/












































































































































































































































Текст
ВВЕДЕНИЕ
Генетика человека изучает явления на-
наследственности и изменчивости у человека
на всех уровнях его организации и существо-
существования: молекулярном, клеточном, организ-
менном и популяционном. Современная ге-
генетика человека базируется на законах клас-
классической генетики, которые имеют универ-
универсальное значение. Так же, как в классической
генетике, появление и становление которой
связано с изучением наследования мутаци-
мутационных изменений в популяциях гороха, дро-
дрозофилы, мыши и других экспериментальных
объектов исследования, основные достиже-
достижения в генетике человека обусловлены анали-
анализом природы и характера наследования мута-
мутационных изменений у человека.
В последние годы выявлено, что спон-
спонтанная наследственная изменчивость весьма
высока — в течение жизни человека при-
приблизительно у 70% людей реализуются те
или иные наследственные болезни. Таким
образом, у большинства людей в течение их
жизни проявляется хотя бы одно серьезное
генетически обусловленное отклонение от
нормы, снижающее продолжительность
жизни человека по сравнению с нормой ли-
либо мешающее его нормальной жизнедея-
жизнедеятельности и работоспособности. Изучение
молекулярной природы таких генетических
изменений, анализ закономерностей их на-
наследования, оценка их распространенности
в различных популяциях человека, изучение
роли мутагенных факторов окружающей
среды в возможном изменении спонтанного
уровня мутагенеза у человека относятся к
наиболее важным направлениям исследова-
исследований в области генетики человека. Опираясь
на эти фундаментальные знания, медицин-
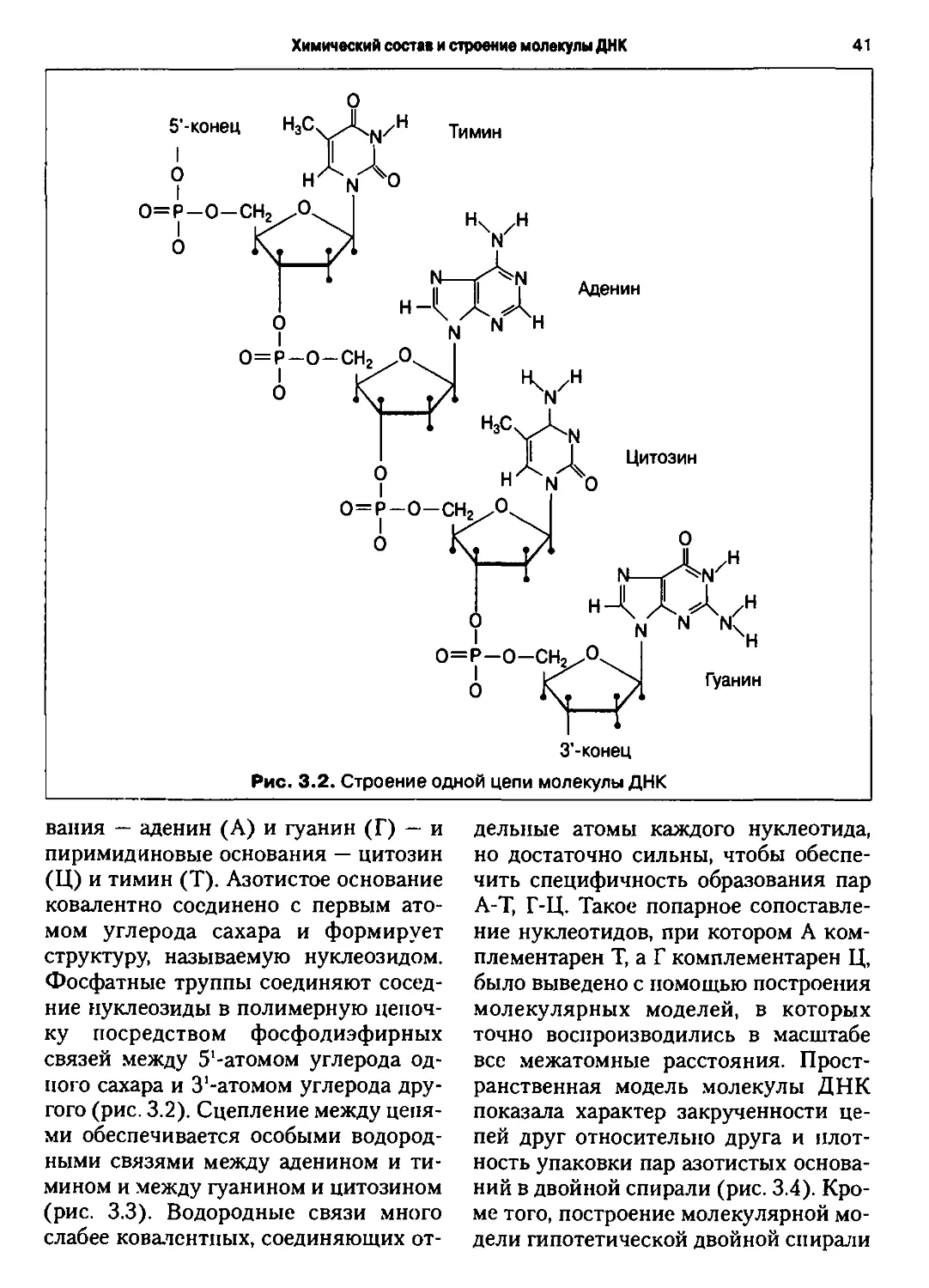
.ская генетика разрабатывает методы диагно-
диагностики, лечения и профилактики наследст-
наследственной патологии, связанной с широким
спектром менделевских, хромосомных и
мультифакториальных наследственных бо-
болезней.
Современный этап развития генетики
человека характеризуется стремительным
прогрессом наших знаний о молекулярном
строении генетического материала и о меха-
механизмах мутагенеза. Наглядным примером
прогресса в области генетики человека явля-
являются успехи реализации международной
программы «Геном человека». Интенсивное
изучение наследственных болезней в клини-
клиниках многих стран увеличило к 1998 г их чис-
число почти до 9000- (в 1966 г было изучено
только около 1500 наследственных болез-
болезней). Для более чем 3900 из этих недугов
изучена локализация мутантных генов в
хромосомах и проведен молекулярный ана-
анализ продуктов их деятельности. Эти дости-
достижения поставили на новую основу разработ-
разработку методов диагностики наследственных бо-
болезней, их профилактики и генотерапии.
В связи с возрастающим загрязнением
окружающей среды, особенно связанным с
радиационным загрязнением целых регио-
регионов в результате Чернобыльской аварии,
ядерных взрывов на полигонах и деятельно-
деятельностью предприятий ядерного топливного
цикла, важное значение приобретает разра-
разработка методов оценки генетических послед-
последствий такого загрязнения для грядущих по-
поколений.
Данный учебник не претендует на исчер-
исчерпывающее изложение материалов по всем
основным разделам генетики человека. По
каждому из обсужденных в книге разделов
имеются монографии ряда исследователей,
из которых наиболее известные и доступные
приведены в списке рекомендуемой литера-
литературы.
Творческие усилия авторов при подготов-
подготовке учебника, состоящего из 10 глав, распреде-
распределились следующим образом. Главы 1,2,8 и 9
написаны НА Топорниной, главы 3, 4, 5 и
10 - Н.С. Стволинской. Глава 6 подготовле-
подготовлена В.А. Шевченко совместно с Г.П. Снигире-
вой. Глава 7 написана В.А. Шевченко совме-
совместно с А.В. Рубановичем.
Учебник предназначен для студентов
университетов, педагогических институтов
и медицинских учебных заведений, для вра-
врачей, генетиков и биологов. Книга написана
достаточно популярно, поэтому она будет
интересна для любого читателя, интересую-
интересующегося вопросами наследственной изменчи-
изменчивости человека.
Успехи генетики человека, ее история
тесно связаны с развитием всех разделов ге-
генетики.
Глава 1. ИСТОРИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Успехи генетики человека, ее исто-
история, тесно связаны с развитием всех
разделов генетики. Задолго до откры-
открытия Г. Менделя различными авторами
были описаны патологические наслед-
наследственные признаки у человека и основ-
основные типы наследования. Первые сведе-
сведения о передаче наследственной патоло-
патологии у человека содержатся в Талмуде D
в. до н.э.), в котором указано на опас-
опасность обрезания крайней плоти у ново-
новорожденных мальчиков, старшие братья
которых или дяди по материнской ли-
линии страдают кровотечением.
К XVIII в. относятся первые описа-
описания доминантного (полидактилии, т.е.
шестипалости) и рецессивного (альби-
(альбинизма у негров) признаков, сделанных
французским ученым П. Мопертюи.
В начале XIX в. несколькими авторами
одновременно было описано наследо-
наследование гемофилии, в результате изуче-
изучения родословных семей, в которых
встречались лица, страдающие этой бо-
болезнью.
В 1814 г. вышла книга лондонского
врача Д. Адамса "Трактат о предполага-
предполагаемых наследственных свойствах болез-
болезней, основанный на клиническом на-
наблюдении." Позже она была переиздана
под названием "Философский трактат о
наследственных свойствах человечес-
человеческой расы". Этот труд стал первым спра-
справочником для генетического консульти-
консультирования. В нем Адаме сформулировал
многие важные принципы медицинской
генетики: "Браки среди родственников
повышают частоту семейных (т.е. рецес-
рецессивных) болезней", "Наследственные
(доминантные) болезни не всегда про-
проявляются сразу после рождения, но мо-
могут развиваться в любом возрасте", "Не
все врожденные болезни являются на-
наследственными, часть из них связана с
внутриутробным поражением плода
(например, за счет сифилиса)".
В середине XIX в. в России над про-
проблемами наследственных болезней и на-
наследственной природы человека работал
В.М. Флоринский. В 1866 г. вышла его
книга "Усовершенствование и вырожде-
вырождение человеческого рода". Наряду с про-
противоречивыми или неверными положе-
положениями, в ней был поднят и правильно
освещен ряд вопросов медицинской ге-
генетики. Среди них: значение среды для
формирования наследственных призна-
признаков, вред близкородственных браков,
наследственный характер многих пато-
патологий (глухонемоты, альбинизма, заячь-
заячьей губы, пороков развития нервной
трубки). Однако этот труд В.М. Фло-
ринского не был оценен в полной мере
его современниками в силу неподготов-
неподготовленности к восприятию этих идей.
В последней четверти XIX в. весо-
весомый вклад в развитие генетики челове-
человека внес английский биолог Ф. Гальтон,
названный К.А. Тимирязевым "одним
из оригинальнейших ученых, исследо-
исследователей и мыслителей." Гальтон впер-
впервые поставил вопрос о наследственнос-
наследственности человека как предмете для изучения
наследственных признаков. Анализи-
Анализируя наследственность ряда семей, Галь-
Гальтон пришел к выводу, что психические
особенности человека обусловлены не
только условиями среды, но и наследст-
наследственными факторами. Кроме того, он
предложил и применил близнецовый
метод для изучения соотносительной
роли среды и наследственности в разви-
развитии признаков. Им же разработан ряд
статистических методов, среди которых
наиболее ценен метод вычисления ко-
коэффициента корреляции. Эти работы
заложили основу для будущего разви-
развития генетики человека. Помимо этого
Гальтон стал родоначальником евгени-
евгеники — науки о наследственном здоровье
человека и путях его улучшения. Одна-
Однако принципиальная ошибка Гальтона
состояла в том, что в практических ме-
мероприятиях евгеники он рекомендовал
не столько избавляться от патологичес-
патологических генов, сколько увеличивать количе-
количество "хороших" генов в человеческих
популяциях путем создания условий
для преимущественного размножения
одаренных людей.
Особого внимания заслуживают ис-
исследования известного английского кли-
клинициста А. Гэррода A857 — 1936 гг.), вне-
внесшего существенный вклад в изучение
проблемы генетики человека. Его работа
"Распространенность алкаптонурии: изу-
изучение химических особенностей" несла
ряд новых идей. Гэррод первым обнару-
обнаружил взаимосвязь между генами и фер-
ферментами, открыл врожденные наруше-
нарушения обмена веществ и положил начало
биохимической генетике. В настоящее
время изучение наследственных болез-
болезней обмена веществ — наиболее актуаль-
актуальный раздел генетики человека.
Труды Гэррода, Адамса и других вра-
врачей — исследователей не были оценены
при их жизни. Биологи обращали мало
внимания на работы медиков. Изучение
наследственности проводилось главным
образом на растениях. К сожалению,
Г. Менделю, как и другим ученым, рабо-
работавшим с растительными объектами, не
были известны данные по генетике чело-
человека. В противном случае открытие зако-
законов генетики могло бы произойти значи-
значительно раньше.
В 1865 г. увидела свет знаменитая ра-
работа чешского ученого Г. Менделя "Опы-
"Опыты над растительными гибридами". Зако-
Законы, открытые им, оставались незамечен-
незамеченными в течение 35 лет и только в 1900 г.
были переоткрыты К. Корренсом (Герма-
(Германия), Э. Чермаком (Австрия) и Г. де Фри-
Фризом (Голландия). С тех пор закономерно-
закономерности наследования, открытые Менделем,
определяют развитие современной гене-
генетики, включая и генетику человека.
Изучая наследования признаков у го-
гороха, Г. Мендель установил три закона:
1. Закон единообразия гибридов пер-
первого поколения;
2. Закон расщепления во втором поко-
поколении по фенотипу 3:1 (при моногибрид-
моногибридном скрещивании);
3. Закон.независимого наследования
признаков.
Успех чешского ученого был связан с
разработкой принципиально нового ме-
методического подхода. Он:
— ввел в науку новый гибридологиче-
гибридологический метод, выбрав для изучения контра-
контрастные пары признаков;
— проводил строгий количественный
учет изучаемых признаков, что позволи-
позволило обнаружить статистические законо-
закономерности наследования;
— анализируя эти закономерности,
пришел к выводу, что зародышевые клет-
клетки несут набор признаков, которые могут
быть определены с помощью скрещива-
скрещиваний.
Опыты Г. Менделя и сделанные из
них выводы стали предпосылкой для со-
создания теории гена — основы современ-
современной генетики, а 1900 г. — год вторичного
открытия законов Менделя — считается
годом рождения генетики. Название но-
новой науке было дано в 1906 г. английским
ученым В.Бэтсоном (от латинского слова
geneo — порождаю), а в 1909 г. датский ге-
генетик В. Иоганнсен предложил такие
важные генетические термины, как ген,
генотип и фенотип.
В 1903 г. американский антрополог
Фараби, изучая родословные в несколь-
нескольких поколениях, впервые установил, что
брахидактилия (короткопалость) у чело-
Глава 1. ИСТОРИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
века наследуется по аутосомно-доми-
нантному типу. Из этой работы следовал
вывод о справедливости менделевских
законов и для человека.
В 1900 г. К Ландштейнер описал сис-
систему групп крови АВО.
В 1924 г. Ф. Бернштейн установил,
что АВО-система групп крови контроли-
контролируется серией множественных аллелей
одного локуса. Спустя 25-30 лет был об-
обнаружен резус-фактор (Rh) и показано,
что гемолитическая желтуха новорож-
новорожденных возникает из-за иммунологичес-
иммунологической несовместимости матери и плода.
Эти открытия также указывали на при-
применимость законов Менделя к наследо-
наследованию признаков у человека.
В 1908 г. Г. Харди и В. Вайнберг неза-
независимо друг от друга пришли к выводу,
что менделевские законы дают возмож-
возможность объяснить распределение частоты
генов из поколения в поколение в попу-
популяциях (от латинского — populus — насе-
население, народ) и условиях генетической
стабильности популяции. Этот закон
был установлен путем анализа наследст-
наследственности человека и лег в основу популя-
ционной генетики.
В 1919 г. Ю.А. Филипченко организо-
организовал кафедру генетики в Петроградском
университете. В это же время Н.И. Вави-
Вавилов сформулировал важнейший генети-
генетический закон — закон гомологических
рядов в наследственной изменчивости.
Одновременно в Москве Н.К. Кольцов
создает свою генетическую школу.
В 20 гг. XX века начала интенсивно
развиваться советская генетика. Под вли-
влиянием идей евгеники, которая получила
широкое распространение в ряде стран
Европы (Англия, Франция, Германия) и
Америке в 1921 г. в Москве Н.К. Кольцо-
Кольцовым было организовано Русское евгени-
евгеническое общество, в 1922 г. в Петрограде
Ю. А. Филипченко создал Бюро по евге-
евгенике.
Эти евгенические организации ориен-
ориентировались на сугубо научные задачи в
отличие от евгенических обществ других
стран. Н.К Кольцов, Ю.А. Филипченко и
другие ученые проводили работы по ге-
генетике одаренности, изучая родословные
выдающихся личностей. Однако эти ис-
исследования грешили методическими
ошибками, противоречиями, определен-
определенным примитивизмом. Вместе с тем были
в евгенических работах и положительные
моменты. Так, Н.К Кольцов и Ю.А. Фи-
Филипченко правильно ставили вопрос о
значении социальных условий в реализа-
реализации индивидуальных особенностей чело-
человека, полностью отвергали насильствен-
насильственный путь улучшения наследственности
человека. Кроме того, силами советских
евгеников были собраны родословные
выдающихся личностей, например,
A.C. Пушкина, Л.Н. Толстого, А.М. Горь-
Горького, Ф.И. Шаляпина и др.
К концу 20-х годов евгенические ис-
исследования в нашей стране были прекра-
прекращены. Падала ее популярность и в других
странах (кроме Германии). Число евгени-
чеких обществ быстро уменьшалось,
журналы закрывались или переименовы-
переименовывались.
Конец 20-х — начало 30-х гг. ознаме-
ознаменовались значительными успехами в раз-
развитии генетики. Родилась и стала обще-
общепризнанной хромосомная теория наслед-
наследственности, было установлено, что на-
наследственность связана с генами, локали-
локализованными в хромосомах клеточных
ядер, что гены в хромосомах расположе-
расположены линейно и образуют группы сцепле-
сцепления.
В этот же период создается популяци-
онная генетика. Большой вклад в разви-
развитие этого раздела внесли С.С. Четвери-
Четвериков, Р. Фишер, Н.П. Дубинин и Д.Д. Ро-
машев, Дж. Е. Холдейн и др.
В ряде стран, в том числе в нашей, на-
начинает развиваться медицинская генети-
ка. С 1932 но 37 гг. работал Московский
медико-биологический институт им.
М. Горького (позднее — Медико-генети-
Медико-генетический институт), возглавляемый
С. Г. Левитом. При нем был организо-
организован Центр близнецовых исследований.
Здесь изучались болезни с наследствен-
наследственным предрасположением — диабет, яз-
язвенная болезнь, аллергия, гипертониче-
гипертоническая болезнь и др. Большой интерес
имели цитогенетические работы по
идентификации первых хромосом чело-
человека. Особого упоминания заслужива-
заслуживают труды талантливого генетика и кли-
нициста-невронатолога С.Н. Давиден-
кова A880-1961). Он первым поставил
вопрос о гетерогенности наследствен-
наследственных заболеваний и начал проводить .ме-
.медико-генетическое консультирование.
К концу 30-х гг. XX в. интерес к гене-
генетике человека начал снижаться. Сократи-
Сократилось и оставалось низким до начала 50-х
гг. количество опубликованных работ.
В Советском Союзе с приходом к вла-
власти в биологической науке Т.Д. Лысенко
все генетические исследования, включая
и исследования по генетике человека, бы-
были запрещены. Генетика была объявлена
"лженаукой". Августовская сессия
ВАСХНИЛ A948 г.) нанесла огромный
вред теоретическим и практическим до-
достижениям генетики, утвердив антинауч-
антинаучные идеи Т.Д.Лысенко. Такое положение
сохранялось до начала 60-х гг.
Возрождение советской генетической
науки началось после разоблачения "уче-
"учения" Лысенко и шло по пути развития
медицинской генетики. В 1964 г. был из-
издан учебник В.П. Эфроимсона по меди-
медицинской генетике, в 1969 г. открыт Ин-
Институт медицинской генетики под руко-
руководством Н.П. Бочкова (в настоящее вре-
время — Научно-исследовательский центр
медицинской генетики РАМН), где нача-
начались широкие исследования по многим
направлениям медицинской генетики.
В 50-х гг. получают широкое развитие
исследования по радиационной генетике
человека. Еще в 1927 г. американский ис-
исследователь Г. Меллер установил силь-
сильное мутагенное действие рентгеновских
лучей. Это открытие показало опасность
облучения половых клеток человека для
последующих поколений, в силу чего че-
человеку как объекту генетических иссле-
исследований стало уделяться больше внима-
внимания.
С 1959 по 1962 гг. количество публи-
публикаций, симпозиумов, конференций по ге-
генетике человека быстро возрастало. Сли-
Слияние генетики, цитологии, цитогенетики,
биохимии способствовало формирова-
формированию клинической генетики.
Усилиями ученых была подтверждена
гетерогенность наследственных патоло-
патологий, когда один и тот же фенотип болезни
обусловлен изменением разных белков.
Трудно переоценить важность этого от-
открытия для диагностики, лечения и ме-
медико-генетического консультирования
наследственных болезней.
В 1944 г. было достоверно установле-
установлено, что передача наследственной инфор-
информации связана с дезоксирибонуклеино-
вой кислотой (ДНК). Это открытие яви-
явилось мощным фактором, стимулирую-
стимулирующим изучение наследственности на мо-
молекулярном уровне. А благодаря созда*
нию в 1953 г. Д. Уотсоном и Ф. Криком
модели макромолекулярной структуры
ДНК, началось углубленное изучение
молекулярной, биохимической и имму-
ногенетики человека.
Убедительный пример значения фун-
фундаментальных исследований для практи-
практического здравоохранения дает история
развития цитогенетики. В 1956 г. X. Тио
и А. Леван установили, что в клетках че-
человека содержится 46 хромосом, а спустя
три года были открыты хромосомные бо-
болезни человека. В 1959 г. Дж. Л ежен ус-
установил цитогенетическую картину воз-
Глава 1. ИСТОРИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
никновения синдрома Дауна (трисомия
по 21-й хромосоме.)- В это же время не-
несколько ученых идентифицировали на
хромосомном уровне синдром Тернера
(ХО) и синдром Клайнфельтера
(XXY). Одновременно была определе-
определена роль Y-хромосомы в определении
пола человека.
В 1960 г. Р. Мурхед с коллегами разра-
разработали метод культивирования лимфо-
лимфоцитов периферической крови для полу-
получения метафазных хромосом человека,
что позволило обнаруживать мутации
хромосом, характерные для определен-
определенных наследственных болезней. Другим
важным открытием для развития цитоге-
нетики человека явилась разработка ме-
методов дифференциальной окраски хро-
хромосом. Благодаря ему стала возможна
идентификация каждой хромосомы че-
человека, а это резко повысило разрешаю-
разрешающую способность цитогенетических ме-
методов.
Еще одним этапом развития совре-
современной генетики человека явилось кар-
картирование и локализация генов в хромо-
хромосомах человека. Достижения цитогенети-
ки, генетики соматических клеток, уве-
увеличение числа генетических маркеров
способствовали успешному изучению
групп сцепления. В настоящее время у
человека установлено 23 группы сцепле-
сцепления. Эти данные нашли непосредствен-
непосредственное применение в диагностике наследст-
наследственных заболеваний и медико-генетичес-
медико-генетическом консультировании.
Тесная связь современной генетики с
химией, физикой, биохимией, физиоло-
физиологией, экологией, фармакологией и други-
другими науками способствовала появлению
новых разделов генетики: цитогенетики,
радиационной генетики, иммуногенети-
ки, фармакогенетики, экологической ге-
генетики.
Во второй половине XX в. начала ин-
интенсивно развиваться молекулярная ге-
генетика и генная инженерия, были разра-
разработаны методы искусственного и фер-
ферментативного синтеза генов. В 1969 г. ин-
индийский ученый Г. Карано впервые осу-
осуществил искусственный синтез гена. С
помощью генной инженерии получены
искусственные гены инсулина, интерфе-
интерферона, соматотропина и др. Эти достиже-
достижения открывают большие перспективы в
диагностике, профилактике и лечении
наследственных болезней человека.
Возможности молекулярной генети-
генетики и развитие современных методов ра-
работы с ДНК нашли применение для ре-
решения практических задач медицинской
генетики.
Конец XX в. ознаменован разработ-
разработкой и началом осуществления грандиоз-
грандиозной международной программы "Геном
человека". Ее задача — изучение генома
человека, включая картирование хромо-
хромосом и секвенирование их ДНК, определе-
определение полной нуклеотидной последова-
последовательности генома, состоящего из трех
миллиардов пар нуклотидов. В рамках
этой программы разрабатываются мето-
методы диагностики и лечения наследствен-
наследственных болезней. В настоящее время уже
возможна ДНК- диагностика более 100
наследственных дефектов. В недалеком
будущем станет реальностью генотера-
пия наиболее распространенных болез-
болезней человека, патогенез которых уже из-
известен.
Вопросы для самоконтроля
1. Какие сведения о наследственных патологиях
были известны в XVHI-XIX вв.?
2. Какова роль классической генетики в развитии
генетики человека?
3. Назовите отечественных ученых, внесших боль-
большой вклад в развитие генетики человека.
4. Когда впервые достоверно описаны хромосомы
человека?
5. Какие науки тесно взаимосвязаны с генетикой
человека на современном этапе? (Объяснить, поче-
почему?)
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Основные закономерности наслед-
наследственности, установленные для живых
организмов, универсальны и в полной
мере справедливы и для человека.
Вместе с тем как объект генетических
исследований человек имеет свои пре-
преимущества и недостатки.
Для людей невозможно планиро-
планировать искусственные браки. Еще в 1923 г.
Н.К. Кольцов отмечал, что "...мы не мо-
можем ставить опыты, мы не можем за-
заставить Нежданову выйти замуж за
Шаляпина только для того, чтобы по-
посмотреть, какие у них будут дети". Од-
Однако эта трудность преодолима благо-
благодаря прицельной выборке из большого
числа брачных пар тех, которые соот-
соответствуют целям данного генетическо-
генетического исследования.
В значительной мере затрудняет
возможности генетического анализа
человека большое число хромосом —
2п=46. Однако разработка новейших
методов работы с ДНК, метода гибри-
гибридизации соматических клеток и неко-
некоторых других методов устраняют эту
трудность.
Из-за небольшого числа потомков
(во второй половине XX в. в большин-
большинстве семей рождалось по 2-3 ребенка)
невозможен анализ расщепления в по-
потомстве одной семьи. Однако в боль-
больших популяциях можно выбрать се-
семьи с интересующими исследователя
признаками. Кроме того, в некоторых
семьях определенные признаки про-
прослежены на протяжении многих поко-
поколений. В таких случаях возможен ге-
генетический анализ. Еще одна труд-
трудность связана с длительностью смены
поколений у человека. Одно поколе-
поколение у человека занимает в среднем 30
лет. И, следовательно, генетик не мо-
может наблюдать более одного-двух по-
поколений.
Для человека характерен большой
генотипический и фенотипический
полиморфизм. Проявление многих
признаков и болезней в сильной степе-
степени зависят от условий внешней среды.
Следует отметить, что понятие "среда"
для человека более широкое по сравне-
сравнению с растениями и животными. На-
Наряду с питанием, климатом и другими
абиотическими и биотическими фак-
факторами, средой для человека являются
и социальные факторы, трудно изме-
изменяемые по желанию исследователя.
Вместе с тем, человека как генетичес-
генетический объект широко изучают врачи
всех специальностей, что нередко по-
помогает установить различные наслед-
наследственные отклонения.
В настоящее время интерес и вни-
внимание к изучению генетики человека
активно возрастают. Так, глобальная
международная программа "Геном че-
человека" имеет своей задачей изучение
генома человека на молекулярном
уровне. Для ее решения используются
все новейшие современные методы ге-
генетики и медицины.
Какими же методами располагает
генетика человека в настоящие дни?
Их немало: генеалогический, близне-
близнецовый, цитогенетический, популяци-
онно-статистический, биохимический,
метод генетики соматических клеток и
молекулярно-генетический. Рассмот-
Рассмотрим подробнее каждый из них.
10
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Генеалогический метод
Относящийся к числу основных в
генетике человека, этот метод опирает-
опирается на генеалогию — учение о родослов-
родословных. Его сутью является составление
родословной и последующий ее ана-
анализ. Впервые такой подход был пред-
предложен английским ученым Ф. Гальто-
ном в 1865 г.
Генеалогический метод широко ис-
используется для решения как научных,
так и прикладных проблем. Он позво-
позволяет выявить наследственный харак-
характер признака и определить тип насле-
наследования. Наряду с этим метод дает
возможность установить сцепленное
наследование, определить тип взаимо-
взаимодействия генов и пенетрантность алле-
аллелей. Генеалогический метод лежит в
основе медико-генетического консуль-
консультирования. Он включает два этапа: со-
составление родословных и их генеало-
генеалогический анализ.
Составление родословной
Сбор сведений о семье начинается с
человека, называемого пробандом.
Обычно это больной с изучаемым забо-
заболеванием. Дети одной родительской
пары называются сибсами (братья-сес-
(братья-сестры). В большинстве случаев родо-
родословная собирается по одному или не-
нескольким признакам. Родословная мо-
может быть полной или ограниченной.
Чем больше поколений прослежено в
родословной, тем она полнее и тем вы-
выше шансы на получение полностью до-
достоверных сведений.
Сбор генетической информации
проводится путем опроса, анкетирова-
анкетирования, личного обследования семьи. Оп-
Опрос начинается обычно с родственни-
родственников по материнской линии: бабушки и
дедушки по материнской линии, с ука-
указанием внуков, детей каждого ребенка
бабушки и дедушки. В родословную
вносят сведения о выкидышах, абор-
абортах, мертворожденных, бесплодных
браках и др.
При составлении родословной ве-
ведется краткая запись данных о каждом
члене рода с указанием его родства по
отношению к пробанду. Обычно ука-
указываются: фамилия, имя и отчество,
дата рождения и смерти, возраст, наци-
национальность, место жительства семьи,
профессия, наличие хронических забо-
заболеваний в семье, причину смерти
умерших и др.
После сбора сведений составляют
графическое изображение родослов-
родословной, используя систему условных обо-
обозначений (рис. 2.1).
Выполняя эту работу, важно соблю-
соблюдать следующие правила:
1. Составление родословной начи-
начинают с пробанда. Братья и сестры рас-
располагаются в порядке рождения слева
направо, начиная со старшего.
2. Все члены родословной распола-
располагаются строго по поколениям в один
ряд.
3. Поколения обозначаются римски-
римскими цифрами слева от родословной
сверху вниз.
4. Арабскими цифрами нумеруется
потомство одного поколения (один
ряд) слева направо.
5. В связи с тем, что некоторые бо-
болезни проявляются в разные периоды
жизни, указывается возраст членов се-
семьи.
6. Отмечаются лично обследован-
обследованные члены родословной.
Графическое изображение родо-
родословной может быть вертикально-го-
вертикально-горизонтальным или расположенным
по кругу (в случае обширных дан-
данных). Схема родословной сопровож-
сопровождается описанием обозначений под
рисунком, которое называется леген-
легендой (рис. 2.2).
Генеалогический метод
11
Q3 -женщина
| | -мужчина
здоровье
—| | -внебрачная
связь
-сибсы
О-
-монозиготные
близнецы
пол неизвестен
-выкидыш
dfadb
-мертворожденный
сю
¦дизиготные
близнецы
-бесплодный
брак
ПЮ
брак
-кровнородственный
брак
С С
Ш 0
¦——¦ ¦
I I!
С_)Ч Ь_)
-повторныйбрак
1уП
-больные
-гетерозиготные
-носители рецес-
рецессивного гена
-лично обсле-
обследованные
-умершие
Рис. 2.1. Символы, используемые при составлении родословной
Генетический анализ родословной
Задача генетического анализа — ус-
тановление наследственного характера
заболевания и типа наследования, вы-
явление гетерозиготных носителей му-
тантного гена, а так же прогнозирова-
ние рождения больных детей в семьях
с наследственной патологией.
Анализ родословной включает сле-
дующие этапы: 1. Установление, явля-
ется ли данный признак или заболева-
ние единичным в семье или имеется
12
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Рис. 2.2. Родословная с концентрическим расположением поколений
несколько случаев (семейный харак-
характер). Если признак встречается не-
несколько раз в разных поколениях, то
можно предполагать, что этот при-
признак имеет наследственную природу.
2. Определение типа наследования
признака. Для этого анализируют ро-
родословную, учитывая следующие мо-
моменты:
1) встречается ли изучаемый при-
признак во всех поколениях и многие ли
члены родословной обладают им;
2) одинакова ли его частота у лиц
обоих полов и у лиц какого пола он
встречается чаще;
3) лицам какого пола передается
признак от больного отца и больной
матери;
Генеалогический метод
13
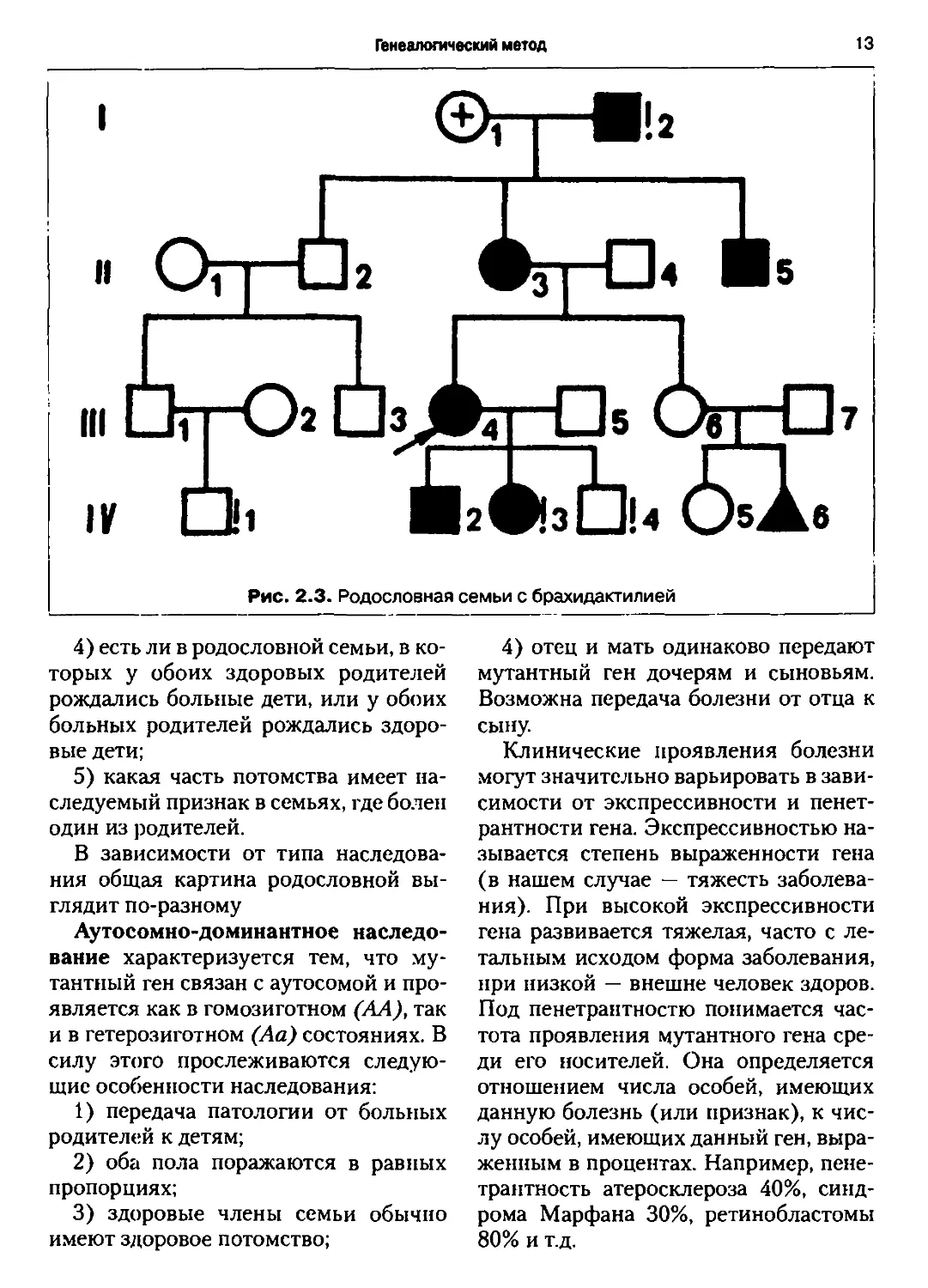
Рис. 2.3. Родословная семьи с брахидактилией
4) есть ли в родословной семьи, в ко-
которых у обоих здоровых родителей
рождались больные дети, или у обоих
больных родителей рождались здоро-
здоровые дети;
5) какая часть потомства имеет на-
наследуемый признак в семьях, где болен
один из родителей.
В зависимости от типа наследова-
наследования общая картина родословной вы-
выглядит по-разному
Аутосомно-доминантное наследо-
наследование характеризуется тем, что му-
тантный ген связан с аутосомой и про-
проявляется как в гомозиготном (АА), так
и в гетерозиготном (Аа) состояниях. В
силу этого прослеживаются следую-
следующие особенности наследования:
1) передача патологии от больных
родителей к детям;
2) оба пола поражаются в равных
пропорциях;
3) здоровые члены семьи обычно
имеют здоровое потомство;
4) отец и мать одинаково передают
мутантный ген дочерям и сыновьям.
Возможна передача болезни от отца к
сыну.
Клинические проявления болезни
могут значительно варьировать в зави-
зависимости от экспрессивности и пенет-
рантности гена. Экспрессивностью на-
называется степень выраженности гена
(в нашем случае — тяжесть заболева-
заболевания). При высокой экспрессивности
гена развивается тяжелая, часто с ле-
летальным исходом форма заболевания,
при низкой — внешне человек здоров.
Под пенетрантностю понимается час-
частота проявления мутантного гена сре-
среди его носителей. Она определяется
отношением числа особей, имеющих
данную болезнь (или признак), к чис-
числу особей, имеющих данный ген, выра-
выраженным в процентах. Например, пене-
трантность атеросклероза 40%, синд-
синдрома Марфана 30%, ретинобластомы
80% и т.д.
14
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Экспрессивность и пенетрантность
колеблются в широких пределах (от О
до 100%) и сильно зависят от условий
внешней среды. По аутосомно-доми-
нантному типу наследуется полидак-
полидактилия (шестипалость), брахидактилия
(короткопалость), ахондроплазия
(карликовость), синдром Марфана
("паучьи пальцы") и другие заболева-
заболевания (рис. 2.3).
При доминантном типе наследова-
наследования, если один из родителей болен
(Аа), вероятность рождения больного
ребенка равна 50% при условии пол-
полной пенетрантности гена. В случае ге-
терозиготности обоих родителей (Аа х
Аа) больные дети могут родиться с ве-
вероятностью 75%. Многие аутосомно-
доминантные заболевания в гомози-
гомозиготном состоянии протекают в более
тяжелой форме, чем у гетерозигот. Од-
Однако в практике нередки случаи, когда
носители доминантного гена остаются
фенотипически здоровыми. В резуль-
результате вид родословной изменяется и по-
появляются пропуски поколений.
Носительство доминантного гена
без фенотипического проявления
можно заподозрить у одного из роди-
родителей, если среди его потомков появи-
появились больные с такой же доминантной
патологией. Когда у здоровых родите-
родителей появился больной ребенок и в ро-
родословной есть другие случаи этой бо-
болезни, то правомочно предполагать,
что у одного из родителей больного
был дефектный ген, который не пенет-
рировал, но был передан потомку.
Доминантный ген может обладать
разной степенью экспрессивности, что
затрудняет установление аутосомно-
доминантного типа наследования. Рас-
Рассмотрим это на примере наследствен-
наследственной патологии соединительной ткани —
синдрома Марфана. Встречаются тя-
тяжелые формы заболевания с классиче-
классическим поражением костной системы
(сколиоз или кифосколиоз, деформа-
деформация грудины, высокий рост), наруше-
нарушением зрения (двуеоронний вывих
хрусталика) и серде*но — сосудистой
системы (расширени г аорты). Наблю-
Наблюдаются и стертые сюрмы синдрома
Марфана, которые ни диагностируют-
диагностируются (астеническое телэсложение, арах-
нодактилия, небольшая миопия). Сла-
Слабо выраженные клинические формы
болезни могут легкэ пропускаться,
тогда родословная тгкже теряет свой
"классический" вид, юявляются про-
пропуски поколений. Поэтому важно про-
проводить личное обслед ование всех чле-
членов семьи.
При аутосомно-ре цессивном типе
наследования мутан'ный ген прояв-
проявляет свое действие только в гомозигот-
гомозиготном состоянии По это л причине в гете-
гетерозиготном состоянии он может суще-
существовать во многих поколениях, не
проявляясь фенотипически.
При данном типе наследования за-
заболевание встречаете! в родословной
редко и не во всех пою >лениях. Вероят-
Вероятность заболевания у дгвочек и мальчи-
мальчиков одинакова. Признак может про-
проявиться у детей, родит ели которых бы-
были здоровы, но являл! сь гетерозигот-
гетерозиготными носителями мутантного гена.
Возможны несколько вариантов таких
браков:
1) мать аа х отец аа — у таких роди-
родителей все дети будут больными (аа);
2) мать Аа х отец аа — 50 % детей бу-
будут больными (генотип аа) и 50 % фе-
фенотипически здоровыми (генотип Аа),
но будут являться гетерозиготными
носителями дефектного гена;
3) мать Аа х отец Аа — 25 % детей
будут больными (генотип аа), 75 % фе-
фенотипически здоровыми (генотипы АА
и Аа), но 50 % из них будут носителя-
носителями мутантного гена (генотип Аа).
Генеалогический метод
15
РГГВ. #!5
Рис. 2.4. Родословная семьи с альбинизмом
Известно, что частота возникнове-
возникновения наследственных рецессивно-ауто-
сомных болезней находится в прямой
зависимости от степени распростра-
распространенности мутантного гена среди насе-
населения. Частота таких болезней особен-
особенно повышается в изолятах и среди на-
населения с высоким процентом кровно-
кровнородственных браков. Такие браки отри-
отрицательно влияют на потомство, на это
указывает тот факт, что умственная от-
отсталость среди детей от родственных
браков в 4 раза выше, чем в семьях с не-
неродственными браками.
При аутосомно-рецессивном типе
наследования (как и при аутосомно-
доминантном) возможны различная
степень экспрессивности и пенетрант-
ности признака. К заболеваниям с ау-
тосомно-рецессивным типом наследо-
наследования относятся многие болезни об-
обмена веществ, среди которых фенил-
кетонурия, галактоземия, альбинизм
(рис.2.4), муковисцидоз и др. Установ-
Установлено, что рецессивные заболевания
чаще диагностируются в раннем воз-
возрасте.
Наследование заболеваний, сцеп-
сцепленных с полом, определяется тем, что
мутантный ген расположен в X или Y-
хромосоме. Известно, что у женщин
имеются две Х-половые хромосомы, а у
мужчин — одна Х- и одна Y-хромосома.
У человека в Х-хромосоме локализова-
локализовано более 200 генов. Гены, расположен-
расположенные в хромосоме X, могут быть рецес-
рецессивными или доминантными.
У женщин мутантный ген может на-
находиться в обеих Х-хромосомах или
только в одной из них; в первом случае
она гомозиготна, во втором — гетеро-
гетерозиготна. Мужчины, являясь гемизи-
готными (имеют только одну хромосо-
хромосому X), передают ее только дочерям и
никогда — сыновьям. Любой ген, как
доминантный, так и рецессивный, ло-
16
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
D-
¦©
i 5
3 4 5 6*78 9 10 11 12 13 14 15 16 17
У
1 2 3
Рис. 2.5. Родословная с Х-сцепленным рецессивным типом наследования (гемофилия)
кализованый в его Х-хромосоме, обя-
обязательно будет проявляться. В этом
главная особенность Х-сцепленного
наследования.
Для Х-сцепленного рецессивного
наследования характерны следующие
особенности:
1) заболевание встречается чаще у
лиц мужского пола;
2) от здоровых родителей могут ро-
родиться больные дети (если мать гете-
гетерозиготна по мутантному гену);
3) больные мужчины не передают
заболевание своим сыновьям, но их
дочери становятся гетерозиготными
носителями болезни;
4) больные женщины могут ро-
родиться только в семьях, где отец бо-
болен, а мать гетерозиготна по мутант-
мутантному гену.
Рассмотрим несколько примеров,
когда в Х-хромосоме локализован ре-
рецессивный ген. Если в брак вступает
здоровая женщина и больной мужчи-
мужчина, то в такой семье все дети будут здо-
здоровыми, а дочери получат от отца одну
Х-хромосому с мутантным геном и бу-
будут гетерозиготными носителями (т.к.
вторую нормальную Х-хромосому они
получат от матери). В том случае, если
в брак вступают здоровый мужчина и
женщина — носительница патологиче-
патологического гена, то вероятность рождения
больного мальчика составит 50% от
всех мальчиков и 25% — от всех детей.
Вероятность рождения больных де-
девочек очень низка и возможна лишь,
если отец болен, а мать гетерозиготна
по мутантному гену. В такой семье по-
половина мальчиков будут больными.
Среди девочек болезнь проявится у
половины, а другая половина будет не-
нести дефектный ген.
Классическим примером рецессив-
рецессивного, сцепленного с полом наследова-
наследования, может служить гемофилия. Боль-
Больные страдают повышенной кровоточи-
кровоточивостью. Причина — недостаточное со-
содержание в крови факторов свертыва-
свертывания крови. На рис. 2.5 изображена ро-
родословная семьи с гемофилией. Ана-
Анализ родословной показывает, что боле-
болеют только мальчики. (II — 1,4; III —
7,15). Отсюда можно предположить,
Герцог Саксои-
Кобург-Готский
Альберт
Q Виктория
T (жена импе-
императора
Фридриха)
Георг III
Эдуард герцог
КентскийA767-1820)
Виктория
A819-1901)
Людовик II, великий
герцог Гессенский
п
I Кайзер
Вильгельм II
VI
VII
VIII
Герцог
Виндзорский
Георг V
Эдуард VII
Георг IV
Граф
-_ Маунттбетон
6 Бирманский
Маргарет
Алиса
Гессенская
1 11 иРина JL -L
Т * Г Генриха)-$рИдр
Ъ 6k"~
Вильгельм
Воль- Генрих Алексей
демор Принц
Сигизмунд
Прусский
Леопольд,
герцог Олбани
Алек-
сандра Л. rt
Федоровна \J " °
(жена
Николая II)
,Леди Мери
Эйбл-Смит
Рулерт.
виконт
Трематон
Альфонс
Беатриса
I I Виктория
I I Евгения
J-. Лужена
1 I VS/Альфонса XIII)
| Леопольд | Морис
i
ф ? ®<Ьи<Ь<Ь\
Гонсалес
Софья
Хуан
Карлос
Рис. 2. в. Наследование, сцепленное с полом
18
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Ш-тО
1 2
< Отв ИгО
у» 5
? оЪ о
Рис. 2.7. Родословная с Y-сцепленным
типом наследования (гипертрихоз).
что ген гемофилии сцеплен с полом.
Больные дети чаще рождаются от здо-
здоровых родителей и, следовательно, ген
болезни рецессивный.
Известно, что гемофилия широко
распространена в королевских семьях
Европы. Это связано с заключением
близкородственных браков. В резуль-
результате возникшие мутации сохранялись
внутри семьи. Королева Виктория в
Англии была носительницей гена ге-
гемофилии. Ее сын Леопольд родился
гемофиликом. Через своих дочерей и
внуков королева Виктория передала
ген гемофилии Вольдемару и Генриху
Прусским, Фридриху Гессенскому, ца-
царевичу Алексею Романову, Рупрехту
Тех-Атлонскому, двум баттенбергским
и двум испанским принцам (рис 2.6).
Кроме гемофилии, в Х-хромосоме ло-
локализованы рецессивные гены, обус-
обусловливающие миопатию Дюшеина, не-
некоторые формы дальтонизма и др. за-
заболевания.
При локализации в хромосоме X до-
доминантного гена тип наследования на-
называется Х-сцепленным доминант-
доминантным. Для него характерны следующие
признаки:
1) болеют как мужчины, так и жен-
женщины, однако больных женщин в два
раза больше, чем больных мужчин;
2) заболевание прослеживается в
каждом поколении;
3) если болен отец, то все его дочери
будут больными, а все сыновья здоро-
здоровыми;
4) если мать больна, то вероятность
рождения больного ребенка равна 50 %
независимо от пола;
5) больными дети будут только тог-
тогда, если болен один из родителей;
6) у здоровых родителей все дети
будут здоровы.
По Х-сцепленному доминантному
типу наследуются фосфатемия (недо-
(недостаток фосфата в крови), коричневая
окраска эмали зубов и др.
Имеет свои особенности и У-сцеп-
ленное наследование.
В Y-хромосоме у мужчин локализо-
локализовано немного генов. Они передаются
только сыновьям и никогда дочерям
(голандрическое наследование). С Y-
хромосомой у мужчин наследуются та-
такие признаки, как гипертрихоз (нали-
(наличие волос по краю ушных раковин),
кожные перепонки между пальцами ног,
развитие семенников, интенсивность
роста тела, конечностей и зубов. Харак-
Характерные особенности наследования с Y-
хромосомой можно видеть на рис. 2.7.
Близнецовый метод
Это метод изучения генетических
закономерностей на близнецах. Впер-
Впервые он был предложен Ф. Гальтоном в
1875 г. Близнецовый метод дает воз-
возможность определить вклад генетиче-
генетических (наследственных) и средовых
факторов (климат, питание, обучение,
воспитание и др.) в развитии конкрет-
Близнецовый метод
19
ных признаков или заболеваний у че-
человека.
При использовании близнецового
метода проводится сравнение :
1) монозиготных (однояйцевых)
близнецов — МБ с дизиготными (раз-
(разнояйцевыми) близнецами — ДБ;
2) партнеров в монозиготных парах
между собой;
3) данных анализа близнецовой вы-
выборки с общей популяцией.
Монозиготные близнецы образуют-
образуются из одной зиготы, разделившейся на
стадии дробления на две (или более)
части. С генетической точки зрения
они идентичны, т.е. обладают одинако-
одинаковыми генотипами. Монозиготные
близнецы всегда одного пола.
Особую группу среди МБ составля-
составляют необычные типы близнецов: двух-
двухголовые (как правило нежизнеспособ-
нежизнеспособные), каспофаги ("сиамские близне-
близнецы"). Наиболее известный случай —
родившиеся в 1811 г. в Сиаме (ныне
Таиланд) сиамские близнецы — Чанг
и Энг. Они прожили 63 года, были же-
женаты на сестрах-близнецах; Чанг про-
произвел на свет 10, а Энг — 12 детей. Ког-
Когда от бронхита умер Чанг, спустя 2 ча-
часа умер и Энг (рис. 2.8) Их связывала
тканевая перемычка шириной около
10 см от грудины до пупка. Позднее
было установлено, что соединявшая
их перемычка содержала печеночную
ткань, связывающую две печени. Лю-
Любая хирургическая попытка разделить
братьев вряд ли в то время была бы ус-
успешной. В настоящее время разъеди-
разъединяют и более сложные связи между
близнецами.
Дизиготные близнецы развиваются
в том случае, если одновременно две
яйцеклетки оплодотворены двумя
сперматозоидами. Естественно, дизи-
дизиготные близнецы имеют различные ге-
генотипы. Они сходны между собой не
Рис. 2.8. Сиамские близнецы Чанг и Энг
более, чем братья и сестры, т.к. имеют
около 50 % идентичных генов. Общая
частота рождения близнецов составля-
составляет примерно 1 %, из них около 1/3 при-
приходится на монозиготных близнецов.
Известно, что число рождений монози-
монозиготных близнецов сходно в разных по-
популяциях, в то время как для дизигот-
ных эта цифра существенно различает-
различается. Например, в США дизиготные
близнецы рождаются чаще среди не-
негров, чем белых. В Европе частота появ-
появления дизиготных близнецов составля-
составляет 8 на 1000 рождений. Однако в от-
отдельных популяциях их бывает больше.
Самая низкая частота рождения близ-
близнецов присуща монголоидным популя-
популяциям, особенно в Японии (Табл. 2.1).
Отмечается, что частота врожденных
уродств у близнецов, как правило, вы-
выше, чем у одиночно рожденных.
20
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Таблица 2.1. Частота многоплодных рождений
Страна
Испания
Португалия
Франция
Австрия
Швейцария
ФРГ
Швеция
Италия
Англия и Уэллс
США (белые)
США (негры)
(Калифорния)
США (китайцы)
США (японцы)
Япония
Период времени
1951-1953
1955-1956
1946-1951
1952-1956
1943-1948
1950-1955
1946-1955
1949-1955
1946-1955
?
1905-1959
9
?
1955-1962
ДЗ/10000 рождений
59
56
71
75
81
82
86
86
89
67
110
22
21
24
МЗ/10000 рождений
32
36
37
34
36
33
32
37
36
39
39
48
46
40
Полагают, что многоплодие генети-
генетически обусловлено. Однако это спра-
справедливо лишь для дизиготных близ-
близнецов. Факторы, влияющие на часто-
частоту рождения близнецов, в настоящее
время мало изучены. Есть данные, по-
показывающие, что вероятность рожде-
рождения дизиготных близнецов повыша-
повышается с увеличением возраста матери, а
так же порядкового номера рождения.
Влияние возраста матери объясняет-
объясняется, вероятно, повышением уровня го-
надотропина, что приводит к учаще-
учащению полиовуляции. Имеются также
данные о снижении частоты рожде-
рождения близнецов почти во всех индуст-
индустриальных странах.
Близнецовый метод включает в себя
диагностику зиготности близнецов. В
настоящее время используются следу-
следующие методы для ее установления.
1. Полисимптомный метод. Он за-
заключается в сравнении пары близне-
близнецов по внешним признакам (форма
бровей, носа, губ, ушных раковин, цвет
волос, глаз и.т.п.). Несмотря на оче-
очевидное удобство, это метод до извест-
известной степени субъективный и может да-
давать ошибки.
2. Иммуногенетический метод. Более
сложный, он основывается на анализе
групп крови, белков сыворотки крови,
лейкоцитарных антигенов, чувстви-
чувствительности к фенилтиокарбамиду и др.
Если у обоих близнецов по этим при-
признакам различий нет, их считают моно-
монозиготными.
Для монозиготных близнецов веро-
вероятность сходства по всем показателям
равна. Вероятность того, что данная
пара близнецов является дизиготной,
равна:
d
а вероятность того, что близнецы мо-
монозиготные, равна Рт=1—Р^, где Рт —
вероятность монозиготности, Pj— веро-
вероятность дизиготности. Предположим,
что P(j = 0,00389, а Рт - 0,99611. Эти
данные позволяют заключить, что дан-
данная пара близнецов является монози-
монозиготной.
3. Достоверным критерием зиготнос-
зиготности близнецов является ириживляемость
Близнецовый метод
21
кусочков кожи. Установлено, что у ди-
зиготных близнецов такая пересадка
всегда заканчивается отторжением, в то
время как у монозиготных пар отмеча-
отмечается высокая приживляемость транс-
трансплантантов.
4. Метод дерматоглифики заключает-
заключается в изучении папиллярных узоров
пальцев, ладоней и стоп. Эти признаки
строго индивидуальны и не изменяются
в течение всей жизни человека. Не слу-
случайно, что эти показатели используются
в криминалистике и в судебной медици-
медицине для опознания личности и установ-
установления отцовства. Сходство дерматогли-
фических показателей у монозиготных
близнецов значительно выше, чем у ди-
зиготных.
5. Близнецовый метод включает так-
также сопоставление групп моно- и дизи-
готных близнецов по изучаемому при-
признаку.
Если какой-либо признак встречает-
встречается у обоих близнецов одной пары, то она
называется конкордантной, если же у
одного из них, то пара близнецов назы-
называется дискордантной. (конкордант-
ность — степень сходства, дискордант-
ность — степень различия).
При сопоставлении моно- и дизигот-
ных близнецов определяют коэффици-
коэффициент парной конкордантности, указыва-
указывающий на долю близнецовых пар, в кото-
которых изучаемый признак проявился у
обоих партнеров. Коэффициент кон-
конкордантности (Кп) выражается в долях
единицы или в процентах и определяет-
определяется по формуле:
п с+д
где С — число конкордантных пар,
Д — число дискордантных пар.
Сравнение парной конкордантности
у моно- и дизиготных близнецов дает
ответ о соотносительной роли наследст-
наследственности и среды в развитии того или
иного признака или болезни. При этом
исходят из предположения о том, что
степень конкордантности достоверно
выше у монозиготных, чем у дизигот-
дизиготных близнецов, если наследственные
факторы имеют доминирующую роль в
развитии признака (табл. 2.2).
Если значение коэффициента кон-
конкордантности примерно близко у моно-
монозиготных и дизиготных близнецов, счи-
считают, что развитие признака определя-
определяется главным образом негенетическими
факторами, т.е. условиями среды.
Если в развитии изучаемого призна-
признака участвуют как генетические, так и не-
негенетические факторы, то у монозигот-
монозиготных близнецов наблюдаются опреде-
определенные внутрипарные различия. При
этом различия между моно- и дизигот-
ными близнецами по степени конкор-
конкордантности будут уменьшаться. В этом
случае считают, что к развитию призна-
признака имеется наследственная предраспо-
предрасположенность.
Для количественной оценки роли на-
наследственности и среды в развитии того
или иного признака используют различ-
различные формулы. Чаще всего пользуются
коэффициентом наследуемости, кото-
который вычисляется по формуле:
н_ КМБ-КДБ
100-КДБ
(в процентах) и:
н _КМБ - КДБ
1-КДБ
(в долях единицы), где Н — коэф-
коэффициент наследуемости. К — коэффи-
коэффициент парной конкордантности в
группе монозиготных (МБ) или дизи-
дизиготных (ДБ) близнецов.
В зависимости от значения Н судят
о влиянии генетических и средовых
факторов на развитие признака. На-
Например, если значение Н близко к 0,
считают, что развитие признака обус-
обусловлено только факторами внешней
22 Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Таблица 2.2. Примеры конкордантности по некоторым признакам
и заболеваниям у МБ и ДБ, %
Признаки
Цвет глаз, волос
Форма губ, ушей
Папиллярные линии
Маниакально-депрессивный психоз
Шизофрения
Эпилепсия
Сахарный диабет
Туберкулез
Ревматизм
Воспаление среднего уха
Косолапость
Врожденный вывих бедра
Корь
Коклюш
Ветряная оспа
Скарлатина
МБ
99,5
97,0
100,0
98,0
92,0
73,1
67,0
60,8
C7,2)
84,0
E8,0)
66,7
47,3
30,1
45,5
41,4
97,4
97,7
92,8
56,4
ДБ
28,0
23.0
65,0
20,0
40,0
15,2
12,1
12,3
A,8)
37,0
B0,0)
23,0
17,3
9,8
18,2
2,8
95,7
92,0
89,2
41,2
среды. При значении Н от 1 до 0,7 —
наследственные факторы имеют доми-
доминирующее значение в развитии при-
признака или болезни, а среднее значение
Н от 0,4 до 0,7 свидетельствует о том,
что признак развивается под действи-
действием факторов внешней среды при нали-
наличии генетической предрасположеннос-
предрасположенности (табл. 2.3).
Рассмотрим несколько примеров.
Как уже было отмечено, группы крови
у человека полностью обусловлены ге-
генотипом и не изменяются под влияни-
влиянием среды. Коэффициент наследуемос-
наследуемости равен 100 %. По некоторым морфо-
морфологическим признакам (форме носа,
бровей, губ и ушей, цвету глаз, волос и
кожи) монозиготные близнецы кон-
кордантны в 97-100 %, а дизиготные в
зависимости от признака — в 70-20 %
случаев. Конкордантность МБ по забо-
заболеваемости шизофренией равна 70 %,
а у ДБ - 13 %. Тогда:
Н =
G0-13)
A00-13)
= 0,65, или 65 %.
В данном случае преобладают гене-
генетические факторы, но существенную
роль играют и условия среды.
С помощью близнецового метода
было выявлено значение генотипа и
среды в патогенезе многих инфекци-
инфекционных болезней. Так, при заболева-
заболевании корью и коклюшем ведущее зна-
значение имеют инфекционные факто-
факторы, а при туберкулезной инфекции —
существенное влияние оказывает ге-
генотип. Исследования, проводимые на
близнецах, помогут ответить на такие
вопросы как: влияние наследствен-
наследственных и средовых факторов на продол-
продолжительность жизни человека, разви-
развитие одаренности, чувствительность к
лекарственным препаратам и др.
В настоящее время близнецовый ме-
метод в генетике человека используется в
Полуляционно-статистический метод
23
сочетании с другими методами генети-
генетического анализа.
Популяционно-
статистический метод
Одним из важных направлений в
современной генетике является попу-
ляционная генетика. Она изучает ге-
генетическую структуру популяций, их
генофонд, взаимодействие факторов,
обусловливающих постоянство и из-
изменение генетической структуры по-
популяций. Под популяцией в генетике
понимается совокупность свободно
скрещивающихся особей одного ви-
вида, занимающих определенный ареал
и обладающих общим генофондом в
ряду поколений. (Генофонд — это вся
совокупность генов, встречающихся
у особей данной популяции).
В медицинской генетике популяци-
онно-статистический метод использу-
используется при изучении наследственных бо-
болезней населения, частоты нормаль-
нормальных и патологических генов, геноти-
генотипов и фенотипов в популяциях раз-
различных местностей, стран и городов.
Кроме того, этот метод изучает законо-
закономерности распространения наследст-
наследственных болезней в разных по строе-
строению популяциях и возможность про-
прогнозировать их частоту в последую-
последующих поколениях.
Популяционно-статистический ме-
метод используется для изучения:
а) частоты генов в популяции, вклю-
включая частоту наследственных болезней;
б) закономерности мутационного
процесса;
в) роли наследственности и среды в
возникновении болезней с наследст-
наследственной предрасположенностью;
г) влияния наследственных и средо-
вых факторов в создании фенотипиче-
ского полиморфизма человека по мно-
многим признакам и др.
Таблица 2.3. Наследуемость
некоторых признаков человека,
определенная близнецовым
методом
Признак
Телосложение
Рост в положении сидя
Вес
Головной индекс
Ментальный возраст по Бинс
IQno Бине
IQ по Отису
Вербальные способности
Арифметические способности
Способности к естественным наукам
Способности к истории и литературе
Орфографические способности
Скорость постукивания ногой
Наследуе-
Наследуемость
0,81
0,76
0,78
0,75
0,65
0,68
0,80
0.68
0,12
0,34
0,45
0,53
0,50
Использование популяционно-ста-
тистического метода включает пра-
правильный выбор популяции, сбор мате-
материала и статистический анализ полу-
полученных результатов.
В основе метода лежит закономер-
закономерность, установленная в 1908 г. англий-
английским математиком Дж. Харди и немец-
немецким врачом В. Вайнбергом для идеаль-
идеальной популяции. Обнаруженная ими
закономерность получила название за-
закона Харди — Вайнберга.
Для идеальной популяции харак-
характерны следующие черты: большая
численность, свободное скрещивание
(панмиксия) организмов, отсутствие
отбора и мутационного процесса, от-
отсутствие миграций в популяцию и из
нее. В идеальной популяции соотно-
соотношение частоты доминантных гомози-
гомозигот (АА), гетерозигот (Аа) и рецес-
рецессивных гомозигот (аа) сохраняется
постоянным из поколения в поколе-
поколение, если никакие эволюционные
факторы не нарушают это равнове-
равновесие. В этом основной смысл закона
24
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Харди-Вайнберга. При изменении
любой из перечисленных черт соот-
соотношение численности генотипов в
популяции нарушается. Факторы,
стимулирующие сдвиг равновесия, —
родственные браки, мутации, дрейф
генов, отбор, миграции и другие. За-
Закон Харди-Вайнберга является осно-
основой при рассмотрении генетических
преобразований, происходящих в ес-
естественных и искусственно создан-
созданных популяциях растений, животных
и человека.
Соотношение численности разных
генотипов и фенотипов в панмиктиче-
ской популяции определяется по фор-
формуле бинома Ньютона:
где р — частота доминантного алле-
аллеля A, q — частота рецессивного аллеля
а, р2 — частота генотипа А А (гомозигот
по доминантному аллелю), q — частота
генотипа аа (гомозигот по рецессивно-
рецессивному аллелю)
И, следовательно, в соответствии с
законом Харди-Вайнберга частота до-
доминантных гомозигот (АА) равна ква-
квадрату вероятности встречаемости до-
доминантного аллеля, частота гетерози-
гот (Аа) - удвоенному произведению
вероятности встречаемости доминант-
доминантного и рецессивного аллелей. Частота
встречаемости рецессивных гомозигот
(аа) равна квадрату вероятности ре-
рецессивного аллеля.
Таким образом, популяционно-ста-
тистический метод дает возможность
рассчитать в популяции человека час-
частоту нормальных и патологических ге-
нов-гетерозигот, доминантных и ре-
рецессивных гомозигот, а также частоту
нормальных и патологических фено-
фенотипов, т.е. определить генетическую
структуру популяции.
Рассмотрим конкретный пример:
В одном из городов при обследова-
обследовании на резус-фактор 16 % людей оказа-
оказалась резус-отрицательными и 84 % —
резус-положительными. Известно, что
резус-положительный фактор обус-
обусловлен доминантным аутосомным ге-
геном С, а резус-отрицательный фактор —
рецессивным геном с. Носители резус-
положительного фактора могут иметь
генотип СС или Сс. Чтобы определить,
какая часть из них гомо- и гетерозигот-
гетерозиготна, используем формулу Харди-Вайн-
Харди-Вайнберга:
Р2СС: 2pq Сс: q'cc = 1
Гомозиготы по рецессивному алле-
аллелю _составляют 16% или 0,16; отсюда
q =~7o7l6 = 0,40 (или 40 %). Итак, час-
частота рецессивного аллеля в популяции
составляет 40%. Как определить часто-
частоту доминантного аллеля? Исходя из
того, что p + q=l,aq = 0,40, то р = 1 —
0,40 = 0,60 или 60 %. Процент в попу-
популяции зигот СС и Сс вычисляем следу-
следующим образом:
СС = р2 = @,60J= 0,36 или 36%; Сс =
2pq = или 2 х 0,60 х 0,40 = 0,48 или 48 %.
Следовательно, среди обследован-
обследованного населения положительный резус-
фактор имели 36 % с генотипом СС и
48 % с генотипом Сс. В итоге 84 % на-
населения были резус-положительными,
а 16% — резус-отрицательными (сс).
Подобные расчеты широко исполь-
используются в медико-генетических иссле-
исследованиях популяций. Вместе с тем
следует отметить, что в малочислен-
малочисленных популяциях человека закон Хар-
Харди-Вайнберга не применим, т.к. стати-
статистические закономерности, на которых
он основан, не имеют значения в слу-
случае малых чисел.
Важным фактором, влияющим на
частоту аллелей в малочисленных по-
Полуляционно-статистический метод
25
пуляциях и в изолятах являются гене-
тико-автоматические процессы или
дрейф генов. Это явление было описа-
описано в 30-хх гг. Н.П. Дубининым и
Д.Д. Ромашовым (СССР) и С.Райтом
и Р.Фишером (США). Оно выражает-
выражается в случайных изменениях частоты
аллелей, не связанных с их селекцион-
селекционной ценностью и действием естествен-
естественного отбора. Из-за дрейфа генов адап-
адаптивные аллели могут быть элиминиро-
элиминированы из популяции, а менее адаптив-
адаптивные и даже патологические (в силу
случайных причин) могут сохраниться
и достигнуть высоких концентраций.
В результате в популяции может про-
происходит!) быстрое и резкое возраста-
возрастание частот редких аллелей.
Генетико-автоматические процессы
наиболее интенсивно протекают при
неравномерном размножении особей в
популяции. Колебания численности
популяции нередко наблюдается у на-
насекомых, грызунов и других животных
в виде т.н. "волн жизни". В отдельные
благоприятные годы численность их
сильно возрастает, а затем резко пада-
падает. Причинами могут быть развитие
эпидемических заболеваний, нехватка
пищи, понижение температуры и др. В
результате спада численности (или в
изолированных популяциях) умень-
уменьшается гетерозиготность и возрастает
генетическая однородность популя-
популяции.
Примером действия дрейфа генов в
человеческих популяциях может слу-
служить "эффект родоначальника". Он
наблюдается, если структура популя-
популяции формируется под влиянием алле-
аллелей ограниченного числа семей. В та-
таких популяциях нередко наблюдается
высокая частота аномального гена, со-
сохранившегося в результате случайно-
случайного дрейфа генов. Возможно, что след-
следствием дрейфа генов является разная
частота резус отрицательных людей в
Европе A4 %) и в Японии A %), не-
неравномерное распространение на-
наследственных болезней по разным
группам населения земного шара. На-
Например, в некоторых популяциях
Швеции широко распространен ген
ювенильной амавротической идио-
идиотии, в Южной Африке — ген порфи-
рии, в Швейцарии — ген наследствен-
наследственной глухоты и др.
Близкородственные браки (инбри-
(инбридинг) значительно влияют на геноти-
пический состав популяции. Такие
браки чаще всего заключаются между
племянницей и дядей, между двоюрод-
двоюродными братом и сестрой. Близкородст-
Близкородственные браки запрещены во многих
странах из-за высокой вероятности
рождения детей с наследственной па-
патологией : родственники, имея общее
происхождение, могут быть носителя-
носителями одного и того же рецессивного па-
патологического гена, и при браке двух
здоровых гетерозигот вероятность
рождения больного ребенка становит-
становится высокой.
Новые гены могут поступать в попу-
популяцию в результате миграции (потока
генов), когда особи из одной популя-
популяции перемещаются в другую и скрещи-
скрещиваются с представителями данной по-
популяции. Реальные популяции редко
бывают полностью изолированными.
Всегда происходит некоторое передви-
передвижение особей из одной популяции в
другую. Такое передвижение может
быть не только активным, но и пассив-
пассивным (перенос семян птицами). Иногда
человек умышленно перемешивает по-
популяции. Например, в Сибири для
улучшения местных соболей в их по-
популяции выпускают баргузинских со-
соболей с очень темной окраской меха,
более ценимой в меховой промышлен-
промышленности. Это приводит к изменению час-
26
тоты аллелей в основной популяции и
среди "иммигрантов". В локальных по-
популяциях частота аллелей может изме-
изменяться, если у "старожилов" и при-
пришельцев исходные частоты аллелей
различны. Аналогичные процессы
происходят и в человеческих сообще-
сообществах.
В США потомство от смешанных
браков между белыми и неграми отно-
относится к негритянскому населению. По
данным Ф. Айала и Дж. Кайгера
A988) частота аллеля, контролирую-
контролирующего резус-фактор у белого населения,
составляет 0,028. В африканских пле-
племенах, от которых происходит совре-
современное негритянское население, час-
частота этого аллеля равна 0,630. Предки
современных негров США были выве-
вывезены из Африки 300 лет (около 10 по-
поколений) назад. Частота аллеля у со-
современного негритянского населения
Америки составляет 0,446. Таким об-
образом, поток генов от белого населе-
населения к негритянскому шел со скоро-
скоростью 3,6 % за 1 поколение. В результа-
результате через 10 поколений доля генов аф-
африканских предков составляет сейчас
0,694 общего числа генов современно-
современного негритянского населения США.
Около 30 % генов американские негры
унаследовали от белого населения.
Очевидно, поток генов между белым и
негритянским населением был значи-
значительным.
Наконец, следует рассмотреть крат-
кратко, как влияют на генетическую струк-
структуру популяций мутационный про-
процесс и отбор. Мутации, как фактор
эволюции, обеспечивают приток но-
новых аллелей в популяцию. По измене-
изменению генотипа мутации подразделяют
на генные (или точковые), внугрихро-
мосомные и межхромосомные, геном-
геномные (изменение числа хромосом.). Ген-
Генные мутации могут быть прямыми
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
(А>а) и обратные (а>А). Частота воз-
возникновения прямых мутаций значи-
значительно выше обратных. Одни и те же
гены могут мутировать многократно.
Кроме того, один и тот же ген может
изменяться в несколько аллельных со-
состояний, образуя серию множествен-
множественных аллелей (A>at, a2, а}... а*). Изуче-
Изучение частоты мутаций у человека, обус-
обусловливающих такие тяжелые болезни,
как гемофилия, ретинобластома, пиг-
пигментная ксеродерма и др. дают основа-
основания полагать, что частота возникнове-
возникновения патологических мутаций отдель-
отдельного гена составляет около 1-2 на сто
тысяч гамет за поколение. Учитывая
общее количество генов у человека
(около 100 тыс.), суммарная мутабиль-
ность — величина немалая.
Частота мутаций может значитель-
значительно возрасти при действии на организм
некоторых физических и химических
факторов — мутагенов. Химические
мутагены обнаружены среди промыш-
промышленных ядов, инсектицидов, гербици-
гербицидов, пищевых добавок и лекарств.
Большинство канцерогенных веществ
также обладают мутагенным действи-
действием. Кроме того, многие биологические
факторы, например, вирусы, живые
вакцины, а также гистамин, стероид-
стероидные гормоны, вырабатываемые в орга-
организме человека, могут вызывать мута-
мутации. Сильными мутагенами являются
различные виды излучений (рентге-
(рентгеновские лучи, гамма — лучи, а и р-час-
тицы, нейтроны и др.), способные про-
продуцировать генные и хромосомные му-
мутации у человека, о чем свидетельству-
свидетельствуют последствия аварии на Чернобыль-
Чернобыльской АЭС (см. главу 6).
К факторам, нарушающим постоян-
постоянство генетической структуры популя-
популяций, относится и естественный отбор,
вызывающий направленное изменение
генофонда путем элиминации из попу-
Цитогенетический метод
27
ляции менее приспособленных особей
или снижения их плодовитости. Рас-
Рассмотрим влияние отбора на примере
доминантной патологии — ахондро-
плазии (карликовости). Эта болезнь
хорошо изучена в популяциях Дании.
Больные имеют пониженную жизне-
жизнеспособность и умирают в детском воз-
возрасте, т.е. устраняются естественным
отбором из популяции. Выжившие
карлики редко вступают в брак и име-
имеют мало детей. Анализ показывает, что
около 20 % генов ахондроплазии не пе-
передаются от родителей детям, а 80 %
этих генов элиминируются из популя-
популяции. Из этих данных следует, что ахон-
дроплазия не оказывает существенно-
существенного влияния на структуру популяции.
Большинство мутантных генотипов
имеют низкую селекционную цен-
ценность и попадают под действие отбора.
По данным В. Маккьюсика A968) сре-
среди мутантов около 15 % плодов поги-
погибают до рождения, 3 % — умирают, не
достигнув половой зрелости, 20 %
умирают до вступления в брак, в 10%
случаев брак остается бесплодным.
Однако не каждый мутантный ген
снижает селективную ценность при-
признака. В ряде случаев патологический
ген в гетерозиготном состоянии может
повышать жизнеспособность особи.
В качестве примера рассмотрим серпо-
видноклеточную анемию. Известно,
что это заболевание распространено в
странах Африки и Азии. У людей, го-
гомозиготных по аллелю HbS, вырабаты-
вырабатывается гемоглобин, отличный от нор-
нормального, обусловленного аллелем
HbS. Гомозиготы HbSHbS погибают, не
достигнув половозрелости. Гетерози-
готы HbAHbS более устойчивы к маля-
малярии, чем нормальные гомозиготы
HbAHbA и HbSHbS. Поэтому в районах
распространения болезни гетерозиго-
ты имеют селективное преимущество.
Отбор работает в пользу гетерозигот.
В районах, где не было малярии, гомо-
гомозиготы HbAHbA обладают одинаковой
приспособленностью с гетерозигота-
ми. При этом отбор направлен против
рецессивных гомозигот. В некоторых
районах Африки гетерозиготы состав-
составляют до 70 %. населения. "Платой" за
приспособленность к условиям суще-
существования служит т.н. генетический
груз, т.е. накопление вредных мутаций
в популяции.
Цитогенетический метод
Основа метода — микроскопическое
изучение хромосом человека. Цитогене-
тические исследования стали широко
использоваться с начала 20-х гг. XX в.
для изучения морфологии хромосом
человека, подсчета хромосом, культи-
культивирования лейкоцитов для получения
метафазных пластинок.
Развитие современной цитогенети-
ки человека связано с именами цито-
цитологов Д.Тио и А.Левана. В 1956 г. они
первыми установили, что у человека
46 (а не 48, как думали раньше) хромо-
хромосом, что положило начало широкому
изучению митотических и мейотичес-
ких хромосом человека.
В 1959 г. французские ученые
Д. Лежен, Р.Тюрпен и М. Готье устано-
установили хромосомную природу болезни
Дауна. В последующие годы были
описаны многие другие хромосомные
синдромы, часто встречающиеся у че-
человека. Цитогенетика стала важней-
важнейшим разделом практической медици-
медицины. В настоящее время цитогенетиче-
цитогенетический метод применяется для диагнос-
диагностики хромосомных болезней, состав-
составления генетических карт хромосом,
изучения мутационного процесса и
других проблем генетики человека.
В 1960 г. в г. Денвере (США) была
разработана первая Международная
28 Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Таблица 2.4. Классификация хромосом человека по размеру
и расположению центромеры
Группа хромосом
АA)
В(Н)
С(Ш)
D(IV)
E(V)
F(VI)
Х-хромосома
(относится
к фуппе III)
Y-хромосома
Номер по кариотипу
1,2,3
4,5
6-12
13-15
16-18
19-20
23
23
Размер, мкм
11-8,3
7,7
7,2-5,7
4,2
3,6-3,2
2,3-2,8
Характеристика хромосом
1 и 3 - метацентрические,
2 - крупная субметацентрическая
Крупные субметацентрические
Средние субметацентрические
Средние акроцентрические
Мелкие субметацентрические
Самые мелкие метацентрические
Средняя субметацешрическая
Мелкая акроцентрическая
классификация хромосом человека. В
ее основу легли размеры хромосом и
положение первичной перетяжки —
центромеры. Все хромосомы по форме
разделены на метацентрические, суб-
субметацентрические и акроцентрические
и подразделены на 7 групп, обозначен-
обозначенных латинскими буквами А, В, С, D, Е,
F и G. Каждая пара хромосом была на-
наделена порядковым номером от 1 до
22, выделены отдельно и поименованы
латинскими буквами — X и Y половые
хромосомы (Табл. 2.4).
В 1971 г. на IV Пражской конферен-
конференции генетиков в дополнении к Денвер-
Денверской классификации были представле-
представлены методы дифференциальной окрас-
окраски хромосом, благодаря которым каж-
каждая хромосома приобретает свой непо-
неповторимый рисунок, что помогает точ-
точной идентификации.
Основные сведения о морфологии
хромосом человека получены при изу-
изучении их в метафазах митоза и профа-
зе-метафазе мейоза. При этом важно,
чтобы количество делящихся клеток,
было достаточно высоко. Важнейшие
цитогенетические работы выполнены
на лимфоцитах периферической кро-
крови, поскольку культивирование лим-
лимфоцитов в течение 2-3 суток в присут-
присутствии фитогемагглютинина позволяет
получить множество метафазных пла-
пластинок для хромосомного анализа.
Цитогенетическому анализу под-
подвергают однослойные метафазные
пластинки с раздельно лежащими хро-
хромосомами. Для этого делящиеся клет-
клетки обрабатывают колхицином и неко-
некоторыми другими химическими веще-
веществами (гипотоническим раствором
солей, метанол-уксусным фиксатором
и др.).
Важным этапом цитогенетического
анализа является окраска полученных
препаратов. Ее проводят простыми,
дифференциальными и флюоресцент-
флюоресцентными методами.
Простая окраска обеспечивает груп-
групповую идентификацию хромосом. Ис-
Используется она для количественного
учета хромосомных аномалий при оп-
определении мутагенности среды (дейст-
(действия радиации, химических мутагенов
и др.). С помощью этого типа окраски
были открыты многие хромосомные
болезни, а также хромосомные аберра-
аберрации), вызывающие самопроизвольные
аборты, врожденные пороки развития,
канцерогенез и т.п.
В 70-е гг. XX в. в медицинской прак-
практике начали применяться методы диф-
Цитогеиетический метод
29
ференциального окрашивания, выяв-
выявляющие структурную разнородность
хромосом по длине, что выражается в
виде чередования светлых и темных
полос (эу- и гетерохроматических рай-
районов). Отмечается, что протяженность
и рисунок полос специфичны для каж-
каждой хромосомы.
Дифференциальное окрашивание
хромосом можно проводить рядом
способов. Первоначально использова-
использовали акрихин- иприт — флюоресцентное
алкилирующее вещество (Q-метод).
Действие его основано на способности
метафазных хромосом дифференци-
дифференциально связывать флюорохромы. После
окрашивания акрихин-ипритом сег-
сегменты приобретают яркое флюоресци-
флюоресцирующее свечение. Для просмотра та-
таких препаратов используют люминес-
люминесцентный микроскоп.
В дальнейшем был разработан спо-
способ окраски хромосом без флюорес-
флюоресцентных красителей. Это — G-окраска
(краситель Гимза). После предвари-
предварительной инкубации в солевом растворе
хромосомы обрабатываются протеа-
зой. В результате они приобретают сег-
сегментированный вид благодаря чередо-
чередованию темно и светлоокрашенных уча-
участков. Механизм образования сегмен-
сегментов пока недостаточно ясен. Предпола-
Предполагается, что окрашенные сегменты —
это гетерохроматиновые участки с по-
повторяющимися последовательностями
ДНК, а неокрашенные — эухроматино-
вые районы с кодирующими последо-
последовательностями ДНК (рис. 2.9).
К разновидностям дифференциаль-
дифференциального окрашивания по методу Гимзы
относятся R-окрашиваемость и С-ок-
рашиваемость. Эти разновидности
дифференциального окрашивания
получают при определенном измене-
изменении времени инкубации препаратов,
окрашенных по методу Гимзы. В пер-
первом случае распределение окрашен-
окрашенных и неокрашенных сегментов будет
обратным тому, что наблюдается при
G и Q-окрашивании. На R-окрашен-
ных хромосомах гетерохроматиновые
и околоцентромерные районы оста-
остаются светлыми. В случае же С-окрас-
ки выявляются районы структурного
гетерохроматина, наиболее устойчи-
устойчивого к химическим и физическим по-
повреждениям. В аутосомах и Х-хромо-
сомах человека эти районы локализо-
локализованы в околоцентромерных участках,
а в Y-хромосоме — в дистальной поло-
половине длинного плеча. Наиболее круп-
крупные блоки С-хроматина имеются в ау-
аутосомах 1, 9 и 16 в области их вторич-
вторичных перетяжек, а также в Y-хромосо-
Y-хромосоме. Самыми мелкими центромерными
блоками обладают Y-хромосома и ау-
тосома 2 (рис. 2.10).
Одной из особенностей хромосом
человека является асинхронность (не-
(неодновременность) репликации по дли-
длине. В каждой хромосоме есть рано и по-
поздно реплицирующиеся участки. Для
выявления последовательности репли-
репликации применяется 5-бромдезоксиури-
дин — аналог тимина. Включившие его
участки окрашиваются слабо.
Применяется 5-бром-дезоксиури-
дин и для дифференциальной окраски
сестринских хроматид, если он вводит-
вводится на полный клеточный цикл. В этом
случае вновь образуемая хроматида,
включит этот аналог тимина и будет
окрашена слабо, а другая (старая) ок-
окрасится интенсивно (рис. 2.11). Этот
метод позволяет выявлять участки об-
обмена между сестринскими хроматида-
ми (СХО).
При воздействии различными мута-
мутагенными факторами число СХО уве-
увеличивается, следовательно, этот метод
пригоден для изучения мутационного
процесса у человека.
30
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
Рис. 2.9. Схематическое изображение хромосом человека
при дифференциальной окраске (G-метод)
Метод генетики соматических клеток
31
1 2 3
4-5
ШИШИ""
е-12
13-15
!6
17-18
19-20 21-22 X Y
Рис. 2.10. Дифференциальное окрашивание хромосом человека по С - технике
Успехи молекулярной цитогенетики
человека позволяют разрабатывать но-
новые методы изучения хромосом. Так,
следует отметить метод флюоресцент-
флюоресцентной гибридизации in situ (FISH), кото-
который дает возможность исследовать
широкий круг вопросов : от локализа-
локализации гена до расшифровки сложных пе-
перестроек между несколькими хромосо-
хромосомами. Метод FISH может применяться
и для диагностики анеуплоидий в ин-
интерфазных ядрах (см. главу 6).
Таким образом, соединение цитоге-
нетических и молекулярно-генетиче-
ских методов в генетике человека де-
делает почти неограниченными воз-
возможности диагностики хромосомных
аномалий.
Метод генетики
соматических клеток
Тот факт, что соматические клетки
несут в себе весь объем генетической
информации, дает возможность изу-
32
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
I .Г
Рис. 2.11. Метафазная пластинка с дифференциальной окраской
сестринских хроматид (Метод СХО)
чать на них генетические закономер-
закономерности всего организма.
Основу метода составляет культиви-
культивирование отдельных соматических кле-
клеток человека и получение из них клонов,
а так же их гибридизацию и селекцию.
Соматические клетки обладают ря-
рядом особенностей:
- быстро размножаются на питатель-
питательных средах;
- легко клонируются и дают генети-
генетически однородное потомство;
- клоны могут сливаться и давать ги-
гибридное потомство;
- легко подвергаются селекции на
специальных питательных средах;
- клетки человека хорошо и долго
сохраняются при замораживании.
Соматические клетки человека по-
получают из разных органов — кожи, ко-
костного мозга, крови, ткани эмбрионов.
Однако чаще всего используют клетки
соединительной ткани (фибробласты)
и лимфоциты крови.
С помощью метода гибридизации
соматических клеток:
а) изучают метаболические процес-
процессы в клетке;
б) выявляют локализацию генов в
хромосомах;
в) исследуют генные мутации;
г) изучают мутагенную и канцеро-
канцерогенную активность химических ве-
веществ.
В 1960 г. было показано, что совмест-
совместно культивируемые клетки различных
линий могут сливаться, образуя гибри-
гибриды, содержащие геномы обеих роди-
родительских форм. Первые такие гибриды
были получены при слиянии клеток
Метод генетики соматических клеток
33
разных линий мышей. Наряду с внут-
внутривидовыми получены и межвидовые
гибриды, например, между клетками
человека и мыши, мыши и хомячка,
мыши и курицы и др. Образование ги-
гибридных клеток происходит чаще, если
в культуру добавлены некоторые веще-
вещества (например, полиэтиленгликоль)
или инактивированный вирус Сендай.
При гибридизации соматических
клеток двух разных линий образуются
гетерокарионы — клетки, которые со-
содержат оба родительских ядра. Затем в
результате митоза и деления образу-
образуются две одноядерные клетки — син-
карионы, имеющие хромосомы обоих
родительских клеток.
В течение первых делений гибрид-
гибридной клетки, не ясно почему, происхо-
происходит потеря хромосом одного из видов.
Так, у гибридов мышь-хомячок элими-
элиминируются хромосомы мыши. Если
присутствие продукта изучаемого гена
коррелирует с наличием определенной
хромосомы в гибриде, то можно пред-
предположить, что этот ген локализован в
данной хромосоме.
Потеря хромосом в гибридных клет-
клетках мышь — человек происходит слу-
случайно. Какая хромосома человека со-
сохранится — предугадать невозможно.
Через определенное число поколений в
гибридной клетке сохранятся все хро-
хромосомы мыши и несколько (в среднем
7) хромосом человека. Однако некото-
некоторые клетки могут содержать одну-две
пары хромосом, другие — 7, а некото-
некоторые — до 20 хромосом человека. Вмес-
Вместе с тем из всей популяции гибридных
клеток можно отобрать стабильные
клоны (клон — потомство одной клет-
клетки), содержащие конкретные человече-
человеческие хромосомы, и провести в них кар-
картирование генов.
Гибридные клетки человека и мыши
имеют 43 пары хромосом: 23 от челове-
человека и 20 от мыши. В дальнейшем проис-
происходит элиминация хромосом того ви-
вида, клетки которого медленнее размно-
размножаются. При этом хромосомы мыши
сохраняются, а хромосомы человека
утрачиваются.
Функционирующие в гибридных
клетках хромосомы синтезируют опре-
определенные белки. Фенотипически хро-
хромосомы мыши и человека отличаются.
Нетрудно определить, какие хромосо-
хромосомы присутствуют в гибриде и выяс-
выяснить, синтез каких белков связан с
данными хромосомами человека.
Обычно гибридные клетки теряют
хромосомы целиком, поэтому, если ка-
какие-либо гены присутствуют или от-
отсутствуют вместе, то они могут быть
отнесены к одной хромосоме. Это поз-
позволяет картировать хромосомы чело-
человека. В ряде случаев для картирования
используют хромосомные перестрой-
перестройки, что дает возможность установить
локализацию генов в определенном
участке хромосомы, определить после-
последовательность их расположения, т. е.
построить карты хромосом человека.
В настоящее время выяснено, что в
Х-хромосоме локализовано 95 генов, в
1-й аутосоме — 24 гена. Ген, определя-
определяющий группы крови по системе АВО,
расположен в 9-й хромосоме, группы
крови по системе MN во 2-й хромосо-
хромосоме, а по системе резус- фактора (Rh) —
в 1-й хромосоме.
Использование метода гибридиза-
гибридизации соматических клеток дает возмож-
возможность изучать механизмы первичного
действия генов и их взаимодействия,
что расширяет возможности точной
диагностики наследственных болезней
на биохимическом уровне.
Использование новых методов и под-
подходов к картированию хромосом позво-
позволило обнаружить в геноме человека
сверхизменчивые участки ДНК (мини-
34
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
сателлиты), характерные для каждого
человека. Одновременно были выделе-
выделены последовательности ДНК, изменяю-
изменяющиеся с повышенной частотой. Они ло-
локализованы по всему геному и имеют
разное число копий. На основе сверхиз-
сверхизменчивых последовательностей ДНК
созданы маркеры или зонды ДНК. Эти
маркеры метят радиоактивным изото-
изотопом и добавляют к ДНК, обработанной
специальным образом. Маркеры "узна-
"узнают" сходные с ними сверхизменчивые
участки на ДНК и присоединяются к
ним. Эти участки становятся радиоак-
радиоактивными и их можно выявить методом
радиоавтографии. Число и распределе-
распределение таких мест присоединения индиви-
индивидуально у каждого человека.
Изучение сверхизменчивых после-
последовательностей ДНК позволяет уста-
установить место гена на хромосоме. Так,
недавно ученые установили, что ген
гиперкератоза (патологическое утол-
утолщение кожных покровов и образова-
образование язв на коже) человека лежит — в
17-й хромосоме, а ген болезни Альц-
геймера (старческое слабоумие) — в
21-й хромосоме.
Помимо этого в настоящее время ак-
активно изучается наследование сахар-
сахарного диабета, шизофрении и ряда дру-
других заболеваний.
Исследование сверхизменчивых
ДНК в клетках опухолей позволяет
обнаружить перестройки хромосом на
ранних стадиях развития опухоли, что
важно для их ранней диагностики.
В последние годы созданы более со-
совершенные генетические карты генома
человека с использованием маркерных
генов и последних достижений моле-
молекулярной генетики.
Биохимический метод
Причиной многих врожденных на-
нарушений метаболизма являются раз-
различные дефекты ферментов, возника-
возникающие вследствие изменяющих их
структуру мутаций. Биохимичские по-
показатели (первичный продукт гена, на-
накопление патологических метаболитов
внутри клетки и во всех клеточных
жидкостях больного) более точно от-
отражают сущность болезни по сравне-
сравнению с показателями клиническими,
поэтому их значение в диагностике на-
наследственных болезней постоянно воз-
возрастает. Использование современных
биохимических методов (электрофо-
(электрофореза, хроматографии, спектроскопии и
др.) позволяют определять любые ме-
метаболиты, специфические для кон-
конкретной наследственной болезни.
Предметом современной биохими-
биохимической диагностики являются специ-
специфические метаболиты, энзимопатии,
различные белки.
Объектами биохимического анализа
могут служить моча, пот, плазма и сы-
сыворотка крови, форменные элементы
крови, культуры клеток (фиброблас-
ты, лимфоциты).
Для биохимической диагностики ис-
используются как простые качественные
реакции (например, хлорид железа для
выявления фенилкетонурии или дини-
трофенилгидразин для выявления ке-
токислот), так и более точные методы.
Например, с помощью тонкослойной
хроматографии мочи и крови можно
диагностировать нарушение обмена
аминокислот, олигосахаридов, мукопо-
лисахаридов. Газовая хроматография
применяется для выявления наруше-
нарушений обмена органических кислот и т.д.
Показаниями для использования
биохимических методов у больных с
наследственным нарушением обмена
веществ являются такие симптомы,
как судороги, кома, рвота, желтуха,
специфический запах мочи и пота, ос-
остановка роста, нарушение физическо-
Молекулярно-генетические методы
35
го развития, непереносимость некото-
некоторых продуктов и лекарств.
Биохимические методы применя-
применяются и для диагностики гетерозигот-
гетерозиготных состояний у взрослых. Известно,
что среди здоровых людей всегда име-
имеется большое число так называемых
носителей патологического гена (ге-
(гетерозиготное носительство). Хотя та-
такие люди внешне здоровы, вероят-
вероятность появления заболевания у их ре-
ребенка всегда существует. В связи с
этим, выявление гетерозиготного но-
сительства - важная задача медицин-
медицинской генетики.
Понятно, что если в брак вступают
гетерозиготные носители какого-либо
заболевания, то риск рождения боль-
больного ребенка в такой семье составит
25%. Шансы на встречу двух носите-
носителей одинакового патологического гена
выше, если в брак вступают родствен-
родственники, т.к. они могут унаследовать один
и тот же рецессивный ген от своего об-
общего предка.
Предположить гетерозиготное но-
носительство у женщины можно, если:
— ее отец поражен наследственной
болезнью;
— у женщины родились больные сы-
сыновья;
— женщина имеет больного брата
или братьев;
— у двух дочерей женщины роди-
родились больные сыновья (или сын);
— у здоровых родителей родился
больной сын, а у матери в родословной
есть больные мужчины.
Выявление гетерозиготных носите-
носителей того или иного заболевания воз-
возможно путем использования биохими-
биохимических тестов (прием фенилаланина
для выявления фенилкетонурии, при-
прием сахара — сахарного диабета и.т.д.),
микроскопического исследования кле-
клеток крови и тканей, определения ак-
активности фермента, измененного в ре-
результата мутации.
Известно, что заболевания, в основе
которых лежит нарушение обмена ве-
веществ, составляют значительную часть
наследственной патологии (фенилке-
тонурия, галактоземия, алкаптонурия,
альбинизм и др.). Так, гетерозиготные
носители фенилкетонурии реагируют
на введение фенилаланина более силь-
сильным повышением содержания амино-
аминокислоты в плазме, чем нормальные го-
гомозиготы (болезнь обусловлена рецес-
рецессивным аллелем).
Биохимический метод широко при-
применяется в медико-генетическом кон-
консультировании для определения риска
рождения больного ребенка. Успехи в
области биохимической генетики спо-
способствуют более широкому внедре-
внедрению диагностики гетерозиготного но-
сительства в практику. Еще недавно
можно было диагностировать не более
10-15 гетерозиготных состояний, в на-
настоящее время — более 200. Однако
следует отметить, что до сих пор име-
имеется немало наследственных заболева-
заболеваний, для которых методы гетерозигот-
гетерозиготной диагностики еще не разработаны.
Молекулярно-
генетические методы
Конечный итог молекулярно-гене-
тических методов — выявление изме-
изменений в определенных участках ДНК,
гена или хромосомы. В их основе ле-
лежат современные методики работы с
ДНК или РНК. В 70-80 гг. в связи с
прогрессом в молекулярной генетике и
успехами в изучении генома человека
молекулярно-генетический подход на-
нашел широкое применение.
Начальным этапом молекулярно-ге-
нетического анализа является получе-
получение образцов ДНК или РНК. Для это-
этого используют геномную ДНК (вся
36
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
ДНК клетки) или отдельные ее фраг-
фрагменты. В последнем случае, чтобы по-
получить достаточное количество таких
фрагментов, необходимо, амплифици-
ровать (размножить) их. Для этого
пользуются полимеразной цепной ре-
реакцией — быстрым методом фермента-
ферментативной репликации определенного
фрагмента ДНК. С его помощью мож-
можно амплифицировать любой участок
ДНК, расположенный между двумя
известными последовательностями.
Анализировать огромные молекулы
ДНК в том виде, в котором они суще-
существуют в клетке, невозможно. Поэтому
прежде их необходимо разделить на
части, обработать разнообразными ре-
стриктазами — бактериальными эндо-
нуклеазами. Эти ферменты способны
разрезать двойную спираль ДНК, при-
причем места разрыва строго специфичны
для данного образца. Расщепление
ДНК рестриктазами дает характерный
набор фрагментов D-6 пар основа-
оснований), отличающихся по длине.
Фракционирование (т.е. разделение)
фрагментов ДНК по размеру и длине
проводится с помощью электрофореза
на поверхности агарозного или поли-
акриламидного геля. Под действием
электрического поля фрагменты ДНК
начинают перемещаться вниз по гелю
со скоростью, зависящей от их длины.
(Чем короче фрагменты, тем быстрее
они движутся). В результате каждый
фрагмент ДНК занимает определенное
положение в виде дискретной полосы в
конкретном месте геля. Длину каждого
фрагмента можно определить путем
сравнения расстояния, пройденного им
и стандартным (с известными размера-
размерами) отрезком ДНК.
Молекулярно-генетическую диа-
диагностику наследственных болезней ис-
используют и для изучения генома чело-
человека. Чтобы выявить необходимые для
этого специфические фрагменты ДНК,
используют блот-гибридизацию по
Саузерну. Сущность этой методики
кратко состоит в следующем: сначала
осуществляют денатурацию ДНК с об-
образованием одноцепочечных фрагмен-
фрагментов, которые переносят на нитроцел-
люлезный или нейлоновый фильтр в
буферном растворе.
Агарозный гель с фрагментами
ДНК помещают на фильтровальную
бумагу, смоченную концентрирован-
концентрированным солевым раствором. На гель на-
накладывают нитроцеллюлезный
фильтр, а сверху помещают сухую
фильтровальную бумагу, в которую
впитывается солевой раствор. ДНК
переносится вместе с раствором, но за-
задерживается фильтром и практически
полностью оказывается на его поверх-
поверхности. Затем одноцепочечные ДНК
фиксируют на фильтре. Расположение
фрагментов на фильтре точно соответ-
соответствует их расположению в геле.
Чтобы выявить нужные фрагменты,
проводят гибридизацию ДНК с ради-
активным ДНК-зондом или клониро-
клонированным фрагментом ДНК. Нуклеотид-
ная последовательность зонда должна
быть полностью или частично компле-
комплементарна изучаемому участку геном-
геномной ДНК.
Результат гибридизации компле-
комплементарных цепей радиоактивного
ДНК-зонда и фрагмента ДНК обнару-
обнаруживают с помощью радиоавтографии:
каждая комплементарная зонду после-
последовательность ДНК проявляется в ви-
виде радиоактивной полосы.
С помощью метода Саузерна можно
составить рестрикционную карту гено-
генома в участке исследуемого гена и уста-
установить, несет ли данный ген какие-ли-
какие-либо дефекты. Так, разработаны эффек-
эффективные методы синтеза искусственных
ДНК-зондов, которые используются в
Молекулярио-генетические методы
37
пренатальной диагностике наследст-
наследственных заболеваний. Для этого из эмб-
эмбриональных клеток, содержащихся в
амниотической жидкости плода, выде-
выделяют ДНК и гибридизируют ее с помо-
помощью Саузерн-блоттинга с радиоактив-
радиоактивным ДНК-зондом. Аномальный эмб-
эмбрион легко распознается, т.к. его ДНК
будет гибридизоваться только с ДНК-
зондом, комплементарным мутантной
последовательности.
В настоящее время имеются различ-
различные методы выявления мутаций. Их
делят на прямые и косвенные. Прямая
диагностика мутаций включает ряд
методов:
1. Определение нуклеотидной по-
последовательности (секвенирование),
дающее возможность выявить замены
оснований, делеции и вставки в изуча-
изучаемом фрагменте.
2. Выявление нарушения места рес-
рестрикции, с помощью блот-гибридиза-
ции по Саузерну. Около 50% нуклео-
тидных замен ведет к изменению сайта
(места) рестрикции. Это делает воз-
возможным выявить мутацию путем рест-
риктного анализа.
3. Проведение аллелоспецифичес-
кой гибридизации с синтетическими
зондами, что позволяет обнаружить
мутации к геномной ДНК. Последова-
Последовательность оснований в зонде может
быть задана по дефектному или нор-
нормальному варианту гена. В обоих слу-
случаях зонд используется для гибриди-
гибридизации с фрагментами ДНК обследуе-
обследуемого индивида.
4. Химическое и ферментативное
расщепление ДНК в местах непра-
неправильной сшивки оснований выявляет
большую группу мутаций, ведущих к
нестабильности ДНК. Метод заключа-
заключается в электрофорезе двухцепочечной
ДНК в нейтральном или равномерно
денатурирующем геле.
5. Регистрация изменения электро-
форетической подвижности мутант-
ных молекул ДНК.
6. Трансляция белкового продукта
осуществляется в системе in vitro на
основе получения специфической
мРНК с добавлением лизата ретикуло-
цитов. Синтезируемый белок анализи-
анализируют с помощью электрофореза. Изме-
Изменение подвижности белка указывает
на наличие мутации.
К косвенному выявлению мутаций
прибегают в тех случаях, когда нуклео-
тидная последовательность гена еще
не расшифрована, но известно его по-
положение на генетической карте. Техни-
Технические приемы такие же, как и в пря-
прямой диагностике, но добавляется мате-
математический анализ.
Диагностике мутаций способствует
нахождение в геноме полиморфных по
длине рестрикционных фрагментов.
Их можно выявить с помощью блот-
гибридизации по Саузерну.
Другим типам полиморфизма ДНК
являются микросателлиты. Это корот-
короткие моно-, ди-, три- и тетрануклеотид-
ные тандемно повторяющиеся после-
последовательности ДНК. Они используют-
используются в качестве маркерных локусов ал-
лельных вариантов гена или маркеров
дефектных мутаций.
В 1993 г. был идентифицирован ген,
ответственный за возникновение тя-
тяжелого заболевания нервной системы
у человека — хореи Гентингтона (ХГ).
Болезнь, проявляющаяся после 40 лет,
выражается в расстройстве движений,
снижении интеллекта, в нарушении
эмоционально-волевой сферы и др.
Этот недуг наследуется по аутосомно-
доминантному типу со 100 % пенет-
рантностью. Ген локализован в корот-
коротком плече 4-й хромосомы.
Оказалось, что ген ХГ содержит об-
область, в которой нуклеотидная после-
Глава 2. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
довательность представлена много-
многократным повторением трех нуклеоти-
дов — ЦАГ (цитозин-аденин-гуанин)
геномной ДНК. В норме количество
таких повторов колеблется от 11 до 34,
а у больных ХГ их 37-86 (в среднем
45). И, следовательно, хорея Гентинг-
тона относится к наследственным за-
заболеваниям, при котором мутация ге-
гена состоит в экспансии (многократном
увеличении числа копий) тринуклео-
тидных ЦАГ-повторов.
Установлено, что форма болезни бо-
более тяжелая, если ХГ проявляется в
молодом возрасте, наследуется по от-
отцовской линии и нарастает в последу-
последующих поколениях. Ученые пришли к
выводу, что число ЦАГ-повторов тесно
связано и со сроком появления первых
симптомов, и с тяжестью заболевания.
В 1992 г. экспансия тринуклеотид-
ных ЦТГ-повторов была обнаружена в
гене, который вызывает миотоничес-
кую дистрофию. Этот ген, названный
ДМ-1, был картирован на 19-й хромо-
хромосоме. Длина последовательности ЦТГ-
повторов весьма различна. Если в нор-
нормальной популяции она колеблется от
5 до 30, то у больных миотонической
дистрофией, количество повторов мо-
может'достигать многих сотен.
Болезнь наследуется по аутосомно-
доминантному типу, обычно начинает-
начинается в зрелом возрасте и проявляется
прогрессирующей мышечной слабос-
слабостью, а в некоторых случаях задержкой
умственного развития, поражением
скелета, сердечно-сосудистой системы
и глаз. Для миотонической дистрофии
характерно возрастание тяжести болез-
болезни на протяжении трех или четырех по-
поколений. Если в первом поколении бо-
болезнь возникает в зрелом возрасте и
проявляется лишь развитием катарак-
катаракты или легким нарушением сократимо-
сократимости мышц, то в последующих поколе-
поколениях болезнь начинается сразу после
рождения ребенка, у которого развива-
развивается выраженная мышечная слабость,
задержка умственного развития.
В последние годы было показано, что
подобный механизм мутаций характе-
характерен и для ряда других наследственных
заболеваний нервной системы челове-
человека: болезни Кеннеди, синдрома фра-
гильной (ломкой) Х-хромосомы и др.
Вопросы для самоконтроля
1. Перечислите методы изучения генетики чело-
человека.
2. Какой из методов является наиболее старым и
в каких случаях он используется?
3. Что такое конкордантность и дискордант-
ность?
4. Какие задачи решаются популяционно-статис-
тическим методом?
5. Каким методом можно определить наличие
хромосомных аномалий?
6. Как выявляется гетерозиготность по ряду бел-
белков?
7. Для чего используются молекулярно-генетичес-
кие методы?
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Основоположник генетики Грегор
Мендель в 1865 г. впервые доказал, что
каждый признак организма определя-
определяется парой наследственных факторов.
В начале XX в. парные наследствен-
наследственные факторы получили название ал-
лельных генов. Примерно в то же вре-
время было выдвинуто предположение,
что гены расположены в хромосомах,
что и положило начало хромосомной
теории наследственности. Впервые эта
теория получила доказательства в ра-
работах Нобелевского лауреата Томаса
Ханта Моргана и его учеников в 1910 г.
В экспериментах на плодовой мушке
Drosophila melanogaster была показана
взаимосвязь между конкретными ге-
генами и конкретными хромосомами.
Долгое время оставалось неизвест-
неизвестным, что представляет собой вещество,
образующее ген, способное к самореп-
саморепликации, мутациям и фенотипическо-
му проявлению. Первые сведения о
физических и химических основах на-
наследственности были получены при
работе с микроорганизмами : бактери-
бактериями, вирусами и бактериофагами. Эти
организмы, ранее изучавшиеся как
возбудители болезней человека и до-
домашних животных, оказались удобны-
удобными объектами для исследования веще-
вещества наследственности и природы ге-
генетического материала.
В 1928 г. бактериолог Ф. Гриффит
изучал пневмококки, вызывающие вос-
воспаление легких у мышей, для получе-
получения вакцины. Выяснилось, что пневмо-
пневмококки бывают двух типов: бескапсуль-
ные и с полисахаридной капсулой, при-
причем возбудителем смертельных форм
воспаления легких являются бактерии
с полисахаридной капсулой. Если мы-
мышам вводили пневмококки без полиса-
полисахаридной капсулы, то они справлялись
с заболеванием и выживали. При инъ-
инъекции убитых бактерий, независимо от
наличия или отсутствия капсулы на их
поверхности, животные вообще не за-
заболевали воспалением легких. Если же
мышам вводили смесь пневмококков:
живых без капсулы и убитых нагрева-
нагреванием, но имеющих капсулу, то живот-
животные погибали. В этом случае из орга-
организмов погибших мышей удалось вы-
выделить бактерии с защитными полиса-
харидными капсулами. Таким образом,
в этих экспериментах бактерии, не
имеющие капсул, приобрели способ-
способность образовывать ее благодаря веще-
веществу наследственности, которое пере-
перешло из убитых бактерий в живые.
Только в 40-х гг. XX в. в другой лабора-
лаборатории была выяснена природа этого за-
загадочного вещества наследственности.
Фактором, превращающим непатоген-
непатогенные бескапсульные пневмококки в па-
патогенные с полисахаридной оболочкой,
оказалась молекула ДНК.
Неопровержимым доказательством
того, что носителем наследственной
информации вирусов и бактериофагов
являются нуклеиновые кислоты, мож-
можно считать демонстрацию их инфекци-
инфекционных свойств. Так, было показано,
что очищенная ДНК некоторых фагов,
из которых наиболее известны <рх174 и
К, может заражать бактерии в отсутст-
отсутствие белковой оболочки.
В 1953 г. Джеймс Уотсон и Френсис
Крик предложили модель структуры
ДНК, которая с тех пор многократно
проверялась и признана правильной.
40
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Дезоксиаденозин
— Нуклеозид —
^^^^^^^^ — ¦ a^i^sawwr *f*^ " " "" 1
Пуриновое основание - аденин
Сахар - дезоксирибоза
Нуклеотид
J
Дезоксиаденозин-5'-фосфат
Дезоксигуанозин
Нуклеозид 1
Пуриновое основание - гуанин
О
Н
Сахар - дезоксирибоэа
Нуклеотид
Дезоксигуанозин 5'-фосфат
Дезокситимидин
Нуклеозид •
Пиримидиновое основание - тимин
I
Сахар - дезоксирибоза
Нуклеотид ¦
Дезокситимидин-5'-фосфат
Дезоксицитидин
Нуклеозид ¦
I
Пиримидиновое основание - Цитозин
ОН
Сахар - дезоксирибоза
Нуклеотид
Дезоксицитидин 5'-фосфат
Рис. 3.1. Нуклеотиды ДНК. Обратите внимание на нумерацию атомов углерода
и азота в пуриновом и пиримидиновом кольцах. Атомы углерода сахара
нумеруются отдельно с использованием штрихованных цифр
Химический состав
и строение молекулы ДНК
Уотсон и Крик предположили, что
природная (нативная) молекула ДНК
представляет собой две полимерные
цепи, соединенные между собой и за-
закрученные в форме двойной спирали.
Основная структурная единица од-
одной цепи — нуклеотид. Он состоит из
трех химически различных частей, со-
соединенных ковалентными связями:
дезоксирибозы, азотистого основания
и фосфатной группы (рис. 3.1). ДНК
содержит нуриновые азотистые осно-
Химический состав и строение молекулы ДНК
Тимин
41
Аденин
З'-конец
Рис. 3.2. Строение одной цепи молекулы ДНК
вания — аденин (А) и гуанин (Г) — и
пиримидиновые основания — цитозин
(Ц) и тимин (Т). Азотистое основание
ковалентно соединено с первым ато-
атомом углерода сахара и формирует
структуру, называемую нуклеозидом.
Фосфатные труппы соединяют сосед-
соседние нуклеозиды в полимерную цепоч-
цепочку посредством фосфодиэфирных
связей между 5'-атомом углерода од-
одного сахара и З'-атомом углерода дру-
другого (рис. 3.2). Сцепление между цепя-
цепями обеспечивается особыми водород-
водородными связями между аденином и ти-
мином и между гуанином и цитозином
(рис. 3.3). Водородные связи много
слабее ковалентных, соединяющих от-
отдельные атомы каждого нуклеотида,
но достаточно сильны, чтобы обеспе-
обеспечить специфичность образования пар
А-Т, Г-Ц. Такое попарное сопоставле-
сопоставление нуклеотидов, при котором А ком-
комплементарен Т, а Г комплементарен Ц,
было выведено с помощью построения
молекулярных моделей, в которых
точно воспроизводились в масштабе
все межатомные расстояния. Прост-
Пространственная модель молекулы ДНК
показала характер закрученности це-
цепей друг относительно друга и плот-
плотность упаковки пар азотистых основа-
оснований в двойной спирали (рис. 3.4). Кро-
Кроме того, построение молекулярной мо-
модели гипотетической двойной спирали
42
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
т Н У Н
Тимин \[/
Цитозин и
н сч о • н н
С С 4f/ Аденин
I
Н С м
Сахар
I I
N N4
' ЧС' ЧН.
II
О
N
С
II С-Н
I I
N N * Н ,СЧ
Сахар V N С
6 I И
о гУанин
I
С-Н
Сахар Н Сахар
Рис. 3.3. Водородные связи между азотистыми основаниями в составе молекулы ДНК.
0.34 нм
О
н
о
С н фосфорфнрных цепях
О
С и N в основаниях
Рис. 3.4. Пространственная модель молекулы ДНК
Химический состаа и строение молекулы ДНК
43
5' конец
Ось спирали
S коней
Рис. 3.5. Две цепи молекулы ДНК расположены "антипараллельно",
так что 5'-конец одной цепи соединен с З'-концом другой
потребовало "антипараллельности"
нуклеотидных цепочек, как это изоб-
изображено на рисунке (рис. 3.5).
Нуклеиновые кислоты — это очень
длинные полимерные цепочки. Ин-
тактные молекулы ДНК содержат в за-
зависимости от вида организмов от не-
нескольких тысяч до многих миллионов
нуклеотидов. Для любой последова-
последовательности азотистых оснований воз-
возможна равная ей по длине комплемен-
комплементарная последовательность, составля-
составляющая вторую цепь двойной спирали.
Конкретная последовательность пар
А-Т и Г-Ц не влияет на структуру мо-
молекулы ДНК, образующей двойную
спираль. Возможное число различных
последовательностей пар оснований в
молекуле ДНК практически бесконеч-
бесконечно и способно кодировать колоссаль-
колоссальное количество информации.
Из модели следует, что физическая
структура природной ДНК может
сильно изменяться при нагревании
или титровании, когда не нарушаются
ковалентные, но разрываются водо-
водородные связи, в результате чего две це-
цепи отделяются друг от друга.
Поскольку цепи ДНК комплемен-
комплементарны, каждая из них при расплетании
двойной спирали способна служить
матрицей для синтеза новой компле-
комплементарной цепи. Последовательность
оснований во вновь синтезируемой це-
цепи будет определяться спецификой во-
водородных связей между азотистыми
основаниями родительской и вновь
синтезируемой цепи (рис. 3.6). Таким
образом, генетическая информация,
содержавшаяся в последовательности
пар оснований родительской молеку-
молекулы, будет полностью воспроизведена в
44
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Родительская
Родительская
Родительская
Рис. 3.6. Предложенная Уотсоном и Криком модель репликации двойной спирали ДНК
двух дочерних молекулах. Более того,
если в процессе удвоения ДНК про-
произошла ошибка и какой-либо нуклео-
тид во вновь образуемой цепи выпал
или оказался некомплементарным ис-
исходному, то это может изменить ин-
информационное содержание молекулы,
причем логично ожидать, что эта
ошибка будет передана дочерним мо-
молекулам ДНК в следующих поколени-
поколениях. Такая замена пары нуклеотидов бу-
будет обладать свойствами генетических
мутаций. Модель структуры ДНК Уот-
сона и Крика объясняет как способ-
Упаковка ДНК ¦ хромосомах
45
H2N-СН2-СНг-СНг -СН2- СН-СООН
L-Аргинин
I
NH2
HN
H,N
^C-NH-CH2-CH2-CH2-
L-Лизин
СН-СООН
I
NH2
Рис. 3.7. Положительно заряженные (основные) аминокислоты гистоновых белков
ность генов к самоудвоению (реплика-
(репликации), так и их информационные свой-
свойства.
Упаковка ДНК в хромосомах
Молекулы ДНК в эукариотических
клетках очень велики. Так, длина мо-
молекул ДНК, выделенных из клеток че-
человека, достигает нескольких сантиме-
сантиметров. Принято считать, что каждая эу-
кариотическая хромосома содержит
одну — единственную непрерывную
молекулу ДНК. Учитывая видовое ко-
количество хромосом у млекопитающих,
можно сказать, что в среднем у них на
интерфазное ядро приходится около
2 м ДНК, находящейся в сферическом
ядре диаметром менее 10 мкм. При
этом в ядре должен сохраняться опре-
определенный порядок расположения мо-
молекул ДНК, чтобы обеспечить ее упо-
упорядоченное функционирование.
Молекулы ДНК в ядрах эукариоти-
эукариотических клеток всегда находятся в ком-
комплексе с белками в составе хроматина,
который образуется из хромосом по-
после окончания деления ядер в резуль-
результате сложного процесса раскручива-
раскручивания (деспирализации) хромосом.
На долю белков приходится около
60% сухого веса хроматина. Белки в
его составе очень разнообразны.
Обычно их разделяют на две группы:
гистоны и негистоновые белки. Имен-
Именно гистоны, характерные только для
эукариотических клеток, осуществля-
осуществляют первые этапы упаковки ДНК, очень
схожие у большинства изученных объ-
объектов. На долю гистонов приходится
до 80% всех белков хроматина. Их вза-
взаимодействие с ДНК происходит за
счет ионных связей и не зависит от по-
последовательности нуклеотидов в со-
составе молекулы ДНК. Гистоны не от-
отличаются большим разнообразием.
Это глобулярные белки, представлен-
представленные 5-7 типами молекул. Наиболее из-
известны следующие классы гистонов:
HI, Н2А, Н2В, НЗ и Н4. Их основные
свойства определяются относительно
высоким содержанием основных ами-
аминокислот: лизина и аргинина (рис. 3.7).
Положительные заряды на аминогруп-
аминогруппах указанных аминокислот обеспечи-
обеспечивают электростатическую связь гисто-
гистонов с отрицательными зарядами на
фосфатных группах ДНК. Из всех
ядерных белков гистоны изучены наи-
наиболее хорошо. Их молекулярная масса
относительно невелика (максималь-
(максимальная — у гистона НЗ — 153 тыс. даль-
тон). Практически у всех эукариот они
обладают сходными свойствами и под-
подразделяются на одни и те же классы.
Из исследованных эти белки наиболее
консервативны: их аминокислотные
последовательности близки даже у от-
отдаленных видов. Исключение состав-
составляют гистоны HI, для которых харак-
характерны значительные межвидовые и
межтканевые вариации. В процессе
жизнедеятельности клеток гистоны
могут подвергаться посттрансляцион-
посттрансляционным модификациям, что изменяет их
свойства и способность связываться с
ДНК. Гистоны синтезируются в цито-
46
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Рис. 3.8. Схема расположения ДНК и гистонов в нуклеосоме
плазме, переносятся в ядро и связыва-
связываются с ДНК во время ее репликации в
S-периоде клеточного цикла. Вклю-
Включившиеся в хроматин гистоны очень
стабильны и имеют низкую скорость
обмена.
Присутствие гистонов во всех эука-
риотических клетках, их сходство да-
даже у очень отдаленных видов, обяза-
обязательность в составе хромосом и хрома-
хроматина — все это говорит о чрезвычайно
важной роли этих белков в жизнедея-
жизнедеятельности клеток. Этапным событием
в изучении упаковки ДНК в составе
хроматина стало открытие нуклеосом —
частиц, в которых происходит первый
этап упаковки ДНК в хроматине.
Сердцевина нуклеосомы всегда кон-
консервативна, содержит восемь молекул:
по две молекулы гистонов Н4, НЗ,
Н2А, Н2В. По поверхности сердцеви-
сердцевины располагается участок ДНК из 146
нуклеотидных пар, образующий 1,75
оборота вокруг сердцевины. Неболь-
Небольшой участок ДНК остается несвязан-
несвязанным с сердцевиной, он называется
линкером (рис. 3.8). В разных объек-
объектах линкерный участок может варьи-
варьировать от 8 до 114 нуклеотидных пар
на нуклеосому. Рассчитано, что на весь
гаплоидный геном человека C х 109
пар оснований) приходится 1,5 х 107
нуклеосом. Общий вид хроматина,
представленного молекулой ДНК,
упакованной с помощью нуклеосом-
ных структур, можно сравнить с буса-
бусами на нитке (рис. 3.9). Нуклеосомы
способны к самосборке при наличии в
пробирке ДНК и гистонов в опреде-
определенном соотношении. Первый нуклео-
сомный уровень компактизации ДНК
увеличивает плотность упаковки ДНК
в 6-7раз.
В следующий этап упаковки нукле-
осомная структура хроматина вовле-
вовлекается с помощью гистона HI, кото-
Упакома ДНК ¦ хромосомах
47
Е^ЗКЙ§3?«»?
Рис. 3.9. Электронная микрофотография эукариотического хроматина,
на которой видны нуклеосомы, соединенные тяжами свободной ДНК
рый связывается с линкерной частью
ДНК и поверхностью нуклеосомы.
Благодаря сложному взаимодействию
всех компонентов возникает упорядо-
упорядоченная структура спирального типа,
которую часто называют соленоидом
(рис. 3.10). Она повышает компакт-
компактность ДНК еще в 40 раз. Поскольку со-
соленоидная структура имеет снижен-
сниженную способность связываться с белка-
белками, обеспечивающими транскрипцию,
то считается, что этот уровень компак-
тизации ДНК может играть роль фак-
фактора, инактивирующего гены. Некото-
Некоторые авторы рассматривают соленоид-
соленоидную структуру как один из возможных
вариантов упаковки хроматина с по-
помощью гистона HI и полагают вероят-
48
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
¦ 100 А
Рис. 3.10. Схематическое изображение
участка соленоида: цепочка нуклеосом
(сферы), каждая из которых обмотана ДНК,
образуют винтовую линию
ным существование и других морфо-
морфологических вариантов, например, нук-
леомер, или сверхбусин (рис. 3.11).
Более высокие уровни компактиза-
ции ДНК в хроматине связаны с неги-
стоновыми белками. На их долю при-
приходится около 20% всех белков хрома-
хроматина. Эту сборную группу белков от-
отличает широкий спектр свойств и
функций. Всего фракция негистоно-
вых белков объединяет около 450 ин-
индивидуальных белков, свойства и кон-
конкретные функции которых еще не до-
достаточно изучены. Выяснено, что не-
некоторые из них специфично связыва-
связываются с определенными участками
ДНК, в результате чего фибриллы хро-
хроматина в местах связывания ДНК с не-
Рис. 3.11. Нуклеомерный тип укладки
нуклеосомной фибриллы
гистоновыми белками образуют петли.
Таким образом, более высокие уровни
упаковки ДНК в составе хроматина
обеспечиваются не спирализациеи ни-
нитей хроматина, а образованием попе-
поперечной петлистой структуры вдоль
хромосомы (рис. 3.12). На всех указан-
указанных этапах компактизации ДНК хро-
хроматин представлен в активной форме,
в нем происходит транскрипция, син-
синтез всех типов молекул РНК. Такой
хроматин называют эухроматином.
Дальнейшая упаковка хроматина ве-
ведет к переходу его в неактивное состо-
состояние с образованием гетерохроматина.
Этот процесс связан со спирализациеи
групп петель и образованием из фиб-
фибрилл хроматина розеткоподобных
Организаций генетического материала в хромосомах человека
49
Рис. 3.12. Петлеобразная и хромомерная компактизация хроматина
I
структур, которые обладают оптичес-
оптической и электронной плотностью и назы-
называются хромомерами (рис. 3.12). Пред-
Предполагается, что вдоль хромосомы рас-
расположено большое количество хромо-
мер, соединенных между собой в еди-
единую структуру участками хроматина с
нуклеосомной или соленоидной упа-
упаковкой ДНК. Каждая пара гомологич-
гомологичных хромосом имеет свой хромомер-
ный рисунок, который можно выявить
с помощью специальных методов ок-
окрашивания при условии сиирализации
хроматина и перехода его в состояние
хромосом.
Петельно-розеточная структура
хроматина обеспечивает не только
упаковку ДНК, но и организует функ-
функциональные хромосом, поскольку в
своих основаниях петли ДНК связаны
с негистоновыми белками, в состав ко-
которых могут входить ферменты репли-
репликации, обеспечивающие удвоение
ДНК, и ферменты транскрипции, бла-
благодаря которым происходит синтез
всех типов РНК.
Участки ДНК, упакованные в виде
гетерохроматина, могут иметь двоя-
двоякую природу. Различают два типа гете-
гетерохроматина: факультативный и кон-
конститутивный (структурный). Факуль-
Факультативный гетерохроматин представля-
представляет собой участки генома, временно
инактивированные в тех или иных
клетках. Примером такого хроматина
служит половой гетерохроматин инак-
тивированной Х-хромосомы в сомати-
соматических клетках женщин. Структурный
гетерохроматин во всех клетках посто-
постоянно находится в неактивном состоя-
состоянии и, вероятно, выполняет структур-
структурные или регуляторные функции.
Организация генетического
материала в хромосомах человека
Общая организация хромосом чело-
человека традиционна: в метафазе хромо-
хромосома состоит из двух сестринских хро-
матид, соединенных между собой в-
районе первичной перетяжки (центро-
(центромеры). Центромера делит хроматиду
на два плеча. Плечи могут быть равны-
равными, тогда хромосома называется мета-
центрической. Если одно плечо немно-
немного короче другого, то хромосомы име-
именуются субметацентрическими. В не-
нескольких парах хромосом человека од-
одно плечо сильно короче другого, такие
50
ГламЗ. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
НН
4 S
—— В —
и п и к п
2 3
А ————•¦——
Ой а о л о
б 12
— с ——
13
16 17 18
—__ Е ———
XX
А А Л
19 20
F ——
21 32
М
X X
9
X Y
Рис. 3.13. 22 пары аутосом и половые хромосомы человека,
составляющие его кариотип
хромосомы носят название акроцент-
рических (рис. 3.13). Тонкая морфоло-
морфология хромосом зависит от фазы митоза.
Наиболее сильно спирализованы хро-
хромосомы в мета- и анафазе.
Вместе с морфологией хромосом из-
изменяется в ходе митозе и морфология
центромеры. Наиболее четко центро-
центромера выражена в виде более тонкого и
светлого участка хромосомы к концу
профазы. Центромеры выполняют в
хромосомах очень важные функции.
Они соединяют две сестринские хро-
матиды, велика также их роль в орга-
Организация генетического материала в хромосомах человека
51
низации веретена деления. В районе
центромеры в профазе формируется
особая белковая структура, имеющая
сродство к белкам микротрубочек ве-
веретена деления. Микротрубочки вере-
веретена деления соединяются с хромати-
дами в районе центромеры так, что на
один центромерный район может при-
приходиться более десяти микротрубочек.
Очень важным моментом в прохожде-
прохождении митоза является синхронность од-
одновременного разделения всех хромо-
хромосом на две хроматиды. Считается, что
ведущая роль в регуляции этого про-
процесса также принадлежит центроме-
центромерам.
Рассматривая общую морфологию
хромосом, нельзя обойти вниманием
их концевые участки, называемые те-
ломерами. Концы хромосом — теломе-
ры, имеют особенности в первичной и
третичной структурах, но об этом речь
пойдет несколько позже. Сначала оз-
ознакомимся с функциями теломерных
районов. Когда деление клетки закон-
закончено и формируются новые клеточные
ядра, то с помощью теломер хромосо-
хромосомы прикрепляются к внутренней ядер-
ядерной мембране, в результате чего каж-
каждая хромосома в деспирализованном
состоянии занимает в ядре строго оп-
определенное место. Помимо этого тело-
мерные районы предотвращают слипа-
слипание хромосом своими концами и пре-
препятствуют образованию дицентриков —
хромосом с двумя центромерами, на-
наличие которых свидетельствует о па-
патологических картинах митоза. Одно-
Одновременно теломеры стабилизируют
хромосомы, защищая их от деградации
клеточными нуклеазами — фермента-
ферментами, катализирующими гидролиз всех
незащищенных ДНК или их фрагмен-
фрагментов. В последнее время стало известно
еще одно назначение теломерных кон-
концов: благодаря им происходит полное
завершение редупликации хромосом
при подготовке клетки к делению.
Среди ферментов, участвующих в уд-
удвоении ДНК, помимо уже известных
ДНК-полимеразы, ДНК-лигазы, гели-
казы, топоизомеразы, а также стабили-
стабилизирующих белков, особое внимание
следует уделить теломеразе, которая
помогает завершить репликацию ДНК
на отстающей цепи.
Принято считать, что каждая хрома-
тида содержит одну из двух идентич-
идентичных дочерних молекул ДНК, образую-
образующихся в процессе репликации. Моле-
Молекула ДНК представляет собой непре-
непрерывную сверхскрученную двойную
спираль, простирающуюся по всей
длине хроматиды. Функционально эта
нить подразделяется на большое число
отрезков, соответствующих отдель-
отдельным генам. Каждый ген несет инфор-
информацию о первичной структуре отдель-
отдельной полипиптидной цепи, рибосомной
РНК, транспортной РНК или выпол-
выполняет регуляторную функцию. Кроме
того, в составе непрерывной нити
ДНК, наряду со смысловыми генами,
находятся многократно повторяющие-
повторяющиеся одинаковые или сходные по составу
нуклеотидные последовательности,
выполняющие, вероятно, регулятор-
ные или структурные функции.
Информация о первичной структу-
структуре полипептидов (последовательности
аминокислот в них) записана в ДНК в
виде трехбуквенного кода, составлен-
составленного из первых букв названий четырех
азотистых оснований, входящих в со-
состав ДНК (АТГЦ). Каждой аминокис-
аминокислоте соответствует определенный
триплет из трех соседних нуклеотидов.
Например, аминокислоте фенилала-
нин в ДНК соответствует кодон ААА, а
аминокислоте серии — АГА. Из 64 воз-
возможных триплетов 61 кодирует 20
аминокислот, обнаруженных в составе
52
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
5Чонец
Инициация транскрипции,
Дистальные кэпировакие
регуляторные ТАТА- I
последовательность I
Сайт полиаденилироеания
Термина ция
транскрипции
J-C
З'-коиец
5'-Фланкирующая последовательность
5'-Кон-
5'-Концевая
неко-
диру-
ющая
после-
дова-
тель-
ность
I Экзон 1 Интрон Экзон21
З'-Кон-
цееая
неко-
диру-
ющая
после-
дова-
тель-
ность
З'-Фланкирующая
' последовательность
Рис. 3.14. Структурная организация типичной транскрипционной единицы
клеточных белков, а 3 кодона являют-
являются стоп-сигналами, прекращающими
синтез полипептидной цепи. Если
триплет, соответствующий метионину,
стоит в начале цепи ДНК, то он выпол-
выполняет функцию возбуждения считыва-
считывания. (Кодоны, выполняющие сигналь-
сигнальные функции, называют нонсенс — ко-
донами). Генетический код вырожден,
т. е. каждая аминокислота может коди-
кодироваться несколькими вариантами
триплетов. Для осуществления синте-
синтеза полипептидов генетическая инфор-
информация, закодированная в ДНК в соста-
составе хроматина, переписывается (про-
(процесс транскрипции) по принципу ком-
плементарности азотистых оснований
на информационную РНК, которая пе-
переходит из ядра в цитоплазму, где при-
принимает участие в процессе трансля-
трансляции: переводе информации с языка
нуклеотидов на язык аминокислот, т. е.
процессе синтеза белка. Каждому дан-
данному кодону соответствует одна и
только одна определенная аминокис-
аминокислота. Процесс считывания генетичес-
генетического кода не допускает возможности
перекрывания кодонов. Начавшись на
определенном кодоне, считывание сле-
следующих идет без знаков препинания и
пропусков вплоть до нонсенс-кодонов.
Положение первого кодона определя-
определяет границы рамки считывания. Генети-
Генетический код человека не отличается по
каким-либо параметрам от генетичес-
генетического кода любых других эукариотичес-
ких организмов.
В пределах одного гена, который ко-
кодирует полипептид, участок молекулы
ДНК подразделяется на функциональ-
функционально различные единицы (рис. 3.14). От-
Отличительная черта строения многих
генов эукариот — прерывистость
структуры смысловой части. Смысло-
Смысловые участки, несущие информацию о
последовательности аминокислот в
белке — экзоны, чередуются с участка-
участками некодирующих последовательнос-
последовательностей — нитронами. Часто интроны по
длине могут превосходить экзоны. На-
Наличие избыточных последовательнос-
последовательностей приводит к тому, что длина гена
может быть в несколько раз больше,
чем требуется для кодирования амино-
аминокислот в белке. Гаплоидный набор9 хро-
хромосом человека содержит 3,5 х 10 нук-
леотидных нар, что но количеству со-
соответствует примерно 1,5 млн. пар ге-
генов. Однако данные по изучению гено-
генома человека показывают, что организм
человека имеет не более 100 тыс. генов.
Это значит, что в клетках человека
только 1% ДНК выполняет кодирую-
кодирующие функции. В отношении оставших-
оставшихся 99% существуют разные гипотезы,
обосновывающие их регуляторные и
Организация генетического материала в хромосомах человека
53
Начало гена
Т Интрон 1
Конец Гена
Цепь ДНК
I
Транскрипция
¦
Пре-м-РНК
I
Сплайсинг
м-РНК
I
Трансляция
Экзон 1 Экзон 2| Экзон 3 i Экзон 4
i Интрон 2 Интрон 3
I I
I I
Экзон \' /' Экзон3 '''
Экзон 2
Экзон 4
Белок
Рис. 3.15. Основные этапы синтеза белка у эукариот
структурные функции. Для человека
данные об экзонно-интронном строе-
строении генов впервые были получены для
гена гемоглобина, а затем и для многих
других генов. Это открытие стало воз-
возможным в конце 70-х гг. благодаря раз-
разработке метода синтеза комплементар-
комплементарных ДНК на матрице м-РНК с помо-
помощью обратной транскриптазы, а также
благодаря освоению целого ряда мето-
методов генно-инженерной техники. В них
входит разрезание генома с помощью
рестриктаз на последовательности, со-
соответствующие отдельным генам, вы-
выделение таких генов, встраивание их в
векторные молекулы, внедрение век-
векторов в клетки бактерий и клонирова-
клонирование там таких рекомбинантных моле-
молекул, что позволяет получать банки ге-
генов.
Процесс транскрипции на ДНК, как
на матрице, связан с синтезом компле-
комплементарной последовательности РНК,
включающей и интроны, и экзоны. За-
Затем в ходе созревания РНК в ядре из
нее удаляются интроны, а концы со-
соседних экзонов сшиваются стык в
стык. Процесс удаления последова-
последовательностей РНК, соответствующих
интронам, и соединение участков с
транскрибируемыми последователь-
последовательностями экзонов называется сплай-
сплайсингом. Кроме того транскрибируемая
молекула модифицируется добавлени-
добавлением метилированного Г-нуклеотида на
5'-конце (кэпирование) и поли-А по-
последовательности на З'-конце. Моди-
Модифицированные участки играют важ-
важную роль в инициации белкового син-
синтеза, защищают транскринт т-РНК от
деградации. Имеются данные, свиде-
свидетельствующие о том, что поли-А конец
участвует в транспорте зрелой м-РНК
из ядра в цитоплазму и продлевает ее
функционирование там. Весь суммар-
суммарный процесс формирования зрелых
молекул РНК из предшественников
называется процессингом. Созревшая
м-РНК выходит в цитоплазму, при-
прикрепляется к рибосоме, где генетичес-
54
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
А У
Г У
Ц У
А А
У Г
Г Ц
м-РНК
Белок,
считанный
по рамке
№1
Белок,
считанный
по рамке
№2
Белок,
считанный
по рамке
№3
II
Аспарагин
Метионин
В алии
I
Цистеин
Терминация
трансляции
Цистеин
Рис. 3.16. Изменения в белке, происходящие при сдвиге рамки считывания
кая информация транслируется в бел-
белковую последовательность (рис. 3.15).
Большинство интронных последо-
последовательностей, по-видимому, не облада-
обладают специфическими функциями. Од-
Однако один и тот же транскрипт РНК
может подвергаться сплайсингу по-
разному, следовательно с одного
транскрипта в ходе сплайсинга способ-
способны образоваться несколько различных
РНК. Такой сплайсинг называется
альтернативным. Альтернативный
сплайсинг сообщает клетке дополни-
дополнительную генетическую пластичность.
Рассмотрим это явление на примере
белка фибронектина.
В плазме крови человека присутст-
присутствует белок фибронектин. Он синтези-
синтезируется клетками печени и выделяется
в кровь. Этот же белок может синтези-
синтезироваться клетками соединительной
ткани, эндотелиальными клетками,
выстилающими кровеносные сосуды,
и некоторыми другими типами клеток.
В этом случае фибронектин в нерас-
нерастворимой форме накапливается в меж-
межклеточном пространстве. Функции
фибронектина многоплановы: он игра-
играет роль в поддержании гомеостаза, в
процессах тромбоза, миграции и диф-
ференцировки клеток сосудов, в раз-
развитии атеросклеротических бляшек в
сосудах. Известно много вариантов
фибронектина, он обладает ткане- и
возрастной специфичностью. Показа-
Показано, что варианты фибронектина обра-
образуются с одного геномного транскрип-
транскрипта за счет альтернативного сплайсинга.
Вариации м-РНК-последовательнос-
тей фибронектина связаны с присутст-
присутствием или отсутствием по крайней мере
двух интронов.
Известные вариации иммуноглобу-
иммуноглобулинов также в значительной мере
обеспечиваются процессом созрева-
созревания новосинтезированных гигантских
м-РНК в ядре.
В начале каждого гена, до его смыс-
смысловой части, представленной экзона-
ми, находятся участки, которые обес-
обеспечивают регуляцию работы гена. К
числу регуляторных участков, одина-
одинаковых для всех генов, относятся ТАТА-
последовательности (рис. 3.14), где че-
чередуются тимин и аденин ("ТАТА-
БОКС"). Этот участок лежит на 30
Организация генетического материала в хромосомах человека
55
нуклеотидов левее места начала счи-
считывания гена. Установлено, что РНК-
полимераза, фермент осуществляю-
осуществляющий транскрипцию, так ложится на
ДНК, что ее опознающая часть закры-
закрывает "TATA-БОКС", а ее активный
центр оказывается над первым считы-
считываемым нуклеотидом. Далее по длине
гена следует промоторный участок, ко-
который способствует правильной уста-
установке рамки считывания нуклеотидов,
поскольку процесс считывания гене-
генетического кода не допускает возмож-
возможности перекрывания кодонов. Иногда
изменения рамки считывания могут
происходить из-за выпадения или до-
добавления одного или нескольких нук-
нуклеотидов, тогда при последующей
сборке белка в нем будет нарушена по-
последовательность аминокислот. Такая
ситуация получила название мутации
со сдвигом рамки (рис. 3.16).
За промоторным участком следует
палиндром ("перевертыш"), или ин-
инвертированный повтор. Этот участок
ДНК одинаково читается в обоих на-
направлениях и имеет центральную точ-
точку, относительно которой последова-
последовательность остается одинаковой в обеих
цепях ДНК. Следовательно, такой уча-
участок ДНК имеет две оси симметрии :
вдоль и поперек. Важное свойство па-
палиндромов — возможность образовы-
образовывать шпильки в РНК или структуры
креста в ДНК за счет комплементарно-
комплементарного взаимодействия не между двумя ни-
нитями ДНК, а между нуклеотидами
каждой цепи. В результате этого па-
палиндром ДНК превращается в крест,
что делает невозможным дальнейшее
продвижение фермента РНК-полиме-
разы, и процесс транскрипции прекра-
прекращается, если рамка считывания уста-
установлена неверно.
В последнее время описаны специ-
специфические регуляторы работы некото-
некоторых генов — энхансеры. Они располо-
расположены впереди гена на расстоянии в
сотни и тысячи нуклеотидных пар от
него. У эукариот существуют специ-
специальные регуляторные белки, опознаю-
опознающие энхансер и присоединяющиеся к
нему, в результате чего происходит ак-
активация работы гена.
По данным разных авторов, содер-
содержание ДНК в диплоидной клетке чело-
человека составляет примерно 7,3 х 10~'2г,
что соответствует 7,1 х 109 нулеотид-
ных пар. Каждая молекула ДНК гете-
рогенна по своему составу. В ней встре-
встречаются участки с уникальной последо-
последовательностью азотистых оснований,
которые несут информацию для боль-
большинства белков клетки. В то же время
в ней встречаются последовательности
нуклеотидов, многократно повторяю-
повторяющиеся в геноме в составе этой же или
других молекул ДНК. Такие повторяю-
повторяющиеся последовательности подразде-
подразделяют на два класса. Первый — умерен-
умеренно повторяющиеся последовательнос-
последовательности с числом повторов от 102 до 105 на ге-
геном. На их долю приходится примерно
четверть ДНК, и они представляют со-
собой блоки истинных генов, как, напри-
например, гены гистонов (рис. 3.17). К этому
же классу умеренных повторов отно-
относятся короткие последовательности,
которые не кодируют белки, они раз-
разбросаны но всему геному, а их длина
соответствует примерно 300 н.п (нук-
(нуклеотидных пар). Второй класс — часто
повторяющиеся последовательности,
или сателлитные ДНК, число повторов
которых на геном превышает миллион
Aх106) раз. Это нетранскрибирующие-
ся участки ДНК, на которых не идет
образование РНК.
В работах разных авторов показано,
что лишь немногим более 50% ДНК ге-
генома человека представлено уникаль-
уникальными фрагментами длиной около 2000
56
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОЮ МАТЕРИАЛА
Мвжгеннын
(спвясврныя)
участок
Нвкодирующнв
последовательности
(жнтровы)
Последовательности
структурного ген*
(зкэоны)
Участок регуляции
активности гава
0-гаобвна
Участок с большим числом
повторов, расположенный
перед структурным геном
(сатвллитиад ДНК)
Двухцепочечная
Рис. 3.17. Сегмент эукариотической ДНК: показаны
различные способы организации гена
н.п., и далеко не все из них представля-
представляют структурные гены. В основном они
распределены в составе длинных моле-
молекул ДНК между короткими, умеренно
повторяющимися последовательнос-
последовательностями, длина которых не превышает
300 н.п. Многие из таких умеренных
повторов имеют сходное строение. Ве-
Вероятно, они выполняют структурные и
регуляторные функции в составе ге-
генов. Умеренные повторы, представля-
представляющие гены, встречаются в каждой
клетке человека, где есть ядро. Они со-
содержат гены, необходимые всем клет-
клеткам в каждой фазе индивидуального
развития. Это гены рибосомной РНК,
гистонов и транспортной РНК.
Гены рибосомной РНК являются ча-
частью района ядрышкового организато-
организатора на хромосомах. Они кодируют ри-
рибосомные РНК, которые синтезируют-
синтезируются в ядрышке. В ядрышке происходит
процессинг рибосомных РНК: из од-
одной гигантской новосинтезированной
молекулы образуются три разные мо-
молекулы рибосомных РНК меньшего
размера, а избыточные последователь-
последовательности удаляются. Зрелые рибосомные
РНК участвуют в сборке субъединиц
рибосом. Таким образом, ядрышко —
это место в ядре, где функционируют
рибосомные гены, оно содержит сово-
совокупность всех рибосомных РНК, нахо-
находящихся на разных стадиях процес-
синга, здесь происходит процесс фор-
формирования субъединиц рибосом. У че-
человека район ядрышкового организа-
организатора расположен в коротких плечах
Организация генетического материала а хромосомах человека
57
St - - it
W
1 2 S 4 5 « 7
01)
10 11
13 14
20
I
Рис. 3.18. Набор хромосом человека, где цветом выделены участки
с рибосомными генами. Из каждой пары хромосом показана только одна
акроцентрических хромосом: 13-й, 14-
й, 15-й, 21-й и 22-й (рис. 3.18). В фор-
формировании ядрышка могут принимать
участие сразу несколько хромосом.
Среднее число копий рибосомных ге-
генов в клетках человека по данным раз-
разных авторов 416-443 на диплоидный
геном.
К классу умеренно повторяющихся
последовательностей у человека мож-
можно отнести семейство многочисленных
генов вариабельных участков имму-
иммуноглобулинов, играющих ключевую
роль в иммунитете. Умеренные повто-
повторы обнаружены во всех хромосомах
человека, где они локализованы по
всей длине плеч.
Часто повторяющиеся последова-
последовательности занимают примерно деся-
десятую часть генома. Участки ДНК с та-
такими повторами выявлены в опреде-
определенных местах хромосом: это около-
центромерные и теломерные районы.
На дифференцированно окрашенных
хромосомах они выявляются в виде ге-
терохроматина. Причем это гетерохро-
матин конститутивный — в нем не
происходит транскрипция. Более того,
конститутивный гетерохроматин мо-
может влиять на близко расположенные
гены, подавляя их активность.
Частые повторы обычно не бывают
длинными. Так, теломерные районы
хромосом человека, ограничивающие
хромосомы с двух концов, содержат
250-1500 повторов ТТАГГ. Эти повто-
повторы играют очень важную роль: пре-
предотвращают слипание хромосом кон-
58
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
цами и их укорачивание при много-
многократных репликациях ДНК в связи с
клеточными делениями. Часто повто-
повторяющиеся последовательности ДНК
иначе называют сателлитной ДНК.
Сателлитная ДНК не всегда имеет ха-
характер видового признака, как это вид-
видно на повторах теломерных районов.
Однако у человека выделены и охарак-
охарактеризованы индивидуальные сател-
литные ДНК, расположенные в раз-
разных хромосомах. Так, известны не-
несколько типов сателлитной ДНК из
Y-хромосомы и 1-й, 9-й и 16-й хромо-
хромосом. Функции сателлитной ДНК во
многом остаются неизвестными. Пред-
Предполагается, например, что сателлитная
ДНК участвует в распознавании гомо-
гомологичных хромосом во время конъюа-
ции в мейозе, рассматривается также
возможность регуляторного участия
сателлитной ДНК в функционирова-
функционировании генов.
Пришедшие в генетику новые мето-
методы позволили расширить знания о
структуре генетического материала.
По современным данным, он оказался
намного менее статичен, чем представ-
представлялось раньше. Так, например, извест-
известны описанные Барбарой Мак-Клинток
мобильные контролирующие генети-
генетические элементы в геноме кукурузы,
способные перемещаться с одного гена
на другой, увеличивая их нестабиль-
нестабильность. Соматическими мутациями,
связанными с присутствием мобиль-
мобильных контролирующих элементов,
обусловлена мозаичная окраска почат-
початков у кукурузы. Найдены мобильные
генетические элементы и у дрожжей.
Позже было выявлено несколько клас-
классов мобильных генетических элемен-
элементов у бактерий и показано, что они мо-
могут встраиваться во многие участки ге-
генома клетки хозяина. В зависимости
от структуры мобильного генетическо-
генетического элемента внедрение бывает строго
специфичным или случайным. Уста-
Установлено, что при внедрении мобильно-
мобильного элемента встраивается не он сам, а
его копия, в то время как исходный
элемент остается на своем месте.
Встраивание мобильного элемента в
структурный ген приводит к мутации.
Кроме того, перемещение мобильных
элементов может стимулировать хро-
хромосомные аберрации. Для выявлен-
выявленных у дрозофилы мобильных генети-
генетических элементов показано, что они
могут образовываться из рассеянных
по геному генных элементов, из повто-
повторяющихся последовательностей кон-
конститутивного гетерохроматина при-
центромерных районов хромосом, а
также иметь вирусную природу. Все
мобильные элементы обладают неко-
некоторыми общими чертами структурной
организации — в частности инвертиро-
инвертированными повторами на концах. Их
репликация независима. Кроме того,
они способны вызывать мутации с вы-
высокой частотой, причем мутации не-
нестабильные, часто ревертирующие к
исходному состоянию. Существуют
предположения, что перенос генов мо-
мобильными элементами является одним
из факторов эволюции. Например, по-
последовательности ДНК, гомологичные
глобиновому гену человека, были об-
обнаружены у бобовых растений. Функ-
Функция такой структуры может заклю-
заключаться в обеспечении кислородом клу-
клубеньковых бактерий. А наличие такого
гена в растениях может быть объясне-
объяснено переносом его от насекомых или
млекопитающих. Следует, однако, за-
заметить, что у человека подобные эле-
элементы в геноме еще не выявлены. Тем
не менее в геноме человека обнаруже-
обнаружены повторяющиеся последовательнос-
последовательности, содержащие палиндромы с инвер-
инвертированными повторами, которые по
Хромосомы человека
59
аналогии напоминают мобильные эле-
элементы. Первым этапом в транспози-
транспозиции некоторой последовательности
ДНК должно быть образование вне-
хромосомных кольцевых копий этого
участка. Подобные кольцевые струк-
структуры обнаружены в стареющих фиб-
робластах в клеточных культурах че-
человека. Кроме того для генома челове-
человека описано явление конверсии глоби-
новых генов, когда один аллель моди-
модифицируется другим, в результате чего
гетерозигота становится гомозиготой.
Новые данные углубляют понима-
понимание структурной организации генети-
генетического материала и механизмов его
работы. Однако по-прежнему верным
остается постулат о стабильности ге-
генетического аппарата, на котором ос-
основаны все закономерности наследо-
наследования признаков. Благодаря стабиль-
стабильности генома, существует наследствен-
наследственность, обеспечивающая преемствен-
преемственность фенотипических признаков из
поколения в поколение.
Хромосомы человека
История развития
цитогенетики человека
Впервые митотические хромосомы
человека были описаны в работах Дж.
Арнольда A879) и В. Флемминга
A882). В последующие годы различ-
различные оценки их количества давали ре-
результаты от 47 до 49 хромосом, причем
у мужчин и женщин находили разное
их число. Эти первые исследования
проводились на гистологических сре-
срезах тестикул или яичников. В то время
техника получения срезов была тако-
такова, что митозы в готовых препаратах
были, как правило, разрушены. Хромо-
Хромосомы на них накладывались одна на
другую, образовывали клубки и плохо
поддавались анализу. Эти трудности
удалось преодолеть только к 50-м гг.
XX в., когда для получения препаратов
хромосом стали использовать суспен-
суспензии клеток, выращенных в клеточных
культурах. Так, первые препараты хро-
хромосом человека с хорошим разрешени-
< ем были получены на клетках фиброб-
ластов эмбриона легкого, выращенных
in vitro. Суспензии клеток промывали
гипотоническим раствором, в резуль-
результате чего клетки набухали и лопались,
а хромосомы свободно распределялись
на стекле. Позже этот способ был усо-
усовершенствован. Перед гипотоничес-
гипотоническим шоком на клетки воздействовали
колхицином — веществом, которое,
разрушая нити веретена деления, оста-
останавливает митоз на стадии метафазы,
когда хромосомы наиболее легко иден-
идентифицировать. Это позволило полу-
получить большое число клеток в метафазе
митоза, и препараты, приготовленные
таким образом были более удобны для
подсчета хромосом. Пользуясь подоб-
подобным методическим подходом, в 1955
году А. Леван и Дж. Тио, изучив 261
метафазную пластинку, пришли к вы-
выводу, что количество хромосом в клет-
клетках человека равно 46, причем как в
мужских, так и в женских клетках. Го-
Годом позже и другие исследователи на
препаратах тестикул трех пожилых
мужчин на стадии метафазы мейоза 1
нашли 23 бивалента, что соответствует
46 хромосомам в диплоидном наборе.
Эти результаты ознаменовали возник-
возникновение новой отрасли исследований —
клинической цитогенетики. В настоя-
настоящее время цитогенетика человека до-
достигла высокого уровня и находится
на переднем крае фундаментальной
цитогенетики.
Нормальный кариотип человека
Препараты хромосом человека мож-
можно приготовить из любых тканей и
60
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
клеточных суспензий, но лишь, если в
них содержатся делящиеся клетки, т. к.
вне деления (во время интерфазы)
хромосомы деспирализуются и пере-
переходят в состояние хроматина. Чаще
всего препараты готовят из клеток ко-
костного мозга, кратковременной куль-
культуры клеток крови или из перевивае-
перевиваемой культуры фибробластов. Наибо-
Наиболее прост и доступен метод культиви-
культивирования клеток крови лейкоцитов и
лимфоцитов. Митозы в таких культу-
культурах стимулируют искусственно. Что-
Чтобы остановить жизненный цикл кле-
клеток в прометафазе, в них подавляют
образование веретена деления, обраба-
обрабатывая веществами с колхициноподоб-
ными свойствами. Для свободного рас-
распределения хромосом на стекле клетки
обрабатывают гипотоническим рас-
раствором, вызывая гипотонический шок.
Затем препарат фиксируют смесью
этанола и уксусной кислоты, высуши-
высушивают и окрашивают. Когда с помощью
стандартных методов хромосомы ок-
окрашиваются целиком, равномерно и
интенсивно, их систематизируют со-
согласно Денверской классификации,
принятой в 1960 г., нумеруя пары хро-
хромосом от 1 до 23.
В кариотипе человека различают
метацентрические, субметацентричес-
кие и акроцентрические хромосомы.
Учитывая относительную длину иле-
чей, положение центромеры и центро-
мерный индекс, который отражает
процентное соотношение длин корот-
короткого плеча и всей хромосомы, 23 пары
хромосом человека разбивают на 7
групп (рис. 3.13). В группу А (№№ 1-
3) входят пары наиболее крупных ме-
тацентрических аутосом. Группа В
(№№ 4-5) объединяет две пары субме-
тацентрических хромосом, неразличи-
неразличимых между собой. Группа С (№№ 6-
12) содержит семь пар аутосом средне-
среднего размера. Размеры и форма этих хро-
хромосом неодинаковы, однако стандарт-
стандартные методы окрашивания не позволя-
позволяют их идентифицировать. В группу D
(№№ 13-15) объединены три пары ак-
роцентрических хромосом среднего
размера, морфологически сходных
между собой. Все хромосомы группы
D содержат спутник, который не все-
всегда выявляется, может быть очень
большим, а иногда и двойным. Длина
короткого плеча этих хромосом также
изменчива. К группе Е (№№ 16-18) от-
относятся три пары почти метацентриче-
ских хромосом, из которых в 16-й паре
центромера наиболее близка к середи-
середине, а две другие пары неотличимы друг
от друга. Группа F содержит мелкие
метацентрические аутосомы (№№ 19-
20), группа G — мелкие акроцентриче-
акроцентрические (№№ 21-22). Внутри групп F и G
пары хромосом неразличимы. Длина
коротких плечей у них изменчива,
как и у хромосом группы D. Короткие
плечи хромосом групп D и G содер-
содержат районы ядрышкового организа-
организатора. Перечисленные 22 пары хромо-
хромосом относятся к аутосомам, одинако-
одинаковым у мужчин и женщин.
Половые хромосомы составляют
23-ю пару. У женщин — это две Х-хро-
мосомы. У мужчин — Х- и Y-хромосо-
мы. Половая Х-хромосома неотличима
от аутосом группы С. При стандарт-
стандартном окрашивании она включается в
состав этой группы: №№ 6-12 и X.
Мужская половая Y-хромосома явля-
является акроцентрической, сходна но мор-
морфологии с хромосомами группы G, но
ее легко отличить по морфологичес-
морфологическим критериям. Длина короткого пле-
плеча Y-хромосомы изменчива и индиви-
индивидуальна, причем варианты длины пле-
плеча наследуются от отца к сыну. Y-хро-
Y-хромосома, в отличие от хромосом послед-
последней группы, не имеет спутников. В ин-
Хромосомы человека
61
терфазных ядрах концевой участок
длинного плеча Y-хромосомы можно
выявить в составе хроматина, пользу-
пользуясь специфическим окрашиванием ак-
акрихин-ипритом : в результате этот уча-
участок выявляется как яркое пятно диа-
диаметром 0,3-1,0 мкм,
Во многих хромосомах человека об-
обнаружены ломкие (фрагильные) уча-
участки, подверженные хромосомным и
хроматидным разрывам. Такие разры-
разрывы легко получить в клеточных куль-
культурах, удаляя фолиевую кислоту из
питательной среды культивируемых
клеток, В настоящее время показано,
что одна из форм умственной отстало-
отсталости человека связана с наличием опре-
определенного фрагильного участка в кон-
концевом районе длинного плеча Х-хро-
мосомы.
Длина одной и той же хромосомы в
разных фазах митоза различна, по-
поскольку конденсация хромосом про-
продолжается до конца метафазы. Кроме
того их конденсация значительно уси-
усиливается в присутствии применяемого
для приготовления препаратов колхи-
колхицина.
Анализ препаратов хромосом чело-
человека показал, что в ряде случаев, как
уже говорилось выше, на некоторых
хромосомах могут существовать вто-
вторичные перетяжки. Спутничными пе-
перетяжками обладают все акроцентри-
ческие хромосомы (пары №№ 13, 14,
15,21,22). Вторичная перетяжка быва-
бывает также в аутосомах пары № 9. В них
она располагается в околоцентромер-
ном районе длинного плеча.
Дифференциальное
окрашивание хромосом
Современные цитогенетические ме-
методики позволяют идентифицировать
по морфологии все пары хромосом на
препарате, а в ряде случаев и хромосо-
хромосомы внутри одной пары. Суть этих ме-
методик состоит в дифференциальном
окрашивании нативных хромосом по
длине, что обеспечивается сравнитель-
сравнительно простыми температурно-солевыми
воздействиями на фиксированные
хромосомы или использованием спе-
специфических красителей (рис. 3.19).
Дифференциальное окрашивание при-
приводит к появлению линейного рисунка
но длине хромосомы.
Несмотря на большое разнообразие
способов обработки хромосомных пре-
препаратов и красителей, выявляемый ли-
линейный рисунок хромосомы всегда
один и тот же. Он меняется только в
зависимости от степени конденсиро-
конденсированное™ хромосомы. Сегмент, види-
видимый как одна полоса в метафазной
хромосоме, в менее конденсированной
прометафазной хромосоме, может
предстать в виде нескольких мелких
полос.
Дифференциальное окрашивание в
зависимости от используемого метода
может охватывать либо всю длину хро-
хромосомы, либо ее центромерный район.
Представление о рисунке диффе-
дифференциально окрашенных по всей дли-
длине хромосом можно получить, окраши-
окрашивая препараты по G-методу с исполь-
использованием красителя Гимзы. В этом
случае хромосомы выглядят состоя-
состоящими из поперечноисчерченных, по-
разному окрашенных сегментов. Каж-
Каждой паре хромосом присущ индивиду-
индивидуальный рисунок исчерченности за счет
неодинаковых размеров сегментов. В
мелких хромосомах рисунок образует-
образуется единичными сегментами, в крупных
хромосомах сегментов много. Общее
для нормального хромосомного набо-
набора число окрашенных и неокрашенных
сегментов в метафазе составляет около
400. В прометафазных хромосомах оно
увеличивается до 850 и более.
62
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
19
Рис. 3.19. Рисунок сегментации дифференциально окрашенных хромосом
в соответствии с Парижской номенклатурой (G-, Q- и R-сегменты). G и Q-сегменты
неокрашены, R-сегменты - черные, вариабельные районы заштрихованы
Разные типы сегментов получили
обозначения G-, Q-, R-, С-, и Т по на-
названиям методов, с помощью которых
они выявляются. Так, для G-сегментов
это метод Гимза после предваритель-
предварительной обработки фиксированных хромо-
хромосом. Q-сегменты после окрашивания
акрихин-ипритом приобретают яркое
флюоресцирующее свечение. Рисунок
Q и G-сегментов полностью иденти-
идентичен. R-сегменты расположены между
Q-сегментами.
Место нахождения С-сегментов
можно обнаружить с использованием
других методов окрашивания. Эти сег-
сегменты связаны с локализацией кон-
конститутивного, или структурного гете-
рохроматина. В разных хромосомах
размер С-сегментов неодинаков. Гете-
рохроматин, обнаруженный по методу
С-окраски, содержится во всех хромо-
хромосомах человека. Он находится в около-
центромерных районах всех хромосом,
а также в дисталыюй части длинного
Хромосомы человека
плеча Y-хромосомы, на длинных пле-
плечах 1-й, 9-й и 16-й пар хромосом. Не-
Небольшие блоки структурного гетеро-
хроматина выявляются в плечах 2-й
пары и Х- хромосом. В акроцентричес-
ких хромосомах гетерохроматин
С-сегментов расположен в коротких
плечах. Накопленные сведения гово-
говорят, что в разных популяциях человека
размеры сегментов гетерохроматина
значительно различаются. Морфоло-
Морфологические особенности сегментов на-
наследуются по законам Менделя (рис.
3.20). Особенно изменчива величина
С-сегментов в аутосомах №№ 1, 4, 9,
13-15, 16, 21-22 и Y-хромосоме. По-
Поскольку у большинства носителей та-
таких особенностей кариотипа отсутст-
отсутствуют фенотипические аномалии, то
подобные особенности в строении хро-
хромосом можно рассматривать как вари-
варианты нормы. Согласно данным общей
цитогенетики, значительное количест-
количественное изменение гетерохроматина в
кариотипе не оказывает сильного от-
отрицательного влияния на развитие ор-
организма человека.
Еще один тип сегментов — Т — вы-
выявляется также с помощью специфи-
специфических методов окрашивания и при-
присутствует в теломерных районах всех
хромосом.
В метафазе митоза может быть вы-
выявлена еще одна характеристика ли-
линейной неоднородности хромосом.
Она связана с асинхронностью репли-
репликации ДНК в их разных участках. Та-
Такой тип окрашивания разбирается по-
подробно в главе 4 в разделе, посвящен-
посвященном клеточному циклу.
Индивидуальная совокупность сег-
сегментов, различающихся по ширине и
интенсивности окрашивания, образует
цитологическую карту каждой хромо-
хромосомы. Основанные на дифференциаль-
дифференциальном окрашивании хромосом цитологи-
63
Рис. 3.20. Наследование хромосомы,
содержащей особенно большой блок
конститутивного гетерохроматина
(С-сегмент), от отца к дочери
ческие карты имеют исключительное
значение для развития цитогенетики
человека. С помощью этих карт стало
реальным выяснить происхождение
аномальных хромосом, вплоть до точ-
точного описания, какие конкретно райо-
районы вовлекаются в ту или иную форму
хромосомного нарушения. На между-
международных совещаниях но номенклатуре
в цитогенетике человека была разрабо-
разработана и введена в практику система обо-
обозначения сегментов нормальных хро-
хромосом и хромосом, подвергшихся тем
или иным структурным перестройкам.
Дальнейшее совершенствование ме-
методов окрашивания хромосом позво-
позволило выявить до 1000 полос на всех
23-х хромосомах человека, характер-
характерных для гаплоидного набора. В сред-
среднем на хромосому при этом приходит-
приходится 50 полос, хотя на некоторых хромо-
хромосомах их можно обнаружить в не-
несколько раз больше, чем на других.
Гаплоидный геном человека состоит из
3x109 пар нуклеотидов, соответственно
каждая полоса содержит в среднем
3x106 пар нуклеотидов, что соответст-
соответствует нескольким сотням генов.
Половой гетерохроматин
В соматических клетках женщин по-
половой хроматин выявляется в виде ге-
гетерохроматина — небольшой хорошо
окрашенной округлой структуры, раз-
64
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Рис. 3.21. Микрофотография. Половой гетерохроматин в ядрах клеток
слизистой полости рта женщин (а, б)
мером 0,8-1,1 мкм, находящейся возле
ядерной мембраны (рис. 3.21). Поло-
Половой хроматин называют также тельцем
Барра, т. к. впервые он был описан
этим ученым в нейронах кошки. Позже
оказалось, что половой гетерохрома-
гетерохроматин присутствует в соматических
клетках всех млекопитающих женско-
женского пола, в том числе и человека. Гомо-
Гомологичные половому хроматину струк-
структуры, так называемые "барабанные па-
палочки" были обнаружены в ядрах по-
лиморфноядерных лейкоцитов. Поло-
Половой гетерохроматин — это одна из
Х-хромосом, которая находится в не-
активном; суперсиирализованном со-
состоянии. Известно, что фенотипичес-
ки пол у человека определяется нали-
наличием или отсутствием Y-хромосомы, а
не количеством Х-хромосом. Если в
кариотипе зиготы присутствует хотя
бы одна Y-хромосома, а количество
Х-хромосом превышает единицу, то по
фенотипу формируется мужчина. Ко-
Количество телец Барра в клетках всегда
на одно меньше, чем число Х-хромо-
Х-хромосом. То есть только одна Х-хромосома
в соматических клетках человека, и
мужчины, и женщины, всегда находит-
находится в активном состоянии. В норме жен-
женщина имеет две, а мужчина одну
Х-хромосому, в связи с чем инактива-
инактивация второй Х-хромосомы у женщин в
виде полового гетерохроматина слу-
служит механизмом компенсации разли-
различий в дозе генов, не оказывающих вли-
влияния на развитие половых признаков и
признаков, сцепленных с Х-хромосо-
мой. Этот же механизм оказался фак-
фактором, благоприятствующим носите-
носителям Х-хромосомных анеуплоидий. Ка-
Какое бы количество Х-хромосом они не
несли, генетически активна только од-
одна. Остальные же Х-хромосомы суще-
существуют в виде факультативного поло-
полового гетерохроматина. Поэтому но ко-
Современные методы картирования хромосом
65
личеству телец Барра в соматических
клетках можно диапюсцировать фор-
форму анеуплоидий. Например, у женщин
с кариотипом 47, XXX обнаруживают-
обнаруживаются два тельца Барра, а с кариотипом 45,
ХО — ни одного. У мужчин с кариоти-
кариотипом XXY — одно.
Образование полового хроматина из
Х-хромосомы происходит на ранних
стадиях эмбрионального развития.
Дробящаяся "женская" зигота млеко-
млекопитающих имеет две функционально
активные Х-хромосомы. У человека
половой хроматин появляется на ста-
стадии развития зародыша в несколько
сотен клеток. В трофобласте Х-хрома-
тин выявлен на 12-й день развития, а в
собственно эмбрионе — на 16-й день.
Половой хроматин образуется сразу во
всех клетках эмбриона. В настоящее
время наиболее приемлемой является
гипотеза, согласно которой в разных
клетках одного организма могут быть
инактивированы разные Х-хромосо-
Х-хромосомы: в одних — отцовская, в других —
материнская. То есть по Х-хромосоме
женщины мозаичны. Это положение
можно рассмотреть на примере актив-
активности фермента глюкозо-6-фосфат де-
гидрогеназы (Г6ФД), ген которого на-
находится в Х-хромосоме. Женщины
имеют два аллеля этого гена, а мужчи-
мужчины только один. Тем не менее средний
уровень активности этого фермента
сходен у представителей обоих полов,
следовательно, должен действовать
механизм дозовой компенсации. У ге-
гетерозиготных женщин с помощью эле-
электрофореза в разных клетках можно
выявить два аллеля Г6ФД. В ряде ра-
работ на эритроцитах in vivo и на фиб-
робластах соединительной ткани в
клеточных культурах у женщин был
выявлен мозаицизм по ГбФД.
Тельца Барра присутствуют не во
всех клетках женщины. Так, обе Х-хро-
Х-хромосомы активны в ооцитах и в клетках
женской половой системы.
Современные методы
картирования хромосом
На рубеже 70-х гг. XX в. молекуляр-
молекулярная генетика достигла определенной
завершенности в своем развитии : бы-
были установлены структура и механизм
репликации ДНК, провозглашена
"центральная догма" экспрессии гена
(транскрипция, трансляция), выясне-
выяснены основные аспекты регуляции ак-
активности гена. Главным объектом ис-
исследования в то время служили мик-
микроорганизмы. Существовавшие в тот
период методы не позволяли серьезно
продвинуться в изучении строения ге-
геномов зукариот, в том числе и генома
человека. Стремительный прорыв в
молекулярной генетике в 70-е гг. стал
возможен благодаря появлению новых
экспериментальных подходов — ис-
использованию рестрикционных эндо-
нуклеаз и становлению нового направ-
направления в молекулярной генетике — ген-
генной инженерии. С помощью этих мето-
методик были открыты совершенно неожи-
неожиданные факты, имеющие теоретичес-
теоретическое и практическое значение в облас-
областях знаний, связанных с действием ге-
генов. Это относится к генетическому
консультированию, включая нрена-
тальную диагностику, к развитию но-
новых подходов в изучении проблем эво-
эволюции и популяционной генетики.
Эти же успехи заставили ученых заду-
задуматься об этической стороне манипу-
манипулирования с человеческим зародышем,
об опасности возникновения возбуди-
возбудителей в процессе генно-инженерных
исследований.
Принципы новых эксперименталь-
экспериментальных подходов должны быть понятны
всем исследователям, однако детали
молекулярно-генетических методов,
66
ГламЗ. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Пяимхда Кмточиая миибраив
Клеточная стайка
Хромосом*
Рис. 3.22. Клетка Е. coli с хромосомой и плазмидой
которые привели к внушительному
прогрессу генетики человека, изложе-
изложены в специальных изданиях. Ниже
рассматриваются основные принципы
этих методов, и в качестве примера
описывается анализ C-глобинового
генного кластера человека с использо-
использованием рестрикционных ферментов,
гибридизации нуклеиновых кислот,
секвенирования ДНК и сортировки
хромосом с помощью цитофлуоромет-
рии.
Анализ р-глобинового
гена человека
Гемоглобин взрослого человека
HbAf состоит из четырех полипептид-
полипептидных цепей: двух а и двух р. Кроме та-
такого гемоглобина, у человека известны
и другие типы этой сложной молеку-
молекулы. Так, у эмбрионов функционирует
фетальный гемоглобин, в состав кото-
которого входят у-цепи, у взрослых людей
в небольшом количестве обнаружива-
обнаруживается гемоглобин НЬА2, в состав кото-
которого входят а-цепи. C-глобиновый кла-
кластер генов в настоящее время полно-
полностью идентифицирован и проанализи-
проанализирован молекулярнотенетическими
методами.
Этапы анализа.
В начале 60-х гг. был завершен ами-
аминокислотный анализ белка гемогло-
гемоглобина. Цепь р состоит из 146 амино-
аминокислот. Вся транскрибируемая часть
гена должна содержать 438 нуклеоти-
нуклеотидов, т. к. генетический код триплетен
A46*3 =438). Этот маленький участок
нужно идентифицировать на одной из
хромосом, в состав которой входит
нить ДНК длиной несколько сантиме-
сантиметров. После того, как этот участок вы-
выявлен, его нужно выделить и накопить
в большом количестве, чтобы опреде-
определить последовательность нуклеотидов
в составе анализируемого отрезка
ДНК-молекулы.
Современные методы картирования хромосом
67
Рестрикционные эндонуклеазы.
Рестрикция (разрезание) выделен-
выделенной из клеток ДНК осуществляется
ферментами — рестрикционными эн-
донуклеазами (рестриктазами). Эти
ферменты вступают в реакцию с опре-
определенными участками ДНК, так назы-
называемыми сайтами узнавания, которые
в клетке обычно бывают защищены
специальными группировками (мети-
(метилированы). После обработки ДНК эн-
донуклеазами фрагменты имеют так
называемые "липкие концы": рестрик-
тазы разрезают две цепи ДНК не в од-
одном месте, а в двух разных местах, в ре-
результате чего одна из цепей оказывает-
оказывается длиннее другой на несколько нукле-
отидов. Свободные нуклеотиды Moiyr
спариваться с комплементарными
нуклеотидами другого фрагмента
ДНК с липкими концами. Благодаря
этому, ДНК из различных источников
может объединяться, образуя реком-
бинантные молекулы. Это свойство
рестрикционных фрагментов ДНК ис-
используется в генной инженерии для
создания искусственных векторов на
основе бактериальных плазмид, или
фагов.
Помимо своей собственной молеку-
молекулы ДНК, замкнутой в кольцо, бакте-
бактерии часто содержат дополнительные
маленькие кольцевые двухцепочечные
молекулы ДНК (рис. 3.22), называе-
называемые нлазмидами. Плазмиды реплици-
реплицируются автономно, независимо от ос-
основной молекулы ДНК. Они могут со-
содержать некоторые гены, например,
определяющие устойчивость бактерий
к антибиотикам или контролирующие
синтез некоторых веществ. Плазмид-
ную ДНК можно выделить и расще-
расщепить подходящей рестриктазой только
в одном месте, при этом кольцевая мо-
молекула превращается в линейную с
липкими концами. Фрагменты чуже-
чужеродной ДНК с такими же липкими
концами, полученные после разреза-
разрезания аналогичной рестриктазой, мож-
можно сшить с плазмидной ДНК с помо-
помощью специального фермента — лига-
зы. Полученную рекомбинантную мо-
молекулу, называемую вектором, можно
ввести в бактерию, где она начнет
многократно реплицироваться. Чуже-
Чужеродный фрагмент ДНК (им может
стать и глобиновый ген) будет много-
многократно амплифицирован вместе с
плазмидой (рис. 3.23). Такая процеду-
процедура называется клонированием генов и
используется для создания банков ге-
генов. Этот методический подход позво-
позволяет разбить геном человека на от-
отдельные фрагменты, каждый из кото-
которых может быть размножен вне орга-
организма человека. Еще одна область при-
применения в молекулярной генетике рес-
трикраз — идентификация генов. Эти
задачи решаются с помощью метода,
разработанного Саузерном в 1975 г.
Суммарную ДНК из клеток челове-
человека гидролизуют эндонуклеазой при-
примерно на 500 000 фрагментов длиной
от 102 до 105 нуклеотидных нар. Затем
фрагменты разделяют по молекуляр-
молекулярной массе с помощью гель-электрофо-
гель-электрофореза на пластинках в агарозе. Следую-
Следующий этап — получение одноценочеч-
ных фрагментов ДНК денатурацией
щелочью прямо в геле. С агарозной
пластинки получают реплику- отпеча-
отпечаток на нитроцеллюлозном фильтре.
Перенесенные на фильтр одноцепо-
чечные фрагменты ДНК можно иден-
идентифицировать путем гибридизации с
радиоактивными ДНК-зондами. Лю-
Любой фрагмент, содержащий последова-
последовательность зондируемого гена, выявля-
выявляется в виде темной полосы на радиоав-
радиоавтографе (фильтре, покрытом фото-
фотоэмульсией и проявленном после взаи-
взаимодействия с радиоактивным зондом).
68
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
клетка
Рис. 3.23. Схема введения чужеродной ДНК в бактериальную плазмиду
с использованием эндонуклеазы RI
Современные методы картирования хромосом
69
Главное условие такого анализа — на-
наличие подходящего радиоактивного
ДНК-зонда.
Создание зонда для гена р-глобина
связано с получением матричной РНК
Р-глобина из созревающих эритроци-
эритроцитов, где активно синтезируется гемо-
гемоглобин. Затем с помощью фермента
обратной транскринтазы, который
обеспечивает считывание нуклеотид-
ной последовательности м-РНК в ком-
комплементарную последовательность
ДНК, получают так называемую к-
ДНК с высокой радиоактивной мет-
меткой. В настоящее время созданы биб-
библиотеки к-ДНК из разных источников.
Имеются геномные библиотеки чело-
человека, полученные генно-инженерными
методами, а также хромосомно-специ-
хромосомно-специфические библиотеки, полученные из
отдельных специфических хромосом
или их участков. Создание таких биб-
библиотек требует выделения отдельных
хромосом. В настоящее время это ста-
стало возможным благодаря сортировке
хромосом цитофлуорометрическим
методом. Для синтеза к-ДНК исполь-
используются также автоматизированные ус-
устройства, в том случае, если произво-
производится синтез к-ДНК гена определен-
определенного белка, аминокислотная последо-
последовательность которого известна.
Гибридизация нуклеиновых кислот
Идентификация фрагментов одно-
цепочечной ДНК на нитроцеллюлоз-
ном фильтре происходит с помощью
гибридизации цепей ДНК. Большинст-
Большинство природных ДНК встречается в виде
двухцепочечных молекул, где азотис-
азотистые основания двух цепей взаимодей-
взаимодействуют по принципу комплементарно-
сти с помощью водородных связей. Во-
Водородные связи в условиях щелочного
гидролиза разрываются, и на фильтре
фиксируются одноцеиочечные фраг-
фрагменты. При добавлении к-ДНК, если
комплементарны отдельные цепи, про-
происходит восстановление двойной спи-
спирали (рис. 3.24). Для выявления реас-
социированных молекул на фильтр на-
наносят фотоэмульсию. Под действием
излучения радиоактивных изотопов в
составе к-ДНК, в фотоэмульсионном
слое восстанавливаются зерна серебра,
которые выглядят после проявления
темными участками.
Метод гибридизации нуклеиновых
кислот можно использовать на препа-
препарате хромосом после их соответствую-
соответствующей обработки. Так, исследуемый геи
можно локализовать в специфическом
хромосомном сегменте, одном из тех
сегментов, которые наблюдаются при
дифференциальном окрашивании.
Поскольку эксперимент проводится с
зондом к-ДНК, синтезированном на
м-РНК, можно утверждать, что иден-
идентифицированный участок хромосомы
содержит активный ген. Известно, что
в геноме помимо активных генов, мо-
могут находиться псевдогены, такие по-
последовательности ДНК, которые гомо-
гомологичны активному гену, но не транс-
транскрибируются из-за отсутствия каких-
то важных последовательностей вне
транскрибируемой части.
Метод гибридизации нуклеиновых
кислот позволил идентифицировать
большое количество генов в хромосо-
хромосомах человека. В настоящее время до-
достоверно картировано около 8000 ге-
генов человека. В большинстве случаев к
идентифицированным генам найдены
альтернативные аллельные формы.
Для нескольких тысяч генов один из
альтернативных аллелей определяет
какое-либо наследственное заболева-
заболевание или аномалии. Другие известные
гены кодируют белки групп крови,
различные антигены, иммуноглобули-
иммуноглобулины, ферменты метаболизма и т.д.
70
ГламЗ. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Рис. 3.24. Принцип РНК или ДНК гибридизации, А - нуклеотидные цепи в растворе.
Б - гибридизация цепей в соответствии с правилами спаривания: тимин образует пару с
аденином, цитозин с гуанином. В - гибридизация может происходить даже
при неполной гомологии, важно, чтобы различия были не слишком велики
Секвенирование ДНК
В последние годы разработаны ме-
методы быстрого секвенирования ДНК.
Для этого длинную молекулу ДНК с
помощью рестрикраз разрезают на
фрагменты удобного размера. Затем
эти фрагменты размножают путем
клонирования и специальными мето-
методами определяют последовательность
нуклеотидов в каждом из фрагментов.
Очередность фрагментов восстанавли-
восстанавливают, используя перекрывающиеся
концы (участки ДНК с одинаковой по-
поел едовательнотью в разных клонах). С
помощью секвенирования получают
точные сведения и о нетранскрибируе-
мых участках ДНК, важных для кон-
контроля транскрипции.
Анализ C-глобинового гена.
Первый этап анализа связан с иден-
идентификацией этого гена в суммарной че-
человеческой ДНК. Для этого ДНК вы-
выделяют из клеток, с помощью эндонук-
леаз получают фрагменты, раздедяют
их в агарозе с помощью гель-электро-
гель-электрофореза, денатурируют на две отдель-
отдельные цепи и создают реплику с препара-
препарата на нитроцеллюлозном фильтре. Сле-
Следующий шаг состоит в том, чтобы най-
найти среди всех фрагментов р-глобино-
вый ген. Для этого необходим радиоак-
радиоактивный ДНК-зонд. Его синтезируют на
р-глобиновой РНК с помощью обрат-
обратной транскриптазы. Далее этот к-ДНК
зонд используют для двух задач. Пер-
Первое — идентифицировать фрагменты
Современные методы картирования хромосом
71
ДНК, соответствующие глобиновому
гену, прямо на фильтре. Задача вторая —
идентифицировать на препарате хро-
хромосом место нахождения этого гена.
Идентификация в обоих случаях про-
происходит с помощью гибридизации ра-
радиоактивной к-ДНК с денатурирован-
денатурированной ДНК препарата. Так, ген гемогло-
бина-р был локализован на коротком
плече 11-й хромосомы.
Для более подробной характеристи-
характеристики глобинового гена необходимо полу-
получить большое количество его ДНК.
Для этого клонируют к-ДНК в каком-
нибудь векторе, например, в бактери-
бактериальной плазмиде. Затем сравнивают
фрагменты геномной ДНК, содержа-
содержащие глобиновый ген, и клонированную
к-ДНК, соответствующую м-РНК.
Оказалось, что фрагменты геномной
ДНК значительно больше, чем к-ДНК.
В р-гемоглобиновом гене были выяв-
выявлены две вставочные последователь-
последовательности (нитроны), которые разделяют
три разные кодирующие области — эк-
зоны. (Исследования многих других
эукариотических генов, показали, что
наличие нитронов является правилом
для большинства из них). Выяснилось
также, что ген р-гемоглобина относит-
относится к уникальным последовательнос-
последовательностям. Кроме того, были найдены псев-
псевдогены, имеющие мутации в кодирую-
кодирующих и фланкирующих, регуляторных
участках. Они называются псевдогена-
псевдогенами, т.к. с них не идет транскрипция.
Сейчас известны последовательнос-
последовательности ДНК различных глобиновых генов.
Их изучение позволило решить много
общих проблем, касающихся организа-
организации и экспрессии генетического мате-
материала в клетке.
Полимеразная цепная реакция
В середине 80-х гг. в молекулярной
генетике был введен в практику совер-
совершенно новый метод, позволяющий
очень быстро размножить следовые
количества молекул ДНК или РНК
Чувствительность его такова, что поз-
позволяет обнаружить в пробе всего одну
присутствующую в ней молекулу. От-
Открытие этого метода позволило много-
многократно ускорить исследования по кар-
картированию генома человека.
Полимеразная цепная реакция была
открыта в 1984 г. К.Б. Мюллисом. Она
основана на том, что новосинтезируе-
мые цепи нуклеиновых кислот могут
служить матрицами в следующих цик-
циклах репликации. Реакция осуществля-
осуществляется последовательными циклами.
Каждый из них начинается с разделе-
разделения двуспиральной молекулы ДНК на
две одноцспочечные. В таком состоя-
состоянии каждая цепочка может служить
матрицей для репликации. Затем од-
ноцепочечные нити ДНК инкубируют
в присутствии ДНК-полимеразы в
растворе со смесью всех четырех нук-
леотидов и специфической последова-
последовательностью ДНК — праймером. При-
Присоединение праймера к одноцепочеч-
ной нити ДНК приводит к образова-
образованию небольшого двуцепочечного от-
отрезка, который удлиняется с помощью
ДНК-полимеразы. Процедуры много-
многократно повторяются, происходит мно-
множественное копирование старых и но-
новых одноцепочечных молекул. От-
Отдельный цикл занимает около 5 мин.,
клонирование одного фрагмента ДНК
происходит всего в течение несколь-
нескольких часов.
Помимо молекулярной генетики,
метод полимеразной цепной реакции
широко используется в практических
исследованиях: пренатальной диагно-
диагностике наследственных болезней, выяв-
выявлении вирусных инфекций, а также в
судебной медицине, поскольку этот
метод позволяет проводить генетичес-
72
Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
кую "дактилоскопию" по одной един-
единственной клетке.
Программа "Геном человека"
С развитием новых технологий мо-
молекулярных исследований, основан-
основанных на быстрых методах работы с
ДНК (клонировании фрагментов
ДНК генноинженерными способами и
с помощью полимеразной реакции, ав-
автоматизированном секвенировании), с
введением в практику молекулярноге-
нетических исследований компьютер-
компьютерных технологий сравнительного ана-
анализа строения геномов представителей
разных систематических групп, с раз-
развитием техники направленного воз-
воздействия на генетический аппарат
клетки и организма в целом и возмож-
возможности создания искусственных фер-
ферментов по нуклеотидной последова-
последовательности фрагмента ДНК темпы раз-
развития молекулярной генетики обрели
стремительный характер и привели к
возникновению в конце 80-х гг. между-
международной программы "Геном челове-
человека". Этот глобальный проект предпо-
предполагает к 2005 г. завершить определение
полной последовательности всех трех
миллиардов нуклеотидных звеньев,
составляющих геном человека. Приня-
Принятие такой программы означает, что ха-
характер развития молекулярной биоло-
биологии достиг совершенно нового уровня.
Произошедший качественный скачок
в технологии позволяет решать прин-
принципиально новые задачи.
Более ранние молекулярногенети-
ческие работы проводились с целью
исследовать строение генов, иденти-
идентифицировать их в целом геноме, изу-
изучить по возможности функцию гена и
уровни его регуляции. Методы совре-
современной молекулярной генетики позво-
позволяют отследить эффект действия того
или иного гена на уровне целого орга-
организма, изучать не отдельные гены, а
структуры и функции целых геномов.
На сегодняшний день изучены: гено-
геномы 141 вируса, более 50 геномов мито-
митохондрий из разных объектов, большое
количество геномов бактерий. Уста-
Установлено, что бактериальный геном со-
содержит 5-6 тыс. генов, из представите-
представителей эукариот наиболее близки к завер-
завершению изучение геномов дрожжей и
нематод. Показано, что геном дрож-
дрожжей имеет в своем составе около 6 тыс.
генов.
Суммарная длина нуклеотидных
последовательностей генома человека
соответствует 3 миллиардам. По дан-
данным разных авторов, такая гигантская
нуклеотидная последовательность мо-
может содержать от 50 до 100 тыс. генов.
В настоящее время известна структура
около 7 тыс. генов. Изучение структу-
структуры генов — не конечная цель програм-
программы. Помимо анализа последовательно-
последовательности нуклеотидов, проводится их кар-
картирование. Каждый ген приписывает-
приписывается к определенной хромосоме в строго
определенное место — локус, устанав-
устанавливается расстояние между генами, со-
составляется карта хромосом человека. В
настоящее время картированы около 8
тыс. генов. Увеличению скорости кар-
картирования генов на хромосомах спо-
способствует выявление маркерных по-
последовательностей для каждой хромо-
хромосомы. Эти маркерные последователь-
последовательности много раз повторяются вдоль
хромосомы и как бы делят ее на огра-
ограниченные участки. Работа с таким не-
небольшим участком хромосомы облег-
облегчает процедуру выделения гена. Благо-
Благодаря существованию маркерных по-
последовательностей, геном человека
разбит на отдельные фрагменты, и
каждый фрагмент в случае необходи-
необходимости может быть легко размножен
вне организма.
Программа "Геном человека"
73
Помимо задачи картирования генов
и установления их структуры, про-
программа "Геном человека" ставит цель
определить структурно-функциональ-
структурно-функциональную взаимосвязь генов. Для решения
этой задачи используются совершенно
новые подходы, которые просто невоз-
невозможно было представить себе несколь-
несколько лет назад. Так, по дефектному фер-
ферменту, который является причиной на-
наследственного заболевания, зная по-
последовательность аминокислот в его
составе, можно искусственно синтези-
синтезировать информационную РНК, а затем
соответствующий участок ДНК, иден-
идентифицировать его на хромосомной
карте, выделить нативный ген и кло-
клонировать его вне организма, чтобы ус-
установить, в чем причина образования
дефектного фермента. Таким способом
были изучены гены дистрофии Дюше-
на, рака молочной железы, мутантной
фенилаланингидроксилазы, являю-
являющейся причиной наследственной фе-
нилкетонурии, и ряда других генов.
Еще один новый методический иод-
ход в изучении функции генов связан с
использованием информационно-ком-
информационно-компьютерных технологий. Этот путь ис-
исследований основан на следующем
предположении : если у представите-
представителей разных систематических групп
имеются одинаковые по структуре ге-
гены, то они выполняют одинаковую
функцию. Таким образом была уста-
установлена причина развития рака тол-
толстой кишки у человека. Методами кло-
клонирования и картирования была изу-
изучена структура генов, отвечающих за
развитие рака толстой кишки. Затем
был проведен поиск в информацион-
информационном поле с помощью компьютерных
технологий. В процессе этого поиска
была предпринята попытка найти в ге-
геноме дрожжей, который уже полно-
полностью расшифрован, гены, сходные по
структуре с исследуемыми генами че-
человека. И они были обнаружены. Ока-
Оказалось, что у дрожжей такие же гены
отвечают за репарацию ДНК. Таким
образом, было установлено, что рак
толстой кишки связан с мутациями ге-
генов, кодирующих ферменты репара-
репарации ДНК.
Важнейшую роль в структурных ис-
исследованиях генома человека играет
изучение его полиморфизма. Популя-
ционный полиморфизм генома чело-
человека является основой для понимания
принципов молекулярной эволюции,
механизмов возникновения патологи-
патологических мутаций, для оценки факторов
риска при воздействии потенциально
токсических агентов окружающей сре-
среды на человеческий организм, нако-
наконец, для понимания основ различной
индивидуальной восприимчивости ле-
лекарств. Эти исследования получили
новый импульс с открытием поли-
полиморфных мини- и макросателлитных
последовательностей ДНК, которые
используют в качестве маркеров при
картировании генома человека.
Новым этапом в изучении структур-
структурно-функциональных связей между ге-
генами в программе "Геном человека" яв-
является возможность клонирования
крупных фрагментов генома в специ-
специальных векторах, способных размно-
размножаться в клетках вместе со встроенны-
встроенными в них фрагментами. В качестве век-
вектора в таких случаях используют ис-
искусственные дрожжевые хромосомы,
появление которых стало возможным
благодаря развитию генетики дрож-
дрожжей. Использование таких векторов
позволяет клонировать фрагменты
ДНК длиной до 106 пар оснований. Это
создает предпосылки для быстрого вы-
выделения нужного фрагмента генома и
использования его для структурного
или функционального анализа.
74 Глава 3. ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Использование новых методов в
изучении генетики человека позволяет
значительно ускорить процесс иссле-
исследовательской работы и накопить ин-
информацию о генах, играющих ключе-
ключевую роль в регуляции жизнедеятель-
жизнедеятельности клетки. К таким генам относятся
гены контроля клеточного цикла, ге-
гены, кодирующие компоненты цепи пе-
передачи сигнала от клеточной поверх-
поверхности к аппарату транскрипции в ядре,
гены, контролирующие развитие эмб-
эмбрионов, гены, ответственные за работу
защитных систем организма, гены им-
иммунной системы. Активно расширяет-
расширяется представление о генах, повреждение
которых приводит к возникновению
раковых опухолей, онкогенах, генах-
супрессорах, генах клеточной смерти и
апоптоза (программируемой гибели
клеток). Все более детальным стано-
становится знание надмолекулярных уров-
уровней организации регуляции экспрес-
экспрессии генов.
Выполнение программы "Геном че-
человека" приближает возможность ис-
использования генной терапии для лече-
лечения патологий, связанных с изменени-
изменением наследственной информации. Ген-
Генная терапия основана на введении в
организм больного искусственных ге-
генетических конструкций. Лечебный
эффект достигается в результате рабо-
работы введенного гена либо за счет подав-
подавления функции "больного" гена. Со-
Современная генная терапия делает пер-
первые шаги и имеет дело с соматически-
соматическими клетками в постнатальном периоде
жизни человека, но в то же время раз-
разрабатываются подходы к генной тера-
терапии клеток эмбриона.
Для создания генетических конст-
конструкций большей частью используются
вирусы. При этом из генома вируса
вырезается часть генов, ответственных
за формирование инфекционных ви-
вирусных частиц и разрушение инфици-
инфицированных клеток. Гены, необходимые
для переноса и функционирования ге-
генетической информации, сохраняют-
сохраняются. К ним добавляются клонированные
гены, оказывающие лечебный эффект.
Одними из наиболее приемлемых для
генной терапии окажутся, видимо, аде-
аденовирусные векторы, поскольку аде-
аденовирусы не встраиваются в геном
клетки хозяина, имеют широкий
спектр поражения тканей человека и
не индуцируют злокачественный рост.
В настоящее время разрабатывают-
разрабатываются способы для целевой доставки ис-
искусственных векторов к тем или иным
органам. Для этого используют ло-
локальные инъекции, аэрозольный ме-
метод, баллистический метод, основан-
основанный на обстреле ткани микрочастица-
микрочастицами тяжелых металлов (золота, вольф-
вольфрама), покрытых векторной ДНК.
Результаты генной терапии можно
рассмотреть на конкретном примере.
У мальчика, больного гемофилией из-
за дефицита девятого фактора сверты-
свертывания крови, с помощью биопсии были
выделены фибробласты кожи и куль-
культивированы в клеточной культуре, В
эти клетки был введен искусственный
вектор, созданный на основе ретрови-
руса и содержащий гены, которые уве-
увеличивают активность девятого факто-
фактора. Инфицированные клетки были
размножены в клеточной культуре и
возвращены в организм мальчика под-
подкожно в районе спины и ягодиц. Клет-
Клетки прижились, в результате была уве-
увеличена активность девятого фактора
свертывания крови, и симптомы гемо-
гемофилии у пациента были сглажены. Та-
Таким образом, генно-инженерные рабо-
работы создают подходы к лечению гемо-
гемофилии человека.
Генная терапия — направление, по-
появившееся на стыке двух наук: биоло-
Программа "Геном человека"
гии и медицины, пока имеет скромные
практические успехи, но с этим на-
направлением связано развитие медици-
медицины XXI-ro столетия.
Перспектива использования дости-
достижений программы "Геном человека"
многопланова: от идентификации ге-
генов, ответственных за возникновение
наследственных и приобретенных за-
заболеваний, до развития систем лече-
лечения, основанных на введении в орга-
организм новой генетической информа-
информации, корректирующей генетические
дефекты (генная терапия), и интенсив-
интенсивных методов диагностики, основанных
на выявлении генетических дефектов,
и перехода в диагностике к наиболее
полному обследованию популяций
для выявления предрасположенности
к болезни.
Вопросы для самоконтроля
1. Какие особенности строения ДНК позволяют
этим молекулам кодировать наследственную инфор-
информацию, самоудваиваться и мутировать?
2. В чем значение упаковки ДНК в хроматине и
хромосомах?
3. Чем гены отличаются от псевдогенов?
4. Сформулируйте понятия: гомологичные и по-
половые хромосомы, аутосомы.
5. Укажите практическое значение выявления по-
полового хроматина.
6. Перечислите современные методы картирова-
картирования хромосом. Поясните значение каждого метода.
7. В чем преимущество полимеразной цепной ре-
реакции?
8. Назовите новые подходы в изучении структур-
структурно- функциональных связей между генами в про-
программе "Геном человека". В чем их практическое зна-
значение?
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Материальной основой биологичес-
биологической преемственности поколений у чело-
человека является процесс оплодотворения:
слияние гамет — яйцеклетки и сперма-
сперматозоида — с образованием зиготы. Каж-
Каждая из гамет приносит равное количест-
количество хромосом. Если число хромосом в га-
гамете обозначить буквой п (гаплоидный
набор), то число хромосом в зиготе бу-
будет равно 2п (диплоидный набор). Об-
Образовавшаяся зигота многократно де-
делится митозом и дает начало новому ор-
организму. В результате каждого митоти-
ческого деления из одной клетки обра-
образуются две дочерние. Число хромосом в
них идентично их числу в родительской
клетке, равно как и качественный набор
генетического материала. Образование
гамет у человека, как и у других много-
многоклеточных эукариотических организ-
организмов, связано с мейотическим делением.
В результате этого деления количество
хромосом в половых клетках уменьша-
уменьшается в 2 раза и становится гаплоидным
(п). Равные по числу хромосом, образо-
образовавшиеся гаметы отличаются друг от
друга по качеству генетического мате-
материала в результате двух видов генетиче-
генетической рекомбинации: независимого рас-
распределения гомологичных хромосом к
полюсам деления и обмена участками
между гомологичными хромосомами в
процессе кроссинговера.
Рассмотрим процессы, лежащие в
основе передачи генетического мате-
материала, с учетом особенностей их про-
протекания у человека.
Митоз. Клеточный цикл
Общепризнано, что митоз — это са-
самый древний способ клеточного раз-
размножения, а все остальные формы де-
деления возникли в процессе эволюции
как его регуляционные или патологи-
патологические изменения. В основе этого
взгляда лежит представление о мито-
митозе как надежном способе распределе-
распределения наследственного материала меж-
между дочерними клетками. Характерной
чертой митоза является авторегуля-
авторегуляция, проявляющаяся в цикличности
протекания событий, связанных с
подготовкой клеток к митозу и самим
процессом деления. Эта цикличность
послужила основанием для возник-
возникновения понятия о митотическом
цикле.
Известно, что делящаяся клетка, а
вместе с ней и ядро, могут находиться
в двух состояниях: в митозе (деление)
и интерфазе (состояние между двумя
митозами). Во время митоза наследст-
наследственная информация, упакованная в
хромосомах, поровну распределяется
между дочерними клетками. В интер-
интерфазе, когда геном находится в "рабо-
"рабочем состоянии", наследственная ин-
информация реализуется. При этом хро-
хромосомы переходят в состояние хрома-
хроматина. Суперспирализованная с помо-
помощью специальных белков ДНК, час-
частично раскручивается, сохраняя
структуру двойной спирали.
На молекулах ДНК как на матри-
матрицах, по принципу комплементарности,
синтезируются все три типа молекул
РНК: информационная, транспортная
и рибосомная. Новосинтезированные
молекулы РНК в комплексе с белками
дозревают и покидают ядро, попадая в
цитоплазму, где с их участием проис-
происходит синтез белков.
Митоз. Клеточный цикл
77
Интерфаза подразделяется на три
периода: G,, S и G2. В период G, дочер-
дочерние клетки вступают после митоза. Ко-
Количество хромосом в них диплоидное,
каждая хромосома состоит из одной
хроматиды. Соответственно у человека
количество двуспиральных молекул
ДНК равно 46, по одной нитевидной
молекуле на хромосому, перешедшую в
состояние хроматина. Объем же клеток,
общее содержание органелл, белков и
РНК вдвое меньше, чем в исходной ро-
родительской клетке. В это время начина-
начинается рост клеток за счет накопления
клеточных белков, мембранных струк-
структур и органелл. Продолжительность пе-
периода G, непостоянна, и, в отличие от
других фаз клеточного цикла, может
изменяться от нулевых значений до
многих часов, (в некоторых случаях да-
даже месяцев) в зависимости от сроков
эмбрио- и онтогенеза, от особенностей
ткани и ряда других факторов.
В S-периоде удваивается ДНК каж-
каждой хроматиновой нити, при этом об-
общее количество ДНК возрастает в 2 ра-
раза (рис. 4.1). В клетках человека, как и в
эукариотических клетках других орга-
организмов, репликация ДНК происходит
одновременно на множестве отдельных
участков вдоль каждой хромосомы с
последующим соединением концов, об-
образовавшихся соседних отрезков. За
счет того, что репликация происходит в
сотнях точек, сокращается время, необ-
необходимое для удвоения молекул ДНК,
длина которых у человека измеряется в
сантиметрах. По данным разных авто-
авторов продолжительность S-периода у
млекопитающих варьирует в пределах
от 7 до 9 ч. Во время и после реплика-
репликации сестринские хроматиды могут об-
обмениваться сегментами: идет процесс
сестринских хроматидных обменов. Так
что обе хроматиды митотической хро-
хромосомы содержат участки другой хро-
ДНК
4с-
2c ¦
10ч
s
9ч
, ¦
'G2 '
4ч
Рис. 4.1. Изменение содержания ДНК
в клеточном цикле
2с — количество ДНК, соответствующее
диплоидному набору хромосом;
4с — удвоенное количество ДНК
матиды. В этом можно убедиться с по-
помощью специального окрашивания
хромосом (рис. 4.2).
Количество сестринских хроматид-
хроматидных обменов в клеточных культурах
человека часто используют в качестве
индикатора мутагенности среды : чем
больше межхроматидных обменов, тем
она выше.
В результате репликации образуют-
образуются две идентичные молекулы ДНК,
упакованные с помощью белков в хро-
хроматине. Эти молекулы отделены друг
от друга, но остаются соединенными в
центромерном районе хромосом. Цен-
Центромера располагается внутри гетеро-
гетерохроматического района. Преднолага-
Рис. 4.2. Сестринские хроматидные
обмены в эпителиальных клетках
перевиваемой клеточной линии V79.
Окрашивание гематоксилином - эозином
78
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
10
12
И И »» У V »
13 14 15
19
20
21
22
XX
Рис. 4.3. Репликационная структура метафазных хромосом, выявляемая
после обработки 5 - бромдезоксиуридином.
Индивидуальная идентификация всех хромосом набора.
Различие гомологичных X - хромосом
ется, что гетерохроматин обеспечивает
ее стабильность. В районе центромеры
расположена сателлитная ДНК, пред-
представленная кластерами высокоповто-
ряющихся последовательностей. Кро-
Кроме того, внутри центромеры находятся
уникальные последовательности ДНК,
которые, как полагают, несут информа-
информацию о расхождении хромосом к проти-
противоположным полюсам клетки.
Наличие множества точек начала
репликации ДНК на каждой хромосо-
хромосоме не означает одновременного начала
этого процесса в каждой точке. Для
репликации в клетках человека харак-
характерна асинхронность его в разных уча-
участках хромосом. В то же время поря-
порядок репликации участков каждой хро-
хромосомы строго постоянен, поэтому
воспроизведение его последовательно-
последовательности служит важнейшей характеристи-
характеристикой каждой хромосомы человека.
Первоначально репликацию ДНК в
хроматине клеток человека изучали с
помощью метода авторадиографии на
клеточных культурах, предварительно
меченых радиоактивным 3Н-тимиди-
ном. Выяснилось, что таким способом
можно легко различить морфологиче-
морфологически сходные хромосомы №№ 4 и 5, 17
и 18, а также акроцентрические хромо-
хромосомы №№ 13,14 и 15. Методом автора-
Митоз. Клеточный цикл
79
диографии было также показано, что в
женских клетках одна из двух Х-хро-
мосом отличается поздним началом
репликации и поздним окончанием
синтеза ДНК. Аналогичными характе-
характеристиками отличаются районы гетеро-
хроматина в X хромосоме человека,
что подтверждает идентичность этих
структур. Кроме того, метод авторади-
авторадиографии позволил улучшить иденти-
идентификацию хромосомных аномалий и
помог выявить несколько новых синд-
синдромов, связанных с патологией в стро-
строении хромосомы.
Новый этап в изучении последова-
последовательности синтеза ДНК по длине каж-
каждой хромосомы человека начался с ис-
использованием в качестве предшествен-
предшественника синтеза ДНК аналога тимидина —
5-бромдезоксиуридина. Включившие
его вместо тимидина участки хромосом
окрашиваются значительно слабее,
следовательно, в светлых участках реп-
репликация ДНК начинается раньше. Та-
Таким образом, цитогенетики получили
возможность изучать хронологию ре-
репродукции хромосом (рис. 4.3).
Каждая хромосома состоит из участ-
участков, реплицированных в разное время.
Районы с ранней и поздней реплика-
репликацией четко чередуютсяч. В метафаз-
ной хромосоме расположение светлых
и темных участков можно видеть с по-
помощью светового микроскопа. Строгая
специфичность картины позволяет
уверенно идентифицировать хромосо-
хромосомы в норме, а также при изменении ка-
риотипа в целом, равно как и отдель-
отдельной хромосомы.
В ^-периоде число молекул ДНК —
удвоенное. Количественное содержа-
содержание ДНК соответствует тетраплоидно-
му Dп) набору хромосом.Уровень син-
синтеза РНК и белка достигает максимума.
Клетки готовятся к вступлению в ми-
митоз, поэтому иначе эта часть клеточного
Рис.4.4. Схема клеточного цикла.
Клеточный цикл включает в себя события:
митотический цикл (М, G^, S, G2)
и состояние пролиферативного покоя
(R1, R2). 2с - количество ДНК
соответствующее диплоидному набору
хромосом, 4с - удвоенное
количество ДНК
цикла называется премитотическим пе-
периодом. Продолжительность G2-nepno-
да у млекопитающих около 4 ч.
В настоящее время клеточный цикл
рассматривают как последователь-
последовательность хронологически связанных со-
событий, происходящих после заверше-
завершения митоза в исходной клетке (Rj)
вплоть до завершения митоза в ее до-
дочерней клетке (R2) (рис. 4.4).
В начале 90-х гг. произошел прорыв
в изучении регуляторных механизмов
размножения клеток. В нормальных
условиях координация событий кле-
клеточного цикла обусловлена регуляци-
регуляцией трех переходов: вступления в митоз,
выхода из митоза и прохождения через
пункт ограничения в периоде Gt, после
которого клетки ориентируются на
синтез ДНК, то есть готовятся всту-
вступить в S-период. Так, идентифициро-
идентифицирован продукт активности гена, которо-
которому принадлежит центральная роль в
регуляции деления. Это белок
р34сас2) отличающийся высокой сте-
степенью филогенетической консерва-
80
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Рис. 4.5. Митоз. Изображены только 2 хромосомы из 46
Митоз. Клеточный цикл
81
тивности в клетках эукариотических
организмов. Показано, что и клетки
человека содержат продукт активнос-
активности данного гена или очень близкий его
гомолог. Наиболее вероятно, что
функциональная роль этого белка свя-
связана с фосфорилированием гистона
Н], участвующего в упаковке ДНК в
составе хроматина.
Процесс митоза в соматических
клетках человека идет стандартно
(рис. 4,5). К концу профазы хромосо-
хромосомы становятся отчетливо видимыми,
причем каждая состоит из двух хрома-
тид. Обе сестринские хроматиды при-
прилежат Q/uia к другой. Центромера во
всех хромосомах представлена неболь-
небольшой светлой кольцеобразной зоной.
Она удерживает две сестринские хро-
хроматиды вместе. Ядрышко исчезает,
ядерная оболочка распадается на
фрагменты. Хромосомы располагают-
располагаются в цитоплазме центральной части
клетки, оттесняя все органоиды к пе-
периферии.
Во время метафазы центромеры
всех хромосом располагаются в эква-
экваториальной плоскости между двумя
полюсами клетки. В это время они ут-
утрачивают вид четко выраженной пере-
перетяжки. Хроматиды каждой хромосомы
начинают отделяться одна от другой,
оставаясь соединенными только в цен-
тромерной области. В районе же цент-
центромер с противоположных сторон при-
прикрепляются нити веретена деления.
Их количество может достигать не-
нескольких десятков в районе каждой
центромеры.
Анафаза начинается с одновремен-
одновременного разделения всех центромер и рас-
расхождения сестринских хроматид каж-
каждой хромосомы к противоположным
полюсам. Утрата синхронности этого
процесса может привести к неправиль-
неправильному их расхождению. Центромеры с
Рис.4.6. Одно из первых изображении
митоза в сперматогониях человека
помощью специальных белков связы-
связываются с нитями веретена деления, ко-
которые и увлекают за собой дочерние
хроматиды к противоположным полю-
полюсам. Анафаза заканчивается с прекра-
прекращением движения хроматид, которые
становятся хромосомами. У каждого
полюса клетки должно оказаться по 46
состоящих из одной хроматиды таких
хромосом.
Телофаза связана с образованием
ядерных оболочек вокруг хромосом на
двух полюсах клетки и началом пере-
перехода хромосом в состояние хроматина.
Завершается телофаза возникновени-
возникновением перетяжки в центральной части де-
делящейся клетки, которая завершает
деление клетки надвое.
Самые ранние описания митоза в
клетках человека сделаны О.Винивор-
тером в 1912 г. Его рисунок с гистоло-
гистологического препарата семенников мож-
можно увидеть на иллюстрации (рис. 4.6).
На нем изображен митоз в сперматого-
нии, и количество хромосом оценива-
оценивалось автором как 48. Прошло более 50
лет, прежде чем появились методы,
позволившие уточнить, что их число в
клетках человека равно 46.
Для эукариот известно, что прохож-
прохождение митоза может быть заблокиро-
заблокировано, физиологически или экспери-
экспериментально, что приводит к развитию
полиплоидных клеток. Системная но-
82
Глава 4. ПЕРЕДАНА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
липлоидия не характерна для челове-
человека. Так, полиплоидные эмбрионы по-
погибают на ранних стадиях развития, и
это — одна из причин спонтанного пре-
прерывания беременности на ранних сро-
сроках. В то же время наличие полипло-
полиплоидных клеток в некоторых органах че-
человека не является патологией. Они
встречаются в сердечной мышце, осо-
особенно в предсердиях, в печени и ряде
желез. При этом клетки могут быть од-
одноядерными и двуядерными, а уровень
их полиплоидии невысок: обычно в
пределах 4 — 8п.
Мейоз
Мейоз — это особый тип клеточного
деления, возникший с появлением по-
полового размножения, при котором два
родителя — отец и мать — дают начало
новому организму. В процессе оплодо-
оплодотворения сливаются гаплоидные ядра
половых клеток родителей, что вдвое
увеличивает количество хромосом в
зиготе. Следовательно, при образова-
образовании половых клеток должно в два раза
уменьшаться количество хромосом, но
таким образом, чтобы совокупность ге-
генетического материала обеспечивала
преемственность поколений. Законо-
Закономерное чередование репликации ДНК
(а соответственно и хромосом), мито-
митозов и мейозов обеспечивает сохране-
сохранение видоспецифического кариотипа
как в индивидуальном развитии — он-
онтогенезе, так и в череде поколений ор-
организмов.
При мейозе из одной диплоидной
клетки образуется 4 гаплоидных. Кро-
Кроме того, в ходе этого деления происхо-
происходит два вида перегруппировки генети-
генетического материала хромосом, т.е. два
вида генетической рекомбинации: 1)
независимое распределение гомоло-
гомологичных хромосом к полюсам деления;
2) кроссинговер — обмен участками
между гомологичными хромосомами.
Эти процессы обеспечивают широчай-
широчайший спектр наследственной изменчи-
изменчивости и генетическую неповторимость
индивидов даже среди потомков одной
пары родителей.
Изучение мейоза у человека связано
с определенными методическими
трудностями из-за недоступности экс-
экспериментального материала. Обычно
мейоз изучают, культивируя in vitro
яичники эмбриона женского пола
старше трех месяцев из материала
аборта. Кроме того, используют мате-
материал биопсии семенников и яичников,
полученный при хирургическом вме-
вмешательстве по медицинским показани-
показаниям, а также трупный материал.
Мейотическое деление у человека
не имеет коренных отличий от мейоза
других эукариот. Есть, однако, некото-
некоторая специфика протекания мейоза в
ходе сперматогенеза и оогенеза у лиц
мужского и женского пола. Эта специ-
специфика затрагивает нюансы процесса,
касающиеся большей частью времен-
временных характеристик и тонкостей диф-
ференцировки. Мы рассмотрим про-
процессы сперматогенеза и оогенеза поз-
позже. Сначала вспомним характерные
этапы мейоза.
Мейоз состоит из двух делений,
следующих друг за другом, между ко-
которыми, что важно, не происходит уд-
удвоение ДНК, а следовательно, и хро-
хромосом. Перед мейозом обязательно
проходит интерфаза, в S-периоде ко-
которой ДНК реплицируется. Следова-
Следовательно, в профазе I мейотического де-
деления нитевидные хромосомы состоят
из двух хроматид. Каждое из двух де-
делений мейоза состоит из про-, мета-,
ана- и телофазы с индексами I или II
(рис. 4.7).
Первое мейотическое деление проте-
протекает значительно дольше, чем второе.
Мвйоз
83
Премейотический
митоз
Поздняя S-фаэа
Репликация ДНК
Лептотеиа
Зиготен!
Пахитвна
Рис. 4.7. Стадии мейоза. Поведение хромосом. Отцовские хромосомы
окрашены в черный цвет, материнские - в белый
84
Рис. 4.8. Синаптонемный комплекс
сперматоцита человека.
К — центромер». Стрелки указывают на
плотные участки. Верхняя и нижняя врез-
врезки: параллельные оси синаптонемного
комплекса. Изгибы могут соответство-
соответствовать местам рекомбинации
Самой длительной фазой первого мей-
отического деления является профаза,
так как именно в ней происходят такие
сложные процессы как образование би-
бивалентов из гомологичных хромосом и
кроссинговер. У женщин профаза мей-
оза I активно протекает в течение не-
нескольких месяцев в период внутриут-
внутриутробного развития, а полностью завер-
завершается к моменту овуляции в половоз-
половозрелом возрасте. Длительность этого пе-
периода у женщин объясняется также од-
одновременным протеканием процесса
дифференцировки и созревания цито-
цитоплазмы будущей яйцеклетки. У муж-
мужчин длительность профазы мейоза I со-
составляет 20 — 25 суток.
Профаза мейоза I подразделяется на
5 подфаз: лептотена, зиготена, нахите-
на, диплотена и диакинез.
Лептотена — стадия тонких нитей.
Ядра клеток, вступающих в мейоз, зна-
значительно крупнее других. В этих ядрах
вместо хроматина выявляются столь
тонкие и длинные нитевидные хромо-
хромосомы, что их трудно проследить по
всей длине. Для лептотены характерно
также появление на тонких хромосо-
Глам4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
мах особых структур, напоминающих
бусины. Это хромомеры — участки бо-
более сильно конденсированного хрома-
хроматина. Число, размер и расположение
хромомер специфичны для каждой
хромосомы. Появление хромомерных
структур отражает постепенный про-
процесс конденсации хромосом из хрома-
хроматина. Каждая хромосома состоит из
двух сестринских хроматид, но их да-
далеко не всегда удается различить под
световым микроскопом, настолько
близко они прилегают друг к другу.
Зиготена — стадия сливающихся
нитей. На этой стадии гомологичные
хромосомы находят друг друга и сли-
сливаются (конъюгируют) с образовани-
образованием бивалента. 46 хромосом человека
конъюгируют в 23 пары гомологичных
хромосом, следовательно, количество
бивалентов равно 23 и соответствует
гаплоидному хромосомному набору.
Бивалент — это стабильная структу-
структура из двух гомологичных хромосом и,
соответственно, из 4 хроматид. Ста-
Стабильность этой структуры поддержи-
поддерживают специфические белки синапто-
синаптонемного комплекса (рис. 4.8). Объеди-
Объединение гомологов чаще всего начинает-
начинается на концах хромосом — в теломерах, а
также в центромерных районах. Позд-
Позднее внутри бивалента по длине соеди-
соединяющихся хромосом формируются
сближающие их белковые тяжи синап-
синаптонемного комплекса.
В настоящее время выявлена специ-
специфичность хромомерного строения ин-
индивидуальных бивалентов человека в
мужском и женском мейозе, т.е. по ри-
рисунку хромомер можно определить, ка-
какая пара хромосом образует тот или
иной бивалент, даже если по размеру и
общей морфологии биваленты одинако-
одинаковы. Конъюгация гомологичных хромо-
хромосом с образованием бивалентов обяза-
обязательна для всех хромосом человека,
Мейоз
85
включая короткие и половые хромосо-
хромосомы. Известно, что конъюгация происхо-
происходит не только между половыми хромо-
хромосомами X и X, но также между X и Y
хромосомами, несмотря на значитель-
значительное превосходство в размерах Х-хромо-
сомы.
В процессе сперматогенеза образо-
образование полового бивалента из X и Y
хромосом начинается раньше других.
При этом конъюгируют между собой
часть короткого плеча Х- и короткое
плечо Y-хромосом (рис. 4.9). Экспери-
Эксперименты по гибридизации ДНК показа-
показали, что эти районы гомологичны меж-
между собой. Негомологичные участки X
и Y хромосом остаются свободными.
Зиготена заканчивается образова-
образованием 23 бивалентов.
Похищена — стадия прохождения
кроссинговера. В пахитене хромосомы
выявляются в виде толстых нитей, по-
поскольку они представлены бивалента-
бивалентами. Именно в бивалентах и происходит
кроссинговер: взаимный обмен иден-
идентичными участками по длине гомоло-
гомологичных хромосом. Генетическим след-
следствием кроссинговера является реком-
рекомбинация сцепленных генов, что обеспе-
обеспечивает широкую генетическую измен-
изменчивость гамет. Морфологически этот
процесс в пахитене уловить нельзя. Для
умозрительного восприятия он изобра-
изображен на схеме (рис. 4.10). Процесс обме-
обмена участками между ДНК несестрен-
ских хроматид в бивалентах можно
представить следующим образом. По
длине хромосомы разбросаны зоны по-
повторяющихся последовательностей
ДНК. С помощью ферментов-рестрик-
таз целостность их может легко нару-
нарушаться, при этом однонитевые участки
молекул ДНК соседних несестринских
хроматид могут образовывать короткие
двунитевые гибриды. Другие, репари-
рующие ферменты способны восста-
Рис. 4.9. Спаривание коротких плеч
хромосом X и Yb раннем мейозе человека
навливать целостность поврежденных
участков. Таким образом, кроссинго-
кроссинговер — это процесс, происходящий со
сложными пространственными измене-
изменениями суперспирализованных участков
молекул ДНК несестренских хроматид
с использованием целого комплекса
ферментов, возможно, объединенных в
специализированную структуру.
Кроссинговер обязательно происхо-
происходит в каждом биваленте. Не исключено,
что отсутствие кроссинговера в каком-
то из бивалентов может быть фактором,
прекращающим течение мейоза.
Позже, в следующей стадии, когда
гомологичные хромосомы в бивален-
бивалентах начинают расходиться, выявляют-
выявляются места, где происходил процесс крос-
кроссинговера. В них гомологичные хромо-
хромосомы во время разрушения бивалентов
соединены дольше всего. Поскольку
морфологически они напоминают гре-
греческую букву "X", их называют хиаз-
хиазмами (рис. 4.10). В зоне хиазм видно,
86
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Рис. 4.10. Схема кроссинговера.
а - парные гомологи, лежащие отдельно;
б - их спирализация в пахитене;
в - расположение хроматид в диплотене;
г - диакинез.
Стрелки указывают на места кроссинго-
кроссинговера
что в перекрест вовлекаются только
хроматиды из четырех: по одной от
каждого гомолога.
Диплотена — стадия двойных ни-
нитей. На этой стадии синаптонемный
комплекс разрушается, и гомологи от-
отталкиваются друг от друга, оставаясь
соединенными только в районе хиазм,
где по-прежнему сохраняется струк-
структура синаптонемного комплекса. По-
Поскольку каждая хиазма соответствует
одному событию кроссинговера, в ко-
котором участвуют две несестренские
хроматиды, то но количеству хиазм
можно судить об интенсивности про-
процесса кроссинговера. По данным рав-
равных авторов, общее число хиазм на
хромосомный набор человека колеб-
колеблется от 35 до 66. Некоторые бивален-
биваленты могут содержать несколько (до 5-
6) хиазм. В среднем на бивалент их
приходится около двух. Анализ гене-
генетического сцепления показал, что
кроссинговер у женщин происходит
Рис. 4.11. Расхождение хромосом
в первом делении мейоза (а) и во втором
делении мейоза (б). 1 - микротрубочки,
2 - хиазма, 3 - сестринские хромзтиды
чаще, чем у мужчин, следовательно, и
хиазм у женщин должно быть больше.
Диакинез — стадия, завершающая
профазу мейоза I. Она является пере-
переходной к метафазе. Число хиазм в ней
уменьшается, биваленты укорачива-
укорачиваются, разрушается ядро, начинает фор-
формироваться веретено деления.
В метафазе I биваленты располага-
располагаются в экваториальной плоскости, в
цитоплазме. Центромеры хромосом
лежат на экваторе, к ним прикреплены
нити веретена деления. Число выстро-
выстроившихся бивалетов соответствует гап-
гаплоидному набору хромосом и у челове-
человека равно 23.
В анафазе I происходит расхожде-
расхождение гомологичных хромосом к проти-
противоположным полюсам клетки. Каждая
хромосома состоит из двух сестрин-
сестринских хроматид (рис. 4.11, а).
Телофаза I. В этой фазе происходит
образование двух дочерних ядер каж-
каждое из которых содержит гаплоидное
Мейоз
87
Рис. 4.12. Схема случайного распределения гомологичных хромосом в анафазе i
первого мейотического деления. Обозначения в тексте I
число хромосом, равное 23. Каждая
хромосома состоит из двух сестрин-
сестринских хроматид.
Промежуток между двумя последу-
последующими делениями мейоза очень неве-
невелик. Почти сразу начинается второе
мейотическое деление. Оно идет по
схеме митоза: 23 хромосомы, состоя-
состоящие из парных сестринских хроматид,
связанных в центромерных участках, в
каждом из двух образованных ядер
проходят профазу и метафазу. В ана-
анафазе они разъединяются и расходятся
к противоположным полюсам (рис.
4.11,6), в результате чего образуется
четыре гаплоидных ядра (рис. 4.7),
различающихся по качеству генетиче-
генетической информации.
Рекомбинация генетического мате-
материала в мейозе осуществляется не толь-
только за счет кроссинговера. В анафазе пер-
первого мейотического деления происхо-
происходит случайное по отношению к полюсам
клетки распределение гомологичных
хромосом из каждого бивалента, что
приводит к возникновению большого
числа комбинаций отцовских и мате-
материнских хромосом в гаметах. Рассмот-
Рассмотрим этот процесс подробнее, проанали-
проанализировав распределение первой и второй
пары гомологичных хромосом в анафа-
анафазе I. Известно, что в каждой паре гомо-
88
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
логичных хромосом зиготы одна хромо-
хромосома приходит из гаметы отца, другая —
из гаметы матери. Обозначим заглавны-
заглавными буквами хромосомы отца, а пропис-
прописными — хромосомы матери (рис. 4.12,
а). А и а — первая пара хромосом, В и b —
вторая пара. В профазе I образуются би-
биваленты. В метафазе I они выстраива-
выстраиваются в экваториальной плоскости (рис.
4.12, б). В анафазе I гомологичные хро-
хромосомы расходятся к противополож-
противоположным полюсам: к одному полюсу пойдут
хромосомы А и В, то есть отцовские, а к
другому а и Ь, то есть материнские (рис.
4.12, в). Но у этого события может быть
и другой исход, когда расположение
хромосом в метафазе на экваторе будет
другим (рис. 4.12, г). Тогда к одному по-
полюсу пойдут хромосомы А и Ь, а к дру-
другому — а и В, то есть сочетание хромо-
хромосом на полюсах будет: одна отцовская,
одна материнская (рис. 4.12, д). Нали-
Наличие двух пар гомологичных хромосом
обеспечивает, как мы видим, образова-
образование четырех типов гамет, качественно
отличающихся друг от друга сочетани-
сочетанием отцовских и материнских хромосом.
У человека 23 пары хромосом, и разно-
разнообразие гамет оценивается как 2м, что
примерно равно 10 миллионам вариан-
вариантов сочетаний отцовских и материнских
хромосом. При оплодотворении прак-
практически равновероятна встреча любого
из сперматозоидов с яйцеклеткой. Это
увеличивает число возможных геноти-
генотипов детей до 223 х 223. Частота генетичес-
генетической рекомбинации в результате незави-
независимого распределения разных пар гомо-
гомологов выше, чем частота рекомбинации
в результате кроссинговера.
Гаметогенез
Гаметогенезом называется процесс
образования зрелых половых клеток-
гамет: мужских — сперматозоидов
(сперматогенез) и женских — яйцекле-
яйцеклеток (оогенез). Гаметогенез происходит
в половых железах (гонадах). Дипло-
Диплоидные клетки зачаткового эпителия
мужских и женских гонад многократ-
многократно делятся митозом, что приводит к
значительному увеличению их коли-
количества, — это фаза размножения. За-
Зачатковые клетки, делящиеся митозом,
называются гониями: мужские — спер-
матогониями, женские — оогониями.
Напомним, что они имеют диплоид-
диплоидный набор хромосом. Фаза размноже-
размножения сменяется фазой роста. Ее обяза-
обязательно проходит каждый гоний. Во
время фазы роста гонии увеличивают-
увеличиваются в объеме,а также осуществляют оп-
определенные этапы дифференцировки,
связанные с изменением структуры
цитоплазмы и ядра. Дифференциров-
ка сиерматогонисв и оогониев идет
различными путями. В течение фазы
роста обязательно имеется S-период, в
котором удваиваются все молекулы
ДНК. В результате каждая хромосома,
находящаяся в состоянии хроматина,
по завершению S-периода будет состо-
состоять из двух хроматид, соединенных
между собой центромерой. Фаза роста
заканчивается, когда гонии преобразу-
преобразуются в качественно новые клетки:
сперматоциты или ооциты I порядка,
готовые вступить в мейоз. Начало мей-
оза открывает новую фазу гаметогене-
за — фазу созревания. Во время фазы
созревания происходят первое и вто-
второе деления мейоза. Из одной дипло-
диплоидной клетки образуется 4 гаплоид-
гаплоидные, отличающиеся друг от друга по
качеству генетической информации.
Помимо мейоза, в фазе созревания
происходит дифференцировка гапло-
гаплоидных клеток и формирование зрелых
гамет.
Половая дифференцировка у эмб-
эмбриона человека начинается очень рано,
в возрасте 5 недель, когда эмбрион
Гаметогенез
89
Рис. 4.13. Схема этапов дифференцировки, наблюдаемых в семенных канальцах.
1 - сперматиды на разных этапах дифференцировки; 2 - сперматоциты второго порядка
в интерфазе; 3 - сперматоциты первого порядка в пахитене; 4 - сперматогонии;
5 - клетка Сертоли
имеет размер 2-3 см. В это время за-
закладываются зачатки половых орга-
органов. Развитие яичников у женщин и у
мужчин основано на митотическом де-
делении клеток и дифференцировке
морфологических структур. Начало
дифференцировки половой системы у
пятинедельного эмбриона связано с
миграцией клеток из желточного меш-
мешка в яичники у девочек и в яички у
мальчиков. Именно мигрирующие
клетки дают начало гониям. Таким об-
образом, первая фаза — фаза размноже-
размножения — начинается у эмбриона человека
на пятой неделе развития. Сперматоге-
Сперматогенез и оогенез у человека идут по-раз-
по-разному. Рассмотрим эти виды гаметоге-
нсза в отдельности.
Сперматогенез
Сперматогенез происходит в изви-
извитых семенных канальцах яичка. Отту-
Оттуда продукты сперматогенеза поступа-
поступают в семявыводящий проток, и далее —
в семяизвергательный канал.
Эпителий зрелого семенного каналь-
канальца состоит примерно из 6 слоев клеток
(рис. 4.13). Самый наружный слой —
клетки зачаткового эпителия, которые
делятся митозом. Образовавшиеся в ре-
результате этого деления клетки переме-
перемещаются внутрь канальца, образуя вто-
второй слой. В нем клетки также сохраня-
сохраняют способность к делению митозом.
Они будут являться сперматогониями.
Фаза размножения в процессе сперма-
сперматогенеза связана именно с этими двумя
слоями клеток. Перемещение размно-
размножающихся сперматогониев в третий
слой связано с началом фазы роста и
превращения сперматогониев в более
крупные сперматоциты 1 порядка. В
этом слое клеток происходит первое
мейотическое деление, и из одного
сперматоцита первого порядка образу-
образуются две клетки с гаплоидным набором
хромосом, которые становятся сперма-
тоцитами второго порядка.
Любой этап клеточного деления со-
сопровождается перемещением клеток
90
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Митохондрии
Зона 10
Гольджи * -
Ядро
Митохондрии
перемещаются
к жгутику
Акросома
системная
Центриоль
Вид
сбоку
Остаточная
зона Гольджи
Д
Формирование
головной
шапочки
акросомы
Хвостовая
трубка
Жгутик
Головка
(ядро, покрытое
головной шапочкой)
с акросомой
Шейка, содержащая
митохондрии
Вид спереди
Рис. 4.14. Схема последовательных этапов превращения
сперматиды (а-г) в сперматозоид (д)
Сперматогенез
91
из более наружного слоя в более внут-
внутренний. Изменяется окружение кле-
клеток, соответственно изменяется и
дифференцировка. Каждый спермато-
цит второго порядка проходит второе
мейотическое деление и дает начало
двум сперматидам с гаплоидным на-
набором хромосом. Вначале сперматиды
являются небольшими клетками с ок-
округлым ядром. Вскоре ядро удлиняет-
удлиняется и располагается у того конца клет-
клетки, который обращен к стенке каналь-
канальца. В каждой сперматиде одна из двух
центриолей образует жгутик, конец
которого обращен к центру канальца
(рис. 4.14). Созревание сперматид
происходит в самых внутренних слоях
семенного канальца. Здесь же сперма-
сперматиды превращаются в зрелые сперма-
сперматозоиды, способные к движению с по-
помощью жгутика и покидают семенной
каналец.
У человека длительность фазы рос-
роста, составляет 14 дней. Мужские поло-
половые клетки не развиваются поодиноч-
поодиночке. Они растут в клонах и объединены
между собой цитоплазматическими
мостиками. Контакты между клетками
разрушаются лишь при формировании
зрелых сперматозоидов. Вспомога-
Вспомогательную функцию выполняют клетки
Сертоли.
Таким образом, в результате процес-
процесса сперматогенеза из одного диплоид-
диплоидного сперматоцита I порядка получа-
получаются 4 зрелых сперматозоида. Общая
схема процесса сперматогенеза пред-
представлена на рисунке (рис. 4.15).
В детстве (от рождения до 10 лет)
семенные канальцы развиты слабо.
Они состоят из клеток зачаткового
эпителия, покрывающих каналец, и
сперматогониев. В подростковом воз-
возрасте (от 10 до 14 лет) сперматогонии
начинают активно делиться, и процесс
сперматогенеза активируется. В даль-
дальнейшем сперматогенез происходит не-
непрерывно, в течение всей жизни муж-
мужчины.
Семенники расположены вне брюш-
брюшной полости, и поэтому спермин раз-
развиваются при температуре на 2-3°С
ниже температуры внутренних облас-
областей тела. У мужчин, принимающих
очень горячие ванны или носящих
слишком тесные трусы, образование
сиермиев снижается, что в конечном
счете может привести к бесплодию. В
норме процесс образования спермия
занимает около 70 суток. На 1 г веса
яичка образуется 107 спермиев в сутки.
Сперматозоиды, или спермин, — это
очень мелкие подвижные клетки. В го-
головке сперматозоида находится ядро,
содержащее гаплоидный набор хромо-
хромосом B3). К периферии от ядра лежит
особая структура — акросома. Функ-
Функционально акросому можно рассмат-
рассматривать как увеличенную лизосому, т.к.
к эта органелла ограничена одной мем-
мембраной и наполнена гидролитически-
гидролитическими ферментами, помогающими про-
проникновению сиермия в ооцит. Округ-
Округлая головка сперматозоида сужается и
переходит в шейку, где иод прямым уг-
углом другу к другу располагается пара
центриолей. Микротрубочки одной из
центриолей являются местом образо-
образования осевой структуры жгутика так,
что система микротрубочек жгутика
проходит от самого начала шейки
сперматозоида до кончика его хвоста.
Более нижняя часть шейки расширена
за счет многочисленных митохондрий,
уложенных в виде спирали вокруг
жгутика. Митохондрии обеспечивают
энергией движение сперматозоида. За-
Заканчивается спермий жгутиком, имею-
имеющим типичное строение.
У человека максимальная дневная
продуктивность составляет 106 спер-
сперматозоидов. Продолжительность су-
92
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Фаза
развития
р
а
з
м
н
о
ж
е
н
и
е
Р
о
Механизм деления
Тип половых клеток Число хромосом
Клетка зачаткового
эпителия
Митоз
'\
Повторные
деления
\
46
Сперматогонии
46
Первое деление мейоза
Второе деление мейоза
Спермогенез
Сперматоцит
первого порядка
46
Сперматоцит
второго порядка
23
Сперматиды
(претерпевающие
изменения)
23
Сперматозоиды
(спермин)
23
Рис. 4.15. Схема сперматогенеза у человека
ществования спермия во влагалище
достигает 2,5 ч, а в шейке матки — 48 ч.
Функция яичек человека не ограни-
ограничена сперматогенезом. Велика их роль
и в обеспечении организма гормонами.
В специальных клетках, находящихся
между семенными канальцами яичка,
синтезируется главный андрогенный
гормон — тестостерон. Он необходим
для образования жизнеспособных
спермиев, а также для развития и под-
поддержания вторичных половых призна-
признаков. К этим признакам относятся: об-
образование наружных мужских поло-
половых органов и придаточных желез ре-
репродуктивной системы, усиленное
развитие мускулатуры, увеличение
гортани и соответственно более низ-
низкий голос, рост и распределение воло-
волосяного покрова, особенности поведе-
поведения, связанные с половой жизнью и
родительскими заботами.
Оогенез
Оогенез
Оогенез происходит в яичниках. У
половозрелой женщины они представ-
представляют собой уплощенные овальные тела
длиной 2,5-5,0 см, шириной 1,5-3,0 см.
Яйцеклетки расположены в корковом
слое яичника в эпителиальных пу-
пузырьках, называемых фолликулами.
Вне беременности примерно каждые
28 дней в одном из яичников созрева-
созревает очередной фолликул, который ло-
лопается, высвобождая ооцит второго
порядка. Этот процесс называется
овуляцией.
Развитие яичников у эмбриона на-
начинается, как было оказано выше, в
возрасте 5 недель. Примерно в то же
время, когда закладываются яичники,
среди клеток коркового слоя выявля-
выявляются первичные половые клетки. Они
мигрируют в развивающийся яичник
из эндодермы желточного мешка.
Женские половые клетки, мигрирую-
мигрирующие в яичник и располагающиеся в его
строме, называют оогониями. В начале
развития яичников эти клетки размно-
размножаются митозом, и число их возраста-
возрастает. Однако еще в пренатальном перио-
периоде большинство первичных половых
клеток погибает. В момент рождения
девочки их количество равно пример-
примерно 2 млн. в обоих яичниках. К моменту
полового созревания большинство из
них дегенерирует, так что в яичниках
их остается только около 40000. Лишь
примерно 450 из них достигают стадии
ооцитов второго порядка и выходят из
яичника в процессе овуляции.
В развивающемся яичнике первич-
первичные полоные клетки покрываются тон-
тонким слоем эпителиальных клеток, об-
образуя фолликулы (рис. 4.16). Клетки,
окружающие ооцит одним слоем, на-
называются фолликулярными. До треть-
третьего месяца жизни плода происходят
только митотические деления. При-
93
мерно к концу третьего месяца оого-
нии, находящиеся в фолликуле, начи-
начинают активно расти и дифференциро-
дифференцироваться, в результате чего каждый оого-
ний превращается в ооцит первого по-
порядка. Ооциты первого порядка всту-
вступают в мейоз. Первое редукционное
деление мейоза в ооцитах первого по-
порядка начинается в пренатальном пе-
периоде к концу третьего месяца жизни
плода. Число ооцитов, вступивших в
это время в мейоз, невелико. На препа-
препаратах их идентифицируют но лептоте-
не и зиготене. Начиная с седьмого ме-
месяца в мейоз вступают новые ооциты
первого порядка. Первые пахитены и
диплотены наблюдаются у семимесяч-
семимесячного плода.
Ооциты первого порядка вступают в
профазу первого деления мейоза, но не
завершают его. Мейоз задерживается в
конце профазы, и клетки вступают в
"фазу покоя" - диктиотену. Ко време-
времени рождения девочки ооциты первого
порядка продолжают находиться на
этой стадии. Их деление завершается
только после наступления половой
зрелости.
Между рождением и наступлением
половой зрелости яичник увеличива-
увеличивается в размере, но лишь отдельные
фолликулы начинают расти и разви-
развиваться. Изменения, которые происхо-
происходят в организме женщины в период по-
полового созревания,обусловлены поло-
половой функцией яичников, которые сти-
стимулируются гонадотропными гормо-
гормонами гипофиза.
Каждый из нескольких тысяч пер-
первичных фолликулов, расположенных в
корковом веществе яичника, ко времени
полового созревания состоит из ооцита
первого порядка диаметром 25-30 мкм,
окруженного одним слоем фоллику-
фолликулярных клеток. Ядро ооцита первого
порядка остается в профазе первого
94
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
12
1 - первичный фолли-
фолликул; 2 - ооцит первого по-
порядка; 3 - фолликулярные
клетки; 4 - полость созре-
созревающего фолликула; 5 -
zona pellucida; 6 - лучис-
лучистый венец (corona radiata);
7 - первое полярное тель-
тельце; 8 - ооцит второго по-
порядка; 9 - метафаза вто-
второго мейотического деле-
деления; 10 - овуляция; 11 -
разрывающийся Граафов
пузырек; 12 - желтое тело
8
Рис. 4.16. Схема последовательных стадий развития, роста и созревания фолликула
и ооцита в яичнике женщины
деления мейоза: ядрышко хорошо вы-
выражено, хромосомы вытянуты в тон-
тонкие нити. В таком виде они остаются,
пока, спустя годы, не завершится мей-
оз. В цитоплазме ооцита можно на-
наблюдать желточные зерна. В электрон-
электронном микроскопе в цитоплазме ооцита
видны обычные органеллы.
Примерно каждые 28 дней в яични-
яичнике половозрелой небеременной жен-
женщины развивается несколько фолли-
фолликулов. Они увеличиваются в размере и
приближаются к поверхности. Однако
обычно только один из них созревает,
прорывает поверхность яичника и вы-
высвобождает ооцит второго порядка.
Развитие зрелого фолликула связа-
связано с двукратным увеличением разме-
размеров ооцита и образованием блестящей
оболочки (zona pellucida) вокруг него.
Стадия роста ооцита сопровождает-
сопровождается активным делением фолликуляр-
фолликулярных клеток и накоплением фоллику-
фолликулярной жидкости внутри фолликула.
Когда фолликул разрывается, находя-
находящийся в нем ооцит второго порядка
окружен фолликулярными клетками.
Эти клетки формируют вокруг ооцита
лучистый венец (corona radiata).
Период роста ооцита сопровождает-
сопровождается образованием большого количества
разных типов РНК. Для этого в клетке
существуют разнообразные механиз-
механизмы. Накопление рибосомной РНК и
образование множества ядрышек, где
происходит сборка и созревание субъе-
субъединиц рибосом, обеспечивается меха-
механизмом амплификации генов. Ампли-
Амплификация рибосомных генов представ-
представляет собой процесс множественной ре-
редупликации участков ДНК, кодирую-
кодирующих рибосомную РНК. В результате
такой локальной редупликации ДНК
количество копий рибосомных генов
увеличивается до 1-2 тысяч, и каждая
из них является районом ядрышкового
Оогмм
95
организатора. В результате образуется
более тысячи дополнительных ядры-
ядрышек, где синтезируются и созревают
рибосомные РНК, необходимые для
сборки рибосом. Рибосомы, в свою оче-
очередь, необходимы для синтеза белка.
Быстрое увеличение количества
разнообразных информационных
РНК происходит за счет активации
транскрипции в локально деспирали-
зованных участках хромосом, когда
хромосомы приобретают структуру
"ламповых щеток". Такая структура
характерна для созревающих ооцитов
у многих видов животных (рис. 4.17).
Известно, что начинает она проявлять-
проявляться в пахитене, а наиболее выражена в
диплотене и диктиотене.
Увеличение количества разнообраз-
разнообразных РНК обеспечивает накопление
питательных веществ в цитоплазме оо-
ооцитов. Этому же процессу способству-
способствуют и фолликулярные клетки, образую-
образующие несколько слоев вокруг ооцита
первого порядка.
Процесс овуляции предваряется бы-
быстрым завершением первого мейотиче-
ского деления в ооците первого поряд-
порядка. В результате образуются две гапло-
гаплоидные клетки, окутанные лучистым
венцом. Распределение цитоплазмы
между ними происходит неравномер-
неравномерно: одна из дочерних клеток — ооцит
второго порядка, — получает почти
всю цитоплазму материнской клетки,
тогда как на долю второй — первого
полярного тельца — не достается поч-
почти ничего. Ооцит второго порядка
вступает во второе мейотическое деле-
деление, но оно доходит только до стадии
метафазы и останавливается до тех
пор, пока не произойдет оплодотворе-
оплодотворение. (Отметим, что вероятность этого
мала). Поскольку во время первого де-
деления мейоза число хромосом сокра-
сокращается вдвое, ооцит второго порядка, а
Рис. 4.17. Микрофотография хромосом
типа "ламповых щеток"
следовательно, и яйцеклетка несет гап-
гаплоидный набор хромосом. Такой же
набор хромосом имеет и первое поляр-
полярное тельце. Общая схема оогенеза
представлена на рисунке (рис. 4.18).
Таким образом, в отличие от образо-
образования спермиев, которое начинается у
мужчин только при наступлении поло-
полового созревания, образование яйцекле-
яйцеклеток у женщин начинается еще до рож-
рождения и завершается для каждой яйце-
яйцеклетки только после ее оплодотворе-
оплодотворения. Созревающие ооциты пребывают в
профазе первого мейоза долгие годы.
Кроме того, у мужчин из одного дипло-
диплоидного сперматоцита первого порядка
образуется 4 гаплоидных сперматозои-
сперматозоида, а у женщин — из одного диплоидно-
диплоидного ооцита первого порядка образуется
лишь одна зрелая гаплоидная яйце-
яйцеклетка. То обстоятельство, что поляр-
полярные тельца почти не получают цито-
цитоплазмы, позволяет ооциту снабдить зи-
зиготу полным набором цитоплазматиче-
ских компонентов, таких, как митохон-
митохондрии и расположенные определенным
образом информационные РНК.
Оплодотворение
Оплодотворение — это сложный
процесс, приводящий к слиянию яй-
яйцеклетки и сперматозоида, объедине-
96
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Фаза
развития
Р
а
з
м
н
о
ж
е
н
и
е
Р
о
с
т
Механизм
деления
С
о
3
р
е
в
а
н
и
е
Митоз
Профаза
МейозаI
Мейоз I
Мейоз II
Стадия
жизненного
цикла
Оогонии
Ооцит
первого
порядка
В
н
У
т
р
и
У
т
р
о
б
н
а
я
До
овуляции
Число
хромосом
46
Ооцит Овуляция
второго
порядка
46
23
23
Полярное
тельце
Оплодо-
Оплодотворение
23 +
23 = 46
Рис. 4.18. Схема оогенеза человека
нию их ядер и образованию диплоид-
диплоидной зиготы, из которой впоследствии
будет развиваться новый организм.
Генетическое значение процесса опло-
оплодотворения состоит в объединении
гаплоидных наборов хромосом отцов-
отцовских и материнских гамет. Отличаю-
Отличающая их высокая степень генетическо-
генетического разнообразия (несмотря на количе-
количественное постоянство хромосомного
набора), обеспечивает генетическое
разнообразие особей. В результате
процесса оплодотворения восстанав-
восстанавливается диилоидность зиготы, что
становится толчком к последующим
митотическим делениям. У человека,
как и у других млекопитающих, про-
процесс оплодотворения, начинающийся
Оплодотворение
97
с проникновения сперматозоида в оо-
цит второго порядка, запускает меха-
механизм созревания яйцеклетки. Как уже
говорилось выше, в ооците второго
порядка второе меиотическое деление
останавливается на стадии метафазы,
а завершение развития яйцеклетки
происходит только после оплодотво-
оплодотворения.
Оплодотворение подразделяется на
три последовательных этапа: в первом
сперматозоид и яйцеклетка сближаются
для возникновения контакта. Второй
этап начинается с прикрепления спер-
сперматозоида к поверхности яйцеклетки и
осуществления контактных взаимодей-
взаимодействий между ними. События третьего
периода разворачиваются после про-
проникновения сперматозоида в яйцеклет-
яйцеклетку и завершается объединением их ядер.
Оплодотворение происходит в
верхних концах фаллопиевых труб,
куда сперматозоиды проходят из вла-
влагалища всего за несколько минут.
Столь высокой скорости их движения
способстнуют активные сокращения
матки и труб, инициируемые биологи-
биологически активными соединениями, вы-
выделяющимися во время полового акта.
Кроме того, на направленность движе-
движения сперматозоидов сильно влияет
градиент pH среды в женских поло-
половых путях.
Жизнеспособность внутри организ-
организма сперматозоиды сохраняют в тече-
течение 1-3 суток, но период высокой фер-
тильности длится лишь 12-24 часа. Са-
Сама способность к оплодотворению оо-
цита появляется только после того, как
он проведет несколько часов в поло-
половых путях. За это время изменяются
структура акросомы и свойства мемб-
мембраны, ее ограничивающей.
Когда спермин скапливаются во-
вокруг ооцита второго порядка, в голо-
головке одного из них происходит очень
быстрая (не более 20 секунд) акросо-
мальная реакция. Она состоит в том,
что мембраны головки спермия и ак-
акросомы разрываются, так что излив-
излившиеся наружу ферменты акросомы
"переваривают" клеточные слои, окру-
окружающие ооцит, включая zona pellucida.
Затем внутренняя мембрана акросомы
выворачивается наизнанку, и спермий
проникает внутрь ооцита. У человека
сперматозоид входит в ооцит целиком
(рис. 4.19).
После того, как один пермий проник
в яйцеклетку, под ее мембраной разры-
разрываются многочисленные вакуоли, ос-
освобождая вещество, под действием ко-
которого zona pellucida утолщается и об-
образует поверх яйцеклетки непроницае-
непроницаемую для спермиев преграду — оболоч-
оболочку оплодотворения. Так предотвраща-
предотвращается проникновение в ооцит других
спермиев. У человека проникновение
сперматозоида в ооцит является сигна-
сигналом для завершения второго мейоти-
ческого деления, ведущего к образова-
образованию гаплоидной яйцеклетки и сопут-
сопутствующих клеток — полярных телец,
впоследствие дегененрирующих. В это
же время в цитоплазме ооцита расса-
рассасывается хвост сперматозоида. Следу-
Следующий этап процесса оплодотворения
связан с объединением генетического
материала яйцеклетки и сперматозои-
сперматозоида в одно диплоидное ядро.
Высококонденсированное ядро
сперматозоида начинает набухать, его
хроматин разрыхляется, и ядро пре-
превращается в структуру, называемую
мужским пронуклеусом. Аналогичные
изменения происходят в женском яд-
ядре, что приводит к образованию жен-
женского пронуклеуса. В процессе форми-
формирования пронуклеусов и в мужском, и
в женском ядре, происходит синтез
ДНК (In 2C), в результате чего каждая
хромосома удваивается и состоит из
98
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Плазматическая}
мембрана
'ертителлин^
о»ое
пространств I
Рис. 4.19. Упрощенная схема оплодотворения
Внехромосомное цитоплазматичвскм наследование
99
двух сестринских хроматид. После то-
того, как репликация ДНК закончена,
пронуклеусы перемещаются к центру
яйцеклетки (рис. 4.19). Постепенно
мембраны пронуклеусов разрушаются,
а отцовские и материнские хромосо-
хромосомы, состоящие каждая из двух хрома-
хроматид, прикрепляются к нитям сформи-
сформировавшегося веретена деления. Затем
хромосомы выстраиваются по эквато-
экватору веретена, создавая характерную для
метафазы митоза картину.
Объединение отцовских и материн-
материнских хромосом в общем митозе перево-
переводит оплодотворенную яйцеклетку в
качественно новое состояние — зиготу.
Зигота проходит стадии анафазы и те-
лофазы и завершает свое первое мито-
тическое деление. В результате образу-
образуются две дочерние диплоидные клет-
клетки — два бластомера. Генетический ма-
материал бластомеров представляет со-
собой совокупность хромосом отца и ма-
матери: в каждой паре гомологичных
хромосом одна хромосома отцовская, а
другая — материнская.
Внехромосомное
цитоплазматическое наследование
Хромосомная теория наследствен-
наследственности Т. Моргана — одна из основопо-
основополагающих теорий генетики, но посте-
постепенно накапливающиеся факты при-
приводили к представлению о существо-
существовании генов и вне хромосом, в цито-
цитоплазме. Такими носителями внехромо-
сомной последовательности оказались
митохондрии. Выяснилось, в частнос-
частности, что цитоплазматическое наследо-
наследование человека связано прежде всего с
митохондриальной ДНК.
В организме человека митохондрии
обеспечивают дыхание всех клеток, за
исключением эритроцитов. Это само-
самовоспроизводящиеся полуавтономные
органеллы содержат кольцевые моле-
молекулы ДНК. Имеют митохондрии и
собственный аппарат белкового синте-
синтеза в виде рибосом, транспортных РНК
и информационных РНК. Установле-
Установлено, что аппарат белкового синтеза ми-
митохондрий отличается от цитоплазма-
тического: он более близок к аппарату
белкового синтеза прокариот.
Как уже говорилось выше, в процес-
процессе оплодотворения человека участву-
участвуют две клетки: яйцеклетка и спермато-
сперматозоид. Яйцеклетка — это крупная клет-
клетка с большим объемом цитоплазмы, в
которой с помощью электронного мик-
микроскопа обнаруживают все органеллы,
в том числе и множество митохондрий.
Сперматозоид же, хотя и имеет упако-
упакованные специальным образом в облас-
области шейки четыре митохондрии, утра-
утрачивает их вскоре после того, как про-
проникает внутрь яйцеклетки, поскольку
его хвостовая часть быстро растворя-
растворяется. Практически, в процесе оплодо-
оплодотворения принимает участие только
ядро сперматозоида. Таким образом,
диплоидная зигота, дающая начало но-
новому организму, несет митохондрии,
полученные только от яйцеклетки, т.е.
все митохондрии во всех клетках лю-
любого индивида имеют материнское
происхождение. Следовательно, гене-
генетический материал, локализованный в
кольцевых молекулах митохондриаль-
митохондриальной ДНК, наследуется по материнской
линии.
В настоящее время полностью рас-
расшифрована последовательность ДНК
и выяснена организация генов в мито-
митохондриях человека. Геном митохонд-
митохондрии представлен кольцевой молекулой
ДНК, содержащей 16569 нуклеопроте-
идов. В сто состав входят гены 12 S- и
16 S-рибосомной РНК, 22 различных
тРНК, субъединицы I, II и III оксида-
зы цитохрома С, шесть субъединиц
АТФ-синтетазы, цитохрома b и девять
100
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
Николай II
I—¦ ^ Принцесса Алиса
| |—г~^Р дочь королевы Виктории
I I—,—Щ
1—' ^
¦ •
Царица
Александра
•-г-П
•*•
Принц Филипп, герцог Эдинбургский
Рис. 4.20. Родство царской семьи Николая II по анализу митохондриальнои ДНК.
Окрашенные кружки и квадраты обозначают родственников женского и мужского пола,
имеющих одинаковую митохондриапьную ДНК, передающуюся по материнской линии
других, пока неизвестных белков. В
транскрибируемых и транслируемых
областях цепей митохондриальнои
ДНК по сравнению с ядерной ДНК
выявлено мало некодируюпих участ-
участков. Установлено также, что по ряду
характеристик генетический код мито-
митохондриальнои ДНК человека отлича-
отличается от универсального.
ДНК в митохондриях является ма-
материнской основой наследственности.
Известно, что генетический материал
в молекулах ДНК, способен мутиро-
мутировать. В митохондриях окислительные
процессы, связанные с синтезом АТФ,
сопровождаются образованием боль-
большого количества свободных радика-
радикалов, которые могут вызывать мутации
в митохондриальнои ДНК, а процесс
репарации митохондриальнои ДНК
развит, вероятно, не так хорошо, как
ДНК ядерной. Так, частота мутаций в
митохондриальнои ДНК примерно в
10 раз выше, чем в ядерной. Для мито-
митохондриальнои ДНК известны точеч-
точечные мутации, делеции, вставки и выпа-
выпадения.
Встает естественный вопрос: может
ли мутация в митохондриальнои ДНК
быть причиной наследственных забо-
заболеваний? Отличительным признаком
такой патологии должна служить ее
передача от матери всем ее детям. Од-
Одни авторы считают, что подобный тип
наследования маловероятен, посколь-
поскольку каждый ооцит содержит множество
митохондрий, и если в одной из них
произошла мутация, то все другие ос-
остаются немутантами, и, следовательно,
не должно быть никакого фенотипиче-
ского эффекта. Но этот вопрос рассма-
рассматривается учеными и с другой сторо-
стороны. Один из подходов к изучению ге-
генома митохондрий основан на обра-
обработке их ДНК рестриктазами и иссле-
исследовании полученных рестриктов —
фрагментов кольцевой молекулы
ДНК. В результате было показано, что
митохондрии одного индивида генети-
генетически однородны. Кроме того, они спо-
способны с большой скоростью обмени-
обмениваться между собой своими митохонд-
риальными ДНК. Причина этого явле-
явления пока до конца не выяснена. Неко-
Некоторыми исследователями установлено,
что для митохондрий характерен дина-
динамический процесс слияния и дробле-
дробления. Как, однако, при фрагментации
митохондрий митохондриальная ДНК
распределяется между фрагментами,
неизвестно. На основе новейших исле-
дований митохондриальнои ДНК в ме-
Тератология
101
дицине ныне формируется целая но-
новая область — митохондриальная ме-
медицина. Ее основатели считают, что
мутации в митохондриальной ДНК
являются причиной более ста болез-
болезней: диабета, некоторых видов миопа-
тии, кардиомиопатии, энцефаломио-
патии, кислотного ацидоза, динамиче-
динамического нарушения мозгового кровооб-
кровообращения, эпилепсии и др.
Недавно обнаружено, что в редких
случаях встречается отцовская наслед-
наследственность митохондриальной ДНК.
По-видимому, это возможно тогда,
когда, после оплодотворения яйце-
яйцеклетки, содержащий митохондрии
хвост сперматозоида не лизируется.
Анализ рестриктов митохондриаль-
митохондриальной ДНК показал, что различные чело-
человеческие популяции отличаются друг
от друга их составом. Это явление по-
получило название полиморфизма мито-
хондриальных ДНК в популяциях че-
человека. Использование еще одного но-
нового метода в исследованиях митохон-
митохондриальной ДНК — ДНК-полимераз-
ной реакции, — позволило использо-
использовать параметры митохондриальной
ДНК для характеристики популяций
человека и изучения филогенетически
закономерностей в процессах разви-
развития. Оказалось также, что митохонд-
митохондриальная ДНК способна сохраняться в
ископаемых останках на протяжении
многих веков. В этом случае ее харак-
характеристики могут служить веским науч-
научным доказательством родства, как
между популяциями, так и между ин-
индивидами. Так, идентификация цар-
царской семьи Николая II по ископаемым
останкам была осуществлена на базе
анализа именно митохондриальной
ДНК (рис. 4.20). В процессе исследо-
исследования установили общность митохон-
митохондриальной ДНК из клеток крови
здравствующего принца Филиппа гер-
герцога Эдинбургского и митохондриаль-
митохондриальной ДНК, выделенной из останков ца-
царицы Александры Федоровны и трех
ее дочерей. Все перечисленные лица
являются родственниками по мате-
материнской линии — потомками принцес-
принцессы Алисы, дочери королевы Виктории.
Родословная этой семьи представлена
на рисунке (рис. 4.20).
Тератология
Индивидуальное развитие организ-
организма начинается с момента оплодотворе-
оплодотворения. Сначала зигота делится митоти-
чески, затем следуют этапы пренаталь-
ного развития: бластула, гаструла, ней-
руляция, имплантация в стенку матки,
закладка и формирование органов и их
систем, рост плода. Пренатальный пе-
период сменяется перинатальным, кото-
который начинается с 28 недели беремен-
беременности, включает роды и заканчивается
через 7 дней после рождения. Далее
идет постнатальный период, вмещаю-
вмещающий в себя всю жизнь человека до
смерти.
В процессе индивидуального разви-
развития имеется ряд критических перио-
периодов, когда организм наиболее уязвим
по отношению к действию различных
факторов. В литературе, посвященной
оценке токсического воздействия ок-
окружающей среды на человека, доста-
достаточно редко рассматривается пробле-
проблема возникновения пороков развития,
связанных с тератогенными фактора-
факторами. Так называются факторы, которые,
действуя в период беременности, при-
приводят к возникновению врожденных
пороков развития у детей, не вызывая
нарушения наследственных структур.
По современным данным, не менее
182 тыс. веществ, искусственно вве-
введенных в окружающую среду челове-
человеком, в том числе и лекарственные пре-
препараты, обладают токсическим эффек-
102
Глава 4. ПЕРЕДАЧА ГЕНЕТИЧЕСКОГО МАТЕРИАЛА
том. Все исследователи единодушны в
том, что беременным женщинам сле-
следует очень осторожно принимать ме-
медикаменты, поскольку большинство
лекарственных препаратов и их мета-
метаболитов могут проходить через барьер
плаценты. Уникальная особенность
системы мать-плацента-плод заключа-
заключается в одновременной циркуляции ле-
лекарственных веществ как в организме
беременной женщины, так и в организ-
организме плода. Наиболее нежелательно ис-
использование лекарств в первой трети
беременности, когда формируются за-
зачатки органов. Возникновение врож-
врожденных пороков — это результат от-
отклонений от нормального развития.
Оно имеет клеточные основы. Разви-
Развитие особи в эмбриогенезе сопряжено с
активным делением клеток, морфоге-
нетическим движением зародышевых
листков, с дифференцировкой и орга-
органогенезом. Тератогенные факторы мо-
могут изменять способность клеток к
размножению и перемещению, нару-
нарушать процесс обретения клетками оп-
определенной специализации. Основны-
Основными механизмами развития пороков на
тканевом уровне являются: гибель от-
отдельных клеточных масс, замедление
распада и рассасывания отмирающих
клеток, нарушение взаимодействия
между тканями. В результате тот или
иной орган недоразвивается или раз-
развивается неправильно. Примерами мо-
могут служить смещение устья аорты, ве-
ведущее к врожденному пороку сердца,
целая группа уродств, связанных с не-
незакрытием нервной трубки.
Для ранних стадий эмбриогенеза из-
известны так называемые критические
периоды, в которых тот или иной раз-
развивающийся орган особо чувствителен
к определенным воздействиям. Крас-
Краснуха женщины между 3-й и 9-й неде-
неделями беременности резко повышает
вероятность развития у плода порока
сердца, катаракты, глухоты. В более
поздние сроки эта болезнь таких по-
последствий не дает. Сходное тератоген-
тератогенное действие предполагается у других
вирусных инфекций: гриппа, оспы, па-
ратита.
Относительно недавно был выяв-
выявлен новый тератоген: — 13-цис-рети-
ноевая кислота (аналог витамина А).
Этот препарат широко использовался
для лечения угрей. Ранее было показа-
показано, что аналоги витамина А могут
вредно влиять на беременность лабо-
лабораторных животных, поэтому этикет-
этикетка препарата предупреждала, что им
не должны пользоваться беременные
женщины. Однако не все будущие ма-
мамы вняли предупреждению, в резуль-
результате из 59 обследованных в группе ри-
риска беременностей 26 плодов роди-
родились здоровыми, 12 были спонтанно
абортированы и 21 из новорожденных
имели уродства. Последующие иссле-
исследования показали, что критическим
периодом для ретиноевой кислоты яв-
являются 20-35 сутки после оплодотво-
оплодотворения.
Другим, пожалуй, самым известным
примером лекарственного химическо-
химического тератогена стал талидомид — широ-
широко используемый как успокоительное
средство в 60-х гг. XX в. транквилиза-
транквилизатор. Для него тератогенным является
период от 20 до 36 суток после оплодо-
оплодотворения. В зависимости от срока, на
котором принимался талидомид, воз-
возникали такие уродства, как отсутствие
или деформация ушей или больших
пальцев на руках, отсутствие или рез-
резкое укорочение рук, ног, смещение бе-
бедра. Трагедия с талидомидом привела
к рождению более 7000 искалеченных
детей. Только после этого жестокого
урока началась разработка схемы про-
проверки лекарственных препаратов на
Тератология
тератогенный эффект. Тем не менее в
интересах здоровья будущего ребенка
следует проявлять крайнюю осторож-
осторожность и, по возможности, вообще отка-
отказаться от приема лекарственных пре-
препаратов, особенно в первом триместре
беременности.
К препаратам, способным вызывать
врожденные пороки развития, отно-
относятся транквилизаторы, противосудо-
рожные, гормональные и противоопу-
противоопухолевые средства. Стероидные гормо-
гормоны, входящие в состав многих контра-
контрацептивов, которые женщины могут
принимать, не зная о наличии бере-
беременности, также относятся к факторам
риска для развивающегося плода.
Использование лекарств на более
поздних сроках развития плода обыч-
обычно не вызывает уродств, но может на-
нарушить процессы приспособления но-
воржденного к новым для него услови-
условиям среды.
Широко применяемые в промыш-
промышленности и сельском хозяйстве бензол,
фенолы, хлоропрен, формальдегид,
ядохимикаты, а также свинец и пары
ртути обладают выраженным токсиче-
токсическим действием. Они могут вызвать
самопроизвольный аборт, внутриут-
внутриутробную смерть плода или рождение
ослабленного ребенка.
Тератогенными могут стать не толь-
только химические и биологические, но и
физические факторы. В том числе раз-
различные излучения, гипо- и гипертер-
гипертермия, гипо- и гипероксия.
Поскольку тератогенные факторы
не нарушают генетических структур,
то такие пороки развития не наследу-
наследуются. Риск повторного рождения боль-
больного ребенка с аналогичным пороком
предельно мал.
Вопросы для самоконтроля
1. В чем значение митоза в передаче наследствен-
наследственной информации?
2. Назовите две причины генетической рекомби-
рекомбинации в мейозе.
3. Укажите отличия спермато- и оогенсза.
4. Поясните значение процесса оплодотворения.
5. Что такое внехромосомная цитоплазматическая
наследственность? В чем ее материальная основа?
6. Что вы знаете о тератогенных факторах? Поче-
Почему они наиболее опасны в критические периоды раз-
развития эмбриона?
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
Менделирующие признаки
Всем эукариотическим организмам
присущи открытые Г.Менделем общие
закономерности наследования призна-
признаков. Для их изучения необходимо
вспомнить основные термины и поня-
понятия, используемые в генетике. Глав-
Главный постулат Менделя, который он
доказал в своих известных экспери-
экспериментах на горохе огородном, состоит в
том, что каждый признак определяется
парой наследственных задатков, позже
получивших название аллельных ге-
генов. С развитием хромосомной теории
наследственности выяснилось, что ал-
лельные гены находятся в одинаковых
локусах гомологичных хромосом и ко-
кодируют один и тот же признак. Пара
аллельных генов может быть одинако-
одинакова (АА или аа), тогда говорят, что
особь гомозиготна по данному призна-
признаку. Если же аллельные гены в паре раз-
разные (Аа), то особь по данному призна-
признаку гетерозиготна. Совокупность генов
данного организма называется геноти-
генотипом. Правда часто под генотипом по-
понимают одну или несколько пар ал-
аллельных генов, которые отвечают за
один и тот же признак. Совокупность
признаков данного организма называ-
называют фенотипом, фенотип формируется
в результате взаимодействия генотипа
с внешней средой.
Г. Мендель ввел понятия доминант-
доминантных и рецессивных генов. Аллель, ко-
который определяет фенотип гетерози-
готы, он назвал доминантным. Напри-
Например, ген А в гетерозиготе Аа. Другой
аллель, не проявляющий себя в гетеро-
гетерозиготном состоянии, назван им рецес-
рецессивным. В нашем случае это ген а.
Основные закономерности наследо-
наследования признаков по Менделю (законы
единообразия гибридов первого поко-
поколения, расщепления на фенотипичес-
кие классы гибридов второго поколе-
поколения и независимого комбинирования
генов) реализуются благодаря сущест-
существованию закона чистоты гамет. Суть
последнего состоит в том, что пара ал-
аллельных генов, определяющая тот или
иной признак: а) никогда не смешива-
смешивается; б) в процессе гаметогенеза расхо-
расходится в разные гаметы, то есть в каж-
каждую из них попадает один ген из ал-
лельной пары. Цитологически это
обеспечивается мейозом: аллельные
гены лежат в гомологичных хромосо-
хромосомах, которые в анафазе мейоза расхо-
расходятся к разным полюсам и попадают в
разные гаметы.
Генетика человека опирается на об-
общие принципы, полученные первона-
первоначально в исследованиях на растениях и
животных. Как и у них, у человека име-
имеются менделирующие, т.е. наследуемые
по законам, установленным Г. Менде-
Менделем, признаки. Для человека, как и для
других эукариот, характерны все типы
наследования: аутосомно-доминант-
ный, аутосомно-рецессивный, наследо-
наследование признаков, сцепленных с поло-
половыми хромосомами, и за счет взаимо-
взаимодействия неаллельных генов. Разрабо-
Разработал Г.Мендель и основной метод гене-
генетики — гибридологический. Он осно-
основан на скрещивании особей одного ви-
вида, обладающих альтернативными при-
признаками, и количественном анализе по-
полученных фенотипических классов.
Естественно, этот метод не может ис-
использоваться в генетике человека.
Менделирующие признаки
105
Умер в детстве
или вероятно
1—1 ч_^ умер в детстве
. Дополнительные
П О сведения
^ ^ к родословной
Рис. 5.1. Родословная с брахидактилией. Обозначения: черные фигуры - пораженные
женщины и мужчины; римские цифры - номера поколений; арабские цифры:
под фигурами - последовательные номера в родословной,
внутри фигур - количество детей
Отчасти в противовес ему в генети-
генетике человека существуют наиболее
давний ее метод — клинико-генеало-
гический анализ наследственности
(см. главу 2).
Первое описание аутосомно-до-
минантного наследования аномалий
у человека дано в 1905 г. Фараби. Ро-
Родословная была составлена для се-
семьи с короткопалостью (брахидакти-
(брахидактилией). У больных укорочены и час-
частично редуцированы фаланги паль-
пальцев рук и ног, кроме того, в результа-
результате укорочения конечностей, для них
характерен низкий рост. На рис. 5.1
показана родословная с 44 поражен-
пораженными, на рис. 5.2 — фенотипическое
проявление признака. Признак пере-
передается от одного из родителей при-
примерно половине детей, независимо от
пола. Анализ родословных других се-
семей свидетельствует, что брахидакти-
лия отстутствует среди потомства ро-
родителей, не являющихся носителями
данного гена. Поскольку признак не
может существовать в скрытом виде,
следовательно, он является доминант-
доминантным. А его проявления, независимо от
пола, позволяют заключить, что он не
сцеплен с полом. На основании изло-
изложенного можно сделать вывод, что
брахидактилия определяется геном,
находящимся в аутосомах, и является
доминантной патологией.
106
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
Рис. 5.2. Фенотипическое проявление
брахидактилии у одной
из представительниц родословной
Фараби
Использование генеалогического
метода позволило выявить доминант-
доминантные, не сцепленные с полом признаки
у человека. Это — темный цвет глаз,
вьющиеся волосы, переносица с гор-
горбинкой, прямой нос (кончик носа смо-
смотрит прямо), ямочка на подбородке,
раннее облысение у мужчин, правору-
кость, способность свертывать язык в
трубочку, белый локон надо лбом, "габ-
"габсбургская губа" — нижняя челюсть уз-
узкая, выступающая вперед, нижняя гу-
губа отвислая и полуоткрытый рот. По
аутосомно-доминантному типу насле-
наследуются также некоторые патологичес-
патологические признаки человека: полидактилия
или многопалость (когда на руке или
ноге имеется от 6 до 9 пальцев), син-
синдактилия (сращение мягких или кост-
костных тканей фаланг двух и более паль-
пальцев), брахидактилия (недоразвитость
дистальных фаланг пальцев, приводя-
приводящая к короткопалости), арахнодакти-
лия (сильно удлиненные "паучьи"
пальцы, один из симптомов синдрома
Марфана), некоторые формы близору-
близорукости. Большинство носителей ауто-
сомно-доминантной аномалии явля-
являются гетерозиготами. Иногда случает-
случается, что два носителя одной и той же до-
доминантной аномалии вступают в брак
и имеют детей. Тогда четверть из них
будут гомозиготами по мутантному
доминантному аллелю (АЛ). Многие
случаи из медицинской практики ука-
указывают на то, что гомозиготы по доми-
доминантным аномалиям поражены тяже-
тяжелее, чем гетерозиготы. Например, в
браке между двумя носителями брахи-
брахидактилии родился ребенок, у которого
не только не доставало пальцев на ру-
руках и ногах, но и имелись множествен-
множественные уродства скелета. Он умер в возра-
возрасте одного года. Другой ребенок в этой
семье был гетерозиготным и имел
обычные симптомы брахидактилии.
Аутосомно-рецессивные мендели-
рующие признаки у человека опреде-
определяются генами, локализованными в
аутосомах, и могут проявиться у по-
потомства в браке двух гетерозигот, двух
рецессивных гомозигот или гетерози-
гетерозиготы и рецессивной гомозиготы. Ис-
Исследования показывают, что большин-
большинство браков, среди потомков которых
наблюдаются рецессивные заболева-
заболевания, происходит между фенотипичес-
ки нормальными гетерозиготами (Аа х
Аа). В потомстве такого брака геноти-
генотипы АА, Аа и аа будут представлены в
соотношении 1:2:1, и вероятность того,
что ребенок окажется пораженным, со-
составит 25%. По аутосомно-рецессивно-
му типу наследуются мягкие прямые
волосы, курносый нос, светлые глаза,
тонкая кожа и резус-отрицательная
первая группы крови, многие болезни
обмена веществ: фенилкетонурия, га-
лактоземия, гистидинимия и др., а так-
также пигментная ксеродерма.
Пигментная ксеродерма — одно из
рецессивных заболеваний — относи-
относительно недавно привлекла внимание
Менделирукнцие признаки
107
g 9
ар
IV
Рис. 5.3. Родословная с пигментной ксеродермой в браке двоюродных сибсов
молекулярных биологов. Эта патоло-
патология обусловлена неспособностью кле-
клеток кожи больного репарировать по-
повреждения ДНК, вызванные ультра-
ультрафиолетовым излучением. В результате
развивается воспаление кожи, особен-
особенно на лице, с последующей атрофией.
Наконец, развивается рак кожи, при-
приводящий в отсутствие лечения к ле-
летальному исходу. На рис. 5.3 представ-
представлена типичная родословная, в которой
родители являются двоюродными сиб-
сами. У больных редким рецессивным
заболеванием степень кровного родст-
родства между родителями обычно значи-
значительно выше среднего уровня в попу-
популяции. Как правило, родители насле-
наследуют этот ген от общего предка и явля-
являются гетерозиготами. Подавляющее
большинство больных аутосомно-ре-
цессивными заболеваниями — это дети
двух гетерозигот.
Помимо аутосомно-доминантного и
аутосомно-рецессивного типов насле-
наследования у человека выявляются также
неполное доминирование, ко домини-
доминирование и сверхдоминирование.
Неполное доминирование связано с
промежуточным проявлением призна-
признака при гетерозиготном состоянии ал-
аллелей (Аа). Например, большой нос
определяется двумя аллелями АА, ма-
маленький нос — аллелями аа, нормаль-
нормальный нос средних размеров — Аа. По
типу неполного доминирования у че-
человека наследуются выпуклость губ и
размеры рта и глаз, расстояние между
глазами.
Кодоминирование — это такое взаи-
взаимодействие аллельных генов, при ко-
котором в гетерозиготном состоянии
оказываются и работают вместе два
доминантных гена одновременно, то
есть каждый аллель детерминирует
свой признак. Наиболее удобно рас-
рассмотреть кодоминирование на приме-
примере наследования групп крови.
Группы крови системы АВО опреде-
определяются тремя аллелями: А, В и О. При-
Причем аллели А и В являются доминант-
доминантными, а аллель О — рецессивным. По-
Попарное сочетание этих трех аллелей в
генотипе дает четыре группы крови.
Аллельные гены, определяющие груп-
группы крови, находятся в девятой паре
хромосом человека и обозначаются со-
соответственно: Г, 1В и 1°. Первая группа
крови определяется наличием в гено-
тие двух рецессивных аллелей 1° 1°.
Фенотипически это проявляется нали-
наличие в сыворотке крови антител аир.
Вторая группа крови может опреде-
108
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
ляться двумя доминантными аллеля-
аллелями 1А1А, если человек гомозиготен, или
аллелями 1А1°, если он гетерозиготен.
Фенотипически вторая группа крови
проявляется наличием на поверхности
эритроцитов антигенов группы А и
присутствием в сыворотке крови анти-
антител р. Третья группа определяется
функционированием аллеля В. И в
этом случае генотип может быть гете-
гетерозиготен AВ1°) или гомозиготен
AВ1В). Фенотипически у людей с треть-
третьей группой крови на поверхности эри-
эритроцитов выявляются антигены В, а
фракции белков крови содержат анти-
антитела а. Люди с четвертой группой кро-
крови сочетают в генотипе два доминант-
доминантных аллеля АВ AА1В), причем оба они
функционируют: поверхность эритро-
эритроцитов несет оба антигена (А и В), а сы-
сыворотка крови во избежание агглюти-
агглютинации соответствующих сывороточ-
сывороточных белков а и р не содержит. Таким
образом люди с четвертой группой
крови являют примеры кодоминиро-
кодоминирования, поскольку у них одновременно
работают два доминантных аллельных
гена.
С развитием методов генетического
анализа на уровне белков у человека
было открыто множество примеров
кодоминирования. Первые случаи
описаны при изучении групп крови
системы MN. В ходе обследования со-
сотен семей проводился статистический
анализ наследования этих групп кро-
крови. Оказалось, что у родителей с груп-
группами крови М рождаются дети только
с такой же группой крови. Аналогичны
закономерности для семей с группой
крови N: дети в них повторяют группу
крови родителей — N. То есть облада-
обладатели групп крови М и N могут быть
только гомозиготными: ММ И NN. В
семьях же, где родители имеют группы
крови М и N, у всех детей группа кро-
крови — MN, причем оба доминантных ал-
аллеля М и N функционируют вместе.
По такому же типу наследуются
многие функциональные белки и фер-
ферментные системы человека. Наследо-
Наследование по типу кодоминирования тесно
связано с проблемой множественного
аллелизма. Фенотипическое проявле-
проявление каждого менделирующего призна-
признака основано на взаимодействии в гено-
генотипе двух аллельных генов. Однако
количество аллелей в человеческих
популяциях далеко не всегда равно
двум. Для групп крови системы MN их
два, а для групп крови системы АВО
существует не два, а три аллеля: А, В и
О. По несколько аллелей известно для
гемоглобина и для многих фермент-
ферментных систем. Сколько бы аллелей ни
существовало в популяции, признак в
конкретном организме определяется
сочетанием только двух из них. В гено-
генотипе они могут взаимодействовать
между собой по типу полного домини-
доминирования; неполного доминирования
или кодоминирования. Явление мно-
множественного аллелизма определяет
фенотипическую гетерогенность чело-
человеческих популяций, это одна из основ
разнообразия генофонда человека. В
основе этой множественности лежат
генные мутации, изменяющие после-
последовательность азотистых оснований
молекулы ДНК в участке, соответству-
соответствующем данному гену. Эти мутации мо-
могут быть нейтральными, полезными,
или вредными. Последние являются
причиной наследственных патологий,
связанных с множественным аллелиз-
мом. Например, известна мутация, из-
изменяющая структуру одной из цепей
белка гемоглобина за счет того, что код
глутаминовой кислоты в концевом
участке гена трансформируется в код
аминокислоты валин. Эта замена ста-
становится причиной возникновения на-
Взаимодействие генов
109
следственои патологии — серповид-
ноклеточной анемии.
Явление сверхдоминирования свя-
связано с тем, что в ряде случаев доми-
доминантные гены в гетерозиготном состо-
состоянии проявляются сильнее, чем в го-
гомозиготном. Это понятие коррелирует
с эффектом гетерозиса и связано с та-
такими сложными признаками, как жиз-
жизнеспособность, общая продолжитель-
продолжительность жизни и др.
Таким образом, у человека, как и у
остальных эукариот, известны все ти-
типы взаимодействия аллельных генов и
большое количество менделирующих
признаков, определяемых этими взаи-
взаимодействиями. Используя мендслев-
ские законы наследования, можно рас-
рассчитать вероятность рождения детей с
теми или иным моделирующими при-
признаками.
Наиболее удобным методическим
подходом к анализу наследования
признаков в нескольких поколениях
является генеалогический метод, осно-
основанный на построении родословных.
Взаимодействие генов
До сих пор мы рассматривали толь-
только признаки, контролируемые моно-
генно. Однако на фенотипическое про-
проявление одного гена обычно влияют
другие гены. Зачастую признаки фор-
формируются при участии нескольких ге-
генов, взаимодействие между которыми
отражается в фенотипе.
Примером сложного взаимодейст-
взаимодействия генов могут служить закономерно-
закономерности наследования системы резус-фак-
резус-фактор: резус плюс (Rh+) и резус минус
(Rh'). В 1939 г. при исследовании сы-
сыворотки крови женщины, родившей
мертвый плод и имевшей в анамнезе
переливание совместимой по АВО
группе крови мужа> были обнаружены
особые антитела, сходные с получае-
получаемыми при иммунизации эксперимен-
экспериментальных животных эритроцитами ма-
каки-резус. Выявленные у больной ан-
антитела получили название резус-анти-
резус-антител, а ее группа крови — резус-отрица-
резус-отрицательной. Группа крови резус-положи-
резус-положительная определяется присутствием на
поверхности эритроцитов особой
группы антигенов, кодируемых струк-
структурными генами, несущими информа-
информацию о мембранных полипептидах. Ге-
Гены, определяющие резус-фактор, на-
находятся в первой паре хромосом чело-
человека. Резус-положительная группа
крови является доминантной, резус-
отрицательная — рецессивной. Резус-
положительные люди могу быть гете-
гетерозиготными ('Rh+/Rh") или гомози-
гомозиготными (Rh /Rh+). Резус-отрица-
Резус-отрицательные — только гомозиготными (Rh"
/Rh").
Позже выяснилось, что антигены и
антитела резус фактора имеют слож-
сложную структуру и состоят из трех ком-
компонентов. Условно антигены резус-
фактора обозначают буквами латин-
латинского алфавита С, D, Е. На основе ана-
анализа генетических данных о наследова-
наследовании резус-фактора в семьях и популя-
популяциях была сформулирована гипотеза о
том, что каждый компонент резус-фак-
резус-фактора определяется своим геном, что эти
гены сцеплены вместе в один локус и
имеют общий оператор или промотор,
который регулирует их количествен-
количественную экспрессию. Поскольку антигены
обозначаются буквами С, D, Е, то таки-
такими же строчными буквами обозначают
гены, отвечающие за синтез соответст-
соответствующего компонента. Генетическая
структура Rh-комплекса показана на
рис. 5.4, где известные к настоящему
времени антигены обозначены буква-
буквами (антиген d пока не обнаружен).
Генетические исследования в семьях
показывают возможность кроссинго-
110
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
D, d С, с Е, е
Рис. 5.4. Гипотетическое
расположение генов,
определяющих резус-фактор
вера между тремя генами в локусе ре-
резус-фактора у гетерозигот. Популяци-
онные исследования выявили разно-
разнообразные фенотипы: CDE, CDe, cDE,
cDe, CdE, Cde, cdE, cde. Взаимодейст-
Взаимодействия между генами, определяющими
резус-фактор, сложные. По всей види-
видимости, главным фактором, определяю-
определяющим резус-антиген, является антиген
D. Он обладает гораздо большей им-
муногенностью, чем антигены С и Е.
Решающая роль D-антигена в форми-
формировании фенотипического проявления
резус-фактора хорошо видна при ана-
анализе родословных (рис. 5.5). Отрица-
Отрицательный резус-фактор выявляется у
людей с генотипом d/d, положитель-
положительный — у людей с генотипом DD и D/d.
У гетерозигот CDe/Cde и Cde/cDe с
сочетанием генов Cde в резус-локусе
экспрессия фактора D изменяется, в
результате чего формируется фенотип
Du со слабой реакцией в ответ на вве-
введение резус-положительных антиге-
антигенов. Следовательно, работа генов в ре-
резус локусе может регулироваться ко-
6
СОе/сОе COe/Cde CDe/COe cde/Cae cOe/cOE cde/cOE cde/Cde
Рис. 5.5. Родословные по резус-фактору.
Обозначения: незакрашенные фигуры - D -
отрицательная кровь; черные фигуры - D -
положительная кровь; заштрихованные
фигуры - слабая реакция (DU - вариант)
личественно, и фенотипическое прояв-
проявление резус-фактора у резус-положи-
резус-положительных людей бывает различным:
большим или меньшим.
Несовместимость по резус-фактору
плода и матери способна стать причи-
причиной развития патологии у плода или
самопроизвольного выкидыша на ран-
ранних сроках беременности. С помощью
специальных чувствительных методов
удалось выявить, что во время родов
около 1 мл крови плода может попа-
попадать в кровоток матери. Если мать —
резус-отрицательная, а плод — резус-
положительный, то после первых ро-
родов мать будет сенсибилизирована к
резус-положительным антигенам. При
последующих беременностях резус-
несовместимым плодом титр анти-Rh-
антител в ее крови может резко возра-
возрасти, и под влиянием их разрушающего
действия у плода возникает характер-
характерная клиническая картина гемолитиче-
гемолитической патологии, выражающейся в ане-
анемии, желтухе или водянке.
В классической генетике наиболее
изученными являются три типа взаи-
взаимодействия неаллельных генов: эпи-
стаз, комплементарность и полимерия.
Они определяют многие наследуемые
признаки человека.
Эпистаз — это такой тип взаимодей-
взаимодействия неаллельных генов, при котором
одна пара аллельных генов подавляет
действие другой пары. Различают эпи-
эпистаз доминантный и рецессивный. До-
Доминантный эпистаз проявляется в том,
что доминантный аллель в гомозигот-
гомозиготном (АА) или гетерозиготном (Аа) со-
состоянии подавляет проявление другой
пары аллелей. При рецессивном эпи-
стазе ингибирующий ген в рецессив-
рецессивном гомозиготном состоянии (аа) не
дает возможность проявиться эписта-
тируемому гену. Подавляющий ген на-
называют супрессором или ингибито-
Взаимодействие генов
111
Рис. 5.в. Родословная по 'бомбейскому феномену". Обратите внимание,
что мать (II,6) с группой крови 0 имеет ребенка с группой крови АВ A11,1)
i
ром, а подавляемый — гипостатичес-
ким. Этот тип взаимодействия наибо-
наиболее характерен для генов, участвую-
участвующих в регуляции онтогенеза и иммун-
иммунных систем человека.
Примером рецессивного эпистаза у
человека может служить "бомбейский
феномен". В Индии была описана се-
семья, в которой родители имели вторую
(АО) и первую @0) группу крови, а
их дети — четвертую (АВ) и первую
@0) (рис. 5.6). Чтобы ребенок в такой
семье имел группу крови АВ, мать
должна иметь группу крови В, но ни-
никак ни О. Позже было выяснено, что в
системе групп крови АВО имеются ре-
рецессивные гены-модификаторы, кото-
которые в гомозиготном состоянии подав-
подавляют экспрессию антигенов на поверх-
поверхности эритроцитов. Например, чело-
человек с третьей группой крови должен
иметь на поверхности эритроцитов ан-
антиген группы В, но эпистатирующий
ген-супрессор в рецессивном гомози-
гомозиготном состоянии (h/h) подавляет
действие гена В, так что соответствую-
соответствующие антигены не образуются, и фено-
типически проявляется группа крови
О. Описанный локус гена-супрессора
не сцеплен с локусом АВО. Гены-су-
нрессоры наследуются независимо от
генов, определяющих группы крови
АВО. Бомбейский феномен имеет час-
частоту 1 на 13 000 среди индусов, говоря-
говорящих на языке махарати и живущих в
окрестностях Бомбея. Он распростра-
распространен также в изоляте на острове Реюнь-
Реюньон. По-видимому, признак детермини-
детерминирован нарушением одного из фермен-
ферментов, участвующих в синтезе антигена.
Комплементарность — это такой
тип взаимодействия, при котором за
признак отвечают несколько неаллель-
ных генов, причем разное сочетание
доминантных и рецессивных аллелей в
их парах изменяет фенотипическое
проявление признака. Но во всех слу-
случаях, когда гены расположены в раз-
разных парах хромосом, в основе расщеп-
расщеплений лежат цифровые законы, уста-
установленные Менделем.
Так, чтобы чтобы человек имел нор-
нормальный слух, необходима согласо-
согласованная деятельность нескольких пар
генов, каждый из которых может быть
представлен доминантными или ре-
рецессивными аллелями. Нормальный
слух развивается только в том случае,
если каждый из этих генов имеет хотя
бы один доминантный аллель в дипло-
диплоидном наборе хромосом. Если хотя бы
одна пара аллелей представлена рецес-
рецессивной гомозиготой, то человек будет
глухим. Поясним сказанное простым
112
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
примером. Предположим, что нор-
нормальный слух формирует пара генов.
В этом случае людям с нормальным
слухом присущи генотипы ААВВ,
ААВЬ, АаВВ, АаВЬ. Наследственная
глухота определяется генотипами:
adbb, Aabb, Aabb, aaBb, aaBB. Исполь-
Используя законы Менделя для дигибридного
скрещивания, легко рассчитать, что
глухие родители (aaBB x AAbb) могут
иметь детей с нормальным слухом
(АаВЬ), а нормально слышащие роди-
родители при соотвтствующем сочетании
генотипов (АаВЬ х АаВЬ) с высокой
долей вероятности (более 40%) — глу-
глухих детей:
Р: АаВЬ х АаВЬ
Fj: 9A-B-: ЪА-bb: ЪааВ-: laabb
слышащие
9
глухие
7
Полимерия — обусловленность оп-
определенного признака несколькими
парами неаллельных генов, обладаю-
обладающих одинаковым действием. Такие ге-
гены называются полимерными. Если
число доминантных аллелей влияет на
степень выраженности признака, по-
полимерия именуется кумулятивной.
Чем больше доминантных аллелей,
тем более интенсивно выражен при-
признак. По типу кумулятивной полиме-
полимерии обычно наследуются признаки,
которые можно выразить количест-
количественно: цвет кожи, цвет волос, рост.
Цвет кожи и волос человека, а также
цвет радужной оболочки глаз обеспе-
обеспечивает пигмент меланин. Формируя
окраску покровов, он предохраняет ор-
организм от воздействия ультрафиолето-
ультрафиолетовых лучей. Существует два типа мела-
меланинов: эумеланин (черный и темно-ко-
темно-коричневый) и феумеланин (желтый и
рыжий). Меланин синтезируется в
клетках из аминокислоты тирозина в
несколько этапов. Регуляция синтеза
осуществляется многими путями и за-
зависит, в частности, от скорости деле-
деления клеток. При ускорении митозов
клеток в основании волоса образуется
феумеланин, а при замедлении — эу-
эумеланин. Описаны некоторые формы
злокачественного перерождения кле-
клеток кожного эпителия, сопровождаю-
сопровождающиеся накоплением меланина (мела-
номы).
Все цвета волос, за исключением
рыжих, составляют непрерывный ряд
от темного до светлого (соответствен-
(соответственно уменьшению концентрации мела-
меланина) и наследуются нолигенно по ти-
типу кумулятивной полимерии. Счита-
Считается, что эти различия обусловлены
чисто количественными изменениями
в содержании эумеланина. Цвет ры-
рыжих волос зависит от наличия феуме-
лаиина. Окраска волос обычно меняет-
меняется с возрастом и стабилизируется с на-
наступлением половой зрелости.
Цвет радужной оболочки глаз опре-
определяют несколько факторов. С одной
стороны, он зависит от присутствия
гранул меланина, а с другой — от ха-
характера отражения света. Черный и ко-
коричневый цвета обусловлены много-
многочисленными пигментными клетками в
переднем слое радужной оболочки. В
светлых глазах содержание пигмента
значительно меньше. Преобладание
голубого цвета в свете, отраженном от
переднего слоя радужной оболочки, не
содержащей пигмента объясняется оп-
оптическим эффектом. Различное содер-
содержание пигмента, определяет весь диа-
диапазон цвета глаз.
По типу кумулятивной полимерии
наследуется также пигментация кожи
человека. На основе генетических ис-
исследований семей, члены которых
имеют разную интенсивность кожной
пигментации, предполагается, что цвет
Наследственность и среда
113
кожи человека определяют три или че-
четыре пары генов.
Полимерные гены, как правило,
обозначают одинаковыми буквами,
чтобы подчеркнуть однонаправлен-
однонаправленность их действия. При формировании
признака не важно, какой паре генов
принадлежат доминантные аллели,
важно их количество. В таком случае
генотип негра условно можно записать
АААЛА4. а генотип белого — аааааа.
Светлокожие негры будут иметь гено-
генотип АААААа, АаАААА, или другие со-
сочетания А и а. Мулатам соответствует
генотип АаАаАа, причем, чем больше
количество а, тем светлее их кожа. Ес-
Если упростить задачу и обозначить ге-
генотип, определяющий цвет кожи чело-
человека не тремя парами генов, а двумя, то
легко убедиться, что в браке людей с
противоположным цветом кожи все
дети будут мулатами:
Р: АААА х аааа
Fj: AaAa - мулаты
В браке двух мулатов AaAa x АаАа
вероятно рождение детей с разным
цветом кожи
- негры {АААА), 1\16
- светлокожие негры (АААа), 4\16
- мулаты (АаАа), 6\1б
- светлокожие мулаты (Аааа), 4\16
- белые (аааа), 1\16.
Таким образом, с определенной до-
долей вероятности родители могут иметь
детей с более светлой или более тем-
темной, чем у них, кожей, более светлыми
или более темными волосами и глаза-
глазами. Поскольку и рост наследуется по
типу кумулятивной полимерии, то де-
дети могут быть и выше, и ниже, чем ро-
родители.
Признание принципа взаимодейст-
взаимодействия генов наводит на мысль о том, что
все гены так или иначе взаимосвязаны
в своем действии. Если один ген ока-
оказывает влияние на работу других ге-
генов, то он может влиять на проявление
не только одного, но и нескольких при-
признаков. Такое множественное действие
гена называют плейотропией. Наибо-
Наиболее ярким примером плейотропного
действия гена у человека является
синдром Марфана, уже упоминавшая-
упоминавшаяся аутосомно-доминантная патология.
Арахнодактилия ("паучьи" пальцы) —
один из симптомов синдрома Марфа-
Марфана. Другими симптомами являются
высокий рост из-за сильного удлине-
удлинения конечностей, гиперподвижность
суставов, ведущий к близорукости,
подвывих хрусталика и аневризм аор-
аорты. Синдром с одинаковой частотой
встречается у мужчин и женщин. В ос-
основе указанных симптомов лежит де-
дефект развития соединительной ткани,
возникающий на ранних этапах онто-
онтогенеза и приводящий к множествен-
множественным фенотипическим проявлениям.
Плейотропным действием обладают
многие наледственные патологии. Оп-
Определенные этапы метаболизма обес-
обеспечивают гены. Продукты метаболиче-
метаболических реакций, в свою очередь регули-
регулируют, а возможно, и контролируют
другие метаболические реакции. По-
Поэтому нарушения метаболизма на од-
одном этапе отразятся на последующих
этапах, так что нарушение экспрессии
одного гена окажет влияние на не-
несколько элементарных признаков.
Наследственность и среда
Фенотиническое проявление при-
признака определяется генами, отвечаю-
отвечающими за этот признак, взаимодействи-
взаимодействием детерминирующих с другими гена-
генами и условиями внешней среды. Сле-
Следовательно, степень фенотипической
выраженности детерминированного
признака (экспрессивность) может
114
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
изменяться: усиливаться или ослаб-
ослабляться. Для многих доминантных
признаков характерно, что ген прояв-
проявляется у всех гетерозигот, но в разной
степени. Многие доминантные заболе-
заболевания обнаруживают значительную
индивидуальную изменчивость и по
возрасту начала, и по тяжести прояв-
проявления, и внутри одной семьи, и в раз-
разных семьях.
В ряде случаев признак может вооб-
вообще не выражаться фенотипически, не-
несмотря на генотипическую предопре-
предопределенность. Частота фенотипического
проявления данного гена среди его но-
носителей называется пенетрантностью
и выражается в процентах. Пенетрант-
ность бывает полной, если признак
проявляется у всех носителей данного
гена A00%), и неполной, если признак
проявляется только у части носите-
носителей. В случае неполной пенетрантнос-
ти иногда при передаче признака одно
поколение пропускается, хотя лишен-
лишенный его индивид, судя по родослов-
родословной, должен быть гетерозиготным. Пе-
нетрантность — это статистическое
понятие. Оценка ее величины часто
зависит от применяемых методов об-
обследования.
Понятия пенетрантности и экспрес-
экспрессивности можно рассмотреть на при-
примере аутосомно-доминантного призна-
признака — полидактилии (многопалости). У
гетерозигот фенотипическое его про-
проявление варьирует. Пенетрантность
может отсутствовать @%), то есть при-
признак не проявляется, и, несмотря на ге-
генетическую предопределенность, ко-
количество пальцев на руках и ногах рав-
равно 5. В то же время у других гетерози-
гетерозигот признак проявляется, но степень
его выраженности у разных лиц раз-
различна. Количество пальцев на руках и
ногах бывает у разных индивидуумов
соответственно: 5,5 — 6,6; 5,6 — 5,7; 6,6 —
6,6 и т.д. В этом случае говорят о варь-
варьирующей экспрессивности.
Иногда тяжелое доминантное забо-
заболевание проявляется только во время
или после репродуктивного периода.
Классический пример — хорея Ген-
тингтона, дегенеративное заболевание
нервных клеток в базальных ганглиях,
приводящее к непроизвольным движе-
движениям, изменениям личности и посте-
постепенно нарастающему слабоумию. Ис-
Исследование 802 случаев хореи Гентинг-
тона в Западной Германии показало,
что возраст начала проявления болез-
болезни варьирует от 6 до 76 лет, но макси-
максимум соответствует возрастным грани-
границам от 26 до 60 лет (93,5%). Таким об-
образом, иенетрантность хореи Гентинг-
тона увеличивается с возрастом.
Учитывая экспрессивность и пенет-
пенетрантность, генетический анализ при-
признака проводят, используя обширные
родословные.
Генетика пола
Из 46 хромосом B3 пары) в кариоти-
пе человека 22 пары одинаковы у муж-
мужчин и женщин (аутосомы), а одна пара,
называемая половой, у разных полов
отличается: у женщин — XX, у муж-
мужчин — XY (см. главу 3). Половые хро-
хромосомы представлены в каждой сома-
соматической клетке индивида. При образо-
образовании гамет во время мейоза гомоло-
гомологичные половые хромосомы расходятся
в разные половые клетки. Следователь-
Следовательно, каждая яйцеклетка помимо 22 ауто-
сом несет одну половую хромосому X
(гаплоидный набор хромосом равен
23). Все сперматозоиды также имеют
гаплоидный набор хромосом, из кото-
которых 22 — аутосомы, а одна — половая.
Половина сперматозоидов содержит X,
другая половина — Y хромосому.
Поскольку женские половые хромо-
хромосомы одинаковы и все яйцеклетки не-
Генетика пола
115
сут Х-хромосому, то женский пол у че-
человека называют гомогаметным. Муж-
Мужской же пол из-за различия половых
хромосом (X или Y) в сперматозоидах
именуют гетерогаметным.
Пол человека определяется в мо-
момент оплодотворения. Женщина имеет
один тип гамет — X, мужчина — два ти-
типа гамет: X и Y, причем, согласно зако-
законам мейоза, образуются они в равной
пропорции. При оплодотворении хро-
хромосомные наборы гамет объединяют-
объединяются. Напомним, что зигота содержит 22
пары аутосом и одну пару половых
хромосом. Если яйцеклетку оплодо-
оплодотворил сперматозоид с Х-хромосомой,
то в зиготе пара половых хромосом бу-
будет XX, из нее разовьется девочка. Ес-
Если же оплодотворение произвел спер-
сперматозоид с Y-хромосомой, то набор по-
половых хромосом в зиготе — XY. Такая
зигота даст начало мужскому организ-
организму. Таким образом, пол будущего ре-
ребенка определяет гетерогаметный по
половым хромосомам мужчина. Соот-
Соотношение полов при рождении, по дан-
данным статистики, соответствует при-
примерно 1:1.
Хромосомное определение пола —
не единственный уровень половой
дифференцировки. Большую роль в
этом процессе у человека играет гор-
гормональная регуляция, происходящая с
помощью половых гормонов, которые
синтезируются половыми железами.
Закладка половых органов человека
начинается у пятинедельного эмбрио-
эмбриона. В зачатки гонад из желточного
мешка мигрируют первичные клетки
зародышевого пути, которые, размно-
размножаясь митозом, дифференцируются в
гонии и становятся предшественника-
предшественниками гамет. У зародышей обоих полов
миграция проходит одинаково. Если
же в клетках зачатков гонад присутст-
присутствует Y-хромосома, то начинают разви-
развиваться семенники, причем начало диф-
дифференцировки связано с функциони-
функционированием эухроматинового района Y-
хромосомы. Если же Y-хромосома от-
отсутствует, то развиваются яичники,
что соответствует женскому типу.
Предполагается, что Y-хромосома не
детерминирует дифференцировку по
мужскому типу, а лишь контролирует
работу соответствующего аутосомного
гена. У индивидов, не имеющих Y-xpo-
мосомы, этот структурный аутосом-
ный ген не активируется.
Человек по своей природе бисексуа-
бисексуален. Зачатки половой системы одина-
одинаковы у зародышей обоих полов. Если
активность Y-хромосомы подавлена,
то зачатки половых органов развива-
развиваются по женскому типу. При полном
отсутствии всех элементов становле-
становления мужского пола формируются жен-
женские половые органы. Их развитие не
нуждается в специальных регулятор-
ных механизмах и является "конститу-
"конститутивным".
Тип вторичных половых признаков
обусловлен дифференцировкой гонад.
Половые органы формируются из
мюллеровых и вольфовых каналов. У
женщин мюллеровы протоки развива-
развиваются в фаллопиевы трубы и матку, а
вольфовы атрофируются. У мужчин
вольфовы каналы развиваются в се-
семенные протоки и семенные пузырьки.
Под влиянием хорионического гона-
дотропина матери лежащие в эмбрио-
эмбриональных семенниках клетки Лейдига
синтезируют стероидные гормоны (те-
(тестостерон), которые участвуют в регу-
регуляции развития особи по мужскому
типу. Одновременно в семенниках в
клетках Сертоли синтезируется гор-
гормон, ингибирующий дифференциров-
дифференцировку мюллеровых протоков. Нормаль-
Нормальные особи мужского пола развиваются
только в случае, если все гормоны,
116
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
Генетический
пол
Ген
Синтезированный
белок
Гонады
зародыша
Зачаток
репродукционной
системы
Плод
6
XV
Тестикулярной
феминизации
Рецептор
андрогена
Ген,
определяющий
развитие
семенников
(Y-хромосома)
Семенники
¦
.Тестостерон
Яичники
Тестикулярной
феминизации
Рецептор
андрогена
Мужская/женская
Мужского пола
Мужская/женская
Женского пола
Рис. 5.7. Схема регуляции дифференцировки репродуктивной системы плода
действующие на зачатки внешних и
внутренних половых органов, "сраба-
"срабатывают" в определенное время в задан-
заданном месте. Схема регуляции развития
репродуктивной системы плода пред-
представлена на рисунке (рис. 5.7).
В настоящее время описано около
20 разнообразных дефектов генов, ко-
которые при нормальном (XY) кариоти-
пе по половым хромосомам приводят к
нарушению дифференцировки внеш-
внешних и внутренних половых признаков,
(гермафродитизму). Эти мутации свя-
связаны с нарушением : а) синтеза поло-
половых гормонов; б) восприимчивости ре-
рецепторов к ним; в) работы ферментов,
участвующих в синтезе регулирующих
факторов и т.д.
Наследование признаков,
сцепленных с полом
Х- и Y-хромосомы гомологичны, по-
поскольку обладают общими гомологич-
гомологичными участками, где локализованы ал-
лельные гены. Однако, несмотря на го-
гомологию отдельных локусов, эти хро-
хромосомы различаются по морфологии
(см. рис. 3.13). Ведь, помимо общих
участков, они несут большой набор раз-
различающихся генов. В Х-хромосоме ле-
лежат гены, которых нет в Y-хромосоме, а
ряд генов Y-хромосомы отсутствуют в
Х-хромосоме. Таким образом, у мужчин
в половых хромосомах некоторые гены
не имеют второго аллеля в гомологич-
гомологичной хромосоме. В таком случае признак
определяется не парой аллельных ге-
генов, как обычный менделирующий
признак, а только одним аллелем. По-
Подобное состояние гена называется ге-
мизиготным (рис. 5.8), а признаки, раз-
развитие которых обусловлено одиноч-
одиночным аллелем, расположенным в одной
из альтернативных половых хромосом,
получили название сцепленных с по-
полом. Она преимущественно развивают-
развиваются у одного из двух полов и по-разному
наследуются у мужчин и женщин.
Генетика пола
117
Г\
14
\J
Гены, определяющие
Х-сцепленные признаки
Гены, определяющие
голандрические признаки
Общая цветовая слепота
Пигментная ксеродерма
X Y
Рис. 5.8. Гипотетическая схема расположения генов в Х- и Y- хромосомах
Признаки, сцепленные с Х-хромосо-
мой, могут быть рецессивными и до-
доминантными. К рецессивным относят-
относятся: гемофилия, дальтонизм (неспособ-
(неспособность различать красный и зеленый
цвета), атрофия зрительного нерва и
миопатия Дюшена. К доминантным —
рахит, не поддающийся лечению вита-
витамином Д, и темная эмаль зубов.
Рассмотрим наследование, сцеплен-
сцепленное с Х-хромосомой, на примере ре-
рецессивного гена гемофилии. У мужчи-
мужчины ген гемофилии, локализованный в
Х-хромосоме, не имеет аллеля в Y-xpo-
мосоме, то есть находится в гемизигот-
ном состоянии. Следовательно, несмо-
несмотря на то, что признак рецессивный, у
мужчин он проявляется:
N — ген нормальной свертываемос-
свертываемости крови,
h — ген гемофилии;
XhY — мужчина с гемофилией;
XNY — мужчина здоров.
У женщин признак определяется
нарой аллельных генов в половых хро-
хромосомах XX, следовательно, гемофи-
гемофилия может проявиться только в гомо-
гомозиготном состоянии:
XNXN - женщина здорова.
XNXb — гетерозиготная женщина, но-
носительница гена гемофилии, здорова,
XhXh — женщина с гемофилией.
Перечислим основные формальные
характеристики Х-сцепленного рецес-
рецессивного наследования. Обычно забо-
заболевание поражает мужчин. Все их фе-
нотипически здоровые дочери являют-
являются гетерозиготными носительницами,
поскольку в процессе оплодотворения
получают от отца Х-хромосому:
гаметы:
Xh
дочерям сыновьям
Среди сыновей гетерозиготных мате-
матерей (XNXb) соотношение больных и здо-
здоровых составляет 1:1, так как гаметы XN
и Xh образуются с равной вероятностью:
vNyn у Y!*Y
гаметы: X",Xh Х*,У
материнские X"
гаметы X"
Отцовские гаметы
Xм
XNXN
XNX"
девочки
здоровы
У
X-Y
X-Y
мальчики
1 : 1
здоровы
больны
Такое наследование получило на-
название крисс-кросс (или крест-на-
118
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
? О
Альберт Виктория
6
О 6
Фридрих III Виктория Людвиг Алиса Леопольд Хелен Беатриче Генрих
D
бйЪ а
Генрих Ирен Фридрих Александра Николай II Алиса Александр Виктория Леопольд Мориц Аль-
I Фоне XIII
Альфонсо
Вальдемар Генрих
Алексей
Руппрехт
Рис. 5.9. Родословная с Х-сцепленной гемофилией А
в европейских королевских домах
крест): сыновья наследуют фенотипи-
ческий признак матери, а дочери —
признак отца.
Законы передачи признаков, сцеп-
сцепленных с Х-хромосомами, были впер-
впервые изучены Т. Морганом.
Наиболее известным примером ста-
стало наследование королевской гемофи-
гемофилии А среди потомков английской ко-
королевы Виктории (рис. 5.9). Королева
Виктория была гетерозиготной и пере-
передала мутантный ген сыну гемофилику
и трем дочерям. (Один из потомков
королевы царевич Алексей в России
также страдал этим недугом). Соглас-
Согласно представленной родословной, как и
следует ожидать при рецессивном X-
сцепленном наследовании, больны ге-
гемофилией мужчины. Однако бывает и
по-другому. Описаны семьи, в родо-
родословных которых наличествуют близ-
близкородственные браки, где гемофилия
средней степени выявляется и у жен-
женщин (рис. 5.10).
I
|66
IV
аа
шшо 6
Рис. 5.10. Родословная с двумя женщинами, гомозиготными по Х-сцепленной гемофилии.
Родители - двоюродные сибсы. Фигуры с точками обозначают гетерозигот
Сцепление генов и карты хромосом
119
Помимо Х-сцепленных, у мужчин
имеются Y-сцепленные признаки. Они
называются голандрическими. Опре-
Определяющие их гены локализованы в тех
районах Y-хромосом, которые не име-
имеют аналогов в Х-хромосомах (рис. 5.8).
Голандрические признаки также опре-
определяются только одним аллелем, а по-
поскольку их гены находятся только в Y-
хромосоме, то выявляются они у муж-
мужчин и передаются от отца к сыну, вер-
вернее — ко всем сыновьям. К голандри-
ческим признакам относятся: волоса-
волосатость ушей, перепонки между пальца-
пальцами ног, ихтиоз (кожа имеет глубокую
исчерчен ность и напоминает рыбью
чешую).
Гомологичные районы Х- и Y-хро-
Y-хромосом содержат аллельные гены, с рав-
равной вероятностью встречающиеся у
лиц мужского и женского пола.
К числу определяемых ими призна-
признакам относятся общая цветовая слепота
(отсутствие цветового зрения) и пиг-
пигментная ксеродерма — заболевание,
при котором под влиянием ультрафио-
ультрафиолетовых лучей на открытых частях те-
тела появляются пигментированные
пятна, которые постепенно преобразу-
преобразуются в папилломы, а затем и в опухо-
опухоли. Оба эти признака являются рецес-
рецессивными. Признаки, связанные с ал-
лельными генами, находящимися в X-
и Y-хромосомах, наследуются по клас-
классическим менделевским законам.
Наследование,ограниченное
и контролируемое полом
Признаки человека, наследование
которых каким-то образом связано с
полом, подразделяются на несколько
категорий.
Одна из категорий — признаки, ог-
ограниченные полом. Их развитие обус-
обусловлено генами, расположенными в
аутосомах обоих полов, но проявляю-
проявляющимися только у одного пола. Напри-
Например, гены, определяющие ширину таза
женщины, локализованы в аутосомах,
наследуются и от отца и от матери, но
проявляются только у женщин. То же
касается возраста полового созревания
девочек. Среди мужских признаков,
ограниченных полом, можно назвать
количество и распределение волосяно-
волосяного покрова на теле.
К иной категории относятся призна-
признаки, контролируемые полом, или зави-
зависимые от пола. Развитие соматических
признаков обусловлено генами, распо-
расположенными в аутосомах, проявляются
они у мужчин и женщин, но по-разно-
по-разному. Например, у мужчин раннее облы-
облысение — признак доминантный, он
проявляется как у доминантных гомо-
гомозигот (АА), так и у гетерозигот (Ли). У
женщин этот признак рецессивный, он
проявляется только у рецессивных го-
гомозигот (аа). Поэтому лысых мужчин
гораздо больше, чем женщин. Другим
примером может служить подагра, у
мужчин ее пенетрантность выше: 80%
против 12% у женщин. Значит, чаще
подагрой болеют мужчины. Экспрес-
Экспрессивность признаков, контролируемых
полом, обусловлена половыми гормо-
гормонами. Например, тип певческого голо-
голоса (бас, баритон, тенор, сопрано, мец-
меццо-сопрано и альт) контролируется по-
половой конституцией. Начиная с перио-
периода полового созревания, признак нахо-
находится под влиянием половых гормо-
гормонов.
Сцепление генов и карты хромосом
Хромосомная теория наследствен-
наследственности была сформулирована и экспе-
экспериментально доказана Т. Морганом и
его сотрудниками. Согласно этой тео-
теории, гены находятся в хромосомах и
расположены в них линейно. Гены, ло-
локализованные в одной хромосоме, на-
120
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
зываются сцепленными, наследуются
вместе и образуют группу сцепления.
Количество групп сцепления соответ-
соответствует числу пар гомологичных хромо-
хромосом. У человека 46 хромосом: 22 пары
аутосом и одна пара половых хромо-
хромосом (XX или XY), следовательно, у
женщин 23 группы сцепления, а у
мужчин — 24, так как половые хромо-
хромосомы мужчины (XY) не полностью го-
гомологичны друг другу. Каждая из по-
половых хромосом мужчины имеет гены,
характерные только для Х- и только
для Y-хромосомы, которым соответст-
соответствуют группы сцепления Х- и Y-хромо-
Y-хромосомы.
Гены, локализованные в одной хро-
хромосоме и образующие группу сцепле-
сцепления, сцеплены не абсолютно. В зиготе-
не профазы первого мейотического де-
деления гомологичные хромосомы сли-
сливаются вместе с образованием бива-
бивалентов, затем в пахитене происходит
кроссинговер-обмен участками между
хроматидами гомологичных хромосом.
Кроссинговер — обязательный про-
процесс. Он осуществляется в каждой па-
паре гомологичных хромосом. Чем даль-
дальше друг от друга расположены гены в
хромосоме, тем чаще между ними про-
происходит кроссинговер. Благодаря это-
этому процессу, возрастает разнообразие
сочетания генов в гаметах. Например,
пара гомологичных хромосом содер-
содержит сцепленные гены АВ и ah. В про-
профазе мейоза гомологичные хромосомы
конъюгируют и образуют бивалент:
АЕ
аЬ
Если кроссинговер между генами А
и В не произойдет, то в результате мей-
мейоза образуется два тина некроссовер-
ных гамет: ДВ_ и аЬ. Если же кроссин-
кроссинговер состоится, то получатся кроссо-
верные гаметы: А?> и_аВ, то есть группы
сцепления изменятся. Чем более уда-
удалены друг от друга гены А и В, тем
больше возрастает вероятность обра-
образования и, соответственно число крос-
соверных гамет.
Если гены в большой хромосоме
расположены на достаточном расстоя-
расстоянии друг от друга и между ними в мей-
озе происходят многочисленные пере-
перекресты, то они могут наследоваться не-
независимо.
Открытие кроссинговера позволило
Т. Моргану и сотрудникам его школы в
первые два десятилетия XX века раз-
разработать принцип построения генети-
генетических карт хромосом. Явление сцеп-
сцепления было использовано ими для вы-
выяснения локализации генов, располо-
расположенных в одной хромосоме, и созда-
создания генных карт плодовой мушки
Drosophila melanogaster. На генетичес-
генетических картах гены располагаются линей-
линейно друг за другом на определенном
расстоянии. Расстояние между генами
определяется в процентах кроссинго-
кроссинговера, или в морганидах A % кроссин-
кроссинговера равен одной морганиде).
Для построения генетических карт у
растений и животных проводят анали-
анализирующие скрещивания, в которых до-
достаточно просто рассчитать процент
особей, образовавшихся в результате
кроссинговера, и построить генетичес-
генетическую карту по трем сцепленным генам.
У человека анализ сцепления генов
классическими методами невозможен,
поскольку невозможны эксперимен-
экспериментальные браки. Поэтому для изучения
групп сцепления и составления карт
хромосом человека используют другие
методы, в первую очередь генеалогиче-
генеалогический, основанный на анализе родо-
родословных. Рассмотрим на конкретном
примере, как можно выявить группу
сцепления генов и констатировать
кроссинговер, анализируя родослов-
родословные.
Сцепление генов и карты хромосом
121
19
IV
о а о
in
ШЙ О
Рис. 5.11. Родословные с цветовой слепотой на красный и зеленый цвет (-),
гемофилией (|) и обоими признаками одновременно (+). А - сцепленное наследование
двух признаков. Б - Выявление кроссинговера между генами, определяющими
гемофилию и дальтонизм
Чтобы выявить кроссинговер, необ-
необходимо исследовать либо большую ро-
родословную, либо несколько небольших.
На рис. 5.11,А приведена родословная, в
которой одновременно наследуются
красно-зеленая слепота (дальтонизм) и
гемофилия. На протяжении трех поко-
поколений пораженными оказываются толь-
только лица мужского пола, следовательно,
признаки являются рецессивными и X-
сцепленными. Гетерозиготные женщи-
женщины A1,1 и 111,2) имеют как здоровых, так
и страдающих дальтонизмом и гемофи-
гемофилией сыновей. Лица женского пола в
данной семье здоровы. Эти факты также
подтверждают Х-сцепленную природу
наследования обоих признаков. По-
Поскольку оба признака определяются ге-
генами, находящимися в Х-хромосоме,
следовательно, гены сцеплены.
Обозначим рецессивные гены, опре-
определяющие дальтонизм и гемофилию, а
и b соответственно, тогда группа сцеп-
сцепления будет X*:
F,:XV
ll.
Гетерозиготная женщина из поколе-
поколения F, (II) выходит замуж за здорового
мужчину (XABY), и имеет двух здоро-
здоровых дочерей, одного здорового сына и
двух сыновей со сцепленным наследо-
наследованием генов гемофилии и дальтониз-
дальтонизма, что соответствует законам наследо-
наследования признаков, сцепленных с Х-хро-
мосомой. В этом случае группа сцепле-
сцепления передается из поколения в поколе-
поколение, и кроссинговер не выявлен.
Если гены, образующие группу
сцепления, находятся на достаточном
расстоянии друг от друга, то между ни-
ними возможен кроссинговер. Известны
родословные, в которых выявляются
кроссоверные особи. Одна из таких ро-
родословных представлена на рис. 5.11,Б.
Проведем генетический анализ этой
родословной. В поколении II женщина
здорова, но является гетерозиготной
носительницей гена гемофилии, полу-
полученного ею от своего отца вместе с X-
хромосомой. Среди сыновей этой жен-
женщины есть не только гемофилик, но и
дальтоник, следовательно мы вправе
предположить, что она является носи-
носительницей ген, определяющего дальто-
дальтонизм, хотя сама здорова. Используя
для обозначения генов, определяющих
развитие гемофилии и дальтонизма,
буквенные символы, указанные выше,
можно записать генотипы вступающих
в брак гетерозиготной женщины и здо-
здорового мужчины (II):
122
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
Х*Ух Х^ХИ
У матери могут образоваться не-
кроссоверныс гаметы: X , X . Но если
между генами, определяющими даль-
дальтонизм и гемофилию, в ооцитах мате-
матери произойдет кророссинговер, тогда
образуются кроссоверные гаметы: X ,
X . При оплодотворении некроссовер-
ных гамет матери гаметами отца X , Y
образуются генотипы, которые опреде-
определяют фенотипическое проявление
дальтонизма и гемофилии у сыновей:
X"Y(III,3) X"Y(IH,2)
дальтонизм гемофилия
При оплодотворении кроссоверных
гамет матери образуются кроссовер-
кроссоверные генотипы, определяющие у сыно-
сыновей фенотипическое проявление двух
признаков одновременно или здоро-
здоровый фенотип:
здоров дальтонизм
гемофилия
Анализируемая родословная невели-
невелика, поэтому нельзя однозначно утверж-
утверждать, то генотип матери именно таков,
каким мы его представили. Веретен и
вариант, что генотип матери X X . Тог-
Тогда фенотип сыновей может оставаться
точно таким же, но 1 и 4 будут некроссо-
верными, а 2 и 3 — кроссоверными.
Точное установление генотипов ро-
родителей при наборе достаточного ко-
количества обследованных семей, позво-
позволяет определить частоту кроссоверных
генотипов. Разработаны сложные ста-
статистические методы для объективной
оценки количества кроссоверных осо-
особей на основе анализа родословных.
Такая оценка дает возможность уста-
установить расстояние между генами, уча-
участвующими в процессе кроссинговера.
В некоторых случаях сцепление генов
может быть выявлено путем обзора об-
обширных родословных. Статистический
анализ большого количества родослов-
родословных конкретизирует параметры сцепле-
сцепления, а компьютерные расчеты уточняют
наиболее вероятные частоты рекомби-
рекомбинаций сцепленных генов. Установлен-
Установленные же частоты рекомбинаций в груп-
группах сцепления позволяют построить
генные карты по группам сцепления.
Пары генов или группы сцепления
аутосомных генов невозможно соотне-
соотнести с конкретными хромосомами, ис-
используя только формально-генетичес-
формально-генетический анализ родословных. Для установ-
установления конкретной локализации генов
использовались морфологические
маркеры хромосом. Например, на
длинном плече первой хромосомы
вблизи центромеры часто обнаружива-
обнаруживается вторичная перетяжка. Морфоло-
Морфология этой перетяжки бывает различной,
а наследуемость определенной морфо-
морфологии прослеживается в череде поко-
поколений. С присутствием слишком тон-
тонкой и длинной перетяжки связано на-
наличие некоторых патологий. Анализ
родословных в связи с морфологией
первой хромосомы выявил группу
сцепления из трех локусов: врожден-
врожденной очаговой катаракты, группы крови
Даффи и локуса Un — 1.
Маркерами при определении групп
сцепления в конкретных хромосомах
могут служить и достаточно крупные,
выявляемые морфологически делеции.
Другой подход в поисках соответст-
соответствия групп сцепления конкретным хро-
хромосомам связан с исследованием уров-
уровня активности метаболических фер-
ферментов. Так, у больного с синдромом
Дауна, причиной которого стала трисо-
мия по 21-й хромосоме, оказалась по-
повышенной активность ряда ферментов,
что может зависеть от увеличения дозы
Сцепление генов и карты хромосом
123
генов за счет увеличения количества
21-х хромосом. Предположение, что эти
ферменты кодируются генами, локали-
локализованными в 21-й хромосоме, позже
подтвердилось. Однако проведение
грубых аналогий всегда связано с неко-
некоторым риском, поскольку хромосом-
хромосомный дисбаланс может приводить к на-
нарушениям регуляции активности генов,
локализованных в разных хромосомах.
Накопление данных о группах сцеп-
сцепления генов у человека продвигалось
очень медленно. Прорыв в этой облас-
области был достигнут с возникновением
новых технологий, основанных на гиб-
гибридизации соматических клеток в кле-
клеточных культурах, в частности, метода
картирования.
В гибридных клетках человек-мышь,
полученных в результате слияния ане-
уплоидных L-клеток мыши и диплоид-
диплоидных эмбриональных фибробластов че-
человека, 75-95% человеческих хромосом
утрачиваются в процессе культивирова-
культивирования, причем их утрата носит случайный
характер. Среди множества разнообраз-
разнообразных гибридов всегда найдется клетка,
сохранившая ту или иную хромосому
человека. После размножения этой
клетки можно провести анализ фермен-
ферментов, активность которых связана с нали-
наличием именно данной хромосомы. Ис-
Использование методов дифференциаль-
дифференциального окрашивания хромосом позволяет
связать гены с определенными локуса-
ми хромосом, так как в гибридных клет-
клетках довольно часты хромосомные раз-
разрывы, перестройки, присутствие не це-
целых хромосом, а отдельных фрагментов.
Современные методы картирования
развивались на основе молекулярной
биологии, генной инженерии, гибриди-
гибридизации на препаратах хромосом с ДНК-
зондами. Новые принципы картирова-
картирования хромосом человека описаны в главе
3. Данные о сцеплении генов в хромосо-
хромосомах, полученные современными мето-
методами картирования, достаточно хорошо
согласуются с первоначальными ре-
результатами, основанными на методах
классической генетики. Так, подтверж-
подтверждено расположение в первой хромосоме
генов группы крови Даффи, локуса Un-
1, и врожденной очаговой катаракты,
картированных ранее. К настоящему
времени эта самая крупная хромосома
человека является наиболее изученной.
В ее составе обнаружены гены, кодиру-
кодирующие белки разных классов. Это и фер-
ферменты, и белки групп крови резус-фак-
резус-фактор, и факторы свертываемости крови.
В популяциях людей отмечен полимор-
полиморфизм генов этой хромосомы. Мутации
в некоторых из них вызывают харак-
характерные генетические заболевания, на-
например, фенилкетонурию.
В последние годы достигнуты боль-
большие успехи в выявлении сцепления и ло-
локализации локусов сцепления в опреде-
определенных хромосомах, изучены все 24
группы сцепления, построены цитологи-
цитологические карты генов хромосом человека, в
которых для каждого сегмента диффе-
дифференциально окрашенной хромосомы по-
показано, какие гены на каком расстоянии
друг от друга там находятся. Ожидается,
что среднее количество генов, соответст-
соответствующих одному сегменту, — несколько
сотен. Сегодня генная карта человека до-
достаточно насыщена; картировано около
8000 генов, и число это быстро растет. В
рамках же международной программы
"Геном человека" к 2005 г. будет картиро-
картировано большинство генов.
Анализ групп сцепления человека
показывает, что в ряде случаев локус
сцепления объединяет родственные ге-
гены. Так, например, сцеплены локусы
гемоглобинов человека: у, 6 и Ь. Имму-
ноглобулиновый район содержит не-
несколько локусов, ответственных за
синтез у-глобулиновых полипеитед-
124
Глава 5. КЛАССИЧЕСКИЕ ТИПЫ НАСЛЕДОВАНИЯ У ЧЕЛОВЕКА
ных цепей. В первой хромосоме лока-
локализовано не менее четырех генов, во-
вовлеченных в контроль процесса глико-
гликолиза. Известны и другие функциональ-
функциональные группы сцепления.
Все тесные группы сцепления назы-
называются кластерами. Считается, что ген-
генные кластеры — результат эволюцион-
эволюционного процесса. Их могут порождать ген-
генные дупликации, неравный кроссинго-
вер, создавая основу для дальнейшей
функциональной специализации генов
в ходе эволюции. В отсутствие хромо-
хромосомных перестроек, разбивающих клас-
кластер, гены остаются тесно сцепленными.
Порядок расположения генов в кла-
кластере может иметь функциональное
значение. Так, в р-глобиновом класте-
кластере человека гены гемоглобина распо-
расположены в той последовательности, в
какой они экспрессируются в онтоге-
онтогенезе человека: е, у, р и а.
Родственные гены не всегда сцепле-
сцеплены в один кластер. Гены а-глобиновой
группы человека, очевидно, родствен-
родственные генам р-глобиновой группы, не яв-
являются сцепленными. Возможно, при-
причину этого явления следует искать в
характере эволюционного процесса,
хотя не исключается и функциональ-
функциональная значимость такой локализации.
Частота кроссинговера зависит не
только от расстояния между генами.
Известны и другие факторы, влияющие
на рекомбинацию сцепленных генов. В
свое время имела место дискуссия от-
относительно влияния возраста родите-
родителей на уровень рекомбинации. Дейст-
Действительно, в ряде локусов частота крос-
кроссинговера в мейозе возрастает с возрас-
возрастом, но для других локусов это утверж-
утверждение неверно. Обсуждается предполо-
предположение о том, что у гомогаметного пола
(XX) кроссинговер происходит чаще.
До недавнего времени исследова-
исследования по сцеплению представляли, в
основном, теоретический интерес.
Ныне же появилась возможность
практически применять полученные
знания. Например, если ген А вызы-
вызывает редкое наследственное заболева-
заболевание, проявляющееся в позднем возра-
возрасте, а ген В тесно сцеплен с геном А и
является генетическим маркером, то
появление в семье признака, опреде-
определяемого геном В, заставляет ожидать
наследственного заболевания, опре-
определяемого геном А. Кроме того, в ран-
раннем возрасте можно предсказать ве-
вероятность наследственной патологии
и скорректировать образ жизни чело-
человека, чтобы уменьшить риск развития
патологии.
Достоверная информация о группах
сцепления важна для дородового гене-
генетического консультирования, посколь-
поскольку точная дородовая диагностика по
любой наследственной натологии осу-
осуществима только тогда, когда возмож-
возможно определенное предсказание на ос-
основе анализа генотипа родителей.
Вопросы для самоконтроля
1 Приведите примеры всех типов взаимодейст-
взаимодействия аллсльных генов: доминирования полного и не-
неполного, ко- и сверхдомннирования.
2. Укажите примеры всех типов взаимодействия
нсаллсльмых генов у человека.
3. Разберите на примерах понятия: экспрессив-
экспрессивность и пенетрантность.
4. Назовите генетические и физиологические
факторы, определяющие пол человека.
5. Как наследуются рецессивные признаки, сцеп-
сцепленные с Х-хромосомой? Пример.
6. Что такое голандрические признаки? Пример.
7. О каких признаках говорят, что они ограниче-
ограничены полом?
8. Сколько групп сцепления у мужчин и жен-
женщин?
9. Перечислите методы, используемые для опре-
определения групп сцепления у человека.
Глава 6. МУТАЦИИ
Классификация мутаций
Термин "мутация" был впервые вве-
введен в 1901 г. голландским ботаником,
автором мутационной теории Гуго Де
Фризом, который занимался изучени-
изучением наследственности энотеры Ламарка
(Oenothera lamarckiana). Наблюдая за
этим растением в условиях экспери-
эксперимента, ученый обнаружил стабильные
фенотипические изменения, которые
назвал мутациями.
Мутации представляют собой вне-
внезапные и устойчивые изменения гено-
генотипа, возникающие иод влиянием фак-
факторов внешней и внутренней среды.
Процесс образования мутаций носит
название мутагенеза, а факторы, вы-
вызывающие мутации, именуются мута-
мутагенами.
Мутации могут возникать в поло-
половых клетках (генеративные мутации).
Такие изменения способны переда-
передаваться следующим поколениям орга-
организмов при половом размножении и
проявляются, как правило, во всех
клетках потомков. Следует подчерк-
подчеркнуть, что, хотя эти мутации возникают
на уровне половых клеток, в их число
входят как аномалии половых хромо-
хромосом, так и аномалии аутосом в этих
клетках.
Мутации, возникающие в соматиче-
соматических клетках (соматические мута-
мутации), наследуются дочерними клетка-
клетками, которые образуются в процессе
митотических делений. Фенотипичес-
Фенотипические последствия таких изменений про-
проявляются только у самой мутантной
особи и только в том случае, если воз-
возникшие мутации препятствуют осуще-
осуществлению специфических функций,
свойственных данной клетке. Сомати-
Соматические мутации могут содержаться не
во всех клетках организма, т.е. нор-
нормальные и мутантные клетки сосуще-
сосуществуют у одного индивидуума, что
приводит к мозаицизму — наличию в
организме клеток, отличающихся по
своему генотипу и его фенотипичес-
ким проявлениям от других клеток
этого же организма. По способу воз-
возникновения различают мутации спон-
спонтанные и индуцированные.
Спонтанные мутации возникают
случайно, т.е. в любой момент любой
ген может претерпеть изменения. При-
Причинами спонтанного мутационного
процесса являются многочисленные
факторы экзогенной и эндогенной
природы, в том числе постоянное воз-
воздействие на организм человека мутаге-
мутагенов химической, биологической и фи-
физической природы (например, естест-
естественный фон облучения, действие ви-
вирусов); ошибки репликации ДНК, ко-
которые копируются и накапливаются в
ряду клеточных поколений; наруше-
нарушение функционирования репаративных
систем; действие экзогенных метабо-
метаболитов; физиологическое состояние и
возраст организма. Спонтанные мута-
мутации могут возникать как в половых,
так и в соматических клетках на ген-
генном, хромосомном и геномном уров-
уровнях.
Индуцированные мутации возника-
возникают в результате направленного воздей-
воздействия на организм мутагенных факто-
факторов различной природы. Различают
физические (радиация, температура,
давление и т.п.), химические (пестици-
(пестициды, тяжелые металлы и пр.) и биологи-
126
Глава 6.
ческие (вирусы, бактерии) факторы
мутагенного действия. Более подробно
вопросы индуцированного мутагенеза
будут рассмотрены ниже.
Мутации возникают независимо от
того, полезны они для организма или
вредны, т.к. носят случайный характер.
В зависимости от действия на орга-
организм принято выделять отрицатель-
отрицательные мутации (летальные, полулеталь-
полулетальные), которые могут приводить либо к
гибели организма, либо к снижению
его жизнеспособности, нейтральные
мутации, не оказывающие существен-
существенного влияния на процессы жизнедея-
жизнедеятельности, и положительные мутации,
повышающие адаптационные способ-
способности организма. Последние встреча-
встречаются достаточно редко, однако играют
существенную роль в процессе биоло-
биологической эволюции. Следует отметить
также, что мутации способны прояв-
проявлять свои отрицательные или положи-
положительные свойства не сами по себе, а
только при определенных условиях.
Так, например, усиление пигментации
может стать полезным признаком для
живущих в Африке, поскольку темная
кожа защищает организм от интенсив-
интенсивного ультрафиолетового излучения, в
северных же странах светлая кожа спо-
способствует синтезу витамина Д при
действии солнечного света.
По локализации в клетке мутации
подразделяют на ядерные и цитоплаз-
матические. Цитоплазматические му-
мутации возникают в результате измене-
изменений генома ДНК-содержащих клеточ-
клеточных органоидов — митохондрий. При-
Принято считать, что митохоиндральная
ДНК контролирует образование мито-
митохондрий, а именно — обеспечивает
внутримитохондриальный синтез не-
небольшого набора белков.
Известны мутации митохондриаль-
ной ДНК у дрожжей. Большое количе-
МУТАЦИИ
ство митохондрий содержится в ооци-
тах, в то время как в спермиях их толь-
только четыре. При оплодотворении мито-
митохондрии спермия не попадают в ооцит,
поэтому все митохондрии во всех
клетках организма имеют материнское
происхождение. Можно предполо-
предположить, что патология, связанная с мута-
мутациями в митохондриальной ДНК,
должна передаваться от индивидуумов
женского пола. Каждый ооцит содер-
содержит множество митохондрий и, если
мутация произошла только в одной из
них, то все остальные остаются нор-
нормальными, и, следовательно, феноти-
пически мутация проявляться не
должна. Правда, существует мнение,
что один из типов атрофии зрительно-
зрительного нерва обусловлен мутациями в ци-
топлазматических генах и наследует-
наследуется, как правило, только по женской ли-
линии.
Изменения в генетическом материа-
материале могут происходить на разных уров-
уровнях. Когда они затрагивают один или
несколько нуклеотидов внутри одного
гена, то возникают генные мутации.
Изменения множества нуклеотидов
или структуры хромосомы в целом на-
называют хромосомными мутациями, а
нарушение численности хромосом от-
относят к геномным мутациям.
Рассмотрим классификацию гене-
генетических изменений с точки зрения
наблюдаемых в результате таких изме-
изменений фенотипов — генетических бо-
болезней. В настоящее время все генети-
генетические болезни человека, учитывая ме-
механизмы их возникновения и характер
наследования, подразделяют на менде-
левские, хромосомные, мультифакто-
риальные, генетические болезни сома-
соматических клеток и болезни генетичес-
генетической несовместимости матери и плода.
Менделевские болезни являются ре-
результатом мутаций в отдельных генах
Генные мутации
127
Тимин
Аденин
[ Гуанин |
Цитозин )
Трансверсии
Транзиции
I Пурин |
Пиримидин )
Рис. 6.1. Механизм возникновения мутаций на нуклеотидном уровне. Возможны четыре
транзиции и восемь трансверсий
и, соответственно, их наследование
подчиняется законам Менделя. Хро-
Хромосомные наследственные болезни
обусловлены изменениями в числе
хромосом в геноме человека либо
структурными перестройками (абер-
(аберрациями) в них. Фенотипическое про-
проявление мультифакториальных болез-
болезней зависит от комплексного взаимо-
взаимодействия между множественными ге-
генетическими факторами и факторами
окружающей среды. Генетические бо-
болезни соматических клеток выделены
в отдельную группу недавно. Их при-
примером могут служить некоторые врож-
врожденные пороки развития, являющиеся
результатом мутации в соматических
клетках в критическом периоде эмбри-
эмбриогенеза. Болезни, возникающие при
несовместимости матери и плода по
антигенам, развиваются из-за иммуно-
иммунологической реакции матери на антиге-
антигены плода (например, гемолитическая
болезнь новорожденных, возникаю-
возникающая в результате несовместимости ма-
матери и плода по Rh-антигену). В неко-
некоторых популяциях такая патология
встречается довольно часто (до 1% но-
новорожденных).
Каждая из групп болезней является
сборной и, в свою очередь, подразделя-
подразделяется на несколько подгрупп. Менделев-
ские болезни включают аутосомные
доминантные, сцепленные с Х-хромо-
сомой и аутосомные рецессивные бо-
болезни. Генетические расстройства
сложной этиологии — мультифактори-
альные болезни, объединяют врожден-
врожденные пороки развития (аномалии), про-
проявляющиеся уже при рождении, и
обычные мультифакториальные бо-
болезни, наблюдаемые в основном в зре-
зрелом возрасте. Для обозначения этих
расстройств часто используют опреде-
определения — "нерегулярно наследуемые",
"полигенные", "расстройства сложной
этиологии".
Предложенной классификацией ге-
генетических изменений у человека мы
будем пользоваться при рассмотрении
как спонтанного, так и индуцирован-
индуцированного мутационного процесса.
128
Глава 6. МУТАЦИИ
СН,
ОН
' II
о
Редкая енольная
форма тимина (Т*)
Редкая иминная
форма аденина (А*)
Редкая иминная
форма цитозина (С*)
Гуанин
Редкая енольная
форма гуанина (G*)
Рис. 6.2. Таутомерные формы оснований ДНК. В центре изображены наиболее
распространенные формы, когда аденин образует водородную связь с тимином,
а гуанин - с цитозином. Сравнительно более редкие таутомеры, образующие
водородные связи по-другому, указаны стрелками. С* образует водородную связь
с A, G*- с Т, Т*- с G, А*- с С
Генные мутации
Генные мутации происходят на мо-
молекулярном уровне и затрагивают,
как правило, один или несколько нук-
леотидов внутри отдельного гена.
Этот тип мутаций можно разделить
на две большие группы. Первую из
них обсусловливает сдвиг рамки счи-
считывания. К второй группе относят
генные мутации, связанные с заменой
пар оснований. Последние составля-
составляют не более 20% спонтанных мутаций,
остальные 80% мутаций происходят в
результате различных делеций и вста-
вставок.
Существуют два типа замены осно-
оснований (рис. 6.1):
1. Транзиции заключаются в замене
одного пуринового на пуриновое осно-
основание или одного пиримидинового на
пиримидиновое основание (А -> G; С
->Т).
2. Трансверсии, при которых пури-
пуриновое основание меняется на пирими-
пиримидиновое или наоборот (А -» С; G -> Т).
Такие изменения происходят как
спонтанно, так и под действием раз-
различных мутагенов.
Спонтанные транзиции возможны
при репликации ДНК, что обусловле-
обусловлено существованием молекулы ДНК в
нескольких таутомерных формах (за
счет изменения положения протона,
меняющего химические свойства мо-
Генные мутации
129
Аденин (А)
5-бромурацил (BU)
(нормальная кетоформа)
Гуанин (G)
5-бромурацил (BU)
' Н ) (редкая енольная форма)
ви
т\
2
\
ви
У
ви
/
\
/
А
BU
А
BU
Ошибка репликации
Ошибка включения
Рис. 6.3. Механизм мутагенного действия 5-бромурацила.
А - спаривание 5-бромурацила (BU) с аденином и Б - с гуанином. Внизу - два
механизма индукции транзиции; В - ошибка репликации, состоящая в том, что BU
включается при репликации в редкой енольной форме, с G B); в третьем цикле
1 репликации C) G нормально спаривается с С, и таким образом завершается переход
АТ-> GC. Г - ошибка включения, состоящая в том, что BU в редкой енольной форме
спаривается с G A), а затем в обычной кетоформе спаривается с А B);
в третьем цикле репликации C) А нормально спаривается с Т,
и таким образом завершается переход GC -» AT
130
Глава 6. МУТАЦИИ
5 AC.AAAAGTCCA
о. I I I I I I I I I I I I I I 1 I I I I I I I I I I I I I
* TG'TTTTCAGGT
5- AC.AAAAGTCCA
11 11 11 11 11 1111 1111111111 11 I I
3' TG'TTTTCA GGT*
I Разрыв
5- AC.AAAAGTCCA
МИНИН II I I ИНИН
3' T TTCA. GGT
A. Исходная молекула ДНК. Б.
Возникновение разрыва.
B. Раскручивающий двойную
спираль белок вызывает рас-
расхождения цепей и их непра-
неправильное воссоединение.
Г. Репаративный синтез запол-
заполняет брешь, возникшую на ста-
стадии В.
Неправильное спаривание
51 AC.AAAAGTCCA
Illlllllli I I I I III
3' t ttcacaggt
g.tt
¦ I i I _^_ Репликаиионная
! ! ! . вилка
Новый синтез и восстановление
двойной спирали
Рис. 6.4. Несколько модифицированная модель мутации сдвига рамки Стрейзингера
лекул). В результате изменяется спо-
способность нуклеотидов образовывать
водородные связи, а аденин приобре-
приобретает свойства гуанина, гуанин — аде-
нина, цитозин — тимина, а тимин —
цитозина. При последующей реплика-
репликации такой таутомерный переход при-
приводит к транзиции. Так, например, в
обычной аминной форме аденин взаи-
взаимодействует с тимином, а в редкой
иминоформе образует пару с цитози-
ном. При последующей репликации
будет иметь место транзиция AT -»GC
(рис. 6.2).
Доказательством связи процесса
репликации с мутагенезом стало от-
открытие мутагенного эффекта аналогов
оснований ДНК — 5-бромурацила и 2-
аминопурина. 5-бромурацил включа-
включается в ДНК вместо тимина и образует
пары с аденином. В дальнейшем при
репликации ДНК возможна мутация
за счет ошибочного спаривания 5-бро-
5-бромурацила с гуанином (ошибка считы-
считывания) либо за счет ошибки при вклю-
включении аналога ДНК (ошибка включе-
включения). Механизм мутагенного действия
5-бромурацила представлен на рис.
6.3. Аналогичные механизмы лежат в
основе мутаций, индуцируемых 2-ами-
нопурином.
Менее ясны механизмы возникнове-
возникновения трансверсий. Одна из причин их
возникновения, по-видимому, заклю-
заключена в механизме репарации. Серпо-
Серповидно-клеточная анемия является яр-
ярким примером молекулярной болезни,
возникающей из-за наследственного
нарушения структуры гемоглобина. У
мутантного гена, кодирующего одну из
цепей гемоглобина, нарушен всего
один нуклеотид, и в матричной РНК
происходит замена аденина на урацил
(ГАА на ГУА). В результате происхо-
происходит изменение биохимического фено-
фенотипа — в р-цепи гемоглобина глутами-
новая кислота замещается на валин.
Это замена изменяет поверхность ге-
моглобиновой молекулы таким обра-
образом, что она вступает в реакцию с со-
соседними молекулами и образует длин-
длинные линейные тяжи, деформирующие
эритроциты. Приобретая серповидно-
клеточную форму, эритроциты либо
закупоривают мелкие сосуды, либо
быстро удаляются из кровообращения,
Генные мутации
131
что может приводить к гемолитичес-
гемолитической анемии.
Мутации со сдвигом рамки считы-
считывания представляют собой вставки
или выпадения одной или нескольких
пар нуклеотидов (рис. 6.4). В зависи-
зависимости от места нарушения изменяется
то или иное количество кодонов. Соот-
Соответственно в белке могут появиться
дополнительные аминокислоты или
измениться их последовательность.
Большая часть мутаций этого типа об-
обнаружена в молекулах ДНК, состоя-
состоящих из одинаковых оснований.
Значимость генных мутаций для
жизнеспособности организма неоди-
неодинакова. Различные изменения в нукле-
отидной последовательности ДНК по-
разному проявляются в фенотипе. Не-
Некоторые "молчащие мутации" не ока-
оказывают влияния на структуру и функ-
функцию белка. Примером такой мутации
может служить замена нуклеотидов,
не приводящая к замене аминокислот.
По функциональному значению
генные мутации можно разделить на 3
класса:
1) ведущие к полной потере функ-
функции;
2) в результате которых происходят
количественные изменения мРНК и
первичных белковых продуктов;
3) доминантно-негативные, изменя-
изменяющие свойства белковых молекул та-
таким образом, что они оказывают по-
повреждающее действие на жизнедея-
жизнедеятельность клеток.
Наибольшим повреждающим дейст-
действием обладают так называемые нон-
нонсенс-мутации, связанные с появлени-
появлением кодонов-терминаторов, вызываю-
вызывающих остановку синтеза белка. Причем,
чем ближе мутации к 5'-концу гена (к
началу транскрипции), тем короче бу-
будут белковые молекулы. Делеции или
инсерции (вставки), некратные трем
нуклеотидам и, следовательно, вызы-
вызывающие сдвиг рамки считывания, мо-
могут также приводить к преждевремен-
преждевременному окончанию синтеза белка или к
образованию бессмысленного белка,
который быстро деградирует.
Миссенс-мутации связаны с заме-
заменой нуклеотидов в кодирующей части
гена. Фенотипически проявляется в
виде замены аминокислоты в белке. В
зависимости от природы аминокислот
и функциональной значимости нару-
нарушенного участка, наблюдается полная
или частичная потеря функциональ-
функциональной активности белка.
Сплайсинговые мутации затрагива-
затрагивают сайты на стыке экзонов и интронов
и сопровождаются либо вырезанием
экзона и образованием делетированно-
го белка, либо вырезанием интронной
области и трансляцией бессмысленно-
бессмысленного измененного белка. Как правило, та-
такие мутации обусловливают тяжелое
течение болезни.
Регуляторные мутации связаны с
количественным нарушением в регу-
ляторных областях гена. Они не при-
приводят к изменениям структуры и
функции белков. Фенотипическое
проявление таких мутаций определя-
определяется пороговым уровнем концентра-
концентрации белка, при котором еще сохраня-
сохраняется его функция.
Динамические мутации или мутации
экспансии представляют собой патоло-
патологическое увеличение числа тринуклео-
тидных повторов, локализованных в ко-
кодирующих и регуляторных частях гена.
Многие тринуклеотидные последова-
последовательности характеризуются высоким
уровнем популяционной изменчивости.
Фенотипическое нарушение проявля-
проявляется в случае превышения определенно-
определенного критического уровня по числу повто-
повторов. Классическим примером мутации
экспансий является синдром ломкой
132
Глава 6. МУТАЦИИ
хромосомы (Fra XA), при которой вы-
выявляется область CGG повторов в регу-
ляторной области FMR-1 гена (Xq
27.3). В норме в этом гене число повто-
повторов варьирует от 6 до 42. Хромосомы,
содержащие 50-200 повторов, считают-
считаются "премутацией". В следующем поко-
поколении число повторов может увели-
увеличиться до 1000 и более, что и обусловли-
обусловливает выраженную клиническую карти-
картину заболевания. Генные мутации иден-
идентифицируют с помощью различных ме-
методов молекулярно-генетического ана-
анализа, в том числе с использованием тех-
техники гибридизации in situ (FISH), ме-
методов PCR (полимеразной цепной реак-
реакции), CMC (химического расщепления
некомплементарных сайтов), SSCP
(анализа конформационного полимор-
полиморфизма однонитевой ДНК), DGGE (де-
(денатурирующего гель-электрофореза).
Хромосомные мутации
Этот тип мутаций объединяет хромо-
хромосомные нарушения, связанные с изме-
изменением структур хромосом. В данном
разделе будут рассмотрены мутации,
наблюдаемые в соматических клетках
на стадии метафазы клеточного цикла.
Такие мутации в научной литературе
обычно именуют хромосомными абер-
аберрациями, подчеркивая при этом их от-
отличие от перестроек, происходящих в
гаметах (хромосомные мутации). Одна-
Однако следует заметить, что принципиаль-
принципиальных морфологических отличий между
перестройками в клетках разного про-
происхождения не имеется.
Классификация хромосомных
аберраций
Хромосомные аберрации можно
классифицировать, используя различ-
различные подходы. В зависимости от того, в
какой момент клеточного цикла — до
или после репликации хромосом воз-
возникли перестройки — выделяют аберра-
аберрации хромосомного и хроматидного ти-
типов. Аберрации хромосомного типа воз-
возникают на предсинтетической стадии —
G, фазе, когда хромосома представлена
однонитевой структурой. Аберрации
хроматидного типа возникают после
репликации хромосом в фазах S и G2 и
затрагивают структуру одной из хрома-
тид. В результате хромосома на стадии
метафазы содержит одну измененную и
одну нормальную хроматиды. Если же
перестройка произошла после реплика-
репликации и затронула обе хроматиды, появ-
появляется изохроматидная аберрация.
Морфологически она неотличима от
аберраций хромосомного типа, хотя по
происхождению относятся к хроматид-
ному типу. Среди аберраций хромосом-
хромосомного и хроматидного типов выделяют
простые и обменные аберрации. В их
основе лежат нарушения одной или не-
нескольких хромосом. Простые аберра-
аберрации — фрагменты (делеции) — возника-
возникают в результате простого разрыва хро-
хромосомы. В каждом случае при этом об-
образуется 2 типа фрагментов — центри-
центрические и ацентрические. Различают тер-
терминальные (концевые) и интерстици-
альные (средних участков хромосом)
делеции или фрагменты.
Обменные аберрации очень разно-
разнообразны. В их основе лежит обмен уча-
участками хромосом (или хроматид) меж-
между разными хромосомами (межхромо-
(межхромосомный обмен) или внутри одной хро-
хромосомы (внутрихромосомный обмен)
при перераспределении генетического
материала. Обменные перестройки
бывают двух типов: симметричные и
асимметричные. Асимметричные об-
обмены приводят к образованию поли-
полицентрических хромосом и ацентричес-
ацентрических фрагментов. При симметричных
же обменах происходит соединение
ацентрических фрагментов с центри-
Хромосомные мутации
133
Аберрации
хроматидного типа
Аберрации
хромосомного типа
Простые
Обменные
Простые
Внутрихромосомные
Межхромосомные
t
Внтуриплечевые
Межплечевые
Полные
Симметричные
Неполные
t
Асимметричные
Рис. 6.5. Общая классификация хромосомных аберраций
ческими, в результате чего хромосомы,
вовлеченные в обменную аберрацию,
остаются моноцентрическими.
Внутрихромосомные обмены могут
происходить как внутри одного (внут-
риплечевой обмен), так и между обо-
обоими плечами хромосомы (межплече-
(межплечевой обмен). Кроме того, обмены могут
быть простыми и сложными, когда в
процесс вовлечены несколько хромо-
хромосом. В результате могут образоваться
необычные и достаточно сложные кон-
конфигурации хромосом. Любой обмен
(симметричный и асимметричный,
межхромосомный и внутрихромосом-
ный) может быть полным (реципрок-
ным) или неполным (нереципрок-
ным). При полном обмене происходит
соединение всех поврежденных участ-
участков, а при неполном обмене часть из
них может остаться с открытым по-
поврежденным участком. На рис. 6.5
представлена классификация основ-
основных типов хромосомных аберраций.
Некоторые хромосомные аберрации
получили название стабильных, по-
поскольку они способны передаваться из
одного клеточного поколения в другое.
Это связано с тем, что при перераспре-
перераспределении генетического материала про-
произошло полное соединение центричес-
центрических и ацентрических фрагментов. Ли-
Лишенная центромер или, напротив,
включающая в себя две или более цен-
центромеры, часть хромосомных аберра-
аберраций может теряться в процессе клеточ-
клеточного деления. Такие структурные абер-
аберрации именуются нестабильными.
Важное значение имеет идентифика-
идентификация хромосомных аберраций по при-
принадлежности к хромосомному или
хроматидному типу, при изучении ин-
индуцированного мутагенеза, т.к. мута-
мутагенные факторы различной природы
(химической, физической, биологиче^
ской) могут вызывать разные тины на-
нарушений. В связи с этим более подроб-
подробно рассмотрим основные типы хромо-
хромосомных аберраций.
Аберрации хромосомного типа
При цитогенетическом анализе
можно различить, в зависимости от
применяемых методов исследования,
несколько типов аберраций этой
группы:
134
Глава 6. МУТАЦИИ
IN"
6.6.1
Itl??
6.6.2
6.6.4
И
< !< X»
6.6.5
I
6.6.6
Рис. 6.6. Аберрации хромосомного типа
6.6.1. Образование парных фрагментов;
6.6.2. Образование центрических колец;
6.6.3. Образование перицентрической ин-
инверсии; 6.6.4. Образование ацентрических
колец; 6.6.5. Образование дицентрической
хромосомы; 6.6.6. Образование реципрок-
ной транслокации
1. Парные (терминальные или ин-
терстициальные) фрагменты, которые
образуются в результате одного или
двух разрывов хромосомы в фазе Gt.
Величина их различна — от очень
длинных, захватывающих почти все
хромосомное плечо, до точечных. Пар-
Парные фрагменты без труда регистриру-
регистрируются при анализе метафаз. Эти аберра-
аберрации относятся к нестабильным. Как
правило, они не проходят через митоз,
т.к., лишенные центромер, не могут
правильно ориентироваться относи-
относительно веретена деления (рис. 6-6.1).
2. Кольцевые хромосомы (центриче-
(центрические кольца) — внутрихромосомные
обмены, при которых интерстициаль-
ный фрагмент хромосомы замкнут в
кольцо. Для образования центрическо-
центрического кольца необходимы два разрыва по
обеим сторонам центромеры, в резуль-
результате чего свободные концы центричес-
центрического фрагмента соединяются между
собой. О полноте обмена можно су-
судить по числу парных фрагментов, со-
сопровождающих кольцевую хромосому.
Один фрагмент характерен для полно-
полного обмена, т.к. концевые фрагменты со-
соединяются между собой. Достаточно
редко кольцевая хромосома сопровож-
дется двумя парными фрагментами
(неполный обмен) — рис. 6-6.2.
3. Инверсии — внутрихромосомные
обмены, при которых может происхо-
происходить переворот сегмента, содержащего
центромеру, на 180°. Такая аберрация
возникает в результате двух разрывов
либо на разных расстояниях от цент-
центромеры, либо на одинаковых (пери-
центрические инверсии). В результате
инверсии сегмента хромосомы внутри
одного плеча хромосомы образуется
парацентрическая инверсия. При ци-
тогенетическом анализе перицентри-
ческую инверсию достаточно легко об-
обнаружить, если разрывы произошли на
Хромосомные мутации
135
разных расстояниях от центромеры.
Другие инверсии (в том числе пара-
парацентрические) с помощью стандарт-
стандартных цитогенетических методов не ре-
регистрируются (рис. 6-6.3).
4. Ацентрические кольца — внутри-
хромосомные обмены, представляю-
представляющие собой замкнутые в кольцо спарен-
спаренные участки хроматид, не содержащие
центромер и сопровождаемые появле-
появлением делетированной хромосомы
(рис. 6-6.4).
5. Дицентрические и полицентриче-
полицентрические хромосомы — это асимметричные
транслокации, возникающие при меж-
межхромосомном обмене. Такие хромосом-
хромосомные аберрации имеют две (дицентри-
ки) или более (полицентрики) центро-
центромер. Наиболее часто встречаются ди-
центрики, образующиеся в результате
одиночных разрывов в двух хромосо-
хромосомах и последующего объединения двух
центромерных фрагментов. Дицентрик
обычно сопровождается одним, реже —
двумя, парнымм фрагментами, что сви-
свидетельствует о неполном обмене. Ди-
Дицентрические хромосомы легко рас-
распознаются с помощью стандартных ци-
цитогенетических методов. Наряду с цен-
центрическими кольцами, этот тип хромо-
хромосомных аберраций широко использует-
используется для количественного изучения му-
мутаций в работах по индуцированному
мутагенезу (рис. 6-6.5).
Симметричные транслокации (рис.
6-6.6) — межхромосомные обмены, ко-
которые возникают без нарушения в рас-
распределении центромер (взаимный об-
обмен ацентрическими участками между
двумя хромосомами). Различают ре-
ципрокные, когда две хромосомы об-
обмениваются участками и нереципрок-
ные транслокации, когда участок од-
одной хромосомы переносится на другую
хромосому. Для осуществления нере-
ципрокной транслокации необходимо
наличие трех разрывов (двух — в од-
одной хромосоме и одного — в другой) и
последующие соединения разорван-
разорванных концов. В особый тип выделяют
так называемые Робертсоновские
транслокации или слияния, которые
приводят к изменению числа хромо-
хромосом. Они образуются при слиянии
двух акроцентрических хромосом в об-
области центромеры, в результате чего
образуется одна метацентрическая
хромосома. Этот тип транслокаций по-
получил свое название по имени иссле-
исследовавшего механизм такого слияния
У. Р. Робертсона.
Стандартное цитогенетическое ис-
исследование позволяет увидеть только
те из симметриччных транслокаций,
которые образовались в результате об-
обмена неравными по длине участками.
При этом морфология хромосом изме-
изменяется: удлинение одной хромосомы
происходит за счет укорочения другой.
Аберрации хроматидного типа
Как и аберрации хромосомного ти-
типа, различают простые (одиночные
фрагменты или делеции) и обменные
хроматидные аберрации.
1. Одиночные фрагменты или хро-
хроматидные делеции образуются при по-
повреждении одной хроматиды. В зави-
зависимости от места разрыва они бывают
различной длины (рис. 6-7.1).
Хроматидные обмены характеризу-
характеризуются многообразием форм, что опреде-
определяется рядом хромосомных факторов:
числом и величиной участков, вовле-
вовлеченных в обмен хромосом и хроматид,
гомологичностью хромосом, участву-
участвующих в обмене, симметричностью и
полнотой обмена (рис. 6-7.2).
Довольно часто межхромосомные
обмены происходят между хроматида-
ми двух хромосом. Их морфология
очень разнообразна. Из-за характерной
им
6.7.1
м
6.7.2
д
6.7.3
. чяч
ччч
6.7.4
Рис. 6.7. Аберрации хроматидного типа
6.7.1. Одиночные фрагменты; различные типы хроматидных обменов: 6.7.2. Межхромосомные хроматидо-хроматидные обмены.
Полные (а, в, е, з) и неполные (б, г, д, ж, и, к), симметричные (а, б, е, ж) и асимметричные (в, г, д, з, и, к). Сложные межхромосомные
хроматидные обмены: в обоих случаях вовлечены четыре хромосомы в результате пяти (л) или шести (м) разрывов. 6.7.3. Хромати-
до-изохроматидные обмены. Локализация точек разрывов (а), конфигурация симметричных (б, в) и асимметричных (г, д) обменов.
6.7.4. Внутрихромосомные межплечевые хроматидные обмены
Геномные мутации
137
Ляплонд ,
Моносомик J
Трисомик
Тетрасоммк
Моноллоид<
Триплонд
Тетрплоид
Анеуплондмя
и
Моиоплокам
Полиплоидия
ы
Рис. 6.8. Типы геномных мутаций
Hi!
конфигурации такие обмены часто на- Геномные мутации
зывают "квадрирадиалы" (рис. 6.7.2). Геномные мутации изменяют число
Как правило, большинство аберраций хромосом. Такие изменения возника-
хроматидного типа достаточно легко ют обычно при нарушении распреде-
идентифицировать с помощью стан- ления хромосом по дочерним клет-
дартных цитогенетических методов. кам.
138
Глава 6. МУТАЦИИ
Различают два основных типа ге-
геномных мутаций (рис. 6.8):
1. Полиплоидия и моноплоидия.
2. Анеуплодия.
При полиплоидии число наборов
негомологичных хромосом в кариоти-
пе отличается от двух (Зп; 4п и т.д.).
Это результат нарушений в митотичес-
ком цикле, когда удвоение хромосом
происходит без последующего деления
ядра и клетки. Одной из причин по-
подобного феномена может быть эндо-
митоз, при котором происходит блоки-
блокирование ахроматического аппарата в
клетке и сохранение ядерной мембра-
мембраны в течение всего митотического цик-
цикла. Разновидностью эндомитоза явля-
является эндоредушшкация — редуплика-
редупликация хромосом, происходящая вне кле-
клеточного деления. При эндоредуплика-
ции как бы повторяются два следую-
следующих друг за другом S периода митоти-
митотического цикла. В результате этого в по-
последующем митозе будет наблюдаться
двойной (тетраплоидный) набор хро-
хромосом. Такие мутации чаще всего при-
приводят к гибели плода еще в эмбриоге-
эмбриогенезе. Триплоидия обнаруживается в
4%, а тетраплоидия приблизительно в
1% всех выкидышей. Для индивидуу-
индивидуумов с такими кариотипами характерны
многочисленные пороки развития, в
том числе асимметричное телосложе-
телосложение, слабоумие, гермафродитизм. Тет-
раплоидные эмбрионы погибают на
ранних сроках беременности, эмбрио-
эмбрионы же с триплоидными клетками из-
изредка выживают, но только если одно-
одновременно с триплоидными содержат
клетки с нормальным кариотипом.
Впервые синдром триплоидии F9,
XXY) был обнаружен у человека в 60-
хх гг. XX в. В литературе описано око-
около 60 случаев триплоидии у детей.
Максимальная продолжительность их
жизни составила 7 дней.
Анеуплоидия — некратное гаплоид-
гаплоидному уменьшение или увеличение чис-
числа хромосом Bп+1; 2п+2; 2п-1 и т.д.) —
возникает в результате ненормального
поведения гомологических хромосом в
мейозе или сестринских хроматид в
митозе.
При нерасхождении хромосом на
одной из стадий гаметогенеза в поло-
половых клетках могут оказаться лишние
хромосомы. В результате при последу-
последующем слиянии с нормальными гапло-
гаплоидными гаметами образуются зиготы
2п+1- или трисомии по какой-либо из
хромосом. Если же в гамете оказывает-
оказывается на одну хромосому меньше, то при
последующем оплодотворении образу-
образуется зигота 2п-1, или моносомик по
одной из хромосом. Нерасхождение
может затронуть не одну, а несколько
пар хромосом, что ведет к трисомии
или моносомии по нескольким хромо-
хромосомам. Часто лишние хромосомы обус-
обусловливают депрессию развития или
гибель особи, их несущей.
Впервые анеуплоидию у человека
обнаружили в 1959 г. Дж. Л еже и
Р. Турпин. Это была трисомия по хро-
хромосоме 21 у больных с синдромом Да-
Дауна — одним из наиболее распростра-
распространенных тяжелых наследственных за-
заболеваний (см. главу 8).
Нерасхождение хромосом в мейозе
может затрагивать любую из 23 пар
хромосом. У человека описаны полные
трисомии по хромосомам №№ 8, 9, 13,
14, 18, 21, X, Y. За редким исключени-
исключением (например, трисомия по 21 хромо-
хромосоме), аутосомные трисомии приводят
к гибели организма на ранних стадиях
эмбриогенеза или в первые дни после
рождения. Особенно тяжелыми случа-
случаями гетероплоидии считаются моносо-
моносомии, при которых происходит утрата
целой группы сцепления генов. При-
Примерно 20% случаев моносомии приво-
Геномные мутации
Таблица 6.1. Частота геномных мутаций
139
Хромосомы
Синдром
Частота среди
новорожденных
Аутосомы
Трисомия 21
Трисомия 13
Трисомия 18
Половые хромосомы (женщины)
ХО, моносомия
XXX трисомия
ХХХХ, тетрасомия
ХХХХХ, пентасомия
Половые хромосомы (мужчины)
XYY трисомия
XXY трисомия
XXYY, тетрасомия
XXXY, тетрасомия
XXXXY, гшнтасомия
XXXXXY, гексасомия
Дауна
Патау
Эдвардса
Тернера
Пониженная плодовитость
Норма
Клайнфсльтера
1/700
1/5000
1/10000
1/5000
1/700
1/1000
1/500
дят к летальному исходу еще в первые
дни эмбрионального развития или за-
заканчиваются гибелью зародыша на бо-
более поздних стадиях. Лишь моносомия
по Х-хромосоме совместима с жизнью.
Например, это больные с синдромом
Шерешевского-Тернера, при котором
2п = 45 D4ХО). Поскольку половые
хромосомы определяют пол, оказывая
влияние на дифференцировку гонад,
индивидуумы ХО — женщины с харак-
характерными нарушениями в развитии
первичных и вторичных половых при-
признаков (см. главу 8).
При иолисомиях по половым хро-
хромосомам встречаются различные ком-
комбинации X и Y-хромосом, а число их с
сохранением при этом жизнеспособно-
жизнеспособности организма может доходить до пя-
пяти. Все подобные изменения кариоти-
па объединяют под общим названием
синдром Клайнфельтера. Больные
мужчины с недоразвитыми мужскими
гонадами, слаборазвитым волосяным
покровом на теле и увеличенными мо-
молочными железами, обычно стериль-
стерильные. Некоторые из них страдают умст-
умственной отсталостью, причем с увели-
увеличением числа Х-хромосом ее вероят-
вероятность растет.
Неправильное расхождение хромо-
хромосом в митозе приводит к возникнове-
возникновению мозаиков — особей, у которых раз-
разные клетки несут различный генотип.
Фенотипически проявление мозаициз-
ма зависит от доли клеток различных
типов, т.е. от того, на какой стадии про-
произошло нарушение в митозе, и от жиз-
жизнеспособности возникающих линий
клеток (рис. 6.9). При недостатке числа
хромосом клетки чаще всего нежизне-
нежизнеспособны. У людей с мозаицизмом
влияние нормальных клеток может
преобладать настолько, что несущие
избыточные хромосомы клетки могут
вообще не находить выражения в фе-
фенотипе или проявляются лишь в виде
отдельных симптомов. Так, около 2%
больных с синдромом Дауна обнару-
обнаруживают мозаицизм трисомных и нор-
нормальных клеток D7.ХХ+21/46ХХ).
При достаточно большой доле нор-
нормальных клеток психическое развитие
больных нередко выше, чем при чис-
чистой трисомии по 21 хромосоме. Чаще
всего встречается мозаицизм по поло-
140
Глава 6. МУТАЦИИ
Яйцеклетка
(xi
Спермий
/ i^ wV
Нерасхождение
хромосом в митозе
(хп
Ч '
{х7)
ПОП
осп
w j
(xxy)
Линия нормальных
клеток
Линия анеуплоидных
клеток
Погибают
Рис. 6. 9. Мозаицизм ХУ/ХХУкак следствие нерасхождения хромосом в митозе
вым хромосомам. Многие индивидуу-
индивидуумы, несущие признаки обоих полов,
являются мозаиками, в клетках кото-
которых половые хромосомы содержатся в
разных комбинаациях, вплоть до кари-
отипа 46.XX/46.XY. Причины такого
мозаицизма различны: оплодотворе-
оплодотворение ооцита двумя различными снер-
миями, слияние до 4-х оплодотворен-
оплодотворенных яйцеклеток или последующие
ошибки в митозе при первом делении
зиготы. У многих женщин с признака-
признаками синдрома Шерешевского-Тернера
кариотин некоторых клеток — 45,Х, а
остальных — 47,ХХХ. Подобный тип
мозаицизма, по-видимому, связан с не-
нерасхождением Х-хромосомы нормаль-
нормальной зиготы в первом после оплодотво-
оплодотворения митотическом делении. Анало-
Аналогичен механизм возникновения из зи-
зиготы мозаицизма XO/XYY.
Известно также довольно много
случаев мозаицизма, при котором в ор-
организме содержится три клеточных
клона - 46,ХХ/45,Х/47,ХХХ, образо-
образованные в результате нерасхождения
хромосом в поздней стадии клеточного
деления.
Приведенные примеры хромосом-
хромосомных болезней человека позволяют в
некоторой степени оценить тяжесть
генетического груза человечества и
сложность организации генома чело-
человека. В целом, по данным Научного
Комитета ООН по действию атомной
радиации за 1988 г., частота естествен-
естественно встречающихся хромосомных бо-
болезней, связанных с аберрациями хро-
хромосом, составляет 400 случаев на 1
млн. новорожденных. Геномные мута-
мутации (изменение числа хромосом)
встречаются с частотой 3400 на 1 млн.
Связь между генотипом и фенотипом
141
10000
8000
6000
4000
2000
8941
6678
2811
3907 4^
111
196619681971 1975 1978 1983 1986 1988 1994 1998
Рис. 6.10 Число менделевских наследственных болезней,
идентифицированных с 1966 по 1998 г.
новорожденных. Вместе такие наслед-
наследственные болезни встречаются у 0,38%
новорожденных.
Связь между генотипом
и фенотипом
Мутационные изменения в любом из
генов человека, равно как и изменения
структуры любой из хромосом, приво-
приводят к тем или иным изменениям в фе-
фенотипе человека. Степень этих измене-
изменений зависит от важности для организма
вовлеченных в мутагенез генов, от мас-
масштабов нарушения генетического мате-
материала и от характера наследования воз-
возникших мутационных изменений.
Большинство экспертов в области
генетики человека считают, что число
генов в геноме человека составляет от
50 000 до 100 000. Размер генов варьи-
варьирует в широких пределах — от 2 до
2000 килобаз (тысяч оснований моле-
молекулы ДНК — kb). Например, согласно
сведениям 1994 г., распределение по
размерам среди 253 генов следующее:
23% генов - менее 10 kb, 36% - между
10 и 25 kb, 20% - между 25 и 50 kb,
13% - между 50 и 100 kb, 7% - между
100 и 500 kb и 1% - более 500 kb.
Успехи в реализации международ-
международной программы "Геном человека" и ин-
интенсивное изучение наследственных
болезней в клиниках многих стран к
1998 г. увеличило их число заболева-
заболеваний до 8941. Поистине стремителен
прогресс в этой области: в 1966 г. было
известно 1487 наследственных болез-
болезней, в 1982 г. - около 4000, а в 1994 -
6678 (рис. 6.10). '
Для многих из этих недугов изучена
локализация мутантных генов и про-
проведен молекулярный анализ продук-
продуктов их деятельности. В специфических
сайтах различных хромосом человека
локализованы более 3900 генов с хоро-
хорошо известными функциями. С другой
стороны, в определенных участках
хромосом картировано более 1100
клинических болезней.
Для многих менделевских наследст-
наследственных болезней характерно большое
число различных аллельных вариан-
вариантов. Например, для гена р-глобина их
известно более 400, для трансмембран-
трансмембранного регулятора кистозного фиброза
(CFTR) — более 500, для гена а-глоби-
на (НВА) - более 100.
Наиболее простую взаимосвязь
между мутациями и обусловленными
ими болезнями представляет концеп-
концепция "одна мутация — одна болезнь".
Постулируя унитарную генетическую
142 Глава 6. МУТАЦИИ
Таблица 6.2.
Молекулярная природа мутаций, приводящих к менделевским болезням
Категории
Аутосомные
доминантные
Сцепленные с
Х-хромосомой
Итого
Аутосомные
рецессивные
Всего
Число менделевских болезней связанных с
Точковыми
мутациями а)
73
16
89 E4%)
Ш
200 F5%)
Точковыми и
протяженными
мутациями б)
18
24
42 B6%)
27
69 B2%)
Протяженными
мутациями и
микроделецион-
ными сишгоомами
25
8
33 B0%)
7
40A3%)
Всего
116
48
164
145
309
а — замены оснований
б — преимущественно делении ДНК
причину для каждой болезни, она на-
нашла подтверждение при описании
многих менделевских болезней. Вмес-
Вместе с тем такая простая взаимосвязь на-
наблюдается далеко не всегда. В настоя-
настоящее время известны примеры других
взаимосвязей между генотипом и фе-
фенотипом:
— мутации в одних и тех же генах
могут быть причиной различных кли-
клинических фенотипов (аллельная гете-
гетерогенность);
— мутации в различных генах про-
проявляются в сходных или очень близ-
близких клинических фенотипах (неал-
лельная гетерогенность).
При анализе аллельных вариантов
767 генов человека было обнаружено,
что 658 генов ассоциированы с какой-
либо одной клинической болезнью, 71
ген — с двумя, 30 генов — с тремя, 5 ге-
генов — с четырьмя, 1 ген — с пятью, 1
ген — с шестью и 1 — с семью болезня-
болезнями. В частности, мутации в генах, кон-
контролирующих рецептор ростового
фактора фибробластов (FGFR) приво-
приводят к различным формам скелетных
нарушений. Точковые мутации в од-
одном из этих генов, а именно в гене
FGFR3 в 4 хромосоме в локусе 4р16.3
были выявлены при изучении случаев
ахондроплазии (АСН), гипохондро-
плазии (НСН) и танатофорной дис-
плазии (TD).
С другой стороны, показано что 26
болезней обусловлены мутациями в
двух различных генах, 11 болезней — в
трех, 4 болезни -в четырех, 4 болезни —
в пяти, 1 болезнь — в шести и 1 бо-
болезнь — в семи и 1 - мутациями в де-
десяти генах. В этих случаях сходные
фенотипы возникают в результате
влияния мутаций многих генов на об-
общие биохимические процессы либо на
одни и те же клеточные структуры.
Например, семь синдромов, связан-
связанных со скелетными нарушениями, а
именно, синдром Аперта (AS), синд-
синдром Кроузона (CS), синдром Джексо-
Джексона-Вейса (JWS), синдром Пфейфера
(PS), ахондроплазия (АСН), танато-
форная дисплазия (TD) и гипохонд-
роплазия (НСН) могут быть результа-
результатом мутаций в трех генах, контроли-
контролирующих рецептор ростового фактора
фибробластов (FGFR).
Согласно результатам молекулярно-
генетических исследований, менделев-
ские болезни могут быть следствием
точковых мутаций, протяженных му-
Мультифакториальные болезни
143
таций и микроделеций. Различия меж-
между этими типами мутационных изме-
изменений связаны с размерами охвачен-
охваченных мутацией фрагментов молекулы
ДНК. В таблице 6.2 представлены со-
соотношения этих типов изменений, по-
полученные при экспериментальном
анализе 309 аутосомно доминатных,
сцепленных с Х-хромосомой и ауто-
аутосомно рецессивных менделевских на-
наследственных болезней.
Видно, что большая часть спонтан-
спонтанных менделевских болезней связана с
точковыми мутациями, т. е. с заменами
отдельных оснований.
В целом частота заболеваний мен-
делевского типа представлена в таб-
таблице 6.3.
Необходимо отметить, что такие ге-
генетические явления как мозаицизм и
экспансия аллелей могут внести допол-
дополнительные сложности в понимание про-
процесса фенотипической реализации му-
мутационных изменений в генах. Напри-
Например, при мозаицизме нормальные и му-
тантные клетки присутствуют у одного
и того же индивидуума. Это может от-
относиться как к соматическим, так и к за-
зародышевым клеткам. В случае, если мо-
мозаицизм в зародышевых клетках затра-
затрагивает доминантную болезнь, она может
не проявиться у данного индивидуума
фенотинически вследствие мозаицизма,
однако в этом случае сохраняется риск
проявления этой болезни у потомков,
унаследовавших мутантный ген.
Мультифакториальные болезни
Мультифакториальные болезни
объединяют широкий класс заболева-
заболеваний, в отношении которых известно,
что, безусловно имея генетический
компонент, они в то же время не под-
подчиняются простому менделевскому
наследованию. К ним относятся неко-
некоторые врожденные аномалии (напри-
Таблица 6.3.
Частота заболеваний
менделевского типа
Типы
менделевских
болезней
Аутосомные
Доминантные
Аутосомные
Рецессивные
Сцепленные
с Х-хромосомой
Всего:
По оценкам 1977
1982, 1986 г.
%
0,95
0,25
0,05
1,25
Оценки
1998 г.
%
1,5
0,75
0,15
2,40
мер, анэнцефалия, мышечно-скелет-
ные аномалии, расщелины неба и верх-
верхней губы), многие болезни взрослых
(диабет, повышенное давление) и
большая часть раков. Возникновение
этих болезней интерпретируются как
результат взаимодействия большого
числа факторов генетической природы
и окружающей среды. Роль последних
наиболее четко прослеживается при
анализе коронарных болезней сердца,
астмы. В связи с широким распростра-
распространением в популяциях людей мульти-
мультифакториальные болезни обусловлива-
обусловливают высокую заболеваемость и смерт-
смертность.
Большинство этих болезней не под-
подчиняются классическим законам на-
наследования, то есть не являются кли-
клиническими проявлениями дефектов
одиночных генов. В то же время среди
родственников больных они встреча-
встречаются с более высокой частотой, чем в
общей популяции. Однако риск забо-
заболеть варьирует среди родственников в
зависимости от особенностей данной
болезни и может быть различным в
разных семьях. Для большинства
мультифакториальных болезней пока
крайне ограничены знания о вовлечен-
вовлеченных в генетический контроль этих бо-
болезней генов, их числа, типа мутацион-
мутационных изменений и характера воздейст-
144
Глава 6. МУТАЦИИ
вующих на их проявление факторов
окружающей среды.
С учетом особенностей фенотипиче-
ского проявления и генотииического
контроля группа мультифакториаль-
ных болезней может быть подразделе-
подразделена на врожденные аномалии и обыч-
обычные мультифакториальные болезни.
Врожденные аномалии представля-
представляют собой структурные дефекты той
или иной степени тяжести, выявляе-
выявляемые при рождении ребенка. Термин
"врожденные" означает лишь присут-
присутствие дефектов при рождении и не
имеет дополнительного этиологичес-
этиологического значения. Врожденные болезни
могут быть обусловлены наследствен-
наследственными и ненаследственными фактора-
факторами. К последним относятся все врож-
врожденные пороки, возникающие за счет
тератогенного действия внешних фак-
факторов, а также врожденные инфекции.
Врожденные аномалии являются ко-
конечным результатом дисморфогенеза
и могут иметь изолированное или мно-
множественное проявление. В отношении
изолированных врожденных анома-
аномалий может быть прослежен путь от ка-
какой-либо одной локальной ошибки
(мутации) в морфогенезе до наблюдае-
наблюдаемого эффекта. В основе множествен-
множественных врожденных аномалий лежат две
или больше различные морфогенети-
ческие ошибки, возникающие во время
внутриутробного развития ребенка.
Наиболее детально спонтанная из-
изменчивость человека изучалась при
проведении трех обширных исследова-
исследований, предпринятых в 70-80е гг. в Кана-
Канаде, в США и в Венгрии. О масштабах
исследований свидетельствует тот
факт, что только в Венгрии было обсле-
обследовано около 2 млн. рождений. Полу-
Полученные результаты показывают, что ча-
частота появления врожденных аномалий
в этих странах сходна и составляет око-
около 6-7%. Из них 2-3% относятся к круп-
крупным врожденным аномалиям, приводя-
приводящим к весьма серьезным дефектам или
к летальному исходу. Частоты врожден-
врожденных аномалий выше у родственников,
носителей этой болезни, чем в целом в
популяции. При этом риски появления
врожденных аномалий варьируют в за-
зависимости от изучаемой болезни и бо-
более высоки у родственников первой
степени родства по сравнению с родст-
родственниками второй и третьей степени.
Обычные мультифакториальные
болезни (такие, как диабет, коронар-
коронарные болезни сердца, эпилепсия, аст-
астма, псориаз и др.) так же, как и рас-
рассматриваемые выше врожденные ано-
аномалии, не подчиняются менделевским
законам наследования. Генетическим
базисом обычных мультифакториаль-
ных болезней являются генетически
восприимчивые индивидуумы, у ко-
которых вероятность реализации той
или иной болезни зависит от присут-
присутствия или отсутствия других факто-
факторов риска генетической или средовой
природы (таких, как действие других
генов, диета, физическая активность,
воздействие различных факторов сре-
среды и др.)
Как показали исследования, прове-
проведенные в Венгрии, общая естественная
(спонтанная) встречаемость обычных
мультифакториальных болезней в по-
популяции (всего около 30 различных
болезней) составляет 60% (включая
все возрастные группы). Эти результа-
результаты хорошо совпали с материалами ис-
исследований в других странах. При
этом отдельные индивидуумы могут
иметь (и реально имеют) более чем од-
одну мультифакториальную болезнь.
Большинство мультифакториальных
болезней проявляются в среднем воз-
возрасте (за исключением некоторых, та-
таких, как инсулинзависимый диабет
Мулыифактории
mellitus, эпилепсия). Для большинства
болезней их возрастное распределение
подчиняется нормальному закону, то
есть в молодом возрасте наблюдается
низкая частота, наибольшая частота
приходится на группу среднего возрас-
возраста и к старости происходит ее сниже-
снижение. Исключение составляет астма,
проявление которой имеет два пика —
первый ранний и второй в среднем
возрасте или позже. В большинстве
случаев различий в заболеваемости те-
теми или иными мультифакториальны-
ми болезнями, например диабет melli-
mellitus, эпилепсия, повышенное давление,
между мужским и женским полом не
наблюдается, хотя некоторые заболе-
заболевания чаще встречаются у мужчин, а
другие — у женщин.
Хотя существует подгруппа мульти-
факториальных болезней, связанных с
мутациями отдельных генов (напри-
(например, мутации в LDLR рецепторном ге-
гене, являющиеся причиной аутосомной
доминантной формы семейной гипер-
холесторомии), для большинства та-
таких болезней характерна генетическая
гетерогенность, то есть к одному и то-
тому же конечному клиническому эф-
эффекту ведут различные генетические
механизмы. Некоторые мультифакто-
риальные болезни возникают в резуль-
результате появления мутаций во множест-
множественных генах (олиго- или полигенах).
Более того, у части генетически пред-
предрасположенных индивидуумов муль-
тифакториальная болезнь может раз-
развиться за счет воздействия факторов
окружающей среды, когда возникают
так называемые фенокопии.
Таким образом, фенотипическое
проявление обычных мультифактори-
альных болезней зависит от сложного
взаимодействия генетических факто-
факторов и факторов среды, что наглядно
представлено на схеме (рис. 6.11).
5 болезни
145
Представленная схема основана на
материалах исследований генетичес-
генетического контроля коронарной сердечной
болезни (CHD). Можно выделить че-
четыре уровня, связывающих генетичес-
генетические изменения с фенотипическим про-
проявлением мультифакториальной бо-
болезни. На рисунке 6.11 представлены
мутации в сцепленных генах (уровень
1), которые через продукты этих генов
(уровень 2) и факторы среды, которые
на уровне 3 вносят вклад в количест-
количественную вариабельность фактора риска
(например, холестерола сыворотки в
случае коронарной болезни сердца) и
таким образом приводят к проявле-
проявлению болезни (уровень 4). На уровне 1
выделяют два класса генов: "полиге-
"полигены", мутантные аллели которых по от-
отдельности только в слабой степени
способны влиять на признаки фактора
риска (обозначены А, В, Z) и "главные
гены", чьи мутантные аллели обладают
сильным эффектом (обозначены как
LDLR мутации).
Если формирование фактора риска
в популяции в целом определяется ал-
лельными вариантами полигенов и
факторами среды, то можно ожидать,
что "созвездие" полигенных аллелей
будет варьировать от одного индиви-
индивидуума к другому. Индивидуальный
риск заболеть данной болезнью опре-
определяется в этом случае особой комби-
комбинацией аллелей в локусах, продукты
которых вовлечены в контроль физио-
физиологических процессов, определяющих
характеристики фактора риска. Варьи-
Варьирование в риске между индивидуума-
индивидуумами того же самого генотипа для этих
полигенов происходит за счет воздей-
воздействия факторов среды.
В противоположность мутациям в
полигенах, мутации в главных генах
связаны с дискретными фенотипичес-
кими категориями, каждая из кото-
146
Глава 6. МУТАЦИИ
Уровень
Здоровье
Болезнь
Другие
факторы
риска
IV Конечное проявление
Фактор риска
I Гены
II Продукты ге>юв
Мутация главного
гена подобно
гену LDLR
Рис. 6.11. Общая схема, иллюстрирующая роль полигенов, больших генов
и факторов среды в этиологии мультифакториальных болезней
рых характеризуется дискретным ри-
риском развития болезни и меньшим
влиянием средовых факторов (напри-
(например, мутации LDLR, представленные
на рис. 6.11). Такие мутации отдель-
отдельных генов, приводящие к мультифак-
ториальной болезни, редки. Хотя они
и приводят к сильным разрушитель-
разрушительным эффектам у индивидуумов, кото-
которые их несут, на популяционном
уровне такие мутации вносят неболь-
небольшой вклад в вариабельность характе-
характеристик фактора риска и, таким обра-
образом, ответственны лишь за неболь-
небольшую долю данной болезни в популя-
популяции.
Общая частота спонтанных мутаций у человека
147
Становится очевидным, что, в отли-
отличие от менделевских болезней, пред-
предсказание на основе генетической ин-
информации будет ли подвержен тот или
иной индивидуум мультифакториаль-
ной болезни, представляет собой
сложную проблему, поскольку в этом
случае наличие генетического дефекта
еще не эквивалентно наличию болез-
болезни. Более того, в случае мультифакто-
риальных болезней не ожидается, что
мутационные дефекты каких-либо от-
отдельных генов послужат причиной для
всех или большинства случаев болез-
болезни. Исходя из этого можно заключить,
что генетическая расшифровка муль-
тифакториальных болезней является
сложной задачей и, судя по всему, бу-
будет представлять главную проблему
медицинской генетики в ближайшем
будущем.
Общая частота спонтанных
мутаций у человека
Оценки частот спонтанных мутаций
основаны, как мы уже указывали, на
обширных исследованиях, проведен-
проведенных в провинции Британская Колум-
Колумбия в Канаде и в Венгрии. Жители
Британской Колумбии являются по-
потомками выходцев из Западной Евро-
Европы, то есть в силу законов популяцион-
ной генетики они в преобладающей
степени имеют генотипы, близкие к ге-
генотипам современных жителей Запад-
Западной Европы. Таким образом, представ-
представленные выше оценки частот менделев-
менделевских, хромосомных и мультифактори-
альных болезней основаны преимуще-
преимущественно на генотипах жителей Запад-
Западной Европы, которое, конечно, далеко
не исчерпывают генетическое разнооб-
разнообразие популяций человека на Земле.
Что это действительно так, свидетель-
свидетельствуют результаты обследования ряда
других популяций и этнических групп:
амиш в Пенсильвании, финнов, фран-
французских канадцев, евреев ашкенази, бу-
буров (потомков датчан) в Южной Афри-
Африке, палестинских арабов, проживаю-
проживающих в Израиле, и ряда других средне-
средневосточных популяций. Показано, что
несколько менделевских болезней, ко-
которые редко встречаются в Западноев-
Западноевропейских популяциях, распростране-
распространены в указанных популяциях и наобо-
наоборот. Например, у женщин-евреек ашке-
ашкенази выявлена высокая частота мута-
мутаций BRCA1 и BRCA2, не наблюдаемая
в других популяциях. Эти мутации
приводят к относительному увеличе-
увеличению частоты рака груди и яичника.
Различия в частотах спонтанных
мутаций отдельных локусов в различ-
различных популяциях человека (особенно в
изолированных) часто связаны с эф-
эффектом основателя и с генетическим
дрейфом. Эффект основателя состоит
в том, что маленькая выборка из боль-
большой популяции, несущая только не-
небольшую часть генетического разнооб-
разнообразия этой популяции, в условиях изо-
изоляции "определяет" генетическую
судьбу своих потомков. Генетическим
дрейфом, который подробно мы будем
обсуждать в главе 7, называют явление
кумулятивных изменений в генных ча-
частотах в результате изменений, связан-
связанных с выборками из небольших по
численности популяций.
Необходимо указать еще на два до-
дополнительных фактора современной
цивилизации, которые потенциально
могут изменить спектр и частоту спон-
спонтанных генетических изменений (и,
соответственно, генетических болез-
болезней) в различных популяциях челове-
человека. Во-первых, такие изменения могут
происходить вследствие увеличения
мобильности смешения популяций.
Во-вторых, частоту заболеваний мо-
может изменить доступность (во всяком
148 Глава 6.
случае, в наиболее развитых странах)
. генетического скрининга, генетическо-
генетического консультирования и пренатальной
диагностики. Как следует из главы 7,
посвященной популяционной генети-
генетике человека, в последнем случае речь
идет, конечно, о воздействии на часто-
частоту заболеваний, а не на частоту кон-
контролирующих данную наследствен-
наследственную болезнь аллелей генов.
Теперь рассмотрим естественную ге-
генетическую изменчивость человека в
целом, включая менделевские, хромо-
хромосомные и мультифакториальные бо-
болезни. Необходимо еще раз подчерк-
подчеркнуть, что в этой области за последние
годы наблюдается стремительный про-
прогресс. Сегодня известно около девяти
тысяч наследственных болезней, и
этот список пополняется ежегодно.
Длительное время основой для оценки
естественной изменчивости человека
служили результаты эпидемиологиче-
эпидемиологического обследования большой популя-
популяции людей в Британской Колумбии
(Канада). На основе этих исследова-
исследований охватывающих возрастную груп-
группу от рождения до 21 года было уста-
установлено, что в этой возрастной группе
наблюдается 10,6 % различных генети-
генетических аномалий.
Недавно законченное обширное об-
обследование в Венгрии расширило
представления о естественной измен-
изменчивости человека. Показано, что в воз-
возрасте до 70 лет, т.е. в течение всей жиз-
жизни человека, выявляется почти на по-
порядок больше наследственных анома-
аномалий, причем значительная часть на-
наследственных болезней проявляется в
среднем и пожилом возрасте. Преобла-
Преобладающую часть выявленной наследст-
наследственной отягощенности человека со-
составляют, как мы уже отмечали, муль-
мультифакториальные болезни (подагра,
язва, диабет, астма и др.) — их абсо-
МУТАЦИИ
лютное число равно 600 тыс. на 1 млн.
людей F0 %). Таким образом, впервые
удалось оценить суммарную (в тече-
течение всей жизни) генетическую отяго-
щенность популяций человека.
Естественная генетическая измен-
изменчивость человека включает в себя так-
также генетическую отягощенность, вы-
выявляемую уже в зародышевых клетках.
В последние годы были разработаны
методы прямого хромосомного анали-
анализа сперматозоидов человека путем оп-
оплодотворения яйцеклеток хомячка.
Показано, что частота аномалий хро-
хромосом в сперматозоидах фертильных
мужчин составляет около 7%. Такие
хромосомные изменения приводят к
раннему прекращению беременности
из-за связанных с аберрациями хромо-
хромосом крупных аномалий развития заро-
зародыша.
Исследования, проведенные в Евро-
Европе, Америке и в Японии в 70-80е гг.,
позволили рассчитать пропорцию ци-
тогенетических нарушений при воз-
возникновении аномалий беременности.
Показано, что спонтанные выкидыши
на сроках от 5 до 28 недель, связанные
с хромосомными аномалиями, состав-
составляют приблизительно 40% от общего
их числа. При этом в ранний период
(от 5 до 7 недель) эта частота ниже и
составляет 17,5%, далее (с 8 до 15 не-
недель) она повышается приблизительно
до 50%, снижается приблизительно до
30% в период от 16 до 19 недель и пада-
падает до 10% в период от 20 до 29 недель.
Общее представление об относи-
относительном вкладе аномалий хромосом
можно получить при анализе соотно-
соотношения частот их встречаемости, заре-
зарегистрированных при исследовании но-
новорожденных, перинатальной гибели и
спонтанных абортов (рис. 6.12). В це-
целом уровень естественной изменчиво-
изменчивости человека весьма высок — он со-
Общая частота спонтанных мутаций у человека
149
1000000 зарегистрированных зачатий
850000 рождений
8333000 детей
17000
850 с
перинатальной гибели
640
трисомиков
5248
¦ 1960
с хромосомными
аномалиями
анеуплоидов
по половым
хромосомам
анормальными
хромосомами
—л—
170
структурных
аберраций
47XYY:
47XXY:
мозаики и др.:
45 X:
47ХХХ:
150000
спонтанных
абортов
полиплоидов 75000 с
хромосомными
1 аномалиями
459
25
298
+13):
трТсомГков
трисомиков
мозаики и др.:
мозаики и др.: J24
37
18): 100
1005
37
769
732
149
1650 сбалансированных .робертсоновские
аутосомных / транслокации
структурных ^—- реципрокные
перестроек \ транслокации
инверсии
459 сбалансированных
структурных [225 других
аберраций
Рис. 6.12. Сводка частоты хромосомных аномалий в случаях спонтанных абортов,
перинатальной гибели и у новорожденных детей
39975
трисомиков
14100 45Х
12300
триплоидов
4200
тетраплоидов
3075
структурных
аберраций
1725мозаиков
ставляет, около 70% различных гене-
генетических отклонений от нормы в тече-
течение 70 лет жизни человека, принятых
условно за среднюю продолжитель-
продолжительность жизни. Эта величина складыва-
складывается из 2,4% менделевских болезней,
0,4% хромосомных болезней, 6% врож-
врожденных аномалий и 60% мультифакто-
риальных болезней. Таким образом, у
70% людей в течение их жизни прояв-
проявляется хотя бы одно серьезное генети-
генетически обусловленное отклонение от
нормы, снижающее продолжитель-
продолжительность жизни человека по сравнению с
нормой, либо мешающее его нормаль-
нормальной работоспособности.
Данные, полученные в Британской
Колумбии, США и Венгрии, легли в
основу обобщенной оценки уровня ес-
естественной изменчивости человека На-
Научным комитетом ООН по действию
атомной радиации, в 1988 г. (см. Табл.
6.4). При составлении данной таблицы
эксперты НКДАР неоднократно под-
подчеркивали, что в будущем оценка уров-
уровня естественной изменчивости может
возрасти за счет уточнений частот от-
отдельных типов наследуемых болезней
150
Глава 6. МУТАЦИИ
Таблица 6.4. Оценка методом удваивающей дозы риска
появления генетических болезней на миллион новорожденных
в популяции, подвергающейся воздействию генетически значимой
дозы, эквивалентной 1 Зиверту на поколение
(при воздействии низких доз, при малой мощности дозы излучения)*
Классификация
болезней
Естественная
частота на
миллион ново-
новорожденных
Эффект воздействия
Поколения
I I II
1 Зв на поколение
Равновесный
уровень
Аутосомные доминантные
и сцепленные с
Х-хромосомой
Аутосомные рецессивные
Хромосомные
— вследствие структурных
аномалий
— вследствие изменения
числа хромосом
Врожденные аномалии
Другие многофакторные
Рано проявляющиеся
доминантные мутации
Наследуемые раки
Общий оцениваемый
риск
10000
2500
400
3400
60000
600000
Неизвестно
Неизвестно
1500 1300 10000
5 5 1500
240 96 400
Возможно очень низкий
Не оценивается
Не оценивается
Не оценивается
Не оценивается
1700 1400 12000
* При расчетах принято, что удваивающая доза равна 1 Зв.
и за счет включения некоторых иных
типов генетической изменчивости че-
человека, по которым пока не произво-
производится количественная оценка, но кото-
которые, безусловно, имеют высокую мута-
мутационную компоненту (например, на-
наследуемые злокачественные опухоли).
В реальности этого прогноза можно
убедиться на примере менделевских
наследственных болезней, оценка час-
частоты которых возросла за последние
10 лет от величины 1,25%, указанной в
данной таблице до величины 2,4%.
Частоты спонтанных мутаций в по-
популяциях человека, представленные в
таблице 4, послужили основой для
расчета генетического риска, связан-
связанного с воздействием ионизирующих
излучений на человека. Эти расчеты, о
которых речь будет ниже, также пред-
представлены в этой таблице. Так как оцен-
оценки генетического риска облучения че-
человека, производимые НКДАР ООН,
не претерпели изменений за последние
10 лет, в дальнейших разделах, посвя-
посвященных рассмотрению индуцирован-
индуцированного мутагенеза у человека, мы будем
придерживаться этих официальных
оценок.
Индуцированный мутагенез
Ныне стало очевидным, что бурное
развитие науки и техники приводит к
интенсивному накоплению в окружа-
окружающей среде разнообразных мутагенов,
способных наносить вред здоровью не
только современного, но и будущих
поколений людей.
Индуцированный мутагенез
151
Воздействие на живой организм
различных мутагенных факторов
внешней среды, в первую очередь, хи-
химических и физических, приводит к
накоплению патологических мута-
мутаций, которые нередко оказывают не-
неблагоприятное влияние на жизнедея-
жизнедеятельность как отдельных клеток, так и
организма в целом. При этом мута-
мутации, возникающие в половых клетках,
могут приводить к спонтанным абор-
абортам, врожденным порокам развития,
мертворождениям, к увеличению час-
частоты наследственных заболеваний.
Мутации, возникающие в соматичес-
соматических клетках, способны инициировать
развитие злокачественных новообра-
новообразований, сокращать продолжитель-
продолжительность жизни, вызывать преждевре-
преждевременное старение, а также неблагопри-
неблагоприятно воздействовать на целый ряд
жизненно важных функций организ-
организма. В связи с этим проблемам, связан-
связанным с индуцированным мутагенезом,
уделяют пристальное внимание. Ос-
Основные закономерности индуциро-
индуцированного мутационного процесса у че-
человека на генном, хромосомном и ге-
геномном уровнях изложены в много-
многочисленных работах, выполненных как
в нашей стране, так и за рубежом.
Большая часть исследований прово-
проводится на соматических клетках, — на-
наиболее доступным и удобным объекте
исследования. При изучении индуци-
индуцированного мутагенеза основное вни-
внимание уделяют следующим вопросам:
спектру мутаций; количественной за-
зависимости частоты мутаций от дозы
мутагена и его качества (т.е. вида); ре-
репарации повреждений ДНК; особен-
особенностям действия малых доз мутагена;
влиянию антимутагенов; индивиду-
индивидуальной чувствительности организма.
Основная масса исследований по
индуцированному мутагенезу посвя-
посвящена радиационному и химическому
мутагенезу.
Мутации, индуцированные
радиацией
Именно при исследовании радиаци-
радиационного мутагенеза была впервые пока-
показана возможность индуцировать мута-
мутации при действии факторов внешней
среды. Основы радиационной генети-
генетики были заложены работами Г.А.Над-
сона и Г.Т.Филиппова в 1925г. в опы-
опытах на плесневых и дрожжевых грибах.
Позже, в 1927г. Г.Д.Меллер, используя
методы количественного учета мута-
мутаций у дрозофилы, обосновал факт му-
мутагенного действия рентгеновских лу-
лучей. В 1928г. Л.Д.Стадлер в опытах на
ячмене и кукурузе показал, что иони-
ионизирующие излучения разных видов
способны вызывать мутации. В после-
последующие два десятилетия происходило
достаточно активное развитие класси-
классической радиационной генегики. Ос-
Основные положения ее изложены в тру-
трудах Д.Ли, Д.Кэтчсайда, Н.В.Тимофее-
Н.В.Тимофеева-Ресовского, К.Г.Циммера, А.Хол-
ландера, А.С.Серебровского, Н.П.Ду-
Н.П.Дубинина, Ядерные взрывы, прогремев-
прогремевшие в Хиросиме и Нагасаки, стимули-
стимулировали бурное развитие работ по изу-
изучению влияния радиации на человека.
Усилия ученых многих стран привели
к разработке современных представле-
представлений о механизмах воздействия иони-
ионизирующих излучений. При этом ос-
основные закономерности воздействия
ионизирующих излучении были
вскрыты в исследованиях, проведен-
проведенных на микроорганизмах, растениях и
животных. Используя принципы экс-
экстраполяции, результаты, полученные
на экспериментальных объектах, ши-
широко используют для оценки генетиче-
генетического риска облучения человека. На-
Например, исследования, проведенные
152
Глава 6.
на мышах, в ходе которых изучали ча-
частоту индуцированных радиацией ка-
катаракт и скелетных аномалий, явились
основой для расчета ожидаемой часто-
частоты индуцированных доминантных му-
мутаций у человека.
Все радиобиологические эффекты,
вызываемые ионизирующими излуче-
излучениями у различных видов живых су-
существ, могут быть подразделены на
стохастические и нестохастические.
Стохастические эффекты характери-
характеризуются линейной беспороговой зави-
зависимостью вероятности их появления от
дозы ионизирующего излучения. При
этом от величины дозы зависит частота
рассматриваемых событий, а не их тя-
тяжесть. К таким эффектам относятся ге-
генетические последствия облучения и
радиационный канцерогенез. Нестоха-
Нестохастические эффекты имеют пороговую
(сигмоидную) зависимость от дозы,
причем с дозой связана как вероят-
вероятность эффекта, так и его тяжесть. При-
Примерами нестохастических эффектов
являются: лучевая болезнь, сокраще-
сокращение продолжительности жизни, смерт-
смертность, индуцированные радиацией по-
пороки развития, поражение иммунной
системы. Следует заметить, что меха-
механизмы возникновения стохастических
и нестохастических эффектов совер-
совершенно различны, поэтому при оценке
рисков появления этих эффектов в ре-
результате облучения недопустимо их
объединение. В этой книге мы будем
рассматривать преимущественно сто-
стохастические эффекты радиации.
Сходство и различие спонтанных
и индуцированных мутаций
В повреждающем действии радиа-
радиации на генетический аппарат клетки
есть несколько основных моментов,
которые имеют важное значение для
оценки последствий облучения.
МУТАЦИИ
1. Как показали многочисленные ис-
исследования, ионизирующие излучения
вызывают все типы мутаций, свойст-
свойственные спонтанному, мутационному
процессу — точковые мутации, аберра-
аберрации хромосом и генные мутации. Од-
Однако следует отметить, что не все типы
спонтанных мутаций с одинаковой ча-
частотой увеличиваются под действием
радиации.
2. Одним из фундаментальных
предложений, на которых основаны
оценки риска облучения человека, яв-
является допущение сходства спонтан-
спонтанных и индуцированных ионизирую-
ионизирующими излучениями мутаций. Предпо-
Предполагая такое сходство, можно оценить
вред, причиненный воздействием ра-
радиации, путем расчета, какую прибав-
прибавку к спонтанному мутационному про-
процессу дает мутагенез, вызванный облу-
облучением. Так производится определе-
определение дозы, удваивающей естественный
мутационный процесс. Однако экспе-
экспериментальные данные молекулярной
генетики демонстрируют различия
между спонтанными и индуцирован-
индуцированными мутациями, вызывающими мен-
делевские болезни. Остановимся на
этом важном вопросе и рассмотрим
различия между этими мутациями:
спонтанные мутации — это чаще всего
точковые мутации и небольшие деле-
ции; большинство же индуцированных
мутации — делеции, затрагивающие
многие гены;
- спонтанные мутации могут вызы-
вызывать как утрату, так и усиление функ-
функции генов, большинство же индуциро-
индуцированных мутаций вызывает потерю
функции;
- происхождение спонтанных мута-
мутаций связано с организацией генов, т.е.
они сайт-специфичны; индуцирован-
индуцированные мутации происходят в результате
случайного попадания энергии излу-
Индуцированный мутагенез
153
чения в генетический материал и мо-
могут затрагивать несколько генов, име-
имеющих разное значение для выживае-
выживаемости организма.
Из этих различий между спонтанны-
спонтанными и индуцированными мутациями
следует важное следствие: вероятность
того, что радиация приведет к возник-
возникновению мутаций, обладающих такой
же специфичностью, какой обладают
спонтанные мутации, очень мала. Дру-
Другими словами, спектры спонтанных и
индуцированных радиацией мутаций,
как следует из молекулярногенетичес-
ких исследований, существенно разли-
различаются.
Ионизирующие излучения в основ-
основном индуцируют микроделеции, поэто-
поэтому важно проанализировать, какими
проявлениями на уровне фенотипа че-
человека сопровождаются такие микроде-
леционные изменения. Поскольку дан-
данные о микроделеционных синдромах,
связанных с воздействием ионизирую-
ионизирующих излучений на человека, отсутству-
отсутствуют, рассмотрим, к каким последствиям
для здоровья человека приводят спон-
спонтанные синдромы, связанные с микро-
делециями. Таких синдромов в настоя-
настоящее время известно около 30. Все они
связаны с микроделециями в разных
хромосомах и обычно сопровождаются
потерей функции нескольких генов.
Фенотипы носителей таких микроделе-
микроделеции зависят от участков хромосом, за-
затронутых микроделециями (например,
хромосомы 19 и 22 изобилуют генами, а
хромосомы 4 и 13 генами обеднены), но
тем не менее разные делеции имеют ряд
общих признаков — они вызывают мно-
многочисленные нарушения развития, ум-
умственную отсталость, замедленный
рост, дисморфныс черты лица. Очевид-
Очевидно, такие же изменения в фенотипе че-
человека будут вызывать микроделеции,
возникающие в результате радиацион-
радиационного воздействия. Основной особенно-
особенностью таких микроделеционных феноти-
фенотипов является несходное с фенотипами
большинства спонтанных мутаций, не-
нечеткое, неясное их проявление.
Различия в клинических фенотипах
спонтанных и индуцированных иони-
ионизирующими излучениями мутаций
имеют принципиальное значение для
оценки риска облучения человека. Де-
Дело в том, что при изучении последст-
последствий воздействия ионизирующих излу-
излучений на популяции человека обычно
проводят анализ социально значимых
отклонений от нормы, которое тради-
традиционно связывают с отклонениями,
подобными фенотипическим проявле-
проявлениям спонтанных мутаций. Измене-
Изменения же, связанные с микроделецион-
ными синдромами, практически оста-
остаются вне поля зрения исследователей
в силу их нечеткого проявления. Та-
Таким образом, большая часть феноти-
пических отклонений, связанных с ми-
микроделециями, индуцированными ио-
ионизирующими излучениями, практи-
практически составляют не учтенный пока
компонент генетического риска облу-
облучения популяций человека.
Дозовые зависимости
частоты мутаций
Частота мутаций зависит от дозы
облучения. Основной закономернос-
закономерностью повреждающего действия радиа-
радиации является бесиороговая линейная
зависимость от дозы облучения при
индукции мутаций, для возникнове-
возникновения которых необходимо только одно
первичное событие (рис. 6.13). Эта за-
закономерность радиационной генетики
имеет исключительно важное значе-
значение, т.к. показывает, что при действии
радиации на наследственные структу-
структуры не существует генетически неэф-
неэффективных доз облучения.
154
Глава 6. МУТАЦИИ
1000 2000 3000
Рис. 6.13. Зависимость частоты
сцепленных с полом летальных мутаций
у дрозофилы от дозы облучения.
По оси абсцисс — доза (рентгены)
По оси ординат — частота сцепленных
с полом леталей (%)
Характер дозовых зависимостей хо-
хорошо изучен на дрозофиле. На рисунке
6.14 приведен пример рассматриваемой
зависимости для точковых мутаций и
хромосомных разрывов у Drosophila
14
12
10
в
2
О
? Тимофеев-Ресовский
^7 Оливер
м Эфроимсон, Шехтман
О ср«
шние вел
°/
f
ж
;
ичины
У
р
У
> ;
Рис. 6.14. Зависимость частоты
точковых мутаций и хромосомных
разрывов у дрозофилы
от дозы облучения
По оси абсцисс - доза (килорентгены)
По оси ординат - мутации (%)
melanogaster. При увеличении дозы об-
облучения, а именно в диапазоне очень
высоких доз, может наблюдаться "эф-
"эффект насыщения", приводящий к от-
отклонению зависимости доза — эффект
от линейной. Это объясняется тем, что
каждое мутационное событие происхо-
происходит за счет однократного попадания в
определенную наследственную струк-
структуру, причем вероятность поражения
этой структуры будет расти линейно с
увеличением дозы до тех пор, пока одна
и та же структура не будет поражаться
более одного раза. При изучении зави-
зависимости частоты индуцируемых мута-
мутаций от дозы мутагенного фактора часто
наблюдают дозовую кривую с максиму-
максимумом. На рисунке (рис. 6.15) представле-
представлена зависимость частоты соматических
мутаций в волосках тычиночных нитей
у традесканции клона 02 от дозы рент-
рентгеновских лучей. При дозе около 2 Гр.
наблюдается максимум числа мутаций.
Более сложная зависимость от дозы
облучения установлена в случае мута-
мутаций, для возникновения которых необ-
необходимо более одного первичного собы-
события. Так, для обменных аберраций, об-
образующихся в результате нескольких
разрывов и последующего воссоедине-
воссоединения разорванных участков хромосом,
были получены экспоненциальные
кривые. Такая зависимость эффекта от
дозы действительно обнаружена во
многих экспериментах. На рисунке
6.16 представлена кривая, полученная
для обменных аберраций у дрозофилы.
Частота индуцированных мутаций
зависит от вида ионизирующего излуче-
излучения (рис. 6.17). Так, если принять гене-
генетическую эффективность гамма-излу-
гамма-излучения за единицу, то рентгеновское из-
излучение при облучении в той же дозе
будет в среднем эффективнее в 2 раза,
медленные нейтроны — в 5 раз, а альфа-
частицы и быстрые нейтроны — в 10 раз.
Индуцированный мутагенез
155
0.1 .
осок
вол
Число
0.001
I I I I I III I I I I I I 11
1 1 1 1 1 1 1 1
0,01 0,1 1,0 10,0
Доза, Гр
Рис. 6.15. Зависимость частоты соматических мутаций в волосках тычиночных нитей
традесканции клона 02 от дозы рентгеновских лучей
12
1
X Бельговский
¦ Хвостова + Гаврилов
¦ Меллер
и Оливер
а Тимофеев-Ресовский
Э
Рис. 6.16. Частота хромосомных аберраций (события с двумя разрывами)
в зависимости oi дозы облучения. Данные получены разными авторами.
По оси абсцисс - доза (килорентгены).
По оси ординат - частота хромосомных аберраций (%)
156
Глава 6. МУТАЦИИ
Рис. 6.17. Зависимость частоты
дицентриков в лимфоцитах
крови человека от вида
ионизирующего излучения.
По оси абсцисс - доза (Грей).
По оси ординат - дицентрики на 1 клетку
1 - быстрые нейтроны, 2 - рентгеновское
излучение, 3 - гамма-облучение "Со
Частота мутаций, возникающих при
облучении, зависит от мощности иони-
ионизирующего излучения (рис. 6.18). Одна
и та же доза, примененная при относи-
относительно низкой мощности, приводит к
возникновению меньшего числа мута-
мутаций, чем та же доза при более высокой
мощности.
Хромосомные нарушения
при действии ионизирующих
излучений
В классических работах по радиаци-
радиационной генетике мутационный эффект
выявляют на фенотипическом уровне,
причем большинство работ выполнено
на растительных объектах, дрозофиле и
других организмах, находящихся в до-
достаточно отдаленном родстве с челове-
человеком. Исследования на человеке начали
активно проводить только после разра-
разработки метода получения препаратов
хромосом из лимфоцитов крови. В на-
настоящее время накоплен огромный
100 200 300 400 500
Рис. 6.18. Зависимость частоты
дицентриков в лимфоцитах
крови человека от мощности
ионизирующего излучения.
По оси абсцисс - доза (рад).
По оси ординат - дицентрики на 1 клетку
практический материал по исследова-
исследованию структурных нарушений хромосом
после облучения in vivo и in vitro. Все
основные положения радиационной ге-
генетики нашли подтверждение при ана-
анализе хромосомных аберраций в клетках
из культуры кожи, костного мозга или
лейкоцитов периферической крови.
Принципиальных морфологических
различий между аберрациями в разных
клетках нет, если исследование прово-
проводят на одной и той же стадии клеточно-
клеточного цикла. Было показано, что в клетках
человека индуцируются те же типы хро-
хромосомных аберраций, что и у других
объектов.
Чаще всего при изучении влияния
облучения на организм человека ис-
используют культуру лимфоцитов пери-
периферической крови. Многочисленные
исследования по радиационной цитоге-
нетике, проведенные на лимфоцитах
крови человека, позволили выявить ос-
основные качественные и количественные
Индуцированный мутагенез
157
закономерности индуцированного му-
мутагенеза. Культура лимфоцитов перифе-
периферической крови представляет собой до-
достаточно простую и в то же время уни-
уникальную по своим возможностям мо-
модель для изучения радиационного мута-
мутагенеза. Для ее получения достаточно
взять всего несколько миллилитров ве-
венозной крови и поместить в питатачь-
ную среду, содержащую стимулятор
роста — фитогемагглютинин (ФГА).
Следует заметить, что в норме лимфо-
лимфоциты периферической крови, как пра-
правило, не делятся, находясь в стадии по-
покоя (интерфазы). Под действием ФГА
происходит их иммунологическая
трансформация и они начинают актив-
активно делиться. Таким образом можно до-
достаточно легко получить большое коли-
количество клеток для цитогенетического
анализа. С помощью специальных мето-
методов обработки и различных способов
окраски клеток готовят препараты для
исследования структурных и количест-
количественных нарушений хромосом.
При индуцированном мутагенезе на-
наблюдаются те же типы хромосомных
аберраций, что и при спонтанном мута-
мутационном процессе. Среди хромосомных
аберраций, выявленных при анализе
лимфоцитов периферической крови по-
после облучения, выделяют стабильные
аберрации (транслокации, инверсии),
которые способны нормально прохо-
проходить через митоз, и нестабильные (ди-
центрики, центрические кольца, парные
фрагменты), элиминирующие при по-
последующих делениях клетки. Особенно
наглядным индикатором радиационно-
радиационного воздействия на организм являются
дицентрики. В норме такие хромосом-
хромосомные нарушения встречаются достаточно
редко — с частотой 3-5 на 10000 проана-
проанализированных клеток. Многочисленны-
Многочисленными исследованиями было показано, что
частота возникновения дицентриков
возрастает пропорционально дозе облу-
облучения. Установлена также зависимость
эффекта от дозы, которая, как правило,
является линейной, но при сравнитель-
сравнительно высоких дозах приобретает экспо-
экспоненциальный характер. Увеличение
числа хромосомных аберраций обнару-
обнаружено при обследовании различных
групп облученных людей, а именно, у
пациентов после рентгенодиагностичес-
ких процедур и терапевтического облу-
облучения, а также у лиц, связанных с про-
профессиональным облучением (работни-
(работники предприятий атомной промышлен-
промышленности, врачи-рентгенологи и др.); у лю-
людей, облучившихся в результате аварий
(например, после аварии на Чернобыль-
Чернобыльской АЭС); у пострадавших в результа-
результате атомной бомбардировки Хиросимы и
Нагасаки.
В настоящее время в связи с широ-
широким использованием атомной энергии
изучение и оценка цитогенетических
эффектов облучения у человека, несо-
несомненно, является важной задачей. Вы-
Выявленные количественные зависимости
частоты хромосомных аберраций от до-
дозы облучения стали основой для разра-
разработки принципов биологической дози-
дозиметрии. При облучении лейкоцитов in
vitro в лабораторных условиях возмож-
возможно получение кривых доза — эффект,
которые могут быть использованы как
калибровочные при количественном
определении полученной дозы. Многи-
Многими исследователями была продемонст-
продемонстрирована одинаковая радиочувстви-
радиочувствительность хромосом при облучении in
vivo и in vitro. Еще в 1966 г. Бендер и Гоч
при обследовании людей, облученных в
результате аварии гамма-лучами и быс-
быстрыми нейтронами, а также при прове-
проведении многочисленных экспериментов
с облучением крови in vitro предложили
формулу для оценки доз облучения. В
последующих работах эта формула не-
158
Глава 6. МУТАЦИИ
\
4
Рис. 6.19. Метафазные пластинки в культуре лимфоцитов крови.
Окраска красителем Гимза
а - норма D6 XX)
б - метафазная пластинка с тремя дицентриками и двумя парными фрагментами (обозна-
(обозначены стрелками) '
в - метафазная пластинка с дицентриком !
г - метафаза с центрическим кольцом и парными фрагментами '
д - метафаза с трицентриком и двумя парными фрагментами I
е - мультиаберрантная клетка с тетрацентриком, дицентриком и многими парными фраг- !
ментами :
Индуцированный мутагенез
159
однократно уточнялась. Чаще всего для
оценки величины поглощенной орга-
организмом дозы используют частоту воз-
возникновения дицентриков и центричес-
центрических кольцевых хромосом. Эти хромо-
хромосомные аберрации легко распознаются
при анализе под микроскопом после
стандартного окрашивания метафазных
препаратов красителем Гимза. На ри-
рисунке 6.19 изображена нормальная ме-
тафазная пластинка и метафазные плас-
пластинки с дицентриками и центрически-
центрическими кольцами. На рисунке представлены
также другие редкие хромосомные абер-
аберрации, наблюдаемые у облученных лю-
людей: трицентрики, тетрацентрики и
мультиаберрантные клетки. К сожале-
сожалению, для целей биологической дозимет-
дозиметрии использование частоты дицентри-
дицентриков и центрических колец ограничено
из-за того, что со временем клетки, несу-
несущие такой тип хромосомных аберраций,
элиминируются из кровяного русла.
Наиболее жрфективен подсчет частоты
дицентриков для оценки дозы облуче-
облучения в ранние сроки (до года) после ра-
радиационного воздействия. В более позд-
поздние сроки, для ретроспективной оценки
дозы облучения, необходима дополни-
дополнительная информация — условия облуче-
облучения, время, прошедшее после радиаци-
радиационного воздействия, скорость элимина-
элиминации клеток с дицентриками и центриче-
центрическими кольцами.
Более перспективным и эффектив-
эффективным методом оценки доз в этом случае
является учет аберраций стабильного
типа — транслокаций, инверсий, частота
которых остается постоянной в течение
длительного времени после облучения.
В отличие от дицентриков, транслока-
транслокации и инверсии не подвергаются селек-
селекции во время клеточного деления. Час-
Частота возникновения аберраций стабиль-
стабильного (транслокации) и нестабильного
(дицентрики) типов сразу после облу-
облучения приблизительно одинакова. До
недавнего времени для анализа частоты
транслокаций использовали дифферен-
дифференциально окрашенные препараты (G-ok-
раска). Под дифференциальной окра-
шиваемостью хромосом понимают их
способность к избирательному окраши-
окрашиванию по длине. При этом каждая хро-
хромосома имеет свой рисунок исчерченно-
сти, позволяющий выявить хромосом-
хромосомные перестройки (например, симмет-
симметричные транслокации), которые невоз-
невозможно идентифицировать при обычном
окрашивании из-за кажущейся морфо-
морфологической однородности хромосом.
Этот метод определения стабильных
хромосомных аберраций весьма трудо-
трудоемкий и требует высокой квалицикации
специалистов для проведения цитогене-
тического анализа.
В 1986 г. в Ливерморской националь-
национальной лаборатории (США) разработан
принципиально новый метод изучения
хромосом — метод флюоресцентного
выявления ДНК хромосом путем гибри-
гибридизации in situ со специфическими мо-
молекулярными зондами (FISH). Он осно-
основан на способности хромосомной ДНК
связываться при определенных услови-
условиях с фрагментами ДНК (ДНК-зонды),
которые включают нуклеотидные по-
последовательности, комплементарные
хромосомной ДНК (рис. 6.20). ДНК-
зонды предварительно метят специаль-
специальными веществами (например, биотином
или дигоксигенином). Меченые ДНК-
зонды наносят на цитогенетические пре-
препараты подготовленных для гибридиза-
гибридизации (денатурированных) метафазных
хромосом. Предварительная обработка
хромосом необходима для облегчения
доступа ДНК-зонда к геномной ДНК.
После того, как произошла гибридиза-
гибридизация, препараты обрабатывают специаль-
специальными флюоресцентными красителями,
конъюгированными с веществами, спо-
160
Глава 6. МУТАЦИИ
Норма
Трансло-
Транслокации
?!-• *'¦
Рис. 6.20. Идентификация транслокаций при помощи FISH метода.
Верхний ряд - нормальные клетки с меченными 1 и 2 хромосомами.
Нижний ряд - транслокации с участием меченых хромосом
собными избирательно присоединяться
к биотину или дигоксигенину. Для био-
биотина таким веществом является стрепта-
видин, а для дигоксигенина — антиди-
гоксигенин. В качестве красителей
обычно используют флюоресцеин-изо-
тиоцианат- FITC; метил-кумарин-3-ук-
сусную кислоту АМСА и др. (Гибриди-
(Гибридизация может проводиться также с зонда-
зондами, имеющими радиоактивную метку).
Цитогенетический анализ проводит-
проводится под люминесцентным микроскопом
в ультрафиолетовом свете. Хромосом-
Хромосомные обмены (транслокации и дицентри-
ки) между разноокрашенными хромо-
хромосомами легко определяются как двух-
двухцветные структуры. Для идентифика-
идентификации транслокаций (с одной центроме-
центромерой) и дицентриков (с двумя центроме-
центромерами) одновременно с хромосомными
ДНК-зондами используют также ДНК-
зонды к центромерным участкам хромо-
хромосом (панцентромерные пробы). При ис-
использовании одного флюоресцентного
красителя для ДНК-зондов обычно ме-
метят около 20% генома, т.е.- не более 3
хромосом, и выявленные таким образом
хромосомные нарушения для анализи-
анализируемой части генома пересчитывают не
весь геном в целом. На рисунке 6.21 изо-
изображены метафазные пластинки, обра-
обработанные с использованием FISH-мето-
да. Современные методические возмож-
возможности позволяют увеличить число цве-
цветов, что, в свою очередь, дает возмож-
возможность увеличить долю окрашенных хро-
хромосом в геноме и повысить точность ис-
исследования.
В настоящее время имеются много-
многочисленные работы по использованию
Индуцированный мутагенез
161
Рис. 6.21.
Метафазные пластинки в культуре
лимфоцитов крови после обработки с
использованием метода FISH.
Представлены нормальная метафаза с
мечеными хромосомами и примеры
транслокаций с участием этих хромосом.
Видны также центромеры, выявляемые при
помощи панцентромерных зондов.
а - нормальная метафаза
б - метафаза с транслокацией
в - матефаза с транслокацией.
традиционных цитогенетических и
FISH-метода для определения частоты
радиационио-индуцированных аберра-
аберраций хромосом в лимфоцитах перифе-
периферической крови. Опубликованы ре-
результаты по ретроспективной биологи-
биологической дозиметрии, полученные с ис-
использованием FISH-метода и других
цитогенетических методов при обсле-
обследовании участников ликвидации ава-
аварии на Чернобыльской АЭС, населе-
населения, проживающего на радиационно-
загрязненных территориях (районы
Семипалатинского полигона, Восточ-
Восточно-Уральского радиоактивного следа),
а также населения, подвергшегося об-
облучению в результате атомных бомбар-
бомбардировок в Японии. Некоторые резуль-
результаты этих исследований будут рассмот-
рассмотрены в следующем разделе.
Цитогенетические эффекты
воздействия ионизирующих
излучений на человека
Анализ частоты аберраций хромо-
хромосом в лимфоцитах периферической
крови при воздействии различных
мутагенных факторов (прежде всего,
162
Глава 6. МУТАЦИИ
3,5
3 --
OK 86 87
90 91 92 93 94 95 96 97
Год проведения анализа
Контроль (82 чел.): 0,2±0,1
1986 D43 чел.): 3,3±0,3
1990B3)чел.):1,0±0,5
1992 A36 чел.): 1,4±0,2
1994 F9 чел.): 1,6±0,3
1996 E3 чел.): 0,7±0,2
1987 B80 чел.): 1,4±0,2
1991A10чел.):0,9±0,2
1993G5чел.):0,9±0,2
1995A10чел.):0,6±0,1
1997D1чел.):1,0±0,2
Рис. 6.22. Частота клеток с дицентриками и центрическими кольцами
в группах ликвидаторов, обследованных в разное время после аварии
ионизирующих излучений) позволя-
позволяет исследовать закономерности мута-
мутагенеза на уровне соматических кле-
клеток. Выявленные при этом законо-
закономерности мутационного процесса
можно в принципе экстраполировать
на половые клетки, менее доступные
для прямого цитогенетического ана-
анализа. В генетических исследованиях
разработаны соответствующие под-
подходы, поэтому изучение закономер-
закономерностей воздействия ионизирующих
излучений на соматические клетки
человека создает основу для анализа
последствий облучения людей в бу-
будущих поколениях. Кроме того, ана-
анализ хромосомных аберраций в лим-
лимфоцитах, как отмечено выше, позво-
Индуцированный мутагенез
163
ляет оценить дозу ионизирующих из-
излучений, полученную человеком в ре-
результате аварийного воздействия ра-
радиации.
Крупномасштабные цитогенетичес-
кие исследования были проведены в
связи с аварией на Чернобыльской
АЭС. Цитогенетические обследования
участников ликвидации последствий
аварии на ЧАЭС были начаты сразу
после аварии в 1986г. и продолжаются
по 1999г. В общей сложности было об-
обследовано около 2000 человек — со-
сотрудников атомной станции, врачей,
дозиметристов, строителей, жителей
г.Припять. Показано, что частота абер-
аберраций хромосом у обследованных в
1986 г. людей значительно (до 17 раз)
превышает контрольный уровень. В
последующие годы частота аберраций
хромосом у обследуемых групп посте-
постепенно снижалась (рис. 6.22). На протя-
протяжении всего времени обследования
(свыше 10 лет) уровень клеток с неста-
нестабильными аберрациями хромосом —
дицентриками и центрическими коль-
кольцами (признанными маркерами радиа-
радиационного воздействия), достоверно от-
отличался от контроля. Таким образом,
несмотря на длительность периода,
прошедшего с момента облучения, в
периферической крови обследованных
людей сохранялся высокий уровень
клеток с нестабильными аберрациями.
Проведено также цитогенетическое
обследование населения Алтайского
края, подвергшегося воздействию вы-
высоких доз ионизирующих излучений в
связи с проведением ядерных испыта-
испытаний в атмосфере на Семипалатинском
полигоне в 1949-1962 гг. Изучение ге-
генетических последствий ядерных
взрывов для населения Алтайского
края начато лишь через несколько де-
десятков лет после радиационного воз-
воздействия. Обследование 226 жителей 7
ю
0.1
4
1
(
i
Контроль
0,01
0.1
Доза(Зе)
Рис. 6.23. Частота клеток
с дицентриками и центрическими
кольцевыми хромосомами
у обследованных групп жителей
Алтайского края в зависимости от
эффективной дозы облучения в
результате ядерного взрыва на
Семипалатинском полигоне в 1949 г. На
оси абсцисс - доза, Зв, на оси ординат -
число клеток с дицентриками и
центрическими кольцевыми
хромосомами на 1000 клеток
населенных пунктов Алтайского края
показало, что частота аберраций хро-
хромосом у подвергшихся облучению лю-
людей достоверно превышает контроль-
контрольный уровень. На рисунке 6.23 пред-
представлена частота клеток с дицентрика-
дицентриками и центрическими кольцевыми хро-
хромосомами у жителей различных насе-
населенных пунктов в зависимости от ве-
величины поглощенных доз. Наблюдает-
Наблюдается статистически достоверная зависи-
зависимость частоты аберраций хромосом от
поглощенной дозы (Р<0,05). Результа-
Результаты исследования наглядно демонстри-
демонстрируют, что даже через несколько деся-
десятилетий после радиационного воздей-
воздействия в периферической крови обсле-
обследуемых жителей наблюдается повы-
повышенная частота клеток с нестабильны-
нестабильными аберрациями хромосом, уровень
164
Глава 6. МУТАЦИИ
0,0 0,2 0.4 0,6 0,8 1.0 1,2 1.4 1,6 1,8 2,0
Рис. 6.24. Зависимость частоты транслокаций на 100 клеток от дозы гамма-излучения
цезия-137. Приведены 95%-ные доверительные интервалы
которых зависит от величины эффек-
эффективной дозы. Можно предположить,
что источником таких клеток, несущих
дицентрики и центрические кольце-
кольцевые хромосомы, являются радиацион-
но пораженные стволовые клетки кро-
кроветворной ткани.
У обследуемых жителей Алтайского
края, подвергшихся облучению, наря-
наряду с аберрантными клетками, содержа-
содержащими 1-2 аберрации хромосом, выяв-
выявлены также мультиаберрантные клет-
клетки, содержащие 3 и более аберраций
хромосом E и более разрывов хромо-
хромосом). Среди мультиаберрантных кле-
клеток выявлены клетки, содержащие ди-
дицентрики, транслокации, центричес-
центрические кольца и даже трицентрики. По-
Последние практически никогда не
встречаются в контрольном материа-
материале. Появление таких клеток скорее
всего связано с воздействием плотнои-
онизирующих излучений, в первую
очередь альфа-частиц плутония-239 и
других радионуклидов, входящих в со-
состав смеси продуктов ядерного взрыва,
поступивших в организм человека по-
после проведения ядерных испытаний.
Мультиаберрантные клетки были
выявлены также у жителей села Мус-
люмово Челябинской области, распо-
расположенного на берегу Теча, загрязнен-
загрязненной радионуклидами в связи с дея-
деятельностью предприятия по производ-
производству плутония "Маяк". Наиболее ин-
интенсивный период загрязнения этой
реки относится к начальному периоду
работы предприятия — 1949-1951 гг.
Несколько тысяч жителей этого села
получили среднюю дозу облучения на
костный мозг около 0,25 Гр, а прибли-
приблизительно у 5% средняя доза на кост-
костный мозг составила 1 Гр. Цитогенети-
ческое обследование более 100 человек
этого села, проведенное в 1994 г., пока-
показало, что уровень нестабильных абер-
аберраций хромосом (дицентриков и цент-
центрических кольцевых хромосом) в 5-10
раз выше, чем в контроле. Наиболее
высокий уровень аберраций хромосом
отмечен у лиц, родившихся до 1949 г.
или в период наиболее высокого за-
Индуцироннный мутагенез
165
s
о
2 1,0.-
л
1,6
1,4
1,2.
1,0
0,8
0,6
0,4
0,2
0
Контроль Ликвидаторы/94 Алтай/94 ТМА/94
Группы
"Ликвидаторы"
Алтайский карай
ТМА (США)
Контроль
Число обсле-
дованных людей
53
24
6
12
Число проа-
нализирован-
нализированных метафаз
45007
13001
3468
13586
Трансло-
Транслокации
166
58
17
13
Частота
транслокаций
на геном
(HG+m)xl0I
1,17+0,05
1,42±0,10
1,54+0,21
0,32+0,05
Рис. 6.25. Частота транслокаций, выявляемых при помощи метода FISH, в лимфоцитах
периферической крови ликвидаторов последствий Чернобыльской аварии, у жителей
Алтайского края, пострадавших от ядерных испытаний на Семипалатинском полигоне,
и у жителей, проживающих в районе атомной станции Три Майл Айленд, США
грязнения р. Теча радионуклидами
A949-1956 гг.). У этих жителей также
были выявлены мультиаберрантные
клетки. Наряду с дицентриками, были
обнаружены также трицентрики и тет-
рацентрики.
Необходимо отметить, что, наряду с
хромосомными аберрациями, у людей,
проживающих вдоль р. Теча, наблюда-
наблюдаются и другие эффекты, свидетельст-
свидетельствующие о значительном повреждаю-
повреждающем действии радиации. Облучение
населения в верховьях этой реки при-
привело к возникновению хронической
лучевой болезни (особенно в селе
Метлино, где хроническая лучевая бо-
болезнь в 1956 г. диагностирована у
64,7% взрослых и 63,2% осмотренных
детей), которая была выявлена у 935
человек. Установлено также увеличе-
увеличение заболеваемости лейкозами у на-
наблюдаемого населения. За 33 года за-
зарегистрировано 52 случая гемабласто-
зов, в том числе 37 случаев лейкозов
среди 17,2 тыс. человек, наблюдаемых
с 1950 г., что на 15 случаев больше
ожидаемого числа заболеваний без об-
облучения. Отмечено также существен-
существенное увеличение заболеваемости раком
кожи, кишечника, печени, желчного
166
Глава 6. МУТАЦИИ
пузыря, шейки матки. Эти эффекты
связаны с тем, что жители 20 сел по р.
Теча G,5 тыс. человек из этих сел были
переселены) получили средние эффек-
эффективные эквивалентные дозы от 3,5 до
170 сЗв. Наибольшие дозы получили
жители выселенного села Метлино
A70 сЗв, численность населения — 1,2
тыс. человек).
Как отмечено выше, частота ста-
стабильных аберраций хромосом, выявля-
выявляемых методом FISH, позволяет оце-
оценить поглощенные дозы ионизирую-
ионизирующих излучений, полученные облучен-
облученными людьми в отдаленные сроки. При
этом для оценки дозы используют ка-
калибровочные кривые, построенные в
тщательных лабораторных экспери-
экспериментах в ходе изучения зависимости
частоты транслокаций от дозы излуче-
излучения (кривые "доза — эффект"). Пример
такой калибровочной кривой представ-
представлен на рисунке 6.24. Зная частоту вы-
выявленных аберраций хромосом у того
или иного человека, можно показать с
определенной степенью достоверности
(на рисунке приведены 95%-ые дове-
доверительные интервалы) полученную им
дозу ионизирующих излучений.
Анализ частоты стабильных аберра-
аберраций хромосом (транслокаций) с ис-
использованием метода FISH проведен у
участников ликвидации последствий
аварии на Чернобыльской АЭС через
8-9 лет после аварии. В обследованной
группе частота клеток с транслокация-
транслокациями превышает этот показатель в кон-
контрольной группе в 4 раза (рис 6.25). На
основе полученных данных с помощью
калибровочной кривой доза — эффект
рассчитана поглощенная доза облуче-
облучения, составившая в среднем 200 мГр.
Для отдельных ликвидаторов погло-
поглощенные дозы достигали 1 Гр.
Применение FISH-метода позволи-
позволило оценить поглощенные дозы для жи-
жителей Алтайского края, подвергшихся
воздействию радиации в связи с испы-
испытанием ядерного оружия. Средняя час-
частота транслокаций для жителей трех
сел более чем в 5 раз превышает кон-
контрольный уровень (рис. 6.25). Средняя
доза для обследованной группы насе-
населения, рассчитанная на основе данных
цитогенетического анализа, составила
около 1 Гр. Такие же результаты полу-
получены при помощи FISH метода при об-
обследовании жителей штата Пенсиль-
Пенсильвания в США, подвергшихся облуче-
облучению в результате аварии на атомной
станции Три Майл Айленд в 1979 г.
(рис. 6.25). Расчетная доза для обсле-
обследованной в 1994 г. группы жителей со-
составила 0,6-0,9 Гр.
Таким образом, анализ аберраций
хромосом в лимфоцитах периферичес-
периферической крови людей, пострадавших от
воздействия ионизирующих излуче-
излучений в рассмотренных ситуациях, поз-
позволил выявить повышенные в не-
несколько раз уровни нестабильных и
стабильных аберраций хромосом по
сравнению контролем. Уровень неста-
нестабильных аберраций хромосом снижа-
снижается во времени. Принято считать, что
в лимфоцитах периферической крови
он уменьшается вдвое в течение 3-4
лет. Однако наблюдаемый через 8 лет
после аварии на ЧАЭС и через 45 лет
после ядерных взрывов на Семипала-
Семипалатинском полигоне повышенный уро-
уровень таких аберраций может быть свя-
связан с постоянным поступлением в
кровь клеток с аберрациями хромосом
вследствие деления несущих аберра-
аберрации хромосом стволовых клеток кро-
кроветворной ткани, тоже пораженной ра-
радиацией.
Истинное представление о масшта-
масштабах первичного поражения клеток, да-
даже десятилетия спустя после облуче-
облучения, дает анализ частоты стабильных
Индуцированный мутагенез
167
аберраций хромосом (транслокаций)
при помощи FISH-метода. Использо-
Использование FISH-метода для реконструк-
реконструкции поглощенных доз дает вполне
удовлетворительные результаты, сов-
совпадающие с оценками произведенны-
произведенными методами физической дозиметрии.
Применение FISH-метода для целей
биологической дозиметрии особенно
перспективно в тех случаях, когда фи-
физическая дозиметрия ограничена. Дру-
Другим важным направлением использо-
использования цитогенетических методов яв-
является оценка генетического риска рб-
лучения популяций человека на осно-
основе данных биологической дозиметрии.
Действительно, если анализ частоты
аберраций хромосом у облученных
людей позволяет определить погло-
поглощенную дозу, то далее, на основе мето-
методологии, разработанной Научным Ко-
Комитетом ООН по действию атомной
радиации, могут быть произведены
расчеты ожидаемых генетических ано-
аномалий у потомков облученных людей.
Оценка генетического риска
облучения человека
Под генетическими рисками облу-
облучения человека понимают ожидаемые
генетические эффекты у потомков об-
облученных людей — у детей, внуков,
правнуков и т.д. Для того, чтобы оце-
оценить эти эффекты в первых поколени-
поколениях после воздействия излучений ис-
используют подходы, основанные на
применении двух методов: метода уд-
удваивающей дозы и прямого метода.
Метод удваивающей дозы базируется
на определении дозы, вызывающей та-
такой же генетический эффект, какой на-
наблюдается в результате естественного
мутационного процесса (удваивает
его). При применении этого метода на-
надо иметь в виду, что уровень естествен-
естественного мутационного процесса в популя-
1 2
Рис. 6.26. Динамика мутационного
процесса (М) в хронически облучаемых
популяциях человека в ряде поколений (п).
1 - уровень естественного мутационного
процесса
2 - хроническое облучение
циях человека — это исторически сло-
сложившийся равновесный уровень, зави-
зависящий от интенсивности естественно-
естественного мутационного процесса, с одной
стороны, и интенсивности отбора про-
против возникших мутаций, с другой. Ин-
Интенсивность отбора в популяциях че-
человека в условиях современной циви-
цивилизации сильно снижена, что и обу-
обусловливает в значительной мере высо-
высокий уровень естественной изменчиво-
изменчивости человека.
При длительном (в течение не-
нескольких поколений) хроническом об-
облучении популяций человека равно-
равновесный уровень мутагенеза за счет вы-
вызванных радиацией наследственных
изменений установится лишь через
семь-десять поколений после начала
облучения (рис. 6.26). При этом инду-
индуцированные ионизирующим излуче-
излучением мутационные изменения соста-
составят определенную дополнительную к
естественной изменчивости человека
компоненту, величина которой зави-
зависит от мощности дозы.
При удваивающей дозе равновес-
равновесный уровень мутагенеза от ионизиру-
ионизирующих излучений сравняется с равно-
равновесным уровнем естественной измен-
168
чивости человека и общий уровень му-
мутагенеза удвоится (рис. 6.26). По оцен-
оценке экспертов НКДАР ООН удваиваю-
удваивающая доза для человека при остром воз-
воздействии ионизирующих излучений
составляет 0,35 Зв на поколение и при
хроническом воздействии редко иони-
ионизирующих излучений при низких
мощностях доз равна 1 Зв на поколе-
поколение. В первом поколении наблюдается
приблизительно 1/10 часть тех мута-
мутаций, которые составят через 7-10 поко-
поколений равновесный уровень.
Необходимо отметить, что все рас-
счеты, связанные с оценкой динамики
мутационного процесса в облучаемых
популяциях человека, получены на ос-
основе экспериментов, проведенных с
хронически облучаемыми популяция-
популяциями модельных объектов исследований
— дрозофилы и лабораторных мышей.
До настоящего времени в мировой на-
научной литературе отсутствуют данные,
позволяющие корректно оценить ди-
динамику мутационного процесса непо-
непосредственно в облучаемых популяци-
популяциях человека, несмотря на наличие по-
популяций, реально подвергшихся облу-
облучению в местах, загрязненных радио-
радионуклидами.
При оценке ожидаемых генетичес-
генетических эффектов облучения в первом по-
поколении экспертами НКДАР, как сле-
следует из таблицы 6.4 (стр. 152), произ-
произведен расчет в отношении не всех ка-
категорий естественной генетической
отягощенности человека, а только их
части: доминантных и сцепленных с
Х-хромосомой, рецессивных и хромо-
хромосомных болезней. Их суммарная ожи-
ожидаемая частота в первом поколении
составляет 17 во втором поколении —
14 и в равновесном состоянии — 120
случаев на 1 млн. новорожденных при
дозе 0,01 Зв. Нетрудно установить,
что категории болезней, по которым
Глава 6. мутации
производится оценка риска облуче-
облучения человека, составляют лишь 2,4 %
от всей выявленной в настоящее вре-
время естественной наследственной отя-
отягощенности человека. По состоянию
вопроса на 1988 г. не была произведе-
произведена оценка риска в отношении врож-
врожденных аномалий и мультифактори-
мультифакториальных болезней, на которые прихо-
приходится 97,6% всей наследственной отя-
отягощенности человека.
Это не было сделано в связи с тем,
что пока неизвестно, можно ли к бо-
болезням сложной этиологии (мульти-
факториальным и врожденным поро-
порокам развития) применять удваиваю-
удваивающую дозу в 1 Зв, и какую величину му-
мутационной компоненты в отношении
этих болезней надо использовать для
расчетов риска облучения. Однако, ес-
если принять, что мутационные компо-
компоненты для этих болезней колеблются
в пределах 5-50 %, как это предполага-
предполагают эксперты НКДАР, то в итоге такого
расчета при дозе 0,01 Зв на миллион
новорожденных в первом поколении
ожидается дополнительно к естест-
естественному уровню изменчивости, рав-
равному, как мы уже отмечали 67,6 % (т.е.
676 000 на миллион новорожденных),
50 случаев наследственных аномалий,
если мутационная компонента для
мультифакториальных болезней будет
равна 5%. В случае, если мутационная
компонента будет равна 50 %, то число
ожидаемых наследственных аномалий
резко возрастет — до 350 случаев.
При хроническом облучении мно-
многих поколений ожидаемый равновес-
равновесный уровень, как следует из расчетов,
составляет в таком случае 450-3400
наследственных аномалий при дозе
0,01 Зв на поколение и на один милли-
миллион новорожденных.
Таким образом, в зависимости от то-
того, в какой степени генетические изме-
Ицдуциромнный мутагенез
169
нения, приводящие к мультифактори-
альным болезням и врожденным ано-
аномалиям, могут индуцироваться иони-
ионизирующими излучениями, будет изме-
изменяться и ожидаемый генетический
риск в следующих поколениях. Если
допустить, что болезни, составляющие
преобладающую часть естественной
наследственной отягощенности попу-
популяций человека, не будут индуциро-
индуцироваться радиацией (что весьма малове-
маловероятно), то генетический риск в пер-
первом поколении составит 17 случаев на-
наследственных болезней на 1 млн. ново-
новорожденных при дозе 0,01 Зв. Если же
допустить, что такие болезни сложной
этиологии (или подобные им) будут
реально индуцироваться радиацией, то
тогда генетический риск облучения
популяций человека составит гораздо
большую величину — от 50 до 350 слу-
случаев на 1 млн. новорожденных при до-
дозе 0,01 Зв.
Все вышеизложенное касалось
оценки риска облучения человека ме-
методом удваивающей дозы. Возможна
оценка риска облучения человека и с
применением прямого метода — путем
анализа частоты индуцированных му-
мутаций отдельных генов, анализа часто-
частоты аберраций хромосом и изменения
числа хромосом у человека и экспери-
экспериментальных объектов. Например, зная
частоту мутаций отдельных генов в
расчете на 0,01 Зв и зная число струк-
структурных генов в геноме человека (около
100 тыс.) можно оценить суммарную
ожидаемую частоту появления генных
мутаций при той или иной дозе. С ис-
использованием прямых методов оценки
генетического риска в настоящее вре-
время экспертами НКДАР ООН произво-
производится оценка частоты индуцирован-
индуцированных доминантных мутаций и реци-
прокных транслокаций. При этом ве-
величины риска для этих типов генети-
генетической изменчивости в принципе сов-
совпадают с теми оценками, которые про-
произведены методом удваивающей дозы.
Несмотря на кажущуюся простоту
подхода, при практическом примене-
применении прямых методов оценки возникает
не меньше сложностей, чем при ис-
использовании метода удваивающей до-
дозы, что связано с ограниченностью на-
наших знаний относительно структуры
генома человека и частот индуциро-
индуцированных мутаций.
Следует отметить, что при исполь-
использовании прямых методов, так же, как и
в случае с методом удваивающей дозы,
основные результаты по частотам ин-
индуцированных мутаций у человека
рассчитаны путем экстраполяции с
данных, полученных на эксперимен-
экспериментальных животных. В частности, для
расчета частоты индуцированных до-
доминантных мутаций у человека ис-
использованы данные по частоте доми-
доминантных мутаций у мышей, приводя-
приводящих к аномалиям скелета, и мутаций,
приводящим к катарактам. В результа-
результате применения ряда переходных коэф-
коэффициентов при экстраполяции данных
на человека, получена частота доми-
доминантных мутаций у мальчиков равная
10-20 случаям, а для девочек — 0-9 слу-
случаев на 1 млн. новорожденных при до-
дозе 0,01 Зв.
Приведенные количественные по-
показатели риска относятся к числу
ожидаемых случаев серьезных гене-
генетических болезней. Термин "серьез-
"серьезная генетическая болезнь", использу-
используемый в документах НКДАР ООН, оз-
означает плохое состояние здоровья,
мешающее нормальной трудоспособ-
трудоспособности, физические и умственные не-
недостатки или нетрудоспособность ге-
генетического происхождения, которые
могут появиться в любом возрасте от
рождения до старости. Разработано
170
Глава 6. МУТАЦИИ
несколько критериев оценки генети-
генетического ущерба от ионизирующих из-
излучений, которое позволяют взвесить
и количественно оценить действи-
действительный груз, налагаемый генетичес-
генетическими болезнями на индивидуальные,
социальные и общественные показа-
показатели здоровья. Для оценки генетичес-
генетического ущерба могут быть использова-
использованы показатели неполноценной жизни
(пребывание в больницах, домашняя
изоляция и т.д.) и сокращение про-
продолжительности жизни.
При допущении, что средняя ожида-
ожидаемая продолжительность жизни со-
составляет 70 лет (и, таким образом, на 1
млн. новорожденных составляет 70
млн. лет) было оценено, что все возни-
возникающие спонтанно генетические бо-
болезни вызывают около 2,3 млн. лет не-
неполноценной жизни на 1 млн. ново-
новорожденных и около 3 млн. лет сокра-
сокращения продолжительности жизни на 1
млн. новорожденных. Для популяции,
подвергшейся действию ионизирую-
ионизирующих излучений в дозе 1 Зв на поколе-
поколение, индуцированные дополнительные
случаи генетических болезней будут
причиной около 50 000 лет неполно-
неполноценной жизни и такого же количества
сокращения продолжительности жиз-
жизни на 1 млн. новорожденных. Эти
оценки произведены без учета болез-
болезней сложной этиологии — врожденных
аномалий и мультифакторных болез-
болезней. При учете последних ожидаемый
генетический ущерб возрастает в 3-20
раз в зависимости от величины мута-
мутационной компоненты этих болезней,
равной, как мы выше отмечали 5-50%.
Представленные оценки генетичес-
генетического риска достаточно приблизитель-
приблизительны, поскольку они не учитывают ряд
важных моментов, способных еще уве-
увеличить предполагаемую опасность об-
облучения (см. таблицу 6.4). К не решен-
решенным вопросам генетики человека отно-
относится оценка влияния на жизнеспособ-
жизнеспособность человека "малых" мутаций (му-
(мутаций, в незначительной степени за-
затрагивающих фенотип человека), ко-
которые при взаимодействии с другими
мутациями могут существенно влиять
на здоровье человека. Роль таких мута-
мутаций в жизнеспособности популяции
показана в экспериментах с дрозофи-
дрозофилой, однако пока полностью отсутст-
отсутствуют подходы к оценке роли таких му-
мутаций у человека.
Многое не ясно и в проблеме насле-
наследуемых раковых заболеваний. В этой
связи важными являются появившие-
появившиеся в последние годы сообщения о рос-
росте злокачественных заболеваний (в
основном, лейкозы) среди групп насе-
населения, проживающих вблизи от ядер-
ядерных установок. В частности, появи-
появились сведения о росте заболеваемости
лейкозом среди детей в деревне Сис-
кейл, находящейся вблизи от Селла-
филда, где расположено основное
предприятие Соединенного Королев-
Королевства Великобритании по переработке
плутония. Отмечено девятикратное
увеличение заболеваемости лейкозом
у детей, матери которых проживали в
этой деревне. Последующие исследо-
исследования вокруг 15 основных ядерных
установок Соединенного Королевства
позволили выявить, что в радиусе 16
км от них смертность от лейкозов
всех видов увеличена на 15% (р—0,01),
от лимфоцитарного лейкоза — на 21%
(р=0,01) и от болезни Ходжкина сре-
среди молодежи в возрасте до 25 лет — на
25% (р=0,05) по сравнению с другими
районами. Анализ полученных ре-
результатов позволил исследователям
сделать вывод, что наблюдаемый эф-
эффект можно объяснить профессио-
профессиональным облучением по месту работы
отцов этих детей, в результате чего
Индуциромнный мутагенез
171
происходит воздействие ионизирую-
ионизирующих излучений на мужские гонады с
последующим возникновением мута-
мутаций в постмейотических половых
клетках. Другими словами, речь идет
о злокачественных заболеваниях, по-
появляющихся у потомков облученных
людей. Такие наследственные заболе-
заболевания составляют часть генетическо-
генетического груза, возникающего при воздейст-
воздействии радиации в последующих поколе-
поколениях.
Важно отметить, что под воздейст-
воздействием высоких доз ионизирующих из-
излучений популяции могут претерпеть
серьезные изменения генетической
структуры, влияющие на формирова-
формирование последующих поколений. При об-
обследовании населенных пунктов Ал-
Алтайского края, подвергшихся воздей-
воздействию высоких доз ионизирующих из-
излучений (от 0,97 до 2,43 Зв) в резуль-
результате ядерного взрыва 1949 г. на Семи-
Семипалатинском испытательном полигоне
показано, что в облученных группах
жителей наблюдается обеднение гено-
генофонда по сравнению с контролем
вследствие снижения генетического
разнообразия аллелей, выявляемых
путем электрофореза белков.
Мало изучена генетическая чувст-
чувствительность половых клеток челове-
человека и ранних эмбриональных этапов
развития. Учет генетического груза и
груза тератогенных изменений, реа-
реализуемых в период развития эмбрио-
эмбрионов человека, по мнению академика
Н.П.Дубинина, должен коренным об-
образом изменить в сторону увеличе-
увеличения оценку генетического риска об-
облучения человека в связи с аварией
на Чернобыльской АЭС и другими
радиационными ситуациями в раз-
различных странах. В этом направлении
важные результаты получены бело-
белорусскими генетиками при изучении
последствий Чернобыльской катаст-
катастрофы.
Сразу после аварии большое коли-
количество женщин стало обращаться в ме-
медицинские учреждения с требованием
прервать беременность, поскольку они
опасались за ее исход. Белорусский
НИИ наследственных и врожденных
заболеваний с целью выяснения врож-
врожденных генетических последствий
сравнил частоту пороков развития, вы-
выявленную у искусственно абортиро-
абортированных плодов до аварии на Черно-
Чернобыльской АЭС (в 1980-1985 гт. были
собраны данные по более чем 10000
случаев прерывания беременности на
сроках 5-12 недель), с той, которая на-
наблюдалась в наиболее загрязненных
радионуклидами районах. Общая час-
частота пороков развития у зародышей в
Минске до аварии составила 5,6%
(табл. 6.5). После аварии не зарегист-
зарегистрировано достоверного увеличения ча-
частоты пороков в Минске, в районах же
Гомельской и Могилевской областей,
подвергшихся наиболее интенсивному
загрязнению в результате аварии, час-
частота пороков развития в 1986-1992 гг.,
A0,53+1,56%) достоверно (Р<0,05)
превышала контрольные показатели.
В загрязненных радионуклидами рай-
районах увеличилась частота всех поро-
пороков, но в наибольшей степени — часто-
частота расщелин губы и неба, удвоение по-
почек и мочеточников, полидактилии и
дефектов нервной трубки.
В Белоруссии проведено также иссле-
исследование динамики частот врожденных
аномалий развития у новорожденных в
периоды до аварии A982-1985 гг.) и по-
после аварии A987-1992 гг.). Анализиро-
Анализировали врожденные пороки развития
(ВПР) строгого учета, т.е. те аномалии,
которые однозначно диагностируются
независимо от уровня подготовки врача
и технического обеспечения медицин-
172
Глава 6. МУТАЦИИ
Таблица 6.5. Частота пороков развития у искусственно
абортированных плодов в Белоруссии до и после аварии на ЧАЭС
Показатели
Количество иссле-
исследованных плодов
Частота пороков к
количеству исследо-
исследованных образцов
но обнаруженным
порокам,%
Достоверность разли-
различий по сравнению
с частотой в
г. Минске в 1986-1990 гг.
Контролируемые регионы
г. Минск
1980-1985 гг.
10168
5,6 ± 0,3
t
Р
B-е полугодие)
1986-1995 гг.
17477
4,7 ±0,2
2,6
<0,05
Зоны первоочередного отселения
B-е полугодие)
1986-1994 гг.*
1243
10,5 ± 1,6
4,7
<0,05
1989-1995
гг."
1352
5,2 ±0.9
0,4
>0,05
Всего
2595
7,4 ± 0,8
2,3
<0,05
* — Хойиикский, Брагинский, Наровлянский, Костюковичский, Климовичский, Славгородский, Чери-
ковский, Краснопольский районы;
** — Буда-Кошелевский и Быховский районы выделены в отдельную строку в связи с тем, что иссле-
исследования в этих районах начаты с 1989 г.
ского учреждения. Сюда входят анэн-
анэнцефалия, спиномозговые грыжи, рас-
расщелины губы и/или неба, полидакти-
полидактилия, редукционные пороки конечнос-
конечностей, атрофия пищевода и ануса, синд-
синдром Дауна и отдельные группы множе-
множественных врожденных пороков разви-
развития (суммарно эти пороки составляют
44-50% от всех манифестных форм
врожденных пороков развития, регист-
регистрируемых в роддомах Белоруссии).
Результаты проведенных исследова-
исследований за 10 лет представлены в таблице
6.6, в которой сопоставлены частоты
пороков строгого учета в трех зонах. В
первую зону входят 15 районов Брест-
Брестской, Гомельской и Могилевской обла-
областей с уровнем загрязнения по Cs-137
1-5 Ku/км^. Во вторую зону включе-
включены 17 районов Гомельской и Могилев-
Могилевской областей с уровнем загрязнения
по Cs-137 15 Ku/км^ и более. В качест-
качестве контроля выбраны незагрязненные
радионуклидами районы Белоруссии.
Количество рождений в первой и кон-
контрольной группах составило до 1986 г.,
соответственно, 22500 и 27100 после
Чернобыльской аварии — 20800 и
27420 в год. Как следует из таблицы,
частота пороков строгого учета возрос-
возросла во всех трех зонах, но особенно зна-
значительно в зонах наибольшего радио-
нуклидного загрязнения. Коэффици-
Коэффициент прироста составил для контроль-
контрольной зоны 1,2, для первой зоны — 1,3 и
для второй зоны — 1,8. Различия сум-
суммарных частот ВПР при сопоставле-
сопоставлении с доаварийным периодом по всем
трем зонам достоверны (Р«0,05). До-
Достоверно различаются между собой
также суммарные частоты за 1986-
1992 гг. по контрольной зоне
E,85±0,17) и по второй, наиболее за-
загрязненной зоне G,09±0,41).
Комбинированное воздействие
радиации и других факторов
окружающей среды
Эффекты ионизирующих излуче-
излучений могут быть сильно модифициро-
Индуцированный мутагенез 173
Таблица 6.6. Абсолютные числа и частоты (на 1000 рождений)
врожденных пороков развития строгого учета
в трех зонах Беларуссии A982—1992 гг.)
Год наблюдения
1982
1983
1984
1985
1982-1985
1987
1988
1989
1990
1991
1992
1987-1992
Коэффициент
прироста
1-5 Ки/км2
170
5,74 ± 0,44
123
3,96 ± 0,36
131
4,32 ± 0,38
135
4,61 ± 0,38
559
4,61 ±0,19
160
5,54 + 0,44
134
4,62 ± 0,40
173
6,32 ± 0,48
199
7,98 ± 0,56
135
5,65 ± 0,49
141
6,22 ± 0,52
942
6,01 ±0,20
1,3
Зоны загрязнения
> 15 Ки/км2
30
3,06 ± 0,56
37
3,58 +0,59
38
3,94 ± 0,64
46
4,76 ± 0,70
151
3,87 ±0,31
62
8,14 ±1,03
73
8,61 ± 1,00
51
6,50 +0,91
40
6,00 ± 0,95
29
4,88 ± 0,90
47
7,77+1,13
302
7,09 ±0,41
1,8
Контрольная группа
196
5,62 ±0,40
167
4,52 ±0,35
150
4,17+0,34
165
4,58 ± 0,36
678
4,72 ±0,18
223
5,94 т. 0,40
190
5,25 + 0,38
196
5,80 ±0,41
221
6,76 +. 0,45
181
5,52 ±0,41
175
5,89 + 0,44
1186
5,85 ±0,17
1,2
ваны естественными или антропоген-
антропогенными факторами среды. Вопросы ком-
комбинированного действия различных
факторов, несмотря на большое вни-
внимание к ним в последние годы, разра-
разработаны пока недостаточно. Вместе с
тем многие аспекты оценки риска об-
облучения человека либо объектов при-
природных сообществ непосредственно
соприкасаются с необходимостью учи-
учитывать то обстоятельство, что радиа-
радиационный мутагенез может быть силь-
сильно модифицирован в своем конечном
выражении в зависимости от темпера-
температуры, воздействия УФ-лучей, магнит-
магнитных и электромагнитных полей, ульт-
ультразвука, промышленной пыли, неорга-
неорганических и органических химических
соединений, антибиотиков и других
лекарственных веществ, канцерогенов,
гормонов, вирусов, бактериальных ин-
инфекций и т.д. Соответственно модифи-
модифицированы могут быть и дозовые зави-
зависимости, о которых шла речь в преды-
предыдущих разделах этой главы.
В настоящее время в окружающую
среду поступает более 4 млн. искусст-
искусственных химических соединений, и
каждый год к этому списку добавляет-
добавляется 300 тыс. новых соединений. Совре-
Современное общество повседневно исполь-
использует около 50 тыс. химикатов, не счи-
174
Глава 6. МУТАЦИИ
m
?
та
Ъ.
о
о
ТО
о.
1
г
A. 20 и более сигарет
Б. Менее 20 сигарет
B. Некурящие
Доза
Рис. 6.27. Смертность от рака органов дыхания как функция дозы облучения,
обусловленной Дочерними продуктами радиоактивного распада радона, для трех групп
рабочих урановых рудников - среди заядлых курильщиков, выкуривающих более
20 сигарет в день (кривая А), среди умеренных курильщиков, выкуривающих менее
20 сигарет в день (кривая Б), и среди некурящих (кривая В)
тая пестицидов, пищевых добавок и ле-
лекарственных препаратов. К категории
широко распространенных следует от-
отнести "энергетические соединения", та-
такие, как окислы азота, углерода, соеди-
соединения серы, ароматические углеводо-
углеводороды. Среди последних 3,4-бензопирен
(БП) часто используют как индикатор
уровня ароматических углеводородов с
канцерогенными свойствами.
Список веществ, которые могут
приводить к отрицательным эффектам
в сочетании с ионизирующими излу-
излучениями, можно пополнить соедине-
соединениями мышьяка, хрома, никеля, мине-
минеральной пылью. В частности, летучая
зола электростанций может обладать
канцерогенными свойствами, служить
в качестве носителя микроэлементов,
радионуклидов или полициклических
ароматических углеводородов.
В последнее время уделяется особое
внимание количественным подходам к
оценке комбинированного действия
радиации и других агентов окружаю-
окружающей среды, основанных на концепциях
аддитивности, синергизма, антагониз-
антагонизма, сенсибилизации и протекции.
Предложено выделить два класса ком-
комбинированных эффектов: случай, ког-
когда ионизирующее излучение и моди-
модифицирующий агент способны вызвать
наблюдаемый эффект, и случай, когда
только ионизирующее излучение мо-
может вызвать наблюдаемый эффект, но
его характер и степень могут модифи-
модифицироваться другим агентом, который
неактивен сам по себе. Соответственно
для первого класса можно выделить
три типа комбинированных взаимо-
взаимодействий. Если результат комбиниро-
комбинированного действия равен сумме эффек-
Индуцированный мутагенез
175
тов каждого из агентов, действующих
в отдельности, то имеет место адди-
аддитивность. В случае, когда результат
взаимодействия оказывается ниже,
чем ожидаемый эффект при независи-
независимом действии агентов, имеет место ан-
антагонизм. Третий вариант, когда ре-
результат комбинированного действия
превосходит сумму эффектов для
агентов, действующих независимо, на-
называют синергизмом.
Для второго класса комбинаций вы-
выделяют два варианта взаимодействия с
противоположными эффектами —
протекцию (защиту) и сенсибилиза-
сенсибилизацию (очувствление).
Естественно, что представленная
классификация не является абсолют-
абсолютной. Учитывая, что многие химические
соединения в зависимости от концент-
концентрации могут вызывать различные эф-
эффекты и тем самым переходить из од-
одного класса взаимодействия в другой,
можно ожидать что при одних дозах
этих факторов будет наблюдаться, на-
например, протекция, при других, более
высоких, — синергизм. Важен сам по
себе факт наличия различных дозовых
зависимостей для ионизирующих из-
излучений и модифицирующих агентов —
реальная картина взаимодействия та-
таких факторов трудно прогнозируема.
Это особенно важно в отношении низ-
низких уровней доз излучений и модифи-
модифицирующих естественных и антропо-
антропогенных факторов среды, оценка эф-
эффективности которых строится в ос-
основном экстраполяционно.
К сожалению, число детально ис-
исследованных примеров взаимодейст-
взаимодействия радиации и других факторов пока
явно ограниченно. Наиболее убеди-
убедительные доказательства взаимодейст-
взаимодействия радиации и других факторов были
получены при обследовании шахтеров
урановых рудников. Оказалось, что
курящие шахтеры урановых рудников
(подвергающиеся воздействию излу-
излучения за счет распада радона) заболе-
заболевают раком органов дыхания гораздо
чаще, чем некурящие (рис. 6.27). На-
Налицо явление синергизма при взаимо-
взаимодействии ионизирующих излучений и
канцерогенных продуктов табачного
дыма.
Вопросы для самоконтроля
1. Какие типы генных мутаций вам известны?
2. Какие изменения генетического материала
можно увидеть под световым микроскопом?
3. Чем отличаются аберрации хромосомного ти-
типа от хроматадных аберраций?
4. Чем отличаются генные мутации от геном-
геномных?
5. К какому виду мутаций относится полиплои-
полиплоидия? 6. Что такое мозаик?
7. Какой набор хромосом встречается при синд-
синдроме Дауна?
8. Чем отличаются менделевские наследствен-
наследственные болезни от мультифакториальных?
9. Что вы знаете об общем уровне спонтанных
мутаций у человека?
10. В чем сходство и различие спонтанных и ин-
индуцированных радиацией мутаций?
11 Какие хромосомные нарушения возникают
при действии ионизирующих излучений?
12. Какие типы генетических изменений позво-
позволяет изучать FISH-метод?
13. Что такое метод удваивающей дозы?
14. Какие примеры реального воздействия ио-
ионизирующих излучений на наследственность чело-
человека вам известна?
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
Популяции
Понуляционная генетика возникла
из попыток соединить дарвиновскую
теорию эволюции и менделевское уче-
учение о наследственности. По этой при-
причине популяционная генетика в пер-
первую очередь изучает наследственные
изменения, происходящие в ряду по-
поколений. Отдельно взятая особь имеет
неизменный на протяжении жизни ге-
генотип, поэтому она не может рассмат-
рассматриваться как элементарная эволюцио-
эволюционирующая единица. В качестве тако-
таковой выступает популяция — совокуп-
совокупность особей одного вида, скрещиваю-
скрещивающихся между собой и имеющих опре-
определенный ареал обитания. Эволюци-
Эволюционные изменения, происходящие в от-
отдельных популяциях, могут распрост-
распространяться по всему ареалу обитания ви-
вида, но не затрагивают популяции дру-
других видов.
Вид — это более высокая таксоно-
таксономическая единица, представленная со-
совокупностью популяций. В основе оп-
определения понятия «вид» лежит воз-
возможность скрещивания. К разным ви-
видам относятся особи, которые полно-
полностью генетически изолированы друг от
друга репродуктивными барьерами. С
этой точки зрения все человечество,
несмотря на межрасовые различия, от-
относится к одному биологическому ви-
виду Homo sapience. Всякая же этнически
однородная, компактно проживающая
группа людей может рассматриваться
как отдельная популяция.
Обычно говоря о популяции, мы
имеем в виду, что ее члены не только в
той или иной степени связаны родст-
родственными узами, но и в значительной
степени доступны друг другу как брач-
брачные партнеры. Так, вряд ли стоит рас-
рассматривать жителей Москвы и Варша-
Варшавы как единую популяцию. Это раз-
различные популяции, связанные незна-
незначительными миграционными потока-
потоками. Частичная репродуктивная изоля-
изоляция отдельных популяций, как прави-
правило, обусловлена их географической ра-
разобщенностью. В единой популяции
всевозможные варианты скрещиваний
должны быть примерно равновероят-
равновероятны. При полной случайности внутри-
популяционных скрещиваний говорят
о панмиксии. Введение этого специ-
специального термина оправдано исключи-
исключительной важностью случайности скре-
скрещиваний для построения теорий эво-
эволюции частот менделирующих генов.
Часто популяция, являясь панмик-
тической по отношению к одной груп-
группе локусов, не является таковой по от-
отношению к другой группе признаков.
Например, для человеческих популя-
популяций характерна положительная ассор-
тивность скрещиваний в отношении
цвета кожи (преимущественное вступ-
вступление в брак людей одной расы). Вме-
Вместе с тем, те же популяции можно счи-
считать панмиктическими по отношению
ко многим другим локусам, например,
определяющим группу крови брачных
партнеров.
Для панмиктических популяций
справедливы определенные соотноше-
соотношения, связывающие частоты аллельных
генов (закон Харди-Вайнберга). Более
того, предположение о случайности
скрещиваний позволяет рассматри-
рассматривать генотипы всех особей, образую-
образующих популяцию, как единую совокуп-
Популяции
177
ность генов, отвлекаясь при этом от их
носителей. Говорят, что панмиктичес-
кая популяция имеет единый гено-
генофонд. Генофонд диплоидной популя-
популяции, насчитывающей N особей, состо-
состоит из 2N гаплоидных геномов, то есть
содержит 2N генов каждого локуса и N
пар гомологичных хромосом.
Исключение составляют половые
хромосомы и локализованные на них
гены, которые представлены в одном
экземпляре.
Изменчивость
и генетический полиморфизм
Любая популяция обнаруживает
внешнюю или фенотипическую из-
изменчивость по большинству качест-
качественных и количественных признаков.
Популяции человека гетерогенны по
росту, пигментации кожи, чертам лица,
группам крови и многим другим при-
признакам. При этом не всегда ясно, какая
доля морфологической изменчивости
обусловлена генетическим разнообра-
разнообразием, а какая возникает за счет измен-
изменчивости внешней среды. Сегодня мы
знаем, что скрытая генетическая из-
изменчивость намного выше, чем можно
заключить из простых наблюдений
морфологической изменчивости. Од-
Однако представления о масштабах гене-
генетической изменчивости не раз подвер-
подвергались пересмотру. Каждый этап раз-
развития генетики сопровождался внед-
внедрением новых методов генетического
анализа, использование которых поз-
позволяло выявить новые пласты скры-
скрытой генетической изменчивости.
Под генетическим полиморфизмом
понимается устойчивое сосуществова-
сосуществование в популяции двух (или более) ге-
нотипически различающихся форм,
причем частота наиболее редкой фор-
формы все же достаточно велика, чтобы ее
поддержание можно было объяснить
мутационным давлением (спонтанным
мутационным процессом). Генетичес-
Генетический полиморфизм — это понятие, ко-
которое конкретизирует термин «генети-
«генетическая изменчивость». Как правило,
феномен полиморфизма связывают с
поддержанием нескольких аллельных
форм гена, что обнаруживается при-
присутствием в популяции гомо- и гетеро-
гетерозиготных генотипов по данному локу-
су. Если же все особи гомозиготны по
одному тому же аллелю, говорят, что
популяция мономорфна по данному
локусу. Соответственно, если в доста-
достаточно большой выборке из популяции
обнаруживается единственный ал-
лельный вариант, локус мономорфен.
В противном случае говорят, что локус
поддерживается в полиморфном со-
состоянии. Очевидные примеры поли-
полиморфизма в популяциях человека —
локусы, контролирующие иммунные
реакции или синтез ферментов.
Для количественной оценки генети-
генетической изменчивости природных по-
популяций используют два показателя:
1. Гетерозиготность — средняя час-
частота особей, гетерозиготных по неко-
некоторым локусам.
2. Полиморфность — средняя доля
полиморфных локусов. Следует под-
подчеркнуть, что конкретные значения
этих показателей зависят от методов
выявления генетической изменчиво-
изменчивости.
В период 1900 — 1930 гг., основным
методом генетического анализа был ме-
метод скрещиваний, в частности, близко-
близкородственные скрещивания, или инбри-
инбридинг. При инбридинге увеличивается
вероятность появления в потомстве ре-
рецессивных гомозигот, у которых прояв-
проявляются рецессивные аллели. Этим ме-
методом было показано, что практически
каждая особь несет несколько рецес-
рецессивных генов, которые фенотипически
178
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
не обнаруживают себя в гетерозигот-
гетерозиготном состоянии. Отсюда возникло пред-
представление, что генотип организмов со-
состоит в основном из гомозиготных ло-
кусов, и лишь редкие локусы гетерози-
гетерозиготны по неким, как правило, вредным
рецессивным аллелям. Случаи поли-
полиморфизма рассматривались, скорее, как
исключения из этого общего правила.
Ситуация изменилась в середине
60-х гг. после внедрения в исследова-
исследовательскую практику метода гель-элект-
гель-электрофореза белков. Суть его заключает-
заключается в разделении в нолиакриламидном
(или крахмальном) геле родственных
белков (изозимов), чьи электрофоре-
тические подвижности различаются
из-за мутационной замены отдельных
аминокислотных остатков. Использо-
Использование этого метода показало, что при-
примерно треть всех локусов, контроли-
контролирующих биохимические реакции, на-
находится в полиморфном состоянии.
При этом средняя гетерозиготность
колеблется от 7% у человека до 42% у
дрозофилы.
Однако изменчивость белков лишь
частично отражает изменчивость пер-
первичного генетического материала —
нуклеотидных последовательностей
ДНК. В конце 80-х гг., благодаря разви-
развитию методов секвенирования ДНК, по-
появилась возможность непосредствен-
непосредственной оценки полиморфизма на уровне
отдельных нуклеотидов. Эти исследова-
исследования подводят черту под давними спора-
спорами о масштабах генетической изменчи-
изменчивости популяций. По современным
представлениям, доля полиморфных
нуклеотидов составляет 1/1000. Учиты-
Учитывая, что обычный ген содержит от одной
до 10 тыс. нуклеотидных пар, получаем,
что почти 100% локусов находятся в по-
полиморфном состоянии, так что каждый
из нас отличается от соседа примерно
по 22 млн. нуклеотидов.
Закон Харди-Вайнберга
Распределение частот генотипов
в равновесной популяции
В любой популяции существует из-
изменчивость в отношении многих при-
признаков и определяющих их локусов.
Например, каждый человек может
иметь один из трех возможных геноти-
генотипов по группе крови MN:
1. Гомозиготный генотип ММ
2. Гетерозиготный генотип MN
3. Гомозиготный генотип NN
Чаще всего встречаются люди с ге-
гетерозиготным фенотипом MN, обла-
обладатели же гомозиготных генотипов
ММ или NN встречаются приблизи-
приблизительно в два раза реже. Вот точные
данные, полученные при обследова-
обследовании белого населения США:
Рмм= 0,292 (или 29,2%),
PMN = 0,496 (или 49,6%),
Рш = 0,212 (или 21,2%).
Здесь и далее мы будем обозначать
буквой Р частоту встречаемости соот-
соответствующего генотипа. Для других
популяций соотношение частот гено-
генотипов может быть совершенно иным,
но отнюдь не произвольным.
Например, вряд ли существует по-
популяция с распределением частот ге-
генотипов 20%, 70%, 10%. Дело в том, что
в популяциях со случайным скрещива-
скрещиванием для любого локуса характерна за-
закономерность:
MN
мм
MN
Действительно, рассматривая рас-
распределение людей по группам крови в
США, мы видим:
Pmn =
= 0,498 *
= 2^0,292*0,212
Это проявление так называемого за-
закона Харди — Вайнберга, согласно ко-
которому при случайном скрещивании
Закон Харди-Вайнберга
179
частоты генотипов неизменны в поко-
поколениях и удовлетворяют соотноше-
соотношению, имеющему вид пропорции:
Рмм : Рмх : Pnn = Р2 : 2pq : q2,
где р и q — частоты встречаемости
генов М и N в генофонде популяции.
Частоты генов (аллелей) можно оце-
оценить по частоте генотипов следующим
образом. Все особи с генотипом ММ
производят гаплоидные половые клет-
клетки, несущие ген М. Кроме них, такие
же половые клетки с вероятностью 1/2
производят гетерозиготные особи MN.
Поэтому частота гена М равна
Р- Рмм + |МЫ = О,292 + ^=0,54.
Частота же гена N равна
|M* = 0,212 + ^=0,46
Правильность вычислений можно
контролировать равенством
p + q-1.
Таковы частоты генов в текущем по-
поколении. При образовании потомства
любая яйцеклетка с вероятностью 0,54
несет ген М. Если скрещивания слу-
случайны, то она с вероятностью 0,54 сли-
сливается с гаметой типа М, и с вероятно-
вероятностью 0,46 с гаметой типа N. Таким об-
образом, частота генотипов ММ в следу-
следующем поколении равна
р2 = о,542 - о,292.
Точно так же частота потомков типа
NN равна
q2 = 0,462 = 0,212.
Гетерозиготы в потомстве образуют-
образуются в двух случаях: яйцеклетка типа М
оплодотворяется сперматозоидом ти-
типа N, либо наоборот. Это соответствует
сумме вероятностей
pq + qp - 2pq - 2» 0,54» 0,46 = 0,496
Обратите внимание, что для рас-
рассмотренной популяции частоты гено-
генотипов потомства в точности равны
частотам генотипов родителей. В
этой ситуации мы будем говорить,
что популяция находится в равнове-
равновесии, имея в виду неизменность частот
генотипов в последующих поколени-
поколениях. Популяция является равновес-
равновесной, если частоты генотипов ММ,
MN и NN находятся в соотношении
р2 : 2pq: q2. Действительно, в этом
случае частоты генов М и N равны,
соответственно
MN
1 NN о 2
а частоты генотипов в следующем
поколении по-прежнему находятся в
соотношении р2: 2pq : q2. На рисунке 1
приведена геометрическая интерпре-
интерпретация соотношений частот генов и ге-
генотипов в равновесной популяции.
Переход к равновесию
в неравновесной популяции
Что произойдет в последующих по-
поколениях с неравновесной популяци-
популяцией? Рассмотрим популяцию, которую
мы приводили в качестве примера «не-
«невозможной» (см. начало раздела).
Пусть в исходном поколении частоты
генотипов MM, MN и NN равны 20%,
70% и 10%, соответственно. Тогда час-
частоты генов М и N равны
р = 0,2+^ = 0,55;
q = 0,1 + Ц = 0,45.
180
Сперматозоиды
м
р = 0,54
N
q = 0,46
М
р = 0,54
ММ
р'= 0,292
ММ
pq = 0,248
N
q = 0,46
MN
pq = 0,248
¦
Рис. 7.1.
Диаграмма, показывающая взаимосвязь
частот генов и генотипов в соответствии
с законом Харди - Вайнберга для
равновесной популяции
Частоты особей с различными геноти-
генотипами в следующем поколении составят
Рмм " Р2 = 0,552 = 0,3025;
Pmn = 2pq = 2 «0,55 «0,45 - 0,495;
Рмм " q2 = 0-452 - 0,2025.
Мы видим, что частоты генотипов в
неравновесной популяции резко изме-
Глава7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
няются за одно поколение (при неиз-
неизменной частоте генов), в результате че-
чего популяция приходит в равновесие.
В дальнейшем частоты генотипов не
изменяются в поколениях (рис.7.2).
Подытоживая сказанное, можно за-
заключить, что менделевское наследова-
наследование само по себе не изменяет частоты
генов и в условиях случайного скре-
скрещивания за одно поколение приводит
популяцию в состояние равновесия.
Отметим, что закон Харди-Вайнбер-
га не является законом в том смысле, в
котором мы говорим о законах Ньюто-
Ньютона или Менделя. Скорее, это неслож-
несложная математическая теорема, которая
вытекает из предположения о случай-
случайности скрещиваний. Этот закон неод-
неоднократно открывался и переоткрывал-
переоткрывался в начале XX в. Помимо немецкого
врача В. Вайнберга и английского ма-
математика Г. Харди, его формулировали
В. Кастл, К. Пирсон и многие другие.
Закон Харди-Вайнберга легко обоб-
обобщается в случаях произвольного числа
аллельных генов. Заметим, что при
двух аллелях частоты генотипов вы-
вычисляются возведением в квадрат сум-
суммы частот генов, которая заведомо рав-
равна единице:
(P+qJ - P2+2pq+q2 - 1
-+¦ мм
-•-MN
-*- NN
Рис. 7.2. Динамика достижения
равновесия частот генотипов при
случайном скрещивании, В исходном
первом поколении популяция находится
в неравновесном состоянии. Равновесие
достигается за одно поколение, после
чего частоты генов не изменяются
Аналогичное соотношение справед-
справедливо для случая трех и большего числа
аллелей. Пусть частоты аллелей А^, А2
и A3 для некоторого локуса равны со-
соответственно р, q и г. Тогда частоты
всех возможных генотипов (AjA^,
А2А2, А3А3, A}A2, А4А3, ^2^3) мож'
но вычислить возведением в квадрат
суммы частот аллелей:
(р + q + гJ = р2 + q2 + Г2 + 2pq +
2pr + 2qr = 1
Этими соотношениями мы восполь-
воспользуемся при анализе полиморфизма в
Закон Харди-Вайнберга
181
популяциях человека по группам кро-
крови системы АВО.
Закон Харди — Вайнберга
для доминантных генов
В предыдущем разделе мы рассмат-
рассматривали ситуацию, когда все генотипы
имеют различное фенотипическое про-
проявление. Это позволяет непосредствен-
непосредственно оценить частоты всех возможных ге-
нотипических классов. Это возможно,
например, при исследованиях поли-
полиморфизма белков методом электрофо-
электрофореза. Однако во многих важных случа-
случаях невозможно отличить гетерозиготы
от гомозигот из-за доминирования од-
одних аллельных генов над другими. В
результате частоты генов нельзя оце-
оценить непосредственно по частоте гено-
генотипов, так что единственной возмож-
возможностью становится применение закона
Харди-Вайнберга. Рассмотрим пример.
Альбинизм у человека обусловлен
редким рецессивным геном, который
мы обозначим как а. Доминирующий
аллель нормальной пигментации есте-
естественно обозначить как А. Тогда гено-
генотип альбиносов будет аа, генотипы
нормально пигментированных — АА
либо аа (гетерозигота неотличима от
гомозиготы). Поданным 1965 г. в евро-
европейских странах на каждые 20 000 но-
новорожденных приходится один случай
появления ребенка — альбиноса. Это
означает, что частота рецессивных го-
гомозигот равна Раа~ 5» 10. Пусть q —
частота гена а. По закону Харди —
Вайнберга в равновесном случае име-
имеем Раа = q2, откуда
q = VPl= V5» 10-5 = 0,007.
Частота нормальных генов равна р =
1 __ а = 1 _ о,ОО7 - 0,993. Теперь мож-
можно вычислить неизвестное соотноше-
соотношение гомо- и гетерозигот среди феноти-
пически нормальных людей.
аа
0,9932 = 0,986;
РАа
0,993 • 0,007 = 0,014.
Обращает на себя внимание, что до-
доля гетерозиготных носителей гена аль-
альбинизма не так уж низка и составляет
1,4% от общей численности популя-
популяции. Это проявление общего правила,
согласно которому редкие аллели при-
присутствуют в популяции главным обра-
образом в гетерозиготном, а не в гомозигот-
гомозиготном состоянии. Это положение под-
подтверждается и другим примером: час-
частота людей, страдающих алькаптону-
рией, равна 1 на 1 млн., в то время как
частота носителей этого гена в гетеро-
гетерозиготном состоянии равна 0,002 (при-
(приблизительно в 1000 раз больше).
Яркий пример генетической измен-
изменчивости — система групп крови АВО.
Полиморфизм по этому признаку был
выявлен в начале нашего века при изу-
изучении реакции агглютинации. Анти-
Антигенные группы крови контролируются
тремя аллельными генами 1А, 1В и 1°.
Обозначим соответствующие аллель-
ные частоты как р, q и г. Гены 1А и 1В ко-
доминантны по отношению друг к дру-
другу, но полностью доминируют над ге-
геном 1°. В результате такого взаимодей-
ствия(см. Табл. 1) шесть возможных
генотипов оказываются распределен-
распределенными по четырем фенотипическим
классам (группам крови) со следую-
следующими частотами.
Таблица 1. Генетическая
изменчивость в системе групп
крови АВО
Группа крови
1(О)
II (А)
П(В)
IV (АВ)
Генотипы
ОО
АА;А0
ВВ;В0
АВ
Равновесные
частоты
фенотипов
р2 + 2рг
q2 + 2qr
2pq
182
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
Предположим, что в некоторой по-
популяции наблюдаются следующие час-
частоты групп крови:
А = 0,45
В =0,13
АВ - 0,06
О = 0,36
Рассчитаем частоты аллелей, пред-
предполагая популяцию равновесной. Так
как частота группы О равна г2, частота
гена 1° равна г = V0.36 = 0,6. Далее за-
заметим, что суммарная частота групп О
и В равна г2 + (q2 + 2qr) = (r + qJ = 0,36
+ 0,13 = 0,49. Откуда г + q = Vo^49 = 0,7.
Таким образом, частота гена 1В равна
q = 0,7 — 0,6 = 0,1. Наконец, частота ал-
леля 1А равна р - 1 — (г + q) - 1 — 0,7 =-
0,3.
Встречаемость гена 1Ь резко разли-
различается в популяциях коренного насе-
населения из разных регионов мира: от
0,02 (американские индейцы, абори-
аборигены Австралии) до 0,24 (жители по-
полуострова Индостан). Существует
предположение, что это связано с по-
повышенной устойчивостью людей с оп-
определенными группами крови к тяже-
тяжелым инфекционным заболеваниям,
характерным для конкретных регио-
регионов.
Условия выполнения
закона Харда — Вайнберга
При всей своей универсальности за-
закон Харди — Вайнберга выполняется
далеко не во всех ситуациях. Различ-
Различные факторы эволюции могут откло-
отклонять популяцию от положения равно-
равновесия и нарушать соотношение гено-
типических частот. Ниже мы перечис-
перечислим условия, которые необходимы для
строго соблюдения закона Харди —
Вайнберга, а в последующих разделах
подробно обсудим каждое из них.
1. Панмиксия, или случайность
скрещиваний. Это самое важное ус-
условие для выполнения закона Хар-
Харди — Вайнберга. Любое предпочтение
при скрещиваниях существенно ис-
искажают соотношения Харди — Вайн-
Вайнберга.
2. Отсутствие отбора. Имеется в ви-
виду, что жизнеспособность и плодови-
плодовитость особей не зависят от их генотипа.
Это требование обеспечивает равную
значимость вклада каждого генотипа в
следующее поколение.
3. Отсутствие мутационного про-
процесса, приводящего к возникновению
новых аллельных вариантов.
4. Отсутствие миграций между
различными независимыми популя-
популяциями.
5. Большая численность популя-
популяции. В популяциях малой численности
возможен так называемый случайный
дрейф генов, который приводит к
флуктуациям частот генов вплоть до
их полного исчезновения.
Естественный отбор
Приспособленность генотипов
и виды отбора
«Естественный отбор — это пережи-
переживание наиболее приспособленных инди-
индивидуумов». Развивая это определение
Ч. Дарвина, можно сказать, что естест-
естественный отбор — это дифференциальное
воспроизведение различных генотипи-
ческих вариантов. Отбор является важ-
важнейшим направляющим фактором эво-
эволюции популяций, выражающим неко-
некоторый интегральный итог взаимодейст-
взаимодействия особей или их групп с окружением.
Количественной мерой интенсивности
процессов отбора может служить отно-
относительная приспособленность, которая
является мерой вклада каждого геноти-
генотипа в следующее поколение.
Понятие приспособленности вклю-
включает в себя:
Естественный отбор
183
1. Жизнеспособность, то есть веро-
вероятность выживания до репродуктив-
репродуктивного периода.
2. Плодовитость, то есть количество
произведенных потомков.
Упрощая ситуацию, можно сказать,
что приспособленность пропорцио-
пропорциональна произведению выживаемости
на плодовитость. Естественный отбор
оценивает лишь суммарную или об-
общую приспособленность, но не ее от-
отдельные компоненты.
При вычислениях приспособлен-
приспособленность принято обозначать буквой w.
По определению она — величина отно-
относительная.
Обычно w наиболее приспособлен-
приспособленного генотипа принимается за едини-
единицу. Приспособленности остальных ге-
генотипов задаются формулой:
w = 1 — s,
где s — коэффициент отбора, опре-
определяющий скорость уменьшения час-
частоты того или иного генотипа.
Рассмотрим несколько случаев, в
которых определение приспособлен-
приспособленности достаточно очевидно.
1. Болезнь Тэя — Сакса, вызывае-
вызываемая накоплением в тканях нервной
системы человека некоторых липидов
(ганглиозидов), приводит к умствен-
умственной отсталости, слепоте и ранней
смерти. Приспособленность индиви-
индивидуумов с этой болезнью равна нулю
(коэффициент отбора равен едини-
единице), поскольку они умирают в раннем
возрасте.
2. Ахондропластические карлики
оставляют в среднем в 5 раз меньше
потомства, чем здоровые люди. В этом
случае приспособленность равна w =
1/5 = 0,2, а коэффициент отбора s = 1 —
0,2 - 0,8.
В случае полиморфизма популяции
но некоторому локусу, мы имеем дело
с тремя генотипами, приспособленнос-
приспособленности которых в общем случае различны:
Генотипы:
Приспособленности:
АА Аа аа
wj W2 W3
Очевидно, что если аллель А доми-
доминирует, то следует положить wj = W2-
Если при этом рецессивная аллель а
является летальной, то W3 = 0, и
W1=W2= 1-
Динамика частот генотипов в по-
последующих поколениях будет цели-
целиком зависеть от соотношения между
присиособленностями генотипов. В
зависимости от того, какой генотип
обладает максимальной приспособ-
приспособленностью, различают три варианта
отбора:
1. Направленный: wl > w2 > w3
Это отбор против гена а во всех его
проявлениях. Результатом будет по-
постепенное вытеснение (элиминация)
аллеля а из популяции.
2. Стабилизирующий: wl < w2 > W3.
В этой ситуации гетерозиготы облада-
обладают селективным преимуществом, что
приводит к поддержанию устойчивого
полиморфизма в популяции.
3. Дизрунтивный: wl > w2 < w3.
Для этого случая характерно вытес-
вытеснение одного из аллелей в зависимос-
зависимости от первоначального соотношения
частот генов.
Ниже мы дадим количественное
описание наиболее важных случаев
отбора в панмиктических популяциях.
Отбор против
рецессивных деталей
Предельным случаем отбора против
рецессивных аллелей является ситуа-
ситуация, когда рецессивные гомозиготы
обладают нулевой приспособленнос-
приспособленностью, то есть погибают до достижения
половозрелости или стерильны. Изве-
184
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
Таблица 2. Изменения частот
генотипов за одно поколение
отбора против рецессивных
гомозигот
Приспосо-
Приспособленность
Частота геноти-
генотипа в исходном
поколении
Вклад генотипа
в следующее
поколение
Частота генотипа
в следующем
поколении
Генотип
АА
I
Р2
Р2
p/0+q)
ПАа
1
2pq
2pq
2q/(l+q)
Гаа
0
q2
0
0
Всего
1
P(l+q)
1
стным примером болезни, обусловлен-
обусловленной рецессивным летальным геном,
может служить фенилкетонурия.
Предположим, что в некотором по-
поколении в равновесной популяции ча-
частота летального рецессивного гена а
равна q. Тогда частота нормального
доминирующего аллеля равна р = 1 —
q. Основные этапы дальнейших рас-
рассуждений представлены в Таблице 2.
В исходном поколении частоты гено-
генотипов соответствуют распределению
Харди — Вайнберга. В следующей
строке приведены частоты генотипов,
умноженные на соответствующие
приспособленности. Тем самым мы
вычисляем относительные вклады
каждого генотипа в следующее поко-
поколение. Эти вклады пропорциональны
частотам генотипов после отбора. Для
того, чтобы они стали частотами гено-
генотипов, их нужно «нормировать», то
есть разделить каждый из них на об-
общую сумму вкладов, равную р2 + 2pq =
р(р + 2q) = рA + q). Результат этой
операции приведен в последней стро-
строке таблицы.
Итак, в следующем поколении час-
частота генотипов Аа равна 2q/(l+q), a
частота самого летального гена равна
половине этой величины, то есть
q/(l+q). Следовательно, за одно поко-
поколение частота рецессивной летали из-
изменяется по формуле:
Обозначим частоты рецессивных
генов в последовательности поколе-
поколений как q0 q,, q2,..., qt. Тогда, если в ис-
исходном (нулевом) поколении частота
деталей равна q0, то в первом поколе-
поколении
„ = 40
M1 1+qo '
во втором поколении —
И Т.Д.
l+qo/(l+qo)
Легко проверить, что эта закономер-
закономерность соблюдается и далее: в поколе-
поколении под номером t
Qt=l+tqo-
Полученная формула точно описы-
описывает динамику снижения частоты ре-
рецессивных деталей в поколениях за
счет отбора. Происходит оно очень
медленно. Вычислим число поколе-
поколений, за которое частота рецессивных
деталей уменьшается вдвое. Имеем
Таким образом, редкая рецессив-
рецессивная деталь, встречающаяся с частотой
10'3 лишь через 1000 поколений бу-
будет встречаться вдвое реже. Если ре-
рецессивная гомозигота выживает, но
имеет пониженную приспособлен-
приспособленность, то скорость выведения из по-
популяции рецессивного гена будет еще
ниже.
Естественный отбор
185
Таблица 3. Изменение частот генотипов
за однопоколение отбора в пользу гетерозигот
Генотип
Всего
Приспособленность
Частота генотипа в исходном поколении
Вклад генотипа в следующее поколение
Частота генотипа в следующем поколении
АА
1-s
Р2
p2(l-s)
p2(l-s)/W
Аа
1
2pq
2pq
2pq/W
Аа
1-t
Я2
q2(l-t)
q2(l-t)/W
1
W
1
Этот вывод полностью перечеркива-
перечеркивает любые евгенические программы
очищения генофонда от вредных ге-
нов.В частности, принудительная сте-
стерилизация лиц, страдающих от наслед-
наследственных заболеваний, абсолютно не
эффективна. Если подобную опера-
операцию проводить на протяжении, ска-
скажем, 20 поколений, то частота вредно-
вредного рецессивного гена уменьшится от
0,001 до 0,001/( 1 +20 • 0,001 )=0,00098.
Но это частоты рецессивных генов.
Доля заболевших равна частоте рецес-
рецессивных гомозигот, то есть квадрату ча-
частоты вредного гена. Таким образом,
заболеваемость уменьшится от 10 6
до 0.000982* 0,96» 10 (не более чем
на 4%).
Отбор в пользу гетерозигот
Отбор в пользу гетерозигот, когда
обе гомозиготы имеют пониженную по
сравнению с гетерозиготой приспособ-
приспособленность, часто называют гетерозисом,
или сверхдоминированием. Этот тип
отбора существенно отличается от рас-
рассмотренного выше направленного от-
отбора: сверхдоминирования приводит к
созданию устойчивого полиморфного
равновесия.
Рассмотрим изменения генотиииче-
ских частот за одно поколение отбора
при сверхдоминировании. Все рассуж-
рассуждения представлены в таблице 3.
В первых двух строчках приведены
приспособленности и частоты геноти-
генотипов в некотором поколении. Вклад в
следующее поколение равен произве-
произведению частоты на приспособленность
(третья строка). Сумма вкладов всех
генотипов называется средней при-
приспособленностью и равна
W = p2(l -s) + 2pq + q2(l-t).
Последняя строка таблицы соответ-
соответствует частотам генотипов в следую-
следующем поколении. Вычислим изменение
частоты аллеля за поколение, опустив
часть простых алгебраических выкла-
выкладок.
1
q-
= q - (.^аа+у^Аа) =
q2(l-t)+pq _ pq(sp - tq)
W
W
Популяция приходит в равновесие,
когда частоты аллелей перестают изме-
изменяться. Тогда при достижении равнове-
равновесия Aq = 0, откуда sp = tq, или s( I — q) =
tq. Окончательное выражение для рав-
равновесной частоты аллеля а имеет вид
s
м t+s •
При этом для аллеля А имеем
s
p"t+s '
В отличие от равновесия в нейтраль-
нейтральной популяции (в отсутствии отбора),
равновесие при сверхдоминировании
является устойчивым. Это означает,
186
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
что при отклонении генных частот от
равновесного значения популяция воз-
возвращается к тому же равновесному
уровню. Напомним, что нейтральная
популяция при отклонении от положе-
положения равновесия за одно поколение пе-
переходит в новое равновесное состояние
(с другими частотами генотипов).
Детально изученным примером
сверхдоминирования в популяциях
человека служит сериовидноклеточ-
ная анемия — болезнь, широко распро-
распространенная в некоторых странах Аф-
Африки и Азии. Анемия обусловлена ано-
аномальным строением гемоглобина
(форма S) и развивается у людей гомо-
гомозиготных по аллелю Hbs. Нормальный
гемоглобин вырабатывается в присут-
присутствии аллеля НЬА в гомо- или гетеро-
гетерозиготном состоянии. Большинство лю-
людей с генотипом HbAHbs погибает до
достижения половозрелости, так что
приспособленность этого генотипа
близка к нулю. Несмотря на это, часто-
частота аллеля достигает довольно высоких
значений в ряде регионов, особенно в
районах распространения малярийно-
малярийного плазмодия. Причина этого состоит в
том, что гетерозиготы НЬЛНЬЛ более
устойчивы к малярии, чем гомозиготы
НЬАНЬА, то есть обладают селектив-
селективным преимуществом по сравнению с
обеими гомозиготами, у которых
смертность от анемии или от малярии
выше, чем у гетерозигот.
Данные, полученные при обследова-
обследовании населения Нигерии, показывают,
что в малярийных районах отбор про-
против гомозигот НЬАНЬА равен s = 0,12, а
против гомозигот HbsHbs : t = 0,87.
Согласно изложенной выше теории
равновесная, частота гена НЬ в этом
случае равна
s 0,12
(s + t) @,12+0,87)
= 0,121,
что весьма близко к фактически на-
наблюдаемой частоте.
Мутации
Мутационное давление из-за низ-
низкой скорости процесса спонтанного
мутирования крайне слабо влияет на
распределение нонуляционных час-
частот аллелей. Под скоростью или час-
частотой мутирования понимается доля
гамет, в которых произошли измене-
изменения данного гена. Для высших орга-
организмов эта величина не превышает
Ю-6 — Ю-5 на локус за поколение. Эво-
Эволюционное значение мутационного
процесса заключается в создании раз-
разнообразия аллелей за счет появлении
новых генов. Однако почти все вновь
возникающие мутантные гены селек-
селективно отрицательны, например, они
обуславливают различные заболева-
заболевания в гомо- или гетерозиготном со-
состоянии.
Необходимо различать три тесно
связанные величины:
1. Скорость мутирования
2. Частота встречаемости мутантно-
го гена в популяции
3. Частота заболеваний, вызванных
мутантным геном.
Первая из них определяется процес-
процессами на клеточном и организменном
уровне. Вторая величина — это равно-
равновесный уровень популяционной часто-
частоты, который пропорционален скорости
мутирования и обратно пропорциона-
пропорционален интенсивности отбора против му-
тантных генов. Третья — определятся
популяционной частотой мутантного
гена и степенью его рецессивности по
отношению к нормальному аллелю.
Между всеми этими величинами су-
существует простая связь, которую мы
обсудим в данном разделе.
Рассмотрим сначала доминантные
летальные мутации. Особи, несущие
Мутации
187
такие гены, не способны к размноже-
размножению, и, следовательно, эти аллели бу-
будут присутствовать в генотипе лишь
тех организмов, у которых они возник-
возникли в результате мутации в данном по-
поколении. Если ц — скорость мутирова-
мутирования, ар — частота мутантного гена, то
предыдущее утверждение можно вы-
выразить равенством: р = ц, то есть рав-
равновесная частота доминантных ле-
летальных мутаций равна скорости му-
мутирования. Однако, частота заболева-
заболеваний при этом будет вдвое выше, так
как заболевания проявляются у всех
гомо- и гетерозигот по мутантному ал-
лелю, что составляет
В качестве доминантных мутаций
можно рассматривать некоторые хро-
хромосомные перестройки. Таковой явля-
является трисомия, вызывающая синдром
Дауна. Так как больные с синдромом
Дауна не оставляют потомства, соот-
соответствующую мутацию можно считать
доминантной леталью. Синдром Дауна
возникает с частотой 1 на 700 ново-
новорожденных, и, значит, мутабильность
для трисомии равна 1/1400.
В случае рецессивных деталей ситу-
ситуация несколько сложнее. Мутацион-
Мутационный процесс постоянно, с малой часто-
частотой ц, индуцирует мутантные аллели,
которые могут сохраняться в популя-
популяции в гетерозиготном состоянии, От-
Отбор элиминирует эти гены лишь при
образовании гомозигот. В результате
частота рецессивных мутаций быстро
достигает равновесного значения, ко-
которое можно оценить с помощью сле-
следующих рассуждений. За одно поколе-
поколение отбора частота рецессивного ле-
летального гена изменяется по правилу:
q
q~* "l+q
Поэтому изменение частоты за одно
поколение составляет
q q2
При малых q снижение частоты за
счет отбора приблизительно равно
Aq * q2. В то же время за счет мутаци-
мутационного процесса происходит увеличе-
увеличение частоты мутаций на величину ц за
поколение. Для поддержания равнове-
равновесия прибыль и убыль частоты должны
быть равны друг другу. Отсюда полу-
получаем равенство q2 = ц, а значит, равно-
равновесный уровень мутантных рецессив-
рецессивных генов равен
q = VjoT
При этом частота соответствующего
заболевания равна частоте мутантных
гомозигот, то есть по-прежнему равна ц.
Частота появления новорожденных
с фенилкетонурией, обусловленной
рецессивным геном, составляет 4 на
100 000, то есть q2= 4» 10. Поэтому ча-
частота этого гена в популяции человека
равна q = V4-105 = 6,3-103. Частота
соответствующих гетерозигот равна
2pq « 2q = 2-6,3-10-з = 1,26-KR Та-
Таким образом, в среднем около 13 чело-
человек из тысячи являются носителями
этого аллеля, хотя частота индивидуу-
индивидуумов, страдающих фенилкетонурией,
составляют всего 4 на 100000.
Приведенные оценки относятся к
случаю полной летальности (или сте-
стерильности) мутантных генотипов. В
некоторых случаях бывает необходи-
необходимо вычислить равновесную частоту
мутаций при неполной элиминации
особей, несущих мутантный ген.
Пусть приспособленность гомозигот
по мутантному гену равна 1-s. Как и
ранее, обозначим буквой ц частоту му-
мутационных превращений нормального
аллеля + в мутантный аллель т. Тогда
188
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
Таблица 4. Равновесные
частоты мутаций
и обусловленных ими заболеваний
Приспособлен-
Приспособленности генотипов
++ +m mm
Скорость
мутирования
+ -ип
Равновесная
частота
аллеля m
Частота
заболеваний
Рецессивные
мутации
1 : 1: 1-s
И
s
Доминант-
Доминантные мутации
1 : 1-s: 1-s
Ц
Ц.
S
2М
S
равновесные частоты мутантного ал-
аллеля определяются формулами, кото-
которые представлены в таблице 4.
Мы опускаем вывод этих формул,
отсылая заинтересованного читателя
к классическому руководству Ч. Ли
"Введение в популяционную генети-
ку".
Приведем один пример из генетики
человека, использующий представлен-
представленные формулы. Ахондроплазия — это
тяжелое заболевание, обусловленное
доминантным аллелем. Частота мута-
мутаций, вызывающих ахондроплазию, со-
составляет 5* 108. Число детей у боль-
больных ахондроплазией в среднем в пять
раз меньше по сравнению со здоровы-
здоровыми людьми. Таким образом,
l08
Равновесная частота мутантного ге-
гена равна
_М__5»10-5
р s 0,8
-6,25 • 10-5.
Частота заболевших при этом со-
составляет 2р = 2«6,25« 10-5 = 1,25» 1(И,
то есть 125 больных на 1 млн. ново-
новорожденных.
Генетический дрейф
Случайным генетическим дрейфом,
или просто, дрейфом генов называет-
называется изменение частот аллелей в ряду
поколений, обусловленное случайны-
случайными причинами. Интенсивность этих
изменений зависит в первую очередь
от численности популяции, точнее, от
числа участвующих в размножении
особей.
Чтобы «прочувствовать» механизм
дрейфа генов, следует мысленно обра-
обратиться к процессу подбрасывания мо-
монетки. Сколько раз выпадает «орел»
при 100 подбрасываниях монеты?
Большинство даст правильный от-
ответ — приблизительно 50. Но далеко
не все понимают, что вероятность вы-
выпадения ровно 50-ти «орлов» довольно
мала — около 7,9%. Хотя с вероятнос-
вероятностью, превышающей 95%, их число бу-
будет попадать в интервал от 40 до 60.
Таким образом, доля «орлов» при
100 подбрасываниях, скорее всего, бу-
будет заметно отличаться от */2> и ока"
жется равной 0,43 или, скажем, 0,56.
Теперь представим себе, что монетка
подбрасывается 1000 раз. В этом слу-
случае вероятность выпадения 430 или
560 «орлов» очень мала. Их доля будет
гораздо ближе к V2> чем Ю0 при под-
подбрасываниях.
Суть этого примера заключается в
том, что, чем больше выборка, тем бли-
ближе соответствие между теоретически
ожидаемой ( V2) и реально наблюдае-
наблюдаемой частотой. В популяциях мы стал-
сталкиваемся с тем же явлением: при не-
небольших численностях теоретически
ожидаемая частота (то есть частота ал-
аллеля в родительском поколении) мо-
может существенно отличаться от реаль-
реально наблюдаемой (то есть от частоты
аллеля у потомства).
Однако между бросанием монеты и
дрейфом гена имеется важное разли-
Генетический дрейф
189
1
0.8
0.6
0.4
0.2
0
А
/ \
1 N=25
N-100 У
/V /
№ ЛГ
' 1
¦
:
N-23OO
•
0
20
40
60
80
Поколения
100
120
140
Рис 7.3. Динамика случайных изменений частоты гена за счет дрейфа
при различной численности популяций B5, 100 и 2500 особей).
Начальная частота гена во всех экспериментах равна 0,5
чие. При бросании монетки вероят-
вероятность выпадения «орла» остается рав-
равной V2 на протяжении всей серии
подбрасываний. Для популяций эта
вероятность изменяется в каждом по-
поколении: частота аллеля в данном по-
поколении представляет собой вероят-
вероятность появления этого аллеля в сле-
следующем поколении. Если, например,
частота аллеля изменилась от 0,5 до
0,6, то вероятность того, что этот ал-
аллель появится в следующем поколе-
поколении равна 0,6. Таким образом, измене-
изменения частот аллелей за счет дрейфа на-
накапливаются в поколениях. Ясно, что
рано или поздно это приведет к тому,
что частота аллеля достигнет значе-
значения, равного нулю (аллель исчезнет)
или единице (исчезнет альтернатив-
альтернативный аллель). В последнем случае го-
говорят о фиксации аллеля. На этом
процесс завершается, так как даль-
дальнейшие изменения частоты аллеля
невозможны.
Случайный дрейф гена легко ими-
имитировать с помощью компьютера,
(рис. 7.3). На нем показаны три слу-
случайные реализации дрейфа гена при
различных численностях популяции.
Из рисунка видно, что при очень ма-
малой численности B5 особей) уже че-
через 40 поколений аллель элиминиру-
элиминируется из популяции по случайным при-
причинам. В другой случайной реализа-
реализации с вероятностью V2 можно наблю-
наблюдать противоположную картину: час-
частота аллеля возрастает до единицы.
Если численность популяции довести
до 100 особей, то для фиксации аллеля
понадобится уже 115 поколений. В по-
популяции большой численности B500
особей) частота аллеля существенно
не изменяется на протяжении 150 по-
поколений. Но это не означает, что в
190
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
этом случае полиморфизм будет под-
поддерживаться сколь угодно долго.
Фиксация аллеля с вероятностью еди-
единица происходит в любых конечных
популяциях при отсутствии источни-
источников новых аллелей (мутации и мигра-
миграции). Однако для это понадобится
число поколений, сравнимое по вели-
величине с численностью популяции.
Влияние генетического дрейфа
можно наблюдать и в изолированных
малочисленных популяциях человека.
При обследовании членов закрытой
секты баптистов в штате Пенсильва-
Пенсильвания (США), основанной в XVIII в.
выходцами из Германии, обнаружено,
что частоты генов групп крови АВО у
членов секты отличаются от таковых у
американцев немецкого происхожде-
происхождения. Особенно разительны эти разли-
различия по частоте гена 1В : 2,5% у членов
секты и 12% у американцев немецкого
происхождения.
В заключение перечислим основные
черты генетического дрейфа.
1. Дрейф приводит к случайным ко-
колебаниям частот аллелей, которые осо-
особенно заметны в малых популяциях.
2. Дрейф неуклонно снижает гене-
генетическую изменчивость популяций,
увеличивая частоту гомозигот. Окон-
Окончательным итогом действия генетичес-
генетического дрейфа является элиминация ли-
либо фиксация аллеля.
3. Число поколений, необходимых
для элиминации (или фиксации) алле-
аллеля за счет дрейфа, сопоставимо по ве-
величине с численностью популяции.
Близкородственные браки
Близкородственные скрещивания,
или инбридинг — одна из форм ассор-
тивности при образовании брачных
пар. При инбридинге скрещивания
между родственными особями проис-
происходят чаще, чем можно было бы ожи-
ожидать, предполагая случайность скрещи-
скрещиваний. Поскольку родственные особи в
генетическом отношении более сходны
между собой, чем не состоящие в род-
родстве, инбридинг ведет к повышению
частоты гомозигот и снижению часто-
частоты гетерозигот по сравнению с ожидае-
ожидаемой но закону Харди — Вайнберга.
Количественной мерой последствий
инбридинга служит коэффициент ин-
инбридинга, который определяется как
вероятность того, что у какой-либо осо-
особи оба гена в данном локусе принадле-
принадлежали одному из предков в каком-то из
предшествовавших поколений. Пусть
F — коэффициент инбридинга. Тогда
можно показать, что частоты генотипов
в инбредной популяции имеют вид:
Раа = Р2 + PQF, Рда - 2pq( 1 - F), Р^ -
q2 + pqF.
Таким образом, доля гетерозигот за
счет инбридинга уменьшается по срав-
сравнению с распределением Харди —
Вайнберга в 1/A-F) раз. При отсутст-
отсутствии инбридинга F = 0, и частоты гено-
генотипов удовлетворяют закону Харди —
Вайнберга.
Вычисление коэффициента инбри-
инбридинга в конкретных ситуациях связано
с анализом родословных и проведени-
проведением сложных комбинаторых рассужде-
рассуждений, поэтому мы будем пользоваться
готовой таблицей коэффициентов ин-
инбридинга в зависимости от системы
скрещиваний (см. Табл. 5).
Инбридинг обычно приводит к по-
понижению приспособленности потом-
потомства вследствие повышения степени
гомозиготности по вредным рецессив-
рецессивным генам. Это явление принято назы-
называть инбредной депрессией.
Рассмотрим рецессивный леталь-
летальный аллель, возникающий мутацион-
мутационным путем с частотой ц = Ю-5. Равно-
Генетический груз популяции
191
весная частота этого гена равна q = VpT
= 0,0032. Заболеваемость равна часто-
частоте гомозигот, то есть составляет q2 =
105. Теперь представим, что в популя-
популяции поддерживается коэффициент
инбридинга, равный F - 1/16, что со-
соответствует скрещиванию двоюрод-
двоюродных сибсов. Тогда частота гомозигот
по рецессивной летали будет равна
q2 + pqF - 10-5 + A-0,0032)»0,0032 «i*
2-Ю'4.
Таким образом, при данной степени
инбридинга частота летальных гомо-
гомозигот возрастает в 20 раз. При скрещи-
скрещивании сибсов (F - V4) вероятность
появления летальных гомозигот воз-
возрастает примерно в 80 раз.
В случае вредных, но не летальных
рецессивных аллелей при коэффици-
коэффициенте отбора s = 0,1 равновесная частота
аллеля равна
Тогда при скрещивании двоюрод-
двоюродных сибсов частота рецессивных гомо-
гомозигот будет составлять
q2 + pqF
7» 10",
+ A -0,01)»0,01<
1
Тб
то есть примерно в семь раз выше,
чем при случайном скрещивании.
Известно, что в потомстве от браков
между двоюродными братьями и сест-
сестрами частота большинства рецессивно
наследуемых дефектов (ихтиоз, альби-
альбинизм, фенилкенотурия) в 5 — 10 раз
выше, чем в потомстве от неродствен-
неродственных браков.
Генетический груз популяции
Как уже было показано в предыду-
предыдущей главе, приблизительно у 70% лю-
Таблица 5. Значения
коэффициентов инбридинга
при разных типах скрещивания
Типы скрещиваний
Самооплодотворение
Сибсы (братья и сестры)
Дядя х племянница, тетка х племянник
Двоюродные сибсы
Двоюродные дядья х племянницы
Троюродные сибсы
Четвероюродные сибсы
F
v4
1/8
1/16
1/32
1/64
1/256
дей в течение жизни проявляются те
или иные наследственные аномалии,
приводящие к серьезным последствиям
для здоровья. Исходя из этого можно
заключить, что популяции человека су-
существенно отягощены различными му-
мутациями, которые либо проявляются
доминантно, либо выщепляются в каж-
каждом поколении благодаря появлению
гомозигот. Однако в большей своей ча-
части, подобно айсбергу, они остаются
скрытыми в генофонде популяции в ге-
гетерозиготном состоянии, составляя ге-
генетический груз популяции;
Термин "генетический груз популя-
популяции" отражает одно из фундаменталь-
фундаментальных понятий нопуляционной генети-
генетики. Впервые генетический груз в попу-
популяциях был выявлен в 20-30-х гг. в ис-
исследованиях природных популяций
дрозофил.
В 1929 гг. выдающийся русский ге-
генетик С. С. Четвериков с группой со-
сотрудников провел генетические иссле-
исследования дрозофил из популяций Кры-
Крыма путем инбридинга потомства отлов-
отловленных самок. В инбредных линиях
были обнаружены разнообразные ви-
видимые мутации, которые у исходных
фенотипически нормальных самок бы-
были скрыты в гетерозиготном состоя-
состоянии. В результате этих работ впервые
была выявлена насыщенность популя-
популяций мутациями, комплекс которых, как
192
Глам7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
предположил С.С. Четвериков, являет-
является эволюционным резервом вида. В
дальнейшем в 1931-1934 гг. Н.П Дуби-
Дубинин с группой сотрудников при иссле-
исследовании генетики природных популя-
популяций дрозофилы обнаружил, что дрозо-
дрозофилы из природных популяций необы-
необычайно часто несут в своем генотипе ре-
рецессивные летальные мутации. Так, в
популяции Кутаиси более 40% дрозо-
дрозофил оказались гетерозиготными по ле-
летальным генам. Каждый из этих генов в
гомозиготном состоянии приводил к
гибели оплодотворенных яйцеклеток.
Открытие отягощенности особей из
природных популяций дрозофилы ле-
летальными мутациями положило начало
учению о генетическом грузе популя-
популяций. В дальнейшем стало ясно, что гене-
генетический груз может быть выявлен
практически в любой популяции раз-
разных видов — будь то растения, живот-
животные или человек.
Американский генетик Г. Меллер и
другие исследователи развили учение
о генетическом грузе, показав, что он
слагается из ряда категорий: леталь-
летальных, полулетальных и субвитальных
изменений. Величина генетического
груза определяется как отношение раз-
разницы между наибольшей приспособ-
приспособленностью (Wmax), что свойственно
особям, обладающим лучшим геноти-
генотипом в популяции, и фактической сред-
средней приспособленностью популяции
(W), отнесенной к величине наиболь-
наибольшей приспособленности.
W -W
Т _ " may "
W
" max
Генетический груз разделяется на
три главных типа:
— сегрегационный груз — выщепле-
ние менее приспособленных гомози-
гомозиготных форм при наличии в популя-
популяции отбора в пользу гетерозигот;
— мутационный груз — результат
появления и накопления в популяци-
популяциях мутаций, которые понижают при-
приспособленность мутантных особей;
— груз дрейфа — результат случай-
случайного увеличения концентрации алле-
аллелей в изолированной популяции. Ча-
Частным случаем этого типа служит по-
повышение доли гомозиготных особей
при инбридинге (инбредный груз; ин-
бредная депрессия).
Объем генетического груза зависит
от мутационного разнообразия, имею-
имеющегося в популяциях. В генетическом
составе популяции широко представ-
представлены рецессивные мутации. Увеличе-
Увеличение концентраций отдельных мутаций
сдерживается отбором, в результате че-
чего каждая рецессивная мутация вклю-
включена в генофонд на уровне низкой кон-
концентрации. Как правило, концентрация
рецессивного аллеля составляет 0,02-
0,03. Появление фенотипических от-
отклонений, что связано с гомозиготное -
тью, происходит в этих условиях с час-
частотой 1 особь на 1000-2500. Многие ре-
рецессивные аллели имеют еще более
низкую концентрацию.
Однако число разных рецессивных
мутаций столь велико, что каждая
особь несет одну или несколько таких
мутаций в гетерозиготном состоянии.
У каждого человека имеются, как по-
полагает академик Н.П. Дубинин, 3-4 эк-
эквивалента летальных мутаций.
Согласно современным оценкам, ча-
частота аутосомных рецессивных мута-
мутаций в популяциях человека составляет
0,75%, причем большая их часть (око-
(около 75%) — следствие точковых мута-
мутаций (см. табл. 6.1).
Влияние отрицательных доминант-
доминантных мутаций в популяциях связано с
прямым фенотипическим проявлени-
проявлением вновь возникающих изменений. У
человека около 60% всех регистрируе-
Генетический груз популяции
193
мых менделевских мутации относятся
к доминантным (абсолютная частота
аутосомных доминантных мутаций со-
составляет 1,5%). В целом различные ти-
типы менделевских наследственных бо-
болезней (аутосомные рецессивные, ау-
тосомные доминантные и сцепленные
с Х-хромосомой) выявляются у 2,4%
людей. Ряд пока не учитываемых до-
доминантных изменений проявляется на
ранних этапах развития зародыша че-
человека в виде серьезных дефектов,
приводящих к прекращению беремен-
беременности.
Преобладающую часть наследствен-
наследственной изменчивости человека составляют
врожденные пороки развития и муль-
тифакториальные болезни (в сумме —
66%), наследование которых не подчи-
подчиняется менделевским законам. Эти за-
заболевания проявляются в результате
сложного взаимодействия генетичес-
генетических изменений и факторов окружаю-
окружающей среды. Из материалов, изложен-
изложенных в главе 6, следует, что генетические
изменения, обуславливающие мульти-
факториальные болезни, составляют
значительную часть генетического гру-
груза популяций человека. Однако эту
компоненту генетического груза пока
трудно оценить количественно в силу
сложности механизмов генетического
контроля таких болезней.
Воздействие мутагенных факторов
должно приводить к увеличению уров-
уровня мутаций в популяциях различных
организмов. Детально закономерности
динамики мутационного процесса в
популяциях исследованы в экспери-
экспериментах с ионизирующими излучения-
излучениями. Первые работы по генетике облу-
облученных популяций выполнены амери-
американским генетиком Б. Уолесом в 1951-
1956 гг. Опыты проводились с экспе-
экспериментальными популяциями D.
melanogaster, созданными из особей,
свободных от леталей и полулеталей
во второй хромосоме. Популяции в
каждом поколении подвергали хрони-
хроническому облучению в дозах 0,9-5,1
сГр/ч. В каждом поколении исследова-
исследовали частоту накопленных летальных
мутаций во второй хромосоме путем
перевода второй хромосомы облучен-
облученных особей в гомозиготное состояние с
помощью специальной системы скре-
скрещивания. Эксперименты продолжа-
продолжались в течение нескольких лет, за это
время в популяциях дрозофилы про-
прошло около 150 поколений.
Результаты анализа по количеству
леталей в облученных и в контрольной
популяциях представлены на рисунке
7.4. В контрольной популяции, свобод-
свободной первоначально от рецессивных ле-
леталей во второй хромосоме, в течение
70 поколений под давлением естест-
естественного мутационного процесса идет
накопление мутаций до определенного
равновесного уровня. Равновесный
уровень естественных мутаций под-
поддерживается в популяции более или
менее постоянно, подвергаясь флюк-
туациям за счет изменения среды и
эволюции генома популяции. В облу-
облученных популяциях концентрация ле-
леталей увеличилась.
Популяция N 1, самцы которой по-
получили единовременную дозу 7 Гр, а
самки 10 Гр, в первых пяти поколениях
имела повышенное количество лета-
леталей по сравнению с контролем. Облу-
Облучение каждого поколения в дозе 0,9
сГр привело к незначительному (по
сравнению с контролем) увеличению в
популяции леталей (популяция N 7), в
то время как облучение в дозе 5,1
сГр/ч на поколение (популяции 5 и 6)
увеличило уровень генетического гру-
груза в несколько раз. Равновесный уро-
уровень концентрации леталей для облу-
облучаемых популяций достигается через
194
Глава 7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
10 20 30 40 СО вО 70 80 90 100 110 120 130 140 160
Поколение
Рис. 7.4. Концентрация летальных мутаций
в облученных экспериментальных популяциях в течение 150 поколений.
Три нижние кривые: популяция № 1 ( ); популяция № 3 ( ); популяция
№ 7( ). Две верхние кривые популяция № 5(—); популяция №6 ( )
60-70 поколений после начала облуче-
облучения.
Рассмотрим более детально, как за-
зависит скорость установления равно-
равновесного уровня мутагенеза в популя-
популяции от интенсивности мутационного
процесса. На рис. 7.5 представлены
расчетные данные, полученные А.В.
чо
Локатнил
Рис. 7.5. Влияние скорости мутагенеза
на величину стационарного уровня
и темп выхода популяции
на стационарный уровень
Рубановичем по величине равновесно-
равновесного уровня и скорости его достижения
при различных гипотетических скоро-
скоростях мутирования A02, 103 и 104),
связанных с предполагаемым воздей-
воздействием различных мутагенных факто-
факторов. Видно, что чем выше темп мути-
мутирования в популяции, тем выше равно-
равновесный уровень и тем быстрее он до-
достигается. Опираясь на эти расчеты,
можно заключить, что при действии
малых доз ионизирующих излучений
равновесный уровень мутаций в попу-
популяциях будет достигаться лишь спустя
весьма значительное число поколений
после начала хронического воздейст-
воздействия ионизирующих излучений.
Результаты, полученные на популя-
популяциях дрозофилы, были в дальнейшем
подтверждены на других эксперимен-
экспериментальных объектах -одноклеточных во-
водорослях, растениях, мышах. Кроме
того, высокий генетический груз в
природных популяциях различных ор-
организмов был выявлен при изучении
генетических последствий ядерной
аварии на предприятии «Маяк», про-
произошедшей в 1957 г, в результате кото-
которой возник Восточно-Уральский ра-
Генетический груз популяции
195
1978 1979 1980 1982 1983 1984 1965 1988 1990 1905
Год
Рис. 7.6. Динамика хлорофильных мутаций в хронически облучаемых
и контрольной популяциях С scabiosa L, произрастающих
при разных концентрациях 90Sr - 90Y в почве
диоактивный след. Например В.А.
Кальченко была прослежена в течение
38 лет динамика хлорофильных му-
мутаций у василька шероховатого
(Centaurea scabiosa L), подвергающе-
подвергающегося хроническому воздействию бета-
излучения стронция-90 и иттрия-90
(рис. 7.6). Видно, что частота выявляе-
выявляемых хлорофильных мутаций (по суще-
существу, летальных и сублетальных мута-
мутаций) поддерживается в хронически об-
облучаемой популяции на высоком рав-
равновесном уровне, значительно превы-
превышающем контрольный. В эксперимен-
экспериментах, проведенных в зоне аварии на
Чернобыльской атомной станции,
В.И. Абрамов изучал динамику гене-
генетического груза в природных популя-
популяция арабидопсиса (Arabidopsis
thaliana), хорошо изученного генети-
генетического объекта. Был проведен анализ
частоты эмбриональных леталей (ре-
(рецессивные летальные мутации), на-
наблюдаемых в стручках этого растения
(регистрировали погибшие зародыши
семян). На рисунке 7.7 видно, что
уровни эмбриональных леталей, на-
наблюдаемые в течение нескольких лет в
облучаемых популяциях, намного пре-
превосходят контрольный уровень, рав-
равный в контрольной популяции 5%. По-
Полученные результаты свидетельствуют
о насыщении генофонда облучаемых
популяций арабидопсиса рецессивны-
рецессивными летальными мутациями.
Возникает вопрос, как скоро после
прекращения воздействия мутаген-
мутагенного фактора популяция может осво-
освободиться от груза индуцированных
мутаций. Этот вопрос интересовал
исследователей с первых шагов за-
зарождения радиационной генетики.
Ответ был найден в экспериментах,
проведенных на дрозофиле Б. Уоле-
сом и Н.В. Тимофеевым-Рессовским,
и на одноклеточных водорослях, про-
196
Глам7. ПОПУЛЯЦИОННАЯ ГЕНЕТИКА
Мутационный груз в популяциях арабидопсиса, произраставших в
30км зоне аварии на ЧАЭС
1986
а Чернобыль
И Шепеличи
D Янов
1987
1990
1991
1992
Рис. 7.7. Мутационный груз в популяциях арабидопсиса,
произраставших в 30 км зоне аварии на ЧАЭС
веденных В. А. Шевченко. Показано,
что уровень мутаций каждого мутант-
ного клона (например, летальных му-
мутаций) снижается после прекращения
облучения в последующих поколени-
поколениях по экспоненциальному закону. Тре-
Требуется несколько десятков поколений
(для популяций дрозофилы — около
30-40), чтобы уровень мутаций в по-
популяции достиг равновесного уровня
контрольных популяций. Однако не-
незначительная часть индуцированных
мутаций остается закрепленной в по-
популяции на более длительное время,
создавая резерв для адаптивной из-
изменчивости популяции при измене-
изменении условий окружающей среды.
В природных популяциях исследо-
исследователь имеет дело с совокупностью ог-
огромного количества различных мута-
мутаций, постоянно возникающих и под-
подвергающихся отбору. Острое облуче-
облучение популяций того или иного вида
индуцирует широкий спектр мутаций,
формирующих мутантные клоны, каж-
каждый из которых обладает своей, прису-
присущей только ему селективной ценнос-
ценностью, своими параметрами отбора.
Популяционные закономерности
едины для любых скрещивающихся
(панмиктических) популяций. Выяв-
Выявленные на экспериментальных объек-
объектах исследования, они имеют прямое
отношение к человеку. Так же, как для
дрозофилы, в облучаемых популяциях
человека при воздействии ионизирую-
ионизирующих излучений в течение многих поко-
поколений предполагается появление рав-
равновесного уровня мутагенеза, характе-
характеризующего накопленный груз индуци-
индуцированных мутаций. Как следует из до-
докладов Научного Комитета ООН по
действию атомной радиации, равно-
равновесный уровень мутаций в облучаемых
популяциях человека, возникающий
через 7-10 поколений после начала
Генетический груз популяции
хронического облучения в дозе 1 Зв на
поколение, приблизительно в восемь
раз превышает эффект облучения, на-
наблюдаемый в первом поколении.
После прекращения воздействия ра-
радиации элиминация индуцированных
мутаций в этой гипотетической популя-
популяции человека до установления равно-
равновесного уровня естественного мутаци-
мутационного процесса, как и в популяциях
дрозофилы, составит многие поколе-
поколения.
Вопросы для самоконтроля
1. В каком случае совокупность особей можно
рассматривать как популяцию?
2. Какие показатели характеризуют генетичес-
генетическую изменчивость популяций?
3. При изучении популяции обнаружены 2 особи
с генотипом АА и 8 особей с генотипом АВ. Соответ-
Соответствуют ли наблюдаемые частоты генотипов закону
Харди — Вайнберга?
4. В каких случаях закон Харди — Вайнберга не
выполняется?
5. Может ли отбор поддерживать генетическое
разнообразие в популяциях?
6. Пусть М — нормальный ген, am — летальный
рецессивный аллель. Каковы приспособленности
всех трех возможных генотипов (MM, Mm, mm)?
7. Какова эффективность отбора против рецес-
рецессивных аллелей?
8. Как связаны частота мутирования и частота за-
заболеваний, вызванных мутантным геном?
9. Как влияет численность популяции на динами-
динамику частот генов?
10. Пусть равновесная частота рецессивного ле-
летального гена равна 10 . Каков генетический груз,
обусловленный данным геном.
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Проблема здоровья людей и генети-
генетика тесно взаимосвязаны. Ученые-гене-
Ученые-генетики пытаются ответить на вопрос, по-
почему одни люди подвержены различ-
различным заболеваниям, в то время как дру-
другие в этих или даже худших условиях
остаются здоровы. В основном это свя-
связано с наследственностью каждого че-
человека, т.е. свойствами его генов, за-
заключенных в хромосомах.
В последние годы отмечаются быст-
быстрые темпы развития генетики челове-
человека и медицинской генетики. Это объ-
объясняется многими причинами и преж-
прежде всего резким увеличением доли на-
наследственной патологии в структуре
заболеваемости и смертности населе-
населения. Статистика показывает, что из
1000 новорожденных у 35-40 выявля-
выявляются различные типы наследственных
болезней, а в смертности детей в возра-
возрасте до 5 лет хромосомные болезни со-
составляют 2-3%, генные — 8-10%, муль-
тифакториальные — 35-40%. Ежегодно
в нашей стране рождается 180 тыс. де-
детей с наследственными заболевания-
заболеваниями. Более половины из них имеют
врожденные пороки, около 35тыс. —
хромосомные болезни и свыше 35
тыс. — генные болезни. Следует отме-
отметить, что число наследственных болез-
болезней у человека с каждым годом растет,
отмечаются новые формы наследст-
наследственной патологии. В 1956 г. было изве-
известно 700 форм наследственных заболе-
заболеваний, а к 1986 году число их увеличи-
увеличилось до 2000. В 1992 количество изве-
известных наследственных болезней и
признаков возросло до 5710.
Все наследственные болезни делят-
делятся на три группы:
1. Генные (моногенные — в основе
патологии одна пара аллельных генов)
2. Хромосомные
3. Болезни с наследственным пред-
предрасположением (мультифакториаль-
ные).
Генные болезни
Генные болезни — это большая груп-
группа заболеваний, возникающих в резуль-
результате повреждения ДНК на уровне гена.
Общая частота генных болезней в
популяции составляет 1-2%. Условно
частоту генных болезней считают вы-
высокой, если она встречается с частотой
1 случай на 10.000 новорожденных,
средней - 1 на 10.000-40.000 и далее -
низкой.
Моногенные формы генных заболе-
заболеваний наследуются в соответствии с
законами Г. Менделя. По типу насле-
наследования они делятся на аутосомно-до-
минантные, аутосомно-рецессивные и
сцепленные с Х- или Y-хромосомами.
Большинство генных патологий
обусловлено мутациями в структурных
генах, осуществляющих свою функцию
через синтез полипептидов — белков.
Любая мутация гена ведет к изменению
структуры или количества белка.
Начало любой генной болезни свя-
связано с первичным эффектом мутант-
ного аллеля. Основная схема генных
болезней включает ряд звеньев: му-
тантный аллель -» измененный пер-
первичный продукт -> цепь последующих
биохимических процессов клетки -»
органы -> организм.
В результате мутации гена на моле-
молекулярном уровне возможны следую-
следующие варианты:
Генные болезни
199
1) синтез аномального белка;
2) выработка избыточного количе-
количества генного продукта;
3) отсутствие выработки первично-
первичного продукта;
4) выработка уменьшенного коли-
количества нормального первичного про-
продукта.
Не заканчиваясь на молекулярном
уровне в первичных звеньях, патогенез
генных болезней продолжается на кле-
клеточном уровне. При различных болез-
болезнях точкой приложения действия му-
тантного гена могут быть как отдель-
отдельные структуры клетки - лизосомы,
мембраны, митохондрии, нероксисо-
мы, так и органы человека. Клиничес-
Клинические проявления генных болезней, тя-
тяжесть и скорость их развития зависят
от особенностей генотипа организма
(гены-модификаторы, доза генов, вре-
время действия мутантного гена, гомо- и
гетерозиготность и др.), возраста боль-
больного, условий внешней среды (пита-
(питание, охлаждение, стрессы, переутомле-
переутомление) и других факторов.
Особенностью генных (как и вооб-
вообще всех наследственных) болезней яв-
является их гетерогенность. Это означа-
означает, что одно и то же фенотипическое
проявление болезни может быть обус-
обусловлено мутациями в разных генах
или разными мутациями внутри одно-
одного гена. Впервые гетерогенность на-
наследственных болезней была выявлена
СН. Давиденковым в 1934 г.
К генным болезням у человека отно-
относятся многочисленные болезни обмена
веществ. Они могут быть связаны с на-
нарушением обмена углеводов, липидов,
стероидов, пуринов и пиримидинов,
билирубина, металлов и др. Пока еще
нет единой классификации наследст-
наследственных болезней обмена веществ. На-
Научной группой ВОЗ предложена сле-
следующая классификация:
1) болезни аминокислотного обмена
(фенилкетонурия, алкаптонурия и
др);
2) наследственные нарушения обме-
обмена углеводов (галаюгоземия, гликоге-
новая болезнь и др.);
3) болезни, связанные с нарушением
линидного обмена (болезнь Ниманна-
Пика, болезнь Гоше и др.);
4) наследственные нарушения обме-
обмена стероидов;
5) наследственные болезни пурино-
вого и пиримидинового обмена (пода-
(подагра, синдром Леша-Найяна и др.);
6) болезни нарушения обмена со-
соединительной ткани (болезнь Марфа-
на, мукополисахаридозы и др.);
7) наследственные нарушения гема-
и порфирина (гемоглобинопатии и
др.);
8) болезни, связанные с нарушением
обмена в эритроцитах (гемолитичес-
(гемолитические анемии и др.);
9) наследственные нарушения обме-
обмена билирубина;
10) наследственные болезни обмена
металлов (болезнь Коновалова-Виль-
Коновалова-Вильсона и др.)
11) наследственные синдромы нару-
нарушения всасывания в пищеваритель-
пищеварительном тракте (муковисцидоз, неперено-
непереносимость лактозы и др.).
Рассмотрим наиболее часто встреча-
встречающиеся и генетически наиболее изу-
изученные в настоящее время генные бо-
болезни.
Наследственные болезни
аминокислотного обмена
Это — самая многочисленная группа
наследственных болезней обмена ве-
веществ. Почти все они наследуются по
аутосомно-рецессивному типу. Причи-
Причина заболеваний — недостаточность то-
того или иного фермента, ответственно-
ответственного за синтез аминокислот. Болезни со-
200
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
провождаются рвотой и обезвожива-
обезвоживанием организма, летаргическим состо-
состоянием или возбуждением и судорога-
судорогами. В позднем возрасте проявляется
угасание умственного и физического
развития.
К наследственным болезням с нару-
нарушенным аминокислотным обменом
относится фенилкетонурия, альби-
альбинизм и др.
Фенилкетонурия (ФКУ) впервые
была описана А. Фелингом в 1934 г. У
больных нарушено превращение ами-
аминокислоты. фенилаланина в тирозин
из-за резкого снижения активности
фермента фенилаланингидроксилазы.
В результате содержание фенилалани-
фенилаланина в крови и моче больных значитель-
значительно возрастает. Далее фенилаланин
превращается в фенилпировиноград-
ную кислоту, которая является нейрот-
ропным ядом и нарушает формирова-
формирование миелиновой оболочки вокруг ак-
аксонов центральной нервной системы.
Фенилкетонурия встречается в
среднем в мировом масштабе с часто-
частотой 1 на 1000 новорожденных. Однако
по этому показателю имеются значи-
значительные различия между популяция-
популяциями: 1:2600 в Турции, 1:4500 в Ирлан-
Ирландии, 1: 30000 в Швеции, 1:119000 в
Японии. Частота гетерозиготного но-
сительства в большинстве европей-
европейских популяций составляет 1:100.
Локус (фенилгидроксилазы) распо-
расположен в длинном плече 12-й хромосо-
хромосомы. В настоящее время для большин-
большинства семей возможна молекулярно-ге-
нетическая диагностика и выявление
гетерозиготного носительства. Бо-
Болезнь наследуется по аутосомно-доми-
нантному типу. Известно несколько
форм фенилкетонурии, которые раз-
различаются по тяжести протекания бо-
болезни. Это связано с наличием 4-х ал-
аллелей гена и их комбинациями.
Ребенок с фенилкетонурией рожда-
рождается здоровым, но в первые же недели
в связи с поступлением фенилалани-
фенилаланина в организм с молоком матери раз-
развивается повышенная возбудимость,
судорожный синдром, склонность к
дерматитам, моча и пот больных име-
имеют характерный "мышиный" запах, но
но главными симптомами ФКУ явля-
являются судорожные припадки и олиго-
олигофрения.
Большинство больных — блондины
со светлой кожей и голубыми глазами,
что определяется недостаточным син-
синтезом пигмента меланина. Диагноз за-
заболевания устанавливается на основа-
основании клинических данных и результа-
результатов биохимического анализа мочи (на
фенилпировиноградную кислоту) и
крови (на фенилаланин). С этой целью
несколько капель крови на фильтро-
фильтровальной бумаге подвергают хромато-
хроматографии и определяют содержание фе-
фенилаланина. Иногда используют про-
пробу Феллинга — в 2,5 мл свежей мочи
ребенка добавляют 10 капель 5% рас-
раствора треххлористого железа и уксус-
уксусной кислоты. Появление сине-зелено-
сине-зеленого окрашивания указывает на наличие
заболевания.
Метод лечения фенилкетонурии в
настоящее время хорошо разработан.
Он состоит в назначении больному дие-
диеты (овощи, фрукты, варенье, мед) и спе-
специально обработанных гидролизатов
белков с низким содержанием фенила-
фенилаланина (лофелак, кетонил, минафен и
др). В настоящее время разработаны ме-
методы дородовой диагностики. Ранняя
диагностика и профилактическое лече-
лечение предупреждают развитие болезни.
Альбинизм (глазо-кожный) описан
в 1959 г. Болезнь обусловлена отсутст-
отсутствием синтеза фермента тирозиназы.
Для нее характерна обесцвеченность
кожи, волос, глаз, независимо от расы
Генные болезни
201
и возраста. Кожа больных розово-
красная, совершенно не загорает. Име-
Имеет предрасположенность к злокачест-
злокачественным новообразованиям. Волосы
белые или желтоватые. Радужка серо-
голубого цвета, но может быть и розо-
розоватая из-за отражения света от глазно-
глазного дна. Больным свойственна сильная
светобоязнь, их зрение снижено и не
улучшается с возрастом.
Альбинизм встречается с частотой 1
на 39.000, наследуется по аутосомно-
рецессивному типу (рис. 8.1). Ген лока-
локализован на длинном плече 11-й хромо-
хромосомы.
Наследственные заболевания,
связанные с нарушением
обмена углеводов
Известно, что углеводы входят в со-
состав ряда биологически-активных ве-
веществ — гормонов, ферментов, муко-
полисахаридов, выполняющих энерге-
энергетическую и структурную функции. В
результате нарушения углеводного об-
обмена развивается гликогеновая бо-
болезнь, галактоземия и др.
Гликогеновая болезнь связана с на-
нарушением синтеза и разложения гли-
гликогена — животного крахмала. Глико-
Гликоген образуется из глюкозы при голода-
голодании; в норме он снова превращается в
глюкозу и усваивается организмо.м.
При нарушении этих процессов у че-
человека развиваются тяжелые заболе-
заболевания — различные типы гликогенозов
К ним относятся болезнь Гирке, бо-
болезнь Помпе и др.
Гликогеноз (I тип — болезнь Гирке).
У больных в печени, почках и слизис-
слизистой кишечника накапливается боль-
большое количество гликогена. Превраще-
Превращение его в глюкозу не происходит, т.к.
отсутствует фермент глюко-6-фосфа-
таза, регулирующий уровень глюкозы
в крови. В результате у больного раз-
вивается гипогликемия, в печени, поч-
почках и слизистой кишечника накапли-
накапливается гликоген. Болезнь Гирке насле-
наследуется по аутосомно-рецессивному
типу.
Сразу после рождения главными
симптомами болезни являются глико-
гемические судороги и гепатомегалия
(увеличение печени). С 1 -го года жизни
отмечается задержка роста. Характерен
вид больного: большая голова, "куколь-
"кукольное лицо", короткая шея, выступающий
живот (рис. 8.2). Кроме того, отмечают-
отмечаются носовые кровотечения, задержка фи-
физического и полового развития, мышеч-
мышечная гипотония. Интеллект при этом
нормальный. В крови повышается уро-
уровень мочевой кислоты, так что с возрас-
возрастом может развиться подагра.
В качестве лечения используется
диетотерапия: частый прием пищи, по-
повышенное содержание углеводов и ог-
ограничение жиров в диете.
202
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
¦ шт —
—r^4tr~
Рис/8.2. Болезнь Гирке. Большая голова, "кукольное лицо",
короткая шея, выступающий живот
Гликогеноз (II тип — болезнь Пом-
Помпе) протекает в более тяжелой форме.
Гликоген накапливается как в печени,
так и в скелетных мышцах, миокарде,
легких, селезенке, надпочечниках,
стенках сосудов, в нейронах.
У новорожденных спустя 1-2 месяца
появляется мышечная слабость, дефи-
дефицит 1,4-глюкозидазы в печени и в
мышцах. В этот же период возникают
кардиомегалия (увеличение сердца) и
макроглоссия (патологическое увели-
увеличение языка). Нередко у больных раз-
развивается тяжелая форма пневмонии
из-за накопления секрета в дыхатель-
дыхательных путях. Дети погибают на первом
году жизни.
Болезнь наследуется по аутосомно-
рецессивному типу. Ген локализован
на длинном плече 17-й хромосомы.
Диагностика заболевания возможна
еще до рождения ребенка. С этой це-
целью определяют активность фермента
1,4-глюкозидазы в амниотической
жидкости и ее клетках.
Известны и другие типы гликогено-
зов (III-VII), которые в общем повто-
повторяют клиническую картину первого
типа. Для их диагностики необходимы
специальные биохимические исследо-
исследования.
Галактоземия. При этом заболева-
заболевании происходит накопление в крови
больного галактозы, что приводит к
поражению многих органов: печени,
нервной системы, глаз и др. Симптомы
болезни появляются у новорожденных
после приема молока, поскольку га-
галактоза — составная часть молочного
сахара лактозы. При гидролизе лакто-
лактозы образуются глюкоза и галактоза.
Последняя необходима для миелини-
зации нервных волокон. При избытке
галактозы в организме она в норме
превращается в глюкозу с помощью
фермента галактоза-1-фосфат-уридил-
трансферазы. При понижении актив-
активности этого фермента происходит на-
накопление галактоза-1-фосфата, ток-
токсичного для печени, мозга, хрусталика
глаза.
Болезнь проявляется с первых дней
жизни расстройствами пищеварения,
интоксикацией (понос, рвота, обезво-
обезвоживание). У больных увеличивается пе-
печень, развивается печеночная недоста-
недостаточность и желтуха. Обнаруживается
катаракта (помутнение хрусталика гла-
глаза), умственная отсталость. У погибших
в первый год жизни детей при вскрытии
обнаруживают цирроз печени.
Наиболее точные методы диагнос-
диагностики галактоземии — определение ак-
активности фермента галактоза-1-фос-
фат-уридилтрансферазы в эритроци-
эритроцитах, а также галактозы в крови и моче,
где уровни ее увеличены. При исклю-
исключении из пищи молока (источника га-
Генные болезни
203
Рис. 8.3. Внешний вид ребенка при болезни Гоше
лактозы) и раннем назначении диеты
больные дети могут нормально разви-
развиваться.
Тип наследования галактоземии —
аутосомно-рецессивный. Ген локали-
локализован на коротком плече 9-й хромосо-
хромосомы. Болезнь встречается с частотой 1
на 16.000 новорожденных.
Наследственные заболевания,
связанные с нарушением
липидного обмена
В число липидов входят холестерол
(холестерин), триглицериды, эфиры
холестерола, фосфолипиды, сфинго-
липиды и др.
Наследственные болезни обмена ли-
липидов (липидозы) подразделяются на
два основных типа : 1) внутриклеточ-
внутриклеточные, при которых происходит накоп-
накопление липидов в клетках различных
тканей и 2) болезни с нарушением ме-
метаболизма липопротеидов, содержа-
содержащихся в крови.
К числу наиболее изученных на-
наследственных заболеваний липидного
обмена первого типа относятся бо-
болезнь Гоше, болезнь Ниманна-Пика и
амавротическая идиотия (болезнь Тея-
Сакса).
Болезнь Гоше характеризуется на-
накоплением цереброзидов в клетках
нервной и ретикуло-эндотелиальной
системы, обусловленным дефицитом
фермента глкжоцереброзидазы. Это
приводит к накоплению в клетках ре-
ретикуло-эндотелиальной системы глю-
коцереброзида. В клетках мозга, пече-
печени, лимфатических узлах обнаружива-
обнаруживаются крупные клетки Гоше. Накопле-
Накопление цереброзида в клетках нервной си-
системы приводит к их разрушению.
Выделяют детскую и ювенильную
формы болезни. Детская проявляется в
первые месяцы жизни задержкой умст-
умственного и физического развития, увели-
увеличением живота, печени и селезенки, за-
затруднением глотания, спазмом гортани.
Возможна дыхательная недостаточность,
инфильтрация (уплотнение легких клет-
клетками Гоше) и судороги. Смерть наступает
на первом году жизни (рис. 8.3).
Наиболее часто встречается юве-
нильная форма болезни Гоше. Она по-
поражает детей различного возраста и
носит хронический характер. Заболе-
Заболевание проявляется, как правило, на
первом году жизни. Возникают пиг-
пигментация кожи (коричневые пятна),
остеопороз (снижение плотности1 кос-
204
Глава 8. НАСЛЕДСТВЕННЫЕ
Рис. 8.4. Болезнь Ниманна-Пика
ти), переломы, деформация костей. В
тканях мозга, печени, селезенки, кост-
костного мозга содержится большое коли-
количество глкжоцереброзидов. В лейко-
лейкоцитах, клетках печени и селезенки
снижена активность глюкозидазы. Тин
наследования аутосомно-рецессив-
ный. Ген локализован на длинном пле-
плече 1-й хромосомы.
Болезнь Ниманн-Пика обусловлена
снижением активности фермента
сфингомиелиназы. В результате про-
происходит накопление сфингомиелина в
клетках печени, селезенке, мозге, рети-
БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
куло-эндотелиальной системе. Вслед-
Вследствие дегенерации нервных клеток
нарушается деятельность нервной си-
системы.
Выделяют несколько форм заболева-
заболевания, различающихся клинически (вре-
(время начала, течение и тяжесть невроло-
неврологических проявлений). Однако имеют-
имеются и общие для всех форм симптомы.
Болезнь чаще проявляется в раннем
возрасте. У ребенка увеличиваются
лимфатические узлы, размеры живота,
печени и селезенки; отмечаются рвота,
отказ от пищи, мышечная слабость,
снижение слуха и зрение (рис. 8.4). У
20-30% детей на сетчатке глаза обнару-
обнаруживается пятно вишневого цвета
(симптом "вишневой косточки"). По-
Поражение нервной системы ведет к от-
отставанию нервно-психического разви-
развития, глухоте, слепоте. Резко снижается
устойчивость к инфекционным забо-
заболеваниям. Дети погибают в раннем
возрасте.
Наследование болезни — аутосом-
но-рецессивное. Ген сфингомиелиназы
картирован на хромосоме 11.
Диагностика болезни Ниманна-Пи-
ка основана на выявлении в плазме
крови и спинномозговой жидкости
повышенного содержания сфингомие-
сфингомиелина. В периферической крови выяв-
выявляются большие зернистые пенистые
клетки Пика. Лечение симптоматиче-
симптоматическое.
Амавротическая идиотия (болезнь
Тея-Сакса) также относится к заболе-
заболеваниям, связанным с нарушением ли-
пидного обмена. Для нее характерно
отложение в клетках мозга, печени,
селезенки и других органах липида
ганглиозида. Причина- снижение ак-
активности фермента гексозаминидазы
А в организме. В результате происхо-
происходит разрушение аксонов нервных кле-
клеток.
Генные болезни
205
Болезнь проявляется в первые меся-
месяцы жизни. Ребенок становится вялым,
малоподвижным, безразличным к ок-
окружающим. Задержка психического
развития приводит к снижению интел-
интеллекта до степени идиотии. Отмечается
мышечная гипотония, судороги, харак-
характерный симптом "вишневой косточки"
на сетчатке глаза. К концу первого го-
года жизни наступает слепота. Причина
— атрофия зрительных нервов. Позд-
Позднее развивается полная обездвижен-
ность. Смерть наступает в 3-4 года.
Тип наследования болезни — ауто-
сомно-рецессивный. Ген локализован
на длинном плече 15-й хромосомы
(рис. 8 5).
Наследственные болезни
соединительной ткани
Роль соединительной ткани в орга-
организме связана с опорной, трофической
и защитной функциями. Важнейшими
компонентами соединительной ткани
являются:
а) клеточные компоненты;
б) коллагеновые, эластические и ре-
ретикулярные волокна;
в) аморфное основное вещество.
Сложная структура соединительной
ткани задана генетически. Патология в
ее системе является причиной различ-
различных наследственных заболеваний и
обусловлена в той или иной степени
нарушениями строения структурных
белков — коллагенов.
Большинство болезней соедини-
соединительной ткани связано с дефектами
опорно-двигательного аппарата и ко-
кожи. К числу их относятся синдром
Марфана, синдром Элерса-Данлоса, а
также мукополисахаридозы.
Синдром Марфана ("паучьи паль-
пальцы") относится к числу наследствен-
наследственных болезней обмена веществ и харак-
характеризуется системным поражением со-
Рис. 8.5. Болезнь Тея-Сакса
единительной ткани. Наследуется по
аутосомно-доминантному типу с высо-
высокой пенетрантностью и различной сте-
степенью экспрессивности. С этим связан
значительный клинический и возраст-
возрастной полиморфизм. Впервые синдром
был описан В. Марфаном в 1886 г.
Причина болезни — мутация в гене, от-
ответственном за синтез белка соедини-
соединительнотканных волокон фибриллина.
Блокирование его сиинтеза приводит
к повышенной растяжимости соедини-
соединительной ткани.
Больных с синдромом Марфана от-
отличают высокий рост, длинные пауко-
206
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Рис. 8.6. Синдром Марфана
а, б - диспропорциональное телосложение, длинные тонкие конечности, узкое лицо,
воронкообразная грудная клетка, плоскостопие; в - арахнодактилия
Генные болезни
207
образные пальцы, деформация груд-
грудной клетки (воронкообразная, киле-
видная, уплощенная), плоскостопие
(рис. 8.6). Нередко имеют место бед-
бедренные и паховые грыжи, гипоплазия
(недоразвитие) мышц, мышечная ги-
гипотония, ухудшение зрения, измене-
изменение формы и размера хрусталика, зна-
значительная миопия вплоть до отслойки
сетчатки, гетерохромия (разное окра-
окрашивание участков радужки); подвы-
подвывих хрусталика, катаракта, косоглазие.
Помимо перечисленного, при синд-
синдроме Марфана характерны врожден-
врожденные пороки сердца, расширение аорты
с развитием аневризмы. Нередко отме-
отмечаются расстройства органов дыхания,
поражения желудочно-кишечного
тракта и мочевыводящей системы.
Лечение в основном симптоматиче-
симптоматическое. Положительное действие оказы-
оказывают массаж, лечебная физкультура, а
в ряде случаев оперативное вмеша-
вмешательство. Большое значение имеет
ранняя диагностика заболевания. Час-
Частота синдрома Марфана в популяции
равна 1:10.0A:15.000).
Следует отметить, что синдромом
Марфана страдали президент США
Рис. 8.7. Мукололисахаридоз, тип I
а - макро- и скалоцефалия (увеличение
размеров и формы черепа), б - внешний
вид
Авраам Линкольн, великий итальян-
итальянский скрипач и композитор Николо
Паганини.
Мукополисахаридозы представле-
представлены целой группой наследственных за-
заболеваний соединительной ткани. Для
них характерно нарушение в организме
метаболизма кислых гликозаминогли-
канов, что связано с недостаточностью
лизосомальных ферментов. В результа-
результате патологические продукты обмена от-
откладываются в соединительной ткани,
в печени, селезенке, роговице и в клет-
клетках центральной нервной системы.
208
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Первые сведения о мукополисаха-
ридозах появились в 1900 г., а затем в
1917-1919 гг. Еще раньше это заболе-
заболевание было известно под названием
"гаргоилизм", поскольку внешне про-
банды напоминали фигуры, украшав-
украшавшие собор Парижской Богоматери.
При мукополисахаридозах поража-
поражаются опорно-двигательный аппарат,
внутренние органы, глаза, нервная си-
система.
Клиническими признаками болезни
служат: замедление роста, короткая
шея и туловище, деформация костей,
снижение интеллекта, грубые черты
лица с крупными губами и языком, пу-
пупочные и паховые грыжи, пороки серд-
сердца, нарушение психического развития
с отставанием от нормы (рис. 8.7).
Тип наследования болезни — ауто-
сомно-рецессивный. Ген картирован на
коротком плече 4-й хромосомы.
Всего выделяют 8 основных типов
мукополисахаридозов в зависимости
от снижения активности разных фер-
ферментов и особенностей клинических
признаков. Для определения типа за-
заболевания исследуются биохимичес-
биохимические показатели кислых гликозаминог-
люканов в крови и моче больных.
Лечение: диетотерапия, физиопро-
физиопроцедуры (электрофорез, магнитотера-
пия, массаж, лечебная физкультура и
др.), гормональные и сердечно-сосуди-
сердечно-сосудистые средства.
Наследственные нарушения
обмена в эритроцитах
К этой группе относятся болезни,
связанные чаще всего с укорочением
срока жизни эритроцитов, а также со
снижением его уровня в крови. Гемо-
Гемоглобин — основной белок эритроци-
эритроцитов. В настоящее время хорошо изуче-
изучена аминокислотная последователь-
последовательность и структура его молекулы. Изу-
Изучение гемоглобина у человека нача-
началось с открытия наследственного забо-
заболевания — серповидноклеточной ане-
анемии. Было показано, что молекуляр-
молекулярная структура серповидноклеточного
гемоглобина отличается от нормаль-
нормального.
По мере совершенствования мето-
методов электрофореза были выявлены бо-
более 400 вариантов гемоглобина и рас-
расшифрована их полная аминокислот-
аминокислотная последовательность. Установлено,
что молекула человеческого гемогло-
гемоглобина состоит из 4-х полипептидных
цепей (а,р,у,6). Большинство разно-
разновидностей гемоглобина человека име-
имеет идентичные а-цепи и различаются
по остальным цепям. К каждой цепи
глобина в специфическом участке при-
присоединяется молекула небелковой
природы — гемогруппа, или гем. Четы-
Четыре глобиновых цепи вместе с темами
образуют функциональную молекулу
гемоглобина, которая переносит кис-
кислород из легких в ткани.
Молекула глобина состоит из 141 —
146 аминокислотных остатков, распо-
расположенных в строго определенном по-
порядке. Последовательность аминокис-
аминокислот в белке называется его первичной
структурой. Пространственное взаи-
взаиморасположение соседних мономеров
в полипептидной цепи именуется вто-
вторичной структурой, а трехмерная кон-
конфигурация белковых субъединиц —
структурой третичной. Четвертичная
же структура гемоглобина реализуется
во взаимной организации четырех
субъединиц в составе функционирую-
функционирующей молекулы.
У детей и взрослых преобладающим
типом гемоглобина является НвА, со-
состоящий из двух а- и двух р-цепей
(а2Рг)> а-цепи у всех гемоглобинов
идентичны, а- и 0-цепи различаются
по многим аминокислотным остаткам.
Генные болезни
209
У всех взрослых есть 2-3 % гемоглоби-
гемоглобина НвА2 (а282). Характерная для него
5-цепь отличается от р-цепи по 10 ами-
аминокислотным остаткам. После рожде-
рождения у всех детей обнаруживается так-
также небольшое количество (меньше
1%) фетального гемоглобина HbF
(а2у2). у-цепь значительно отличается
от р-цепей. Синтез у-цепей у эмбриона
происходит в основном в печени и се-
селезенке, но могут они синтезироваться
и в клетках костного мозга. А р-цепи,
наоборот, синтезируются главным об-
образом в клетках костного мозга.
Все нормальные гемоглобины чело-
человека имеют идентичную трехмерную
структуру, оптимальную для переноса
кислорода.
Аминокислотная последователь-
последовательность каждой глобиновой цепи коди-
кодируется своим геном, равно как и синтез
небелковой гемогрунпы.
Гены глобинов у человека образуют
мультигенное семейство и расположе-
расположены на хромосомах двух кластеров (Кла-
(Кластеры — ipymta генов, расположенных
в определенных локусах, объединен-
объединенных общими функциями.) — а-кластср
занимает 25000 нар нуклеотидов в ко-
коротком плече 16-й хромосмы. Семейст-
Семейство у-р-8-генов расположено в коротком
плече 11-й хромосомы F0 000 п.н.).
Все глобиновые гены имеют сход-
сходную функциональную организацию.
Они содержат 3 кодирующие последо-
последовательности (экзоны) и 2 интрона, ко-
которые транскрибируются вместе с эк-
зонами и вырезаются в ходе процес-
синга. Варианты гемоглобинов возни-
возникают в результате мутаций в конкрет-
конкретном глобиновом гене. Эти варианты
(чаще всего) отличаются одной амино-
аминокислотой в глобиновой цепи. Описано
более 350 таких единичных замен. За-
Замены аминокислот влияют на сродст-
сродство молекулы гемоглобина кислорода.
Нарушение же его функций ведет к
возникновению заболеваний. К дру-
другим типам мутаций, изменяющим ге-
гемоглобин, относятся: делении, дупли-
дупликации, мутации сдвига рамки считыва-
считывания, мутации, нарушающие процес-
синг РНК и др.
В результате мутаций в эритроцитах
и гемоглобине возникают наследст-
наследственные болезни человека — гемолити-
гемолитические анемии и гемоглобинопатии.
Гемолитические анемии включают
заболевания, обсусловленные сниже-
снижением уровня гемоглобина и укороче-
укорочением срока жизни эритроцитов. Кроме
того, причиной болезни могут быть:
1) Нарушение мембраны эритроци-
эритроцитов.
2) Нарушение активности фермен-
ферментов эритроцитов (ферментов, гликоли-
гликолиза пентозофосфатного цикла и др.).
3) Нарушение структуры или синте-
синтеза гемоглобина.
Рассмотрим кратко наиболее распро-
страненныую форму наследственной
гемолитической анемии у человека.
Наследственный микросфероцитоз
— гемолитическая анемия Минков-
ского-Шоффара. Болезнь описана в
1900 г. Примерно в половине случаев
она проявляется у новорожденных.
Диагноз ставится в возрасте 3-10 лет.
Заболевание обусловлено генетичес-
генетическими аномалиями эритроцитов и свя-
связано с врожденной недостаточностью
липидов их оболочки. В результате по-
повышения проницаемости мембраны в
клетку проникают ионы натрия и те-
теряется АТФ. Эритроциты принимают
сферическую форму. Измененные эри-
эритроциты разрушаются в селезенке с
образованием токсического белка —
билирубина.
При данном заболевании отмечают
желтуху, анемию, спленомегалию (раз-
(разрыв селезенки), изменения скелета.
210
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Болезнь может протекать в двух фор-
формах — хронической и острой, при кото-
которой усиливается гемолиз, обусловли-
обусловливающий анемию.
У детей в первые месяцы жизни час-
часто возникает "ядерная желтуха". При-
Причина — поражение ядер головного моз-
мозга за счет высокого содержания били-
билирубина. В более старшем возрасте вы-
высокий уровень билирубина приводит к
образованию камней и развитию жел-
чекаменной болезни.
Для больных характерно увеличение
селезенки и печени, деформация скеле-
скелета; башенный череп, готическое небо,
нарушенное расположение зубов.
Для диагностики заболевания ис-
исследуют кровь. Характерными призна-
признаками являются обнаружение микро-
сфероцитов, снижение осмотической
стойкости эритроцитов и др.
Тип наследования — аутосомно-до-
минантный с неполной пенетрантнос-
тью. Ген картирован на коротком плече
8-й хромосомы.
Наследственные аномалии
циркулирующих белков.
Гемоглобинопатии
Гемоглобинопатии — это болезни,
связанные с наследственным наруше-
нарушением синтеза гемоглобина. Выделяют
количественные (структурные) и каче-
качественные их формы. Первые характе-
характеризуются изменением первичной
структуры белков гемоглобина, что мо-
может приводить к нарушению его ста-
стабильности и функции. При качествен-
качественных формах структура гемоглобина ос-
остается номрмальной, снижена лишь
скорость синтеза глобиновых цепей.
Талассемии. Данная патология обус-
обусловлена снижением скорости синтеза
полипептидных цепей нормального ге-
гемоглобина А. Впервые болезнь описана
в 1925 г. Ее название происходит от гре-
греческого "Таласса" — Средиземное море.
Полагают, что со средиземноморским
регионом связано происхождение боль-
большинства носителей гена талассемии.
Талассемия встречается в гомо- и ге-
гетерозиготных формах. По клинической
картине принято различать большую,
промежуточную, малую и минималь-
минимальную формы. Остановимся на одной из
них.
Гомозиготная (большая) талассе-
талассемия, она же — анемия Кули вызывает-
вызывается резким снижением образования ге-
гемоглобина НЬА( и увеличением коли-
количества гемоглобина F.
Клинически болезнь проявляется к
концу первого года жизни ребенка.
Для нее характерны монголоидность
лица, башенный тип черепа, отстава-
отставание физического развития. При дан-
данной патологии в крови больного обна-
обнаруживаются мишеневидные эритроци-
эритроциты с низким содержанием Нв, укоро-
укороченной продолжительностью жизни и
повышенной осмотической стойкос-
стойкостью. У больных отмечается увеличе-
увеличение селезенки и, реже, печени.
По тяжести заболевания выделяют
несколько форм талассемии. Тяжелая
талассемия заканчивается быстрой ги-
гибелью в первые месяцы жизни ребен-
ребенка. При хронической — больные дети
доживают до 5-8 лет, а при легких фор-
формах больные доживают до взрослого
возраста.
Серповидноклеточная анемия —
наиболее часто встречаемое наследст-
наследственное заболевание, вызванное изме-
изменением структуры молекулы гемогло-
гемоглобина. Люди, страдающие серповидно-
клеточной анемией, в большинстве
случаев погибают, не достигнув зрело-
зрелого возраста. В условиях низкого пар-
парциального давления кислорода их эри-
эритроциты приобретают форму серпа. У
родителей больного эритроциты име-
Хромосомные болезни человека
211
ют несколько измененную форму, но
они не страдают анемией.
Это заболевание впервые обнару-
обнаружил в 1910 г. Дж. Херрик у студента-
негра, страдавшего тяжелой формой
анемии. В крови больного он выявил
эритроциты необычной серповидной
формы. Оказалось, что это заболева-
заболевание не редкое явление у американских
негров (рис. 8.8).
В 1946 г. Нобелевский лауреат Л.
Полинг с коллегами провели биохи-
биохимический и генетический анализ гемо-
гемоглобина больных и здоровых людей и
показали, что гемоглобины нормаль-
нормальных и серповидноклеточных эритро-
эритроцитов различаются по подвижности в
электрическом ноле и растворимости.
Оказалось, что гемоглобин у людей с
признаками серповидноклеточности
представляет смесь равных количеств
и нормального и мутантного гемогло-
гемоглобина. Стало ясно, что мутация, вызы-
вызывающая серповидноклеточную ане-
анемию, связана с изменением химичес-
химической структуры гемоглобина.
Дальнейшие исследования показа-
показали, что в случае серповидноклеточной
анемии происходит замена глютами-
новой кислоты на валин в шестой паре
нуклеотидов гена, кодирующего C-
цепь гемоглобина человека. У гетеро-
зигот измененный гемоглобин состав-
составляет 20-45 %, у гомозигот - 60-99 %
общего гемоглобина.
При данной патологии отмечают
бледность кожи и слизистых оболочек,
желтушность. У 60% детей увеличена
печень. Отмечается также шумы в об-
области сердца и др. Болезнь протекает в
виде чередования кризов и ремиссий.
Специальных методов лечения нет.
Важное значение имеет предохранение
больного от воздействия факторов, про-
провоцирующих развитие болезни (гипо-
(гипоксия, обезвоживание, холод и др.).
Рис. 8.8. Сканирующая электронная
микрофотография эритроцитов
а - нормальная форма эритроцита;
б и в - эритроциты больного, гомозигот-
гомозиготного по гену серповидноклеточной ане-
анемии, в условиях гипоксии напоминают
I по форме серп
I " , i
Хромосомные болезни человека
К хромосомным относятся болезни,
обусловленные геномными мутация-
мутациями или структурными изменениями
отдельных хромосом.
Хромосомные болезни возникают в
результате мутаций в половых клетках
одного из родителей. Из поколения в
поколение передаются не более 3-5 %
из них. Хромосомными нарушениями
обусловлены примерно 50 % спонтан-
спонтанных абортов и 7 % всех мертворожде-
ний. По данным 1987 г. из 1000 ново-
новорожденных 7 имели различные хромо-
хромосомные аномалии.
Все хромосомные болезни принято
делить на две группы.
Связанные с аномалиями
числа хромосом
В эту группу входят три подгруппы:
1. Болезни, обусловленные наруше-
нарушением числа аутосом.
2. Болезни, связанные с увеличени-
увеличением или уменьшением числа половых
Х- и Y-хромосом.
212 Глава В. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Таблица 8.1. Частота рождения детей
с хромосомными болезнями в зависимости от возраста матери
Возраст матери, годы
20
25
30
35
40
45
49
Частота рождения детей
с болезнью Дауна
1:1667
1:1250
1:952
1:378
1:106
1:30
1:12
с любой хромосомной болезнью
1:526
1:476
1:385
1:192
1:66
1:21
1:8
3. Болезни, причиной которых явля-
является полиплоидия — кратное увеличе-
увеличение гаплоидного набора хромосом.
Связанные со структурными
нарушениями (аберрациями)
хромосом
Их причинами бывают:
1. Транслокации — обменные пере-
перестройки между негомологичными хро-
хромосомами.
2. Делеции — потери участка хромо-
хромосомы.
3. Инверсии — повороты участка
хромосомы на 180 фадусов.
4. Дупликации — удвоения участка
хромосомы.
5. Изохромосомия — хромосомы с
повторяющимися генетическим мате-
материалом в обоих плечах.
6. Возникновение кольцевых хромо-
хромосом (соединение двух концевых деле-
ций в обоих плечах хромосомы).
В настоящее время у человека изве-
известно более 700 таких заболеваний, вы-
вызванных структурными нарушениями
хромосом. Имеющиеся данные пока-
показывают, что около 25% приходится на
аутосомные трисомии, 46% — на пато-
патологию половых хромосом. Структур-
Структурные перестройки составляют 10,4%.
Среди хромосомных перестроек наи-
наиболее часто встречаются транслокации
и делеции.
Рассмотрим наиболее часто встреча-
встречаемые хромосомные болезни.
Синдром Дауна (трисомия по 21-й
хромосоме). Данное заболевание от-
относится к числу наиболее распрост-
распространенных патологий человека. Встре-
Встречается у новорожденных с частотой 1:
700-800. Частота рождения детей с
болезнью Дауна зависит от возраста
матери и в меньшей степени от возра-
возраста отца. Риск рождения больного ре-
ребенка матерью в 40-46 лет в 16 раз
Таблица 8.2 Частота рождения детей с болезнью Дауна
Автор
Jenkins A933)
MalpasA937)
Parker A950)
HugA951)
Carter, Me
Капу A951)
Collman,
StollcrA961)
Местность
Чикаго
Ливерпуль
Вашингтон
Цюрих
Лондон
Австралия
(штат Виктория)
Частота
1:636
1:776
1:873
1:520
1:666
1:688
Автор
OsterA953)
Schul, Neel
A962)
Wagner^ 1962)
Jaworska
A962)
E. Ф. Давиденкова
A966)
Местность
Копенгаген
Хиросима и
Нагасаки
Гонолулу
Польша
Ленинград
Частота
1:765
1:785
1:478
1:575
1:912
Хромосомные болезни человека
213
Дерматоглифика при болезни Дауна,
а — ладонь нормального субъекта, б — ладонь при болезни Дауна
выше, чем в 20-24 года. Заметных
различий по частоте заболевания бо-
болезнью Дауна в разных географичес-
географических и расовых популяциях (Табл. 8.1.
и 8.2) не отмечается. Обычно трисо-
мия но 21-й паре хромосом составля-
составляет около 95% от общего числа боль-
больных с синдромом Дауна. Примерно
4% больных страдает транслокацион-
транслокационной формой и около 2% — мозичны-
ми. При транслокационном варианте
в кариотипе наблюдается 46 хромо-
хромосом, а лишняя 21-я хромосома транс-
лоцирована чаще всего на хромосому
из групп D или G.
Лежащее в основе синдрома Дауна
нерасхождение 21-й нары хромосом
происходит либо в яйцеклетке во вре-
время мейоза, либо на ранних стадиях
дробления зиготы.
Соотношение больных мальчиков и
девочек среди новорожденных состав-
составляет 1:1. Признаки синдрома заметны
уже при рождении и позднее проявля-
i
ются более четко. Больные обычно не-
невысокого роста, отличаются слабоуми-
слабоумием. У них характерная внешность: мон-
монголоидный разрез глаз, круглое упло-
уплощенное лицо, короткий нос, плоская
переносица, эпикант (полулунная вер-
вертикальная складка у внутреннего угла
Рис. 8.9. Дети с болезнью Дауна
214
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Таблица 8.3. Наиболее частые внешние признаки синдрома Дауна
Порок или признак
Мозговой череп и лицо
брахицефалия
монголоидный разрез глазных щелей
эпикант
плоская спинка носа
узкое небо
деформированные ушные раковины
Костно-мышечная система
короткие и широкие кисти
клинодактилия мизинца
деформация грудной клетки
Глаза
помутнение хрусталика
Встречаемость, % от общего числа больных
98,3
81,1
79,8
51,4
65,9
58,8
43,2
80,0
64,4
56,3
26,9
72,1
32,2
Таблица 8.4. Врожденные пороки развития внутренних органов
у больных с синдромом Дауна
Пораженная система и порок
Сердечно-сосудистая система
дефект межжелудочковой перегородки
дефект межпредсердной перегородки
аномалии крупных сосудов
Органы пищеварения
атрезия или стеноз двенадцатиперстной кишки
атрезия пищевода
атрезия прямой кишки и ануса
Мечевая система (гипоплазия почек,
гидроуретер, гидронефроз)
Встречаемость, %
от общего числа больных
53,2
31,4
24,3
23,1
15,3
6,6
0,9
1,1
5,9
глаза), маленькие деформированные
уши, полуоткрытый рот со слегка вы-
высунутым языком и выступающей ниж-
нижней челюстью (рис. 8.9), (Табл. 8.3).
За счет сильной мышечной гипото-
гипотонии объем движений в суставах увели-
увеличен. Дети с синдромом Дауна в первый
год жизни заметно отстают в психомо-
психомоторном развитии. Они позже начина-
начинают ходить и сидеть. Нередко у них от-
отмечаются врожденные пороки сердца,
характерные изменения дерматогли-
дерматоглифики (на ладони имеется глубокая
"обезьянья борозда").
Умственное развитие больных с
синдромом Дауна отстает; если не при-
применяются специальные методы обуче-
обучения возможно, развитие тяжелой иди-
идиотии. Болезнь сопровождается расст-
расстройством эндокринных желез, нару-
нарушением обмена веществ, снижением
иммунитета. По этой причине они час-
часто болеют пневмонией, инфекционны-
инфекционными заболеваниями. (Табл.8.4).
Продолжительность жизни больных
ограничена. Однако с помощью специ-
специальных методов обучения, укрепления
физического здоровья, правильного
питания и ухода, применения лекарст-
лекарственных препаратов и др. ее можно зна-
значительно продлить. Многие люди с
синдромом Дауна способны жить са-
Хромосомные болезни человека
215
мостоятельно, овладевать несложны-
несложными профессиями, создавать семьи.
Синдром Патау (трисомия по 13-й
хромосоме) описан в 1960 г. у детей с
множественными врожденными поро-
пороками развития. Встречается у ново-
новорожденных с частотой 1:5000 — 1:7000.
Болезнь обсуловлена трисомией по 13-
й хромосоме у 80-85% больных с синд-
синдромом Патау. Нерасхождение хромо-
хромосом в мейозе происходит чаще всего у
матери. Другие случаи (мозаицизм,
изохромосома, транслокация и др.)
встречаются очень редко (рис. 8.10).
Мальчики и девочки с синдромом Па-
Патау рождаются с одинаковой частотой.
При рождении больные дети отлича-
отличаются малым весом, хотя рождаются в
срок. Для беременных женщин типич-
типично многоводие. При синдроме Патау
Рис. 8.10. Синдром Патау
а - аномалии лица, б - полисиндактилия обеих стоп
216
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
v ч i б
Рис. 8.11. Синдром Эдвардса
черепно-лицевые аномалии и характерное
расположение пальцев кисти того же больного (а, б)
характерны различные врожденные
пороки развития лица и головного моз-
мозга. Определяют типичный внешний
вид больного: окружность черепа
уменьшена, низкий скошенный лоб, уз-
узкие глазные щели, запавшая переноси-
переносица, ушные раковины низко расположе-
расположены и деформированы. Часто встречает-
встречается расщелина верхней губы и неба. Из
аномалий костно-мышечной системы
характерны полидактилия (шестипа-
(шестипалость), повышенная подвижность сус-
суставов. Среди патологии внутренних
органов всегда отмечаются врожден-
врожденные пороки сердца (дефекты межжелу-
дочковой и межпредсердной перегоро-
перегородок), органов пищеварения, кисты по-
почек и др.). Кроме того, в большинстве
случаев поражены гениталии: у дево-
девочек — это удвоение матки и влагалища,
у мальчиков — крипторхизм (задержка
яичка при спускании в мошонку). У
80-85% больных выявляется глухота.
В связи с тяжелыми пороками раз-
развития 95% больных детей умирают в
возрасте до одного года. Практически
все дети с синдромом Патау страдают
глубокой идиотией.
Синдром Эдвардса (трисомия по
18-й хромосоме). Описан в 1960 г. Эд-
вардсом. Частота больных среди ново-
новорожденных равна 1:5000 — 1:7000. Со-
Соотношение мальчиков и девочек с син-
синдромом Эдвардса составляет 1:3. При-
Причины преобладания больных девочек
пока не известны.
Наиболее характерными особеннос-
особенностями синдрома являются изменения
мозгового черепа и лица, дефекты
опорно-двигательного аппарата, сер-
Хромосомные болезни человека
217
дечно-сосудистой системы и половых
органов.
Внешняя картина нарушений мно-
многообразна : маленький рот, узкие и ко-
короткие глазные щели, косоглазие, низ-
низкорасположенные и деформирован-
деформированные уши, вывернутая нижняя губа, ко-
короткая и складчатая шея, выступаю-
выступающий затылок, удлиненный черен.
Отмечаются также флексорное по-
положение кистей, аномально развитые
стопы, косолапость и др. Для синдрома
характерны дефекты мочевой систе-
системы, пороки сердца (рис. 8.11).
90% детей с синдромом Эдвардса
погибают до 1 года. Причиной смерти
становятся пневмонии, кишечная не-
непроходимость, сердечно-сосудистая
недостаточность.
Синдромы, обусловленные
внутрихромосомными
перестройками
К этому тину перестроек хромосом
(наряду с делениями, дупликациями
и инверсиями) относятся частичные
трисомии и моносомии аутосом. Об-
Обнаружение указанных хромосомных
нарушений стало возможным после
разработки методов дифференциаль-
дифференциальной окраски хромосом, позволяющих
не только идентифицировать каждую
хромосому набора, но и судить о про-
происхождении аномальной хромосомы,
ее дополнительного или утраченного
участка. В настоящее время для боль-
большинства хромосом человека обнару-
обнаружены частичные дупликации или де-
делении. Они могут затрагивать как ко-
короткое или длинное плечо хромосо-
хромосомы, так и одновременно оба плеча.
Для многих хромосом выявлена от-
относительная клиническая специфич-
специфичность фенотипических отклонений
при аутосомных частичных трисоми-
ях или моносомиях, однако клинико-
цитогенетические синдромы пока вы-
выделить не удалось.
Приведем два примера синдромов,
обусловленных внутрихромосомной
патологией.
Синдром "кошачьего крика" связан
с дслецией короткого плеча 5-й хромо-
хромосомы. Впервые описан Дж.Леженом в
1963 г. Признаком его служит необыч-
необычный плач детей, напоминающий мяука-
мяуканье или крик кошки. Это связано с пато-
патологией гортани или голосовых связок.
Однако с возрастом этот крик исчезает.
Клиническая картина синдрома зна-
значительно варьирует. Наиболее типич-
типичным, помимо "кошачьего крика", явля-
является умственное и физическое недо-
недоразвитие, микроцефалия (аномально
уменьшенная голова).
Своеобразен внешний вид больных:
лунообразное лицо, микрогения (ма-
(маленькие размеры верхней челюсти),
эпикант (вертикальная складка кожи у
внутреннего угла глазной щели), высо-
высокое небо, плоская спинка носа, косо-
косоглазие. Ушные раковины расположены
низко и деформированы. Отмечаются
также врожденные пороки сердца, па-
патология костно-мышечной системы,
синдактилия стоп (полное или частич-
частичное сращение соседних пальцев), плос-
плоскостопие, косолапость и др.), мышеч-
мышечная гипотония (рис. 8.12).
Большинство детей умирает в ран-
раннем возрасте. Вместе с тем известны
описания больных старше 50 лет. По-
нуляционная частота синдрома "коша-
"кошачьего крика" 1:40000 - 1:50000 ново-
новорожденных. Размер делеции в разных
случаях различен.
Синдром Вольфа-Хиршхорна впер-
впервые описан в 1965 г. У 80 % страдаю-
страдающих им новорожденных цитологичес-
цитологическую основу данного синдрома состав-
составляет делеция короткого плеча 4-й хро-
хромосомы. Размеры делеции колеблются
218
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Рис. 8.12. Синдром "кошачьего крика"
а - внешний вид больного; б - кариотип больного
от небольших терминальных до зани-
занимающих около половины дистальной
части короткого плеча. Отмечается,
что большинство делеций возникает
заново, около 13 % происходит в ре-
результате транслокаций у родителей.
Реже в геноме больных, помимо
траснлокации, имеются и кольцевые
хромосомы. Наряду с делениями хро-
хромосом, патология у новорожденных
может быть обусловлена инверсиями,
дупликациями, изохромосомами.
Болезнь характеризуется многочис-
многочисленными врожденными пороками раз-
развития, задержкой умственного и пси-
психомоторного развития.
У новорожденных небольшой вес
при нормальной продолжительности
беременности. Среди внешних призна-
признаков отмечаются: микроцефалия, клю-
клювовидный нос, эпикант, антимонголо-
антимонголоидный разрез глаз (опущение наруж-
наружных углов глазных щелей), аномаль-
аномальные ушные раковины, расщелина
верхней губы и неба, маленький рот,
деформация стоп и др.
Дети с синдромом Вольфа-Хирш-
хорна маложизнеспособны, как прави-
правило умирают в возрасте до одного года.
Частота этого синдрома низкая — 1 :
100000 рождений.
Синдромы с числовыми
аномалиями половых хромосом
Синдром Шерешевского-Тернера
впервые описан Н.А. Шерешевским в
1925 г., а позднее, в 1938 г. Х.Х. Терне-
Тернером.
Причиной болезни является наруше-
нарушение расхождения половых хромосом.
Болеют только женщины, у них отсут-
отсутствует одна Х-хромосома D5 ХО).
Частота встречаемости синдрома 1 :
3000 новорожденных девочек. Отмеча-
Хромосомные болезни человека
219
ется, что только у 20% женщин бере-
беременность больным плодом сохраняет-
сохраняется до конца и рождается живой ребе-
ребенок. В остальных случаях происходит
самопроизвольный аборт или мертво-
рождение.
Для синдрома характерны: низко-
рослость, половой инфантилизм, со-
соматические нарушения. У детей уже
на первом году жизни отмечается от-
отставание в росте, что становится наи-
наиболее четко заметно к 9-10 годам.
Средний рост больных взрослых жен-
женщин составляет в среднем 135 см. У
них имеются аномалии в развитии
скелета: короткая шея с боковыми
кожными складками, короткая и ши-
широкая грудная клетка, чрезмерная по-
подвижность локтевых и коленных сус-
суставов, укорочение 4-5-го пальцев на
руках. Характерен внешний вид боль-
больных: микрогнатия (недоразвитие ниж-
нижней челюсти), эпикант, низкопосажен-
ные деформированные уши, высокое
твердое небо и др. (рис. 8.13). Нередко
отмечается косоглазие, катаракта, де-
дефекты слуха, аномалии мочевой сис-
системы (удвоение почек, мочевыводя-
щих путей).
Важной особенностью этого заболе-
заболевания является половой инфантилизм.
Внутренние и наружные гениталии не-
недоразвиты, в период полового созрева-
созревания вторичные половые признаки от-
отсутствуют или развиты слабо, недораз-
недоразвиты влагалище и матка, менструаций
нет, больные бесплодны. Однако в лите-
литературе существуют данные о рождении
детей у женщин с синдромом Шерешев-
ского-Тернера.
В 50 % случаев больные страдают ум-
умственной отсталостью, они пассивны,
склонны к психогенным реакциям и
психозам.
Продолжительность жизни близка к
норме. Лечение направлено на стимуля-
Рис. 8.13. Внешний вид больной
, с синдромом Шерешевского -Тернера |
цию роста и уменьшение полового ин-
инфантилизма (длительные курсы поло-
половых гормонов и др.).
Синдром полисемии по Х-хромосо-
ме у женщин. Синдром включает три-
сомию (кариотии 47, XXX), тетрасомию
D8, ХХХХ), пентасомию D9, ХХХХХ).
Наиболее часто встречается трисомия —
1 на 1000 родившихся девочек. Клини-
Клиническая картина довольно разнообраз-
разнообразная. Отмечается незначительное сниже-
снижение интеллекта, повышенная вероят-
вероятность развития психозов и шизофрении
с неблагоприятным типом течения.
Плодовитость таких женщин страдает в
меньшей степени.
При тетра- и нентасомии — X повы-
повышается степень умственной отсталости,
отмечаются соматические аномалии, не-
недоразвитие гениталий. Диагностика
синдрома полисомии X включает опре-
определение полового хроматина и исследо-
исследование кариотина больного. Рациональ-
Рационального лечения нет (Табл. 8.5).
220
Глава 8. НАСЛЕДСТВЕННЫЕ
Синдром Клайнфельтера описан в
1942 г. Н. Клайнфельтером. Болеют
только мальчики. Частота встречаемос-
встречаемости — 2 из 1000 новорожденных мальчи-
мальчиков. Установлено, что больные имеют
лишнюю Х-хромосому (кариотип 47,
XXY, вместо 46, XY). Наряду с этим,
встречаются варианты полисомий с
большим числом Х- и Y-хромосом, ко-
которые также относят к синдрому Клайн-
Клайнфельтера (Табл. 8.5).
До рождения заболевание клиничес-
клинически не диагностируется. Генетические
аномалии проявляются в период поло-
полового созревания в виде недоразвития се-
семенников и вторичных половых при-
признаков.
Для мужчин с синдромом Клайн-
Клайнфельтера характерны высокий рост, ев-
нухоидный тип сложения (широкий
" таз, узкие плечи), гинекомастия (раз-
(развитие грудных желез больше, чем в
норме), слабый рост волос на лице, в
подмышечных впадинах и на лобке.
Яички уменьшены в размерах, отмеча-
отмечается половой инфантилизм, склон-
склонность к ожирению. При этом у боль-
больных нарушен сперматогенез и они бес-
бесплодны. Их умственное развитие от-
отстает, однако иногда интеллект нор-
нормальный.
Увеличение числа Х-хромосом в ге-
генотипе сопровождается усилением ум-
умственной отсталости, нарушением пси-
психики, антисоциальными поступками и
алкоголизмом (рис. 8.14).
Синдром дисомии по Y-хромосоме
D7, XYY) описан в 1961 г. Встречается с
БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
частотой 1 на 1000 новорожденных
мальчиков.
Мужчины с набором хромосом 47
XYY не отличаются от нормы по физи-
физическому и умственному развитию. От-
Отмечается небольшое увеличение роста —
около 185 см. Иногда наблюдается не-
незначительное снижение интеллекта,
склонность к агрессивным и антисоци-
антисоциальным поступкам. По некоторым дан-
данным, в местах заключения мужчин с ге-
генотипом XYY в 10 раз больше, чем муж-
мужчин с нормальным генотипом.
Болезни, причиной которых
является полиплодия
Полинлодия — связана с кратным уве-
увеличением гашюдиного набора хромосом.
Причной образования полиплоидов яв-
является нарушение процесса мейоза
вследствие мутации. В результате дочер-
дочерняя половая клетка получает вместо гап-
гаплоидного B3) диполидный D6) набор
хромосом, т.е. 69 хромосом (у мужчин ка-
кариотип 69, XYY, у женщин - 69, XXX).
Рождение детей с полиплоидией на-
наблюдается очень редко. Около 22,6%
всех спонтанных абортов обусловлены
полиплоидей. Следует отметить, что
триплодия встречается в три раза чаще
по сравнению с тетраплоидией.
При триплоидии беременность
протекает с осложнениями (сильный
токсикоз, повышение уровня хорио-
нального гонадотропина и др.) Ос-
Основными пороками развития являют-
являются : расщелина губы и неба, низко рас-
расположенные ушные раковины, сраще-
Таблица 8.5. Типы полисомий по половым хромосомам у человека
Х-полисомии при
отсутствии
У-хромосомы
47, XXX
48, ХХХХ
49, ХХХХХ
Х-полисомии с
одной У хромосомой
47, ХХУ
48. ХХХУ
49. ХХХХУ
У-полисомии с одной
Х-хромосомой
47, ХУУ
48, ХУУУ
49, ХУУУУ
Полисомия по обеим
хромосомам
48, ХХУУ
49, ХХХУУ
Факторы, повышающие риск рождения детей с хромосомными болезнями
221
ние соседних пальцев кисти или сто-
стопы, аномалии в развитии всех внут-
внутренних органов и др. Дети с синдро-
синдромом триплоидии практически нежиз-
нежизнеспособны и погибают в первые дни
после рождения. Биопсия разных уча-
участков кожи у таких детей показала,
что часть тканей у них триплоидная,
часть — диплоидная.
Факторы, повышающие риск
рождения детей
с хромосомными болезнями
Причины возникновения хромосом-
хромосомных болезней до настоящего времени
недостаточно изучены. Имеются экспе-
экспериментальные данные о влиянии на му-
мутационный процесс таких факторов,
как: действие ионизирующих излуче-
излучении, химических веществ, вирусов. Дру-
Другими причинами нерасхождения хромо-
хромосом могут быть: сезонность, возраст от-
отца и матери, порядок рождения детей,
х прием лекарств во время беременности,
гормональные нарушения, алкоголизм
и др. Не исключается до определенной
степени и генетическое детерминирова-
детерминирование нерасхождения хромосом. Повто-
Повторим, однако, что причины образования
геномных и хромосомных мутаций на
ранних стадиях развития зародыша до
сих пор окончательно не раскрыты.
К биологическим факторам повыше-
повышения риска рождения детей с хромосом-
хромосомными аномалиями может быть отнесен
возраст матери. Как видно из таблицы
8.1, риск рождения больного ребенка
особенно резко возрастает после 35 «чет.
Это характерно для любых хромосом-
хромосомных болезней, но наиболее четко наблю-
наблюдается для болезни Дауна.
В медико-генетическом планирова-
планировании беременности особое значение уде-
уделяется двум факторам — наличию ане-
уплоидии по аутосомам у ребенка и воз-
возрасту матери старше 35 лет.
Рис. 8.14. Внешний вид больного
с синдромом Клайнфельтера
К кариотипическим факторам риска
у супружеских пар относятся: анеупло-
идия (чаще в мозаичной форме), ро-
бертсоновские транслокации (слияние
двух телоцентрических хромосом в об-
области деления) кольцевые хромосомы,
инверсии. Степень повышения риска
зависит от типа хромосомных наруше-
нарушений.
В настоящее время оценка степени
риска уступает более точной пренаталь-
ной цитогенетической диагностики эм-
эмбриона или плода.
Болезни с наследственной
предрасположенностью
(мультифакториальные)
Болезни с наследственной предрас-
предрасположенностью, в отличие от генных
болезней, обусловлены как наследст-
222 Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Таблица 8.6. Наиболее частые болезни с наследственной
предрасположенностью
Группы и нозологические формы
Врожденные пороки развития:
расщелина губы и неба
спинномозговая грыжа
стеноз привратника
анэнцефалия и черепно-мозговая грыжа
вывих бедра
косолапость
Психические и нервные болезни:
шизофрения
эпилепсия
маниакально-депрессивный психоз
рассеянный склероз
Соматические болезни среднего возраста
псориаз
бронхиальная астма
язвенная болезнь желудка и двенадцатиперстной кишки
коронарная болезнь сердца
гипертоническая болезнь
диабет
Распространенность на 1000 человек
(в соответствующей возрастной группе)
1-2
1
0,5-3
1
2-5
5
10-20
8-10
2-5
0,02 - 0,7
10-20
2-5
20-50
50 - 100
100 - 200
10-20
венными, так и, в значительной степе-
степени, факторами внешней среды. Эта
группа болезней в настоящее время со-
составляет 92% от общего числа наслед-
наследственных патологий человека. С воз-
возрастом частота заболеваний возраста-
возрастает. В детском возрасте процент боль-
больных составляет не менее 10 %, а в по-
пожилом - 25-30 %.
К наиболее часто встречающимся
мультифакториальным болезням от-
относятся: ревматизм, ишемическая
болезнь сердца, гипертоническая и
язвенная болезни, цирроз печени,
сахарный диабет, бронхиальная аст-
астма, псориаз, шизофрения и др.
(Табл. 8.6).
Болезни с наследственным предрас-
предрасположением связаны с действием мно-
многих генов, поэтому их называют также
мультифакториальными.
Являясь многофакторными система-
системами, они сложны для генетического ана-
анализа. Лишь в последнее время успехи в
изучении генома человека и картирова-
картировании его генов открывают возможности
Таблица 8.7. Наследственная предрасположенность
к некоторым мультифакториальным болезням
Показатель
Частота в обшей популяции, %
Частота повторных случаев среди
родственников 1-й сте пени
родства, %
Конкордантность близнецов
монозиготных
дизиготных
Ишмичес-
кая болезнь
сердца
БС
19
30-60
67
43
Ревматизм
Р
2
10
37
7
Сахарный
диабет
СД
0,6
10
42
12
Язвенная
болезнь
ЯБ
0,6
8
50
14
Шизо-
Шизофрения
Ш
1
14
67
18
Болезни с наследственной предрасположенностью (мультифакториальные)
Таблица 8.8. Эмпирический риск при некоторых
мультифакториальных болезнях
223
Nn/n
1.
2.
3.
4-
5.
6.
7
8.
9
10.
И.
12.
13.
Заболевания, пороки
Анэнцефалия
Врожденные пороки сердца
Косолапость
Опухоль Вильмса
Рак молочной железы
Эпилепсия
Шизофрения:
если болен один из родителей;
если больны оба родителя;
для сибсов в спорадических случаях
I Геосложненная миопия высокой степени
Ревматоидный артрит
Псориаз
Язвенная болезнь желудка
Атонический дерматит
бронхиальная астма
Риск для сибсов
(и в отдельных случаях для потомства)
2-5%
2 - 4 % (в зависимости от формы)
2%
5%
6-7%
3- 12%
10%
40%
12,5 - 20%
10 - 15 % (для детей и сибсов)
5%
16% (для сибсов)
20 % (для детей пробаида)
7,5%
16%
8-9%
выявления генетической предрасполо-
предрасположенности и основных причин развития
мультифакториальных заболеваний,
(Табл. 8.7).
Наследственная предрасположен-
предрасположенность может иметь моно- или поли-
полигенную природу. В первом случае она
обусловлена мутацией одного гена,
для проявления которой необходим
определенный внешний фактор, а во
втором случае — сочетанием аллелей
нескольких генов и комплексом фак-
факторов внешней среды.
Клиническая картина и тяжесть тече-
течения мультифакториальных болезней че-
человека в зависимости от пола и возраста
очень различны. Вместе с тем, при всем
их разнообразии, выделяют следующие
общие особенности:
1. Высокая частота заболеваний в
популяции. Так, шизофренией болеют
около 1% населения, сахарным диабе-
диабетом — 5%, аллергическими заболева-
заболеваниями — более 10%, гипертонией —
около 30%.
2. Клинический полиморфизм забо-
заболеваний варьирует от скрытых суб-
субклинических форм до ярко выражен-
выраженных проявлений.
3. Особенности наследования забо-
заболеваний не соответствуют менделев-
ским закономерностям.
4. Степень проявления болезни зави-
зависит от пола и возраста больного интен-
интенсивности работы его эндокринной сис-
системы, неблагоприятных факторов
внешней и внутренней среды, напри-
например, нерационального питания и др.
Генетический прогноз при мульти-
мультифакториальных заболеваниях зависит
от следующих факторов:
1) чем ниже частота болезни в попу-
популяции, тем выше риск для родственни-
родственников пробанда;
2) чем сильнее степень выраженнос-
выраженности болезни у пробанда, тем больше риск
развития болезни у его родственников;
3) риск для родственников пробанда
зависит от степени родства с поражен-
пораженным членом семьи;
224
Глава 8. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
4) риск для родственников будет вы-
выше, если пробанд относится к менее по-
поражаемому полу;
Для оценки риска при мультифакто-
риальной патологии собирают эмпири-
эмпирические данные о популяционной и се-
семейственной частоте каждого заболева-
заболевания или порока развития (Табл. 8.8).
Полигенная природа болезней с на-
наследственной предрасположенностью
подтверждается с помощью генеалоги-
генеалогического, близнецового и иопуляционно-
статистического методов. Достаточно
объективен и чувствителен близнецо-
близнецовый метод. При его использовании про-
проводят сравнение конкордантности мо-
но- и дизиготных близнецов или срав-
сравнение конкордантности выросших вме-
вместе или порознь монозиготных близне-
близнецов. Было показано, что конкордант-
конкордантность монозиготных близнецов выше,
чем дизиготных по ряду болезней сер-
сердечно-сосудистой системы (гиперто-
(гипертонии, инфаркту миокарда, инсульту, рев-
ревматизму). Это указывает на генетичес-
генетическую предрасположенность к указанным
заболеваниям. Изучение природы зло-
злокачественных новообразований у моно-
монозиготных близнецов показало невысо-
невысокую конкордантность A1 %),но вместе
с тем, она в 3-4 раза превышает таковую
для дизиготных близнецов. Очевидно,
что значение внешних факторов (осо-
(особенно канцерогенных) для возникнове-
возникновении рака намного больше наследствен-
наследственных.
С помощью близнецового метода по-
показана наследственная предрасполо-
предрасположенность к некоторым инфекционным
заболеваниям (туберкулез, полиомие-
полиомиелит) и многим распространенным бо-
болезням (ишемическая болезнь сердца,
ревматизм, сахарный диабет, язвенная
болезнь, шизофрения и др.) (Табл. 8.7).
В заключение следует отметить, что
распространение мультифакториаль-
ных болезней в разных популяциях че-
человека может значительно варьировать,
что связано с различием генетических и
средовых факторов. В результате гене-
генетических процессов, происходящих в
человеческих популяциях (отбор, мута-
мутации, миграции, дрейф генов), частота ге-
генов, определяющих наследственную
предрасположенность, может возрас-
возрастать или уменьшаться вплоть до полной
их элиминации.
Успехи программы "Геном человека",
выделение и расшифровка молекуляр-
молекулярной организации генов, изучение при-
причин их патологии несомненно будут
способствовать разработке профилак-
профилактических мероприятий и выявлению
групп людей, склонных к мультифакто-
риальным заболеваниям.
Вопросы для самоконтроля
1. Каковы подходы к классификации генных бо-
болезней?
2. Почему генные болезни называют болезными
обмена веществ или энзимопатиями?
3. В чем сущность фенилкетонурии? Возможно ли
в настоящее время вылечить больных с фенилкетону-
рией?
4. Опишите наследственные болезни соединитель-
соединительной ткани?
5. В чем сущность классификации хромосомных
болезней?
6. Опишите синдромы Дауна, Патау, Эдвардса.
7. В чем сущность синдромов, связанных с измене-
ним числа половых хромосом? Дайте им характерис-
характеристику.
8. Какие генетические закономерности можно вы-
выявить у ультифакториальных болезней?
9. В чем сущность синдрома Лежена ("кошачьего
крика")?
10. Выберите один правильный ответ. Укажите
правильную формулу кариотипа при синдроме Патау:
а) 47 XX; 18+; б) 46 XY, 13+; в) 46 XX 5р-; г) 47
ХХУ;дL7 45,ХО.
Глава 9. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
В настоящее время число детей с тя-
тяжелыми наследственными заболевани-
заболеваниями в странах бывшего СНГ превышает
один миллион. На их лечение расходу-
расходуются огромные материальные средства.
В связи с этим диагностика, профилак-
профилактика и лечение наследственных и врож-
врожденных заболеваний у детей приобрета-
приобретает большое значение.
Наиболее эффективным методом
профилактики наследственной патоло-
патологии является медико-генетическое кон-
консультирование, главная цель которого
состоит в определении прогноза рожде-
рождения больных детей в семье, а также —
консультирование по вопросам даль-
дальнейшего планирования семьи.
Первая медико-генетическая кон-
консультация была организована в конце
20-х гг. в Москве крупнейшим отече-
отечественным неврологом и генетиком
С.Н. Давиденковым при институте
нервно-психиатрической профилак-
профилактики.
Первый кабинет по медико-генетиче-
медико-генетическому консультированию был организо-
организован в 1941 г. Дж. Нилом в Мичиганском
университете (США). В России в 1932 г.
под руководством С. Г.Левита был со-
создан медико-генетический институт.
Иинтенсивное развитие медико-гене-
медико-генетической помощи в нашей и других
странах началось в 60-70 гг. XX в., что
было связано как с ростом удельного ве-
веса наследственных заболеваний, так и с
достижениями в изучении хромосом-
хромосомной патологии и болезней обмена ве-
веществ. По данным 1995 г., на террито-
территории Российской Федерации существо-
существовало 70 медико-генетических учрежде-
учреждений, услугами которых пользовались
около 80 тыс. семей.
Цели, задачи и методы
медико-генетического
консультирования (МГК)
Основная цель медико-генетического
консультирования — предупреждение
рождения больного ребенка. Главными
задачами МГК являются:
1. Установление точного диагноза на-
наследственной патологии.
2. Пренатальная (дородовая) диагно-
диагностика врожденных и наследственных
заболеваний различными методами
(ультразвуковыми, цитогенетическими,
биохимическими, молекулярно-генети-
ческими).
3. Определение типа наследования
заболевания.
4. Оценка величины риска рождения
больного ребенка и оказание помощи в
принятии решения.
5. Пропаганда медико-генетических
знаний среди врачей и населения.
Поводом для медико-генетического
консультирования могут быть:
1. Рождение ребенка с врожденными
пороками развития, умственной и фи-
физической отсталостью, слепотой и глу-
глухотой, судорогами и др.
2. Спонтанные аборты, выкидыши,
мертворождения.
3. Близкородственные браки.
4. Неблагополучное течение беремен-
беременности.
5. Работа супругов на вредном пред-
предприятии.
6. Несовместимость супружеских пар
по резус-фактору крови.
7. Возраст женщины старше 35 лет, а
мужчины — 40 лет.
Медико-генетическая консультация
включает 4 этапа: диагноз, прогноз, за-
заключение и совет.
226
Глава 9. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
Работа начинается с уточнения диа-
диагноза заболевания. Точный диагноз —
необходимое условие для любой кон-
консультации. В некоторых случаях диа-
диагноз наследственной патологии мо-
может быть установлен врачом еще пе-
перед направлением в консультацию.
Это относится к хорошо изученным и
довольно часто встречаемым наслед-
наследственным болезням, например, болез-
болезни Дауна, сахарному диабету, гемофи-
гемофилии, мышечной дистрофии и др. Чаще
же диагноз неясен.
В медико-генетических консульта-
консультациях диагноз уточняется благодаря
использованию современных генети-
генетических, биохимических, иммуногене-
тических и других методов.
Одним из основных методов явля-
является генеалогический метод, т.е. со-
составление родословной для супруже-
супружеской пары, обратившейся в консуль-
консультацию. В первую очередь это относит-
относится к тому из супругов, в родословной
которого имелась наследственная па-
патология. Тщательный сбор родослов-
родословной дает определенную информацию
для постановки диагноза болезни.
В более сложных случаях, напри-
например, при рождении ребенка с множе-
множественными пороками развития, пра-
правильный диагноз может быть постав-
поставлен лишь при использовании специ-
специальных методов исследования. В про-
процессе диагностики нередко возникает
необходимость обследования не толь-
только пациента, но и других членов се-
семьи.
После установления диагноза опре-
определяется прогноз для потомства, т.е.
величина повторного риска рождения
больного ребенка. Основой для реше-
решения этой задачи являются теоретичес-
теоретические расчеты с использованием мето-
методов генетического анализа и вариаци-
вариационной статистики или таблиц эмпи-
эмпирического риска. Это входит в функ-
функции врача-генетика.
Передача наследственных заболева-
заболеваний возможна несколькими путями в
зависимости от особенностей переда-
передачи наследственной патологии. Напри-
Например, если у ребенка имеется заболева-
заболевание, как у одного из родителей, это
указывает на доминантный тип насле-
наследования. В таком случае при полной
пенетрантности гена больные члены
семьи передадут заболевание полови-
половине своих детей.
Наследственная патология у ребенка
здоровых родителей указывает на ре-
рецессивный тип наследования. Риск
рождения больного ребенка у родите-
родителей с рецессивным заболеванием со-
составляет 25%. По данным 1976 г. у чело-
человека было известно 789 рецессивно на-
наследуемых заболеваний и 944, наследуе-
наследуемых по доминантному типу.
Наследственная патология может
быть сцеплена с полом (Х-сцепленный
тип наследования). В этих условиях
риск заболевания у .мальчиков и носи-
тельства у девочек составляет 50%. Та-
Таких заболеваний в настоящее время из-
известно около 150.
В случае мультифакториальных бо-
болезней генетическое консультирова-
консультирование является достаточно точным. Эти
болезни обусловлены взаимодействи-
взаимодействием многих генов с факторами внешней
среды. Число патологических генов и
их относительный вклад в заболева-
заболевание в большинстве случаев неизвест-
неизвестны. Для расчета генетического риска
используются специально разработан-
разработанные таблицы эмпирического риска
при мультифакториальных заболева-
заболеваниях (см. гл. 8 и табл. 8.8).
Генетический риск до 5% считается
низким и не является противопоказани-
противопоказанием к повторному рождению ребенка в
семье. Риск от 6 до 20% принято считать
Цели, задачи и методы медико-генетическо консультирования (МГК)
227
средним, и в этом случае для дальней-
дальнейшего планирования семьи рекомендует-
рекомендуется всестороннее обследование. Генети-
Генетический риск свыше 20% принято отно-
относить к высокому риску. Дальнейшее де-
деторождение в данной семье не рекомен-
рекомендуется.
При хромосомных болезнях вероят-
вероятность повторного рождения больного
ребенка крайне низка и не превышает
1% (при отсутствии других факторов
риска).
Для транслокационной формы болез-
болезни Дауна при вычислении риска важно
определить, кто из родителей несет сба-
сбалансированную транслокацию. Напри-
Например при гранслокации A4/21) величи-
величина риска равна 10%, если носителем яв-
является мать, и 2,5% — если носитель
отец. При транслокации 21-й хромосо-
хромосомы на ее гомолог, риск рождения боль-
больного ребенка составляет 100%, незави-
независимо от того, кто из родителей является
носителем транслокации.
Для определения риска повторного
рождения ребенка с патологией важно
установить гетерозиготных носителей
мутантного гена. Особое значение это
имеет при аутосомно-рецессивном типе
наследования, при наследовании, сцеп-
сцепленном с полом, и близкородственных
браках.
В ряде случаев гетерозиготное носи-
тельство устанавливается при анализе
родословной, а также путем клиничес-
клинических и биохимических анализов. Так, ес-
если у отца имеется рецессивное заболе-
заболевание, сцепленное с Х-хромосомой (на-
(например, гемофилия), то с вероятностью
100% его дочь будет гетерозиготна по
данному гену. Наряду с этим, снижение
антигемофильного глобулина в сыво-
сыворотке крови у дочерей отца-гемофилика
может служить вполне убедительным
доказательством гетерозиготного носи-
тельства гена гемофилии.
В настоящее время некоторые на-
наследственные заболевания устанавли-
устанавливаются с помощью ДНК-диагностики.
Гетерозиготным носителям дефект-
дефектных генов следует избегать близкород-
близкородственных браков, заметно увеличиваю-
увеличивающих риск рождения детей с наследст-
наследственной патологией.
Заключение медико-генетического
консультирования и советы родителям
(два последних этапа) могут быть объе-
объединены. В результате проведенных ге-
генетических исследований врач-генетик
дает заключение об имеющейся болез-
болезни, знакомит с вероятностью возникно-
возникновения болезни в будущем, дает соответ-
соответствующие рекомендации. При этом учи-
учитывается не только величина риска по-
появления больного ребенка, но и тяжесть
наследственного или врожденного забо-
заболевания, возможности пренатальной
диагностики и эффективности лечения.
Вместе с тем, все решения по дальней-
дальнейшему планированию семьи принимают-
принимаются только супругами.
Современные методы
пренатальной диагностики
наследственных заболеваний
Эффективность медико-генетическо-
медико-генетического консультирования значительно воз-
возрастает благодаря использованию со-
современных методов пренатальной (до-
(дородовой) диагностики. Она позволяет
задолго до рождения ребенка опреде-
определить заболевание и, если необходимо,
прервать беременность. Такая ситуация
возникает в случае наследственных за-
заболеваний, лечение которых в настоя-
настоящее время не дает нужных результатов.
Основными показателями к проведе-
проведению дородовой диагностики являются:
1). Наличие в семье точно установ-
установленного наследственного заболевания.
2). Наличие в семье заболевания,
сцепленного с полом.
228
Глава 9. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
3). Возраст будущей матери от 35 лет,
а отца — от 40 лет.
4). Гетерозиготность обоих родителей
по одной паре аллелей при аутосомно-
рецессивном заболевании
5). Наличие структурных перестроек
хромосом (особенно транслокаций и
инверсий) у одного из родителей.
6). Наличие в анамнезе беременной,
длительной работы на вредных для здо-
здоровья производствах, проживание в зо-
зонах с повышенным радиационным фо-
фоном и др.
К основным методам пренатальной
диагностики относятся:
1. Определение альфа-фетопротеина.
2. Ультразвуковое исследование пло-
плода (УЗИ).
3. Биопсия хориона и плаценты.
4. Амниоцентез (прокол плодного пу-
пузыря для получения околоплодной
жидкости).
5. Кордоцентез (взятие крови из пу-
пуповины).
6. Фетоскопия (введение зонда и ос-
осмотр плода).
Определение альфа-фетопротеина
(АФП) в крови и амниотической жид-
жидкости беременной получило большое
распространение. АФП — это белок, вы-
вырабатываемый клетками печени плода.
Оптимальным сроком для его определе-
определения в сыворотке крови матери является
15-16-я неделя беременности. Установ-
Установлено, что концентрация АФП в крови
беременной может существенно повы-
повышаться при некоторых аномалиях пло-
плода, например, спиномозговой грыже,
врожденном нефрозе, дефектах нервной
трубки и брюшной стенки. В случае
уточнения диагноза аномалии нервной
трубки проводится детальное ультра-
ультразвуковое обследование плода и содер-
содержание белка в амниотической жидкос-
жидкости. Следует отметить, что концентрация
АФП в крови беременных может быть
повышена и при других заболеваниях
матери: опухоль печени, хронический
гепатит, цирроз и др.
В крови женщин, вынашивающих
плод с хромосомными аномалиями
(синдром Дауна, Эдвардса и др.) уро-
уровень АФП снижен. Если концентрация
АФП мала, назначается цитогенетичес-
кий анализ клеток плода.
Ультразвуковое
исследование (УЗИ)
Метод используется главным обра-
образом для выявления врожденных поро-
пороков развития и основан на способности
ультразвуковой волны отражаться от
поверхности двух сред с различной
плотностью. Это позволяет получить
изображение на экране монитора.
Оптимальные сроки проведения
УЗИ — 17-23 недели беременности. Од-
Однако при определенных показаниях (ре-
(редукция конечностей, задержка роста эм-
эмбриона или плода) УЗИ проводят в бо-
более ранние сроки.
С помощью ультразвукового иссле-
исследования можно исследовать строение
плода (его головки, туловища, конечно-
конечностей, половых органов), выявить пора-
поражение головного мозга, пороки разви-
развития костей скелета и внутренних орга-
органов, задержку роста эмбриона или пло-
плода и др. Накопленные данные показыва-
показывают, что УЗИ не приносит вреда развива-
развивающемуся плоду. В некоторых странах
эту процедуру проводят всем беремен-
беременным, что позволяет предупредить рож-
рождение детей с пороками развития. Пере-
Перечень врожденных дефектов развития,
диагностируемых с помощью УЗИ, до-
достаточно широк (таб. 9.1).
Биопсия хориона и плаценты
К числу перспективных методов
нренатальной диагностики относится
Современные методы пренатальной диагностики наследственных заболеваний
Таблица 9.1. Врожденные пороки развития,
диагностируемые с помощью УЗИ
229
Система или орган
ЦНС
Конечности
Сердце
Почки
Желудочно-кишечный тракт
Пороки
Грубые пороки мозга, черепно- и спинномозговые
грыжи, недоразвитие передних отделов мозга и др.
Редукционные пороки, тяжелые формы
карликовости с укорочением конечностей,
ломкость костей и др.
Тяжелый порок сердца
Дефекты развития почек, поликистоз почек
Аномалии развития двенадцатиперстной кишки,
дефекты передней брюшной стенки
биопсия плода и плаценты. Она прово-
проводится на более ранних, чем УЗИ, сро-
сроках беременности G-9 недель). Вор-
Ворсинки хориона берут особым шприцом
с помощью гибкого катетера через
шейку матки (рис. 9.1, 9.2). Затем их
подвергают лабораторной диагностике
с помощью цитологических, биохими-
биохимических, молекулярно-генетических ме-
методов. В случае выявления наследст-
наследственных заболеваний у плода беремен-
беременность прерывают. Одним из осложне-
осложнений хорионбиопсии является относи-
относительно высокая частота спонтанных
абортов (выкидышей). Общие потери
плода после хорионбиопсии в среднем
составляют 2,5-3%, а в некоторых слу-
случаях и выше, что ограничивает приме-
применение этого метода. Каких-либо нару-
нарушений плаценты, роста плода, появле-
появлений пороков развития и увеличения
перинатальной смертности после хо-
хорионбиопсии не отмечается. Однако,
существуют данные, что ранняя хори-
онбиопсия может вызывать попереч-
поперечные врожденные апмутации конечнос-
конечностей (редукционные пороки). По этой
причине в последнее время хорионби-
опсию не рекомендуется проводить ра-
ранее 8-й недели, а плацентобиопсию —
12-й недели беременности.
I
Рис. 9.1. Плацентобиопсия
230
Глава 9. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
Матка
Шприц
Рис. 9.2. Хорионбиопсия
Амниоцентез
Это — прокол плодного пузыря для
взятия 8-10 мл околоплодной жидко-
жидкости с находящимися в ней слущенны-
ми клетками амниона и плода. По-
Последние являются основным субстра-
субстратом для цитологических и биохими-
биохимических исследований. Этот метод яв-
является наиболее распространенным и
доступным. Проводится на 15-18-й
неделе беременности. Риск осложне-
осложнения ее течения незначителен @,2%).
Амниоцентез делают чрезбрюшинно
под контролем УЗИ, чтобы не повре-
повредить плаценту (рис. 9.3). Чрезвлага-
лищный амниоцентез применяется
редко.
С помощью этого метода диагности-
диагностируют многие хромосомные нарушения,
болезни, сцепленные с полом, болезни
обмена веществ (болезнь Тея-Сакса,
мукополисахаридозы, гликогенозы,
фенилкетонурия и др.).
Современные методы пренатальной диагностики наследственных заболеваний
231
Рис. 9.4. Кордоцентез
Кордоцентез
В этом случае берутся образцы
крови (лейкоциты) из пуповинных
сосудов плода, для цитогенетическо-
го, молекулярно-генетического и би-
биохимического анализов крови. Кровь
более удобна для исследования, чем
клетки амниотической жидкости, т.к.
лимфоциты быстрее и надежнее
культивируются.
Кордоцентез проводится под кон-
контролем УЗИ на 18-22-й неделе бере-
беременности (рис. 9.4). Его используют
для диагностики хромосомных болез-
болезней, таких, как: гемоглобинопатии,
энзимопатии, различные иммуноде-
фицитные аномалии, синдром фра-
гильной (ломкой) Х-хромосомы и др.
Фетоскопия
Данный метод основан на прямом
рассмотрении плода через специаль-
специальный прибор — фетоскоп (тонкий эла-
эластичный зонд со специальной опти-
оптической системой). Зонд вводится в
плоскость амниона через брюшную
систему. Проводится на 18-23-й неде-
неделе беременности. Фетоскопия ис-
используется редко и только при осо-
особых показаниях, поскольку вхожде-
вхождение зонда в амниотическую жидкость
может вызвать осложнение беремен-
беременности. Выкидыши отмечаются в 7-8%
случаев фетоскопии. Проще и безо-
безопаснее использовать УЗИ, способное
выявить большинство пороков разви-
развития.
Методы нренатальной диагности-
диагностики постоянно совершенствуются и
все шире применяются в профилак-
профилактике наследственных заболеваний.
Дальнейшее совершенствование и
расширение применения этих мето-
методов — одна из задач современной ме-
медицинской генетики.
Вопросы для самоконтроля
1. Основная цель и задачи медико-генетического
консультирования (МГК).
2. Перечислите показания для направления на ме-
медико-генетическое консультирование; основные эта-
этапы МГК.
3. Что такое прогноз потомства?
4. Что имеется в виду под термином "пренатальная
диагностика"? Назовите основные показатели для
проведения пренатальной диагностики.
5. Перечислите важнейшие методы дородовой диа-
диагностики.
6. Выберите два правильных ответа.
Состояния, диагностируемые у плода с помощью
УЗИ; 1) синдром Марфана; 2) редукционные пороки
конечностей; 3) фенилкетонурия; 4) нарушения в раз-
развитии мозга.
7. Какой из указанных методов дает наибольший
процент осложнений: а) фетоскония; б) хорионбиоп-
сия; в) кордиоцентез; г) УЗИ.
Глава 10. ПРОБЛЕМЫ КАНЦЕРОГЕНЕЗА
Одно из тяжелейших заболеваний че-
человека и животных — рак — занимает
второе место после сердечнососудистых
заболеваний среди причин смертности
населения Земли. В развитых странах от
него умирает каждый пятый. Известно
более ста видов раковых заболеваний,
для каждого из которого характерны
свои особенности. Наиболее распростра-
распространенными являются пять видов рака —
легких, молочной железы, толстой киш-
кишки, простаты и матки. Им соответствует
более 50% случаев всех первичных онко-
онкологических диагнозов.
Ныне под термином рак объединяют
группу родственных заболеваний, в ос-
основе которых лежат нарушения фунда-
фундаментальных законов поведения клеток в
многоклеточном организме. В процессе
роста раковой опухоли отдельные клет-
клетки, бесконтрольно делясь, стремятся к
собственному процветанию в ущерб со-
соседям, но в конце концов разрушают все
клеточное сообщество и погибают вмес-
вместе с ним. От 80 до 90% опухолей челове-
человека развивается в тех органах и тканях,
где состав клеток постоянно обновляет-
обновляется, и клетки, соответственно, находятся в
митотическом цикле.
Несмотря на большое разнообразие
раковых заболеваний, клетки, всех рако-
раковых опухолей имеют общие свойства и
значительно отличаются от здоровых.
Одно из главных их отличий — спо-
способность к бесконечному делению. Изве-
Известно, что в каждом организме клетки мо-
могут делиться лишь определенное число
раз.
Например, для одного из видов нема-
нематод установлено, что оплодотворенное
яйцо может совершить столько делений,
что у взрослой особи в соматических
клетках в сумме насчитывается только
959 ядер. Общее количество ядер в клет-
клетках человека подсчитать невозможно, но
существуют другие методы, позволяю-
позволяющие учитывать способность клеток к де-
делению, в частности, метод клеточных
культур.
В искусственных условиях культиви-
культивирования клетки человека прикрепляют-
прикрепляются к поверхности сосуда с питательной
средой и начинают активно делиться.
Нормальные клетки, полученные из раз-
разных типов тканей, делятся до тех пор, по-
пока не покроют поверхность сосуда пол-
полностью и не начнут контактировать меж-
между собой. То есть нормальные клетки об-
образуют однослойные культуры. Опухо-
Опухолевые же клетки преодолевают барьер
контактного торможения и могут обра-
образовывать многослойные культуры.
Чтобы пересеять клеточные культуры
из одного флакона в несколько сосудов,
их снимают со стекла и переносят в пи-
питательную среду в несколько чистых
флаконов, где клетки вновь прикрепля-
прикрепляются к стеклу и начинают делиться, по-
покрывая всю поверхность. Нормальные
клетки могут выдержать несколько та-
таких пассажей, а опухолевые, в силу спо-
способности к бесконечному делению, со-
сохраняют устойчивость к пересеву очень
длительное время. Существуют переви-
перевиваемые клеточные опухолевые линии
человека, полученные в середине 50-х гг.
XX в., и успешно культивируемые до
сих пор.
Несмотря на приобретенную способ-
способность к бесконечному делению, опухоле-
опухолевые клетки частично сохраняют свою
специализацию, т.е. остаются частично
дифференцированными. Например,
клетки меланомы синтезируют гранулы
пигмента, клетки карциномы частично
сохраняют синтез промежуточных фила-
ментов цитоскелета дифференцирован-
дифференцированных клеток-предшественников.
История вопроса
233
Часто морфологические изменения
опухолевых клеток выражены не четко.
Одно из наиболее характерных для всех
опухолевых клеток морфологическое от-
отклонение — изменение формы ядра и
числа ядер. Нередко также в делящихся
опухолевых клетках изменены число и
структура хромосом. Так, для
хронического миелоидного лейкоза ха-
характерна определенная транслокация в
клетках миелоидного ряда: перенос
фрагмента длинного плеча 22-й хромо-
хромосомы на длинное плечо 9-й хромосомы.
Нередко в опухолевых клетках присут-
присутствуют небольшие добавочные хромосо-
хромосомы, образующиеся за счет амплифика-
амплификации какого-либо участка одной из хро-
хромосом. Еще одно распространенное на-
нарушение — измененная плоидность ядер.
В последние годы большое внимание
уделяется изучению цитоскелета опухо-
опухолевых клеток. Показано, что в опухоле-
опухолевых клетах спектр синтеза цитоскелет-
ных белков изменяется, с чем связывают
большую подвижность опухолевых кле-
клеток и нарушение их контактов с соседни-
соседними клетками и субстратом.
Все раковые опухоли подразделяют на
две большие категории: доброкачествен-
доброкачественные и злокачественные. Рассмотренные
выше свойства опухолевых клеток ха-
характерны для обеих категорий. Однако
клетки злокачественных опухолей обла-
обладают дополнительными свойствами.
Они способны освобождаться от меж-
межклеточных контактов с другими клетка-
клетками, перемещаться и проходить путь до
кровеносного или лимфатического сосу-
сосуда, проникать через базальную мембрану
кровеносного сосуда и слой эндотели-
альных клеток, попадая в просвет сосуда
и, путешествуя но нему, проходить
сквозь стенку сосуда в обратном направ-
направлении, обосновываться на новом месте,
размножаться и давать начало новой
опухоли — метастазу.
История вопроса
В конце XIX в. была высказана идея
о том, что нарушения в ядре клетки иг-
играют главную роль в процессе канцеро-
канцерогенеза. Изучая протекание митоза,
Хансеманн предположил, что нормаль-
нормальные клетки приобретают способность к
злокачественному росту из-за наруше-
нарушении структуры хроматина. В начале
XX в. основоположник мутационного
учения Гуго де Фриз высказал мнение,
что рак развивается в результате сома-
соматических мутаций. К 30-м гг. с развити-
развитием теории индуцированного мутагене-
мутагенеза формируется гипотеза о том, что рак —
это производное точковых соматичес-
соматических мутаций, возникающих на генном
уровне.
В 50-е гг. одновременно с общей тео-
теорией мутационного процесса развивает-
развивается и мутационная теория раковых забо-
заболеваний. Рождается идея, что в основе
злокачественности лежит не одномо-
одномоментное появление отдельной мутации,
а более сложный процесс, например, по-
последовательные мутации ряда генов в од-
одной клетке. В частности, было показано,
что возникновение ретинобластомы в
глазу человека является двухступенча-
двухступенчатым процессом. Предрасположенность к
этой форме рака зависит от одного из ге-
генов, передающихся по наследству. Раз-
Развитие же опухоли связано с дополни-
дополнительной соматической мутацией в дру-
другом независимом гене. Клетка, несущая
двойную мутацию, трансформируется,
приобретая способность к злокачествен-
злокачественному росту.
В 80- гг. идея о многоступенчатости
мутагенеза, лежащего в основе злокаче-
злокачественного перерождения клеток, разви-
развивается дальше. Формируется представ-
представление о том, что канцерогенез связан с
мутациями достаточно узкой группы ге-
генов, отвечающих за дифференцировку
клеток.
234
Глава 10. ПРОБЛЕМЫ КАНЦЕРОГЕНЕЗА
Параллельно с развитием мутацион-
мутационной теории канцерогенеза шло изучение
канцерогенов химической и физической
природы.
К физическим канцерогенам относят
рентгеновское, гамма- и ультрафиолето-
ультрафиолетовое излучения. Они оказывают как пря-
прямое, мутагенное и канцерогенное дейст-
действие на структуру ДНК, так и непрямое,
вызванное повреждением клеточных ма-
макромолекул свободно-радикальными
формами кислорода, которые образуют-
образуются в тканях под действием облучения.
Именно лучевой канцерогенез был глав-
главной опасностью для первых радиологов
и рентгенологов, работавших с радием и
лучами рентгена без защиты от облуче-
облучения. Из-за него у многих из них разви-
развивался рак кожи. При общем облучении
организма чаще всего развиваются лей-
лейкозы, реже — опухоли костей из-за на-
накопления в них радиоактивного строн-
стронция, являющегося аналогом кальция, и
рак щитовидной железы, провоцируе-
провоцируемый накоплением в ней радиоактивного
йода. Для шахтеров урановых рудников,
вдыхающих радиоактивную пыль, ха-
характерен рак легких.
Химические канцерогены объединя-
объединяют широкий круг веществ: от простых,
таких, как четыреххлористый углерод,
до весьма сложных полициклических и
гетероциклических соединений. Хими-
Химические канцерогены вызывают сходные
биологические эффекты, которые выра-
выражаются в стимуляции неограниченного
размножения клеток-предшественников
опухоли.
Хрестоматийными примерами форм
рака, индуцированных химическими
канцерогенами, являются плоскоклеточ-
плоскоклеточная карцинома легких, развивающаяся у
курильщиков, мезотелиома плевры, сти-
стимулированная асбестовой пылью, рак
мошонки у трубочистов, рак мочевого
пузыря у работников химического пред-
предприятия, контактирующих с 2-нафтила-
мином.
По мере развития теории мутационно-
мутационного процесса стало понятно, что все хими-
химические и физические канцерогены обла-
обладают общим свойством: они являются
мутагенами. Таким образом, химический
и физический мутагенез лежит в основе
опухолевой трансформации клеток.
Наряду с мутационной теорией канце-
канцерогенеза, долгие годы существовала так-
также и вирусная теория, поскольку экспе-
экспериментально было доказано, что опухо-
опухоли могут индуцироваться как ДНК-, так
и РНК-содержащимися вирусами. Наи-
Наиболее изучены ДНК-содержащий вирус
SV40, выделенный из клеток обезьяны, и
вирус саркомы Рауса — куриный ретро-
вирус, часто приводящий к развитию ра-
раковых опухолей животных. Однако сре-
среди множества причин, приводящих
практически ко всем известным видам
раковых заболеваний человека, вирусы
не фигурируют. Возможно, иммунная
система человека разрушает клетки, ин-
инфицированные вирусом, и защищает лю-
людей от возникновения раковых опухолей
вирусной природы.
Обобщение данных о поведении виру-
вирусов в клетках хозяина позволило устано-
установить, что, воздействуя на клетку, они сти-
стимулируют ее к вступлению в S-фазу кле-
клеточного цикла и заставляют проходить
митотический цикл снова и снова. Одна
из причин такого воздействия — наличие
в вирусном геноме онкогенов, способных
регулировать митотический цикл клетки
хозяина. Другая причина состоит в нару-
нарушении контроля экспрессии встроенных
в геном клетки хозяина вирусных генов,
регулирующих репликацию ДНК, и за
счет того, что при встраивании в хромо-
хромосому хозяина в результате случайного со-
события вирусный геном оказывается в
ином окружении. Таким образом, посте-
постепенно вирусная теория опухолевой
Факторы, способствующие юзниюмммию опухоли
235
трансформации превратилась в вирусо-
генетическую, в основе которой — изме-
изменение работы генов, регулирующих ми-
тотический цикл клеток.
В конце 90-х гг. мутационная и виру-
со-генетическая теории развития опухо-
опухолей объединились в одну. Показано, что
трансформация нормальной клетки в
опухолевую сопровождается сложным
мутационным процессом, который за-
затрагивает несколько (от 3 до 7) генов,
контролирующих пролиферативную ак-
активность клеток. Мутации в каждом гене
происходят независимо, и каждая из му-
мутаций имеет очень низкую вероятность
возникновения. Причины же мутацион-
мутационного процесса могут быть различны: как
физический и химический мутагенез,
так и мутагенез вирусной природы, од-
одновременно нельзя не учитывать воз-
возможность спонтанного мутагенеза.
За два последние десятилетия выяв-
выявлено значительное число генов, обеспе-
обеспечивающих трансформацию нормальных
клеток в опухолевые. Условно их можно
подразделить на несколько групп. К пер-
первой относятся протоонкогены, актив-
активность которых провоцирует пролифера-
пролиферацию клеток. Ко второй группе относятся
гены-супрессоры, активность которых
подавляет клеточные деления. В третью
группу можно объединить выделенные в
последнее время несколько генов, обес-
обеспечивающих стабилизацию структуры,
размеров и упорядоченность вторичной
структуры ДНК. К этой группе относят-
относятся гены теломеразного комплекса и сис-
системы репарации ДНК. Нарушение их
функциональной активности играет
важную роль в возникновении и разви-
развитии опухолей.
Факторы, способствующие
возникновению опухоли
Заболеваемость большинством видов
рака резко увеличивается с возрастом.
Например, смертность от рака толстого
кишечника среди двадцатилетних не
превышает десяти человек на 1 млн на-
населения за год, среди шестидесятилет-
шестидесятилетних — около ста человек, а среди восьми-
восьмидесятилетних эта цифра приближается к
400. Повышение вероятности ракового
заболевания с возрастом тем, что в тече-
течение жизни человек многократно контак-
контактирует с мутагенами разной природы.
Известно, что стимулируют мутаци-
мутационный процесс длительность воздейст-
воздействия и сила мутагена. Так, частота воз-
возникновения рака легкого резко возрас-
возрастает лишь после 10-20 лет курения, а му-
мутагеном в этом случае является бензпи-
рен-компонент угольной смолы и табач-
табачного дыма. Частота возникновения лей-
лейкоза в Хиросиме и Нагасаки оставалась
низкой в первые 5 лет после атомной
бомбардировки и достигла пика только
через 8 лет после нее.
Роль сопутствующего фактора, уско-
ускоряющего развитие опухолевых заболе-
заболеваний, играет раневой процесс. Хорошо
известно, что вслед за образованием ра-
раны наступает период ее заживления, ко-
который всегда связан с делением клеток.
Интенсивное деление клеток любой тка-
ткани сопровождается изменением регуля-
регуляции клеточного цикла. Значительная
часть находящихся вне митотического
цикла клеток должна вновь войти в фа-
фазу Gj, чтобы пройти синтетический пе-
период, связанный с удвоением ДНК, под-
подготовиться к митозу и поделиться. Такая
функциональная перестройка связана с
изменением активности генов, ре1ули-
рующих клеточный цикл. В ходе перест-
перестройки клеточной активности в работу
включаются новые гены, которые до
этого момента длительное время не
функционировали. Некоторые гены,
участвующие в регуляции клеточного
цикла соматических клеток, могут ока-
оказаться дефектными и этап их включения
236
Глава 10. ПРОБЛЕМЫ КАНЦЕРОГЕНЕЗА
способен стать пусковым моментом в
развитии опухолевого перерождения.
Как говорилось выше, вирусные ин-
инфекции не являются причиной развития
раковых заболеваний у человека, однако
некоторые из них стимулируют разви-
развитие опухоли. Так, вирус гепатита в соче-
сочетании с другими факторами риска, таки-
такими, как алкоголь, курение, употребление
пищевых продуктов, зараженных гриб-
грибками, может стимулировать развитие
рака печени. Вирус иммунодифицита
человека (HIV-1) в условиях иммунной
недостаточности или заражения други-
другими вирусами может стимулировать раз-
развитие саркомы Капоши, рака эпители-
эпителиальных клеток кровеносных сосудов.
Причины такой стимуляции до конца не
выяснены. Предполагается, что сами ви-
вирусы могут мутировать и становиться
онкогенными, или изменяется актив-
активность генов вируса, встроенного в геном
клетки хозяина, из-за изменения при-
привычного окружения генов.
Предполагается, что значительную
роль в регуляции работы генов, являю-
являющихся причиной опухолевой трансфор-
трансформации клеток, играет окружающая сре-
среда. Известно, что почти в каждой стране
есть местности, где очень низок уровень
заболевания тем или иным видом рака.
То есть образ жизни людей способен
предотвращать развитие того или иного
вида или формы рака. Например, у жи-
жителей окрестностей Бомбея (Индия) в
200 раз ниже, чем в других местах, уро-
уровень заболевания раком кожи. В Ниге-
Нигерии в 300 раз ниже уровень заболева-
заболевания раком пищевода, в Англии — в 100
раз ниже уровень заболевания раком
печени.
Таким образом, раневой процесс, ви-
вирусные инфекции и образ жизни оказы-
оказываются важными факторами, влияющи-
влияющими на вероятность развития раковых
опухолей.
Развитие раковой опухоли
В настоящее время считается, что опу-
опухоль развивается из одной единственной
клетки, несколько мутантных генов ко-
которой нарушают регуляцию клеточного
деления. Поскольку эта мутировавшая
клетка несет наследственные изменения,
все ее потомки будут иметь аналогичные
мутации. К моменту обнаружения опу-
опухоли (когда она выявляется на рентгено-
рентгенограмме) в ее составе насчитывается до
108 родственных клеток. Диаметр опухо-
опухоли в это время составляет лишь 1 мм. К
моменту пальпирования опухоль разрас-
разрастается до 10 мм в диаметре, а количество
клеток достигает 109. Летальный исход
приближается при достижении опухо-
опухолью диаметра 10 см, количество клеток в
ней при этом достигает 1012.
Все опухолевые клетки несут наслед-
наследственные изменения генов социального
контроля. В них наблюдается частичная
утрата черт клеточной дифференциров-
ки. Кроме того, злокачественное пере-
перерождение опухолевых клеток сопровож-
сопровождается секрецией в окружающую среду
протеолитических ферментов, что спо-
способствует образованию метастазов.
Развитие раковой опухоли связано с
рядом общих процессов, закономерности
которых сходны для всех видов и форм
рака, хотя каждая конкретная форма это-
этого заболевания имеет собствешгую спе-
специфику.
Вопросы для самоконтроля
1. Назовите отличительные свойства клеток рако-
раковых опухолей.
2. Отличительные особенности доброкачествен-
доброкачественных и злокачественных опухолей.
3. Как понимать идею о многоступенчатости мута-
мутагена?
4. В чем взаимосвязь мутационной и вирусной те-
рии канцерогенеза?
5. Назовите факторы, способствующие возникно-
возникновению опухоли.
Литература
1. Албертс Б., Брей Д, Льюис Дж.,
Рэфф М., Роберте К., Уотсон Дж. Мо-
Молекулярная биология клетки: В 3 т. М.:
Мир, 1994.
2. Алтухов Ю.П. Генетические про-
процессы в популяциях. М.:Наука, 1989.
3. Айала Ф., Кайгер Д. Современная
генетика.М.: Мир, 1987, т. 1.
4. Баев А.А. Программа "Геном чело-
человека", ее возникновение, содержание и
развитие. Итоги науки и техники. ВИ-
ВИНИТИ. Сер. Геном человека. — 1990.
5. Бочков Н.П. Клиническая генети-
генетика. М.: Медицина, 1997.
6. Бочков Н.П., Захаров А.Ф., Ива-
Иванов В.И. Медицинская генетика. М.:
Медицина, 1984.
7. Бочков Н.П., Чеботарев А.Н. На-
Наследственность человека и мутагены
внешней среды. М.: Медицина 1989.
8. Дубинин Н.И. Эволюция популя-
популяций и радиация. Москва: Госатомиз-
дат. 1966, 743с.
Э.Дубинин Н.П. Генетика. Кишинев,
1985.
10 Дубинин Н.П. Некоторые про-
проблемы современной генетики. М.: На-
Наука, 1994.
11. Захаров А.Ф. Хромосомы чело-
человека (Проблемы линейной организа-
организации). М.: Медицина, 1977.
12. Инге-Вечтомов СТ. Генетика с
основами селекции. — М.: Высшая
школа, 1989.
13. Козлов СИ. и др. Наследствен-
Наследственные синдромы и медико-генетическое
консультирование. Л., 1987.
14. Ли Ч. Введение в популяцион-
ную генетику. М.: Мир, 1978.
15. Льюин Б. Гены. М.: Мир, 1987.
16. Мак-Кьюсик В.А. Генетика чело-
человека. "Мир", 1967.
17. Наследственность человека и ок-
окружающая среда. — М.: Наука, 1992.
18. Орехова В.А., Лашковская Т.А.,
Шейбак М.П. Медицинская генетика.
Минск: Вышэйшая школа, 1998.
19. Последствия Чернобыльской ка-
катастрофы: Здоровье человека. Центр
экологической политики России.
Москва, 1996.
20. Приходченко Н.Н, Шкурат Т.П.
Основы генетики человека. Ростов-на-
Дону: Феникс, 1997.
21. Ратнер В.А. Математическая ио-
пуляционная генетика. Новосибирск:
Наука, 1977.
22. Фогель Ф., Мотульски А. Генети-
Генетика человека: В 3 т. М.: Мир, 1989,1990.
23. Хесин Р. Б. Непостоянство гено-
генома. М.: Наука, 1984.
24. Шевченко В А Померанцева М.Д.
Генетические последствия действия
ионизирующих излучений. М.: Наука,
1985.
25. Штерн К. Основы генетики чело-
человека. М.: Мир, 1987.
26. Эфроимсон В.П. Введение в ме-
медицинскую генетику. Изд. 2-е, М.: Ме-
Медицина, 1968.
27. McKusick V.A. Mendelian
Inheritance in Man. XI edition, Volumes
1-2. Johns Hopkins University Press,
Baltimore, 1994.