа-
нием кристаллизационной воды. С повышением температуры
растворимость селенитов р.з.э. уменьшается. Некоторые дан-
ные о селенитах р.з.э. приведены в табл. 1.
В литературе описано получение селенатов р.з.э. раство-
рением соответствующих гидроокисей или карбонатов в селе-
новой кислоте с последующим упариванием полученного
раствора [1].
Нами разработана методика получения селенатрв р.з.э.
путем растворения их окислов в селеновой кислоте (исклю-
чение составляют селенаты церия, празеодима и тербия,
окислы которых нерастворимы в селеновой кислоте; селена-
ты этих трех элементов получают путем растворения в селе-
новой кислоте соответствующих карбонатов).
СХЕМА СИНТЕЗА СЕЛЕНАТОВ Р.З.Э.
R2O3 + 3H2SeO4 - Rj(SeO4)s + ЗН2О;
R2(CO3)s + 3H2SeO4 — R2 (SeO4)3 + 3H2O + 'CO,,
где R—La, Ce, Pr, Nd, Sm, Eu, Qd, Tb, Dy, Ho, Er, Tu, Yb, Lu.
Характеристика основного сырья
Лантан окись, РЭТУ 96—59.
Церий углекислый, МРТУ 6-09-250—63.
Празеодим углекислый, РЭТУ 1075—63.
120
ээя и ян
-йввКяэкодо
Формула ОО « ся XX ю ю и—? Л do О 4» (Л СЛ « С4 сс <U -ю о О О о о 2.0 О о ° о О u5l?Q0<Dq?’—'ООС^аЗ^ООсЛ *•» oOqoOqOoo п О <? U .¥ 4» V $ V Ф> u О О СП СЛ ^,сл ««У -4-Д* 44 Л* N’4*' 2-’Оез’ОХ)>»0*Г -?х> o-z “монаги£>- = О С4 Г о 1» сл м S сс с 4) Ч CJ S X
Наименование соединения Лантан селеновокислый 1 Церий селеновокислый Празеодим селеновокислый Неодим селеновокислыи Самарий селеновокислый Европий селеновокислый Гадолиний селеновокислыи Тербий селеновокислый Диспрозий селеновокислый Гольмий селеновокислый Эрбий селеиовокислый Тулий селеновокислый Иттербий селеновокислый Лютеций селеновокислый Иттрий селеновокислый | * Данные относятся к безво
121
Неодим окись, РЭТУ 1076—63.
Самарий окись, РЭТУ 1077--63.
Европий окись, РЭТУ 1078—63.
Гадолиний окись, РЭТУ 1079—63.
Тербий углекислый, РЭТУ 1080—63
Диспрозий окись, РЭТУ 1081—63.
Гольмий окись, РЭТУ 1082—63.
Эрбий окись, РЭТУ 1083—63.
Тулий окись, РЭТУ 1084—63.
Иттербий окись, РЭТУ 1085—63.
Лютеций окись, РЭТУ 1086—63.
Иттрий окись, РЭТУ 1072—63.
Селеновая кислота, ч., Р>ТУ РУ 2100—54.
Диэтилдитиокарбамат натрия, ч., ГОСТ 8864—58.
Условия получения
Окись р.з.э. помещают в фарфоровую чашу, смачивают
дистиллированной водой в соотношении 1 : 2 и при нагрева-
нии реакционной смеси до 80—90° обрабатывают 24%-ной
селеновой кислотой уд. веса 1,20. Кислоту приливают неболь-
шими порциями при перемешивании. Полученную массу при
периодическом перемешивании упаривают досуха и сухой
остаток растворяют при комнатной температуре в дистилли-
рованной воде (из расчета 400 мл воды на 100 г получаемого
продукта). Остаток должен полностью раствориться, после
чего в растворе устанавливается pH 2,5—3.
Если полного растворения не происходит и pH реакцион-
ной массы меньше 2,5, операцию упарки досуха и растворе-
ние в дистиллированной воде необходимо провести повторно.
Затем раствор отфильтровывают, разбавляют до уд. ве-
са 1,1 (50 г/л R2O3) и при температуре не выше 25° осаж-
дают тяжелые металлы и железо 3%-ным раствором диэтил-
дитиокарбамата натрия из расчета 30 мл на 100 г получаемо-
го продукта. Раствор диэтилдитиокарбамата натрия прили-
вают по каплям при энергичном перемешивании, а затем
смесь периодически перемешивают в течение часа и отстаи-
вают 6 часов. Отстоявшийся раствор фильтруют. Осадок
промывают 2—3 раза дистиллированной водой. Основной
раствор и промывные воды объединяют и упаривают досуха.
С момента появления на поверхности раствора пленки необ-
ходимо раствор периодически перемешивать.
Выход металла в соль составляет 93%.
Расход основного сырья на получение 100 г соли приве-
ден в табл. 2.
По данной методике получают продукты следующего ка-
чества:
а) содержание основного вещества не менее 97—99%;
122
Таблица 2
Наименование соли Расход сырья
100%-ной окиси р.з.э. 80 %-ной i селеновой КИСЛОТЫ i диэтилди- тиокарба- 1 мата натрия
теоретиче- ски факти- чески
Лантан селеновокислый 40,0 43,1 72,0 1,о
Церий селеновокислый 45,0* 46,3 73,0 1,0
Празеодим селеновокислый 42,5** 45,7 73,0 1,0
Неодим селеновокислый 42,7 45,0 72,0 1,0
Самарий селеновокислый 40,0 43,0 67,0 1,0
Европий селеновокислый 42,0 45,0 70,0 1,0
Гадолиний селеновокислый 41,0 44,0 66,0 1,0
Тербий селеновокислый 39,0*** 42,0 61,0 1,0
Диспрозий селеновокислый 41,5 45,0 65,0 1,0
Гольмий селеновокнслый 42,0 45,0 65,0 1,0
Эрбий селеновокислый 42,2 45,4 64,0 1,0
Тулий селеновокнслый 36,6 39,4 55,4 1,0
Иттербий селеновокислый 43,0 46,0 64,0 1,0
Лютеций селеновокнслый 38,0 41,0 56,0 1,0
Иттрий селеновокислый 30,1 32,4 78,0 1,0
* Расход карбоната церия приведен в пересчете па двуокись церия.
* * Расход карбоната празеодима приведен в пересчете на окись
празеодима. ,
* ** Расход карбоната тербия приведен в пересчете на окись тербия.
б) Содержание примесей, не более: железо — 1 • Ю~2—
— 5-10“3; тяжелые металлы — 1 • 10*2—5,10“3; кальций —
5-10-2—3-10-2%; примеси других р. з. э. -- 1,0—0,1 %.
ЛИТЕРАТУРА
1. В. В. Серебренников. Химия редкоземельных элементов,
т. 1. Томск, ТГУ, 1959, стр. 329.
2. Handbook of Chemistry and Physics 37th ed., v. 1, 1955—56.
Поступила в декабре 1965 г.
ЦЗЛ
УДК 546.651/.659'131.074 546.633'131.07
БЕЗВОДНЫЕ ХЛОРИДЫ РЕДКОЗЕМЕЛЬНЫХ
ЭЛЕМЕНТОВ И СКАНДИЯ
Г. Е. РЕВЗИН
Безводные хлориды трехвалентных редкоземельных эле-
ментов (р.з.э.) общей формулы LnCl-з, где Ln — лантанид,
представляют собой весьма гигроскопичные кристаллические
вещества, хорошо растворимые в воде и низших спиртах
метиловом и этиловом-—и окрашенные в различные цвета в
зависимости от цвета входящего в соединение катиона.
Во влажном воздухе безводные хлориды энергично по-
глощают влагу, переходя в кристаллогидраты с различным
содержанием молекул кристаллизационной воды [1, 2].
Основные физико-химические свойства безводных хлори-
дов р.з.э. и скандия приведены в таблице *.
В литературе [1, 4] описан ряд методов, приводящих к
получению чистых безводных хлоридов редкоземельных эле-
ментов и скандия, которые условно можно подразделить на
следующие группы:
1. Обезвоживание гидратированных хлоридов р.з.э.
2. Конверсия окислов в хлориды.
3 Некоторые специальные методы.
Последняя группа способов относится к случаям, когда
редкоземельный элемент или скандий находятся в иных
(кроме окислов или хлорида-гидрата) химических формах.
Безводные хлориды р.з.э. и скандия могут быть получены пу-
тем взаимодействия редкоземельных металлов с хлором [5],
сульфидов, карбидов, нитридов или гидридов р.з.э. с хлори-
стым водородом или хлором [6—9], обезвоживанием бензо-
атов р.з.э. с последующим пропусканием хлористого водорода
в суспензию бензоатов в эфире {10].
На практике для получения безводных хлоридов р.з.э.
наиболее широко используются методы, основанные на обез-
воживании их кристаллогидратов или конверсии окислов в
хлориды [1, 2, 4, 11 — 17]
* Приведенные величины физико-химических характеристик хлоридов
р.з.э., иттрия и скандия взяты из литературных источников [1—3].
124
Хлорид Молекул яр-i ный вес I Цвет Удельный вес 2/С.ч3 Температу- ра плавле- ния,"С Температура кипения, °C Теплота об- разования ккал!моль
LaCl3 245,269 Белый 3,79-3,94 852 1750 255,91
СеС13 246,479 Белый 3,92-3,97 802 1720 252,84
РгС1, 247,266 Светло-зеле- ный 4,02—4,15 776 1700 259,09
NdCls 250,599 Сиреневый 4,14-4,19 760 1680 245,61
SmCl3 256,709 Желтый 4,27—4,46 728 Разлагается —
EuCl3 258,319 Желто-зеле- ный — 623 —»— 239,0
GdCl3 263,61'9 Белый 4,52 600 15S0 240,09
TbCl3 265,283 Белый 4,35 588 1550 —
DyCl, 268,859 Желтовато- белый 3,60—3,67 654 1560 —
I IoCl3 271,289 Темно-жел- тый 4,25 718 1500 —
ErCl., 273,619 Розовый — 774 1500 229,07
TuCl3 275,293 Зеленый 4,34 821 1490 —
YbCl3 279,399 Белый — 854 Разл. 230
L11CI3 281,329 Белый — 892 1480 —
YCI3 195,264 Белый 2,81 - 2,67 700 1503 2 52,69
ScCl, 151,715 Белый 2,39 940 800-850 215,0
При обезвоживании кристаллогидратов имеет место гид-
ролиз хлоридов с образованием нерастворимых в воде окси-
хлоридов [4], поэтому обезвоживание проводят в условиях,
предотвращающих гидролиз. Для этого используют хлори-
стый водород хлористый тионил, четыреххлористый углерод,
хлористый аммоний, монохлорид серы и другие.
Наиболее удобным методом синтеза безводных хлоридов
является конверсия окислов в хлориды, так как окислы яв-
ляются той формой, в которой р.з.э. выделяются из природ-
ного сырья. Конверсию окислов в хлориды осуществляют с.
помощью восстановителей и хлорирующих агентов, например
нагреванием смеси угля с окислом р.з.э. в токе хлора [11],
нагреванием смеси окисла с. хлористым аммонием [12], на-
греванием окислов р.з.э. в парах хлора и однохлористой серы
[13], в парах четыреххлористого углерода и другие [14, 15].
В силу различия свойств редкоземельных элементов и их
безводных хлоридов условия получения безводных хлоридов
заметно изменяются в ряду лантанидов от лантана до лю-
теция.
Нами разработаны и уточнены способы и оптимальные
условия процесса получения безводных хлоридов лантана,
церия, празеодима, неодима, самария, европия, диспрозия,
эрбия, иттербия, лютеция, а также иттрия п скандия.
125
СХЕМА СИНТЕЗА БЕЗВОДНЫХ ХЛОРИДОВ РЕДКОЗЕМЕЛЬНЫХ
ЭЛЕМЕНТОВ И СКАНДИЯ
2М2О3 4- ЗСС14 — 4МС13 + ЗСО,
МС13-/гН2ОСС^МС13+ «НгО ]
Характеристика основного сырья
Лантан окись, РЭТУ 96—59.
Церий углекислый, МРТУ 6-09-250--63.
Празеодим окись. РЭТУ 1075—63.
Неодим окись, РЭТУ 1076—64.
Самарий окись. РЭТУ 1077—63.
Европий окись, РЭТУ 1078—63.
Гадолиний окись. РЭТУ 1079—63.
Тербий окись, РЭТУ 1080—63.
Диспрозий окись, РЭТУ 1081—63.
Гольмий окись, РЭТУ 1082—62.
Эрбий окись. РЭТУ 1083—63.
Тулий окись, РЭТУ 1084—63.
Иттербий окись, РЭТУ 1085-63.
Лютеций окись, РЭТУ 1086—63.
Иттрий окись, РЭТУ 1072—63.
Скандий окись, ЦМТУ 4854—57.
Соляная кислота, х.ч., ГОСТ 3118—46.
Углерод четыреххлористый, ч., ГОСТ 5827 51.
Условия получения безводных хлоридов р.з.э. иттриевой
группы и скандия
Безводные хлориды иттриевой группы наиболее легко
могут быть синтезированы непосредственной конверсией
окислов парами четыреххлористого углерода [15].
Синтез осуществляется на установке, изображенной на
рисунке. Навеску окиси р.з.э. помещают в кварцевую или
фарфоровую лодочку слоем не более 1,5 см. В баллончик-
испаритель помещают четыреххлористый углерод и, подо-
гревая испаритель, подают пары четыреххлористого углеро-
да в кварцевую трубу, вставленную в печь, с такой ско-
ростью, чтобы в холодильнике конденсировалась ~1 капля
четыреххлористого углерода в секунду. Одновременно вклю-
чают обогрев печи, в течение 1,5—2 часов поднимают темпе-
ратуру печи до заданного уровня и выдерживают при этой
температуре определенное время. Затем отключают обогрев
и охлаждают печь, продолжая пропускать пары четыреххло-
ристого углерода. По достижении температуры 100° прекра-
щают подачу паров четыреххлористого углерода, разгружают
126
печь и помещают лодочку с продуктом в сухую камеру, где
в дальнейшем и производят расфасовку продукта.
ЛАТР1
Схема установки для хлорирования окисей р.з.э. и скандия парами
четыреххлористого углерода
1 — баллончик-испаритель; 2—трубчатая печь; 3 — лодочка; 4— квар-
цевая труба; 5 — термопара; 6—воздушный холодильник; 7 — колба-
приемник; 8— затвор; 9— хлоркальциевая трубка; 10— электроплитка
Единовременная загрузка окиси р.з.э. зависит от габари-
тов печи и может составлять от нескольких граммов до 0,5—
1,0 кг и более.
Полученные безводные хлориды представляют собой слег-
ка сцементированные твердые вещества, растворяющиеся в
воде с выделением тепла и дающие прозрачные растворы,
иногда со слабой опалесценцией.
Температура, при которой проводится конец хлорирова-
ния, и время выдержки для различных окислов представле-
ны ниже.
Окисел Температу- ра, °C Время, час
Dy2O, 620-640 2
Ег,О, 700 2
ТтцОз 700 2
Yb2Os 700 2
Lu2O 700 2
Y2O3 700 4
SCgOg 700 6
Расход четыреххлористою углерода зависит от количест-
ва загружаемого окисла и составляет от 2 до 5 кг на 1 кг
безводного хлорида.
Выход хлоридов р.з.э. равен 97—98% и может быть уве-
личен до 99—100% за счет сведения к минимуму механиче-
ских потерь при загрузке и выгрузке продукта.
127
Количество нерастворимого в воде остатка (оксихлорида)
не превышает 0,2—0,5%; содержание иных примесей зависит
только от чистоты исходных окисей р.з.э., поскольку в про-
цессе синтеза внесение примесей исключено. Загрязнения
хлоридов продуктами пиролиза четыреххлористого углерода
не наблюдается.
Условия получения безводных хлоридов цериевой группы
и хлоридов гадолиния, тербия и гольмия
Безводные хлориды р.з.э. цериевой группы также могут
быть получены хлорированием их окислов, однако этот про-
цесс занимает большее время по сравнению с получением
хлоридов иттриевой группы.
Кроме того, хлориды самария, европия, гадолиния и тер-
бия имеют низкую температуру плавления и образующийся
на поверхности окисла плав хлорида препятствует дальней-
шему хлорированию, а снижение температуры вызывает сни-
жение скорости реакции настолько что хлорирование окислов
этих элементов приходится проводить в течение 12—24 часов
[16, 17].
Поэтому для получения безводных хлоридов этих элемен-
тов наиболее удобным является способ обезвоживания их
кристаллогидратов при нагревании в парах четыреххлористо-
го углерода [15].
Процесс осуществляется на той же установке (см. рис. 1).
Кристаллогидрат хлорида р.з.э., полученный растворе-
нием окиси в соляной кислоте до pH 1—1,5 и упариванием
раствора на водяной бане досуха, загружают в лодочку.
Обезвоживание хлоридов р.з.э. производят, пропуская через
кварцевую трубу пары четыреххлористого углерода вначале
при температуре 100° в течение 1 часа, затем поднимают тем-
пературу до 200—250° и выдерживают продукт при этой тем-
пературе еще 1 час, после чего температуру поднимают до
450—500° и выдерживают продукт еще 1—1,5 часа. Хлориды
европия, самария, гадолиния, тербия и гольмия легко окис-
ляются кислородом воздуха до оксихлоридов, поэтому обез-
воживание их кристаллогидратов проводят в атмосфере азо-
та, для чего азот из баллона барботируют через баллончик-
испаритель, и насыщенный парами четыреххлористого
углерода подают в трубчатую печь. После охлаждения печи
до 100° лодочку с хлоридом помещают в сухую камеру, где
затем расфасовывают полученный продукт.
Расход четыреххлористого углерода зависит от количест-
ва загружаемого водного хлорида и составляет от 2 до 5 кг
на 1 кг безводного хлорида.
Выход хлоридов р.з.э. равен 95—96%.
128
Содержание нерастворимого в воде остатка (оксихлорида)
составляет 0,5 н- 1,0%.
ЛИТЕРАТУРА
1. В. В. Серебренников. Химия редкоземельных элементов, т. 1,
Томск, Изд. ТГУ, 1959, стр. 196.
2. В. В. Серебренников, Л. А. Алексеенко. Курс химии
редкоземельных элементов, Томск, ТГУ, 1963, стр. 62.
3. М. X. Кара петьяиц, М. Л. Карапетьянц. Таблицы неко-
торых термодинамических свойств различных веществ, Тр. хнм.-технол.
ин-та им. Д. И. Менделеева. 34 (1961).
4. М. D. Taylor. Chem. Revs., 62, 503, 513 (1962).
5. J. J. Berzelius. Ann. de Poggend, 15, 385 (1829),
6. H. Moissan. Compt. rend, 4, 1 (1893).
7. H. Moissan. Compt. rend. 131, 595, (1900).
8. O. Peterson. Ber., 28, 2419 (1895).
9. C. Mattgnon, M. Dele pine. Compt. rend., 132, 36 (1901).
10. P. Braumann, S. Takvorian. Compt. rend., 194, 1579 (1932).
11. H. Oersted. Over. D. vid. Selk. Forts., 251 (1824).
12. B. S. Hopkins. 1. B. Reed, L. F. A u d 1 e t h. I. Amer. Chem.
Soc., 57, 1159 (1935).
13. С. M a 11 g п о n, F. В о u r i о n. Compt, rend. 138, 760 (1904),
14. P, Camboulivc. Compt. rend., 150, 175 (1910).
15. С. А. Репин. Редкоземельные элементы, M„ Изд. АН СССР,
1963, стр. 71.
16. J. Е. Miller, S. Е. Miller, R. С. Himes. J. Amer. Chem.
Soc., 81, 449 (1959).
17. С, T. Stubblefield, L. E у r i n g. J. Amer. Chem. Soc., 77,
3004 (1955).
Поступила в декабре 1965 г, ЦЗЛ
УДК 546.681.3'141.07
ГАЛЛИЙ ТРЕХБРОМИСТЫЙ
О. Н. БРЕУСОВ, В. Г. ЛАВРЕНТЬЕВА
GaBrs
М.в. 309,44
Галлий трехбромистый (трибромид галлия) представляет
собой бесцветные, чрезвычайно гигроскопичные кристаллы,
сильно дымящие на воздухе; т. пл. 122.5° и т. кип. 279°.
В литературе указывается на возможность прямого син-
теза трехбромистого галлия из элементов с последующей
очисткой получаемого при этом продукта от свободного бро-
ма перегонкой в вакууме или в токе индифферентного газа
[1-3].
Нами проверена и уточнена методика проведения этого
процесса.
СХЕМА СИНТЕЗА ТРЕХБРОМИСТОГО ГАЛЛИЯ
2Ga 4- Вг2 —+ 2GaBt
2GaBr Вг3 —> 2GaBr3
2GaBr3 + Вг2 — 2GaBr3
Характеристика основного сырья
Галлий металлический, Гл-1, РЭТУ 851—61; содержание
галлия не менее 99,5%.
Бром жидкий, ч.д.а., Гост 4109—48.
Условия получения
Синтез трехбромистого галлия проводят путем взаимо-
действия металлического галлия с углекислотой, насыщенной
парами брома, при температуре 300— 350°. Углекислоту пред-
варительно очищают от примеси кислорода над металличе-
130
ской медью при 600° и от влаги над серной кислотой и фос-
форным ангидридом.
В колбу, обогреваемую электрической спиралью по всей
своей поверхности, помешают 50 г металлического галлия.
Колба должна быть снабжена впаянной трубкой для подачи
газов к поверхности металлического галлия и отводом для
образующегося продукта. Пары образующегося трехброми-
стого галлия конденсируются в охлаждаемом водой стек-
лянном приемнике, соединенном со стеклянным реактором —
колбой — при помощи шлифа. Перед началом процесса, а
также после окончания бромирования реактор и приемник
продувают очищенной углекислотой.
Выход трехбромистого галлия равен 209 г, что составляет
94% от теоретического.
Расход брома не превышает 500 г и расход углекислоты
10 ж3. Непрореагировавшие, газы поглощаются раствором
щелочи.
Данная методика позволяет получать препарат, удовлет-
воряющий требованиям РЭТУ 945—62.
ЛИТЕРАТУРА
1. Gmellns Handbuch der anorganischen Chemie, 8 Aufl., 36, 80
(1936).
2. Руководство по препаративной неорганической химии, Под ред.
Г. Брауера М., ИЛ, 1956, стр. 399.
3. W. К 1 е m m, W. Til k. Z. anorgan. und allgem. Chemie, 207, 161
(1932).
Поступила в декабре 1965 г.
ЦЗЛ
УДК 546.682.3'137.07
ИНДИЙ ХЛОРНОКИСЛЫЙ
И. М. СУХАНОВА, В. Д. ЗАМЕДЯНСКАЯ
1п(С1О4)2-8Н2О М.в. 557,29
Индий хлорнокислый представляет собой белое кристал-
лическое вещество, расплывающееся на воздухе, хорошо ра-
створимое в воде и абсолютном спирте; т. пл. 80°; т. разл.
200° [1].
Для получения индия хлорнокислого можно использовать
нейтрализацию хлорной кислоты гидроокисью индия с по-
следующим упариванием полученного раствора.
СХЕМА СИНТЕЗА ИНДИЯ ХЛОРНОКИСЛОГО
1п(ОН)3+ЗНСЮ4^1п(С1О4)з+ЗН2О
Характеристика основного сырья
Индий гидрат окиси, ч., РЭТУ 944—62.
Хлорная кислота, ч.д.а., ТУ МХП ОРУ 87—57.
Условия получения
Нагревают до 70—80° 320 мл 10%-ной хлорной кислоты и
небольшими порциями прибавляют 16,6 г гидроокиси индия
(в пересчете на 100%-ный продукт). Полученный раствор,
имеющий pH 1 —1,5, отфильтровывают от механических при-
месей на воронке Бюхнера через хлорвиниловую ткань. Про-
зрачный раствор упаривают на водяной бане при периодиче-
ском перемешивании до получения сухой рассыпчатой мас-
сы кристаллов.
Выход равен 51,35 г, что составляет 95% от теоретиче-
ского.
132
Препарат хранят в запарафинированных склянках с при-
тертыми пробками.
Данная методика позволяет получать индий хлорнокис-
лый, соответствующий требованиям РЭТУ 1050—63: 1) со-
держание индия в пересчете на 1п(СЮ4)3-8 Н3О — не ме-
нее 97%; 2) контролируемые примеси (содержание в %, не
более): железо—1 1б~2; натрий — 2-10-2; сульфаты —
3 • 10 “2; хлориды — 1 • 10'-.
ЛИТЕРАТУРА
1. Справочник химика, т. II, М., Госхнмиздат, 1963, ст*р. 70.
Поступила в декабре 1965 ЦЗЛ
УДК 546.131.07
БЕЗВОДНЫЕ ХЛОРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Типовой метод
Л. М. ПЕТРОВА, О. Н. БРЕУСОВ
1пС13
ZrCl4
ТеС16
МоС16
ТеС14
WC16
ReCk
Индий треххлористый
Цирконий четыреххлористый
Тантал пятихлористый
Молибден пятихлористый
Теллур четыреххлористый
Вольфрам шестихлористый
Реиий пятихлористый
М. в. 221,18
М. в. 233,03
М. в. 358,21
М. в. 273,20
М. в. 269,41
М. в. 396,57
М. в. 363,46
Получение хлоридов переходных металлов связано с ря-
дом трудностей, которые обусловлены большой гигроскопич-
ностью этих соединений, склонностью их к гидролитическо-
му разложению, а также способностью образовывать низшие
хлориды.
По литературным данным [1—6], безводные хлориды мож-
но получать хлорированием переходных металлов газообраз-
ным хлором с последующей возгонкой образующихся хло-
ридов.
В настоящем сообщении приводится типовой метод полу-
чения реактивной чистоты безводных хлоридов циркония,
рения, тантала, индия, вольфрама, теллура, молибдена выс-
ших валентностей с применением лабораторного оборудо-
вания.
СХЕМА СИНТЕЗА БЕЗВОДНЫХ ХЛОРИДОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ
2Ме 4- п С12 = 2МеС1„
Характеристика основного сырья
Хлор, техн , в баллонах, ГОСТ 6718—53.
Цирконий, РЭТУ 604—59.
134
Рений, РЭТУ 88—59.
Тантал, РЭТУ 715—61.
Индий, ЦМТУ 4783—56.
Вольфрам ТУ ВМ 155—54.
Тёллур, ГОСТ 9514-60.
Молибден, ТУ ВМ, 7-153—54.
Условия получения
Безводные хлориды получают путем взаимодействия ме-
таллов с газообразным хлором при повышенной температуре
на установке лабораторного типа, представленной на ри-
сунке.
Схема установки для получения хлоридов переходных металлов
1— баллон с азотом; 2— баллон с хлором; 3— гидрозатвор; 4 — элект-
ропечь; 5 — трубка с активированным углем; 6 — осушитель с H2SO4;
7 — осушитель с PjOs; 8 — хлоратор; 9 — электропечь; 10— конденсатор;
11 — электроспираль, 12—поглотитель с NaOH; 13 — потенциометры
Вначале система очищается от воздуха азотом. Давление
азота и хлора в системе поддерживается с помощью гидро-
затвора, наполненного насыщенным раствором хлористого
натрия, установленного непосредственно после баллонов. Ве-
личина давления определяется при этом высотой столба ра-
створа хлористого натрия, составляющего 400—500 мм.
Продолжительность продувки азотом 1 час.
Азот и хлор, поступающие в систему, очищают от кисло-
рода с помощью активированного угля, нагретого до 700°, а
от влаги — с помощью оеушителей с серной кислотой и фос-
форным ангидридом.
135
Навеску металла от 0,1 кг до 2 кг (в зависимости от того,
какое количество хлорида нужно получить) помещают в го-
ризонтальную Г-образную кварцевую трубу диаметром 45 мм
и длиной 1000 мм. В качестве конденсатора используется
стеклянная колба емкостью 5 л, присоединяемая к хлорато-
ру посредством шлифа. Шлифовое соединение обогревают
электрической спиралью. Горизонтальную трубчатую элект-
ропечь подключают к сети через электронный
ский потенциометр ЭПД-12 с автоматическим
температуры, электроспираль—через ЛАТР типа
250-2.
Температурные параметры процессов хлорирования ме-
таллов представлены в таблице 1.
автоматиче-
регулятором
PHO
Таблица 1
Физические коистаиты Параметры процессов хлорирования
Хлориды металлов температу- температу- температу температу-
ра плавле- ра кипения, ра хлориро- ра обогре-
ния. °C °C вания, °C ваемой спи-
рали, °C
Цирконий четырех- 437 (25 атм) Сублимиру- 500 400
хлористый ет
Рений пятихлорис- тый 260 330 600 400
Тантал пятихлорис- тый 220 242 400 300
Индий треххлорис- 585 (под Сублимиру- 500 500
тый б/в давл.) ет
Вольфрам шести- хлористый 275 346 800 400
Теллур четырех- хлористый 224 390 400 500 400
Молибден пятихло- ристый 194 268 300
Окончание процесса хлорирования легко определяется по
прекращению выделения паров хлоридов. Избыточный хлор
поглощается 10%-ным раствором едкого натра.
Ввиду сильной гигроскопичности безводных хлоридов нх
расфасовка и отбор проб для анализа проводятся в герметич-
ной камере, осушенной с помощью азота и фосфорного ан-
гидрида.
Анализ полученных хлоридов представлен в таблице 2.
На основании данных анализа составлены технические
условия на безводные хлориды переходных металлов реак-
тивной чистоты.
Выход безводных хлоридов составляет 87—90% от теоре-
тического.
136
137
ЛИТЕРАТУРА
1. Э. И. к р е ч. ж. прим. химии, 10, 1938 (1937).
2. Н. Rem у., Lehrbuch der anorganischen Chemie, II, 1959, стр. 92.
3. Н. В. Барышников, А. Н. 3 е л и к м а и. Изв. вузов, «Цветная
металлургия», 5, 6, 98 (1962).
4. О. П. Колчнн. Извлечение и очистка редких металлов. М„ Атом-
издат, i960, стр. 46.
5. О. А. Сонги н а. Редкие металлы. М., Металлургиздат, 1964,
стр. 62, 402.
6. Д. М. Ю х т а н о в. Производство селена и теллура, М„ Метал-
.тургнздат, 1955, стр. 20.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.289'221.07
ГЕРМАНИЙ ДИСУЛЬФИД
О. Н. БРЕУСОВ, М. Н. КОРОТКЕВИЧ
GeS2 М.в. 136,71
Дисульфид германия (германий двусернистый) представ-
ляет собой белое или желтовато-белое вещество, кристалли-
зующееся в ромбической сингонии или рентгеноаморфное;
его плотность равна 2,94; т. пл. 825°; т. кип. 1530°. В холод-
ной воде растворимость GeS2 составляет 0,45 г на 100 мл
воды, в горячей воде растворимость незначительно увеличи-
вается. Германий дисульфид растворим в щелочах и раство-
рах щелочных сульфидов, нерастворим в толуоле, спирте и
эфире [1, 2].
Получают GeS2 различными методами:
1) взаимодействием элементарных германия и серы при
нагревании в кварцевой ампуле [2, 3];
2) взаимодействием газообразных четыреххлористого
германия и сероводорода [2];
3) взаимодействием смеси паров сероводорода и серы с
металлическим германием [2];
4) осаждением дисульфида германия из солянокислых
растворов германия действием сероводорода [2].
Последний метод нельзя рекомендовать для препаратив-
ных целей, так как получающийся продукт всегда содержит
значительные количества хлора (до 7%), который полностью
невозможно удалить.
Часто описываемый в литературе способ, основанный на
осаждении дисульфида германия из сернокислых растворов
германия, также нельзя считать удовлетворительным из-за
малой растворимости соединений германия в сернокислых
растворах.
Описанный в настоящей работе метод получения этого
вещества основан на кислотном разложении предварительно
получаемого раствора тиогерманата аммония [4].
139
СХЕМА СИНТЕЗА ДИСУЛЬФИДА ГЕРМАНИЯ
Основные реакции:
GeO2 + 3(NH4)2S + Н2О — (NH4),GeS3 4 4NH4OH
(NH4)2GeS3 + H2SO4 — (NH4)2SO4 4- GeS2| + H2S
Побочная реакция:
3H,S 4- H2SO4 — 4S| + 4H2O
Характеристика основного сырья
Двуокись германия, техн., ЦМТУ 3431—53.
Аммоний односернистый, ч.д.а., ГОСТ 3767—56.
Серная кислота, ч., ГОСТ 4204—48.
Условия получения
В платиновой чашке нагревают 100 г двуокиси германия
до плавления (1200°) и быстро охлаждают на воздухе. Полу-
ченную стеклообразную двуокись германия растворяют в
500 мл раствора односернистого аммония (уд. вес 0,9 г/сж3).
Полученный раствор тиогерманата аммония осторожно при
непрерывном перемешивании вливают в 945 мл разбавлен-
ной (1:5) серной кислоты. Выпавший осадок дисульфида
германия оставляют под раствором на 2 часа, отфильтровы-
вают и отмывают от сульфат-иона сероводородной водой до
отрицательной реакции на SO4 (проба с ВаС12). Высушенный
на воздухе продукт переносят в аппарат Сокслета и отмы-
вают от серы толуолом. Затем его промывают диэтиловым
эфиром и высушивают при 100°.
Выход дисульфида германия равен 179 г, что составляет
95% от теоретического.
Обработку продукта толуолом и эфиром можно заменить
отгонкой серы в вакууме (1 -т-5 мм рт. ст.) при температу-
ре 400—500" в течение 1 часа. Сплавлением получающегося
продукта в запаянной ампуле можно получить кристалличе-
ский дисульфид германия.
Данная методика позволяет получать препарат, удовлет-
воряющий требованиям технических условий РЭТУ 1018—62.
ЛИТЕРАТУРА
1. Справочник химика, 2-е изд., т. II, М.—Л., Тосхпмиздат, 1963,
стр. 55.
2. Н. С. Николаев, С. Б. Петрова. Успехи химии, 25 (1), 105
(1956).
3. Н. Remy. Treatise on Inorganic Chemistry, London -N-V, 1, 1956
стр. 522.
4. Л. M. Петрова, О. H. Бреусов, М. Н. К о р о т к е в и ч,
В. Г. Лаврентьева. Авт. свид. 163166; Бюлл. изобр., № 12 (1964).
Поступила в декабре 1965 г. ЦЗЛ
140
УДК 546.289.4'131.07
ГЕРМАНИЙ ЧЕТЫРЕХХЛОРИСТЫЙ
О. II. БРЕУСОВ, Л. М. ПЕТРОВА
GeCl4 М. в. 214,40
Четыреххлористый германий (тетрахлорид германия) яв-
ляется важнейшим соединением германия и широко исполь-
зуется в препаративной химии и технологии этого элемента.
В литературе описано несколько способов его получения.
Чаще других применяют: а) хлорирование металлического
германия газообразным хлором при температуре 500—600°
[1] и б) взаимодействие двуокиси германия с соляной кисло-
той в толстостенной запаянной склянке при температуре
170 -180° [2, 3].
Нами [4] предложен способ получения тетрахлорида гер-
мания, основанный на повышенной реакционной способности
стеклообразной двуокиси германия.
СХЕМА СИНТЕЗА ГЕРМАНИЯ ЧЕТЫРЕХХЛОРИСТОГО
GeO2 + 4НС1 GeC14 + 2Н2О
Характеристика основного сырья
Двуокись германия, техн., ЦМТУ 3431—53.
Соляная кислота, ч. д.а., ГОСТ 3118—46.
Условия получения
В платиновой чашке нагревают до плавления (1115°)
100 г двуокиси германия и резко охлаждают на воздухе.
Образовавшуюся двуокись германия дробят и помешают в
колбу с 4,5 л соляной кислоты уд. веса к 1,19 г!см3. После
настаивания в течение 2—3 дней на дне колбы образуется
тяжелый слой четыреххлористого германия, который отде-
141
ляют от соляной кислоты на делительной воронке и перего-
няют. Собирают фракцию, кипящую при 83—85°.
Выход тетрахлорида германия равен 270 г, что состав-
ляет 95% от теоретического.
Данная методика позволяет получать препарат квалифи-
кации «ч», удовлетворяющий требованиям технических ус-
ловий СТУ-38—4, 2 р-19—63.
ЛИТЕРАТУРА
1. Г. Брауэр. Руководство по препаративной неорганической хи-
мии, М., ИИЛ, 1956, стр. 342.
2. Gtnelins Handbuch der anorganischen Chem e, SN 45, 1958, стр 498
3. Г. А. Меер сон, А. И. Зеликман. Металлургия редких метал-
лов, Металлургиздат, 1955, стр. 541.
4. Л. М. Петрова, О. Н. Б р е у с о в, М. Н. К о р о т к е в и ч,
В. Г. Лаврентьева. Авт. свид. 163166; Бюлл, изобр., № 12 (1964).
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.289.4'141.07
ГЕРМАНИЙ ЧЕТЫРЕХБРОМИСТЫЙ
О. Н. БРЕУСОВ, В. Г. ЛАВРЕНТЬЕВА
GeBr4 М. в. 392,22
Германий четырехбромистый (тетрабромид германия)
представляет собой весьма гигроскопичное дымящее на воз-
духе желтовато-белое вещество с относительной плотностью
3,13 г/см.3-, т. пл. 26,1°, т. кип. 186,5° [1]. Применяется для
получения высоко чистого металла и в элементоорганическом
синтезе.
Получают германий четырехбромистый взаимодействием
металлического германия с элементарным бромом [2], а так-
же взаимодействием стеклообразной или растворимой форм
двуокиси германия с концентрированной бромистоводород-
ной кислотой [3]. Последний метод предпочтительнее, так
как используют более дешевое исходное сырье — двуокись
германия.
СХЕМА СИНТЕЗА ТЕТРАБРОМИДА ГЕРМАНИЯ
GeO2 + НВг = GeBr4 + 2Н3О
Характеристика основного сырья
Двуокись германия, техн., ЦМТУ 3431—53; содержание
GeO2 не менее 99%.
Бромистоводородная кислота, ч., ГОСТ 2062—43.
Условия получения
В платиновой чашке нагревают 100 г двуокиси германия
до плавления (1200°) и расплав быстро охлаждают на воз-
духе. Полученную стеклообразную GeOa мелко измельчают
и нагревают в 2 л концентрированной бромистоводородной
143
кислоты до температуры 110—120° в колбе с обратным холо-
дильником. По мере образования тяжелого желтоватого ма-
слянистого слоя германия четырехбромистого его периодиче-
ски удаляют из сферы реакции, для чего удобнее пользовать-
ся колбой, снгтбженной выпускным краном. Затем полученный
продукт отделяют от бромистоводородной кислоты и перего-
няют.
Выход четырехбромистого германия равен 260 г, что со-
ставляет 85% от теоретического.
Данная методика позволяет получать препарат, удовлет-
воряющий требованиям технических условий РЭТУ 743—61.
ЛИТЕРАТУРА
1. Справочник химика, М., Госхимиздат, т. 2, 1963, стр. 54.
2. Gmelins Handbuch der anorganichen Chemie, SN 45. Germanium
1958, стр. 82.
3. Л. M. Петрова, О. H. Бреусов, М. Н. К о р о т к е в и ч,
В. Г. Лаврентьева. Авт. свид. 163166; Бюлл. изобр., № 12 (1964).
Поступила в декабре 1у65 г. ЦЗ.i
УДК 546.289.4'151.07
ГЕРМАНИЙ ЧЕТЫРЕХЙОДИСТЫИ
О. Н. FРЕЙСОВ, Л. М. ПЕТРОВА
GeJ4 М. в. 580,20
Четырехйодистый германий (тетрайодид германия) нахо
дит применение в лабораторной практике для синтеза соеди-
нений германия.
По литературным данным, продукт получают путем ра-
створения двуокиси германия в йодистоводородной кислоте
[1]. Однако неполнота прохождения реакпии потребовала
уточнения условий процесса.
Нами определены оптимальные соотношения компонентов,
продолжительность синтеза, а также предложено аппаратур-
ное оформление процесса очистки германия четырехйоди-
стого.
СХЕМА СИНТЕЗА ГЕРМАНИЯ ЧЕГЫРЕХЙОДИСТОГО
GeO2 + 4HJ — GeJ4 Д- 2Н2О
Характеристика основного сырья
Двуокись германия, техн., ЦМТУ 3431—53,
Иодистоводородная кислота, ч.д.а., ГОСТ 4200—48.
Хлороформ, ч., ГОСТ 3160—51.
Условия получения
Процесс получения германия четырехйодис.того провидят
в стеклянном перегонном аппарате. Навеску двуокиси гер-
мания (100 г) засыпают в колбу и заливают 1,5 кг йодисто-
водородной кислоты (в трехкратном избытке против стехио-
метрически рассчитанного количества).
Температуру в аппарате поднимают до кипения смеси и
кипятят 4 часа. Двуокись германия растворяется, из раство-
10 Зак. 1060 145
ра выделяется масса, состоящая из ярко-красных кристаллов
тетрайодида германия. Полученную массу отфильтровывают
на воронке Бюхнера от оставшейся йодистоводородной кис-
лоты. Тетрайодид германия очищают перекристаллизацией из
хлороформа, которую проводят в аппарате Сокслета. Для
экстрагирования 1 кг продукта необходимо 3 л хлороформа.
Раствор четырехйодистого германия в хлороформе, со-
бранный в колбе от аппарата Сокслета, переливают в фар-
форовую чашу и выпаривают на водяной бане досуха.
Выход германия четырехйодистого равен 630 г, что со-
ставляет 90% от теоретического.
Данная методика позволяет получать продукт, соответ-
ствующий РЭТУ 813—61.
ЛИТЕРАТУРА
1. Руководство по препаративной неорганической химии, Под ред.
Г. Брауера. М„ ИЛ, 1956, стр. 344.
Поступили в декабре 1965 г. ЦЗЛ
УДК 546.818-31.07
СВИНЕЦ ЗАКИСЬ-ОКИСЬ
Сурик свинцовый, ортоплюмбат свинца
С. Д. К.УТОЛИН. А. И. ВУЛИХ
/°\ /°\
рь( )Рь; ;рь
ХО' "О'
Pb3O4 М. в. 685,56
Свинец закись—-окись по внешнему виду представляет со-
бой оранжево-красный порошок, в воде почти не растворяет-
ся, в кислотах разлагается или растворяется.
Свинец закись-окись обычно получают длительным (15—
20 часов) нагреванием на воздухе моноокиси или карбоната
свинца [1. 2]. Описан также метод, основанный на взаимо-
действии в водной среде плюмбита и плюмбата калия [3].
По данным работы [4], окисление моноокиси с образованием
сурика протекает более интенсивно при повышении давления
кислорода и температуры. Однако авторами эта зависимость
была прослежена лишь до температуры 560° и давления
кислорода, равного 1 атмосфере.
Нами разработана методика, основанная на окислении
моноокиси под повышенным давлением кислорода, что поз-
воляет резко сократить продолжительность процесса и по-
высить содержание РЬ3Ол в продукте до 97—98% против
92% при окислении на воздухе [1].
СХЕМА СИНТЕЗА СВИНЦА ЗАКИСИ-ОКИСИ
6РЬО + О2 — 2РЬ3О4
10*
147
Характеристика основного сырья
Свинец окись, ч. или ч.д.а. ГОСТ 9199—59.
Кислород газообразный, компримированный, ГОСТ 5583—
5<8, или жидкий, ГОСТ 6331 — 52.
Условия получения
В платиновую чашку помещают 500 г окиси свинца. Чаш-
ку устанавливают в реторте из нержавеющей стали, рассчи
танной на давление до 10 атмосфер. Реторту помещают в
печь, соединяют с кислородным баллоном через двухступен-
чатый кислородный редуктор и продувают кислородом в
течение нескольких минут. Затем повышают давление кисло-
рода в реторте до 4—7 атмосфер и температуру до 650—700°
и в этих условиях дают выдержку в течение 3 часов. Затем
реторту охлаждают (под давлением кислорода! см. примеча-
ние) и выгружают продукт, соответствующий квалификации
«ч.» или д.а.» в зависимости от квалификации исходной
окиси свинца.
Выход закиси-окиси свинца равен 500—510 г, что состав-
ляет 97—99% от теоретического. Фактическое содержание
РЬ3О4 — не менее 97%; препарат соответствует ВТУ УХКП
40—60.
Примечание. При температуре выше 550° па воздухе сурик дис-
социирует на окись свинца и кислород, поэтому при охлаждении реторты
с препаратом необходимо поддерживать в ней давление кислорода, пока
температура не понизится до 500’.
ЛИТЕРАТУРА
1. Ю. В. Карякин, И. И, Ангелов. Чистые химические реакти-
вы, М„ Гоехимиздат, 1955, стр. 510.
2. Е. Ф. Беленький, И. В. Рискни. Химия и технология пиг-
ментов, Л., Гоехимиздат, 1960, сгр. 505.
3. Руководство по препаративной неорганической химии, Под ред.
Г. Брауэра, М.. ИЛ, 1956, стр. 361.
4. А. А. Ра вдел ь, Н. В, Огарев. Ж. прикл. химии, 33, 70 (I960).
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.882.3'131.07
НИОБИЙ ТРЕХХЛОРИСТЫЙ
Л. м ПЕТРОВА, Б. Г. КОРШУНОВ
NbCla М. в. 199,26
В литературе описан метод получения трихлорида нио-
бия [1], основанный на восстановлении пятихлористого ниобия
металлическим ниобием. Реакция проводилась в вакуумиро-
ванной запаянной ампуле при 400° в течение 80 часов. Оста-
точное давление составляло 1 • 10 ’ мм.
Нами уточнены условия этого синтеза и определена воз-
можность получения продукта достаточно высокого качества
при 320° в течение 12 часов при 1 • 10-2 мм, что значительно
упрощает процесс.
СХЕМА СИНТЕЗА ТРИХЛОРИДА НИОБИЯ
3NbCl5 + 2Nb = 5NbCl8
Характеристика основного сырья
Ниобий пятихлористый, ч., РЭТУ 683—61.
Ниобий металлический, техн., РЭТУ 62—58.
Условия получения
Исходные вещества берут в стехиометрическом соотноше-
нии. Навески пятихлористого ниобия 34 г и металлического
порошкообразного ниобия 7,8 а загружают в ампулу из тер-
мостойкого стекла (диаметр ампулы 60 мм, длина 300 мм).
После загрузки смесь тщательно перемешивают путем
встряхивания содержимого ампулы.
Процесс загрузки смеси в ампулу проводят в «сухой»
камере для предотвращения взаимодействия пятихлористого
ниобия с влагой воздуха.
149
Ампулу с загруженной смесью присоединяют к вакуум-
ному насосу РВН-20 и откачивают до остаточного давления
0,01 мм. Вакуум измеряют с помощью вакуумметра ВИТ-1А
и термоэлектрической лампы ЛТ-2. После вакуумирования
ампулу запаивают. Запаянную ампулу помещают в трубча-
тую электропечь и выдерживают при температуре 320° в те-
чение 12 часов. Температуру трубчатой электропечи регули-
руют с помощью автоматического потенциометра ЭПД-12.
После охлаждения электропечи с ампулой до комнатной
температуры ампулу помещают в «сухую» камеру, разби-
вают и выгружают темно-коричневый порошок трихлорида
ниобия, тщательно его растирают и фасуют в ампулы.
Выход трихлорида ниобия равен 37 г, что составляет 90%
от теоретического.
Продолжительность цикла не превышает 18 часов.
Найдено, % Nb—46,9,0; Cl—52,00.
NbCl,. Вычислено, % Nb—49,62; Cl—53,38.
ЛИТЕРАТУРА
I. H. Schafer, L. Bauer. Z. anorgan. und allgem. Chem., 277. 140
(1950).
Поступила в марте I9<'6 г. цЗЛ
УДК 546.883.4'131.07
ТАНТАЛ ЧЕТЫРЕХХЛОРИСТЫЙ
Л. М. ПЕТРОВА, Ь. Г. КОРШУНОВ
ТаСЦ М. в. 322,76
В литературе описан метод получения тетрахлорида тан-
тала [1], основанный на восстановлении пятихлористого тан-
тала металлическим алюминием. Для проведения этой реак-
ции авторы помещали раздельно алюминиевую фольгу и
избыток цятихлористого тантала в стеклянную трубку с тре-
мя температурными зонами (400°, 230°, 200°), вакуумирова-
ли до остаточного давления 1 • 10-4 мм и нагревали в тече-
ние 70 часов. Образующийся газообразный тетрахлорид
тантала осаждался на стенках средней части ампулы.
Нами были проверены и упрощены условия синтеза. Пред-
лагается при процессе восстановления смешивать исходные
вещества, что сокращает продолжительность процесса вос-
становления до 12 часов, снижает температуру восстановле-
ния до 220°, дает возможность применять менее глубокий
вакуум, порядка 1• 10-2 мм.
СХЕМА СИНТЕЗА ТЕТРАХЛОРИДА ТАНТАЛА
ЗТаС16 + А1 = ЗТаС14 + А1С13
Характеристика основного сырья
Тантал пятихлористый, ч., ВТУ ГКХ 1590 61.
Алюминий металлический, ч., порошок, класс А-2 ТУ
МХП 3568-52.
Условия получения
Тантал пятихлористый берут в Ю°/о-ном избытке от сте-
хиометрически рассчитанного количества. Навеска тантала
пятихлористого составляет 39,4 г, а алюминия 0,9 г. Компо-
151
ненты загружают в ампулу из термостойкого стекла (рис. 1).
Загрузку материалов в ампулу необходимо проводить в «су-
хой» камере для предотвращения окисления пятихлористого
тантала. Компоненты тщательно перемешивают путем встря-
хивания содержимого ампулы.
мест» перепайки
Рис. I. Ампула для получения
тантала четыреххлористого
Ампулу с загруженной смесью присоединяют к вакуумно-
му насосу РВН-20 и откачивают до остаточного давления
0,01 мм. Вакуум измеряют с помощью вакуумметра ВИТ-1А
и термоэлектрической лампы ЛТ-2. По окончании вакууми-
рования, которое длится 3 часа, ампулу запаивают. Запаян-
ную ампулу помещают в трубчатую электропечь и выдержи-
вают при 220° в течение 12 часов. Температуру в трубчатой
электропечи регулируют с помощью автоматического потен-
циометра ЭПД-12.
По окончании процесса восстановления электропечь с ам-
пулой охлаждают до комнатной температуры, ампулу выни-
мают, помещают в «сухую» камеру, разбивают и полученную
смесь четыреххлористого тантала, треххлористого алюминия
и избытка пятихлористого тантала помещают в стеклянную
ампулу с перетяжкой (рис. 2).
Перетя/нка
Пеапо перепайки
гоо
j _ к вакуун-
! «I ногу
т . I
Рис. 2. Ампула для очистки тантала четыреххлористого
Эту ампулу также откачивают до остаточного давления
0,01 мм, запаивают и нагревают в электропечи при темпера-
туре 220° в течение 6 часов.
В электропечи ампулу с перетяжкой располагают таким
образом, чтобы одна ее часть находилась в зоне комнатной
температуры (в этой части ампулы собирается алюминий
треххлористый и тантал пятихлористый).
После охлаждения электропечи с ампулой до комнатной
температуры ее помещают в «сухую» камеру, разбивают и
152
выгружают тантал четыреххлористый. тщательно его расти-
рают и фасуют в ампулы.
Выход тетрахлорида тантала равен 29 г, что составляет
85% от теоретического.
По внешнему виду тетрахлорид тантала представляет со-
бой порошок темно-зеленого цвета.
Продолжительность цикла не превышает 27 часов.
Найдено, %: Та—54,20; С1—42,00.
ТаС14. Вычислено, %: Та-56,06; С1 -43,94.
ЛИТЕРАТУРА
1. Н. Schafer, L. Grau. Z. anorgan. und allgem. Chem.. 275, 198
(1954).
Поступила в декабре 1965 г. ЦЗЛ
УД К 546.882.5' 141.07 + 546.883.5' 141.07
НИОБИЙ ПЯТИБРОМИСТЫЙ И ТАНТАЛ
ПЯТИБРОМИСТЫЙ
О. И. БРЕУСОВ, В. Г. ЛАВРЕНТЬЕВА, А. И. ТОЛКАЧЕВ
NbBr5 М. в. 492,45
TaBr6 М. в. 580,49
Ниобий пятибромистый представляет собой кристаллы
пурпурно-красного цвета, легко разлагающиеся во влажном
воздухе с образованием оксибромида NbOBr3 (желтого цве-
та); физические константы: т. пл. 267,5°, т. кип. 361,6°, ра-
створим в спирте [1]; плотность — 4,36 г/слт3 [2].
Тантал пятибромистый представляет собой кристаллы
желто-оранжевого цвета, которые разлагаются при взаимо-
действии с водой или влажным воздухом; физические кон-
станты: т. пл. 267°, т. кип. 345°, начинает интенсивно возго-
няться при 320°, растворим в абсолютном спирте, эфире [1];
плотность — 5,0 г!см? [2].
Легколетучие бромиды ниобия и тантала, как и броми-
ды других тугоплавких металлов, применяются при нанесе-
нии покрытий ниобия и тантала на другие металлы и мате-
риалы [3].
Для получения пентабромидов ниобии и тантала предло-
жено бромирование металла при повышенных температурах,
бромирование смеси пятиокиси ниобия или тантала с углем,
взаимодействие окислов с четырехбромистым углеродом
[2—5]. При небольших масштабах синтеза удобнее первый из
перечисленных способов.
Нами проверен и уточнен режим синтеза по этому спо-
собу.
СХЕМА СИНТЕЗА ПЕНТАБРОМИДОВ НИОБИЯ И ТАНТАЛА
Nb + 2’/2Br2 — NbBr5
Та + 21/2Вг2 -* ТаВг5
154
Характеристика основного сырья
Ниобий металлический (штабики), РЭТУ 75—58.
Тантал металлический (жесть), ТУ 5 и 8 СУО 021016.
Бром жидкий, ч.д.а., ГОСТ 4109—48.
Условия получения
Синтез пентабромида ниобия (тантала) проводят в гер-
метичной, тщательно осушенной аппаратуре (см. рисунок).
Аргон из баллона (У) через гидравлический затвор (2),
предохранительную склянку (<?) и склянку Мюнке (4) с
серной кислотой поступает в систему. Одновременно вклю-
чают обогрев трубчатой печи (5). Пропускание аргона в си-
стему (см. примечание) продолжают в течение 1 часа после
нагрева печи до температуры 750°, затем начинают барбо-
таж аргона через бром, залитый в склянку (6). Смесь брома
с аргоном проходит через серную кислоту в склянке Мюн-
ке (7) и трубку (8) с фосфорным ангидридом, где освобож-
дается от остатков влаги и поступает в кварцевую трубу (9)
на бромирование загруженных в нее ниобия металлического
или тантала металлического.
Конденсация тяжелых паров пятибромистого ниобия или
пятибромистого тантала происходит в двух колбах с отрост-
ками (10) и в стеклянном стакане со шлифом (11).
Непрореагировавший бром поступает на поглощение в
сосуд (12) с 10°/о-ным раствором едкого кали. После окон-
чания бромирования (прекращение образования паров
NbBr5 и ТаВг5) обогрев трубчатой печи прекращается и си-
155
стема охлаждается в токе аргона. Приемники с пятиброми-
стым ниобием или танталом переносят в бокс, где и произво-
дят расфасовку бромидов.
При загрузке кварцевой трубки 50 г металлических нио-
бия или тантала получают 238 г ниобия пятибромистого и
144 г тантала пятибромистого.что составляет 90% от теоре-
тического выхода. Расход брома равен 0,5 кг, аргона —
10 м3.
Описанная методика позволяет получать препараты, удов-
летворяющие требованиям РЭТУ 741—61 для пентабромида
ниобия и РЭТУ 1029—62 для пентабромида тантала.
Пр и м е ч а н и е. Можно проводить бромирование ниобия и тантала
в инертной среде азота или углекислоты. В этом случае проводится
очистка газов от кислорода, как описано в статье «Молибден трехбро-
мистый» в настоящем сборнике.
ЛИТЕРАТУРА
1. Справочник химика, т. 2, М._ Гоехимиздат, 1963, стр. 156, 220.
2. Я. Г. Горощепко. Химия ниобия и тантала, Киев, Изд. «Науко-
ва думка», 1965, стр. 329.
3. Краткая химическая энциклопедия, т. 3, М.. изд. «Советская эн-
циклопедия», 1964, стр. 485.
4. Руководство по препаративной неорганической химии, Иод. ред.
Г. Брауэра, М„ ИЛ, 1956, стр. 602.
5. С. А. Щука рев, Е. К. Смирнова, И. В. Василькова,
А. И. Л е п п о. Вести. Ленингр. ун-та, 16, 113 (1960).
6. С. А. Щ у к а р е в, Е. К. Смирнова, И. В. Василькова.
Н. И. Боровкова. Ж. неоргап. химии, 6, 1213 (1962)
Поступила в декабре 1965 г.
УДК 546.865'221.07
СУРЬМА ПЯТИСЕРНИСТАЯ
И. А. ШИШЕНКОВА, Р. С. БЕСИКОВ, А. И. ВУЛИХ
s = Sb-S-Sb = S
II II
S S
Sb2S6 М. в. 403,82
Сурьма пятисериистая находит возрастающее применение
в качестве красителя, пирофорного вещества, ветеринарного
средства, компонента резиновых смесей и др. [1, 2]; изучают-
ся также возможности применения этого продукта во многих
отраслях новой техники.
Обычный способ получения пятисернистой сурьмы заклю-
чается в разложении раствора тиоаитимоната натрия разбав-
ленными серной и соляной кислотами [1—4]. Основная труд-
ность при этом состоит в том, что осадок пятисернистой
сурьмы весьма активно сорбирует из раствора ион натрия и
анионы кислот-осадителей, а длительная промывка, необхо-
димая для очистки продукта, вызывает его разложение.
Разработанный нами способ получения пятисернистой
сурьмы заключается в обработке раствора тиоантимоната
натрия катионитом в водородной форме. Благодаря связыва-
нию катионов ионитом и отсутствию каких-либо посторонних
анионов, вносимых кислотами при известных методах, дости-
гается получение препарата более высокой чистоты и ста-
бильности.
СХЕМА СИНТЕЗА СУРЬМЫ ПЯТИСЕРНИСТОЙ
3NaaS + Sb2Ss+ 2S — 2Na3SbS4;
2Na3SbS4 + 6RH -> Sb2S61 + 6RNa + 3HaS
где R -- радикал функциональной группы катионита КУ-2.
157
Характеристика основного сырья
Сурьма трехсернистая, ч., ТУ МХП 2717—51.
Натрий сернистый, ч., ГОСТ 2053—43.
Сера черенковая, ч., ТУ МХП ОРУ 83—57.
Условия получения
Приготовление раствора сульфосурьмянокислого натрия.
Растворяют 275 г сернистого натрия в 660 мл горячей дистил-
лированной воды. В полученном растворе последовательно
растворяют при нагревании и энергичном перемешивании
20 г тонкоизмельчснной серы, смоченной спиртом, и НО г
трехсернистой сурьмы. Образовавшийся раствор сульфо-
сурьмянокислого натрия разбавляют дистиллированной во-
дой до объема 4 л (удельный вес 1,10 г/с.и3, концентрация
сульфосурьмянокислого натрия 50 г[л) и отфильтровывают
от нерастворимого остатка и механических примесей.
Получение пятисернистой сурьмы. Раствор сульфосурь-
мянокислого натрия, содержащий 50 г/л вещества, пропу-
скают снизу вверх через колонку, наполненную не более чем
на половину (по высоте.) катионитом КУ-2 в Н-форме (см.
примечание 1). Количество катионита, необходимое для об-
работки 4 л раствора указанной концентрации, составляет
500 г. Скорость пропускания раствора сульфосурьмянокисло-
го натрия устанавливают таким образом, чтобы получить
взвешенный слой катионита в колонке. При этом образую-
щаяся пятисернистая сурьма в виде тонкой суспензии выно-
сится из колонки током раствора.
Пропускание раствора Na3SbS4 через колонку с катиони-
том продолжают, пока в фильтрате на выходе из колонки не
будет достигнуто значение pH 5 (по универсальной индика-
торной бумаге). Осадок пятисернистон сурьмы отделяют от
раствора на воронке Бюхнера, промывают на фильтре не-
большим количеством дистиллированной воды и высушивают
до постоянного веса при температуре 50—60°.
Маточный раствор, содержащий до 5 г/л сульфосурьмя-
нокислого натрия, используют для приготовления раствора
тиоантимоната натрия вместо воды в следующем цикле про-
цесса.
Катионит регенерируют (переводят из натриевой формы
в водородную) 5—10%-ной серной или соляной кислотой.
Выход пятисернистой сурьмы равен 110 г, что составляет
85% от теоретического. Общий выход с учетом возврата в
процесс тиоантимоната, остающегося в маточнике, дости-
гает 95%.
Указанный метод позволяет получать продукт, удовлет-
воряющий требованиям РЭТУ 925—62 на сурьму пятисерни-
стую «чистую» (см. примечание 2).
158
Пятисернистая сурьма по внешнему виду представляет
собой темно-оранжевый порошок. Выше 130° разлагается иа
серу и трехсернистую сурьму; под влиянием света и влаги
частично разлагается; нерастворима в воде, спирте, разбав-
ленных кислотах; хорошо растворяется в щелочах и щелоч-
ных карбонатах.
Примечания:
1. Работу следует проводить при хорошей вытяжной вентиляции, так
как в процессе, особенно па стадии взаимодействия раствора сульфо-
сурьмянокислого натрия с Н-катионитом, интенсивно выделяется серово-
дород. Основное количество сероводорода следует поглощать раствором
щелсчч
2. При получении сурьмы пятисернистой ионообменным методом со-
держание свободной серы в продукте не превышает 3% (по ТУ допу-
скается 8%). Кроме того, происходит дополнительная очистка от таких
примесей, как железо и мышьяк. Особо следует отметить очистку от
мышьяка, так как содержание его в исходных веществах значительно и
колеблется в пределах 0,05—0,1%.
ЛИТЕРАТУРА
1. Сб. «Сурьма», М„ ИЛ, 1954, стр 68.
2. Н.П. Сажин. Сурьма, М., Металлургиздат, 1941, стр. 122.
3. Ю. В. Карякин, И. И. А пгело в. Чистые химические реакти-
вы, М„ Госхимиздат, 1955, стр. 513.
4. Г. Реми. Курс неорганической химии, т. 1, М„ ИЛ, 1963, стр. 274.
Поступила в декабре 1965 г. ЦЗЛ
УДК 546.863'32'16’
КАЛИЙ-СУРЬМА ФТОРИСТАЯ
Калий пентафторантимонат
О. Н. БРЕУСОВ, В. Г. ЛАВРЕНТЬЕВА
K2SbF5 М.в. 294,94
Калий-сурьма фтористый (пентафторантимонат калия)
представляет собой маслянистый наощупь белый кристалли-
ческий порошок, хорошо растворимый в воде, нерастворимый
в спирте и эфире; при температуре выше. 120° плавится с раз-
ложением [1]. Применяется в лабораторной практике.
Пентафторантимонат калия получают путем взаимодейст-
вия фтористого калия и трехфтористой сурьмы в водном ра-
створе [I].
Нами проверены и уточнены условия проведения процесса.
СХЕМА СИНТЕЗА ПЕНТАФТОРАНТИМОНАТА КАЛИЯ
КОНф HF -> KF-4-H2O
SbF3+2KF -> KsSbFe
Характеристика основного сырья
Сурьма трехфтористая, ч., МРТУ 6-09-2605—65.
Кали едкое, ч., ГОСТ 4203—48.
Кислота фтористоводородная, техн., ГОСТ 2567—54.
Условия получения
Растворяют 42 г едкого кали в 210 мл дистиллированной
воды. Раствор фильтруют и после охлаждения (растворение
КОН в воде сопровождается большим выделением тепла)
нейтрализуют 37 мл 40%-ной фтористоводородной кислоты.
По окончании нейтрализации pH раствора должно быть 5—6.
160
Растворяют 62 г сурьмы трехфтористой в 150 мл дистил-
лированной воды. Механические примеси отфильтровывают.
Приливают раствор трехфтористой сурьмы к раствору
фтористого калия и упаривают полученный раствор калия-
сурьмы фтористого под инфракрасной лампой досуха при
перемешивании.
Выход вещества равен 96 г, что составляет 95% от теоре-
тического.
Данная методика позволяет получать продукт, соответ-
ствующий МРТУ 6-09-1885—64 (содержание сурьмы в пере-
счете на KsSbFj-- 99 4-101 %).
ЛИТЕРАТУРА
1. Omelins Handbuch der anorganischen Chemie. 8- Aufl.. SN 34,
1958, стр. 792.
Поступила в декабре 1965 г. цзЛ
УДК 546.863'141.07
СУРЬМА ТРЕХБРОМИСТАЯ
Г. Е. РЕВЗИН, С. М. АРХИПОВ, В. Д. ЗАМЕДЯНСКАЯ
SbBr3 ЛА.в. 301,48
Трехбромистая сурьма находит применение, как и трех-
хлористая, в органическом синтезе в качестве катализатора
реакций алкилирования (ацилирования) ароматических сое-
динений по Фриделю — Крафтсу [1—3] и в лабораторной
практике.
Трехбромистая сурьма способна образовывать комплекс-
ные соединения с бромидами одновалентных металлов и ам-
мония [4, 5]. Методы получения трехбромистой сурьмы, опи-
санные в литературе, заключаются во взаимодействии сурьмы
с бромом [4—6].
Синтез осуществляется либо внесением небольшими пор-
циями измельченной сурьмы в охлаждаемую колбу с бромом,
либо пропусканием насыщенного парами брома тока азота
над порошкообразной сурьмой [4—6].
Эти методы, несмотря на свою простоту, обладают рядом
недостатков. Основным их недостатком является то, что
получаемая трехбромистая сурьма загрязнена непрореагиро-
вавшими исходными веществами, и продукт необходимо под-
вергать очистке перегонкой или перекристаллизацией из
сероуглерода [6]. Кроме того, оба эти способа малопроизво-
дительны, так как бромирование сурьмы протекает очень
бурно, с большим выделением тепла, и процесс невозможно
проводить в больших масштабах.
Трехбромистая сурьма по внешнему виду представляет
собой белое гигроскопичное кристаллическое вещество; ее
плотность 4,148 г/см? [7]; т. пл. 96 + 0,5° [8, 9], т. кип. 280—
288° [6, 7]. Существует в двух модификациях — а—SbBr3 и
р—SbBr3 [8, 9]. Водой гидролизуется с образованием окси-
бромида. В соляной и бромистоводородной кислотах раство-
ряется без выделения оксибромида [4—7].
162
Трехбромистая сурьма хорошо растворима в органиче-
Нами разработан метод получения трехбромистой сурь-
мы путем взаимодействия металлической сурьмы и брома в
растворе четыреххлористого углерода [11]. Метод прост,
удобен и может быть воспроизведен в промышленном мас-
штабе. Проведение реакции в инертном растворителе снижает
скорость реакции, а изменение температуры делает ее легко-
управляемой.
СХЕМА СИНТЕЗА ТРЕХБРОМИСТОЙ СУРЬМЫ
2 Sb + 3 Вг2=2 SbBr3.
Характеристика основного сырья
Сурьма металлическая, Су—О, ГОСТ 1089—41.
Бром, ч., ГОСТ 4109—48.
Углерод четыреххлористый, ч._ ГОСТ 5827--51.
Условия получения
В круглодонную колбу емкостью 500 мл, снабженную об-
ратным холодильником, присоединяемым к колбе при помо-
щи шлифа, помешают 20 г металлической сурьмы, измель-
ченной до крупности 0,5—1, мм; сюда же заливают раствор
39,4 г брома в 420 г четыреххлористого углерода. Реакцион-
ную массу нагревают при перемешивании до начала слабого
кипения, поддерживая его далее в течение 1 часа до обес-
цвечивания раствора.
Затем отсоединяют обратный холодильник и горячий ра-
створ быстро декантируют с осадка неирореагировавшей
металлической сурьмы в сухой стеклянный стакан. Раствор
охлаждают до 103. Выпавшие игольчатые кристаллы трех-
бромистой сурьмы отфильтровывают, промывают на фильт-
163
ре 25 мл охлажденного до 0 5° четыреххлористого углерода,
отжимают и сушат в вакуум-эксикаторе.
Выход трехбромистой сурьмы в виде снежно-белых иголь-
чатых кристаллов равен 49,1 г, что составляет 85% от теоре-
тического. Из маточника упариванием можно дополнительно
выделить еще некоторое количество трехбромистой сурьмы
или использовать этот маточник для синтеза в следующем
цикле. Кроме того, из остатка в реакционной колбе утилизи-
руется 2,2 г металлической сурьмы.
Нами определена растворимость и теплота растворения
трехбромистой сурьмы в четыреххлористом углероде. Ра-
створимость трехбромистой сурьмы составляет (в весовых %)
при 0°—0,58, при 25°—1,76; при 50°—4,44.
Теплота растворения трехбромистой сурьмы в четырех-
хлористом углероде при 25° равна 7,1 ккал/моль.
Описанный метод синтеза позволяет получать продукт
квалификации «чистый», удовлетворяющий требованиям ТУ
ГКХ РУ 1867—62, с содержанием трехбромистой сурьмы не
менее 99%.
Л И Т Е Р А Т У Р А
1. А. С е р р е й. Справочник по органическим реакциям, М., Госхим-
издат, 1962, стр. 253.
2. П. Каррер. Курс органической химии, М., Госхимиздат, 1962,
стр. 485.
3. А. Е. Ч и ч и б а б п н. Основные начала органической химии, т. 1,
М., Госхимиздат, 1963, стр. 213.
4. Б. В. Некрасов. Курс общей химии, М„ Госхимиздат, 1962,
стр. 404.
5. Г. Реми. Курс неорганической химии, М., ИЛ, 1963, стр. 720.
6. Руководство по препаративной неорганической химии, под ред.
Г. Брауэра, М., ИЛ, 1956, стр. 298.
7. Справочник химика, т. II, М., Госхимиздат. 1963. стр. 214.
8. D. W. Cushen, R. Hui me. J. Chem. Soc., 1962, 2218.
9. D. W. Gushes, R. Hulme. J. Chem. Soc., 1964, 4162.
10. Справочник по растворимости, т. I, книга I. M.—Л., изд. AH
СССР, 1962, стр. 932.
11. Г. E. Ревзин, С. М. Архипов, В. Д. Замедяиская. Авт.
свид. 169087; Бюлл. изобр., № 6 (1965).
Поступила в декабре 1965 г.
УДК 546.863'151.07
СУРЬМА ТРЕХЙОДИСТАЯ
Г. £ РЕВЗИН, И. А. ШИШЕНКОВА
SbJj М.в. 502,46
Трехйодистая сурьма — кристаллическое вещество, в за-
висимости от способа получения существует либо в виде ру-
биново-красных пластинок с перламутровым блеском, либо
в виде буро-фиолетовых кристаллов.
Существует в двух модификациях—гексагональной и
ромбической [1—3]. Ромбическая модификация — желтого
цвета, гексагональная — рубиново-красного. Плотность ром-
бической модификации — 4,77 [1, 3], плотность гексагональ-
ной—4,85 [3]; т. пл. 167-171° [1 3, 4]; т. кип. 397—401°
1.1, 3, 4].
Трехйодистая сурьма во влажном воздухе медленно раз-
лагается с образованием основных солей. Водой гидролизует-
ся с образованием оксийодида [2, 4]. В растворе соляной,
йодистоводородной кислот и йодистого калия растворяется
без выделения оксийодида. Сурьма трехйодистая хороню
растворима в спиртах, сероуглероде, ацетоне, ароматических
углеводородах (бензоле и толуоле), нерастворима в четырех-
хлористом углероде и хлороформе [1—5].
Сурьма трехйодистая способна образовывать комплекс-
ные соединения с йодидами щелочных металлов и аммония
[2—4], а также может присоединять йод, образуя твердый
раствор, из которого йод легко удаляется подходящими ра-
створителями, например эфиром [3].
Сурьма трехйодистая находит применение в лаборатор-
ной практике.
Методы получения сурьмы трехйодистой, описанные в ли-
тературе, заключаются во взаимодействии сурьмы с йодом
[2—6], осуществляемом либо пропусканием паров йода над
нагретой порошкообразной сурьмой [2], либо сплавлением
165
элементов, либо взаимодействием сурьмы с йодом в толуоле
[4, 5] или сероуглероде [3].
Эти методы обладают рядом недостатков. Процесс полу-
чения сурьмы трехйодистой взаимодействием сурьмы с пара-
ми йода малопроизводителен и приводит к получению за-
грязненного исходными веществами продукта. Аналогичные
результаты дает также и сплавление сурьмы с йодом. Кроме
того, при механическом смешении сурьмы с йодом в ступке
иногда по неизвестным причинам начинается спонтанное
взаимодействие сурьмы с йодом, протекающее с большой
скоростью (подобно взрыву) и сопровождающееся выделе-
нием большого количества тепла и паров йода.
Синтез сурьмы трехйодистой в толуоле осложняется тем,
что при температуре кипения толуола (110,6°) из раствора
выделяется в виде паров некоторая часть йода. Синтез в
сероуглероде неприемлем вследствие высокой его токсич-
ности.
Нами разработаны два метода получения сурьмы трех-
йодистой, представляющие собой усовершенствованные ва-
рианты двух выше описанных.
СХЕМА СИНТЕЗА СУРЬМЫ ТРЕХЙОДИСТОЙ
2 Sb + 3 J2—2 SbJ3.
Характеристика основного сырья
Сурьма металлическая, СУ-0, ГОСТ 1089—41.
Йод металлический, ч.д.а., ГОСТ 545—41.
Бензол, ч.д.а., ГОСТ 5955—51.
Условия получения
1. В фарфоровый стакан помешают 57 г порошкообразной
металлической сурьмы, смачивают ее 20 мл четыреххлористо-
го углерода и добавляют 180 г йода. Смесь тщательно пере-
мешивают, при этом обеспечивается большая однородность
реакционной массы и исключается возможность ее саморазо-
гревания.
Фарфоровый стакан с реакционной смесью помещают на
закрытую листом асбеста электроплитку, накрывают фарфо-
ровой чашкой и нагревают до получения гомогенного плава,
который сразу же переливают в чистую сухую фарфоровую
чашку; остаток непрореагировавшей сурьмы остается при
этом на дне того стакана, в котором проводилось плавление.
После остывания плав трехйодистой сурьмы измельчают
в фарфоровой ступке, дважды промывают 50 мл четырех-
хлористого углерода для удаления свободного йода и высу-
шивают в вакууме.
166
Получают 230 г трехйодистой сурьмы темно-бурого цвета
с фиолетовым оттенком и содержанием основного вещества
99%, что соответствует выходу по сурьме 97% от теоретиче-
ского.
2. В реакционную колбу емкостью 5 л, снабженную обрат-
ным холодильником со шлифом, засыпают 57 а металличе-
ской сурьмы, заливают 3 л бензола и засыпают 112 г йода.
Реакционную смесь кипятят на водяной бане до исчезновения
фиолетовой окраски йода, после чего добавляют еще 56 г
йода и кипятят с обратным холодильником в течение 1 часа.
Горячий раствор фильтруют через воронку горячего
фильтрования и охлаждают до температуры 10—15°. При
этом из раствора выделяется трехйодистая сурьма в виде
кирпично-красных чешуйчатых кристаллов с фиолетовым пер-
ламутровым блеском.
Выход трехйодистой сурьмы после сушки в вакууме равен
200 г с содержанием основного вещества 99,0—99,5%, что
соответствует прямому выходу 85% по сурьме от теоретиче-
ского. Кроме того, из осадка утилизируют 3 г непрореагиро-
вавшей металлической сурьмы, а из бензольного раствора
выделяют при упаривании дополнительно около 15 г вещест-
ва, содержащего примесь йода, легко удаляемую четырех-
хлористым углеродом.
Указанные методы синтеза позволяют получать продукт,
удовлетворяющий требованиям ВТУ МРУ 538—52.
ЛИТЕРАТУРА
1. Справочник химика, II, М.—Л., Госхимиздат, 19G3, стр. 214.
2. Ф. Эфраим. Неорганическая химия, часть II, Л., Госхимиздат,
1933, стр. 206.
3. Г. Реми. Курс неорганической химии, М., Инлитиздат, 1963,
стр. 721.
4. Б. Б. Некрасов. Курс обшей химии, М., Госхимиздат, 1962,
стр. 441.
5. Н. Г. Ключников. Руководство по неорганическому синтезу, М,
Изд. «Химия», 1965, стр. 203.
6. W. Biltz, Sapper. Z. anorg. Chem., 203, 277 (1931).
Поступила в марте 1966 г.
1Ш1
УДК 546.244 = 31.07
ТЕЛЛУР ДВУОКИСЬ
Г. Е. РЕВЗИН
ТеО2 М.в. 159,59
Двуокись теллура представляет собой белое негигроско-
пичное вещество, кристаллизуется независимо от способа
получения в тетрагональной модификации по типу С 4. Его
плотность, рассчитанная по рентгенографическим данным,
составляет 6,02 ti'civ' [1, 2]. Температура плавления 732,6—
733° [1, 2], теплота плавления 60,1 ±0,6 ккал'моль [1], теплота
образования из элементов 87,1 ккал!моль [3].
Двуокись теллура плавится без разложения; в расплав-
ленном состоянии окрашена в темно-желтый цвет.
Зависимость давления насыщенного пара вещества от
температуры описывается уравнением
мм рт. ст.—---—-----и 12,257 [4].
Двуокись теллура, будучи амфолитом, растворяется как
в кислотах, так и в щелочах [1]. Растворимость в воде чрез-
вычайно мала и составляет 0,00067% [5].
Изоэлектрическая точка, соответствующая минимальной
растворимости вещества, имеет pH 3,8 [5].
Для двуокиси теллура более характерны окислительные
свойства, чем восстановительные: она легко восстанавливает-
ся до теллура даже такими слабыми восстановителями, как
виноградный сахар, тогда как окисление ее до шестивалент-
ного состояния происходит лишь под действием таких силь-
ных окислителей, как двухромовокислый калий, марганцово-
кислый калий, хлорноватая кислота, перекись водорода и т. п.
[!, 6].
Двуокись теллура находит применение в лабораторной
практике.
168
В литературе [1, 2, 5—8] описайы способы получения дву-
окиси теллура путем термического разложения основного
нитрата теллура при 400—450° [1, 2, 8]; термического разло-
жения теллуровой кислоты при температуре около 600°
[1, 2, 8]; окисления теллуроводорода [1]; сжигания теллура в
токе воздуха или кислорода [1]; гидролиза солянокислых ра-
створов теллура; осаждения аммиаком из раствора теллура
в царской водке; осаждения азотной кислотой из раствора
теллурита калия или натрия [7] и ряд других методов, пред-
ставляющих собой вариации вышеперечисленных.
Анализ указанных методов получения двуокиси теллура
показывает, что первые три нз них связаны с необходимостью
получения промежуточных соединений — основного нитрата,
теллуровой кислоты или теллуроводорода, причем получение
их сопряжено с определенными трудностями, а теллурово-
дород, кроме того, чрезвычайно токсичен.
Окисление теллура на воздухе или в токе кислорода тре-
бует применения высоких температур и не удается осу-
ществить количественно; получаемый продукт загрязнен как
неокисленным теллуром, так и примесями, содержащимися в
исходном теллуре.
•При гидролизе растворов теллура в соляной кислоте или
царской водке получается аморфный осадок теллуристой
кислоты, загрязненный трудноудаляемыми хлор-ионами.
Наиболее простым и удобным методом получения чистой
двуокиси теллура является осаждение ее азотной кислотой
из раствора теллурита калия или натрия; при этом попутно
возможна дополнительная очистка двуокиси теллура от ряда
примесей, в частности, от примесей свинца и меди [9].
Нами уточнены условия получения чистой двуокиси тел-
лура окислением металлического теллура азотной кислотой
с последующей очисткой вещества через теллурит натрия.
СХЕМА СИНТЕЗА ДВУОКИСИ ТЕЛЛУРА
Fe4-4HNO8=TeO2+4NO2l+2H2O
ТеО2 + 2 N'aOH = Na2TeO3-l-H2O.
Na2TeO34-2 HNO3 = Te02+2 NaNO3+H2O,
Характеристика основного сырья
Теллур металлический, Т-1, ГОСТ 9514—60.
Азотная кислота, ч.д.а., ГОСТ 4461—48.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Условия получения
К ЮО г порошкообразного теллура, помещенным в стакан
из термостойкого стекла емкостью 500 мл, небольшими пор-
169
циями приливают 250 мл азотной кислоты уд. веса 1,35 при
периодическом перемешивании. По окончании приливания
азотной кислоты реакционную смесь нагревают на электро-
плитке в течение 1 часа до удаления основной массы окис-
лов азота при периодическом перемешивании. Затем реак-
ционную смесь охлаждают, отфильтровывают серый осадок
двуокиси теллура и промывают его на фильтре дистиллиро-
ванной водой до нейтральной реакции промывной жидкости.
Отфильтрованный осадок двуокиси теллура, содержащей
неокисленпый теллур, помещают в стакан емкостью 1 л, при
постоянном перемешивании заливают туда 800 мл 10%-ного
раствора едкого натра и нагревают 30 минут при темпера-
туре 90—100°.
Образовавшийся раствор теллурита натрия отфильтровы-
вают от нерастворившегося в едком натре осадка теллура.
Для получения кристаллического, легко фильтрующегося
осадка двуокиси теллура ее осаждение проводят из горяче-
го раствора, для чего фильтрованный раствор теллурита
натрия нагревают до температуры 100° и при постоянном
перемешивании небольшими порциями добавляют азотную
кислоту уд. веса 1,18—1,20.
После каждого добавления очередной порции азотной
кислоты проверяют pH раствора. Осаждение заканчивают,
когда pH раствора над осадком достигнет 3,5—4 и останется
неизменным при интенсивном перемешивании и температуре
100° в течение 15—20 минут. Затем реакционную смесь ох-
лаждают, осадок двуокиси теллура отфильтровывают от
маточного раствора, промывают на фильтре горячей дистил-
лированной водой до отсутствия МО'з-иона в фильтрате (про-
ба с дифениламином) и сушат в сушильном шкафу под пере-
вернутым стаканом (во избежание восстановления ее орга-
нической пылью) при температуре 150—170" в течение
2—3 часов. С вышеуказанной загрузки получают 113,2 г чи-
сто-белого цвета двуокиси теллура с содержанием основного
вещества 99,8%, что соответствует 91% от теоретического
выхода.
Из азотнокислого маточника дополнительно может быть
выделено при упаривании и прокаливании 8,5 г двуокиси тел -
лура, имеющей желтовато-зеленый цвет, обусловленный при-
месями меди и железа.
Остаток нерастворенного в едком натре теллура состав-
ляет 2,2 г.
Указанный метод синтеза позволяет получать продукт,
удовлетворяющий требованиям МРТУ 6-09-686—63.
ЛИТЕРАТУРА
1. А. А, Кудрявцев. Химия и технология селена и теллура, М.,
Изд. «Высшая школа», 1961, стр. 85.
170
2. Руководство по препаративной неорганической химии, Под ред.
Г. Брауэра, М., ИЛ, 1956, стр. 227.
3. А. И. Крестовников, А. С. Шахов. Физико-химические и
термодинамические свойства редких элементов, М., Металлургиздат,
1943. стр. 147.
4. В. П. 3 л о м а н о а, А. В. Новоселова, А. С. П а ш и н к и н,
1О. П. Симонов, К. Н. Семененко. Ж. неорган. .химии, 3, 7 (1958).
5. J. Issa, S A w a d, J. Phys. Chem.’ 58, 948 (1954).
6. Б. В. Некрасов. Курс общей химии, М., Госхимиздат, 1963,
стр. 381.
7. 1. Kasarnowsky. Z. phys. Chemie, 109, 287 (1925).
8. Н. Г. Ключников. Руководство по неорганическому синтезу,
М., Изд. «Химия», 1965, стр. 130.
9. О. И. Воробьева, А. Л Мсньков. Ж. неорган. химии, 2,
2526 (1957).
Поступила к декабре 1966 г. ЦЗЛ
УДК 546.246 = 325.04.07
ТЕЛЛУРОВАЯ КИСЛОТА
Ортотеллуровая кислота
Г. Е. РЕВЗИН
Н2ТеО4 • 2 Н2О М.в. 229,65
Теллуровая кислота представляет собой кристаллическое
вещество белого цвета, хорошо растворима в воде и щелочах,
малорастворима в азотной кислоте и нерастворима в органи-
ческих растворителях. Кристаллизуется в виде кристаллов
моноклинной или кубической сингонии. Плотность моноклин-
ной модификации — 3,071 г/см3, кубической модификации —
3,17 г/см3.
Растворимость теллуровой кислоты в воде составляет [1]:
Теплота растворения теллуровой кислоты в воде равна
3,35 ккал/моль.
Теллуровая кислота является слабой кислотой; кислотные
свойства ее характеризуются следующими величинами кон-
стант диссоциации; 7(i=2-10~8; /<2 = 3.10 11 [1].
Теллуровая кислота образует соли, в которых атомы во-
дорода полностью или частично замещены атомами метал-
лов. Из солей теллуровой кислоты только соли щелочных ме-
таллов и аммония хорошо растворимы в воде.
Проявляя слабые окислительные свойства, теллуровая
кислота при взаимодействии с сильными восстановителями
(гидразином, сернистым газом и т. п.) восстанавливается до
172
элементарного теллура, а слабыми восстановителями (Hg
и др.) —до четырехвалентного теллура [2].
Характерным свойством теллуровой кислоты является
образование комплексных гетерополикислот с кислотными
ангидридами (например, вольфрамовым и молибденовым).
При нагревании до температуры 100—220э теллуровая
кислота, обезвоживаясь, переходит в метателлуровую кисло-
ту (Н2ТеО4), которая выше 220э переходит в трехокись тел-
лура (теллуровый ангидрид), а при температуре выше 400э
разлагается на двуокись теллура и кислород [2].
Теллуровая кислота находит применение в лабораторной
практике.
В литературе описаны следующие способы получения
теллуровой кислоты:
1. Окисление металлического теллура раствором хлорно-
ватой кислоты [2—6]
5 Те + 6 НСЮз+12 Н2О = 5 Н6ТеО6 + 3 С12.
2. Окисление двуокиси теллура перманганатом калия в
растворе азотной кислоты [2, 3, 7, 8]
5 ТеО24-2 KMnO4 + 6 HNO3+12 Н2О = 5 Н6ТеО6 + 2 KN'O3 +
+ 2 Mn(NO3)2.
3. Окисление двуокиси теллура в щелочном растворе
30%-ным раствором перекиси водорода с последующим вы-
саливанием концентрированной азотной кислотой [2, 3]
Na2TeO3+H2O2+H2O—Na2TeO4 • 2 Н2О
Na2TeO4 • 2 Н2О + 2 HNO3=H6TeO6 + 2 NaNO3.
4. Окисление элементарного теллура хромовым ангидри-
дом и азотной кислотой [2, 3, 10]
2 Те + 3 СгО3 + 12 HNO3=2 Н6ТеО6 + 3 Cr(NO3)s + 3 NO2.
5. Взаимодействие аморфного теллура с перекисью водо-
рода [2, 11, 12]
Те + 3 Н2О2=Н6ТеО6.
6. Окисление водной суспензии теллура газообразным
хлором [13]
Те+6 Н2О + 3 С12=Н6ТеО6 + 6 НО.
7. Получение теллурата кальция прокаливанием смеси
гидроокиси кальция с двуокисью теллура на воздухе при
975—1075° с последующим выделением Н6ТеО6 действием
HNO3 [14]
2 Са(ОН)2 + 2 ТеО2 + О2=2 СаТеО4 + 2 Н2О,
СаТеО4 + 2 HNO3 + 2 H2O=H6TeOe+Ca(NO8)2.
173
8. Окисление теллура или двуокиси теллура перекисью
водорода в сернокислом растворе [2, 3, 9].
H2SO4 + H2O2=H2SO6 + HaO,
TeO2 + H2SO5 + 3 H2O=H6TeOe + H2SO4.
Сравнение методов получения теллуровой кислоты пока-
зывает, что наиболее простым является метод, основанный на
окислении аморфного теллура перекисью водорода, однако
необходимость применения аморфного теллура и концент-
рированной (80—85%-ной) перекиси водорода делает его
нетехнологичным.
Остальные методы неудобны либо необходимостью при-
менения дорогих и дефицитных окислителей (НСЮз, КМпО4,
СгОз), либо сложностью очистки теллуровой кислоты от при-
месей побочных продуктов реакции.
Нами разработан метод получения теллуровой кислоты
путем окисления двуокиси теллура 30%-ным раствором пере-
киси водорода без применения каких-либо других окислите-
лей и реагентов [15], что выгодно отличает его от всех выше-
перечисленных.
СХЕМА СИНТЕЗА ТЕЛЛУРОВОЙ КИСЛОТЫ
Te+4HNO3 -> TeO2+4NO2+2H2O
ТеО2ф-Н2О2+2Н2О -> Н2ТеО4-2НаО
Характеристика основного сырья
Теллур металлический, Т-1, ГОСТ 9514—60.
Азотная кислота, ч.д.а., ГОСТ 4461—48.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Перекись водорода, мед., ГОСТ 177—55.
Условия получения
Навеску 100 г металлического теллура помешают в стек-
лянный термостойкий стакан, смачивают небольшим коли-
чеством дистиллированной воды до образования густой ка-
шицы и окисляют 300 мл концентрированной азотной кисло-
ты удельного веса 1,35—1,37. Кислоту приливают небольши-
ми порциями во избежание бурной реакции и вспенивания
реакционной массы. После приливания всего количества
азотной кислоты реакционную массу прогревают на электри-
ческой плитке в течение 0,5—1 часа до прекращения выде-
ления окислов азота. Затем полученную грязную двуокись
теллура отфильтровывают на воронке Бюхнера, промывают
дистиллированной водой и растворяют в щелочи для отделе-
ния двуокиси теллура от непрореагировавшего теллура.
174
Для растворения берут раствор 65 г едкого натра в
500 мл дистиллированной воды. Растворение ведут в течение
0,5 часа при подогревании раствора до температуры 70—80°
и постоянном перемешивании.
По окончании растворения, когда в осадке не остается
белых частиц двуокиси теллура, раствор охлаждают, фильт-
руют и нейтрализуют азотной кислотой при перемешивании
до pH 6. При этом осаждается гидратированная двуокись
теллура в виде белого хлопьевидного осадка. Двуокись тел-
лура отфильтровывают, отжимают на фильтре и переносят в
колбу емкостью 3 л. Туда же заливают 300 мл 30%-ной пе-
рекиси водорода, реакционную смесь подогревают до начала
кипения и колбу переносят с нагревателя на лист асбеста.
Начавшаяся при подогревании реакция в дальнейшем
идет за счет тепла, выделяющегося при окислении двуокиси
теллура, и заканчивается в течение 0.5 часа.
Теллуровую кислоту выделяют из раствора добавлением
к нему двойного объема концентрированной азотной кисло-
ты. Выделившуюся в виде белого мелкокристаллического
осадка теллуровую кислоту отфильтровывают на воронке
Бюхнера, отжимают на фильтре, промывают 50 мл спирта и
сушат на фильтре в течение 1 часа.
Выход теллуровой кислоты 160 г, что составляет 85% от
теоретического.
Чистота полученной по описанной методике теллуровой
кислоты 99,5—99,9%.
Описанный метод синтеза позволяет получать продукт,
удовлетворяющий требованиям ТУ ГКХ РУ 1683—61, а имен-
но: содержание теллуровой кислоты не менее 98,5%; наи-
большие количества допустимых примесей (в %): сульфа-
ты — 1 • 10% железо — 1 • 10~2.
Л ИГЕРАТУРА
1. А. Н. Крестовников, А. С. Шахов. Физико-химические и
термодинамические свойства редких элементов, М., Металлургиздат,
1943, стр. 147.
2. А. А. Кудрявцев. Химия и технология селена и теллура, М.,
Высшая школа, 1961, стр. 90.
3. Руководство по препаративной неорганической химии, под род.
Г. Брауэра, М., 1956, стр. 228.
4. J. М е у е г. Н. Moldenhouer. Z. anorg. Chem. 119. 132 (1921).
5. J. Meyer, W. Franke. Z. anorgan und allgem. Chem. 193, 191
(1930).
6. J. Meyer, M. H о I о w a t у i. Chem. Ber., 81, 119 (1948).
7. F. Mathers, C’ Rise, 11. 13 r'o d e г i c k. R. Forney. Proc. Indi-
ana Acad. Set., 52. 114, (1942), C.A., 38: 1800.
8. Inorganic Syntheses, III N. Y. London, 1950, стр. 145
175
9. L. О i I b c r t s о n. J. Amer. Chem. Soc., 55, 1460 (1933).
10. L. Staudenmaier. Z. anorg. Chem., 10. 189 (1895).
11. Б. В. Некрасов. Курс общей химии, М_, Госхимиздат, 1963,
стр. 322.
12. F. Fouasson. Ann. Chlm. [12] 3, 594 (1948); С.А.. 43; 2534с.
13. Р. Browning, Н. М I n n i n g. Amer, J. Sci., 36, 72, C.A., 7: 3809.
14. F. Mathers. G, Bradbury. J. Amer. Chem. Soc.. 51,3229 (1929).
15. Г. E. Ревзин. Авт. свид. 167834. Бюлл. изобр., № 3 (1965).
Поступила в декабре 1965 г.
ЦЗЛ
УДК 546.773'141.07
МОЛИБДЕН ТРЕХБРОМИСТЫЙ
В. Г. ЛАВРЕНТЬЕВА, О. Н. БРЕЪСОВ
МоВг3 М.в. 335,66
Молибден трехбромистый (трибромид молибдена) пред-
ставляет собой темно-зеленые кристаллические иглы, разла-
гающиеся при нагревании выше 60°, нерастворимые в воде,
кислотах и в обычных органических растворителях, но легко-
растворимые в кипящем безводном пиридине. Может найти
применение для получения тугоплавких покрытии.
В литературе описывается прямой синтез трс.хбромистого
молибдена из элементов [1, 2].
Нами была проверена и уточнена методика проведения
этого процесса.
СХЕМА СИНТЕЗА ТРЕХБРОМИСТОГО МОЛИБДЕНА
2Мо4-ЗВг3 -> 2МоВг3
Характеристика основного сырья
Молйбденовые штабики сварные, ТУ ВМ 7-153—54.
Молибденовая жесть, МРН, ТУ 5 и 8 СУО 021 001.
Бром жидкий, ч.д.а., ГОСТ 4109—48.
Условия получения
Синтез трехбромистого молибдена проводят путем взаи-
модействия металлического молибдена с парами брома при
температуре 850°.
Отходы молибденовой жести являются наиболее дешевым
сырьем для получения бромида молибдена, однако эта жесть
содержит до 0,07% кремния, и при получении из нее бромида
12 Зак. 1060 1 77
молибдена содержание кремния в готовом продукте превы-
шает допустимые нормы. Поэтому применяют смесь молиб-
деновой жести и стружки из чистых молибденовых штабиков.
Молибден металлический (полоски, стружки) отмывают
от железа кипячением с концентрированной соляной кисло-
той (Т : Ж=1 : 2), промывают дистиллированной водой и су-
шат при температуре 100°.
Азот из баллона (/) через гидравлический затвор (2) и
предохранительную склянку (3) поступает в кварцевую тру-
бу (4), находящуюся в трубчатой печи (5), нагретой до тем-
пературы 600°.
В кварцевую трубу (4) помещают металлическую медь,
которая при температуре 600° реагирует с кислородом, со-
держащимся в азоте по реакции 2 Сп + Ог—-2 СнО.
Затем азот осушается от влаги в склянке (7) с серной
кислотой и в склянке (8) с пятиокисью фосфора. Систему
продувают азотом в течение 1 часа. Затем включают обогрев
печи (10), продолжая продувание азота. Когда температура
печи (10) поднимется до 850°, начинают бромирование мо-
либдена, для этого открывают зажим на пути тока азота
через склянку Л^юнке (6) с бромом и закрывают зажим на
обводном шланге для азота.
Азот и пары брома проходят через серную кислоту, нали-
тую. в склянку Мюнке (7) и склянку (8) с фосфорным ан-
гидридом, где освобождаются от остатков влаги и попадают
в реакционную трубу (9) с помещенным в нее металличе-
ским молибденом.
Пары азота и брома после бромирования проходят через
склянку (11) с 10%-ным раствором едкой щелочи для улав-
ливания брома. Бромирование продолжают в течение 6 часов.
178
Полученный трибромид молибдена отмывают дистиллирован-
ной водой до отсутствия хлора и брома в пробе фильтрата с
азотнокислым серебром. Готовый продукт просушивают при
температуре не выше 50°.
При загрузке 30 г металла выход трехбромистого молиб-
дена равен 73 г, что составляет 70% от теоретического.
Расход брома не превышает 0,5 кг, азота 1,5 кг.
Данная методика позволяет получать трехбромистый мо-
либден, удовлетворяющий требованиям РЭТУ 1022—62.
Поступила в декабре 1965 г.
ЛИТЕРАТУРА
1. Руководство по неорганической препаративной химии, Под редак-
цией Г. Брауэра, М.—Л., ИЛ, 1956, стр. 645.
2. С. А. Щук а рев, И. В. Василькова, Д. В. Корольков,
С. С. Никольский. Вести. Лепипгр. ун-та, 4, 150 (1962).
цзл
УДК 546.719.7'221.07
РЕНИЙ СЕМИСЕРНИСТЫЙ
О. Н. БРЕУСОВ, В. Г. ЛАВРЕНТЬЕВА
Re2S7 4 х
Рений семисернистый (гептасульфид рения) является од-
ним из важнейших соединений рения и обычно представляет
собой твердый раствор серы в гептасульфиде рения, при на-
гревании разлагается; плотность около 4,’8 г/см3. Может най-
ти применение в качестве катализатора в органическом син-
тезе.
В литературе описано два основных метода получения
этого соединения: а) путем осаждения из кислых растворов
перренатов сероводородом [1—4] и б) взаимодействием кис-
лых растворов перренатов с тиосульфатом натрия [1]. В дан-
ной работе уточнены условия синтеза по второму из этих
методов, который представляется более предпочтительным,
так как позволяет избежать применения сероводорода, дает
лучший выход и приводит к получению более чистого про-
дукта.
СХЕМА СИНТЕЗА РЕНИЯ СЕМИСЕРНИСТОГО
. Основная реакция:
2 KReO4+7Na2S2O3+2HCl -> Re2S, + 7 Na2SO4+2 KC1+H2O
Побочные реакции:
Na2S2O3+2HCl -> 2NaCl+SO2 f-S+H2O
Re2S7+A'S -> ReaS,+x
Характеристика основного сырья
Перренат калия, ч., ВТУ МГУХП 201—58, содержание ос-
новного вещества не менее 96%.
Натрий серноватистокислый, ч., ВТУ МХП 4134—58.
180
Соляная кислота, ч., РОСТ 3118—46.
Толуол, ч.д.а., ГОСТ 5789—51.
Условия получения
К горячему раствору 90 г перрената калия в 1 л 12 %-ной
соляной кислоты медленно, при постоянном перемешивании
и нагревании, приливают раствор 250 г гипосульфита натрия
в 1 л горячей дистиллированной воды. Смесь кипятят в тече-
ние 2 часов. Затем для растворения свободной серы от-
фильтрованный осадок кипятят в течение 3 часов с 0,4 л то-
луола. Гептасульфид рения отфильтровывают на воронке
Бюхнера и сушат на фильтре в токе воздуха. Затем осадок
промывают дистиллированной водой до полного удаления
хлор-иона и сушат при температуре не выше 120". Промыв-
ные воды собирают и после подкисления выделяют из них
рений действием гипосульфита натрия.
Выход гептасульфида рения равен 80 г, что составляет
85% от теоретического.
Данная методика позволяет получать препарат, удовлет-
воряющий требованиям РЭ ТУ 873—62.
ЛИТЕРАТУРА
1. И. Д р у ц е. Рений, М., ИЛ, 1951, стр. 80.
2. W. Blitz, F. Welbke. Z. anorg. Chem., 203,3 (1931).
3. Б. В. Некрасов. Курс общей химии, М„ Госхимиздат, 1961,
стр. 277.
4. J. К. F а 1 tn n I, G. В S S а 1 а г i a. Analyt. chim acta, 12,519 (1955).
Поступила в декабре 1965 г.
цзл
УДК 546.732 = 31.05
КОБАЛЬТ ЗАКИСЬ
Л. И. ВУЛИХ, Д. А. ПАХОМОВ, С. А. КУТОЛИН
СоО
М.в. 74,93
Закись кобальта — одно из важнейших соединений этого
металла, применяемое непосредственно (катализ, производ-
ство стекла и керамики) или являющееся- полупродуктом для
синтеза других соединений кобальта [1, 2].
В литературе описаны методы получения закиси кобаль-
та термическим разложением солей или гидроокиси двухва-
лентного кобальта, а также путем нагревания высших бкис-
лов кобальта в присутствии твердого или газообразного вос-
становителя [1 — 5]. Известно [6 8], что при температуре
выше 900° высшие окислы кобальта (Со3О3, Со3О4) разла-
гаются и в отсутствие восстановителя с образованием закиси
кобальта, но при последующем охлаждении на воздухе за-
кись вновь окисляется. В связи с этим рекомендуют получе-
ние закиси кобальта из высших окислов в атмосфере азота
[6].
Нами разработана методика получения закиси кобальта,
основанная на термической диссоциации окиси или закись-
скиси кобальта в вакууме и последующем охлаждении заки-
си в вакууме.
СХЕМА СИНТЕЗА КОБАЛЬТА ЗАКИСИ
СоаО3 2 СоО+1/2 Оа
СовО4 -> ЗСоО-ф1;’2Оа
Характеристика основного сырья
Кобальт окись, ч. или ч.д.а., ГОСТ 4467—48.
Кобальт закись-окись, ч., ВТУ МХП У 295—51.
182
Условия получения
В фарфоровый тигель загружают 112 г окиси кобальта
(в расчете на Со2Оз) или 109 г закись-окиси кобальта (в рас-
чете на CQ3O4) и помещают в вакуумную электропечь. Повы-
шают температуру в реторте печи до 850° и одновременно
откачивают воздух из реторты форвакуумным насосом до
0,2—0,5 мм. При этих условиях выдерживают навеску в те-
чение 60—90 минут. Затем обогрев печи отключают и, не
прекращая откачивания, охлаждают реторту до температуры
<50°. В обычных условиях закись кобальта практически не
окисляется воздухом и разгрузка печи может производиться
без особых предосторожностей.
Выход закиси кобальта равен 100 г, что составляет 98%
от теоретического. Вещество представляет собой серо-зеле-
ный порошок; содержание в нем СоО в пределах от 99,7 до
99,9% (см. примечание). Закись кобальта нерастворима в
воде, разбавленными кислотами разлагается с образованием
солей кобальта; т. пл. закиси кобальта ~ 1800° плотность—
6,4.
Примечание. Определение содержания высших окислов в закиси
кобальта проводится методом, описанным в монографии [2]: навеску ве-
щества обрабатывают ~ 10 М уксусной кислотой и кипятят с обратным
холодильником в течение 1 часа; высшие окислы при этом не раство-
ряются.
ЛИТЕРАТУРА
1. Ф. М. Перельман, А. Я. Зворыкин, Н. В. Гудима, Ко-
бальт, М.—Л., Изд. АН СССР, 1949.
2. Cobalt, its Chemistry. Metallurgy and. Uses. Ed. by R. Young, N.-
— Y.—London, 1960, pp. 78, 394.
3. S. S. В h a t n a g e r, B. Prakash, M. A. Qu yum. J. Indian
Chem. !-oc„ 18, 540 (1941).
4. J. Racine. Compt. rend., 220, 823 (1945).
5. A. H. Кузнецов, А. А. Шестопалова. H. Ф. Кулиш. Ж.
физ. химии, 32, 73 (1958).
6. Г. И. Чуфаров, ЛА. Г. Журавлева, Е. П. Татиевская
Докл. АН СССР, 73, 1209 (1950).
7. Т. М. Овчинникова, Э. Ш. Иоффе, Л. Л. Р о т и н я н Докл.
АН СССР, 100, 469 (1955).
8. В. И. Смирнов, М. А. Авдеев. Изв. АН Каз. ССР, серия гор-
ного дела, 1, 97 (1957).
Поступила в декабре 1965 г, цзд
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ,
ОПИСАННЫХ В НАСТОЯЩЕМ ВЫПУСКЕ
Бромистоводородная кислота .................................... 5
Бромноватая кислота............................................ 5
Вольфрам шестихлористый ......................................134
Гадолииий селеновокислый.....................................122
Гадолиний углекислый..........................................107
Гадолиний хлористый безводный.................................124
Галлий трехбромистый..........................................130
Германий дисульфид............................................139
Германий четырехбромистый.....................................143
Германий четырехйодистый......................................145
Германий четыреххлористый.................................... 141
Гольмий селеновокислый .......................................122
Гольмий углекислый............................................108
Гольмий хлористый безводный...................................124
Диспрозий селеновокислый .....................................122
Диспрозий углекислый..........................................108
Диспрозий хлористый безводный.................................124
Европий селеновокислый ...................................... 122
Европий сернокислый закисный..................................112
Европий углекислый............................................107
Европий хлористый безводный...................................128
Индий треххлористый...........................................134
Индий хлорнокислый............................................132
Иттербий селеновокислый.......................................122
Иттербий углекислый...........................................108
Иттербий хлористый безводный.................................126
Иттрий селеновокислый....................................... 122
Иттрий углекислый.............................................108
Иттрий хлористый безводный...................................126
Йодистоводородная кислота..................................... 5
Йодная кислота................................................ 5
Йодноватая кислота.........................•.................. 5
Калий манганит............................................... 24
Калий ниобат (мета).......................................... 17
Калий-сурьма фтористый.......................................150
Калий танталат (мета)........................................ 21
Калий титанат (мета) ....••.................................. 14
184
кобальт закись...............................................182
Лантан углекислый............................................107
Лантан селенистокислый...................................... 116
Лантан селеновокислый........................................122
Лантан хлористый безводный...................................124
Литий алюмогидрид . . . •................................... 38
Литий борогидрид............................................ 27
Литий гидрат окиси безводный................................ 54
Литий карбид (ацетиленид).................................... 43
Литий манганит............................................... 24
Литий ниобат (мета).......................................... 17
Литий нитрид................................................. 52
Литий окись ................................................. 54
Литий силикат (мета)......................................... 48
Литий силикат (орто)......................................... 48
Литий танталат (мета)........................................ 21
Литий теллуристокислый....................................... 57
Литий тетрафенилборат........................................ 33
Литий тетрафторборный........................................ 31
Литий титанат (мета)......................................... 14
Литий фенолят................................................ 45
Литий феррит................................................. 59
Литий цирконат............................................... 50
Лютеций селеиовокислый.......................................122
Лютеций углекислый ..........................................108
Лютеций хлористый безводный..................................126
Молибден пятихлористый.......................................134
Молибден трехбромнетый.......................................177
Мышьяковая кислота............................................ 5
Натрий манганит ............................................. 24
Натрий ниобат (мета).............•........................... 17
Натрий танталат (мета)...................................... 21
Натрий титанат (мета)...................................... 14
Неодим селенистокислый ......................................116
Неодим' селеиовокислый.......................................122
Неодим сульфид полуторный..................'..................ПО
Неодим углекислый............................................107
Неодим хлористый безводный...................................124
Ниобий пятибромистый.........................................154
Ниобий треххлористый....................................... 149
Празеодим селенистокислый....................................116
Празеодим селеновокнслый.....................................122
Празеодим сульфид полуторный ......................• .... НО
Празеодим углекислый........................................ 107
Празеодим хлористый безводный •..............................124
Рениевая кислота ............................................. 5
Рений пятихлористый..........................................134
Рений семисернистый..........................................180
Роданистоводородная кислота .................................. 5
Рубидий борогидрид........................................... 61
Рубидий бромистый ........................................... 88
Рубидии бромноватокислый ос.ч............................... 77
Рубидий гидрат окиси ........................................ 70
Рубидий йодистый............................................. 91
Рубидий йодноватокислый ос.ч................................. 77
Рубидий марганцовокислый..................................... 72
185
Рубидий ниобат (мета)......................... ........ 17
Рубидий мышьяковокислый однозамещенный ос.ч............ 67
Рубидий танталат (мета)................................ 21
Рубидий титанат (мета)................................. 14
Рубидий углекислый..................................... 74
Рубидий фенолят ....................................... 64
Рубидий хлорноватокислый ос. ч. ........................ 77
Самарий селеновокнслый........................................122
Самарий сульфид полуторный..................-..................НО
Самарий углекислый............................................107
Самарий хлористый безводный...................................124
Свинец закись-окись.......................................... 147
Скандий оксигидрат ............................................ 101
Скандий хлористый безводный.....................................121
Сурьма пятисернистая ........................................ . 157
Сурьма трехбромистая .......................................... 162
Сурьма трехйодистая ........................................... 165
Тантал пятибромистый.........................................154
Тантал пятихлористый ....................................... 134
Тантал четыре- хлористый.....................................151
Теллур двуокись ............................................ 168
Теллур четыреххлористый......................................134
Теллуровая кислота .... ...................................... 5
Тербий селеновокнслый........................................122
Тербий углекислый . ................ . •................107
Тербий хлористый безводный...................................124
Тулий селеновокислый ..................................... 122
Тулий хлористый безводный....................................126
Хлорноватая кислота........................................ 5
Хроморубидиевые квасцы.................................... 85
Хромоцезиевые квасцы....................................... 85
Цезий азотнокислый выс. ч.................................... 94
Цезий борогидрид......................................• . . . 61
Цезий бромистый.............................................. 88
Цезий бромноватокислый ос. ч................................ 77
Цезий гидрат окиси........................................... 70
Цезий железистосинеродистый.................................. 99
Цезий йодистый .................. . . .................... 91
Цезий йодноватокислый ос.ч................................... 77
Цезий мар1 анцовокислый . . . •.............................. 72
Цезий мышьяковокислый однозамещенный ос.ч................67
Цезий ниобат (мета) ........................................ 17
Цезий титанат (мета)......................................... 14
Цезий углекислый . . •...................................... 74
Цезий фенолят................• ........................... 64
Цезий фосфорноватистокислый ................................. 97
Цезий хлорноватокислый ос ч.................................. 77
Церий селепистокнслый........................................ 116
Церий селеновокислый ........................................ 122
Церий углекислый..............................................107
Церий хлористый безводный....................................124
Цирконий четыреххлористый..................................... 134
Эрбий селеновокислый..........................................122
Эрбий углекислый.......................................• . . . 108
Эрбий хлористый безводный.....................................126
186
Редакционная коллегий
Е. А. Божевольнов, А. В. Бромберг.
В. Г. Брудзь, В. М. Дзиомко, И. А.
Красавин, Р. П. Ластовский (гл. редактор),
Г. И. Михайлов, Г. А. Певцов.
Замеченные опечатки
Стр. Строчка Напечатано Следует читать
34 2 снизу RLI- ... RU +...
38 15 сверху активированный активный
67 2 снизу ПА] [1,3]
8 снизу [3] [2]
12, 13, 18 И
снизу
68 13 сверху К- Ь 10“3 К—1.10~з%
Редактор Б. Г. Козлов
Техн, редактор А. И. Пирожкова
Корректоры: Л. Г. Черкасова, Л. А. Климанова
Сдано в набор 8.10. 1966 г. Подп. к печати 18.7.1967 г.
Л-55881 Формат бум. 60X90’/и Печ. л. 11.75 Уч-изд. л. 8.»
Зак. 1060 Тираж 1С00 экз. Цена 61 кои-
Типография ВАХЗ